
Federal Court of Australia
Pharmacia LLC v Juno Pharmaceuticals Pty Ltd [2022] FCA 92
ORDERS
DATE OF ORDER: |
THE COURT ORDERS THAT:
1. Pursuant to s 37AI of the Federal Court of Australia Act 1976 (Cth) and on the ground that it is necessary to prevent prejudice to the proper administration of justice, the text of the reasons for judgment published today may be published and disclosed only to the persons listed in order 1(a) of the orders made on 26 April 2021.
2. The parties confer and, by 1 March 2022, provide to chambers:
(a) an agreed form of the reasons for judgment that is suitable for publication, with redactions noted; and
(b) short minutes of order giving effect to these reasons marked-up to indicate any areas of disagreement.
3. The proceedings be listed for a case management hearing at 9.30am on 17 March 2022.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
[1] | |
[1] | |
[8] | |
[14] | |
[15] | |
[16] | |
[24] | |
[31] | |
[32] | |
[53] | |
[76] | |
[76] | |
[106] | |
[107] | |
[107] | |
[111] | |
[117] | |
[117] | |
[122] | |
[136] | |
[136] | |
[148] | |
[154] | |
[170] | |
[174] | |
[175] | |
[175] | |
[182] | |
[186] | |
[190] | |
[197] | |
[199] | |
[215] | |
[219] | |
[219] | |
[223] | |
[227] | |
[232] | |
[232] | |
[235] | |
[257] | |
[277] | |
[277] | |
[283] | |
7.6 Step 3: identification of parecoxib sodium and intravenous administration | [293] |
[297] | |
[298] | |
[306] | |
7.8 Step 5: testing parecoxib sodium and decision to formulate as a lyophilised composition | [322] |
[322] | |
[325] | |
[351] | |
[351] | |
[354] | |
[383] | |
[385] | |
[385] | |
[388] | |
[393] | |
[408] | |
[418] | |
[420] |
BURLEY J:
1 In these proceedings Pharmacia LLC and Pfizer Australia Pty Ltd (the applicants) seek relief from Juno Pharmaceuticals Pty Ltd and Neo Health (Australia) Pty Ltd (the respondents) for the infringement of patent no. 2002256031, which is entitled “Reconstitutable parenteral composition containing a COX-2 inhibitor”. The respondents deny infringement and advance a cross-claim alleging that the asserted claims in the patent are invalid.
2 Pharmacia and Pfizer, both related entities of Pfizer Inc., are respectively the owner and exclusive licensee in Australia of the patent. The patent relates to the formulation and administration of COX-2 inhibitors. Pharmacia is the developer of products that are marketed in Australia under the Dynastat brand for the management of post-operative pain. Pfizer markets and supplies the Dynastat products in Australia and is the sponsor of the registrations for those products on the Australian Register of Therapeutic Goods (ARTG).
3 The active ingredient in Dynastat is parecoxib, which is a “prodrug”, being a molecule that is converted into a pharmacologically active agent after administration to a patient. Parecoxib converts to the selective cyclooxygenase-2 (COX-2) inhibiting drug valdecoxib following administration to a patient. The inhibition of COX enzymes, of which one of the isoforms is COX-2, is believed to be a mechanism by which nonsteroidal anti-inflammatory drugs, or NSAIDs, exert anti-inflammatory, antipyretic (anti-fever) and analgesic effects.
4 The patent has a priority date of 3 April 2001 and was filed on 2 April 2002.
5 Juno offers for sale and supplies parecoxib products in Australia and, until August 2020, Neo supplied those products to Juno for sale on the Australian market. The applicants contend that both have infringed each of claims 1, 4, 5, 7, 11, 14, 15, 17-21, 24, 26-28, 30 and 34-42 (the relevant claims) of the patent. Juno has obtained ARTG registration for formulations of parecoxib (as sodium) for the indication “a single peri-operative dose for the management of post-operative pain” (Juno products). The applicants seek a declaration of infringement, injunctive relief, delivery up of infringing product and promotional materials, damages and other ancillary relief.
6 The applicable form of the Patents Act 1990 (Cth) is the form that it took following the Patents Amendment Act 2001 (Cth), but before the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth).
7 On 18 December 2019 orders were made that issues of pecuniary relief (including liability for and quantum of that relief) be heard and determined separately from and after the hearing of all other issues in the proceedings. Accordingly, this judgment concerns liability for non-pecuniary relief.
8 In broad terms the issues for determination by the court are as follows.
9 First, whether the Juno products fall within the relevant claims. The applicants have tendered records concerning the method of production and characteristics of six separate batches of the Juno products. They agree that the resolution of the identified disputes arising from those batches will be able to be applied across other batches produced by or for Juno.
10 Secondly, whether the relevant claims are invalid for lack of inventive step within s 18(1)(b)(ii) of the Patents Act. The respondents rely upon the common general knowledge as at the priority date and two pieces of prior art information that they contend fall within the prior art base. First, a paper by Kewal K Jain in the “Expert Opinion on Investigational Drugs” journal entitled “Evaluation of intravenous parecoxib for the relief of acute post-surgical pain” (Jain). Secondly, a paper by John J Talley, et al, in the “Journal of Medicinal Chemistry” entitled “N-[[(5-Methyl-3-phenylisoxazol-4-yl)-phenyl]sulfonyl]propanamide, Sodium Salt, Parecoxib Sodium: A Potent and Selective Inhibitor of COX-2 for Parenteral Administration”, pp 1661-1663 (Talley).
11 Thirdly, whether the relevant claims are invalid for lack of definition and/or clarity within ss 40(2)(b) and 40(3).
12 Fourthly, whether the applicants are entitled to injunctive relief if they are successful in establishing infringement of a valid claim.
13 In their amended particulars of invalidity the respondents additionally pleaded lack of novelty, lack of utility and lack of fair basis. However, in their opening submissions at the hearing they indicated that they did not press the first of these and part of the second and in closing submissions they shed the balance.
14 For the reasons set out in more detail below, I have found that three of the exemplar batches of the Juno products fall within the scope of claims 1, 4, 5, 11, 14, 15, 17, 18, 19, 20, 21, 24, 34, 35, 36, 37, 38, 39, 40 and 41 but not claims 7, 26, 27, 28, 30 or 42. I have found that the other three exemplar batches do not fall within the scope of any of the relevant claims. I have rejected the challenges to the validity of the relevant claims. I have also determined that the applicants are entitled to injunctive relief to restrain infringement.
15 Seven witnesses gave evidence for the purposes of the proceedings, six of whom were expert witnesses. Five of the expert witnesses were cross-examined. No challenges were made to the credit of any of the witnesses and I generally found them to be good witnesses who gave evidence in accordance with their obligations as experts to assist the court. In general, the expert witnesses were well qualified to give evidence on the subjects that they addressed. The parties made submissions as to the relative weight to be given to their evidence from time to time. I address specific aspects of those submissions, where relevant, during the course of my reasons.
16 Gerhard Johannes Winter is a professor and chair of Pharmaceutical Technology and Biopharmaceutics in the Department of Pharmacy at Ludwig-Maximilians-University, Munich. He studied pharmacy at the University of Heidelberg and graduated in 1982. He was awarded a PhD by the University of Heidelberg in 1987. From 1987 to 1988 he worked for Merck KGaA leading a laboratory for solid dosage form development, after which he worked at Boehringer Mannheim (which later became Roche) from 1988 until 1999. There he was responsible for parenteral (including freeze-dried parenteral) and liquid dosage form development across every step in the development process from pre-formulation through to the completion of the product for regulatory submission. During this period he led a large scale aseptic pilot plant to supply worldwide clinical studies with parenteral lyophilisates (freeze-dried products) and sterile injectable solutions. He was also involved in drug delivery research. From 1993, he was promoted to head the department for parenteral and liquid dosage form development and became responsible for overseeing a group of about five PhD level scientists who each oversaw their own teams. In 1997 he was promoted to become the deputy head of pharmaceutics development where he oversaw the formulation department, a group working on solid dosage forms and a group dealing with clinical supplies, manufacturing and managing the packaging of supplies to support clinical studies. While with Boehringer Mannheim/Roche he was involved in the development of a number of major drug products, including erythropoietin (EPO), reteplase (rPA), various antibodies and small molecular weight drugs such as ibandronate (a bisphospohonate), all of which became marketed formulations. In addition, Professor Winter worked on over 50 formulation candidates that did not make it to market, including small and large molecular weight drugs and liquid and lyophilised parenteral dosage forms.
17 In 1999, Professor Winter was appointed as a full professor and the chair of Pharmaceutical Technology and Biopharmaceutics in the Department of Pharmacy at Ludwig-Maximilians-University, Munich. Between 1999 and 2001, the focus of his academic work and research was on drying technology, including freeze-drying and new methods of freeze-drying. Since his appointment he has worked as a consultant for a number of companies, including Boehringer Ingelheim, Roche and MediGene. Professor Winter is a named author of numerous peer-reviewed publications, about 90% of which he estimates relate to formulation. Of those he estimates that at least 80% relate to parenteral formulations, of which about 30% concern freeze-dried parenteral formulations.
18 In his first affidavit, Professor Winter responds to an overview of the drug development process given by Dr Robertson in his first affidavit. The uncontroversial aspects of these affidavits were distilled into a formulation primer, to which I refer in section 3.1 below. In his second affidavit, Professor Winter was asked to address aspects of the patent and then give his opinion as to whether the Juno products fall within the relevant claims. In his third affidavit, Professor Winter identifies that, before he was given the patent and complied with the instructions to which he refers in his second affidavit, he was provided with the Talley article, and then the Jain article, and commented on their disclosure. He records that he was then asked what steps he would have taken, as at 3 April 2001, to formulate a parecoxib drug product and who he would have expected to have been in the team given that task. He was instructed to have regard to information which he regarded as well-known and generally accepted by pharmaceutical formulators as at 3 April 2001 as well as the information set out in Talley and Jain. After responding to this task he then responds to the second affidavit of Dr Robertson. Professor Winter joined with Dr Robertson in the preparation of a joint expert report and gave concurrent evidence with Dr Robertson during which he was cross-examined.
19 Charles Roger Goucke is a clinical associate professor at the Faculty of Health Sciences and Medicine at the University of Western Australia. He also practices as a specialist pain medicine physician. He obtained a Bachelor of Medicine and Bachelor of Surgery from the University of Liverpool, United Kingdom in 1972 and worked in various different countries after the completion of his training, working as a general practitioner from 1976 until 1981. He then trained as an anaesthetist and worked in Australia, the United Kingdom and Saudi Arabia before commencing work as a staff specialist in the Departments of Anaesthesia and Pain Management at Sir Charles Gairdner Hospital in Perth, Western Australia. In 1992 he was awarded fellowship of the Australian and New Zealand College of Anaesthetists and in 1994 he became the head of the Department of Pain Management at the Sir Charles Gairdner Hospital. Since the early 1990s his commitments have become increasingly focussed on pain management. In addition, he has held roles at the University of Western Australia as an honorary lecturer from 1989 to March 2005, as a clinical senior lecturer between 2005 and 2008, and since 2008 as a clinical associate professor.
20 Professor Goucke was asked to give evidence about his drug development experience, the hypothetical drug development team, literature searches that he would conduct and various hypothetical search strategies that he was shown. He gave one affidavit and was cross-examined.
21 Rodney Ian Lindsay Cruise is an IP researcher and patent attorney and a partner of Phillips Ormonde Fitzpatrick. He has over 31 years of experience in intellectual property research in the area of patents, trade marks and designs. He was awarded a Bachelor of Science (Applied Chemistry) degree by RMIT in 1987 and worked as a trainee patent attorney and then patent attorney from 1989 until he became a partner in 1999. Since 2000, he has managed IP Organisers Pty Ltd, an intellectual property research service company which specialises in conducting research in relation to intellectual property including patents.
22 Mr Cruise gives evidence that as at April 2001 he was experienced in using medical databases, having had experience using these databases since at least the early 1990s. He explains his understanding of using the medical database PubMed and the Medical Subject Headings, or MeSH terms, in that database as at April 2001. He gives evidence of various searches he conducted on PubMed as if he were doing so in April 2001. He also responds to parts of the affidavit evidence of Professor Scott and Dr Mokdsi, and gives evidence regarding the likelihood of Jain being located using MeSH terms. He gave one affidavit and was cross-examined.
23 Nicholas Alexander Tyacke is a partner at DLA Piper Australia and the solicitor with conduct of the proceedings on behalf of the applicants. In his affidavit, he annexes product information about the Juno products and confidential documents produced by the respondents relating to the development and manufacture of the Juno products (Neo confidential documents). Those confidential documents comprise batch records for six batches of Juno products and other documents regarding the manufacture and composition of the Juno products. Mr Tyacke was not cross-examined.
2.2 The respondents’ witnesses
24 Alan Duncan Robertson is a medicinal chemist who completed a Bachelor of Science degree in 1978 and a PhD in synthetic organic chemistry in 1981 at the University of Glasgow, Scotland. He is now the chief executive officer and managing director of Alsonex Pty Ltd, a biopharmaceutical company developing new treatments for neurodegenerative diseases. From 1984 until 1992 Dr Robertson worked at the Wellcome Foundation Ltd in the United Kingdom in the roles of senior chemist, medicinal chemist and senior research scientist. As a senior research scientist he was appointed as leader of a project investigating potential new drugs for the treatment of migraines. That project led to the development of a new drug marketed as Zomig and included the formulation of an oral presentation of the drug. In July 1992, Dr Robertson moved to Australia and worked as product development manager at David Bull Laboratories, a division of F.H. Faulding and Co Ltd in Melbourne, where he was responsible for running the new product development department that included a team of formulation scientists responsible for the development of injectable drugs. There he managed a team of about 12 scientists, including several at PhD level. His work included the development of injectable formulations for generic products, some of which were lyophilised (freeze-dried) products. During this time he directed and supervised formulators in their work and from time to time did benchtop formulation work, although he was mainly in a supervisory position. He was present on a day-to-day basis during the development of lyophilisation processes. From 1994 until 1999, Dr Robertson worked as pharmaceutical development manager at Amrad Operations Pty Ltd. Whilst there he oversaw the building of a drug development laboratory as well as work on new drug formulations, including for a potential HIV treatment known as conocurvone. That compound had significant solubility issues which needed to be addressed by a new formulation. He also worked on formulating Propofol, an anaesthetic product. Another project that he worked on whilst at Amrad was a collaboration with the University of Queensland which targeted pain management and involved synthesising the peptide conotoxin (taken from the cone snail) and formulating a stable solution of the product to be used in clinical trials. In his time at Amrad his role insofar as it concerned the task of formulation was to direct, supervise and formulate work that needed to be done by the formulation scientists. From 1999 until 2002 he worked as an independent contractor for several companies and research organisations in which his roles involved the formulation of new chemical entities for administration to humans and animals.
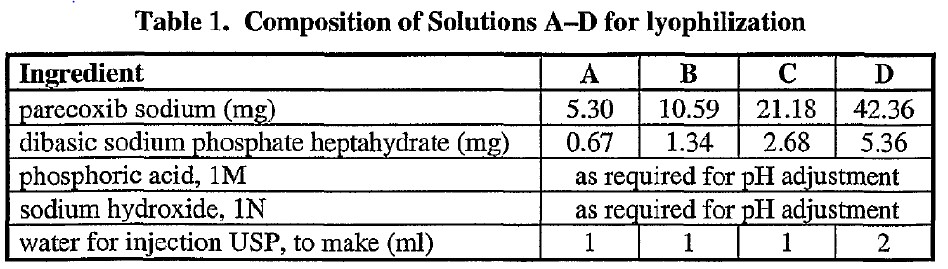
25 In his first affidavit, Dr Robertson first gives an overview of the drug development process, parts of which form the basis for the formulation primer. In his second affidavit, Dr Robertson was provided with a copy of the Jain article and a copy of the first affidavit of Professor Scott. He was given instructions to consider the task as at 3 April 2001 of formulating a pharmaceutical composition for administration to patients in which parecoxib is the active pharmaceutical ingredient, having regard only to the information contained in the first Scott affidavit, the Jain article, the information in his first affidavit and the common general knowledge of the person skilled in the art as at 3 April 2001. He proceeds to address that task. He then gives evidence about the disclosure of the patent, which was provided to him after he had reached his views regarding the formulation task. In his third affidavit, Dr Robertson responds to the evidence of Professor Winter in relation to his earlier evidence, and takes issue with aspects of Dr Winter’s evidence on the question of infringement.
26 Dr Robertson joined with Professor Winter in the preparation of a joint expert report, gave concurrent evidence with Professor Winter and was cross-examined during the course of the concurrent evidence session.
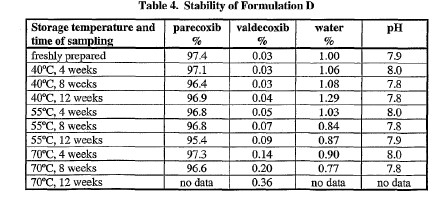
27 David Anderson Scott has been a specialist anaesthetist since 1985. He was the founder and head of the Acute Pain Service at St Vincent’s Hospital in Melbourne from 1989 until 2009. He has a professional interest in acute pain medicine and acute postoperative pain in particular and, in 1998, undertook a PhD in the neuropharmacology of neuropathic pain, which he completed in 2004. He lectures regularly on the subject of acute pain management and in 2005 was awarded a Fellowship of the Faculty of Pain Medicine, Australian and New Zealand College of Anaesthetists.
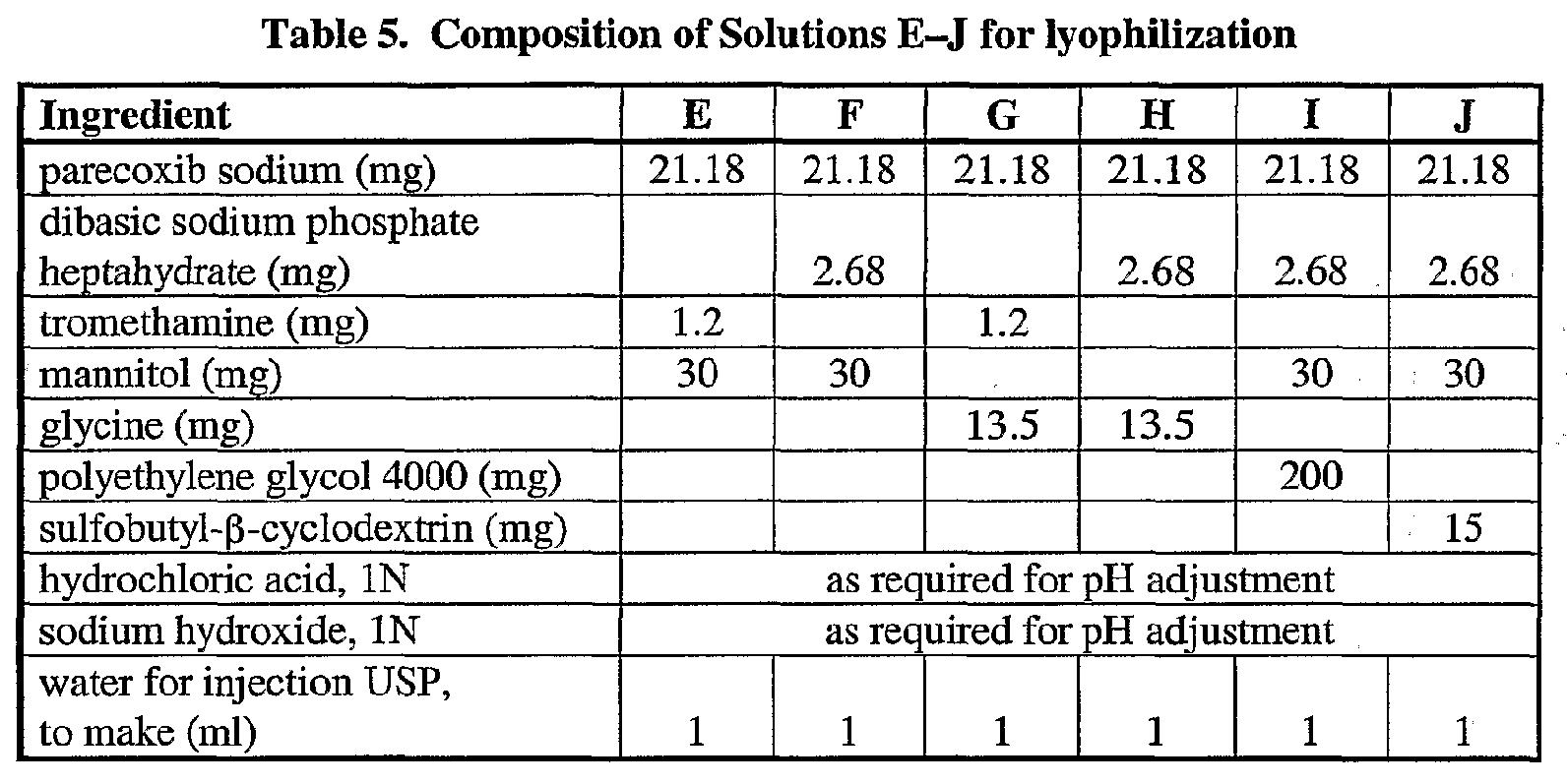
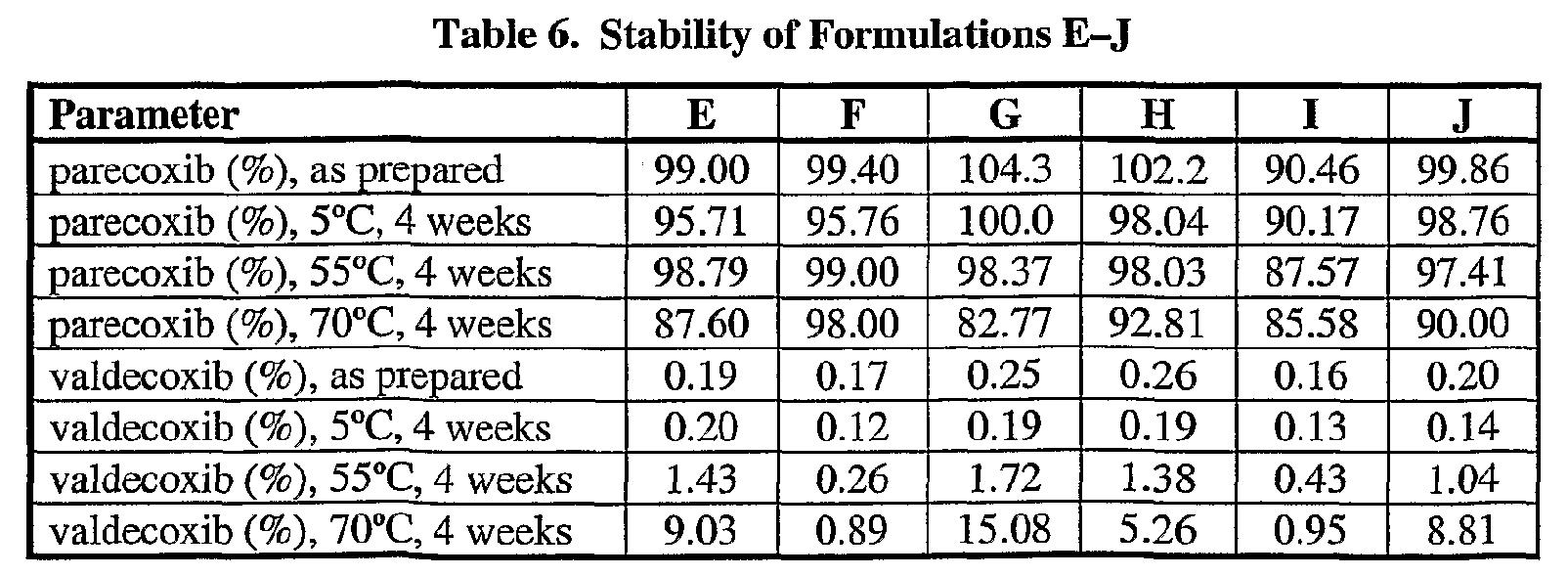
28 In his first affidavit, Professor Scott provides an overview of the management of acute post-surgical pain as at 3 April 2001 which forms the basis for the pain primer to which I refer in section 3.2 below. In his second affidavit, Professor Scott responds to an instruction to assume that he is part of a notional research group, including one or more additional experts playing different roles such as a pharmaceutical formulation specialist, which has been asked to identify and formulate an analgesic for the Australian market which would be better than, or at least a useful alternative to, the drugs then available for use for the management of acute post-surgical pain. He gives evidence of the strategy that he would adopt to conduct a literature search to address this subject as at the priority date, and exhibits his results. Amongst the materials he locates is the Jain article. After affirming his second affidavit, Professor Scott was then provided with two affidavits given by Dr Mokdsi and one by Mr Cruise. In his third affidavit, Professor Scott confirms his view that Dr Mokdsi correctly implemented his search strategy. Professor Scott was cross-examined.
29 George Mokdsi is a patent searcher and a director of The Patent Searcher Pty Ltd, a company he founded in 2019 that performs patent searches. He was awarded a Bachelor of Science (Hons) and a PhD in chemistry from the University of Sydney in 1996 and 2000 respectively. Since 2000, he has worked as an intellectual property analyst, specialising in patent analysis and research for the duration of his career.
30 In his first affidavit, Dr Mokdsi reports that he conducted a search using the PubMed online database as if it were April 2001 according to a search strategy provided to him by the solicitors for the respondents, and exhibits the results. In his second affidavit, Dr Mokdsi responds to the evidence of Mr Cruise. He refers to receiving supplementary instructions for the conduct of a search and annexes the results of that search, both of which he had omitted to include in his first affidavit. He responds to the criticisms made by Mr Cruise by conducting further searches. Dr Mokdsi was not cross-examined.
3. BACKGROUND COMMON GENERAL KNOWLEDGE
31 The following background information is extracted from the formulation primer and the pain primer. The parties agree that it forms part of the background common general knowledge as at the priority date. In the case of the formulation primer, there were some limited areas of disagreement or differences in emphasis between Professor Winter and Dr Robertson (the formulation experts). To the extent necessary, in what follows I have included my findings as to the (limited) areas of dispute.
32 In the pharmaceutical industry, as the drug molecule moves through its development it is often described as the active ingredient, the drug substance or active pharmaceutical ingredient (API) and these terms are often used interchangeably. The API may be defined as the therapeutically active ingredient in a pharmaceutical drug product and is the chemical entity that provides the clinical benefit.
33 One of the important preclinical aspects of testing a new drug molecule is to determine its ADMET properties. ADMET refers to Absorption, Distribution, Metabolism, Excretion and Toxicity. The ADMET team generally consists of experts in drug metabolism, pharmacokinetics and toxicology and involves the analysis of the drug in various biological matrices. The purpose of ADMET testing is to obtain information that will allow the API to proceed into clinical trials in humans (hence the name "preclinical development stage"). ADMET testing provides an initial view of the API’s toxicity compared with its efficacy. It therefore serves as a means of selecting the starting dosage in healthy volunteers in Phase I clinical trials. The objective of a Phase I clinical trial is to determine if the drug can be delivered safely and engage with its target.
34 The formulation used in the Phase I clinical trials is preferably, but not invariably, the same as that used in the Phase II and Phase III clinical trials and in the commercial product. Because of this, the formulator is typically involved in the preclinical stage. The formulator's role is to formulate the API into a commercially viable formulation that can be used in clinical trials and then to market. The formulation stage is described further below.
35 There is generally a considerable amount of time between the Phase I trials and the commercial product being finalised and changes in the formulation are commonly made during that time due to various reasons. For example, a liquid formulation might be changed to a lyophilisate due to a realisation that the liquid formulation does not remain stable for a sufficient amount of time for a commercial product.
36 A drug development team will consider the optimum route of delivery for the drug, which should be convenient for the person administering the drug and convenient for the person receiving the drug. The selected route of administration of the drug will significantly affect how the drug is formulated.
37 Oral administration is the most desirable and the most common route of drug delivery, however, alternative routes are often used, including, but not limited to, injectable, inhalation, rectal, sublingual and transdermal. Routes of administration other than oral are often used if, for example, the patient is unable to or has difficulties in swallowing oral medication, the patient is unable to or has difficulties in absorbing the oral medication through the gastrointestinal (GI) tract or a faster-onset of action is required.
38 Injectable routes of administration typically include, but are not limited to, the subcutaneous (under the skin), intramuscular and intravenous. Reasons for choosing an injectable route over an oral route for example include drugs with low oral bioavailability, patients who are unable to take the drug by mouth (e.g., it irritates the GI tract), the need for immediate effect (e.g., emergency situations), the patient is unconscious or otherwise not able to swallow or there is a desire to control the rate of absorption and duration of effect.
