
FEDERAL COURT OF AUSTRALIA
Sanofi v Amgen Inc. (No 3) [2025] FCA 387
File number: | NSD 876 of 2022 |
Judgment of: | NICHOLAS J |
Date of judgment: | 23 April 2025 |
Catchwords: | PATENTS – appeal from decision of Delegate of Commissioner of Patents holding oppositions to patent applications unsuccessful – applications for patents including claims to isolated monoclonal antibodies that bind to epitopes on PCSK9 that include one or more specified amino acid residues and which block binding of PCSK9 to LDLR and claims to isolated monoclonal antibodies that bind to at least one specified amino acid on PCSK9 and which block binding of PCSK9 to LDLR – where opposition based on (inter alia) alleged failure to meet requirements of s 40 of the Patents Act 1990 (Cth) in the form it took before amendment by the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth) – whether claims would be clearly invalid if Applications proceeded to grant – whether claims fairly based on the matter described in the specifications – whether claims fail to define the invention – whether claims fail to describe the invention fully –whether claims to a single invention comprising a class of antibodies or a multiplicity of different inventions not fully described – whether claims for a manner of manufacture – whether claims entitled to priority date of 23 August 2007 based on provisional application filed on that date – whether claims involve an inventive step – whether notional skilled team would have been directly led to try to generate antibodies in the expectation that they may well block binding between PCSK9 and LDLR – consideration of state of the art at the priority date PATENTS – whether claims to isolated monoclonal antibodies that compete with specified reference antibodies for binding to PCSK9 lack clarity by failing to specify a numerical value or otherwise failing to provide a workable standard with respect to such competition Held: Appeal dismissed |
Legislation: | Evidence Act 1995 (Cth) s 57 Patents Act 1990 (Cth) ss 18(1)(a), 40, 60 Patents Act 1952 (Cth) s 40 Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth) Patents Act 1949 (UK) s 4(3)(c) Patents Act 1977 (UK) s 14 |
Cases cited: | Aktiebolaget Hässle v Alphapharm Pty Ltd (2002) 212 CLR 411 Albany Molecular Research Inc v Alphapharm Pty Ltd (2011) 90 IPR 457 AMP Inc v Utilux Pty Ltd (1971) 45 ALJR 123 Anaesthetic Supplies Pty Ltd v Rescare Ltd (1994) 50 FCR 1 Ariosa Diagnostics, Inc v Sequenom, Inc (2021) 391 ALR 473 Aristocrat Technologies Australia Pty Ltd v Commissioner of Patents (2022) 274 CLR 115 AstraZeneca AB v Apotex Pty Ltd (2014) 226 FCR 324 Atlantis Corporation Pty Ltd v Schindler (1997) 39 IPR 29 Austal Ships Pty Ltd v Stena Rederi Aktiebolag (2005) 66 IPR 420 British Thomson-Houston Company Ld. v Corona Lamp Works Ld. (1922) 39 RPC 49 Burroughs Corp (Perkins’) Application [1974] RPC 147 CCOM Pty Ltd v Jiejing Pty Ltd (1994) 51 FCR 260 Commissioner of Patents v Microcell Ltd (1958) 102 CLR 232 Coopers Animal Health Australia Ltd v Western Stock Distributors Pty Ltd (1987) 15 FCR 382 D’Arcy v Myriad Genetics Inc (2015) 258 CLR 334 Electric & Musical Industries Ltd v Lissen Ltd (1938) 56 RPC 23 Eli Lilly and Co v Pfizer Overseas Pharmaceuticals (2005) 218 ALR 408 Encompass Corporation Pty Ltd v InfoTrack Pty Ltd (2019) 372 ALR 646 Evans Medical Ltd’s Patent [1998] RPC 517 F Hoffman-La Roche AG v New England Biolabs Inc (2000) 99 FCR 56 Genentech Inc.’s Patent [1987] RPC 553 General Tire & Rubber Company v Firestone Tyre & Rubber Company Limited [1972] RPC 457 Gilead Sciences Pty Ltd v Idenix Pharmaceuticals LLC (2016) 117 IPR 252 GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Generic Partners Pty Ltd (2018) 264 FCR 474 Grant v Commissioner of Patents (2006) 154 FCR 62 ICI Chemicals and Polymers Ltd v Lubrizol Corporation Inc (1999) 45 IPR 577 Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 Jupiters Ltd v Neurizon Pty Ltd (2005) 222 ALR 155 Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 Kirin-Amgen Inc v Hoechst Marion Roussel Ltd) [2005] RPC 169 Leonardis v Sartas No 1 Pty Ltd (1996) 67 FCR 126 Lockwood Security Products Pty Limited v Doric Products Pty Ltd (2004) 217 CLR 274 Meat & Livestock Australia Ltd v Cargill, Inc (2018) 354 ALR 95 Merck & Co Inc v Arrow Pharmaceuticals Ltd (2006) 154 FCR 31 Merck Sharp & Dohme Corporation v Wyeth LLC (No 3) (2020) 155 IPR 1 Minnesota Mining and Manufacturing Company v Beiersdorf (Australia) Limited (1980) 144 CLR 253 Monsanto Co v Commissioner of Patents (1974) 48 ALJR 59 Mullard Radio Valve Co Ltd v British Belmont Radio Ltd (1938) 56 RPC 1 National Resource Development Corporation v Commissioner of Patents (1959) 102 CLR 252 Neurim Pharmaceuticals (1991) Ltd v Generic Partners Pty Ltd (No 5) [2024] FCA 360 NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1995) 183 CLR 655 Olin Corporation v Super Cartridge Co Pty Ltd (1977) 180 CLR 236 Pfizer Overseas Pharmaceuticals v Eli Lilly & Co (2005) 225 ALR 416 Rehm Pty Ltd v Websters Security Systems (International) Pty Ltd (1988) 81 ALR 79 Research Affiliates and Commissioner of Patents v RPL Central Pty Ltd (2015) 238 FCR 27 Research Affiliates LLC v Commissioner of Patents (2014) 227 FCR 378 Reynolds v Herbert Smith & Co Ltd (1902) 20 RPC 123 Sequenom, Inc v Ariosa Diagnostics, Inc (2019) 143 IPR 24 Shave v H V McKay Massey Harris Pty Ltd (1935) 52 CLR 701 Stauffer Chemical Co’s Application [1977] RPC 33 ToolGen Incorporated v Fisher (No 2) [2023] FCA 794 Tramanco Pty Ltd v BPW Transpec Pty Ltd (2014) 105 IPR 18 Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610 Bodkin C, Patent Law in Australia (4th ed, Law Book Co, 2024) Terrell T, Terrell on the Law of Patents (13th ed, Sweet & Maxwell, 1982) |
Division: | General Division |
Registry: | New South Wales |
National Practice Area: | Intellectual Property |
Sub-area: | Patents and associated Statutes |
Number of paragraphs: | 470 |
Date of last submissions: | 16 February 2024 |
Date of hearing: | 6-17 November and 11-13 December 2023 |
Counsel for the Appellant: | Mr D Shavin KC with Ms K Beattie |
Solicitor for the Appellant: | Jones Day |
Counsel for the Respondent: | Mr T Cordiner KC with Ms C Cunliffe and Ms M McGrath |
Solicitor for the Respondent: | Wrays Lawyers Pty Ltd |
ORDERS
NSD 876 of 2022 | ||
| ||
BETWEEN: | SANOFI Appellant | |
AND: | AMGEN INC. Respondent | |
order made by: | NICHOLAS J |
DATE OF ORDER: | 23 April 2025 |
THE COURT ORDERS THAT:
1. The appeal be dismissed.
2. The decision of the Delegate of the Commissioner of Patents made on 26 September 2022 be affirmed.
3. Each of Australian Patent Applications AU 2013203677, AU 2013203748, AU 2013203685, AU 2013203689 and AU 2013203751 proceed to grant.
4. The time within which any application for leave to appeal must be made is extended to 21 May 2025.
5. Order 3 is stayed subject to any further order:
(a) until 21 May 2025; or
(b) if any application for leave to appeal is filed by that date, until the determination of that application or the determination of any appeal if leave to appeal is granted.
6. The appellant pay the respondent’s costs of the appeal.
7. Exhibits GAP-A7 and GAP-A8 be returned to the respondent’s solicitors.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
REASONS FOR JUDGMENT
INTRODUCTION | [1] |
GENERAL PRINCIPLES | [23] |
WITNESSES | [28] |
SCIENTIFIC BACKGROUND | [54] |
THE COMMON SPECIFICATION | [119] |
SECTION 40 | [176] |
PRIORITY DATE | [344] |
THE NOTIONAL PERSON SKILLED IN THE ART | [368] |
COMMON GENERAL KNOWLEDGE | [373] |
INVENTIVE STEP | [391] |
LACK OF CLARITY | [428] |
SUFFICIENCY | [455] |
ADDITIONAL MATTERS | [467] |
DISPOSITION | [470] |
NICHOLAS J:
INTRODUCTION
1 The appellant (“Sanofi”) appeals from a decision of the Delegate of the Commissioner of Patents made on 26 September 2022 dismissing Sanofi’s opposition of the grant of five patent applications in the name of the respondent (“Amgen”): Sanofi v Amgen Inc. [2022] APO 67. The appeal is brought under s 60 of the Patents Act 1990 (Cth) (“the Act”).
2 The patent applications (collectively, “the Applications”) are:
(1) AU 2013203677 (“677”);
(2) AU 2013203748 (“748”);
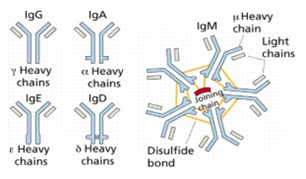
(3) AU 2013203685 (“685”);
(4) AU 2013203689 (“689”); and
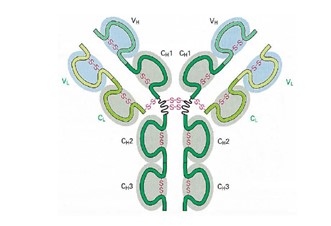
(5) AU 2013203751 (“751”).
Each of the Applications is entitled “Antigen Binding Proteins to Proprotein Convertase Subtilisin Kexin Type 9 (PCSK9)”.
3 The Applications were filed on 11 April 2013. The earliest asserted priority date for each of the relevant claims is 23 August 2007 (“the priority date”).
4 Each of the Applications claims divisional status from Application No. 2008288791 (“the Parent Application”) filed on 22 August 2008 under the provisions of the Patent Cooperation Treaty as PCT/US2008/074097. The Parent Application was first published on 26 February 2009.
5 Low density lipoprotein receptor (“LDLR”) on the surface of the liver takes up low density lipoprotein (“LDL”) which is then transported to lysosomes for degradation. The LDLR is then returned to the liver surface to take up more LDL. High levels of LDL cholesterol are associated with increased risk of cardiovascular and related diseases. Reduction in LDL cholesterol concentrations may therefore lead to improved health outcomes.
6 PCSK9 refers to proprotein convertase subtilisin kexin Type 9. PCSK9 is a protein involved in the degradation of LDLR. The Applications are concerned with isolated monoclonal antibodies (“isolated MAbs”) that target PCSK9 (the antigen). The MAbs block or inhibit the activity of PCSK9 by binding to it, thereby increasing the level of LDLR available to clear LDL and reduce LDL cholesterol levels.
7 The binding of an antibody to its antigen occurs via non-covalent interactions between amino acid residues of the antibody and the antigen.
8 The Applications have a Common Specification (“CS”), which differs as between the Applications in the consistory-like clauses and some figures. Each of the independent claims of the Applications is to an isolated MAb. The term “isolated” means the MAb exists separate from the other components with which it would usually be found, and the term monoclonal describes uniform antibodies of the same specificity and comprising the same heavy and light chains, with some minor variations to their amino acid sequences to be expected. Most relevantly, MAbs will bind to the same epitope, a term which is defined immediately below.
9 The CS and the claims of some of the Applications refer to amino acid residues on PCSK9. The claims identify residues by reference to (inter alia) “SEQ ID NO: 1” and SEQ ID NO: 3”, both being amino acid sequences of PCSK9 set out in Figures 1A and 1B to the CS, respectively. The term “epitope” refers to the specific region on the antigen to which the antibody binds. The amino acid sequences in Figures 1A and 1B are relevantly the same, except that Figure 1B includes the signal sequence, whereas Figure 1A does not. The specification teaches that “any amino acid position in SEQ ID NO: 1, will correspond to an amino acid position 30 amino acids further into the protein in SEQ ID NO: 3”.
10 The parties have agreed upon a set of exemplary claims in each of the Applications (“the Exemplary Claims”). Those claims can be divided into the “Epitope Claims” and the “Residue Claims” of 677, 689 and 751, and the “Competition Claims” of 677, 685 and 748.
11 677 includes independent claims 1 and 5. Claim 1, an Epitope Claim, is for:
1. An isolated monoclonal antibody that binds an epitope on hPCSK9 comprising one or more of amino acid residues 207, 208, 162, 164, 167, or 123 of SEQ ID NO: 1, and wherein the monoclonal antibody blocks binding of PCSK9 to LDLR.
Claim 5, a Residue Claim, is for:
5. An isolated monoclonal antibody that binds to at least one of amino acid residues 207, 208, 162, 164, 167, or 123 of SEQ ID NO: 1, and wherein the monoclonal antibody blocks binding of PCSK9 to LDLR.
12 677 also includes a Competition Claim:
23. The isolated monoclonal antibody according to any one of claims 1-20, wherein the isolated human monoclonal antibody competes for binding to PCSK9 with an antibody comprising a heavy chain amino acid sequence comprising an amino acid sequence of SEQ ID NO: 49 and a light chain amino acid sequence comprising an amino acid sequence of SEQ ID NO: 23.
13 689 includes independent claim 1, a Residue Claim for:
An isolated monoclonal antibody, wherein, when bound to PCSK9, the monoclonal antibody binds to at least one of the following residues: S153, I154, P155, R194, D238, A239, I369, S372, D374, C375, T377, C378, F379, V380, or S381 of SEQ ID NO: 3, and wherein the monoclonal antibody blocks binding of PCSK9 to LDLR.
14 751 includes independent claims 1, 2, 10, and 13. Claims 1 and 10 are Epitope Claims. Claims 2 and 13 are Residue Claims. These claims provide:
1. An isolated monoclonal antibody that recognizes an epitope on human PCSK9 comprising amino acid residues: S153, R194, D238, D374, T377, and F379 of SEQ ID NO: 3, wherein the monoclonal antibody reduces binding between PCSK9 and EGFa domain of LDLR.
2. An isolated monoclonal antibody that binds to amino acids in human PCSK9 of SEQ ID NO: 1, wherein the amino acids comprise S153, R194, D238, D374, T377, and F379 of SEQ ID NO: 3, and wherein the monoclonal antibody reduces binding between PCSK9 and an EGFa domain of LDLR.
…
10. An isolated monoclonal antibody that recognizes an epitope on human PCSK9, wherein the epitope comprises at least ten of the following residues: S153, I154, P155, R194, D238, A239, I369, S372, D374, C375, T377, C378, F379, V380, or S381 of SEQ ID NO: 3, wherein the monoclonal antibody reduces binding between PCSK9 and an EGFa domain of LDLR.
…
13. An isolated monoclonal antibody that binds to amino acids in human PCSK9, wherein the amino acids comprise ten or more of the following amino acids: S153, I154, P155, R194, D238, A239, I369, S372, D374, C375, T377, C378, F379, V380, or S381 of SEQ ID NO: 3, and wherein the monoclonal antibody reduces binding between PCSK9 and an EGFa domain of LDLR.
Claims 2 and 13 do not use the word “residue”, however, as with claims 1 and 10, they identify particular amino acid residues by reference to their place in the sequence.
15 Claim 1 of 677 refers to a MAb that binds to an epitope of PCSK9 so as to block binding of PSK9 to LDLR. Claims 1 and 10 of 751 adopt the same approach as claim 1 of 677, although using slightly different terminology. Claims 5 of 677, 1 of 689 and 2 and 13 of 751 refer to MAbs that bind to particular residues of PCSK9, rather than epitopes. The “EGFa domain” of LDLR refers to a section of LDLR, which is the region of LDLR that binds to PCSK9.
16 Claim 23 of 677, and claims 1 of 685 and 748, introduce the concept of competition in referring to a MAb that competes for binding to PCSK9 with another antibody with a particular amino acid sequence.
17 685 includes claim 1, a Competition Claim for:
An isolated monoclonal antibody that binds to human PCSK9 and reduces binding between human PCSK9 and an EGFa domain of LDLR, wherein said monoclonal antibody competes for binding to PCSK9 with an antibody that comprises a heavy chain variable region of the amino acid sequence in SEQ ID NO: 67; and a light chain variable region of the amino acid sequence in SEQ ID NO: 12.
18 748 includes claim 1, another Competition Claim, which provides:
An isolated monoclonal antibody that binds to human PCSK9 and is neutralizing in that an excess of said antibody reduces the quantity of human PCSK9 bound to LDLR in an in vitro competitive binding assay, wherein said monoclonal antibody competes for binding to PCSK9 with an antibody that comprises: a heavy chain variable region of the amino acid sequence in SEQ ID NO: 49; and a light chain variable region of the amino acid sequence in SEQ ID NO: 23.
19 The antibodies identified in claims 23 of 677, 1 of 685 and 1 of 748 by reference to their heavy and light chain regions are identified in the CS as either of antibodies 31H4 and 21B12, which are proffered as benchmarks against which other MAbs may be assessed for the purpose of determining whether they have the relevant binding effect. It is convenient to refer to these two antibodies as the “reference antibodies”.
20 CS [0607] includes the following definition of “comprise” and “comprising”
Throughout the specification and claims, unless the context requires otherwise, the word “comprise” or variations such as “comprises” or “comprising”, will be understood to imply the inclusion of a stated integer or group of integers but not the exclusion of any other integer or group of integers.
21 Applying this definition to the Epitope and Residue Claims, the MAb need only bind to, or bind to an epitope that includes, one or more (or in the case of claims 10 and 13 of 751, at least 10) of the specified amino acid residues. For the Epitope Claims, there does not need to be a non-covalent interaction between the MAb and any particular residue, provided that the specified residue is part of the epitope on PCSK9 bound by the MAb.
22 Sanofi contended that any patent granted on any of the Applications would be invalid on various grounds. Sanofi summarised its opposition to the Applications as follows:
(a) Failure to define the invention – The claims of each of the Applications fail to define the invention in that they pertain to any “isolated monoclonal antibody” having some claimed functional features only. The “blocking” function of an antibody, knowledge of the epitope or a residue or residues on an antigen to which an antibody binds, or reference to another antibody with which an antibody competes, does not define the structure of, or characterise the construction of, an antibody as claimed.
(b) Not a manner of manufacture – The claims of each of the Applications fail to define a manner of manufacture because they are for no more than a mere desideratum, namely, any MAb providing particular functional outcomes.
(c) Lack of fair basis – The claims travel well beyond any invention described in the specification and omit essential features of that invention and are therefore not fairly based.
(d) Lack of clarity – The Competition Claims lack clarity because it is not clear whether “competes” means any degree of competition or some particular numerical degree of competition and, in any event, there is no workable standard to determine the exact boundaries of the claims. Various alternative competitive binding assays described in the specification can produce different results.
(e) Failure to describe the invention fully – The claims of each of the Applications are properly characterised as claims to a multiplicity of inventions, each a different antibody with a unique amino acid sequence and binding to a different epitope on PCSK9. The Applications are therefore insufficient because the person skilled in the art (“PSA”) would need to engage in the research project described in Examples 1 to 3 of the Applications in an attempt to produce one embodiment of each such invention.
(f) Lack of inventive step – The use of an antibody to inhibit PCSK9 had been postulated in well-known, peer-reviewed literature. LDLR residues in the PCSK9 and LDLR EGFa domain interface (“the PCSK9/EGFa interface”) were known. It was “logical” that an antibody that blocks the binding of PCSK9 to LDLR (to any degree) would do so by binding to an epitope that included any one of the residues in the PCSK9/EGFa interface. It follows that the inventions do not involve any inventive step.
GENERAL PRINCIPLES
Standard of Proof
23 It is common ground that the Applications should proceed to grant unless the Court is satisfied that any such grant would be clearly invalid. Sanofi, the opponent, bears the onus of showing that the grant of a standard patent based on the Applications would be clearly invalid.
24 Although contested issues of fact are to be decided on the balance of probabilities, the ultimate question raised under each of the relevant grounds of opposition, typically involving mixed issues of fact and law, is to be decided by reference to the higher standard. This has important implications for the present case in which the relevant grounds of opposition are the subject of significant differences of opinion between expert witnesses. As Bennett J observed when deciding an opposition based on an alleged lack of inventive step in Austal Ships Pty Ltd v Stena Rederi Aktiebolag (2005) 66 IPR 420 (“Stena”) at [12]:
I can accept that a lower standard may apply to proof of evidence such as whether a document has been published or, indeed, whether a prior art vessel was well-known. I do not accept that it properly applies to the factual question that itself is the test for obviousness or lack of inventive step. Where the factual question is itself the legal test, as set out in s 7(3) of the Act, it seems to me that it should be determined at the higher standard. That means that where there are two opposing expert views that are conclusive on obviousness, both presented bona fide by witnesses of accepted expertise, unless one set of views can be rejected on proper grounds, the legal burden to establish a ground of opposition is not discharged; the court cannot be practically certain that obviousness or lack of inventive step is established.
25 Her Honour’s approach to the expert evidence was followed by Beach J in Meat & Livestock Australia Ltd v Cargill, Inc (2018) 354 ALR 95 (“Meat & Livestock v Cargill”) at [11], although his Honour favoured the language of “clearly invalid” used by Emmett J in F Hoffman-La Roche AG v New England Biolabs Inc (2000) 99 FCR 56 at [67], rather than that of “practically certain”. I have adopted the same approach as Beach J.
Principles of Construction
26 There was no dispute between the parties as to the principles governing the construction of a patent specification or, in this case, a patent application. These principles were summarised by the Full Court in Jupiters Ltd v Neurizon Pty Ltd (2005) 222 ALR 155 (Hill, Finn and Gyles JJ) as follows at [67]:
…
(i) the proper construction of a specification is a matter of law: Décor Corporation Pty Ltd v Dart Industries Inc (1988) 13 IPR 385 at 400 [(Décor Corporation Pty Ltd)];
(ii) a patent specification should be given a purposive, not a purely literal, construction: Flexible Steel Lacing Co v Beltreco Ltd (2000) 49 IPR 331; [2000] FCA 890 at [81] (Flexible Steel Lacing); and it is not to be read in the abstract but is to be construed in the light of the common general knowledge and the art before the priority date: [Kimberly]-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1; 177 ALR 460; 50 IPR 513; [2001] HCA 8 [(Kimberly-Clark v Arico)] at [24];
(iii) the words used in a specification are to be given the meaning which the normal person skilled in the art would attach to them, having regard to his or her own general knowledge and to what is disclosed in the body of the specification: Décor Corporation Pty Ltd at 391;
(iv) while the claims are to be construed in the context of the specification as a whole, it is not legitimate to narrow or expand the boundaries of monopoly as fixed by the words of a claim by adding to those words glosses drawn from other parts of the specification, although terms in the claim which are unclear may be defined by reference to the body of the specification: [Kimberly]-Clark v Arico at [15]; Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610; Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 at 478; the body of a specification cannot be used to change a clear claim for one subject matter into a claim for another and different subject matter: Electric & Musical Industries Ltd v Lissen Ltd [1938] 4 All ER 221 at 224–5; (1938) 56 RPC 23 at 39;
(v) experts can give evidence on the meaning which those skilled in the art would give to technical or scientific terms and phrases and on unusual or special meanings to be given by skilled addressees to words which might otherwise bear their ordinary meaning: Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479 at 485–6 (Sartas No 1 Pty Ltd); the court is to place itself in the position of some person acquainted with the surrounding circumstances as to the state of the art and manufacture at the time ([Kimberly]-Clark v Arico at [24]); and
(vi) it is for the court, not for any witness however expert, to construe the specification; Sartas No 1 Pty Ltd at 485–6.
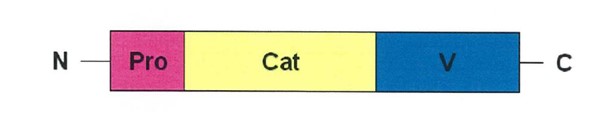
27 The notion of purposive construction requires that the specification be read through the eyes of the PSA with practical knowledge and experience in the field of work in which the invention was intended to be used, and with the proper purpose of the invention in mind: GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Generic Partners Pty Ltd (2018) 264 FCR 474 (“GSK v Generic”) at [106].
WITNESSES
Sanofi’s Witnesses
Professor Jay D. Horton
28 Professor Jay Horton is a Professor in the Department of Internal Medicine and the Department of Molecular Genetics, University of Texas Southwestern Medical Center (“UT Southwestern”), in Dallas. He holds an undergraduate degree in Zoology and a Doctor of Medicine (“MD”) from the University of Iowa. After obtaining his MD, he completed an internal medicine residency and gastroenterology fellowship at UT Southwestern. He then completed a post-doctoral fellowship in the Department of Molecular Genetics at UT Southwestern. The work he undertook subsequent to his fellowship, as a junior member of the Department of Molecular Genetics, led to the identification of novel proteins regulated by sterol regulatory element binding proteins, one of which was PCSK9. Following that, his group undertook research into the function and biology of PCSK9.
29 Professor Horton gave evidence about PCSK9 and the therapeutic modalities used to inhibit it. Professor Horton provided consultancy services to a number of pharmaceutical companies before the priority date in relation to PCSK9. This work, which commenced early 2005, involved consulting on projects investigating PCSK9 as a possible drug target for the treatment of high cholesterol. He provided services to Merck, Pfizer, Amgen, Eli Lily, Alnylam, Bristol-Myers Squibb (“BMS”), Novartis, Regeneron and Schering-Plough in relation to PCSK9. There appeared to be no dispute that he was, at the relevant times, one of the world’s leading experts on PCSK9. Professor Horton and his team were not directly involved in drug discovery or drug development.
30 As I will later explain, I found that some of Professor Horton’s evidence as to the state of the art at the priority date was most likely affected by hindsight. That said, Professor Horton impressed me as a highly knowledgeable expert on PCSK9 as is evident from his enormous contribution to the scientific literature on that topic.
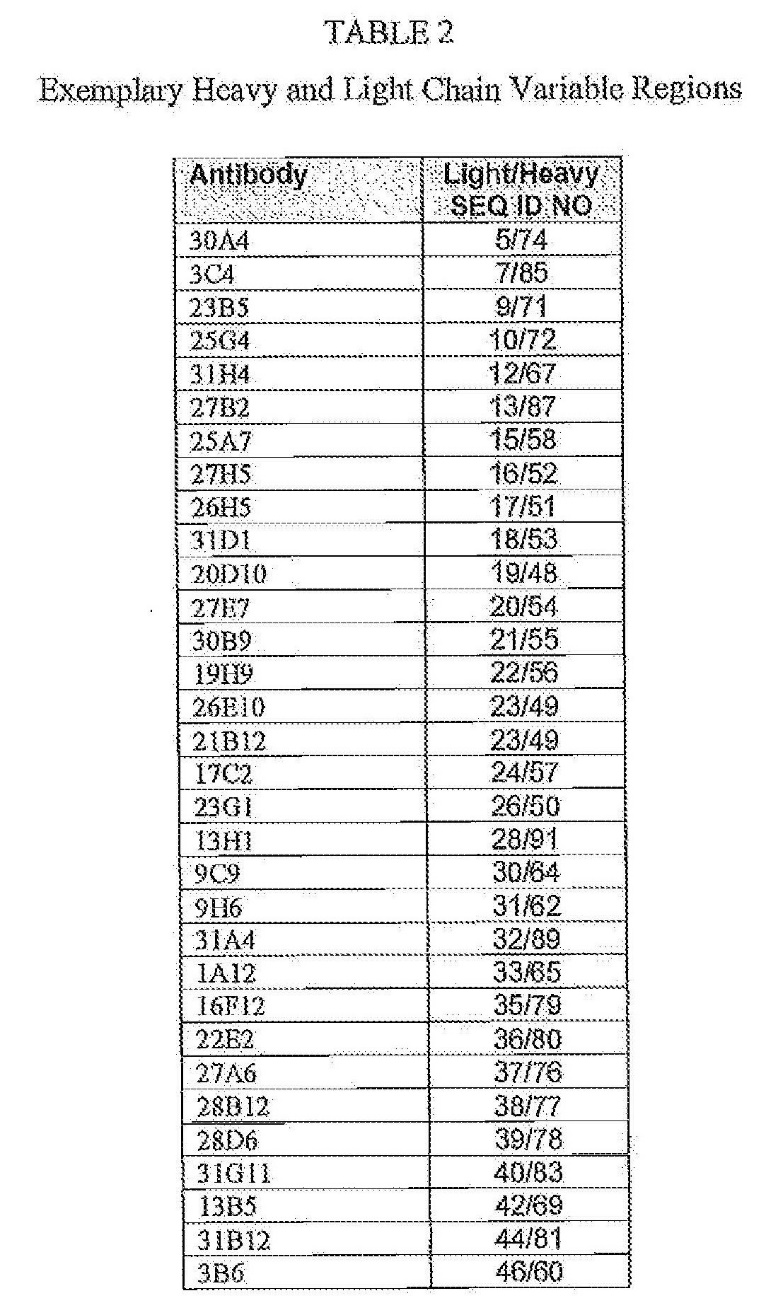
Professor Stephen Michael Mahler
31 Professor Stephen Mahler is an Emeritus Professor at the Australian Institute for Biotechnology at the University of Queensland, and the Global Chief, Science and Technology at the Aegros Group. He holds a Bachelor of Science (Honours) from the University of Sydney and a Doctor of Philosophy in Biochemistry from the University of Queensland.
32 Professor Mahler primarily gave evidence about monoclonal antibodies, including antibody production and analysis of binding activity. He has more than 35 years of experience in the antibody field, including the production, discovery, and development of monoclonal antibodies for therapeutic and diagnostic purposes. He has an extensive academic career focused on monoclonal antibody technology.
33 Professor Mahler was criticised by Amgen on the ground that he considered it his role as an expert witness for Sanofi to find fault with the CS wherever possible. Examples of this tendency identified by Amgen included evidence given by him concerning “antibody excess” which I refer to in more detail later in these reasons. Apart from that example, the other matters upon which Amgen relied in support of its attack on Professor Mahler’s reliability as an expert witness were to my mind relatively inconsequential. I considered Professor Mahler an impressive witness and I found much of his evidence helpful.
Professor Michael Parker
34 Professor Michael Parker is the Head of the Structural Biology Unit at St Vincent’s Institute of Medical Research, in Melbourne, and a Professorial Fellow and Professor at the Department of Biochemistry and Molecular Biology and Director of the Bio21 Institute at the University of Melbourne. He holds a Bachelor of Science in Chemistry with Honours from the Australian National University, and a Doctor of Philosophy from the University of Oxford.
35 Professor M Parker’s evidence was directed to crystallography and analysis of antibody-antigen interactions. He is a structural biologist, and expert in X-ray crystallography including the use of X-ray crystallography to solve the atomic structure of proteins. He has more than 40 years of experience in this field.
36 Amgen was critical of Professor M Parker’s evidence as “overly pedantic” and as expecting the CS to meet standards of disclosure that were not always adhered to in his own publications. Professor M Parker asserted in his written evidence that certain data files (the PDB files used in the PyMOL analysis) were not available to him, but Amgen submits that he did not request that they be provided to him. Amgen described his failure to request this data so that he could conduct his own analysis of it (rather than merely relying on Professor Petsko’s analysis) as “inexplicable”.
37 There is considerable force in Amgen’s submission, though ultimately the validity of Professor M Parker’s criticisms of the PyMOL analysis relied on by Amgen to establish certain non-covalent interactions need not be addressed, since my conclusions with respect to the fair basis challenge do not depend on it.
Other witnesses
38 Sanofi also relied on affidavit evidence of Anthony Muratore, Samin Raihan, Lisa Taliadoros and Maria Garcia of Jones Day. They were not cross-examined and their affidavit evidence was uncontroversial.
Amgen’s Witnesses
Dr Rex Arnold Parker
39 Dr Rex Parker was a Director and Senior Research Fellow of the Hopewell Biology Discovery Department at BMS. He holds undergraduate degrees in chemistry and biology, and a PhD in biochemistry, from Indiana University. After completing his doctoral research, he joined BMS's cardiovascular and metabolic diseases research department looking at ways to regulate LDL cholesterol and sterol metabolism.
40 Dr R Parker gave evidence about PCSK9 and the therapeutic modalities to inhibit it. Between 2002 and 2014, he was responsible for BMS’s lipid-lowering and anti-atherosclerotic drug discovery, and was co-leader of BMS's PCSK9 inhibitor discovery research program. In 2009, he was promoted to the position of Senior Research Fellow for Cardiovascular Drug Discovery at BMS, which was the position he held until he retired in 2014. He has many years’ experience in the field of drug discovery and drug development.
41 Sanofi was critical of Dr R Parker’s evidence on the basis that he was a “contrarian”, and that he was excessively critical of all scientific literature supporting the view that PCSK9 had an extracellular mode of action. I do not accept Sanofi’s criticisms of Dr R Parker as a witness, which I deal with in further detail below when considering the parties’ submissions on the topic of inventive step. Generally speaking, I considered Dr R Parker an impressive witness whose evidence was of considerable assistance to me on that topic.
Professor Angel Francisco Lopez
42 Professor Lopez holds a degree in medicine from the School of Medicine at the University of Rosario, Argentina, and a PhD from the National Institute for Medical Research, University of London. He has over 40 years of experience working in the field of antibodies and has created antibodies within his laboratory for the treatment of acute myeloid leukaemia. He is currently the Head of the Cytokine Receptor Laboratory and head of the Division of Human Immunology at SA Pathology in Adelaide, an Adjunct Clinical Professor of the Faculty of Medicine at the University of Adelaide and an Adjunct Professor at the University of South Australia. Professor Lopez gave evidence about antibody generation using hybridoma technology, immunisation and screening protocols.
43 Professor Lopez was criticised in Sanofi’s submissions on the grounds that his evidence was often unresponsive and that, particularly in relation to crystallography, he volunteered opinions that travelled outside his area of expertise. Sanofi also referred to what it said was “the tendentious approach he took when he perceived his evidence to be unhelpful to Amgen’s case”. I took that to mean that Professor Lopez was biased against Sanofi. There is nothing in Sanofi’s submissions which justifies that very serious attack upon Professor Lopez’s credit. I found Professor Lopez’s evidence concerning antibody development helpful in understanding the technology and in assessing the difficulties that may be experienced by the PSA in generating an anti-PCSK9 antibody without the benefit of the CS. I reject Sanofi’s submission that he was biased or that he sought to argue Amgen’s case in a manner that was inconsistent with his duty as an independent expert.
Professor Peter John Hudson
44 Professor Peter Hudson has expertise and extensive experience in the field of antibody structure, function and development, and in the area of X-ray crystallography. He currently provides scientific consultancy services in the development of antibodies as clinical products as the Managing Director of Diantron Biomed Pty Ltd, and also serves as the Chief Scientific Officer of AviPep Pty Ltd and as Senior Scientific Advisor to CarTherics Pty Ltd (for the development of T-cell therapies). He holds a Bachelor of Science (Honours) from the Department of Biochemistry, University of Adelaide and a PhD from the MRC Laboratory of Molecular Biology, University of Cambridge. Professor Hudson gave evidence as to antibody technology and structural biology (including crystallography).
45 As with Professor Lopez, Sanofi submitted that Professor Hudson gave evidence which displayed what it said was a “disturbing tendency” to argue Amgen’s case. The evidence to which Sanofi points in support of this submission does not support it. For example, it criticised Professor Hudson for having by his own admission speculated about the inventors’ reasons for having followed an immunisation protocol involving more than 10 immunisations of mice in the process of generating antibodies. But he freely admitted that this was his own interpretation of why the inventors may have proceeded as they did. Later in his evidence he agreed that he was mistaken in suggesting in earlier evidence that mice were given a “massive double boost” of 10mg of antigen as opposed to 5mg split across two different injections.
46 I do not consider this or any other evidence given by Professor Hudson as reflecting a tendentious approach to the giving of evidence. Nor do I think that any other inconsistency to which Sanofi pointed in his evidence diminishes the quality of his evidence in areas where it most matters. I generally found Professor Hudson to be an impressive witness whose evidence was quite helpful.
