
FEDERAL COURT OF AUSTRALIA
Merck Sharp & Dohme Corporation v Wyeth LLC (No 3) [2020] FCA 1477
Table of Corrections | |
In paragraph 439, the phrase “[check]” has been removed from the final sentence | |
1 June 2021 | In paragraphs 527, 531 and 532 “Hoffman LJ” has been changed to “Lord Hoffmann” |
1 June 2021 | In paragraphs 168 and 537-540 “Lord Hoffman” has been changed to “Lord Hoffmann” |
ORDERS
BURLEY J | |
DATE OF ORDER: | 14 October 2020 |
THE COURT ORDERS THAT:
1. The parties provide short minutes of order to chambers as to the appropriate form of orders giving effect to these reasons and costs, with any areas of disagreement marked up, by 4 November 2020.
2. The interlocutory application filed by the applicants/cross-respondents on 24 July 2020 be dismissed, with no order as to costs.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
BURLEY J:
1 It is perhaps not inappropriate that, at a time when the world is affected by the COVID-19 pandemic, the present dispute concerns attempts to improve disease immunity. Two pharmaceutical companies are in the race to develop better forms of immunisation against Streptococcus pneumoniae, which is a leading cause of meningitis, pneumonia and severe invasive disease in people, especially infants and young children, throughout the world. These proceedings concern an aspect of that race.
2 Merck Sharp & Dohme Corporation and Merck Sharp & Dohme (Australia) Pty Ltd (collectively, MSD) contend that three patents owned by Wyeth LLC are invalid. Australian Patents No. 2006235013 (013 patent) and No. 2013206844 (844 patent) are entitled “Multivalent pneumococcal polysaccharide-protein conjugate composition” and concern a multivalent immunogenic composition comprising 13 distinct polysaccharide-protein conjugates. The priority date of their claims is 8 April 2005. They are referred to below as the composition patents. The third is Australian Patent No. 2012216628 (container patent) which is entitled “Novel Formulations which Stabilize and Inhibit Precipitation of Immunogenic Compositions”. It concerns a siliconised container means whereby polysaccharide-protein conjugates may be stabilised. The priority date of the claims is 26 April 2006.
3 Wyeth in its Amended Notice of Cross-Claim of 12 November 2019 seeks declaratory, injunctive and other relief against MSD on the basis that MSD will infringe claims 1 – 8, 10 – 13 and 16 – 17 of the 013 patent (asserted 013 patent claims) and claims 1 – 6 and 11 – 14 of the 844 patent (asserted 844 patent claims) (collectively, the asserted composition patent claims) by the launch of a 15-valent vaccine (MSD’s 15-valent vaccine) that it intends to sell in Australia. At the commencement of the trial Wyeth contended that MSD will infringe claims 1 – 8, 16 – 18 and 20 – 23 of the container patent. Somewhat after the trial, Wyeth applied to re-open its case to add an allegation that claim 9 of the container patent will also be infringed. MSD initially opposed that course, but after some considerable delay, following a contested hearing on the subject, MSD changed its position. As a consequence, Wyeth’s case was re-opened and more than a year after the initial hearing had concluded, a further day of hearing concerning allegations of infringement and invalidity of claim 9 was conducted. Accordingly, the container patent claims asserted against MSD are claims 1 – 9, 16 – 18 and 20 – 23 (the asserted container patent claims).
4 In this judgment I first address questions of validity and infringement in relation to the composition patents before turning to the same questions as they arise in relation to the container patent. The law that applies to the 013 and container patents is the form of the Patents Act 1990 (Cth) (or pre-RTB Patents Act) amended by the Patents Amendment (Innovation Patents) Act 2000 (Cth) but prior to the changes implemented by the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth) (RTB Act). The post RTB Act version of the Patents Act (post-RTB Patents Act) applies to the 844 patent.
1.1 Issues arising in relation to the composition patents
5 In relation to the question of infringement, there is no dispute as to the make-up of MSD’s 15-valent vaccine. The only issue is whether, as a matter of construction, the claims include that product within their scope. The primary construction issue arises from the use of the term “comprising” in the claims, in circumstances where the claim identifies 13 nominated serotypes, and the alleged infringing product includes those 13 plus two more. If that construction question is resolved adversely to MSD it accepts that its 15-valent vaccine will infringe, but it contends that the asserted composition patent claims are invalid on a number of bases.
6 As to invalidity, MSD first contends that, regardless of the construction adopted, the claims are not novel in the light of the publication authored by C de la Peña et al called “Presente y futuro de la vacunación antineumocócica” published in 2004 by Pediátrika (Volume 24(4)), either read alone or read with the publication authored by S K Obaro et al called “Safety and immunogenicity of a nonavalent pneumococcal vaccine conjugated to CRM197 administered simultaneously but in a separate syringe with diphtheria, tetanus and pertussis vaccines in Gambian infants” published in 2000 by the Pediatric Infectious Disease Journal (Volume 19(5)).
7 MSD next contends that the claims lack an inventive step either in the light of the common general knowledge alone or in the light of a number of other pieces of prior art information falling within s 7(3) of the Patents Act.
8 MSD further contends that the invention claimed in the composition patents is not a manner of manufacture within s 18(1)(a) of the Patents Act, is not useful within s 18(1)(c) and was obtained by a false suggestion or misrepresentation within s 138(3)(e). MSD further contends that the claims lack fair basis (or, in the case of the 844 patent, support) within s 40(3) of the Patents Act, and lack clarity within s 40(2)(b).
1.2 Issues arising in relation to the container patent
9 Wyeth contends that MSD threatens to make its 15-valent vaccine available in a siliconised container means that falls within the scope of the asserted container patent claims. Substantially the same non-infringement argument arises as for the composition patents, although only in respect of claim 18.
10 In its challenge to the validity of the container patent, MSD contends that the asserted claims (with the exception of claim 9) are not fairly based in accordance with s 40(3) of the Patents Act and are not clearly defined within s 40(2)(b). It also contends that all of the asserted container patent claims lack novelty in light of International Patent Application No. PCT/IB02/03495 published as WO 03/009869 on 6 February 2003 and entitled “Vaccines Comprising Aluminium Adjuvants and Histidine” (Chiron patent), and lack an inventive step in the light of the common general knowledge alone or in the light of prior art information within s 7(3) of the Patents Act. MSD also contends that the invention claimed in the container patent is not to a manner of new manufacture within s 18(1) of the Patents Act and is not useful within the requirements of s 18(1)(c).
11 For the reasons set out in further detail below, I have determined that:
(1) Wyeth has established that MSD’s 15-valent vaccine will infringe all of the asserted composition patent claims and the asserted container patent claims, subject only to my findings as to validity;
(2) the novelty, inventive step, manner of manufacture, clarity, fair basis, inutility and false suggestion challenges to the asserted 013 patent claims fail;
(3) the lack of support challenge to the asserted 844 patent claims succeeds, but that the novelty, inventive step, manner of manufacture, clarity, inutility and false suggestion challenges to that patent fail; and
(4) the inventive step challenge to the asserted container patent claims succeeds, but the novelty, manner of manufacture, fair basis, clarity and inutility challenges to that patent fail.
12 The result is that Wyeth has established that the asserted 013 patent claims are valid and infringed. MSD has established that the asserted 844 patent claims and the asserted container patent claims are invalid. The only orders that I make are that the parties confer and provide to my chambers proposed short minutes giving effect to these reasons by 4 November 2020, noting any differences in approach in mark up. A case management hearing will be conducted at a convenient date shortly after that.
13 In the reasons that follow, I first consider the composition patents before moving to the container patent.
2. COMPOSITION PATENTS: THE WITNESSES
14 The composition patents concern multivalent pneumococcal polysaccharide-protein conjugate compositions used for vaccinating an individual to trigger an adaptive immune response. The aim of such a vaccination is to protect the host against the consequences of subsequent exposure to a pneumococcal pathogen bearing one or more antigens contained in the vaccine preparation and/or to provide indirect protection to the community by interrupting transmission of the pathogen. The product claims are for polysaccharide-protein conjugate vaccines, which are to be distinguished from three other forms of vaccines known in April 2005, being whole-cell vaccines, polysaccharide vaccines and protein vaccines. The field of the invention in the composition patents concerns microbiology and immunology. Before addressing the terms of the specification, I first refer to the witnesses relied upon by the parties and then set out parts of a substantial technical primer.
15 James Cleland Paton has since March 2013 been a Professor of Microbiology in the School of Biological Sciences and Director of the Research Centre for Infectious Diseases at the University of Adelaide. Prior to holding this role, from September 2000 he was Professor of Microbiology in the Discipline of Microbiology and Immunology within the School of Molecular and Biomedical Science at the same university. He gained his PhD from the University of Adelaide in 1979 and since then his research has focussed on the study of the molecular basis for bacterial pathogenesis as well as disease prevention and therapeutic strategies for the control of bacterial infectious diseases, including vaccine development. His research has encompassed the pathogenesis of disease caused by Streptococcus pneumoniae, and is aimed at understanding key events in the host-pathogen interaction and identifying and evaluating novel drug targets and vaccine antigens.
16 In the 1980s, Professor Paton’s research focus was on protein antigen vaccines. He began to work on polysaccharide-protein conjugate proteins for Haemophilus influenzae type b (Hib), which was a very common cause of meningitis and other life-threatening diseases. The Hib vaccine was very successful and spurred activity into the development of pneumococcal polysaccharide-protein conjugate vaccines. In 2004 he wrote a review in a book edited by E I Tuomanen entitled The Pneumococcus (ASM Press, Washington D.C.) in which he discussed the developments in pneumococcal vaccines (Paton Review) and where he observed that the development of pneumococcal polysaccharide-protein conjugate vaccines had been “considerably more complex” than was the case with Hib owing to the multiplicity of disease-causing serotypes. In his first affidavit he observes that the development work used established carrier proteins (such as diphtheria toxoid, tetanus toxoid and cross-reacting material 197 (CRM197)), which were favoured by vaccine developers because they were known to be safe in children and had already been approved by regulators, thereby avoiding the need to test the safety of a previously uncharacterised alternative carrier protein.
17 Professor Paton affirmed several affidavits in these proceedings. In his first affidavit he outlines his background and experience relevant to the technology in issue and responds to the following question asked of him (the MSD Problem):
How would you have gone about developing a polysaccharide-protein conjugate pneumococcal vaccine that was an improvement on Prevnar 7 before April 2005?
18 I set out his response in more detail later in these reasons. After providing his answer, Professor Paton then reviewed the composition patents. He was next provided with and reviewed various prior art publications. He gives evidence that he would have expected to find these from a literature search conducted before April 2005.
19 In his second affidavit Professor Paton responds to evidence given on behalf of Wyeth concerning: immune interference and carrier induced epitope suppression (or CIES); which serotypes he would have selected if developing a polysaccharide-protein conjugate pneumococcal vaccine to improve on Prevnar 7 before the priority date; the composition of the skilled team; and example 16 in the composition patents. In his third affidavit he adduces evidence about the 5th International Symposium on Pneumococci and Pneumococcal Disease held in Alice Springs in April 2006.
20 Dennis Lee Kasper has since 1997 been Professor of Medicine and Professor of Microbiology and Immunobiology at Harvard Medical School in Boston in the United States. He completed a degree in Medicine at the University of Illinois in Chicago in 1967 and since 1973 has run his own research laboratory, studying the capsular polysaccharides of several extracellular bacteria, including their interaction with the immune system. A major focus of his work has been the development of human vaccines, including polysaccharide-protein conjugate vaccines. He is the named inventor on dozens of patents and patent applications worldwide. He was elected to the United States National Academy of Medicine in 2001 and was elected a member of the National Academy of Sciences in April 2018. He has consulted for many pharmaceutical and biotechnology companies. Since 1975 his primary interest has been in immunology, immunochemistry and genetics of bacterial polysaccharides and their production, in the context of polysaccharide-containing vaccines.
21 Professor Kasper affirmed two affidavits in these proceedings. In his first, which is the only one of present relevance, he notes that he gave evidence before the United States Patent Trial and Appeal Board in various proceedings between MSD and Wyeth concerning similar patents to the composition patents in suit. Professor Kasper gives evidence in reply to that of Professor Dagan in relation to the subjects of CIES, and the cross-protection between serotypes 6B and 6A, and 19F and 19A.
22 Alison Margaret Jones is a solicitor at Corrs Chambers Westgarth, the solicitors representing MSD. She gives evidence of electronic searches that she conducted in September 2018 using various search terms.
23 Richard Anthony Strugnell has since 2001 been a Professor of Microbiology at the University of Melbourne. He obtained a PhD from Monash University in 1985 and his postdoctoral research first focussed on Treponema pallidum and the pathogenesis of syphilis, and then later on a recombinant Salmonella vaccine. Since 1995 he has conducted research into the underlying immunopathogenesis of Salmonella infections. His research work was re-focussed in 2005, when he was involved in research to develop recombinant Salmonella vaccines expressing pneumococcal proteins. He has also initiated studies into the nosocomial pathogen Klebsiella pneumoniae, which is a major cause of morbidity in the world as it has become very drug resistant. That work involved testing to see whether Klebsiella pneumoniae could usefully be a target of a conjugate vaccine.
24 From 1998 until about 2011 Professor Strugnell held the position of Regional Editor of Vaccine, a peer reviewed medical journal targeted towards medical professionals who are interested in vaccines. He is the named inventor on three patents and two patent applications. Since 1992 Professor Strugnell has regularly lectured undergraduate students at the University of Melbourne in the areas of microbiology and immunology. His more recent undergraduate teaching responsibilities have been in the areas of bacterial pathogenesis, host/pathogen relationships, vaccine development and medical bacteriology.
25 In his first affidavit Professor Strugnell responds to the question of how he would go about solving the problem of developing an improved pneumococcal vaccine on the basis of what was known to him and what he understands to have been well-known to others working in the field of immunology and microbiology, including as it relates to vaccine development and particularly pneumococcal vaccines, as at April 2005 (the Wyeth Problem). He was asked to describe the steps that he would have taken, as a matter of routine, at April 2005, to address the Wyeth Problem with a reasonable expectation of success. I address his answer in more detail later in these reasons. Professor Strugnell then addresses the content of the composition patents, the Peña and Obaro publications and the other prior art documents relied upon by MSD in support of its obviousness case. He then responds to the evidence of Professor Paton.
26 In his second affidavit Professor Strugnell responds to the second affidavit of Professor Paton and the first affidavit of Professor Kasper.
27 Ron Dagan has since 1992 been a Professor of Pediatrics and Infectious Diseases at the Ben-Gurion University of the Negev in Israel, and has since 2015 held the position of Distinguished Professor at the University. He is also an Emeritus Director of the Pediatric Infectious Disease Unit of the Soroka University Medical Centre in Israel. He obtained a medical degree from the Hebrew University in Jerusalem in 1974 and from 1987 until 2014 he was the Director of the Pediatric Infectious Disease Unit at the Soroka University Medical Centre. His research has focussed on pneumococcal vaccines, the epidemiology and introduction of hepatitis A vaccines, the epidemiology of vaccine-preventable diseases, the pathology of otitis media and prediction of its bacteriological and clinical response to various antibiotics, and the epidemiology and prevention of enteric and invasive infections in young children.
28 Professor Dagan gives evidence that he has been a leader in clinical development of many pneumococcal conjugate vaccine candidates and was one of the first to describe the importance of carriage in indirect protection (or herd immunity) and the potential for serotype replacement as a result of pneumococcal vaccination.
29 Professor Dagan has in the past been engaged by Wyeth to give evidence in the European Patent Office and the Intellectual Property Tribunal in the Republic of Korea in relation to patents related to the composition patents. He has been and remains a consultant, adviser and researcher for numerous pharmaceutical companies.
30 Professor Dagan was asked by Allens, the solicitors for Wyeth, to describe the field of pneumococcal vaccines, including what he and others in the field considered as at April 2005 to be future options for pneumococcal vaccinations. He gives evidence that pneumococcal polysaccharide vaccines have been used widely since the late 1970s, and that as at April 2005, a 23-valent pneumococcal polysaccharide vaccine was in use throughout much of the world. However, these were considered to be poorly immunogenic in infants and young children. As at April 2005 the only commercially available pneumococcal conjugate vaccine was Prevnar 7. Professor Dagan identifies other known pneumococcal conjugate vaccines that were also being tested in clinical trials.
31 Professor Dagan addresses an article that he and others wrote in 2004 entitled “Reduction of Antibody Response to an 11-Valent Pneumococcal Vaccine Coadministered with a Vaccine Containing Acellular Pertussis Components” (Dagan 2004). He explains that his view in 2004 was that existing adjuvants and carrier protein technologies were not the solution to providing improved pneumococcal vaccines, and that he thought that novel approaches would be needed.
32 Professor Dagan gives evidence about the difficulties that he expects would have been encountered if he had been asked to increase the coverage of pneumococcal conjugate vaccines by adding more serotypes to existing formulations in April 2005, and he also addresses the Wyeth Problem. I address his evidence in relation to this in more detail later in these reasons.
33 Professor Dagan then reviewed the compositions patents, the prior art information relied upon by MSD in its novelty and obviousness cases, and responded to parts of the evidence given by Professor Paton.
34 Thomas Kis-Major is a professional translator. He reviewed the English translation of Peña provided by MSD and criticised its accuracy. He was not cross-examined.
2.3 The composition patents joint expert report and concurrent evidence
35 Professors Paton, Kasper, Dagan and Strugnell joined in the preparation of a joint expert report (composition JER) in which they confronted their many differences of opinion. For the most part they adhered to their differences. They gave concurrent evidence during which they were cross-examined.
36 As a general matter, I have found that the experts gave evidence to the best of their ability in an attempt to assist the Court. Each is distinguished in his field. I consider in section 7.4 below their respective experience and qualifications in the context of their ability to assist the Court in assessing whether or not the composition patents claim an invention which involves an inventive step. Except where otherwise noted, I reject the assertions made by each side that the opposing witnesses were not prepared or able to give objective or credible evidence.
3. COMPOSITION PATENTS: BACKGROUND PRIMER
37 The parties cooperated to produce a detailed primer of background information relevant to the composition patents. They accept that the material in it forms part of the common general knowledge before 8 April 2005. What follows in this section has been extracted from the primer.
The immune system
38 The immune system protects the body against infections that might be caused by exposure to pathogens. There are two major interconnected immune responses in humans – the innate immune response and the adaptive immune response. The innate and adaptive immune responses do not operate in isolation. The adaptive immune response is dependent on, and enhanced by, elements of the innate immune response.
The innate immune response
39 The innate immune response is the first line of defence against pathogens. In many cases, an infection is completely controlled by innate immune mechanisms before adaptive immunity is triggered. The most virulent pathogens, however, usually have ways to overcome the innate defences. The innate system is not augmented by previous exposure to the same pathogen. Should the same pathogen infect on a subsequent occasion, the innate immune system will respond in the same way as during the first encounter.
40 The cells of the innate immune system include a diverse range of leukocytes, also known as white blood cells. These cells can, individually or in combination, identify and eliminate pathogens.
41 Some white blood cells are capable of killing pathogens by engulfing them, a process called phagocytosis. Others express a set of receptors which recognise and bind to molecular patterns on pathogens. This binding activates the release of signalling proteins, defensins and other anti-bacterial peptides, and degradative enzymes.
42 Inflammation is an important feature of the innate immune response to a bacterial infection, such as an infection by pneumococcus. An infection-driven inflammatory response is characterized by redness, heat and swelling at the site of the infection. Cytokines mediate the inflammatory response, increasing the permeability of blood vessels to fluid and proteins. This leads to local swelling and an accumulation of proteins that assist in eliminating pathogens. Cytokines may also stimulate other cells in order to attract, and facilitate the movement of, leukocytes to the site of the infection.
43 The innate immune system includes the complement system, which consists of over 20 interacting proteins. These complement proteins enhance other parts of the immune system. The complement proteins are activated in a cascade, where the activation of one protein leads to the activation of the next. The individual proteins are given designations C1 to C9, and the sub-fragments of these proteins are given designations such as C3a, C3b and C5a. The complement system can be activated directly, by a pathogen, or indirectly, by pathogen-bound antibodies produced during an adaptive immune response. The complement system assists the immune system in various ways: by attachment of C3b to the surface of pathogens, to mark them and make them susceptible to phagocytosis; by promoting an inflammatory response (through C3a and C5a) and thereby bringing more phagocytes and lymphocytes to the site of infection; and by direct killing of some types of bacteria by rupturing the bacterial membrane (through a complex comprising C5b, C6, C7, C8 and C9).
The adaptive immune response
44 Many pathogens have developed features that enable them to evade the innate immune response. Some bacteria (including pneumococci) have evolved a polysaccharide coating, or “capsule”, which is not recognised as a pathogen by the body’s receptors and which may inhibit the deposition of the complement protein fragment, C3b. Pathogens that are able to evade the innate immune response can multiply rapidly and cause disease or death.
45 Adaptive immunity is typically triggered when an infection eludes the innate defence mechanisms and reaches a threshold level. The adaptive immune response involves recognition and actions that are specific to features of the pathogen. Adaptive immunity takes days to weeks to become fully established; much longer than the innate immune response. However, the adaptive immune response can learn from previous encounters with specific pathogens and then destroy them more quickly and effectively if they are encountered again, through a process called immunological memory (a phenomenon which is the target of most vaccines).
46 There are two broad classes of adaptive immune responses – humoral immune responses and cell-mediated immune responses. As humoral immunity is particularly important in the defence against infection caused by bacteria such as the pneumococcus, it merits some further explanation.
47 There are two main types of lymphocytes: B-cells and T-cells. These make and secrete antibodies, also known as immunoglobulins, which mediate the humoral immune response. The five major types of antibodies are IgM, IgD, IgG, IgA and IgE. Antibodies are proteins that bind specifically to a particular antigen. An antigen is a molecule that is capable of being recognised by the immune system. B-cells and T-cells carry receptors of only one specificity; that is, they only carry receptors for one antigen. B-cells and T-cells that have not interacted with their specific antigen are known as naïve B-cells or T-cells. Antigen recognition by mature B-cells involves the binding of a B-cell receptor to a binding site – or epitope – on an antigen. Antigens can have multiple epitopes, each recognised by a different receptor on a different B-cell. Some antigens, including polysaccharides, can have the same epitope repeated multiple times.
48 While B-cell receptors can recognise and bind to virtually any structure, the receptors on conventional T-cells recognise only antigenic peptides that are bound to major histocompatibility (MHC) molecules that are displayed on the surfaces of antigen-presenting cells.
49 The humoral immune response is initiated when naïve B-cells bind to their specific epitope on an antigen and become activated. The activation of a naïve B-cell may depend on various factors, including:
(1) the strength of the association, or affinity, of the B-cell receptor for a particular epitope on an antigen;
(2) competition with other B-cells or antibody for binding with the epitope on an antigen;
(3) the abundance of the antigen and the epitope on that antigen;
(4) the duration over which the antigen is present;
(5) how and where the epitope on an antigen is encountered by the B-cell;
(6) the magnitude, duration and frequency of B-cell receptor signalling; and
(7) a range of regulatory factors influenced by matters such as the presence or absence of cytokines, including those produced by the innate immune response and by regulatory T-cells.
50 Humoral immune responses can be either T-cell-dependent (TD response) or T-cell-independent (TI response). In a TD response, a T-cell-dependent antigen (TD antigen) binds to the antigen receptor on the B-cell. The B-cell internalises and processes the bound antigen, which causes it to present certain peptides and MHC molecules on its surface. It is typically trapped in the lymph node and then migrates to the zone in the lymph node where it can interact with activated helper T-cells which bind to that particular peptide-MHC complex (as noted above, T-cells recognise peptides bound to MHC molecules). The T-cells may directly bind to the B-cell and may secrete cytokines. The combination of these factors can stimulate activation and proliferation of the B-cells. Some of the proliferating B-cells immediately secrete antibodies that provide some measure of short-term protection to the host. Other proliferating B-cells migrate to a different part of the lymph node and form a germinal centre where they can rapidly proliferate.
51 The B-cells produced undergo differentiation into either plasma cells or memory B-cells. Most become plasma B-cells, which are the main antibody-secreting cells. Some of these plasma B-cells migrate to the bone marrow, where they can live for months or years and continue to secrete antibodies. This provides for the longevity of vaccine responses. The memory B-cells do not secrete antibodies. They are long-lived and can continue to live for the lifetime of the host. The role of memory B-cells is to be activated by a later encounter with the same antigen in what is known as a secondary immune response. The secondary immune response is more rapid, stronger and of higher affinity than the primary immune response. A secondary immune response is characterised in its first few days by the production of large amounts of certain immunoglobins: IgG antibody, with some IgA and IgE. In the absence of a memory response, repeated exposure to an antigen does not provide a secondary immune response and instead simply replicates, at best, the primary immune response.
52 Some antigens, particularly polysaccharides, are generally not capable of inducing a TD response because they are not processed and presented to T-cells as a peptide-MHC complex. However, some of these antigens can still produce an adaptive immune response because they have repeated epitopes which bind to multiple B-cell receptors on the same B-cell, bypassing the need for the helper T-cells. This is a TI response, generated by T-cell-independent antigens (TI antigens).
53 Capsular polysaccharides, like those which encapsulate pneumococcus bacteria, have long repeating structures with many copies of the same epitope. They belong to the TI-2 group of antigens. These repetitive antigens are capable of delivering prolonged and repetitive signaling to a specific B-cell by simultaneously binding and cross-linking a critical minimum number of B-cell receptors which can induce a TI (specifically, TI-2) response.
54 Compared with TD responses, TI responses are relatively rapid and elicit the transient production of antibodies of low affinity, usually without substantial affinity maturation and usually without inducing immunological memory. Although there is no binding between B-cells and helper T-cells in response to a TI antigen, if TD antigens are also present, nearby T-cells may still release cytokines in response to the TD antigens, which increase the magnitude of the response. Additionally, these T-cells may induce some degree of isotype switching (particularly from IgM to IgG), some low-level affinity maturation and some low-level memory B-cell generation.
55 Importantly, the human response to TI-2 antigens usually develops only after the age of 2 years. Children do not generally make fully effective immune responses against some polysaccharide antigens until about 5 years of age. However, antibody responses to pneumococcal polysaccharides can vary depending on the age of the person to whom it is administered and the pneumococcal serogroup. The typically poor response in infants and young children to polysaccharide antigens renders them particularly susceptible to infections with encapsulated bacteria, where the body relies on a TI response.
Antibodies
56 Antibodies are of great importance to the adaptive immune system.
57 The three main functions of antibodies are: neutralisation of pathogens by binding to them; opsonisation of pathogens (binding them so that they can be engulfed by phagocytes); and activating the complement system to destroy pathogens.
58 An antibody can theoretically bind to any epitope for which it has affinity and with which it comes into contact. Different molecules closely related in shape or chemical sequence may all bind to a given antibody with varying degrees of strength. This means that an antibody can bind to epitopes which are similar but not identical to the epitope which originally induced its production. This can lead to cross-protection from antibodies that have specificity for different antigens which bear sufficiently similar epitopes.
59 The pneumococcus is an infectious bacterium. Pneumococcal infections are a major cause of morbidity and mortality in humans of all ages but particularly in the very young, the very old and individuals with specific immunodeficiencies. Under certain conditions, the pneumococcus can generate a protective polysaccharide covering or capsule that provides protection against phagocytosis.
60 The pneumococcal cell wall is a complex structure. Relevantly, it contains a variety of surface proteins, including pneumococcal surface protein A (PspA) and pneumococcal surface adhesin A (PsaA), which are associated with virulence. These proteins are involved in direct interactions with host cells or in concealing the bacterial surface from the host defence mechanisms. The pneumococcus also expresses non-surface proteins, such as pneumolysin, which is a pore-forming toxin.
61 The polysaccharide capsule of pneumococci is variable. In clinical practice, polysaccharide variants – called serotypes – are identified by their reactions with type-specific antisera.
62 About 90 different capsular polysaccharide serotypes had been described as at April 2005. Further serotypes continue to be found. The “Danish system” is the most widely used system for classifying pneumococcal serotypes. This system allocates new serotypes with a sequential number. Serotypes which are cross-reactive with known serotypes (that is, the antigen of that serotype can combine with the antibody for a closely related antigen of another serotype) are not given a new number but are allocated a letter. For example, when a new serotype which was cross-reactive with serotype 7 was found, serotype 7 was renamed 7F (F for 'first') and the related serotype was named 7A.
63 Factors relating to the polysaccharide capsule that are likely to influence the virulence of different serotypes include:
(a) the molecular mass, charge and hydrophobicity of the capsular polysaccharide;
(b) the number, shape and form of specific epitopes within the polysaccharide;
(c) their accessibility; and
(d) the length of the polysaccharide chains.
64 The polysaccharide capsule is one important virulence factor for pneumococci, but there are other very important factors which vary between different clones of any one serotype, including: their ability to adhere to, and penetrate, mucosal and other membranes; their ability to express enzymes capable of degrading complement proteins; and their ability to resist killing after phagocytosis. Expression of these factors is variable and depends upon intrinsic bacterial genetic factors, as well as the environmental conditions to which the bacterium is exposed.
65 The distribution of serotypes common in carriage and disease varies over time and by geographical region, and by age within geographical regions.
66 The pneumococcus has not traditionally been a bacterium associated with high levels of clinical antibiotic resistance compared with other pathogens, but clinical resistance is nevertheless a relevant issue. As at April 2005, antibiotic-resistant pneumococci had been found throughout the world.
Pneumococcal disease
67 Colonisation of the nasopharynx by the pneumococcus in humans is common and most humans are colonised at least once early in life. This is not usually symptomatic, although it can be associated with low-level inflammation. However, the movement of pneumococci from the nasopharynx into other sites in the body can cause serious, and potentially life-threatening, disease. The detailed mechanisms that allow transition from carriage to onset of disease were not fully understood as at April 2005, nor are they fully understood today. At a general level, however, the development of pneumococcal disease results from disturbance of the balance between host and pathogen. This can occur, for example, through concurrent viral infection, malnutrition, exposure to cold, immune deficiency, or the arrival of a new, more pathogenic, clone or serotype.
68 The pneumococcus can cause pneumonia (infection of the lung), as well as invasive diseases such as meningitis (infection of the tissue covering the brain and spinal cord), which can cause death or permanent disability, and sepsis (bacteria growing in the blood) and bacteremia (bacteria in the blood), which are also potentially fatal.
69 The pneumococcus is also a major cause of otitis media (middle ear infection) and sinusitis. Otitis media is one of the most common causes of visits to doctors by infants and children. Otitis media therefore places a high economic burden on health care systems. Although otitis media is usually not life-threatening, untreated infections may cause damage to the structures of the middle ear that can result in permanent hearing loss.
70 The pneumococcus is one of the most important bacterial pathogens that affects humans. As at April 2005, global pneumococcal infections were estimated to cause around one to two million childhood deaths per year, and a similar number of adult deaths.
Host defence against the pneumococcus
71 Colonising pneumococci can be removed from the nasopharynx by innate immune mechanisms. However, if the innate immune system is avoided or overwhelmed, the adaptive immune system becomes of critical importance. In the context of pneumococcal infections, adaptive immunity is largely mediated by antibodies. These antibodies can be directed to the capsular polysaccharide or to other components of the bacterium.
72 Capsular pneumococcal polysaccharides are TI-2 antigens. As discussed above, TI-2 antigens can stimulate antibody responses in adults without T-cell help. As the TI-2 response is generally poor in young children and infants, they are particularly susceptible to pneumococcal infections (although in the first few months of life they may be protected by maternal antibodies).
73 As noted above, antibodies are crucial to the adaptive immune response to pneumococcal infections. Antibodies can bind to the polysaccharide capsule around the pneumococcus, allowing for phagocytosis. For some types of antibodies, binding to the surface of the pneumococcus can also lead to the activation of the complement system. Antibodies can also neutralise pneumococci by blocking attachment to host surfaces. Some antibodies are less effective at opsonisation and therefore do not make the pneumococci susceptible to phagocytosis. An antibody that binds to the pneumococcus but does not promote effective clearance of it is called a non-functional antibody.
74 A vaccine is a preparation containing one or more antigens which is intended to trigger an adaptive immune response in the host to whom the vaccine is administered. The intention of vaccination is to protect the host against the consequences of subsequent exposure to a pathogen bearing that antigen, or to provide indirect protection to the community by interrupting transmission of a pathogen, or both.
75 As at April 2005, the following types of pneumococcal vaccines had been or were being developed, and had been tested in either or both of animals and humans:
(1) whole-cell vaccines;
(2) polysaccharide vaccines;
(3) polysaccharide-protein conjugate vaccines; and
(4) protein vaccines.
76 Only polysaccharide and polysaccharide-protein conjugate pneumococcal vaccines were approved and in commercial use at April 2005. This is still the case today.
77 The claims of the composition patents are directed to polysaccharide-protein conjugate vaccines where 13 serotypes are capsular polysaccharide antigens each conjugated separately to the carrier protein CRM197.
78 A whole-cell vaccine is an inactivated vaccine which contains whole pathogens that have been killed or inactivated by irradiation or chemical treatment, so that they can no longer cause disease. Whole-cell vaccines have been highly effective and are used to protect against important pathogens (e.g. influenza and polio). Whole-cell pneumococcal vaccines were the first type of pneumococcal vaccines, and were marketed in the United States from around 1900. The first large-scale clinical trial of any pneumococcal vaccine was a trial of a crude whole-cell vaccine conducted in South Africa in 1911. These early vaccines were developed without regard to serotype.
79 By the 1940s, an increased understanding of serotype specificity led to whole-cell pneumococcal vaccine development giving way to the development of polysaccharide vaccines (and later conjugate vaccines). By April 2005, some researchers were also working on whole-cell vaccines in animal models. Whole-cell vaccines are among the simplest and cheapest vaccines to produce and because whole-cell killed pneumococci contain many non-capsular antigens that are common to all strains and serotypes of pneumococci, they have the potential to provide a level of serotype-independent immunity. Since April 2005, the development of a whole-cell pneumococcal vaccine has continued.
80 Polysaccharide vaccines contain purified capsular polysaccharides as antigens. Polysaccharide vaccines are relatively simple, stable and cheap to produce. They are intended to provide protection against the specific serotypes included in the vaccine.
81 A 14-valent polysaccharide vaccine was marketed by Merck in the US from 1977. It contained capsular polysaccharides from serotypes 1, 2, 3, 4, 5, 6A, 7F, 8, 9N, 12F, 18C, 19F, 23F and 25F. From 1983, the 14-valent polysaccharide vaccine was replaced by two 23-valent polysaccharide vaccines, one marketed by Merck under the brand name “Pneumovax 23”, and the other marketed by Lederle Laboratories.
82 As capsular polysaccharides are TI-2 antigens, antibody responses to pneumococcal polysaccharide vaccines are usually characterised by a failure to induce significant and sustained amounts of antibodies in children under 2 years of age. Antibody responses to pneumococcal polysaccharides vary depending on the age of the person to whom they are administered and the serogroups of the polysaccharide.
83 In adults, antibody levels decrease rapidly in a few months after vaccination with a polysaccharide vaccine and a repeated vaccination does not typically result in a secondary immune response. Further polysaccharide vaccines do not reduce nasopharyngeal carriage of vaccine serotypes by children or by adults, and so their use does not confer indirect protection (“herd protection”) on the population.
84 Protein vaccines are composed of purified or recombinant protein antigens from a pathogen. Protein vaccines are typically used for pathogens which have exposed external proteins, as the protein antigen(s) selected must be readily accessible to antibody in order to provide effective protection against subsequent exposure to the pathogen.
85 For many years before April 2005, some researchers had been working on a vaccine approach based on immunity against non-capsular antigens common to all pneumococcal serotypes to avoid the issues of serotype specificity and limits to the number of serotypes that could be covered with polysaccharide vaccines or pneumococcal conjugate vaccines, on the one hand, and lack of efficacy in children, in particular, with polysaccharide vaccines, on the other.
86 Pneumococcal proteins that had been considered or trialed as potential vaccine candidates at April 2005 included pneumolysoid (genetically modified pneumolysin), PspA and PsaA. As at April 2005, it was proposed that pneumococcal proteins could potentially be used as stand-alone vaccines, or in combination with pneumococcal conjugate vaccines, or as carrier proteins for polysaccharide antigens in pneumococcal conjugate vaccines.
3.4 Polysaccharide-protein conjugate vaccines
87 A polysaccharide-protein conjugate vaccine contains purified capsular polysaccharides as antigens, each of which are covalently bonded (conjugated) to a carrier protein. By conjugating a capsular polysaccharide to a carrier protein, a stronger antibody response to the polysaccharide is obtained.
88 The coupling of a capsular polysaccharide to a protein carrier is intended to improve the immunogenicity of the vaccine by inducing a TD response, rather than a TI response, to the polysaccharide antigens in the conjugates.
89 The conjugation process involves a chemical reaction between the carrier protein and the polysaccharide antigen. The efficiency of this reaction may vary, depending upon: the polysaccharide antigen, in particular its constituent sugars; the carrier protein, in particular its amino acid content; and the conjugation chemistry that is used.
90 The first commercially available conjugate vaccine – a Hib conjugate vaccine – was marketed in the US from 1987. Formulations of Hib conjugate vaccines which had been licensed before April 2005 used different carrier proteins, known generally to be safe for human use, including the following:
(a) Tetanus toxoid.
(b) Diphtheria toxoid.
(c) CRM197 – which is a non-toxic form of diphtheria toxin that contains a single amino acid substitution. This single amino acid substitution removes its enzymatic activity, making CRM197 non-toxic without the further chemical modification required for diphtheria toxin and tetanus toxin.
(d) Outer membrane protein complex of Neisseria meningitidis serogroup B (OMPC).
91 A simplified representation of the immune response to polysaccharide vaccines and polysaccharide-protein conjugate vaccines described above is shown below:
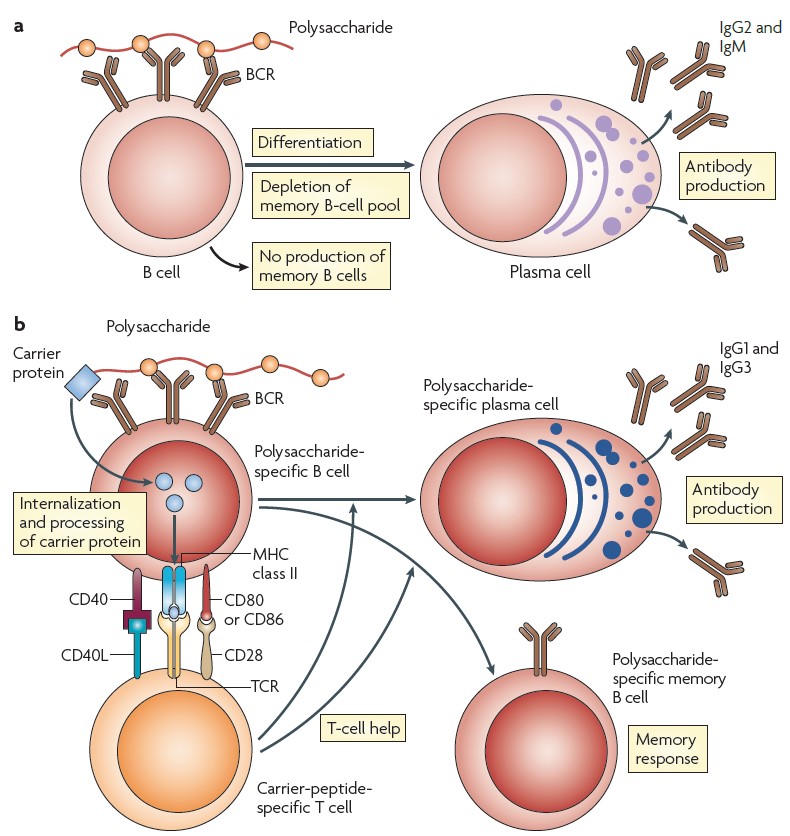
Figure 4. The immune response to (a) polysaccharide vaccines (TI-2 response) and (b) conjugate vaccines (TD response)
92 As explained above, the failure to generate memory cells from a polysaccharide vaccine means that a secondary immune response cannot usually be elicited by immunisation with a further dose of the polysaccharide vaccine. The generation of memory B-cells from a conjugate vaccine allows a secondary immune response to be elicited upon immunisation with a booster dose.
93 The first commercial conjugate vaccine, directed against Hib, involved only conjugates of a single serotype. The development of pneumococcal conjugate vaccines was more complex than in the case of Hib because of the need to provide protection against multiple serotypes.
94 As at April 2005, the following pneumococcal conjugate vaccines had been developed, or had been or were being tested in clinical trials:
(1) A 7-valent vaccine, developed by Wyeth, containing polysaccharides of serotypes 4, 6B, 9V, 14, 18C, 19F and 23F, each conjugated to CRM197. This vaccine, marketed as ‘Prevnar’ (or ‘Prevenar’ in some countries, including Australia, and later called ‘Prevnar 7’), was the only pneumococcal conjugate vaccine licensed anywhere in the world at April 2005.
(2) A 7-valent vaccine, developed by MSD, containing polysaccharides from the same serotypes as Prevnar 7, each conjugated to OMPC.
(3) A 9-valent vaccine, developed by Wyeth, containing polysaccharides of serotypes 1 and 5 in addition to the seven serotypes included in the Wyeth 7-valent vaccines, each conjugated to CRM197 (Prevnar 9).
(4) An 11-valent vaccine, developed by Aventis-Pasteur (the predecessor of Sanofi), containing polysaccharides of serotypes 3 and 7F in addition to the nine serotypes included in the Wyeth 9-valent vaccine, with the polysaccharides from serotypes 3, 6B, 14 and 18C conjugated to diphtheria toxoid and the polysaccharides from serotypes 1, 4, 5, 7F, 9V, 19F and 23F conjugated to tetanus toxoid.
(5) An 11-valent vaccine, developed by GlaxoSmithKline (GSK), containing polysaccharides of serotypes 3 and 7F in addition to the nine serotypes included in the Wyeth 9-valent vaccine, each conjugated to Haemophilus influenzae protein D.
95 The introduction of routine use of Prevnar 7 in the United States, in about 2000, resulted in a significant decline in the rates of invasive pneumococcal disease, not only among vaccinated individuals but also among the population more generally, and especially in the elderly, indicating a substantial indirect protection effect.
96 There was evidence before April 2005 that pneumococcal conjugate vaccines were having the effect of reducing nasopharyngeal carriage of pneumococci of the same serotype as those included in the pneumococcal conjugate vaccines (i.e., vaccine serotypes). The mechanisms by which pneumococcal conjugate vaccines could interrupt nasopharyngeal carriage were not completely understood as at April 2005. The interruption of carriage of vaccine serotypes had the potential to leave a niche for carriage of, and infection by, non-vaccine serotypes, a phenomenon known as serotype replacement. These new serotypes might have been as virulent, or more virulent, or less virulent, than the vaccine serotypes.
3.5 Vaccine formulation and development
97 An adjuvant is a substance that, when mixed with an antigen, increases its ability to provoke an adaptive immune response.
98 Adjuvants typically serve at least two functions. First, they may provide the signals discussed above that induce low-level local inflammation, drawing immune system cells to the site of injection. This enhances and accelerates the adaptive immune response to the vaccine antigens. Secondly, by adsorbing or trapping the vaccine antigen, adjuvants may provide a depot at the site of injection, which releases vaccine antigen more slowly after administration. The sustained release of vaccine antigen assists in maintaining vaccine antigen presence during the development of an adaptive immune response and, as a result, helps to promote B-cell activation.
99 One class of adjuvants is aluminium adjuvants. Aluminium-containing adjuvants include aluminium salts, most commonly aluminium hydroxide and aluminium phosphate.
100 Some vaccines are presented in a freeze-dried (lyophilised) form, which must be reconstituted with a liquid (diluent) before administration. There is a preference for vaccines to be presented in a liquid ready-to-use form, if a stable and effective liquid formulation can be made, optimally pre-loaded into single use syringes.
101 Vaccines may contain additional components, or excipients. An excipient is a substance other than the active substance, included for various purposes such as improving stability of the active ingredients, appearance of the vaccine, and patient tolerability.
102 Vaccines are administered in the expectation that they will be effective in protecting at least some people to whom they are administered or, depending on the vaccine, the community as a whole, from disease. Vaccines do not prevent disease in 100% of vaccinated individuals in diverse populations such as human populations, given the variability between individuals of the immune responses generated.
103 Vaccines undergo pre-clinical testing in animals to demonstrate that they are suitable for testing in humans. Several animal species have been used in pre-clinical trials including mice, rats, rabbits, chinchillas and monkeys. These trials are designed to detect evidence of local or systemic toxicity that might indicate a potential safety issue in humans. They also assess immunogenicity and experimental efficacy in animal models (including challenge studies) and the effects of administering multiple doses. In challenge studies using animal models, vaccinated and unvaccinated animals are compared after direct challenge (infection) with the target pathogen under controlled experimental conditions.
104 Animal models are typically used to assess: vaccine safety and toxicity; vaccine dose and formulation; the nature, magnitude and duration of the immune response; protection against challenge infection and cross-protection from the pathogen of interest; and the potential for preventing disease transmission within a specific population.
105 The predictive value of animal models for immunogenicity and vaccine efficacy in humans depends on the pathogen, the robustness of the animal model, and the correlates of protection.
106 Ordinarily, to evaluate the immunogenicity of a vaccine, animals are injected with the candidate vaccine. The animals are bled before and after vaccination to obtain sera for in vitro analysis. In the context of pneumococcal vaccines, assays which are commonly used in these analyses include those which detect and quantify the level of antibody (e.g. enzyme-linked immunosorbent assay (ELISA)) and which measure the opsonophagocytic activity of antibody (e.g. opsonophagocytic assay (OPA)).
107 The ELISA is the most common method used to detect the presence of specific antibodies in serum. An ELISA uses enzyme to cause a colour change to indicate that the relevant antibody has been detected. The amount of antibody in serum (expressed in terms of titre) may be quantified in an ELISA. Titres are typically measured on a logarithmic scale. A higher titre means there is a greater concentration of antibodies in serum.
108 An ELISA can identify the presence and quantity of antibodies in a sample but cannot determine whether the antibodies are functional (i.e. whether they effectively opsonise or neutralise the pathogen). The OPA is used to test the opsonophagocytic activity of vaccine-induced antibodies in vitro. The classic OPA determines the titres of sera that, when incubated with the bacteria of interest, reduce the number of live bacteria (or colony-forming units) by more than half.
109 ELISAs and OPAs can both be performed using sera from various test animals or humans.
110 The next stage in vaccine development is to carry out clinical trials in humans. These are classified into four phases: Phase I through to IV. In Phase I, small numbers of human volunteers are given the vaccine to assess the safety of the vaccine preparation. In Phases II and III, the vaccine is tested on larger groups of volunteers in order to confirm the proposed dose, assess immunogenicity and collect additional safety data.
111 Following regulatory approval of a vaccine, Phase IV studies are used to monitor effectiveness of the vaccine in the general population and to collect information about any low frequency adverse effects associated with widespread use of the vaccine in larger cohorts, over longer periods of time.
4. COMPOSITION PATENTS – SPECIFICATION AND CLAIMS
4.1 The specification of the 013 patent
112 The 013 patent is entitled “Multivalent pneumococcal polysaccharide-protein conjugate composition”. The Field of the Invention is said to relate generally to medicine and specifically to microbiology, immunology, vaccines and the prevention of infection by bacterial pathogen by immunisation. The patent often refers to “Prevnar”, which I refer to in this judgment as Prevnar 7.
113 The “Background of the Invention” commences by noting that Streptococcus pneumoniae is a leading cause of meningitis, pneumonia and severe invasive disease in infants and young children throughout the world. It says that multivalent pneumococcal polysaccharide vaccines have been licensed for many years and have proved valuable in preventing pneumococcal disease in elderly adults and high-risk patients, but not infants and young children. It says:
The 7-valent pneumococcal conjugate vaccine (7vPnC, Prevnar) was the first of its kind demonstrated to be highly immunogenic and effective against invasive disease and otitis media in infants and young children. This vaccine is now approved in many countries around the world. Prevnar contains the capsular polysaccharides from serotypes 4, 6B, 9V, 14, 18C, 19F and 23F, each conjugated to a carrier protein designated CRM197. Prevnar covers approximately 80-90%, 60-80% and 40-80% of invasive pneumococcal disease (IPD) in the US, Europe and other regions around the world respectively [1,2]. Surveillance data gathered in the years following Prevnar’s introduction has clearly demonstrated a reduction of invasive pneumococcal disease in US infants as expected (FIG. 1) [3,4].
(parenthetical references “[ ]” are to cited publications)
114 I refer below to the capsular polysaccharides from serotypes 4, 6B, 9V, 14, 18C, 19F and 23F each conjugated to carrier protein CRM197 as the Prevnar 7 serotypes. The Background continues by referring to the effect of particular additional serotypes on the prevalence of invasive pneumococcal disease (page 1 line 25 – page 2 line 5):
Surveillance of IPD conducted in US infants prior to the introduction of Prevnar demonstrated that a significant portion of disease due to serogroups 6 and 19 was due to the 6A (approximately one-third) and 19A (approximately one-fourth) serotypes [5,6]. Pneumococcal invasive disease surveillance conducted in the US after licensure of Prevnar suggests that a large burden of disease is still attributable to serotypes 6A and 19A (FIG 1) [3]. Moreover, these two serotypes account for more cases of invasive disease than serotypes 1, 3, 5 and 7F combined (8.2 vs 3.3 cases/100,000 children 2 years and under). In addition, serotypes 6A and 19A are associated with high rates of antibiotic resistance (FIG 2) [7,8,9]. While it is possible that serogroup cross-protection will result in a decline of serotype 6A and 19A disease as more children are immunized, there is evidence to suggest that there will be a limit to the decline, and a significant burden of disease due to these serotypes will remain (see below).
115 The Background concludes (page 2 lines 7 – 12):
Given the relative burden and importance of invasive pneumococcal disease due to serotypes 1, 3, 5, 6A, 7F, and 19A, adding these serotypes to the Prevnar formulation would increase coverage for invasive disease to >90% in the US and Europe, and as high as 70%-80% in Asia and Latin America. This vaccine would significantly expand coverage beyond that of Prevnar, and provide coverage for 6A and 19A that is not dependent on the limitations of serogroup cross-protection.
116 It is apparent that a problem to which the specification is directed is that of increasing coverage of the existing Prevnar 7 vaccine by the addition of further nominated serotypes.
117 The “Summary of the Invention” then provides a series of statements as to what is said to be the invention, the first of which is (page 2 lines 15 – 20):
Accordingly, the present invention provides generally a multivalent immunogenic composition comprising 13 distinct polysaccharide-protein conjugates, wherein each of the conjugates contains a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, together with a physiologically acceptable vehicle. Optionally, an adjuvant, such as an aluminium-based adjuvant, is included in the formulation.
118 Where this paragraph refers to a generally multivalent immunogenic composition, it is apparent that the multivalent composition of the patent is specific, insofar as it concerns the choice of serotypes included. As Professor Paton says, and the specification confirms, it is not possible to extrapolate the data in the 013 patent to other serotypes beyond the 13 claimed.
119 The next statement identifies the 13-valent conjugate, entitled 13vPnC, by reference to the seven serotypes from Prevnar 7 with the addition of six further serotypes, being 1, 3, 5, 6A, 7F, and 19A (I refer to these as the 13 chosen serotypes):
More specifically, the present invention provides a 13-valent pneumococcal conjugate (13vPnC) composition comprising the seven serotypes in the 7vPnC vaccine (4, 6B, 9V, 14, 18C, 19F and 23F) plus six additional serotypes (1, 3, 5, 6A, 7F and 19A).
120 The Summary of the Invention next identifies that the carrier protein may be CRM197 (page 2 lines 30 – 33):
The present invention also provides a multivalent immunogenic composition, wherein the capsular polysaccharides are from serotypes 1, 3, 4, 5, 6A, 7F, 9V, 14, 18C, 19A, 19F and 23F of Streptococcus pneumonia, the carrier protein is CRM197…
121 The specification then states that an aluminium-based adjuvant may be added to this combination, before including the following, which appears to be a statement of a broad invention involving two or more serotypes, one of which must be serotype 3 (page 3 lines 4 – 9):
The present invention also provides a multivalent immunogenic composition, comprising polysaccharide-protein conjugates together with a physiologically acceptable vehicle, wherein each of the conjugates comprises a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, and the capsular polysaccharides are prepared from serotype 3 and at least one additional serotype.
122 The next paragraph describes one embodiment of the composition wherein an additional serotype is selected from the remaining 12 chosen serotypes. Another embodiment involves this composition with an aluminium-based adjuvant. The Summary of the Invention next states that the invention provides a multivalent immunogenic composition comprising polysaccharide-protein conjugates together with a physiologically acceptable vehicle (page 3 lines 21 – 24):
...wherein each of the conjugates comprises a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, and the capsular polysaccharides are prepared from serotypes 4, 6B, 9V, 14, 18C, 19F, 23F and at least one additional serotype.
123 The next embodiment provides that the additional serotype is selected from the group consisting of serotypes 1, 3, 5, 6A, 7F and 19A, while the next two embodiments add CRM197 as the protein carrier and an aluminium-based adjuvant respectively.
124 The “Detailed Description of the Invention” then proceeds from page 4 until page 10 to explain, by reference to published data, how the 13 chosen serotypes came to be selected.
125 It begins by stating that data from invasive pneumococcal disease surveillance between 1995 and 1998 indicated the success of Prevnar 7, such that there could be no justification for the removal of any of the Prevnar 7 serotypes from the next generation of pneumococcal conjugate vaccines. Thereafter, the specification reviews published and unpublished literature for the balance of the six additional serotypes making up the 13 chosen serotypes, concluding in respect of each that coverage against invasive pneumococcal disease will increase if it is included in a conjugate vaccine.
126 For example, in respect of serotypes 1 and 5, the specification states at page 5 lines 2 – 8:
In the US, the rate of IPD caused by serotype 1 in children under the age of 5 is <2%, about the same as for each of types 3 and 7F [1,6]. Serotypes 1 and 5 account for higher rates of IPD in US populations at high risk for invasive pneumococcal disease. Specifically, serotype 1 causes 3.5% of IPD in Alaskan native children <2 years of age, and 18% in children 2-4 years of age [11]. Both serotype 1 and serotype 5 significantly cause disease in other parts of the world and in indigenous populations in developed countries [12,13,14].
127 Parenthetical reference [1] refers to an article authored by W P Hausdorff et al entitled “Which pneumococcal serogroups cause the most invasive disease: implications for conjugate vaccine formulation and use, part I” published in 2000 by the Clinical Infectious Diseases Journal (Volume 30) (Hausdorff 2000). It and the other 33 cited articles are incorporated into the specification by reference. MSD relies on some of these in its validity challenges.
128 The following passage appears in relation to serotype 3. The final sentence in particular is relied upon by MSD in its inutility challenge to the 013 patent (page 6 lines 5 – 22) (emphasis added):
However, attempts to produce a multivalent pneumococcal conjugate vaccine that exhibits significant immunogenicity with respect to serotype 3 polysaccharides have been unsuccessful. For example, in a study of the immunogenicity and safety of an 11-valent pneumococcal protein D conjugate vaccine (11-Pn-PD), no priming effect was observed for serotype 3 in infants who had received three doses of the vaccine followed by a booster dose of either the same vaccine or a pneumococcal polysaccharide vaccine (Nurka et al. (2004) Ped. Inf. Dis. J., 23:1008-1014...[further studies are then identified]...Accordingly, a pneumococcal conjugate vaccine comprising capsular polysaccharide from serotype 3 and capable of eliciting an immunogenic response to serotype 3 polysaccharides provides a significant improvement of the existing state of the art.
129 The inclusion of serotypes 6A and 19A is the subject of particular attention in submissions in relation to the lack of inventive step challenge. The specification at pages 6 – 10 refers to the reasons for including these serotypes, based primarily on published literature, which is cited, and also internal data generated by the patentee. This part of the specification commences by stating that surveillance data in the literature suggests that serotypes 6A and 19A account for more invasive pneumococcal disease in US children less than 2 years of age than serotypes 1, 3, 5 and 7F combined, and that 6A and 19A are commonly associated with antibiotic resistance and play an important role in otitis media. It says that “[t]he ability of the current Prevnar vaccine to protect against disease due to 6A and 19A is not clear” (emphasis added).
130 The specification first discusses the immune responses to serotypes 6A and 19A that have been induced by the inclusion of the 6B and 19F polysaccharides in Prevnar 7. This is a reference to cross-protection induced by 6B and 19F. The specification states that the data from several trials suggest that IgG (that is, a particular type of antibody) responses to 6A are induced by 6B antigens, but that they are generally lower, and that the OPA activity with 6A organisms is different than with 6B organisms. The specification also refers to low levels of cross-reactive IgG and OPA responses to serotype 19A after immunisation with 19F polysaccharide. Internal Wyeth data concerning OPA responses are reported in the specification to be consistent with the published findings of others, and demonstrate “induction of cross-reactive functional antibody to 6A polysaccharide after immunization with 6B polysaccharide, although at a lower level, and very little functional antibody to 19A after immunization with 19F”. The specification then looks at the impact of serotypes 6B and 19F on 6A and 19A immunisation in animal models and efficacy/effectiveness trials on humans. It concludes (page 10 lines 17 – 24):
The post-marketing surveillance data and the case-control study results noted in FIG. 1 and Table 2 with the 7vPnC vaccine suggest that, consistent with the other information on immune responses and performance in the animals models described above, there may be some cross-protection against 6A disease, but to a lesser extent than 6B disease. Furthermore, it appears the protection against 19A is limited. Therefore, a 13vPnC vaccine containing serotypes 6A and 19A provides coverage that is not dependent on the limitations of serogroup cross-protection by serotypes 6B and 19F.
131 After providing a justification for the inclusion of each of the chosen 13 serotypes, the specification then repeats at page 11 lines 1 – 9 the statement of one embodiment of the invention, being a multivalent immunogenic composition comprising the 13 chosen serotypes, together with a physiologically acceptable vehicle, wherein each of the conjugates contains a different capsular polysaccharide conjugated to a carrier protein, one such carrier protein being CRM197, and optionally having an adjuvant.
132 The specification then describes how to make the 13-valent vaccine. It says (page 11 line 11 – page 12 line 2):
Capsular polysaccharides are prepared by standard techniques known to those skilled in the art. In the present invention, capsular polysaccharide are prepared from serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F and 23F of Streptococcus pneumoniae. These pneumococcal conjugates are prepared by separate processes and formulated into a single dosage formulation. For example, in one embodiment, each pneumococcal polysaccharide serotype is grown in a soy-based medium. The individual polysaccharides are then purified through centrifugation, precipitation, ultra-filtration, and column chromatography. The purified polysaccharides are chemically activated to make the saccharides capable of reacting with the carrier protein.
Once activated, each capsular polysaccharide is separately conjugated to a carrier protein to form a glycoconjugate. In one embodiment, each capsular polysaccharide is conjugated to the same carrier protein. In this embodiment the conjugation is effected by reductive amination.
The chemical activation of the polysaccharides and subsequent conjugation to the carrier protein are achieved by conventional means. See, for example, [US patents are identified].
Carrier proteins are preferably proteins that are non-toxic and non-reactogenic and obtainable in sufficient amount and purity. Carrier proteins should be amenable to standard conjugation procedures. In a particular embodiment of the present invention, CRM197 is used as the carrier protein.
133 Other carrier proteins that the specification says can be used are then identified, including OMPC. The specification says that after the individual glycoconjugates are purified, they are compounded to formulate the immunogenic composition of the invention using “art-recognised methods”. It then describes the use of adjuvants. An “adjuvant” is defined as “a substance that serves to enhance the immunogenicity of an immunogenic composition of this invention” (page 13 line 5).
134 The specification then identifies in general terms dosing levels for the serotypes to be used in a vaccine. At page 16 reference is made to the use of additional antigens against otitis media caused by infection from other (non-Streptococcus pneumoniae) bacteria:
The compositions of this invention may further include one or more additional antigens for use against otitis media caused by infection with other bacteria. Such bacteria include nontypable Haemophilus influenza, Moraxella catarrhalis...and Alloiococcus otitidis.
...
The compositions of this invention may also include one or more proteins from Streptococcus pneumoniae.
135 The specification then notes that the disclosure given so far “generally describes the present invention” (page 17 line 1). A more complete understanding is said to be able to be obtained by reference to the examples which follow.
136 Examples 1, 3, 5, 7, 9, and 11 respectively refer to the preparation of serotypes 1, 3, 5, 6A, 7F and 19A, while examples 2, 4, 6, 8, 10 and 12 refer respectively to the preparation of the same serotypes but conjugated to CRM197. Example 13 refers to the preparation of each of serotypes 4, 6B, 9V, 14, 18C, 19F and 23F (that is, the Prevnar 7 serotypes) and example 14 refers to the preparation of CRM197 conjugates for each of those serotypes.
137 Detail is given in example 1 of the preparation of the master and working cell banks for serotype 1, the fermentation and harvesting required, the purification of the pneumococcal polysaccharide and then the characterisation of the serotype.
138 An issue arises in the context of the inventive step challenge concerning the method used in the specification to effect the conjugation of the 13 purified polysaccharides individually to CRM197. In this regard, notwithstanding the observations made in the introduction to the examples that the polysaccharides are prepared by “standard techniques” and that chemical activation and subsequent conjugation to the carrier protein are achieved by “conventional means”, particular ingredients, steps and ratios of polysaccharide to CRM197 are identified and explained in the specification. They are different for each serotype. I describe some of the differences below.
139 Example 2 addresses the preparation of the activation and conjugation of the purified polysaccharide for serotype 1. It provides that pH 9.0 was added for partial deacetylation for 3 hours at 50 degrees to a vessel containing the polysaccharide. The reaction was cooled to 20 degrees and neutralized. Oxidation in the presence of sodium periodate was performed by incubation at 2 – 8 degrees, and the mixture stirred for 15 – 21 hours.
140 The mixture was concentrated and diafiltered and the activated saccharide was filled into 100 mL glass lyophilisation bottles and “shell-frozen” at -75 degrees and lyophilized. The process of shell-freezing is described.
141 Bottles of lyophilized material were then brought to room temperature and resuspended in CRM197 solution at a saccharide/protein ratio of 2:1. 1M buffer was added at a nominated ionic strength and pH, and sodium cyanoborohydride was added. The reaction was incubated at 23 degrees for 18 hours, followed by a second incubation at 37 degrees for 72 hours. The mixture was diluted with cold saline followed by the addition of 1M of sodium carbonate to adjust the pH level. Unreacted aldehydes were quenched by addition of sodium borohydride at a specified temperature for 3 – 6 hours. The reaction mixture was then diluted with saline, diafiltered with phosphate buffer and saline and filtered again. It was diluted to a target of 0.5mg/mL in 0.9% saline and then filtered into final bulk concentrate and stored. It was then characterised.
142 In example 4, the preparation of the activation and conjugation of serotype 3 is described. The details of the process described are somewhat different compared with serotype 1. Containers of purified serotype were thawed and WFI and 2M of acetic acid added to a final concentration of 0.2M and 2mg/mL of saccharide. The temperature was raised to 85 degrees for one hour to hydrolyze the polysaccharide. The reaction was cooled to less than or equal to 25 degrees and 1M of magnesium chloride added to a final concentration of 0.1M. Oxidation in the presence of sodium periodate was performed for 16 – 24 hours at 23 degrees. The activation reaction mixture was concentrated and diafiltered with WFI and further filtered. For compounding, 0.2M of sodium phosphate, pH 7.0, was added to the activated saccharide into a final concentration of 10mM and a pH of 6.0 – 6.5. CRM197 was mixed with it to a ratio of 2g of saccharide per 1g CRM197. The combined solution was shell-frozen, then brought to room temperature and resuspended in 0.1M of sodium phosphate buffer of pH 7.0 to a final saccharide concentration of 20mg/mL. A molar equivalent of sodium cyanoborohydride was added. The reaction was incubated at 37 degrees for 48 hours. The reaction mixture was diluted, quenched, and then incubated at 23 degrees for 3 – 6 hours. It was then diafiltered and further filtered and then characterised.
143 In example 6 the preparation of the activation and conjugation serotype 5 is explained. The ration of activated serotype to CRM197 is 0.8:1. The concentrations, pH levels and steps are different to those described for serotypes 1 and 3.
144 In example 8 the preparation of the activation and conjugation of serotype 6A is described. It commences by noting that the serotype 6A polysaccharide is a high molecular weight polymer that had to be reduced in size prior to oxidation, a step not taken in the earlier examples. The steps and concentrations used are also different. The saccharide/protein ratio is also different, being 1:1.
