
Federal Court of Australia
Boehringer Ingelheim Animal Health USA Inc v Zoetis Services LLC [2023] FCA 1119
ORDERS
BOEHRINGER INGELHEIM ANIMAL HEALTH USA INC. Appellant | ||
AND: | Respondent | |
AND BETWEEN: | Cross-Appellant | |
AND: | BOEHRINGER INGELHEIM ANIMAL HEALTH USA INC. Cross-Respondent | |
DATE OF ORDER: | 21 SEPTEMBER 2023 |
THE COURT ORDERS THAT:
1. If the parties agree on the appropriate orders to be made by the Court reflecting these reasons for judgment, the parties file a minute of proposed orders on or before 5 October 2023.
2. If the parties are unable to agree on the orders which should be made, each of the parties file and serve on or before 5 October 2023:
(a) A minute of the orders that the party proposes; and
(b) Any outline of submissions in support of the proposed orders (limited to three pages).
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
ROFE J:
[1] | |
[10] | |
[11] | |
[28] | |
[81] | |
[85] | |
[88] | |
[90] | |
[95] | |
[99] | |
[103] | |
[110] | |
[112] | |
[118] | |
Regard to confidential information not part of common general knowledge | [130] |
Not representative of non-inventive person skilled in the art | [147] |
[154] | |
[158] | |
[175] | |
[184] | |
[189] | |
[211] | |
[224] | |
[235] | |
[259] | |
[261] | |
[263] | |
[267] | |
[277] | |
[284] | |
[292] | |
[294] | |
[306] | |
[320] | |
[345] | |
[360] | |
[370] | |
[372] | |
[378] | |
[384] | |
[389] | |
[393] | |
[409] | |
[417] | |
[421] | |
[425] | |
[425] | |
[437] | |
[447] | |
[455] | |
[472] | |
[476] | |
[477] | |
[489] | |
[494] | |
[496] | |
[519] | |
[525] | |
[532] | |
[556] | |
[584] | |
[599] | |
[610] | |
[612] | |
[618] | |
[618] | |
[636] | |
[647] | |
[650] | |
[656] | |
Dr Nordgren’s evidence as to the examples in the Applications | [656] |
[684] | |
[697] | |
[716] | |
[725] | |
[744] | |
[769] |
INTRODUCTION
1 These proceedings are an appeal pursuant to s 60(4) of the Patents Act 1990 (Cth) (the Patents Act or Act) from a decision of a delegate of the Commissioner of Patents by the opponent, Boehringer Ingelheim Animal Health USA Inc, and a cross-appeal by the patent applicant, Zoetis Services LLC, to the grant of three related patent applications: Boehringer Ingelheim Animal Health USA Inc. v Zoetis Services LLC [2020] APO 40 (Delegate’s decision).
2 The three patent applications were each filed on 3 April 2013, and their claim of a priority date of 4 April 2012 was not challenged. The three Australian Patent applications are the:
(a) 535 Application, entitled “Mycoplasma hyopneumoniae vaccine”;
(b) 537 Application, entitled “PCV/Mycoplasma hyopneumoniae combination vaccine”; and
(c) 540 Application entitled “PCV/Mycoplasma hyopneumoniae/PRRS combination vaccine”.
3 The delegate found claims 1–2, 4–6, 9–10 and 12–18 of the 535 Application did not involve an inventive step. The oppositions to the 537 and 540 Applications were unsuccessful.
4 Boehringer filed a notice of appeal and notice of contention. Zoetis filed a notice of cross-appeal and a notice of contention.
5 Ultimately on appeal, the following grounds of opposition were pressed for each Application:
(1) That the invention claimed is not supported by the matter disclosed in the specification within s 40(3) of the Act;
(2) That the complete specification does not disclose the invention in a manner which is clear enough and complete enough for it to be performed by a person skilled in the relevant art within s 40(2)(a) of the Act;
(3) That the complete specification does not disclose the best method known to Zoetis within the requirements of s 40(2)(aa) of the Act;
(4) That the invention claimed does not involve an inventive step because it was obvious in light of the common general knowledge considered together with information in any one or more of the Okada papers (defined later); and
(5) That the invention claimed in certain “kit” claims is not for a “manner of manufacture” within s 18(1)(a) of the Act.
6 Although styled as an appeal, the present proceeding is in the original jurisdiction of this Court and involves a hearing de novo on the grounds and evidence before the Court. As the opponent to the grant of a patent on each of the Applications, Boehringer bears the onus in relation to each ground of opposition raised.
7 Section 60(3A) of the Act, which was introduced with the passage of the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth) (RTB Act), provides that the Commissioner may refuse an application if satisfied, on the balance of probabilities, that a ground of opposition exists. That is the standard by which the present proceeding is to be judged.
8 The Patents Act that applies to the present dispute is in the form as amended by the introduction of the RTB Act.
9 For the reasons that follow, I have concluded that the opposition to the grant succeeds in respect of the following claims:
(a) 535 Application:
(i) Claims 1, 3, 7–8, 11–12, 16–17
(b) 537 Application:
(i) Claims 1 to 24
(c) 540 Application:
(i) Claims 1 to 25
10 Before going further, I will set out a brief background to vaccines, taken principally from uncontroversial parts of the first affidavits of Professor Chase and Dr Nordgren and supplemented with extracts from Professor Browning’s affidavit.
11 A pathogen is an organism that can infect and cause disease to a host such as a pig or a human. There are four main types of pathogens:
(a) bacteria;
(b) viruses;
(c) fungi; and
(d) parasites, such as protozoa and helminths (eg nematodes or roundworms).
12 The immune system protects its host from infection by pathogens. The immune system is a complex system comprised of different types of cells, tissues and chemotypic messengers, each of which have specific roles, and which work together to protect and heal the host from damage caused by foreign invading pathogens.
13 There are three main types of immunity:
(a) innate immunity (also known as the “innate immune system”, “innate immune response”, or “primary immune response”);
(b) adaptive immunity (also known as the “adaptive immune system”, “adaptive immune response”, or “secondary immune response”); and
(c) passive immunity (also known as the “passive immune system” or “passive immune response”).
14 Each pathogen has unique lifecycles and components, typically proteins or carbohydrates, which stimulate the immune response of a host. The components of pathogens which are capable of being recognised by the immune system and generating a response are known as “antigens”.
15 The innate immune response is the first line of defence against infection. Most components of innate immunity are present before the onset of infection and consist of a set of disease-resistant mechanisms that are not specific to a particular pathogen. These include physical barriers such as skin and the mucosal membranes, as well as phagocytic cells such as macrophages. The innate system is able to recognise a given class of molecules but does not change its response based on its previous exposure to the same pathogen.
16 Macrophages can engulf, or “phagocytize”, invading pathogens and foreign particles and expose them to other intracellular compounds involved in the innate immune response, such as lysozymes (enzymes which can cleave bacterial cell wall membranes). Macrophages, and other components of the innate immune response such as dendritic cells, also stimulate the adaptive immune response, by displaying antigens on their cell surface.
17 The adaptive immune response is typically triggered when there is a recognised antigenic challenge to a host organism. Adaptive immunity provides a second, specific and comprehensive line of defence that eliminates pathogens that evade the innate responses or persist in spite of them. An important consequence of adaptive immune response is memory. If the same pathogen infects the body a second time, memory cells provide the means for the adaptive immune system to make a rapid and often highly effective attack on the invading pathogen.
18 Two characteristics of the adaptive immune system that are crucial to a successful immune response are its antigenic specificity and its diversity. The antigenic specificity of the adaptive immune response enables it to recognise subtle differences among antigens, such as a difference of one amino acid in two otherwise identical proteins. The diversity of the adaptive immune response allows it to recognise billions of unique structures on foreign antigens.
19 The major agents of adaptive immunity are lymphocytes and the antibodies that they produce. Lymphocytes are a class of white blood cells that are produced in the bone marrow, and that produce and display antibodies. The two major types of lymphocytes are B lymphocytes (or B cells) and T lymphocytes (or T cells).
20 Antibodies, also known as immunoglobulins (or Ig), are proteins that mediate the humoral immune response by recognising and binding to antigens.
21 Antibodies bind to specific portions of an antigen. Each antibody-binding site of an antigen is called an “epitope”. The combined strength of the interactions between a single antigen-binding site on an antibody and a single epitope is referred to as the affinity of the antibody for that epitope.
22 There are five major antibody classes, or immunoglobulins: IgG, IgM, IgD, IgA and IgE, each of which plays a specific role in the immune response of a host.
23 Antibodies consist of “heavy” polypeptide chains and “light” polypeptide chains. Each chain comprises a “constant” region and a “variable” region. The constant region is common across all antibodies of a class, while the variable region of each antibody is specific to the antigen against which it was generated.
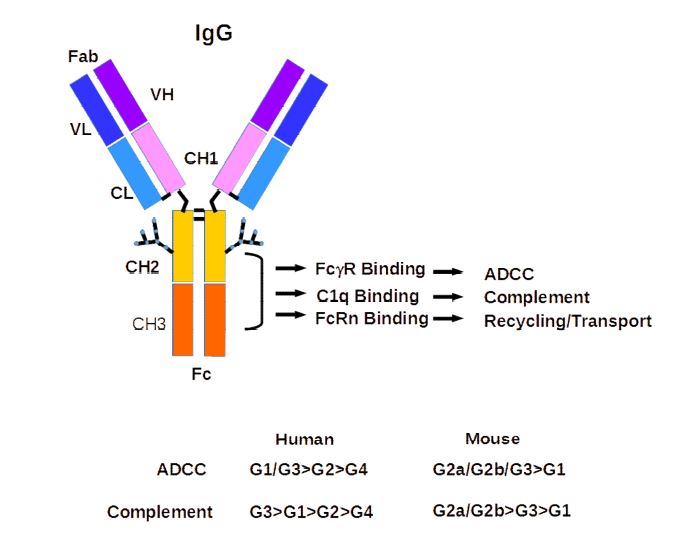
24 The most abundant immunoglobulin is immunoglobulin G (IgG), which is released by Plasma B cells. There are several sub-classes of IgG. IgG consists of two “heavy” polypeptide chains and two “light” polypeptide chains in a “Y” shape:
25 A vaccine is a preparation containing one or more antigens which is intended to trigger an adaptive immune response, but not cause disease, or at least not cause severe symptoms of disease, associated with that pathogen. The vaccine is intended to generate memory cells against a pathogen. This protects the host against the consequences of subsequent exposure to a pathogen bearing that antigen.
26 Vaccines involve presenting a killed or weakened version of a pathogen (or an antigen or antigens, or specific parts of an antigen or antigens of a pathogen) to the immune system, in a form suitable for the immune system to recognise the antigen or antigens and stimulate the body to produce antigen-specific antibodies and related memory cells in response. If the vaccinated individual is exposed to the pathogen again after vaccination, the immune system will be primed and able to rapidly produce antigen-specific antibodies in order to neutralize and prevent the pathogen from reproducing and the disease from developing.
27 Pathogens often have more than one antigen against which hosts will generate antibodies and memory cells that are capable of providing adaptive immunity. This means that, for a given pathogen, a number of different antigenic compositions may be able to confer acceptable levels of immunity.
28 There are three patent applications in issue.
29 The Applications have substantially the same specification, save for different introductory statements as to the field of the invention and consistory statements, and different examples being included in the 537 and 540 Applications.
30 The 535 Application is entitled “Mycoplasma hyopneumoniae vaccine” and the invention is said to relate to Mycoplasma hyopneumoniae (M. hyo). More particularly, the invention relates to the soluble portion of an M. hyo whole cell preparation and its use in a vaccine for protecting pigs against enzootic pneumonia.
31 The 537 Application is entitled “PCV/Mycoplasma hyopneumoniae combination vaccine”, and the invention is said to relate to porcine circovirus and M. hyo. More particularly, the invention relates to a multivalent immunogenic composition including a soluble portion of an M. hyo whole cell preparation and a PCV-2 antigen and its use in a vaccine for protecting pigs against enzootic pneumonia and Post-weaning Multisystemic Wasting Syndrome (PMWS).
32 The 540 Application is entitled “PCV/Mycoplasma hyopneumoniae/PRRS combination vaccine” and the invention is said to relate to porcine circovirus, M. hyo and porcine reproductive and respiratory syndrome (PRRS) virus. More particularly, the invention relates to a trivalent immunogenic composition including a soluble portion of an M. hyo whole cell preparation, a PCV-2 antigen and a PRRS virus antigen and its use in a vaccine for protecting pigs against enzootic pneumonia and PMWS.
33 Relevant parts of the specification (of all three Applications), referred to throughout these reasons for judgment, include the following.
34 The background of the invention notes enzootic pneumonia in swine, also called mycoplasmal pneumonia, is caused by M. hyo. Whilst infected pigs show only mild cough and fever symptoms, the disease has significant economic impact due to reduced feed efficiency and reduced weight gain. The primary M. hyo infection may be followed by secondary infection by other mycoplasma species as well as other bacterial pathogens.
35 The specification describes M. hyo as a small, prokaryotic microbe capable of a free living existence, although it is often found in association with eukaryotic cells because it has absolute requirements for exogenous sterols and fatty acids. These requirements generally necessitate growth in serum-containing media. M. hyo is bounded by a cell membrane, but not a cell wall.
36 After describing in more detail the mechanism and effect of an M. hyo infection in a pig, and the characteristic lung lesions found in infected pigs, the specification observes (at page 1a, line 3) that there is a great need for effective preventative and treatment measures.
37 The specification continues (at page 2, lines 5–11):
Vaccines containing preparations of mycoplasmal organisms grown in serum-containing medium have been marketed, but raise concerns regarding adverse reactions induced by serum components (such as immunocomplexes or non-immunogenic specific proteins) present in the immunizing material. Other attempts to provide M. hyo vaccines have been successful, but the disease remains widespread.
38 The specification then explains that M. hyo and porcine circovirus type 2 (PCV-2) are the two most prevalent pathogens that are encountered in the pig industry. Swine infected with PCV-2 exhibit a syndrome commonly referred to as Post-weaning Multisystemic Wasting Syndrome (defined above as PMWS). In addition to PMWS, PCV-2 has been associated with several other infections, one of which is PRRS.
39 At page 2a, lines 1–2, the specification states that it is an object of the present invention to overcome or ameliorate at least one of the disadvantages of the prior art, or to provide a useful alternative. An example given is an improved vaccine against mycoplasma infection in swine.
40 The specification continues (at page 3, lines 1–4):
M. hyo vaccine will be compatible with other porcine antigens, such as PCV2 and PRRS virus, whether they are given concurrently as separate single vaccines or combined in a ready-to-use vaccine. It would be highly desirable to provide a ready-to-use, single-dose M. hyo/PCV2 combination vaccine.
41 Under the heading “summary of the invention”, the specification describes four aspects of the invention, the first aspect being (page 3, lines 7–11):
According to a first aspect, the present invention relates to an immunogenic composition comprising the supernatant of a Mycoplasma hyopneumoniae (M.hyo) culture, wherein the supernatant of the M.hyo culture has been separated from insoluble cellular material by centrifugation, filtration, or precipitation and is substantially free of both (i) IgG and (ii) immunocomplexes comprised of antigen bound to immunoglobulin.
42 The second aspect of the invention relates to a kit for use in carrying out the invention, the third aspect, a method for preparing an immunogenic composition, and the fourth, a method for immunising a pig against M. hyo.
43 At page 3a, lines 10–12, the specification states that in one aspect, the soluble portion of the M. hyo whole cell preparation has been treated with Protein A or Protein G prior to being added to the immunogenic composition.
44 The specification then describes some embodiments of the invention (at page 3a, line 14–page 5, line 13):
In one embodiment, the soluble portion of the M. hyo preparation includes at least one M. hyo protein antigen. In another embodiment, the soluble portion of the M. hyo preparation includes two or more M. hyo protein antigens.
In some embodiments, the immunogenic composition of the present invention further includes at least one additional antigen. In one embodiment, the at least one additional antigen is protective against a microorganism that can cause disease in pigs.
In one embodiment, the microorganism includes bacteria, viruses, or protozoans. In another embodiment, the microorganism is selected from, but is not limited to, the following: porcine circovirus type 2 (PCV2), porcine reproductive and respiratory syndrome virus (PRRSV), porcine parvovirus (PPV), Haemophilus parasuis, Pasteurella multocida, Streptococcum [sic] suis, Staphylococcus hyicus, Actinobacilllus [sic] pleuropneumoniae, Bordetella bronchiseptica, Salmonella choleraesuis, Salmonella enteritidis, Erysipelothrix rhusiopathiae, Mycoplama [sic] hyorhinis, Mycoplasma hyosynoviae, leptospira bacteria, Lawsonia intracellularis, swine influenza virus (SIV), Escherichia coli antigen, Brachyspira hyodysenteriae, porcine respiratory coronavirus, Porcine Epidemic Diarrhea (PED) virus, rotavirus, Torque teno virus (TTV), Porcine Cytomegalovirus, Porcine enteroviruses, Encephalomyocarditis virus, a pathogen causative of Aujesky's [sic] Disease, Classical Swine fever (CSF) and a pathogen causative of Swine Transmissable [sic] Gastroenteritis, or combinations thereof.
In certain embodiments, the at least one additional antigen is a porcine circovirus type 2 (PCV2) antigen, a PRRS virus antigen or a combination thereof. In one embodiment, the composition elicits a protective immune response in a pig against both M. hyo and PCV2. In another embodiment, the composition elicits a protective immune response in a pig against M. hyo, PCV2 and PRRS virus.
In one embodiment, the PCV2 antigen is in the form of a chimeric type-1-type 2 [sic] circovirus, the chimeric virus including an inactivated recombinant porcine circovirus type 1 expressing the porcine circovirus type 2 ORF2 protein. In another embodiment, the PCV2 antigen is in the form of a recombinant ORF2 protein. In still another embodiment, the recombinant ORF2 protein is expressed from a baculovirus vector.
In some embodiments, the composition of the present invention further includes an adjuvant. In one embodiment, the adjuvant is selected from, but is not limited to, the following: an oil-in-water adjuvant, a polymer and water adjuvant, a water-in-oil adjuvant, an aluminum hydroxide adjuvant, a vitamin E adjuvant and combinations thereof. In another embodiment, the composition of the present invention further includes a pharmaceutically acceptable carrier.
In certain embodiments, the composition of the present invention elicits a protective immune response against M. hyo when administered as a single dose administration. In further embodiments, the composition elicits a protective immune response against M. hyo and at least one additional microorganism that can cause disease in pigs when administered as a single dose administration. In still further embodiments, a composition of the present invention elicits a protective response against both M. hyo and at least one additional microorganism that causes disease in pigs when administered as a two dose administration.
The present invention also provides a method of immunizing a pig against M. hyo. This method includes administering to the pig an immunogenic composition including a soluble portion of an M. hyo whole cell preparation, wherein the soluble portion of the M. hyo preparation is substantially free of both (i) IgG and (ii) immunocomplexes comprised of antigen bound to immunoglobulin. In one embodiment, the soluble portion of the M. hyo preparation of the administered composition includes at least one M. hyo protein antigen.
In one embodiment of the method of the present invention, the composition is administered intramuscularly, intradermally, transdermally, or subcutaneously. In another embodiment of the method of this invention, the composition is administered in a single dose. In yet another embodiment of the method of this invention, the composition is administered as two doses.
In a further embodiment of the method of the present invention, the composition is administered in conjunction with at least one additional antigen that is protective against a microorganism that can cause disease in pigs, such as one or more of the microorganisms described above. Such other antigens can be given concurrently with the M. hyo composition (i.e., as separate single vaccines) or combined in a ready-to-use vaccine.
45 There follows a brief description of the drawings and the sequences.
46 At page 8, lines 15–24, there is the following detailed description of the invention:
The present invention provides an immunogenic composition including a soluble portion of an M. hyo whole cell preparation, wherein the soluble portion of the M. hyo preparation is substantially free of both (i) IgG and (ii) antigen-bound immunocomplexes. Applicants have surprisingly discovered that the insoluble fraction of the M. hyo whole cell preparation is non-immunogenic. In contrast, the IgG-free M. hyo soluble preparation is immunogenic and can be effectively combined with antigens from other pathogens, such as PCV2, without analytical or immunological interference between the antigens. This makes the M. hyo soluble preparation of this invention an effective platform for multivalent vaccines, including one-bottle, ready-to-use formulations. Applicants have also surprisingly discovered that removing the immunoglobulin and the insoluble cell debris enhances the safety of the immunogenic composition.
47 The specification then includes (at page 8, line 26–page 13, line 4) a number of definitions of terms used in the specification, including “comprising” at page 8, lines 30–2:
As used herein, the term “comprising” is intended to mean that the compositions and methods include the recited elements, but do not exclude other elements.
48 At page 9, lines 1–9, the specification states that, as defined, a soluble portion of an M. hyo whole cell preparation refers to a soluble liquid fraction of an M. hyo whole cell preparation after separation of the insoluble material and substantial removal of IgG and antigen-bound immunocomplexes. The specification continues:
The M. hyo soluble portion may alternatively be referred to as the supernatant fraction, culture supernatant and the like. It includes M. hyo expressed soluble proteins (M. hyo protein antigens) that have been separated or isolated from insoluble proteins, whole bacteria, and other insoluble M. hyo cellular material by conventional means, such as centrifugation, filtration, or precipitation. In addition to including M. hyo-specific soluble proteins, the soluble portion of the M. hyo whole cell preparation also includes heterologous proteins, such as those contained in the culture medium used for M. hyo fermentation.
The term “antigen” as used in the specification refers to a compound, composition, or immunogenic substance that can stimulate the production of antibodies or a T-cell response, or both, in an animal, including compositions that are injected or absorbed into an animal. The immune response may be generated to the whole molecule, or to a portion of the molecule (e.g., an epitope or hapten).
49 The term an “immunogenic or immunological composition”, as used in the specification, refers to a composition of matter that comprises at least one antigen which elicits an immunological response in the host of a cellular and or antibody-mediated immune response to the composition or vaccine of interest.
50 At page 9, lines 20–30 the specification continues:
The term "immune response" as used in the specification refers to a response elicited in an animal. An immune response may refer to cellular immunity (CMI); humoral immunity or may involve both. The present invention also contemplates a response limited to a part of the immune system. Usually, an “immunological response” includes, but is not limited to, one or more of the following effects: the production or activation of antibodies, B cells, helper T cells, suppressor T cells, and/or cytotoxic T cells and/or yd T cells, directed specifically to an antigen or antigens included in the composition or vaccine of interest. Preferably, the host will display either a therapeutic or protective immunological response such that resistance to new infection will be enhanced and/or the clinical severity of the disease reduced. Such protection will be demonstrated by either a reduction or lack of symptoms normally displayed by an infected host, a quicker recovery time and/or a lowered viral titer in the infected host.
51 The term “adjuvant”, as used in the specification (at page 10, line 5), means:
[A] composition comprised of one or more substances that enhances the immune response to an antigen(s). The mechanism of how an adjuvant operates is not entirely known. Some adjuvants are believed to enhance the immune response by slowly releasing the antigen, while other adjuvants are strongly immunogenic in their own right and are believed to function synergistically.
52 The term “multivalent”, as used in the specification, means a vaccine containing more than one antigen whether from the same species (ie different isolates of Mycoplasma hyopneumoniae), from a different species (ie isolates from both Pasteurella hemolytica and Pasteurella multocida), or a vaccine containing a combination of antigens from different genera (eg a vaccine comprising antigens from Pasteurella multocida, Salmonella, Escherichia coli, Haemophilus somnus and Clostridium).
53 The term “vaccine composition”, as used in the specification, includes at least one antigen or immunogen in a pharmaceutically acceptable vehicle useful for inducing an immune response in a host.
54 At page 13, lines 5–9, the specification states:
All currently available M. hyo vaccines are made from killed whole cell mycoplasma preparations (bacterins). In contrast, the present invention employs a soluble portion of a Mycoplasma hyopneumoniae (M. hyo) whole cell preparation, wherein the soluble portion of the M. hyo preparation is substantially free of both (i) IgG and (ii) immunocomplexes comprised of antigen bound to immunoglobulin.
55 The specification then discusses the growth media requirements specific to M. hyo at page 13, lines 11–23:
M. hyo has absolute requirements for exogenous sterols and fatty acids. These requirements generally necessitate growth of M. hyo in serum-containing media, such as porcine serum. Separation of the insoluble material from the soluble portion of the M. hyo whole cell preparation (e.g., by centrifugation, filtration, or precipitation) does not remove the porcine IgG or immune complexes. In one embodiment of the present invention, the M. hyo soluble portion is treated with protein-A or protein-G in order to substantially remove the IgG and immune complexes contained in the culture supernatant. In this embodiment, it is understood that protein A treatment occurs post- M. hyo fermentation. This is alternatively referred to herein as downstream protein A treatment. In another embodiment, upstream protein A treatment of the growth media (i.e., before M. hyo fermentation) can be employed. Protein A binds to the Fc portion of IgG. Protein G binds preferentially to the Fc portion of IgG, but can also bind to the Fab region. Methods for purifying/removing total IgG from crude protein mixtures, such as tissue culture supernatant, serum and ascites fluid are known in the art.
56 At page 13, lines 25–27, the specification notes that in some embodiments, the soluble portion of the M. hyo preparation includes at least one M. hyo protein antigen. In other embodiments, the soluble portion of the M. hyo preparation includes two or more M. hyo protein antigens.
57 The specification discusses (at page 13, line 29–page 14, line 25) at least one M. hyo antigen included in the M. hyo soluble portion. In one embodiment, the M. hyo supernatant fraction is said to include one or more of the following M. hyo protein specific antigens: M. hyo proteins of approximately 46kD (p46), 64kD (p64) and 97kD (p97); kD being molecular weight expressed in kilo Daltons. In another embodiment, the M. hyo culture supernatant may include further M. hyo specific antigens such as, but not limited to, proteins of approximately 41 kD (p41), 42kD (p42), 89kD (p89), and 65kD (p65). The specification cites Okada et al, “Protective effect of Vaccination with Culture Supernate of M. hyopneumoniae against Experimental Infection in Pigs” (2000) Journal of Veterinary Medicine, Series B 47(7) 527–533 (Okada 2000a) following the list of specific protein antigens.
58 At page 15, lines 18–22, the specification details another embodiment:
In one embodiment, M. hyo soluble p46 antigen is included in the compositions of the invention at a final concentration of about 1.5 µg/ml to about 10 µg/ml, preferably at about 2 µg/ml to about 6 µg/ml. It is noted that p46 is the protein used for the M. hyo potency test (see example section below). In another embodiment, the M. hyo antigen can be included in the compositions at a final amount of about 5.5% to about 35% of the M. hyo whole culture protein A-treated supernatant.
59 At page 15, lines 24–30, the specification states that the M. hyo soluble preparation of the present invention is both safe and efficacious against M. hyo and is suitable for single dose administration. In addition, it is said that it has been surprisingly discovered that the M. hyo soluble preparation can be effectively combined with antigens from other pathogens without immunological interference between the antigens. That is said to make the M. hyo soluble preparation of the invention an effective platform for multivalent vaccines. The additional antigens may be given concurrently with the M. hyo composition (ie as separate single vaccines) or combined in a ready-to-use vaccine.
60 At page 16, the specification provides details of other embodiments, including where:
(a) The immunogenic composition includes at least one M. hyo soluble antigen and at least one additional antigen; or
(b) The immunogenic composition includes the combination of at least one M. hyo soluble antigen (eg two or more) and a PCV-2 antigen.
61 See also page 19, lines 23–26, which describes further embodiments, including a combination of at least one M. hyo soluble antigen (eg two or more), a porcine circovirus type 2 (PCV-2) antigen and a PRRS virus antigen.
62 Page 19, lines 9–21, includes the following examples of absolute concentration:
In one embodiment, a chimeric PCV1-2 virus is included in the compositions of the invention at a level of at least 1.0 < RP< 5.0, wherein RP is the Relative Potency unit determined by ELISA antigen quantification (in vitro potency test) compared to a reference vaccine. In another embodiment, a chimeric PCV1-2 virus is included in the composition of the invention at a final concentration of about 0.5% to about 5% of 20-times (20X) concentrated bulk PCV1-2 antigen.
In another embodiment, the PCV2 ORF2 recombinant protein is included in the compositions of the invention at a level of at least 0.2 µg antigen/ml of the final immunogenic composition (µg/ml). In a further embodiment, the PCV2 ORF2 recombinant protein inclusion level is from about 0.2 to about 400 µg/ml. In yet another embodiment, the PCV2 ORF2 recombinant protein inclusion level is from about 0.3 to about 200 µg/ml. In a still further embodiment, the PCV2 ORF2 recombinant protein inclusion level is from about 0.35 to about 100 µg/ml. In still another embodiment, the PCV2 ORF2 recombinant protein inclusion level is from about 0.4 to about 50 µg/ml.
63 Examples of suitable adjuvants for use in the compositions of the invention are listed at page 24.
64 The specification notes at page 26, lines 24–27 the methods used to separate the M. hyo whole cell preparation from the insoluble cellular material. These conventional methods include filtration, centrifugation and precipitation across various embodiments.
65 The 535 Application has 13 examples. Example 1 provides a high level description of a method by which an M. hyo cell culture is fermented and inactivated for the purposes of producing a PCV-2 combinable M. hyo antigen. Example 2 provides a description of methods by which chimeric porcine circovirus (cPCV)1-2 can be produced. Examples 1 and 2 are replicated in the 537 and 540 Applications.
66 Example 3 (in each of the Applications) is entitled “Down Stream Processing of M. hyo antigens and Analytical Testing of these Processed Antigens”. This example describes how a number of M. hyo preparations prepared as described in example 1 were treated and then analysed for M. hyo specific p46 antigen. The processed M. hyo antigens were then employed in example 4 (in each of the Applications) to prepare M. hyo vaccine formulations. Formulations relevant to BI’s validity case, which are discussed later, include:
T03: (10X UF (ultra filtration) concentrated) Concentrated by tangential flow filtration via a 100KDa molecular weight cut-off membrane (hollow fibre). Final volume reduction was equal to 10X.
T04 and T05: (10X UF concentrated and centrifuged) Concentrated mycoplasma cells (from T03) were collected and washed one time with PBS via centrifugation at ~20,000xg (Sorvall model RCB).
T06 and T07: (10X centrifuged) Inactivated fermentation fluid was centrifuged at ~20,000xg (Sorvall RC5B) and washed one time by resuspending the cells in PBS followed by additional centrifugation. Final volume reduction was equal to 10X.
T08: (10X centrifuged and heated) Mycoplasma cells were concentrated and washed per T06 and heated to 65°C for 10 minutes.
T09: (cell-free supernatant) Supernatant collected from the first centrifugation as described for T06 was filter sterilized through a 0.2 micron filter (Nalgene).
T10: (cell-free-supernatant-Protein-A treated) Sterile supernatant (prepared per T09) was mixed with Protein A resin (Protein A Sepharose, Pharmacia Inc) at a 10:1 volume ratio for 4 hours. Resin was removed and sterile filtration and filtered fluid was stored at 2-8˚C. This process uses post-fermentation “downstream” Protein A treatment to remove antibodies and immunocomplexes.
67 The specification notes (at page 32) that, although the present invention does not preclude upstream Protein A treatment, the present inventors have found that, in the case of M. hyo, upstream processing of the growth media led to P46 results which were lower and inconsistent as compared to untreated media.
68 The specification continues at page 33, line 13:
Since it is known in the art that Protein A binds IgG, it is understood by those of ordinary skill in the art that not only PCV2 antibody, but other swine antibodies, including PRRS antibody, HPS antibody, and SIV antibody will be effectively removed by the Protein-A treatment. This makes the Cell-free Protein-A treated M. hyo supernatant of this invention compatible not only with PCV2 antigen, but also with other porcine antigens due to the lack of immunological interference between the antigens. Additionally, the removal of the non-protective cell debris and removal of the immunoglobulin and antigen/immunoglobulin complexes is reasonably expected to make a safer vaccine.
69 The differences between the three specifications as far as the examples are concerned can be summarised as follows:
(a) Examples 12 and 13 of the 537 Application are not present in the 535 or 540 Applications. These examples describe two studies, the first designed to evaluate the efficacy of the PCV1-2 chimera, killed virus fraction of an experimental 1-bottle PCV-2/M. hyo combination vaccine, administered once to piglets, and the second to evaluate the efficacy of the M. hyo fraction of an experimental PCV1-2 chimera.
(b) Examples 14 and 15 of the 537 Application are labelled example 12 and 13, respectively, in the 535 and 540 Applications.
(c) The 540 Application contains three additional examples not present in the 535 or 537: examples 14 to 16. Examples 14 to 16 describe various further studies designed to evaluate the efficacy of the M. hyo, PCV-2 and PRRS components respectively of a trivalent vaccine of the invention.
70 The 535 Application’s specification ends with 18 claims. Claim 1 of the 535 Application claims:
An immunogenic composition comprising the supernatant of a Mycoplasma hyopneumoniae (M. hyo) culture, wherein the supernatant of the M. hyo culture has been separated from insoluble cellular material by centrifugation, filtration, or precipitation and is substantially free of both (i) IgG and (ii) immunocomplexes comprised of antigen bound to immunoglobulin.
71 Claim 2 of the 535 Application is dependent upon claim 1, and adds “wherein the soluble portion has been treated with Protein A or Protein G prior to being added to the immunogenic composition”.
72 Claim 3 of the 535 Application is dependent upon claims 1 or 2, and claims:
The composition of claim 1 or claim 2, wherein the composition further comprises at least one additional antigen which is protective against a microorganism selected from the group consisting of porcine reproductive and respiratory syndrome virus (PRRSV), porcine parvovirus (PPV), Haemophilus parasuis, Pasteurella multocida, Streptococcum [sic] suis, Staphylococcus hyicus, Actinobacilllus [sic] pleuropneumoniae, Bordetella bronchiseptica, Salmonella choleraesuis, Salmonella enteritidis, Erysipelothrix rhusiopathiae, Mycoplama [sic] hyorhinis, Mycoplasma hyosynoviae, leptospira bacteria, Lawsonia intracellularis, swine influenza vims (SIV), Escherichia coli antigen, Brachyspira hyodysenteriae, porcine respiratory coronaviruses, Porcine Epidemic Diarrhea (PED) vims, rotavims, Torque teno virus (TTV), Porcine Cytomegalovirus, Porcine enterovimses, Encephalomyocarditis virus, a pathogen causative of Aujesky’s [sic] Disease, Classical Swine fever (CSF) and a pathogen causative of Swine Transmissable [sic] Gastroenteritis, or combinations thereof.
(Emphasis added.)
73 The bolded references above are to the “5 Pathogens” which were the subject of discussion by the experts and which are relevant to Boehringer’s validity case. Four of the 5 Pathogens are viruses, and one, Brachyspira hyodysenteriae, is a bacteria.
74 Claim 16 of the 535 Application is a “kit” claim, which claims:
A kit for use in carrying out the method of claim 9 comprising:
a bottle comprising an immunogenic composition including the supernatant of a Mycoplasma hyopneumoniae (M. hyo) culture, wherein the supernatant of the M. hyo culture has been separated from insoluble cellular material by centrifugation, filtration, or precipitation and is substantially free of both (i) IgG and (ii) antigen/immunoglobulin immunocomplexes.
75 The 537 Application contains 24 claims, the broadest of which is claim 1, which claims:
A multivalent immunogenic composition comprising the supernatant of a Mycoplasma hyopneumoniae (M. hyo) culture; and a porcine circovirus type 2 (PCV2) antigen, wherein the supernatant of the M. hyo culture has been separated from insoluble cellular material by centrifugation, filtration, or precipitation and is substantially free of both (i) IgG and (ii) immunocomplexes comprised of antigen bound to immunoglobulin.
(Emphasis added.)
76 Claim 2 of the 537 Application is dependent on claim 1 and claims where the soluble portion of the M. hyo preparation has been treated with Protein A or Protein G prior to being added to the immunogenic composition.
77 Claim 3 claims the composition of claim 1 or 2 wherein the composition is in the form of a ready-to-use liquid composition.
78 Dependent claim 8 of the 537 Application claims the composition of any one of claims 1 to 7, further comprising at least one additional antigen which is protective against a microorganism selected from the same group listed in claim 3 of the 535 Application with the exception of PRRSV which is in the list of antigens in claim 3 of the 535 Application but is not found in the list in claim 8 of the 537 Application.
79 The 540 Application contains 25 claims. Claim 1 of the 540 Application claims:
A trivalent immunogenic composition comprising the supernatant of a Mycoplasma hyopneumoniae (M. hyo) culture; a porcine circovirus type 2 (PCV2) antigen; and a porcine reproductive and respiratory syndrome (PRRS) virus antigen, wherein the supernatant of the M. hyo culture has been separated from insoluble cellular material by centrifugation, filtration, or precipitation and is substantially free of both (i) IgG and (ii) immunocomplexes comprised of antigen bound to immunoglobulin.
