
FEDERAL COURT OF AUSTRALIA
Sun Pharma ANZ Pty Ltd v Otsuka Pharmaceutical Co Ltd [2025] FCA 44
ORDERS
DATE OF ORDER: |
THE COURT ORDERS THAT:
1. Pursuant to ss 37AF(1)(b) and 37AG(1)(a) of the Federal Court of Australia Act 1976 (Cth), access to and disclosure (by publication or otherwise) of the unredacted text of the reasons for judgment delivered today be restricted to the external legal representatives of the parties until further order of the Court.
2. By 7 February 2025, the parties confer to determine whether any paragraphs of these reasons require redaction before being publicly released and provide a draft order to the chambers of Downes J concerning the publication of these reasons.
3. The parties are directed to confer and provide a draft order giving effect to these reasons to the chambers of Downes J by 12 February 2025.
4. If the parties are unable to reach agreement about the form of orders, then the parties are to notify chambers and provide their own version of a draft order, with the areas of disagreement set out in mark-up.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
DOWNES J:
1. OVERVIEW
1 The first respondent/cross-claimant, Otsuka Pharmaceutical Co., Ltd (Otsuka Japan), is the patentee of Australian Patent No. 2004285448, entitled “Controlled release sterile injectable aripiprazole formulation and method” (Patent) which has a priority date of 23 October 2003. The Patent was filed on 18 October 2004.
2 The second respondent/cross-claimant is an exclusive licensee, and the third and fourth respondents/cross-claimants are sub-licensees, pursuant to a deed dated 7 May 2024. In these reasons, I will describe the respondents as Otsuka unless it is necessary to distinguish between them.
3 The Patent describes and claims controlled release formulations which contain aripiprazole as the active pharmaceutical ingredient (API). In particular, the therapeutic effect (or mechanism of action) of the API in the formulations is the binding of the aripiprazole molecules to receptors (primarily D2 receptors) in the brain, which is useful in treating schizophrenia and bipolar I disorder.
4 On 13 August 2014, Otsuka Japan sought an extension of the term of the Patent (Request). The Request was based on a single alleged “pharmaceutical substance”, predicated on part of claim 16 of the Patent, and two products listed on the Australian Register of Therapeutic Goods (ARTG) on 25 July 2014. Both of these ARTG products are kits, comprising (inter alia) aripiprazole (as monohydrate) 300 [or 400] mg powder and solvent for prolonged release suspension for injection vial [or pre-filled syringe], named ABILIFY MAINTENA (ARTG Goods).
5 On 30 September 2014, IP Australia rejected the Request, stating that (inter alia) the “substance” in the ARTG Goods was aripiprazole itself, which was first included in the ARTG on 21 May 2003 (being Otsuka’s earlier aripiprazole product, the ABILIFY tablet product). After further correspondence, the Request was granted, so that the Patent presently expires on 25 July 2029 (Extension).
6 In this proceeding, the applicant/cross-respondent, Sun Pharma ANZ Pty Ltd (Sun Pharma), contends that the Extension was wrongly granted and/or is wrongly existing in the Patent Register and should be removed. There is no dispute that Sun Pharma has standing to seek rectification of the Register by removal of the Extension under s 192 of the Patents Act 1990 (Cth), as a “person aggrieved”.
7 In response, Otsuka relies on eight claims of the Patent (which the parties described as the PTE Claims), or parts thereof, and ten asserted pharmaceutical substances per se.
8 The PTE Claims are exemplified by two different types of claims: (i) those involving controlled release liquid (i.e. ready to use) injectable formulations (being claims 1, 3, 6 and 14) (Controlled Release Injectable Formulation Claims); and (ii) those involving freeze-dried (i.e. lyophilised) controlled release formulations (being claims 16, 19, 21 and 25) (Freeze-dried Controlled Release Formulation Claims). The formulations which are referred to in these claims are the Controlled Release Injectable Formulations and the Freeze-dried Controlled Release Formulations respectively.
9 Otsuka relies on the following pharmaceutical substance(s) per se for the purposes of this proceeding (which I will describe as the asserted pharmaceutical substances per se):
(I) a controlled release sterile aripiprazole injectable formulation, comprising aripiprazole having a mean particle size of about 1 to 10 microns or alternatively about 2 to about 4 microns;
(II) further or in the alternative, (I) above, wherein the aripiprazole is in the form of a monohydrate or alternatively Aripiprazole Hydrate A;
(III) further or in the alternative, (I) or (II) above, also comprising sodium carmellose (carboxymethyl cellulose), mannitol, monobasic monohydrate sodium phosphate and sodium hydroxide;
(IV) further or in the alternative, (III) above, also comprising water for injection;
(V) further or in the alternative, (IV) above, which upon injection releases aripiprazole over at least about one week;
(VI) a sterile lyophilised/freeze-dried aripiprazole formulation comprising aripiprazole having a mean particle size of about 1 to 10 microns or alternatively within the range from about 2 to about 4 microns;
(VII) further or in the alternative, (VI) above, wherein the aripiprazole is in the form of a monohydrate or alternatively Aripiprazole Hydrate A;
(VIII) further or in the alternative, (VI) or (VII) above, also comprising sodium carmellose (carboxymethyl cellulose), mannitol, monobasic monohydrate sodium phosphate and sodium hydroxide;
(IX) further or in the alternative to (VIII) above, which upon constitution with water forms a sterile injectable formulation;
(X) further or in the alternative to (IX) above, which upon injection releases aripiprazole over a period of at least about two weeks.
10 The lack of reference to “controlled release” in substance VI (and accordingly for substances VII–X) is an obvious typographical error, given that it was included in substance I. As this proceeding was brought on an urgent basis, and no point was taken by Sun Pharma in relation to it, I will proceed on the basis that the words “controlled release” are to be inserted before the word “sterile” in substance VI.
11 Sun Pharma contends that the Extension is invalid because none of the asserted pharmaceutical substances per se, nor the PTE Claims (or parts thereof) on which they are predicated, meet the requirements of s 70 of the Patents Act. It advances eight discrete bases for its contention that the Extension is invalid. Those were summarised in an aide memoire which was handed up during opening submissions, and which document became the focus of the parties’ closing submissions. That is so notwithstanding that the parties had filed an Agreed Statement of Issues on 21 October 2024. As to this, certain of the agreed issues no longer required resolution by the time of hearing (namely those in [1], [3] and [8]).
12 The aide memoire identified the following bases:
(1) The Freeze-dried Controlled Release Formulations (relevantly, claims 16, 19, 21 and 25) do not satisfy s 70(2)(a) of the Patents Act because such formulations do not meet the definition of “pharmaceutical substance”.
(2) Insofar as the Controlled Release Injectable Formulations (relevantly, claims 1, 3, 6 and 14) are capable of being a “pharmaceutical substance” (which is denied), the ARTG Goods do not contain or consist of such substance, because the ARTG Goods comprise a freeze-dried powder in a vial (cf s 70(3) of the Patents Act).
(3) Insofar as the PTE Claims claim anything other than the active ingredient aripiprazole simpliciter, such claims are not claims to a “pharmaceutical substance”, as the definition of “pharmaceutical substance” (properly construed) is confined to active ingredients and does not include formulations with excipients.
(4) If the Court determines that the definition of “pharmaceutical substance” does include formulations, neither the Freeze-dried Controlled Release Formulations nor the Controlled Release Injectable Formulations qualify, because the excipients within those formulations are not for a “therapeutic use” as defined and/or do not have an “application (or one of whose applications) [which] involves a chemical interaction, or physico-chemical interaction, with a human physiological system”.
(5) The majority of the asserted pharmaceutical substances per se impermissibly do not take all of the integers of the PTE Claims, and thus cannot be relied upon for the purposes of s 70.
(6) Relatedly, when regard is had to all of the integers of the PTE Claims, they include process features and/or features which limit the use of the formulations, contrary to Full Court authority.
(7) Contrary to s 70(3) of the Patents Act, the ARTG Goods do not take the Relevant Features [which term is defined below].
(8) Each of the PTE Claims is invalid and should be revoked for lack of clarity and/or lack of definition pursuant to s 40(3) and/or s 40(2)(b) of the Patents Act. Accordingly, none of the PTE Claims is available to form the basis for an extension of term under s 70 of the Patents Act.
13 Items 3 and 4 in the aide memoire require a particular construction of the Patents Act to be accepted. Whether that construction is correct was also in issue in Cipla Australia Pty Ltd v Novo Nordisk A/S [2024] FCA 1414, which decision was delivered during the trial of this case. The impact of Cipla on the issues in this case was addressed by the parties, both in writing and in oral closing submissions. Notably, although Sun Pharma’s written submissions contended that aspects of Cipla were plainly wrong such that I should decline to follow it, those submissions were withdrawn. Instead, Sun Pharma reserves its position in any appeal to contend that Cipla was wrongly decided.
14 Sun Pharma has applied to register and intends to sponsor and supply a product registered on the ARTG which is a generic version of the ABILIFY MAINTENA (400 mg) powder and solvent for injection. It wishes to launch that product on 1 April 2025 and requested that this decision be delivered as soon as possible. There was therefore some urgency in having this proceeding heard and determined.
15 Otsuka cross-claimed for threatened infringement in respect of Sun Pharma’s proposed generic product. In an effort for this proceeding to be heard and determined expeditiously, Sun Pharma did not contest the claim of threatened infringement (if the Extension and the PTE Claims were found to be valid) by making certain admissions for the purposes of this proceeding. Those admissions were made without prejudice to Sun Pharma’s position on the proper construction of each of the claims, and in circumstances where it maintained its position concerning the invalidity of the PTE Claims and the Extension: see, in particular, [25]–[30], [35], [37], [39]–[41] and [46] of the Amended Defence to Cross-Claim. Contrary to Otsuka’s submission, the admissions by Sun Pharma do not assist Otsuka’s case for this reason.
16 Otsuka also cross-claimed for threatened infringement of s 18(1) of the Australian Consumer Law (ACL), which is Schedule 2 to the Competition and Consumer Act 2010 (Cth), which claim was denied.
17 For the reasons which follow, I have determined that the PTE Claims and Extension are invalid, and that the Cross-Claim should be dismissed. That is because:
(1) Substances I–IV and VI–IX do not in substance fall within the scope of the claim or claims as required by s 70(2)(a) of the Patents Act.
(2) The PTE Claims are invalid as they fail to comply with ss 40(2)(b) and 40(3) of the Patents Act.
18 As many aspects of the written and oral evidence were restricted by reasons of confidentiality, orders will be made which restrict access to these reasons and provide the parties with an opportunity to address which aspects should remain confidential.
2. WITNESSES CALLED BY THE PARTIES
19 Sun Pharma adduced expert evidence from two experts:
(1) Professor Gerhard Johannes Winter, a Professor and former Chair of the Pharmaceutical Technology and Biopharmaceutics group of the Department of Pharmacy at Ludwig Maximillians University of Munich. Professor Winter affirmed two affidavits, dated 19 July 2024 (Winter 1) and 15 October 2024 (Winter 2) although Sun Pharma did not read [38]–[45] of Winter 2;
(2) Professor Guy Manning Goodwin, psychiatrist and Emeritus Professor in the Department of Psychiatry at the University of Oxford and the Chief Medical Officer at Compass Pathways. Professor Goodwin swore two affidavits dated 19 July 2024 (Goodwin 1) and 9 October 2024 (Goodwin 2) although Sun Pharma did not read [19]– [25] of Goodwin 2.
20 Sun Pharma also relied on an affidavit of its solicitor, Ms Nina Fitzgerald, dated 19 July 2024.
21 Otsuka adduced expert evidence from Professor Allan Mark Evans, a pharmaceutical scientist and a clinical pharmacologist, and an Adjunct Professor at the University of South Australia. Professor Evans affirmed two affidavits, dated 20 September 2024 and 26 November 2024. All references in these reasons to the affidavit evidence of Professor Evans is to the affidavit dated 20 September 2024.
22 The experts prepared a joint expert report dated 5 November 2024 (JER).
23 Of the witnesses, only Professor Winter and Professor Evans were required for cross-examination, and they gave concurrent expert evidence on 2, 3 and 4 December 2024.
24 Of these two experts, Professor Winter was, by far, the more impressive expert, both on paper and in person. Professor Winter has a vast amount of experience in the formulation of drug products, including lyophilised products, and he is by far the more qualified of the two experts. I also found that Professor Winter’s evidence was more direct and on point, whereas the evidence of Professor Evans often avoided dealing with an issue squarely. Some of his affidavit evidence is, in particular, obfuscatory insofar as it purports to respond to the evidence of Professor Winter. That is because, while it refers to particular aspects of Professor Winter’s evidence expressly, it does not identify any disagreement with it or explain the reasons for that disagreement.
25 Further, in addition to having superior qualifications and experience to that of Professor Evans, Professor Winter displayed such a depth of knowledge and understanding of the topics being addressed that I found his views to be more compelling, well-reasoned, consistent and logical than those of Professor Evans. Further, all of Professor Winter’s oral answers to questions were dispassionate and he made appropriate concessions. He was an exemplary expert witness.
26 Otsuka was critical of Professor Winter and contended that he did not approach and interpret the Patent in the same way as a person skilled in the art would have engaged in this task. However, I reject all of these criticisms. Generally, Professor Winter raised genuine concerns about lack of data in the Patent, which concerns should have been able to be addressed by Professor Evans if they were excessive or unduly critical, but that did not occur.
27 By contrast to Professor Winter, Professor Evans did, on occasion, become antagonistic when giving his answers, and at times it seemed that his answers were intended to protect the PTE Claims from the invalidity attack.
28 More significantly, it did not appear that the affidavit evidence of Professor Evans provided the full picture of the work which he had done, and a full statement of the reasons for the opinions expressed by him.
29 Professor Evans states that his analysis in respect of Example 4 and Figure 3 of the Patent was “based only on the information provided in the 448 patent, in particular, Example 4 and Figure 3”: Evans, [117]. However, it emerged during the hearing that the analysis was not so limited, particularly as his analysis was heavily predicated on a 28-day “proposed clinical use”. During the concurrent evidence session, Professor Evans accepted that “the 28-day dosing interval was not disclosed in example 4 or figure 3” and could not recall whether it was disclosed or suggested to him by Otsuka’s legal representatives to use the 28-day dosing interval in his calculations based on ABILIFY MAINTENA.
30 It also transpired that Professor Evans had performed calculations using “multiple dosing at a range of doses” using Microsoft Excel, but this was not contained in his affidavits, or referred to by him as forming part of his reasons for his conclusions, contrary to clauses 3(d) and 3(e) of the Harmonised Expert Witness Code of Conduct which is Annexure A to the Expert Evidence Practice Note (GPN-EXPT). As recognised by [2.4] of that Practice Note, such matters affect the weight to be attached to this expert’s opinion evidence.
31 For these reasons and for further reasons given below, I have preferred the evidence of Professor Winter to that of Professor Evans where the two experts disagreed as I attach less weight to the evidence of Professor Evans.
3. THE PATENT
3.1 Release from the depot and absorption into central compartment
32 The following matters formed part of the common general knowledge in Australia as at 23 October 2003.
33 In general terms, one can differentiate drug release profiles in the following way:
(1) immediate release: where a drug substance is released almost immediately upon administration; and
(2) controlled release: any formulation wherein the release of the drug substance differs from an immediate release formulation in that the release of the drug substance is retarded, for example due to the construction of the formulation (like a coated tablet or polymer-based formulations).
34 A type of controlled release formulation is an extended release formulation wherein the drug is released over time, rather than immediately. By extending the release profile of a drug, a formulator can (amongst other things) reduce the frequency of dosing required.
35 Aqueous suspensions are typically made up of a poorly soluble drug substance, one or more excipients, and water. Upon combination with the water, the drug substance forms a suspension whereby the particles of drug substance do not dissolve but exist as (undissolved) solid particles within the liquid. The formulation is then administered to a patient by injection.
36 Since the drug substance is poorly soluble in physiological conditions, it forms a physical “depot” at the injection site.
37 A depot is an injectable formulation that releases a given amount of drug substance slowly over time. The depot, when used in the context of an aqueous suspension formulation, is the collection of particles of drug substance that are trapped at the site of administration. Over time, molecules of the drug substance dissolve from the particles of drug substance in the depot and, depending on the site and route of administration, must cross a number of biological membranes before entering the “central compartment”, which is the blood plasma and organs (such as the liver) that receive a very rich blood supply and turnover of blood.
38 The process of dissolution of a drug substance from a depot is often referred to as “release”, whereas the entry of the drug substance into the central compartment is often referred to as “absorption”.
39 For aqueous suspension formulations, the process of dissolution (or release) of a particulate drug substance is dictated by the following (non-exhaustive) factors:
(1) The size of the drug substance particles: the larger the size of a drug particle, the longer it will take for all of the drug substance to dissolve (or release) from the depot.
(2) The physico-chemical properties of the drug: for example, drugs with lower aqueous solubility (and, more specifically, with lower solubility at physiological conditions) will typically take longer to dissolve upon administration, and so will release drug molecules over a longer period than drugs with higher aqueous solubility.
(3) Any modifications made to the drug substance to decrease its solubility at physiological conditions.
(4) The site of administration of the formulation. This is because of reasons associated with the amount of mechanical force to which a site is subjected and the tissues neighbouring the injection site. Suspension formulations are more prone to be affected by these factors than other extended release formulations, which can impact the time that it takes for a drug substance in particulate form to completely dissolve upon administration.
(5) The volume of formulation being injected. The pressure associated with the administration of larger volumes (2 ml or more) of formulation can cause the formulation to disperse over a greater volume of space inside the body, which can decrease the time that it takes for the drug to completely dissolve from the formulation.
(6) The shape and nature of the drug substance particles, which can stimulate an immune response and delay the time that it takes for the drug to dissolve and be transported away from the site of the injection.
(7) The concentration and dosage of the drug substance.
40 The process of absorption can occur in three main ways:
(1) Often when a formulation is administered by injection, the process of injection causes capillaries at the site of injection to rupture. Due to the body’s self-healing processes, these ruptures will be sealed off quickly. Nevertheless, a small amount of the drug substance may be transported from the site of injection immediately through these ruptured capillaries.
