
FEDERAL COURT OF AUSTRALIA
Sandoz AG v Bayer Intellectual Property GmbH [2023] FCA 1321
ORDERS
DATE OF ORDER: |
THE COURT DECLARES THAT:
1. The Cross-Respondent has threatened to infringe claims 3 and 4 of Australian Patent AU2006208613 entitled “Prevention and treatment of thromboembolic disorders” (the 613 Patent).
THE COURT ORDERS THAT:
1. The First and Second Applicants’ application be dismissed.
2. The First and Second Cross-Claimants’ cross-claim be allowed in-part.
3. The Cross-Respondent, whether by itself, its directors, officers, servants, agents, Related Bodies Corporate (as defined in the Corporations Act 2001 (Cth)), Associated Entities (as defined in the Corporations Act) or howsoever otherwise (together, Sandoz), during the term of the 613 Patent be restrained from infringing claims 3 and 4 of the 613 Patent, including (without limitation) by engaging in the following acts within the patent area (as that term is defined in the Patents Act 1990 (Cth)), without the licence or authority of the Cross-Claimant:
(a) importing, selling, supplying or otherwise disposing of the Sandoz Rivaroxaban Products;
(b) offering to sell, supply or otherwise dispose of the Sandoz Rivaroxaban Products;
(c) authorising other persons to use the Sandoz Rivaroxaban Products or engage in any of the acts described in subpara (a) and (b) above;
(d) procuring or inducing, whether by instructions or otherwise, any of the acts described in subpara (a) and (b) above; or
(e) otherwise infringing the 613 Patent.
4. The 613 Patent be amended in the manner described in the Annexure hereto.
5. The First Applicant and Second Applicant/Cross-Respondent pay the Respondent/Cross-Claimants’ costs of the application and the cross-claim with a 15% reduction for the Applicants succeeding on the meaning of “in hydrophilized form” issue. Such costs are to be taxed if not agreed.
6. The Cross-Claimants pay the costs of the amendment application.
7. If any party seeks a variation of the costs orders in paragraphs 5 and 6 above, it may, within seven days, file and serve a written submission (of no more than two pages) and any affidavit in support. In that event, the other party may file and serve a responding written submission (of no more than two pages) and any affidavit in support within a further seven days, and the issue of costs will be determined on the papers.
THE COURT CERTIFIES THAT:
8. Pursuant to s 19 of the Patents Act, the validity of each of the claims of the Australian Patent AU2004305226 entitled “Method for the production of a solid, orally applicable pharmaceutical composition” and the 613 Patent were questioned in this proceeding and found to be valid.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
Insert the number “7” immediately after the incomplete PCT number PCT/04/01289 and insert
the country code "EP" before the application date code of "04" on page 10, line 13 of the 613 Patent, as shown below.

REASONS FOR JUDGMENT
ROFE J:
[1] | |
[8] | |
[13] | |
[69] | |
[93] | |
[104] | |
[104] | |
[125] | |
[143] | |
[143] | |
[144] | |
[149] | |
[163] | |
[183] | |
[184] | |
[194] | |
[199] | |
[219] | |
[237] | |
[238] | |
[241] | |
[244] | |
[244] | |
[261] | |
[273] | |
[280] | |
[289] | |
[301] | |
[302] | |
[306] | |
[311] | |
[314] | |
[321] | |
[328] | |
[335] | |
[341] | |
[352] | |
[358] | |
[361] | |
[367] | |
10.1.13 Unmet need for safe and effective new oral anticoagulant | [374] |
[383] | |
[417] | |
[423] | |
[431] | |
[443] | |
[449] | |
[467] | |
[472] | |
[497] | |
[516] | |
[524] | |
[529] | |
[540] | |
[579] | |
[591] | |
[624] | |
[627] | |
[628] | |
[631] | |
[638] | |
[653] | |
10.5.5.1 Evidence on the information disclosed in the Abstracts | [656] |
[666] | |
[672] | |
[680] | |
[688] | |
[693] | |
[707] | |
12.3 Patent prosecution history: Overseas equivalent patents and applications of the 613 Patent | [723] |
[724] | |
[730] | |
[735] | |
[738] | |
[741] | |
[748] | |
[752] | |
[756] | |
[767] | |
[773] | |
[774] | |
[787] | |
[789] | |
[792] | |
[799] |
1 Bayer Intellectual Property GmbH is the patentee of the two patents in suit:
(a) Australian Patent No. 2004305226 entitled “Method for the production of a solid, orally applicable pharmaceutical composition” (the 226 Patent); and
(b) Australian Patent No. 2006208613 entitled “Prevention and treatment of thromboembolic disorders” (the 613 Patent).
2 Bayer Australia Ltd is, and has been since 14 February 2023, the exclusive licensee of the 226 Patent and the 613 Patent in Australia. Bayer Australia markets and sells Pharmaceutical Benefits Scheme listed products comprising rivaroxaban in Australia under the trade name XARELTO.
3 Sandoz AG and Sandoz Pty Ltd (Sandoz Australia) (together, Sandoz) seek to revoke the 226 and 613 Patents. It contends that the claims of each patent are invalid on several bases.
4 By way of cross-claim, Bayer seeks permanent injunctive relief against Sandoz Australia for threatened infringement of claims 8 to 22 of the 226 Patent and claims 3 and 4 of the 613 Patent (asserted claims).
5 Subject to the validity of the 613 Patent, Sandoz Australia admits infringement of the claims asserted against it. One issue remains for determination on the infringement of the 226 Patent: the construction of the term “in hydrophilized form”.
6 By an interlocutory application dated 27 June 2022, Bayer seeks to amend the 613 Patent to correct what it terms “a mistake”. The reference to “PCT/04/01289” on page 10, line 12, of the specification should read “PCT/EP04/012897” (the mistake). No amendments are sought to the claims. Sandoz opposes the amendment on a number of grounds.
7 In summary, and for the reasons that I explain in more detail below, I find that both the 226 Patent and the 613 Patent are valid. However, on my construction of “in hydrophilized form”, I do not consider that the Sandoz rivaroxaban products infringe the 226 Patent. Therefore, I only find that Sandoz has threatened to infringe claims 3 and 4 of the 613 Patent. I will also exercise my discretion to grant Bayer’s amendment to the 613 Patent.
8 The 226 Patent relevantly relates to orally administrable pharmaceutical compositions comprising rivaroxaban “in hydrophilized form” and the use of those pharmaceutical compositions for the prophylaxis and/or treatment of a thromboembolic disease.
9 The 613 Patent relates to a method of treatment of thromboembolic disorders by once daily administration of a rapid-release tablet comprising rivaroxaban.
10 Rivaroxaban itself had, as one of a class of factor Xa inhibitors, been disclosed and claimed in an earlier Bayer patent application, International Patent Publication No. WO 01/47919 (WO 919), which is a key piece of prior art in relation to each Patent. The validity of the Australian patent resulting from that application, Australian Patent No. 775126 (the Compound Patent), is not challenged by Sandoz in this proceeding. The Compound Patent, as extended, is due to expire on 24 November 2023.
11 Sandoz Australia is the sponsor on the Australian Register of Therapeutic Goods of the following pharmaceutical compositions, each comprising rivaroxaban (the Sandoz products):
(a) ARTG ID 335415: Rivaroxaban Sandoz rivaroxaban 10 mg film-coated tablet blister pack.
(b) ARTG ID 335420: Rivaroxaban Sandoz rivaroxaban 15 mg film-coated tablet blister pack.
(c) ARTG ID 335496: Rivaroxaban Sandoz rivaroxaban 20 mg film-coated tablet blister pack.
12 Sandoz Australia intends, without Bayer’s authorisation, to exploit the Sandoz products in Australia after 24 November 2023.
13 The parties filed an agreed glossary and technical primer prior to the commencement of the trial. This section includes extracts from the glossary of the agreed technical primer.
14 Absorption refers to the process by which an API (defined below) moves from the site of administration into a site in the body, usually the bloodstream. Absorption may be defined in terms of its rate and extent. For an orally administered API, the administration site is the gastrointestinal (GI) tract. In the case of a solid oral dosage form, the total extent of absorption may be determined by a number of factors, including the extent of release, solubility, permeability, local and pre-systemic metabolism, stability characteristics of the API in the GI tract and GI tract transit times. In the case of a solid oral dosage form, the rate of absorption may be determined by factors including the rate of release (including disintegration), dissolution rate, solubility, permeability, local and pre-systemic metabolism, rate of degradation (if any) of the API in the GI tract and the presence of food in the GI tract.
15 Absorption half-life is the time it takes for the absorption of 50% of the dose that was absorbed (eg that moved from the administration site into the blood stream) when absorption is a first-order process.
16 Absorption profile of an API refers to both its rate and extent of absorption.
17 Activated Partial Thromboplastin Time (APTT or aPPT) is an assay used to monitor heparin treatment and is also known as PTT or Partial Thromboplastin Time. APPT is sensitive to the heparin-induced inhibition of factors IIa, IXa, Xa and XIa. It is performed on citrated plasma, which has been processed to remove the platelets, using an APTT reagent and a recalcification step.
18 Anticoagulants are a class of antithrombotic drugs that inhibit the formation and/or propagation of coagulation, and/or the formation of fibrin, and are used in the treatment of thromboembolic disorders. By inhibiting the formation or growth of a clot, the anticoagulant allows the body time to break down the clot. Anticoagulants are also used prophylactically and chronically to decrease the risk of a new clot forming.
19 API means active pharmaceutical ingredient. The words “API” and “drug” can be used interchangeably.
20 Atrial fibrillation is a disease in which the heart beats irregularly. The irregular heart beat can cause clots in the heart or arterial system because the blood flow is not smooth, and thus is subject to turbulence, which results in blood clotting in areas of slow flow (most frequently the left atrial appendage).
21 Arterial thromboembolism occurs where a blood clot forms in the arterial system. Generally, the most common pathologies are atrial fibrillation, peripheral vascular disease or coronary artery thrombosis. Clots formed due to atrial fibrillation and peripheral vascular disease can result in a stroke, whereas clots formed due to coronary artery thrombosis can result in heart attacks.
22 AUC, or area under the curve, refers to the area under the concentration versus time curve. The “concentration” refers to the concentration of a drug, biomarker or clotting factor. When measuring the concentration of a drug in plasma, AUC is a measure of the extent of absorption of a drug. AUC may be defined according to a particular time period.
23 Bioavailability refers to the extent and rate of absorption of the API from a dosage form into a body site. Bioavailability is determined by, among other things:
(a) the absorption profile of the API;
(b) the dosage form within which it is administered to the patient; and
(c) its first pass extraction (by metabolism or excretion).
The fraction of API absorbed from a dosage form is usually estimated by comparing AUC of the test dosage form or dosage amount, and a reference dosage form.
24 Clearance (CL) refers to a measure of the efficiency with which an API is eliminated from blood or plasma, and encompasses the API’s metabolism and excretion. Clearance is defined as the "volume of blood or plasma cleared of drug per unit time", and is thus expressed in units of volume per time (eg L/hr).
25 Clot or thrombus consists of a mesh of fibrin that embeds activated platelets and red and white blood cells.
26 Cmax is the peak concentration of the API, biomarker or a clotting factor(s) in a biological matrix after a treatment. In the case of an API, Cmax is the peak plasma concentration achieved following administration of a single dose of the API or during repeated dosing.
27 Cmin is the minimum concentration of the API, biomarker or a clotting factor(s) in a biological matrix after multiple treatments. In the case of an API, Cmin is the minimum plasma concentration achieved during the time interval between administration of two doses of an API during repeated dosing.
28 Coagulation cascade refers to a series of activation steps of various enzymes (factors) involved in coagulation. Under normal physiological conditions, it is only upon an injury that the coagulation system should rapidly and locally respond at an injured blood vessel wall to generate a clot.
29 Coronary artery thrombosis can occur where a clot is formed in a blood vessel in the heart.
30 Deep Vein Thrombosis (DVT) refers to the formation of a blood clot in a deep vein, usually in the leg. If DVT is left untreated, the blood clot can break off from the formation site and travel to the lungs causing PE. PE occurs where there is a blockage of the pulmonary artery in the lung and can be life-threatening.
31 Direct compression is a tablet manufacturing process in which the API is blended with excipients and compressed into finished tablets without modifying the physical nature of the blended material itself.
32 Dissolution rate refers to the rate at which a solid or liquid (eg a solid or liquid form of a drug) dissolves in a solvent.
33 Distribution refers to both the rate and extent to which an API moves into, and out of, different body components, including movement between plasma and blood cells or between the bloodstream and tissues. For APIs that become systemically available via the blood, the extent of the API’s distribution can be quantified by the term “apparent volume of distribution” (V), which is a proportionality between the total amount of the API in the body and the concentration of the API in the plasma: 
For example, if 100 mg of an API is in the body at a particular time and, at that same time, the plasma concentration is 5 mg/L, the volume of distribution will be 20 L.
34 Dosage form refers to the form in which the API is administered to the patient, such as tablets, capsules, solutions, suspensions, nasal sprays etc.
35 Dosage regimen refers to the regimen by which an API is administered, and includes the dose (quantity of API administered), dosage form, route (eg oral, intravenous, subcutaneous) and frequency of administration.
36 Elimination refers to the irreversible removal of an API from the body by metabolism (conversion to a different chemical species) and/or excretion.
37 Elimination half-life refers to the time taken for the concentration of the API in blood, plasma or serum to fall by half during the elimination phase of a pharmacokinetic profile.
38 Excretion refers to the removal of the API from the body via an excretory process, such as filtration across the kidneys and removal via the urine, breath, sweat and faeces.
39 Factors refers to the main enzymes involved in the coagulation cascade.
40 Fibrin is the fibrous protein that makes up a clot or thrombus.
41 First order elimination occurs where the rate of elimination of a drug is directly proportional to drug concentration.
42 Granulation involves the combination of powder particles, often with a binder, to form granules. Granules comprise multiple different particles. These granules are an intermediate product, which are compressed into a tablet or filled into a capsule. Granulation methods can be broken into two categories: wet and dry. Wet granulation involves the combination of drug particles with a binder and other excipients into granules in the presence of a solvent, typically water. The parties agreed that moist granulation and wet granulation refer to the same process. Dry granulation is a similar process conducted in the absence of the solvent.
43 In vitro refers to analysis, tests or evaluations conducted outside a living organisation (eg in a test tube).
44 In vivo refers to analysis, tests or evaluations conducted in a living organism.
45 International Normalized Ratio (INR) is a corrected ratio that expresses the patient’s prothrombin time (PT) over the mean normal PT, adjusted by the thromboplastin responsiveness value (known as the international sensitivity index (ISI), which is specific to each thromboplastin reagent).
46 Log P is the logarithm of the octanol-water coefficient of a molecule. That is, it is the logarithm of the ratio of a compound’s concentration in octanol (which is an organic solvent) and water.
47 Melting point is the temperature at which a compound will change from solid to liquid state.
48 Metabolism refers to the process by which enzymes in the body change the chemical structure of the API, thereby converting it into a different chemical form. Metabolism of an API occurs primarily in the liver, but can occur to varying extents in other organs. The extent of metabolism and the type of metabolism of an API depends on its interactions with the many metabolic enzymes in the body, and is thus highly dependent on the API’s chemical structure.
49 Metabolites are the products of metabolism. As metabolites have a different chemical structure to the parent API, they can have different pharmacokinetic and pharmacodynamic properties to the parent API. Metabolites can be inert, can contribute to the improvement in the condition (efficacy) or produce side effects.
50 Off-target side effects refers to the consequences that can occur when a drug affects targets or tissues other than targets with which the drug was meant to bind.
51 Parenteral administration refers to administration other than by the GI tract (eg mouth), generally injection or infusion.
52 Peripheral vascular disease refers to where the arteries develop atherosclerosis, which is the thickening or hardening of arteries caused by the build-up of plaque in the inner lining of an artery. Rupture of the plaque results in a highly thrombotic surface, resulting in blood clot formation.
53 Permeability refers to the ability of an API to move across cell membranes such as those lining the GI system. Permeability encompasses passive movement (eg through or between the cells of the GI lining) and active movement (eg via transporters within the GI lining). Permeability is also affected by efflux transporters which restrict movement across the GI lining.
54 Pharmacokinetics refers to the study of the time course of API in the body. The pharmacokinetic profile of an API depends on its:
(a) absorption;
(b) distribution;
(c) metabolism; and
(d) excretion.
“Elimination” is the combination of metabolism and excretion. Pharmacokinetics is sometimes referred to as PK or by using the acronym ADME.
55 Pharmacodynamics refers to the time course of the intensity of the pharmacological effect(s) that an API has in the body. Pharmacodynamics incorporates both (i) the therapeutic effect of the API (ie treating or improving the relevant condition) and (ii) any side effects caused by the API. Pharmacodynamics may be distinguished from pharmacokinetics in that pharmacodynamics describes how an API affects the body, whereas pharmacokinetics describes how the body affects an API. Pharmacodynamics is sometimes referred to as PD.
56 Plasma refers to the liquid component of blood after it has undergone centrifugation to separate the cellular components such as red and white blood cells and platelets.
57 Pre-formulation typically includes experimental, in vitro investigations into at least the stability, dissolution and solubility of the drug (in various potentially relevant biological and environmental conditions), its crystal form and polymorphism, as well as its compatibility with any excipients that might be selected for use in the formulations used in clinical trials. To assess such matters, particularly for potentially sold oral dosage forms, in vitro solubility and dissolution testing are often performed.
58 Prothrombin time (PT) test is a clotting test that was used to measure the effects of warfarin treatment before the priority dates. It was known to respond to reductions in factors II, VII, IX and X during warfarin therapy. It was performed by adding thromboplastin (generally a phospholipid-protein tissue extract) and calcium to plasma, which has been treated with citrate, to prevent it from clotting.
59 Pulmonary embolism occurs where there is a blockage of the pulmonary artery in the lung.
60 Solubility refers to the maximum amount of a compound that will dissolve in a given volume of specified fluid at a particular temperature and pH. Solubility is distinct from dissolution, which refers to the rate at which particles dissolve in solution.
61 Steady-state occurs during repeated (or constant) administration once there is no further accumulation of a drug. Once a drug reaches steady-state, values of Cmax and Cmin remain constant and plasma levels remain within what is defined as a “steady-state range”.
62 Stroke can be caused by clots occluding blood vessels or from bleeding outside blood vessels. Clots can be formed from atrial fibrillation (with the clot forming in the heart), or in cerebral blood supply vessels (both large and small calibre), or from thrombosis in the deep veins of the legs that crosses into the arterial system through a hole in the heart. Some clots may move further after they have detached from the initial location (eg embolized) and travelled to the brain.
63 Terminal half-life is the time taken for a drug concentration to decrease by half across the final portion of a drug concentration versus time profile.
64 Tmax is the time at which Cmax occurs.
65 Therapeutic index or therapeutic window in anticoagulants refers to the range of anticoagulant effect, spanning a lower limit where the risk of thrombosis is unacceptably increased, to an upper limit where unwanted bleeding occurs with unacceptable frequency. A narrow or low therapeutic index is to be distinguished from a high or wide therapeutic index. A higher or wider therapeutic index is one where a broader range of doses of the compound can be given to a person to achieve a desired effect without having adverse effects. A low therapeutic index will mean a risk of toxicity if levels are too high and therapeutic failure if levels are too low.
66 Thromboembolism is a general term describing the combined processes of thrombosis (where a vessel is occluded by clots in part, or in entirety), or embolism (when a clot breaks off from the formation site and lodges elsewhere in the body). Thrombotic or thromboembolic disorders involve thrombosis and/or thromboembolism, which may develop in the venous or arterial circulatory systems.
67 Thrombosis is a pathological clot formation resulting in the obstruction of blood flow through a blood vessel. The primary mechanisms for thrombosis are the improper regulation of the coagulation cascade, and/or excessive platelet activation.
68 Venous thromboembolism (VTE) refers to a clot that develops in the venous system. Common manifestations of VTE are DVT and pulmonary embolism.
69 Bayer relies on the evidence of Professor James Polli in support of its cross-claim for infringement. Professor Polli is a Professor and Ralph F. Shangraw/Noxell Endowed Chair in Industrial Pharmacy and Pharmaceutics at the University of Maryland, Baltimore. He obtained a PhD in pharmaceutics from the University of Michigan in 1993. Professor Polli made two affidavits dated 6 October 2022 and 29 January 2023.
70 Professor Polli has published approximately 100 scientific articles in the areas of dissolution, drug intestinal permeability, transporter substrate requirements, prodrug design, oral bioavailability, in vitro-in vivo correlation and bioequivalence. His two primary research interests are improving oral bioavailability of drugs through formulation and chemical approaches, and developing public quality standards for oral dosage forms.
71 Bayer relies on the evidence of Professor Polli, Professor Allan Evans and Professor Mark Crowther in relation to the validity of the 613 and 226 Patents.
72 Professor Evans is a pharmaceutical scientist and pharmacologist, with extensive teaching, research and industry experience in drug discovery and development. He is a Professor of Pharmaceutical Science at the University of South Australia. He obtained a PhD in pharmacokinetics at the University of Adelaide. Professor Evans made one affidavit dated 8 February 2023.
73 At the relevant dates, Professor Evans was Head of the School of Pharmacy and Medical Sciences at the University of South Australia. He was also lecturing in pharmacokinetics, biopharmaceutics, clinical pharmacology and pharmacy practice.
74 Professor Evans has acted as primary investigator, investigator or consultant in at least 50 clinical trials studying the pharmacokinetics, safety and/or efficacy of APIs. A focus of Professor Evans’ work was to understand whether doses were safe and therapeutically effective in humans and the relationship between safety and therapeutic effectiveness.
75 Professor Crowther is a haematologist specialising in the treatment of thromboembolic disorders with extensive clinical, research, teaching and industry experience. He is a Professor and Chair of the Department of Medicine and Leo Pharma Chair in thromboembolism research at McMaster University in Canada. Professor Crowther made one affidavit dated 7 February 2023.
76 Professor Crowther obtained a Doctor of Medicine from the University of Western Ontario in 1990 and completed a residency in internal medicine and haematology. He went on to become a research fellow in thromboembolism at McMaster University and the Medical Research Council of Canada, as well as a clinical fellow in internal medicine. Professor Crowther also completed a Master of Science in epidemiology and biostatistics from McMaster University in 1998. He is listed as an author on over 600 peer-reviewed articles.
77 Professor Crowther has consulted extensively with the pharmaceutical industry. He has training in clinical trial design and has been engaged in a number of clinical studies including as principal or co-principal investigator to determine the most appropriate dose to initiate and maintain the therapeutic effect of warfarin. Professor Crowther has also been engaged as the principal investigator of an international project for approximately 10–15 studies in the field of coagulation. Most of Professor Crowther’s clinical trial experience has been in Phase II and III clinical trials, but he has also been involved in the early phase discussions as a consultant when defining the therapeutic objectives of a trial program.
78 Bayer also relies on an affidavit from a patent analyst, Dr Louisa King, made on 30 January 2023.
79 Dr King is a director of IP searching and an IP analyst at Aperture Insight. Dr King gave evidence reflecting the results of searches she carried out to locate “PCT/04/01289” referred to on page 10 of the 613 Patent.
80 Sandoz relies on the evidence of Professor Michael Roberts. Professor Roberts is currently the Adjunct Professor of Therapeutics & Pharmaceutical Science at the University of South Australia, an Emeritus Professor of Clinical Pharmacology & Therapeutics and Director of Therapeutics Research Centre at the Diamantina Institute, University of Queensland.
81 Professor Roberts made three affidavits dated 3 October 2022 in support of Sandoz’s case on invalidity, 30 January 2023 in answer to Bayer’s evidence on infringement and 3 March 2023 in reply to Bayer’s evidence on validity.
82 Professor Roberts gave evidence of his active research in areas of pharmacology and toxicity. He has authored more than 578 peer reviewed articles and contributed chapters or other sections to more than 75 books.
83 Professor Roberts was involved in University of Tasmania projects from 1978–1984 developing a slow release oral dosage form of aspirin for use in the prevention of stroke and heart attacks. The teams on these projects conducted in vitro and human dose ranging studies on the antithrombotic effect of aspirin with enteric coating. From 1986–1989, in his capacity as head of the University of Otago Department of Pharmacy, Professor Roberts was involved in conducting clinical trials, including early-phase clinical trials or later stage comparative trials comparing innovator and generic products.
84 Bayer accepts that Professor Roberts is suitably qualified to express an opinion as to matters of formulation and clinical pharmacology in this proceeding.
85 Prior to the hearing, Professor Polli and Professor Roberts prepared a joint report for the Court in which they gave their responses to 11 questions posed by the parties, and indicated where they agreed or disagreed in their response (JER1). The two experts gave concurrent oral evidence at the hearing as a joint expert session (JES1). The parties rely on JER1 primarily in relation to the validity of the 226 Patent. During the course of JES1, it became clear that Professor Roberts had a broader experience in pharmaceutics than Professor Poli.
86 Sandoz also relies on the evidence of Professor Ross Baker, a senior consultant haematologist and clinician scientist, in support of its case of invalidity. Professor Baker made one affidavit dated 3 October 2022.
87 Professor Baker has over 30 years’ experience as a senior consultant haematologist, has published over 138 peer reviewed papers in journals and acted as principal investigator in over 133 international Phase II and Phase III clinical trials. He has worked as principal investigator and in an advisory capacity for pharmaceutical companies in the development of various anticoagulants (eg rivaroxaban, ximelagatran, fondaparinux, dabigatran and apixaban).
88 Professor Baker has also acted as a peer reviewer for scientific abstracts submitted to the International Society on Thrombosis and Haemostasis Annual Congress.
89 Sandoz also relies on the evidence of a patent attorney, Mr Rodney Cruise, in support of its case in relation to best method and ascertainment. Mr Cruise made one affidavit in the proceeding on 30 September 2023.
90 Mr Cruise is a principal of the intellectual property firm, Phillips Ormonde Fitzpatrick, and the manager of the intellectual property research company, IP Organisers Pty Ltd. Mr Cruise’s evidence related to searches for patent and non-patent literature on the Chemical Abstracts database.
91 Prior to the hearing, Professors Roberts, Baker, Crowther and Evans prepared a second joint report for the Court in which they gave their responses to 10 questions posed by the parties, and indicated where they agreed or disagreed in their response (JER2).
92 The experts gave concurrent oral evidence in two groups at the hearing: Professors Polli and Roberts in the first session, and Professors Roberts, Baker, Crowther and Evans in the second session.
93 The person skilled in the art is not a reference to a specific person but is a legal construct drawn by reference to the available evidence. As French CJ explained in AstraZeneca AB v Apotex Pty Ltd (2015) 257 CLR 356 at [23] (AstraZeneca HC):
The notional person is not an avatar for expert witnesses whose testimony is accepted by the court. It is a pale shadow of a real person — a tool of analysis which guides the court in determining, by reference to expert and other evidence, whether an invention as claimed does not involve an inventive step.
94 The legal construct may not be a single person but may be a team of persons “whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those which are contained in the document to be construed”: General Tire & Rubber Co v Firestone Tyre & Rubber Co Ltd [1972] RPC 457 at 485 (per Sachs LJ, Buckley and Orr LJJ agreeing).
95 The person skilled in the art is likely to have a “practical interest in the subject matter … and may often work in the art in which the invention is connected”: KD Kanopy Australasia Pty Ltd v Insta Image Pty Ltd (2007) 711 IPR 615 at [16] (per Kiefel J). They are unimaginative and without inventive capacity.
96 In relation to the 226 Patent, Sandoz says that the skilled team includes a haematologist, a formulator and a pharmacologist.
97 Bayer considers the person skilled in the art possesses a mixture of attributes drawn from a team of skilled scientists, including a medicinal chemist, a clinician (being a haematologist), a formulator and a clinical pharmacologist.
98 The parties agreed that the question of construction for the 226 Patent is informed by the views of a formulator, and that Professors Roberts and Polli were appropriately qualified to give such evidence. However, Sandoz expressed some doubt regarding Professor Polli’s experience as a formulator and his expertise in pharmaceutics, pointing to the evidence he gave that he had only taught compounding and physical pharmacy, not tabletting, for the past 30 years.
99 The experts agreed that a drug company’s clinical trial program is very carefully constructed and will require the input of all team members, including pharmacologists and clinicians. The experts agreed that clinical trials involve a broad range of specialists for Phase I and II studies. A clinician will be involved in the conduct of clinical trials and will have input, with others, into their design, analysis and interpretation. Professor Baker considered that the input from a clinician would be particularly relevant in developing anticoagulant drugs given the complexity of the coagulation system. Other specialists that may be included in the team as required include pharmaceutical scientists, toxicologists, statisticians, analysts and database specialists, as well as experts in regulatory requirements as these studies will form a seminal component of a regulatory filing for the product. The roles of the various members of the skilled team and their involvement during the stages of the drug development process are discussed further below in section 10.1.14 “Stages of drug development”.
100 Professor Crowther noted that the term “clinician” is quite broad, which may help explain some of the differing views of the experts, particularly with respect to the role of a clinician in the early stages of drug discovery and development. Professors Evans and Crowther considered that the term applies to a health professional (a medical practitioner or specialist) providing day-to-day care (directly or indirectly) in a clinical setting such as a hospital. Professors Roberts and Baker took the view that the “clinician” was the person providing a clinical perspective, and that may be, for instance, in providing advice on an aspect of patient care or providing advice to other clinicians working directly with patients, as may arise for clinicians working in anatomical and chemical pathology, clinical pharmacology, public health and laboratory medicine. They gave as an example a haematologist specialising in laboratory medicine who will frequently have postgraduate fellowship and/or PhD qualifications and whose main responsibility is to supervise routine clinical, as well as translational, research laboratories. The haematologist’s duties may include laboratory and clinical research, pathology testing and/or patient care.
101 The experts agreed that there should be a clinical perspective in a drug development program. In this case, I consider that the “clinician” would be a practising haematologist with experience in treating patients with thromboembolic disorders and diseases, and who may also have some laboratory experience.
102 The clinician may be involved at the outset of a drug discovery program in terms of providing input as to desirable drug properties and, crucially, desirable improvements over current therapies (the existing standard of care). Clinicians are not generally involved in the drug discovery aspect of the program, or selecting the drug or defining the development program, but they play a critical part in the design, conduct and evaluation of the clinical studies themselves. A clinician’s involvement in design may be limited in the case of multi-centre clinical trials (usually Phase III) but there is usually at least one clinician who has a central role in leading the entire clinical trial program.
103 The roles of the various members of the skilled team and their involvement during the stages of the drug development process are discussed further below in section 10.1.14 “Stages of drug development”.
104 Sandoz alleges that the claims of the 226 Patent are invalid for lack of clarity, by reason of their use of the unfamiliar term “in hydrophilized form”. This term is not defined in the 226 Patent and neither Professor Polli nor Professor Roberts had heard of the term “hydrophilization” before.
105 The 226 Patent claims an earliest priority date of 27 November 2003 (First Priority Date).
106 The 226 Patent was amended on 18 May 2010 to introduce seven new consistory clauses on page 2a described as further aspects of the invention, to delete claim 21 and insert new claims 21 to 23. Bayer does not rely on the added consistory clauses to provide fair basis for the claimed invention.
107 The invention described in the 226 Patent is said to relate to a process for the preparation of a solid, orally administrable pharmaceutical composition, comprising 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-1,3-oxazolidin-5-yl}-methyl)-2-thiophenecarboxamide (compound I) in hydrophilized form, and “its” use [ie use of the pharmaceutical composition comprising compound I in hydrophilized form] for the prophylaxis and/or treatment of diseases.
108 Compound I is now known as rivaroxaban. It is a direct factor Xa inhibitor.
109 The specification of the 226 Patent explains that rivaroxaban is a low molecular weight, orally administrable inhibitor of blood clotting factor Xa, which can be employed for the prophylaxis and/or treatment of various thromboembolic diseases.
110 The 226 Patent does not otherwise describe rivaroxaban in detail, instead referring the reader to WO 919 and incorporating the disclosure of WO 919 by reference (page 1, lines 6–12). More will be said about WO 919 later, as Sandoz relies on WO 919 in its inventive step challenge to both the 226 and 613 Patents.
111 The specification of the 226 Patent explains that rivaroxaban has a relatively poor water solubility and that, therefore, difficulties with oral bioavailability and an increased biological variability of the absorption rate can result. The specification observes that various concepts have been described in the past to increase oral bioavailability. Three concepts for increasing bioavailability are discussed. It is in relation to the third, “a process for hydrophilization”, that a reference is made to two papers (the Lerk papers) (at page 1, line 17 to page 2, line 4):
Thus, solutions of active compounds are frequently used which can be filled, for example, into soft gelatine capsules. On account of the poor solubility of the active compound (I) in the solvents used for this purpose, this option is not applicable, however, in the present case, since, in the necessary dose strength, capsule sizes would result which are no longer swallowable.
An alternative process is the amorphization of the active compound. Here, the solution method proves problematical, since the active compound (I) is also poorly soluble in pharmaceutically acceptable solvents such as ethanol or acetone. Amorphization of the active compound by means of the fusion method is also disadvantageous because of the high melting point of the active compound (about 230°C), since an undesirably high proportion of breakdown components is formed during the preparation.
Furthermore, a process for the hydrophilization of hydrophobic active compounds as exemplified by hexobarbital and phenytoin has been described (Lerk, Lagas, Fell, Nauta, Journal of Pharmaceutical Sciences Vol. 67, No. 7, July 1978, 935 – 939: “Effect of Hydrophilization of Hydrophobic Drugs on Release Rate from Capsules” [Lerk 1978]; Lerk, Lagas, Lie-A-Huen, Broersma, Zuurman, Journal of Pharmaceutical Sciences Vol. 68, No. 5, May 1979, 634-638: “In vitro and In vivo Availability of Hydrophilized Phenytoin from Capsules” [Lerk 1979]). The active compound particles are blended here in a mixer with a methyl- or hydroxyethylcellulose solution with extensive avoidance of an agglomeration step and then dried. The active compound thus obtained is subsequently filled into hard gelatine capsules without further treatment.
112 The Lerk papers are described in more detail below in section 6.5.
113 Immediately following the discussion of the three concepts the specification states:
Surprisingly, it has now been found that a special treatment of the surface of the active compound (I) in the course of the moist granulation brings about improved absorption behaviour. The use of the active compound (I) in hydrophilized form in the preparation of solid, orally administrable pharmaceutical compositions leads to a significant increase in the bioavailability of the formulation thus obtained.
114 The present invention is said at page 2, line 10, to relate to a process for the preparation of a solid, orally administrable pharmaceutical composition comprising rivaroxaban in hydrophilized form, in which:
(a) first granules comprising the active rivaroxaban in hydrophilized form are prepared by moist granulation; and
(b) the granules are then converted into the pharmaceutical composition, if appropriate with addition of pharmaceutically suitable additives.
115 The specification states that the moist granulation in step (a) can be carried out in a mixer (mixer granulation) or in a fluidized bed (fluidized bed granulation). Fluidized bed granulation is said to be preferred.
116 From page 2a, line 24, some preferred embodiments are described by reference to the particular form of the active compound (crystalline or micronized), and the specification then moves on at page 3 to a more detailed description of the process of the invention. The granulating liquid “used according to the invention” is said to contain a solvent, a hydrophilic binding agent and, if appropriate, a wetting agent. The hydrophilic binding agent is, in this case, dispersed in the granulating liquid or preferably dissolved therein.
117 From page 3, line 4 onwards, the specification discusses solvents used for the granulating liquid, hydrophilic binding agents employed for the granulating liquid, hydrophilic polymers (including hydroxypropylmethylcellulose (HPMC)) and optionally present wetting agents.
118 The specification describes that the granules obtained in process step (a) are subsequently converted into the pharmaceutical composition according to the invention in process step (b). At page 4, lines 20–22, a number of options are described for step (b) of the process, including tabletting, filling into capsules and filling as sachets.
119 At page 5, lines 3–12, the specification discusses the solid, orally administrable pharmaceutical composition of the invention:
The present invention further relates to a solid, orally administrable pharmaceutical composition, comprising 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-l,3-oxazolidin-5-yl}- methyl)-2-thiophenecarboxamide (I) in hydrophilized form.
The solid, orally administrable pharmaceutical composition according to the invention by way of example and preferably comprises granules, hard gelatine capsules or sachets filled with granules, and tablets releasing the active compound (I) rapidly or in a modified (delayed) manner. Tablets are preferred, in particular tablets rapidly releasing the active compound (I). In the context of the present invention, rapid-release tablets are in particular those which, according to the USP release method using apparatus 2 (paddle), such as described in the experimental section in chapter 5.2.2., have a Q value (30 minutes) of 75%.
120 The specification further describes at page 5, lines 28–32, that the invention relates to the use of the pharmaceutical composition according to the invention for the prophylaxis and/or treatment of various thromboembolic diseases such as cardiac infarct, angina pectoris (including unstable angina), reocclusions and restenoses after an angioplasty or aortocoronary bypass, cerebral infarct, transitory ischemic attacks, peripheral arterial occlusive diseases, pulmonary embolisms or deep venous thromboses.
121 The invention is illustrated by means of preferred exemplary embodiments, each of which involves wet granulation (page 6, line 1 to page 11, line 8). The specification notes at page 6 that the examples are “preferred exemplary embodiments” and non-limiting.
(a) Example 1 describes the preparation of a tablet comprising compound I in hydrophilized form by a fluidized bed granulation process (a type of wet granulation).
(b) Example 2 describes the preparation of a tablet comprising compound I in hydrophilized form via a “high speed granulation process”.
(c) Examples 3 and 4, respectively, describe preparation of sachets, and preparation of gelatine capsules, each containing compound I in hydrophilized form. In both cases, the drug is prepared using a method similar to Example 1.
(d) Example 5 compares the tablet properties and improved bioavailability of formulations comprising hydrophilized compound I prepared by the fluidised bed process described in Example 1, with a tablet comprising micronized compound I in non-hydrophilized form, prepared by direct tabletting without granulation.
122 The specification ends with 23 claims. Claims 1 to 7 claim a process for the preparation of a solid, orally administrable pharmaceutical composition comprising compound I in hydrophilized form, using the two-step process described above at [114].
123 Claims 8 to 22 claim a pharmaceutical composition of rivaroxaban “in hydrophilized form”, and methods involving the use of such a composition. These claims are asserted against Sandoz.
124 Claim 23 is an omnibus claim which is not asserted.
125 The 613 Patent relates to a method of treatment of thromboembolic disorders by once daily administration of a rapid-release tablet comprising rivaroxaban.
126 The 613 Patent claims an earliest priority date of 31 January 2005 (Second Priority Date).
127 The background of the invention identifies the blood coagulation mechanism, the blood coagulation cascade, the fact that there are numerous factors and intrinsic and extrinsic pathways, as well as the fact that activated serine protease Xa cleaves prothrombin to thrombin, allowing a further step in the cascade to occur. Following a discussion of haemostasis, coagulation and thromboembolic disorders, there is then a discussion of the existing anticoagulants (heparin, low molecular weight heparin (LMWH) and warfarin) and their “often severe disadvantages” which make the effective treatment of thromboembolic disorders “very difficult and unsatisfactory”.