(Emphasis added.)
80 Claims 2 to 13 of the 540 Application are dependent claims that also define trivalent immunogenic compositions, adding various characteristics to claim 1. In particular, claim 3 defines a “ready-to-use” composition containing the M. hyo and PCV-2 antigens but not the PRRS virus antigen. Claims 24 and 25 are claims to methods of preparing an immunogenic composition comprising the M. hyo supernatant, and PCV-2 and PRRS antigens.
81 The invention described and claimed in the specification of each Application may be broadly summarised as the provision of an immunogenic composition being an M. hyo supernatant platform which may itself be used as a vaccine, or which can be combined with other vaccines to form a preferably single dose ready-to-use vaccine. Relevant to each application, the nature of the M. hyo supernatant platform is such that immunogenic interference when combined with other antigens is absent due to the removal of IgG and immunocomplexes comprised of antigen bound to immunoglobulin (immunocomplexes) in the production of the M. hyo supernatant.
82 The 535 Application claims an immunogenic composition being an M. hyo supernatant platform, which may itself be used as a vaccine, or which can be combined with other vaccines to form a preferably single dose ready-to-use vaccine.
83 The invention as claimed in the 537 Application is a multivalent immunogenic composition being an M. hyo supernatant platform combined with a PCV-2 antigen, which may be combined with other vaccines to form a preferably single dose ready-to-use vaccine.
84 The invention as claimed in the 540 Application is a trivalent immunogenic composition being an M. hyo supernatant platform combined with a PCV-2 antigen and a PRRS antigen.
85 The statutory provisions of particular relevance to this matter are as follows.
86 Section 7 of the Act states:
7 Novelty, inventive step and innovative step
Novelty
(1) For the purposes of this Act, an invention is to be taken to be novel when compared with the prior art base unless it is not novel in the light of any one of the following kinds of information, each of which must be considered separately:
(a) prior art information (other than that mentioned in paragraph (c)) made publicly available in a single document or through doing a single act;
(b) prior art information (other than that mentioned in paragraph (c)) made publicly available in 2 or more related documents, or through doing 2 or more related acts, if the relationship between the documents or acts is such that a person skilled in the relevant art would treat them as a single source of that information;
(c) prior art information contained in a single specification of the kind mentioned in subparagraph (b)(ii) of the definition of prior art base in Schedule 1.
Inventive step
(2) For the purposes of this Act, an invention is to be taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed (whether in or out of the patent area) before the priority date of the relevant claim, whether that knowledge is considered separately or together with the information mentioned in subsection (3).
(3) The information for the purposes of subsection (2) is:
(a) any single piece of prior art information; or
(b) a combination of any 2 or more pieces of prior art information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have combined.
87 Section 40, which deals with sufficiency, support and best method, states:
…
Requirements relating to complete specifications
(2) A complete specification must:
(a) disclose the invention in a manner which is clear enough and complete enough for the invention to be performed by a person skilled in the relevant art; and
(aa) disclose the best method known to the applicant of performing the invention; and
(b) where it relates to an application for a standard patent—end with a claim or claims defining the invention; and
(c) where it relates to an application for an innovation patent—end with at least one and no more than 5 claims defining the invention.
(3) The claim or claims must be clear and succinct and supported by matter disclosed in the specification.
(3A) The claim or claims must not rely on references to descriptions, drawings, graphics or photographs unless absolutely necessary to define the invention.
(4) The claim or claims must relate to one invention only.
88 Four expert witnesses gave evidence at trial. Boehringer engaged Dr Nordgren and Professor Chase, and Zoetis engaged Professor McVey and Professor Browning.
89 Prior to the trial, the experts prepared a Joint Expert Report (JER). They were each cross-examined in the course of a joint session over 3 days. Dr Nordgren was also cross-examined in a separate session which followed the completion of the joint session.
90 Dr Nordgren has more than 35 years of experience in vaccine development and formulation for animals and has been involved in the registration of over 80 animal health products. He has a Bachelor of Science, a Master of Science (Veterinary Parasitology) and a Doctorate of Philosophy in Veterinary Immunology.
91 Dr Nordgren has worked for leading animal healthcare companies, including Solvay Animal Health, Boehringer Ingelheim Vetmedica and Merial Ltd. Dr Nordgren has managed global teams which have been responsible for over 80 unique animal products, including vaccines. A significant number of the vaccines for which he was responsible were developed before 2012.
92 While working at BIV in approximately 1998, Dr Nordgren was involved in a vaccine development project which created a one-dose inactivated whole cell bacterin vaccine for M. hyo in pigs. During the joint expert session Dr Nordgren agreed with senior counsel for Zoetis, Mr Flynn, that bacterins were a state of the art product and the best available option by 1998.
93 While working at Merial from 1999, Dr Nordgren was responsible for reviewing patents, and on occasion gave evidence in patent proceedings for Merial. His role at Merial also required the surveillance of products entering the market and acquisition of innovative competitor products.
94 Zoetis strongly challenged Dr Nordgren’s independence and ability to give impartial and relevant expert evidence on four grounds which I consider at the end of the evidence section.
95 Professor Chase has over 30 years of experience in veterinary immunology, including experience developing and researching vaccines for the treatment of diseases in swine. He is currently a Professor in the Department of Veterinary and Biomedical Science at South Dakota University. He holds a professional doctorate degree in Veterinary Medicine and a Master of Science and PhD.
96 During the period before the priority date, Professor Chase was involved in swine and ruminant disease diagnostics while teaching virology and immunology and undertaking research in infectious disease and immunology. Since 1 June 1998, Professor Chase has also held the position of President at his contract research company that undertakes, amongst other research, swine vaccine studies.
97 No challenge was made to Professor Chase’s independence. Professor Chase answered questions directly and engaged with the views expressed by his colleagues on technical matters. I consider that Professor Chase was a helpful witness who did his best to assist the Court throughout his evidence.
98 Zoetis made a hindsight challenge in relation to some of Professor Chase’s evidence. This challenge was based on the manner in which Professor Chase was briefed with the prior art and the questions put to him in the preparation of his evidence for the opposition proceeding. I deal with this challenge later where it is relevant to the issue of inventive step.
99 Professor McVey has around 40 years’ experience in animal health and veterinary microbiology. He has worked with both academic institutions and pharmaceutical and animal health companies in the field of vaccine formulation and development. Professor McVey holds a Doctor of Veterinary Medicine degree and a PhD in Veterinary Microbiology. He also has experience in the field working as a veterinarian, including treating endemic diseases of commercial farm animals, and has been employed by Merial from 1995–1998, and Pfizer Animal Health from 1998–2006.
100 Professor McVey is currently a Professor and Director of the School and Veterinary Medicine and Biomedical Sciences at the University of Nebraska-Lincoln and the Associate Dean of the Nebraska/Iowa Program for Veterinary Medicine.
101 Professor McVey has particular expertise in the immunology of infectious diseases of livestock, including diseases associated with M. hyo, PCV-2 and PRRS, and associated control measures including vaccine development.
102 No challenge was made to Professor McVey’s independence as an expert witness and he gave helpful affidavit and oral evidence.
103 Professor Browning is a Professor of Veterinary Microbiology in the Faculty of Veterinary and Agricultural Sciences at the University of Melbourne and has been since 2003. He has more than 35 years’ experience in veterinary microbiology, including the pathogenesis and epidemiology of infectious diseases and related animal vaccines.
104 Professor Browning’s research focuses on vaccines to control bacterial and viral respiratory diseases in swine and other livestock. He has particular expertise in relation to mycoplasmas including M. hyo.
105 Professor Browning has some industry experience, having collaborated on research projects on vaccine development and diagnostics with a range of industry partners including Pfizer, Bioproperties Pty Ltd, Grange Laboratories and BioBest Laboratories.
106 From 1998 to 2017, Professor Browning advised the Australian Pesticides and Veterinary Medicines Authority on the safety, efficacy, chemistry and manufacture of veterinary vaccines.
107 During oral evidence, Professor Browning described himself as “a bit pedantic”. This characteristic made him consider carefully all propositions put to him, or comments made by the other experts before accepting or rejecting them. Professor Browning was not afraid to depart from the comments of the other three expert witnesses where he felt it appropriate.
108 Boehringer pursued a line of questioning during the joint expert session which called Professor Browning’s independence into question. Professor Browning has worked on a number of projects that had received industry funding, including from Pfizer Animal Health (now Zoetis). As a result of these research projects, he is a named inventor on a number of patents co-owned by Zoetis and the University of Melbourne. The other named inventors on at least one of those patents are Zoetis employees. Professor Browning confirmed that he is entitled to a share of the royalties from this patent.
109 All of the research projects funded by Zoetis were declared in Professor Browning’s extensive CV, along with any patents on which he is a named inventor. I consider that Professor Browning was an independent, careful expert who gave considered and useful evidence.
110 Mr Eichmeyer is Boehringer’s Director of International Project Management – Vaccines. He has been employed by Boehringer, and its predecessors, since 2001, and is responsible for leading teams involved in vaccine registration across the world. Mr Eichmeyer was the head of a research project undertaken by Boehringer to develop a ready-to-use bivalent M. hyo/PCV-2 vaccine combination product. This project did not lead to the development of a bivalent, ready-to-use vaccine that was superior to Boehringer’s monovalent offerings.
111 Mr Eichmeyer swore one affidavit in the proceeding but was not called by either party. Zoetis tendered part of his affidavit
Zoetis’ challenge to Dr Nordgren
112 Zoetis challenged Dr Nordgren’s independence and ability to give impartial and relevant expert evidence on four grounds.
113 First, as a result of his lengthy and ongoing consulting arrangements with Boehringer and its predecessors, Zoetis contends that Dr Nordgren cannot be considered to be an independent witness.
114 Second, Zoetis submits that Dr Nordgren took into account significant non-common general knowledge in forming his opinions. Dr Nordgren failed to disclose his involvement in highly relevant confidential in-house M. hyo research work during his time at Merial. This, combined with a failure in his evidence to differentiate his use of the knowledge derived from his involvement with in-house research work from information generally known to all in the field in the formation of his opinions, means that the Court cannot disentangle the confidential in-house information and the common general knowledge used to form the basis of his opinions.
115 Third, Zoetis submits that Dr Nordgren is not representative of the hypothetical non-inventive person skilled in the art. Zoetis contends that Dr Nordgren’s inventorship on relevant patents and patent applications, and his participation in inventive research work relevant to the subject matter of the Applications, before the priority date, means that he is particularly ill-equipped to opine on the knowledge or approach of an uninventive person of ordinary skill in the field of swine vaccine development.
116 Fourth, Zoetis submits that Dr Nordgren’s role at Merial and Boehringer was not limited to undertaking research and experiments in the development of animal vaccine products. His ongoing consultancy role at Boehringer and its predecessors required him to review, and be involved in (including by providing expert witness testimony), challenges to competitors’ patents and patent applications relating to vaccine products. Zoetis submits that, in giving evidence in this proceeding, Dr Nordgren was acting under an obligation to act in the best interests of Boehringer and was not acting as an independent expert.
117 Accordingly, Zoetis submits that the Court should be reluctant to rely on Dr Nordgren’s evidence other than where it has been corroborated by the other experts.
Ongoing consulting role with Boehringer
118 Zoetis submits that Dr Nordgren’s relationship with Boehringer, its predecessor companies, and related entities is unique by reason of its longevity and, in particular, having regard to the fact that the services provided by Dr Nordgren relate not only to technical matters, but also to patent reviews, strategy and providing expert evidence.
119 Over a 20-year period from 1996 to 2016, Dr Nordgren held senior positions in Boehringer’s predecessor companies, including Vice President, Global Biologics Research & Development at Boehringer Ingelheim Vetmedica (1996-1999), Executive Director, Global Biologics Research & Development at Merial Ltd (1999-2001), Head of Research & Technology Acquisition at Merial Ltd (2001-2006), Vice President & Global Head of Biologics Research & Development at Merial Ltd (2006-2013) and Vice President & Global Head of External Innovation at Merial Ltd (2013-2016).
120 In his first affidavit, Dr Nordgren stated in paragraph 3 that “[a]part from my role as an expert witness, I have no direct relationship with either the Boehringer Ingelheim or Zoetis groups of companies”.
121 Dr Nordgren’s third affidavit was filed the day before the trial commenced. In his third affidavit, Dr Nordgren was asked to “describe in greater detail any work that I undertake for the Boehringer Ingelheim group of companies, referred to in paragraph 3 of my First Affidavit”. Dr Nordgren clarified that his reference in his first affidavit to his role as an expert witness was intended to encompass his role as an expert witness in this proceeding and other cases, and stated:
I undertake consulting work for the Boehringer Ingelheim group of companies on matters including contract disputes and patent litigation. This work includes acting as an independent expert witness for the Boehringer Ingelheim group of companies. However, the work is broader than being an expert witness. This is because, from time to time, I review and provide comments to the Boehringer Ingelheim group of companies on patents and patent applications in matters where I am not also asked to act as an expert witness. I wish to correct paragraph 3 of my First Affidavit to the extent it suggested that I am always formally engaged by the Boehringer Ingelheim group of companies as an expert witness in matters where I provide consulting services.
(Emphasis added.)
122 As revealed in his third affidavit, after ceasing his employment with Merial in 2016, Dr Nordgren was engaged by Merial as a consultant pursuant to a confidential agreement. Sometime later, Dr Nordgren entered into a “very similar” agreement pursuant to which he provides consulting services for the Boehringer Ingelheim group of companies (which includes Boehringer). The consultancy services provided by Dr Nordgren under the terms of that agreement do not relate to vaccine development, but rather to “contract disputes and patent litigation”.
123 Dr Nordgren agreed that part of his Boehringer consultancy role required him to review, and be involved in (including by providing expert witness testimony) challenges to competitors’ patents and patent applications relating to vaccine products. Since November 2015, Dr Nordgren has been called by Merial and Boehringer as an expert witness in eight domestic animal vaccine related patent proceedings, including in the European Patent Office, the US Patent Office, this Court and the Australian Patent Office. Dr Nordgren did not recall being aware of, or exposed to, the Applications prior to being asked about them in the course of preparing his evidence in this proceeding.
124 The existence of Dr Nordgren’s consulting agreements with Merial and Boehringer was disclosed for the first time in his third affidavit. In cross-examination, Dr Nordgren confirmed that his work for the Boehringer group of companies and their predecessors under those agreements is “wider than acting as an independent expert witness”.
125 The first approach to Dr Nordgren to act as an expert in this proceeding was made by Boehringer, rather than Boehringer’s instructing solicitors in this proceeding.
126 In the course of cross-examination, the following exchange took place between Zoetis’ senior counsel and Dr Nordgren:
[Counsel]: And under that contract [i.e., the Boehringer Consultancy Agreement], although we don’t have it, you tell us in paragraph 5 [of your third affidavit], that under that contract, you undertake consulting work from BI group of companies on matters including contract dispute and patent litigation; correct?
DR NORDGREN: That’s correct.
[Counsel]: And under that contract, you’re required to act in the best interests of BI; correct?
DR NORDGREN: On those cases that I’m working on; yes.
[Counsel]: And there’s no requirement in that contract that you act independently of BI if you’re acting as an expert witness; do you agree?
DR NORDGREN: I would agree.
[Counsel]: And in these proceedings, you are engaged, pursuant to that contract with BI, aren’t you?
DR NORDGREN: Yes, sir.
127 Zoetis submits that Dr Nordgren’s answers in the exchange set out above make clear that, in giving evidence in this proceeding, Dr Nordgren understood himself, first, to be under an obligation to act in the best interests of Boehringer and, secondly, to be under no obligation to act independently, an approach which is inconsistent with the Court’s Expert Evidence Practice Note and the Harmonised Expert Witness Code of Conduct.
128 Boehringer sought to explain the reason for Dr Nordgren’s third affidavit as being to clarify that Dr Nordgren’s reference to his “role as an expert witness” was intended to encompass his role as an expert witness in other proceedings in addition to the present. Boehringer says that it became apparent at a hearing before the Registrar that Zoetis had interpreted the words as only referring to this proceeding. However, the third affidavit goes beyond clarifying Dr Nordgren’s role as an expert witness.
129 Dr Nordgren’s third affidavit makes plain that his role with Boehringer is broader than his being engaged as an expert witness. His statement in his first affidavit: “Aside from my role as an expert witness, I have no direct relationship with either Boehringer Ingelheim or Zoetis groups of companies” (emphasis added), was not correct when the first affidavit was made. If Dr Nordgren’s third affidavit had not been filed on the eve of the trial, the Court would not have been informed of Dr Nordgren’s ongoing consulting role with Boehringer.
Regard to confidential information not part of common general knowledge
130 In his first affidavit Dr Nordgren mentioned two instances of experience with M. hyo: he oversaw or supervised the development of “an industry-first, one-dose inactivated whole cell vaccine, also known as a bacterin for M. hyo in pigs”, and whilst at Merial, his involvement in “the development of a significant number of vaccine products, including for the following diseases of swine: (a) M. hyo; (b) PRRS; and (c) Porcine circovirus 2 (PCV-2), including the first PCV-2 vaccine, which was released first in Europe and subsequently in other countries”. No further details of his prior experience with M. hyo were provided.
131 It emerged in the course of the concurrent evidence session that, before the priority date, in approximately 2008 or 2009, Dr Nordgren had led a team of researchers at Merial engaged in experimental work aimed at combining a PCV-2 vaccine with, amongst others, an M. hyo vaccine.
132 Dr Nordgren’s experimental M. hyo and PCV-2 work included efforts aimed at addressing impediments the M. hyo formulation may present to combining it with a PCV-2 antigen. Strategies investigated by Dr Nordgren and his colleagues at Merial included attempting to source serum with a reduced likelihood of containing anti-PCV-2 antibodies, as well as attempting to clarify serum of anti-PCV-2 antibodies. The research work was done in an effort to develop a process that would eliminate to a substantial degree the antibodies from the serum to enable Merial to formulate a vaccine (the in-house Merial M. hyo research work).
133 According to Dr Nordgren, the M. hyo experimental work was regarded by Merial as being highly confidential and a trade secret that Merial was concerned to ensure that its competitors did not find out. The confidential information obtained by Dr Nordgren as a result of the Merial M. hyo experimental work was not publicly disclosed. The confidential in-house knowledge gained by Dr Nordgren in the course of the Merial M. hyo experimental work did not form part of the common general knowledge of the person skilled in the art.
134 Boehringer submitted that it was “unsurprising” that Dr Nordgren did not expressly mention his prior work at Merial on combination vaccines, even after reviewing the Applications. According to Boehringer, the Applications concern the use of an M. hyo supernatant, whereas Dr Nordgren’s work at Merial involved its existing bacterin product and seeing if they could be improved with a goal to potentially combining them with PVC-2.
135 Boehringer also submitted that Dr Nordgren’s first affidavit “does impliedly” refer to his in-house M. hyo/PCV-2 research work. Boehringer explained that part of that work involved looking for PCV-2 antibody free serum, and that Dr Nordgren discussed his experience in trying to source PCV-2 antibody free serum in his affidavit, but was not taken to that in cross-examination.
136 However, in Dr Nordgren’s first affidavit, he stated that in April 2012 there was a need for bivalent M. hyo/PCV-2 and trivalent M. hyo/PCV-2/PRRS combination vaccines. There was no requirement or limitation that the M. hyo be in the form of a supernatant.
137 Dr Nordgren conceded that he did not refer to the in-house Merial M. hyo research work in any of his affidavits filed in the proceeding, including his third affidavit filed on the eve of the trial.
138 Dr Nordgren agreed that it would have been “highly relevant” and “important” to disclose this work to the Court. In particular, Dr Nordgren agreed that the fact he had engaged in the in-house Merial M. hyo research work was material to assessing the difference between his own knowledge and the knowledge of others in the field.
139 Dr Nordgren agreed in cross-examination that he took information from the confidential in-house Merial M. hyo research work into account in reaching the views he expressed in his affidavits in these proceedings.
[Counsel]: And to the extent that you had held any view as at April 2012 about the potential preparations based on the supernatant of M. hyo vaccines, that view was based on matters including your particular knowledge regarding experimental vaccines. Correct?
DR NORDGREN: That’s correct.
140 Zoetis submits that Dr Nordgren’s failure to disclose his involvement in the in-house Merial M. hyo research work, or the fact that he took this work into account when preparing his affidavits, presents insurmountable difficulties for the evaluation of Dr Nordgren’s evidence. According to Zoetis, it is impossible to determine which of the opinions expressed by Dr Nordgren in his affidavits and the joint expert session are based upon material which formed part of the common general knowledge of those skilled in the art, and those which are infected with the confidential in-house Merial information.
141 Zoetis contends that the effect of Dr Nordgren’s failure to disclose his involvement in the in-house Merial M. hyo research work is compounded by the absence of any instruction to Dr Nordgren in the course of preparing his affidavits to differentiate between his own particular knowledge (such as that obtained from confidential in-house research projects) and matters he considered to be generally well known and accepted by those skilled in the area.
142 Dr Nordgren was aware in April 2012 of studies demonstrating that specific enrichments of different culture fractions can improve the effectiveness of a vaccine response. He provided no details of the studies but observed that the exact compositions of commercially available M. hyo vaccines were most often kept confidential, as a trade secret.
143 Before the priority date, Dr Nordgren had collaborated with Professor Ross (the author of the Ross 1984 paper referred to in the Okada papers discussed below in the inventive step section) to research an experimental subunit vaccine against M. hyo using the 97kD antigen. The 97kD antigen research program was not pursued further as the 97kD antigen on its own did not produce sufficient protection. The collaborative work with Professor Ross was not published. Dr Nordgren agreed that he knew from the Ross paper and his teams’ own experiments that the combination of different antigens in supernatant based M. hyo vaccines were immunogenic and provided an adaptive immune response.
144 In addition, Dr Nordgren was involved, before the priority date, in the development of a first-in-class one-dose inactivated whole-cell (bacterin) vaccine against M. hyo that was commercially released around 1998, the first commercially available vaccine against PRRS virus, and the first PCV-2 vaccine to be released.
145 Before the priority date, Dr Nordgren worked with and tested experimental M. hyo vaccine preparations made from M. hyo cell culture supernatant fractions in the course of his work at Solvay and in the course of supervising the work of his team at Boehringer. In both cases, the project did not result in the launch of a commercial product and the information concerning those M. hyo supernatant was never made public.
146 I consider that the in-house work done by Dr Nordgren on a bivalent M. hyo/PCV-2 combination vaccine, the choices made by his research team and the reasons for those choices, and the success or failure of that work may well have had a role in the formation of the opinions which Dr Nordgren was being asked to give when addressing the development of a new or improved bivalent M. hyo/PCV-2 vaccine.
Not representative of non-inventive person skilled in the art
147 During the concurrent session, Dr Nordgren agreed that he is “inventive” in relation to M. hyo/PCV-2 combinations, and that he “knew more than most” people who participate in project teams associated with vaccines.
148 Dr Nordgren is named as an inventor on a number of patents and patent applications. Several examples were tendered during the hearing. That Dr Nordgren is listed as an inventor on patents and patent applications is not apparent from his first affidavit (or subsequent affidavits) as he did not annexe a curriculum vitae (CV) listing the patents and patent applications on which he is listed as an inventor.
149 Boehringer submits that Dr Nordgren’s CV was omitted from his affidavits simply because the information in his CV was already summarised at paragraphs 6 to 20 of his first affidavit, and that no inference can be drawn from his failure to do so as the Expert Evidence Practice Note does not require an expert to annexe a CV. Whilst that may be correct, the inference is not sought to be drawn from the absence of a CV per se, rather from the absence of detailed information that would usually be found in a CV, such as inventorship on patents and published papers.
150 Being listed as an inventor on a patent itself is unsurprising in an expert witness in a patent case. However, it is relevant when assessing the evidence of an expert, for the purpose of determining by reference to the non-inventive person skilled in the art whether an invention as claimed involves an inventive step, to know whether the expert is listed as an inventor on one or multiple patents and patent applications. It will also be relevant if the invention claimed in the patent or patent application is in the same or a related field to the patent in suit.
151 At least one family of Dr Nordgren’s patents and patent applications were directed to subject matter bearing a relationship to the subject matter of the Applications, including the WO 462 application in the name of Merial Limited (WO 2005/009462), filed on 26 July 2004 (and the Australian patent granted on the WO 462 application: AU 2004259034). Dr Nordgren’s inventorship on the WO 462 application or its Australian progeny was not disclosed in any of his affidavits.
152 The WO 462 application is entitled “Vaccine formulations comprising an oil-in-water emulsion”. Claims 26 and 27 of the WO 462 application are directed to a novel M. hyo/PCV-2 combination vaccine, including a one-dose regimen. Although Dr Nordgren initially sought to suggest that the claims directed to M. hyo/PCV-2 combination vaccines were not adequately supported by experimental work and may have been dropped after the application was filed, he subsequently accepted that those claims were retained in an Australian patent granted on the WO 462 application.
153 Dr Nordgren agreed that it would be relevant for the Court to take the WO 462 application into account in determining the difference between his knowledge and the knowledge of those of ordinary skill in the art.
154 Dr Nordgren’s first affidavit omitted relevant information including, in particular, his prior in-house experience with an M. hyo/PCV-2 vaccine and his ongoing consultancy with Boehringer. Details only emerged subsequently due to the tenacity of Zoetis’ legal team.
155 I do not consider that Dr Nordgren engaged in a deliberate practice to hide the extent of his prior knowledge of M. hyo by omitting his in-house Merial research experience from his affidavit. I consider that Dr Nordgren did his best to give independent evidence and answer the questions put to him by his instructors. However, the absence of a detailed description of that earlier confidential research work is unfortunate. That absence, combined with a lack of any explanation by Dr Nordgren as to how he was able to put any knowledge derived from that work aside in formulating his opinions, undermines the weight that would otherwise be given to his evidence as to what would be obvious to the non-inventive person skilled in the art, armed only with the common general knowledge and the Okada documents (separately or in combination) seeking to develop a new or improved M. hyo/PCV-2 vaccine, or trivalent M. hyo/PCV-2/PRRS vaccine.
156 Dr Nordgren agreed that he had used information that was not part of the common general knowledge in formulating his opinions. I am unable to disentangle the parts of Dr Nordgren’s evidence which are infected with information not generally known to others in the field of vaccine development from those that are based on solely common general knowledge. Where, particularly in matters relating to inventive step, Dr Nordgren’s evidence is not corroborated by the evidence of the other experts, particularly Professor Chase and Professor McVey, I will not accept that evidence. I note, however, that in large part, Dr Nordgren’s evidence was corroborated by the other experts.
157 Dr Nordgren’s evidence on the best method issue is in a different category. Dr Nordgren gives evidence as to what is disclosed in the examples of the Applications. This evidence was unchallenged, and I do not consider it to be undermined by his failure to disclose the information about his prior research and ongoing consultancy.
158 In Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1, Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ said at [24]:
It is well settled that the complete specification is not to be read in the abstract; here it is to be construed in the light of the common general knowledge and the art before 2 July 1984, the priority date; the court is to place itself “in the position of some person acquainted with the surrounding circumstances as to the state of [the] art and manufacture at the time”.
159 For the purposes of the Joint Expert Report, the parties defined the relevant field as relating to animal vaccine development.
160 Zoetis submits that the technical field of the invention described and claimed in the Applications is animal vaccine development, in particular, vaccines for swine. According to Zoetis, the notional skilled team to which the Applications are directed would include microbiologists (including bacteriology and virology) together with individuals having expertise in the development and evaluation of vaccine candidates.
161 Boehringer contends that the skilled person to whom the Applications are directed is a team with experience in the field of animal health. That team consists of a vaccinologist, with expertise in vaccine development and formulation for animals, and an expert in the field of immunology and the development of vaccines for swine.
162 Where the parties differ is that Zoetis submits that the Applications are directed to a team with specific expertise in, and a deep understanding of, M. hyo (or mycoplasmas generally), and that depending on the combination of antigens in the immunogenic composition, the skilled team would collaborate with, or consult, scientists with a deep understanding of each target pathogen or disease (the list in claim 3 of the 535 Application and claim 8 of the 537 Application). Boehringer disagrees.
163 Each of the Applications disclose immunological compositions (including vaccines) being M. hyo platforms which may be combined with other antigens such as PCV-2 and those listed in claim 3 of the 535 Application and claim 8 of the 537 Application. Zoetis submits that the team should be differently constituted for each combination of antigens to be included in the immunogenic composition, and that the team would collaborate with academics or specialists with a deep understanding of each of the target pathogens and who had familiarity with the state of research and the literature published in relation to their particular pathogen.
164 Zoetis’ purported “skilling up” of the skilled addressee has particular relevance in the context of the assessment of the challenges for want of disclosure and support made in respect of the multiple antigen claims (for example, claim 3 of the 535 Application and claim 8 of the 537 Application).
165 Boehringer submits that defects in the support and disclosure requirements cannot be solved by adding specialists to the team such that the common general knowledge is expanded to include their specialised knowledge.
166 Boehringer submitted that a similar argument was considered and rejected by Jagot J in Gilead Sciences Pty Ltd v Idenix Pharmaceuticals LLC (2016) 117 IPR 252, in relation to the old (pre-RTB Act) test for sufficiency. Idenix contended that the relevant area of discourse may be characterised as the development of nucleoside analogues for use as antiviral drugs for Flaviviridae infections, including HCV. Gilead described that characterisation as so specific that it “is almost to require a person to have worked on the development of the invention in suit”. At [207], Jagot J observed:
Given the terms of the Idenix patent I do not consider Idenix’s characterisation unreasonable. This does not mean, however, that every expert must have expertise across the entire spectrum of the area of discourse in order to give evidence relevant to the common general knowledge. Nor does it mean that, insofar as the chemistry is concerned, the skilled addressee is a person with specialist, even unique (as in the case of Dr Borthwick), experience in the fluorination of a tertiary hydroxyl. As Idenix no doubt intended, to adopt this approach would pitch the debate at a level of specificity ensuring Professor Meier and Dr Borthwick alone could be representative of the skilled addressee. I do not accept this and I also do not accept that the evidence given by Professor Furneaux and Dr Lambert is immaterial to the common general knowledge of the skilled addressee.
167 Her Honour held at [209] that it was immaterial what label was placed on the notional skilled addressee, and continued:
The issue is the knowledge which the skilled addressee is taken to have, which is the common general knowledge of the skilled addressee of documents dealing with the development of nucleoside analogues for use as antiviral drugs for Flaviviridae infections, including HCV. As Gilead submitted, the need for inferences about the common general knowledge to be based on the evidence:
… cannot be avoided by artificially constructing a skilled team to include a person …with highly specific and unusual knowledge gained through literature searches or idiosyncratic experience not shared by others in the field. Nor can it be avoided by artificially excluding others who happen not to possess that knowledge. The common general knowledge is a general body of knowledge known and used by all those in the field.
168 The skilled addressee “is not an avatar for expert witnesses whose testimony is accepted by the court”, but a tool for analysis: AstraZeneca AB v Apotex Pty Ltd; AstraZeneca AB v Watson Pharma Pty Ltd; AstraZeneca AB v Ascent Pharma Pty Ltd (2015) 257 CLR 356 at [23] (AstraZeneca HCA). The issue is what common general knowledge is to be attributed to the skilled addressee for the purposes of assessing the grounds of invalidity alleged.
169 After reviewing the principles applicable to assessing the common general knowledge at [210] to [214], Jagot J concluded in Gilead at [219]:
In other words it is apparent from his evidence that Professor Meier had a particular expertise in the fluorination of nucleoside analogues by reason of work he had done in 1999 as part of which he had read and assimilated lots of papers about fluorination reactions. While some familiarity with fluorination reactions might be imputed to the skilled addressee of the 350 application and the Idenix patent it would be wrong to conceive of the skilled addressee as having specialist experience and expertise in that regard by reason of personal involvement in the fluorination of nucleosides (or, in the case of Dr Borthwick, carbocyclic nucleosides). Neither the 350 application nor the Idenix patent is directed towards such a narrow topic as the fluorination of nucleosides. I do not accept that the skilled addressee can be treated as having available as part of the common general knowledge all of the expertise and experience in the fluorination of nucleosides that Professor Meier and Dr Borthwick had by reason only of their involvement in particular projects for that purpose.
(Emphasis added.)
170 The field of the invention does not change. The characteristics attributed to the hypothetical person skilled in the art do not change according to the ground of invalidity which they are used to assess. In other words, the skilled addressee is not to be “skilled up” in relation to the support and disclosure requirements, and dumbed down for inventive step.
171 Neither side led evidence as to what additional matters might constitute the common general knowledge of specialists in the 5 Pathogens or those pathogens listed in claim 3 of the 535 Application. Boehringer, because it did not consider such specialist knowledge appropriate to the skilled addressee, and Zoetis, as it submitted that the onus was on Boehringer to establish the specialist knowledge of the skilled addressee.
172 Professor Browning, who came from a more academic than commercial background, had a greater understanding of mycoplasmas than the other experts. However, none of the experts were a specialist with a deep understanding of any of the 5 Pathogens relied on by Boehringer in relation to its disclosure and support grounds of challenge, which is considered later.
173 None of the Applications is directed towards the identification of the particular antigens protective against the range of potential microorganisms, which may be combined with the M. hyo supernatant platform. Rather, the 535 and 537 Applications claim the combination of the M. hyo supernatant platform combined with at least one additional antigen protective against a microorganism selected from the specified list, once that antigen or antigens have been identified.
174 I consider that persons skilled in the art, those with a practical interest in the subject matter of the invention, will include veterinary microbiologists (with expertise in bacteriology and virology) and immunologists with skill and expertise in the development and formulation of vaccines for animals, in particular swine. Each of the expert witnesses called in the present matter have experience in the development of animal vaccines against a range of pathogens, and are qualified to provide evidence in this proceeding.
175 There is one principal issue of construction in this case, Boehringer having abandoned its challenge to “substantially free”. That is, what was called by Zoetis, “the antigen issue”, which primarily arises from the use of the term “comprising” in claim 1 of the Applications. The resolution of this construction issue determines the extent of the invention required to be disclosed and supported for the purposes of s 40 of the Act.
176 Before addressing “the antigen issue”, it is necessary to construe the other integers in the claim to provide context. The construction of the remaining integers of the claim was not a matter of great dispute.
177 First, the principles of construction of patent claims are well settled and established. See Jupiters Ltd v Neurizon Pty Ltd (2005) 65 IPR 86 at [67] (per Hill, Finn and Gyles JJ).
178 I highlight the following principles which are of particular relevance to this case:
(a) The Court is to read the specification through the eyes of the skilled addressee, with practical knowledge and experience in the field of work in which the invention was intended to be used, and a proper understanding of the purpose of the invention: GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Generic Partners Pty Ltd (2018) 2131 IPR 384 at [106] (per Middleton, Nicholas and Burley JJ).
(b) In construing the claims, a generous measure of common sense should be used: Ranbaxy Laboratories Ltd v AstraZeneca AB (2013) 101 IPR 11 at [108] (per Middleton J); and Streetworx Pty Ltd v Artcraft Urban Group Pty Ltd (2014) 110 IPR 82 at [58]–[69] (per Beach J).
(c) A construction that gives the claims a sensible and workable meaning is to be preferred: Asahi Kasei Kogyo Kabushiki Kaisha v WR Grace & Co (1991) 22 IPR 491 at 515 per Heerey (not disturbed on appeal: WR Grace & Co v Asahi Kasei Kogyo Kabushiki Kaisha (1993) 25 IPR 481 at 500–501 (per Northrop, Lockhart and Cooper JJ)). A construction that would lead to an absurd result is to be avoided: NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1993) 26 IPR 513 at 287 (per Northrop, Lockhart and Burchett JJ).
(d) Although the claims are to be construed in the context of the specification as a whole, it is not legitimate to narrow or expand the boundaries of monopoly as fixed by the words of a claim by adding to those words glosses drawn from other parts of the specification: Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610 (per Menzies J).
(e) Claims are to be construed by the Court through the eyes of the skilled addressee of the particular patent and without regard to the alleged infringing article: Fresenius Medical Care Australia Pty Ltd v Gambro Pty Ltd (2005) 67 IPR 230 at [95] (per Wilcox, Branson and Bennett JJ).
(f) Claims should not be construed by applying “the kind of meticulous verbal analysis in which lawyers are too often tempted by their training to indulge”: Catnic Components Ltd v Hill & Smith Ltd [1982] RPC 183 at 243 per Lord Diplock, quoted with approval in Artcraft Urban Group Pty Ltd v Streetworx Pty Ltd (2016) 117 IPR 210 at [79] (per Greenwood J).
