
FEDERAL COURT OF AUSTRALIA
Newron Pharmaceuticals S.p.A v Arrotex Pharmaceuticals Pty Ltd (Application for Preliminary Discovery) [2025] FCA 1321
File number(s): | NSD 1860 of 2025 |
Judgment of: | NEEDHAM J |
Date of judgment: | 29 October 2025 |
Catchwords: | PRACTICE AND PROCEDURE – urgent application for preliminary discovery pursuant to r 7.23 of the Federal Court Rules 2011 (Cth) – potential action for threatened infringement of patent – where prospective respondent intends to list generic version of prospective applicant’s pharmaceutical product on the Pharmaceuticals Benefits Scheme – whether prospective applicants hold a reasonable belief that they may be entitled to relief for patent infringement as required by r 7.23(1)(a) of the Federal Court Rules 2011 (Cth) – principles to be applied in assessing the reasonableness of the belief – whether application was conducted as a “mini-trial” – where scientific evidence adduced by prospective respondent – whether discretion to order preliminary discovery under r 7.23(2) of the Federal Court Rules 2011 (Cth) should be exercised |
Legislation: | Evidence Act 1995 (Cth) s 136 Patents Act 1990 (Cth) ss 13, 120 Federal Court Rules 2011 (Cth) rr 7.23, 8.05, 14.01, 16.01 Explanatory Memorandum, Therapeutic Goods Legislation Amendment (Copyright) Bill 2011 (Cth) |
Cases cited: | Actavis UK Limited v Eli Lilly and Company [2017] UKSC 48 Commonwealth Scientific and Industrial Research Organisation v Urrbrae Foods Pty Ltd [2023] FCA 504; (2023) 174 IPR 377 Fresenius Medical Care Australia Pty Ltd v Gambro (2005) 224 ALR 168 Olin Corporation v Super Cartridge Co Pty Ltd & Anor (1977) 180 CLR 236 Pfizer Ireland Pharmaceuticals v Samsung Bioepis AU Pty Ltd [2017] FCAFC 193; (2017) 257 FCR 62 |
Division: | General Division |
Registry: | New South Wales |
National Practice Area: | Intellectual Property |
Sub-area: | Patents and associated Statutes |
Number of paragraphs: | 103 |
Date of hearing: | 22 October 2025 |
Counsel for the Prospective Applicants: | Mr C. Dimitriadis SC with Mr B.A Mee |
Counsel for the Prospective Respondent: | Mr N. Murray SC with Mr M. Fleming |
ORDERS
NSD 1860 of 2025 | ||
| ||
BETWEEN: | NEWRON PHARMACEUTICALS S.P.A First Prospective Applicant ZAMBON S.P.A Second Prospective Applicant | |
AND: | ARROTEX PHARMACEUTICALS PTY LTD (ACN 605 552 234) Prospective Respondent | |
order made by: | NEEDHAM J |
DATE OF ORDER: | 29 OCTOBER 2025 |
THE COURT ORDERS THAT:
1. Until further order, pursuant to s 37AF of the Federal Court of Australia Act 1976 (Cth) these reasons will be disclosed to the following persons only:
(a) any judge, employee or other personnel of the Court;
(b) the prospective applicants, including its external legal representatives;
(c) the prospective respondent, including its external legal representatives; and
will not be open to public inspection or disclosed in open Court or disclosed in the
open part of any Court transcript.
2. The parties to provide draft orders to chambers for preliminary discovery in accordance with these reasons to be given by close of business 31 October 2025.
3. The matter be listed for case management hearing at 2.15pm on 31 October 2025.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
REASONS FOR JUDGMENT
NEEDHAM J:
1 Newron Pharmaceuticals S.p.A. and Zambon S.p.A, the prospective applicants, commenced proceedings against Arrotex Pharmaceuticals Pty Ltd, the prospective respondent, by way of Originating Application for an order for preliminary discovery pursuant to r 7.23 of the Federal Court Rules 2011 (Cth).
2 The proceedings came before me as Duty Judge on 22 October 2025, after being listed for hearing by Kennett J on 13 October 2025. Kennett J set a timetable for the filing of affidavits and submissions which was generally complied with (apart from a late flurry of affidavits, a matter to which I will return). I am grateful to counsel and their instructors for the efficient manner in which this urgent hearing has been prepared and argued.
3 At the close of the hearing I made confidentiality orders as requested by the parties restricting access to and disclosure of certain documents to certain recipients (there being two categories each of documents and recipients). At the end of the hearing the parties had not reached agreement as to the terms of any orders which would be appropriate were an order for preliminary discovery to be made, including the confidentiality regime which should apply, but were hopeful of restricting the areas of disagreement.
4 Given the imminence of the proposed listing of the prospective respondent’s products on the Pharmaceutical Benefits Scheme (PBS), these reasons may not be as finely wrought as they would be without time constraints. Because of the confidentiality issues, I will restrict publication of these reasons until the parties have had a chance to consider them and make any application for redactions or amendments, and will adjourn the matter to a time convenient to the parties for any applications as to the making of final orders including any amendments to the confidentiality orders, and a notation as to the agreed position as to security for costs (each of the prospective applicants being Italian companies).
An application for preliminary discovery
5 The prospective applicants are, respectively, the patentee and exclusive licensee of two patents which relate to both the product and the process of producing high purity safinamide, which (as safinamide mesilate, a salt of safinamide) is the active ingredient in Xadago, a treatment for moderate- to late-stage Parkinson’s Disease. Xadago is made by Zambon and is marketed in Australia and world-wide. Xadago is the only safinamide product which is currently on the market and available in Australia. The prospective respondent has four Australian Register of Therapeutic Goods (ARTG) registrations for safinamide products which have been approved as generic versions of Xadago (the Arrotex products) and it expects those products to be listed on the PBS (the specific date is part of the confidential information in these proceedings) and to commence supply of the Arrotex products in the near future.
6 The relevant patents are:
Australian Patent No. 2007263328 titled “Process for the production of 2- [4 - (3- and 2-fluorobenzyloxy) benzylamino] propanamides” (328 Patent); and
Australian Patent No. 2008334778 titled “Process for the production of 2-[4-(3- or 2- fluorobenzyloxy)benzylamino]propanamides with high purity degree” (778 Patent).
7 The patents relate to both safinamide and ralfinamide (a structurally similar compound but which is not relevant to these proceedings), and their salts, with “high yields and high enantiomeric and chemical purity”. The level of each of Xadago’s and the Arrotex products’ purity of safinamide is a contested factual issue in these proceedings.
8 The patents protect both the product of high purity safinamide and the process by which that is made. In other words, (relevantly) safinamide which contains two particular impurities at a level less than 0.03% by weight is covered by the patent, by whichever process it may be made, and they also protect the particular processes which result in that high purity safinamide.