145 Similar differences in the method of activation and conjugation may be perceived from the other examples.
146 Example 15 is entitled “Formulation of a multivalent Pneumococcal Conjugate Vaccine” and describes the formulation of the final vaccine. The final bulk concentrates of the 13 chosen serotypes are prepared in a manner set out in some detail.
147 Example 16 provides results of experiments concerning the immunogenicity of the 13-valent conjugate vaccine. It states that to date, the preclinical studies performed on the 13vPnC vaccine have been in rabbits. The results were characterised by antigen-specific ELISA for serum IgG concentrations and for antibody function by OPA. Study #HT01-0021 examines the ability of the 13vPnC vaccine with AIPO4 adjuvant to elicit vaccine serotype-specific immune responses.
148 In relation to study #HT01-0021, the results for functional responses were assessed in rabbits following immunisation with two 13vPnC formulations, one with AIPO4 adjuvant and the other without the adjuvant, but both with the 13 chosen serotypes. Secondary objectives included an evaluation of the kinetics and duration of the antibody response. When comparing vaccine formulations, the one with the adjuvant had higher OPA geometric mean titers (GMT). For the majority of the serotypes, OPA titers measured at week 4 were at least four times higher than those at the baseline week zero. The kinetic responses were also evaluated for each of the serotypes from serum pools of both treatment groups. The specification reports that with the exception of serotype 1, antibody responses were superior for rabbits receiving the vaccine with adjuvant added. It says (page 44 lines 4 – 9):
Overall, the data indicate that the 13vPnC vaccine formulated with aluminium phosphate is immunogenic in rabbits, eliciting substantial antibody responses to the pneumococcal capsular polysaccharide contained in the vaccine and these responses are associated with functional activity. The responses observed to the seven core serotypes following immunization with 13vPnC + AIPO4 are consistent with historical responses of rabbits to the heptavalent formulation.
149 In this passage the seven “core” serotypes are the Prevnar 7 serotypes which make up the heptavalent formulation mentioned in the final line of the passage.
150 Table 4 then sets out the results of 13vPnC with and without AIPO4 as an adjuvant.
151 Study #HT01-0036 is described as comparing rabbit immune responses to the polysaccharides contained in the vaccine after immunisation with the 13vPnC vaccine, and with or without conjugation to the CRM197 protein. Immune responses were evaluated using an IgG ELISA and complement-mediated OPA measuring functional antibody. The data is reported to indicate that the conjugate vaccine elicited higher serum IgG titers than free polysaccharide or free polysaccharide mixed with unconjugated CRM197 vaccine. The specification states (page 46 line 21 – page 47 line 2):
With the exception of S. pneumoniae type 14, the 13vPnC vaccine was able to induce functional antibodies to the representative strains of S. pneumoniae in an OPA (Table 6).
152 MSD relies on this statement, and the results reported in Table 6, in its inutility challenge.
153 The specification then makes four general statements to which the parties refer in their submissions. The first is to the effect that the foregoing discussion and examples are merely present to provide a detailed description of certain embodiments. The second is that all journal articles, other references, patents and patent applications that are identified in the 013 patent are incorporated by reference in their entirety. In this regard, the specification lists 35 references, some of which are identified by number in passages quoted above. The third general statement may be regarded as a boilerplate reference, but is nevertheless important in the context of the construction arguments. I refer to it below as the comprising passage. It is as follows:
Throughout this specification and the claims which follow, unless the context requires otherwise, the word “comprise”, and variations such as “comprises” and “comprising”, will be understood to imply the inclusion of a stated integer or step or group of integers or steps but not the exclusion of any other integer or step or group of integers or steps.
154 Finally, the specification states that the reference in the specification to any prior publication or information derived from a publication, or to any matter which is known, is not and should not be taken as an acknowledgement or admission that such publication or information forms part of the common general knowledge in the field.
4.2 The claims of the 013 patent
155 The asserted claims are as follows:
1. A multivalent immunogenic composition, comprising: 13 distinct polysaccharide-protein conjugates, together with a physiologically acceptable vehicle, wherein each of the conjugates comprises a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, and the capsular polysaccharides are prepared from serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F and 23F and wherein said carrier protein is CRM197.
2. The immunogenic composition of claim 1, further comprising an adjuvant.
3. The immunogenic composition claim 2, wherein the adjuvant is an aluminum-based adjuvant.
4. The immunogenic composition of claim 3, wherein the adjuvant is selected from the group consisting of aluminum phosphate, aluminum sulfate and aluminum hydroxide.
5. The immunogenic composition of claim 4, wherein the adjuvant is aluminum phosphate.
6. The immunogenic composition of any one of claims 1 to 5 comprising 1 to 5 μg of each polysaccharide.
7. The immunogenic composition of any one of claims 1 to 5 comprising 2 μg of each polysaccharide, except for 6B at 4 μg.
8. The immunogenic composition of claim 6 or 7 comprising 0.125 mg of elemental aluminum (0.5 mg aluminum phosphate) adjuvant.
9. The immunogenic composition of any one of claims 1 to 5 wherein each 0.5 mL dose is formulated to contain: 2 μg of each polysaccharide, except for 6B at 4 μg; approximately 29 μg CRM197 carrier protein; 0.125 mg of elemental aluminum (0.5 mg aluminum phosphate) adjuvant; and sodium chloride and sodium succinate buffer as excipients.
10. A method of inducing an immune response to a Streptococcus pneumoniae capsular polysaccharide conjugate, comprising administering to a human an immunologically effective amount of the immunogenic composition of any one of claims 1 to 9.
11. A sterile liquid formulation comprising pneumococcal capsular polysaccharides of serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F individually conjugated to CRM197.
12. The sterile liquid formulation of claim 11 comprising 1 to 5 μg of each polysaccharide.
13. The sterile liquid formulation of claim 11 comprising 2 μg of each polysaccharide, except for 6B at 4 μg.
14. A sterile liquid formulation of pneumococcal capsular polysaccharides of serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F individually conjugated to CRM197 wherein each 0.5 mL dose is formulated to contain: 2 μg of each polysaccharide, except for 6B at 4 μg; approximately 29 μg CRM197 carrier protein; 0.125 mg of elemental aluminum (0.5 mg aluminum phosphate) adjuvant; and sodium chloride and sodium succinate buffer as excipients.
15. A sterile liquid formulation of pneumococcal capsular polysaccharides of serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F individually conjugated to CRM197 wherein each 0.5 mL dose is formulated to contain: 2.2 μg of each polysaccharide, except for 6B at 4.4 μg; approximately 29 μg CRM197 carrier protein; 0.125 mg of elemental aluminum (0.5 mg aluminum phosphate) adjuvant; and sodium chloride and sodium succinate buffer as excipients.
16. A method of protecting a human susceptible to pneumococcal infection, by means of administering the composition of anyone of claims 1 to 9 or 11 to 15 via a systemic route.
17. The method of claim 16 wherein said administration is via the intramuscular, route.
18. The immunogenic composition of claim l and the sterile liquid formulation of any one of claims 11, 14 and 15, substantially as hereinbefore described and with reference to any of the Examples and/or figures.
4.3 The specification and claims of the 844 patent
156 The specification of the 844 patent is substantially the same as that of the 013 patent. No party relies on any difference in the text of the 844 patent for the purpose of considering its disclosure.
157 The 844 claims are as follows:
1. A multivalent immunogenic composition, comprising polysaccharide-protein conjugates together with a physiologically acceptable vehicle, wherein each of the conjugates comprises a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, and the capsular polysaccharides are prepared from serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F and 23F, wherein the carrier protein is CRM197 for use as a vaccine to protect or treat a human susceptible to pneumococcal infection.
2. The immunogenic composition of claims 1, wherein conjugation is effected by reductive amination.
3. The immunogenic composition of claim 1 or claim 2, further comprising an adjuvant.
4. The immunogenic composition of claim 3, wherein the adjuvant is an aluminum-based adjuvant.
5. The immunogenic composition of claim 5, wherein the adjuvant is selected from the group consisting of aluminium phosphate, aluminium sulfate and aluminium hydroxide.
6. The immunogenic composition of claim 4, wherein the adjuvant is aluminium phosphate.
7. The immunogenic composition according to any one of claims 1 to 6 which further comprises one or more antigens for use against otitis media caused by infection with other bacteria.
8. The immunogenic composition of claim 7 wherein said other bacteria is selected from the group consisting of nontypable Haemophilus influenza, Moraxella catarrhalis and Alloiococcus otitidis.
9. The immunogenic composition of any one of claims 1 to 8 which further comprises one or more proteins from Streptococcus pneumoniae.
10. The immunogenic composition of any one of claims 1 to 9 which further comprises one or more proteins from Neisseria meningitidis type B.
11. The immunogenic composition of any one of claims 1 to 10 for use in vaccination wherein following an initial vaccination, subjects receive one or several booster immunizations adequately spaced.
12. The immunogenic composition of any one of claims 1 to 10 for use in the vaccination of infants or toddlers, wherein the vaccination schedule is 2, 4, 6 and 12-15 months of age.
13. The immunogenic composition of any one of claims 1 to 10 for use in the vaccination of adolescents or adults.
14. The immunogenic composition of any one of claims 1 to 10 for intramuscular administration.
158 The invention described and claimed in the specification may be broadly summarised as the idea to add serotypes 1, 3, 5, 6A, 7F and 19A to the Prevnar 7 serotypes and to conjugate each to CRM197 to yield an immunogenic composition, coupled with the disclosure of the practical means by which this is achieved.
159 The specification and claims are directed to pharmaceuticals products for the purpose of achieving vaccines.
4.5 The person skilled in the art
160 As I have noted, the composition patents are entitled “Multivalent pneumococcal polysaccharide-protein conjugate composition”. The invention is described to relate generally to the field of medicine, and in particular to microbiology, immunology, vaccines and the prevention of infection by a bacterial pathogen by immunisation. However, the particular focus of the invention described in the specification is the development and use of multivalent pneumococcal polysaccharide conjugate vaccines. The person skilled in the art is likely to have a practical interest in this subject matter and this is the field of the invention.
161 Persons with a practical interest in the subject matter of the invention will include microbiologists and immunologists. There is no real dispute that all of the expert witnesses called in the present have such an interest. The parties agree that for the purposes of considering the question of inventive step within s 7(2) the hypothetical person skilled in the art will be a team. They agree that the team will include persons skilled in the disciplines of microbiology and immunology. MSD contends that such persons will preferably have experience working with pneumococcal vaccines. Wyeth contends that such persons will have experience in particular with encapsulated bacteria. The divide reflects the differing levels of experience between Professors Kasper and Paton on the one hand, whose experience before the priority date was substantially in working with pneumococcal vaccines, and the experience of Professor Strugnell, whose experience was in encapsulated bacteria more generally, but mostly not in pneumococcal vaccines. In my view all of these experts are qualified to assist the Court in ascertaining the approach of the hypothetical skilled team. I refer below, where necessary, to the effect of the relatively lesser experience of Professor Strugnell in the central aspects of the subject matter of the inventive step enquiry, and make allowance for it.
162 There was some debate as to whether an epidemiologist would also participate in the team and whether a person in that discipline would be a clinician. Having regard to the disclosure of the specification, in my view there is little doubt that a person skilled in reviewing data about demographics and populations at risk would provide the scientific members of the team with what is expected from the improved vaccine. This was the view of Professors Kasper, Strugnell and Dagan, with whom I agree. In my view all of the experts were sufficiently experienced and qualified to opine on this subject. Professor Dagan, as a clinician whose experience is primarily in relation to clinical matters, is in a lesser position to give authoritative evidence about other aspects of vaccine development.
163 Where I refer below to the skilled team I refer to persons with the particular characteristics in the two above paragraphs.
5. COMPOSITION PATENTS: CONSTRUCTION ISSUES
164 The following issues arise between the parties in relation to the construction of terms in the composition patents:
(1) Whether the asserted composition patent claims include within their scope one or more Streptococcus pneumoniae serotypes in addition to the 13 serotypes. This focusses attention on the meaning of the words “comprises” and “comprising” where they appear in the asserted claims (the comprising issue).
(2) What is the proper construction of “immunogenic” as it appears in claim 1 of the 013 patent?
5.1 The principles of patent construction
165 The principles of construction are not in dispute. It is for the Court to determine and characterise the invention having regard to the principles of construction that are now well settled. Many are summarised in Jupiters Ltd v Neurizon Pty Ltd [2005] FCAFC 90; 65 IPR 86 (Hill, Finn and Gyles JJ) at [67]:
There is no real dispute between the parties as to the principles of construction to be applied in this matter although there is some difference in emphasis. It suffices for present purposes to refer to the following:
(i) the proper construction of a specification is a matter of law: Décor Corp Pty Ltd v Dart Industries Inc (1988) 13 IPR 385 at 400;
(ii) a patent specification should be given a purposive, not a purely literal, construction: Flexible Steel Lacing Company v Beltreco Ltd (2000) 49 IPR 331 at [81]; and it is not to be read in the abstract but is to be construed in the light of the common general knowledge and the art before the priority date: Kimberley-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [24];
(iii) the words used in a specification are to be given the meaning which the normal person skilled in the art would attach to them, having regard to his or her own general knowledge and to what is disclosed in the body of the specification: Décor Corp Pty Ltd at 391;
(iv) while the claims are to be construed in the context of the specification as a whole, it is not legitimate to narrow or expand the boundaries of monopoly as fixed by the words of a claim by adding to those words glosses drawn from other parts of the specification, although terms in the claim which are unclear may be defined by reference to the body of the specification: Kimberley-Clark v Arico at [15]; Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610; Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 at 478; the body of a specification cannot be used to change a clear claim for one subject matter into a claim for another and different subject matter: Electric & Musical Industries Ltd v Lissen Ltd [1938] 56 RPC 23 at 39;
(v) experts can give evidence on the meaning which those skilled in the art would give to technical or scientific terms and phrases and on unusual or special meanings to be given by skilled addressees to words which might otherwise bear their ordinary meaning: Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479 at 485-486; the Court is to place itself in the position of some person acquainted with the surrounding circumstances as to the state of the art and manufacture at the time (Kimberley-Clark v Arico at [24]); and
(vi) it is for the Court, not for any witness however expert, to construe the specification; Sartas No 1 Pty Ltd, at 485–486.
166 It is important to emphasise the need to read a patent specification as a whole and in the light of the common general knowledge and to give it a practical and common sense construction that is “purposive”. Such an approach requires the Court to read the specification through the eyes of the skilled addressee, with practical knowledge and experience in the field of work in which the invention was intended to be used, and a proper understanding of the purpose of the invention: GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Limited v Generic Partners Pty Limited [2018] FCAFC 71; 264 FCR 474 at [106] (Middleton, Nicholas and Burley JJ).
167 As Lord Hoffmann explained in Kirin-Amgen Inc v Hoechst Marion Roussel Ltd [2004] UKHL 46; [2005] 1 All E.R. 667 at [34]:
“Purposive construction” does not mean that one is extending or going beyond the definition of the technical matter for which the patentee seeks protection in the claims. The question is always what the person skilled in the art would have understood the patentee to be using the language of the claim to mean. And for this purpose, the language he has chosen is usually of critical importance. The conventions of word meaning and syntax enable us to express our meanings with great accuracy and subtlety and the skilled man will ordinarily assume that the patentee has chosen his language accordingly. As a number of judges have pointed out, the specification is a unilateral document in words of the patentee’s own choosing. Furthermore, the words will usually have been chosen upon skilled advice. The specification is not a document inter rusticos for which broad allowances must be made. On the other hand, it must be recognised that the patentee is trying to describe something which, at any rate in his opinion, is new; which has not existed before and of which there may be no generally accepted definition. There will be occasions upon which it will be obvious to the skilled man that the patentee must in some respect have departed from conventional use of language or included in his description of the invention some element which he did not mean to be essential. But one would not expect that to happen very often.
168 Lord Hoffmann was referring here to the meaning conveyed to the skilled addressee by the language used and was not directing himself to a situation in which the skilled addressee deduced that the language of the claim, although conveying to him or her a particular meaning, could never have been intended to mean what it conveyed.
169 Wyeth submits that the position is straightforward. It contends that “comprising” in the claims, read in the context of the specification, is used in an inclusive sense (“including”), as opposed to an exhaustive sense (“consisting of”). This is said to accord with the comprising passage set out at [153]. It submits that MSD has not established otherwise, and therefore it has not rebutted that express presumption. Due to this inclusive construction, each of the asserted composition patent claims includes within its scope any immunogenic composition which has the integers of the claim, including polysaccharide-protein conjugate that includes each of the chosen 13 serotypes, notwithstanding such an immunogenic composition may also contain other integers, such as polysaccharide-protein conjugates of one or more Streptococcus pneumoniae serotypes in addition to the 13 chosen serotypes.
170 The primary construction advanced by MSD is that the claimed compositions are only to polysaccharide-protein conjugates of each of the chosen 13 serotypes and do not encompass immunogenic compositions which also have conjugates from any other Streptococcus pneumoniae serotypes.
171 MSD submits that the language used in the claim mandates the conclusion that the asserted claims are limited to a composition with polysaccharide-protein conjugates from the chosen 13 serotypes. MSD submits that its construction is the only one that is consistent with the description in the specification of the “invention”. It submits that the language of each of the independent claims makes clear that they are limited to the 13 serotypes specified and that where, as here, one or more things in a class are expressly mentioned, other serotypes are excluded.
172 Furthermore, MSD submits that the use of the words “comprising” and its accompanying definition in the specification do not assist Wyeth. MSD submits that the comprising passage requires the word “comprises” in the third line of claim 1 to be understood in an exhaustive sense, unlike its counterpart “comprising”, because of various contextual matters, an understanding of the disclosure of the specification as a whole, the language of the consistory clause for claim 1 and the language of the dependent claims.
173 MSD additionally submits that the construction that it proposes yields a common sense and practical result. By the priority date of April 2005 around 90 different serotypes had been described and since then that number has risen to 98. MSD submits that if Wyeth’s construction is accepted then the claims will necessarily extend to an immunogenic composition of 90 or more serotypes and could cover millions of different combinations. Such a construction would be untenable and the Court would not interpret a claim in that way: Abbott Laboratories v Corbridge Group Pty Ltd [2002] FCAFC 314; 57 IPR 432 at [30] – [35] (Lee, Emmett and Hely JJ). Furthermore, such an invention may not work, because MSD submits it was common general knowledge as at April 2005 that such a large increase in the number of serotypes would require a greater amount of carrier protein to be included in the vaccine. As the amount increases, there is a risk that this will impede the operation of the vaccine by causing aggregation and/or leading to adverse reactions. It may also lead to immune interference, if, contrary to MSD’s contention, that was a realistic concern to the person skilled in the art.
174 Finally, MSD submits that the position taken by Wyeth in advancing its present construction argument is contrary to that which it adopted in correspondence with the Commissioner of Patents, where in a letter dated 20 April 2016 its patent attorneys stated that the claimed composition “is 13-valent” without any suggestion that the claimed invention covers a valency of 13 or more.
175 I first address claim 1 of the 013 patent, upon which the parties focussed in the course of argument. It is repeated below, with integer numbers added for convenience:
(1) A multivalent immunogenic composition, comprising:
(2) 13 distinct polysaccharide-protein conjugates,
(3) together with a physiologically acceptable vehicle,
(4) wherein each of the conjugates comprises a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, and the capsular polysaccharides are prepared from [the chosen 13 serotypes] and
(5) wherein said carrier protein is CRM197.
176 It may be seen that claim 1 is for a multivalent immunogenic composition (integer (1)) with three constituents. The first is 13 distinct polysaccharides (integer (2)), each of which comprises a capsular polysaccharide from a different serotype conjugated to a carrier protein (integer 4). The second is a physiologically acceptable vehicle (integer (3)). The third is the carrier protein CRM197 (integer (5)).
177 MSD contends that claim 1 is limited to a multivalent immunogenic composition with polysaccharide-protein conjugates from the 13 chosen serotypes in it. Put another way, it submits that any combination that includes more than the nominated 13 serotypes cannot infringe. Wyeth submits that any multivalent immunogenic composition that has integers (2) – (5) within it will infringe, even if additional components are included.
178 The question is to be resolved by consideration first of the language used in the claim. In this regard there is no ambiguity. The words “comprising” and “comprises” are clearly defined in the specification in an inclusive sense, or in other words, “including” and “includes”. The only exception is whether the context requires otherwise.
179 There can be no doubt that where in integer (1) the claim refers to “[a] multivalent immunogenic composition comprising...” the word “comprising” means “including”. If the definition of the term “comprising” in the body of the specification is not sufficient to establish this point, as much is confirmed as a matter of context, when one considers the claims dependent upon claim 1. For instance, in accordance with orthodox claim drafting, claim 9, which narrows the scope of claim 1, identifies amongst other things additional excipients to be included in the form of the excipients sodium chloride and sodium succinate buffer. If “comprising” was not inclusive, then claim 9 would make little sense.
180 Accordingly, the scope of claim 1 is apparently defined by reference to what the multivalent immunogenic composition under consideration for the purposes of infringement includes, not by reference to what it consists of. If it includes all of the elements of integers (2) – (5), then it is likely to infringe, even if it also contains things additional to the elements so identified (I say “likely” because that is subject to exceptions not presently relevant, such as whether or not the allegedly infringing composition is properly characterised as a different combination of elements: see Fresenius Medical Care Australia Pty Limited v Gambro Pty Limited [2005] FCAFC 220; 224 ALR 168 at [70] and [92] (Wilcox, Branson and Bennett JJ), an approach that MSD eschews (transcript 1193)).
181 The question then posed by MSD’s submissions, is whether any aspect of the language of the claim supports a construction that the composition is limited to containing the 13 polysaccharide-protein conjugates nominated in integer (4).
182 MSD does not provide a satisfactory answer to this question.
183 First, it submits that a proper understanding of the “invention” as described in the specification compels this conclusion. I shall return to this argument, but it does not grapple with the question of construction of the words used in the claim. It is trite to observe that the monopoly is set out by the patentee in words of its own choosing. It is those words that must first be construed. Contrary to the suggestion made by MSD, I do not understand Welch Perrin & Co Pty Ltd v Worrel [1961] HCA 91; 106 CLR 588 at 609 – 617 (Dixon CJ, Kitto and Windeyer JJ) to suggest otherwise.
184 Secondly, MSD submits claim 1 is expressly limited to the 13 chosen serotypes by the inclusion in integer (4) of the words “... are prepared from [the chosen 13 serotypes]”, which must be understood to mean that the polysaccharide-protein conjugates must be prepared from those 13 serotypes, and no others. However, while there is no doubt that to be within the scope of the claim the 13 chosen serotypes must be present, the question is whether any language precludes the inclusion of further serotypes, having regard to the conceded meaning of the word “comprising” in the first line of claim 1. The words singled out in MSD’s submission do not advance the debate. Nor do I consider that reliance on the maxim expressio unius exclusio alterius (“the express inclusion of one thing is the exclusion of another”) assists, for much the same reason.
185 Thirdly, MSD relies on various aspects of the claim set, when read as a whole, to influence the construction of claim 1. None of these matters address the real issue of the language used in the claim itself. In one such argument MSD contends that because all of the independent claims identify a specific number of polysaccharide-protein conjugates to be included, it would be “absurd” for the claim to be construed as applicable to a variable number of serotypes, extending potentially to 90 or more serotypes. However, the question at this point is what the claim under consideration means. If it is ambiguous, a construction that is not absurd might be chosen, but the first question is to identify ambiguity. This argument does not do so.
186 In another contention MSD observes that there is no dependent claim that expressly allows for any further serotypes, such that it may be assumed that the inclusive meaning of the word “comprising” does not apply to additional serotypes. However, this point does not address the breadth of the meaning of “comprising” having regard to the definition in the specification.
187 In another argument, MSD states that if Wyeth had wanted to claim immunogenic compositions with more than the 13 chosen serotypes, it could have done so by using the words “at least” before listing the serotypes”, but it did not. In this regard the claims must be understood on the basis of the proposition that what is not claimed is disclaimed (citing Nichia Corporation v Arrow Electronics Australia Pty Ltd [2019] FCAFC 2 at [48] – [49] (Jagot J, with whom Besanko and Nicholas JJ agreed)). However, one is here fixed with the task of construing the particular claim before the Court – it is of no particular assistance to cast about wistfully for other words that were not chosen.
188 Perhaps MSD’s most persuasive argument is that whereas the word “comprising” in the first line of claim 1 should be construed as meaning “including”, the word ”comprises” in the third line of claim 1 should be understood to mean “consists of” and accordingly be exhaustive of the serotypes included. In this regard, MSD emphasises that the body of the specification is directed towards an invention that specifically identifies and nominates the 13 chosen serotypes. By identifying in integer (4) that each of the conjugates comprises a particular capsular polysaccharide, the patentee is making express that the composition must only include those 13 polysaccharide-protein conjugates.
189 However, the difficulty with this argument is that the context does not “require” such a construction. Indeed the comprising passage makes plain that there is a bias towards reading the word as meaning “includes”. In my view the words emphasised above do not do so. Furthermore, it is unlikely that the same root word used twice in the same claim would be construed to have different meanings.
190 In this regard MSD places reliance on the decision of the Full Court in Actavis Pty Ltd v Orion Corporation [2016] FCAFC 121 (Allsop CJ, Nicholas and Yates JJ), where the Full Court overturned the decision at first instance insofar as it construed the word “comprise” and its variants to contort the claimed process into one different to that described in the specification (at [176] – [181]). However, I do not consider that the present position is relevantly similar to that which was considered by the Full Court in Actavis, which concerned the construction of a method claim in quite different circumstances. The position is perhaps more analogous to that which was considered by the Full Court in Bitech Engineering v Garth Living Pty Ltd [2010] FCAFC 75; 86 IPR 468 (Sundberg, Bennett and Yates JJ) where an apparatus claim also included the word “comprising” (at [6]). There, the Court found that the primary judge fell into error by failing to conclude that because the impugned device contained all of the integers of the claim, the device infringed the claim (at [26]). The Court found that the claims in that case did not expressly or impliedly exclude the presence of features additional to those identified in the claim, and so long as an apparatus possessed the features of the claim, it would infringe the patent: see [27] – [29] and [32].
191 Accordingly, in my view the language of claim 1 is to be understood to have the meaning contended for by Wyeth. I do not consider that this language is ambiguous having regard to the definition in the specification.
192 However, a patent specification should be given a purposive, not a purely literal, construction: Flexible Steel Lacing Co v Beltreco Ltd [2000] FCA 890; 49 IPR 331 at [81] (Hely J); and it is not to be read in the abstract but is to be construed in the light of the common general knowledge and the art before the priority date: Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd [2001] HCA 8; 207 CLR 1 at [24] (Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ); Neurizon at [67]. Accordingly, I now turn to the specification to ascertain whether it mandates a different outcome.
193 I have in section 4.1 referred to the disclosure of the specification in the 013 patent. In the Background of the Invention the patentee refers to Prevnar 7, and to surveillance reported in various publications of the effects on invasive pneumococcal disease before and after the introduction of Prevnar 7, to demonstrate that adding serotypes 1, 3, 5, 6A, 7F and 19A to the Prevnar 7 serotypes “would increase coverage for invasive disease” in the US, Europe, Asia and Latin America, significantly expand coverage beyond that of Prevnar 7, and provide coverage for serotypes 6A and 19A that is not dependent on the limitations of cross-protection.
194 The Summary of the Invention states that the invention provides generally a multivalent composition comprising 13 serotypes, each conjugated to a carrier protein, with a physiologically acceptable vehicle and optionally with an adjuvant (page 2 lines 20 – 23):
More specifically, the invention provides a 13-valent pneumococcal conjugate (13vPnC) composition comprising the seven serotypes in the 7vPnC vaccine...plus six additional serotypes...
195 The Summary of the Invention also refers to the invention providing a multivalent composition comprising polysaccharide-protein conjugates, together with a physiologically acceptable vehicle, with each conjugate comprising a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, and the capsular polysaccharides being “prepared from serotype 3 and at least one additional serotype”. No maximum number of potential serotypes is given. Another embodiment identifies that the additional serotype is selected from one of the 13 chosen serotypes (other than serotype 3).
196 The Summary of the Invention refers to another embodiment, being a multivalent composition comprising polysaccharide-protein conjugates, together with a physiologically acceptable vehicle, with each conjugate comprising a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, and the capsular polysaccharides are prepared from the Prevnar 7 serotypes and at least one additional serotype. Another embodiment identifies that the additional serotype is selected from the group consisting of 1, 3, 5, 6A, 7F and 19A.
197 The Detailed Description of the Invention identifies the benefits, apparently based on cited articles cited in the specification, of inclusion of each of the chosen serotypes conjugated to a carrier protein. On page 11 at lines 1 – 6 the specification reinforces the focus on the inclusion of the chosen 13 serotypes by stating:
Accordingly, the present invention provides a multivalent immunogenic composition comprising 13 distinct polysaccharide-protein conjugates, wherein each of the conjugates contains a different capsular polysaccharide conjugated to a carrier protein [prepared from the chosen 13 serotypes] together with a physiologically acceptable vehicle...
198 The examples address various matters including the preparation of the chosen 13 serotypes and CRM197. The experiments address their efficacy in certain environments.
199 There can be little doubt that the invention disclosed and described in the specification is for a combination that involves a multivalent immunogenic composition of the 13 chosen serotypes conjugated to CRM197 in combination with a physiologically acceptable vehicle and optionally an adjuvant. The invention as disclosed is the careful nomination of six serotypes in addition to the Prevnar 7 serotypes that have been used in a multivalent immunogenic composition as claimed. That is the focus of the passages in the Summary of the Invention and also the Detailed Description and the examples. However, in my view nothing within the body of the specification indicates that a composition that includes those serotypes and also other matters, including further serotypes conjugated to CRM197, could not fall within the scope of the invention described.
200 Accordingly, the view that I have taken as to the construction of the claims, having regard to the inclusive meaning of “comprises”, is not in discord with the disclosure of the specification as a whole.
201 At this point I note that the language of the second independent claim, claim 11 of the 013 patent, is less favourable to MSD. It is (emphasis added):
A sterile liquid formulation comprising pneumococcal capsular polysaccharides of serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F individually conjugated to CRM197.
202 In submissions MSD emphasises that the formulation is of serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F individually conjugated to CRM197 and submits that as a matter of construction the formulation must be only of those serotypes. However, having regard to the construction to be given to “comprising” with respect to claim 1, I cannot see how the context could be understood as requiring that comprising be given an exhaustive meaning in this claim.
203 Finally, MSD submits that Wyeth adopted a different position as to the construction of the claims in its correspondence with the Commissioner of Patents to that which it now asserts. It submits that Wyeth’s willingness to alter its position on the construction of the composition patents, as suits its need, does not reflect well on Wyeth and the cogency of the construction in these proceedings. I return to this subject in the context of allegations of false suggestion, in section 13.4 below. It suffices to say for present purposes that, as the authorities note, the question of construction is a matter of law which is for the Court to determine. The submissions made by the patent attorneys representing Wyeth in the course of prosecution do not preclude the Court from arriving at the correct construction.
5.2.3 Infringement of the asserted composition patent claims
204 I have resolved the comprising issue in favour of Wyeth. The parties agree that the consequence of this is that MSD’s 15-valent vaccine that it proposes to launch will infringe the asserted composition patent claims.
5.3 The meaning of “immunogenic”
205 The term “immunogenic” appears in claim 1 of the each of the composition patents. MSD contends that it means that the composition elicits an immune response, but at no particular level. Wyeth contends that the term appears in both claims as part of a composite expression “A multivalent immunogenic composition...” It contends the phrase means that the composition must be capable of being administered with the intention that it will elicit an adaptive immune response that is effective against disease or carriage of each of the 13 chosen serotypes (that is, a protective response), drawing on the meaning of the word “multivalent”.
206 I prefer the construction offered by MSD.
207 The word “immunogenic” is the adjective of that noun “immunity” and is defined to mean “causing immunity”: Macquarie Dictionary (Rev. 3rd ed, 2001, Sydney). “Immune” is defined in the Macquarie Dictionary to mean “protected from a disease or the like, as by inoculation”. However, it is apparent from the expert evidence that those in the art understand that there may be levels of immunity that fall short of protective immunity. In the light of the expert evidence, immunogenic is used in the composition patents as a relative term. In his oral evidence Professor Dagan said that he did not understand claim 1 to require a particular level of immunogenicity. In his affidavit evidence he gives evidence that an “immunogenic” composition according to claim 1 may not result in protective immunity in the recipient. Protective immunity is the ability of the immune system to protect the host against future infections by the same pathogen. As Professor Paton explains, it results from the body’s learned response to pathogens through acquired memory of specific antigens, resulting in adaptive immunity. He understands that there may be a level of immunity that falls short of adaptive immunity, and that claim 1 refers to a composition that produces an immune response, which may be short of an adaptive immune response.
208 In my view the construction propounded by MSD is also supported by the language in the specification, which discusses relative levels of immunity. In the Background of the Invention, Prevnar 7 is referred to as the first pneumococcal conjugate vaccine of its kind to be “highly immunogenic and effective against invasive disease and otitis media...” (page 1 line 15 – 17), implying that a lower level of immunogenicity may be ineffective against disease. In the Summary of the Invention a method is identified for inducing an “immune response”, comprising administering to a human “an immunologically effective amount of any of the immunogenic compositions just described”, which includes the composition of claim 1 (page 4 lines 1 – 4). Such language distinguishes an immunologically effective composition from a merely immunogenic composition. Later, in the Detailed Description of the Invention, a pneumococcal conjugate vaccine comprising capsular polysaccharides from serotype 3 is said to be “capable of eliciting an immunogenic response to serotype 3 polysaccharides” (page 6 line 19 – 23). The efficacy of the immunogenic response is left open; it may or may not be protective.
209 Finally, it may be noted that claim 10 in the 013 patent refers directly to a method of inducing an immune response by administering an “immunologically effective amount of the immunogenic composition” of any of claims 1 – 9, thereby distinguishing between simply immunogenic on the one hand and immunologically effective on the other. As I have noted, that distinction is also made in the body of the specification at page 4 lines 1 – 4.
210 Contrary to the submission advanced by Wyeth, I am not persuaded that the definition of “multivalent” operates to require that the immunogenic composition elicit an adaptive immune response that is effective against disease or the carriage of disease of each of the named serotypes. In the context of the phrase “[a] multivalent immunogenic composition”, the word “multivalent” refers to the fact that more than one serotype is in the composition.
6. COMPOSITION PATENTS: LACK OF NOVELTY
211 Under each of the pre-RTB and post-RTB versions of the Patents Act, an invention is taken to be novel when compared to the prior art base unless it is not novel in light of, amongst other things, prior art information made publicly available in a single document pursuant to s 7(1)(a), or prior art information made publicly available in two or more related documents, if the relationship between the documents is such that a person skilled in the art would treat them as a single source of that information pursuant to s 7(1)(b).
212 MSD pleads that the invention claimed in the composition patents was not novel as at the priority date of 8 April 2005 when compared with the prior art base which, for the purpose of s 18(1)(b)(i) of the Patents Act, was:
(1) Peña; and
(2) Peña considered together with Obaro.
213 There is no dispute that Peña is prior art information made publicly available in a single document within s 7(1)(a) of the Patents Act. Nor is there any dispute that Peña and Obara together constitute prior art information made publicly available in two or more related documents such that a person skilled in the relevant art would treat them as a single source of that information within s 7(1)(b) of the Patents Act.
214 There was a controversy at trial about the translation of Peña from Spanish to English, which was resolved by the provision of a substitute translation, to which reference is made below. In English, the title to Peña is “Present and future of the anti-pneumococcal vaccination”. Beneath the title, the authors are identified in conjunction with the words “Commercial Department of Wyeth Farma, S.A. Madrid (Spain)”.
215 The Summary in the article provides:
Pneumococcal infections are a significant cause of morbidity, hospitalization and mortality worldwide; are one of the ten main causes of mortality; and represent 40% of pneumonia deaths in subjects less than five years of age. Universal vaccination would have a significant impact on the community. Currently, there are two available vaccines to prevent invasive pneumococcal illness in Spain: 23-valent polysaccharides (VNP-23V) and 7-valent conjugated (VNC-7V).
There are other conjugated vaccines for 9, 11 and 13 serotypes, although they have not yet been marketed and are in a very advanced study phase.
216 Under the heading “Vaccination”, the article identifies details of the 23-valent pneumococcal capsular polysaccharide vaccine and then says:
The 7-valent pneumococcal conjugate vaccine contains the purified saccharides of the capsular antigens of seven serotypes of Streptococcus pneumoniae (4, 6B, 9V, 14, 18C, 19F and 23F) conjugated individually with a protein, a nontoxic mutant of the diphtheria toxin, CRM197, and forming [sic: forms] glycoconjugates.
217 There is no doubt that the 7-valent vaccine so identified is Prevnar 7.
218 After addressing matters concerning the 23-valent polysaccharide vaccine and Prevnar 7, the following passage appears:
Other pneumococcal vaccines
As we know, there are currently two vaccines available for the prevention of invasive pneumococcal disease: the 23-valent polysaccharide (VNP-23V) and the 7-valent conjugate vaccine (VNC-7V).
There are other pneumococcal conjugate vaccines that have not yet been marketed and that are in advanced phases of study:
- The vaccine of 9- serotypes (adds 1 and 5), which increases the coverage up to 87% in children less than two years of age and in children between two and five years of age.
- Of 11- serotypes (adds 3 and 7F). Serotype 3 is the most likely to cause invasive disease in adults in Spain; therefore, the use of these vaccines could have a favorable impact on the incidence of the infection by this serotype.
- Of 13- serotypes (adds 6A and 19A).
219 Immediately following is a commentary on the 9-valent vaccine which refers to several studies conducted with that vaccine that assessed its safety and immunogenicity. Reference is made to the 9-valent vaccine being administered simultaneously with a DTP vaccine, but in separate syringes, to Gambian children. A footnote then refers the reader to Obaro. There is no dispute that a skilled worker in the field would have concluded that the 9-valent vaccine referred to is the vaccine referred to in Obaro, which is the 9-valent Wyeth composition.
220 On the same page, a little after the heading “The future of pneumococcal vaccination”, the following paragraphs appear (emphasis added):
The geographic variability of pneumococcal serotypes represents a problem when developing a vaccine with worldwide coverage. We would almost have to design a specific vaccine for each geographic area, conducting a prior epidemiological study of the most common serotypes, which would only be possible in developed countries.
Furthermore, we know that the spectrum of serotypes widens with advancing age, which complicates the acquisition of vaccines for age groups other than children, although children are the group at greatest risk and for whom the current vaccine is most effective. In this respect, work is being conducted to incorporate new serotypes to the 7-valent conjugate vaccine, with the 9-valent (which incorporates the serotypes 1 and 5), 11-valent (adding 3 and 7F) and 13-valent (6A and 19a) vaccines in various stages of research. This could broaden the spectrum of ages and countries, although we will continue to have much diversity in coverage. In addition, attempts are being made to incorporate the pneumococci that show the greatest resistance to antibiotics.
221 MSD submits that the disclosure of Peña anticipates a number of claims of both the 013 patent and the 844 patent. It submits that the only integer of claims 1 and 11 of the 013 patent and claim 1 of the 844 patent that is not expressly referred to is that each serotype of the 13-valent vaccine is conjugated to CRM197. It submits that the person skilled in the art would perceive that Peña implicitly discloses that the carrier protein for the 13-valent vaccine is CRM197. That is because first, Peña states that the additional serotypes were “incorporated” into the 7-valent conjugate vaccine, Prevnar 7, which (as Professors Paton and Kasper understood) implies that the 13-valent vaccine would also use CRM197 as the carrier protein. Secondly, the person skilled in the art would assume that the 13-valent vaccine was being developed by Wyeth because Peña “is a Wyeth publication and its authors worked for Wyeth in Spain”, and because Peña does not refer to any non-Wyeth vaccines. Accordingly, the person skilled in the art would assume that the 13-valent vaccine had the same carrier protein as used by Wyeth in previous vaccines, which was disclosed as CRM197.
222 Wyeth submits that to anticipate the patentee’s claim, the prior publication must contain clear and unmistakable directions to do what the patentee claims to have invented, citing AstraZeneca AB v Apotex Pty Ltd [2014] FCAFC 99; 226 FCR 324 (AstraZeneca (FC)) at [293] (Besanko, Foster, Nicholas and Yates JJ, with whom Jessup J agreed). Further, the disclosure must be enabling in that it must allow the skilled reader to create what is disclosed using only the disclosure, common general knowledge and routine trial and experimentation, citing Olin Corporation v Super Cartridge Co Pty Ltd [1977] HCA 23; 180 CLR 236 at 260 – 261 (per Stephen and Mason JJ). It submits that at most Peña discloses that a pneumococcal conjugate vaccine with 13 serotypes was under development. No other information about the composition of the 13-valent vaccine is disclosed. It does not disclose the carrier protein or proteins to which the serotypes were conjugated. Even if the use of CRM197 was disclosed, no directions are stated to make the claimed product or perform any method in respect of that product, and as such any disclosure was not enabling.
223 To say that a claim of a patent is not novel in the light of prior art information is to say that it adds nothing new to that with which it is compared. In the case of a paper anticipation, it assumes that the product described is made, or the process outlined is followed. Each integer of the claimed invention must be disclosed in the prior publication. The reverse infringement test is a practical way of detecting whether that is so in most cases: Meyers Taylor Pty Ltd v Vicarr Industries Ltd [1977] HCA 19; 137 CLR 228 at 235 (Aickin J).
224 That is not to say that the person skilled in the art cannot exercise some degree of skill and experience in considering a prior disclosure. In C. Van der Lely N.V. v Bamfords Ltd [1963] RPC 61 Lord Reid considered whether a skilled reader would infer from photographs of a machine in magazine articles that the rake wheels of the alleged anticipating machine were ground driven rather than gear driven. He said at 72 (emphasis added):
The appellants’ first argument is that it is not enough that the photograph should disclose a probability that the wheels were ground driven. There cannot be anticipation, they say, unless this is shown clearly and unmistakably. I cannot accept that argument. I would agree that the anticipating material must be as good for practical purposes as the material in the appellants' specification. But I can see no practical difference between a definite statement of fact and material from which the skilled man would clearly infer its existence.
So the question is whether the typical skilled man would infer ground drive from the photograph. It would not be enough that a quick-witted man would guess that. But in this case I think that it is proved that the ordinary skilled man would have had good grounds for reaching the conclusion that these wheels were ground driven. He could not have been certain, but practical men act on something less than absolute certainty.
225 The Full Court in AstraZeneca (FC) at [293] noted that the touchstone for determining whether a prior publication anticipates a claimed invention is stated in General Tire & Rubber Co Ltd v Firestone Tyre & Rubber Co Ltd [1971] 7 WLUK 130; [1972] RPC 457 at 485 – 486 (Sachs, Buckley and Orr LJJ) as follows:
When the prior inventor’s publication and the patentee’s claim have respectively been construed by the court in the light of all properly admissible evidence…the question whether the patentee’s claim is new…falls to be decided as a question of fact. If the prior inventor’s publication contains a clear description of, or clear instructions to do or make, something that would infringe the patentee’s claim if carried out after the grant of the patentee’s patent, the patentee’s claim will have been shown to lack the necessary novelty, that is to say, it will have been anticipated. The prior inventor, however, and the patentee may have approached the same device from different starting points and may for this reason, or it may be for other reasons, have so described their devices that it cannot be immediately discerned from a reading of the language which they have respectively used that they have discovered in truth the same device; but if carrying out the directions contained in the prior inventor’s publication will inevitably result in something being made or done which, if the patentee’s patent were valid, would constitute an infringement of the patentee’s claim, this circumstance demonstrates that the patentee’s claim has in fact been anticipated.
If, on the other hand, the prior publication contains a direction which is capable of being carried out in a manner which would infringe the patentee’s claim, but would be at least as likely to be carried out in a way which would not do so, the patentee’s claim will not have been anticipated, although it may fail on the ground of obviousness. To anticipate the patentee’s claim the prior publication must contain clear and unmistakable directions to do what the patentee claims to have invented…A signpost, however clear, upon the road to the patentee’s invention will not suffice. The prior inventor must be clearly shown to have planted his flag at the precise destination before the patentees.
226 In AstraZeneca (FC) the Full Court makes plain that the limited role that common general knowledge may play in the context of novelty:
[352] Although the common general knowledge can be used in a limited way to construe a prior art document, s 7(1) does not permit the common general knowledge to be used as a resource that can be deployed complementarily to arrive at a disclosure which the document alone, properly construed, does not make. If it were otherwise, the separate requirement of an inventive step to support a patentable invention (see s 18(1)(b)(ii) of the Act) would be otiose. The test of novelty would encompass the test for inventive step, without the need to satisfy the threshold requirements of s 7(3) (as it then stood) that the information in the document be information that the person skilled in the art could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant to work in the relevant art in the patent area. All that would be required is that the information in the prior art document be publicly available.
227 The question of lack of novelty was recently revisited by an expanded Full Court in Mylan Health Pty Ltd v Sun Pharma ANZ Pty Ltd [2020] FCAFC 116; 380 ALR 582 (Middleton, Jagot, Yates, Beach and Moshinsky JJ). After considering relevant authorities, the Full Court re-emphasised the longstanding test in simple terms at [104]:
We do not accept that a documentary disclosure containing an hypothesis cannot be an anticipatory disclosure that deprives an invention of novelty. In such a case the question, simply put, remains: what does the prior document disclose? The occasion on which, or the context in which, a particular documentary disclosure is made may well inform the interpretation of the document’s content. But if, as a matter of interpretation, the document nonetheless discloses that which is later claimed as an invention, that disclosure will anticipate the invention and deprive it of novelty.
228 In that case, the argument on appeal was whether a document anticipated the claims because it advanced no more than a reasoned hypothesis for treatment, not a method of treatment as such ([66]). The primary judge found that it did so anticipate because he found that the document clearly disclosed a method within the claims. The Full Court upheld that conclusion, noting at [82]:
The combined passages from Hill v Evans speak of the need for a prior documentary disclosure to provide information that is equal to the invention that is claimed, if the prior documentary disclosure is to be anticipatory and thereby deprive the invention of novelty. As Hill v Evans makes clear, equality in this context refers to both the specificity of the information and its completeness. Unless these twin qualities are present, the prior disclosure will not be sufficient to deprive the invention of novelty.
229 Having said this, I do not accept Wyeth’s submission that in respect of a product claim, the prior disclosure must do more than describe the product the subject of the claim.
230 Nevertheless, I consider that the approach taken by MSD to the construction of Peña is in error. It exceeds the permissible use of the common general knowledge, which is to be used to understand the disclosure of the prior art document, not to supplement it with matters not disclosed but instead drawn from the common general knowledge. Instead, it relies on inference and assumption, not upon disclosure, as the means by which it arrives at a novelty defeating disclosure. The crucial passage in the article upon which MSD relies, “[i]n this respect, work is being conducted to incorporate new serotypes to the 7-valent conjugate vaccine, with the 9-valent (which incorporates the serotypes 1 and 5), 11-valent (adding 3 and 6F) and 13-valent (6A and 19a) vaccines in various stages of research”, makes no reference to the company that is said to be developing the vaccines. Nor does the article as a whole. MSD relies on an inference that all of these products are being developed by Wyeth. The article does disclose that the 7-valent conjugate vaccine is Prevnar 7 (a Wyeth product). But it does not identify to what carrier protein or proteins the 9-, 11- or 13-valent vaccines under development are conjugated, or who is developing them.
231 Professor Paton gives evidence that he would have assumed that the 13-valent vaccine referred to was being developed by Wyeth. That inference is based on the fact that no other reference is made to any other company or publication in relation to the vaccines under development. Considering the statement in the Summary that the other vaccines “have not yet been marketed and are in a very advanced study phase”, he would have assumed that only a company developing a vaccine would have detailed information about that vaccine. He would also have assumed that the 9- and 11-valent vaccines to which the article refers were those under development by Wyeth. He was aware of the 9-valent pneumococcal conjugate vaccine under development by Wyeth and knew that it used CRM197 as the protein carrier. Based on these several assumptions, Professor Paton considers that although Peña does not explicitly say that each serotype of the 13-valent vaccine under development was conjugated to CRM197, it implied that this was the case. In that sense, he draws attention to the language Peña uses that the additional serotypes were being “incorporated” into the pre-existing vaccines. Furthermore, the only carrier protein mentioned in Peña is CRM197. These matters implied to him that the 13-valent vaccine would also use CRM197.
232 Professor Strugnell notes that Peña does not state to which carrier protein the polysaccharides from each serotype included in the 9-, 11- and 13-valent vaccines are conjugated. Had he read Peña at April 2005, he would have assumed that the 9-valent vaccine is a reference to the Wyeth 9-valent CRM197 conjugate vaccine which is referred to in Obaro, and that the 11-valent vaccine is a reference to either the Aventis 11-valent bi-carrier vaccine or the GSK 11-valent vaccine, or both. He was not aware of any 13-valent pneumococcal conjugate vaccine at the time and would not have known what carrier protein or proteins would be used. Furthermore, he notes that Peña does not disclose a pneumococcal conjugate vaccine that includes polysaccharide-protein conjugates prepared using polysaccharides from each of the 13 serotypes in claim 1, where these are all individually conjugated to CRM197.
233 Professor Dagan takes the same approach as Professor Strugnell.
234 Having regard to the assistance provided by these witnesses, it is for the Court to construe the prior art document. The relevant language is in ordinary English.
235 I consider that the assumptions made by Professor Paton demonstrate that he goes beyond the disclosure of the document in arriving at the conclusion that it teaches a composition in accordance with claim 1 of the composition patents. Whilst it is plain enough to the skilled reader from the text of the article that the 7-valent pneumococcal conjugate vaccine referred to is that developed by Wyeth, and accordingly is conjugated to CRM197, I do not think that it is legitimate to assume that the 9-, 11- and 13- valent vaccines are also conjugated CRM197. There is simply no disclosure of the carrier protein used. In my view it reads too much into the words “work is being conducted to incorporate new serotypes to the 7-valent conjugate vaccine”, to find that this is a disclosure that the serotypes are necessarily being conjugated to CRM197. To reach that conclusion, it would be necessary to assume that because the Prevnar 7 serotypes are being incorporated, so too is the carrier protein. Furthermore, nowhere does Peña state that it refers exclusively to compositions under development by Wyeth. Professor Paton assumes this as a fact. His assumption is in the nature of the quick-witted guess of the type that Lord Reid identified in Van der Lely. The same may be said of a similar conclusion reached by Professor Kasper.
236 Furthermore, if one were to stray from the disclosure of the article, and draw on the common general knowledge to supplement it (which is impermissible: AstraZeneca (FC) at [352]), then the person skilled in the art would have knowledge that a 9-valent pneumococcal conjugate vaccine was under development by Wyeth conjugated to CRM197, that 11-valent pneumococcal conjugate vaccines were under development by Aventis and GSK using different protein carriers, and that a Wyeth 11-valent pneumococcal conjugate vaccine was under development, but with an unidentified protein carrier (see section 8.1). Accordingly, one would remain uncertain as to what carrier protein was used in the 13-valent vaccine mentioned in Peña. That uncertainty cannot be remedied by inference or intelligent guesswork.
237 Accordingly, I find that the disclosure of Peña is not sufficient to defeat the novelty of the claims of the composition patents. Given my findings with respect to the CRM197 integer, it unnecessary to address the rest of MSD’s s 7(1)(a) challenges to the balance of the asserted composition patent claims.
6.6 The disclosure of Peña when read with Obaro
238 There is no dispute that by reason of the cross-reference in the footnote to Peña, Obaro is a document within s 7(1)(b) that a person skilled in the art would treat as a single source of information with Peña.
239 Obaro states its objective is to evaluate the safety and immunogenicity of a nonavalent pneumococcal conjugate vaccine and the antigenic interaction when administered simultaneously with diphtheria, tetanus and pertussis vaccines. It does so by reference to results of a study on Gambian infants. Under the heading “Vaccines” the article says:
The nonavalent pneumococcal conjugate vaccine (PnCV) used in the study was manufactured by Wyeth-Lederle Vaccines and Pediatrics, Rochester NY...It is prepared in a lyohpilized form and contains 2 μg of types 1, 4, 5, 9V, 14, 19F and 23F pneumococcal polysaccharides, 2 μg of type 18C oligosaccharide and 4 μg of type 6B polysaccharide. Each polysaccharide or oligosaccharide is coupled independently to CRM197, a nontoxic mutant of diphtheria toxoid, to give a total of ~20 μg of CRM197 per dose. The vaccine was prepared in single dose vials, reconstituted before injection with 0.75 ml of a saline diluent containing aluminium phosphate. Each 0.5-ml dose contained 0.5 mg of aluminium phosphate.
240 MSD does not rely on Obaro to supplement the disclosure of Peña by reference to the use of a 13-valent vaccine; only a nonavalent vaccine is disclosed. Whilst the serotypes in the 9-valent vaccine are conjugated to CRM197, the deficiencies that I have addressed in terms of the 13-valent vaccines are not resolved. Obaro is relied upon to provide a disclosure of the inclusion and quantity of an aluminium adjuvant, the physiologically acceptable vehicle, and other aspects of the dependent claims.
241 Having regard to my findings in relation to the disclosure of Peña, Obaro does not assist MSD further in relation to its lack of novelty allegations, which must accordingly be rejected.
7. COMPOSITION PATENTS: LACK OF INVENTIVE STEP – INTRODUCTION
242 MSD pleads that the invention claimed in the asserted 013 patent claims was obvious and did not involve an inventive step for the purposes of s 18(1)(b)(ii) and s 7(2) of the Patents Act as at the priority date. It relies for this purpose on the common general knowledge before 8 April 2005. Having regard to the form of the Patents Act that applies to the 013 patent, being the pre-RTB Patents Act, the common general knowledge is to be considered as it existed in the “patent area”, defined in Schedule 1 to mean Australia, as at the priority date: s 7(2). MSD also relies in its closing submissions on the single pieces of prior art information listed below, which it contends the skilled person in the art could reasonably be expected to have ascertained, understood and regarded as relevant within s 7(3)(a) of the Patents Act, and in the case of two or more publications itemised together below multiple pieces of prior art information which the skilled person could before 8 April 2005 be reasonably expected to have combined within s 7(3)(b):
(a) Peña alone or with Obaro;
(b) Hausdorff 2000 alone or with another article by Hausdorff et al entitled “Multinational study of pneumococcal serotypes causing acute otitis media in children” published in 2002 by the Pediatric Infectious Disease Journal (Volume 21(11)) (Hausdorff 2002);
(c) an article by X Yu et al entitled “Immunity to Cross-Reactive Serotypes Induced by Pneumococcal Conjugate Vaccines in Infants” published in 1999 by the Journal of Infectious Diseases (Volume 180);
(d) chapter 23 of the fourth edition of a book edited by S A Plotkin and W A Orenstein entitled Vaccines published in 2004 by Elsevier Inc.;
(e) an article by C G Whitney et al entitled “Decline in Invasive Pneumococcal Disease after the Introduction of Protein-Polysaccharide Conjugate Vaccine” published in 2003 by the New England Journal of Medicine (Volume 348(18)) (Whitney 2003); and
(f) an article by J Eskola et al entitled “Efficacy of a Pneumococcal Conjugate Vaccine Against Acute Otitis Media” published in 2001 by the New England Journal of Medicine (Volume 344(6)).
243 Other publications identified in the particulars to MSD’s lack of inventive step case were not advanced in closing submissions.
244 MSD also contends that the invention claimed in the asserted 844 patent claims was obvious and did not involve an inventive step for the purposes of s 18(1)(b)(ii) and s 7(2) and (3) of the post-RTB Patents Act. It relies on the same pieces of prior art information and the same combinations of prior art information as those identified in relation to the 013 patent.
245 As it turns out, one aspect of the distinction between the pre- and post-RTB forms of s 7(2) and 7(3) is not material to the present case. Whilst the pre-RTB form of s 7(2) confines the common general knowledge to that information as it existed at the priority date within the patent area and the post-RTB form does not, I have no difficulty in finding that the field of the art is an international one, where the common general knowledge in Australia (the patent area) is the same as that outside the patent area.
246 Section 18(1)(b)(ii) of the pre-RTB Patents Act provide that an invention is a patentable invention for the purposes of a standard patent if the invention, so far as claimed in any claim, involves an inventive step when compared with the prior art base as it existed before the priority date of that claim.
247 Sub-sections 7(2) and (3) provide:
(2) For the purposes of this Act, an invention is to be taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed in the patent area before the priority date of the relevant claim, whether that knowledge is considered separately or together with the information mentioned in subsection (3).
(3) The information for the purposes of subsection (2) is:
(a) any single piece of prior art information; or
(b) a combination of any 2 or more pieces of prior art information;
being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood, regarded as relevant and, in the case of information mentioned in paragraph (b), combined as mentioned in that paragraph.
248 By s 7(2) an hypothetical person skilled in the art, notionally possessed with the common general knowledge as it existed before the priority date, must find the invention to be obvious, whether or not the common general knowledge is supplemented by prior art information within s 7(3): AstraZeneca AB v Apotex Pty Ltd [2015] HCA 30; 257 CLR 356 (AstraZeneca (HC)) at [18] (per French CJ).
249 The law concerning the requirement for an inventive step reflects a balance of policy considerations in patent law of encouraging and rewarding inventors without impeding advances and improvements by skilled, non-inventive persons: Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) [2007] HCA 21; 235 CLR 173 (Lockwood No 2) at [48] (Gummow, Hayne, Callinan, Heydon and Crennan JJ). The cases over the years have made a number of statements as to what is required to answer the “jury question” of whether or not an invention is obvious. It is a question of fact. The question is not what is obvious to a court, but depends on analysis of the invention as claimed having regard to the state of the common general knowledge, any information relied upon for the purpose of s 7(3), and the approach taken to it by the person skilled in the art: Lockwood No 2 at [51].
250 As a basic premise, the question is always “is the step taken over the prior art an ‘obvious step’ or an ‘inventive step’”? This is often an issue borne out by the evidence of the experts: Lockwood No 2 [52]. Whilst the question remains one for the courts to determine, the courts do so by reference to the available evidence, including that of persons who might be representative of the skilled person in the art: AstraZeneca (HC) at [70] (Kiefel J, as her Honour then was). Various formulations of the question have been set out in the cases. In R D Werner & Co Inc v Bailey Aluminium Products Pty Ltd [1989] FCA 57; 25 FCR 565 at 574 Lockhart J said that there must be “some difficulty overcome, some barrier crossed”. A “scintilla of invention” is sufficient to support the validity of a patent: Aktiebolaget Hässle v Alphapharm Pty Limited [2002] HCA 59; 212 CLR 411 at [48] (per Gleeson CJ, Gaudron, Gummow and Hayne JJ). In Allsop Inc v Bintang Ltd [1989] FCA 428; 15 IPR 686 at 701 the Full Court (Bowen CJ, Beaumont and Burchett JJ) noted that for the invention to be inventive, it must be “beyond the skill of the calling”.
251 Although identified as a single person, it is established that the person skilled in the art may be a composite or team of persons: General Tire at 485. The hypothetical construct represented by that notional team is intended as an aid to the Court in addressing the “hypothetical question of whether a person, with the same knowledge in the field and aware of the problem to which the patent was directed, would be led directly to the claimed invention”: AstraZeneca (HC) at [70].
252 In AstraZeneca (HC) French CJ noted at [15] that relevant content was given to the word “obvious” by Aickin J in Wellcome Foundation Ltd v VR Laboratories (Aust) Pty Ltd [1981] HCA 12; 148 CLR 262 at 286, where Aickin J posed the test:
whether the hypothetical addressee faced with the same problem would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not.
253 At [15] French CJ (with whom Gageler and Keane JJ and Nettle J agreed) (citations omitted and square brackets in the original) explained:
The idea of steps taken "as a matter of routine" did not, as was pointed out in AB Hässle, include "a course of action which was complex and detailed, as well as laborious, with a good deal of trial and error, with dead ends and the retracing of steps". The question posed in AB Hässle was whether, in relation to a particular patent, putative experiments, leading from the relevant prior art base to the invention as claimed, are part of the inventive step claimed or are "of a routine character" to be tried "as a matter of course". That way of approaching the matter was said to have an affinity with the question posed by Graham J in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd. The question, stripped of references specific to the case before Graham J, can be framed as follows:
"Would the notional research group at the relevant date, in all the circumstances, which include a knowledge of all the relevant prior art and of the facts of the nature and success of [the existing compound], directly be led as a matter of course to try [the claimed inventive step] in the expectation that it might well produce a useful alternative to or better drug than [the existing compound]?"
That question does not import, as a criterion of obviousness, that the inventive step claimed would be perceived by the hypothetical addressee as "worth a try" or "obvious to try". As was said in AB Hässle, the adoption of a criterion of validity expressed in those terms begs the question presented by the statute.
254 The approach proposed by Graham J in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd [1970] RPC 157 to which French CJ refers is often referred to as the “modified Cripps question”. The application of the modified Cripps question has been the subject of recent consideration. In Generic Health Pty Ltd v Bayer Pharma Aktiengesellschaft [2014] FCAFC 73; 222 FCR 336, the Full Court said at [71] (Besanko, Middleton and Nicholas JJ) (emphasis added):
We do not think that the plurality in Alphapharm were saying that the reformulated Cripps question was the test to be applied in every case. Rather, it is a formulation of the test which will be of assistance in cases, particularly those of a similar nature to Alphapharm. The plurality did not reject as an alternative expression of the test the question whether experiments were of a routine character to be tried as a matter of course (The Wellcome Foundation Limited v VR Laboratories (Aust) Proprietary Limited (1981) 148 CLR 262, at 280-281, 286, per Aickin J). We do not think there is a divide here in terms of whether an expectation of success is relevant between a test which refers to routine steps to be tried as a matter of course and the reformulated Cripps question. It is difficult to think of a case where an expectation that an experiment might well succeed is not implicit in the characterisation of steps as routine and to be tried as a matter of course. On the other hand, we think a test formulated in terms of worthwhile to try was firmly rejected by the High Court in Alphapharm (see also Pfizer, at 476, [287], per French and Lindgren JJ [Pfizer Overseas Pharmaceuticals v Eli Lilly and Co (2005) 225 ALR 416]). The fact (if it be the fact) that the position in the United States may have shifted does not affect the binding nature of what the plurality said in Alphapharm.
255 In Nichia the Full Court picked up on the emphasised passage in concluding that, in finding that there were “a number of unknowns” and that the patentee “did not know” that a combination would produce a satisfactory result within the claim, the primary judge strayed from “the test of steps taken in an expectation that they might well produce the invention or a useful result towards a test of an expectation of knowing that steps will produce a useful result based on predictive capacity” (emphasis added) (at [88] – [89]). The relevant test is expecting that the steps may well work, rather than knowing that steps will or would or even may well work (at [99]).
256 In relation to having multiple avenues to try, in Nichia the Full Court adopted as orthodox the statement of Laddie J in Brugger v Medic-Aid Ltd [1996] WLUK 122; RPC 635 at 661:
…if a particular route is an obvious one to take or try, it is not rendered any less obvious from a technical point of view merely because there are a number, and perhaps a large number, of other obvious routes as well. If a number of obvious routes exist it is more or less inevitable that a skilled worker will try some before others. The order in which he chooses to try them may depend on factors such as the ease and speed with which they can be tried, the availability of testing equipment, the costs involved and the commercial interests of his employer. There is no rule of law or logic which says that only the option which is likely to be tried first or second is to be treated as obvious for the purpose of patent legislation.
257 In AstraZeneca (FC) Jessup J considered the approach to be taken by a primary judge in characterising an invention. Jessup J recorded that in that case the primary judge reviewed the specification and found that it made plain that the inventors had come up with the administration of a particular dose or dosage range of rosuvastatin but not rosuvastatin itself, which had previously been invented (at [462]). One of the relevant claims was for a method of treating a patient with a particular dose range of rosuvastatin (at [463]). The primary judge approached the question of inventive step by characterising the inventive concept as lying in the dosage range alone. The primary judge found that knowledge of rosuvastatin was not part of the common general knowledge at the priority date. However, having regard to the disclosure of the specification, the specification itself made the existence of rosuvastatin “a given”, and located the inventive concept in the discovery of a dosage range (at [468]). Accordingly the primary judge considered that the relevant problem was the dosage range of rosuvastatin to achieve the objective of lowering cholesterol without significant side effects. It was not, as the patentee, submitted, finding dosages of alternative statins of rosuvastatin in general (on the basis that rosuvastatin could not be considered to form part of the relevant knowledge).