(2) After the suspension formulation has been injected and some of the particulate drug substance has dissolved, the dissolved molecules will diffuse from the tissue or interstitial tissue across one or more biological membranes into surrounding capillaries, and then be transported into the central compartment.
(3) The dissolved drug molecules can also diffuse into the lymphatic tubes that are present in skeletal muscles, and enter the lymphatic transport system, from where they are eventually transported to and absorbed into the central compartment.
41 A drug may be released from the depot but not absorbed due to physico-chemical reasons. For example, it is energetically preferable for drugs that are lipophilic to remain in the fatty tissue. Therefore, a lipophilic drug substance may be in solution at the site of administration but, if administered subcutaneously, it will be energetically favourable for that drug substance to remain in the fatty tissue rather than diffusing across biological membranes to enter the bloodstream.
42 The mechanisms and timeframes involved in the process of absorption of a drug substance (once released from the depot) will vary depending on a range of factors, including the following:
(1) The chemical composition of the drug substance and any modifications made to it to increase its lipophilicity or ionisation at physiological conditions.
(2) The site and route of administration and the pathway to the central compartment. The time taken for molecules of a drug substance to enter the central compartment will depend on the length of the pathway to the central compartment (including the number and types of membranes that it needs to pass), which in turn depends on the site and route of administration (as well as the physical and chemical characteristics of the drug substance). The time taken for the drug substance molecules to enter the central compartment will also depend on the presence or absence of phagocytotic cell types, and the velocity of any transport fluids that the drug substance is exposed to, such as the velocity of blood in the blood stream or the velocity of the lymphatic flow. Typically, formulations that are administered intramuscularly will be absorbed faster than if injected subcutaneously because the diffusion path length from the site of administration to the central compartment is shorter for drugs administered intramuscularly.
43 Several factors can affect the amount of a drug in the blood plasma. In particular, the blood plasma levels of a drug substance will depend on (inter alia) the amount of the drug administered, the bioavailability of the a drug substance, the site of administration, the time it takes for the drug molecules to dissolve, and then to be absorbed, upon administration, and the rates at which the drug is metabolised, distributed and excreted upon administration.
3.2 The specification
44 Aripiprazole is an antipsychotic agent useful in treating schizophrenia. Aripiprazole itself is not the invention claimed in the Patent, nor was it a “new” substance as at the filing date – rather, it was the subject of US Patent No. 5,006,528 (a compound patent) granted over a decade earlier in 1991. Rather, the Patent is directed to controlled release aripiprazole formulations and methods for preparing and using such formulations.
45 The Field of the Invention relevantly states:
The present invention relates to a controlled release sterile freeze-dried aripiprazole formulation, an injectable formulation which contains the sterile freeze-dried aripiprazole and which releases aripiprazole over at least a one week period, a method for preparing the above formulation, and a method for treating schizophrenia and related disorders employing the above formulation.
46 In the Background of the Invention, the following is stated:
Aripiprazole which has the structure
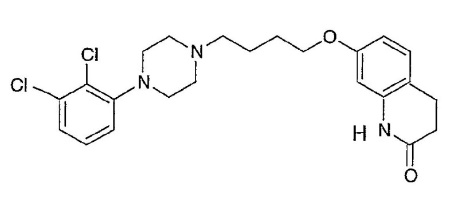
is an atypical antipsychotic agent useful in treating schizophrenia. It has poor aqueous solubility (<1μg/mL at room temperature).
47 The Background also states, “A long-acting aripiprazole sterile injectionable formulation has merit as a drug dosage form in that it may increase the compliance of patients and thereby lower the rate of relapse in the treatment of schizophrenia”.
48 The formulations described and claimed in the Patent include (a) freeze-dried controlled release aripiprazole formulations that need to be reconstituted with water to form an injectable formulation before administration; and (b) controlled release injectable formulations that are administered as aqueous “ready-to-use” suspensions.
49 A Brief Description of the Invention includes the following statements about the freeze-dried controlled release aripiprazole formulations:
Disclosed herein is a sterile freeze-dried aripiprazole formulation which upon constitution with water for injection releases aripiprazole, in therapeutic amounts, over a period of at least about one week, and preferably over a period of two, three or four weeks and up to six weeks or more. The freeze-dried aripiprazole formulation may include:
(a) aripiprazole, and
(b) a vehicle for the aripiprazole,
which formulation upon constitution with water forms an injectable suspension which, upon injection, preferably intramuscularly, releases therapeutic amounts of aripiprazole over a period of at least one week, preferably two, three or four weeks, and up to six weeks or more.
50 After referring to five different aspects of the invention, the following is stated:
A mean particle size of the freeze-dried aripiprazole formulation within the range from about 1 to about 30 microns is essential in formulating an injectable which releases aripiprazole over a period of at least about one week and up to six weeks or more, for example up to 8 weeks.
It has been found that the smaller the mean particle size of the freeze-dried aripiprazole, the shorter the period of extended release. Thus, in accordance with the present invention, when the mean particle size is about 1 micron, the aripiprazole will be released over a period of less than three weeks, preferably about two weeks. When the mean particle size is more than about 1 micron, the aripiprazole will be released over a period of at least two weeks, preferably about three to four weeks, and up to six weeks or more. Thus, in accordance with the present invention, the aripiprazole release duration can be modified by changing the particle size of the aripiprazole in the freeze-dried formulation.
The term “mean particle size” refers to volume mean diameter as measured by laser-light scattering (LLS) methods. Particle size distribution is measured by LLS methods and mean particle size is calculated from the particle size distribution.
51 The specification then refers to the controlled release injectable formulations as follows:
Also disclosed herein is a controlled release sterile injectable aripiprazole formulation in the form of a sterile suspension, that is, the freeze-dried formulation of the invention suspended in water for injection, is provided which, upon injection, preferably intramuscularly, releases therapeutic amounts of aripiprazole over a period of at least one week, which includes:
(a) aripiprazole,
(b) a vehicle therefor, and
(c) water for injection.
The controlled release sterile injectable formulation of the invention in the form of a sterile suspension allows for high drug loadings per unit volume of the formulation and therefore permits delivery of relatively high doses of aripiprazole in a small injection volume (0.1 - 600 mg of drug per 1 mL of suspension).
52 The Brief Description also states:
As an unexpected observation, it has been discovered that a suspension of aripiprazole suspended in an aqueous solvent system will maintain a substantially constant aripiprazole drug plasma concentration when administered by injection; preferably as an intra-muscular injection. No large “burst phenomenon” is observed and it is considerably surprising that a constant aripiprazole drug plasma concentration can be maintained from one (1) to more than eight (8) weeks employing the aripiprazole suspension of the invention. The daily starting dose for an orally administered aripiprazole formulation is fifteen (15) milligrams. In order to administer a drug dose equivalent to one (1) to more than eight (8) weeks of the oral dosage quantity requires the administration of a very large amount of the drug as a single dose. The aqueous aripiprazole injectable formulation of the invention may be administered to deliver large amounts of the drug without creating patient compliance problems.
The aripiprazole injectable formulation of the invention may include anhydrous or monohydrate crystalline forms of aripiprazole or an admixture containing both. If the monohydrate is used, the maintenance of an extended drug plasma concentration is possible.
The aripiprazole injectable formulation of the invention can be administered as an aqueous ready-to-use suspension; however, by freeze-drying this suspension a more useful drug product can be supplied.
53 In the Detailed Description, the following is stated:
The controlled release sterile injectable aripiprazole formulation of the invention will include aripiprazole in an amount within the range from about 1 to about 40%, preferably from about 5 to about 20%, and more preferably from about 8 to about 15% by weight based on the weight of the sterile injectable formulation.
As indicated, desired mean particle size of the aripiprazole is essential in producing an injectable formulation having the desired controlled release properties of the aripiprazole. Thus, to produce desired controlled release, the aripiprazole should have a mean particle size within the range from about 1 to about 30 microns, preferably from about 1 to about 20 microns, and more preferably for about 1 to about 10 to 15 microns.
Where the desired controlled release period is at least about two weeks, up to six weeks or more, preferably about three to about four weeks, the aripiprazole will have a mean particle size within the range from about 1 to about 20, preferably from about 1 to about 10 microns, more preferably from about 2 to about 4 microns, and most preferably about 2.5 microns.
54 It also stated that:
The aripiprazole formulation of the invention will preferably be formed of:
A. aripiprazole,
B. a vehicle therefor, which includes:
(a) one or more suspending agents,
(b) one or more bulking agents,
(c) one or more buffers, and
(d) optionally one or more pH adjusting agents.
The suspending agent will be present in an amount within the range from about 0.2 to about 10% by weight, preferably for about 0.5 to about 5% by weight based on the total weight of the sterile injectable formulation. Examples of suspending agents suitable for use include, but are not limited to, one, two or more of the following: sodium carboxymethyl cellulose, hydroxypropyl cellulose, carboxymethyl cellulose, hydroxypropylethyl cellulose, hydroxypropylmethyl cellulose, and polyvinylpyrrolidone, with sodium carboxymethyl cellulose and polyvinylpyrrolidone being preferred. Other suspending agents suitable for use in the vehicle for the aripiprazole include various polymers, low molecular weight oligomers, natural products, and surfactants, including nonionic and ionic surfactants, such as cetyl pyridinium chloride, gelatin, casein, lecithin (phosphatides), dextran, glycerol, gum acacia, cholesterol, tragacanth, stearic acid, benzalkonium chloride [etc]…
Carboxymethyl cellulose or the sodium salt thereof is particularly preferred where the desired mean particle size is about 1 micron or above. …
The bulking agent (also referred to as a cryogenic/lyophilize protecting agent) will be present in an amount within the range from about 1 to about 10% by weight, preferably from about 3 to about 8% by weight, more preferably from about 4 to about 5% by weight based on the total weight of the sterile injectable formulation. Examples of bulking agents suitable for use herein include, but are not limited to, one, two or more of the following: mannitol, sucrose, maltose, xylitol, glucose, starches, sorbital, and the like, with mannitol being preferred for formulations where the mean particle size is about 1 micron or above. It has been found that xylitol and/or sorbitol enhances stability of the aripiprazole formulation by inhibiting crystal growth and agglomeration of drug particles so that desired particle size may be achieved and maintained.
The buffer will be employed in an amount to adjust pH of an aqueous suspension of the freeze-dried aripiprazole formulation to from about 6 to about 8, preferably about 7. …Examples of buffers suitable for use herein include, but are not limited to, one, two or more of the following: sodium phosphate, potassium phosphate, or TRIS buffer, with sodium phosphate being preferred.
The freeze-dried formulation of the invention may optionally include a pH adjusting agent which is employed in an amount to adjust pH of the aqueous suspension of the freeze-dried aripiprazole within the range from about 6 to about 7.5, preferably about 7 and may be an acid or base depending upon whether the pH of the aqueous suspension of the freeze-dried aripiprazole needs to be raised or lowered to reach the desired neutral pH of about 7. …
The freeze-dried aripiprazole formulations may be constituted with an amount of water for injection to provide from about 10 to about 400 mg of aripiprazole delivered in a volume of 2.5 mL or less, preferably 2 mL for a two to six week dosage.
55 Reference is also made to the “desired crystalline form of the aripiprazole” which exists in monohydrate form (Aripiprazole Hydrate A) as well as in a number of anhydrous forms, namely Anhydride Crystals B–G. The specification identifies methods of manufacturing Aripiprazole Hydrate A and preparing Aripiprazole Anhydride Crystals B, and then identifies preferred injectable formulations in the form of aqueous suspensions. It is then stated that:
The aripiprazole formulations of the invention are used to treat schizophrenia and related disorders such as bipolar disorder and dementia in human patients. The preferred dosage employed for the injectable formulations of the invention will be a single injection or multiple injections containing from about 100 to about 400 mg aripiprazole/mL given one to two times monthly. The injectable formulation is preferably administered intramuscularly, although subcutaneous injections are acceptable as well.
56 Four examples are then set out which are said to represent preferred embodiments of the invention.
57 In Examples 1 and 2, there is a description of two ways to prepare an aripiprazole injectable (IM Depot) aqueous suspension (200 mg aripiprazole/2 mL, 200 mg/vial), one by media milling (Example 1) and the other by impinging jet crystallisation (Example 2).
58 Example 3 (Animal PK Data) relates to injecting the formulation in Example 1 into 15 rats and 5 dogs. Reference is made to Figures 1 and 2 as showing “mean plasma concentrations vs. time profiles”. Reference is then made to “PK profiles” (with PK being a reference to pharmacokinetics) and it is stated that:
Mean aripiprazole rats’ serum concentration-time profiles are shown graphically in Fig.1. Aripiprazole aqueous suspensions showed steady serum concentration for at least 4 weeks in the rats’ model.
Mean aripiprazole dogs’ serum concentration-time profiles are shown graphically in Fig.2.
Aripiprazole aqueous suspensions showed steady serum concentration for 3-4 weeks in the dogs’ model.
59 Example 4 (Human PK Data) relates to a single-dose IM Depot study. The following is the entirety of the information relating to Example 4:
Aripiprazole I.M. depot formulation prepared in Example 1 was administered intramuscularly to patients diagnosed with chronic, stable schizophrenia or schizoaffective disorder at [sic]. The study design included administration of a 5-mg dose of aripiprazole solution to all subjects followed by a single dose of IM depot at 15, 50, and 100 mg per patient. Samples for PK analysis were collected until plasma concentrations of aripiprazole were less than the lower limit of quantification (LLQ) for 2 consecutive visits.
Figure 3 shows mean plasma concentrations vs. time profiles of aripiprazole in subjects 2 and 3 dosed with 15 mg of IM Depot, and subjects 4 and 5 who received 50 mg of IM Depot. In all cases aripiprazole plasma levels showed a fast onset of release and sustained release for at least 30 days.
60 Figure 3 (which appears after the claims) shows the following:
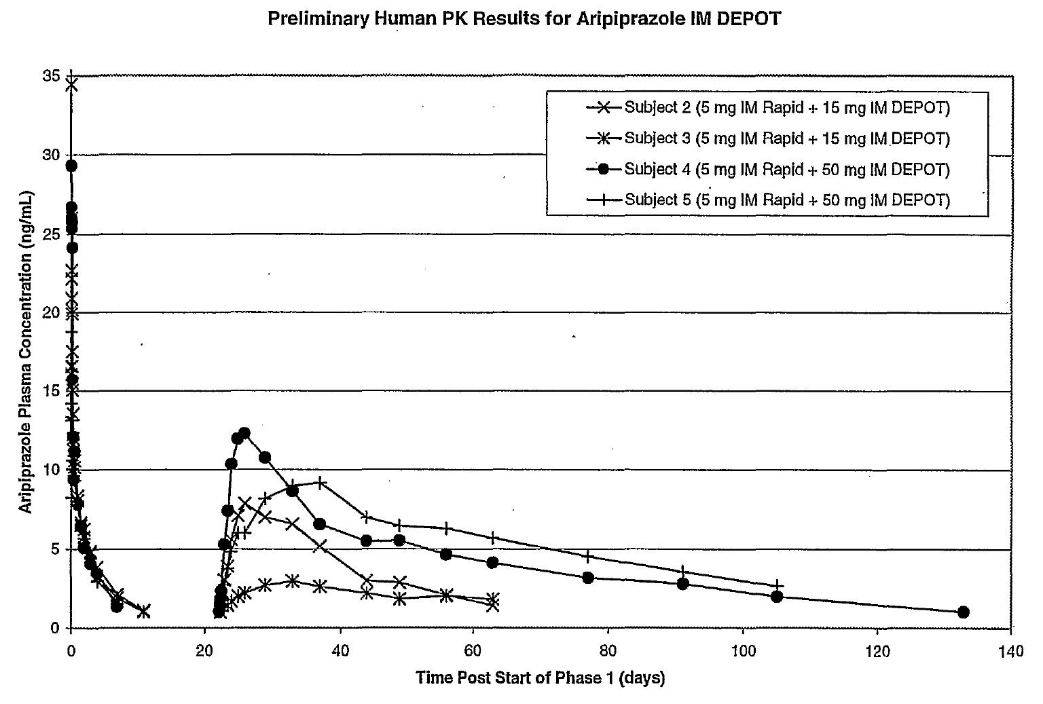
61 The reference to IM Rapid appears to be a reference to the 5-mg dose of aripiprazole solution referred to in Example 4.
62 There are 74 claims, but only eight of these matter for this case (being the PTE Claims).
63 The Controlled Release Injectable Formulation Claims are exemplified by claim 1, which is as follows:
1. A controlled release sterile aripiprazole injectable formulation which upon injection releases aripiprazole over a period of at least one week, which comprises:
(a) aripiprazole having a mean particle size of about 1 to 10 microns,
(b) a vehicle therefor, and
(c) water for injection.
64 The other Controlled Release Injectable Formulation Claims are as follows:
3. A controlled release aripiprazole injectable formulation which upon injection releases aripiprazole over a period of at least one week, which comprises:
(a) aripiprazole having a mean particle size of about 1 to 10 microns, and
(b) a vehicle therefor, said vehicle comprising:
(1) one or more suspending agents,
(2) optionally one or more bulking agents, and
(3) optionally one or more buffering agents, and
(c) water for injection.
…
6. The formulation as defined in any one of claims 3 to 5 wherein the aripiprazole has a mean particle size within the range from about 2 to about 4 microns.
…
14. The formulation as defined in any one of claims 1 to 13 wherein the aripiprazole is in the form of a monohydrate.
65 The Freeze-dried Controlled Release Formulation Claims are exemplified by Claim 16, which is as follows:
16. A sterile freeze-dried controlled release aripiprazole formulation which comprises:
(a) aripiprazole having a mean particle size of about 1 to 10 microns, and
(b) a vehicle therefor,
which formulation upon constitution with water forms a sterile injectable formulation which upon injection releases aripiprazole over a period of at least about two weeks.
66 The other Freeze-dried Controlled Release Formulation Claims are as follows:
19. The freeze-dried formulation as defined in any one of claims 16 to 18 wherein the aripiprazole has a mean particle size within the range from about 2 to about 4 microns.
…
21. The freeze-dried formulation as defined in any one of claims 16 to 20 wherein said vehicle comprises:
(a) one or more suspending agents,
(b) one or more bulking agents, and
(c) one or more buffering agents.