39 When commencing a formulation task, the formulator seeks to understand the properties of the API (having regard to the selected route of administration).
40 Where the drug development involves the formulation of a known drug molecule, the formulator will acquire publicly available information about the relevant properties of the API, for example, from a search of relevant medical or chemical databases. Where the drug is a new drug molecule the formulator will attempt to acquire any information about the relevant properties of similar APIs from a search of relevant medical or chemical databases. In both instances, the formulator will give consideration to the chemical structure of the drug molecule.
41 Properties of the API in which the formulator will be interested include the solubility of the API in aqueous solutions, dissolution rate (which is the speed at which the API can go into dissolution), ionisation potential (which is relevant to the ability of the API to form salts and the pH of the resulting drug substance solution, which may require adjustment of buffering), stability of the API and its salts if any and any excipient interactions with the API.
42 It is generally critical that injectable formulations, particularly formulations prepared for intravenous use, do not contain any particulate matter, and that precipitation of either the API or added excipients does not occur during or following injection. Solid particulate matter can in many cases cause thrombosis and the formation of blood clots that can be dangerous to the subject receiving the injection.
43 For any dosage form and route of administration, a minimum desired stability for a commercial pharmaceutical product is generally about two years. For some vaccines, for example, it can be shorter.
44 Excipients are substances other than the API that may be included in a formulation. Ideally, excipients have no biological activity in their own right and are only included in any given formulation of an API if they are required to solve a particular problem.
45 Consideration which may lead to the use and types of excipients will vary depending upon the form of the drug product (e.g. solid or liquid) and its route of administration. A particular excipient will only be added to a formulation if it achieves a particular purpose. The purposes of adding an excipient to a formulation may include, for example:
(a) to enhance stability, bioavailability or patient acceptability;
(b) to assist in the effectiveness of delivery; and
(c) to assist in maintaining the integrity of a drug product during storage.
46 Compounds used as excipients are intended to be inert, that is, they are intended not to react with the API. In approaching the formulation of an API, formulators will seek to avoid any non-reversible reactions between the API and potential excipients.
47 Excipients are generally selected from a list of Generally Regarded as Safe (GRAS) substances. GRAS substances are those which are already well accepted by government drug regulators, including the Therapeutic Goods Administration (TGA) in Australia and the Food and Drug Administration in the United States. There is a very large number of commonly used excipients.
48 Excipients commonly used in pharmaceutical tablets and capsules include diluents or fillers, binders, disintegrants, lubricants, colouring agents and preservatives.
49 Excipients may be required to ensure a satisfactory production process.
50 Fillers, binders and disintegrants may be added to the drug substance to increase its bulk and impart desirable properties lacking in the drug substance alone. Other common excipients for injectable pharmaceutical solutions may include antioxidants and metal ion chelators.
51 pH is a measure of hydrogen ion concentration of a solution. The term translates the values of the concentration of the hydrogen ion – which ordinarily ranges between about 1 and 10-14 gram-equivalents per litre – into numbers between 0 and 14. Aqueous solutions at 25°C with a pH less than 7 are acidic, while those with a pH greater than 7 are basic or alkaline. It is a logarithmic scale based on the number of hydrogen ions (H+) in solution which can be expressed as pH = -log[H+]. The pH changes by 1 for every power of 10 change in hydrogen ion concentration. For example, a solution with a pH of 3 has 10 times the concentration of hydrogen ions than a solution with a pH of 4 and 100 times the concentration of hydrogen ions than a solution with a pH of 5.
52 A physiologically acceptable pH of a pharmaceutical solution for an intravenous injection needs to take into account the pH of blood which is 7.4. Generally, a pH value of no more than plus or minus about 1.0 log unit from 7.4, so a pH of about 6.4 to 8.4, is considered acceptable. Formulations outside this range run the risk of causing irritation and inflammation at the site of injection, although such formulations do exist.
53 Pain is a distressing feeling often caused by intense or damaging stimuli. Post-surgical pain refers to pain experienced immediately after, and as a consequence of, surgery. Post-surgical pain, or post-operative pain (also referred to as peri-operative pain), is an example of acute pain. Almost all surgical procedures are associated with some form of acute post-surgical pain.
54 Acute postoperative pain can limit recovery from the procedure and may lead to the development of chronic pain. Chronic post-surgical pain is typically defined as pain still present three months after surgery.
55 Acute post-surgical pain is managed, variously, by different classes of analgesics (drugs that relieve pain) in addition to other techniques to manage pain. For example, physical and physiotherapy techniques. The use of a range of drugs and treatment techniques is called multi-modal analgesia.
56 The classes of analgesic drugs used to manage acute post-surgical pain include opioids, non-steroidal anti-inflammatory drugs (NSAIDs), other non-opioid analgesics and adjuvants such as clonidine and ketamine. Other analgesic techniques, often used in combination with these analgesic drugs, include the use of local anaesthetic techniques.
57 A multi-modal approach to managing acute post-surgical pain by treating a patient with a range of analgesic medications is commonly used to reduce the total dose of individual analgesic medications required and to treat pain by targeting different mechanisms and neuronal pathways. This helps to manage acute pain while reducing medication-related side effects.
58 Opioids have long been considered the gold standard for post-surgical analgesia, as they are highly effective for the treatment of acute pain, and able to be administered by a number of routes, including parenterally.
59 Opioids are morphine-like drugs that work by binding to opioid receptors, which are found principally in the central nervous system (spinal cord and brain), and also the gastrointestinal tract. The term opioid encompasses both naturally occurring opiates (e.g. morphine and codeine) derived from the resin of the opium poppy, semi-synthetic (e.g. oxycodone) and synthetic opioids (e.g. fentanyl).
60 Opioids are used primarily for their strong analgesic effect. However, opioid analgesics may have adverse effects such as respiratory depression, sedation, cough suppression, nausea and vomiting and impaired bladder and bowel function. With longer term use, the development of tolerance to their effects, and also opioid-induced hyperalgesia, limit their effectiveness. The onset of opioid dependence and addiction is also a risk factor in a minority of cases.
61 When opioids are used as part of a multi-modal analgesic approach, the dose of opioid required is often able to be decreased resulting in fewer opioid-related side effects (“opioid sparing” benefits).
62 The most common non-opioid analgesics for treating acute post-operative pain are NSAIDs and acetaminophen (paracetamol). Following some types of surgery, these analgesics are used in conjunction with local anesthetic techniques and also adjuvant medications, depending on the circumstances.
63 For milder forms of acute post-operative pain, NSAIDs and paracetamol can each be used alone or in combination for the treatment of acute post-operative pain. Where stronger analgesia is required, other agents (such as opioids) or techniques (such as with local anaesthetics) can be added to these to treat the post-operative pain.
64 Paracetamol is a weak non-opioid analgesic that lacks anti-inflammatory properties, but reduces fever and has good gastrointestinal tolerability. It has minimal side effects. As at April 2001, it could only be administered in Australia by enteral routes (i.e., orally or suppository).
65 NSAIDs are class of drugs that reduce pain and fever and decrease tissue inflammation. They work primarily by inhibiting the activity of cyclooxygenase enzymes (COX-1 and/or COX-2). In cells, these enzymes are involved in the synthesis of key biological mediators, including prostaglandins, which are involved in inflammation, and thromboxanes, which are involved in blood clotting. They are also involved in pain processing in the central nervous system. COX-2 enzymes are increased in activity in response to triggers such as tissue injury.
66 There are two types of NSAIDs available: non-selective and COX-2 selective.
67 Most NSAIDs are non-selective and inhibit the activity of both COX-1 and COX-2. These NSAIDs, while reducing inflammation, also inhibit platelet aggregation and may increase bleeding following certain operations. Such NSAIDs also increase the risk of gastrointestinal ulcers and bleeds as a result of decreased gastric cytoprotection. This is because non-selective NSAIDs inhibit COX-1 which ordinarily serves a homeostatic role in the function of kidney, gut mucosa, smooth muscle, platelets and endothelium.
68 The most commonly used non-selective NSAIDs are aspirin, indomethacin, ibuprofen, naproxen and diclofenac all of which are available over the counter (OTC) in tablet form in most countries. In acute post-surgical pain management, the non-selective NSAID ketorolac can be administered orally and parenterally (intravenous or intramuscular).
69 COX-2 selective inhibitors (or Coxibs) preferentially inhibit COX-2 and do not significantly inhibit COX-1 at therapeutic concentrations. They, therefore, have a superior safety and tolerability profile by causing less gastrointestinal and bleeding side effects, whilst providing effective analgesia. As of 3 April 2001 COX-2 selective inhibitors were of considerable interest for use as part of multi-modal analgesia in post-operative acute pain management instead of non-selective NSAIDs.
70 As at 3 April 2001, celecoxib (Celebrex) and rofecoxib (Vioxx), both COX-2 selective inhibitors, were registered on the Australian Register of Therapeutic Goods and were available in Australia. Rofecoxib in particular was used for the management of acute post-operative pain. Celecoxib was then being used for a number of indications which also included the treatment of acute post-operative pain. These drugs were only available in oral tablet form.
71 As at 3 April 2001, certain analgesics for the management of acute post-operative pain were available in oral or rectal dosage forms (e.g. tablets/capsules, liquids, suppositories) whereas other analgesics were available to be administered parenterally. A parenteral route of administration is typically one given by injection.
72 The mode of delivery of medication for the treatment of acute post-surgical pain is an important consideration. When managing acute post-operative pain perioperatively, parenteral analgesics are often preferred over oral analgesics, particularly if the analgesic needs to be administered when the patient is under anaesthesia, when rapid onset analgesia is required or when the patient is unable to swallow medications or is unable to absorb medications in the gastrointestinal (GI) tract (e.g. following bowel surgery).
73 As at 3 April 2001, the majority of opioid analgesics, when used for the management of acute post-operative pain, were administered parenterally. These included, for example, morphine (available in parenteral and oral forms), pethidine (parenteral) and fentanyl (parenteral). Some opioid analgesics for the treatment of acute post-operative pain were only available in enteral dosage forms such as oxycodone.
74 As at 3 April 2001, most of the non-selective NSAIDs could only be administered enterally. Ketorolac (a non-selective NSAID) was available for intramuscular use (or intravenously, off-label) for acute post-surgical pain management. However, as with other non-selective NSAIDs, it was known to have been associated with complications including operative site bleeding, GI bleeding and ulceration, and renal failure. These complications could sometimes be severe or even fatal.
75 As at 3 April 2001, all of the COX-2 selective inhibitors available in Australia were only available in enteral dosage forms.
76 The patent is entitled “Reconstitutable parenteral composition containing a COX-2 inhibitor”. It commences with a description of the field of the invention which is said to relate to water-soluble selective COX-2 inhibitory drugs and salts and prodrugs thereof, and in particular to parecoxib, such as in the form of its sodium salt (parecoxib sodium). It notes that parecoxib is a water-soluble prodrug of the selective COX-2 inhibitory drug valdecoxib. It goes on to say (page 1 lines 6 – 13):
More particularly, the invention relates to parenterally deliverable, for example injectable, formulations of water-soluble selective COX-2 inhibitory drugs and salts and prodrugs thereof. Even more particularly, the invention relates to such formulations that are prepared as powders for reconstitution in an aqueous carrier prior to parenteral administration. The invention also relates to processes for preparing such reconstitutable formulations, to therapeutic methods of use of such formulations and to use of such formulations in manufacture of medicaments.
77 The background of the invention then provides some information about COX inhibitory drugs. It states that inhibition of cyclooxygenase enzymes is believed to be at least the primary mechanism by which NSAIDs exert their anti-inflammatory, antipyretic (anti-fever) and analgesic (pain relieving) effects, through inhibition of prostaglandin synthesis. It refers to “conventional” NSAIDs such as ketorolac, diclofenac, naproxen and salts thereof as inhibiting both COX-1 and the inflammation-associated or inducible COX-2 isoforms of cyclooxygenase at therapeutic doses. A disadvantage with this is that the inhibition of COX-1, which produces prostaglandins that are necessary for normal cell function, appears to account for adverse side effects associated with the use of conventional NSAIDS (page 1 lines 15 – 23). These side effects are identified later in the background as including (page 2 lines 15 – line 20):
...upper gastrointestinal tract ulceration and bleeding, particularly in elderly subjects; reduced renal function, potentially leading to fluid retention and exacerbation of hypertension; and inhibition of platelet function, potentially predisposing the subject to increased bleeding, for example during surgery. Such side effects have seriously limited the use of parenteral formulations of non-selective NSAIDs.
78 The background states that, by contrast, selective inhibition of COX-2 without substantial inhibition of COX-1 leads to anti-inflammatory, antipyretic, analgesic and other useful therapeutic effects while minimising or eliminating such adverse side effects. Examples are given of COX-2 inhibitory drugs such as celecoxib and rofecoxib, said to have been first commercially available in 1999, that have represented a major advance in the art (page 1 lines 23 – 28).
79 The background then observes that the selective COX-2 inhibitors mentioned are formulated in a variety of orally deliverable dosage forms. It goes on (page 1 line 30 – page 2 line 11):
Parenteral routes of administration, including subcutaneous, intramuscular and intravenous injection, offer numerous benefits over oral delivery in particular situations, for a wide variety of drugs. For example, parenteral administration of a drug typically results in attainment of a therapeutically effective blood serum concentration of the drug in a shorter time than is achievable by oral administration. This is especially true of intravenous injection, whereby the drug is placed directly in the bloodstream. Parenteral administration also results in more predictable blood serum concentrations of the drug, because losses in the gastrointestinal tract due to metabolism, binding to food and other causes are eliminated. For similar reasons, parenteral administration often permits dose reduction. Parenteral administration is generally the preferred method of drug delivery in emergency situations, and is also useful in treating subjects who are uncooperative, unconscious, or otherwise unable or unwilling to accept oral medication.
80 The specification identifies the area of the present invention saying (page 2 lines 21 – 23):
It would therefore represent a further significant advance in the art if a parenterally deliverable formulation of a selective COX-2 inhibitory drug could be provided.
81 The specification continues (page 2 line 24 – page 3 line 3):
It is known to prepare parenteral formulations by a process of lyophilization (freeze-drying) of an aqueous solution of the therapeutic agent. See for example Remington: The Science and Practice of Pharmacy, 19th edition (1995), Mack Publishing, pp.1544-1546. According to Remington, excipients often are added to the therapeutic agent to increase the amount of solids, so that the resulting powder is more readily visible when the amount of the therapeutic agent is very small. “Some consider it ideal for the dried-product plug to occupy essentially the same volume as that of the original solution. To achieve this, the solids content of the original product must be between approximately 5 and 25%. Among the substances found most useful for this purpose, usually as a combination, are sodium or potassium phosphates, citric acid, tartaric acid, gelatin and carbohydrates such as dextrose, mannitol and dextran.” Remington, loc. cit.
82 The specification then states that parecoxib was disclosed in US Patent No. 5,932,598 (598 patent) and is one of a class of water-soluble prodrugs of selective COX-2 inhibitory drugs. It says of the characteristics of the prodrug (page 3 lines 5 – 15):
Parecoxib rapidly converts to the substantially water-insoluble selective COX-2 inhibitory drug valdecoxib following administration to a subject. Parecoxib also converts to valdecoxib upon exposure to water, for example upon dissolution in water. The high water solubility of parecoxib, particularly of salts of parecoxib such as the sodium salt, by comparison with most selective COX-2 inhibitory drugs such as celecoxib and valdecoxib, has led to interest in developing parecoxib for parenteral use. Parecoxib, having the structural formula (I) below, itself shows weak in vitro inhibitory activity against both COX-1 and COX-2, while valdecoxib (II) has strong inhibitory activity against COX-2 but is a weak inhibitor of COX-1.
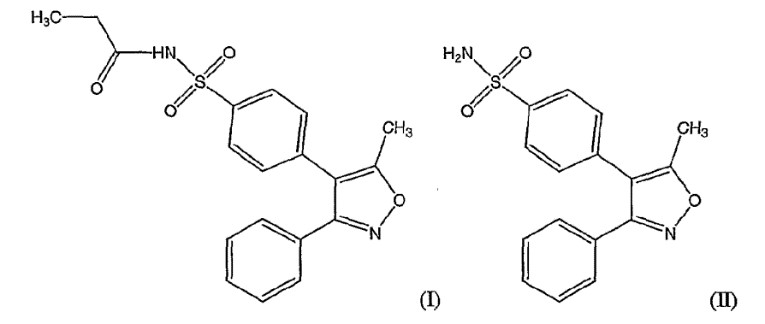
83 The specification then goes on to identify other known water-soluble selective COX-2 inhibitory drugs as including a series of water-soluble benzopyrans, and provides a structural formula for a particular compound within that class.
84 The background concludes with what the applicants characterise as a statement of the problem sought to be solved by the inventors (page 4 lines 2 – 7):
While these and other selective COX-2 inhibitory drugs and prodrugs have been proposed in general terms for parenteral administration, no pharmaceutically acceptable injectable formulation of such drugs or prodrugs has hitherto been described. As will be clear from the disclosure that follows, numerous problems have beset the formulator attempting to prepare such a formulation, illustratively of parecoxib. The present invention provides a solution to these problems.
The invention is thus said to involve a formulation that overcomes particular problems.
85 The specification then has a section entitled “Summary of the Invention”. It commences with what the parties accept is, broadly, a consistory clause for claim 1 (page 4 lines 9 – 17):
There is now provided, in one embodiment, a pharmaceutical composition comprising, in powder form, (a) at least one water-soluble therapeutic agent selected from selective COX-2 inhibitory drugs and prodrugs and salts thereof, in a therapeutically effective total amount constituting about 30% to about 90% by weight, (b) a parenterally acceptable buffering agent in an amount of about 5% to about 60% by weight, and optionally (c) other parenterally acceptable excipient ingredients in a total amount not greater than about 10% by weight, of the composition. The composition is reconsitutable in a parenterally acceptable solvent liquid, preferably an aqueous liquid, to form an injectable solution.
This embodiment is said to include a COX-2 inhibitor in an amount of about 30% to about 90%, a buffer in an amount of about 5% to about 60% and other excipients in up to about 10% by weight of the powder composition.
86 The specification refers to other embodiments of the invention including a process comprising a step of lyophilisation, an injectable solution prepared by the reconstitution of the lyophilised composition, an article of manufacture being a vial containing the lyophilised composition and a method of treatment using the composition.
87 The specification includes a section entitled “Detailed Description of the Invention”. It commences (page 5 lines 25 – page 6 line 8):
A pharmaceutical composition of the present invention comprises as the therapeutic agent:
(a) a water-soluble selective COX-2 inhibitory drug;
(b) a water-soluble salt of a selective COX-2 inhibitory drug, whether or not such drug is itself water-soluble;
(c) a water-soluble prodrug of a selective COX-2 inhibitory drug, whether or not such drug is itself water-soluble; or
(d) a water-soluble salt of a prodrug of a selective COX-2 inhibitory drug, whether or not such drug is itself water-soluble.
More than one such therapeutic agent can be present, but in general it is preferred to include only one such selective COX-2 inhibitory drug or prodrug or salt thereof in the composition.
(emphasis added)
88 The specification gives a definition of “water soluble” and provides, from page 6 to page 11, a lengthy list of selective COX-2 inhibitory drugs that may be used in the formulation of the invention, by reference to registered patents. Parecoxib is identified as a particularly useful prodrug of valdecoxib and more particularly a water-soluble salt of parecoxib, such as parecoxib sodium, is said to be preferred (page 13 lines 8 – 11). Parecoxib is said “illustratively” to be prepared (i.e., synthesised) in the manner set out in the 598 patent (page 13 lines 12 – 13).
89 The specification then addresses the component parts of the invention and their preferable relative amounts. It refers to the therapeutic agents and then the buffering agent, noting that an especially preferred buffering agent is dibasic sodium phosphate (page 14 lines 20 – 22). It states (page 14 lines 23 – 27):
In one embodiment, the pH of the composition upon reconstitution is about 7 to about 9, preferably about 7.5 to about 8.5, for example about 8. If desired, pH can be adjusted by including in the composition, in addition to the buffering agent, a small amount of an acid, for example phosphoric acid, and/or a base, for example sodium hydroxide.
90 The expert evidence and the parties’ submissions use the term pH adjusters to describe the use of a small amount of acids or bases to adjust pH in the formulation.
91 In a passage that is relevant to a number of issues, the specification provides (page 14 line 28 – page 15 line 10):
Excipients other than the buffering agent, if present, constitute not more than about 10%, preferably not more than about 5%, by weight of the composition prior to reconstitution. The term “excipient” herein embraces all non-therapeutically active components of the composition except for water. In one embodiment of the invention, no excipients other than the buffering agent are substantially present.
Surprisingly, it has been found important to include in the composition no more than about 10% by weight, preferably no more than about 5% by weight, and most preferably substantially no amount, of ingredients commonly used as bulking agents in reconstitutable parenteral formulations, other than buffering agents. In particular, the widely used bulking agent mannitol is preferably excluded from the composition, or if included, is present at no more than about 10%, preferably no more than about 5%, by weight of the composition. According to the present invention, it is believed that by minimizing the amount of, or excluding altogether, such bulking agents, especially mannitol, as components of the composition, acceptable chemical stability of the therapeutic agent can be assured.
(emphasis added)
92 The specification then refers to the residual water in the composition of the invention at page 15 lines 14 – 22:
A reconstitutable powder composition of the invention preferably contains less than about 5%, more preferably less than about 2%, and most preferably less than about 1%, by weight of water. Typically the moisture content is about 0.5% to about 1% by weight. It is especially important to keep the amount of water to such a low level where the therapeutic agent has a tendency to degrade or convert to a less soluble form in presence of water. Powder compositions of the invention exhibit acceptable chemical stability of the therapeutic agent for at least about 30 days, preferably at least about 6 months, most preferably at least about 2 years, when stored at room temperature (about 20-25°C) in a sealed vial.
93 The specification then defines what is said to be “acceptable chemical stability”. It then refers further to the circumstance where parecoxib is the therapeutic agent. It notes that partial conversion to valdecoxib can occur in a composition over time, and because valdecoxib is itself therapeutically active, such conversion does not result in a loss of therapeutic effect. However, as valdecoxib has extremely low solubility in water, the specification provides that it is desirable to minimise such conversion prior to reconstitution so that complete dissolution of the therapeutic agent is assured. Furthermore, it notes that particulates in the form of significant quantities of valdecoxib are undesirable. In this context, the specification notes that conversion from parecoxib to valdecoxib in a reconstitutable powder composition can be reduced by the reduction or elimination of bulking agents such as mannitol, as illustrated in examples 1 and 2 (page 16 lines 8 – 11).
94 The specification further addresses the undesirability of the conversion from parecoxib to valdecoxib in the powder composition, and notes that in an aqueous medium such conversion can be greatly reduced by maintaining the medium at a pH of about 7 or higher (page 16 lines 32 – 33). It also notes that aqueous solubility of parecoxib sodium itself is strongly affected by pH.
95 The specification goes on to note that a powder composition of the invention preferably will have sufficient porosity to permit rapid dissolution of the therapeutic agent upon reconstitution in the solvent liquid. It describes a process to prepare the powder by which this can be achieved, by reference to parecoxib sodium (page 17 line 32 – page 18 line 7):
In this process, parecoxib sodium and dibasic sodium phosphate heptahydrate as buffering agent are dissolved in water to form an aqueous solution. Preferably water for injection is used as the solvent. Parecoxib sodium and the buffering agent are present in the solution at concentrations relative to each other consistent with the desired relative concentrations of these ingredients in the final composition. Absolute concentrations of these ingredients are not critical; however, in the interest of process efficiency it is generally preferred that the concentration of parecoxib sodium be as high as can be conveniently prepared without risking exceeding the limit of solubility...
96 The specification then describes how the solution is placed into sterilised vials, each vial receiving “a measured volume of solution having a desired unit dosage amount of parecoxib sodium”. They are stoppered (having an opening for sublimation to occur) and placed in a lyophilisation chamber to be lyophilised, preferably in a three-phase cycle involving:
(1) the solution in each vial being frozen to a temperature below the glass transition temperature of the solution;
(2) freeze-drying by drawing a vacuum in the lyophilisation chamber during which ice sublimates from the frozen solution, forming a partially dried cake. During this phase the temperature is raised from freezing temperature (for example about -40°C to about 0°C over a period of hours) and then held at 0°C for a prolonged period; and
(3) completing the drying under a vacuum during which the temperature is further raised to about 40°C “to drive off remaining moisture and provide a powder having a moisture content of less than about 5%, preferably less than about 2%, more preferably less than about 1%, by weight”.
97 The specification refers in more detail to other embodiments of the invention, including the article of manufacture (a glass vial containing a powder composition), disorders that can be treated and methods of treatment.
98 The specification provides four examples, which are said to illuminate aspects of the invention but “are not to be construed as limitations” (page 29 lines 12 – 13).
99 Example 1 identifies four reconstitutable powder compositions A, B, C and D containing, respectively, 5, 10, 20 and 40 mg dosage amounts of parecoxib in the form of parecoxib sodium. It describes that solutions for lyophilisation were prepared having compositions set out in Table 1, as follows:
100 The preparation of the solutions is described as follows. Dibasic sodium phosphate heptahydrate was dissolved in a suitable volume of water for injection and pH of the resulting solution adjusted to 8.1 using 1M phosphoric acid. Parecoxib sodium was then dissolved in this solution and the pH checked and readjusted “if necessary” with 1M phosphoric acid or 1N sodium hydroxide and the volume adjusted to a target volume by adding water. The example reports that after being put into stoppered vials and undergoing lyophilisation, the resulting formulations formed cakes in the vials showing good appearance (no cracking or collapsing of the cake). Formulations A, B and C were analysed for residual water content and for valdecoxib. The results are set out in Table 3:

101 Formulation D (40 mg parecoxib) was tested for pH and residual water content and analysed for parecoxib and valdecoxib when freshly prepared and following 4, 8 and 12 weeks storage at various temperatures. The results of tests are shown in Table 4:
102 Example 2 provides Formulations E – J, each containing 20 mg parecoxib in the form of parecoxib sodium. The specification notes that solutions E – J, corresponding to formulations E – J, were prepared and lyophilised using a similar procedure to formulations A – D. However, it notes that formulations E – J contain more than about 10% of excipient ingredients other than the buffering agent, and that the formulations are presented for comparative purposes. Table 5 sets out the composition of solutions E – J for lyophilisation as follows:
103 The specification then sets out table 6 which contains data regarding the percentages of parecoxb and valdecoxib, expressed on an excipient-free basis, in formulations E – J when freshly prepared and after four weeks storage at various temperatures.
104 The specification then notes (at page 32 line 15 – page 33 line 10):
It will be noted that Formulations E-J exhibited poorer chemical stability than Formulations A-D of the invention. Formulations F and I, each of which contained 30 mg mannitol in addition to dibasic sodium phosphate, exhibited the greatest stability of the formulations tested in this Example, but nonetheless showed a much greater degree of conversion of parecoxib to valdecoxib than did Formulations A-D after storage for 4 weeks at 55°C or 70°C. Chemical stability of Formulations E, G, H and J was unacceptably poor.
Furthermore, none of formulations E-J exhibited instantaneous dissolution upon reconstitution. Formulation I, which contained 200 mg polyethylene glycol 4000 in addition to mannitol and dibasic sodium phosphate, proved especially slow and difficult to dissolve in attempts to reconstitute the formulation.
105 Example 3 describes a pharmacokinetic study of blood plasma concentration of valdecoxib in human subjects given either an intravenous dose of parecoxib sodium or oral dose of valdecoxib. Example 4 describes a clinical study performed on patients undergoing dental surgery regarding the effectiveness of parecoxib as pain relief.
106 The relevant claims are set out below:
1. A pharmaceutical composition comprising, in powder form:
(a) at least one water-soluble therapeutic agent selected from selective COX-2 inhibitory drugs and prodrugs and salts thereof, in a therapeutically effective total amount constituting about 30% to about 90% by weight,
(b) a parenterally acceptable buffering agent in an amount of about 5% to about 60% by weight, and
(c) other parenterally acceptable excipient ingredients in a total amount of zero to about 10% by weight of the composition;
said composition being reconstitutable in a parenterally acceptable solvent liquid to form an injectable solution.
…
4. The composition of Claim 1, wherein the therapeutic agent comprises parecoxib or a salt thereof.
5. The composition of Claim 1, wherein the therapeutic agent comprises parecoxib sodium.
…
7. The composition of any one of Claims 1 to 6, wherein the therapeutic agent is present in an amount of about 40% of about 85%.
…
11. The composition of any one of Claims 1 to 6, that consists essentially of the therapeutic agent and the buffering agent.
…
14. The composition of any one of Claims 1 to 6, wherein the buffering agent is dibasic sodium phosphate.
15. The composition of any one of Claims 1 to 6, that, upon reconstitution, has a pH of about 7 to about 9.
…
17. An injectable solution prepared by reconstituting a composition of any one of Claims 1 to 16, in a parenterally acceptable solvent.