128 Heparin is said to be administered parenterally (intravenously or subcutaneously) and to have a non-selective action as it inhibits a plurality of factors of the blood coagulation cascade at the same time. Warfarin is described as a vitamin K antagonist with a slow onset of action (latency to the onset of action 36 to 48 hours). Owing to the high risk of bleeding and the narrow therapeutic index, time-consuming individual adjustment and monitoring of the patient are said to be required.
129 The specification then describes at page 2, lines 20–29, a novel therapeutic approach for the treatment of thromboembolic disorders which aims to inhibit factor Xa.
130 The specification sets out that oral application is the preferable route of administration, and that once daily oral application is preferred for convenience and compliance. However, it says this can be “difficult to achieve depending on the specific behaviour and properties of the drug substances, especially its plasma concentration half-life”.
131 The 613 Patent then states at page 3, lines 5–10:
When the drug substance is applied in no more than a therapeutically effective amount, which is usually preferred in order to minimize the exposure of the patient with that drug substance in order to avoid potential side effects, the drug must be given approximately every half live [sic] (see for example: Malcolm Rowland, Thomas N. Tozer, in “Clinical Pharmacokinetics, Concepts and Applications”, 3rd edition, Lea and Febiger, Philadelphia 1995, pp 83).
132 Following this, at page 3, line 17, the invention in the 613 Patent is identified:
Surprisingly, it has now been found in patients at frequent medication that once daily oral administration of a direct factor Xa inhibitor with a plasma concentration half lifetime of 10 hours or less demonstrated efficacy when compared to standard therapy and at the same time was as effective as after twice daily (bid) administration.
133 From page 3a, line 18, a preferred embodiment of the invention is said to be:
In a preferred embodiment, the present invention relates to [rivaroxaban], a low molecular weight, orally administrable direct inhibitor of blood clotting factor Xa (see WO-A 01/47919, whose disclosure is hereby included by way of reference) as the active ingredient.
134 On page 4, lines 6–9, the plasma concentration half-life of rivaroxaban is described:
For [rivaroxaban] a plasma concentration half life of 4-6 hours has been demonstrated at steady state in humans in a multiple dose escalation study (D. Kubitza et al, … [Blood Abstract 3004]).
135 Page 4 of the 613 Patent discusses a clinical study, set out in greater detail on pages 11 to 14, that compared rivaroxaban administered once and twice daily with LMWH in patients undergoing orthopaedic surgery.
136 There is then discussion of various other (unclaimed) compounds which are said to be preferred embodiments of the invention, each of which appear to have been the subject of a publication. These are other factor Xa inhibitors, in addition to rivaroxaban. They are not within the scope of the claims (the claims having been amended to narrow them to rivaroxaban, after the application for the 613 Patent was filed). For example:
AX-1826 [S. Takehana et al. Japanese Journal of Pharmacology 2000, 82 (Suppl. 1), 213P; T. Kayahara et al. Japanese Journal of Pharmacology 2000, 82 (Suppl. 1), 213P]
HMR-2906 [XVIIth Congress of the International Society for Thrombosis and Haemostasis, Washington D.C., USA, 14-21 Aug 1999; Generating greater value from our products and pipeline. Aventis SA Company Presentation, 05 Feb 2004]
137 On page 10 of the 613 Patent, tablets rapidly releasing the active compound are said to be preferred, in particular those with a particular release rates defined by reference to a specified USP release method. Then follows the passage relevant to the best method case, which Bayer seeks to amend:
Very particularly preferred are rapid-release tablets containing 5-Chloro-N-({(5S)-2 oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-1,3-oxazolidin-5-yl}-methyl)-2 thiophenecarboxamide as active ingredient. Preparation of such tablets is for example described in PCT/04/01289, whose disclosure is hereby included by way of reference.
138 The 613 Patent then discusses the clinical trial study (comprising 642 patients) that was referred to on page 4, which involved orally administering to different groups of patients different doses of rivaroxaban as rapid-release tablets — 2.5 mg, 5 mg, 10 mg, 20 mg and 30 mg twice daily, and 30 mg once a day. The data on page 14, lines 5–7, demonstrates the safety and efficacy of such once daily dosing of rivaroxaban.
139 The 613 Patent has four claims. Claims 1 and 3 are independent. Claim 1, a Swiss style claim to the use of rivaroxaban to manufacture a medicament, and claim 2, which claims the use of that medicament to treat thromboembolic disorders, were not pressed at the hearing.
140 Claim 3 provides:
A method for the treatment of a thromboembolic disorder in a patient said method comprising administration to the patient of a rapid-release tablet comprising 5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-l,3-oxazolidin-5-yl}methyl)-2-thiophenecarboxamide once daily for at least five consecutive days, and wherein the 5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-l,3-oxazolidin-5-yl}methyl)-2-thiophenecarboxamide has a plasma concentration half life of 10 hours or less when orally administered to a human patient.
141 Claim 4 is to the method of claim 3, wherein the thromboembolic disorder is selected from a specified group.
142 Claims 3 and 4 are asserted against Sandoz.
143 Subject to the validity of the 613 Patent, Sandoz Australia admits the direct and indirect infringement of each of the asserted claims of the 613 Patent.
144 The only issue for determination on infringement arises in respect to the 226 Patent. The term “in hydrophilized form” forms an essential part of the definition of the invention in all of the claims (either directly, or via dependency) of the 226 Patent. Bayer’s infringement case turns upon the construction of the term “in hydrophilized form” in each of the asserted claims of the 226 Patent.
145 Sandoz’s primary submission is that the term “in hydrophilized form” is unclear, such that the claims of the 226 Patent are unclear and are liable to be revoked on that basis. It follows that there can be no infringement. Sandoz submits that Bayer’s attempt to give meaning to “in hydrophilized form” by resorting to the Lerk papers — two prior art papers cited in the background section of the Patent, each dated more than 24 years before the asserted priority date — is both illegitimate, in that it is plainly wrong as a matter of construction, and unsuccessful, in that it fails to give any clear meaning to the term even if that impermissible process of construction is adopted.
146 If, contrary to that submission, the term “in hydrophilized form” can be given any meaning, Sandoz submits that it must refer to a composition that is produced using a wet granulation technique having regard to the disclosure of the 226 Patent. As Bayer concedes, Sandoz’s rivaroxaban products are not produced by such a process, and therefore it follows that there can be no infringement on that construction.
147 If, however, the term “in hydrophilized form” is given a broader meaning to encompass a composition made by a process not involving wet granulation, Sandoz submits the asserted claims of the 226 Patent are not fairly based, or not entitled to their claimed priority date, and are liable to be revoked as a result. It follows again that there can be no infringement. Sandoz also challenges the validity of the asserted claims on other bases, not related to the use of the term “in hydrophilized form”.
148 The parties came to an agreement on day one of the trial confining the scope of the 226 Patent infringement case as follows:
1. The Applicants/Cross-respondent (Sandoz) accept that, if, contrary to its submission, the Court finds that:
a. the asserted claims of the 226 Patent are valid;
b. the term "in hydrophilized form" in the asserted claims of the 226 Patent is clear; and
c. on its proper construction, that term encompasses a composition in which the surface of the hydrophobic active pharmaceutical ingredient (API) has been rendered hydrophilic as a result of close physical interactions between rivaroxaban and hydrophilic excipients without the use of any liquid,
then the Sandoz Rivaroxaban Products (as defined in the Amended Statement of Cross-claim) have the features of the asserted claims.
2. Bayer accepts that, if the term "in hydrophilized form" in the asserted claims of the 226 Patent, on its proper construction, only refers to a composition which has been made with the use of wet or moist granulation, or a liquid, then the Sandoz Rivaroxaban Products do not have the features of the asserted claims.
6.3 Principles of construction and clarity
149 A patent is construed through the eyes of the person skilled in the art: GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Limited v Generic Partners Pty Limited (2018) 131 IPR 384 at [106] (per Middleton, Nicholas and Burley JJ) (GSK FC).
150 The principles governing claim construction are well established and have been recently set out by Rares, Moshinsky and Burley JJ in Airco Fasteners Pty Ltd v Illinois Tool Works Inc [2023] FCAFC 7 at [48], citing Jupiters Ltd v Neurizon Pty Ltd (2005) 65 IPR 86 at [67] (per Hill, Finn and Gyles JJ):
(i) the proper construction of a specification is a matter of law: Décor Corp Pty Ltd v Dart Industries Inc (1988) 13 IPR 385 at 400;
(ii) a patent specification should be given a purposive, not a purely literal, construction: Flexible Steel Lacing Company v Beltreco Ltd (2000) 49 IPR 331 at [81]; and it is not to be read in the abstract but is to be construed in the light of the common general knowledge and the art before the priority date: Kimberley-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [24];
(iii) the words used in a specification are to be given the meaning which the normal person skilled in the art would attach to them, having regard to his or her own general knowledge and to what is disclosed in the body of the specification: Décor Corp Pty Ltd at 391;
(iv) while the claims are to be construed in the context of the specification as a whole, it is not legitimate to narrow or expand the boundaries of monopoly as fixed by the words of a claim by adding to those words glosses drawn from other parts of the specification, although terms in the claim which are unclear may be defined by reference to the body of the specification: Kimberley-Clark v Arico at [15]; Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610; Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 at 478; the body of a specification cannot be used to change a clear claim for one subject matter into a claim for another and different subject matter: Electric & Musical Industries Ltd v Lissen Ltd [1938] 56 RPC 23 at 39;
(v) experts can give evidence on the meaning which those skilled in the art would give to technical or scientific terms and phrases and on unusual or special meanings to be given by skilled addressees to words which might otherwise bear their ordinary meaning: Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479 at 485-486; the Court is to place itself in the position of some person acquainted with the surrounding circumstances as to the state of the art and manufacture at the time (Kimberley-Clark v Arico at [24]); and
(vi) it is for the Court, not for any witness however expert, to construe the specification; Sartas No 1 Pty Ltd, at 485–486.
151 To those, I would also add:
(a) when construing claims, “a generous measure of common sense should be used”: Product Management Group Pty Ltd v Blue Gentian LLC (2015) 116 IPR 54 at [36] (per Kenny, Nicholas and Beach JJ); and
(b) a construction that would lead to an absurd result is to be avoided: NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1993) 26 IPR 513 at 559 (per Northrop, Lockhart and Burchett JJ).
152 It is impermissible for either the court or the witnesses to approach the issues of construction with any regard to the alleged infringing articles: Fresenius Medical Care Australia Pty Ltd v Gambro Pty Ltd (2005) 67 IPR 230 at [95] (per Wilcox, Branson and Bennett JJ). As observed by Heerey J in Welcome Real-Time SA v Catuity Inc (2001) 51 IPR 327 at [21]:
All that needs be added is the perhaps trite observation that the alleged infringement is to be ignored when construing the patent. Although the forensic contest will throw up the particular construction issues to be resolved, a patent must, as the saying goes, be construed as if the infringer had never been born.
153 Although it is preferred that claims should be construed in a manner that avoids redundancy, Full Courts of this Court have indicated that where the plain meaning of a claim has the effect that one or more dependent claims are redundant, then the consequence of redundancy would not drive the construction of the claim itself: Nichia Corporation v Arrow Electronics Australia Pty Ltd [2019] FCAFC 2 at [41] (per Jagot J, Besanko J agreeing at [1] and Nicholas J agreeing at [126]); Davies v Lazer Safe Pty Ltd [2019] FCAFC 65 at [65] (per Greenwood, White and Burley JJ).
154 Claims should be construed in the context of the specification, even where there is no ambiguity: Product Management at [37] (per Kenny, Nicholas and Beach JJ), cited in Esco Corporation v Ronneby Road Pty Ltd (2018) 131 IPR 1 at [144] (per Greenwood, Rares and Moshinsky JJ). Reference is made to the specification to understand the background and context of the claims and to ascertain the meaning of technical terms: Minnesota Mining and Manufacturing Co v Beiersdorf (Aust) Ltd (1980) 144 CLR 253 at 271–287 (per Aickin J). Terms in the claim which are unclear may be clarified or defined by reference to the body of the specification: Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [15] (per Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ). However, the plain and unambiguous meaning of a claim cannot be varied or qualified by reference to the body of the specification: Kimberly-Clark at [15].
155 The requirement of clarity is also well-established. Relevantly, s 40(3) of the Patents Act 1990 (Cth) requires that the claims of a patent specification be clear. In Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610, Dixon CJ, Kitto and Windeyer JJ said:
It is, however, fitting that we remind ourselves of the criterion to be applied when it is said that a specification is ambiguous. For, as the Chief Justice pointed out in Martin v Scribal (1954) 92 CLR 17, at p 59 … we are not construing a written instrument operating inter partes, but a public instrument which must, if it is to be valid, define a monopoly in such a way that it is not reasonably capable of being misunderstood. Nevertheless, it is to be remembered that any purely verbal or grammatical question that can be resolved according to ordinary rules for the construction of written documents, does not, once it has been resolved, leave uncertain the ambit of the monopoly claimed (see Kauzal v Lee (1936) 58 CLR 670, at p 685).
156 Sandoz submits that given the unfamiliar and ambiguous nature of the term “in hydrophilized form”, this is not a case of a “purely verbal or grammatical question that can be resolved according to ordinary rules for the construction of written documents”.
157 As Bayer submits that it is legitimate to have regard to the disclosure in the Lerk papers in order to properly understand the term “in hydrophilized form”, it is also necessary to refer to the principles that bear upon the extent to which, if at all, a document cited in a patent specification may be treated as part of its disclosure for the purpose of resolving any issue of clarity or construction.
158 The specification is to be construed in the light of the common general knowledge before the priority date. However, the common general knowledge is limited to information which had entered the common stock of knowledge and was well known to, and generally accepted by, the bulk of those in the field at that time. Sandoz submits that documents, such as the Lerk papers, which do not form part of the common general knowledge cannot be taken into account via that route.
159 For a cited document which did not form part of the common general knowledge, it is necessary to have regard to the nature and purpose of the reference to the document in the specification in question. A cited document may, for example, be said to be incorporated by reference into the specification. In such a case, it may be legitimate to have regard to its contents. However, the authorities demonstrate that, even in such cases, it remains necessary to have regard to the nature and purpose of the reference to the document, and that it may be legitimate to use the contents of the document for some purposes but not others: Idenix Pharmaceuticals LLC v Gilead Sciences Pty Ltd (2017) 134 IPR 1 at [165] (per Nicholas, Beach and Burley JJ).
160 Sandoz submits that where (as in the 226 Patent) the cited document is not incorporated by reference, the prima facie position is that its contents cannot be referred to, or treated as part of the disclosure of the specification. Sandoz accepts that it is not strictly necessary to use the incantation of “incorporation by reference” in order to ensure that a document, or part of its contents, which is cited in the specification is incorporated into, or forms part of, the disclosure of the specification. However, regard must be had to the nature and purpose of the reference in considering whether or not the patentee intends to incorporate some or all of its contents as part of the disclosure. The citation of a document as prior art in the background section of the specification is unlikely to entail such an indication. Further, where the material details of the prior art process or the product are themselves described in the specification, Sandoz submits that it is likely that the reference is to be understood as providing background only, or further information, rather than evincing an intention to incorporate additional content into the disclosure of the specification.
161 Bayer submits that Sandoz’s approach to the Lerk papers is erroneous and should be rejected. Bayer submits that the correct approach is that, where the patentee demonstrates an intention that words used in the specification have a particular meaning, effect should be given to such a “dictionary”: see Austal Ships Sales Pty Ltd v Stena Rederi Aktiebolag (2008) 77 IPR 229 at [13] (per Heerey, Finn and Dowsett JJ) quoting Flexible Steel Lacing Co v Beltreco Ltd (2000) 49 IPR 331 at [76]–[77] (per Hely J).
162 Bayer submits that the correct approach to construction by reference to a document referred to in a specification is that described by Greenwood J in Uniline Australia Ltd v SBriggs Pty Ltd (2009) 81 IPR 42 at [73]–[75].
6.4 Meaning of “in hydrophilized form”
163 The term “in hydrophilized form” does not have an ordinary meaning. Neither of the very experienced formulation experts were familiar with the term “in hydrophilized form”. Both experts agreed that a formulator would probably not have heard the term before reading the 226 Patent.
164 As Sandoz points out, the Lerk papers are not said to be incorporated by reference, nor do they form part of the common general knowledge. That being so, the correct approach according to Sandoz is to not read them. Sandoz submits that if there is any permissible use to be made of the Lerk papers in construing the 226 Patent, which Sandoz disputes, that use is very limited. At best, they might give some context to the particular discussion of the Lerk papers by the authors of the 226 Patent: see Uniline at [75] (per Greenwood J). They are not put forward as, and do not stand as, a means of defining the term “in hydrophilized form”, which is unclear to the skilled reader. They are not referred to for that purpose, or as part of a definition of that term.
165 Sandoz submits that the construction of claims that use an unfamiliar term to describe an essential characteristic of the invention is not to be determined by “plumbing the depths” of the disclosure of papers cited as prior art in the background section of the patent and then adding common general knowledge. Relying on Welch Perrin at 610 (per Dixon CJ, Kitto and Windeyer JJ), Sandoz submits that a patent is a public instrument “which must, if it is to be valid, define a monopoly in such a way that it is not reasonably capable of being misunderstood”. Sandoz submits that the Lerk papers do not remedy the lack of definition in the 226 Patent.
166 If, contrary to its primary submission, the person skilled in the art would have regard to the Lerk papers, Sandoz submits that the term is still unclear as the Lerk papers do not purport to define the term and do not provide any clear disclosure of, or the boundaries of the concept of, “in hydrophilized form”.
167 If, contrary to those submissions, it is possible to discern any meaning of the term “in hydrophilized form”, Sandoz submits that it is not the all-encompassing meaning contended for by Bayer. The only process disclosed in the 226 Patent for preparing a composition comprising rivaroxaban “in hydrophilized form” involves wet granulation with a hydrophilic binder such as HPMC, a standard technique for formulating solid oral dosage forms before the priority date, and an excipient commonly used in wet granulation. Similarly, Sandoz submits that the only process disclosed in the Lerk papers for “hydrophilizing” a hydrophobic active compound involves blending particles of the compound in a mixer with a solution of a hydrophilic excipient. In both cases, the production of a compound “in hydrophilized form” requires the use of a liquid with a hydrophilic excipient. On this construction, Sandoz submits that there is no infringement because the Sandoz rivaroxaban products are not made using wet granulation or a liquid.
168 Sandoz submits that, following the joint session, there was no consensus between the experts on what was taught by the Lerk papers, rather the entire discussion tended to underscore the ambiguity in the term.
169 Bayer submits, employing the language in Uniline, that the person skilled in the art would read the Lerk papers to give context to, and to understand, the words “hydrophilization” and “in hydrophilized form” in light of the particular discussion in the 226 Patent of “hydrophilization” and “a process for the hydrophilization of hydrophobic active compounds” the subject of the Lerk papers.
170 Bayer contends that it does not seek to refer to the Lerk papers beyond that permitted context and understanding. In that context, Bayer submits that the person skilled in the art would read and understand the words “in hydrophilized form” in the context of the whole of the specification to describe the surface of a hydrophobic drug particle which has been rendered hydrophilic as a result of close physical interactions with a hydrophilic excipient.
171 Bayer contends that, consistent with the evidence of Professors Roberts and Polli, the Lerk papers would be understood by the person skilled in the art to disclose a principle for improving dissolution beyond the precise experiments conducted in those papers. Having read the Lerk papers, Bayer submits that the person skilled in the art would understand that “in hydrophilized form”, which requires achieving close physical interactions between a hydrophobic drug particle and a hydrophilic excipient, might be achieved in a number of different ways, including intensive mixing without the use of a liquid in a confined space or a lower energy process with water.
172 As I explain below, I consider that the person skilled in the art would have recourse to the Lerk papers. Thus, it is appropriate to briefly outline the manner in which the Lerk papers arise in the specification and their contents.
173 The reference to the Lerk papers arises in the section of the 226 Patent which discusses three concepts to increase oral bioavailability which have been discussed in the past.
174 The first involves solutions of active compounds being filled into gelatine capsules. That option is said to be unavailable in the present case as the capsule size required in order to provide the necessary dose would not be swallowable.
175 Second is a process of amorphization of the active compound which Professor Polli described as being a process wherein a crystalline drug is converted to a non-crystalline, amorphous drug form in the hope that the amorphous form will have higher solubility. The “solution method” of amorphization is said by the specification to be problematical as the active compound, as well as being relatively poorly soluble in water, is also poorly soluble in pharmaceutically acceptable solvents such as ethanol or acetone. The “fusion” method is also said to be disadvantageous due to the high melting point of the active compound, which would lead to an undesirably high proportion of breakdown components forming during the preparation.
176 Third is “a process for the hydrophilization of hydrophobic active compounds as exemplified by hexobarbital and phenytoin [which] has been described [in the Lerk papers]”. The specification describes how the active compound particles are blended in a mixer with methyl- or hydroxyethylcellulose solution with extensive avoidance of an agglomeration step and then dried. The active compound thus obtained is subsequently filled into hard gelatine capsules without further treatment.
177 Immediately following the discussion of the three past concepts for increasing oral bioavailability is the “surprisingly, it has now been found” paragraph (extracted above at [113]). This paragraph refers to a “special treatment of the surface of the active compound (I) in the course of the moist granulation [which] brings about improved absorption behaviour”. That special treatment results in the active compound (I) being “in hydrophilized form” and leads to a “significant increase in the bioavailability of the formulation thus obtained”.
178 Before considering the Lerk papers, it is useful to summarise the state of the common general knowledge about wet and dry granulation as at the First Priority Date. As at that date, there were two commonly used production methods for the manufacture of a solid oral dosage form, being:
(a) granulation (encompassing wet and dry granulation); and
(b) direct compression.
179 Granulation involves the combination of powder particles with a binder to form granules. The rationale for granulation includes: to increase the bulk density of the powder, to improve the flowability of the powder, to improve mixing homogeneity, and to affect the dissolution process for hydrophobic poorly soluble particles. The granules are compressed into a tablet or filled into a capsule.
180 Wet granulation involves the combination of drug particles with a binder and other excipients into granules in the presence of a granulating fluid, usually water. The granulation liquid may be used alone, or as a solvent containing a binding agent. Organic solvents, rather than water, may be used if the drug is water sensitive or rapid drying is required. At the First Priority Date, wet granulation was considered an effective means, in terms of production and cost, to prepare good-quality granulation.
181 Wet-granulation techniques were widely used in the pharmaceutical industry to make tablets to achieve a number of objectives, including homogeneity, improved flowability, compactibility and stability, and to cause the production of hydrophilic surfaces. Wet granulation is not suitable for drugs that are sensitive to moisture or heat.
182 Dry granulation is a similar process, but carried out in a dry environment in which no liquid is used (eg using a dry binder). Dry granulation is generally suitable for drugs that are sensitive to moisture and heat. It involves less processing steps than wet granulation but generally does not result in the advantages of wet granulation in terms of improved flowability and compactibility. Therefore, dry granulation can generally only be used where the tablet ingredients have sufficient inherent binding or cohesive properties.
183 The first thing to note is that, at the First Priority Date, the Lerk papers were over 24 years old. They are clearly not common general knowledge, nor had the term “hydrophilized” been embraced by formulators after the publication of the Lerk papers as neither expert had come across the term “hydrophilized” prior to reading the 226 Patent in 2022, over 40 years after publication of the Lerk papers.
184 Lerk 1978 is entitled “Effect of Hydrophilization of Hydrophobic Drugs on Release Rates from Capsules”.
185 The abstract states that the release of poorly soluble hydrophobic drugs from capsules can be improved significantly by the creation of a hydrophilic surface by intensive mixing of the hydrophobic drug with a small amount of a solution of a hydrophilic excipient. The abstract continues:
The data presented indicate that the hydrophilic material is mechanically distributed over the hydrophobic surface. The creation of hydrophilic capillaries in a capsule or tablet allowed the rapid penetration of the dissolution fluid, resulting in a dispersion of well-wetted particles, so that the maximum surface area of the powder was exposed to the dissolution medium.
186 The last sentence of the abstract concludes that “hydrophilization of hydrophobic drugs has the important benefit that the release rate from capsules is independent of the surface tension of the dissolution medium”. This is to be contrasted with non-hydrophilized, hydrophobic drug samples in which the release rate varied with the surface tension properties of the dissolution medium.
187 The body of the article begins by discussing several methods for increasing the dissolution rate of poorly water-soluble drugs, and the problems associated with those methods. The article continues:
In an ideal situation, the drug would be released from the dosage form as discrete, well-wetted particles, so that the maximum surface area afforded by the powder would be exposed to the dissolution medium. For hydrophobic materials, such a situation may be difficult; but it may be possible to achieve if the surface properties of the material are changed from hydrophobic into hydrophilic by coating the hydrophobic particles with a hydrophilic material. Such a coating should aid in the penetration of fluid and the wetting of individual particles but should not unduly retard the dissolution rate of the drug.
188 Lerk 1978 states that the coating of particles may be achieved by fluidisation techniques (endnoting four articles) but notes that problems may arise if the powder is cohesive. The authors note that, in their study, a technique described by de Jong (a further endnoted article) was used, in which a powder (the hydrophobic API hexobarbital) was intensively mixed with a small amount of hydrophilic binder in solution in a high-speed mixer so that the hexobarbital was rendered hydrophilic. The rigorous mixing is said to inhibit agglomeration, resulting in a fine free-flowing powder. The authors note that, although this method was originally developed for microgranule production, it is claimed that the powder is coated with the binding agent so hydrophobic substances can be rendered hydrophilic. The authors state “[t]his study applied this technique to increase the dissolution rate of a hydrophobic, relatively insoluble drug from hard gelatin capsules”.
189 The materials section records that the mean particle sizes of the milled and classified hexobarbital, the methylcellulose and milled HPMC were 17, 30 and 32 µm respectively.
190 The methods section also records that dispersion of the binder over the drug surface was carried out in a small high-speed mixer (two or four minutes at ~9000 rpm). Dry-mixed powders of hexobarbital and binder were prepared using a tumbler mixer running at 90 rpm for 30 minutes.
191 Lerk 1978 compared the dissolution rates of the different forms of hexobarbital: intensively mixed (treated with 1%, 3% or 5% binder solution), physically mixed (dry mixed) and pure, each loosely packed into hard gelatin capsules.
192 The results and discussion section reported:
(a) The physical mixes exhibited release rates of the same order of magnitude as the pure drug.
(b) The results indicate that the degree of distribution of a hydrophilic binder over the surface of a hydrophobic drug affects the release rate from capsules.
(c) The input energy of the mixer can promote spreading of the liquid. The success of this operation depends, first, on factors affecting the spreading such as mixer efficiency, contact angle and viscosity, and second, whether the film is stable when formed.
(d) High-speed mixing of hexobarbital with methylcellulose or HPMC solutions resulted in decreased contact angles.
(e) The release rate of a strongly hydrophobic drug like hexobarbital from capsules can be increased considerably by rendering the hydrophobic surface of the drug hydrophilic.
193 Lerk 1978 concludes with the statement that the results show the release rate of the treated drug to be independent of the surface tension of the dissolution medium. Because there are differences in surface tension of human gastric juice, the realisation of a release rate from capsules that is independent of the surface tension of the medium is of practical importance, especially where absorption is dissolution rate limited.
194 Lerk 1979 is entitled “Availability of Hydrophilized Phenytoin from Capsules”.
195 It explains that the purpose of the study’s investigation was to examine the penetration rate effect on the availability of poorly soluble, hydrophobic drugs from capsules. To this end, the effect of phenytoin hydrophilization on the liquid penetration rate into prepared plugs, on the disintegration time, on the in vitro release rate and on in vivo absorption in seven healthy volunteers was studied.
196 The abstract of Lerk 1979 states that “[h]ydrophilization was performed by intensive mixing of the hydrophobic drug [phenytoin] with a small amount of methylcellulose solution.” The intensive mixing was carried out in a small high-speed mixer which was run for six minutes at ~9000 rpm. A bioavailability study in seven healthy volunteers showed immediate absorption of the treated (hydrophilized) drug, but a one hour absorption lag time for the pure, untreated drug.
197 The authors of Lerk 1979 note that when a disintegratable tablet or capsule is administered orally to humans, gastric fluid must penetrate the porous mass before disintegration and absorption can occur. Any factor influencing the penetration rate may influence the absorption rate. The authors note that the liquid penetration rate into a porous mass is given by the “Washburn equation”.
198 Citing Lerk 1978, the authors note that the effect on the capsule release rate of a surface hydrophilic binder distribution over a hydrophobic drug had been the subject of recent study. The increased hydrophilized hexobarbital release rate (observed in Lerk 1978) was said to be independent of the dissolution medium wetting potency, in contrast to the untreated drug.
199 Professor Roberts’ evidence was that he came to the case not knowing what “hydrophilized” meant. Professor Polli was also not familiar with the term “in hydrophilized form” at the First Priority Date.
200 Professor Roberts’ instinctive reaction after reading the term in the 226 Patent was that it was reminiscent of “lyophilized” and “lyophilisation” — a freeze drying process in which water is removed from a product.
201 Professor Roberts agreed that in the normal course of reading the 226 Patent at the priority date he would, upon reading the reference to the Lerk papers, get copies of the papers and read them with the objective of understanding what the patentee meant by the term “hydrophilized”. Contrary to his usual practice, and over his specific request to see the papers, Professor Roberts was instructed to provide a meaning for “in hydrophilized form” without recourse to the Lerk papers. He arrived at two possible definitions in the context of the 226 Patent and in the absence of the Lerk papers:
(a) a hydrophobic drug which has been subjected to a rudimentary wet granulation process which is mixed with a solution binder and then dried; or
(b) a micronized drug that has been suspended in a solution with a binder and surfactant and added as granulating liquid to a powder mixture of at least a diluent and a disintegrant in the course of wet granulation.
202 Professor Polli’s evidence was that, in accordance with his usual practice, when he encountered the reference to the Lerk papers in the context of a term with which he was not familiar, he requested copies of the Lerk papers in order to understand the exemplified “process for hydrophilization” in the 226 Patent.
203 In JER1, both experts considered the Lerk papers “highly relevant” to the meaning of “in hydrophilized form” and agreed that a formulator would carefully read the two articles with the objective of understanding the meaning of the term used by the patentee.
204 Professor Polli had regard to the Lerk papers which he said described a process for the hydrophilization of hydrophobic active compounds. Professor Polli’s evidence in his first affidavit was that, having read the specification of the 226 Patent and the Lerk papers, he understood the term “in hydrophilized form” as used in the 226 Patent to describe that the surface of the hydrophobic API had been rendered hydrophilic as a result of close physical interactions with a hydrophilic excipient. He understood that the “hydrophilized form” referred to in the 226 Patent might be achieved in a number of different ways at the First Priority Date. The way in which the Lerk papers described that an API could be rendered “in hydrophilized form” was by high-speed mixing which, as Professor Polli described, achieves intensive mixing of, and consequently close physical interactions between, the API and the hydrophobic excipients. Professor Polli was aware of different processes utilizing various equipment to achieve high-speed or intensive mixing at the First Priority Date.
205 Before he was given access to the Lerk papers, Professor Roberts was asked to comment on Professor Polli’s understanding of “in hydrophilized form”. Not unsurprisingly, Professor Roberts focussed on the disclosure in the 226 Patent to take issue with Professor Polli’s opinion that “in hydrophilized form” might be achieved in a number of different ways. In particular, Professor Roberts noted the 226 Patent only disclosed “hydrophilization” in the course of moist or wet granulation. Any other method by which “hydrophilization” might be achieved was, in his view, inconsistent with the preparation of example 1. Professor Roberts’ evidence was that he did not understand from the 226 Patent that intensive or high-speed mixing was required.
206 After his first affidavit was finalised and he had commented on Professor Polli’s understanding (reached with the benefit of reading the Lerk papers), Professor Roberts was given access to the Lerk papers, and asked whether he agreed with Professor Polli’s understanding having regard to the Lerk papers and the 226 Patent.
207 Professor Roberts maintained his disagreement with Professor Polli’s understanding – he did not understand the 226 Patent to be claiming a concept described in the Lerk papers. He gave three reasons for his disagreement:
(a) Professor Polli’s definition omits the requirement that the drug is mixed with a binder in the presence of a solution in circumstances where the Lerk papers do not provide a basis to do so.
(b) The definition appears to be limited to surface interactions or surface effects which Professor Roberts considered was not involved in the experiments described in the Lerk papers.
(c) The definition appears to extend to all surface interactions that render the surface of the hydrophobic API hydrophilic. Professor Roberts considered this statement to be contrary to the disclosure in the 226 Patent.
208 In his second affidavit, Professor Roberts observed that the Lerk papers did not describe any examples of “hydrophilization” in which the hydrophilic excipient was not “in solution”. Professor Roberts considered that, in some cases, the cellulose polymers would technically be present as a colloidal solution or suspension, so he preferred to say “in the presence of a liquid”.
209 Professor Roberts considered that the process described in the Lerk papers was a “rudimentary wet granulation process”. He understood the reference to “hardly any granulation” occurring during preparation in Lerk 1979 to refer to agglomeration – the combination of two or more drug particles, not the creation of a mass larger than the drug alone.
210 Professor Roberts observed that he did not expect high-speed mixing of dry powders to achieve the same reduction in surface tension and contact angle of the drug-particle interface as mixing in the presence of a liquid. Professor Roberts’ evidence was that he did not understand from Lerk 1978 that “hydrophilization” could take place with intensive, high-speed dry mixing in the absence of a solution.
211 In JER1, Professor Polli disagreed with Professor Roberts’ definition of “hydrophilization” as he did not agree that a formulator would consider a definition which did not consider the main findings from the two Lerk articles and which focussed on a process. He disagreed that “hydrophilization” necessarily involves wet granulation as he considered that Lerk 1978 purposely avoids agglomeration (eg avoids an agglomeration step; avoids making granules). The powder in Lerk is barely wet and not indicative of wet granulation. The main reasons to employ wet granulation concern manufacturing (eg flow, dusting) which are not issues in the Lerk papers. Also, in Professor Polli’s opinion, a formulator would see that the 226 Patent, in summarising the Lerk papers, emphasises the avoidance of an agglomeration step.
212 In JER1, the experts disagreed in relation to the information that a formulator might derive from the Lerk papers read in the context of common general knowledge. Professor Polli considered that a formulator would understand from Lerk 1978 that the improved dissolution from the hydrophilized drug was achieved via intensive mixing of drug with a hydrophilic excipient, modifying the drug surface:
A formulator would take special note of Figure 2 in Lerk 1978, which concerns release rate from capsule, or more specifically percent drug dissolved versus time (or dissolution profile or dissolution curve) from capsules. Figure 2 plots dissolution curves of drug hexobarbital that were untreated, treated, and mixed with methylcellulose. Methylcellulose (and hydroxyethylcellulose) are denoted to be binders. From methods, untreated is pure drug packed into capsules. Treated is hydrophilized drug (using term in 226 patent) packed into capsules. Mixed with methylcellulose is “dry mixed powders of hexobarbital and binder” packed into capsules, respectively. Mixed is also sometimes denoted as physically mixed or physical mixes.
Figure 2 shows treated dissolution profile was significantly higher than each untreated and physically mixed. Dissolution profiles of physical mixes were about the same (or lower) than untreated (i.e. pure drug). That is, the composition of drug and the binder methylcellulose alone itself (physically mixed, but without intensive mixing) did not enhance dissolution at all.
…
Hydrophilized drug was produced using a high-speed mixer (about 9000rpm). Physical mixes were produced using a tumbler mixer (90rpm), which was not intensive mixing. In describing the low dissolution of the physical mixes, the manuscript states that results indicate that the degree of distribution of a hydrophilic binder over the surface of a hydrophobic drug affects the release rate from capsules. The manuscript further indicates that the input of energy from the mixer can promote spreading of the liquid (i.e. promote wettability of the powder, as assessed by contact angle measurement). Contact angle measurement involves the surface of the powder. I disagree (with Roberts 1, 48) that the most significant difference between the treated drug and physical mix is the presence of water during mixing.
213 In contrast, Professor Roberts considered that the most significant difference between the “treated drug” and the “physical mix” in Lerk 1978 was the presence of water during mixing (rather than the 100 fold difference in mixing speed). He noted that Lerk 1978 did not describe any examples of “hydrophilization” in which the hydrophilic excipient was not in the form of a solution, and indicated that where the drug is dry mixed with excipients the results are worse than both the wet mixed samples, and the drug alone without any binder.
214 In the joint session, Professor Roberts referred to the observation in Lerk 1978 that the release rate of the treated hexobarbitol was independent of the surface tension of the dissolution medium. He mused that the results in figure 12 of Lerk 1978 for the dissolution rate in a dissolution medium with varying concentrations of polysorbate 80 demonstrated that the increased dissolution rate involved more than just coating of the surface of the hydrophobic active compound, possibly wicking via capillaries. He referred to the discussion of the Washburn equation in Lerk 1979 to support his theory.
215 It was suggested to Professor Polli in cross-examination that making a hydrophilized form of a hydrophobic active compound via direct compression after dry granulation with a hydrophilic excipient only occurred to him during oral evidence, as he had not expressly mentioned dry granulation in his affidavit. That suggestion was rejected by Professor Polli. His written evidence was that a range of techniques could be used to render a hydrophobic API surface hydrophilic, and he did not consider that he needed to expressly discuss dry compression or dry granulation as one of the range of available techniques in that context.
216 In the joint session, Professors Polli and Roberts agreed the following:
(a) that the fluid penetration in the Lerk papers is being promoted by the API being coated with hydrophilic excipient;
(b) the process described in the Lerk papers to render the surface of the hydrophobic active hydrophilic used:
(i) intensive mixing (of active compound and solution); and
(ii) a very small amount of liquid;
(c) there were no examples in the Lerk papers where the hydrophilic excipient is not in the form of a solution;
(d) the treated drug in the Lerk papers had a better dissolution rate than the untreated drug; and
(e) other ways to reduce particle size via high-speed mixing existed at the First Priority Date, such as a hammer mill.
217 Ultimately, the experts reached agreement that in a “hydrophilized form”, the hydrophobic surface of an active compound is rendered hydrophilic by being at least partially coated with a hydrophilic excipient.
218 However, the experts disagreed as to whether the process via which the result of hydrophilization of the hydrophobic active was achieved was limited to the process described in the Lerk papers (ie with some, but minimal, liquid or solvent) or by a variety of methods (including those without liquid or solvent), of which the one described in the Lerk papers was just one example.
219 The only construction issue on the 226 Patent is the meaning of the term “in hydrophilized form”. Prior to this proceeding, neither expert had encountered the term.
220 The evidence of both experts was that they would in their normal practice refer to the Lerk papers when seeking to understand what was meant by “hydrophilized”. Professor Roberts’ definitions reached in the absence of the Lerk papers can be put to one side.
221 It is true that the Lerk papers are not common general knowledge or expressly incorporated by reference, but the patentee has made reference to them for a reason.