258 The primary judge then proceeded to consider inventive step on the basis that rosuvastatin was known to the notional skilled team. That approach met with the problem that rosuvastatin, as noted above, was not found to be part of the common general knowledge as at the priority date. The primary judge answered that by finding that the skilled addressee must be imputed with a knowledge of rosuvastatin and of its membership of the known class of statins useful for a known purpose, being the treatment of hypercholesterolemia (at [471]).
259 That approach was rejected by Jessup J and by the balance of the Full Court. The latter definitively concluded that the Patents Act does not expressly or impliedly contemplate that the body of knowledge and information against which the question of inventive step is to be determined may be enlarged by reference to the content of the specification including, in particular, any problem that the invention is explicitly or implicitly directed at solving (at [202]). The High Court did not criticise this reasoning in dismissing the appeal.
260 In Insta Image Pty Ltd v KD Kanopy Australasia Pty Ltd [2008] FCAFC 139; 239 FCR 117 at [80] (Lindgren, Bennett and Logan JJ) the Full Court considered lack of inventive step in the context of the Patents Act. It held at [80] that, in determining the issue of obviousness it was necessary:
(1) to identify the invention “so far as claimed in any claim”;
(2) to identify the “person skilled in the relevant art”;
(3) to identify the common general knowledge as it existed in Australia before the priority date;
(4) to inquire under s 7(2) whether the invention referred to in (1) above would have been obvious to the person referred to in (2) above in light of the knowledge referred to in (3) above; and
(5) to inquire whether that invention would have been obvious to that person in the light of that knowledge when that knowledge is considered together with either kinds of information mentioned in s 7(3) (additional prior art information).
261 MSD contends that an affirmative answer should be given to the question:
At April 2005, would the person skilled in the art be directly led as a matter of course to try the claimed invention in the expectation that it might well produce a useful alternative to or better vaccine than existing pneumococcal vaccines, particularly Prevnar 7?
262 MSD submits that all of the 13 chosen serotypes in the composition patents were “a given”. Other vaccines had already included 11 of these, and serotypes 6A and 19A would have been included based on publicly available epidemiological data – as Wyeth itself did and as explained in the composition patents. MSD submits that the common general knowledge included knowledge of Prevnar 7 and also of vaccines under development as at April 2005, being the Wyeth 9-valent vaccine, and also two 11-valent vaccines, one being developed by GSK and the other by Aventis. MSD submits that an effective vaccine is one which can elicit an antibody response that protects the recipient from disease caused by each serotype in it and that the ELISA and OPA tests were conventionally used to determine the presence and quantity of antibodies in a sample. The OPA test was preferred because it determined whether antibodies are functional. Insofar as the claims include adjuvants, it submits that the adjuvant in Prevnar 7 was aluminium phosphate, and that it would be the natural choice to enhance immunogenicity. MSD submits that there was a clear advantage in using a single carrier for making conjugates and that the obvious choice was to use CRM197, which was used in Prevnar 7. It submits that the approach of Wyeth to contending that CIES/immune interference would have deterred the skilled team from using CRM197 is a false issue.
263 Wyeth submits that: the number of serotypes, and the particular choice of the 13 chosen serotypes, was not a given; immune interference was a concern with the addition of serotypes; serotype 3 had problems; serotypes and 19A and 6A were unnecessary and undesirable; and CRM197 was one option which others had eschewed in favour of other carriers even though it had been freely available since the 1970s. Further, it submits that making such a conjugate would have been technically difficult to achieve.
264 Wyeth submits that MSD must establish that the skilled team would have been directly led as a matter of course to try the composition with a reasonable expectation of success. In this regard, it relies on its construction of “immunogenic” to mean that the composition will elicit an adaptive immune response that is effective against disease or carriage of each of the 13 chosen serotypes. I have addressed and rejected Wyeth’s submission on the construction of this term in section 5.4 above.
7.4 The approach taken to inventive step by the experts
265 Professor Paton’s evidence relevant to the question of inventive step began with a summary of his involvement and recollection of matters concerning immunisation against pneumococcal disease in the 1990s through to the priority date. Set out below is a summary of his evidence in chief.
266 In the 1990s Professor Paton’s research involved the study of pneumococcal pathogenesis in order to identify the disease-causing components of Streptococcus pneumoniae and the identification of vaccine antigens that provide non-serotype dependent protection against pneumococcus. His focus was on creating a universal vaccine that was capable of covering all serotypes, such that the vaccine would provide non-serotype dependent protection. He says in the Paton Review that in the mid to late 1990s there was an increased prevalence in vaccinated populations of non-vaccine serotypes, which had the potential to result in increased spread of disease caused by those serotypes. He also notes that there were limits to cross-protection between serotypes and that an assumption of cross-protection may be incorrect, referring to studies where there was weaker than expected cross-protection.
267 In 2000, Wyeth released Prevnar 7. Professor Paton read an article about it shortly after its publication. It was based on serotypes which were prevalent in developed countries, where there had been active surveillance of serotypes. He knew that it was typically administered as a course of three injections at two, four and six months of age, followed by a booster at 12 – 15 months. He knew that Prevnar 7 was very successful in inducing an immune response in children under five and also resulted in a significant decrease in infection among the elderly. In the Paton Review, he says that:
Conjugate vaccines developed to date by various manufacturers are either 7-, 9- or 11- valent, using different cross-linking chemistries, and employ a range of protein carriers such as tetanus or diphtheria toxoids, the diphtheria toxin derivative CRM197, or outer membrane proteins from Neisseria meningitidis group B or non typeable H. influenzae. The 7-valent formulation includes types 4, 6B, 9V, 14, 18C, 19F and 23F...The 9-valent formulation includes these same types with the addition of types 1 and 5, which although uncommon in Europe and North America are important causes of invasive pediatric disease in other geographic regions...Types 3 and 7F were also included in the 11-valent formulation.
268 In his affidavit, Professor Paton says that the reference in this article to 9- and 11-valent vaccines was to experimental vaccines of those valencies that Professor Paton understood were being developed by Wyeth and an 11-valent vaccine being developed by GSK. Professor Paton’s reference to an 11-valent Wyeth vaccine provoked some controversy at trial. It became apparent in cross-examination that the other 11-valent vaccine to which he refers in the Paton Review was that of Aventis, which used the bi-carrier approach, and which was discontinued in 2002, rather than that of Wyeth. I refer to this further below.
269 Professor Paton identifies that before April 2005 he was aware that there were two main types of conjugation chemistries: reductive amination and carbodiimide chemistry. He had experience using the latter in his own laboratory and was aware that Prevnar 7 used the former when he wrote his Review Article.
270 In his first affidavit he outlines his background and experience relevant to the technology in issue and responds to the MSD Problem, set out again below:
How would you have gone about developing a polysaccharide-protein conjugate pneumococcal vaccine that was an improvement on Prevnar 7 before April 2005?
271 MSD advances its case on lack of inventive step from Professor Paton’s answer to this question, which I set out below:
85. The starting point when developing a new conjugate vaccine is the existing conjugate vaccine because, as I explained above, a new vaccine must not be inferior compared with the existing vaccine.
86. As I have discussed above, the aim of developing an improved polysaccharide-protein conjugate vaccine was (and still is) to increase coverage against additional prevalent serotypes.
87. As I have discussed above, it was well known which serotypes were prevalent and issues with cross-protection were also well known.
88. I would have searched the literature for studies and recommendations on serotypes to include in a vaccine and I would have adopted the same strategy for searching that I had already been using, as I have described above. However, given that the specific task related to a polysaccharide-protein conjugate pneumococcal vaccine, I would have added the search term "conjugate". I would have considered the most recent publications first. Later publications may well have included summaries or references to earlier work of relevance.
89. Having identified the additional serotypes to include in the conjugate vaccine, I would have obtained purified polysaccharides from the additional serotypes using standard extraction and purification techniques (e.g., those used by previous manufacturers of polysaccharide vaccines). I would have conjugated those purified polysaccharides to the same carrier protein, in the same way as Prevnar 7. I then would have performed the stock standard experimental assays to test the immunogenicity of the new vaccine components (individually and then in combination with each other and with the serotypes of the original 7 valent vaccine). This would involve immunisation of laboratory animals with the new formulation followed by standard serological analysis (i.e., an ELISA) of blood serum collected after the course of immunisation. This would have given me an indication of whether the new vaccine components elicited an immune response.
272 Professor Strugnell was asked to address the Wyeth Problem by Allens, set out again below:
…how I would go about solving the problem of developing an improved pneumococcal vaccine…on the basis of what was known to me and what I understand to have been well-known to others working in the field of immunology and microbiology, including as it relates to vaccine development and particularly pneumococcal vaccines, as at April 2005.
273 Professor Strugnell says that in developing any new pneumococcal vaccine he would consider its purposes. Having done so, one strategy that was consistent with his work before the priority date was to develop a recombinant Salmonella vaccine expressing pneumococcal proteins. He notes that he commenced work of this type in 2005, but that although protein vaccine trials are still being conducted today, no pneumococcal protein vaccine has yet been marketed.
274 Another option would have been to add one of the protein vaccine candidates to an existing pneumococcal conjugate vaccine, such as Prevnar 7, which would have the benefit of conferring serotype specific protection. Professor Strugnell says that as at April 2005 and today, it was not possible to predict whether such a combination would work.
275 A third option would have been to try to expand the serotype coverage of a pneumococcal conjugate vaccine by adding further serotypes in order to expand the protection afforded by the existing formulations. He notes that he would only have regarded such a vaccine as acceptable if the protection afforded was not diminished by reason of the inclusion of additional serotypes. One approach in this regard could have included researching combinations of serotypes specific to particular geographic regions or countries, enabling a smaller number of serotypes to be chosen. Another approach would have been researching and adding important serotypes that are prevalent globally.
276 Professor Strugnell gives evidence that in order to pursue the third option, he would have confirmed the status of pneumococcal conjugate vaccines on the market and in development as at April 2005 and, if necessary, conducted specific searches to obtain this information. He would also have obtained the product information for Prevnar 7. He then gives evidence concerning the pneumococcal conjugate vaccines that he knew were in development before April 2005. He says that it was his understanding as at April 2005 that the number of serotypes that could be included in a conjugate vaccine was limited, citing Plotkin and another article in support, and states that there was no expectation that a pneumococcal conjugate vaccine with more than 10 serotypes could be successfully achieved, but that it would be a major research project to attempt to do so. Moreover, he expresses the view that there would be a number of further questions to ask, including:
(a) Which serotypes to include? This he considers is a question for clinicians and epidemiologists in the team, and would involve balancing a number of considerations, which he addresses in detail.
(b) Whether to use short lengths of saccharide (oligosaccharides) or longer lengths (polysaccharides)?
(c) Which protein or proteins to use as carriers? He would have expected to use multiple carrier proteins rather than a single one for all serotypes to develop a high valency pneumococcal conjugate vaccine, because of the teaching of Plotkin.
(d) Whether immune interference issues would impair the value of adding further serotypes and require the use of different/multiple carrier proteins or a different conjugation method.
277 Professor Strugnell expresses the view that these issues would lead him away from trying to add further serotypes to the Prevnar 7 pneumococcal conjugate vaccine, and instead to try one of the other options.
278 Professor Dagan gives evidence that had he been asked to increase the coverage of pneumococcal conjugate vaccines by adding more serotypes to existing formulations, he would have expected a number of difficulties.
279 One is immune interference, a term that he uses to describe all types of interference phenomena which may occur in the immune system. One concern with respect to immune interference is that pre-existing immunity to a carrier protein may suppress the immune response to a polysaccharide linked to the same carrier protein, thus jeopardising the polysaccharide immune responses. He calls this form of carrier-mediated interference “carrier-induced epitope suppression” or CIES. He considered that CRM197 was no different to any other carrier protein in its likelihood of being affected by immune interference generally or CIES. Professor Dagan identifies a number of other difficulties with pneumococcal conjugate vaccines that he says as at April 2005 were well-known, being that there were drawbacks with the complexity of the manufacturing process for pneumococcal conjugate vaccines which increased their cost, the limited number of capsular polysaccharides that was considered could be included in them, and the potential for replacement disease as a result of serotype replacement, because not all serotypes were covered. These concerns, he considers, were driving research efforts before April 2005 to develop a “universal” pneumococcal vaccine based on immunity against non-capsular antigens common to all serotypes. He also refers to other potential approaches that were being explored at the time.
280 Professor Dagan considers that in April 2005 there were various options for research and investigation in relation to pneumococcal conjugate vaccines, including:
(a) researching into protein vaccines, either as stand-alone vaccines or as components to be added to pneumococcal conjugate vaccines – he considered this the area of greatest interest;
(b) increasing the coverage of pneumococcal conjugate vaccines by adding more serotypes to existing ones, using more and different carrier proteins, or using pneumococcal proteins as carrier proteins; and
(c) developing country-specific pneumococcal conjugate vaccines containing the most prevalent serotypes for particular countries.
281 Professor Dagan was also asked to comment specifically on how he would go about solving the Wyeth Problem if he were directed to try to develop a new pneumococcal conjugate vaccine based on Prevnar 7. He gives evidence that he would have expected that the polysaccharides from the nine serotypes in the Wyeth 9-valent pneumococcal conjugate vaccine could be conjugated to CRM197 to produce a vaccine that could protect recipients against disease caused by those nine serotypes, but he would not have known whether more serotypes could be added. He says that if they were all conjugated to CRM197, he would have expected it to interfere with the antibody responses to one or more of the serotypes in the vaccine, compromising the protection afforded. To try to avoid the risk of CIES he would instead have considered conjugating some of the serotypes to one carrier protein and others to a different carrier protein. This, he says, was the approach adopted by Aventis for its 11-valent pneumococcal conjugate vaccine and he would have considered protein D (the carrier protein used in the GSK 11-valent pneumococcal conjugate vaccine) an attractive option because it was not present in any co-administered vaccines. He would not have known without experimentation whether any expanded pneumococcal conjugate vaccine would solve the Wyeth Problem.
282 Professor Kasper gives evidence, in response to the evidence given by Professor Dagan, about his knowledge of immune interference and CIES before April 2005, the state of knowledge as to cross-protection between serotypes 6B and 6A, and 19F and 19A, and whether he would have included serotypes 6A and 19A in a new multivalent immunogenic pneumococcal conjugate vaccine before April 2005. He gives evidence that he would not have been deterred from pursuing a 13-valent pneumococcal conjugate vaccine with CRM197 as a single carrier protein because of immune interference or CIES. He says that CRM197 was first developed in the 1970s and has been used as a carrier protein since the 1980s. Before April 2005 it had been shown to be a good carrier protein in a number of approved conjugate vaccines, including in Prevnar 7 and against meningococcus. Prevnar 7 is itself an expanded form of earlier lower-valency compositions using CRM197 as the only carrier protein. He considers that there was typically a preference for particular carrier proteins for which there was prior successful experience and know-how within the vaccine development industry, and clear advantages to using a single carrier protein in a multivalent conjugate vaccine in terms of efficiency, costs, simplicity and minimisation of the risk of adverse reactions. He considers that a multi-carrier composition is inherently inefficient and expensive. Given what he considers to be the strong immunogenicity reported for all nine pneumococcal serotypes of the Wyeth 9-valent CRM197 conjugate vaccine, he would have a reasonable expectation that a 13-valent CRM197 vaccine would be immunogenic as well. Expanding from 9-valent composition to a 13-valent composition does not require adding large amounts of carrier protein, and he would expect it to work.
283 In relation to serotype selection, Professor Kasper was asked to comment on the state of knowledge as to cross-protection between serotypes 6B and 6A, and 19F and 19A. He considers that the literature, before the introduction of Prevnar 7, identified serotypes 6A and 19A as clinically relevant serotypes for which there was no conjugate vaccine. He cites a number of documents relied upon by MSD in support of its case under s 7(3) of the Patents Act. He gives the opinion that based on the literature, he would not have assumed that serotypes 6B and 19F of Prevnar 7 would provide sufficient cross-protection to serotypes 6A and 19A respectively.
7.5 The relative expertise of the experts
284 The parties made various submissions going to the expertise and relevance of the experts. In my view Professor Paton had relevant experience in the field before the priority date. He wrote articles relevant to that area and was involved in research and development work that qualified him as a person to speak about the subject matter. I consider that his expertise is squarely within the field of the composition patents and that before the priority date he was working in that field. His work encompassed the pathogenesis of disease caused by Streptococcus pneumoniae, aiming to understand key events in the host-pathogen interaction and to identify and evaluate novel drug targets and vaccine antigens that provide non-serotype dependent protection against pneumococcus.
285 Professor Strugnell is an expert in microbiology and immunology, including encapsulated bacteria, who has worked as part of several groups that developed vaccines against several pathogens, and who has familiarised himself with the literature in the field of pneumococcal vaccines as at April 2005. He had not before the priority date worked on multivalent pneumococcal polysaccharide conjugate vaccines or formulated any vaccines for use in humans and had no experience in making glycoconjugates. His evidence in answer to that of Professor Paton was based on many publications that he read after April 2005 for the purposes of preparing his affidavit evidence. Nevertheless, his evidence as to the approach that he would have taken before April 2005 was informed by his prior training and experience.
286 Wyeth contends that the microbiologist and immunologist should have familiarity with encapsulated bacteria generally. It submits that Professor Strugnell is sufficiently expert in the field to assist the Court, given the matters outlined in the paragraph above. MSD submits that the relevant experience of that member of the team should be more specific to the patent at hand, and should include experience in pneumococcal vaccines. MSD submits that as Professor Strugnell had no such experience, he is not sufficiently expert in the field.
287 This dispute does not go to the identification of the skilled team, but rather the relative weight to be given to the evidence of the experts, and in particular Professor Strugnell, a matter that I address in relation to specific aspects of the evidence further below. It is certainly the case that in relative terms Professor Paton had more pre-priority date experience in the field. He had direct experience in making conjugates, had published on the subject of pneumococcal conjugate vaccines, attended relevant conferences in the field and had experience in vaccine formulation. Professor Strugnell’s experience was more limited, and it would appear that much of his reading of the literature on the subject was undertaken after the priority date and for the purposes of the preparation of his evidence. These matters cause me to consider that Professor Strugnell’s evidence does not always carry with it the colour of pre-priority date practical experience directly referable to the subject matter of the patents, a matter which I take into account in considering the weight to be given to some of his opinions. However, I accept that Professor Strugnell is able to give cogent and admissible evidence.
288 Professor Dagan is a highly qualified and experienced clinician. He has consulted many companies in relation to the development of products. From time to time his evidence strayed into areas beyond his expertise, insofar as he gave opinions going to the means by which vaccines may be made (which he has never done himself). He is also the progenitor of the CIES theory, having written on the subject since 1998. His very extensive experience, and numerous consulting positions with drug companies cause me to consider that Professor Dagan’s knowledge in relation to immune interference and CIES exceeded that of the person of ordinary skill in the art at the priority date.
289 Professor Kasper’s experience is squarely within the field of the composition patents. He distinguished within that field and has a wealth of experience, however to some extent I consider that the length and breadth of his experience and his obvious inventiveness (reflected in the large number of patents to his name) indicate that his knowledge exceeds that likely to be possessed by the inventive hypothetical skilled worker in the field.
290 I take these matters into consideration when evaluating the evidence of the experts.
8. COMPOSITION PATENTS: ASPECTS OF THE COMMON GENERAL KNOWLEDGE
291 Before turning to the analysis of the issue of whether or not the claimed invention involves an inventive step, it is first necessary to address some contentious aspects of what MSD submits formed part of the common general knowledge as at April 2005.
292 The notional team possesses the common general knowledge in the relevant field in so far as it is relevant to the subject matter of the patents. This includes the background knowledge and experience available to all those persons engaged in the relevant field within the patent area, and includes publications to which they would refer as a matter of course: Minnesota Mining and Manufacturing Company v Beiersdorf (Australia) Limited [1980] HCA 9; 144 CLR 253 at 292 (per Aickin J, with whom Barwick CJ and Stephen, Mason and Wilson JJ agreed). The High Court emphasised in Alphapharm at [31] that information cannot be treated as part of the common general knowledge in the absence of evidence of its general acceptance and assimilation by persons skilled in the art.
293 In this respect my findings are as follows.
8.1 Known pneumococcal conjugate vaccines
294 The first commercial conjugate vaccine, directed against Hib, involved only conjugates of a single serotype. The development of pneumococcal conjugate vaccines was more complex than in the case of Hib because of the need to provide protection against multiple serotypes.
295 As at April 2005, the following pneumococcal conjugate vaccines were known.
296 Prevnar 7 was the only commercially available pneumococcal conjugate vaccine as at the priority date and was known to be highly successful in reducing disease caused by pneumococcus. It was known that Prevnar 7 used serotypes 4, 6B, 9V, 14, 18C, 19F and 23F each conjugated to CRM197 as the sole carrier protein. It was also known that the method of conjugation was reactive amination and that the quantity of CRM197 included per serotype was 2 μg for each serotype other than 6B, which had 4 μg, and that the total amount of CRM197 was about 20 μg. It was also known that the formulation used aluminium adjuvant. Many other details of the formulation have not been established to form part of the common general knowledge.
297 The introduction of routine use of Prevnar 7 in the US in about 2000 resulted in a significant decline in the rates of invasive pneumococcal disease (IPD), not only among vaccinated individuals but also among the population more generally, and especially in the elderly, indicating a substantial indirect protection effect.
298 There was evidence before April 2005 that pneumococcal conjugate vaccines were having the effect of reducing nasopharyngeal carriage of pneumococci of the same serotypes included in the pneumococcal conjugate vaccines. The mechanisms by which pneumococcal conjugate vaccines could interrupt nasopharyngeal carriage were not completely understood as at April 2005. The interruption of carriage of vaccine serotypes had the potential to leave a niche for carriage of, and infection by, non-vaccine serotypes, a phenomenon known as serotype replacement. These new serotypes might have been equally, more, or less virulent than the vaccine serotypes.
8.1.2 Merck’s 7-valent vaccine
299 A 7-valent pneumococcal conjugate vaccine, developed by Merck, containing polysaccharides from the Prevnar 7 serotypes each conjugated to OMPC, formed part of the common general knowledge.
8.1.3 Wyeth’s 9-valent pneumococcal conjugate vaccine
300 The experts agree that it was common general knowledge that Prevnar 9, a 9-valent pneumococcal conjugate vaccine developed by Wyeth, containing polysaccharides of serotypes 1, 4, 5, 6B, 9V, 14, 18C, 19F and 23F, each conjugated to CRM197, had been used in clinical trials before 8 April 2005 and that the vaccine was safe and was seen as immunogenic.
301 The parties disagree as to how much else was known about Prevnar 9. Obaro discloses that Prevnar 9 has the same amount of carrier protein as Prevnar 7 (being a total of 20 μg), even though two additional serotypes are deployed. However, whilst Obaro may be incorporated as a piece of information pursuant to s 7(3), a subject to which I return later in these reasons, that information is not part of the common general knowledge.
302 Professors Kasper and Paton express the view that it is likely that Wyeth used the same process of conjugation as for Prevnar 7. Insofar as that evidence is at the level of generality that the process involved first activating the polysaccharide by reaction with sodium periodate and then coupling the carrier protein directly to the polysaccharide through reductive amination, that evidence may be accepted. It is, as those witnesses say, a logical deduction from the fact that Wyeth used the same technique for its Prevnar 7 vaccine. It was considered by Professor Strugnell to be likely. It is also suggested in Plotkin.
303 The experts agree that it was known that an 11-valent pneumococcal conjugate vaccine, developed by GSK, containing polysaccharides of serotypes 1, 3, 4, 5, 6B, 7F, 9V, 14, 18C, 19F and 23F (the 11-valent vaccine serotypes) each conjugated to protein D had been used in clinical trials.
304 Wyeth contends that it was common general knowledge that GSK adopted the new protein D carrier to avoid immune interference observed in relation to existing carrier proteins. Wyeth relies on an article by A Nurkka et al entitled “Immunogenicity and Safety of the Eleven Valent Pneumococcal Polysaccharide-Protein D Conjugate Vaccine in Infants” published in 2004 by the Pediatric Infectious Disease Journal (Volume 23(11)). However, this article has not been established to form part of the common general knowledge and I am not satisfied that GSK’s reason for adopting protein D was generally known.
305 Wyeth also contends that it was common general knowledge that GSK was having technical problems with hyporesponsiveness to serotype 3. Wyeth relies on Nurkka, an article by Whitney entitled “The potential of pneumococcal conjugate vaccines for children” which was published in 2002 by The Paediatric Infectious Diseases Journal (Volume 21(10)) (Whitney 2002) and an article by F M Russell and E K Mulholland entitled “Recent advances in pneumococcal vaccination of children” published in 2004 by the Annals of Tropical Paediatrics (Volume 24). To the extent that Professor Dagan gives evidence on the subject, he relies on Whitney 2002 and Russell as the basis for his knowledge, articles which have not been shown to be common general knowledge. No other expert gave evidence of knowledge of these matters. Professor Paton gave evidence that he does not know why GSK changed its formulation. I am not satisfied that technical problems experienced by GSK in relation to serotype 3 formed part of the common general knowledge.
8.1.5 Aventis’s 11-valent vaccine
306 The experts agree that an Aventis 11-valent pneumococcal conjugate vaccine, with serotypes 3, 6B, 14 and 18C conjugated to diphtheria toxoid, and the polysaccharides from serotypes 1, 4, 5, 7F, 9V, 19F and 23F conjugated to tetanus toxoid, was known to have been used in clinical trials. The use of two carrier proteins is referred to below as a bi-carrier approach. Furthermore, it was common general knowledge that Aventis decided to stop development of its vaccine in 2002 – a matter that is noted in Chapter 23 of Plotkin which is accepted to form part of the common general knowledge.
307 Wyeth submits that the reason that Aventis had adopted the bi-carrier approach was concerns about immune interference, and that this approach was taken despite the fact that it was likely to be more complicated and expensive than using a single carrier. It cites Dagan 2004 and another article by Professor Dagan et al entitled “Reduced Response to Multiple Vaccines Sharing Common Protein Epitopes that are Administered Simultaneously to Infants”, published in 1998 by Infection and Immunity (Volume 66(5)) (Dagan 1998). It secondly submits that the reason for the discontinuance of the vaccine was immune interference, citing Russell. These matters, Wyeth submits, were within the common general knowledge. I do not agree.
308 In relation to the first submission, Professor Dagan gives evidence that in the late 1990s, Aventis had adopted the bi-carrier approach for its 11-valent pneumococcal conjugate vaccine after a study that he had led. The study tested two 4-valent pneumococcal conjugate vaccines containing polysaccharide antigens from serotypes 6B, 14, 19F and 23F conjugated to either a tetanus toxoid or diphtheria toxoid carrier protein. The study showed that the anti-Hib polysaccharide (polyribosylribitol phosphate) and anti-tetanus antibody responses were reduced when the 4-valent tetanus toxoid pneumococcal conjugate vaccine (as opposed to the 4-valent diphtheria toxoid pneumococcal conjugate vaccine) was administered together with a Hib tetanus toxoid conjugate vaccine and diphtheria-tetanus-pertussis vaccine. The magnitude of the reduction in response was dependent on the total dose of tetanus toxoid being administered. The results were published in Dagan 1988, which, Professor Dagan says, was the first report that raised concerns about loading infants with proteins (for example, the tetanus toxoid) in the form of carriers for conjugate vaccines. He gives evidence that in Dagan 2004 he reported that “this observation [i.e. CIES] led to the development of a candidate 11-valent [pneumococcal conjugate vaccine] in which the antigenic load of any single carrier is minimized by producing a bi-carrier glycoconjugate”.
309 Professor Dagan’s own knowledge as to why Aventis decided to adopt a bi-carrier approach is plainly not of itself part of the common general knowledge. He gives evidence that the results of Dagan 1998 led to Aventis changing its policy with respect to the bi-carrier approach; however, he knew that from his involvement in confidential discussions of that nature with Aventis while it was developing its 11-valent vaccine. There is no convincing evidence that the information contained in Dagan 1998 or Dagan 2004 formed part of the common general knowledge. Infection and Immunity is a “first rate” publication to which Professor Paton and Professor Strugnell subscribed, but not all the contents of all articles published over the years by those publications will necessarily form part of the common general knowledge for that reason alone.
310 Professor Paton does not recall reading Dagan 2004 before April 2005. Professor Kasper did not have the same understanding as Professor Dagan to the reason why the bi-carrier approach was adopted. He understood in 2005 that it was based on a study conducted in Iceland published by Sigurdardottir that showed that certain serotypes gave advantageous enhanced responses depending on the carriers used. Professor Strugnell was not working in the field of pneumococcal conjugate vaccines before the priority date. Although he also subscribed to Infection and Immunity, he gives no evidence that he read the Dagan articles before the priority date.
311 In relation to the second submission, I am not satisfied that it was common general knowledge that the reason why Aventis discontinued its 11-valent pneumococcal conjugate vaccine was immune interference. Wyeth contends that the results reported in Dagan 2004 were the reason for the discontinuance, but that fact is not stated in Dagan 2004. Wyeth also relies on Russell. No evidence indicates that either of Professors Kasper or Paton was aware of the article before the priority date or was subscribed to the journal it was published in. Professor Strugnell does not recall reading it before the priority date. As I have noted, Professor Dagan’s knowledge on the subject of the Aventis’s work is idiosyncratic and cannot be relied upon as representative of the common general knowledge.
8.1.6 Wyeth’s 11-valent vaccine
312 MSD contends that part of the common general knowledge of the skilled team was an understanding (rather than the fact itself) that Wyeth was developing an 11-valent pneumococcal conjugate vaccine that included the 11-valent vaccine serotypes each conjugated to CRM197. Surprisingly, given the identity of the patentee, this was a matter of considerable controversy. At trial Wyeth submitted that it not part of the common general knowledge that Wyeth was developing such a vaccine, and further that the very existence of such a product was a “phantom”.
313 Professor Paton gives evidence in chief that the reference in the Paton Review to two 11-valent pneumococcal conjugate vaccines was referring to one being developed by GSK and another being developed by Wyeth. In cross-examination he accepted that the reference to a Wyeth pneumococcal conjugate vaccine was a mistake. Having regard to an article footnoted to the Patent Review, he accepted that his article was referring to the Aventis 11-valent vaccine and not a Wyeth vaccine. However, he maintained his evidence that it was his understanding, prior to April 2005, that Wyeth was developing an 11-valent pneumococcal conjugate vaccine using CRM197 as the sole carrier protein. Although his affidavit in chief reflected error in this respect, I do not accept Wyeth’s submission that his answers reflect adversely on his credit or the clarity of his recollections. Despite sustained cross-examination on this point, I accept that it was Professor Paton’s understanding before April 2005 that Wyeth was developing an 11-valent vaccine.
314 In his written evidence, Professor Dagan responded to Professor Paton’s evidence about Wyeth developing an 11-valent pneumococcal conjugate vaccine by saying “I am not aware, and was not aware before April 2005, that such a vaccine had been developed”, and that as far as he was aware no 11-valent vaccine had reached the clinical trial stage. This seems to me to have been something of an elision. In his oral evidence he confirmed that before April 2005 he was aware of and had read three articles in which reference is made to a Wyeth 11-valent vaccine under development at a pre-clinical stage: one by K L O’Brien and M Santosham entitled “Potential Impact of Conjugate Pneumococcal Vaccines on Pediatric Pneumococcal disease” published in 2004 as a review article in the American Journal of Epidemiology (Volume 159(7)); another by D L Klein and Eskola entitled “Development and testing of Streptococcus pneumoniae conjugate vaccines” published in 1999 in Clinical Microbiology and Infection (Volume 5); and another by G D Overturf entitled “Pneumococcal Vaccination of Children” published in 2002 in Seminars in Pediatric Infectious Diseases (Volume 13(3)). Each refers to a Wyeth pneumococcal conjugate vaccine under development with 11 serotypes, which match those of the GSK and Aventis vaccines, each conjugated to CRM197 as the carrier protein. The Klein article refers to conjugation of the serotypes by a process of reductive amination.
315 It is apparent to me from his oral evidence that Professor Dagan had a clear understanding that prior to April 2005 there were publications that suggested that Wyeth had been developing an 11-valent vaccine. He nonetheless declined to engage in his affidavit evidence with Professor Paton’s evidence on the point, despite referring directly to the paragraph in Professor Paton’s affidavit where the subject is raised. In his oral evidence, Professor Dagan was somewhat evasive on this point but ultimately accepted that his understanding was that a Wyeth 11-valent vaccine was in pre-clinical trials being tested on animals. It was his view that in the period of time from 1999 when the vaccine was first mentioned (in Klein) until 2004, when it was mentioned in O’Brien, the vaccine appeared not to have developed, because so far as he could tell the reported information had not developed beyond brief tabular references and had not advanced beyond the preclinical stage. He considered that due to the passage of time between the references, it was likely that the vaccine development had failed, and O’Brien had simply taken the data from the Klein article and mentioned it again. Accordingly, it was his view that a vaccine had not been “developed”.
316 Professor Kasper gave evidence that the literature indicated to him before the priority date that Wyeth was expanding its 9-valent Prevnar 9 to an 11-valent iteration with CRM197 as the sole carrier protein, citing Overturf at page 158. It was suggested in cross-examination that Professor Kasper did not read the article before April 2005, but the upshot of his evidence, which I accept, was that he was “quite sure” that he had done so.
317 Professor Strugnell gives evidence that he would not necessarily have been aware of a vaccine in pre-clinical development by Wyeth, and was not aware with respect to any 11-valent vaccine. That is no doubt because Professor Strugnell was not involved directly in the field of pneumococcal conjugate vaccines at the time.
318 Having regard to the totality of the evidence, I consider that the common general knowledge included an understanding that Wyeth had been working on an 11-valent pneumococcal conjugate vaccine before April 2005. However, the evidence does not support a finding beyond this level of knowledge.
319 At trial Wyeth contended that there is no evidence to support a contention that Wyeth was actually working on an 11-valent vaccine, as opposed to people thinking it was doing so. That, however, is not to the point. The common general knowledge is the skilled person’s notional intellectual equipment and general understanding of the relevant area of discourse. A general understanding may form part of that relevant knowledge, even if it is not proved to be true, or is ultimately found not to be true. An example would be a widely accepted scientific hypothesis that is subsequently proved to be incorrect. No doubt at one stage the theory that the earth was flat likely formed part of the common general knowledge of astronomers.
8.1.6.1 MSD’s application to re-open of 24 July 2020
320 After reaching the conclusions expressed above in relation to Wyeth’s 11-valent vaccine, on 24 July 2020, MSD filed an interlocutory application supported by an affidavit sworn by its solicitor, seeking leave to re-open its evidence for the purpose of tendering a document. The document is a submission dated 8 July 2020 filed by Wyeth in the United Kingdom trial equivalent to the container patent trial in Australia. The only aspect of the submission relied upon is a statement that “By 2006, Wyeth had worked on a 11v vaccine. It was never tested in clinical trials [Khandke 1 §22] and is mentioned in the O’Brien paper as being pre-clinical work”. I invited the parties to make written submissions on the application, which they have done, and I indicated that I would dispose of it on the papers in my final judgment, which I now do.
321 The relevant issue concerning the Wyeth 11-valent vaccine is whether or not knowledge about it formed part of the common general knowledge. Whether or not Wyeth actually worked on such a vaccine is not wholly irrelevant to that question, because if it were accepted, it could rationally affect (directly or indirectly) the assessment of the probability of the existence of the fact in issue, namely whether it was common general knowledge that Wyeth had worked on the vaccine. However, as I have noted, a finding that knowledge of the Wyeth 11-valent vaccine forms part of the common general knowledge would not be precluded if Wyeth had in fact not worked on such a vaccine.
322 MSD submits that the trial was conducted on the basis of an inadvertent error, namely that Wyeth had not worked on an 11-valent vaccine, when in fact it had done so. MSD seeks leave to re-open to avoid the possibility that the Court proceeds on a misapprehension and accordingly, in weighing the balance of probabilities, fails to take into account a relevant fact. It also submits that the evidence sought to be adduced shows that Wyeth’s challenge to the credit of Professor Paton based on his evidence concerning his knowledge of the Wyeth 11-valent vaccine under development was misguided.
323 In the present case, Wyeth undoubtedly advanced the submission at trial that the 11-valent Wyeth vaccine was a “phantom”. One may assume that this submission was made on instructions. The proposed further evidence appears to contradict it, and is relied upon as an admission. However, Wyeth opposes the re-opening. It does so first on the basis that the document is irrelevant, secondly because the present case is not within the classes of cases appropriate for re-opening and thirdly because the tender would prejudice Wyeth.
324 The principles governing the re-opening of a party’s evidence are summarised in F.Y.D Investments Pty Ltd v Promptair Pty Ltd [2017] FCA 1097 at [30] – [33] (White J). The overriding principle is whether it is in the interests of the administration of justice, having regard to the circumstances of the case, to permit the re-opening.
325 In my view the evidence proposed to be adduced is relevant, but only slightly so, for the reason explained. It addresses a point in contest at the hearing that Wyeth could, and in my view should, have not left in issue having regard to the obligations on a party imposed by s 37M of the Federal Court of Australia Act 1976 (Cth). However, the submission sought to be tendered is at a high level of abstraction and provides no information that materially aids the consideration of whether it was common general knowledge that the Wyeth 11-valent vaccine existed. There has been no real delay in bringing the material before the Court, but having regard to all relevant considerations, including the conclusions that I have reached regarding the common general knowledge, in my view it is not in the interests of justice to permit MSD to re-open its case. The interlocutory application will be dismissed, with no order as to costs.
326 The information contained in Chapter 23 entitled “Pneumococcal conjugate vaccines” in Plotkin is accepted to form part of the common general knowledge.
327 The following passage at pages 595 – 596 adopted some significance in the course of the evidence and I return to it during the course of my consideration of some of the disputed areas identified below:
Vaccines
Over the past 15 years, several manufacturers have developed pneumococcal conjugate vaccines through different approaches. These vaccines differ in the included serotypes, the carrier proteins, and their conjugation chemistry (Table 23-3).
The number of pneumococcal serotypes included in current vaccine candidates in clinical development range from 7 to 11. Serotypes 4, 6B, 9V, 14, 18C, 19F, and 23F are common to all vaccines. The Wyeth 9-valent candidate vaccine includes serotypes 1 and 5 in addition to the previous seven, while GlaxoSmithKline and Aventis Pasteur have included serotypes 1, 3, 5 and 7 in their 11-valent vaccines.
The amount of polysaccharide for each conjugate also differs between the vaccines, ranging from 1 to 10 μg per serotype. In general, pneumococcal conjugates contain less polysaccharide per serotype than the licensed Hib conjugate vaccines. This most likely reflects both the desire to reduce the total amounts of carrier protein and polysaccharide in the vaccine and a perceived greater immunogenicity for pneumococcal serotypes compared with Hib. Although it would be preferable to include a larger number of different polysaccharides in a conjugate vaccine, technically this becomes challenging. In addition, incremental benefits in coverage from increasing the number of serotypes remain low after the standard 11 serotypes have been included. Moreover, the total amount of carrier protein in the final vaccine may need to be limited because too much carrier protein can impair the antibody response to the polysaccharide antigen.
Several different carriers have been used, four being the same proteins used in Hib conjugates. Wyeth continues using CRM197, a nontoxic mutant of diphtheria toxin, in their pneumococcal vaccine. The Aventis Pasteur vaccine candidate has two different carriers: pneumococcal polysaccharides from serotypes 3, 6B, 14, and 18C are conjugated to diphtheria toxoid and those from serotypes 1, 4, 5, 7F, 9V, 19F, and 23F to tetanus protein. Merck uses OMPC, modified from N. meningitidis, as in their Hib conjugate. GlaxoSmithKline has decided to conjugate pneumococcal polysaccharides to the H. influenzae protein D. Other potential carrier proteins that have been tested in animal studies include bovine serum albumin, human immunoglobulin G (lgG), complement C3d, keyhole limpet hemocyanin, flagellar protein of Salmonella, pertussis toxoid, and pneumolysin toxoid.
Conjugates between the carrier and hapten parts [i.e. polysaccharide serotypes] have been made using a variety of procedures. Vaccine manufacturers have in general adapted their Hib conjugation technology to covalently link the carrier and hapten parts together. The basic procedure used by Wyeth first activates the polysaccharide by reaction with periodate. The carrier protein is then coupled directly to the polysaccharide through reductive amination...
9. COMPOSITION PATENTS: ANALYSIS OF INVENTIVE STEP IN LIGHT OF COMMON GENERAL KNOWLEDGE ALONE
328 MSD relies on Professor Paton as a proxy for the hypothetical but uninventive worker in the field. In his first affidavit, before he reviews the content of the composition patents, Professor Paton sets out the aim of his task in addressing the MSD problem, which was to increase the coverage of immunity against additional serotypes.
329 MSD submits that the pathway commences with the existing conjugate vaccine. In April 2005, that was Prevnar 7, which was a pneumococcal conjugate vaccine containing the Prevnar 7 serotypes, each conjugated to 2 μg of CRM197, with the exception of 6B, that had 4 μg, with a total of about 20 μg. Prevnar 7 was known to be formulated with an aluminium adjuvant. It was known that the serotypes selected in the 11-valent pneumococcal conjugate vaccines under development before April 2005 were 1, 3, 4, 5, 6B, 7F, 9V, 14, 18C, 19F and 23F. From there, MSD submits, the skilled team would move to recognise that a pneumococcal conjugate vaccine with the 13 chosen serotypes should be developed by adding serotypes 6A and 19A, all conjugated to CRM197.
330 In my view MSD over simplifies the process, and the obviousness of the task facing the skilled team at the priority date.
331 Prevnar 7 was the only publicly available pneumococcal conjugate vaccine at the time. Several others had been under development, but none were on the market. Scant details of the way in which Prevnar 7 was made were part of the common general knowledge. Less information was available about the other pneumococcal conjugate vaccines under development to which I refer in section 8.1 above.
332 The evidence convincingly establishes that those in the field knew, as a matter of common general knowledge, that significant work was being done to develop an 11-valent pneumococcal conjugate vaccine and that the best serotypes to include were the 11-valent serotypes. Those 11 serotypes were indeed a given.
333 However, it is considerably less clear where the skilled team would choose to go from there. Difficult choices demanding decisions, often based on complex research, were required. I address the issues raised by reference to the sub-headings below.
9.2 Moving to a 13-valent pneumococcal conjugate vaccine
334 MSD submits that the uninventive skilled team would as a matter of course move to a 13-valent pneumococcal conjugate vaccine to answer the MSD Problem, but no evidence indicates that any other work was being done on a vaccine with 13 serotypes at the time. It was plainly known to be advantageous to develop a vaccine against as many antigens as possible, which meant adding further polysaccharide serotypes to a conjugate vaccine. Yet Plotkin counselled caution. After referring to the Wyeth 9-valent vaccine, the GSK and Aventis 11-vaccines and the Merck 7-valent vaccine in a table at page 596, it said (emphasis added):
...Although it would be preferable to include a larger number of different polysaccharides in a conjugate vaccine, technically this becomes challenging. In addition, incremental benefits in coverage from increasing the number of serotypes remain low after the standard 11 serotypes have been included...
335 Professor Paton was an author of an article entitled “Additive Attenuation of Virulence of Streptococcus pneumoniae by Mutation of the Genes Encoding Pneumolysin and other Putative Pneumococcal Virulence Proteins” published in 2000 by Infection and Immunity (Volume 68). The introductory paragraph refers to the fact that polyvalent pneumococcal vaccines based on purified capsular polysaccharides have been available for decades but that they have, inter alia, a problem with poor vaccine immunogenicity in children. This, the article says, is addressed by conjugation of the polysaccharides to protein carriers. However, “serotype coverage will be more limited, as it is unlikely that more than 11-serotypes will be included in such conjugate formulations” (emphasis added).
336 In the Paton Review at page 127, Professor Paton said (emphasis added):
Development of pneumococcal PS-protein conjugate vaccines has been considerably more complex than was the case with Hib, owing to the multiplicity of disease-causing serotypes. A number of parameters which influence immunogenicity of conjugate antigens need to be optimized for each type, including the molecular size of the PS [i.e. polysaccharide] component, the carrier protein, the PS/carrier ratio, and the method used to covalently link the two components. In view of this developmental complexity, the number of serotypes that can be included is by necessity less than in the PS vaccine. However, the conjugate vaccines are principally designed to prevent invasive disease and otitis media in young children, for whom the range of disease-causing serotypes is more restricted than in adults.
337 This passage suggests that it was Professor Paton’s view that there were complexities involved in the development of pneumococcal conjugate vaccines where there is an increasing number of serotypes, and a need to optimise multiple parameters including the molecular size of the polysaccharide component, the carrier protein, the polysaccharide/carrier ratio, and the method used to covalently link the two components. Professor Paton seems to have changed his view on this since writing the article, and sought to distance himself from that view in cross-examination, but in my view the meaning is clear. There were thought to be technical challenges and impediments to moving beyond 11 serotypes including a requirement that the immunogenicity of conjugate antigens needed to be optimised for each serotype.
338 The “PS vaccine” referred to in the emphasised sentence is a drug called Pneumovax 23, which has 23 serotypes. Professor Paton explains in his evidence in chief that he considers that a conjugate vaccine having as many as 23 serotypes would not be commercially viable at the priority date. Later in the Paton Review Article he says (emphasis added):
…Inclusion of additional conjugated polysaccharides in the formulation may be required if nonvaccine types become too prevalent.
There are limits, however, on just how many capsular types can be accommodated. Polyvalent PS-protein conjugate vaccines are very expensive to produce, and addition of further PS types or periodic reformulation to take account of altered serotype prevalence will add further to this cost.
339 Further, Professor Paton is a co-author of a chapter in a book edited by V A Fischetti et al entitled Gram-Positive Pathogens and published in 2006 (2nd ed, ASM Press, Washington D.C.). The chapter is entitled “Pneumococcal Vaccines”. Under the heading “Noncapsular PS Vaccines” the authors refer to the clinical efficacy of polysaccharide vaccines being limited by poor immunogenicity in high-risk groups, particularly young children, and that this is being addressed by conjugation of the polysaccharides to protein carriers, thereby converting the polysaccharides from T-cell-independent to T-cell-dependent antigens. The authors say at page 291 (emphasis added):
However, serotype coverage will be limited, as it is unlikely that more than 11 serotypes will be included in such conjugate formulations.
340 The passages from his work in 2000, 2004 and 2006 tend to contradict MSD’s submission to the effect that it was obvious that a 13-valent pneumococcal conjugate vaccine would solve the MSD Problem. To the extent that Professor Paton’s oral evidence tended to support MSD’s submission, he was discomforted in cross-examination on the subject, and his attempts to distance his view from that of his co-authors on this point in the 2006 article were not convincing. I am not satisfied that in his later article he was having regard only to questions of cost. Further, despite the fact that his 2000 article was published in Infection and Immunity before the advent of Prevnar 7, and the fact that there had been considerable work in the field from then until April 2005, the consistency of his view expressed over the period from 2000 until 2004 and thereafter in 2006 suggests to me that before April 2005, it was Professor Paton’s view that there were technical challenges to proceeding towards a pneumococcal conjugate vaccine that used more than 11 serotypes. I consider that, to the extent that he expressed the view in his evidence that there were no such challenges, that view was tainted by hindsight.
341 Professor Kasper did not provide evidence in chief on this subject. He was asked by the solicitors acting for MSD whether he would have been “deterred” from pursuing a 13-valent conjugate vaccine with CRM197 as a single carrier protein because of immune interference or CIES. Plainly, that was an invitation to hindsight.
342 Professor Strugnell disagrees with Professor Paton’s approach. His view is that there was no expectation at the time that a high valency pneumococcal conjugate vaccine with more than 10 serotypes could be achieved, quoting the passage from Plotkin to which I have referred above at [334]. He considers that the development of any higher valency pneumococcal conjugate vaccine would entail a major research project with an uncertain outcome.
343 Further, the assumption tied up in MSD’s approach to the problem is that there existed already a successful 11-valent vaccine and that one simply needed to proceed from that to a 13-valent version. The state of developments in the field at the time, considering the GSK, Aventis and Wyeth products that were or had been under development, indicates that there was no fixed approach to an 11-valent pneumococcal conjugate vaccine, other than the serotypes to be selected. Different carrier proteins had been proposed, and experimental work had been tried and abandoned in respect of the bi-carrier approach used by Aventis. The state of the evidence is such that the common general knowledge does not include the fact that an 11-valent vaccine had been successfully made.
344 Accordingly, I do not accept that the uninventive skilled team would have concluded that extending coverage beyond 11 serotypes before the priority date was an idea that was likely to succeed without encountering significant technical difficulties. Indeed, it is to be noted that nowhere in Professor Paton’s evidence did he state, unprompted, that he would have moved to a 13-valent pneumococcal conjugate vaccine.
345 Professor Paton says that after identifying the additional serotypes to include in the conjugate vaccine, he would have obtained purified polysaccharides from the additional serotypes using “standard” extraction and purification techniques (e.g. those used by previous manufacturers of polysaccharide vaccines). He would have conjugated those purified polysaccharides to the same carrier protein, in the “same way” as Prevnar 7. He then would have performed the “stock standard” experimental assays to test the immunogenicity of the new vaccine components, both individually and then in combination with each other and with the Prevnar 7 serotypes.
346 Professor Paton’s evidence is that there were routine ways of conducting experiments to establish the impact of varied conditions on the immunogenicity of CRM197. Although they were not trivial, he considers that they were “well within” the capability of well-resourced persons in the field as at the priority date. However, he provides no evidence of how the process is to be achieved.
347 The only published work in evidence on the subject that also forms part of the common general knowledge is the passage in Plotkin, set out above, which relevantly provides: “The basic procedure used by Wyeth first activates the polysaccharide by reaction with periodate. The carrier protein is then coupled directly to the polysaccharide through reductive amination”.
348 Professor Kasper did not address the subject in his written evidence, but in his oral evidence he was taken to the following statement, which he agrees he gave in a declaration in the United States:
The conjugation reaction conditions must strike a delicate balance. The conditions must be robust enough to ensure that a sufficient number of the polysaccharide sugars are conjugated but mild enough to maintain a sufficient number of native unconjugated sugars and to minimise the alteration of the polysaccharide structure and consequently its immunogenicity at the site of conjugation.
349 To Professor Kasper these steps are “routine”. He gives evidence that they could be achieved within a range. His evidence is that the methods of making conjugate vaccines were well known and involved no more than routine experiments and optimisation. He has had experience in conjugating antigens to protein carriers. His evidence is that the optimisation process involves setting up a series of reactions under known controlled conditions, usually comparing several different conjugates then testing them in animals. He notes that the structures of the pneumococcal polysaccharides had been discovered and were known, which is the most technically challenging aspect, and so did not need to be done again.
350 However, even knowing this, he accepted in his oral evidence that the following conjugation reaction conditions must be considered and allowed for in producing a conjugate:
(a) whether and how hydrolysis is performed to reduce the size of the polysaccharide;
(b) the concentration of sodium periodate;
(c) the temperature and time of oxidative reaction;
(d) the temperature and time of conjugation; and
(e) the process conditions for separating conjugated an unconjugated polysaccharides.
351 During his oral evidence the following exchange took place:
MR BANNON: And what I’m putting to you, Professor, is that somebody would have to go through the process of trial and error, necessitating lengthy complex development efforts of the type you referred to in the paper in front of you.
PROF KASPER: I am saying that, yes, one would have to go through a certain number of experiments in order to get the patent – the patent – the conjugate that you would like to get. It’s not something you would do in a day, but it’s something that is easily achievable and takes no unique thinking. It’s all routinely laid out. You vary certain things in order to achieve it. You run them on columns, like you said, to know the molecular size. This is all routine in a laboratory that does this.
352 Professor Kasper accepts in his evidence that the way in which a conjugate was made could affect its immunogenicity. If one did not achieve the desired level of immunogenicity in animals, one would have to go back and try again with the conjugate by changing the parameters. While he considered the process “routine” at this point of his oral evidence, it sits uncomfortably with his statement at another point that optimising the carrier protein for each serotype in a multivalent vaccine “would be an enormous and extremely costly” task.
353 Professor Strugnell had before the priority date never prepared an antigen for use in a vaccine and had no experience in making glycoconjugates. For the purpose of preparing his affidavits he conducted a literature search and failed to find any assistance from publicly available sources as to the method of conjugation used for Prevnar 7. He anticipates the task to involve considerable trial and error, and that it is quite complicated. He does, however, defer to Professor Kasper, who had experience in actually making conjugates before the priority date, whereas his knowledge was confined to the literature.
354 The parties put diametrically opposed submissions in relation to this issue. Wyeth submits that the absence of evidence going to the method of conjugation shows that it was common general knowledge that the manufacture of conjugate vaccines was technically challenging. Adding more serotypes would have been complex, not least because the result of each conjugation may affect the immunogenicity of the vaccine and would require testing in animals. Each iteration would take three or four weeks.
355 MSD relies on the evidence of Professor Kasper and Paton that the process is “routine”. That, it submits, is supported by the statement in the composition patents that “capsular polysaccharides are prepared by standard techniques known to those skilled in the art” (page 11 lines 11 – 12) and “[t]he chemical activation of the polysaccharides and subsequent conjugation to the carrier protein are achieved by conventional means (page 11 lines 27 – 28).
356 Having regard to the whole of the evidence, I do not consider that the evidence supports the submission that the process of conjugating serotypes to a carrier protein was routine or that in embarking on the development of a 13-valent pneumococcal conjugate vaccine the skilled team would have had the relevant expectation of success. The evidence given by Professor Kasper demonstrates that there were many experimental steps required to yield a conjugation that would provide an immunogenic composition, which included effectively conjugating each of the serotypes to the carrier protein.
357 In my view the absence of evidence on the subject makes it impossible to determine that the non-inventive hypothetical skilled team would not have to undertake a course of action which was complex and detailed, as well as laborious, with a good deal of trial and error, with dead ends and the retracing of steps. Whilst Professor Paton says that it was routine, his publications that are more or less contemporaneous with the priority date indicate that there were technical challenges, and that is supported by Plotkin. There was no published information in evidence addressing the process. Professor Kasper’s experience on the subject exceeds that of the hypothetical skilled team, and his eminence and skills are such that I am not convinced that he would be a proxy for an uninventive person in the field. This blunts the impact of his statement that the process is “routine”.
358 The reference in the composition patents to the achievement by “conventional means” of chemical activation of the polysaccharides and their subsequent conjugation to the carrier protein does not assist MSD. As noted in AstraZeneca (FC), disclosures in the specification have a limited role in the inventive step enquiry. In any event, the words “conventional means” are followed by a reference, by way of example, to two patents addressing this subject. Whilst it may be the case that the broad process of conjugation using reductive amination is known to those in the art, I do not read the references to “conventional” to amount to an admission of common general knowledge of all of the steps taken. Indeed, read in the context of the subsequent examples, to which I have referred in some detail in section 4.1 above, it is apparent that the specification is at pains to identify the particular steps for the preparation and purification of the serotypes, and their subsequent activation and conjugation to CRM197. Read as a whole, I understand the specification to teach that the means of achieving the composition is of some complexity, and that the examples in the patent provide explicit guidance for each serotype to protein conjugation.
359 I accept the evidence given by Professor Strugnell to the effect that such detail is desirable to inform the reader of how the polysaccharide of each serotype and the conjugates of each serotype have been made, because the immunogenicity of conjugates can be influenced by the conjugation process. Although the claims of the composition patents are not for the process by which the multivalent immunogenic compositions are made, it is material to the consideration of inventive step that there were considerable hurdles in the way for the skilled team seeking to develop such a composition.
360 At one level it may be said that Wyeth did not in an evidentiary sense put in issue the question of whether there was a technical challenge involved in producing a 13-valent pneumococcal conjugate vaccine conjugated to CRM197. It called no witness with relevant experience before the priority date, and no person from within Wyeth who worked on the development of its patented vaccine came forward to give evidence. What challenges did those people overcome to achieve the invention?
361 However, the obligation to demonstrate a lack of inventive step lies on MSD. Its evidence in chief refers only in the most general terms to the technical process involved, with conclusory statements as to how “routine” the task was. The likelihood that the hypothetical skilled team may succeed in arriving at an immunogenic composition conjugated to CRM197 cannot be entirely divorced from consideration of what, if any, challenges may lie in its path in the formulation of the product. In the present case, I am not satisfied that MSD has established that challenges of conjugating 13 serotypes to CRM197 to which I have referred did not present to the skilled team a task requiring trial and error, the retracing of steps and an absence of confidence that they may succeed in achieving an immunogenic conjugate vaccine.
362 I am fortified in this view by the evidence that, as at April 2005, there was in fact no common general knowledge that any team had produced a 13-valent pneumococcal conjugate vaccine. The evidence of the work of Aventis and GSK indicates that whilst an 11-valent solution was technically feasible using the 11-valent vaccine serotypes, no research group had been known to advance beyond that point to a 13 serotype solution. This suggests that, despite the theoretical advantages of greater coverage of immunity, it was not a “given” that it was technically feasible to achieve that outcome.
9.4 Choice of additional serotypes
363 There is a contest between the parties as to whether the hypothetical skilled team would have selected and added serotypes 6A and 19A to the 11-valent vaccine serotypes used in other pneumococcal conjugate vaccines under development by April 2005. The issue concerns whether or not the hypothetical skilled team would understand that there was a need to add those serotypes, notwithstanding that it was well reported that cross-protection was supplied against those antigens by the inclusion of serotypes 6B and 19F in Prevnar 7.
364 MSD submits there was a prevalence of invasive pneumococcal disease caused by serotypes 6A and 19A and that by April 2005 it was known that it was highly unlikely that serotypes 6B and 19F used in Prevnar 7 would be able to provide sufficient cross-protection against 6A and 19A. It relies on the interpretation of Whitney 2003 offered by Professors Paton and Kasper. Wyeth accepts as common ground that serotypes 6A and 19A were important causes of diseases associated with the pneumococcus. However, it submits that it was common general knowledge that the degree of cross-protection afforded by serotypes 6A and 19A in Prevnar 7 was not insufficient so as to make it necessary to take the expensive, inconvenient and technically difficult step of including serotypes 6A and 19A in any new improved vaccine.
365 The parties do not disagree as to the disclosure of Whitney 2003, but disagree as to its effect. It is a case-control study examining surveillance data collected from 1998 to 2002 disclosing statistically significant reductions in invasive pneumococcal disease caused by vaccine serotypes and vaccine-related (that is, cross-reactive) serotypes following the introduction of Prevnar 7 in the United States. It reports that the rate of disease caused by vaccine and vaccine-related serotypes declined by 78% and 50% respectively in children under two years of age. In Table 1 the outcomes for specific serotypes are given.
366 In the “Results” section on the front page, the paper says:
The rate of invasive disease dropped from an average of 24.3 cases per 100,000 persons in 1998 and 1999 to 17.3 per 100,000 in 2001. The largest decline was in children under two years of age. In this group, the rate of disease was 69 percent lower in 2001 than the base-line rate (59.0 cases per 100,000 vs. 188.0 per 100,000, P<0.001); the rate of disease caused by vaccine and vaccine-related serotypes declined by 78 percent (P<0.001) and 50 percent (P<0.001), respectively...
367 In the “Conclusions” section of the front page, the paper observes that (emphasis added):
The use of pneumococcal conjugate vaccine is preventing disease in young children, for whom the vaccine is indicated, and may be reducing the rate of disease in adults. The vaccine provides an effective new tool for reducing disease caused by drug-resistant strains.
368 Whilst Whitney 2003 reports Prevnar 7 as being effective, Professor Paton notes that, in relation to children under two years of age, the decline in the estimated rate for serotype 6B reported in Whitney 2003 is 65%, whereas the decline in the estimated rate for serotype 6A is lower, at 45%. Similarly, the decline in the estimated rate for 19F is 83% whereas the decline in serotype 19A is lower, at 40%. The decline for 19A did not reach statistical significance. Professor Kasper makes similar observations. These results indicate to the Professors that there was an obvious need to add these two to the suite of serotypes in order to provide better protection for those antigens.
369 Professor Dagan sees the data in Whitney 2003 as showing that were statistically significant reductions in invasive pneumococcal disease both for the vaccine serotypes (6B, 19F) and the vaccine-related serotypes (6A, 19A). Professor Strugnell’s evidence is that it was known at April 2005 that serotypes 6A and 19A accounted for a substantial proportion of disease within their serogroups, and that these were not included in Prevnar 7. However, Professors Strugnell and Dagan both express the view that the skilled team would only add a new serotype if there was a demonstrated need for it. In their view, Whitney 2003 indicated that serotypes 6B and 19F in the existing pneumococcal conjugate vaccines, and particularly Prevnar 7, were providing cross-protection against serotypes 6A and 19A. Based on that, they would not have been led to add serotypes 6A and 19A in an improved pneumococcal conjugate vaccine.
370 Eskola, which is a 2001 paper and concerns the efficacy of a pneumococcal conjugate vaccine against acute otitis media, provides similar results. It too formed part of the common general knowledge.
371 The interpretation offered by Professors Paton and Kasper is that the lesser reduction in the rate of disease in respect of the vaccine-related serotypes amounts to a sub-optimal level of cross-protection provided to them by the vaccine serotypes. Given this, and in the interest of adding serotypes that cause the most disease that were not included in previous formulations, it was plain serotypes 6A and 19A should be included. Furthermore, these Professors convincingly point out that the issue of concern for them was not only the degree of cross-protection but also the prevalence of the disease for which the inclusion of serotypes 6A and 19A would afford protection. As Professor Paton said:
PROF PATON: The issue of cross-protection between 6A and 6B and 19A and 19B has been a matter for discussion by those in the pneumococcal vaccine field for now a very long time, over 30 years. I recall a paper by Robbins et al in 1983 where they were considering that very issue when determining the formulation of the 23 valent polysaccharide vaccine and I note that both serotypes 19A and 19F were included in that formulation. This vaccine, whilst not particularly effective in young children and not recommended for young children, is nevertheless very important in protecting older adults from pneumococcal disease. So it has been an issue around for a long time.
To me, there is clear evidence that there is – there is evidence in some studies of statistically significant reduction of 6A after immunisation with 6B, and some – even some studies also for 19F and 19A after immunisation with 19F, you know, in the case of Prevnar. But I’m not aware of any study where the extent of reduction and disease of the heterologous serotype has matched that of the homologous serotype.
MS HOWARD: Yes, thank you. Professor Strugnell.
PROF PATON: Just one other point I would make - - -
MS HOWARD: Yes.
PROF PATON: - - - is the fact that notwithstanding- quite apart from the cross-protection issue types 19A and 6A are highly prevalent causes of disease. And so in the context of formulating a higher valent vaccine, then if there was not complete cross-protection of – against 19A and 6A, then it would just as important to include these in a vaccine as a new serotype that might be 15 or something like that, which is a much less frequent cause of disease. Many, many studies show that both 6A and 19A are among the most high – you know, among the highest causes of disease. So unless your cross-protection is almost perfect, then you will have a disappointing outcome in terms of protection.
372 Professor Kasper offers further information, by referring to the structure of the polysaccharides and how single linkage differences between them make for different immune responses. He expresses the view, based in part on his own research, that single linkage differences may lead to no cross-protection. However, that level of knowledge was not shown to form part of the common general knowledge. In this respect Professor Kasper’s knowledge, and his approach, may be considered to exceed that which I would attribute to the uninventive hypothetical skilled team.
373 In an article by Professor Dagan and others entitled “Acute Otitis Media Caused by Antibiotic-Resistant Streptococcus pneumoniae in Southern Israel: Implication for Immunizing with Conjugate Vaccines” published in 2000 in the Journal of Infectious Diseases (Volume 181), the authors refer to serotypes 6A and 19A as being absent from the vaccines the subject of the study, and being “significant in terms of both prevalence and antibiotic resistance rate”. The article refers to an expectation of partial cross-protection for serotype 6A by serotype 6B, but that data suggests that none exists for serotype 19A by 19F. It proposes that if the conjugate vaccines the subject of the study prove efficacious against acute otitis media, “the addition of at least 19A and, if needed, 6A to the vaccines should be considered to further extend the antipneumococcal coverage”. Wyeth submits that this article should be placed in context, as it was published before the data in Whitney 2003 and Eskola became available and before the impact of Prevnar 7 was understood. However, whilst Whitney 2003 and Eskola identify some cross-protection, it is far from complete. In my view the Dagan article tends to support the opinions expressed by Professors Paton and Kasper.