18. The solution of Claim 17, wherein the solvent is an aqueous solvent.
19. The solution of Claim 17 or 18, having pH of about 7.5 to about 8.5.
20. The solution of any one of Claims 17 to 19, wherein the aqueous solvent contains dextrose and/or sodium chloride.
21. An article of manufacture comprising a sealed vial having contained therewithin a unit dosage amount of a composition of any one of Claims 1 to 16, in a sterile condition.
…
24. The article of manufacture of Claim 21, wherein the composition comprises as the therapeutic agent parecoxib sodium in a dosage amount of about 1 mg to about 200 mg, more preferably about 10 mg to about 100 mg.
…
26. A process for preparing a reconstitutable selective COX-2 inhibitory composition, the process comprising a step of lyophilizing an aqueous solution that comprises:
(a) at least one therapeutic agent selected from selective COX-2 inhibitory drugs and prodrugs and salts thereof, in a therapeutically effective total amount constituting about 30% to about 90% by weight,
(b) a parenterally acceptable buffering agent in an amount of about 5% to about 60% by weight, and
(c) other parenterally acceptable excipient ingredients in a total amount of zero to about 10% by weight of the composition, excluding water;
said lyophilizing step resulting in formation of a readily reconstitutable powder.
27. The process of Claim 26, wherein the therapeutic agent is parecoxib sodium.
28. The process of Claim 26 or 27, wherein the buffering agent is dibasic sodium phosphate.
…
30. The process of Claim 29, wherein, in the step of preparing the solution, the parecoxib sodium is added last.
…
34. A method of treating and/or preventing a COX-2 mediated disorder in a subject, the method comprising reconstituting a unit dosage amount of a composition of any one of Claims 1 to 16, in a physiologically acceptable amount of a parenterally acceptable solvent liquid to form an injectable solution, and administering the solution parenterally to the subject.
35. The method of Claim 34, wherein the parenteral administration is by intradermal, subcutaneous, intramuscular, intravenous, intramedullary, intra-articular, intrasynovial, intraspinal, intrathecal or intracardiac injection or infusion.
36. The method of Claim 34 or 35, wherein the parenteral administration is by intravenous injection or infusion.
37. The method of Claim 34 or 35, wherein the composition is injected intravenously as a bolus.
38. Use of a reconstituted unit dosage amount of a composition of any one of Claims 1 to 16, in a physiologically acceptable amount of a parentally acceptable solvent liquid to form an injectable solution for treating and/or preventing a COX-2 mediator disorder in a subject.
39. The use of Claim 38, wherein the parental administration is by intradermal, subcutaneous, intramuscular, intravenous, intramedullary, intra-articular, intrasynovial, intraspinal, intrathecal or intracardiac injection or infusion.
40. The use of Claim 38 or 39, wherein the parental administration is by intravenous injection or infusion.
41. The use of Claim 38 or 39, wherein the composition is injected intravenously as a bolus.
42. A pharmaceutical composition as defined in Claim 1, an injectable solution prepared by reconstituting said composition, an article of manufacture comprising said composition, a process for preparing a reconstitutable selected COX-2 inhibitory composition, or a method of treating and/or preventing a COX-2 mediated disorder in a subject, substantially as herein described with reference to any one of the Examples.
107 The characteristics of the person skilled in the art, whose knowledge may be relevant in the context of the construction of the patent and also for the purposes of the lack of inventive step case, are in dispute, but only to a limited extent. The parties agree that one person skilled in the art to whom the patent is addressed would have experience in pharmaceutical formulation and development, with a PhD or Masters level degree plus relevant experience in the discipline and would be familiar with chemical principles. There is no dispute that the formulation experts, Dr Robertson and Professor Winter, have these skills.
108 The disagreement between the parties as to the identity of the skilled addressee arises primarily in the context of the respondents’ challenge to the patent on the ground of lack of inventive step. However, it is convenient to address the arguments raised at this point.
109 The applicants submit that it is not necessary to resort to the concept of a notional team because in this case Professor Winter and Dr Robertson were able to understand the invention and put it into effect. They submit that the invention of the patent does not lie in the selection of COX-2 inhibitors useful in the treatment of COX-2 mediated disorders, which the patent recites as being well-known, but rather in formulation. They further submit that even if the notional skilled addressee is best conceptualised as a team, it would not include a clinician such as a pain management specialist, thereby excluding the evidence of Professor Scott and Professor Goucke. The applicants adopt the evidence of Professor Winter in contending that the others who would have an interest in formulating parecoxib would likely include an analytical expert, a chemical expert and a pre-clinical development expert who would be someone with expertise in pharmacology and toxicology who would provide information about biological activity of the compound, preliminary doses and toxicological profile.
110 The respondents submit that a conceptual team is appropriate. They generally agree with Professor Winter’s characterisation but submit that the team would include, either in lieu of or in addition to the pre-clinical development expert, a pain expert who is likely to be able to speak to the administration of the formulation under development and the use of the method claimed in the method claims. Such a person would have skills similar to those of Professor Scott. The respondents posit that when seeking to develop an analgesic for the Australian market which would be better than, or at least a useful alternative to, the drugs currently on the market for the management of acute post-surgical pain, the hypothetical team would include not only a formulator but also a pain clinician.
111 The construction of the patent is a function of the court, being a matter of law, but since patents often contain technical subject material, the court must, by evidence, be put in the position of a person of the kind to whom the document is addressed, being a person skilled in the relevant art at the relevant date: General Tire & Rubber Co v Firestone Tyre & Rubber Co Ltd [1972] RPC 457 at 485 (Sachs LJ); Commissioner of Patents v Rokt Pte Ltd [2020] FCAFC 86; 277 FCR 267 at [73] (Rares, Nicholas and Burley JJ). Skilled addressees are those likely to have a practical interest in the subject matter of the invention: Catnic Components v Hill & Smith Ltd [1982] RPC 183 at 242 (Diplock LJ). There may be more than a single person with such an interest, and the notional skilled reader to whom the document is addressed may not be a single person but a team, whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those which are contained in the document to be construed: General Tire at 485. Put another way, the skilled addressee is a notional person who may have an interest in using the products or methods of the invention, making the products of the invention, or making products used to carry out the methods of the invention either alone or in collaboration with others having such an interest: Aristocrat Technologies Australia Pty Limited v Konami Australia Pty Limited [2015] FCA 735; 114 IPR 28 at [26] (Nicholas J); Hood v Bush Pharmacy Pty Ltd [2020] FCA 1686; 158 IPR 229 at [59] (Nicholas J).
112 I disagree with the applicants’ submission that no team is likely to be involved. The task of formulation appears to me to be likely to require a team effort. The formulation experts’ evidence is to the same effect. Furthermore, in my view, the applicants’ characterisation of the invention fails to take into account its breadth which, the patent reveals, concerns not only formulation but also methods of treatment by parenteral administration of COX-2 inhibitors: see claims 34 – 42. Indeed, the background of the patent addresses the clinical advantages of the parenteral administration of drugs for pain relief (page 1 line 30 – page 2 line 11) and the preferred use of selective COX-2 inhibitors in injectable form for that purpose (page 2 lines 22 – 24). The use of COX-2 inhibitors in the treatment of pain, including postoperative pain, is identified later in the patent (for example, page 23 lines 19 – 26) and the preferred uses for compositions of the invention specifically include pain management, particularly for post-surgical pain (page 25 lines 4 – 8). Example 4 addresses the clinical use of a COX-2 inhibitor discussing the results of a clinical trial concerning the benefits of the use of an intravenous dose of a parecoxib sodium formulation administered to patients following dental surgery.
113 Moreover, it was common general knowledge by the priority date that selective COX-2 inhibitors were considered to be superior to non-selective NSAIDs and were of considerable interest for use in the treatment of acute pain, particularly post-operative pain, instead of non-selective NSAIDs. Rofecoxib and celecoxib, both selective COX-2 inhibitors, were being used for the management of post-operative pain and were available only in oral tablet form. However, no selective COX-2 inhibitors were available for injection, which was a preferred form of administration when treating acute pain. There was accordingly a known interest in an injectable COX-2 inhibitor for the treatment of acute pain. That interest was particularly known to persons involved in post-surgical pain. The evidence of Professor Scott provides independent support to the content of the patent as to the relevance of COX-2 inhibitors to pain experts.
114 These matters clearly place as persons with a practical interest in the subject matter of the patent those with an interest in the treatment of pain. Furthermore, it is difficult to imagine that a formulator could develop an injectable formulation of a selective COX-2 inhibitor without an understanding of its likely clinical effects. Whilst Professor Winter considered that a pre-clinical expert would provide information about the biological activity of the compound, preliminary doses and toxicological profile, it is in my view likely that a formulation team would, either in addition to or in the alternative to a pre-clinical expert, have access to the skills of a person with experience in the administration of the type of drug under development, such as a pain clinician. In this regard, Professor Scott gave evidence that he had undertaken work in the course of undertaking his PhD which involved looking at novel analgesic drugs which might be useful in the treatment of pain. Dr Robertson gave evidence, which I accept, that generally a drug development program commences with the identification of a condition to be treated, generally in consultation with a medical specialist with expertise in the relevant condition to gain an understanding of relevant matters such as existing molecules used to treat the condition and the benefits or side effects of those molecules that might be desirable to target or avoid, as well as any requirements or advantages as to the route of administration to treat the target condition.
115 Of course, the notional person is not an avatar for expert witnesses whose evidence is accepted by the court. It is a tool of analysis which guides the court in determining, as relevant here, whether an invention as claimed does not involve an inventive step: AstraZeneca AB v Apotex Pty Ltd [2015] HCA 30; 257 CLR 356 at [23] (French CJ). The evidence from expert witnesses must be understood on that basis.
116 In my view, people with a practical interest in the invention the subject of the patent would include those who are experienced in: (1) pharmaceutical formulation; (2) analytical methods required to characterise the formulation; (3) chemistry and being able to provide information about the synthesis, safety, reactivity and the impurity profile of the drug compound; and (4) the clinical application and use of the drug compound. In my view, the notional team would include a person with clinical knowledge about the treatment of pain, including acute pain, and that person would be someone with clinical experience in that field.
117 Several issues arise concerning the meaning of the claims. For the most part they arise in the context of the infringement case and relate to attempts by the parties to either maximise or minimise (according to their interests) the weight of the components, and the total weight, of the pharmaceutical composition of interest to navigate the boundaries of the monopoly claimed. It is appropriate to consider questions of construction shorn of the distraction of the forensic interests of the parties or, as if the infringer had never been born: CCOM Pty Ltd v Jeijing Pty Ltd [1994] FCA 396; 51 FCR 260 at 267-268 (Spender, Gummow and Heerey J). Accordingly, in these reasons I first consider questions of construction before turning to whether or not the respondents’ products may be said to infringe the claims.
118 The construction disputes concern:
(a) whether residual water is to be considered to form part of the “composition” of claim 1 and claim 26;
(d) the identity of the “at least one water soluble therapeutic agent” in claims 1 and 26 (the water soluble therapeutic agent argument);
(e) the meaning of “about” as it appears in the context of specified percentage weight ranges in claims 1 and 26; and
(f) the meaning of “essentially” in claim 11.
119 Claims 1 and 26 are independent claims upon which a number of the relevant claims depend. Unless otherwise stated, my findings as to construction and infringement in relation to those independent claims will also apply to the relevant claims dependent upon them.
120 It may be noted that the specification provides that except where the context requires otherwise, the word “comprise” or variations of that word is used in an inclusive sense (page 35 lines 8 – 9).
121 The principles of claim construction are not in dispute. They are conveniently set out in Jupiters v Neurizon [2005] FCAFC 90; 65 IPR 86 at [67] (Hill, Finn and Gyles JJ) as follows:
(i) the proper construction of a specification is a matter of law: Décor Corp Pty Ltd v Dart Industries Inc (1988) 13 IPR 385 at 400;
(ii) a patent specification should be given a purposive, not a purely literal, construction: Flexible Steel Lacing Company v Beltreco Ltd (2000) 49 IPR 331 at [81]; and it is not to be read in the abstract but is to be construed in the light of the common general knowledge and the art before the priority date: Kimberley-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [24];
(iii) the words used in a specification are to be given the meaning which the normal person skilled in the art would attach to them, having regard to his or her own general knowledge and to what is disclosed in the body of the specification: Décor Corp Pty Ltd at 391;
(iv) while the claims are to be construed in the context of the specification as a whole, it is not legitimate to narrow or expand the boundaries of monopoly as fixed by the words of a claim by adding to those words glosses drawn from other parts of the specification, although terms in the claim which are unclear may be defined by reference to the body of the specification: Kimberley-Clark v Arico at [15]; Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610; Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 at 478; the body of a specification cannot be used to change a clear claim for one subject matter into a claim for another and different subject matter: Electric & Musical Industries Ltd v Lissen Ltd [1938] 56 RPC 23 at 39;
(v) experts can give evidence on the meaning which those skilled in the art would give to technical or scientific terms and phrases and on unusual or special meanings to be given by skilled addressees to words which might otherwise bear their ordinary meaning: Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479 at 485-486; the Court is to place itself in the position of some person acquainted with the surrounding circumstances as to the state of the art and manufacture at the time (Kimberley-Clark v Arico at [24]); and
(vi) it is for the Court, not for any witness however expert, to construe the specification; Sartas No 1 Pty Ltd, at 485–486.
122 Claim 1 is a claim for a “pharmaceutical composition” which comprises, “in powder form” the constituents (a) to (c) within certain percentage ranges, each measured “by weight of the composition”. The evidence makes tolerably clear that the process of lyophilisation, although designed to remove all water from the composition during the freeze-drying process, in practice will likely leave some residual water in the lyophilisate. There is a dispute as to how to treat this residual water in the lyophilised powder in the calculation of the percentages of the constituents by weight as identified. As the claimed ranges of the individual constituents (a) – (c) are expressed as percentages of the total weight of the composition, the total weight of the composition is directly relevant to the question of whether the individual constituents fall within the claimed ranges.
123 The applicants contend that any residual water must be taken into account when calculating the total weight of the composition. They submit that this is a natural consequence of the defined meaning of “comprising” as “including”, as well as the language used in the specification. The respondents submit that residual water is not to be taken into account in calculating the total weight of the composition. They contend that residual water is an artefact of the process of lyophilisation that cannot be controlled, and therefore must be excluded. They submit that residual water has no therapeutic or formulation benefit and should be understood implicitly to be excluded from consideration when determining the weight of the composition as a whole.
124 For the reasons set out below, I consider that any residual water in the composition must be taken into account when considering the total weight of the composition of claim 1 but not claim 26.
125 First, the language of claim 1 supports this construction. Claim 1 is for a “pharmaceutical composition” comprising (that is, including but not necessarily consisting wholly of), the constituent components (a), (b) and (c) which are, respectively, therapeutic agent, buffering agent and “other parenterally acceptable excipient ingredients”. The expert evidence of Professor Winter and Dr Robertson confirms that it was usual for lyophilised powder compositions to contain certain amounts of water. No language suggests that if water is present it is not to be regarded as part of the composition or that “comprising” is not to be understood in an inclusive sense. While residual water is not part of any of the constituents identified in (a)-(c), the inclusive definition of the word “comprising” allows for it to form part of the composition in powder form.
126 In this context it may be noted that the language in the specification supports the view that the person skilled in the art would understand that the composition of the invention is likely to include residual water. It provides, at page 14 lines 30 – 31, that the term “excipient” embraces “all non-therapeutically active components of the composition except for water”. This indicates that in the discourse of the specification, water is to be understood to be a non-therapeutically active component of the composition, albeit not an excipient.
127 The specification also provides, at page 15 lines 14 – 19:
A reconstitutable powder composition of the invention preferably contains less than about 5%, more preferably less than about 2%, and most preferably less than about 1%, by weight of water. Typically the moisture content is about 0.5% to about 1%, by weight. It is especially important to keep the amount of water to such a low level where the therapeutic agent has a tendency to degrade or convert to a less soluble form in presence of water.
128 The percentage of residual water in exemplar formulations A, B and C is shown in Table 3 of example 1 (set out at [100] above). The percentage of residual water in formulation D at various times during stability testing is shown in Table 4 (set out in [101] above). Plainly the specification contemplates that the powder composition of the invention will include within it residual water.
129 Secondly, reading the claims as a whole it may be seen that claim 26 provides some limited support for this construction. Claim 26 is for a process for preparing a reconstitutable composition by lyophilising an aqueous solution comprising ingredients (a), (b) and (c). The “composition” is accordingly in the form of a powder. Components (a)-(c) are described as follows:
(a) at least one therapeutic agent selected from selective COX-2 inhibitory drugs and prodrugs and salts thereof, in a therapeutically effective total amount constituting about 30% to about 90% by weight,
(b) a parenterally acceptable buffering agent in an amount of about 5% to about 60% by weight, and
(c) other parenterally acceptable excipient ingredients in a total amount of zero to about 10% by weight of the composition, excluding water…
130 It may be seen from the emphasised words in (c) that the percentage weight ranges are expressed by reference to the “composition” (i.e., the reconstitutable powder) rather than the aqueous solution. In that circumstance the patentee prescribes that the weight measurements are “excluding water”. Understood in this way, the effect of the words “excluding water” could be understood to mean that no water is to be taken into account in calculating the weight of the composition (which will include residual water) or alternatively that the excluded water consists of the water component of the aqueous solution and it is that water which is not to be taken into account in calculating the weight of the composition.
131 Professor Winter took the latter view. He gave evidence that the percentage weight ranges in (a)-(c) were expressed as “excluding water” to ensure that the weights for the therapeutic agent, buffering agent and other parenterally acceptable excipient ingredients identified in (a), (b) and (c) are not impractically small, because the starting components of the process in claim 26 are in an aqueous solution and the overwhelmingly dominant part of that solution would, without the exclusion, be water. However, that evidence does not approach the claim from the perspective of the language that it uses. As I have noted, the explicit reference in (c) to the “weight of the composition” draws attention to the final lyophilised powder. Furthermore, the patentee has chosen to use the broad words “excluding water” rather than “excluding the water in the aqueous solution”. In that circumstance, I would not read down the clear words of the claim by reference to the gloss proposed by Professor Winter. Indeed, in the body of the specification, the patentee uses the term “water” broadly including when describing residual water: see for example page 15 lines 14 – 22 set out at [127] above. In those circumstances, in my view, claim 26 excludes from its calculation of the weight of the composition all water, including residual water. This conclusion points in favour of the notion that where the patentee excludes water from the weight of the composition (as it does in claim 26), it does so expressly. Consequently, claim 26 provides some support for the construction of claim 1 that I have preferred that the language of claim 1 does not also exclude water from the weight of the composition.
132 Thirdly, the respondents submit that water is simply an unwanted artefact of lyophilisation, that it has no therapeutic or formulation role and that experts in the field would know to discount such water as is found in a powder. They submit that the amount of residual water can vary from batch to batch as much as in the range from 0-6% and point to the evidence of Dr Robertson that water is an “uncontrolled variable” in a lyophilised formulation. They submit that on the construction proposed by the applicants, it would not be possible to ascertain whether or not a formulation falls within the scope of the claims until after a given batch is made and tested, and that infringement may vary from batch to batch, depending on the efficacy of the lyophilisation process to eliminate water. Further, they submit that lyophilisation is a manufacturing process that is not part of claim 1, and that the skilled addressee would not understand the breadth of the claim to vary depending on the efficiency of that process.
133 However, these points cannot be used to distort the clear language of the claim, when read in the context of the specification as a whole which, for the reasons given above, require residual water to be included. Nor, in my view, do they have quite the dramatic practical consequences that the respondents’ submissions suggest. As the experts noted in their evidence, residual water must be controlled as part of a validated manufacturing process within set limits. A Karl Fischer titrator, a piece of equipment able to analyse the amount of water in a sample by chemical reaction, would typically be used to analyse water content in samples and batches would be controlled for consistency. Whilst there may well be variability, that is expected to be within a tight range of variables that the person skilled in the art is able to moderate.
134 Finally, the respondents also seek to support their construction of claim 1 by reference to claim 32. Relevantly, claim 32 relates to a process which must “result in a powder having a moisture content of less than about 2% by weight”. The respondents submit that this demonstrates that “when the patentee intends residual water to be treated as a material component of the powder, it explicitly says so”. I disagree. Claim 32, understood in the context of the claims upon which it is dependent, is to a particular lyophilisation cycle to be performed on an aqueous solution comprising the therapeutic agent, buffering agent and other excipient ingredients. The parameters chosen in claim 32 accord broadly with those disclosed in the specification at page 18 line 19 – page 19 line 31. The specification also discloses that a lyophilisation cycle can be used to achieve a powder with a low moisture content which it discloses is preferable to improve the stability of the composition. In my view, claim 32 simply claims one such process. Its language does not demonstrate that only when the claims refer to residual water is it to be treated as a material component of the composition. To the contrary, it recognises that a powder composition will contain water, and that this may be minimised by using a particular process of lyophilisation.
135 Accordingly, in my view, residual water must be taken into account when considering the weight of the composition of claim 1 (and its dependent claims), but not when considering claim 26 (and its dependent claims).
5.3 The water-soluble therapeutic agent argument
136 The parties are in dispute about the meaning in claim 1 of:
(a) at least one water-soluble therapeutic agent selected from selective COX-2 inhibitory drugs and prodrugs and salts thereof…
This dispute concerns the identity of the therapeutic agent when a salt of a prodrug is included in the composition. More specifically it concerns whether both the prodrug and the salt thereof can be characterised as the therapeutic agent for the purposes of the claim.
137 The applicants submit that subparagraph (a) is to be understood to mean that either parecoxib or parecoxib sodium (the sodium salt of parecoxib) may be the therapeutic agent within a given composition for the purpose of the claim. They submit that this is consistent with the way that the patent uses the term “parecoxib” to include parecoxib salts. They provide examples from the specification to support their argument. The applicants’ position is that claim 1 should be construed in a way such that when the composition in powder form includes a selective COX-2 inhibitory prodrug in the form of a salt (such as parecoxib sodium), then for the purpose of considering the weight of the “water-soluble therapeutic agent”, one may take into account either the weight of the “parent” compound (e.g. parecoxib) or the salt (e.g. parecoxib sodium).
138 The respondents dispute this construction. They contend that an ordinary reading of claim 1 is that where a salt of a prodrug is used as the water-soluble therapeutic agent in formulating a powder composition, it is the salt rather than the acid component of the salt that must be considered when assessing whether or not the composition has the claimed percentage weights of ingredients. The respondents also draw on aspects of the specification to support their construction. Both parties refer to evidence given by Professor Winter and Dr Robertson.
139 The specification, on page 3 lines 5 – 11, provides some relevant background information as follows:
Parecoxib rapidly converts to the substantially water-insoluble selective COX-2 inhibitory drug valdecoxib following administration to a subject. Parecoxib also converts to valdecoxib upon exposure to water, for example upon dissolution in water. The high water solubility of parecoxib, particularly of salts of parecoxib such as the sodium salt, by comparison with most selective COX-2 inhibitory drugs such as celecoxib and valdecoxib, has led to interest in developing parecoxib for parenteral use.
140 The expert evidence indicates that the sodium component of parecoxib sodium does not have a therapeutic effect. Nonetheless, it has a formulation purpose. The use of the sodium salt form of parecoxib is a means by which parecoxib is rendered more conveniently soluble for administration to the subject. In solution, the sodium disassociates from the parecoxib. Upon injection into the subject, parecoxib is later converted to valdecoxib which has the desired therapeutic effect. It is the conversion from parecoxib to valdecoxib that identifies parecoxib as a prodrug.
141 For the following reasons, I reject the applicants’ argument.
142 First, claim 1 refers to a composition in powder form. In parecoxib sodium’s solid state, the sodium cation and the parecoxib anion are paired to each other by an ionic bond. When in powder form, parecoxib sodium is a single chemical entity that is aptly described as “a water soluble therapeutic agent”.
143 Secondly, the dependent claims support this construction. Claim 4 is to the composition of claim 1 “wherein the therapeutic agent comprises parecoxib or a salt thereof”. Claim 4 demonstrates that the patent draws a distinction between parecoxib (or what the applicants might term the “parent” compound) and salts thereof, for example, parecoxib sodium. This tends to indicate that if a salt of a drug or prodrug is selected for inclusion in the composition, it is considered as a distinct component from its corresponding drug or prodrug. This notion is also supported by claim 5 which is to the composition of claim 1 wherein the therapeutic agent comprises parecoxib sodium. Although the term “comprises” is used in the inclusive sense, here leaving open the possibility that other therapeutic agents in addition to parecoxib sodium are included in the composition, claim 5 indicates that parecoxib sodium is to be understood in its own right to be a therapeutic agent.
144 Thirdly, the specification supports this construction. For instance, in the passage from the patent at page 5 line 25 – page 6 line 2, set out in [87] above, the specification identifies that “[a] pharmaceutical composition of the present invention comprises as the therapeutic agent” one of four alternative options. Option (c) is a “water-soluble prodrug” of a selective COX-2 inhibitor. Option (d) is a water-soluble salt of a prodrug. Option (c) is apt to describe parecoxib alone, and option (d) is apt to describe parecoxib sodium. The distinction between them is clear. In his evidence, Professor Winter agreed that (c) would include parecoxib and that (d) would include parecoxib sodium.
145 In several other places, the specification distinguishes between parecoxib per se, and parecoxib in the form of a salt. An example of this appears at page 13 lines 8 – 11:
A particularly useful prodrug of valdecoxib for use in compositions of the invention is parecoxib, more particularly a water-soluble salt thereof, for example parecoxib sodium.
146 These references make plain that despite the fact that parecoxib is the part of the compound parecoxib sodium that is eventually converted into valdecoxib which has the desired therapeutic effect, nevertheless for the purpose of the discourse of the patent, compositions of parecoxib are distinguished from, and referred to differently to, compositions of parecoxib sodium. However, both are identified as distinct therapeutic agents. In that context, I do not accept it to be determinative, as the applicants submit, that some in the industry more generally refer to a “parent” compound (e.g. parecoxib) as the “therapeutic agent”, even if it is in the form of a salt (e.g. parecoxib sodium). Here, it is the clear choice of language used by the inventors in the specification that is most relevant.
147 Accordingly, where claim 1 provides for a “pharmaceutical composition comprising, in powder form: (a) at least one water-soluble therapeutic agent selected from selective COX-2 inhibitory drugs and prodrugs and salts thereof, in a therapeutically effective total amount constituting about 30% to about 90% by weight”, the “at least one water-soluble therapeutic agent” may be either a drug, a prodrug or a salt thereof, depending on which is added to the formulation. If the salt of a prodrug is included in the composition, that compound will be regarded as the therapeutic agent.
148 In the context of this debate, the parties again place some reliance on claim 26. It will be recalled that claim 26 is for a process. Unlike claim 1 which is for a composition in powder form, claim 26 focusses on the step of lyophilisation which is performed on an aqueous solution comprising (including): (a) at least one therapeutic agent; (b) buffering agent; and (c) other excipients. These are to undergo a lyophilising step to result in the reconstitutable powder.
149 The applicants contend that in relation to claim 26 there is an additional reason why it is appropriate to consider the prodrug (here, parecoxib) as the therapeutic agent rather than the salt (here, parecoxib sodium). They submit that in solution most of the parecoxib sodium is dissociated into parecoxib anions (negatively charged ions) and sodium cations (positively charged ions). This argument was primarily based on the evidence of Professor Winter, who considered that when parecoxib sodium is in solution, the therapeutic agent must be understood to be the free acid of parecoxib or alternatively the parecoxib anion. However, this argument does not withstand scrutiny.
150 First, although claim 26 is for a process for preparing a reconstitutable composition comprising a step of lyophilising an aqueous solution including at least one therapeutic agent selected from selective COX-2 inhibitory drugs and prodrugs and salts thereof, there is no suggestion in the specification that the reference to “prodrugs and salts thereof” should be construed any differently in claim 26 to how that phrase is used in claim 1. It is to be assumed that those words mean the same thing in both claims unless there is a reason to consider otherwise.
151 Secondly, the aspects of the specification to which I have referred above in [144]-[146] tend to support the same construction. Furthermore, where referring to the ingredients dissolved to form the pre-lyophilisation aqueous solution, the specification identifies the ingredient in solution as being parecoxib sodium (rather than parecoxib anions and sodium cations): see, for example, page 18 lines 2 – 3 “[p]arecoxib sodium and the buffering agent are present in the solution”; page 18 lines 11 – 14 “[t]he solution…is then metered into one or more vials. Each vial receives a measured volume of solution having a desired unit dosage amount of parecoxib sodium”; see also the ingredients listed in the compositions of solutions A-D for lyophilisation in Table 1 in example 1 (page 29).