222 In Uniline, the specification of the patent in suit recited that the problem of “stairstepping” and “excessive torsional loading” was discussed in another patent specification, AP825. The experts in that case were not familiar with the term “stairstepping” or the AP825 patent. Both experts obtained a copy of AP825 and had regard to its contents to assist them in understanding the description of the prior art disclosed in AP825, and the term “stairstepping”. In that context, Greenwood J stated at [75]:
The correct approach to the use of AP825 for the purposes of construction, is not to read the document and substitute into the patent in suit the content of the prior art disclosures made by the document. The prior patent may be read by a person skilled in the art of the patent in suit to give context to and better understand the particular discussion selected by the author of the patent in suit in the specification, concerning the device or method the subject of the prior patent.
(Emphasis in original.)
223 In Idenix, Nicholas, Beach and Burley JJ observed at [164] that the Emory patent referred to in the patent in suit was “merely one of more than 200 documents referred to in the Idenix patent”. They further noted that it was referred to in the background section for a purpose unrelated to the synthesis of the compound under consideration and, contrary to the evidence in the present case, it was apparent from the evidence that none of the experts had regard to the Emory patent. Their Honours commented that the boilerplate-provision to the effect that all publications mentioned in the specification were incorporated by reference was not of assistance. The Full Court at [166] rejected Idenix’s attempt to finesse the relevant disclosure by reference to the Emory patent.
224 In Becton Dickinson Pty Ltd v B Braun Melsungen AG [2018] FCA 1692, the parent patent of the patent in suit referred to a “device of this kind” said to be known from a European patent. The parent patent was incorporated by reference into each of the three patents in suit. All of the patents in suit expressly referred to the European patent. As in this case, the European patent was neither incorporated by reference (in that case into the parent) nor shown to be common general knowledge. At [79], Nicholas J considered that it was not permissible to have regard to the European patent for the purpose of construing the claims of the patents in suit.
225 A process for the “hydrophilization” of hydrophobic active compounds is first mentioned as the third of three processes discussed in a passage detailing concepts “described in the past” to increase oral bioavailability. Hydrophilization follows after “amorphization”, which is explained above at [175].
226 Like the process of amorphization produces an active compound in an amorphous form, hydrophilization would be understood by the person skilled in the art as a process which results in a drug in hydrophilized form.
227 The question is: what does the person skilled in the art understand by the term “in hydrophilized form”?
228 The specification of the 226 Patent states from page 1, line 28:
a process for the hydrophilization of hydrophobic active compounds as exemplified by hexobarbitol and phenytoin has been described [in the Lerk papers]. The active compound particles are blended here in a mixer with a methyl- or hydroxyethylcellulose solution with extensive avoidance of an agglomeration step and then dried. The active compound thus obtained is subsequently filled into hard gelatine capsules without further treatment.
229 From the reference to “hydrophilization” in this passage, which occurs in the section discussing methods to increase oral bioavailability of active compounds, it is apparent that hydrophilization is a process performed on hydrophobic active compounds (such as hexobarbitol) in order to increase their oral bioavailability.
230 I consider that the approach outlined by Greenwood J at [75] in Uniline is appropriate. The person skilled in the art may read the prior art Lerk papers to give context to, and better understand, the discussion in the 226 Patent, commencing at page 1, line 28, and the claims. This accords with the evidence as to what the experts would do in the normal course of reading the 226 Patent. Both experts agreed they would read the Lerk papers to glean an understanding of what the patentee meant by the term “hydrophilization” and “in hydrophilized form” in the specification and the claims.
231 The person skilled in the art reading the 226 Patent specification and wanting further understanding of the terms “hydrophilization” and “in hydrophilized form” would obtain and read copies of the Lerk papers. The person skilled in the art turning to the Lerk papers would begin by reading the abstracts of the papers. The abstracts of both papers begin in a similar way, by describing hydrophilization, or the creation of a hydrophilic surface on a hydrophobic drug “by intensive mixing of the hydrophobic drug with a small amount of” a solution of a hydrophilic excipient or a small amount of methylcellulose solution. The only process of “hydrophilization” described in the Lerk papers involves the intensive mixing of the hydrophobic drug in the presence of a small (or minimal) amount of hydrophilic excipient in solution.
232 Bayer accepts that the specific exemplifications of “hydrophilization” in the 226 Patent, including by reference to the Lerk papers, take place in the presence of liquid (in the case of the Lerk papers, the amount of liquid is “very small”). However, Bayer submits that the meaning of “in hydrophilized form” is not confined to a product of the specific exemplifications of the processes for hydrophilization in the 226 Patent or the Lerk papers. The 226 Patent expressly states at page 6 that the invention is not restricted to the preferred exemplary embodiments. Bayer submits that the experts understood (and would have understood at the First Priority Date) that liquid would not be required to achieve “hydrophilization”. The surface of hydrophobic API could also be rendered hydrophilic by high-speed mixing without liquid in a confined space.
233 It must be recalled that the person skilled in the art, turning to the Lerk papers for an understanding of the terms “hydrophilization” and “in hydrophilized form”, has not encountered the terms before. They have no understanding of the terms and are not seeking clarification or a more detailed understanding of the terms. They are simply looking for a meaning. It must also be recalled that the person skilled in the art is not mining the depths of the discussion in the Lerk papers by examining in minute detail the graphs and figures found therein; they are looking to the Lerk papers to give context to, and better understand, the use of the terms in the 226 Patent’s specification and claims. That context is obtained from reading the abstract and the discussion of the processes used in the Lerk papers, each of which involved intensive mixing with a small amount of solution.
234 The person skilled in the art reading the 226 Patent would be well familiar with wet granulation. They would understand that the patentee must intend “hydrophilization” to be something other than what they understand to be ordinary wet granulation. If ordinary wet granulation was intended, there is no need for the patentee to resort to the unfamiliar term “hydrophilization” to describe the intended concept.
235 The process by which a hydrophobic API is hydrophilized in the Lerk papers involves intensive mixing of the API with a hydrophilic excipient in a small amount of solution. Lerk 1978’s mention of “fluidisation techniques” and citation of three papers obliquely suggests that there are other methods by which the coating of the particles with hydrophilic material may be achieved. However, it notes that problems may be experienced if the powder is cohesive. The only hydrophilization process actually discussed in Lerk 1978 is the technique described by de Jong (endnote 12) which was used in the study, in which a powder and a small amount of binder solution are mixed intensively in a kneading machine or high-speed mixer.
236 I therefore consider that a person skilled in the art reading the specification of the 226 Patent, including the Lerk papers, would construe the term “in hydrophilized form” to describe where the hydrophobic surface of an active compound is rendered hydrophilic by being at least partially coated with a hydrophilic excipient through a process of intensive mixing of the hydrophobic active compound with the hydrophilic excipient in a small amount of solution. The process of “hydrophilization” is the process whereby the surface properties of a hydrophobic API are rendered hydrophilic by coating the hydrophobic particles with a hydrophilic material via a process of intensive mixing of the hydrophobic active compound with the hydrophilic excipient in a small amount of solution. In short, these processes necessarily include the use of a liquid.
237 On the basis of the construction of “in hydrophilized form” that I have adopted above, Bayer accepts that the Sandoz Rivaroxaban Products do not infringe the asserted claims of the 226 Patent.
238 Sandoz advances a number of substantive grounds of invalidity against all of the claims in each of the 226 and 613 Patents. Sandoz also says the asserted claims of the 226 Patent are not entitled to a priority date earlier than 18 May 2010.
239 In relation to the 226 Patent, Sandoz submits that, first, the asserted claims are not fairly based. Second, the 226 Patent is anticipated by WO 940 if the claims have the deferred priority date and therefore lacks novelty. Third, the invention claimed in each of the claims of the 226 Patent is obvious at the First Priority Date in light of the common general knowledge together with WO 919. Alternatively, if the priority date is shifted to the date of the amendments to the specification to incorporate consistory clauses, the invention claimed in the asserted claims are obvious in light of WO 940. In either case, the 226 Patent lacks an inventive step. Lastly, the claims of the 226 Patent lack clarity.
240 Sandoz seeks to invalidate the claims of the 613 Patent because it submits the 613 Patent does not disclose the best method known to Bayer for making a rapid-release tablet containing rivaroxaban and therefore does not comply with s 40(2)(a) of the Act. Second, Sandoz contends that the 613 Patent is obvious in light of the common general knowledge considered together with either WO 919 or the Blood Abstracts, and therefore lacks an inventive step. Lastly, Sandoz submits that each of the claims are not clear and do not comply with s 40(3) of the Act.
8. FAIR BASIS AND PRIORITY DATE (226 PATENT)
241 Sandoz submits that if the term “in hydrophilized form” is given a broader meaning, such as to encompass a composition made by a process not involving wet granulation, or otherwise not involving the use of a liquid with a hydrophilic excipient, then the asserted claims of the 226 Patent are not fairly based. This because there is no real and reasonably clear disclosure of such an invention in the body of the specification (whether or not regard is had to the contents of the Lerk papers).
242 Sandoz’s priority date, novelty, and fair basis grounds only arise if, contrary to Sandoz’s submission, the consistory clauses on page 2, line 23 to page 2a, line 21 of the 226 Patent provide fair basis for the asserted claims. Throughout the hearing and in closing submissions, Bayer made a point of not placing any reliance on the seven aspects of the invention added by the 18 May 2010 amendments in support of its construction of “in hydrophilized form”.
243 On the basis of the construction of “in hydrophilized form” that I have adopted above, it is not necessary to consider Sandoz’s novelty case, fair basis case, or its priority date case.
244 Section 40(2)(a) of the relevant version of the Act imposes two distinct but closely related requirements. First is the sufficiency requirement. The second requirement is that the complete specification disclose the best method known to the applicant of performing the invention at the filing date: GSK FC at [184] (per Middleton, Nicholas and Burley JJ).
245 The requirement to disclose the best method is not met simply by satisfying the sufficiency requirement: Les Laboratoires Servier v Apotex Pty Ltd (2016) 117 IPR 415 at [135] (per Bennett, Besanko and Beach JJ); BlueScope Steel Ltd v Dongkuk Steel Mill Co, Ltd (No 2) (2019) 152 IPR 195 at [839] (Beach J).
246 The key to understanding the obligation of the patentee is to understand that the section is directed to the method of performance of the invention. In the context of the best method requirement, “the ‘invention’ is the embodiment which is described and around which the claims are drawn”: Sandvik Intellectual Property AB v Quarry Mining & Construction Equipment Pty Ltd (2017) 126 IPR 427 at [94] (per Greenwood, Rares and Moshinsky JJ) referring to the High Court in Kimberley-Clark at [21] (per Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ). As Bennett, Besanko and Beach JJ stated in Servier 2016 at [124], “[t]he monopoly is circumscribed by the claims but the nature of the invention is as described in the whole of the specification”.
247 The Full Court in Servier 2016 had regard to the definitions of “perform” at [123]:
Perform is relevantly defined in the Macquarie Dictionary to include: “to carry out; execute, do”; and “to carry into effect; fulfil”. The meanings of “perform” in the Shorter Oxford English Dictionary are relevantly “execute, accomplish, do, (any action, operation or process undertaken or ordered)” and “make or construct (an object)”.
248 It also had regard to the definition of “method” at [125]: “Method is relevantly defined in the Macquarie Dictionary as: ‘a mode of procedure’ and ‘a way of doing something’”.
249 The best method requirement applies to all patents, whether they contain product claims or method claims, or both. No distinction is drawn in the language of the statute between a product and a process in providing for the obligation to provide the best method of performing the invention. In Servier 2016, Bennett, Besanko and Beach JJ at [108] observed that “a distinction between products and processes that ignores the specific features of the invention claimed is unhelpful”.
250 In Sandvik, Greenwood, Rares and Moshinsky JJ summarised the key principles with respect to best method at [115]:
(a) The purpose of the requirement in s 40(2)(a) to disclose the best method known to the applicant of performing the invention is to allow the public the full benefit of the invention when the monopoly expires: Servier at [108].
(b) Although a patentee might not be explicitly required to act in good faith, principles of good faith underlie the best method requirement: Servier at [108].
(c) The nature and extent of the disclosure required to satisfy the best method requirement will depend on the nature of the invention itself: Firebelt at [52]–[53]; Servier at [108].
(d) The key to understanding the obligation of the patentee is to understand that the section is directed to the method of performance of the invention. The monopoly is circumscribed by the claims, but the nature of the invention is as described in the whole of the specification: Servier at [124].
(e) The requirement to describe the best method of performing the invention is ordinarily satisfied by including in the specification a detailed description of one or more preferred embodiments of the invention: Firebelt at [53]; Servier at [104]. See also Blanco White (5th ed) at [4-516]; Bodkin C, Patent Law in Australia (2nd ed, Thomson Reuters, 2014) at [5280].
(Emphasis in original.)
251 The best method does not need to be identified as such in the specification: Pfizer Overseas Pharmaceuticals v Eli Lilly & Co (2005) 68 IPR 1 at [374] (per French and Lindgren JJ).
252 In Servier 2016, Bennett, Besanko and Beach JJ stated at [103] and [104]:
[103] The authorities that have dealt with s 40(2)(a), its precedents and equivalents, must be understood in context. The first, and most important, factor is the nature of the invention being described and claimed. Servier divides this simply into products and processes but that is not sufficient. It is necessary to understand the invention itself in order to appreciate what is required of an inventor by way of disclosure in the specification in order to secure a monopoly from the public. In some cases, the claim to a product will require no description of the method of obtaining it and it can be left to the skilled worker (as in AMP v Utilux). In other cases, the product claim, properly understood, will require sufficient directions in order to obtain the monopoly.
[104] The Full Court in Firebelt observed that the statutory obligation was an obligation to disclose the best method of performing the invention. Their Honours also said (at [53]), referring to the requirement of s 40(2) of the Act (in its totality), that the patentee is required to give the best information in his (or now her) power as to how to carry out the invention (emphasis in the original). The Full Court was of the view that the requirement is ordinarily satisfied by including a detailed description of one or more preferred embodiments of the invention and concluded in that case that, by taking account of the rest of the specification together with the figures, an embodiment was depicted of the claimed device.
253 Their Honours further observed at [135]: “The patentee has an obligation to include aspects of the method of manufacture that are material to the advantages it is claimed the invention brings”.
254 In that case, Servier did not disclose its specific method of making perindopril arginine. It simply referred to using a “classical salification method”. However, it argued the invention was to a product and the process for making the product was not part of the inventive step, therefore its specific method did not need to be disclosed: see at [145]. Servier pointed to the fact that the primary judge did not find that Servier “knew that the classical method of salification was inferior to either of the two methods that it did utilise”. It also claimed that there was no evidence that classical salification would fail to produce the patented product with the same quality, stability and utility: see at [145]–[147]. Servier ultimately argued that a method was disclosed in the specification, “it employed two versions of a method of classical salification and that there was no obligation actually to employ the best method identified”. This argument was rejected by the Court. The evidence showed that there may be different approaches and different parameters to “a classical salification” method. Their Honours observed at [173] and [177]:
[173] The disclosure of the 1986 or 1991 methods would have saved the skilled addressee the possible dead ends and false starts that would otherwise have been risked in attempting to make the salt. It would have disclosed certainty of method rather than leaving the skilled addressee at risk of failure and of choosing from all of the parameters that may be used in classical salification.
…
[177] In describing only the general method of classical salification rather than a specific method, such as the known 1986 and 1991 method, which would have provided the information to the skilled reader of a method for obtaining a form of perindopril arginine which met the characteristics of the claimed invention, Servier failed to describe the best method known to it of performing the invention.
255 Further, and relatedly, Beach J stated in BlueScope at [836]:
A patent will satisfy the best method requirement if the skilled addressee can arrive at the best method known to the patent applicant of performing the invention by some routine experimentation, but without ingenuity or undue experimentation. A patent applicant does not need to disclose details of the method which would be well-known and understood by the skilled addressee such as well-known analytical agents, commonly used methods, well-known terms of art, or a description of machinery in standard use. But a complete specification must disclose each essential element or feature for performing the invention, even if a skilled addressee would know or could readily ascertain that element. Where the best performance of the invention requires a step that is omitted by the complete specification, even if it could be readily ascertained by a skilled addressee, that will not meet the best method requirement.
256 Justice Beach went on to say at [840] that “[t]he patentee was not entitled to leave the public with the risk of the non-ascertainment of this step, since it had practical relevance to whether the benefit of the invention was obtained”.
257 Both the Full Courts in Servier 2016 and Sandvik considered that principles of good faith underlie the best method obligation. The Servier 2016 Full Court explained at [64] the proposition underlying the best method requirement in s 40(2)(a) as follows:
The proposition underlying a separate and additional obligation on the part of the inventor filing a complete specification is that where an inventor in fact knows of a method at the time of filing the complete patent application, which has taken the methodology to a more satisfactory stage or provides more certainty so that the public may more quickly and easily utilise the invention for which a monopoly is granted, the inventor is under an obligation to disclose that method.
(Emphasis added.)
258 The obligation for the patentee to disclose the best method known to them is longstanding. As their Honours explained in Servier 2016 at [72]:
[I]n Edison and Swan United Electric Light Company v Holland (1889) 6 RPC 243 at 279, the [English] Court of Appeal said that the patentee must state “in what manner the patented invention is to be performed” so that others know how practically to avail themselves of the invention when the patent is expired: “how they are to do what is necessary to carry out the new invention”, that is, how the invention is to be performed.
259 Both the Servier 2016 (at [73]) and the Sandvik (at [100]) Full Courts referred to the fact that the English Court of Appeal in Vidal Dyes Syndicate Ltd v Levinstein Ltd (1912) 29 RPC 245 differentiated between the two duties incumbent upon a patentee. First, the obligation by which the patentee must “particularly describe and ascertain the nature of the invention” and, second, that by which the patentee must “particularly describe and ascertain ‘in what manner the same is to be performed’”. In relation to the second duty, the Court of Appeal in Vidal said at 269:
It is settled law that a patentee must act towards the public uberrima fide, and must give the best information in his power as to how to carry out the invention. He is therefore bound to tell the public all the steps that can advantageously be taken in carrying out the invention. But he is not limited to claiming only the best way of carrying it out.
260 The disclosure of the method must be such as to relieve the skilled addressee seeking to implement the invention, including achieving the advantages associated with the invention, as at the expiry of the patent from being faced with undertaking a potentially extensive course of trial and error experiments to overcome the blind alleys and pitfalls which the patentee had already navigated or overcome as at the filing date.
261 Sandoz submits that the 613 Patent does not disclose the best method of working the invention because it does not disclose the best method known to Bayer for making a rapid-release tablet containing rivaroxaban, which Bayer has admitted was that described in WO 940. Sandoz submits that it is the invention as described in the whole of the specification for which the best method must be provided, not necessarily the invention claimed.
262 Bayer admits that the best method known to it of making a rapid-release tablet containing rivaroxaban is the wet granulation method disclosed in the international publication of the 226 Patent.
263 Sandoz submits that claims 1 and 2 of the 613 Patent (which are Swiss style claims) involve the manufacture of a rapid-release tablet. Therefore, even if the best method requirement were limited to the invention as claimed (which Sandoz submits is contrary to principles articulated in Sandvik), that invention involves the manufacture of a rapid-release tablet. The best method in relation to a Swiss style claim would always have included the manner of making the medicament. Even asserted claims 3 and 4, which are to a method, require a rapid-release tablet to be administered as part of the method.
264 Sandoz submits that the only disclosure of the “rapid-release tablet” feature is that at page 10, lines 10–13, of the 613 Patent:
Very particularly preferred are rapid-release tablets containing [rivaroxaban] as active ingredient. Preparation of such tablets is for example described in PCT/04/01289, whose disclosure is included by way of reference.
265 Sandoz submits that “PCT/04/01289” is meaningless. Although it appears to the person skilled in the art to be a patent reference, a search for that reference on Google, Chemical Abstracts, or Patentscope does not produce any results, as a document with that reference does not exist. Sandoz accepts that an expert patent searcher would be able to investigate and determine that “PCT/04/01289” is an incorrect reference to the patent application number “PCT/EP2004/012897”. However, according to Sandoz, the skilled addressee of the 613 Patent does not include an expert patent searcher. At its highest, Sandoz submits that the evidence in support of Bayer’s case is that the skilled addressee could engage a patent searcher to supply the correct reference. Sandoz submits this is not sufficient to satisfy the best method requirement because it does not have the effect of disclosing the best method, and common general knowledge cannot fill the gap.
266 Bayer submits the invention claimed in asserted claims 3 and 4 is a method of treatment of a thromboembolic disorder, not the underlying dosage form.
267 Bayer submits that an unintentional and inadvertent error in the course of drafting the priority document (omitting the number “7” and the country code “EP”) should not invalidate the entire patent. Bayer says this sentence is not required to understand the best method.
268 Bayer further submits that the preferred embodiment of the invention, being a method of treatment, is disclosed. Bayer submits that the best method requirement is satisfied as the particulars for the best embodiment are given at page 10, lines 7–12, including “[v]ery particularly preferred are rapid-release tablets containing [rivaroxaban] as active ingredient”.
269 Bayer put its position in its closing submissions as follows.
(a) A rapid-release tablet is an essential integer of the invention claimed in the 613 Patent.
(b) The best method known to Bayer at the filing date of the 613 Patent for making the rapid-release tablet containing rivaroxaban described in the claims of the 613 Patent, was described in PCT/EP04/012897 (also known as WO 2005/060940); this application became the 226 Patent.
(c) As to the preparation of rapid-release tablets containing rivaroxaban, the 613 Patent states: “Preparation of such tablets is for example described in PCT/04/01289, whose disclosure is hereby included by way of reference”.
(d) The reference to “PCT/04/01289” was an unintentional and inadvertent mistake. It should read PCT/EP04/012897.
(e) The skilled team, either through the inclusion of individuals with expertise in intellectual property, or by assistance from a patent searcher, would have been able to find the correct PCT reference in 15 minutes or less.
(f) Alternatively, given the 613 Patent (disclosing rivaroxaban and no formulation information), the person skilled in the art could, by way of common general knowledge and permissible routine experimentation, make a rapid-release tablet comprising rivaroxaban.
270 Bayer contends that it is not required to disclose the method of making a rapid-release tablet containing rivaroxaban in order to satisfy the requirement for it to disclose the best method known to it of performing the invention, which it says is a method of treatment.
271 Bayer submits that if the best method of performing the invention in the 613 Patent requires disclosure of the separate and distinct invention in the 226 Patent (which relevantly includes a process for preparing rivaroxaban “in hydrophilized form”), that should be rejected as it is not the statutory requirement. Section 40(2)(a) is directed towards the best method of performing the invention described in the patent, not a separate and distinct invention claimed in a separate patent (the 226 Patent).
272 Bayer submits that if, contrary to its contention, it was obliged to disclose a method of making a rapid-release tablet containing rivaroxaban (“in hydrophilized form”) in the 613 Patent to satisfy the best method requirement, the mistake in the PCT reference does not vitiate the disclosure of that method because:
(a) the mistake could be rectified (and the best method, thus, disclosed) without the exercise of any inventive faculty or prolonged research; or
(b) Sandoz’s evidence demonstrates that the person skilled in the art could, by way of common general knowledge and routine experimentation without undue burden, make a rapid-release tablet comprising rivaroxaban.
273 The best method requirement is separate to the sufficiency requirement, and it is not sufficient to satisfy the best method requirement to simply satisfy the sufficiency requirement.
274 An assessment of the best method requirement begins with the proper identification of the invention described in the 613 Patent. I consider that invention to be a method of treatment of thromboembolic disorders by administering a single rapid-release, oral tablet comprising rivaroxaban to be taken once per day.
275 The specification of the 613 Patent begins by stating that the invention relates to the field of blood coagulation, more specifically a method of treatment of a thromboembolic disorder by administering a direct factor Xa inhibitor once daily.
276 According to Bayer’s submissions in support of the inventive step in the 226 Patent, the hydrophilized form of rivaroxaban is not obvious. Thus, the person skilled in the art trying to make the invention described in the 613 Patent upon its expiry (without access to the hydrophilized form patent) will be required to embark on undertaking a potentially extensive course of trial and error experiments to overcome the blind alleys and pitfalls which the patentee had already navigated or overcome as at the filing date.
277 It is irrelevant that the best method itself might include an invention which may be the subject of an earlier patent. The patentee cannot avoid satisfying its obligation of good faith by hiding behind a separate patent if the invention described therein is necessary for the performance of the best method of the invention of the patent in suit.
278 I accept that the mistake in the 613 Patent was unintentional. I do not consider that Bayer has engaged in a scheme to deprive the public of the best way to make the rapid-release form of rivaroxaban tablet by hiding it in plain sight in a published patent specification under an erroneous patent number.
279 The patentee has not deliberately failed to fulfil its good faith duty to disclose the best method of performing the invention. The method has been made available to the public. The evidence establishes that the correct reference can be found by the person skilled in the art engaging a patent searcher or searching the patent databases themselves. Therefore, the crucial question in determining whether Bayer has satisfied the best method requirement for the 613 Patent turns on the relevant principles regarding rectifying mistakes in patent specifications.
280 Bayer relies on the following “true rule” stated by Romer LJ in No-Fume Ltd v Frank Pitchford & Co Ltd (1935) 52 RPC 231 at 243:
Specifications very frequently contain mistakes; they also have omissions. But if a man skilled in the art can easily rectify the mistakes and can readily supply the omissions, the patent will not be held to be invalid. The test to be applied for the purpose of ascertaining whether a man skilled in the art can readily correct the mistakes or readily supply the omissions, has been stated to be this: Can he rectify the mistakes and supply the omissions without the exercise of any inventive faculty? If he can, then the description of the specification is sufficient. If he cannot, the patent will be void for insufficiency.
281 This passage was quoted with approval by McTiernan J in AMP Inc v Utilux Pty Ltd [1971] 45 ALJR 123 at 128.
282 Sandoz submits the “true rule” is not applicable in every circumstance. The error or omission must be at once discoverable on the face of the specification, without the need for experimentation or further enquiry, nor can it be an erroneous statement amounting to a false suggestion, even if the error would be at once observed by the skilled reader: Valensi v British Radio Corporation [1973] RPC 337 at 375 (per Russell, Buckley and Cairns LJJ).
283 To similar effect is the passage from the judgment of Lord Westbury LC in Simpson v Holliday (1865) 30 LR 1 HL 315, quoted in Vidal by Fletcher-Moulton LJ at 271, which distinguishes between errors on the face of the specification which any “workman of ordinary skill and experience would perceive and correct”, and errors which are discoverable only by experiment and further inquiry.
284 In GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Limited v Generic Partners Pty Limited (2016) 119 IPR 1, Beach J held at [353] (in a judgment upheld by the Full Court in GSK FC) that the reference in claim 1 to a “basket” would have been understood by the skilled addressee as an error and that “the skilled addressee would read claim 1 as referring to the compendial USP type III apparatus with reciprocating cylinder, understanding the reference to ‘basket’ as being an error”. Justice Beach refused to interpret “basket” to mean “cylinder” because he considered that this would be tantamount to “re-writ[ing] the claim under the guise of construction”: at [382]. In support of his conclusion, Beach J observed:
(a) that the word “basket” had a clear and well understood meaning in relation to USP dissolution testing, namely as part of the USP type I apparatus;
(b) the evidence was that it was known that compendial USP apparatuses were modified to overcome issues experienced in testing;
(c) it was part of the common general knowledge that, as at the priority date, it may have been possible and potentially practicable to adapt a USP type III apparatus to fit a (USP type I) basket in substitution for a cylinder (at [313]); and
(d) if “basket” means “basket”, the invention still works.
285 I consider that the mistake in the 613 specification is an unintentional “typo”, which the person skilled in the art can readily recognise from the face of the specification. The person skilled in the art can readily supply the missing numerals and letters, either by conducting a search themselves or with the assistance of a patent searcher, without the exercise of any inventive faculty.
286 For the reasons set out in the amendment section below, I reject Sandoz’s submission that the only possible inference is that Bayer deliberately maintained the mistake even as it amended the claims to introduce the rapid-release integer as an integer of each claim.
287 Bayer does not seek to maintain an unfair advantage by hiding the best method in plain sight via an incomplete reference to the specification which contains the best method. That specification is a published patent specification in the name of Bayer.
288 I consider that, from a practical point of view, the public is fairly given possession of the invention described in the 613 Patent: see Adhesives Pty Ltd v Aktieselskabet Dansk Gaerings-Industri (1935) 55 CLR 523 at 546 (per Evatt J).
289 An invention is a patentable invention if the invention, when compared with the prior art base, involves an inventive step: see s 18(1)(b)(ii) of the Act. Sections 7(2) and (3) of the relevant version of Act identify the nature of the enquiry. They provide:
(2) For the purposes of this Act, an invention is to be taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed in the patent area before the priority date of the relevant claim, whether that knowledge is considered separately or together with the information mentioned in subsection (3).
(3) The information for the purposes of subsection (2) is:
(a) any single piece of prior art information; or
(b) a combination of any 2 or more pieces of prior art information;
being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood, regarded as relevant and, in the case of information mentioned in paragraph (b), combined as mentioned in that paragraph.
290 The onus is on Sandoz to establish that the claimed invention is obvious. In AstraZeneca AB v Apotex Pty Ltd (2014) 107 IPR 177 (AstraZeneca FC), Besanko, Foster, Nicholas and Yates JJ said at [200]:
Put another way, the invention is deemed to involve an inventive step when compared with the prior art base unless it would be obvious to the hypothetical skilled addressee having regard to such knowledge and information.
(Emphasis in original.)
291 When assessing obviousness, it is to be remembered that the inventive element needed under s 7(3) is “very small”: Ranbaxy Laboratories Ltd v AstraZeneca AB (2013) 101 IPR 11 at [213] (per Middleton J). Only a “scintilla” of invention is required: Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) (2007) 235 CLR 173 at [52] (per Gummow, Hayne, Callinan, Heydon and Crennan JJ).
292 In Aktiebolaget Hässle v Alphapharm Pty Ltd (2002) 212 CLR 411 at [50] (AB Hässle), Gleeson CJ, Gaudron, Gummow and Hayne JJ referred to the formulation of the relevant test by Aickin J in Wellcome Foundation Ltd v VR Laboratories (Aust) Pty Ltd (1981) 148 CLR 262 at 286:
The test is whether the hypothetical addressee faced with the same problem would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not.
293 Their Honours observed at [51]:
What Aickin J had in mind as “routine” appears from an earlier passage in his judgment in which he was discussing the question whether evidence of the steps taken by the patentee was relevant and therefore admissible in a revocation action. His Honour said:
“Evidence of what he did by way of experiment may be another matter. It might show that the experiments devised for the purpose were part of an inventive step. Alternatively it might show that the experiments were of a routine character which the uninventive worker in the field would try as a matter of course. The latter could be relevant though not decisive in every case. It may be that the perception of the true nature of the problem was the inventive step which, once taken, revealed that straightforward experiments will provide the solution. It will always be necessary to distinguish between experiments leading to an invention and subsequent experiments for checking and testing the product or process the subject of the invention.”
(Emphasis added in AB Hässle.)
294 Their Honours then sought to unravel the “distinct strands of thought” in Aickin J’s reasoning, concluding at [53] that the correct approach is the reformulated “Cripps questions” by Graham J in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd [1970] RPC 157 at 187-188: whether the person skilled in the art at the relevant date with the relevant knowledge would directly be led, as a matter of course, to try the claimed invention in the expectation that it might well produce a useful alternative to, or a better drug than, the existing compound.
295 Their Honours warned against “the twin traps of hindsight and oversimplification” and stated that routine steps taken as a “matter of course” did not include “a course of action which was complex and detailed, as well as laborious, with a good deal of trial and error, with dead ends and the retracing of steps”: AB Hässle at [58]. See also AstraZeneca HC at [15] (per French CJ); Hanwha Solutions Corp v REC Solar Pte Ltd [2023] FCA 1017 at [419]–[421], [429] (per Burley J). It is also apparent from the plurality’s discussion in AB Hässle at [70]–[76] that “the fact that the person skilled in the art would consider a step or series of steps worthwhile to try or worth a try does not mean that the resulting invention is obvious or lacks an inventive step”: see Australian Mud Company v Globaltech Corporation Pty Ltd (2018) 138 IPR 33 at [452] (per Besanko J).
296 The test for obviousness set out in AB Hässle is not “the test to be applied in every case” but is the appropriate test to be applied in this case, which is of a similar nature to AB Hässle: see Generic Health Pty Ltd v Bayer Pharma Aktiengesellschaft (2014) 106 IPR 381 at [71] (per Besanko, Middleton and Nicholas JJ).
297 Sandoz submits that the invention claimed in all of the claims of the 226 Patent is obvious in light of the common general knowledge together with WO 919. Alternatively, if the priority date is shifted, the asserted claims are obvious in light of WO 940. Lastly, the 226 Patent is anticipated by WO 940 if the claims have the deferred priority date.
298 Sandoz also seeks to invalidate the invention claimed in all claims of the 613 Patent as being obvious in light of the common general knowledge, considered together with the information disclosed in WO 919 or the Blood Abstracts, but not a combination of them.
299 A preliminary question is whether WO 919 and the Blood Abstracts are indeed s 7(3) documents for the purposes of the inventive step analysis.
300 Before considering the s 7(3) issues, I will set out what I consider constitutes the common general knowledge of the person skilled in the art at the priority dates.
301 The parties agreed the terms of a technical primer, which they accepted formed part of the common general knowledge of those skilled in the art as at 27 November 2003 and 31 January 2005. The discussion of the common general knowledge below includes extracts from the technical primer. It also includes evidence from the experts in JER1 and JER2, and evidence from the experts given during the joint sessions.
10.1.1 Haemostasis and thrombosis
302 Haemostasis is a physiological process that stops bleeding at the site of an injury to a blood vessel. This is done by the creation of a sealing clot or “thrombus”.
303 In healthy humans, there is a haemostatic balance between procoagulant forces and anticoagulant forces. Procoagulant forces promote the formation and/or propagation of coagulation, and/or the formation of fibrin.
304 Under normal physiological conditions, intravascular blood should flow freely and not clot. It is only upon an injury that the coagulation system should rapidly and locally respond at the injured blood vessel wall to generate a clot.
305 The clot is created by a mesh of fibrin that embeds activated platelets and red and white blood cells.
306 The coagulation cascade refers to the activation and sequential amplification of many inert plasma coagulation factors in an orderly fashion. The coagulation cascade results in the formation of a clot.
307 The main enzymes involved in the coagulation cascade are termed “factors” and are referred to (non-sequentially) by the Roman numerals I to XIII. An “a” is used to indicate if the factor is in its active form.
308 There are more than 12 different coagulation factors in blood plasma, each of which interacts stepwise with another coagulation factor. The interaction changes the next target coagulation factor from an inactive form to an active form, which then interacts with a subsequent coagulation factor.
309 Below is a diagrammatic representation of the coagulation cascade.
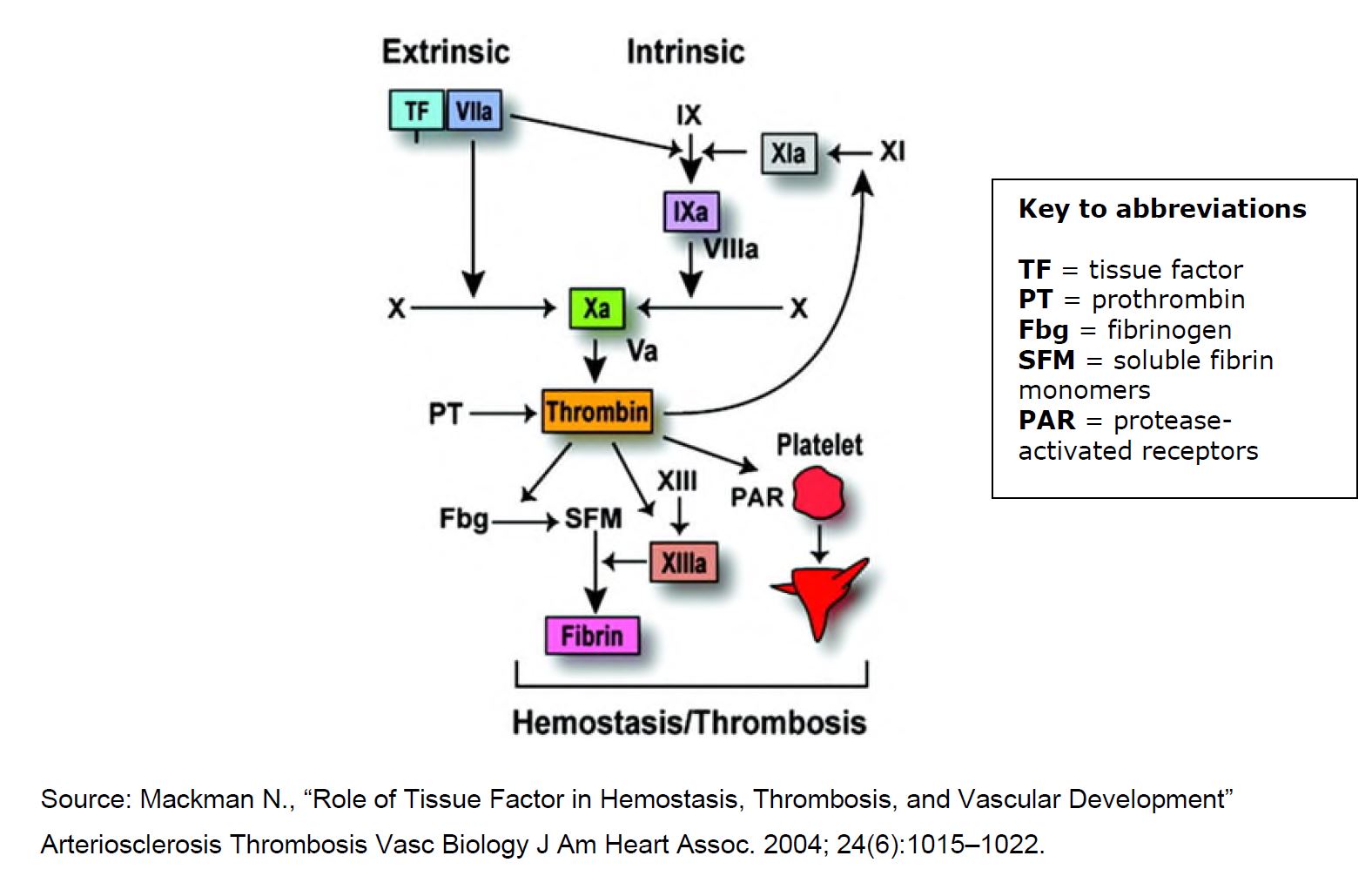
310 Professor Baker agreed that the coagulation cascade is complex and understanding its complexity is still evolving.
311 Anticoagulants are substances that inhibit blood coagulation. Bleeding risk is a feature of all anticoagulants by design and is the trade-off for reducing the risk of having a thrombotic event. All the experts agreed that special care needs to be taken with the administration of anticoagulants: too much and there is a risk of bleeding, and too little and there is a risk that clots might form.
312 At the priority dates, there were numerous parenteral and oral anticoagulants at various stages of research and development, clinical use and regulatory approval.
313 The main anticoagulants in clinical use were orally administered warfarin and parenterally administered heparins, which inhibited more than one factor in the coagulation cascade and acted indirectly.