…
25. The freeze-dried formulation as defined in any one of claims 16 to 24 wherein the aripiprazole is in the form of a monohydrate.
67 The PTE Claims do not claim (a) any specified dosages; (b) that such dosage be of a “therapeutic amount”; nor (c) any dosage interval (frequency). Nor do they refer to, or identify, a claimed blood plasma concentration.
3.3 Relevant Feature
68 Having regard to the letter of instructions to the experts for the purposes of preparing the JER, the reference in each of the PTE Claims to “which upon injection releases aripiprazole over a period of [a specified time]” (or equivalent) is referred to as the Relevant Feature.
69 Professor Evans and Professor Winter agreed that the Relevant Feature in each of the PTE Claims follow a common structure and all refer to three elements:
(1) a process of injection (i.e. the injection of the claimed formulation; whether it is a subcutaneous or intramuscular injection is not specified). (Professor Evans notes that the Patent states that the route of administration is preferably intramuscularly and the Examples provided relate to intramuscular administration);
(2) a parameter for the release of aripiprazole (which is the dissolution of aripiprazole molecules from the aripiprazole particle depot at the injection site); and
(3) a defined discrete boundary for release expressed in weeks (e.g. in multiples of seven days, plus/minus 10% where the term “about” is used).
70 It is apparent from this evidence that the Relevant Feature is not to be construed as “mean plasma concentration” or some other drug concentration metric.
71 Further, by this evidence, it is apparent that a person skilled in the art would understand that “release”, as it is to be understood in the context of the PTE Claims and the specification as a whole, means the dissolution of aripiprazole molecules from the aripiprazole particle depot at the injection site.
4. ISSUES OF STATUTORY CONSTRUCTION
72 Before dealing with the contentions by Sun Pharma concerning the validity of the Extension, certain issues of statutory construction which arose in this case will be addressed.
4.1 The Patents Act
73 Section 70 of the Patents Act requires three conditions to be satisfied by a patentee seeking a term extension, each of which must be satisfied in respect of a “pharmaceutical substance”.
74 Schedule 1 of the Patents Act defines “pharmaceutical substance” as follows:
pharmaceutical substance means a substance (including a mixture or compound of substances) for therapeutic use whose application (or one of whose applications) involves:
(a) a chemical interaction, or physico-chemical interaction, with a human physiological system; or
(b) action on an infectious agent, or on a toxin or other poison, in a human body;
but does not include a substance that is solely for use in in vitro diagnosis or in vitro testing.
75 Schedule 1 defines “therapeutic use” as follows:
therapeutic use means use for the purpose of:
(a) preventing, diagnosing, curing or alleviating a disease, ailment, defect or injury in persons; or
(b) influencing, inhibiting or modifying a physiological process in persons; or
(c) testing the susceptibility of persons to a disease or ailment.
76 The first condition operates to identify a candidate patent for extension and is set out in s 70(2) as follows:
Either or both of the following conditions must be satisfied:
(a) one or more pharmaceutical substances per se must in substance be disclosed in the complete specification of the patent and in substance fall within the scope of the claim or claims of that specification;
(b) one or more pharmaceutical substances when produced by a process that involves the use of recombinant DNA technology, must in substance be disclosed in the complete specification of the patent and in substance fall within the scope of the claim or claims of that specification.
77 Sun Pharma alleges that the first condition was not satisfied by reference to s 70(2)(a). It was common ground that s 70(2)(b) did not apply in this case; however, as will be seen, that section is relevant to the proper construction of s 70(2)(a).
78 The second condition (relevantly to this case) is whether the pharmaceutical substance is contained in a good on the ARTG. In this regard, s 70(3) provides:
Both of the following conditions must be satisfied in relation to at least one of those pharmaceutical substances:
(a) goods containing, or consisting of, the substance must be included in the Australian Register of Therapeutic Goods;
(b) the period beginning on the date of the patent and ending on the first regulatory approval date for the substance must be at least 5 years.
79 Sun Pharma alleges that the second condition was not satisfied because s 70(3)(a) was not satisfied. Sun Pharma also submits that s 70(3)(b) was not satisfied only if, contrary to the Full Court authorities, Otsuka is permitted to rely upon aripiprazole alone as satisfying the requirements of s 70(2)(a).
80 The third condition is that the term of the patent must not have been previously extended: s 70(4). It is common ground that the third condition was satisfied.
81 Pursuant to s 78 of the Patents Act, following the grant of an extension under s 70, it is not an infringement of the patent to exploit any form of the invention other than a pharmaceutical substance per se that is in substance disclosed in the complete specification of the patent and in substance falls within the scope of the claim or claims of that specification. The operation of s 78 has been recognised as narrowing a patentee’s monopoly in the extended term: see, for example, Merck Sharp & Dohme Corp v Sandoz Pty Ltd (2022) 291 FCR 26; [2022] FCAFC 40 at [109] (Allsop CJ, Yates and Burley JJ). As will be seen, this is relevant to the proper construction of s 70.
4.2 Meaning of “pharmaceutical substance per se”
82 Section 70(2) refers to “one or more pharmaceutical substances per se”. The meaning of pharmaceutical substance per se has been addressed in a number of decisions of this Court, and in passing by the High Court.
83 The term “pharmaceutical substance per se” means the pharmaceutical substance “by or in itself, intrinsically, essentially”, or “taken alone; essentially; without reference to anything else”: Boehringer Ingelheim International GmbH v Commissioner of Patents (2001) 112 FCR 595; [2001] FCA 647 at [34], [37] (Wilcox, Whitlam and Gyles JJ) (Boehringer FC).
84 The Full Court in Boehringer FC stated at [37] that the decision of Heerey J, the primary judge, was “correct, substantially for the [following] reasons”, which reasons were reproduced at [17] of its decision:
The 1990 Act in its present form manifests a policy which draws a distinction between, on the one hand, a pharmaceutical substance that is the subject of a patent claim and, on the other hand, a pharmaceutical substance that forms part of a method or process claim. The specific exception to the latter (an exception which proves the rule) is the provision for recombinant DNA technology in s 70(2)(b).
Broadly speaking, a claim in relation to a pharmaceutical substance can be made in three ways:
(i) a new and inventive product alone;
(ii) an old or known product prepared by a new and inventive process;
(iii) an old or known product used in a new and inventive mode of treatment.
What is clear in s 70 is that only the first type of claim to a pharmaceutical product is to be subject to extension rights. So far as a new process is concerned, it is only when the new process answers the particular description in s 70(2)(b) (recombinant DNA process) that it can be the subject of an extension. As counsel for the Commissioner submitted, the policy to be deduced in the light of the legislative history is that Parliament has decided that what is intended to be fostered is primary research and development in inventive substances, not the way they are made or the way they are used, with the sole (and important) exception of recombinant DNA techniques, this being an area particularly worthy of assistance for research and development.
In the light of this history, the relevance of the expression ‘per se’ becomes clear. Section 70(2)(a) is only to make extension rights available when the claim is for a pharmaceutical substance as such, as opposed to a substance forming part of a method or process.
(Emphasis added.)
85 The Full Court in Boehringer FC continued at [38]–[40]:
There are serious difficulties about the appellant’s argument. One is that it effectively reads out of s 70(2)(a) the words “per se”. On the appellant’s argument, it is enough that the complete specification disclose one or more pharmaceutical substances, whether as the sole element in an invention or in combination with other elements. If that had been the legislative intention, the paragraph could have read: “one or more pharmaceutical substances must in substance be disclosed”. There would have been no need for “per se”.
Second, the Second Reading Speech and the Explanatory Memorandum provide no support to the appellant’s argument; quite the contrary. The Second Reading Speech speaks about the “development of a new drug” and the research and testing required before “the product” can enter the market. This is plainly a reference to the drug itself; not to the drug in combination with other elements.
Similarly, the Explanatory Memorandum says that claims to “pharmaceutical substances per se, would usually be restricted to new and inventive substances”. The Explanatory Memorandum excludes the application of the new provisions to “new processes of making pharmaceutical substances or new methods of using pharmaceutical substances, where the substances themselves are known”.
(Emphasis original.)
86 In Prejay Holdings Ltd v Commissioner of Patents (2003) 57 IPR 424; [2003] FCAFC 77, the Full Court concluded that it is not enough to constitute a claim for a pharmaceutical substance per se that a pharmaceutical substance “appears in a claim in combination with other integers or as part of the description of a method (or process) that is the subject of a claim”: see [24] (Wilcox and Cooper JJ) and [35]–[36] (Allsop J). At [40] and [42], Allsop J added the following:
… The definition refers to a substance, which must have a purpose or use — therapeutic use, and whose application involves the other matters identified in the definition. The definition is of a particular kind of substance, but it is of a substance, and only a substance.
…
While the secondary materials are not entirely internally consistent, the above result and the above construction accord with what appears to me to be the burden of the secondary materials, exemplified by the following part of the explanatory memorandum, referred to by the Full Court in Boehringer at [24]:
The extension of term provisions will be available for patents that include claims to pharmaceutical substances per se provided the other criteria are met. These claims to pharmaceutical substances per se, would usually be restricted to new and inventive substances. Patents that claims [sic], will not be eligible unless the process involves the use of recombinant DNA technology. Claims which limit the use of a known substance to a particular environment, for example claims to pharmaceutical substances when used in a new and inventive method of treatment, are not considered to be claims to the pharmaceutical substance per se…
(Emphasis original.)
87 In Pharmacia Italia SpA v Mayne Pharma Pty Ltd (2006) 69 IPR 1; [2006] FCA 305, Weinberg J stated at [96]:
…If a claim, properly understood, is, in effect, intended to protect some novel process or method, no extension can be granted. The claim, when read sensibly, and as a whole, must be to a “pharmaceutical substance per se”. At least in circumstances where the substance itself is known, the claim must not be, in essence, to a new process of making that substance, or a new method of using it.
88 His Honour also observed at [98] and [101]:
The matter must largely be one of impression, and degree. Reasonable minds may differ as to whether the legitimate boundaries of a “pharmaceutical substance per se” have been crossed.
…
Patent rights, and in particular the right to an extension, are very much dependent upon the language of the claim which defines the invention. When construing a claim, in order to determine whether the requirements set out in s 70(2)(a) are satisfied, it is appropriate to have regard to the reason why any reference to process has been included in the claim as formulated. There is a difference between seeking to protect a process (which can be the subject of a patent, but cannot be the subject of an extension), and merely referring incidentally to some elements of process, that are not themselves novel, in order to better describe the new and inventive substance. Section 70(2)(a) provides that a new and inventive substance that is a “pharmaceutical substance per se” can be the subject of an extension. …
89 In Spirit Pharmaceuticals Pty Ltd v Mundipharma Pty Ltd (2013) 216 FCR 344; [2013] FCA 658 at [45], Rares J approved the approach taken in Pharmacia at [101].
90 In H Lundbeck A/S v Alphapharm Pty Ltd (2009) 177 FCR 151; [2009] FCAFC 70 (Alphapharm FC), Bennett J (with whom Middleton J agreed) referred to the requirements of s 70 at [242] and stated at [243] that:
A patent may be obtained if it discloses and claims, for example, a new method of preparation, or purification that results in greater efficacy, or improvement in the delivery of a known pharmaceutical substance already listed on the ARTG. The scheme of the 1990 Act does not provide for the extension of term of each such patent which relates to the same substance.
91 In Commissioner of Patents v AbbVie Biotechnology Ltd (2017) 253 FCR 436; [2017] FCAFC 129 (Besanko, Yates and Beach JJ), the Full Court at [28] cited the Explanatory Memorandum for the Intellectual Property Laws Amendment Bill 1997 (Cth), which inserted the current s 70 into the Patents Act as follows:
The extension of term provisions will be available for patents that include claims to pharmaceutical substances per se (provided that the other criteria are met) [i.e. a product claim]. These claims to pharmaceutical substances per se would usually be restricted to new and inventive substances. Patents that claim pharmaceutical substances when produced by a particular process (product by process claims) will not be eligible unless that process involves the use of recombinant DNA technology. Claims which limit the use of a known substance to a particular environment, for example claims to pharmaceutical substances when used in a new and inventive method of treatment, are not considered to be claims to pharmaceutical substances per se.
92 The Full Court in AbbVie emphasised at [49] that the “cases have recognised that the [concern of s 70(2)(a)] is with inventions that are products, not inventions that are methods or processes” and referred to Boehringer Ingelheim International v Commissioner of Patents (2000) AIPC 91-670; [2000] FCA 1918, before stating at [53]–[57]:
In Prejay Holdings Ltd v Commissioner of Patents (2003) 57 IPR 424 (Prejay), Wilcox and Cooper JJ commented on the Full Court’s observation in Boehringer Ingelheim [International GmbH v Commissioner of Patents (2001) 112 FCR 595], saying (at [24]):
As is apparent from the context of these words, especially the Full Court’s references to the legislative history of s 70, the Full Court was saying that, for a substance to fall within s 70(2)(a) it must itself be the subject of a claim in the relevant patent. It is not enough that the substance appears in a claim in combination with other integers or as part of the description of a method (or process) that is the subject of a claim. The policy adopted in s 70 was to confine extensions to patents that claim invention of the substance itself.
Once again, their Honours were addressing one policy objective. Importantly, their Honours went on to consider the significance of s 70(2)(b) in that regard, saying (at [25]):
This conclusion is not negatived by the terms of s 70(2)(b) of the Act … that paragraph does not require disclosure of a process. Rather, it requires the disclosure of “one or more pharmaceutical substances” that are produced by a particular process.
(Emphasis in original.)
This observation is significant because it acknowledges that s 70(2)(a) and s 70(2)(b) address the same concern — extensions of term in relation to claims directed to pharmaceutical substances, not methods or processes involving pharmaceutical substances. The only exception is the one specifically acknowledged by s 70(2)(b), where pharmaceutical substances can be produced by a process that involves recombinant DNA technology. But, even so, the matter claimed must be the pharmaceutical substance or substances so produced, not other methods or processes involving those substances.
…Nonetheless, each provision’s concern is with pharmaceutical substances, not additional or other matter concerning or involving the use of pharmaceutical substances. In this way, s 70(2)(b) can be construed conformably with s 70(2)(a), and both provisions can be given an harmonious operation, directed to the same end.
This understanding is consistent with the passage in the Explanatory Memorandum quoted at [28] above. The passage emphasises that the extension of term provisions are directed to new and inventive substances — not the method or process by which they are produced (other than involving recombinant DNA technology). Of particular significance is the specific acknowledgement that claims to pharmaceutical substances when used in new and inventive methods of treatment are not intended to be part of the extension of term regime. This passage in the Explanatory Memorandum refers to “pharmaceutical substances per se”, but its reference to product by process claims, and to recombinant DNA technology in particular, signifies that both limbs of s 70(2) are being discussed: see also Section 8 on p 9 of the Explanatory Memorandum, and [7] and [10] of the Notes on Clauses in Sch 1 thereto.
93 In Cipla, Perram J summarised the effect of the Full Court authorities, stating at [5]:
…Although not directly relevant to this case, it is useful to know for some of the arguments to be considered that the words ‘per se’ have been held by the Full Court of this Court to entail that an extension cannot be granted for a patent which discloses claims for a method and is confined (subject to presently immaterial exceptions) to patents claiming products: Boehringer Ingelheim International GmbH v Commissioner of Patents [2001] FCA 647; 112 FCR 595 (‘Boehringer (FC)’) at [37] per Wilcox, Whitlam and Gyles JJ, affirming the decision of Heerey J in Boehringer Ingelheim International v Commissioner for Patents [2000] FCA 1918 (‘Boehringer’); Commissioner of Patents v AbbVie Biotechnology Ltd [2017] FCAFC 129; 253 FCR 436 at [49] per Besanko, Yates and Beach JJ…
94 In the High Court decision of Alphapharm Pty Ltd v H Lundbeck A/S (2014) 254 CLR 247; [2014] HCA 42 (Alphapharm HC at [23], Crennan, Bell and Gageler JJ stated:
A little more needs to be said about the Escitalopram Patent. Lundbeck, a Danish pharmaceutical company, applied for the Escitalopram Patent on 13 June 1989 (the expiry date of which became 13 June 2009), for an invention entitled “(+)-Enantiomer of citalopram and process for the preparation thereof”. There are six claims – claims 1 to 5 are product claims and claim 6 is a method claim, which, for present purposes, can be put to one side. Claim 1 claims a compound (an enantiomer) known as “(+)-citalopram” and its non-toxic acid addition salts, and claims 3 and 5 claim a pharmaceutical composition comprising, as an active ingredient, that compound. The pharmaceutical substance disclosed in the complete specification, (+)-citalopram, is used to treat depression.
(Emphasis added; footnotes omitted.)
95 The emphasised sentence in [23] of Alphapharm HC was accompanied by footnote 40:
Relevantly, the extension of term scheme under the Act covers standard patents for pharmaceutical substances per se pursuant to s 70(2)(a), hence patents for pharmaceutical methods or tablets do not fall within the scheme. It can be noted that pharmaceutical substances produced by a process that involves the use of recombinant DNA technology, the subject matter of s 70(2)(b), are not relevant to this case.
96 About these aspects of the reasons in Alphapharm HC, Perram J stated the following in Cipla at [175]–[176]:
The statement that patents for pharmaceutical methods cannot be a patent for a pharmaceutical substance per se accords with the manner in which the words ‘per se’ has been interpreted in this Court in the authorities noted at [5] above…
…The statement is that tablets cannot be a pharmaceutical substance per se. Since this appears immediately after an explanation of the uncontroversial proposition that a patent for a pharmaceutical method cannot be a pharmaceutical substance per se and since there is no reference to tablets in s 70(2) at all, I read this footnote as exhibiting a conclusion that a patent for a tablet is a patent for a pharmaceutical method of delivery. If not read that way, the reference to ‘tablet’ seems to come from nowhere although an alternative view may be that it derives from the passage in explanatory memorandum for the Intellectual Property Laws Amendment Act 2006 set out above which also refers to tablets. But wherever the reference to a tablet comes from, it is clear that the conclusion in footnote 40 is about the proposition that a method patent will not disclose a pharmaceutical substance per se within the meaning of s 70(2).