152 Thirdly, claim 27, which is dependent on claim 26, specifically identifies that the therapeutic agent is parecoxib sodium, despite the fact that it is in aqueous solution. This is a further clear indication that in the discourse of the patent, parecoxib sodium is regarded as a therapeutic agent despite the fact that in an aqueous solution it dissociates to form parecoxib anions and sodium cations. Similarly, claim 29, which is also dependent on claim 26, refers to the process “prior to the lyophilizing step, [wherein] the solution is prepared by dissolving the parecoxib sodium and the dibasic sodium phosphate in water for injection, sterilized and then metered into vials, each containing a volume of solution having a unit dosage amount of parecoxib sodium...”. This is a still further indication that the patent regards parecoxib sodium to be the therapeutic agent before and after it is dissolved to form an aqueous solution.
153 Accordingly, for each of claims 1 and 26 (and the claims dependent on these claims) a drug, prodrug or salt thereof may be considered the “water-soluble therapeutic agent” for the purpose of considering the weight of the (a) component of the composition. Where the composition is formulated using parecoxib sodium as the selective COX-2 inhibitory salt of the prodrug parecoxib, parecoxib sodium is the therapeutic agent the weight of which is to be taken into account in assessing whether the composition falls into the claimed ranges.
154 The next construction question concerns the meaning of “about” as it appears in the context of claimed percentage weight ranges of the components of the claimed compositions. The parties based their arguments on claim 1, however, the outcome of the dispute concerning the meaning of “about” was not said by the parties to be any different with respect to any of the relevant claims.
155 The applicants submit that the word should be understood to allow for a 5% margin of error, so that “about 90%” in paragraph (a) of claim 1 extends to 94.5% as its upper limit (5% of 90% being 4.5%). The respondents contend that “about” allows for rounding to the nearest whole number, such that “about 90%” extends to 90.49% as its upper limit.
156 The parties accept that the word “about” is one of ordinary English and is not a term of art, and the formulation experts agree in their joint report that it is “difficult to impossible to define the term” which is “mostly avoided in natural sciences”. In their joint report the formulation experts also agree that the meaning of the term is not one that they can answer well on the basis of science. Nevertheless, the experts both attempt to supply a meaning for the word, and each party adopted the meaning proposed by its expert.
157 I accept that the word must be understood to extend the ranges set out in the various claims. In claim 1 it applies to provide that the therapeutic agent of (a) is to be in a total amount constituting “about 30% to about 90% by weight”, the buffering agent of (b) is to be in amount of “about 5% to about 60% by weight” and the other excipients are to be in an amount of “zero to about 10% by weight of the composition”. The question of how far the range is to be extended is by no means easy to answer. However, for the reasons set out below, I consider that extension should be no more than to allow for rounding of the percentages in the manner contended for by the respondents.
158 First, the evidence makes plain that in field of pharmaceutical sciences, and specifically in relation to the substances with which the present invention is concerned, those in the art typically deal with precise measurements. Pharmaceutical substances are produced in accordance with close tolerances and sophisticated measuring systems. Indeed, the fact that, as the experts say, the imprecise term “about” is mostly avoided in the natural sciences is no doubt because those engaged in the field are accustomed to working within precise parameters.
159 In this regard, I note that, for the most part, the measurements supplied in the patent are expressed to two decimal places, suggesting that it is of utility to the skilled reader to know the amount of a measurement to the hundredth part. This is particularly apparent from example 1 which provides quantities of parecoxib sodium and dibasic sodium phosphate heptahydrate in mg to two decimal places. The applicants point out that some measurements in the specification are given to one decimal place, including the percentages of parecoxib in formulation D set out in Table 4. However, the bulk of the measurements given in the examples are more precise. In my view, that provides an insight that the scope for variance in the percentages of the components of claim 1 is not intended to be large.
160 Secondly, the claims themselves provide a range of variance. As I have noted, in claim 1, (a) has a range of 30% to 90%, for (b) it is 5% to 60% and for (c) it is zero to 10%. The inventors have gone to the trouble to specify the scope of their monopoly by reference to the range between these specific figures. As a matter of construction, the word “about” should not be understood to supply any significant extension of the ranges nominated. Any allowance for errors in measurement, upon which the applicants placed some reliance, may be understood to have been accommodated within these ranges.
161 Thirdly, the word “about” means, as a matter of ordinary English, “near; close to”: Macquarie Dictionary (5th ed, Macquarie Dictionary Publishers, Sydney, 2009). A construction that gives effect to the ordinary English meaning of the word is to be preferred in circumstances where it is not a term of art. In this context, the applicants submit that a construction of “about” that simply allows for rounding tends to introduce redundancy, because the skilled addressee would understand the claims to allow for this in any event. They rely on Professor Winter’s evidence to the effect that such rounding would take place in any event, applying normal scientific practice. However, I am not satisfied that the claims would be interpreted in this manner if “about” had been omitted. Indeed, absent an approximating term, and where specific ranges are specified within the claim, it is by no means apparent that a variance beyond the claimed range would fall within the scope of the monopoly.
162 Fourthly, the specification itself provides no basis for construing “about” to extend beyond the narrower construction posited by the respondents. The metrics deployed by the applicants to justify that “about” should be understood to permit an extension of the range by 5% are opaque. They rely on the evidence of Professor Winter for the proposition that such an extension is justified because of the teaching in the patent as to:
(a) the so-called “5% total, 1% single impurity rule”;
(b) residual water;
(c) variations in the amount of pH adjusters; and
(d) tolerances allowed for in the Juno products and other pharmaceutical compositions.
163 In relation to (a), the specification provides at page 15 lines 23 – 29:
“Acceptable chemical stability” herein means that the composition, following the defined time period (e.g., about 30 days, about 6 months or about 2 years), passes a standard test for chemical purity of the therapeutic agent, for example as may be required for approval by a regulatory authority. An example of such a test is the “5% total, 1% single impurity rule”, whereby a preparation of a candidate drug must contain not more than 5% total impurities, and not more than 1% of any single impurity.
164 It is not apparent how this statement assists the applicants. It provides a metric for evaluating impurities over time ranging from 30 days to two years. In his oral evidence, Dr Robertson rejected the proposition that the exemplified rule referred to in the passage above provided a basis for the proposition that it was generally considered to be acceptable that the amount of therapeutic agent in a lyophilised product to be 5% higher or lower than the labelled amount. Professor Winter conceded his unfamiliarity with the rule. In my view, the test is not concerned with, and has no bearing on, the construction of the upper and lower bounds of the claimed percentage weights of the components of the composition. Rather, it is posited by the patentee as a means of determining the acceptable level of stability of a therapeutic agent, having regard to the teaching of the specification that the therapeutic agent degrades over time, which may be acceptable to a regulator.
165 In relation to (b), the specification provides, at page 15 lines 14 – 22 (see [92] above), that “[a] reconstitutable powder composition of the invention preferably contains less than about 5%, more preferably less than about 2% and most preferably less than about 1%, by weight of water”. It also provides that “typically” the moisture content is about 0.5% to about 1% by weight. The applicants contend that this passage supplies a rationale for attributing the figure of 5% to “about”. However, it is difficult to understand why the person skilled in the art would seize upon 5% rather than the more “typical” 0.5% to 1%. Furthermore, the word “about” appears in claim 26, where water is specifically excluded from the weight of the powder composition. Professor Winter accepted in cross-examination that such a rationale could not apply there. I find that the passage relied upon provides little basis for selecting a range of 5% for the word “about”.
166 In relation to (c), the applicants’ reliance on the fact that pH adjusters are used in small amounts to modify the pH of a given formulation similarly provides little insight or assistance in determining the appropriate meaning to be attributed to “about”. There is no reasoning in the specification to suggest that the ranges given in the claims do not provide for small variations in those amounts.
167 In relation to (d), in closing submissions the applicants referred to the tolerances specified for various components and impurities in the Juno products in the certificates of analysis provided by the respondents. As an example, the applicants draw upon a particular tolerance which is stated to be “plus/minus 5% for the assay (= the amount of parecoxib)”. The content of Juno’s documents, as a matter of principle, can add little to the manner in which the word “about” as used in the patent is to be construed. It may be true that tolerances are expressed in those documents going to a weight (or percentage) range of given ingredients to be added to, or found in, a composition. However, the purpose of the claim is to stake out the metes and bounds of the monopoly claimed. It is not a list of ingredients, but a means of determining whether the impugned product infringes. The evidence adduced does not support the proposition that the components of a given composition cannot be ascertained with considerable accuracy. One cannot move from the certificates of analysis to the construction of the relevant claims.
168 Finally, the applicants contend that the word “about” cannot refer to a simple rounding of figures because otherwise formulation D in example 1 would not fall within the claims. They submit that because formulation D is one example said to fall within the scope of the invention and because formulation D contains around 92.79% of parecoxib sodium, the word “about” cannot be confined to a rounding of figures to the nearest whole number because, if it were so construed, formulation D would fall outside the scope of claim 1, 92.79% being greater than “about 90%”. This argument is flawed in several respects. First, it relies on the evidence of Professor Winter in order to get to the figure of 92.79%. He in turn makes an assumption as to the amount of residual water in the powder (he says 1%) and that dibasic sodium phosphate heptahydrate will become anhydrous in the post-lypophilisation composition and be present in the amount of 2.84 mg. Neither proposition is set out in the example in the patent. Indeed, based on the figures in the specification (which give, for formulation D, figures of 42.36 mg of parecoxib sodium and 5.36 mg of dibasic sodium phosphate heptahydrate) Dr Robertson, using only information contained in Table 1, arrived at the conclusion that parecoxib sodium was present in formulation D at approximately 88.8% by weight. On Dr Robertson’s calculations, formulation D falls within claim 1 irrespective of the meaning to be attributed to “about”. Secondly, I do not consider that the inventors intended that the meaning of the word “about” should be mined from the depths of one aspect of an example in the patent, gold only being apparent after digging in such an obscure (and apparently assumption laden and contestable) place. Even if they did so intend, such obscurity provides no sound basis for construing the claim.
169 In the result, I consider that the word “about” should be understood as allowing a small amount of variance from the ranges specified in claim 1 (and each of the other claims), but confined to the rounding to the nearest whole percentage point.
5.5 The construction of “essentially” in claim 11
170 Claim 11 is as follows:
The composition of any one of Claims 1 to 6, that consists essentially of the therapeutic agent and the buffering agent.
171 It will be recalled that claim 1 specifies, in summary, a pharmaceutical composition comprising, in powder form, three ingredients: (a) at least one therapeutic agent in a therapeutically effective total amount constituting about 30% to about 90% by weight; (b) a buffering agent in an amount of about 5% to about 60% by weight; and (c) other excipient ingredients in a total amount of zero to about 10% by weight of the composition.
172 In the joint expert report, the formulation experts agreed that in the context of claim 11 “essentially” means “mostly” such that the composition of the claim contains mostly, but not exclusively, the therapeutic agent and a buffering agent, but other things (such as water) could also be present. Equating “essentially” with “mostly” accords with the description of the invention, which, after stating (at page 14 lines 6 – 8) that the buffering agent is present in an amount of about 5% to about 60% and is typically the predominant excipient ingredient, provides that in one embodiment “the reconstitutable powder composition consists essentially of the therapeutic agent and the buffering agent” (page 14 lines 9 – 10).
173 The word “essentially”, like “mostly”, is a relative term that by itself is of no definite meaning. However, taken in the context of the specification as a whole, it is apparent that the description of the embodiment as consisting “essentially” of the therapeutic agent and buffering agent does no more than identify a composition that contains something within the upper range of the percentages of therapeutic agent and buffering agent that are specified in claim 1. Given that claim 11 is dependent on claim 1, it is to be read such that the invention claimed in claim 11 is a subset of the composition of claim 1 (it is “the composition of any of claims 1 to 6”). Claim 1 prescribes that the total amount of excipient ingredients is to amount to no more than about 10% by weight of the composition. Accordingly, compositions of claim 11 must have at least about 90% by weight of therapeutic agent and buffer. This accords with the ordinary meaning that the composition consists “essentially” (or mostly) of therapeutic agent and buffer.
5.6 Conclusions in relation to the construction issues
174 I have resolved the construction issues as follows:
(a) for claim 1 and its relevant dependent claims, residual water is to be included in measurement of the total weight of the composition;
(b) for claim 26 and its relevant dependent claims, residual water is not to be so included;
(c) a “water soluble therapeutic agent” within claim 1 or claim 26 may be in the form of the salt of a COX-2 inhibitory drug or prodrug, for example, parecoxib sodium, if that is the ingredient added to the formulation;
(d) the word “about”, insofar as it refers to relative weight percentages in the relevant claims, means rounding up or down to the nearest whole number percentage; and
(e) the use of the word “essentially” in claim 11 does not extend the scope of the monopoly claimed beyond the limits of claim 1.
175 The parties helpfully cooperated in the preparation of the case to ensure that the matters in dispute were confined to the essentials. The respondents made admissions as to their conduct that enabled the characteristics of the Juno products to be clearly identified. In particular, they admit that as a result of an application made by Juno to the TGA, the Juno products were listed on the ARTG on or around 4 April 2018 with the indication of “a single peri-operative dose for the management of post-operative pain”. They further admit that Juno offers for sale and supplies the Juno products in Australia for use by persons for the management of post-operative pain, in accordance with that indication and that until August 2020 Neo supplied the Juno products to Juno for sale on the Australian market.
176 In the course of the preparation of the matter for trial the respondents supplied documents to the applicants identifying the constituent features of the ingredients of the Juno products and the means by which those products were made (defined earlier in these reasons as the Neo confidential documents). The documents largely related to six batches of the Juno products. Although those particular batches were not in fact ever brought into Australia, the trial proceeded on the basis, and the parties agreed, that resolution of the disputed issues of construction, and in particular the meaning of the word “about” and the infringement issues (to which I refer below) with respect to those batches will be sufficient for the result to be applied to all subsequent batches of the Juno products that have been brought into Australia and distributed.
177 Within the Neo confidential documents, the following are of particular relevance:
(a) certificates of analysis for six batches of the Juno products numbered 31601 to 31606, three being for the 40 mg product and three for the 20 mg product;
(b) an extract entitled “batch formula” taken from the common technical documents lodged with the ARTG providing the formula for a batch of 22,000 vials of the 40 mg powder for injection;
(c) an extract entitled “description and composition of the drug product” (Product Description), also from the common technical documents, setting out the composition of the dosage form of the 40 mg product; and
(d) an extract entitled “Description of Manufacturing process and process controls” (Manufacturing Process Description) also from the common technical documents.
178 The parties provided, in confidential exhibit 3, agreed calculations of the percentage, by weight, of the therapeutic agent in each of the six batches for which certificates of analysis were provided, calculated in several different ways to allow for the potential outcomes of the matters in dispute arising from the construction issues, and also for the different approaches to calculation that the experts have taken, and to which I refer in more detail below.
179 The remaining disputes between the parties arise from disagreements between the experts as to the factors to take into account in calculating the weight of the composition for the purpose of assessing whether the weight of the therapeutic agent included in the six batches falls within the percentage ranges set out in the claims.
180 The relevant infringement matters in dispute remaining for determination are as follows:
(1) whether the weight of parecoxib sodium should be determined from the batch formula and Product Description, or whether the assay (the results of a high-performance liquid chromatography (HPLC) analysis) in the certificates of analysis should be taken into account. I refer to this below as the assay dispute;
(2) whether to include a weight for the pH adjusters referred to in the batch formula (O-phosphoric acid and/or sodium hydroxide). I refer to this as the pH adjusters dispute;
(3) whether the Juno products comprise “essentially” parecoxib sodium and dibasic sodium phosphate such that claim 11 is infringed; and
(4) whether the Juno products are compositions substantially described in the examples of the patent such that claim 42 is infringed.
181 In the passages that follow, details of the Juno products have been generalised to preserve confidentiality. Where the context requires explicit disclosure, the confidential information will be redacted in the publicly available version of these reasons.
182 The batch formula provides a list of ingredients and the quantities included in the bulk batch for the manufacture of several thousand vials of the 40 mg product. The Product Description repeats the list of ingredients, but provides the quantity in milligrams for each of the ingredients with the exception of two ingredients being the pH adjusters O-Phosphoric acid or Sodium hydroxide, the quantity for each of which is expressed as “q.s.” meaning quantum satis, “the amount which is enough”.
183 The Manufacturing Process Description states that the quantities of active substance and excipients are weighed in suitable containers, vials are prepared and other equipment is readied. The description then states:
4. Collect 70% of the amount of [water for injection] into the vessel. [Buffer] Disodium Phosphate Anhydrous is added under continuous stirring until complete dissolution. Check the pH of the solution. Adjust the pH of the solution to [redacted text] with addition of sufficient quantity of [pH adjusters] 0.1 N O-Phosphoric acid or 0.1 N Sodium hydroxide as required. Then Parecoxib Sodium, under stirring, is added and stirred until complete dissolution. Check the pH of the solution. Adjust the volume of solution with [water for injection] and stir the solution for 10 min in compounding vessel.
184 Following this step, the bulk solution is filtered and placed into vials. The vials are then loaded into the lyophilisator and lyophilised.
185 The certificate of analysis for each batch identifies the name of the product as “Parecoxib”, the dosage form (e.g. 40 mg/vial powder for injection), the batch size, the date of manufacture and storage temperature and other details. It lists a number of tests performed on the product which include:
(a) a description of the product based on a visual inspection;
(b) pH;
(c) water content;
(d) particulate contamination;
(e) assay (HPLC); and
(f) container/closure integrity test.
For each listed test, the certificate identifies specifications to be achieved in order for the batch to pass.
186 In calculating the amount of parecoxib sodium in each impugned batch Dr Robertson and Professor Winter took different approaches. Professor Winter took as accurate the figure set out in the Product Description for the total amount of parecoxib sodium and divided this by the total number of vials to determine the amount in each vial. Dr Robertson corrected that amount by reference to the result of the assay as indicated in the certificates of analysis which records the percentage of the labelled amount of parecoxib in the analysed sample. The difference in outcome between the two approaches is relatively minor, there being less than 1% difference between the numbers put forward by Professor Winter and Dr Robertson.
187 The expert evidence reveals that the purpose of the assay in the certificate of analysis is to verify that the amount that the batch formula dictates should go into the batch is the amount that actually goes in. The certificates of analysis indicate that the manufacturer may produce a batch that complies with regulatory requirements if the content of the API is between 95% and 105% of the labelled amount. If the batch is not within the range, it will fail. As such, the assay is a means by which the accuracy of the manufacturing process is checked to ensure that it is within an acceptable range of deviation from the batch formula.
188 Professor Winter expressed the view that the means by which the samples are prepared for the assay can produce error. This led him to the view that it was preferable to take as correct the amount said by the manufacturer to have been put in the batch as disclosed in the batch formula. As he put it, the assay procedure requires certain assumptions to be made and the taking of steps prone to introduce error. He gave evidence that the accuracy of the assay depends on: the preparation of the product solution using a microliter pipette; the precision of the fill volume of the vial before the lyophilisation; and knowing how the amount of the reconstitution solution has been calculated. These matters indicate to Professor Winter that, despite the good level of precision of the HPLC process, the assay value itself may not be accurate, and that it is preferable to accept the stated value in the Product Description.
189 However, the purpose of the assay is to verify how much parecoxib sodium ended up in the vials of a particular batch. The experts agree that at least three vials would be used from each batch to test the amount which, Dr Robertson posited, would “iron out any inconsistencies between the vials”. Dr Robertson said, without demur from Professor Winter, that for pharmaceutical products of this nature, the assay would be validated, and thorough precision would be part of that validation. I accept that evidence. Furthermore, I do not accept the logic of Professor Winter’s evidence that inaccuracies in the preparation of the samples for the assay analysis outweigh the benefit of taking the result of the assay into account. This is because Professor Winter accepted that the process of preparing the batches themselves using the batch formula, and then dividing the batch precisely into many thousands of vials, is also prone to error. Indeed in his written evidence, he volunteered that there may be errors in weighing in the solid ingredients and in filling the batch vessel up to the required volume weight with water. Given that the purpose of the assay is to verify, to one decimal point of a percentage value, that what went into the batch was sufficiently accurate to enable the product to be described as having the dose listed, in my view, it is appropriate to take into account the assay result in measuring the amount of parecoxib sodium in the impugned batches. This view accords with the approach taken by Dr Robertson.
190 There is a dispute between the parties as to how to treat the pH adjusters in calculating the weight of the composition of claim 1. It will be recalled that the component identified in (c) of claim 1 provides that “other parenterally acceptable excipients” must be in a total amount of zero to about 10% by weight of the composition. There is no dispute that pH adjusters fall within the description of “other parenterally acceptable excipients”. The applicants rely on the evidence of Professor Winter and submit that pH adjusters must be taken into account in calculating the total weight of the composition. They submit that they are part of the composition and that the batch formula identifies them by amount. They also rely on a consumer medicine information document entitled “Parecoxib JPL Powder for Injection”, tendered as exhibit 4 at the hearing, which states that the formulation contains the pH adjusters. The respondents do not disagree that the pH adjusters are excipients whose weight might be taken into account. They rely, however, on the evidence of Dr Robertson to demonstrate that no calculation is possible on the basis of the information provided in the Neo confidential documents. They contend that no weight should be allowed for in the composition for the pH adjusters as this approach is preferable to “guessing” the amounts actually used.
191 The experts agree that the weight of the pH adjusters cannot be exactly measured. There was no assay for pH adjusters in the certificates of analysis and no document in evidence provides the amount included in each batch.
192 Professor Winter contends, however, that as the batch formula makes provision for two pH adjusters, specifically, 3.723g of O-phosphoric acid and 4.000g of sodium hydroxide, it is appropriate to include the total amount of both in the calculation of the total weight of the composition. In the joint expert report he says that: (1) it is “not helpful” to say that the amount is “not known”; and (2) the batch formula provides a definite number for both compounds which is the “most plausible” and appropriate number to be used.
193 Dr Robertson disputes this approach because, in his view, the pH adjusters are to be added to the batch “as required” as is explained in paragraph 4 of the Manufacturing Process Description (see [183] above). This accords with his understanding of the purpose of these excipients which is to adjust the pH of the solution where necessary. One pH adjuster, being acidic, lowers the pH and the other, being alkaline, raises it. To Dr Robertson, the “q.s.” notation in the Product Description confirms that, for each batch, an amount of one or the other of the pH adjusters may or may not be added. His view is that based on the information that the experts had been provided, it is not possible to know whether any pH adjuster was added or not.
194 The manufacturing process description indicates in step 4 that there are two points at which a pH adjustment may be required. The first is after the buffer is dissolved in water to form a solution. It is specified that either one or the other of the pH adjusters could be added as required to adjust the pH to be within the desired range. However, there is no dispute that each pH adjuster serves a counteracting purpose, to either raise or lower the pH. Only one adjuster should be required unless too much of one adjuster is used such that the target pH is overshot and the other adjuster is then needed to push the pH in the other direction. Dr Robertson considered needing to use both adjusters in this way would amount to “very bad practice”. Professor Winter accepted that in the majority of cases there would be no overshoot. Indeed, in the joint expert report the experts agreed that although it is possible in the case of a pH adjustment that has gone too far in one direction that the other reagent “could” be used to re-adjust the pH into the target range, if an overshoot had not occurred, then only one of the pH adjusters would be used.
195 The second point at which the pH adjustment may occur is after the addition and dissolution of parecoxib sodium has occurred. At this point, the Manufacturing Process Description instructs to “[c]heck the pH”. Professor Winter accepted that the general assumption would be that no further adjustment would be required, although he considered that it was possible.
196 In the result, I consider that there is no evidentiary basis for me arrive, on the balance of probabilities, at a figure as to the amount of pH adjusters used in any of the six batches. I accept it likely that one or other of the adjusters would be used, but the evidence does not supply a basis for concluding what the amount should be. In light of the directions in the Product Description and Manufacturing Process Description to add the pH adjusters in quantities as required, and the expert evidence that generally only one of the two pH adjusters would be required in any given batch, in my view, the applicants position that the total amounts of both pH adjusters disclosed in the batch formula should be included in the calculation of the total weight of the composition is unsustainable. The onus lies on the applicants to establish infringement. The respondents urge that the appropriate course is to conclude that no weight for pH adjusters should be included in the calculation. I accept that this is the appropriate course to take.
197 The applicants rely on the evidence of Professor Winter to support the proposition that claim 11 is infringed. As I have found in section 5.5 above, the words “[t]he composition of any one of Claims 1 to 6, that consists essentially of the therapeutic agent and the buffering agent” do no more than situate the scope of the claim within the upper reaches of the percentages specified in paragraphs (a) and (b) of claim 1. It does not preclude the inclusion of a small weight of other excipients consistent with paragraph (c) of claim 1, but limits this component to a range that is closer to zero than 10% of the composition by weight.
198 In the present case, the relevant dispute concerns whether the maximum amount of therapeutic agent exceeds the upper limit of “about 90% by weight” that is stipulated in claim 1. Accordingly, my findings of infringement in relation to claim 1 will apply equally to claim 11.
6.6 Infringement of omnibus claim 42
199 The applicants rely on claim 42, when read as follows:
A pharmaceutical composition as defined in Claim 1…substantially as herein described with reference to any one of the Examples.
200 The applicants contend that claim 42 is infringed because each Juno product is a pharmaceutical composition as claimed in claim 1 substantially as described in example 1, particularly formulation D (set out at [99] above).
201 The focus of the dispute between the parties lies on the effect to be given to the words “substantially as herein described”, which frequently appears in omnibus-style claims.
202 In Britax Childcare Pty Ltd v Infa-Secure Pty Ltd [No 3] [2012] FCA 1019, Middleton J said:
[29] Without more and in the absence of evidence to the contrary, references in an omnibus claim to claims as “substantially as described herein” are typically construed as being more or less limited to the preferred embodiment described in the specification: see Townsend Controls Pty Ltd v Gilead (1989) 16 IPR 469 at 489; Rescare Ltd v Anaesthetic Supplies Pty Ltd (1992) 111 ALR 205 at 241. Aspects of Justice Gummow’s decision in Rescare were reversed on appeal, but the principles his Honour set out regarding the construction of omnibus claims were not the subject of comment by the Full Court (see Anaesthetic Supplies Pty Ltd v Rescare Ltd (1994) 50 FCR 1).
[30] “Substantially” in this context simply means ‘in substance’: see Deere & Co 82 RPC 461 at 463; Townsend Controls 16 IPR 469 at 489. In Raleigh Cycle Co 65 RPC 141 at 159 Lord Morton rejected an argument that the use of the word “substantially” in an omnibus claim would “introduce uncertainty as to the scope of the monopoly”. His Lordship held that:
[t]he word merely indicates that the patentees are not limiting their monopoly to an electric generator which corresponds in every detail with the generator shown in the drawings, but claim the right to object to the manufacture or sale of an electric generator which is in substance the same as the generator so shown. It may be said with some force that the rights of the patentees would have been the same if the word “substantially” had been omitted from the claim. Even so, its presence cannot render the claim invalid. It may be a matter of some difficulty, in some cases, for the Court to decide whether an alleged infringement is or is not substantially the same as the electric generator shown in the drawings, but the Court does not shrink from such a task.
203 It is apparent that the words “substantially as herein described” are to be understood having regard to the nature of the description of the example nominated in the context of the specification as a whole, in the same manner as other claims are construed. However, when an omnibus claim is in issue, greater attention is generally placed by the words of the claim on the body of the specification to provide the necessary definition of the invention as required by s 40(2)(b) of the Patents Act: see also GlaxoSmithKline Australia Pty Ltd v Reckitt Benckiser Healthcare (UK) Ltd [2016] FCAFC 90; 120 IPR 406 at [69] (Allsop CJ, Yates and Robertson JJ).
204 The applicants accept that there are differences between the composition described in example 1 formulation D in the specification, and the Juno products, but contend nevertheless that infringement is made out.
205 The respondents rely on four differences between the Juno products and the composition identified by reference to formulation D in example 1. They submit that: (1) the buffer is different; (2) the pH is different; (3) the concentration of pH adjusters is different; and (4) that the amount of residual water is different. There is force in the respondents’ submissions.
206 First, the respondents identify a difference of pH, noting that formulation D has a pH of 8.1 whereas the range of pH for the Juno products is [redacted text] to [redacted text]. There is no dispute as to this difference.
207 Secondly, the concentrations of the pH adjusters in the Juno products differ from the concentrations used in formulation D.
208 The respondents refer to the evidence given by Professor Winter that the pH adjuster in the Juno products is 0.1 N O-Phosphoric acid, which is about 0.03 M O-Phosphoric acid, compared to the 1 M O-Phosphoric acid used in formulation D. Furthermore, Professor Winter accepted that the concentration of the sodium hydroxide, being the pH adjuster used if the solution becomes too acidic, is ten times higher in formulation D than in the Juno products.