314 Warfarin is administered orally. It has a slow onset of action, taking five to 10 days for its therapeutic effects to fully manifest.
315 Warfarin is a vitamin K antagonist. Warfarin, originally used commercially as rat poison (causing the rats to bleed to death), was first approved for medical use in the 1950s and yet warfarin and other vitamin K antagonists were still the only oral anticoagulants approved in Australia as at the Second Priority Date.
316 Warfarin is an indirect and non-specific anticoagulant. It exerts its clinical effect by inhibiting an enzyme in the liver which, in turn, blocks the recycling of vitamin K in the body. This results in a relative vitamin K deficiency, which then reduces the production of normally functioning blood clotting factors II, VII, IX and X in the liver. Warfarin has a narrow therapeutic index.
317 Warfarin also has a pro-coagulation effect. It inhibits the normal function of protein C and protein S, which are vitamin-K-dependent inhibitors of coagulation.
318 On initiation of warfarin therapy, frequent dose adjustment is required with laboratory monitoring of warfarin’s effect. The dose is adjusted using the international normalised ratio (INR) which is based on a standardised PT. PT is a measure of the time it takes a clot to form via the extrinsic pathway when activated by the tissue factor/factor VII complex.
319 Although commonly used over many decades, the experts agreed that warfarin was a “problematic drug”. Warfarin has the following limitations:
(a) It has a relatively narrow therapeutic window, and the potential to cause bleeding.
(b) The slow onset of action of warfarin means that it is not suitable for the treatment of an acute thromboembolic event, unless it is initially co-administered with another drug.
(c) Warfarin does not have a predictable dose-response, due in part to genetic variability in patient metabolism. As a result, there is unpredictable inter-patient and intra-patient variability in warfarin’s dose effect.
(d) Warfarin also has a propensity for food and drug interactions.
(e) The unpredictable dose-response of warfarin necessitates regular monitoring, which involves INR testing to be undertaken in patients at least once a month, but often more frequently.
320 There was a desire to overcome at least some of the shortcomings of warfarin. The experts agreed that, as at the priority dates, the replacement of warfarin with newer medications was a highly desirable outcome.
321 Heparin is a naturally occurring polysaccharide. As discussed in the 613 Patent, heparin is administered parenterally, usually by intravenous infusion or subcutaneously in hospital. Heparin is administered twice or three times daily.
322 Heparin is an indirect anticoagulant. It exerts its clinical effect by binding to a protein in the blood called antithrombin III. The heparin-antithrombin complex is able to inhibit specific coagulation factors II and X (and to a lesser extent IX and XI). The avidity with which these heparins bind to these factors depends on the form of the heparin. Heparin, as a part of the heparin-antithrombin complex, can inhibit factors IIa and Xa (and to a lesser extent IXa and XIa), without selectivity for factor Xa.
323 Heparin has a fast onset of action and a short elimination half-life. It has a narrow therapeutic window. Heparin can be used for emergency acute treatment of thrombotic events when given by a continuous intravenous infusion, typically in a hospital setting.
324 The heterogeneity of heparin compounds leads to significant variation in anticoagulation effect in patients. As a result, much like warfarin, heparin does not have a predictable dose-response. For this reason, the effect of heparin must be monitored in patients. Professor Baker explained that in order to administer LMWH, a heparin derivative discussed further below, nurses would have to visit patients at their homes to administer the agent and draw blood for monitoring the INR.
325 Intravenous heparin treatment requires patients to remain in hospital, and blood tests need to be performed as often as every six to eight hours.
326 Heparin treatment is generally monitored using APTT.
327 There was a desire to overcome at least some of the shortcomings of heparin at the priority dates.
10.1.6 Low molecular weight heparin (LMWH)
328 LMWH was introduced into clinical practice in the 1990s. Enoxaparin and dalteparin are commonly used LMWHs.
329 LMWH is derived from heparin that has been processed to yield smaller polysaccharide fragments than those found in heparin.
330 LMWHs are administered by subcutaneous injection, which can cause pain and bruising.
331 LMWH has enhanced factor Xa inhibition in comparison to heparin, although it does still inhibit factor IIa and to a lesser extent factors IXa and XIa.
332 LMWHs are administered once or twice daily. It is not administered once every half-life.
333 LMWHs produce a more predictable anticoagulant effect than heparin. Unlike heparin, the anticoagulant effect of a fixed dose of LMWH can be predicted based on weight.
334 Due to the more predictable dose-response, laboratory monitoring of LMWHs is not generally required. In addition, LMWH treatment is generally followed by prophylactic warfarin therapy and there is a cross-over period when both drugs need to be administered and patients require a blood test to measure and monitor the INR and adjust the dose of warfarin if necessary.
10.1.7 Hirudin and Bivalirudin
335 Hirudin is a potent and specific thrombin (factor IIa) inhibitor, derived from the saliva of the medicinal leech.
336 Hirudin inhibits coagulation by forming a stoichiometric, slowly reversible complex with thrombin. The hirudin-thrombin complex is almost irreversible.
337 By the Second Priority Date, hirudin had been used successfully by injection to prevent and treat arterial or venous thromboembolic complications of heparin-induced thrombocytopenia, a very rare allergic reaction to heparin. Hirudin was neither available nor approved for use in Australia.
338 Bivalirudin is a synthetic congener of hirudin and has a similar mechanism of action to hirudin.
339 Bivalirudin is administered as an intravenous injection.
340 Clinical trials in relation to bivalirudin were ongoing in North America. It had been approved in North America for certain indications by the Second Priority Date, but not in Australia.
341 Professor Baker agreed that, as at the Second Priority Date, ximelagatran was the most clinically advanced direct-acting single point (factor IIa) inhibitor. It had been provisionally approved in some European countries for the prevention of blood clots in patients undergoing hip and knee replacement surgery and initial treatment of AF, DVT and PE, but not anywhere else in the world, including Australia.
342 Ximelagatran is a prodrug form of melagatran. Melagatran is not reliably absorbed after oral administration. Ximelagatran undergoes rapid biotransformation in the liver to melagatran.
343 Ximelagatran (as melagatran) is a direct and specific inhibitor of thrombin. Melagatran has an elimination half-life of three to four hours.
344 Ximelagatran has a rapid onset of action after oral administration.
345 Ximelagatran was administered orally, twice daily. It was therefore not administered once every half-life.
346 Ximelagatran has a predictable anticoagulant response after oral administration and it was being studied with administration in fixed doses without laboratory monitoring.
347 In an article co-authored with Professor Weitz — Crowther M and Weitz J, “New Anticoagulants: An Update” (2004) 2(11) Clinical Advances in Hematology & Oncology 743 — and annexed to his affidavit, Professor Crowther compared the potential advantages of ximelagatran over warfarin. The advantages included a rapid onset of action, no food, drug or alcohol interactions and a wide therapeutic window.
348 The most concerning side effect of ximelagatran was an elevation of liver enzymes, which could increase the risk of liver failure. Concerns with respect to liver toxicity appeared to be benign and it was considered that the issue could be addressed by routine liver function monitoring during the initial phase of treatment.
349 However, more research and development were required in relation to the long-term use of ximelagatran as there was insufficient data in relation to the liver toxicity safety issue.
350 Before the Second Priority Date (but not the First Priority Date), following concerns with respect to liver toxicity, a United States Food and Drug Administration (FDA) panel had announced that it was deferring the approval of the drug in the United States pending further data. This ultimately led to the termination of the ximelagatran development program and its withdrawal from the market in the limited number of jurisdictions where it had been provisionally approved. Ximelagatran was never approved in Australia.
351 As at the Second Priority Date, the hepatoxicity concerns with ximelagatran would have been part of the common general knowledge.
352 Fondaparinux is a synthetic anticoagulant that is identical to the heparin pentasaccharide (a five-sugar sequence), being a small component of the structure of LMWH and heparin.
353 Fondaparinux is a specific inhibitor of factor Xa that works indirectly through the facilitation of antithrombin activity. Fondaparinux is not a direct factor Xa inhibitor. Fondaparinux is administered once daily by subcutaneous injection. It has an elimination half-life of about 17 hours.
354 Phase II and Phase III studies had been undertaken for the initial treatment of DVT/PE, thromboprophylaxis in orthopaedic surgery, medical and general surgery patients, and treatment of arterial thrombosis.
355 Potential disadvantages of fondaparinux include that it must be administered by injection and, if a patient suffers excessive bleeding as an effect of fondaparinux, it is more difficult to reverse with protamine sulfate than other drugs. With a 17-hour half-life, this could be problematic in a patient with life-threatening bleeding.
356 By the Second Priority Date, fondaparinux had been approved for thromboprophylaxis in high-risk orthopedic patients and treatment of DVT/PE in many jurisdictions, including in Australia.
357 Fondaparinux was superior to LMWH for hip fracture surgery and was used in preference to LMWH in Australia after approval in 2002. There were wide differences in clinician comfort with the use of fondaparinux in patients as well as delays in approval for reimbursement.
10.1.10 Razaxaban (aka DPC-906)
358 DPC-906 (also known as razaxaban) was a specific, oral inhibitor of factor Xa that was administered twice daily and that was in early stage phase development in 2003. Professor Baker only became aware that DPC-906 was razaxaban in the course of preparing for this proceeding.
359 As at the Second Priority Date, Professor Baker was aware from reading Weitz J I, Hirsh J and Samama M M, “New Anticoagulant Drugs: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy” (2004) 126(3 Suppl) Chest 265S (Chest paper), a widely read paper annexed to both his and Professor Crowther’s affidavits, that the clinical trials of DPC-906 had been stopped. At that time, the reason that the trials had been stopped — because DPC-906 caused excess bleeding in the high-dose arms — was not widely known. The source data had not been published, and knowledge of this complication was not widely disseminated.
360 As such, I do not consider that it was common general at the priority dates that DPC-906 was an available and clinically appropriate oral inhibitor of factor Xa.
361 There was research and development being conducted in relation to argatroban, idraparinux, and DX 9065, the details of which were summarised by Professor Baker in the following table, which was Appendix 1 to JER2 (with whom Professor Crowther agreed). As at the Second Priority Date, Professor Baker’s evidence was that there was substantial and ongoing research and development in relation to new and improved parenteral and oral anticoagulants targeting many aspects of the coagulation cascade, including factors VII, IXa, IIa.
362 Other than the provisional licensing of ximelagatran in Europe, Professor Baker was unaware of any other novel oral agent that was even close to receiving approval in 2005.
363 Professor Baker’s table summarised what was generally known and accepted at the Second Priority Date in relation to the following anti-coagulants. Professors Evans and Roberts had “comfort” in the information in the table.
Coagulation factor target | Mechanism of action | Half-life | Frequency and route of administration | Clinical development Status Jan 2005 | |
Warfarin | II, VII, IX and X | Vit K reductase inhibitor | Oral, once daily | Discovered 1941 established | |
Heparin | IIa, Xa (and to a lesser extent XIa and IXa) | Indirect binds to ATII | ~ 30 min after IV administration | IV/SC twice, three time a day | Discovered 1916 established |
LMWH | Xa>IIa | Indirect binds to ATII | 4 hours | SC once or twice daily | Discovered 1980’s Established (daily dose AUS approved 9/5/2000) |
Fondaparinux | Xa only | Indirect binds to ATIII | 17 hours | SC once daily | Registered for DVT/PE prevention (approved AUS 20/3/2002) and treatment (approved AUS 29/11/2004) |
Idraparinux | Xa only | Indirect binds to ATIII | 130 hours | SC weekly | Phase III DVT/PE trials commenced May 2003 and AF trials September 2003 |
Hirudin and Bivalirudin | IIa only | Direct | 1–2 hours | IV/SC twice, a day | Established in North America, not approved in Australia |
Ximelagatran | IIa only | Direct | 3–4 hours | Oral, twice a day | Phase III completed. Registration for AF/VTE in France 2003 and VTE prevention in Europe 2004. FDA deferred approval Sept 2004 until further LFT data provided |
Argatroban | IIa only | Direct | 45 min | IV | Established for HIT |
DX 9065 | FXa | Direct | 40 min | IV | Discovered 1993 Up to Phase II trials in coronary artery disease |
DPC-906 “razaxaban” | FXa | Direct | Not known | Oral, twice a day | Phase II trials |
364 Professor Crowther and Professor Baker agreed that idraparinux was completing Phase III trials at the Second Priority Date for long term treatment of DVT/PE and atrial fibrillation.
365 Although the DX 9065 program commenced in 1993, the drug had a very short half-life and was administered intravenously, which Professors Backer and Crowther agreed did not stack up well in a comparison with heparin.
366 Although argatroban was approved in Australia in 2003 and in Canada in 2005, it was not widely used. It demonstrated that a parenteral and specific thrombin inhibitor was effective for treating and preventing thrombosis.
10.1.12 Single point inhibition and factors IIa and Xa
367 The standard of care at the priority dates was multipoint inhibitors, such as warfarin, heparin and LMWH. Multipoint inhibitors, which inhibit more than one factor in the coagulation cascade and act indirectly, had been widely used (for over 50 years) by the First Priority Date.
368 As at the Second Priority Date, it was known that the targeting of a single factor could produce a safe and effective anticoagulation. There was interest in the development of new anticoagulants directed to the specific and non-specific inhibition of factors IIa (thrombin) and Xa, as promising results had been achieved, particularly with ximelagatran.
369 It was also known that direct factor inhibitors inactivated platelet-bound factor, as well as free factor Xa. Professor Crowther agreed this was the case for direct thrombin inhibitors as at the Second Priority Date, and it was believed to be the case for direct factor Xa inhibitors at that time. He observed that the fact that the direct factor inhibitor could get into the clot and “effectively sterilise the clot” was seen as an advantage.
370 Professor Crowther agreed that the following quote, taken from the summary at the end of the Chest paper, was a fair reflection of the state of knowledge and interest in relation to the development of new anticoagulant agents, particularly oral ones as at the Second Priority Date:
With a large number of new anticoagulant agents in advanced stages of development, our armamentarium of treatment options is likely to soon be expanded. Particularly promising are new oral anticoagulant agents because they have the potential to streamline the long-term prevention and treatment of patients with venous and arterial thrombosis.
371 The apparently unqualified optimism of this quote needs to be understood in the context in which it appears in the article, namely, at the end of two pages discussing the difficulties and challenges associated with the development of new anticoagulant agents headed “Conclusions and Future Directions” which commences with the following paragraph:
The development of new anticoagulant agents is challenging. Adequately powered phase II clinical trials are needed to identify the optimal anticoagulant dose, and large and expensive phase III programs are necessary to compare the benefit-to-risk profiles of new agents with those of conventional treatment regimens. Even if new anticoagulant agents prove to be superior to the currently available agents, their advantages have to be substantial to offset the additional cost. The challenges for the development of new anticoagulant agents are different for the prevention and treatment of venous thrombosis than they are for those of arterial thrombosis.
372 Not only are there challenges with the development of new anticoagulant agents, as the above paragraph highlights, there are different challenges in developing anticoagulant agents for the prevention and treatment of venous thrombosis and for those of arterial thrombosis. There would also be different challenges and concerns in developing an oral anticoagulation agent for short-term use after orthopaedic surgery, or an agent for extended prophylactic use such as the prevention of strokes in patients with atrial fibrillation.
373 The Chest paper noted that many of the new anticoagulants were initially evaluated as thromboprophylaxis in high-risk orthopaedic patients. This approach was chosen because the risks of DVT remain high in this patient population several weeks after surgery despite currently accepted thromboprophylaxis regimens, and regulatory agencies are willing to accept this end point as a surrogate for clinically important venous thromboembolism. As a result, antithrombotic efficacy can be established in relatively small numbers of patients. In contrast, demonstrating efficacy in acute coronary syndromes requires a much larger sample size.
10.1.13 Unmet need for safe and effective new oral anticoagulant
374 It is clear that, at the priority dates, there was an unmet need for developing a safe and effective new oral anticoagulant to replace warfarin or be an oral substitute for LMWH.
375 Professor Baker described a safe and effective orally administered anticoagulant as being “particularly desirable”. By “safe”, Professor Baker meant a drug that had an anticoagulant effect but did not cause unacceptable side effects such as excessive bleeding. It is generally more convenient for patients to take an anticoagulant orally, rather than having an intravenous or subcutaneous injection. Intravenous administration requires assistance from a healthcare professional. Subcutaneous injections may also require assistance from a healthcare professional, depending upon the patient. Injections can cause bruising, pain and discomfort, and there are risks of infection and bleeding at the injection site. Ideally, the oral anticoagulant would be taken once a day.
376 In a note published in January 2002 in the Medical Journal of Australia under the heading, “Updates in Medicine”, Professor Baker wrote in relation to potential new anticoagulants:
Each drug must show a positive benefit-to-risk profile, and particularly cost effectiveness, in the face of the marginal therapeutic advantage over established agents. The new drugs are likely to avoid the serious non-anticoagulant side effects of heparin, such as thrombocytopenia, and perhaps osteoporosis. With the trend for reduced hospital stay and evidence suggesting that the risk of venous thrombosis remains high for several months after orthopaedic surgery, oral agents are likely candidates for improving care. The oral thrombin inhibitor H376/95 is arousing most interest, as it produces predictable anticoagulant response without laboratory monitoring. It is currently being evaluated in phase III trials as a possible substitute for warfarin in venous disease and atrial fibrillation.
377 Professor Baker explained in the Joint Session that H376/95 is what is now known as ximelagatran.
378 The experts agreed that there was substantial research and development ongoing in relation to new and improved parenteral and oral anticoagulants targeting many aspects of the coagulation cascade, including factors IIa, VII, IXa. However, as Professor Crowther observed in his 2004 article, discussed above at [347], “[a]lthough several promising new anticoagulants are in development, few have been approved”.
379 There were various potential methods by which to improve the field of anticoagulant therapy, including:
(a) an anticoagulant that did not require monitoring, even if it had to be taken more than once a day. This would represent a significant improvement in treatment options for both patients and healthcare providers; and
(b) an oral anticoagulant with a faster onset of action than warfarin for patients who would only be taking the drug in the short-term (eg as a result of an orthopaedic surgery); and
(c) ideally, an oral anticoagulant that could be taken once per day and did not require monitoring.
380 These methods could potentially be achieved by:
(a) making improvements to an existing molecule, for example an oral form of LMWH; or
(b) developing a new molecule.
381 According to Professor Baker, as at the Second Priority Date, there was particular interest in the development of new thrombin and factor Xa inhibitors. His January 2002 note, discussed above, referred to research underway at that time:
Most focus has been on factor X and thrombin, but there are new anticoagulants for almost every coagulant factor. Three direct thrombin inhibitors (hirudin, bivalirudin, and argatroban) are approved for clinical use in the United States. Four other anticoagulants (activated protein C, tissue factor pathway inhibitor, synthetic pentasaccharide [fondaparinux], and the oral thrombin inhibitor H376/95 [ximelagatran]) are undergoing or have completed phase III evaluation studies.
382 As at the First Priority Date, LMWH and warfarin were well established as safe and effective agents for thromboprophylaxis in high-risk patients and for the treatment of venous thromboembolism. To gain widespread acceptance, any new anticoagulant agent would therefore need to have a benefit/risk ratio that was at least as good as these existing agents and at a comparable cost.
10.1.14 Stages of drug development
383 It was common ground that the drug development process can be complex, time consuming, expensive and iterative. Statistically, it is not uncommon for drug development projects to fail if, for example, the drug product is considered unsafe, lacking efficacy or not commercially viable.
384 The experts noted in JER2 that they had provided in their affidavits a “common overview of the drug development process and the information that is required in order to make a submission for regulatory approval for the use of a new drug in a specific indication”. The drug development process was well defined at the priority dates and can be generally split into a drug discovery process and a drug development process, the stages of which are discussed below.
385 The experts agreed that the research and development process in relation to a new chemical entity (NCE) for small molecules is different from the development of new, improved or generic formulations of pharmaceutical products that have already obtained marketing authorization. A starting point for generic drug formulation development includes knowing the active pharmaceutical ingredient (including form), dose, dosage form and route of administration as well as the safety and efficacy information. For a NCE, there is no reference product that provides confidence that the drug can be safe and efficacious.
386 The research and development process in relation to an NCE is also different to the development process for extending or “repurposing” a known drug for a new indication or for a drug that is a member of a class of drugs where some members are already on the market. Professor Evans contrasted the risks inherent in trying a NCE for the first time in humans, with a drug which is the third or fourth generation of a chemical class. Less unusual or unpredictable toxicities are likely when another member of a known class of drugs was tested in humans for the first time.
387 The experts were largely in agreement as to the nature of the distinct steps involved in a new drug development program (rather than the development of a generic drug) and the information that is required in order to make a submission for regulatory approval for the use of a new drug in a specific indication. The steps of the drug development program follow an established pattern, which is in large part dictated by the requirements of the relevant regulatory bodies, although every drug development program will have its nuances as to how the steps unfold.
388 The stages of the drug development process for a NCE (and first in its class) identified by the experts include the following:
(a) Discovery or identification of the NCE (including its chemical structure) and selection and optimization of a lead compound or compounds;
(b) Pre-clinical collection of data as to the chemistry, pharmacology, toxicology, physicochemical properties, pharmacokinetics and biostatistics of the new compound;
(c) Designing and conducting comprehensive in silico and in vitro studies to provide an indication as to the drug’s potential pharmacokinetics and biological activity in humans;
(d) Phase I clinical studies, also known as “first-in-human” studies, in which the drug is given to a small number of typically healthy participants;
(e) Phase II clinical studies, whereby the dose or dose regimen is administered to a small number of participants, typically patients who have the relevant condition being treated;
(f) Phase III clinical studies, in which the drug is administered on a large scale to patients; and
(g) Phase IV studies, which are ongoing and in which post-approval marketing surveillance is conducted to monitor the use of the product in the market.
389 Each step of the drug development program provides new data that can halt the program, lead to revision of the program, modify the development of the program or provide confidence to pursue a predetermined plan.
390 The first step is to identify and select a lead candidate or candidates. According to Professor Roberts, this involves a medicinal chemist synthesising or discovering a range of drugs for testing. There will also be input from a pharmacologist. The product development team then conducts tests on the drugs to characterise their physicochemical properties. Large numbers of drug candidates may be tested in animal models during the screening phase of drug discovery, and it is not unusual for multiple (six to 12) drug candidates to be investigated during such discovery studies. Such in vivo animal models may provide a rudimentary indication of pharmacological activity, but not clinical efficacy. Professor Evan observed that “there are things that you can [do] in an animal study to investigate whether or not a drug is orally absorbed that you wouldn’t necessarily progress to a human study”.
391 If the compound is known (its specific chemical structure and physicochemical form has been published), the development team would need to begin with an in silico evaluation of the compound, which could give an indication of physicochemical and some pharmacological properties. This analysis may define whether or not the compound is suitable for oral administration. If the in silico evaluations suggest that it is not suitable for oral administration, the team may decide not to proceed further.
392 Prior to clinical studies being undertaken in humans, and before a drug can be administered to a human for the first time, data needs to be collected for the drug from pre-clinical studies spanning chemistry, pharmacology, toxicology, pharmaceutical science and biostatistics. This information would be assembled for consideration and interrogation by a regulatory body (eg as a New Drug Application in the United States), as well as human research ethics committees.
393 The experts explained that there are two categories of pre-clinical tests in new drug development.
(a) mandatory tests that are required by regulatory bodies for the development of all drugs as the routine requirements ahead of use in humans and which are essential for selecting doses for initial human testing (first time in man). These include a large number of single and multiple dose safety, toxicity, pharmacokinetics (absorption, distribution, metabolism, excretion (ADME or LADMET)) studies in multiple animal species; and
(b) “proof-of-concept” type tests that are specific to the particular drug class being investigated (in this case, tests of antithrombotic properties) using in vitro and animal models, which may or may not be established in the literature.
394 The mandatory type tests will reveal information on safety and possible pharmacokinetic properties in humans. The proof-of-concept type studies can give an early indication of relative efficacy. Pharmacologists play an important role in these studies. A clinician may have input into this phase in some circumstances. For example, a clinician with expertise in the particular research area may be involved in advising on the specific proof-of-concept studies that may be most relevant to the clinical setting (eg selection of suitable animal models or advising on the most appropriate surrogate pharmacodynamic end points of efficacy).
395 The results from pre-clinical studies would be used to predict the initial safe starting dose for a first time in human Phase I study. In most instances, this prediction work normally falls to pharmacokineticists and pharmacologists. The experts differed in their views on the role of a clinician in this process, but they all agreed that the level of involvement of the clinician will depend on the drug, the program, the therapeutic area and market, the clinical need, and the resources available to the company undertaking the drug development program.
396 The “prediction” work undertaken ahead of Phase I clinical testing would provide an indication (although not definitive) of how much of the drug might need to be given in the very first human study and what route of administration might be used (eg there may be a mg per kg oral dose range predicted from animal studies). The pre-clinical data may also help to include or exclude certain routes of administration (eg oral route of administration).
397 The selection of the dosage form also requires information on the drug itself and various aspects of chemistry, manufacturing and controls. This work would primarily be carried out by a pharmaceutical scientist(s) and would not generally involve a clinician.
398 The pharmaceutical scientist would conduct a wide range of in vitro tests to verify suitable quality features even for a simple formulation. For example, in the case of a simple tablet, it would be tested for disintegration, dissolution, hardness, friability, water content and stability. In terms of stability, the pharmaceutical scientist would be concerned not just with the possible degradation of the drug but also the appearance of degradation products (which also need to be identified and tested) and comprehensive packaging data (because often there is considerable time between making the product and administering it).
399 A pharmaceutical scientist would lead the selection of a formulation for a Phase I study. The pharmaceutical scientist would need to give attention to the quality and stability of the formulation or the range of formulations that need to be developed. Not only does a suitable formation need to be produced and assessed for performance, but it needs to be demonstrated to be stable in the packaging material and containers used to store it ahead of a clinical study. For an initial Phase I study, stability data does not have to exceed a couple of months, but for longer term Phase II studies, which can last many months, extensive stability data is required.
400 Professor Evans agreed that models for assessing antithrombotic activity existed as at the Second Priority Date. He distinguished between models to establish pharmacological activity, which he said were much more common, and models to show clinical efficacy.
401 Phase I clinical studies are the first time the drug is given to a human and involve the administration of the drug to a small number of healthy volunteers with the aim of identifying the maximum safe tolerated dose of the drugs. An early Phase I study would usually involve a wide range of single doses (starting with low doses and scaling up). Simple dosage forms, such as a solution, that can be easily scaled up or down are often used at this early stage. This minimises the complexities that come with other formulations.
402 A clinician would generally not have the expertise to be involved in formulation development work but they would be involved in the planning, execution, and evaluation of all clinical studies, and a clinical perspective (possibly from a practising clinician) would be required in the discussions that define the dosage form that might best meet clinical need.
403 Professor Evans noted that historically most Phase I studies are conducted using male volunteers, and (refreshingly) acknowledged that safety in healthy young males does not guarantee safety in females in Phase II studies.
404 Phase II clinical trials (sometimes called dose ranging studies) typically involve administering a number of different dosages of the drug, or different formulations, to a number of patients (usually between 20 and 100) who have the relevant condition being treated. The goal is to identify the most efficacious dose and formulation and confirm its safety.
405 For the early patient Phase II clinical trials, the information from all the Phase I trials is absolutely critical, particularly in terms of safety, biomarkers and pharmacokinetics. This information will ideally permit the development of pharmacokinetic-pharmacodynamic (PK-PD) models that can assist in predicting the performance of different dosage regimens in future studies. The experts observed that this requires the input of a clinician in advising on, and evaluating the efficacy and safety of, “instruments” to be used, as well as a pharmacologist with particular expertise in pharmacokinetics and statistical modelling.
406 If a suitable PK-PD model has been developed, it can be used to predict, for example, how plasma levels of the drug will vary during a dosage interval, whether the plasma level will fall within a particular range, and for how long plasma levels will fall into, and out of, the desired concentration range (if that is known).
407 Professors Evans and Roberts observed that knowledge of the pharmacokinetics of the drug from Phase I clinical trials (including its elimination half-life, if known) is of absolute importance for dose regimen design. A clinician’s input into the PK-PD modelling would not be significant, except to provide guidance on target pharmacodynamic outcomes.
408 Professors Roberts and Baker noted that while pharmacokinetic parameters are important, the pharmacodynamic properties of the drug are equally important, if they can be measured. Indeed, in some circumstances, pharmacodynamics can be dominant in determining outcomes for indirect, delayed or irreversible acting drugs, including the thromboembolic and antiplatelet drugs used at that time, such as warfarin and aspirin. They considered that a clinician could play a major role at this stage, especially in the assessment of in vitro efficacy tests when that aspect was within their expertise, but also in both the planning and evaluation of efficacy and safety aspects of dose-ranging Phase II studies.
409 In developing an anticoagulant drug, the dose and dosage regimen involves striking a balance between administering enough of the drug to give a good anticoagulant effect, and not administering too much of the drug, which may cause excessive (otherwise avoidable) bleeding.
410 According to Professors Baker and Crowther, as at the Second Priority Date, there was an established pathway for conducting Phase II clinical trials of new anticoagulants, in the form of prophylaxis testing in orthopaedic surgery patients as a first step. This was a well-defined model with a well-defined outcome. After orthopaedic surgery, patients are at risk of bleeding or DVT. Anticoagulant thromboprophylaxis after major orthopaedic surgery was the standard of care as at the priority dates as it substantially reduces the risk of serious thromboembolism, compared to the minimal risk of increased bleeding, and bleeds in orthopaedic surgery patients are rarely difficult to control or fatal.
411 Professor Crowther’s evidence was that the bleeds following orthopaedic surgery tend to be at the surgical site. The rate of fatal bleeding after major orthopaedic surgery is very low. Rather, the rate of bleeding into the joint, which impacts long-term joint function, is the concern. Professor Crowther agreed that the risks of major or fatal bleeding following orthopaedic surgery were significantly lower than, for example, those for atrial fibrillation patients, who generally had higher rates of comorbid conditions.
412 According to Professor Crowther, orthopaedic surgery was chosen as the established pathway for testing anticoagulant drugs due to the “event rate” experienced from such surgery, rather than the lower risk of fatal bleeding. Smaller studies could be undertaken because the outcome event rate, which is venous thromboembolism, was much higher. In comparison, proving that an oral anticoagulant was effective for prevention of stroke requires studies that involve tens of thousands of patients, whereas an orthopaedic surgery study could be successfully completed in the correct circumstances with a few hundred patients.
413 Professor Baker noted that in Phase II clinical trials for anticoagulants, it is typical to test different doses and dosage regimens, including both once and twice daily dosage regimens, so that the effects can be compared with established anticoagulants.
414 Phase III studies typically involve very large scale administration of the drug to patients who are ideally from a diverse range of populations. The goal is to establish the efficacy of the drug, often relative to an existing product used for the same indication (the established standard of care).
415 Professor Evans noted that, not infrequently, it was not a direct journey from Phase II to Phase III. Results emerging from the Phase II studies may reveal issues requiring a change of direction. For example, there may be issues that require the formulation to be re-examined or modified, in which case it will be necessary to go back and conduct additional Phase I studies. If the Phase II studies showed a lack of efficacy, this may require a dose increase and additional Phase I studies using the higher dose.
416 Professor Crowther observed that in Phase III trials in high-risk clinical situations like thromboembolic disorders, the team is looking for efficacy. However, they also spend a lot of time looking at toxicity and safety because anticoagulants are high-risk medications and, as a result, there are issues with respect to causing bleeding as well as preventing thrombosis.
417 It is not uncommon for drug development projects to fail, for example, if the drug product is considered unsafe, lacking efficacy or not commercially viable. It is also very difficult, if not impossible, to identify from a chemical structure (without more) whether a product will succeed through to marketing approval.
418 The majority of lead drug candidates that make it to pre-clinical studies never advance to clinical trials. Most drugs that enter clinical trials do not progress to receive regulatory approval. According to Venkatesh S and Lipper R A, “Role of the Development Scientist in Compound Lead Selection and Optimization” (2000) 89 (February) Journal of Pharmaceutical Sciences 145, 146, which was cited by the experts in JER1, this attrition rate is about 90%. That same article notes that the historical success rate for compounds going from pre-clinical trials into Phase I trials in humans is around 40%.
419 Professor Roberts agreed with the figure of 90% across the whole drug development process, observing that only about 30% of compounds make it from Phase I into Phase II clinical trials, and around 30% of compounds make it from Phase II into Phase III studies. According to Professor Roberts, when these chances of success are multiplied together, that gives a 90% chance that a compound entering Phase I trials will not proceed to market. Professor Roberts said that this is because all sorts of things could go wrong along the steps of the development process.
420 To give a drug the best chance of getting approval, it may be necessary in some cases for the medicinal chemist to change the structure of the compound along the way, change the dosage form, and/or change the formulation, such as to make changes as between immediate release, sustained release or delayed release dosage forms. Most of these potential problems cannot be anticipated at the time a compound candidate is selected as the lead compound.
421 These risks of failure are particularly acute for NCEs, for many of the reasons described above, particularly because the development team is starting with a “blank slate” compared to developing a generic drug, which means there is simply no way of anticipating what will be encountered in the development process.
422 The substantial risks and uncertainties associated with drug development are particularly problematic in the high risk field of thrombosis involving a novel, first in class drug. For example, as Professor Roberts recognised, there are “so many failures” and “no one knew what was going to work”. Further, Professor Baker agreed that the refusal of the FDA panel to recommend the use of ximelagatran in 2004 confirmed that the task of developing an anticoagulant drug that was a selective inhibitor, in solid oral dosage form and that was going to be safe and effective, was full of risk.
423 Sandoz’s inventive step case is based on s 7(3) of the Act. It does not rely on common general knowledge alone.
424 On appeal to the High Court in AstraZenca HC, Kiefel J (as her Honour then was) explained at [85] the purpose of s 7(3) as being to identify documents which may be used for the s 7(2) question of obviousness, observing that:
The sub-section does so by reference to attributes of a document and the information contained in it, as seen through the eyes of the skilled person. So long as the information in the document has those attributes stipulated by s 7(3) it may be used for the purposes of the inquiry under s 7(2). This is essentially a question of evidence, not statutory construction.
425 There are three requirements for purported prior art to qualify under s 7(3) (as in effect at the relevant time): that a person skilled in the art could be reasonably expected to have: 1) ascertained; 2) understood; and 3) regarded as relevant the information.
426 First, “ascertained” simply means “discovered” or “found out”: Lockwood No 2 at [132] (per Gummow, Hayne, Callinan, Heydon and Crennan JJ). As Beach J explained in Sequenom Inc v Ariosa Diagnostics Inc (2019) 143 IPR 24 at [598], a “document could be ascertained if it was published in such a manner or form that it could reasonably have been expected to be found by a person skilled in the art”. It is essential to both inventive step cases that the s 7(3) document would be ascertained by the hypothetical person skilled in the art.
427 Second, “understood” in s 7(3) means that, “having discovered the information, the addressee would have ‘comprehended it’ or ‘appreciated its meaning or import’”: Lockwood No 2 at [132].
428 Third, the meaning of the phrase “regarded as relevant” was addressed in Lockwood No 2 at [152]–[153] in the following terms:
[152] Given the history, context, purpose and specific words of limitation in s 7(3), all of which were addressed by this Court in Firebelt, the phrase “relevant to work in the relevant art” should not be construed as meaning relevant to any work in the relevant art, including work irrelevant to the particular problem or long-felt want or need, in respect of which the invention constitutes an advance in the art. The phrase can only be construed as being directed to prior disclosures, that is publicly available information (not part of common general knowledge) which a person skilled in the relevant art could be expected to have regarded as relevant to solving a particular problem or meeting a long-felt want or need as the patentee claims to have done. Otherwise the words of limitation in the last forty words of s 7(3) would have no role to play. Any piece of public information in the relevant art would be included, as is the case with the much broader and quite different formulation in the cognate provisions in the United Kingdom, which do not depend on the standard of a skilled person’s opinion of the relevance of the information.
[153] The question of what a person skilled in the relevant art would regard as relevant, when faced with the same problem as the patentee, is to be determined on the evidence. The starting point is the subject matter of the invention to be considered together with evidence in respect of prior art, common general knowledge, the way in which the invention is an advance in the art, and any related matters. It should be mentioned that the starting point is not necessarily the inventive step as claimed, or even agreed between parties, because the evidence, particularly in respect of a combination of integers, may support a different inventive step.
(Citations omitted.)
429 Finally, with respect to the qualifying phrase “reasonably expected”, Heerey, Kiefel and Bennett JJ observed in Commissioner of Patents v Emperor Sports Pty Ltd (2006) 67 IPR 488 at [31]:
Section 7(3) does not assume an ascertainability by any and all skilled persons, of whatever description, of all publicly available prior art documents anywhere in the world. Nor does it assume that the skilled person has found the document in question, so that the only question is whether he or she has understood it and regarded it as relevant. Such a construction ignores the elements of expectation and reasonableness, as applied to the particular skilled person.
430 Further, Beach J observed in Sequenom at [599] that:
The expectation as to what the skilled addressee “could” ascertain is a reasonable expectation, not a fanciful one. That reasonable expectation is to be assessed in light of the characteristics of the relevant hypothetical skilled person and the particular problem faced, the overcoming of which is said to involve an inventive step. The inquiry is hypothetical.
431 Sandoz relies on a single piece of alleged prior art information, WO 919, with respect to the 226 Patent. Sandoz also relies on WO 919, in addition to other prior art information, in relation to the 613 Patent. WO 919 is entitled “Substituted oxazolidinones and their use in the field of blood coagulation” and was published on 5 July 2001.
432 For the reasons below, I do not consider that Sandoz has established that WO 919 satisfies the requirements to be a s 7(3) document.
433 Professor Roberts (wearing his pharmaceutical scientist hat) was asked by Sandoz’s instructors to undertake the following hypothetical task:
Ashurst asked me to assume:
(a) I am a member of a pharmaceutical product development team … at the Relevant Date;
(b) the product development team is seeking to develop a product for the treatment of thromboembolic diseases containing a Factor Xa inhibitor as the active pharmaceutical ingredient; and
(c) I am tasked with developing a formulation for an immediate release solid oral dosage form that is suitable to take forward through clinical trials ([First] Hypothetical Task).
(Italics added.)
434 Bayer took issue with the italicised parts of the First Hypothetical Task.
435 Early in his first affidavit, Professor Roberts described the literature searches he would undertake to find any information about a target drug in order to supplement the information in the product profile that he had been given by a research team. Typically, Professor Roberts would conduct searches for the target drug by its chemical name, code name and/or any other reference names. He would conduct his searches on a number of databases, including SciFinder (which includes Chemical Abstracts), which he described as the primary database he searched, the Merck Index, Martindale, the USPTO patent database, and search engines such as Google and Yahoo to identify any publicly available records including press releases or annual reports.
436 Professor Roberts considered that the First Hypothetical Task was directed towards a formulator with a pharmaceutical science skill set (like himself) rather than a straight formulator, such as Professor Poli.