97 Because it was irrelevant to the issue in dispute between the parties in that case, Perram J stated at [178] of Cipla that:
…It is not necessary to consider whether footnote 40 is a seriously considered dicta of the High Court which I am bound to follow (see Hill v Zuda Pty Ltd [2022] HCA 21; 275 CLR 24 at [26] per the Court) or just an obiter dictum made in passing. Given its presence in a footnote in a section headed ‘The background facts’ in a case having nothing to do with this issue, I would tend to favour the latter characterisation…
98 His Honour concluded that it was not a considered obiter dictum and that he was not legally required to follow it, but noted at [181] that, “[o]bviously, anything said by the High Court, even if a footnote, must be taken into account”.
99 The following emerges from these authorities for the purposes of identifying a claim for a pharmaceutical substance per se (which are not mutually exclusive):
(1) only a claim for a pharmaceutical substance as such or alone will qualify;
(2) a pharmaceutical substance which forms part of a method or process does not qualify;
(3) an existing pharmaceutical substance prepared by a new and inventive process does not qualify;
(4) a pharmaceutical substance when produced by a particular process (product by process claim) does not qualify;
(5) a new and inventive method of using an existing pharmaceutical substance (such as in a new method of treatment) does not qualify. This could extend to a new and inventive pharmaceutical method of delivery.
4.3 Meaning of “in substance fall within the scope of the claim[s]”
100 Sun Pharma submits that the expression “in substance fall within the scope of the claim[s]” in s 70(2)(a) requires that the pharmaceutical substance per se must take all of the essential integers of the claim. Otsuka submits to the contrary and contends that, provided the pharmaceutical substance per se is included amongst the things claimed, that will suffice.
101 In Boehringer FC, the appellant urged the Full Court to only focus on part of the claim that was directed to the “substance”. In doing so, the appellant argued that s 70(2)(a) means “no more than that the pharmaceutical substance must be a specific claimed feature of the invention”, and that it “does not matter that the substance appears only as one element in a combination of elements”: see [26]. It also argued that “[t]he expression mandates nothing more than that the claims include as an essential feature the pharmaceutical substance”: see [27] (emphasis original). In response, the respondent submitted (see [31], [35]):
The effect of s 70(2)(a) insisting that substances fall within the scope of the claims of the specification is to prevent any extensions of term from having a broadening effect on the claims of the extended patent. This policy of not allowing any broadening by extensions is demonstrated by the new s 78. To extend the patent in this case would have that very effect of widening when it did not, during its primary term, include protection for the substance alone. This would make an infringement something which was not previously an infringement…
…
...the words “in substance fall within the scope of the claim or claims” of the specification, refer to the elements that make up the claim or claims. They point out that, if one essential integer is removed from a patent claim, the effect is to broaden the claim. They note their opponents concede that the use of the pharmaceutical substance itself would not be an infringement; it would be necessary for the infringer also to replicate the additional elements of any claim that was said to be infringed. It follows, according to counsel, that the pharmaceutical substance, standing alone, does not “in substance fall within the scope of” any claim of the specification.
102 As to this, the Full Court stated at [37]–[38] that:
The submissions made on behalf of the respondent are clearly to be preferred… [and there] are serious difficulties about the appellant’s argument. One is that it effectively reads out of s 70(2)(a) the words “per se”. On the appellant’s argument, it is enough that the complete specification disclose one or more pharmaceutical substances, whether as the sole element in an invention or in combination with other elements. If that had been the legislative intention, the paragraph could have read: “one or more pharmaceutical substances must in substance be disclosed”. There would have been no need for “per se”.
103 Further, the Full Court relied on the effect that the respondent’s construction of s 70(2)(a) would have on the monopoly of a patentee in the extended term by reason of s 78, with the result that it regarded the appellant’s construction of s 70 as “anomalous”: at [41]. At [42], the Full Court concluded that:
The appellant’s best point is that in ordinary usage a necessary integer of a whole would be regarded as falling within the scope of that whole. However, in the context of s 70(2)(a), we think that falling within the scope of a claim in a patent specification means included amongst the things claimed. Here, the substance, in itself, is not a thing claimed in the patent sense.
104 Pausing there, it is apparent that the precise submission advanced by Otsuka in this case was also advanced by the appellant in Boehringer FC and was rejected by the Full Court. At [42] of that decision, the Full Court referred to what it considered was the appellant’s “best point”, stating that it “is that in ordinary usage a necessary integer of a whole would be regarded as falling within the scope of that whole”. The Full Court then rejected that “best point”, stating that “[h]owever, in the context of s 70(2)(a), we think that falling within the scope of a claim in a patent specification means included amongst the things claimed. Here, the substance, in itself, is not a thing claimed in the patent sense” (emphasis added). In other words, the Court was referring to a claim which was comprised of the pharmaceutical substance per se with no other integers. When the reasons of the Full Court are read in context, that is the correct representation of the Full Court’s finding.
105 The reasoning in Boehringer FC was followed by the Full Court in Prejay at [23]–[24], which stated at [24] that:
…the Full Court [in Boehringer FC] was saying that, for a substance to fall within s 70(2)(a) it must itself be the subject of a claim in the relevant patent. It is not enough that the substance appears in a claim in combination with other integers or as part of the description of a method (or process) that is the subject of a claim. The policy adopted in s 70 was to confine extensions to patents that claim invention of the substance itself.
106 The reasoning in Boehringer FC and Prejay was adopted by the Full Court in AbbVie at [52]– [54].
107 Between Prejay and AbbVie, the decision of Alphapharm FC was handed down. In that case, the Court was addressing the question of whether Cipramil (being the goods on the ARTG) “contains or consists of” the (+)-enantiomer molecule, which was the pharmaceutical substance for the purposes of s 70(2)(a): see [231], [239]. At [240], Bennett J stated that “[w]hat is required is an analysis of whether there are goods containing or consisting of (+)-citalopram”. In that same paragraph, her Honour also observed that “[t]here is no suggestion from the words of s 70(3) that the relevant “goods” could or must include no more than one pharmaceutical substance”. It was in that context that Bennett J stated at [242] that “[i]n my view, s 70 recognises that the pharmaceutical substance per se may not equate with the subject matter of the claim of the patent in terms”.
108 Otsuka relies upon the statement in Alphapharm FC at [242]. However, once again, when read in context, the statement at [242] in Alphapharm FC was not addressing the construction of the expression “in substance fall within the scope of the claim[s]” in s 70(2)(a) and nor was it purporting not to follow Boehringer FC and Prejay. Rather, the Full Court considered that, provided the pharmaceutical substance per se was contained in the goods listed on the ARTG, whether alone or with other substances, that would suffice for the purposes of s 70(3): see [240]–[244].
109 For these reasons, Sun Pharma’s position is the correct one, namely that the expression “in substance fall within the scope of the claim[s]” in s 70(2)(a) requires that the pharmaceutical substance per se must take all of the essential integers of the claim.
4.4 Whether the definition of “pharmaceutical substance” excludes formulations
110 In Cipla, it was held that “pharmaceutical substance” includes formulations: see, for example, the findings at [38], [183]–[186]. The reasons for doing so included that both Pharmacia and Spirit have as part of their ratio decidendi that a pharmaceutical substance may include a formulation: see [181].
111 In closing, Sun Pharma submits that the decision in Cipla on this issue of construction is incorrect but does not seek a finding that it was “plainly wrong”. Sun Pharma expressly reserves its position (in any appeal from this decision) to argue that Cipla was wrongly decided.
112 Although it provided a non-exhaustive list of the reasons why it contended that the construction in Cipla was incorrect, I did not understand it to be necessary that I address those submissions, especially as they were not the subject of oral submissions. In any event, addressing such arguments lacks utility in circumstances where no party urges that I reach a conclusion that Perram J was plainly wrong such that I ought not follow Cipla.
113 This has the consequence that I should, and will, follow it for the reasons given by French J in Hicks v Minister for Immigration & Multicultural & Indigenous Affairs [2003] FCA 757 at [75]–[76].
114 For these reasons, the definition of “pharmaceutical substance” includes formulations.
4.5 Whether formulations can only include substances which have therapeutic use
115 In Cipla, Perram J characterised Cipla’s “alternative case” as follows ([187]–[188]):
…It becomes necessary in light of this conclusion to consider Cipla’s alternative case that, even if ‘pharmaceutical substance’ is capable of encompassing formulations, it does not encompass either of the relevant formulations claimed by the 862 Patent.
The pith of Cipla’s alternative case is that here, unlike in Spirit, the excipients do not individually or together have a therapeutic use in the formulations separate from the liraglutide itself. As already noted, there are two limbs to this point: first, somewhat obscurely, whether it is necessary that the excipients in a formulation themselves be for a therapeutic use in order that the formulation be a ‘pharmaceutical substance’; and secondly, if so, whether the excipients in the formulations claimed by the 862 Patent are for a therapeutic use.
(Emphasis added.)
116 His Honour then addressed the “first limb” of that case at [189]–[191]:
The reader may be forgiven for struggling to understand this submission. As I understood it, the submission emerged as an illustration derived from the judgment of Rares J in Spirit and went as follows: in Spirit, the active ingredient was oxycodone but it was formulated as a controlled release tablet. This outcome was achieved by including the oxycodone in a substance (a deliberately neutral word) which resisted the process of digestion in the gastrointestinal tract and slowed the release of the oxycodone. Rares J held that the formulation was a pharmaceutical substance. Cipla’s point, which was very briefly, perhaps vanishingly, expressed, seemed to be that if, contrary to its primary submission, a formulation including excipients could be a pharmaceutical substance then, as a matter of law, such a situation was confined to the circumstances obtaining in Spirit, that is to say, the situation where the excipient was for therapeutic use.
Unpicked in this way, this submission is revealed as a repetition of Cipla’s primary argument that a formulation can never be a pharmaceutical substance. The effect of Rares J’s conclusion that the controlled release excipient was for a therapeutic use inevitably entails that the controlled release excipient satisfied the definition of a pharmaceutical substance. Thus, whilst Cipla glancingly posits this as an alternative legal case to its broad contention that every element in a formulation must be for a therapeutic use, the facts of Spirit show that it was a case where every element of the formulation did have that quality.
Thus, when the fog lifts on this legal contention, it turns out to be the same as Cipla’s primary submission and is to be rejected for the same reasons. If the submission means anything else, then I do not understand it and reject it for that reason. Having cleared this out of the way, the remaining issues are the factual ones raised by Novo Nordisk on the assumption, contrary to my conclusion, that each excipient must be for therapeutic use in its own right.
117 By its written closing submissions, Sun Pharma submits that, to the extent that the decision in Cipla is a holding that the excipients in a formulation need not have a “therapeutic use”, then it is plainly wrong and should not be followed. During oral submissions, Sun Pharma retreated from this position, instead submitting that Perram J misunderstood Cipla’s argument and so had failed to address it, no relevant holding was made by his Honour and so it is not necessary to conclude that his Honour was plainly wrong.
118 I do not accept this characterisation of the reasons in Cipla.
119 Once Perram J found that a formulation including excipients or non-active ingredients could be a pharmaceutical substance, a submission that all excipients must be for a therapeutic use is (in truth) a repetition of the primary argument, as his Honour found at [190].
120 Further and as Otsuka submits, Perram J did hold that the excipients in a formulation need not have or “be for” a “therapeutic use in its own right” in order for the formulation to be a pharmaceutical substance. That is apparent from, at the least, the final sentence of [191].
121 Nor do I accept that such a finding was plainly wrong for the following reasons.
122 Firstly, the conclusion reached in Cipla accords with the decision of Pharmacia at [107], in which Weinberg J stated that:
…Almost every pharmaceutical product will consist of a combination of individual substances, some of which may be intrinsically therapeutic (active ingredients), while others serve different, albeit essential, roles. To restrict the capacity to extend a patent to those unlikely cases where every component of the compound is itself therapeutically useful would be to deprive [s 70] of any real utility, and largely defeat the purpose of its enactment.
123 Secondly, at [125]–[126] of Cipla, Perram J addressed the ordinary meaning of the text of the statutory definition of pharmaceutical substance, stating that:
…If the words in the first pair of parentheses were omitted from the definition then the word ‘substance’ would be construed to mean an element, a compound, or a mixture of elements or compounds. Whilst it is possible to imagine other substances, such as dark matter and ectoplasm, I do not think that the word ‘substance’ in a definition concerned with pharmaceutical substances would embrace these. It follows that if the words in parentheses were omitted from the definition then a formulation as a mixture of compounds would be a pharmaceutical substance so long as it satisfied the remaining requirements of the definition (namely that it be for therapeutic use and that its ‘application’ (scil. ‘use’) involve a chemical interaction with a human physiological system). Whether it is the mixture as a whole or each of its constituent parts which must be for a therapeutic use is the subject of Cipla’s alternative case discussed below.
The words in parentheses in the definition clarify that a substance includes a mixture or a compound of substances. Whilst I do not think this clarification is at all necessary, it is scarcely plausible that they were inserted to narrow the range of substances that could qualify as pharmaceutical substances. Yet this is the effect of Cipla’s submission. If narrowing in that fashion was intended, there were plenty of other, much clearer ways of indicating that such a limitation was intended. Another way of putting this is to observe that Cipla’s construction means that the word ‘including’ where it appears in the parenthetic phrase operates not to ‘include’ certain substances but, in fact, to exclude them. I am reluctant to interpret ‘include’ in this fashion.
124 Thus, his Honour considered that:
(1) the word “substance” (if it appeared on its own and without the words in parentheses) would be construed to mean an element, a compound, or a mixture of elements or compounds;
(2) in that event, a formulation would be a pharmaceutical substance so long as it satisfied the remaining requirements of the definition;
(3) the words in parentheses in the definition clarify that a substance includes a mixture or a compound of substances.
125 Once it is accepted that the words in parentheses in the definition of pharmaceutical substance clarify that the “substance” can be constituted by more than one substance, the focus of the text is then upon that “substance” (as a whole and howsoever formulated). To put it another way, once one accepts that a pharmaceutical substance includes a formulation, it necessarily follows from the text that the focus of the statutory definition is upon whether that formulation (as a whole) is for therapeutic use and whether that formulation (as a whole) is one whose application involves one of the actions or interactions in (a) or (b) of the statutory definition. There is nothing in the text of the statutory definition which requires one to also analyse whether, and be satisfied that, each constituent part of the formulation satisfies both of those requirements before the formulation itself can be found to be a pharmaceutical substance. Such additional requirements are neither expressed nor implied by the words used.
126 For these reasons, there is no ambiguity; rather, there is one clear interpretation based on the statutory text, being that stated above.
127 That more than suffices to be satisfied that Perram J was not plainly wrong in holding that each of the excipients in a formulation need not have a therapeutic use in its own right in order for the formulation to be a pharmaceutical substance, such that I should not depart from his Honour’s construction of the statutory definition of “pharmaceutical substance”. However, even if Sun Pharma is correct and Perram J did not determine the issue, my view of the proper construction of the legislation is that stated above.
5. VALIDITY OF EXTENSION
5.1 Whether the Freeze-dried Controlled Release Formulations meet the definition of “pharmaceutical substance”
128 Sun Pharma contends that the Freeze-dried Controlled Release Formulations do not satisfy s 70(2)(a) because they do not meet the requirements of the definition of “pharmaceutical substance” including “whose application (or one of whose applications) involves ... a chemical interaction, or physico-chemical interaction, with a human physiological system”.
129 It submits that is because the Freeze-dried Controlled Release Formulations are not (themselves) “applied” so as to “involve a chemical interaction, or physico-chemical interaction, with a human physiological system” because they need to first be reconstituted with water to form injectable (liquid) formulations before those formulations (i.e. different substances) are “applied” so as to “involve a chemical interaction, or physico-chemical interaction, with a human physiological system”.
130 Sun Pharma submits that the effect of the words “whose application involves a chemical interaction, or physico-chemical interaction, with a human physiological system” is of critical importance to a proper understanding s 70 and the definition of “pharmaceutical substance” (emphasis in submissions). It submits that the word “whose” indicates that the substance has to be the thing that is applied to the human body so as to involve a chemical interaction, or physico-chemical interaction, with a human physiological system.
131 In Cipla at [124], Perram J concluded that “application” in the definition of pharmaceutical substance refers to a use to which the substance is put, which construction is, in my respectful view, correct. His Honour also recognised that the fact that the substance may have uses which do not involve the interactions and actions in the subclauses of the provision was unremarkable: at [123].
132 The conclusion that Sun Pharma urges me to reach contradicts the construction of “application” in Cipla but it does not submit that this construction was plainly wrong.
133 In substance, Sun Pharma presses an interpretation of the word “application” to mean a physical application when arguing that the freeze-dried substance itself is not applied to the body. However, the conclusion reached by Perram J is that “application” is the use to which the substance is put, and generally its purpose, not its physical application. In this case, the “application” is the treatment of schizophrenia and related disorders. When being put to use to treat schizophrenia and related disorders, the aripiprazole molecules contained within the freeze-dried substance cause a physico-chemical interaction when they bind with receptors in the brain. That the interaction only takes place after reconstitution and injection does not mean that it is not an application of that freeze-dried substance, given that ultimately it is the substance which enables the treatment to occur, albeit with the involvement of additional elements.
134 It has been recognised by this Court that the pharmaceutical substance per se claimed in a patent “may not have the necessary therapeutic use or enable the required physico-chemical interaction unless formulated”: Alphapharm FC at [242]. The need to reconstitute a freeze-dried formulation is no different to the need to formulate if the pharmaceutical substance per se was a single chemical compound; and is in fact anticipated by the statements in the Freeze-dried Controlled Release Formulation Claims that reconstitution with water is required prior to injection.
135 For these reasons, the first contention advanced by Sun Pharma must fail.
5.2 Whether the Controlled Release Injectable Formulations satisfy s 70(3)
136 This aspect of Sun Pharma’s case proceeds on the assumption that the Controlled Release Injectable Formulations meet the definition of a “pharmaceutical substance” for the purposes of s 70(2)(a). It also proceeds on the basis that the pharmaceutical substance for the purposes of s 70(2)(a) and s 70(3) must be the same substance, which Otsuka accepts and which is correct: see, for example, Novartis AG v Pharmacor Pty Limited (No 3) [2024] FCA 1307 at [292] (Yates J).