209 The applicants respond by relying on the evidence of Professor Winter to the effect that to accommodate the differences in concentration, he would simply use more of the pH adjusters when making the Juno products compared to when making formulation D, and that using less concentrated pH adjusters is understandable as a matter of industrial manufacturing process. The applicants also submit that because water is removed (except for residual water) from the composition of the final product, it makes no difference whether more dilute forms of the pH adjusters are used.
210 Thirdly, the respondents rely on the fact that the residual water content of formulation D is identified as being 1% when the residual water in the Juno products, depending on the relevant batch, is between 2.48% and 4.37%. There is no dispute as to this difference, however the applicants seek to minimise the significance of this difference by submitting that the patent generally teaches that compositions of the invention may contain up to 5% residual water.
211 Fourthly, as to the buffer, the applicants contend that although formulation D uses dibasic sodium phosphate heptahydrate as the buffer, and the Juno products use dibasic sodium phosphate anhydrous as the buffer, the distinction is without a difference, because after lyophilisation the form of dibasic sodium phosphate used will be the same. However, the evidence was not as unequivocal as to this. The experts agreed in their joint report that it is possible that the heptahydrate would become anhydrous upon lyophilisation, but that depends on whether the residual water in the composition is bound or adsorbed to the sodium phosphate or parecoxib in the composition, or both. In oral evidence, Dr Robertson clarified that only when there is no residual water could one be confident that the buffer is in its anhydrous form. Professor Winter did not disagree. Dr Robertson also gave evidence that if residual water is present, then there are a number of different hydrated forms of dibasic sodium phosphate, including monohydrate, diydrate as well as the heptahydrate, and it is not possible to say whether and which form will be present in the powder form without further analysis. I accept this evidence.
212 As noted, claim 42, as relied upon by the applicants, is for a pharmaceutical composition “as defined in Claim 1…substantially as herein described with reference to any one of the Examples”. The words “as defined in claim 1” make clear that a composition not falling within the scope of claim 1 will not infringe claim 42.
213 Formulation D is described in example 1 as including five specified ingredients in various amounts and, in the case of the pH adjusters, at particular concentrations. The difference in the amount of residual water in formulation D as compared to the Juno products is of some significance. I have addressed considerations relevant to residual water in section 5.2 above. The applicants point out that the patent teaches, at page 15 lines 14 – 17, that compositions of the invention may contain up to 5% residual water, and that the Juno products are within that range. However, as noted in section 5.2, the amount of residual water in a composition is of significance to the scope of claim 1. Indeed, the applicants have successfully emphasised the importance of including residual water in calculations for determining whether a composition contains the proportions of components specified in claim 1 such that it infringes. It seems to me that it would be inconsistent if one were to take into account residual water as a relevant aspect of the composition for claim 1, and then discount a difference in the amount of residual water between an alleged infringing product, and that specified within formulation D, when considering dependent claim 42. Of note, but perhaps of less significance, is that one of the five specified ingredients, the buffer used in the respective solutions prior to lyophilisation is different in the Juno product, the relevant difference being that the buffer used in formulation D is dibasic sodium phosphate heptahydrate whereas the buffer added to the Juno product is dibasic sodium phosphate anhydrous. I am not persuaded by the evidence that after lyophilisation the composition of the formulation D will contain the same buffer as that in the Juno products. On the other hand, I do not consider that the difference in concentrations of the pH adjusters is likely to be significant for substantially the reasons articulated by Professor Winter. Nor do I consider that the difference in pH identified is significant.
214 However, having regard to the identified differences between formulation D and the Juno products, I consider that the composition of the Juno products is not “substantially as herein described” by reference to formulation D in example 1 of the patent. Accordingly, the Juno products do not fall within the scope of that claim.
6.7 Resolution of questions of infringement
215 The findings set out in sections 5 and 6 above enable a determination as to infringement to be made in relation to each of the six exemplar batches of the Juno products that have been considered.
216 In summary, I have resolved the construction and infringement disputes relevant to whether the relevant claims are infringed as follows:
(a) the therapeutic agent in the Juno products is parecoxib sodium;
(b) “about”, as that word is used in claims 1 and 26 (and their dependent claims) in relation to weight percentage ranges of components in the composition, allows for rounding to the nearest whole percentage such that “about 90%” extends to 90.49% at its upper bound;
(c) the total weight of parecoxib sodium in each batch is that set out in the batch formula as adjusted by the assay values in the corresponding certificates of analysis;
(d) no weight is to be attributed to the pH adjusters in the total weight of the samples; and
(e) residual water is to be included in the total weight of the samples for the assessment of claim 1 and its dependent claims, but not for claim 26 and its dependent claims.
217 Confidential Annexure A to these reasons sets out the agreed calculations relevant to each of the batches. In conclusion I have found that:
(1) batches 31601, 31602 and 31603 do not fall within the scope of claim 1 or any of its relevant dependent claims; and
(2) batches 31604, 31605 and 31606 fall within the scope of claim 1 and also claim 11. Having regard to the admissions made by the respondents regarding the features of the Juno products, these three batches also fall within the scope of claims 4, 5, 14, 15, 17, 18, 19, 20, 21, 24, 34, 35, 36, 37, 38, 39, 40 and 41. These batches do not fall within claim 7; and
(3) none of the six representative batches fall within the scope of claim 26 or any of its dependent claims.
218 As I understand the agreed position at trial, the conclusions set out above are sufficient when coupled with the admissions made by the respondents to enable the parties to draw up short minutes of orders giving effect to these reasons. These will include the making of relevant orders in relation to authorisation, joint tortfeasorship and also infringement by supply of the Juno products pursuant to s 117 of the Patents Act.
219 The respondents contend that the relevant claims are not for a patentable invention within the meaning of s 18(1)(b)(ii) of the Patents Act in that, when compared with the prior art base as it existed before the priority date, the alleged invention does not involve an inventive step. In their amended particulars of invalidity the respondents contend that the invention would have been obvious to the person skilled in the art in the light of the common general knowledge alone or in the light of three nominated prior art documents. Prior to the hearing, the prior art documents were reduced to two in number, being Jain and Talley.
220 Section 7(2) of the Patents Act provides that an invention is taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed in the patent area before the priority date of the relevant claim, whether that knowledge is considered separately or together with the information in s 7(3).
221 The applicable version of s 7(3) provides:
The information for the purposes of subsection (2) is:
(a) any single piece of prior art information; or
(b) a combination of any 2 or more pieces of prior art information;
being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood, regarded as relevant and, in the case of information mentioned in paragraph (b), combined as mentioned in that paragraph.
222 In closing submissions the respondents did not advance a case based on common general knowledge alone. Instead they contend that Jain and Talley constitute two pieces of prior art information that would be combined in accordance with the requirements of s 7(3).
223 In closing submissions the respondents contended that the lack of inventive step of the invention claimed may be demonstrated by reference to a straightforward “development pathway” of eight steps which, it submits, are as follows:
(1) Jain would be identified by a pain specialist such as Professor Scott, who would be a member of the notional formulation team;
(2) Jain would then be supplied to a formulation expert such as Dr Robertson who would obtain Talley, which is footnoted in Jain;
(3) upon reading Jain and Talley, the formulation expert would decide to attempt to formulate parecoxib sodium as an intravenous formulation;
(4) the formulation expert would obtain parecoxib sodium, either from a commercial supplier or by synthesising it;
(5) the formulation expert would then conduct testing on parecoxib sodium and decide to formulate it as a reconstitutable lyophilised powder composition (and not a ready-to-use injectable composition);
(6) the expert would decide upon the composition of the pre-lyophilisation solution;
(7) a routine lyophilisation process would be used to prepare the reconstitutable powder composition; and
(8) the lyophilised drug product would then be reconstituted using a solution of 0.9% sodium chloride.
224 The respondents contend that the question of whether a claimed invention involves an inventive step may be assessed in a number of ways. A party need not lead expert evidence that echoes the particular test. Rather, the question of obviousness is one for the court which is to be answered in light of the whole of the evidence. In support of this proposition, the respondents cite AstraZeneca at [43] (French CJ). They submit that, in cases involving pharmaceutical patents, it is not uncommon to pose the reformulated Cripps question endorsed by the High Court in Aktiebolaget Hässle v Alphapharm Pty Ltd [2002] HCA 59; 212 CLR 411 at [53] (Gleeson CJ, Gaudron, Gummow and Hayne JJ) which they propose should take the following form:
Would the notional research group as at April 2001, in all the circumstances, which would include the CGK and Jain and Talley, directly be led as a matter of course to formulate a lyophilized powder (such as in a sealed vial in a sterile condition) comprising parecoxib sodium (in a dosage amount of between about 1mg to about 200mg and when preparing the aqueous solution before lyophilisation it is to be added last), a buffering agent (such as dibasic sodium phosphate) and other excipients (if any), within the relative weight percentage ranges claimed in the asserted claims; said composition being reconstitutable in an aqueous solvent (such as sodium chloride) to form an injectable solution (with a pH of between about 7.5 to about 8.5) and for use in a method of treating and/or preventing a COX-2 mediated disorder (such as wherein the administration is by intravenous bolus); in the expectation that it might well produce a useful result.
225 The respondents submit that it is not necessary for there to be certainty of outcome, but that the skilled addressee be directly led as a matter of course to try the claimed invention in the expectation that a particular research path “might well produce” a useful result, citing Mylan Health v Sun Pharmaceutical Ltd [2020] FCAFC 116; 279 FCR 354 at [502] (Middleton, Jagot, Yates, Beach and Moshinsky JJ). They submit that whether or not the analysis is conducted by reference to this question, or at the more general level of whether or not there is a difficulty overcome or barrier crossed, the invention claimed in the relevant claims lacks an inventive step.
226 The respondents advance an alternative case based on the prospect that I find some of the broader relevant claims to be obvious but not all of the dependent claims. For narrower claims 15, 17, 19 and 21 the respondents contend that particular features are “descriptive” or “a form of parameteritis” that are not of themselves sufficient to confer or amount to an inventive step.
227 The applicants contend that the evidence adduced by the respondents does not satisfy the requirements of s 7(3) of the Patents Act such that Jain or Talley would not have been ascertained by the skilled addressee. They contend that Professor Scott would not form part of the skilled team, that the search approach and terms that he used were not typical of the hypothetical worker and that the search terms used were infected by hindsight bias because of the nature of the task that Professor Scott was asked to undertake. They contend that the simpler keyword searches conducted by Professor Goucke were more representative, and they did not turn up Jain or Talley.
228 The applicants contend that the first step in the inventive step analysis is to consider whether the problem addressed by the patent itself was common general knowledge or disclosed in s 7(3) information before the priority date. They submit that the problems addressed by the patent were not common general knowledge or disclosed in Jain or Talley. In particular, the applicants contend that three problems are identified and addressed in the patent:
(1) the conversion of parecoxib to valdecoxib in solution (the conversion problem). The applicants contend that this was not part of the common general knowledge and is not disclosed in Jain or Talley;
(2) the instability of parecoxib in powder form is such that parecoxib converts to valdecoxib when in a lyophilised powder (the stability problem). The applicants contend that this was not part of the common general knowledge. The data in the patent teaches that this can be reduced to acceptable levels by reducing or eliminating other excipients, including bulking agents such as mannitol; and
(3) the reconstitution time of the powdered formulation (the reconstitution problem). The patent teaches that the parecoxib formulations containing mannitol and other excipients (in formulations E – J of example 1) did not dissolve instantly. The applicants submit that this was also not common general knowledge or disclosed in the prior art.
229 The applicants submit that the person skilled in the art would prefer to develop an injectable formulation that is ready to use as a liquid. They submit that as the conversion problem was not common general knowledge, the formulator would only turn to the development of a reconstitutable lyophilised powder after trying to make the ready to use option work. The applicants submit that it would take a significant period of time for the formulator to discover the conversion problem. They submit that if Jain was found using MeSH search terms (which was the approach adopted by Professor Scott), it could only have been found shortly before the April 2001 priority date (when Jain was indexed on the PubMed database using MeSH terms), with the result that the conversion problem could not have been identified, let alone solved, by April 2001. This gives rise to what I refer to in section 7.7 as the timing argument.
230 The applicants next contend that even after realising the need for a lyophilised formulation to overcome the conversion problem, the stability problem and the reconstitution problems would not have been apparent and did not form part of the common general knowledge. They submit that a key aspect of the common general knowledge was that bulking agents were very common and popular in 2001 and that a person seeking to make a lyophilised product would likely add a bulking agent to the formulation. A common bulking agent was mannitol. They submit that the formulation would not simply lyophilise the ready to use intravenous solution that was developed, but would reformulate for lyophilisation. That, the applicant’s submit, would involve applying a common general knowledge rule of thumb to increase solids content to about 10% in the pre-lyophilised solution before lyophilising the formulation. Following the rule of thumb would then cause complications, being the stability and reconstitution problems, that the skilled formulator would not foresee, which would ultimately need to be overcome.
231 Before addressing the ultimate issue, I commence by addressing the disputes that emerge by reference to the eight steps that the respondents rely upon.
7.4 Step 1: the skilled team and ascertaining Jain
232 The respondents rely on a combination of the evidence of Professor Scott and Dr Mokdsi to support the submission that the person skilled in the art could, before the priority date, be reasonably expected to have ascertained, understood and regarded Jain as relevant within s 7(3). They also rely on evidence given by Professor Goucke in cross-examination to support the appropriateness of the search technique used by Professor Scott.
233 In answer, the applicants first submit that the notional addressee is a formulation scientist alone and, accordingly, searches conducted by Professor Scott do not meet the threshold of relevance. I have rejected that submission in section 4.3 above. Next, they submit that if a notional skilled addressee is to be conceived of as a team, the team would not include a pain management physician such as Professor Scott. I have also rejected that submission in section 4.3.
234 The applicants next contend that the evidence does not establish that the notional skilled addressee would use MeSH terms in conducting searches. In this respect they rely on the evidence of Professor Goucke and Mr Cruise. They submit that the search strategies proposed by Professor Scott are unusually complex and affected by hindsight because the initial task that Professor Scott was asked to perform “inevitably directed” him towards Jain. They rely on Professor Goucke’s evidence that he would not use MeSH terms but rather keyword searches and, deploying search terms chosen by him, he would not have found Jain. The applicants submit that reliance by the respondents on the cross-examination of Professor Goucke is misplaced and that the evidence does not establish that the notional skilled addressee would have ascertained Jain or Talley. They also submit that Professor Scott in any event does not represent the hypothetical skilled worker because he is too experienced in conducting academic work in pain management.
235 Professor Scott was asked to assume that he was part of a notional research group, including one or more additional experts playing different roles such as a pharmaceutical formulation specialist, which has been asked:
…to identify and formulate an analgesic for the Australian market which would be better than or at least a useful alternative to the drugs then currently in use for the management of acute post-surgical pain.
(the initial task)
236 He notes that he discussed analgesics used for the management of acute post-surgical pain in his first affidavit, which formed the basis of the pain primer to which I have referred in section 3.2 above. He gives evidence that before 3 April 2001 he would have approached the task by conducting a literature search using the online database PubMed utilising a strategy of starting with a high-level search and then progressing down into more detail with more specific terms. His preferred approach was to use search strategies such as MeSH term classifications and keywords with wildcards and if the results were too large, he would limit the searches further by adding more specific or limiting terms. He would confine his search to results more recent than 1996. Professor Scott gave evidence of five sets of words and search strategies that he would have used including broad terms and limiting words designed to narrow the search results.
237 The evidence indicates that PubMed is an online database that comprises millions of citations for biomedical literature from MEDLINE, life science journals and online books. PubMed was a very commonly used database for biomedical research in April 2001. The MeSH search term approach involves using search terms following a system promoted and controlled by the publisher of PubMed and organised in a particular hierarchy. MeSH terms that can be used to search PubMed are, and were before 3 April 2001, on a database made available by the publisher. MeSH terms had been available to those conducting PubMed searches for decades prior to 2001. MeSH terms were tools available to be used by persons wishing to search the PubMed database well before the priority date.
238 Although Professor Goucke did not use MeSH terms in framing his searches, he accepted in his oral evidence that he was “possibly” aware of their availability before April 2001. Professor Scott, Mr Cruise and Dr Mokdsi were all aware of MeSH terms before the priority date.
239 I am comfortably satisfied that the availability and use of MeSH search terms generally formed part of the common general knowledge of medical researchers, including pain specialists, as well as those likely to assist such researchers in the conduct of PubMed searches, including librarians, as at the priority date. They were tools supplied by the publisher of the database to aid in its search.
240 There is no dispute that it is possible closely to simulate a search on PubMed now as if it were being conducted on 3 April 2001.
241 Professor Scott gave evidence that he would conduct a literature search on PubMed, using MeSH term classifications and keywords with wildcards to ensure his searches captured other forms of the target words, to produce a list of potential papers identified by title, author, journal, date and abstract. If the results of the search were too large, he would limit the searches further by adding additional, more-specific or limiting terms. He would also limit his search to results more recent than 1996.
242 Professor Scott identified at the outset five kinds of search strategies. Search 1 was: Analgesics, non-narcotic [MeSH Major Topic] AND pain, postoperative [MeSH Major Topic] as the broadest search further limited by the addition of filters: (a) anti-inflammatory agents, non-steroidal/pharmacology* [MeSH term]; (b) anti-inflammatory agents, non-steroidal/pharmacology* [MeSH term] AND (intravenous OR parenteral OR intramuscular) [Text word]; (c) cyclooxygenase 2 inhibitors [MeSH term]; and (d) cyclooxygenase 2 inhibitors [MeSH term] AND (intravenous OR parenteral OR intramuscular) [Text word]. Search 2 was: Analgesics, non-narcotic [MeSH Major Topic] AND pain/drug therapy* [MeSH subheading] further limited by: (a) anti-inflammatory agents, non-steroidal/pharmacology* [MeSH term]; (b) anti-inflammatory agents, non-steroidal/pharmacology* [MeSH term] AND (intravenous OR parenteral OR intramuscular) [Text word]; (c) cyclooxygenase 2 inhibitors [MeSH term]; and (d) cyclooxygenase 2 inhibitors [MeSH term] AND (intravenous OR parenteral OR intramuscular) [Text word]. Search 3 was: Analgesics, opioid [MeSH Major Topic] AND pain, postoperative [MeSH Major Topic] further limited by: (a) (intravenous OR parenteral OR intramuscular) [Text word]. Search 4 was a slight permutation of search 3, substituting the “pain, postoperative” MeSH Major Topic with the “pain/drug therapy*” MeSH subheading, and search 5 was (Cox-2 inhibitors OR coxibs) [All Fields] AND (postoperative OR post-operative) [All Fields] AND acute pain [All Fields].
243 Professor Scott explained that each search involves searching against the first topic and then “drilling down” from an initial high level search to include more specific terms as filtered by (a), and then the first search as filtered by (b), and so on. He explained that in normal practice if he were performing the searches himself he would enter the high level search terms, see whether the search produced too many or too few results, and then apply further filters.
244 Dr Mokdsi was then provided with Professor Scott’s search strategy and asked to conduct searches as they would have been done as at April 2001. He did this with the five search strategies and recorded his results. Professor Scott then reviewed the affidavit of Mr Cruise and identified some search terms that Dr Mokdsi had misspelled, or had been incorrectly transcribed when he gave his instructions. Despite several criticisms advanced by the applicants, I consider that the errors were simple mistakes and do not reflect adversely on the task performed. The results using the correct search terms produced only marginally different results, none of which are in my view material.
245 The outcome of the corrected search conducted by Dr Mokdsi using the first search framework proposed by Professor Scott for “analgesics, non-narcotic” [MeSH Major Topic] AND “pain, postoperative” [MeSH Major Topic], using PubMed sources available from the beginning of 1996 until 30 April 2001 yielded 348 publications. Adding to the first search the filters respectively of: (a) “anti-inflammatory agents, non-steroidal/pharmacology” [MeSH Terms] reduced the number to 171; (b) “anti-inflammatory agents, non-steroidal/pharmacology” [MeSH Terms] AND (“intravenous” or “parenteral” or “intramuscular”) [Text Word] reduced the number to 68; (c) “cyclooxygenase 2 inhibitors” [MeSH Terms] yielded only 5 publications; and (d) “cyclooxygenase 2 inhibitors” [MeSH Terms] AND (“intravenous” or “parenteral” or “intramuscular”) reduced the number of publications to 1.
246 The search results for search 2 using the second search strategy provided by Professor Scott commenced by yielding 705 publications and, by the addition of the same refinements as (a) – (d) in search 1, the results were reduced respectively to 314, 58, 26 and 1.
247 It is notable that the results of the highest level of searches 1 and 2 resulted in respectively 348 and 705 publications. The evidence of Professor Goucke was that, in his view, it was reasonable when addressing the task for the reviewer to read the text of anything up to 1100 publications.
248 The results obtained by Dr Mokdsi were then provided to Professor Scott. He confirmed that the searches undertaken were of the kind that he would have done and considered the results. He added an additional search that he conducted himself.
249 Professor Scott reviewed the results and identified an aggregate of 253 articles of initial interest by reference to their titles and abstracts. After further review he reduced that number to 114 articles of potential interest. He identified four categories of drug candidates for potential use in the management of acute post-operative pain: (1) novel systemic analgesics (which include selective COX-2 inhibitors); (2) stereo-isomer racemic formulations; (3) novel routes including parenterals of “conventional therapies” (and others); and (4) novel “antagonist roles” such as mu-antagonists. He was then asked to refine the 114 citations to identify which of those citations, and the corresponding compounds, he would have given preference to for further consideration by the team. He identified 28 articles, highlighting 11 of those that he would have prioritised. The Jain article was one of those 11 prioritised.
250 Professor Scott explained in his oral evidence that Jain stood out because of its title and its abstract indicating that it was a human study of a parenterally administered drug of interest to him, being a non-steroidal COX-2 inhibitor. He classed it as one of the “highly relevant” articles to the task.
251 Professor Scott identified as the date range for his searches articles published between 1 January 1996 and 30 April 2001, thereby potentially including articles only able to be found after the priority date in his search results, the priority date being 3 April 2001. However, the evidence demonstrates that it is possible to identify the date upon which an article is able to be located using MeSH terms.
252 The evidence indicates that the MeSH terms associated with Jain were added to the PubMed database at 10.01am on 27 March 2001, being about one week before the priority date (depending upon in which time zone the MeSH terms were added). I find that a search using the MeSH terms proposed by Professor Scott, conducted as at 3 April 2001, would have produced Jain as a result.
253 Mr Cruise made several criticisms of the searching approach adopted by Professor Scott and also picked up errors in Dr Mokdsi’s execution of the search strategy. As I have noted, Dr Mokdsi then gave a second affidavit, conducting the searches again but correcting the errors identified by Mr Cruise. Having corrected these errors, Dr Mokdsi’s searches returned, for the most part, fewer results. However, Jain was identified by both the initial and corrected searches. In my view, the errors detected by Mr Cruise in Dr Mokdsi’s initial searches do not detract from the reliability of the exercise conducted. Nor, in my view, does Mr Cruise’s observation that the search instructions provided by Professor Scott are more specific than he had seen before. That, no doubt, was in part a result of the necessarily artificial exercise that Professor Scott was asked to perform, which included anticipating filters that may be useful to narrow down search results of the broader searches, a process that would normally take place iteratively.
254 Professor Goucke gives evidence that he would have used keyword searches if asked to perform the same initial task as Professor Scott. He would have used different words to those chosen by Professor Scott and searched in the title and abstract fields. He would then review the titles of the search results and classify them by relevance for further review. He would then review the abstracts of those articles nominated for further review to identify articles of interest. Unlike Professor Scott, he would not have restricted his results by reference to a date.
255 Professor Goucke gives evidence that he would not have used MeSH terms, a technique that he did not know how to use in April 2001. His keyword search terms were implemented by Mr Cruise. They did not yield Jain. His first search term was (analgesic OR analgesia) AND human AND (medication OR drug), which produced 640 results. He devised five further searches which returned, respectively, 1,790, 10,992, 260, 152 and 653 results. None produced Jain or Talley.
256 Professor Goucke gives evidence that he has never been involved in a research group asked to identify and formulate an analgesic. He considered that it would be unusual for anaesthetists or specialist pain medicine physicians to be involved in a research group early in the drug development process. He reviewed Professor Scott’s resume and considered that Professor Scott is different from a typical pain medicine physician because: (a) he had undertaken a PhD focussing on neuropharmacology; (b) he had an unusual level of experience in designing and evaluating trials and research; and (c) he had written articles related to early stage drug development and testing work. Professor Goucke gives evidence that, unlike Professor Scott, his involvement in the drug development process has always been after a particular drug has been developed and shown to be safe for use in animals and humans, at which point a pharmaceutical company will seek out clinicians and recruit them to assist in phase II and III clinical studies.
257 The challenge to step 1 broadly concerns whether or not Jain could reasonably be expected to have been ascertained within s 7(3) of the Patents Act having regard to: (1) the relevance and appropriateness of the initial task posed to Professor Scott; (2) the use by Professor Scott of MeSH terms; and (3) the qualifications and experience of Professor Scott.
258 The legal context of the dispute arises under s 7(2) and, more particularly, s 7(3) of the Patents Act.
259 In AstraZeneca AB v Apotex Pty Ltd [2014] FCAFC 99; 226 FCR 324 the majority (Besanko, Foster, Nicholas and Yates JJ) said at [203]:
If the problem addressed by a patent specification is itself common general knowledge, or if knowledge of the problem is s 7(3) information, then such knowledge or information will be attributed to the hypothetical person skilled in the art for the purpose of assessing obviousness. But if the problem cannot be attributed to the hypothetical person skilled in the art in either of these ways then it is not permissible to attribute a knowledge of the problem on the basis of the inventor’s “starting point” such as might be gleaned from a reading of the complete specification as a whole.
260 However, before a document containing prior art information can be used along with the common general knowledge for the purposes of the s 7(2) inquiry, it is necessary that it meets the requirements of s 7(3). The High Court in Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) [2007] HCA 21; 235 CLR 173 (Lockwood (No 2)) at [127] explained that, in answering the question of obviousness, the information referred to in s 7(3), like that part of the prior art base which is the common general knowledge, is considered for a particular purpose. That purpose is to look forward from the prior art base to see what the skilled person is likely to have done when faced with a problem similar to that which the patentee claims to have solved with the claimed invention.
261 It is in that context that one considers the appropriateness of the initial task posed to Professor Scott.
262 The agreed pain primer indicates that, before April 2001, acute post-operative pain was most commonly treated by the use of classes of analgesics including opioids and NSAIDs. A multi-modal approach of using a range of analgesic medications to treat acute post-surgical pain was most often used. Opioids were the gold standard drugs for this purpose because of their effectiveness in treating pain, but they came with adverse effects such as respiratory depression, sedation, cough suppression, nausea and vomiting and impaired bowel and bladder function. NSAIDs (inhibitors of COX-1 and/or COX-2) and paracetamol were the most common alternatives to opioids. However, non-selective COX-2 inhibitors were known to cause a number of undesired side effects including inhibiting platelet aggregation and increasing bleeding following certain operations. They were also known to increase the risk of gastrointestinal ulcers and bleeds as a result of decreased gastric cytoprotection. This is because non-selective NSAIDs inhibit COX-1 which ordinarily serves a homeostatic role in the function of kidney, gut mucosa, smooth muscle, platelets and endothelium.
263 On the other hand, it was common general knowledge that COX-2 selective inhibitors did not significantly inhibit COX-1 at therapeutic concentrations. As a result, they were understood to have a superior safety and tolerability profile by causing less gastrointestinal and bleeding side effects whilst providing effective analgesia. Accordingly, as at 3 April 2001, COX-2 selective inhibitors were of considerable interest to those skilled in the art for use as part of multi-modal analgesia in post-operative acute pain management instead of non-selective NSAIDs and opioids.
264 The initial task may be said to arise from the common general knowledge. It is broadly expressed and concerns the identification and formulation of an analgesic that would be better than, or a useful alternative to, available drugs for a particular use, namely, the treatment of acute post-surgical pain. The identification of “acute post-surgical pain” is not a “nudge” or leading prompt.
265 The invention is directed towards the development of a formulation of COX-2 selective inhibitors for use (amongst other things) in the treatment of post-operative pain. As the pain primer demonstrates, that was a known problem. The patent provides in its background at pages 1 – 2 that the invention is directed towards a parenterally deliverable formulation of a selective COX-2 inhibitory drug, the uses of which are described later in the specification to include the treatment of acute post-surgical pain. In my view, the initial task does not “lead” the skilled person in the art to that problem. Rather, it poses an open textured question that is supported by the common general knowledge.