437 Professor Roberts’ evidence was that he would begin the First Hypothetical Task by conducting literature searches of the kind just described for a factor Xa inhibitor. He would also conduct broader searches using the terms “anti-thrombotic” and “anti-clotting” in case there were publications or documents about products in development where it had not been disclosed that the relevant product worked via factor Xa inhibition.
438 No search results in relation to the First Hypothetical Task were annexed to Professor Roberts’ affidavit. After his description of his likely searches, he was provided with a copy of WO 919, and asked what information, if any, WO 919 disclosed that he considered relevant to the First Hypothetical Task. Professor Roberts then spent six pages of his affidavit discussing the disclosure of WO 919 and his choice of a preferred compound. Bayer submits that the irresistible inference to any reasonable person undertaking the First Hypothetical Task would be that they were given WO 919 because it was relevant to the task.
439 Following his discussion of WO 919, Professor Roberts was provided with a 71-page “redacted” spreadsheet of search results (containing limited bibliographic information) and asked to assume that the spreadsheet reflected the results of a Chemical Abstracts search for patent literature relating to factor Xa inhibitors. Professor Roberts’ evidence was that he did not ask for the search to be limited in that manner, and that he would not normally limit a search to only patent literature. Professor Roberts was never given the results for the searches that he identified that he would have carried out across all the databases he identified. Even if his search was limited to the Chemical Abstracts database alone, his typical approach would include searching peer-reviewed literature, patents, citations and general search engines, not just patent literature. Professor Roberts agreed that he did not instruct Ashurst to limit the search to a single database (Chemical Abstracts) and to further limit the search on that database to patents only, thereby excluding these other resources which, in the normal course, he would examine. The obvious inference is that it was Ashurst, not Professor Roberts, that so limited the search results in this manner.
440 Mr Cruise conducted the search of Chemical Abstracts limited to only patents using the search term “factor Xa inhibitors”, not the term “factor Xa inhibitor” proposed by Professor Roberts. Mr Cruise’s search for “factor Xa inhibitors” produced about 177 results and 1,200 pages of results. Mr Cruise agreed that it was important to his search whether “factor Xa inhibitor” or “factor Xa inhibitors” was used, and that if the search was not limited to patent literature, the results would have been “significantly greater” than 177. Mr Cruise’s 1,200 pages of results were reduced (presumably by the instructors) to a 71-page spreadsheet which was provided to Professor Roberts.
441 Professor Roberts only had a limited time (of about two hours) to review the spreadsheet of results. Following a “rapid scan” of the results, he classified the results as “top priority”, “yes”, “potentially relevant” and “no”, which he explained reflected his “initial view”. Professor Roberts marked WO 919 as a “top priority” document. Professor Roberts’ evidence was that he would have proceeded to retrieve the full document for each of the results he identified as "top priority", "yes" or "potentially relevant" (151 documents). Professor Roberts observed that his initial ranking was just his initial view and not absolute and that he may change his mind having reviewed a paper “because sometimes abstracts don’t tell you the full story”. Professor Roberts was not provided with copies of any of the 151 documents, other than WO 919 which he had been given earlier. Before making a final selection, Professor Roberts said that he would have carried out further searching over additional databases, including a citation search.
442 Professor Polli’s evidence was that a formulator would not be responsible for selecting a lead drug candidate to take forward for clinical trials, so he would not be carrying out a search process to find a potential candidate.
443 Bayer submits that it is not in dispute that the intellectual exercise that s 7(3) requires is providing an expert with a statement regarding the nature of the “task, problem or suboptimal situation” that must be addressed: AstraZeneca FC at [524] (per Jessup J). However, it takes issue with the “task” given to Professor Roberts in the First Hypothetical Task. According to Bayer, Professor Roberts was given a starting point which included aspects of the invention, and that the “formulation of the ‘task’ amounted to leading Professor Roberts to the invention”.
444 Bayer submits that Sandoz has not established that WO 919 is a s 7(3) document. Not only was Professor Roberts given the wrong starting point, but he was then directed to find WO 919. Professor Roberts was provided with a copy of WO 919, and asked to discuss what it disclosed in relation to the First Hypothetical Task, before he was given the limited Chemical Abstracts patent results spreadsheet, rather than the full results of his proposed search strategy across multiple databases.
445 Sandoz rejects Bayer’s criticism of its s 7(3) evidence, noting that the s 7(3) exercise is a threshold requirement, not a mini-trial in and of itself. Sandoz submits that its evidence follows the “pattern” that was laid out in AstraZeneca. Relying on the evidence of the search processes in Otsuka Pharmaceutical Co Ltd v Generic Health Pty Ltd (No 4) (2015) 113 IPR 191 and Merck Sharp & Dohme Corp v Wyeth LLC (No 3) (2020) 155 IPR 1, Sandoz submits that the search process outlined in AstraZeneca is not prescriptive, and that an absence of such search evidence is not fatal.
446 Sandoz submits that it is not to the point that Professor Roberts would not have limited his search to patents, and that a search of other databases would have identified more results. The fact that additional documents may have met the threshold requirements of s 7(3) does not undermine the proposition that WO 919 meets those requirements and, in particular, that WO 919 could be reasonably expected to have been ascertained by the skilled team.
447 According to Bayer, the First Hypothetical Task did not reflect the situation in which the person skilled in the art would engage in the intellectual exercise for which s 7(3) provides, including because:
(a) The safety and effectiveness of oral single point inhibitors directly targeting factor Xa and other enzymes higher in the coagulation cascade was treated with some caution and scepticism.
(b) Professor Baker, for Sandoz, would have focused on at least thrombin and factor Xa inhibitors. Professor Crowther’s main focus would have been on the most clinically advanced thrombin inhibitor, ximelagatran.
(c) The only factor Xa inhibitor that was available for any indication anywhere in the world (fondaparinux) was administered by subcutaneous injection. Two of the three main anticoagulants were administered parenterally.
(d) The dosage form and release rate of any new compound which had not been previously formulated into any pharmaceutical composition is informed by physiochemical, pharmacokinetic and pharmacodynamic considerations. A clinician may express a preference in that regard, but it begs belief to suggest that the formulator in the notional team would be instructed as to the release rate and dosage of such a “target drug” in circumstances where, as in this case:
(i) the target drug had not been identified;
(ii) the target drug was a newly synthesised compound that had never been formulated into an approved pharmaceutical product; and
(iii) the experimental information regarding that compound did not include information on the formulation of that compound.
448 To properly test the proposition of ascertainment and relevance, the most prudent approach, and Bayer contends the necessary approach in the contested circumstances in the present case, would have been for Sandoz’s lawyers to test whether Professor Roberts would have actually identified WO 919 as a document he would read from the results of his own search strategy. It did not do so. The failure to do so cannot be rectified and, Bayer submits, is fatal to Sandoz’s case that Professor Roberts would have ascertained and selected the WO 919 patent.
449 The gold standard exemplar for the evidence required to establish that a document satisfies s 7(3) is the search process set out in AstraZeneca FC and endorsed by the High Court in AstraZeneca HC (see, eg at [20]–[21] (per French CJ), [75]–[80], [83]–[86] (per Kiefel J), [103]–[107], [111] (per Gageler and Keane JJ)). As Sandoz seeks to draw parallels between this case and the invention claimed in the patent in suit (051 Patent) in AstraZeneca, it is worth setting out the substance of that case in some detail.
450 The primary judge in the proceeding — Apotex Pty Ltd v AstraZeneca AB (No 4) (2013) 100 IPR 285 — was assisted by the expert evidence of Professor O’Brien and Dr Reece. They gave evidence of searches they carried out leading to identification of the two pieces of prior art information invoked against the patent; the Watanabe Article (published as Watanabe et al, “Synthesis and Biological activity of Methansulfonamide Pyrimidine- and N-methanesufonyl pyrrole-substituted 3,5-dihtyroxy-6-heptenoates, A Novel Series of HMG-CoA Reductase Inhibitors” (1997) 5(2) Bioorganic & Medicinal Chemistry 437) and Patent 471. In the course of identifying that prior art information, the experts had regard to other prior art publications disclosed by their searches.
451 Chief Justice French at [21] (Kiefel J, Gageler and Keane JJ and Nettle J agreeing) upheld the remarks by Jessup J in AstraZeneca FC at [530]:
It is, in my view, wholly within the scheme of the subsection that [the skilled person] might well sort through all manner of information with a view to finding something that is “regarded as relevant”. There is nothing in the provision which would place an embargo upon the skilled person using combinations of sources of information along the road to that destination.
452 By way of background to the inventive step case in AstraZeneca, the 051 Patent specification referred to a number of statins, and identified that the level of efficacy and safety achieved at the recommended dosages of currently marketed statins was less than optimal. A solution was then set out:
Surprisingly it has now been found that when dosed orally to patients with hypercholesterolemia at particular dosages or in a particular dosage range the Agent lowers total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-C) by an unexpected degree, and without any significant adverse side effects.
453 The invention claimed in Patent 051 ultimately related to the use of a chemical compound, rosuvastatin, at low dosage levels (5-10 mg daily) for the treatment of hypercholesterolemia.
454 As French CJ observed at [28]–[30], the primary judge found that the “existence, the name and the chemical structure of rosuvastatin and the fact that it had been subject to clinical trials were not part of the common general knowledge before the priority date of the 051 Patent”. However, the compound and its chemical structure were disclosed in the 471 Patent and the Watanabe Article.
455 The primary judge’s findings as to the common general knowledge at the priority date were not challenged. These included that statins had for some time been prescribed by medical practitioners for the treatment of conditions such as hypercholesterolemia and for lowering low density lipoprotein (LDL) cholesterol. Statins were typically taken once a day as tablets. According to the MIMS Annual 1998, recommended starting doses of statins were typically 10, 20 or 40 mg. Medical practitioners usually prescribed the lowest dose of statins to begin with to minimise the risk of adverse side effects associated with statins. If the target level of LDL cholesterol was not met and the dose had been well tolerated, it would be increased (dose titration) over months.
456 The first piece of prior art, the 471 Patent, was summarised by French CJ at [31]–[33], including that the compound of the patent was rosuvastatin, although that name was not used. The specification set out methods for its preparation and stated that it could be administered orally in a variety of forms. The specification also mentioned dosages but there was no reference to clinical trials in relation to the compound. One of the listed inventors was the first-named author of the Watanabe Article.
457 Chief Justice French then summarised the disclosure in the Watanabe Article at [34]. The article reported on the synthesis and biological activity of particular compounds, including rosuvastatin, although it was referred to as S-4522. The enzyme inhibitory effect of S-4522 was said to have been studied in hepatocytes isolated from rat livers, and found to be 100-fold more potent than a statin compound widely used clinically. The article noted that a clinical trial of S-4522 was in progress.
458 Both experts had been instructed with a hypothetical task. Dr Reece was instructed as follows:
You are given a new statin and are told that it is useful in the treatment of hypercholesterolemia, hyperlipoproteinemia and atherosclerosis. HMG-CoA reductase inhibitors are the most widely used prescription medication for the treatment of hypercholesterolaemia. A number of HMG-CoA reductase inhibitors are marketed, namely lovastatin, pravastatin, simvastatin, fluvastatin, atorvastatin and cerivastatin, and are collectively referred to as “statins”. Despite the benefits of statin therapy, less than optimal results may be achieved in patients, due to the level of efficacy and safety achieved at the recommended dosages of the currently marketed statins. Accordingly it is important to find dosages of alternative statins which beneficially alter lipid levels to a significantly greater extent than similar dosages of currently used statins and which have a similar or improved safety profile.
(Emphasis added.)
459 Professor O’Brien’s instructions set out only the section in italics. In AstraZeneca FC, Jessup J held at [522] that the utility of Dr Reece’s evidence was not compromised because of the premise in his instructions that there was a new statin in existence.
460 As can be seen from the statement of instructions, one significant difference between this case and AstraZeneca is that in AstraZeneca there were already other statins on the market, and the person skilled in the art was looking to find dosages of alternative statins to those currently available on the market.
461 The primary judge described the search process of the two experts as “routine and conventional literature searches” (at [328]). Dr Reece’s search process was set out at [37] of French CJ’s reasons:
In response to the problem set for him, Dr Reece searched for published articles which had the potential to provide information about a new statin with the characteristics referred to in his instructions. He acquired nineteen abstracts of articles that appeared to fall within his terms of reference. He obtained the full texts of the articles concerned. He concluded that only three were relevant to his terms of reference. The lead authors were respectively Aoki, Watanabe and Thompson. The Aoki Article referred to an HMG-CoA reductase inhibitor called NK-104. The Watanabe Article, as already outlined, contained a report on a series of compounds including rosuvastatin which Dr Reece recognised as “a very potent inhibitor of cholesterol biosynthesis” and as “definitely a candidate for further development”. The Thompson Article referred to a number of “the more promising looking compounds in the pipeline”, including NK-104 and rosuvastatin. Doctor Reece’s assessment of the relevance of the Watanabe Article involved his assumption that the unknown compound with the characteristics for which he was searching was at the stage of phase II trials at least. As noted above, the Watanabe Article stated that rosuvastatin was in clinical trials. Doctor Reece took that to mean either phase I or phase II trials. The Thompson Article told him that rosuvastatin was in phase II trials. Consistently with his approach to the construction of s 7(3), which has been accepted as correct earlier in these reasons, Jessup J, in effect, concluded that Dr Reece’s use of the Thompson Article did not prevent his evidence from supporting a finding that the Watanabe Article met the relevance requirement.
(Citations omitted.)
462 His Honour then described Professor O’Brien’s search process at [38]:
Professor O’Brien’s search and elimination process led him to the Watanabe and Aoki Articles and, via a footnote in Watanabe, to a Japanese patent equivalent to Patent 471. He was subsequently provided with a copy of the equivalent United States patent. He identified five abstracts of articles, two of which related to the statin cerivastatin. Those two abstracts suggested it was less efficacious than atorvastatin. Another article by Betteridge was a general reference which did not describe any new statin. Professor O’Brien preferred rosuvastatin, designated as S-4522 in the Watanabe Article, over NK-104, the subject of the Aoki Article. Rosuvastatin appeared to be more effective than pravastatin, fluvastatin and lovastatin and had progressed to clinical trials. The Watanabe Article also indicated that it had high potency with reduced side effects in clinical use. It appeared to be further along the line of development than NK-104. Professor O’Brien therefore regarded rosuvastatin as the most relevant compound in relation to the defined problem.
463 The 471 Patent or the Watanabe Article armed the person skilled in the art with rosuvastatin (designated as S-4522 in the Watanabe Article). The only remaining issue was whether the person skilled in the art would be directly led as a matter of course to try the dosage claimed in the 051 Patent (a dosage of 5-10 mg daily). Justice Jessup observed at [544] that “[e]ven before [Dr Reece] had reviewed the Watanabe article and the 471 Patent — that is to say, working only with the instructions given to him that there was a new statin at least as efficacious and safe as others with which he was familiar — Dr Reece expressed the expectation that the dose sizes used in the hypothetical Phase II trials would be 5 mg, 10 mg and 20 mg”. Thus, after reviewing the 471 Patent and the Watanabe article, Dr Reece simply followed his earlier evidence.
464 The evidence in AstraZeneca was that the person skilled in the art would never have chosen the dose to be tested simply by trying the doses that worked for other statins. However, Jessup J observed at [547] that, although neither the Watanabe article nor the 471 Patent contained safety data from animal or human trials, the evidence of Dr Reece made it quite clear that such trials would conventionally be carried out based on the information disclose in the Watanabe Article (see also Kiefel J at [94] on appeal). Such trials “would fall within the concept of working towards the invention with an expectation of success” referred to in AB Hässle at [53]–[54] (per Gleeson CJ, Gaudron, Gummow and Hayne JJ).
465 In contrast to the present case, rosuvastatin was not a first in class to be registered molecule. Statins (or HMG-CoA reductase inhibitors) were commonly used and were regularly prescribed. They were known to be a safe and effective once per day oral treatment for reducing LDL cholesterol in patients with, or at risk of, cardiovascular disease or with elevated cholesterol levels.
466 Both experts in AstraZeneca were tasked with finding an alternative statin to those statins already commonly used and prescribed. They were not looking for a new class of non-statin lipid lowering compounds that worked via a different mechanism in the body (ie other than HMG-CoA reductase inhibitors) that would be a better alternative to the existing statins for the treatment of hypercholesterolaemia.
467 In Otsuka, the skilled team, which included clinical psychiatrists, was looking for new drugs to treat the negative symptoms of, and cognitive impairment associated with, schizophrenia. The publications in question (Saha and Petrie 1998) were published in Schizophrenia Research, which according to the evidence was one of the two premier journals for clinical psychiatrists who wished to keep up to date with developments in the treatment of schizophrenia. In cross-examination, Professor Singh said that, as at the priority date, he “read the journal diligently” and he agreed that it was one of his “stock-in-trade” resources: at [409]. Another expert, Dr O’Dea, described Schizophrenia Research as “one of the most reputable schizophrenia journals in Australia and the world”, calling it a “key publication for psychiatrists who are interested in new developments in the treatment of schizophrenia”: at [412]. The evidence of a third expert, Professor McGorry, was that he “kept up-to-date with developments in the field of schizophrenia by regularly reading journals, including Schizophrenia Research”: at [413].
468 At [414] of Otsuka, Yates J observed that “there was no evidence concerning the nature of any hypothetical literature search that the person skilled in the art would have undertaken at the priority date”. Nevertheless, on the basis of the evidence before him as to the repute of Schizophrenia Research and the reading habits of psychiatrists wanting to keep up to date, Yates J was satisfied that, at the priority date, the person skilled in the art would have consulted editions of Schizophrenia Research published in the years proximate to that date. Whilst this finding has relevance to whether the Blood Abstracts satisfy the requirements of s 7(3), a subject to which I will return, it does not assist Sandoz with respect to WO 919.
469 In Merck, Burley J considered whether a prior art patent, known as the Chiron patent, satisfied the requirements of s 7(3). His Honour at [840] characterised the problem posed to the person skilled in the art as being whether a stable formulation could be achieved. This was a broader characterisation than that propounded by Wyeth which was whether “the resolution of a detected problem arising from silicone oil induced aggregation”.
470 The evidence in Merck indicated that the Chiron Corporation was known to be working in the field of vaccines before April 2006 as a research institute, a formulator and as a world leader in conjugate vaccine formulation. Each of the two experts was aware of the work of Chiron in the field. Professor Petrovsky’s evidence was that he would have searched for terms including “vaccine”, “adjuvant”, “conjugate”, “polysaccharide” and “stability”. He would have actively searched for publications by Chiron, having regard to their reputation in the field. In his oral evidence, Professor Dalby accepted that if he had been looking to create a vaccine formulation with a stable pH, he would have been looking for documents from Chiron and would have used the word “vaccine” as a search term and would have found the Chiron patent.
471 In the absence of evidence of literature searches, Burley J was satisfied on the basis of the evidence that the Chiron patent would have been located, particularly having regard to its title (“Vaccines Comprising Aluminium Adjuvants and Histidine”), and the high likelihood that the notional skilled formulator would have been interested to know of the work done by Chiron in relation to vaccines. At [846], Burley J cited Otsuka (at [414]) for the proposition that the “absence of evidence concerning the nature of any hypothetical literature search is not fatal to the correct application of s 7(3)”. Ultimately, both Yates J in Otsuka (at [399]) and Burley J in Merck (at [846]) concluded that it is a question of fact on the evidence whether or not the person skilled in the art could reasonably be expected to have ascertained, understood and regarded as relevant a particular item of prior art information.
472 The absence of evidence concerning the nature of any hypothetical literature search is not fatal to the correct application of s 7(3). Indeed, sometimes such searches, particularly in relation to articles in highly regarded scientific journals to which all the experts subscribe and in all likelihood publish in, spawn an enquiry that “adds unnecessary expense and argument to an already time consuming aspect of a validity challenge”: Hanwha at [367] (per Burley J). Each case depends on its facts.
473 Rather than rely on evidence as to each step of the search process, Sandoz seeks to take a short cut, relying on the absence of search evidence in Merck and Otsuka, to jump straight to the assertion that the person skilled in the art would ascertain WO 919, without establishing the foundation for such an assertion via evidence of the search process the person skilled in the art would undertake.
474 This is not a case such as Otsuka where the purported s 7(3) document is an article published in a premier journal regularly read by those in the field to keep up to date with developments. Nor was there evidence, unlike in Merck, that Bayer was known to be actively researching and developing factor Xa inhibitors.
475 WO 919 is a patent specification. Whilst the experts might have regard to patent literature if it turns up in a search, a patent specification is significantly different to an article in a high impact premier journal which is read by all those in the field wanting to keep up to date with the latest developments. It must be found. Therefore, there needs to be evidence to establish that the patent specification would, in fact, be found.
476 I consider that the WO 919 s 7(3) case bears more similarity to the Kazakov document in Sequenom than any of the cases relied upon by Sandoz. In that case, the prior art comprised an obscure Russian article which Beach J found was only identified using “peculiar search terms” (at [628]) by a search strategy that was formulated by lawyers, not the experts.
477 I do not consider that the evidence establishes that the person skilled in the art could reasonably be expected to have ascertained, understood and regarded WO 919 as relevant so as to satisfy the s 7(3) requirements. The evidence demonstrates that WO 919 could have been returned by a search of relevant patent databases but fails to demonstrate that it would have been reasonable to expect a person skilled in the art to have ascertained it.
478 At the outset, Professor Roberts was directed by the First Hypothetical Task to focus on factor Xa inhibitors. That hypothetical task was narrower than the problem found in the common general knowledge and skewed to finding the answer. The result — focussing on factor Xa inhibitors and ignoring factor II inhibitors — is a narrower search than would actually be carried out by the person skilled in the art.
479 Sandoz impermissibly sought to support its narrowing of the task to only factor Xa inhibitors by referring to a contemporaneous statement from Bayer that factor Xa was a “particularly promising target”. The citation given for the statement was a 2004 abstract which was not part of the common general knowledge or established as a s 7(3) document. Professor Baker’s evidence in relation to the later priority date of the 613 Patent was that there was also interest at that time in at least another inhibitor – thrombin inhibitors. He gave evidence that, had he been given the First Hypothetical Task, he would have focussed on both factor Xa inhibitors and thrombin inhibitors, and Professor Crowther said he would have just focussed on thrombin inhibitors (particularly ximelagatran) had he been given the task.
480 As is apparent from the discussion of the common general knowledge above, there is no evidence to support such a narrow focus which ignores other potential antithrombotic candidates. The common general knowledge was simply that there was an unmet need for a safe and effective new oral anticoagulant to replace warfarin and LMWH.
481 There was no evidence that the focus of the person skilled in the art would be only on factor Xa inhibitors and nothing else. Professor Roberts’ evidence was that the general area of antithrombotics was a very active area in which a lot of people were looking for possibilities. Professor Baker, commenting on the field at the Second Priority Date, observed that there was a lot of excitement about new anticoagulants, in particular thrombin and factor Xa inhibitors.
482 The First Hypothetical Task differed from Professor Roberts’ usual literature search experience, wherein he would search for information about a target drug in order to supplement the information about the drug that he would have received from the other members of the team. Professor Roberts explained that, as a formulator, he was typically not the member of the research team that would identify the target drug. The search process that he outlined was one that he would undertake as part of a drug development, rather than a drug discovery, team where his role as formulator only arose after the target drug had been identified.
483 Professor Roberts typical search practice is set out above at [435]. As I noted above at [440], the literature search undertaken by Mr Cruise was contrary to Professor Roberts’ instructions as to his usual search process. In short, instead of searching all the databases that Professor Roberts said that he would typically search, using the terms Professor Roberts suggested, the search was run for “factor Xa inhibitors” across one database within Chemical Abstracts — the patent records.
484 I do not disagree that the patent database would have been one of the databases searched. However, it would not have been the only database searched. Searches of the other databases, such as MedLine, would have produced peer-reviewed articles and review articles. In practical terms, seven other databases were not included and two other search terms (“anti-thrombotic” and “anti-clotting”) were not included.
485 The limited search conducted by Mr Cruise produced 1,200 pages of results. The raw results of the limited Chemical Abstracts patent search were not given to Professor Roberts. Before the search results were given to him, the results were winnowed down from 1,200 pages to a 71-page spreadsheet. However, no details were provided as to the winnowing process.
486 The omission of the other databases and sources of information from the search vastly reduced the volume of the search results that could be expected had the search contemplated by Professor Roberts been carried out. The volume of results was also likely further reduced to a significant, but unascertainable, extent by only focussing on factor Xa inhibitors.
487 Prior to his being given the patent results spreadsheet, a copy of WO 919 was provided to Professor Roberts and he was asked to describe what information, if any, WO 919 disclosed that he considered relevant to the First Hypothetical Task. Professor Roberts read WO 919 and his detailed discussion of what relevant information was disclosed by WO 919 occupied nine pages of his first affidavit.
488 After his consideration of WO 919, Professor Roberts then reviewed the patent results spreadsheet and, perhaps unsurprisingly, he identified WO 919 as one of the nine documents which appeared to him most relevant and most likely to contain information of use for the First Hypothetical Task. Yes, Professor Roberts was able to justify his selection of WO 919 as a “top priority” document from the results in the spreadsheet. However, there was no evidence that Professor Baker, who was actively researching antithrombotics at the priority dates, was aware of WO 919 at that time. In these circumstances, the question of whether Professor Roberts would have selected WO 919 if he was provided with the full suite of results from searches undertaken across all his suggested databases, and with broader search terms than just “factor Xa inhibitors”, cannot be answered on the evidence before me. There was no evidence as to how Professor Roberts would prioritise between different peer-reviewed journal articles, how he would prioritise as between journal articles and patents, or whether he might prioritise other antithrombotics over factor Xa inhibitors on the basis of the peer-reviewed articles located.
489 I do not consider that the evidence establishes that it would be reasonable for the person skilled in the art undertaking a search of the kind described by Professor Roberts, but not restricted to factor Xa inhibitors, to have found WO 919.
490 Sandoz’s obviousness case in relation to the 226 Patent is dependent upon it establishing that WO 919 would reasonably be expected to be ascertained by the person skilled in the art. As discussed above, the High Court in AstraZeneca HC set out with approval the search strategy adopted by Professor O’Brien and Dr Reece. These search strategies were not dissimilar to what Professor Roberts described as his usual search process. There was no evidence of any impediment to conducting the search process in accordance with Professor Roberts’ usual search strategy. In light of the importance of WO 919 to its obviousness case, I am left to wonder why Sandoz chose to short cut the search evidence, narrowing the target and the database searched, and providing a copy of WO 919 to Professor Roberts before he was given the winnowed spreadsheet of results. The only inference that I can draw is that the reason is because they were not confident that WO 919 would be found without these interferences.
491 Sandoz’s answer to the defects in the search evidence is that the fact that WO 919 was ascertained in a search, and Professor Roberts identified it in the list of patent results, is sufficient to establish that it would have been reasonable to expect a person skilled in the art to have found it. It contended that “there is no reason to conclude a different outcome would have followed had he reviewed other documents as well” but offered no support for this bare assertion.
492 Sandoz observes that the High Court in AstraZeneca HC (per French CJ at [24]) firmly rejected the proposition that s 7(2) does not allow the Court to decide the question of want of inventive step on the basis that “the only course available to the skilled person [was] that identified in the s 7(3) document [a]dded to the prior art base”. Sandoz submits that it is clear following that decision that if a document meets the requirements of s 7(3), it is not to the point that a search may have turned up other relevant documents. Sandoz relies on the observation of Gageler and Keane JJ at [115]:
Section 7(2) does not contemplate that a choice between apparently effective solutions must be attributed to the notional skilled addressee, much less that the notional skilled addressee might be so befuddled by an embarrassment of choices as to cease pursuit of the solution.
493 Something more is required to establish that it would have been reasonable to expect a person skilled in the art to have found a document for the purposes of s 7(3) than just showing that the document turns up amongst thousands of results in a broad search of all the relevant databases.
494 The Watanabe Article and the 471 Patent in AstraZeneca were the product of an iterative search process during which the experts used their expertise to devise search terms to narrow the search and used information in documents to arrive at their selections. More was required in AstraZeneca to satisfy s 7(3) than a search for “statins” across all possible databases.
495 In Sequenom at [668], Beach J rejected the evidence from the patent searcher that the Russian Kazakov article was indexed and searchable in English on MedLine as sufficient to establish that it would have been reasonable to expect a person skilled in the art to have found it. There was no evidence that a search using the keywords suggested by the experts would find the document. Justice Beach observed at [663] that there was “little to support the suggestion that the idiosyncratic and hindsight affected search strategy relied on by Ariosa could have been reasonably employed by the person skilled in the art undertaking the relevant project without knowledge of the invention”. Justice Beach also noted at [658] that many of the search terms proposed by Ariosa’s expert would be “expansively broad if used in isolation, or additively, and likely to return hundreds if not thousands of mostly irrelevant results”. There is no evidence to refute the possibility that the same outcome would have occurred had Professor Roberts’ search process been employed in this case.
496 Accordingly, I find that the skilled person could not be reasonably expected to have ascertained WO 919.
497 The Blood Abstracts were published in the November 2003 supplement of the journal Blood (the Abstract Book), in advance of the annual American Society of Haematology (ASH) conference held in December 2003 in San Diego, California. The three specific abstracts in question — Abstracts 3003, 3004 and 3010 — are described in more detail below at [628]–[630].
498 In contrast to WO 919, I consider that Sandoz has established that the Blood Abstracts satisfy the requirements to be s 7(3) documents.
499 Professor Baker described the task he was given by Ashurst as follows:
Ashurst asked me to assume that, in January 2005, I was part of a product development team working at or with a pharmaceutical company on a project to identify a drug and develop it into a new or improved pharmaceutical product to treat thromboembolic disorders. Asked to assume that the project did not involve the discovery of a new drug, but rather involved identifying a drug from the literature, formulating it (or instructing a pharmaceutical formulator on the team to formulate it) into a suitable dosage form, and then developing it into a pharmaceutical product.
(Second Hypothetical Task).
500 In describing his usual search process, Professor Baker said that he would search and review the literature and clinical trials databases to identify thrombin and factor Xa inhibitors in development. He would search publications using databases such as PubMed, and he would also search patent databases. His search terms would include “factor IIa inhibitors” and “factor Xa inhibitors”. Professor Baker would also review copies of the journals that he subscribed to (one of which was Blood), in order to identify thrombin and factor Xa inhibitors in development. He would also review conference abstracts and presentations to identify thrombin and factor Xa inhibitors that are in development, because he considered them to be a useful source of information. Professor Baker noted that his practice was to pay particular attention to conference presentations and abstracts because he was interested in whether his laboratory could make a research contribution. As he explained, “that’s what you need to do to connect from Australia … [to] the world, to really make a contribution”.
501 In the second joint session, Professors Baker and Crowther agreed that, in response to the Second Hypothetical Task, they would have reviewed the papers presented at the plenary and oral sessions of the 2003 ASH conference. As at the Second Priority Date, they would have reviewed the relevant Abstract Book from at least 2002, 2003 and 2004.
502 Professor Crowther described the annual ASH conference as “the major” international conference in the field of haematology, and more prominent than any other. He was a regular attendee at the ASH conferences since attending his first in 1991. Professor Crowther estimated that there would have been around 10,000 physicians, research scientists and others who attended the 2003 ASH conference. The Abstract Book states that there were approximately 300 pharmaceutical, research-based and other companies in attendance at the 2003 ASH conference.
503 Professor Baker had been a member of ASH since 1993, Professor Crowther since 1992. As part of their membership, ASH members have a subscription to Blood. ASH distributes copies of each conference Abstract Book to its members in order to provide them with a resource to keep abreast of current and recent developments in the field of haematology. Professors Baker and Crowther both received the 2003 Abstract Book.
504 Professors Baker and Crowther agreed that the ASH conference abstracts could be categorised into four “tiers”. Abstracts of the plenary session where the top eight to 10 abstracts from the entire meeting were presented were considered to be in “tier 1”, the top tier. “Tier 2” abstracts were those selected for oral presentations. “Tier 3” were abstracts of poster presentations. A poster is prepared and put up in an enormous hall for a defined time during which conference attendees are able to walk around and view the posters. “Tier 4” abstracts were those abstracts not accepted for poster presentation which were just published in the conference abstract volumes. The Blood Abstracts were tier 3 abstracts of poster presentations.
505 The Abstract Book is an “enormous tome”, often extending into multiple volumes (to avoid a single book being “many kilograms”), containing “many thousands” of abstracts. Professor Baker said that as many as 8000 abstracts may be included across the 2002, 2003 and 2004 editions of the Abstract Book. In these circumstances, Professors Baker and Crowther agreed that they would both approach the Abstract Book by first reviewing the “top line results” consisting of tier 1 and tier 2 abstracts. Professor Crowther’s evidence was that he would also focus on tier 3 abstracts that showed “mature results”, such as Phase III trials.
506 However, both experts outlined that, in response to the task to identify a suitable drug to treat thromboembolic disorders, they would adopt a targeted approach to searching the Abstract Book. Professor Baker identified a number of sections of the Abstract Book that he would review in the context of the Second Hypothetical Task, including the “Antithrombotic Therapy I” and “Antithrombotic Therapy II” poster sessions, as well as the entry for “Factor Xa inhibitor” in the subject index. Professor Crowther ultimately agreed that in response to the Second Hypothetical Task (which assumed he would not have actually attended the ASH conference), it would be reasonable to use the headings at the beginning of the Abstract Book, identify the sessions in which abstracts were presented according to their subject matter (such as “Antithrombotic Therapy”) and then look at the abstracts collected under that heading. Professor Crowther agreed that using the subject matter index was another option. In this case, the Blood Abstracts are three of only four abstracts that are listed under the keyword “factor Xa inhibitor” in the subject index.
507 The Blood Abstracts are from poster presentations at the “Antithrombotic Therapy II” poster session at the ASH conference. The abstracts for that session can be located via the table of contents of the 2003 Abstract Book, under the entry “Antithrombotic Therapy II”. There are 27 abstracts for posters in the “Antithrombotic Therapy II” session, three of which (the Blood Abstracts) concern BAY 59-7939. Further, the titles of each of the Blood Abstracts highlight BAY 59-7939 as an “oral, direct Factor Xa inhibitor”.
508 Professor Roberts and Professor Baker’s evidence was that the appearance of the three Abstracts together with a common title theme of “Oral, Direct Factor Xa Inhibitor” in human subjects would have drawn them both to read the Blood Abstracts further because (a) there was much activity in trying to find an oral replacement for warfarin and LMWH at that time; (b) there was a suggestion of direct activity on the late stage of the coagulation cascade — in contrast to both warfarin and LMWH; and (c) they were presenting Phase I human data.
509 Bayer pointed to the fact that more prominent and clinically advanced drug candidates were described in the 2003 Abstract Book which makes it less likely that a person skilled in the art could reasonably be expected to identify the Blood Abstracts. For example, Abstract 41 was the subject of an oral presentation, and therefore a tier 2 abstract that would have generated significant interest and excitement. Abstract 41 discussed a Phase II randomised double-blind study for a factor Xa inhibitor called razaxaban. The study was ultimately dropped due to increased reports of bleeding at higher doses but reported otherwise promising results. The compound razaxaban was also discussed in another abstract, Abstract 3011, which immediately followed Abstract 3010 (one of the Blood Abstracts) in the Book. Professor Baker agreed that if he had read Abstract 3010, he would have also read Abstract 3011 and realised that the compound was the subject of an oral presentation (Abstract 41). Professor Baker also agreed that he would have looked at the abstracts relating to ximelagatran and razaxaban, such as Abstract 41, with comparable interest to the Blood Abstracts.
510 Bayer’s reliance on Abstract 41 relates to its allegation that Professor Baker was “led” to the Blood Abstracts by Sandoz’s lawyers because he was asked to comment on the Blood Abstracts before undertaking the Second Hypothetical Task and, therefore, must have assumed that these abstracts were relevant to that task. I agree, for essentially the reasons I gave above in relation to WO 919 and by reference to the search process endorsed in AstraZeneca, the search process employed by Ashurst with respect to Professor Baker and the Blood Abstracts was not ideal.
511 However, I am nonetheless satisfied that a person skilled in the art could reasonably be expected to have ascertained the Blood Abstracts and understood them as relevant.
512 First, Professor Baker’s evidence was that the Blood Abstracts “would have stood out” to him because:
specific factor X inhibition, and its effect on these coagulation perimeters and thrombin generation, would have been particularly interesting. We didn’t know …what the effect these agents would have on thrombin generation, and whether they would be the similar to the effects that a person with haemophilia may have with a bypassing agent.
…
[T]hese abstracts, looking at a new agent with factor X, with some laboratory aspects, and particularly thrombin generation, was particularly interesting to me.
513 Second, the experts in JER2 also agreed that the Blood Abstracts were relevant to the work of a clinician or pharmacologist in relation to the Second Hypothetical Task. The experts observed that researchers are always interested in what else is happening internationally within their field and the information in the Abstracts would contribute to the overall body of knowledge that would help to guide their own work.
514 Third, Professor Crowther’s evidence, which was not so affected by any allegation of being “led” to the Blood Abstracts, was that he would have identified them in the manner described above.
515 Accordingly, I find that, before the Second Priority Date, the Blood Abstracts could be reasonably expected to have been ascertained, understood, and regarded as relevant by the person skilled in the art.
516 Bayer observes that both the 226 and 613 Patents encompass inventions which include identifying, selecting, formulating and synthesising compound I (rivaroxaban), and then using that compound for the treatment of thromboembolic diseases and disorders.
517 According to Bayer, Sandoz seeks to characterise the inventions without regard to, or to at least diminish the relevance of, the importance of identifying and selecting compound I as part of the invention to be assessed for the purpose of s 7(2).
518 Section 7(2) is a deeming provision. Absent a positive conclusion on the proposition following the word “unless”, the invention is taken to involve an inventive step for the purposes of the Act. Section 7(3) expands the information that can be considered for the purposes of assessing obviousness. The purpose of s 7(3) was explained by the High Court in Lockwood (No 2) at [49]:
Previously, only common general knowledge was taken into account when assessing an inventive step. Now, additional information which was publicly available as at the priority date must also be taken into account. Broadly speaking, s 7(3) has as its purpose the specification of the additional publicly available information (s 7(3) information) which must be added to common general knowledge for the purposes of deciding whether an alleged invention is obvious when compared with the prior art base.
519 The Court further explained at [127] that, in answering the question of obviousness, the information referred to in s 7(3), like that part of the prior art base which is the common general knowledge, is considered for a particular purpose. That purpose is to look forward from the prior art base to see what the skilled person is likely to have done when faced with a problem similar to that which the patentee claims to have solved with the claimed invention.
520 The question is whether the invention claimed in the 226 and 613 Patents would have been obvious to the skilled addressee in the light of the common general knowledge when considered together with the additional information brought in under s 7(3), namely, the information to be found in either WO 919 (in the case of the 226 and 613 Patents) and the Blood Abstracts (in the case of the 613 Patent).