Professor Gregory Anthony Petsko
47 Professor Petsko is a Professor of Neurology at Harvard Medical School and Brigham Women’s Hospital, Adjunct Professor of Biomedical Engineering at Cornell University and Emeritus Professor in Biochemistry and Chemistry at Brandeis University. He holds a Bachelor of Chemistry from Princeton University and a PhD in Molecular Biophysics from Oxford University. He is an elected member of the National Academy of Sciences (1995) and the American Academy of Arts and Sciences (2002). He has been recognised by the latter for his pioneering work in the field of X-ray crystallography. In 2023 he was awarded a National Medal of Science by the President of the USA.
48 Professor Petsko has expertise in various related fields including, most relevantly, structural biochemistry, and X-ray crystallography. He has authored or co-authored over 300 peer reviewed articles mostly detailing the relationship between protein structure and function. Much of his work has included the use of X-ray crystallography to solve the atomic structure of proteins either alone or in complex with other molecules. He is a co-author (with Professor Dagmar Ringe) of a textbook first published in 2003 concerning basic principles of protein structure and function entitled “Primers in Biology: Protein Structure and Function”.
49 Sanofi submitted that Professor Petsko, while highly qualified, was often loquacious in his answers and “passionately committed to Amgen’s case”. Sanofi was also critical of Professor Petsko for not disclosing in his written evidence that he prepared or relied on an electron density map in respect of residue S153 on PCSK9.
50 Professor Petsko made clear in his oral evidence that his analysis of S153 relied only on the structural information that would be available to Professor M Parker and did not rely on the electron density map for S153. Professor Petsko’s cross examination revealed he had seen the electron density map for S153 “a number of years ago” and that he could not “unsee” this information. When Professor Petsko was pressed on the topic of S153, he made clear he did not use the electron density map as part of his analysis, but instead used the B-Factors provided in Table 35.3 and the rotamer for S153 recorded in the coordinates in Table 35.3.
51 There are a few points I would make with regard to the suggestion that Professor Petsko was “loquacious” and “passionately committed to Amgen’s case”. First, much of Professor Petkso’s oral evidence was directed to explaining the relevant science and the CS, often in answer to questions from the Court. In this regard, the impression I formed of Professor Petsko was not that he was loquacious, but that he is an excellent communicator with an impressive ability to explain the relevant science pellucidly. I reject Sanofi’s submission that Professor Petsko’s evidence was tendentious or affected by bias. I regard him as a very impressive witness whose evidence I found helpful.
Other witnesses
52 Amgen also relied on affidavit evidence of Louisa King, Bindhu Holavanahalli, and Mark O’Leavey, none of whom were cross-examined. Some of this evidence was contentious in that it related to 3D models created by Mr O’Leavey, an engineer with expertise in production of 3D models of molecules of interest using crystallographic data. These 3D models produced by Mr O’Leavey formed part of the “Modelling Evidence” the subject of objection by Sanofi and a ruling on evidence recorded in MFI-10. I note that it was not necessary for me to refer to the Modelling Evidence for the purpose of deciding any issue in the case.
Joint Expert Reports
53 Several expert conclaves were conducted, and four separate joint expert reports (“JERs”) were produced. Dr R Parker and Professor Horton produced a JER regarding knowledge as to the mechanism of action of PCSK9 reducing LDLR levels and approaches to the inhibition of PCSK9. Professors Petsko, Hudson, Lopez, M Parker and Mahler (“the Antibody and Crystallography Experts”) jointly produced a JER documenting the results of a conclave dealing with both antibody and crystallography related issues. Professors Mahler, Lopez and Hudson (“the Antibody Experts”) also produced a JER dealing with further antibody issues, and Professors M Parker, Petsko and Hudson (“the Crystallography Experts”) produced a JER dealing with crystallography issues.
SCIENTIFIC BACKGROUND
54 The parties have filed an “Agreed Primer”, which contains some scientific background relevant to the invention and the CS. The following background information has been extracted from the Agreed Primer, the CS, or evidence otherwise agreed by the relevant experts.
Antibody structure
55 An antibody (also known as an immunoglobulin) is a type of protein. Proteins are complex molecules made up of amino acids (often referred to as “amino acid residues” or “residues”).
56 The sequence of amino acids is referred to as the “primary structure” of the protein. There are 20 naturally occurring amino acids, and in sequences they are represented by either a one-letter or a three-letter abbreviation. For example, aspartic acid is represented by the three-letter abbreviation “asp”, and the one-letter abbreviation “D”. A specific amino acid residue on a protein can be referred to using the abbreviated name and its position number in the protein. For example, D238 means the aspartic acid residue at position number 238 of the protein.
57 Stretches of protein will typically arrange into distinct local conformations, referred to as “secondary structures”. The secondary structure of a protein is determined by the primary structure. The secondary structure of the protein will assemble into a 3D structure, the overall shape of which is referred to as the “tertiary structure” of the protein.
58 Antibodies can be represented two dimensionally as having a Y shaped structure, comprised of heavy and light chains of amino acids, as shown below:
In reality, antibodies have extremely complex three-dimensional (“3D”) shapes. The biological function of a protein is dependent on its 3D shape, because the shape of a protein affects how it interacts with other molecules.
59 As depicted above, there are five different classes of antibodies in mammals (IgA, IgD, IgE, IgG and IgM). Each of the classes is made up of various sub-classes (or isotypes). For example, the IgG class has four subclasses (IgG1, IgG2, IgG3 and IgG4 antibodies).
60 The light and heavy chains are made up of different regions, also referred to as “domains”. Each light chain consists of one variable domain, and one constant domain. Each heavy chain consists of one variable domain and three constant domains. The constant domains are referred to as such because the amino acid sequence in these domains is highly similar in all antibodies of the same class. The light and heavy chains are held together by disulfide (-S-S-) bonds, as shown below:
The “constant end” of an antibody (in the antibody depicted above, the two heavy chain CH2-CH3 regions) is called the Fc region.
61 The variable domains of the light and heavy chains contain the amino acid residues that contribute to the binding of the antibody to the antigen. Variable domains of the antibody comprise conserved regions referred to as “framework regions”, and hypervariable regions referred to as “complementarity determining regions” (“CDRs”). CDRs are typically around four to 20 amino acids in length. CDRs interact with the target protein (i.e. the antigen) and make up the functional binding site of the antibody. The antigen-binding site on the antibody is referred to as the paratope.
Antibody-antigen interactions
62 Each antibody typically binds to a particular antigen. The site on the antigen to which the antibody binds is referred to as the epitope. The CS refers to an epitope at [0233] as:
… a region of an antigen that is bound by an antigen binding protein [i.e., antibody] that targets that antigen, and when the antigen is a protein, includes specific amino acids that directly contact the antigen binding protein.
63 The CS distinguishes between structural epitopes and functional epitopes as follows at CS [0571]:
Epitopes can be further defined as structural or functional. Functional epitopes are generally a subset of the structural epitopes and have those residues that directly contribute to the affinity of the interaction (e.g. hydrogen bonds, ionic interactions). Structural epitopes can be thought of as the patch of the target which is covered by the antibody.
64 The Antibody and Crystallography Experts agreed that the term “structural epitope” refers to the region of the target antigen covered by the antibody that binds it. They also agreed that the term “functional epitope” refers to the region of the target antigen that is directly involved in non-covalent interactions with the antibody that contribute to the affinity the two proteins have for each other.
65 The CS also distinguishes between linear and conformational epitopes. Amgen defines a conformational epitope as one “composed of discontinuous amino acid sequences that are brought together by protein folding in the antigen”. Sanofi’s definition, which varies only slightly, draws a distinction between conformational epitopes which “[conform] with the native three-dimensional structure of the antigen” and linear epitopes, composed of residues which are contiguous. I do not consider that there is a meaningful distinction between the two proposed definitions, but in any event, it is sufficient to refer to Amgen’s definition.
66 The measure of the strength of binding between the antibody and antigen is referred to as affinity. The affinity of the binding between an antibody and its antigen is the sum of all the non-covalent interactions between the antibody and the antigen.
67 The specificity of an antibody is the ability of the antibody to distinguish between a particular molecular structure and all other structures. Specific binding occurs when an antibody binds preferentially to its antigen over other proteins. Non-specific binding occurs when an antibody binds to an epitope that is common across a range of proteins, including its antigen and unrelated proteins. Specificity is measured through assays that may examine, for example, whether an antibody binds to proteins other than the target protein.
68 Protein-protein (including antibody-antigen) binding occurs when the 3D shape and contours of one protein allow some of its amino acid residues to align with that of another protein, such that one or (typically) multiple individual non-covalent interactions between those residues can take place. The three major types of non-covalent interactions that may contribute to binding of an antibody to its antigen are:
(a) Salt bridge (ionic bond) – an interaction between the positive charge of an atom of one amino acid side chain in the first protein with the negative charge of an atom of another amino acid side chain in the second protein. This is the strongest of the non-covalent interactions;
(b) Hydrogen bond – an interaction between the hydrogen atom attached to a highly electronegative atom in an amino acid residue of one protein, and a highly electronegative atom in an amino acid residue of another protein; and
(c) Van der Waals interactions – these include a range of different types of typically very weak interactions that arise due to a temporary redistribution of electrons of atoms.
69 Protein-protein interactions may also include other interactions involving ligands (“ligand mediated interactions”) such as water, where, for example, an atom in an amino acid residue in a first protein forms a hydrogen bond with a water molecule, and the same water molecule also forms a hydrogen bond with an atom in an amino acid residue in a second protein.
70 Typically, approximately 15 to 20 amino acid residues are involved in the binding of an antibody to its antigen. Although each individual interaction may be weak, the combination of interactions can result in strong binding. Put another way, although a relatively small amount of energy is needed to overcome (or “break”) an individual non-covalent interaction, a relatively large amount of energy may be needed to “break” all the non-covalent interactions and separate the antibody from its antigen.
71 The binding of an antibody to an antigen is a dynamic process in which the antibody rapidly binds to and dissociates from the antigen over time. In general, the higher the affinity that the two proteins have for one another, the more time the proteins will spend in their bound state. The lower the affinity that two proteins have for one another, the more time they will spend in their unbound state.
72 The binding of an antibody to its antigen is a dynamic process in which the antibody rapidly binds to and dissociates from the antigen over time. The antigen-antibody complex is in equilibrium with free antigen and free antibody, which can be represented by the following equation:
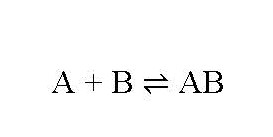
73 In general, the higher the affinity two proteins have for one another, the more time the proteins will spend in the “bound” state (i.e. the right-hand side of the equation: “AB”), and the lower the affinity two proteins have for one another, the more time the proteins will spend in the “unbound” state (i.e. the left hand side of the equation: “A + B”).
74 The dissociation constant (“KD”) is a quantitative estimation of the strength of binding of two proteins (e.g., antigen-antibody). For example, in an antibody-antigen interaction, a KD of 10-9 to 10-10 moles (“M”) is considered a strong interaction, whereas a KD of between 10-6 and 10-7 M is considered a relatively weak interaction. The higher the affinity, the smaller the dissociation constant.
75 The binding of an antibody to its antigen does not necessarily result in a biological outcome. Whether or not a biological outcome is produced may depend on the strength of the binding (the affinity), which is particularly important in the case of therapeutic antibodies. Typically, though not necessarily, a therapeutic antibody has a KD of 10-9 M.
Antibody production
76 In the human body, hematopoietic stem cells give rise to B cells, which produce antibodies.
77 Two methodologies were developed in the early 1990s for creating new fully human antibodies against a particular antigen, namely, transgenic mice and phage display. With the advent of these technologies, it was routine by the priority date, to produce fully human antibodies in high yield cell lines. Further, by the priority date, the methods, and processes for the manufacture of antibodies were mature and well established.
Transgenic mice
78 Transgenic mice are mice that have been genetically modified, in this context to produce fully human antibodies. When a transgenic mouse is immunised (i.e. injected with) the antigen of interest, B cells are created, some of which produce antibodies that bind to the antigen. These B cells are “remembered” by the immune system and undergo random mutations, which may lead to antibodies having increased or reduced binding affinity.
79 After the spleens of the mice are removed, the B cells are isolated. Isolation of a single B cell from this sample and clonal expansion results in a colony of cells arising from that single “parent” cell. Antibodies produced from a unique parent cell (and its cloned colony) are termed “monoclonal antibodies” (i.e., MAbs).
80 B cells must be “immortalised” to survive outside the body. To do so, a B cell is fused with a myeloma cell to create a “hybridoma”, which has the characteristics of both the B cell (in that it expresses and secretes a MAb) and the myeloma cell (in that it can grow and divide indefinitely ex vivo).
81 The hybridoma thus provides a cell “factory” that can be used in the large-scale production of MAbs. The hybridoma is cultured and MAbs are secreted to the outside of the cell, accumulating in the surrounding media. After an appropriate time in the cell culture, the media is harvested, and the antibody is purified from the media. The panel of antibodies produced by these hybridomas can then be screened to identify those with the desired attributes including, for example, their ability to bind to a target antigen.
82 The recovery of B cells from mice and their immortalisation, and the techniques for screening the binding of antibodies to a target antigen, as a general matter, were known procedures as at the priority date.
83 The Antibody Experts agreed that, once a mouse has been injected with the selected protein or part thereof which is the intended target antigen, the researcher has no control over how the mouse’s immune system will react. As Professor Mahler said, once injection has occurred, “[t]he mouse immune system then takes over, and whatever antibodies are raised are raised…and you get what you get”. I accept that evidence.
Phage Display
84 Phage display is a technique involving the use of certain types of viruses (phage) that use the cells they infect as a “factory” to produce viral particles.
85 Phage display also utilises an immunoglobulin gene library, which is a large collection of DNA sequences that encode the variable regions of antibodies. These libraries were available for purchase by the priority date. This DNA is inserted into ‘phagemids’ and cultured in bacteria, resulting in the production of a library of phage with the antibody variable regions ‘displayed’ on their surface.
86 The phage library is then screened against an antigen of interest, in a process called “bio-panning”. This results in a sub-library of phage antibodies which bind to the antigen, as well as some non-specific binders. The process is then repeated using this sub-library of phage antibodies (and subsequent sub-libraries) to increase the ratio of phage antibodies that bind specifically to the antigen.
87 The resultant sub-library of phage antibodies can then be screened to identify those with desired attributes. Preparation of MAbs by screening phage display libraries was well known by the priority date.
Techniques to analyse antibodies and antibody-antigen interactions
88 The following information is relevant to the techniques employed by the PSA in their analysis of individual antibodies and the interactions as between antibodies and antigens.
X-ray Crystallography
89 Atoms within chemical molecules exist in a 3D conformation. X-ray crystallography (or simply, crystallography) enables determination of that 3D arrangement (x, y and z coordinates). From well before 2007/2008, crystallography has been universally accepted as the ‘gold standard’ technique for studying the precise manner of binding between two proteins.
90 The technique involves shining an X-ray beam onto a sample of the protein or protein complex. Some of the X-rays interact with electrons of the atoms in the sample, resulting in the production of new X-rays that are emitted in many directions (“diffracted”).
91 However, when a beam of X-rays is directed at a single protein molecule, the scatter of the X-ray beam will be too small to detect. Instead, a sample of the protein (or protein complex) in the form of a 3D crystal lattice consisting of a regular, repeating arrangement of the protein molecule (or protein complex) is required. A typical crystal has tens of thousands of identical, repeating “unit cells” which amplify the X-ray beam, creating a signal that has sufficient intensity to be detected. Obtaining crystals of a protein or protein complex relies on various factors (such as the stability of the protein complex) and is usually the most rate-limiting step of this process.
92 Using a protein (or protein complex) crystal, the ‘diffraction pattern’ of the scattered X-rays can be collected and then analysed mathematically to construct an initial 3D image of the electron clouds of the protein, referred to as an “electron density map”. The electron density map is then compared to the amino acid sequence of the protein, and, based on knowledge of the chemical structure of each amino acid, the electron clouds observed can be assigned to particular amino acid residues. This process is called ‘refinement’ and involves considering all possibilities until the best consensus fit with the amino acid sequence is reached. It is done using a computer program. The overall percentage disagreement between the model’s calculated diffraction pattern and the observed diffraction data is referred to as the “R factor”.
93 The process of fitting amino acid residues in a protein to the electron density map makes it possible to identify 3D atomic coordinates for atoms in a protein (or protein complex), represented as x, y and z values.
94 Subject to the resolution of the crystallography experiment, the 3D coordinates of ligands, such as water, will be identified separately. Resolution can be thought of as the extent to which it is possible to identify atoms or groups of atoms. The higher the resolution, the greater the level of detail of the map and the greater the extent to which it is possible to distinguish one atom from another in the electron density map. Resolution is typically measured in angstroms (“Å”), which is 1×10-10 m (or 0.0000001 mm). Even at a high resolution, it may still be difficult to detect the position of certain side chains, if they are too mobile (or not well ordered enough).
95 Two relevant crystallography parameters are temperature (or ‘B’) factor, which is a measure of the spread of electron density, and occupancy, which is the fraction of an atom present within a particular site.
96 Computer programs can be used to generate depictions of the structure of a protein or a protein-protein complex from the atomic coordinates. These can be viewed in 3D on a computer. One such computer program, which was widely available as at 23 August 2007, is known as PyMOL.
Mutagenesis
97 Mutagenesis studies are another means by which the PSA may assess whether a particular residue is involved in a protein-protein interaction. In such studies, one or more mutated versions of the antigen are prepared whereby, in each mutated form of the antigen, one residue is mutated from the ‘wild-type’ residue to a different residue. The degree of binding of the antibody to the mutated antigen is then compared to the degree of binding of the antibody to the wild-type antigen.
Assays which measure binding, competition and blocking
98 The Agreed Primer sets out the details of various assays that the PSA can use to measure the binding of an antibody to an antigen, competition for binding as between two antibodies and whether an antibody blocks another molecule from binding with the target antigen.
ELISA
99 ELISA is a technique designed to detect and quantify a protein of interest (e.g., an antigen or antibody). The technique was first designed in the early 1970s.
100 A sample of one protein is immobilised onto the surface of the well in a micro-plate, and then a sample containing another protein is added to the well. Detection of any binding between the two proteins is achieved in a number of different ways. The immobilisation of the first protein can also be achieved a number of different ways, in order to orient the antigen differently.
101 ELISA can be used to measure the binding of an antibody to an antigen or to detect competition between two or more antibodies binding the same antigen. For example, ELISA can be used to determine if an antibody blocks the binding of an antigen (e.g., PCSK9) to its target (e.g., LDLR).
102 There are different forms of ELISA, depending on the purpose of the assay to be undertaken. Direct ELISA involves the immobilisation of an antigen to the surface of a well, and the addition of an antibody which has been conjugated with a detection label. The antibody and antigen are incubated for a period of time to allow for binding, after which point, the well is washed to remove any unbound antibody. A substrate is added, which reacts with the detection label. If any antibody (now bound to the antigen) is present, the substrate reaction with the detection label results in a detectable colour. Typically, the detection methodology involves a colorimetric reaction that can be read by a spectrophotometer. The spectrophotometer detects the intensity of the colour, and the readout is measured in optical density (“OD”) units. A higher OD indicates more antibodies are bound to the antigen in the well. Generally, an antibody that has a stronger affinity for the antigen will have a higher OD than an antibody with a weaker affinity.
103 Indirect Detection ELISA similarly involves the immobilisation of an antigen to a well, the addition of an antibody (“the primary antibody”) and incubation to allow for binding. The well is then washed to remove the unbound primary antibody, and a “secondary antibody” is added and incubated for a period of time to allow the secondary antibody to bind to the primary antibody. The secondary antibody typically carries a label, which generates a colour, typically following a chemical reaction. The colour can be detected and measured, and the binding between the antigen and primary antibody can be calculated from the OD.
104 Sandwich ELISA is an alternative format of Indirect Detection ELISA. It can be used in competition assays, to detect whether an antibody can compete with the binding of another antibody to the same antigen. When used in this way, the surface of the well is coated with the “capture antibody”, and the antigen is added to the well and captured. A “detection antibody”, which carries a label that generates a colour, is then added. According to the Agreed Primer, the detection antibody can bind the antigen:
if the epitope on the antigen to which it binds does not overlap with the epitope which binds to the …capture antibody. If the epitopes overlap, the antibodies compete for binding and the …detection antibody will not bind.
105 Sandwich ELISAs can also be performed using a “premix” protocol, where the capture antibody is immobilised to the well, but the detection antibody is premixed or prebound with the antigen before being added to the well. Again, the primer explains:
Binding of the…detection antibody indicates that the…capture antibody and…detection antibody do not compete because they can bind to the…antigen at the same time. However, if the…detection antibody is not bound, this indicates that the two antibodies are not capable of binding to the…antigen at the same time.
Surface Plasmon Resonance (“SPR”)
106 SPR is used to monitor the interaction between two proteins without the use of labels and colour reactions. Biacore is the trade name for a bench top instrument that measures SPR.
107 In this assay, an automatic sampler injects samples through flow cells on a sensor chip. The sensor chip provides the site where the interaction between an antibody and antigen takes place, measures the mass change on the surface over time and converts it into an SPR signal.
108 The method is used to detect the binding of a soluble antigen to an antibody. The antibody is immobilised onto the sensor chip surface. Next, the antigen is added to the surface of the sensor chip. The binding interaction that takes place between the antibody and the antigen (if there is one) is represented as an increase in response units (“RU”) on a graph (referred to as a “sensorgram”).
109 SPR can also be used to test whether two antibodies compete for binding to a given antigen. One way of detecting competition involves immobilising a first antibody to the sensor chip. The antigen is then flowed over the surface of the chip, and the sensorgram shows an increase in RU that corresponds to the mass of the antigen which is now bound. Then, a second antibody is flowed over the sensor chip. If the first and second antibodies do not compete for binding to the antigen, the sensorgram will show another increase in RU that corresponds to the mass of the second antibody, reflecting that the two antibodies are capable of binding to the antigen at the same time. However, if the first and second antibodies compete for binding to the antigen, there will be no increase in RU when the second antibody is flowed over the sensor chip.
Kinetic Exclusion Assay (“KinExA”)
110 KinExA is a technique for measuring the binding affinity between two proteins, for example, an antigen and antibody. The antigen and antibody are mixed together to form an equilibrium solution, which is then passed through a column to which the antigen has previously been immobilised. Any free antibody that binds to the column is measured by a detection antibody, which carries a fluorescent label. Fluorescence is continuously monitored and represented graphically.
Luminex
111 Luminex is a form of multiplex bead array assay designed to rapidly test a large number of antibody candidates for their binding to a protein target of interest. In one variation of this method, target competition between a capture antibody and a detection antibody is measured. In this assay, target proteins are captured onto spherical beads in suspension. Different coloured beads are premixed with different antibody candidates, such that each coloured bead is coated with a different antibody. The beads are then mixed together with a protein target (e.g., antigen). The ability of the individual antibody candidate to bind to the protein target is measured by a detection antibody carrying a label, using a flow cell to separate the beads. As with the ELISA method, the amount of binding of an antibody to its target can be measured on each bead by fluorescence.
112 In an inverted format, mutated target proteins (e.g., different mutated forms of the antigen) can be bound to the coloured beads, and the assay measures the ability of an antibody to bind to the mutant target protein.
The cholesterol pathway and PCSK9
113 Serum cholesterol levels in humans predict the risk of atherosclerotic cardiovascular disease (“ASCVD”), coronary heart disease, cerebral ischemia and peripheral arterial disease.
114 Serum cholesterol is insoluble in water and cannot be transported in isolation. It is transported in blood as a component of water-soluble lipoproteins. Serum cholesterol is found in all the lipoproteins in the circulatory system. These lipoproteins are termed very low density lipoprotein, low density lipoprotein (referred to above as “LDL”) and high density lipoprotein (“HDL”). LDL cholesterol is responsible for the epidemiological association between ASCVD and serum cholesterol.
115 The uptake of LDL takes place primarily via coated pit regions of the plasma membrane of hepatocytes (liver cells), by the pathway of receptor-mediated endocytosis (internalisation). In the 1980s, Brown and Goldstein identified LDL receptor (referred to above as “LDLR”) and showed it is responsible for removing LDL from the blood.
116 The binding between LDLR and LDL takes place on the outer surface of the plasma membrane of the cell. With or without LDL bound to it, cell surface LDLR is reinternalized by endocytosis. It can either be recycled back to the cell surface or routed to the lysosome, where the LDL is degraded. After LDL degradation, LDLR remains intact and is trafficked back to the cell surface where the cycle is repeated.
117 As I have explained above, PCSK9 is a protein involved in the degradation of LDLR. The antibodies the subject of the Applications target PCSK9, blocking or inhibiting its activity degrading LDLR, thereby increasing the level of LDLR available to degrade LDL.
118 PCSK9 consists of three regions or domains; starting from the N-terminus of the protein there is a “prodomain” followed by a “catalytic domain”, followed by a C-terminal domain (also referred to as the V domain). The two-dimensional primary structure of PCSK9 can be represented as shown below.
THE COMMON SPECIFICATION
Introduction
119 According to the CS, the field of the invention relates to antigen binding proteins (“ABPs”, which include antibodies) that bind to PCSK9 and methods of making and using them. The CS provides some background to PCSK9 including its role in regulating the levels of LDLR.
120 The CS includes a section entitled “Summary of Various Embodiments” which includes a large number of consistory statements that mirror many of the claims, including the Exemplary Claims. These statements include that appearing at CS [0009] which states that “the invention comprises an isolated antigen binding protein that specifically binds to an epitope that is bound by any of the ABPs disclosed herein”. The consistory clauses vary amongst the Applications in accordance with the claims.
121 The various figures included in the CS are identified at CS [0063]-[0159]. These figures include various figures depicting:
(a) the amino acid sequence of PCSK9 (Figures 1A and 1B);
(b) the amino acid sequences of parts of various antibodies (Figures 2A – 3JJJ); and
(c) structural models of PCSK9 and the reference antibodies.
122 The section of the CS entitled “Detailed Description of Certain Exemplary Embodiments” includes definitions of many terms used throughout the CS. I have previously referred to the definition of “epitope” at CS [0233]. I will refer to other definitions in the course of these reasons when addressing the specific topics.
123 The CS refers at [0245] to the amino acid sequence for PCSK9 presented in Figures 1A and 1B, noting that the structure of the protein had recently been solved by two groups. The CS states at [0246] that the ABPs that bind PCSK9 “are provided herein”. According to CS [0250], these ABPs can be used in a variety of therapeutic applications including for treating cholesterol related disorders including hypercholesterolemia, and also in the diagnosis of such conditions.
124 The CS at [0259] describes the general structure of the variable regions of light and heavy chains which include “conserved framework regions (FR)” and CDRs. The CS relevantly states at [0259]:
The CDRs from the two chains of each heavy chain/light chain pair mentioned above typically are aligned by the framework regions to form a structure that binds specifically with a specific epitope on the target protein (e.g., PCSK9).
Production and selection of Table 2 Antibodies
125 CS [0260]-[0262] describes by reference to various figures 32 different examples of antibodies and the amino acid sequences for the variable regions of their light and heavy chains. The MAbs referred to in Table 2 (“the Table 2 Antibodies”), at CS [0261], are a selection of those generated in XenoMouse mice which can produce fully human antibodies (Example 1). The mice were injected with PCSK9 eleven times. Selected mice were then sacrificed and their lymph nodes were used to create hybridomas to produce MAbs capable of binding to PCSK9 (Example 2).
126 Following the production of the hybridomas, a screening process was undertaken (Example 3). The first “primary screen” identified 3104 PCSK9 specific hybridomas of interest. This number was reduced to 2441 following a “confirmatory screen”. Further screening identified 579 MAbs that bound to both mouse and human PCSK9. These MAbs were then screened for binding to a mutant variant of PCSK9 (“D374Y mutant”). This mutant was used because it has a higher binding affinity to LDLR. Over 96% of those antibodies identified as binding to wild-type PCSK9 (i.e., non-mutated) were found to bind to the D374Y mutant. A further assay then tested whether the MAbs could block the D374Y mutant from binding to LDLR, The D374Y screen identified a subset of 384 MAbs that blocked binding between D374Y and LDLR “well”, and 100 which blocked the interaction “strongly”.
127 This assay was repeated and 85 antibodies were found to block the interaction between the D374Y mutant and LDLR greater than 90%. A screen was also performed on those antibodies found to bind to wild-type PCSK9 but not the D374Y mutant, for their ability to block binding between wild-type PCSK9 and LDLR.
128 CS [0422] explains that based on the results of the assays described, “several hybridoma lines were identified as producing antibodies with desired interactions with PCSK9”, and “[l]imiting dilution was used to isolate a manageable number of clones from each line” which were given line numbers (e.g., 21B12) and clone numbers (e.g., 21B12.1). The clones of each line generally behaved the same. The isolated clones were then expanded in hybridoma media, and the concentration and potency of the antibodies to PCSK9 in the supernatants of those cultures were tested. Those hybridomas with the highest titer of antibodies (i.e., highest detected amount) were identified. The selected hybridomas are listed in Table 2. Relevantly, CS [0260]-[0262] sets out:
[0260] Various heavy chain and light chain variable regions are provided herein and are depicted in FIGs. 2A-3JJ and 3LL-3BBB. In some embodiments, each of these variable regions can be attached to the above heavy and light chain constant regions to form a complete antibody heavy and light chain, respectively. Further, each of the so generated heavy and light chain sequences can be combined to form a complete antibody structure.
[0261] Specific examples of some of the variable regions of the light and heavy chains of the antibodies that are provided and their corresponding amino acid sequences are summarized in TABLE 2.
[0262] Again, each of the exemplary variable heavy chains listed in Table 2 can be combined with any of the exemplary variable light chains shown in Table 2 to form an antibody. Table 2 shows exemplary light and heavy chain pairings found in several of the antibodies….
129 As I have alluded to above, the amino acid sequences for the variable regions of the light and heavy chains of the Table 2 Antibodies are included in the figures to the specification. The nucleic acid sequences for the light and heavy chains were first determined by nucleotide sequencing, from which their amino acid sequences were then deduced (Example 9, “Sequence Analysis of Antibody Heavy and Light Chains”). Each of the numbers in Table 2 corresponds to a particular sequenced variable region. For example, the sequence for the light and heavy chain variable regions for 30A4 are found in SEQ ID NO: 5 and SEQ ID NO: 74, respectively, both of which are reproduced as Figure 3EE.
130 The CS also explains that, in addition to combining the light and heavy chains identified in Table 2, other pairings and combinations can be made. For example, CS [0262] continues:
In some instances, the antibodies include at least one variable heavy chain and one variable light chain from those listed in Table 2. In other instances, the antibodies contain two identical light chains and two identical heavy chains. As an example, an antibody or antigen binding protein can include a heavy chain and a light chain, two heavy chains, or two light chains. In some embodiments the antigen binding protein comprises (and/or consists) of 1, 2, and/or 3 heavy and/or light CDRs from at least one of the sequences listed in Table 2 (CDRs for the sequences are outlined in FIGs. 2A-3D, and other embodiments in FIGs. 3CCC-3JJJ and 15A-15D). In some embodiments, all 6 CDRs (CDR1-3 from the light (CDRL1, CDRL2, CDRL3) and CDR1-3 from the heavy (CDRH1, CDRH2, and CDRH3)) are part of the ABP. In some embodiments, 1, 2, 3, 4, 5, or more CDRs are included in the ABP. In some embodiments, one heavy and one light CDR from the CDRs in the sequences in Table 2 is included in the ABP (CDRs for the sequences in table 2 are outlined in FIGs. 2A-3D). In some embodiments, additional sections (e.g., as depicted in FIG. 2A-2D, 3A-3D, and other embodiments in 3CCC-3JJJ and 15A-15D) are also included in the ABP. Examples of CDRs and FRs for the heavy and light chains noted in Table 2 are outlined in FIGs. 2A-3D (and other embodiments in FIGs. 3CCC-3JJJ and 15A-15D). Optional light chain variable sequences (including CDR1, CDR2, CDR3, FR1, FR2, FR3, and FR4) can be selected from the following: 5, 7, 9, 10, 12, 13, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 26, 28, 30, 31, 32, 33, 35, 36, 37, 38, 39, 40, 42, 44, and 46. Optional heavy chain variable sequences (including CDR1, CDR2, CDR3, FR1, FR2, FR3, and FR4) can be selected from the following: 74, 85, 71, 72, 67, 87, 58, 52, 51, 53, 48, 54, 55, 56, 49, 57, 50, 91, 64, 62, 89, 65, 79, 80, 76, 77, 78, 83, 69, 81, and 60.
131 The antibodies described in these paragraphs of the CS (including Table 2) are not said to be embodiments of the invention as claimed. Table 2 identifies what are called “Exemplary Heavy and Light Chain Variable Regions” of 32 antibodies. Which of those antibodies falls within the claims depends on, in the case of claim 1 of 677, whether they bind to an epitope on PCSK9 that includes one or more of the relevant residues and also block binding of PCSK9 to LDLR. This must also be true of the various antibodies generally described in CS [0262] with different arrangements of the exemplary heavy and light chain variable regions.
Residues bound on PCSK9
132 The CS then shifts to a discussion of antibody binding to PCSK9 and the site at which it may bind. It does so by reference to (inter alia) the amino sequence for PCSK9 disclosed in SEQ ID NO: 1 (Figure 1A) and the three domains of the antigen. The CS states at [0264]:
In some embodiments, the ABP binds selectively to the form of PCSK9 that binds to LDLR (e.g., the autocatalyzed form of the molecule). In some embodiments, the antigen binding protein does not bind to the c-terminus of the cataylytic [sic] domain (e.g., the 5. 5-10, 10-15, 15-20, 20-25, 25-30, 30-40 most [sic] amino acids in the c-terminus). In some embodiments, the antigen binding protein does not bind to the n-terminus of the catalytic domain (e.g., the 5. 5-10, 10-15, 15-20, 20-25, 25-30, 30-40 most [sic] amino acids in the n-terminus). In some embodiments, the ABP binds to amino acids within amino acids 1-100 of the mature form of PCSK9. In some embodiments, the ABP binds to amino acids within (and/or amino acid sequences consisting of) amino acids 31-100, 100-200, 31-152, 153-692, 200-300, 300-400, 452-683, 400-500, 500-600, 31-692, 31-449, and/or 600-692. In some embodiments, the ABP binds to the catalytic domain. In some embodiments, the neutralizing and/or non-neutralizing ABP binds to the prodomain. In some embodiments, the ABP binds to both the catalytic and pro domains. In some embodiments, the ABP binds to the catalytic domain so as to obstruct an area on the catalytic domain that interacts with the pro domain. In some embodiments, the ABP binds to the catalytic domain at a location or surface that the pro-domain interacts with as outlined in Piper et al … In some embodiments, the ABP binds to the catalytic domain and restricts the mobility of the prodomain. In some embodiments, the ABP binds to the catalytic domain without binding to the pro-domain. In some embodiments, the ABP binds to the catalytic domain, without binding to the pro-domain, while preventing the pro domain from reorienting to allow PCSK9 to bind to LDLR. In some embodiments, the ABP binds in the same epitope as those surrounding residues 149-152 of the pro-domain in Piper et al. In some embodiments, the ABPs bind to the groove (as outlined in Piper et al.) on the V domain. In some embodiments, the ABPs bind to the histidine-rich patch proximal to the groove on the V domain. In some embodiments, such antibodies (that bind to the V domain) are not neutralizing. In some embodiments, antibodies that bind to the V domain are neutralizing. In some embodiments, the neutralizing ABPs prevent the binding of PCSK9 to LDLR. In some embodiments, the neturalizing [sic] ABPs, while preventing the PCSK9 degradation of LDLR, do not prevent the binding of PCSK9 to LDLR (for example ABP3 1A4). In some embodiments, the ABP binds to or blocks at least one of the histidines depicted in Figure 4 of the Piper et al. paper. In some embodiments, the ABP blocks the catalytic triad in PCSK9.
133 The various antibodies (or ABPs) are described by reference to the domain of PCSK9 and the amino acids on PCSK9 to which the antibody binds or does not bind. Looking at the various ranges of residues specified in CS [0264], it is apparent that every residue of PCSK9 is mentioned, not merely those referred to in the claims.