(g) A patent may claim less than it teaches or enables: Kirin-Amgen v Hoechst Marion Roussel (2004) 64 IPR 444 at [33] (per Lord Hoffmann).
179 Claim 1 of the 535 Application claims an “immunogenic composition”, claim 1 of the 537 claims a “multivalent immunogenic composition”, and claim 1 of the 540 claims a “trivalent immunogenic composition”.
180 The term “immunogenic composition” is defined in the body of each specification to mean “a composition of matter that comprises at least one antigen which elicits an immunological response”. An “immunological response” is said in the specification to include, but not be limited to, effects such as the production or activation of antibodies, B cells and helper T cells, amongst others, directed specifically to an antigen or antigens included in the composition. As defined in the specification, an “immunogenic composition” is not required to elicit a protective immunological response which provides enhanced resistance to new infection, and/or reduction of the clinical severity of the disease caused by the antigen or antigens.
181 An immunological composition may or may not elicit a protective response. That the immunological response need not be protective for claim 1 of the Applications is confirmed by the existence of dependent claims which introduce such a requirement. For example, claim 7 in the 535 Application claims the composition of any of claims 1 to 6, “wherein the composition elicits a protective immune response against M. hyo”.
182 In the 537 and 540 Applications, the immunogenic composition claimed in claim 1 is a “multivalent” or “trivalent” immunogenic composition respectively. The term “multivalent” is defined in the specifications as meaning a vaccine (the term “composition” is not used in the definition) containing more than one antigen, whether from the same species or from different species, or a vaccine containing a combination of antigens from different genera. Again, the reference to an “immunogenic composition” does not require that each antigen provide any particular level of immunological response, such as a response which is protective in relation to a particular condition.
183 The 540 Application claims differ to those of the 535 and 537 Applications, in that claim 1 expressly claims (and is limited to) a trivalent composition. There is no dependent claim listing additional antigens as there is in both the 535 and 537 Applications. A trivalent composition does not admit the inclusion of more than three antigens, whether from the same species or from different species.
184 The experts agreed that “substantially free” means “close to pure of a specific component, but not absolutely or completely free” such that the supernatant may still contain trace amounts of IgG and/or immunocomplexes. They also accepted that the claim does not define or specify any quantitative threshold or require that any particular test be used, and that the assessment of whether a composition is free of the relevant components might depend on factors such as the technique used to detect them and the conditions under which the test is conducted.
185 In the context of Table 3 (of Example 3), Professor Browning suggested a numerical threshold S/P ratio (sample to positive ratio) of less than 0.099 as a reasonable threshold for what he considered would be “substantially free”. He agreed that the figure was not drawn from the specification, and that the S/P ratio was expressed to be relative to a positive control, which, in the case of Table 3, he agreed that he did not know how much antibody was in the positive control.
186 Professor Browning explained that the maximum possible optical density was two, so a ratio of below 0.099 was a significant reduction from the maximum value. Professor Browning agreed that his threshold ratio would allow for some anti-PCV-2 antibodies to be present and cause a degree of interference with the PCV-2 antigen.
187 The term “supernatant” is a technical term which the experts agreed meant the liquid component obtained from a whole bacterial culture that has been separated from substantially all of the bacterial cells and insoluble materials. The separation step would typically occur using centrifugation to separate the insoluble components from the soluble components in commercial vaccine processes, although filtration could also be used.
188 Claim 1 states that the supernatant has been “separated from insoluble cellular material”. The experts agreed that this expression means that the supernatant has been separated from the insoluble cellular fraction. During the expert evidence session, Professor McVey stated this does not require removal of absolutely all insoluble cellular material, some trace amounts may remain. Professor Browning did not consider the words of the specification to provide a threshold or indicate whether some trace amounts of insoluble cellular material could be present in the supernatant. The other experts agreed with this characterisation.
189 Claim 1 of the 535 Application claims an “immunogenic composition comprising the supernatant of a Mycoplasma hyopneumoniae (M hyo) culture”. Claim 1 of the 537 Application claims a “multivalent immunogenic composition comprising the supernatant of a Mycoplasma hyopneumoniae (M hyo) culture; and a porcine circovirus type 2 (PCV-2) antigen”. Claim 1 of the 540 Application claims a “trivalent immunogenic composition comprising the supernatant of a Mycoplasma hyopneumoniae (M hyo) culture; and a porcine circovirus type 2 (PCV-2) antigen; and a porcine reproductive and respiratory syndrome (PRRS) virus antigen”.
190 Claim 3 of the 535 Application and claim 8 of the 537 Application, claims the composition of claim 1, further comprising at least one additional antigen which is protective against a microorganism selected from a specified list of microorganisms.
191 Each Application purports to provide a definition of the term “comprising” as used in the specification:
“[C]omprising” is intended to mean that the compositions and methods include the recited elements, but do not exclude other elements.
192 On Zoetis’ construction of the claims, antigens that are not expressly mentioned in a claim (either in terms or by reference to another claim) are not elements of the invention defined by that claim. Those additional elements are not expressly included in the claim, but neither are they excluded.
193 Zoetis refers to the definition of “comprising” in the specification and says that it means that the recited integers (the expressed elements of the claim) are included in the claim, but that the claim is neutral as to other elements that may be present (such as additional antigens, or adjuvants). They are not excluded from the composition, but neither are they required nor the subject of the claim. The claim does not define the invention by reference to the presence, or absence, of that (unidentified) integer.
194 Zoetis submits it is plain that a composition having additional integers not included in the claim can still infringe a claim. But that consequence does not mean that those additional integers must be regarded as part of the invention defined by the claim. Zoetis submits that the use of an immunogenic composition containing an additional antigen would infringe such a claim, but that such a composition did not need to be disclosed or supported by the description in the specification in order for the claim to be valid. This is illustrated by the example of a claim which claims a product comprising elements A+B+C, but which claim is infringed by a product which has elements A+B+C+D. Element D is not expressly claimed, but the product will infringe the claim because A, B and C are present. Element D does not need to be disclosed or supported in the specification for the claim to the product comprising A+B+C to be valid.
195 Similarly, Zoetis submits that the immunogenic composition of claim 1 so defined may be combined with other optional elements, such as a suitable adjuvant, or a pharmaceutically acceptable carrier. Those other elements are not integers of the composition defined by claim 1, but may still be present in the immunogenic composition. According to Zoetis, the person skilled in the art would understand that other elements of vaccines such as an adjuvant may be present in the product of the immunogenic composition claim for it to work, even if it is not expressly mentioned in the claim.
196 Zoetis refers to Lindgren J in General Clutch Corporation v Sbriggs Pty Ltd (1997) 38 IPR 359 at [376]–[377], in which his Honour stated that a construction that would result in non-compliance with s 40 of the Act should be avoided where possible, as a reason to prefer its construction.
197 Zoetis submits that its construction should be preferred as it is the construction that best accords with the language of the claims (considered in their context), gives coherence to the claim set, and is consistent with the nature of the invention as disclosed in the specification as a whole.
198 As it states in its closing submissions, Zoetis relies in particular on:
(a) the absence in claim 1 of any reference to antigens other than “the supernatant of a Mycoplasma hyopneumoniae (M. hyo) culture, wherein the supernatant of the M. hyo culture has been separated from insoluble cellular material by centrifugation, filtration, or precipitation and is substantially free of both (i) IgG and (ii) immunocomplexes comprised of antigen bound to immunoglobulin” (M. hyo soluble preparation) (535 Application), the M. hyo soluble preparation and a PCV-2 antigen (537 Application) and the M. hyo soluble preparation, a PCV-2 antigen and a PRRS antigen (540 Application);
(b) the use of the words “further comprises” or “in conjunction with” in all claims that identify additional antigens. It says that “further” would be otiose on Boehringer’s construction;
(c) the use of the words “additional antigen” in claims identifying antigens other than M. hyo (535), M. hyo and PCV-2 (537), or M. hyo, PCV-2 and PRRS (540), again would be otiose on the Boehringer construction;
(d) the dependencies of claims that refer to any “additional antigen” claim;
(e) separately, and despite the extensive discussion about PCV-2 in the 535 specification, the deliberate absence of any reference to PCV-2 in the claims of the 535 Application;
(f) similarly, the absence of any reference to PRRS in the claims of the 537 Application, despite it being discussed in the 535 specification; and
(g) the invention as disclosed in the specification, which indicates that it is concerned with the ability to create particularly useful combinations of antigens, rather than combinations of any and all antigens.
199 In contrast, Boehringer propounded an inclusive construction of “comprising” and the claim, which it submits accords with Burley J’s construction of a closely analogous claim containing the term “comprising” in Merck Sharp & Dohme Corporation v Wyeth LLC (No 3) (2020) 155 IPR 1.
200 Given the disclosure in the body of the specification, Boehringer submits that the skilled person would understand that an immunogenic composition of the kind claimed in claim 1 may contain components other than those specifically identified in the claim, including additional antigens of the kind described in the specification (and claimed in claim 3), and other components such as adjuvants and pharmaceutically acceptable carriers (claim 6). It follows that the immunogenic composition is not limited to the features expressly referred to in the claim but may include additional antigens (such as those listed in claim 3 of the 535 Application) and other components.
201 On Boehringer’s proposed construction, by reason of the word “comprising” in claim 1 of the 535 and 537 Applications, Boehringer contends that:
(a) every claim of the 535 and 537 Applications; and
(b) claims 24 and 25 of the 540 Application;
“encompass” immunogenic compositions comprising antigens protective against microorganisms in addition to those specifically identified in the claim.
202 Boehringer submits that in accordance with orthodox claim construction, the claim set starts with a broad claim, the scope of which is then narrowed by the introduction of dependencies. Boehringer submits that its construction of claim 1 is confirmed by the features of the dependent claims of the 535 Application, which specify, by way of limitation, specific kinds of components that may be included in the immunogenic composition. These include additional antigens (claim 3), an adjuvant (claims 4 and 5) and a pharmaceutically acceptable carrier (claim 6). These claims, being dependent on claim 1, are narrower in scope by reason of the requirement to include the additional components. Their presence emphasises that claim 1 is broader, and encompasses compositions in which those components are present, as well as compositions in which they are not.
203 Boehringer submits that Zoetis’ approach to construction is wrong in principle and contrary to authority. The only relevant distinction on the question of construction is between something that is within the scope of a claim, on the one hand, and something that is outside its scope, on the other. There is no intermediate third position of the kind contended for by Zoetis.
204 Zoetis’ senior counsel, Mr Flynn, accepted that the Merck definition of “comprising” could not be distinguished from the definition in the Applications (see Merck at [179]–[180] (per Burley J)). Rather, he sought to distinguish Merck from the present case. First, on the basis that it was a different patent. Second, the Merck claim used “comprising” twice in the claim, and the only “further comprising” claims added the “further” integer of an adjuvant. Third, on the basis that the Court in Merck was plainly in error to the extent that it interpreted words similar to “does not exclude” to mean “includes”, and to the extent the Court in Merck apparently relied on an analysis which wrongly equated (at [549]):
(i) all the elements of compositions which would infringe a claim (“fall within the monopoly of the claims”); with
(ii) all the elements of a composition which are included in a claim for the purposes of identifying the invention defined by the claim within the meaning of s 40.
205 Zoetis submits that is wrong to speak of the “scope of the claim” in the context of construction. It points to the wording of s 40(2)(b) which requires the specification to end with a claim or claims defining the invention, and does not speak of the scope of the claim.
206 Zoetis submits that it is necessary and important to differentiate between the process of construction (which must occur without regard to an alleged infringing article) and the principles concerning infringement, including those concerning infringement of combination claims despite the addition of integers: see Nichia Corporation v Arrow Electronics Australia Pty Ltd [2019] FCAFC 2 at [45] (per Besanko, Jagot and Nicholas JJ). Zoetis submits that whilst claim 1 would be infringed by an immunogenic composition that might have additional antigens in addition to the M. hyo supernatant, those additional antigens are not elements of the composition claimed in claim 1.
207 As the specification describes, the immunogenic composition so defined may be combined with other optional elements, such as a suitable adjuvant, or a pharmaceutically acceptable carrier, or additional antigens. Those other elements are not integers of the composition defined by claim 1.
208 Zoetis submits that “comprising” in this instance should be understood as a practical means of drafting. If claim 1 defined an “immunogenic composition comprising of the M. hyo Soluble Portion and nothing else” then infringement could be avoided by adding any manner of additional things, for example, blue food colouring, or an M. hyo insoluble fraction, or water, or a preservative, and so on. Zoetis submits that its construction, which relates to the particular issue of “additional antigens” is not at odds with the “definition” of “comprising” set out in the specification.
209 Zoetis rejected Boehringer’s suggestion that claim 1 of the 535 Application could be rewritten as “monovalent” (or claim 2 of the 537 Application as “bivalent”). Zoetis submits that the claims of the 535 and 537 Applications include claims that do claim (“further comprise”) additional antigens protective against another pathogen, and which depend for their meaning on the terms of claim 1. Those claims would not make sense if the composition referred to in claim 1 was limited to a monovalent (or, for the 537 Application, bivalent) composition.
210 As Zoetis submitted, Boehringer’s attempt to rely on the inventor’s use of “trivalent” in claim 1 of the 540 Application to construe the claims of the 535 and 537 Applications, was not a legitimate approach to claim construction.
Merck and the meaning of “comprising”
211 In Merck, Burley J considered the meaning of “comprising” in a claim in a similar form to claim 1, involving an immunogenic composition, which used both “comprising” and “comprises” and claimed:
A multivalent immunogenic composition, comprising: 13 distinct polysaccharide-protein conjugates, together with a physiologically acceptable vehicle, wherein each of the conjugates comprises a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, and the capsular polysaccharides are prepared from serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F and 23F and wherein said carrier protein is CRM197.
(Emphasis added.)
212 Like the present case, the specification in Merck also included a definition of “comprising”, in the following terms:
Throughout this specification and the claims which follow, unless the context requires otherwise, the word “comprise”, and variations such as “comprises” and “comprising”, will be understood to imply the inclusion of a stated integer or step or group of integers or steps but not the exclusion of any other integer or step or group of integers or steps.
213 Like the present specifications, the specifications in Merck included dependent claims which used the phrase “further comprising”, albeit to claim an immunogenic composition of claim 1 “further comprising an adjuvant” (claim 2 of the 013 patent, claim 3 of the 844 patent), or which “further comprises one or more antigens for use against otitis media caused by infection with other bacteria” (claim 7 of the 844 patent): Merck at [155] and [157].
214 The relevant issue for determination in Merck was whether the asserted composition patent claims included within their scope one or more Streptococcus pneumoniae serotypes in addition to the 13 serotypes listed. This focussed attention on the meaning of the words “comprises” and “comprising” where they appeared in the asserted claims (“the comprising issue”).
215 In the context of the specification, the respondent, Wyeth, propounded an inclusive meaning for “comprising”, as opposed to an exhaustive meaning (“consisting of”), such that the claim included within its scope any immunogenic composition which had the integers of the claim. This included polysaccharide-protein conjugate which included each of the chosen 13 serotypes, notwithstanding such an immunogenic composition may also contain other integers, such as polysaccharide-protein conjugates of one or more Streptococcus pneumoniae serotypes in addition to the 13 chosen serotypes. Wyeth contended that any multivalent immunogenic composition that had the integers (2) to (5) within it, would infringe, even if additional components (such as additional Streptococcus pneumoniae serotypes) were included.
216 It is to be noted that the additional serotypes submitted by Wyeth to fall within the claim on the construction propounded by it were limited to Streptococcus pneumonia serotypes beyond the 13 listed (ie the 98 serotypes discovered by the time of the trial), and not serotypes at large unrelated to Streptococcus pneumoniae.
217 Merck argued that the claimed compositions were only to polysaccharide-protein conjugates of each of the chosen 13 serotypes and did not encompass immunogenic compositions which also have conjugates from any other Streptococcus pneumoniae serotypes. Merck submitted that the language of the claim made clear that it was limited to the 13 serotypes specified and that where one or more things in a class are expressly mentioned, other serotypes are excluded.
218 Merck submitted that its construction yielded a common sense and practical result. By the priority date, around 90 different serotypes had been described (and since then 98). On Wyeth’s construction, the claims would necessarily extend to an immunogenic composition of 90 or more serotypes and could cover millions of different combinations. Merck described such a construction as “untenable” and “absurd”.
219 Merck’s construction required the word “comprises” to be understood in an exhaustive sense, unlike “comprising” in the same claim.
220 At [178], Burley J held that there was no ambiguity in the use of the word “comprising” in the claim, given the clear definition in the specification. He observed that the question was to be resolved by, first, consideration of the language used in the claim. In this regard, there was no ambiguity. The words “comprising” and “comprises” were clearly defined in the specification in an inclusive sense, or in other words, “including” and “includes”. The only exception is whether the context required otherwise.
221 At [179], Burley J preferred Wyeth’s construction and found:
There can be no doubt that where in integer (1) the claim refers to “[a] multivalent immunogenic composition comprising…” the word “comprising” means “including”. If the definition of the term “comprising” in the body of the specification is not sufficient to establish this point, as much is confirmed as a matter of context, when one considers the claims dependent upon claim 1. For instance, in accordance with orthodox claim drafting, claim 9, which narrows the scope of claim 1, identifies amongst other things additional excipients to be included in the form of the excipients sodium chloride and sodium succinate buffer. If “comprising” was not inclusive, then claim 9 would make little sense.
(Emphasis in original.)
222 At [180], Burley J observed that the scope of claim 1 was apparently defined by reference to what the multivalent immunogenic composition under consideration for the purposes of infringement includes, not by reference to what it consists of. Thus, subject to some exceptions which he noted were not relevant to the case, Burley J held that an immunogenic composition that included all the elements of the integers of the claim would infringe, even if it also contained serotypes in addition to the 13 serotypes identified in the claim.
223 In answer to Merck’s contention that such a construction was “absurd”, Burley J observed at [185]:
In one such argument [Merck] contends that because all of the independent claims identify a specific number of polysaccharide-protein conjugates to be included, it would be “absurd” for the claim to be construed as applicable to a variable number of serotypes, extending potentially to 90 or more serotypes. However, the question at this point is what the claim under consideration means. If it is ambiguous, a construction that is not absurd might be chosen, but the first question is to identify ambiguity. This argument does not do so.
Further authorities on the meaning of “comprising”
224 In Abbott Laboratories v Corbridge Group Pty Ltd (2002) 57 IPR 432, Lee, Emmett and Hely JJ construed “comprising” in the sense of “consisting of”, contrary to the inserted inclusive definition of comprising. The Full Court stated at [31] that if it was construed in an inclusive sense, “then the scope of the claim might be enormous”.
225 The meaning and effect of “comprising” in a patent claim is to be determined having regard to the description of the invention in the whole of the specification: Abbott Laboratories at [34]. The term may bear different meanings in different parts of the specification depending upon the context, and “comprising” may be exhaustive of certain things without being exhaustive of others: Gambro Pty Ltd v Fresenius Medical Care South East Asia Pty Ltd (2004) 61 IPR 442 at [115]–[120] (per Allsop J, as his Honour then was) (not disturbed on appeal: Fresenius at [64] (per Wilcox, Branson and Bennett JJ)).
226 An indication in the specification that “comprises” is “not intended to exclude” other integers or steps may need to refer to the context (which includes the other claims and specification itself) in which the word “comprises” or “comprising” appears: see, for example, Actavis Pty Ltd v Orion Corporation [2016] FCAFC 121 at [168]–[181] (per Allsop CJ, Nicholas and Yates JJ); Apotex Pty Ltd v ICOS Corp (No 3) (2018) 135 IPR 13 at [563]–[578] (per Besanko J); ToolGen Incorporated v Fisher (No 2) [2023] FCA 794 at [159] (per Nicholas J).
227 In Actavis, the relevant claim, claim 17 claimed:
A method for preparing an oral solid composition comprising entacapone, levodopa, and carbidopa, or a pharmaceutically acceptable salt or hydrate thereof, wherein the method comprises
a) mixing pharmacologically effective amounts of entacapone and levodopa, or a pharmaceutically acceptable salt or hydrate thereof, with at least one pharmaceutically acceptable excipient and a disintegrant to obtain a first mixture;
b) granulating the first mixture to obtain a granule batch;
c) adding a pharmacologically effective amount of carbidopa, or a pharmaceutically acceptable salt or hydrate thereof, optionally a lubricant, and optionally one or more pharmaceutically acceptable excipients to the granule batch to obtain a second mixture;
d) formulating the second mixture into a plurality of dosage forms.
228 The specification in Actavis also included the following explanation:
Throughout the description and claims of the specification the word “comprise” and variations of the word, such as “comprising” and “comprises”, is not intended to exclude other additives, components, integers or steps.
229 In Actavis, the appellants submitted that the primary judge’s construction of claim 17 was based on the word “comprises” having an inclusive meaning where used in the chapeau to the claim. The appellants submitted that, although the specification stated that the word “comprises” is “not intended to exclude other additives, components, integers or steps”, the word “comprises” cannot function to enable the addition of steps or features that are otherwise excluded by the claim itself. The appellants submitted that, if that were not the case, any words in the claim after the word “comprises” would not provide any limitation upon the claimed monopoly, and the claim would be unclear or fail for want of definition
230 The Full Court in Actavis (per Allsop CJ, Nicholas and Yates JJ) overrode the definition the primary judge had given “comprise”, stating at [178] and [179]:
178 Undoubtedly, this indication in the specification must be borne in mind when construing claim 17. It cannot, however, contort the claimed process into a substantively different process. Put another way, the indication cannot give the word “comprise” and its variants an unbridled operation, when the relevant description and claims themselves specify, with appropriate precision, the step or steps to be taken that will provide the promised advantage.
179 For this reason, the better reading of claim 17 is that the word “comprises” must yield to the direction that the intended pharmacologically effective amounts of each active agent must be added at the step in the process that is specified for that addition. In our respectful view, the primary judge erred in construing claim 17 otherwise.
231 The 946 Patent in ICOS included a definition of “comprising” as follows:
Throughout the specification, unless the context requires otherwise, the word “comprise” or variations such as “comprises” or “comprising”, will be understood to imply the inclusion of a stated integer or group of integers but not the exclusion of any other integer or group of integers.
232 In ICOS, a number of the relevant claims in the 946 Patent referred to a dosage form or method of treatment comprising identified elements. Justice Besanko observed at [563] that it is clear from the definition of the word “comprising” in the 946 Patent that the claims are not restricted to the identified elements. Apotex submitted that the relevant claims of the 946 Patent were not confined to monotherapy, the 946 Patent “contemplates” the co-administration of tadalafil and nitrates. It further submitted, by way of example, that claim 1 would include a composition containing both tadalafil and testosterone and, indeed, the claims would encompass any composition which included the relevant quantity of tadalafil.
233 At [577], Besanko J observed that to construe the 946 Patent as extending beyond monotherapy by reason of the definition of the word “comprising” would be to impermissibly change the nature of the invention. He noted that the Full Court in Actavis had considered a similar definition of “comprise” and rejected the suggestion that the definition was decisive: Besanko J in ICOS at [577] referring to Actavis at [178].
234 These authorities establish that a definition of “comprise” in a specification “cannot give the word ‘comprise’ and its variants an unbridled operation” to contort a claimed invention into a substantially different invention to that described in the specification: Actavis at [178] (per Allsop CJ, Nicholas and Yates JJ). Whether a definition of “comprise” may need to yield to the context of the description of the invention in the specification will be specific to the particular specification in each case.
235 I consider that claim 1 of the 535 Application claims an immunogenic composition including at least one M. hyo antigen (one or more). Claim 1 is not limited to one M. hyo protein antigen, but it is limited to M. hyo antigens, ie those found in the M. hyo supernatant.
236 Embodiments of the immunogenic composition as being just the M. hyo supernatant (without the addition of other antigens) described in the specification support this construction. At page 3a, the specification describes embodiments where the soluble portion of the M. hyo preparation “includes at least one” or “two or more M. hyo protein antigens”. The specification explains at p13 that the M. hyo supernatant fraction includes one or more of the identified M. hyo specific protein antigens, such as P46. The description of embodiments at page 16 speak of “at least one M. hyo soluble antigen”.
237 I do not consider that the use of “comprising” extends the scope of claim 1 to include antigens to other diseases such as, but not limited to, those listed in claim 3, nor to a universe of antigens beyond those.
238 The 535 Application is entitled “Mycoplasma hyopneumoniae vaccine”, and the invention is said to relate to the soluble portion of an M. hyo whole cell preparation and its use in a vaccine for protecting pigs against enzootic pneumonia (also called mycoplasmal pneumonia) which is caused by M. hyo.
239 Claim 1 of the 535 is to an “immunogenic composition” comprising the supernatant of an M. hyo culture, wherein the supernatant has been separated from insoluble cellular material by one of three methods so as to be substantially free of both (i) IgG and (ii) immunocomplexes comprised of antigen bound to immunoglobulin. The specification defines an “immunogenic composition” as a composition of matter that comprises at least one antigen which elicits an immunological response.
240 Whilst an “immunogenic composition” may elicit an immunological response which is not sufficient to be considered protective, a composition which elicits a protective response is clearly an “immunogenic composition”. This is also supported by the claim structure where the protective response claims (claims 7 and 8) are dependent upon claim 1.
241 At page 13, the specification describes embodiments of the invention in which the soluble portion of the M. hyo preparation includes at least one M. hyo protein antigen or two or more M. hyo protein antigens. Example M. hyo protein antigens are described in the specification, including: p41, p42, p46, p64, p89, p97, p102 and p216.
242 The immunogenic composition including at least one M. hyo antigen from the soluble portion of the M. hyo preparation described in the specification and claimed in claim 1 is what Zoetis calls an “M. hyo platform”.
243 After a detailed description of the M. hyo immunological composition, the specification describes further embodiments in which the immunological composition of the invention includes at least one M. hyo soluble antigen and at least one additional antigen protective against a microorganism (bacteria, virus or protozoan) which can cause disease in pigs.
244 The person skilled in the art would not understand the invention defined by claim 1 to include a universe of antigens not mentioned in the specification, or even those listed in claim 3. The common sense result is a construction that the invention is limited by what is listed in the specification, and not the universe of other possible antigens that could be included with the M. hyo composition.
245 The composition in claim 3 states the invention “further comprises at least one additional antigen”. Such antigen or antigens is to be selected from a list of microorganisms that cause diseases in pigs.
246 The specifications of the three Applications are very similar. These later embodiments which include additional antigens (ie antigens other than M. hyo) protective against microorganisms that cause diseases in pigs are picked up as the subject of the claims in the 537 and 540 Applications. A specification may describe more than one invention — it does not need to claim everything described in the specification.
247 The immunological composition of claim 1 may also include inessential elements such as excipients and adjuvants. The definition of comprising ensures that the immunological composition of the invention is not limited to only the essential integers listed, but also includes elements which the skilled person would understand to be required for an immunological composition, but not to be an essential part of the invention.
248 Claim 1 of each Application does not expressly refer to an adjuvant being comprised in the immunogenic composition, although some of the dependent claims require one. Nonetheless, the experts explained that most vaccines (other than attenuated vaccines) require an adjuvant. In light of this fact, it is apparent that the skilled addressee would not consider that the immunogenic composition only consists of the supernatant of M. hyo (in the case of the 535 Application) or M. hyo and a PCV-2 antigen (in the case of the 537 Application). However, this same approach would not apply to other antigens not listed in the claims.
249 Claim 3 of the 535 Application claims the composition of claim 1 (or claim 2) wherein the composition further comprises at least one additional antigen which is protective against a microorganism selected from a list of microorganisms. Properly construed, the claim includes within its scope an immunogenic composition of claim 1 which includes one additional antigen selected from the list as well as one which includes many additional antigens selected from the list. No upper limit for the number of additional antigens is expressed in the claim. The language of “comprises at least one additional antigen” indicates that the claim covers the spectrum between these two possibilities. I do not consider that the claim extends to additional antigens beyond those listed in claim 3.
250 Claims 4 and 6 of the 535 Application define a combination of the composition of claim 1 with a further element (an adjuvant or a pharmaceutically acceptable carrier respectively).
251 Zoetis observes that PCV-2 is not included in the list of microorganisms in claim 3. A construction of claim 1 which included a universe of antigens beyond those listed in claim 3, gives the selection identified in claim 3 no work to do, and would be inconsistent with the patent applicant’s deliberate omission of PCV-2 from claim 3.
252 This construction is consistent with the steps of the method set out in claim 18 of the 535 Application.
253 Analogous reasoning applies to the claims of the 537 Application, save that the claims define a “multivalent” immunogenic composition that includes the M. hyo supernatant (including any number of M. hyo antigens) and a PCV-2 antigen in combination.
254 “Multivalent” is defined at page 10 of the specification as meaning a vaccine containing more than one antigen from the same species or a different species, or a vaccine containing a combination of antigens from different genera.
255 In the 537 Application, the list of microorganisms is found in claim 8. Again, the claim includes within its scope a multivalent immunogenic composition which includes the M. hyo supernatant, a PCV-2 antigen, and further comprising at least one additional antigen which is protective against a microorganism selected from the group listed, or many additional antigens selected from the group. In this case, the PRRS antigen is not included in those listed in claim 8.
256 Properly construed, claim 8 defines a multivalent immunogenic composition of claim 1 (or of any of claims 1–7) which includes one additional antigen selected from the list as well as one which includes many additional antigens selected from the list. No upper limit for the number of additional antigens is expressed in the claim. The language of “comprises at least one additional antigen” indicates that the claim covers the spectrum between these two possibilities. I do not consider that the claim extends to additional antigens beyond those listed in claim 8.
257 The claims of the 540 Application are different. Claim 1 claims a trivalent immunogenic composition and expressly refers to the M. hyo supernatant, a PCV-2 antigen and a PRRS antigen.
258 There is no claim in the 540 Application which features a “shopping list” of additional antigens in the 540 Application. Boehringer sought to suggest that the method claims 24 and 25 were broader than claim 1 because they did not specify that the composition is trivalent, and thus Boehringer contended that the claims encompassed immunogenic compositions with more than the three specified antigens. Claims 24 and 25 are to a method of preparing an immunogenic composition containing the M. hyo supernatant, a PCV-2 antigen and a PRRS virus antigen. I do not consider that claims 24 and 25 claim an invention which is broader than an immunogenic composition containing the antigens specified: the M. hyo supernatant, PCV-2 and PRRS.
OVERVIEW OF VALIDITY CHALLENGES
259 There is no challenge to the validity of any of the claims of the three Applications on the grounds of lack of novelty or inutility. Each claim of each of the Applications is thus taken to disclose a new and useful product or method: Sunbeam Corporation v Morphy Richards (Aust) Pty Limited (1961) 180 CLR 98 at 111 (per Windeyer J).
260 Boehringer challenges the validity of the Applications on the following grounds:
(a) Lack of inventive step;
(b) Lack of disclosure (s40(2)(a));
(c) Lack of support (s40(3));
(d) Lack of best method (s40(2)(aa); and
(e) Lack of manner of manufacture.
261 The content of common general knowledge was explained by Aickin J in Minnesota Mining & Manufacturing Co v Beiersdorf (Australia) Ltd (1980) 144 CLR 253 (in the context of the Patents Act 1952 (Cth)) at 292, as:
[T]hat which is known or used by those in the relevant trade. It forms the background knowledge and experience which is available to all in the trade in considering the making of new products, or the making of improvements in old, and it must be treated as being used by an individual as a general body of knowledge.
262 In addition to the matters set out in the background section above, the following matters were widely and generally known by those in the field of animal vaccine development and formed part of the common general knowledge of the person skilled in the art as at the priority date.
263 Animal vaccination raises very different financial and economic considerations from human vaccination. Animal vaccine production (particularly, in the case of commercially farmed animals) must be cost-effective. In particular, because of the cost of production and because the profits per unit sold are often much lower than a human vaccine.
264 Most commercially reared pigs are only expected to live for approximately 6 months, and to be an economically viable product to the farmer, the pigs needed to reach market weight in that time period in a reasonably healthy condition without high medical overheads from, for example, vaccination costs.
265 Animal vaccination programs impose a significant cost, which varies depending on the method of vaccination. Certain important diseases are almost always present in swine herds, particularly in intensively raised herds, causing chronic economic losses.
266 Swine vaccines are usually administered through intramuscular administration to piglets from three weeks of age.
Types of commercially available vaccines
267 The main types of vaccines for animals that were commercially available before April 2012 included bacterins, inactivated viral vaccines, recombinant sub-unit vaccines and modified live vaccines.
268 Bacterins are inactivated or killed bacterial cells, typically prepared by a fermentation process. The pathogen may be inactivated by various methods including formaldehyde, binary ethanolamine or temperature. Bacterins can consist of inactivated whole cells including at least some of the culture medium in which they were grown (that is, culture supernatant).
269 Modified live vaccines are also known as live attenuated vaccines. Modified live vaccines contain whole pathogens such as viruses that have been attenuated (altered) so that they stimulate an adaptive immune response, but either do not cause disease or cause only a mild or attenuated version of the disease. Modified live virus vaccines suffer from stability issues. For this reason, as at the priority date, most modified live virus vaccines were freeze-dried, or “lyophilized”, to increase storage stability, and were sold in “pellets” of freeze-dried materials. Freeze-dried virus vaccines must be rediluted or resuspended immediately prior to administration using a pharmaceutically acceptable diluent.
270 Sub-unit vaccines are made from antigenic proteins or other suspended cellular components derived from the pathogenic organism of interest. Where the sub-unit of interest is a protein, for example a membrane lipoprotein, it (in some cases) can be recombinantly produced in vitro in a high volume.
271 Adjuvants are compositions containing certain substances that enhance the immune response to an antigen(s). There were many types of adjuvants available by the priority date, including oil or polymer-in-water, water-in-oil or aluminium hydroxide adjuvants. The type of adjuvant used was important in improving the immunogenic response to an antigen, and in improving the duration of immunity.
272 Most vaccines, except attenuated vaccines and some inactivated viral vaccines, (and mRNA vaccines, which were not commonly known or used at the priority date), require an adjuvant.
273 Autogenous vaccines are essentially personalized inactivated whole cell vaccines, which are developed from isolates of a pathogen taken from samples of infected animal(s), for use on the same farm as the animal(s) from which samples were taken.
274 Vaccines include monovalent vaccines, which are vaccines that contain antigen from a single species of microorganism, and multivalent or combination vaccines, which are vaccines that contain antigens from multiple species of microorganisms.
275 Supernatant vaccines were known, but not widely employed, at the priority date. Professor Browning gave evidence that he was aware of toxoid supernatant vaccines, which were directed to pathogens that secrete a toxin, such as Clostridium tetani and Clostridium perfringens. As the toxin is secreted into the culture medium during bacterial culture, toxoid vaccines can be prepared from the bacterial culture medium in which a culture of the bacteria is grown. The bacterial cells of the culture are separated from the culture medium containing the secreted toxin, for example, by centrifugation. The retained supernatant, which contains the toxins, is then treated to prepare the toxoid vaccine. This type of vaccine does not contain cells.
276 Professor Chase was aware of an E. coli supernatant vaccine in which the fimbriae (hair like appendages) of the E. coli cells are broken off and released into the liquid fraction of the cell culture. The cell culture is purified to remove cellular material and other unwanted proteins and impurities, leaving the liquid supernatant fraction.
277 The most damaging pathogens of concern to swine producers included M. hyo, PCV-2 and PRRS virus. These three pathogens were known to be the most important contributors to Porcine Respiratory Disease Complex (PRDC) which was of economic concern to swine producers.
278 M. hyo was known to be the primary pathogen responsible for enzootic pneumonia. It was known that M. hyo was highly prevalent worldwide, endemic in almost all commercial swine herds and an important factor in economic losses in the pig industry. Infections with M. hyo are highly prevalent in almost all swine producing areas, and they cause significant economic losses due to increased medication use and decreased performance of the pigs.
279 M. hyo infection enhances the susceptibility of infected pigs to other respiratory pathogens. In addition to causing a range of respiratory complications on its own, M. hyo infection exacerbates secondary infections that result in poor pig performance (eg a reduction in daily weight gain and inefficient feed conversion) and additional healthcare-related costs.
280 Mycoplasmas are single celled prokaryotes which lack a cell wall and only have a cytoplasmic membrane. They lack a nuclear membrane. Mycoplasmas are the smallest free-living self-replicating microorganisms. M. hyo is a relatively fragile organism that is prone to break into multiple membrane fragments during the processing steps involved in vaccine production.
281 PCV-2 is a small virus (a circovirus in the family circoviridae) endemic in swine herds that causes respiratory disease. Symptoms of PCV-2 infection include lung lesions, fever, wasting (ie weight loss), and related economic issues, such as requiring a greater amount of time to reach market weight.