137 Based on these premises, Sun Pharma contends that s 70(3) is not satisfied because the ARTG Goods comprise a freeze-dried powder in a vial together with a separate vial containing the water for injection amongst other things. It submits that this has the consequence that the ARTG Goods do not contain or consist of the same pharmaceutical substances which are relied upon by Otsuka as satisfying s 70(2)(a), which are injectable formulations.
138 Section 70(3) directs attention to the goods listed on the ARTG. As stated by the Full Court in Commissioner of Patents v Ono Pharmaceutical Company Ltd (2022) 291 FCR 1; [2022] FCAFC 39 at [121] (Allsop CJ, Yates and Burley JJ), the relevant inquiry is a matter of objective determination and “concerns the state of the ARTG”.
139 In Alphapharm FC, the product containing the racemate, Cipramil, was registered on the ARTG. In that case, the pharmaceutical substance for the purposes of s 70(2)(a) was (+)-citalopram. The question was whether Cipramil was a good that contained the pharmaceutical substance (+)-citalopram.
140 At [230], Bennett J recorded the submissions of Lundbeck concerning the “correct approach to s 70” as follows:
(a) First, identify the “pharmaceutical substance”, which is the pharmaceutical substance per se of s 70(2)(a). Plainly, that substance is (+)-citalopram, not part of a racemate.
(b) Secondly, identify “the pharmaceutical substance” contained in the relevant goods which are included in the ARTG. Plainly, in the case of Cipramil, that substance is racemic citalopram.
(c) Thirdly, ask whether they are each the same “pharmaceutical substance”…
141 At [231], her Honour accepted the first step, but stated that “the remaining approach suggested by Lundbeck finds no support in the language of s 70, particularly once s 70(3) is considered in context”. At [233]–[235], Bennett J continued:
The next level of inquiry is under s 70(3) where one asks: are there goods on the ARTG that contain (+)-citalopram, the (+)-enantiomer?
This raised the other aspect of the semantic argument referred to by the primary judge, the meaning of “contain” in s 70(3)(a). The question was whether Cipramil, the racemate, was a good that contained the pharmaceutical substance (+)-citalopram (at [509]). The primary judge recognised that the racemate is not one and the same thing as the enantiomer (at [529]), and that the racemate and enantiomer are involved in different physico-chemical interactions manifested in different pharmacodynamics and pharmacokinetics (at [530]). However, his Honour held that the word “contain” is a word of plain meaning, used in the 1990 Act in conjunction with the alternative “consist of”, and held that Citalopram contains the molecule that is identified as (+)-citalopram.
In my view, the primary judge’s reasoning at [484] and following is compelling… The racemate is made up of equal parts of the (+) and (–) enantiomers and (+)-citalopram is the major, if not the only, active ingredient in Cipramil.
(Emphasis added.)
142 After referring to the primary judge’s reasons in further detail, Bennett J stated at [238]–[240]:
The definition of pharmaceutical substance focuses on the ingredient for therapeutic use that involves the relevant type of interaction. It is the (+)-enantiomer molecule that has the relevant therapeutic effect. Indeed, as his Honour observed at [512], the therapeutic benefit of citalopram comes from the presence in it of (+)-citalopram.
The level of the inquiry required by s 70(3) does not look to the therapeutic effect of the pharmaceutical substance. Rather, it is a simple comparison of the pharmaceutical substance with the “ingredients” of the goods on the ARTG. The question is whether Cipramil contains or consists of the (+)-enantiomer molecule. Given that the racemic mixture includes both the (–)-enantiomer and the (+)-enantiomer in equal parts, Cipramil must “contain” the (+)-enantiomer. The third step posed by Lundbeck is, therefore, redundant.
There is no requirement in s 70(3) to compare or contrast the effectiveness of the pharmaceutical substance with the goods with ARTG approval, nor to consider the purity of the pharmaceutical substances. There is no suggestion from the words of s 70(3) that the relevant “goods” could or must include no more than one pharmaceutical substance. What is required is an analysis of whether there are goods containing or consisting of (+)-citalopram and, if so, which of those goods had the first regulatory approval date.
(Emphasis added.)
143 Importantly, her Honour observed at [242] that “[f]or example, a chemical compound claimed in a patent may not have the necessary therapeutic use or enable the required physico-chemical interaction unless formulated. It is sufficient if the pharmaceutical substance is in substance disclosed in the specification and in substance falls within the scope of the claim or claims. At that point in the scheme of s 70, the satisfaction of s 70(2), the necessary definition of the pharmaceutical substance by reference to the patent ceases”.
144 At [244], her Honour stated that “the pharmaceutical substance per se is the molecule, the (+)-enantiomer. The racemic mixture is a solution that contains both (+) and (–) enantiomers in equal proportions. That (+)-enantiomer molecule per se fulfils the requirements of s 70(2). It is the molecule that “works” as a pharmaceutical substance alone, or together with other substances, in goods listed on the ARTG. These other substances may be components of a formulation or may otherwise be described as impurities, such as the (–)-enantiomer”.
145 More recently in Novartis at [258]–[259], Yates J observed:
As regards s 70(3)(a), the inquiry is the factual one of whether the nominated goods, included in the ARTG, contain or consist of the disclosed and claimed “pharmaceutical substance per se”—in other words, is the “pharmaceutical substance per se” an “ingredient” of the nominated goods? The inquiry does not look to the therapeutic effect of the pharmaceutical substance: [Alphapharm FC] at [239]. Nor is the factual inquiry bounded by what the ARTG, itself, discloses about the goods.
Section 70(3)(a) looks, firstly, to whether goods contain or consist of the pharmaceutical substance(s) identified in the s 70(2) inquiry. If the answer is “yes”, the next question is whether those goods are included in the ARTG. The answer to this question is a simple “yes” or “no”. No further interrogation of the ARTG is necessary.
146 In this case, the Australian Product Information for ABILIFY MAINTENA (Aripiprazole (as monohydrate)) Prolonged-Release Suspension for Injection (Therapeutic Kit) (KIT PI) identifies that the ARTG Goods are kits comprising (inter alia) a vial of freeze-dried powder (aripiprazole and vehicle) and a separate vial of diluent.
147 Sun Pharma submits that such kits neither contain, nor consist of, an injectable formulation, and it submits that the substances are therefore different. It relies upon the evidence of Professor Winter that:
[F]or indisputable physical reasons, the product described in the KIT PI cannot have the features of the claims (like e.g. claim 1-15 and others) that refer to a solution/suspension because the claimed formulation is a liquid, the product is a solid (lyophilisate).
(Emphasis original.)
148 Sun Pharma notes that Professor Evans agreed with this, and that he accepted that there were differences between the freeze-dried formulation and the injectable formulation, including that:
(1) one is liquid and one is a solid;
(2) the freeze-dried formulation does not have a pH or viscosity;
(3) the freeze-dried formulation has several melting points, one for each ingredient within the freeze-dried formulation, whereas the injectable (ready to use) formulations do not have a melting point.
149 Sun Pharma also relies upon the fact that ready-to-use injectable formulations are capable of ARTG registration separately to freeze-dried formulations that require reconstitution. It relies upon the agreement of Professor Evans that it was not unusual for such ready-to-use injectable formulations to be registered, and that Otsuka has registered its own ready-to-use extended-release injectable suspension product, ABILIFY ASIMTUFII. As to these extraneous facts, I do not accept that they are relevant to the question of whether s 70(3) is satisfied.
150 The approach posited by Sun Pharma is somewhat beguiling. Instinctively, one accepts that a liquid differs from a solid and so, in colloquial and also scientific terms, it is correct to say that they are not the same “substance” as such. Further, it must be accepted that a liquid will have different properties to a solid.
151 However, the relevant authorities do not support an approach where the form of the goods registered on the ARTG has any impact upon the question whether s 70(3) is satisfied. Nor do they require consideration as to whether the goods on the ARTG require formulation, or reconstitution or some other step to be taken for the purposes of administration of the product to a patient before a determination can be made as to whether s 70(3) is satisfied. Instead, the authorities make plain that the correct approach is a simple comparison of the pharmaceutical substance identified in the s 70(2) inquiry with the ingredients of the goods on the ARTG and, if the pharmaceutical substance is an ingredient, “[n]o further interrogation of the ARTG is necessary”: Novartis at [259].
152 In this case, Sun Pharma did not submit that the ingredients of ABILIFY MAINTENA differed from the Controlled Release Injectable Formulations. Indeed, as referred to above, the Patent itself contemplates that the injectable formulations can be freeze-dried, which supports a conclusion that the injectable and freeze-dried formulations have the same ingredients albeit that water is required to reconstitute the freeze-dried formulations.
153 Thus, to answer the questions posed in Novartis at [259] by reference to this case:
(1) Are there goods which contain or consist of the pharmaceutical substance(s) identified in the s 70(2) inquiry? Yes.
(2) Are those goods included in the ARTG? Yes.
154 It follows that the second contention advanced by Sun Pharma must fail.
5.3 Whether asserted pharmaceutical substances per se qualify being formulations
155 Sun Pharma’s third contention is that, as a matter of statutory construction, the definition of “pharmaceutical substance” does not extend to formulations (i.e. API(s) together with excipients). Applying that reasoning, it submits that none of the asserted pharmaceutical substances per se can give rise to a valid patent term extension as all are formulations.
156 For the reasons given above concerning the proper construction of the legislation, the fact that the asserted pharmaceutical substances per se are formulations does not disentitle them to being the basis for a valid patent term extension, and Sun Pharma’s third contention must fail.
5.4 Whether asserted pharmaceutical substances per se qualify if they include excipients which are not pharmaceutical substances
157 Sun Pharma’s fourth contention is that, even if the definition of “pharmaceutical substance” can include formulations, neither the Freeze-dried Controlled Release Formulations nor the Controlled Release Injectable Formulations qualify, because the excipients in these formulations are not for a “therapeutic use” (as defined) and/or they do not have an “application (or one of whose applications) [which] involves a chemical interaction, or physico-chemical interaction, with a human physiological system”.
5.4.1 Conclusion based on construction of legislation
158 On the proper construction of the legislation and for the reasons explained above, the fourth contention advanced by Sun Pharma must fail.
5.4.2 Therapeutic effect as disclosed by the Patent
159 On the assumption that my construction is incorrect, it is necessary to determine whether, on the facts of this case, each of the excipients in the asserted pharmaceutical substances per se qualify as a “pharmaceutical substance” within the meaning of the Patents Act.
160 The relevant statutory definitions are set out above. In Cipla at [198]–[201] and after referring to these definitions, Perram J stated:
In my view, whether a substance is for therapeutic use is to be determined by reference to the terms of the patent in which the substance appears. The word ‘for’ in the definition of ‘pharmaceutical substance’ and the explicit use of the word ‘purpose’ in the definition of a ‘therapeutic use’ show that the inquiry posited by both definitions focuses on the purpose for which the substance is used. The inquiry posited by the two definitions is objective and does not turn on the subjective state of mind of the patentee or, for that matter, anyone else.
The relevance of the two definitions is that they provide a gateway into the extension provisions in Part 3 and the springboarding provision in Part 11, s 119A. The former turns on a patent claiming a pharmaceutical substance per se, the latter on the presence of a pharmaceutical patent, i.e., a patent claiming a pharmaceutical substance; or, a method, use or product relating to a pharmaceutical substance.
What both have in common is a pharmaceutical substance. The concept of a pharmaceutical substance lies therefore at the core of Part 3 and s 119A. It must be known at the time that a patent is granted whether these provisions apply to the patent. The operation of the extension and springboarding provisions cannot be contingent on what may, at some later date, be shown about the therapeutic effects of a substance claimed in it. Were it otherwise, a patent claiming a substance for which it made no claim for therapeutic use could afterwards become a patent claiming a pharmaceutical substance on the adduction of evidence that the substance had a therapeutic use, even though no such claim was made in the patent. This would lead to a chaotic operation of the extension regime which can scarcely have been contemplated.
Thus, not only is the purpose inquiry posited by the two definitions objective in nature but the circumstances from which that objectively determined purpose are to be ascertained are limited to the terms of the patent construed in the orthodox way.
161 As occurred in Cipla, the parties adduced evidence from their experts concerning the therapeutic effects of the excipients in the formulations other than the API (i.e. aripiprazole). No party submits that the approach taken in Cipla was wrong, or that it did not apply in this case. This has the consequence that much of the expert evidence concerning the excipients was not relevant.
162 Applying the approach taken in Cipla to the Patent in this case, none of the excipients has any therapeutic effect. That is because, in short, there is no statement anywhere in the Patent that mannitol, carboxymethylcellulose (CMC or its sodium salt Na-CMC), sodium phosphate, sodium hydroxide (NaOH) or the water for injection, or any of the excipients in the asserted pharmaceutical substances per se, has any therapeutic use or therapeutic effect.
163 That is not only apparent from the Patent itself but was also the subject of evidence by Professor Winter (which evidence I accept) as follows:
(1) The 448 Patent does not suggest that any of the excipients contribute to, or otherwise affect, aripiprazole’s therapeutic effect in the body upon injection of the formulations: Winter 2, [14(c)].
(2) The vehicle described and claimed in the 448 Patent does not impact the activity of aripiprazole following administration (including its absorption, its distribution throughout the body, its metabolism, excretion or, importantly, its interaction with receptors in the brain): Winter 2, [16].
(3) The 448 Patent does not contain any information that suggests that any of the excipients described or claimed in the aripiprazole injectable formulations impact the rate, duration or extent of release of aripiprazole, and therefore contribute to its therapeutic effect. Rather, the therapeutic effect of the aripiprazole injectable formulations disclosed in the 448 Patent is solely the interaction of aripiprazole molecules with the relevant receptors in the brain to treat schizophrenia: Winter 2, [18].
(4) He does not agree [with Professor Evans] that CMC has a role in keeping aripiprazole particles well dispersed and in forming a “suitable depot”. The 448 Patent does not describe what constitutes a “suitable depot” and nor does it describe how the aripiprazole particles must be dispersed after administration: Winter 2, [33(b)].
(5) The description of the 448 Patent and the relevant claims do not provide any information that the excipients would have such effects of any kind [being an effect on release, absorption, distribution, metabolism, or elimination of aripiprazole from disclosed and claimed formulations]: JER, page 21, lines 4–8 (Winter).
(6) It has to be decided on common general knowledge and on the description and claims of the 448 Patent by an experienced formulator on a scientific basis whether the inactive ingredients in the vehicle would have a relevant effect on the release of aripiprazole at the site of injection or not: JER, page 21, lines 18–22 (Winter).
164 For these reasons, having regard to the terms of the Patent, none of the excipients in the asserted pharmaceutical substances per se is for “therapeutic use” (as defined) whose application involves a chemical or physico-chemical interaction with a human physiological system, with the consequence that they are not “pharmaceutical substances” within the meaning of the Patents Act.
5.4.3 Factual findings
165 In the event that I am permitted to step outside of the bounds of the disclosure in the Patent, I would reach the same conclusion as in section 5.4.2 above.
166 That is for the following reasons.
167 Firstly, Professor Goodwin’s evidence was that the therapeutic effect of the Controlled Release Injectable Formulations is the binding of molecular aripiprazole with receptors in the brain (and more particularly, the partial agonism of the D2 receptor in the brain): Goodwin 1, [121]. Professor Goodwin also opined that the other claimed features of the Controlled Release Injectable Formulation Claims (i.e. the excipients, the water for injection, the duration of release and the aripiprazole crystal polymorph) do not themselves have any therapeutic effect on the body and do not affect the therapeutic effect of the molecular form of aripiprazole: Goodwin 1, [122]. Professor Goodwin was not required by Otsuka to attend the hearing, and so this evidence was not challenged. Further, no submission was made by Otsuka that this evidence should not be accepted, or why that was the case.
168 Secondly, as is recorded in the JER, the experts (being Professor Evans and Professor Winter) agreed that mannitol, Na-Phosphate, NaOH and water for injection have no effect on the release of aripiprazole from the depot: JER, pages 19–20.
169 Thirdly, although the experts did not reach agreement about the impact of Na-CMC on the release of aripiprazole from the depot, I prefer the evidence of Professor Winter to that of Professor Evans, for the reasons already explained. Professor Evans’ position in the JER was that he could not “rule out a possible impact of Na-CMC on the release of aripiprazole from the depot” and could not “guarantee” a different outcome if it was absent or replaced by a different suspending agent (emphasis added). Such evidence was vague and somewhat speculative. It is also difficult to place much weight on this evidence in circumstances where Professor Evans conceded during the hearing that, despite his review of hundreds of pages of Otsuka’s internal documents and clinical study documents, he did not see any evidence that the excipients, individually or collectively, have an effect on the release of aripiprazole other than in one instance in rats, which supporting data is not identified in his affidavit evidence. By contrast, Professor Winter’s position in the JER was that Na-CMC has no relevant effect on the release of aripiprazole from the depot and described Professor Evans’ “speculations” about how it could affect the release from the depot as “non-specific and [which] find no support in the Patent”, which was consistent with his affidavit evidence and oral evidence during the hearing. Further, contrary to Otsuka’s submissions, Professor Winter did consider the question as a matter of scientific fact: see, for example, Winter 2 at [14(d)], [18] (second and third sentences), [20], [32] and [33(a)].
170 Fourthly, Otsuka seeks a finding that CMC has an “indirect effect on the duration of release by reducing agglomeration prior to injection”. However, the legislation requires that the pharmaceutical substance be for therapeutic use whose application involves a chemical or physico-chemical interaction with a human physiological system. The word “involves” in the definition relates to the application of the substance or mixture: Spirit at [64]. Any pre-administration role that CMC or any of the other excipients may play in the manufacturing and pre-administration stages has no application involving the necessary chemical or physico-chemical interaction with a human physiological system.
5.5 Whether PTE claims qualify if they include process features and/or limitations by result
171 Sun Pharma contends that none of the PTE Claims claim a pharmaceutical substance per se within the meaning of s 70(2)(a) because, when regard is had to all of the integers of the PTE Claims, they include process features or “in use” features and/or features that limit the use of the products as follows:
(1) Each of the Controlled Release Injectable Formulations includes at least one or more process features and/or features that limit the use of the formulations by reason of one or more of the following features or parameters: (a) “controlled release”; (b) “injectable formulation”; and/or (c) “which upon injection releases aripiprazole over a period of [a specified period of time]”.