9 The preliminary discovery application seeks production of documents and samples relating to and of the Arrotex products, for the purpose of deciding whether to commence a proceeding for actual or threatened patent infringement. During argument the prospective applicants foreshadowed that the application would be restricted in the first instance to documents, with any production of samples pursuant to r 14.01 of the Rules being sought after consideration of the documents if required (as per the approach taken in Commonwealth Scientific and Industrial Research Organisation v Urrbrae Foods Pty Ltd [2023] FCA 504; (2023) 174 IPR 377 (Moshinsky J) at [4]).
Matters not in contest on this application
10 There are a number of matters which are uncontested. I set these out below.
11 It is not contested that Xadago contains high purity safinamide in accordance with the patents. Nor is it in dispute that the Arrotex products contain safinamide, and that they have been approved as generic versions of Xadago.
12 It was further common ground that a generic version is not necessarily exactly equivalent to the originator drug, nor does bioequivalence require exact equivalence. Those concepts were discussed by Professor Roberts, who gave evidence for the prospective respondent.
13 The Australian Product Information document that accompanies the Arrotex products “recites, relies on, and contains statements based on the clinical trials which were conducted for Xadago using high purity safinamide”. Arrotex did not itself conduct any of its own clinical trials.
14 The question of process was not agitated in this application. The prospective applicants relied on the fact that the Arrotex products contained safinamide. The prospective respondent did not put forward any evidence as to the process by which the Arrotex products were produced.
15 Section 13(1) of the Patents Act 1990 (Cth) gives the patent owner the exclusive rights, during the terms of the patents, to exploit the invention (which, relevantly, includes licensing the patents). As the owner of the patents, and the exclusive licensee under the patents, each of Newron and Zambon respectively has the right to commence proceedings for infringement of the patents (s 120(1) of the Patents Act). Arrotex does not hold a licence to exploit the product, or the processes, protected by the patents.
16 The parties were generally ad idem as to the principles relating to preliminary discovery. They each relied on the reasons of the Full Court (Allsop CJ, Perram and Nicholas JJ) in Pfizer Ireland Pharmaceuticals v Samsung Bioepis AU Pty Ltd [2017] FCAFC 193; (2017) 257 FCR 62 (especially at [4]-[8], and [98]-[126]) to establish the following principles:
(a) rule 7.23 is a “beneficial provision, the purpose of which is to enable a person who believes he, she or it may have a right to seek relief to obtain information to make a responsible decision as to whether to commence proceedings” (Allsop CJ at [4]);
(b) the words of the relevant rule, r 7.23(1)(a), are focused on what “may” be the position (Allsop CJ at [8]);
(c) “One must keep the words of the rule firmly in mind in examining the material that exists in order to come to an evaluation as to whether the relevant person reasonably believes that he or she may have a right to relief. That evaluation may well be one about which reasonable minds may differ” (Allsop CJ at [8]);
(d) “… the rule has two features:
• there must be a reasonable belief as to a particular state of affairs; and
• that state of affairs consists of the possibility (required by the word “may”) that the prospective applicant has a right to obtain relief from the prospective respondent.”
(Perram J at [101]);
(e) “The following propositions about preliminary discovery applications should be accepted:
(i) the prospective applicant must prove that it has a belief that it may (not does) have a right to relief;
(ii) it must demonstrate that the belief is reasonable, either by reference to material known to the person holding the belief or by other material subsequently placed before the Court;
(iii) the person deposing to the belief need not give evidence of the belief a second time to the extent that additional material is placed before the Court on the issue of the reasonableness of the belief. That belief may be inferred;
(iv) the question of whether the belief is reasonable requires one to ask whether a person apprised of all of the material before the person holding the belief (or subsequently the Court) could reasonably believe that they may have a right to obtain relief; and
(v) it is useful to ask whether the material inclines the mind to that proposition but very important to keep at the forefront of the inclining mind the subjunctive nature of the proposition. One may believe that a person may have a case on certain material without one’s mind being in any way inclined to the notion that they do have such a case.”
(Perram J at [120]);
(f) In order to defeat a claim for preliminary discovery, the prospective respondent will need to show, either that the subjectively held belief does not exist (which is not put by the prospective respondent in this case), or, “if it does, that there is no reasonable basis for thinking that there may be (not is) such a case” (Perram J at [121]). That may be done by demonstrating that no reasonable person, faced with the evidence relied on by the prospective applicant, would think that a right to relief might exist: Perram J at [125].
17 Given the last principle, and the difficulties faced by responding to an application for preliminary discovery which is founded on a subjective belief, which needs only to be reasonably held, the Full Court in Pfizer cautioned against a prospective respondent conducting their case as if it were a “mini-trial” (Allsop CJ at [2]) or a “dress rehearsal for a trial” (Perram J at [126]).
18 I turn now to the evidence brought by the respective parties.
The prospective applicants’ evidence
19 The prospective applicants relied on an affidavit of Kent Teague, solicitor, dated 8 October 2025, who annexed the patents and various correspondence from his firm and the prospective applicants’ former solicitors seeking documents from various sources including from Arrotex and its solicitors, and alerting them to the imminence of this litigation. He also gave evidence about the Common Technical Document, which is a document containing information relating to the quality, safety and efficacy of products in a standardised format. He noted that the document must be submitted to the Therapeutic Goods Administration (TGA) as part of any application to include products on the ARTG.
20 A second affidavit by Mr Teague of 21 October 2025 dealt with correspondence about any confidentiality orders which may be appropriate should an order for preliminary discovery be made. Complaints about the breadth of the personnel of the prospective applicant to whom disclosure was proposed were made by the prospective respondent, and as to the exclusion of the confidential annexures of Ms Smith’s affidavit (as to which see below). My earlier reference to a “flurry” of affidavits is explained by the rapid succession of correspondence, and Mr Teague’s introductory words to one paragraph being “Moments later …”. Correspondence on these topics continued to 5.16pm on 21 October 2025, the evening before the hearing.
21 The prospective applicants primarily relied on an affidavit dated 9 October 2025 by Zambon’s General Counsel, Rosella De Dominicis, who has been in that position since 2012. Ms De Dominicis is the person “primarily responsible for making legal decisions on Zambon’s behalf relating to its concerns” about the possible infringement of the patents. In making those decisions she is informed by, inter alia, Roberta Morelli, Patent Director at Zambon.
22 Each of the patents discusses an impurity (Impurity IIa) and its methanesulfonate salt (Impurity IIc) and contends that safety issues arise where, as in prior art methods, impurity levels are found at greater than 0.03% by weight. The safety concerns include inhibition of what were referred to as CYP 450 cytochromes and HERG channel blocking, which in patients who genetically have poorly functioning metabolisms, is claimed to cause an adverse effect.
23 The patents contend (as set out by Ms De Dominicis at [16] of her affidavit):
(a) Impurity IIa as the methanesulfonate salt (which is referred to in the Patents as Impurity IIc) strongly inhibits in the micro- and submicro-molar range CYP3A4, CYP2D6, CYP2C19, CYP2C9 (four cytochrome enzymes of the CYP450 system) and HERG currents and is highly cytotoxic, compared with safinamide methanesulfonate (referred to as “Ic” in the Patents) with high purity degree: (see page 4 of the 328 Patent and page 5 of the 778 Patent).