282 The PCV-2 capsid protein “ORF-2” was known to be antigenic, and suitable for inducing immunity in vaccinated animals. As at the priority date, the ORF-2 protein was routinely produced using a modified baculovirus vector containing the gene encoding the PCV-2 ORF-2 protein. This virus was used to infect an insect cell line and cause it to produce the ORF-2 protein. The insect cells were then cultured to the required volume and the ORF-2 protein isolated and mixed with an adjuvant. This composition was then used for the final vaccine product.
283 PRRS virus is an arterivirus and is somewhat more complex than PCV-2. As at the priority date, the primary antigenic components of this virus were not definitively elucidated and understood. There were no successful purified sub-unit vaccines against PRRS virus available by that date. Instead, vaccines against PRRS virus were generally made from modified live PRRS viruses, which were lyophilized for stability, and reconstituted immediately prior to administration.
284 M. hyo is relatively fragile and prone to breaking into multiple membrane fragments during the processing steps involved in vaccine manufacture.
285 M. hyo has fastidious growth requirements. M. hyo has absolute requirements for exogenous sterols and fatty acids found in porcine serum in order to grow. These requirements generally necessitate growth of M. hyo in media containing animal serum, such as porcine serum. Few bacterial pathogens other than M. hyo require serum in their culture media.
286 Professor McVey described M. hyo as an extremely fastidious and slow growing organism, which must be grown in an expensive, highly enriched laboratory media which, by April 2012, usually included serum from the host animal and/or other tissue components. Culture of M. hyo for study or commercial use was therefore a long and onerous process requiring a specialized laboratory and specialized media. M. hyo growth medium typically includes 10–30% serum.
287 It was known in April 2012 that it was possible to grow M. hyo in synthetic media, but growth yields tended to be far lower and less consistent when working in this way.
288 Growth media containing animal serum could lead to adverse reactions in the vaccinated animal because of the presence of unaccounted for serum components (such as lipids, proteins, immunocomplexes or live microorganisms) in the immunising material.
289 Animal serum, and any other components used in the growth process that are derived from animals, are known as substances of animal origin, or SAO. SAO may include non-immunogenic proteins, lipids, immunocomplexes and sugars. SAO may also include antibodies against pathogens endemic to that animal, to which the animal is highly likely to have been exposed. These antibodies are known as serum-derived antibodies. For example, swine serum may contain serum-derived antibodies against PCV-2, ie antibodies that bind to PCV-2 or an antigen of PCV-2 (PCV-2 antibodies).
290 It was appreciated at the priority date that serum-derived antibodies and other components might reduce efficacy, affect uniformity of batches of vaccine because of analytical interference, and cause adverse reactions in vaccinated animals.
291 As at the priority, date, commercial supplies of PCV-2 antibody-free serum had become increasingly scarce, inconsistent and, for the purposes of commercial production, prohibitively expensive and difficult to source.
292 As at the priority date, it was known that maternal antibodies (transmitted to the piglet via the colostrum) might interfere with a vaccine’s effectiveness in the piglet. PCV-2 antibodies strongly interfered whereas PRRS antibodies did not. Professor Browning explained that PCV-2 being a small, very regular virus induced a very high antibody response.
293 The maternal antibodies in piglets are relatively short lived (persisting in the piglet for around 3-4 weeks) so the timing of the piglet vaccination is a way to mitigate interference from maternal serum antibodies. The technique of timing vaccination to avoid maternal serum antibodies was also used for companion animals such as dogs, cats and horses.
294 The experts agreed in the JER that, as at the priority date, there were many different antigens and processes examined experimentally for potential use in immunising swine against diseases associated with M. hyo, including: live attenuated vaccines, purified subunit vaccines, freeze-thaw saline solution extracts, supernatants of culture, whole cells, detergent extracts from cells, saline washed whole cells, adjuvants, and centrifuged clarified supernatant.
295 M. hyo vaccines were commercially available as at the priority date. In the JER, the experts noted that trade names vary and change by country and distributor, so they gave three examples of the types of M. hyo vaccine preparations commercially available at the priority date:
(a) a monovalent M. hyo whole cell bacterin;
(b) a “rough filtrate” vaccine known as “Mycobuster”; and
(c) a live attenuated M. hyo vaccine delivered intrathoracically (in China).
296 In the joint session, the experts gave evidence that they knew of the following commercially available M. hyo vaccines at the priority date: Ingelvac CircoFLEX and MycoFLEX from Boehringer, RespiSure-ONE, and Suvaxyn MH1 from Fort Dodge/Zoetis.
297 The vast majority of the commercially available M. hyo vaccines were bacterins. The experts identified only two M. hyo vaccines commercially available at the priority date that were not bacterins: Mycobuster and a live attenuated vaccine administered intrathoracically that was available in China.
298 The M. hyo component of the commercially available monovalent and combination vaccines was based on a bacterin suspension of killed M. hyo cells. These were developed from killed M. hyo cultures, grown in media containing swine serum, and contained the whole cell bacterin in the medium in which it was grown.
299 Bacterins commonly include non-antigenic components. In addition to containing inactivated whole cells, bacterin vaccines may also include cell fragments, cellular membranes and cellular components from whole cells that have lysed as a consequence of the culturing, inactivating or processing steps used to create the vaccine (eg centrifugation and washing steps in the case of washed cell vaccines).
300 The bacterin was the state of the art for M. hyo vaccines in swine as at the priority date. Professor McVey confirmed that bacterins remained “the most suitable format because they were able to provide effective protection against M. hyo infection and were simple and economically efficient to produce”.
301 Professor Chase, Professor McVey and Dr Nordgren were aware of variability issues with the M. hyo bacterin vaccines on the market. Professor McVey described bacterins as “inherently crude compositions”.
302 As at the priority date several surface proteins of M. hyo had been characterised, including p65, p97, p46 and NrdF, and had been shown to induce an adaptive immune response to M. hyo. The fact that something elicits an adaptive immune response does not mean that it will provide protective efficacy, nor that it will provide adequate protection in the sense of being as good as, or better than, available vaccines. Lipoproteins were also suspected to induce an adaptive immune response, but their identity was not well defined.
303 No single characterised M. hyo protein antigen was known to provide adequate protection on its own. Membrane antigens were thought to provide protection. The experts agreed that p46 was characterised as a surface protein. The experts agreed that, during processing steps, membrane fragments including the p46, p65, p97 and NrdF antigenic proteins could break apart from the cells. Professor Chase, Professor McVey and Dr Nordgren considered that the antigen containing membrane fragments could be soluble, and that the antigen proteins themselves could be referred to as a membrane fragment of M. hyo. Professor Browning agreed with the other experts that p46 was a protein that might be contained in a membrane fragment.
304 The article by Simionatto et al, “Mycoplasma Hyopneumoniae: From Disease to Vaccine Development” (2013) 165 (No 3-4) Veterinary Microbiology 234 was published in in April 2013. The Simionatto article conducted a review of the literature on M. hyo vaccine development as at around the priority date. Although published shortly after the priority date, the published studies reviewed by the article were published prior to the priority date and provide a summary of the common general knowledge as to M. hyo vaccines at the priority date.
305 Simionatto confirmed that bacterins formed the basis of currently used commercial M. hyo vaccines and noted that bacterins had high production costs due to the difficulty of cultivating M. hyo in vitro, thus there was a need to develop more effective and lower cost vaccines. Simionatto summarised the investigative work into new vaccines to offer better protection against M. hyo as at just before the priority date. This investigative work included studies evaluating recombinant proteins of M. hyo in various forms of administration and formulations. Dr Nordgren agreed that Simionatto did not suggest that any viable alternative to commercial vaccines had actually been developed as at the date it was published.
Commercially available vaccines for swine pathogens
306 The M. hyo vaccine, “Mycobuster”, was available commercially in Japan as at the priority date. The experts agreed that the “rough filtrate” vaccine, Mycobuster, could also be described as a supernatant vaccine. Mycobuster was prepared by filter-separating the supernatant from some of the whole cells/insoluble material, and using the supernatant-containing filtrate.
307 The concentration of M. hyo is typically measured in colour changing units (CCU), which is a colorimetric test based on the change in pH in the medium resulting from acidic byproducts of growth that indicates the number of live M. hyo bacteria present. During M. hyo culturing, it is typically measured per mL of culture. CCU is a common method for measuring the amount of viable organisms that are in a sample (live bacteria). This method does not measure dead bacteria, insoluble proteins or fragments of membrane.
308 The Mycobuster product information sheet states that the amount of viable bacteria per filtration is 2.5 x 1010 CCU, which reduced to 2.5 x 107 CCU after the filtration step. Professor Chase observed that there had been a reduction of over 1000 fold in the number of viable bacteria in the preparation of the Mycobuster supernatant. The experts agreed that the final Mycobuster vaccine was a supernatant.
309 A one-dose bacterin was the state of the art vaccine for M. hyo. One-dose vaccination is preferable as it requires less labour and can be implemented more easily in routine management practices on the farm.
310 There were PCV-2 vaccines commercially available at the priority date. Examples included Merial’s Circoflex, Porcilis PCV-2 (Intervet/Schering Plough), Ingelvac Circoflex (Boehringer Ingelheim), and Suvaxyn PCV-2 (Fort Dodge/Zoetis). The majority of the PCV-2 vaccine doses sold were recombinant sub-unit vaccines.
311 There were several combination vaccines for protection against M. hyo and PCV-2 available at the priority date. An M. hyo bacterin was commonly included as the base or diluent for other swine vaccines, or was bundled together with other swine vaccines
312 One such product was Ingelvac’s FLEXCombo (from Boehringer Ingelheim). Ingelvac FLEXCombo consisted of two components: an M. hyo component (Ingelvac MycoFLEX) consisting of an inactivated M. hyo bacterin; and a PCV-2 component (Ingelvac CircoFLEX) consisting of inactivated baculovirus-expressed PCV-2 ORF2. This product required only one dose, but was not provided in a single “ready-to-use” pre-mixed vial. Instead, it was supplied in two separate bottles and required mixing of the two components from the separate bottles immediately before injection.
313 The term “ready-to-use” in the vaccine context means that the immunogenic composition is already premixed and is ready to be applied to the target animal without further mixing or preparation steps being required.
314 Another M. hyo/PCV-2 combination vaccine product was Circumvent PCV-M from Intervet/Merck Animal Health. Circumvent PCV-M also consisted of two components: an M. hyo component consisting of an M. hyo bacterin; and a PCV-2 component consisting of baculovirus-expressed PCV-2 antigen. Circumvent PCV-M was supplied in a single bottle ready-to-use format but required two doses.
315 There was no one-dose one-bottle ready-to-use M. hyo/PCV-2 vaccine available as at the priority date. Such a product was considered desirable and animal health companies were attempting to develop such a product.
316 Although Professor Chase, Dr Nordgren and Professor McVey agreed that sometimes two doses of a product are needed to boost or extend immunity for the life of the pig, none of them disagreed with Professor Browning’s observation that “all other things being equal, a one-dose vaccine is more flexible than a two-dose vaccine”.
317 Unlike PCV-2, the antigenic component(s) of PRRS were not well defined. Several proteins encoded by a number of genes are required for an adequate immune response. For this reason, the majority of PRRS vaccines on the market as at the priority date were lyophilized modified live PRRS vaccines, the first being introduced on the market in approximately the mid-1990s.
318 There was combination M. hyo, PCV-2 and PRRS vaccine available: Boehringer’s 3FLEX. 3FLEX was supplied in three vials and prepared for administration by rehydrating the lyophilised PRRS virus component with the M. hyo and PCV-2 components.
319 The experts agreed that, as at the priority date, there was a desire for a ready-to-use single dose M. hyo/PCV-2 combination vaccine and also a desire for a ready-to-use single dose combination M. hyo with PCV-2 and PRRS vaccine.
Immunological and assay interference
320 The presence of serum-derived antibodies in a vaccine preparation can be problematic if the serum-derived antibodies are able to bind to the antigen or antigens included in the preparation. This is because the binding of serum-derived antibodies to the antigen or antigens can inhibit or block their presentation to the immune system. This can prevent the antigen or antigens from being presented in a form suitable for the antigen or antigens to be recognised by the immune system and sufficiently establish adaptive immunity. Immunological interference can affect the efficacy of the vaccine preparation.
321 For example, if a PCV-2 antigen or PCV-2 antigen-containing preparation (ie a vaccine against PCV-2, also referred to as a PCV-2 vaccine) is added to or mixed with a second preparation containing serum-derived PCV-2 antibodies, the antibodies may bind to the PCV-2 antigen and neutralise it, or otherwise mask the PCV-2 antigen from the immune system. This interference reduces the amount of PCV-2 antigen in the vaccine preparation that is available to be recognised by the immune system and generate an immune response. This in turn reduces the efficacy of the vaccine.
322 The amount of serum-derived PCV-2 antibodies in each individual batch of swine serum varies, depending on the health status of the swine herd from which each batch is sourced. Therefore, in the case of combination or co-administered vaccines that include a PCV-2 antigen and a second preparation containing serum-derived PCV-2 antibodies, the extent to which the PCV-2 antigen will be neutralised or otherwise masked will vary from batch to batch.
323 Assay interference occurs when accurate measurement of the amount of one of the components present in the final multicomponent vaccine is prevented by one or more of the other components.
324 Both immunological and assay interference was well known (with multicomponent vaccines) as at the priority date, and it was necessary to test for it. As Professor Browning observed, “you can’t take two different vaccines, mix them together and assume they’re going to work … as they would as individual vaccines”. As well as being common sense, it was also a regulatory requirement to demonstrate whether or not there was interference. Where present, interference needed to be addressed, whether the combination vaccine was ready-to-use or mixed at the point of administration.
325 In the context of the development and manufacture of an M. hyo/PCV-2 combination vaccine, one form of interference that could arise would be from anti-PCV-2 antibodies present in the M. hyo component and derived from the serum containing growth medium in which M. hyo was grown.
326 In the context of the development and manufacture of a combination M. hyo/PCV-2 vaccine, the experts agreed in the JER that:
Interference always needs to be addressed with multicomponent vaccines. Immunological interference occurs when one vaccine is combined with another vaccine and interferes with the immune response to one or both of the components. Assessment of immunological interference (components in each vaccine) is a regulatory requirement. Assay interference occurs when accurate measurement of the amount of one of the components present in the final multicomponent vaccine is prevented by one or more of the other components. Assay interference can be a problem when quality control assays are performed on the final product, as it prevents accurate determination of the amount of each component present in the final multicomponent vaccine, which is also a regulatory requirement. Assay interference is the reason that some multicomponent vaccines are supplied in multiple vials.
327 The experts agreed that, as at the priority date, immunological interference could be overcome by using a variety of techniques or a combination of them. The two primary techniques given by the experts included removal of the interfering component or adding additional amounts of the component that is interfered with (the antigen). The experts also agreed that:
To counter the interference by antibodies in pig serum with the immunological response to PCV-2, techniques to reduce the amount of antibodies might have included column chromatography of the serum, use of serum with low levels of antibodies against PCV-2, use of serum from pigs from countries free of PCV-2, use of serum from other animal species, or reducing the concentration of pig serum in the growth medium for M. hyo.
328 Professor Chase identified the use of adjuvants as another technique that was available at the priority date to overcome immunological interference, particularly given the availability and low cost of adjuvants. Professor Browning and Professor McVey agreed that adjusting the amount or changing the adjuvant or a combination of adjuvants might overcome the interference problem. Professor McVey also suggested that changing the order in which components were mixed together might also work.
329 The experts considered the addition of more antigen was the less preferable of the two techniques proposed by them to counter immunological interference. Serum antibody levels are likely to vary between serum batches so different amounts of antigen would need to be added. Professor Browning observed that:
The difficulty with adding additional amounts [of antigen] is that that will work where the interference is – is predictable, and, so if it’s an interaction between two specific proteins in the mix, then that’s possible. It’s much harder to predict that when you’re talking about antibody in serum that has been derived from pigs in the field where the level of that may go up and down depending on the batch of serum that has been used.
…
[A] regulator is not going to allow you to vary the amount of antigen from batch to batch of the vaccine.
…
It’s … not a viable approach in the circumstances.
330 Professor Chase noted that the antigen is the most expensive component of a vaccine, so removing the interfering component rather than adding more of the expensive antigen typically made more sense.
331 Dr Nordgren and Professor Chase explained that, even if techniques were employed to overcome immunological interference, assay interference may still occur. The only strategies identified by the experts to deal with assay interference were the use of a different assay or releasing a two-bottle product.
332 The experts agreed that M. hyo vaccines could be made by either treating the serum containing growth media prior to use in the M. hyo cell culture process to remove serum-derived antibodies from the serum or growth medium, or treating the cell culture itself after M. hyo cell growth to remove serum-derived antibodies. The former treatment technique is “upstream processing”, and the latter is “downstream processing”. Upstream and downstream processing in relation to cell cultures were well-known as at the priority date.
333 The experts gave examples of the “variety of techniques” to counter interference by antibodies in pig serum with the immunological response to PCV-2 by reducing the amount of PCV-2 antibodies in the pig serum (containing growth media) in their joint JER response:
(a) Column chromatography of the serum;
(b) Use of serum with low levels of antibodies against PCV-2;
(c) Use of serum from another species; or
(d) Reducing the concentration of pig serum in the growth medium for M. hyo.
334 In his affidavit, Professor Chase added that filtration, centrifugation and precipitation were also well-known methods as at the priority date for removing antibodies from compositions containing serum. Professor Chase gave the further example of salting out the antibodies with ammonium sulphate.
335 The experts identified three difficulties involved with each of the serum-based techniques they had identified.
336 First, it was difficult to source swine serum with low levels of PCV-2 antibodies. Professor McVey’s evidence was that it is difficult to find such herds. By the priority, date PCV-2 had become endemic in many areas and countries which were previously PCV-2 free, such as New Zealand. Further, a large volume of serum is required to support a large-scale fermentation and there can be issues with batch consistency. Continual testing would be required to ensure that the level of antibody stayed the same from one batch to the next. Professor Browning also suggested the use of fetal serum (which does not contain antibodies) in M. hyo growth media to attempt to avoid some of these issues.
337 Second, there were difficulties involved with using serum from another species. Non-swine serum does not provide the level of growth support in the fermentation that swine serum provides, so that time to peak titer is likely to be extended. Professor McVey described using serum from another species as “in one sense, trading one problem for another”.
338 Third, a reduced concentration of swine serum in the growth medium would also lead to a slower growth of M. hyo. Professor Browning’s evidence was that not only would the growth rate be slower, but “you may not get the same levels of growth and it may decrease viability”.
339 In the JER, the experts observed that column chromatography could be used to avoid antibody-antigen interference in an M. hyo/PCV-2 vaccine by treating the serum used in the M. hyo culture process, or the serum-containing growth media used in the M. hyo culture process. Professor Browning expressed doubts as to the practicality of passing serum through the column due to its high viscosity.
340 The experts confirmed that they had in mind two column chromatography techniques in their JER response, both of which were available as at the priority date namely: (a) protein A/G affinity chromatography; and (b) size exclusion chromatography.
341 Professor Browning’s evidence was that he only had in mind size exclusion chromatography of the growth medium. He also noted that the use of size exclusion chromatography would need to be tested as it might also remove some of the protective antigens, depending on their size. Professor Browning agreed with the proposition put to him in the joint session that Protein A and Protein G chromatography would be less likely to remove the antigens, but noted it would still need to be tested. He also agreed that the testing he referred to was available at the priority date.
342 The experts agreed that, as at the priority date, themethods for substantially removing total IgG from crude protein mixtures such as tissue culture supernatant, serum containing growth media and ascites fluid were known in the art.
343 I note that Zoetis contended that not all the techniques suggested by the experts would result in a composition “substantially free” of immunocomplexes of all types, not just PCV-2 antibodies specifically. For example, using serum from disease free herds.
344 Professor Chase agreed that it was not possible to predict what the outcome of any particular technique or combination of techniques would be without doing experimental work to determine whether the technique or combination of techniques chosen in fact reduced the immunological interference. Professor Chase described combining the available techniques as being like moving chess pieces around. He agreed that it would also be necessary to take into account the effect of the chosen technique or combination of techniques on assay interference.
Protein A/G affinity chromatography
345 Protein A is a cell wall component produced by several strains of Staphylococcus aureus. Protein G is a bacterial cell wall protein isolated from group G streptococci.
346 Protein A has high affinity for the constant region of the heavy chain of antibodies (known as the Fc region), and specifically binds to the Fc region of IgG. Protein G is another protein that binds to the Fc portion of IgG. The Fc region is the area of the antibody that binds to cellular receptors to induce immune signalling, and the Fab regions of antibodies are the antigen-binding regions. Binding through the Fc region is not affected by the antigen specificity of the antibody, whether or not the Fab region is bound to antigen, nor by whether the antibody has formed an immune complex by binding with its specific antigen (an antibody-antigen immunocomplex).
347 Chromatography columns with pre-loaded Protein A and Protein G resins were commercially available as at the priority date. The experts agreed that the properties of Protein A and Protein G were well known before the priority date, and that Protein A and Protein G based affinity chromatography for use in capturing and retaining antibodies was also well known.
348 Professor Browning was familiar with the use of Protein A and G based affinity chromatography to purify or enrich antibodies (by capturing and retaining them on the column) from a solution or serum. Following the purification or enrichment, the antibodies can be eluted from the column, and the liquid fraction that passed through the column is discarded.
349 Professor McVey’s evidence was that Protein A and G worked because they would bind to the antibodies present in a preparation, so that when the preparation was passed through a column, the antibodies in the preparation would be bound to the column and the liquid fraction would pass through the column and be discarded.
350 The experts agreed with the proposition put to them by BI’s senior counsel in the joint session, that if one wanted to do something with the liquid fraction that had passed through the column it could be retained and be the subject of investigations “if that was what you were interested in”. The experts also agreed with the suggestion put to them in the joint session that two possible uses to which Protein A and G could be put were to purify the antibody (positive selection) or to remove antibodies from a sample which you want to remove them from (negative selection).
351 The experts also agreed that a technique to counter the interference by antibodies in pig serum (or pig serum containing growth media) with the immunological response to PCV-2 may have included column chromatography of the growth medium. Professor Browning limited his response to only size exclusion chromatography but agreed that some of the antigens may be removed by that technique. He also stated that Protein A and G column chromatography would be less likely to remove antigens.
352 Professor Chase gave one example in oral evidence of using Protein A chromatography in a non-vaccine context for “both” purposes of capturing and removing immunoglobulin in the particular context of “preparing” (capturing) a desired immunoglobulin and then separating it from other immunoglobulins to be eliminated. This isolated example was not used to purify a supernatant in the vaccine development context and does not form part of the relevant common general knowledge.
353 In the course of being asked about the disclosure of the 535 Application in the joint session, Professor McVey was taken to the statement “that methods for purifying/removing total IgG from crude protein mixtures, such as tissue culture supernatant serum and ascites fluid, were known in the art”. He was asked whether that reflected his understanding at the priority date. Professor McVey agreed, adding “especially experimentally”. Professor McVey’s response said nothing about the methods by which, the purpose for which, or the extent to which, IgG was purified or removed from crude protein mixtures before the priority date. These matters were canvassed with the experts in the joint session as I have outlined above.
354 Dr Nordgren explained in oral evidence that, in around 2008 or 2009, his team at Merial carried out research directed at addressing any impediments the M. hyo formulation might impose to combining it with a PCV-2 antigen. He did not suggest that he or his team considered, or evaluated, the use of Protein A or Protein G at all, and his agreement that his “work with Merial demonstrated a different route to attempt to solve the same problem” makes clear that he considered the route chosen by him and his colleagues at Merial to be different from that disclosed in the Applications. Further, Dr Nordgren regarded the information obtained from his Merial work to be confidential.
355 The only pre-priority date, publicly available document in evidence that described the potential use of Protein A or Protein G to deplete antibodies from a vaccine was a PCT application WO 2012/025612 entitled “The Potency Test for Vaccine Formulations” (the 612 Application), published one month before the priority date. Boehringer abandoned its s 7(3) inventive step case based on the 612 Application during the hearing.
356 The experts agreed that the 612 Application was focussed on treatment of antibodies in the pig serum in the M. hyo component to enable in vitro testing to accurately measure the amount of the PCV-2 component in an M. hyo-PCV-2 combination vaccine to enable release of the product for sale. They also agreed that the 612 Application was not critical to the development of an efficacious two component vaccine.
357 The invention described in the 612 Application is a method of determination of antigen content of a first antigen in a mixture containing two or more antigens. The 612 Application contains only a single reference to Protein A and Protein G on page 8. It suggests the “possible” separation of the second antigen from antibodies capable of binding with the first antigen in a composition comprising the second antigen and the antibodies, as part of an invention relating to the measurement of antigen levels (ie an assay). Protein A and Protein G are said to be ligands commonly used for such a purpose, and usually they are bound to a solid phase that facilitates the separation of the antigen from the antibodies. The 612 Application does not give any examples of how to do so or suggest that it has been tried. Protein A and Protein G are not mentioned in the examples of the 612 Application.
358 None of the experts gave evidence that they had ever used Protein A or Protein G “in a reverse manner” to deplete a vaccine composition of IgG and immunocomplexes before the priority date. None of the experts were aware of Protein A and/or G being used in a negative selection application to remove unwanted IgG or immunocomplexes by binding them to the column and collecting the “purified” liquid fraction. Nor had any of the experts retained the liquid fraction that had passed through a column for the purposes of further investigation.
359 Professor Chase was briefed with the Okada papers together with the 612 Application at the same time. Professor Chase agreed in the joint session that “the idea of using Protein A and Protein G on the supernatant would never have occurred to [him] had [he] not at the same time as [he was] reviewing Okada, reviewed the 612 Application”.
Pierce Biotechnology catalogue
360 Boehringer tendered a number of manufacturer’s catalogues from before the priority date to show that Protein A and G were commercially available in kit forms as at the priority date.
361 The high point of the various catalogue disclosures relied upon by Boehringer in relation to Protein A and G was the following statement, contained at page 56 of the 70-page 2004 Pierce Biotechnology catalogue entitled “Products for Affinity Purification” in introduction to the section entitled “Affinity Procedures for Contaminant Removal”:
Following the primary purification procedure to obtain a sample of interest, secondary purification steps to remove contaminants may be required. The contaminants can be inhibitors, interfering substances or inappropriate buffers. A number of available affinity supports allow researchers to either specifically purify a protein of interest away from a complex mixture of biological molecules (positive selection) or remove specific contaminants from a sample containing a protein of interest (negative selection). Nearly any given affinity purification system can be used for either positive or negative selection, depending on whether the non-bound or eluted fraction is recovered. For example, immobilized Protein A can be used for general affinity purification of antibodies (positive selection), but it can also be used to selectively remove immunoglobulins from a sample in which they are considered a contaminant (negative selection).
362 The Pierce Biotechnology catalogue has an extensive earlier section entitled “Affinity Purification of Antibodies”. The section discusses the use of Proteins A, G and A/G to purify antibodies by capturing the desired antibodies on the column and then eluting the bound antibodies.
363 The statement at page 56 of the Pierce catalogue on which Boehringer relies is in general terms, referring to the use of Protein A to “selectively remove immunoglobulins from a sample in which they are considered a contaminant”. It makes no reference to vaccine manufacture, the removal of immunocomplexes, or achieving a final composition that is “substantially free” of IgG and immunocomplexes.
364 Professor McVey agreed with the proposition that the statement was:
[A] description of two possible ways in which antibody purification products, including Protein A can be used. They can be used to purify the antibody, positive selection, or to remove antibodies from a sample which you want to remove them from, which is called negative selection
365 However, Professor McVey and the other experts were not taken to the discussion in the section that related to the example given in the introduction which related to the removal of mixtures of immunoglobulins from preparations of polyclonal antibodies, a different use to that in the Applications. This stated:
Removal of Nonspecific Antibodies
Because they contain mixtures of immunoglobulins, preparations of polyclonal antibodies from serum or other samples often have cross-reactivity to unintended targets in the sample being probed in Western blot or ELISA. It is important to purchase or prepare antibodies that do not cross-react in this way. For example, by passing a rabbit anti-mouse IgG polyclonal antibody sample over a column of immobilized human serum proteins, one can ensure that the resulting antibody will react only to mouse IgG primary antibody without cross-reacting with human immunoglobulins in the sample. Pierce offers selected antibodies that have been pre-adsorbed in this way to prevent cross-reactivity to unintended immunoglobulin species. Such pre-adsorption of antibodies is a form of negative selection affinity purification wherein the immobilized ligand is a mixture of proteins. For more information on pre-adsorbed antibodies, order literature #1600923 from Pierce customer service.
Cross-reactivity of antibodies to bacterial proteins is a common problem for researchers investigating recombinant proteins prepared in Escherichia coli. The possibility of such cross-reactivity can be reduced or eliminated by removing immunoglobulins in the polyclonal antibody sample that bind to proteins from E. coli. Pierce offers the Immobilized E.coli Lysate Kit for this purpose. Proteins from total lysate of E.coli strain BMH 71-18 have been immobilized onto a 4% cross-linked agarose support.
366 The Pierce Biotechnology catalogue was not the subject of any written evidence by Boehringer’s experts. Selected extracts from the catalogue were put to the experts during the joint session. The experts agreed that the general information in the catalogue about Protein A and Protein G was typical of the information that was available concerning the binding characteristics of Protein A and Protein G. The single statement extracted above does not form part of the common general knowledge of those skilled in the field. It does not accord with the experts’ evidence as to what was known at the priority date as to the uses of Proteins A and G.
367 Agreement by some or all of the experts that they were aware of the companies named in the Pierce Biotechnology or other catalogues, or that certain information contained in those catalogues was typical of information “available” before the priority date, falls far short of establishing that any particular information contained in any of those documents was common general knowledge before the priority date.
368 I consider that the properties of Protein A and Protein G, the existence of commercially available Protein A and Protein G column chromatography kits, and their use to purify or enrich antibodies (by capturing and retaining them on the column) was well known and part of the common general knowledge as at the priority date.
369 However, I am not satisfied that the use of Protein A and Protein G in the “reverse manner” — to remove IgG or immunocomplexes from a preparation by running the preparation through a column to remove IgG and immunocomplexes from the liquid and retaining the liquid — was part of the common general knowledge at the priority date.
370 Boehringer contends that the invention claimed in each of the claims of the 535, 537 and 540 Applications would have been obvious to a person skilled in the relevant art in light of the common general knowledge before the priority date considered together with information in any one or more of the three prior art publications which were referred to as the Okada papers. The three Okada papers are discussed in detail below.
371 The inventions described and claimed in the Applications are not alleged to be obvious in light of the common general knowledge alone at the priority date.
372 Section 18(1)(b)(ii) of the Act requires that the invention involve an inventive step when compared with the prior art base as it stood before the priority date. This is to be assessed in accordance with s 7(2) of the Act, which asks whether the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge before the priority date of the relevant claim, considered separately or together with information of the kind in s 7(3) of the Act.
373 One test for obviousness is the “Cripps question” re-stated by the High Court in Aktiebolaget Hässle v Alphapharm Pty Ltd (2002) 212 CLR 411 at [53] (per Gleeson CJ, Gaudron, Gummow and Hayne JJ). On this approach, if the skilled addressee would, in light of the prior art base and the common general knowledge, directly be led as a matter of course to try the invention in the expectation that it might well produce a useful or better alternative, the invention will lack an inventive step. Another way of approaching the issue is the inquiry in Wellcome Foundation Ltd v VR Laboratories (Aust) Pty Ltd (1981) 148 CLR 262 at 286 (per Aickin J), which asks whether the claimed invention is of a routine character, to be tried as a matter of course.
374 The Full Court in Generic Health v Bayer Pharma Aktiengesellschaft (2014) 106 IPR 381 considered both approaches, observing at [71] (per Besanko, Middleton and Nicholas JJ):
We do not think there is a divide here in terms of whether an expectation of success is relevant between a test which refers to routine steps to be tried as a matter of course and the reformulated Cripps question. It is difficult to think of a case where an expectation that an experiment might well succeed is not implicit in the characterisation of steps as routine and to be tried as a matter of course.
375 Neither approach requires certainty of outcome.
376 In Alphapharm at [58], Gleeson CJ, Gaudron, Gummow and Hayne JJ found that the idea of steps “taken as a matter of routine” did not include “a course of action which was complex and detailed, as well as laborious, with a good deal of trial and error, with dead ends and the retracing of steps”.
377 In AstraZeneca AB v Apotex Pty Ltd (2014) 107 IPR 177, Besanko, Foster, Nicholas and Yates JJ addressed the question of whether the problem was itself part of the common general knowledge, stating at [203]:
If the problem addressed by a patent specification is itself common general knowledge, or if knowledge of the problem is s 7(3) information, then such knowledge or information will be attributed to the hypothetical person skilled in the art for the purpose of assessing obviousness. But if the problem cannot be attributed to the hypothetical person skilled in the art in either of these ways then it is not permissible to attribute a knowledge of the problem on the basis of the inventor’s “starting point” such as might be gleaned from a reading of the complete specification as a whole.
378 In Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) (2007) 235 CLR 173, Gummow, Hayne, Callinan, Heydon and Crennan JJ observed at [122] that s 7(2) contemplates that the standard by which obviousness is to be judged, namely a person skilled in the relevant art, would possess common general knowledge and have regard to any s 7(3) information.
379 The applicable form of s 7(3) is that introduced by the RTB amendments to the Act. The changes to s 7(3) brought about by the RTB amendments modified the question of availability, removing the qualifying requirement that the prior art be “ascertained, understood and regarded as relevant” but did not alter the correct approach to the evaluative question.
380 In Lockwood (No 2), the High Court observed at [127] that by enlarging the prior art base through including relevant prior disclosures beyond those disclosures proven to be part of the common general knowledge, these provisions raise the threshold for inventiveness. The Court explained that in answering the question of obviousness, the prior disclosures to be taken into account as enlarged by s 7(3), like that part of the prior art base which is the common general knowledge, is considered for a particular purpose. That purpose is to look forward from the prior art base to see what the skilled person is likely to have done when faced with a problem similar to that which the patentee claims to have solved with the claimed invention.
381 As Laddie J observed in Inhale Therapeutic Systems Inc v Quadrant Healthcare Plc [2002] RPC 21 at [47] (in relation to the UK legislation), although the skilled reader is assumed to have read and understood the contents of the prior art, that does not mean that all prior art will be considered equally interesting. The notional skilled person is not assumed to know or suspect in advance of reading it that any particular piece of prior art has the answer to a problem or is relevant to it. The skilled reader comes to the prior art without any preconceptions and, in particular, without any expectation that it offers a solution to any problem.
382 The skilled addressee is assumed to be in possession of the pleaded s 7(3) document and to have read it with interest, but is not assumed or compelled to use any information contained within it and, when that information is considered together with the common general knowledge, may well disregard it or consider that it adds nothing of significance to it.
383 Sections 7(2) and 7(3)(a) do not permit the person skilled in the art to combine non-common general knowledge information with the contents of the “plus one” document. As observed by Nettle J in AstraZeneca HCA at [120], it is evident from the plain and ordinary meaning of s 7(2) that “the single document referred to in s 7(3)(a) must be capable of standing alone without interpretative or corroborative assistance from another document or other source of information apart from common general knowledge”. See also AstraZeneca AB at [209] (per Besanko, Foster, Nicholas and Yates JJ).
384 As a general comment on the Okada papers, the experts agreed in the JER that the studies described in the three papers: Okada 2000a, Okada 2000b and Okada 1999, were conducted on insufficient numbers of animals to enable statistically significant results to be generated and meet normal standards of scientific publication.
385 The experts also agreed that none of the Okada papers made reference to the issue of interference of antibodies with other components in a multiple component vaccine, as none of the papers considered or made reference to an M. hyo combination vaccine.
386 Zoetis submitted that there was no evidence to support the combination of “subsets” of the Okada papers. Okada 2000a does not refer to either Okada 1999 or Okada 2000b, and Okada 1999 does not refer to the two later Okada papers. Zoetis submits that Boehringer asked no questions in the concurrent evidence session to seek to establish the availability of any such combination of “subsets”.
387 Zoetis accepted that if the skilled person was motivated to investigate Okada 2000b further, then, based on Okada 2000b considered together with the common general knowledge, the combination of Okada 2000a, 2000b and 1999 would be available as a s 7(3) combination, but not otherwise.
388 However, Zoetis’ primary position is that the combination step is not reached because:
(a) the only evidence supporting the combination of the Okada papers is based on the assumption that the skilled person reading Okada 2000b would be motivated to investigate Okada 2000b further;
(d) to the contrary, the skilled person would not be motivated to investigate Okada 2000b further, and thus would not “combine” it with any other document it refers to;
(e) Okada 1999 does not refer to either of the 2000 papers; and
(f) Okada 2000a does not refer to either Okada 1999 or Okada 2000b.