521 First, it is necessary to identify (or characterise) the invention “so far as claimed in any claim” of the 226 and 613 Patents: AstraZeneca FC at [498] (per Jessup J); Insta Image Pty Ltd v KD Kanopy Australasia Pty Ltd (2008) 78 IPR 20 at [80] (per Lindgren, Bennett and Logan JJ). Justice Jessup observed in AstraZeneca FC at [499] that in order to gain the kind of understanding of the claim that is necessary for the purpose of identifying or characterising the invention claimed, “each claim must be read practically and sensibly having regard to the audience to whom it is addressed and allowing for the realistic prospect that members of that audience would naturally read the specification”.
522 Like the specification in AstraZeneca, there is no suggestion in the specification of the 226 and 613 Patents that the inventors had invented rivaroxaban: indeed, it is explicitly said that the compound (that is now known as rivaroxaban) was disclosed in WO 919. What the inventors say they had done was to have invented a pharmaceutical composition with identified features (hydrophilized form or rapid release) that would enable rivaroxaban to be practically used in an orally administrable form suitable for the treatment or prophylaxis of thromboembolic disorders. Rivaroxaban is an essential integer in each of the claims of the 226 and 613 Patents.
523 The Act does not contemplate, either expressly or impliedly, that the scope of prior art against which obviousness is assessed may encompass the inventor’s (or patent applicant’s) description in the specification of the invention, including any problem that the invention is directed at solving: AstraZeneca FC at [202] (per Besanko, Foster, Nicholas and Yates JJ). Further, earlier work done by the inventors which does not form part of the common general knowledge or the additional s 7(3) information is not relevant, and knowledge of that work cannot be ascribed to the inventor: AstraZeneca FC at [209] (per Besanko, Foster, Nicholas and Yates JJ). As Jessup J observed in AstraZeneca FC at [487]:
In other words, it should not matter whether earlier parts of the inventive journey had been travelled by the particular inventor, by some other inventor, or by no one. The question which arose in Insta Image would have been exactly the same (as would the answer) if there had been no earlier versions of the framework structure then invented. It would also have been exactly the same (and so would the answer) if the earlier versions had been invented by another person.
524 Each of the asserted claims of the 226 Patent claim a “pharmaceutical composition”. Claims 19 to 22 claim the use of the pharmaceutical composition for use in the treatment or prophylaxis of a thromboembolic disease. The phrase “pharmaceutical composition” imports, at a minimum, an expectation that the pharmaceutical composition for use in human patients is safe and efficacious in order for it to be useful: see Boehringer Ingelheim Animal Health USA Inc v Intervet International BV (2022) 166 IPR 468 at [96] (per Perram, Nicholas and Burley JJ). In the case of claims 19 to 22, the expectation is that the pharmaceutical composition will be safe and effective for use in humans for the treatment of thromboembolic disease. Claims 8 to 18 do not claim a treatment aspect, but instead concern the form of the pharmaceutical composition.
525 Sandoz does not allege that the invention claimed in the 226 Patent is obvious in light of the common general knowledge alone. It relies on WO 919 as a piece of prior art information pursuant to s 7(3) of the Act, which is to be taken into account in conjunction with the common general knowledge.
526 For the reasons set out above, I do not consider that Sandoz has established that WO 919 satisfies the requirements of s 7(3) and the obviousness case cannot succeed on that basis. In case I am wrong in relation to whether the person skilled in the art could reasonably be expected to ascertain WO 919, I now consider the Sandoz obviousness case based on the common general knowledge plus the information contained in WO 919.
527 Sandoz’s high-level inventive step case is that the skilled person would simply put the disclosure in WO 919 into practice by taking the most preferred compound and using standard techniques to formulate it, resulting in the claimed invention. According to Sandoz, it does not matter whether some skilled persons might consider other members of the class, in addition to rivaroxaban, and investigate their properties as well. There was no suggestion on the evidence that such inquiries, if conducted, would lead in a different direction (eg away from the development of rivaroxaban).
528 In contrast, Bayer’s case is that the research described in WO 919 is “very very early” drug discovery, that the comments made in the specification are speculative at best, and that it is just the start of a process to select a potential lead compound for further pharmaceutical development, requiring a research project to get to the claimed invention.
529 WO 919 identifies the field of the invention as being blood coagulation, and states that the invention relates to novel oxazolidinone derivatives and processes for their preparation and their use as active compounds in medicaments. It describes the background to blood coagulation and existing anticoagulants, including heparin, LMWH and vitamin K antagonists, such as warfarin. The discussion of prior art anticoagulants is preceded by the statement at page 2, lines 9–12:
The anticoagulants, i.e. substances for inhibiting or preventing blood coagulation, which are known from the prior art have various, often grave disadvantages. Accordingly, in practice, an efficient treatment method or prophylaxis of thromboembolic disorders is very difficult and unsatisfactory.
530 At page 3, the specification states that recently a novel therapeutic approach for the treatment and prophylaxis of thromboembolic disorders has been described. This novel therapeutic approach aims to inhibit factor Xa. WO 919 also discloses that it is an object of the invention in the patent to provide novel substances with a wide therapeutic window for controlling disorders. It states that the compounds which are disclosed should be suitable for more efficient prophylaxis and treatment of a range of thromboembolic disorders, and that another object is to provide novel anticoagulants which inhibit factor Xa with increased selectivity.
531 WO 919 discloses a large class of novel oxazolidinone derivatives of the general formula (I). On page 14, line 29, WO 919 states that “very particular preference” is given to the compound with the following formula:
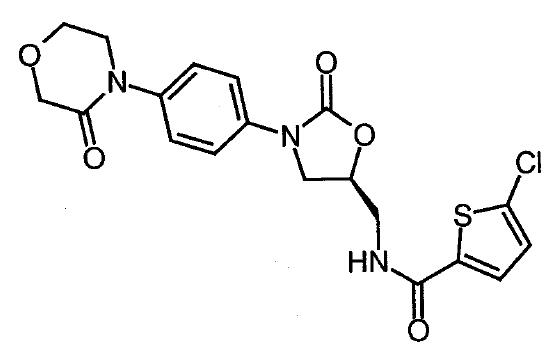
532 This compound is the only individual compound which is specifically called out in this way (said to be “very particularly preferred”) and claimed individually in claim 7 of WO 919. This compound is rivaroxaban.
533 WO 919 provides a detailed description of chemical synthetic processes for making the compounds. WO 919 discloses that the compounds have an unforeseeable useful pharmacological activity spectrum and are particularly suitable for the prophylaxis or treatment of disorders. It also discloses that they act as anticoagulants and can therefore be employed in medicaments for the prophylaxis or therapy of thromboembolic disorders. Further, it discloses that the compounds act as selective inhibitors of factor Xa as compared with other serine proteases. After a discussion of the measure of selectivity, the following statement appears at page 39, lines 21–24:
The present Invention thus provides oxazolidinones of the formula (I) effecting in particular an unexpected, strong and selection inhibition of factor Xa
534 All customary forms of administration are said to be suitable for administration of the compounds. Oral administration is said to be very particularly preferable. The compounds are said to be able to be administered as tablets or granules, using pharmaceutically acceptable excipients or solvents. After a passage which provides ranges of amounts of active compound that can be present, the following statement appears at page 40, lines 26–33:
In spite of this, if appropriate, it may be necessary to depart from the amounts mentioned … in some cases, it may be adequate to manage with less than the above mentioned minimum amount, while in other cases the upper limit must be exceeded. In the case of the administration of relatively large amounts, it may be advisable to divide these into several individual administrations over the course of a day.
535 Sandoz submits that this disclosure proceeds on the basis that the default position will be to administer once a day, unless there is a reason for doing otherwise, noting that this is unsurprising, and reflects the common general knowledge position that once daily dosing is most desirable, and should be adopted if the properties of the drug and the condition being treated allow.
536 WO 919 goes on to explain at page 41, lines 24–33, that the compounds have advantages over conventional preparations because a greater therapeutic range is achieved by the selective inhibition of factor Xa, which means that there is a lower risk of bleeding for the patient, easier adjustment for the physician and a more rapid onset of action.
537 WO 919 then discloses a series of examples which can be summarised as in vitro blood work in human blood samples and in vivo tests in rats. The first group of these (A) evaluate the physiological activity of some of the compounds, including rivaroxaban, through the use of in vitro methods to determine factor Xa inhibition, selectivity and anticoagulant action, as well as in vivo testing of antithrombotic activity in three rat models. The second group (B) provide methods for synthesis of the compounds.
538 In the first group of examples, there is a report on the results of testing in relation to a number of the compounds, including rivaroxaban (listed as Example 44). Based on oral administration to an unspecified number of anaesthetised fasted male rats using a pharyngeal tube, rivaroxaban is said to have an ED50 (a measure of the dose that provides the desired pharmacological effect in 50% of a study population) of 3 mg/kg. Rivaroxaban is one of only two compounds for which results from oral administration to rats are reported. The other, Example 123, is also reported to have an ED50 of 3 mg/kg. The ED50 of six other compounds was determined for intravenous administration to rats via their tail vein.
539 In the second group of examples, synthesis and structure information and other data is given for compounds, including rivaroxaban (Example 44), such as its melting point (indicating the drug is pure and crystalline), its mass, its optical rotation and its IC50. According to Professor Roberts, the IC50 for rivaroxaban is very low and, assuming that the IC refers to specific factor Xa inhibition, this indicates the preferred compound is quite potent.
540 The experts agreed that, as at the First Priority Date, there was interest in seeking to develop a safe and effective oral dosage anticoagulant that improved on warfarin and LMWH.
541 The experts agreed WO 919 would be of interest and value to a drug development team looking at developing a new antithrombotic drug and that, on reading WO 919, the skilled reader would focus on the compounds within the general formula (I) described by the patentee as “very preferred”. In particular, they would focus on Example 44 because it was listed as a particular chemical, discussed in examples and the only compound the subject of a claim.
542 As WO 919 is a patent specification, the data in the specification has not been peer reviewed, and there is much less data provided in WO 919 about the “very preferred” compound in Example 44, than would be expected by the experts to be contained in a scientific paper.
543 Professors Roberts and Baker described Example 44 as “exciting” because, according to the information in WO 919, it was a highly potent factor Xa inhibitor. Professor Roberts observed that an orally administered anticoagulant would be a huge advantage and if there was a fairly safe tolerance in terms of therapeutic range, that would be a “gamechanger” compared to what was available at the time.
544 Professor Baker agreed that he had been directed to discuss Example 44, and that there was a compound disclosed in WO 919 that had an even better IC50 value than Example 44, which he had not mentioned in his affidavit. He also agreed that there might be other compounds that had an IC50 of a similar magnitude which were not highlighted in WO 919. Professor Roberts observed that whilst Example 123 also had a low ED50, he would avoid it due to safety concerns he held about the molecule based on it being a heterocyclic aromatic amine, some of which were known to be carcinogenic. He would not proceed with a compound for which he perceived a safety risk. Professor Evans commented that whilst an IC50 value was important, it was not the sole factor on which to choose a drug for development.
545 Professor Baker noted that WO 919 did not contain any clinical data in support of the advantages mentioned in the specification so he considered the statements to be “speculative”. Professor Evans also considered the statements in WO 919 to be speculative. He noted that there was no evidence in the specification to support the assertion that the compounds disclosed had a wider therapeutic window than the conventional preparations on the basis of selective inhibition of factor Xa. Professor Roberts called the specification’s statement that the various compounds listed were suitable for treating a range of diseases, including thromboembolic disorders, “ambitious” and a “statement of hope”.
546 Professor Roberts qualified the ED50 figures in the examples in WO 919 by observing that in addition to not being taken in vivo in humans, no detail was provided as to the sample size, replicates or variability in the figures so that it was not possible to know if the results were statistically significant.
547 Professor Roberts considered that there was sufficient information disclosed about the “very preferred compound” rivaroxaban, including its low IC50 value and selective inhibition of factor Xa, for him to select it to be the lead candidate to take forward through the process of testing as a drug to treat thromboembolic disorders. Professor Evans disagreed. On his view, there was not sufficient information in WO 919 to do a comparative analysis of the various compounds that showed potential. The small number of tests undertaken by the patentee did not give Professor Evans the confidence that they had selected the appropriate compound from the large number in the class.
548 Professor Roberts said that he could obtain the following further information about Example 44 from the discussion provided in WO 919 and from looking at its structure and making estimates based on that information. Example 44 was likely to:
(a) be somewhat polar and moderately hydrophobic;
(b) be poorly water soluble (due to the absence of OH and COOH groups);
(c) have a high melting point, and therefore be crystalline;
(d) have a high PKa, as there was nothing ionisable to any great extent; and
(e) to satisfy Lipinski’s rule of 5.
549 Lipinski’s rule of 5 was described by Professor Roberts as a “yardstick” or routinely used rule of thumb for considering whether a compound will be orally absorbed across the gastrointestinal membrane. Lipinski’s rule of 5 applies the following four criteria:
(a) the compound has no more than 5 hydrogen bond donors;
(b) the compound has no more than 10 hydrogen bond acceptors;
(c) the compound has a molecular mass of less than 500 Daltons; and
(d) the compound’s log P (the logarithm of the octanol-water partition coefficient of the molecule) is less than or equal to 5.
550 Professor Roberts’ evidence was that he could make a rough estimate of an initial starting dosage range for use in humans based on the narrowest range of 0.5 to 8 mg/kg given in WO 919. Professor Roberts agreed the “narrowest” range given in WO 919, which covered a 16 fold variance, was a broad range. To calculate the starting dose range in humans, Professor Roberts would use a rough scale up of a power of about 0.7 from the doses given to the rats. Professor Polli did not consider the scale up exercise to be quite so straightforward to translate the rat dose into a human dose, noting that there were other factors to be considered such as stability and metabolic stability. Professor Roberts’ evidence was that he would also carry out tests, such as some human microsomal incubation, to understand how the compound would be metabolised, how long it would hang around in the body and what metabolites would be produced.
551 Professor Roberts explained that Lipinski’s rule of 5 is a way to estimate whether drugs will be orally absorbed across the gastrointestinal membrane. The criteria are not absolute. A drug may satisfy three of the four rules and still be generally suitable for formulation into an oral dosage form. Professors Polli and Roberts agreed that Lipinski’s rule of 5 is a method for avoiding things that will not work.
552 Professors Roberts and Polli discussed the Yalkowsky equation as a method to estimate the aqueous solubility of a drug. In his first affidavit, Professor Roberts described that he often used the Yalkowsky equation to calculate a “very approximate” molar aqueous solubility of a drug. The equation requires two parameters: the melting point and log P. Professor Roberts calculated an estimated log P for Example 44 based on its chemical structure, to use in a calculation to predict drug solubility. Professor Roberts noted that it was a theoretical estimate which could differ significantly from the value obtained through experimental testing.
553 Professor Roberts observed that, at best, estimates give ballpark figures, “they’re not precise – they are in the ballpark. They give you just a range”. Both experts agreed that estimates and rules of thumb, such as Lipinski’s rule of 5, are useful tools to see if something should not be taken forward, but they do not provide a guarantee of success going forward.
554 Professor Roberts also considered that synthetic schemes for synthesising Example 44 gave him information about the purity level, the absence of contaminants, and the absence of compounds of concern forming during synthesis. Professor Roberts acknowledged that, although WO 919 makes general propositions about therapeutic benefits with respect to the class of compounds as a whole, the patent discloses some data regarding certain compounds being used for preventing coagulation ex vivo in humans, for example, for banked blood and biological samples. Professor Roberts considered that you could extrapolate from this ex vivo data to in vivo information about the effect of the compounds.
555 In JER2, Professor Roberts and Professor Baker described the steps that they would take once they had chosen the preferred compound. They would begin by conducting an in silico evaluation to confirm the possibility of delivering the compound orally to humans. Professor Roberts observed that, at the priority dates, there was lots of software available that could facilitate an in silico analysis on the compound (and related compounds) to provide information on the likely activity, absorption, distribution, metabolism, excretion, toxicity, and other aspects for its potential use in humans.
556 Professors Polli and Roberts also discussed the existence and use of predictive algorithms, such as GastroPlus, which could be used to simulate the gastrointestinal environment to predict the physicochemical properties such as absorption and metabolism of molecules. Professor Polli noted that, given the lack of information about Example 44 in WO 919, he did not think such algorithms would be of much assistance.
557 If the predictions and estimates did not raise any red flags, Professor Roberts’ and Professor Baker’s evidence was that they would seek all the available information before seeking to synthesise the compound and, if necessary, purify it to pharmaceutical grade.
558 Professor Roberts agreed that the limited information provided as to Example 44 in WO 919 was not the sort of picture he would have if he had done the full clinical work up himself. Professor Roberts agreed that it was important to know as much as possible about a compound before it was put into humans and that, before putting a compound into humans, he would conduct a full suite of pre-clinical tests on the compound himself rather than just relying on data included in the specification. On the basis of the information on Example 44 in WO 919, Professor Roberts considered it would nevertheless be “worth taking a punt” on undertaking pre-clinical work to find out more about the compound.
559 Professor Roberts’ evidence was that he would probably start conducting tests with a solution rather than using an oral dosing form at the outset. However, the First Hypothetical Task specified an oral dosage form. He agreed that he would not make an oral dosage form without having undertaken a great deal of studies.
560 Professor Roberts and Professor Evans agreed that WO 919 did not provide the following specific data about Example 44:
(a) stability;
(b) selectivity;
(c) solubility;
(d) most physicochemical characteristics;
(e) pharmacokinetics;
(f) pharmacodynamic parameters – absorption, distribution, metabolism and excretion;
(g) formulation;
(h) clinical safety and efficacy;
(i) manufacturability; and
(j) drug to drug interactions.
561 Professor Roberts’ evidence was that by the time the research team selected a lead candidate, they would have a large amount of information about the biopharmaceutics, or LADMET, of the compound, which encompasses its:
(a) Liberation – the release of the active drug from the formulation;
(b) Absorption – the absorption of the active drug in the body, usually into the blood stream, which is sometimes described as systemic absorption (as distinct from local absorption to the site of application, which occurs with, for example, an eye drop);
(c) Distribution – the extent to which the active drug distributes throughout the body (and the areas of the body in which the drug concentrates) as well as the rate of distribution;
(d) Metabolism – how the drug is processed in the body, usually in the liver, kidney and lung, but sometimes in the blood and other organs, and which involves the conversion of the active drug into another form;
(e) Excretion – the removal of the active drug, or its remnants, from the body, often through the kidney as urine, lungs as breath or liver as bile or, less commonly, excretion by sweat or in the milk of a lactating person; and
(f) Toxicology – the adverse effects of the active drug and pharmaceutical product.
562 Professor Roberts agreed that the decision of what molecule would be taken forward would likely be made by the medicinal chemist and pharmacologist in the team. The clinician would be involved in the choice of dosage form with the aim being to best meet the indication and to ensure patient adherence. The clinician would not be involved in considerations relating to the composition, manufacturing method and evaluation of the dosage form.
563 The experts agreed that Example 44 was a “first in class” compound, in that, at the First Priority Date, there were no direct factor Xa inhibitors on the market or known to be the subject of any clinical trials.
564 Professor Baker agreed that, as at the Second Priority Date, the safety and effectiveness of an oral single point inhibitor targeting factor Xa was a particularly novel and nascent area of investigation. Although a great deal of effort was being put into identifying new anticoagulants, safety concerns were being discovered — such as with respect to ximelagatran — and often at an advanced stage in the clinical trial process that could undermine their safety and effectiveness. None had been approved in Australia as at the Second Priority Date. Other than the provisional licensing of ximelagatran in Europe, Professor Baker was unaware of any other oral agent that was even close to approval in 2005.
565 Professor Baker’s evidence was that he was not involved in selecting lead candidates or pre-clinical animal trials. He would take the identified compounds, noting the preference for oral administration revealed in WO 919, to a pharmacologist to confirm the suggestion that this was a compound that inhibited factor Xa directly and could be given orally. He agreed that before the compound went anywhere near a human, a “wealth of studies” would need to have been undertaken such as a research project with all the risks inherent in a research project. Professor Baker also agreed that:
The prospect of getting [a] member of a class in a patent like this to a new and improved medicament that can be administered safely and efficaciously to a human is very small.
566 Professor Baker said that the development team would not proceed to test Example 44 in Phase I human clinical trials until they had the full suite of information from the pre-clinical testing. On the basis that there was nothing adverse in that information, they would proceed to design a Phase I study for testing the compounds in humans for the first time. The information gathered from the Phase I studies would then be used to design the Phase II studies.
567 In JER2, the experts agreed that:
The results from initial Phase I studies will then provide information on how the drug behaves (its pharmacokinetics and pharmacodynamics) and from this the formulation for the next study may need to be modified, with patient safety and efficacy being paramount. If the modifications involve developing a dosage form that reproduces the performance of the initial forms (eg the development of a coated tablet instead of an uncoated tablet), this work would primarily be performed by a pharmaceutical scientist. If the modification needs to be more significant, the pharmacologists and clinicians may need to be involved to signal what the target outcomes of the modification are (eg improved bioavailability, sustained release profile, methods to achieve faster, slower or more extensive absorption) and the pharmaceutical scientists would set about this task.
568 In light of the dose-associated risks of bleeding and clotting, Professor Baker advocated adopting a cautious approach in the clinical trials of anticoagulants. He also noted the unpredictable nature of a new compound that may activate other mediates like allergy, anaphylaxis and other adverse effects that may appear in a Phase I study and which would stop a drug development program very quickly.
569 The experts agreed in the joint session that the drug development process is not linear. There are uncertainties inherent in the drug development process and problems or issues may arise at any stage during the process requiring a change in project direction. The experts observed that whilst the steps of the drug development program may at first glance appear to be routine, as they are a particular series of tests that must be undertaken for a drug candidate to proceed to the next step, the reality is that the results of the tests are far from routine as each step of the drug development program can provide new and unexpected data that can halt the program, lead to the revision of the program, modify the development plan or provide confidence to pursue a predetermined plan.
570 Professor Evans observed that none of the compounds in WO 919 had ever been administered to a human being. He was concerned that there may be other adverse events or toxicities that may make them far worse than warfarin in terms of their effects on people. Professor Evans gave the examples of a hypersensitivity reaction or gastrointestinal erosion causing ulcers. A drug that might cause gastrointestinal erosion that also blocks coagulation of blood would cause disastrous gastrointestinal bleeds. Professor Roberts agreed that there were issues of potential toxicity which would have to be worked out. Professor Evans further stated that it was not possible to predict before testing that these risks would eventuate, and that the only way to find out was to conduct the usual process of selecting a compound and conducting the appropriate tests, a process which might take three to four years.
571 As to the rat tests in the examples in WO 919, Professor Evans considered that, at best, they showed that “plausibly” there was absorption of the drug in animals after oral administration. He noted that the drugs were administered by a tube directly into the rat stomach, and that WO 919 gave no information as to what the drug was dissolved in, how much was absorbed and whether there was any toxicity to the rats’ stomachs. In discussion with Professor Roberts, both experts speculated that the drug may have been dissolved in dimethyl sulfoxide, a chemical not administered to humans. Professor Evans considered that the limited information derived from the rat tests was not sufficient to conclude that the drug was suitable for oral administration in humans.
572 In the context of discussing the stopped clinical trials of an orally active agent, DPC-906, reported in the Chest paper, Professor Baker agreed that the task of developing a product that was a solid oral dosage form, and that was a selective inhibitor that was going to be both safe and effective, was “full of risk”.
573 Professor Roberts acknowledged that even when a lead compound has been selected, there is “absolutely” an enormous amount that can go wrong and, if it did, the consequence for a human with this class of compounds can be “catastrophic”. These concerns were particularly acute with respect to direct factor inhibitors as the community were aware in 2005 of the FDA’s refusal to recommend the use of ximelagatran due to concerns regarding liver toxicity, as explained above at [350].
574 Although WO 919 contains no information about the solubility of Example 44, Sandoz relies on Professor Roberts evidence that the person skilled in the art would identify by in silico analysis that it was poorly soluble. It therefore contends that it would have been obvious to address the solubility of Example 44, as was done by the 226 Patent.
575 Professor Roberts gave the following evidence regarding solubility in his first affidavit:
Solubility and dissolution rate in water are especially important characteristics for solid oral dosage forms. In order to be absorbed in the gastrointestinal tract, the drug has to dissolve in the fluid of the stomach, which is primarily water and other things such as hydrochloric acid. In addition, the drug may need to be in solution in the intestine (which typically is an environment with a pH of 6 to 8).
Solubility refers to the measure of the weight of a compound that will dissolve in a particular volume of solvent (typically water) at a particular temperature and pH. The ratio of weight to volume is referred to as equilibrium solubility. Dissolution rate refers to the rate at which a certain weight of compound will dissolve into a solvent. The two concepts are closely related — solubility is the extent to which the drug will dissolve, dissolution rate is the rate at which that occurs.
A hydrophobic compound is usually poorly soluble in water. A hydrophilic compound is usually highly soluble in water.
576 Professor Roberts then outlined common strategies that he adopted (alone or in combination) to facilitate drug dissolution into water and absorption into the body:
(a) Salt forms – using a different salt form of a drug, although this is not always the best solution and the drug’s counter-ions may actually be less soluble;
(b) Excipients – adding excipients, or changing the selection of excipients to improve solubility, dissolution rate and absorption;
(c) Micronizing – processing the drug to achieve the smallest possible particle size (eg, via grinding or particular types of crystallisation), because smaller particles generally have a higher dissolution rate due to the improved surface area to volume ratio;
(d) Molecular form – processing the drug into an amorphous form or a crystalline form, for example:
(i) Crystalline form – arranging the drug from an amorphous form (ie, no crystalline structures) into a crystalline habit that is more soluble; or
(ii) Solid dispersion – melting the drug and mixing it with another molten drug (so that both drugs are in amorphous form), and allowing the mixture to dry into a solid dispersion; and
(e) Macromolecules – binding the drug with, or otherwise inserting the drug into, a macromolecule (eg, cyclodextrin).
577 Professor Roberts also outlined further strategies he commonly adopts for drugs that are very poorly water soluble. For example, for drugs that only need to be present in a low dose, he commonly pre-dissolves the drug into a small volume of a suitable solvent (sometimes described as an auxiliary solvent) and then incorporates that solution into the product formulation (for example, a tablet where the volume of the auxiliary solvent can be absorbed by the tablet ingredients).
578 Professor Roberts’ evidence was that if he was asked to formulate an oral dosage form of Example 44 for use in clinical trials he would formulate a tablet using wet granulation. He explained that wet granulation was his preferred approach for the production of tablets and with respect to rivaroxaban:
(a) there is nothing in WO 919 to suggest the drug is unstable and needs to be formulated in the absence of a solvent or solution binder; and
(b) since the estimated log P of the drug indicates that the drug is hydrophobic, wet granulation would aid in achieving a more intimate mixing and coating of the drug with the hydrophilic binder.
10.4.3 The Sandoz inventive step case
579 Sandoz submits that once WO 919 is ascertained, there is really no scope for argument on the evidence that the invention claimed in the claims of the 226 Patent was otherwise than obvious. Sandoz submits that the person skilled in the art would simply put Bayer’s disclosure in WO 919 into practice by taking the most preferred compound and using standard techniques to formulate it. On the basis of the information disclosed in WO 919, a formulator would formulate a rivaroxaban tablet using wet granulation with a hydrophilic binder, which Sandoz submits satisfies any definition of “in hydrophilized form”.
580 Sandoz submits that the person skilled in the art reading WO 919 would select the “very preferred” compound in Example 44 (rivaroxaban) as their lead candidate on the basis that WO 919 discloses (and claims) rivaroxaban by reference to its chemical name as:
(a) the most preferred compound of the class;
(b) being suitable for the treatment of thromboembolic disorders;
(c) having a wide therapeutic spectrum or index;
(d) being suitable for oral administration;
(e) being suitable for formulation into standard dosage forms such as tablets or in granules; and
(f) being suitable for once daily administration unless there is a reason to do otherwise.
581 WO 919 provides the structure of rivaroxaban and a method for its synthesis. Sandoz submits that based on the information given in WO 919, the skilled team could calculate the molecular weight and estimate the pKa of rivaroxaban. These would be used to predict that rivaroxaban will be somewhat polar, moderately hydrophobic and poorly water soluble, but well absorbed through the gastrointestinal tract.
582 According to Sandoz, the evidence makes it clear that wet granulation with a hydrophilic excipient, as disclosed and claimed in the 226 Patent, was an obvious way to formulate rivaroxaban in light of the disclosure in WO 919. Wet granulation was a common general knowledge technique identified in standard texts for addressing the very issue that is said to have arisen in the 226 Patent (eg poor solubility leading to poor bioavailability). Professor Polli’s evidence was that wet granulation was the most widely used and general method of tablet preparation at the First Priority Date, due to the greater probability that the granulation would meet all physical requirements for the compression of good tablets, and that granulation with the judicious selection of excipients would do no harm to a poorly soluble drug.
583 Sandoz relied on Professor Roberts’ evidence that it was possible for the pharmacologist or clinician to scale up from the rat doses used in WO 919 to a starting dose for human trials. Normal pre-clinical work could be done to “confirm” the data.
584 Sandoz submitted that although Professor Polli was not familiar with the model by which human doses could be scaled up from rat doses, he confirmed that he would be able to obtain information to take the formulation further through pre-clinical studies, that this was work he would ordinarily have to do before formulating any compound, and that the steps that he would take would reflect his standard approach to formulation.
585 Based on the disclosure of WO 919, Sandoz submits that the skilled team would proceed with rivaroxaban as the lead candidate (or a lead candidate) given its low IC50 value, selective inhibition of factor Xa and the fact that it is singled out by the authors of WO 919. The skilled team would characterise the physicochemical properties of the compound and formulate a dosage form for use in clinical trials. In doing so, they would apply common general knowledge regarding standard and conventional formulation techniques, and then clinical studies would be carried out as a matter of course.
586 Even if the formulation used for Phase I studies was a simple one, the skilled team would typically formulate a more optimal dosage form in Phase II that more closely resembles the desired final product as claimed in the claims of the 226 Patent.
587 Sandoz submits that the unchallenged evidence of Professor Roberts as to micronization of the active or crystalline form establishes that the invention claimed in the dependent claims, which variously claim the active compound in a crystalline (claim 9) or micronized (claim 10) form, a particular concentration of the active compound (claim 11), or the use of a particular wetting agent (claim 13), was obvious. Sandoz submits that Bayer did not suggest that the claimed features were relevant to inventive step. Finally, Sandoz contends that the other dependent claims, whilst not the subject of specific submissions, were also obvious for the same reasons.
588 Sandoz submits that the facts of this case are similar to those in AstraZeneca and that this Court should follow the same approach as that taken by the High Court which leads to the conclusion that the claimed invention in the 226 Patent was obvious. In particular, Sandoz points to the reasoning of Kiefel J at [93]–[95] in AstraZeneca HC:
The final submission was that, armed with the Watanabe article and the common general knowledge, the skilled person would not have been led directly to the invention because the starting dosages are an essential element of the invention and were not revealed by that article or by any other prior art document.
This may be accepted. The dosages were revealed to AstraZeneca by clinical trials on humans, as inevitably they would be if undertaken. The point is that the Watanabe article contained sufficient information, including as to the results of the pre-clinical trials on animals, for Professor O’Brien to consider that clinical trials were warranted. Dr Reece was of the same view.
Professor O’Brien’s evidence about what the right dosages might be, although accurate, was largely speculative and this was not his area of expertise. However, Dr Reece, who has a background in clinical pharmacology and research, gave evidence that dose sizes which would be trialled could be expected to start from 5 mg, 10 mg and 20 mg. I do not understand AstraZeneca to contend that the starting dosages disclosed in the Patent would not be identified by normal clinical trials, which utilise certain standards and procedures. The evidence therefore shows that the skilled person would be led to the invention.
(Emphasis added.)
589 Applying that reasoning to this case, Sandoz submits that WO 919 “contained sufficient information, including as to the results of the pre-clinical trials on animals, for [the experts] to consider that clinical trials were warranted”. Further, the skilled team did not have to know or expect the correct dosages in advance, as the appropriate dosages would be revealed by the routine course of clinical trials using standard procedures. Therefore, the skilled person would be led to the invention in the 226 Patent, just as the Court found was the case in AstraZeneca.
590 Bayer submits that Sandoz’s inventive step case for both Patents rises no higher than that it would be worthwhile to take Example 44 in WO 919 into a further research and development project, including carrying out routine pre-clinical and Phase I, Phase II and Phase III tests.
591 The submissions of Sandoz and Bayer proceeded on the basis that the claimed invention of the 226 Patent was directed to an improved treatment for thromboembolic diseases over the existing treatments, warfarin and LMWH.
592 The evidence was that, at the First Priority Date, there was an unmet need for a safe and effective new oral anticoagulant to replace warfarin or to be an oral substitute for LMWH that had a wider therapeutic index than these existing drugs and did not require monitoring. A once per day dose was considered ideal, but any dose regime for oral dosing was considered better than the current treatments.
593 None of the oxazolidine derivatives described in WO 919 (including rivaroxaban) or their chemical structures were part of the common general knowledge as at both priority dates.
594 I accept that, given the description of (the compound now known as) rivaroxaban as the “very preferred” compound, its low IC50 value, its selection as one of the few compounds used in the rat test examples and the patentee’s selection of that compound as the only one to be individually claimed, the skilled person reading WO 919 is likely to select rivaroxaban as a lead candidate to take into further drug development work, if not the lead candidate. By further drug development, I mean the full suite of pre-clinical tests that Professors Roberts, Evans and Baker said they would carry out to in order to consider whether Example 44 was suitable to take into Phase I, first in human trials.
595 However, choice of the lead candidate to take into the pre-clinical testing stage is not the end of the story. As Professor Roberts acknowledged, even when a lead compound has been selected, there is “absolutely” an enormous amount that can go wrong and, if it did, the consequence for a human with this class of compounds can be “catastrophic”.
596 The identification of a potential lead candidate to take into pre-clinical testing, which far precedes trials in humans, is a very early step along the drug development pathway. There are many further steps along the way to achieving a safe and efficacious antithrombotic pharmaceutical composition, each of which presents an opportunity for failure or change in direction, including changes to chemical structure, formulation, release rate and dosage form. The risks of failure or a change in direction are even more likely when, as in this case, the compound is a first in class NCE, the side effects are entirely unknown, other unrelated compounds in the relevant field are known to have failed in early phase studies, and where the therapeutic window lies between uncontrolled bleeding and the formation of clots which can cause strokes or heart attacks.
597 At first glance, there may appear to be superficial similarities between the facts of this case and those of AstraZeneca which might therefore suggest a similar result. Namely, the selection of a lead candidate molecule from a piece of prior art — in this case, from the WO 919 Patent, in AstraZeneca, from the Watanabe Article — routine dose ranging trials and then, as a matter of obviousness and inevitability, the patented invention. However, there are at least the following important differences between the facts of this case and those in AstraZeneca. As at the AstraZeneca priority date:
(a) statins were well established and well known in Australia as a safe and effective treatment for reducing LDL cholesterol in patients with, or at risk of, cardiovascular disease or with elevated cholesterol levels;
(b) statins were a known chemical class about which predictions could safely be made about safety and efficacy;
(c) the side effects of statins were known;
(d) there was a known dose-efficiency-side effect relationship;
(e) rosuvastatin was not the first statin, or even amongst the first few statins developed — a number of statins were available on the market and being prescribed in Australia;
(f) the prior art documents, the 471 Patent and Watanabe Article, identified the structure of rosuvastatin (only relevant for the Blood Abstracts); and
(g) predictions could be made about the safety and efficacy of rosuvastatin on the basis of the other statins already approved and on the market.
598 In contrast to the facts of AstraZeneca, as at the priority date of the 226 Patent:
(a) there were no factor Xa inhibitors on the market;
(b) there were no approved direct factor inhibitors on the market;
(c) there had been mixed results for other new anticoagulant agents, such as ximelagatran and razaxaban, in clinical trials;
(d) rivaroxaban was an NCE, first in class molecule; and
(e) rivaroxaban was not a member of a known class of drugs about which predictions could reliably be made about efficacy, side effects and safety.
599 In summary, in contrast to the situation in AstraZeneca, the drug in this case was a new compound, from a new class of compounds to those anticoagulant agents being prescribed (warfarin and LMWH) and with a different mechanism of action (factor Xa inhibition) to those existing agents. In AstraZeneca, rosuvastatin was one of a class of statins which were commonly being prescribed as once a day oral treatment for hypercholesteremia, and rosuvastatin worked via the same mechanism of action in the human body as the currently prescribed statins. The only purported inventive step of the 051 Patent was the low dosage level — and therefore reduced side effects and need for monitoring patients — of rosuvastatin compared to existing statins: see AstraZeneca HC at [2] and [27] (per French CJ).
600 The experts in this case considered the statements in the WO 919 specification to be speculative and aspirational general statements for the class of compounds disclosed. Whilst the information in WO 919 might cause the person skilled in the art to choose Example 44 as a starting point, the speculative statements of the patentee could not be relied upon as the sole basis for putting Example 44 into Phase I trials in humans. Professor Roberts would conduct his own pre-clinical tests to get the full suite of information about Example 44 before proceeding further with the compound.
601 The experts considered that there were risks involved in testing a new anticoagulant for the first time in humans and that there may be potentially risky side effects. They were also aware that other anticoagulants (such as ximelagatran in late 2004) had not progressed along the drug development pathway.
602 WO 919 does not provide information as to many relevant matters which are important to considering whether to take Example 44 forward in the drug development process. These matters are listed earlier, and include, but are not limited to: the LADMET properties, toxicology and stability.
603 Some predictions, such as the initial starting dose, could be made on the basis of the rough estimates calculated by Professor Roberts, as discussed above at [548]–[550], but the experts agreed that these predictions were not indicative of success, rather they gave an indication of whether a molecule should be rejected before going further in the testing process.
604 Once all the data from the pre-clinical testing was obtained, the team would then consider whether Example 44 was suitable to progress into Phase I clinical trials in humans.
605 On Sandoz’s case, once the lead candidate has been selected, the drug development process moves along the conveyor belt of pre-ordained routine steps. These steps start with pre-clinical testing and conclude with Phase III trials. As Sandoz submits, “if the data from one step in the drug development process is positive, it is routine to progress to the subsequent step in the process”. The process continues along the set path until either the project is stopped due to a problem experienced in one of the routine clinical testing steps, or a regulatory authority approves the drug for administration to patients. In the case of rivaroxaban, there were no difficulties to overcome and no severe concerns regarding safety or side effects (such as occurred with ximelegatran or razaxaban). Thus, the compound proceeded along the well-established dug development path moving from pre-clinical testing to Phase III clinical trials without hiccups until it was approved.
606 According to Sandoz, nothing about the journey other than the choice of compound has the potential to be inventive. Given that the lead candidate is suggested by WO919, Sandoz submits that there is nothing inventive in the choice of Example 44 in the 226 Patent. That may well have been the case if there were factor Xa inhibitors already being commonly prescribed as a once a day anticoagulant, and therefore rivaroxaban would be the fifth or so factor Xa inhibitor on the market if approved – as was the case in AstraZeneca. But that is not the case here.
607 At one level, all steps along the research and development path of a new drug are well established and follow a routine, or well known, path. The successful completion of one step, and the information obtained from that step, leads to the next step in the series. However, I do not consider that the passage of a drug along a well-known path for drug discovery, including the range of pre-clinical testing and phases of clinical trials, constitutes “routine steps” in the way Aickin J intended in Wellcome.