(2) Each of the Freeze-dried Controlled Release Formulations includes at least one or more process features and/or features that limit the use of the formulations by reason of one or more of the following features or parameters: (a) “freeze-dried controlled release”; (b) “which [freeze-dried] formulation upon constitution with water forms a sterile injectable formulation”; and/or (c) “which [injectable formulation] upon injection releases aripiprazole over a period of [a specified period of time]”.
172 Sun Pharma submits that the Full Court authorities (and the High Court in Alphapharm HC) make clear that a pharmaceutical substance per se must not include process features and/or features that limit the use of products. It submits that the High Court’s statement in Alphapharm HC is consistent with its position that none of the PTE Claims are to a pharmaceutical substance per se, and thus – to use the words of the High Court – “do not fall within the scheme”. It submits that, analogously with a patent for a tablet, the Patent claims both Freeze-Dried Controlled Release Formulations (which are in a powder form), but also must be reconstituted prior to injection, and then administered via injection, and then release over a certain period of time. It submits that each of these features or characteristics take such formulations outside the “scheme”, and that the same can be said for the Controlled Release Injectable Formulations. Sun Pharma submits that each of the PTE Claims are akin to a “pharmaceutical method of delivery”, in that they claim injectable or freeze-dried formulations that assist in the delivery of the aripiprazole to be administered to the body.
173 Sun Pharma also submits that the Patent merely claims a monopoly over a process or method of using a previously known substance (for which there were products already on the ARTG) via a particular delivery system or form of administration, the scope of such monopoly being defined (or sought to be defined) by the relevant Release Features post-administration.
174 Otsuka submits that the relevant question is whether the claim read as a whole is to a product, rather than a process or method. It relies upon the manner of analysis undertaken in Pharmacia by Weinberg J at [99]–[101] in relation to the claims which were before his Honour, which approach was approved by Rares J in Spirit as noted above. Otsuka also submits that the presence of a result in a product claim does not prevent there being a pharmaceutical substance per se within the meaning of s 70(2)(a).
175 Adopting the approach in Pharmacia at [101], the PTE Claims refer incidentally to methods of process, but they do so in order to better describe the new and inventive substance, being the claimed injectable or freeze-dried formulations.
176 Further, the PTE Claims do not claim a process or method of delivery of aripiprazole as such – there is no reference, for example, to dosage amounts, dosage frequency or site of injection, such as intramuscular or subcutaneous administration. Further, that the PTE Claims have process or in-use features does not have the consequence that the claims are not to a new and inventive substance (being a formulation which is able to be injected or reconstituted then injected), rather than a process or method.
177 In addition, Sun Pharma did not identify any authority in support of its submission that a claim which contains a limitation by result cannot be a pharmaceutical substance per se within the meaning of s 70(2)(a). To the contrary, in Spirit, the contention that certain claims were limited by result did not prevent a finding by Rares J that the formulation was a pharmaceutical substance per se: see [72], [73] and [75].
178 Finally, that the claimed formulations are substances which are claimed to be able to be injected does not transform the PTE Claims to claims for pharmaceutical methods of delivery or claims for a particular delivery system or form of administration which fall outside the scope of the definition of pharmaceutical substance per se.
179 For these reasons, the fifth contention advanced by Sun Pharma must fail.
5.6 Whether asserted pharmaceutical substances per se fall within the scope of PTE Claims
180 Sun Pharma contends that the majority of the asserted pharmaceutical substances per se do not “fall within the scope of” any of the PTE Claims because they only take part of PTE Claims.
181 As addressed above, a pharmaceutical substance must take all of the integers of a claim to “fall within the scope” of the claim within the meaning of s 70(2) of the Patents Act.
182 The asserted pharmaceutical substances per se listed at (I) to (IV) and (VI) to (IX) do not take all of the integers of any of the PTE Claims.
183 Accordingly, they do not “fall within the scope of” any of the PTE Claims for the purposes of s 70(2)(a) and cannot be relied upon by Otsuka for the purposes of justifying the Extension.
184 For these reasons, the sixth contention advanced by Sun Pharma succeeds.
5.7 Whether ARTG Goods take each feature of the PTE Claims
185 This aspect of Sun Pharma’s case depended upon Professor Winter’s expressed doubts that the ABLIFY MAINTENA product met the Relevant Features even though the product has been approved by the Therapeutic Goods Administration for once-monthly injections. Such a contention can therefore only apply to asserted pharmaceutical substances per se V (i.e. release period of at least one week) and X (i.e. release period of at least two weeks).
186 By its closing submissions, Sun Pharma relied upon the following evidence in support of Professor Winter’s expressed doubts:
(1) as noted in the KIT PI, oral medication of aripiprazole is given concomitantly for the first 14 days (suggesting that the aripiprazole depot formulation may not release in the first 14 days). Professor Winter also expressed concern that the feature would not be met in respect of “[e]very unit that is produced and sold and potentially injected”;
(2) as shown in a confidential study conducted by Otsuka which is summarised at Annexure AME-17 (CB 1499 and 1500) (246 Study), there is a real likelihood that, for certain subjects being administered with ABLIFY MAINTENA (after reconstitution), no aripiprazole will release from the depot within the first two weeks and/or the aripiprazole will have completely released within two weeks. It relies upon the fact (as is the case) that the PTE Claims do not specify a “mean” release over a patient population; and
(3) oral evidence given by Professor Winter concerning the graph in the 246 Study.
187 However, that oral medication is given for the first 14 days was the subject of an adequate explanation by Professor Evans in the JER, who stated that:
…when commencing a drug/formulation with a long half-life (especially a product such as Abilify Maintena which has a terminal half-life of one month or more) it is usual in clinical practice to expect that therapeutic levels cannot be attained immediately. Sometimes this initial period is managed clinically through the use of a loading dose (for example, where the first dose is higher than subsequent doses). For a person already stabilised on Abilify Maintena tablets, continuing to take these tablets for 14 days while the drug levels from the IM depot continue to build, seems sensible.
(Emphasis original.)
188 Further, notwithstanding his reservations and doubts, Professor Winter concluded in the JER that “it is likely that the product described in the KIT PI after reconstitution and injection releases aripiprazole over a period of at least one week, also over a period of at least two weeks and also over a period of at least three weeks. It thereby would fulfil results features for some of the relevant 448 Patent claims”: JER, page 30 at [5].
189 In the face of the conclusion reached by Professor Winter in the JER, which Sun Pharma did not address or seek to overcome in its closing submissions, the seventh contention raised by Sun Pharma must fail.
6. VALIDITY OF PTE CLAIMS
190 Section 70(2)(a) requires (inter alia) that a pharmaceutical substance per se must in substance “fall within the scope of the claim or claims of that specification”. Sun Pharma contends that this requirement is not met because the PTE Claims are invalid because they do not satisfy s 40(3) and/or s 40(2)(b) of the Patents Act.
191 On the premise that each of the PTE Claims claim the Relevant Feature i.e. “which upon injection releases aripiprazole over [a specified period of time]” (or equivalent), Sun Pharma contends that the person skilled in the art would not be able to determine whether or not all aripiprazole formulations (i.e. all embodiments) that otherwise meet the requirements of the PTE Claims fall within or outside the Relevant Feature based upon the information in the Patent and the common general knowledge, even allowing for routine experimentation.
192 This gives rise to two questions:
(1) whether (as Sun Pharma submits) the PTE Claims contain limitations by result, or whether (as Otsuka submits) the specification teaches that the Relevant Feature (insofar as it is confined to the PTE Claims) is an inherent property of formulations prepared according to the invention;
(2) whether the PTE Claims satisfy s 40(3) and/or s 40(2)(b) of the Patents Act.
193 As to s 40(3), the requirement that claims be clear exists because “readers should be able to ascertain the precise extent of monopoly claimed”: Nesbit Evans Group Australia Pty Ltd v Impro Ltd (1997) 39 IPR 56 at 94 (Lindgren J, with whom Hill J agreed). Thus, the requirement for clarity is not satisfied where the person skilled in the art cannot objectively ascertain whether what he proposes to do falls within the claim’s ambit: BlueScope Steel Ltd v Dongkuk Steel Mill Co, Ltd (No 2) (2019) 152 IPR 195; [2019] FCA 2117 (Beach J) at [705] and [710].
194 In Austal Ships Sales Pty Ltd v Stena Rederi Aktiebolag (2008) 77 IPR 229; [2008] FCAFC 121 (Heerey, Finn and Dowsett JJ) at [13]–[14], the Full Court cited the decision of Hely J in Flexible Steel Lacing Company v Beltreco Ltd (2000) 49 IPR 331; [2000] FCA 890 with approval. In the latter decision at [80], Hely J quoted Mr Blanco White QC in Patents for Inventions and the Protection of Industrial Designs (5th ed, The Law Book Company, 1983) at 4-701 (which appears at [14] of Austal Ships):
Thus a claim is bad if no reasonably certain construction can be given to it, or it is fairly and equally open to diverse meanings. But the rule goes further than this. A court is not bound to find a meaning for a claim, nor to approach a claim with the “conviction that its language is capable of a reasonable construction when carefully examined” that is the due of an Act of Parliament. Thus a claim may be bad for uncertainty although the court could find its true meaning (and would do so if the words concerned appeared in a commercial contract) if it is so obscure that “its proper construction must always remain a matter of doubt”. On the other hand, what matters is not the grammar of the claim, but whether a reader would be left in doubt whether any given apparatus or method fell within the claim or not; the purpose of the rule is to enable the public to rely upon the words of the claim as defining the rights of the patentee. Accordingly, a mere grammatical ambiguity, not affecting the scope of the monopoly, will not invalidate…
(Emphasis added.)
195 To similar effect, Besanko J stated in Vehicle Monitoring Systems Pty Limited v SARB Management Group Pty Ltd trading as Database Consultants Australia (No 8) [2023] FCA 182 at [666] that “lack of clarity is concerned with the certainty surrounding the scope of the monopoly claimed” and “a claim will not fail for lack of clarity if it identifies a workable standard suitable to the intended use”.
196 Section 40(2)(b) requires that, for a standard patent, a complete specification must “end with a claim or claims defining the invention”. Claims must “mark out the monopoly operating to disclaim what is not specifically and definitely claimed… to ensure that the public and specifically a manufacturer will not have difficulty being satisfied that a claim is not infringed”: see Pharmacia Italia SpA v Mayne Pharma Pty Ltd (2005) 66 IPR 84; [2005] FCA 1078 at [29] (Crennan J).
197 In BlueScope at [715], Beach J cited General Tire & Rubber Co v Firestone Tyre and Rubber Co Ltd (1971) 1A IPR 121 and stated:
…[In that case, the] Court also observed that allowances should be made for any difficulties of the case, so that an alleged issue of want of definition should always be considered in relation to the particular facts. It concluded (at IPR 167-8; RPC 513-4):
It is clear in our judgment that the question whether the patentee has sufficiently defined the scope of his claims is to be considered in relation to the facts of each case, that allowance is to be made for any difficulties to which the circumstances give rise, and that all that is required of the patentee is to give as clear a definition as the subject matter admits of. It is also clear in our judgment that, while the court is to have regard to all the relevant facts, the issue of definition is to be considered as a practical matter and little weight is to be given to puzzles set out at the edge of the claim which would not as a practical matter cause difficulty to a manufacturer wishing to satisfy himself that he is not infringing the patent. We accept also that definition of the scope of a claim is not necessarily insufficient because cases may arise in which it is difficult to decide whether there has been infringement or not provided the question can be formulated which the court has to answer in the issue of infringement.
198 In Albany Molecular Research Inc v Alphapharm Pty Ltd (2011) 90 IPR 457; [2011] FCA 120 at [174], Jessup J observed that “[a] claim which is a model of verbal or grammatical clarity may none the less fail the test of this requirement if it leaves the definition of the boundaries of the invention uncertain or variable”. To similar effect, Beach J stated in Meat & Livestock Australia Ltd v Cargill, Inc (2018) 129 IPR 278; [2018] FCA 51 at [940]: “[a] claim will be bad if it fails to define the monopoly claimed so that the skilled addressee of the patent can know the exact boundaries of the area within which they will be trespassers”.
199 Particular issues arise in the case of claims which are limited by result.
200 In Rescare Ltd v Anaesthetic Supplies Pty Ltd (1992) 25 IPR 119 at 125–126, Gummow J quoted Patents for Inventions and the Protection of Industrial Designs (op. cit.) at 4-413:
To amount to a limitation by result, what is in the claim must at least be a limitation: something that draws a line between two classes of things that would otherwise fall within the claim: with the implication that conditions of the manufacture can be adjusted, by the reader of the specification, to secure the specified result. It is, of course, a matter of construction to determine whether words in the claim effect a limitation or merely assert that complying with the claim will secure a certain result…
See also BlueScope at [718]; Axent Holdings Pty Ltd t/a Axent Global v Compusign Australia Pty Ltd (2020) 154 IPR 431; [2020] FCA 1373 at [262]–[263] (Kenny J), which was cited with approval in Vald Pty Ltd v KangaTech Pty Ltd (No 5) [2024] FCA 333 at [148] (Downes J).
201 In Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth (2011) 119 IPR 194; [2011] FCAFC 132, Yates J (with whom Nicholas J agreed) referred at [259] to a contention that a claim lacks clarity as a “criticism that can be made of many claims limited by result” but that such claims “do not fail for lack of clarity simply because further steps must be taken by the person skilled in the art, in the nature of trial and error or routine testing not involving invention, in order to understand when the result will be achieved…The necessity for, or extent of, the work required in that regard may point to the possibility of insufficient description…”.
202 In BlueScope at [719], Beach J quoted Patents for Inventions and the Protection of Industrial Designs (op. cit.) at 4-703:
…particular care must be taken when claims [limited by result] are adopted to give in the specification all possible assistance in determining what does, and what does not, give the useful result concerned: clarity of claim and sufficiency of description go together here. Thus a claim limited by result has been held bad for ambiguity where the instructions for attaining the result were meaningless to those in the art. In addition, it must not be forgotten that there is no authority for putting upon the reader of the specification the burden of making any but “simple” experiments…
203 Further, in GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Apotex Pty Ltd (2016) 119 IPR 1; [2016] FCA 608, referring to Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 at 480 at [700], Beach J said in relation to claims which were “akin to a limitation by result claim”:
[i]n such a case, it is said that the specification must not only enable the addressee to carry out the invention claimed in the form of any particular embodiment but also enable the addressee of the Patent to ascertain whether the particular result concerned is or is not secured by forms of the invention lying near the boundaries of the claim. It is said that such a limitation is permissible only if it is sufficient to characterise the construction of the article claimed…
6.1 Whether PTE Claims are claims limited by result
6.1.1 Known factors that affect release of aripiprazole
204 It is trite that a claim is construed from the perspective of a person skilled in the relevant art as to how such a person would have understood the patentee to be using the words of the claim in the context of the specification as a whole: see, for example, BlueScope at [691].
205 By the JER, Professor Winter and Professor Evans agreed that an element of the Relevant Feature in the PTE Claims was a defined discrete boundary for release (which is the dissolution of aripiprazole molecules from the aripiprazole particle depot at the injection site) expressed in weeks (e.g. in multiples of seven days, plus/minus 10% where the term “about” is used).
206 A person skilled in the art would be aware, when reviewing the PTE Claims, that there are a number of variable factors that affect the length of time of the release of aripiprazole from the depot in each of the formulations that otherwise fall within each of the PTE Claims. These factors were known and formed part of the common general knowledge, as addressed above. The following identifies particular factors relating to the release of aripiprazole which were the subject of evidence.
Dosage
207 The dosage (i.e., how much aripiprazole is contained in a given formulation) affects how long the formulation releases in vivo: see, for example, Winter 1 at [155(c)]. Professor Winter’s evidence was that concentration and dosage amount of aripiprazole will affect (in some way) the release of a depot aripiprazole formulation: see Winter 1, [64], [152]; Winter 2, [17].
208 However, there is nothing in the PTE Claims that limits the dosage amount in the claimed formulations. Indeed, the Patent refers to, and expressly allows for, a dosage of aripiprazole between 0.1 to 600 mg per 1 mL.
209 Some of the claims in the Patent refer to dosage expressly (see, for example, claims 47 and 48). However, the PTE Claims do not refer to dosage, and Professor Evans accepted during cross-examination that the PTE Claims do not specify a particular dosage or dosage range.
210 Some of the claims in the Patent require that the aripiprazole be released from the depot in a “therapeutic amount” (see, for example, claims 57 and 58). However, the PTE Claims do not require this.
211 Some of the claims in the Patent refer to dosage interval (frequency of doses) (see, for example, claims 47 and 48). However, the PTE Claims do not refer to dosage interval, and Professor Evans also accepted this during cross-examination.
212 Relatedly, certain claims of the Patent (but not the PTE Claims), include both the Relevant Feature and a dosing frequency requirement. Professor Evans agreed that this demonstrates that the release period is not the same as the dosing interval.
213 Moreover, Professor Evans agreed that “a formulator has to consider a number of factors in respect of dosing frequency of a drug in addition to its release” such as the therapeutic window of the drug substance and steady state concentration.
Particle size
214 As already observed, it was common general knowledge that particle size (and the associated surface area of the particles) has an impact on the release: see, for example, Winter 1, [64(a)].
215 Nevertheless, the Patent does not describe the mathematical relationship between particle size and absorption/release.
Volume
216 Professor Winter’s evidence was that the pressure associated with the administration of larger volumes (2 ml or more) of formulation can decrease the time that it takes for the drug to completely dissolve from the formulation: Winter 1, [64(e)], [155(d)]; also Winter 2, [37(c)]. Professor Evans agreed that volume could affect absorption: Evans, [50(i)].
217 However, the PTE Claims do not refer to or contain any volume measurements.
Other factors
218 There are other factors which affect the release of aripiprazole from the depot which are not referred to in the PTE Claims:
(1) whether the formulation is injected subcutaneously or intramuscularly: see, for example, Evans, [50(e)]; Winter 2, [17(d)];
(2) if injected intramuscularly, the site of administration (deltoid or gluteal muscle): Winter 2, [17(d)]. A related consideration is blood flow to and from the injection site: see, for example, Evans, [52]; Winter 2, [37(b)].