389 Okada 1999 is cited as Okada et al, “Evaluation of Mycoplasma hyopneumoniae Inactivated Vaccine in Pigs under Field Conditions” (1999) 61 (10) Journal of Veterinary Medical Science 1131.
390 The paper describes a study involving 212 pigs from three herds. The herds were: a specific pathogen free herd, a high health status herd with a history of M. hyo, and a low health status herd with a history of M. hyo. Pigs from each herd were randomly assigned into two groups: one group was injected twice at four-week intervals with an “inactivated vaccine prepared from broth culture supernatant of M. hyo”, and the other group served as the control. The vaccinated pigs were not exposed to M. hyo but were monitored in the field.
391 The vaccine used in the study was the commercially available “Mycobuster” vaccine, described as an inactivated vaccine prepared from broth culture supernatant of M. hyo with an aluminium hydroxide adjuvant. One of the authors was employed by the manufacturer of the Mycobuster vaccine.
392 The paper reported that clinical evidence from the vaccination of all three herds showed that the inactivated vaccine against M. hyo infection proved to be safe under field conditions. In the conventional pig herds (high and low health status herds), vaccination resulted in a significant reduction of lung lesions in vaccinated pigs and a significant reduction in the extent of the lung lesions observed in the vaccinated pigs. The pathogen free pigs which had not previously been exposed to M. hyo were not infected with M. hyo. The vaccinated pathogen free pigs had an increased antibody titer against M. hyo four weeks after the second vaccination. There was no M. hyo antibody titer increase observed in the unvaccinated pigs.
393 Okada 2000a is cited as Okada et al, “Protective effect of Vaccination with Culture Supernate of M. hyopneumoniae against Experimental Infection in Pigs” (2000) 47 (7) Journal of Veterinary Medicine B 527. The peer-reviewed Journal of Veterinary Medicine is now Zoonoses and Public Health.
394 The summary to the paper states that the protective activity of M. hyo inactivated vaccine prepared from whole cells and cell-free culture supernates was evaluated experimentally using hysterectomy-produced, colostrum deprived pigs (HPCD pigs) in which mycoplasmal pneumonia had been induced. The summary concludes that vaccination with culture supernate of M. hyo can provide protection against M. hyo infection and that these antigens in the culture supernate may be closely related to the reduction of lung lesions.
395 The introduction refers to the observation by the authors in Ross et al, “Characteristics of protective activity of Mycoplasma hyponeumoniae vaccine” (1984) 45 (10) American Journal of Veterinary Research 1899, recorded in their earlier paper that M. hyo culture supernate induced protection against the development of pneumonia in swine. However, Okada 2000a noted that centrifugation used in the preparation of the supernate in Ross does not remove all of the mycoplasma cells or membranes; therefore, it is possible that whole cells or membranes also contributed to the protection. The introduction outlines that the Okada 2000a study used inactivated vaccines from sedimented whole cells and cell-free culture supernates and evaluated their effects on lung lesions, isolation of M. hyo, and on serum antibody production in experimentally infected pigs.
396 The three preparations used in Okada 2000a were prepared as follows:
M. hyopneumoniae strain 1986-1 was grown in BHL medium by rotating the medium continuously at 37°C for 96h. The BHL medium was prepared using 10% swine serum instead of horse serum (Yamamoto et al., 1986). The number of viable organisms was titrated by serial 10-fold dilutions in BHL broth and expressed as colour-changing units (CCU). Cultures were processed as follows for the three inactivated vaccine preparations (Table 1): (1) sedimented whole cells were washed twice with 50mM Tris-HCl buffer containing 250mM NaCl, pH7.2 (Tris buffer) following centrifugation at 28000 g for 30min, then resuspended with the same buffer to a final concentration of 1010 CCU/0.2 ml; (2) culture supernate containing less than 101 CCU/0.2 ml were collected from the centrifuged (28000 g for 30min) cultures; (3) a control vaccine was prepared containing only the BHL medium instead of the vaccine preparation. These preparations were treated overnight with 0.2% formalin at room temperature. Formalin-treated antigens were subcultured to ensure that the organism was killed. Equal volumes of aluminium hydroxide gel were mixed with each formalin-treated antigen and used as a vaccine.
397 Twenty four-week-old HPCD pigs, reared together and allowed to mingle in a pathogen-free environment, were randomly allocated into three groups. Each pig was intramuscularly inoculated twice with 2 ml doses of different vaccines at 2 week intervals. Group A received sedimented whole cells, Group B received supernate of culture (cell free) and Group C (the control) received only BHL medium. Four weeks after their first vaccination, all pigs were challenged intranasally with M. hyo broth culture.
398 The experts agreed that the use of HPCD piglets reduces confounding variables in the study such as the interference from the presence of maternally derived serum antibodies.
399 The experts agreed that the centrifugation used in Ross did not remove all the mycoplasma cells or membranes. Thus, it was possible that there were whole cells in the Ross supernatant and that they contributed to the protective efficacy of the supernatant.
400 The experts also agreed that the purpose of the study conducted in Okada 2000a was to confirm that the protective efficacy of the supernatant in Ross was provided by the membrane fragments, rather than any whole cells that remained in the supernatant.
401 The M. hyo supernatant used in Okada 2000a was subjected to more intense centrifugation than the one used in Ross. Professor McVey, Professor Chase and Dr Nordgren agreed that the centrifugation step in Okada 2000a substantially removed the whole cells from the supernatant. The experts agreed that there was a separation of the supernatant from the viable whole cells to a greater degree in the Okada 2000a study than in Ross. The experts described the centrifuged M. hyo preparation of Okada 2000a as a supernatant of culture and a centrifuged clarified supernatant. The centrifuged clarified supernate of Okada 2000a was not a bacterin.
402 The group of five pigs that received the centrifuged clarified supernatant had a lower extent of lung lesions than the other two groups, and the lowest mean percentage of lung lesions of the three groups.
403 Immunoblotting was performed on the sera from pigs immunised with vaccines before they were challenged with exposure to M. hyo. The immunoblotting identified that the major M. hyo antigens (97, 89, 65, 46, 42 and 41kDa) were present not only in whole cells but also in the supernate of cultures.
404 The final paragraph of the Okada 2000a conclusion states:
In the present study, vaccination with the supernate of M. hyopneumoniae cultures protected against M. hyopneumoniae infection. Our results indicate that vaccination with culture supernate is a good tool for the control of MPS and offers a foundation for the further investigation of protective antigens.
405 Professor McVey and Professor Browning considered that Okada 2000a did not provide any useful information over that which was already known from Ross. However, Professor McVey’s later evidence in the joint session was that the Okada 2000a vaccines were more intensely centrifuged than those in Ross and were a viable cell-free supernatant, or at least greatly cell-reduced. He also said that the clinical data was not present in Ross, and that Okada 2000a discloses an immunoblotting assay which identified M. hyo antigens in the supernatant, which was also not in Ross.
406 Professor McVey, Professor Chase and Dr Nordgren agreed in the joint session that the following information from Okada 2000a was not in Ross:
(a) The identity of the M. hyo antigens and their presence in the supernatant (and whole cells);
(b) the Western blot which shows that the pigs responded by producing antibodies to the proteins (antigens) present in the supernatant preparation;
(c) the results of the study on the pigs (the reduction in lung lesions, which Professor McVey described as a trend); and
(d) the conclusion that the antigens in the supernate may be closely related to the reduction in lung lesions.
407 Professor Browning generally agreed with the above, but qualified his response in relation to the Western blot results, noting that the results gave no information as to the concentration (absolute or relative) of the antigens present.
408 Professor McVey, Professor Chase and Dr Nordgren agreed that the additional information over Ross had some utility. Each agreed that they would take the above information at face value with the other information in the Okada 2000a paper and combine it with their generally known knowledge as at the priority date in deciding what to make of Okada 2000a.
409 Okada 2000b, cited as Okada et al, “Cytological and Immunological Changes in Bronchoalveolar Lavage Fluid and Histological Observation of Lung Lesions in Pigs Immunised with Mycoplasma hyopneumoniae Inactivated Vaccine Prepared from Broth Culture Supernate” (2000) 18 Vaccine 2825. Vaccine is a peer reviewed, high-impact, leading journal published by Elsevier in the area of vaccine development and vaccine research.
410 The paper describes M. hyo challenge studies in which two groups were compared: the first group of five piglets which were not immunised and the second group of five piglets which were immunised with an M. hyo vaccine prepared from M. hyo cell culture supernate. The purpose of the study was stated to be to investigate the effect of the vaccination through sequential analyses of cytological and immunological changes in bronchoalveolar lavage fluid.
411 The vaccine used in the study was described as being that marketed as “Mycobuster”. However, it appeared that it was a further processed version of the commercially available Mycobuster, as the culture supernate collected from the centrifuged cultures was said to contain less than 101 CCU/ml of organisms. The Mycobuster Product Information describes Mycobuster as containing less than 2.5 x 107 or more to 2.5 x 109 viable bacteria per 100 ml post filtration. The broth culture supernate was then treated overnight with 0.2% formalin to ensure that the organisms were killed.
412 That the composition of the vaccine used in the study was unclear was demonstrated by Professor Nordgren’s first thought, expressed in his first affidavit, that the studies in Okada 2000b were using the commercial version of the Mycobuster vaccine. He later realised his error and changed his evidence in his second affidavit to indicate that it was actually a refined, experimental, and further processed version of the Mycobuster vaccine.
413 The two groups of pigs used in the Okada 2000b study were four week old specific pathogen free (SPF) piglets. During the study, the pigs of both groups were housed separately in a pathogen free environment. SPF pigs were used to reduce confounding effects such as the interference from the presence of pre-existing serum antibodies.
414 Okada 2000b demonstrates that the M. hyo supernatant vaccine was effective in stimulating an adaptive immune response in the vaccinated pigs and providing protection from M. hyo related diseases, because pigs in the vaccinated group produced a significantly faster immune response than pigs in the control group (demonstrated by the greater amounts of IgG in the bronchoalveolar lavage fluid of the vaccinated pigs post-inoculation, the trends in the M. hyo levels in vaccinated pigs over the four week time period, and the reduction of IgG levels of the vaccinated groups from three weeks after inoculation) and that the vaccine provided effective protection from damage to the respiratory tract caused by M. hyo.
415 The primary purpose of Okada 2000b was to investigate the mode of action of the vaccine studied by analysing the cytology of bronchoalveolar lavage fluid and the pathology of lung lesions. The paper notes that the major specific antigens (97, 89, 65, 46, 42 and 41kD) were present in the culture supernate. Okada 2000a is cited as support for that statement.
416 Okada 2000b concludes that “consequently, vaccination may be an effective method to improve the economic losses resulting from [Mycoplasmal pneumonia of swine]”.
Statistical issues with Okada papers
417 As noted earlier, the experts agreed in the JER that the studies described in Okada 1999, Okada 2000a and Okada 2000b were conducted on insufficient numbers of animals to enable statistically significant results to be generated and meet normal standards of scientific publication.
418 Professor McVey’s evidence was that he would often have regard to preliminary data that would not meet the standards of statistical significance, and that the data in the Okada papers was preliminary data, and provided support in understanding the potential value of an M. hyo supernate vaccine. He agreed that he saw some protective effect in the papers, and there was a trend to reduction in lung lesions in Okada 2000a. Professor McVey described the studies in the Okada papers as a pilot or proof of concept. In the joint session, Professor McVey added that his response after reading the Okada papers was that he would want to conduct tests in more pigs.
419 Both the studies the subject of Okada 2000a and 2000b used disease free pigs, which Professor McVey and Dr Nordgren agreed reduced the confounding factors present in the studies. Professor Chase explained that the use of disease-free pigs removed maternal antibody interference effects.
420 Professor McVey, Professor Chase and Dr Nordgren agreed that the papers provide useful information, notwithstanding their lack of statistical significance. Professor Browning disagreed. He considered that the statistical methods used in each of the Okada studies was not appropriate for the data that were generated from the studies and thus that the validity of the conclusions were not able to be established from the papers. Professor Browning’s evidence was that the lack of appropriate statistical support for the conclusions would have caused him to reject all three of the Okada studies as not demonstrating efficacy.
Expert comments on Okada papers
421 Professor McVey was the only one of the experts that had read the Okada papers before the priority date.
422 The experts agreed that none of the Okada papers compared the efficacy of the Mycobuster vaccine (or the refined version of Mycobuster used in Okada 2000a) with a commercially available bacterin. Dr Nordgren agreed that there was also no comparison of the efficacy of Mycobuster with the efficacy of the supernatant studied in Ross.
423 Professor McVey, Professor Chase and Dr Nordgren considered that it is standard practice for vaccine development teams to assess the totality of information available to allow them to make determinations regarding vaccine development and to determine the value of a vaccine program for field use. In the context of previous data that was generally known to them (including Ross), they considered that the clinical data presented in each of the Okada papers would have provided support in the understanding of the potential value of an M. hyo supernate derived vaccine. Professor McVey’s evidence was that he would want to conduct tests in more pigs.
424 In addition to the statistical difficulties he identified, Professor Browning considered that the description in the Okada papers of the methods used for culture of M. hyo and the method of preparation of the supernatant was insufficiently precise to enable him to replicate the studies. Professor McVey and Dr Nordgren considered that adequate information was supplied in the Okada papers in comparison to typical antigen description sections in publications of this nature. Professor McVey and Dr Nordgren considered that the description of the vaccine preparation in the Okada papers would have provided suitable guidance in the preparation of test vaccines (similar to the vaccines of the Okada publications) in a commercial development laboratory. Professor Chase agreed.
425 Boehringer contends that the alleged invention in each Application was obvious in light of the common general knowledge combined with the Okada publications (considered together or separately), each of which is prior art information available under s 7(3) of the Act. Under s 7(3)(b), pieces of prior art information may be combined if the skilled person could, before the priority date of the relevant claim, be reasonably expected to have combined them.
426 Boehringer submits that there was a motivation to try a supernatant, because it was known that bacterins had variable efficacy.
427 Based on Okada 2000a, a peer reviewed journal, the skilled person would conclude that the supernate preparation might well provide an advantage over the whole cell preparation, because it can overcome the inconsistent or varied levels of protection experienced with whole cell preparations/bacterins.
428 Further, Boehringer submits that the skilled person armed with Okada 2000a would consider an M. hyo culture supernatant vaccine to be a promising candidate for a new or improved M. hyo vaccine, as a monovalent vaccine and as part of a bivalent and trivalent vaccine. Okada 2000a provided more information than the earlier Ross paper, including the identification of the protein antigens in the supernate. The person skilled in the art would use the immunoblotting assay in Okada 2000a to compare whether their supernate preparation contained the protein antigens shown to be present in the supernate preparation used in Okada 2000a and associated with the reduction in lung lesions.
429 On the basis of Okada 2000a, Boehringer argues that the person skilled in the art would have made a supernatant and removed antibodies with an expectation of success. Professor McVey’s evidence in the joint session was that if he had been tasked to look at making a combination vaccine with Okada 2000a in hand, he would have applied a technique such as Protein A or G to remove anti-PCV-2 antibodies from the M. hyo preparation with an expectation that it may well remove enough antibodies for him to address the issue of interference. He would expect that it may work but he would need to test it to confirm. Professor Chase and Dr Nordgren agreed. Any means of removing antibodies from serum via upstream or downstream processing using size exclusion or Protein A/G column chromatography, or by using serum from disease-free herds or from another species, would produce a supernatant which was substantially free of IgG and immunocomplexes within the meaning of claim 1 of each of the Applications.
430 Boehringer submits that Professor McVey, Professor Chase and Dr Nordgren would similarly have had an expectation of success in developing an M. hyo/PCV-2 and/or PRRS vaccine. In the latter case, the PRRS antigen would be in the lyophilised form to be reconstituted at the time of administration in the M. hyo/PCV-2 solution.
431 Boehringer also submits that it is not to the point that Okada 2000a did not provide a comparison between the M. hyo supernatant and a commercially available bacterin. It is enough that the supernatant vaccine might provide a useful alternative to bacterins. Okada 2000b refers to both Okada 2000a and Okada 1999, and the skilled person could reasonably be expected to combine Okada 2000b with either one or both of Okada 2000a and Okada 1999.
432 Boehringer submits that the skilled addressee, armed with the common general knowledge and Okada 2000b, would prepare an M. hyo supernatant vaccine as described in Okada 2000b, and evaluate whether the six major specific antigens listed in Okada 2000b were present and the efficacy of the vaccine against other pre-existing vaccine production processes.
433 Because the skilled addressee would be developing an M. hyo vaccine with a view to combining it with other vaccines or antigens, and PCV-2 antigens in particular, they would consider it important to be free of serum-derived antibodies that could interfere with the other vaccines or antigens. This would most likely be achieved by including a purification step as part of the method for preparing the vaccine, using a technique suitable for soluble preparations such as Protein A or Protein G based affinity chromatography, or any of the other means of removing antibodies from serum set out above. As a result, the skilled addressee would produce a supernatant which was substantially free of IgG and immunocomplexes within the meaning of claim 1 of each of the Applications.
434 Boehringer further submitted that the skilled person would also conclude that the results reported in each of Okada 2000a and Okada 2000b support and add weight to each other.
435 Okada 1999 describes the clinical, commercial and economic benefits of using the Mycobuster culture supernatant vaccine. Based on Okada 1999, Boehringer submits that:
the skilled person would conclude that an M. hyo culture supernate vaccine can induce an adaptive immune response and provide protection from M. hyo related damage in high health and low health herds;
the use of an M. hyo culture supernate vaccine can provide significantly increased pig performance in herds of low health status; and
in addition to reducing the damage caused by M. hyo infection, the use of a culture supernate vaccine may provide protection against M. hyo infection in pigs in herds of high health status.
436 Okada 1999 therefore provides further support for the results in Okada 2000a and 2000b.
437 Zoetis contends that the “problem/solution” approach is inappropriate in this case. First, citing Gummow, Hayne, Callinan, Heydon and Crennan JJ in Lockwood (No 2) at [65], Zoetis submits that such an approach is unfair because:
[T]he Applications record an elegant and insightful, but not technically complex, solution associated with the issue of avoiding interference issues associated with combination vaccines, in circumstances where no expert witness called before the Court, even an inventive one such as Dr Nordgren, had reached that solution before the Priority Date.
438 Zoetis submits that the idea for overcoming the issue of both assay and immunological interference by applying Protein A or Protein G to a supernatant was an ingenious way to overcome a practical difficulty, and created a new and useful M. hyo “platform” which was substantially free of not only anti-PCV-2 antibodies, but substantially free of IgG and immunocomplexes generally. They submit it possessed elegant simplicity. However, once the invention is known, its very elegance and simplicity is apt to mislead through the impossibility of excluding hindsight if a problem/solution approach is adopted.
439 Second, Zoetis submits that the problem/solution approach is inapt where, as in this case, there are three problems solved by the Applications, not one rolled-up problem. According to Zoetis, Boehringer rolls up three problems (the need for improved M. hyo vaccines, a need for M. hyo combination vaccines, and a need for “platform” vaccines), each of which it asserts is common general knowledge and then seeks to apply the problem/solution approach to that rolled-up problem, not to each separate problem. Zoetis submits that this rolled-up problem infected Boehringer’s briefing of its expert witnesses.
440 Zoetis observes that each of the three problems was distinct and that there was no evidence that there was any common general knowledge problem of the nature of a need for “platform vaccines”. Zoetis submits that combining the three problems into one rolled-up problem deprives the inventions in the present case of one of their key features: an insight which solved at least three problems (or, more accurately, solved at least two problems and met an unfelt want) simultaneously and thus led to the multiple inventions claimed across the three Applications. Zoetis further submits that the “litigious happenstance” of the three Applications being considered together in the present case provides no warrant for collapsing the distinct problems solved by the inventions in those three Applications into one compound problem.
441 As to the Okada papers, Zoetis submits that the deficiencies in them are such that the skilled reader would likely say “I have read the Okada papers with interest, but I am not interested”, and put them aside as being of no assistance. Zoetis observes that this approach is borne out by the evidence of Professor McVey, the only one of the experts who had read the Okada papers before the priority date, and who did nothing with them.
442 Zoetis submits that the nature of the questions asked of Professor Chase in putting together his declaration in the Patent Office, and the questioning of the experts in the joint session, funnelled their responses towards the desired result. Zoetis gave two examples of what it considered the “funnelling" questions put to Professor Chase in the course of giving his first round of evidence in the Patent Office. That is, before he saw the Applications, and before he was given the Okada papers. First, Professor Chase was specifically asked to expand upon the process of preparing a supernatant vaccine and “how I might go about preparing a supernatant vaccine”. That instruction came immediately after he had listed both inactivated whole cell preparations (or parts thereof) or supernatant from culture as available approaches, without expressing any preference for a supernatant (or indeed even being asked which he preferred). Thus, he had no opportunity to say he preferred a bacterin but was instead directed to expand upon the preparation of a supernatant.
443 Second, Professor Chase was specifically asked to expand upon the use of affinity chromatography in the context of vaccine development. Again, that instruction came immediately after he had listed a large number of available techniques for the removal of antibodies from compositions, including filtration, precipitation, centrifugation, column based techniques such as sepharose/sephadex size-exclusion, ion-exchange chromatography and affinity chromatography, as the last listed. Again, Professor Chase was not asked to, and did not, express any particular preference for affinity chromatography, rather he was directed to expand upon only that technique.
444 Zoetis submits that Professor Chase was given two very large hints as to the nature of the invention: prepare a supernatant vaccine and do something with affinity chromatography, before he was given the Okada papers and the 612 Application. After being given copies of the Okada papers and the 612 Application, Professor Chase was specifically “tasked with adding another antigen to a M. hyo culture supernatant vaccine” to produce a multivalent immunogenic composition. He was therefore given the final piece of the puzzle. That is, knowing that he was to prepare a supernatant vaccine, knowing that he was being asked to expand upon the use of affinity chromatography out of the long list of options he had provided and knowing the non-common general knowledge contents of the 612 Application, he was told that he needed to add another antigen to the supernatant to make a multivalent vaccine.
445 Zoetis submits that by the time he came to prepare his affidavit in this proceeding, Professor Chase had read the Okada papers and the 612 Application and knew of the invention described and claimed in the Applications. In that context, Professor Chase was asked by Boehringer’s instructors to explain, using only his knowledge (as opposed to what was common knowledge) before April 2012:
(a) the process of preparing a monovalent vaccine based on a cell culture supernatant; and
(b) the process of preparing a multivalent vaccine based on the supernatant of an M. hyo culture.
446 Zoetis also relies on the “objective evidence of inventiveness”, Boehringer’s failure to produce a ready-to-use M. hyo/bacterin product, and Zoetis’ commercial success with the commercial embodiments of the inventions claimed in the Applications.
447 Both parties referred to confidential in-house research and development work done by the other, as secondary indicia to support their respective cases on inventive step.
448 Zoetis referred to Lockwood (No 2) at [118], wherein the Court noted that the primary judge considered it “of particular relevance … that no non-inventive persons skilled in the art who were called as witnesses came up with the combination … before the priority date, and that Doric … copied the ‘bare idea’ of Lockwood’s improved lock”. Zoetis submits that the High Court found no error in that approach. Zoetis contends that the secondary evidence in the present case is even more striking and provides an additional reason to be cautious of Boehringer’s approach.
449 Such in-house research work is typically confidential and as such will not form part of the relevant common general knowledge. The test for whether the invention is obvious is a hypothetical one, so the actual work undertaken by the parties is of limited, if any, relevance to the hypothetical question.
450 Boehringer observed that the Zoetis combination vaccine development team had access to Okada 2000a (although not the other Okada documents). Boehringer submitted that the use the Zoetis combination vaccine team made of Okada 2000a was “instructive” as to what the person skilled in the art in possession of Okada 2000a would do. Boehringer submitted that Okada 2000a was the inspiration for Zoetis including an M. hyo supernatant in its testing program, and that, on reading the paper, the team immediately proceeded to test the supernatant, along with Protein A treatment and obtained a positive result.
451 Zoetis referred to Dr Nordgren’s confidential in-house work leading a team at Merial undertaking experimental work on combining a PCV-2 vaccine with an M. hyo vaccine. As discussed earlier, this work was not disclosed in Dr Nordgren’s affidavits. That experimental work included efforts aimed at addressing impediments the M. hyo formulation may present to combining it with a PCV-2 antigen. Strategies investigated by Dr Nordgren and his colleagues at Merial included attempting to source swine serum with a reduced likelihood of containing anti-PCV-2 antibodies, as well as attempting to clarify serum of anti-PCV-2 antibodies. Zoetis observed that, in contrast to his evidence as to what he would have done if tasked with developing a bivalent M. hyo/PCV-2 vaccine, neither Dr Nordgren nor his team considered applying Protein A or Protein G to the M. hyo vaccine or Mycobuster.
452 Zoetis also referred to other combination vaccine development work done by Boehringer’s own in-house research team between 2009 to 2014 to develop a new ready-to-use bivalent PCV-2 and M. hyo combination vaccine product to supersede its then product “FLEXCombo”.
453 Zoetis also observed that Professor McVey, who did not possess the additional information possessed by the Zoetis team but was in possession of all the Okada papers before the priority date, found no interest in them.
454 Zoetis submits that Boehringer’s failed M. hyo/PCV-2 combination vaccine project, and Zoetis’ asserted rapid success in gaining market share, are strong secondary indicators of the inventiveness of Zoetis’ inventions as set out in the Applications.
455 Boehringer submits that Zoetis’ use of Okada 2000a provides strong support for Boehringer’s case. Zoetis had access to Okada 2000a (although not the other Okada documents). Boehringer contends that Okada 2000a was the inspiration for Zoetis including an M. hyo supernatant in its testing programme. Upon reading the paper, the Zoetis team immediately proceeded to test the supernatant, along with Protein A treatment, and obtained a positive result.
456 Zoetis submits that in every case which does not involve a s 7(3) document, an inventor is presumed to be in possession of the common general knowledge and arrives at the invention. That fact alone says nothing about whether the invention is inventive over common general knowledge. Similarly with a s 7(3) document, the fact that the inventor arrives at the invention when they are in possession of the s 7(3) document says nothing about whether the invention is inventive over the common general knowledge considered together with the s 7(3) document. Furthermore, the inventors are, by definition, unlikely to be representative of the non-inventive person skilled in the art.
457 In 2010, Zoetis’ predecessor, Pfizer Animal Health, had a vaccine development team looking to develop a one-bottle, M. hyo/PCV-2 combination vaccine. At that stage the Zoetis research team was looking at a combination of an M. hyo (bacterin) and a PCV-2 antigen.
458 A final or near final draft of a poster entitled “Process Alternative for PCV-2 Compatible Mycoplasma hyopneumoniae Antigen” reporting on the state of the Pfizer/Zoetis team’s work (authored by a team including four of the inventors), circulated to the authors of the poster via email on 16 September 2010, provides a contemporaneous record of the Zoetis combination vaccine team’s ongoing work to produce a second generation M. hyo /PCV-2 combination vaccine product. The poster highlights the following findings:
(a) Identification of the potency of M. hyo antigen P46;
(b) Ubiquitous exposure of serum producing swine herds to PCV-2 resulting in antibody tainted serum supplies;
(c) Recognition that downstream processing (centrifugation) which effectively removed PCV-2 also resulted in a loss of P46 antigen;
(d) Consideration of how to produce the desired high P46/low PCV-2 antibody product;
(e) Use of Protein A upstream to remove PCV-2 antibodies from the growth medium without retarding M. hyo growth support;
(f) Observation that P46 antigen was being lost in the waste stream; and
(g) Investigating recovery of supernatant antigen using Protein A to remove PCV-2 antibodies followed by sterile filtration.
459 This work, conducted prior to the team being in receipt of Okada 2000a, shows that the team was already using Protein A to remove PCV-2 antibodies, albeit upstream.
460 In January 2011, results of tests on piglets using a combination of the centrifuged (insoluble) M. hyo antigen and PCV-2 antigen showed it not to be efficacious in reducing lung lesions after M. hyo challenge. The testing used a sufficient population of piglets (20 in each group, aside from one group of 19) to give statistically valid results.
461 Within a week of the results of the study, the team began troubleshooting the M. hyo lack of efficacy results exercise. Amongst a list of “items still to be discussed” in an updated “brainstorming” document, attached to an email of 18 January 2011 summarising the team’s discussions, item 10 referred to the application of Protein A/G upstream to purify the M. hyo growth media prior to cultivating M. hyo. An email later on the same day stated “[f]uture or current, consult Bob Garlick and evaluate Protein A treatment of 1x or concentrated supernatant for removal of aPCV [sic]”.
462 Later the same day, 16 January 2011, Okada 2000a appeared, attached to an email from one of the inventors, Jeffrey Galvin:
See below for another group for the next Mhyo study?
Keith has done his homework. Look at the attached paper [Okada 2000a]…2000 publication showing sedimented (centrifuged) cells do not work as a vaccine.
463 Okada 2000a confirmed the results the team had seen in their recent failed tests in piglets.
464 A post priority date Zoetis technical update in relation to its (single dose) Fostera™ PCV MH product stated:
During cell growth of mycoplasmas, immunogenic components are released into the culture broth. This has been demonstrated in previous research where protection against M. hyo was achieved when using the cell-free supernatant of an M. hyo culture as compared to using sedimented, whole M. hyo cells.
465 The technical update attributed the previous research to the study reported in Okada 2000a. The technical updated continued:
Simply removing the M. hyo cells does not render the M. hyo antigens compatible with PCV2 antigens. Zoetis scientists introduced a step that removes the troublesome anti-PCV antibodies from the M. hyo containing fluid: ‘Protein A’ chromatography.
…
For production of Fostera PCV MH, cell-free M. hyo culture fluid (containing critical M. hyo antigens) is passed through a Protein A chromatography column to remove the antibodies. The resultant fluid is a purified and substantially IgG antibody-reduced M. hyo fraction (Figure 1).
By using Protein A to help remove PCV antibodies (and other irrelevant IgG antibodies) from the M. hyo fraction, the PCV antigens in the combination vaccine remains fully potent. In fact, research evaluating multiple combinations of whole-cell and fluid-only versions of M. hyo antigen confirmed that the call-free, Protein A-treated M. hyo antigen was highly efficacious.
466 As to Protein A, the technical update stated:
Notably, Protein A is commonly used in human medicines for its ability to purify IgG immunoglobulins (Table 1 shows some examples of Protein A use in monoclonal antibody therapy, including treatments for human cancer and autoimmune disorders). Thus, Protein A acts like a biomolecular ‘sponge’ that selectively binds and removes antibodies.
467 Table 1 of the technical update set out seven examples of human monoclonal antibody therapies that utilize Protein A technology. An Amersham Biosciences “Antibody Purification Handbook” (2002), annexed to Professor Chase’s first affidavit, describes the use of Protein A and Protein G for the purification of monoclonal IgG type antibodies (and their isolation from ascites fluid, cell culture supernatants and serum). This is an application of Protein A to purify antibodies in accordance with the well-known uses of Protein A that formed part of the common general knowledge.
468 Zoetis submits that its combination vaccine development team was well progressed along the inventive journey by the time the team had Okada 2000a in hand, to a level beyond the common general knowledge. In particular, prior to obtaining the Okada 2000a paper, the Zoetis team were applying Protein A treatment to serum culture media (upstream) to produce a compatible M. hyo antigen (ie an M. hyo antigen substantially free of anti-PCV-2 antibodies). The team that was investigating the use of Protein A had the realisation that applying Protein A to the supernatant (downstream) would provide another means of producing a compatible M. hyo antigen. Albeit, by way of the P46 antigen salvaged from the waste stream being added back to the whole cell bacterin to increase antigenicity.
469 Prior to the circulation of Okada 2000a, Zoetis had completed a study of its own showing that centrifuged insoluble M. hyo vaccines were not efficacious in reducing lung lesions due to M. hyo challenge. Accordingly, Zoetis has independent non-common general knowledge information relating to the lack of efficacy of the insoluble preparations at the time Okada 2000a was circulated. Zoetis was aware of antigen P46 and that it was in the “waste stream” to such a level that it was investigating salvaging it from the waste stream.
470 I find that Zoetis possessed information in addition to the common general knowledge by the time the team had access to Okada 2000a. Zoetis was applying Protein A upstream to remove PCV-2 antibodies from the serum containing growth media. This was not a common general knowledge use of Protein A. Further, Okada 2000a makes no reference to the application of Protein A or Protein G upstream or downstream.
471 The work done and the knowledge possessed by the Zoetis research team prior to it obtaining Okada 2000a put it in a position far beyond that of a person skilled in the art armed only with the common general knowledge.
472 Zoetis submits that Boehringer never arrived at the invention, despite having a team working to develop a ready-to-use M. hyo/PCV-2 vaccine for five years. Further, even though, according to its case now, Okada 2000b was published in a “high impact journal” some nine years before Boehringer’s ready-to-use project began, Mr Eichmeyer’s evidence was that he was not aware of the Okada papers as at the priority date, nor was he, as project lead, aware of any of his colleagues being aware of the Okada papers either.
473 Boehringer submits that this is not a surprising result as the purpose of the project was to combine Boehringer’s existing products, which included an M. hyo bacterin, not to investigate alternative production processes. Thus, the success or failure of this work is irrelevant to the question of inventive step.
474 There is no challenge to inventive step based on the common general knowledge alone. Thus, Zoetis submits that the idea of an advantageous “platform” M. hyo vaccine, in the sense described in the Applications (ie one which is substantially free of IgG and immunocomplexes [of all kinds] and thus compatible with other antigens) was not common general knowledge.
475 Zoetis observes that Professor Chase did not use the term “platform” in the Patent Office until after he had read the Applications, and only referred to an “advantageous platform” for the first time in reply to Professor Browning. Professor Chase agreed that he had never had the idea to use a M. hyo supernatant to which Protein A had been applied as a “platform” for a bivalent or trivalent vaccine before he read the Applications.
476 Other than Zoetis’ assertions in submissions as to the commercial success of its commercial vaccine, there was no evidence as to the market share or annual sales or profits associated with the Zoetis commercial embodiment of the inventions claimed in the Applications.
Hindsight and Boehringer’s inventive step case
477 The authorities are replete with warnings of the danger of hindsight in assessing whether an invention is obvious, particularly where the invention is a combination of integers. In Alphapharm at [21], Gleeson CJ, Gaudron, Gummow and Hayne JJ repeated Lord Diplock’s warning in Technograph Printed Circuits Ltd v Mills & Rockley (Electronics) Ltd [1972] RPC 346 at 362:
Once an invention has been made it is generally possible to postulate a combination of steps by which the inventor might have arrived at the invention that he claims in his specification if he started from something that was already known. But it is only because the invention has been made and has proved successful that it is possible to postulate from what starting point and by what particular combination of steps the inventor could have arrived at his invention. It may be that taken in isolation none of the steps which it is now possible to postulate, if taken in isolation, appears to call for any inventive ingenuity. It is improbable that this reconstruction a posteriori represents the mental process by which the inventor in fact arrived at his invention, but, even if it were, inventive ingenuity lay in perceiving that the final result which it was the object of the inventor to achieve was attainable from the particular starting point and in his selection of the particular combination of steps which would lead to that result.
478 Aspects of the questions put to the experts in the preparation of their evidence and in the joint session involved an element of hindsight.
479 I referred earlier to Zoetis’ submissions in relation to Boehringer’s “funnelling” of Professor Chase via the line of questioning put to him in the course of preparing his evidence. Each time that Professor Chase, in the course of preparing his declaration, gave an answer that included options, only the option that would lead to the invention was selected for further explanation.
480 In the course of giving his first round of evidence in the Patent Office, that is, before he saw the Applications, and before he was given the Okada papers, Professor Chase was specifically asked to expand upon the process of preparing a supernatant vaccines and “how I might go about preparing a supernatant vaccine”. That instruction came immediately after he had listed both inactivated whole cell preparations (or parts thereof) or supernatant from culture as available approaches, without expressing any preference for a supernatant (or indeed even being asked which he preferred). Thus, he had no opportunity to say he preferred a bacterin but was instead led to a supernatant.
481 Later in the same evidence, Professor Chase was specifically asked to expand upon the use of affinity chromatography in the context of vaccine development. Again, that instruction came immediately after he had listed a large number of available techniques for the removal of antibodies from compositions including filtration, precipitation, centrifugation, column based techniques such as sepharose/sephadex size-exclusion, ion-exchange chromatography and affinity chromatography, as the last listed. Professor Chase was not asked to, and did not, express any particular preference for affinity chromatography, rather he was directed to expand upon only that technique.