374 Taking the evidence as a whole, I am persuaded that the idea of adding serotypes 6A and 19A to the 11-valent vaccine serotypes was ultimately one of balancing the prevalence of the disease against the degree of cross-protection available. I prefer the evidence of Professors Paton and Kasper in this regard and accept that it was obvious to select serotypes 6A and 19A for use in any 13-valent conjugate formulation.
375 MSD submits that the skilled team would as a matter of course elect to conjugate the 13 chosen serotypes to a single carrier protein, and that this would be CRM197. It relies first on the fact that Prevnar 7 was the only vaccine on the market and it used CRM197, and secondly on the fact that Prevnar 9, which was shown to have been safe and immunogenic, also used CRM197.
376 The Aventis 11-valent vaccine using a bi-carrier approach had been abandoned in 2002. The GSK 11-valent vaccine was produced using protein D as the carrier protein. Both had been shown to produce antibody concentrations worthy of note by Plotkin.
377 Professor Paton and Professor Kasper would both have first used a single carrier protein approach, which they consider would have been simpler. I accept that evidence. Although Professor Dagan disagrees that there was a general rule that it was simpler to use a single carrier, I prefer the evidence of Professor Paton and Professor Kasper who, unlike Professor Dagan, had the benchtop experience to comment on the subject. I accept that having regard to the established history of use of CRM197 as a carrier protein, including its successful use in Prevnar 7, it was a straightforward and logical decision to try it in the development of higher valency pneumococcal conjugate vaccines.
378 I now turn to consider the role of immune interference in the analysis. Wyeth submits that the term “immune interference” refers broadly to interference with the responses to conjugated and/or co-administered antigens following administration of conjugate vaccines. The interference with the immune response may be positive, in that the immune response to a particular antigen is enhanced, or negative, in that the response is suppressed. It submits that it was common general knowledge that immune interference, including but not limited to CIES, was a concern for conjugate vaccines, especially as the number of serotypes and carrier protein increased above the amounts in Prevnar 7. Because of this, a pneumococcal conjugate vaccine that involved the addition of substantially more serotypes to Prevnar 7 using the same carrier protein would likely not provide at least the same level of protection as Prevnar 7 in respect of the existing Prevnar 7 serotypes and so not provide a practical and ethical alternative or improved vaccine to Prevnar 7. It submits that this concern operated to direct the skilled team away from the inclusion of more serotypes in pneumococcal conjugate vaccines generally and to adopt approaches to avoid those anticipated problems, such as using multiple carrier proteins or new carrier proteins. Those approaches would have led the skilled team to be reluctant to add further serotypes to the existing known pneumococcal conjugate vaccines. MSD disputes that considerations concerning immune interference would have deflected the skilled team from arriving at the claimed invention.
379 Generally, Wyeth’s submissions on this subject were perhaps a little diffuse, but insofar as one can tell, tended to put forward three supposed causes of immune interference: (1) the amount of carrier protein; (2) the addition of serotypes; and (3) the co-administration of multiples vaccines. As will be seen below, the evidence indicates that the relevance of points (2) and (3) is that they may vary according to the amount of carrier protein or proteins (point (1)) administered to a person at the time of vaccination. As MSD submits, the upshot of the evidence discussed below is that the total amount of carrier protein in a final vaccine may need to be limited because too much carrier protein could impair the antibody response to the polysaccharide antigen. However, MSD submits that this was within the skill of those in the field.
9.6.2 The expert evidence concerning immune interference
380 Professor Strugnell in his evidence in chief addresses immune interference. One form, sometimes referred to as “bystander interference”, occurs when the co-administration and or combination of vaccines containing a given carrier protein in a conjugate induces interference that extends to unrelated non-conjugated antigens that are part of the combinations in use. As Professor Dagan explains, another form, CIES, arises from pre-existing immunity to a carrier protein, which may suppress the immune response to a polysaccharide linked to the same carrier protein, thus jeopardising the polysaccharide immune responses. Professor Strugnell says that if the conjugates are predominantly captured and internalised by carrier-specific B-cells rather than polysaccharide-specific B-cells, then a predominantly anti-carrier antibody response will ensue, thereby suppressing the immune response to the polysaccharide antigens in the conjugates. Alternatively, pre-existing antibodies directed at the carrier protein can sometimes increase the antibody response to the polysaccharide antigens by increasing binding to the conjugates. Professor Strugnell gives evidence that at another level, there is competition between the different polysaccharide antigens of different serotypes in the conjugates in a multivalent conjugate vaccine in circumstances where the polysaccharide antigens of different serotypes are each conjugated to a carrier protein of the same type. This may result in the immunogenicity of one or more of the individual conjugates being impaired or suppressed. He says that one explanation for this is that the serotype specific B-cells are competing for a finite number of carrier-primed helper T-cells, and this finite number may be less than required for full T-cell ‘help’ for each of the conjugates of a different serotype in the vaccine. Professor Dagan generally agreed with this description.
381 I am not satisfied that the matters to which Professors Dagan and Strugnell refer, at the level of detail which I have summarised, formed part of the common general knowledge. Professor Strugnell’s evidence in chief did not contain references to published materials. He did not work in the field of pneumococcal conjugate vaccines at the time and was not familiar in April 2005 with the Wyeth 9-valent pneumococcal conjugate vaccine or the GSK and Aventis 11-valent pneumococcal conjugate vaccines, although Wyeth accepts that these formed part of the common general knowledge of those in the field. While Professor Strugnell refers to being “familiar with the concept of prior immunity affecting subsequent immunity” at the priority date, he agrees that his evidence in chief is based on publications provided to him by Allens and others that he found via searches for the purposes of the proceedings.
382 I also do not consider that the knowledge of Professor Dagan concerning CIES may be taken to reflect the common general knowledge. I summarise aspects of his evidence about immune interference below.
383 Professor Dagan explains that Dagan 1998 (cited in Plotkin) reported a study in which he was involved conducted by Aventis, where two separate 4-valent pneumococcal conjugate vaccines containing polysaccharide antigens from serotypes 6B, 14 19F and 23F were conjugated to tetanus toxoid and diphtheria toxoid carrier proteins respectively. The study examined the potential interference with the immune response of several co-administered vaccines containing the same protein component, being tetanus toxoid or diphtheria toxoid. The study showed that when the tetanus toxoid pneumococcal conjugate vaccine was administered to infants together with a Hib tetanus toxoid conjugate vaccine and also a diphtheria-tetanus-pertussis vaccine, the anti-diphtheria and anti-tetanus antibody responses were reduced compared with those administered with any of those alone, or with a placebo. A dose range study showed that the antitetanus and anti-polysaccharide antibody concentrations were inversely related to the amounts of tetanus toxoid administered, in that the antibody concentrations lowered as the amount of tetanus toxoid administered was increased. The text to the article then refers to the first conjugate vaccines which were directed against Hib, where a polysaccharide was covalently conjugated to a protein carrier. The article goes on to say (citations omitted):
The same technology is now used to widen the range of conjugate vaccines against invasive organisms such as pneumococci and other encapsulated organisms. Multiple vaccines based on the same protein carrier and thus having common antigenic epitopes might be available soon, and the possibility of their interactions must be considered. The simultaneous administration of several conjugate vaccines sharing the same protein carrier and the carrier itself may be associated with the suppression of the response to polysaccharides through various mechanisms.
384 Accordingly, Dagan 1998 refers to immune interference arising from the co-administration of several vaccines each sharing the same carrier protein, specifically tetanus toxoid. As the Abstract to the article states, “This phenomenon, which we believe derives from interference by a common protein carrier, should be taken into account when the introduction of an immunization program including multiple conjugate vaccines is considered”.
385 Reported evidence of immune interference is that immune responses were sometimes reduced (and sometimes not) when multiple different vaccines were co-administered.
386 Immune interference in a more general sense was known to those skilled in the art. Professor Dagan relies heavily on his own publication, Dagan 2004. It reports that immune response between seven pneumococcal conjugates conjugated to tetanus toxoid in the Aventis-Pasteur 11-valent pneumococcal conjugate vaccine (the serotypes being 1, 4, 5, 7F, 9V, 19F and 23F) was significantly reduced after primary and booster immunisation, when the vaccine was given concurrently with an acellular pertussis-containing vaccine (being a combination of diphtheria toxoid, tetanus toxoid, acellular pertussis, inactivated poliovirus and Hib conjugate vaccines). He concluded that this result was due to interference between the vaccines. He and his co-authors postulated that the ratio of tetanus-specific T-cells relative to polysaccharide-specific B cells may have been reduced, or cells suppressed, by the relatively large amount of carrier protein in the 11-valent pneumococcal conjugate vaccine and co-administered combination vaccine. Reference is made to the observed results in Dagan 1998.
387 Professor Dagan gives evidence that the results led to the development of a candidate 11-valent pneumococcal conjugate vaccine in which the antigenic load of any single carrier is minimised by producing a bi-carrier glycoconjugate. That candidate drug was, according to Professor Dagan, the Aventis 11-valent vaccine.
388 Dagan 1998 and Dagan 2004 have not been shown to form part of the common general knowledge. Professor Dagan’s emphasis on them, and concerns in relation to the addition of serotypes, were no doubt elevated because he was directly involved in research in the subject. As noted in my summary of his evidence in section 7.4.3 above, Professor Dagan regarded immune interference as a real concern. He considered that there was no evidence that increasing the serotype coverage to higher than seven would not cause interference, in circumstances where it was documented that, at a certain point, the addition of serotypes could result in a reduction in immunogenicity, and such results were unpredictable. He gives evidence of Aventis’s bi-carrier approach which is derived from his participation in its research, and he was apparently privy to its decision-making processes. CIES was the subject of Professor Dagan’s specific research and he had pioneered developments in understanding it and immune interference. These matters would appear to have elevated his knowledge, and perhaps his concerns, about the likely effect of CIES upon the development of a multivalent pneumococcal conjugate vaccine, and in particular whether or not a single carrier protein would be effective, to a level somewhat beyond that which may be expected of the person skilled in the art in the field.
389 Professor Kasper considered that the phenomenon had not yet been proven, but was attributed by some to the immune system. He considered that there was no data concerning immune interference that would have prevented him from moving forward to use CRM197 as a carrier protein conjugated to 13 serotypes. The evidence indicated to him that CRM197 was not associated with any decreased responses when used as a carrier for a polysaccharide, that there was improved immunogenicity when vaccines were combined, and they always gave responses well above protective levels, citing, amongst others, Obaro. Dagan 2004 and Dagan 1998 did not show enough of a decrease in immunogenicity to raise concerns about moving forward with conjugate vaccines, and neither showed an issue with adding serotypes to CRM197.
390 Wyeth’s closing submissions refer to a number of articles co-authored by Professor Kasper in order to challenge the views to which he adhered in cross-examination, and to establish that Professor Kasper considered immune interference was a concern to be taken into account, especially as the number of serotypes and carrier protein increased. However, the articles relied upon are not directed to equivalent subject matter. In one (exhibit G) the researchers were testing for immune responses to see whether a new carrier was effective. The remark relied upon by Wyeth is a general statement going to the risk of immune interference where previous exposure to a certain carrier results in immunisation to that carrier, but it does not refer to issues concerning the amount of carrier in one vaccine. In another (exhibit J), the question concerned whether two Group B streptococcal polysaccharides with overlapping structures generated an immune response when administered simultaneously via two different vaccines, which is different to issues of immune interference caused by a protein carrier.
391 Professor Paton was aware before April 2005 of the general possibility of immune responses being reduced as a result of over-administration of a vaccine antigen, this being what he terms immune interference. He was also aware that immune interference was said by some in the field to be an issue associated with the development of multivalent polysaccharide-protein conjugate vaccines. He understands the form of immune interference identified by Professor Strugnell as CIES to be a theoretical mechanism proposed to account for the possibility of a reduced immune response to a polysaccharide antigen in a conjugate vaccine where multiple polysaccharides are conjugated to the same carrier protein. Before April 2005, Professor Paton did not consider that there was a clear mechanistic explanation underlying the concept. He considered that it had not risen beyond the level of a theoretical issue, and was not aware of any evidence that demonstrated before April 2005 that CIES was a real phenomenon in the context of multivalent pneumococcal conjugate vaccines. It would not have deterred him from proceeding to develop a 13-valent pneumococcal conjugate vaccine.
392 In the Paton Review Article, Professor Paton identifies that the use of pneumolysoid, or other suitable pneumococcal proteins, “as carriers for [polysaccharides] in conjugate vaccines may also minimise any problems associated with overuse of existing carrier proteins” (emphasis added). I do not consider that this reference tends to contradict his evidence on the subject of immune interference and those vaccines. In closing submissions Wyeth suggested, by reference to some of Professor Paton’s articles published in 1994, 1996 and 2001, that immune interference was considered by him then to be of concern. However, that suggestion does not survive scrutiny. None rise above the statement in Plotkin that the amount of carrier used “may need” to be limited. None refer to the use of CRM197.
393 Ultimately, I consider that the likely approach of the skilled team is to be understood having regard to the content of Chapter 23 of Plotkin. The experts were asked about the following passage in Plotkin at page 596 under the heading “Vaccines” (emphasis added):
The amount of polysaccharide for each conjugate also differs between the vaccines, ranging from 1 to 10 μg per serotype. In general, pneumococcal conjugates contain less polysaccharide per serotype than the licensed Hib conjugate vaccines. This most likely reflects both the desire to reduce the total amounts of carrier protein and polysaccharide in the vaccine and a perceived greater immunogenicity for pneumococcal serotypes compared with Hib. Although it would be preferable to include a larger number of different polysaccharides in a conjugate vaccine, technically this becomes challenging. In addition, incremental benefits in coverage from increasing the number of serotypes remain low after the standard 11 serotypes have been included. Moreover, the total amount of carrier protein in the final vaccine may need to be limited because too much carrier protein can impair the antibody response to the polysaccharide antigen.
394 Professors Strugnell and Dagan agree that the passage represented the state of the art. Professors Kasper and Paton consider that the Plotkin passage indicates that there were concerns within the field about immune interference, but that there was no definitive evidence presented as to whether those concerns were justified.
395 In concurrent evidence the distinction in the views was clarified. It appears that Professor Dagan and Professor Kasper agreed that by April 2005 there was not enough information to enable one fully to understand CIES. Professor Kasper’s view, reiterating that expressed in his affidavit evidence, was that there was no convincing data supporting the existence of CIES as applied to polysaccharide glycoconjugates before 2005. Professor Dagan’s evidence is that CIES is a “phenomenon”, but the explanation for it is not yet known.
396 As I have noted, the information in in Chapter 23 of Plotkin has been shown to be part of the common general knowledge. As the experts said in the composition JER, it provides the relevant state of the art for both experimental and licenced pneumococcal vaccines. In that chapter the authors also relevantly said in a passage on page 604 under the heading “Variables Influencing Immunogenicity of Conjugate Vaccines” (emphasis added, citations omitted):
Several factors affect the magnitude of the antibody response. In addition to the composition of the vaccine, these include the number of vaccine doses, the vaccination schedule, and possible interference with simultaneously administered vaccines. Clear-cut differences exist in the antibody responses to different capsular-type polysaccharides in the conjugate vaccines. Polysaccharide-based PncCRM vaccines (bi-, penta-, or heptavalent) have been shown to be immunogenic in infants, whereas the antibody responses to the corresponding oligosaccharide formulations have remained relatively modest. Inclusion of additional serotypes does not seem to significantly affect the immunogenicity of each individual conjugated polysaccharide. Serotypes 6B and 23F appear to be poor immunogens in spite of conjugation, whereas serotype 19F induces a relatively high antibody concentration. Serotypes such as 3 and 18C are usually satisfactory immunogens even after the first dose. A significant increase in anti-6B lgG can be seen only after the third dose. For serotype 4, there is already a marked response after the first dose, and a further increase after the second dose, but not after the third dose. Serotype 19F induces an antibody response after the second dose, and the third dose does not increase the mean antibody concentrations.
…
Immunity to carrier proteins is beneficial for the improved immunogenicity of conjugate vaccines. Early priming with carrier proteins enhances the immune response to polysaccharides in subsequent immunizations with conjugates. Although this T-cell immunity against carrier proteins is essential for responses to polysaccharides, B-cell immunity to carrier protein may become detrimental on induction of anti-carrier antibodies. In a situation where polysaccharide- and protein-specific B cells compete to capture conjugates, high amounts of antibodies to carrier proteins may result in a suppressed immune response to polysaccharides. This argues for minimizing the amount of antigen and implementing mixed carrier vaccines.
397 The second last quoted sentence includes a footnoted reference to Dagan 1998. The emphasised passage in the first paragraph quoted indicates that inclusion of additional serotypes does not significantly affect immunogenicity of each individual conjugated polysaccharide.
398 CIES is also addressed in Chapter 29 of Plotkin which is entitled “Combination Vaccines”, under the heading “Carrier-Induced Epitopic Suppression”. It is described as the phenomenon where antibody responses to haptens presented on a carrier are inhibited by prior immunisation with the specific carrier. It states that concurrent administration of two conjugate vaccines employing the same carrier may lead to interference. After referring to some studies, it says that data “make it clear that the effect of prior or concomitant administration of proteins used in conjugate vaccines is unpredictable and must be evaluated for each vaccine combination”. Whilst Wyeth submits that this chapter formed part of the common general knowledge, no evidence supports that proposition. The agreement of the experts on this subject was confined to Chapter 23. Accordingly, for present purposes it must be set to one side.
9.6.3 Findings in relation to immune interference
399 Having regard to all of the relevant evidence, including Plotkin, in my view the position insofar as the common general knowledge is concerned is that the skilled team before April 2005 was aware that the total amount of carrier protein in a final vaccine may need to be limited because too much carrier protein could impair the antibody response to the polysaccharide antigen. However, there is no evidence to support, specifically, the proposition advanced that CIES had been reported in relation to a single multivalent conjugate vaccine after the addition of serotypes to an earlier vaccine of the same type, or, generally, that by increasing the serotype coverage to valencies above 7 with CRM197 as the carrier, the amount of carrier protein would increase to a level to make the occurrence of CIES likely.
400 I prefer to rely on the objective evidence represented by the passages in Plotkin to which I have referred and the practical evidence given by Professors Paton and Kasper, rather than Professor Dagan, concerning the level to which issues arising from immune interference would affect consideration of the development of a new composition. In my view the skilled team would not have been directed away from the inclusion of more serotypes in a pneumococcal conjugate vaccine by reason of concerns about immune interference or have considered that by reason of immune interference (however defined) a 13-valent pneumococcal conjugate vaccine would not work. Taking Plotkin as an objective touchstone on the subject, it is apparent that at page 596 the concerns raised are muted. It suggests at 596 “the total amount of carrier protein in the final vaccine may need to be limited because too much carrier protein can impair the antibody response to the polysaccharide antigen” (emphasis added). At page 604 it suggests that “Inclusion of additional serotypes does not seem to significantly affect the immunogenicity of each individual conjugated polysaccharide.” Accordingly, Wyeth’s reliance on immune interference to strengthen its inventive step case fails.
9.7 Primary conclusions in relation to lack of inventive step in the light of the common general knowledge
401 For the reasons developed above, in my view MSD has not established lack of inventive step on the basis of common general knowledge alone. In particular I am not persuaded that the hypothetical skilled but uninventive team would have considered it obvious, in seeking to improve Prevnar 7, to move from the common general knowledge to the claimed invention, in circumstances where there were acknowledged technical challenges in doing so, and no clear means by which the skilled team would address and answer them as set out in sections 9.2 and 9.3 above.
402 The common general knowledge indicates that only one multivalent immunogenic pneumococcal conjugate vaccine was on the market, being Prevnar 7. Little was known about how the polysaccharides were prepared, purified, activated and conjugated. Less was known about Wyeth’s 9-valent composition. The skilled team may well have wished to consider moving from that to an 11-valent, rather than a 13-valent, pneumococcal conjugate vaccine conjugated to CRM197, but the common general knowledge provided no information as to how either of those options may be achieved. The process would be challenging, requiring extensive work with unpredictable and uncertain results. Even if a decision had been made to move beyond 11 serotypes, whilst as a matter of theory the addition of serotypes 6A and 19A to the 11-valent vaccine serotypes was an attractive (and obvious) option, the means by which that was to be achieved was not.
9.8 Secondary indicia of inventiveness
403 Wyeth submits a secondary indication of non-obviousness is the fact that MSD, Aventis and GSK were pursuing solutions to the problem of developing a vaccine that was an improvement over Prevnar 7 and took a different route to that which is now said to be obvious by MSD. It submits that it may be inferred that each of those companies had access to a relevant team of experts, armed with at least the common general knowledge and access to CRM197, and that they pursued alternative approaches, none arriving at a 13-valent solution, and none conjugating to CRM197. Wyeth relies on the observations made by the High Court in Lockwood No 2, to the effect that Australian courts should be slow to ignore secondary evidence: [115] – [119].
404 MSD submits that Wyeth is precluded from raising this argument, because Federal Court Practice Note IP-1 (the practice note) required it first to be pleaded. The practice note states that where an invalidity claim is raised on the basis of lack of inventive step, the patentee may wish to rely on secondary indicia, for instance commercial success. If it seeks to do so, it should inform the Court of that fact “at the earliest opportunity”. The Court may require that relevant facts or matters be pleaded or particularised “so that the other party is provided with an adequate opportunity to address the issue by evidence”. MSD submits that in the absence of pleading the subject, Wyeth may not raise the argument.
405 Three points may be made in rejecting MSD’s position. First, the practice note does not require a pleading but instead notice to the Court, where appropriate. Secondly, MSD’s case relied upon the prior art pneumococcal conjugate vaccines. MSD put on evidence to address these vaccines. Its inventive step challenge involved knowledge of the serotypes used in those vaccines and knowledge that vaccines with a greater number of serotypes than Prevnar 7 were being developed. Thirdly, plainly enough, Wyeth was entitled to respond to that evidence. It did so in its affidavit material in chief in some detail, drawing attention to the differences between the various prior-art vaccines. There can be no doubt, having regard to that evidence, that Wyeth gave sufficient notice of its intention to rely on secondary evidence.
406 Next, MSD submits that where a patentee relies upon secondary considerations as an indicium of inventive merit, it bears the evidentiary onus, citing Garford Pty Ltd v DYWIDAG Systems International Pty Ltd [2015] FCAFC 6; 110 IPR 30 at [84] (Dowsett, McKerracher and Nicholas JJ). That proposition is of course correct. MSD submits that Wyeth has failed to discharge its onus by establishing that the attempts of the other manufacturers did not fail for reasons irrelevant to the inventive step inquiry, such as commercial reasons or other reasons.
407 However, Wyeth’s submission is not put at the level that the other companies “failed”, but rather as an indication that the solution that was said by MSD to be very plain (or obvious) was not pursued by the several teams that were, before April 2005, attempting to develop an improved form of pneumococcal conjugate vaccine. It is true that there could be many reasons as to why they took approaches that did not involve the use of 13 serotypes or that involved (variously) a bi-carrier approach (Aventis), the use of protein D (GSK) or OMPC (MSD) as a protein carrier. But at a high level, it is not irrelevant to add to the inventive step calculus the following question: if in April 2005 it was obvious to conceive of the idea to develop a 13-valent pneumococcal conjugate vaccine and then put it into practice, why did no one else who, it may be inferred, was working to produce a viable alternative or better pneumococcal conjugate vaccine to Prevnar 7, adopt that approach? In this respect, secondary evidence is just that. It does not, and has not in the present case, provided the basis for a primary view. It indicates that the primary view formed is supported by a relevant inference that is available on the evidence.
408 Wyeth submits that a further indication of inventiveness is that MSD has copied the invention claimed in the composition patents. It submits that MSD’s sole purpose in commencing this proceeding is to make sure that the way is clear for it to launch its 15-valent vaccine, a matter that it pleaded in its Statement of Cross-Claim. Wyeth submits that MSD admits that the 15-valent vaccine includes pneumococcal polysaccharide-protein conjugates of each of the 13 serotypes identified in the asserted claims, all conjugated to CRM197. This, together with the fact that it abandoned the development of its 7-valent vaccine using OMPC as the carrier protein, supports an inference of copying, and that the invention is inventive.
409 In the present case I give this submission no weight. First, Wyeth has not given notice of reliance on the point. An allegation of patent infringement does not equate to an allegation of copying and it is unfair to cast these allegations properly for the first time in closing submissions without specific notice. The position is factually quite different to that in Lockwood No 2 where Mr Alchin, the inventor of the infringing product, was cross-examined on the subject. Secondly, no evidence discloses when or how MSD developed its 15-valent vaccine, or when it made a decision to develop such a vaccine. The onus lies on Wyeth to establish the details of those events, which it has not done.
10. COMPOSITION PATENTS: ANALYSIS OF INVENTIVE STEP IN LIGHT OF THE PRIOR ART INFORMATION WITHIN S 7(3)
410 MSD submits that if the asserted composition patent claims are not obvious having regard to the common general knowledge alone, then they are obvious having regard to the common general knowledge in conjunction with prior art information pursuant to s 7(3). The additional information is:
(a) Peña and Obaro;
(b) Hausdorff 2000 and 2002; and
(c) Yu.
411 MSD also relied upon Plotkin, Whitney 2003 and Eskola for the purposes of s 7(3), but these have been determined to form part of the common general knowledge in any event.
412 For the reasons set out below, in my view the additional information contained in those publications does not serve to render obvious that which was not obvious having regard to the common general knowledge alone.
413 MSD relies on Peña alone or read with Obaro pursuant to s 7(3) of the Patents Act. The pre-RTB Patents Act applies to the 013 patent, and the post-RTB Patents Act applies to the 844 patent.
414 In relation to the 013 patent, MSD submits that Peña and Obaro are pieces of prior art information that the skilled person in the field could, before the priority date, be reasonably expected to have ascertained, understood, regarded as relevant and, where Peña and Obaro are combined, to have combined. Wyeth disputes that Peña would have been ascertained. In relation to the 844 patent, there is no dispute that both pieces of prior art information are to be made available to the notional hypothetical skilled team, the post-RTB Patents Act version of s 7(3) no longer having the requirement that the prior art be ascertained, understood, and regarded as relevant.
415 MSD submits that Peña refers to work being done and studies performed on a 13-valent pneumococcal conjugate vaccine with each of the 13 claimed serotypes, and that the inclusion of serotypes 6A and 19A in that composition provides a basis for the hypothetical skilled team to include them in its 13-valent vaccination. It also submits that the hypothetical skilled team would have understood that Peña implicitly discloses CRM197 as the carrier for the serotypes, or alternatively, that it would have chosen to use CRM197 based on Peña because it emphasised the success of Prevnar 7 and the Wyeth 9-valent vaccine, both of which had used CRM197, and because CRM197 was a known and safe carrier protein.
416 In my view the disclosure of Peña provides some limited assistance to MSD, insofar as it clearly indicates that the 13-valent vaccine under development involves the use of serotypes 6A and 19A and discloses that those serotypes would be worth trying in a future composition. However, it does not provide sufficient assistance to overcome the difficulties that I have identified in MSD’s lack of inventive step case. The invention claimed is not the mere idea to develop a 13-valent pneumococcal conjugate vaccine. It is the idea coupled with the execution of making all immunogenic compositions with the claims. As I have noted in section 9, I am not satisfied that the skilled team would have had the relevant expectation of success in making a composition within the claims. Peña does not resolve the difficulties to which I have referred in sections 9.2 and 9.3 above. In short, whilst suggesting that a 13-valent pneumococcal vaccine conjugated to CRM197 was at some stage of development, it provides no information to indicate how that desirable but difficult goal may be achieved.
417 In my view, combining Obaro with Peña does not advance the position any further. It refers to the Wyeth 9-valent pneumococcal conjugate vaccine as being safe and immunogenic. It does not advance any information going to difficulties that may arise in relation to the development of a 13-valent conjugate vaccine.
418 Having regard to these conclusions, it is not necessary to address in detail Wyeth’s submission that Peña would not have been ascertained for the purposes of the 013 patent. My preliminary view, is that, having regard to the evidence of Ms Jones, and the search terms used, Peña would have been ascertained, understood and regarded as relevant. Nonetheless, by combining the common general knowledge with the disclosure of Peña and Obaro, I do not consider that the invention claimed in the composition patents would have been obvious to the skilled but uninventive hypothetical team.
10.3 Hausdorff 2000 and Hausdorff 2002
419 MSD relies separately, and as a combination, on Hausdorff 2000 and Hausdorff 2002 as pieces of prior art information within s 7(3) of the Patents Act. There is no dispute that they are available for review within the requirements of that section under the pre-RTB Patents Act or post-RTB Patents Act. Neither advances MSD’s case very far.
420 MSD contends that a person skilled in the art would have understood both Hausdorff 2000 and 2002 to be recommending a 13-valent vaccine that includes serotypes 6A and 19A and CRM197 as the protein carrier. Wyeth submits that the person skilled in the art upon reading Hausdorff 2000 would have concluded that whether or not those serotypes would be included was dependent on the degree of cross-protection provided by related serotypes, and that further trials and investigation was required. Wyeth accepts that upon reading Hausdorff 2002 a person skilled in the art would have considered serotypes 6A and 19A to be important serotypes in respect of any vaccine intended to combat otitis media. However, that does not make it a matter of routine to include those serotypes in any pneumococcal conjugate vaccine, given the issue of cross-protection, in respect of which Hausdorff 2000 had directed the person skilled in the art to monitor ongoing efficacy trials.
421 Hausdorff 2000 provides an analysis of approximately 70 data sets to compare the serotypes causing invasive pneumococcal disease with those represented in 7-, 9- and 11-valent conjugate vaccine formulations. I consider it likely that the hypothetical skilled team would have formed an understanding that the vaccines mentioned in the article were those known to have been or were being developed at the time, namely Prevnar 7, the Wyeth 9-valent vaccine, and the Aventis and GSK 11-valent vaccines.
422 The article mentions the importance of serotypes 6A and 19A. Under the heading “Results” the paper describes the studies forming the basis of the data sets, and their wide geographical spread. It says:
Unfortunately, very few study reports from outside the Western Hemisphere, Oceania, and Europe included serotype-specific information, precluding detailed global analyses. However it is apparent that nonvaccine serotypes 6A and 19A account for a substantial portion of disease within their serogroups.
423 The paper considers disease-causing serotypes by region and by age group. Under the heading “Discussion”, it includes the following sentence:
To maximize coverage of [invasive pneumococcal disease] in younger children, for example, future vaccines may need to include 6A and 19A, depending on the cross-protection seen in ongoing efficacy trials with the current vaccine formulations (containing 6B and 19F). To optimize global coverage, future vaccines should also include representatives of serogroups 12 and 15.
424 The person skilled in the art would have understood that serotypes 6A and 19A were emerging as additional serotypes of interest. In my view that would have indicated that it was obvious to select serotypes 6A and 19A as candidate serotypes for inclusion in a vaccine, provided that a decision was made to move from an 11-valent pneumococcal conjugate vaccine to a 13-valent pneumococcal conjugate vaccine.
425 The abstract to Hausdorff 2002 states that the authors analysed nine datasets comprising pneumococcal isolates from middle ear fluid samples collected from 1994 until 2000 to examine the distribution of pneumococcal serotypes in relation to several demographic and epidemiologic variables, including gender, age, antibiotic resistance and source of culture material. The results identify major serotypes and their prevalence. The conclusions report that analysis of several geographically diverse datasets indicates that “a limited number of serotypes, largely represented in [Prevnar 7], accounted for the majority of episodes of pneumococcal [acute otitis media] in children between 6 and 59 months of age. Certain serotypes appeared to be relatively more significant in children <6 months or >59 months of age”.
426 It is apparent that Hausdorff 2002 was not a study of the efficacy of existing vaccines, or of cross-protection afforded between serotypes. After looking at the datasets, the paper refers to the pneumococcal conjugate vaccines available, being what the hypothetical skilled team would have understood to be the same 7-, 9- and 11-valent vaccines identified in Hausdorff 2000. In the Discussion section, the paper states that one of its aims is to identify the pneumococcal serotypes “most responsible for [acute otitis media] in children and relate those to specific vaccine formulations”. The findings include the statement:
It appears that the serotypes represented in PCV-11, plus 6A and 19A, comprise all major serotypes in each age group studied.
427 In my view this would have further conveyed to the skilled team that serotypes 6A and 19A were of interest, and it would have been obvious reading these two articles to select serotypes 6A and 19A as candidate serotypes for inclusion in a 13-valent pneumococcal conjugate vaccine. However, for the reasons set out in relation to the Peña and Obaro publications, the provision of this information does not advance MSD’s inventive step case any further.
428 The abstract to Yu explains that infants were immunised with one of the three experimental pneumococcal conjugate vaccines containing serotypes 6B and 19F but not 6A or 19A. Their sera were studied for the capacity to opsonise Streptococcus pneumoniae 6A, 6B, 19A and 19F serotypes. The article discloses that two of the experimental vaccines were prepared by Wyeth and one by MSD.
429 MSD relies on the disclosure of Yu as yet a further publication demonstrating the obviousness of the choice of serotypes 6A and 19A in a 13-valent pneumococcal conjugate vaccine. Having regard to my conclusions in this respect in relation to the Peña, Obaro and Hausdorff publications, it is unnecessary to address the subject any further.
11. COMPOSITION PATENTS: AN EVIDENTIARY RULING
430 At the hearing objection was taken by MSD to the following part of the first affidavit of Professor Strugnell, and the article to which it refers (at [264]):
I am now aware that, after April 2005, GSK has trialled a combined pneumococcal conjugate and protein vaccine. The inclusion of the protein antigens did not provide any benefit in terms of reducing nasopharyngeal carriage of pneumococci in infants. See Odutola et al, 'Efficacy of a novel, protein-based pneumococcal vaccine against nasopharyngeal carriage of Streptococcus pneumoniae in infants: A phase 2, randomized, controlled, observer-blind study', Vaccine, 2017; 35:2531-2542.
431 Wyeth contended that it is relevant to the likely success of alternative routes of enquiry available to the person skilled in the art. The passage was provisionally admitted. No submission was made as to its content in closing addresses. Nor was I taken to the Odutola article to which it refers. The article was published after the priority date. The evidence can have at best only a most tangential bearing on the inventive step enquiry. No relevance of the objected to portion has been established. I reject it.
12. COMPOSITION PATENTS: INUTILITY
432 Section 18(1)(c) of the Patents Act provides that an invention is a patentable invention for the purposes of a standard patent if the invention, so far as claimed in any claim, is useful. Until recently, the requirement that an invention so far as claimed be “useful” within s 18(1)(c) was defined solely by reference to the common law development of that concept. The words in Lane Fox v Kensington and Knightsbridge Electric Lighting Co [1892] 3 Ch. 424 at 431 of Lindley LJ (with whom Lopes LJ agreed) set the scene (emphasis added):
The utility of the alleged invention depends not on whether by following the directions in the complete specification all the results now necessary for commercial success can be obtained, but on whether by such directions the effects which the patentee professed to produce could be produced, and on the practical utility of those effects.
433 What the patentee “professed to produce” is to be ascertained by having regard to what is now routinely referred to as the “promise of the invention” being the promise that the specification is said to make of the invention claimed: Rehm Pty Limited v Websters Security Systems (International) Pty Limited [1988] FCA 232; 81 ALR 79 at 84 and 96 – 97 (Gummow J); Décor Corp Pty Ltd v Dart Industries Inc [1988] FCA 682; 13 IPR 385 at 394 (per Lockhart J). This is assessed as a matter of construction of the specification: see generally ESCO Corporation v Ronneby Road Pty Ltd [2018] FCAFC 46; 358 ALR 431 at [182] – [239] (Greenwood, Rares and Moshinsky JJ).
434 For post-RTB Patent Act patents, such as the 844 patent, s 7A provides a further definition:
Meaning of useful
(1) For the purposes of this Act, an invention is taken not to be useful unless a specific, substantial and credible use for the invention (so far as claimed) is disclosed in the complete specification.
(2) The disclosure in the complete specification must be sufficient for that specific, substantial and credible use to be appreciated by a person skilled in the relevant art.
(3) Subsection (1) does not otherwise affect the meaning of the word useful in this Act.
435 MSD does not rely on s 7A.
436 In each case it is necessary to consider the nature of the promise of the invention by reference to the specification and also whether that promise is met by that which is the subject of the claims. Often that enquiry gives rise to a question of claim construction: if a broad claim includes something that does not meet the promise of the invention, will it be invalid for want of utility? In this context at first blush there appears to be some tension in the authorities.
437 One relevant principle of claim construction is that it is not legitimate to narrow or expand the boundaries of the monopoly as fixed by the words of a claim by adding to those words glosses drawn from other parts of the specification: see Welch Perrin at 610. In H Lundbeck A/S v Alphapharm Pty Ltd [2009] FCAFC 70; 177 FCR 151 at [81] Emmett J said that:
A claim is bad if it covers means that will not produce the desired result, even if a skilled person would know which means to avoid. That is to say, everything that is within the scope of a claim must be useful, otherwise the claim will fail for inutility (see William WM Wrigley Junior Company v Cadbury Schweppes Pty Ltd (2005) 66 IPR 298 at [138]).
438 However, that is not to say that for the purposes of the inutility ground of revocation claim construction should be approached mechanistically. The passage from WM Wrigley Jr Co v Cadbury Schweppes Pty Ltd [2005] FCA 1035; 66 IPR 298 at [90] (Heerey J) at [138] to which Emmett J referred was as follows:
A claim is bad if it covers means that will not produce the desired result even if a skilful person would know which means to avoid: Norton and Gregory Limited v Jacobs (1937) 54 RPC 271 at 276, Welch Perrin & Co Pty Limited v Worrel (1961) 106 CLR 588 at 601, Martin Engineering Co v Trison Holdings Pty Ltd (1989) 14 IPR 330 at 337. Menzies J pointed out in Welch Perrin (at 602) that this does not mean that a specification should be construed in a way that any sensible person would appreciate would lead to unworkability when by construction it could be given a more limited meaning. However, in the present case the claims read in the light of the specification as a whole distinguish between those claims which specify a particular amount of hydrogenated isomaltulose (2, 3 and 15) and the remainder, which do not. To imply a minimum of 50 per cent in the latter claims would be, in the words of Lord Greene MR in Norton (at 276) not to construe the specification but to amend it. I therefore uphold this ground.
439 From here it may be seen that the question of whether a broad claim is construed to lack utility is governed by whether a sensible person (in the art) would realise that a claimed approach would lead to unworkability, and not apply that. In Lundbeck, the relevant claim under consideration was claim 5, which provided for a pharmaceutical composition according to claim 3 or 4, with an active ingredient present in an amount from 0.1 to 100 milligrams per unit dose (at [44]). Emmett J did not find it necessary to reach a conclusion in respect of the utility argument advanced, because he found the invention claimed not to be novel. Bennett J found claim 5 to lack utility, with whom Middleton J agreed (at [218]), whilst claims 3 and 4 were found not to lack utility.
440 It is apparent that a claim will lack utility if, by its proper construction, the skilled person is compelled to make something that does not meet the promise, or otherwise fails to work. This was explained by Bennett J in Austal Ships Pty Ltd v Stena Rederi Aktiebolag [2005] FCA 805; 66 IPR 420 (bold emphasis added, italics in the original):
[235] In Welch Perrin at 602, the alleged lack of utility was that the claims were so general that an unworkable machine could be made in conformity therewith, although a most useful machine could also be made within the claim. Menzies J, at 601, considered the principle that all within the scope of the claim must be useful if the claim is not to fail for inutility. His Honour refined the principle in Norton and held that a specification should not be ‘construed in a way that any sensible person would appreciate would lead to unworkability when by construction it could be given a more limited meaning’ (at 602).
[236] It is apparent that in Washex Machinery at 18, Stephen J was of the view that the claim did not need to specify a limitation that was common knowledge in the art for that limitation to apply. Further, to postulate “a quite purposeful adoption” of a form which would obviously malfunction was “not an appropriate mode of testing validity of a patent specification”.
[237] In Martin Engineering Co v Trison Holdings Pty Ltd (1989) 14 IPR 330 at 336-338 (‘Martin Engineering’), Burchett J discussed lack of utility, both in the sense of the claims asserting a monopoly, over the useful and the non-useful and also in the failure of the range of claimed devices to fulfil the promise of the specification, to overcome the identified problem. As to the former, Burchett J accepted that if, on its correct construction a claim asserts a monopoly in respect of something useful and also in something not useful, the patent is bad. However, his Honour observed that Norton was decided on the proper construction of the claims. Burchett J distinguished the reasoning [in] Norton in cases where the words of the claim were not “clear words” (at 337 – 338). Rather than adopt Lord Greene’s concept of a rigid separation of claim and body of the specification, his Honour said that the claims are not to be construed without regard to the specification of which they form part. He also affirmed the necessity to consider the claims as would the person skilled in the art desirous of making use of the invention. This included ‘limitations dictated by common sense after a perusal of the whole of the specification including the claims’ (at 338). That approval is, in my opinion, consistent with proper claim construction in Australia.
441 In Sandvik Intellectual Property AB v Quarry Mining & Construction Equipment Pty Ltd [2017] FCAFC 138; 348 ALR 156 the Full Court (Greenwood, Rares and Moshinsky JJ) endorsed the above reasoning as correct (at [192]). The outcome of Sandvik on its facts illustrates the approach. There, the Court was concerned with claims to an extension drilling system that used extension rods and included a drive chuck for driving the outside surface of a coupling. Claims 1 – 3 included no limitation as to the means by which the rods were driven. Claim 4 was construed to require that the rods had a hexagonal or round cross-section. The primary judge had found on the evidence that a rod with a round profile would not work in a chuck. The consequence was that claim 4 was correctly found by the primary judge to lack utility (at [201] – [202]). However, claims 1 to 3 did not lack utility. Even though the invention claimed in claim 4 fell within the scope of those claims, the Full Court found that the skilled addressee would not read claims 1 to 3 as including extension rods with a round end (at [203]).
442 MSD pleads that the composition patents expressly promise that the immunogenic compositions of the invention will, first, increase coverage for invasive pneumococcal disease due to serotypes 1, 3, 5, 6A, 7F and 19A to greater than 90% in the United States and Europe and as high as 70% – 90% in Asia and Latin America (asserted promise 1), and secondly, provide coverage for serotypes 6A and 19A that is not dependent on the limitations of serogroup cross-protection. The content of this asserted promise was modified as the case developed in submissions to be that the immunogenic compositions of the invention will provide an effective immune response for each of the 13 chosen serotypes that is not dependent on the limitations of serogroup cross-protection (asserted promise 2). Furthermore, an additional promise is said to be that the immunogenic compositions of the invention will not cause significant adverse effects and will be safe (asserted promise 3). I refer to the three collectively as the asserted promises.
443 MSD relies in its submissions on specific passages drawn from the specification to support its contentions. MSD contends that none of the asserted promises is met by the invention claimed in the asserted claims because the specification discloses that no functional antibodies are produced in response to the presence of serotype 14 in the claimed compositions. It also contends that the asserted promises will not be met because the scope of the invention claimed includes a composition with serotypes in addition to those 13 serotypes specifically identified.
444 Wyeth disputes that the asserted promises are made, disputes that the asserted promises have been shown not to have been met, and contends that any argument based on asserted promise 2 is precluded, because it has not been sufficiently pleaded.
12.3 Were the asserted promises made?
445 In relation to asserted promise 1, MSD relies on the following passage in the Background of the Invention (page 2 lines 7 – 12) (emphasis added):
Given the relative burden and importance of invasive pneumococcal disease due to serotypes 1, 3, 5, 6A, 7F, and 19A, adding these serotypes to the Prevnar formulation would increase coverage for invasive disease to >90% in the US and Europe, and as high as 70%-80% in Asia and Latin America. This vaccine would significantly expand coverage beyond that of Prevnar, and provide coverage for 6A and 19A that is not dependent on the limitations of serogroup cross-protection.
It will be recalled that the Prevnar 7 serotypes are 4, 6B, 9V, 14, 18C, 19F and 23F. Ultimately, the submission is that this promise is not met because the 13vPnC composition in the specification does not induce functional antibodies to serotype 14.
446 The extract is set out in the Background of the Invention, before the invention is introduced. Prior to it, the specification refers to the overall burden of disease caused by the listed serotypes in certain geographical regions. The extract does not promise that the composition of the invention will cause a particular clinical result in a particular individual. Rather it is that an overall population effect “would” increase coverage. It amounts to a forecast, based on statistical information, of the likely effect of the inclusion of the additional serotypes.
447 Asserted promise 2 is that immunogenic compositions of the invention will induce an “effective immune response” against each of the 13 serotypes claimed. This is said to arise implicitly from the disclosure of the specification. MSD draws on:
(a) The description of the Field of the Invention at page 1 lines 5 – 7 as including “the prevention of infection by a bacterial pathogen by immunization.
(b) The reference in the Background of the Invention at page 1 lines 15 – 17, where Prevnar 7 is mentioned as being “highly immunogenic and effective against invasive disease and otitis media”.
(c) The justification provided from page 4 line 22 to page 10 line 24 for including each serotype in the claimed compositions. A summary of this part of the specification is set out in section 4.1.
(d) The statements at page 14 lines 24 – 25 that “the vaccine formulations of the present invention can be used to protect or treat a human susceptible to pneumococcal infection” and at page 15 lines 1 – 2 that “the amount of conjugate...induces an immunoprotective response…”
448 Wyeth contends that asserted promise 2 is not sufficiently pleaded and the ground of inutility based on it may not be advanced. Certainly paragraph 17 of the Third Further Amended Consolidated Statement of Claim is somewhat Delphic. However, particular (b) to that paragraph, when read with (c), indicates that one promise said to have been made in the specification is that some immunogenic compositions will not meet a promise of inducing an immune response. That provides some notice of the argument to Wyeth. Furthermore, the subject matter of the debate was canvassed in the expert evidence and was raised prior to the trial as an issue for determination, albeit with Wyeth contending that it had not been adequately pleaded. The parties led evidence and made opening and closing submissions on the issue. In my view Wyeth has had sufficient notice of the case advanced, and opportunity to respond to that case, to warrant it being advanced in closing submissions: see Banque Commerciale S.A., En Liquidation v Akhil Holdings Ltd [1990] HCA 11; 169 CLR 279 at 286 – 287 (per Mason CJ and Gaudron J); Stefanovski v Digital Central Australia (Assets) Pty Ltd [2018] FCAFC 31; 368 ALR 607 at [63] and [65] (McKerracher, Robertson and Derrington JJ).
449 There is no dispute that a promise may be implied from language used in the specification.
450 However, I am not persuaded that a promise is made that an effective immune response in respect of each serotype will be provided. Rather, I consider that the promise implied is that the claimed compositions are capable of achieving an immunogenic response in relation to each serotype in a percentage of the population (modified promise 2) (having regard to my finding on the meaning of “immunogenic” in the specification: see section 5.4).
451 Referring to the aspects of the specification upon which MSD relies, the part of the Field of the Invention identified in [447(a)] above, and the Background of the Invention in [447(b)], represent prefatory statements of high generalisation that serve to set the scene for the invention.
452 The words in [447(c)] identify, by reference to the cited literature, the deductions that have caused the inventors to select and include the 13 chosen serotypes. These are based on population-wide studies indicating invasive pneumococcal disease rates amongst children in various parts of the world. The introductory words of the Detailed Description of the Invention on page 4 of the specification observe that since the introduction of Prevnar 7 in 2000, there has been a “significant decrease in the overall [invasive pneumococcal disease rates] due to a decrease in disease due to the vaccine serotypes” (emphasis added). Accordingly, the Prevnar 7 serotypes have been retained. The specification here speaks of disease rates in a population. Similarly, in relation to serotypes 1, 3, 5 and 7F, the specification draws conclusions from the available data to extrapolate the benefits to be accrued from providing a conjugate vaccination that includes those serotypes. The prevalence of disease caused by different serotypes varies according to location. For example on page 5 lines 28 – 29, the specification postulates that the addition of serotypes 3 and 7F “will increase coverage against [invasive pneumococcal disease] in most areas of the world by approximately 3%-7% and in Asia by around 9%”. Statements as to the “likely” effects on a population cannot be understood to imply that the composition of the invention claimed will yield specific therapeutic effect in respect of each individual. The specification here is postulating at the level of likelihood, depending on the population concerned.
453 Finally, MSD relies on the passages in [447(d)]. The passages relied upon from pages 14 – 15 of the specification are emphasised below:
The vaccine formulations of the present invention can be used to protect or treat a human susceptible to pneumococcal infection, by means of administering the vaccine via a systemic or mucosal route. These administrations can include injection via the intramuscular, intraperitoneal, intradermal or subcutaneous routes; or via mucosal administration to the oral/alimentary, respiratory or genitourinary tracts. In one embodiment, intranasal administration is used for the treatment of pneumonia or otitis media (as nasopharyngeal carriage of pneumococcal can be more effectively prevented, thus attenuating infection at its earliest stage).
The amount of conjugate in each vaccine dose is selected as an amount that induces an immunoprotective response without significant adverse effects. Such amount can vary depending on the pneumococcal serotype. Generally, each dose will comprise 0.1 to 100 μg of polysaccharide, particularly 0.1 to 10 μg, and more particularly 1 to 5 μg.
454 This extract, taken with the other passages upon which MSD relies, and read in the context of the specification as a whole, does not result in asserted promise 2, but rather modified promise 2 to which I have referred above at [450], namely that the claimed compositions are capable of achieving an immune response in relation to each serotype in a percentage of the population. This accords with the understanding of that the person skilled in the art has when reading the specification, namely that vaccines do not prevent disease in 100% of vaccinated individuals in diverse populations such as human populations, given the variability between individuals of the immune responses generated. The person skilled in the art would realise that where the specification refers to there being an immunogenic response, it is referring to populations as a whole, rather than each individual person.
455 Asserted promise 3 is that the immunogenic compositions of the invention claimed will be safe, in that they will not have significant adverse effects. MSD relies on the second paragraph set out above at [453] from pages 14 – 15 of the specification. In my view this passage amounts to a statement, in the form of a promise, that amounts of conjugate used will ensure that there are not significant adverse effects (modified promise 3).
12.4 Consideration of whether modified promise 2 is met – serotype 14
456 MSD submits that the asserted promises are not met by reference to the reported results in the specification concerning serotype 14, although it advances no written or oral submissions as to how asserted promise 3 is said to be relevant or not met on this basis. My findings rejecting MSD’s contentions as to whether the asserted promises were made perhaps render otiose consideration of whether all of the asserted promises are met; however, for completeness I now address the question of whether modified promises 2 and 3 are met.
457 MSD contends that the evidence in the specification demonstrates that there was no functional antibody reaction produced for serotype 14. It relies solely on the following passage given in the context of example 16, at pages 46 – 47 (emphasis added):
Table 5 presents GMT data obtained from week 4 bleeds analyzed in antigen specific IgG ELISAs. Additional analyses show the ratio of GMT values at week 4 to week 0. The data indicate that the conjugate vaccine preparation elicited greater serum IgG titers than free PS or free PS + CRM197 vaccine. With the exception of S. pneumoniae type 14, the 13vPnC vaccine was able to induce functional antibodies to the representative strains of S. pneumoniae in an OPA (Table 6).
458 MSD submits that the common general knowledge was that the tests conventionally used by April 2005 were OPA, which measures functional antibodies, and ELISA, which measures quantity of antibodies, but not their functionality. The parties agree that this was common general knowledge at the priority date. MSD relies on the oral evidence of Professor Dagan who said “[w]hen there is no functional antibody you would not expect that the vaccine would be effective” and on Professor Paton’s evidence indicates that “at least by the OPA criteria in Table 4” in example 16, serotype 14 is non-immunogenic. Thus, he said, the vaccine would not be expected to be effective against that serotype. MSD submits that where there is a failure to demonstrate functional antibodies, as demonstrated in Tables 4 and 6, the result must be that the vaccine is not expected to provide any protection against disease caused by that serotype. Accordingly, MSD submits that the specification discloses on its face to the person skilled in the art that there is no effective immune response for each serotype, which represents a failure to meet either asserted promise 1 or 2.
459 Several propositions stand in the way of a conclusion that MSD should succeed.
460 First, the specification states, by reference to study #HT01-0021 (page 44 lines 4 – 9) (emphasis added):
Overall, the data indicate that the 13vPnC vaccine formulated with aluminium phosphate is immunogenic in rabbits, eliciting substantial antibody responses to the pneumococcal capsular polysaccharides contained in the vaccine and these responses are associated with functional activity. The responses observed to the seven core serotypes following immunization with 13vPnC + AIPO4 are consistent with historical responses of rabbits to the heptavalent formulation.
461 The heptavalent formulation referred to is Prevnar 7. There is no evidence to suggest that Prevnar 7 was not immunogenic in relation to serotype 14. To the contrary, Whitney 2003 is cited in the composition patents at footnote 3, and incorporated by reference. It indicates that Prevnar 7 produced an immune response for serotype 14.
462 Secondly, the results reported for example 16 in the specification contrast the from OPA test results with the ELISA test results. The difference between the two is explained in the agreed primer:
Ordinarily, to evaluate the immunogenicity of a vaccine, animals are injected with the candidate vaccine. The animals are bled before and after vaccination to obtain sera for in vitro analysis. In the context of pneumococcal vaccines, assays which are commonly used in these analyses include those which detect and quantify the level of antibody (e.g. enzyme-linked immunosorbent assay (ELISA)) and which measure the opsonophagocytic activity of antibody (e.g. opsonophagocytic assay (OPA)).
The ELISA is the most common method used to detect the presence of specific antibodies in serum. An ELISA uses enzyme to cause a colour change to indicate that the relevant antibody has been detected. The amount of antibody in serum (expressed in terms of titre) may be quantified in an ELISA. Titres are typically measured on a logarithmic scale. A higher titre means there is a greater concentration of antibodies in serum.
An ELISA can identify the presence and quantity of antibodies in a sample but cannot determine whether the antibodies are functional (i.e. whether they effectively opsonise or neutralise the pathogen). The opsonophagocytic assay or OPA is used to test the opsonophagocytic activity of vaccine-induced antibodies in vitro. The classic OPA determines the titres of sera that, when incubated with the bacteria of interest, reduce the number of live bacteria (or colony-forming units) by more than half.
ELISAs and OPAs can both be performed using sera from various test animals or humans.
463 Professor Dagan gives evidence, which I accept, that in 2003 a World Health Organisation working group of which he was a participant discussed recommendations for the evaluation of new pneumococcal conjugate vaccines following licensure of Prevnar 7. They were to be licensed on the basis of non-inferiority compared with Prevnar 7, as determined by immunogenicity studies. The primary means for evaluation was to consider the proportion of subjects attaining serotype-specific anti-polysaccharide IgG antibody concentrations of greater or equal to 0.35 μg/ml one month after a three-dose priming series, as measured using ELISA, using Prevnar 7 as a control. The antibody threshold was to serve as a correlate for protection against invasive pneumococcal disease. As a secondary endpoint, OPA was to measure opsonophagocytic activity after a three-dose priming series.
464 This evidence indicates that whilst ELISA test results do not measure functional antibody response, they were used by those in the art as a means of predicting a correlation for protection against invasive pneumococcal disease. The ELISA test results reported in the specification do not demonstrate an absence of immunogenic response for serotype 14.
465 Accordingly, I consider, having regard to the matters to which I have just referred, that the extract at pages 46 – 47 of the specification upon which MSD heavily relies is to be understood as a statement that for the particular analysis conducted in relation to study #HT01-0036, all but serotype 14 induced functional antibodies. I would not construe that passage as a general statement that a composition of the claims including serotype 14 is not functional, or that the composition yields no immune response in relation to serotype 14.
466 Thirdly, MSD has by no means established that the conclusion identified at page 44 of the specification, quoted above at [460], is incorrect. It is notable that MSD does not seek to establish through evidence that the invention is not useful. Rather, it relies on the disclosures on the face of the specification. In so doing it was exposed to the prospect that the specification does not yield sufficient information for the case to be established to the satisfaction of the Court. The onus was on MSD to establish that the composition of the claims is not immunogenic (as I have defined that term) in relation to serotype 14, and in my view it has not done so.
12.5 Consideration of whether modified promise 3 is met – inutility consequence of the resolution of the comprising issue
467 Next, MSD contends that the invention asserted in the claims is not useful because none of the asserted promises are met if the word “comprising” is given an inclusive meaning, such that a composition including more than 13 serotypes conjugated to CRM197 falls within the claims. This is particularly relevant to asserted promise 3. Whilst claim 1 of the 013 patent (as an example) specifies that the immunogenic composition includes the 13 identified serotypes conjugated to CRM197, MSD submits that it is possible that a composition including many more serotypes (about 90 had been described by April 2005) may fall within the scope of the claim. Were some or all of those additional serotypes to be included, then the composition would not be useful because, inter alia, there would be too much carrier protein, with the consequence that the vaccine would not: provide the coverage benefits and meet asserted promise 1; elicit an effective immunogenic response with respect to all serotypes included in the composition and meet asserted promise 2; or be safe for administration and meet asserted promise 3.
468 The utility of the alleged invention depends on whether, by following the directions in the complete specification, the effects which the patentee professed to produce could be produced, and on the practical utility of those effects: Lane Fox at 431. MSD has not demonstrated the contrary with respect to the complete specification. The specification does not direct the skilled addressee to produce a composition containing more than 13 serotypes.
469 In Welch Perrin, the lack of utility alleged was based on the contention that the claims were so general that an unworkable machine could be made in conformity with them, although a most useful machine could also be made within them. Menzies J held that a specification should not be “construed in a way that any sensible person would appreciate would lead to unworkability when by construction it could be given a more limited meaning” (at 601 – 602). The approach of the Full Court in Sandvik demonstrates that a claim is not rendered inutile where the person skilled in the art knows how to produce a workable result, unless the construction of the claim compels an unworkable result. That is apposite in the present case.
470 Accordingly, the inutility challenge to the claims of the compositions patents fails.
13. COMPOSITION PATENTS: FALSE SUGGESTION
471 A patent may be revoked either in whole or in part in so far as it relates to a claim if the complete specification was obtained by fraud, false suggestion or misrepresentation: s 138(3)(e) of the Patents Act.
472 MSD relies on two grounds in this regard. The first concerns both composition patents. It contends that on page 6 lines 5 – 22 of the specification the patentee represented to the Commissioner of Patents that the invention “solved the problem of producing a multivalent pneumococcal conjugate vaccine that exhibits significant immunogenicity with respect to serotype 3 polysaccharides” (the asserted serotype 3 representation). It contends that this representation was false or was a misrepresentation because the data provided in Tables 4 and 6 of the composition patents show that there was no, or no significant, improvement in immunogenicity with respect to serotype 3 provided by the invention.
473 The second ground concerns only the 844 patent. MSD contends that in a letter from its patent attorneys to the Commissioner dated 20 April 2016, the patentee represented that:
(1) the claimed composition is 13-valent;
(2) the claimed composition has been clinically confirmed and today it is marketed as Prevnar 13; and
(3) the inventors chose a vaccine design which is a 13-valent pneumococcal conjugate vaccine;
(together, the asserted 13-valent representation).
474 That representation is said to be false or involve a misrepresentation in that each claim encompasses immunogenic compositions that have more than 13 serotypes.
475 MSD contends that each of the asserted representations were material to the grant of the patent to which it related.
476 The relevant principles to be applied were summarised by the Full Court in Ranbaxy Australia Pty Ltd (ACN 110 781 826) v Warner-Lambert Company LLC [2008] FCAFC 82; 77 IPR 449 (Emmett, Weinberg and Bennett JJ):
[82] If a representation that was false or misleading materially contributed to the Commissioner’s decision to grant a patent, even if other circumstances or causes also played a part in the making of that decision, it may be said that the patent was obtained by a false suggestion or misrepresentation (or on a false suggestion or representation, to use the language of the 1952 Act). It is sufficient if the representation materially contributed to the Commissioner’s decision to grant the patent or was a material, inducing factor, which led to the grant. However, it is not necessary to establish that the representation was material in the sense that, without it, the patent would not have proceeded to grant (Pfizer Overseas Pharmaceuticals v Ely Lilly & Co (2005) 225 ALR 416 at 495). It is not necessary to show that, but for the suggestion or representation, no grant would have been made (Prestige Group (Australia) Pty Limited v Dart Industries Inc (1990) 95 ALR 533 at 537-538).
[83] Bearing in mind that the grant of a patent is a right in rem, the Commissioner could be expected to take a position if a misrepresentation did in fact play a part in the decision to grant a patent and it is a relevant factor that the Commissioner chooses not to give evidence (ICI Chemicals & Polymers Limited v The Lubrizol Corporation Inc (2000) 106 FCR 214 at 244-245). In the absence of such evidence, it is for the Court to make a finding, based on the evidence before it. In the absence of explicit evidence that the Commissioner, or the Commissioner’s delegate, was in fact misled, it may nevertheless be inferred that a representation in fact contributed to the decision to grant a patent, if the representation was objectively likely to contribute to such a decision and the patent was in fact granted (see Synthetic Turf Development Pty Limited v Sports Technology International Pty Limited [2004] FCA 1179 at [2], and WM Wrigley Jr Co v Cadbury Schweppes Pty Limited (2005) 66 IPR 298 at 321).
13.3 The asserted serotype 3 representation
477 The specification states at page 5 line 28 to page 6 line 22 (emphasis added, citations omitted):
The addition of serotypes 3 and 7F will increase coverage against IPD in most areas of the world by approximately 3%-7%, and in Asia by around 9%. Thus, an 11-valent vaccine would cover 50% in Asia and around 80% of IPD in all other regions. These serotypes are also important with respect to otitis media coverage. In a multinational study of pneumococcal serotypes causing otitis media, Hausdorff et al found serotype 3 to be the 8th most common middle ear fluid isolate overall. Serotype 3 accounted for up to 8.7% of pneumococcal serotypes associated with otitis media. Thus, the importance of types 3 and 7F in otitis media, as well as in IPD, warrants their inclusion in a pneumococcal conjugate vaccine.
However, attempts to produce a multivalent pneumococcal conjugate vaccine that exhibits significant immunogenicity with respect to serotype 3 polysaccharides have been unsuccessful. For example, in a study of the immunogenicity and safety of an 11-valent pneumococcal protein D conjugate vaccine (11-Pn-PD), no priming effect was observed for serotype 3 in infants who had received three doses of the vaccine followed by a booster dose of either the same vaccine or a pneumococcal polysaccharide vaccine. In yet another study, which assessed the efficacy of an 11-Pn-PD in the prevention of acute otitis media, the vaccine did not provide protection against episodes caused by serotype 3. Accordingly, a pneumococcal conjugate vaccine comprising capsular polysaccharides from serotype 3 and capable of eliciting an immunogenic response to serotype 3 polysaccharides provides a significant improvement of the existing state of the art.
478 This passage appears under the heading “Detailed Description of the Invention” and the sub-heading “Inclusion of Serotypes 1, 3, 5 and 7F” (which are not included within the Prevnar 7 serotypes).
479 Wyeth submits that the asserted serotype 3 representation cannot be gleaned from these passages and that there has been no material misrepresentation. I agree.
480 In my view the patentee in this passage represents that, in contrast to the previous attempts to produce a multivalent pneumococcal conjugate vaccine exhibiting significant immunogenicity with respect to serotype 3, it has developed a pneumococcal conjugate vaccine comprising capsular polysaccharides from serotype 3 that is capable of eliciting an immunogenic response to serotype 3 polysaccharides. I do not consider that the statement amounts to a representation that there will be “significant immunogenicity” with respect to serotype 3 polysaccharides. Rather, the representation is that by producing a pneumococcal conjugate vaccine that is capable of eliciting an immunogenic response, a “significant improvement” is achieved over the existing state of the art.
481 Accordingly, there is no statement to the effect that the invention produces a significant immunogenic response in respect of serotype 3; rather, it is stated that the invention produces an immunogenic response.
482 All of MSD’s submissions on the asserted serotype 3 promise were directed at proving that serotype 3 did not provide significant immunogenicity. As MSD has failed to establish that the asserted serotype 3 representation was made, its first ground of revocation based on false suggestion fails. However, for completeness, I now turn to consider whether MSD has established that the asserted representation was false.
483 MSD relies on the evidence of Professor Paton, who reviews the results set out in Table 4, which is part of example 16. He notes that in Table 4, the specification says that “OPA titers were detected in week 4 serum pools to all vaccine serotypes in both groups. For the majority of the serotypes, OPA titers measured at week 4 were at least 4-fold higher than those at week 0 (baseline)”. Professor Paton’s evidence is that despite this statement (emphasis added):
...the response to serotype 3 without adjuvant was negligible. At week 0 the GMT is given as “<8” and at week 4 the GMT is given as 8. The ratio is said to be 2, though one can immediately see that it could be closer to 1. This indicates that the immune response to serotype 3 was not significantly better after vaccination than without any vaccination at all. Put another way, the vaccine appeared to make little or no difference to the host’s immune response to serotype 3.