Antibody variants
134 The CS describes further variants of ABPs at CS [0280]-[0295]. These paragraphs describe the ABPs by reference to their amino acid sequences when compared to those of the Table 2 Antibodies and sequences identified in various figures in the CS. The CS includes the following disclosures at [0280]-[0294]:
[0280] Other antibodies that are provided are variants of the ABPs listed above formed by combination or subparts of the variable heavy and variable light chains shown in Table 2 and comprise variable light and/or variable heavy chains that each have at least 50%, 50-60, 60-70, 70-80%, 80-85%, 85-90%, 90-95%, 95-97%, 97-99%, or above 99% identity to the amino acid sequences of the sequences in Table 2 (either the entire sequence or a subpart of the sequence, e.g., one or more CDR). In some instances, such antibodies include at least one heavy chain and one light chain, whereas in other instances the variant forms contain two identical light chains and two identical heavy chains (or subparts thereof). In some embodiments, the sequence comparison in FIG. 2A-3D (and 13A-13J and other embodiments in 15A-15D) can be used in order to identify sections of the antibodies that can be modified by observing those variations that impact binding and those variations that do not appear to impact binding. For example, by comparing similar sequences, one can identify those sections (e.g., particular amino acids) that can be modified and how they can be modified while still retaining (or improving) the functionality of the ABP. In some embodiments, variants of ABPs include those consensus groups and sequences depicted in FIGs. 13A, 13C, 13F, 13G, 13H, 131 and/or 13J and variations are allowed in the positions identified as variable in the figures …
[0281] In certain embodiments, an antigen binding protein comprises a heavy chain comprising a variable region comprising an amino acid sequence at least 90% identical to an amino acid sequence selected from at least one of the sequences of SEQ ID NO: 74, 85, 71, 72, 67, 87, 58, 52, 51, 53, 48, 54, 55, 56, 49, 57, 50, 91, 64, 62, 89, 65, 79, 80, 76, 77, 78, 83, 69, 81, and 60. In certain embodiments, an antigen binding protein comprises a heavy chain comprising a variable region comprising an amino acid sequence at least 95% identical to an amino acid sequence selected from at least one of the sequences of [the previously mentioned SEQ IDs]. In certain embodiments, an antigen binding protein comprises a heavy chain comprising a variable region comprising an amino acid sequence at least 99% identical to an amino acid sequence selected from at least one of the sequences of [the previously mentioned SEQ IDs].
[0282] In some embodiments, the antigen binding protein comprises a sequence that is at least 90%, 90-95%, and/or 95-99% identical to one or more CDRs from the CDRs in at least one of sequences of [the previously mentioned SEQ IDs]. In some embodiments, 1, 2, 3, 4, 5, or 6 CDR (each being at least 90%, 90-95%, and/or 95-99% identical to the above sequences) is present.
[0283] In some embodiments, the antigen binding protein comprises a sequence that is at least 90%, 90-95%, and/or 95-99% identical to one or more FRs from the FRs in at least one of sequences of [the previously mentioned SEQ IDs]. In some embodiments, 1, 2, 3, or 4 FR (each being at least 90%, 90-95%, and/or 95-99% identical to the above sequences) is present.
[0284] In certain embodiments, an antigen binding protein comprises a light chain comprising a variable region comprising an amino acid sequence at least 90% identical to an amino acid sequence selected from at least one of the sequences of SEQ ID NO: 5, 7, 9, 10, 12, 13, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 26, 28, 30, 31, 32, 33, 35, 36, 37, 38, 39, 40, 42, 44, and 46. In certain embodiments, an antigen binding protein comprises a light chain comprising a variable region comprising an amino acid sequence at least 95% identical to an amino acid sequence selected from at least one of the sequences of [the previously mentioned SEQ IDs]. In certain embodiments, an antigen binding protein comprises a light chain comprising a variable region comprising an amino acid sequence at least 99% identical to an amino acid sequence selected from at least one of the sequences of [the previously mentioned SEQ IDs].
[0285] In some embodiments, the antigen binding protein comprises a sequence that is at least 90%, 90-95%, and/or 95-99% identical to one or more CDRs from the CDRs in at least one of sequences of [the previously mentioned SEQ IDs]. In some embodiments, 1, 2, 3, 4, 5, or 6 CDR (each being at least 90%, 90-95%, and/or 95-99% identical to the above sequences) is present.
[0286] In some embodiments, the antigen binding protein comprises a sequence that is at least 90%, 90-95%, and/or 95-99% identical to one or more FRs from the FRs in at least one of sequences of [the previously mentioned SEQ IDs]. In some embodiments, 1, 2, 3, or 4 FR (each being at least 90%, 90-95%, and/or 95-99% identical to the above sequences) is present.
[0287] In light of the present disclosure, a skilled artisan will be able to determine suitable variants of the ABPs as set forth herein using well-known techniques. In certain embodiments, one skilled in the art can identify suitable areas of the molecule that may be changed without destroying activity by targeting regions not believed to be important for activity. In certain embodiments, one can identify residues and portions of the molecules that are conserved among similar polypeptides. In certain embodiments, even areas that can be important for biological activity or for structure can be subject to conservative amino acid substitutions without destroying the biological activity or without adversely affecting the polypeptide structure.
[0288] Additionally, one skilled in the art can review structure-function studies identifying residues in similar polypeptides that are important for activity or structure. In view of such a comparison, one can predict the importance of amino acid residues in a protein that correspond to amino acid residues which are important for activity or structure in similar proteins. One skilled in the art can opt for chemically similar amino acid substitutions for such predicted important amino acid residues.
[0289] One skilled in the art can also analyze the three-dimensional structure and amino acid sequence in relation to that structure in similar ABPs. In view of such information, one skilled in the art can predict the alignment of amino acid residues of an antibody with respect to its three dimensional structure. In certain embodiments, one skilled in the art can choose not to make radical changes to amino acid residues predicted to be on the surface of the protein, since such residues can be involved in important interactions with other molecules. Moreover, one skilled in the art can generate test variants containing a single amino acid substitution at each desired amino acid residue. The variants can then be screened using activity assays known to those skilled in the art. Such variants can be used to gather information about suitable variants. For example, if one discovered that a change to a particular amino acid residue resulted in destroyed, undesirably reduced, or unsuitable activity, variants with such a change can be avoided. In other words, based on information gathered from such routine experiments, one skilled in the art can readily determine the amino acids where further substitutions should be avoided either alone or in combination with other mutations.
135 The CS describes at [0292]-[0293] further embodiments in which the ABPs include glycosylation and cysteine variants involving the substitution of amino acid residues. The CS also discloses embodiments with variations to the nucleic acid sequences encoding the disclosed ABPs at [0294]:
[0294] In some embodiments, the variants are variants of the nucleic acid sequences of the ABPs disclosed herein. One of skill in the art will appreciate that the above discussion can be used for identifying, evaluating, and/creating ABP protein variants and also for nucleic acid sequences that can encode for those protein variants. Thus, nucleic acid sequences encoding for those protein variants (as well as nucleic acid sequences that encode for the ABPs in Table 2, but are different from those explicitly disclosed herein) are contemplated. For example, an ABP variant can have at least 80, 80-85, 85-90, 90-95, 95-97, 97-99 or greater identity to at least one nucleic acid sequence described in SEQ ID NOs: 152, 153, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151 or at least one to six (and various combinations thereof) of the CDR(s) encoded by the nucleic acid sequences in [the previously mentioned SEQ IDs].
The figures 80-85, 85-90, 90-95, 95-97 and 97-99 in [0294] above appear to be percentage variations (though this is not explicit) from the nucleic acid sequences.
Antibody production techniques
136 The CS also includes a lengthy discussion of the process by which antibodies may be produced, including by immunisation with an antigen such as PCSK9. These techniques are utilised in Examples 1 and 2, leading to the production of the Table 2 Antibodies, although other techniques are also described. The CS includes working examples showing how antibodies 31H4 (Example 4.1), 21B12 (Example 5) and 16F12 (Example 7) were produced by from hybridoma cell lines.
Identification of epitopes on PCSK9
137 The CS includes details of what it refers to as “Exemplary Epitopes”. The CS states at [0334]:
Epitopes to which anti-PCSK9 antibodies bind are provided. In some embodiments, epitopes that are bound by the presently disclosed antibodies are particularly useful. In some embodiments, antigen binding proteins that bind to any of the epitopes that are bound by the antibodies described herein are useful. In some embodiments, the epitopes bound by any of the antibodies listed in Table 2 and FIGs. 2 and 3 are especially useful. In some embodiments, the epitope is on the catalytic domain PCSK9.
138 The CS describes the process of identifying PCSK9 residues involved in or affecting binding with ABPs using arginine/glutamic acid scanning. The technique is described as follows at CS [0338]:
In some embodiments, the domain(s)/region(s) containing residues that are in contact with or are buried by an antibody can be identified by mutating specific residues in PCSK9 (e.g., a wild-type antigen) and determining whether the antigen binding protein can bind the mutated or variant PCSK9 protein. By making a number of individual mutations, residues that play a direct role in binding or that are in sufficiently close proximity to the antibody such that a mutation can affect binding between the antigen binding protein and antigen can be identified. From a knowledge of these amino acids, the domain(s) or region(s) of the antigen that contain residues in contact with the antigen binding protein or covered by the antibody can be elucidated. Such a domain can include the binding epitope of an antigen binding protein. One specific example of this general approach utilizes an arginine/glutamic acid scanning protocol … In general, arginine and glutamic acids are substituted (typically individually) for an amino acid in the wild-type polypeptide because these amino acids are charged and bulky and thus have the potential to disrupt binding between an antigen binding protein and an antigen in the region of the antigen where the mutation is introduced. Arginines that exist in the wild-type antigen are replaced with glutamic acid. A variety of such individual mutants are obtained and the collected binding results analyzed to determine what residues affect binding.
139 Reference is then made to Example 39 which describes one such experiment. In the context of Example 39 the CS states at [0567] that arginine/glutamic acid scanning:
… determines if a residue is part of the structural epitope, meaning those residues in the antigen which contact or are buried by the antibody. Arginine and glutamic acid sidechains are charged and bulky and can disrupt antibody binding even if the mutated residue is not directly involved in antibody binding.
140 The CS states at [0348]-[0350]:
[0348] As noted above, residues directly involved in binding or covered by an antigen binding protein can be identified from scanning results. These residues can thus provide an indication of the domains or regions of SEQ ID NO: 1 (or SEQ ID NO: 303 or SEQ ID NO: 3) that contain the binding region(s) to which antigen binding proteins bind. As can be seen from the results summarized in Example 39, in some embodiments an antigen binding protein binds to a domain containing at least one of amino acids: 207, 208, 185, 181, 439, 513, 538, 539, 132, 351, 390, 413, 582, 162, 164, 167, 123, 129, 311, 313, 337, 519, 521, and 554 of SEQ ID NO: 1 or SEQ ID NO: 303. In some embodiments, the antigen binding protein binds to a region containing at least one of [the above mentioned amino acids] of SEQ ID NO: 1 or SEQ ID NO: 303.
[0349] In some embodiments, the antigen binding protein binds to a region containing at least one of amino acids 162, 164, 167, 207 and/or 208 of SEQ ID NO: 1 or SEQ ID NO: 303. In some embodiments, more than one (e.g., 2, 3, 4, or 5) of the identified residues are part of the region that is bound by the ABP. In some embodiments, the ABP competes with ABP 21B12.
[0350] In some embodiments, the antigen binding protein binds to a region containing at least one of amino acid 185 of SEQ ID NO: 1 or SEQ ID NO: 303. In some embodiments, the ABP competes with ABP 31H4.
141 The amino acid residues referred to in [0348]-[0350] include some of the residues referred to in the Exemplary Claims. As is apparent from what is extracted above, the CS asserts that in some embodiments the MAbs (or ABPs) bind to a region containing at least one of those amino acids. It also asserts that those MAbs compete with the reference antibodies.
142 Antibodies 21B12 and 31H4, the reference antibodies, may be used to determine whether other antibodies bind to the same epitope on PCSK9. This is done by way of binning experiments such as those described in Example 10, the results of which are set out in Table 8.3 at CS [0452]. In these experiments antibodies with the same “binding profile” were grouped together in the same “epitope bin”. As CS [0454] explains, if two MAbs (MAb1 and MAb2) have “a similar epitope on the antigen”, MAb1 will not show binding to the antigen already bound by MAb2, whereas if they each have different epitopes on the antigen, both will still bind. A further “binning” experiment is Example 37, which explains that “[a]s in Example 10, ABPs that compete with each other can be thought of as binding to the same site on the target and in common parlance are said to ‘bin’ together” (at CS [0560]).
143 The results of Example 10 showed that a total of 18 MAbs binned with 21B12 (specifically, clone 21B12.2) and a total of 6 MAbs binned with 31H4.
Neutralisation
144 As previously mentioned, the Exemplary Claims include a claim in which the MAb binds to PCSK9 “and is neutralizing in that an excess of said antibody reduces the quantity of human PCSK9 bound to LDLR in an in vitro competitive binding assay”. The claim does not use the term “neutralising antibody”. Nevertheless, the following discussion in CS [0229] is still relevant to understanding the invention and the mechanism of action of the MAbs described:
The term “neutralizing antigen binding protein” or “neutralizing antibody” refers to an antigen binding protein or antibody, respectively, that binds to a ligand and prevents or reduces the biological effect of that ligand. This can be done, for example, by directly blocking a binding site on the ligand or by binding to the ligand and altering the ligand's ability to bind through indirect means (such as structural or energetic alterations in the ligand). In some embodiments, the term can also denote an antigen binding protein that prevents the protein to which it is bound from performing a biological function. In assessing the binding and/or specificity of an antigen binding protein, e.g., an antibody or immunologically functional fragment thereof, an antibody or fragment can substantially inhibit binding of a ligand to its binding partner when an excess of antibody reduces the quantity of binding partner bound to the ligand by at least about 1-20, 20-30%, 30-40%, 40-50%, 50-60%, 60-70%, 70-80%, 80-85%, 85 90%, 90-95%, 95-97%, 97-98%, 98-99% or more (as measured in an in vitro competitive binding assay). In some embodiments, in the case of PCSK9 antigen binding proteins, such a neutralizing molecule can diminish the ability of PCSK9 to bind the LDLR. In some embodiments, the neutralizing ability is characterized and/or described via a competition assay. In some embodiments, the neutralizing ability is described in terms of an IC50 or EC50 value. In some embodiments, ABPs 27B2, 13H1, 13B5 and 3C4 are non-neutralizing ABPs, 3B6, 9C9 and 31A4 are weak neutralizers, and the remaining ABPs in Table 2 are strong neutralizers. In some embodiments, the antibodies or antigen binding proteins neutralize by binding to PCSK9 and preventing PCSK9 from binding to LDLR (or reducing the ability of PCSK9 to bind to LDLR). In some embodiments, the antibodies or ABPs neutralize by binding to PCSK9, and while still allowing PCSK9 to bind to LDLR, preventing or reducing the PCSK9 mediated degradation of LDLR. Thus, in some embodiments, a neutralizing ABP or antibody can still permit PCSK9/LDLR binding, but will prevent (or reduce) subsequent PCSK9 involved degradation of LDLR.
145 A distinction is drawn here between neutralising antibodies, non-neutralising antibodies and weak neutralisers. There are four Table 2 Antibodies identified as non-neutralising (27B2, 13H1, 13B5 and 3C4) and another three are identified as weak neutralisers (3B6, 9C9 and 31A4). All other Table 2 Antibodies are said to be strong neutralisers of PCSK9. It is also evident, from CS [0229], that blocking the binding site on PCSK9 to which LDLR would bind is not the only means of neutralising PCSK9 activity.
Further experiments undertaken
146 The CS includes as examples further work undertaken to characterise the binding profile of MAbs 16F12, 21B12 and 31H4. Various experiments were undertaken to measure the affinity of each of those MAbs to PCSK9. Measures of affinity using an affinity analysis (BIACore) are presented in Tables 7.1 and 7.2. A different affinity analysis (KinExA) was undertaken on MAbs 16F12 and 31H4 with results presented in Tables 8.1 and 8.2. MAbs 31H4 and 21B12 were also tested in Example 11 for efficacy in blocking D374Y mutant binding to LDLR.
147 A number of the examples in the CS (Examples 13-17) are concerned with the effect of MAb 31H4 and 16F12 on LDLR, serum cholesterol, and HDL using various experiment protocols in mice. Example 18 is concerned with “Epitope Mapping of Human Anti PCSK9 Antibodies”. It describes methods that could be used to determine which residues in PCSK9 are involved in forming, or part of, the epitope of the ABPs described. The methods identified are SPOTs peptide array, combinatorial alanine scanning and “a combination of alanine scanning and/or arginine scanning, antibody FAB/PCSK9 co-crystallization, and limited proteolysis/LC-MS (liquid chromatography mass spec.)”. This example does not contain any results of such experiments.
148 Examples 19-21 are concerned with the use of antibodies, including 31H4, 21B12 and 16F12, as monotherapy or in combination with a statin. Example 22 is concerned with the use of such antibodies as a diagnostic agent. Examples 23-24 are also concerned with the use of antibodies either as monotherapy or in combination with a statin.
149 Example 25 provides details of consensus sequences determined through analyses of CDRs corresponding to the heavy chain and light chain variable regions (VH and VL) of anti-PCSK9 antibodies. CS [0496] asserts the results in Figures 13A-13J “present a large amount of guidance as to the importance of particular amino acids … and which amino acid positions can likely be altered ”.
150 Example 26 provides results of testing the ability of antibodies to lower LDL in mice. The tests were conducted using antibodies 16F12, 21B12 and 31H4. Both 31H4 and 21B12 were said to show significant reductions in LDL cholesterol levels. Antibody 16F12 was less effective. The data is said to be consistent with binding data which shows near equivalent binding affinity between 31H4 and 21B12, and a lower affinity of 16F12 to human PCSK9.
151 Example 27 (read with Figures 16A and 16B) show that antibodies 31H4 and 21B12 bind to the ProCat domain of PCSK9. This was determined through combining either the ProCat or V domains of PCSK9 with 31H4 and 21B12 and analysing whether binding had occurred using Native PAGE for complex formation.
152 Example 28 presents the solved crystal structure of the PCSK9 ProCat domain bound to the LDLR EGFa domain. CS [0504] explains that this interaction “appears to occur across a surface of PCSK9 between residues D374 and S153” as depicted in Figure 17 reproduced below (identified according to their position in SEQ ID NO: 3).
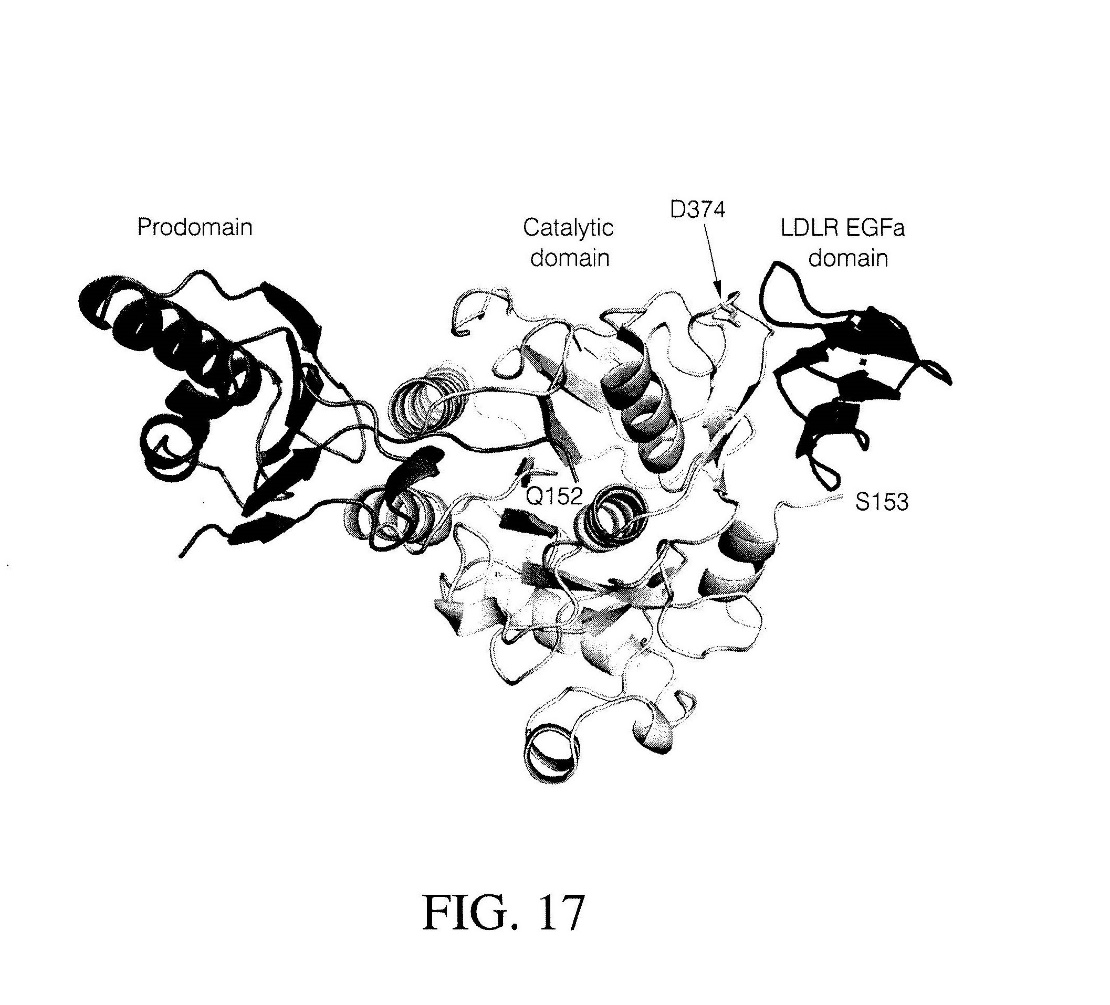
153 The CS continues at [0505]-[0507]:
[0505] Specific core PCSK9 amino acid residues of the interaction interface with the LDLR EGFa domain were defined as PCSK9 residues that are within 5 Å of the EGFa domain. The core residues are as follows: S153, I154, P155, R194, D238, A239, I369, S372, D374, C375, T377, C378, F379, V380, and S381.
[0506] Boundary PCSK9 amino acid residues of the interaction interface with the LDLR EGFa domain were defined as PCSK9 residues that are 5-8 Å from the EGFa domain. The boundary residues are as follows: W156, N157, L158, E159, H193, E195, H229, R237, G240, K243, D367, I368, G370, A371, S373, S376, and Q382. Residues that are underlined are nearly or completely buried within PCSK9.
[0507] As will be appreciated by one of skill in the art, the results from this example demonstrate where PCSK9 and EGFa interact. Thus, antibodies that interact with or block any of these residues can be useful as antibodies that inhibit the interaction between PCSK9 and the EGFa domain of LDLR (and/or LDLR generally). In some embodiments, antibodies that, when bound to PCSK9, interact with or block any of the above residues or are within 15-8, 8, 8-5, or 5 angstroms of the above residues are contemplated to provide useful inhibition of PCSK9 binding to LDLR.
154 Specific core residues of PCSK9 are said to be within 5 Å (i.e. within 5 x 10-10 m) of the EGFa domain of LDLR. Boundary residues are said to be PCSK9 residues within 5-8 Å from the EGFa domain.
155 Example 29 is concerned with interaction between antibody 31H4 and PCSK9, and presents the solved crystal structure of the full length of PCSK9 and a fragment of 31H4. Binding is shown in Figures 18A and 18B, as occurring in the region of the catalytic domain of PCSK9 with contacts said to be occurring with amino acids in both the prodomain and catalytic domain. Various core and boundary residues on PCSK9 of the “interaction interface” between PCSK9 and 31H4 are identified.
156 Example 30 provides the same type of information for antibody 21B12, and presents the solved crystal structure of PCSK9 ProCat domain bound to fragments of 31H4 and 21B12. The crystal structure, depicted in Figures 19A and 19B, is said to show that 21B12 and 31H4 have distinct binding sites and that both can bind to PCSK9 simultaneously. CS [0514]-[0515] identifies the specific core and boundary PCSK9 residues for the interaction interface with 21B12. These include the six residues identified in claims 1 and 5 of 677. CS [0516] also refers residues within 5 Å or 5-8 Å of the PCSK9 protein for the heavy and light chains of antibody 21B12. CS [0517] states:
As will be appreciated by one of skill in the art, the results from Example 30 demonstrate where antigen binding proteins to PCSK9 can interact on PCSK9 and still block PCSK9 from interacting with EGFa (and thus LDLR). Thus, antigen binding proteins that interact with any of these PCSK9 residues or that block any of these residues can be useful as antibodies that inhibit the interaction of PCSK9 and EGFa (and LDLR accordingly). Thus, in some embodiments, antibodies that interact with any of the above residues or interact with residues that are within 5 Å of the above residues are contemplated to provide useful inhibition [of] PCSK9 binding to LDLR. Similarly, antigen binding proteins that block any of the above residues (which can be determined, for example, via a competition assay) can also be useful for inhibition of PCSK9/LDLR interaction.
157 Although Example 30 identifies some residues of antibody 21B12, I understand the residues referred to in CS [0517] are the PCSK9 core residues identified in CS [0514] and the boundary residues identified in CS [0515].
158 Example 31 is an examination of the PCSK9/31H4/21B12 complex overlaid on the PCSK9/ EGFa structure. It is said to show that antibody 21B12 directly interacts with a subset of amino acid residues specifically involved in binding to the LDLR EFGa domain. Both 21B12 and 31H4 are said to partially overlap with the position of the EGFa domain of LDLR, and sterically interfere with its binding to PCSK9. Table 12 identifies various residues for PCSK9 “from interfaces in common in both the core region and boundary region for the different binding partners”.
159 Example 32 is directed to the structural interaction between full length PCSK9 and a full length representation of LDLR. The investigation described in this example was said at CS [0523] to show that:
… antigen binding proteins that bind to PCSK9 can also inhibit the interaction between PCSK9 and the LDLR by clashing with various regions of the LDLR (not just the site at which LDLR and PCSK9 interact).
160 Examples 33-35 discuss how the various crystal structures and complexes examined in prior examples were made, and how data was collected and structural determinations made. Example 36 examines the crystal structure of the 31A4/PCSK9 complex. 31A4 was determined to bind to the PCSK9 V domain. CS [0551] states:
Analysis of the structure shows where this antibody interacts with PCSK9 and demonstrated that antibodies that do not bind to the LDLR binding surface of PCSK9 can still inhibit the degradation of LDLR that is mediated through PCSK9 (when the results are viewed in combination with Example 40 and 41 below) …
161 Examples 37-39 provide details of “Epitope Mapping”. As alluded to above, Example 37 describes further binning experiments in addition to Example 10. Although a different experimental protocol was followed, the object of the experiment was again to determine which antibodies “bin” together. The data from the experiment is reproduced in Table 37.1. It identifies 18 MAbs that bin with 21B12 (Bin 1), nine MAbs that bin with 31H4 (Bin 3), and three MAbs that bin with both 21B12 and 31H4 (i.e., compete with bins 1 and 3). 39 MAbs were analysed for the purpose this example.
162 The Example 37 discussion refers to a number of factors that influence binding and the outcome of competition assays. It notes that the order in which the MAbs are deployed can affect the outcome of the competition assay. This can be because the binding of a first MAb to an antigen (e.g. PCSK9) can produce conformational changes in the antigen which affect binding of the second MAb. CS [0565] also notes that:
… epitopes which overlap but do not completely occlude each other may allow for the second ABP to still have enough high-affinity interactions with the target to allow binding. ABPs with a much higher affinity may have a greater ability to bump a blocking ABP out of the way. In general, if competition is observed in either order the ABPs are said to bin together, and if both ABPs can block each other then it is likely that the epitopes overlap more completely.
163 Example 38 uses “Western Blot” analysis to demonstrate whether the epitopes for 13H1, 3C4 and 31A4 are linear or conformational. It was concluded that 13H1 bound to a linear epitope on the prodomain of PCSK9 whereas the other two antibodies appeared to bind to conformational epitopes. It is not apparent from the CS if any other MAbs were tested other than the three to which I have referred.
164 In Example 39, five MAbs representative of each “bin” were selected from Example 37 for further epitope analysis using arginine/glutamic acid scanning (a form of mutagnesis), as described above. In Example 39, comparisons are made between binding of the selected antibodies to the PCSK9 mutants (i.e. those with modified sidechains) and wild-type PCSK9. The mutated residues are identified in Table 39.1 and Fig 26. Both “EC50” and “Bmax” values are used in Example 39 to assess whether a particular residue is part of the structural epitope for the relevant MAb.
165 EC50 is a measurement of the concentration of the protein required to achieve 50% of the maximum binding signal over a range of concentrations tested. The lower the EC50 (i.e., the lower the antibody concentration required to achieve 50% of the maximum binding signal), the stronger the binding. An EC50 “hit” was recorded when the EC50 for binding to the wild-type PCSK9 was significantly lower than the EC50 for binding to the mutant PCSK9.
166 Bmax is a measurement of the maximum binding signal of the test antibody to PCSK9. The Bmax of the test antibody when bound to wild-type PCSK9 was compared to the Bmax of the test antibody when bound to the mutant form of PCSK9. A residue was registered as a Bmax “hit” when the Bmax of the test antibody bound to mutant PCSK9 was at least 70% lower than the Bmax of the test antibody bound to wild-type PCSK9. Alternatively, a hit was registered where the Bmax of the test antibody binding to the mutant PCSK9 was at least 50% lower than the Bmax of other antibodies to the same mutant, when all other antibodies had a Bmax of at least 40% to wild-type PCSK9.
167 Professor Petsko stated:
In my opinion, these are stringent cut-offs (set to require a large change in binding) that are likely to miss “real” hits (false negatives) but minimize false positives. Although the EC50 and Bmax are different measurements, they are obtained by analyzing the same data sets. I note that the residues identified through the Bmax methodology are different from those identified through the EC50 methodology. This is not surprising to me. Both methods measure different parts of the data curve. When analyzing the results of arginine scanning, it is important to look at the results of both of these methods in order to get a complete list of hits.
168 The EC50 and Bmax “hits” are recorded in Table 39.5, reproduced below, for each of the five test antibodies and mapped onto crystal structures depicted in Figures 27A-27E. From Table 39.5 the amino acid substitution can be identified together with the location of the mutated residue on SEQ ID NO: 1. For example, the Bmax results for antibody 12H11 show that the serine at position 123 on SEQ ID: NO 1 (equivalent to position 153 on SEQ ID NO: 3) has been replaced with arginine. This is one of the residues referred to in the Epitope and Residue Claims.
169 The CS states at [0594]:
While there were approximately a dozen mutants that could have been expected to have an effect on binding (based upon the crystal structure), the present experiment demonstrated that, surprisingly, they did not. As will be appreciated by one of skill in the art, the results presented above are in good agreement with the crystal structures and PCSK-9's [sic] binding of these antibodies. This demonstrates that the provided structural and corresponding functional data adequately identifies the key residues and areas of interaction of the neutralizing ABPs and PCSK9. Thus, variants of the ABPs that possess the ability to bind to the above noted areas are adequately provided by the present description.
170 There was a debate between the experts as to the reliability of the EC50 and Bmax data and its usefulness in mapping PCSK9 epitopes. Most of the experts agreed that arginine scanning can provide data that identifies residues in the structural epitope. Professor Lopez considered it a very useful method, particularly when used in combination with crystallography. Professor Petsko’s evidence was to the same general effect, though, in his view, the results of this technique must always be viewed in light of the crystal structure and other available information. Professor Hudson considered that the arginine scanning methodology used in Example 39 was a valid method of determining residues of the structural and functional epitope of an antigen bound by an antibody. In particular, he agreed with the statement appearing at CS [0348] (set out above) that residues directly involved in binding or covered by an antigen binding protein can be identified from scanning results and that these residues can thus provide an indication of the domain or regions of PCSK9 that contain the binding regions to which an MAb binds. He agreed with Professor Lopez that arginine scanning is a useful screen to determine interactions between residues when used in combination with crystal structures.
171 Professor M Parker drew attention to what he considered to be inconsistencies in the EC50 and Bmax data, and poor correlation between the results of the arginine scanning experiment with the results of the crystallography experiments recorded in the CS, as well as Professor Petsko’s analysis. Professor Mahler drew attention to a number of limitations with respect to arginine scanning which he identified in the JER as follows:
Firstly, the mutation might disrupt the direct physico-chemical interaction between residues which contributes to the binding.
Secondly, the mutation may be proximal to the residues contributing to the direct physico-chemical interaction but change the juxtaposition of the two residues such that they no longer interact.
Thirdly…the mutation may be distal to the residues contributing to the direct physico-chemical interaction, but might affect the native structure of PCSK9 such that the juxtaposition of residues that are involved indirect [sic] physico-chemical interaction, is altered.
Fourthly, the mutation may be advantageous and actually enhance the binding affinity; for example, an arginine may be able to form a salt bridge with a negatively charged residue on the antibody or produce subtle changes in protein structure, leading to changes in interactions at a number of residues.
172 However, in Professor Hudson’s view, the CS provides techniques to avoid false negative and positive hits, although he pointed out that the inconsistency between the EC50 and Bmax results indicated some false negatives.
173 I accept that there are limitations with arginine scanning when used for epitope mapping. However, no expert suggested that it was perfect or that it did not have limitations. Professors Petsko, Lopez and Hudson considered that it was one of several techniques that can be employed in combination to identify residues of an epitope. I accept that evidence which is consistent with the approach taken in the CS and the statement appearing in CS [0348].
174 Example 40 is titled “PCSK9 Domain Binding Assay”. The results of the assay are presented in Figures 28A and 28B, which show the domain on PCSK9 to which each of 31 different MAbs bind (i.e., the Table 2 Antibodies, excluding 25A7). Professor Lopez’s evidence explains that of those 31, 28 bound to the ProCat domain. He also observed that, consistent with the data obtained from Example 37, all of the MAbs shown to bin with either 21B12 or 31H4 (in the experiment conducted as Example 10, as I understand it) were found to bind to the ProCat domain of PCSK9.
175 Example 41 demonstrates how to identify and characterise an antibody that is non-competitive with LDLR for binding with PCSK9 but is still neutralising toward PCSK9 activity. The experiment was performed on MAb 31A4 which was found to display significant PCSK9 neutralising ability without blocking the PCSK9/LDLR binding interaction.
SECTION 40
176 It was common ground that the provisions of the Act as they stood prior to amendment by the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth) (“the RTB Act”) applied in this proceeding, because the requests for examination for all five Applications were filed on 11 April 2013.
177 Section 40 of the Act as it stood prior to amendment by the RTB Act provides:
40 Specifications
(1) A provisional specification must describe the invention.
(2) A complete specification must:
(a) describe the invention fully, including the best method known to the applicant of performing the invention; and
(b) where it relates to an application for a standard patent–end with a claim or claims defining the invention; and
(c) where it relates to an application for an innovation patent–end with at least one and no more than 5 claims defining the invention.
(3) The claim or claims must be clear and succinct and fairly based on the matter described in the specification.
(4) The claim or claims must relate to one invention only.
FAILURE TO DEFINE THE INVENTION
The function of the claims
178 Sanofi’s principal attack upon the Applications was based on what it says is the failure of the claims to define the invention. It relied on s 40(2)(b) of the Act as imposing a specific requirement that the claims define the invention. Sanofi says that lack of clarity and lack of definition often intersect and overlap but that lack of definition is a conceptually distinct ground of invalidity and should be treated as such.
179 Another view of s 40(2)(b), preferred by Dr Bodkin in Patent Law in Australia 4th ed, Thomson Reuters, 2024 (“Bodkin”) at [22910], is that the purpose of that provision “is purely to ensure that the specification includes at least one claim and to set out the function of each claim, namely to define what it is that the patentee has exclusive right to, during the term of the patent”. The learned author’s opinion is based on the view that “[i]t is difficult, if not impossible, to conceive of a situation in which a claim could fail to define the described invention but nevertheless be clear, succinct and fairly based on … the disclosure”.
180 The overall effect of s 40(2)(b) and (3) is clear: a standard patent must end with at least one claim “defining the invention” in a manner that is clear and succinct and fairly based on the matter described in the specification. The function of a claim was identified by McTiernan J in AMP Inc v Utilux Pty Ltd (1971) 45 ALJR 123 as follows at 128:
The description of the invention is not the definition of it. “A claim is a portion of the specification which fulfils a separate and distinct function. It, and it alone, defines the monopoly; and the patentee is under a statutory obligation to state in the claims clearly and distinctly what is the invention which he desires to protect”: see Lord Russell of Killowen in E.M.I. Ltd. v. Lissen Ltd (1939), 56 R.P.C. 23, at p. 41.
181 The distinction between the requirement that a claim define the invention and that it be clear was recognised by Jessup J in Albany Molecular Research Inc v Alphapharm Pty Ltd (2011) 90 IPR 457 (“AMR v Alphapharm”). As his Honour observed at [174]:
…I accept that, from a verbal or grammatical viewpoint, the expression “substantially pure” is not unclear. I accept also that “any purely verbal or grammatical question that can be resolved according to ordinary rules for the construction of written documents, does not, once it has been resolved, leave uncertain the ambit of the monopoly claimed”: Welch Perrin and Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610. But the concern of s 40(3), in my view, is not confined to such matters. The purpose of a claim is to define the invention (s 40(2)(b)), and it is in that context that the requirement of clarity is to be considered. In my view, that requirement is that a claim must clearly define the invention, so far as it relates to the matter claimed. A claim which is a model of verbal or grammatical clarity may none the less fail the test of this requirement if it leaves the definition of the boundaries of the invention uncertain or variable.