(b) Compounds with strong inhibition of drug-metabolizing enzymes, in particular CYP 450 enzymes, and HERG channel blocking properties have a high probability to be toxic and that their development has to be stopped at an early stage: (see page 4 of the 328 Patent and page 5 of the 778 Patent)
(c) Safinamide and its salts produced using prior art methods contain Impurity IIa, or its methanesulfonate salt IIc, higher than 0.03% by weight, and are therefore unsuitable for safe therapeutical applications: (see page 8 of the 328 Patent and page 10 of the 778 Patent).
(d) Pharmaceutical preparations containing safinamide or its salt where the content of IIa, or its salts is not lower than 0.03%, and preferably not lower than 0.01% by weight, with respect to safinamide or its salt containing Impurity IIc are not suitable as medicaments: (see page 8 of the 328 Patent and page 10 of the 778 Patent).
24 Ms De Dominicis set out, on information from Elena Barbanti, IP Director for Newron (and who is apparently listed as one of the inventors of the patents), a number of claims made in the patents as set out at paragraphs 19-24 of her affidavit. In summary, the product claim is for high purity safinamide, being safinamide with impurities lower than 0.03% by weight, and the process claim is for the way in which that high-purity safinamide is produced (see, for example, Claim 1 in the 328 Patent, and Claim 1 of the 778 Patent). Xadago is itself a pharmaceutical formulation protected by the 328 Patent (see Claim 44 of the 328 Patent).
25 Xadago is approved by the ARTG and distributed under the PBS. Ms De Dominicis says that the processes described in the patents are the “only publicly disclosed methods of which Zambon is aware for producing high purity degree safinamide within the meaning of, at least, claim 36 and claim 37 of the 328 Patent”. Those claims are as follows (as set out in paragraph 19 of Ms De Dominicis’ affidavit):
36. High purity degree safinamide or ralfinamide or a salt thereof with a pharmaceutically acceptable acid with a content of the respective impurity (S)-2-[3-(3- fluorobenzyl)-4-(3-fluorobenzyloxy)benzylamino]propanamide (IIa) [Impurity IIa] or (S)-2-[3-(2-fluorobenzyl)-4-(2-fluorobenzyloxy)benzylamino]propanamide (IIb)
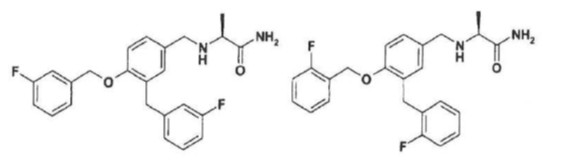
or a salt thereof with a pharmaceutically acceptable acid which is lower than 0.03% (by weight).
37. High purity degree safinamide or ralfinamide salt with a pharmaceutically acceptable acid as in claim 36 wherein the pharmaceutically acceptable acid is methanesulfonic acid and the content of the respective impurity of formula (IIa) or (IIb) as the salt with methanesulfonic acid is lower than 0.01% (by weight).
26 Ms De Dominicis says that the Xadago Product Information contains a statement, referred to as the Enzyme Statement, as follows:
Safinamide does not appear to significantly induce or inhibit enzymes at clinically relevant systemic concentrations. In vitro metabolism studies have indicated that there is no meaningful induction or inhibition of cytochrome P450, CYP2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2El and 3A3/5 at concentrations which are relevant (Cmax of free safinamide 0.4 μM at 100 mg/day) in man. Dedicated drug-drug interaction studies performed with ketoconazole, L-dopa and CYPlA2 and CYP3A4 substrates (caffeine and midazolam), did not detect any clinically significant effects on the pharmacokinetics of safinamide, or L-dopa, caffeine and midazolam.
(prospective applicants’ emphasis added)
27 The studies referred to were carried out using the high purity safinamide protected by the patents. There have been four clinical trials carried out on Xadago (and therefore, Ms De Dominicis says, in relation to the therapeutic safety and tolerability of high purity safinamide).
28 To the best of Zambon’s knowledge, as Ms De Dominicis was informed by Ms Barbanti:
(a) There are no studies capable of supporting the statements in the Xadago Product Information extracted in paragraph 31 above, other than studies carried out using high degree purity safinamide (as mesilate) with Impurity IIc lower than 0.03% (by weight).
(b) No clinical information is available regarding the safety or potential drug – drug interactions of any formulation of safinamide other than studies carried out using high degree purity safinamide (as mesilate) with Impurity IIc lower than 0.03% (by weight).
(c) There are no studies capable of supporting the statements in the Xadago Product Information extracted in paragraph 31 above in respect of safinamide (as mesilate) with Impurity IIc higher than 0.03% (by weight).
29 The Arrotex products:
(a) have the same indications (ie, use for Parkinson’s Disease) as Xadago;
(b) were approved as generic versions of Xadago;
(c) will contain the same strengths of safinamide as Xadago; and
(d) are accompanied by Product Information documents which (as noted above) contain the Enzyme Statement in the same terms as the Xadago Product Information document, and thus refer to the same studies in each case.
30 At paragraph 38 of her affidavit, Ms De Dominicis says:
Having regard to [the matters quoted in [28] above], I would not expect a generic pharmaceutical company to seek approval of a safinamide mesilate medicine with an Impurity Ila level of above 0.03%.
31 In relation to the application for preliminary discovery, Ms De Dominicis says that she infers from the above that the Arrotex products must have been approved on the basis of bioequivalence with Xadago, and there is nothing in the material available to her which would lead her to believe that the Arrotex products differed from Xadago in terms of efficacy and safety. She believes, but does not know, that the Arrotex products may contain high purity safinamide, and/or may be made using the processes in the 328 Patent or the 778 Patent.
32 Taking all of the above factors into account, she says that she is “not in possession of sufficient information to form a view about whether Zambon should commence a proceeding against Arrotex”. She believes that “Zambon may have a right to obtain relief against Arrotex in respect of threatened infringement of one or more claims of the patents”. I note here that it was not contested by the prospective respondent that Ms De Dominicis does in fact hold that belief. Mr Weber, CEO of Newron, has informed her that he holds the same belief. He is the decision-maker for Newron.
The prospective respondent’s evidence
33 Arrotex relied on three affidavits of Ms Kellech Smith, a solicitor who holds a BSc majoring in chemistry. Ms Smith’s evidence included a number of statements which were objected to but which were either not read, or were limited by reason of s 136 of the Evidence Act 1995 (Cth) to use as submissions. Ms Smith, in her first affidavit of 17 October 2025, reviewed the prior art processes for the preparation of safinamide compounds cited by the patents, and made observations that they revealed that “toxicity of the compounds … is negligible” and [the patent] “does not refer to Impurity IIa or IIb” (Australian Patent 1990057299). Ms Smith observed that a paper referred to in the 328 and 778 Patents also “does not disclose any impurities that may arise … and does not refer to Impurity IIa or IIb” (Pevarello P et al, “Synthesis and Anticonvulsant Activity of a New Class of 2-[(Arylalkyl)amino]alkanamide Derivatives” (1998) 41 (No 4) Journal of Medicinal Chemistry 579-590). Other articles relating to prior art processes concerning safinamide revealed “no safety problem” (Marzo A et al, “Pharmacokinetics and pharmacodynamics of safinamide, a neuroprotectant with antiparkinsonian and anticonvulsant activity” (2004) 50 Pharmacological Research 77-85, 84) and “does not refer to … any impurities” (Bialer M et al, “Progress report on new antiepileptic drugs: a summary of the Fifth Eilat Conference (EILAT V)” (2001) 43 Epilepsy Research 11-58).