484 It is apparent from this passage that Professor Paton, reading the specification as a person skilled in the art, concludes that the results reported lead to “negligible” immune response, apparently on the assumption that the ratio would be closer to 1 than 2. Plainly, he accepts that there has been some level of response, but plainly, “negligible” does not equate to “significant”. Professor Paton’s evidence is not answered. That may be sufficient to cast doubt on the accuracy of the pleaded representation had I found it to be have been made. However, I am not satisfied that the ground would in any event cross the materiality threshold. That is because there is an inherent difficulty in advancing a case of false suggestion or representation where the representation, and also the means of ascertaining that it is false, are both included in the specification. Both are likely to be apparent to the person skilled in the art upon reading the document as a whole. The Commissioner effectively stands in the shoes of the person skilled in the art when considering whether or not to accept the patent. In so doing, she may be expected to adopt the same construction of the specification as the person skilled in the art. In such a circumstance, the representation is unlikely to be material to the grant, because the data that is said to falsify it is available to be seen in the same document, and by necessity the Commissioner may be assumed to have read and understood it.
485 In the present case, if one takes Professor Paton to be representative of the person skilled in the art, he read both the passage at pages 5 – 6 of the specification, and interpreted the results set out in Table 4 in the manner set out above. He was in a position to evaluate the representation by reference to the results. So too was the Commissioner, but she has not come forward to suggest that she misunderstood something with respect to this representation. These matters give me some confidence in concluding that no material misrepresentation has been established in relation to the first ground relied upon.
13.4 The asserted 13-valent representation
486 This ground concerns only the 844 patent.
487 On 15 October 2015, the Commissioner wrote to the patent attorneys representing Wyeth and indicated her view that the invention as claimed lacks novelty and an inventive step over certain items of prior art, including Peña and a number of other documents that disclose 11-valent pneumococcal conjugate vaccines. The attorneys responded at length on 20 April 2016. Their letter commences by attaching a proposed amended claim set for the 844 patent, which is the same as the claims that proceeded to grant. The following passages suffice to illustrate the context upon which MSD relies. In one passage of the letter the attorneys say (emphasis in the original):
With respect to the Examiner’s inventive step objection, we submit that the present application discloses, for the first time, a composition of a second-generation pneumococcal conjugate vaccine (PCV). Unlike other next-generation pneumococcal conjugate vaccines that had been tested at the time of the invention, the claimed composition is 13-valent (the highest valency tested prior to the invention was 11-valent), and it includes two serotypes, 6A and 19A, which had not been used in prior art multivalent conjugate vaccines...
In another the attorneys say (emphasis in the original):
The present inventors chose a vaccine design which is a 13-valent PCV; that is, a higher valency than so far had been tested for any vaccine…
488 MSD submits that in making these (and similar) statements the attorneys made the asserted 13-valent representation. It submits that the attorneys failed to put forward the construction that Wyeth now relies upon which, MSD submits, has the effect that the claim covers a valency of 13 or more. In so doing, the 13-valent representation involves a false suggestion or misrepresentation as to the scope of the monopoly that the patentee sought to cover by the claims.
489 I am unable to accept MSD’s submissions for three reasons. First, the Commissioner was possessed of a copy of the specification and the claims for the 844 patent. It was a matter for her to construe that document and form a view as to the scope of the claims and she no doubt did so in accordance with the principles of construction that I have found applied in section 5.1 above. Secondly, taken in context, the statements in the attorneys’ letter draw attention to the fact that the invention as claimed is for a 13-valent pneumococcal conjugate vaccine by way of distinguishing that claim from prior vaccines of 11 or fewer serotypes. In that context it was correct to state that the composition the subject of the claim is 13-valent, because that was the number of particular serotypes identified in the claims. The letter is not directed towards other aspects of the construction of the claims. Thirdly, even were the first two propositions to be incorrect, and the letter to be construed as a statement that the subject of the claims was to an invention consisting of 13 serotypes and no more conjugated to CRM197, it is settled law that an applicant for a patent can make submissions to the Patent Office as to the proper construction or effect of a claim, and this will not be found to have been a false suggestion or representation simply because such a submission may later be held to be incorrect: NSI Dental Pty Ltd v University of Melbourne [2006] FCA 1216; 69 IPR 542 at [206] (Tamberlin J); ICI Chemicals & Polymers Ltd v The Lubrizol Corporation Inc [2000] FCA 1349; 106 FCR 214 at [91] (Lee, Heerey and Lehane JJ).
490 Accordingly, the second ground must also be dismissed.
14. COMPOSITION PATENTS: LACK OF CLARITY
491 A claim must be clear and succinct: s 40(3) of the Patents Act. MSD contends that the claims of the composition patents are not clear and succinct, because the meaning of the term “comprising”, where it appears in the claims in relation to the 13 serotypes of Streptococcus pneumoniae, is unclear.
492 This contention must be rejected. There is no lack of clarity arising from the word “comprising” the meaning of which is explained in the body of the specification, and apposite in the context of the claims.
15. 013 PATENT: LACK OF FAIR BASIS
493 The claims of a patent to which the pre-RTB Patents Act applies must be fairly based on the matter described in the specification: s 40(3).
494 MSD submits that the disclosure in the specification of the 013 patent is of a composition with the 13 specified serotypes and no more. It submits that there is no real and reasonably clear disclosure of an immunogenic composition with more than 13 serotypes, and that accordingly the claims lack fair basis, citing Lockwood Security Products Pty Ltd v Doric Products Pty Ltd [2004] HCA 58; 217 CLR 274 (Lockwood No 1) at [1] and [69] (Gleeson CJ, McHugh, Gummow, Hayne and Heydon JJ).
495 Wyeth submits that MSD misconceives the correct construction of the claims. It submits that they are not for a monopoly over vaccines with up to 90 serotypes or more, or for an immunogenic composition with more than 13 serotypes. They are for a multivalent immunogenic composition of 13 specified serotypes conjugated to CRM197. It submits that a composition with more than 13 serotypes can, on a correct construction of the word “comprises”, fall within the scope of the claims. That does not mean that Wyeth claims all of the serotypes in such a composition, but if all of the integers of the claims are present, even if more than 13 serotypes are included in the composition, the composition will infringe.
496 Fair basis involves a comparison between the claims made in the patent and what is disclosed in the specification in order to determine whether there is a real and reasonably clear disclosure of the invention claimed, or whether the invention claimed travels beyond the invention described: Bitech at [39]. The enquiry as to the disclosure of the body of the specification is whether broadly, in a general sense, it describes the invention as claimed: Lockwood No 1 at [69]. This involves consideration of what the specification discloses as a matter of substance.
497 In Lockwood No 1 the requirement for fair basis, often repeated since, was set out at [69]:
“Real and reasonably clear disclosure". Section 40(3) requires, in Fullagar J's words, "a real and reasonably clear disclosure." But those words, when used in connection with s 40(3), do not limit disclosures to preferred embodiments.
"The circumstance that something is a requirement for the best method of performing an invention does not make it necessarily a requirement for all claims; likewise, the circumstance that material is part of the description of the invention does not mean that it must be included as an integer of each claim. Rather, the question is whether there is a real and reasonably clear disclosure in the body of the specification of what is then claimed, so that the alleged invention as claimed is broadly, that is to say in a general sense, described in the body of the specification."
Fullagar J's phrase serves the function of compelling attention to the construction of the specification as a whole, putting aside particular parts which, although in isolation they might appear to point against the "real" disclosure, are in truth only loose or stray remarks.
498 In the present case there is no dispute that the invention as claimed is described in the body of the specification. As MSD’s submission reflects, it is accepted that a composition with the 13 serotypes in the combination of the claims is disclosed. The complaint advanced is that if the scope of the claim extends to MSD’s 15-valent vaccine, or indeed to a product having any further serotypes in addition to the 13 in the combination as claimed, then there is no real and reasonably clear disclosure of that combination.
499 It seems to me that this submission distracts from the real enquiry, which is not whether the infringing article is disclosed in the specification, or whether a particular embodiment is disclosed, but whether the invention claimed is disclosed: Bitech at [40].
500 The features of claim 1 of the 013 patent are set out earlier in these reasons. Unless there is not a real and reasonably clear disclosure of a multivalent composition comprising the 13 chosen serotypes conjugated to CRM197 together with a physiologically acceptable vehicle within that claim, it will be fairly based. In the present case, as much is correctly conceded. The Summary of the Invention contains a statement that the invention does consist of these features. No other aspect of the disclosure of the specification suggests otherwise: cf Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth [2011] FCAFC 132; 119 IPR 194 at [242] (per Yates J).
501 In these circumstances, the fair basis challenge to the claims of the 013 patent must fail.
16. 844 PATENT: LACK OF SUPPORT
502 MSD contends that the asserted 844 patent claims lack support and are accordingly invalid for failure to comply with s 40(3) of the post-RTB Patents Act, which provides (emphasis added):
(3) The claim or claims must be clear and succinct and supported by matter disclosed in the specification.
503 The requirement that the claims be supported by matter disclosed in the specification is new to the Patents Act, and replaces the requirement under the pre-RTB Patents Act that the claims be fairly based on the matter disclosed in the specification. I refer to it below as the claim support obligation.
504 The RTB amendments also introduced a new obligation under s 40(2)(a) as follows:
(2) A complete specification must:
(a) disclose the invention in a manner which is clear enough and complete enough for the invention to be performed by a person skilled in the relevant art…
505 This replaces the requirement under the former s 40(2)(a) that the complete specification must describe the invention fully. I refer to the new requirement below as the disclosure obligation. MSD does not rely on a failure by the 844 patent to satisfy the disclosure obligation.
506 MSD contends in its closing submissions that, in the event that I reject its arguments in relation to the comprising issue (which I have), the claim support obligation is not met because there is no description or disclosure in the specification of any pneumococcal polysaccharide-protein vaccine other than one having pneumococcal polysaccharides from the 13 chosen serotypes identified in the specification and claims. It also pleads various other matters, which were not relied upon in its closing submissions.
507 The case advanced by both parties was confined in closing submissions to only a handful of paragraphs and little in the way of oral address, despite the fact that the claim support obligation is new to the law of Australia. As noted below, the secondary materials relevant to the amendments made to s 40 of the Patents Act point to the relevance of United Kingdom and European law, which the parties drew on this in support of their arguments. After argument was complete, the decision of the United Kingdom Supreme Court in Regeneron Pharmaceuticals Inc (Respondent) v Kymab Ltd (Applicant) [2020] UKSC 27; Bus LR 1394 (per P Reed, Hodge, Briggs and Sales LJJ) was delivered. The parties were invited to supply further written submissions concerning the applicability of the Supreme Court’s decision, which they have done.
508 MSD submits that the claim support obligation requires an enquiry into whether the claims are supported by the description in the specification, citing Schering Biotech Corp’s Application [1993] RPC 249 at 252 (Aldous J). It submits that to satisfy the obligation, the scope of the claims “should correspond to the technical contribution to the art” provided in the specification, an expression drawn from the often cited decision of the European Patent Office’s Technical Board of Appeal in Exxon/Fuel Oils (T 409/91) [1993] 3 WLUK 282; [1994] EPOR 149 at [3.3].
509 Wyeth in its submissions also places reliance on the requirement set out in Exxon/Fuel Oils. It submits that the specification clearly provides support for the claimed invention in respect of the 13 chosen serotypes conjugated to CRM197. It submits that the specification does not need to provide support for a composition comprising more than 13 serotypes “because any composition comprising the 13 chosen serotypes conjugated to CRM197 would nonetheless embody the technical contribution made by the invention with respect to those named serotypes”. Wyeth further submits that MSD’s argument is wrong as a matter of law. It would mean, Wyeth submits, that any product claim, particularly one that uses “comprising” or “including”, is liable to be revoked for lack of support on the basis that the claimed product could be produced with an additional integer not mentioned in the specification. It also seeks to distinguish the present case from the United Kingdom Supreme Court’s decision in Regeneron.
510 I return to address in more detail the arguments raised by the parties below.
511 In his Second Reading Speech introducing the Intellectual Property Laws Amendment (Raising the Bar) Bill 2011 [2012] (Cth) (RTB Bill), the Hon. Mark Dreyfus QC MP observed that the RTB Bill sought to raise patent standards to address concerns that Australia’s standards are lower than elsewhere, which discourages export of technology developed in Australia, and inhibits the growth of Australia business. He said:
A theme in this bill is one of recalibrating and raising Australian standards to align them more closely with those of other major trading partners around the world. Some patent standards around the world have developed to the point where there is an aligned approach internationally. In such cases...the bill raises Australian patenting standards to that common approach.
The bill raises patent standards in three important areas. First, it raises standards with respect to the information provided in patent applications and specifications. Disclosure is a cornerstone of the patent system. Patents are an exchange between inventors and the public. In exchange for a 20-year monopoly on commercialisation, the innovator must tell the public how their idea works. This disclosure allows for potential follow-on investors to build on initial work to produce even better innovations.
The bill raises standards to require that the patent right is consistent with the information provided in the patent specification. There must be enough information disclosed for the public to make and use the invention. Also, a specific substantial and credible use for the invention must be also disclosed and the scope of the claims for patent protection must not extend beyond what has been disclosed.
Importantly, the amended provisions mirror similar provisions in the United Kingdom and Europe. It is intended that Australian courts will have regard to developments in the law in the courts of those other jurisdictions when interpreting the new provisions and will develop Australian law in a consistent fashion…
512 The Explanatory Memorandum to the RTB Bill also makes plain that the intention of the changes was to bring the law into conformity with the laws of Australia’s major trading partners. In relation to the new disclosure obligation, it states that this is intended to align the disclosure requirement with that applying in other jurisdictions, with the effect that sufficient information must be provided to enable the whole width of the claimed invention to be performed by the skilled person without undue burden, or the need for further invention. This, it says, more clearly reflects a fundamental principle of the patent system: in exchange for the exclusive rights given to the patentee, the patentee must share with the public the information necessary to make and use the invention.
513 In relation to the claim support obligation, the Explanatory Memorandum notes that despite the underlying concept and policy between fair basis and support being similar, the different terminology has produced different substantive law in different countries, which has produced unnecessary complexity and uncertainty for those seeking protection in Australia and in other jurisdictions. The change is intended to align Australian requirements with those of overseas jurisdictions, such as the United Kingdom. It says (citations omitted):
Overseas law generally requires there to be a relationship between the claims and the description, and between the claims and any document from which priority is being claimed. This is expressed by the requirement that a claim be ‘supported by’ or ‘fully supported by’ the description. Broadly speaking, the terms ‘support’ and ‘full support’ pick up two concepts:
• there must be a basis in the description for each claim; and
• the scope of the claims must not be broader than is justified by the extent of the description, drawings and contribution to the art.
...
Overseas case law and administrative decisions in respect of the ‘support’ requirement will be available to Australian courts and administrative decision makers to assist in interpreting the new provision.
514 Having regard to the content of the secondary materials, there can be little doubt that Parliament considers that it is appropriate for the Court to have regard to the law in the European Union and the United Kingdom in considering the scope of the requirement for “support”: Acts Interpretation Act 1901 (Cth) s 15AB.
16.3 The law in Europe and the United Kingdom
515 The Explanatory Memorandum refers to the Convention on the Grant of European Patents, opened for signature 5 October 1973, 1065 UNTS 199 (entered into force 7 October 1977) (EPC), and the Patents Act 1977 (UK) (UK Act).
516 Art 83 of the EPC is entitled “Disclosure of the invention” and provides:
The European patent application must disclose the invention in a manner sufficiently clear and complete for it to be carried out by a person skilled in the art.
517 The UK Act provides for the disclosure obligation in s 14(3), and a corresponding ground of invalidity in s 72(1)(c). Section 14(3) provides:
The specification of an application shall disclose the invention in a manner which is clear enough and complete enough for the invention to be performed by a person skilled in the art.
518 Art 84 of the EPC, entitled “Claims”, sets out the European equivalent of the claim support obligation:
The claims shall define the matter for which protection is sought. They shall be clear and concise and be supported by the description.
519 The UK Act provides for the claim support obligation in two ways. First in relation to a requirement applicable to the breadth of the claims at s 14(5)(c):
The claim or claims shall...(c) be supported by the description.
Secondly, in relation to the allocation of a priority date, s 5(2)(a) provides:
[I]f an invention to which the application in suit relates is supported by matter disclosed in the earlier relevant application or applications, the priority date of that invention shall instead of being the date of filing the application in suit be the date of filing the relevant application in which that matter was disclosed, or, if it was disclosed in more than one relevant application, the earliest of them…
520 In the United Kingdom, the term “support” is considered to have the same meaning and requirements in both s 5(2)(a) and s 14(5)(c): Asahi Kasei Kogyo KK’s Application [1991] 5 WLUK 114; RPC 485 at 536 (per Oliver LJ, with whom Keith, Brandon and Ackner LJJ agreed).
521 It will be seen immediately that the s 40(2)(a) Patents Act disclosure obligation is expressed in terms that are virtually the same as in s 14(3) of the UK Act. The language of the claim support obligation in s 40(3) (“...claims must be...supported by matter disclosed in the specification”) is very similar to that in s 14(5)(c) of the UK Act.
522 It may be noted that in the United Kingdom, the highest courts have emphasised the influential effect of the European Patent Office Technical Board on the development of the law in that Country: Generics (UK) Ltd v H Lundbeck A/S [2009] UKHL 12; RPC 13 (Generics UK (HL)) at [86] (per Neuberger LJ).
16.3.1 The disclosure obligation: “classical insufficiency”
523 Under the UK Act, the disclosure obligation has been interpreted to require the teaching of the specification to enable the skilled addressee to perform the invention. For reasons explained below, the failure to meet this requirement is often referred to in the cases as “classical insufficiency”. It requires an assessment by the court of the steps which it would be necessary for the skilled reader or team to take in following the teaching of the specification and in order to arrive within the claim: Zipher Ltd v Markem Systems Ltd [2009] EWHC 1379; FSR 1 at [362] – [363] (Floyd J).
524 In Mentor Corp v Hollister Inc (No 2) [1992] 7 WLUK 465; [1993] RPC 7 at 14, the Court of Appeal (per Lloyd LJ, with whom Stuart-Smith and Scott LJJ agreed) endorsed a passage from Aldous J at first instance (Mentor Corp v Hollister Inc [1991] 3 WLUK 167; FSR 557 at 562) who said:
The section requires the skilled man to be able to perform the invention, but does not lay down the limits as to the time and energy that the skilled man must spend seeking to perform the invention before it is insufficient. Clearly there must be a limit. The subsection, by using the words “clearly enough and completely enough”, contemplates that patent specifications need not set out every detail necessary for performance, but can leave the skilled man to use his skill to perform the invention. In so doing he must seek success. He should not be required to carry out any prolonged research, enquiry or experiment. He may need to carry out the ordinary methods of trial and error, which involve no inventive step and generally are necessary in applying the particular discovery to produce a practical result. In each case, it is a question of fact, depending on the nature of the invention, as to whether the steps needed to perform the invention are ordinary steps of trial and error which a skilled man would realise would be necessary and normal to produce a practical result.
525 In Novartis AG v Johns & Johnson Medical Ltd [2010] EWCA Civ 1039; [2011] E.C.C. 10, Jacob LJ said at [74]:
The heart of the test is: “Can the skilled person readily perform the invention over the whole area claimed without undue burden and without needing inventive skill?”
526 In Terrell on the Law of Patents (19th ed, Sweet & Maxwell, London, 2020), at page 404 the learned editors (Sir Colin Birss et al) propose as a convenient summary of the elements of this aspect of classical insufficiency the following passage provided by Kitchin J (as his Lordship then was) in Eli Lily v Human Genome Sciences [2008] 7 WLUK 978; RPC 29 at [239]:
The specification must disclose the invention clearly and completely enough for it to be performed by a person skilled in the art. The key elements of this requirement which bear on the present case are these:
(i) the first step is to identify the invention and that is to be done by reading and construing the claims;
(ii) in the case of a product claim that means making or otherwise obtaining the product;
(iii) in the case of a process claim, it means working the process;
(iv) sufficiency of the disclosure must be assessed on the basis of the specification as a whole including the description and the claims;
(v) the disclosure is aimed at the skilled person who may use his common general knowledge to supplement the information contained in the specification;
(vi) the specification must be sufficient to allow the invention to be performed over the whole scope of the claim;
(vii) the specification must be sufficient to allow the invention to be so performed without undue burden.
527 “Classical insufficiency” is to be distinguished from “Biogen insufficiency” which is also considered under United Kingdom law to form part of the disclosure obligation. That overlap may be considered to be confusing at first, because Biogen insufficiency draws on the law of support, identified in s 14(5)(c) of the UK Act. However, as the cases in that jurisdiction explain, the reason for this is because the UK Act contains a “logical gap” arising from its drafting, in that whilst s 14(5)(c) imposes the claim support obligation as a statutory requirement for the grant of a patent, there is no concomitant provision whereby a granted patent that fails to satisfy the claim support obligation may be revoked. That gap was plugged when the House of Lords resolved that the claim support obligation fell under the umbrella of the requirement that the patent specification contain an enabling disclosure: Biogen Inc v Medeva Plc [1996] 10 WLUK 486; [1997] RPC 1 at 47 (Lord Hoffmann, with whose reasons the other members of the House of Lords agreed). Accordingly, in the context of revocation actions, the UK courts sometimes (but not always) refer to a distinction between classical insufficiency and Biogen insufficiency, the former arising from s 14(3) and the latter arising from s 14(5)(c), but both falling within the unifying requirement that there be an enabling disclosure, and both being available as a ground of invalidity within s 14(3).
528 The main difference between the two is that the disclosure obligation under s 14(3) relates to the specification as a whole whereas the claim support obligation under s 14(5)(c) relates to the claims which define the invention: Generics UK (HL) at [19]. As Walker LJ said in Generics (UK) at [20]:
Ss 14(3) and (5)(c) operate together, as EPC Arts 83 and 84 operate together, to spell out the need for an “enabling disclosure”, which is central to the law of patents…The disclosure must be such as to enable the invention to be performed (that is, to be carried out if it is a process, or to be made if it is a product) to the full extent of the claims. The question whether there is sufficient enabling disclosure often interacts with a question of construction as to the extent of the claims...
529 In Terrell the learned editors summarised the distinction between classical sufficiency and Biogen sufficiency in the following terms at page 403:
The self-standing objection that a claim is broader than the technical contribution of the patent, even when it can be performed, is sometimes referred to as “Biogen insufficiency”. It is to be contrasted with “classical insufficiency” which is concerned with whether or not embodiments within the claim can be performed. Thus peculiarly under English law it is said that at patent can be insufficient even if it is possible to make everything within the scope of the claim, if the scope of the claims exceed the technical contribution.
16.3.2 The claim support obligation: “Biogen insufficiency”
530 The content of the claim support obligation was identified by Walker LJ in Generics (UK) at [19], by reference to the Exxon/Fuel Oils decision which said at [3.3]:
Furthermore, Article 84 EPC also requires that the claims must be supported by the description, in other words it is the definition of the invention in the claims that needs support. In the Board’s judgment, this requirement reflects the general legal principle that the extent of the patent monopoly, as defined by the claims, should correspond to the technical contribution to the art in order for it to be supported, or justified…
531 It may be seen that for the claim breadth to be supported (or justified), it must correspond to the “technical contribution to the art”. In Biogen, the House of Lords was concerned with whether or not a claim was entitled to priority from an earlier application within s 5(2)(a) of the UK Act, and so the claim support obligation was clearly in issue. It was considering a complicated claim to a product, being a molecule identified partly by the way in which it had been made, and partly by what it did. On the facts, the patentee could not claim either the product (a recombinant DNA molecule involving fragments of the “Dane particle”), as it had already been made, or the process involved (recombinant DNA technology enabling expression in a cell), as it had already been invented. Lord Hoffmann said at 50 – 51 that the question:
...is not whether the claimed invention could deliver the goods, but whether the claims cover other ways in which they might be delivered: ways which owe nothing to the teaching of the patent or any principle which it disclosed.
It will be remembered that in Genentech I/Polypeptide expression the Technical Board spoke of the need for the patent to give protection against other ways of achieving the same effect “in a manner which could not have been envisaged without the invention”. This shows that there is more than one way in which the breadth of a claim may exceed the technical contribution to the art embodied in the invention. The patent may claim results which it does not enable, such as making a wide class of products when it enables only one of those products and discloses no principle which would enable others to be made. Or it may claim every way of achieving a result when it enables only one way and it is possible to envisage other ways of achieving that result which make no use of the invention.
532 Earlier in his reasons (at 48), Lord Hoffmann had said that if the invention disclosed in the specification is “a principle of general application, the claims may be in correspondingly broad terms”. This seems to have set the foxes running, and later was further developed by Lord Hoffmann in Kirin-Amgen Inc v Hoechst Marion Roussel Ltd [2004] UKHL 46; [2005] 1 All ER 667 (with whom Hope, Rodger, Walker and Brown LJJ agreed). His Lordship said (emphasis added):
[112] This gave rise to a good deal of argument about what amounted to a “principle of general application”. In my opinion there is nothing difficult or mysterious about it. It simply means an element of the claim which is stated in general terms. Such a claim is sufficiently enabled if one can reasonably expect the invention to work with anything which falls within the general term. For example, in Genentech/Polypeptide expression (T 292/85) [1989] O.J. EPO 275, the patentee claimed in general terms a plasmid suitable for transforming a bacterial host which included an expression control sequence to enable the expression of exogenous DNA as a recoverable polypeptide. The patentee had obviously not tried the invention on every plasmid, every bacterial host or every sequence of exogenous DNA. But the Technical Board of Appeal found that the invention was fully enabled because it could reasonably be expected to work with any of them.
[113] This is an example of an invention of striking breadth and originality. But the notion of a “principle of general application” applies to any element of the claim, however humble, which is stated in general terms. A reference to a requirement of “connecting means” is enabled if the invention can reasonably be expected to work with any means of connection. The patentee does not have to have experimented with all of them.
533 So expressed, the claim support obligation is based on a requirement that the technical contribution to the art disclosed by the specification justifies the breadth of the monopoly claimed. As the learned authors of Terrell put it at page 212:
Furthermore, in the Biogen case the House of Lords decided that what constitutes a sufficiently enabling disclosure to support a claim will depend on the breadth of the claim itself. Thus a disclosure of a principle capable of general application may support claims in correspondingly general terms, such that a patentee will not need to show that they have proved the application of the principle in every individual instance. However, if the claims include a number of discrete methods or products the patentee must enable the invention to be performed in respect of each of them.
534 In Regeneron the Court considered two patents which sought to confer a monopoly over the creation of a range of types of transgenic mice. The insufficiency challenge to the patents in the Court of Appeal (in Regeneron Pharmaceuticals, Inc v Kymab Ltd [2018] EWCA Civ 671; RPC 14) and the Supreme Court involved an analysis of what may be considered to be an “enabling disclosure” as developed in the authorities, which I have noted is a concept that encompasses the disclosure obligation (or classical insufficiency) and the claim support obligation (or Biogen insufficiency).
535 The relevant claim was for a range of transgenic mice (which were referred to as “products”) answering a certain description. The Court of Appeal held that the teaching in the patent coupled with the available common general knowledge as at the priority date enabled some, but not all, types of mice within the claimed range to be made (Regeneron at [4]). Nevertheless, the claim support obligation was met because the invention claimed was for an inventive, ground-breaking principle, such that every type of mouse within the specified range that could now be made or would in the future be made would display the benefits which the invention was designed to achieve (at [4]). The central issue on appeal accordingly arose because although the beneficial effects of the principle disclosed in the patent would apply to every mouse within the range claimed, the specification did not disclose how to make every mouse in the range (at [11]).
536 The Supreme Court considered the question of the validity of the claim within the general concept of “sufficiency”, as I have described above. In so doing, the claim support obligation and the disclosure obligation were considered under the general rubric of “enablement” whereby, as Briggs LJ said, “the essential patent bargain is not satisfied in relation to products in that part of the range which cannot be made, using the teaching in the patent” (at [25] – [26]).
537 Lord Hoffmann’s reference to a principle of general application in Biogen arose as a focal point of the distinction between the approach taken by the Court of Appeal and that of the Supreme Court. As summarised by Briggs LJ, in relation to the concept of enablement the Court of Appeal:
…concluded that it would defeat the implementation of the essential patent bargain if applied to a case in which the invention amounted to a principle of general application, which would yield the relevant increase in efficiency or usefulness across a range of potential product types if they incorporated the invention, as and when they could be made, even if only a few could be made as at the priority date by using the teaching in the patent. In bare outline their reasoning was as follows. The patent bargain requires that the reward given to the patentee should be commensurate with the contribution which the invention makes to the art. An invention which consists of a new generally applicable principle may contribute to the art by its use, not only in products which can currently be made, but equally in products which will only be capable of being made in the future, after further inventive research and development. To limit the patentee strictly to a monopoly over the products which can immediately be made would be to deprive the patentee of any reward for the public benefit which will be derived from the use of that same invention in future types of product. In a fast-moving field, where new products quickly outperform their predecessors so as to render them obsolete, the reward of a monopoly limited to those immediately capable of being made would be short-lived and illusory. Accordingly the invention should be regarded as sufficiently enabled across the range if it can be seen that it will in due course benefit all products in the range, provided that, as at the priority date, the teaching in the patent enables at least one type to be made immediately…
538 In so reasoning, the question of the breath of the claim arose, and whether (to use the definitions I have applied) the claim support obligation was met. The Supreme Court reviewed European and UK law on this broad subject of enablement, but picked up the claim support obligation by reference to Biogen. Most particularly, at [53] the Supreme Court returned to Lord Hoffmann’s clarification in Generics (UK) Ltd v H Lundbeck A/S [2008] EWCA Civ 311; RPC 19 (Generics UK (CoA)) of the meaning to be given to his earlier reference to “a principle of general application” in Kirin-Amgen (to which I have referred at [532] above).
539 To explain what follows it is necessary to follow a short tributary for a moment. After Kirin-Amgen was decided in 2004, Biogen insufficiency was further considered in Generics UK (CoA). Unusually, Lord Hoffmann sat on the Court of Appeal (stepping down to that Court from the House of Lords) and delivered the judgment of the court (with whom Jacob and Smith LJJ agreed). The case then went to the House of Lords on appeal, which upheld Lord Hoffmann’s decision, but for different reasons: Generics UK (HL).
540 Lord Hoffmann in Generics UK (CoA) considered the finding of the trial judge that the claim in suit lacked Biogen insufficiency because the relevant “technical contribution” to the art was the inventive step. Lord Hoffmann found that to be the incorrect approach. The technical contribution to the art for a product clam is the product, and not the process by which it was made (at [30]).
541 In Regeneron, the Supreme Court endorsed this approach (at [54]). In so doing, it successfully bypassed confusion arising from the House of Lords decision in Generics UK (HL), which had upheld the decision of the Court of Appeal but, confusingly, appears nonetheless to have accepted that the technical contribution to the art was somehow linked to the inventive step rather than being the contribution to the art disclosed in the specification: see Walker LJ at [34]; Mance LJ at [45]; Phillips LJ at [1].
542 The Supreme Court concluded at [56]:
Reflection upon those European and UK authorities yields the following principles:
i) The requirement of sufficiency imposed by article 83 of the EPC exists to ensure that the extent of the monopoly conferred by the patent corresponds with the extent of the contribution which it makes to the art.
ii) In the case of a product claim, the contribution to the art is the ability of the skilled person to make the product itself, rather than (if different) the invention.
iii) Patentees are free to choose how widely to frame the range of products for which they claim protection. But they need to ensure that they make no broader claim than is enabled by their disclosure.
iv) The disclosure required of the patentee is such as will, coupled with the common general knowledge existing as at the priority date, be sufficient to enable the skilled person to make substantially all the types or embodiments of products within the scope of the claim. That is what, in the context of a product claim, enablement means.
v) A claim which seeks to protect products which cannot be made by the skilled person using the disclosure in the patent will, subject to de minimis or wholly irrelevant exceptions, be bound to exceed the contribution to the art made by the patent, measured as it must be at the priority date.
vi) This does not mean that the patentee has to demonstrate in the disclosure that every embodiment within the scope of the claim has been tried, tested and proved to have been enabled to be made. Patentees may rely, if they can, upon a principle of general application if it would appear reasonably likely to enable the whole range of products within the scope of the claim to be made. But they take the risk, if challenged, that the supposed general principle will be proved at trial not in fact to enable a significant, relevant, part of the claimed range to be made, as at the priority date.
vii) Nor will a claim which in substance passes the sufficiency test be defeated by dividing the product claim into a range denominated by some wholly irrelevant factor, such as the length of a mouse’s tail. The requirement to show enablement across the whole scope of the claim applies only across a relevant range. Put broadly, the range will be relevant if it is denominated by reference to a variable which significantly affects the value or utility of the product in achieving the purpose for which it is to be made.
viii) Enablement across the scope of a product claim is not established merely by showing that all products within the relevant range will, if and when they can be made, deliver the same general benefit intended to be generated by the invention, regardless how valuable and ground-breaking that invention may prove to be.
543 It is true, as Wyeth submits, that the decision in Regeneron concerns the broader concept of enablement and sufficiency as it is applied in the United Kingdom. However, as the reasoning in the United Kingdom authorities makes clear, classical insufficiency and Biogen insufficiency are really two sides of the same coin. Both concern the essential patent bargain, and both require consideration, as a matter of substance, of the scope of the disclosure of the specification when read against the scope of the claims. Insofar as the support obligation is concerned, in the United Kingdom it is not enough for the “inventive step” to be applicable to the full range of products falling within the scope of the claims. It is the full range of products that must be able to be made on the basis of the disclosure in the specification. As the Supreme Court said at [58]:
A comparison between those principles and those applied by the Court of Appeal reveals that they did not correctly apply the law as it stands, for the following reasons. First, I cannot accept their summary of the essential patent bargain. In the case of a product claim, the contribution to the art is the product which is enabled to be made by the disclosure, not the invention itself. Patents are about products and processes, not pure ideas. Secondly, I do not accept their conclusion that an invention may be “enabled” in relation to a particular type of product falling within the scope of the claim even if it does not permit the skilled person to make it. They thought it was enough that the benefits which the invention unlocked (in terms of preventing murine immunological sickness) would in due course be realised over the whole range, if and when all embodiments within the range could be made. In practical terms they upheld a monopoly over that part of the range of products answering the broad description in Claim 1 which was likely to be of most benefit to medical genetic engineering, at a time when the disclosure in the patent only enabled the skilled person to make products over a very small part of the range, and at the least beneficial end of the range denominated by the amount of the human variable region gene locus incorporated in the hybrid gene structure. It is now known that the type of mouse fitted with a Reverse Chimeric Locus which actually does serve as the gold standard in the art has the whole of the human variable region gene locus as part of its hybrid antibody gene structure. Yet the Court of Appeal would have upheld a monopoly for its manufacture and exploitation when the disclosure in the patent, coupled with the common general knowledge, would not have enabled a skilled person to make such a mouse at all. The ability of both the appellant and the respondent to make such a mouse now depends upon further (and different) inventions separately made by each of them some years after the priority date.
16.4 The law of support in Australia
544 It is apparent from the language adopted in the sections and also from the Second Reading Speech and the Explanatory Memorandum that the intention of parliament in amending s 40(2)(a) and s 40(3) of the Patents Act was to align the law in relation to these requirements with that of the United Kingdom and Europe. That is not to say that all aspects of the approach adopted in the United Kingdom are to be adopted here. In particular, there is no warrant provided in the language of s 40(2)(a) to incorporate within the disclosure obligation a separate claim support obligation, in addition to the one within s 40(3). Nor is there any need to do so: failure to comply with either ss 40(2)(a) or 40(3) provides a basis upon which a granted patent may be revoked: s 138(3)(f). There is no gap in the Patents Act akin to the one in the UK Act referred above, and each ground is to be considered separately. Nevertheless, the law as it has developed in the United Kingdom and Europe in relation to the support obligation, when disentangled from classical insufficiency, provides guidance as to how s 40(3) should be approached.
545 The theme common to each ground, however, reflects what the Minister described in his Second Reading Speech as the cornerstone of the patent system, namely that in exchange for a monopoly on commercialisation, the patentee must tell the public how their idea works. As said in the Explanatory Memorandum, in exchange for the exclusive rights given to the patentee, the patentee must share with the public the information necessary to make and use the invention. This is the essential exchange between inventors and the public which has long been a feature of patent law in Australia: see Lockwood No 1 at [57].
546 In CSR Building Products Ltd v United States Gypsum Company [2015] APO 72, Dr S D Barker adopted the summary provided by Aldous J in Schering Biotech at 252 – 253, which has been often followed in the United Kingdom (emphasis added):
…to decide whether the claims are supported by the description it is necessary to ascertain what is the invention which is specified in the claims and then compare that with the invention which has been described in the specification. Thereafter the court’s task is to decide whether the invention in the claims is supported by the description. I do not believe that the mere mention in the specification of features appearing in the claim will necessarily be a sufficient support. The word “support” means more than that and requires the description to be the base which can fairly entitle the patentee to a monopoly of the width claimed.
547 That approach encapsulates broadly the claim support obligation under s 40(3). To it may be added the requirement that the technical contribution to the art must be ascertained. Where it is a product, it is that which must be supported in the sense that the technical contribution to the art disclosed by the specification must justify the breath of the monopoly claimed.
548 It will be recalled that claim 1 of the 844 patent is as follows:
A multivalent immunogenic composition, comprising polysaccharide-protein conjugate together with a physiologically acceptable vehicle, wherein each of the conjugates comprises a capsular polysaccharide from a different serotype of Streptococcus pneumoniae conjugated to a carrier protein, and the capsular polysaccharides are prepared from [the 13 chosen serotypes], wherein the carrier protein is CRM197 for use as a vaccine to protect or treat a human susceptible to pneumococcal infection.
549 I have concluded in section 5.3.2 above that “comprising” is to be construed inclusively. A multivalent immunogenic composition including the integers of this claim, but also possessing additional features, will fall within the monopoly of the claim. The example embraced by Wyeth (obviously enough) is MSD’s 15-valent vaccine. But as Wyeth submits, any composition comprising the 13 chosen serotypes would infringe the claim. That is not limited to a formulation containing 15 serotypes, but may go beyond.
550 I have in section 4.1 reviewed in some detail the specification of the composition patents. There is no doubt that the disclosure of the specification focusses on the 13 chosen serotypes. As I have found, it discloses the idea to add serotypes 1, 3, 5, 6A, 7F and 19A to the Prevnar 7 serotypes and to conjugate them to CRM197, coupled with the practical means by which this is achieved. There is no disclosure of the conjugation of any additional serotypes to CRM197. The evidence of Professor Paton is that he does not consider that it is possible to extrapolate the data in the composition patents to other serotypes, because it is focussed on the serotypes covered by the patents. That evidence is uncontested. My findings as to the common general knowledge reveal that adding serotypes to the composition claimed would be a complex and difficult process.
551 The idea underlying the invention, which is described in the Background to the Invention, is that the protection against Streptococcus pneumoniae provided by Prevnar 7 can be improved by the addition of serotypes 1, 3, 5, 6A, 7F and 19A. The addition of those serotypes will provide, inter alia, coverage that is not dependent on the limitations of cross-protection for serotypes 6A and 19A. The solution then described and put into effect by reference to the examples is a 13-valent immunogenic composition using the 13 chosen serotypes. The selection of the particular additional serotypes is a matter of analysis and consideration in the specification, and an enabling disclosure is provided such that the person skilled in the art is able to perform the preparation, purification, and conjugation to CRM197, for each of the 13 chosen serotypes. For serotype 3, the specification identifies that the selection of CRM197 as the carrier protein is an advantage, and notes (page 6 lines 5 – 23) that previously tried compositions, such as an 11-valent composition conjugated to protein D, were not successful.
552 The technical contribution to the art as described in the specification lies in the identification of the additional chosen serotypes, the choice of CRM197 as the protein carrier and the provision to the skilled reader of a means by which the claimed 13-valent composition may be made. The disclosure of the specification is not for a principle of general application beyond the product.
553 I consider that MSD has established that the asserted 844 patent claims do not satisfy the claim support obligation under s 40(3) and as consequence they are invalid for want of support. The patentee has established in its specification that it has hit upon a new product which has a beneficial effect, but it has claimed a monopoly that includes compositions that are not the product of the technical contribution to the art provided by the specification. The inclusively worded claims do not correspond to the technical contribution to the art. The claims cover products that the specification does not enable, and the specification discloses no principle that would enable others to be made: Biogen at 50 – 51.
554 Contrary to the submission advanced by Wyeth, the decision of the Supreme Court in Regeneron tends to demonstrate the difficulty with its position, rather than the opposite. In that case variant mice falling within the range claimed were predictably able to provide the benefits achieved by the invention, however not all variants claimed were able to be made by the person skilled in the art having regard to the disclosure of the specification. The submission advanced by Wyeth is that because any composition comprising the 13 named serotypes conjugated to CRM197 would nonetheless embody the technical contribution made the invention with respect to those serotypes, the claim support obligation is met. However, in my view there is no material distinction between claiming a range that is not able to be made (as in Regeneron) and claiming a specific composition which is left open by inclusive langue so that additional serotypes may be included in the composition (as here). In both cases, the claim breadth exceeds the technical contribution provided by the disclosure of the relevant specification.
555 Wyeth submits that the factual circumstances are such that MSD “cannot avoid” a finding of infringement by adding integers to Wyeth’s claimed product. It also submits that to find in favour of MSD is contrary to patent law, because every product claim using the words “comprising” or “including” is liable to be revoked for lack of support. Neither proposition is correct. The question concerns the breadth of the claims drafted by the patentee, and their intersection with s 40(3), not the alleged infringing conduct of MSD. Here, the patentee chose to use inclusive and non-exhaustive language to encompass the components of the composition. It no doubt did so to spread the net of its monopoly wide. It could have chosen to do otherwise. Whether other claims are invalid will depend on the particular language they use, and the scope of the disclosure of the relevant specification.
556 I accept that the requirement that the claims should correspond with the technical contribution to the art does not mean that everything falling within the scope of the claims must be enabled. The requirement is one of substance and de minimis differences are to be set to one side. However, in the present case the identification and enablement of the 13 chosen serotypes, each conjugated to CRM197, is central to the disclosure.
557 Finally, contrary to Wyeth’s submission I do not consider that the invention may be characterised as a principle of broad application. It is a product that encompasses the particular embodiment of the 13-valent composition.
17. COMPOSITION PATENTS: MANNER OF MANUFACTURE
558 MSD pleads that the asserted composition claims are not to a manner of new manufacture within the meaning of s 6 of the Statute of Monopolies (21 Jac, c 3) for the purposes of s 18(1)(a) of the Patents Act. In its closing submissions it advances three bases:
(1) The composition patents admit on the face of their specifications that there is no invention, citing various passages upon which it relies for such admissions (the face of the specification argument).
(2) The claims of the composition patents include within their scope compositions which have polysaccharide-protein conjugates of Streptococcus pneumoniae serotypes in addition to the 13 chosen serotypes. Accordingly, the claims are either to mere desiderata or compositions that are yet to be identified or discovered (the more than 13 serotypes argument).
(3) The grant of a patent with respect to the claimed immunogenic compositions is generally inconvenient because the indeterminate scope of the claims will prevent or hinder research and development of immunogenic compositions for use as vaccines against Streptococcus pneumoniae, contrary to the public interest (the generally inconvenient argument).
559 Wyeth contends that MSD’s case in each respect is misconceived. I address each in turn below.
17.2 The face of the specification argument
560 MSD submits that there is no “inventiveness” on the face of the specification of the composition patents and that accordingly, the claims are not to a manner of manufacture. Whilst there are numerous ways in which the word “invention” may be understood (see, for instance, Kimberly-Clark at [18]), MSD advances this ground on the basis that there is no inventive step apparent on the face of the specification, and accordingly the specification fails. Given the presence of 18(1)(b)(ii) and s 7(2) of the Patents Act, which provide for a separate ground of lack of inventive step, the face of the specification argument is an odd ground. Wyeth submits that it does not exist however, there is authority to suggest that the ground is available.
561 Section 18(1)(a) of the Act relevantly provides:
…an invention is a patentable invention for the purposes of a standard patent if the invention, so far as claimed in any claim:
(a) is a manner of manufacture within the meaning of s 6 of the Statute of Monopolies…
562 Section 6 of the Statute of Monopolies declared all monopolies to be void except for:
Letters Patents and Grants of Privilege for ... the sole working or making of any manner of new Manufactures within this Realm, to the true and first Inventor and Inventors of such Manufactures, which others at the time of making such Letters Patents and Grants shall not use, so as also they be not contrary to the Law, nor mischievous to the State, by raising prices of Commodities at home, or hurt of Trade, or generally inconvenient...
563 In D’Arcy v Myriad Genetics Inc [2015] HCA 35; 258 CLR 334, French CJ, Kiefel, Bell and Keane JJ said at [12] (emphasis added, citations omitted):
The term "patentable invention" is defined in the Dictionary in Sched 1 to the Act as "an invention of the kind mentioned in section 18." The term "invention" is defined as:
"any manner of new manufacture the subject of letters patent and grant of privilege within section 6 of the Statute of Monopolies, and includes an alleged invention."
It is not clear, and was not debated in this appeal, how the expression "manner of manufacture" differs from the expression "manner of new manufacture". The definition of "invention" has been used in Commonwealth patent statutes since federation. It allows for exclusion from the class of "invention", and therefore from the class of "patentable invention", anything which is not, on the face of the specification, a proper subject of letters patent according to traditional principles. That anterior exclusion may be based upon an admission, on the face of the specification, which makes clear that the invention claimed is not novel or does not involve an inventive step. This appeal, however, collapses the anterior and subsequent questions – "Is there an invention?" and "Is there a patentable invention?" – into one inquiry. That inquiry requires a definition of the allegedly patentable invention. That definition depends upon the construction of the impugned claims read in the light of the specification as a whole and the relevant prior art. The prior art in this case was reflected in expert evidence at trial and set out in the scientific primer agreed between the parties and summarised later in these reasons.
564 In relation to the emphasised passage, the plurality cited N V Philips Gloeilampenfabrieken v Mirabella International Pty Ltd [1995] HCA 15; 183 CLR 655 at 665 and 667 (per Brennan, Deane and Toohey JJ).
565 Wyeth submits that there is no separate threshold requirement for patentability that inventiveness be shown on the face of the specification. I am unable to agree. In Philips the plurality affirmed that the necessary quality of inventiveness on the face of the specification remains a threshold requirement for patentability, notwithstanding the inclusion in s 18 of the separate grounds of invalidity of lack of inventive step and lack of novelty. Their Honours considered that the legislative intent evinced by the language of s 18(1)(a) read with the definition of “invention” contained in the Dictionary was that there should remain a basis for excluding inventions from patent protection that, on the face of the specification, claim nothing more than “the use of a known material in the manufacture of known articles for the purpose of which its known properties make that material suitable”, quoting Commissioner of Patents v Microcell Ltd [1959] HCA 71; 102 CLR 232 at 251 (per Dixon CJ, McTiernan, Fullagar, Taylor and Windeyer JJ).
566 The plurality at 664 (which the Court in Lockwood No 2 cited at [106]) further held that the threshold requirement that there be an “alleged invention” will remain unsatisfied if it is apparent on the face of the specification that the subject matter of the claim is, by reason of the absence of the necessary quality of inventiveness, not a manner of new manufacture. This does not mean, the Court said, that the threshold requirement of “an alleged invention” corresponds with or renders otiose the more specific requirements of inventive step or novelty, but that “[i]t simply means that, if it is apparent on the face of the specification that the quality of inventiveness necessary for there to be a proper subject of letters patent under the Statute of Monopolies is absent, one need go no further” (emphasis added). The Court developed this a little later at 664 – 665 (citation omitted):
It is true that it can be argued that there is internal tension in an overall legislative scheme which imposes a threshold requirement of inventiveness reflecting the effect of the saving clause in s 6 of the Statute of Monopolies and then proceeds, if that threshold requirement be satisfied, to impose more specific requirements of novelty and inventive step. It seems to us, however, that there are several answers to that argument…it seems to us to be highly unlikely that it was the legislative intent that there should be a significant alteration of the law as explained in Microcell by extending the ambit of a patentable invention so as to include what is “nothing more” than “the use of a known material in the manufacture of known articles for the purpose of which its known properties make that material suitable”.
567 Successive courts have grappled with the precise meaning of Philips in this regard. The Full Court in Bristol-Meyers Squibb Company v F H Faulding & Co Limited [2000] FCA 316; 97 FCR 524 at [30] (Black CJ and Lehane JJ, with whom Finkelstein J agreed generally at [162]) said that Philips stands for the proposition, as a matter of construction of the Patents Act, that if, on the basis of what was known as revealed on the face of the specification, the invention claimed was obvious or did not involve an inventive step, the threshold requirement of inventiveness is not met. The Court considered that this is the only approach which does not render the separate requirement of inventive step in s 18(1)(b)(ii) otiose. It seems to me that I am bound by this construction, which has been followed by the Full Court in AstraZeneca (FC) at [391]. The reasoning of the Court in Lockwood No 2 is not inconsistent with this approach. It observes, in obiter dicta, that Microcell stands for the narrow proposition that a Commissioner of Patents (or, one may infer, the Court) may refuse an application for patent protection (or revoke a subsequently granted patent) where a specification “on its face” shows the invention is not a manner of new manufacture. The example given is that this may arise from admissions concerning novelty. However, having regard to the reasoning in Philips in the passage cited, it is apparent that this may apply to a case where lack of inventive step is apparent on the face of the specification. Having regard to the more recent apparent endorsement of Philips by the High Court in D’Arcy, I am not disposed to accept the submission advanced by Wyeth that since Lockwood No 2 there is no separate threshold requirement. In this regard, I regretfully disagree with Beach J’s reasoning in Sequenom, Inc. v Ariosa Diagnostics, Inc. [2019] FCA 1011; 143 IPR 24 at [392] and [393]. His Honour does not appear to have been taken to the Full Court cases to which I have referred.
568 Even so, it is apparent that the Full Court in Bristol-Meyers considered the retention of a form of vestigial ground of lack of inventive step within the ground of manner of manufacture with caution. As it notes at [27], Philips does not provide a comprehensive answer to the question of: by what body of knowledge is that inventiveness to be judged? The Full Court decided that this question was to be resolved by confining consideration of inventive step to the face of the specification. This raises a high bar to those seeking to establish lack of inventiveness in this way. In November 2011, in its response to the Senate Community Affairs Reference Committee Gene Patents Report of November 2010, the Federal Government addressed concerns held by the Advisory Council on Intellectual Property and the Australian Law Reform Commission by saying that it intended to “develop legislation to define patentable subject matter using clear and contemporary language”. However, no changes have yet been made.
569 Accordingly, I proceed on the basis that this sub-category of absence of manner of manufacture is available as a ground of revocation.
570 MSD relies on several propositions, drawn from the face of the specification of the composition patents, to piece together the conclusion that the invention claimed is not to manner of manufacture. The first is that the composition patents admit that the 13 chosen serotypes were selected based on publicly available data and literature. The second is that the specification admits that the production of the compositions was “routine”. Consideration of these points is sufficient to reject the argument advanced by MSD.
571 In relation to the first proposition, it may be recalled that the specification refers to the Prevnar 7 serotypes and develops in the course of the detailed description of the invention how the additional six serotypes were selected. Whilst the specification does refer to and incorporate by reference numerous articles such as Hausdorff 2000 and Whitney 2003, none disclose a composition that falls within the claims, or contain a clear statement that the 13 chosen serotypes be included in a composition.
572 In relation to the second proposition, I have considered in some detail in section 4.1 the disclosure of examples 1 – 14. The reference to “standard techniques” for the preparation of the capsular polysaccharides, and “conventional means” for the chemical activation of the polysaccharides and subsequent conjugation to the carrier protein (page 11 lines 11 – 25), is to a very high level abstraction of knowledge that may be imputed to the person skilled in the art. However, the descriptions in examples 1 – 14 go into the detail necessary to enable that person to make the invention. The procedures are detailed and varied. It is not open, having regard to the disclosure set out in those examples, to conclude that the means by which the compositions were produced in accordance with those examples was “routine”. Indeed, as the inventive step analysis explains, MSD has not discharged its onus of establishing that it was routine.
573 Furthermore, as the Full Court said in Bristol-Meyers at [45], there is an air of unreality about posing a question of whether the invention claimed lacks the necessary quality of inventiveness in a case, such as the present, where the question of lack of inventive step pursuant to s 7(2) has been considered at length. Its comments as follows are apposite here:
This is not a case, like Philips, where there was no attack on the patents on the ground of obviousness. It was, instead, a case where expert evidence, including evidence as to common general knowledge, was available (and was given). Where the Court has evidence on the basis of which it can make a finding about common general knowledge, and the other information referred to in s 7(2) and (3), and about what would or would not have been obvious to persons skilled in the relevant art, it must be only rarely that it will be appropriate to find (by resort to a “threshold test”) lack of inventiveness on the face of a specification. In our opinion this is not a case where such a finding is justified.
574 The same may be said of the present case. To this it might be noted that the present case is not analogous to Merck & Co Inc v Arrow Pharmaceuticals Limited [2006] FCAFC 91; 154 FCR 31 (Heerey, Kiefel and Dowsett JJ).
17.3 The more than 13 serotypes argument and the generally inconvenient argument
575 MSD submits that because the asserted composition patent claims include within their scope any composition that includes the 13 chosen serotypes, the claims incorporate within the monopoly immunogenic compositions with potentially more than 90 known pneumococcal serotypes. The composition patents may therefore claim many separate inventions and accordingly not be the proper subject of letters patent pursuant to factor 3.1 identified at [28] of D’Arcy.
576 This ground of invalidity draws on a different thread to that relied upon for the face of the specification argument.
577 In D’Arcy the plurality said at [28]:
A number of factors may be relevant in determining whether the exclusive rights created by the grant of letters patent should be held by judicial decision, applying s 18(1)(a) of the Act, to be capable of extension to a particular class of claim. According to existing principle derived from the NRDC decision, the first two factors are necessary to characterisation of an invention claimed as a manner of manufacture:
1. Whether the invention as claimed is for a product made, or a process producing an outcome as a result of human action.
2. Whether the invention as claimed has economic utility.
When the invention falls within the existing concept of manner of manufacture, as it has been developed through cases, they will also ordinarily be sufficient. When a new class of claim involves a significant new application or extension of the concept of "manner of manufacture", other factors including factors connected directly or indirectly to the purpose of the Act may assume importance. They include:
3. Whether patentability would be consistent with the purposes of the Act and, in particular:
3.1. whether the invention as claimed, if patentable under s 18(1)(a), could give rise to a large new field of monopoly protection with potentially negative effects on innovation;
3.2. whether the invention as claimed, if patentable under s 18(1)(a), could, because of the content of the claims, have a chilling effect on activities beyond those formally the subject of the exclusive rights granted to the patentee;
3.3. whether to accord patentability to the invention as claimed would involve the court in assessing important and conflicting public and private interests and purposes.
…
Factors 3, 4 and 6 are of primary importance. Those primary factors are not mutually exclusive. It may be that one or more of them would inform the "generally inconvenient" limitation in s 6 of the Statute of Monopolies. It is not necessary to consider that question given that no reliance was placed upon that proviso. They are nevertheless also relevant to the ongoing development of the concept of "manner of manufacture".
578 I cannot see that factor 3.1 arises in the present case. That is because the commencement of the enquiry is, as the High Court said in D’Arcy, consideration of the invention claimed: at [12]. Then one considers whether it is for an artificially created state of affairs of economic utility.
579 In my view it is plain enough that the asserted claims are for pharmaceutical compositions of the type long recognised to fall within the class of patentable inventions as identified in National Research Development Corporation v Commissioner of Patents [1959] HCA 67; 102 CLR 252 (Dixon CJ, Kitto and Windeyer JJ). The claims and the disclosure of the specification in the present case may be distinguished from those in D’Arcy. In that case, the Court was not persuaded that the impugned claims were for a chemical compound. Rather, despite the language used in the claim, the substance was information embodied in arrangements of nucleotides. The information was not “made by human action”. It was “discerned” (at [6]). The impugned product claims did not fall within the established boundaries, and “wider considerations” came into play (at [27]). Viewed as a matter of substance, the invention was characterised as “information” and not merely a product (at [89]).
580 However, here the claims are for a composition that falls within the existing concept of manner of manufacture, as it has been developed through the cases. It is a pharmaceutical product in the form of an immunological composition. As the Court in D’Arcy noted at [28], that will also ordinarily be sufficient. This ground must be rejected.
581 Nor do I accept the submission, fleetingly put, that the asserted claims constitute a “mere desideratum”: cf Grant v Commissioner of Patents [2006] FCAFC 120; 154 FCR 62 at [18] (Heerey, Kiefel and Bennett JJ).
582 MSD next submits that the content of the claims, if valid, would have a “chilling effect” on activities beyond those formally the subject of the exclusive rights granted to Wyeth, and accordingly this informs the “generally inconvenient” limitation in s 6 of the Statute of Monopolies. MSD relies on factors 3.1 and 3.2 in D’Arcy, set out above. MSD submits that for a new conjugate vaccine with expanded coverage, in order to satisfy non-inferiority requirements, one must start with the 13 chosen serotypes. It submits that if the asserted composition patent claims cover conjugate vaccines of serotypes beyond the 13 specified, this would deter research.
583 The same arguments arise in relation to factors 3.1 and 3.2 as those to which I have referred above. Furthermore, D’Arcy at [28] is not directly applicable in relation to this argument. There may perhaps be a policy argument available were it to be established that the only manner in which expanded protection against pneumococcal disease beyond the existing coverage was by the chosen serotypes. But that is not the case here. The asserted claims are in each case limited by the requirement that the serotypes be conjugated to CRM197. The evidence indicates that other carrier proteins are available, such as protein D. It also indicates that a bi-carrier approach may be used, or that research to expand the coverage against pneumococcal disease could head in the direction of protein vaccines. No evidence suggests that the only realistic avenue of research available is to advance the development of coverage by reference to the 13 chosen serotypes using CRM197 as the carrier protein. I am not satisfied that this ground is made out.
18. CONTAINER PATENT: INTRODUCTION
584 The container patent concerns the use of components in a formulation which stabilise a composition including one or more polysaccharide-protein conjugates. The priority date for the claims is 26 April 2006, about a year later than the priority date applicable to the claims of the compositions patents. Furthermore, as I note below, whilst the skilled addressees of the container patent remain involved in the field of immunology, they are mostly interested as formulators of polysaccharide-protein conjugate vaccines rather than the developers of such conjugates. As a result of these differences, the pleadings, and the cast of witnesses and arguments presented by the parties were different to those advanced in respect of the composition patents.
585 Wyeth alleges in its Amended Statement of Cross-Claim filed on 12 November 2019 that MSD intends to import and offer to sell, sell, keep and market in Australia a 15-valent pneumococcal polysaccharide-protein conjugate vaccine together with a physiologically acceptable vehicle in which each of the 13 chosen serotypes plus serotypes 23F and 33F is conjugated to CRM197. It alleges that by doing so MSD will infringe each of asserted container patent claims. One feature of each of those claims is “[a] siliconized container means”. MSD admits that it intends to import such a product, but denies that it threatens to infringe on two bases. First, because some of the products that that it intends to import do not use a siliconised container means. Wyeth submits that having regard to the manner in which the case has been conducted, including by the pleadings and evidence, MSD is precluded from raising this issue (siliconised container dispute). Secondly, MSD contends that its 15-valent vaccine does not infringe claim 18 of the container patent, for substantially the same reasons that it contends that the claims of the composition patents do not extend to a monopoly beyond the 13 chosen serotypes (container patent construction dispute).
586 Otherwise, MSD’s defence to Wyeth’s infringement allegations lies in its contention that the asserted container patent claims are invalid for the following reasons:
(a) Lack of novelty in light of the Chiron patent.
(b) Lack of an inventive step in light of the common general knowledge alone pursuant to s 7(2) of the Patents Act or in light of the common general knowledge plus certain documents pursuant to s 7(3). In the particulars of invalidity set out in its Third Further Amended Consolidated Statement of Claim MSD pleaded over 15 documents upon which it relied. Ultimately in closing submissions MSD pressed its case on the basis of the common general knowledge plus six additional documents for the purposes of s 7(3), namely: (i) the Chiron patent; (ii) an article by G Kanra et al entitled “Safety, Tolerability and Immunogenicity of a Haemophilus Influenza Type b vaccine containing Aluminium Phosphate Adjuvant Administered at 2, 3 and 4 Months of Age” published in 1999 in the Turkish Journal of Pediatrics (Volume 41(4)); (iii) an article by D M Katkocin et al entitled “Characterization of Multivalent Pneumococcal Conjugate Vaccines” published 2000 in Developments in Biologicals (Volume 103); and for claim 18 only (iv) Hausdorff 2000, (v) Hausdorff 2002 and (vi) abstracts presented at the 5th International Symposium on Pneumococci and Pneumococcal Disease held in Alice Springs from April 2 – 6 in 2006 (ISPPD abstracts).
(c) Lack of manner of manufacture.
(d) Lack of fair basis.
(e) Lack of definition.
(f) Lack of utility.
19. CONTAINER PATENT: THE WITNESSES
587 Nikolai Petrovsky has since 2004 been the Director of the Department of Diabetes and Endocrinology at Flinders Medical Centre and Professor of Medicine at Flinders University in South Australia. He was awarded a PhD in 1998, and from then until 2004 was the Director of the National Health Sciences Centre, a clinical research organisation in Canberra. During that period he was also a Senior Endocrinologist at the Canberra Hospital. From 2003 to 2004 he was also Associate Professor at the Australian National University Medical School conducting immunology research projects. Professor Petrovsky has been involved in vaccine research since 1998. In particular, he has been involved in research related to the development of novel vaccines and vaccine adjuvants, including for protein vaccines, polysaccharide vaccines and polysaccharide-protein conjugate vaccines, including against pneumococcus. He has been aware of Prevnar 7 since it was first released in 2000, was aware of its formulation, and did some research on it in the early 2000s. The research included combining Prevnar 7 with an inulin adjuvant to determine whether it enhanced immune responses in mice, and also, in 2005, to see if formulating Prevnar 7 with different adjuvants had any impact on its immunogenicity and stability. He noted at the time that even though Prevnar 7 already contained an aluminium adjuvant, the addition of some other adjuvants was able to enhance immunogenicity in mice.
588 In 2002 Professor Petrovsky founded Vaxine Pty Ltd, an Australian biotechnical company specialising in vaccine development and formulation, vaccine adjuvants, vaccine clinical trials and immunology. He, together with a collaborator, developed Vaxine’s inulin polysaccharide adjuvant technology. He has been involved with Vaxine in the development and formulation of vaccines for a range of pathogens, including pneumococcus, influenza virus, hepatitis B virus, malaria, Japanese encephalitis virus, rabies virus and Human Immunodeficiency Virus. He has also coordinated clinical trials for new vaccine formulations which were sponsored by Vaxine.
589 In 2004 Professor Petrovsky co-authored a highly cited review article on vaccine adjuvants entitled “Vaccine adjuvants: Current state and future trends”, published in Immunology and Cell Biology (Volume 82) (Petrovsky Review).
590 In his first affidavit, Professor Petrovsky describes his work in developing novel vaccines and formulations, including work done on pneumococcal vaccines. He also gives evidence about how he kept up to date with developments in vaccine formulation, and describes the types of factors that were thought to affect protein stability in a vaccine formulation. Professor Petrovsky first worked with MSD’s Pneumovax 23, a mixture of pure polysaccharides taken from different pneumococcal serotypes, with which his group tested its inulin adjuvant. As I have noted, inulin was also tested with Prevnar 7. In 2004 Professor Petrovsky began to work with Professor Paton on testing Professor Paton’s pneumococcal vaccines with inulin adjuvant. In about 2006 Professor Petrovsky collaborated with Matrivax on research into whether the immune response to Prevnar 7 could be improved by combining the polysaccharides with a carrier protein that did not involve chemical conjugation of the polysaccharide to the protein. Professor Petrovsky describes his work on other vaccines, including projects on Hib and hepatitis B vaccines that he worked on in 2002 and 2003. In about 2004 he began collaborating with Dr Neil Ravenscroft on matters relating to the formulation of polysaccharide-protein conjugate vaccines, including on the development of an inulin polysaccharide adjuvant.