608 Sandoz’s routine step argument relies on comments by Heerey J in Eli Lilly & Co v Pfizer Overseas Pharmaceuticals (2005) 64 IPR 506 at [193] to the effect that tests for oral bioavailability and toxicity are “essentially routine” for those skilled in the area (cited with approval in Apotex Pty Ltd v Warner-Lambert Co LLC (No 2) (2016) 122 IPR 17 at [261]–[274] (per Nicholas J) and Warner-Lambert Co LLC v Apotex Pty Ltd (No 2) (2018) 129 IPR 205 at [134] (per Jagot, Yates and Burley JJ)). These comments were made in the context of considering whether a person skilled in the art could work the claimed invention for the purposes of the sufficiency requirement, without the need for inventive ingenuity. The question whether a skilled person can work a claimed invention using standard or routine steps is very different to whether an invention results from the person skilled in the art taking routine steps taken as a “matter of course”. In the case of sufficiency, the invention has been made and the skilled addressee is seeking to make the invention, knowing the endpoint and filling in the gaps in the description of the invention in the specification, if any. That is a very different perspective to the person skilled in the art looking forward from the common general knowledge and embarking on their voyage of discovery with no known endpoint.
609 Not only must the person skilled in the art take the steps in question as a matter of routine, but they must also be carried out with an expectation of success. As was explored in the second joint session with the experts, there are risks inherent at each step along the drug development path. There is only around a 10% chance that a candidate drug will progress the entire way along the drug development path from pre-clinical testing to reach the approval stage. It is only from the successful endpoint looking back that it can be said in the case of a particular drug that there were no hurdles or obstacles that had to be overcome along the drug development pathway. This is the opposite of the s 7(2) test for inventive step which is a prospective test, looking forward from the prior art. Whether or not Bayer’s actual development path of the drug the subject of the Patent claims was straightforward and “routine” cannot be taken into account in an assessment of obviousness for the purposes of s 7(2).
610 At the First Priority Date, rivaroxaban was a new chemical entity. The experts accepted that there was vast difference between the expectations for a first in class drug selected as a lead candidate and a fifth in class molecule. For an NCE, particularly where the NCE is the first in class, there is a much higher risk of failure compared to a research and development process for a generic drug. There is also less confidence with a NCE than with a third or fourth (or more) generation of a chemical class used in other drugs. With a third or fourth generation drug (such as the statins at issue in AstraZeneca), the team would have more confidence that there will be less unpredicted adverse events and toxicities than for a first in class candidate drug. Where the other earlier members of the class are already on the market, there would be information as to side effects, toxicities and stability, which would likely form part of the common general knowledge.
611 Therefore, in the drug development project to develop Example 44, the development team had no guidance from other compounds of the same class or with the same mechanism of action that have successfully passed through the drug development pathway to be approved for use as a once a day anticoagulant agent in humans. Namely, there was no body of knowledge from which predictions as to side effects could be reliably made.
612 As I said above, the well-known standard series of steps of a drug development pathway are not the “routine steps” contemplated by Aickin J in Wellcome at 286. They are beyond tests carried out merely to confirm that a formulation works, or for regulatory purposes: Boehringer at [110] (per Perram, Nicholas and Burley JJ).
613 The steps foreshadowed by Professors Roberts and Baker form part of a new, arduous, comprehensive, risky and unpredictable research project for a first in class NCE. Each test or step is undertaken with the purpose of trying to obtain information and to explore possibilities with the aim of ascertaining a full suite of information to enable a determination of whether to proceed with the compound into the next phase of testing. The pre-clinical tests are not undertaken with the requisite expectation of developing a safe and effective drug to treat thromboembolic disorders. Rather, the information gathered at each stage of the drug development process (cumulatively with the information gathered at the previous stages) will be used by the team to decide whether or not to proceed to test the compound in the next phase of clinical studies. As such, I do not consider that a person skilled in the art, equipped with the common general knowledge and the information in WO 919, would have the requisite expectation of success at the time of selecting Example 44 that the chosen compound would pass through all the drug development stages, from pre-clinical testing to successful completion of Phase III trials, to be approved for use in humans as a safe and effective once per day treatment for thromboembolic disorders.
614 Sandoz’s “routine steps” argument is somewhat reminiscent of that run by Apotex in Apotex Pty Ltd v ICOS Corporation (No 3) (2018) 135 IPR 13. In that case, Besanko J at [346] summarised Apotex’s argument as “the proposition that, providing one could characterise a particular trial, test or experiment as routine or conventional, then it satisfied the ‘requirement’ that it be carried out with an expectation of success”. In response to that proposition, his Honour observed at [347] and [348]:
[347] It is not easy to grasp the full extent of Apotex’s argument. It cannot be that all the Court is required to do is to identify the test or experiment which led to the invention and then ask whether a test or experiment of that nature has been carried out many times before and is, in that sense, routine. A dose ranging study involving amounts of 1-20 mg of a compound might be a study that is carried out in the case of many clinical trials involving pharmaceutical compounds, but that does not make it routine in this case. …The step must be one that the development team is directly led to and would carry out as a matter of course. It is at least implicit that that step would be taken because it may well succeed.
[348] In this context, it is important to remember that in this country the fact that the development team would consider a step or series of steps worthwhile to try or worth a try does not mean that the resulting invention is obvious or lacks an inventive step.
615 Justice Besanko then referred to the High Court’s rejection of the “worthwhile to try” test in AB Hässle and observed at [349] that the Court in that case endorsed a requirement that the step or series of steps be carried out with an expectation of success.
616 Further, as set above at [293], the plurality in AB Hässle considered at [51] what Aickin J had in mind by his use of “routine” by referring to an earlier passage of Wellcome (at 280-281):
Evidence of what he did by way of experiment may be another matter. It might show that the experiments devised for the purpose were part of an inventive step. Alternatively it might show that the experiments were of a routine character which the uninventive worker in the field would try as a matter of course. The latter could be relevant though not decisive in every case. It may be that the perception of the true nature of the problem was the inventive step which, once taken, revealed that straightforward experiments will provide the solution. It will always be necessary to distinguish between experiments leading to an invention and subsequent experiments for checking and testing the product or process the subject of the invention. The latter would not be material to obviousness but might be material to the question of utility.
617 The plurality referred, without determining the point, to the working trials the subject of Dr Cederberg’s evidence, and mused at [52] that those trials might be an example of the “subsequent experiments for checking and testing” to which Aickin J referred. The evidence in AB Hässle was that Dr Cederberg’s trials in humans were conducted to confirm that various formulations proposed in the laboratory actually worked in the body: see at [25].
618 In Eli Lilly and Co Ltd v Apotex Pty Ltd (2013) 100 IPR 451, Middleton J at [554] distinguished the trials for “checking and testing” in AB Hässle from the clinical trials of olanzapine, as follows:
In [AB Hässle], the High Court acknowledged the possibility (but did not ultimately decide) that the evidence of the clinical trials given by Dr Cederberg was “checking and testing” (and thus, not evidence that was material to the question of obviousness). However, the difference between [AB Hässle] and the present case is that in [AB Hässle], the compound omeprazole was already known to be safe and effective. With olanzapine, it could not be known that the inventors had developed a safe and effective drug until after the clinical trials. There was no stopping of the inquiry at the time of synthesis of the compound in April 1982.
619 As explained by Middleton J in Eli Lilly v Apotex, the experiments for “checking and testing” contemplated by the High Court in AB Hässle are different to the skilled team undertaking a suite of pre-clinical trials to decide whether or not to put a compound into humans for the first time. Having made the decision to proceed to trials in humans, the Phase I clinical trials are carried out to ascertain whether the compound is safe in humans and to obtain information to support the next decision in the process — whether to proceed with the compound into Phase II trials. As also observed by Middleton J in Eli Lilly v Apotex, there is no stopping the inquiry at the point of synthesis of the compound (or its selection from a s 7(3) document) for a new compound from a novel class of compound that has not been previously tested in humans.
620 In AB Hässle, the plurality at [58] distinguished the tracing of a course of action which was complex and detailed, as well as laborious, “with a good deal of trial and error, with dead ends and the retracing of steps” from the taking of routine steps to which the hypothetical formulator was taken as a matter of course.
621 As Professor Evans observed, none of the compounds on WO 919 had ever been administered to a human being. There was the possibility that the compounds, including Example 44, may have adverse events or toxicities that may make them far worse than warfarin in terms of their side effects on people. Professor Evans gave as an example gastrointestinal erosion causing ulcers. A drug that might cause gastrointestinal erosion that also blocks coagulation of blood would cause disastrous gastrointestinal bleeds. Professor Roberts agreed that there were issues of potential toxicity with Example 44 which would have to be worked out. It is not possible to predict before testing that these risks of side effects and toxicities would eventuate.
622 In AB Hässle, the plurality referred at [67] to Buckley LJ’s “voyage of discovery” passage from his judgment in Re Beecham Group Ltd’s (Amoxycillin) Application [1980] RPC 261 at 296, describing it as the “vital passage”:
I am fully prepared to assume on the evidence before the court that [prior patent] 978, should be regarded as having made clear to one skilled in the field of penicillins that the epimers of the para-hydroxy and the meta-hydroxy compounds were likely to prove fruitful avenues of research, possibly the most promising avenues known to exist. I accept that the lines which that research would follow would be what [the opponent’s] witnesses described as ‘routine’, ie well-known. I accept that anyone experienced in penicillin research who pursued research along those avenues would probably have found what Beecham found. But with great deference to the learned judge, I do not agree that this is enough to constitute the claim to Amoxycillin as a penicillin for administration to humans obvious for the purposes of section 14(1)(e) of the [1949 UK Act]. To reach the discovery of the particular characteristics of Amoxycillin and its suitability for treating humans the research worker would have had to embark upon a voyage of discovery. It is possible now to see that his voyage would have been short and perhaps uneventfully straightforward, but where each of his two, or possibly more, vessels would make landfall and what those places would be like would not have been obvious to him at the outset. The voyage might have been clearly worth trying but not as a means of reaching a specific hoped-for destination.
(Emphasis added.)
623 In my view, Bayer’s drug development journey is more akin to the voyage of discovery described by Buckley LJ than the “working towards the invention with an expectation of success” referred to in AB Hässle. Although I am loathe to put a figure on the expectation of success, it seems to me that a 90% (or thereabouts) chance of Example 44 not making it through Phase III clinical trials is not in the realms of an “expectation of success”.
624 Sandoz submits that the invention claimed in the 613 Patent does not involve any inventive step when compared to the common general knowledge in combination with: 1) WO 919 as a s 7(3) document; or 2) the Blood Abstracts as s 7(3) documents.
625 Sandoz does not press the inventive step case based on the combination of WO 919 and the Blood Abstracts. Sandoz does not rely on each of the three Blood Abstracts individually but in combination together.
626 According to Sandoz, the adoption by Bayer of a once daily dosage regimen for rivaroxaban is incapable of supporting an inventive step. Once daily was the obviously most desirable dosing frequency which was commonly used in practice at the Second Priority Date, including for other anticoagulants. Sandoz submits that the suitability of rivaroxaban for such a dosing regimen is due to the inherent properties of the compound, not anything Bayer did to make it suitable. Moreover, the means for confirming the appropriateness of such a regimen were routine and would conventionally be carried out.
627 For the reasons set out above in relation to WO 919 in respect of the 226 Patent, I do not consider that the claimed invention in the 613 Patent is obvious in light of the common knowledge in combination with the information disclosed in WO 919.
10.5.2 Overview of the Blood Abstracts
628 Abstract 3003 reports a Phase I clinical trial in which the participants, who were healthy volunteers, were administered a 5 mg dose and a 30 mg dose of “BAY 59-7939”. Abstract 3003 states that BAY 59-7939 was effective at inhibiting thrombin generation. It states that a single 30 mg dose of BAY 59-7939 “exerted a sustained effect in some assays of thrombin generation for up to 24 hours”.
629 Abstract 3004 reports that in a “parallel-group, randomized, single-blind, placebo controlled trial, 64 subjects received multiple oral doses of BAY 59-7939: 5 mg od [once daily], bid [twice daily], or tid [three times daily], or 10 mg, 20 mg, or 30 mg bid for 5 days with food.” Abstract 3004 concludes that “BAY 59-7939 has predictable dose-dependent pharmacodynamics and pharmacokinetics without signs or symptoms of bleeding”, and that “oral administration of BAY 59-7939 was safe and well tolerated in doses up to 30 mg bid”.
630 Abstract 3010 describes a single dose escalation Phase I study in which 103 male participants received a single dose of BAY 59-7939. Tablets containing doses from 1.25 mg to 80 mg, and 5 mg and 10 mg oral solutions, were administered. Abstract 3010 concludes that “BAY 59-7939 was safe and well tolerated across a wide range of oral doses (1.25-80 mg) with predictable dose-dependent pharmacodynamics and pharmacokinetics”, and that “BAY 59-7939 offers predictable anticoagulation with an excellent safety profile”.
10.5.3 Disclosure of Blood Abstracts
631 It was common ground that if the Blood Abstracts could reasonably have been expected to be ascertained by a person skilled in the art at the Second Priority Date, a person skilled in the art could reasonably be expected to combine each of those abstracts. Bayer also accepted that the Blood Abstracts might be said to “contribute to the overall body of knowledge” in the field of thrombosis and, in that sense, be relevant if they satisfied the requirements of s 7(3).
632 This section summarises matters that the experts agreed were disclosed in the Blood Abstracts, and the matters not disclosed.
633 All the experts agreed that the Blood Abstracts in combination show that the compound BAY59-7939 had demonstrable effects in various surrogate tests of thrombosis over a dose range that seems safe. They further agreed that the Abstracts disclose that the compound BAY59-7939:
(a) is a direct factor Xa inhibitor;
(b) is pharmacologically active (on the clotting tests described), including after oral administration;
(c) has a relatively short half-life (4–6 hours) in the population tested;
(d) has a rapid onset of action with a Tmax of 2.5 to 4 hours according to Abstract 3003, and 2 hours according to Abstract 3010;
(e) has a reversible mechanism of action;
(f) has an absorption profile that is dependent on how the drug is administered (solution or tablet; fasting or fed);
(g) shows some sign of non-linear absorption at higher doses;
(h) exhibits maximum drug effects early in drug cycle where plasma concentrations tend to be at their highest and, as time progresses, the effects of the drug diminish;
(i) displays a close correlation between pharmacokinetic and pharmacokinetic profiles, wherein:
(i) peak pharmacodynamic effects occur around the same time as pharmacokinetic peaks (about 2 hours after administration, although not at “identical” times); and
(ii) any pharmacodynamic effects will diminish as the plasma levels decline;
(j) exhibits an apparent maximal pharmacodynamic response at about 30 mg.
634 The experts also agreed that there is limited information provided in the Abstracts, and while that was not unexpected for conference abstracts which are constrained by a prescribed word count, it means that the authors have to be scrupulous in including certain information and excluding other information. The authors of the Abstracts would have had significantly more information about BAY 59-7939 than they disclosed in the Abstracts. All experts agreed that Bayer, as a large pharmaceutical company, would have a data set supporting the Phase I study (including all of the chemical, pharmaceutical and pre-clinical data that is required prior to conducting a Phase I study), and for that reason they would accept what Bayer said in the Blood Abstracts at face value.
635 The Blood Abstracts do not disclose any safety concerns with BAY 59-7939. Professor Evans said that he would expect the authors to mention in the Abstracts if they had any major concerns about BAY 59-7939.
636 The Blood Abstracts do not disclose the identity or structure of compound BAY 59-7939, or even its chemical class. Nor was that information part of the common general knowledge. All that is disclosed is its pharmacological class – that it is a direct factor Xa inhibitor. Sandoz speculated that Bayer might disclose the structure of BAY 59-7939 to a person skilled in the art if asked. There was no evidence to support that proposition and Professor Crowther considered it unlikely.
637 The Blood Abstracts also do not disclose the following information for BAY 59-7939:
(a) the form of the compound used in the studies (whether it was the hydrate, a sulphate or a prodrug);
(b) toxicity details;
(c) formulation details;
(d) chemical form (eg polymorph, crystalline, salt);
(e) excipient details (which play an important role in absorption); or
(f) the therapeutic window.
638 The fundamental issue with Sandoz’s inventive step case based on the disclosure in the Blood Abstracts is that none of the Blood Abstracts identify the structure of compound BAY 59-7939, or even its chemical class.
639 If Sandoz was able to establish that it was obvious to formulate an oral dose of an unknown compound “BAY 59-7939” which could be taken once a day to treat a thromboembolic disorder, would that be sufficient to establish that the invention claimed in the 613 Patent was obvious?
640 Sandoz sought to suggest that as part of the usual routine process of testing and checking, the person skilled in the art would learn that BAY 59-7939 was rivaroxaban, as its structure had been published in an article (referred to as MFI-1 later in these reasons) four days before the Second Priority Date. I rejected the tender of MFI-1 during the hearing, for reasons explained in the next section. Sandoz continued to press the inventive step case based on BAY 59-7939 in the absence of MFI-1.
641 Sandoz considered that its inventive step argument was not harmed by not knowing the structure of BAY 59-7939. According to Sandoz, it is routine for pharmaceutical companies to use code names for compounds in development, and the experts were clearly able to assess the potential utility of the compounds without the structure, and to arrive at a conception of the invention without these details. Sandoz noted that Bayer itself in the 613 Patent refers to a host of preferred compounds by code name, without providing chemical names or structures.
642 Sandoz submits that it is apparent from JER2 and the joint session that the experts (and, inferentially, the skilled team) had no difficulty understanding and interpreting the information in the Blood Abstracts (and other abstracts which referred to compounds by code name) without knowing the identity of the compound. The fact that, in the real world, the next steps would be to check and test the compound to confirm the promise of the Blood Abstracts, which would require having the compound, is not to the point, as the question posed by s 7(2) involves a hypothetical task, not a real world one.
643 Bayer submitted that Sandoz’s contention that the structure of BAY 59-7939 is irrelevant is incorrect as a matter of law, citing Nettle J in AstraZeneca at [120] in support:
The plain and ordinary meaning of the words in s 7(2) — “each of which must be considered separately” — is that the single document referred to in s 7(3)(a) must be capable of standing alone without interpretative or corroborative assistance from another document or other source of information apart from common general knowledge.
644 Bayer distinguishes the hypothetical situation where the person skilled in the art lacks knowledge of the structure of BAY 59-7939 from that where the person skilled in the art is given information such that they could synthesize a compound, but not in the time available. The time available to carry out a task being an example of one of the “factual contingencies” or practicalities the Full Court considered not relevant to the hypothetical enquiry in Pharmacia LLC v Juno Pharmaceuticals Pty Ltd (2022) 168 IPR 431 at [124] (per Jagot, Yates and Downes JJ).
645 I consider that ascertaining the structure of BAY 59-7939 is more than a matter of “checking and testing the product or process of the invention” after the invention has been made by “straightforward experiments” of the kind described by Aickin J in Wellcome.
646 The identity and structure of BAY 59-7939 goes to the heart of the disclosure and information in the Blood Abstracts. Without further information, the person skilled in the art in possession of the Blood Abstracts would have no way of synthesizing compound BAY59-7939, and could not test that compound in pre-clinical testing and, depending on the results of those trials, in Phase I and II studies in humans. As Professor Roberts explained: “You can’t do anything without a structure” and “without the precise compounds in the abstract, you can’t use the data in the abstract” or “take them into a phase I or phase II clinical trial”.
647 With only the information from the Abstracts, Professor Baker similarly “would not dream” of going to an ethics committee for approval for a Phase II study and would not know if the development project would fail or succeed. He also accepted that without further, undisclosed information from Bayer regarding its Phase I study program, he “wouldn’t be able to take it forward to a Phase II study”.
648 If they had the structure of BAY 59-7939, the experts explained the logical next step as conducting an in silico analysis then, depending upon the outcome of that analysis, they would obtain and evaluate the compound, conduct pre-clinical safety, toxicity and pharmacological testing and then develop a formulation for a Phase I study ahead of seeking approval to run such a study. The experts further explained that the results of the Phase I studies reported in the Blood abstracts were encouraging in terms of the development of a safe and potentially effective compound.
649 Sandoz submits that Bayer’s argument is unattractive as a matter of policy. If accepted, it would encourage re-patenting of the prior art by the withholding, rather than disclosure, of information and would also encourage applications for patents for routine and uninventive follow-on work in relation to previously disclosed compounds. That may well be a valid policy concern. However, the High Court is clear that for the purposes of assessing whether an invention lacks an inventive step, the only information that may be considered is that which is part of the common general knowledge, or which is permitted pursuant to s 7(3); AstraZeneca HC at [21] (per French CJ), [61]–[69] (per Kiefel J).
650 Although Professor Crowther may have been aware that BAY 59 7939 was in Phase II trials in January 2005, I do not consider that information was part of the common general knowledge. Professor Crowther acquired his knowledge by reason of his being part of a team involved in a series of studies which included the early clinical trials of the relevant compound. That information would have been confidential to the members of the team and not known more broadly. That information cannot be taken into consideration in considering whether the invention is obvious for the purposes of s 7(2).
651 In AstraZeneca FC, Jessup J stated at [497]:
But the result yielded by the application of s 7(2) of the Patents Act to the requirement set out in s 18(1)(b)(ii) does not depend upon the fortuitous circumstance of what the particular inventor had done on a previous occasion, or what he or she knew. The question is to be addressed by reference to the conclusion which would have been reached by the notional “person skilled in the relevant art”. In answering that question, he or she is assumed to be equipped with the common general knowledge.
652 Without the structure of BAY 59-3959, or a sample of the compound from which its structure could be discerned, I do not consider that the skilled team would (or could) proceed any further. For example, the skilled team could not formulate it into an oral dose for administration to humans, carry out any confirmatory testing, predict any side effects or test it in Phase II trials in humans.
653 If I am wrong on the fundamental issue above, I do not, in any event, consider that the invention claimed in the 613 Patent is obvious in light of the common general knowledge and the information disclosed in the combined Blood Abstracts.
654 With respect to the common general knowledge, there are two points worth noting. First, the experts were in agreement that, as at the Second Priority Date, investigation was ongoing as to whether the conjecture that compounds that were specific inhibitors had a wide therapeutic index was correct.
655 Second, one addition to the common general knowledge in the intervening years between the First and Second priority dates is the hepatoxicity concerns with ximelagatran and the deferral of the approval by the FDA of the drug in the United States pending further data. As already discussed, ximelagatran is a direct and specific inhibitor of thrombin with a rapid onset of action, which was administered twice daily and has an elimination half-life of three to four hours. However, more research and development was required in relation to the long-term use of ximelagatran as there was insufficient data in relation to the abovementioned safety issue.
10.5.5.1 Evidence on the information disclosed in the Abstracts
656 Professor Roberts agreed that without knowing the identity and structure of the compounds in the Blood Abstracts, you cannot use the data in the Abstracts. Nor can you take the compounds into a Phase I or II clinical trial.
657 The Blood Abstracts measured the pharmacodynamic effects of BAY 59-7939 using various assays.
658 Abstract 3003 reported the use of three assays:
(1) endogenous thrombin potential (ETP);
(2) platelet-induced clotting time (PICT), which was also referred to as a Russell viper venom time assay; and
(3) platelet-induced thrombin generation time (PITT).
659 Professor Baker’s evidence was that, as at the Secondary Priority Date, each of the three assays had been the subject of peer-reviewed literature. Abstracts 3004 and 3010 each used the PT, PTT and HepTest assays, each of which were standard assays.
660 Professor Baker’s evidence was that the ETP assay was used in the clinical laboratory in haemophilia care to try and guide therapy. It was a commercially available assay with standard reagents and the instrument to detect thrombin generation. Professor Baker’s laboratory ran the ETP assay as a routine test for investigation in research. Professor Baker considered that if he had BAY 59-7939, he would be able to confirm the results reported in the Abstract 3003 ETP assay in his laboratory. Professor Baker and the other experts agreed that the ETP assay was not validated for factor Xa.
661 The experts also agreed that the three assays reported in Abstract 3003 were not commonly used assays for factor Xa as at the Second Priority Date. Professor Baker expected that, as good scientists, the authors of the Abstract would standardise the assay used in their laboratory so that they could report and reproduce their findings reliably. However, he agreed that none of the detail that would enable him to reproduce the assay in the internally standardised form was present in Abstract 3003.
662 Professor Baker considered that he would not perform the PITT assay as there were “so many variables” and it may not be as useful as the other two assays. Professor Baker said that the use of platelets adds another dimension to the complexity of the PITT assay “because everyone’s platelets are different, that the processing of the blood sample can activate platelets, so the reproducibility of that assay isn’t as reliable as the other assays”. However, he considered that he would be able to replicate the ETP and PICT assays in his laboratory on the basis of the information provided in the Abstracts. Professors Evans and Crowther disagreed; they did not consider that there was sufficient data for them to reproduce the assays reported in the Blood Abstracts in ways that would be reliable.
663 The experts considered that the results from the Phase I studies in the Blood Abstracts were “overall encouraging” in terms of the development of a safe and potentially effective compound. Professor Evans’ view was that the results were promising and might suggest that the compound is worth investigating further.
664 Professor Evans noted that, although there was a statement in the Blood Abstracts that there was a correlation between the plasma concentrations and the inhibition of the clotting factors, there was no reported pharmacokinetic data.
665 Professor Evans observed that Abstract 3003 referred to the fact that the maximum inhibition occurs two hours following drug administration. That suggested that the maximum effect of the drug is occurring early in the administration cycle where the plasma concentrations tend to be at their highest. The other information provided in Abstract 3003 is that as time progresses, the effects of the drug diminish, and that is evidenced by the fact that 12 hours after dosing the effects are less than two hours after dosing. Professor Evans said this conclusion accorded with the information in the other two Abstracts, which signalled to him that the half-life of the drug is about four to six hours. Professor Evans considered that if the information from the Abstracts was combined together, it suggests that there is a reasonably close correlation between pharmacokinetics and pharmacodynamic effect. However, Professor Evans said that he would need the full data suite in order to validate that hypothesis.
666 Sandoz submits that the information in the Blood Abstracts, taken together, provides compelling reasons to take BAY 59-7939 forward to Phase II clinical trials.
667 Sandoz submits that the skilled team would conclude based on the Blood Abstracts that BAY 59-7939 has demonstrable effects in various surrogate tests of thrombosis over a dose range that seems safe, and that the results do not disclose any safety concerns. The skilled team would understand that doses of 5 mg to 30 mg seem safe in healthy male volunteers and that 30 mg is the top of the dose-response curve and showed pharmacodynamic effects at 24 hours. They would understand that absorption could be delayed by administering the drug as a tablet with food. On this basis, it was routine to trial doses of up to 30 mg once and twice daily in a Phase II trial.
668 Bayer submits that the Blood Abstracts do not disclose important matters such as physicochemical properties, formulation, pharmacokinetics or pharmacodynamics. Thus, even if the structure of BAY 59-7939 was disclosed (which it is not in this hypothetical scenario), there is insufficient information to try to take BAY 59-7939 forward at all, let alone into high-risk Phase II trials in patients, or to have any expectation that the compound might well constitute an improved safe and effective treatment for thromboembolic disorders. I agree.
669 The evidence from the experts was that they would not proceed further down the drug development pathway without knowing the identity and structure of the compound BAY 59-7939. For the reasons set out below, I have rejected the tender of MFI-1 via which Sandoz purported to include the structure of rivaroxaban in the Second Hypothetical Task as merely “checking and testing”. There was no evidence that a large pharmaceutical company such as Bayer would provide details of BAY 59-7939. Professor Crowther’s evidence was that he had frequently made requests to drug companies for access to confidential data about their drug products but these requests were routinely rejected, and that pharmaceutical companies “jealously guarded” such information.
670 As at the Second Priority Date, BAY 59-7939 was still a first in class NCE. Even if the person skilled in the art obtained a sample or details of its structure (which, on the evidence, they would not), the evidence of the experts was that they would still need to conduct pre-clinical testing and, depending on the results, then Phase I trials before conducting Phase II trials. Although the Blood Abstracts’ results might be “encouraging”, there was still much of the voyage of discovery to complete. The predictions that might be made in the case of a later molecule in the class are not available for the first in class NCE.
671 I therefore do not consider that such a person equipped with the common general knowledge and the information in the Blood Abstracts would have the requisite expectation of success that BAY 59-7939 would pass through all the drug development stages to successful completion of Phase III trials and ultimately be approved for use in human as a safe and effective once per day treatment for thromboembolic disorders.
10.5.6 Evidentiary ruling – MFI 1
672 On the final day of the second joint session, Sandoz sought to tender a copy of an article, cited as E Perzborn et al, ‘In vitro and In Vivo Studies of the Novel Antithrombotic Agent BAY 59-7939 — an Oral, Direct Factor Xa Inhibitor’ (2005) 3(3) Journal of Thrombosis and Haemostasis 514, published four days before the 613 Patent priority date (MFI-1). Bayer objected to the tender. MFI-1 was not pleaded as a s 7(3) document, nor was it discussed by any of the experts in their affidavits, or suggested that it was part of the common general knowledge. Reference is made to MFI-1 at page 3a, line 23, to page 4, line 1, of the 613 Patent.
673 Sandoz sought to tender MFI-1, not as a s 7(3) document, but to prove that the chemical name and structure of BAY59-7939 was published before the priority date of the 613 Patent, and thus the structure of BAY59-7939 was not confidential information of Bayer at the priority date.
674 As the tender arose in the presence of the expert witnesses during the last day of the joint session, the parties agreed to make submissions on whether the tender should be accepted at a later time. I noted that I would be assisted by an affidavit outlining when the Sandoz instructors first obtained a copy of MFI-1 ahead of the parties’ submissions. Sandoz ultimately withdrew the tender on the same day.
675 Prior to the cross-examination of Dr Handke-Ergueden, Sandoz sought to draw attention to two matters in her evidence. The first was that in its submissions to the Board of Appeal of the European Patent Office (EPO), Bayer referred to document D16, which was MFI-1. The second piece of evidence referred to by Sandoz was the decision of the Board itself, which refers to Bayer’s submissions and states “D16 indicates the chemical name and structure of BAY 59-7939”. Sandoz contends that the submissions and Board decision establish that the chemical name and structure of BAY 59-7939 was public at the relevant time.
676 On the first day of closing submissions, Sandoz sought to re-tender MFI-1 for a limited purpose and subject to a limitation under s 136 of the Evidence Act 1995 (Cth). Sandoz accepted that it had been aware of MFI-1 for a considerable period of time before the proceeding, and noted that the document had been raised in the EPO proceeding. Bayer again objected to the tender of MFI-1, noting that if it had received prior notice of the document it would have made submissions as to the relevant grace period provision, s 24 of the Act.
677 I rejected the re-tender of MFI-1. The document was not put forward as a s 7(3) document, nor was there any evidence to support such a proposition. There was no evidence as to the document being part of the common general knowledge. As such, I do not consider that it contains information that can be considered for the purposes of considering whether the invention claimed in the 613 Patent is obvious: AstraZeneca HC at [21] (per French CJ), [61]–[69] (per Kiefel J).
678 Sandoz had been aware of the document for a considerable amount of time before the hearing and had remained silent in its evidence and submissions on how it proposed to deploy it. Although it was raised in the EPO proceeding, it was not raised in this proceeding until the last day of expert evidence, on day seven of an 11-day trial. With hindsight, Sandoz’s reference to the “checking and testing” proposition could be seen as an oblique reference to the purpose for which MFI-1 is intended to be used. However, there was no express mention of MFI-1 and its purpose sufficient to put Bayer on notice as to what was being alleged. I consider that if Sandoz wanted to rely on MFI-1 as part of its “checking” of the drug development project argument, it should have been raised expressly, preferably in the pleadings, so it could have been discussed in the expert evidence, and Bayer could have had the chance to properly respond.
679 Its failure to raise the document earlier, either in pleadings or by notice before the trial, effectively ambushed Bayer. Further, when it first sought to tender MFI-1, I requested that Sandoz provide an affidavit saying when it first had a copy of the document. Instead of providing such an affidavit, Sandoz withdrew the tender of the document without any explanation, only to produce the document again in closing oral submissions. Sandoz’s course of conduct with respect to MFI-1 is entirely inconsistent with its overarching obligations in s 37M and s 37N of the Federal Court of Australia Act 1976 (Cth) (FCA Act) to conduct proceedings as “quickly, inexpensively and efficiently as possible”.
680 Sandoz submits that each of the claims of the 613 Patent are not clear because the expression “a plasma concentration half life of 10 hours or less when orally administered to a human patient” in each of claims 1 and 3 of the 613 Patent is not clear.
681 Sandoz contends that the “plasma concentration half life” integer is an artefact of the 613 Patent application as filed. In that application, claims 1 and 3 were directed to a “factor Xa inhibitor” with a half-life of 10 hours or less, not limited to rivaroxaban. Bayer amended the claims in 2011, inter alia, to limit them to rivaroxaban, but did not delete the “plasma concentration half life” integer.
682 Bayer’s position is that the claims are clear: the plasma concentration is an inherent property of rivaroxaban and “10 hours or less” refers to an average. I agree.
683 The 613 Patent specification notes at page 3, lines 1–2, that “‘half-life’ is the time that it takes for the plasma concentration or the amount of drug in the body to be reduced by 50%” (citing a textbook by Goodman and Gillmans in support).
684 The experts did not have trouble understanding what was meant by the expression “a plasma concentration half life of 10 hours or less”. In the second joint session, the experts agreed that:
(a) “plasma concentration half life” refers to the elimination half-life of the drug;
(b) the elimination half-life of a drug is an inherent property of the drug in the sense that it is not affected by how the drug is formulated or administered;
(c) the elimination half-life of a drug can differ between patients based on a number of factors, including age, renal function, dose administered and ethnicity; and
(d) in the absence of any qualification, a single figure given for a half-life is the average half-life (although Professor Roberts took a slightly different view).
685 In putting together the table which formed appendix 1 to JER2 (set out at [363] above), Professor Baker reported the half-life of the anticoagulants in the table. For example, the half-life of fondaparinux is given in the table as “17 hours”. Professor Baker agreed that the half-life figure listed in the table was the average half-life of the anticoagulant listed. Professor Crowther reviewed the table and agreed with its contents, and did not raise any issue with understanding the half-life figures set out in the table. Professors Roberts and Evans noted that they had “comfort in the information provided” in the table.
686 The authorities provide that a claim must be construed in a practical, common sense manner with an eye to the utility of the claimed invention, and that absurd constructions should be avoided: Glaxosmithkline Australia Pty Ltd v Reckitt Benckiser Healthcare (UK) Ltd (2016) 120 IPR 406 at [63] (per Allsop CJ, Yates and Robertson JJ); Austal Ships Sales v Stena Rederi Aktiebolag (2008) 77 IPR 229 at [81] (per Heerey, Finn and Dowsett JJ); Product Management at [36] (per Kenny, Nicholas and Beach JJ); NV Philips at 559 (per Northrop, Lockhart and Burchett JJ)
687 Construing the claims of the 613 Patent with a modicum of common sense, I consider that the person skilled in the art would understand the phrase “a plasma concentration half life of 10 hours or less” as referring to the average elimination half-life of rivaroxaban.
688 Bayer seeks by its 27 June 2022 interlocutory application to amend page 10, line 13 of the 613 Patent by inserting the number “7” immediately after the incomplete PCT number PCT/04/01289 and insert the country code “EP” before the application date code of “04”.
689 The amendment application has been advertised in accordance with r 34.41 of the Federal Court Rules 2011 (Cth) (FCA Rules). On 6 April 2022, the Commissioner advised that she was prima facie satisfied that the proposed amendment met the requirements of s 102 of the Act and that she did not seek to be heard in relation to it. The proposed amendment was advertised in the Official Journal of Patents Supplement dated 21 April 2022. No third parties lodged an objection to the amendment.
690 No objection has been raised by Sandoz that the amendment does not satisfy the requirements of s 102 of the Act. The only issue for determination is whether the Court ought to exercise its discretion to allow the amendment pursuant to s 105 of the Act favourably to Bayer.
691 Bayer relies on the following evidence in support of the application:
(a) affidavit of Aaron Yates dated 6 July 2022;
(b) affidavit of Dr Gabriele Handke-Ergueden dated 5 July 2022;
(c) affidavit of Dr Stefan Beyreuther dated 28 February 2023;
(d) affidavit of Dr Alan Graff dated 23 March 2023; and
(e) affidavit of Dr Louisa King dated 30 January 2023.
692 Sandoz relies on the affidavit of Mr Cruise dated 30 September 2022.
693 The 613 Patent is directed to the administration of a “rapid-release tablet” containing rivaroxaban to treat a thromboembolic disorder in a once daily dosage regimen for at least five days, the rapid-release tablet being an essential integer of the claims. The specification purports to disclose a way of preparing rapid-release tablets containing rivaroxaban as the active ingredient, which is said to be very particularly preferred.
694 The document which is said to be incorporated by reference for the purpose of disclosing a method of preparing such rapid-release tablets, “PCT/04/01289”, does not exist. The erroneous reference to “PCT/04/01289” was in the priority document filed on 31 January 2005.
695 Bayer seeks to cure the mistake by amending the reference at page 10, line 13 of the 613 Patent to read “PCT/EP04/012897”. The proposed amendments are underlined.
696 PCT application PCT/EP04/012897 was published as WO 2005/060940 A2 on 7 July 2005. The description and claims of both are identical. In Australia, the PCT application became the 226 Patent. The PCT application describes a method of producing a rapid-release tablet comprising the compound now known as rivaroxaban. Bayer admits that the best method known to Bayer at the filing date of the 613 Patent application for making a rapid-release tablet comprising rivaroxaban was described in the PCT application. Bayer submits that the reference to PCT/04/01289 in the 613 Patent is a mistake. The parties disagree as to whether the mistake is an invalidating mistake.
697 What potentially elevates this amendment beyond the correction of a “mistake” is Sandoz’s best method challenge. Sandoz submits that the best method for performing the invention of the 613 Patent involves the use of the rapid-release tablets containing rivaroxaban prepared according to the method disclosed in PCT/04/01289. By reason of the non-existence of PCT/04/01289, Sandoz submits the best method is not disclosed in the 613 Patent. Bayer’s reason for seeking the amendment is to address the alleged failure to satisfy what it describes as the “uniquely Australian” best method requirement. In applying for the amendment, Bayer made clear its position that it does not concede that, without the amendment, the 613 Patent would not disclose the best method known to Bayer of performing the invention. As I have found above, I am satisfied that the 613 Patent discloses the best method of performing the invention in its unamended form.
698 Sandoz submits that Bayer has nonetheless failed to make a full and frank disclosure, unreasonably delayed in seeking amendment, and obtained unfair advantage from the 613 Patent while on notice of the mistake. As such, the proposed amendment should be denied.
699 Bayer does not dispute that it had at least constructive knowledge of the mistake in overseas counterparts of the 613 Patent from 27 April 2009, when the mistake was first raised in Canada and the Canadian equivalent of the 613 Patent was amended to comply with a Canadian Patent Rule relating to cross-referencing requirements.