34 Ms Smith’s affidavit contained some confidential annexures. The following paragraph is a summary and will be redacted in any published version of these reasons.
35 [REDACTED]
36 The second affidavit of Ms Smith, dated 21 October 2025, dealt with confidentiality concerns and in particular the issues which may arise should confidential trade information of Arrotex be given to its trade rivals, the prospective applicants. Ms Smith also deals with the confidential nature of the product launch.
37 The prospective respondent primarily relied on an expert report of Professor Michael Roberts of 17 October 2025. Professor Roberts has an impressive CV and publications list, and no objection was taken to his evidence on grounds of his lack of qualifications to give the opinions in his report. He is, in brief, an Emeritus Professor of Clinical Pharmacology and Therapeutics at the University of Queensland, and Emeritus Professor of Therapeutics and Pharmaceutical Science at UniSA Clinical and Health Sciences. He has been involved in academia, teaching and publishing, government and regulatory work, and has also acted as a consultant to the pharmaceutical industry since around 1983.
38 Professor Roberts gave evidence on the following matters, and I summarise his opinions very briefly:
(a) Bioequivalence and bioavailability
(i) Drugs to be sold in Australia must be approved by the TGA by submitting a Common Technical Document and the TGA will then assess them so as to be satisfied that they are “efficacious and safe”.
(ii) For new drugs, this must be done through sufficiently large clinical trials. For generic products, “efficacy can alternatively be established through bioequivalence to an originator drug product”, with quality and safety still independently considered.
(iii) Bioequivalence can be achieved if products are “pharmaceutically equivalent or pharmaceutical alternatives and their bioavailabilities (rate and extent) after administration in the same molar dose lie within acceptable predefined limits” (see European Medicines Agency, Guideline on the investigation of bioequivalence, (Doc. Ref: CPMP/EWP/QWP/1401/98 Rev. 1/Corr**) (Bioequivalence Guidelines) at [1.1]).
(iv) Bioavailability refers to the amount of active ingredient that becomes available to be absorbed, concentrated in the blood, metabolised and then excreted. Concentration builds up and is absorbed over time, and ideally reaches peak concentration early and stays within the therapeutic range.
(v) In considering whether a drug is bioequivalent, the blood concentration-time parameters “do not need to be identical to the originator drug product, but instead must be within acceptable limits (those limits being noted by the Bioequivalence Guidelines as 80% to 125%).
(b) Pharmaceutical equivalents and alternatives: Pharmaceutical equivalents contain the same active substances in the same dosage forms that meet the same or comparable standards. Pharmaceutical alternatives are products with different salts, esters, ethers (and so on) or which differ in dosage form or strength.
(c) Impurities: The Bioequivalence Guidelines do not require a determination of whether two drug products “have the same, or even similar, levels of impurities”. In fact, they do not refer to “impurity” at all. As bioequivalence is focused on the factors set out in (a)(iii) above, Professor Roberts says that “the TGA is not seeking to identify relative similarity in other aspects of formulation”.
(d) Safety and quality: To consider safety and quality, the TGA will assess the information in the Common Technical Document modules, and ask any follow-up questions, and “will assess the presence of any impurities against specified and regulatory limits”, relevantly the International Conference on Harmonisation Guidelines (ICHG) (ICHG Q3A(2) was tendered as Exhibit 2). The assessment is done as an independent process, not “by comparison to the impurity profile of the originator drug product”. Professor Roberts notes the processes by which new drugs are assessed against impurity standards, being the ICHG or, where available, monographs (of which there appear to be none for safinamide). Where a new generic drug product falls outside the standard the ICHG allows, or there is no monograph available, then reference will be made to the originator drug product for a new drug product.
(e) The patents:
(i) Professor Roberts observes that the Relevant Impurity (being IIa) is a “synthesis impurity” which arises during the process of production of the active ingredient. He notes that he understands “high purity degree” safinamide to mean less than 0.03% of the Relevant Impurity. He was asked:
whether the information provided in the Patent [being the 328 Patent]:
(a) provides a basis for the statements on page 8, lines 19 to 31 to the effect that, unless safinamide contains the Relevant Impurity at an amount below the Impurity Threshold, it will be unsafe for use in a drug product; and/or
(b) provides a basis for 0.03% (by weight) as the appropriate threshold above which the relevant drug product will be unsafe.
(ii) He said that in his view, “the information in the Patent does not provide a basis for either proposition”.
(iii) The basis for this view was derived from his interpretation of the data in tables 1-4 on pages 4 to 6A of the 328 Patent, which (in summary):
(A) Do not provide any human in vivo safety data;
(B) Two are in vitro tests, and report on relative inhibition of various substances, but do not provide sufficient information to identify the human in vivo threshold which would render the product unsafe;
(C) Compare high purity safinamide with (tables 1, 3 and 4) 100% relevant impurity, and (table 2) 0.3% relevant impurity (10x the impurity threshold); and
(D) Do not compare the high purity safinamide with the percentages of relevant impurity produced by prior art synthesis methods in other examples in the patent.
(iv) In relation to the CYP 450 enzyme system and HERG channels, he notes that the statement as to their toxicity in the patent (at page 4) is “an overstatement, because it ignores the role of the dose”.
(f) The opinions in (e)(iii) and (iv) above are expanded upon in some detail in paragraph 67 of Professor Roberts’ affidavit onwards, noting that the 328 Patent does not provide information on what level of inhibition of CYP 450 enzymes or HERG channels would be unsafe. Tables 3 and 4 were mice mortality tests, which he said were not pointed to potential toxicity, but instead to whether the relevant impurity had the pharmacological activity of safinamide (relevantly).
(g) Basis for Impurity Threshold: Professor Roberts was not able to identify a scientific basis for 0.03% being the relevant threshold, and says that the 328 Patent notes that prior art methods resulted in higher amounts.
39 Professor Roberts was also asked to review the Product Information for ARX-Safinamide, one of the Arrotex products, and noted that it does not disclose the synthetic method by which the safinamide was created, nor does it disclose the amount of relevant impurity.
40 His review of the Product Information for ARX-Safinamide caused him to take the view that it:
… does not provide any information which indicates that that ARX-Safinamide product may contain the Relevant Impurity at a percentage below the Impurity Threshold. Further, it enables calculations as to the estimated plasma concentration of Relevant Impurity at steady state, at various assumed concentrations, which demonstrates that there is no reason to believe the amount of Relevant Impurity may be below the Impurity Threshold.