482 Professor Chase was provided with the 612 Application, described by Zoetis as “the final piece of the puzzle”, together with the Okada papers. Professor Chase was specifically “tasked with adding another antigen to a M. hyo culture supernatant vaccine” to produce a multivalent immunogenic composition. That is, he was given the 612 Application and the Okada papers and told that he needed to add another antigen to the supernatant to make a multivalent vaccine, knowing that he was to prepare a supernatant vaccine; knowing that he was being asked to expand upon the use of affinity chromatography out of the long list of options he had provided; and knowing the non-common general knowledge contents of the 612 Application, including the references to Protein A and Protein G.
483 By the time Professor Chase came to give his affidavit evidence in this proceeding, he had read the Applications in the course of giving his evidence in the Office. But the question Professor Chase was asked in his affidavits, even after he already knew what the invention was from seeing the Applications in the Patent Office, was itself leading him to the invention. It included the following:
Ashurst asked me to explain, using only my knowledge before April 2012:
…
(b) the process of preparing a monovalent vaccine based on a cell culture supernatant;
(c) the process of preparing a multivalent vaccine based on the supernatant of an M. hyo culture.
484 Boehringer’s questioning in the joint session also had the effect of moving the experts towards the desired outcome. By focussing at each stage only on an option known to lead to the invention and ignoring the other options the experts said were available, Boehringer sought to create the impression that the invention is merely the endpoint of following a path of obvious or routine choices.
485 In the JER, the experts agreed that there were a “variety of techniques” that could be used to overcome immunological interference that may occur in a combination vaccine, and gave the examples of removing the interfering component or adding more of the component that is interfered with. The experts gave four possible techniques to reduce the amount of PCV-2 antibodies in pig serum to counter interference from antibodies in serum, one of which was “column chromatography”.
486 Before the variety of other techniques that could be used to reduce interference was exposed, Boehringer’s funnelling of the invention continued by questioning in the joint session which focussed:
(a) only on the option to remove antibodies;
(b) in relation to removing antibodies, only on one option of those listed by the experts as a way to remove antibodies, namely column chromatography;
(c) only on affinity chromatography to the exclusion of size exclusion chromatography (the only form of column chromatography contemplated by Professor Browning); and
(d) focussed only on one option encompassed by affinity chromatography, namely the use of Protein A and Protein G in the reverse manner as per the invention.
487 There then followed a long sequence of questions directed at establishing that Protein A and Protein G were known techniques used to purify antibodies that could also be used to obtain a solution which had a reduced number of antibodies, by reference to catalogues such as the Pierce Biotechnology catalogue, after the experts were taken to the 535 Application.
488 Boehringer’s case in relation to the use of Protein A or Protein G affinity chromatography succumbs to hindsight, assuming at the outset that a person skilled in the art wishes to employ that technique for a non-common general knowledge purpose (namely, to make an immunogenic composition that is substantially free of IgG and immunocomplexes of all kinds), and then reasoning that the person skilled in the art could engage in experimentation to see whether or not Protein A or Protein G affinity chromatography would be effective for that purpose.
Dr Nordgren’s work on an M. hyo/PCV-2 combination vaccine
489 Zoetis submits that Dr Nordgren (who admits that he is inventive) and his colleagues at Merial (now Boehringer) knew details of the Mycobuster product in 2008-2009 equivalent to the information provided by Okada 1999 (ie that it involved some separation of soluble and insoluble cellular material and that it was efficacious and on the market), as well as the details disclosed by Ross. And yet, the thought to use Protein A or Protein G on Mycobuster did not occur to him or his colleagues in attempting to develop a combination M. hyo/PCV-2 vaccine.
490 Dr Nordgren contended in his first affidavit that after reading both Okada 1999, which studied Mycobuster, and Okada 2000b (which at the time of making his first affidavit Dr Nordgren considered to be referring to the commercially available Mycobuster) he would have arrived at the invention.
491 However, this contention was in contrast to his evidence about what happened in the real world That is, neither he nor his colleagues thought to apply Protein A or Protein G to Mycobuster in their investigation of developing a combination M. hyo/PCV-2 vaccine in 2008–2009.
[Counsel]: And at about that time, indeed, during the currency of that project you learnt about Mycobuster. Correct?
DR NORDGREN: That’s correct.
[Counsel]: And you didn’t say to your colleagues, “Let’s achieve the combination by applying protein A to the Mycobuster vaccine as a first step”, did you?
DR NORDGREN: No, I did not.
[Counsel]: And that’s because neither you nor your colleagues thought of it. Correct?
DR NORDGREN: To apply that to Mycobuster? No.
…
DR NORDGREN: To add protein A to MycoBuster to improve it any further? No, I didn’t think of that.
492 Zoetis’ counsel criticised Dr Nordgren on the basis that he suggested changes based on Okada 1999 which he did not make when he was investigating Mycobuster. Dr Nordgren explained that he did not have access to Mycobuster; it was a product that was registered and licensed in Japan, and Merial wished to co-market it in Japan and perhaps take it to Korea. Merial did not have an active combination vaccine program at that stage, because its PCV-2 vaccine in Europe was a vaccine for sows, not piglets. To the extent that Merial was considering a combination vaccine, it intended to use its licensed vaccine (a bacterin).
493 Dr Nordgren repeatedly explained he was not considering developing a bivalent or trivalent vaccine at the priority date.
Consideration
494 Boehringer does not assert that the inventions claimed in each of the Applications are obvious on the basis of common general knowledge alone. Boehringer relies on the common general knowledge plus one of the Okada papers, or a combination of those papers.
495 The question is whether the invention as claimed in the claims of the 535 Application (and the 537 and 540 Applications) would have been obvious to the skilled addressee in light of the common general knowledge considered together with information brought in under s 7(3), namely the information to be found in the individual Okada papers (or a combination of the Okada papers). The question must be asked separately for each of the Applications.
496 I will start with claim 1 of the 535 Application. Claim 1 of the 535 Application claims an immunogenic composition (not a vaccine) comprising a supernatant of an M. hyo culture, wherein the supernatant of the M. hyo culture has been separated from insoluble cellular material by centrifugation, filtration or precipitation and is substantially free of both IgG and immunocomplexes comprised of antigen bound to immunoglobulin. The use of Protein A or Protein G is not introduced until claim 2.
497 I consider that, as at the priority date, there was a motivation for the skilled person to look for an improved M. hyo vaccine, or an alternative to a bacterin, because the existing state of the art M. hyo vaccines were bacterins, and bacterins were known to have variable efficacy. Dr Nordgren explained that bacterins suffered from variability. Professor McVey agreed that there was a need for an improved M. hyo vaccine as the existing vaccines on the market at the priority date were not perfect. The existing vaccines had variable efficacy and issues with stability, because bacterins are inherently crude. Professor Chase agreed. Professor Browning was not aware of the variable performance of bacterins.
498 Each s 7(3) document must be considered separately, unless the person skilled in the art would combine them. Boehringer puts forward its inventive step case based on Okada 2000a. There is no evidence that the person skilled in the art would combine Okada 2000a with the other Okada documents. However, when Okada 2000b is considered, that document does refer to the other two Okada documents. Zoetis accepted that Okada 2000b might be combined with the other Okada documents, if the person skilled in the art were motivated to investigate Okada 2000b further (which Zoetis submits they would not).
499 Okada 2000a, which was published in a peer reviewed journal, describes a comparison of the effectiveness of two candidate M. hyo vaccines: one prepared from M. hyo cell culture supernate which is described as a “cell-free culture supernate” and the other a pelleted vaccine from sedimented whole cells. Okada 2000a reports that the supernate vaccine had superior and more consistent efficacy than the pelleted vaccine or the non-vaccinated control group.
500 Professor McVey, Professor Chase and Dr Nordgren agreed that the centrifugation step in Okada 2000a substantially removed the whole cells from the supernatant. The experts agreed that the centrifuged M. hyo vaccine in Okada 2000a could be described as a supernatant of culture, or a centrifuged clarified supernatant, and that it was not a bacterin.
501 Professor Chase, Professor McVey and Dr Nordgren agreed that the intensity of the centrifugation step in Okada 2000a was more intense than the examples in the Applications and would provide at least the same, if not a greater, degree of separation of insoluble cellular material from the supernatant than the examples in the Application.
502 Even though the studies in the Okada papers were small scale and conducted on insufficient numbers of pigs to provide statistically significant results, Professor Chase, Professor McVey and Dr Nordgren agreed in the JER, in relation to the Okada publications generally, that: “the clinical data presented … in the context of previous data that were generally known, would have provided support in the understanding of the potential value of an M. hyo supernate derived vaccine”. In explaining their conclusion, they said that “it is standard practice for vaccine development teams to assess the totality of information available to allow them to make determinations regarding vaccine development and to determine value of vaccine program for field use”.
503 Further, Professor McVey and Dr Nordgren (the only experts with experience in a commercial development laboratory) agreed that the description of the vaccine preparation would provide suitable guidance in preparing test vaccines (similar to the Okada vaccines) in a commercial development laboratory.
504 Professor McVey gave evidence that he would often have regard to preliminary data that would not meet the standards of statistical significance, and that the data in the Okada papers was preliminary data, which he considered showed a trend and provided support in understanding the potential value of an M. hyo supernate vaccine. He agreed that he saw some protective effect in the papers.
505 Professor Chase’s evidence was that studies with small numbers of animals may be done as pilots to decide, on the basis of the clinical impact, whether to move forward or not. Professor McVey described the Okada studies as a pilot or proof of concept and said that he would like to carry out more studies on more pigs.
506 Professor McVey and Professor Chase agreed that, as a result of the use of HPCD piglets, variables in the study including maternal antibody interference was reduced, rendering the data in the Okada 2000a paper more meaningful than if such piglets were not used.
507 I consider that the skilled person armed with Okada 2000a would consider an M. hyo culture supernatant vaccine to be a promising candidate for a new or improved M. hyo vaccine, and a useful alternative to a bacterin.
508 I also consider that a skilled person armed with Okada 2000a would have prepared an M. hyo supernatant, as set out in Okada 2000a, with the expectation that it might well provide a useful alternative to a bacterin, an improved M. hyo vaccine. The M. hyo supernatant prepared by the person skilled in the art on the basis of the Okada 2000a paper would be separated from insoluble cellular material by centrifugation.
509 Professor McVey’s evidence was that if he was interested in combining vaccines to make a combination vaccine, he would expect interference would be likely to occur to some extent, and that it would need to be dealt with. He would conduct standard tests to assess the nature and extent of the interference. The other experts agreed.
510 On the basis of Okada 2000a, Professor McVey, Professor Chase and Dr Nordgren would have made a supernatant and, conscious to reduce interference, would have subjected it to a further technique to remove serum sourced antibodies (and immunocomplexes) with an expectation of success. Professor McVey’s evidence was that if he had been tasked to look at making a combination vaccine with Okada 2000a in hand, he would have wanted to remove anti-PCV-2 antibodies from the M. hyo preparation.
511 Any of the means of removing antibodies from serum by the variety of techniques contemplated by the experts would be used with an expectation that that it may well remove enough antibodies to address the issue of immunological interference from antibodies in the serum used in the growth serum. I do not include the use of Protein A and Protein G among the techniques that may be adopted by the experts as the use of Protein A or Protein G in the reverse manner was not part of the common general knowledge.
512 Professor McVey expected to produce a supernatant which was substantially free of IgG and immunocomplexes. Professor McVey considered that such a supernatant would be less crude than a bacterin. He would expect that it may work but he would need to test it to confirm whether it did. Professor McVey agreed that such tests would be of a routine nature.
513 Professor McVey agreed that if he had been tasked with making a combination vaccine of M. hyo with another antigen, such as PCV-2, as at April 2012, and he was given the Okada 2000a paper to consider as part of that task, he would have recognised that one thing he would need to do would be to address the likelihood of interference occurring between an M. hyo supernatant preparation of the kind described in Okada 2000a and the PCV-2 antigen. Professor McVey agreed that he would use one or more of the techniques to remove any PCV-2 antibodies present in the M. hyo supernatant. Professor McVey’s evidence was that he would expect the application of the techniques would remove enough of the antibody to address the interference, but that he would need to confirm that with testing using standard tools and techniques. Professor Chase and Dr Nordgren agreed.
514 I consider that the invention claimed in claim 1 of the 535 Application is obvious in light of the common general knowledge and the information in Okada 2000a. It is unnecessary to consider Okada 1999 or Okada 2000b or any combination of the Okada papers.
515 Claim 2 claims the composition of claim 1, wherein the soluble portion has been treated with Protein A or Protein G prior to being added to the immunogenic composition.
516 The application of Protein A and Protein G in the “reverse manner” to remove unwanted antibodies and immunocomplexes from a supernatant was not part of the common general knowledge of the person skilled in the art. None of the Okada papers makes any reference to Protein A or Protein G or the application of Protein A or Protein G to the supernatant for any reason.
517 None of the experts had used Protein A or Protein G in the reverse manner prior to the priority date. Professor Chase agreed that the idea of using Protein A and Protein G on the supernatant only occurred to him because he was given the 612 Application at the same time as the Okada papers.
518 I consider that the use of Protein A or Protein G in the reverse manner to treat a supernatant to remove antibodies and immunocomplexes, and thereby overcome the issue of both assay and immunological interference, was inventive. The invention claimed in claim 2 of the 535 Application involves an inventive step.
519 Claim 1 of the 537 Application claims a multivalent immunogenic composition comprising the supernatant of an M. hyo culture and a PCV-2 antigen, wherein the supernatant of the M. hyo culture has been separated from insoluble cellular material by centrifugation, filtration or precipitation and is substantially free of both IgG and immunocomplexes.
520 There were several M. hyo/PCV-2 combination vaccines commercially available at the priority date. The available M. hyo/PCV-2 combination vaccine products were either one dose, two vial vaccines, or one ready-to-use vial which required two doses. There was no one-dose, one-bottle, ready-to-use M. hyo/PCV-2 vaccine available as at the priority date. Such a product was considered desirable.
521 Professor McVey’s evidence was that if he was interested in combining vaccines to make a combination vaccine, he would expect that interference would be likely to occur to some extent, and that it would need to be dealt with. He would conduct standard tests to assess the nature and extent of the interference. The other experts agreed.
522 Professor McVey agreed that if he had been tasked with making a combination vaccine of M. hyo with another antigen, such as PCV-2, as at April 2012, and he was given the Okada 2000a paper to consider as part of that task, he would have recognised that one thing he would need to do would be to address the likelihood of interference occurring between an M. hyo supernatant preparation of the kind described in Okada 2000a and the PCV-2 antigen. Professor McVey agreed that he would use one or more of the techniques to remove any PCV-2 antibodies present in the M. hyo supernatant. Professor McVey’s evidence was that he would expect the application of the techniques would remove enough of the antibody to address the interference, but that he would need to confirm that with testing using standard tools and techniques. Professor Chase and Dr Nordgren agreed. As with the 535 Application discussion above, I do not include the use of Protein A and Protein G among the techniques that may be adopted by the experts as the use of Protein A or Protein G in the reverse manner was not part of the common general knowledge.
523 I consider that the invention claimed in claim 1 of the 537 Application is obvious.
524 For the same reasons as set out above in relation to claim 2 of the 535 Application, I consider that the use of Protein A or Protein G in the reverse manner to treat a supernatant to remove antibodies and immunocomplexes, and thereby overcome the issue of both assay and immunological interference as claimed in claim 2 of the 537 Application, was inventive.
525 Claim 1 of the 540 Application claims a trivalent immunogenic composition comprising the supernatant of an M. hyo culture, a PCV-2 antigen, and a PRRS antigen wherein the supernatant of the M. hyo culture has been separated from insoluble cellular material by centrifugation, filtration or precipitation and is substantially free of both IgG and immunocomplexes.
526 The majority of the commercially available PRRS vaccines at the priority date were lyophilized modified live PRRS vaccines. An example was 3FLEX, which was supplied in three vials and prepared for administration by rehydrating the lyophilised PRRS virus component with the M. hyo (as a bacterin) and PCV-2 components.
527 For the reasons I discussed above in relation to claim 1 of the 535 Application, I consider that, as at the priority date, there was a motivation for the skilled person to look for an improved M. hyo vaccine, or an alternative to a bacterin, because the existing state of the art M. hyo vaccines were bacterins, and bacterins were known to have variable efficacy.
528 Once arriving at an improved M. hyo vaccine, it would be obvious to use it in the existing commercial combination vaccines available at the priority date.
529 Professor McVey gave evidence that if he was going down the route of developing an M. hyo supernatant vaccine in combination with a PCV-2 antigen and a PRRS antigen, he would have an expectation that dealing with a PRRS virus antigen in the same way as the 3FLEX product might well produce a useful result. Professor Chase and Dr Nordgren agreed. Professor Browning agreed that it was a reasonable approach if the M. hyo supernatant route was being pursued.
530 I consider that the invention claimed in claim 1 of the 540 Application is obvious.
531 For the same reasons as set out above in relation to claim 2 of the 535 Application, I consider that the use of Protein A or Protein G in the reverse manner to treat a supernatant to remove antibodies and immunocomplexes, and thereby overcome the issue of both assay and immunological interference as claimed in claim 2 of the 540 Application, was inventive.
532 The support requirement is found in s 40(3) of the Act which provides that the claims must be supported by matter disclosed in the specification.
533 As Burley J noted in Merck, the claim support obligation was introduced as a replacement for the former requirement of fair basis. The purpose of this change is explained in the Explanatory Memorandum to the Intellectual Property Laws Amendment (Raising the Bar) Bill 2011 (Cth). Justice Burley summarised the requirements of s 40(3) in Merck at [545]–[547]:
545 The theme common to each ground, however, reflects what the Minister described in his Second Reading Speech as the cornerstone of the patent system, namely that in exchange for a monopoly on commercialisation, the patentee must tell the public how their idea works. As said in the Explanatory Memorandum, in exchange for the exclusive rights given to the patentee, the patentee must share with the public the information necessary to make and use the invention. This is the essential exchange between inventors and the public which has long been a feature of patent law in Australia: see Lockwood (No 1) at [57].
546 In CSR Building Products Ltd v United States Gypsum Company [2015] APO 72, Dr S D Barker adopted the summary provided by Aldous J in Schering Biotech at 252–253, which has been often followed in the United Kingdom (emphasis added):
…to decide whether the claims are supported by the description it is necessary to ascertain what is the invention which is specified in the claims and then compare that with the invention which has been described in the specification. Thereafter the court’s task is to decide whether the invention in the claims is supported by the description. I do not believe that the mere mention in the specification of features appearing in the claim will necessarily be a sufficient support. The word “support” means more than that and requires the description to be the base which can fairly entitle the patentee to a monopoly of the width claimed.
547 That approach encapsulates broadly the claim support obligation under s 40(3). To it may be added the requirement that the technical contribution to the art must be ascertained. Where it is a product, it is that which must be supported in the sense that the technical contribution to the art disclosed by the specification must justify the breath of the monopoly claimed.
534 This statement of the position has been adopted without criticism in other cases in this Court: Rakman International Pty Ltd v Trafalgar Group Pty Ltd (2022)166 IPR 264 at [120] (per Yates J); Jusand Nominees Pty Ltd v Rattlejack Innovations Pty Ltd (2022) 167 IPR 1 at [370]–[372], [481] (per Rofe J); TCT Group Pty Ltd v Polaris IP Pty Ltd (2022) 170 IPR 313 at [241] (per Burley J); ToolGen at [395] (per Nicholas J).
535 In his discussion of the requirements of s 40(3) in TCT Group, Burley J also referred at [243] to the following passage from the EPO Boards of Appeal decision Fuel Oils/Exxon (T409/91) [1994] OJ EPO 653 at 3.3, which has been often cited, including by Lord Neuberger in Generics (UK) Ltd v H Lundbeck A/S [2009] 2 All ER 955 at [97]:
[there is a] general legal principle that the extent of the patent monopoly, as defined by the claims, should correspond to the technical contribution to the art in order for it to be supported, or justified… This means that the definitions in the claims should essentially correspond to the scope of the invention as disclosed in the description. In other words,... the claims should not extend to subject-matter which, after reading the description, would still not be at the disposal of the person skilled in the art.
(Emphasis in original.)
536 In TCT Group, Burley J clarified at [244] that the phrase “at the disposal of the person skilled in the art” should be understood to mean that the patent must enable the invention to be performed by such a person.
537 In ToolGen, Nicholas J referred at [394] to Burley J’s analysis of the European and UK law of support in Merck and observed that the effect of that analysis is that the “support” requirement requires that the technical contribution to the art disclosed by the specification justify the breadth of the claim, stating at [396]:
The question whether a disclosure in a patent application fairly entitles the patentee to a monopoly of the width claimed calls for an assessment of the patentee’s contribution to the art, which must be weighed against the scope of the patentee’s monopoly as defined by the claims. The monopoly, according to UK and European authorities, must be justified by the technical contribution to the art that arises from the disclosure of the specification.
538 Justice Nicholas noted in ToolGen at [397] that difficulties can arise in applying the test he had set out at [396], including determining the patentee’s technical contribution to the art. The technical contribution may not always correspond with the inventive step. For example, where the invention claimed involves a very significant inventive step and yet, by reason of some deficiency in the disclosure of the specification, the public is deprived of its side of the patent bargain.
539 Justice Nicholas further observed at [400] of ToolGen that the concept of “plausibility” had been developed in the UK authorities as a check on the practice of “speculative claiming”: claiming inventions that are not shown to have a sufficiently plausible or credible justification or support (sometimes also referred to as “armchair” claiming), to ensure that the monopoly claimed was no more extensive than the contribution to the art made by the relevant disclosure.
540 In Warner-Lambert LLC v Generics (UK) Ltd t/a Mylan [2018] UKSC 56, Lord Sumption set out at [37] a number of propositions relevant to the concept of plausibility. Justice Nicholas in ToolGen observed at [402] that whilst some of those propositions were expressed in terms most relevant to claims for methods of treatment, they were not confined to such claims and were relevant to the concept of support more generally. Lord Sumption observed in Warner Lambert that the proposition that a product is efficacious for the treatment of a particular condition is not made plausible by a bare assertion to that effect. He went on to consider what information may render an assertion that a product is efficacious plausible at [37]:
the claimed therapeutic effect may well be rendered plausible by a specification showing that something was worth trying for a reason, ie not just because there was an abstract possibility that it would work but because reasonable scientific grounds were disclosed for expecting that it might well work. The disclosure of those grounds marks the difference between a speculation and a contribution to the art. This is in substance what the Technical Board of Appeal has held in the context of article 56, when addressing the sufficiency of disclosure made in support of claims extending beyond the teaching of the patent
541 As I have construed the claims above:
(a) Claim 1 of the 535 Application claims an immunogenic composition of M. hyo antigens (the M. hyo platform);
(b) Claim 1 of the 537 Application claims a multivalent composition of the M. hyo platform and the PCV-2 antigen; and
(c) Claim 1 of the 540 Application claims a trivalent composition of the M. hyo platform and the PCV-2 antigen and the PRRS antigen.
542 On the proper construction of the claims of the three Applications, the only claims that define an invention that includes, as an essential element, any antigen other than the M. hyo platform, a PCV-2 antigen and/or a PRRS antigen are:
(a) claims 3, 8 and 12 of the 535 Application (and claims dependent thereon); and
(b) claims 8, 13 and 17 of the 537 Application (and claims dependent thereon).
543 Claim 3 of the 535 Application defines an invention which covers a spectrum of immunogenic compositions from the M. hyo platform, with one additional antigen to the M. hyo platform combined with all the antigens in the shopping list (but not beyond those) and all combinations thereof in between. There is no information in the specification which enables the person skilled in the art to make an immunogenic composition consisting of the M. hyo platform with multiple additional antigens other than PCV-2 or PRRS, selected from those listed in claim 3 or combinations of those, and in particular one which elicits a protective effect.
544 Claim 8 of the 535 Application defines an invention which is the immunogenic composition of claim 3, wherein the composition elicits a protective immune response against M. hyo and at least one additional antigen with the further qualification that the composition be administered as a single dose administration.
545 The detailed description of the 535 and 537 Applications states that the immunogenic composition of those Applications includes the soluble portion of the M. hyo whole cell preparation, wherein the IgG free M. hyo soluble preparation can be effectively combined with antigens from other pathogens, such as PCV-2, without analytical or immunological interference between the antigens, and that this makes the M. hyo soluble preparation an effective platform for multivalent vaccines.
546 Each of the specifications of the Applications disclose that the M. hyo soluble preparation can be combined with antigens from other pathogens “without immunological interference between the antigens”. The 535 Application discloses at page 15 that:
the M. hyo soluble preparation can be effectively combined with antigens from other pathogens, such as PCV-2, without immunological interference between the antigens. This makes the M. hyo soluble preparation of this invention an effective platform for multivalent vaccines.
547 See also the disclosure at page 16, lines 4–9, of the 547 Application.
548 It is clear from the detailed description of the 535 and 537 Applications that the immunogenic composition may include at least one additional antigen, and each Application explains that the additional antigen component may be protective against bacteria, viruses, or protozoans that are known to infect pigs. Each Application (including the 540 Application) goes on to explain that examples of such microorganisms include, but are not limited to, a shopping list, including Brachyspira hyodysenteriae (a bacterial pathogen) and porcine respiratory coronavirus, Porcine Cytomegalovirus, Torque Teno Virus and Encephalomyocarditis virus (Brachyspira and the four viruses or the 5 Pathogens).
549 The immunogenic composition of claim 3 of the 535 Application may be comprised only of M. hyo supernatant plus one antigen or it may consist of any number of other antigens (at least one) from the shopping list, including each of the 5 Pathogens, all of the 5 Pathogens, or combinations of the 5 Pathogens (and the others listed).
550 For the purposes of its support and disclosure cases, Boehringer focussed its arguments on a subgroup of the shopping list of antigens, being the 5 Pathogens. Boehringer submits that the Applications do not provide a clear and complete disclosure of any immunogenic composition comprising, or a method involving the administration of, an immunogenic composition which is protective against one or more of the 5 Pathogens.
551 I interpolate from Boehringer’s submissions that it accepts that the Applications do provide sufficient disclosure of any immunogenic composition comprising, or method involving the administration of, an antigen which is protective against M. hyo, PCV-2 or PRRS virus (claim 1 of each of the 535 and 537 Applications and all the claims of the 540 Application).
552 First, Boehringer submits that the experts agreed that, as at the priority date, there were no vaccines available against many swine pathogens, including the 5 Pathogens. Nor were the experts aware of research reporting on antigenic compositions of those pathogens that could elicit an effective immune response. The Applications provide no guidance as to how to identify vaccine/antigens for the 5 Pathogens. Thus, Boehringer submitted that the skilled addressee would first need to identify a vaccine/antigen for the pathogen/s to be targeted.
553 The identification of the vaccine would need to proceed empirically because it was necessary to identify the protective immunogens, the mechanisms and protective immunity and the key factors involved in virulence. The evidence was that each of the four viruses (one of the 5 Pathogens being a bacteria) had its own sets of challenges associated with being able to develop a vaccine.
554 Second, even where an antigen for each of the 5 Pathogens was identified, it would still not be a straightforward exercise to develop a combination vaccine with M. hyo (and PCV-2). A range of problems may arise with the combination. For example, the adjuvants used with one vaccine may have an adverse effect on the other immunogens in the combination. There is also a possibility of antigenic interference. Differences in pH, osmolality and other factors might also affect the antigen. The Applications do not disclose how to avoid the problems that may be encountered when making the combinations contemplated (for example, by claim 3 of the 535 Application). The process to develop a combination vaccine would still involve a substantial research project.
555 In response, Zoetis submits that Boehringer’s support (and disclosure – discussed below) cases fail for at least four reasons. As discussed above, Zoetis’ response to Boehringer’s case depends on the skilled team and whether it is open to that team to “skill up” by collaborating with experts in the particular pathogens of interest. Zoetis submits that:
(a) Boehringer has not adduced evidence to support any finding as to the sufficiency of the disclosure through the eyes of a skilled team (including the additional member/s with specialised knowledge about the additional pathogen) making a combination vaccine against M. hyo and any of the 5 Pathogens (or any other pathogen).
(b) The evidence Boehringer has adduced is directed to the ability of a skilled person to make a vaccine against the 5 Pathogens or other pathogens, and not to the sufficiency of the disclosure in the Applications as to the use of the M. hyo platform (or soluble preparation) in combination, or in conjunction, with additional antigens.
(c) Even if Boehringer had adduced evidence from an expert in one of the 5 Pathogens to demonstrate a lack of an “available input” for a claim, that would not support a finding of insufficient disclosure. Asking whether the particular input is enabled is the wrong question. The question is whether the invention as claimed — which in every case makes use of the M. hyo platform — is enabled. In the case of at least one of the antigen claims, it is the combination of the M. hyo soluble preparation with an antigen protective against at least one of the claimed additional antigens that must be enabled, not the protective antigen itself, which is not the invention.
(d) The evidence does not give the skilled reader any reason to be sceptical about whether the M. hyo platform could be combined with an antigen protective against any other pathogen. The experts agreed that it was “likely” that they could be combined without interference, and any “scepticism” appears to be the result of a lack of market demand for viruses against some of the less prevalent pathogens. That lack of demand has nothing to do with the M. hyo platform or the general principle disclosed in the Applications.
556 The questions to the experts in the joint session, and their responses and ensuing cross- examination, focussed on the 5 Pathogens.
557 As at the priority date, commercial vaccines against the four viral pathogens of the 5 Pathogens were not available.
558 The experts agreed in the JER that a skilled vaccine development team would not be able to make specific immunogenic compositions comprising the M. hyo supernatant and antigen(s) protective against one or more of the four viruses using only the information in the 535 Application. The experts further agreed that it may have been possible to prepare an immunogenic composition comprising the M. hyo supernatant and antigen(s) protective against Brachyspira. However, they noted that it was not clear whether an immunogenic composition (particularly one that could elicit a protective response) comprising M. hyo supernatant and antigen(s) protective against Brachyspira could be developed.
559 The experts agreed in the JER that a skilled animal vaccine development team would not be able to make specific immunogenic compositions comprising M. hyo supernatant and PCV-2 antigen and antigen(s) protective against one or more of the four viruses using only the information in the 537 Application. The experts further agreed that it may have been possible to prepare an immunogenic composition comprising M. hyo supernatant and PCV-2 and antigen(s) protective against Brachyspira. However, it is not clear whether an immunogenic composition comprising M. hyo supernatant and PCV-2 and antigen(s) protective against Brachyspira could be developed.
560 Each of the experts agreed that they gave their answers above by reference to what they considered to be generally known and accepted in the field of animal vaccine development as at the priority date. Professor Browning added to his answer by noting that, in his view, the skilled vaccine development team would probably include specialists in particular pathogens, rather than being a “group of generalists”.
561 Professor McVey explained, and each expert agreed, that they were unable to determine whether a composition containing M. hyo and Brachyspira could be developed, because:
With brachyspira there was some information relative to antigens. So there was some immunologic data available for evaluation. It was not clear or known by me as to how that could be formulated into a vaccine.
562 In cross examination, junior counsel for Boehringer, Ms Cunliffe, asked the experts questions as to porcine respiratory coronavirus, an example taken from the 5 Pathogens.
563 Professor McVey and Professor Browning agreed that, as at the priority date, a vaccine for porcine respiratory coronavirus was desirable. As at that date, none of the experts were aware of any experimental vaccines for porcine respiratory coronavirus, or that any antigens for porcine respiratory coronavirus had been identified.
564 Even though viruses closely related to the four viruses had been more carefully studied than the four viruses as at the priority date, the experts gave evidence that developing a vaccine for each of the four viruses has its own set of challenges. The challenges to the development of a vaccine included the following:
(a) The choice of vaccine type: a bacterin, live attenuated, killed virus, subunit, gene deleted whole virus;
(b) Many pathogens have high antigenic diversity and may be capable of evading or misdirecting the host immune response;
(c) Method of inactivating the virus as some forms of inactivation may negatively affect the antigen and the immune response;
(d) Producing the antigen in sufficient quantities;
(e) The challenge model;
(f) Dose finding studies to quantify amount of antigen required to elicit a protective response (in the absence of any comparator vaccine);
(g) Selection of an appropriate adjuvant (other than for attenuated virus vaccine);
(h) In vivo testing looking at the acute immune response of the target animal in disease-free herds;
(i) Longevity of protection (some exceptions — diseases which occur only immediately post weaning);
(j) Immunological interference with other antigens if a combination (especially if different kinds — ie live attenuated or purified protein);
(k) pH and osmolality; and
(l) Safety and efficacy.
565 The experts’ evidence was that vaccine development was not predictable and that trial and error experiments are required because it is not possible to predict that an experimental composition which develops an antibody response in the target animal will be safe and efficacious. The source of the antigen, the formulation and type of adjuvant, the presence of additional antigens and the age of the intended target animal are significant variables that must be evaluated in order to produce a safe and efficacious vaccine.
566 Professor McVey’s evidence was that where a vaccine (or antigen) had not been identified, the identification of the vaccine would need to proceed empirically, because it was necessary to identify the protective immunogens, the mechanisms of protective immunity and the key factors involved in virulence. Although Professor Browning initially suggested that there were close parallels for each of the viruses which meant that the skilled team would not necessarily follow a purely empirical path, he later confirmed that this evidence did not cause him to change his evidence in the JER.
567 Professor McVey’s evidence was that many pathogens have high antigenic diversity and may be capable of evading and misdirecting the host immune response. Dr Nordgren and Professor Chase agreed that this could occur.
568 The experts agreed that, even when an antigen for each of the four viruses was identified, challenges remained. It would be necessary to consider what type of vaccine to develop: for example, a live attenuated vaccine, a bacterin, a fractionated bacterin, a killed virus vaccine or a subunit vaccine. For more complex viruses, of which the antigenic components have not been properly elucidated or understood, it may not be possible to use a subunit vaccine.
569 In the case of an inactivated virus vaccine, it would also be necessary to identify the appropriate means of inactivation of the virus. The experts agreed that some types of inactivation might negatively affect the antigen and the immune response.
570 It would be also be necessary to develop processing parameters by which the antigen for the vaccine could be produced. Each of the four viruses was difficult to grow. Dr Nordgren gave evidence of his involvement in an attempt to develop a vaccine for porcine respiratory coronavirus. The project did not ultimately proceed as the team was unable to grow the virus to sufficient titres that a typical inactivated vaccine would be expected to work.
571 I accept that the development of an antigen for each of the 5 Pathogens (and the others listed)is not part of the claimed invention of the Applications. However, the extensive nature of the process of work involved to identify and produce an appropriate vaccine for each antigen, and the unpredictable nature of the vaccine ultimately produced, is highly relevant to whether there is support in the specifications for the claimed combination of such antigens with the M. hyo platform (and PCV-2 and/or PRRS).
572 Where there is a combination of antigens, there is the prospect of interference between those antigens leading to a poor immune response in respect of the “overpowered” antigen. Antigen interference (also known as efficacy interference or vaccine interference) can occur in a number of ways when antigenic compositions are combined. A dominant antigen could distract the immune system from raising a response to another antigen of interest in the multivalent vaccine.
573 Professor Browning’s evidence was that the chance of antigenic interference was less likely if the antigens involved were purified proteins, but that it would be “quite a different matter” if the antigen was a live attenuated virus.
574 The experts agreed that it was not possible to say definitively ahead of time whether there would be interference between previously uncombined antigens and that it would be necessary to test the combination of antigens to determine whether there was interference.
575 The experts agreed that there are some antigens that cannot be stably combined at all. Professor Browning gave as an example the case where one live attenuated virus was immunosuppressive.
576 In most cases (other than RNA and attenuated virus vaccines), it is necessary to select an adjuvant to use in the vaccine. Professor McVey’s evidence was that not all adjuvants work with all antigens and that it may require a process of trial and error to find the appropriate adjuvant for a given antigen. For example, some adjuvants may be viricidal, which is an issue for modified live virus vaccines.
577 Further issues may arise with a combination of antigens. The adjuvant used with one vaccine may have an adverse effect on the additional antigen/s. There could also be incompatibility between antigen emulsions or components. Differences in pH, osmolality and other factors might also affect the antigen/s in the combination.
578 It would be necessary to do in vivo tests on the combined vaccine to assess immunogenicity and ensure a robust and sustained antibody response or other immune response and to assess the safety and efficacy of the combination.
579 It would then be necessary to determine the correct dosage, which would involve iterative dose finding studies. Professor McVey’s evidence was that the task of dose finding studies would be complex, because there would be no comparator products. Dr Nordgren and Professor Chase explained that it would be challenging to determine the dose for each of the four viruses because there was a poorly understood challenge model.
580 Once dose had been determined, it would be necessary to undertake in vivo tests on animals in controlled conditions to determine safety and efficacy of the vaccine. These would usually involve looking at the acute immune response of the animal over weeks, and comparing the increase in antibody titres in the vaccinated animals against control animals. It would be desirable to also assess the longevity of immunity or protection provided by the vaccine. The experts agreed that the testing is likely to involve the use of expensive trials using HPCD pigs, less expensive (but difficult to source) disease free pig herds or groups large enough to obtain results which have statistical significance.