591 Professor Petrovsky describes how he would have gone about developing a formulation that reduces aggregation or inhibits precipitation of a polysaccharide-protein conjugate before April 2006. He also reviews the container patent and addresses the Chiron patent. In an annexure to his second affidavit he provides a schedule of responses to Professor Dalby’s evidence in answer. I address Professor Petrovsky’s evidence in chief in further detail in relation to the inventive step arguments.
592 In October 2016 Professor Petrovsky was retained by patent attorneys to give evidence in relation to an opposition to Australian Patent Application No. 2014268186 in the name of Wyeth, which had substantially the same specification as the container patent but claims relating to the Neisseria meningitidis 2086 protein. He exhibits a copy of his declaration to his affidavit. In it, he referred to being informed that the opposition related to the field of vaccines and vaccine formulation, but was not informed of the parties involved, nor of the patent application. He then provided background information known to him in the field before being provided with, and reviewing, the patent application. He reproduces in his affidavit evidence aspects of this background information, supplemented by further comments and addressing additional specific topics in response to requests from Corrs.
593 Christopher Jones is an analytical chemist who from 1991 until his retirement in 2015 was Head of Division at the Laboratory for Molecular Structure at the National Institute for Biological Standards and Control (NIBSC), which is now a centre of the UK Medicines and Healthcare Products Regulatory Agency. NIBSC is a non-departmental government body that supports regulation of biological medicines in the UK, including with respect to licensing procedures, statutory lot release of materials coming to market and relevant research. It is required by the UK government to work with the World Health Organization (WHO) on production, value assignment, and distribution of WHO international standards. Dr Jones was heavily involved with WHO expert groups and pharmacopeias, and routinely discussed existing and novel products with manufacturers. His group carried out detailed physicochemical analysis of biologics, including with respect to at least eight polysaccharide and polysaccharide-protein conjugate vaccines that included pneumococcal, meningococcal, and Hib polysaccharide-protein conjugate vaccines. This involved the use of nuclear magnetic resonance and optical spectroscopy, mass spectroscopy and specialist chromatography for glycan and protein analysis. Dr Jones is an expert on the characterisation and quality control of polysaccharide and polysaccharide-protein conjugate vaccines.
594 Dr Jones gave evidence in the United States Patent Trial and Appeal Board in various proceedings between MSD and Wyeth concerning the United States equivalent of the container patent. He was asked to give evidence in reply to that of Professor Dalby and did so, limited to three topics regarding the state of knowledge in the field of vaccine formulation before 26 April 2006:
(a) the extent to which the polysaccharide component and the protein component of pneumococcal polysaccharide-protein conjugates contribute to the aggregation of pneumococcal-protein conjugates, including by reference to: an article by B Bolgiano et al entitled “Effect of physico modification on the immunogenicity of Haemphilus influenza type b oligosaccharide-CRM197 conjugate vaccines”, published in 2001 in Vaccine (Volume 19); an article by M M Ho et al entitled “Solution stability studies of the subunit components of meningococcal C oligosaccharide-CRM197 conjugate vaccines”, published in 2001 in Biotechnology and Applied Biochemistry (Volume 33) (Ho 2001); and an article by F Berti et al entitled “Water Accessibility, Aggregation, and Motional Features of Polysaccharide-Protein Conjugate Vaccines”, published in 2004 in the Biophysical Journal (Volume 86);
(b) whether the inclusion of an aluminium-based adjuvant in the form of an aluminium salt in a pneumococcal polysaccharide-protein conjugate affects aggregation of the conjugate; and
(c) silicone oil-induced aggregation, including consideration of an article by Latoya S. Jones et al entitled “Silicone Oil Induced Aggregation of Proteins”, published in 2005 in the Journal of Pharmaceutical Sciences (Volume 94(4)) (Jones 2005), and silicone oil-induced aggregation in polysaccharide-protein conjugate vaccines.
595 Paul Dalby has since 2013 been a Professor of Biochemical Engineering and Biotechnology at University College London (UCL) and since 2016 also the Deputy Head (Research) of the department of Biochemical Engineering. He is also the Director of the EPSRC Centre for Doctoral Training in Innovative Manufacturing in Emerging Macromolecular Therapies. Professor Dalby has given evidence in the United States before the Patent Trial and Appeal Board between MSD and Wyeth in relation to similar patents. In the current proceeding, he was asked to describe his qualifications and experience, including his experience with vaccine formulation. In answering, he gives evidence that during his doctorate, which he completed in 1998, he studied: protein folding mechanisms; the use of protein engineering to alter the relative stabilities of proteins in native, denatured, intermediate and transition states; and the impacts of changing temperature, pH and viscosity on formulation. From 2000 to 2006 he was a lecturer in the Department of Biochemical Engineering at UCL where his research focussed on a number of matters that he says are of relevance to the formulation of biopharmaceuticals, including vaccines, being: (a) developing new strategies for evolutionary protein and enzyme design; (b) investigating the factors affecting protein stability during biopharmaceutical downstream processes; (c) and developing methods of stability analysis to optimise the process and maintain a stable and proper protein formulation.
596 From 2004 to 2006 he was the principal investigator on a research project to establish protein, process and formulation engineering methods for novel protein fusions. Since 2000 he has supervised numerous projects in the area of protein stability.
597 Professor Dalby gives evidence about how he kept up to date with the field of vaccine formulation before April 2006. He provides the technical background to certain relevant scientific concepts, parts of which are included in the primer that is set out in section 20.1 below. He was asked to describe how he would go about improving the formulation for a polysaccharide-protein conjugate vaccine against Streptococcus pneumoniae as at April 2006. He was then asked to consider specifically how he would have gone about formulating a conjugate vaccine with the Prevnar 7 serotypes, plus six additional serotypes of Streptococcus pneumoniae, all conjugated to CRM197. His answer to this question is summarised in the inventive step analysis below.
598 Professor Dalby then reviews the container patent and addresses the potential ingredients in vaccine formulation referred to in it. He gives evidence in this context of his awareness of silicone oil induced aggregation as at April 2006 and explains how he would have tested for it. He then addresses the claims of the container patent, before addressing the novelty and inventive step citations, and then commenting at length on the first affidavit of Professor Petrovsky, and in less detail on the first affidavit of Professor Paton.
599 In paragraphs 138 – 142 of his affidavit, Professor Dalby refers to an article published in 2018 by V Saller et al entitled “Influence of particle shedding from silicone tubing on antibody stability” in Journal of Pharmacy and Pharmacology (Volume 70). Objection was taken to these paragraphs. Wyeth relies on them in response to the inventive step case advanced by MSD and also in relation to allegations of lack of utility. In closing submissions no reference was made to the relevance of this material in the context of the utility argument. I address its relevance, and admissibility, below in the course of my consideration of the inventive step case.
19.3 The container patent joint expert report and concurrent evidence
600 The experts collaborated in the preparation of a joint expert report concerning issues arising in respect of the container patent (container JER). They also gave concurrent evidence, during which they were cross-examined. In closing submissions the parties made a number of submissions going to the relevance and weight to be ascribed to the evidence of the particular witnesses. On the whole, subject to the particular reservations that I express during the course of the reasons that follow, I found that each of the experts was able to provide a contribution to the knowledge required to consider the various issues in the case.
20. CONTAINER PATENT: BACKGROUND COMMON GENERAL KNOWLEDGE
601 In this section I set out parts from the agreed primer and parts from the container JER that may be taken to be within the common general knowledge in the relevant field before April 2006.
Vaccines
602 It was well understood and accepted by those working in the field of vaccine formulation well before April 2006 that a vaccine should be safe, and be sufficiently efficacious to result in reduction of disease in the individual or in a population, recognising that no vaccine will result in the complete prevention of infection by subsequent challenge with the wild type pathogen. There are a number of additional secondary matters of relevance, including the cost per dose of the vaccine, and the route and ease of administration.
Proteins
603 Proteins are involved in many biological processes. They can function as enzymes that catalyse important chemical reactions which enable life to occur, or as regulators of chemical reactions in the form of chemical messengers or hormones. Proteins also behave as transporters across membranes, receptors to receive stimuli, effectors to trigger downstream effects, and also have structural functions. Immunoglobulins are a class of proteins.
604 Amino acids are the monomeric sub-units of proteins. Amino acids form proteins. There are 20 standard amino acids, and a number of other non-standard amino acids.
605 Proteins consist of chains of amino acid residues linked by peptide bonds. The formation of a peptide bond is shown in Figure 1 below.
606 Dipeptides consist of two amino acid residues and polypeptides consist of many amino acid residues linked in a linear chain. A protein consists of one or more polypeptide chains. There are many naturally occurring and synthetic proteins.
607 Proteins have four levels of structure – primary, secondary, tertiary and quaternary:
(a) The primary structure of a protein is the amino acid sequence of its polypeptide chain(s).
(b) The secondary structure of a protein refers to the local conformation of the backbone of the polypeptide chain.
(c) The tertiary structure of a protein refers to the folded polypeptide in which secondary structures are arranged in a particular orientation in 3-dimensions.
(d) The quaternary structure of a protein refers to the arrangement of multiple polypeptide subunits in a multi-subunit complex.
608 The levels of structure of a protein are shown in Figure 2 below.
609 The properties and function of a protein are often determined by its structure. The ultimate tertiary structure of the protein is determined by a combination of the sequence of amino acids of the polypeptide chain, the secondary structures, and the balance of countervailing forces acting on and from within the protein. Those forces include electrostatic forces, which include the association of two ionic protein groups of opposite charge (also known as salt bridges), and dipole-dipole interactions, which are non-covalent associations between electrically neutral molecules.
610 Polar molecules typically dissolve readily in water, as they can replace water-water interactions with more energetically favourable water-solute interactions. Polar molecules are therefore often hydrophilic – or water loving. Non-polar molecules, however, are unable to form water-solute interactions, and are therefore poorly soluble in water. Non-polar molecules are therefore hydrophobic – or water fearing. In an aqueous solution, more hydrophobic regions tend to avoid contact with water by clustering together, and the more hydrophilic regions surround the hydrophobic regions so as to maximise the polar interactions with water, as shown in Figure 3 below.
611 Variations in temperature, ionic strength of the solution, redox potential of the solution, solute concentration, light, shear stress, pH, presence of metal ions, catalysts, chaotropes, or detergents can all cause changes to a protein's conformation. Changes in protein conformation play a necessary role in the function of certain proteins, such as enzymes or transporter proteins that must change conformation in order to perform their physiological function. Where, as a result of unfolding (usually permanent unfolding), the protein loses its specific biological activity, it is said to be denatured.
Saccharides
612 Saccharides or 'sugars' are essential components of all living organisms and are the most abundant class of biological molecules. Saccharides are molecules comprised predominantly of carbon, hydrogen and oxygen atoms. The monosaccharide is the basic unit of all saccharides. Glucose, shown in Figure 4 below, is an example of a monosaccharide.
613 A disaccharide consists of two joined monosaccharides.
614 A polysaccharide may consist of repeating units of the same monosaccharide, or may consist of more than one type of monosaccharide unit.
615 Polysaccharides may form branched structures as well as linear structures.
Polysaccharide-protein conjugates
616 Polysaccharide-protein conjugates consist of purified polysaccharide chains chemically conjugated to a carrier protein.
617 There are a number of methods of conjugating polysaccharide chains to the carrier protein, including:
(a) reductive amination, which is a method of attaching the polysaccharide directly to the protein or via a linker;
(b) alkylation of a sulphydryl group, whereby sulphydryls are randomly introduced onto the surface of a carrier protein which are then allowed to react with bromoacyl groups on the polysaccharide;
(c) cyanylation of the polysaccharide followed by coupling to the carrier protein; and
(d) random activation of the polysaccharide with cyanogen bromide, addition of a linker, and attachment of carrier proteins.
Stability and degradation
618 The importance of obtaining a formulation that remains stable over the shelf life of the vaccine was well understood in the field of vaccine formulation before April 2006. It was also known that the chemical and physical stability of a vaccine formulation can potentially be influenced by a number of degradation reactions, which are described below.
619 The mechanisms of degradation may be broadly classed into chemical degradation and physical degradation. Chemical degradation involves the breaking of covalent bonds or forming of new covalent bonds, whereas physical degradation involves changes to the physical state of the conjugate, such as aggregation or phase transition. These two broad classes of degradation are interrelated.
620 Examples of mechanisms of chemical degradation of a saccharide-protein conjugate, each of which was known in the field of vaccine formulation before April 2006, are described below.
(a) Protein deamidation occurs in proteins when an amide group is removed from the side chains of the amino acids glutamine or asparagine, via a succinimide intermediate, and then typically hydrolysed to form aspartate or iso-aspartate. Deamidation can de-stabilise the conjugate leading to a reduction in immunogenicity.
(b) Disulphide exchange refers to the scrambling of disulphide bonds between sulphide bridges in proteins.
(c) Protein oxidation refers to oxidation reactions between amino acid residues and reactive oxygen species.
(d) Depolymerisation of saccharide chains refers to reactions which break the chemical bonds within the saccharide chains of the conjugate, causing saccharide oligomers and polymers to be freed from the conjugate.
(e) Cleavage of saccharide chains from the carrier protein usually occurs via a hydrolytic reaction.
(f) Migration or loss of O-acetyl groups from polysaccharide chains may occur.
621 Examples of mechanisms of physical degradation of a saccharide-protein conjugate, each of which was known in the field of vaccine formulation before April 2006, are listed below:
(a) Aggregation refers to the formation of aggregates of the conjugate, or of another component in a conjugate formulation such as an aluminium salt adjuvant. Visible aggregation, or aggregation which causes a reduction in immunogenicity of the vaccine, is likely to be a problem in a formulation.
(b) Denaturation may be induced by a range of factors including changes to temperature, pressure and pH, and the presence of other chemicals.
(c) Adsorption of the conjugate to a container surface is not, in the strict sense, a mechanism of degradation of a component of the conjugate biomolecule or the overall conjugate. However, adsorption of the conjugate to the container surface affects the concentration of the conjugate in solution and therefore the effective dose. Adsorbed conjugates can also form aggregates. Factors such as the temperature, ionic strength, pH and surface tension of the solution influence the adsorption of a conjugate to the container surface. The concentrations of the conjugate and excipients in solution, and the nature of the container surface, will also influence the propensity for adsorption.
622 Chemical and physical mechanisms of degradation are more likely to occur at an accelerated rate at interfaces such as the liquid-solid interface with the container wall, or the liquid-gas interface at the headspace in the container. This was known in the field of vaccine formulation before April 2006.
623 Some vaccines before April 2006 were supplied in lyophilised (freeze dried) form. However, ready-to-use liquid formulations are more convenient and avoid a potential source of administration error.
Excipients generally
624 Before April 2006 it was known that there was a set of excipients that were “generally regarded as safe" (GRAS) by regulatory bodies, and these were already being used in pharmaceutical products licensed for human use. These products included chemical pharmaceuticals, vaccines, protein biopharmaceuticals, and nucleic acid-base pharmaceuticals. The list of known GRAS excipients formed a “toolbox” of components from which vaccine formulators would first draw, as required, to address problems that may be encountered during vaccine formulation, such as a stability or aggregation issue.
Saline
625 Isotonicity was a desirable, though not essential, feature of an injectable vaccine formulation in order to minimise the pain and discomfort to the recipient.
Adjuvants
626 To enhance the response to the vaccine antigen(s), a formulator could use an adjuvant from the toolbox, such as aluminium phosphate or hydroxide. As Prevnar 7 already contained an aluminium phosphate adjuvant, inclusion of aluminium phosphate as an adjuvant would be an obvious starting point for a formulator preparing a 13-valent pneumococcal conjugate vaccine. I return to the subject of adjuvants later in these reasons.
The requirement for vaccine stability
627 Before 26 April 2006, it was critical for a vaccine formulation to demonstrate long-term stability under storage conditions, in order to be licensed for use. Forced degradation studies were performed by formulators as part of the early stages of development of a formulation. For a vaccine to be licensed, real-time stability data on the final vaccine formulation would be gathered under expected long-term storage conditions in order to set the shelf life of the vaccine. A vaccine that demonstrated unexpected aggregation or cloudiness would not be approved for use as at April 2006. Aggregation would be harder to detect visually in the presence of an aluminium adjuvant as this is made up of particles and makes the vaccine solution cloudy.
21. CONTAINER PATENT: THE SPECIFICATION AND CLAIMS
628 The container patent is entitled “Novel Formulations which Stabilize and Inhibit Precipitation of Immunogenic Compositions”. The invention is said generally to relate to the fields of immunology, bacteriology, vaccine formulation, protein stability and process development. More particularly, the invention is said to relate to “novel formulations which inhibit precipitation of immunogenic compositions”.
629 The Background of the Invention provides an important basis for understanding the invention that is described. It refers to the general acceptance in the bio-pharmaceutical arts that improving the stability of an immunogenic composition is a necessary and highly desirable goal. It gives two examples of an “immunogenic composition” being a protein immunogen and a polysaccharide-protein conjugate. The Background goes on to say (emphasis added):
...an immunogenic composition must appear fresh, elegant and professional when administered to a patient. Any changes in stability and/or physical appearance of the immunogenic composition, such as color change, clouding or haziness, may cause a patient or consumer to lose confidence in the product. Furthermore, because many immunogenic formulations are dispensed in multiple-dose containers, uniformity of dose content of the active ingredient (e.g., a polysaccharide-protein conjugate) over time must be assured (e.g., a cloudy solution can lead to a non-uniform dosage pattern). Additionally, the immunogenic composition must be active throughout its “expected” shelf life, wherein any breakdown of the immunogenic composition to an inactive or otherwise undesired form (e.g., an aggregate) lowers the total concentration of the product.
630 The Background refers to several reports in the literature suggesting that the stability of a particular immunogenic composition is at least in part dependent upon the specific protein or carrier protein. Reference is made to three publications, being Bolgiano, Ho, and an article by Ho et al entitled “Physico-Chemical and immunological examination of the termal stability of tetanus toxoid conjugate vaccines” published in 2002 in Vaccine (Volume 20) (Ho 2002). These, and about 16 other publications, are identified by their full titles and publication details at the conclusion of the specification. They are all said to be incorporated by reference into the specification. The specification later states that the reference to any publication or information in a publication, or to any matter which is said to be known, is not to be taken as an acknowledgement or admission that such material forms part of the common general knowledge.
631 The Background continues by saying that by way of example, stability analysis in Ho 2002 of meningococcal C (MenC) polysaccharides and Haemophilus influenzae type b polysaccharides, conjugated either to a tetanus toxoid or a CRM197 carrier protein, revealed different stability profiles dependent on the carrier protein. In Ho 2001, MenC-CRM197 conjugates from two different manufacturers were analysed where the MenC-CRM197 conjugates differed in their conjugation chemistry and length of conjugate polysaccharide (both having the same carrier protein, CRM197). The Background goes on (page 2 lines 7 – 11):
Data from this study further indicated that factors such as conjugation chemistry (e.g. reductive amination either directly or via a chemical spacer group), number of conjugation sites, polysaccharide chain length, pH, storage buffer, storage temperature(s) and freeze/thaw cycles also influence the stability of an immunogenic composition.
632 The Background then says that when developing a formulation for an immunogenic composition, many factors must be considered to ensure that it is safe, stable, robust and cost effective. These include (page 2 lines 14 – 23):
...chemical stability of the immunogenic composition (e.g. hydrolysis of saccharides, de-polymerization of polysaccharides, proteolysis or fragmentation of proteins), physical/thermal stability of the immunogenic composition (e.g. aggregation, precipitation, adsorption), compatibility of the immunogenic composition with the container/closure system, interactions between immunogenic composition and inactive ingredients (e.g. buffers, salts, excipients, cryoprotectants), the manufacturing process, the dosage form (e.g. lyophilized, liquid), the environmental conditions encountered during shipping, storage and handling (e.g. temperature, humidity, shear forces), and the length of time between manufacture and usage.
633 The Background then refers to the problem caused by silicone oil (page 2 lines 24 – 33):
It has been suggested in the art, that silicone oil, which induces protein secondary and tertiary conformational changes, might be responsible for the aggregation/precipitation seen in certain protein pharmaceutical preparations (Jones et al., 2005). For example, several reports in the 1980s implicated the release of silicone oil from disposable plastic syringes as the causative agent in the aggregation of human insulin [six references are then given]. Chantelau et al. (1986) observed that after three or more withdrawals from a ten-dose preparation of insulin (using a siliconized disposable syringe), the vial would begin clouding due [to] silicone oil contamination, thereby resulting in aggregation and deactivation of the insulin (Chantelau et al., 1986). Paradoxically, silicone oil is a necessary component of plastic syringes, as it serves to lubricate the rubber plunger and facilitate transfer of the plunger down the syringe barrel (i.e., silicone oil improves the syringeability of the formulation).
634 The specification later notes that the words “precipitation”, “aggregation”, “clouding” and “particulate formulation” may be used interchangeably and are meant to refer to any physical interaction or chemical reaction which results in the “aggregation” of a polysaccharide-protein conjugate or a protein (or polypeptide) immunogen.
635 The Background continues by noting that the use of silicone oil is not limited to syringes, as it is used as a coating for glass vials to minimise adsorption, as a lubricant to prevent conglomeration of rubber stoppers during filing procedures, as a lubricant critical to the processability/machinability of glass and elastomeric closures and as a lubricant to ease needle penetration of vial rubber stoppers. It notes that the siliconisation of such containers is not well controlled or standardised and that there is a high degree of variability of the silicone oil content from one lot to another.
636 The Background concludes:
There is therefore an ongoing need in the art for formulations which enhance stability and inhibit precipitation of immunogenic compositions.
637 It is apparent that the focus of the Background is upon the formulation of immunogenic compositions to ensure that they are stable. There are said to be many considerations relevant to producing a suitable formulation, but a particular problem is identified as arising from the use of silicone oil which might be responsible for aggregation. The literature cited is said to indicate that cloudiness results.
638 The specification then provides a Summary of the Invention which extends from page 3 to page 12. It begins (page 3 lines 12 – 17):
The present invention broadly relates to novel formulations which stabilize and inhibit precipitation of immunogenic compositions. More specifically in certain embodiments, the present invention is directed to novel formulations which inhibit precipitation of immunogenic compositions comprised in container means. In one specific embodiment, the invention is directed to novel formulations which stabilize immunogenic compositions against silicone oil interactions, shear forces, shipping agitation, and the like.
639 There follows a consistory statement for claim 1:
Thus, in certain embodiments, the present invention is directed to a siliconized container means filled with a formulation comprising (i) a pH buffered saline solution, wherein the buffer has a pKa of about 3.5 to about 7.5, (ii) an aluminium salt and (iii) one or more polysaccharide-protein conjugates wherein the polysaccharide-protein conjugate comprises one or more pneumococcal polysaccharides.
640 In this form, the invention is essentially for any formulation of one or more pneumococcal polysaccharide-protein conjugates in a siliconised container that includes pH buffered saline in the range specified and an aluminium salt. The claim is not limited to any particular protein carrier. As the expert evidence reveals, the pH buffered saline solution is to improve the stability of the formulation. The expert evidence also indicates that the pKa range of 3.5 to 7.5 is broad. As a rule of thumb, a buffer is effective within one pH unit of its pKa value. Applying the rule of thumb, Professor Petrovsky considers that the range would include pH of 2.5 to 8.5. The aluminium salt is an adjuvant that is said in the specification to enhance the immune response of the vaccine antigen.
641 The Summary of the Invention is also (emphasis added):
...directed to formulations which stabilize a polysaccharide-protein conjugate, the formulation comprising (i) a pH buffered saline solution, wherein the buffer has a pKa of about 3.5 to about 7.5, (ii) a surfactant and (iii) one or more polysaccharide-protein conjugates.
(page 3 lines 22 – 26)
642 A surfactant is a class of detergent that acts as a surface-active agent, being a compound that lowers the surface tension between either two liquids, a liquid and a solid, or a gas and a liquid. The specification later says that a surfactant is generally defined as a molecule or compound comprising a hydrophilic group or moiety and a liphoilic (hydrophobic) group or moiety, and/or a molecule, substance or compound that lowers or reduces surface tension of a solution. It is said that any surfactant or any combination of surfactants which stabilises and inhibits aggregation of an immunogenic composition may be used in the invention. It further says (page 17 lines 33 – 36):
A person of skill in the art may readily determine a suitable surfactant or surfactant combination by measuring the surface tension of a particular immunogenic composition formulation in the presence and absence of the surfactant(s).
643 Numerous other embodiments are described in the Summary of the Invention.
644 The specification then provides a Brief Description of the Figures. Figure 2 is described as showing the total antigenicity loss of the 13-valent pneumococcal conjugate vaccine (with the 13 chosen serotypes) formulated with AIPO4 (0.25mg/ml) and filled in a BD Hypak syringe after two hours, eight hours and 24 hours of agitation at 500 rpm and 2 – 8 degrees Celsius.
645 The Detailed Description of the Invention commences on page 13 and includes sections entitled Surfactants, Container Means, Adjuvants and Pharmaceutical Carriers/Excipients, Immunogens, and Examples.
646 In the introductory part, the Detailed Description provides the following broad characterisation of the invention (page 13 lines 5 – 14):
The present invention addresses an ongoing need in the art to improve the stability of immunogenic compositions such as polysaccharide-protein conjugates and protein immunogens. Thus the present invention broadly relates to novel surfactant formulations and/or novel aluminium salt formulations which stabilize and inhibit precipitation of immunogenic compositions. More particularly, the invention described hereinafter, addresses a need in the art for formulations which stabilize and inhibit particulate formation (e.g., aggregation, precipitation) of immunogenic compositions which are processed, developed, formulated, manufactured and/or stored in container means such as fermenters, bioreactors, vials, flasks, bags, syringes, rubber stoppers, tubing and the like.
647 The specification then repeats what is set out in the Background concerning the various factors that influence the stability of immunogenic compositions. It then says (page 13 lines 24 – 26):
The stability of an immunogenic composition of the invention is readily determined using standard techniques, which are well known and routine to those of skill in the art.
648 Further down the same page it provides (page 13 lines 32 – 35) (emphasis added):
As set forth in detail herein, the present invention relates to the unexpected and surprising results that formulating an immunogenic composition with a surfactant such as Tween 80 significantly enhances the stability and inhibits precipitation of an immunogenic composition.
649 The specification then says that for example, it was observed in example 1 (the text reads “example 2” but is in error) that a 13-valent pneumococcal conjugate formulated in buffered saline and filled in a single dose syringe would begin precipitating out of solution within 10 minutes at 2 – 8 degrees Celsius upon gentle agitation. The Detailed Description says (page 14 lines 5 – 14) (emphasis added):
However it was surprisingly observed that the 13vPnC, formulated in buffered saline and 0.001% Tween 80, filled in a single dose syringe and gently agitated at 2-8 degrees°C, was stable for twenty-five days with no visible signs of precipitation (data not shown). Thus, this data demonstrated that the addition of a surfactant (e.g., Tween 80) to an immunogenic composition formulation enhances the stability of the immunogenic composition.
A second stability study of the 13vPnC further confirmed that the addition of a surfactant to the formulation significantly enhanced the stability of the 13vPnC...
650 The specification then says that in other experiments it was demonstrated that the stability of an immunogenic streptococcal C5a peptidase composition was greatly enhanced when formulated with a surfactant such as Tween 80. It also notes that a 13-valent pneumococcal conjugate immunogenic composition of the invention may also be formulated with or without an adjuvant, such as aluminium phosphate. Example 4 is said to be experiments formulated with a buffer, salt and AIPO4, without the addition of a surfactant. The specification observes that in an experiment within example 4, where immunogenic compositions were formulated with and without AIPO4 in identical syringes, it was observed that the 13-valent pneumococcal conjugate, formulated without the adjuvant, sustained greater antigenicity losses than with it, referring to Figures 6 and 7.
651 The specification then provides a further statement of the invention (page 15 lines 18 – 25):
Thus, the invention as set forth herein, is directed to novel formulations which stabilize and inhibit aggregation or precipitation of immunogenic compositions such as polysaccharide-protein conjugates (e.g., a 13vPnC) and protein immunogens (e.g., a streptococcal C5a peptidase...), against the various factors which influence the stability of immunogenic compositions (e.g., shear forces, shipping agitation, silicone oil interactions, adsorption, manufacturing processes, temperature, humidity, length of time between manufacture and usage, etc.).
652 Later the specification states that “in certain other embodiments, the invention is directed to a formulation which inhibits silicone oil induced precipitation of a polysaccharide-protein conjugate comprised in a siliconized container means”. The specification goes on (page 16 lines 29 – 34):
The process of aggregation (e.g., protein aggregation) is well known (but not well understood) and described in the art, and is often influenced by numerous physicochemical stresses, including heat, pressure, pH, agitation, shear forces, freeze-thawing, dehydration, heavy metals, phenolic compounds, silicon oil, denaturant and the like.
653 Later, the Detailed Description supplies a definition of an adjuvant (page 19 lines 15 – 16):
An adjuvant is a substance that enhances the immune response when administered together with an immunogen or antigen.
654 The specification then makes reference to the carrier. It says (page 20 line 28 – page 21 line 2):
In certain embodiments, the immunogenic composition formulations comprise a pharmaceutically acceptable diluent, excipient or a pharmaceutically acceptable carrier...As used herein the language “pharmaceutically acceptable carrier” is intended to include any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like, compatible with administration to humans or other vertebrate hosts. The appropriate carrier is evident to those skilled in the art and will depend in large part upon the route of administration.
655 It later says (page 21 lines 18 – 24):
The immunogenic compositions of the present invention, are not limited by the selection of conventional, physiologically acceptable carriers, diluents and excipients such as solvents, buffers, adjuvants, or other ingredients useful in pharmaceutical preparations of the types described above. The preparation of these pharmaceutically acceptable compositions, from the above-described components, having appropriate pH isotonicity, stability and other conventional characteristics is within the skill of the art.
656 The specification then addresses immunogens, and defines the term “polysaccharides” to include any antigenic saccharide element commonly used in the immunologic and bacterial vaccine arts, including, but not limited to, a “saccharide”, an “oligosaccharide”, a “polysaccharide”, a “glycoconjugate” and others. Polysaccharides are said to be “prepared by standard techniques known to those skilled in the art”. The chemical activation of the polysaccharides and subsequent conjugation to the carrier protein (that is, a polysaccharide-protein conjugate) are said to be achieved “by conventional means”.
657 The Detailed Description then moves to the examples, which are said to be presented for illustrative purposes only. I have summarised the effect of example 1 above. Example 2 concerns a formulation using the protein streptococcal C5a peptidase. Example 3 concerns the 13-valent pneumococcal conjugate considered in example 1, but is said to be focussed on the influence of the siliconised container means on stability. Example 4 repeats the experiments performed in example 3, with the exception that the antigenicity of the 13-valent pneumococcal conjugate antigen was tested using a methodology similar to that described in example 1. Example 5 describes an experimental analysis of a composition including the 2086 protein, which is not presently relevant.
658 It is apparent that the disclosure of the specification is to container means whereby stability issues arising in the formulation of immunogenic compositions for use as vaccines are addressed. One aspect of the invention, emphasised in the specification, is the use of surfactants to address aggregation that arises during the course of formulation, also described as precipitation arising from silicone oil induced aggregation.
659 The claims of the container patent are as follows:
1. A siliconized container means filled with a formulation comprising (i) a pH buffered saline solution, wherein the buffer has a pKa of about 3.5 to about 7.5, (ii) an aluminum salt and (iii) one or more polysaccharide-protein conjugates wherein the polysaccharide-protein conjugate comprises one or more pneumococcal polysaccharides.
2. The siliconized container means of claim 1, wherein said formulation the pH buffered saline solution has a pH of 5.5 to 7.5.
3. The siliconized container means of any one of claims 1 to 2, wherein in said formulation the buffer is phosphate, succinate, histidine or citrate.
4. The siliconized container means of any one of claims 1 to 3, wherein in said formulation the salt in the pH buffered saline solution comprises magnesium chloride, potassium chloride, sodium chloride or a combination thereof.
5. The siliconized container means of any one of claims 1 to 4, wherein in said formulation the aluminum salt is aluminum hydroxide, aluminum phosphate or aluminum sulfate.
6. The siliconized container means of any one of claims 1 to 5, wherein in said formulation the aluminum salt is aluminum phosphate.
7. The siliconized container means of claim 1, wherein in said formulation the buffer is histidine, the salt in the pH buffered saline solution is sodium chloride and the aluminum salt is aluminum phosphate.
8. The siliconized container means of claim 1, wherein in said formulation the buffer is histidine at pH 5.8, the salt in the pH buffered saline solution is sodium chloride and the aluminum salt is aluminum phosphate.
9. The siliconized container means of any one of claims 1 to 8, wherein said formulation further comprises a surfactant selected from the group consisting of polysorbate 20 (Tween™20), polysorbate 40 (Tween™40), polysorbate 60 (TweenTM60), polysorbate 65 (Tween™65), polysorbate 80 (Tween™80), polysorbate 85 (Tween™85), Triton™ N-1 01, Triton™ X-100, oxtoxynol 40, nonoxynol-9, triethanolamine, triethanolamine polypeptide oleate, polyoxyethylene-660 hydroxystearate (PEG-15, Solutol H15), polyoxyethylene-35-ricinoleate (Cremophor EL™), soy lecithin and a poloxamer.
10. The siliconized container means of any one of claim 1 to 8, wherein said formulation further comprising polysorbate 80.
11. The siliconized container means of claim 10, wherein the final concentration of the polysorbate 80 in the formulation is at least 0.01 % to 10% polysorbate 80 weight/volume of the formulation.
12. The siliconized container means of any one of claim 1 to 6, wherein in said formulation the buffer is succinate at a final concentration of 1 mM to 10 mM and pH 5.8 to 6.0.
13. The siliconized container means of claim 12, wherein in said formulation the succinate buffer is at a final concentration of 5 mM.
14. The siliconized container means of any one of claim 1 to 12, wherein said formulation further comprises one or more meningococcal polysaccharides, one or more meningococcal antigenic proteins, or a combination thereof.
15. The siliconized container means of any one of claim 1 to 12, further comprising one or more streptococcal polysaccharides, one or more streptococcal antigenic proteins, or a combination thereof.
16. The siliconized container means of claim 1, wherein in said formulation the polysaccharide-protein conjugate comprises one or more pneumococcal polysaccharides, the buffer is histidine, the salt in the pH buffered saline solution is sodium chloride and the aluminum salt is aluminum phosphate.
17. The siliconized container means of claim 1, wherein in said formulation the polysaccharide-protein conjugate comprises one or more pneumococcal polysaccharides, the buffer is histidine at pH 5.8, the salt in the pH buffered saline solution is sodium chloride and the aluminum salt is aluminum phosphate.
18. The siliconized container means of any one of claims 1 to 16, wherein said one or more polysaccharides comprise a S. pneumoniae serotype 4 polysaccharide, a S. pneumoniae serotype 6B polysaccharide, a S. pneumoniae serotype 9V polysaccharide, a S. pneumoniae serotype 14 polysaccharide, a S. pneumoniae serotype 18C polysaccharide, a S. pneumoniae serotype 19F polysaccharide, a S. pneumoniae serotype 23F polysaccharide, a S. pneumoniae serotype 1 polysaccharide, a S. pneumoniae serotype 3 polysaccharide, a S. pneumoniae serotype 5 polysaccharide, a S. pneumoniae serotype 6A polysaccharide, a S. pneumoniae serotype 7F polysaccharide and a S. pneumoniae serotype 19A polysaccharide.
19. The siliconized container means of any one of claim 1 to 16, wherein the polysaccharide-protein conjugate formulation is a 13-valent pneumococcal conjugate (13vPnC) formulation comprising a S. pneumoniae serotype 4 polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 6B polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 9V polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 14 polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 18C polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 19F polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 23F polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 1 polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 3 polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 5 polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 6A polysaccharide conjugated to a CRM197 polypeptide, a S. pneumoniae serotype 7F polysaccharide conjugated to a CRM197 polypeptide and a S. pneumoniae serotype 19A polysaccharide conjugated to a CRM197 polypeptide.
20. The siliconized container means of any one of claims 1 to 19 selected from the group consisting of a vial, a syringe, a flask, a fermentor, a bioreactor, tubing, a pipe, a bag, a jar, an ampoule, a cartridge and a disposable pen.
21. The siliconized container means of claim 20 wherein said container is made of glass, metals or polymers.
22. The siliconized container means of claim 20 wherein said container is a syringe.
23. The siliconized container means of claim 20 wherein said container is a glass syringe.
24. A siliconized container means according to claim 1 substantially as hereinbefore described with reference to any one of the Examples and/or Figures.
21.3 The field of the invention, the person skilled in the art and the experts
660 The container patent concerns the formulation of compositions intended to trigger an adaptive immune response, that is, vaccines. The body of the specification is focussed upon “immunogenic compositions” that are described in the Background as including protein immunogens and polysaccharide-protein conjugates, and the focus of the invention then described, including the embodiments, is upon those two forms of immunogen. All of the claims are directed to compositions of polysaccharide-protein conjugates, which are defined to include saccharides and glycoconjugates. This subject matter may be considered to be the field.
661 The person skilled in the art is one who has a practical interest in the subject matter of this field, and may be summarised as a person with an interest in the formulation of such immunogenic compositions as vaccines. The evidence indicates that this includes a formulator of protein vaccines or polysaccharide-protein conjugate vaccines who typically will have a PhD and a number of years of experience in the relevant field. The PhD qualification signifies an ability to conduct original research. The person skilled in the art will have a good understanding of development in the field by keeping up to date with internationally published literature. None of the witnesses is an avatar for the hypothetical formulator. In reaching the views I express below, I have synthesised and weighed the evidence of each witness. I refer to the person skilled in the art as the skilled formulator. The skilled formulator would collaborate from time to time in the development of a vaccine formulation with an immunologist or clinician familiar with the active components and their purpose.
662 For the purpose of considering the patent specification and the grounds of invalidity each of Professor Petrovsky and Professor Dalby are able to give admissible evidence. Each has the relevant skills, training and experience in the relevant field.
663 The parties disagree as to the weight to be given to the evidence of each. Wyeth submits that the person skilled in the art is a formulator of biological medicines including vaccines such as polysaccharide conjugate vaccines. Professor Dalby is a formulator of biological medicines. Wyeth accepts that he had never formulated a polysaccharide-protein conjugate vaccine before April 2006, but submits he was experienced in the development of biologics. Accordingly, Wyeth submits, his evidence is relevant. Before the priority date Professor Dalby had read the literature on vaccine formulation and on polysaccharide-protein conjugates insofar as it related to formulation. Professor Dalby gave oral evidence that he had “developed a vaccine relatively recently”, upon which Wyeth relies. Furthermore, Wyeth submits that Professor Dalby was devising techniques such as high-throughput screening and formulation screening techniques that were applicable to all biological medicines, including vaccines.
664 MSD submits that as a vaccine formulator with experience in formulating polysaccharide-protein conjugate vaccines before the priority date, Professor Petrovsky is more representative of the person skilled in the art than Professor Dalby. It correctly points out that since 1998, Professor Petrovsky has been actively involved in research relating to the development and formulation of vaccines, including conjugate vaccines, against a range of pathogens including pneumococcus. In 2002 he founded Vaxine, a company specialising in vaccine development and formulation, including the development of vaccine adjuvants. He also co-authored the Petrovsky Review Article. The evidence also demonstrates that he was in the team “doing the vaccine formulation” before the priority date, putting him in the position of being actively involved. By contrast, MSD submits that the evidence reveals that Professor Dalby was not engaged in any formulation of vaccines before the priority date. His expertise in formulating biologics does not extend to vaccine formulation, he was accordingly not representative of the person skilled in the art and his evidence should be given little, if any, weight.
665 There is force in the submissions advanced by MSD, although they go too far in submitting that Professor Dalby’s evidence should be given no weight. His evidence is useful in considering the appropriate construction of the specification and the claims, and the grounds of invalidity advanced. Plainly his knowledge and experience qualify him to give such evidence. When it comes to consideration of the ground of lack of inventive step, his lack of experience as a vaccine formulator causes me to place greater emphasis upon the evidence of Professor Petrovsky. Professor Dalby’s experience before April 2006 in relation to vaccine formulation is limited to the more theoretical matters that he knew as a result of his work concerning biologics that at a general level included vaccines, but he was not directly involved in their development. Unlike Professor Petrovsky, he had not published any papers on vaccine formulation before the priority date, and, importantly in my view, he had never formulated a vaccine or worked on polysaccharide-protein conjugate vaccines before or after April 2006. Whilst he gives evidence that before April 2006 he was speaking to people who were working in vaccine formulation, he was not involved in it himself. He lacks the practical knowledge that Professor Petrovsky supplies. That had the consequence that in his evidence he tended to overcomplicate the degree of difficulty and complexity facing the work of the skilled formulator.
666 Dr Jones’ experience as an analytical chemist lies in the physicochemical analysis, characterisation and quality control of conjugate vaccines, including in relation to their structure and stability. He worked in regulatory matters directly related to vaccine formulations. His knowledge does not equate to that of a vaccine formulator, but his knowledge of technical matters relevant to the requirements that polysaccharide-protein conjugate vaccines must satisfy in order to meet regulatory standards is such that there is a clear overlap between his knowledge, and the matters that one might expect a formulator to have. I do not consider that his evidence serves as a close proxy to the knowledge of the skilled formulator, but in my view he is qualified to give evidence in respect of the subjects traversed in his evidence, and I take it into account as one of a number of sources of information.
22. CONTAINER PATENT: INFRINGEMENT
22.1 Container patent construction dispute
667 MSD submits that, largely for the reasons given in relation to the composition patents, claim 18 of the container patent does not extend to a formulation with 15 (or more than 13) serotypes.
668 I have considered the equivalent argument in relation to the claims of the composition patents in section 5.3.2 above. For substantially the same reasons as set out in that section, the answer is the same, and the argument advanced by MSD fails.
669 Claim 18 is for:
18. The siliconized container means of any one of claims 1 to 16, wherein said one or more polysaccharides comprise a S. pneumoniae serotype 4 polysaccharide, a S. pneumoniae serotype 6B polysaccharide, a S. pneumoniae serotype 9V polysaccharide, a S. pneumoniae serotype 14 polysaccharide, a S. pneumoniae serotype 18C polysaccharide, a S. pneumoniae serotype 19F polysaccharide, a S. pneumoniae serotype 23F polysaccharide, a S. pneumoniae serotype 1 polysaccharide, a S. pneumoniae serotype 3 polysaccharide, a S. pneumoniae serotype 5 polysaccharide, a S. pneumoniae serotype 6A polysaccharide, a S. pneumoniae serotype 7F polysaccharide and a S. pneumoniae serotype 19A polysaccharide.
I continue to refer to the serotypes listed as the 13 chosen serotypes.
670 The words “comprise” and “comprising” are defined in the body of the specification of the container patent to mean includes and including, unless the context requires otherwise. MSD accepts, as must be the case, that in claim 1, upon which claim 18 is dependent, the word “comprising” must be understood in its inclusive sense. The body of the specification envisages embodiments of the composition comprising “one or more” pneumococcal polysaccharides, as does claim 1. The specification also lists certain pneumococcal polysaccharide serotypes that might be included in that composition, being the 13 chosen serotypes.
671 MSD relies on the fact that only the 13 chosen serotypes are listed and the subject of the examples, however, unsurprisingly, the examples are expressly stated to be just that. They do not purport to be an expression of the totality of the invention.
672 In my view, nothing in the arguments advanced by MSD persuades me that I should arrive at any different conclusion in relation to this issue to that which I reached in relation to the equivalent argument in respect of the asserted composition patent claims.
22.2 The siliconised container dispute
673 In order to explain this dispute it is necessary briefly to refer to the history of these proceedings. The hearing of the proceedings was conducted on the basis of MSD’s Second Further Amended Consolidated Statement of Claim which was filed on 21 November 2018, and Wyeth’s Statement of Cross-Claim, which was filed on 13 November 2017.
674 The Cross-Claim relevantly pleaded as follows:
(a) On 14 August 2017 MSD informed Wyeth that it intends to import into and offer to sell in Australia a 15-valent polysaccharide-protein conjugate vaccine together with a physiologically acceptable vehicle.
(b) On 13 October 2017 Wyeth invited MSD to indicate when it intends to launch that product, and whether MSD’s non-infringement case extends beyond its contentions that the 15-valent vaccine falls outside the claims of the composition patents and that those patents are invalid. MSD did not provide the requested indications.
(c) MSD has threatened to infringe the asserted container patent and the asserted composition patent claims.
675 In its Defence to the Statement of Cross-Claim, filed on 11 December 2017, MSD relevantly:
(a) Admitted that it threatened to import and sell a 15-valent vaccine as defined in the Cross-Claim.
(b) Pleaded its non-infringement argument based on the contention that claim 18 of the container patent does not extend to a formulation of 15 serotypes.
(c) Otherwise admitted that its 15-valent vaccine “possesses all of the integers of the claims”.
(d) Relied on its contention that the asserted claims are not valid and are liable to be revoked.
(e) Otherwise denied infringement.
676 Closing submissions in the proceedings were made in February 2019.
677 On 25 June 2019 Wyeth filed an interlocutory application seeking leave to re-open its case of infringement of the container patent to add an allegation that claim 9 had been infringed. It is unnecessary to go into detail about that application. Initially it was contested, and a hearing was conducted on 10 October 2019. However, the hearing was adjourned after cross-examination and sometime later the parties resolved their differences in relation to the application. Liberty to re-open the evidence and to rely on claim 9 was granted by consent. Evidence was filed, and additional closing submissions were made on the subject of the validity, and MSD’s alleged infringement, of claim 9 on 16 March 2020.
678 As part of that process, the parties were directed to file and serve amended pleadings. Relevantly, Wyeth filed an Amended Statement of Cross-Claim (Amended Cross-Claim). This relevantly included the following:
(1) An allegation that Wyeth intends to import and sell in Australia a 15-valent polysaccharide-protein conjugate vaccine with 15 identified serotypes conjugated to CRM197.
(2) Additional particulars in [6] – [9] addressing integers of the asserted claims and alleging that MSD’s 15-valent vaccine possesses each integer.
(3) A specific allegation as follows:
10. The 15-valent vaccine will be contained in a siliconized container means.
Particulars to paragraphs 7 to 10
(i) Confidential Annexure CA-3 to the affidavit of Dr Chitrananda Abeygunawardana dated 5 November 2019.
679 MSD responded to [10] of the Cross-Claim in its Amended Defence in the following terms (emphasis added):
10. In answer to paragraph 10 of the statement of cross-claim [MSD]:
(a) admit[s] that:
(i) [MSD] intends that the specified single dose pre-filled syringe will be one type of container means for the 15-valent vaccine;
(ii) the specified single dose pre-filled syringe is a “siliconized container means” within the meaning of the Asserted Container Patent Claims;
(b) otherwise:
(i) say that [MSD] intend[s] that types of container means other than the specified single dose pre-filled syringe will be used for the 15-valent vaccine;
(ii) deny that the types of container means referred to in (i) are “siliconized container means” within the meaning of the Asserted Container Patent Claims.
Particulars
(i) Confidential particulars will be provided by letter to [Wyeth’s] solicitors.
680 Wyeth contends that in MSD’s Defence, it admitted that its threatened future launch of its 15-valent vaccine, by whatever means, in whatever container, will infringe the asserted composition patent claims, save for claim 18. It does not consent to the withdrawal of the admission.
681 MSD contends that no such admission has been made. It submits that properly characterised, paragraph 12(d) of its Defence is an admission that its 15-valent vaccine possesses each of the integers of the claims (subject to the claim 18 issue) in a global sense, just as the Cross-Claim alleges infringement of all of the asserted claims in a global sense. That is factually correct, because MSD intended, and intends, to sell its vaccine in a siliconized container. It submits, however, that one would not read into that admission a further admission that MSD only intended to sell its 15-valent vaccine in a siliconized container. When Wyeth pleaded in its Amended Cross-Claim the detail of each of the integers said to be possessed by the 15-valent vaccine and its container, MSD then made plain that it also intended to sell that vaccine in containers that are not siliconised.
682 In my view it was open to MSD to proceed in the way that it did. Its general admission in the Defence was to the effect that it proposed to import and sell in Australia a product which had all of the integers of the asserted claims. It does not seek to resile from that admission. However, the admission did not address a matter irrelevant to the pleaded case, namely that MSD intended to import a product that was not siliconised. It has now clarified that it also intends to import and sell its 15-valent vaccine in a non-siliconised container. It did not make any admission in its Defence that it would not do so. No leave is required to plead [10(b)] in its Amended Defence.
23. CONTAINER PATENT: LACK OF NOVELTY
683 MSD pleads that the invention claimed in the asserted container patent claims was not novel as at 26 April 2006 when compared with the prior art base for the purpose of s 18(1)(b)(ii) of the Patents Act in light of the disclosure of the Chiron patent. In its closing submissions it accepts that it does not anticipate claim 18, but contends that otherwise all of the asserted container patent claims are anticipated.
23.2 The disclosure of the Chiron patent
684 The Chiron patent is entitled “Vaccines comprising aluminium adjuvants and histidine”. The word “comprising” is defined to mean “including”. The abstract states:
To improve the stability of vaccines comprising aluminium salt(s), the invention uses the amino acid histidine. This can improve pH stability and adjuvant adsorption and reduce antigen hydrolysis. Histidine is preferably presen[t] during adsorption to the aluminium salt(s). The antigen in the vaccine may be a protein or a saccharide and is preferably from N. meningitidis.
685 The field of the invention is vaccine formulation. The section entitled “Background Art” describes that adjuvants traditionally used in human vaccines have been aluminium salts such as aluminium hydroxide and aluminium phosphate. These may not always be compatible with particular antigens. In addition, it is necessary to consider vaccine stability when using aluminium salts. These issues in general only arise when it comes to formulating an antigen for clinical use, and may not be appreciated during the development of the antigen itself. It is said to be an object:
...to provide improvements in the stability of vaccines which include aluminium salts and, in particular, improvements in pH stability (buffering) and adjuvant adsorption at various temperatures and/or improvements in antigen stability (e.g. reduction in hydrolysis).
686 The invention is said to be based on the surprising discovery that the amino acid histidine (a buffer) enhances the stability of vaccines which include aluminium salt adjuvants: “[t]his has been found both for saccharide antigens and for protein antigens”. The invention is said to provide a composition providing an antigen, an aluminium salt and histidine as well as a process for producing this composition by mixing them together.
687 The Chiron patent then proceeds to make specific reference to each of these components. In relation to the antigen, it provides that it “is preferably a protein antigen or a saccharide antigen (optionally conjugated)”. There follows a list of 18 specific bacterial antigens for use with the invention, the fourth of which is a saccharide antigen from Streptococcus pneumoniae. In addition, 10 viral antigens for use with the invention are listed and three further antigens (a prion protein, an amyloid protein and a cancer antigen) are listed.
688 The Chiron patents states that where a saccharide or carbohydrate antigen is used, it is “preferably conjugated to a carrier protein in order to enhance immunogenicity [references]...The CRM197 diphtheria toxoid is particularly preferred”.
689 Other suitable carriers are listed. The antigen is said preferably to be adsorbed to the aluminium salt. The typical concentration will be at least 1 μg/ml each.
690 In relation to the aluminium salt the specification refers to a preference for an aluminium hydroxide or an aluminium phosphate, but says that any other suitable salt may be used. The use of histidine in combination with an aluminium phosphate is said to be “particularly advantageous for acidic antigens”.
691 There is no dispute that capsular polysaccharides are on the whole in the class of acidic antigens. Professor Petrovsky understands the disclosure to mean that the pH achieved by the use of histidine as a buffer improves the adsorption of specific conjugates (the antigens) to aluminium phosphate. He says that histidine would be his choice of buffer for an acidic antigen. Professor Dalby contends in his written evidence that Chiron does not suggest the use of histidine with aluminium phosphate adjuvant as a buffer; however, that observation cannot be reconciled with a statement on line 15 of page 5 that histidine indeed “preferably acts as a buffer”.
692 In relation to the histidine component, the Chiron patent notes that histidine is a standard amino acid that is readily available for use with the invention and may be ionised within the composition. It says (page 5 lines 17 – 20):
The composition preferably has enhanced pH stability and/or reduced antigen hydrolysis when compared to an equivalent composition in which histidine buffer system is either replaced with a sodium phosphate buffer system or in which no buffer system is included. Reduced hydrolysis may be a consequence of enhanced pH stability.
693 The specification then addresses “Further characteristics of the composition” and refers to a number of preferences and options. In one, the specification states (page 6 lines 7 – 9):
The pH of the composition is said to be preferably between 6 and 7 (e.g. between 6.3 and 7.0). The pH may be maintained by the use of a buffer. This will typically be achieved inherently by the histidine of the composition.
694 Another provides for the addition of a surfactant (page 6 lines 14 – 15):
The composition may comprise a detergent (e.g. a Tween, such as Tween 80) in order to minimise adsorption of antigens to containers.
695 In a section entitled “Immunogenic compositions and medicaments” the Chiron patent refers to the invention as being typically a vaccine composition which is of use as a medicament. It describes that its use is preferably for the prevention of disease caused by Neisseria (e.g. meningitis, septicaemia, gonorrhoea etc.), by H. influenzae (e.g. otitis media, bronchitis, pneumonia, cellulitis, pericarditis, meningitis etc.) or by pneumococcus (e.g. meningitis, sepsis, pneumonia etc.).
696 Under the heading “further components of the composition” the document refers to a number of other ingredients, including carriers (which are listed) and an adjuvant in addition to the aluminium salt.
697 The invention is said to be useful for treating children and teenagers and is typically prepared as an injectable.
698 The patent later describes the step of admixing antigen, aluminium salt and histidine. It provides that to make a composition of the invention these three must be combined, with the preference that when the antigen and aluminium salt are mixed, the histidine should be present. There follows a description of the drawings.
699 Under the heading “Modes for carrying out the invention” are a number of examples. In each of examples 7, 8 and 9 the formulations include a polysaccharide-protein conjugate, an aluminium adjuvant, a pH buffered saline solution (with histidine) and Tween 80 (a surfactant).
700 In example 8, reference is made to considering the stability of the formulation either in the bulk mixture or after packaging into vials.
701 The claims describe various alternative formulations. Claims 2 to 22 are dependent on claim 1, which is for a composition comprising an antigen, an aluminium salt and histidine. Claim 3 defines the antigen to be bacterial and selected from one of a group of 17 alternatives, the fourth of which is Streptococcus pneumoniae. Claim 14 identifies that the pH of the composition is “between 6 and 7”. Claim 15 provides that the composition includes a pharmaceutically acceptable carrier.
702 I have summarised aspects of the relevant law in relation to novelty in section 6.4 above.
703 There is no issue that the Chiron patent does not expressly disclose the integer (present in all of the asserted claims of the container patent) of a siliconised container. MSD, however, contends that, in light of the common general knowledge, this may nevertheless be taken to be implicitly disclosed. I address this argument below.
704 The question arises as to whether or not a “siliconized container means” may be implied, as MSD submits, in light of the common general knowledge. MSD points out that vials closed with rubber stoppers treated with silicone oil and syringes containing siliconised rubber plungers were known and part of the common general knowledge. It contends that there is an implicit disclosure of the “siliconized container means” integer, because a vaccine formulator would have perceived that when Chiron refers to the formulation being “injectable”, it is a reference to syringes that have siliconised rubber plungers and vials that had siliconised rubber stoppers.
705 Wyeth submits that the Chiron patent does not anticipate any of the claims, first because there is no disclosure of a siliconised container, which is a feature of all claims. It submits that the common general knowledge cannot be used to supplement the disclosure, but even if it was, at best, the common general knowledge was that silicone coatings were sometimes applied to containers and that laboratory glassware was usually not siliconised. Secondly, Wyeth contends that the Chiron patent does not contain any clear and unmistakable direction to make any particular combination, but instead provides a “laundry list” of what could be included in a vaccine formulation. Finally, it submits that the Chiron patent claims provide no relevant direction. They only mention the pneumococcus antigen as one in a list of 17 other potential bacterial antigens and not as the preferred one. Furthermore, they refer to an oligosaccharide conjugated antigen, not a polysaccharide conjugated antigen, and the inclusion of salt is optional.
706 Wyeth relies on Professor Dalby’s evidence to the effect that whilst silicone was used for lubrication in syringes and in the rubber stoppers and rubber part of the plungers in some syringes, different containers can have different amounts of silicone oil and that some had none. Professor Dalby also gives evidence that the glassware used in his laboratory was routinely not siliconised before April 2006 and that both siliconised and non-siliconised glass vials and syringes were available and used at the time.
707 Professor Petrovsky’s evidence indicates that it was routine at the time to use siliconised containers for vaccines. I address this further in my findings going to the common general knowledge. In short, I accept that it may be considered to be typical for the skilled formulator of protein vaccines, including polysaccharide-protein conjugate vaccines, to have used siliconised containers.
708 However, that is not sufficient to achieve the result that the claims of the container patent are anticipated. It is necessary for there to be equivalence of disclosure. The person skilled in the art reading the Chiron patent may make an educated guess that a siliconised container is used, and may even be right, but he or she cannot know and the Chiron patent does not say. The question of whether the skilled formulator would make an educated guess that a siliconised container means was used cannot be answered by determining that that was probable based on the common general knowledge. Disclosure is the key. As the Full Court said in AstraZeneca (FC) at [352]:
Although the common general knowledge can be used in a limited way to construe a prior art document, s 7(1) does not permit the common general knowledge to be used as a resource that can be deployed complementarily to arrive at a disclosure which the document alone, properly construed, does not make. If it were otherwise, the separate requirement of inventive step to support a patentable invention (see s 18(1)(b)(ii) of the Act) would be otiose.
709 Accordingly, none of the asserted container patent claims are anticipated.
710 If I am wrong in reaching that conclusion, then I would conclude that in respect of almost all of the asserted container patent claims, with the exception of claims 8, 17 – 18 and 20 – 23, MSD had made out its novelty challenge. While MSD appears to submit that the Chiron patent anticipates claims 20 – 23, I consider that they are not anticipated when they are dependent on claim 18, which MSD accepts is not anticipated. I consider that, leaving aside the siliconised container integer, there is disclosure of the remaining elements of those claims. This may most briefly be identified by reference to the claims in Chiron, which form part of the disclosure, although the body of the specification of that document also provides the relevant teaching:
(1) In relation to claims 1 – 3 of the container patent, Chiron discloses the use of a histidine buffer, sodium salt and an aluminium adjuvant in a vaccine. The disclosure of a saccharide antigen being an oligosaccharide antigen is equivalent to a polysaccharide in claim 1 of the container patent, given that “polysaccharide” is defined in the container patent to include oligosaccharides. Claim 5 of the Chiron patent discloses a “conjugated oligosaccharide antigen”, which is an equivalent to the polysaccharide-protein conjugate in claim 1 of the container patent, having regard to the express preference of conjugating the saccharide antigen to a carrier protein at page 3 line 20 of the Chiron patent. The preferred pH range of between 6 and 7 set out on page 6 of Chiron is within the range given in claims 1 of the container patent.
(2) In relation to claim 4 of the container patent, the Chiron patent teaches that the salt may, for example, be a sodium salt such as sodium phosphate or sodium chloride (page 5 line 28).
(3) In relation to claims 5 and 6 of the container patent, the Chiron patent discloses that the aluminium adjuvant may be “preferably an aluminium hydroxide or an aluminium phosphate” (page 4 lines 19 – 20), and the antigen is preferably adsorbed to the aluminium salt (page 4 line 5).
(4) In relation to claims 7 and 16 of the container patent, the Chiron patent discloses this combination in claim 12, dependent on claims 8, 5 and 1.
(5) In relation to claims 8 and 17 of the container patent, there is no specific disclosure of the buffer histidine being at a pH of 5.8. In the Chiron patent the disclosed range is between 6 and 7.
(6) In relation to claim 9 of the container patent, the Chiron patent discloses the use of a surfactant in its formulation, including in examples 7, 8 and 9, which include Tween 80.
24. CONTAINER PATENT: LACK OF INVENTIVE STEP
711 MSD challenges each of the asserted container patent claims on the basis that they lack an inventive step. Wyeth accepts that the inventiveness of these claims may be assessed against independent claim 1 and dependent claims 9 and 18. It accepts that the combinations represented by dependent claims 2 – 8, 16 – 17 and 20 – 23 confer no independent inventiveness over claims 1, 9 and 18.
712 MSD submits that the only question is whether it was obvious to include a buffer and an adjuvant (for claims 1 and 18) and a surfactant (for claim 9) in a pneumococcal conjugate vaccine presented in a siliconised container means (e.g. a syringe or vial). It submits that the selection of the 13 chosen serotypes for claim 18 confers no separate inventiveness on an otherwise uninventive combination. MSD builds its case around the proposition that in April 2006 Prevnar 7 and its constituent parts formed part of the common general knowledge, and submits that a convenient way to approach the question is by considering the addition of six serotypes to the formulation of Prevnar 7. It submits that nothing in the container patent suggests that the use of a buffer or surfactant was something special. The container patent describes the use of a buffer as “conventional”, states that “a person of skill in the art may readily determine a suitable surfactant”, and states that achieving stability was conventional. MSD submits that there is no gap between the common general knowledge and the invention as claimed. Accordingly, the invention is nothing more than a conventional vaccine formulation using common excipients that are routinely tested and used in such formulations.
713 MSD submits that if an aggregation problem was encountered during formulation before the priority date, the approach of the formulator would have been to seek to resolve it based on using excipients known to be useful, and by testing them on a “checker board” to resolve it. It submits that the evidence discloses that is not beyond the skill of the calling, and there was no difficulty overcome, or barrier crossed, citing Lockwood No 2 at [52]. The process simply involved the application of routine steps that led to the invention, citing AstraZeneca (HC). MSD puts the relevant question in two ways: first, would the person skilled in the art have taken, as a matter of routine, steps which might lead from the common general knowledge, including the Prevnar 7 formulation, to the formulation of a 13-valent pneumococcal conjugate vaccine including the claimed components? Secondly, would the person skilled in the art as at April 2006, in all the circumstances including the common general knowledge, have been directly led as a matter of course to add a buffer and/or a surfactant to the Prevnar 7 formulation, but with the 13 chosen serotypes, in the expectation that they might well produce a useful alternative or better formulation than the Prevnar 7 formulation? It submits that the answer to both is “yes”.
714 Wyeth submits that an invention may lie in or be based on the discovery of a previously unknown problem. It submits that whether the inventor in fact stumbled upon the claimed invention as a matter of chance, or arrived at the invention as the result of a great intellectual effort, is irrelevant to determining whether the invention is obvious. The claimed combination is based on the discovery for the first time of the problem of silicone oil induced aggregation, and the resolution of that problem.
715 Wyeth submits that the person skilled in the art is unlikely to have encountered precipitation of the kind observed in example 1 of the container patent. MSD has put forward no evidence that precipitation of the kind described would have been observed at all, in particular using Professor Petrovsky’s “checker board” approach. If no problem was found, then the person skilled in the art would have had no reason to introduce an unnecessary ingredient into the vaccine formulation (in this case, a buffer). Wyeth submits that even if the person skilled in the art had encountered precipitation of the kind described in the specification, he or she would not have expected to observe silicone oil induced aggregation in a conjugate vaccine formulation because silicone coatings were only sometimes applied to containers, in varying amounts, and the laboratory glassware was usually not siliconised. It next submits that even if the person skilled in the art appreciated the problem of silicone oil induced aggregation, he or she would not have arrived at the claimed combination as a matter of course with a reasonable expectation of success. It was common general knowledge that changing the formulation by the addition of a buffer may not solve the aggregation problem, and might equally create a new stability problem or an immunogenicity problem, potentially resulting in a formulation that was not safe, chemically and physically stable and immunogenic.
716 Significantly, there is no dispute between the parties that one way to test the inventiveness of the claims is to consider the approach that the notional person skilled in the art would have taken to the task of formulating a 13-valent polysaccharide-protein conjugate vaccine from the starting point of the Prevnar 7 formulation. This was the approach taken by the inventors, as seen in example 1 of the container patent, and the agreed approach adopted by the experts in the container JER.
717 In his first affidavit Professor Petrovsky gives evidence that he kept up to date with developments in vaccine formulation, matters that he considered were commonly known in the field of vaccine formulation in Australia before April 2006. By April 2006 he knew of several commercial polysaccharide-protein conjugate vaccines, including against Hib, pneumococcus and meningococcus. In the context of Hib, he was aware of the work of Chiron from the early 1990s, which was responsible for developing CRM197 as a carrier protein. Professor Petrovsky explains the criteria by which vaccines were evaluated for safety, efficacy and stability. He also identifies aggregation as a known and undesirable aspect of formulation development, and also refers to excipients used in vaccine formulation.