41 Professor Roberts performed calculations on the available data, on stated assumptions, which gave rise to his opinion that:
there is no reason to believe the percentage of Relevant Impurity in the ARX-Safinamide products may be below the Impurity Threshold, because even amounts of Relevant Impurity far above that threshold would still result in blood plasma concentrations so low as to be therapeutically meaningless.
42 In relation to the Enzyme Statement which appears in the Product Information for the Arrotex products in identical terms to that in the Xadago Product Information (as set out at [26] above), Professor Roberts noted that this was a “statement about safinamide generally” and that the Product Information for ARX-Safinamide does not state that the studies were conducted using the safinamide in the Arrotex products. Relying on his conclusion in the paragraph above, “the percentage of the Relevant Impurity could be well above the Impurity Threshold without giving rise to any meaningful inhibition of the relevant enzymes”.
Questions to be determined
43 As I am reminded by the former Chief Justice in Pfizer, to “keep the words of the rule firmly in mind in examining the material that exists in order to come to an evaluation as to whether the relevant person reasonably believes that he or she may have a right to relief” (at [8]), I set out r 7.23:
7.23 Discovery from prospective respondent
(1) A prospective applicant may apply to the Court for an order under subrule (2) if the prospective applicant:
(a) reasonably believes that the prospective applicant may have the right to obtain relief in the Court from a prospective respondent whose description has been ascertained; and
(b) after making reasonable inquiries, does not have sufficient information to decide whether to start a proceeding in the Court to obtain that relief; and
(c) reasonably believes that:
(i) the prospective respondent has or is likely to have or has had or is likely to have had in the prospective respondent’s control documents directly relevant to the question whether the prospective applicant has a right to obtain the relief; and
(ii) inspection of the documents by the prospective applicant would assist in making the decision.
(2) If the Court is satisfied about matters mentioned in subrule (1), the Court may order the prospective respondent to give discovery to the prospective applicant of the documents of the kind mentioned in subparagraph (1)(c)(i).
44 The questions to be asked, assisted by the formulation of Perram J in Pfizer at [120], are the following:
(a) Does the prospective applicant believe that it may have a right to relief? (Noting that the belief is not whether there is a cause of action; rather, it is a belief in the possibility of relief).
(b) Is that belief reasonable? Or, turning the question around given the way the proceedings unfolded, with expert evidence, has the prospective respondent succeeded in showing that there is no reasonable basis for that belief?
(c) Have they made sufficient inquiries to obtain sufficient information to decide whether to start a proceeding in the Court to obtain that relief?
(d) If so, do they reasonably believe that the prospective respondent has documents directly relevant to the question of whether the prospective applicant has a right to obtain the relief?
(e) Would inspection of the documents assist?
(f) What are the confidentiality provisions which should be imposed on any discovery?
45 I will deal with questions (b) and (f), as the main areas of contest, last.
Do the prospective applicants believe that they may have a right to relief?
46 This question is uncontroversial, since it is not contested that, for the purposes of this application, the prospective applicants hold a belief that they may have a right to relief. Each of Ms De Dominicis, for Zambon, and Mr Weber, for Newron, holds that belief.
Inquiries and documents
47 I can deal with questions (c), (d), and (e) together. These matters are not significantly in contest and now, after much correspondence, the prospective respondent does not take issue with these. Ms De Dominicis’ evidence as to her inability to form a view on whether to commence proceedings was given after the efforts detailed by Mr Teague in his first affidavit, which demonstrated efforts to obtain documents and samples that would enable her to be able to make that decision.
Is the belief reasonable?
48 The prospective applicants contend that, contrary to the strictures of the Full Court in Pfizer, the prospective respondent has, by relying on expert evidence from Professor Roberts, sought to conduct a “mini-trial” or a dress rehearsal for the final hearing. Senior counsel for the prospective applicants, Mr Dimitriadis SC (who appeared with Mr Mee), stressed that r 7.23 envisages a summary or interlocutory procedure. Consistently with that, there were no applications to cross-examine the witnesses, and I was given assistance by counsel in interpreting the scientific evidence as summarised (however briefly) above.
49 The evidence brought by the prospective respondent was scientific; both Ms Smith’s analysis of prior art processes which are different from those described in the patents, and Professor Roberts’ evidence which seeks to challenge the validity of the claims made in the patents.
50 The prospective respondent submitted that technical aspects of proceedings do not change the test for preliminary discovery under the Rules, and that if the question is whether a subjective belief is reasonably held in a case involving technical aspects of pharmaceuticals, some consideration of technical factors is unavoidable. The prospective applicants contended that the invitation to make what were effectively final rulings on the likelihood of Arrotex products infringing the patents was not appropriate at this interlocutory hearing.
51 Mr Dimitriadis submitted that the threshold for an order for preliminary discovery was low; the relevant belief may be reasonable even if a different or contrary view, or range of views, is open, or, even if the belief is wrong. He relied on the principles in Pfizer as summarised above, and noted that, similarly to the present case, the prospective respondent in Pfizer sought to demonstrate the lack of reasonableness of the relevant belief by way of expert evidence. He submitted that the highest use of Professor Roberts’ report was to show that there might be a contest at any trial about whether the claims in the patents are correct, but that it did not rise to a conclusive undermining of Ms De Dominicis’ belief.
52 The prospective respondent submitted that Professor Roberts’ evidence demonstrated that the matters on which the reasonable belief was said to be based do not “meet the threshold under the Rules” because “the evidence on which the prospective applicants rely is not appropriately attuned to the right questions”.
53 In weighing Professor Roberts’ evidence, which, as noted above, was both untested and unanswered, I need to keep in mind the statutory purpose of r 7.23 and the subjective test to be applied to the prospective applicants’ state of mind. Mr Murray submitted that the only witness qualified to give the scientific evidence which goes to, in particular, the purity and enzyme aspects of safinamide and its regulation in Australia, is Professor Roberts.
54 As r 7.23 is a beneficial provision, it must be applied in a way which gives full weight to its purpose, which is to allow a person to make up their mind whether to commence proceedings. The provision plays an important role in the administration of justice in enabling parties to satisfy themselves as to the proper basis of any proceedings brought before the Court, in particular, in ensuring that practitioners are able to fulfil their professional obligations pursuant to r 16.01 of the Rules to certify that there is a proper basis “for each allegation in the pleading”. If a party has a sufficient level of certainty about the claim, based on information available to them, to be able to file a pleading (or affidavit supporting an originating application: r 8.05(2) of the Rules) then preliminary discovery should not be ordered. It is not contended by the prospective respondent in this case that the prospective applicants do, in fact, have enough information to make a decision to commence.
55 Allsop CJ in Pfizer at [69] commented that the question of whether a belief was reasonable was one about which minds may reasonably differ, and went on to say that in order to succeed, the prospective respondent must provide evidence at a level which would demonstrate that the prospective applicant’s belief was based on “unreasonable, untenable, irrational or baseless” considerations or views. On an interlocutory hearing, it is “generally inapt” to make final judgments on contested scientific evidence, particularly where no cross-examination is undertaken (in Pfizer, as here).