700 Bayer submits that it first became aware of the mistake in Australia in late February 2022, when its lawyers informed it of Sandoz’s best method challenge in this proceeding based on the non-existence of PCT/04/01289. Bayer submits that it then immediately sought the proposed amendment to the 613 Patent out of an abundance of caution.
701 It is common ground that the best method known to Bayer for making a rapid-release tablet containing rivaroxaban at the relevant time was that described in PCT/EP04/012897.
702 Sandoz submits Bayer has been on notice of the mistake since at least 2009. Bayer failed to take any action to correct the mistake, or even consider the significance of the mistake under Australian law, even after it was put on notice, despite the fact that Bayer amended the 613 Patent in 2011 to include a rapid-release tablet as an integer of every claim. Following the amendment of the 613 Patent in 2011, Sandoz submits that maintaining the mistake can no longer be characterised as a mistake.
703 Bayer submits there is no relevant delay or taking unfair advantage of the 613 Patent. Bayer submits it did not know, nor was there any basis on which it ought to have known, that an amendment of the 613 Patent should have been sought prior to 28 February 2022.
704 Bayer submits that it did not conceal or keep private the relevant method. That method was, and at all material times has been, publicly available by way of PCT/EP04/012897 published as WO 940. Bayer further points to Professor Roberts’ evidence (on the inventive step case) that he is “confident [he] would be able to formulate an immediate (or rapid) release tablet comprising rivaroxaban”.
705 Bayer submits that it is only after the Full Court decision in Servier 2016 that it became apparent that the method of making of a rapid-release tablet might form part of the best method of performing the invention of the 613 Patent. Bayer submits that had it turned its mind to the mistake prior to the 2011 amendment, it would not have considered any amendment necessary. Bayer submits that there is no continuing obligation to consider whether to amend.
706 Bayer further submits that there is no suggestion that Sandoz or any other party ever intended to exploit rivaroxaban before the expiry of the compound patent on 23 November 2023, thus no party has been disadvantaged by the existence of the mistake. Nor was there any evidence that anyone was disadvantaged by reason of the mistake. Rather, Sandoz’s own documents show that it was aware of the correct specification.
707 Bayer relied on evidence from Dr Alan Graff, Bayer’s then in-house patent attorney, who prepared and filed the priority document. Dr Graff observed that the draft of the specification of the priority document that he was shown referred to the Bayer internal reference to the family of patents relating to PCT/EP04/012897. Dr Graff had no specific recollection of preparing the priority document, but he considered that the mistake was unintentional and an inadvertent error made in the course of drafting the priority document. Dr Graff was not cross-examined.
708 Dr Gabriele Handke-Ergueden, a qualified European Patent Attorney and Senior Patent Counsel at Bayer, gave evidence as to the prosecution of the international patent family of which the 613 Patent is a member. Since about 2006, Dr Handke-Ergueden has been the person responsible for managing and providing instructions on behalf of Bayer in relation to that patent family. Bayer requested national phase processing for the patent family in around 25 jurisdictions, including Australia, and advised that Bayer has filed corresponding applications in Argentina, Hong Kong and Chinese Taipei (meaning that the patent family has been filed in around 28 jurisdictions). Dr Handke-Ergueden agreed that she relied on her local advisers for advice on the laws of each jurisdiction.
709 Dr Handke-Ergueden was unable to recall when she first became aware of the mistake. It is fair to say that she had no independent recollection of any of the prosecution events or requests for amendment, or what motivated them, and relied on the Bayer documents retrieved during her investigation to establish what had occurred. However, as put by Bayer’s counsel in oral closing, it is unsurprising that Dr Handke-Ergueden could not recall the correction of a typographical error in two patent applications in a family of around 28 applications that occurred 13 years ago.
710 In the course of investigating the origin of the mistake and how it may have occurred, Dr Handke-Ergueden became aware that the mistake had been raised in the prosecution of the equivalent patent application to the 613 Patent in Canada and Israel in 2009 and 2010, respectively. The Canadian and Israeli amendments are discussed further below. She did not recall the mistake being identified in these jurisdictions prior to making her investigation. To the best of Dr Handke-Ergueden’s recollection, aside from Canada, Israel and now Australia, none of the other members of the patent family have been the subject of an informal or formal request for an amendment to correct the mistake.
711 Dr Handke-Ergueden’s evidence was that it was not her practice to automatically seek to amend all patents and patent applications in a family in other jurisdictions when an amendment is made in one jurisdiction (such as Canada or Israel). Whether she considers making a corresponding amendment in other jurisdictions depends on a number of factors including the nature of the amendment and the reasons why the amendment was made in the first jurisdiction. Dr Handke-Ergueden’s evidence was that if Bayer was considering filing an infringement lawsuit, the relevant patents it intended to enforce would be looked at before proceedings were issued.
712 Dr Handke-Ergueden also gave evidence that two of 13 opponents to the European equivalent to the 613 Patent raised an issue related to the mistake in early 2016 in the European opposition. According to Dr Handke-Ergueden:
(a) the mistake and the issues related to it did not play a prominent role in the EPO’s findings or decision;
(b) the mistake and the issues raised by the two opponents did not impact the validity of the European equivalent patent; and
(c) no amendment was proposed to be made, and the European patent has not been amended, to correct the mistake.
713 Dr Beyreuther, Bayer’s Senior Patent Litigation Counsel, is responsible for the enforcement, amongst others, of granted patents in the same family as the 613 Patent around the world. Dr Beyreuther provides instructions to Bayer’s Australian lawyers in respect of Bayer’s Australian rivaroxaban patents, including in respect of this proceeding.
714 Dr Beyreuther gave evidence that he was first made aware of the mistake in the 613 Patent, and the possibility and desirability of making an amendment, in a Microsoft Teams conference with Bayer’s Australian lawyers on 28 February 2022. His evidence was that, to the best of his recollection, he had not considered the possibility or desirability of making an amendment to correct the mistake in the 613 Patent prior to it being raised by Bayer’s Australian lawyers.
715 Dr Beyreuther’s evidence was that, prior to this time, Bayer had given no consideration to whether amendments should be made to the 613 Patent. It was Dr Beyreuther’s evidence that it is not his practice to consider the desirability or possibility of amendments to Australian patents in view of developments overseas, especially where amendments to claims have not been proposed or made.
716 Dr Beyreuther’s evidence was that he was involved in providing instructions to Bayer’s United States lawyers in legal proceedings involving the United States equivalent of the 613 Patent, which commenced in about April 2017. He became aware that the mistake was in the United States equivalent patent in the course of those proceedings. According to Dr Beyreuther, on 2 April 2019, the United States District Court found, in connection with a dispute concerning the construction of the claims of the United States equivalent patent, that the mistake was an obvious typographical error and that a person skilled in the art would understand the reference to refer to “PCT/EP04/012897”. No amendment was proposed or made to the United States equivalent patent to correct the mistake.
717 Dr Beyreuther did not recall considering the possibility or desirability of seeking to amend any patent in the family in any other jurisdiction in view of the European opposition or the United States proceedings. Sandoz relies on this fact as an element of delay that should disentitle Bayer to a favourable exercise of discretion under s 105 of the Act.
718 Bayer’s Australian solicitor, Mr Aaron Yates, a principal lawyer in the firm of Davies Collison Cave Law (DCCL), also gave evidence. Davies Collison Cave (DCC) is DCCL’s associated patent and trade mark attorney firm. Mr Yates was not required for cross-examination.
719 Mr Yates’ evidence was that it was not immediately apparent to him from Sandoz’s particulars of invalidity that the mistake lay at the heart of the best method challenge. Sandoz’s particulars referred to the best method known to Bayer for making a rapid-release tablet containing rivaroxaban described in “international Publication No. WO 2005/060940”. Mr Yates stated:
In late January 2022, I became aware that “International Publication No. WO2005/060940 A2” is the publication number for, and corresponds to, the Rivaroxaban Formulation PCT (i.e., PCT/EP04/012897) and that page 10 of the 613 Patent referred to “PCT/04/01289”. As a result of this, I became aware of the Error in the 613 Patent.
From this, I understood that Sandoz’ lack of best method allegations (and related lack of sufficiency allegations) very likely rely on the existence of the Error in the 613 Patent.
720 Mr Yates then informed Dr Beyreuther of the mistake on 28 February 2022. Mr Yates explained that Bayer could apply to correct the mistake to address Sandoz’s allegation that the 613 Patent fails to disclose the best method described in WO 940 and Sandoz’s related allegation that the 613 Patent specification does not describe the invention as claimed in each of the claims fully.
721 Mr Yates considered that there would be no basis for Sandoz to continue to pursue its allegations of insufficiency and failure to disclose the best method if the amendment to cure the mistake was made to the 613 Patent.
722 To the best of Mr Yates’ recollection, his law firm did not consider the possibility of amending the 613 Patent to correct the mistake prior to late January 2022.
12.3 Patent prosecution history: Overseas equivalent patents and applications of the 613 Patent
723 The following matters are taken from the affidavits of Dr Handke-Ergueden and Dr Beyreuther and their annexures (and documents tendered during the hearing).
724 In 2009, in the course of prosecuting the application for the Canadian equivalent of the 613 Patent, Bayer’s patent attorneys filed a voluntary amendment prior to any examination report to comply with Rule 81 of the Canadian Patent Rules. The amendment arose in the context of Canadian specific cross-referencing requirements. In a letter dated 27 April 2009 to Dr Handke-Ergueden, Bayer’s Canadian attorneys wrote:
We also propose removing incorporations by reference from the description in accordance with Canadian practice. We also propose inserting the publication number for the application mentioned on page 10. Please confirm that this application is PCT/EP2004/012897, published as WO 2005/060940.
725 Bayer concedes it had at least constructive knowledge of the mistake from this date.
726 By letter dated 20 July 2009, Dr Handke-Ergueden, amongst other things, confirmed that application PCT/EP2004/012897 was published as WO 2005/060940.
727 On 18 August 2009, Bayer’s Canadian patent attorneys filed the voluntary amendment, which removed the incorporation by reference and corrected the mistake.
728 Dr Handke-Ergueden’s investigations of Bayer’s records unearthed an Examination Report dated 20 December 2011 from the Canadian Patent Office which stated:
The description includes reference to a document that is not fully identified and does not comply with subsection 81(3) of the Patent Rules. The patent documents referred to on page 10, line 12 should be identified by country and either patent or publication number. Furthermore, said document citation appears to be wrong.
729 The Canadian equivalent application was subsequently amended to refer to the WO publication number only “in response to the objection under Rule 81(3)”. The Canadian equivalent application to the 613 Patent was abandoned on 25 November 2013.
730 In late December 2009, in the course of prosecuting the application for the Israeli equivalent of the 613 Patent, the examiner raised an objection in relation to “an overlap between claims”. Bayer provided instructions to its Israeli attorneys via letter on 9 April 2010. In that letter, signed by Dr Achten “for Dr. Handke-Ergueden”, Bayer notified the Israeli patent attorneys of the mistake, stating:
One feature of the new claims is the rapid-release tablet. The use of a rapid-release tablet once daily for at least five consecutive days is not mentioned in the claims of the above mentioned applications. Support how to prepare the rapid-release tablet is given in the description of the present application on page 10 line 10 to 13 (foreign countries text). Unfortunately, there is a mistake in the reference. The reference must read PCT/04/012897.”
(Underlining in original.)
731 Sandoz relies on the Bayer instruction letter of 9 April 2010 to show that, as at that date, Dr Handke-Ergueden was aware that the mistake was relevant to whether there was disclosure to support the preparation of the rapid-release tablet.
732 On 28 June 2010, Bayer’s Israeli attorneys filed a response to the examiner’s objection, which followed the form of Bayer’s instructions and included a request to amend the mistake.
733 The Israeli patent office responded by letter on 31 August 2011. The letter stated:
The passage on page 10 line 13 does not fulfill [sic] the provisions of Section 12(a) of the Law as the PCT document disclosed therein is inconcise [sic]; the country is not specified in the number (PCT/04/012897). It is my understanding that the number should be corrected as follows: PCT/EP04/012897.
In accordance with the stipulations of Section 12 of the Law and Patent Regulations 20(a)(1) and 20(a)(2) the term “incorporated by reference” is unclear and is not allowable.
(Emphasis in original.)
734 The Israeli equivalent application of the 613 Patent was abandoned by Bayer on 23 May 2013.
735 On or around 22 July 2015, two of 13 opponents to the application for the European equivalent of the 613 Patent raised the mistake as part of their oppositions (both in the context of the lack of sufficiency objection pursuant to Art 83 of the European Patent Convention). Bayer considered, as submitted in the EPO opposition on 16 November 2016, that the person skilled in the art would recognise and resolve the mistake and would have no difficulty to unambiguously identify and correct the reference.
736 Bayer submitted at the EPO opposition hearing that a simple search in a patent database program such as THOMSON INNOVATION® for patent applications with the country code WO (PCT applications) filed in the year of 2004 and containing the chemical entity “1,3-oxazolidin-5-yl” returned only six patent families, of which only two related to rivaroxaban and only one contained in its application the digits “1289”.
737 On 30 April 2018, the EPO held that the requirements of Art 83 were fulfilled. On 16 May 2022, that decision was affirmed on appeal.
738 On 2 April 2019, a United States District Court found in relation to the United States equivalent of the 613 Patent that “a person of ordinary skill in the art would understand there to be a typographical error”.
Here, there is no debate that the reference to “PCT/04/01289” in the ’218 patent is an obvious typographical error and that it should be “PCT/04/012897.” The published version of international application PCT/04/012897 (i.e., WO 2005/060940) is listed on the face of the ’218 patent under “Foreign Patent Documents.” There is no international application PCT/04/01289, so a person of ordinary skill in the art would understand there to be a typographical error.
739 No amendment was made to the United States patent.
740 Other than the Canadian and Israeli equivalents, no amendment has been proposed or made to any patent equivalent to the 613 Patent in any of the 26 other jurisdictions where a corresponding application was filed.
12.4 Prosecution of application which became the 613 Patent
741 Examination was requested of the Australian application for the 613 Patent on 6 November 2009. An adverse examination report was issued on 31 May 2010. This was sent to Dr Handke-Ergueden by DCC by letter dated 25 June 2010. Among other things, the examination report stated that the claims of the 613 Patent lacked novelty and an inventive step in light of Abstract 3004, and that two claims lacked novelty and an inventive step in light of the United States equivalent of WO 919. The examiner made no reference to the mistake in the examination report.
742 On 12 May 2011, Dr Handke-Ergueden gave instructions to DCC to respond to the adverse examination report. Her instructions were very similar to the instructions provided to the Israeli attorneys. Dr Handke-Ergueden’s evidence was that she often used letters from one jurisdiction as templates for other jurisdictions, and she agreed that it appeared that she had used the letter of 9 April 2010 to the Israeli attorneys as a template for the letter of instructions to DCC. However, as Sandoz points out, the letter to DCC omitted the statement (quoted above) that the correct PCT reference provided support for how to prepare the rapid-release tablet, and also omitted the request to correct the mistake.
743 In accordance with Bayer’s instructions, DCC responded to the examination report on 31 May 2011. The response included a request that amendments (unrelated to the mistake) be made to the specification and claims of the application (that was ultimately granted as the 613 Patent).
744 The application for the 613 Patent was accepted on 6 June 2011. There being no oppositions to the grant, the 613 Patent was granted on 29 September 2011.
745 In late January 2022, Mr Yates became aware of the mistake and therefore understood for the first time that Sandoz’s best method argument relied on the existence of the mistake. He informed Dr Beyreuther of the mistake on 28 February 2022, and explained that Bayer could apply to correct the mistake in the 613 Patent to address Sandoz’s best method allegation. Although Bayer denies that any claims of the 613 Patent are invalid without the proposed amendment, Mr Yates considered that there would be no basis for Sandoz to continue to pursue its allegations of failure to disclose best method and insufficiency if the proposed amendment was made.
746 Later on 28 February 2022, Mr Yates received instructions from Dr Beyreuther to inform the Court of Bayer’s intention to apply to make the proposed amendment.
747 After complying with the advertising requirements prescribed by the FCA Rules, Bayer applied to amend the 613 Patent by interlocutory application dated 27 June 2022.
12.5 Evidence of the skilled reader
748 Professor Roberts’ evidence was that he recognised “PCT/04/01289” as a patent reference number and would search for it as a keyword on Google or Yahoo. If that did not work, Professor Roberts would search for “01289” and “5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-l,3-oxazolidin-5-yl}- methyl)-2-thiophenecarboxamide” on Google and Yahoo.
749 Professor Roberts expected that his searches on Google and Yahoo would return the relevant patent. In the event that they did not, he would then search for “PCT/04/01289” on Chemical Abstracts and, if that failed, other databases.
750 In other words, Professor Roberts was not put off looking for the patent by the mistake, and he was confident that he would be able to locate it with some searching.
751 Whilst Professor Roberts’ Google searches did not locate the patent, Dr King’s search on Chemical Abstracts (along the lines suggested by Professor Roberts) did, in around 15 minutes.
12.6 Evidence of professional searchers
752 Bayer submits that the skilled team would include individuals with expertise in intellectual property, who would have been able to find the correct PCT reference in 15 minutes or less. Alternatively, if the skilled team did not include an intellectual property specialist, the person skilled in the art would have obtained the correct PCT reference from a patent searcher.
753 Dr King, a qualified Patent Information Professional with over 20 years patent searching experience, gave evidence as to the steps she would have taken at the filing date to obtain a copy of the document “PCT/04/01289”. Dr King understood the document to be a PCT application from the “PCT” reference. She knew the filing date from the 613 Patent and (correctly) assumed that, if the relevant document was filed by Bayer, it would have been filed in Europe or Germany and therefore would either have the EP or DE country code. With these assumptions, she conducted a search of the PATENTSCOPE database using “PCT/EP2004*” and “Bayer” and the keywords “blood”, “coagulation”, “thromb*”, “factor” or “5-chloro”. Such a search returned six results, the first being PCT/EP2004/012897. Dr King’s evidence was that she was able to find the correct reference using this process in less than 10 minutes.
754 An alternate search of the PATENTSCOPE database conducted by Dr King without any assumptions found the correct reference in about 15 minutes. In that search, Dr King used the term “morpholinyl*” being what Dr King considered to be a relatively unique chemical fragment or functional group in the chemical compound name of the preferred compound.
755 Sandoz adduced evidence from an independent patent searcher, Mr Cruise, who said that it would have similarly taken him about 10 to 15 minutes to locate the correct reference. Mr Cruise confirmed that he would have known that a search of the incorrect PCT reference on the Chemical Abstracts database would not work because it was missing a country code and because the number was only five digits.
12.7 Principles relating to the exercise of the discretion
756 Section 105 of the Act, as in force at the relevant date, provided:
105 Amendments directed by court
(1) In any relevant proceedings in relation to a patent, the court may, on the application of the patentee, by order direct the amendment of the patent, the patent request or the complete specification in the manner specified in the order.
(2) An order may be made subject to such terms (if any) as to costs, advertisements or otherwise, as the court thinks fit.
(3) The patentee must give notice of an application for an order to the Commissioner, who is entitled to appear and be heard, and must appear if the court directs.
(4) A court is not to direct an amendment that is not allowable under section 102.
(5) The patentee must file a copy of an order within the prescribed period.
(6) On the filing of a copy of an order, the patent, patent request or complete specification is to be taken to have been amended in the manner specified in the order.
757 There was no dispute between the parties concerning the general principles that inform the approach that the court should take in considering whether the discretion under s 105 of the Act should be exercised in favour of permitting the amendment that Bayer has sought.
758 In Les Laboratoires Servier v Apotex Pty Ltd (2010) 89 IPR 219, Emmett J identified at [59] that the “underlying purpose of the power conferred by s 105 is to allow a patentee to validate what would otherwise be an invalid or partially invalid patent”. His Honour observed that “[i]t is a power conferred on the Court for the benefit of the patentee”, noting that “the exercise of power in favour of the patentee is an indulgence”. As the Full Court went on to observe, the rationale underlying the court’s discretion to refuse permission to amend is the duty of the court to protect the public interest, in particular the protection of the public from abuse of the monopoly conferred by the grant of a patent: at [57]–[59] (per Emmett J), [76] (per Kenny and Stone JJ).
759 In Servier 2016, Bennett, Besanko and Beach JJ summarised a non-exclusive list of principles relevant to the Court’s discretion at [242]–[245]:
[242] While the power to amend should in appropriate circumstances be exercised in favour of the patentee, it bears mentioning that it will not always be possible to overcome a ground of revocation by an amendment. Accordingly, the ground of revocation sought to be overcome is also relevant to the way in which the discretion should be exercised. In the case of a failure to comply with the best method requirement, it would be necessary to take into account, in the exercise of the discretion, the reason for that obligation and the time at which it is meant to be fulfilled.
[243] A number of principles have been established and discussed in detail in the authorities in the United Kingdom and in this Court with respect to amendment of granted patents, relevantly and in summary:
• The discretion exists for the benefit of the patentee.
• The onus to establish that amendment should be allowed is on the patentee.
• Generally, a permissible amendment (ie one which is permitted under the Act) will be allowed unless there are circumstances which would lead the court to refuse amendment.
• The patentee must make full disclosure of all relevant matters.
• The Court’s focus is on a patentee’s conduct, not the merit of an invention.
• Amendment should be sought promptly and where a patentee delays for an unreasonable period, the patentee has the onus of showing that it delayed on reasonable grounds, such as a belief, on reasonable grounds, that an amendment was not necessary.
• Unreasonable delay is a circumstance likely to lead to refusal of the amendment.
• In assessing delay, the time when the patentee was unaware and reasonably did not know of the need for amendment is not taken into account. The relevant delay is from when the patentee knows of the likely invalidity, or has its attention drawn to a defect in the patent, or is advised to strengthen the patent by amendment. That is, amendment will not be permitted in cases where a patentee knows or ought to know that amendment should be sought and fails to do so for a substantial period of time. Thus the reasonableness of the conduct of the patentee is a relevant consideration when assessing delay.
• Mere delay is not, of itself, sufficient to refuse to exercise the discretion to amend. The fact of delay is, however, relevant to whether the respondent or the general public have suffered detriment.
• If a patentee seeks to take unfair advantage of the unamended patent, knowing that it requires amendment, then refusal of the amendment is likely.
• The proportionality of the asserted culpability of the patentee as compared with the effect of loss of protection for the invention should be considered.
…
[245] We would add that the nature of the amendment and the ground of invalidity sought to be overcome by it are important matters to be taken into consideration when exercising the discretion. In the present case, the amendment is not a narrowing amendment to defeat a piece of prior art. It is to remedy the failure to comply with an obligation that crystallised on the filing of the patent application or, in the alternative, on the grant of the patent.
760 The discretion whether to allow the amendment is a broad one. The discretionary factors identified in the cases do not constitute a set of rigid rules to be inflexibly applied: Meat & Livestock Australia Ltd v Cargill, Inc (2019) 139 IPR 47 at [462] (per Beach J). As Nicholas J observed in Neurim Pharmaceuticals (1991) Ltd v Generic Partners Pty Ltd (No 2) (2019) 139 IPR 424 at [107]:
The discretion arising under s 105 of the Act … is generally exercised by reference to a number of guiding principles that seek to balance the right of the patentee to apply to amend its patent in an appropriate case and the public interest in ensuring that an amendment application is made promptly, for proper purposes, and not so as to allow the patentee to obtain an unfair advantage were it to be allowed.
761 Further, as Yates J observed in Bayer Pharma Aktiengesellschaft v Generic Health Pty Ltd (2012) 99 IPR 59 at [162], what is essentially at issue with respect to s 105 of the Act is “whether, by its conduct, the patentee has abused the monopoly that has been conferred upon it by the grant of the patent in its unamended form”.
762 In CSL Ltd v Novo Nordisk Pharmaceuticals Pty Ltd (No 2) (2010) 89 IPR 288, Jessup J at [75] described the circumstances in which delay might be considered to amount to an abuse of the monopoly:
Clearly “mere delay” is not disqualifying, and neither is delay accompanied by knowledge of the relevant prior art. But when the circumstances are such that the patentee not only knows of the prior art but has been exposed to the view that the prior art would, or might well, be fatal to the validity of the claims in question, the onus, as it were, effectively shifts to him or her (the patentee) to bring forward a reasonable explanation why inaction was preferred to amendment at that stage. The patentee is, of course, under no obligation to accept the view to which he or she is exposed, but if the view comes from a credible source the patentee will be best placed later to justify his or her inaction by evidence of serious attention having been given to the matter, ideally in the form of professional advice.
763 Further, even where no public or private detriment is apparent, a reasonable explanation is required were there has been a delay: Novartis AG v Bausch & Lomb (Australia) Pty Ltd (2004) 62 IPR 71 at [135] (per Merkel J). A patent applicant’s relevant knowledge of risk may be informed by its overseas experiences, with the genesis being a PCT application, and its experiences thereafter being seen not as “a collection of isolated events, but as a continuum against which the applicants’ actual or constructive knowledge of the need for an amendment might be inferred ”: CSL at [88] (per Jessup J); BlueScope at [1356] (per Beach J).
764 In Novartis, Merkel J recognised that there appeared to be a distinction in the exercise of the discretion in the authorities between validating amendments to cure otherwise invalid claims, and other amendments such as deletion amendments involving deleting invalid claims, or the correction of errors or mistakes. At [66], Merkel J observed that a more lenient approach may be adopted for the latter type of amendments, noting that:
The reason for the leniency was explained by Pearson LJ in Van der Lely at 74–5 where his Lordship observed that a court is prima facie disposed to the deletion of invalid claims because if such claims were to remain in the patent they would constitute a potential “nuisance to industry”. A similar observation can be made in respect of the mere correction of errors or mistakes which are not sought to cure any actual or potential invalidity.
765 In BlueScope, the proposed amendment was intended to overcome a best method challenge. At [1485], Beach J outlined several ways in which the public had been disadvantaged by the patentee’s failure to disclose the best method of performing the invention. The public had been deprived of the knowledge of what was the best method, which in turn impeded persons from:
(a) lawfully exploiting the patent during its term, such as acts for experimental purposes (s 119C of the Act);
(b) conducting research in other ways that do not constitute exploitation of the invention; and
(c) legitimately seeking to exploit commercially what the patentee has learned whilst avoiding the forbidden territory of the claims.
766 His Honour held that it was not necessary to demonstrate that a member of the public had in fact suffered detriment, it was sufficient to infer detriment from the patentee’s failure to comply with its obligations under s 40(2)(a) of the Act.
12.8 Significance of amendments
767 Sandoz does not dispute the evidence of Dr Graff that the incorrect reference was most likely inserted into the priority document of the patent family of the 613 Patent by inadvertent error.
768 Bayer seeks to amend the 613 Patent to forestall any argument that the best method of preparing the rapid-release tablet known to Bayer is not described in the 613 Patent. There is no suggestion that Bayer sought the amendment for another purpose, such as to avoid prior art to defeat a novelty or inventive step attack.
769 In the course of cross-examining Dr Handke-Ergueden, Sandoz suggested that the amendments made to the Israeli equivalent were relevant to whether there was disclosure to support the rapid-release tablet integer thereby “fixing Bayer with knowledge of a potential problem”. I note that Sandoz makes no support or sufficiency challenge to the 613 Patent in this case.
770 In Pfizer at [374], French and Lindgren JJ explained that:
The requirement that an applicant disclose the best method known to the applicant of performing the invention safeguards against an applicant’s holding back with a view to getting the benefit of the patent monopoly without conferring on the public the full consideration for the grant of that monopoly: compare Firebelt.
771 In BlueScope, Beach J at [1512] found that BlueScope had, at the very least, constructive knowledge that it needed to amend the specification well before it actually sought to do so, and “simply made a calculated decision to take its chances with the disclosure it had made, whilst simultaneously benefiting commercially from the best method to the detriment of the public”.
772 Here, whilst Bayer may have had constructive knowledge of the mistake from 2009, I do not consider that it made a calculated decision to take its chances and benefit from the mistake once it became aware of its existence. There was no benefit to Bayer to be had: the mistake could simply be remedied by the skilled reader either conducting a search themselves or engaging a searcher. The intended reference patent was publicly available and could by located within 15 minutes of searching. Therefore, the public were not being sent off on a “wild goose chase” whilst Bayer kept the best method of performing the invention a secret.
12.9 Grounds of opposition to the amendment
773 Sandoz advances three discretionary grounds in support of its submission that, while the amendment sought is allowable in accordance with s 102 of the Act, the Court should exercise its discretion under s 105 of the Act to refuse Bayer’s application. It is convenient to deal with the discretionary grounds in the sequence of:
(a) unreasonable delay in making the amendment application;
(b) Bayer has obtained an unfair advantage by failing to disclose the best method of performing the invention to the disadvantage of the public; and
(c) Bayer has failed to satisfy the good faith disclosure obligation.
774 It is clear from Dr Handke-Ergueden, Dr Beyreuther and Mr Yates’ evidence that after the grant of the 613 Patent and until the time that the particulars of invalidity were considered in late January 2022, Bayer gave no consideration as to whether the 613 Patent should be amended. Once the Sandoz invalidity case was appreciated, Bayer promptly informed the Court of its intention to amend the 613 Patent. This is not a case where the patentee has deliberately delayed in seeking the amendments after having determined that they should be made.
775 Instead, Sandoz relies on the amendment of the mistake in the Canadian equivalent patent to submit that Bayer was on notice for 12 years and failed to disclose critical information. Consequently, Sandoz submits the Court should infer that, had Bayer sought local advice earlier, for example in 2011 (when it gave instructions to amend the 613 Patent application), DCC (and any other Australian patent attorney) would have advised Bayer to amend the mistake.
776 Sandoz relies on the amendment to the Israeli equivalent of the patent to fix Bayer with knowledge of a potential support problem as to the rapid-release tablet, and to submit that, by failing to make appropriate enquiries as to whether the mistake should be amended in Australia, Bayer had at least constructive knowledge of the “validity risks”.
777 Sandoz submits that Bayer knew from the problems in Canada and Israel that the document said to be incorporated by reference did not exist and that the best method of performing the invention was not disclosed. According to Sandoz, Bayer decided to wait for any issues to be raised (either in adverse examination reports in Australia, or by DCC, or in the course of proceedings) before it amended the 613 Patent to correct the mistake and disclose the best method. Sandoz submits that the fact that Bayer made amendments to the 613 Patent application during the Australian prosecution, but did not correct the mistake at that time despite being on at least constructive notice of its existence, means that the mistake cannot be characterised as a mere mistake. Rather, it constitutes an unreasonable delay that disentitles Bayer from amending the patent.
778 Bayer submits that there has been no culpable delay in seeking the amendment on its part. It has not persisted with claims that it knew to be unjustifiably broad or invalid. Bayer concedes that it has had constructive notice of the existence of the mistake since April 2009. However, it submits that prior to the Full Court’s decision in Servier 2016, which held at [109] that the best method of performing the invention required more than sufficient instruction to work the invention, no Australian patent attorney would have advised Bayer to amend the 613 Patent.
779 As Yates J said in Bayer Pharma at [163], citing Servier 2010, among other authorities:
Delay in seeking an amendment may constitute disentitling conduct, although mere delay is unlikely to be so. If a patentee delays in seeking an amendment for an unreasonable period of time, that circumstance may be disentitling unless the delay can be reasonably explained. Generally, delay needs to be coupled with improper conduct or resulting detriment to a particular person or the public more generally. Thus the significance of delay must be considered in the context of all the circumstances of a given case.
(Citations Omitted.)
780 Dr Handke-Ergueden’s evidence was that, absent unusual circumstances, it was not her practice to routinely raise a matter that arose in one jurisdiction with all those responsible for prosecution of the patent family in the other jurisdictions. Nor was it her practice to automatically seek to amend all patents and patent applications in a family in other jurisdictions when an amendment was made in the course of a prosecution in one jurisdiction (such as Canada or Israel). Consistent with her practice, on becoming aware of the mistake in the Canadian equivalent application, Dr Handke-Ergueden did not notify or send out for local advice as to whether to amend the relevant patent in each of the other roughly 27 jurisdictions in which an equivalent patent was being prosecuted.
781 Bayer and its patent attorneys in each jurisdiction responded to the issues that were raised in the examination reports in each such jurisdiction.
782 Of the around 28 jurisdictions in which applications were filed for the equivalent of the 613 Patent, the mistake was only raised in four jurisdictions (other than Australia). In two, Europe and the United States, patent office or court proceedings confirmed that the mistake did not necessitate amendment of the application. In Canada and Israel, the mistake was raised in the context of local laws.
783 The prosecution of patent applications is jurisdiction specific given the different laws and procedures that apply and therefore the “the relevant lens for the inquiry is the knowledge of risk under Australian law”: BlueScope at [1356] (per Beach J); CSL at [88] (per Jessup J). See also Neurim at [46]–[48] and [52] (per Nicholas J); Bayer Pharma at [210] and [213] (per Yates J).
784 In this sense, the patentee’s assessment of an amendment of a typo-style mistake is unlike the assessment of an amendment to avoid prior art which may well raise issues of novelty or inventive step that require consideration in all jurisdictions in which an application is being prosecuted. It would be expected that any amendment to overcome potentially invalidating prior art would be assessed for applications across all jurisdictions. However, the best method requirement is a peculiarly Australian ground of invalidity. It is understandable that for patent applications being prosecuted around the world from other than in Australia, an error picked up in a foreign jurisdiction, which may be relevant only to an Australian specific requirement such as best method, may not be recognised by overseas attorneys as requiring advice from Australian attorneys. I consider that it would impose an unrealistic burden to expect a patentee to circulate to its patent attorneys in every jurisdiction each and every issue that arises in the course of the prosecution of any application in a patent family around the world in case it raises a jurisdiction specific problem.
785 The amendments that were made in Australia in May 2011 were made by Bayer in response to the Australian examiner’s objections. The mistake was not raised at all by the Australian patent office in the course of prosecution of the 613 Patent application.
786 Bayer has explained the delay from the time the mistake was first raised in Canada and Israel to the filing of the amendment application in this proceeding. I am satisfied that there has been no culpable delay.
12.9.2 Full and frank disclosure
787 Sandoz submits that Bayer did not attach all of the relevant documents and correspondence from the United States, Canada and Israel to its evidence in relation to these issues, despite filing evidence which purported to provide full and frank disclosure. In particular, it did not include:
(a) the letter dated 6 August 2009 from Fetherstonhaugh to Dr Handke-Ergueden proposing amendments to the Canadian equivalent;
(b) the letter dated 12 August 2009 from Dr Handke-Ergueden to Fetherstonhaugh agreeing to the proposed amendments;
(c) the letter dated 31 August 2011 from Reinhold Cohn to Dr Handke-Ergueden enclosing an adverse examination report; and
(d) the Joint Claim Construction Brief filed by Bayer in the United States proceedings against Teva, which was subsequently annexed to Dr Beyreuther’s affidavit.
788 However, all of the relevant material (including the above documents) was either annexed to the Bayer witnesses’ affidavits or provided in Bayer’s discovery as part of the discoverable categories to which I found Sandoz was entitled.
789 Sandoz submits that while the 613 Patent still contained the mistake eight years after Bayer first became aware of the issue, Bayer asserted the 613 Patent in correspondence against Apotex Pty Ltd in 2017, and announced its intention to enforce its patents against Apotex. Two years later, Bayer asserted the 613 Patent against Apotex in correspondence prior to Sandoz commencing the revocation action.
790 There was no evidence that either Apotex or Sandoz intended to launch a rivaroxaban product prior to the expiry of the compound patent on November 2023. There was no evidence that either Apotex or Sandoz had refrained from conducting experiments or research in relation to rivaroxaban by reason of the 613 Patent and, in particular, the mistake.
791 Furthermore, Bayer has not sought to hide the best method for making a rapid-release rivaroxaban tablet from the skilled reader by maintaining the mistake uncorrected for as long as possible. As I have said, the relevant specification was published and publicly available. The evidence was that it took little effort on the part of the person skilled in the art to locate the correct patent.
792 I consider that, on the facts, Bayer’s application to amend falls at the more lenient end of the discretion spectrum contemplated by Merkel J in Novartis.
793 The proposed amendment is not a validating amendment made to avoid prior art of which the patentee was on notice for a lengthy period of time. No amendment is sought to the claims of the 613 Patent. Nor has Bayer engaged in improper conduct and persisted with claims that it knew were unjustifiably wide, or taken unfair advantage of the unamended patent.
794 At best, the criticism that can be made of Bayer is that in a perfect world, with the benefit of hindsight of the now known facts and law, the mistake would have been picked up and corrected during prosecution of the Australian patent application. Failing to reach that “counsel of perfection” is not a basis upon which to disallow this application.
795 Whilst Bayer had knowledge of the mistake since 2009, mere delay without more is not sufficient. I do not consider that Bayer’s delay in making the amendment application to be a culpable delay. I consider that Bayer has provided a satisfactory explanation for the delay in seeking the amendment.
796 The factors affecting the exercise of the discretion in this case are quite different to the amendment application in Servier 2016, where the patentee had been advised 10 years earlier that an amendment ought to be made to include a method for manufacture of the arginine salt of perindopril (the “Harris letter”) but chose to ignore that advice. The factors are also quite different from that in BlueScope, where the Australian patent office had raised a best method objection against the use of the term “special operational measures”: see at [1394] (per Beach J). In that case, the amendment was sought “years after the filing date” to supplement the disclosure of the best method by saying that “special operational measures” were not special but were known to skilled persons.
797 Too often it has become the practice in patent amendment cases in this Court for opponents to amendments to seek access to the prosecution files of all equivalent patents and patent applications in a patent family in all jurisdictions in order to trawl through them to find a loose thread that may, with the aid of hindsight, be used to found a suggestion of ongoing improper conduct sufficient to disentitle the patentee of the amendment to the exercise of the court’s discretion to amend. This practice results in delay and adds significantly to the costs of patent amendment applications, the costs of which are usually borne by the patentee seeking the indulgence of an amendment. Of course, opponents are entitled to oppose patent amendments on discretionary grounds. However, the practice of trawling through documents I have described, which often necessitates complex and lengthy discovery processes to identify those documents, is contrary to the overarching obligations imposed by s 37M and s 37N of the FCA Act. Where the amendment is not amending the claim, or amending to avoid prior art, and where the patentee does not appear to be acting from improper motives, the opponent should think twice before proceeding down this path.
798 I will exercise my discretion to allow the amendment to the 613 Patent.
799 For the reasons given above, I find that both the 226 Patent and the 613 Patent are valid.
800 Bayer accepts that, on my construction of the term “in hydrophilized form”, the Sandoz Rivaroxaban products do not infringe the claims of the 226 Patent.
801 I will exercise my discretion to amend the 613 Patent in the form sought. I will also grant the injunction sought by Bayer to prevent Sandoz’s threatened infringement of the 613 Patent.
802 Accordingly, I reject Sandoz’s application and accept Bayer’s cross-claim in part. I will make orders to reflect these reasons.
I certify that the preceding eight hundred and two (802) numbered paragraphs are a true copy of the Reasons for Judgment of the Honourable Justice Rofe. |
Associate:
SCHEDULE OF PARTIES
VID 683 of 2021 | |
BAYER AUSTRALIA LIMITED |