266 It is not to the point (as the applicants submit) that the patent is also directed towards the formulation for use of selective COX-2 inhibitors in the treatment of other conditions. One pathway to obviousness will be sufficient.
267 The debate concerning the use by Professor Scott of MeSH terms also arises in the context of s 7(3).
268 The term “ascertain” within s 7(3) of the Patents Act means “to find”: Commissioner of Patents v Emperor Sports Pty Ltd [2006] FCAFC 26; 149 FCR 386 at [30] (Heerey, Kiefel and Bennett JJ). Whether or not the prior art information can be reasonably expected to have been found depends on what the skilled person is likely to have done when faced with a problem similar to that which the patentee claims to have solved with the claimed invention. Section 7(3) requires more than a possibility. It requires a prediction as to the events that would have taken place as at the priority date. The prediction must be sufficiently reliable to be regarded as reasonable: Aspirating IP Ltd v Vision Systems Limited [2010] FCA 1061; 88 IPR 52 at [457] (Besanko J).
269 The relevant question is whether, having regard to the evidence, it may reasonably be predicted that the hypothetical non-inventive skilled worker faced with the task before April 2001 would have ascertained Jain.
270 I am satisfied that the task nominated for Professor Scott was appropriate and provides the foundation for his determination of the search methodology that he adopted.
271 Further, in my view, there is a logical connection between the task that Professor Scott was set and the broader searches that he proposed. For instance, the first: Analgesics, non-narcotic [MeSH Major Topic] AND pain, postoperative [MeSH Major Topic] sits conformably with the task of identifying an analgesic for the management of post-operative pain. The nomination of “non-narcotic” in searches 1 and 2 reflects the accepted common general knowledge that opioids produced adverse side effects. The subsidiary search terms identified in those searches all reflect aspects of the accepted common general knowledge concerning the use of NSAIDs, and the preferred position that the analgesic be administered parenterally. Searches 3 and 4 focus on opioids, then the gold standard drug for the management of acute post-surgical pain.
272 Professor Goucke’s evidence was that he would use the same database (PubMed) but would not use MeSH terms to conduct his searches. Instead, he would rely on keyword searches. He was not aware in 2001 of how to use MeSH terms, but accepted that in addition to conducting a search himself, he may also have sought the assistance of a librarian. He also accepted that other pain management experts acting reasonably may have chosen to use the MeSH terms, if they understood the system. He accepted that he would defer to Professor Scott as to appropriate terms to use in MeSH searches.
273 I consider that the use by Professor Scott of MeSH terms, which are provided by the publisher of the PubMed database to assist users in the conduct of searches on that database, to be conventional and appropriate. I have found that the availability of MeSH terms formed part of the common general knowledge. A person wishing to search the PubMed database as at the priority date had available to them the publisher’s MeSH search terms which the publisher used to code or tag articles in the PubMed database. As the evidence of Professor Scott indicates, the use of those terms could ensure that articles, coded by the publisher as being relevant to certain topics, are identified. I do not accept that persons such as the pain experts would not use such tools. The evidence discloses that PubMed and MeSH terms had been available and used for decades before April 2001. Although Professor Goucke did not himself use them at this point in time, it is apparent that Professor Scott’s normal approach in 2001 was to use such terms to conduct literature searches. They have advantages over simple keyword searches and, in my view, at least one known and available approach to take as at the priority date was to use them. Furthermore, Professor Goucke could quite easily have asked a librarian to assist him, which was something that he did from time to time. I find that those with expertise in searching were likely to be familiar with conducting literature searches using MeSH term which, as I have noted, was a tool made available by the publisher of PubMed to assist in using its database.
274 I do not consider that the fact that Professor Goucke, using a less refined form of searching, failed to find Jain leads to the conclusion that the path chosen by Professor Scott was not one that would routinely be taken by the person skilled in the art.
275 Moreover, I do not accept the applicants’ submission that Professor Scott is not representative of a pain expert likely to be found in the hypothetical team. In this context, Professor Goucke’s evidence was that because Professor Scott had a PhD in neuropharmacology, developed experience in designing and evaluating trials and published on the topic of early stage drug development, he would not be representative of a pain expert likely to be in the hypothetical team. In my view, these matters tend to support the proposition that Professor Scott is the type of person likely to be engaged in the early stages of drug formulation rather than Professor Goucke, whose experience as at April 2001 was as a clinician responsible for conducting phase II and III clinical trials for drugs that had already been identified and formulated. I have indicated earlier in my reasons that the skilled team would include in addition to or in the alternative to a pre-clinical development expert, a pain expert able to speak to the administration of the formulation under development. Professor Scott appears to fit that bill more readily than Professor Goucke, having regard to his study and experience.
276 Accordingly, in my view, Jain is a piece of prior art information for the purposes of s 7(2) that the skilled person could, before April 2001, be reasonably expected to have ascertained, understood and regarded as relevant within s 7(3).
7.5 Step 2: Jain and identifying Talley
277 The respondents’ second step involves the supply of Jain to a formulation expert, such as Dr Robertson, who would then obtain Talley, which is footnoted in Jain.
278 In his first affidavit Dr Robertson gives evidence that a drug development project usually commences with identifying a target disease or condition which is generally done in consultation with a medical specialist with expertise in the relevant disease or condition to gain an understanding of relevant matters such as the available drugs to treat that condition, any benefits of existing drug classes, any drug classes or molecules that may be desirable to target, and side effects of existing drug classes or molecules that may be desirable to avoid, including as to the route of administration. He also provides a general description of the process of drug development from his perspective as a formulator, the content of which is set out above as the formulation primer in section 3.1.
279 In his second affidavit Dr Robertson refers to being supplied with Jain by the solicitors for the respondents and asked to consider the following formulation task:
…the task as at 3 April 2001 of formulating a pharmaceutical composition for administration to human patients in which parecoxib is the active pharmaceutical ingredient (API)…
Dr Robertson was instructed to have regard only to the information set out in Professor Scott’s first affidavit (which forms the basis for the pain primer), the matters set out in Jain, the content of his first affidavit (which contains information used in the formulation primer) and any other information that he regarded as well-known and generally accepted by himself and other formulators in the field in Australia as at 3 April 2001.
280 Dr Robertson gave evidence that he would immediately identify and obtain a copy of Talley which is footnoted in Jain and, for reasons that he develops in his affidavit, would have proceeded to formulate parecoxib having the following characteristics:
(a) for intravenous administration;
(b) a homogeneous aqueous solution comprising phosphate buffered saline;
(c) at a concentration less than or equal to 22 mg/ml; and
(d) at a volume of 2 mL or less.
281 Professor Winter was provided with similar information to that provided to Dr Robertson. He was given copies of Talley and Jain and the information set out in Dr Robertson’s first affidavit. He was asked how he would go about formulating a parecoxib drug product having regard to Talley and Jain, to what he regarded as well-known and generally understood by himself and other formulators before 3 April 2001 and to the general process of drug development by formulators described by Dr Robertson in his first affidavit (to the extent that he agreed with Dr Robertson’s views).
282 The formulation experts did not agree in the result. In order to explain some of their differences it is necessary first to consider each of Jain and Talley.
7.5.2 The disclosure of Jain and Talley
283 Jain is entitled “Evaluation of intravenous parecoxib for the relief of acute post-surgical pain”. It commences:
Parecoxib is a prodrug of valdecoxib, which is a potent and selective inhibitor of COX-2. Intravenous preparation of parecoxib is in Phase III clinical trials for the management of acute and severe post-surgical pain. It is the only COX-2 inhibitor that is available in a parenteral formulation. Clinical results compare parecoxib with ketorolac, a NSAID, which is the only non-narcotic analgesic available in parenteral formulation that can be administered for the relief of moderate to severe acute pain. Pharmacokinetic studies have shown that parecoxib is converted to valdecoxib within a short time following administration by im. or iv. injection. In clinical trials, parecoxib compares favourably with ketorolac and produces less gastric or duodenal ulcers, the predominant adverse effect, than ketorolac. Parecoxib thus fulfils some of the desirable characteristics of an ideal non-narcotic analgesic for severe post-surgical pain and has application in other acutely painful conditions. Parecoxib is expected to be filed for approval before the end of 2000 and is expected to be introduced in the market in 2001. It has favourable prospects for a fair share of the post-surgical pain relief market which is valued at approximately US $1billion for the year 2000.
284 The introduction to the article then identifies that its focus is the use of parenteral formulations of parecoxib (a prodrug of valdecoxib) for the treatment of acute post-surgical pain, valdecoxib belonging to the NSAIDs class and undergoing clinical trials for the treatment of arthritis and pain (Talley is footnoted at this point). These drugs are described as inhibiting the synthesis of prostaglandins and thromboxane by inhibiting the enzyme COX-1 or COX-2. It says that COX-2 selective drugs, such as celecoxib and rofecoxib (approved in the US for acute pain indications), have resulted in NSAIDs with good analgesic and anti-inflammatory effects, without the gastrointestinal and antiplatelet side effect profile of older drugs. It notes that few NSAIDs can be administered parenterally and that there has been interest in the development of a safe non-narcotic parenteral analgesic for post-surgical pain, moving away from the dominant drug morphine and its derivatives.
285 Later, the article identifies products in development, including parecoxib which is described as a “[p]rodrug of valdecoxib, a COX-2 inhibitor/iv injection” under Phase III development. The article then sets out details regarding parecoxib including the following:
5.1 Chemistry
Parecoxib [chemical name supplied] is an injectable sulphonamide-based prodrug of the COX-2 inhibitor valdecoxib. Valdecoxib has poor water solubility, but parecoxib is rapidly cleaved to form valdecoxib in minutes. The chemical structure is shown in Figure 1. Acylation of isoxazole sulphonamide with an anhydride in the presence of triethylamine leads of the formation of the corresponding acylated sulphonamide.
(emphasis added)
286 The article refers to the pharmacodynamics of valdecoxib and the pharmacokinetics of parecoxib. Under the heading “Clinical trials” the article refers to the results of a study being presented at a 2000 conference. It says that the efficacy of parecoxib to combat post-surgical dental pain was evaluated in a double-blind, randomised study and:
Analgesic efficacy of iv. doses of 1, 2, 5, 10, 20, 50 and 100mg parecoxib was compared with 30mg of ketorolac and placebo.
287 The article notes that all active treatments were superior to placebo, and that a phase III study of the efficacy of parecoxib 20 mg and 40 mg intravenous doses given post-surgically was in progress for determining the narcotic-sparing effectiveness in relieving pain following general surgery.
288 The article provides an expert opinion that is favourable to the use of parecoxib as a prodrug. It concludes:
From the available information, iv formulation of parecoxib appears to be a promising drug for the relief of severe post-surgical pain. It fulfils some of the desirable characteristics of an ideal analgesic for this indication and is safe and effective. Superiority of COX-2 inhibitors over NSAIDs, in terms of adverse effect profile, has not been established as yet. Publication of detailed results of Phase III studies will help in further assessment of this drug. It has a wider application in the relief of acute pain besides the post-operative pain and is likely to replace ketorolac, which is the current standard iv. non-narcotic analgesic for post-surgical pain.
(emphasis added)
289 Talley is entitled N-[[(5-Methyl-3-phenylisoxazol-4-yl)-phenyl]sulfonyl]propanamide, Sodium Salt, Parecoxib Sodium: A Potent and Selective Inhibitor of COX-2 for Parenteral Administration. There is no dispute between the formulation experts that they would obtain Talley after reading the cross-reference to it in the first footnote of Jain.
290 Talley notes the benefits of selective inhibitors of COX-2 as being widely recognised as offering promise of treatment of inflammatory conditions without the side effects associated with non-selective inhibitors, and notes that relatively few NSAIDs may be administered parenterally for the treatment of pain and inflammation. It says:
In general, COX-2 inhibitors of the diarylheterocycle class such as 1 and 2 possess modest aqueous solubility. This physicochemical characteristic restricts the dosing options available for this class of drug. In considering the development of a COX-2 inhibitor for parenteral administration, we wondered if a prodrug of a sulphonamide-based inhibitor could be designed which would possess the appropriate combination of aqueous solubility and in vivo antiinflammatory activity. Herein we describe our efforts that culminated in the identification of the injectable COX-2 inhibitor parecoxib sodium.
291 Talley describes the process of synthesis of parecoxib sodium and then refers, amongst other things, to the authors’ strategy to develop an injectable COX-2 inhibitor commencing with the idea of identifying a water-soluble prodrug that would undergo biotransformation in vivo (i.e., in the patient). It found that its preferred form, which was parecoxib sodium (being identified as 5b) had solubility of 22 mg/ml in phosphate-buffered saline at 25°C. Intravenous administration of this form was said to show “considerable activity in an acute analgesic assay”. It concluded:
The availability of a safe and efficacious injectable COX-2 inhibitor for acute pain management, particularly postsurgical pain, constitutes an important unmet medical need. Parecoxib sodium, 5b, a water-soluble prodrug of valdecoxib, 4, was identified as a highly potent and selective inhibitor of PGs from COX-2. In a therapeutic model of acute pain, parecoxib sodium showed excellent efficacy and a rapid onset of action comparable with the most potent analgesic NSAID ketorolac. Parecoxib sodium is currently in clinical evaluation for the management of acute pain.
292 The formulation experts agreed that Jain is a review article and that the author had not conducted his own scientific studies. As such, Talley is the only source of a statement in Jain that “parecoxib is rapidly cleaved from valdecoxib in minutes”. The reference to “cleaved” indicates to them that an enzymatic process is being identified (i.e., in the body). The formulation experts agreed that nothing is disclosed in Talley or Jain about the conversion of parecoxib to valdecoxib in aqueous solution in vitro (i.e., outside the body).
7.6 Step 3: identification of parecoxib sodium and intravenous administration
293 The experts agreed that, faced with the formulation task and the content of Jain and Talley, they would consider it appropriate to proceed towards the development of a parecoxib sodium formulation for intravenous administration. They also agree that neither Jain nor Talley identify or refer to a lyophilised formulation.
294 Professor Winter gives evidence that based on the descriptions in Jain and Talley he would have assumed that the parecoxib formulation described was a “ready to use” liquid formulation.
295 Based on Jain and Talley, he would have attempted to formulate a ready to use formulation of parecoxib sodium with the following features:
(a) a bolus injectable solution for intravenous and intramuscular application;
(b) including a phosphate based buffer with a pH similar to phosphate buffered saline (PBS) (i.e., pH 7.4), although he would have tried different pH levels because he would expect the solubility of parecoxib to be affected by pH;
(c) a concentration of “significantly less than 22 mg/ml parecoxib” based on the teaching in Talley that the maximum solubility of parecoxib in PBS at 25°C was 22 mg/ml; and
(d) doses in amounts of 1, 2, 5, 10, 20, 40, 50 and 100 mg for the intravenous product and 1, 2, 5, 10 and 20 mg for the intramuscular product.
296 Dr Robertson would have taken a similar approach, although he would target a concentration for the liquid solution of less than or equal to 22 mg/ml at a total volume of 2 mL or less.
7.7 Step 4: obtaining parecoxib sodium
297 Neither formulation expert could proceed with their hypothetical task without having parecoxib sodium. Two issues arise. The first concerns whether or not the skilled team would obtain valdecoxib or parecoxib. The second concerns the significance of the time that it would take to obtain valdecoxib or parecoxib (and to perform other notional steps) which gives rise to the timing argument.
7.7.1 Obtaining parecoxib sodium, valdecoxib and parecoxib
298 In relation to obtaining valdecoxib and parecoxib, the respondents first submit that the notional team would have been able to obtain parecoxib sodium as at 3 April 2001. They rely for this purpose upon the evidence of Professor Winter who said in his oral evidence that he may have been able to obtain it from a pharmaceutical company in April 2001. However, Professor Winter’s evidence does not rise above speculation as to what he may have been able to acquire had it been available. Indeed, in his written evidence, Professor Winter expressed the view that he believed parecoxib would not have been available commercially in 2001 because it was a relatively new compound and it was uncommon for originators to make compounds available commercially before they had commercialised the compound for sale as a pharmaceutical product. I do not consider the evidence to provide any evidentiary basis for a finding that parecoxib sodium would have been available to the person skilled in the art at the priority date, and I reject the submission.
299 The respondents next submit that the skilled team would have no difficulty synthesising parecoxib sodium based on the information in Jain and Talley. The applicants submit that the respondents’ case must fail at this point. They submit that there is no evidence that the experts would have been able to synthesise parecoxib without access to valdecoxib which, they submit, Dr Robertson acknowledged he would only know how to do if he had read the “valdecoxib patent”.
300 Dr Robertson’s evidence in chief was that he would have been able to synthesise parecoxib in good yield and high purity by following the directions supplied in either Jain or Talley. He said that parecoxib could be readily prepared from valdecoxib using standard and basic chemistry reactions that were well known to him, and explained the process. It is apparent that Dr Robertson considered that he would be able to obtain valdecoxib as the starting point for the synthesis. In response, Professor Winter agreed that it appeared that parecoxib could be synthesised from valdecoxib as described in Talley and as Dr Robertson had said. He said that if he could not obtain valdecoxib commercially, he expected that it would have taken weeks or months for an experienced chemist with the requisite equipment and funding and support team to synthesise parecoxib.
301 It may be noted that the focus of Dr Robertson’s written evidence is on his ability to synthesise parecoxib based on the directions in Jain and Talley, which proceeded on the assumption that it could be readily prepared from valdecoxib. He did not appear to consider the steps necessary to synthesise valdecoxib.
302 The formulation experts were asked in question 29 of the joint expert report whether it would be a difficult task for an experienced chemist to synthesise valdecoxib or parecoxib. They were also asked what steps would be involved and approximately how long it would take. In their joint answer they said:
We agree that the synthesis as such would not be a difficult task for the experienced chemist, when the precursor materials are all available. According to [Dr Robertson], the synthesis takes about 4 major steps and can be carried out in a non complicated way, as there were no unusual reagents. [Professor Winter] accepts that expert statement as he is not a synthetic chemist. Although the net time to carry out the synthetic steps could only be a few days, we agree that overall, with supplies management, sourcing, analytics, small scale purification etc. the entire task will take at least several weeks…
303 In cross-examination, Dr Robertson was asked about the four major steps to which he had referred, and he volunteered that they were simple steps that he had seen in “the original valdecoxib patent” but could not recall what they were. In further evidence on the point it transpired that he had independently located that patent from the United States Patent and Trademark Office. The valdecoxib patent was not demonstrated to form part of the prior art base for the purpose of s 7(3) of the Patents Act, was not a pleaded piece of prior art and cannot be taken into consideration.
304 Dr Robertson’s views, based on his after-acquired knowledge of the valdecoxib patent, cast doubt on his evidence that valdecoxib or parecoxib could easily have been synthesised. Indeed his apparent reliance on the valdecoxib patent exposes an ambiguity in the answer to question 29 of the joint expert report, namely, whether the synthesis of valdecoxib and parecoxib would not be a difficult task once the steps referred to were known (having consulted the valdecoxib patent), or whether the identification of the steps and their carrying out would not be difficult and formed part of the common general knowledge. This ambiguity is not resolved by having regard to Dr Robertson’s written evidence which addresses only the synthesis of parecoxib using valdecoxib as the precursor, and does not explain how valdecoxib is to be synthesised in its own right. I am left in a state of uncertainty as to whether the skilled team would, on the basis of the common general knowledge, have been able to synthesise parecoxib sodium, without first having access to valdecoxib, before the priority date. This exposes a forensic weakness in the respondents’ case to which it will be necessary to return.
305 In closing submissions, the respondents sought to address this weakness by tendering and referring to exhibit E, which is an article footnoted in Talley. They submit that this evidence demonstrates that valdecoxib was straightforward to synthesise before 3 April 2001. However, exhibit E was not pleaded as an item of prior art information for the purpose of s 7(3) of the Patents Act or addressed in the evidence. In my view, it may not be taken into account for that purpose.
306 In relation to the timing argument, the applicants submit that the respondents’ development pathway could never lead to a conclusion of obviousness, because there is not sufficient time for the development steps to take place between the day when Jain could first be found and the priority date.
307 The applicants contend that the problems associated with formulating a stable and readily reconstitutable lyophilised formulation of a COX-2 inhibitor would not have been apparent before the priority date. They submit that because the problems that are identified in the patent were not part of the common general knowledge or disclosed in Jain and Talley, they were problems that would not be immediately apparent to the skilled person. They submit that a search using Professor Scott’s search strategy likely could only have located Jain on or after 28 March 2001, which is the date when MeSH terms were added to Jain’s record in the PubMed database. That is only one week before the priority date. They submit that the steps involved in formulating a ready to use injectable formulation of parecoxib would take much longer than a week, and that the experts agree that the stability tests required to make the determination that a ready to use formulation was unsuitable would take up to two years. Accordingly, they submit that the skilled formulator could not have known that a lyophilised formulation would be necessary. Nor would the skilled formulator know that a lyophilised formulation containing significant quantities of excipients other than the buffering agent would likely be unstable. As a result, the applicants submit that the inventive step case is defeated by time. As they put it in closing submissions:
We submit on the question of inventive step where one has to ask would it have been obvious – would the invention have been obvious in the light of the common general knowledge and any section 7(3) information if a critical element or input, being the identification of the problem or problems where – plural – that would then need to be addressed is not something that is immediately apparent. It is necessary to consider what would need to be done by the skilled person before that problem or those problems could be identified and then addressed, so the timing is relevant for that particular reason, your Honour.
308 The respondents submit that the timing argument is misconceived having regard to the hypothetical nature of the test for inventive step. In the alternative, they contend that even if the argument is accepted, experienced formulators would have had sufficient time after ascertaining Jain and Talley to complete the necessary steps for the formulation of parecoxib sodium.
309 Consideration of this question concerns the language of ss 7(2) and 7(3) of the Patents Act. Section 7(2) provides:
For the purposes of this Act, an invention is to be taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed in the patent area before the priority date of the relevant claim, whether that knowledge is considered separately or together with the information mentioned in subsection (3).
310 Section 7(3) provides that for the purposes of s 7(2), information is any single piece of prior art information or a combination of two or more pieces of prior art information being information that the skilled person could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant.
311 In AstraZeneca French CJ considered the history of these provisions. His Honour noted, at [12], that the test for want of inventive step has long been an overtly qualitative test rather than a quantitative one and, at [15], cited Aickin J (with whom Gibbs ACJ, Stephen, Mason and Wilson JJ agreed), who in Wellcome Foundation Ltd v VR Laboratories (Aust) Pty Ltd [1981] HCA 12; 148 CLR 262 at 286 posed the test as it applied under the Patents Act 1952 (Cth) (the 1952 Act) as being:
whether the hypothetical addressee faced with the same problem would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not.
312 Chief Justice French noted that in Hässle at [58] (Gleeson CJ, Gaudron, Gummow and Hayne JJ), the High Court found that the idea of steps “taken as a matter of routine” did not include “a course of action which was complex and detailed, as well as laborious, with a good deal of trial and error, with dead ends and the retracing of steps”. The distinction between identifying work of a routine nature, on the one hand, and work that involves an inventive step, on the other, may, as French CJ observed at [15] in AstraZeneca, also be approached by having regard to the modified Cripps question endorsed in Hässle:
Would the notional research group at the relevant date, in all the circumstances, which include a knowledge of all the relevant prior art and of the facts of the nature and success of [the existing compound], directly be led as a matter of course to try [the claimed inventive step] in the expectation that it might well produce a useful alternative to or better drug than [the existing compound]?
313 The Patents Act involved raising the threshold of inventiveness for patents to that set by the 1952 Act by allowing more information to be taken into account in addition to the common general knowledge for the purpose of deciding whether an alleged invention is obvious when compared with the prior art base. However, the qualitative test remains that posed by Aickin J in Wellcome Foundation in terms of the hypothetical addressee taking, as a matter of routine, steps which might lead from the prior art to the invention: AstraZeneca at [17] (French CJ).
314 Section 7(2) is a deeming provision whereby a claim for want of inventive step will be defeated unless one of the alternative conditions in s 7(2), read with s 7(3), is satisfied. Relevantly for present purposes, this involves an hypothetical person skilled in the relevant art notionally possessed of the common general knowledge as at the priority date and being provided with prior art information made publicly available in two or more related documents, finding the invention to be obvious. Chief Justice French observed in AstraZeneca at [18]:
The judicial determination whether want of inventive step is established pursuant to s 7 is mediated through the legal construct of the hypothetical person skilled in the relevant art. The construct is of a kind well-known to the law and used for setting parameters for evaluative judgments. It is a tool of analysis and is given statutory recognition, for that limited purpose, in s 7.
315 However, in a well-known passage, French CJ noted that the person skilled in the relevant art is not an avatar for expert witnesses whose testimony is accepted by the court. It is a tool of analysis which guides the court in determining, by reference to expert and other evidence, whether an invention does not involve an inventive step: AstraZeneca at [23].
316 The language of s 7 of the Patents Act indicates that the enquiry is whether an invention is obvious in light of relevant information (that is, common general knowledge and “prior art information”) existing and publicly available before the priority date. No temporal requirements are imposed by the section, other than that the common general knowledge must exist and the prior art information must be publicly available before the priority date.
317 Section 7(2) provides that the question is whether the invention “would have been obvious to a person skilled in the relevant art in light of the common general knowledge as it existed in the patent area before the priority date”. Similarly, s 7(3) focusses on the information that the person skilled in the relevant art could have reasonably ascertained before the priority date. Notably, nothing in s 7 requires consideration of whether or not the person skilled in the relevant art had sufficient time after being equipped with that information to determine the problem which needed to be overcome and complete, as a practical matter, the claimed invention before the priority date.
318 In my view, the observations of French CJ in AstraZeneca (with whom Gageler and Keane JJ at [97] and Nettle J at [118], [120] generally agreed), confirm that the task of the court involves an inquiry that is explicitly hypothetical. It is not concerned with what has happened, or what could have happened, but what would have happened had information of the character described been available to the person skilled in the relevant art. The statutory language does not invite a drift from the hypothetical to the practical in terms of timing.
319 In this regard I accept as cogent the submission advanced by the respondents that the approach encouraged by the applicants is similar to that which was rejected by the Full Court in Novozymes A/S v Danisco A/S [2013] FCAFC 6; 99 IPR 417. That case concerned a novelty challenge based on an inherent disclosure. The question was whether certain claims were anticipated by the application of the test in General Tire. The primary judge found that even if it had been established that the skilled addressee, following the instructions in the prior art document in question, would have chosen to use the relevant enzyme and that such use would inevitably result in the process of the claims before the priority date, the prior art document would not amount to an anticipation. That was said by the primary judge to be because the hypothetical skilled addressee had only 32 days in which to complete the disclosure by following the directions, and the party asserting lack of novelty had not demonstrated by evidence how the skilled addressee would do so before the priority date: Novozymes at [173]. The submission that the primary judge accepted was that the General Tire test was not available unless, as a practical matter, the example could in fact have been worked by someone whose first exposure to the subject had been on a reading of the prior art document on the day of its publication: Novozymes at [175].
320 In rejecting this submission Jessop J said at [177] (Greenwood and Yates JJ agreeing):
Although General Tire is part of Australian jurisprudence in the relevant area, it must be accommodated to the terms of s 7(1) of the Patents Act, upon which everything depends. That is to say, the inevitability of outcome to which General Tire refers must be such as would arise from recourse to the information referred to in the section. At the expense of repetition, it is here useful to remind ourselves of what the Court of Appeal said in that case ([1972] RPC at 485-486):
… if carrying out the directions contained in the prior inventor's publication will inevitably result in something being made or done which, if the patentee's patent were valid, would constitute an infringement of the patentee's claim, this circumstance demonstrates that the patentee's claim has in fact been anticipated. [Emphasis added]
This proposition is explicitly hypothetical. It is concerned not with what has happened or with what could have happened, but with what would have happened if the directions were carried out. As such, the proposition is in complete harmony with s 7(1), and with every other presently relevant aspect of the jurisprudence in this area. It is not the doing of it, nor even the ability to do it, that amounts to anticipation: it is the content of the information. If the information contains directions which, if carried out, would constitute an infringement of the patent in suit, the invention under the latter is not novel.
(emphasis in original)
321 Similar reasoning may be applied to the hypothetical task under s 7(2). It too is directed towards the availability of information before the priority date. The question is whether, armed with that information, the invention claimed would have been obvious to a person skilled in the relevant art. In the way it is expressed in Wellcome Foundation, the inquiry is whether armed with that information the hypothetical addressee faced with the same problem would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not. To add to this hypothetical task questions of how long it might take for a particular worker in the field to proceed from the prior art base to the claimed invention, and whether by the priority date that might have been achieved, confuses the tool for analysis with a concept of practical application. Yet the person skilled in the relevant art is not an avatar but a tool of analysis, the prior art base a construct of the legislation and the common general knowledge an assumed state of knowledge that no single individual need possess. This reasoning applies, whether or not the routine steps involve undertaking experimental work which leads to the identification of a problem to be solved or to what may be characterised as the inventive step alone.