56 With those principles in mind, I turn to the scientific evidence and to the submissions of the prospective respondent. Its written submissions contend, without incorporating the subjunctive “may”, that “the prospective applicants’ belief regarding a right to relief is not reasonable”. It submitted that the prospective applicants’ case that it had a reasonable belief could be distilled to three core propositions, summarised below:
(a) Ms De Dominicis understands that the Arrotex products contain safinamide (the product argument);
(b) She understands that the Arrotex products are registered on the basis of bioequivalence to Xadago (the bioequivalence argument); and
(c) She understands that the Product Information documents for the Arrotex products include the Enzyme Statement, and believes that the TGA would not register an unsafe product (the safety argument).
57 These grounds were refined in oral submissions. Senior counsel for the prospective respondent, Mr Murray SC (who appeared with Mr Fleming) submitted that the three grounds above which the prospective applicants relied upon to form their subjective belief, did not meet the relevant threshold under r 7.23, and there were key matters, such as the subtleties of bioequivalence which were not addressed by the prospective applicants in their evidence in chief.
58 Mr Dimitriadis submitted that Ms De Dominicis’ views were not incapable of being held. While that may seem to be in the category of “damning with faint praise”, that would be a sufficient standard to base a reasonable belief that the prospective applicants may have a right to relief. The reasonableness of the prospective applicants’ belief requires a focus on the basis of that belief, which is a relatively low bar, with the prospective respondent having to leap the much higher bar of saying that, based on that material, no reasonable person could think that a right to obtain relief might exist.
The product argument
59 In relation to the product argument, the prospective applicants submitted that “the dispute is narrowly focused on one integer” which is the “purity integer”. As high purity safinamide, however produced, is the invention protected by the patents (along with safinamide produced by the processes protected by the patents), the prospective applicants contend that, as the Arrotex products contain safinamide, there is a reasonable ground for a belief that there may be a right to relief on the basis of that fact alone.
60 The following three paragraphs are based on confidential material and will be redacted in the published version of these reasons.
61 [PARAGRAPHS 61-63 REDACTED]
62 [PARAGRAPHS 61-63 REDACTED]
63 [PARAGRAPHS 61-63 REDACTED]
64 It is noted that the prospective applicants’ decision makers were not given access to Ms Smith’s confidential annexures, and so, while it is appropriate for the Court to consider material which was not before the decision-maker in assessing reasonableness (see Pfizer at [120(iii)-(iv)]), it was submitted that I should be slow to do so in this case.
65 It was suggested by the prospective respondent in correspondence, sent the day before the hearing, that the confidential annexures could be provided to Ms Barbanti and Ms Morelli, who are IP directors of the prospective applicants respectively, and that they could then discuss particular matters with the decision makers; this was not a regime which was taken up (whether that was for practical reasons including Australia/Italy time zones, or whether it was otherwise not a satisfactory proposal).
66 The prospective applicants further submitted that in determining reasonableness of the belief, I should take into account that Professor Roberts was asked questions upon which he gave his opinion, rather than commenting on Ms De Dominicis’ affidavit, causing, as it were, a disconnect between the evidence of the two sides. The prospective applicants contend that this is a further factor which goes to determining the reasonableness of the belief. The prospective respondent submitted that Professor Roberts’ review of the way in which the TGA regulates drugs in Australia, rather than having regard to the claims in the patents, is the proper way to approach the matter.
67 The prospective respondent, while accepting that the Arrotex products contain safinamide, in the same dosage strengths and indications as Xadago, submitted that those facts tell the reasonable person looking to form a relevant belief nothing about impurities. It submitted that, as this is not an infringement trial, the Court need not make a finding of fact that the Arrotex products do not infringe; however, it contends that the evidence brought by the prospective respondent demonstrates that the purity feature is not present. This submission was not made so that I can make a finding of fact to that effect, but to function as material which points away from the required state of mind on the part of the prospective applicants.
68 In reply, the prospective applicants submitted that they did not seek to make a case that the TGA did in fact make an assessment of impurities. But they submitted that the fact that the TGA has approved the Arrotex products is entirely consistent with the proposition that they may contain safinamide at the same purity level as Xadago. They make this point also with regard to the bioequivalence argument, to which I now turn.
The bioequivalence argument
69 The prospective applicants rely on the fact that Arrotex has been approved as a generic product for Xadago. They do not assert that mere bioequivalence, in and of itself, is determinative of the Arrotex products having the same level of purity as Xadago, but say that it is “certainly consistent with that”. As the Product Information documents for Arrotex refer to the same clinical trials and data which were obtained for Xadago, they say that reliance on that data is a basis for the relevant reasonable belief. The submission was that reliance on the high purity trials was another piece of the “reasonable belief” puzzle; while it is not determinative that the Arrotex products contain the high purity safinamide protected by the patents, it is not inconsistent with that, and forms the basis for a relevant reasonable belief. Mr Dimitriadis referred to the inclusion of these clinical trials and data as “holding out” the Product information relevant to Xadago as being comparable to the Arrotex products.
70 The bioequivalence argument includes the presence of the Enzyme Statement in the Product Information documents for the Arrotex products. The prospective applicants say that the inclusion of the Enzyme Statement is entirely consistent with the Arrotex products using high purity safinamide.
71 The prospective respondent submitted that any bioequivalence of the products is not an integer of impurities. Rather, bioequivalence relates only to whether the active ingredient in the generic product provides a sufficiently similar dose of that active ingredient to the originator product, by way of measuring how much is absorbed into the blood over time. Nor do patents form any part of the TGA’s reference points when assessing safety of products.
72 It was submitted by Mr Murray that the explanation of bioequivalence not being a direct comparison of the originator and generic drug was not provided by the prospective applicant in its evidence, and so the evidence from Professor Roberts would assist in giving more background to that question. It was submitted that the evidence on the meaning of bioequivalence would reinforce the submission that, while the Arrotex products are generic versions of Xadago, this “tells [the Court] absolutely nothing that would incline the mind towards accepting that the product may infringe” or about the impurity level.
73 In reply, the prospective applicants submitted that the prospective respondent’s position on the bioequivalence argument is misplaced, as even taking Professor Roberts’ views and how the TGA assesses bioequivalence as correct, it does not demonstrate that the views held by the prospective applicants are views that no reasonable person could hold.
The safety argument
74 Ms De Dominicis relies on information in the patents, which supports her belief, that a product which contains greater than 0.03% by weight of impurity would not be suitable for safe therapeutic applications. She relies on the recorded adverse effects, particularly in patients with poorly functioning metabolisms, and who are taking a combination of medicaments which may contribute to the inhibition of CYP 450 enzymes and the blocking of HERG channels.