6.1.2 Proper construction of the PTE Claims
219 The PTE Claims have a release period of at least one week (claims 1, 3, 6 and 14) or two weeks (claims 16, 19, 21 and 25).
220 However, although they claim different release periods, claims 1 and 16 have the same range of mean particle size of about 1 to 10 microns. Similarly, the range of mean particle size of 2 to 4 microns is the same in claims 6 and 19, but they claim different release periods.
221 Other claims of the Patent have identical particle size ranges, yet different release periods. For example, claim 18 depends on claim 16, but includes the limitation that the aripiprazole must be released after constitution and injection “over a period of at least about three weeks” (instead of “at least about two weeks” in claim 16). Claim 24 also depends on claim 16 but includes a release period of “about a four week period”. Further, claims 58 and 59 each depend on independent claim 57, and the only feature that varies is the length of time over which the aripiprazole must release from the depot (i.e. “at least about two weeks” in claim 57, “at least about three weeks” in claim 58 and “about a four week period” in claim 59). There are other claims of the Patent which provide an upper and lower limit (e.g. claims 8 and 11), and claims that provide a specific time for release (e.g. “over about a four week period” in claim 24).
222 Having regard to the variations in the release periods within the claims, it is apparent that the same particle size alone will not provide the same release period. The same conclusion would be reached even if one had regard to the PTE Claims in isolation.
223 That is not a surprising outcome, as it accords with the common general knowledge being that other factors in addition to particle size will play a role in the duration of the release of the aripiprazole, as has already been addressed.
224 Further, each of the PTE Claims claim many different embodiments within each claim – i.e. they allow for a variation in mean particle size of aripiprazole, variations of dosage and variations in respect of the vehicle – but each of those embodiments must meet the stated release period. The person skilled in the art is therefore directed to select the particle size (within the stated range), excipients and water but must also make other choices concerning matters which are not referred to in the PTE Claims (such as dosage concentration, dosage frequency, volume and site of administration) by reference to the result to be achieved, being the stated release period. This provides strong support for a conclusion that the PTE Claims are limited by result.
225 In support of its contention that the Relevant Feature (insofar as it is confined to the PTE Claims) is an inherent property of formulations prepared according to the invention, Otsuka relies upon particular statements in the specification apart from the claims. Its primary argument is that the Patent teaches that the release period is an inherent feature of the particle size alone.
226 The following is stated in the specification in the section “Brief Description of the Invention”:
Disclosed herein is a sterile freeze-dried aripiprazole formulation which upon constitution with water for injection releases aripiprazole, in therapeutic amounts, over a period of at least about one week, and preferably over a period of two, three or four weeks and up to six weeks or more…
227 After referring to five aspects of the invention, the specification continues:
A mean particle size of the freeze-dried aripiprazole formulation within the range from about 1 to about 30 microns is essential in formulating an injectable which releases aripiprazole over a period of at least about one week and up to six weeks or more, for example up to 8 weeks.
It has been found that the smaller the mean particle size of the freeze-dried aripiprazole, the shorter the period of extended release. Thus, in accordance with the present invention, when the mean particle size is about 1 micron, the aripiprazole will be released over a period of less than three weeks, preferably about two weeks. When the mean particle size is more than about 1 micron, the aripiprazole will be released over a period of at least two weeks, preferably about three to four weeks, and up to six weeks or more. Thus, in accordance with the present invention, the aripiprazole release duration can be modified by changing the particle size of the aripiprazole in the freeze-dried formulation.
…
Also disclosed herein is a controlled release sterile injectable aripiprazole formulation in the form of a sterile suspension, that is, the freeze-dried formulation of the invention suspended in water for injection, is provided which, upon injection, preferably intramuscularly, releases therapeutic amounts of aripiprazole over a period of at least one week…
(Emphasis added.)
228 Pausing there, the specification purports to teach that the “at least about two weeks” release period is an inherent property of the formulations according to the invention with an aripiprazole particle size of more than about 1 micron. According to Professor Winter and Professor Evans, the phrase “more than about 1 micron” would be understood as more than 1.1 microns.
229 This is inconsistent with claim 16, for example, which has a release period of “at least about two weeks” and a mean particle size of about 1 to 10 microns (which range includes about 1 micron, which is not more than about 1 micron).
230 Further, when the mean particle size is about 1 micron, the specification teaches that the aripiprazole “will be released over a period of less than three weeks, preferably about two weeks”. However, it does not state how much is “less than three weeks”. For example, two days is a period of less than three weeks. Such imprecise wording does not teach that the “at least one week” release period is an inherent property of such formulations according to the invention with an aripiprazole particle size of about 1 micron. The only reference to one week is earlier in this passage of the specification, as set out above, and there is no articulated or even implied correlation in that passage between any particular particle size in the range “from about 1 to about 30 microns” and “a period of at least about one week and up to six weeks or more, for example up to 8 weeks”.
231 Under the heading “Detailed Description of the Invention”, the following is stated:
The controlled release sterile injectable aripiprazole formulation of the invention will include aripiprazole in an amount within the range from about 1 to about 40%, preferably from about 5 to about 20%, and more preferably from about 8 to about 15% by weight based on the weight of the sterile injectable formulation.
As indicated, desired mean particle size of the aripiprazole is essential in producing an injectable formulation having the desired controlled release properties of the aripiprazole. Thus, to produce desired controlled release, the aripiprazole should have a mean particle size within the range from about 1 to about 30 microns, preferably from about 1 to about 20 microns, and more preferably for about 1 to about 10 to 15 microns.
Where the desired controlled release period is at least about two weeks, up to six weeks or more, preferably about three to about four weeks, the aripiprazole will have a mean particle size within the range from about 1 to about 20, preferably from about 1 to about 10 microns, more preferably from about 2 to about 4 microns, and most preferably about 2.5 microns.
(Emphasis added.)
232 By contrast with what is stated in the Brief Description, this part of the specification purports to teach that the “at least about two weeks” release period is an inherent property of the formulations according to the invention with an aripiprazole particle size within a range from about 1 micron, not more than about 1 micron (as referred to in the Brief Description).
233 These inconsistencies have the consequence that the Relevant Feature in the PTE Claims is not an inherent property of formulations prepared according to the PTE Claims – it is self-evident that other factors must come into play.
234 As there is a range of factors which will affect the release of the aripiprazole, including (but not confined to) particle size, and as choices must be made concerning these factors in order to achieve the minimum release period which is stated in the PTE Claims, it cannot be concluded that all of the formulations which otherwise meet each of the PTE Claims will release for more than one or two weeks.
235 It follows that the PTE Claims do not assert that complying with them will secure the stated result (being the release period): see Rescare at 125–126.
236 For these reasons, the PTE Claims are claims limited by reference to result with the implication that conditions of the manufacture of the formulations can be adjusted by the reader of the specification to secure the specified result, being the Relevant Feature.
6.2 Whether PTE Claims satisfy s 40(3) and/or s 40(2)(b) of the Patents Act
237 In this case, the person skilled in the art must be able to adjust the formulations disclosed and claimed in the Patent to ensure that they satisfy the Relevant Feature (the claimed result), or, in other words, ascertain whether or not a formulation that otherwise falls within the claims satisfies the Relevant Feature.
6.2.1 Significance of the term “release” in the PTE Claims
238 The Relevant Feature in the PTE Claims is directed to the “release” of aripiprazole over a stated period of time, that is, a “discrete boundary for release expressed in weeks”.
239 Professor Winter gave evidence that, in order to identify formulations comprising aripiprazole that fall inside or outside these boundaries, it is necessary to precisely determine the duration of release of aripiprazole: see Winter 2, [100]. This evidence was not contradicted by Professor Evans.
240 As observed above, the experts agree that “release” of aripiprazole is the dissolution of aripiprazole molecules from the depot at the injection site.
241 Having regard to the common general knowledge, “release” is not the same thing as “absorption”, with the latter being entry into the central compartment (which includes the blood plasma). As Professor Evans stated, “[o]nce the drug has been dissolved within bodily fluid (released), it can be absorbed by the human body, by passing through membranes to enter the bloodstream…”: Evans, [43].
242 Further, “release” is not the same as blood plasma concentration.
243 Professor Winter’s evidence was that blood plasma measurement of a drug is not equivalent to the duration of release of a drug from the depot or the site of the injection; rather, the blood plasma level of a drug provides information about the concentration of the drug substance in the blood at a particular point in time: Winter 1, [152], [304].
244 Professor Evans appeared to accept that the concentration of aripiprazole in the blood plasma is not the same as the release (dissolution) of aripiprazole from the depot: Evans, [49], [185]. For example, in the JER, Professor Evans posited a scenario where the release of aripiprazole was to cease at any time (input becomes zero) and stated that, “[i]f this was to happen, one would expect the plasma levels of aripiprazole to decrease according to the inherent terminal half-life of the drug…”. By that evidence, Professor Evans recognises that, while release has an effect upon blood plasma levels, blood plasma levels are not the same as the release of the aripiprazole from the particle depot at the injection site, and there can be a delay between the cessation of the release and the disappearance of the drug from the blood plasma.
245 Professor Winter gave evidence to the same effect in Winter 1 at [175]:
A blood plasma profile is determined by a range of factors, including the time and extent to which drug molecules are absorbed, the distribution kinetics within the body, the speed at which the drug is metabolised, and the time it takes for the drug to be eliminated from the body. As a result, depending on the rates of each of these factors, an active ingredient can be circulating in the blood for significant periods after the last of the drug has been released from a formulation.
(Emphasis original.)
246 Further, the reference to “release” in the PTE Claims is not, as Otsuka submits, “release as measured by blood plasma concentration of aripiprazole”. Blood plasma concentration is not referred to in the PTE Claims, and the experts did not define “release” by reference to blood plasma concentration or refer to blood plasma concentration in their description of the three elements of the Relevant Feature.
247 Nor is it correct that the Patent is using blood plasma concentration to refer to extended release, as Otsuka submits. Having regard to the common general knowledge, a person skilled in the art would understand that the reference to “release” in the PTE Claims is a different process to entry of the drug substance into the blood plasma, and would not conflate these concepts.
6.2.2 Example 4 and Figure 3
248 Other than the bare assertions in the specification concerning the relationship between mean aripiprazole particle size and particular release durations as identified above, the only manner by which release might be determined which is disclosed in the Patent is the blood plasma concentration data and descriptions contained in Examples 1–4 and Figures 1–3.
249 Sun Pharma contends that the blood plasma concentration data in the Patent does not provide a workable standard to identify whether all formulations that otherwise meet any of the PTE Claims fall inside or outside the Relevant Feature.
250 Otsuka submits that the teaching of the Patent cannot be departed from and quotes the statement of Aickin J in Minnesota Mining and Manufacturing Company v Beiersdorf (Australia) Limited (1980) 144 CLR 253 at 274 that “[l]ack of precise definition in claims is not fatal to their validity so long as they provide a workable standard suitable to the intended use”. It submits that the person skilled in the art would be able to determine whether the Relevant Feature is present in a formulation otherwise possessing the features of the PTE Claims by taking blood plasma concentration measurements of aripiprazole at various times following injection, that the Patent teaches that blood plasma concentrations can and are to be used to determine release, and that the Patent indicates that analysis of such concentrations acts as a surrogate for determining the release period.
251 Otsuka relies upon the evidence of Professor Evans to the effect that:
(1) Plasma concentration measurements can be used to determine whether a particular formulation releases aripiprazole for the minimum period after injection specified in the Relevant Features; if the aripiprazole stops being released, it would drop substantially within days. If the aripiprazole is continuing to be released, the plasma concentrations will be maintained or steadily trend downwards.
(2) Although Professor Evans does not see the need to do so, if it were necessary, blood samples could be collected at more frequent intervals either side of the “boundary” to assist in determining whether the Relevant Feature is satisfied.
252 The blood plasma concentration data which is provided in the Patent is somewhat limited.
253 Example 4 (Human PK Data) relates to a single-dose IM Depot study. For convenience, it is set out again (in its entirety):
Aripiprazole I.M. depot formulation prepared in Example 1 was administered intramuscularly to patients diagnosed with chronic, stable schizophrenia or schizoaffective disorder at [sic]. The study design included administration of a 5-mg dose of aripiprazole solution to all subjects followed by a single dose of IM depot at 15, 50, and 100 mg per patient. Samples for PK analysis were collected until plasma concentrations of aripiprazole were less than the lower limit of quantification (LLQ) for 2 consecutive visits.
Figure 3 shows mean plasma concentrations vs. time profiles of aripiprazole in subjects 2 and 3 dosed with 15 mg of IM Depot, and subjects 4 and 5 who received 50 mg of IM Depot. In all cases aripiprazole plasma levels showed a fast onset of release and sustained release for at least 30 days.
254 Figure 3 (which shows aripiprazole plasma concentration on the y axis and time on the x axis) is also shown again:
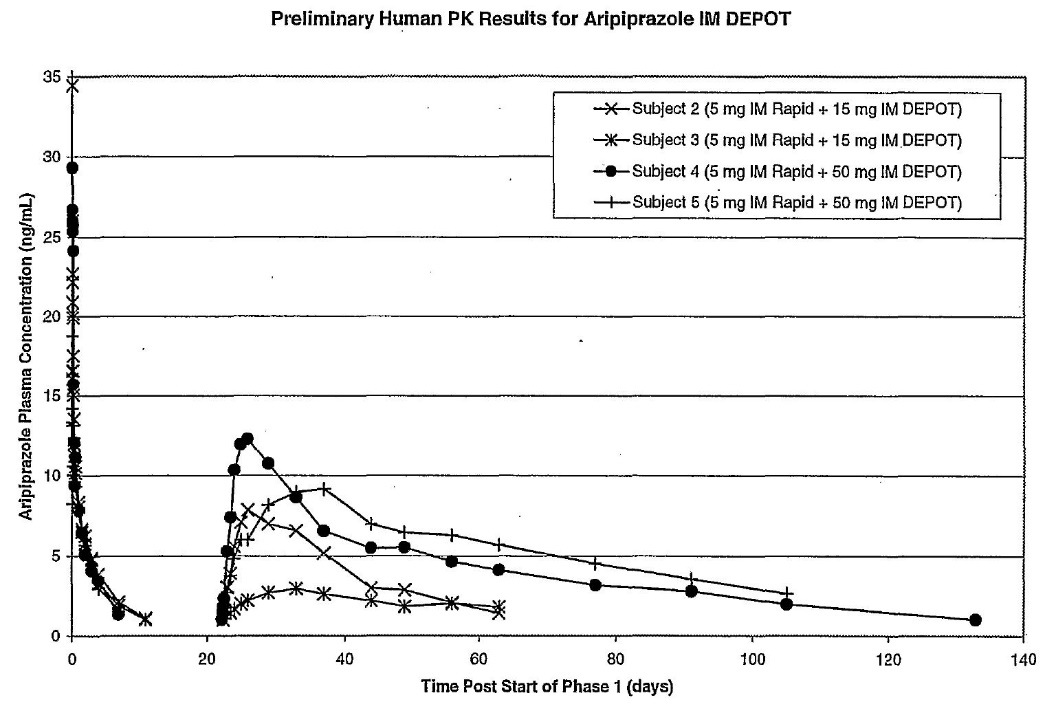
255 Figure 3 shows significant variability across limited data points (such as, for example, the differences in blood plasma concentration between subjects 4 and 5 both in terms of plasma concentration and duration). Further, the lines for subjects 2 and 5 are difficult to distinguish from each other as they each appear to have a cross at various locations along the line (with the cross being designated for subject 2 only). The x axis is not divided into seven day increments (which would be aligned with the release boundaries in the PTE Claims), subject 1 has not been included in Figure 3 without explanation and there is no reference in Figure 3 to the results obtained from the single dose of 100 mg IM depot which is referred to in Example 4. It is also headed “Preliminary Human PK Results etc” (emphasis added).
256 Importantly, Figure 3 does not purport to depict or even refer to “release” (the meaning of which was agreed by the experts), which is different to blood plasma concentration (which is shown). In the JER and by reference to Figure 3, Professor Winter further explained that “release is nowhere measured” in the Patent (including the Examples), and the approach of “looking at only 4 individual blood plasma level curves with about 12 to maximally 20 data points each… using the unaided eye” is prone to error.
257 In the JER, Professor Winter also gave this evidence, which evidence I accept:
Even today with very modern and very expensive methods, release from an intramuscular depot in vivo in humans is practicably not measurable with a precision that allows determination of whether a formulation would fall in or outside the boundaries of the 448 Patent. With this in mind it is not at all plausible that the single in vivo study in the 448 Patent with 4 subjects and nothing more than about 50 blood plasma values (surrogate data) could provide evidence that all formulations within each of the relevant claims would fall within the boundaries of each of the relevant claims.
…
When contemplating the many different formulations one must recall that the dose, the crystal form, the site or type of injection, the volume, the exact particle size and other parameter options are all left open, providing a wide array of many formulations and to predict their behaviour on the basis of the extremely little technical information contained in the 448 Patent is impossible.
258 In Example 4, it is stated by reference to Figure 3 that, “[i]n all cases aripiprazole plasma levels showed a fast onset of release and sustained release for at least 30 days”. However, as release (within the meaning of the PTE Claims) is not shown in Figure 3, only blood plasma concentration is shown, there will be at least some delay between release and entry of the drug substance into the plasma and as the drug substance can remain in the blood plasma after the release has ceased, the basis for asserting that Figure 3 “shows” release (whether fast or sustained) is neither apparent nor explained, and there must have been some intermediate analysis performed which underlies this statement.
259 As Professor Winter explained in the concurrent expert session, while blood plasma concentration data can provide some information, it cannot provide an actual duration of release or be used to measure the release. Professor Winter also gave this evidence, with which I agree:
--I understand that the authors of the patent want to – they try to use the data in figure 3, which we have looked at a moment ago, would show a fast onset of release and sustained release. In fact, they do not – silently, the authors of the patent apply some thought of additional thinking that one might apply to go from the one to the other.