718 Professor Petrovsky was asked by Corrs to discuss the types of factors that were thought to affect protein stability in a vaccine formulation, including in a polysaccharide-protein conjugate, before April 2006. He describes the factors likely to influence protein stability, and says that in many cases, problems with protein stability and aggregation are associated with protein concentration and can be mitigated by reducing protein concentration. He says that his approach to addressing aggregation and stability in general was to find a solution by experimenting with the obvious choices of vaccine components, rather than diagnosing the cause of the problem at a molecular level. By way of example, he describes working on an aggregation problem in the early 2000s with a hepatitis B vaccine. He did not attempt to diagnose the cause, but added a surfactant (polysorbate) and an aluminium adjuvant (aluminium hydroxide), which resolved the problem. He says that when dealing with polysaccharide-protein conjugates, a factor to bear in mind is that under certain conditions, such as when an aluminium adjuvant is present, cleavage of the polysaccharide from its carrier protein, or depolymerisation of the polysaccharide chain, may occur. Further, different polysaccharides have different optimal pHs. An optimal polysaccharide-protein conjugate will avoid polysaccharide hydrolysis by paying careful regard to the choice of pH and making sure to take into account the choice and effect of the adjuvant as part of the formulation.
719 Professor Petrovsky was asked how he would have gone about developing a formulation that reduces aggregation or inhibits precipitation of a polysaccharide-protein conjugate before April 2006. He gives evidence that he would have searched the literature to see if there were any descriptions of formulations of other polysaccharide-protein conjugate vaccine formulations or other formulations that he considered relevant. He would have searched various literature and patent databases using the words “vaccine” and “adjuvant” and various other search words.
720 Professor Dalby gives evidence that he would have first looked at existing polysaccharide-protein conjugate vaccines against Streptococcus pneumoniae to see if they could be improved. By April 2006 he was only aware of one approved product, Prevnar 7. He notes that the published information about Prevnar 7 in the June 2006 edition of “MIMS Annual – Australian Edition” reveals:
(a) it was a 7-valent pneumococcal conjugate vaccine;
(b) each 0.5 mL dose contained 2 micrograms of each of the antigens except for serotype 6B, which had 4 micrograms;
(c) it contained sodium chloride, water for injections and 0.5 mg of aluminium phosphate adjuvant per 0.5mL dose, and had no other ingredients;
(d) the saccharide antigens were each conjugated to the CRM197 protein;
(e) it presented as a liquid suspension in single dose glass vials and pre-filled syringes;
(f) it must be shaken before administration as an intramuscular injection;
(g) the most commonly reported adverse reactions were injection site reactions and fever;
(h) it involves one to four 0.5mL doses, depending on the age of the infant; and
(i) it is to be stored at 2 to 8 degrees Celsius.
721 He concludes, based on this information, that nothing stands out to him initially as needing improvement from a formulation perspective, and he would have little reason to reformulate it. However, if pushed, he notes that the liquid vaccine must be stored at 2 to 8 degrees Celsius, and he may have considered trying to find a formulation that was stable at room temperature, but that would be a major research project of unknown outcome. He postulates that another option would be to produce a lyophilised presentation that would be more suitable for locations where cold chain transport and storage requirements could not be met.
722 He then says that if he were told that the vaccine he was to formulate was to contain the Prevnar 7 serotypes plus six additional serotypes of Streptococcus pneumoniae (of any identity), all conjugated to CRM197, then his starting point would likely have been the Prevnar 7 formulation. He says that if adding conjugates with polysaccharides from the new serotypes did not affect the stability or immunogenicity of the formulation, which would need to be evaluated through testing, then he would not have changed anything about the Prevnar 7 formulation. He considers that adding six new serotypes would almost double the amount of conjugate in the formulation, but the concentration of protein would still be in the range of micrograms per millilitre, which is very low by formulation standards and he would not have expected that any change in the formulation would be required. However, any formulation change may have unpredictable effects on stability and a broad range of testing and research would be required to make sure that the new formulation was not subject to any of a number of potential forms of chemical or physical degradation being: (i) protein deamination; (ii) disulphide exchange; (iii) protein oxidation; (iv) depolymerisation of saccharide chains; (v) cleavage of saccharide chains from the carrier protein; (vi) migration or loss of O-acetyl groups; (vii) aggregation; (viii) denaturation caused by changes to temperature, pressure and pH and the presence of other chemicals; and (ix) adsorption of the conjugate to the container surface. He considered that if the introduction of additional serotypes caused problems with the safety, efficacy or stability of the formulation, then he would undertake further testing and experimentation to identify the causes and mechanisms of that instability and to find a solution.
24.4 Findings of common general knowledge and other relevant matters
723 Before turning to consider the question of inventive step in the context of the combination of each of the claims, I first make findings in relation to some disputed aspects of the common general knowledge before April 2006 and other matters of dispute between the parties.
24.4.1 The formulator’s approach to reformulation
724 Professor Petrovsky’s experience before April 2006 was that when formulating a new vaccine, companies typically formulated multiple different liquid formulations with variations of a few standard excipients that were typically included in vaccine formulations to test for these matters, and then considered all of the stability data when selecting the candidate for clinical trials. This is what the experts referred to as the “checker board” approach. The experts agreed in their oral evidence that industry formulators would take the antigen and put it in plates or in large numbers of tubes. As an example given by Professor Petrovsky, on the X axis they will have a range of pH levels, on the Y axis the tubes will go from water to increasing concentrations of salt, and on the Z axis they may have other excipients, such as increasing doses of surfactant. In that way the formulator can map out the whole antigen, or the zone within which the antigen is stable. Stability testing would be conducted from an early stage, by adding heat to accelerate thermodynamic reactions.
725 Professor Dalby agrees that this approach was taken in the industry, but considers that some forms of visual detection of reactions are not amenable to such screening. In this regard he singled out the ability to detect the formation of precipitation in the presence or absence of aluminium phosphate. Professor Petrovsky explained that it was standing operating procedure to perform visual inspection against a black background, even if using a checker board of 100 or 200 samples. Having regard to the fact that aggregation is a common problem in formulation (as to which see section 24.4.6 below), I accept that it is likely that any visible precipitation would have been observed in adopting this approach.
726 The experts agree that it was known to the person skilled in the art before April 2006 that GRAS-listed excipients were typically used at first in formulation. They refer to this list by the metaphor of a “toolbox” from which excipients could be drawn to address a vaccine stability or presentation problems. A large number of such excipients, something in the region of 370, were available generally. However, there is no doubt that the person skilled in the art would choose from this list selectively, based on experience and scientific knowledge. For instance, if a particular pH was required, a buffer would be chosen, of which, as Professor Petrovsky says, only two or three had been used in vaccines.
727 In his oral evidence, Professor Dalby took what he described as a “combinatorial” approach, and considered that there was an “astronomical number of variables” when one considered all of the 370 potential GRAS-listed excipients if trying to develop a formulation. Professor Petrovsky considered that of those 370 excipients, only about 20 were used and generally available to formulators at the priority date. Professor Dalby responded to this in the following manner:
MR BANNON: Is there some limitation within that 370?
PROF DALBY: There is some limitation because I wouldn’t be using an antimicrobial to solve the problem of silicone induced aggregation, for example. But I would be able to use polyols, amino acids, potentially surfactants, buffers, change in pH. And then also be looking at the adjuvant, amounts of all those things as well. So they are all intricately interacting in ways you can’t predict so you are left with a combinatorial problem still.
MR BANNON: Is there a number of variables – I think you have mentioned a couple of things – adjuvant and different levels of adjuvant. You mentioned buffer. Is there a number of variables, a finite number of variables with a number of options, or is there – can you perhaps - - -
PROF DALBY: Well, there’s an astronomical number of variables when you look at all combinations. Even if you limited it to 20 options, 20 to the power of 20 is huge so - - -
MR BANNON: Could you just explain that, what you mean by that.
PROF DALBY: If you looked at all combinations, mathematic - - -
HIS HONOUR: You don’t need to explain it for me.
PROF DALBY: Mathematically the number of combinations when you have 20 different things you can add, you can add them all on their own, all their pairs, all as three, all as four, all as five and it’s an astronomical number. So you practically cannot screen that. So you have to define in some way your choice down to a practical number, and the best way to do that is to understand the problem and then pick your excipients based on the problem.
728 Wyeth sought to make much of this numerical approach. It submits that formulation components interact in intricate, complex and unpredictable ways. It submits that even with 20 excipients available, Professor Dalby gave evidence that the solution to any issue identified in the context of formulation problem with an “astronomical number of variables”, quoting the above passage of Professor Dalby’s evidence. As explained below, I do not think that this is what Professor Dalby is saying here, although his evidence did show a tendency to overcomplicate matters. If it is, then it is in my view far-fetched and I reject it.
729 However, in Professor Dalby’s favour, I do not consider that the passage quoted above supports Wyeth’s submission. The final sentence indicates that Professor Dalby would have taken the approach of defining the choice of solution to a given problem by first understanding what it was, and then selecting excipients likely to resolve it. At a high level, the difference between Professor Petrovsky and Professor Dalby lies in the degree of confidence each would have had in selecting a proposed formulation, or excipient to use in a formulation. Professor Dalby, who had no experience in polysaccharide-protein conjugate formulation before the priority date, took a theoretical approach that before proceeding with any formulation he would have conducted extensive tests.
730 However, in my view the skilled formulator would not approach a formulation problem in ignorance and from a standing start, but rather with an understanding of the basic interactions and reactions likely to take place within the active ingredient of interest. As I have noted, the person skilled in the art is a person likely to have a PhD and years of experience in the field of vaccine formulation. Those qualifications and experience would be brought to bear on the educated selection of excipients likely to be used.
731 Professor Petrovsky would have approached a stability issue encountered in the formulation of the polysaccharide-protein conjugate vaccine in a similar way to that with a protein vaccine. Any factor which influences the chemical or physical properties of a protein has, in his view, the potential to affect protein stability. Some of the factors would have included storage conditions, light, temperature, formulation pH, vaccine constituents, protein concentration and choice of formulation packaging. Solving issues relating to formulation stability generally came down to making choices of vaccine components based on their well understood mechanisms of action. I discuss relevant aspects of these later in these reasons.
732 I accept the explanation that Professor Petrovsky gave in oral evidence:
So as we have discussed, to solve any problem in vaccine manufacture or pharmaceuticals is impossible if you assume that there’s 20 to the power of 20 combinations. That simply isn’t how, you know, the game operates. So essentially what we do when we encounter a problem is that we look for a short list of examples of excipients that have previously been found to address a similar problem. So if we have aggregation of our vaccine we ask, well, are there other vaccines that we know about or are in the literature or we have heard about where they have solved a problem of aggregation and if so what was their solution. And you will find that Tween 80 has been used by multiple manufacturers in the past to stabilise their vaccines. So suddenly rather than saying we will select from the 50 surfactants that are possibly on the excipient list, the obvious place to try first is to select Tween 80 and see if it helps or if it doesn’t help...So the idea that I would test 20 to the 20 combinations, it simply isn’t, one, realistic nor is it how industry operates.
733 I consider this further in the context of the issue arising in relation to precipitation and aggregation in section 24.4.6 below.
24.4.2 Containers and silicone
734 In the container JER the experts agree that before 26 April 2006 a variety of containers were available for use by vaccine formulators including glass vials, and less commonly, pre-filled glass syringes and plastic syringes, and that these included containers treated with silicone oil or which used rubber stoppers or plungers treated with silicone oil. They agree that given the increasing awareness of the leachability of silicone oil within containers having siliconised containers and rubber stoppers, some manufacturers had started to address this problem by baking the silicone oil onto the glass surface. They also agreed that before 26 April 2006 containers available for use by vaccine formulators included:
(a) vials closed with rubber stoppers treated with silicone oil; and
(b) syringes containing siliconised rubber plungers, but that their use was not universal.
735 There was a difference of view as to the degree to which siliconised containers were used in the context of vaccine formulation. As noted previously, Professor Dalby’s evidence was that silicone was used for lubrication in syringes and in the rubber stoppers and rubber part of the plungers in some syringes, but that different containers could have different amounts of silicone oil, and that some could have none and those non-siliconised syringes were available. Professor Dalby also gave evidence that the glassware used in his laboratory was usually not siliconised before April 2006.
736 Professor Petrovsky’s evidence indicates that it was routine at the time to use siliconised containers for vaccines. In his oral evidence, he explained his perspective in contrast to that of Professor Dalby:
So maybe an important point to understand – and the difference between, I guess, vaccines, which I work in, and biologicals, which Professor Dalby works in, is how important siliconisation is to a vaccine formulation as opposed to a biological formulation. In biologicals, typically, the biological is present at very high concentration, even gram amounts, in the glass vial, so a little bit of adsorption of that biological and loss of the biological on the glass vial doesn’t impact on its availability when you inject it. With a vaccine, we’re dealing with very tiny amounts of antigen, as we’ve heard, with, you know, anywhere from five to 60 micrograms. So 1000 to 10 thousand times less protein. So if any of that antigen sticks to a vessel wall – a glass wall, which it does if you don’t siliconise it, then you have no active vaccine in the formulation. And so to suggest that the vaccines are not in siliconised containers or not, you know – the fact is that since we’ve had silicone, we’ve been siliconising the containers because we have to avoid that tiny amount of sticking, otherwise we have no antigen left in the vaccine. And that’s very different to biologicals, which I accept can be formulated in non-siliconised containers.
737 This explanation provides a rational basis for the distinction between the position of Professor Dalby, which is based on his experience with biological medicines, and that of Professor Petrovsky, whose experience in the field of vaccine formulation, and particularly conjugate vaccine formulation, is considerably deeper than that of Professor Dalby. Professor Dalby did not disagree. I accept that a skilled formulator working on the development of a vaccination as at April 2006 would be likely to use a siliconised container, for the reasons given by Professor Petrovsky.
738 Furthermore, the experts agreed in the container JER that it would be usual to test any new vaccine formulation in a variety of containers to assess suitability for future storage and delivery. In my view such testing would include siliconised containers, or syringes with siliconised components.
739 Wyeth submits that the content of a text called Remington: The Science and Practice of Pharmacy published in 2000 (20th ed, Lippincott Williams & Wilkins, Washington) at page 789 is inconsistent with the proposition that the skilled formulator was using siliconised containers as a matter of course for vaccines. The information in the passage was not shown to form part of the common general knowledge. Nor does it on its face establish the proposition for which the submission contends. It simply suggests that to reduce the problem of leachables coatings have been applied, the most successful being Teflon. It provides no relevant context for protein or polysaccharide vaccines or polysaccharide-protein conjugate vaccines. The reference is in any event some 6 years before the priority date.
740 Having regard to these matters, in my view a skilled formulator engaged in the formulation of a 13-valent pneumococcal vaccine using Prevnar 7 as a starting point would be likely to use a siliconised container for the formulation task.
741 The goal of vaccination is the generation of a strong immune response to the administered antigen to provide long-term protection against infection. This is the same for protein, polysaccharide and polysaccharide-protein conjugate vaccines. Proteins and polysaccharides were by April 2006 the most commonly used antigen components of bacterial vaccines.
742 Polysaccharide-protein conjugates were introduced into commercial vaccines in the 1980s. The attraction of conjugation to a carrier protein derives from the fact that the polysaccharides by themselves are T-cell-independent antigens and in the absence of T-cell help, the B-cell immune response against the polysaccharide is very weak and does not elicit a long-term memory response (that is, an adaptive immune response). It was found that conjugating a polysaccharide to an independent immunogenic carrier protein, able to be recognised by T-cells, provided T-cell help to polysaccharide-specific B-cells, thereby allowing them to form long-term memory responses.
743 Professor Petrovsky gives evidence, which I accept, that as all polysaccharide-protein conjugate vaccines function in this manner, developments in the formulation of polysaccharide-protein conjugate vaccines for one pathogen are relevant of interest in considering the development of others for different pathogens. If the skilled formulator encountered a problem in developing a formulation for a polysaccharide-protein conjugate, he or she would first consider known solutions specific to that type of vaccine, but would also consider solutions for protein and/or polysaccharide vaccines. Professor Dalby disputes, in particular, the relevance of prior experience in relation to protein vaccines, which I address further in section 24.4.6 below.
744 The experts agree that Prevnar 7 was a licensed, stable, pneumococcal polysaccharide-protein conjugate vaccine formulation that was available and used in Australia as at April 2006. The constituents of Prevnar 7 were known to include its 7 different capsular polysaccharides each conjugated to CRM197 carrier protein, saline and an aluminium phosphate adjuvant, but with no buffer or other excipients. It was available in pre-filled syringes or single-dose vials, and was stable within its stated shelf life and storage conditions. The experts agree that Prevnar 7 would be a good starting position for development of a 13-valent pneumococcal vaccine. The particular microgram amounts of the polysaccharide serotypes conjugated to CRM197 were also known to the person skilled in the art and formed part of the common general knowledge.
745 There is no dispute that although Prevnar 7 would be the starting point for formulation of a 13-valent pneumococcal-protein conjugate vaccine, its stability could not be predicted from the Prevnar 7 formulation. Dr Jones and Professor Petrovsky consider that as the complexity and total combined concentration of the conjugates would be increased in a 13-valent vaccine, different behaviours could be expected and that as a consequence the formulation would need to be subjected to stability testing. Dr Jones also noted that every individual polysaccharide was known to behave quite differently, with particular polysaccharides, for instance 6A, l9A, and 19F, being known to be of relatively low stability. His view is that any sensible formulator would have assumed that the new formulation might be unstable and hence would have routinely performed stress testing during development to assess its stability. In the container JER Professor Dalby stated that as Prevnar 7 was a stable commercial product, he would not expect to see instability or aggregation in a 13-valent pneumococcal polysaccharide-protein conjugate formulation. Nevertheless, he accepted that when a formulation is varied, it will need to be tested for stability, and if any form of instability is located from the various tests, then one of the things that one might start to look at is alternative pHs. In my view the skilled formulator would expect that adding any serotypes to the Prevnar 7 formulation could well lead to instability and that this would have to be tested.
746 The main criteria considered and evaluated during vaccine development and formulation were safety, efficacy and stability. Stability studies were routinely conducted throughout formulation development to ensure that the physical and chemical properties of an antigen in the formulation do not significantly change in the presence of additional vaccine components. Whether the vaccine was a protein, polysaccharide, or polysaccharide-protein conjugate vaccine, it was critical to the efficacy of a vaccine formulation that the antigen was capable of eliciting an effective adaptive immune response against the native form of the protein or polysaccharide expressed by the pathogen. It was accordingly important to produce a formulation in which the integrity and physico-chemical properties of an antigen were maintained in the presence of other vaccine constituents and under various conditions to which the formulation is likely to be exposed, such as during manufacture, storage, handling and ultimately administration to a patient. It was a regulatory requirement for vaccine formulations to be tested in the containers intended to be used to store and administer the formulation.
747 Immunogenicity studies were routinely performed during formulation development to assess the efficacy of candidate antigens in eliciting an adaptive immune response in a suitable subject. Furthermore, it was routine to test vaccines for aggregation. Those tests were expected to identify any visible aggregation, regardless of its cause.
748 Accelerated stability studies replicated real world conditions by using higher temperatures for shorter periods. These were common in the industry and accepted by regulators during clinical development.
749 As I have noted, there is no dispute that when antigens were formulated with additional components those formulations were typically tested to ensure that immunogenicity of the antigen was maintained and the immunogenicity of the formulation was optimised.
24.4.6 The problem of aggregation generally for polysaccharide conjugate vaccines and protein vaccines
750 Aggregation is a generally undesirable outcome in formulation. It is one of the two very common problems that is likely to be encountered when formulating any vaccine (the other being hydrolysis of the antigen). It affects the appearance of a vaccine and may suggest a safety or efficacy problem. Unexpected aggregation or cloudiness would not be approved for use. Typically, the release criteria for vaccines before April 2006 involved prior visual inspection to identify particulates, which may indicate that matter has precipitated out of solution, which is the result of a high level of aggregation, and instability.
751 Generally, the structure of a protein is influenced by the arrangement of its hydrophobic and hydrophilic regions. Hydrophobic regions are those that tend to repel or fail to mix with water, whereas hydrophilic regions tend to mix with or dissolve in water. In order to avoid water, hydrophobic portions of a protein often position themselves in the core of a protein. In contrast, in order to be close to water, hydrophilic portions of a protein often position themselves at the surface of the protein.
752 Since at least the 1980s, when people started expressing and purifying recombinant proteins, it was known that some proteins had a tendency to aggregate – stick to each other – and to stick to hydrophobic surfaces and interfaces (adsorption) such as glass vials. Aggregation may be visible to the eye in the form of precipitation.
753 Professor Petrovsky considers that the carrier protein of polysaccharide-protein conjugates was likely to have been a major contributor to aggregation in much the same way that it was for protein formulations. When the hydrophobic regions of a carrier protein are exposed, those regions will tend to adhere to other hydrophobic regions, such as on other proteins. These hydrophobic forces were a major driver of aggregation. He considers that the presence of polysaccharides in the conjugate were unlikely to interfere with the hydrophobic interactions to the extent that aggregation would have been inhibited in a polysaccharide-protein conjugate. Furthermore, he considers that the act of conjugation would have opened up the carrier protein’s structure in a way that made aggregation more likely.
754 Professor Petrovsky refers to several articles that he had read before April 2006, being Ho 2001, Ho 2002, Bolgiano, and Berti, and exhibits these articles as support for the view that he held at the time, that under increased temperature, the carrier protein of a conjugate unfolded and triggered aggregation of the conjugates. He considers that as a result of the protein being a contributor to aggregation, before April 2006 he would have expected that the same issues with respect to aggregation could have arisen with polysaccharide-protein conjugates as with protein vaccines and that the same solutions for overcoming aggregation would apply.
755 Professor Dalby does not agree that issues encountered for protein vaccines were relevant before the priority date to the formulation of polysaccharide-protein conjugate vaccines, because he considers that the polysaccharide chains, being hydrophilic, would have surrounded the more hydrophobic protein. As a result, he would have expected that the influence of the polysaccharide component of the conjugate would dominate the interaction between the conjugates and the solution, as well as the interaction between the individual conjugates. Professor Dalby rejects Professor Petrovsky’s interpretation of Ho 2001, Ho 2002, Bolgiano and Berti concerning protein aggregation, and considers that they are irrelevant, on several bases, including that: they show only small conformation change; they are mostly directed to oligosaccharide-protein conjugates rather than polysaccharide-protein conjugates; and the temperatures used in the studies which caused unfolding of the carrier protein were excessive. His view is that whether or not the results could have been applied to polysaccharide-protein conjugates would need to have been evaluated.
756 I prefer the view of Professor Petrovsky for the following reasons.
757 First, the view of Professor Petrovsky was held before April 2006, based on what he then knew and understood. I accept his evidence that this was the view he then held, and take his reaction to the articles cited to be untainted by knowledge of the issues in the litigation and based on his view as a skilled formulator directly in the field. Professor Dalby does not give evidence that he had read these articles before the priority date. Nor does he cite any publication known in the field before the priority date tending to support his view.
758 Secondly, the temperature conditions under which these articles considered the conformation of the protein conjugates were in accordance with the recommended testing pursuant to the Australian Regulatory Guidelines for Prescription Medicines. Professor Dalby’s criticism of such accelerated stability studies on this basis appears also to reflect his lack of experience in the field.
759 Thirdly, the fact that the Ho 2001 and Bolgiano studies do not concern polysaccharide-protein conjugates but oligosaccharide-protein conjugates does not mean that there is not a relevant predictive content to them. As Professor Petrovsky convincingly explains, oligosaccharides are a subset of polysaccharides and they can form the basis for consideration of the behaviour of proteins in a polysaccharide-protein conjugate. Further, as Professor Dalby himself notes, Berti, which does concern polysaccharide-protein conjugates, concludes “that the polysaccharide-protein conjugates have a strong tendency to aggregate”.
760 Fourthly, as a matter of science and knowledge at the time, Dr Jones’s evidence supports that of Professor Petrovsky. In 1997 he published an article reporting that CRM197 was particularly susceptible to unfolding due to external stresses such as changes in temperature and pH, and that an unconjugated CRM197 was less likely to unfold. His view at the time that Bolgiano was published in 2001, which he had read a number of times before 2006, was that CRM197 conjugates would form aggregates that could affect vaccine immunogenicity, and that this was attributed to unfolding and subsequent aggregation of the CRM197 carrier protein component, not the polysaccharides. This was observed in Bolgiano in stability studies considering incubation in storage of 20 degrees Celsius for 5 weeks, 37 degrees for 2 weeks and 55 degrees for 1 one week. The authors reported “unfolding and subsequent self-association or aggregation” of conjugated CRM197 and noted the “extreme aggregation of the carrier protein” as a possible cause for the observed reduction in the immunogenicity of the vaccine stored at 55 degrees Celsius. Whilst Bolgiano addresses the saccharide component, at no point does it suggest that polysaccharide aggregation (as opposed to protein aggregation) was a concern. In relation to Ho 2001, Dr Jones notes that the conclusion of the paper, which addressed similar issues, emphasised the “the stability of the properties of the carrier protein as significant factors in the stability of the conjugate”. As in Bolgiano, the authors of Ho 2001 did not observe any evidence of polysaccharide aggregation, or suggest that polysaccharide aggregation was a concern. Dr Jones considers that the temperatures described in Bolgiano and Ho 2001 are consistent with those used during normal accelerated stability studies conducted to investigate any potential change in stability over a vaccine formulation’s anticipated shelf-life of two years. Although Dr Jones is not representative of the skilled formulator, he was plainly working in the field of polysaccharide-protein conjugate vaccines before April 2006. I consider his evidence to be corroborative of the views expressed by Professor Petrovsky, both as a matter of the understanding of the science and as to the likely knowledge of the skilled person.
761 Fifthly, I also accept that it was a matter of common general knowledge that it was likely that aggregation would be a problem for conjugate vaccines, just as it was for protein vaccines, and that as a consequence the skilled formulator would have approached the problem in the same way as for protein vaccines. This is the evidence of Professor Petrovsky and Dr Jones, which I accept. Professor Petrovsky explains how he understood the hydrophobic regions to be exposed when the protein of a polysaccharide-protein conjugate is unfolded:
762 Finally, it seems to me that Professor Dalby’s response to the question of aggregation reflects the difference between him and Professor Petrovsky in their approach to formulation to which I have referred. Professor Petrovsky would have developed a theory of the mechanism of any aggregation and approached formulation of a solution with that theory in mind. Professor Dalby’s evidence indicates that he would not have moved beyond the point of identifying aggregation in a formulation without first conducting a research project until he learnt the root cause of the problem. Only then would he have continued formulating. Until he had conducted the research project he would have had no expectation that anything that he used to solve the problem would succeed. In my view, having regard to his deeper experience in the field before the priority date, Professor Petrovsky is in a better position to give persuasive evidence on this point, and I accept his approach, as set out in section 24.4.1 above.
24.4.7 Silicone oil induced aggregation
763 As I have found, it was common general knowledge before April 2006 that proteins could stick (adsorb) to glass vials, and that formulators of protein or polysaccharide-protein conjugate vaccines typically used siliconised glass vials and rubber stoppers in their work to avoid adhesion. I have also found that protein aggregation was a major concern for vaccine formulators.
764 A further issue arises between the parties as to whether or not the issue of silicone oil induced aggregation was known to the hypothetical skilled formulator. This is important to the case advanced by Wyeth, because it relies on the evidence of Professor Dalby to the effect that he would not have taken steps to reformulate Prevnar 7 using additional serotypes if he had encountered aggregation, until he had first conducted a research project to understand the mechanisms of such aggregation. In that regard, he says that he would not have tested for silicone oil induced aggregation if he had encountered aggregation and if he had, through research and testing, determined that the formation of particulates in a formulation was the result of the presence of silicone oil, he would have sought to understand the mechanism by which the silicone oil was causing the particulates to form. Alternatively, he may have used containers that were not siliconised, taken the recommendation in Jones 2005 and reduced the content of silicone oil, or baked the silicone onto the glass to reduce the amount of free silicone oil in the solution. He would not have known whether the last two approaches would work, because it would depend on whether the mechanism by which the particulates were forming involved protein to protein or conjugate to conjugate interactions occurring at a siliconised surface (which would mean that pre-baking to the surface would not work), or whether the mechanism involved silicone oil droplets. Finding solutions to the problem would have involved a research project into poorly understood interactions.
765 MSD relies on two answers to this evidence. First, it contends that the skilled formulator was well aware before April 2006 of the problem of aggregation that routinely occurred in the course of vaccine formulation, and that known techniques would be applied to resolve it, regardless of its cause. Those techniques typically involved the adjustment of pH by the addition of buffers and surfactants. Secondly, it contends that in any event, aggregation caused by silicone oil in vaccine formulation had been known to those in the art since well before April 2006. In relation to both points, it relies on the evidence of Professor Petrovsky.
766 Professor Petrovsky gives evidence that silicone oil induced aggregation was generally known to be a problem for pharmaceutical formulations containing hydrophobic proteins in the absence of alum (that is, any aluminium salt). He explains that the common use of alum in non-live vaccines reduced this problem, because most of the antigen present in the vaccine formulation (including in a polysaccharide-protein conjugate vaccine) is not free, but is bound to the alum. As a result, the bound antigen cannot stick to other antigens or to the glass surface of the vial. Nevertheless, issues of silicone contamination and silicone oil’s role in protein aggregation were frequent in the field.
767 Professor Petrovsky was also aware since the 1980s of reports of silicone oil interacting with insulin and denaturing it, based on the work of Chantelau which he had read at that time. He gave the opinion in his oral evidence that Chantelau demonstrated that silicone oil interacted with the insulin and denatured it, causing cloudiness in the formulation, with the result that international advisories were put out around the world for patients not to use insulin if it became cloudy. He considers that this was a major finding in the insulin industry.
768 Dr Jones gives evidence that he was aware of silicone oil induced aggregation being a general topic of conversation with his colleagues at NIBSC before April 2006, and was aware that silicone oil could lead to product problems, including aggregation. At the time, he understood that aggregation might occur in a protein vaccine which came into contact with silicone oil, and that such aggregation was mediated through silicone oil induced unfolding of the proteins. He expected that polysaccharide-protein conjugate vaccines would be affected by silicone oil in the same way as pharmaceutical protein preparations were.
769 The following passage from the oral evidence of Professor Petrovsky, with whom Dr Jones agreed (and I accept), addresses the underlying mechanism that they understood before April 2006 to apply to aggregation, including silicone oil induced aggregation:
I think, in terms of the question, “Do we need to understand silicone aggregation as a phenomena”, what we knew pre-2006 – and this goes back decades in scientific literature – is that aggregation is triggered by the creation of surfaces in the formulation. So it doesn’t matter whether it’s a contact with a glass surface – and, in fact, one of the reasons we siliconised vials was to reduce surface contact of the product with the surface of the glass, because this triggered aggregation. Similarly we know that if you simply shake up a vial very aggressively, that this creates air-water interfaces – and they are, again, surfaces. And wherever you create surfaces in any way, you run the risk of triggering aggregation. So the idea that it was novel – that adding an oil particle to a liquid solution, which, again, is an interface and a surface between water and, in this case, oil – that that was surprising that triggered aggregation, I think, is incorrect. We knew the mechanism, which is anything that creates a surface interaction between two interfaces will trigger aggregation.
So – and the solution, again, that we knew to that is to avoid those interfaces, whether it’s a solid-liquid interface or a liquid-liquid interface or an air-liquid interface, we use a surfactant because a surfactant, essentially, inserts itself at the interface between those two surfaces and stops the protein interacting with that surface domain, which is the domain that triggers aggregation. And that has been known for 20 or 30 years – that surfactants will reduce aggregation if it’s driven by an interface between two different media. And so you can put silicone oil into that understanding and say, “We already understood the mechanism, even if not of the individual molecular level”.
770 In the container JER the experts agree that as at April 2006 there was an increasing awareness of the leachability of silicone oil within containers having siliconised containers and rubber stoppers.
771 I am satisfied that there was a general awareness amongst formulators of vaccines that one cause of protein aggregation could be silicone oil in containers. I am also satisfied that there was a general awareness that silicone oil could induce protein aggregation in a polysaccharide-protein conjugate vaccine, having regard to the protein carrier component of those vaccines.
772 A buffer is a mixture of a weak acid, or a weak base, that is added to a solution following the addition of small amounts of acid (hydrogen ions) or base (hydroxide ions) in order to prevent the solution from becoming more acidic or basic and thereby moving out of an optimal pH range. The structure and function of proteins are sensitive to pH as this influences the degree of ionisation of their weakly acidic and basic groups. With a protein vaccine, maintaining the conformation (that is, the three-dimensional arrangement) of the protein is important in order to maintain the relevant conformational epitopes. Changes in pH can cause changes in conformation. This is important when considering problems encountered in the form of aggregation. As I have noted in section 24.4.6 above, an understood cause of aggregation in protein vaccines and also polysaccharide-protein conjugate vaccines was the unfolding of the protein when exposed to increased temperature in stability testing.
773 The optimal pH was known to help to maintain the native conformation of the antigen. As Professor Dalby accepted, the goal with any formulation before the priority date was to keep the pH within an optimum pH for the shelf life of the product. Care was needed when determining the appropriate pH of a formulation of a polysaccharide-protein conjugate, as a non-ideal pH could lead to cleavage (by hydrolysis) of the polysaccharide.
774 Before April 2006 it was known that the stability of antigens was affected by pH and that buffers were available to regulate the pH of a vaccine to a particular level to optimise its stability. The experts agree that the first choice of buffers would be selected from phosphate, citrate, acetate, malate, succinate, histidine, borax and bicarbonate buffers.
775 Furthermore, there is no disagreement that as a matter of formulation buffers could be used to regulate the pH, if a formulation was considered to be unstable at a particular pH, or that having the formulation at a particular pH would improve stability. Nor is there any dispute that polysaccharides were known to be unstable at alkaline pH levels.
776 There was a limited number of buffers that were used in vaccine formulations, and phosphate buffers, such as phosphate buffered saline, were a preferred choice because: (a) they are very biocompatible (with a pH not far from neutral, in ranges of 6 – 8.5); (b) they are stable over time and have powerful buffering capabilities; (c) they are relatively inexpensive; (d) they exhibit little or no toxicity; and (e) they possess a pH range which makes them suitable for most applications. Phosphate buffers were before April 2006, and remain, the most commonly used buffers in biology.
777 Professor Petrovsky had buffers available in his lab which he used before April 2006 that provided different optimal pH ranges. He routinely used bicarbonate and phosphate buffers in adjuvanted vaccine formulations, including histidine which he had used in a formulation of a vaccine antigen. It was known to him that the selection of a buffer to match the optimal pH of the antigen allows the buffer to be used at a lower concentration, which was considered good practice.
778 Professor Petrovsky considered before April 2006 that as the optimal pH would help to maintain the native conformation of the antigen, it would also help prevent aggregation. His view was that a physiological pH was 7.4 and that an acceptable range was between 4 and 9. He considered that it was critical during the early stages of vaccine development to determine the optimal pH range for the antigen of interest to inform the choice of appropriate buffer. That involved characterising the behaviour of the protein antigen, looking at the protein folding and aggregation under different pH conditions and testing different buffers suspected of having suitable buffering properties.
779 There was no dispute between the experts that buffers were well known to be used before April 2006 in vaccine formulation, or as to the purpose of that use, which is summarised above. The point of departure is that Professor Dalby disputes that it was known that buffers were commonly used in polysaccharide-protein conjugate vaccines. He also disputes that the use of buffers in protein vaccines provides a basis for predicting that a buffer in a polysaccharide-protein conjugate vaccine will have a similar effect. In my view Professor Petrovsky’s view that the protein carrier in a polysaccharide-protein conjugate would have behaved in a similar way to a protein in a protein vaccine is to be accepted, and forms part of the common general knowledge.
780 Furthermore, the evidence includes a number of examples of relevant vaccines that had been formulated with a buffer. Professor Petrovsky gives several examples. One is the original Chiron Hib vaccine which he became aware of around 2003 to 2004 when working with a Cuban institute on a number of projects, including its Hepatitis B and Hib vaccines. This was a polysaccharide-protein conjugate vaccine conjugated to CRM197 that included a buffer, salt, aluminium adjuvant and surfactant. Another was the Japanese encephalitis virus (JEV) that he worked on in 2002. It was an inactivated virus vaccine that also contained a buffer, salt, and surfactant. The antigenic component of the inactivated virus was a glycosylated envelope protein (a natural glycoconjugate), which had naturally conjugated polysaccharides on the surface of the protein antigens. The lyophilised form of this vaccine was available in Australia before April 2006. Authors at the Australian National University the University of Queensland had published an article about the JEV before April 2006. Professor Dalby disputes that this vaccine can be classed as a polysaccharide-protein conjugate vaccine. However, as Professor Petrovsky points out, the JEV vaccine was made up of surface proteins that are glycoproteins, that is, proteins conjugated to polysaccharides, which he considers to be naturally occurring polysaccharide-protein conjugates. These glycoproteins have essentially the same properties as artificially created glycoconjugates. I accept this explanation.
781 Another is Gardasil, which Professor Petrovsky provided an expert opinion about (comparing the formulations of Gardasil and Cervarix for an independent consulting firm). This was a protein vaccine which was formulated with a buffer, salt aluminium adjuvant and surfactant. As a result of his work he was familiar with the HPV technology that led to Gardasil. In this context he also refers to a paper by L Shi et al entitled “Stabilization of Human Papillomavirus Virus-Like Particles by Non-Ionic Surfactants” published in 2005 in the Journal of Pharmaceutical Sciences (Volume 5(7)), which Professor Petrovsky read shortly after it was published.
782 In addition, Professor Dalby gives evidence of his awareness that there were three saccharide-protein conjugate vaccines in Australia by April 2006 containing a buffer.
783 Having regard to these matters, I am persuaded that Professor Petrovsky’s view before April 2006 that it was known that in the formulation of a polysaccharide-protein conjugate vaccine a buffer could be used for its usual function – to adjust the pH in a formulation – was not (as Wyeth submits) idiosyncratic, but rather represents the understanding and knowledge of those in the field before April 2006.
784 Since the 1920s aluminium salt adjuvants were known and used in non-live vaccines and only a few rare examples of non-live vaccines did not include an adjuvant. The only classes of vaccine that did not typically include an adjuvant before April 2006 were pure polysaccharide (non-conjugated) and live viral vaccines. Adjuvants were known to enhance the immunogenicity of the formulation by promoting an inflammatory response in the vaccine recipient at the site of injection and providing sustained release of the antigen once the vaccine was administered. The Petrovsky Review states that “with few exceptions, alum [a term he generally uses for any aluminium salt] remains the sole adjuvant approved for human use”.
785 An adjuvant, such as aluminium phosphate, when formulated with an antigen, could adsorb the protein to its surface.
786 Professor Petrovsky considered before April 2006 that when such adsorption took place, it could not only be important to the adjuvant action in enhancing immunogenicity, but also important in stabilising the antigens, thereby preventing degradation during storage. His view is that adsorption was known before April 2006 to inhibit intermolecular aggregation of protein, particularly for hydrophobic proteins which are prone to aggregation in aqueous solutions. He says that whilst he knew this to be true for vaccine formulations involving proteins, he also understood at the time that it would also apply to the use of an adjuvant for polysaccharide-protein conjugate vaccine formulations. Namely, when an aluminium adjuvant was added to a formulation containing a polysaccharide-protein conjugate, the conjugate adsorbed to the aluminium salt. In both cases, the process of adsorption reduced the likelihood of the antigens self-aggregating. In this regard, he explains that before April 2006 it was his understanding that proteins were biomolecules that interacted with and responded to their surroundings by changing their conformation. When a protein was bound to an aluminium adjuvant, however, the protein became fixed on the surface of the aluminium adjuvant particle and less able to change its conformation over time. He gives evidence of two publications to support this proposition, being a chapter by S L Hem and Joe L. White called “Structure and Properties of Aluminium-Containing Adjuvants” published in 1995 in Vaccine Design: The Subunit and Adjuvant Approach (Volume 5, Springer, New York), and an article by A Alam et al entitled “Stability of an antifertility vaccine consisting of gonadopropin subunits linked to tetanus toxoid” published in 1989 in Vaccine (Volume 7). Professor Petrovsky had read the first before April 2006 and gives evidence, which I accept, that he likely read the second before then.
787 Professor Dalby disagrees that aluminium salt adjuvants were added to formulations to provide a benefit of increased stability. He considers that they were not known to create more favourable polar environments for proteins by adsorption, and considers that there was no expectation that adsorption would have a stabilising effect. He considers that these matters were not generally known before April 2006 and nor was the general principle regarding stability to which Professor Petrovsky refers.
788 Professor Petrovsky was briefly challenged on his understanding of Hem and Alam during the course of the concurrent evidence, but he persuasively adhered to his views. In relation to Hem, he said:
PROF PETROVSKY: Again, you know, my interpretation – and, you know, this is complex science, but that, in effect, if you adsorb a protein to alum, then there – it obviously reduces the propensity to aggregation, if we call it propensity. And Stanley Hem largely, then, focuses on the problem of desorption, which is the trigger of – obviously, of aggregation. And so that’s why if – once you have adsorbed your protein to alum, if then the protein comes off the alum, it’s actually much more prone to aggregation, and that’s why regulators set limits on the level of both the adsorption, but also on desorption. So I think, again, the inference is high levels of adsorption to alum are stabilising, providing the antigen doesn’t then desorb from the alum. And I think, again, this comes down to, you know, work which shows that the protein after adsorption to alum is – may be changed in some way. So if it then desorbs, it’s much more prone to aggregation. So that was the science as I understood it before 2006.
789 Professor Dalby responded by contending that the section of Hem to which Professor Petrovsky was referring does not mention protein aggregation. It refers to the impact of binding the proteins on the colloidal interactions of the alum. His view was that “colloidal interactions” mean whether they are repelling or attracting. The nature of binding the protein onto the adjuvant changes its net charge and therefore it changes whether there is a colloidal interaction between the alum particles.
790 Professor Petrovsky picked this up in his response, to observe that the point being made is the same:
PROF PETROVSKY: So, in fact, this is exactly – it’s a good point because it’s exactly what’s happening in the container patent when they add silicone oil. So it’s the same process. When you’re seeing these big visible aggregates in the presence of alum, then these are colloidal aggregates. And so the trigger, I think, in the container patent is that you’ve got aggregates, including of aluminium-bound conjugates. And so they’re trying to solve that problem. So it’s a colloidal problem and Stanley Hem here is saying that adsorption to alum, you know, reduces these problems of colloidal aggregation of alum-bound antigens or proteins in this case. So I would say this goes directly to the problem in the container patent, of colloidal aggregation.
791 Similarly, the oral evidence in relation to the Alam reference tended to reinforce, rather than erode, my confidence in the accuracy of Professor Petrovsky’s explanation. In this regard Hem and Alam are not part of the common general knowledge, and I do not take them as such. Rather, they provide support for the evidence given by Professor Petrovsky as to the matters to which he refers.
792 Furthermore, Dr Jones gives evidence that before April 2006 he expected a polysaccharide-protein conjugate to adsorb to the aluminium salt. Since the process of aggregation of a polysaccharide-protein conjugate, which leads to precipitation, involves the coming together of conjugates that are free in solution, there would be less opportunity for such aggregation to occur in the presence of an aluminium salt. That is because a significant proportion of the conjugates will be adsorbed on the surface of the aluminium salt and will therefore have less opportunity to interact with other conjugates in solution. Before April 2006, Dr Jones considered that the addition of aluminium salt to a formulation containing a polysaccharide-protein conjugate was likely to reduce the occurrence of aggregation between the conjugate molecules, and accordingly increase stability as well as enhance immunogenicity.
793 In my view the evidence of Professor Petrovsky, as supported by that of Dr Jones, is to be preferred to that of Professor Dalby in this respect. I consider that their knowledge is more likely to reflect the understanding of the notional skilled formulator. Having regard to the oral evidence of Professor Dalby and Professor Petrovsky in relation to the issue, in my view the publications to which Professor Petrovsky refers support the view that he expresses.
794 Surfactants are a general class of detergent that act as surface-active agents. They are usually made up of a hydrophilic head region and a hydrophobic tail region. The hydrophobic region of the surfactant binds to the hydrophobic regions of the protein, shielding them to make them hydrophilic. Surfactants may exert their effect at solid-liquid, liquid-liquid and liquid-air interfaces. The use of surfactants in vaccines, and protein formulations generally, was to assist with solubility, particularly for hydrophobic proteins which are poorly soluble in aqueous solution. A surfactant may be included in a parenteral formulation to increase protein solubility. It was known that proteins in a formulation could adsorb to the container wall, and surfactants were used in vaccines to overcome this problem in protein formulations.
795 There is no dispute that surfactants were known to be used for particular purposes in protein vaccines. However, there is a dispute between the experts as whether it was known that surfactants could be used for the purpose of assisting with the stability of proteins, particularly for hydrophobic proteins. Professor Petrovsky considers surfactants were known to reduce aggregation, while Professor Dalby considers that this opinion conflates solubility with aggregation because aggregation is not caused only by the hydrophobic regions of a protein.
796 Professor Petrovsky provides the following diagram to explain how he understood (before April 2006) a surfactant to operate to address aggregation:
797 As an example of a publication supporting the view that he held before April 2006 as to the action of surfactants, he refers to what he describes as a frequently cited article that he read during his PhD, by Bam et al entitled “Stability of Protein Formulations: Investigation of Surfactant Effects by a Novel EPR Spectroscopic Technique” published in 1995 in Pharmaceutical Research (Volume 12(1)). Professor Petrovsky considers that surfactants were known as agents that could be used to address aggregation observed during formulation.
798 Professor Dalby accepts that surfactants were known to improve the solubility of proteins, and that proteins in solution could be more stable, but in the present case, the objective is for the conjugates to be adsorbed to the adjuvant, and not to be in solution. Too much surfactant disrupts the bonds within a protein and will have a de-stabilising effect instead. Furthermore, he contends that Bam is not relevant because it concerns only the effect of surfactants on protein formulations, not polysaccharide-protein conjugates. Professor Petrovsky joins issue on these points. First, he disagrees that Bam is not relevant because it concerns protein vaccines. For the reasons I have explained above in section 24.4.6, I accept that the skilled formulator would have had regard to the behaviour of proteins in protein vaccines, as described in Bam, as relevant in estimating the likely behaviour of a polysaccharide-protein conjugate.
799 Secondly, Professor Petrovsky agrees that surfactants were known to improve solubility and thereby stability, but explains further:
However, prior to 2006 it was known that adsorption to aluminium phosphate is rarely complete and a fraction of the vaccine antigen remains in solution. It is the role of the surfactant in an aluminium adsorbed vaccine to keep this non-adsorbed antigen fraction soluble to prevent aggregation and loss of stability.
For a surfactant to stabilise an aluminium adjuvant bound polysaccharide-protein conjugate, the surfactant must be added to the formulation after the aluminium adjuvant bound conjugate is performed. This is because aluminium adjuvant is “sticky” and will bind to any charged component of a formulation. If surfactant is added to a formulation before the aluminium adjuvant is bound to the conjugate, the aluminium adjuvant and surfactant will interact, blocking aluminium adjuvant from fully absorbing to the conjugate.
800 Professor Petrovsky gives evidence that since he first started working in a laboratory in 1994, he knew that surfactants could be used alone or in conjunction with other methods to prevent aggregation. He says that there was a range of surfactants commonly used before April 2006, including Triton X, a harsh surfactant, and three main types of Tween, being Tween 20, 40 and 80, which are mild surfactants and were more commonly used. His selection of which Tween to use before April 2006 was a Tween in a concentration of between 0.02% and 1%. He was aware that polysorbate 80 was the most commonly used surfactant in vaccines, and gives examples of nine commercial vaccines where that surfactant was used before April 2006. Professor Petrovsky considers that the principles upon which he used surfactants in protein and polysaccharide vaccines would have applied to the use of a surfactant for a polysaccharide-protein conjugate vaccine, and he would have expected a surfactant to work in the same manner in the experimental 13-valent formulation.
801 Professor Petrovsky gives evidence that if a stability issue (such as aggregation) were to have been detected in a polysaccharide-protein conjugate vaccine formulation before April 2006 that contained an aluminium adjuvant (such as in a modified Prevnar 7 formulation), then he would have assumed that the adsorption of the protein to the aluminium phosphate was not complete. The role of the surfactant was to keep the non-adsorbed antigen fraction soluble, to prevent aggregation and loss of stability. He would have added a surfactant to the formulation after the aluminium adjuvant (in the event that the aluminium adjuvant did not sufficiently prevent aggregation), to ensure that the adjuvant had first bound to the conjugate and would not interact adversely with the surfactant.
802 In his oral evidence Professor Dalby accepted that surfactants were known to be one option for surface-based suppression of interaction with surfaces, such as would possibly lead to aggregation. He mentioned that there are several different surfactants, and he could probably list “about 20”. Equally, he said, albumin or polyols may instead be added to coat surfaces to avoid other proteins absorbing into them. However, he said there is a known danger of surfactants destabilising the protein:
So they are actually not a choice that you would make just because you see them somewhere else. You would need to test different options to make sure you have got the best option for your molecule. As for available surfaces, there are, of course, the adjuvant as well – itself is a surface. Potentially, you could be causing destabilisation by binding to the adjuvant itself, and so one choice is to change the adjuvant. And there are other options; there are oil-based adjuvants – as one option, by squalene – so you can change something, create a new problem and you keep having to test and fix that problem.
803 In response Professor Petrovsky said (emphasis added):
So, again, in terms of the formulation, I’m not disagreeing that limited testing doesn’t occur. In fact, as I explained before, you know, chequerboard testing is routine with ingredients such as surfactants, which, you know, for those of us – and, in fact, you know, we have – I have examples in our own formulation. The first thing we did when we encountered a problem of protein aggregation is we added a small amount of surfactant, in fact, Tween 80, which is the – exactly the surfactant used in this patent and, in fact, in a lot of prior vaccine products, including conjugate vaccines.
So it’s a, sort of, yes, you could – there are alternatives, but given that you have a regulatory environment that’s conservative, you would never use a surfactant that, you know, isn’t already used in an existing vaccine if you can avoid it. So all I’m saying is it’s – essentially, there’s some obvious choices, and the Tweens are obviously choices because we all use them for this purpose and they are in approved vaccines. We certainly know not to use them at too high a concentration, because I agree with Professor Dalby that, you know, if you use a 10 per cent surfactant it would denature your protein and aggravate the problem. So we all know to minimise the amount of surfactant to a level that is sufficient to overcome the problem, but not so much that it might aggravate the problem.
So I guess that was known well before 2006. And, as I say, it was one of the obvious things you would go to first if you have got evidence of an interface problem that is causing aggregation. Admittedly, the other – you may add other components, and so you may, as I said, explore the effect of pH at the same time, you may look at the ionic strength and you may add in peptides, as alluded to by Professor Dalby, which, again, further make your protein comfortable in its environment. But surfactant is the number 1 in industry to solve the problem of things sticking to other things, because it’s a detergent, so it removes the potential for things to stick together. So I still think it is the number 1 thing I would go to if I had an aggregation problem.
804 I accept the evidence of Professor Petrovsky, and prefer it to that of Professor Dalby. I accept that surfactants were known to be used by formulators of vaccines for the purpose of addressing problems arising from aggregation in protein vaccines and that they represented one of a number of known means by which problems arising from aggregation encountered in the formulation of a polysaccharide-protein conjugate could be addressed before the priority date. In this regard I am not persuaded by the submission advanced by Wyeth that the fact that there was no commercial conjugate vaccine on the market in Australia by April 2006 is a relevant consideration. Five protein vaccines were available containing surfactants to which Wyeth refers in its submissions: Boostrix, Kinrix, Inferix, Quadracel and Pediarix. Having regard to the understanding of the skilled formulator about the mechanism of action of surfactants, in my view knowledge of these uses would be considered by the skilled formulator to be analogous. I accept the evidence of Professor Petrovsky in this regard.
25. CONTAINER PATENT: ANALYSIS OF LACK OF INVENTIVE STEP IN THE LIGHT OF THE COMMON GENERAL KNOWLEDGE ALONE
805 For the purposes of the Patents Act, an invention is taken to involve an inventive step when compared with the prior art base, unless the invention would be obvious to a person skilled in the art in the light of the common general knowledge, either with or without the information to be considered under s 7(3). The several ways that the question of what is “obvious” may be tested have been considered in section 7.2 above. In each case it is necessary to look from the prior art base to see what the person skilled in the art (the skilled formulator) is likely to have done when faced with a similar problem to that which the patentee claims to have solved with the claimed invention: AstraZeneca (HC) at [69] (Kiefel J); Lockwood No 2 at [127].
806 In the present case two forms of analysis were proposed. In the first, MSD submits that there was no relevant distance between the state of the art in the common general knowledge and the invention claimed. It submits that it was obvious to formulate a polysaccharide-protein conjugate in a siliconised container with an adjuvant and a buffer in accordance all of the asserted container patent claims.
807 Wyeth contends that this approach does not sufficiently postulate the forecast required. It characterises the “problem” that the patentee sought to solve as silicone oil induced aggregation caused by the unexpected interaction between a siliconised container and the immunogenic formulations the subject of the claims. Wyeth submits that the skilled formulator would not have identified the problem, and if he or she did, would have paused there to identify its cause. It submits that there was no common general knowledge about silicone oil induced aggregation and the skilled formulator would not have arrived at the invention with any expectation of success. MSD in turn joins issue with Wyeth, by proposing, as an alternative approach, that if a problem/solution analysis is to be adopted, then the problem posed in the container patent is to produce a stabilised vaccine formulation, one aspect of which is to avoid aggregation, whether induced by silicone oil or otherwise.
808 As I have noted, the parties accept that a convenient framework of analysis is to consider the approach likely to be taken by the skilled formulator prior to April 2006 to the formulation of a polysaccharide-protein conjugate vaccine that has additional serotypes to Prevnar 7. In the evidence, the experts agreed that Prevnar 7, which formed part of the common general knowledge, would be a good starting point to the formulation of a 13-valent polysaccharide-protein conjugate vaccine. The parties based their submissions around this approach, each contending for the opposite conclusion, and it is a useful basis for analysis.
809 Three points may immediately be noted. First, as the experts agreed in the container JER, the stability of a 13-valent polysaccharide-protein conjugate formulation could not be predicted simply because the Prevnar 7 formulation was stable. Stability testing, which was an inevitable part of the formulation process for any skilled formulator, would have been required during the process of development.
810 Secondly, the skilled formulator would have had an understanding of the way in which the protein in the polysaccharide-protein conjugate was likely to behave. Despite Wyeth’s submissions to the contrary, I accept that the skilled formulator’s approach would be guided by experience not only in relation to other polysaccharide-protein conjugate vaccines but also protein vaccines. I have addressed this issue in more detail in section 24.4.6 above.
811 Thirdly, the “problem” is not one of silicone oil induced aggregation, as submitted by Wyeth, but to formulate a suitably stable vaccine. In this regard I accept, on the balance of probabilities, that the skilled formulator is likely to test the formulations in siliconised containers (see section 24.4.2) and that stability studies would involve the detection of aggregation. Wyeth in its written closing submissions at various points suggested that the skilled formulator would not detect instability, and that part of the invention involved the discovery, for the first time, of silicone oil induced aggregation in conjugate vaccine formulations, although the latter position was correctly eschewed in oral closing submissions. I have found above that silicone oil induced aggregation was a known problem (see section 24.4.7). But perhaps more relevantly, instability in the form of aggregation generally was a familiar difficulty encountered in formulation. It was looked for, and expected. There may have been different reasons for aggregation, but the skilled formulator, whose task it was to produce a stable and immunogenic formulation, would have looked to see if there was a practical solution to the problem. Inventive step does not lie in a discovery, but a particular means by which a discovery is harnessed; an idea of itself cannot be the subject of a patent, it must end in a manner of new manufacture: Lockwood No 2 at [59] – [60]. The final position of Wyeth, as I understand it, is that the invention lay in the identification of the problem of silicone oil induced aggregation and then resolving that by way of the claimed formulation. In this regard, the repeated submission that the skilled formulator would have been surprised to find that the formulation tested in example 1 of the container patent precipitated so quickly goes nowhere. Inventive step, if there be one, lies in the steps taken from that point. It follows that I reject the submission advanced by Wyeth that the skilled formulator is unlikely to have encountered precipitation of the kind observed in the experiment of example 1 in the container patent. As I have noted, the role of the Court is to consider what the notional skilled addressee would have done faced with a similar problem to which the patentee claims to have solved. As a matter of fact, I find, in any event, that the problem of visible precipitation would have been discovered by the skilled formulator.
812 There was some difference between the experts in the approach to the formulation task, which I have considered in section 24.4.1 above. I am satisfied that the skilled formulator prior to April 2006 would have, in formulating a 13-valent polysaccharide-protein conjugate vaccine using Prevnar 7 as a starting point, first characterised the suitable pH for the formulation, having regard to the fact that the addition of serotypes conjugated to CRM197 was likely to have an effect on its acidity. I am also satisfied that a range of pH levels would have been tested in parallel, as modified by a buffer, together with the other contents of Prevnar 7, being the aluminium phosphate adjuvant and saline.
813 I have noted that instability, including aggregation, was expected by the skilled formulator, and looked for. It was one of the two most likely irritants with which a formulator had to deal during development. As I have found in section 24.4.6, aggregation was understood by the skilled formulator often to be specifically protein aggregation. The structure of a protein is influenced by the arrangement of its hydrophobic and hydrophilic regions. The interaction of the hydrophobic regions in the carrier protein in a polysaccharide-protein conjugate was known to be a likely major contributor to aggregation, in the same way that it was known that in protein vaccines the proteins have a tendency to aggregate. Indeed the process of conjugation was known to tend to open up the structure of the protein and to enhance the prospect of aggregation. Furthermore, the addition of further serotypes increases the concentration of protein and adds to that propensity.
814 One known factor affecting the conformation of proteins (that is, their three-dimensional arrangement) is pH. The addition of a buffer was well known in the art to regulate the pH of a vaccine in order to optimise its stability. By adjusting the pH, the conformation of a protein can be regulated to avoid changes in acidity or alkalinity that would promote aggregation. I am persuaded that the skilled vaccine formulator would have in the early stages of formulation determined the optimal pH range for the antigen of interest. In the event of encountering aggregation, the addition of a suitable buffer would have as a matter of course been tried, with the expectation that it would assist in retaining the conformation of the protein and thereby avoid or minimise aggregation. The buffer would have been used within the range prescribed in claim 1, which is very broad and encompasses physiologically acceptable amounts. In this regard, there was no dispute in the evidence that buffers were known and used in other vaccine formulations before April 2006.
815 The invention claimed in claim 1 is limited to the inclusion, in combination, of a siliconised container means filled with a formulation comprising a pH buffered saline solution with a pKa of about 3.5 to about 7.5, an aluminium salt, and one or more polysaccharide-protein conjugates, where the conjugate comprises one or more pneumococcal polysaccharides. The Prevnar 7 formulation included all but the buffer. The inclusion of aluminium salt adjuvant was known to enhance the immunogenicity of the polysaccharide-protein conjugates (see section 24.4.9 above) and also, by means of its mechanism of action, to provide a stabilising effect. In my view, the hypothetical skilled but uninventive formulator would have been directly led as a matter of course to try the addition of a buffer in the expectation it may well have produced a useful result. The evidence establishes convincingly that there is no barrier crossed or hurdle overcome for the skilled formulator to have conceived of and implemented the addition of a buffer to this formulation to achieve a suitable pH for the Prevnar 7 formulation where additional serotypes are added.
816 I reject the submission advanced by Wyeth that the root cause of any aggregation, whether it be caused by silicone oil or otherwise, would first need to have been determined by the uninventive but skilled formulator prior to continuing with the formulation. To the extent that a correlation was drawn between aggregation and the presence of silicone oil in the container used, the person skilled in the art would have understood that the mechanism of action may have been because of an unfolding of the protein in the presence of the silicone. However, the precise reasons why would not have been of concern. Aggregation was a common problem. The modification of pH levels and the addition of surfactants were familiar solutions in the field.
817 In this regard I am not persuaded by the evidence of Professor Dalby, who, as I have noted, was not closely representative of the hypothetical uninventive but skilled formulator. I do not accept that the hypothetical skilled formulator would have paused work upon discovering aggregation and then undertaken a research project to determine its cause. I prefer the approach explained by Professor Petrovsky, to the effect that if a stability problem was encountered, the formulator would have looked to bring his or her experience to bear to address the problem. In this regard, it is perhaps telling that Professor Dalby accepted in his oral evidence that if any instability was identified in testing a pneumococcal conjugate vaccine with serotypes added to Prevnar 7, “one of the things you might start to look at is alternative pHs along with other variables, in terms of excipients”.
818 As an alternative tool of analysis, one may consider the two aspects of the modified Cripps question. In relation to the invention claimed in claim 1, the first aspect is: in the light of the common general knowledge, would the person skilled in the art at the priority date be directly led as a matter of course to try adding a buffer (in the range of about pKa 3.5 to 7.5) to the combination of a siliconised container, adjuvant and 13 polysaccharide-protein conjugates, in the event that a stability issue was encountered? The second aspect is: would that person have done so in the expectation that the addition of the buffer might well be useful in producing a stable and effective vaccine? Having regard to the matters to which I have referred, the answer to each is affirmative.
819 In relation to claim 9, I have discussed in section 24.4.10 above the use of surfactants. The mechanism by which surfactants worked in protein and polysaccharide-protein conjugate vaccines was known to those skilled in the field. They are a general class of detergents that assist with the stability of proteins in formulation, particularly for hydrophobic proteins that are prone to aggregation. They were known to be used alone or in conjunction with other excipients (such as buffers and adjuvants) to prevent or reduce aggregation. They were known to work by inserting themselves between solid-liquid, liquid-liquid or air-liquid interfaces and in so doing preventing proteins from interacting with such interfaces, such as container walls, or the surface domains of other proteins. The Tween surfactants were well known to be useful, having regard to different effects. Tween 80 is the first surfactant that Professor Petrovsky would have thought of using.
820 In my view the hypothetical skilled but uninventive formulator would have been directly led as a matter of course to try, in the event that he or she had encountered a stability issue with the formulation such as visible aggregation, a buffer and a surfactant to address the problem, with an expectation that they would be successful in addressing the problem. I have no doubt that they would have been tried separately and together. That is because they both have independent actions, but both could be expected to deal with problems of aggregation.
821 In relation to the invention claimed in claim 9, the first aspect of the modified Cripps question is: in the light of the common general knowledge would the person skilled in the art at the priority date have been directly led as a matter of course to add a buffer (within the required pKa range) and a surfactant (within the surfactants identified in the claim) to the combination of a siliconised container, adjuvant and 13 polysaccharide-protein conjugates, in the event that a stability issue was encountered? Secondly, would that person have done so in the expectation that the addition of each might well be useful in producing a stable and effective vaccine? Again, having regard to the matters to which I have referred, the answer to each is affirmative.
822 It is true that a degree of experimentation would have been required. But the question of obviousness is not to be answered on the basis only of whether or not an outcome can be predicted at the outset. The fact that standard stability testing is conducted to ensure that a formulation under development is stable does not yield a conclusion that there is an inventive step. Trial and error are normal, everyday parts of laboratory work and non-inventive laboratory experiments: “[t]hat is what the hypothetical skilled worker in a laboratory does – if the outcomes of experiments were known, there would be little point in doing them. That is the nature of everyday, non-inventive, research”: Apotex Pty Ltd v Sanofi-Aventis [2009] FCAFC 134; 82 IPR 416 at [177] (Bennett and Middleton JJ), and to like effect AstraZeneca (FC) at [547] (Jessup J). The trial and error required to adjust the pH of the formulation to ensure that it is stable is, in my view, utterly mundane for a person with the education and experience of the hypothetical skilled formulator. As the Full Court noted in Mylan at [502]:
The reformulated Cripps question does not require certainty of outcome. It requires that the skilled addressee be directly led as a matter of course to try the claimed invention in the expectation that a particular research path “might well produce” a useful result (Hässle v Alphapharm at [53]). It does not require the skilled addressee to know that the steps will produce a useful result.