75 The prospective applicants relied on the statements in the claims in the patents as to the relative safety of high purity safinamide as being an important part of the reasonable belief held by them, and the investigations and experimental studies referred to in the patents as being grounds for that belief. They submitted that the safety profile of Xadago was based on clinical trials of over 3000 subjects, some of whom were treated for a period of over two years. The Product Information documents in each case have a section numbered 4.8 which is headed “Adverse Effects (Undesirable Effects)”. The same clinical data for safinamide is provided in relation to the Arrotex products, which the prospective applicants say is a ground for a reasonable belief that the Arrotex products may contain high purity safinamide, as this is the product upon which the clinical trials were conducted.
76 The prospective respondent took issue with the patents as forming the basis of a reasonable belief, and instead noted that the TGA does not require that safinamide have a purity level lower than 0.03%. The ICHG, which has been adopted by the TGA as its approach to impurities, sets out in Attachment 1 the various reporting thresholds for levels of impurity. For a maximum daily dose of less than or equal to two grams a day (the relevant dose for both Xadago and the Arrotex products) it states:
(a) A reporting threshold at 0.05%, defined as the limit above which an impurity should be reported;
(b) An identification threshold at 0.1%, defined as the limit above which an impurity should be identified; and
(c) A qualification threshold at 0.15%, defined as the limit above which an impurity should be qualified and particular further measures must be taken.
77 In submissions, Mr Murray pointed to the fact that:
There are no British Pharmacopoeia (BP) or USA Pharmacopeia (USP) monographs for the drug substance safinamide mesilate or the finished product
as appears in Exhibit 1 (Australian Government Department of Health, Australian Public Assessment Report for Safinamide (Therapeutic Goods Administration, Canberra, 2019)). Mr Murray asked, possibly rhetorically in the absence of evidence as to the kind of matters which give rise to pharmacopeia monographs:
… if some of the things said in the patent about the impurity levels for this drug are true, one would expect there to be a monograph telling the world … and importantly regulators, “you should be applying this threshold”.
78 Professor Roberts noted that “where there is a monograph in a major pharmacopoeia, it is the quality standard for that drug substance, and includes defined impurity tests”.
79 Given that the enzyme studies were performed on safinamide with impurity levels less than 0.03%, and the TGA impurity qualification limit is 0.15%, the prospective respondent submitted that there was nothing which would incline a reasonable person to think that the impurity level of the Arrotex products was less than 0.03%. Mr Murray submitted that the effect of the Enzyme Statement is that studies with less than 0.01% impurity have not caused any problems with enzyme inhibition, and it “says nothing about the level at which the impurity must be present in order to make the statement about enzyme inhibition … untrue”. He characterised the position of the prospective applicants on the safety argument as “an unreasonable and baseless position based on the material that has been identified”.
80 In discussion during the hearing, Mr Murray submitted that the prospective applicants’ case relied on safety as claimed in the patents, and in order for that claim to be reasonable, it would need to be true that safinamide was unsafe at a level higher than 0.03% impurity. He submitted that the regulatory regime in Australia does not recognise that 0.03% is a safety threshold for the regulation of safinamide, and what the patent says cannot contribute to any inference that the product may infringe because it has been approved by the TGA for sale in the Australian market.
81 The prospective respondent relied on a statement in the Product Information documents that “Safinamide does not appear to significantly induce or inhibit enzymes at clinically relevant systemic concentrations” but noted that in neither the Xadago version nor the Arrotex products’ version was the high purity aspect of it referred to. In relation to the similarity of the wording between the products’ Product Information documents, the prospective respondent referred to the Explanatory Memorandum to the Therapeutic Goods Legislation Amendment (Copyright) Bill 2011 (Cth) which provides that it is standard practice for the TGA to approve the text of Product Information for generic versions that are “essentially the same” as the approved Product Information of the original medicine, so as to ensure that health professions “receive the same information about a medicine, regardless of the brand, thus avoiding any perception that differences in the text reflect clinical or pharmacological differences”.
82 This submission, and reliance on the Explanatory Memorandum was made to demonstrate that the prospective applicants could not rely on the respective Product Information documents as a “marketing” exercise or “holding out” of qualities of the Arrotex products or their levels of impurity or otherwise.
83 Further, Mr Murray sought to rely on Professor Roberts’ analysis of the studies underlying the data in the patents. I have referred above to four studies, and I was taken in detail to the way in which the tests undertaken compared high purity safinamide, on the one hand, with a dose which comprised 100% of the relevant impurity in three of the four tables of data. This was submitted to be “an extreme comparison” and not relevant to the question of whether 0.03% was the relevant threshold. (See also the Table 3 method of 3 mg dose of either high purity safinamide or 100% impurity safinamide, per kilogram of the weight of the unfortunate mouse assisting in these scientific endeavours).
84 The prospective respondent noted that the evidence from Zambon was from its in-house lawyer, and that Zambon had only been the exclusive licensee for six weeks, each of which should go to the weight of the evidence for the prospective applicants. On that basis, Mr Murray questioned why Ms De Dominicis’ expectation (set out at [30] above), that an approval of a safinamide product with more than 0.03% would not be sought, should be given any weight as an objectively reasonable statement, based on her apparent lack of expertise and the dearth of any expression of familiarity with the workings of the TGA.
85 In response to this point, the prospective applicants relied on the various parts of Ms De Dominicis’ affidavit setting out her consultations with various persons. At paragraph 9 of her affidavit she says that she had been informed by Ms Morelli, Patent Director at Zambon, of the “scientific concepts discussed in each of the Patents and in this affidavit” which Mr Dimitriadis says is sufficient to satisfy the Court that she is speaking from a position of both knowledge and informed belief.
86 More generally, the prospective applicants submitted that Ms De Dominicis, referred to other studies which have been carried out, including the in vitro metabolism studies and drug-drug interaction studies, and the four clinical trials. It was submitted that to engage in a mini-trial as to the accuracy of the statements in the patent is not only inappropriate on an interlocutory application, but also not relevant to the question at hand, which is the question of a reasonable basis for a subjective opinion. In relation to the ICHG, Mr Dimitriadis took me to page 11 of that document which indicates that “Lower thresholds can be appropriate if the impurity is unusually toxic” to ground a submission that the ICHG provisions do not require a finding that safinamide is safe at impurity levels greater than 0.03%.
Determination
87 Is the belief held by the prospective applicants one which is based on “unreasonable, untenable, irrational or baseless” considerations or views? (see Allsop CJ in Pfizer at [69]).
88 Some degree of speculation, or reliance on circumstantial evidence, is almost always part of the factual architecture which goes to make up a belief in the possibility of relief, because it is the reasonableness of the belief being tested, not the truth of the facts alleged (see Perram J in Pfizer at [154]). The prospective applicant does not need to establish a prima facie case of patent infringement (see Nicholas J in Pfizer at [180]). Instead, I need to determine whether the expressed belief was reasonable as against the background of the evidence before me, even if that evidence has not been considered by the prospective applicants in coming to their belief, as is the case with the confidential exhibits KNS-8 and KNS-9.