7.8 Step 5: testing parecoxib sodium and decision to formulate as a lyophilised composition
322 The respondents submit that armed with Jain, Talley and parecoxib sodium, the skilled addressee would have been motivated to investigate the stability of parecoxib sodium as part of the routine formulation process. Testing would have revealed that parecoxib sodium converts rapidly to valdecoxib in an aqueous solution. The experts diverge from this point.
323 The respondents rely on the evidence of Dr Robertson to the effect that he would be conscious that parecoxib is a prodrug of valdecoxib and because of its poor water solubility he would be concerned about the potential for particulate valdecoxib to form if parecoxib sodium is formulated as a ready to use injectable solution. He was asked to assume that routine analytical stability tests would show that there is a partial conversion of parecoxib to valdecoxib in aqueous solution over time. He gave evidence that upon learning this, the formulator would make the decision to formulate parecoxib sodium as a reconstitutable lyophilised powder to limit the time that the parecoxib sodium would be in a solution prior to administration to the patient thereby reducing the prospect of significant conversion to valdecoxib.
324 The applicants challenge this approach. They note that both experts agreed that they would first attempt a ready to use liquid formulation of parecoxib sodium and that it was only upon performing stability tests on that formulation that it would become apparent that the formulation was unstable, a process that would take at least about 26 weeks. They submit that the skilled worker, faced with the stability problem, would first attempt various measures such as adjusting pH or adding a cosolvent, such as polyethylene glycol or propylene glycol, in order to formulate a ready to use solution that was sufficiently stable. Only after these attempts failed would the skilled worker be motivated to try to develop a reconstitutable powder, which was the approach described by Professor Winter in his evidence.
7.8.2 Consideration of the evidence
325 There is no dispute that the skilled worker would first attempt to formulate parecoxib sodium for intravenous administration. Dr Robertson understood that this was the form that Jain reported was in Phase III clinical trials. He also noted that Jain disclosed that no other COX-2 inhibitor was available for intravenous administration. It was also known to be generally preferable for an injectable formulation to be formulated as a ready to use solution, including because administration is more straightforward.
326 Dr Robertson gives evidence that in order to determine the stability of parecoxib sodium in solution he would conduct routine stability tests by storing samples at appropriate temperatures over time that would translate to a shelf life of two years at room temperature. He would use higher temperatures for accelerated stability testing. He would analyse for degradation products such as valdecoxib at appropriate intervals. In his written evidence Professor Winter broadly agreed with this approach, noting that accelerated stability testing according to international guidelines involves storing samples for six months at around 40°C at around 75% relative humidity.
327 Dr Robertson was asked by the solicitors for the respondents to assume that the stability tests that he conducted would show that there was a partial conversion of parecoxib to valdecoxib in aqueous solution over a period of time. Dr Robertson said that this assumption would cause him to formulate parecoxib sodium as a reconstitutable freeze-dried (lyophilised) powder. There is no dispute that the process of lyophilisation was well-known in the art of formulation before the priority date as a suitable means by which an active compound which is susceptible to degradation in the presence of water could be formulated into a more stable composition. It serves to limit the amount of time the active pharmaceutical ingredient containing the molecule is in solution (i.e., until shortly before administration) thereby reducing the prospect of significant degradation or conversion. Dr Robertson explains that this approach would allow parecoxib sodium to be administered as an intravenous solution, which has been reconstituted into a liquid from the powder shortly before administration.
328 In his written evidence Professor Winter took issue with two aspects of the approach so described by Dr Robertson. First, as to the clarity of the assumption Dr Robertson was asked to make about the stability of parecoxib in aqueous solution. Professor Winter notes that the assumption says nothing about how quickly the conversion from parecoxib to valdecoxib would take place, or whether the conversion resulted in unacceptable stability. Secondly, Professor Winter said that even if stability studies showed unacceptable amounts of conversion, he would not immediately have moved to try a lyophilised formulation, but rather would have continued to pursue a ready to use formulation, which would have been cheaper, more convenient to administer and more competitive. He says that he would have attempted to obtain a stable ready to use formulation by adding a parenterally acceptable non-aqueous organic solvent, such as polyethylene glycol or propylene glycol, and adjusting the pH of the formulation.
329 The dispute between the experts about the outcome of stability tests was resolved in the oral evidence.
330 Professor Winter readily agreed that upon obtaining parecoxib sodium he would immediately run analytical stability tests on it. These, he accepted, are routinely performed by formulators in the process of development. Those tests are generally performed over a shorter time period to stability tests performed on final product formulations.
331 Professor Winter accepted that he would have read in Jain and Talley that valdecoxib has poor water solubility. His first approach would have been to formulate a ready to use liquid formulation of parecoxib sodium in a solution that included a phosphate buffer, because Talley indicated to him that parecoxib sodium is compatible with that buffer. He would test the stability of parecoxib sodium in water for injection at a range of pHs. Part of this testing would have involved routine stress tests at elevated temperatures because the elevated temperatures would most likely accelerate the rate of any degradation. There is no dispute that had he performed such tests, parecoxib sodium would very rapidly convert to valdecoxib. He accepted that by that process he would find out that parecoxib sodium was unstable if left in solution over a period of time.
332 I am satisfied that routine testing of the type that would have been conducted by the skilled formulator before April 2001 would have demonstrated that parecoxib sodium was unstable in aqueous solution such that the formulator would have been obliged to take steps to address that problem. Put another way, although the conversion problem was not disclosed in Jain or Talley and did not form part of the common general knowledge, it would certainly have been discovered upon the conducting of routine tests.
333 The next question concerns how the skilled formulator would address that problem. The respondents submit that the evidence supports the conclusion that a routine and logical next step would be for the skilled worker to proceed to lyophilisation of the candidate formulation in order to avoid the problem of parecoxib sodium converting to valdecoxib in solution.
334 Professor Winter and the applicants resist that proposition, contending that the skilled formulator would instead prefer to attempt to create a stable, ready to use liquid formulation by adjusting the pH of the formulation and using a non-aqueous organic solvent to solubilise any unwanted valdecoxib that did not remain in solution. The respondents characterise this as “highly speculative”.
335 The evidence demonstrates that there would be reasons for the skilled formulator to target a ready to use liquid formulation over a lyophilised formulation. It was known (and common knowledge in the field) that lyophilisation increased expense and resulted in a formulation that had to be reconstituted prior to administration which imposed a burden on clinicians administering the drug. These factors would have disposed the skilled formulator to prefer a ready to use solution.
336 Professor Winter considered that in April 2001 he would have tried to add an organic solvent like polyethylene glycol or propylene glycol to the formulation if he encountered the stability issue of valdecoxib precipitating out of the buffered parecoxib sodium solution. The purpose would be to solubilise unwanted valdecoxib precipitates that developed. He considered that replacing 20-30% of the water with one of these solvents may assist. He would also have tried adjusting the pH of the solution in the hope that this would render any valdecoxib that formed more soluble.
337 There was no dispute that these solvents were known to be useful in the formulation of injectable drugs prior to April 2001, although they were not perfect or without issues. The experts agreed in their joint report that side effects and allergy considerations had to be weighed against the disadvantages of lyophilisation.
338 The range of side effects was explored in oral evidence. Professor Winter was queried about whether thrombophlebitis (an inflammatory process causing blood clots) and cardiotoxicity (damage to the heart muscle) may result from the use of these organic solvents. He gave evidence that he did not recall that these were known side-effects, but explained that he did not consider them to be a significant risk given that he would be careful to dilute them to between 20-30% in water such that only very small amounts of the proposed solvents would be included in the formulation. He noted that the use of all excipients involved a balance between their benefits and potential side effects. He was also aware that formulations containing higher concentrations of cosolvents could cause pain upon injection or haemolysis (destruction of red blood cells). This provided a further incentive to maintain the concentration at between 20-30%.
339 Dr Robertson preferred to avoid the use of the organic solvents because he was not optimistic that they would work to achieve a stable ready to use formulation. He was sceptical that they would solve the problem of valdecoxib precipitating from parecoxib sodium in a ready to use liquid solution, and expected that rather than stabilise, an organic solvent would lead to instability of the parecoxib sodium in solution. He would therefore go straight to lyophilisation after the stability tests revealed the instability of parecoxib sodium in aqueous solution.
340 The position between the experts was explained in the following passage of their concurrent evidence:
MR DIMITRIADIS: And do you accept that another consideration that a formulator, as at April 2001, might reasonably take into account is that the addition of a nonaqueous solvent, such as polyethylene glycol or propylene glycol, may have an effect in reducing the conversion of parecoxib to valdecoxib in the solution?
DR ROBERTSON: No, I think it would be converse. I think it would accelerate the conversion of parecoxib to valdecoxib.
HIS HONOUR: Could you explain why that would be?
DR ROBERTSON: Well, because propylene glycol has a primary alcohol, so not dissimilar to mannitol. So I would expect that that, in basic solution, that primary alcohol would be deprotonated to the sodium cation, and it would attack parecoxib and cleave the proton back to valdecoxib.
HIS HONOUR: And, Professor Winter, do you agree with that summary of the chemistry?
PROF WINTER: I agree that this is theoretically possible, but I disagree that in 2001, and with the information at hand, we would be able – we as the formulator community – to predict that this would happen and accelerate the degradation. Not at all. And, your Honour, I might also point your attention to – even if, theoretically, taking this assumption that I do not believe into account, polyethylene glycol would slightly accelerate the degradation ..... conversion from parecoxib to valdecoxib. If on the other side it would make valdecoxib more soluble, we would even have a win situation there, because this conversion is not a problem. The problem is that the valdecoxib may precipitate, and if the we have one, two, three, four, five per cent of valdecoxib, this product would be perfect for the patient. He would not have any harm, would be perfectly active. And as long as this is not precipitating, a perfect product would be in our hand.
HIS HONOUR: Dr Robertson, do you wish to respond to that last observation?
DR ROBERTSON: Well, that’s – that is true, but it’s an area that we have no information on. Valdecoxib is extremely insoluble in – in water, as we’ve just been discussing, so I don’t know whether it would be one per cent, five per cent or many more per cent, and I don’t even know whether it propylene glycol or polyethylene glycol would solubilise the valdecoxib. It has got a hard job to do. So we – we don’t know. If it did that, that – that would be helpful, but again, you’ve got to balance that against the pharmacological downsides of introducing these co-solvents into your formulation.
(emphasis added)
341 It would appear that upon encountering the conversion problem the hypothetical formulator reaches a fork in the road. The choice is whether to persist with a “ready to use” approach or move to developing a lyophilised formulation. In advance, neither presents a clear answer.
342 One difficulty with the view taken by Professor Winter is whether the organic solvents could achieve the result of dissolving any precipitating valdecoxib at low concentrations. He agreed in cross-examination that valdecoxib was reported in Jain and Talley to have poor water solubility. The formulator would need to select an appropriate concentration of solvent within the solution such that it would dissolve any precipitated valdecoxib and yet not exceed the range that Professor Winter nominated of between 20-30%. Professor Winter disagreed that the low solubility of valdecoxib would make him disregard the use of a solvent at the outset.
343 Professor Winter gave evidence that the solubility of valdecoxib is pH dependent, and so a further dynamic in the formulation was to choose a pH to improve the solubility of any valdecoxib that may have precipitated out of the solution. The range of physiologically acceptable pHs for intravenous injection was known to be between about 6.4 to 8.4.
344 Dr Robertson expected, based on his understanding of the chemistry, that as valdecoxib is a sulphonamide, in order to make it more soluble, the pH would have to be increased to about 12 or 14 or a “very high number, anyhow”. That view is to some extent supported by a post-priority date publication, tendered at the hearing as exhibit D, that indicates that valdecoxib is freely soluble in organic solvents and alkaline (pH=12) aqueous solutions.
345 As a result of his views as to the chemical interactions involved, Dr Robertson rejected the view propounded by Professor Winter that adjusting the ready to use formulation by adding the organic solvents would be likely to succeed. He considered that whilst adjusting the pH may have an effect, the process of trying organic solvents and adjusting the pH would not address the real problem, which was that valdecoxib would precipitate out of the solution of parecoxib sodium, water and buffer. He thought that the solvents would accelerate the precipitation and be unlikely to solubilise the valdecoxib.
346 The applicants criticise Dr Robertson’s approach to the organic solvents as being tainted by hindsight. In particular, they submit that his reference to mannitol (referred to in the passage quoted above at [340]) could only have arisen from his ex post facto knowledge of the content of the patent (which discloses, broadly, that mannitol has a negative effect on the stability of a lyophilised parecoxib sodium formulation). However, whilst I accept that the reference to mannitol was possibly influenced by his reading of the patent, I am not persuaded that his understanding of the underlying chemistry was based on his reading of the patent. It seems to me that Dr Robertson based his opinion on the chemical structure of the organic solvents of which he was aware before April 2001, and the chemical structure of parecoxib sodium which was supplied in Jain. He then used mannitol in his oral evidence as an example familiar to those listening to illustrate their likely effects. I note that whilst Professor Winter agreed that Dr Robertson’s explanation of the science was theoretically possible, he disagreed that, as at April 2001, a formulator would have been able to predict the interaction between an organic solvent and parecoxib sodium.
347 Professor Winter summarised his position as to the interaction of the solvent and pH with parecoxib sodium and valdecoxib as follows:
… it is my impression that the pH has a complex effect on the entire scenario. The pH will affect the solubility of valdecoxib to an extent I do not know; maybe more, maybe less. The pH will for sure affect the solubility of parecoxib and parecoxib sodium, and the pH will eventually – I do not know – affect the conversion rate of parecoxib to valdecoxib. So – and if we consider an organic solvent in the background, the situation becomes even more complex, because an organic solvent now interferes with the dissociation of water and the pH. And that altogether creates a very complex situation where we are not able, in my opinion, to know what or which of those effects dominate. This is my comment.
348 It is relevant to note at this point that Professor Winter accepted that, although his preference was to proceed in this manner, other formulators acting reasonably in the same position before April 2001 could proceed instead to development of a lyophilised formulation in the expectation that it may produce a stable product. He agreed that lyophilisation was a commonly used technique for formulating small-molecule pharmaceuticals that are unstable in aqueous media.
349 In relation to the process of drug development, Professor Winter also agreed that pharmaceutical research groups were typically time driven in their approach to formulation development and that they might reasonably, depending on their resources, have sought to develop a ready to use and lyophilised formulation in parallel. However, as will be seen in my consideration of the respondents’ step 6, once the decision is made to move to a formulation that involves lyophilisation, further choices are presented.
350 I return to this evidence in my analysis of the ultimate issue.
7.9 Step 6: composition of the pre-lyophilisation solution
351 The respondents submit that the notional formulator would have been led as a matter of course to formulate parecoxib sodium as a lyophilised powder and would do so by lyophilising a solution of parecoxib sodium and an appropriate buffer. Through routine testing the formulator would determine the appropriate relative weights of each component that would fall within the claims of the patent. They submit that using this approach the formulator would encounter no difficulty in achieving an elegant cake (being a powder with desirable properties) that could be reconstituted. They rely on the evidence of Dr Robertson to the effect that this is the approach that he would take.
352 The applicants disagree. They submit that the experts agreed that a formulator would likely add a bulking agent to the pre-lyophilisation formulation (that is, the solution prior to lyophilisation) to increase the amount of solid material in the pre-lyophilisation solution to about 10%. This, they submit, is a rule of thumb that the skilled formulator would apply in the present case because the pre-lyophilisation formulation would otherwise contain a low percentage of solids. The applicants submit that the formulator would be most likely to choose to use mannitol as a bulking agent, not only because it was very well-known and used in the field for that purpose, but also because it would ensure that the powder could be reconstituted with water for injection and still maintain isotonicity. They submit that by adopting this conventional approach to formulation the hypothetical formulator would have run into the stability problem (parecoxib sodium converting to valdecoxib when in powder form) and the reconstitution problem (the lyophilisate not dissolving satisfactorily). The applicants submit that neither of those problems formed part of the common general knowledge. Nor was understanding the causes of those problems straightforward.
353 In reply, the respondents contend that while the experts agreed that a bulking agent may be added “at some stage in the development” of a freeze-dried product, the contest on the evidence was as to when it may be attempted. They submit that the evidence of Dr Robertson demonstrates that the skilled formulator would lyophilise a formulation of parecoxib sodium using dibasic sodium phosphate as the buffer. They submit that this formulation would then be tested to ensure it had sufficient elegance, shape and structure. The cake would be tested for the presence of residual water. If routine tests showed that there were issues with the lyophilisate’s structure that may impact on its stability or reconstitution time, then Dr Robertson may consider adding a bulking agent, but only if there were a need. The respondents dispute that any rule of thumb as to a minimum amount of bulking agent to be added to the pre-lyophilisation composition has relevant application. The respondents make three further submissions in this regard. First, that a 100 mg formulation of parecoxib, one of the dosages they assert is proposed in Professor Winter’s evidence, would not require a bulking agent even under the rule of thumb. Secondly, that the alleged risks of not using a bulking agent identified by Professor Winter were irrelevant to the formulation process. Thirdly, they submit that there were specific risks associated with using mannitol as a bulking agent such that a skilled formulator would not use it.
7.9.2 Consideration of the evidence
354 Dr Robertson’s evidence is that Jain discusses the administration of 20 mg and 40 mg doses of parecoxib in a Phase III double-blind placebo-controlled randomised study. Whilst it does not provide the administration volume for intravenous administration in the case of post-surgical pain, generally, his view is that a small volume is preferred when being administered by a medical professional, and volumes such as 1-2 mL can be administered conveniently using a syringe. He notes that in Talley, parecoxib sodium is described as having a solubility of 22 mg/mL which, in his experience, is sufficient for formulating an intravenous formulation of 1 or 2 mL. He would accordingly formulate parecoxib with injection volumes of 1 mL for 20 mg of parecoxib, and 2 mL for 40 mg of parecoxib.
355 Dr Robertson gave evidence that his preferred formulation would include parecoxib sodium and a phosphate buffer in a solution of around 20 mg/mL at a pH of 7.4 before lyophilisation. He said that in doing so he would ensure that the majority of the composition was comprised of the active ingredient and that the amount of buffer and any other excipients was kept low because, in his view, excipients are only included in any given formulation if they are required to solve a particular problem. He gave evidence that his preference for reconstituting the lyophilised powder was to use a diluent of 0.9% w/w solution sodium chloride, to ensure that the reconstituted solution was isotonic.
356 Professor Winter challenged that approach. Whilst he accepted as a matter of principle that unnecessary excipients would not be added to a formulation, his evidence was to the effect that an experienced formulator would typically consider how to formulate the pre-lyophilised solution by having regard to a well-known consideration in the field that it is desirable for the dried product resulting from lyophilisation to occupy the same volume as the original solution. He referred to the text book Alphonso R. Gennaro et al, Remington: The Science and Practice of Pharmacy (19th ed, Mack Publishing, Easton, 1995), which is quoted in the patent (page 2 lines 29 – 32) as saying that in order to achieve this volume “the solids content of the original product must be between approximately 5 and 25%” of the total original solution.
357 Professor Winter gave evidence that a formulator with experience in lyophilisation would also consider:
(a) limiting the amount of excipient salts, as products which contain salts are difficult to dry;
(b) the potential need to add a bulking agent such as mannitol, which was the most common bulking agent before April 2001, to achieve an elegant cake;
(c) the need to ensure that the reconstituted solution would be isotonic, which is commonly achieved with a bulking agent;
(d) the need to form a cake which is porous and dissolves quickly and completely, which is commonly achieved by adding a bulking agent; and
(e) the need to add stabilisers, such as mannitol (the most common stabiliser used at the time) to ensure that the active ingredient could be stored in a stable condition while dry.
358 The disagreement between Dr Robertson and Professor Winter was explored in the joint expert report and in their oral evidence. Much apparent disagreement was resolved.
359 In their joint report, the experts considered the passage in Remington. They agreed in answer to question 17 as follows:
Bulking agents were very common and were immensely popular in 2001. A person seeking to make a lyophilised product would likely add a bulking agent at some stage in the development of the freeze dried product; an experienced formulator would make the decision on a rule of thumb that ca. 10% solid material would likely be appropriate. The experience formulator would not necessarily need to read Remington.
(emphasis added)
360 The experts also discussed the use of mannitol. In the joint expert report they agreed:
Mannitol was very popular in the pharmaceutical industry as a bulking agent for freeze dried products. It wasn’t the only carbohydrate used for that purpose but the one most often used. A lyophilisate (cake) containing mannitol mostly had particularly good appearance, but not always. One had to consider the risk of re-crystallization in different modifications and vial cracking…One key feature of mannitol was the belief that it was a non-reactive, inert excipient.
(emphasis added)
361 Furthermore, the oral and written evidence supports a finding that it was known by formulators before April 2001 that the addition of mannitol may have been useful for at least the following purposes:
(a) to increase the mass of the lyophilised product and to disburse the active ingredient, reducing the likelihood of any residual water, for example from the rubber stoppers used to close the vial, coming into contact with the active ingredient;
(b) to aid in the drying of the active ingredient by providing a matrix together with the active ingredient and other excipients that is porous and stable and allows moisture to be removed;
(c) to guard against the risk of collapse of the cake;
(d) to guard against the risk of the cake being blown out of the vial by water vapour convection;
(e) to reduce the risk of poor dissolution upon reconstitution; and
(f) to assist in ensuring that the reconstituted solution is isotonic.
362 The experts agreed that the formulations of the patent fell outside the rule of thumb because the content of solids in the pre-lyophilisation solution as a percentage of the amount of water added before lyophilisation is less than about 10%. Accordingly, were the rule of thumb to be applied, the use of bulking agent would normally be required.
363 Both experts were skilfully cross-examined about the approaches that they would take. In answer to a general question, without parecoxib sodium as the target active ingredient, they said:
MR COOKE: I will ask another question, then. Professor, you would agree, wouldn’t you, that when formulating a lyophilised pharmaceutical before April 2001, if you used no excipients, you would not be certain that you would produce a useful result. But you might just, might well just do that, mightn’t you? And that would justify you, first, trying to lyophilise the formulation with no excipients?
…
PROF WINTER: Yes. I understand the question in a way that this hypothetical formulation without excipients would be one of the first to start with. And I totally disagree with. The ones I would start, first, with is those that give me the – let’s say, “highest probability of success”. I think, this is the term you’re familiar with, and this is also a term that the formulator, let’s say, uses in the back of his head. And from there, he might walk into several directions and variations, and one of them could also be to try a formulation without an excipient, and therefore it is one of many choices, but surely not the starting one or the preferred one for theoretical reasons.
364 Asked to comment on Professor Winter’s answer, Dr Robertson said:
DR ROBERTSON: Yes. Well, I say from – you know, for APIs of a – of a suitable bulk, I suppose. When we – when we started off, you know, with an API that we knew nothing about, first of all, you’ve got to gather a lot of information, and certainly can you – can it be freeze-dried at all is the very first question that we ask. So – so generally while – this is not, maybe – this is on the way to a formulation. We would generally attempt to freeze-dry the API on its own, so that we could at least understand that, and then – that would not – that would [not] necessarily be the final formulation, but part of our understanding on the way to a final formulation. And then we would add, if that was the – if that didn’t work out, then we might add other excipients; if that did work out, we would feel gratified, and then add excipients as we felt the need. But this is, you know – I don’t know. Simplest is always the best and we – we start – we used to start from no excipients and then add them as required, if that – if that makes sense, rather than use a standard formulation and take them away as required. So that may be two different ways of doing things, but as I’ve described, it is the way that we did it.
(emphasis added; the word “not” in brackets is a correction added after reviewing the video file)
365 It is notable here that Professor Winter made clear that he would not attempt a formulation that he did not consider had a probability of success and, consequently, he would not try a formulation that did not involve using bulking agent.
366 I understand from his reference to “for API’s of a suitable bulk” that Dr Robertson agreed that where the active ingredient was of a suitable bulk he might try formulating the pre-lyophilisation solution with no excipients. It is a little unclear in the latter part of his answer whether, and in what circumstances, freeze-drying the active ingredient on its own would be simply part of a development path alone, or whether it may be the solution and represent a potential final formulation. Later, Dr Robertson sought to clarify his evidence. He said that where he was starting with a parecoxib sodium formulation with a solids content of considerably lower than 10%, being about 3% (as in the patent):
I think I said earlier that, you know, when you’re starting out in the formulation you would determine whether the API could be freeze-dried at all. Then you would see whether it could be freeze-dried with an excipient which was of interest if you’re to get the formulation, and if you failed in all of these, then you might consider – in fact, you would consider adding a bulking agent, but, you know, I think it’s a different approach that I have, whereas I would start with an understanding of how the API could be lyophilised on its own, and add bulking agents or excipients as required.
…. My approach would not be to add a bulking agent just to get up to 10 per cent because that’s the rule of thumb, and then subtract bulking agents as necessary ..... the other way around.
(emphasis added)
367 However, Dr Robertson subsequently accepted that, having regard to the rule of thumb, his approach would not likely be the approach taken by other formulators in the field:
MR DIMITRIADIS: …Dr Robertson, I suggest that having regard to your agreement in question 17 of the joint report, you would accept that an experienced formulator seeking to make a lyophilised product would add a bulking agent in order to increase the solids content up to about 10 per cent solid material in the circumstances that I was putting to you. Do you accept that?
DR ROBERTSON: I think what we agreed was that it would likely be appropriate. That doesn’t mean that it’s essential.
MR DIMITRIADIS: And if the experienced formulator applied the rule of thumb, that’s what they would do. Correct?
DR ROBERTSON: If they were to apply the rule of thumb, but an experienced formulator would not necessarily take the rule of thumb as gospel.
(emphasis added)
368 Returning to Professor Winter, later in his oral evidence, a more concrete proposition was put to him. He was asked to assume that he was endeavouring to make a lyophilised form of parecoxib sodium for injection, and had formulated a pre-lyophilisation formulation consisting of parecoxib sodium with a phosphate buffer at a pH of 8.1. It was suggested to him that “a logical approach” would be to prepare the lyophilised formulation without a bulking agent. He responded:
MR COOKE: Now, you would agree, wouldn’t you, that although the formulator would not be certain that the formulation would produce a suitable cake, a logical approach to prepare the lyophilised formulation would be without a bulking agent because it may well produce a useful result.
PROF WINTER: Well, you asked me a similar question earlier and I said no. The standard approach would be to go with a bulking agent. I agreed and I still agree that trying it out without a bulking agent would be one of a number of, let’s say, experiments I would conduct. I cannot deny that. I cannot exclude that.
MR COOKE: Okay. And you would run that at the same time as your experiment with the bulking agent if you’re in a pharmaceutical company, you’re pressed for time. You would run the experiments together.
PROF WINTER: This is a matter of other circumstances like how much material you have, how – are the freeze dryers booked out and so on. Maybe not at exactly same time, but in the course of a formulation package or a set of experiments, this could be done. Whether it’s done immediately, this is a matter of, let’s say, organisation, logistics and - - -
369 Professor Winter was pressed on his evidence about trying a formulation without bulking agent. He was asked to assume formulations without bulking agents containing 20 mg or 100 mg of parecoxib sodium, a disodium sodium phosphate buffer, water for injection and with or without a pH adjuster. It was suggested to him that after these formulations were lyophilised the next step would be to test the lyophilised cakes to see whether they had sufficient structure:
PROF WINTER: Yes, that would include this – analysing the quality of what you have achieved, which is, as for that structure, it is residual water, it is quality of the drug, of course. Yes, that’s true. And if this is not what you like to see, then you have to do it again, and eventually again, and again. This is then always taking one more cycle, until you are satisfied, or do you not reach the target. And this is, in a way, also the reason why I say, well, let us go with a bulking agent first, because then, the probability of success to reach a good cake that is dissolves fast, is very high. And leaving that out, I might need to go in several circles that may take time, and – to block the machine, and so on, and so on.
370 Having regard to this evidence, I am not persuaded that the skilled formulator would take the path proposed by Dr Robertson in his oral evidence. In my view, the evidence of the common general knowledge both as to the use of bulking agents in lyophilised formulations and the frequent use of mannitol is compelling. Dr Robertson’s frank acceptance that other formulators would be likely to consider it appropriate to apply the rule of thumb provides support to the evidence of Professor Winter as to the likely approach.
371 The evidence set out above makes plain that the use of bulking agents in the course of lyophilising active ingredients where they fell outside of the rule of thumb formed part of the common general knowledge. As Dr Robertson accepted, whilst adding a bulking agent was not the course that he would have taken, it is the course that he would expect other formulators to have taken before April 2001. Furthermore, it was common general knowledge that mannitol would likely be used as the bulking agent.