823 Wyeth submits that of the nine commercial protein vaccines that use a surfactant together with an aluminium adjuvant to which Professor Petrovsky refers, none also contain a buffer. To the extent that this submission suggests that inventiveness lay in the use of a buffer, together with a surfactant and an adjuvant in combination, I reject it. It is quite apparent that the skilled vaccine formulator was familiar with the modes of action of each of these known excipients and it was well within his or her skill to use them in combination, expecting that they would have combined to produce a stable and immunogenic formulation.
824 Wyeth also submits that a conclusion of obviousness would be flawed as a matter of law, citing SNF (Australia) Pty Limited v BASF Australia Ltd [2019] FCA 425; 140 IPR 276 at [510] and [513] (Beach J). The first passage cited states: “...it is erroneous to characterise as obvious the variation of all parameters or the trying of all choices until one proves successful, where the prior art did not point to it”. However, in my view the prior art, and the common general knowledge understanding of the science, did point to it, in the manner that I have described. The decision to add a buffer was based on the understanding of the skilled formulator of the underlying science. The same may be said of the use of a surfactant. That understanding not only gives the formulator an appreciation for the likely causes of aggregation, but also the likely means by which it can be addressed in the context of the formulation as a whole.
825 The second passage states: “[i]t is simply impermissible to take any one integer or take each integer seriatim and ask whether each integer involved an inventive step”. That statement is, of course, axiomatic. It is the whole combination that must be considered, and the question is not whether each integer is obvious, but whether the combination as claimed is obvious. However, by considering the starting point of Prevnar 7, the addition of serotypes to that combination, and the consideration of the responses to any stability issues identified, one is able to navigate that problem and arrive not at each separate integer, but the combination claimed.
826 Wyeth also relies on the evidence of Professor Dalby that there was a number of alternative approaches that could have been taken to formulating a 13-valent conjugate vaccine using Prevnar 7 as a starting point. One may have been to cease using siliconised containers, or to reduce the amount of silicone in them. Another may have been to use different types of excipients, or successively remove excipients to determine the cause of the precipitation described in the container patent.
827 However, the case advanced by MSD focusses in the first instance on steps that include the addition of a buffer and a surfactant to the Prevnar 7 formulation. Other routes may have been more or less obvious than that; indeed, two or more routes to the claimed invention may be obvious. The question is not whether it would have been obvious to choose one option over another. Section 7(2) does not contemplate that the person skilled in the art must choose between apparently effective solutions: AstraZeneca at [115]. In Nichia Corporation v Arrow Electronics Australia Pty Ltd (No 4) [2017] FCA 864 (Nichia (Yates J)) Yates J said at [275] – [276]:
[275] The respondent criticised the applicant’s case as presenting the false image of a bewildering range of choices for the person skilled in the art. Whilst I do not accept that the person skilled in the art would have been likely to pursue all the options presented in the applicant’s evidence (for example, the use of defect emission bands, as discussed by Dr Butcher), I am satisfied that the person skilled in the art would have had available the broad options I have described above and that these would have presented the person skilled in the art with a number of realistic choices to pursue that might yield effective solutions.
[276] However, the question of obviousness is not directly concerned with choices of this kind. The question is whether the pathway chosen by the inventor is one which the person skilled in the art, equipped with the common general knowledge before the priority date, would have been directly led as a matter of course to try. If obvious, a solution does not become less obvious simply because the person skilled in the art might have been presented with other possibly effective solutions of the kind I have described: see, for example, the related comments made by Gageler and Keane JJ in AstraZeneca AB v Apotex Pty Ltd (2015) 323 ALR 605; [2015] HCA 30 at [115]. Still less will that be the case where these other possibly effective solutions are said to be more obvious.
828 The Full Court in Nichia at [93] accepted the statements at [276] as orthodox. In the present case, in my view the addition of a buffer with or without a surfactant to the formulation of a 13-valent pneumococcal conjugate vaccine, when using the Prevnar 7 formulation as a starting point, was one that the person skilled in the art, equipped with the common general knowledge before the priority date, would have been directly led as a matter of course to try with the expectation that it might well produce a useful result.
829 Finally, I refer to three points made by Wyeth in its submissions. First, it submits that protein to protein aggregation is now “known” not to be the mechanism that causes silicone oil induced aggregation. That submission is based on [138] to [142] of Professor Dalby’s affidavit. A relevance objection was taken to these paragraphs, and I deferred ruling on it until now. The evidence refers to Saller. The article itself was not put into evidence.
830 In [138] – [142] Professor Dalby gives evidence that the article “suggests” a mechanism causing silicone oil induced aggregation which is not protein-protein aggregation but has to do with the way proteins interact with small silicone oil droplets. He considers this theory is “plausible” and is better than the theory proposed by Professor Petrovsky. Accordingly, based on Professor Dalby’s own characterisation of the article, it rises no higher than postulating a theory of action. In my view this evidence is of marginal, if any relevance. Having regard to the date of publication, the article plainly does not have a bearing on the state of common general knowledge before April 2006. Furthermore, Professor Dalby’s evidence provides no basis upon which it might be said that the approach of the skilled formulator that I have described would fail, or be expected to fail, to yield a successful outcome.
831 Secondly, Wyeth submits that the views of Professor Petrovsky were tainted by hindsight, and in particular his contention that Tween 80 was an obvious choice of surfactant. The danger of a hindsight analysis must certainly be taken into account, and I have done so in considering the weight to give to Professor Petrovsky’s evidence, including by having regard to the evidence of Professor Dalby and also Dr Jones. I accept that the witnesses were, by the time that they gave their evidence, familiar with the container patent and the solution that it proposed. However, Professor Petrovsky was able to support his own recollection of pre-April 2006 knowledge by reference to publications, examples and scientific knowledge well prior to that date. This provides substantial corroboration, and ‘carbon dating’ for his views. The ultimate analysis does not depend on the evidence of one expert, and in these reasons Professor Petrovsky is not adopted as an avatar for the skilled formulator. His evidence contributes to the conclusions, but has not been determinative of them. Furthermore, my conclusions are guided not by conclusory statements by the experts of what steps they would have taken, but their explanations as to why, as a matter of logic, they would have taken them. I am satisfied that in so doing the insidious influence of hindsight has been avoided.
832 Thirdly, Wyeth points out that MSD in its own patent, filed in 2011 after the priority date of the container patent and entitled “15-valent pneumococcal polysaccharide-protein conjugate vaccine composition”, did not include a surfactant in the patent’s combination of components which did amongst other things include 15 serotypes conjugated to CRM197, an adjuvant and a buffer. Yet, MSD’s 15-valent vaccine the subject of Wyeth’s infringement claim includes a surfactant. From this, Wyeth submits, the Court should infer that the claim 9 combination is valuable and important. However, in my view this cuts both ways, and is remote from the legal test to be applied. The absence of a claim to a combination for the surfactant may be understood to reflect a view that there is nothing inventive in the addition of a surfactant. Indeed, the independent claim in MSD’s patent is confined to a particular multivalent composition with a pharmaceutically acceptable carrier. A better inference may well be that beyond the identification of the particular polysaccharide-protein conjugate in the independent claim, no feature of the carrier confers inventiveness.
833 For these reasons I consider that the invention claimed in claim 1 does not involve an inventive step.
834 No independent inventive step is asserted by Wyeth in claims 2 – 8 and they too lack an inventive step.
835 For the reasons given above, I also consider that claim 9 also lacks an inventive step.
836 I do not consider that claim 18 has any independent inventive step, when considered together with the combination of claim 1 or claim 9. That claim adds to prior claims the 13 chosen serotypes. The analysis above takes into account the evidence of the experts where they considered the formulation of a 13-valent pneumococcal conjugate vaccine. Whilst that evidence proceeded on the basis that the formulation task used Prevnar 7 as a starting point for adding additional serotypes, there is no suggestion in the expert evidence that the particular identity or composition of the 13 serotypes to be formulated was material to the task. The challenges that faced the skilled formulator arose from the stability issues to which I have referred, and were not specific to particular serotypes or a particular group of serotypes. The container patent does not suggest otherwise. Accordingly, in my view the combination of claim 18, whether dependent on claim 1 or claim 9, is no less obvious than those claims.
837 If I am wrong about that, then there is a further basis upon which I consider that that claim 18 lacks an inventive step. I have found that the skilled formulator would have collaborated with an immunologist or clinician familiar with the active ingredients in developing the formulation of a pneumococcal conjugate vaccine. Indeed the evidence indicates that Professor Paton worked with Professor Petrovsky before the priority date in developing pneumococcal vaccines using inulin. In section 8.1 above I have found that all but serotypes 6A and 19A of the 13 chosen serotypes were well understood by immunologists in the field to be the likely candidates for development of a new vaccine. Indeed the common general knowledge was that they were the serotypes used in the GSK, Aventis and Wyeth 11-valent formulations under development. I have also found in section 9.4 that the idea of adding serotypes 6A and 19A to the 11-valent vaccine serotypes was an obvious choice, having regard to the common general knowledge. For these reasons, I am further satisfied that the selection of the 13 chosen serotypes included within claim 18 lacks an inventive step.
26. CONTAINER PATENT: LACK OF INVENTIVE STEP IN LIGHT OF THE S 7(3) PRIOR ART INFORMATION
838 In AstraZeneca (HC) Kiefel J said at [68]:
[68] Before a document containing prior art information can be used along with the common general knowledge for the purposes of the s 7(2) inquiry, it is necessary that it meet the requirements of s 7(3). In Lockwood Security Products Pty Ltd v Doric Products Pty Ltd [No 2] it was explained that prior art information which is publicly available in a single document is “ascertained” if it is discovered or found out, and “understood” means that, having discovered the information, the skilled person would have comprehended it or appreciated its meaning or import. The Court also explained that the phrase “relevant to work in the relevant art” is directed to publicly available information, not part of the common general knowledge, which the skilled person could be expected to have regarded as relevant to solving a particular problem, or meeting a long-felt want or need, as the patentee claims to have done.
[69] Lockwood [No 2] also explains that, in answering the question of obviousness, the information referred to in s 7(3), like that part of the prior art base which is the common general knowledge, is considered for a particular purpose. That purpose is to look forward from the prior art base to see what the skilled person is likely to have done when faced with a problem similar to that which the patentee claims to have solved with the claimed invention. It is this aspect of the s 7(2) inquiry which assumes particular importance on these appeals.
[70] In addressing s 7(2), it is to be borne in mind that the skilled person is an artificial construct, intended as an aid to the courts in addressing the hypothetical question of whether a person, with the same knowledge in the field and aware of the problem to which the patent was directed, would be led directly to the claimed invention. The statute’s creation of the skilled person construct for this purpose is not to be taken as an invitation to deal with the question posed by s 7(2) entirely in the abstract. Whilst the question remains one for the courts to determine, the courts do so by reference to the available evidence including that of persons who might be representative of the skilled person.
(footnotes omitted)
839 In its closing submissions MSD relies on six separate items of prior art information: the Chiron patent: Kanra; Katkocin; ISPPD abstracts; Hausdorff 2000; and Hausdorff 2002.
840 Wyeth contends that MSD has not established that the Chiron patent would have been ascertained or regarded as relevant within s 7(3). It submits that the Chiron patent does not address the problem of silicone oil induced aggregation. However, as I have noted, the problem posed to the person skilled in the art was not the resolution of a detected problem arising from silicone oil induced aggregation, but whether a stable formulation could be achieved. The search under s 7(3) is necessarily broader than that characterised by Wyeth.
841 Wyeth also submits that MSD has failed to prove by evidence that the Chiron patent would have been ascertained or regarded as relevant.
842 In this regard the evidence indicates that Chiron was known to be working in the field of vaccines before April 2006. The Hib vaccines became available in Australia in the early 1990s and were produced by Chiron, as well as other companies. CRM197 was later used in Hib conjugate vaccines. Chiron was known as a research institute as well as a formulator and as a world leader in conjugate vaccine formulation. Each of Professor Petrovsky and Professor Dalby was aware of the work of Chiron in the field.
843 If the skilled formulator were embarking on a project to formulate a 13-valent pneumococcal conjugate vaccine using the Prevnar 7 formulation as a starting point, he or she would have commenced by conducting a literature review, including a search of patent databases. Professor Petrovsky gives evidence, which I accept, that he would have searched for terms including “vaccine”, “adjuvant”, “conjugate”, “polysaccharide” and “stability”. He would have actively searched for publications by Chiron, having regard to their reputation in the field. Professor Dalby says in his written evidence that he would not have actively searched for publications by Chiron, not because of any view that they may not have been of assistance, but because they were working in other areas as well as vaccine formulations and so the search may have brought up many irrelevant documents. However, in his oral evidence he accepted that if he had been looking to create a vaccine formulation with a stable pH, he would have been looking for documents from Chiron and would have used the word “vaccine” as a search term and would have found the Chiron patent.
844 The fact that the search may have turned up multiple documents, some of which were more relevant than others, is not the question. The question is whether the Chiron patent would have been ascertained within s 7(3). Professor Dalby’s reservation that too many documents originating from Chiron may have been found is beside the point, and may be set to one side.
845 Having regard to the evidence, I consider it likely that the skilled formulator would have searched the relevant patent databases, and would have found the Chiron patent. It is entitled (emphasis added) “Vaccines Comprising Aluminium Adjuvants and Histidine”. The person skilled in the art knew that Prevnar 7 contained an aluminium adjuvant and that Chiron was active in the field. I do not accept the submission advanced by Wyeth, as amplified in the evidence of Professor Dalby, that the only search conducted would be by reference to silicone oil induced aggregation. The skilled formulator knew before the priority date that it was necessary to attempt to obtain a stable formulation and would as a matter of course have considered formulations developed by others in the field, without confining the direction of the task to the particular problem of silicone oil induced aggregation.
846 It is correct to say, as Wyeth points out, that there is no evidence of online searches conducted to demonstrate that the Chiron patent would have been included in the results. Nevertheless, having regard to the size and scope of the notional task, and the expertise of the hypothetical formulator, I am satisfied on the basis of the evidence that the Chiron patent would have been located, not least having regard to its title, and the high likelihood, which I find to be established, that the notional skilled formulator would have been interested to know of the work done by Chiron in relation to vaccines. In this regard I note the analysis of the requirements of s 7(3) set out by Yates J in Otsuka Pharmaceutical Co., Ltd v Generic Health Pty Ltd (No 4) [2015] FCA 634; 113 IPR 191 at [399] – [418]. The absence of evidence concerning the nature of any hypothetical literature search is not fatal to the correct application of s 7(3) (at [414]). In each case, whether or not the person skilled in the art could reasonably expected to have ascertained, understood and regarded as relevant a particular item of prior art information is to be determined on the evidence as a question of fact (at [399]). Patent databases were readily searchable before April 2006, and it is apparent from the face of the Chiron patent that its international publication date was 6 February 2003. Its inclusion within international patent databases available for searching before the priority date may be accepted.
847 I now turn to consider the disclosure of the Chiron patent. It is summarised in section 23.2 above.
848 The evidence of Professor Petrovsky is that the Chiron patent discloses formulations that have been found to work. Necessarily the formulations described are stable, and included within that concept is that there is no unacceptable aggregation. In my view the language of the Chiron patent supports this view. The disclosure is not, as Professor Dalby contends, confined to pH stability. It expressly concerns the preparation of vaccine formulations for use as medicaments, which must be stable, and I consider that the skilled addressee would have reached that conclusion. One aspect of the disclosure is directed to pH stability, but as a whole it reports the results of experimental work and conclusions drawn in respect of a broad range of formulations suitable for administration to patients. Within that disclosure, as one of three preferred options, is the treatment of pneumococcus.
849 Whilst various alternatives are identified in the body of the specification, the claims make explicit the combinations that the authors of the document considered to be useful for preparing effective formulations for the use in the treatment of patients.
850 I consider that the skilled formulator, possessed with the common general knowledge and engaged in the task of formulating a 13-valent conjugate vaccine using Prevnar 7 as a starting point, would have found relevant and useful the Chiron patent’s disclosure of a stable formulation to be used as a medicament, in circumstances where:
(1) there is a preference for the use of a polysaccharide conjugated to a carrier protein, where CRM197 is “particularly preferred”;
(2) there is a combination of that conjugate with an adjuvant, a buffer (histidine) and a surfactant (for example a Tween, such as Tween 80);
(3) there is a description that a buffer improves the adsorption of conjugates to the adjuvant;
(4) a buffer enhances the pH stability of the composition within a preferable range of between 6 and 7;
(5) the composition may also include a surfactant in order to minimise adsorption of antigens to the containers; and
(6) examples 7, 8 and 9 formulations include polysaccharide-protein conjugates with an aluminium adjuvant, pH buffered saline solution and surfactant (Tween 80).
851 I consider that the skilled formulator would regard this disclosure as relevant within s 7(3).
852 If I am incorrect in my conclusion that claims 1 and 9 are invalid for lack of inventive step based on the common general knowledge alone, in my view the disclosure of the Chiron patent provides a basis whereby it may be concluded that those claims are nonetheless obvious. The skilled formulator would have been likely to use siliconised containers in his or her formulation efforts. That decision would have, upon reading Chiron, led directly to trying the use of a histidine buffer in conjunction with an adjuvant, reinforced by the understanding that it was preferable to keep the pH of the acidic polysaccharide-protein conjugate to a pH range of between 6 to 7. The disclosure of Chiron patent provides a further basis upon which the skilled formulator would expect such a combination to produce a stable formulation. The Chiron patent demonstrates that concerns (as expressed by Professor Dalby in his evidence) of unknown interactions between the buffer, when added, or the surfactant, when added, to the other ingredients in the Prevnar 7 formulation with additional serotypes would not have been likely to eventuate. If they had eventuated, they could have been addressed by no more than routine adjustments.
853 The summary of Kanra states that the primary aim of the study was to assess the tolerability and immunogenicity of a new Haemophilus influenzae type b (Hib)/AIPO4 (Chiron, SpA) vaccine in two-month-old healthy infants. It concludes that the new Hib vaccine “is safe and well tolerated, and induced a good PRP antibody response...”.
854 The article relevantly states:
Some years ago, Chiron Vaccines in Siena, Italy developed a HbOC-like vaccine (VaxemHib) consisting of oligomers of capsular PRP of Haemophilus influenzae type b conjugated to CRM197.
The vaccine is presented in two vials for mixing before injection: one containing the Hib conjugate to CRM197 protein and the other aluminium hydroxide as adjuvant. In view of the inconvenience of mixing the two vials before injection, an improved formulation has been recently developed at Chiron Vaccines in Siena. The adjuvant has been replaced by aluminium phosphate which permits formulation of the vaccine in a stable solution presented in a ready-to-use vial.
As preclinical studies on stability and immunogenicity showed promising results, an expanded pilot study has been carried out.
We present here results of a clinical pilot study to assess the safety of the new Hib formulation in two-month-old healthy infants, and to obtain preliminary data on immunogenicity.
855 The vaccine used in the trial is described under the heading “Materials and Methods” as follows:
The Hib/AIPO4 vaccine under investigation (Chiron Vaccines SpA, Siena Italy, lot number N32P18H1) contained 10 μg CRM197. Hib conjugate as oligosaccharide, 0.3mg aluminium phosphate, 0.01% thimerosal and 0.005% polysorbate (Tween) 80 and qs 0.5 ml phosphate buffered saline. It was provided as a single dose pre-filled syringe and was administered as an intramuscular injection in the left thigh.
856 Under the heading “Discussion” the article says:
...The most commonly used adjuvants for human vaccines are aluminium hydroxide, aluminium phosphate and calcium phosphate. In animals it has been shown that aluminium phosphate may be a more potent adjuvant than aluminium hydroxide for several antigen [sic]. In addition, the aluminium hydroxide may lead to catalytic depolymerization of PRP. The substitution of the aluminium hydroxide adjuvant has also led to a more convenient presentation of the vaccine which can be formulated in a single container.
857 Kanra describes a formulation of an oligosaccharide conjugate vaccine produced by Chiron Vaccines that was used in the trial to which the article refers. It was a Hib conjugate, and not a pneumococcal protein conjugate. The paper describes a previous formulation, in which the conjugate and the aluminium hydroxide adjuvant were in separate containers, as an “inconvenience”, and so the improved formulation incorporated the adjuvant together with the conjugate. It does not disclose one way or another whether its formulation was contained in a siliconised container. Professor Petrovsky assumes that it was, but could not be certain.
858 Professor Petrovsky understands the movement from the two vial approach to the single vial approach to reflect an improvement made to the formulation to overcome an unstated stability issue arising from combining the ingredients in the separated vials. The addition of aluminium phosphate in place of aluminium hydroxide in the single vial approach permitted the formulation of the vaccine in a stable solution presented in a ready-to-use vial. That is an inference that was available to the skilled formulator. The apparent purpose of the article was to consider the effectiveness of the changed, single vial formulation. Whilst Professor Petrovsky was challenged on his view in cross-examination, in my view it is a sensible construction of the disclosure of the purpose of the article. Contrary to the submission advanced by Wyeth, no credit point arises. Professor Petrovsky correctly infers that the resulting formulation was stable.
859 The new single vial formulation is described to include the Hib oligosaccharide conjugate, aluminium phosphate, thimerosal, a Tween 80 surfactant and phosphate buffered saline.
860 In his oral evidence Professor Dalby considered that the presence of thimerosal, an antimicrobial agent, may have had a relevant effect, such that its addition may have potentially had an impact on the other excipients, and that it was “possible” that it may have affected the stability of the composition. Professor Petrovsky gave evidence that the addition of an antimicrobial agent is standard for any multi-dose vaccine, because once the rubber stopper on the vial is punctured, sterility issues may arise. Many vaccine formulations are prepared without thimerosal for single doses, and with it for multiple doses. Professor Jones agreed that it was a standard inclusion in multi-dose vials. They both considered that there would have been no expectation that the addition of the antimicrobial agent would have an effect on the stability and efficacy of the vaccine. An attempt to challenge the views of Professor Petrovsky and Dr Jones by reference to the Remington text failed. The passage relied upon, which was said by Wyeth to indicate that stability concerns arose upon the addition of an antimicrobial agent, was accepted by all the experts to be directed at the effectiveness of an agent in performing its role, rather than suggesting that the addition of an antimicrobial agent may adversely affect the efficacy of the composition. Although its inclusion would have to have been tested, it was no more than routine.
861 In my view, the initial concern expressed by Professor Dalby about thimerosal reflected his lack of experience in the formulation of vaccines and demonstrated his more theoretical, rather than practical, approach. To the extent that the experts ended up in disagreement on this point, I prefer the evidence of Professor Petrovsky and Dr Jones.
862 Having said that, I am not satisfied that Kanra would have been ascertained by the hypothetical formulator within s 7(3), even though, for the reasons that I have stated, I consider that it would have been understood and regarded as relevant. That is because there is no evidence that The Turkish Journal of Pediatrics was a searchable journal as of April 2006. Professor Petrovsky was simply provided with the publication by the solicitors for MSD.
863 Accordingly, it is not available for consideration in the context of s 7(2). Were I to be incorrect on this point, I would consider that the publication would separately buttress the fact that the hypothetical formulator skilled in the art would have had an expectation that the combinations contained in claim 1 and claim 9 would be achieved.
864 The Katkocin publication concerns the Prevnar 7 formulation, and was published by a person working in Wyeth-Lederle Vaccines in the USA. There is no dispute that a person skilled in the art would have ascertained, understood and regarded it as relevant within s 7(3) of the pre-RTB Patents Act. There is little dispute as to its disclosure.
865 Katkocin states that formulation and characterization during the scale-up of the new Prevnar 7 vaccine was successfully tested in infants and received CDC and FDA endorsement. Under the heading “Results”, the author identifies that the kinetics of binding pneumococcal conjugates to aluminium phosphate were determined in two different formulations, one with a phosphate buffer to test whether phosphate can modify adsorption, and the second without a buffer. The results are given in Table 3, by reference to the percentage binding of the polysaccharide-protein conjugate of serotype 9V in the 7-valent conjugate vaccine. Those results show that after 7 days, the adsorption for the composition including the phosphate buffer was 87%, and without the buffer was 94%. The article states that long-term binding studies (through 36 months) also show no change in adsorption. The results for the 9V serotype are considered to be representative of results generated for the other six serotypes.
866 Professor Petrovsky considers that the results serve to confirm his view that the addition of a buffer to a proposed composition of a polysaccharide-protein conjugate vaccine is no more than a standard process. The results for compositions with and without a buffer were both within regulatory requirements (which require adsorption of over 85%), both over the short term and long term. Professor Dalby does not disagree, but notes that he would agree with the approach of the formulators of Prevnar 7 and elect not to add a buffer, because he would not add in something that was not necessary to add. He agrees with Professor Petrovsky that there is no reason based on stability not to choose either of the two formulations considered in the article.
867 Wyeth submits that the effect of Katkocin is to teach away from the use of a buffer, because of the difference between the 87% and 94% levels of adsorption. However, the fact that both were within the acceptable regulatory range would, in my view, have taught the skilled formulator that a buffer was available for use, should it be necessary to add one for its normal purposes (see section 24.4.8), without a relevantly adverse impact on stability or immunogenicity.
868 In the context of the s 7(2) enquiry, in my view the disclosure of Katkocin would have taught the skilled formulator that the addition of a buffer to the Prevnar 7 formulation was unlikely to result in any adverse stability effects. Having regard to the common general knowledge of the usefulness of buffers, it teaches that a buffer may well be used to regulate pH and have an effect on adsorption, a matter that I have found forms part of the common general knowledge in any event.
26.4 Hausdorff 2000 and Hausdorff 2002
869 I have reviewed the Hausdorff disclosures in section 10.3 above. They concern the identification of the 13 chosen serotypes and are accordingly relevant only to the claim 18 inventive step analysis. To the extent that I am wrong about the selection of serotypes 6A and 19A being obvious in light of the common general knowledge alone, the disclosure of these articles confirms that there is no inventive step in the selection of the 13 chosen serotypes and no independent inventive step in the combination of claim 18 beyond the combinations of claims 1 and 9.
870 In his third affidavit, affirmed on 17 October 2018, Professor Paton gives evidence that he was supplied by Corrs with the ISPPD abstracts. The 5th International Symposium on Pneumococci and Pneumococcal Disease was held shortly before the 26 April 2006 priority date of the container patent. He gives no evidence of having read those abstracts at the time.
871 MSD faintly relies on these documents for the purposes of s 7(3) of the pre-RTB Patents Act. It provides no evidence as to how the skilled formulator would have ascertained them, or whether they could be obtained in a search. Nor does the evidence suggest that a person in the position of the skilled formulator would have attended the symposium. I am not satisfied that the requirements of the term “ascertained” in s 7(3) have been met.
27. CONTAINER PATENT: MANNER OF MANUFACTURE
872 MSD pleads that the asserted container patent claims are not to a manner of new manufacture within the meaning of s 6 of the Statute of Monopolies for the purposes of s 18(1) of the Patents Act for two reasons. First, that it is apparent on the face of the specification that the invention does not satisfy the threshold for patentability set out in Philips (Philips ground). Secondly, that the asserted claims are to nothing but a known container including known components, the known properties of which made them suitable for inclusion in vaccine formulations, within Microcell (Microcell ground). For both grounds in closing submissions MSD relies on identified passages in the specification, which are said to contain the following admissions:
(1) Improving the stability of an immunogenic composition was a known goal (page 1), and it was also known to be desirable to inhibit precipitation/aggregation of immunogenic compositions (page 2 lines 12 – 17).
(2) The use of polysaccharide-protein conjugates in vaccines was known (page 1 line 32 – page 2 line 11).
(3) The use of pH buffered saline was known. Factors including pH and storage buffer were known to influence stability (page 2 lines 7 – 11). The specification does not suggest that the addition of a buffer had any unexpected or surprising result.
(4) The use of aluminium adjuvants in vaccine formulations was known (e.g. page 11 lines 13 – 14, page 36 lines 10 – 13);
(5) The use of surfactants was known, they were commercially available (page 26 line 26), their properties were known (page 17 lines 12 – 15), and it was known how to determine a suitable surfactant (page 17 line 32 – page 18 line 2).
(6) Siliconised containers were known (page 2 line 24 – page 3 line 7).
873 Furthermore, MSD submits that for all the asserted container patent claims but claim 9, there is nothing in the specification that puts forward a discovery, citing Wrigley. MSD emphasises that the claims are not limited to formulations that reduce silicone oil induced aggregation, and submits that by incorporating Jones 2005 into the specification, Wyeth has also admitted on the face of the specification that by changing the pH of the composition, one can control silicone oil induced aggregation. In relation to claim 9, the specification asserts “unexpected and surprising results” of the use of a formulation with surfactant such as Tween 80, but MSD submits that claim 9 is still, at most, to an invention that realises advantages inherent in an existing substance and practice, citing Arrow Pharmaceuticals.
874 Wyeth resists both grounds. It submits that the first ground is not available as a matter of law, repeating its submissions to which I have referred (and rejected) in section 17.2. In relation to both, it contends that the disclosures on the face of the specification relied upon by MSD do not support the conclusion for four reasons. First, the statement on page 42 of the container patent makes plain that none of the references in the specification may constitute an admission as to what was known. Secondly, most of the references relied upon by MSD form part of the disclosure as to how to work the claimed invention. The reliance on these by MSD forms a hindsight analysis, and they cannot be characterised as admissions as to what was known having regard to the fact that the invention as claimed is to a combination. Third, Wyeth submits that the container patent is directed to the discovery of the problem of silicone oil induced aggregation when pneumococcal polysaccharide-protein conjugates were stored in siliconised containers, and to providing a solution to that problem. MSD is unable to point to any admissions on the face of the specification where the materials are being used “for the purpose [here, to solve the problem of silicone oil induced aggregation when polysaccharide-protein conjugates were stored in siliconised containers] of which [their] known properties make that material suitable”.
875 Finally, Wyeth contends that the submission advanced by MSD that nothing in the asserted claims is put forward as a discovery is irrelevant, citing Nichia (Yates J) at [392] to the effect that there is no legal requirement for the specification to make any positive affirmation of inventiveness.
876 I have addressed manner of manufacture in the context of the composition patent in section 17 above. For this ground to be made out, it must be established that the lack of inventiveness was admitted on the face of the specification: Lockwood No 2 at [106]. The position was succinctly summarised by Yates J in Nichia (Yates J) at [386]:
In Advanced Building Systems Pty Limited v Ramset Fasteners (Aust) Pty Limited (1998) 194 CLR 171; [1998] HCA 19 (ABS), the High Court explained (at [36]-[40]), with reference to Philips, that the notion that a “new use of an old substance” cannot be patentable subject matter was a 19th century development of the law on obviousness and lack of inventive step. Where this lack of inventive step was admitted on the face of the specification, the grant of a patent could be refused in the first instance on the basis of the admission made. The admission disentitled the patent applicant to argue that even an alleged invention was disclosed. It should be noted here that Microcell makes clear (at 246) that an express admission is not necessary. What is important is the state of affairs shown on the face of the specification itself.
877 MSD relies on Microcell. At page 249 the High Court said:
Many valid patents are for new uses of old things. But it is not an inventive idea for which a monopoly can be claimed to take a substance which is known and used for the making of various articles, and make out of it an article for which its known properties make it suitable, although it has not in fact been used to make that article before.
878 The alleged invention in that case was to a self-propelled-rocket projector comprising a tube of synthetic resinous plastic material reinforced with mineral fibres. With respect to that alleged invention, the High Court said at 251:
We have in truth nothing but a claim for the use of a known material in the manufacture of known articles for the purpose of which its known properties make that material suitable. A claim for nothing more than that cannot be subject matter for a patent, and the position cannot be affected either by the fact that nobody thought of doing the thing before, or by the fact that, when somebody did think of doing it, it was found to be a good thing to do.
879 The essence of the submission advanced by MSD is that the claims are to known siliconised containers, which include known components (ph buffered saline, aluminium adjuvant, surfactant), the known properties of which made them suitable for a vaccine formulation.
880 In my view the claims are for a combination of several elements that interact with each other to produce a combination whereby each of the integers operates in mutual relation to the others in the sense described by Dixon J in Palmer v Dunlop Perdriau Rubber Company Limited [1937] 43; 59 CLR 30 at 73. They are not merely separate known elements where each continues to operate as it did before it was combined.
881 The question in the present case is not whether particular integers of the claims were known individually for their inherent characteristics, but whether it may be concluded, having regard only to the face of the specification, that in truth the combination of integers making up the asserted invention is nothing but a claim for the use of a known material in the manufacture of known articles for the purpose of which its known properties make that material suitable.
882 In my view the specification falls short of this, from which it is apparent that there is a working interrelationship between the components in what is asserted to be a new combination. It is not sufficient for each component to be recited to be known. The point may be demonstrated by consideration of the passages emphasised in MSD’s submissions. In the reasoning that follows, I refer to page and line numbers from the specification. The passages so identified are mostly set out in section 21.1 above, and, for brevity, they are not repeated here.
883 In relation to passage (1), on page 1 it is the case that it was a known goal to seek to improve the stability of an immunogenic composition. However, the words page 2 lines 12 – 17 of the specification must be read with the whole paragraph of page 2 lines 12 – 23, which points to complexities involved in achieving such a goal. This paragraph indicates that in developing an immunogenic composition, the chemical stability of the immunogenic composition, the physical/thermal stability of the composition (for example, aggregation, precipitation, adsorption), the compatibility of the composition with the container, the interactions between the immunogenic composition and inactive ingredients, the manufacturing process, the dosage form, the environmental conditions encountered during shipping, storage and handling, and the length of time between manufacture and usage all must be considered.
884 It is in this context that one considers the description in the specification of the particular components of the composition claimed. The Background suggests that aggregation/precipitation presents a problem for the stability of the composition when formulated in such a container, but in the context of the variety of other considerations that must be taken into account that are set out above at [883]. Overall, the Background states that there is a need for formulations to enhance stability and inhibit precipitation, but it says nothing as to how the complexities to which it refers are resolved.
885 It is true that the Detailed Description includes references to individual excipients. An adjuvant, described as a substance that enhances the immune response when administered together with an immunogen or antigen (page 19 lines 14 – 16), is known. At page 21 lines 18 – 24 the patentee describes a number of ingredients to compositions of the invention as “conventional” including excipients such as buffers, adjuvants or solvents. Furthermore, the specification states that the preparation of these compositions from those components “having appropriate pH isotonicity, stability and other conventional characteristics is within the skill of the art”. However, nowhere does the specification identify or suggest that the combination chosen falls within the skill of those in the art. This applies to each of the passages relied upon by MSD. The use of polysaccharide protein-conjugates (passage 2), pH buffered saline (passage 3), aluminium adjuvants (passage 4), surfactants (passage 5), and siliconised containers (passage 6) are all recited to be known, but the patentee does not say, or imply, that the particular combination of the ingredients was so known.
886 Indeed, having regard to the passages in the specification to which I have referred, and the examples given, the patent discloses, as a matter of substance, that the alleged invention is one that involves arriving at a new combination of integers, and that the interaction between them to solve stability problems, including that arising from silicone oil induced aggregation, is new. The evidence as to the manner in which the components interact supports this conclusion.
887 Two further points should be made.
888 First, the asserted lack of a specific statement of inventiveness in the specification is not determinative of the question. There is no requirement in the Patents Act that the patentee identify in the specification the particular invention, or inventive step that it has achieved. MSD’s case cannot be made out on the basis of what the specification does not state – it must be on the basis of what it does state: Nichia (Yates J) at [391].
889 At this point it is perhaps apposite to emphasise the distinction between the ground presently relied upon and that of lack of inventive step. In the latter, relying only upon the common general knowledge, I have been able to conclude that the combination claimed is not inventive. However, manner of manufacture is circumscribed: see Bristol-Myers at [30] (and AstraZeneca (FC) at [384]).
890 Secondly, MSD relies on the incorporation by reference of Jones 2005 as comprising admissions on the face of the specification that silicone oil induced aggregation was known, and that the means of preventing it was also known.
891 Wyeth submits that Jones 2005 is to be understood as not being incorporated by reference in the specification in its entirety, but for the limited purpose identified on page 2 lines 24 – 27 of the specification, namely to identify a reference where it had been suggested in the art that silicone oil, which induces protein secondary and tertiary conformational changes, might be responsible for aggregation/precipitation seen in protein pharmaceutical preparations. I disagree that the available use of Jones 2005 is so limited. At page 25 lines 25 – 26 the specification states that all patents and publications cited are incorporated by reference. I consider that the description of Jones 2005 represents an invitation to the person skilled in the art to consider Jones 2005 generally for the purpose of understanding the problems caused by silicone oil. That is different to the more limited context of the Emory patent which the Full Court considered was not incorporated by reference in its entirety: Idenix Pharmaceuticals LLC v Gilead Sciences Pty Ltd [2017] FCAFC 196; 134 IPR 1 at [165] (Nicholas, Beach and Burley JJ).
892 The disclosure of Jones 2005 was the subject of expert evidence and may be summarised as follows.
893 It is entitled “Silicone Oil Induced Aggregation of Proteins”. The Abstract states that prior to delivery to the patient, protein pharmaceuticals often come in contact with a variety of surfaces (e.g. syringes and stoppers), which are treated to facilitate processing or to inhibit protein binding. One such coating, silicone oil:
...has previously been implicated in the induction of protein aggregation. We have investigated the propensity of model proteins to aggregate when silicone oil is present in solution and find significant induction of aggregation in four proteins of various molecular weights and isoelectric points in the presence of 0.5% oil. The ability of silicone oil to induce conformational changes that might be responsible for this aggregation was also examined...Neither method produces evidence of large conformational changes or alterations in thermal stability although in a limited number of cases some small changes suggest the possibility of minor structural alterations. The most probable explanation for silicone oil induced aggregation is that the oil has direct effects on intermolecular interactions responsible for protein association through interaction with protein surfaces or indirectly through effects on the solvent.
894 The text begins with an observation that silicone oil contamination “has long been suspected of being responsible in some cases for the aggregation seen in certain protein pharmaceutical preparations”. It refers to several publications from the 1980s. It also refers to the use of silicone oil not being limited to syringes, but also as a coating for porous glass vials to minimise protein adsorption and as a lubricant to prevent the conglomeration of rubber stoppers during filling procedures. The paper continues:
In addition, it is the author’s experience that questions of silicone oil contamination and its potential role in protein aggregation arise frequently during the pharmaceutical development of proteins generally, although little information about this potential problem is available in the scientific literature.
895 The purpose of the investigation reported in the paper is said to have been to assess the ability of silicone oil to induce aggregation of a variety of proteins over a range of pH and to investigate whether several biophysical techniques that are sensitive to changes in protein secondary and tertiary structure can detect silicone oil induced conformational changes that might be responsible for aggregation. Four proteins are chosen. The paper states “the choice of buffer pH was based on…pharmaceutical relevance” and that the study examined several pH levels.
896 Under the heading “Methods” the article states that three buffers (sodium phosphate, sodium chloride, and sodium acetate) were used. A stock solution (suspension) of 1% w/v silicone oil in buffer was prepared by combining silicone oil and buffer. Protein solutions were prepared in each buffer by adding buffer to an appropriate amount of lyophilized protein to obtain a protein concentration between 1 and 2 mg/mL.
897 In the Results and Discussion section the article states (emphasis added):
The solution parameters for the aggregation study were selected to permit detection of protein aggregates due to the presence of silicone oil over a relatively short time...Although we have attributed the increases in turbidity to protein aggregation, it is possible that the observed increases are caused, at least in part, by the effect of the protein on the silicone oil dispersion itself. Unfortunately, there is no obvious experimental method to easily distinguish between this and turbidity increases due to protein aggregation. Our assumption that protein association is responsible for turbidity is based on the fact that aggregated protein can be separated by centrifugation from the protein/silicone oil emulsions and directly identified in the pelleted material.
898 Professor Petrovsky accepted the emphasised passage as indicating that the authors demonstrated aggregation in the manner described. Professor Dalby was more sceptical, and considered that because the methodology was not set out, he could not be comfortable that protein aggregation was the cause of detected turbidity. He considered that the cause could be protein, both protein and silicone oil, or just silicone oil. However, the statement of fact in the emphasised passage – which I understand to mean that aggregated protein was separated by centrifugation – indicates that this is the finding of the authors. Whilst there is no report showing the data from the centrifugation, I share the view expressed by Professor Petrovsky that in a peer reviewed article the clear statement that the authors had centrifuged the products should be accepted. As Dr Jones explained, if he were the editor of the journal, he would want peripheral material, such as data not key to the main argument, excluded.
899 The authors then go on to characterise the aggregation from the centrifugation in Figure 1.
900 The results of the study are discussed at page 922 (emphasis added):
The results of this silicone oil-induced aggregation study of several proteins reveal only limited information regarding general trends. The most obvious one is that the more hydrophobic proteins, BSA (classified as hydrophobic based on the well known presence of its apolar binding sites) and ConA, have a greater tendency to aggregate than the relatively more hydrophilic ones (lysozyme and Rnase A). This result was not unexpected and suggests that the interactions are at least in part apolar in nature. All proteins exhibited a pH-dependence in their tendency to aggregate in the presence of the oil. There was, however, no clear trend...to this dependence....
901 Professor Petrovsky and Dr Jones considered that the emphasised statement was supported by Figure 1, which shows that there is a pH-dependent effect on the aggregation. Professor Dalby accepted that the authors saw a difference in aggregation that was pH-dependent.
902 The Conclusions state that in general, methods that are commonly used to monitor changes in protein structure do not provide consistent evidence that silicone oil induces major structural changes that might be responsible for inducing aggregation. Aggregation is most commonly thought to arise from molten globule like states of proteins. Such states are usually detected by structure alterations, but that was not the case here. It is possible that more subtle structural changes may be involved in the aggregation processes. It says:
Most importantly, a direct effect of the oil on the interactions that mediate protein/protein interactions responsible for aggregation seems likely. Whatever the molecular basis for the observed aggregation behaviour, however, it can clearly be minimized by reducing the content of silicone oil in protein pharmaceutical formulations.
903 Certainly it is the case that Jones 2005 teaches not only the fact of protein aggregation, but also that altering the pH by using a buffer can affect the degree of protein aggregation. However, this does not materially alter the calculus in relation to the manner of manufacture ground. The particular combination of claim 1 – the broadest claim – shows that the invention is a combination of a pneumococcal polysaccharide-protein conjugate where the composition is not only buffered, but also contains an aluminium salt adjuvant. The disclosure of Jones 2005 does teach that using a buffer is likely to have an effect on aggregation. But the specification, on its face, does not disclose the combination including an adjuvant (claim 1). Nor does it disclose the effects or use of a surfactant (claim 9).
904 The admissions on the face of the specification are insufficient to yield the conclusion that the claims are in truth nothing more than for the use of a known article for the purpose for which its known properties make that material useful, within Microcell. Nor, having regard to the disclosure of the identified passages, and to the statement on page 42 of the specification, is the Philips ground established.
28. CONTAINER PATENT: LACK OF FAIR BASIS AND LACK OF CLARITY
905 MSD pleads that the asserted container patent claims are not fairly based on the matter described in the specification within s 40(3) of the Patents Act on the three bases set out below:
(1) the asserted claims, other than claim 9, travel beyond the matter described in the specification in that they do not include a surfactant;
(2) the asserted claims travel beyond the matter described in the specification in that there is no real and reasonably clear disclosure in the specification of a formulation which has polysaccharide-protein conjugates of Streptococcus pneumoniae serotypes in addition to the 13 chosen serotypes; and
(3) claims 8 and 17 travel beyond the matter described in the specification in that there is no real or reasonably clear disclosure in the specification of histidine at pH 5.8 as a buffer.
906 I address each of the bases separately below.
907 MSD correctly submits that, with the exception of claim 9, the asserted container patent claims do not require a surfactant. The consequence, it submits, is that the claims are not fairly based because there is no real and reasonably clear disclosure of an invention without a surfactant. In particular, MSD submits that a critical passage in the specification is page 13 lines 32 – 25, which states:
…the present invention relates to the unexpected and surprising results that formulating an immunogenic composition with a surfactant such as Tween 80 significantly enhances the stability and inhibits precipitation of an immunogenic composition.
It submits that this passage demonstrates that the asserted invention is the use of the surfactant, and any combination that does not require the inclusion of a surfactant is not fairly based.
908 I do not consider that this ground is made out.
909 Neither the Patents Act nor the statement of principle in Lockwood No 1 at [69] imposes an obligation upon the patentee to disclose or identify an asserted inventive step in the specification. In any event, the specification makes a number of statements as to what the patentee considers to be the invention. In the Summary of the Invention is a consistory clause that matches the broadest claim, being claim 1. No surfactant is included. Furthermore, on page 13 lines 5 – 9, prior to the passage emphasised by MSD is the statement (emphasis added):
The present invention addresses an ongoing need in the art to improve the stability of immunogenic compositions such as polysaccharide-protein conjugates and protein immunogens. Thus, the present invention broadly relates to novel surfactant formulations and/or novel aluminium salt formulations which stabilize and inhibit precipitation of immunogenic compositions.
910 The second sentence quoted makes plain that the patent asserts as the invention, not only a novel surfactant formulation, but also other novel aluminium salt formulations. In this regard the position in Sigma at [242] is not analogous. In that case, there was a clear statement of the impossibility to achieve the invention with technology that fell within the claims.
28.2 Disclosure of serotypes in addition to the 13 chosen serotypes
911 MSD submits that the specification uses the 13 chosen serotypes in the examples to demonstrate a group of antigens that can be used in the claimed formulation, but does not disclose any formulation with conjugates of any additional serotypes to the 13 chosen serotypes. Due to the “one or more” phrasing used in claim 1 (and dependent claims), it has no upper limit, and accordingly encompasses formulations which have more than 13 serotypes. On this basis, it submits that all claims except for claim 18 include formulations in addition to the 13 chosen serotypes and so are not fairly based.
912 For similar reasons to those set out in the previous section, this fair basis challenge fails. The consistory clause for claim 1 (page 3 lines 18 – 22) makes clear that the patentee considers that an aspect of the invention is any formulation comprising the integers of a siliconised container, a pH buffered solution with a pKa of about 3.5 to about 7.5, an aluminium salt together and any number of polysaccharide-protein conjugates of one or more pneumococcal polysaccharides.
913 In relation to claim 18, MSD submits that it must be read as limited to the 13 chosen serotypes, such that MSD’s 15-valent vaccine cannot infringe. I have rejected that argument. The fall back argument advanced by MSD is that if the claim is not so limited, the claim must lack fair basis, for substantially the same reasons as set in section 15. For substantially the same reasons as set out in that section, this argument must also be rejected.
28.3 Histidine buffer at pH 5.8
914 MSD submits that the specification contains no real and reasonably clear disclosure of the integer contained in claims 8 and 17 that “the buffer is histidine at pH 5.8”. In particular, it argues that while the specification discloses in several places that histidine may be used as the buffer, there is no disclosure of its use at pH 5.8.
915 The specification includes several passages where the use of a histidine buffer is described. An example of such a disclosure is at page 3 lines 32 – 34:
In certain embodiments, the pH buffered saline solution of the formulations has a pH of 5.5 to 7.5. In other embodiments, the buffer is phosphate, succinate, histidine or citrate. In certain embodiments, the buffer is succinate at a final concentration of 1mM to 10mM and pH 5.8 to 6.0.
916 In a number of other places in the specification, the patentee provides first a pH range for the buffer of 5.5 to 7.5 and for a range for a succinate buffer of between pH 5.8 to 6.0. Accordingly, the disclosure of the specification is that the histidine buffer may be within the range of 5.5 to 7.5. The selection of a pH from that range for the purposes of claims 8 and 17 is not explained anywhere in the specification. It might be considered it represents a narrow point within the available range that the patentee has chosen for the purpose of the claim. I was directed to no expert evidence on the subject in closing submissions to suggest that this selection was not available within the range that the skilled formulator might chose. I am conscious that in considering lack of fair basis, the Court should not make a meticulous verbal analysis. The question is whether the invention as claimed is disclosed in a “general sense”: Lockwood No 1 at [69]. I am not satisfied that MSD has established that it is not disclosed in the requisite sense. This challenge fails.
917 MSD relies on the same three bases discussed above in support of a submission that the claims do not sufficiently or clearly define the alleged invention within s 40(2)(a) of the Patents Act.
918 MSD directed none of its closing submissions towards explaining why it relies upon the first and third bases with respect to the lack of definition ground. For similar reasoning as above, these challenges fail.
919 In relation to the second basis, MSD submits that a patent claim must define the monopoly in a way that is not reasonably capable of being misunderstood, citing Welch Perrin at 610. It submits that the claims lack clarity because it is unclear whether the proper construction of the claims is that put forward by MSD, Wyeth or otherwise.
920 Whilst not clearly identified, this argument can only apply to claim 18 as there is no relevant construction dispute arising from the other asserted container patent claims. However, the argument cannot succeed. The invention as defined by claim 18 is for the composition there defined, including the 13 serotypes identified.
29. CONTAINER PATENT: INUTILITY
921 MSD contends that the invention claimed in any of the asserted container patent claims is not useful within s 18(1)(c) of the Patents Act in that the claims include siliconised containers filled with vaccine formulations that do not achieve the promises of the specification. MSD contends that there are two relevant promises, namely that the formulation of the claims:
(a) will be stable; and
(b) will inhibit silicone oil induced aggregation sufficiently to result in a stable vaccine.
922 MSD next contends that neither of the asserted promises is met by formulations within the asserted container patent claims. It couches its arguments by reference to four points, which I describe in more detail below as: the pH range argument; the protein concentration argument; the surfactant argument; and the absence of surfactant argument.
923 Wyeth disputes that the stability promise is made in the specification and contends that the only promised result made in respect of silicone oil induced aggregation is for a formulation that stabilises the immunogen to an acceptable level against silicone oil induced aggregation, being a level where it no longer presents a problem in the form of visible precipitation, and ensuring that silicone oil induced aggregation does not increase above that level over the stated shelf life of the vaccine. Wyeth characterises the absence of surfactant argument as the primary ground upon which MSD relies, and contends that it has not been pleaded. Wyeth disputes that there has been any failure to meet the only promise made, which it contends is the asserted silicone oil induced aggregation promise, as qualified by it above.
924 The relevant law relating to this ground is summarised in section 12.1 above.
29.2 Were the asserted promises made?
925 Although framed by MSD as involving two separate promises, in my view the single promise of the invention is that the formulations described and claimed will provide a stable formulation. The promise includes that, to the extent necessary, the stable formulations described and claimed will also inhibit precipitation of immunogenic compositions. That is the natural meaning to be attributed to the words first appearing in the Summary of the Invention (page 3 lines 12 – 13):
The present invention broadly relates to novel formulations which stabilize and inhibit precipitation of immunogenic compositions.
926 The context provided by the Background of the Invention serves to confirm that this is correct, given that attention is paid not only to the background art and knowledge in relation to silicone oil induced aggregation, but also the challenges arising in relation to the stability of an immunogenic composition generally.
927 The point is reinforced in other passages, such as in the Detailed Description of the Invention where the patentee states (page 13 lines 5 – 9) (emphasis added):
The present invention addresses an ongoing need in the art to improve the stability of immunogenic compositions such as polysaccharide-protein conjugates and protein immunogens. Thus, the present invention broadly relates to novel surfactant formulations and/or novel aluminium salt formulations which stabilize and inhibit precipitation of immunogenic compositions.
928 Having regard to the whole of the disclosure of the specification, aspects of which I have addressed in more detail in section 21.1 above, I consider that the promise made is that the formulation described and claimed will be stable and includes that in achieving stability, precipitation and aggregation will be inhibited, including silicone oil induced aggregation.
29.3 Consideration of whether the promise is met
929 MSD first advances the pH range argument, which applies to all of the asserted container patent claims. Claim 1 includes a pH buffered saline solution within a pKa range of about 3.5 to about 7.5. Claim 2 includes a pH from 5.5 to 7.5. MSD’s argument runs as follows.
930 Professor Dalby gives evidence that as a general rule of thumb, a buffer is effective within one pH unit of its pKa value. He says that “accordingly” for the pKa range of 3.5 to 7.5, he would expect solutions “with pH of around 4 to 8 to be covered” (this appears to involve a typographical error). Professor Petrovsky agrees with the rule of thumb, but says that in relation to claim 1, the pH range would therefore extend from 2.5 to 8.5. Professor Petrovsky gives evidence that he “would not expect” a conjugate at pH 2.5 to be stable. He also gives evidence that formulations that have a pH of around 4 to 5, or a pH of around 7 to 7.5, “may hydrolyse the polysaccharide”. No evidence in the way of experimentation or otherwise supports this assertion. Conversely, no evidence contradicts either proposition, and Professor Petrovsky was not taken to either in cross-examination.
931 Although there is no requirement to prove inutility by experiment (Idenix at [257]), it is nevertheless necessary for MSD to establish to the requisite standard that the claimed combination is not useful as a stable formulation.
932 The statement to the effect that formulations within the range claimed in claim 2 “may hydrolyse” in my view is not sufficient to warrant a conclusion that a formulation within the claim would not work. Put another way, the strength of this evidence is not sufficient to yield the conclusion that, on the balance of probabilities, the claimed formulation is not stable.
933 In relation to claim 1, Professor Petrovsky’s statement that he would not expect a conjugate of pH 2.5 to be stable is somewhat stronger. However, without further development by explanation, that evidence amounts to a theoretical statement of expectation that may or may not prove to be correct. It is an hypothesis yet to be tested, and accordingly is unlike the position accepted in Alphapharm at [470], where Lindgren J was able to conclude, on the basis of the unequivocal evidence of Professor Montgomery, that the dosage range claimed included quantities well below the useful minimum and well above the useful maximum.
934 Accordingly, the pH range argument does not succeed.
935 MSD secondly advances what it calls the protein concentration argument. It notes that claim 1 does not limit the protein concentration in the formulation of claim 1. The argument runs as follows.
936 Professor Dalby gives evidence that as a rule of thumb, problems may arise with protein aggregation in solution where there is a protein concentration above 1000 μg/ml. Example 3 of the container patent concerns a formulation within the scope of the asserted container patent claims, namely, a 13-valent polysaccharide-protein conjugate with a buffer, salt and adjuvant. An experiment was conducted whereby serotypes 4 and 6B were formulated with protein concentrations ranging from 25 μg/ml to 200 μg/ml in the absence and presence of AIPO4 (aluminium adjuvant) in containers using siliconised stoppers. Where the concentration of serotypes 4B and 6B was 100 μg/ml and 200 μg/ml respectively, fibre-like particulates were observed for both the adjuvanted and the unadjuvanted formulations. These results are displayed in table 5 of the container patent.
937 MSD submits that, by applying the rule of thumb that the experts adopt of 1000 μg/ml, the person skilled in the art may have been led, in the absence of a limitation of the protein concentration to be used in claim 1, to adopt a protein concentration that produced visible precipitate. In short, the results in example 3 demonstrate that visible precipitation may be caused by the use of 100 μg/ml or 200 μg/ml. This shows that the person skilled in the art could have used what he or she considered to be a normal amount of protein concentration for different serotypes in a formulation within the claims which would not have achieved the promise of the invention.
938 The decision of the Full Court in Sandvik demonstrates that where a claim is broad in compass, it is not to be applied in such a way that it is to be made unworkable. In that case, claims 1 to 3 did not lack utility, even though they included within their scope the breadth of claim 4, because the skilled addressee would understand that a drill rod with a round end cannot be driven by a drive chuck or an adaptor and would therefore not use a rod with a round end (at [201]). Accordingly, it was only claim 4, which upon its proper construction required the rod profile to be round, that was rendered inutile (at [202]).
939 The present case is somewhat different. MSD submits here that by applying a sensible construction to claim 1, the person skilled in the art could select a protein concentration of up to 1000 μg/ml and not expect protein aggregation. Yet at much lower concentrations, specifically at 100 or 200 μg/mL, visible aggregation was not only encountered, but was not resolved by the use of the formulation claimed in claim 1. Put another way, by adopting an entirely orthodox approach to formulating a polysaccharide-protein conjugate within a formulation, within the range of protein concentration usual in the art, the person skilled in the art would have found that the formulation of claim 1 did not avoid visible precipitation.
940 It may be thought that this leads to the conclusion that claim 1 accordingly lacks utility, but two points stand in the way of that conclusion. First, claim 1 is silent as to protein concentration. Nothing compels a conclusion that any particular protein concentration must be selected. It is well within the skills of the person skilled in the art to determine the appropriate protein concentration to avoid visible precipitation, using the techniques and applying the principles to which I have referred in my consideration of the common general knowledge in section 25.2.
941 Secondly, example 3, including table 5, provides a clear disclosure that fibre-like white particulates were visible when concentrations of 100 μg/mL and 200 μg/mL were used for serotypes 4 and 6B respectively. The argument advanced by MSD skilfully seeks to avoid the difficulty that a person would not construe a claim in a way that is not sensible. However, it does not overcome the fact that the person skilled in the art also has the benefit of the disclosure of the specification when applying the invention. The utility of the alleged invention depends on whether by following the directions in the complete specification, the effects which the patentee proposed to produced could be produced: Lane Fox at 431. It appears to me that a skilled reader would understand from example 3 that, whatever the rule of thumb may be, low concentrations of protein may be necessary, at least for serotypes 4 and 6B, to avoid visible precipitation.
942 In a final thread to this argument, MSD contends that claim 1 contains no limitation on the protein concentration in the formulation. There is no limit to the number of conjugates that may be included. As a result, protein amounts that do not work will fall within the claims. However, I am satisfied that a person skilled in the art would understand to limit the amount of protein in a formulation to a workable level.
943 Accordingly, the protein concentration argument does not establish that the claims lack utility.
944 The third argument advanced by MSD is called the surfactant argument and concerns all of the asserted container patent claims, because MSD submits each claim may include a surfactant. MSD notes that none of claims 1 – 8 require the presence of a surfactant. Claim 9 then adds an integer specific to surfactants. Claim 11 then identifies that the final concentration of the polysorbate 80 surfactant in the formulation is at least 0.01% to 10% polysorbate 80 weight/volume of the formulation. This, MSD submits, is a range of polysorbate of 1000 fold. Claim 18 is dependent on claim 11. As MSD puts it, claims 1 – 9 necessarily include within their scope claim 11, with the consequence that the broad range of 0.01% to 10% weight/volume of polysorbate 80 to the formulation is included within their scope. This, it submits, is an unworkable range, on the basis of the evidence of the experts. Professor Dalby’s opinion is that surfactants in high concentrations can reduce the stability of proteins by disrupting bonds within the protein. Professor Petrovsky agrees, and says that he would have selected a surfactant such as Tween 80 and used it at a concentration, as a general rule of thumb, of between 0.02% and 1%. He gives evidence that if one uses a surfactant at a concentration of 10%, it would denature the protein, which, as the primer notes, is a known form of physical degradation of a saccharide-protein conjugate.
945 However, it seems to me that this argument fails for the same reason that the argument in respect of claims 1 – 3 failed in Sandvik. Claim 11 is not one of the asserted container patent claims and its validity is not challenged. If it were, and the evidence remained unchanged, it may well be that the broad range presents a difficulty. However, it is not. It is apparent that the skilled formulator who approaches the construction of the asserted container patent claims would not select Tween 80 at the ends of the spectrum that were unworkable. To the contrary, as I have found, the normal skills in the art would be applied to select a workable amount of Tween 80.
946 The fourth point MSD refers to as the absence of surfactant argument, which in oral submissions MSD re-labelled as concerning the presence of adjuvants rather than the absence of surfactants. The challenge does not concern claim 9, or claims dependent on claim 9. The argument is as follows.
947 Example 4 in the container patent includes an experiment using a siliconised, commercially available container (BD Hypak syringes capped with West 4432 ready to use plungers) containing polysaccharide-protein conjugates of the 13 chosen serotypes together with salt (sodium chloride) and succinate buffer at a pH of 5.8. A similar experiment was tested with and without an adjuvant. The reported results of the formulation with the adjuvant (relevant to all claims except claim 9), using that commercially available container with a high level of silicone, agitated in controlled conditions, are that:
(a) there is a significant loss of antigenicity; and
(b) that loss is reported in figure 2 to be above 30% for 3 serotypes.
948 The evidence of the experts is that antigenicity can be a measure of stability. Professor Petrovsky gives evidence that the figure 2 formulation is an example of a failed vaccine. Professor Dalby accepts that the formulation did not completely stabilise the vaccine. Accordingly, MSD submits that claim 1, and the claims dependent on it that do not involve the use of a surfactant, are invalid for want of utility.
949 Wyeth first submits that example 4 does not establish that the claimed combination without a surfactant fails to reduce the silicone oil induced aggregation to an acceptable level, being one where it no longer presents a problem in the form of visible precipitation. However, this submission depends on acceptance of Wyeth’s interpretation of the promise of the invention, which I have rejected. It next submits that figure 2 of the specification does not provide an appropriate comparison, because it was the positive control, and the specification also includes a negative control, demonstrating that the patentee is attempting to determine the nature of the problem. It submits that without the buffer and adjuvant, the loss of antigen would be worse and, having regard to the teaching of example 4, the skilled reader would know to avoid containers with a high silicone content.
950 Wyeth further contends that this argument is not available to MSD, because it was not sufficiently pleaded. However, having regard to the content of the Third Further Amended Consolidated Statement of Claim at [12(c)(xi)], the opening submissions advanced by MSD, and the evidence adduced, in my view the argument is adequately pleaded and notified to Wyeth, and I propose to address it.
951 I have described the disclosure of the specification in section 21.1 above. The examples are presented for illustrative purposes (page 25 line 31). Example 4 is entitled “Aluminium adjuvants inhibit the formation of 13vPnC particulates in the presence of siliconized container means”. It involves the use of liquid formulations of the 13 chosen serotypes in a buffer of pH 5.8, with and without aluminium adjuvant. A number of different containers are used. One, identified as the “positive control” is a “BD Hypak syringe” and is said in the specification to be available from a catalogue, and purchased by the patentee. Another, identified as a “negative control” is an unsiliconised syringe.
952 The containers were subject to controlled agitation conditions, and the total antigenicity of each serotype was measured. The experts agree that antigenicity tests are a form of stability testing.
953 The positive control was reported in figure 2 of the specification to have antigenicity losses of over 30% for three serotypes after agitation of 8 and 24 hours. Professor Petrovsky gave evidence that in his view this is “a perfect example of a failed vaccine” that was “highly unstable”. Professor Dalby agrees that in this container the formulation failed to stabilise the vaccine.
954 However, in his oral evidence, Professor Dalby contended that the conditions applied by the patentee to the samples were not representative of appropriate stability testing. He says, referring to page 38 of the specification, that the agitation conditions were optimised based on antigenicity loss for the two controls, and that the agitation in the system of 500 rpm was not of the type used in a long-term stability test.
955 Professor Dalby’s evidence does not withstand scrutiny. The whole of the passage on pages 38 – 39 of the specification is as follows:
Prior to the study, the agitation conditions were optimized based on the antigenicity loss of the two controls: (1) the worst-case control (positive control, high silicone; FIG 2) and (2) the best-case control (negative control, no silicone; FIG 3). The conditions were then optimized such that the antigenicity loss was low in positive control, yet detectable in the negative control. This was to ensure that the agitation was neither too weak to produce precipitation in the syringes; nor too strong, such that the precipitation might be caused by factors other than the silicone interaction (e.g., by shear forces). Thus, agitation at 500 rpm (pause mode) for twenty-four hours was chosen as the most suitable agitation condition, while a temperature of 2-8 degrees°C and a horizontal position were used to simulate the conditions in real time product shipping and handling.
956 The final sentence makes plain that the experiment was designed as an accelerated stability study. That is what Professor Petrovsky understood the patentee to intend. In my view that is plainly the preferable construction of the specification. I reject Professor Dalby’s contrary view.
957 The consequence is that example 4 provides a clear teaching that the positive control produces an unstable formulation. However, that does not yield the result that the ground of inutility succeeds. The specification teaches in no uncertain terms that the positive control failed because of the high amount of silicone used. Containers with lesser amounts significantly reduced the antigenicity losses to minor amounts. The experiment showed something of the parameters of the invention. The skilled reader is able to understand and interpret the results, and then apply them to produce something within the claims. Put another way, by following the directions in the complete specification, the effects which the patentee professed to produce could be produced. Accordingly, this final ground of inutility must also fail.
958 For the reasons set out perhaps far too fully above, I have found that the asserted 013 patent claims are valid and will be infringed by MSD’s 15-valent vaccine. I have found that the asserted 844 patent claims would have been infringed, but are invalid because they lack support within s 40(3) of the post-RTB Patents Act. I have also found that the asserted container patent claims would have been infringed, but that they are invalid for want of inventive step.
959 I will direct that the parties confer and propose short minutes of order giving effect to this judgment.
I certify that the preceding nine hundred and fifty-nine (959) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Burley. |
Associate:
Dated: 14 October 2020