89 I turn, yet again, to Allsop CJ in Pfizer at [69] where the former Chief Justice said:
The relevant question posed by the rule is whether the applicant reasonably believes that it may have the right to obtain relief. It is not whether one scientific view is more or less persuasive than another. In the field of science, expert views may differ about important scientific aspects that can be seen to bear upon a question.
90 Giving the views of Professor Roberts the appropriate weight, in the context of this interlocutory hearing, it appears to me that the evidence brought by the prospective applicants does not demonstrate affirmatively that the safety level of impurities in safinamide is lower than 0.03%, nor does it demonstrate affirmatively that the bioequivalence of the generic Arrotex products mean that they are the same high purity formulation as Xadago. I do not however need to be persuaded to that level.
91 I take the view that, given the range of scientific views expressed in the patents and the evidence for the prospective applicants, and the fact that Arrotex does indeed contain safinamide, there is a reasonable basis for the belief that the prospective applicants may have a right to relief. I do not consider that the confidential exhibits KNS-8 and KNS-9 demonstrate that such a belief is “unreasonable, untenable, irrational, or baseless”.
92 I am particularly persuaded by two factors; one is that the patents would be infringed by products containing high purity safinamide, however produced, and the second is that I am not persuaded that the safety argument is as one-sided as the prospective respondent contends. As agreed during the hearing, I do not need to determine, even on an interlocutory or preliminary basis, whether safinamide containing greater than 0.03% of Impurities IIa or IIc is suitable for medicament use; but I do need to determine whether the beliefs expressed by Ms De Dominicis are reasonable. It seems to me that the prospective respondent’s contention that as the TGA does not recognise the 0.03% impurity level as a safety threshold, her belief is unreasonable, does not necessarily follow. Nor does a submission that bioequivalence of the prospective parties’ products, and reliance on the same testing for each products’ Product Information, does not give rise to a belief that the impurity factor may be present, assist. It does not rule out that the protected impurity level may be present.
93 I agree with the prospective applicants’ submissions that the totality of the evidence must be dealt with, rather than picking off a number of areas of scientific disagreement. When all of the evidence is considered, a belief that the prospective applicants may be entitled to relief appears to be reasonable; that is, it is not a view that could not be held by any reasonable person.
94 I come to this view not only on the grounds put forth by the prospective applicants but also by areas not dealt with by the evidence. The fact that there is no evidence of Impurity IIc being tested for by the prospective respondent, for example, leaves a lacuna in the evidence. Mr Murray sought to downplay this by noting that if there were amounts of Impurity IIc disclosed, it would only add to the impurity level and thereby take the impurity level higher (making it less high purity); however, given the limited testing (and the redactions to KNS-8 and KNS-9) it is impossible to know what the levels are, and because of that, a belief as to the right to relief seems not unreasonable.
95 Professor Roberts does not deal in detail with the issue of a safety threshold in relation to drug interactions between safinamide and other medicaments (an issue of concern to Ms De Dominicis expressed in her affidavit, and arising from the claims in the patents). Professor Roberts does quote a paragraph from the Product Information for ARX-Safinamide which refers to the drug-drug interaction studies but was only asked about whether that paragraph “provides any reason to believe that the safinamide used in … [the Arrotex products] may contain the Relevant Impurity at a percentage below the Impurity Threshold”. In relation to the “dose is the poison” analysis that the patents overstate the risk of negative drug interactions (referred to at [38(e)(iv)] above), Professor Roberts said:
There are a large number of substances that inhibit CYP 450 enzymes (e.g., grapefruit juice inhibits CYP3A4) and are not considered highly toxic. The issue is whether the amount of inhibition is of concern, taking into account the potency of the inhibitor, the likely concentrations of the inhibitor in the body, and the specific substance being inhibited.
96 At paragraph 100 of his affidavit, Professor Roberts said:
As a result [of Professor Roberts’ calculations], taking into account the likely blood plasma concentrations of Relevant Impurity in vivo, there is no reason to believe the percentage of Relevant Impurity in the ARX-Safinamide products may be below the Impurity Threshold, because even amounts of Relevant Impurity far above that threshold would still result in blood plasma concentrations so low as to be therapeutically meaningless.
97 I take the view that this reasoning is somewhat circular; because his calculations are that safinamide with higher impurity levels are not toxic, then there is no reason to believe that the Arrotex products are high purity products. It is equally open to believe that they may be high purity products.
98 There is no further analysis in Professor Roberts’ affidavit of the way in which other inhibitors combined with safinamide (at any level of purity) could be, or could not be, highly toxic. Professor Roberts’ analysis is based on his calculations of toxicity by reason of blood plasma concentrations in vivo. I am not able, on this application, to accept that the calculations on the various assumptions are unassailable. Where I have only one scientific view from that particular perspective, and Ms De Dominicis’ views obtained from various other sources on the other but looking at the question from a different perspective, it is difficult for me to say that her views are unreasonable.
99 This is indeed a question of “reasonable minds reasonably differing” and while I generally accept Professor Roberts’ evidence, the two sides do not so nearly overlap to enable me to say that there is no reasonable basis upon which Ms De Dominicis’ belief can be held. None of the evidence rules out the possibility that the Arrotex products may contain high purity safinamide so that the relevant belief can be said to be baseless or unfounded.
Proposed orders
100 Accordingly, I am prepared to order preliminary discovery.
101 Mr Mee and Mr Fleming were attempting, at the end of the hearing, to come to an agreed position as to discovery categories and confidentiality regimes in the event an order for preliminary discovery were made. I will publish these reasons, and stand the matter over to a convenient date to the parties to provide a version, or competing versions in markup, of final orders. Those orders should include the agreement the parties have reached as to the position as to security for costs and as to the costs of these proceedings.
The ‘doctrine of equivalents’ argument
102 The prospective applicants invited me, as an alternative position, to grant preliminary discovery on the basis that they are entitled to seek to understand how close to the high purity threshold the Arrotex products fall, on the basis of the doctrine of equivalents as expressed – or “re-enlivened” – by the UK Supreme Court in Actavis UK Limited v Eli Lilly and Company [2017] UKSC 48. The submission was that an infringement case could be brought where there was not a strict infringement of a patent but the product or process has “an immaterial variation”. Given the determination to which I have come, I do not need to determine whether this argument would have succeeded, but I note that the prospective respondent relied on the High Court’s statement in Olin Corporation v Super Cartridge Co Pty Ltd & Anor (1977) 180 CLR 236 where Gibbs J said (at 245) that to demonstrate infringement it must be shown that the respondent’s process took “each and every one of the essential integers of the appellant’s claim” (see also Fresenius Medical Care Australia Pty Ltd v Gambro (2005) 224 ALR 168 at [49]-[50]).
103 I raised with counsel for the prospective applicants whether, on this application for preliminary discovery, it would be appropriate to revisit the position as expressed by the High Court. Happily, given my determination of this application, I need not do so.
I certify that the preceding one hundred and three (103) numbered paragraphs are a true copy of the Reasons for Judgment of the Honourable Justice Needham. |
Associate:
Dated: 29 October 2025