372 Having regard to the whole of the evidence, I am not satisfied that hypothetical skilled formulator would have approached the formulation of parecoxib sodium in the manner described by Dr Robertson. I consider that his approach is idiosyncratic having regard to the common general knowledge as at the priority date. I consider that the most likely path that the skilled team would take would be to use a bulking agent, most likely mannitol, to produce a lyophilised formulation of parecoxib sodium. The common general knowledge points to this path as being the one with a reasonable prospect of success.
373 I accept that at times in his oral evidence Professor Winter accepted after some prompting that he might try to lyophilise a buffered solution containing the active ingredient without a bulking agent, but he was resolute that this was not his preference. His preference was to choose a formulation that he considered was likely to succeed and that would involve applying the rule of thumb such that a bulking agent would be included to bulk out the solid content. Moreover, Professor Winter’s expectation was that if he deviated from that rule he would simply be reiteratively experimenting and going round in circles. I do not accept that this evidence amounts to an acceptance that he would have an expectation of success had he resorted to trying a formulation that did not include a bulking agent.
374 A debate emerged between the experts as to whether a predicted interaction between mannitol and parecoxib would deter a formulator from taking Professor Winter’s approach of adding mannitol as a bulking agent. I do not consider that the hypothetical skilled formulator would have foreseen in advance that the use of mannitol as a bulking agent (capable of providing the other benefits to which I have referred in [361] above) would have caused difficulties or diminished the likely success of the preferred formulation.
375 Dr Robertson gave evidence that he would not use mannitol in a pre-lyophilisation solution because mannitol appeared to accelerate the decomposition of parecoxib to valdecoxib. However, as he explained in his oral evidence, that view was expressed on the basis of the results set out in example 2 of the patent. Dr Robertson noted from those results that the inventors backtracked from the use of mannitol and considered that there had to be a reason why mannitol facilitates the accelerated decomposition of parecoxib to valdecoxib. Although he noted that “it may not be that the…formulator thought of this up front”, he hypothesised that mannitol, as a weak acid, may deprotonate in the presence of a base and become unstable such that it reacts with parecoxib. In this way he opined that it would catalyse the undesirable conversion of parecoxib to valdecoxib. He and Professor Winter agreed that substantial deprotonation of mannitol would not occur except at a very high pH (in the joint expert report, they considered a pH of above 12). Professor Winter thought that any deprotonation would be negligible at a pH of 8 and would not affect the stability of parecoxib sodium. Dr Robertson thought that there would be minimal deprotonation at a lower pH but that this would nonetheless be sufficient to affect the stability of the parecoxib sodium.
376 The explanation offered by Dr Robertson was plainly given in hindsight, he having had the benefit of the results of the experiments reported in the patent. Furthermore, Professor Winter disagreed with Dr Robertson’s theory that deprotonation would be likely to cause instability at a pH of about 8 and disagreed that other formulators would perceive this to be a cause for instability. Having regard to the hindsight built into Dr Robertson’s analysis, in this regard I prefer the evidence of Professor Winter.
377 Nor am I persuaded by the three remaining arguments advanced by the respondents to the effect that the rule of thumb would not arise.
378 The first is a submission that, armed with knowledge: (a) that a lyphophilised powder formulation of parecoxib sodium does not collapse; (b) that there was found to be no need to add a bulking agent to aid in the dissolution of parecoxib sodium given its solubility; and (c) that there was no information to give reason to believe that water is unevenly distributed throughout the freeze-dried parecoxib sodium formulation; it is apparent that the skilled formulator would not have encountered any of the risks that would motivate the addition of a bulking agent. This argument is based wholly on hindsight and must for that reason be rejected.
379 Secondly, the respondents contend that the evidence supports the proposition that excipients should only be used where appropriate and where they are “inert” and “safe”. They contend that Dr Robertson explained that mannitol is neither.
380 However, in my view, this argument cannot go far in light of the agreement of the experts that mannitol was very popular in the pharmaceutical industry as a bulking agent for freeze-dried products. It is plain that the skilled formulator, who will have significant scientific qualifications and experience, would be aware that they must be familiar with the properties of excipients used in formulating pharmaceutical products and account for likely chemical and other interactions with the other components in a formulation. One example acknowledged by the experts is the likely effect (mostly beneficial) of mannitol on the crystal structure of the “cake”. Another is mannitol’s ability to affect the tonicity of the powder composition. I consider that the accepted popularity of mannitol amongst those in the field may be taken to indicate a general positive disposition towards its use in formulating freeze-dried products. In this context, in my view, the debate between the experts as to whether mannitol is “inert” was largely a side wind.
381 The third involves the somewhat convoluted proposition that one of the dosage amounts proposed by Professor Winter in his written evidence (being 100 mg parecoxib for intravenous use) falls outside the rule of thumb, and inside the scope of the asserted claims in the patent. Accordingly, the respondents submit that, taking the applicants’ case at its highest, the skilled worker would not add a bulking agent and that therefore there is no inventive step.
382 This submission falls at the first hurdle. The evidence given by Professor Winter did not propose a dosage amount for a lyophilised solution. Nor did Dr Robertson. Rather, the amounts proposed were for a ready to use dosage form. As noted above, Professor Winter’s evidence was that it was commonly necessary to change the formulation if a product previously being prepared as a ready to use formulation is instead formulated as a lyophilised product. For example, as I have discussed above, it may be necessary to add a bulking agent to a lyophilised formulation in order to achieve certain objectives not relevant to ready to use formulations.
383 There was little debate between the parties about the process of lyophilisation. Whilst the patent, at pages 18 – 20, describes a staged process of lyophilisation, none of the relevant claims is for an invention requiring particular steps to be taken in the course of freeze-drying the pre-lyophilisation solution. The broadest asserted process claim, claim 26, does not limit the process to any particular manner by which lyophilisation is performed. Professor Winter accepted that if a formulator did not apply the stepwise process described in the specification, nonetheless a successful formulation could still be achieved. Furthermore, the experts agreed that step-wise freezing was a known procedure before April 2001.
384 I find that the process of lyophilisation required to achieve a freeze-dried product formed part of the common general knowledge and was within the skills of the formulator before April 2001.
385 Prior to administration, a lyophilised powder formulation needs to be reconstituted with a solvent (also known as a diluent) in order for it to be injected into the subject.
386 The respondents submit that it was known to those in the art that the preferred solvents for reconstituting a lyophilised drug product were aqueous-based solvents. They refer to the patent, which they contend admits that “water for injection” and “aqueous liquid[s] containing a solute such as dextrose or sodium chloride [including] 0.9% sodium chloride injection USP” were known parenterally acceptable solvents as at the priority date. They submit that the skilled addressee would have been motivated to add a diluent that was itself isotonic. This is because the composition of parecoxib sodium and a phosphate buffer does not have “built-in” isotonicity, and it is critical that the reconstituted formulation be isotonic. This would lead the notional formulator to use 0.9% sodium chloride rather than water for injection when reconstituting the lyophilised formulation for administration.
387 The applicants contend that the skilled formulator would add an excipient to the pre-lyophilised formulation in order to achieve isotonicity. They submit that this would provide a further motivation to use a bulking agent such as mannitol, which was known to achieve “built-in” isotonicity such that an additional excipient, such as sodium chloride, did not need to be added upon reconstitution. Instead, the lyophilised powder composition could be reconstituted using water for injection, which was a convenient and readily-accessible solvent. They submit that this was another motivation for the choice of mannitol.
388 The term “tonicity” refers to the ability of an extracellular solution to make water move into or out of a cell by osmosis. The experts agreed that “isotonicity” describes a solution with the same tonicity as a relevant body fluid which, for present purposes, is either blood plasma or subcutaneous tissue fluid depending on the route of administration. The goal in developing a drug for intravenous injection is to have a solution which is isotonic with blood plasma and thereby unlikely to cause significant change in the overall tonicity of a patient’s blood plasma. A non-isotonic injectable product is undesirable because it may destroy red blood cells if injected intravenously and, if injected subcutaneously or intramuscularly, it may cause pain or irritation as it causes cell damage. Accordingly, it is important that the reconstituted solution be isotonic.
389 Professor Winter favoured achieving isotonicity by the use of mannitol in the pre-lyophilisation formulation because it would achieve “built-in” isotonicity of the lyophilised powder composition with the result that water for injection, which does not affect the overall tonicity, could be used for reconstitution. Dr Robertson agreed that this was a known property of mannitol and its use would avoid the need for another excipient to be used upon reconstitution.
390 Both parties rely on the answer to question 27 in the joint expert report where the position was set out as follows:
What were the preferred solvents for reconstituting a lyophilised drug product? Why was each of these preferred?
Preferred solvents are aqueous based solvents. It can be water for injection, if the lyophilized formulation has “built in” isotonicity by the necessary amount of API plus excipients. It can be 0.9% NaCl solution, which is by itself isotonic, if the formulator has not put something into the lyophilized formulation to increase tonicity, and if the tonicity of the API alone is low. Solvents with e.g. a different, non-standard concentration of NaCl (e.g. like 0.7%) are not desired at all.
391 Dr Robertson gave evidence in chief that his preference would be to use a formulation of parecoxib in 0.9% w/w solution of sodium chloride for reconstitution, which approximates the isotonicity of blood. This would be required to ensure isotonicity if the lyophilisate was made up of parecoxib sodium and a phosphate buffer alone. If water for injection were used to reconstitute such a lyophilisate, the resulting solution would by hypotonic.
392 There can be no real doubt that the skilled formulator would ensure that the reconstituted formulation when ready for injection would be isotonic. Nor is there any doubt that the formulator would expect that the use of mannitol as a bulking agent would assist in ensuring that water for injection could be used to reconstitute the lyophilised powder because it provides the lyophilisate with “in-built” isotonicity. I also accept that if the skilled formulator decided simply to use parecoxib sodium and a phosphate buffer in the formulation at the pre-lyophilisation stage, then it would be straightforward to select 0.9% w/w sodium chloride as the solvent used to reconstitute the lyophilisate to ensure that the final dose had the appropriate tonicity. However, I consider that the hypothetical formulator, before April 2001, would most likely have used mannitol in the pre-lyophilisation solution. This would have provided the multiple benefits that mannitol was known to confer (to which I refer at [361] above), one of which was that the lyophilisate could be reconstituted using water for injection, which was a more convenient diluent for clinicians to use.
7.12 Analysis of inventive step
393 I have set out earlier in these reasons ss 7(2) and (3) of the Patents Act and aspects of the law relevant to their application. The law concerning the requirement for an inventive step reflects a balance of policy considerations in patent law of encouraging and rewarding inventors without impeding advances and improvements by skilled, non-inventive persons: Lockwood (No 2) at [48] (Gummow, Hayne, Callinan, Heydon and Crennan JJ). The cases over the years have made a number of statements as to what is required to answer the “jury question” of whether or not an invention is obvious. It is a question of fact. The question is not what is obvious to a court, but depends on analysis of the invention as claimed having regard to the state of the common general knowledge, any information relied upon for the purpose of s 7(3), and the approach taken to it by the person skilled in the art: Lockwood (No 2) at [51].
394 As a basic premise, the question is always “is the step taken over the prior art an ‘obvious step’ or an ‘inventive step’”? This is often an issue borne out by the evidence of the experts: Lockwood (No 2) at [52]. Whilst the question remains one for the courts to determine, the courts do so by reference to the available evidence, including that of persons who might be representative of the skilled person in the art: AstraZeneca at [70] (Kiefel J, as her Honour then was). Various formulations of the question have been set out in the cases. In R D Werner & Co Inc v Bailey Aluminium Products Pty Ltd [1989] FCA 57; 25 FCR 565 at 574 Lockhart J said that there must be “some difficulty overcome, some barrier crossed”. A “scintilla of invention” is sufficient to support the validity of a patent: Hässle at [48] (Gleeson CJ, Gaudron, Gummow and Hayne JJ). However, the reference to “scintilla” is to some degree circular because, however small it may be, the claim must still possess the quality of inventiveness. In Allsop Inc v Bintang Ltd [1989] FCA 428; 15 IPR 686 at 701 the Full Court (Bowen CJ, Beaumont and Burchett JJ) noted that for the invention to be inventive, it must be “beyond the skill of the calling”.
395 As I have noted above, in AstraZeneca French CJ noted at [15] that relevant content was given to the word “obvious” by Aickin J in Wellcome Foundation at 286, where Aickin J posed the test:
whether the hypothetical addressee faced with the same problem would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not.
396 At [15] French CJ (with whom Gageler and Keane JJ, and Nettle J agreed) explained:
The idea of steps taken "as a matter of routine" did not, as was pointed out in AB Hässle, include "a course of action which was complex and detailed, as well as laborious, with a good deal of trial and error, with dead ends and the retracing of steps". The question posed in AB Hässle was whether, in relation to a particular patent, putative experiments, leading from the relevant prior art base to the invention as claimed, are part of the inventive step claimed or are "of a routine character" to be tried "as a matter of course". That way of approaching the matter was said to have an affinity with the question posed by Graham J in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd. The question, stripped of references specific to the case before Graham J, can be framed as follows:
"Would the notional research group at the relevant date, in all the circumstances, which include a knowledge of all the relevant prior art and of the facts of the nature and success of [the existing compound], directly be led as a matter of course to try [the claimed inventive step] in the expectation that it might well produce a useful alternative to or better drug than [the existing compound]?"
That question does not import, as a criterion of obviousness, that the inventive step claimed would be perceived by the hypothetical addressee as "worth a try" or "obvious to try". As was said in AB Hässle, the adoption of a criterion of validity expressed in those terms begs the question presented by the statute.
(citations omitted and square brackets in the original)
397 In the present case the respondents propose an eight step pathway that, they submit, leads to the claimed invention. As the review of the evidence and submissions above indicates, a number of the steps along that pathway are fairly straightforward. For the reasons set out in sections 7.4 to 7.6 above, I am satisfied that the hypothetical skilled team would readily navigate each of steps 1, 2 and 3.
398 Step 4 presents the hurdle of obtaining parecoxib sodium. For the reasons set out in section 7.7.1 above, I am not satisfied that the respondents have discharged the onus that lies upon them with regard to this aspect of the development pathway. It has not been shown by the respondents that the skilled team armed only with the common general knowledge and information in the pleaded s 7(3) documents could have obtained valdecoxib before the priority date. Against the prospect that I am incorrect in that analysis, I proceed now on the assumption that step 4 is surmounted.
399 In relation to step 5, as I have noted in section 7.8, there is no doubt that the conversion problem would have been encountered by the skilled formulator. The experts diverged as to their approach to the next steps. The applicants characterise the evidence of Professor Winter, who proposed that he would use organic solvents and the adjustment of pH levels to address any unwanted valdecoxib that precipitated out of the solution, as a blind alley or complicated step that the hypothetical skilled addressee would encounter. The respondents submit that the skilled formulator would perceive the difficulties likely to arise with that approach and, as Dr Robertson said, proceed directly to a formulation that involved lyophilisation.
400 It is apparent that Professor Winter’s approach is one that involved trying quite hard to continue with the use of a ready to use formulation, consistent with his view as to the cost consequences and other downsides of using a lyophilised formulation. Dr Robertson took the view that he could not see much advantage in persisting with developing a ready to use formulation because of the difficulties that he foresaw.
401 Two routes were available. One involved persisting with a ready to use formulation and dealing with the conversion problem by the use of organic solvents and making adjustments to the pH in the manner described in section 7.8. That itself was fraught with uncertainty. Another approach was to move away from a ready to use formulation to try the well-known technique of lyophilising a parecoxib sodium formulation. I am satisfied that had they moved to lyophilisation the most likely next step would be to use mannitol as part of the formulation. That is also a step away from the invention claimed. It was by no means obvious to select a formulation with minimal components claimed in the patent. As I have set out more fully in section 7.9, the probable course that the formulator would take would be to note that the appropriate amount of parecoxib sodium used to treat acute post-surgical pain was very low such that the rule of thumb, which dictates that the pre-lyophilisation solution should have a solids content of around 10%, would apply. They would turn to mannitol as a suitable bulking agent which had several known formulation advantages. They would expect to succeed in that approach but would be frustrated when they encountered the stability problem and the reconstitution problem. This is precisely the type of blind alley or dead end that bespeaks inventiveness in the solution adopted by the patentee: Hässle at [58]; see also Merck Sharp and Dohme Corporation v Wyeth LLC (No 3) [2020] FCA 1477; 155 IPR 1 at [253] and [357] (Burley J); NSI Dental Pty Ltd v University of Melbourne [2006] FCA 1216; 69 IPR 542 at [152] and [179] (Tamberlin J).
402 Furthermore, having regard to the whole of the evidence, I am not satisfied that even upon encountering problems with the use of mannitol as a bulking agent, the hypothetical skilled formulator would move to the invention claimed. As I have noted, the experts agreed that the formulations of the patent fell outside the rule of thumb because the content of solids in the pre-lyophilisation solution as a percentage of the amount of water added before lyophilisation is less than about 10%. Consequently, were the rule of thumb to be applied, a bulking agent would be required. Yet, upon encountering the stability and reconstitution problems, the hypothetical skilled formulation could have taken a number of alternative routes. One could be to try other bulking agents. Another could be to revert to a ready to use formulation and perhaps struggle on with the use of organic solvents. Another, perhaps, would be to abandon the rule of thumb, but for the reasons given, I am not persuaded that Dr Robertson’s approach as described in his evidence is representative of the hypothetical skilled formulator in this respect.
403 Accordingly, in my view the notional research group would not be directly led as a matter of course to formulate a lyophilised powder within the relevant claims, or to try the claimed invention with the relevant expectation of success. The hurdles presented by steps 5 and 6 (with or without step 4) to which I have referred demonstrate that the respondents have failed to establish that the invention as claimed is obvious.
404 I am to some extent fortified in this conclusion by the content of exhibit C, parts of which were tendered by the applicants and the respondents. It is a Pharmacia document relating to Dynastat, the applicants’ commercial parecoxib sodium product. It bears a copyright notice “2004” and includes the following statement:
Different formulations containing buffer/bulking agent/solubiliser were tested for stability. The commercial composition, containing dibasic sodium phosphate heptahydrate as the only excipient, was the most stable formulation investigated. Moreover, this product reconstituted faster than the early formulation containing mannitol (which had been used in the Phase I/Phase II trials).
405 The applicants contend that it would not be until a formulator had attempted a lyophilised formulation including mannitol as a bulking agent that either the stability problem or the reconstitution problem would have become apparent. It would appear from exhibit C that the initial steps taken by the patentee, which apparently included using mannitol in the formulation as a bulking agent, reflect the approach that I have found would have been taken by the uninventive hypothetical formulator.
406 Finally, for completeness I note that my findings that the invention claimed in all of the relevant claims has not been shown to lack an inventive step for the reasons set out above means that the respondents’ alternative argument based on parameteritis cannot succeed.
407 Accordingly, the inventive step case as advanced must fail.
8. LACK OF CLARITY AND DEFINITION
408 Section 40(2)(b) of the Patents Act provides:
A complete specification must…where it relates to an application for a standard patent – end with a claim or claims defining the invention.
409 Section 40(3) relevantly provides:
The claim or claims must be clear and succinct…
410 In their amended particulars of invalidity, the respondents contend that each of the relevant claims do not comply with s 40(2)(b) of the Patents Act in that they do not define the invention and, further or in the alternative, do not comply with s 40(3) in that they are not clear and succinct. In this context the respondents plead, first, that the use of the term “about” in each of the relevant claims is indefinite, unclear and does not provide a workable standard and, secondly, that the use of the term “essentially” in claim 11 suffers from the same defects.
411 In closing submissions, the respondents clarified their position in relation to “about”, contending that only insofar as the applicants’ posited construction of that term is accepted, then it lacks clarity. In section 5.4 above I have rejected the construction advanced by the applicants. Consequently this ground of invalidity is no longer relevant.
412 The sole remaining contention under these grounds of invalidity is that claim 11 lacks definition within s 40(2)(b) and is not clear and succinct within s 40(3) of the Patents Act.
413 The requirement that a claim be clear and succinct within s 40(3) must be understood in the context of the task that the claim performs, which is to define the invention as required by s 40(2)(b). As noted in Albany Molecular Research Inc v Alphapharm Pty Ltd [2011] FCA 120; 90 IPR 457 at [174] (Jessup J), a claim which is a model of verbal or grammatical clarity may nonetheless fail the test of this requirement if it leaves the definition of the boundaries of the invention uncertain or variable.
414 In Martin v Scribal Pty Ltd [1954] HCA 48; 92 CLR 17 Dixon CJ said at 59, “the principles governing the definition of a monopoly operating over the public at large require a description which is not reasonably capable of misunderstanding”. However, it is to be noted that in Minnesota Mining and Manufacturing Co v Beiersdorf (Australia) Ltd [1980] HCA 9; 144 CLR 253 at 274, Aickin J said that the principle with respect to clarity is that “[l]ack of precise definition in claims is not fatal to their validity so long as they provide a workable standard suitable to the intended use”.
415 Apparently imprecise terms are frequently acceptably used in claims, examples including “about”, “approximately”, “substantially” and “relatively”.
416 In Flexible Steel Lacing Co v Beltreco Ltd [2000] FCA 890; 49 IPR 331 at [81], Hely J said:
It is permissible for an invention to be described in a way which involves matters of degree. Lack of precise definition in claims is not fatal to their validity, so long as they provide a workable standard suitable to the intended use. The consideration is whether, on any reasonable view, the claim has meaning. In determining this, the expressions in question must be understood in a practical, commonsense manner. Absurd constructions should be avoided and mere technicalities should not defeat the grant of protection.
(citations omitted)
417 As I have noted, it is apparent that claim 11 does not extend the scope of the monopoly beyond the reaches of claim 1. It may be said that the little work that claim 11 does is to emphasise that the invention so claimed requires that the ingredients specified in (a) and (b) of claim 1 make up most of the composition. It is difficult to conceive of a composition falling within claim 1 which would not be made up mostly of therapeutic agent and buffer, the claim requiring that other excipient ingredients be kept to less than about 10% by weight. Having regard to the conclusion that I have reached that the scope of claim 11 does not exceed the limits of the claim 1, I do not consider the claim to be invalid for failure to satisfy the requirements of either ss 40(2)(a) or 40(3) of the Patents Act.
418 The respondents submit that, even in the event that I find valid claims to be infringed, it is appropriate in the present case for the court to decline to grant injunctive relief. They submit that such relief is discretionary and governed by equitable principles. They submit that in the present case such relief should be refused on the bases: (a) that the Juno products were registered on the ARTG and launched on the Australian market in April 2018 and yet proceedings were not commenced until 22 August 2019; (b) that the patent will expire on 2 April 2022, after which Juno will be free to exploit the claimed invention in any event; and (c) in those circumstances it would not be a suitable case for the exercise of injunctive powers which would involve the disruption of supply lines to hospitals when the patent only has a number of weeks to run in any event. In those circumstances, the respondents submit that injunctive relief should be declined.
419 In the usual course, upon a finding of an infringement of a valid claim of a patent, the patentee will be granted an injunction to restrain such breach: Calidad Pty Ltd v Seiko Epson Corporation (No 2) [2019] FCAFC 168; 147 IPR 386 at [28]-[30] (Greenwood, Jagot and Yates JJ); Merck Sharp & Dohme Corp v Wyeth LLC (No 4) [2020] FCA 1719; 157 IPR 1 at [8] (Burley J). I am not satisfied that the factors identified by the respondents justify a departure from that course. In relation to (a), it is apparent from documents tendered in evidence that the parties engaged in correspondence from May 2018, after the Juno products were registered on the ARTG, continuously until the commencement of proceedings such that I do not consider the delay to be at all significant. In relation to (b), whilst it is true enough that the patent will soon expire, while the patent is valid and infringed the patentee is prima facie entitled to enforce its rights under it. The fact that they will cease shortly does not of itself diminish the entitlement of the patentee to the rights that accrue to it. Finally, the argument raised in (c) is not supported by evidence. Taken together, in my view these matters provide an insufficient basis to decline to grant injunctive relief.
420 I have found that certain of six exemplar batches of the Juno products would, had they been made or imported into Australia, have infringed each of claims 1, 4, 5, 11, 14, 15, 17, 18, 19, 20, 21, 24, 34, 35, 36, 37, 38, 39, 40 and 41 but not claims 7, 26, 27, 28, 30 and 42 and that the challenge to the validity of the relevant claims fails. At trial the parties agreed that such findings will be sufficient for them to reach agreement as to the scope of the infringement by the respondents with regard to the Juno products sold in Australia. I will make directions for the parties to endeavour to agree to the form of short minutes of order giving effect to these reasons and list the matter for case management. It is not unusual for cases of this sort to involve a dispute as to the appropriate costs order to make. My preliminary view is that the applicants have succeeded in all but relatively limited respects and that the result should be the respondents pay the applicants’ costs of the claim and the cross-claim. However, the parties may make provision for the filing of short submissions on the subject of costs should they be unable to agree on that subject.
I certify that the preceding four hundred and twenty (420) numbered paragraphs are a true copy of the Reasons for Judgment of the Honourable Justice Burley. |
Associate:
CONFIDENTIAL ANNEXURE A
At the hearing, the parties tendered a table of agreed calculations setting out the percentage of parecoxib sodium present in the representative six batches of the Juno products in the event that I came to the conclusions on construction that I have set out above. The relevant calculations are extracted in the table below. My findings regarding which batches are within the scope of the claims are based on these agreed calculations.
% weight parecoxib sodium | ||
Batch No. | Including residual water in total weight of composition | Excluding residual water from total weight of composition |
31601 | [redacted text] | [redacted text] |
31602 | [redacted text] | [redacted text] |
31603 | [redacted text] | [redacted text] |
31604 | [redacted text] | [redacted text] |
31605 | [redacted text] | [redacted text] |
31606 | [redacted text] | [redacted text] |
Batches 31601, 31602 and 31603
The parties agreed that, if residual water is included in the total weight of the composition, batch 31601 contains [redacted text]% parecoxib sodium. The amount of the therapeutic agent parecoxib sodium in batch 31601 is therefore outside the “about 30% to about 90%” range specified in claim 1, as that range is extended having regard to the preferred construction of “about”. Consequently, batch 31601 does not fall within the scope of claim 1 or any of the relevant claims dependent on it. For the same reasons, batches 31602 and 31603 do not fall within the scope of claim 1 or any of its dependent claims. Further, having regard to my observations above regarding the differences between the Juno products and the example 1 formulation D composition in the patent, none of the batches fall within the scope of claim 42.
The parties agreed that, if residual water is excluded from the total weight of the composition as I have found is required by claim 26, batch 31601 contains [redacted text]% parecoxib sodium. Consequently, the amount of therapeutic agent included is outside the claimed range such that batch 31601 does not fall within the scope of claim 26 or any of the relevant claims dependent on it. For the same reasons, batches 31602 and 31603 do not fall within claim 26 or any of its relevant dependent claims.
Batches 31604, 31605 and 31606
The parties agreed that, if residual water is included in the total weight of the composition, batch 31604 contains [redacted text]% parecoxib sodium. The amount of the therapeutic agent parecoxib sodium in batch 31604 is therefore within the “about 30% to about 90%” range specified in claim 1, given the preferred construction of “about” effectively extends the upper bound of the range to 90.49% to allow for rounding to the nearest whole percentage. Consequently, and having regard to the admissions made by the respondents with respect to the remaining integers, batch 31604 falls within the scope of claim 1. It is also within claim 11. It is not, however, within the scope of claim 7 because it is a composition that contains more than “about 85%” parecoxib sodium.
Having regard to further admissions made by the respondents, batch 31604 is also within the scope of dependent claims 4, 5, 14, 15, 17, 18, 19, 20, 21, 24, 34, 35, 36, 37, 38, 39, 40 and 41.
For the same reasons, batches 31605 and 31606 are also within the scope of claim 1 and dependent claims 4, 5, 11, 14, 15, 17, 18, 19, 20, 21, 24, 34, 35, 36, 37, 38, 39, 40 and 41.
Further, having regard to my observations above regarding the differences between the Juno products and the example 1 formulation D composition in the patent, none of the batches are within the scope of claim 42.
The parties agreed that, if residual water is excluded from the total weight of the composition as I have found is required by claim 26, batch 31604 contains [redacted text]% parecoxib sodium. Consequently, the amount of the therapeutic agent parecoxib sodium included in batch 31604 is outside the claimed range such that batch 31604 is not within the scope of claim 26 or any of the relevant claims dependent on it. For the same reasons, batches 31605 and 31606 are not within the scope of claim 26 or any of its dependent claims.
NSD 1370 of 2019 | |
NEO HEALTH (AUSTRALIA) PTY LTD ACN 605 322 763 | |
PFIZER AUSTRALIA PTY LTD ACN 008 422 348 |