182 It is the function of the claims to fix the boundaries of the patentee’s monopoly: Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610. As Beach J observed in Meat & Livestock v Cargill at [940]:
A claim will be bad if it fails to define the monopoly claimed so that the skilled addressee of the patent can know the exact boundaries of the area within which they will be trespassers.
183 In the context of s 40 of the Patents Act 1952 (Cth) (“the 1952 Act”), Barwick CJ and Mason J appear to have accepted that the requirement that a claim define the invention was distinct from the requirement that the claim be clear: see Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 (“Interlego”) at 475 where their Honours refer to two grounds of invalidity upheld by the trial judge (Stephen J) based on the failure of the claim to comply with s 40 “in that it did not define the invention and was not clear”. See also, Atlantis Corporation Pty Ltd v Schindler (1997) 39 IPR 29 (“Atlantis”) at 36 per Lockhart J and at 50 per Wilcox and Lindgren JJ, Terrell on the Law of Patents (13th ed, Sweet & Maxwell, 1982) at 3.12 referring to s 14 Patents Act 1977 (UK), s 4(3)(c) of the Patents Act 1949 (UK) which required that every complete specification “shall end with a claim or claims defining the scope of the invention claimed” and s 32(1)(i) of that act allowing for revocation if “the scope of any claim of the complete specification is not sufficiently and clearly defined ”.
184 The requirement that the scope of a claim be “sufficiently and clearly defined” was considered by the English Court of Appeal in General Tire & Rubber Company v Firestone Tyre & Rubber Company Limited [1972] RPC 457 (“General Tire”). The Court said at 515-516:
It is clear in our judgment that the question whether the patentee has sufficiently defined the scope of his claims is to be considered in relation to the facts of each case, that allowance is to be made for any difficulties to which the circumstances give rise, and that all that is required of the patentee is to give as clear a definition as the subject matter admits of. It is also clear in our judgment that, while the court is to have regard to all the relevant facts, the issue of definition is to be considered as a practical matter and little weight is to be given to puzzles set out at the edge of the claim which would not as a practical matter cause difficulty to a manufacturer wishing to satisfy himself that he is not infringing the patent. We accept also that definition of the scope of a claim is not necessarily insufficient because cases may arise in which it is difficult to decide whether there has been infringement or not provided the question can be formulated which the court has to answer in deciding the issue of infringement.
Claims limited by result
185 Each of the Exemplary Claims is limited by an “ultimate result”, wherein the isolated MAb:
(1) “blocks binding of PCSK9 to LDLR” (claims 1 and 5 of 677, claim 1 of 689);
(2) “competes for binding to PCSK9 with an antibody” comprising certain features (claim 23 of 677);
(3) “binds to human PCSK9 and reduces binding between human PCSK9 and an EGFa domain of LDLR, wherein said monoclonal antibody competes for binding to PCSK9 with an antibody” comprising certain features (claim 1 of 685); and
(4) “binds to human PCSK9 and is neutralizing in that an excess of said antibody reduces the quantity of human PCSK9 bound to LDLR in an in vitro competitive binding assay, wherein said monoclonal antibody competes for binding to PCSK9 with an antibody” comprising certain features (claim 1 of 748);
(5) “reduces binding between PCSK9 and [an] EGFa domain of LDLR” (claims 1, 2, 10 and 13 of 751).
186 The Exemplary Claims which are Epitope and Residue claims are limited by a further “functional requirement”, that the MAb:
(a) in the case of the Epitope Claims, binds to (or recognises) an epitope on PCSK9 that includes one or more of the identified amino acid residues; and
(b) in the case of the Residue Claims, binds to one or more of the identified amino acid residues.
187 Both the “ultimate result” limitation and “functional requirement” limitation are limitations by result. Purpose and result (or effect) can enter into a claim to limit its scope or to assist in defining the physical attributes of the product the subject of the claim. The claims in issue are all concerned with physico-chemical or biological results or effects. Bodkin sets out the following at [21980] regarding inventions that are defined by a result or effect rather than a physical structure:
It is not an uncommon practice to define either a feature in a claim or the whole combination claimed, by defining the result or effect that the feature or combination is to create rather than by defining the physical arrangement or structure of the feature. Indeed defining a feature by reference to a result may in some circumstances represent the only, or the only practicable, way of defining a structure or composition. There is no doubt that claiming by reference to a result to be achieved may be a legitimate way of describing the physical characteristics of the claimed invention or some part of it, even if it is not clear from the claim what is the purpose to be served by the specified result. Defining an invention, or some part of it, by reference to a result to be obtained will seldom lead to a lack of clarity in a claim, provided that in the case of an article the defined result introduces a limitation into the claim that is sufficient to characterise the construction of the article: that is, the claim will not lack clarity provided it is possible to distinguish clearly articles in which the defined result is obtained from those in which it is not.
(Footnotes omitted)
As Dr Bodkin recognises, the authorities establish that a claim may be made by reference to a result if, in the case of an article, the limitation is sufficient to characterise the construction of the article claimed. Dr Bodkin suggests that a claim will meet this requirement where one is able to distinguish between those items which fall within the patentee’s monopoly, and those which do not. Although that understanding is expressed in the context of the clarity requirement, the authorities discussed below indicate that it is equally apposite to claim definition.
188 Interlego was concerned with the validity of a claim to toy building blocks. The relevant claim included a requirement that “the positions and dimensions of the secondary projections [on the toy blocks] relatively to the positions and dimensions of the primary projections [on the toy blocks] are such that, in a pair of assembled blocks the lateral face or faces of at least one primary projection of one block will be clamped against the lateral face or faces of at least one secondary projection of the adjacent block”. Justice Stephen (the trial judge) concluded that the claim did not comply with s 40 of the 1952 Act in that it did not define the invention and was not clear. His Honour found that the claim had failed to identify “the exact boundaries of the area over which monopoly is claimed” because the claim only identified two of the four surfaces involved in the clamping action. His Honour observed at 471:
… No doubt, as was exemplified in No-Fume Ltd. v. Frank Pitchford & Co. Ltd. [(1935) 52 RPC 231], a claim may validly be limited merely by the result to be attained but, as is said in Terrell on the Law of Patents, 11th ed., (1965), p. 96, this may only be done if “the limitation is ‘sufficient to characterise the construction of the article claimed’ Mullard Radio Valve Co. Ltd. v. British Belmont Radio Ltd. [(1938) 56 RPC 1, at p 16].” Here the limitation, by reference to the result, the attaining of a clamping effect between two projections, does not characterize the construction of the toy building blocks.
In Raleigh Cycle Co. Ltd. v. H. Miller & Co. Ltd. [(1946) 63 RPC 113] Vaisey J., at first instance, gathered together a number of passages from the cases defining the function of a claim and certain of these, culled from the judgment of Lord Russell of Killowen in Electric & Musical Industries Ltd. v. Lissen Ltd. [[1938] 4 All ER 221; 56 RPC 23], Lord Normand described, on appeal [[1948] 1 All ER 308 at p 319] as an authoritative exposition of the function of the claim in a specification. It suffices, for my purpose, to refer specifically to one of these passages, in which it is said [[1938] 4 All ER at p 224; 56 RPC at p 39]:
“The function of the claims is to define clearly and with precision the monopoly claimed, so that others may know the exact boundaries of the area within which they will be trespassers."
In the present case claim 1 fails in this respect, it nowhere defines with clarity or precision, or indeed at all, the exact boundaries of the area over which monopoly is claimed. On the contrary the matter is left quite indefinite …
189 Chief Justice Barwick and Justice Mason came to a different view concluding that the claim, properly construed, required that the clamping of the unidentified surfaces be provided by the walls of the block and/or another secondary projection. Their Honours observed at 480:
The claim asserts a monopoly limited by reference to the result of incorporating in the construction of the hollow blocks the suggested relationship between primary and secondary projections, the result being that at least one primary projection is “clamped against” at least one secondary projection. It is permissible to limit a claim by reference to result (see No-Fume Ltd. v. Frank Pitchford & Co. Ltd. [(1935) 52 RPC 231]), so long as, in the case of an article, the limitation is “sufficient to characterize the construction of the article claimed” (Mullard Radio Valve Co. Ltd. v. British Belmont Radio Ltd. [(1938) 56 RPC 1 at p 16]). As we have observed, the claim is in our opinion sufficiently limited.
190 The word “construction” in the phrase “sufficient to characterize the construction of the article claimed” is used as a noun not a verb. The question is whether the claim sufficiently describes the properties of the claimed article such that the boundaries of the monopoly are clearly defined.
191 Mr Shavin KC, who appeared for Sanofi, submitted that each of the Exemplary Claims did not sufficiently define the structure of a MAb either by reference to its amino acid sequence or otherwise. He also submitted that the claims did not permit others to know whether a MAb that they proposed to make was either inside or outside the claims in advance of making it. Referring to Lord Russell of Killowen’s well-known statement in Electric & Musical Industries Ltd v Lissen Ltd (1938) 56 RPC 23 (“EMI v Lissen”) at 39, Mr Shavin submitted that his Lordship was there referring to the need for claims to define the monopoly claimed so that others may know in advance whether what they propose to do will trespass on the patentee’s monopoly.
192 In support of the latter submission Mr Shavin KC also referred to the statement of Stephen J in Monsanto Co v Commissioner of Patents (1974) 48 ALJR 59 (“Monsanto”) at 60. The issue in that case was whether the claim language lacked clarity (rather than definition) due to the use of the adjective “substantial” in the phrase “any substantial effect as a cooling medium” which was said by the respondent in that case to make it impossible to determine the limits of the claim. Stephen J rejected that argument, holding (at 60) that “[t]here will … be no difficulty in a third party ascertaining whether or not what he proposes to do falls within the ambit of the claim …”. Mr Shavin seized on his Honour’s use of the words “what he proposes to do” in support of his argument that the claim must define the monopoly in terms which permit others to know whether a product will infringe in advance of it being made.
193 In its opening submissions Sanofi submitted that the claims do not define any invention:
… because they do not define…“an…antibody”. They do not elucidate the amino acid sequence (i.e., the structure) … of any antibody or any fragment thereof …Therefore, the claims fail to define the structure of the very thing that is said to have been invented, and by which the desired result might be achieved.
Ultimately, Sanofi contended in its closing submissions that none of the Exemplary Claims defined any MAb by reference to the amino acid sequence of its variable domain, nor did any aspect of the Exemplary Claims enable the PSA to discern the amino acid sequence of any claimed antibody. Instead, the claimed MAbs are, Sanofi asserted, defined solely and indirectly by reference to the antigen PCSK9 (in the case of the Epitope Claims), or residues on PCSK9 (in the case of the Residue Claims), or both PCSK9 and a reference antibody (in the case of the Competition Claims).
194 Putting aside Sanofi’s clarity complaint in relation to the Competition Claims, Sanofi has not submitted that the PSA is unable to determine, by completing experiments after the generation of the antibody, whether such an antibody falls within the Exemplary Claims. While it was open to Sanofi to make such an argument based on its Amended Notice of Appeal (“ANOA”), the relevant paragraphs in the ANOA are not those relied upon or pressed in its closing written submissions.
195 Amgen does not accept that the claims must define the scope of the monopoly so that others will know in advance whether what they propose to make will fall within the scope of the claims. Amgen explained its position in written submissions as follows:
Ultimately, the question is a simple one. Can a person skilled in the art determine whether the thing they have or the act they have done, falls within the claim, including by, if necessary, routine experimentation? Any specific structural or functional limitation should allow the person skilled in the art to sufficiently “characterise the construction of the article claimed” by reference to its inherent characteristics.
196 On Amgen’s case, a claim defines the invention sufficiently if it permits the skilled addressee to ascertain whether the product they have made is inside or outside the claims. Amgen says that it is not necessary that this can be done before the product is made. Nor does Amgen accept the basic premise of Sanofi’s submission which requires that any claimed MAb be defined by reference to its amino acid sequence or a part thereof.
Consideration
197 Section 13(1) of the Act confers on the patentee the exclusive right to exploit the invention during the term of the patent. The claim defines the scope of the invention and, therefore, the scope of the patentee’s exclusive rights or, as it is often described, the patentee’s monopoly. A claim defines the scope of the monopoly if, with the requisite degree of clarity, it defines the invention. In the language of Lord Russell in EMI v Lissen at 39, which I have referred to above:
The function of the claims is to define clearly and with precision the monopoly claimed, so that others may know the exact boundaries of the area within which they will be trespassers. Their primary object is to limit and not to extend the monopoly. What is not claimed is disclaimed.
198 Mr Shavin KC’s argument relies on the practical difficulties that may face a person wishing to make a MAb, without knowing in advance of undertaking the work necessary whether what they ultimately make will be inside or outside the claims. However, in my opinion, those difficulties are not attributable to a lack of claim definition. Rather, the difficulties relied on by Sanofi arise from the fact that it may not be possible to determine whether a particular MAb is within the scope of the claims until it has been fully ascertained and tested to determine where it binds to the antigen. The person will know from their reading of the claims that a MAb having the specified properties will infringe the claims. They may not know whether a particular MAb will infringe until after it has been generated and tested. But it does not follow the scope of the invention has not been defined or that the PSA does not know the boundaries of the forbidden territory.
199 Relevantly, at [21980] of Bodkin, Dr Bodkin expresses the view that:
…it is not objectionable for a claim to include reference, implicitly or explicitly, to a method of testing which is needed to demonstrate that the result defined in the claim is achieved, provided the test method is adequately described in the body of the specification. The same applies to a feature of a claim whose properties are defined by reference to the result of conducting a specified test …
Mr Shavin KC’s argument under this ground is not that the method of testing is unclear or ambiguous but instead that defining the MAb by reference to results obtained through testing after generation is impermissible. In my opinion Mr Shavin’s argument is contrary to authority.
200 With regard to Stephen J’s observation in Monsanto at 60, his Honour was concerned in that case with an issue as to the meaning of a particular phrase which was said to be insufficiently precise, at least at the margins. The question in Monsanto was whether the claim lacked clarity. Notwithstanding the way in which Stephen J expressed himself, I do not understand his Honour to be saying that a claim does not define the invention unless it can be said in advance of making a product whether or not it will infringe. The point on which Sanofi’s argument relies was neither argued nor determined in Monsanto.
201 Mr Shavin KC’s submissions focused on the statement from Interlego at 480 to which I have referred adopting language found in Mullard Radio Valve Co Ltd v British Belmont Radio Ltd (1938) 56 RPC 1 (“Mullard”) at 16 per Sir Wilfrid Greene MR. Mullard concerned an application to amend (inter alia) a claim for an apparatus (claim 2) which had been held invalid in previous litigation. The proposed amendment was allowed by the trial judge (Morton J) but disallowed by the Court of Appeal. The proposed new claim read:
A discharge tube for amplifying electric oscillations comprising a screening grid between the control grid and the anode, characterised in that an auxiliary electrode is placed between the screening grid and the anode and next to the latter and is directly connected to the cathode so as to be maintained continuously at the cathode potential whereby the increase of the screening grid current at the expense of the anode current will be substantially avoided.
202 The reasons given by the three members of the Court are diverse, but the judgments of Sir Wilfrid Greene MR and Clauson LJ both show they considered the new claims to be bad for want of subject matter. The Master of the Rolls held at 10-11:
Unlike Claim 1, which is a process claim, the amended Claim 2 is, as was the original Claim 2, an apparatus claim and it must stand or fall as such. In such a claim it is permissible in a proper case to define the physical characteristics of the article by a suitable reference to the results which it is to achieve. It is no objection to such a method of definition in cases where it is appropriate that experiment may be required in order that a worker in the art may know whether an article which he has made does or does not infringe the patent. A permissible reference to results is in truth as much a description of the physical characteristics of the article as a reference to its size or strength or chemical composition would be, and in many cases such a reference is the only practical method of describing the article or some characteristic which it possesses. On the other hand, it is not permissible to import into an apparatus claim a method of definition which is appropriate only to a process claim. Thus an article which is not in itself patentable cannot be made the subject of a good apparatus claim merely by pointing out that, if it is used in a particular way or in a particular collocation, it will produce novel and useful results.
203 In this passage his Lordship endorses three important propositions. First, it is permissible in an appropriate case to define the physical characteristics of an article by reference to the results it is to achieve. Secondly, there can be no objection that some experimentation may be required to determine whether the article has such characteristics. Thirdly, in many cases it may not be practical to define the physical characteristics of an article except by reference to a result which it is intended to achieve. Sir Wilfrid Greene MR’s analysis is inconsistent with Sanofi’s interpretation of Stephen J’s judgment in Monsanto. Mullard was not cited by Stephen J in Monsanto, and it therefore seems unlikely that his Honour intended to hold that a claim must define an invention in terms which make it possible to know whether an article will infringe in advance of it being made.
204 Sir Wilfrid Greene MR also observed in Mullard at 15 that “the words ‘control’ and ‘screening’ postulate nothing in the way of physical characteristics, except that the grids … must be capable of acting in the manner indicated”. His Lordship considered that this language was wide enough to cover grids capable of being used in a manner wholly unrelated to the true invention. His Lordship continued (in a passage including what is cited in Interlego) at 16:
Out of respect for the learned [trial] judge, I will explain in a few words the considerations which, in my opinion, make it impossible to support what is the basic finding in his judgment. He holds … that at the date of the Patent anyone skilled in the art would have understood the phrases “screening grid” and “control grid” and that the Specification and drawings would have enabled a competent workman to manufacture a discharge tube having an anode and a control grid with a screening grid placed between them. That such a workman could have constructed a discharge tube with grids capable by their physical construction of acting as screening and control grids I agree; but the difficulty is that a discharge tube could have been constructed in accordance with the directions given in which the same grids were also capable of acting in a different way; in other words, the learned Judge appears to assume in this passage that the words “control” and “screening” are sufficient to confine the grids in question to grids with specific physical characteristics sufficient to differentiate them, and in this I cannot agree with him. That this was his view is, I think, shown by his reference to the words “of large diameter” contained in the Specification in [British Thomson-Houston v. Corona Lamp Works, Ld. (1922) 39 RPC 49]. Those words were words of physical description, and it was held that they were sufficient to characterise the construction of the article claimed. In the present case, if my own reasoning is correct and my view of the evidence well founded, the words "screening" and "control" necessarily import a reference to the manner in which the grids are used and are not sufficient, without the importation of such a reference, to characterise the valve.
205 In relation to the requirement “whereby the increase of the screening grid current at the expense of the anode current will be substantially avoided”, Clauson LJ said at 20:
Since the decision of this Court in the case of No-Fume, Ld., v. Frank Pitchford & Company, Ld. [(1935) 52 RPC 231], it must be taken, at all events in this Court, that an article which is differentiated from articles of a similar type by the requirement that its proportions should be so adjusted as to produce in use a stated result may well be patentable. The problem raised by the words under consideration does not seem to me to be concluded by this consideration.
206 The problem with the language of the claim which his Lordship went on to identify at 20-21 was that it extended to not only new, but also existing configurations of the relevant article, which anyone was free to make. The words of the claim were therefore insufficient to confine the article in question to those which were new.
207 Both the Master of the Rolls and Clauson LJ regarded claim 2 as a claim to an article which could be distinguished from the prior art only by the new use to which the claim contemplated it could be put. That the inventors had conceived a new use to which a known article might be put was not sufficient to make the known article patentable. The references to “screening grid” and “control grid”, were understood to be words referring to the manner in which these components were arranged, but which did not sufficiently characterise the physical characteristics of the article claimed to render it patentable. Since those words did not define the article, but instead its use, and the article itself was not patentable, the claim was bad. But the claim might also have been disallowed for the simple reason that its language was unclear.
208 The decision in Mullard may be contrasted with the decision of the House of Lords in British Thomson-Houston Company Ld. v Corona Lamp Works Ld. (1922) 39 RPC 49. In that case, the Court of Appeal had upheld the trial judge’s conclusion that a claim to an electric lamp consisting of (inter alia) a metal filament of large diameter or cross-section lacked definition. The claim was additionally limited by result “that the filament may be raised to a much higher temperature than is practicable in a vacuum lamp without prohibitive vaporisation or deterioration or excessive shortening of useful life”. The complaint pressed was that there was insufficient definition to determine what constituted a large diameter because “the only way in which a lamp-marker can ascertain whether he is infringing [i.e., whether the largeness of the filament fell within the claim]… is by making experiments [to determine whether the result was obtained]”: per Viscount Finlay at 77, quoting the Master of the Rolls (Lord Sterndale) upholding the reasoning of the trial judge. Viscount Finlay went on to say at 77:
I am unable to agree with the view taken upon this point in the Courts below. The fact that the directions are admitted to be sufficient to enable any competent workman to make a lamp according to the Patent seems to me to be inconsistent with the contention that the ambit of the invention is not sufficiently defined, inasmuch as, in order to carry out the invention by making a lamp, the workman must be able to attach a meaning to the words “large diameter” occurring in the Specification. But, apart from this consideration, it appears to be that there is no ambiguity about the words…The workman is working to get the result which is described in the later lines of Claim 1, and in choosing the degree of largeness he will be guided by the results yielded by particular sizes.
209 The Antibody and Crystallography Experts agreed that:
… the Claims themselves set forth the properties (features) that an antibody must satisfy. In doing so, to someone with our knowledge and experience, the Claims suggest the strategy for determining or the type of experiments needed to determine if an antibody satisfies the Claims.
210 Professors Petsko, Hudson and Lopez agreed that the CS identifies methods to determine whether an antibody had each of the features identified within, inter alia, the Exemplary Claims. Although Professors Mahler and M Parker did not agree with that proposition, their evidence does not persuade me that the PSA would not be able to determine whether a MAb falls within the claims without undue difficulty or invention.
211 First, as to the Epitope and Residue Claims, the Antibody and Crystallography Experts (aside from Professor Parker) agreed that arginine scanning, the method described in Example 39 of the CS, “can provide data that identifies residues in the structural epitope”. Professors Lopez, Petsko and Hudson considered that the technique was useful when viewed in light of X-ray crystallography data. Professor Mahler qualified his answer with reference to the “limitations” of the technique and described instances where the results may correlate to other unintended structural changes or interactions, rather than showing which residues are part of the structural epitope. In oral evidence, he stated he did not agree with the conclusions reached in the CS about the results of Example 39. In the JER prepared by the Antibody and Crystallography Experts, he stated that “crystallography doesn’t identify the functional or structural epitope”.
212 Professor M Parker considered that, while the CS used arginine scanning mutagenesis to identify “residues that might interact”, the results of that experiment poorly correlated with the predictions of interactions obtained from crystallographic analysis. However, in response to a question about arginine scanning, he explained the importance of combining arginine scanning with X-ray crystallography “to reach a convincing conclusion about residue interactions”. Like Professor Mahler, he referred to evidence he had given that was critical of the use of arginine scanning as a “standalone approach” due to unintended results of residue substitution.
213 CS [0594] is consistent with the use of arginine scanning and crystallography together to identify the structural and functional epitopes. In this regard, the CS does not rely on a “standalone approach”. Professor Hudson’s responses to questions raised as part of the JER also indicate that the specification includes methods to account for false positives and negatives obtained through arginine scanning.
214 In relation to the Residue Claims specifically, Professor M Parker considered that X-ray crystallography could be used to predict non-covalent interactions, but that “complementary methods” are required to be convinced of such interactions (like mutagenesis) and the “robustness” of the data must also considered. Although the Crystallography Experts had differing views as to the parameters they would employ when conducting such an analysis, those differences do not suggest that the PSA would be unable to carry out experiments to determine whether an antibody fell within the Residue Claims without undue difficulty or invention.
215 Finally, as to the Competition Claims, Sanofi’s complaint (which arises under the ground of lack of clarity) is not that the PSA would not have known how to conduct a competition assay, but instead that there are several different competition assays which it asserts may produce different results, coupled with a general lack of clarity as to the meaning of the word competes, leading to the absence of a “workable standard”. Putting that complaint aside, the evidence does not indicate that the PSA would not know how to perform blocking and competition assays at the priority date in order to determine whether a particular antibody fell within the Competition Claims. The Antibody Experts all gave evidence indicating their familiarity with such techniques, including Professor Hudson who gave a detailed overview of the competition assays he was aware of in 2007.
216 The Exemplary Claims are not to any therapeutic agent that blocks or inhibits the activity of PCSK9. Implicit in the term “isolated monoclonal antibody” are specific physical characteristics common to all MAbs, including long strings of amino acid residues with distinct physical characteristics (e.g., heavy and light chains arranged in a ‘Y’-shaped structure). Beyond this, the MAbs of the claims are defined by their function including, in the case of the Epitope Claims, their capacity to bind to a structural epitope which includes one or more identified amino acid residues or, in the case of the Residue Claims, to one or more identified residues (which are part of the functional epitope).
217 Leaving aside the lack of clarity from which the Competition Claims are said to suffer, it is not alleged by Sanofi that any of the other claims lack clarity. Nor did Sanofi contend in its closing submissions that the PSA would not be able to determine whether a MAb was within the scope of any of the Exemplary Claims. In any event, the evidence does not persuade me that any of the Exemplary Claims would for that reason be “clearly invalid” if the Applications were to proceed to grant.
218 I am not persuaded that it is necessary for the claims to define the MAbs by their amnio acid sequences or some fragment thereof. What is essential is that the PSA be able to determine whether a particular MAb is within or outside the claims. In this regard, it is important not to conflate the requirement that the claims define the invention with the requirement that the CS describe the invention fully (i.e. sufficiency) arising under s 40(2)(a) of the Act before amendment by the RTB Act. The functional limitations have not been shown to be insufficient to characterise the construction of the relevant MAbs so as to enable the PSA to determine whether any particular MAb falls within the scope of the claims. Sanofi’s opposition based on lack of definition fails.
MANNER OF MANUFACTURE
219 For an invention to be patentable, under s 18(1)(a) of the Act it must be a manner of manufacture within the meaning of s 6 of the Statute of Monopolies. The question that arises under s 18(1)(a) is: Is this a proper subject of letters patent according to the principles which have been developed for the application of s 6 of the Statute of Monopolies?: Aristocrat Technologies Australia Pty Ltd v Commissioner of Patents (2022) 274 CLR 115 at [21]. In addressing that question it is necessary to look to the “substance” of the claimed invention in order to determine whether it is to patentable subject matter: D’Arcy v Myriad Genetics Inc (2015) 258 CLR 334 (“Myriad”) at [87]-[88] per French CJ, Kiefel, Bell and Keane JJ, and [145] per Gageler and Nettle JJ, citing Research Affiliates LLC v Commissioner of Patents (2014) 227 FCR 378 (“Research Affiliates”) at [107].
220 Where the invention claimed falls within an established category of patentable subject matter, it will generally be a manner of manufacture if: (a) the invention claimed is for a product made, or a process producing an outcome, as a result of human action; and (b) the invention claimed has economic utility. However, broader considerations may become relevant where the subject-matter claimed is not within the established categories of what is patentable: Myriad at [28]. These considerations may include potential “chilling effects” on activities outside of those the subject of the exclusive rights of the patentee, the coherence of the law of patentability, and the patent laws of other countries.
221 Sanofi submitted that it is not possible to identify the structure of any putative “isolated monoclonal antibody” within the claims of the Applications. It submitted that the substance of the invention as claimed is not a MAb and is instead no more than an abstract idea or a “mere desideratum” of any antibody that has the desired functional outcome. By this I understood Sanofi to mean that each of the claims was, in substance, to no more than a desired result or outcome whereby the binding of PCSK9 to LDLR is blocked or inhibited.
222 This point was articulated by Sanofi in its written submissions as follows:
6.10 The issues raised by the claims of the [Applications] are whether the claims, properly characterised, disclose proper subject matter for the grant of letters patent or, in the context of this ground of opposition, disclose a mere desideratum.
6.11 A claim will fail to constitute a manner of manufacture if it does not identify something that extends beyond an abstract idea or mere desideratum. Put another way, the invention claimed must involve the practical application to a useful end.
223 The expression “mere desideratum” was used by the Full Court in Grant v Commissioner of Patents (2006) 154 FCR 62 (“Grant”) at [18], citing Burroughs Corp (Perkins’) Application [1974] RPC 147 at 160. In Grant the Full Court held that a method of protecting assets from creditors was not a manner of manufacture, observing at [18] that “[t]here must be more than ‘a mere method or a mere idea or mere desideratum’”.
224 Sanofi also relied on the judgment of the Full Court in Encompass Corporation Pty Ltd v InfoTrack Pty Ltd (2019) 372 ALR 646 (“Encompass”). That case was concerned with a patent for a method for displaying information derived from electronic searches across multiple databases for the purpose of providing “business intelligence”. The method utilised a “suitably programmed” processing system to implement the claimed method. The Full Court said at [100]:
In oral submissions, the appellants sought to make much of the fact (if it be a fact) that the claimed method cannot be implemented using “generic software”. The difficulty with this submission is that the claims in suit do not secure, as an essential feature of the invention, any particular software or programming that would carry out the method. It is left entirely to those wishing to use the method to devise, and then to implement, a suitable computer program for that purpose. As we have said, all that the specification teaches is that the processing system be “suitably programmed”.
225 The Full Court following Research Affiliates and Commissioner of Patents v RPL Central Pty Ltd (2015) 238 FCR 27 (“RPL Central”), held at [99] that the invention of the claims was “in truth, no more than an instruction to apply an abstract idea (the steps of the method) using generic computer technology”. Sanofi submitted that the same reasoning applies to the Exemplary Claims because each of them properly understood is a claim to an abstract idea for a desirable antibody that will provide an identified result.
226 Sanofi also referred here to the process which must be undertaken to generate antibodies, and submitted:
the claims do not identify the structure of the isolated monoclonal antibodies claimed and the expert evidence establishes that it was not at any relevant date (or now) possible to reverse engineer a particular antibody from one of the functional attributes identified in the claims. It can be seen that the claims do not encompass proper subject matter for the grant of letters patent.
227 Sanofi also submitted that even if the Exemplary Claims encompass proper subject matter for the grant of letters patent, a closer analysis of the science demonstrates that the claims should be rejected under the following principles articulated by the plurality (French CJ, Kiefel, Bell and Keane JJ) in Myriad at [28]:
A number of factors may be relevant in determining whether the exclusive rights created by the grant of letters patent should be held by judicial decision, applying s 18(1)(a) of the Act, to be capable of extension to a particular class of claim. According to existing principle derived from [National Resource Development Corporation v Commissioner of Patents (1959) 102 CLR 252], the first two factors are necessary to characterisation of an invention claimed as a manner of manufacture:
1. Whether the invention as claimed is for a product made, or a process producing an outcome as a result of human action.
2. Whether the invention as claimed has economic utility.
When the invention falls within the existing concept of manner of manufacture, as it has been developed through cases, they will also ordinarily be sufficient. When a new class of claim involves a significant new application or extension of the concept of “manner of manufacture”, other factors including factors connected directly or indirectly to the purpose of the Act may assume importance. They include:
3. Whether patentability would be consistent with the purposes of the Act and, in particular:
3.1 whether the invention as claimed, if patentable under s 18(1)(a), could give rise to a large new field of monopoly protection with potentially negative effects on innovation;
3.2 whether the invention as claimed, if patentable under s 18(1)(a), could, because of the content of the claims, have a chilling effect on activities beyond those formally the subject of the exclusive rights granted to the patentee;
3.3 whether to accord patentability to the invention as claimed would involve the court in assessing important and conflicting public and private interests and purposes.
4. Whether to accord patentability to the invention as claimed would enhance or detract from the coherence of the law relating to inherent patentability.
5. Relevantly to Australia’s place in the international community of nations:
5.1 Australia’s obligations under international law;
5.2 the patent laws of other countries.
6. Whether to accord patentability to the class of invention as claimed would involve law-making of a kind which should be done by the legislature.
Factors 3, 4 and 6 are of primary importance. Those primary factors are not mutually exclusive. It may be that one or more of them would inform the “generally inconvenient” limitation in s 6 of the Statute of Monopolies. It is not necessary to consider that question given that no reliance was placed upon that proviso. They are nevertheless also relevant to the ongoing development of the concept of “manner of manufacture”.
228 Sanofi referred to factor 3.2, and relied on what it contended would be the “chilling effect” of the Exemplary Claims. It submitted:
Any company or person seeking an improved method of blocking the binding of PCSK9 to LDLR by way of an antibody would have to undertake a full research project to create new monoclonal antibodies and would only know, after having made the monoclonal antibodies and tested them, whether they fall within the bounds of any claim. Thus, any company seeking to develop new monoclonal antibodies to inhibit the binding of PCSK9 to LDLR would have to incur the full cost of that research project before being able to ascertain if it might fall within the scope of any of the claims. This position is analogous to that discussed by the plurality in [Myriad] at [93]. Claims of such breadth will have a chilling effect on legitimate competition and innovation in this field.
Sanofi placed no reliance on factors 4, 5 or 6.
229 In paragraph [93] of Myriad, which Sanofi also relied on, the plurality said:
When proper regard is paid to their emphasis on genetic information, the subject matter of the claims lies at the boundaries of the concept of “manner of manufacture”. That it does lie at the boundaries is further evidenced by the odd consequence that if the claims are properly the subject of a patent, the patent could be infringed without the infringer being aware of that fact. That consequence coupled with the very large, indeed unquantified size of the relevant class of isolated nucleic acids, all of which bear the requisite information, raises the risk of a chilling effect upon legitimate innovative activity outside the formal boundaries of the monopoly and risks creating a penumbral de facto monopoly impeding the activities of legitimate improvers and inventors.
(Footnote omitted)
230 Finally, Sanofi submitted that the significance of the residues and epitopes in the Exemplary Claims lies only in the fact that Amgen considers that these epitopes and residues inform the region where an antibody binds on PCSK9 in order inhibit or block binding of PCSK9 to the EGFa domain of LDLR. Sanofi further submitted that the true significance of the subject matter claimed was its informational character. Amgen submitted that this point was first raised by Sanofi in its closing submissions, and was not addressed in expert evidence. While this point was, in fact, raised by Sanofi in its opening submissions, it was not explored in evidence or developed in any detail in Sanofi’s closing submissions.
Consideration
231 I understood Sanofi to accept that a particular MAb (e.g., 21B12) characterised in the CS by reference to its amino acid sequence would constitute patentable subject matter. What renders the Exemplary Claims unpatentable on Sanofi’s approach is the fact that the MAbs are insufficiently characterised, whether by their amino acid sequence or otherwise, and that the class of MAbs within the Exemplary Claims are instead defined solely by reference to the functional requirements of the claims. The matters relied on in support of this submission were essentially the same as those relied on by Sanofi in support of its contention that the Exemplary Claims do not define the invention.
232 In considering Sanofi’s submissions it is useful to begin with a reference to the fundamental distinction between mere discovery and an invention. As Buckley J said in Reynolds v Herbert Smith & Co Ltd (1902) 20 RPC 123 at 126:
Discovery adds to the amount of human knowledge, but it does so only by lifting the veil and disclosing something which before had been unseen or dimly seen. Invention also adds to human knowledge, but not merely by disclosing something. Invention necessarily involves also the suggestion of an act to be done, and it must be an act which results in a new product, or a new result, or a new process, or a new combination for producing an old product or an old result.
233 The same point was made by Whitford J in Genentech Inc.’s Patent [1987] RPC 553, in a statement expressly approved by the House of Lords in Kirin-Amgen Inc v Hoechst Marion Roussel Ltd [2005] RPC 169. His Lordship said at 566:
It is trite law that you cannot patent a discovery, but if on the basis of that discovery you can tell people how it can be usefully employed, then a patentable invention may result. This in my view would be the case, even though once you have made the discovery, the way in which it can be usefully employed is obvious enough.
234 In National Resource Development Corporation v Commissioner of Patents (1959) 102 CLR 252 (“NRDC”) the High Court referred to the distinction between discovery and invention as follows at 264:
The truth is that the distinction between discovery and invention is not precise enough to be other than misleading in this area of discussion. There may indeed be a discovery without invention-either because the discovery is of some piece of abstract information without any suggestion of a practical application of it to a useful end, or because its application lies outside the realm of “manufacture”. But where a person finds out that a useful result may be produced by doing something which has not been done by that procedure before, his claim for a patent is not validly answered by telling him that although there was ingenuity in his discovery that the materials used in the process would produce the useful result no ingenuity was involved in showing how the discovery, once it had been made, might be applied. The fallacy lies in dividing up the process that he puts forward as his invention. It is the whole process that must be considered; and he need not show more than one inventive step in the advance which he has made beyond the prior limits of the relevant art.