[T81.34–38]
260 It is this “additional thinking” that is not disclosed or described in the Patent.
261 When Professor Winter was asked about the conclusions reached in Example 4 concerning release period, he explained that the Patent does not inform the reader how release is determined. He said that the authors of the Patent did not use terms like “deconvolution” to explain how those conclusions were reached; rather, it is left “to the reader, to think about”. He accepted that the authors of the Patent wanted to support those conclusions with Example 4 and Figure 3, but, in his opinion, they “fail to do so”.
262 In my view, it is not the case that Professor Winter approached the Patent with the eyes of a “sceptical expert who does not believe what he reads”, as Otsuka contends, nor is it the case that he adopted an approach which was not the practical and commonsense approach of the person skilled in the art. Instead, it was apparent that Professor Winter strived to comprehend how the sparse information provided in the Examples and the crude depiction of blood plasma concentration data in Figure 3, with its 20 day time increments and four subjects, could be utilised by a person skilled in the art to determine whether a given formulation that otherwise satisfies any of the PTE Claims falls inside or outside the release boundary in those claims. In circumstances where the invention constitutes formulations which are designed to be injected into humans, form a depot in the muscle and be released over specified time periods for the purposes of treating serious psychiatric conditions, Professor Winter’s explanations as to how this could not be done based on the information in the Patent were neither overly critical nor unduly sceptical.
263 To the extent that Professor Evans disagreed with the evidence of Professor Winter in the JER concerning these issues, Professor Evans accepted that he improperly answered Question 3 in the JER on the basis that he was only required to consider whether a single product met the Relevant Features, while Question 3 required the experts to determine whether or not all aripiprazole formulations within each of the PTE Claims fall within or outside the boundaries of each of the Relevant Features. It is therefore not necessary to address the evidence of Professor Evans in the JER. I otherwise prefer the evidence of Professor Winter for the reasons already explained.
264 Further, even if it could be said that the evidence established that some formulations, such as that in Example 1, would achieve minimum periods of release, that is not the end of the matter. That is because the issue is whether the person skilled in the art is able to determine whether other formulations which lie closer to the boundaries of the PTE Claims, such as with a mean particle size that may or may not achieve a specified minimum release period depending on the formulation, will fall within the PTE Claims. For example, a formulation with a mean particle size of about 1 micron for release over a period of at least two weeks, such as that contemplated by claim 16, would require the day on which the formulation stops releasing (rather than being measurable in blood plasma) to be measured with precision in order to determine whether it released for less or more than two weeks.
265 In summary, the Patent does not show the person skilled in the art how to determine whether or not each formulation that otherwise satisfies any of the PTE Claims falls inside or outside the release boundary specified in the Relevant Feature. The Patent is silent as to how the person skilled in the art is to extrapolate from the disclosed blood plasma concentration data to a determination of the release of aripiprazole from the depot. As the person skilled in the art is not given that information, the person skilled in the art is not able to use that information to enable them to determine whether all formulations which otherwise fall within any of the PTE Claims take the Relevant Features.
266 For this reason, the PTE Claims are bad for ambiguity because the instructions in the Patent for attaining the Relevant Feature are meaningless to those skilled in the art: BlueScope at [719]. Contrary to Otsuka’s submissions, these matters would cause difficulty to a manufacturer wishing to satisfy himself that he is not infringing the PTE Claims.
267 This conclusion is fortified by the content of the Otsuka confidential documents which, like Figure 3, show significant inter-patient variability such that a person skilled in the art would not consider that blood plasma concentration data alone will be sufficient to determine if the Relevant Feature in the PTE Claims has been met in relation to any given formulation.
6.2.3 Confidential Otsuka documents
268 The first document is Confidential Annexure GJW-9, being a copy of a document titled “Aripiprazole, Final Clinical Study Report for Study CN138020, Assessment of the in vivo release characteristics and safety of an intramuscular depot formulation of aripiprazole in subjects with schizophrenia or schizoaffective disorder” from the Australian regulatory dossier for ABILIFY MAINTENA (Study Document).
269 The Study Document is a clinical study report [REDACTED]. It encompassed both compartmental and non-compartmental PK analysis, and deconvolution analysis for the purposes of analysing the results of the study.
270 Table 9.2A at page 52 of the Study Document contained a summary table of aripiprazole pharmacokinetic parameters. For the [REDACTED], the letter N represented the number of people who received that dose [REDACTED]: see, generally, T207.39–48 and T209.21. In respect of the difference in the results for [REDACTED] in Table 9.2A, Professor Evans agreed that there was “significant variability” in blood plasma metrics between [REDACTED]: T209.18–44. Indeed, Professor Evans said that “generally when I’m looking at inter-patient variability, I don’t just look at [REDACTED] because [REDACTED] can be quite different” ... “to look at the variability between people within a population, you would look at more [REDACTED]”: T211.5–16. His evidence in this regard is to be contrasted with his confident answers concerning the sufficiency of the information given in Example 4 and Figure 3 in the Patent (involving two sets of two subjects, with each set receiving a different dose).
271 At page 61 of the Study Document, it was stated that there appeared to be a:
[REDACTED]
272 As to this passage, Professor Evans agreed that the authors of the study did not know, even after the study had been carried out, the “specific relationship between ... the composition of the muscle tissue and the relative differences in the release”.
273 The authors also noted on page 61 of the Study Document that [REDACTED]. Professor Evans agreed that this same comment must apply to the small sample size in Figure 3 of the Patent.
274 Professor Winter gave evidence about the Study Document at Winter 2, [141(a)]:
The Study Document does not report the results of measurements on the release of aripiprazole from the depot at the site of injection. Rather, the Study Document includes certain information regarding the absorption of aripiprazole from the Study Document Preparations. Absorption is a different process to release… [and] in any event, the information in the Study Document does not provide a sufficiently accurate measure of absorption so as to use this information to precisely determine the duration of release of aripiprazole from the depot at the site of administration.
275 Professor Winter also gave evidence that not only did the Study Document provide “no direct calculation of the duration of release”, but its analysis is “impacted by significant variability, a small sample size, the use of an intramuscular immediate release comparator that has not been completely analysed and the simplistic pharmacokinetic model employed based on unvalidated assumptions”: Winter 2, [155].
276 Professor Winter also stated in the JER that:
Accepting the data in [the Study Document] to be technically correct, it becomes clear that a formulation according to example 1 in the 448 Patent cannot fulfill claims 8, 11, 24, 59 due to a calculated release being longer than four weeks or about four weeks.
277 Otsuka relies upon this evidence in support of its contention that Professor Winter accepted that the formulation in Example 1 would be longer than four weeks and therefore achieve the release periods in the PTE Claims. Even if that is the case, it does not assist Otsuka for the reasons explained in [264] above.
278 The second document is the summary of the 246 Study which was described as follows:
[REDACTED]
279 The document includes a table which depicts plasma concentrations of aripiprazole over time (measured in weeks) as follows:
[REDACTED]
280 Notably, this study involved [REDACTED]. The table above shows significant inter-patient variation of blood plasma concentration of aripiprazole between patients who were administered with the same 400mg IM Depot formulation.
281 The following further salient features are worth highlighting:
(1) the 246 Study was performed by Otsuka [REDACTED];
(2) patient blood plasma concentration results ranged [REDACTED]. Professor Evans agreed that this is a “large interpatient variation”, and that there “were some patients among them that have minimum plasma levels”. Professor Evans also agreed that this means that “if you’re using blood plasma as a proxy, then what this shows is that the patients had either stopped releasing or had never started releasing”;
(3) some subject(s) obtained readings of [REDACTED];
(4) the 246 Study has many more data points than Figure 3;
(5) Professor Evans agreed that “this [REDACTED] study, [REDACTED], demonstrates that, even if you could have done a human study at the relevant date, then that’s also likely to have shown a huge variability in blood plasma concentrations”.
282 A further Otsuka study was summarised in Confidential Annexure AME-18 being a copy of a document titled “Clinical Study Report: Assessment of the Safety, Tolerability, and Pharmacokinetics of Aripiprazole IM Depot Formulation by Single Administration in Patients with Schizophrenia (study-31-07-002- report)” from the Australian regulatory dossier for ABILIFY MAINTENA. This was a [REDACTED]. In the JER, Professor Evans agreed with Professor Winter that the “results of this study show high inter-individual variability”. Professor Winter gave evidence that “[t]his is yet another example of the significant variability in blood plasma concentrations observed following administration of the injectable aripiprazole formulations”: Winter 2, [179].
6.2.4 Nature of any experimentation required
283 For the reasons given above, blood plasma profiles for a given dose of a given formulation administered to a particular sample of subjects does not provide sufficient information for the purposes of working out whether that same formulation or other formulations fall inside or outside the release boundaries in the PTE Claims, and the person skilled in the art cannot determine the release of aripiprazole in a given formulation from a depot upon administration simply by testing the amount of the drug in a person’s blood plasma at different time points. Further, there has been shown to be huge inter-patient variability in respect of a given aripiprazole depot formulation, both in Figure 3 and in Otsuka’s own documents. Thus, contrary to Otsuka’s submission, the use of blood plasma concentration to determine release is not a “workable standard”.
284 As such, it must be determined whether other routine methods known by the person skilled in the art could be used to determine release, or whether inventive faculty or burdensome experimentation is required.
285 In this case, the person skilled in the art would need to experiment with (at least) dosage concentration and frequency, volume, site of administration and particle size in order to determine whether any given formulation falls within or outside the boundaries of any of the PTE Claims. However, as explained by Professor Winter, “it is not possible to determine through routine testing available at the Relevant Date whether or not the aripiprazole particles have completely dissolved from a site of administration upon injection into the body”: Winter 1, [168].
286 Further:
(1) as there is “no direct method to measure the release of aripiprazole from the site of injection, it would be necessary to consider a combination of methods in order to determine whether an aripiprazole formulation satisf[ies]” the Relevant Features;
(2) such combination of methods is a complicated process such that “reasonable minds may differ about what would be the appropriate model to apply, and/or about what assumptions to rely on for using that model (thereby, arriving at different results)”; and
(3) the person skilled in the art would need to also deal with issues in respect of sample size, large inter-patient variability, formulating an appropriate intravenous comparator, applying appropriate PK analysis techniques, deploying deconvolution studies, and so forth.
(Winter 2, [116]; see also Winter 1, [165]–[181]; Winter 2, [110]–[116].)
287 Otsuka relies upon the following evidence to contend that the experiments would be routine (with the following transcript references taken from the submissions):
(1) Plasma concentrations are used to design dosage regimens: T173.36–174.4.
(2) Dosage regimens are necessarily based on the duration of release of the drug: e.g., T280.44-46. [However, the evidence cited for this proposition was not to this effect].
(3) Professor Winter was well aware of how to measure blood plasma concentration over a period of time: T174.18–21. In order to determine how long the drug is present in the plasma, one takes blood samples over the period of time for which one hopes to have the dosage frequency: T177.19–24.
(4) Professor Winter taught his students how to do in vivo blood plasma concentration over time profiles: T174.47–175.7. He gave them the “basic instruction” that “the important aspect is to take enough blood samples to get enough information for typically the different phases of a blood plasma curve”: T175.9–24.
(5) As part of a design process for a one weekly dosage of a drug, the blood plasma concentration for more than a week after administration would be used; the same applies for a fortnightly drug: T174.6–16. As a “rough idea”, if looking for a once weekly dosage regimen, one could take 15 samples: T176.6–177.6. This would be a practical way to determine whether a formulation has a sufficient blood plasma concentration: T177.1–11.
288 However, as Sun Pharma submits, this evidence relates to the use of blood plasma concentration data to determine dosage of a formulation, which is not the relevant question (or, at best for Otsuka, is only one part of any process to determine whether the minimum release period has been achieved).
289 Professor Evans gave evidence in the JER that “[t]he 448 Patent claims also allow for different forms of aripiprazole (hydrates and polymorphs) and different formulation excipients” and oral evidence that “[o]ne cannot rule out an impact of different formulation aspects on the manufacturability, the physical stability, the user acceptance and the pharmaceutical performance of every single permutation of the variables” and in respect of the release of aripiprazole from the depot.
290 During the hearing, Professor Evans agreed that, within the PTE Claims, there included many different permutations of excipients, hydrates and polymorphs. He also agreed with the following matters:
(1) the Patent includes a long list of suspending agents;
(2) each of the PTE Claims include formulations with one, two or more of these dozens of suspending agents;
(3) the PTE Claims can include formulations with one or more of these suspending agents in varying amounts.
(4) certain of the suspending agents in the Patent are “different in nature from each other” including “fatty esters” and are in contrast to CMC in the asserted “pharmaceutical substances”;
(5) if the formulations contained one or more of the suspending agents in the Patent (other than CMC), then he could not rule out those suspending agents having an effect on the release of aripiprazole from the depot and the person skilled in the art would need to test these formulations;
(6) further, the Patent includes a list of bulking agents;
(7) each of the PTE Claims include formulations with one, two or more of these bulking agents;
(8) the PTE Claims can include formulations with one or more of these bulking agents in varying amounts;
(9) the Patent informs that two of these bulking agents can affect crystal growth and agglomeration, namely, xylitol and/or sorbitol;
(10) if the formulations contained one or more of the bulking agents in the Patent (other than mannitol), then he would need to test the formulation in order to work out the extent that they (amongst other factors) affect the release of aripiprazole from the depot.
291 Professor Evans also accepted that if he was to even attempt to determine whether a formulation would fall within or outside the boundaries in any of the PTE Claims, he would need to experiment with such formulations and would need to “design and conduct a human study”, to collect and analyse blood plasma data.
292 Professor Evans further agreed that:
(1) such human study would require ethical approval and regulatory approval (or at least, regulatory “acknowledgement” in the case of Australian studies);
(2) such human study would also need an immediate release comparator;
(3) “it would be necessary to formulate and experiment in relation to any suitable immediate release formulation [that] could be used”;
(4) he would need to “administer the comparator formulations and the depot formulations to the patient population under controlled conditions”;
(5) he would then need to analyse the data to determine the most appropriate pharmacokinetic analysis to adopt to interpret the study’s results.
293 After performing that process, Professor Evans would need to repeat it for each formulation, after adjusting variables to obtain the desired result.
294 The confidential Otsuka documents demonstrate that if the person skilled in the art was to try to use the blood plasma profiles as a proxy to identify whether a formulation that otherwise meets the PTE Claims falls inside or outside the release boundaries, such experiments, modelling and deconvolution studies are not routine or straightforward, and instead are onerous, time consuming and would provide greatly variable and unreliable results.
295 For example, Professor Evans agreed that the Study Document summarised a study that took over two years; involved three study centres; involved compartmental and non-compartmental analysis; involved deconvolution analysis; involved creating an IM Rapid control formulation; involved enrolling subjects with chronic stable schizophrenia or schizoaffective disorder who are otherwise considered healthy as determined by medical history, physical examination, 12-lead ECG and clinical laboratory evaluations; administering the immediate release and the depot formulations; obtaining and analysing the PK data; and theorising about the reasons and variability in that data between patients, including the lack of trends in relation to dose proportionality. He agreed that the experiment disclosed in the Study Document was not a routine experiment and required considerable analysis, testing and theorising, and that he would have obtained different results even if he tried to conduct the same test.
296 For these reasons, even if the person skilled in the art was able to use blood plasma concentrations as an indirect means to identify whether a given formulation that otherwise meets the PTE Claims falls inside or outside the release boundaries, experiments would need to be performed. Such experiments are not simple, routine or straightforward, but would be onerous, time-consuming, and would provide variable and unreliable results such that the person performing the experiments would be left in doubt as to whether any given formulation falls within the scope of the PTE Claims or not: Austal Ships at [14].
297 It follows that the person skilled in the art would not be able to determine whether or not all aripiprazole formulations (that is, all embodiments) that otherwise meet the requirements of the PTE Claims fall within or outside the Relevant Feature based upon the information in the Patent and the common general knowledge, even allowing for routine experimentation.
6.3 Conclusion on validity of PTE Claims
298 For these reasons, the PTE Claims are invalid because they fail to define the invention and lack clarity, as required by ss 40(2)(b) and 40(3) of the Patents Act. It follows that they should be revoked.
7. CONCLUSION ON VALIDITY OF EXTENSION
299 In summary, the asserted pharmaceutical substances per se do not meet the requirements of s 70 of the Patents Act for the following reasons:
(1) Substances I–IV and VI–IX do not contain the Relevant Feature which is contained in each of the PTE Claims with the consequence that they do not in substance fall within the scope of the claim or claims as required by s 70(2)(a) of the Patents Act.
(2) Substances I–X do not in substance fall within the scope of the claim or claims as required by s 70(2)(a) because the PTE Claims are invalid as they fail to comply with ss 40(2)(b) and 40(3) of the Patents Act.
8. CROSS-CLAIM
300 As observed, Sun Pharma has applied to register and intends to sponsor and supply products registered on the ARTG which are a generic version of the ABILIFY MAINTENA (400 mg) powder and solvent for injection, and it wishes to launch those products on 1 April 2025.
301 By reference to the proposed Sun Pharma products, it is common ground that the Cross-Claim based on threatened infringement of the Patent must succeed in the event that the Patent and the Extension are found to be valid to any extent. As that has not occurred, this claim must fail.
302 Otsuka also claims that Sun Pharma has threatened to contravene s 18(1) of the ACL. Otsuka’s case relies on the Court’s finding of threatened infringement of the Patent, at least in part. As no finding of threatened infringement of the Patent has been made, this claim must also fail.
9. DISPOSITION
303 I will direct the parties to confer and either provide my chambers with an agreed form of order which reflects these reasons, or with their own version of a proposed order.
I certify that the preceding three hundred and three (303) numbered paragraphs are a true copy of the Reasons for Judgment of the Honourable Justice Downes. |
Associate:
SCHEDULE OF PARTIES
NSD 172 of 2024 | |
OTSUKA AUSTRALIA PHARMACEUTICAL PTY LTD (ACN 601 768 754) | |
H. LUNDBECK A/S | |
Third Cross-Claimant: | LUNDBECK AUSTRALIA PTY LTD (ACN 070 094 290) |
Fourth Cross-Claimant: | OTSUKA AUSTRALIA PHARMACEUTICAL PTY LTD (ACN 601 768 754) |