581 In light of these matters, the process of developing a vaccine for a single virus in combination with the M. hyo platform, even when an antigen had been identified and developed, would involve a substantial research project, with empirical work involving trial and error.
582 Professor McVey gave evidence that the Applications gave no assistance with that task in relation to the porcine respiratory coronavirus.
583 Professor Browning’s, Dr Nordgren’s and Professor Chase’s evidence was that the specifications give details of an adjuvant that could be used as a starting point. However, it would be necessary to test that adjuvant for each of the additional antigens listed in claim 3, and the combinations thereof. The experts agreed that the specifications provided no assistance with dose, type of vaccine or developing a challenge model.
Consideration
584 Zoetis submits that all the experts were generalists (aside from Professor Browning who had expertise with respect to M. hyo) who had no specialist knowledge about the 5 Pathogens or any research work done in respect of them as at the priority date.
585 Boehringer rejected Zoetis’ contention that the skilled team for the purpose of s 40(2)(a) includes an expert in each of the pathogens in relation to which a composition may be developed (and therefore, presumably, that the common general knowledge includes their specialised knowledge). There was no evidence as to the nature of this additional knowledge, and the question of whether it would have made the task any easier.
586 This “skilling up” approach to the person skilled in the art team for the purposes of considering the s 40 requirements should be rejected.
587 Justice Jagot in Gilead discussed this issue in relation to the old test for sufficiency. Her Honour held at [209] that the need for inferences about common general knowledge to be based on the evidence cannot be avoided by artificially constructing a skilled team to include a person with highly specific and unusual knowledge gained through literature searches or idiosyncratic experience. The common general knowledge is a general body of knowledge known and used by all those in the field. At [219] her Honour rejected the view that the common general knowledge included specialist experience and expertise about the relevant field (see also [607]–[612]). Her Honour’s reasoning was upheld in Idenix Pharmaceuticals LLC v Gilead Sciences Pty Ltd (2017) 134 IPR 1 at [192]–[200] (per Nicholas, Beach and Burley JJ).
588 Furthermore, even if it were possible to draw additional specialists into the team, specialists in a particular virus or antigen would not be a specialist in how that antigen will interact with any of the other antigens in the list, or combinations of them or with various adjuvants.
589 As I have found, claim 3 of the 535 Application is not limited to an immunogenic composition consisting of the M. hyo platform and one additional antigen from the list. It covers the spectrum from the one additional antigen to the M. hyo platform, plus all those antigens in the list, and all combinations thereof, with and without adjuvants. It also includes immunogenic compositions which elicit a protective effect – ie vaccines.
590 The 535 and 537 Applications do not disclose how to ascertain and identify the antigens for each of the micro-organisms listed in the claim shopping list. However, I accept that the ascertainment of a particular antigen against one of the microorganisms listed in the shopping list is not part of the invention claimed. Claim 3 of the 535 Application claims the immunogenic composition of the M. hyo combined with other antigens, once those antigens are identified.
591 However, the specification does not teach the reader how to make an immunogenic composition (with or without a protective effect) for combinations of the M. hyo platform with multiple antigens from the list without immunological interference between the antigens. I do not consider that it is a straightforward exercise to combine the additional antigens with the M. hyo platform, to produce an immunogenic composition including a vaccine. A research project would be required.
592 As the experts confirmed, the 535 and 537 Applications would give them no assistance with developing a vaccine against porcine respiratory coronavirus (save, perhaps, for the choice of adjuvants). The same is equally true of each of the viruses, and Brachyspira.
593 The specifications do not provide an enabling disclosure of all the things that fall within the scope of the claims that claim the M. hyo platform with a combination of antigens from the shopping list.
594 In light of this evidence, the specification does not provide an enabling disclosure of all the things that fall within the scope of claim 3 of the 535 Application (or claim 8 of the 537 Application). A research project would be involved in developing a vaccine for any of the viruses or Brachyspira, and there would be very considerable work in determining whether and how a vaccine against the viruses or Brachyspira could be combined with the compositions disclosed in the Applications.
595 It is not plausible that the invention can be worked across the full scope of the claims on the basis of the disclosure provided in the specifications. In light of the difficulties in developing vaccines for some of the viruses, and the difficulties in combining vaccines, the claimed monopoly is more extensive than the technical contribution to the art disclosed in the specifications. The technical contribution disclosed is the removal of immunological interference by removal of IgG and immunocomplexes comprised of antigen bound to immunoglobulin. It does not extend beyond that to removal of antigenic interference or adverse interaction with adjuvants.
596 In any event, the invention could not be performed across the full scope of the claims without undue experimentation. There is a research project involved in developing a vaccine for each virus, and very considerable work involved in combining them with the compositions disclosed in each Application.
597 I consider that the breadth of claim 3 of the 535 Application exceeds the technical contribution of the specification because the claim covers ways of achieving a result, being the spectrum of possible antigen combinations, which owe nothing to the disclosure of the specification.
598 I consider that the breadth of claim 8 of the 537 Application exceeds the technical contribution of the specification because the claim covers ways of achieving a result, being the spectrum of possible antigen combinations, which owe nothing to the disclosure of the specification.
599 Section 40(2)(a) of the Act provides that the complete specification “must disclose the invention in a manner which is clear enough and complete enough for the invention to be performed by a person skilled in the relevant art”.
600 The Raising the Bar Bill explains at pp 46-47 that this amendment was intended to align the disclosure requirement with that applying in other jurisdictions so that sufficient information must be provided to enable “the whole width of the claimed invention” to be performed by the skilled person without undue burden or further invention.
601 At [143]–[146] of Cytec Industries Inc v Nalco Company (2021) 162 IPR 202, Burley J endorsed the three steps set out by Dr S D Barker in CSR Building Products Ltd v United States Gypsum Co [2015] APO 72 at [95] and the two further questions from Re Evolva SA (2017) 133 IPR 147 at [45], and observed that the steps represented a significant shift from the previous law as set out by the High Court in Kimberley-Clark at [25] (per Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ). In Cytec at [146], Burley J observed that the steps set out in the CSR and Evolva decisions of the Commissioner was the correct approach for s 40(2)(a).
602 The five steps for determining compliance with s 40(2)(a) are:
(a) construe the claims to identify the invention and determine x as claimed;
(b) construe the description to determine what it discloses to the person skilled in the art;
(c) decide whether the specification provides an enabling disclosure of all the things that fall within the scope of the claims;
(d) consider whether is it plausible that the invention can be worked out across the full scope of the claims; and
(e) consider if the invention can be performed by the person skilled in the art across the full scope of the claims without undue experimentation.
603 Zoetis notes that it is the invention claimed, not what might infringe the claim (or what the claim might "catch" or “encompass”), that is the focus of the s 40 obligations (see s 40(2)(b) and s 40(3)). Section 40(4), which provides that the claim or claims must relate to one invention only, does not obviate the need to assess disclosure or support by reference to the invention as claimed in each claim. Sufficiency of disclosure is to be assessed against the invention as claimed: ToolGen at [175] (per Nicholas J).
604 The skilled addressee may supplement the information contained in the specification with their common general knowledge as at the priority date. Zoetis submits that different aspects of the hypothetical skilled addressee may be different for different claims or different grounds of invalidity. In the present case, this is of particular significance for the “additional antigen” claims (such as claim 3 of the 535 Application) where, as discussed earlier, Zoetis seeks to supplement the skilled team via collaboration with academics or specialists with a deep understanding of each particular pathogen or pathogens under consideration to provide a “skilled up” skilled addressee team.
605 Zoetis submits that Boehringer bears the onus to satisfy the Court that the specifications do not satisfy s 40(2)(a) in respect of each claim. Zoetis submits that it is for Boehringer to prove, via expert evidence, that the skilled addressee is unable to perform the invention (of each claim) across the full scope of the claim without further experimentation. Zoetis contends that Boehringer has failed in that task as it has led no evidence from academics or specialists with a deep understanding of the microorganisms/pathogens listed in, for example, claim 3 of the 535 Application.
606 Boehringer’s alleged “failure” to lead evidence from such specialists follows from its rejection of Zoetis’ “skilled up” skilled team as the appropriate hypothetical skilled addressee for the purposes of assessing compliance with s 40(2)(a). Boehringer has constructed its support and disclosure cases on the basis of the skilled addressee team discussed earlier, without collaboration with specialist academics and scientists.
607 As discussed above, I do not consider that the composition of the skilled team expands to include specialists in each particular antigen under consideration, nor would such specialists be specialised in how their antigen interacts with other antigens when put together in a combination of the kind claimed in claim 3 of the 535 Application (or claim 8 of the 537 Application).
608 Boehringer contends that the Applications do not provide a clear and complete disclosure of any immunogenic composition comprising, or method involving the administration of, an antigen which is protective against a microorganism other than M. hyo, PCV-2 or PRRS virus. Relatedly, the Applications do not provide a clear and complete disclosure of an immunogenic composition comprising, or a method involving the administration of, an antigen or antigens which are protective against one or more of the 5 Pathogens.
609 Zoetis submits, and I accept, that it is implicit from Boehringer’s case that the specifications of the Applications disclose the invention, as claimed in each claim, in a manner clear enough and complete enough for the skilled addressee to perform the invention with antigens protective against M. hyo, PCV-2 and PRRS: claim 1 of each of the 535, 537 and 540 Applications. The Court must therefore proceed on the basis that there is an enabling disclosure of, and support for, combinations with PCV-2 and/or PRRS across the breadth of those claims.
Scope of the invention as claimed
610 All of the claims of the 535 and 537 Applications include as an essential integer an immunogenic composition. That immunogenic composition is not limited to a monovalent composition (for the 535 Application) or a bivalent composition (in the case of the 537 Application). All of the claims of the 535 Application and the 537 Application include within their scope an immunogenic composition comprising an additional antigen protective against Brachyspira and the viruses. Such a composition is specifically claimed by claims 3, 8 and 12 of the 535 Application and claims 8, 13 and 17 of the 537 Application.
611 Boehringer submits the following:
The descriptions do not disclose immunogenic compositions with antigens against Brachyspira and the viruses. As the experts confirmed, the Applications would give them no assistance with developing a vaccine against porcine respiratory coronavirus (save, perhaps, for the choice of adjuvants). The same is equally true of each of the viruses, and Brachyspira.
The specifications do not provide an enabling disclosure of all the things that fall within the scope of the claims. In light of this evidence, the specification does not provide an enabling disclosure of all the things that fall within the scope of the claims. A research project would be involved in developing a vaccine for any of the viruses or Brachyspira, and there would be very considerable work in determining whether and how a vaccine against the viruses or Brachyspira could be combined with the compositions disclosed in the Applications.
It is not plausible that the invention can be worked across the full scope of the claims. In light of the difficulties in developing vaccines for some of the viruses, and the difficulties in combining vaccines, it is not plausible that the invention can be worked across the full scope of the claims. The experts unanimously considered that although the invention might be able to be worked in respect of immunogenic compositions containing antigens to the viruses, it is possible that it would not work.
The invention cannot be performed across the full scope of the claims without undue experimentation. In any event, the invention could not be performed across the full scope of the claims without undue experimentation. There is a research project involved in developing a vaccine for each virus, and very considerable work involved in combining them with the compositions disclosed in each Application.
(Emphasis in original.)
612 I do not consider that ascertaining the relevant antigen to the 5 Pathogens (and challenges inherent in that process) and the other antigens listed is part of the invention disclosed in the specifications. The invention disclosed in the specifications is the combination of the M. hyo platform with the “new” antigen with, and in combination with, other antigens, to produce an immunogenic composition. This term is sufficiently broad to include a vaccine, in which immunogenic interference with serum antibodies is minimised by removal of the IgG in the production of the M. hyo platform.
613 The specifications provide an enabling disclosure of, and support for, claims to an immunogenic composition consisting of the M. hyo platform alone, or in combination with PCV-2 and/or PRRS. Although, as discussed below, the specification does not provide any absolute concentration of antigens, I consider that the person skilled in the art would, through a program of routine experimentation, be able to perform the invention across claims 1 and 2 of the 535 Application, claims 1–7 of the 537 Application and the claims of the 540 Application.
614 However, I do not consider that the specifications of the 535 and 537 Applications provide an enabling disclosure of the claims which include an immunogenic composition consisting of the M. hyo platform in combination with the 5 Pathogens individually, combinations of the 5 Pathogens, all of the 5 Pathogens or in combination with any, or all, of the other antigens listed in claim 3 of the 535 Application and claim 8 of the 537 Application.
615 Whilst the invention results in the removal of serum IgG so that interference with serum antibodies is minimised in respect of the new antigen/s, there remains the issue of antigenic interference between the antigens in the combination, and also how the new antigen/s will react with particular adjuvants.
616 In light of these matters, the process of developing a vaccine for a combination of the M. hyo platform with a 5 Pathogen virus or viruses, even when an antigen had been developed for that virus or viruses, would have involved undue burden, requiring a substantial research project.
617 Therefore, neither of the 535 or 537 Applications provide an enabling disclosure for the claims to combinations. Further, it is not plausible that the invention could be worked across the scope of the claims and, working the invention across the scope of the claims would require undue burden, requiring a significant research project. Claims 3, 8 and 12 of the 535 Application and claims 8, 13 and 17 of the 537 Applications fail to comply with s 40(2)(a) on that basis.
Principles
618 Section 40(2)(aa) of the Act provides that the specification must disclose the best method known to the applicant of performing the invention.
619 In the context of the pre-RTB Act best method requirement in 40(2)(a), the “invention” is the embodiment which is described and around which the claims are drawn (cf the invention so far as claimed in any claim): see Kimberly-Clark at [21] (per Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ); Sandvik Intellectual Property AB v Quarry Mining & Construction Equipment Pty Ltd (2017) 348 ALR 156 at [94] (per Greenwood, Rares and Moshinsky JJ).
620 The Full Court in Sandvik reviewed the authorities relating to best method, paying close attention in particular to the Full Court’s decision in Les Laboratoires Servier v Apotex Pty Ltd (2016) 117 IPR 415, which also reviewed the authorities from [96] to [114] (per Bennett, Besanko and Beach JJ).
621 The Full Court stated in Sandvik at [115] that the following key principles may be stated on the basis of the authorities:
(a) The purpose of the requirement in s 40(2)(a) to disclose the best method known to the applicant of performing the invention is to allow the public the full benefit of the invention when the monopoly expires: Servier at [108].
(b) Although a patentee might not be explicitly required to act in good faith, principles of good faith underlie the best method requirement: Servier at [108].
(c) The nature and extent of the disclosure required to satisfy the best method requirement will depend on the nature of the invention itself: Firebelt at [52]–[53]; Servier at [108].
(d) The key to understanding the obligation of the patentee is to understand that the section is directed to the method of performance of the invention. The monopoly is circumscribed by the claims, but the nature of the invention is as described in the whole of the specification: Servier at [124].
(e) The requirement to describe the best method of performing the invention is ordinarily satisfied by including in the specification a detailed description of one or more preferred embodiments of the invention: Firebelt at [53]; Servier at [104]. See also Blanco White (5th ed) at [4–516]; Bodkin C, Patent Law in Australia (2nd ed, Thomson Reuters, 2014) at [5280].
(Emphasis added.)
622 To these principles I would also add that where a specification describes more than one embodiment of the invention, it is not necessary for the specification to state which of the embodiments is the “best”; Sandvik at [113] (per Greenwood, Rares and Moshinsky JJ) citing Alphapharm at [210] (per Lehane J).
623 The circumstance that something is a requirement for the best method of performing an invention does not make it necessarily a requirement for all claims: Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (2004) 217 CLR 274 at [69] (per Gleeson CJ, McHugh, Gummow, Hayne and Heydon JJ); Sandvik at [95] (per Greenwood, Rares and Moshinsky JJ).
624 The requirements of pre-RTB Act sufficiency and best method are separate and different requirements: Servier at [134] (per Bennett, Besanko and Beach JJ). Post-RTB Act, there can be no doubt: the requirements are contained in separate subsections of s 40 and are clearly separate. A claimed invention may be sufficiently enabled so as to satisfy the requirement of sufficiency (disclosure and support) but still fail best method.
625 Both the Full Courts in Servier and Sandvik considered that principles of good faith underlie the best method obligation. At [64], the Servier Full Court explained the proposition underlying the best method requirement in s 40(2)(a) as follows:
The proposition underlying a separate and additional obligation on the part of the inventor filing a complete specification is that where an inventor in fact knows of a method at the time of filing the complete patent application, which has taken the methodology to a more satisfactory stage or provides more certainty so that the public may more quickly and easily utilise the invention for which a monopoly is granted, the inventor is under an obligation to disclose that method.
(Emphasis added.)
626 The obligation for the patentee to disclose the best method known to them is longstanding. As their Honours explained in Servier at [72], the English Court of Appeal said in Edison and Swan United Electric Light Company v Holland (1889) 6 RPC 243 at 279 that the patentee must state “in what manner the patented invention is to be performed” so that others know how practically to avail themselves of the invention when the patent is expired; “how they are to do what is necessary to carry out the new invention”, that is, how the invention is to be performed.
627 Both Full Courts referred to the fact that the English Court of Appeal in Vidal Dyes Syndicate Ltd v Levinstein Ltd (1912) 29 RPC 245 differentiated between the two duties incumbent upon a patentee. First, the obligation by which the patentee must “particularly describe and ascertain the nature of the invention” and, secondly, that by which the patentee must “particularly describe and ascertain ‘in what manner the same is to be performed’”. In relation to the second duty, the Court of Appeal said (at 269):
It is settled law that a patentee must act towards the public uberrima fide, and must give the best information in his power as to how to carry out the invention. He is therefore bound to tell the public all the steps that can advantageously be taken in carrying out the invention. But he is not limited to claiming only the best way of carrying it out.
628 At [94], the Servier Full Court referred to Firebelt Pty Ltd v Brambles Australia (2000) 51 IPR 531, noting that Spender, Drummond and Mansfield JJ in that case had explained (at [48]), in relation to the requirement to disclose the best method of performing the invention, that:
[This] requirement is to ensure good faith on the part of the patentee, and to protect the public against a patentee who deliberately keeps to himself something novel and not previously published which he knows of or has found out gives the best results, with a view to getting the benefit of monopoly without giving to the public the corresponding consideration of knowledge of the best method of performing the invention.
629 This passage was also quoted by the Full Court in Sandvik at [105].
630 The Full Courts in Servier (at [98]–[100]) and Sandvik (at [106]) referred to the observation of French and Lindgren JJ (Crennan J agreeing) in Pfizer Overseas Pharmaceuticals v Eli Lilly & Co (2005) 68 IPR 1 at [374]:
The requirement that an applicant disclose the best method known to him or her of performing the invention safeguards against an applicant’s holding back with a view to getting the benefit of a patent monopoly without conferring on the public the full consideration for the grant of that monopoly.
631 The Servier Full Court at [101] endorsed the observation of Yates J in DSI Australia (Holdings) Pty Ltd v Garford Pty Ltd (2013) 100 IPR 19 at [327] that the patentee’s obligations under s 40(2)(a) do not extend to describing or cautioning against something that will not work.
632 The Servier Full Court also referred at [105] to a passage from Bodkin C, Patent Law in Australia (2nd ed, Thomson Reuters, 2014), which was cited by the Sandvik Full Court at [107]:
In Patent Law in Australia Dr Bodkin says (at [5270]) that the requirement to describe the best method known to the Patentee:
is to supplement the necessity for a full description by requiring the patentee to disclose additional information which, if not available to potential users of the invention, could place the patentee in a stronger competitive position even though no patent protection existed [ie when the patent ceases to be in force]. It is included to help ensure good faith on behalf of the patentee.
633 At [106], the Servier Full Court cited with approval the author’s opinion that the requirement is to disclose the most effective means of carrying out the invention known to the patentee at the relevant time — the filing date.
634 In discussing the nature of the disclosure required, the Servier Full Court stated at [134]:
Perindopril arginine is generally a more stable product than perindopril erbumine and the claim is not to a specific form of the arginine salt. If Servier knew of a method that provides a form of the salt with the characteristics exemplified in the Patent, which characteristics provided the stated advantages of the invention over the prior art, it was incumbent on it to provide that method. This would relieve the skilled worker from making the choices within those necessarily made or available in a classical salification. The disclosure of the method known to Servier would not only have relieved the skilled addressee of confronting blind alleys and pitfalls which may not be uncommon in a general sense but also, and importantly, would tell the skilled addressee the methodology to achieve the form that obtains the result which constitutes the invention, that is increased stability and storage length. While claim 1 does not refer to any particular form of perindopril arginine, crystalline or otherwise, if Servier had a method that produced a product that was at least sufficiently crystalline or in a sufficiently good form so that it could be used in the API for the tablets used in the stability study described in the specification, that is precisely what should have been disclosed.
(Emphasis of the Sandvik Full Court at [112].)
635 There was no allegation of lack of (pre-RTB Act) sufficiency in Servier. The Full Court observed at [135] that the mere fact that a specification described a method that which conveyed sufficient information to a skilled addressee to enable them to work the invention does not necessarily satisfy the patentee’s additional obligation to describe the best method. The Full Court held (and the Full Court in Sandvik emphasised at [112]) that the patentee has an obligation to include aspects of the method of manufacture that are material to the advantages it is claimed the invention brings.
636 The disclosure of the method must be such as to relieve the skilled addressee seeking to implement the invention, including achieving the advantages associated with the invention, as at the expiry of the patent from being faced with undertaking a potentially extensive course of trial and error experiments to overcome the blind alleys and pitfalls which the patentee had already navigated or overcome as at the filing date.
Boehringer’s pleaded best method case
637 Boehringer was given leave fairly late in the pre-trial process to amend its grounds of appeal to include a narrow best method case. This amendment did not result in any additional evidence being filed by either party.
638 Zoetis was relentless in holding Boehringer to its narrow best method case, and not allowing it to stray from that case. Zoetis was careful to monitor throughout the trial when it considered Boehringer stepped outside the pleaded case. This continued in its closing written and oral submissions.
639 Boehringer contends that the specification of the 535 Application does not disclose the best method known to Zoetis of performing the invention claimed in each of claims 3, 7–8 and 11 of the 535 Application as at the filing date of the application, being 3 April 2013. Boehringer relied upon the following particulars to paragraph 8 in its second further amended notice of appeal:
(a) At 3 April 2013, the best method known to the Respondent of formulating a composition comprising the supernatant of an M. hyo culture involved using the concentration of M. hyo p46 antigen contained in one of the following experimental vaccines, each of which is relied upon in the alternative: L100211J (T10 in Examples 3-6); L0411RK08 (T02 in Examples 7-8); L0711RK13 and L0711RK14 (T07 and T08 in Example 9). That method is not disclosed in the specification, because the specification describes the M. hyo p46 antigen content of the experimental vaccines in relative units of measurement ("reference units" and "relative potency"), and the absolute concentration of M. hyo p46 antigen cannot be derived, or derived without undue experimentation, from the information in the specification. In particular, the Appellant relies on the matters set out in paragraphs [115]-[132], [134]-[139], [142], [144]-[146], [148]-[151], [153]-[155], [157], [159], [161], [163], [165], [167], [169], [171] and [173]-[176] of the Second Affidavit of Robert Martin Nordgren dated 20 December 2021.
(b) Further and in the alternative, at 3 April 2013, the best method known to the Respondent of formulating a composition comprising the supernatant of an M. hyo culture and a PCV-2 antigen involved using the concentration of PCV-2 antigen contained in one of the following experimental vaccines, each of which is relied upon in the alternative: L0411RK08 (T02 in Examples 7-8); L0711RK13 and L0711RK14 (T07 and T08 in Example 9). That method is not disclosed in the specification, because the specification describes the PCV-2 antigen content in relative units of measurement ("relative potency"), and the absolute concentration of PCV-2 antigen cannot be derived, or derived without undue experimentation, from the information in the specification. In particular, the Appellant relies on the matters set out in paragraphs [122]-[125], [134]-[139], [142], [144]-[146], [148]-[151], [153]-[155], [157], [159], [161], [163], [165], [167], [169], [171] and [176] of the Second Affidavit of Robert Martin Nordgren dated 20 December 2021.
…
(d) Further and in the alternative, at 3 April 2013, the best method known to the Respondent of making and administering an immunogenic composition comprising the supernatant of an M. hyo culture was the formulation and administration of one of the following experimental vaccines, each of which is relied upon in the alternative: L1211RK09 and L1211RK15 (T02 and T03 in Examples 12-13 of the 537 Application). That method is not disclosed in the specification, because the specification omits some or all of the information in Examples 12 and 13 of the 537 Application concerning the M. hyo and PCV-2 antigen content of the experimental vaccines tested in those Examples.
(e) Further and in the alternative, at 3 April 2013, the best method known to the Respondent of making and administering an immunogenic composition comprising the supernatant of an M. hyo culture was the formulation and administration of one of the following experimental vaccines referred to in the specification of the 540 Application, each of which is relied upon in the alternative: L0912RK08 and L0912RK09 (T02 and T03 in Example 14 of the 540 Application); L0712RK33 (T03 in Example 15 of the 540 Application); L0912RK12 (T02, T03 and T04 of Example 16 of the 540 Application). That method is not disclosed in the specification, because the specification omits some or all of the information in Examples 14, 15 and 16 of the 540 Application concerning the M. hyo and PCV-2 antigen content of the experimental vaccines tested in those Examples.
640 A similar best method case was alleged in respect of claims 1–24 of the 537 Application which repeated the particulars at paragraphs 8(c) and (e) and added:
(a) Paragraphs 8(a) and 8(b) above are repeated in relation to the experimental vaccines described therein and also in relation to L1211RK09 and L1211RK15 (T02 and T03 in Examples 12-13 of the 537 Application).
641 Boehringer also contended that the 540 Application did not disclose the best method known to Zoetis of performing the invention claimed in each of claims 1–25 of the 540 Application as at the filing date of the application, 3 April 2013. Particulars 8(c) and (d) were repeated and the following added:
(a) Paragraphs 8(a) and 8(b) above are repeated in relation to the experimental vaccines described therein and also in relation to L0912RK08 and L0912RK09 (T02 and T03 in Example 14 of the 540 Application); L0712RK33 (T03 in Example 15 of the 540 Application); L0912RK12 (T02, T03 and T04 of Example 16 of the 540 Application).
642 Boehringer contends that it has identified in the pleading of its best method case 10 experimental vaccines or investigational veterinary products (IVPs), each of which it says, in the alternative, reflect the best method known to Zoetis for performing the invention in the Applications.
643 In each instance, the M. hyo or M. hyo and PCV-2 antigen concentration or content of the particular IVP is said to form part of that best method. The particulars identify, in each instance, the reason why that best method is not disclosed: this is either because relative units are used for the antigen content or concentration, or because the examples in which the IVP is described are not present at all in the particular Application.
644 Zoetis contends that Boehringer’s pleaded case distils down to two limbs.
645 First, the alleged inability to derive the “absolute” concentrations of M. hyo p46 antigen or PCV-2 antigen of ten specified experimental vaccines (the 10 IVPs) mentioned in the Applications from the “relative” concentrations or to do so without undue burden.
646 Secondly, the alleged omission from the Applications of “some or all of the information in [Examples disclosed in one or two of the Applications] concerning the M. hyo and PCV-2 antigen content of [specific IVPs] tested in those Examples”.
647 Zoetis says that the pleading of the second limb is clear in indicating that the deficiency alleged is the non-inclusion of information concerning antigen content that is included in one or other of the 537 and 540 Applications but not included in the other Applications. That is, it is a case confined to omission of information already in one or two of the Applications regarding the M. hyo and PCV-2 antigen content of the relevant IVPs.
Boehringer’s submissions
648 Boehringer submits the following:
(a) The Applications are directed to an immunogenic composition having the features described, which is suitable for use in a vaccine. The Applications describe preferred forms of such a composition by setting out certain details in the examples of specific IVPs made by Zoetis. On the face of the Applications, these IVPs are preferred embodiments of the invention, and one of them must reflect the best method known to Zoetis of performing the invention.
(b) Zoetis has failed to comply with the best method requirement, because the Applications do not disclose the M. hyo and PCV-2 antigen concentration or content of the preferred immunogenic compositions of the invention. The Applications either specify the potencies of those compositions in relative units, without sufficiently defining the relevant reference or comparator, or in certain cases, do not describe the preferred compositions at all.
(c) Further, it is apparent that the concentration or content of M. hyo and (if present) PCV-2 antigen in the IVPs is critical to their performance, and thus the performance of the invention. As such, these matters form part of the best method of performing the invention, and must be disclosed.
(d) The Applications do not disclose these matters, either because they use relative units for the antigen concentration or content without identifying a reference product or providing enough information to allow a skilled addressee to ascertain the concentration or content in absolute terms, or, in some instances, because the IVPs are not described at all.
(e) The skilled addressee of the Applications is not given the information necessary to put the best method of performing the invention into practice. Boehringer says that it is no answer to this for Zoetis to rely on the broad ranges given in the description in the specification (eg 535 Application at page 15, lines 18–22 and page 19, lines 9–21) for the concentrations of the M. hyo and PCV-2 antigens in absolute units. Those ranges do not disclose the antigen concentration or content of any particular composition, and the description makes it clear that this is a critical aspect of the invention.
649 Accordingly, Boehringer submits that the nature of the invention is to be ascertained from the specification as a whole, and it is no answer to an allegation of failure to disclose the best method that the feature not disclosed is not an integer of the claims.
650 Further, where the specification purports to address the best method requirement by providing a detailed description of preferred embodiments, it is incumbent on the applicant to describe the best embodiment known to it at the time of filing the application.
Zoetis’ submissions
651 Zoetis complained of Boehringer advancing “chameleonic” positions as to which of the 10 IVPs it was alleging to be “the best”, and as to the criterion by which “best” ought to be assessed.
652 Zoetis submits that at the heart of Boehringer’s case are twin fallacies that:
(a) one example or embodiment in a patent specification must itself be the best method of performing the invention in every material respect; and
(b) s 40(2)(aa) requires every detail of every such example or embodiment, material or not, to be disclosed so that the example can be replicated precisely.
653 Zoetis submits that the best method of performing the invention in the Applications is the manufacture of compositions that achieve the asserted advantages of combinability and compatibility with other porcine antigens, while retaining immunogenicity. To be clear, Zoetis submits that the best method of performing the invention is not achieving a particular level of efficacy, or stability, or any other criterion.
654 Zoetis submits that the best method of performing the invention in claim 1 of:
(a) the 535 Application is the making of the M. hyo soluble preparation (which is separated from insoluble cellular material and substantially free of IgG and immunocomplexes);
(b) the 537 Application is the making of compositions comprising the M. hyo soluble preparation (which is separated from insoluble cellular material and substantially free of IgG and immunocomplexes) and a PCV-2 antigen; and
(c) the 540 Application is the making of compositions comprising the M. hyo soluble preparation (which is separated from insoluble cellular material and substantially free of IgG and immunocomplexes), a PCV-2 antigen and a PRRS antigen.
655 The Applications disclose extensive detail as to how the M. hyo soluble preparation was prepared, in particular, the processes by which the supernatant was separated, and the use of Protein A to substantially remove IgG and immunocomplexes. According to Zoetis, the disclosure of the processing steps and the use of Protein A to substantially remove IgG satisfies Zoetis’ obligations to disclose the best method. The Applications identify a preferred strain of M. hyo for use in the invention. The Applications also disclose detail as to the selection of a PCV-2 and PRRS antigen.
656 Finally, Zoetis submits that, as Boehringer submits the best method is a particular IVP, the best method must comprise every element of an IVP, not only its antigen concentration or information concerning its antigen content, but also including adjuvants, excipients and the like, notwithstanding that Boehringer pleads no material facts in relation to adjuvants or excipients. It follows, according to Zoetis, that Boehringer cannot prove any particular IVP is the best based solely on matters relating to antigen content, because the characteristics of any particular IVP is a function of all of its elements. There is therefore a fundamental gap at the heart of Boehringer’s case — it alleges that the best method is a particular embodiment, being an IVP, but it seeks to plead nothing about the role of any element comprising that embodiment other than antigen concentration or information concerning antigen content. Therefore, the case that any particular IVP is the best fails because Boehringer has no case based on all the elements of any particular IVP.
Dr Nordgren’s evidence as to the examples in the Applications
657 Boehringer’s particulars refer to the evidence of Dr Nordgren in his second affidavit. In these paragraphs, Dr Nordgren discusses certain examples in the Applications. For each example, Dr Nordgren then responds to the question of whether he could “replicate” the IVPs utilised in that example. Examples 1 to 11 are the same in each of the Applications.
658 Dr Nordgren’s evidence on this topic was unchallenged. Zoetis’ experts, Professor McVey and Professor Browning, did not comment on this part of Dr Nordgren’s evidence. I do not consider that Dr Nordgren’s evidence on this topic is subject to the reservations that I expressed earlier.
659 Dr Nordgren’s evidence is that if the antigen content of a formulation is described in terms of relative potency, it is not possible to replicate that formulation without details of the reference product against which the relative potency was measured. Usually, the relative potency of an experimental vaccine refers to the potency of that vaccine as measured against a “reference product”. The reference product is typically a vaccine preparation that has been demonstrated to provide the minimum level of protection that is considered to be adequate.
660 Dr Nordgren described the manner in which relative potency is established. The immunogenicity of a reference product is established in the target species. The reference product immunogenicity is then standardised by way of in vivo or in vitro assay.
661 Dr Nordgren’s evidence was that the results of a relative potency assay are specific to the particular conditions of the relevant in vivo (ie animal model) and in vitro (ie ELISA) assay(s), including the protein(s) being measured, as well as the reference product against which the experimental vaccine candidates were compared.
662 Dr Nordgren’s evidence was that the ranges of the M. hyo antigen concentration or content given in the Applications are not meaningful for the purpose of disclosing the details of any particular formulation, because the ranges are so broad. In his opinion, the ranges given in the Applications of the PCV-2 antigen concentration or content are not meaningful either.
663 Professor McVey’s evidence was that the immune response relates to the antigen concentration or content of the specific protective antigens in the vaccine: a greater titre of specific protective antigens is likely to lead to a greater duration of immunity.
664 Dr Nordgren commenced his discussion of the examples with Example 9: “Evaluation of M. hyo Efficiency of a 1-Bottle PCV-2/M. hyo Combination Vaccine in 10% SP-oil”. It will be recalled that examples 1 to 11 are the same across all three Applications. The Applications describe the study in Example 9 as a proof of concept designed to evaluate the M. hyo fraction efficacy of four experimental PCV-2/M. hyo vaccines prepared by different manufacturing processes which utilise Protein A for IgG removal compared to control vaccines prepared with the standard M. hyo manufacturing process.
665 Dr Nordgren observed that Example 9 of the Applications describes the M. hyo antigen present in the four experimental vaccines: T05 (IVP Serial L0711RK11), T06 (IVP Serial L0711RK12), T07 (IVP Serial L0711RK13) and T08 (IVP Serial L0711RK14), in terms of relative potency. Dr Nordgren further observed that Example 9 also describes the PCV-2 antigen present in the IVPs in terms of relative potency.
666 Dr Nordgren noted that the Applications do not provide the details of the reference product or products against which the relative potencies in Example 9 were tested, nor provide any details regarding the amounts of those antigens in the IVPs, such as by way of μg/mL.
667 Dr Nordgren’s evidence was that without details of the reference product against which the relative potencies were tested, or details regarding the amounts of the antigens in the IVP’s, he would not be able to replicate the IVPs of Example 9 (T05, T06, T07 and T08).
668 Dr Nordgren then considered Example 12 of the 535 and 540 Applications (Example 14 of the 537 Application); “Evaluation of Virucidal Activity Against PRRS Virus”. The Applications describe the studies set out in Example 12 (and 14 of the 537 Application) as being designed to evaluate the various adjuvant platforms for virucidal activity against PRRS virus.
669 Dr Nordgren observed that the adjuvant compatibility examples of Example 12 describe five additional IVPs, with serial numbers L0912RK12, L0912RK10, L0912RK11, L0912RK08 and L0912RK09. Table 14 of the 535 and 540 Applications (Table 25 of the 537 Application) describes the M. hyo and PCV-2 antigens present in these IVPs in terms of relative potency, relative to IVP L1211RK15.
670 The 535 and 537 Applications state that the antigenic potency of the vaccine serials in Table 14 (and Table 25 in the 537) were compared to a “Reference PCV/M. hyo vaccine serial (L1211RK15)”. The 540 Application simply refers to “Reference L1211RK15” in Table 14.
671 The 535 Application states that L1211RK15 contained 0.688% of a 20x concentrate of PCV-2 antigen (prepared as described in Example 2); and 9.4% of M. hyo antigen prepared as described in Example 11.