235 The Full Court in Ariosa Diagnostics, Inc v Sequenom, Inc (2021) 391 ALR 473 (Middleton, Nicholas and Burley JJ) (“Ariosa”) referred to three principles that emerge from NRDC. The Full Court said at [114]:
… First, the distinction between mere discovery and an invention lies in its practical application to a useful end. Secondly, it is important that the invention be considered as a unitary concept, not segregated artificially into parts. The invention may arise from an idea and then be applied in a perfectly well known way, and yet the combined effect of the idea and its application may result in patentable subject matter, as arose from the example described in Hickton’s Patent Syndicate v Patents and Machine Improvements Co Ltd (1909) 26 RPC 339...The approach of the appellants in the present appeal, which seeks to disaggregate the discovery of cffDNA in maternal plasma or serum from the method used to harness that discovery, tends to overlook these matters. Thirdly, an invention may reside in an abstract idea, such as the condensation of steam that is then put to a useful end, even though the way of putting it to that end can be carried out in many useful ways, all of which are otherwise known.
236 The Full Court in CCOM Pty Ltd v Jiejing Pty Ltd (1994) 51 FCR 260 (“CCOM”) considered a submission that the claim in issue in that case was not for a manner of manufacture because it merely identified “basic characteristics” or “desiderata” and claimed all ways of achieving them. The Full Court rejected this submission at 294-295:
… it was submitted that all that had been done was to select a desirable characteristic of a computer program, the ability to search, in the manner described, a data base of the type described, and “to claim all computers present and future possessing that characteristic”.
That submission should not be accepted. It may be that such a claim lacks novelty, is obvious, or lacks utility, or there is a failure to comply with one or other of the limbs of s 40 because, for example, the invention is not fully described or the claim is not clear and succinct. But if such hurdles all are surmounted, then in our opinion in a case such as the present there does not remain an independent ground of objection as to patentability, within the sense of s 18(1)(a) of the 1990 Act.
237 Similarly, in Eli Lilly and Co v Pfizer Overseas Pharmaceuticals (2005) 218 ALR 408 (“Eli Lilly”), Heerey J rejected an argument that the following claim was unpatentable as mere desideratum, that is to say, something which is disclosed as no more than a wished for result:
A method of orally treating man to cure or prevent erectile dysfunction in man in need of such treatment, which comprises treatment with an orally effective amount of cGMP PDEV inhibitor, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition containing either entity.
238 His Honour (whose reasoning on this issue was not disturbed on appeal) held at [213]:
There can be no doubt that oral treatment of impotence was seen as desirable and much preferable to the existing alternatives of intracavernosal injection, surgical implantation, vacuum construction devices and reconstructive surgery. However, the alleged invention disclosed in the patent goes further. The patent specification tells the reader the manner in which the desired object is to be achieved, namely by the use of an orally effective cGMP PDEV inhibitor. This cannot be characterised, as Lilly says, as a “bare idea”.
The claim was not to a “bare idea” or “mere desideratum”, because it required treatment with an orally effective amount of a cGMP PDEv inhibitor, rather than merely stating a wished for result (i.e., the oral treatment of impotence).
239 Similarly, Jessup J in AMR v Alphapharm rejected an argument that a claim to “substantially pure piperidine derivative compounds” of a particular formula was not a manner of manufacture. His Honour said at [198]:
… I consider that the specification in the present case, on its face, goes further than to disclose “a wished for result”. It asserts that the prior art does not permit the synthesis of the compounds of interest in substantially pure form and, therefore, that those substantially pure compounds did not previously exist. It “tells the reader the manner in which the desired object is to be achieved” (Eli Lilly at [213]).
240 While the claim in that case was, in effect, to the “wished for result” of a substantially pure form of the compounds falling within the claimed formula, the invention was not to a mere desideratum. Nor did it matter that the claim encompassed formulations made using any method.
241 I do not accept that the Exemplary Claims are to abstract information or a mere idea or desideratum. The claimed MAbs are chemical compounds defined by their binding characteristics and their ability to block or inhibit interactions between LDLR and PCSK9. They are a product made through human action beginning with the immunisation of transgenic mice with PCSK9 so as to generate an immune response followed by the creation of hybridomas.
242 On the face of the specification, the substance of the claimed invention is MAbs that block PCSK9 binding to LDLR and: (a) have a structural epitope which includes one or more specific residues on PCSK9; (b) have a functional epitope which includes one or more of those specific residues; or (c) compete for binding to PCSK9with a reference antibody. They are physical molecules capable of binding to PCSK9 and blocking its interaction with LDLR.
243 Sanofi does not allege that the MAbs are not novel, and although Sanofi alleges that they do not involve an inventive step, it does not suggest that any such lack of inventiveness is apparent on the face of the specification: see Myriad at [130]-[131] per Gageler and Nettle JJ citing (inter alia) Commissioner of Patents v Microcell Ltd (1958) 102 CLR 232 and NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1995) 183 CLR 655 at 663-664; Merck & Co Inc v Arrow Pharmaceuticals Ltd (2006) 154 FCR 31 at [63] per Heerey, Kiefel and Dowsett JJ.
244 The MAbs of the Exemplary Claims are suitable for use in a variety of therapeutic treatments that are described in detail in the CS either as a monotherapy or in combination with other therapeutics: see CS [0358]-[0370]. Sanofi does not dispute that they have economic utility. Clearly, they are products that have been shown to be useful in the treatment of various cardiovascular and related diseases.
245 Although Research Affiliates, RPL Central and Encompass contain a discussion of relevant principes, they are decisions on their facts concerned with a particular class of subject matter which has been held to be unpatentable. I do not find those decisions of assistance in determining whether the Exemplary Claims are for a manner of manufacture. The invention that is claimed in this case is not at all comparable to a claim for an idea as to how to manage and display information utilising “suitably programmed software” or “generic computer technology”.
246 As mentioned, Sanofi submitted that even if the Exemplary Claims may appear to encompass proper subject matter for the grant of letters patent, a closer analysis of the science demonstrates that the claims should be rejected under the principles considered in Myriad at [28] (i.e., the “chilling effect”). I do not accept that submission. The economic significance of the isolated nucleic acid claimed in Myriad resided in the genetic information (the nucleotide sequence) encoded on the BRCA1 and BRCA2 genes, and not in “chemical, structural or functional differences” between those nucleic acids and the isolated nucleic acids. That was, as the plurality in Myriad observed at [90], the focus of the claims. For that reason, the subject matter of the claims was at the boundaries of the concept of manner of manufacture.
247 In the present case, the Exemplary Claims are, in substance, to MAbs that block or inhibit the binding of PCSK9 to LDLR. The focus of the claims is on the effect that the MAbs of the claims have on the interaction of those molecules. That is not a focus on the genetic information conveyed by the MAbs. I do not consider the Exemplary Claims to be a new class of claim involving a significant new application or extension of the concept of manner of manufacture. It follows that there is no need to consider factors 3-6 set out by the plurality in [28] of Myriad, including whether allowing a claim will have a “chilling effect”.
248 In any case, Sanofi’s contention that the Exemplary Claims would, if allowed, have a chilling effect is by no means self-evident, and is not supported by the evidence. While it may be accepted that a company or person seeking to develop a MAb that blocks binding of PCSK9 to LDLR may not know whether any particular MAb is within the claims until after it has been produced and tested, that is not an uncommon circumstance in the case of claims limited by result. The plurality in Myriad, at [93], emphasised that the claims were to, essentially, the genetic information that the isolated nucleic acids the subject of the claims embodied and conveyed. That, coupled with the very large class of such compounds bearing that information, were factors which the plurality found raised the risk of a chilling effect.
249 The MAbs of the Exemplary Claims do not embody or convey information. While the claims convey information concerning the structural and functional epitopes to which those MAbs bind, this information is not embodied in or conveyed by the MAbs of the invention. Nor do the claims give rise to the problem identified in Myriad by Gordon J at [259]-[260]. This case therefore is not analogous to Myriad.
250 I accept, as Sanofi submitted, that pharmaceutical companies other than Amgen were engaged in research into various methods of inhibiting PCSK9. However, there is no evidence from which it can be inferred that any of those companies’ research work was inhibited by the publication in 2009 of the Parent Application or any related patent application published any time thereafter. Nor is there any other evidence from which it may be inferred that the grant of a patent arising out of any of the Applications would have a chilling effect on activities involving the development or production of MAbs beyond the scope of the Exemplary Claims.
251 The Applications do not claim all the methods available for achieving the result of inhibiting PCSK9 activity to reduce LDL levels. The evidence shows that various companies filed patent applications after the publication of the Parent Application which were concerned with the inhibition of PCSK9 including by means involving anti-PCSK9 antibodies.
252 Finally, to the extent that any company or person runs the risk of making antibodies which fall within the scope of the Exemplary Claims in the course of research and development, the experimental purpose exemption found in s 119C of the Act provides them with very broad protection. Section 119C provides:
119C Infringement exemptions: acts for experimental purposes
(1) A person may, without infringing a patent for an invention, do an act that would infringe the patent apart from this subsection, if the act is done for experimental purposes relating to the subject matter of the invention.
(2) For the purposes of this section, experimental purposes relating to the subject matter of the invention include, but are not limited to, the following:
(a) determining the properties of the invention;
(b) determining the scope of a claim relating to the invention;
(c) improving or modifying the invention;
(d) determining the validity of the patent or of a claim relating to the invention;
(e) determining whether the patent for the invention would be, or has been, infringed by the doing of an act.
This exemption would extend to any experimental work directed to the development of any new MAb or improvements to any of the MAbs disclosed in the CS.
253 I am not persuaded that any of the Applications is for a class of invention that would, if it was the subject of a grant, have a chilling effect on activities outside the scope of the claims. In the result, I am not persuaded that any of the Exemplary Claims is not for a manner of manufacture.
FAIR BASIS
254 Prior to its amendment by the RTB Act, s 40(3) of the Act required the claims of a patent to be “fairly based on the matter described in the specification”. Section 40(3) now requires that the claims be “supported by matter disclosed in the specification”. Establishing “support” requires that the technical contribution to the art disclosed by the specification justify the breadth of the claim: see ToolGen Incorporated v Fisher (No 2) [2023] FCA 794 (Nicholas J) (“Toolgen”) at [391]-[396] following Merck Sharp & Dohme Corporation v Wyeth LLC (No 3) (2020) 155 IPR 1 (Burley J) at [546]-[547].
255 Fair basis, on the other hand, is not concerned with the technical contribution to the art or the inventive merit of the invention. In Olin Corporation v Super Cartridge Co Pty Ltd (1977) 180 CLR 236 (“Olin”) Barwick CJ said at 240:
The question whether the claim is fairly based is not to be resolved . . . by considering whether a monopoly in the product would be an undue reward for the disclosure. Rather, the question is a narrow one, namely whether the claim to the product being new, useful, and inventive, that is to say, the claim as expressed, travels beyond the matter disclosed in the specification.
That statement was approved by the High Court in Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 (“Kimberly-Clark”) at [15] and again in Lockwood Security Products Pty Limited v Doric Products Pty Ltd (2004) 217 CLR 274 (“Lockwood No 1”) at [57].
256 In Lockwood No 1 the High Court made clear that neither the technical contribution to the art nor the inventive merit of an invention is relevant to fair basis. The High Court said of s 40(3) at [54]:
… the language points to a comparison between the claims and what is described in the specification only, and again it does not call for any inquiry into an ‘‘inventive step’’, or inventive ‘‘merit’’ or a ‘‘technical contribution to the art’’.
257 Fair basis requires that the body of the specification contain “a real and reasonably clear disclosure” of what is claimed: Lockwood No 1 at [69]. This requires that “the alleged invention as claimed is broadly, that is to say in a general sense, described in the body of the specification”: Rehm Pty Ltd v Websters Security Systems (International) Pty Ltd (1988) 81 ALR 79 at 95, per Gummow J, in a passage approved in Lockwood No 1 at [69]. In determining whether there has been a real and reasonably clear disclosure, the Court should eschew “an over meticulous verbal analysis”, and should not seek to “isolate in the body of the specification ‘essential integers’ or ‘essential features’ of an alleged invention and…ask whether they correspond with the essential integers of a claim in question”: Lockwood No 1 at [68].
258 A claim is not fairly based if it “travels beyond the matter disclosed in the specification”: Olin at 240 per Barwick CJ. While the Court will generally have regard to the “consistory clauses” or like statements which appear in a specification, the mere “coincidence of language” between a consistory clause or like statement in the body of the specification and the claim is not enough to establish fair basis if it is apparent that the language does not reflect the invention described in the specification when read as a whole: Lockwood No 1 at [87], citing Atlantis at 50. As the High Court said in Lockwood No 1 at [99]:
… the correct position is that a claim based on what has been cast in the form of a consistory clause is not fairly based if other parts of the matter in the specification show that the invention is narrower than that consistory clause. The inquiry is into what the body of the specification read as a whole discloses as the invention. An assertion by the inventor in a consistory clause of that of which the invention consists does not compel the conclusion by the court that the claims are fairly based nor is the assertion determinative of the identity of the invention. The consistory clause is to be considered by the court with the rest of the specification.
(Footnote omitted)
259 Determining whether a claim travels beyond the matter disclosed in the specification may involve determining whether the claim is to a class of articles broader than a more limited subset of that class described in the body of the specification: see, for example, Pfizer Overseas Pharmaceuticals v Eli Lilly & Co (2005) 225 ALR 416 at [276] per French and Lindgren JJ. Further, a claim may also travel beyond the matter disclosed in the body of the specification if it omits an essential element of the invention disclosed such that the invention described in the specification is fundamentally different from the invention claimed: see, for example, AstraZeneca AB v Apotex Pty Ltd (2014) 226 FCR 324 at [254]-[255] (“AstraZeneca FFC”).
260 In considering a claim for a pharmaceutical composition, the Full Court (Middleton, Nicholas and Burley JJ) in GSK v Generic observed at [156] that the invention as claimed need not be limited to preferred embodiments exemplified in the body of the specification. In that case, the sole independent claim was to a pharmaceutical composition comprising a bilayer tablet with certain formulation and pharmacokinetic properties including a dissolution profile determined by a particular method. The invention was exemplified by reference to two particular formulations. The Full Court held that the invention was not limited to those two formulations. Further, the Full Court did not accept the submission that the specification had to provide a “predictive connection” which would enable one to understand how a particular formulation would behave in comparison to the exemplified formulations. The Full Court said at [167]-[168]:
[167] Apotex also submitted that even if the disclosure in the specification is not limited to Formulations C and D, the ranges are not fairly based in the absence of an explanation of the mechanism or reasoning founding the relationship between those ranges and the pharmacokinetic parameters. It argued that for the claims to be fairly based, the specification must disclose an established [in-vitro in-vivo correlation] to provide a “predictive connection”. We do not accept this submission.
[168] The specification does not need to identify the reasoning or the “theoretical basis” underpinning the selection of content, ratio and dissolution limits referred to in the claims. Similarly, for the purpose of providing fair basis, the specification does not need to describe the invention in a different or more detailed way than the claims …
261 The Full Court also noted at [164] that the question whether the claim met the promise of the invention is not a relevant consideration for fair basis. A claim will not fail to be fairly based because it includes within its scope embodiments that do not meet the promise of the patent or that are not useful.
The Parties’ Submissions
262 Sanofi submitted that none of the claims of the Applications are fairly based, because the claimed invention is different from the invention described in the CS.
Sanofi’s characterisation of the invention
263 Sanofi sought to characterise the invention disclosed in the specification by reference to the priority documents. It says that the invention disclosed in the first priority document, US Provisional Patent Application 60/957,668 (P1), is:
…isolated monoclonal antibodies 21B12, 31H4 and, perhaps, 16F12 with specific amino acid sequences with respect to the CDRs of each such amino acid and specific permitted substitutions to such CDRs.
264 The second priority document, US Provisional Patent Application 61/008,965 (P2), discloses (inter alia) figures containing representations of various solved crystal structures of PCSK9 in complex with 21B12, 31H4 and the EGFa domain of LDLR, whether in full or parts thereof. In its written submissions, Sanofi drew attention to the agreement amongst some of the experts that these figures “[did] not yield any extra information” over and above P1. In the examples accompanying those figures, P2 also identifies “core” and “boundary” residues on PCSK9 for each interaction. Core and boundary residues are discussed above in relation to the Common Specification.
265 Sanofi submitted that the disclosure of core and boundary residues did not change the true characterisation of the invention. It submitted that this is also true of the disclosure of atomic coordinates for some of the solved crystal structures in the third priority document, US Provisional Patent Application 61/010,630 (P3), and the disclosure of additional experiments, including a binning experiment and scanning mutagenesis, in US Provisional Patent Application 61/086,133 (P4). Sanofi’s fair basis submissions proceeded on the basis that the CS discloses the invention as “at best three isolated monoclonal antibodies (21B12, 31H4 and, perhaps, 16F12)”.
266 I should say at the outset that the issue of fair basis is to be considered based on the complete specification read as a whole without regard to the timing or sequence by which content came to be incorporated into the document. For present purposes it is therefore necessary to focus on the disclosure of the CS as a whole rather than any priority document.
Epitope and Residue Claims
267 Sanofi submitted that the Epitope and Residue Claims are to:
(1) “hundreds, if not thousands or many more, antibodies”
(2) “each epitope to which each (and any) antibody binds, provided it includes one (or some set of) the residues stated in the claim”; (emphasis original)
(3) “antibodies that ‘block’ binding of the PCSK9/LDLR interaction to any degree”; and
(4) “antibodies that reduce total cholesterol level by any degree or not at all”.
Sanofi advanced four arguments in support of its fair basis attack on the Epitope and Residue Claims, which it said were based on different constructions of the CS. The first of these, it says, is based on the construction least favourable to its case. As I understood Sanofi’s submission, what it refers to as the “least favourable construction to its case” is a construction that equates the disclosure of core and boundary residues of the interaction interfaces of PCSK9 and 21B12 or 31H4 in the Applications to a description of the functional and structural epitopes of those antibodies.
Sanofi’s first argument
268 Sanofi said that even if I accept that the disclosure of “core” and “boundary” residues of the interaction interfaces for 21B12 and 31H4 in the Applications is a sufficient description of residues within the structural and functional epitopes of those antibodies, Amgen’s fair basis case relies on “an invention ‘reasonably extrapolated from’ or ‘a sound prediction’ based on a single neutralising antibody (21B12) or two single neutralising antibodies (21B12 and 31H4)”. That process of extrapolation, it says, may be likened to that rejected by the Full Court in GSK v Generic at [156]-[171].
269 In the course of developing its first argument Sanofi submitted:
(1) The CS does not disclose the Table 2 Antibodies as “the invention”.
(2) Even if some subset of those competing Table 2 Antibodies have overlapping epitopes with 21B12 or 31H4, the extent of the overlap is not identified nor is any structural or functional epitope of the antibody identified.
(3) It is “farfetched” to suggest that overlapping epitopes identify an antibody that is “likely” or “more likely than not” to have an epitope comprising one of the six residues identified in claim 1 of 677 (or, it follows, any of the residues identified in the other Exemplary Claims).
(4) Even if the PSA were permitted to engage in some “prediction” or “extrapolation” of the invention described in the specification to “demonstrate a ‘principle’”, the predictions made by Professor Petsko about those antibodies which co-binned with 21B12 were made on the premise that those antibodies were “strong blockers”, but the claims are not so confined. In any event, the “principle” of an antibody that binds to PCSK9 to block LDLR was common general knowledge and incorporated into the specification in its entirety.
(5) The PSA would not assume that changes to the amino acid sequences of antibodies would result in antibodies that would have the same properties, and the CS does not disclose proof of this principle.
270 Sanofi also submitted that, on this construction of the CS, the claims still omit essential elements of the invention. This submission was developed with reference to the concept of the “sweet spot” of PCSK9, a term referred to by the United States Supreme Court in Amgen Inc. v. Sanofi, 598 US 594 (2023) at 4:
The sweet spot is a sequence of 15 amino acids out of PCSK9’s 692 total amino acids ... By binding to the sweet spot, scientists found, an antibody could prevent PCSK9 from binding to and degrading LDL receptors.
271 Sanofi contended that the Epitope and Residue claims omit several residues within the sweet spot, and other residues identified by Amgen for the interaction interface between PCSK9 and 21B12. Sanofi also contended that many of the claims describe antibodies for which no particular antibody is identified in the CS. For example, it submitted:
Claim 10 of the 751 Application claims “an…antibody that recognizes an epitope …comprising at least ten of the following residues”. Claim 13 of the 751 Application claims “an…antibody that binds to… ten or more” amino acid residues identified in claim 13 of the 751 Application. The Applications do not describe any antibody that does so.
272 Claim 1 of 689 was said by Sanofi to refer to a subset of the core PCSK9/EGFa interface interaction residues identified in CS [0505]. However, Sanofi submitted that the CS does not describe “any single antibody (or even any combination of antibodies) that binds to all of the claimed residues, or even a significant subset thereof”. For that reason, Sanofi says this claim cannot be fairly based on either 21B12 or 31H4. Generally, Sanofi’s submissions on this point take issue with the Exemplary Claims including certain residues, where no specific (i.e., generated and tested) antibody has been identified in the CS as binding to that residue.
Sanofi’s second alternative argument
273 Sanofi’s second alternative argument concerns Amgen’s alleged reliance on the results of a of a PyMOL analysis of the crystallographic coordinates disclosed in the CS. “PyMOL analysis” here refers to the analysis undertaken by Professor Petsko, which involved inputting the crystal coordinates provided in the CS into the PyMOL program.
274 In his declaration, Professor Petsko explained that the first step in this process involves PyMOL identifying PCSK9 residues that are within a certain distance from the relevant heavy or light chain of the antibody. He explained that:
Structural biologists understand, consistent with the … disclosure [at 0525 of the CS], that ‘the distance between the [antibody] and PCSK9 is the shortest distance between the covalently bonded atom on PCSK9 and the covalently bonded atom of the [antibody] that are the closest atoms of PCSK9 and the [antibody].’
Next, the structural biologist will identify the type of non-covalent interaction that may form between the atoms of the amino acids that are involved in the binding:
[B]ased upon the known chemistry of those atoms, a structural biologist concludes that a certain type of noncovalent interaction (or interactions) has formed between them.
The second step of this process involves a manual determination by the expert as to the existence of non-covalent interactions.
275 According to Sanofi, Amgen should not be permitted to rely on such extraneous analysis in proving that the Exemplary Claims are fairly based, but even if it is permitted to do so, that analysis is open to criticism because Professor Petsko has improperly identified a non-covalent interaction between 21B12 and residue S153 on PCSK9. As part of that criticism, Sanofi drew attention to the fact that Amgen failed to produce an electron density map for S153 and submitted that the Court should infer from “Amgen’s conduct in suppressing that evidence [that] the electron density map would not have assisted Amgen’s case as to whether S153 forms a noncovalent interaction with 21B12 or with any other antibody”.
276 S153, also identified as 123 in SEQ ID NO: 1, appears in each of the Exemplary Epitope and Residue Claims. Senior Counsel for Sanofi, Mr Shavin KC, argued that “if there is no involvement of S153 [in binding with 21B12] … that is fatal to the claim[s]”.
Sanofi’s alternative argument
277 Sanofi’s third alternative argument proceeds on the construction of the specification that it said most closely aligned with the evidence. It says that on this construction, the residues identified as core and boundary do not equate to residues within the structural and functional epitopes of 21B12 and 31H4. Sanofi referred to the absence of certain claimed residues from the arginine scanning data contained in Example 39, and said that the arginine scanning data only demonstrates:
a small number of residues in the structural epitope of a small number of antibodies – including 21B12 and 31H4, which the experts agree does not constitute the entire epitope (which would be 15-20 residues in the functional epitope).
Sanofi emphasised the fact that there is no arginine scanning hit for S153 in regard to 21B12, and that some of the data recorded was, in the opinion of Professor M Parker, the sort that would be “thrown out”.
278 Sanofi also refers to evidence given by some of the experts to the effect that the distance of a residue from a binding partner “can only predict and not enable designation of non-covalent interactions in the residues of the functional epitope” and that core and boundary residues don’t define any “physical chemical interactions…some of the [core and boundary] residues may or may not be [in] the structural epitope”.
279 Sanofi also referred to Professor Petsko’s evidence with which Professor Hudson agreed that he would “never” use distance alone to draw conclusions about an interaction. Sanofi says that the PSA would not understand that the structural epitope includes all residues within the “interaction interface”.
280 Sanofi further contended that:
(1) “The PSA would not understand the designation of ‘core’ and/or ‘boundary’ residues to define the structural epitope of an antibody. At its highest, the PSA would understand that the ‘core’ residues and some ‘boundary’ residues may form part of the structural epitope”; and
(2) “Nor would the PSA understand the designation of ‘core’ and ‘boundary’ residues as a reference to PCSK9 residues with which any atoms on any residues of 21B12, 31H4 or LDLR-EGFa formed non-covalent interactions. At best, the PSA would understand the designation of ‘core’ residues as a convenient way of providing a list of residues to consider analysing to determine whether or not they are involved in non-covalent interactions.”
As a result, Sanofi says, the designation of core and boundary residues is not a real and reasonably clear disclosure of the invention of the Epitope and Residue Claims.
281 As part of this third alternative argument, Sanofi again took issue with the use of a PyMOL analysis to determine whether or not a non-covalent interaction exists, because (it submitted) reasonable, expert minds may differ as to whether an interaction exists based on that analysis, and PyMOL only serves to measure the distance between two atoms.
Sanofi’s fourth argument
282 Sanofi’s fourth alternative argument was put very briefly in its written submissions:
The fourth argument is that already considered with respect to claim definition: i.e., none of the claims of the Applications characterise the construction of “an isolated monoclonal antibody” across the breadth of the claims.
283 Sanofi referred to Olin at 263-264, per Stephen and Mason JJ, in support of its argument that this requirement (i.e., that the specification “describes distinctive characteristics of the result or product and in the case of an article the limitation is ‘sufficient to characterize the construction of the article claimed’”) should be considered in the context of fair basis.
Additional criticisms raised by Sanofi
284 Separate from the above arguments, but as part of its fair basis attack, Sanofi advanced several criticisms of Professor Petsko’s interpretation of the CS. Sanofi said that Professor Petsko has “engaged in a tremendous effort to find support for the breadth of the claims in the Applications”, but that his approach is inconsistent with what would be disclosed to the PSA upon a reading of the document.
285 Sanofi also submitted that Professor Petsko’s predictions about the binding of Table 2 Antibodies were “impermissibly infected by, and inextricably linked to, information extraneous to the specification, i.e., the crystal structures of the so called ‘Later Crystallised Antibodies’.” Amgen prepared crystal structures for the Later Crystallised Antibodies, 8A3, 11F1, 1A12 and 25G4, after the filing of the Applications. According to Professor Petsko, those structures “show that the epitopes for the Later Crystallized Antibodies were distributed across the PCSK9/LDLR interface”.
286 Sanofi also criticised Professor Petsko’s use of the standard “more likely than not” when describing his level of certainty as to whether certain matters could be deduced from crystal structures. Sanofi contended that this same standard was applied by Professors Hudson and Lopez in oral evidence. Sanofi highlighted Professor Petsko’s observation that:
Crystallography is one area about which people can look at a structure and sometimes disagree about what the structure is really trying to say …
287 Sanofi pointed to the differences between the residues Professor Petsko identified as being involved in non-covalent interactions in his declaration, compared with those he identified in his affidavit. Sanofi says that where data provided in a document is capable of two equally valid interpretations, there can be no real and reasonably clear disclosure in the specification. In support of this, it relies on the observations of Laddie J in Evans Medical Ltd’s Patent [1998] RPC 517. Those observations, made in the course of considering whether the body of a specification sufficiently enabled the PSA to produce the claimed invention, include the following at 536-537:
The reality may be that the document is so obscure that different results can be achieved by trying to follow its teaching. It is not enough if the instructions are such that a number of equally qualified notional addressees can arrive at completely different end points, some within the scope of the claimed invention and some not. If reasonable addresses can come to different conclusions there is a conundrum as to which is right. That is not enablement.
Amgen’s submissions on the Epitope and Residue claims
288 Amgen contended that there is a real and reasonably clear disclosure of the invention referred to in the consistory statements, appearing in the summary section of each Application. In the case of the three Applications containing the Epitope and Residue Claims, those statements are as follows:
677 | [007c] In one aspect of the present invention, there is provided an isolated monoclonal antibody that binds an epitope on hPCSK9 comprising one or more of amino acid residues 207, 208, 162, 164, 167 or 123 of SEQ ID NO: 1, and wherein the monoclonal antibody blocks binding of PCSK9 to LDLR. |
689 | [007b] In one aspect of the present invention, there is provided an isolated monoclonal antibody, wherein, when bound to PCSK9, the monoclonal antibody binds to at least one of the following residues: S153, I154, P155, R194, D238, A239, I369, S372, D374, C375, T377, C378, F379, V380, or S381 of SEQ ID NO: 3, and wherein the monoclonal antibody blocks binding of PCSK9 to LDLR. |
751 | [007b] In one aspect of the present invention, there is provided an isolated monoclonal antibody recognizes an epitope on human PCSK9 comprising amino acid residues: S153, R194, D238, D374, T377, and F379 of SEQ ID NO: 3, wherein the monoclonal antibody reduces binding between PCSK9 and EGFa domain of LDLR. |
Further, Amgen said that each such statement is supported by the balance of the CS.
289 Attention should also be drawn to paragraphs [007c], [007d] and [007e] of 751 which state:
[007c] In another aspect of the present invention, there is provided an isolated monoclonal antibody that binds to amino acids in human PCSK9 of SEQ ID NO: 1, wherein the amino acids comprise S153, R194, D238, D374, T377, and F379 of SEQ ID NO: 3, and wherein the monoclonal antibody reduces binding between PCSK9 and an EGFa domain of LDLR.
[007d] In a further aspect of the present invention there is provided an isolated monoclonal antibody that recognizes an epitope on human PCSK9, wherein the epitope comprises at least ten of the following residues: S153, I154, P155, R194, D238, A239, I369, S372, D374, C375, T377, C378, F379, V380, or S381 of SEQ ID NO: 3, wherein the monoclonal antibody reduces binding between PCSK9 and an EGFa domain of LDLR.
[007e] In yet another aspect of the present invention there is provided an isolated monoclonal antibody that binds to amino acids in human PCSK9, wherein the amino acids comprise ten or more of the following amino acids: S153, I154, P155, R194, D238, A239, 1369, S372, D374, C375, T377, C378, F379, V380, or S381 of SEQ ID NO: 3, and wherein the monoclonal antibody reduces binding between PCSK9 and an EGFa domain of LDLR.
290 Amgen contended that Sanofi has mischaracterised the invention of the CS (in relation to the Epitope and Residue Claims), which it described as follows:
With respect to the epitope claims, the CS describes a class of blocking antibodies that bind epitopes comprising the recited residues. With respect to the residue claims, the CS describes a class of blocking antibodies that bind the recited residues. The CS also exemplifies antibodies within these classes.
291 Amgen drew attention to Examples 28 to 32, the disclosure of the core and boundary residues identified therein, and the following statements in CS [0524]:
As will be appreciated by one of skill in the art, Examples 28-32, and their accompanying figures, provide a detailed description of how and where EGFa interacts with PCSK9 and how two representative neutralizing antigen binding proteins, 211B12 and 31H4 interact with PCSK9 and produce their neutralizing effect. As such, one of skill in the art will readily be able to identify antigen binding molecules that can similarly reduce the binding between EGFa (including LDLR) and PCSK9 by identifying other antigen binding molecules that bind at or near at least one of the same locations on PCSK9 …
292 The crux of Amgen’s submission about fair basis, with regard to the Epitope and Residue Claims, was as follows:
… Sanofi ignores the fact that the description of the invention is not limited to 21B12 and 31H4. Rather, the CS discloses that any antibodies that have an epitope that includes an interaction with the Nominated Residues will be useful. 21B12 and 31H4 are examples of such antibodies. The CS discloses antibodies (not a single antibody) binding “at least one” or “one or more” of the identified residues and teaches that such antibodies can block PCSK9 from binding to LDLR. The claimed antibodies thus are fairly based on the description in the CS.
(Emphasis original)
The “Nominated Residues” are defined by Amgen as the specific PCSK9 amino acid residues located in the “small and specific region” on PCSK9 to which the class of claimed antibodies bind.
293 As to the omission of essential elements of the invention, Amgen said Sanofi’s assertion that the Epitope and Residue Claims must correspond to those residues in the “sweet spot” does not reflect the language of the CS, which Amgen says discloses “useful antibodies [which] will bind to one or more of the core residues for 21B12 or 31H4, in addition to one [or] more core residues for EGFa”.
294 Amgen said it does not need to rely on the PyMOL analysis performed by Professor Petsko, which it said was led in response to Sanofi’s sufficiency attack that was largely abandoned. However, Amgen contended that the inputting of data of the kind contained in Table 35 of the CS (containing atomic coordinates) into PyMOL was routine at the priority date. It said that Sanofi’s attack on Professor Petsko’s analysis is misconceived, because Amgen does not need to prove that certain non-covalent bonds exist in order for the claims to be fairly based.
295 Amgen also submitted that Professor M Parker largely agreed with Professor Petsko’s analysis of existing non-covalent interactions, and that no additional information outside of the CS is required to view, analyse and form conclusions about the residues on PCSK9 and their binding capabilities.
296 With respect to Sanofi’s focus on S153, Amgen submitted again that it does not need to prove the existence of any particular non-covalent interaction (via arginine scanning mutagenesis or otherwise), but that, in any case, the evidence demonstrates that S153 is at least part of the structural epitope of 21B12. S153 is a core residue in the interaction interface with the EGFa domain of LDLR and was identified as an arginine scanning “hit” for antibody 12H11.
297 With respect to whether or not claims that specify residues in the structural and functional epitopes (i.e., the Epitope and Residue Claims) are fairly based on the designation of “core” and “boundary” residues in the CS, Amgen said that it did not need to prove that the core and boundary residues are involved in the structural or functional epitope of any antibody, as their disclosure alone provides a sufficient basis for the claims. However, Amgen submitted that based on the disclosures in the CS, this equivalency can be drawn.
298 Amgen submitted that the term “interaction interface” as used in the CS is a reference to the structural epitope, which includes the functional epitope. “Interaction interface”, it says, includes both core and boundary residues, which are nominated in the claims. Amgen also says that the evidence does show that at least the core residues are “more likely than not to be involved in non-covalent interactions with the antibody”. Although Professors M Parker and Mahler disagreed with this statement, Amgen said that “their evidence rose no higher than to speculate that there was a ‘possibility’ that there are core residues that don’t interact”. Amgen refers to CS [0542] which explains the basis for the selection of the 5 Å “cutoff”. That paragraph states:
Core interaction interface amino acids were determined as being all amino acid residues with at least one atom less than or equal to 5 Å from the PCSK9 partner protein. 5 Å was chosen as the core region cutoff distance to allow for atoms within a van der Waals radius plus a possible water-mediated hydrogen bond. Boundary interaction interface amino acids were determined as all amino acid residues with at least one atom less than or equal to 8 Å from the PCSK9 partner protein but not included in the core interaction list. Less than or equal to 8 Å was chosen as the boundary region cutoff distance to allow for the length of an extended arginine amino acid.
299 With respect to the additional criticisms made by Sanofi regarding Professor Petsko’s evidence, Amgen submitted that these arguments disregard the true content and meaning of Professor Petsko’s evidence. In particular, it said that the Later Crystallised Antibodies merely “reinforced” his view rather than impermissibly infected his evidence. Amgen also submitted that Sanofi has ignored the agreement in relation to many matters amongst Professors Petsko, Hudson and M Parker. It also said there was no inconsistency in evidence Professor Petsko gave in relation to the existence of non-covalent interactions as between his declaration and affidavit and, in any case, such inconsistency was never put to him.
Consideration of the Epitope and Residue Claims
300 The key consistory clauses in relation to the Epitope and Residue Claims are set out at above. Those consistory clauses refer to antibodies that bind to particular epitopes comprising particular residues, or antibodies that bind directly to particular residues.
301 Starting with 677, the content of paragraph [007c] mirrors claims 1 and 5. Paragraph [007c] describes an isolated MAb that binds to an epitope of hPCSK9 comprising one or more of amino acid residues 207, 208, 162, 164, 167 or 123 of SEQ ID NO: 1, which blocks the binding of PCSK9 to LDLR. Claim 1 of 677 is to an isolated MAb that binds to an epitope which includes one or more of those residues (i.e., the residue is part of the structural epitope). Claim 5 is to an isolated MAb that binds to one or more of those residues (i.e., the residue is part of the functional epitope). Both claims 1 and 5 are limited by the secondary requirement that the MAb blocks the binding of PCSK9 to LDLR.
302 Paragraph [007c], in referring generally to an “epitope”, encompasses both a structural and functional epitope to which the antibody binds. Any doubt about this is resolved by reference to CS [0571], which explains “[e]pitopes can be defined as structural or functional. Functional epitopes are generally a subset of the structural epitopes and have those residues that directly contribute to the affinity of the interaction…”.
303 Paragraph [007b] of 689 similarly mirrors the content of claim 1 of 689. Paragraph [007b] describes an isolated MAb that, when bound to PCSK9, binds to at least one of the following residues of SEQ ID NO: 3: S153, I154, P155, R194, D238, A239, I369, S372, D374, C375, T377, C378, F379, V380, or S381. Paragraph [007b] also requires that the MAb blocks binding of PCSK9 to LDLR. Those requirements are replicated in claim 1 of 689.
304 The same can be said of paragraphs [007b] and [007c] and claims 1 and 2 of 751. Paragraph [007b] of 751 describes an isolated MAb that recognises an epitope of human PCSK9 comprising the following amino acid residues: S153, R194, D238, D374, T377, and F379 of SEQ ID NO: 3, and reduces binding between PCSK9 and the EGFa domain of LDLR. Paragraph [007c] describes an isolated MAb that binds to those same amino acids and reduces binding between PCSK9 and the EGFa domain of LDLR. Claim 1 is a claim to an isolated MAb that recognises an epitope comprising those residues, which reduces binding between PCSK9 and the EGFa domain of LDLR. Claim 2 is to an isolated MAb which binds to those residues (i.e., the residues are in the functional epitope) and achieves the same function.
305 Claims 10 and 13 of 751 correspond to consistory clauses [007d] and [007e], which describe an isolated MAb that either recognises an epitope on human PCSK9 which comprises at least ten of the following residues of SEQ ID NO: 3: S153, I154, P155, R194, D238, A239, I369, S372, D374, C375, T377, C378, F379, V380, or S381, or one which binds to at least ten or more of those residues, and which reduces binding between PCSK9 and the EGFa domain of LDLR. Claim 10 is to an isolated MAb that binds to an epitope comprising ten or more of those residues, while claim 13 is to an isolated MAb that binds to ten or more of those residues. Again, each antibody reduces binding between PCSK9 and the EGFa domain of LDLR. With respect to 751, it appears to be uncontroversial that “recognises an epitope” equates to “binds an epitope”.
306 The various consistory clauses show that the invention the subject of each of the Applications is a class of antibodies which bind to structural or functional epitopes comprising one or more nominated residues, which either block or reduce binding between PCSK9 and LDLR (or more specifically, the EGFa domain of LDLR). The question is then whether the remainder of the CS describes the same invention as the consistory clauses.
Examples 28-30
307 It is apparent from the CS that the residues referred to in each of the Exemplary Epitope and Residue Claims have been drawn from the core and boundary residues identified through analysis of various solved crystal structures. That analysis is presented in Examples 28-30. Each of these examples identifies core and boundary residues of the “interaction interface” between PCSK9 and either the EGFa domain of LDLR, 31H4, or 21B12.
308 Example 28 presents “the solved crystal structure of PCSK9 ProCat…bound to the LDLR EGFa domain”. The structure is represented in Figure 17 and is said to reveal that “the interaction of PCSK9 and EGFa appears to occur across a surface of PCSK9 that is between residues D374 and S153”. The final three paragraphs provide:
[0505] Specific core PCSK9 amino acid residues of the interaction interface with the LDLR EGFa domain were defined as PCSK9 residues that are within 5 Å of the EGFa domain. The core residues are as follows: S153, I154, P155, R194, D238, A239, I369, S372, D374, C375, T377, C378, F379, V380, and S381.
[0506] Boundary PCSK9 amino acid residues of the interaction interface with the LDLR EGFa domain were defined as PCSK9 residues that are 5-8 Å from the EGFa domain. The boundary residues are as follows: W156, N157, L158, E159, H193, E195, H229, R237, G240, K243, D367, I368, G370, A371, S373, S376, and Q382. Residues that are underlined are nearly or completely buried within PCSK9.
[0507] As will be appreciated by one of skill in the art, the results from this example demonstrate where PCSK9 and EGFa interact. Thus, antibodies that interact with or block any of these residues can be useful as antibodies that inhibit the interaction between PCSK9 and the EGFa domain of LDLR (and/or LDLR generally). In some embodiments, antibodies that, when bound to PCSK9, interact with or block any of the above residues or are within 15-8, 8, 8-5, or 5 angstroms of the above residues are contemplated to provide useful inhibition of PCSK9 binding to LDLR.
(Emphasis added)
309 Example 29 presents “the crystal structure of full length PCSK9 … bound to the Fab fragment of 31H4”. The structure is depicted in Figures 18A and 18B. The text of the example provides:
[0509] The depicted structure also allows one to identify specific core PCSK9 amino acid residues for the interaction interface of 31H4 with PCSK9. This was defined as residues that are within 5 Å of the 31H4 protein. The core residues are as follows: W72, F150, A151, Q152, T214, R215, F216, H217, A220, S221, K222, S225, H226, C255, Q256, G257, K258, N317, F318, T347, L348, G349, T350, L351, E366, D367, D374, V380, S381, Q382, S383, and G384.
[0510] The structures were also used to identify boundary PCSK9 amino acid residues for the interaction interface with 31H4. These residues were PCSK9 residues that were 5-8 Å from the 31H4 protein. The boundary residues are as follows: K69, D70, P71, S148, V149, D186, T187, E211, D212, G213, R218, Q219, C223, D224, G227, H229, L253, N254, G259, P288, A290, G291, G316, R319, Y325, V346, G352, T353, G365, 1368, 1369, S372, S373, C378, F379, T385, S386, and Q387. Amino acid residues completely buried within the PCSK9 protein are underlined.
…
[0512] As will be appreciated by one of skill in the art, the results from Example 29 demonstrate where antibodies to PCSK9 can interact on PCSK9 and still block PCSK9 from interacting with EGFa (and thus LDLR). Thus, antigen binding proteins that interact with any of these PCSK9 residues, or that block any of these residues (e.g., from other antigen binding proteins that bind to these residues), can be useful as antibodies that inhibit the interaction of PCSK9 and EGFa (and LDLR accordingly). Thus, in some embodiments, antigen binding proteins that interact with any of the above residues or interact with residues that are within 5 Å of the above residues are contemplated to provide useful inhibition [of] PCSK9 binding to LDLR. Similarly, antigen binding proteins that block any of the above residues (which can be determined, for example, via a competition assay) can also be useful for inhibition of the PCSK9/LDLR interaction.
(Emphasis added)
310 Example 30 presents “the crystal structure of PCSK9 ProCat … bound to the Fab fragments of 31H4 and 21B12”, which is depicted in Figures 19A and 19B. The text of the example provides:
[0514] Specific core PCSK9 amino acid residues of the interaction interface with 21B12 were defined as PCSK9 residues that are within 5 Å of the 21B12 protein. The core residues are as follows: S153, S188, I189, Q190, S191, D192, R194, E197, G198, R199, V200, D224, R237, D238, K243, S373, D374, S376, T377, and F379.
[0515] Boundary PCSK9 amino acid residues of the interaction interface with 21B12 were defined as PCSK9 residues that were 5-8 Å from the 21B12 protein. The boundary residues are as follows: I154, T187, H193, E195, I196, M201, V202, C223, T228, S235, G236, A239, G244, M247, I369, S372, C375, and C378. Amino acid residues nearly or completely buried within the PCSK9 protein are underlined.
…
[0517] As will be appreciated by one of skill in the art, the results from Example 30 demonstrate where antigen binding proteins to PCSK9 can interact on PCSK9 and still block PCSK9 from interacting with EGFa (and thus LDLR). Thus, antigen binding proteins that interact with any of these PCSK9 residues or that block any of these residues can be useful as antibodies that inhibit the interaction of PCSK9 and EGFa (and LDLR accordingly). Thus, in some embodiments, antibodies that interact with any of the above residues or interact with residues that are within 5 Å of the above residues are contemplated to provide useful inhibition [of] PCSK9 binding to LDLR. Similarly, antigen binding proteins that block any of the above residues (which can be determined, for example, via a competition assay) can also be useful for inhibition of PCSK9/LDLR interaction.
311 Each of the residues listed in the Exemplary Epitope and Residue Claims is referred to in one or more of these examples, as the table below demonstrates:
Residue (SEQ ID NO: 1/ SEQ ID NO: 3) | Example |
123/S153 (Claims 1 and 5 of 677, 1 of 689, 1, 2, 10 and 13 of 751) | Example 28 (core), example 30 (core) |
124/ I154 (Claims 1 of 689, 10 and 13 of 751) | Example 28 (core), example 30 (boundary) |
125/ P155 (Claims 1 of689, 10 and 13 of 751) | Example 28 (core) |
162/ D192 (Claims 1 and 5 of 677) | Example 30 (core) |
164/ R194 (Claims 1 and 5 of 677, 1 of 689, 1, 2, 10 and 13 of 751) | Example 28 (core), example 30 (core) |
167/ E197 (Claims 1 and 5 of 677) | Example 30 (core) |
207/ R237 (Claims 1 and 5 of 677) | Example 28 (boundary), example 30 (core) |
208/ D238 (Claims 1 and 5 of 677, 1 of 689, 1, 2, 10 and 13 of 751) | Example 28 (core), example 30 (core) |
209/ A239 (Claims 1 of 689, 10 and 13 of 751) | Example 28 (core), example 30 (boundary) |
339/ I369 (Claims 1 of 689, 10 and 13 of 751) | Example 28 (core), example 29 (boundary), example 30 (boundary) |
342/ S372 (Claims 1 of 689, 10 and 13 of 751) | Example 28 (core), example 29 (boundary), example 30 (boundary) |
334/ D374 (Claims 1 of 689 Application, 1, 2, 10 and 13 of 751) | Example 28 (core), example 29 (core), example 30 (core) |
345/ C375 (Claims 1 of 689, 10 and 13 of 751) | Example 28 (core), example 30 (boundary) |
347/ T377 (Claims 1 of 689, 1, 2, 10 and 13 of 751) | Example 28 (core), example 30 (core) |
348/ C378 (Claims 1 of 689, 10 and 13 of 751) | Example 28 (core), example 29 (boundary), example 30 (boundary) |
349/ F379 (Claims 1 of 689, 1, 2, 10 and 13 of 751) | Example 28 (core), example 29 (boundary), example 30 (core) |
350/ V380 (Claims 1 of 689, 10 and 13 of 751) | Example 28 (core), example 29 (core) |
351/ S381 (Claims 1 of 689, 10 and 13 of 751) | Example 28 (core), example 29 (core) |
Sanofi’s submissions
312 I now turn to my consideration of Sanofi’s first alternative argument with respect to the Epitope and Residue Claims. At the outset, I find that, the invention described in the CS is a class of antibodies which bind to particular structural or functional epitopes, identified by reference to certain residues on PCSK9.
313 After identifying certain residues on PCSK9 within 5 Å and 5-8 Å of the EGFa domain of LDLR (in the relevant interaction interface), the CS explains that the results “demonstrate where PCSK9 and EGFa interact”, and that antibodies that interact with or block any of those residues “can be useful as antibodies that inhibit the interaction between PCSK9 and the EGFa domain of LDLR”. Examples 29 and 30 identify residues in the interaction interface between PCSK9, 21B12 and 31H4. Those examples end with statements explaining that antibodies which interact with the identified residues or block the identified residues are anticipated to be useful in inhibiting the interaction of PCSK9 and the EGFa domain of LDLR. CS [0512] explains that “blocking” a residue in this context means blocking a residue “from other antigen binding proteins that bind to these residues”.
314 In my view, the terms “interact” and “block”, used in these examples, are wide enough to encompass situations where the identified residues fall within structural or functional epitopes of the relevant antibody. While it is clear that residues “interacting” must necessarily capture residues in the functional epitope (i.e., those involved in chemical interactions that contribute to affinity), residues interacted with or blocked by the antibody must also include residues in the structural epitope, which is “the patch of the target… covered by the antibody” (CS [0571]).
315 Accordingly, the Epitope and Residue Claims can be fairly based, even if the CS does not state that the residues within those claims are part of structural or functional epitopes for either of the reference antibodies, or for any other antibody. It is enough that the CS explains that antibodies which interact with, or block the residues identified in examples 28-30 will be useful in the inhibition of PCSK9 binding to LDLR. It is not necessary for the CS to provide scientific proof of the relationship between core and boundary residues and the functional and structural epitopes of any antibody.
316 A large part of Sanofi’s first argument relied on the proposition that the claims cover many more antibodies than the true invention, which should be understood to be 21B12 and 31H4 (and perhaps 16F12). Sanofi has not pointed to any statement in the CS which shows that the invention disclosed is so limited. The CS describes a class of antibodies, in both the consistory clauses and elsewhere including in the discussion of Examples 28-32. In particular, CS [0524] states:
As will be appreciated by one of skill in the art, Examples 28-32, and their accompanying figures, provide a detailed description of how and where EGFa interacts with PCSK9 and how two representative neutralizing antigen binding proteins, 21B12 and 31H4 interact with PCSK9 and produce their neutralizing effect. As such, one of skill in the art will readily be able to identify antigen binding molecules that can similarly reduce the binding between EGFa (including LDLR) and PCSK9 by identifying other antigen binding molecules that bind at or near at least one of the same locations on PCSK9.
(Emphasis added)
317 It is true that the list of claimed residues is generated with reference to 21B12 and 31H4 (as well as the EGFa domain of LDLR). However, various parts of the CS describe a class of antibodies which block or interact with the residues identified in Examples 28-30. The CS does not need to exemplify every antibody the subject of the claims in order for the claims to be fairly based. Nor does the CS require the PSA to engage in impermissible extrapolation. The disclosures in the CS demonstrate that the reference antibodies exemplify the invention, but do not limit it.
318 Sanofi’s comparison between the exemplified antibodies and the potential total number of antibodies falling within the class (i.e., “hundreds, if not thousands or many more”) does not assist it once it is accepted that the invention described is not limited to 21B12, 31H4 and 16F12. While this kind of analysis might well be relevant to the support requirement now found in s 40(3) of the Act, it has no role to play in assessing fair basis.
319 In oral submissions, Sanofi also placed heavy emphasis on [59] of Lockwood No 1 and the following statement from Shave v H V McKay Massey Harris Pty Ltd (1935) 52 CLR 701 at 709, per Rich, Dixon, Evatt and McTiernan JJ:
When a combination claim states an invention which gives an old result by a new means, the monopoly is limited, at any rate prima facie, to the new means. But when by a new application of principle the inventor has obtained a new result or thing, even when it be done by a combination, he may claim all the alternative means by which the thing or result may be achieved.
Sanofi submitted that the CS describes a new application of a known principle, rather than the application of a new principle.
320 At face value, that submission does not fit easily into the consideration of fair basis. Putting aside issues of construction where reference to the common general knowledge is permitted, fair basis is essentially concerned with what the specification discloses to the PSA. In order for Sanofi’s submission to succeed, it would have to appear from the specification that the invention was an old result obtained by new means. Sanofi has not identified any matter which supports that characterisation of the invention. As Mr Cordiner KC, for Amgen, submitted:
There’s nothing in the specification which tells you that antibodies binding to PCSK9 are an old result. There’s nothing, actually, even going so far as saying antibodies which bind and block PCSK9 binding to LDLR is an old result, and certainly not one which is limited to the target regions or areas which these antibodies must bind in order to have the relevant properties of the claims.
321 The remainder of Sanofi’s first argument is that the claims omit essential elements of the inventions. The crux of this argument is that certain residues, identified as part of the “sweet spot” or as core residues in certain interaction interfaces, do not appear in the claims.
322 There is no basis for this submission. A claim which lacks fair basis because it omits a particular element is one that, by that omission, becomes a claim to a fundamentally different invention. That is to say, an element of an invention will not be “essential” in this context unless, by failing to include that element, the invention becomes a quite different one. That was the case in AstraZeneca FFC, where the omission of a certain requirement from the claims would have had the effect that the invention claimed was fundamentally different from that described in the priority document (in the context of external fair basis). That is plainly not the case here.
323 Sanofi also submitted that, for a number of claims, the CS does not identify a specific antibody (i.e., by name or sequence, as has been done for the Table 2 Antibodies) that falls within the claim. In support of that submission, Sanofi relied on a concession by Professor Petsko that, in an analysis performed of the crystal structures of 21B12, 31H4, and the Later Crystallised Antibodies, he did not identify any antibody that had non-covalent interactions with more than nine amino acid residues, meaning he could not identify an antibody covered by claim 13 of 751. Sanofi’s reliance on this concession is contrary to Sanofi’s insistence that Amgen should not be allowed to rely on evidence about the Later Crystallised Antibodies. Nevertheless, as I have explained, the CS does not need to prove that any of the exemplified antibodies fall within the claims (i.e., form non-covalent interactions with the relevant residues or bind to epitopes containing the relevant residues) in order for the claims to be fairly based.
324 Sanofi’s second alternative argument only arises if I accept that it is permissible to rely on the results of a PyMOL analysis. As set out above, this argument hinges on a criticism of Professor Petsko’s analysis of whether a non-covalent interaction exists between 21B12 and S153.
325 Putting aside the question of whether or not it is permissible to rely on an analysis performed by Professor Petsko of the data contained in Table 35, Amgen is correct that it does not have to prove a non-covalent interaction between the residues in the claims and 21B12 or 31H4. That is not required in order to establish that the claims are fairly based, for the reasons given when addressing Sanofi’s first argument. As such, Amgen does not need to rely on the PyMOL analysis.
326 Sanofi’s third alternative argument was, in essence, that the Epitope and Residue claims are not fairly based because the CS does not actually disclose that the residues referred to in the Epitope and Residue Claims are part of the structural or functional epitopes for any antibodies. The crux of this argument is that the structural and functional epitopes for antibodies the subject of the invention are not actually identified in the CS. This argument can be rejected for the same reasons as Sanofi’s first alternative argument. The disclosures in Examples 28-30, when read with the consistory clauses, provide a fair basis for the Epitope and Residue Claims.
327 Finally, Sanofi’s fourth alternative argument seems to be a restatement of its claim definition argument. As I understand it, Sanofi’s complaint here is that the CS does not sufficiently characterise, by reference to their amino acid sequences, all the antibodies that fall within the scope of the Epitope and Residue Claims. For the reasons set out above, this argument must fail. It is not necessary that the amino acid sequences for the entire class of antibodies be set out in order for the Epitope and Residue Claims to be fairly based.
328 The additional criticisms raised by Sanofi, including its criticism of aspects of Professor Petsko’s evidence, address matters outside the CS and are irrelevant to fair basis. Amgen does not need to prove the correctness of Professor Petsko’s analysis with respect to the crystal structures in order to establish fair basis.
329 In the result, I am not persuaded that any of the Epitope and Residue Claims would be clearly invalid for lack of fair basis.
Competition claims
330 Sanofi’s submissions on fair basis with respect to the Competition Claims were more limited. It repeats its submission that the invention as described in the specification is not the Table 2 Antibodies that compete with 21B12, 31H4 or both. It said that the “suggestion that an applicant invented both the invention described in the specification, together with anything that may be later demonstrated to compete with that thing at a later point in time, must be wrong as a matter of principle”. It then submitted that the competing antibodies must be limited to “antibodies defined by way of their unique, specific amino acid sequence in Figures 1A-3JJ and 3LL to 3BBB of the specification”. It said that the Competition Claims, contrastingly:
seek a monopoly in regard to any antibody that competes with 21B12 or 31H4 (as tested in any order under any conditions in any competition binding assay) and, on Amgen’s case, competes to any degree and reduces binding of PCSK9/LDLR to any degree.
331 Sanofi also submitted that if the Competition claims are understood to include those antibodies which compete to any degree, then the competing antibodies “are not described in the Applications by reference to any degree of competition”, and referred to evidence given regarding antibody 30A4, which was designated by Professor Petsko as falling within claim 1 of 748 and 685, and said by Professor Hudson to be “competing but not completely competing”.
332 Amgen submitted that the CS describes a class of antibodies that block PCSK9 binding to LDLR and compete with the reference antibodies. It drew attention to the following consistory clauses in 748 and 685:
748 | [007d] In another aspect of the present invention, there is provided an isolated monoclonal antibody that binds to human PCSK9 and is neutralizing in that an excess of said antibody reduces the quantity of human PCSK9 bound to LDLR in an in vitro competitive binding assay, wherein said monoclonal antibody competes for binding to PCSK9 with an antibody that comprises: a heavy chain variable region of the amino acid sequence in SEQ ID NO: 49; and a light chain variable region of the amino acid sequence in SEQ ID NO: 23. |
685 | [007d] In a further aspect of the present invention, there is provided an isolated monoclonal antibody that binds to human PCSK9 and reduces binding between human PCSK9 and an EGFa domain of LDLR, wherein said monoclonal antibody competes for binding to PCSK9 with an antibody that comprises a heavy chain variable region of the amino acid sequence in SEQ ID NO: 67; and a light chain variable region of the amino acid sequence in SEQ ID NO: 12. |
Attention should also be drawn to references to competition within the consistory clauses of 677, such as [0015]:
In some aspects, the invention comprises an isolated antigen binding protein that competes for binding to PCSK9 with an antigen binding protein disclosed herein.
333 Amgen said the CS provides detailed disclosure of multiple antibodies that bind, block, and bin/compete. Further, it said that Sanofi’s submissions also ignore evidence that the most likely cause of competition is an overlapping epitope.
Consideration of the Competition Claims
334 The key consistory clauses relevant to the Competition Claims are set out above. There does not appear to be any dispute that claims 1 of 748 and 685 correspond with paragraph [007d] as it is set out in each of those Applications.
335 Claim 1 of 748 is to an isolated MAb that, inter alia, competes with 21B12. Claim 1 of 685 is to an isolated MAb that, inter alia, competes with 31H4. The light and heavy sequence IDs referred to in each of the relevant claims and consistory clauses can be matched to those appearing in the Figures 3JJ (for 21B12) and 3E (for 31H4).
336 I am also satisfied that paragraph [0015] of 677 describes the invention of claim 23 of 677, which is a general claim to the isolated MAb according to any one of claims 1-20 (which includes the Exemplary Epitope or Residue Claims of that Application) and which competes with 21B12.
337 The question is then whether the remainder of the CS is consistent with those consistory clauses. Sanofi’s submissions on this question were very brief. Sanofi did not point to any particular statement in the CS that teaches against the invention being antibodies that compete with 21B12 and 31H4. The submissions instead again took issue with the breadth of the monopoly falling within the Competition Claims, and repeated some of the arguments previously considered.
338 At a general level, CS [0248] provides:
…In some embodiments, the ABP binds to any one of the epitopes bound by the antibodies discussed herein. In some embodiments, this can be determined by competition assays between the antibodies disclosed herein and other antibodies.
339 CS [0349]-[0350] explicitly contemplates the preparation of antibodies which compete with 21B12 and 31H4:
[0349] In some embodiments, the antigen binding protein binds to a region containing at least one of amino acids 162, 164, 167, 207 and/or 208 of SEQ ID NO: 1 or SEQ ID NO: 303. In some embodiments, more than one (e.g., 2, 3, 4, or 5) of the identified residues are part of the region that is bound by the ABP. In some embodiments, the ABP competes with ABP 21B12.
[0350] In some embodiments, the antigen binding protein binds to a region containing at least one of amino acid 185 of SEQ ID NO: 1 or SEQ ID NO: 303. In some embodiments, the ABP competes with ABP 31H4.
(Emphasis added)
That is a disclosure of the invention of a class of antibodies that compete with each of the reference antibodies referred to in the Competition Claims.
340 The nature of the examples, including Examples 10 and 37 which rely on competition assays, suggests that the invention described in the CS is, in addition to antibodies which bind to particular residues or epitopes, a class of antibodies that compete with the reference antibodies or other antibodies described in the CS. The text contained in Examples 28-30 (reproduced above) refers to antibodies which “block” the specified residues as being useful, wherein the term “block” is used to describe one ABP blocking another from binding to said residues. That is also consistent with the description of a class of antibodies which compete with the reference antibodies including by the blocking of certain residues.
341 Sanofi also referred to the degree of competition required for an antibody to fall within the claims. There is nothing in the CS which would suggest that a claim to antibodies which compete could not be fairly based because it did not specify any particular percentage or degree of competition.
342 As to claim 23 of 677, given that the dependent elements of that claim are fairly based (as explained above in relation to the Epitope and Residue Claims), and there is clear disclosure of a class of antibodies that compete with antibodies within the CS, this claim is also fairly based on the CS.
343 I am not persuaded that any of the Competition Claims would be clearly invalid for lack of fair basis.
PRIORITY DATE
344 As set out above, each of the Applications claims an earliest asserted priority date of 23 August 2007 based on the filing of US 60/957,668 (P1). Other potential priority dates are 21 December 2007 based on the filing of US 61/008,965 (P2), 9 January 2008 based on the filing of US 61/010,630 (P3) and 4 August 2008 based on the filing of US61/086,133 (P4). All four documents are United States provisional patent applications.
345 Relevantly, and as touched upon above in relation to fair basis, P1 includes Examples 1 to 26 (including many of the relevant antibody sequences and blocking and binning studies). P2 includes the additions of Examples 27 to 35 (including crystal structures of PCSK9 in complex with LDLR, 31H4 and 21B12, and the accompanying analysis). P3 includes the additions of paragraphs at CS [0555], [0556] and [0557] (contained in P3 [0461]) and Tables 35.1, 35.2 and 35.3. P4 includes the additions of Examples 36 to 41 (except for the parts of those examples added by P3).
346 Section 40(1) of the Act as it stood before amendment by the RTB Act requires that a provisional specification “describe the invention”. It does not require that a provisional specification fully describe the invention or provide an enabling disclosure: cf. 40(1) after amendment by the RTB Act. As a result of the difference between these two standards, the requirement of external fair basis (i.e., the requirement that the claims are fairly based upon the relevant priority document) calls for something less than the requirement of internal fair basis, which I have dealt with above.
347 As to the description of the invention required of a provisional application under s 40(1) before its amendment, Lockhart J (with whom Wilcox J agreed) said in Anaesthetic Supplies Pty Ltd v Rescare Ltd (1994) 50 FCR 1 at 20:
All that the provisional specification needs to do is describe generally and fairly the nature of the invention, and not to enter into all the minute details as to the manner in which the invention is to be carried out. It is a mode of protecting an inventor until the time of filing the final specification. It is not intended to be a complete description of the invention, but simply to disclose the invention fairly, though in its rough state. The interval of time between the provisional and the final is intended to provide an opportunity for the development and precise expression of the invention foreshadowed in the provisional.
348 The Full Court in CCOM at 280-281, and the Full Court in Leonardis v Sartas No 1 Pty Ltd (1996) 67 FCR 126 at 139-140, referred with approval to Buckley LJ’s judgment in Stauffer Chemical Co’s Application [1977] RPC 33 (“Stauffer”) in which his Lordship said at 54:
If a new feature were a development along the same line of thought which constitutes or underlies the invention described in the earlier document, it might be that that development could properly be regarded as fairly based on the matter disclosed in the earlier document, and that the new process described in the later document which incorporates that development could as a whole be regarded as fairly based upon the matter disclosed in the earlier document. If, on the other hand, the additional feature involves a new inventive step or brings something new into the combination which represents a departure from the idea of the invention described in the earlier document, it could not, I think, be properly described as fairly based upon the earlier document.
349 The Full Court in CCOM also referred with approval to the following observation of Fox J in Coopers Animal Health Australia Ltd v Western Stock Distributors Pty Ltd (1987) 15 FCR 382 at 389:
Where the holder of the provisional specification proceeds with a complete specification with a view to the grant of a patent, it is recognised that greater definition, as a result of further experimentation or otherwise, may be achieved before the later step is taken and the result expressed therein. Some generality of expression in the provisional specification is accepted.
P1
350 P1 does not include any specific description or disclosure of any epitope or residue to which any antibody binds. It does not include the results of experiments conducted using X-ray crystallography, or arginine scanning, which I have referred to above in dealing with internal fair basis. P1 does, however, include Figures 3E-3JJ, which include the amino acid sequences for the variable regions of the heavy and light chains of the Table 2 Antibodies (including 21B12 and 31H4). It will be recalled here that a large proportion of those Table 2 Antibodies are strong neutralizers: see CS [0229], P1 [0145].
351 The consistory clauses of P1 include the following:
[0006] In some embodiments, the invention comprises an antigen binding protein to PCSK9.
[0007] In some aspects, the invention comprises an isolated antigen binding protein that binds PCSK9 comprising…[reference is made here to various combinations of the heavy chain CDRs and light chain CDRs identified by amino acid sequence in P1].
[0008] In some aspects, the invention comprises an isolated antigen binding protein that specifically binds to an epitope that is bound by any of the ABPs disclosed herein.
…
[0012] In some aspects, the invention comprises an isolated antigen binding protein that competes for binding to PCSK9 with an antigen binding protein disclosed herein.
352 Reference is also made to an ABP which binds to various domains of PCSK9. For example, P1 [0164] states:
In some embodiments, the ABP binds to the catalytic domain of PCSK9. In some embodiments, the ABP binds to the mature form of PCSK9. In some embodiments the ABP binds in the prodomain of PCSK9. In some embodiments, the ABP selectively binds to the mature form of PCSK9. In some embodiments, the ABP binds to the catalytic domain in a manner such that PCSK9 cannot bind or bind as efficiently to LDLR…In some embodiments, the ABP binds to any one of the epitopes bound by the antibodies discussed herein. In some embodiments, this can be determined by competition assays between the antibodies disclosed herein and other antibod[i]es. In some embodiments, the ABP binds to an epitope bound by one of the antibodies described in Table 2. In some embodiments, the antigen binding proteins bind to a specific conformational state of PCSK9 so as to prevent PCSK9 from interacting with LDLR.
See also P1 [0184] and [0251].
Priority date of the Epitope and Residue Claims
353 Sanofi submitted that the priority date of the Epitope and Residue Claims could not be the date of filing of P1 (23 August 2007) because there is no description or disclosure in P1 of any epitopes or any residues to which any antibody binds. I do not accept that submission.
354 I accept that those parts of P1 to which I have referred do not identify specific epitopes or residues on PCSK9 to which antibodies disclosed in P1 bind. P1 does not describe the class of antibodies falling within the claims by reference to a set of residues to which the antibodies bind directly, or residues within the epitopes to which the antibodies bind. Instead, the language of P1 is much more general.
355 In its written submissions Sanofi characterised the further work recorded in P2 to P4 as no more than “routine experiments” directed to elucidating the properties of the antibodies previously made by the inventors. I accept that this further work was routine and non-inventive. Importantly, however, the Epitope and Residue Claims do not introduce any feature involving a new inventive step (beyond anything already disclosed in P1) or any other matter which represents a departure from the more general description of the invention in P1.
356 In my opinion, the CS follows “the same line of thought” disclosed in P1. Applying the test enunciated by Buckley LJ in Stauffer, I am not persuaded that P1 does not confer priority on the Epitope and Residue claims.
357 Although it is not necessary to do so, I will refer briefly to some submissions made by Sanofi in relation to P2 in case my conclusion in relation to the 23 August 2007 priority date is wrong.
358 Sanofi submitted that the “core” and “boundary” residues described in P2, and the atomic coordinates provided in P3, do not provide a real and reasonably clear disclosure of the residues within the epitopes for 21B12 and 31H4, or the residues to which those antibodies bind.
359 As I have set out above in the context of fair basis, the disclosure of core and boundary residues obtained through a crystallographic analysis of various interaction interfaces in Examples 28-30 is enough to fairly base the Epitope and Residue Claims. The core and boundary residues are described as being useful in the inhibition of PCSK9’s interaction with LDLR. It follows that in the event that the disclosure in P1 is insufficient to confer priority from 23 August 2007, the disclosure of these residues in P2 is sufficient to confer priority on the Epitope and Residue Claims.
360 I note as well that the residues referred to in the claims of 677 identified by reference to SEQ ID NO: 1 (residues 207, 208, 162, 164, 167 and 123) are identified by reference to SEQ ID NO: 3 in P2 (although it is referred to as SEQ ID NO: 1 in that document) at [0025], which corresponds with CS [0040]. That paragraph describes embodiments of the invention in which the antibody binds to PCSK9 and is positioned 8 Å or less from at least one of the of the identified residues. Those same paragraphs also identify the other residues referred to in the Exemplary Epitope and Residue Claims.
361 As to P4, Sanofi also contended that the CS teaches that predictions based on X-ray crystallography need to be confirmed by other experimental approaches, such as arginine scanning. It referred to CS [0479] which states:
In the alternative, given that it is possible that the epitope is conformational, a combination of alanine scanning and/or arginine scanning, antibody FAB/PCSK9 co-crystalization [sic], and limited proteolysis/LC-MS (liquid chromatography mass spec.) can be employed to indentify [sic] the epitopes.
According to Sanofi’s submission, it follows from this paragraph that the priority date of each of the Epitope and Residue Claims can be no earlier than the disclosure of the arginine scanning experiments upon the filing of P4.
362 I do not accept that the priority date depends on the disclosure of the arginine scanning experiments. That submission can firstly be rejected for the same reasons I have rejected Sanofi’s submission that interactions between PCSK9 and the residues the subject of the Epitope and Residue Claims must be proven via arginine scanning for the claims to be fairly based. Additionally, Sanofi’s submission based on CS [0479] misstates the effect of that paragraph. CS [0479] merely describes the means by which conformational epitopes may be identified. It does not disclose the invention or limit the disclosures of the invention appearing elsewhere in the CS. It indicates that a combination of mutagenesis and crystallography can be used, not that it is required. At no point does CS state or imply that arginine scanning is required to establish epitopes or residues.
363 In the result, Sanofi’s contention that the Epitope and Residue claims are not entitled to a priority date any earlier than the date on which P4 was filed (4 August 2008) is rejected.
Priority date of the Competition Claims
364 As to the Competition Claims, Sanofi’s submissions on priority date were brief. Its written submissions merely referred to submissions made by Sanofi in relation to the asserted lack of internal fair basis for the Competition Claims, which were broadly to the effect that the Table 2 Antibodies are not the invention described in the CS, and that at best the competing antibodies must be the limited number of antibodies defined by way of their amino acid sequences in the figures of the CS.
365 Both reference antibodies 21B12 and 31H4 were described in Table 2 of P1, and the sequences for the light and heavy chain variable regions for those antibodies appear in Figures 3JJ and 3E of P1, respectively. P1 [0012] provides that “in some aspects, the invention comprises an isolated antigen binding protein that competes for binding to PCSK9 with an antigen binding protein disclosed herein.” P1 also includes at [0147] a definition of the term “competes” and a description of various assays that can be used to assess competition. At P1 [0351] to [0353] the results of Example 10 are provided, including the identity of 18 antibodies that competed with 21B12 and 6 antibodies that competed with 31H4. Even though the Table 2 Antibodies are not the invention, P1 nevertheless describes the invention the subject of the Competition Claims in 685 and 748 generally and fairly.
366 The disclosures I have identified above in P1 are also sufficient to confer priority on Claim 23 of 677, which is also directed to antibodies that compete with 21B12, although Sanofi’s submissions on the priority date of the Competition Claims are directed only to those claims in 748 and 685.
367 In the result, Sanofi’s contention that the Competition claims are not entitled to a priority date based on P1 is also rejected.
THE NOTIONAL PERSON SKILLED IN THE ART
The Skilled Addressee
368 The notional skilled addressee, or the PSA, is a legal construct and a tool of analysis framed by reference to the available evidence. This will include the patent application and evidence of persons with knowledge and experience in the field of the invention. The skilled addressee brings to the reading of the relevant document the background knowledge and experience available to those working in that field. The legal construct may not be a single person but may be a team of persons “whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those which are contained in the document to be construed”: General Tire at 485.
369 It is common ground that the PSA in this case will be a team. Amgen submitted that the PSA in this case was a team led by a senior biochemist/biologist and/or medicinal/organic chemist, which has expertise in discovering and developing drug therapies, and which would be able to call upon, inter alia, an antibody expert and a structural biologist with relevant crystallographic expertise. The team would have access to an expert on PCSK9, who would act as a consultant. Sanofi submitted that the PSA in this case is a notional team of skilled scientists with knowledge and experience in the field of antibodies, protein crystallography and PCSK9.
370 There is little difference between the parties’ descriptions of the PSA except in so far as Amgen seeks to characterise various experts, including the PCSK9 expert, as sitting outside the core team. Thus, on Amgen’s formulation, the PCSK9 expert is not a member of the team, but is instead a person with whom the team may consult.
371 Dr R Parker and Professor Horton were asked about the qualifications and experience of those with an interest in PCSK9 and its inhibition. Sanofi’s submission as to the composition of the notional team broadly reflects the agreement reached between Dr R Parker and Professor Horton in the JER. Dr R Parker, who previously worked for BMS, provided some additional commentary which I found helpful:
Industry scientists with interests in PCSK9 and its inhibition were biomedical researchers with experience in lipid/cholesterol metabolism and cardiovascular and metabolic diseases. PCSK9 was considered among the highest priority targets at Bristol-Myers Squibb (“BMS”) and was resourced accordingly (involvement expanded as work progressed). The BMS PCSK9 drug discovery team comprised scientists with the following backgrounds. The team was led by a senior PhD biochemist/biologist with 20 years of experience (i.e., myself), and co-led by a senior PhD medicinal/organic chemist with ~15 years of experience. The team included other PhD and master level scientists with 5-10 or more years of experience. Together, team and leaders had expertise and experience in the following disciplines: molecular biology, cell biology, immunochemistry, fluorescence microscopy and instrumentation, in vivo physiology, applied genomics/ bioinformatics, high-throughput screening/instrumentation technology, protein expression and purification, computer-aided drug design (CADD)/protein in silico modelling, protein biophysics, protein crystallography, nuclear magnetic resonance (NMR), pharmacokinetics, and toxicology. Each senior scientist had a lab group with bachelor/master level associate scientists. The team also had connections with clinical researchers (MD and associates) and the global marketing group for relevant advice. Smaller biotechnology companies would obviously be more limited in scope and scale of the group interested in PCSK9 and its inhibition.
372 Dr R Parker’s opinion recognises that the composition of the team may vary in the case of smaller biotechnology companies. However, based on his evidence of his experience at BMS, I find that the PSA comprises a team of researchers led by an experienced PhD biochemist/biologist and PhD medicinal chemist. The team would have expertise and experience in (inter alia) molecular biology, protein biophysics, protein modelling and crystallography. The team also includes researchers with expertise in lipid/cholesterol metabolism, cardiovascular diseases and PCSK9.
COMMON GENERAL KNOWLEDGE
373 The relevant principles relating to common general knowledge were summarised by me as follows in ToolGen at [97]-[98]:
[97] The common general knowledge is a general body of knowledge attributed to the hypothetical non-inventive skilled person or team. In Minnesota Mining and Manufacturing Company v Beiersdorf (Australia) Limited (1980) 144 CLR 253, Aickin J at 292 referred to common general knowledge as “… the background knowledge and experience which is available to all in the trade in considering the making of new products, or the making of improvements in old, and it must be treated as being used by an individual as a general body of knowledge”. It is knowledge actually known and used by skilled persons in the relevant field generally or accepted by the bulk of those who are engaged in the particular art: Ranbaxy Laboratories Ltd v AstraZeneca AB (2013) 101 IPR 11, Middleton J at [215] (“Ranbaxy Laboratories”) citing British Acoustic Films Ltd v Nettlefold Productions (1936) 53 RPC 221 at 250 (“British Acoustic Films”). Knowledge is not common general knowledge unless it is sufficiently widely known or used to become generally accepted and assimilated into the minds of people skilled in the relevant art and therefore part of the common general knowledge: see Gilead Sciences Pty Ltd v Idenix Pharmaceuticals LLC (2016) 117 IPR 252 at [210]-[214] (“Gilead”) where Jagot J referred to some of the leading authorities including, Aktiebolaget Hässle v Alphapharm Pty Ltd (2002) 212 CLR 411 at [31]. It will include publications to which the skilled addressee would refer as a matter of course: ICI Chemicals and Polymers Ltd v Lubrizol Corporation Inc (1999) 45 IPR 577 at [112] per Emmett J.
[98] However, information does not become common general knowledge merely because it might appear in a journal even if it is one read by persons in the art: Ranbaxy Laboratories at [217] per Middleton J citing (inter alia) Eli Lilly & Co Ltd v Apotex Pty Ltd (2013) 100 IPR 451 at [468]; Wake Forest University Health Sciences v Smith & Nephew Pty Ltd (No 2) (2011) 92 IPR 496 at [96], General Tire at 482-3. In the latter case, the English Court of Appeal referred with approval to the following passage in the judgment of Luxmoore J in British Acoustic Films at 250:
In my judgment it is not sufficient to prove common general knowledge that a particular disclosure is made in an article, or series of articles, in a scientific journal, no matter how wide the circulation of that journal may be, in the absence of any evidence that the disclosure is accepted generally by those who are engaged in the art to which the disclosure relates. A piece of particular knowledge as disclosed in a scientific paper does not become common general knowledge merely because it is widely read, and still less because it is widely circulated. Such a piece of knowledge only becomes general knowledge when it is generally known and accepted without question by the bulk of those who are engaged in the particular art; in other words, when it becomes part of their common stock of knowledge relating to the art.
While the relevant principles were not in dispute, there was significant disagreement between the parties as to how they apply in this case.
374 Section 7(2), before its amendment by the RTB Act, refers to the common general knowledge “as it existed in the patent area”. The limitation imposed by those words was removed by the RTB Act amendments. For the purposes of this case the limitation still applies.
375 Amgen submitted that Sanofi failed to establish what the common general knowledge was in Australia as at the priority date. In particular, it was submitted that there was no evidence from which it could be inferred that there was any common general knowledge in Australia relating to PCSK9 and its role as a potential therapeutic target. So, while Amgen accepted that the PSA would include the expertise of a PCSK9 expert, it also submitted that the common general knowledge as it existed in Australia at the priority date would include little if any information as to PCSK9 and its role as a potential therapeutic target.
376 There was little direct evidence as to the extent to which there were any researchers studying PCSK9 and its relationship to LDL in Australia at the priority date. The evidence touching on that question included affidavit and oral evidence from Dr R Parker and a scientific paper entitled “Identification and characterization of two non-secreted PCSK9 mutants associated with familial hypercholesterolemia in cohorts from New Zealand and South Africa” by Vivienne M Homer et al published in Atherosclerosis in 2008 (“Homer 2008”) which was accepted for publication on 11 July 2007 and available online from 31 August 2007.
377 The lead author of Homer 2008, Vivienne Homer, is associated with Canterbury Health Laboratories in Christchurch, New Zealand, which is where I infer she conducted research work with four of her co-authors. Three other co-authors, Francesca Charlton, Fabien Mangilia and Phillip Barter, are associated with The Heart Research Institute, Camperdown, Sydney, NSW, which is where I infer they worked. Another author, David R Sullivan, is shown as associated with the Royal Prince Alfred Hospital in Sydney, NSW, where I infer he worked. Another author, Gilles Lambert, is shown as associated with both the Heart Research Institute in Sydney and the Université de Nantes Inserm in Nantes, France.
378 Homer 2008 includes the results of a study of patients with familial hypercholesterolemia (“FH”) and the subsequent investigation of mutations in PCSK9 associated with FH. The study concludes, inter alia, “[b]ecause PCSK9 inhibition provides long-term protection against cardiovascular diseases … the development of a pharmacological inhibitor of PCSK9 seems promising”. The literature cited in Homer 2008 includes important papers by Lagace 2006 and Horton 2007 referred to in more detail later in these reasons. Publication details for each paper I refer to by the name of the lead author and publication year appear in the bibliography in Annexure A.
379 Dr R Parker’s affidavit evidence was that, to the best of his recollection, none of his colleagues at BMS were from Australia, or left to work in Australia, and he was not aware of any Australian scientists or researchers with a knowledge or interest in PCSK9 prior to January 2008. However, he gave oral evidence in which he acknowledged having become aware of Homer 2008 at or about 31 August 2007. He also gave some oral evidence as to his expectations concerning common practice in the industry. As to that evidence, Dr R Parker was referred in his cross-examination to various publications including that of Lagace 2006 and other publications in “high-impact peer review journals”. Dr R Parker gave the following evidence:
MR SHAVIN: And people in the field would look to those journals as journals that had a strong peer review process and produced high-quality papers.
DR PARKER: As did I.
MR SHAVIN: Yes. And so to understand what is happening, you would expect that your colleagues, like you, would have been looking for publications, be that online or ultimately in print.
DR PARKER: They were, certainly. We discussed them.
MR SHAVIN: Yes. And you discussed them at conferences.
DR PARKER: No. I mean, internally in our team.
MR SHAVIN: In your team. And you would have expected that that was common practice in the industry.
DR PARKER: I would.
MR SHAVIN: Yes. Including in Australia.
DR PARKER: Definitely.
HIS HONOUR: Sorry. Did you say, “definitely”?
DR PARKER: Definitely, I said. Yes. Definitely. Yes.
380 Amgen sought to answer this evidence by referring to Dr R Parker’s affidavit evidence. It submitted that his oral evidence must have been speculation because he knew no Australian colleagues. I do not accept that submission. I consider it likely that there were researchers working on PCSK9 and exploring its relationship to FH in Australia before the priority date. I also infer that those researchers were, like Dr R Parker and others working in the field, staying abreast of developments by reading high-quality journals such as that in which Lagace 2006 appeared at the time of or shortly after their online publication. The evidence in my view supports the inference, and I find, that PCSK9 including its relationship to LDL cholesterol and the potential role of PCSK9 inhibitors in the reduction of LDL cholesterol levels, occupied a field of research that was essentially global in nature, and that what was known to, and accepted by, researchers in the United States, New Zealand, France and other countries, would have also been known and accepted by researchers working in the field in Australia.
381 Subject to there being a finding that the common general knowledge in the field was the same in Australia and overseas, Amgen accepted (for the purpose of this proceeding only) that, as at 23 August 2007, the common general knowledge included various matters set out in a document filed on 24 November 2023 (“the Amgen CGK Admissions”). I will refer to these admissions in more detail where necessary when considering the question of obviousness. Some of the Amgen CGK Admissions are cross-referenced to the publications from which they were drawn and include information drawn from various scientific papers relating to PCSK9, including Benjannet 2004, Rashid 2005, Maxwell 2005, Cohen 2006 and Lagace 2006.
382 Other matters admitted by Amgen to be common general knowledge in its submissions included:
Genetic studies clearly established that the genetic loss of PCSK9 resulted in lower plasma LDL cholesterol levels and a reduction of cardiovascular events. It was clearly desirable to seek a PCSK9 inhibitor to reduce LDL levels (CGK1).
The site at which PCSK9 functioned to lower LDL cholesterol and potentially the mechanism by which it worked would direct the approach used to identify and develop a PCSK9 inhibitor (CGK2).
The literature reported studies investigating both an extracellular and intracellular pathway for PCSK9. Both pathways may have co-existed. The quantitative contributions of extracellular and intracellular pathways in the overall physiological action of PCSK9 were not known and were difficult to determine (CGK3).
While those matters are covered by Amgen’s express admissions, I should also record that they are in my opinion established by the evidence of Professor Horton and Dr R Parker.
383 Other matters asserted by Amgen to be common general knowledge at the priority date included:
LDL competes with PCSK9 for binding to LDLR extracellularly (i.e. outside the cell) (CGK4).
PCSK9 binds LDLR with significantly higher affinity (leading to tighter binding) at acidic endosomal pH (inside the cell) when compared to neutral pH (outside the cell). Binding affinity data and plasma concentration levels of PCSK9 suggested that PCSK9 would not bind LDLR to any appreciable degree at neutral pH (outside the cell) (CGK5).
There was no information specifying which residues of PCSK9 interacted with the EGFa domain of LDLR (CGK6).
The following approaches to developing a PCSK9 inhibitor were “agnostic” to the cellular site at which PCSK9 operated in vivo, meaning that they could inhibit PCSK9’s activity regardless of whether PCSK9 functioned intracellularly, extracellularly or both:
(i) Antisense oligonucleotides (“ASOs”) or RNA interference (e.g. small interfering RNA or siRNA) to degrade the PCSK9 mRNA in the liver (if the mRNA is degraded no protein is ever made and thus this approach would not depend on where PCSK9 functions);
(ii) small molecules that inhibited the autocatalytic cleavage of PCSK9 in the endoplasmic reticulum (“ER”) of the liver cell (it had been demonstrated before the priority date that PCSK9 must cleave itself in the ER for it to fold normally and be secreted and thus this approach would also not depend on where PCSK9 functions); and
(iii) small molecules that selectively and specifically blocked PCSK9 secretion from the cell (all studies suggested that the activity of PCSK9 occurs in a post-ER compartment).
Because these approaches aimed to inhibit PCSK9 before its secretion from the ER, they worked inside the cell (i.e. intracellularly) (CGK7).
Potential therapeutic agents to inhibit PCSK9 outside of the cell included antibodies, small molecules, and peptides. Antibodies can only work to bind PCSK9 outside the cell. For antibodies to be effective as a PCSK9 inhibitor, PCSK9 had to function primarily outside the cell (extracellularly), i.e. at least 50% (CGK8).
384 I did not understand CGK4, CGK6, CGK7 or CGK8 to be contentious. I accept they were common general knowledge at the priority date. As to CGK5, I accept that some studies (including Fisher 2007 and Cunningham 2007) showed that PCSK9 bound with significantly higher affinity at the acidic endosomal pH compared to neutral extracellular pH, which suggested that a small portion of LDLR was bound by PCSK9 extracellularly. However, the evidence cited by Amgen in support of CKG5 (including Cunningham 2007 and Zhang 2007, both of which I accept were CGK) does not establish it was known and accepted by persons skilled in the art that PCSK9 would not bind LDLR to “any appreciable degree” outside the cell at neutral pH.
385 I also accept Amgen’s submission that:
The first and only publication before the priority date showing in vivo proof of concept of a PCSK9 therapeutic was Graham 2007, which reported that ASOs suppressed PCSK9 mRNA gene levels in the liver and generated cholesterol lowering effects in vivo in a mouse model without adverse effects.
The evidence does not disclose the existence at the priority date of any publicly available information indicating that any person or any company was developing an antibody as a therapeutic PCSK9 inhibitor.
386 Amgen did not accept that at the priority date PCSK9 was “one of the most promising pharmaceutical targets since the development of statins”. However, those words reflect the opinion expressed by Dr R Parker in the JER in which he said:
I considered that PCSK9 was the one of the most promising pharmaceutical targets since the development of statins … We (meaning I and my colleagues in the BMS PCSK9 drug discovery team) also recognized that it would be challenging. We saw that effective inhibition or suppression of PCSK9 expression had the potential to lower LDL levels even more than statins, and that this would have beneficial effects on “hard” CV clinical endpoints including mortality. It could be additive or synergistic with statins and they could potentially be used together in some patients … We saw that a range of therapeutic approaches might be able to accomplish this. The unknowns about how and where PCSK9 functioned meant that multiple approaches and modalities were called for, and a lot of resources were warranted …
Dr R Parker’s evidence indicates that he considered PCSK9 to be a promising but challenging target which might be addressed by a range of approaches including its inhibition or suppression.
387 Dr R Parker and Professor Horton agreed that the site at which PCSK9 functioned to lower cholesterol, and potentially the mechanism by which it worked, would direct the approaches used to identify and develop an inhibitor. As at the priority date, the site at which PCSK9 functioned to lower LDL cholesterol had not been conclusively determined.
388 Of the different approaches to PCSK9 inhibition available, there were some, which I have set out above, that were agnostic as to where PCSK9 functioned. Dr R Parker and Professor Horton disagreed as to the overall enthusiasm and feasibility of the various approaches at the priority date. Whether antibodies were feasible depended on the extent to which PCSK9 functioned extracellularly, something which had not been established at the priority date.
389 Professor Horton suggested that he placed little weight on the literature which suggested that PCSK9 functioned to reduce LDLR levels intracellularly for various reasons, including some relating to the design of the reported studies and the fact that in his opinion this hypothesis did not make biological sense. However, in my opinion this evidence is very likely the product of hindsight. His more contemporaneous statements on the topic appear in papers he co-authored. For example, in Horton 2007 he stated (at p 367) that “[t]he major cellular site at which PCSK9 acts has not been established”. In a presentation Professor Horton gave on 21 December 2007 he (and his co-authors/co-presenters) stated that “[c]urrent evidence suggests that PCSK9 may work at two cellular sites”. I find that it was common general knowledge at the priority date that the major cellular site at which PCSK9 degraded LDLR had not been established and that it was possible that it had both intracellular and extracellular activity. However, the extent of any extracellular activity had not been established. In this regard, I find that the state of the common general knowledge as at the priority date was as summarised by Lagace 2006 as follows:
Considered together, the available data now suggest that PCSK9 can function both extra- and intracellularly, but we do not know which pathway predominates under normal and/or pathologic conditions.
This statement is discussed in more detail below, but it is important to note that it is expressed rather tentatively (i.e., “the available data now suggest”) which is consistent with Dr R Parker’s view that this was an “emerging hypothesis”.
390 As to other matters, I note that both parties accepted that antibody engineering and protein science is an international field and that there were many leading scientists working in this field in Australia at the priority date. Amgen submitted, and I accept, that the procedures and technologies available for making and screening MAbs were by this time reasonably mature. Those techniques are described in these reasons in the discussion of the scientific background and the CS. They would have been well-known and routinely used by members of the notional team at the priority date engaged in the making and screening of MAbs intended to target PCSK9.
INVENTIVE STEP
391 The principles relevant to lack of inventive step (obviousness) were not in dispute. It is sufficient for my purposes to refer to the following summary of some of the key principles in Neurim Pharmaceuticals (1991) Ltd v Generic Partners Pty Ltd (No 5) [2024] FCA 360:
[384] An invention is a patentable invention if the invention, when compared with the prior art base, involves an inventive step: see s 18(1)(b)(ii) of the Act. Section 7(2) of the Act uses the word “obvious” in the course of describing what must be established before an invention can be held not to involve an inventive step. Something may be “obvious” in light of the common general knowledge, or the common general knowledge coupled with relevant s 7(3) information.
[385] An invention may be obvious in light of the common general knowledge if the person skilled in the art faced with the same problem as the inventor would have taken, as a matter of routine, whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not. If the person skilled in the art would be directly led as a matter of course to take such steps in the expectation that doing so might well produce a useful or better alternative to the prior art then the invention will be obvious: Wellcome Foundation Limited v VR Laboratories (Aust.) Proprietary Limited (1981) 148 CLR 262 (“Wellcome”) at 286 per Aickin J, [Aktiebolaget Hässle v Alphapharm Pty Limited (2002) 212 CLR 411 (“Alphapharm”)] at [50]-[53]. In Alphapharm at [53] the plurality approved what is commonly referred to as the reformulated or modified Cripps question as providing an acceptable approach to obviousness.
[386] A claimed invention is not obvious merely because the person skilled in the art would consider that it was “worthwhile to try”. However, there will be some cases in which the person skilled in the art will be directly led, as a matter of course, to try a number of different alternatives in the expectation that each may well produce a useful alternative. Merely because one pathway to an invention is shown to be obvious, does not mean that another such pathway might not also be obvious. Two or more pathways may be obvious, even though some of them might be more obvious than others.
[387] Further:
• The question is not whether the claimed invention is obvious to the Court, but whether it would be obvious to the person skilled in the art who is taken to be equipped with the relevant common general knowledge as it stood at the priority date together with any relevant s 7(3) information but who lacks any capacity for inventiveness.
• A “scintilla of invention” can sustain a valid patent, but there must be “some difficulty overcome, some barrier crossed” or something “beyond the skill of the calling”: Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) (2007) 235 CLR 173…at [52]; RD Werner & Co Inc v Bailey Aluminium Products Pty Ltd (1989) 25 FCR 565 at 574; Allsop Inc v Bintang Ltd (1989) 15 IPR 686 at 701.
• The court must be wary of the misuse of hindsight or ex post facto analysis in deciding whether a claimed invention lacks any inventive step: see Aicken J in [Minnesota Mining and Manufacturing Company v Beiersdorf (Australia) Limited (1980) 144 CLR 253] at 293-294 and Wellcome at 286, and the plurality in Alphapharm at [21].
392 Sanofi’s primary case was that each of the Exemplary Claims lacked any inventive step based on the common general knowledge as it stood at the priority date. Sanofi submitted that this case was strongly supported by the evidence of Professor Horton and Dr R Parker together with information appearing in (inter alia) Lagace 2006 and Zhang 2007. Each of Lagace 2006 and Zhang 2007 was relied on by Sanofi as common general knowledge or, alternatively, as a document to which regard could be had under s 7(3) of the Act. I accept that Lagace 2006 and Zhang 2007 were both important papers which would have been well known to the PSA before the priority date. I accept that they are relevant to the inventive step analysis as common general knowledge or, alternatively, as information to which regard may be had pursuant to s 7(3) of the Act.
393 Lagace 2006 describes experimental work in which TgPCSK9 mice (transgenic mice that expressed human PCSK9 in liver) were parabiosed with wild-type mice, to determine whether secreted PCSK9 derived from the TgPCSK9 mice was able to decrease LDLR levels in liver when transferred via shared circulation to wild-type mice. Lagace 2006 includes in its abstract the following statement:
We conclude that secreted PCSK9 [i.e., extracellular PCSK9] associates with the LDLR and reduces hepatic [i.e., liver] LDLR protein levels.
394 The “Discussion” in Lagace 2006 includes the following at page 3002:
Considered together, the available data now suggest that PCSK9 can function both extra- and intracellularly, but we do not know which pathway predominates under normal and/ or pathologic conditions. Currently, all studies suggesting that the protein functions intracellularly have been performed using PCSK9 overexpression via a strong CMV promoter. Overexpression may permit association of PCSK9 and the LDLR in an intracellular compartment that does not occur physiologically. In the current studies, we were able to demonstrate that physiologically relevant concentrations of PCSK9 could significantly reduce the number of cell-surface LDLRs when added to HepG2 cells … Additional studies to determine the relative contribution of extracellular PCSK9 in the regulation of LDLR protein levels will require determining whether the infusion of PCSK9 into the circulation of PCSK9-knockout mice will decrease the number of LDLRs in liver and raise plasma cholesterol levels.
…
The genetic data from humans with loss-of-function mutations in PCSK9 combined with the studies in knockout mice that lack PCSK9 clearly indicate that inhibitors of the protease would be of therapeutic benefit for the treatment of hypercholesterolemia. Inasmuch as overexpression of the catalytically inactive form of PCSK9 in mice did not alter LDLR protein levels, an inhibitor of PCSK9's protease activity in the ER [i.e., intracellular activity] should be sufficient to block its ability to reduce LDLR protein levels. If PCSK9 functions as a secreted factor [i.e., extracellularly] as suggested by the current data, then additional approaches to neutralize its activity, including the development of antibodies to block its interaction with the LDLR or inhibitors to block its action in plasma, can be explored for the treatment of hypercholesterolemia.
(Footnotes omitted)
395 There are three points to make about Lagace 2006. First, the extracted passage states that the available data suggests PCSK9 can function both intracellularly and extracellularly but that it is not known which pathway predominates under normal conditions. The authors were of the view that additional studies were required to determine the relative contribution of extracellular PCSK9 to the regulation of LDLR levels. Secondly, the suggestion that antibodies might be developed to block or inhibit PCSK9’s interaction with LDLR extracellularly is expressed to be dependent on PCSK9 functioning “as a secreted factor as suggested by the current data”. Thirdly, the authors state that “if” that is the case then additional approaches (i.e., non-intracellular approaches) including the development of antibodies “can be explored”.
396 While I accept that Lagace 2006 suggests that PCSK9 inhibiting antibodies might be developed, that suggestion is in my opinion highly qualified. The relevant discussion indicates that further research is required to confirm whether, and if so to what extent, PCSK9 functions extracellularly. I do not understand Lagace 2006 to be suggesting that antibodies are likely to be effective in blocking or inhibiting the PCSK9 and LDLR interaction. I consider the PSA reading Lagace 2006 would have understood that the potential for antibodies to be effective in that role was a matter of considerable uncertainty and dependent on the outcome of further research into the extracellular activity of PCSK9.
397 Zhang 2007 shows that PCSK9 bound to the EGFa domain of LDLR. That is to say, Zhang 2007 identified the site on LDLR to which PCSK9 bound (not the site on PCSK9 to which LDLR bound, or the site on PCSK9 to which an inhibitor could bind to prevent the LDLR/PCSK9 interaction). Even though Zhang 2007 determined that PCSK9 binds to the EGFa domain of LDLR, the binding site on PCSK9 was not defined. Zhang 2007 expressly recognised this at page 18602 where the authors state “the site of binding of PCSK9 to the extracellular domain of the LDLR has not been defined”. Zhang 2007 also suggests that there may be substantially less binding affinity between PCSK9 and LDLR outside the cell compared to the acidic environment inside the cell, because binding between PCSK9 and LDLR at a pH of 5.2 (acidic) was 50-fold higher than at a pH of 7.0 (neutral). This observation is referred to by Amgen in its submissions as forming the basis for CGK5.
398 Sanofi also asserted that Kwon 2008 (a paper which published the residues of PCSK9 that bind to the EGFa domain of LDLR) formed part of the common general knowledge, or alternatively, could be relied upon under s 7(3). However, Kwon 2008 was published on 12 February 2008 (i.e., after the priority date) and is therefore not relevant to inventive step as CGK or s 7(3) information.
399 In its written submissions Sanofi emphasised Professor Horton’s view that the literature had conclusively demonstrated the existence of an extracellular pathway for PCSK9 before the priority date of 23 August 2007. For reasons previously explained, I do not accept that this had been “conclusively” established as at that date. Professor Horton’s response to the JER question that Sanofi relies upon in support of this submission as to his view does not use the word “conclusively” in his reference to several papers which had demonstrated the extracellular pathway. Earlier, in response to the same question, the JER records:
[Professor Horton] thought the preponderance of experimental evidence supported PCSK9 functioning in blood to destroy LDLRs thus the development of antibodies to block the PCSK9-LDLR …[was] the preferred route given industries experience in making therapeutic antibodies, the speed at which this can be accomplished -both to obtain an affective [sic] blocking antibody and to get an antibody to clinic, and the safety and efficacy record of antibodies already used in the clinic at the time of all three relevant dates for chronic diseases.
In my view, that evidence indicates that Professor Horton’s view as to the existence of the extracellular pathway was much less definitive than Sanofi has portrayed it in its submissions.
400 I have referred above to the acknowledgement in Horton 2007, a review article co-authored by Professor Horton and published in early 2007 that “[t]he major cellular site at which PCSK9 acts has not been established”. Horton 2007 referred to data that suggested PCSK9 might promote degradation of LDLR as it migrates from the ER to the cell membrane. An alternative hypothesis, also referred to by the authors, was that PCSK9 might act on LDLR only after it is secreted. The text accompanying Figure 2 in Horton 2007 states that “[c]urrent evidence indicates that PCSK9 might work at two cellular sites”. In their concluding remarks, the authors stated that “several important mechanistic and clinical questions remain” about PCSK9 and LDL in metabolism, and that “[f]urther studies will be required to identify the specific site(s) in the cell at which PCSK9 functions and to determine whether catalytic activity is required for LDLR degradation”.
401 Dr R Parker gave evidence, which I accept, that as at the priority date, there was what he termed “an emerging hypothesis” that PCSK9 functioned extracellularly but which had not yet displaced the “prevailing notion that an intracellular mechanism of action of PCSK9 on LDLR functioned under usual physiological conditions”. Similarly, Professor Seidah, who Dr R Parker described as a leading researcher in the field whose team first characterised PCSK9 (originally named NARC-1) in 2003, referred in a paper published in March 2007 (Seidah 2007) to “a yet unknown mechanism [by which] high levels of PCSK9 lead to a faster rate of degradation of cell surface LDLR, resulting in increased circulating LDL [cholesterol] …”.
402 Even if it was common general knowledge that PCSK9 had both an intracellular and extracellular pathway, Professor Horton explained in cross-examination that for an antibody to work against PCSK9, PCSK9 would need to have extracellular activity of at least 50%. While Professor Horton said that it was possible that lower levels of extracellular activity might have been effective, the 50% figure was based on “what we knew for a fact”. This is consistent with the following statement appearing in Kwon 2008, a paper whose co-authors included Professor Horton and Dr Thomas Lagace:
If PCSK9 functions primarily as a secreted protein in plasma, agents that interfere with the PCSK9:LDLR interaction at the cell surface also have the potential to lower plasma LDL[ cholesterol] levels in individuals with hypercholesterolemia.
403 In Horton 2009, Professor Horton and his co-authors discussed possible therapeutic approaches to inhibiting PCSK9 activity. This review article (submitted in November 2008, after the priority date) referred to three options including, first, the intracellular inhibition of PCSK9 catalytic activity necessary to facilitate PCSK9 secretion from the ER and, second, the inhibition of PCSK9 synthesis by degrading PCSK9 mRNA using ASOs or siRNA. It then referred to a third option as follows:
A third option to inhibit PCSK9 activity is to prevent binding of PCSK9 to LDLRs at the cell surface with small molecules, peptides, or antibodies directed against PCSK9. Overexpressing soluble LDLR or adding EGF-A fragments to cultured cells inhibits the ability of exogenously added PCSK9 to mediate LDLR degradation. The success of this approach depends on PCSK9 functioning primarily at the cell surface.
(Footnotes omitted)
404 Neither article states nor implies that PCSK9 functions primarily outside the cell. Rather, they indicate that this was still an open question. There was a clear difference of opinion between Professor Horton and Dr R Parker on this point as to the state of the common general knowledge at the priority date. In my opinion Dr R Parker’s opinion is to be preferred.
405 Sanofi submitted that there was a great deal of evidence given by Dr R Parker as to uncertainties or questions regarding the literature that was said to elucidate the extracellular pathway, despite the relevant studies having been published in peer reviewed journals by multiple groups including major innovator pharmaceutical companies. Sanofi’s written submissions on this point referred to some highly generalised cross-examination of Dr R Parker in which he was referred to several papers published before the priority date, but in the context of general industry interest in PCSK9. These papers included Cunningham 2007, Piper 2007 and Nassoury 2007. Sanofi also referred to an agreement between Dr R Parker and Professor Horton in the JER that “several papers reported the binding of extracellular PCSK9 to the LDLR leading to their destruction via internalization into the endosome/lysosome”, including Cameron 2006, Lagace 2006, Holla 2007, Fisher 2007, Qian 2007, McNutt 2007 and Li 2007.
406 I do not accept Sanofi’s criticism of Dr Parker’s evidence. The articles to which it refers were all published prior to Kwon 2008 and Horton 2009, both of which leave open the question whether PCSK9 functions primarily outside the cell. For example, Piper 2007 states:
The specific interactions between PCSK9 and the LDLR remain to be elucidated, and it is likely that knowledge of this interaction will be beneficial in completing the picture of how PCSK9 regulates extracellular levels of LDLR.
407 While at BMS, Dr R Parker occupied a very senior research position in charge of a team he headed investigating PCSK9 inhibitors. In the JER, he identified the approach of suppressing PCSK9 gene expression at the mRNA level through the use of ASOs or siRNA as most promising. An important factor influencing this approach was the fact that it was agonistic as to PCSK9’s mechanism of action in that it would suppress PCSK9 levels inside and outside the hepatocyte surface. Professor Horton’s response to Dr R Parker’s answer to this effect was that such approaches were not commonly taken, and the development and clinical use of these technologies was in its “infancy”, although he agreed that they were completely agnostic as to where or how PCSK9 functioned.
408 There are several points to note about Dr R Parker’s approach. First, it was consistent with the view that PCSK9’s mechanism of action had yet to be established, and it sought to accommodate that unknown. Secondly, this approach was recognised and apparently favoured not only by Dr R Parker but by other researchers as well. Homer 2008 suggested that antisense inhibition (i.e. using ASOs) may provide a promising approach to PCSK9 inhibition. In addition to intracellular inhibition of PCSK9 catalytic activity, ASOs were also identified as an option in Horton 2009.
409 The reference to ASOs in Horton 2009 is significant, as it indicates that as at 2009, Professor Horton was aware of, and had contemplated, the use of ASOs in the inhibition of PCSK9. While Horton 2009 also refers to the possibility of using antibodies to inhibit PCSK9 activity, it does so subject to the express proviso that the success of the approach will depend on PCSK9 functioning primarily at the cell surface. Although Horton 2009 post-dates the priority date, it does not support the inference that as at the priority date, the PSA would have been likely to try antibodies far ahead of ASOs.
410 In its submissions, Sanofi characterised Dr R Parker as a contrarian whose approach was idiosyncratic and not representative of the PSA. Sanofi referred to evidence given by Dr R Parker as to his employer’s approach to research. He said:
…we’re not just trying to follow what everybody is doing. We’re not just following the horse that got out of the gate first… we had our own approaches and it’s a highly competitive industry. I think we all accept that. This is especially on a target like – that had the potential that PCSK9 had. Hugely competitive. So we’re going to take approaches that we think we can succeed at. We’re going to take approaches that we think we may be right about and, if our ideas turned out to be correct, we’re going to be at the head of the field and everybody else is going to be looking at us.
411 Sanofi submitted that the desire to come up with a novel approach to better competitors rather than “jumping on the bandwagon” was understandable, but it was not the approach of the PSA. It submitted that Dr R Parker’s approach was based on the significant resources which he had to develop a PCSK9 inhibitor.
412 The notion of “jumping on the bandwagon” is not apposite. Although the evidence indicates that patent applications claiming antibodies to PCSK9 had been filed by different pharmaceutical companies (including Novartis, Merck and also BMS), these were provisional applications, none of which were publicly available at the priority date. They were filed during the course of what I would infer to be confidential research programs that were in progress and not yet complete at the priority date. In any event, I do not think the existence of such applications (none of which form part of the relevant prior art base) is of any real assistance in addressing the question of obviousness and, in particular, the modified Cripps question.
413 The BMS application lists Dr R Parker as an inventor and was relied upon by Sanofi to show “the true state of acceptance in the art of the prospects of developing an antibody” to PCSK9. Dr R Parker identified the application in his affidavit as being directed to novel polypeptides (being truncated forms of PCSK9 which were missing large pieces of the protein). I understand, from Dr Parker’s evidence, that the antibody covered in claim 20 of that application was not to the native form of PCSK9 (i.e., the form which was being researched primarily by his group). In his affidavit, he clarified that:
…at the time of filing WO 797, we had not used the PCSK9-b and PCSK9-c polypeptide sequences to generate antibodies, and I had no intention to do so. The generality of these passages is consistent with the fact that we had not done any antibody experiments and the WO 797 only relates to the cloning of two PCSK9 variants. We never developed antibodies to these variants.
The application filed by BMS appears to be directed to variants of PCSK9 and methods of producing them. The abstract states:
The present invention provides novel polynucleotides encoding PCSK9b and PCSK9c polypeptides, fragments and homologues thereof. Also provided are vectors, host cells, antibodies, and recombinant and synthetic methods for producing said polypeptides. The invention further relates to diagnostic and therapeutic methods for applying these novel PCSK9b and PCSK9c polypeptides to the diagnosis, treatment, and/or prevention of various diseases and/or disorders related to these polypeptides. The invention further relates to screening methods for identifying agonists and antagonists of the polynucleotides and polypeptides of the present invention.
414 I am not persuaded that the BMS application indicates that the “true state” of the common general knowledge at the time of the priority date is at odds with the approach Dr R Parker took in his evidence.
415 In answer to a question from the Court, Dr R Parker explained that he did not make a conscious decision to not follow the crowd:
I mean, we are competitive, … but we don’t know what the others are doing, that’s a key point here. I mean, I knew that Merck was working on small molecules because they reported in March at a conference, and I think Dr Horton was there…I didn’t know others were working on antibodies. I don’t know how it would be that I would have known that; I didn’t have access to that kind of infiltration or spies, or whatever it would have taken. I didn’t. I wish I had but I didn’t.
416 In answer to a further question concerning the difficulties associated with the intracellular versus the extracellular approach, Dr R Parker indicated (as I have explained above) that siRNA, ASOs and approaches that inhibited PCSK9 self-autocatalytic cleavage or blocked secretion would address a situation where PCSK9 activity was intracellular or extracellular. Professor Horton agreed, but drew attention to what he regarded as problems with intracellular inhibitors. He said:
Then and now we still have no intracellular inhibitor. And so the extracellular pathway, that’s why it was so exciting because it provided an easy and quick method to move a drug forward. It, of course, did depend upon it being correct. But developing antibodies that block the interaction of PCSK9 and the LDL receptor can be done relatively rapidly and inexpensively. And the assay was already in the literature for them to use because we had already proven that PCSK9 and the LDL receptor were bound together. And so it was simply a function of generating antibodies and looking for one that would block their interaction and then test it in a cultured cell or an animal to see if it worked. And so it wasn’t a huge outlay in expense and the time is relatively short and it could be done very quickly by pharmaceutical companies.
417 Amgen submitted that, viewed as a whole, Dr R Parker’s evidence shows, not that he was a contrarian, but that he was trying to make sense of the science in a rapidly changing field to make the decisions that were most likely to produce a useful outcome. As Dr R Parker explained, PCSK9:
… was a high stakes, high difficulty target … And I have to say, your Honour, you can perhaps imagine it’s difficult for me here today, being a – a careful scientist at the time and working in the environment I’m working in, and yet standing here today and telling you the story with full knowledge that antibodies work. And I would say that in some sense I have to accept that I was wrong – that we were wrong, not necessarily on the antisense approach, but wrong on thinking that monoclonal antibodies were not the way to go. That’s a difficult position to be in. But I’m trying to put myself back in the period of time. It’s pretty difficult for me to assess degrees of improbability or – or risk or likelihood of success. I – I think it was a difficult target in all respects. There was nothing easy here.
418 I found Dr R Parker’s evidence persuasive, while Professor Horton’s evidence struck me as heavily influenced by hindsight. While Professor Horton considered the extracellular pathway exciting, his reasoning appears to have more to do with the ease with which antibodies could be developed and their established usage in other areas, rather than whether they would work. Whether they would work was something which depended on whether PCSK9’s activity occurred primarily outside the cell.
419 Sanofi placed reliance on Dr R Parker’s evidence concerning a technology known as Adnectins which BMS commenced to explore in the context of PCSK9 in early 2008, which followed on from BMS’s acquisition of Nexus Therapeutics in September 2007. Notably, Adnectins (like antibodies) target PCSK9 outside the cell, but they are distinct from antibodies and the product of a proprietary technology. Antibodies are 12 times the size of Adnectins. Dr R Parker explained in his written evidence:
In 2008, we judged that taking an additional approach that might be able to target PCSK9 outside the cell, which we believed was the surface of the plasma membrane, was now worth taking as complimentary to the other therapeutic modalities in which we were deeply engaged. This led to the beginnings of the Adnectin program in mid-2008. The potential advantages of Adnectins over antibodies and other lgG molecules including lower cost of goods and the ability to formulate high doses were attractive and added to our decision making to begin working in this area. Further, by engineering the Adnectin with protein domains that promote hepatic internalization, I and my BMS Research Team speculated that an intracellular locus of interaction might be possible.
420 According to Dr R Parker, advantages of this technology were that the discovery process was faster and less costly than antibody technology, and resulted in biologics smaller in molecular size than antibodies with potentially faster tissue penetration. As I understand it, the smaller size was attractive primarily because it allowed for a higher concentration of therapeutic to be administered in a lower dose, which alleviated some of the perceived issues with antibodies based on Dr R Parker’s understanding at the time of the extracellular environment.
421 Research into the development of these biologics as PCSK9 inhibitors began at BMS in mid-2008. Dr R Parker described it as a complementary program that provided an alternative to other research approaches including the development of PCSK9 inhibiting ASOs, which BMS had been pursuing since early 2007. Dr R Parker provided the following evidence about the diversity of approaches to finding a PCSK9 inhibitor at BMS
245. The diversity of approaches that we attempted at BMS to finding a PCSK9 inhibitor therapeutic reflects, in my opinion, the fact that the pathway to identifying a PCSK9 inhibitor was not clear or straightforward. There were many uncertainties that I had to confront in relation to the mode of action of PCSK9 within the body and it was not evident at [the priority date] which drug (inhibitor) modality would succeed.
246. The range of options pursued by BMS did not include an antibody approach. This was not simply a reflection of the types of therapeutic candidates that BMS had focused on traditionally. BMS had antibody expertise, and in any event, the collaborations and acquisitions described above make it clear that my team under my supervision was prepared to pursue all therapeutic options that appeared viable, based on what was known about PCSK9 at the time. I and my BMS Research Team considered the possibility of using specialized antibody technology but concluded that an antibody was not a promising option to pursue.
422 That evidence is consistent with Dr R Parker’s oral evidence and is in my view persuasive. In particular, I accept that he and his research team at BMS did not consider antibody technology as a promising option to pursue in the search for an effective PCSK9 inhibitor.
423 Sanofi’s antibody expert, Professor Mahler, agreed that “before you read the common specification, you would not know … if it was possible to make antibodies that inhibit the binding interaction of PCSK9 and LDLR”. Similarly, Professor Hudson’s evidence, with which Professor Lopez agreed, suggested that the chances of finding an antibody that inhibited PCSK9 (or which competed with 21B12 or 31H4) were “pretty low”. Professor Hudson said:
… without the [CS], you’re flying blind and, with it, you know exactly what you’re going to do. So what I mean by flying blind is, if you imagine yourself in the cockpit of an – flying a plane. You’re the pilot. At night, you can’t see the runway. You can’t see the airport because it’s not lit up. You’ve got a panel of instruments in front of you, which are the techniques you’ve got available to you. You – these are not calibrated and anyone can fail.
You don’t know if they’re going to work. So what’s my chances of hitting the deck, landing on one of those two epitope surfaces, meaning the runways? Pretty low. I wouldn’t like my chances …
424 Professor Hudson further explained:
The issue that we have here, your Honour, is that in any one of those techniques … There’s a significant chance of failure and, if any one fails, you don’t get anything out of 10 step screening process, starting with the immunisation to achieving the final antibodies. And so your chances of getting through those 10 steps with an antibody is incredibly low in many cases and as many people – my colleagues have found, there are some antigens you can’t raise an antibody to … I think it’s a complex process with limited chances of success without the teachings of the [CS] … I really think that, without the [CS], it would be – chances of success are very low, given the complexity of the individual steps.
425 I am not persuaded that Professor Hudson’s evidence on this topic should be discounted as reflecting anything less than the bona fide view of a highly qualified expert with considerable experience in developing MAbs. Professor Horton’s evidence suggesting that at the priority date the development of a PCSK9 inhibiting antibody would be straightforward or quick or easy is in my view the product of hindsight and over-simplification. In this context, it is important to note that Professor Horton is not an antibody expert and, unlike Professor Hudson, he had no expertise in the generation of antibodies. Nor did Professor Horton’s team work directly on drug discovery or development.
426 It is clear that the notional skilled team would have considered the possibility of generating PCSK9 inhibiting monoclonal antibodies as a potential treatment for hypercholesterolemia and related cardiovascular diseases. However, the question is whether it would have been directly led to try to generate such antibodies as a matter of course in the expectation that they may well block or inhibit binding between PCSK9 and LDLR. Having regard to the evidence, especially that of Dr R Parker and Professor Hudson, I am not persuaded that the modified Cripps question should be answered in the affirmative. In particular, I accept that the pathway to the generation of anti-PCSK9 antibodies was not clear or straightforward and was the subject of considerable uncertainty relating to PCSK9’s mechanism of action (i.e. whether it functioned primarily extracellularly) and whether it would even be possible to generate an antibody to PCSK9. I am therefore not satisfied that the notional skilled team would have been directly led to try an anti-PCSK9 antibody in the expectation that it may well succeed.
427 In my opinion Sanofi’s evidence falls short of establishing that any of the Exemplary Claims would, if the Applications proceeded to grant, be clearly invalid as not involving an inventive step.
LACK OF CLARITY
428 In its written closing submissions, Sanofi submitted that if the Court accepts Amgen’s contention that the ground of claim definition rises no higher than that of clarity, Sanofi relies on the same matters in support of its clarity attack on which it relied in support of its claim definition attack. That submission was not developed in either Sanofi’s written or oral closing submissions. In Sanofi’s oral submissions I was told that the lack of clarity ground related only to the Competition Claims. In any event, I am not persuaded that any of the Epitope or Residue Claims suffer from a lack of clarity.
429 With regard to the Competition Claims, the word “competes” refers to competition between two or more ABPs for binding to another protein (including an antigen such as PCSK9). No witness had any difficulty with that term as it would generally be understood by the PSA. Sanofi submitted that the Competition Claims are unclear because:
(a) they do not define whether a numerical threshold applies to “competes” (and if so, what numerical threshold); and
(b) they fail to provide a “workable standard” to determine whether an antibody as claimed in the Competition Claims, “competes for binding to PCSK9”.
430 While these arguments were expressed disjunctively, they are both directed to the same question, which is whether the Competition Claims fail to comply with s 40(3) of the Act because they do not provide a workable standard suitable to the intended use by which the PSA can determine whether an antibody competes for binding with either of the reference antibodies: Minnesota Mining and Manufacturing Company v Beiersdorf (Australia) Limited (1980) 144 CLR 253 at 274 per Aickin J.
431 Sanofi pointed to discussion in the CS concerning the term “competes” which states at CS [0231]:
The term "compete" when used in the context of antigen binding proteins (e.g., neutralizing antigen binding proteins or neutralizing antibodies) that compete for the same epitope means competition between antigen binding proteins as determined by an assay in which the antigen binding protein (e.g., antibody or immunologically functional fragment thereof) being tested prevents or inhibits (e.g., reduces) specific binding of a reference antigen binding protein (e.g., a ligand, or a reference antibody) to a common antigen (e.g., PCSK9 or a fragment thereof). Numerous types of competitive binding assays can be used to determine if one antigen binding protein competes with another, for example: solid phase direct or indirect radioimmunoassay (RIA), solid phase direct or indirect enzyme immunoassay (EIA), sandwich competition assay (see, e.g., Stahli et al., 1983, Methods in Enzymology 9:242-253); solid phase direct biotin-avidin EIA (see, e.g., Kirkland et al., 1986, J. Immunol. 137:3614-3619) solid phase direct labeled [sic] assay, solid phase direct labeled [sic] sandwich assay (see, e.g., Harlow and Lane, 1988, Antibodies, A Laboratory Manual, Cold Spring Harbor Press); solid phase direct label RIA using I-125 label (see, e.g., Morel et al., 1988, Molec. Immunol. 25:7-15); solid phase direct biotin-avidin EIA (see, e.g., Cheung, et al., 1990, Virology 176:546-552); and direct labeled [sic] RIA (Moldenhauer et al., 1990, Scand. J. Immunol. 32:77-82). Typically, such an assay involves the use of purified antigen bound to a solid surface or cells bearing either of these, an unlabelled test antigen binding protein and a labeled [sic] reference antigen binding protein. Competitive inhibition is measured by determining the amount of label bound to the solid surface or cells in the presence of the test antigen binding protein. Usually the test antigen binding protein is present in excess. Antigen binding proteins identified by competition assay (competing antigen binding proteins) include antigen binding proteins binding to the same epitope as the reference antigen binding proteins and antigen binding proteins binding to an adjacent epitope sufficiently proximal to the epitope bound by the reference antigen binding protein for steric hindrance to occur. Additional details regarding methods for determining competitive binding are provided in the examples herein. Usually, when a competing antigen binding protein is present in excess, it will inhibit (e.g., reduce) specific binding of a reference antigen binding protein to a common antigen by at least 40-45%, 45-50%, 50-55%, 55-60%, 60-65%, 65-70%, 70-75% or 75% or more. In some instances, binding is inhibited by at least 80-85%, 85-90%, 90-95%, 95-97%, or 97% or more.
432 It was submitted by Sanofi that CS [0231] defines “compete”. I agree that it defines that term, in its first sentence, in the context of antibodies competing for the same epitope, to mean competition determined by an assay in which a “test” ABP prevents or inhibits a “reference” ABP from binding to a “common antigen”. That definition does not exclude the possibility that competition may occur between antibodies for other reasons. The skilled addressee reading the CS and the Competition Claims would understand the reference ABP to relevantly include MAbs 31H4 and 21B12 and the reference to a common antigen to refer to PCSK9.
433 CS [0231] refers (non-exhaustively) to the types of competition which might occur (e.g., the test ABP competes with the reference ABP to bind to the same epitope or an adjacent epitope “sufficiently proximal” so that steric hindrance occurs). Later, in CS [0565], reference is made to competition observed due to “conformational changes”, meaning that the ABPs need not compete for the same or adjacent epitopes, but the binding of the test ABP causes a conformational change (i.e., change in shape) to the antigen which impedes binding of the reference ABP. This is discussed in more detail below. CS [0229] also contemplates the concept of competition by means other than competition for the same or adjacent epitopes:
The term "neutralizing antigen binding protein" or "neutralizing antibody" refers to an antigen binding protein or antibody, respectively, that binds to a ligand and prevents or reduces the biological effect of that ligand. This can be done, for example, by directly blocking a binding site on the ligand or by binding to the ligand and altering the ligand's ability to bind through indirect means (such as structural or energetic alterations in the ligand)…
(Emphasis added)
434 In my view, “competes” as used in the CS and the Competition Claims can refer to any scenario in which the test ABP prevents or inhibits the reference ABP from binding to the common antigen. While Sanofi’s closing written submissions suggested that Amgen attempted to confine “competes” to ABPs competing for the same epitope, Amgen’s submissions are agnostic to the mechanism of competition. It explained:
Sanofi addresses the various causes of competition that are posited in the [CS]… For the purposes of construction, these matters are irrelevant. Competition is determined by an assay, and it does not matter what causes the competition.
I agree with that interpretation of the CS and the Competition Claims.
435 CS [0231] identifies various types of assays that may be used to determine whether the test ABP competes with a reference ABP, but it does not require that they compete to any particular degree. The range of percentages provided in the paragraph is not exhaustive, and is prefaced with the word “usually”. The claims require that the MAb competes with a reference antibody so as to prevent or inhibit the latter binding to PCSK9. Some MAbs will compete strongly, while others will compete less strongly. Either way, on what I regard as the proper construction of the Competition Claims, if there is any competition between the relevant MAb and the reference antibody, this requirement of the claim will be met.
436 My construction of the Competition Claims broadly accords with Professor Hudson’s and Professor Lopez’s evidence of their understanding of the term. Although Professor Hudson did, at one point, suggest that for antibodies to be competing within the meaning of the specification there had to be “significant competition”, his response indicates that he was focussed on CS [0231] and was qualified by his clarification, “[a]nd I think you’re asking me is 45 per cent significant. Yes. If that was the question.” His responses to other questions during cross-examination support the view that he understood competition to be a binary concept, in particular his statement that “competition is competition” and “[i]t’s a sliding scale from 100 per cent down to zero”. His evidence, and Professor Lopez’s evidence, indicates that competition which was less than “complete” would still be understood to fall within the Competition Claims. Contrary to Sanofi’s submission, I do not accept that the Competition Claims lack clarity because they fail to impose a numerical threshold for the purpose of determining whether there is competition.
437 CS [0231] does not mandate the use of any particular type of competitive binding assay but merely describes a number of different types that can be used. It follows that there is nothing in the CS which would preclude the skilled addressee from ascertaining whether a MAb competes with a reference antibody for binding to PCSK9 using a different type of competition assay to those referred to in CS [0231].
438 Professor Hudson stated that it was not unusual to get different results from different competitive binding assays. In this regard, Professor Hudson said:
I think, whenever you do experiments using different technologies or analytical techniques, you can end up with different results. I mean, not – they’re usually in the same order, if I’m making myself clear … from a high binding antibody to a weak antibody or a weak blocker to a high blocker, but the actual numerical value may differ.
I take from this evidence that while the numerical values produced by different assays may vary, they will usually produce consistent results in terms of identifying which antibodies compete and whether they do so strongly or weakly.
439 Professor Mahler gave evidence that the CS did not teach the PSA how to produce antibodies falling within the Competition Claims. In the course of giving that evidence he did not suggest that he would have any difficulty determining whether a particular antibody competed with either of the reference antibodies. Asked whether the CS assists in generating antibodies within the claims, Professor Mahler said:
PROF MAHLER: So what it means is that – what it means is that when you – when you go through immunisation protocol and raise antibodies, you don’t have any control over what you get. You can’t – it’s not as though you have a preconceived idea that, “I’m going to make antibodies that compete and therefore I’m going to do this” … You don’t know what you’re going to get and so you’re going to immunise with PCSK9, you’re going to generate a panel of antibodies, and then you will test them, and then you will know whether they compete or not.
But you can’t have a preconceived idea about – about what you want and translate that into some protocol that gives you something that – that the animal does…You have no control over which antibodies are going to be raised. Everything happens downstream with the testing of what you have got so – so what I meant by that is that you – you – you can’t have a preconceived idea that the application is going to assist me to make antibodies that compete, because it – it doesn’t. That – that’s the way I read the question.
HIS HONOUR: Well, it assists you to determine whether or not they compete at the very end of your program.
PROF MAHLER: It’s after you – it’s after you let the animal do the work. You – you screen the antibodies, you end up with a panel, then you can say whether they compete. But the fact that you want – you want antibodies that compete – well, there’s nothing you can do. The application doesn’t assist or give you any – it can’t. It – it’s not possible. It can’t.
HIS HONOUR: Well - - -
PROF MAHLER: It can’t tell you how to make antibodies that compete.
HIS HONOUR: What about the assistance it gives at that very last step … though?
PROF MAHLER: Which last step is that, sorry?
HIS HONOUR: Determining whether or not they do compete.
PROF MAHLER: Of course, because you have got – right at the end you have got the antibodies that they compete with so then you can test them. But that’s totally independent of do these antibodies assist me in generating antibodies that compete. They – they don’t.
440 In fairness to Professor Mahler, in this part of his evidence, he was not addressing the reliability of competition assays specifically. In his extensive written evidence he asserted that:
… the different methodologies in the different types of assays discussed in paragraph [0231] are likely to yield different results. Therefore, it is typical in the industry when reporting results, to identify the assay by which the results were obtained.
However, I understood this evidence to be referring to potential numerical differences in assay results. He did not say that any of the assays identified in CS [0231] would be likely to produce either a false positive or false negative.
441 Sanofi drew attention to the treatment of MAb 30A4 in the CS which was allocated to bin 1 in the competition assay used in Example 10 and bin 5 in the different competition assay used in Example 37. As I have explained above, antibodies that compete with each other were grouped together in the same bin in each example. Table 8.3 shows 30A4 as binning with 21B12, whereas in Table 37.1, antibody 30A4 is shown in bin 5 when 21B12 is in bin 1. This indicates that in the competition assay used in Example 10, 30A4 competed with 21B12, but the same could not be said of the competition assay used in Example 37. CS [0564] describes bin 5 as “a catch all” bin for antibodies that do not fit into other bins. It is not clear from CS [0564] what results were observed to warrant placement in this bin, as opposed to bin 4, which was for antibodies that did not compete with either 21B12 or 31H4.
442 Importantly, the Antibody and Crystallography Experts agreed in the JER that “the results from Examples 10 and 37 were generally consistent and complementary”. In the JER Professor Mahler also specifically observed that the Example 37 data was quite consistent with that of Example 10 and that there was no added teaching from Example 37. He said:
Examples 10 and 37: There were some discrepancies in the binning for antibodies listed in Tables 2 and 8.3 of the Application, from Examples 10 and 37, respectively, as pointed out in Paragraph 167 of my Declaration dated 28th [February] 2018. Overall there was consistency in the binning of antibodies with a few differences. It is my experience that different methods can yield different results in some circumstances.
443 In the declaration to which Professor Mahler refers he noted that in Example 37 antibody 30A4 was listed in bin 5. Although he noted that antibodies 31D1, 25A7 and 20D10 were not listed in Example 37, he did not identify any other inconsistency in the binning results. It is not apparent from Professor Mahler’s evidence whether he had regard to the binning data for Example 37 in the tables reproduced in Figures 23A-23D to the CS.
444 In the JER, Professor Hudson said that 30A4 had been incorrectly allocated to bin 5 rather than bin 1. When cross-examined about the binning of 30A4 in Table 37.1, he referred to the data in Figures 23A-23D, which he said showed that the binding of 30A4 is particularly weak, but that it did compete with some antibodies in bin 1 and there was a “slight overlap” with bin 3 antibodies. He suggested that the inventors may have put 30A4 in bin 5 because they were uncertain as to whether it should be put in bin 1 (i.e., competing with 21B2) and bin 2 (i.e., competing with both 21B2 and 31H4) and therefore put it in the “catch-all” bin. That is a reasonable hypothesis, given that it was not put into bin 4 (i.e., the bin for antibodies that did not compete with bins 1 and 3). Professor Hudson was not referred to any other example of what Sanofi submitted are inconsistent test results based on the use of different competition assays.
445 Professor Hudson described 30A4 as “an unusual antibody” based on its binning data and the different results as “anomalous”. Professor Petsko said in the JER:
There are a few minor discrepancies between the results … All antibodies that were tested in both Example 10 and 37 and were found in Example 10 to co-bin with 31H4 were still found to do so in Example 37. All but one of the antibodies that co-binned with 21B12 in Example 10 were found to do so in Example 37; the exception was 30A4, which co-binned with 21B12 in Example 10 but was put in the “catch-all” bin in Example 37. However, when I examine the Example 37 data in Figures 23A-23C of the 677 Application, I conclude that 30A4 in fact DOES co-bin with 21B12. The apparent discrepancy may therefore be a clerical error.
446 I accept that there is good reason to believe that 30A4 has been incorrectly assigned to bin 5 rather than bin 1. I am not persuaded that the different binnings for 30A4 in Example 10 and Example 37 demonstrate that different competition assays will give different answers to the binary question of whether two antibodies compete for binding to PCSK9.
447 As to the order in which the antibodies are tested, the CS makes clear that the order in which the ABPs are employed can be important (see CS [0565] reproduced below). In this regard, Professor Hudson gave this evidence:
MR SHAVIN: The order in which the ABPs are employed can be important - - -
PROF HUDSON: Yes.
MR SHAVIN: - - - and you accept, don’t you, that if you’re doing a competitive binding assay, you may get different results depending upon the sequence that you use.
PROF HUDSON: Correct.
MR SHAVIN: Yes. And if ABP(a) is employed as the reference antibody and blocks the binding of ABP(b), the converse is not always true, and you accept that.
PROF HUDSON: Correct.
448 Although the order in which the competition assay is run (i.e. which antibody is added first) may affect the results, this is something that would be known to the skilled addressee. CS [0565] states:
As will be appreciated by one of skill in the art, if the reference ABP prevents the binding of the probe ABP then the antibodies are said to be in the same bin. The order in which the ABPs are employed can be important. If ABP A is employed as the reference ABP and blocks the binding of ABP B the converse is not always true: ABP B used as the reference ABP will not necessarily block ABP A. There are a number of factors in play here: the binding of an ABP can cause conformational changes in the target which prevent the binding of the second ABP, or epitopes which overlap but do not completely occlude each other may allow for the second ABP to still have enough high-affinity interactions with the target to allow binding. ABPs with a much higher affinity may have a greater ability to bump a blocking ABP out of the way. In general, if competition is observed in either order the ABPs are said to bin together, and if both ABPs can block each other then it is likely that the epitopes overlap more completely.
This paragraph of the CS teaches the skilled addressee that if competition is observed in either order then the test antibody and the reference antibody will bin together, thereby implying that they will compete for binding to PCSK9.
449 Sanofi also submitted that the Competition Claims lacked clarity because the CS did not disclose how much excess antibody should be used in a competition assay. Claim 1 of 685 does not require that any competitive assay utilise an “excess” of an antibody. However, claim 1 of 748 is for a MAb that:
… binds to human PCSK9 and is neutralizing in that an excess of said antibody reduces the quantity of human PCSK9 bound to LDLR in an in vitro competitive binding assay, wherein said monoclonal antibody competes for binding to PCSK9 with [21B12].
I note, though, that in this claim the requirement for an “excess” of antibody in an in vitro competitive binding assay appears to relate to the assay used to determine if the MAb competes with PCSK9, rather than the assay used to determine if the MAb competes with 21B12.
450 In any event, Sanofi’s submission on this point appears to relate to both 748 and 685. It submitted that the amount of “excess” antibody affects the results of a competitive binding assay, and too little excess (and, I infer, the absence of an excess) can result in a false negative. The concept of an “excess” is referred to in CS [0231], which explains that when conducting a competitive binding assay, “[u]sually the test antigen binding protein is present in excess”.
451 Professor Mahler gave evidence concerning the term “excess” and suggested, at least at some points in his evidence, that it was unclear to him what amount of excess needed to be used. However, I ultimately understood him to accept that he could determine how much antibody would be required to give “an excess of said antibody”, something which his evidence does not suggest would be beyond the capacity of the PSA. Professor Mahler agreed that, if necessary, the amount of antibody to be used in the competition assay could be determined by carrying out a separate assay for that purpose.
452 Professor Hudson said that “excess means excess” and that an assay could be used to identify a relevant excess. Professor Lopez said that “when we talk about excess antibody … [we are] trying to make sure we have got enough antibody … to be able to detect an inhibitory effect”. Both Professors Hudson and Lopez agreed that the results of a competitive binding assay would be the same regardless of whether a small or large excess was used, because in each case the excess would by definition be more than the amount required. It was not put to either of Professors Hudson Lopez that it would be beyond the skill of the PSA to determine an appropriate concentration of antibody to provide an excess in a competition assay.
453 Professor Mahler is the co-inventor of a patent published in 2014 entitled “Anti-CD83 antibodies and use thereof”. Claim 15 of that patent is for a CD83 binding protein which (inter alia) competitively inhibits binding of a reference antibody or which binds to the same epitope in CD83 as that antibody. The specification includes a statement that “[a]ssays for determining a CD83 binding protein that competitively inhibits binding of an antibody will be apparent to the skilled artisan”. Professor Mahler accepted that the specification did not include any description of any competitive binding assay. In particular, he agreed that there was no description in the specification of the level of excess antibody required to perform the competition assay. That evidence strongly suggests to me that Professor Mahler would accept that ascertaining how much antibody to use when conducting a competition assay is something that the PSA could do using standard and well-known techniques.
454 I am not persuaded that the Competition Claims do not provide a workable standard due to a failure to identify a particular competition assay. In particular, I am not satisfied that the use of different assays, to determine whether two antibodies compete for binding to the same antigen, would be likely to produce materially different results. I am also satisfied that the skilled addressee could determine how much antibody to use in a competition assay to ensure that there was an excess of the relevant antibody. In the result, I am not persuaded that any of the Competition Claims lack clarity.
SUFFICIENCY
455 A complete specification must “describe the invention fully”: s 40(2)(a) of the Act pre-amendment by the RTB Act. In Kimberly-Clark, the High Court identified the relevant question under s 40(2)(a) as follows at [25]:
… [W]ill the disclosure enable the addressee of the specification to produce something within each claim without new inventions or additions or prolonged study of matters presenting initial difficulty?”
(Footnote omitted)
456 Sanofi accepted that if the invention in each claim of the Applications is properly characterised as a single invention, namely a class of monoclonal antibodies, then on the standard of proof required in this appeal, the claims are fully described under the principles in Kimberly-Clark. That is because Sanofi accepted that the PSA could generate at least one antibody within each such claim.
457 Sanofi submitted that where a claim is in effect a claim to more than one invention, the specification must describe how to perform an embodiment of each invention within the claim. It says that the claims of the 677, 689 and 751 are not claims to a class of antibodies, but are claims to a very large number of individual, monoclonal antibodies, each binding to its own particular epitope on PCSK9. In support of its submission Sanofi relied on Tramanco Pty Ltd v BPW Transpec Pty Ltd (2014) 105 IPR 18 (“Tramanco”).
458 Tramanco was concerned with a method for logging the performance of a vehicle suspension system, by which method one or more of three different performance parameters was determined. It was conceded by the patentee in that case that the claim would be invalid for lack of sufficiency if the complete specification did not enable the PSA to determine all three parameters using the method. The Full Court accepted (obiter) that the concession was properly made.
459 Sanofi relies on the following statement in Tramanco at [207] (per Nicholas J, Allsop CJ at [53] and Greenwood J at [164]) agreeing):
In Kimberly-Clark and [Lockwood No 1] the High Court was concerned with various product claims each of which consisted of a number of features all of which were essential (in the sense of non-optional) integers of the claim. However, there are some types of claim that may need to be approached slightly differently including, in particular, claims to methods for producing one or more specified results. For example, a claim for a method of producing one or more of outcomes A, B or C might be infringed if the alleged infringer uses the method to produce outcome A, but not outcome B or C…it would seem to me to be wrong in principle to hold that the description of the invention is sufficient if the specification enables the use of the method to achieve outcome A, but not outcomes B or C. It would be inconsistent with the purposes of the Act to confer a monopoly on a patentee for a method of producing any of outcomes A, B or C, if the patentee’s disclosure only enabled the use of the method to produce some of those outcomes.
460 The question arising from the cited passage of Tramanco is whether there is “more than one invention around which the claims are drawn”: Ariosa at [198]. In Ariosa the Full Court observed at [201]:
The Court in Tramanco did not eschew the requirement set out in Kimberly-Clark at [25] that the disclosure enable the addressee to produce something within each claim. Rather it considered that if a claim properly construed had the effect of claiming three distinct alternative outcomes, the position would not be any different to a case where each outcome was expressed in a separate claim. If the disclosure of the specification did not enable at least one method falling within each of the alternatives, then it would not conform with the requirement of s 40(2)(a) …
461 In the same proceeding at first instance, the trial judge, Beach J, referred to Tramanco in some detail: Sequenom, Inc v Ariosa Diagnostics, Inc (2019) 143 IPR 24 at [827]-[856]. In particular, his Honour said at [854]-[855]:
[854] … in my view the peculiar claims at issue in Tramanco were to a method for logging the performance of a vehicle suspension and determining one or more parameters selected from the group consisting of A, B, and C (ie the claim included methods for determining only one parameter out of A, B, C, combinations of two parameters A+B, A+C, B+C and all three parameters A+B+C).
[855] But leaving aside special classes of claims to methods producing one or more specified optional outcomes, it is well settled that only one embodiment within each claim need be enabled for sufficiency purposes. An invention is sufficiently disclosed if a skilled person could make a single embodiment of the invention which falls within the scope of the claims …
462 In Meat & Livestock v Cargill, Beach J at [906] also observed that the claim under consideration in Tramanco at [207] “was in form and in substance a claim to a method of producing alternative results or outcomes”. His Honour distinguished the claims in the case before him on that basis, finding that the claims were sufficiently enabled because the PSA could readily perform an embodiment of the invention within the scope of each claim.
463 The claims in the present case do not claim distinct alternative outcomes. The outcome postulated in claims 1 and 5 of 677 and claim 1 of 689 is one in which “the monoclonal antibody blocks binding of PCSK9 to LDLR”. Claim 23 of 677 necessarily describes that same outcome, as it is dependent on earlier claims, but through an antibody which also competes for binding to PCSK9 with 21B12. In the case of claims 1, 2, 10 and 13 of 751, the outcome is that “the monoclonal antibody reduces binding between PCSK9 and an EGFa domain of LDLR”. It follows that Tramanco is distinguishable.
464 Nor am I persuaded that each of the claims is for a number of different inventions. When considering the issue of fair basis I concluded that the each of the specifications for 677, 689 and 751, which include various consistory clauses and mutual examples, describe a class of antibodies which bind either directly to certain nominated residues, or to epitopes containing certain nominated residues. That conclusion was based not merely on matters of form, but also the substance of the relevant disclosures. That characterisation of the invention is at odds with Sanofi’s argument (which I reject) that each of the claims of 677, 689 and 751 include many different inventions.
465 In the result, I am not persuaded that any of the claims of 677, 689 or 751 would be clearly invalid for non-compliance with s 40(2)(a) of the Act.
466 Although Sanofi’s submissions on this point focussed on the Epitope and Residue Claims, it also submitted that the Competition Claims in 685 and 748 had a “similar vice” and were also properly understood to be claims to a multiplicity of inventions. This submission can be rejected for the same reasons given in relation to the Epitope and Residue Claims. The Competition Claims are to a class of antibodies defined by competition and to one singular outcome (i.e., reduction of binding between PCSK9 and the EGFa domain of LDLR in the case of 685, and neutralisation of PCSK9 as observed in a competitive binding assay in the case of 751).
ADDITIONAL MATTERS
467 The evidence of the Later Crystallised Antibodies (i.e., the crystal structures for antibodies 8A3, 11F1, 1A12 and 25G4) was admitted by me under s 57 of the Evidence Act 1995 (Cth). Evidence regarding the crystal structures of the Later Crystallised Antibodies that is not included in the CS is not relevant to fair basis. Given the limited way in which Sanofi argued the insufficiency ground, evidence of the Later Crystallised Antibodies is also not relevant to that ground. I therefore give that evidence no weight.
468 The parties disagreed as to whether the Applications as filed included colour versions of the crystal structures reproduced in Figures 27A-27E. Amgen informed the Court that for the purposes of this proceeding only it did not rely on the colour versions of the Figures to address any of the grounds of invalidity asserted by Sanofi. I have only had regard to the black and white reproductions of the relevant figures as they appear in the Court Book.
469 The parties also disagreed as to whether a revised written closing submission filed by Sanofi was outside the scope of the leave given to the parties, in that it did more than make corrections to the original version of that document. I have had regard to the revised document filed by Sanofi even though some material included in it was outside the leave granted. I do not consider that this will have caused Amgen any real prejudice.
DISPOSITION
470 For Sanofi to succeed in this appeal it is necessary that it satisfy me that the Applications include one or more claims which would be clearly invalid if the relevant application proceeded to grant. I am not so satisfied. In those circumstances there will be an order dismissing the appeal together with orders affirming the decision of the Delegate and directing that each of the Applications proceed to grant. There will also be an order that Sanofi pay Amgen’s costs of the appeal.
I certify that the preceding four hundred and seventy (470) numbered paragraphs are a true copy of the Reasons for Judgment of the Honourable Justice Nicholas. |
Associate:
Dated: 23 April 2025
Annexure A
Bibliography
Benjannet 2004: Suzanne Benjannet et al, (2004) “NARC-1/PCSK9 and Its Natural Mutants” The Journal of Biological Chemistry 279(47):48865-48875.
Cameron 2006: Jamie Cameron et al, (2006) “Effect of mutations in the PCSK9 gene on the cell surface LDL receptors” Human Molecular Genetics 15(9):1551-1558.
Cohen 2006: Jonathan C Cohen et al, (2006) “Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease” The New England Journal of Medicine 354(12): 1264-1272.
Cunningham 2007: David Cunningham et al, “Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia” Nature Structural Molecular Biology 2007 May;14(5):413-9.
Fisher 2007: Timothy S Fisher et al, “Effects of pH and Low Density Lipoprotein (LDL) on PCSK9-dependant LDL Receptor Regulation” The Journal of Biological Chemistry 282(28):20502-20512.
Graham 2007: Mark J Graham et al, (2007) “Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice” Journal of Lipid Research 48:763-767.
Holla 2007: Øystein L Holla et al, (2007) “Degradation of the LDL receptors by PCSK9 is not mediated by a secreted protein acted upon by PCSK9 extracellularly” BMC Cell Biology 8:9.
Homer 2008: Vivienne M Homer et al, (2008) “Identification and characterization of two non-secreted PCSK9 mutants associated with familial hypercholesterolemia in cohorts from New Zealand and South Africa” Atherosclerosis 196:659-666.
Horton 2007: Jay D Horton et al, (2007) “Molecular biology of PCSK9: its role in LDL metabolism” TRENDS in Biochemical Sciences 32(2):71-77.
Horton 2009: Jay D Horton et al, (2009) “PCSK9: a convertase that coordinates LDL catabolism” Journal of Lipid Research April Supplement S172-S177.
Kwon 2008: Hyock Joo Kwon et al, “Molecular basis for LDL receptor recognition by PCSK9” PNAS 105(6):1820-1825.
Lagace 2006: Thomas A Lagace et al, (2006) “Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice” The Journal of Clinical Investigation 116(11): 2995-3005 and Supplementary data.
Li 2007: Jun Li et al, (2007) “Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity” Biochem. J 406:203-207
Maxwell 2005: Kara N Maxwell et al, (2005) “Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment” PNAS 102(6):2069-2074.
McNutt 2007: Markey C McNutt et al, (2007) “Catalytic Activity Is Not Required for Secreted PCSK9 to Reduce Low Density Lipoprotein Receptors in HepG2 Cells” The Journal of Biological Chemistry 282(29):20799-20803.
Nassoury 2007: Nasha Nassoury et al, (2007) “The Cellular Trafficking of the Secretory Proprotein Convertase PCSK9 and Its Dependence on the LDLR” Traffic 8:718-732.
Piper 2007: Derek E Piper et al, (2007) “The Crystal Structure of PCSK9: A Regulator of Plasma LDL-Cholesterol” Structure 75:545-552.
Qian 2007: Yue-Wei Qian et al, (2007) “Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis” Journal of Lipid Research 48:1488-1498.
Rashid 2005: Shirya Rashid et al, (2005) “Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9” PNAS 102(15):537.
Seidah 2007: Nabil G Seidah and Annik Prat (2007) “The proprotein convertases are potential targets in the treatment of dyslipidemia” J Mol Med 85:6.
Zhang 2007: Da-Wei Zhang et al, (2007) “Binding of Proprotein Convertase Subtilisin/Kexin Type 9 to Epidermal Growth Factor like Repeat A of Low Density Lipoprotein Receptor Decreases Receptor Recycling and Increases Degradation” The Journal of Biological Chemistry 282(25):18602-18612.