672 The 537 Application refers to the serial L1211RK15 being “described above”. “Above” is example 12 of the 537 Application which first refers to L1211RK15 as T03, an experimental preparation of high passage PCV Type1-Type2 chimera, killed virus (0.688%-Medium) and M. hyo antigen (9.4%-Medium). At page 51, still in the description of Example 12, under the heading “Vaccine Potency Testing”, the specification states that L1211RK15 “was considered to be the reference candidate. Consequently, relative potency for both the M. hyo fraction and the PCV2 fraction were determined versus this candidate reference”. Example 12 concludes “All vaccines proved to be efficacious and vaccine serial L1211RK15 was selected as a reference candidate”.
673 According to Dr Nordgren, IVP L1211RK15 is not described anywhere else in the 535 or 540 Applications. There are no details of the amounts of M. hyo or PCV-2 antigen in the reference product, or the other IVPs in Example 12. Dr Nordgren’s evidence is that without this information he would be unable to reproduce the IVPs in Example 12.
674 Dr Nordgren noted that IVP L1211RK15 is described in Example 14 of the 537 Application. However, he further noted that Example 14 provides no absolute measurements of the M. hyo or PCV-2 antigen content of IVP L1211RK15. As a result of not being able to determine the amount of M. hyo or PCV-2 antigen in IVP L1211RK15, Dr Nordgren considered that he was also unable to determine the amounts of M. hyo or PCV-2 antigen present in the IVPs described in Table 25 of Example 14 that would enable him to reproduce these IVPs.
675 Dr Nordgren then discussed Example 12 of the 537 Application: “Efficacy of the PCV1-2 Chimeric Fraction Following Intramuscular Administration of a 1-bottle PCV2/M. hyo Combination Vaccine in 10% SP-oil”. The 537 Application describes the study in this example as being designed to evaluate “the efficacy of the PCV1-2 chimera killed virus fraction of an experimental 1-Bottle PCV2/M. hyo combination vaccine, administered once to pigs of 21+/- 3 days of age and challenged with a virulent PCV2 isolate at approximately 6 weeks of age”.
676 Dr Nordgren observed that Example 12 of the 537 Application describes five IVPs, namely, four bivalent vaccines (T02, T03, T04 and T05) and a control vaccine of M. hyo antigen only, prepared as described in Example 11 (T01). T03 is serial L1211RK15. Lines 20–21 of page 49 of the 537 Application state that the IVPs “are described below and in Table 13, where the antigen dose (% of the PCV2 and M. hyo antigen lots) is provided”.
677 Dr Nordgren understood “antigen lots” to mean antigen stock preparations. He noted that Example 12 of the 537 Application does not provide any details of the antigen content of the PCV-2 and M. hyo antigen lots that were included in the IVPs (for example, by way of concentration, μg/mL). Dr Nordgren did not consider that the antigen dose information expressed as a % of the antigen lots to be useful. He considered that Example 12 does not disclose the amount of target PCV-2 and M. hyo antigens that are included in T01, T02, T03, T04 and T05, and that, because he was unable to determine the amounts of PCV-2 and M. hyo antigens in those IVPs, he would be unable to replicate those formulations, which included L1211RK15 (T03).
678 Dr Nordgren observed that Example 11 of the Applications provided the most detailed description of a process to make a Protein A treated M. hyo supernatant. Example 11 (see, for example, page 49, lines 3–5 of the 540 Application) states that “Anti-PCV2 antibodies and M. hyo antigen levels were measured in the final antigen fluid via PCV2 specific antibody ELISA and p46 antigen quantification ELISA, respectively”. However, Example 11 does not provide any details regarding:
(a) the amount of M. hyo antigen present in the supernatant pre- and post- Protein A treatment, such as by way of RU/mL (including reference measurement details) or by way of p46 concentration in μg/mL; or
(b) the anti-PCV-2 antibody levels, such as by way of μg/mL.
679 Dr Nordgren also discussed Example 15 of the 540 Application: “Evaluation of M. hyo efficacy of a PCV2/M. hyo/PRRS combination vaccine followed by M. hyo Challenge”. The 540 Application describes the study the subject of the example as being designed to evaluate the efficacy of the M. hyo fraction of an experimental PCV-2/M. hyo/PRRS combination vaccine, administered intramuscularly in susceptible pigs at three weeks of age and challenged with a virulent M. hyo isolate three weeks post vaccination.
680 Dr Nordgren observed that the study involved three IVPs, namely two experimental vaccines (T02 and T03) and a control vaccine containing PCV-2 and PRRSV antigen only (T01). The example states that the M. hyo supernatant used in T03 was that described in Example 11 with an additional step to concentrate the antigen 20x by molecular filtration. Table 14 shows the PCV-2/M. hyo element of T03 comes from serial L0712RK33. Example 15 concludes by stating that “the results in the present example demonstrate that an experimental trivalent vaccine formulation (T03 treatment) used in this study produced significant efficacy against M. hyo challenge”.
681 Dr Nordgren noted that Example 11 does not provide any details regarding the amount of M. hyo antigen present in the supernatant pre- and post-Protein A treatment.
682 Dr Nordgren observed that Table 19 of Example 15 of the 540 Application describes the PCV-2 and M. hyo antigens present in IVPs T02 and T03 in terms of relative potency (3.4 and 2.7 “relative potency units”). Example 15 does not provide any details of the reference products against which the relative potencies were tested.
683 Dr Nordgren observed that Example 14 of the 540 Application describes three IVPs, namely two experimental vaccines (T02 and T03) and a control vaccine containing M. hyo and PRRS antigen only (T01). Table 15 describes the amount of PCV-2 antigen added to IVPs T02 and T03 as a percentage, but this information is not useful because the 540 Application does not provide any other details about the PCV-2 antigen stock preparation that was included in IVPs T02 and T03, such as the concentration of PCV-2 antigen in the stock preparation. Table 15 of the 540 Application describes the amount of M. hyo antigen in RU/mL, but it is not clear whether this is a reference to the amount of M. hyo antigen in the M. hyo antigen stock solution, or in the final IVPs. If it is the former, the 540 Application does not provide any details of the volume of M. hyo antigen stock solution added to the final IVPs. The 540 Application also does not provide any details of the standard curve and reference measurement details used to calculate the RU/mL of the M. hyo antigen.
684 Example 16 of the 540 Application describes four IVPs, namely, three trivalent IVPs (T02, T03 and T04) and a control vaccine containing M. hyo and PCV-2 antigen only (T01). Example 16 describes the amount of PCV-2 antigen as a percentage, but Dr Nordgren did not consider this information useful because the 540 Application does not provide any other details about the PCV-2 antigen stock preparation that was included in IVPs T01, T02, T03 and T04. The 540 Application (at page 56, lines 22–23) states that the M. hyo supernatant used in T01, T02, T03 and T04 was prepared as described in Example 11.
685 Boehringer tendered a numbers of Zoetis (and Pfizer, its predecessor) documents in support of its best method case. Many of these documents contained information claimed to be confidential to Zoetis.
686 Boehringer relied upon Zoetis’ in-house documents to support its case that L1211RK15 and L0912RK08 were the best method known to Zoetis of implementing the invention in each of the Applications at the filing date. In summary, the documents establish the following matters.
687 Zoetis had a team looking to develop an M. hyo/PCV-2 one-bottle vaccine. In the course of developing its proposed M. hyo/PCV-2 combination one-bottle vaccine, Zoetis developed two M. hyo/PCV-2 qualifying “vaccine serials”: L1211RK15 (low) and L1211RK09 (high).
688 The two qualifying serials, L1211RK09 and L1211RK15, were both subjected to a clinical MID (Minimum Immunising Dose) study, together with three other IVPs. This study became Example 12 of the 537 Application.
689 In an email to the Zoetis vaccine development team dated 20 April 2012, the principal scientist sent the mean p46 values for serials LK1211RK09 and L1211RK15 in micrograms per ml, as measured on day zero, noting that serial LK1211RK09 was formulated at 1.5x that of serial L1211RK15.
690 In September 2012, Mr Nitzel (one of the inventors) reporting to the M. hyo/PCV-2 One Bottle team, informed his team that they had obtained the “best case” scenario (ie confirmation of efficacy for both potential qualifying serials) for the M. hyo fraction of the qualifying serials. The email noted that the team already had efficacy for the PCV-2 fraction.
691 In October 2012, the Zoetis M. hyo/PCV-2 One Bottle team recommended L1211RK15 as the qualifying serial, the reference serial against which the relative potencies of other serials were determined.
692 By late March 2013, L1211RK15 had been selected as the single qualifying reference serial for PCV-2 and M. hyo. L1211RK15’s status as the qualifying serial is confirmed by the 537 Application, which refers to it as the reference candidate at page 51 line 7. By late March 2013, Zoetis had 15 months of data about L1211RK15.
693 Zoetis tendered a copy of the 12 April 2012 meeting minutes of the One Bottle assay sub team, which it submitted showed there was a discrepancy in quantification of the antigen concentrations of the serials. On their first page, the minutes discuss the coefficient of variation percentages (no comment is made on the results themselves). It is also reflected in the way the data are presented in document AT112: pages 1526–1527 present the data by test vial, and pages 1528–1529 present the same data by scientist (presumably to investigate variability between scientists). Section 4 of the meeting minutes discusses variability between scientists, and concludes with the statement that one scientist plans to be the only analyst for Day 0. Section 5 of the meeting minutes then refers to the “Day 0 testing” for M. hyo p46 and PCV-2 antigens.
694 However, by the time L1211RK15 was selected as the qualifying serial in late March 2013 and it was being used as the reference against which the relative potencies of other samples were being tested against, there were accepted values for the antigen concentration of samples at T0 (day zero).
695 A March 2013 Zoetis report entitled “PCV/M. hyo One Bottle Formulation Technical Transfer Report”, noted that serial L1211RK15 was the single reference for PCV-2 and M. hyo, and formulation target values were given on a relative potency basis, relative to L1211RK15. The same report reported that “pivotal L1211RK15 MID vaccine” was used in the antigen stability studies.
696 Zoetis also had a Trivalent Project team working on developing a trivalent combination vaccine against PCV-2, PRRS and M. hyo, by building on the one-bottle formulation PCV-2/M. hyo vaccine. The study tested potency of the M. hyo and PCV-2 antigens of two experimental trivalent serials (one of which was L0912RK08) against the reference serials L1211RK09 and L1211RK15.
697 Serial L0912RK08 was formulated to have substantially the same M. hyo and PCV-2 potency levels as L1211RK15 (see Table 14 of the 540 Application) but was prepared from an M. hyo antigen lot that used 10x filter concentrated M. hyo. The antigen content of the antigen lot used in L0912RK08 was concentrated in order to enable its use in a trivalent composition. In Example 14 of the 540 Application, the trivalent vaccine formulated using L0912RK08 as diluent was proven to have significant protective effect.
Boehringer’s detailed submissions
698 Boehringer highlights that single dose administration is an important benefit of the alleged invention. In this regard, the experts understood that the Applications teach that an M. hyo supernatant can be effective when administered in a single dose. As noted, the dependent claims include claims to a composition that elicits a protective immune response when administered as a single dose. The immune response relates to the antigen concentration or content of the specific protective antigens in the vaccine: a greater titre of specific protective antigens is likely to lead to a greater duration of immunity.
699 Boehringer submits that the expert evidence confirms that the ranges of the M. hyo antigen concentration or content given in the Applications are not meaningful for the purpose of disclosing the details of any particular formulation, because the ranges are so broad. The ranges of the PCV-2 antigen concentration or content are not meaningful for similar reasons.
700 The potency of the IVPs in the examples is expressed in relative potency (RP) terms or reference units per mL (RU/mL), but in each case the reference or comparator vaccine is not sufficiently described.
701 Even when a comparator vaccine is identified, in general terms, as a commercial vaccine (eg expired Respisure One), that does not assist with making an IVP of similar potency. That is because the potency of a commercial vaccine is not public information. In any case, the potency of a commercial vaccine may vary over time and between batches, and the use of relative potency referable to a comparator vaccine gives rise to inconsistent testing values.
702 Potency information in the Applications is given by reference to a reference vaccine for which no absolute measurements of the antigen content are given and does not assist with reproducing an IVP of similar potency. Potency information given as a percentage of an antigen lot is also unclear and does not assist with reproducing an IVP of similar potency.
703 L1211RK15 and L0912RK08 are two of the 10 pleaded IVPs upon which Boehringer focussed in submissions.
(a) Serial L1211RK15, a bivalent (M. hyo and PCV-2) IVP, is the subject of Examples 12 and 13 of the 537 Application (and used as a reference vaccine in Table 14 of Example 12 of the 535 Application and 540 Applications and Table 25 of the 537 Application).
(b) Serial L0912RK08, a bivalent (M. hyo and PCV-2) IVP, is the subject of Example 14 of the 540 Application (which also appears in Table 14 of the 535 and Table 25 of the 537 Application).
Boehringer submits that these two IVPs are good candidates for reflecting the best method known to Zoetis at the time of filing, and that the M. hyo and PCV-2 antigen concentration or content were not disclosed for both IVPs in the Applications.
704 As at the filing date, Zoetis knew the concentration of antigens in its preferred M. hyo and PCV-2 reference and qualifying sample; L1211RK15 (certainly on Day 0). Thus, Boehringer submits that the relative potencies in the Applications were relative to a sample with known concentration, making the relative potency meaningful only to a skilled person with the knowledge of the concentration of the reference sample.
705 The Applications do not provide sufficient information to the skilled addressee to make a formulation with the potency of L1211RK15. The 537 Application describes L1211RK15 by way of a percentage of “antigen lot” (or antigen stock preparation) that was included in the overall IVP, but does not provide any information about the antigen content of the antigen lot that was used to make L1211RK15 (or L1211RK09). The relative potency of L1211RK15 is also given relative to itself, which does not assist. The information in the 537 Application is not sufficient to enable a skilled person to know the amount of M. hyo or PCV-2 antigens included in L1211RK15, and a skilled person would be unable to replicate that formulation.
706 Boehringer also submits that the 540 Application does not provide any information about the potency of L1211RK15. Further, no information about M. hyo potency is provided in the 535 Application for L1211RK15, and the limited information about PCV-2 potency in the 535 Application does not assist.
707 Similarly, none of the Applications provide sufficient information to the skilled addressee to make a formulation with the potency of L0912RK08: only the 540 Application mentions it, and the information provided is insufficient. For example, Table 14 of the 540 Application describes the potency of L0912RK08 relative to L1211RK15, however, as submitted, the 540 Application does not provide any information about the potency of L1211RK15.
708 Finally, Boehringer notes that none of the Applications describe a “qualifying serial” for the purposes of a monovalent M. hyo vaccine. This is no doubt because each of the Applications is directed towards a multivalent immunogenic composition comprising at least M. hyo and PCV-2 antigens, and in the case of the 540 Application, at least M. hyo, PCV-2 and PRRS virus antigens.
709 Boehringer then addressed a third pleaded IVP: L0712RK33, which was the subject of Example 15 of the 540 Application (which is not present in the 535 or 537 Applications). L0712RK33 is a bivalent (M. hyo and PCV-2) IVP used as the diluent for a hydrophilised PRRS vaccine. Boehringer submits that L0712RK33 is the IVP which has a relative potency that most closely resembles the target relative potency of Zoetis’ commercial product (which had been determined by the filing date). According to Boehringer, insufficient information is provided in the 540 Application for the skilled addressee to reproduce an IVP of the same or similar potency as L0712RK33. This is because the 540 Application provides no details regarding the reference vaccine against which the relative potency of the M. hyo or PCV-2 components of L0712RK33 was measured.
710 Boehringer submits that Zoetis’ documents indicate that the M. hyo and PCV-2 of L0712RK33 were set and tested relative to L1211RK15. However, that fact is not disclosed in the 540 Application: that is, L1211RK15 is not identified as the reference or comparator for the relative units used. Further, even if the 540 Application did disclose that the M. hyo and PCV-2 potencies of L0712RK33 were relative to L1211RK15, Boehringer submits that this would not have assisted the skilled person because the 540 Application does not provide any information about the potency of L1211RK15.
711 Boehringer observes that L0712RK33 is not disclosed at all in the 535 Application or the 537 Application, so those Applications also do not provide any information to the skilled addressee to reproduce a formulation with the potency of L0712RK33.
712 In a table annexed to its submissions, Boehringer gave details as to the remainder of its pleaded IVPs. L100211J was the sole M. hyo-only IVP. It is described in Table 4 of the 535 Application as having an M. hyo antigen content of 452 units (unspecified) per dose, and in Table 5 the target/observed relative potency is given as 13/12.1. No details of the reference vaccine used to calculate the relative potency is provided.
713 In response to Boehringer’s submissions as to the absence of any concentration information, particularly of the reference serial, Zoetis points to the ranges of concentration given in the specifications and says that those provide a sufficient disclosure of the absolute concentration for M. hyo p46 antigen and PCV-2 antigen in each Application. Zoetis submits that the absolute concentration range combined with the relative potencies provided in the examples is a disclosure of the best method of performing the invention known to the patent applicant.
714 Zoetis submits that the absolute concentration of p46 M. hyo antigen or PCV-2 antigen is not material having regard to the nature of the invention described and claimed in the Applications. Even if the absolute concentration was held to be material, Zoetis submits that Boehringer has not shown that Zoetis’ disclosure of the ranges of absolute concentrations is a deficient disclosure. Dr Nordgren was not asked to conduct a study similar to those reported in the Applications or to make a commercial product. Nor was he asked how he would go about using the ranges in the specification or the information about relative potency to make a composition for any particular purpose.
715 Zoetis observes that the “preferred” range of about 2 µg/mL to about 6 µg/mL aligns almost exactly with the range of values recorded in a document tendered by Zoetis, for L1211RK09 and L1211RK15. Zoetis observed that all the 10 pleaded IVPs fall within the preferred range.
716 Finally, Zoetis attacks Boehringer’s pleaded best method case, asserting that there was no challenge pleaded as to the combination of absolute M. hyo p46 and PCV-2 antigen levels in any particular IVP or otherwise.
Nature of the invention in each Application
717 First, and most importantly, it is necessary to ascertain the nature of the invention, being the embodiment which is described by the specification as a whole and around which the claims are drawn (cf the invention so far as claimed in any claim): Sandvik at [94] (per Greenwood, Rares and Moshinsky JJ).
718 The invention in the 535 Application and around which the claims are drawn may be described as an improved immunogenic composition or vaccine to elicit a protective response in pigs against the disease caused by M. hyo, utilising an M. hyo soluble preparation which can also be used as a base or platform for a combination vaccine with at least one additional antigen protective against selected microorganisms (other than PCV-2, as there are no claims to an M. hyo/PCV-2 combination).
719 Whilst claims 1 to 6 are to an immunogenic composition, a close reading of the whole of the specification shows that the invention is concerned with eliciting a protective immune response to protect pigs from disease caused by M. hyo, (ie a vaccine) which is within the specification’s definition of an immunogenic composition. The later claims are to methods of eliciting a protective response, and immunising a pig utilising the immunogenic composition (ie vaccines). The title of the 535 Application is “Mycoplasma hyopneumoniae vaccine” and the field of the invention relates to a vaccine. The 535 Application states that what is needed is an improved vaccine, and that it would be highly desirable to provide a ready-to-use, single dose M. hyo/PCV-2 combination vaccine. The prior art discussed in the specification are vaccines.
720 The invention described in the 537 Application and around which the claims are drawn may be described as an improved vaccine to protect pigs against the diseases caused by M. hyo and PCV-2, utilising an M. hyo soluble preparation as a base or platform for a combination vaccine with PCV-2 antigen and potentially at least one additional antigen protective against selected microorganisms (other than PRRS, as there are no claims to an M. hyo/PCV-2/PRRS combination).
721 Again, whilst the early claims of the 537 are to a multivalent immunogenic composition, a close reading of the whole of the discussion in the specification shows that the invention is concerned with the protection of pigs from mycoplasmal pneumonia and Post-weaning Multisystemic Wasting Syndrome caused by the microorganisms M. hyo and PVC-2 respectively (ie a combination vaccine against M. hyo and PCV-2). The title “PCV/Mycoplasma hyopneumoniae combination vaccine” to the field of the invention, the background of the invention, the summary of the invention, the detailed description and the examples all support the invention being an improved vaccine to protect pigs against the diseases caused by M. hyo and PCV-2.
722 The benefits said to accrue to the 537 Application invention are the same as for the 535 Application. Although given that PCV-2 is ubiquitous, more so than PRRS, the removal of the serum derived antibodies, likely to be PCV-2 and potentially others, is more important in the M. hyo/PCV-2 combination vaccine.
723 The invention described in the 540 Application and around which the claims are drawn may be described as an improved trivalent vaccine to protect pigs against the diseases caused by M. hyo, PCV-2, and PRRS, utilising an M. hyo soluble preparation as a base or platform for the combination vaccine with PCV-2 and PRRS antigens.
724 Again, whilst the early claims of the 540 Application are to a trivalent immunogenic composition, a close reading of the whole of the discussion in the specification shows that the invention is concerned with the protection of pigs from mycoplasmal pneumonia, Post-weaning Multisystemic Wasting Syndrome and PRRS virus caused by the microorganisms M. hyo, PVC-2 and PRRS respectively (ie a combination vaccine against M. hyo, PCV-2 and PRRS). The title “PCV/Mycoplasma hyopneumoniae/PRRS combination vaccine” to the field of the invention, the background of the invention, the summary of the invention, the detailed description and the examples all support the invention being an improved vaccine to protect pigs against the diseases caused by M. hyo, PCV-2 and PRRS.
725 The question is whether each of the Applications describe the best method known to Zoetis of performing each of the inventions at their time of filing.
Consideration
726 Boehringer makes no best method challenge in relation to the invention claimed in claims 1 and 2 of the 535 Application: the M. hyo platform. Boehringer alleges that the 537 and 540 Applications do not disclose a best method of performing the invention claimed in each of the claims of those Applications.
727 An M. hyo/PCV-2 combination immunogenic composition which is suitable for use in a vaccine is central to the invention in both the 537 and 540 Applications. The Applications describe preferred forms of such a composition by setting out certain details in the examples of specific experimental vaccines or IVPs. The concentration or content of M. hyo and PCV-2 antigen in the IVPs is critical to their performance, and thus the performance of the invention.
728 As at the filing date of each of the Applications, the Zoetis team was using the bivalent (M. hyo/PCV-2) IVP, L1211RK15 as the reference sample against which the relative potency of other IVPs was being tested. L1211RK15 had been chosen following a minimum immunising dose clinical study in pigs. At that time, the Zoetis team knew the composition of L1211RK15, including the absolute concentration of the M. hyo and PCV-2 antigens in that IVP.
729 Zoetis chose to keep the absolute concentration of the M. hyo and PCV-2 antigens in L1211RK15 — the key to unlocking the IVPs in the examples — to itself, disclosing only the relativities of other IVPs to the reference sample in the Applications.
730 Rather than providing the key, the Applications instead refer to a broad range of the antigen concentrations, expressed as absolute concentrations (in µg antigen/ml), within which the concentration preferably lies. Dr Nordgren described the range as being so broad as to be meaningless.
731 The Applications either specify the potencies of the IVPs in the examples in relative units, without defining the relevant reference or comparator, or in certain cases, do not describe the preferred compositions at all. The examples describe particular IVPs and evaluate their performance by comparing them against each other. Various passages in the specifications indicate that some IVPs performed better than others, for example in terms of efficacy. There are also references to the need to “balance” the antigens, which the experts confirm as being important.
732 The three Applications claim to have developed an immunogenic composition that provides protective efficacy after a single dose. The experts understood single dose efficacy to be an important aspect of the invention. The potency of, and balance between, the antigens in such a composition is critical to its ability to meet that objective. Similarly, it is relevant that the Applications claim to have solved the problem of interference, including not only interference by serum-derived antibodies, but also antigen-antigen interference. Again, the potency of, and balance between, the antigens in such a composition is critical.
733 The Applications do not provide sufficient information to the skilled addressee to make a formulation with the potency of L1211RK15 (or any other of the IVPs). The 537 Application describes L1211RK15 by way of a percentage of “antigen lot” (or antigen stock preparation) that was included in the overall IVP, but does not provide any information about the antigen content of the antigen lot that was used to make L1211RK15 (or L1211RK09). The relative potency of L1211RK15 is also given relative to itself, which does not assist. The information in the 537 Application is not sufficient to enable a skilled person to know the amount of M. hyo or PCV-2 antigens included in L1211RK15, and a skilled person would be unable to replicate that formulation. The 540 Application does not provide any information about the potency of L1211RK15.
734 Even though the 535 Application does not claim an M. hyo/PCV-2 combination, it still uses L1211RK15 as the reference vaccine against which other IVPs are compared. See for example, Example 12.
735 Zoetis was not obliged to identify the best method as such in the Applications. I am prepared to assume that Zoetis considers that including IVPs in the examples in the Applications, in combination with the disclosed ranges, satisfy its obligation to include the best method known to it at the time of filing.
736 However, I consider that Zoetis has chosen to hide the best method in plain sight in each of the Applications. At first glance it appears that details, including concentration, are provided for the IVPs discussed in the examples. It is only when the skilled reader goes looking for the concentration information for the reference sample against which all the relative concentrations are given that it is apparent that information — the key to the relative concentrations — is not provided anywhere in the Applications.
737 Keeping the key to itself is contrary to the obligation of good faith which, as the authorities emphasise, is fundamental to the best method requirement.
738 Zoetis’ response, that the concentration ranges provided in the Applications are sufficient, is not an answer. The person skilled in the art seeking to make the invention at the expiry of the patents granted on the Applications would be forced to expend time and money embarking on a program of work to find the best starting point in that range – information that was known to the patentee when the Applications were filed, and information which it kept to itself.
739 Zoetis’ focus on the precision of the pleadings and requiring Boehringer to nominate its IVP choice as the “best” as its answer to the best method challenge is at odds with the obligation of good faith. By doing so, the best method case became more akin to a game of battleship, wherein the challenger, not blessed with the patent applicant’s full knowledge of the in-house research and development work and context leading to the choice of the commercial product, is forced to nominate IVP’s as the “best method” known to the Patent Applicant. Ultimately, this was irrelevant as, whichever of the IVPs nominated, absolute concentration details were not provided for any of the antigens in them.
740 Zoetis’ demand that Boehringer define “best” was also not in keeping with its good faith requirement. When it filed the Applications, Zoetis knew the “key”, the absolute concentration of the reference IVP, and chose to keep it to itself.
741 I consider that Boehringer’s no absolute concentration best method challenge was adequately pleaded. There was no surprise to Zoetis in the case that was put in relation to the absence of any absolute concentration for the M. hyo and PCV-2 antigens. Boehringer made it abundantly clear in its opening and closing that its primary best method case was based on the absence of any absolute antigen concentration of M. hyo and/or PCV-2 antigens.
742 In GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Generic Partners Pty Ltd (2018) 2131 IPR 384 at [106] (per Middleton, Nicholas and Burley JJ), the missing information as to the grade of HPMC was tailored to optimising the commercial process on the patent applicant’s manufacturing machinery/process. The concentration details (particularly of the reference IVP) are fundamental to the working of the invention, not the optimisation of the commercial product in the specific circumstances of the patent applicant’s own lab.
743 The disclosure of the best method known to the patentee tells the skilled worker, seeking to make the invention on the expiry of the patent, the methodology to achieve the form of the invention that obtains the result (including touted benefits) which constitutes the invention, and relieves them from having to confront the blind alleys and pitfalls (ie undertake the routine trial and experiments and animal studies otherwise involved in making the invention,) and already overcome by the patentee by the time the Application was filed.
744 By keeping to itself the concentration of M. hyo and PCV-2 antigens in its selected reference sample L1211RK15, Zoetis has denied the person skilled in the art the best method of performing the invention claimed in claims 3, 7–8 and 11 of the 535 Application, claims 1 to 24 of the 537 Application and claims 1 to 25 of the 540 Application.
745 Boehringer alleges that certain of claims of the Applications are claims to “mere collocations” of integers rather than a combination, and as such are not patentable subject matter pursuant to s 18(1) of the Act.
746 Both parties dealt with the ground of manner of manufacture only briefly in their oral and written submissions.
747 Where a claimed “combination” consists only of integers which do not interact with each other to produce a new result or product, it is not a manner of manufacture. Rather, it is a “mere collocation of separate parts”, each performing its own separate function, which is unpatentable; Firebelt at [21] (per Spender, Drummond and Mansfield JJ); Smith & Nephew Pty Ltd v Wake Forest University Health Sciences (2002) 82 IPR 467 at [16] (per Finn, Bennett and Middleton JJ).
748 The actual claim under consideration in Wake Forest, claim 49, was as follows:
An apparatus for applying negative pressure to a wound beneath a fluid-impermeable seal comprising:
a screen means for positioning beneath said seal for preventing overgrowth of tissue in the wound, …;
a flexible tube having an inlet and inserted into said open cell polymer foam section and an outlet end for extending from beneath said seal and for supplying said negative pressure; and
wherein said apparatus is in an aseptic package.
(Emphasis added.)
749 The aseptic package was held by the Full Court to not be a patentable combination.
750 The patentee in Wake Forest referred to Pugh v Riley Cycle Co Ltd (1914) 31 RPC 266 to submit that the only effect of including the additional integer, the aseptic packaging, was to limit the claim, without affecting the invention in the claimed combination.
751 The Full Court observed at [18] that the patentee did not support a construction of the claim as a limitation of the scope of the claim. Rather, the aseptic bag was contended by the patentee to be a feature of the apparatus in a kit form. As a matter of construction, the contention was that the aseptic package was an essential integer of the combination the subject of the claim, not an optional additional limitation on the scope of the monopoly.
752 The Full Court in Wake Forest observed at [33] that once the aseptic package is said to be an essential integer and part of the invention claimed, it must interact purposefully and functionally with the other integers to produce the negative wound pressure. The Full Court distinguished the case of Pugh on the basis that the aseptic package was not an optional extra merely assisting in the presentation of the apparatus, or limiting or adding to the claim.
753 The claim itself specified the one “desired effect” of the apparatus; to apply negative pressure to a wound. The Full Court contrasted claim 49 with claim 36 which talked in terms of an apparatus for facilitating the healing of wounds. The Full Court observed at [26] that the aseptic package may facilitate the healing of wounds, but it had nothing to do with the application of negative pressure, the function of the apparatus itself. Given that the aseptic package was discarded prior to use, it had no role in achieving the sole desired effect of negative wound pressure specified in the claim. The terms of the claim itself made it clear that the “in an aseptic package” integer was not part of the claimed “apparatus”.
754 The Full Court observed at [33] that the aseptic packaging may be said to have a purpose — to ensure sterility in treating the wound — but it lacked any desirable functional result in the way described by Dixon J in Palmer v Dunlop Perdriau Rubber Co Ltd (1937) 59 CLR 30 and Aickin J in Minnesota Mining: the essential interaction of the integers of the combination to produce a new result.
755 The invention claimed in each of claims 16 and 17 of the 535 Application, claims 21–23 of the 537 Application and claims 19–23 of the 540 Application includes as an integer a bottle (535 and 537 Applications) or bottles (540 Application) containing an immunogenic composition. Further, some of the claims (claim 17 of the 535 Application, claim 22 of the 537 Application and claims 22 and 23 of the 540 Application) also include an instruction manual “containing information to administer the immunogenic composition” with the claimed kit.
756 An example “kit” claim is claim 21 of the 537 Application:
A kit for use in carrying out the method of claim 14 [a method of immunising a pig against M. hyo and PCV2] comprising: a bottle comprising an immunogenic composition including both a PCV2 antigen and the supernatant of [M. hyo] culture, wherein the supernatant of the M. hyo culture has been separated from insoluble cellular material by centrifugation, filtration, or precipitation and is substantially free of both (i) IgG and (ii) antigen/immunoglobulin immune complexes.
757 Claim 22 of the 537 Application is an example instruction manual claim:
The kit of claim 21, further including an instruction manual which contains the information to administer the immunogenic composition.
758 Boehringer submits the “kit” claims of the Applications are mere collocations of separate parts and that, as such, none of these claims are directed to a manner of manufacture. Although the bottle may provide a functional purpose (containing the composition) it plays no part in achieving the immunogenicity of the composition. Boehringer submits that the bottle of the kit claims is analogous to the aseptic packaging in claim 49 in Wake Forest.
759 Zoetis submits that unlike the aseptic packaging claims in Wake Forest that played no part in achieving the desired effect, the relevant claims in this case are not to “immunogenic compositions”, rather they are directed to kits for use in a method of immunisation.
760 Zoetis contends that Boehringer’s assertion that the desired effect of the invention is the “immunogenicity of the composition” does not reflect a fair reading of the Applications, which disclose at least the following additional effects that are material to methods of immunisation and kits for use therein:
(a) suitability for singe-dose administration;
(b) an M. hyo vaccine compatible with other porcine antigens, such as PCV-2 and PRRS virus, whether they are given concurrently as separate single vaccines or combined in a ready-to-use vaccine;
(c) the M. hyo soluble preparation as a “platform” for multivalent vaccines (without interference);
(d) single dose, ready-to-use, one bottle administration for a combination M. hyo/PCV-2 vaccine, which requires no mixing of separate vaccines, so there is no risk of contamination or additional labour associated with mixing and no requirement to use the mixture within a few hours;
(e) a one bottle combination vaccine, which cuts waste and refrigerated storage space in half, and eliminates the labour associated with administering a second dose to the animal; and
(f) a single dose trivalent vaccine, where the M. hyo/PCV-2 component is provided as a ready-to-use in one bottle liquid composition which can be easily combined with the PRRS component in a second bottle or container such that all antigens can be administered to the pig simultaneously.
761 According to Zoetis, those desired effects in relation to the vaccines are specifically contrasted in the specification (see, eg 535 Application, page 16, line 23 to page 17, line 2) with the prior art combination vaccines, such as Circumvent PCVM (a two-dose ready-to-use vaccine), or a single-dose, 2-bottle vaccine which requires the simultaneous administration of separate vaccines (eg Boehringer’s CircoFLEX and MycoFlex). The prior art M. hyo/PCV-2 combination vaccines are disadvantageous because they either require two dosing occasions, or one dosing occasion with two bottles.
762 Zoetis submits that the “single bottle” or “two bottles” integers of the combinations contribute to the achievement of one or more of these effects because they are the means by which a ready-to-use single composition, or a ready-to-use M. hyo/PCV-2 component, is provided to the user for carrying out the method of immunising a pig by administering the composition/s so provided (and in accordance with instructions, including as a single dose ready-to-use vaccine).
763 I consider that these asserted effects of the one-bottle kit inventions, which are described in the specifications, are akin to the benefits ascribed to the aseptic packaging. In Wake Forest, the aseptic packaging had a purpose: the specification stated that the apparatus was preferably packed in a sterile condition to avoid the need for sterilisation of the apparatus prior to use. That the aseptic packaging fulfilled its purpose and contributed to the achievement of the effect of sterile conditions was not sufficient for it to be found to interact purposefully and functionally with the other integers to produce negative wound pressure.
764 Zoetis further submitted that the storage of the antigens for a combination vaccine in separate bottles prior to administration limited assay interference as potency tests would be conducted on each of the separate bottles. This argument can only apply to the kit claims of the 540 Application, as it is the only Application with claims to a kit with two bottles.
765 Zoetis offered no separate argument in defence of the instruction manual claims.
766 Claim 16 of the 535 Application claims a kit for use in carrying out the method of claim 9. Claim 9 claims a method of immunising a pig against M. hyo via administering a composition of claim 1. The single bottle in this claimed invention has no role other than containing the solution prior to administration. There is no purposeful and functional interaction between the bottle and the immunogenic composition contents to immunise a pig against M. hyo.
767 Claim 21 of the 537 Application is also a claim to a single bottle including a combination vaccine, so there is no avoidance of assay interference via separate bottles for separate antigens in this case. There is no purposeful and functional interaction between the bottle and the immunogenic composition contents needed to immunise a pig against M. hyo and PCV-2.
768 Claims 19 and 20 of the 540 Application claim a kit comprising a first bottle, wherein the first bottle is provided as a ready-to-use liquid composition. Claim 22 makes express reference to the second bottle, wherein the PRRS virus antigen is in the form of a lyophilized composition. I accept that the two bottles may limit interference prior to administration of the trivalent vaccine, and thereby have a functional interaction with the other integers of the claimed invention.
769 The instruction manual claims add nothing but information. The instruction manual does not act purposefully or functionally with the other integers of the claims. The claims to the instruction manual are claims to a mere collocation.
770 For the reasons set out above, I consider that other than claim 2 of the 535 Application, all of the claims of the Applications are invalid.
I certify that the preceding seven hundred and seventy (770) numbered paragraphs are a true copy of the Reasons for Judgment of the Honourable Justice Rofe. |
Associate:

