
FEDERAL COURT OF AUSTRALIA
Gill v Ethicon Sàrl (No 5) [2019] FCA 1905
ORDERS
NSD 1590 of 2012 | ||
| ||
BETWEEN: | KATHRYN GILL First Applicant DIANE DAWSON Second Applicant ANN SANDERS Third Applicant | |
AND: | ETHICON SÀRL First Respondent ETHICON INC. Second Respondent JOHNSON & JOHNSON MEDICAL PTY LTD (ACN 000 160 403) Third Respondent | |
JUDGE: | KATZMANN J |
DATE OF ORDER: | 21 november 2019 |
THE COURT ORDERS THAT:
1. By 20 December 2019, each applicant is to notify the respondents and the Court of her election as to whether she will accept an award of damages under the relevant provisions of the Trade Practices Act 1974 (Cth) or at common law, as modified by the statutory scheme, if any, that operates in the State in which she lives.
2. By 14 February 2020, the parties bring in short minutes giving effect to these reasons.
3. In the event that the parties are unable to agree on the form of orders or all of the orders, including the form of the common questions and the answers to them for the purposes of s 33ZB of the Federal Court of Australia Act 1976 (Cth), a timetable for the filing and exchange of submissions be forwarded to chambers by 17 January 2020, with a view to a hearing in the week commencing 10 February 2020.
4. Liberty to apply be granted on two (2) days’ notice.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
REASONS FOR JUDGMENT
Table of contents
KATZMANN J:
1 This is a representative action concerning nine medical devices, all made using knitted polypropylene, a thermoplastic polymer. The devices were designed to be surgically implanted in women to alleviate either stress urinary incontinence or pelvic organ prolapse. The action calls into question the safety of those devices. Its outcome will affect thousands of women who were implanted with any of the devices in Australia and who suffered any complications caused by those devices at any time before 4 July 2017. A number of the final orders that will be made in the proceedings will bind all such women except for those who have exercised their rights to opt out of the action.
2 The medical devices used for the treatment of stress urinary incontinence which are the subject of this action are known by the trade names Gynecare Tension-free Vaginal Tape System (TVT), Gynecare TVT Obturator System (TVT-O), Gynecare TVT Secur System (TVT Secur), Gynecare TVT Exact Continence System (TVT Exact) and Gynecare TVT Abbrevo Continence System (TVT Abbrevo). When I refer to them collectively in this judgment, I use the shorthand expression “the SUI devices”.
3 The medical devices used for the treatment of pelvic organ prolapse which are the subject of this action are known by the trade names Gynecare Gynemesh Prolene Soft (Gynemesh PS), Gynecare Prolift Pelvic Floor Repair System (Prolift), Gynecare Prolift+M Pelvic Floor Repair System (Prolift+M) and Gynecare Prosima Pelvic Floor Repair System (Prosima). I refer to them collectively as “the POP devices”.
4 When I refer to the SUI devices as well as the POP devices, I call them “the Ethicon devices”.
5 In a nutshell, the applicants’ case is that all the Ethicon devices cause a number of potentially serious complications but the respondents were so driven by commercial interests that they neglected to undertake the investigations reasonably necessary to inform themselves and the community of the extent, if not also the nature, of the risks posed by the devices and to take appropriate or sufficient remedial action. To the extent that they were aware, they failed to make adequate disclosure. As the applicants put it in their opening submissions:
[A]t all times from the development of the first TVT products, to the commencement of proceedings, the Respondents were motivated by a sales-driven culture, with an emphasis on the perceived urgent need to get products to doctors before competitors. There was a focus on profit and maintaining market share, without adequate focus on safety, efficacy and potential complications. In the race to sell as many products as possible, the Respondents failed to undertake proper or adequate evaluations or [disclose] known complications and risks. 1
6 Since the commencement of this proceeding, all the POP devices have been removed from the market but, with the exception of TVT Secur, all the SUI devices are still available for sale.2
7 The devices in question were made by two foreign companies: the first respondent, Ethicon Sàrl (Société Anonyme à Responsabilité Limitée or Limited Liability Company) and; the second respondent, Ethicon Inc., both members of the Johnson & Johnson group of companies.
8 Ethicon Sàrl is a Swiss corporation and the manufacturer of a variety of polypropylene implantable medical devices, including all but one of the devices the subject of this litigation. Ethicon Inc. is an American corporation and the manufacturer of the remaining one, Gynemesh PS.3 It was also involved in the marketing of all the relevant devices through its Gynecare division. Ethicon Inc. is a subsidiary of Johnson & Johnson, an American public company, listed on the New York Stock Exchange. Ethicon Sàrl is a subsidiary of another Swiss company but the ultimate parent company is Johnson & Johnson. In these circumstances, unless it is necessary for a particular reason to distinguish between them, I will generally refer to both Ethicon companies simply as “Ethicon”.
9 Ethicon Sàrl and Ethicon Inc. supplied the Ethicon devices to the third respondent, a related Australian company, Johnson & Johnson Medical Pty Limited (JJM). JJM promoted and supplied the Ethicon devices to Australian hospitals and doctors.
10 The respondents were represented by the same lawyers and, through their senior counsel, made it clear that no point would be taken that the knowledge of one of the companies in the group was not shared by another.4
11 According to statements made on the Australian Register of Therapeutic Goods (ARTG), the purpose of each of the SUI devices is to, by way of a sling, treat stress urinary incontinence and female urinary incontinence resulting from urethral hypermobility and/or intrinsic sphincter deficiency.5 The purpose of the POP devices was to provide “tissue reinforcement and long-lasting stabilisation of fascial structures of the pelvic floor in vaginal wall prolapses”.6
12 The devices were promoted as effective in restoring normal anatomy and sexual function, as not subject to degradation or weakening by tissue enzymes, and as having high patient satisfaction. Instructions for use (IFU) supplied with the devices warned of some possible adverse reactions and contraindications. Yet, a number of potential complications, known to the respondents from the time the devices were first supplied, were not disclosed and those that were disclosed were often minimised. Some of the potential complications could occur with any form of pelvic surgery. Others were unique to implantable synthetic mesh devices.
PART II: OVERVIEW OF THE PROCEEDING
13 This action is brought under Pt IVA of the Federal Court of Australia Act 1976 (Cth) (FCA Act) by three applicants, both on their own behalf and on behalf of other women who claim to have suffered complications from the implantation of one or other of the Ethicon devices during the relevant period. I was informed at the beginning of the trial that there are some 700 registered group members but as the class is an open one, and more than 90,000 Ethicon devices have been supplied in Australia,7 it is highly likely that there are many more.
14 The three applicants are Kathryn Gill, Diane Dawson, and Ann Sanders. Mrs Gill had pelvic organ prolapse and was implanted with a Prolift device on 12 January 2007. Mrs Dawson also had pelvic organ prolapse and was implanted with Gynemesh PS on 8 May 2009. Mrs Sanders suffered from stress urinary incontinence and was implanted with TVT on 12 March 2001.8
15 The applicants allege that the respondents contravened various provisions of the Trade Practices Act 1974 (Cth) (TPA) and the Competition and Consumer Act 2010 (Cth) (CCA). The statutory claims are based on:
s 74B of the Trade Practices Act, because it is alleged that the Ethicon devices were not reasonably fit for the particular purpose for which they were acquired by the group members and not fit for the disclosed purpose under s 55 of Sch 2 to the Competition and Consumer Act — the Australian Consumer Law (ACL);
s 74D of the Trade Practices Act, because it is alleged that the Ethicon devices were not of merchantable quality and did not comply with the guarantee given by s 54 of the ACL in that they were not of acceptable quality;
s 75AD of the Trade Practices Act in that the Ethicon devices were allegedly supplied with a “defect” and s 138 of the ACL because they were allegedly supplied with “a safety defect”; and
s 52 of the Trade Practices Act and s 18 of the ACL in that the information the respondents released in connection with the Ethicon devices, including the IFUs accompanying the products, and the way in which they were marketed and promoted was misleading or deceptive or likely to mislead or deceive consumers.
16 The applicants also allege that the respondents are liable to the applicants in negligence for:
failing to undertake any, or any adequate, clinical or other evaluation of the Ethicon devices before releasing them in Australia;
failing to conduct any, or any adequate, evaluation of safety and effectiveness of the Ethicon devices after their release in Australia; and
failing to inform them, their treating doctors, and/or the hospitals in which the treatments were administered, of the inadequate evaluations about, and the risks of, or susceptibilities to, complications of the kinds from which they suffered.
17 The respondents deny liability and, in the cases of Mrs Gill and Mrs Sanders, they contend that, in any event, their actions are statute-barred.
18 An Originating Application and Statement of Claim were filed on 15 October 2012 naming a single applicant, Mrs Julie Davis. On 6 April 2016, by orders of the Court, Mrs Gill, Mrs Dawson, and Mrs Sanders were substituted for Mrs Davis, and two sub-groups were created, one referred to in the pleading as the “Mesh Sub-Group”, mesh being a reference to the POP devices, the other as the “Tape Sub-Group”, tape being a reference to the SUI devices.
19 The trial commenced at the beginning of July 2017 and did not conclude until the end of February 2018. Evidence was adduced from 48 witnesses, 35 of whom gave oral evidence. Of the 48 witnesses, 37 were experts hailing from nine different disciplines. Each witness gave evidence individually, as my proposal that experts in the same discipline prepare joint reports and give concurrent evidence was opposed by both parties. Extensive written and oral submissions were made.
20 More than 5,500 documents were tendered, running to over 164,000 pages. Mercifully, the trial was conducted electronically, which, to some extent, eased the burden on the Court.
21 Each of the applicants gave evidence on affidavit and was cross-examined. Affidavits were also read from their husbands, only one of whom, Steven Gill, was required for cross-examination.
22 The applicants adduced evidence from four urogynaecologists. They were Andrew Korda, Wael Agur, Jerry Blaivas, and Michael Thomas Margolis. Professor Korda is an Australian. Dr Agur practises in Scotland and was described by one of the respondents’ experts as an opinion leader in Great Britain.9 Professor Blaivas and Assistant Professor Margolis hail from the United States. Urogynaecology, I should explain, is a surgical subspecialty of urology and gynaecology, which involves the diagnosis and treatment of female pelvic floor disorders.10
23 After the respondents had indicated they would not be calling him, the applicants tendered a report prepared for the respondents by Malcolm Frazer, another urogynaecologist.
24 The applicants also tendered reports and read affidavits from a number of other expert witnesses in a range of disciplines. They included the following people who, unless otherwise indicated, are based in Australia: Bilal Chughtai, a urologist, from the United States; Uwe Klinge, a general surgeon and biomaterials researcher who lives in Belgium; Bernd Klosterhalfen from Germany and Vladimir Iakovlev from Canada, both pathologists; two biomechanical engineers, Russell Dunn and Scott Guelcher from the United States; four regulatory experts, Derrick Beech, Bryan Allman from the United Kingdom, Peggy Pence and Anne Holland from the United States; three epidemiologists, Howard Hu from Canada, Cara Krulewitch from the United States, and Mark Woodward; Ian Gordon, a biostatistician; two colorectal surgeons, Alan Meagher and Anthony Eyers; Patricia Jungfer, a psychiatrist; Joseph Slesenger, a specialist in occupational medicine; and Lindy Williams and Timothy Walsh, both occupational therapists.
25 Of these witnesses, only Dr Beech, Professor Woodward, Ms Williams, and Mr Walsh were not required for cross-examination.
26 Affidavits were also read from Robyn Leake, an obstetrician and gynaecologist who treated Mrs Gill for a number of years, James Swan, an obstetrician and gynaecologist who treated Mrs Dawson, and Sandra McNeill, an obstetrician and gynaecologist who assisted in the operation in which Mrs Sanders was implanted with TVT.
27 The respondents adduced evidence from six urogynaecologists: Piet Hinoul from the United States, Pierre Collinet from France, Jan Deprest from Belgium, Alan Lam, Jan-Paul Roovers from the Netherlands, and Anna Rosamilia; an engineer, Steven McLean from the United States; Paul Santerre, a professor of biomaterials from Canada; Thomas Wright, a pathologist from the United States; three psychiatrists, Lisa Brown, Anthony Samuels, and Rosalie Wilcox; and an occupational therapist, Susan Borthwick. All of these witnesses, except Dr Wilcox, were required for cross-examination.
28 Dr Hinoul was the only witness from any of the three respondents to give evidence. Dr Hinoul is a urogynaecologist born in Belgium and educated in the United States, Belgium, and the Netherlands. He holds a PhD in bio-medical sciences from the University of Amsterdam. Since June 2014 he has held the position of Vice President–Medical Affairs at Ethicon Inc., based in Somerville, New Jersey, USA.11 He gave evidence about the Ethicon devices and their development and Ethicon’s conduct. Some of that evidence was based on company records. Not all of it concerned matters of fact.
29 Dr Hinoul joined Ethicon in 2008, when he was appointed Director of Medical Affairs – Europe, Middle East and Africa (Women’s Health and Urology), based in Paris, France.12 According to Dr Hinoul, the Director of Medical Affairs is responsible for “generating evidence for new medical devices”, “for ensuring that there is sufficient evidence to enable products to be released onto the market” and for products that are already on the market, and for “monitoring the literature and the various studies involving the products”.13 The Director of Medical Affairs is also responsible for assessing their risks and benefits. As Director of Medical Affairs, Dr Hinoul said that he was responsible for Ethicon’s pelvic floor repair and incontinence repair products. His duties included undertaking risk/benefit analyses of devices, drafting clinical evaluation reports, assessing clinical literature, presenting at panel meetings, and reviewing adverse events. He also contributed to various Ethicon documents which required “medical input”. They included IFUs supplied with the devices, patient brochures, marketing, design verification, and professional education materials.14
30 In December 2010 Dr Hinoul became the Worldwide Director of Medical Affairs (Women’s Health and Urology) for Ethicon Inc., based in Somerville, New Jersey. In June 2012 his responsibilities increased to cover other aspects of the Ethicon business and from April 2013, until his promotion to Vice-President–Medical Affairs, he held the position of Worldwide Director of Medical Affairs (Ethicon Endo-Surgery (Energy Franchise)).15
31 Dr Hinoul presented as a company spokesman. His affidavit was lengthy (363 pages) but not full and frank. It cast Ethicon’s conduct in the most favourable light. In cross-examination, Dr Hinoul was inclined not to give responsive answers to potentially uncomfortable questions and tended to be evasive where direct answers would not suit the respondents’ interests. At times he steadfastly defended the indefensible.
32 Of the Medical Affairs Directors who preceded him, only his immediate predecessor, Dr Axel Arnaud,16 remains with Ethicon.17 Dr Arnaud was a key figure in the development and post-market surveillance of a number, if not all, of the Ethicon devices. In particular, he investigated the TVT procedure in 1996 and TVT-O in 2002 and was a moving force in the development of the POP devices. No explanation was provided to the Court for his absence nor for the absence of the other Medical Affairs Directors. Presumably the respondents were content to rely solely on their reports, despite the criticisms made of them by the applicants’ experts and others.
33 The rest of the evidence consisted of documents. One legacy of the parties’ approach to tendering documents is that documents referenced by multiple witnesses or submissions were tendered multiple times. Sometimes the same document was tendered by both parties. Sometimes the same document was tendered multiple times by the same party. The result was that one document might have two or more document identification (ID) numbers. Rather than refer to multiple document ID numbers for the same documents, I have elected to use one only, and where a document is mentioned more than once in these reasons, I have endeavoured to refer to the document by the same ID number throughout. No significance should be attached to the choice of ID tender number. Moreover, a source cited in a footnote should not be regarded as the only source upon which I relied to form the view expressed in the relevant sentence or paragraph. The ID numbers have been included in the footnotes to assist the parties to identify source material. They are otherwise of no consequence.
34 In a case of this magnitude, it is impossible to refer to all the evidence and it is unnecessary to do so. My failure to refer to any particular item of evidence, however, ought not to be taken as an indication that I have not considered it. Having regard to the length of the judgment and the connection between the subject-matter of its various parts, a degree of repetition is unavoidable. Indeed, it is often necessary. I have tried to keep repetition to a minimum, although some readers may consider that I did not try hard enough.
35 At this point I also wish to make some remarks about the conduct of the trial. An extraordinary amount of work goes into a trial of this size and nature and the stakes are high. No doubt these pressures take their toll on lawyers and their clients. Despite these pressures, and notwithstanding the zealous prosecution of their clients’ interests, counsel conducted the trial with a good deal of equanimity and courtesy. This made the herculean task that confronted me far less daunting and much more tolerable than it might otherwise have been. For that, I am truly grateful.
36 On the other hand, the case could certainly have been conducted with greater efficiency.
37 It is questionable, for example, whether it was necessary for the applicants to plead so many causes of action. The applicants’ pleadings were convoluted and, owing to cumbersome cross-referencing, often difficult to follow. The parties quarrelled about points upon which they should have been able to reach agreement. Insufficient judgment was exercised about the number of experts that should be retained in each discipline, the way in which the evidence should be elicited, and the documents that should be tendered. Sources for some submissions were not always provided, which meant that I had to trawl through the evidence to find them for myself. I appreciate that the litigation was a substantial logistical exercise and that it is easy to be wise after the event. I do hope, however, that when the dust settles, both sides reflect on what could have been done better, as I have done.
PART III: THE HISTORY AND DEVELOPMENT OF THE ETHICON DEVICES
38 Despite some sharp and often irreconcilable differences of opinion, there was a good deal of common ground. Unless otherwise indicated, my account of the facts is based on agreed facts, and evidence which was not contradicted or challenged.
The use of synthetic mesh and polypropylene in surgery
39 Prolene is a polypropylene resin first made by Ethicon in the 1960s.18 Subject to certain conditions, it was approved by the Food and Drug Administration (FDA) in the United States in April 1969 for use in non-absorbable surgical sutures.19 Ethicon Inc. had sought approval for its product as a new “drug”. Strict conditions had to be satisfied before approval could be given. Ethicon was required to submit detailed reports of preclinical investigations, including studies on laboratory animals. In order for an animal study to be considered appropriate, proper attention had to be given to the conditions of use recommended in the proposed labelling, including whether the “drug” was intended for short or long-term administration.20 Ethicon conducted studies of tissue reactions to the sutures in rats, rabbits, and dogs. It conducted studies of the tensile strength of the sutures when implanted in rats. Ethicon was also required to submit reports of all clinical testing and all information relating to the evaluation of the safety and effectiveness of the sutures.21 After approval had been granted, Ethicon continued to test its sutures and submitted quarterly reports to the FDA.22
40 Prolene sutures continue to be widely used, including in pelvic reconstructive surgery. Dr Hinoul described them as “functional, safe and effective”.23 The applicants did not suggest otherwise.
41 Synthetic mesh was first used in the repair of hernias in the abdominal wall. Francis Usher was the pioneer. In 1958 he published on the use of synthetic mesh in six dogs to reinforce the abdominal wall and close the abdominal wall and thoracic tissue defects. Dr Usher initially used a woven material made of polyester but rapidly changed to a knitted fabric made of a high-density polyethylene known as Marlex.24 Marlex became increasingly stiff after implantation and at times caused considerable local wound complications, seroma formation, infection, and “stiff belly”.25 At some later point in time, another polymer, polypropylene, was used in the manufacture of Marlex.
42 In the early 1970s, Ethicon developed Prolene sutures into a knitted flat mesh26 and, in 1997, into a three-dimensional form known as the “Prolene Hernia System”.27 Ethicon conducted a good deal of research on Prolene sutures, both before and after it had obtained regulatory approval for their use. But the Prolene Hernia System was cleared for sale on the back of the approval for Prolene sutures, based on their supposed “substantial equivalence”. The first of the Ethicon devices was cleared for sale on the back of the regulatory approval of Prolene sutures and, in part, because of its supposed “substantial equivalence” to the Prolene Hernia System, despite the differences in design, use, anatomy, and site-specific considerations.
Stress urinary incontinence and its treatments
43 Stress urinary incontinence is the involuntary leakage of urine during activities such as coughing, sneezing, lifting, laughing or exercising.28 It is to be contrasted with urge urinary incontinence which is involuntary leakage of urine accompanied by a compelling urge or need to urinate.29 For completeness, overactive bladder is a symptom complex characterised by urgency (the compelling need to urinate), usually with urinary frequency and nocturia (rising at night to urinate) and, sometimes, with urge incontinence.30 Mixed urinary incontinence is a combination of stress urinary incontinence and urge urinary incontinence.31
44 The only symptom of stress urinary incontinence is leakage of urine during activities accompanied by increased abdominal pressure like coughing, sneezing, lifting, running, and various other forms of physical exercise. It does not affect any aspect of the anatomy. It affects the quality of the sufferer’s life. It is undoubtedly distressing. But it is never life-threatening.
45 The urethra (the duct or tube extending from the bladder down to the wall of the upper vagina through which urine is carried out of the body) and the bladder are supported by pelvic floor muscles, which contract during coughing, sneezing and exercise to prevent leakage of urine.
46 Whereas urge incontinence is a “bladder problem”, caused by involuntary contraction of the detrusor muscle), stress urinary incontinence is a “sphincter problem”.32 The urinary sphincter is an arrangement of muscles situated closest to the bladder. Ordinarily, voluntary urination causes the sphincter to relax and the detrusor muscle (the smooth muscle in the wall of the bladder) to squeeze or contract, resulting in the expulsion of urine from the bladder down the urethra and out of the body. Weakness in the muscles or damage to the bladder neck support can result in leakage.
47 There are two types of stress urinary incontinence: urethral hypermobility and intrinsic sphincter deficiency. Urethral hypermobility describes the situation in which the urethra has moved outside the pelvis and activities such as coughing or sneezing (known as Valsalva activities) put added pressure on the bladder, causing leakage. Intrinsic sphincter deficiency simply refers to weak urethral sphincter muscles or poor urethral closure function.33
48 Risk factors include pregnancy and vaginal birth and other conditions which cause an increase in abdominal pressure, such as obesity, chronic cough, chronic heavy lifting and constipation.
49 Treatment is always elective.
Traditional treatments for stress urinary incontinence
50 There are surgical and non-surgical options.
51 Non-surgical (or conservative) treatments include general lifestyle changes, pelvic floor exercises, and the use of continence devices such as a pessary. Conservative treatments are not always successful. Some women experience alleviation of their symptoms; some do not and elect to have surgery.
52 Traditional surgical treatment options include:
(1) Burch colposuspension, named after Dr John Burch who first described it in 1961,34 which is used to correct urodynamic stress incontinence.
(2) needle suspension procedures;
(3) sling procedures using either the patient’s own connective tissue (fascia) (known as autologous slings) or foreign graft material; and
(4) use of urethral bulking agents, involving injection of a variety of different substances around the bladder neck and into the urethral sphincter, to thicken the urethral wall so as to provide greater urethral resistance during increases in abdominal pressure.35
53 In Australia, at least until the late 1990s, the Burch colposuspension was described as “the gold standard” surgical treatment for stress urinary incontinence. Associate Professor Rosamilia said that it had an 85% cure rate at five years, although she did not cite a source.36
54 The Burch colposuspension is performed either through abdominal incision (open abdominal surgery) or laparoscopically (keyhole surgery) in which the retropubic space (the space between the pubic bone and the bladder) is dissected and the neck of the bladder is elevated (or suspended) by sutures passed through the Cooper’s ligament, which borders the femoral ring, and attached to the pubic bone.37 The effect on the anatomy is to prevent or minimise descent of the proximal urethra during increases in abdominal pressure and to create a sling of tissue against which the urethra is compressed;38
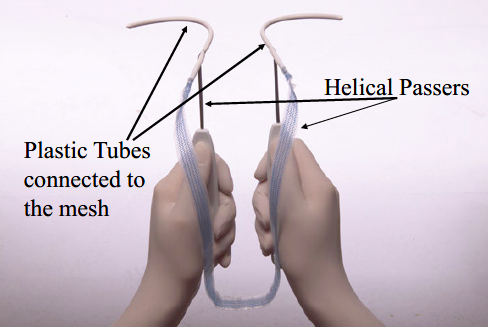
55 The object of autologous slings is to cause the urethra to hit against something during a Valsalva activity so that the urethra momentarily shuts off. A Valsalva manoeuvre, named after an Italian anatomist, Antonio Valsalva, is the action of attempting to exhale with a closed mouth and nostrils. It increases pressure in the middle ear and chest and is used as a means of equalising pressure in the ears.39 The slings are placed under the neck of the bladder and are commonly referred to as “pubovaginal slings”.
56 The autologous fascial sling procedure was first described in 1907.40 Although it was a mode of treatment favoured by Professor Blaivas, it never achieved widespread popularity, because of its high complication rates, particularly in inexperienced hands.41 The complications with which it was associated included increased rates of urinary tract infection, urge incontinence, voiding dysfunction, erosion, and, occasionally, the need for surgical revision to improve voiding.42 It is not in dispute that the complications include continued incontinence, voiding dysfunction, urinary retention, pain, and dyspareunia (pain with sexual intercourse).43 Historically, the procedure was reserved for cases of intrinsic sphincter deficiency or prior surgical failure but there was evidence that it was an effective treatment for all types of stress incontinence with acceptable “long-term” efficacy.44 In 1998, Chaikin et al published the results of a prospective and retrospective study of 251 women with all types of stress incontinence who underwent pubovaginal fascial sling surgery by a single surgeon, and were followed up after a median period of three years (with a range of one to 15 years). Chaikin et al (1998) claimed that they had “demonstrated that the procedure [could] be performed in a reproducible fashion with minimal morbidity”.45 They said that postoperative urinary retention should be minimal if the sling is not tied with excessive tension. They acknowledged, however, that persistent and de novo urge incontinence remained “a vexing problem”. In cross-examination, Professor Blaivas, who was a co-author of the Chaikin article, suggested that in 1998 a five year follow-up would have been considered “long term”, although he would not consider it to be long term now.46
57 The SUI devices are midurethral slings. Midurethral slings may be surgically implanted in a number of ways. They may pass under the midurethra, run behind the pubic bone through the retropubic space (the cavity between the urethra and the muscles above it, the rectus muscles and the peritoneum) and exit above the pubic bone. This type of sling is known as a retropubic sling. Then there are those which are also placed under the midurethra but, unlike the retropubic slings, they pass through the obturator space and exit in the groin. They are called transobturator slings.
58 There are also single-incision slings, sometimes called “mini-slings”, which also pass under the midurethra, but are typically shorter in length than the multi-incision midurethral slings, and can be placed via either the retropubic or the transobturator route.
59 Despite the variety of approaches and materials used, it is widely accepted that all these slings should be placed under “minimal tension” to prevent the development of additional voiding dysfunction, such as obstruction with incomplete voiding.47
60 Every one of these surgical treatment options is attended by risks, although the nature and extent of the risks vary from procedure to procedure. The complications associated with the Burch colposuspension, for example, include urinary tract infection, urinary retention, urethral obstruction, de novo overactive bladder, haematuria, neurologic symptoms, pelvic pain, dyspareunia, and death. The most common complications associated with the other traditional forms of surgery are voiding dysfunction and urethral and urinary retention. They are managed conservatively with either intermittent self-catheterisation or an indwelling catheter. Refractory pain, that is pain that is resistant to treatment, is exceedingly rare after any of these procedures.48 The complications of treatment with the SUI devices are discussed below.
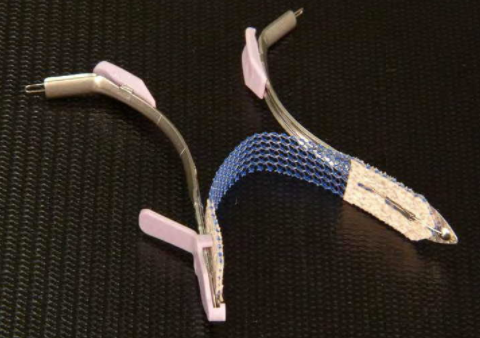
61 Regardless of the nature of the surgery, clinical outcomes appear to be worse for patients who have had previous surgery for stress urinary incontinence.49
62 It is common ground that, before deciding on the most appropriate course or method of treatment, a treating surgeon would consult with the patient, obtain her medical and surgical history, and assess her clinical needs.
The development of midurethral synthetic slings
63 As I have noted, the SUI devices are midurethral slings. The object of midurethral and pubovaginal slings is similar, but, unlike pubovaginal slings, midurethral slings are placed under the midurethra. Presumably for this reason, they are sometimes referred to as “suburethral slings”. The SUI devices are designed to be inserted without tension.
64 Tension-free vaginal tape was the brainchild of an Australian pelvic floor surgeon, Peter Petros. Its story begins in 1986 with two unrelated observations, which I gather were made by Dr Petros. The first was that a haemostat (an instrument for preventing blood flow by compression of a blood vessel) applied immediately behind the pubic symphysis (the immovable joint between the pubic bones in the centre of the pelvis), at the level of the midurethra, controlled urine loss on coughing. The second was that, on implantation, polytetrafluoroethylene (PTFE, such as Teflon) tape created a collagenous tissue reaction around it. These observations generated two hypotheses: that a loose pubourethral ligament causes stress urinary incontinence and that a tape implanted in the exact position of the damaged pubourethral ligament would create a collagenous “neoligament” to reinforce it, restoring function and continence.50
65 Dr Petros began animal studies at the Royal Perth Hospital in 1987 using a tape made from Mersilene, a polyester material produced by Ethicon.51 The tape was inserted in the vaginal cavities of 13 dogs for periods from six to 12 weeks. Apart from a sticky yellow vaginal discharge, there were apparently no “ill effects”.
66 Human studies began over the following two years and continued into the 1990s. Between 1998 and 1999, 30 women underwent a “combined intravaginal sling and tuck” procedure, which involved creating an artificial pubourethral ligament and tightening the suburethral vagina. At the end of six weeks, all patients remained continent.52 Within two weeks of the tape being removed, however, 50% of the patients reported recurrent incontinence.53
67 In the early 1990s, doctors from the Department of Obstetrics and Gynaecology at the University Hospital in Uppsala, Sweden, developed a technique for treating female urinary incontinence under local anaesthetic. Their objective was to restore the pubourethral ligament and the suburethral vaginal hammock. The technique was described in a paper by Professor Ulmsten and Dr Petros entitled “Intravaginal Slingplasty (IVS): an ambulatory surgical procedure for treatment of female urinary incontinence” published in 1995. 54 It involved placing under the urethra a tape-like strip of mesh in a U-shape sling formation using a specially designed instrument referred to as a “tunneller” introduced through paraurethrally dissected canals. The technique was used on 50 patients and drew on previous experimental and clinical studies. Thirty-eight of the 50 patients suffered from genuine stress incontinence, objectively verified, and 12 had symptoms and signs of both urge and stress incontinence. The mesh used was Mersilene (in 37 patients), Goretex (in five), Teflon (in six), and Lyodura (in two). Patients were evaluated post-operatively at intervals of one month, six months, and one to two years.55
68 The work of Ulmsten and Petros appears to have been driven by the prevalence of urinary leakage in post-menopausal women and the fact that the surgical treatment was generally extensive and required general anaesthesia. Ulmsten and Petros considered that there was “a strong need for more precise and simple ambulatory surgical methods to treat female urinary incontinence”. They specified the following criteria for these procedures:
(1) the type of defect causing the patient’s leakage must be defined and described in the work-up, as should the manner in which surgery can restore the dysfunction;
(2) the procedure should allow the patient to return to work shortly after the operation;
(3) the operation should be carried out under local anaesthesia, enabling the patient’s cooperation during the procedure and reducing costs and the number of services otherwise required; and
(4) both subjective and objective evaluation of the patient should be carried out, “recognizing her symptoms and the quality of life situation both before and after the operation”.56
69 Early results appeared promising. The authors wrote that 39 patients (78%) were completely cured of their stress incontinence and another six (12%) reported a considerable improvement of their urinary incontinence, leaking only occasionally. They also wrote that there were no intra or post-operative complications. They acknowledged, however, that, since the technique was new, no long-term results were available, although they speculated that the long-term results of their prosthetic sling would be similar to those involving “conventional” sling procedures. Ulmsten and Petros concluded that if their results were to be reproduced in ongoing follow-up studies including more patients (how many more they did not say) their method “should be considered as a promising new ambulatory procedure for treatment of female urinary incontinence”.57
70 It transpired, however, that, upon surgical insertion, the Goretex and Mersilene tapes were rejected in eight to 10 percent of patients. Further work followed, and in 1996 the results of another study by Ulmsten and others were published.58 This time the tapes were fashioned from Prolene and the technique was said to have been improved. I return to this study later in these reasons. It is sufficient at this point to note that the work of Professor Ulmsten and his team culminated in the production of the Ethicon midurethral sling known as TVT.
71 As noted above, the SUI devices the subject of the current litigation are TVT, TVT-O, TVT Secur, TVT Exact and TVT Abbrevo.
72 Each of the SUI devices consists of a sterile, single use device comprising polypropylene mesh in the form of a tape and a set of instruments to facilitate mesh implant placement. Each was designed for the treatment of stress urinary incontinence resulting from urethral hypermobility or intrinsic sphincter deficiency. TVT and TVT Exact were designed for use as pubourethral slings. TVT-O, TVT Secur and TVT Abbrevo were designed for use as suburethral slings.
The mesh used in the SUI devices
73 The mesh used in the SUI devices is made from Prolene, which, as I mentioned earlier, was developed by Ethicon in the 1960s as a suture material. Prolene consists of a non-absorbable polypropylene base resin and a number of additives. Polypropylene is a polymer made from the monomer propylene, a component of natural gas made from carbon and hydrogen.59 A polymer, I interpolate, is a material composed of long chains of chemical building blocks, called monomers, bonded together. Propylene is polymerised into polypropylene in a chemical reaction in which propylene molecules (monomers) are combined together in a step-wise fashion to ultimately form linear, chain-like macromolecules60 which turn into flakes, chips or pellets.61 In the manufacturing process, efforts are made to control the lengths of the individual chains (referred to as polymer molecular weight) and the variation in lengths between the different chains (referred to as polydispersity).62 The mechanical properties of polypropylene are directly influenced by its molecular weight and/or molecular weight distribution, which is the name given to the range of polymer chain lengths.63
74 Prolene fibres are manufactured by melting polypropylene flakes or pellets in a heated extruder which exit through a spinneret (a metal end cap on the extruder with holes of various shapes) forming strands or filaments.64 During this process, various additives are added to the polypropylene, including:
(1) two antioxidants: dilauryl thiodipropionate (or DLTDP), to improve long-term storage of the resin and fibre and to reduce the potential oxidative reaction with UV light, and Santonox R, to promote stability during compounding and extrusion;
(2) two lubricants: calcium stearate and Procol LA-10 (previously Luberol), to help reduce tissue drag and promote tissue passage; and
(3) a colourant: copper phthalocyanate (or CPC), to enhance visibility when it is desired to have a blue dyed monofilament fibre rather than a clear monofilament fibre.65
75 The additive package in use in 2003 was the same as that used in the original 1991 formulation save that the Santonox levels were reduced slightly (by 0.05%).66 There has been no change since. Neither side contended that the reduction in Santonox was of significance.
76 The single strands of Prolene (referred to as monofilaments) were then knitted into a specific pattern to form the mesh and the mesh was then scoured and annealed.67
77 Finally, the mesh was cut into various sizes. The mesh was originally mechanically cut and sealed using a hot knife.68 From June 2006, laser cutting was introduced after Ethicon was inundated with complaints from surgeons about particle loss. 69 In a PowerPoint presentation from August 2006,70 no doubt designed to showcase the benefit of laser-cut over mechanically cut mesh, samples of mesh that had been laser-cut were compared with samples that had been mechanically cut after both sets had been pulled to 50% elongation and then relaxed.71 The mechanically cut (MCM) samples were described as follows:
The MCM samples show the degradation of the structure of the mesh in certain areas where, because of particle loss, the knit has opened and a portion of the construction has been lost. The area may also be stretched and narrowed resulting in roping due to this occurrence.
78 Accompanying photographs illustrated the description and also showed fraying of the mesh, a phenomenon described by Carol Holloway, Product Complaint Analyst, Worldwide Customer Quality, for Gynecare, in a letter dated 12 October 2005 as “inherent in the product based on the mesh construction”.72
79 In contrast, the laser-cut samples showed no degradation of the structure of the mesh, because no, or nearly no, particles were lost. The knit construction stayed intact and there was no roping. Although the mesh could be stretched and narrowed it was “generally less than the [mechanical cut mesh]”.73
80 The TVT system (sometimes referred to as “TVT Classic” or “TVT Retropubic”) is a sterile, single patient use device made up of one piece of undyed or blue Prolene, in the form of a tape, covered by a plastic (polyethylene) sheath (cut and overlapping in the middle) and held between two curved stainless steel needles (trocars) that are bonded to the mesh and sheath with plastic collars. The trocars (also called introducers) have two parts: a handle and an inserted threaded metal shaft designed to facilitate the passage of the tape from the vagina to the skin of the abdomen.74
81 The tape is 1.1cm wide and 45cm long.75 According to the description in the TVT technical file, maintained by Ethicon, it is about 0.7mm thick.76 It is inserted at the midurethral region, to create a sling on which the urethra can rest when there is a sudden increase in abdominal pressure.77
82 When the device was first supplied, however, it appears that the tape was shorter. It was 40cm, not 45cm, in length.78
83 The images below show the tape (left) and the tape together with the trocars (right):

84 TVT requires the surgeon to use a retropubic (bottom to top) transvaginal surgical approach. The sling is inserted by means of the trocar through a small vaginal incision under the midurethra, passes vertically behind the pubic bone through the retropubic space, and exits through the skin in the lower abdomen. This is known as a “U” shaped sling orientation. The surgeon is unable to see where the trocar goes. It is not in dispute that the blind passage of the trocar creates a risk of perforation of the bladder and urethra and of damage to blood vessels and nerves in the retropubic space.79 Serious complications due to perforation of the large vessels and intestinal viscera have also been reported.80
85 TVT gained regulatory approval in Europe in 199781 and was cleared by the FDA on 28 January 1998.82 On 21 July 1998 it was approved by the Therapeutic Goods Administration (TGA) as a class IIB device for supply in Australia. It was first sold here in October 1999.83
86 Dr Hinoul deposed that TVT was cleared by the FDA on the basis that it was substantially equivalent to the ProteGen sling manufactured by Boston Scientific, one of Ethicon’s competitors, which was already on the market.84 Both TVT and ProteGen were intended for use as pubourethral slings for the treatment of stress urinary incontinence but, as Dr Bryan Allman, a regulatory expert who worked for Boston Scientific at the time, observed, the two devices were very different.85 For a start, they were made of different materials. TVT was made of knitted filaments of extended polypropylene (Prolene). ProteGen was made of woven polyester impregnated with bovine collagen. TVT was entirely non-absorbable. ProteGen had an absorbable collagen coating. The tools were also different. TVT was supplied with a reusable introducer and rigid catheter guide. ProteGen was supplied with disposable single use instruments: a bone locator, a suture anchor system, a suture passer, and a suture spacer. Both were intended for anterior fixation, but TVT was to be fixed to abdominal skin by friction at first and later by tissue ingrowth, whereas ProteGen was fixed by a suture anchored to bone.86
87 Certain modifications were made over time. I have already mentioned the change in the cutting technique. In addition, in 1999 the diameter of the needles used with the device was reduced from 6mm to 5mm.87 A blue version of the TVT tape was developed in October 2001, by incorporating a pigmented blue fibre into the knitted mesh. The stated purpose of this development was to enhance visibility in the surgical field.88
88 Fundamentally, however, the tape did not change. It has always been made from Prolene and the composition of Prolene has not altered in any material respect.
89 TVT-O is also a sterile, single patient use device, which consists of a piece of undyed or blue Prolene mesh tape covered by a plastic sheath overlapping in the middle. Plastic tube receptacles are attached at each end. TVT-O is made from the same Prolene mesh used in TVT and its dimensions are identical. It differs from TVT in only two respects: the tools or instruments supplied with the device and the method of attachment. The tools or instruments consist of two helical passers, shown in the photograph below taken from Dr Hinoul’s affidavit, and a winged guide, which facilitates the passage of the helical passers through the obturator membrane.
90 The helical passers are inserted through the vagina but then pass through the obturator foramen, a large opening between the ischium (the curved bone forming the base of each half of the pelvis and one of the three bones that fuse to form the hip) and the pubic bones, rather than the retropubic space as with TVT and, unlike TVT, exit through the upper legs, rather than the abdomen.89
91 Like TVT, TVT-O was developed by a surgeon. As we shall see, Ethicon’s interest in such a device was largely, if not entirely, motivated by its concern about increasing competition in the marketplace that followed the early success of TVT.
92 TVT-O received regulatory approval in Europe and the United States in December 2003.90 It was first supplied in Australia in March 2004.91
93 TVT Secur was indicated for use in women as a suburethral sling for the treatment of stress urinary incontinence resulting from urethral hypermobility and/or intrinsic sphincter deficiency.92 But it was significantly different from its predecessors in composition, size, and method of fixation.
94 The “Gynecare TVT Secur System” consisted of the implantable device together with a set of instruments to facilitate the placement of the device in the pelvis.
95 The device was a piece of blue Prolene mesh in the form of a tape (8cm long and 1.1cm wide) sandwiched between layers of absorbable fleece (2cm long) made from Vicryl (polyglactin 910) and PDS (poly-p-dioxanone) undyed yarn. The instruments were two metal inserters, a finger pad, a protective cover, and a release wire.93
96 This is what it looked like, with the instruments:
97 The tape was inserted via a single, small vaginal incision. It could be implanted in either a “U” position (which is comparable to a retropubic position) or a “hammock” position (which is comparable to the transobturator sling).94 In the former case, the mesh is implanted towards the retropubic space but not through it. In the latter case, the tape is implanted towards the obturator opening but does not exit the opening or the externus muscle. In neither case is there an exit site.95 Unlike TVT and TVT-O, the mesh in TVT Secur was not covered with a plastic sheath96 and the mesh used to make the tape was always laser-cut rather than mechanically cut.97
98 TVT Secur was the only one of the SUI devices that required a single incision.
99 A single-incision sling is defined as a sling that does not involve either a retropubic or transobturator passage of the tape or trocar and involves only a single vaginal incision so there are no exit wounds in the groin or lower abdomen.98 The background to the development of single-incision slings was described by Nambiar et al (2014):
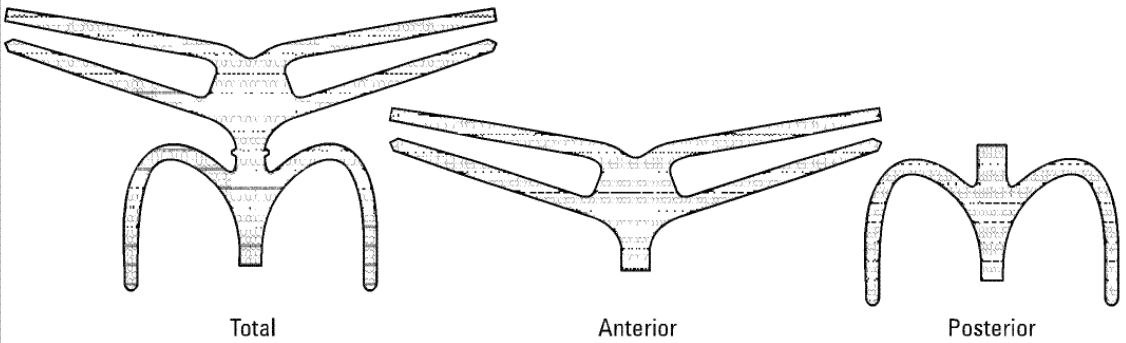
Historically many types of surgery have been performed to treat women with stress urinary incontinence. Over the past 10 years, the accepted standard technique has been the mid-urethral sling operation, whereby an artificial tape or mesh is placed directly beneath the urethra and is anchored to the tissues in adjacent parts of the groin or just above the pubic bone. Examples of such slings that are commonly used are tension-free vaginal tape (TVT™) and transobturator tape (TOT). These operations are usually quite successful, with success rates approaching 80%or 90%. However, they have been shown to result in significant side effects, which can be bothersome and sometimes even dangerous, such as damage to the bladder caused by tape insertion, erosion of the tape into the urethra during the healing period or chronic thigh/groin pain.
In an effort to maintain efficacy while eliminating some of the side effects, a new generation of slings has been developed, called ’single incision slings’ or ’mini-slings’ (sic); these slings are the subject of this review. They are designed to be shorter (in length) than standard mid-urethral slings and do not penetrate the tissues as deeply as standard slings. It was therefore thought that they would cause fewer side effects while being no less effective…99
100 TVT Secur was cleared for sale in the United States on 28 November 2005 and in Europe on 4 May 2006.100 It was launched in Australia in April 2007,101 but sold for less than a year. Sales were halted in March 2008 and its registration was cancelled by the TGA in June 2012.102
101 According to a Clinical Expert Report signed by two of Ethicon’s medical directors on 28 February 2006,103 TVT Secur was intended to address the complication of bladder perforation (with the retropubic approach) and thigh pain (with the obturator approach) using a less invasive procedure.
102 TVT Exact is a retropubic sling made of Prolene mesh, just like TVT, and, despite the respondents’ submission to the contrary,104 at 1.1 x 45cm, the dimensions of its sling are identical to the TVT sling.105 Unlike TVT, however, all the instruments supplied with the device are fully disposable. The mesh is covered by a clear plastic implant sheath and held between two white trocars which are bonded to the implant and the implant sheath. The trocar is slightly thinner (at 4.2mm) than the TVT trocar (at 5.0mm).106
103 TVT Exact was released to the Australian market in July 2010.107
104 TVT Abbrevo gained regulatory clearance in the United States on 1 July 2010, and in Europe in September 2010. It was first supplied in Australia in October 2010.108
105 It is a transobturator sling like TVT-O.109 It is made up of a single piece of laser-cut blue Prolene mesh tape covered by clear polyethylene sheaths and was supplied with a set of instruments to facilitate placement of the device: a placement loop, a pair of helical passers, and a winged guide.110 The mesh used in the system, however, is considerably shorter than TVT and TVT-O (12cm as against 45cm111) and has sometimes been called a mini TVT-O. Unlike TVT-O, the mesh does not need to be trimmed on either end once it has been implanted.112
106 According to a clinical evaluation report dated 17 August 2010 and signed by Dr Hinoul, TVT Abbrevo was designed to reduce the amount of mesh left behind in the body, to reduce pain possibly caused by the presence of the tape in the adductor muscles, and to improve the ergonomics of the TVT-O procedure. Dr Hinoul claimed that this modified TVT-O device “leads to reduction of tissue trauma, and reduction in the total length of mesh left behind in the body to a total of 12cm”.113
Pelvic organ prolapse and its treatments
107 Pelvic organ prolapse is the downward displacement of a pelvic organ, which, in the case of a woman means the uterus, the different vaginal compartments or neighbouring organs such as the bladder, rectum or bowel.114 As Professor Deprest stressed, it is an anatomical finding, that is, a change from the normal anatomy, and it may not be associated with any symptoms.115
108 Prolapse occurs when the muscles, ligaments and fascia (the network of supporting tissues that hold the organs in their correct positions) fall or slip out of place.
109 Pelvic organ prolapse is defined as an anatomical change in which there is downward displacement of a pelvic organ.116 There are three different types of pelvic organ prolapse: uterine prolapse in which the uterus descends, cervical prolapse involving the descent of the cervix (the neck of the uterus) and vaginal prolapse involving the descent of one or more of the compartments of the vagina.117 In a prolapse of the anterior compartment of the vagina, either the bladder or uterus (or, in the absence of a uterus the vaginal vault) bulges into the front wall of the vagina.118 This is also referred to as a cystocoele (from the Greek kustis meaning bladder and kele meaning “tumour”) or urethrocoele. In an apical compartment prolapse (sometimes called a vault, uterine, or middle compartment prolapse), the uterus descends or herniates into or outside the vagina or, if the uterus has been removed, the vaginal vault descends.119 In a posterior compartment prolapse, the rectum (the lower part of the large bowel) or part of the small intestine bulges into the upper part of the back wall of the vagina. The former is known as a rectocoele and the latter as an enterocoele.120
110 Diagnosis is based on a combination of information provided by a patient to her doctor about her medical history and symptoms, and a medical examination. The major cause is vaginal childbirth, which requires the levator ani muscle to substantially distend.121 A levator muscle is a muscle whose contraction causes a part of the body to be lifted. The levator ani muscle supports the pelvic organs and helps to prevent urinary incontinence. Professor Korda explained that, when a woman gives birth vaginally, the levator ani muscle in the average woman stretches by 150%. It is known that if that muscle is stretched in a laboratory setting beyond 150% it will never be able to return to normal tensile strength or length. This overstretching causes the natural “hammock” support of the pelvic floor to disappear and the downward descent of the pelvic organs to occur.122
111 Prolapse is described in stages, indicating the extent of the descent. Different staging or grading systems have been devised, including the Baden-Walker classification and the Pelvic Organ Prolapse Quantification (POP-Q) classification.
112 Risk factors for pelvic organ prolapse include pregnancy and childbirth; ageing and menopause; and conditions that cause excessive pressure on the pelvic floor, such as obesity, chronic cough, and chronic constipation.
113 Other factors which have been implicated in pelvic organ prolapse are asthma, genetic predisposition, and metabolic disorders. Age is universally cited as an established risk factor and uterine prolapse appears to be age-related. But Professor Korda’s evidence was that the general proposition was not supported by unexpectedly high rates of mild or moderate prolapse in young or asymptomatic women and increased tissue stiffness after menopause. He said that anterior and central compartment prolapse appear to deteriorate up until the age of 55 but thereafter improvement occurs.123
114 Symptoms of pelvic organ prolapse in women can include a heavy dragging feeling in the vagina or lower back; the feeling of a lump or bulge in or outside the vagina; urinary symptoms such as slow urinary stream, a feeling of incomplete bladder emptying, urinary frequency (needing to pass urine eight or more times a day), 124 or urgent desire to pass urine; 125 bowel symptoms such as difficulty moving the bowel or a feeling of incomplete bowel evacuation, or needing to press on the vaginal wall to empty the bowel; and pain, including lower back pain.126
115 Lower urinary tract symptoms, such as incontinence, are sometimes associated with prolapse but are not caused by it. Recurrent urinary tract infections can be caused by prolapse due to incomplete bladder emptying.
116 Prolapse can affect sexual function and cause dyspareunia, inability to penetrate the vagina due to obstruction, vaginal laxity, and loss of libido.
117 Depending on its severity or extent, while pelvic organ prolapse is not a life-threatening illness, as Associate Professor Rosamilia observed it can have a drastic effect on a woman’s quality of life.127
Traditional treatments for pelvic organ prolapse
118 As with stress urinary incontinence, there are both non-surgical and surgical treatment options.
119 Non-surgical treatments include lifestyle interventions, pessaries and pelvic physiotherapy. Symptoms may be improved with the use of vaginal oestrogen to improve the epithelium in post-menopausal women, avoidance of constipation and chronic cough, and training the pelvic floor muscles.128
120 Surgical treatment options include reconstructive surgery and vaginal closure or removal surgery (also known as obliterative procedures). The objective of all these treatments is to correct the prolapse while maintaining or improving vaginal sexual function and remedy lower urinary tract symptoms and disorders. Although it is not always feasible, their broad purpose is to restore the so-called normal pelvic anatomy.129
121 Reconstructive surgery may be accomplished using vaginal or abdominal approaches.
122 One of the forms of vaginal closure is colporrhaphy (literally, suturing the vagina in place). This is a procedure used to repair an anterior or posterior vaginal wall prolapse. It involves an incision into the relevant compartment of the vagina and plication (folding) of the pubocervical fascia (connective tissue forming layers between the vagina and bladder) with sutures. The objective is to repair the midline fascial defects by using and tightening up the stretched-out fascia.130
123 Apical prolapse can be repaired vaginally using sacrospinous ligament fixation (also called colpopexy), which involves suspension of the apex of the vagina to the sacrospinous ligament by means of (usually absorbable) sutures or uterosacral ligament suspension, which involves suturing the vagina to the uterosacral ligaments.131
124 A culdoplasty is performed to correct an apical prolapse after hysterectomy. Sutures are used to suspend the vaginal vault at the origin of the uterosacral ligaments, which support the vagina, and to close off the pouch of Douglas (a small area between the uterus and the rectum).132
125 The abdominal procedures include sacrocolpopexy (also called “sacral colpopexy”), which can be performed laparoscopically, robotically or by open surgery via an abdominal approach in the lower abdomen. This procedure is used to correct apical prolapse. The apex of the vagina is stitched or fixed to the sacrum by means of sutures and a small amount of surgical mesh.133 The procedure is also used to correct a combination of apical, anterior and posterior prolapse. It is only available for women who have undergone a hysterectomy. Often the two procedures are performed in the same operation. Another option is an abdominal hysteropexy, which is also used to correct apical prolapse but in which the uterus is preserved. As with a sacrocolpopexy, the apex of the vagina is stitched or fixated to the sacrum by means of a small quantity of surgical mesh, and the surgery can be performed either laparoscopically or by open surgery via an abdominal approach in the lower abdomen.134
126 Obliterative procedures include vaginal closure surgery (colpocleisis), which involves stitching the vaginal walls together to create a barrier in order to prevent the prolapse re-occurring,135 or total colpectomy, which involves the total excision of the vagina in a woman with no uterus and vaginal eversion (that is, where the vagina is turned outwards or inside out).
127 Surgery which uses the patient’s own tissue is commonly referred to as “native tissue repair”.
128 Prosthetic material used during surgery may be made from absorbable material such as animal tissue, non-absorbable synthetic material, or a combination of absorbable and non-absorbable material.
129 There is no single best approach for all patients. Treatment is individualised according to each patient’s symptoms.
130 All surgical treatment options are associated with risks, although the evidence indicates that complications from native tissue repair are generally short-lived and treatable.136 Professor Korda listed them as failure; injury to adjacent and contiguous organs, such as the bladder, urethra, ureters, bowel, major blood vessels, and nerves; the development of lower urinary tract symptoms such as urinary incontinence, narrowing of the vagina, and dyspareunia.137
131 The medical devices used for the treatment of prolapse which are the subject of the current litigation are Gynemesh PS, Prolift, Prolift+M and Prosima.
132 Prolift, Prolift+M, and Prosima were intended for tissue reinforcement and long-lasting stabilisation of fascial structures of the pelvic floor in vaginal wall prolapse where surgical treatment was intended, either as mechanical support or bridging material for the fascial defect. Gynemesh PS had the same indication until March 2013, when its indication for use was changed, as discussed below at [139].
133 Before any of these devices was available, however, it appears that surgeons concerned about the failure rate with native tissue repairs experimented with synthetic hernia mesh to treat pelvic organ prolapse. Dr Hinoul was one of these surgeons. He said he began to use a polypropylene hernia mesh to treat pelvic organ prolapse in the late 1990s or early 2000s. He used a transvaginal approach, inserting the mesh through the obturator foramen using needle drivers and anchoring it in the muscles of the groin using a technique similar to that later used to implant the Prolift device.138
The meshes used in the POP devices
134 The meshes used in all these devices were made from non-absorbable polypropylene (Ethicon’s Prolene Soft, except for Prolift+M, which is made from Gynemesh M.
135 Prolene Soft mesh was developed by Ethicon in the late 1990s for use in hernia repair and approved by the FDA for that purpose. Like Prolene it was and is made from knitted filaments of polypropylene.139 Prolene Soft was described in a memorandum from Dr Ning Wang of Corporate Product Characterization at Ethicon Inc. as “a revised construction [Prolene] mesh, fabricated of knitted filaments of natural color and blue pigmented polypropylene identical in composition to that used in [Prolene]”.140 It differs from Prolene, however, in two respects. It has a smaller filament diameter (at 3.5mm as against 6mm), which is said to give it a softer feel,141and it has a different knit design.142
136 Gynemesh M is a partially absorbable mesh consisting of approximately equal parts of Prolene Soft and Monocryl, an absorbable poliglecaprone-25 monofilament fibre.143
137 Gynemesh PS received regulatory clearance in the United States on 8 January 2002, in Europe on 20 March 2003, and in Australia on 26 May 2003. It was first supplied to the Australian market in July 2003.144
138 Gynemesh PS is made from Prolene Soft mesh.145 The “PS” in its name is an acronym for Prolene Soft. It consists of rectangular sheets of mesh that may be cut as desired by the surgeon. This is what it looks like.

139 According to Dr Hinoul, the only difference between Prolene Soft and Gynemesh PS was that Gynemesh PS was cleared for the specific indication for use in “tissue reinforcement and long-lasting stablization of the pelvic floor in vaginal prolapse”, whereas Prolene Soft was cleared for the general indication for use in “repair of hernia or other fascial defects that require the addition of a reinforcing or bridging material”.146 Ethicon treated the change of indication as a matter of no consequence. Its position was that the change was not a new intended use, which might have led to additional regulatory obligations, but a subset of the previous one, which enabled Ethicon to rely on earlier regulatory approval or clearance. In her 2005 Clinical Expert Report on Prolift, Dr Charlotte Owens, then Medical Director for Gynecare, claimed that this approach arose from the “[r]ealization that pelvic floor disorders are physiologic and functional fascial hernias”.147
140 Between March 2003 and March 2013, Gynemesh PS was indicated for tissue reinforcement and long-lasting stabilisation of fascial structures of the pelvic floor in vaginal wall prolapse where surgical treatment was intended, either as mechanical support or bridging material for the fascial defect. On 16 March 2013, however, the indication for use of Gynemesh PS was narrowed to “a bridging material for apical vaginal and uterine prolapse where surgical treatment (laparotomy or laparoscopic approach) is warranted”.148 In other words, it was no longer indicated for transvaginal use but only for prolapse repair using an abdominal approach. The reasons for the change of indication are discussed later in these reasons.
141 On 17 August 2017, JJM notified the TGA that it would be discontinuing Gynemesh effective immediately and on 22 August 2017 the TGA cancelled its entry on the ARTG.149
142 Prolift was cleared for sale in Europe on 2 March 2005, around the same time in the United States, and in Australia on 30 March 2005. It was supplied in this country in and from June 2005 until 15 August 2012.150 Registration was cancelled on 21 April 2015.151
143 The Prolift pelvic floor repair system consisted of a pre-cut Gynemesh PS mesh implant (featuring a central mesh body and mesh arms) and included surgical tools used to facilitate insertion of the mesh fabric through the vaginal area (the Prolift Guide, the Prolift cannula and the Prolift retrieval device).
144 There were three different Prolift systems: anterior, posterior and apical, and total vaginal repairs (the “Anterior”, “Posterior” and “Total” pelvic floor repair systems respectively). Each system was sold separately and included the same guides, cannulas, and retrieval devices but the numbers of accessories varied. The mesh fabric was pre-cut into different shapes to accomplish the different types of repair, but a surgeon could choose to trim the mesh to suit the patient’s anatomy.
145 “Prolift Anterior” consisted of a piece of Prolene Soft with four arms secured through a transobturator approach.152 Self-evidently, it was designed for the repair of anterior vaginal defects. “Prolift Posterior” consisted of a piece of Prolene Soft with two arms secured using a transgluteal approach.153 It was designed to repair posterior and/or apical vaginal vault defects. “Prolift Total” was a combination of a Prolift Anterior and a Prolift Posterior.154 It was designed for a total vaginal repair. This is how the mesh implants were depicted in the IFU:
146 All three devices were developed for the surgical management of pelvic organ prolapse via a vaginal, as opposed to an abdominal, approach.
147 Dr Arnaud explained the background to the development of Prolift in a memorandum he prepared in January 2007:
In the mid 2000, at a time Gynecare [a division of Ethicon Inc.] was enjoying a great monopolistic position on the market for slings, I thought that the next opportunity for the company could be the development of meshes for pelvic organ prolapse repair. Unfortunately, no one such as Ulmsten had come to me with a great solution and I had to be proactive.155
It was quite obvious that no advance could be made in this area as long as a standardized procedure would not be described.
Thus, I took the initiative of setting up a group of experts in order to work on the development of a standardized procedure which would make sense and would be reproducible by the average gynecologist.
148 Dr Arnaud said that he visited Professor Bernard Jacquetin, whom he described “an indisputable [key opinion leader] in the area of prolapse repair” and invited him to become “the ‘Ulmsten’ of POP repair”. Professor Jacquetin took up the invitation and the two of them chose a number of other French experts to work on the project. Their objectives, according to Dr Arnaud, were twofold: to work on developing a standardised technique for the surgical management of pelvic organ prolapse with mesh via a vaginal approach and to try to better understand the mechanism of vaginal erosion which was associated with the use of synthetic materials in order to reduce its occurrence. The group of experts, which became known as the “TVM Group”, met for the first time in June 2000. Gynecare France was placed in charge of its logistic and material coordination.156
149 Before going any further, I should explain what is meant by “erosion” in this context. Like a good deal of the nomenclature in this area, different terms are sometimes used to mean the same thing and there is a lack of precision in the terminology. Some of the professional associations have tried to do something about it, but the evidence in the present case suggests that their efforts have not had any significant effect.
150 The medical definition of an erosion is the “state of being worn away, as by friction or pressure”.157 In 2011 the Working Group on Complications Terminology, established by the International Urogynecological Association (IUGA) and the International Continence Society (ICS), considered that this definition was not necessarily apt to describe the clinical process and recommended that the term be abandoned and replaced with two new terms:
Exposure: A condition of displaying, revealing, exhibiting or making accessible … (e.g. vaginal mesh visualized through separated vaginal epithelium …)
Extrusion: Passage gradually out of a body structure or tissue … (e.g. a loop of tape protruding into the vaginal cavity …)158
151 Sometimes, the evidence indicates, the term “erosion” is used to refer to the migration of mesh into an organ, like the bladder or the urethra, which is an extension of the process of extrusion described in the IUGA/ICS lexicon and “exposure” to refer to anything short of that. Often, “erosion” is used in the literature, and also by some of the expert witnesses in this case, as a synonym for exposure and/or extrusion. Other commentators used the term “protrusion” in lieu of extrusion.159 Generally speaking, in this judgment I use the terms used by the witnesses when I refer to their evidence and the terms used in the scientific literature when I refer to that evidence. Otherwise, I may use the term “erosion” to capture any or all of these events.
152 I now return to the narrative.
153 Once he was convinced of the viability of the TVM project, Dr Arnaud set out to persuade others within Ethicon. In his memorandum on the development of Prolift to which I referred above, Dr Arnaud confessed to having had a hard time persuading some of “the marketing people” of the wisdom of the project. He said that they “wanted to kill [the] project as they found the procedure was too difficult and could only be a procedure for some happy few”. By early 2005, however, Dr Arnaud had prevailed160and, once Ethicon agreed to proceed, the evidence indicates that the marketing people enthusiastically embraced the project. It was only a matter of a few months before regulatory approval was obtained and the Prolift device was released to the market.
154 I hasten to add that the fact that this initiative was taken to exploit a marketing opportunity does not mean that advances in surgical treatment for pelvic organ prolapse were not desirable. There was a common perception that the rate of recurrent prolapse after native tissue repair was too high. The TVM Group put it at 20 to 30%. Surgeons had experimented with mesh as a means of reducing the rate, but the Group noted that “[a]lthough meshes … have been used to repair type 3 or 4 prolapse recurrence, authors still do not recommend their use for primary repair because of reported complications”.161 The desire to improve the status quo through innovation and to commercialise the resulting technology was not in itself problematic. It is, however, part of the context in which to consider the more critical question, which is whether the clinical data available to Ethicon were sufficient to support the conclusions expressed in its pre-market clinical evaluation report for Prolift and the decision to take the device to market, a question to which I will come in due course.
155 It will be recalled that one of Dr Arnaud’s two objectives in developing meshes for the repair of pelvic organ prolapse was to come up with a standardised procedure which the average gynaecologist would understand and could reproduce. Ethicon described the objective of the Prolift procedure as the achievement of “a complete anatomic repair of pelvic floor defects in a standardized way”.162
156 The technique for inserting the Prolift prostheses is described in a 30-page illustrated guide entitled “Gynecare Prolift – Total Pelvic Floor Repair, Anterior Pelvic Floor Repair, Posterior Pelvic Floor Repair – Surgical Technique” (the Guide).163 The metadata in the electronic court book indicates that this document was completed in early January 2005. There is no evidence as to its distribution but Dr Hinoul deposed that the Prolift IFU advised surgeons to review the Guide because it instructed them to refer to “the recommended surgical technique” for “further information” on the Prolift procedures.164 There was no relevant mention of a guide or guidebook in the Prolift IFU. It was mentioned in the Prolift+M IFU, which instructs surgeons to contact their “company sales representative” to obtain the Guide.165
157 A summary of the surgical technique, explained in greater length in the Guide, appears in the first publication on the technique. That was an article by Fatton et al (2007) entitled “Transvaginal repair of genital prolapse: preliminary results of a new tension-free vaginal mesh (Prolift™ technique) – a case series multicentric study”.166 The authors explained that the technique had been refined over a five-year period through more than 600 surgical interventions by nine French skilled vaginal surgeons. They described the technique in this way:
The synthetic material is a pre-cut non-absorbable monofilament soft prolene mesh. The mesh has three distinct parts … The anterior part is inserted between the bladder and the vagina and secured bilaterally by two arms through each obturator foramen. The posterior part is placed between the rectum and the vagina and is secured bilaterally by one arm passing through each ischiorectal fossa and sacrospinous ligament. The intermediate section corresponding to the vaginal apex separates the anterior and posterior parts and can be cut if needed. Instruments were designed to facilitate proper implant placement … Cannula-equipped guides were used to optimize the passage through the tissues, preventing muscle trauma and disruption of the arcus tendinous fascia pelvis (ATFP) and to allow the placement of the retrieval device. Retrieval devices were provided to easily catch and smoothly pass each prosthesis strap through the pelvis …
This procedure may be performed under spinal or general anesthesia. All patients were placed in the lithotomy position with thighs flexed at approximately 90°. After cleaning the entire surgical area with antiseptic, a urine culture is performed and an in-dwelling catheter is placed. All patients had an intravenous perioperative antibiotic prophylaxis.
158 More detail is provided in the article. It is unnecessary to repeat it here. It is sufficient to make the following observations.
159 First, is common ground that the technique is difficult and that it involved complex pelvic surgery.167
160 Associate Professor Rosamilia described Prolift surgery as “technically challenging”. She believed it required “considerable skill with sacrospinous colpopexy and deeper dissection”.168 She said that surgery with anchored mesh kits (like Prolift or Prolift+M) is “more complex” than native tissue repair.169
161 Second, although Fatton et al stated that “[p]rospective evaluation of the functional outcome [was] necessary to support the widespread use of this technique and to recommend it to young women with weakened tissues”,170 no such evaluation took place before Prolift was released to the market.
162 Prolift+M obtained regulatory clearance in Europe on 18 March 2008, in the United States on 15 May 2008, and was first supplied in Australia in December 2009.171
163 As I mentioned earlier, Prolift+M was made from Gynemesh M.
164 Like Prolift, there were three different Prolift+M systems: for anterior, posterior and apical, and total vaginal repairs (Prolift+M Anterior, Posterior and Total). Each system was sold separately and included the same guides, cannulas and retrieval devices supplied with Prolift. Like Prolift, the mesh was pre-cut into different shapes according to the different types of repair. The surgical technique was the same. A surgeon could also choose to trim the mesh to the patient’s anatomy.
165 This is what it looked like:
166 A is Total, B Anterior, and C Posterior.
167 Prolift+M was designed to address problems with Prolift. Yet, when Prolift+M was launched, Ethicon did not withdraw Prolift from sale and continued to promote it. In fact, all the devices remained in the Australian marketplace until all the POP devices except for Gynemesh PS were “decommercialised” on 15 August 2012, a little over three months after they were “decommercialised” in the United States. Registration was cancelled on 21 April 2015. 172
168 Prosima was first supplied in Australia in April 2010. It was not supplied after 15 August 2012 and, along with the other mesh kits, the TGA cancelled its registration on 21 April 2015.173
169 The Prosima system was the brainchild of an Australian surgeon, Marcus Carey, with whom Ethicon entered into a consultancy agreement in September 2004.174 The agreement offered Dr Carey incentives in the form of milestone payments which included: payment of USD 100,000 to be paid after publication of clinical study results in an internationally recognised, peer reviewed journal; and payment of USD 400,000 after the device became available for sale in the United States or the European Union in accordance with the terms of the agreement. Dr Carey was also entitled to royalties on the net sales of the product. This incentive arrangement meant that Dr Carey’s research, discussed below, was potentially tainted by bias or, at least, the perception of bias.
170 The development of Prosima was known internally at Ethicon as “Project Mint”. Like Prolift+M, Prosima was conceived and designed to improve perceived deficiencies in predicate devices.
171 “Mint” was regarded as an opportunity to expand the customer base. A PowerPoint presentation dated May 2006 entitled “Project Mint: Volume Estimate Summary”175 by Ethicon Women’s Health & Urology stated that “Mint [had] potential to be positioned: … [f]or patients whose prolapse is not severe enough to warrant Prolift” and was seen as an “[o]pportunity to expand [the] patient base” to include women with stage 2 and 3 prolapses.
172 Notwithstanding the safety concerns in the medical community related to the use of Prolift, the document identified a “[n]eed to find opportunities for Mint that do not cannibalize Prolift”. As always, Ethicon did not contemplate replacing a device because of safety issues, only introducing an additional one which might attract new customers or customers who were turning away from the old one.
173 Like the other mesh kits, there were three different Prosima systems: for anterior, posterior and apical, and combined vaginal repairs (Prosima Anterior, Posterior, and Total). Like Prolift, Prosima was made from Gynemesh PS. But the device was a different size and shape from Prolift and it could be placed either in the anterior or posterior position or, using the combined system, in both, as required. This is what the mesh looked like:
174 Two different inserter tools to facilitate placement anteriorly and posteriorly were included with the Prosima Anterior, Posterior, and Total systems as appropriate.
175 Unlike the Prolift and Prolift+M systems, the Prosima system provided maintenance of the vaginal canal during the period of healing following surgical repair of vaginal wall prolapse with a silicone vaginal support device (VSD) and a balloon. A surgeon could choose to trim the mesh or the VSD to suit the patient’s anatomy. Dr Hinoul explained that the VSD was designed to maintain the mesh in place as tissue ingrowth occurs.176
176 The VSD and balloon were inserted in the vaginal canal by the surgeon following implantation of the mesh fabric. The balloon was then inflated using a syringe which was supplied with the VSD. This replaced the need for gauze packing of the surgical area and provided support to the patient’s vaginal tissues post-operatively. The balloon was designed to be deflated and removed after 24 hours. The VSD could be left in the patient for up to four weeks post-operation in order to provide further post-operative support to the patient’s vaginal tissues before being removed by the patient’s surgeon.
177 In the case of Prosima, for implanting the anterior prosthesis, an incision was first made similar to that which was made to implant Prolift. Next, a full thickness dissection was undertaken with two channels created on either side of the bladder to the pelvic sidewall in the direction of the ischial spine.177 The arms were then introduced by means of an instrument called an introducer, so that the distal part of the arm was flush with the parietal fascia of the obturator internus muscle. The body of the device had a proximal tab which allowed for the mesh to be tacked to the apex of the vagina during placement of the arms.178
178 For the posterior prosthesis, a full thickness vaginal wall dissection was again carried out. The dissection was then extended on either side of the rectum to the levator ani muscle at the level of the ischial spine. After this extension, the distal parts of the straps or arms were placed up to and abutting the sacrospinous ligaments using an instrument called an inserter. Then the VSD was inserted to provide support for the vaginal tissues in the post-operative period.179
179 Professor Collinet maintained that the technique was standardised and reproducible.180 But the applicants argued that standardisation was never likely to succeed because of the known variations in surgeon technical skills as well as patient tissue and anatomy, the tendency for surgeons to modify their technique, the desire of surgeons to be able to individualise the technique to individual patients, and the unpredictable nature of the tissue response, including contraction.
180 The applicants’ argument was supported by Professor Korda, whose evidence I accept. Despite Professor Collinet’s position, it was not seriously challenged. What is more, it accords with common sense. Professor Korda stated:
Given the variability of tissue thickness, compliance and vaginal anatomy in individual patients, the difference in surgical training, skill and ability of individual surgeons and the level of precision required to place the mesh at a depth so it does not erode, it is almost impossible to get the mesh placement in a perfect position in the hands of ordinary skilled gynaecologists, urogynaecologists and urologists.
It is my view that given the inconsistency of the skill level of ordinary skilled surgeons, the variation of the vaginal anatomy in individual patients and the level of precision required for mesh placement, mesh implant surgery is not within the skill base of the average surgeon and as a result a standardised technique cannot realistically be achieved. I do not believe that it is ever possible to achieve a “reliably safe result” as asserted by Professor Collinet.181
181 Dr Agur expressed a similar opinion. He acknowledged that the TVM studies attempted to standardise the surgical techniques but said it was inevitable that variations would occur in the hands of ordinary surgeons. He referred in particular to variations in the nature of dissection, in the precise placement of the mesh, in the amount of tension applied, and in the use of transfixing sutures. He also said that expanding the use of mesh to general gynaecologists and urologists with variable technical and patient selection skills inevitably introduced significant variations. He said that the large surface area of the mesh resulted in the presence of mesh in in the lateral pelvic wall close to nerves and muscles, where it was not needed, () and that the use of trocars to guide placement was inevitably associated with the increased risk of organ damage. His evidence was that the so-called standardised technique was “not reliably safe” and its long-term efficacy was not yet established, not even in the hands of sub-specialist urogynaecologists and experienced pelvic floor surgeons.182 He noted that, when a group of experienced surgeons embarked on testing the reproducibility of the results of the TVM study in their own hands, within a research context and independent of the manufacturer, they stopped the trial prematurely due to the high incidence of adverse events.183 This was a reference to the Iglesia study published in 2010,184 discussed below.
182 Professor Korda’s view was that, before transvaginal mesh was released to the market, studies should have been published demonstrating the reproducibility of the technique with a cohort of competent ordinary skilled surgeons who would ultimately use it. As we shall see, no such studies were published.
PART IV: THE RISKS POSED BY THE USE OF THE ETHICON DEVICES
183 In this Part of the judgment, I identify and describe the adverse events the applicants alleged could be caused by the Ethicon devices. In Part VI, I discuss the evidence about the prevalence of the complications and their clinical significance. Later, in Part XI, I look at the warnings and other information supplied by the respondents about these matters.
184 The applicants pleaded that the SUI devices could cause the following complications, referred to in the pleadings as “the Tape Complications”:
a chronic inflammatory reaction of the tissues surrounding or attached to the implants;
extrusion or erosion of the mesh into surrounding organs, including the vaginal wall, bladder or urethra;
infection;
chronic pain;
dyspareunia and/or apareunia (avoidance of sexual intercourse);
difficulty voiding;
offensive vaginal discharge;
de novo or recurrent urinary incontinence;
damage to surrounding organs, nerves, ligaments, tissue and/or blood vessels;
haemorrhage;
leg weakness;
reoperation or revision surgery associated with complications; and
psychiatric injury.185
185 The applicants alleged that the POP devices could cause the same complications with the addition of difficulty defecating and recurrence of prolapse. The chronic inflammatory reaction was described slightly differently, as “a chronic inflammatory reaction, or an inflammatory reaction which is not minimal or transient in nature, of the tissues in which the mesh implants were implanted, attached and in addition, or alternatively, the surrounding tissues”.186 I do not take the applicants, however, to have intended to make any distinction. In other words, this was a distinction without a difference. It is apparent that the applicants’ intention was to refer to the foreign body response which inevitably occurs when a medical device is implanted and which is described in detail in Part V of these reasons.
186 In relation to the POP devices, they also listed as potential complications “protrusion” and “exposure”, but in the end no distinction was made between the SUI and POP devices in this regard. Likewise, the applicants pleaded that one of the complications attending the POP devices was the need for surgery to remove the mesh implant as a consequence of those particular complications, but this would have been required for the SUI devices too and, in any event, was covered by other alleged complications, such as “reoperation or revision surgery associated with complications”.
187 The applicants further alleged that both the SUI and the POP devices were difficult, if not impossible, to remove safely from patients suffering from one or more of the pleaded complications, that one or more surgical procedures might be required, and that removal carried the risk of new complications or of aggravating existing complications. In the pleadings, these matters were referred to as the “Tape Removal Complications” or the “Mesh Removal Complications”, as the case may be.
188 In their defence, the respondents asserted that “all surgical procedures present risks” but otherwise denied these allegations. The basis for the respondents’ denial was obscure. It did not emerge in evidence and early in the cross-examination of Dr Hinoul it quickly became clear that the denial was unsustainable.
189 In cross-examination, Dr Hinoul acknowledged that, from the time each of the devices was first supplied anywhere in the world, Ethicon knew of its potential to cause each of the pleaded complications. He conceded that, from the time of first supply, Ethicon was aware that a foreign body reaction to surrounding tissue would create a scar, that the mesh could be subjected to a contracting force applied by surrounding scar tissue, that the response of the host tissue was variable, and that any significant degree of contraction could lead to pain as could the scarring itself.187 Furthermore, he admitted that from that time Ethicon knew that there was a risk of mesh exposure and extrusion into the vaginal canal or another organ, that mesh exposure or extrusion could be difficult to treat, and that it could cause pain or discomfort.188 He also admitted that at the date of first supply Ethicon knew that both mesh erosion or extrusion and pain could occur many years after any of the devices had been implanted.189 Moreover, he admitted that Ethicon knew at that time that implantation carried a lifelong risk of erosion and pain,190 as well as risks of: dyspareunia and, as a consequence, apareunia; difficulty voiding; difficulty defecating; offensive discharge; leg weakness; and damage to surrounding organs, ligaments, tissues, and blood vessels.191
190 Dr Hinoul also conceded that Ethicon knew at that time that both acute and chronic pain could be caused by each of the devices, that chronic pain could be very damaging and debilitating, indeed “life altering”, and that multiple operations might be necessary to attempt to alleviate the pain. He agreed that the mesh could be difficult, if not impossible, to remove safely or without complications and that, in the case of Prolift, it could be disastrous. He said that Prolift could be removed safely but admitted that there was always a risk in so doing of causing damage to surrounding structures.192 He also admitted that, at the time each of the Ethicon devices was launched, Ethicon knew that, in the event of complications, the original condition (stress urinary incontinence or pelvic organ prolapse) could recur.193
191 Ultimately, then, there was no dispute that all of the complications could be caused by implantation of the Ethicon devices. What is more, lead counsel for the respondents, Mr Finch SC, told the Court in closing argument that the respondents accepted that each of the pleaded complications was clinically significant.194
192 These concessions were consistent with what appeared in Ethicon’s internal documents, which makes the respondents’ pleading all the more puzzling. In a Health Hazard Evaluation, for example, completed on 10 January 2014, Dr Hinoul acknowledged that “[n]o new adverse events [had] been identified since the initial launch of Ethicon Inc’s mesh based solutions…”.195
193 During cross-examination, the only qualifications Dr Hinoul made were that many of the pleaded complications could occur with any kind of surgery or pelvic floor surgery and/or that, at least some of them, were “small”, “very”, “highly” or “extremely” unlikely, “rare” or “extremely rare”.196 Dr Hinoul said that he was relying on reports in the literature and his personal experience.197 Both were unreliable foundations for an opinion as to the likelihood of complications and particularly at the time the devices were launched, having regard to the absence of data from long-term studies and properly conducted randomised controlled trials.
194 Be that as it may, in the Health Hazard Evaluation to which I referred above, Dr Hinoul said that:
Ethicon Inc. does acknowledge that in a small, yet significant subgroup of patients, these complications are to be considered serious.
195 Still, Dr Hinoul’s evidence exposed the real dispute. There was no issue that the devices could cause the pleaded complications. The dispute largely turned on the magnitude of the risks, the gravity of those risks, and the extent to which the respondents were obliged to disclose them.
196 After the trial concluded, the applicants applied for, and were granted, leave to amend their pleadings so as to add an allegation that the respondents failed to give any or any sufficient information or warning that the chronic inflammatory response to the implants could be affected by conditions which affect the immune response and healing, including autoimmune and connective tissue disorders. My reasons for doing so are set out in Gill v Ethicon SÀRL [2018] FCA 470 at [81]–[107] (Gill (No 1)). For convenience, I will refer to these conditions as “autoimmune disorders”.
197 From now on I will refer to all the risks discussed above, including the heightened risk of a chronic inflammatory response for women with autoimmune disorders, as “the pleaded complications”.
The nature of the pleaded complications and the circumstances in which they can arise
198 Notwithstanding the concessions made by Dr Hinoul, it remains necessary to refer to other evidence about the risks. There is a large body of such evidence, a good deal of which was doubtless generated by the respondents’ defence.
199 The respondents emphasised that several of the complications could occur regardless of the use of mesh. But the weight of the evidence indicates that the use of mesh causes complications of a kind, degree or rate different from or greater than those associated with traditional methods of pelvic floor repair. These complications cannot wholly be explained by insufficient surgical training or experience. Other factors, including product design, mesh porosity, the quantity of mesh used, the route and methods of implantation and patient-specific factors cause or contribute to the development of adverse reactions following the implantation of synthetic mesh, including the various Ethicon devices.
200 In this section of the judgment, I discuss the nature of these complications.
201 The evidence on this question was given largely by the urogynaecologists, gynaecologists, and urologists.
202 It is convenient at this point to introduce them and to provide a snapshot of their qualifications and experience.
203 Professor Korda is an Australian clinician and academic. The bulk of his career was spent at the King George V and Royal Prince Alfred Hospitals in Sydney either on staff or as a Visiting Medical Officer. From 1991 to 1997 he was the Head of the Department of General Gynaecology and Urogynaecology at those hospitals and from 1996 to 2013 he was the Chairman of the Pelvic Floor Unit at Royal Prince Alfred and Concord Hospitals. He was also a Visiting Gynaecologist at the Mater Misericordiae Hospital in Sydney from 1986 to 2001. He was a clinical lecturer in obstetrics and gynaecology at the University of Sydney from 1975 to 2013. In 2008 he was appointed Foundation Professor of Obstetrics and Gynaecology at Western Sydney University and in 2015 Conjoint Professor.
204 Dr Agur is a consultant gynaecologist and obstetrician and lead urogynaecologist at the University Hospital Crosshouse in Kilmarnock, an Honorary Clinical Senior Lecturer at the University of Glasgow and a Career Fellow with NHS Research in Scotland. He is considerably younger than Professor Korda. He did not become a member of the Royal College of Obstetricians and Gynaecologists until May 2003 and did not attain subspecialty accreditation in urogynaecology until 2009.
205 Professor Blaivas is Adjunct Professor of Urology at SUNY Downstate Medical School in Brooklyn New York, a position he has held since 2008, and has practised as a urologist since the late 1970s. He has also published widely in his areas of expertise.
206 Dr Margolis is Assistant Clinical Professor in the Department of Obstetrics & Gynaecology at the David Geffen School of Medicine at the University of California, Los Angeles and also works in private practice. He has held a variety of academic positions over the years at a number of prestigious American universities in the fields of obstetrics and gynaecology, including urogynaecology and pelvic surgery.
207 Professor Collinet is a professor of medicine, consultant gynaecologist, and head of the gynaecologic surgical unit of the Jeanne de Flandre Hospital in Lille, France. He obtained his qualifications in gynaecology and obstetrics in 2003. After the launch of Prolift, he was engaged on several occasions by Ethicon to take part in and lead training sessions around the world in surgical techniques and, in particular, the implantation of Prolift. His work in urogynaecology, both clinical and research, was mostly confined to a period of seven years, ceasing in 2012. During that period he undertook hundreds of prolapse operations using vaginal meshes and treated what he referred to in his report as “specific complications” identified as prosthetic erosions and retraction.198 Since then he has conducted about 20 surgical prolapse procedures a year.199
208 Professor Deprest is a consultant urogynaecologist, a professor of obstetrics and gynaecology at the Catholic University in Leuven, Belgium and an honorary professor at University College, London. He is also the co-director of the multidisciplinary pelvic floor unit at Universitair Ziekenhuis (UZ) Leuven, the largest university hospital in Europe.200 He has a research interest in pelvic floor dysfunction, including aetiology, secondary prevention and treatment. Since 2002 he has been involved in translational research in urogynaecologic surgery and has studied the host response to biological graft materials and synthetic mesh implants. His clinical research has mainly focussed on the outcomes of specific operations including laparoscopic sacrocolpopexy, native tissue repair of anterior vaginal wall prolapse, vaginal mesh surgery and tape insertion. He is the author of over 470 peer-reviewed articles published in the international literature and over 60 book chapters. At the time he prepared his report, he was a member of the board of the Group of Biomedical Sciences and he has held various positions with a number of professional organisations including the IUGA, the European Uroyanecological Association (EUGA), the European Society of Gynaecological Endoscopy, and the Flemish Society of Obstetrics and Gynaecology. He was also involved in the IUGA working party on meshes in prolapse and incontinence.201
209 Professor Roovers is a Dutch urogynaecologist. He completed his training as a gynaecologist in 2006 and the same year undertook an eight-month fellowship in urogynaecology at the UZ in Leuven. Since he has been head of the gynaecology department at the Academical Medical Centre in Amsterdam and was appointed the chair of gynaecology at the University of Amsterdam in 2014. Since 2016 he has also been an honorary professor at the University of Cape Town in South Africa. He was a supervisor of Dr Hinoul’s PhD thesis.202 He is a former chair of the education committee of the IUGA and is currently the chair of the scientific committee of the EUGA.203
210 Associate Professor Lam and Associate Professor Rosamilia are both Australian practitioners.
211 Associate Professor Lam is the director of the Centre for Advanced Reproductive Endosurgery in Sydney. He currently holds appointments as clinical associate professor at Sydney Medical School, gynaecological endoscopic surgeon at Royal North Shore Hospital, The Mater Hospital, and St George Private Hospital. He is a board member of the World Endometriosis Society and the International Society for Gynecologic Endoscopy and a former president of the Australasian Gynaecological Endoscopy and Surgical Society.204
212 Adjunct Associate Professor Rosamilia is a urogynaecologist. Since 2008 she has been the head of the pelvic floor clinic at Monash Medical Centre, one of three tertiary obstetrics and gynaecology teaching hospitals in Melbourne.205 She has been heavily involved in urogynaecology education and has held a number of leadership roles in various professional societies, including the IUGA and the Australasian Gynaecological Endoscopy and Surgical Society. Since 2010 she served as a urogynaecology member of the Urogenital Prostheses Clinical Advisory Group to the Department of Health.
Complications directly associated with the use of mesh
213 Professor Deprest described a number of complications directly associated with the use of mesh (including the slings made from the mesh), sometimes called “graft associated complications” or “graft related complications”.206 They included erosion; contraction or shrinkage; infection; pain; and reintervention.
214 Mesh can extrude, or “erode”, into the bladder and/or the urethra. Professor Chughtai described mesh erosion as “a debilitating complication of transvaginal mesh placement, which can require a myriad of surgeries to repair” but which may have no remedial effect.207 Professor Deprest said that long-term extrusions into the bowel are rare with stress urinary incontinence devices but more common in mesh repair for prolapse, particularly if the device is placed between the rectum and vagina.208
215 Erosion (commonly referred to as exposure) refers to the exposure of the foreign or graft material to the vaginal lumen (canal). Frequent symptoms of exposure are vaginal bleeding, pain, and dyspareunia.209 Recurrent urinary tract infections, haematuria (blood in the urine), and irritative bladder symptoms can also be symptoms of erosion. Exposure of the mesh graft through the vaginal epithelium can also be associated with vaginal discharge, pain and recurrent infections.210
216 Mesh exposure can occur immediately or soon after surgery. But it can also occur much later, including years after surgery, and away from the incision line.
217 It appears that the mechanism by which mesh erodes (extrudes or becomes exposed) is not known.211 Professor Deprest said that several mechanisms have been suggested including thinning of the epithelium on account of aging or menopause, for example, and an active process occurring underneath the epithelium, such as folding of the mesh or blood collection.212 He said that erosion which occurs sometime after surgery is likely to be due to either migration of the mesh or secondary loss of tissue integrity over the area of the implant. Consequently, he attributed late onset exposure to erosion of the covering epithelium (the membranous lining).213 Although Dr Hinoul was quite insistent that the mesh does not migrate or move,214 the weight of the evidence is against him.
218 Professor Korda considered that mesh erosion occurs because the mesh is a rigid substance and during movement, such as sitting or standing or sexual activity, the vagina stretches, but the mesh cannot stretch at the same rate or time. The result, he said, is that the vaginal tissue abrades against the mesh, which can cause the mesh to erode.215 While he deferred to the biomedical scientists and pathologists in this respect, his evidence was supported by Professor Klosterhalfen. Of particular relevance to this matter is the evidence of Professor Klosterhalfen about the “mechanical mismatch” between polypropylene mesh and vaginal tissue, discussed in Part V below.
219 Professor Klinge said that erosion can be caused by contraction of the mesh (strictly contraction of the mesh/tissue scar complex).216 Professor Klosterhalfen said that it could be caused by folds and curls in the mesh due to bridging of the scar tissue (bridging fibrosis).217 Both these matters are discussed at length in Part V.
220 Professor Blaivas said that erosion can be caused by surgical error where the mesh is inadvertently passed through the lower urinary tract or vagina at the time of surgery. But he discounted this as a factor in “the modern era” since almost all patients undergo cystoscopy and vaginal inspection at the time of implantation. He also said that erosion can be caused during surgery if the mesh is placed too close to the vaginal epithelium or too close to the lower urinary tract or in its wall. Finally, he said that erosion may occur because of the acute and chronic inflammatory process, sometimes associated with bacterial infection, much in the same way that a seroma (a collection of fluid under the surface of the skin) or subcutaneous abscess (a collection of pus under the skin) can burst through the skin.218
221 Notes from a “mesh exposure meeting” produced by Ethicon, identified a number of potential causes of mesh exposure, including mesh folding, mechanical irritation, infection, surface biocompatibility, the use of the transvaginal route, as well as patient factors such as a thin vaginal wall and poor collagen in scar tissue.219
222 It was common ground that patient factors, such as obesity, poorly-controlled diabetes (associated with more infections), vaginal atrophy and smoking, may all impede wound healing220 and that delayed or inadequate healing has been identified as a cause of mesh exposure.221
223 Notwithstanding the attempt by Dr Hinoul to minimise its significance, exposure may have serious consequences. Professor Korda explained that, since the vagina is never free of organisms (bacteria), once the mesh is exposed or erodes, the vagina “inevitably” becomes infected and the nidus (focus) of the infection is virtually impossible to treat because antibiotics have great difficulty penetrating through the mesh.222 He explained that antibiotics could not get to the bacteria sitting on the implant as there are no blood vessels in the mesh fibres. Professor Korda also said that chronic inflammation is the result of exposed mesh and exposure may also aggravate chronic inflammation, particularly when bacteria colonise the exposed mesh.223
224 Professor Deprest made the same point. He reported that when the mesh is exposed to the vaginal lumen, “it will almost certainly become infected” as resident bacteria will attach to the implant. Infection can also occur, he added, as “a primary process” and when it does it can cause mesh exposure.224
225 Professor Korda pointed out that the relationship between inflammation and pain has been known since the first century AD. Along with redness, heat and swelling, he said that pain is one of the classic signs of inflammation.225
226 None of this evidence was apparently controvercial. Professor Korda was not cross-examined on these points. It was logical and persuasive. There is no good reason why I should not accept it and I do. I will return to the question of infection a little later.
227 The next graft-related complication to which Professor Deprest referred was pain, which can occur with or without mesh exposure. Professor Deprest said that pain can be felt along the entire trajectory of the implant, which in the case of retropubic tapes can be around the urethra and in the case of transobturator tapes through the muscles. Sometimes it is difficult for patients to localise their pain. Professor Deprest explained:
It is believed that this process starts locally, yet when the problem of pain is long standing (more than 3 to 6 months), the pain may become “centralized”. This means the process becomes less dependent on what happens locally, yet with the unfortunate side effect that even local removal of the causative factor may not solve the problem. However, this is phenomenon is seen in the absence of implants, and is sometimes referred to, without any criticism of the patient, as ghost [phantom] pain.226
228 The evidence indicates that pain can be both more severe and more enduring after repair with procedures involving the use of mesh than after procedures which do not. Professor Korda said that dyspareunia is more severe after mesh repair than after traditional prolapse surgery.
229 Professor Korda said that, while prolapse surgery with or without mesh can cause damage to the urethra, the bladder and the rectum, there is a greater incidence of damage to pelvic nerves and major vessels with mesh surgery.227 He also said that, for anterior compartment repair, the use of mesh is associated with longer operating time, greater blood loss; increased incidence of bladder damage; more severe dyspareunia;228 de novo urinary incontinence;229 chronic and severe pain; and repeat surgery as a result of mesh erosion or contraction.230 He added that mesh surgery in the posterior compartment is associated with a higher incidence of rectal damage and a significant mesh erosion rate (up to 10%).231
230 Professor Chughtai, whose evidence was confined to the POP devices, said that the most commonly reported complications amongst women with transvaginal mesh used to treat pelvic organ prolapse are erosion into the vagina and mesh contraction or shrinkage, both of which can cause severe side effects including pain in the pelvis and dyspareunia.232 He said that even if the mesh is removed, patients may likely have residual pelvic pain and dyspareunia.233
231 Professor Deprest also gave evidence that there is a greater problem with pain in patients who have been implanted with mesh than patients who have native tissue repairs.234
232 Moreover, in contrast with native tissue repair, pain after mesh repair can arise well after surgery, sometimes years later.
233 Professor Korda said that in his long period of clinical practice in urogynaecology (around 30 years) during which he had performed thousands of native tissue repairs, the severe, chronic and intractable pain that can occur after mesh surgery was “virtually non-existent” after native tissue repair.235 He was adamant that, while pain and dyspareunia can occur after native tissue repair, they are never of the same magnitude, duration or gravity as the pain and dyspareunia that occur after mesh repair.236 He said that the incidence of pain after native tissue repair is “extremely low” and is usually related to vaginal incisional pain which resolves within four to six weeks of the operation.237 He elaborated on this evidence in his oral testimony:
If you do a simple vaginal operation, and you don’t – with native tissue repair and you don’t actually incorporate nerves inadvertently into the repair, then usually there is pain immediately after the operation and perhaps for two to three weeks or maybe four weeks until the sutures dissolve. And then the pain disappears, and, you know, I’ve been operating on people for a long time, and it’s virtually never seen as an ongoing, prolonged chronic problem … [W]hat we are seeing in these cases in this case, in this matter, is that some of these ladies have suffered chronic intractable pain.238
234 The probable explanation for this chronic intractable pain, he added, is that it is “due to shrinkage of vaginal tissue around the tape or around the mesh which sets up an inflammatory reaction …”.
235 In an article published in 2010, Dr Donald Ostergard attributed the enduring nature of the pain to both mesh shrinkage or contraction and tension on the arms of the mesh with neuroma formation. As he explained it, since mesh is anchored in tissue, its shrinkage will increase the tension on the anchoring tissue with resulting pain. He said that “[n]o mesh seems to be immune from this process”.239
236 The main difference between mesh repair and native tissue repair, according to Professor Korda, was that mesh surgery carried “new and unique” risks likely to be permanently incapacitating, such as chronic and severe pain, mesh contracture, and mesh erosion.240 He said that complications such as mesh erosion, mesh contraction and bunching, severe chronic pelvic pain, severe dyspareunia and pain on movement, sitting, and standing were not encountered in pelvic floor surgery before the introduction of synthetic mesh.241 He also said that, with the exception of sacrospinous ligament suspension, nerve injuries are virtually unknown after native tissue repairs but are “reasonably frequent” after repair of prolapse using mesh kits. The reason for this, he explained, was that the fixation arms of the kits are often hooked to the coccygeus sacrospinous ligament complex which is very close to the sciatic nerve and the pudendal nerve and its branches, and are inserted through the obturator foramen and can damage the obturator nerves.242
237 Further, mesh implant surgery can cause hispareunia (severe pain in the male partner during sexual intercourse) usually due to contact with the sharp fibres of eroded mesh poking through the vaginal mucosa.243
238 Dr Blaivas, whose evidence was largely confined to the SUI devices, said that there were two complications that were virtually unique to mesh implants: erosion into the lower urinary tract, vagina, or bowel; and refractory pain (pain that does not yield to treatment). He said that before the introduction of mesh surgeries, they were “almost never” mentioned in the peer-reviewed literature.244 There is evidence, however, that intractable suprapubic pain has been described after colposuspension.245
239 In a position statement on midurethral slings issued in May 2017, the Royal Australian and New Zealand College of Obstetricians and Gynaecologists (RANZCOG) observed that “sling insertion can cause pain and dyspareunia”, that transobturator slings can cause groin as well as pelvic pain, that the pain can persist and in some cases become intractable, and that removal surgery may be required, but that complete removal can be difficult, can cause complications of its own, and, in any case, may not completely resolve chronic pain or other adverse symptoms.246
240 Professor Korda expressed the view that the complications of mesh surgery for prolapse are generally long-term and sometimes incurable. He listed these as:
reoperation after vaginal mesh surgery at statistically significant higher rates of 8.5% to 22% compared to native tissue repair (3.2% to 9.7%);
reoperation after laparoscopic sacrocolpopexy (5.9%, 2.9% being for specific complications and 1.7% for prolapse recurrence);
vaginal mesh exposure (mean rate of exposure of 13.1% based on the literature), much lower at 3% and 2.5% after open and laparoscopic sacrocolpopexy respectively;
late visceral (bladder and rectum) mesh exposure after laparoscopic sacralcolpopexy (0.5% to 1%) and also vaginal surgery (rate not given);
infection (the rate of which is unknown, with some series reporting 80% bacterial mesh colonisation), abscess, cellulitis, spondylodiscitis;
painful mesh contraction (involving vaginal pain on movement, dyspareunia and localised tenderness over prominent and tense portions of the mesh), with an incidence rate of between 4 and 11%; and
chronic pelvic pain, which has been poorly studied, but occurs in an estimated 0% to 30% of patients following transvaginal mesh placement.247
241 Professor Korda went on to say that there were few procedures available that could permanently reverse or treat the complications of mesh implant surgery.248
242 Evidence to similar effect was given by Professor Chughtai.
243 Professor Chughtai noted that women who underwent mesh repair for pelvic organ prolapse tended to have a higher retreatment rate within two years of surgery, and a higher reoperation rate, in comparison to those undergoing native tissue repairs. He attributed the difference to “major complications” of mesh repair surgery, such as dyspareunia and chronic pelvic pain caused by mesh shrinkage or extrusion into the vagina.249 Professor Collinet described Professor Chughtai as an opinion leader because of his participation in many clinical trials and his scientific output (in excess of 100 scientific papers on the care of vaginal prolapse and stress urinary incontinence).250
Reoperation or revision surgery
244 Removal of the mesh (which may be required in cases of mesh exposure, extrusion, erosion, chronic pain, or recurrent prolapse) presents additional problems. Mesh exposure through the vaginal wall can often be treated with topical oestrogen or local antibiotics, especially if the exposure is small. But if conservative treatment is unsuccessful, the exposure is not small, or there is extensive pain in the operation area, then surgical excision will be required and it may be necessary to remove the mesh.251
245 In an article published in the International Urogynecology Journal in 2006, members of the TVM Group noted that when the mesh becomes exposed, “the complete and rapid removal of the material” is warranted “in order to prevent serious surgical sequelae”.252
246 Yet the evidence on both sides was that the mesh is difficult, if not impossible, to remove entirely as it is or becomes integrated in the connective tissue, and that removal surgery may not relieve pain. 253
247 In her evidence in chief, Associate Professor Rosamilia’s said that most patients with mesh exposure require one operation only to manage the problem.254 She acknowledged, however, that more than one procedure can often be required to manage chronic pain.255
248 Professor Korda accepted that small areas of the mesh can be excised in many patients and the vagina “appropriately” closed afterwards. But he said that “in a large percentage of patients mesh exposure and erosion creates intractable pain and offensive continuous blood stained vaginal discharge, dyspareunia, difficulty sitting or moving and in such patients removing the exposed part of the mesh and closing the epithelium does not resolve their problem”.256
249 The process of excising mesh is, as Professor Korda put it, “technically difficult and fraught with danger”257. Professor Korda explained that the mesh becomes intimately incorporated into the vaginal epithelium, the underlying fascia, and the adjacent and contiguous organs (such as the urethra, bladder, and small and large bowel) and dissection can cause inadvertent injury to those structures. It can also cause the formation of fistulas (abnormal communications between two hollow organs or between a hollow organ and the exterior).258
250 At a meeting with, amongst others, Laura Angelini and Brigitte Hellhammer from Ethicon, held on 20 December 2000, Dr (later Professor) Klinge, who was experienced in explanting meshes after abdominal wall and inguinal hernia repair, described it as “a demanding task only to be performed by an experienced surgeon”.259 Associate Professor Lam testified that this reflected his own view in relation to explanting Prolift or Gynemesh.260
251 Both Associate Professor Lam and Associate Professor Rosamilia insisted that it is not usually necessary to remove all the mesh.261 Professor Korda accepted that in many patients in whom mesh exposure occurs small areas of the mesh can be excised and the vagina can be closed appropriately afterwards. He observed, however, that inability to remove the mesh entirely often leaves patients with unresolved and continuing symptoms.262
252 Professor Chughtai,263 Dr Agur,264 and Professor Deprest gave evidence to the same effect.265 Associate Professor Lam said that removal of mesh via the transvaginal route may be impossible if surgical access is poor due to extensive vaginal scarring either from the mesh itself or from previous pelvic floor operations.266
253 Professor Korda said that, in a large percentage of patients, mesh exposure and erosion create intractable pain and offensive continuous blood-stained vaginal discharge, dyspareunia, and difficulty sitting or moving. In those patients, these problems are not solved by removing the excised portion of the mesh and closing the epithelium because “the host response to the mesh is one of an inflammatory nature and smoulders on”. Further, if the mesh becomes infected while exposed, the inflammatory process and infection spread along the fibres of the mesh underneath the vaginal skin, causing inflammation and a severe host response leading to fibrosis.267
254 Dr Agur said that the retropubic devices were generally easier to remove in their entirety, regardless of the time of implantation. This was because the anatomy of the route they traverse is well understood by pelvic surgeons and they pass through fewer muscles. On the other hand, he said that the transobturator tape traverses more muscles (in the groin and thigh) to which it becomes densely adherent and the route is unfamiliar to pelvic surgeons. In general, surgery aiming at complete mesh removal is required for patients suffering from implant-related chronic pain and surgery of this kind is complex and highly specialised. He said it poses a unique technical challenge and is associated with a relatively high risk of injury to nearby pelvic organs, surrounding nerves and blood vessels. He said that complete surgical removal is often impossible. It requires extensive surgery with local damage to tissues and pelvic organs causing scar tissue which is usually permanent and commonly chronic pain persists even if all the implanted material is removed.268
255 Complete removal is technically very difficult or impossible for most of the POP devices due to the large surface area, dense adherence to native tissues and proximity to pelvic organs. Dr Agur said that safe removal is impossible in the majority of cases unless the device was implanted within a period of two to four weeks before removal is attempted.269 The same is true of the transobturator SUI devices.270 Speaking of the POP implants, Professor Korda stated:
It is my view that the mesh implant usually becomes attached and densely adherent to underlying structures.
In the case of the anterior compartment the mesh is attached to the urethra and the bladder, in case of the apex of the vagina the mesh is adherent to the vagina, sometimes the small bowel and the ligamentous structures such as the sacrospinous ligament and in the case of the posterior compartment the mesh is densely adherent not only to the vagina but to the rectum.
Removal of the mesh requires the dissection of the mesh from both the overlying vaginal epithelium, the fascia below the epithelium and the underlying structures mentioned above. The adherence of the mesh to these structures is such that during even the most careful dissection it is possible to damage the urethra, the bladder, the ureters, the pudendal nerve, the pudendal artery and the rectum.
It is my view that it is often impossible to remove the mesh in its entirety without causing life threatening damage.271
256 Professor Korda added that it was particularly difficult to excise and remove mesh in its entirety when the fixation arms went through the obturator canal because of the prospect of bleeding and damage to the obturator nerve.272
257 Associate Professor Rosamilia agreed that the mesh is difficult to remove completely as it becomes integrated in the connective tissue.273
Risks attributable to the recommended techniques for implantation
258 Although limited vaginal dissection is required for insertion of the SUI devices compared with other surgical procedures for stress urinary incontinence, the venous plexus (a complex network of veins) in the retropubic space is vulnerable to damage during the blind passage of the introducer involved in the implantation of the retropubic devices. Striking pelvic vessels with a needle will cause bleeding from the retropubic space, which may present as increased intra-operative blood loss or later as a retropubic haematoma. Major vascular injury can occur and is potentially fatal. 274 Haemorrhage can be “dramatic” and is difficult to manage.275 The symptoms of retropubic haematoma include pain, voiding difficulty, pelvic mass and a drop in haemoglobin.276
259 Bladder perforation is a well-recognised risk of the retropubic procedures277 because of the blind passage of the needle in the retropubic space. Professor Deprest said that this occurs in up to 5% of cases, more frequently during “the learning curve” and where there has been previous retropubic surgery.278
260 Obstructive voiding symptoms (including, rarely, retention) can also occur, possibly in association with a “higher than desirable tension”.279
261 The introduction of the transobturator tape (including TVT-O) dramatically lowered the rate of bladder perforations but gave rise to different risks. Professor Deprest explained the position well. He wrote:
TOT essentially also provides mid-urethral support, yet the tape is not directed upwards between the bladder and the pubis, however lateral to the obturator membrane. The tape trajectory is continued through a number of muscles going to the medial side of the thigh.
…
On that trajectory one can be close to or on small nerves which run in unpredictable locations. The piercing or vicinity of permanent tape material may cause specific pain at these locations, unique to this approach.280
262 Injuries to the bladder, urethra, rectum and pelvic nerves may arise from the technique used to implant the mesh kits (Prolift, Prolift+M, and Prosima).281 Professor Korda explained that:
Injury to the bladder, urethra and rectum can occur during dissection and are usually inadvertent.
With the exception of sacrospinous ligament suspension, nerve injuries are virtually unknown after native tissue repairs but are reasonably frequent after mesh kits as the fixation arms of the mesh kits are often hooked to the coccygeus sacrospinous ligament complex which is in very close anatomical proximity to the sciatic nerve and pudendal nerve and its branches[.]
263 It is common ground that all meshes used in the Ethicon devices must be inserted without tension. Professor Korda said that if the mesh tension is too tight “all complications are more likely to occur”.282 I did not understand this evidence to be controversial. He was certainly not cross-examined on it.
264 Yet achieving the requisite amount of tension is not easy. Experience helps, but even the most experienced surgeons can occasionally fail. Professor Korda pointed out, for example, that there is a significantly greater incidence of voiding problems after retropubic TVT surgery because of difficulty in getting the right amount of tension.283 He also pointed out that the introduction of trocars into the various anatomical spaces to retrieve the arms of the mesh kits (Prolift, Prolift+M and Prosima) is “essentially a blind procedure”, conducted by touch, and requires a thorough knowledge of the bony structures of the pelvis. The tension is achieved by pulling on the arms of the devices. Since some of the anatomical spaces through which the arms are placed are very narrow, Professor Korda said that it was often difficult to be accurate or consistent in achieving the right amount of tension.284 He said that the Prolift procedure (also used to implant Prolift+M) was not reproducible by the average, reasonably competent gynaecologist in order to achieve reliably safe results.285
265 It was common ground that the use of both the TVM technique and the use of Prolift could cause hardening of the vagina and vaginal, perineal and anal pain. It was also common ground that insertion of a vaginal prosthesis can have a deleterious effect on sexuality and sexual function and cause de novo dyspareunia. Professor Collinet conceded as much.286
The significance of surgical training and the learning curve
266 Professor Collinet noted that, irrespective of the device used, procedures involving the use of vaginal mesh required “special surgical training, and therefore new surgical expertise” due to the need for particular dissections not previously called for in pelvic floor surgery. He observed and said that all surgical innovations require special training and involve a learning curve and he attributed the “sizable numbers” of mesh complications, to the widespread adoption of the mesh kits and the lack of adequate initial training.287 Evidence to like effect was given by Associate Professor Rosamilia and Professor Roovers.288
267 Associate Professor Rosamilia relied on two articles in support of the notion that surgeon experience is a significant factor in mesh exposure rates.289 One, by Dwyer and O’Reilly, published in August 2004, a retrospective study of 97 women, related to the use of Atrium polypropylene mesh in the repair of large or recurrent anterior and posterior prolapse.290 The study began in February 1999 and finished in May 2002. The women were followed up at six weeks, six months, 12 months, and two years. Erosion was the most common complication. Over the study period the rate of mesh erosion decreased from 19% in 1999 to 13% in 2000 and 4% in 2001/2002. The authors concluded that the incidence of this complication was related to surgical experience. Associate Professor Rosamilia emphasised this point in her report. She omitted to point out, however, that the differences were not statistically significant.
268 The second article upon which Associate Professor Rosamilia relied was by Achtari et al, published online in January 2005. This was a retrospective review of the records of 198 women who had undergone transvaginal mesh repair surgery for pelvic organ prolapse using either Atrium or Vypro II. Vypro II was a composite mesh made from polypropylene and polyglactin 910 mesh. The authors detected a correlation in the differences in erosion rates according to experience but in cross-examination Associate Professor Rosamilia conceded that they were not statistically significant. 291
269 In both cases, Associate Professor Rosamilia described the omission to refer to these qualifications as an oversight. 292
270 Notwithstanding the successful assault on Associate Professor Rosamilia’s evidence, I accept that inexperience and lack of adequate training in the surgical techniques for implanting the various devices contributed to the incidence of complications. Professor Korda’s evidence on this question was not materially different.293 Moreover, it stands to reason that complications would be higher in less experienced hands. As Dr Agur observed, however, there is no good evidence that the learning curve accounts to any significant extent for “the inherent mesh complications”.294 Even in the best of hands, using “great surgical skill”, mesh erosion can occur and has been reported.295
271 A report on the outcomes of 917 consecutive transvaginal mesh procedures conducted at the Centre for Advanced Reproductive Endosurgery in Sydney where Associate Professor Lam works, and of which he was a co-author, showed an erosion rate for Prolift of 46 out of 232 cases (20.4%).296 When his attention was drawn to these figures during cross-examination,297 he tried to distance himself from the evidence, claimed it was an error, and that the real figure was “around 10 percent”.298 Although he had referenced the report in his written evidence, he later insisted it was a draft but was unable to produce the final version.299 Whether or not the document was a draft, his attempts to resile from the higher erosion rate were quite unconvincing. What is more, regardless of the true figure, his evidence supports the applicants’ case that mesh erosion can occur even in the best of hands.
272 Dr Leake, a gynaecologist who operated on Mrs Gill for one of her mesh erosions, deposed that transvaginal mesh kits like Prolift were “notoriously difficult for a surgeon to get right every time” even for a surgeon skilled in prolapse surgery.300 According to Dr Leake, tension was the critical factor in the success of Prolift and if implants were put in too tightly patients often experienced pain and dyspareunia.
273 Dr Leake said that mesh may also become too tight because of variations in the patient’s response to it. She explained that “[s]ome patients scar a lot more vigorously than others” and she had found that some ligaments could not withstand the trauma associated with insertion or fixation of mesh to them, which also affected the outcome from mesh surgery. For some patients, the deepest anchor point for the anterior part of the device (the anchor point closest to the patient’s head) was not adequate to hold the prolapse up and the mesh sometimes rolled forwards, predisposing the patient to erosion and the feeling of recurrent prolapse. Further, if the anchor points sagged, this led to inversion of the top of the vagina, which could lead to excessive tension elsewhere, particularly where the mesh rolled over on itself, and cause lower back pain, chronic pelvic pain, and dyspareunia.301
274 If a patient developed a haematoma from the surgery, a not uncommon occurrence in vaginal surgery with or without mesh, Dr Leake found, through experience, that healing was often impaired and erosions could occur. She said that the development of a haematoma after mesh implantation can become a far greater problem than a haematoma after native tissue repair because of healing difficulties in the presence of mesh.
275 Dr Leake was of the opinion that the “pulley system” used for fixing the upper ends or arms of the Prolift contributed to the problem of excessive tensioning. The Prolift technique involved “a blind approach”, which made it difficult to determine whether or not there would be sufficient or excessive tension after the operation was complete.302
276 Like the other witnesses, Dr Leake also testified that excising mesh from the body is very difficult. She said that, although it is possible to excise those parts of the mesh which have eroded through the vagina, if there is tension in the mesh arms the vaginal skin can get scarred and, once the erosion has been repaired, it can be hard to stretch again.303
277 Dr Leake was not required for cross-examination and I accept her evidence.
The significance of the immune response and other patient-specific factors
278 In Gill (No 1), I gave the respondents leave to make further submissions on the matters raised by the applicants’ allegation that the chronic inflammatory response to the implants could be affected by conditions which affect the immune response and healing, including autoimmune disorders. In response to this invitation, the respondents filed supplementary submissions on 14 June 2018. They argued that there were three evidentiary gaps in the applicants’ case. The first was that the allegation was not put to Dr Hinoul so that, to the extent that the applicants claim that the proposition was within Ethicon’s knowledge, they needed to prove that independently. The second was that the applicants had not adduced evidence from any of the surgeons with whom the discussions about implant surgery had taken place. The third was that the applicants had not demonstrated the clinical significance of autoimmune disorders and the chronic inflammatory response to the Ethicon devices.
279 At this point, I will deal only with the first and third arguments. I will deal with the second argument in Parts XIII and XIV when I discuss the applicants’ case concerning the failure of the respondents to provide adequate warnings and other information about the risks posed by the Ethicon devices.
280 The applicants relied on the concessions from Dr Hinoul that when the devices were supplied to the market, the respondents knew that the foreign body response to the mesh was chronic, that mesh exposure was a risk, that there was a lifelong risk of erosion, that mesh exposure or extrusion could cause pain, and that there was a lifetime risk of pain, including life-altering chronic pain. The applicants also submitted that the respondents had known for “many years” that Prolene produced a chronic inflammatory response. And the applicants referred to the evidence that inflammation can be associated with pain.
281 Dr Hinoul’s concessions and this evidence are beside the point. The point the respondents were making was that the substance of the allegation about the potential effect of the chronic inflammatory response on women with these conditions was not put to Dr Hinoul. Of that there can be no doubt. Nevertheless, there was a wealth of other evidence, discussed below, to prove that immunocompromised patients were at particular risk of harm from the chronic inflammatory response and that the respondents either knew or ought to have known that was so at least from the time the very first Ethicon device was launched. The proposition that this was not a matter of clinical significance must be rejected.
282 The immune response to any foreign material has been described as “complex, dynamic, and patient-specific”.304 It was uncontroversial that the individual host response is unpredictable.305 The evidence established that a heightened chronic inflammatory response to implantation with non-absorbable polypropylene may occur in patients who have autoimmune disorders or have been using immune-suppressants for a long time and that Ethicon knew this at the time the first of the Ethicon devices was released to the market.
283 On 12 October 1990 the FDA notified Ethicon that it had reclassified non-absorbable polypropylene sutures from class III to class II and enclosed a letter dated 5 July 1990 to the Vice-President of the United States Surgical Corporation (USSC), another manufacturer of non-absorbable polypropylene surgical sutures.306 USSC had apparently petitioned the FDA to reclassify the non-absorbable polypropylene suture, the FDA’s decision to reclassify the suture followed a review of its petition, and this letter advised them of the outcome of that review and the reasons for the FDA’s decision. That letter, which was tendered by the respondents, included the following passage:
The patient’s health and response to the suture material may affect wound healing (Refs. 11, 17, 18, 48, 66, 74, 80, 81, 87, 107, 108, 109 and 121). Patients whose health has been compromised or weakened by poor nutrition, advanced age, obesity, uncontrolled diabetes, infection, anemia, or with certain forms of cancer, may exhibit delayed wound healing (Refs. 11, 17, 18, 48, 66, 74, 80, 81, 87, 107, 108, 109 and 121) which may increase the likelihood of suture failure.307
284 The letter also stated:
Also, many of the above-identified performance parameters and risks can be adequately controlled by labeling disclosures which may be incorporated into a class II standard or required by the class I misbranding controls, which include, among other things, the requirement of adequate directions for use.
285 This letter put Ethicon on notice that patients whose health had been compromised or weakened by a variety of factors could exhibit delayed wound healing in response to non-absorbable polypropylene and that the risk to their health could be adequately controlled by labelling disclosures, including adequate directions for use. This letter did not directly address the applicants’ allegation. Nevertheless, it was sufficient to alert the respondents that implanting non-absorbable polypropylene, a key material used in the manufacture of all the Ethicon devices, could pose a greater risk of harm to women who were immunocompromised — at least a greater risk of delayed wound healing. It also advised them of action that could be taken to address the risk.
286 Professor Santerre, who, it will be recalled, was a biomaterials expert called by the respondents, stated in his first report that the response to the implantation of a foreign body (like the Ethicon devices) will vary from patient to patient but that polypropylene mesh “would likely be the wrong choice of a material by the surgeon for a patient with any compromised immune system which could delay tissue integration with the mesh”.308 Earlier in his report he explained:
Immune response will depend on a patient’s genetics (inherently has an active or compromised, or moderate immune reactivity), disease state (e.g. diabetes, connective tissue disorders, blood disorders), age (young people typically have much more active and accommodating wound healing than older patients), gender, extent of injury (e.g. more damage to local tissues from the surgical process will generate a greater inflammatory reaction and more challenging host response), environmental conditions (smoking, diet, stress, etc.) and sensitivity to biomaterial chemistry (non-common patient allergies) … If new tissue cannot integrate fast through a porous biomaterial then the material can possibly dislocate, and in some cases ultimately migrate and protrude from the unresolved surgical insertion site.
Hence, it follows on to say that not all patients will achieve a successful integration of biomaterials with new tissue because it is well known that the above parameters compromise wound closure and healing for some patients. If wound healing is different from one human to another, then it also follows that wound healing differs between species. Hence, animal models are not absolute substitutes for human clinical trials and pre-market evaluation.309
287 As I observed in Gill (No 1), several sources were given for the statement in the first sentence. The earliest was a 1997 publication at which time none of the Ethicon devices was available for sale in Australia. None of the sources Professor Santerre cited was a publication one might reasonably expect gynaecologists or urogynaecologists to read, at least not routinely. But as manufacturers of biomaterials, even if it was not actually aware that “wound closure and healing” would be compromised for patients with immunosuppressed conditions, it should have been. Moreover, they should also have been aware that these patients were also at a higher risk than others of migration, extrusion, and erosion of their implants.
288 Professor Santerre discussed the relationship between immune response and infection. He stated:
In addition, if the immune response for a specific patient is not compromised then macrophage activity will be very elevated early on and may contribute to controlling bacteria contamination at the site until healing is finished and new-blood vessel formation is complete (note macrophages have an inherent role of digesting bacteria). If the thickness of the fibrous capsule around the implant is thin and macrophages can easily penetrate the mesh structure from the new blood vessels, then there will remain a relatively low level (e.g. an active basal level) of macrophage activity contributing in part to keeping bacteria contamination in check within the mesh’s integrated tissues.
However, in patients with a compromised immune response (for any of the reasons discussed below in section 4.3), then infection will be a challenge, no matter what synthetic biomaterial will be used in this application. 310
(Emphasis added)
289 Both paragraphs in the above extract were supported by references to a 2001 article on the in vitro interaction of bacteria with polypropylene and polyester prostheses published in the journal Biomaterials in 2001.311
290 In cross-examination, Professor Santerre said that “high responders” were likely to have an adverse outcome from a mesh implant. He agreed, in effect, that the high responders included patients who had autoimmune disorders or were undergoing long-term immunosuppressant therapy.312
291 Professor Santerre considered that the early pre-market studies conducted by Ethicon between 1991 and 1997 “were not immune to the existence of patient to patient variability”, noting that it was observed in early mesh testing and could reasonably be anticipated.313
292 The effect of Professor Santerre’s evidence was that patients who are immunocompromised for one reason or another are likely to experience a more intense inflammatory reaction than patients who are not. It follows that they are and have always been at higher risk of developing complications, such as erosion/exposure, infection, and chronic pain.
293 In the chapter on mesh infection and migration in the book Meshes: benefits and risks, edited by Professor Volker Schumpelick and published in 2004, it was noted that “[i]mmune-suppressed patients are very susceptible to infections …”.314 The book contained the papers presented to the third Suvretta conference in Switzerland in January 2003, which was sponsored by Ethicon315 and attended by a number of Ethicon personnel, including Brigitte Hellhammer and Boris Batke.316
294 In their article on the conclusions of the IUGA Roundtable published online on 6 May 2006, Davila et al noted that the occurrence of healing abnormalities after graft implantation, such as erosion, rejection, and infection, is becoming increasingly recognised as a potentially serious problem.317 They compiled a list of “[c]urrently accepted relative contraindications to the use of biomaterials”. One of those contradictions was “[i]mmunosuppressed patients”. The full list was:
1. History of previous pelvic radiation
2. Severe urogenital atrophy
3. Immunosuppressed patient
4. Presence of active pelvic or vaginal infection
5. Patient currently on systemic steroids
6. Host factors including:
a. Poorly controlled diabetes
b. Morbid obesity
c. Heavy smokers
295 Davila et al (2006) wrote that contributing factors to healing abnormalities like erosion, rejection, and infection were likely to include immune reaction to the graft and individual host factors including patients with sensitive skin and/or multiple allergies. They emphasised the importance of integration of a graft into the host tissue:
Integration of a graft into host tissue is important. The implanted graft should allow for prompt collagen in-growth and neovascularization. This should occur without the occurrence of infection or significant inflammatory reaction. A limited amount of inflammatory reaction is necessary to promote neovascularization and collagen in-growth. Grafts that are poorly integrated include microporous synthetic grafts and biologic grafts treated with chemical cross-linking. These grafts may become encapsulated, leading to hardening, shrinkage, and other graft changes, which may subsequently lead to dyspareunia, alteration of normal anatomy, and increased risk of failure.
296 In 2012 the results were published of a study by the Nordic Transvaginal Mesh Group aimed at identifying risk factors for mesh exposures after anterior pelvic organ prolapse repair using Prolift.318 Elmér et al (2012) wrote:
Somatic inflammatory disease showed a significant, albeit weak, association with mesh exposures. We recognize that the term “somatic inflammatory disease” entails several conditions with a more or less heterogeneous pathoetiology. However, inflammatory diseases such as rheumoid (sic) arthritis, Sjögreńs disease, and lupus erythematosus, also share characteristics such as multiple organ involvement, afflicted connective tissues, and underlying autoimmune mechanisms. These disease traits may by themselves influence wound healing but treatment is often accompanied by immunosuppressant medications which may prolong wound healing. Despite the overall large number of patients in the study, the number of cases with somatic inflammatory disease was few (indicated by the wide confidence intervals) and the association is in need of corroboration in further studies.
297 They concluded that, along with smoking and multiparity (two or more births), somatic inflammatory diseases were possible risk factors for mesh exposures after implantation with Prolift.
298 Ethicon and Gynecare Scandinavia were among the sponsors of the study. Two of the authors, Drs Altman and Falconer, declared that they were advisers to Ethicon and/or Gynecare Scandinavia.
PART V: BIOCOMPATIBILITY ISSUES
299 It was common ground that the biomechanical properties of the mesh and mesh stiffness in particular are amongst the main reasons for the post-surgery complications of mesh implantation.319 Witnesses on both sides testified that exposure of polypropylene mesh, including the mesh used in the various Ethicon devices, can cause infection. As Professor Deprest put it in remarks expressly approved by Professor Korda,320 “when a material is exposed to the vaginal lumen it will almost certainly become infected: resident bacteria will attach to the implant”.321 Infection may also be a primary process, of course. In such a case it was also common ground that it can cause exposure of the mesh.
300 A good deal of evidence was led by the applicants to explain the mechanisms by which the admitted complications arise or might arise. The evidence was very interesting and some of it was controversial. As a matter of law, however, having regard to Dr Hinoul’s concessions that the devices can cause the pleaded complications, it was unnecessary to prove the mechanism(s) by which they may occur. The respondents accepted as much during closing argument.322 For this reason, my discussion will not reflect all the evidence or all the submissions made in relation to it. Above all, I do not propose to decide disputes simply to satisfy someone’s intellectual curiosity. There is enough to be done without that.
Some reflections on the witnesses
301 The evidence about these issues was given for the applicants by Professors Klosterhalfen and Klinge, Associate Professor Guelcher, and Dr Dunn, and for the respondents by Professors Deprest, Wright and Santerre, and Dr MacLean.
302 I was impressed by the evidence of Professors Klosterhalfen and Klinge. They both struck me as honest and trustworthy. Their knowledge of, and experience with, the use of polypropylene implants, in general, and the POP devices, in particular, was far superior to that of any other witness. Their evidence was informed by their extensive and profound experience.
303 On the other hand I was not impressed with a number of the respondents’ witnesses.
304 I begin with Professor Wright.
305 In contrast to the generally favourable impression I formed of Professors Klinge and Klosterhalfen, I found Professor Wright to be generally unimpressive. He appeared too eager to please the respondents. In his first report to the Court, for example, he wrote that the inflammatory response to the implants was transitory (resolving within to two to three weeks), just as the respondents had written in all the instructions for use (IFUs) supplied with the Ethicon devices.323 In cross-examination, however, he conceded that the inflammatory response was chronic.324 The report cited passages in references in support of his conclusions without referring to passages in the same references that detracted from them. Indeed, his review of the literature appeared to be selective. He was coy about referring to scar tissue. He defined fibrosis as the “[f]ormation of excess fibrous connective tissue in a damaged organ or tissue”.325 But Professor Klinge gave unchallenged evidence that only in certain rare primary diseases does fibrosis occur without any previous tissue injury. Otherwise, he deposed, “fibrosis means scar”.326 Professor Wright also stated that scar contraction typically occurs in burn patients or on the palms, soles, and thorax, and is not normally seen at other anatomical sites.327 But Professor Klinge pointed out that it has been known for over a hundred years that scar contraction occurs everywhere in the body.328 Not only was Professor Klinge’s evidence in this regard not challenged, but there was no dispute between the parties that scars contract around meshes.
306 Professor Wright’s experience and expertise in the area of polypropylene meshes is inconsequential in comparison with that of Professors Klosterhalfen and Klinge. In contrast to the extensive research undertaken by them, Professor Wright admitted to not having conducted independent research on the host response to polypropylene meshes or the biocompatibility of polypropylene meshes for the treatment of pelvic organ prolapse. His experience in the area is recent and confined to the litigation context. It involved looking at “over 50 explants”, about 75% of which, the cross-examination revealed, were only tissue biopsies which included partially excised mesh from SUI or POP devices.329
307 In cross-examination Professor Wright refused to answer certain questions claiming that he did not have expertise in biomaterials or urogynaecology, despite having opined on matters falling within these fields in his two reports. On a number of occasions, he drew distinctions that were unconvincing or unclear (such as the point at which fibrous connective tissue becomes dense enough to be characterised as scar tissue). He ridiculed the concept of bridging fibrosis, discussed below, but when asked whether he could identify an article that was critical of it, he said he could not.330 Some of his positions proved to be inconsistent.
308 Overall, he struck me as arrogant and partisan.
309 For all these reasons, where their opinions conflict, I prefer the evidence given by Professors Klosterhalfen and Klinge to that of Professor Wright.
310 Professor Santerre’s reports read like an unqualified endorsement of Ethicon’s products. He cited a number of papers to support his favourable opinions of Ethicon’s conduct without referring to important parts of those papers that were critical of them. That was most evident in his selective use of chapter 13 of Host Response to Biomaterials, which was written by Barone et al (2015) and titled “Host Response to Biomaterials for Pelvic Floor Reconstruction”.331 Professor Santerre’s reports were also peppered with selective and incomplete citations of complication rates and statements from clinical studies.332
311 In a number of instances Professor Santerre’s evidence was internally inconsistent. For example, it was put to Professor Santerre in cross-examination that “agitation of vaginal tissue which contains an implant could have an effect on the tissue reaction to that implant”. He replied: “The answer is yes, if there is poor integration of that implant around”. He later changed this to: “particularly if there is poor integration of that implant around”. It was then put to him, “So even after full integration it’s wise not to engage in too much physical activity, at the risk of a negative reaction to the implant?” He responded, “Everybody – everybody will be different, and I will leave that to a clinician’s call”.333
312 During cross-examination, Professor Santerre often failed to respond directly to questions.
313 When pressed to answer questions directly, however, he made a number of important concessions. For example, he accepted that, for as long as a foreign body (such as a mesh implant) is present in the human body, there is likely to be an ongoing or chronic inflammatory response.334 In addition, he accepted that it was likely the antioxidants in the Prolene meshes would be depleted and oxidation would occur at the surface of the implants, which could account for the appearance of cracks and flaking observed microscopically by Professor Iakovlev and others.335 In fact, as the applicants submitted, he ultimately agreed with each of the central contentions of the applicants’ biomaterials case.336
314 At first, Professor Deprest appeared to be a model witness. Unlike Professor Santerre and some of the other witnesses, he gave direct and responsive answers to questions put to him. I found him generally impressive, although I became troubled by some aspects of his report. For one thing, I was very concerned about his unqualified statement that “the recent SCENIHR [Scientific Committee on Emerging and Newly Identified Health Risks] (2015) report recommends that PP [polypropylene] should be the only product used in vaginal implants”.337 As far as I can tell, the report itself makes no such recommendation, at least not expressly. Be that as it may, it appeared from his evidence in chief that Professor Deprest endorsed such a recommendation. The cross-examination revealed that nothing could be further from the truth. Professor Deprest was a co-author, with Drs Slack and Davilla, on behalf of the board of the IUGA, of a submission made in response to a draft of the SCENIHR report made available for public comment. The conclusion of the IUGA submission was completely at odds with such a recommendation. Indeed, the submission described the recommendation as indefensible. It reads:
In conclusion we reject the recommendation that Polypropylene is safe for vaginal use… as this is not based on any translational or clinical evidence, and therefore cannot be defended.338
315 Professor Deprest agreed in cross-examination that this was his personal opinion at the time and that he remained of this opinion.339
316 When asked whether it occurred to him that the Court might like to know that his opinion differed from the SCENIHR recommendation, he said that it had.340 His attempt to justify the omission from his report of any reference to that opinion was incomprehensible. At first, he said he was quoting from “the official [SCENIR] report” when he was not. Even if he were, that was no reason not to venture his own opinion. After all, one of the critical matters with which his report was concerned was his opinion on the safety of Ethicon’s urogynaecological polypropylene meshes. Citing the recommendation of the SCENIHR report without mentioning his own opinion was apt to mislead the Court. It amounted to a representation that he shared the view of the Committee or, at least, that he did not disagree with it. Why he put himself in this position is a mystery. It leads me to believe that he was not as impartial as I first thought.
317 Other matters of significance emerged in cross-examination.
318 When asked why he did not include a reference to a paper he had co-authored,341 which contained statements apparently favourable to the applicants’ case, Professor Deprest replied: “I didn’t add it to my evidence because we usually don’t cite ourselves”.342 I must say I found this reply difficult to believe, especially since he had cited in his report some 19 articles upon which his name appeared as an author.
319 A party may only call an expert to give expert evidence at a trial if the party has served a report from the expert which, amongst other things, contains an acknowledgment that the expert has read, understood and complied with the practice note on expert evidence: Federal Court Rules 2011 (Cth) (Rules), r 23.11, read with r 23.13.
320 Professor Deprest claimed to have read and complied with the Expert Evidence Practice Note (GPN-EXT). Moreover, he agreed to be bound by it. Clause 5.2 of the Practice Note requires that the expert attach or exhibit to his or her report documents that record any instructions given to him or her. Professor Deprest admitted to receiving a letter from Clayton Utz, the respondents’ solicitors, but did not attach that letter to his report. He also said that he had had numerous teleconferences with the lawyers. It is inconceivable that no notes were taken at those teleconferences. Later, however, in response to a call for documents recording instructions, including notes of conversations, the respondents’ lead counsel, Mr Finch SC informed the Court that there were no such documents. 343 The absence of any such document is troubling. While it may not be a breach of the letter of the Practice Note, it constitutes a failure to adhere to its spirit.
321 In BrisConnections Finance Pty Limited (Receivers and Managers Appointed) v Arup Pty Limited (2017) 252 FCR 450, Lee J considered the importance of a letter of instruction in providing clarity to the Court about the questions the expert has been asked and the assumptions he or she was asked to make. His Honour’s remarks arose in the context of a letter of instruction that was apparently provided (almost) contemporaneously with the finalisation of the expert report. His Honour stated at [71]:
The point of a letter of instruction being annexed to a report is not to act out a stylised ritual, but to provide to (scil) the Court with a transparent indication of what has been provided to the expert and the questions that the expert was actually asked to address. It should be able to be read literally without being silly. As is (at the very least) implicit in FCR 23, the work of the expert is to attend to the questions “the expert was asked to address”, not to invert the process by using the expert’s specialised knowledge in order to identify the questions that should have been asked and the assumptions that should have been given. … The integrity of the expert evidence process and the independence of experts is best facilitated by transparency in what is being asked of experts prior to, or at the time, they are forming their opinions and, if the questions need to change because they are misdirected, a record being made by way of supplementary instructions as to what has changed.
(Original emphasis in bold, italicised emphasis added)
322 I respectfully agree. The lack of transparency means that the Court may never know the extent to which the witness’s opinion has been influenced by the party retaining the witness.
323 No doubt for good reason, Professor Deprest was not asked in cross-examination what oral communications he had had with the respondents’ lawyers. For a start, the cross-examiner would not know the answer he might receive to such a question. In any case, he might well not have remembered. Even if he purported to have a memory of them, it would in all likelihood be unreliable. Finally, any answer he might have given could not be tested.
324 Dr MacLean, for the most part, responded to questions with what appeared to be well-reasoned answers. But his failure to fully disclose the relationship with Ethicon of his company (Exponent) and his colleagues (Drs White and Ong) raised doubts about his independence and the weight to be given to one of the studies on which he relied (the Thames study).344 This was particularly so as he was given an opportunity during examination-in-chief to disclose any past relationships with Ethicon.
325 The absence of any letter of instructions to Dr MacLean (from either Clayton Utz, which represent the respondents in the present proceeding or the US firm, Butler Snow, which represented Ethicon in US litigation) is also of note. When asked to describe his interactions with Chris Hutchison of Butler Snow, Dr MacLean was not precise in his responses, giving me cause for concern around the content of his instructions.
The nature and extent of the dispute
326 There is no dispute that “the ideal mesh” is yet to be found and that the optimal biomechanical properties for urogynaecological meshes remain unknown.345 This was a point the respondents made repeatedly as if it were a mitigating, if not exculpatory, factor.
327 Moreover, the respondents accepted that pore size is a critical component of the biocompatibility of any mesh used in vivo and that pores must be of sufficient size to allow the entry of macrophages “and other processes” to ensure that bacteria are not housed in the pores of the mesh.346 Critically, they also accepted that the pore sizes change in vivo and that the mesh contracts.347
328 There were seven broad issues in contention:
(1) whether the changes to pore sizes in vivo are significant;
(2) whether the classification system upon which many of the respondents’ witnesses relied is still applicable;
(3) whether data derived from the pelvis data pool compiled by Professor Klosterhalfen and others at the University of Aachen is unreliable and, if so, the extent to which that affects his and Professor Klinge’s opinions;
(4) whether bridging fibrosis can occur with any of the Ethicon devices and, if so, whether it is of clinical significance;
(5) whether mesh contraction is of clinical significance;
(6) whether chronic pain is attributable to nerve entrapment in the contracted mesh or scar tissue; and
(7) whether the polypropylene used in the Ethicon devices degrades in vivo and, if so, whether the degradation is clinically significant.
329 Before dealing with these issues, however, it is necessary to provide some context.
330 All the Ethicon devices are made from biomaterials. A biomaterial is a material used in a medical device which is intended to interact with biological systems.348 Professor Wright said that a biomaterial is regarded as biocompatible if it has the ability to perform with an appropriate host response in a specific situation. This definition of biocompatibility was drawn from the writings of Professor David Williams, a noted expert in the field, and has been current since 1987.349 It is not materially different from the definition given by the Second Consensus Conference on Definitions in Biomaterials in Chester, UK, in 1991 (“the ability of the material to perform with an appropriate host response to the application”) to which Associate Professor Guelcher, one of the applicants’ biomaterials experts, referred.350 It drew criticism, however, from Professor Klosterhalfen:
In my opinion, the “Williams definition” is unclear: I do not understand the term “appropriate host response in a specific situation”. Who defines what is appropriate? Further, this definition only relates to the host response (in particular, tissue response), but not to the function of the material or device.
Moreover, the definition is only appropriate for short-term applications, such as preclinical animal studies. For permanent implanted devices (like surgical meshes) the definition should be modified as follows: the biocompatibility of a permanent implantable medical device refers to the ability of the device to perform its intended function, with the desired degree of incorporation in the host, without eliciting any undesirable local or systemic effects in that host.351
331 I doubt, however, that there is much difference between the views of Professors Klosterhalfen and Williams on this point. Indeed, Professor Klinge cited Professor Williams in support of the definition given by Professor Kosterhalfen.352 In another article, published in the journal Biomaterials in 2008, Professor Williams wrote:
This definition, which clearly places the word in the category of a concept rather than a practical descriptor of a process, is based on the three tenets that a material has to perform and not simply exist in the tissues, that the response which it evokes has to be appropriate for the application, and that the nature of the response to a specific material and its appropriateness may vary from one situation to another. 353
332 In an earlier article, published in the Journal of Applied Polymer Science in 1994, of which he was a co-author, Professor Williams made it clear that to be biocompatible a permanent implant should not have any harmful effect on human tissue.354
333 As I mentioned earlier, Professor Williams defined biocompatibility as “the ability to perform with an appropriate host response in a specific situation”. In a chapter entitled “General Concepts of Biocompatibility” published in Black J and Hastings G (eds), Handbook of Biomaterial Properties (Springer Science+Business Media, 1998) he also made the following pertinent observation:
To recognize the very effective performance of a material under one set of conditions but then to assume that the same material can perform equally well under entirely different circumstances is inherently dangerous since it takes into account neither the variations one might expect to see in the host response from site to site nor the fact that what is appropriate for one situation may not be appropriate for another.355
334 Yet, in 2003, six years after TVT was launched and many years after Prolene had first been used for hernia repair, Professor Klosterhalfen and others wrote in an article on “Biological Response to Mesh” published in the journal, European Surgery, that “[o]ur knowledge about the long-term biocompatibility and tissue response of mesh in humans is still poor, although a few reports exist”.356 In September of the same year, Drs Arnaud and Robinson, in a joint presentation for Ethicon, observed that “the scientific knowledge about the use of meshes in surgery is still in its infancy, at least for pelvic floor applications”.357 They emphasised the necessity for the mesh to resist infection and the high risk of infection for pelvic meshes, observing that “[t]he vaginal approach is a rather unique situation in surgery as a prosthetic material is placed through a septic cavity” and that before TVT, apart from the mouth and ENT surgery, “meshes [were] never used in such a condition”.
335 In the chapter to which I referred, Professor Williams went on to point out that for a material to be biocompatible “the interaction between the material and the tissues is one which leads to an acceptable balance between inflammation and repair”. This is presumably what he meant by “an appropriate host response”. Professor Williams emphasised that “host variables are as important as material variables in the determination of biocompatibility”. Not all tissues, he added, not even all tissues of the same variety, will be able to respond in the same way and host variables like age and overall health status will have a major effect.358 At the same time, he noted that there are significant regional and tissue-specific variations in the local host response.359
336 One of the other points Professor Williams made in this chapter was that it was necessary to recognise that, if there is an interfacial reaction (that is, a reaction at the place where the foreign body is in contact with the tissue), there is no reason why the products of that reaction and their effects have to be confined to the locality of the interface. He said that the presence of a benign local response does not necessarily mean there will be no systemic or remote side effects.360
337 In an article published in 1982 in the Journal of Materials Science, Professor Williams also wrote:
Although the specific materials requirements will differ according to the nature of the application, it is a fundamental requirement in each and every case that the polymer should display adequate biocompatibility. This implies that, for permanent implant applications, the material should not degrade within the physiological environment, nor should it have any adverse effect on the tissue …361
(Emphasis added)
338 To similar effect, Professor Klinge’s evidence was that the biocompatibility of a long-term implantable medical device can be defined as the ability of the device to perform its intended function, with the desired degree of incorporation in the host, without eliciting any undesirable local or systematic effects in that host.362
339 In an article tendered by the respondents, published in the International Urogynecology Journal in July 2003, Dr Michel Cosson and his fellow authors reviewed the literature on all types of synthetic implants used in both prolapse repair and the treatment of stress urinary incontinence and analysed the mechanical properties of, and tolerance to, the various devices. They concluded:
This review of existing prosthetic products demonstrates that no perfect product currently exists. Two categories of product seem to us to have promising properties with regard to their use in transvaginal surgery for restoring pelvic function. On the one hand are synthetic implants with mechanical properties of strength and elasticity, essentially made of polypropylene. Their strength is unchallenged, but it remains to be established whether they are well tolerated when introduced by the vaginal route.363
(Emphasis added)
340 In these circumstances, it is difficult to understand how the Ethicon devices could have been considered biocompatible or suitable for release to the market without first testing their performance in vaginal tissue in a wide selection of patients and over a sufficient period of time.
341 When a biomaterial such as polypropylene is implanted in the body, it provokes an inflammatory response in the host tissue. This inflammatory response is known as the “foreign body reaction” or “foreign body response”. It causes a layer of scar tissue to form around the implant or the pores of the implant, which is weaker and more rigid than normal healthy tissue.364 Indeed, synthetic non-degradable meshes are believed to work by inducing this foreign body reaction and the consequential fibrosis which enables the foreign material to adhere to the host tissue to provide support to weakened support structures.365
342 The scarring is apparent after about two to three weeks and certainly after about six weeks.366 The extent of the foreign body response (and therefore the extent of the inflammation) is influenced by a number of factors to which I will come in due course.
343 The inflammatory response takes place in stages. First proteins are adsorbed. In the early stages, mononuclear cells (monocytes) migrate to the surface of the biomaterial, where they can adhere and participate in the foreign body reaction.367 Monocytes, produced by the foreign body reaction, can differentiate into macrophages and foreign body giant cells, which are a collection of used macrophages.368 Macrophages (literally “large eaters” or “feeders”) are specialised phagocytes (literally “feeder cells”). They remove dying or dead cells and cellular debris and clear bacteria.369 Associate Professor Guelcher described the space between the surface of the biomaterial and the adhered macrophage as a “privileged microenvironment” where these types of cells release reactive molecular species called “reactive oxygen intermediates” or “reactive oxygen species”, as well as acids and enzymes. 370 Reactive oxygen species are chemical compounds containing oxygen (such as hydroxyl radicals and hydrogen peroxide) that are secreted by the inflammatory cells while they are adhering to the foreign body.371 I will return to the subject of the reactive oxygen species later in these reasons when discussing the applicants’ case that the Ethicon devices are subjected to oxidative degradation after implantation.
344 Macrophages then fuse and become multinucleated foreign body giant cells, which adhere to the surface of the mesh. They attract and activate fibroblasts (the main cells that form scar tissue) and angiogenesis (the formation of new blood cells). A major effect of the foreign body reaction is that the implant is covered by, or embedded in, a granuloma (a mass of granular tissue) with an inner layer of macrophages and foreign body giant cells and an outer layer of fibrotic (scar) tissue.372
345 The foreign body reaction to an implant can continue over an extended period of time. Unlike normal wound healing, where the number of inflammatory cells decreases over time leaving only the scar tissue, in a foreign body reaction the inflammatory cells may persist for years, even decades, becoming “a chronic inflammatory response”.373 For a permanent implant, like each of the Ethicon devices, the response endures for the duration of the time the implant remains in the body. As Professor Deprest put it in his first report:
Between the openings (pores) present in the implant, the body “grows” scar tissue, so that the implant eventually integrates or becomes part of the body, and ideally exerts its supportive function … When an implant is not degradable the remodelling and inflammation process is permanent …374
(Original emphasis)
346 The persistence of the foreign body reaction is important, especially in younger patients in whom the mesh will remain for several decades. As Professor Klinge observed, it means that in many clinical studies with short periods of follow-up, the morbidity rates are likely to be underestimated.375
347 The foreign body reaction can also vary in intensity, and the intensity of the foreign body reaction, more particularly the extent of the inflammatory response, correlates with the extent of the fibrosis.376 Professor Klinge said that histopathological investigations have shown that the amount of fibrosis is directly related to the amount of the inflammatory, cellular foreign body reaction induced at the interface of the biomaterial and the host tissues. The Aachen Group directed its early mesh research to reducing the amount of foreign material used, its surface area, as well as the density of the mesh primarily in order to minimise the degree of inflammation.377
348 Professor Klosterhalfen’s evidence was that the intensity of the foreign body reaction to polypropylene can be explained by the failure of macrophages to resolve the inflammation in the initial period following implantation,378 the characteristics of the surface area of the mesh in contact with the host tissues,379 differences in the surrounding tissues and the mechanical loads and stresses to which the implant is subjected,380 and the genetic background of the particular individual.381
349 The greater the surface area of the device, both in terms of the amount of fibre length and the density and diameter of the fibres, the greater the foreign body reaction and the inflammatory response.382 Studies have demonstrate that the extent of the reaction is dose-dependent.383
350 Furthermore, the respondents recognised that decreasing the biomaterial content of polypropylene meshes reduces the inflammatory response.384
351 Professor Klosterhalfen also deposed that mechanically cut mesh was “particularly problematic” because it frayed and pieces of polypropylene could break off during implantation. These characteristics, he said, created a greater inflammatory response leading to “greater fibrosis and scarring”.385 He also said that mechanically cut mesh also creates sharp edges that can cut tissues, which can cause pain and increase the likelihood of erosion, and cutting mesh mechanically can cause the mesh to rope.386 He was not cross-examined about any of this evidence and I did not understand it to be controversial.
352 As I indicated earlier, studies demonstrated that the foreign body reaction in response to the implantation of polypropylene mesh continues until the mesh is removed. Despite that, until relatively recently, in the instructions for use issued for all the Ethicon devices the foreign body reaction was described as “transitory”. Notwithstanding what they said in the instructions for use, Ethicon was well aware of that.
353 Ultimately, despite what the respondents were telling consumers, it was common ground that as long as the implant remains in the body, the foreign body or inflammatory response to polypropylene implants, including the devices in question, is not “transient” or “transitory” but chronic and permanent.387 Indeed, this was an intended outcome, as some fibrosis (scarring) is required to enable the device to adhere to the tissue and remain in place. The problem is when the inflammatory response is greater than necessary to create the desired level of fibrosis. Professor Klinge said in his affidavit that “incorporation of the mesh into tissue is not the problem; it is the incorporation of mesh into scar tissue that is the issue”.388 Scar tissue is non-functional tissue and the amount of scar tissue around the mesh is responsible for the stiffness and “functional outcome of the implanted tissue area”.389
354 The respondents submitted that, given the consensus about the duration of the foreign body reaction, the question for determination is whether the applicants have proved that the foreign body reaction has a clinically significant effect and causes the asserted complications.390 The answer to the question is that they did prove that the foreign body reaction is clinically significant and can cause many, if not all, of the pleaded complications.
355 Cobb et al (2005), for example, wrote that the intensity and extent of the foreign body reaction was the most important factor influencing the biocompatibility or tolerance of a subject to the foreign body.391
356 Deprest et al (2006) said that local complications in synthetic meshes, including low-weight, large pore, monofilament materials, seem to be related to an increased foreign body reaction.392
357 Moreover, as the applicants pointed out, the study by Elmer et al on the histological inflammatory response to Prolift, published in the Journal of Urology in 2009, and relied upon by both Professors Santerre and Wright, suggested a relationship between the inflammatory response to polypropylene mesh and complications, such as erosions.393 It found that eight out of 10 patients (80%) had mild granular formation and two erosions one year after surgery. The authors emphasised that a larger study population of patients who had undergone transvaginal mesh surgery “would undoubtedly generate an increased number of mesh complications”. They added that clinicians and patients should be aware of the possibility of late onset “mesh related inflammatory reactions” when using a large polypropylene mesh (like Prolift).394
358 In cross-examination, Dr Hinoul acknowledged that “the mesh tissue interaction leads to scar contraction”.395 Professor Roovers also testified that mesh shrinkage is caused by the fibrotic reaction and that this can cause the scar to contract and the mesh.396 For the reasons given below, scar contraction can cause a number of complications.
359 Professor Klosterhalfen said that an intense foreign body reaction with bridging fibrosis (discussed below) can cause chronic pain.397 He noted that, while fibrosis is useful for the integration of the mesh, “excessive fibrosis with bridging is fatal for mesh function and the long-term outcome”.398
360 Dr Daniela Ulrich and others (including Associate Professor Rosamilia) wrote in an article published in 2012 that, depending on the nature and extent of the response, this inflammatory response is primarily responsible for the significant complications arising from the use of synthetic meshes, including mesh exposure/erosion, mesh contraction, infection, and pain.399
361 Ethicon told the FDA in February 2008 that the benefit of Prolift+M over Prolift was its ability to reduce the amount of foreign body remaining in the patient after implantation.400 If the foreign body response was not clinically significant, there would be no need to reduce the amount of the foreign body.
362 In March 2009, Dr Hinoul and his collaborators from Ethicon wrote that the foreign body reaction induced by the mesh used in Prolift+M (both acute and chronic) can lead to both exposure and shrinkage.401
363 A good deal of evidence on the biocompatibility of the Ethicon devices and the complications that can be attributed to the use of polypropylene mesh derived from the work of a multi-disciplinary group of doctors and scientists based in Aachen in northwest Germany. Professors Klosterhalfen and Klinge were members of that group.
364 Professor Klosterhalfen is a surgical pathologist who has worked in the field of anatomical and clinical pathology for over 25 years. He has devoted much of his career to the study of the body’s response to implanted devices, including polypropylene meshes, and how the design of those devices influences biocompatibility.402 He claims to be the first pathologist to describe in detail the foreign body reaction to polypropylene mesh.403
365 As a resident in the Department of Surgery at the Medical University in Aachen, Professor Klosterhalfen met Dr Uwe Klinge (now Professor Klinge), an abdominal surgeon who was then specialising in general surgery.
366 Professor Klosterhalfen became a consultant in general pathology in 1998, was appointed an associate professor of surgical pathology the following year and in 2003, only five years after he had completed his training in pathology, was appointed to his current chair.404 In 2003 he established the Institute of Pathology in Düren, Germany, which he still heads. It is one of the top three institutes of pathology in Germany. It processes over 150,000 tissue specimens a year, including about 20,000 tissue samples from the female pelvic floor and genital tract. Professor Klosterhalfen is personally involved in the study of about 50,000 samples each year and, since the Institute was established, he estimated, he has had responsibility for the analysis of over 600,000 tissue specimens.405
367 Both he and Professor Klinge are prolific researchers and have published widely on the effect of biomaterials used in both abdominal and pelvic surgery.
368 Professor Klosterhalfen is a highly respected pathologist. He described himself as a world leader in understanding the mechanical properties of polypropylene mesh and the response of human tissue to it.406 The respondents did not quarrel with this description. His opinions on these matters are regularly sought by scientists, physicians, and industry.407 Those people who have had professional dealings with him have the utmost respect for his opinions and the quality of his work. That was apparent from a host of Ethicon documents. Amongst others, Professor Deprest sent explants to him. In cross-examination Professor Deprest said that he did not question Professor Klosterhalfen’s analyses of the explants and did not doubt his knowledge.408 Dr Hinoul described him in an email to colleagues at Ethicon as “the god of surgical pathology on the subject of textile implants in this solar system”.
369 JJM or another company in the Johnson & Johnson group (the evidence does not permit me to say) engaged an external consulting firm, PA Consulting Group, to investigate mesh erosion in pelvic floor repair. In particular, PA Consulting was asked to review the literature and conduct “a broad analysis of the problem”, which was to include an informal meta-analysis of the literature, interviews of both internal and external experts, and consideration of potential animal models. The Group reported on 18 May 2011.409 Professor Klosterhalfen was the only pathologist who was interviewed.
370 While working as a general surgeon Professor Klinge performed thousands of abdominal operations, many of which involved the use of synthetic surgical mesh. He became a specialist in abdominal or visceral surgery in 2004. In 2005 he was appointed professor of surgery at Aachen University.410 He stopped operating in 2006 to concentrate on research and for the last decade he has been the principal scientific investigator in the university’s surgical department.411 Although he has never performed surgery for the repair of stress urinary incontinence or pelvic organ prolapse, Professor Klinge has used meshes manufactured by Ethicon in the course of hernia repairs.412
371 In the early 1990s, as a result of observing and performing revision operations on patients who had been implanted with surgical mesh for the treatment of hernias, amongst other things, Professor Klinge became aware of complications related to the use of mesh including the build-up of wound fluid around the mesh, pain, infection, and fistula formation into the bowels. Keen to learn how to avoid those complications, he started research in the area of biomaterials science.413
372 At that time Professor Klinge invited Professor Klosterhalfen to become part of an interdisciplinary working group at the University of Aachen. The Interdisciplinary Centre for Biomaterials Research or IZKF BIOMAT was thereby established.414 Its object was to bring together specialists from multiple disciplines, including chemistry, medicine, engineering, physics, and biology for the purpose of researching and developing biomaterials, including surgical meshes.415 The sub-group examining surgical meshes (the Aachen Group as it was sometimes known) aimed to find a textile construction that was best adapted to the physiological requirements of the abdominal wall or groin so as to improve the biocompatibility of mesh used in hernia repair surgery.416 Before long, however, the work of the group expanded to include pelvic mesh. It was in the early 2000s that Professor Klosterhalfen began examining tissue samples from female pelvises which had been implanted with synthetic mesh.417
373 The pelvis data pool was created following an approach in 2003 by Dr Peter Meier on behalf of Ethicon. Professor Klosterhalfen understood that Dr Meier wanted him to assess tissue reaction (including the foreign body reaction) to pelvic meshes in the same way he had done with hernia meshes. The first 172 explants were sent to him by Ethicon.418 Later, clinicians from around the world started sending him mesh explants for analysis directly.
374 In the course of this work, Professor Klosterhalfen has examined thousands of tissue samples containing mesh and has built two data pools, one relating to hernia mesh (the hernia data pool) and another relating to pelvic mesh (the pelvis data pool).419 At the time he swore his first affidavit in early October 2016, the combined data pool, which he referred to as his data pool, contained more than 15,000 explanted polypropylene mesh tissue samples. Professor Klosterhalfen believes the data pool to be the largest such data pool in the world. Of the 15,000 explanted polypropylene mesh tissue samples, approximately 5,000 are human explants. The rest are preclinical animal explants. Of the 5,000 human explants, approximately 4,300 are hernia mesh explants and 700 are pelvic mesh explants.420 Of the 700 pelvic mesh explants, approximately 490 are sling explants and the remaining 210 are prolapse mesh explants. The data pool contains explants from each of the nine Ethicon devices, the subject of the proceeding.421 Some 1,000 of the 4,300 hernia mesh explants are full explants; that is to say that in each case the entire implant and tissue was removed for analysis.422
375 The year 1998 saw the publication in the European Journal of Surgery of an article by Professors Klinge, Klosterhalfen and others, including Dr Oblenski of the Institute for the Research and Development of Textile Materials in Aachen, entitled “Modified Mesh for Hernia Repair that is Adapted to the Physiology of the Abdominal Wall”.423 The article reported on the results of a rat study, the objective of which was the development of a new mesh for hernia repairs. The authors determined that the tensile strengths of meshes used for hernia surgery were far beyond what was necessary for fascial closure. Adjusting the meshes to the physiologically-required forces allowed for a considerable reduction in the amount of material. The authors postulated that reducing the material might reduce the rate of local wound complications and the degree of restriction of mobility of the abdominal wall caused by the then current meshes. The textiles they analysed were Mersilene (a polyester mesh made by Ethicon), Prolene, and the new combination of polypropylene with polyglactin 910, which they called Soft Hernia Mesh, which became Prolene Soft, and which was used to make UltraPro hernia mesh and Prolift+M. The effect of the addition of polyglactin was to reduce the non-absorbable component and therefore the amount of mesh after implantation.
376 This was one of the first papers to describe how the structure and mechanics of mesh affect its biocompatibility. Until then, according to Professor Klosterhalfen, the general view was that any complications with mesh were due to the surgery and not the product.424
377 Professor Klosterhalfen also demonstrated that a reduction in the amount of material reduced the foreign body response.425 That same year, Professor Klosterhalfen published the results of a dog study which showed a reduction in shrinkage of mesh with the reduction of polypropylene content.426 By 2000, if not earlier, Ethicon accepted that the amount of implanted material affects the extent of “mesh-induced inflammation”.427
378 Professor Klinge had a long-standing relationship with Ethicon. In December 1993, while he was preparing for a talk about mesh at a conference on inguinal hernia supported by various mesh manufacturers, he met one of Ethicon’s engineers, Dr Boris Obolenski.428 In February the following year Professor Klinge attended a meeting at the Suvretta House Hotel in St Moritz, Switzerland, to discuss hernia repair surgery and, in particular, the use of mesh. In his presentation he stressed that there was insufficient data to relate the properties of mesh to the biochemical demands and argued that it was necessary to look for textile constructions designed specifically for the purpose or reinforcing the abdominal wall.429 This was the first of five meetings which became known as the Suvretta meetings or conferences. Professor Klinge attended them all. So did Professor Klosterhalfen.430 Ethicon was one of a number of sponsors of the 1994 meeting but the sole sponsor of all subsequent meetings.431
379 After the 1994 Suvretta meeting, Professor Klinge prepared a draft protocol for a project the main objective of which was to find a textile construction better adapted to the physiological requirements of the abdominal wall or groin and so improve the biocompatibility of hernia meshes. Ethicon agreed to join the project and provide the filaments for the necessary tests and the currently available mesh constructs for comparison.432
380 Between 1994 and 2000 the Aachen Group met regularly with Ethicon scientists and other personnel. Aachen University entered into a contract with Ethicon in 1995 and the project continued for 10 years, supported by research grants.433 During this time, Professor Klinge said that the Group learned a great deal about the textiles, defined some standard biomechanical characterisation for better comparison, established models for testing the tissue response in animals, looked for parameters that reflected the inflammatory and fibrotic activity of the foreign body reaction, developed a technique to quantify the biomechanical impact of the meshes on the stiffness of the abdominal wall, and measured the biomechanical properties of tissues.434
381 In 1999, the year after the second Suvretta meeting, a textbook entitled Incisional Hernia was published.435 Professors Klinge and Klosterhalfen contributed chapters on experimental and histological aspects of biomaterials. There, they emphasised the importance of biocompatibility and called for an examination of whether meshes were necessary at all and whether they were “specifically suitable”. They pointed out that, “in contrast to normal suture material, it is necessary to choose the material very carefully”.436 Amongst “the important points” of their histological studies on surgical meshes, they noted that surgical meshes are not inert, even years after implantation; that they lead to chronic irritation of the host tissues; that the tissue reaction is dose-dependent (the greater the weight and surface area in contact with the mesh the greater the irritation of the tissues); and that the use of “heavy-weight meshes such as Prolene” should be avoided.
382 In May 2000, Professors Klinge and Klosterhalfen attended a meeting in Hamburg with various Ethicon personnel including Dr Holste. There, amongst other things, they discussed reducing mesh material by between 5% and 10%.437
383 In November 2001, Ethicon engaged Professor Klosterhalfen as a consultant.438 Professor Klosterhalfen also participated in studies of mesh implants, including those sent by Ethicon for histopathological examination and analysis, and reported periodically to Ethicon on his observations.439 For the next ten years he shared the results of his studies and research with Ethicon. He provided advice to Ethicon about how it could improve the design of its mesh so as to achieve better tissue integration and reduce mesh complications, including the foreign body reaction, infection, erosion, and contraction.440 In August 2002, for example, he sent Ethicon a copy of a paper he had written on the biological responses to mesh which pointed out that, while modern biomaterials including polymers are physically and chemically inert, they are not biologically inert. To the contrary, he wrote, they trigger a wide variety of adverse responses in vivo including inflammation, fibrosis, calcification, thrombosis, and infection.441
384 He also discussed his research results during regular visits to Aachen by members of Ethicon’s Research and Development team, especially Dr Engel and Dr Holste, and he would attend Ethicon’s Hamburg premises where he would discuss his research and give presentations.442
385 The work of the Aachen Group led to the launch by Ethicon in 1999 of Prolene Soft mesh (used in Gynemesh PS, Prolift, and Prosima) and of a number of hybrid meshes, made of a combination of polypropylene and an absorbable polymer to improve handling and reduce the amount of polypropylene. One of those hybrid meshes was UltraPro, launched in 2002, which was the mesh used in Prolift+M.443 All the tests the Group conducted on these meshes were premised on their use in the abdominal wall or the groin (the area of the hip on each side of the body located in the folds where the abdomen joins the upper thigh, also known as the inguinal area).
386 In July 2007 Professor Klosterhalfen met with Drs Meier and Holste. At that meeting Professor Klosterhalfen discussed the need to design a monofilament, very large pore mesh that is elastic and adapts to the biomechanics of the vagina.444 At the same meeting, Professor Klosterhalfen suggested that Ethicon contact Professor Prescher in Aachen about the morphology of the pelvic floor.445 I was not taken to any evidence to indicate that this suggestion was taken up.
387 In October 2007, after examining about 20 pelvic floor meshes received from Professor Deprest, Professor Klosterhalfen concluded that nearly all exhibited (signs of) erosions and demonstrated the same tissue reaction as hernia meshes but with greater incidence of infection.446
388 By early April 2008 Professor Klosterhalfen had evaluated about 100 explanted pelvic mesh samples. He deposed that his analysis showed that mesh erosion was typically accompanied by mesh infection and that mesh shrinkage and contraction was more apparent than in hernia explants.447 In June 2009 he prepared an intermediate report for Ethicon based on his analysis of 172 pelvic mesh explants. He found that the tissue reaction to pelvic and hernia mesh was the same but that folding occurred more frequently in the pelvic mesh explants which he attributed to “the mechanical mismatch” (discussed below) between the implant and the pelvic tissue.448
389 Professor Klosterhalfen’s consultancy agreement with Ethicon, signed on 26 November 2001,449 was renewed on several occasions. In December 2011, however, Professor Klosterhalfen advised Dr Holste that he would not be renewing his contract.450 In his affidavit he stated that his relationship had become strained during 2009 when he felt that Ethicon was no longer interested in collaborating with him but wanted him as an opinion leader, to become more involved in marketing and promoting its products. Professor Klosterhalfen went on to explain:
Ethicon wanted me to say that Prolift + M and UltraPro solved all of the problems which I had identified with mesh. However, I did not believe that. I considered that the fundamental problem with all polypropylene mesh was that it did not have the mechanical characteristics suitable for the female pelvis.451
This evidence was not challenged in cross-examination.
The opinions of Professors Klosterhalfen and Klinge about the Ethicon devices
390 Professors Klosterhalfen and Klinge expressed strong views about the unsuitability of the Ethicon devices for the purpose for which they were supplied and the deficiencies in the IFUs accompanying them.
391 Based on their research, testing conducted by others, including Ethicon’s own scientists, and their examination of explanted meshes, Professors Klosterhalfen and Klinge concluded that pore size and geometry are the most important influences on the outcome of tissue repair using a synthetic material. They concluded that pore sizes change in vivo due to the biomechanical forces to which the meshes are exposed so that, contrary to what had earlier been assumed, a “large-pore” mesh, as the respondents contended both Prolene and Prolene Soft were, can deform under load. If that occurs, Professors Klosterhalfen and Klinge said, there is an increased risk of injury by scarring.452 What matters, they argued, is not just porosity, but “effective porosity”.
392 Professor Klinge explained that, except for some cells in the heart and the brain, cells in the body are permanently subjected to remodelling; they die and are renewed or replaced by other cells. Cells in soft tissues die within weeks or months and are replaced by local stem cells. Scars, on the other hand, are always replaced by scar tissue, fat tissue by fat tissue, and fascial tissue by fascial tissue. One effect of this permanent cellular turnover is that any strain imposed on a foreign body (including a mesh implant) will cause the foreign body to migrate to escape the stress and an area of “enhanced remodelling” will be preserved at the interface as the inflammatory cells will be replaced after a shorter period than scar or collagen.453 The relevant forces include those exerted during ordinary activities such as straining on the toilet, exercise, sexual intercourse, even just standing up. The degree of risk increases with the degree of strain.454 These matters are discussed at greater length below.
393 Professor Wright said that it was difficult to envisage a mechanism that would allow mesh to migrate once fibrous connective tissue has grown into the pores and anchored the mesh in the adjacent vaginal tissue.455 This evidence reflected his lack of relevant clinical expertise.
394 Ethicon considered that insufficient integration of fascial tissue caused migration of mesh.456 Members of the TVM Group described migration of mesh prostheses as a complication of the use of mesh in the surgical treatment of vaginal prolapse.457
395 Professor Klosterhalfen said that all the studies undertaken by the Aachen Group indicate that even decades after implantation surgical meshes show a tissue and cell turnover at the mesh interface. Consequently, there is never complete integration or an endpoint to integration of the mesh.458
396 Professor Klinge pointed out that migration of foreign bodies is an everyday experience in orthopedic surgery and also in general surgery for the treatment of anal fistula. He went on to say that:
Remodelling of the soft tissues is a fact, and the more inflammation, the more proliferation, the more remodelling, and the higher the risk for migration. The presence of granulomas always means chronic inflammation, which always favours migration in case of mechanical strain (as in the pelvic floor area).
397 Professors Klosterhalfen and Klinge referred to the different forces at work in the abdomen and the pelvic floor and the unique nature of the vaginal environment.459 In its pre-market notification to the FDA for Prolene Soft, Ethicon claimed that the process by which the mesh is knitted, interlinking each fibre junction, provides for elasticity in both directions and this “bi-directional elastic property allows adaption to various stresses encountered in the body”.460 The representation of elasticity in both directions was incorporated into the instructions for use for all the POP devices. Professor Klinge considered that, while this constituted an acknowledgment that elastic elongation occurs as a result of mechanical stress and that the devices are subjected to strain in vivo, the concept of bi-directional elasticity does not account for the anisotropic behaviour of pelvic tissues. “Anisotropic” derives from the Greek words anisos meaning unequal and tropos meaning turn. Anisotropic objects or substances have a physical property which has a different value when measured in different directions. Bilateral elasticity is not enough to allow the mesh to adapt to the various stresses encountered in the body.461 Moreover, the reference to elasticity is misleading. An elastic material is one that spontaneously resumes its normal bulk or shape after having been contracted, dilated, or distorted by external force, whether after a short or a long interval.462 Both professors pointed out that polypropylene is not an elastic polymer since, once stretched, it does not return to its original state. In cross-examination, Professor Klinge explained that deformation of the pores of the mesh stretches the small loops connecting the fibres and, over time, causes both to elongate with use.463
398 Professor Klinge said that the Prolene mesh in the SUI devices contracts or shrinks by 30-50% after implantation, citing studies of the mesh after implantation in dogs conducted by the Aachen Group in 1998, before the launch of TVT.464 He also said:
A knitted surgical mesh device like the TVT that is permanently implanted in human tissue must be designed in such a manner that the pores of the mesh do not collapse and deform upon the expected forces of implantation as well as the expected in vivo forces. Under minimal strain, the TVT mesh pores deform and collapse thereby increasing the risk of injury to patients in which it is implanted and is a less safe design than products that better withstand these conditions and do not display these poor outcomes. It is my opinion that permanent deformation and pore collapse of the TVT mesh leads to fibrotic bridging, scar plate formation, excessive scarring through and around the mesh and a host of tissue complications that can lead to chronic pain, recurrence, erosions, dyspareunia and need for reoperation, to name a few, making it unnecessarily unsafe for its intended purpose of being permanently implanted in a woman’s pelvic tissue.
He added that shrinkage of the mesh after implantation, caused by fibrosis, also leads to such complications.465
399 Professor Klinge deposed that when tension is placed on the mesh, it curls and ropes causing increased scarring between the fibres and its frayed, un-bordered edges shed particles of polypropylene before, during, and after surgery. He said that the release of particles into the surrounding tissue leads to an increased inflammatory response, erosion, chronic pelvic pain, implant failure, chronic sexual dysfunction and dyspareunia, organ damage, urinary dysfunction, inability to remove the device and the need for surgical intervention.466
400 He said that the Prolene mesh used in the SUI devices was over-engineered in that it was many times stronger than necessary for its intended purpose and that it leaves far more polymer material behind than is necessary. He expressed the view that “any pelvic mesh designed with this much excess surface area and weight unreasonably and unnecessarily increases the risk of injury to the patient …”.467
401 He regarded as unreasonable Ethicon’s failure to make critical design changes to each of its SUI devices before launch. He considered that the use of Prolene in the SUI devices unnecessarily compromised patient safety, leading to such complications as chronic inflammation, excessive scarring through and around the mesh, nerve entrapment, chronic pain, dyspareunia, erosion, recurrence, and the need for corrective surgery.468
402 Professor Klinge expressed similar opinions with respect to Prolene Soft (Gynemesh PS) used in the various POP devices. He said that “[i]t is impossible to establish reliable parameters for the design of a device for use in POP repair surgery” without understanding the biomechanics of the pelvic floor. Yet he said that Ethicon had “a very poor understanding” and apparently still does. In this respect, he was supported by Professor Deprest who had written that the mechanical requirements for implants in pelvic surgery had not been defined by 2006 and the mechanisms causing local complications in urogynaecology were poorly understood, then and still.469
403 Professor Klinge deposed that the POP devices were not specifically designed to function in the pelvic floor; they were over-engineered (that is, they were much stronger than necessary); they will create “an intensified and chronic [foreign body reaction]”; they will have pores that are too small to resist fibrotic bridging and the formation of scar plates; and they will curl, rope and fray causing particle loss and sharp edges. All these “design failures”, he added, “cause an unnecessary risk of patient complications and injuries”, including (amongst other things) chronic pain, dyspareunia, erosion, exposure, infection, and the recurrence of the condition which the implantation of the device was designed to treat.470 He said that it was unreasonable of Ethicon not “to properly study and/or make the necessary design changes” to avoid the “known and serious complication” of mesh shrinkage and the other safety hazards already mentioned. This opinion, he said, applied to all the POP devices that used Prolene Soft mesh.471
404 Professor Klosterhalfen’s opinions were to the same effect.
405 On the face of things, having regard to the extensive work undertaken in this area by both Professors Klinge and Klosterhalfen and their close collaboration with Ethicon over a substantial period of time, their opinions were compelling. On the other hand, the logical consequence of the opinions they expressed, at least on one construction, was that every woman in whom these devices was implanted would suffer from the pleaded complications at least if she lived long enough. While it is possible that this is so, the evidence does not establish that every woman with an Ethicon device will more probably than not experience all or even any of those complications. Nonetheless, the evidence does establish on the balance of probabilities that she could.
The attack on the Aachen Group’s pelvis data pool
406 Since the evidence of Professors Klosterhalfen and Klinge was so powerful, before going any further it is necessary to address the respondents’ attempt to undercut its force.
407 It is common ground that, until the hearing in this case, the respondents never questioned the reliability of the information derived from the pelvis data pool or the information provided by Professor Klosterhalfen in his reports to Ethicon about his findings.472 Yet, drawing upon answers derived from their cross-examination of the Professor about data recorded in some spreadsheets relating to the data pool, the respondents submitted that it was “plain” that there was “substantial doubt as to the accuracy and reliability of the theories developed by Professors Klosterhalfen and Klinge”. The reason the respondents gave was that “they are reliant on the data from the Pelvis Data Pool” and, to the extent that the data could be verified, “significant errors (acknowledged by Professor Klosterhalfen) have been shown”.473 They made this submission, I should add, while at the same time acknowledging that the “theories” and work of the two men draw on both the hernia and the pelvis data pool.474 In particular, the respondents submitted that the Court should be cautious about coming to conclusions solely on the basis of the data pool, particularly where those conclusions conflict with other evidence. They urged the Court to be mindful of its “inherent limitations”.475
408 I will come to the matters upon which the respondents rely shortly but one point should be made at the outset. That is this. No conclusion I reach could ever be based solely on the data pool. If the respondents mean that I should be cautious about accepting opinions reached solely on the basis of the data pool, that will depend on the extent to which the particular shortcomings raised by the respondents affect the opinions of the witnesses that were derived from the data pool.
409 I now turn to the respondents’ complaints.
410 First, the respondents pointed to errors in an Excel spreadsheet relating to the pelvis data pool.476 The spreadsheet, which was cited in Professor Klosterhalfen’s second affidavit, contained four worksheets, entitled “Raw data”, “Final I”, “Final II statistics” and “Final PP (J&J)”.477 The respondents submitted that there were errors and inconsistencies in the first three worksheets. They also put weight on the fact that these worksheets were prepared by a student of Professor Klosterhalfen and not the Professor himself. It should be noted, that the student who collected the “raw data” was a medical graduate, although not yet a pathologist.478
411 In cross-examination Professor Klosterhalfen explained that tissue samples were sent by clinicians to his centre together with background information. It was then analysed by the student.479 The student’s analysis, together with the additional information from the clinician, were recorded in the first worksheet (“Raw Data”). For each sample, information was recorded under the following headings: infection, FBR (foreign body reaction), fibrosis, polymer, mono/multi, other (which recorded information about observable adverse events such as erosion and shrinkage), age (of the patient), and mesh class (usually by reference to the weight and size of the pores). Additional information was recorded without any headings, most notably about the identity of the devices (such as TVT-O or Prolift), the condition for which the device was implanted, and adverse events reported by the clinician (such as pain).
412 Professor Klosterhalfen described the compilation of the information in the first worksheet as follows:
She [the student] was looking for – or responsible for this data pool. She made the first analysis. She made the macroscopy and she collected all the slides and tried to get out – to collect the clinical information, and that’s why the spreadsheet is called raw data. These are uncontrolled data. It’s her view what she was collecting there.480
413 The second worksheet (“Final I”) replicated some of the headings and added new ones. Importantly, there was a category entitled “mesh class”, which categorised the device as either “1” (for large pore mesh) or “2” (for small pore mesh).481 The characterisation was made by the student.482 Professor Klosterhalfen accepted that any errors in the first worksheet would “infect” the second worksheet, and because of the errors present, one would not rely on the mesh classification in the second sheet.483 The same was the case for the third worksheet.484
414 Professor Klosterhalfen was cross-examined about some of these errors.485 He was taken to a couple of entries, for example, in which Vypro II was described as a heavyweight small pore mesh, when it is actually a lightweight large pore mesh (before implantation at least).486 Professor Klosterhalfen said that the error could have been in the identification of the mesh rather than the description of its characteristics or vice versa.487 The respondents claimed that there were numerous occasions where an explant was incorrectly described although they did not take the Court to those occasions. But these were the student’s errors, not the Professor’s.
415 Importantly, Professor Klosterhalfen testified that he worked with the student on the fourth and final worksheet (“Final PP (J&J)”) and double-checked the data.488 As I understood the evidence, this was the data upon which Professor Klosterhalfen relied, not the data in the earlier worksheets.489 He explained that the data pools are “a scientific work tool”. It was only after the data were checked with the clinicians and the slides were checked, that he accessed the data pool.490
416 Nevertheless, because the original analysis was undertaken by the student, the respondents submitted that there was room for error in both the identification of the type of mesh explanted as well as its characterisation (as a large or small pore mesh). Yet, as to errors in identifying the type of mesh, Professor Klosterhalfen explained that clinical data including the type of implant was checked two or three times with the treating clinician before being included in the data pool and any mesh that could not be confirmed with the treating clinician was not included in the pool.491 As to any errors in the original characterisation by the student while collecting the raw data, in cross-examination Professor Klosterhalfen readily acknowledged that errors would arise at that point: “You will find a lot of that, most probably” and “You can go through all 500; you will find mistakes”.492 While a couple of column headings in the final worksheet were misleading or inapt (“PP” instead of “small or large pore” and “J&J” when not all the explants were Ethicon explants), Professor Klosterhalfen did not accept that errors in earlier worksheets made their way into the final worksheets.493 Indeed, he insisted that the data in those spreadsheets were accurate:
MR FINCH: …You will accept this part of the spreadsheet, that the student one is full of errors of allocation, both as to – possibly as to the nomination of what product it was and certainly as to the equation of the products with the size pores?---Yes. But the next step is that you control that and that you have to get in contact with the doctors, that you have to get the history of the patient, and the - - -
Okay?--- - - - how do you say, the protocols of the operation. And then finally you will find these correct data in the last spreadsheets.494
(Emphasis added)
417 Still, the respondents argued that, because the doctor who implanted the mesh was very often not the doctor who explanted it, there was room for error on that account in the identification of the mesh. While Professor Klosterhalfen acknowledged, in effect, that there was room for error on the part of the clinician who provided the explant, the evidence indicates that any such error would not find its way into the final spreadsheets. He explained:
See, basically what you have to know, you see: you get these examples and the clinical colleague is writing what he believes what have been implanted. And it’s very often that doctor who has implanted this mesh is not the doctor who explants the mesh finally, especially after eight years. And all this clinical data, you have to check it twice or third.495
…
You see, what is relevant here, if you ask me what is the definite data, the last spreadsheet, for instance. So – and basically, as I told you before, for me these data pools are a scientific work tool. And when all these data are checked with the clinicians and the slides are checked, I go into this data pool and basically if you look at the last spreadsheet you see only one and two, one and two, yes, no, yes no. So a statistical analysis is possible. That’s what we – what is interesting for me.496
418 Professor Klosterhalfen said that the complication rates he mentioned in his presentations, including his presentation in Istanbul on the “Pathology of Meshes in the Pelvic Floor: Actual Data” given on 6 September 2015 (the Istanbul presentation),497 were drawn from the last of the four worksheets (or a version of it).498 He stated that he had personally checked the data he used for the presentation. Where, for example, there was a discrepancy between what the student had recorded and the information provided by the clinician, and he was able to do so, he double-checked the information including, it appears, by examining the sample microscopically.499 I accept that he did check the data. His credit was never impugned and it was not suggested that this evidence was unreliable.
419 Second, although the respondents accepted that the fourth and final worksheet was double-checked by Professor Klosterhalfen, they complained that they had “no way to test its contents” because they had not had access to the underlying data used to prepare the spreadsheet. In a footnote to their closing submissions they asserted that a notice to produce “the documents or things which Professor Klosterhalfen created, considered or relied upon in relation to his examination of the Pelvis Data Pool” (to which the applicants had taken objection) was served on the applicants on 3 August 2017.500
420 The respondents argued that the applicants’ failure to produce the data was important in a context where there were differences in the conclusions reached by Professor Klosterhalfen and the clinicians who provided the original data. They pointed to instances where Professor Klosterhalfen said he could not identify an erosion from an explant or slide although the clinician who provided it had recorded one. They submitted that this necessarily casts doubt upon the reliability of the data provided by the clinicians (or, arguably, on Professor Klosterhalfen) which in turn casts doubt upon the reliability of the data pool, particularly when the record of the reasons for explanation, such as chronic pain, dyspareunia, discomfort/pain, recurrence, migration of mesh, and fistula/abscesses, was derived from calls to the clinicians, calls made by the student and not verified by the Professor through a second call.501
421 Quite apart from the inappropriateness of serving the notice to produce on the applicants when the material to which it referred would obviously not have been in their possession or control, the notice to produce was not in evidence and was never called upon. In those circumstances, the complaint about the failure to produce the underlying data goes nowhere. In the first place, in the absence of a call on the notice there was no failure to produce the documents. In any case, the evidence was that not all notes were kept and it was not suggested that there was anything untoward about that. Although the slides were retained, Professor Klosterhalfen observed that they were the patients’ property and could only be released with their permission.502 This observation was not challenged.
422 Finally, to the extent that the respondents claimed that they were deprived of an opportunity to verify the information, that complaint is difficult to take seriously when they had a longstanding contractual relationship with Professor Klosterhalfen. It will be recalled that it was Ethicon (through Dr Meier) suggested the type of information to be collected and understood the way it would be collected and could at any time have asked to review the Professor’s data and methods, but never did so.503
423 The applicants rightly took issue with the characterisation of Professor Klosterhalfen’s concessions as an acknowledgement of “significant errors” in the pelvis data pool. As they argued, the respondents’ submission was not accompanied by a transcript reference and no errors were acknowledged or disclosed in the final worksheet, which was the one upon which the Professor relied.504
424 As to the admitted errors, I fail to see how they matter — even if they can be said to be “significant”. There will always be errors in working documents. It is the final document that counts.
425 Third, the respondents argued that there were inherent limitations in the conclusions that can be reached on the basis of the pelvic data pool because the explants necessarily represent cases with “a symptomatic complication”, which is why they were explanted, and so the complication rates drawn from the data pool represent a percentage of that cohort (with a symptomatic complication) rather than a broader, representative population of patients including those with no symptoms. The respondents submitted that the data pool does not provide evidence of “the pathological processes associated with a successful (or asymptomatic) outcome”. They argued that “the Data Pools comprise solely of explants, which represent bad cases (or they would not have been explanted)”.505
426 It is not strictly true to say that the data pools are comprised solely of explants representing “bad cases”. This was an assumption. The same assumption was made by Professor Deprest.506 It was an understandable assumption, but it was wrong. Professor Klosterhalfen pointed out in his second affidavit that the data pool does have some internal controls. 507 He explained:
It is true that the structure of a database sometimes lacks appropriate controls. Therefore human mesh samples or biopsies of meshes without any complication have to be included. In my Hernia Data Pool, mesh samples explanted in the context of other surgical procedures (e.g. large bowel cancer, prostate cancer or bladder cancer) serve as an internal control. In total, my Hernia Data Pool includes 112 mesh biopsies and 33 full explants, serving as controls.
427 Professor Klosterhalfen’s opinion about the significance of nerve entrapment in explants was influenced by the comparison with the controls. He said that in the hernia data pool it was rare to find entrapped nerves in mesh explanted other than for pain but that they could often be seen in cases where mesh was explanted for chronic pain.508
428 Nevertheless, I accept that the complication rates mentioned in the Istanbul presentation are percentages of complications revealed in the explants provided to Professor Klosterhalfen and are not representative of the complication rates in all patients implanted with polypropylene mesh. I did not understand Professor Klosterhalfen to have represented that they were.
429 Fourth, the respondents contended that the results Professor Klosterhalfen recorded in the Istanbul presentation cast doubt on his own thesis. That is because slide 44 records an erosion rate of 64.5% for large porous meshes (on average 24.7 months after implantation) and 40.8% for small porous meshes (on average 39.4 months after implantation).509 The respondents argued that the data is either inconsistent with the Professor’s “theory” or the data is wrong because of error in the collection or recording of it.
430 I reject the argument. I accept the applicants’ submission that the data is not wrong and the figures are consistent with Professor Klosterhalfen’s theory of effective porosity or, at least, not inconsistent with it. “Effective porosity”, discussed in more detail below, is the percentage of the area of mesh that is filled only by sufficiently large (effective) pores (to allow for the ingrowth of healthy tissue)510 and is concerned with the size of the pores after implantation and under load.511 The concept was first referred to publicly in a 2007 article by the Aachen Group: “New objective measurement to characterize the porosity of textile implants” (Mühl et al (2007)512. As a result of the studies carried out by the Aachen Group, it was concluded that to allow for the ingrowth of healthy tissues, the pores in the mesh need to be greater than 1000 microns (micrometres or μm and the equivalent of 1mm) in all directions.513 In his testimony in this case, Professor Klinge explained that, if you apply tension to a large pore mesh (UItraPro was the example he gave), small pores can be created, removing the advantages of the large pore mesh and, in effect, transforming it into a small pore mesh.514
431 Fifth, the respondents complained that only some of the complications were independently checked by Professor Klosterhalfen and, when they were checked, there were differences between the Professor’s views and those of the clinicians who provided the initial data.515
432 This was at best a simplistic representation of the effect of the evidence.
433 The evidence was that, if the student had recorded an erosion when the clinician had not, Professor Klosterhalfen, himself, checked the slide or slides.516 He said that you can see an erosion in the microscope. He added, however, that sometimes the clinician did not record an erosion when it was clearly visible in the slides. He said that the same was true for infection. As he explained later in his evidence, certain cells are markers of infection. They are the polymorphonuclear granulocytes (PMNs) and he had seen those cells in explants.517 On the other hand, he was reliant on the clinicians for information as to other complications like pain, migration, and recurrence. It is inconceivable that Ethicon would not have been aware of this at the time Dr Meier asked Professor Klosterhalfen to collect the information.
434 The mere fact that a student collected and checked the information from the clinicians does not make the information unreliable. Given the size of the data pools, I am not persuaded that any such errors are likely to have had any significant effect or that they detract from the force of the opinions expressed by Professors Klosterhalfen and Klinge, even if, contrary to my understanding of the evidence, one or more of them did creep into the final iterations of the spreadsheets.
435 Sixth, the respondents submitted that Professor Klosterhalfen’s evidence should not be accepted because the “theories” he and Professor Klinge propounded were inconsistent with Professor Wright’s experience and “the rigorous testing [by Ethicon] of the meshes used in the Implants”.518
436 This submission must be rejected.
437 For the reasons given above, Professor Wright’s experience is not comparable to the experience of Professors Klinge and Klosterhalfen. In the circumstances, any difference of opinion between Professor Wright on the one hand and Professors Klosterhalfen and Klinge on the other cannot be taken as a valid criticism of the data pool.
438 Moreover, the evidence does not establish that Ethicon subjected the meshes used in the Ethicon devices, particularly the pelvic meshes, to “rigorous testing” — far from it in fact.
439 In any case, the conclusions Professor Klosterhalfen reached and the opinions he formed, like those of Professor Klinge, were not based solely on the data in the data pool, let alone the pelvis data pool. Much of their thinking was informed by their animal studies, their observations of the tissue in which the mesh was embedded or encased, their tests, their extensive reading and research, and the studies of hernia explants.
440 The concept of bridging fibrosis, for example, was first described in reports and articles published before the pelvis data pool was set up.519 It was described, for example, in the report of a study of the foreign body reaction to meshes used for the repair of abdominal wall hernias by Professors Klinge and Klosterhalfen and others, published in the European Journal of Surgery in 1999: Klinge et al (1999).520
441 It was in the early 2000s that they recognised the connection between bridging fibrosis and pore size. On 6 March 2002 the results were published from the Aachen Group’s study of the impact of pore sizes in rats.521 That study compared Marlex (made by the UK firm Bard) and Vypro (made by Ethicon). In the body of the article the authors, who included Professors Klinge and Klosterhalfen, wrote:
[T]he monofilament mesh with its smaller pores almost exclusively is embedded into granulomas and scar tissue, which bridges the whole pore diameter of less than 1 mm. In the case of small pores the extent of the activated foreign body reaction does not permit the ingrowth or persistence of local tissue. A large distance between the single filaments not only guarantees the maintenance of elasticity but will hamper the bridging of the inflammation across the pore.
442 Six months later, in September 2002, the results of another rat study by the group comparing Prolene hernia explants to explants of another material, Vypro II (made of polypropylene and polyglactin multifilaments), found that the Prolene mesh was entirely encapsulated in a scar plate. The authors, who again included Professors Klinge and Klosterhalfen, wrote that the data confirmed that a reduction in the total amount of implanted material and an increased pore size have a favourable impact on integration of the mesh with fewer undesirable side effects. They concluded on the basis of the results, that for proper incorporation of healthy tissue pore sizes should be “at least 1000 µm”, in contrast to the then prevailing wisdom that 75 to 100µm was sufficient.522
443 An article published the following month on mesh implants in hernia repair reported on a rat study comparing the inflammatory cell response to Prolene (according to the abstract, Marlex according to the body of the article), Mersilene, and Vypro after 7 and 90 days of implantation. The authors, all from Aachen University and who included Professors Klinge and Klosterhalfen, wrote that the findings underlined “a more pronounced inflammatory reaction and cell turnover around heavy-weight and small-pored [polypropylene] mesh implants”, consistent with other investigations describing “an accented foreign body reaction and fibrosis for heavy-weight polypropylene meshes”.523
444 Professor Klosterhalfen stated at various points in his evidence that he had consistently observed in histopathology of explanted meshes “contraction of tissue” and “shrinkage”, folding and curling of mesh along with dense scarification, chronic inflammatory responses, and nerve entrapment.524 He also noted that those “in vivo conditions” had been widely reported in the published literature.525 He said that the opinion he expressed that those pathological findings were consistent with, and directly related to, the pain and contraction experienced by women who had received the implants was based on his examination of over 600 samples of histopathology as well as his own testing, research, review of medical and scientific literature and work he had done with experts across the world.526 He was not challenged about this in cross-examination.
445 A number of the opinions Professor Klosterhalfen said were informed by the pelvic data pool were not seriously disputed. They include his opinions about the foreign body reaction to polypropylene mesh implants, which I consider next, the extent to which the mesh shrinks or contracts in vivo, the occurrence of infection with erosion, and the possibility of late infections.
446 More particularly, the respondents did not point to any specific opinion given by either Professors Klosterhalfen or Klinge that was founded on, or undermined or weakened by, the errors in the worksheets. Indeed, the respondents made no attempt to explain the connection, if there was one, between the errors and the witnesses’ opinions. Professor Klosterhalfen’s evidence was that he relied on the final spreadsheet and there was no evidence of error in that document.
447 Furthermore, it was not put to Professor Klosterhalfen that the errors in the worksheets undermined his opinions and Professor Klinge was not even cross-examined on the subject. Since it was not put to either witness, the submission that errors made by the student cast doubt on the accuracy or reliability of their opinions ought not to have been made: Browne v Dunn (1893) 6 R 67 (HL).
448 Finally, it will be recalled that the two data pools included over 15,000 explants. The vast majority of human explants were hernia explants. In other words the hernia data pool was much larger than the pelvic data pool. No attack was made on the hernia data pool. No questions were asked of either Professor Klosterhalfen or Professor Klinge about that data pool. It was not suggested that the conclusions derived from the data collected from hernia explants or explants from animals were unreliable. Yet it is abundantly clear that, to the extent that the opinions of the professors were based on what they had learned from their studies of explants, those opinions were derived from the entirety of the information in both data pools.
449 For all these reasons, I reject the submission that there is substantial doubt about the reliability of the theories developed by Professors Klosterhalfen and Klinge because of “significant errors” in three of the four worksheets.
450 The pore of a textile is the area or space between filaments.527 As the applicants submitted, none of the respondents’ witnesses seriously questioned the importance of pore size to the biocompatibility of the devices and the relationship between pore size and the inflammatory response to polypropylene mesh.528 The respondents accepted that pore size is a critical component of the biocompatibility of any mesh used in the body.529
451 By 1998, when Professor Klosterhalfen first published on the subject, it was known that there were higher complication rates when small pore mesh was used.530 It was well accepted, for example, that large pore meshes had significantly lower infection rates than small pore meshes.531
452 The debate in this case turned on the appropriate pore size, including the classification of pore sizes, and the concept of bridging fibrosis.
453 The specific dispute concerns whether the classification of biomaterials by Professor Parviz Amid in his seminal article, “Classification of biomaterials and their related complications in abdominal wall hernia surgery”, published in 1997 in the journal Hernia, has any continuing relevance, particularly in relation to pelvic meshes.532 The respondents maintained that it has and that it should be applied, doubtless because of what Professor Amid said about Prolene. The answer to the question is that the Amid classification is of limited utility, certainly in relation to meshes in the pelvic floor, although many of the observations made by Professor Amid in that article remain important.
454 In his 1997 article Professor Amid classified the hernia meshes then on the market according to pore size. He also discussed the importance of pore size, molecular permeation, and shrinkage of biomaterials, the role of biomaterials in increasing the risk of infection, the formation of seromas and fistulas, and the failure of repair due to “shrinkage” of the mesh. Professor Amid said that “[p]revention of biomaterial-related complications requires in-depth knowledge and understanding of the physical properties of prostheses, of which, the porosity and the pore size of the materials are of paramount importance”. None of this was controversial.
455 Professor Amid divided meshes into four types. Type 1 included meshes with pores greater than 75μm (0.075mm). He referred to them as “totally macroporous” prostheses. Type II included meshes with pores less than 10μm (0.01mm). He described type III as “[m]acroporous prosthesis with multifilamentous or microporous components”. Type IV were films with submicronic pores of less than 1μm. The difficulty with Professor Amid’s classification, at least for the purpose of classifying pelvic meshes, is that it assumes that the pore size does not change after implantation. The evidence before the Court shows that this assumption was incorrect.
456 Professor Amid classified both Marlex and Prolene as type I or macroporous (large pore) meshes. He wrote:
These prostheses contain pores larger than 75 microns, which is the required pore size for admission of macrophages, fibroblasts (fibroplasia), blood vessels (angiogenesis) and collagen fibers into the pores ...533
457 Professor Amid explained that surgical infection, promoted by implantation of biomaterials, is caused by infiltration and proliferation of bacteria into and within the pores and interstices of these synthetic materials. When interstices or pores are less than 10μm in each of their three dimensions, he wrote, bacteria averaging 1μm cannot be eliminated by macrophages and neutrophilic granulocytes, which are too big to enter the space. Consequently, small pore prostheses harbour bacteria and can promote their growth resulting in “biomaterials-related infection”.
458 The TVM Group emphasised the problem about the interstices. According to the English translation, they wrote:534
The concept of interstice must be clearly understood of the risk of infection associated with endovaginal surgery (sic).
Porosity is also a major mesh characteristic. Porosity will promote colonization of the prosthetic tissue “like a lattice by ivy”.
Fibroblasts, collagen fibers, and neo-vessels will be incorporated if they can circulate easily between the stitches, provided the pores are large enough.
459 Until the Aachen Group’s research, however, it seems that little, if any, thought was given to the possibility that the size of the pores would be affected by the forces operating in the body after implantation.
460 Professor Klosterhalfen explained what should have been obvious from the outset:
[M]eshes are knitted and therefore have numerous large and small pores. It is not enough to simply look at the largest pores. Rather, it is necessary to look at the distribution of pores (porosity), the effective pore size under load and after tissue integration, and the pore geometry (the smallest diameter of the pore determines the risk of bridging fibrosis). All these factors need to be taken into account when looking at the pore size of the [SUI devices] and the [POP devices]. Experimental studies have shown that the effective pore size of [polypropylene] meshes is less than 1mm …535
461 While Professor Amid’s classification is still used by many hernia surgeons to assess the risk of infection, Professors Klosterhalfen and Klinge considered it was not appropriate for the assessment of tissue integration in mesh.536 Professor Klosterhalfen pointed out that it only takes into account meshes produced and used for hernia repair before 1998 and it fails to take into account what happens to the mesh after implantation and under load. 537 Moreover, as Professor Klinge observed, Professor Amid’s focus was on the risk of bacterial infection and not on biocompatibility, the foreign body reaction, or the risk for excessive scar formation.538 These matters were the focus of subsequent research, including but by no means confined to that conducted by the Aachen Group. 539
462 In 2011, based on an analysis of 1,000 explanted meshes, Professors Klinge and Klosterhalfen modified the classification of surgical meshes for hernia repair. They came up with six different classes. Under this modified system, described in an article in Hernia published in May 2012, they placed UltraPro in the large pore class (characterised by a textile porosity of greater than 60% or an effective porosity greater than 0%) and Prolene and Marlex as small pore meshes (characterised by a textile porosity of less than 60% and without any effective porosity).540
463 This classification covers particular mesh modifications, including mesh for laparoscopic and intraperitoneal incisional hernia repair, which were not covered by Professor Amid’s classification. As Professor Klosterhalfen explained in evidence, it was based on increased knowledge about the significance of pore size, pore geometry, and pore stability under mechanical load.541
464 Despite the increase in knowledge over the last two decades and their acknowledgment that pore sizes change in vivo, the respondents maintained that the Amid classification “remains applicable for classifying meshes”, including meshes used in the treatment of stress urinary incontinence and pelvic organ prolapse. They also contended that a minimum pore size of 75μmis sufficient to avoid the formation of a scar plate. They offered four reasons: first, there are examples of scientific literature that positively refer to the Amid classification; second, the majority of the scientific literature refers to the classification without comment; third, there are significant limitations to the reliability of the data from Professor Klosterhalfen’s data pools; and fourth, the effective porosity “theory” of Professors Klosterhalfen and Klinge is inconsistent with Professor Wright’s experience.542
465 I have already addressed the question of the reliability of Professor Klosterhalfen’s data. I set out below my response to the other three reasons.
466 Dealing with the last point first, it is difficult to understand what the respondents meant by Professor Wright’s “experience” in this context. While he disagreed with a statement in an Ethicon document that fibrotic bridging “is usually observed in all modifications with pore sizes of less than 1 millimetre”, he conceded he had undertaken no research of his own. He said he disagreed because “the limit” was based on an extensive study of the literature, a study conducted for the purpose of US and Australian litigation. He acknowledged that (in contrast) Professors Klosterhalfen and Klinge had spent most of their professional lives conducting independent research on this very topic.543
467 In any case, as I have already said, Professor Wright’s experience is inconsequential in comparison with the experience of Professors Klosterhalfen and Klinge.
468 Second, the fact that a number of scientific papers have uncritically adopted the Amid classification does not prove its continuing utility, particularly in the area of pelvic floor meshes. To enable any weight to be placed on the reliance by the authors of the papers on the use of the classification, it would be necessary to know whether they were aware of the research of the Aachen Group and had considered the matters that Professors Klosterhalfen and Klinge raised in their evidence in this case, including the important point they make that pore sizes change in vivo. I was taken to no such evidence.
469 Third, Professor Klosterhalfen gave unchallenged evidence that the validity of the Amid classification has never been proven in a clinical study.544
470 Fourth, the Amid classification was devised for synthetic mesh used in abdominal wall hernia surgery. Consequently, it did not take into account the different environment of, or the forces at work in, the female pelvic floor.
471 Fifth, Barone et al (2015) observed that the Amid classification did not consider the spaces created due to the methods of construction of the mesh. They noted that knit construction, for example, “creates small voids around the location at which filaments are joined”, which “provide microporous elements even in large pore meshes”. 545
472 Sixth, since the Amid classification was devised, there has been an increase in knowledge about pore geometry and the effect of mechanical loading on the pores. There is nothing to indicate, for example, that Professor Amid was cognisant of the fact that pore sizes change in vivo or, if he was, that he took it into account.
473 The research conducted by Barone et al demonstrated what happens to the pores under mechanical load. They cited with evident approval the work of Professors Klinge and Klosterhalfen, but not just their work. Of particular relevance in the present context is the following passage:
Notably, it has been shown that effective tissue in-growth with polypropylene mesh, characterized by the quality of the tissue which forms around mesh fibers, only occurs in mesh pores with a diameter of 1000μm or greater (Klinge and Klosterhalfen, 2011). Pore sizes less than 1000μm have greatly enhanced inflammatory and fibrotic responses (Weyhe et al., 2006; Bellon et al., 2002). While a pore size of 1000μm appears to be the threshold for polypropylene, the thickness of fibrous encapsulation is expected to vary depending on the polymer used as that (sic) the degree of fibrous connective tissue deposition is believed to be dependent on protein interaction with the fiber surface and related to hydrophobicity of the polymer (Klinge et al., 2002d). It is important to note that these findings on pore size were all determined using an abdominal wall model. Though the general foreign body response to mesh should consist of similar mechanisms in the vagina and adjacent supportive tissues, there are distinct differences in the biology of these sites, which likely impacts the host response to SUI, and POP meshes. Therefore, the critical pore diameter for urogynecological meshes to minimize scar plate formation may be distinct from that found in abdominal wall studies.
(Emphasis added)
474 Seventh, much earlier the following observations were made at the IUGA Grafts Roundtable and published online in an article written by Professor Deprest and others on 6 May 2006 (Davila, 2006): 546
Currently existing graft classification systems do not apply to the use of grafts in the pelvis. The Amid classification published in 1997 is frequently quoted as the accepted classification system for synthetic grafts (see Dwyer and Deprest contributions). As only Type l mesh is recommended for use in the pelvis, this classification does not, in general, apply to the use of synthetic grafts in the pelvis. There are currently multiple subtypes of Type l mesh, with markedly variable physical characteristics, including softness, weave, elasticity, and pore size, among others. There are, thus, great and clinically significant differences among Type l synthetic mesh materials. To improve clarity when referring to Type l mesh, other physical characteristics of the mesh should be described. Clear examples of these characteristics include directionality and distortion with the stretch, which can play a significant role in the outcomes of a suburethral sling procedure.
(Emphasis added)
475 Eighth, by 2008, if not earlier, Ethicon itself recognised the shortcomings of the Amid classification.
476 Terri Dunn of Ethicon Women’s Health & Urology, for example, wrote in an email dated 24 June 2008:
The AMID chart is truly out of date in relationship (sic) to what pelvic floor mesh needs to be in pore size.547
477 In October 2008 Dr Trezwik and Dr Meier, both Ethicon employees, submitted to Johnson & Johnson Medical GmbH an “invention disclosure” for a vaginal implant for pelvic floor reconstruction adapted to the biomechanical environment of the pelvic floor. In their description of the benefits of the invention. They wrote:
A reduced mesh pore size (< 1mm) is identified as a major cause of “bridging fibrosis” causing reduced tissue compliance in the area of the mesh implants. The temporary stress shielding of the mesh during the period of implantation circumvent to problem of collapsing mesh pores under load during implantation.548
478 I discuss “bridging fibrosis” below. “Stress shielding” has been well described by the Moalli group. It occurs when a stiffer material (here the mesh) bears the bulk of the load and buffers or shields surrounding tissues from these forces: Barone et al (2015).549 The less stiff material (the soft tissue), now shielded from the load it normally carries, undergoes “a maladaptive remodelling response characterized by degeneration and atrophy (loss of collagen, elastin, and skeletal/smooth muscle)”.550
479 A 2010 PowerPoint presentation by Dr Boris Batke, then Ethicon’s Assistant Director of Research and Development, entitled “Mesh Innovation for Pelvic Floor Repair”, classified a small pore size polypropylene mesh as one which was less than 1mm.551 Dr Batke contrasted mesh of this kind with partially absorbable lightweight meshes. He treated only the latter as macroporous.552 Incidentally, he stated that “[c]linical data have shown more chronic pain, testicular pain and foreign body sensation” with microporous meshes than with partially absorbable macroporous meshes. The note below this slide (slide 10) reads:
Today we have clinical evidence that the mesh construction, the amount of foreign material, the pore size and filament thickness do play a role in post operative pain, and plenty of studies on animal models, explanted meshes etc can explain this by looking at how the body’s immune system reacts to the mesh implant.
480 Three references were given. None was to the work of the Aachen Group. That is not to say that Dr Batke was not influenced by the work of the group. Elsewhere in his presentation he referred to the article by Klosterhalfen et al “The lightweight and large porous mesh concept for hernia repair”.553 But it suggests that his conclusions were informed by other research and independent thought.
481 Dr Batke also asserted that there was no place for the microporous (which he referred to as “[h]eavyweight”) mesh in modern pelvic organ prolapse repair — in other words, there was no place for mesh with pores of less than 1mm.554
482 On 27 June 2011, in a response to a request from a urogynaecologist for some images — “[e]ven the old slides that have the old AMID (sic) classification” — for a presentation he was giving, Brian Luscombe, Ethicon’s US Product Director, Female Pelvic Medicine & Reconstructive Surgery, observed:
As I am sure you know, the world you knew 7-10 years ago on AMID classification is no longer even relevant to some extent. What people think they know about mesh is changing. … In the next year or two it is going to change further with more discussion on topics like partially absorbable mesh, bridging fibrosis, and stress shielding.555
483 Although he had steadfastly refused to accept that it was out of date, when confronted with the Luscombe email in cross-examination, Professor Wright agreed that the Amid classification was no longer relevant “to some extent” “in terms of biocompatibility and biomaterials science”.556
484 Four months earlier, in February 2011, a confidential memorandum under the joint letterheads of Ethicon Women’s Health & Urology and JJM contained a graphic illustration of how pore sizes are affected by load. It also stated:
For mesh strips, even a mild loading of 1N [one newton] has a dramatic impact on the resulting pores size as far as soft meshes like Ultrapro are concerned. Under this loading the initial pore size of between 3–4 mm decreases to values down to 0,3mm.557
485 I interpolate that it was common ground that 1N is the amount of force necessary to keep an apple weighing about 100g from falling.558 One acquires a good idea of just how mild that load is when one sees from a letter Ethicon wrote to the FDA in February 2008 that the force required to pull an arm of a Prolift+M implant through a cannula is 0.73lbf (pounds of force), which equates to 3.24N, and that 12lbf (53.38N) is the figure given for the forces applied to Prolift during insertion.559
486 The February 2011 confidential memorandum went on to say that “[t]he phenomena (sic) of pore size variation as a function of the applied load is related to kinematical effects based on textile mesh design” and “is closely related to the angle geometry of the pore forming filaments”.560
487 The memorandum also contained the following candid admission:
The development of knowledge to understand the mechanics of pelvic floor disorders is imperative; yet, we are only just beginning to determine the necessary criteria on which to base design for pelvic floor implants.
488 The author of this document is not certain. Professor Klinge said that it was written by Jürgen Trzewik, an Ethicon senior scientist, and Christophe Vaihe, principal scientist. This may well have been an assumption, albeit a reasonable one. But no-one suggested otherwise. The document was unsigned but their names appeared on the first page under the heading “Approvals”. Although the document was unsigned, there is no reason why it should not be given weight. Certainly the respondents did not proffer one.
489 Similarly, the effect of applying a mechanical load to polypropylene mesh was graphically illustrated in a slide featuring in a PowerPoint presentation on the importance of mesh properties prepared by Dr Meier, then Ethicon’s Principal Scientist R&D:561
490 The text appearing under the images reads:
Figure 1 illustrate[s] the pore size dependency of Ultrapro for two load cases. Microscopic view on individual pores at zero tension state 0 N (left) and under uni-axle [(scil) Axial] load of 1 N (middle). The right image shows the macroscopic view [of] an Ultrapro strip elongated at a uniaxial (scil) load of 1N.
491 Similarly, in another PowerPoint presentation, Dr Holste, then Ethicon’s Senior Research Fellow, deviated from the Amin classifications. He referred to a mesh of less than 800μm (0.8mm) in diameter (that is, a mesh about 10 times larger than the 75+ μm in Amid’s Type 1 classification) as a small pore mesh, stating that fibrosis is more pronounced in meshes with pores of these dimensions “forming scar plates”.562
492 The unchallenged and uncontradicted evidence of Professor Klosterhalfen is that, after the publication of the Aachen Group’s findings on bridging fibrosis in pores less than 1mm, which followed numerous experiments with experimental polypropylene mesh modifications with different and defined pore sizes, “all companies modified their [polypropylene] mesh portfolio adding [polypropylene] meshes with pores greater than 1mm” (original emphasis).563
493 There was a dispute in this case about the size of the pores in the Ethicon devices
494 Even before implantation the pore sizes of the mesh used in each of the relevant Ethicon devices are not uniform. According to a table in the Gynemesh PS technical file, Prolene mesh (used in all the SUI tapes) has a maximum pore size of 0.37mm2 and a minimum of 0.24mm2. Its average porosity is said to be 53.1%. Prolene Soft mesh, the mesh used in Gynemesh PS and all the other POP devices, is said to have a maximum pore size of 2.38mm2 and a minimum pore size of 0.29mm2. Its average porosity is recorded as 65.6%.564 Pore sizes for Prolift+M (based on figures for UltraPro) range from 3–5mm.565
495 The figures given in the Gynemesh PS technical file or figures like it appear in other Ethicon internal documents as well, including documents prepared by members of its research and development team. For example, in his 2010 PowerPoint presentation Dr Batke said that the pore size of Gynemesh PS was 2.44mm2 and the pore size of UltraPro was 4mm.566 Presumably these were references to the largest pore sizes. Dr Batke did not specifically refer to the other Ethicon meshes.
496 The applicants relied on the figures in the Gynemesh PS technical file. The respondents, on the other hand, relied on measurements in a report by an organisation called LNE, dated 28 November 2008, whose figures were related by Dr Hinoul in his affidavit. LNE had been asked by Ethicon to evaluate the properties of TVT and Prolift. LNE said that it randomly defined 10 zones of fabric and in each zone measured the height and width of five pores randomly selected.567 It reported a mean width pore size of 1.06mm (with a standard deviation of 0.05mm) and a mean height pore size of 1.55mm for one SUI tape and, for one POP device, a mean width pore size of 2.34mm (with a standard deviation of 0.07mm) and a mean height of 1.39mm (and a standard deviation of 0.08mm).568
497 Professor Klosterhalfen challenged these figures. He said that TVT has a number of different size pores and these figures measure the length and width of the largest pores. Professor Klinge said, with a mean pore size of 1.06mm, a figure he also disputed, at least 50% of the pores would be smaller than 1mm.569
498 The respondents did not account for the differences between the LNE figures and those they had recorded themselves. In any case, the decision to provide mean figures obscures the full extent of the differences between sizes of the pores and masks the problem of the interstices and the small voids around the location at which filaments are joined.
499 That is apparent from an Excel spreadsheet dated 24 October 2013 prepared by Daniel Burkley, Ethicon’s principal scientist, which documented pore sizes for Prolene Soft mesh based on two image files taken on 26 March 2006 and 10 July 2010. Three sets of measurements were taken from each image and also averaged. Pore sizes ranged from 0.04mm² to 3.14mm². In total, 54 measurements were taken. As Professor Klosterhalfen pointed out, the vast majority of the pores were well under 1mm. Eighteen were below 0.28mm, the minimum figure given in the Gynemesh PS file. Dr Burkley indicated that some variation in pore size from lot to lot was expected.570
500 As I have already observed, regardless of the true range of pore sizes in the pristine product, it was common ground that pore sizes will reduce after implantation. Professor Klosterhalfen’s evidence was that the pores can collapse altogether. This was how he explained it.
501 Once mesh is implanted in the body, it moves and will deform with bodily movement. Mechanical stresses placed on the mesh affect pore size. In the pelvic floor there are numerous sources of mechanical stress. Even defaecation and urination, for example, place tension on the mesh.571 If the textile structure is not stable, mechanical load can cause pores to collapse. After implantation, mesh fibres become embodied in the granuloma and this process significantly reduces the pore size of the pristine mesh. Consequently, the size of the pores before implantation is not the size of the pores after implantation. Whether the mesh is sufficiently porous depends on the effective size of the pores. Effective porosity is what you get after implantation and growth of fibrotic tissue.572 Effective pores and high porosity limit fibrotic bridging, encapsulation and contraction of mesh. To be effective, it is necessary that the pore size be stable and maintained consistently under load or stress.573 The collapse of pores under mechanical load results in pore sizes of less than 1mm (1,000µm) with subsequent fibrotic bridging and mesh encapsulation.574
502 The problem of collapsing pores under load was accepted by Ethicon. It was one of the reasons behind Project Thunder, discussed below. It was identified by Drs Trezwik and Meier in their invention disclosure.575
503 Moalli et al (2008) noted that one of the primary problems of using TVT is that the mesh easily deforms when tensioning under the urethra. They conducted mechanical testing on six commonly used midurethral polypropylene slings, including two made from Prolene: TVT and TVT-O.576 Moalli et al developed a cyclical testing protocol to measure permanent elongation resulting from repetitive loading within a range of forces relevant for in vivo loading: cough, sneeze, straining, and moving from a sitting to a standing position. Cyclical loading induced permanent elongation in all six slings. After 10 cycles of between 0.5 and 5N (about 0.1 and 1.1lbs), the authors reported that the “Gynecare samples had permanently elongated by 17.5±4.2%, indicating that although very little force applied, there is irreversible deformation of the TVT™”. After 10 cycles between 0.5 and 15N (about 0.1 and 3.4lbs), Gynecare samples permanently elongated 42.3±3.2%. “Importantly”, the authors observed, “elongation after cyclical loading was not reversible, indicating that once a sling deforms, it does not revert to its initial form”. They noted that their results were similar to those obtained for the Gynecare in a previous study published in 2003.577
504 The following images of TVT taken from the article, clearly illustrate the collapse of pores under load. The image marked “a” shows the Ethicon sling before loading. The other images show the sling at different stages of loading. The authors commented that, in image “d”, “the sling has elongated more than 70% of its original length and has lost its structural integrity such that it resembles a string”.

505 Moalli et al (2008) identified their “most important finding” in the following paragraph:
The most important finding of the paper is that Gynecare TVT™ mesh has a unique tensile behavior which is characterized by an initial region of very low stiffness in which the mesh easily elongates in response to small changes in force. This is followed by a transition period (inflection point) and an area of high stiffness. As a result of this behavior, after cyclical loading at low loads (0.5 to 5 N or 0.1 to 1.1 lbs), Gynecare mesh permanently elongated by more than 10% of its initial length, confirming the easy permanent deformability of this mesh that is observed clinically during placement. The mesh with mechanical behavior most similar to Gynecare was that supplied by AMS [Amercian Medical Systems].
506 They indicated that the behavior of these slings in vivo and after incorporation into host tissue could be inferred from their study findings but that rigorous in-vivo studies were the next logical next step for determining the relationship between textile and tensile properties of polypropylene slings, tissue behavior, efficacy, patient morbidity, and patient satisfaction.
507 In his evidence in the present case, Professor Klosterhalfen confirmed that Prolene in general (by which he meant both Prolene and Prolene Soft) has no effective pores because, under mechanical load, its pores effectively disappear.578 The collapse of the pores, he added, is likely to occur during or shortly after implantation.579
508 Professor Klinge gave evidence to similar effect. He pointed to tests of Prolene and Prolene Soft under load that he had conducted in 2014 with Professor Thomas Mühl in the context of US litigation. Those tests showed that under 4.9N/cm of strain (which Professor Klinge described as “minimal force”) the textile porosity of Prolene decreased to 50% if the strain was applied perpendicular to the warp course and to 57% if the strain was applied in line with the warp fibres and that any large pores completely disappeared if the structure was stressed at right angles to the warp fibres. The figures were different for the three Prolift arms. Textile porosity of arms 1 and 2 at rest was 58 to 58.9% and decreased to 10% for arm 3. Effective porosity disappeared, however, at a strain of 4.9N/cm in arms 1 and 2 and at a lower strain of 2.5N/cm in arm 3.580
509 While the textile porosity of Prolene Soft was 63% in comparison to 51% for Prolene in the Klinge/Mühl study, the effective porosity of Prolene Soft was 26% and, in the arms, decreased rapidly under strain, dropping to 0% at a strain of 4.9N/cm.581
510 Those tests also showed that under minimal strain the Prolift arms curled (or roped) under minimal load (5.2538N/cm) and did not return to their original shape. Professor Klinge said that, as strips of mesh begin to curl, the pores reduce in size as the fibres “become situated too close together”, enhancing the chronic inflammatory response, and leading to bridging fibrosis.582
511 Professor Deprest expressed similar opinions. He wrote that after implantation, all meshes are “loaded by physiologic forces”, which may cause “tremendous geometrical changes” at the pore level. He added that, if the implant is deformed by these “loading forces” and does not return to its initial state, “the integration process into the body may be compromised as well”.583
512 Furthermore, as Professor Klosterhalfen observed, Professor Deprest’s opinion was supported by numerous studies, which he cited in his report. He went on to refer in particular to the conclusions of Barone et al in their article, “Textile properties of synthetic prolapse mesh in response to uniaxial loading”, published in the American Journal of Obstetrics & Gynecology in September 2016, which Professor Deprest also cited.
513 This was a report on a study conducted by Dr William Barone and his fellow authors from the University of Pittsburgh, Professor Pamela Moalli, and Dr Steven Abramowitch, who had participated in the study of the slings reported in Moalli et al (2008). This later study was of four synthetic meshes with distinct pore geometries to determine the effect of tensile loading and pore orientation on mesh porosity and pore dimensions. One of the meshes was Gynemesh PS. Barone et al (2016) wrote:
In this study, it was shown that the pore size of current prolapse meshes dramatically decreases in response to mechanical loading. These findings suggest that prolapse meshes, which are more likely to experience tensile forces in vivo relative to hernia repair meshes, have pores that are unfavorable for tissue integration after surgical tensioning and/or loading in urogynecologic surgeries. Such decreases in pore geometry support the hypothesis that regional increases in the concentration of mesh leads to an enhanced local foreign body response. Although pore deformation in transvaginal meshes requires further characterization, the findings presented here provide a mechanical understanding that can be used to recognize potential areas of concern for complex mesh geometries. Understanding mesh mechanics in response to surgical and in vivo loading conditions may provide improved design criteria for mesh and a refinement of surgical techniques, ultimately leading to better patient outcomes.584
514 They concluded that “all current prolapse meshes will likely experience a complete collapse of pores at some load”.
515 A major finding of the study was that the porosity of nearly all tested products approached 0% in response to just 10N of applied force, which is within the expected physiologic range and consistent with the forces they said they would have expected meshes to experience before tissue incorporation during implantation and in vivo.
516 Professor Wright agreed that the pore size of a mesh before implantation might not be the pore size of the mesh after implantation. He assigned two possible causes for the reduction in pore size. One was the possibility of stretching during implantation. The other was that shortly after implantation “chronic inflammation will surround the mesh fibers”, causing the mesh to shrink somewhat. Despite this, however, Professor Wright said that the pore sizes in vivo were large enough in all Ethicon meshes used in gynaecological surgery to allow good incorporation of the mesh into the host tissue. He said that, once fibrous ingrowth occurs, he could not envisage a potential mechanism by which the various loads and stresses placed on the mesh would affect its pore size without also tearing the fibrous connective tissue around the mesh fibres.585 But as Professor Klinge pointed out, Professor Wright did not consider the physiological forces placed on the mesh after implantation, nor did he consider the remodelling of soft tissue as a mechanism for pore deformation that also occurs after implantation.586Professor Klosterhalfen responded to these opinions of Professor Wright at [120]–[123] of his second affidavit. He wrote:
120 Dr. Wright has not understood the concept of “effective pores”. The concept of effective porosity includes that no fibrotic tissue is formed by the FBR and that the granuloma does not fill out the pores, ideally the whole mesh indicates only a perifilamental fibrosis. If the fibrotic reaction leaves no space in the pore and the mesh is encapsulated by a fibrotic layer on the top, the reaction is called bridging fibrosis …
121 Prolene in general has no effective pores, the minimum size required for effective pores in PP meshes is greater than 1mm. PS [Prolene Soft] has effective pores (26%), however under mechanical load the number drops to 0% ... Therefore, Dr. Wright’s statement that “the size of the pore in vivo remains large enough with all of the Ethicon meshes used in gynaecological surgery to allow good incorporation of the mesh into the host tissue” is incorrect.
122 The collapse of the pores of PS is likely to occur during or shortly after the implantation due to the mechanical load. Later, after mesh integration, mechanical loads lead to an increased cell turnover in the mesh interface to compensate for mechanical stresses. Cell turnover due to mechanical loads or stress again is the basis of tissue remodelling; for example, the correction of tooth position by braces. The same is true for meshes under mechanical load. The phenomenon of a chronic inflammatory reaction with consecutive and persisting cell turnover and tissue remodelling explains changes of the mesh configuration or shape after implantation and explains late complications like erosion or chronic pain.
123 Contrary to Dr. Wright’s opinion, studies indicate that tissue integration after mesh implantation has no “endpoint”, as tissue remodelling can be observed even years after implantation.587
517 Once implanted, Professor Klosterhalfen said, to avoid fibrotic bridging and permit appropriate ingrowth of fatty tissue or loose connective tissue (rather than dense scar tissue) pore sizes need to be greater than 1mm between two polypropylene filaments.588 Professor Klinge’s evidence was to the same effect.589
518 Since Professor Amid did not consider the impact of mechanical load on mesh after implantation or the risk of excessive scar formation, let alone what pore sizes might be necessary in mesh used in the repair of the female pelvic floor, I am persuaded that his classification is not useful for present purposes. I am also persuaded that under mechanical load the pores of polypropylene mesh, including Prolene and Prolene Soft, will reduce in size and can, indeed, collapse. It is unnecessary to decide, as the respondents invited me to do, whether a pore size of more than 1mm is required in order to minimise complications. Having said that, given that I accept the evidence of Professors Klosterhalfen and Klinge. It follows that I accept their evidence about the problems of meshes with smaller pore sizes.
519 Professor Klinge’s evidence was that the failure to account for the phenomenon of pore size variations as a function of the applied load was an unacceptable design failure of Prolift.590 He argued that a knitted surgical mesh device that is permanently implanted in human tissue must be designed in such a way that its pores do not collapse and deform when subjected to the expected forces both during implantation and after implantation. Yet, the tests he and Professor Mühl performed on TVT, the results of which were replicated by Professor Moalli’s group, showed that under minimal strain its pores deform and collapse. “Permanent deformation and pore collapse”, he continued, “leads to fibrotic bridging, scar plate formation, excessive scarring through and around the mesh and a host of tissue complications that can lead to chronic pain, recurrence, erosions, dyspareunia and need for reoperation, to name a few, making it unsafe for its intended purpose of being permanently implanted in a woman’s pelvic tissue”.591
520 Professor Klosterhalfen said that, if the implant is integrated into the body it is covered by fibrotic (scar) tissue, which may encapsulate the mesh in part or in whole. Whether it is completely encapsulated depends on the structure of the mesh. Foils and microporous meshes, he said, are always encapsulated in that the fibrotic reaction forms a complete capsule around the mesh. Professor Klosterhalfen said that this phenomenon is called “bridging fibrosis”.592 Yet, Professor Klinge made it clear that there is a difference between encapsulation and fibrotic bridging. He clarified the position at [79] of his affidavit:
The encapsulation of a mesh implant is not the same as fibrotic bridging. Pure encapsulation without ingrowth into the mesh refers to the formation of a capsule that surrounds the entire mesh. In the case of encapsulation, the entire mesh can usually be surgically removed from the capsule without any problems as the mesh is not incorporated into the tissue. This type of encapsulation occurs with some meshes, for example, GoreTex mesh. Conversely, “bridging” means that the pores are filled by scar tissue, and that the scar bridges the distance between two filaments, thereby surrounding and encapsulating the entire mesh. This type of encapsulation is different from pure encapsulation as the formation of scar within the mesh makes it very difficult to extract the mesh from the tissue.593
521 Later in his affidavit at [113], Professor Klinge said that fibrotic bridging occurs when the fibrotic capsule of the foreign body granulomas side by side, crosses the pore, filling out the entire distance between fibres by scar tissue forming a rigid “scar/mesh compound” or “scar plate”, enclosing the mesh and leaving no room for further tissue ingrowth. He said that the formation of a scar plate leads to a number of complications including loss of elasticity and pain associated with the rigidity, shrinkage or contraction of the mesh, mesh erosion, nerve entrapment, bacterial encasement, chronic pain and dyspareunia (in the case of pelvic mesh).
522 These opinions were based on the numerous studies conducted by members of the Aachen Group. As the applicants submitted, they were also supported by the findings of Dr William Cobb and others published in 2005.594
523 Professor Moalli’s group have also reported on this phenomenon. They wrote that “[f]ibrous encapsulation and its potential contraction by resident myofibroblasts may induce pain after mesh implantation”: Barone et al (2016).595
524 In Mühl et al (2008) stated that bridging will occur with polypropylene filaments of less than 600–800 microns in diameter and will form a dense scar plate around the entire mesh.596
525 At least two of the respondents’ witnesses were familiar with the concept of bridging fibrosis.
526 Professor Deprest described bridging fibrosis in his report. He said he had observed that smaller pore materials with closer filaments experience bridging fibrosis “where the fibrous tissue around on (sic) filament comes in contact with the fibrous tissue around the next one”. He illustrated his point with copies of histologic slides from his own research.597
527 Although he did not discuss the matter in his evidence in chief, in cross-examination Professor Santerre agreed that a permanent mesh implant is surrounded with a fibrotic capsule and (for hernia meshes at least) if the fibres are too close, the fibrotic response to neighbouring fibres overlaps or bridges, resulting in bridging fibrosis or encapsulation of the mesh.598 He also agreed that bridging fibrosis leads to the formation of a continuous scar plate and prevents tissue from growing into the mesh structure.599
528 On the other hand, Professor Wright ridiculed the concept of bridging fibrosis. In cross-examination he referred to it as a “silly term”. He insinuated that the concept was recognised by only a small minority of people.600 He claimed that no pathologist in the United States would diagnose bridging fibrosis, although he never identified a reason or basis for that claim.601 Nevertheless, he said he knew of no article which criticised the term.602 Moreover, in cross-examination he appeared to accept the existence of the concept. He agreed that chronic inflammation adjacent to the mesh filaments can lead to increased fibrosis adjacent to the filaments and that, depending on the size of the pores, the fibrosis can fill the whole of the pores or the spaces between the individual filaments. He agreed that it can surround the mesh and can change its surface area. He also agreed that some meshes can become encapsulated.603
529 Despite Professor Wright’s contempt for the term, the respondents did not deny that bridging fibrosis can occur.604 Rather, for the reasons they gave in support of the continuing relevance of the Amid classification, the respondents argued that a minimum pore size of 75µm was enough to avoid it.
530 I have already discussed the problems with the respondents’ argument. It is sufficient to record in this context that Professor Amid did not deal with the question of the sufficiency of the pore size to avoid bridging fibrosis. Indeed, he did not address bridging fibrosis. Nor did many of the articles upon which the respondents relied in defence of the 75µm threshold. To the extent that they commented at all on the rationale, they were concerned with the pore sizes necessary to prevent the growth of bacteria and the risk of infection. While the concepts are related, they are not the same.
531 I also reject any suggestion that bridging fibrosis has not been documented to occur with the Ethicon devices. Professor Klinge said that clinical studies of explanted meshes as well as all animal experiments showed that Prolene mesh bridges, regardless of the site of implantation, and that Prolene Soft (Gynemesh PS) and UltraPro (used in Prolift+M) show bridging when explanted from areas of tension as in the pelvic floor.605 In a report of a study of local tissue reactions to a number of non-absorbable meshes used in abdominal wall hernia repairs (including Marlex, Prolene and Vypro) published in 1999, to which Ethicon provided financial support,606 Professor Klinge and his fellow researchers reported that both the polyester and polypropylene meshes showed pronounced chronic inflammation and “a strong interlinking formation of connective tissue [scar tissue] through the mesh-pores”. The authors went on to say that the embedded connective tissue forms a rigid scar plate, which is responsible for substantial mesh shrinkage,607 a subject to which I will come shortly. They wrote:
All the polypropylene meshes [Marlex, Prolene, and Atrium] (implantation time 3-24 months) had similar histological patterns after explantation.
Commonly, there was a predominant foreign body reaction with typical foreign body granulomas including epithelioid cells and giant cells. However, and contrary to ePTFE, polyester, and the reduced polypropylene mesh there was persistent acute inflammation with varying amounts of CD15-positive PMN and focal fibrinoid necrosis in most cases. The inflammatory process was accompanied by pronounced perifilamentous fibrosis with an extensive amount of deposited collagen fibres… Adjacent to the mesh the fibres were mainly orientated parallel to the polypropylene threads. In the periphery, connective tissue with numerous collagen fibres formed a thick capsule in which the whole mesh was integrated. These mesh modifications were characterised by complete penetration of connective tissue into the pores.
532 Further, in the 2007 Ethicon swine study all three meshes in the subcutaneous model (Prolene, Prolene Soft, and UltraPro) were reported as exhibiting “thicker connective tissue that ‘bridged’ between mesh nodes”. The report also noted that “[c]onnective tissue bridging between nodes has often been observed in meshes experiencing contraction”.608
533 I find that bridging fibrosis has been documented to occur with Prolene, Prolene Soft, and UltraPro., which means that it can occur following implantation of all the Ethicon devices.
534 As to whether bridging fibrosis is of clinical significance, the respondents’ position is inconsistent with the position taken by its own scientists. For example, in a 2001 Ethicon report of a rat study of the tissue reaction over 21 days to various polypropylene meshes, including Prolene, Prolene Soft, and various versions of Vypro, the authors (Dr Richard Hutchinson, Principal Scientist, Corporate Product Characterization and Dr Thomas Barbolt, a pathologist and Senior Research Fellow, Corporate Product Characterization) wrote that:
Integration of the mesh by surrounding connective tissue plays a major role in the ability of the mesh to confer supporting strength to the clinical repair, i.e., the weak link is the interface between the device and living tissue. Thus, it is critical that the mesh allows for this tissue ingrowth and of sufficient magnitude to provide this important function. However, it has been recognized that excessive formation of scar tissue (fibrosis), and subsequent contraction as it matures, can result in discomfort for the patient.609
535 In a white paper on early clinical experience with Gynemesh PS produced in May 2004 (said to reflect the clinical experience of a number of surgeons, including Dr Lucente and Dr Kirkemo),610 Ethicon stated that the formation of rigid scar plates around the mesh could cause deformation of the repair site, bunching of the mesh, and decrease mobility of the repaired tissue.611
536 In a PowerPoint presentation prepared in 2007 by Kerstin Spychaj, a senior scientist in Ethicon’s Research and Development team,612 which reviewed both the literature and internal studies, small porous meshes (defined as less than 1mm) were said to lead to fibrotic bridging which, in turn, resulted in increased shrinkage.613
537 I accept this evidence. I find that bridging fibrosis is, indeed, of clinical significance.
538 There is a wealth of evidence that contraction, often called shrinkage and “retraction” by the French, is a complication of the use of polypropylene meshes. Almost invariably this phenomenon is referred to in the literature as either “mesh contraction” or “mesh shrinkage”. As both Professor Klinge for the applicants and Professor Deprest for the respondents explained, however, the mesh does not contract or shrink as a pullover might in the washing machine. What happens is that there is a reduction in the surface area originally covered by the mesh brought about by the retraction of the fibrotic tissues around the mesh. Contraction of scar tissue is a physiological reaction of maturing scar characterised by a constant water loss.614 The greater the area of scarring, the greater the degree of contraction.615
539 Professor Klinge said that, as a hernia surgeon he had removed “a lot of contracted [polypropylene] meshes from patients”, which exhibited “a huge amount of scar tissue”. He has published over 30 peer-reviewed publications on the subject of mesh contraction including the contraction of polypropylene meshes.616
540 The applicants’ case is that contraction leads to folding, stiffening, and loss of tissue function,617 extrusion, erosion, and infection, and can cause other complications such as chronic pain, dyspareunia, and a recurrence of the disorder for which the device was implanted.618 Professor Chughtai said that mesh contraction can lead to vaginal pain, vaginal shortening and/or tightening which, in turn, can cause severe side-effects, most commonly pelvic pain and dyspareunia.619 Professor Klinge explained that:
Contraction not only leads to poor coverage leading to recurrence, but will also increase the amount of [foreign body reaction] due to pore collapse. This phenomenon then leads to additional complications, including pain, dyspareunia, nerve entrapment, increased inflammation, urinary and faecal incontinence, urinary retention, blood vessel injury, chronic pelvic pain, erosions, an inability to remove the device and the need for painful and, at times, dangerous revision surgery.
541 Dr Donald Ostergard wrote in 2010 that:
Because the vagina is a tubular structure, a decreased calibre and shortening are the inevitable results [of mesh shrinkage]. Dyspareunia can be explained by such mesh shrinkage, as well as by tension on mesh arms with neuroma formation. Because the mesh is anchored in tissue, its shrinkage will put more and more tension on the anchoring tissue, with resulting pain. No mesh seems to be immune from this process … Unfortunately, this fibrous tissue will continue to contract regardless of what the surgeon trying to remove the mesh is able to do. The more surgery, the more scar tissue that will form.620
542 I referred above to the study findings of the Aachen Group on mesh contraction or shrinkage. Fatton et al (2007) also noted shrinkage of mesh in 18 out of 110 patients (17%) three months after implantation of Prolift in a retrospective multicentric study.621
543 In his affidavit Dr Hinoul asserted that the applicants’ experts were incorrect to say that mesh contraction was a cause of dyspareunia and expressed the view that mesh does not contract but that during wound healing tissues may contract, regardless of whether mesh is present. He added that, since studies indicate no difference in total vaginal length or caliber with Gynemesh PS and Prolift in comparison to native tissue repair, if contraction does occur, it is no different than that which is seen with native tissue.622
544 These statements were not only at odds with the overwhelming weight of the evidence, they were also disingenuous. I shall deal first with the issue of whether tissue contraction has an effect on the size of the mesh.
545 In 1997 Professor Amid reported findings of mesh shrinkage of 20% in hernia explants 10 months after implantation.623
546 In 1998 Professor Klinge and his colleagues reported on the shrinkage of polypropylene mesh in vivo in an experimental animal study comparing Marlex to Vypro. They found that polypropylene meshes (Marlex) shrank to about 30 to 50% of their original size within two to six months of implantation.624
547 In a PowerPoint presentation on Gynecare’s pelvic floor strategy produced in November 2004, Zenobia Walji noted:
We have experienced 20–40% of contraction using mesh. This requires a larger piece to cover the prolapse and using a “tension-free approach” – Dr. Mauro Cervigni (Italy), TVM Group (France)625
(Original emphasis.)
548 The TVM Group reported 80 cases of mesh shrinkage in its study of 684 patients (a rate of 11.7%) at seven centres in France during the period November 2002 to December 2004. Eighteen of 110 Prolift patients (16.9%) studied over a shorter period (March to October 2005) were found to have mesh shrinkage. 626
549 A prospective study of 125 Prolift patients operated on at the University Hospital in Clermont-Ferrand in France between March 2005 and August 2006 revealed a rate of “[p]ainful mesh shrinkage” of 19.6% at 18 months follow-up.627
550 In 2005 Cobb et al reported that all meshes, regardless of their composition undergo a 20 to 50 percent reduction in their initial size.628 The following year saw the publication of their findings from an animal study on shrinkage in three different meshes which showed no significant difference in the degree of contraction in the three meshes tested: UltraPro, Prolene Soft and Marlex. UltraPro shrank by 29%, Prolene Soft by 33% and Marlex by 28%.629
551 A 2007 study of mesh contraction and tissue integration in swine models over a 13 week period conducted by Ethicon Inc.,630 which Dr Hinoul neglected to mention, showed that Prolene Soft (the mesh used in Gynemesh PS, Prolift, and Prosima) contracted by 27 to 35% and UltraPro (the mesh used in Prolift+M) by about 14%. Furthermore, gross observations of the mesh showed folding, rippling, and distortion of the meshes, greater at the subcutaneous site than in the pre-peritoneum site, with Prolene Soft and UltraPro exhibiting the greater incidence of folding, rippling, and distortion in comparison with Prolene.631 Importantly, the Ethicon swine study also revealed that all meshes were stiffer than native tissue and that Prolene mesh was stiffer than the other meshes.632
552 In a paper on the subject of “mesh shrinkage” prepared for the 2008 IUGA annual meeting in Taipei, Professor Bernard Jacquetin and Dr Luka Velemir noted that mesh retraction occurs during the scarring and remodelling process and is related to the extent of the tissue inflammation around the mesh after implantation which induces the wound contraction.633
553 On 18 January 2011 Professor Klosterhalfen told consultants engaged by Ethicon and Michel Richter of Ethicon at a three-hour meeting that:634
At the high level, there are two classes of “shrinkage” observed with mesh implants (Note: the term “shrinkage” is a misnomer. Tissue reaction over time encapsulates the mesh with connective tissue and effectively ‘crushes’ the mesh into a ball (like crushing a sheet of paper); the mesh does not truly shrink):
– The first is in the immediate short term following implant; the implant is observed to lift and may ‘roll up’ from its position. This occurs as a result of poor positioning, placement and/or suturing of the implant by the clinician.
– The second class of shrinkage is the formation of scar tissue; observed in the longer term (months) following implantation. This scar tissue can reduce and compact, causing the mesh to crumple up.635
554 The minutes of that meeting also record a note “that all implanted meshes will show surface area reduction with time and this was up to 50% with older, small pore meshes”.
555 Professor Klinge deposed that Professor Klosterhalfen merely restated what was widely known in the scientific community and the manufacturing industry in the 1990s, namely, that older, heavyweight small pore meshes like Prolene used in Ethicon’s SUI devices increase the risk of shrinkage or contraction of the mesh by up to 50% of its area.636
556 Professor Wright’s evidence was that it was impossible to observe contraction or folding.637 I reject this evidence. Professor Klinge’s experience, which is far greater than Professor Wright’s, proves otherwise. Professor Klinge deposed that shrinkage is apparent when you measure the distance between two filaments of a pristine mesh and compare them to the distance between filaments after implantation, shrinkage. He said that folding (where there are two or more doubled layers of mesh) is also clearly seen in explants and is not affected by the orientation in vivo.638 Furthermore, in 2008 Hinoul et al reported that shrinkage of the mesh had been observed in a patient six months after surgery.639 In cross-examination, Dr Hinoul said that when the mesh-tissue construct shrinks, the mesh folds.640 He said that when he carries his procedures he always tries to avoid getting folds in the mesh by flattening it out.641
557 In cross-examination of the applicants’ experts, it was not suggested that shrinkage did not occur. Rather, it was put to Professor Klinge that more intense shrinkage with heavyweight small poor mesh was not established by 2005. Without hesitation, he repudiated the notion.642
558 Professor Wright admitted in cross-examination that the meshes used in all the Ethicon devices could “show shrinkage”, that shrinkage can reduce the size of the pores, and that the number of pores within a mesh that are greater than 1mm might decrease as a result of shrinkage and fill with “fibrous connective [scar] tissue”.643 As a matter of logic, it seems to me, this would increase the chances of infection and pain.
559 Professor Deprest said that it was initially thought that contraction was an active process resulting from contracting fibrotic scar. I interpolate that in 2007 Klinge et al had written that “[w]ound contraction of the fibrous integration leads to shrinkage of the implant area …”.644 More recently, however, Professor Deprest said that it has been shown to occur very early after implantation. He explained that what is thought to be happening is that, due to load on the implant when a woman awakes or starts to move around, the implant undergoes deformation and does not return to its original state. His opinion was that both processes are likely to be in play.645 That opinion is supported by the work of Professor Moalli’s group which identified two potential mechanisms: mechanical loading and fibroblast-induced contraction as part of the foreign body response. Barone et al wrote that both these mechanisms are likely to be related to the geometry of the mesh, the loading environment in which the mesh is placed, and the characteristics of the mesh such as pore size.646
560 Professor Klosterhalfen said that mesh shrinkage is closely related to “bridging” or “bridging fibrosis”. He explained that in all mesh implants with a granuloma size around each mesh fibre of more than half the pore size of the mesh the granuloma of one fibre starts to blend with the granulomas formed on neighbouring fibres increasing the area of granuloma. He continued:
As a result, the outer fibrotic layer of each granuloma around each polypropylene fibre form an outer fibrotic capsule, forming a fibrotic tissue layer in parallel to the surface to the mesh. Accordingly, the outer fibrotic tissue layer covers not only single fibres of the mesh, but larger areas of whole mesh. Finally, if this fibrotic capsule starts to retract or shrink, the mesh has to follow and adapt to the reduced surface area. As a result the mesh wrinkles and buckles, forming folds. Ordinarily, bridging occurs in small, porous meshes.647
561 Dr Hinoul’s statement that, if contraction occurs, it is no different for native tissue, makes no sense. As Professor Klinge explained:
In general contraction is an active process, the cause of which may be the changed configuration of the collagen molecules by dehydration. While the wound area gets smaller by growing cells and regeneration from the border, an area may be “reduced” but will not actively shrink. Correspondingly, tissue may grow over a mesh if it is visible in the subcutaneous space. In contrast, a wound made of collagen will develop shrinkage, as collagen contracts, and therefore it will push the mesh together with folds, and this happens in all parts of the body. A reduction in the area is a positive sign of healing if it is done by regeneration, whereas scar contraction often causes functional problems by increased stiffness, reduced stretchability, forming edges with local accumulation of material with its subsequent foreign body inflammation …648
562 Professor Klinge said that the greater the scarring the greater the degree of contraction. Furthermore, a mature scar without a foreign body usually does not show chronic inflammation.649
563 Notwithstanding the evidence given in chief by Dr Hinoul that suggested otherwise, as I have already indicated there was ultimately no issue that contraction occurs with the mesh used in all the Ethicon devices. The only dispute concerned its clinical significance. Despite conceding that there was a potential link between pain (including dyspareunia) and mesh contraction,650 Dr Hinoul maintained that mesh contraction had no clinical significance651 and the respondents argued that the applicants have not been able to establish that mesh deforms in the human body, with clinically significant effects.652
564 I reject the respondents’ argument. The evidence establishes that contraction does have clinical significance.
565 The respondents relied on an article about a study using a rabbit model published in 2009 which tested four types of polypropylene prostheses, all of which contracted.653 The severity of the shrinkage was unpredictable for any of them. For this reason, the authors wrote, “this could be a problem when prostheses are inserted into the vagina; [i]t is possible that the retraction produced pain and dyspareunia”. The respondents submitted that the authors’ failure to express a concluded view was “consistent with the uncertain clinical significance of contraction”.654
566 Since this was a study of hernia mesh and it involved a rabbit, the inconclusive nature of the authors’ remarks is neither here nor there. For a start, pain and dyspareunia are subjective. It is difficult to know how the rabbit would communicate her feelings to the researchers. Besides, none of the meshes were made by Ethicon.
567 The respondents also relied to a study of Prolift mesh by Svabik et al (2010)655 in which the authors found that, despite shrinkage of approximately 15%, the data did not allow for “any conclusions as to whether intraoperative folding or postoperative mesh shrinkage is of clinical significance”.656
568 This was a small study involving only 36 patients, comparing pre-implantation of the mesh with ultrasound appearances four days and three to five months after surgery. It was not designed to determine whether intraoperative folding or postoperative mesh shrinkage was of clinical significance. As the authors themselves stated, it is “only plausible to use ultrasound imaging for follow-up after mesh implantation to gain information about placement and extent of the mesh and, ultimately, to correlate such findings with clinical success and failure”.
569 The respondents also relied on evidence from Associate Professor Rosamilia that she did not see any evidence that contraction is a clinical problem for slings for incontinence procedures657 and Professor Wright’s evidence that he had not seen any pathological consequence of contracted meshes.658
570 This evidence was at odds, not only with the applicants’ evidence, but also with evidence adduced given by other witnesses called by the respondents.
571 Professor Roovers confirmed in cross-examination that mesh contraction can cause pain and dyspareunia.659
572 Professor Collinet, another of the respondents’ urogynaecological experts, agreed in cross-examination that mesh contraction can cause dyspareunia.660
573 Associate Professor Lam accepted that mesh contraction may lead to “painful scars”.661
574 Professor Deprest’s evidence on this subject was also instructive, although the respondents neglected to refer to it. It also supported the applicants’ case.
575 In his first report, Professor Deprest deposed that “[a] physically deformed implant may cause pain or other discomfort, both at rest as well as on effort or during intercourse”, even without exposure, and that the pain or other discomfort can be felt along the entire trajectory of the implant.662
576 Moreover, the respondents’ own records show that they accepted that contraction was clinically significant.
577 In the minutes of a meeting held on 4 April 2007 Dr Meier, from Ethicon R&D noted that the main customer unmet needs included “[l]ess mesh shrinkage and tissue contraction (to reduce vaginal stiffness and pain)”.663 There is no suggestion that anyone at the meeting questioned the connection between shrinkage and tissue contraction on the one hand and vaginal stiffness and pain on the other.
578 A document produced on 6 September 2007 by Ethicon R&D on the clinical strategy for Prolift+M described mesh shrinkage as a more serious complication than mesh exposure:
Mesh retraction (“shrinkage”) is less common but it is considered more serious. It can cause vaginal anatomic distortion, which may eventually have a negative impact on sexual function. Its treatment is difficult. Additionally, the scar plate that forms with in-growth of tissue into the mesh can cause stiffness of the vagina that further impacts sexual function in a negative manner.664
579 Indeed, the stated rationale for developing Prolift+M was to minimise these complications.665
580 In an article published in 2011, of which Dr Hinoul was the second-named author, describing the rationale for the development of Prolift+M, the following statements appear:
Among the most prevalent complications are mesh exposure and shrinkage of tissue around the mesh. These may result in pelvic pain and dyspareunia.
…
One of the key rationales for adopting a new, lighter-weight mesh with improved directional elastic properties was to minimize tissue shrinkage, which may lead to dyspareunia.666
581 These statements that shrinkage or contraction may result in pelvic pain and dyspareunia were not intended to signify that the connection was theoretical, only that shrinkage or contraction did not invariably have these consequences.
582 The respondents also argued that the first published study to report evidence of a correlation between contraction and complications was one by Rogowski et al published in 2013 (discussed below) and later studies were inconclusive.667
583 I reject this argument, too. There were studies reporting a correlation well before 2013 and later studies were not inconclusive.
584 In as early as 1981, mesh contraction was cited as one of two factors responsible for mesh extrusion in a paper by Randle Voyles et al (“Emergency Abdominal Wall Reconstruction with Polypropylene Mesh: Short-term Benefits Versus Long-term Complications”).668 That paper reported on surgery using Marlex.
585 An article by Clarke et al in the Journal of Surgical Research published in 1996, and referred to in Ethicon’s technical file on Prolene, reported on a study of 12 dogs into whose abdominal walls both Prolene mesh and xenogeneic (porcine) small intestine submucosa (SIS) was implanted. The Prolene mesh, in contrast to the control, was found to be wrinkled in all dogs. The authors thought that the wrinkling of polypropylene mesh after implantation could be caused by contraction of the original defect during wound healing and/or muscle relaxation from anaesthesia. They also considered that the wrinkling could contribute to complications such as seroma visceral erosion, skin graft erosion, and infection.669
586 In October 2010 an article on vaginal mesh contraction by two Australian surgeons, Drs Benjamin Feiner and Christopher Maher, was published in the journal Obstetrics and Gynecology.670 The article referred to “a number of patients who developed substantial morbidity related to mesh contraction”.
587 Feiner and Maher reported on a case series of 17 patients who had been implanted with transvaginal mesh: six with Prolift Total; four with Prolift Anterior; four with Perigee, a device made by American Medical Systems Inc. (AMS); the remaining three with Apogee-Perigee (both AMS products, analogues of Prolift Anterior and Posterior).671 The focus of the study was “the clinical expression of mesh contraction”. All 17 of the patients had vaginal mesh contraction and severe vaginal pain, aggravated by movement; all of those in the cohort who were sexually active (14) experienced severe dyspareunia. The authors reported that:
On a thorough vaginal examination localized areas of prominent tense mesh were noticed under the vaginal epithelium in all the patients. Palpation of the contracted mesh reproduced the pain these women experienced with movement and sexual intercourse. After primary surgical intervention to release the tension caused by the contracted mesh, 88% of patients had resolution or substantial reduction of the vaginal pain. All women had resolution of the pain if including the three who underwent further excision of the entire accessible mesh.
588 They concluded that vaginal mesh contraction was a serious complication after pelvic organ prolapse using polypropylene mesh, characterised by severe vaginal pain and dyspareunia and on vaginal examination focal tenderness over contracted portions of the mesh. They defined vaginal mesh contraction as an adverse outcome from prolapse repair with armed polypropylene mesh in women who experience vaginal pain with movement and dyspareunia and on examination have localised areas of prominent tense and tender mesh under the vaginal epithelium.
589 In cross-examination, Associate Professor Lam said that he had made similar findings in patients with painful mesh contraction and agreed with the authors’ definition of vaginal mesh contraction.672
590 Withagen et al wrote in 2011 that both “[m]esh exposure and shrinkage of fibrous tissues around the mesh may result in pelvic pain and dyspareunia”.673
591 In his 2012 review article, Dr Daniel Elliott of the Department of Urology, Female Urology and Voiding Dysfunction at the prestigious Mayo Clinic, wrote that “[w]hen vaginal mesh contracts, it causes vaginal fibrosis, infection, chronic vaginal pain, chronic pelvic pain, vaginal shortening, vaginal narrowing, vaginal extrusion, adjacent organ erosion, and dyspareunia”.674
592 In its 2015 opinion on the safety of urogynaecological meshes, SCENIHR noted that “excessive fibrosis may lead to mesh contraction resulting in increased pull on adjacent tissues leading to complications such as voiding dysfunction, pain and painful intercourse”.675
593 The respondents’ submissions were also inconsistent with their own documents, the opinions of the TVM Group, and the position of the FDA.
594 The minutes of a meeting of a scientific advisory panel on pelvic floor repair held in Chicago on 22 June 2001, apparently constituted by Ethicon and attended by a number of Ethicon employees, including Dr Martin Weisberg, Dr Brigitte Hellhammer, and Zeonbia Walji, record that contraction is a contributing factor to erosion and it is necessary to avoid contraction of the tissue in order to avoid erosion.676
595 The “main concern” of Professor Jacquetin and Dr Cosson, two members of the TVM Group, in 2004, according to an email sent on 10 May 2004 by Ophélie Berthier, Gynecare Marketing Manager France, to Zenobia Walji of Ethicon US, was “the shrinkage of the mesh which may lead to pain [and] dyspareunia”.677
596 In their November 2004 article, the TVM Group described mesh contraction and its after-effects as “much more worrying” than erosion and referred to dyspareunia as “the symptomatic manifestation” of contraction.678
597 In their 2008 paper on mesh shrinkage, Professor Jacquetin and Dr Velemir wrote that over time it appeared to the TVM Group that “mesh retraction or shrinkage (reduction of the mesh area and loss of compliance after tissue incorporation) was probably the most contributing factor to recurrences, postoperative pain and dyspareunia” (original emphasis).679Jacquetin and Velemir found shrinkage of between 15 and 25% of patients operated on between March 2005 and August 2006 and said that the retraction (contraction) was associated with mesh thickening on ultrasound.680 They provided advice on how to manage the complications. They emphasised that:
When a mesh procedure seems indicated, it is important to remember that severe mesh retraction may result in severe complications including dyspareunia, pain and recurrence; unfortunately, risk factors for these cannot always be identified.
(Original emphasis.)
They said that better understanding, assessment and prevention of mesh retraction at the time of augmented reconstructive pelvic surgery remained their “principal challenge” for the years to come.
598 This document was apparently discovered by Ethicon and cited by Professor Klinge in his affidavit, but none of the respondents witnesses referred to it.
599 Contrary to the respondents’ submission, largely based on Professor Wright’s evidence, that contraction occurs (only) within the first few weeks of implantation,681 the TVM Group described it as a “frequent late complication”.682
600 On 13 April 2005 Gene Kammerer sent an email to his colleague Dr Barbolt at Ethicon US, copied to numerous others in Ethicon’s R&D department, on the subject of “ULTRAPRO vs PROLENE Soft Mesh” in which he said (without alteration):
Regarding which attributes to investigate to show a difference between materials, I have this input. The issue which I am trying to investigate/solve is one of scar contracture around the mesh. In pelvic floor repair even with the PSM [presumably Prolene Soft Mesh], we have seen some scar contracture which translates into procedural complications. I don’t want to state % here because the situation which produces the complication is in itself complicated and specific to each patient. Also, most of the data comes from VOC [voice of the customer, that is, feedback from surgeons] and not our documented studies. However, it is important to know that the surgeons who are our consultants on the ProLift product are asking for a mesh which is better than PSM in this area.
The complications which are identified in the market are 1) recurrence of the prolapse 2) pain 3) stiffness 4) erosion and 5) discomfort during sex. The surgeons attribute these conditions to scar contracture. If we could find a way to reduce the scar formation by some % and subsequently the contracture it would give us a significant advantage over the competition as well as make the procedure better for the patient …683
(Emphasis added)
601 Dr Linda Cardozo, a British urogynaecologist under whom Dr Hinoul trained and whom he told the Court he holds in high regard, informed Ethicon in June 2005 in an interview relating to mesh in prolapse repair that mesh folding would increase complications.684
602 Amongst the “highlights” noted from the discussion at an Ethicon expert meeting on 2 June 2006 attended by (amongst others) Professors Cosson, Klosterhalfen, Deprest, Jacquetin, Lucente, and, from Ethicon, Ms Berthier, Ms Hellhammer, Dr Meier and Dr Holste, were that “shrinkage is not controlled by ‘softness’ of mesh” and that the top priorities included the elimination of “shrinkage”.685 The position had not changed by 23 February 2007 when a similar meeting took place.686 It is inconceivable that elimination of shrinkage would be a top priority unless shrinkage was clinically significant.
603 The Prolift Surgeon’s Resource Monograph issued in 2007 contained the observation that:
Contraction of the mesh and/or reduction in the vaginal epithelial dimension is the primary exam finding in a subset of patients with dyspareunia.687
604 In its clinical strategy for Prolift+M, dated 6 September 2007, Ethicon stated that mesh contraction “can cause vaginal anatomic distortion, which may eventually have a negative impact on sexual function”, and is difficult to treat.688
605 The confidential memorandum bearing the joint letterheads of Ethicon Women’s Health & Urology and JJM, dated 16 February 2011, which I mentioned earlier, includes the following statement:
[T]here is evidence that meshes shrink in vivo leading to increased stiffness, pain, and poor restoration of the normal properties of the vagina compliance (Jones et al., 2009).689
The reference was to an article by Keisha Jones, Andrew Feola, Leslie Meyn, Steven Abramowitch, and Pamela Moalli entitled “Tensile properties of commonly used prolapse meshes” (2009) 20(7) Int Urogynecol J Pelvic Floor Dysfunct 847–853.
606 Christophe Vailhé, Mathew Krever, and Suzanne Landgrebe of Ethicon wrote in December 2011 that “[a] good graft design will require resistance to folding, or “balling” which could be due, amongst other things, to “excessive tissue contraction”, since it is believed to be a “significant factor in promoting exposure of the implant”.690
607 In 2008 the TVM Group reported that in their retrospective study of 684 patients there was “a significant link” between “prosthetic retractions” (contractions) and vaginal “expositions” (exposure) and granulomas. The Group also reported that retractions and relapse of prolapse were “significantly linked”.691 They postulated that retraction “favored” relapse because it creates non-reinforced zones692. In addition the Group wrote that prosthetic retractions can cause pelvic pain, dyspareunia, and dyschaesia (painful defaecation). Professor Collinet was a co-author of this article.
608 A Safety Communication issued by the FDA in 2011 relevantly stated:693
Reports in the literature associate mesh contraction with vaginal shortening, vaginal tightening and vaginal pain.
Both mesh erosion and mesh contraction may lead to severe pelvic pain, painful sexual intercourse or an inability to engage in sexual intercourse…
609 In 2013 a group of Polish researchers published the results of a study on possible correlations between mesh retraction (contraction) after anterior vaginal mesh repair with Prolift and de novo stress urinary incontinence, overactive bladder (OAB), and vaginal pain symptoms: Rogowski et al (2013).694 They found that mesh retraction was “significantly larger” in a subgroup of patients who reported postoperative vaginal pain compared with those who did not and that “[a] significant correlation was found between mesh retraction and the severity of vaginal pain”. They also found that mesh retraction was “significantly greater” in the subgroup reporting de novo vaginal pain at follow-up compared to those without de novo vaginal pain. And they said that mesh retraction was also “significantly larger” in patients with OAB than in patients who did not present with this complication at follow-up. Furthermore, the authors reported that the proportion of patients with postoperative vaginal pain and the proportion with postoperative OAB symptoms were in each case significantly higher in the high mesh retraction group.
610 Taking into account all the evidence on the subject, I am not only satisfied that polypropylene mesh, including those used in the Ethicon devices, contracts in the way Professors Klinge, Klosterhalfen and Deprest explained, but I am also satisfied that contraction of the mesh has clinical significance. It can increase the risk of recurrence of the condition which it was designed to arrest and it can cause a number of complications such as mesh exposure/erosion and chronic pain, including at rest and with sexual intercourse.
611 Professor Klosterhalfen’s evidence was that polypropylene mesh products are incompatible with the female pelvis because, while the meshes are flexible, they are not elastic. He said that flexible structures like polypropylene meshes are only able to elongate in one direction but an elastic structure like the vagina can stretch in all directions.695 He explained that the mesh acts as an inelastic material in an area needing elasticity and motion.696 He referred to this as a “mechanical mismatch”:697 He said that the mechanical mismatch is responsible for the main complications of polypropylene meshes in the pelvic floor. He explained:
From a mechanical point-of-view, a harder material in contact with a softer one can induce erosion in the latter [and] from a biological point-of-view, mechanical mismatch induces inflammation and tissue remodelling”.
612 Professor Klosterhalfen said that the mesh is “overstretched” and remains elongated, forming folds and wrinkles. 698
613 A PowerPoint presentation prepared by Ethicon Women’s Health & Urology for an Ethicon meeting in May 2008 about the development of a new device for pelvic floor repair, noted that “there is still NO evidence of a Device created specifically for the female pelvis”, “no patient-centric PF material”, pelvic floor materials were “still over-engineered”, less foreign body material was required, materials should “correlate to measured female pelvic physiological characteristics”, and the elasticity of the implant had to “differ according to direction”.699
614 An Ethicon PowerPoint presentation from March 2009, which compared the elasticity of the vagina with the elasticity of Prolift and Prolift+M before and after Monocryl absorption, showed that neither mesh matched the elasticity of the vagina and that Prolift was the least elastic.700 Professor Klosterhalfen said that this was particularly important because Prolift+M has Monocryl present for at least 84 days and that is “the most crucial implantation time period for a mesh with respect to tissue ingrowth”.
615 Professor Klosterhalfen agreed with the findings reported in this presentation. They accorded with his own experience, including his examination of explanted meshes. His opinion was that the mesh used in the Ethicon devices is “too stiff, inelastic, has too much material and inadequate pore size”. He also expressed the view that these characteristics cause excessive inflammatory response and excessive contraction which, in turn results in pain in the pelvis including dyspareunia. He was adamant that none of the Ethicon meshes used in the devices has the elasticity and “physiologic biomechanics” that are specifically adapted for surrounding tissues in the female pelvis. He reminded us that the meshes were all created for the purpose of hernia repair without regard to the biomechanics of the female pelvis.701
616 Similarly, Professor Deprest said that the main reasons for post-surgery complications and with complications associated with erosion included the biomechanical properties of the mesh, especially “mesh stiffness”.702
617 This concept of mechanical mismatch may well have been drawn from Dietz et al (2003). 703 In their paper, published in the International Urogynecology Journal in August 2003, 17 months before Prolift was launched, Professor Hans Peter Dietz and others from the Royal Hospital for Women in Sydney observed that, although tissue reaction is clearly an important factor, “common sense would suggest that any differential between the biomechanical properties of implant and surrounding tissue is likely to influence the likelihood of erosion”. They added:
This introduces the concept of the tissue-implant interface, which plays an important role in all biomaterial interactions. A mismatch between implant and tissue properties does not allow for the appropriate transmission of loads at the interface and may attribute to the poor clinical results observed with some of these materials.
618 They pointed out that in vivo clinical performance will depend on a range of factors, including surgical technique (especially the extent of the dissection), the mechanical properties of the host tissue, “biological ingrowth/ongrowth of the implants, and not least on in-vivo loading characteristics”.
619 Professor Wright’s opinion, however, was that, because Prolene Soft and TVT use the same synthetic polypropylene as Prolene used in sutures and hernia mesh, the host tissue responses to the individual fibres are expected to be the same.704 He then proceeded to cite three tissue studies which he said “clearly document that Prolene Soft usually evokes an initial minimal to mild acute inflammatory response, followed by a minimal to mild chronic inflammation that is accompanied by a minimal to mild fibrosis”.
620 There are several problems with this opinion.
621 First, two of the three tissue studies were animal studies, involving rat and rabbit models, over relatively short periods of time. These were small scale studies with short periods of follow-up. Professor Wright neglected to mention the sites of implantation, the size of the studies, or the length of follow-up. One, by Ethicon researchers (2001), was a 91-day rat study in which the meshes were implanted under the skin over the sacral vertebral column.705 The other, by Boulanger et al (2005), was a study of five mesh types in 12 sows over 72 days and only 10 prostheses were available for study.706 The meshes were implanted in the peritoneum (the lining of the abdomen).
622 Second, while the Boulanger study showed that tissue integration was best with polypropylene meshes and that Prolene and Prolene Soft induced the least severe inflammatory reactions, Professor Wright failed to refer to the cautionary note in the last sentence of the article in which the authors stated:
The role of atrophy, bacteriological environment and devascularization of the vagina in complications like erosion or infection must be explored, and the long-term efficiency and tolerance evaluated before a wide diffusion and promotion of these surgical techniques.
623 Third, Professor Wright did not take heed of the observations made in one of the articles he cited (by Elmer et al, 2009)707 that “rodent immunology may differ substantially from that in humans and the appropriateness of transferring data on biocompatibility between species is questionable”.708
624 Fourth, Professor Wright also cited Elmer et al (2009) in support of the proposition that Prolene Soft usually evokes an initial minimal to mild acute inflammatory response, followed by a minimal to mild chronic inflammation accompanied by a minimal to mild fibrosis.709 But he did not refer to the qualification made by the authors themselves that:
[I]t is important to recognize that a larger study population with transvaginal mesh surgery would undoubtedly generate an increased number of mesh related complications. Clinicians and patients should be aware of the possibility of late mesh related inflammatory reactions when using a large polypropylene mesh.710
625 Professor Wright went on to say that studies that used other forms of polypropylene mesh have shown that with longer implantation times the host tissue response includes moderately dense fibrous connective tissue.
626 One was a study by Pierce et al (2009)711 of 45 rabbits, 22 of whom were implanted with polypropylene mesh in the abdomen and most also had mesh placed in the posterior vagina. The control group received biologic graft materials. The period of follow-up was nine months. Notably, there were significant differences in results for the abdominal and vaginal implants. Erosion occurred in 6/22 (27%) of rabbits with vaginal polypropylene implants and those with longer grafts (2.9cm as opposed to 2.0cm) suffered vaginal erosions at a much higher rate (5/12 or 42%) compared with 1/10 or 10% in those who received the shorter pieces. No erosions occurred at the abdominal sites.
627 On the face of things, this study did not support Professor Wright’s assertion that the host tissue response should be the same for hernia meshes and pelvic meshes. The authors themselves, in a passage to which Professor Wright did not refer, stated:
This study importantly provides data to substantiate prior speculation that implantation of graft materials in the vagina elicits a different host tissue response than abdominal implantation.712
The authors also emphasised the need for additional research “to evaluate the long-term safety and efficacy of specific graft materials used in pelvic reconstructive surgery”. This was a decade after the release in Australia of TVT and six years after the release of Gynemesh PS.
628 The second of those studies was a 2002 study of 76 hernia explants conducted by Professors Klosterhalfen and Klinge. 713 At this point neither of them had begun to examine pelvic explants. The pelvis data pool was not even in contemplation. It should have been obvious to Professor Wright that the views of Professors Klosterhalfen and Klinge have evolved since then.
629 Professor Klosterhalfen said that it was an over-simplification to say that the host tissue responses to the individual fibres in Prolene Soft and TVT are expected to be the same as seen with Prolene sutures and hernia mesh. While the quality and nature of the tissue reaction is generally the same at the level of each individual fibre, it is necessary to consider the integration of the mesh as a whole. Differences are apparent when the integration of the entire mesh is considered. He said that Professor Wright neglected to take into account the importance of textile structure to the biocompatibility and function of the mesh. Professor Klosterhalfen explained that the intensity of the foreign body reaction depends on the surface area of polypropylene in contact with the host tissues; and the size of this area mainly depends on the porosity of the mesh or distribution of the pores. If the pores are too small, granulomas (small masses of inflamed granular tissue) have to fuse together and the fibrosis (scarring) can encapsulate the entire mesh causing the mesh to become stiff and lose function. Since the meshes are knitted, there are numerous large and small pores and it is not enough to just look at the large pores.714 That is obvious in an image of Prolene Soft reproduced in Professor Klosterhalfen’s second affidavit:
630 Professor Klosterhalfen explained that it is necessary to look at the distribution of the pores, the effective pore size under load and after tissue integration, and the pore geometry. The smallest diameter of the pore determines the risk of bridging fibrosis. He said that Prolene as used in the TVT devices has a porosity of 50.1% and an effective porosity of 0%.715 Similarly, Prolene Soft has a porosity of 62% which drops to 10% under load and an effective porosity of 26%, which falls to 0% under load. Measurements of pore sizes confirmed that the majority of pores are much smaller than 1mm.716
631 For all these reasons, as Professor Klosterhalfen deposed, it is inappropriate to transfer results of experimental studies on polypropylene fibres or sutures to polypropylene meshes.
632 Furthermore, almost all the following propositions are uncontroversial. Many of them are self-evident.
633 First, a suture is a single fibre with only one compartment (loop).717 All the meshes in question consist of a considerably larger amount of foreign material and multiple compartments (pores).718 Consequently, the nature and extent of the reaction to a mesh implant will be different from the nature and extent of the reaction to a suture or sutures. Professor Deprest and his fellow authors explained in a 2006 article that “[t]he nature and extent of the inflammatory reaction [generated by synthetic mesh implants] is regulated by the chemical and physical structure of the implant, the amount of material, and surface of the contact-area with the host”.719
634 Second, hernia mesh is used as a flat mesh in largely tension-free conditions whereas the pelvic floor is an area subject to a great deal of stress and strain. Professor Klinge explained the position this way in re-examination:
The – the introduction of meshes for hernia repair, it was related to the term “tension free”. That means that you have a – a huge flat mesh that reinforces the tissue. And it was – it was considered to be in contrast to the suture repair which causes some – some tension to the tissue, with all the problems of cutting through the tissue or creating pain. So this tension free is a general principle and the major advantage of using meshes for hernia repair, and so later on it – it became evident that if you’re using these hernia meshes in – in an area where you applied some tension to it, that this will create some problems. We have it in the abdominal cavity as well, when we are placing or when we tried to place these meshes close to the oesophagus in hiatal area, you have movement of the diaphragm, you have movement of the muscles, you applied some tension to the meshes, and then we have complications such – of erosion, migration, serious complications in this area. So the application of – of meshes that are designed for mesh – for a tension free application, in an area where you applied some tension, is not a good idea, or is – makes it dangerous to the patient and, from my point of view, a similar thing happens in the pelvic floor where you have to consider forces that are changing the appearance of the meshes in a completely different way than in the tension free procedures of – of the abdominal wall.720
635 Third, the clinical studies conducted by the Aachen Group confirmed that the more foreign body implanted or surface area covered, the greater the degree of inflammation.721
636 Fourth, the effect of polypropylene mesh on tissue differs according to the environment in which it is implanted.
637 As Professor Iakovlev pointed out, the mesh cannot expand or contract and, in order to function properly, several organs in the pelvis need to expand and contract. The mobility and function of the anterior abdominal wall, on the other hand, are much more limited.722 Further, the Ethicon devices are designed to be implanted superficially under sensitive mucosa and can easily erode through the mucosa. In contrast, erosions of hernia meshes are rare.
638 Notwithstanding the approach taken in the present case and in the development of the Ethicon devices, there is some evidence that the respondents recognised this.
639 In one of the slides in a PowerPoint presentation bearing his name dated 17 November 2004, Dr David Robinson, then an Ethicon medical director, emphasised that “[t]he Vagina is NOT the Abdomen (nor similar to any other surgical environment)”.723 Slide 17 identified the following “[u]nique issues to the vagina in the placement of large meshes”:
- Cannot be sterilized
- Relatively thin overlay with no real fascial layer
- Attachment sites are difficult to access
- The three dimensional architecture and various vector forces are complex
- Subject to great forces with little or no bony (and often pelvic floor muscle) reinforcement
- Must remain pliable for filling and emptying of pelvic organs and maintain the capacity for sexual function.
640 A “confidential” PowerPoint presentation prepared by Ethicon’s Women’s Health & Urology Research and Development team acknowledged that:
Today’s vaginal implants do not consider the patients’ biomechanical needs.724
It recognised that “unmet biomechanics” lead to “misfunction, pain & shrinkage”, which in turn, leads to a “handicapped patient”. The document is undated but, since it refers to a 2006 article, was obviously created after the launch of Prolift.
641 At some stage during the first decade of this century, Ethicon began work on designing a mesh that was intended to be adapted to the pelvic floor and that would resist pore collapse. The project was originally referred to as Project T-Pro and later Project Thunder. Nothing eventually came of it, however, and it appears that it was abandoned in about 2011 following budgetary cuts.725
642 In their Invention Disclosure, Drs Trezwik and Meier identified the problem the proposed mesh was designed to solve in the following way:
Each implant introduced into the human body has to mimic the mechanical properties of the tissue it should support, no matter if it is a total hip replacement, vascular graft or mesh for soft tissue reinforcement. In any of these areas the tissue properties are not uniform (isotropic) but change continuously within different areas of the respective anatomical region. Often these variations correlate with the specific mechanical load that occurs in these tissues anisotropy). Furthermore different kinds of involved biologic structures (e.g. Fascia, ligaments, muscle, tendons etc.) have different biomechanical properties. The ideal implant must take these variations into consideration and minic the different mechanical tissue properties …
Mesh based implants which are currently used in pelvic floor reconstruction are based on mesh constructions originally designed for the treatment of hernias in the abdominal wall region. It is important to understand that the biomechanical properties of the abdominal wall and the pelvic floor differ especially in regard of elasticity and anisotropic material behaviour. To fulfil the desired biomechanical compatibility of mesh based implants for pelvic floor reconstruction, it is important to take the biomechanical properties of the implantation site into consideration.726
643 At one point, and despite their reliance on Professor Wright, the respondents submitted that it was apparent that the forces in the female pelvis are not the same as those in the abdominal wall. They contended that the forces are unlikely to be unidirectional and more likely to be multidirectional, and in the case of POP mesh, there are differences between the forces applied to the arms and the body of the mesh.727 These submissions should be accepted. They reflect the evidence given by Professors Klosterhalfen and Klinge.
644 The opinions of Professor Klosterhalfen and Klinge were also supported by the work of Professor Moalli and her colleagues.
645 Barone et al (2015) (Professor Moalli included) noted that literature and marketing pamphlets distributed by manufacturers of urogynaecological mesh demonstrate (or perhaps more accurately seek to demonstrate) biocompatibility of SUI and POP products via implantation studies in the abdominal wall. They accepted that these studies were necessary “to initially demonstrate the ability for mesh designs to be implanted in a host withut overt rejection”. At the same time, however, they emphasized that “the abdominal wall and the pelvic floor are quite distinct in terms of both biological factors and the structural functions that a mesh device is intended to withstand”. Consequently, they wrote that “current urogynaecological mesh designs are more similar to a prototype solution rather than an optimal one”.728
646 Barone et al explained that the environment of the pelvic floor is much more complicated than the abdominal wall from both a biological and mechanical perspective.729
647 For a start, they pointed out, hernia repair meshes are only in direct contact with the abdominal fascia. Pelvic meshes, on the other hand, are placed in an environment with a wide range of soft tissues, including smooth and striated muscle, various kinds of connective tissue, and specialised organs.
648 What is more, the vagina is regarded as a “clean-contaminated” field. When polypropylene is implanted transvaginally, it is seeded with bacteria which contribute to infection and inflammation in the tissues.
649 Professor Klosterhalfen said that even a low-grade infection can enhance the foreign body reaction, contraction, and encapsulation, and that infection occurs with erosion of polypropylene mesh. In 90% of the cases of erosion seen in Professor Kosterhalfen’s pelvis data pool, there were histological signs of infection.730
650 Professor Klinge deposed that the implantation of mesh can result in a biofilm which will make it difficult for the host cells to kill the infection and the development of a biofilm will protect the harmful bacteria the host cells set out to kill.731 A biofilm is an assembly of colonies of bacteria fixed upon a support and locked into an encapsulating matrix which is resistant to both stress and antimicrobials.732
651 Professor Klinge said that one of the major causes of mesh-related infections in patients who have been implanted with pelvic floor meshes transvaginally is the formation of a biofilm.733 I accept this evidence. In a 2007 article relating to the use of synthetic mesh in the treatment of stress urinary incontinence, Klinge et al wrote that infections are usually caused by an intraoperative colonisation of biofilm-forming bacteria that may reactivate in the patient’s later life and present as late abscesses or fistula. They said that sterile handling of the implant and the use of intravenous antibiotics during surgery significantly reduces infection rates, but pointed out that late-onset infections are mainly dependent on the patients’ individual immune system.734
The TVM Group were alive to this. Berrocal et al (2004) said that acute infection was “very rare”, “undoubtedly decapitated by prophylactic antimicrobial therapy” presumably during implantation, but “the real problem associated with the placement of [mesh prostheses]”, including those made of Prolene and Prolene Soft, was chronic infection. 735
652 The respondents take issue with the applicants’ case that infections may occur because of bridging fibrosis or mesh deformation causing a reduction in pore size effected by either phenomenon.736 To the extent that the reduction in pore size prevents the macrophages and neutrophils from doing their work, the evidence is persuasive that, if the pore size is reduced at least to 75µm or below (the figure given in 1997 by Professor Amid), then anything that causes the pore sizes to diminish or, worse still, collapse can increase the risk of infection. It is unnecessary to resolve the question, however, because there is no dispute and no doubt on the evidence that the mesh can both cause an infection and exacerbate an existing infection.
653 In numerous other respects the environment of the pelvic floor is very different from the wall of the abdomen or, for that matter, the groin.
654 Barone et al (2015) pointed out that, “[i]n general, the soft tissues of the pelvic floor are metabolically active, with compositions that have been shown to change dramatically with normal aging and in response to hormone-driven events such as pregnancy, menstrual cycle, and menopause”. They referred to a number of animal studies in which “markedly different host responses” had been demonstrated to abdominal hernia mesh and POP mesh.737
655 Then there is the mechanical environment. Barone et al noted that the structures making up the pelvic floor, like most biological tissues, respond to the presence or absence of mechanical loads.738 They regarded the most significant consideration for pelvic meshes to be the fact that they are subjected to “predominantly uniaxial tensile loading conditions”, an environment, they wrote, which is “quite different” from the loading conditions to which abdominal hernia mesh is subjected. They explained:
During a hernia repair, the mesh is placed within the abdominal wall, fixed along its entire perimeter. Since the abdominal cavity can be thought of as a pressurized vessel, the mesh graft must function as the wall of a pressurized vessel. Under the pressure exerted within the abdominal cavity, the abdomen expands or is “inflated,” and the abdominal wall resists and limits this expansion. Given this loading condition, hernia mesh is placed in tension along all axes simultaneously, much like the surface of a balloon upon inflation. This loading environment helps the mesh maintain its original geometry and pore sizes … However, incontinence meshes are used as suspension structures for slings in the retropubic or transobturator space. Similarly prolapse meshes act as suspension cables, attaching the vagina to the sacrum or pelvic sidewall. These configurations load the mesh, particularly the fixation arms of these devices, primarily in a tensile uniaxial fashion. Even while the name TVT suggests that the device does not experience tension, its intended function is to have the urethra compressed along the sling, which would place the mesh in tension. The tension-free component is thus more representative of the surgical technique rather than the long-term in vivo function of the device.
656 Importantly, Barone et al referred to a number of studies, including one of their own published in 2008, which showed a dramatic reduction in maximum pore size and porosity of “nearly all synthetic meshes” under uni-axial load. In many cases, they said, all the pores in the tensioned mesh were smaller than 1mm in diameter, “diminishing the potential for tissue ingrowth and promoting bridging of fibrous encapsulations such that long-term tissue incorporation is compromised” (citations omitted). In these studies forces of 5 and 10N were shown to cause the dramatic reductions in pore size. It will be recalled that Ethicon’s own research showed that a force of merely 1N could have this effect. It will also be recalled that Professor Moalli’s team established in the study published a year later (Barone et al (2016), that all current prolapse meshes were likely to experience a complete collapse of pores “at some load”.
657 In cross-examination Professor Wright revealed that he had not come across the Barone et al (2015) chapter (chapter 13).739 He said that it would not show up in standard literature reviews.740 Yet, the book from which it was extracted (Host Response to Biomaterials) was described by Professor Santerre as “a pretty comprehensive encyclopaedia (scil) in the international biomaterials world”.741 Professor Santerre volunteered that chapter 13 was “an important chapter to read because it was a compendium review from a key opinion leader in the field”.
658 In cross-examination Professor Wright agreed that uni-axial loads could behave as Barone et al described but, despite the opinions he had expressed in his report about the suitability of the Ethicon devices, admitted he knew nothing about “the forces which are involved with mesh in the vagina”.742
659 Barone et al (2015) also observed that there was “significant evidence” to show that the stiffness of an implantable device can change tissue remodelling and response. They wrote that, in general, “implant stiffness is believed to induce a maladaptive remodelling response through a phenomenon known as stress shielding”. They regarded the concepts of “force distribution” and “stress shielding” as “extremely important” considerations since it has been shown that the vagina responds to mechanical stimuli.743
660 Furthermore, the female genital area has a much higher nerve density in comparison with the anterior abdominal wall and the groin. Professor Iakovlev said that the scar inhabiting and surrounding transvaginal mesh has the highest nerve density of all explanted surgical meshes he has examined as a pathologist.744 Consequently, placement of mesh implants in the female pelvis carries a higher risk of chronic pain than the placement of mesh for hernia repair, whether ventral or in the groin.745 Professor Wright quibbled with aspects of this evidence. There was also a dispute about whether the nerves in question are sensory (rather than motor) and whether, even if they are (or include) sensory nerves, this is the cause of chronic pain. Still, in cross-examination Professor Wright accepted that there was a vastly higher rate of mesh exposure/erosion with vaginal implants compared to abdominal wall implants. In cross-examination he said that he “did not expect abdominally-implanted mesh to have [an] erosion rate of any significance”.746 In his view this was explicable by anatomic differences in the thickness of the overlying tissue. 747
661 There are other differences of significance that are likely to account for the higher rate of complications in the pelvic floor with the use of polypropylene mesh in general, and the Ethicon meshes in particular.
662 The differences between the anterior abdominal wall and the female pelvis were well summarised by Professor Iakovlev in his second report:
a. There are no sharply separated parallel anatomical planes, but rather several round mobile organs in a narrow pelvic space. There is also no real fascia and no subcutaneous fat.
b. There are several organs in the pelvis that need to expand and contract for their function. The mesh cannot contract or expand; moreover, together with the encapsulating scar it provides non-physiological connections to the mobile organs. In comparison, the mobility and the function of the anterior abdominal wall are much more limited.
c. The devices are designed to be implanted superficially under sensitive mucosa and can easily become eroded through the mucosa. Erosions of hernia meshes are rare.
d. The devices are designed to correct the anatomical position of the organs and therefore press against the organs. This pressure can force the mesh to migrate into the organs. In contrast, a hernia mesh would have a much larger area of pressure distribution than a Tape Implant.
e. Vaginal mesh devices cross paths of normal innervation and vascular supply to the mucosa, the bladder and the rectum. After implantation, the innervation and vascular supply need to be restored either through or around the mesh. In comparison, hernia mesh is placed parallel to the anatomical layers and parallel to the nerve and vessel direction.
f. The female genital area is one of the most sensitive areas in the female body. It is much more sensitive than the anterior abdominal wall.
g. The arms of the [POP devices] and the ends of the [SUI devices] cross the muscles and innervation network of the soft tissues and skin. Hernia meshes are placed parallel to the muscular layers, nerves and vessels.748
663 Dr Iakovlev explained the effect of these differences on the incidence and severity of complications as follows.749
664 First, erosion through the vaginal mucosa is one of the most common complications of implantation with polypropylene mesh but it is rare to encounter mesh erosion through abdominal skin or into internal organs with hernia mesh.
665 Second, chronic pain is a complication of both hernia and vaginal mesh. The differences are mainly in the distribution and pattern of radiation. Since the arms of the POP devices and the ends of the SUI devices cross many structures in the pelvis, “pain distribution can involve areas from suprapubic to vaginal, introital, deep pelvic/vaginal, obturator/hip/groin, and into the buttock”. Pain can also radiate into the medial thigh.
666 Third, with hernia mesh, pain on intercourse can occur in cases of mesh migration into the spermatic cord, but dyspareunia is more prevalent as a complication of vaginal mesh.
667 Fourth, urinary obstruction is a complication “almost unique to vaginal implants”.
668 Fifth, other de novo urinary symptoms, such as overactive bladder and urge incontinence, are much more frequent for vaginal mesh implants.
669 Sixth, mesh excision is much more problematic in the case of vaginal implants as it is difficult to readily access the obturator and other “deep parts”. In addition, removal of mesh that has migrated into the bladder or rectum poses a risk of fistulas.
670 In his 2012 article, Dr Elliott observed that it was unsurprising that the same kind of complications arising from the use of synthetic mesh to repair abdominal wall hernias arise after transvaginal repair of pelvic organ prolapse.750 At the same time, however, he observed that a question or concern which should have been raised and thoroughly studied before widespread industry promotion and surgeon acceptance of transvaginal mesh kits is that “the vagina is not the abdominal wall”. As he explained:
The physical angles of support are different. The shape is different. The bacterial flora is different. The anatomic function is different. The pathophysiology of the problems is different. The reparative procedures are different. The volume, quality, and physiologic composition of the tissue coverage overlying the mesh is different. The surgical alternatives and options to treat the underlying problem are different. This list goes on and on. The only significant similarity between abdominal wall hernias and POP is that something is protruding where it should not. Yet, despite all these differences, the actual synthetic meshes used for both abdominal and vaginal defects are strikingly similar, if not nearly identical.
671 The respondents privately acknowledged in February 2011 in a draft internal discussion paper on the biomechanical considerations for pelvic floor design that “pain and discomfort can result from stiff mesh[es] that were originally designed for hernia surgery and ‘over-engineered’ to exceed the burst strength of the abdominal wall at the cost of losing compliance …”.751 This paper, written by two Ethicon scientists, also noted that the vaginal tissue to be augmented by the mesh is often structurally compromised, atrophic, and devascularised. They asserted that the poor quality of the tissue increases the risk of poor tissue incorporation into the mesh potentially resulting in suboptimal healing and mesh exposure or erosion.
672 Professor Korda said that “the mechanism of pain” (by which I understood him to mean chronic pain) following mesh implantation is not well understood. Professor Klosterhalfen gave evidence to the same effect.752
673 Professor Korda postulated that scarring, mesh exposure, and complete contraction of scar tissue may be contributing factors.753 Later, he said that chronic pain is due to an excessive inflammatory reaction against the mesh.754
674 In a 2005 study of 347 polypropylene mesh specimens, including Prolene, obtained from hernia explants, nerve fibres were found in more than 60% of all mesh specimens removed due to chronic pain. In their report on that study, Klosterhalfen, Junge, and Klinge wrote:
In the postretrieval study, most explants from all the patients with chronic pain in their medical history, indicate nerve fibers and fascicles in the interface of the mesh. Today, immunohistochemical stains allow the detection of even the smallest nerve structures that are mainly found in or around the foreign body granuloma. Due to the nature of the granuloma as a chronic inflammation, it may be speculated that these nerve structures are irritated by the inflammation and cause the sensation of pain. In some cases real traumatic neuroma can be found at the interface of the mesh-recipient tissues, an indicator of the mechanical destruction of the nerve by the mesh.755
675 In the present case Professor Klosterhalfen deposed that the chronic pain experienced by women who had mesh, including the Ethicon devices, implanted in their vaginas can be explained in part by entrapped, severed or partially severed nerves (neuromas or pseudo-neuromas). He said that he had observed damaged and entrapped nerves in vaginal explants and the surrounding tissue submitted for histopathological examinations he has undertaken. He also pointed out that hernia meshes explanted for pain have been found to have a statistically significantly higher nerve density than those partially removed for recurrence.756
676 In cross-examination Professor Klosterhalfen was asked to explain what he meant by the mesh entrapping nerves in scar tissue:
[O]ne of the key elements in your chain of reasoning is that nerves can and do, and you can see them, grow into scar tissue?---Excuse me?
Nerves can and do, and you have seen them, grow into scar tissue?---Yes, they can be entrapped in scar tissue, whatever is the mechanism.
When you say entrapped, that’s what I wanted to drill in. Are they already there and the scar tissue grows up around them or does the nerve grow into the nerve tissue as well? That’s what I’m - - -?---They are both theories. I’m a great believer that these are basically nerves which are already present which get into contact with the mesh, and if you take this prolapse meshes, this is a real large area you have to implant, so the probability of – that the mesh comes in contact with these nerves is pretty high. But there are a couple of other people say that you really have new nerve proliferations there.
It’s possible and not yet resolved fully; is that fair?---Yes. I never tested it, so - - -
And it’s possible that both processes might be true?---Yes, what you see that they are still there.757
677 The respondents argued that the effect of this evidence was that Professor Klosterhalfen conceded that nerve entrapment was just a theory. They submitted that the science of nerve entrapment was still developing.758
678 It matters not, however, because Professor Klosterhalfen’s evidence was that, even without entrapment, mesh implants can induce neuropathic pain:
The principle why these nerves send wrong signals which finally end in some type of pain is simply that the nerve is not able to glide within his axons, that it is simply sticking to something. And, of course, if you have a mesh which doesn’t meet or which is not adapted to the elasticity models the tissue is in and it’s rough and if you have sheer stretches, then the mesh itself without entrapment can, of course, induce this type of neuropathic pain. But basically all what we know from other clinical fields, in particular from traumatology and orthopaedic surgery, from reconstructive surgery from burns, if a nerve fibre is sticking into this scar tissue, yes, this is generally accepted as a reason for this pain.759
679 There are, of course, many other mechanisms through which mesh may cause or contribute to pain. Professor Iakovlev gave evidence of some of them which was not the subject of debate and was not challenged. They were irritation of pain receptors through inflammation, erosion, scarring, tissue distortion, oedema, and vascular thrombosis.760
680 The respondents accepted that small nerve branches are invariably severed during implantation of the devices and that these nerve branches will attempt to regenerate and form nerve twigs. They maintained, however, based on Professor Wright’s evidence, that there is no histological basis for attributing pain to the presence of nerves near the mesh because the stains used to visualise the nerves under microscopy do not allow one to distinguish between motor and sensory nerves.761 They also pointed to the following evidence from Professor Wright casting doubt on whether the nerve endings would be sensory:
Although the clitoris is heavily innervated (about 6,000 to 8.000 nerve endings most of which are sensory) and the introitus has touch sensitive nerve endings, the vaginal sheath beyond the first inch has almost no touch sensitive nerve endings. In fact we rarely need to use anesthesia prior to taking a superficial vaginal biopsy since there are so few pain fibers.762
681 There are a number of problems with this evidence. Professor Iakovlev pointed out that the arms of the POP devices and the ends of the SUI devices cross many structures in the pelvis so that pain distribution can involve areas from the suprapubic to the vaginal, introital, deep pelvic, vaginal, obturator, hip, groin and buttock. The pain can also radiate into the medial thigh.763 There are several sensory nerves in the female pelvis, including the pudendal nerve. The chance of mesh coming into contact with sensory nerves is high, particularly if it moves or erodes.
682 In any case, Professor Iakovlev pointed out, by reference to a textbook on neuropathology, that peripheral nerves are predominantly mixed, that is both motor and sensory. Nerve roots exit the spinal cords as motor and sensory roots and immediately merge to form mixed nerves. Closer to the surface, where motor functions are limited, the nerves contain more sensory fibres and some are purely sensory nerves, especially immediately under the skin and mucosa. He said that the majority of peripheral nerves are positive for S100 and most of them contain sensory fibres. He added:
The general rule is that location defines the function and function defines the type of innervation. The main function of superficial tissues is sensory, including pain. The deeper locations, especially within bladder and rectal walls will contain more motor fibers and the damage of these nerves will affect function of the organs or skeletal muscle.
683 Professor Iakovlev went on to explain that the observation of innervation within the mesh indicates that two mechanisms of pain are possible:764
i. The tissue is able to generate sensation of pain in response to noxious stimuli (nociceptive pain). The additional factors for the noxious stimuli can be scar related tissue distortion and tensioning, inflammation lowering the pain threshold, tissue damage by mesh migration etc. This mechanism is equivalent to any normal tissue subjected to inflammation and mechanical factors (pinching, pressure, stretching etc.)
ii. The ingrown nerves are at risk of entrapment, distortion and neuroma formation (neuropathic pain). From my experience, these lesions can be focal and may not be easy to find in limited specimens. Larger specimens usually show several severely distorted nerves. In this mechanism of pain there are also several contribution factors. Additional forces of scarring, mesh deformation, and mesh migration would be additional factors to cause nerve damage.
684 For all these reasons I favour the opinions of the applicants’ experts. But since it is uncontentious that all the Ethicon devices may cause pain, including chronic pain, and that there are various mechanisms that may be responsible for it, it is unnecessary to choose between them.
Oxidation or oxidative degradation
685 Oxidation (also referred to as “oxidisation” and “oxidative degradation”) is a chemical reaction in which oxygen in the air or any other chemical substance that contains oxygen in its molecular structure chemically reacts with the polymer.765 There is no dispute that polypropylene is subject to oxidation. Nor is there any dispute about the effects of oxidation.
686 The instructions for use provided with all the Ethicon devices, however, stated that Prolene is not subject to degradation or weakening by the action of tissue enzymes. The applicants argue otherwise. If the applicants are correct, these statements are erroneous and for that reason misleading. If the mesh does degrade or weaken in this way, it may affect the safety of the various devices.
687 Oxidation in polypropylene can lead to a reduction in molecular weight. It can also change the physical properties and appearance of the polymer. It can turn the polypropylene a yellowish brown. It can also cause pitting, flaking and cracking of the polymer. Generally the material becomes more brittle and loses tensile strength.766 The embrittlement of polypropylene by oxidation or other chemical degradation is known as “corrosion stress cracking”.767 Associate Professor Guelcher’s evidence, which I did not understand to be disputed, was that when polypropylene is exposed to strong oxidising agents, such as reactive oxygen species, it can undergo corrosion stress cracking and that mechanical stress (by tensile, bending, or compressive forces) can also cause or accelerate the process.768 Associate Professor Guelcher said that, because of differences in these environmental factors from one anatomic site in the body to another, it is important to test the polypropylene device in an anatomically relevant preclinical model in order to evaluate its biocompatibility.769
688 As long as there is a source of oxygen, all polypropylene is susceptible to oxidative changes, whether stored at room temperature or implanted.770 Polypropylene is highly susceptible to oxidation because of its molecular structure.771
689 Polypropylene can oxidise as a result of exposure to heat, acidic and “basic” environments, amongst other things. The energy associated with these environments can cause the molecular chain to break because oxidative attack occurs at the bonds between a tertiary carbon atom and a neighbouring hydrogen atom. This results in the formation of free radicals. Free radicals are generally reactive. In the presence of oxygen, they can form hydroperoxides which can decompose to form a hydroxyl free radical and a carbon oxygen free radical.772 A free radical is defined in the Oxford Dictionary of Science as an atom or group of atoms with an unpaired valence electron, that is, an electron in one of the outer shells of an atom that takes part in forming chemical bonds: Oxidation can also cause a loss in average molecular weight and shifts in molecular weight distribution.773
690 In 1994, an article on the mechanisms of oxidative degradation of biomechanical polymers by free radicals was published in the Journal of Applied Polymer Science. The authors, one of whom was Professor Williams, wrote:
The process of phagocytosis, in which cells ingest and attempt to digest extraneous matter … is accompanied by a set of biochemical events, including a sharp increase in oxygen uptake followed by the formation of a number of highly reactive oxygen reduction products …
These highly reactive radicals generated by cellular mechanisms at or near the surface of implanted polymers may contribute to damage of the polymer surface in the same fashion as established polymer degradation reactions by reactive radicals.774
691 Indeed, it is because polypropylene is subject to oxidation that Ethicon added antioxidants to it in the manufacture of Prolene (and Prolene Soft). Antioxidants are chemical compounds that will inhibit or retard the oxidation of polypropylene.775 It will be recalled that two antioxidants were added to polypropylene in the manufacture of Prolene and Prolene Soft. They were Santonox R and DLDTP. The purpose of adding Santonox R was to prevent thermal oxidation during the extrusion process. DLDTP was added to prevent oxidation as a result of long-term storage. It follows that, neither antioxidant was added to protect the implants from reactive oxygen species in the body. Studies show that the hydroxyl free radical is likely to be one of the main causes of degradation in some medical polymers.776
692 There were two issues:
(1) whether the applicants have proved that polypropylene (and Prolene in particular) is subject to oxidation in vivo; and
(2) if so, whether, if it does, it has any significant effect.
693 The weight of evidence indicates that polypropylene is subject to oxidation in vivo. With the exception of Dr MacLean, all the experts who gave evidence on the subject considered that, despite the addition of antioxidants during the manufacturing process, Prolene was no exception.
694 Both Dr Dunn and Associate Professor Guelcher said that the antioxidants used to stabilise Prolene may delay oxidation (and degradation) but do not prevent it.777 Once the antioxidants are depleted the polymer is unprotected from oxidation.778 Dr Dunn said that the precise time that antioxidants are expended is “patient and condition specific”.779 No doubt that is because the foreign body reaction which is ultimately responsible for the release of free radicals will vary from one patient to another. Associate Professor Guelcher observed that the antioxidant package used in Prolene pre-dated the discovery of oxidative degradation of biomaterials in response to the foreign body reaction.780
695 In his evidence in chief, Professor Santerre said it was his opinion that polypropylene materials like Prolene will be subject to oxidative biodegradation. He claimed that the reason Ethicon used antioxidants was to protect their products from oxidation in vivo but he quickly resiled from that position in cross-examination, conceding that he had no basis for making the claim.781 During cross-examination he agreed that the Ethicon devices would undergo oxidation and produce oxidation products, despite the addition of antioxidants to the Prolene fibres during manufacture.782 He also testified that it was most likely that the antioxidants would respond to reactive oxygen species during the course of the foreign body reaction on implantation.783 He said that oxidation was an “outer surface” phenomenon and that it could explain the presence of flakes and cracks. Furthermore, he agreed that the presence of flakes and cracks would create more surface area and could expose a new area of the polymer to inflammatory activity in the absence of antioxidants, including several years after implantation.784
696 Moreover, there was support in the scientific literature for the proposition that polypropylene undergoes oxidative degradation in the body.
697 Mary et al (1998) reported on an investigation in the relative in vivo biocompatibility and biostability of PVDF (polyvinylidene fluoride) monofilament sutures and Prolene sutures.785 This was an animal study conducted in Quebec by a multidisciplinary group. It involved 20 adult female mongrel dogs. Healing behaviour was measured by tissue reaction using light and scanning electron microscopy (SEM) and Fourier transform infrared spectroscopy (FTIR).
698 After cleaning to remove adhering tissue, their relative biostability was assessed by examining under the microscope their chemistry and surface morphology (form and structure). Interestingly, the study confirmed that both PVDF and Prolene sutures contained some background level of oxidative species even before implantation. The authors found that “both types of sutures experience[d] a similar healing sequence, with short-term deposition of fibrin, platelets, and thrombi giving way to the growth of a surrounding collagen capsule between 1 and 3 months postoperatively”. During that period, the authors wrote, both also exhibited “surface oxidation”, and surface degradation could be seen one and two years later in the Prolene sutures.
699 A later study was conducted by a group of biological engineers and surgeons in the United States (Costello et al (2007)) to test the hypothesis that oxidation could be responsible for changes in material properties of polypropylene hernia meshes and for some of the complications associated with the use of mesh.786 The Costello study examined the polypropylene components of 14 polypropylene/expanded polytetrafluoroethylene composite hernia meshes (made by manufacturers other than Ethicon) explanted from patients requiring revision surgery due to chronic pain, recurrence, infection, adhesions, or other complications and compared them to pristine polypropylene mesh materials. Several techniques were used, including SEM, differential scanning calorimetry (DSC), and thermogravimetric analysis.
700 The results of the study were said to support their hypothesis and indicated that the explanted meshes did undergo degradation in vivo, “most likely due to oxidation”. The authors noted that, as a result of the chronic inflammatory response to the foreign body, the mesh material is exposed to “a continuous bath of oxidants”. The authors also commented:
While the analysis of the physiochemical factors of the explanted and pristine specimens provided evidence of oxidation, there are many factors that will affect the severity of oxidation. Since the explants were removed from the patients due to complications of the mesh, the explanted materials have a variety of implantation times ranging from months to years. The severity of the oxidation of the polypropylene material is likely affected by the implantation time, as well as other unique, patient factors such as age, history of smoking, body mass index (BMI), pulmonary disease, diabetes, and potential genetic variation, including variable inflammation and collagen defects.
701 They said that chemical analysis using FTIR would provide information about the chemical species present on the surface of the materials, “further strengthening the evidence of oxidation”. FTIR is a widely used technique to facilitate determination of chemical functional groups by their absorption frequency.787
702 2010 saw the publication of the results of a French study by Clavé and others.788 This was a study of 100 polypropylene and polyester meshes explanted from patients who had experienced complications after transvaginal placement of the meshes. The control group consisted of pristine samples of two Ethicon polypropylene meshes, Prolene and Prolene Soft (Gynemesh PS and Parietex (a Covidien product)). It was said to be the first such study. Its purpose was to evaluate the relative degradation characteristics of polypropylene and polyester prosthetics. The explants were subjected to histological, microscopic (SEM), differential scanning calorimetry (DSC), and chemical analyses (FTIR).
703 For the purpose of the SEM analysis, an explant was considered degraded if it showed morphological differences in comparison to the corresponding pristine implants. Analysis of different mesh explants was reported as showing evidence of damage including superficial degradation, which appeared as a peeling of the surface of the fibre, transverse cracks in the threads, significant cracks with disintegrated surfaces and partially detached material, and superficial or deep flaking. Forty-two percent of the implants were degraded. Degradation was only seen in samples implanted for at least three months. All types of polypropylene implants were affected in this way but none of the polyester implants found to have been altered or degraded.789
704 The authors wrote that in vivo oxidation should create carboxyl groups on the material which can be detected by FTIR analysis. But the results of that analysis in this case were equivocal. It revealed absorption levels in the polypropylene explants that could be attributed to either carboxylate carbonyl or to residual products of biological origin.
705 The DSC thermograms of both low and high density polypropylene explants were similar to the pristine Prolene and Prolene Soft samples. The authors concluded that, if oxidation occurs in those meshes, it happens “in the amorphous zones and crystallinity is preserved”.
706 In the result, the only conclusion the authors drew was that polypropylene was not inert and, although they found evidence of superficial degradation, consistent with oxidation, they did not conclude that the meshes were subjected to oxidation in vivo, as the applicants submissions suggested.790
707 Imel et al, however, in another study of polypropylene pelvic mesh explants, did find “clear signs of oxidation”.791 In the article reporting the study’s results, published in the journal Biomaterials in September 2015, the authors wrote that all explanted samples showed the presence of oxygen. They considered that the results could only be explained by in vivo oxidative degradation of polypropylene. They went on to say:
Oxidative degradation of the polypropylene results in a decrease in polypropylene molecular weight, which is known to reduce the strength of the fibers and can eventually lead to fiber failure. The degradation of the structure of the fibers due to oxidation and reduction in molecular weight eventually results in the appearance of horizontal cracking on the fiber surfaces, which becomes more severe at longer implantation times. This cracking is consistent with weakening of the implanted fibers. This overall picture of oxidative degradation of implanted PP fibers is consistent with previous studies on explanted PP fibers using the same and similar analytical techniques, as reviewed above.
708 This was a much smaller study. Explants were taken from 11 patients. The meshes were not made by Ethicon but by Boston Scientific Corporation. The study was initiated during litigation against Boston Scientific and was sponsored by patients who were suing the company.
709 Iakovlev, Gulecher, and Bendavid examined 164 mesh explants using conventional microscopy and four were also examined by transmission electron microscopy to search for signs of polypropylene degradation. The meshes were made by a number of different manufacturers. They included 28 Prolene explants (from TVT and TVT-O) and four Gynemesh PS explants (from Prolift). Based on the ability of the material to absorb dyes, the authors concluded that the meshes had degraded. The authors also concluded that the degradation layer had been formed in vivo because of “inflammatory cells trapped within fissures, melting cause by cautery of excision surgery, and gradual but progressive growth of the degradation layer while in the body”.792
710 In 2016 Talley et al (including Iakovlev, Dunn, and Guelcher) conducted in vitro experiments on several midurethral slings designed to simulate the foreign body reaction.793 Their hypothesis was that polypropylene would oxidise under those conditions causing degradation of the polymer. Three slings were selected: Ethicon’s TVT and Boston Scientific’s Advantage and Lynx. All three were monofilament type 1 meshes according to the Amid classification and all were said to have had antioxidants added to the polypropylene during the manufacturing process. Thirteen specimens were incubated at temperatures of 37°C for up to five weeks in oxidative media simulating the privileged microenvironment between an adherent macrophage and the surface of the polypropylene — an environment, the authors explained in their article on the study, in which hydrogen peroxide (H2O2) and cobalt ions react to form hydroxyl radicals. Specimens were removed at intervals, washed in deionised water and dried. FTIR analysis was deployed to test for the presence of hydroxyl groups in the hydroperoxide intermediate and for terminal carbonyl end groups. At five weeks one specimen was removed, washed in deionised water and dried for SEM analysis to identify evidence of oxidative degradation such as pitting, flaking or cracking.
711 FTIR spectra of all three meshes showed the presence of hydroxyl and carbonyl groups after five weeks incubation, indicating that the polypropylene had oxidised. No significant differences were observed at any point in time between the three meshes. The authors concluded that these results confirmed their hypothesis that polypropylene oxidises in response to reactive oxygen species such as hydroxyl radicals.
712 In contrast to pristine mesh, SEM images reportedly revealed evidence of “large-scale pitting, flaking, and peeling associated with surface degradation”. Medium magnification SEM images of the meshes revealed evidence on the surface of pits greater than 1µm deep, peeling flakes greater than 10µm in length, and shallow craters also longer than 10µm. High magnification images showed small pits of less than 5µm in diameter and greater than 1µm in depth had also formed on the surface of the mesh. These observations were said to be consistent with their hypothesis that polypropylene oxidises in response to radical oxygen species and results in embrittlement of the polypropylene and surface degradation. The authors concluded that antioxidants added to the polypropylene to protect it during processing and long-term storage do not prevent eventual oxidation in response to reactive oxygen species. They wrote that recent attempts to explain the presence of carbonyl peaks on the surface of explanted polypropylene fibres as a cross-linked protein-formalin complex, biofilms, or residual antioxidants, could not account for the increase in carbonyl and hydroperoxide groups they had observed after in vitro incubation in oxidative medium in the absence of proteins.
713 In addition, Ethicon’s own scientists concluded that Prolene is subject to oxidative degradation in vivo.
714 Ethicon was aware of surface cracking in explanted Prolene sutures used in cardiovascular and ophthalmic surgery at least since the early 1980s. It formed a committee (the Prolene Microcrack Committee) to investigate the responsible mechanism. 794
715 In March 1983 Barbara Matlaga of the Ethicon Research Foundation reported to Dr Anthony Lunn (also of Ethicon) on a review she had carried out of histological preparations from the most recent explants, which had also shown surface microcracking. She reviewed the slides by light microscopy using polarised light to help identify the cracking. She detected cracking in two of five samples taken from vascular grafts after six (sample #1) and four and a half years (sample #2) respectively.795 In the absence of segments of sufficient length from sample #1, tests were carried out only on sample #2. They revealed a breaking strength of 54%.796 But the Ethicon scientists considered that it was not possible to determine on the basis of their examinations whether the surface cracking contributed to the loss of breaking strength.797
716 Dr Emil Borysko, an Ethicon scientist, found severe cracking in specimens taken from sample #1 and concluded that the severity of the cracking was obviously related to implantation time.798
717 Another 1983 study, this time of six formalin-fixed tissue human cardiovascular explants containing Prolene sutures supplied by other doctors, revealed surface cracking in the sample taken after the longest period since implantation (seven years). The depth of the cracking in this sample was 3 to 4.5 microns, which was said to be consistent with other specimens from previous samples up to six years after surgery. The cracked layer appeared blue in gross specimens and particles of blue dye were evident in histological sections of the layer. The Ethicon scientists concluded from these observations that “the layer is dyed PROLENE polymer and not an isolated protein coating on the strands”. The significance of this finding will become apparent in due course. An average of the tensile strength measurements of the samples in this study disclosed a loss of strength for all materials tested. But the scientists considered that the breaking strength data should be viewed with caution as damage to strands could have occurred during removal notwithstanding efforts taken to prevent it.799
718 On 5 November 1984 Dr Peter Moy, an Ethicon scientist, summarised the findings from the studies in a memorandum prepared for Dr Melveger.800 He said that sutures from both cardiovascular and ophthalmic sites had been shown to exhibit surface microcracks. The cracks were predominantly transverse and perpendicular to the axis of the fibre. They were not uniformly distributed along suture lengths either in severity (depth and density) or location. Nor was there any obvious correlation with areas of high stress, such as in the region of the suture knot. In severe cases, the cracks produced a separated layer of apparently uniform thickness and a relatively clean under-surface and secondary longitudinal cracks gave rise to what Dr Moy referred to as “‘brick’ like structures”.
719 Experiments were carried out to determine whether the microcracking had a physical or chemical cause, that is whether it was attributable to environmental stress cracking or oxidative degradation (both of which could lead to transverse cracking) and whether the discrete thickness of the cracked layer arose from a natural separation point in the fibre. Dr Moy explained that “[e]nvironmental stress-cracking refers to the cracking of polymeric parts (usually under some sort of stress, either internally or externally applied) when exposed to a sensitizing agent that is not an active solvent”. On the subject of oxidation, he wrote:
Transverse cracks form as a result of structural reorganization of oxidized polymer that has already undergone significant drops in the molecular weight of the polymer. Chain-scission initiated by the incorporation of oxygen in the polymer takes place primarily in the amorphous phase of the polymer due to oxygen solubility and mobility (packing) considerations. Cracking only occurs when stress-bearing tie molecules in the amorphous regions are severed and retraction of the molecules into the crystalline regions takes place under the internal stress of the fiber. For this reason oxidized polypropylene generally exhibits an increase in density with a concomitant increase in the degree of crystallinity of the polymer. The oxidized polymer however is embrittled with losses in tensile strength and elongation … [S]tudies show that such surface cracking generally does not appear untill (sic) the latter stages of the oxidation process.
720 Various tests were carried out. The thermal characteristics of the crack layer on explants suggested to Dr Moy that “the layering effect may be due to a biological deposit” (original emphasis). IR spectroscopy studies of flakes of the surface material from another explant showed the presence of both polypropylene and protein. He and his team concluded that:
1. The layer does not germinate from an inherent morphological feature (skin/core) in the fiber, as indicated by the 1ack of significant orientational or crystalline differences between the skin region and the core region.
2. Transverse cracking in PROLENE fibers may be induced by physical (ESC) [environmental stress cracking] and chemical (oxidation) processes. Laboratory cracking of PROLENE by ESC do not approach the severity of cracks observed in explanted samples. The relatively high strength retention of explants and the absence of large concentrations of oxidative end products (by spectroscopy) are inconsistent with a strictly oxidative mechanism.
3. The thermal stability of the crack layer (on explant 83-165) suggests that a significant proportion of the crack layer by volume is proteinaceous.
…
721 Dr Moy said that the evidence tended to favour a biological origin for the microcrack layer. He suggested, however, that an additional study be carried out to establish or disprove this hypothesis. He said that could be done by similar transmission electron microscopic examinations on known oxidised Prolene samples “to determine whether a different skin/core morphology could be generated by oxidation”.
722 Consequently, Mr Burkley carried out infrared examinations of explanted Prolene sutures to determine the composition of the exterior cracked surface. First, surface spectra were obtained using attenuated total reflection (ATR). Then particular areas and pieces of the explants were examined using an FTIR microscope. Last, polypropylene films were intentionally oxidised and coated with protein which were then treated like the explants to see if artefacts were produced. They were examined also using FTIR. Mr Burkley set out the results of his examination in a memorandum dated 13 November 1984.801 He found that the outer cracked surface was a composite of protein and partially oxidised polypropylene. He said that, although surface protein is efficiently removed with Soluene treatment, residual protein remains within the cracked material. He considered that this suggested “a stronger, (perhaps chemical) interaction of protein and oxidized polypropylene”. He also noted that the Soluene treatment not only removes surface protein but also altered the oxidised polypropylene surface by removing lower molecular weight species of the oxidised polypropylene with the protein.
723 On 11 March 1985 Dr Moy wrote again to Dr Melveger.802 Although studies on Prolene microcracks had been conducted “for some time”, Dr Moy was concerned about limitations in the laboratory experiments around which the bulk of the work had centred. He said that “[a]dditional quantifiable data” was necessary to satisfactorily resolve some fundamental issues about Prolene microcracking. He recommended that additional tests be carried out to:
measure the thickness of the microcracked layer on microtomed samples to provide quantitative information on the growth of the microcrack layer;
analyse the same samples by FTIR for “possible oxidation species” and for the presence and level of residual stabilisers in order of implantation time and severity of microcracking; and
staining the explant sample sections with protein stains to visualise the microcrack-Prolene interface, then examine them with TEM to establish the degree and nature of dispersion of the protein coating on the Prolene and differentiate from three different protein deposition mechanisms.
724 On 22 September 1987 Mr Burkley examined samples of Prolene sutures carefully removed from washed and dried specimens received from the laboratory of Professor R Guidon803 against a Prolene suture control using SEM.804 Samples of sutures removed two years after implantation showed no cracking. Samples removed eight years after implantation showed “severe cracking”.805 Some of the eight year samples (numbered 83D035) were examined optically and, using a needle, the cracked surfaces were easily scraped off. The surface scrapings were melted on the Mettler hot stage (which enables samples to be observed under the microscope while it is heated and cooled) and melted at a range previously observed for oxidatively degraded polypropylene. He reported on his findings in a memorandum to Dr McIveger on 30 September 1987:
1. The amount of DLTDP is reduced in the explanted sutures. No DLTDP is observed in the surface scraped (cracked regions) of 83D035. The observed DLTDP decreases with implant time.
2. No protein is observed in any spectra of the explanted sutures.
3. The surface scraped material from the cracked regions of 83D035 has a melting range indicative of degraded polypropylene. The IR spectra of this scraped material is clearly polypropylene, but it appears to be degraded in an oxidative fashion. There are a number of degradation species possible from the IR data. Hydroxyl and acid/ester functionality are definitely present. Ketone and/or unsaturated species are suggested, but not verified.
4. The degraded portion of the 8-year explant makes up only a minor portion of the entire suture.
725 Mr Burkley’s findings concerning the reduction in the antioxidant DLTDP in the explanted sutures and its absence from the cracked regions support the evidence given by the applicants’ witnesses that the antioxidants used in the manufacture of Prolene do not provide permanent protection from oxidation. His findings as a whole support the applicants’ case that polypropylene degrades in vivo.
726 Minutes of an Ethicon meeting on 8 October 1987 convened to discuss the status of the study referred to Mr Burkley’s findings and conclusions.806 The meeting decided that the next step was to do electron spectroscopy for chemical analysis and some microscopy work to study the depth profiles of the cracks. One of the attendees, Mr F Schiller, suggested some drying experiments to see if the drying procedure played a significant role in cracking.
727 On 30 May 1985 Ethicon circulated a protocol for the study in beagles of subcutaneous implantation of Prolene and other sutures (PVDF, Ethilon, and Novafil) which was to begin on 3 June 1985 (dog study). The stated purpose was to assess breaking strength and other parameters of the sutures (monofilament size 5–0) after they had been in the bodies of the dogs for 10 years and with baseline testing at intervals of two, five, and seven years of “unimplanted sutures”.807
728 The study began in November 1985. Twenty-four dogs were implanted with sets of Ethilon, Prolene, PVDF, and Novafil sutures. In 1990, after five years, explants from five dogs were described. One dog died prematurely after six years and 10.5 months implantation time — before the seven year results were reviewed.808 In June 1992 sutures were explanted from another group of four dogs.809 Despite the original intention, the study did not continue past the seven year mark.
729 At the two year interval seven or eight sutures of each product were evaluated. Save for one suture, Prolene displayed no discernible cracking, according to the report of FD Schiller.810
730 The five year report delivered in 1990, compiled by Dr Elke Lindemann, relevantly concluded as follows.811 After five years in vivo the PVDF 5-0 suture was the only explanted material from five dogs which did not show any surface damage due to degradation. Two of the seven Prolene explants, two revealed cracking. The Prolene surface, intact at the two year point, showed signs of degradation at five years.
731 At seven years, degradation in the Prolene continued to increase and PVDF was considered to be “by far the most surface resistant in-house made suture in terms of cracking”. Cracking was evident in the Prolene suture explants (examined by light microscopy and SEM) at three out of seven sites. 812
732 In his first report Professor Santerre cast doubt about the findings of these studies, however, suggesting that the use of Clorox bleach to clean the samples may have compromised the conclusions reached in at least one investigation, since Clorox bleach contains hypoclorous acid, which he said was a potent oxidant:
This compromises the study’s conclusions because we do not know whether the cleaning process may have initiated the generation of changes in the chemistry (i.e. infrared spectroscopy data) and/or the surface morphology (as observed by surface cracking phenomena seen in the Scanning Electron Microscopy images), or whether the in vivo system generated those observations, or whether those observations were a combination of both.813
733 The report to which Professor Santerre referred did not state that the samples were cleaned with Clorox bleach. This was put to Professor Santerre during cross-examination and he was not able to locate a reference to it. There is, however, a reference in Mr Burkley’s notes of his examination of the explants supplied by Professor Guidoin to samples (sutures 83D035) being cleaned by Professor Guidoin with “a Clorox-like chemical”.814
734 Professor Santerre confessed during cross-examination that he could not say “with absolute certainty” that the chemical did or did not have an effect, but he insisted that its potential effect should be considered.815 He added:
I put it forward to the court that consideration of using a very powerful oxidant on materials that had been in the body for a long period of time, that have the potential for being a mixture of the polymer, the antioxidants and protein and lipids grown up that it is important to consider this is a factor influencing what those individuals were looking at in that time.816
735 There are a number of difficulties with this evidence.
736 First, although he was in a position to do so, Professor Santerre did not test his hypothesis by carrying out any experiments of his own.817
737 Second, Professor Santerre’s hypothesis was inconsistent with Dr MacLean’s evidence. When asked about the effect of bleach on oxidation during cross-examination (in relation to the Thames paper818), Dr MacLean was adamant that it would not have any effect:
Just cleaning it with bleach is not going to cause it to oxidise?---No, I’ve actually done some work that shows that it doesn’t in the confines of this type of cleaning experiment.819
738 Third, Professor Santerre’s hypothesis was also inconsistent with the protocols for analysing explanted biochemical devices contained in ISO12891 which state:
While no method is listed for cleaning PP [polypropylene] explants, the standard recommends cleaning with sodium hypochlorite bleach solution for the closely related polyolefin, ultrahigh molecular weight polyethylene.820
739 It was for this reason that Imel et al used sodium hypochlorite to clean their samples.821
740 Since the International Standards encourage the use of bleach to clean polypropylene, it is unlikely that the consequences Professor Santerre feared would result merely from the use of bleach.
741 Fourth, unlike Professor Santerre, Imel et al did test whether cleaning the Pinnacle explants with bleach did cause oxidation of the specimens. For this purpose they carried out FTIR analysis of non-implanted Pinnacle. They reported:
Consistent with published literature and the known chemical compatibility of polypropylene, no evidence of oxidation is seen and the spectrum is essentially identical to the spectrum obtained for non-implanted Pinnacle prior to the bleach treatment.822
742 Fifth, in any case, oxidation was found to have occurred when samples were cleaned by other methods. Mary et al, for example, used an enzyme incubation technique which they developed in their laboratory (described at pp 200–201 of their report).823 In order to eliminate organic residue, Clavé et al used a sodium chloride solution to treat their explants, then washed them with deionised water, before extracting samples with pure cyclohexane.824
743 In their submissions the respondents did not rely on Professor Santerre’s hypothesis. Their arguments were as follows.825
744 First, while Liebert et al reported in 1976 on oxidation of unstabilised polypropylene, they also reported that infrared spectra and mechanical testing of both implanted and non-implanted filaments containing an antioxidant showed no changes in either chemical or physical properties as a result of implantation. I note that in their reports both Dr MacLean and Professor Santerre relied on the Liebert study as evidence that the antioxidants in Prolene protect it from oxidative degradation.826
745 Second, there is still a lively academic debate as to whether degradation of polypropylene implants occurs in vivo and Dr MacLean’s experience was that his multistep cleaning process demonstrates that Prolene does not oxidise in vivo. Rather, a layer of biological materials is deposited on the fibres, following implantation, and the removal of that material from explanted mesh demonstrates that the mesh fibre surfaces were not oxidised. Further the resultant cleaned fibres exhibited extrusion lines associated with the fibre manufacturing process, similar to those found on pristine exemplar mesh fibres.
746 Both these arguments have limitations. For the following reasons the material upon which they rest does not undermine the weight of evidence to the contrary.
747 The respondents are correct in their summary of the Liebert study. As the applicants submitted, however, it did not establish that antioxidants prevent oxidative degradation. What it did prove is that polypropylene degrades in vivo as a result of oxidation.827
748 Liebert et al reported on a study conducted to examine the role of oxygen in the reaction between a typical hydrocarbon-type polymer (polypropylene) and the body. Polypropylene was selected for a number of reasons but primarily because in its pure form it is easily oxidised. Extruded filaments of unmodified polypropylene with and without antioxidant were implanted subcutaneously in Syrian golden hamsters and remained undisturbed for periods as long as the life-span of the animal. Increases in hydroxyl or carbonyl were measured by infrared spectroscopy. Molecular weights were measured by gel-permeation chromatography, which separates polymers in solution based on their size.828
749 The infrared spectra showed that the pure polypropylene filaments degraded by an oxidation mechanism similar to that which has been found by others by the autoxidation of polypropylene at elevated temperatures. Carbonyl groups were observed to form after 99 days of implantation. The induction time, that is the time oxidative changes become measurable and beyond which the rate of change dramatically increases, was determined to be approximately 108 days.829
750 No changes, however, in mechanical or chemical properties were observed for the stabilised filaments, that is, those to which antioxidant had been added. The authors wrote:
These results support the view that the changes observed for pure implanted filaments are due to oxidation rather than diffusional or other unknown effects, since the antioxidant specifically inhibits and/or retards oxidation.830
751 The authors did not conclude that polypropylene treated with an antioxidant does not undergo oxidation. It is likely that the antioxidant would at least delay oxidation and the authors were at pains to point out that long-term effects were not studied. Moreover, as Associate Professor Guelcher pointed out, the last point at which data was collected from the hamster in whom the stabilised filaments had been implanted was at about 95 days (because, he inferred, the hamster died at this time) before carbonyl groups were observed to form in the unstabilised filaments and well before the induction time commenced, whereas the last point for the collection of data from the untreated filaments was at 150 days.831
752 The “lively debate” to which the respondents referred in their submissions took place in the pages of the International Urogynecological Journal. More accurately, it was a heated exchange of opinions. It began with the publication of the article in 2017 by Thames et al somewhat provocatively entitled “The myth: in vivo degradation of polypropylene-based meshes”.832
753 Thames et al (2017) maintained that “properly formulated [polypropylene] with high performance additives is stable in oxidizing media, including elevated temperatures, in vivo applications”. They asserted that Liebert et al “established the profound stabilizing effects of antioxidants”. As I have already pointed out, however, the Liebert study only established the stabilising effects of antioxidants for a period of about 95 days.
754 Thames et al also referred to two studies by Professor Williams to support their hypothesis that stabilised polypropylene does not degrade in vivo. They suggested that in one article published in 1982,833 Williams had “referenced Liebert” in concluding that since high activation energies are necessary for the degradation of high molecular weight polymers, then “no such degradation should occur within the confines of the human body”. In fact, this statement in the Williams article came before any discussion of the Liebert study. Moreover, Thames et al omitted the qualification made by Williams that followed:
While this prediction is largely borne out in practice, there is some evidence that other factors are involved and that unexpected degradation mechanisms operate within the body.
755 Thames et al additionally cited a statement from the second Williams article, also published in 1992, that “hydrophobic homochain polymers should be stable under in vivo conditions”. But the statement was taken out of context.834 This is what Williams wrote in context:
There is little doubt that there is one overriding, fundamental mechanism which controls much of the degradation observed in vivo. This is hydrolysis, a mechanism that can, under appropriate conditions, readily result in molecular fragmentation. It is especially seen in heterochain polymers … Hydrophilic polymers with ester groups are going to be quite susceptible; on the other hand, hydrophobic homochain polymers (e.g. PTFE) should be stable under in-vivo conditions.
It was common ground that polypropylene is a hydrophobic homochain polymer.835 Williams continued:
Since the aqueous phase of the body remains reasonably constant (i.e. pH and temperature do not change) it should be possible to predict accurately the susceptibility to, and the kinetics of, in-vivo degradation, if aqueous hydrolysis was the only mechanism available.
This is clearly not the case, for it is certainly known that rates of degradation of some materials vary widely, depending upon the circumstances of implantation, and that degradation may well be occurring with certain polymers in vivo which cannot be attributed to hydrolysis.
756 Furthermore, in his 1982 review article, entitled “Biodegradation of surgical polymers”, Professor Williams wrote that all polymers are susceptible to degradation under physiological conditions.
757 Thames et al claimed that the studies in which researchers had concluded that polypropylene oxidises in vivo failed to consider the natural adsorption of biological material when medical devices come into contact with bodily fluids and the reaction of fixatives with biological materials coating the devices and creating “a polymerized proteinaceous layer covering the fiber”. They stated that these researchers either did not try to remove or did not adequately remove biological materials from the devices. They also argued that formalin fixation could affect chemical analysis. Importantly, however, they did not refer to the studies conducted by Ethicon’s own researchers. Yet, the manuscript for the article was said to be based on findings from earlier work they had undertaken that was financed by Ethicon, Professor Thames had provided “litigation consulting services” to Ethicon, and at least one of the two other authors also provided consulting services to Ethicon.
758 Thames et al conducted a study of a number of Prolene explants (both SUI and POP devices) which they first cleaned in a multi-stage process they described as “novel and effective”. The result was that they found no evidence of degradation or oxidation. It was certainly thorough, perhaps too thorough.
759 In a letter to the editor of the International Urogynecological Journal published later in 2016, Dr Margaret Thompson, Associate Professor Guelcher, Professor Iakovlev, Dr Bendavid, and Dr Donald Ostergard took Thames et al to task. They levelled the following criticisms at the study and the article.836
760 First, they drew attention to the authors’ affiliations to Ethicon and other defendants in mesh litigation, although those affiliations had been disclosed in the article.
761 Second, they contended that, in reaching their conclusions, Thames et al had ignored “the vast body of peer-reviewed literature” (estimated at over 100 articles) accepting or describing the degradation of polypropylene in variable conditions and the degradation in the body of other implantable polymers. They cited the Liebert, Imel, and Mary articles.
762 Third, they said they were unaware of any other peer-reviewed journal article supporting the notion that polypropylene does not degrade in the body.
763 Fourth, they contended that Thames et al failed to address the histological findings reported by other researchers. They included the appearance of surface cracking in fresh explanted meshes before formalin exposure, the birefringence (brightness under polarised light) of the degraded layer (the “bark”), the failure of the bark to stain for proteins, the ability of the degraded layer to melt and meld with the non-degraded core with surgical cautery, the presence of free particles of the “bark” in the tissue with associated foreign body response, and the entrapment of inflammatory cells and tissue matrix in the “bark” fissures, and the presence of blue granules in the bark layer.
764 Fifth, they contended that the study used “an unproven and unvalidated methodology”, apparently designed to remove from the surface of the mesh fibres “all detachable material” (including protein and degraded polypropylene) without discrimination. They criticised Thames et al for “fail[ing] to acknowledge that the aggressive cleaning, which included shaking and sonication for long periods, can have an abrasive effect capable of removing all detachable materials nonspecifically”. They stated that, while sonication (using ultrasonic waves to agitate particles in a liquid medium) is used to clean metals and jewels, to remove plaque during dental cleaning, to pulverise renal calculi, and as an industrial process for liquefying solids, it has not been reported as a proper method for evaluating degradation in a polymer.
765 Sixth, observing that Thames et al did not conduct a qualitative evaluation of the removed layer, they argued that a more rigorous protocol would have analysed the cleaning solution effluent for both degraded polypropylene and protein. They said that Thames et al merely assumed that the surface layer was a cross-linked protein on the basis that “the chemistry of formaldehyde fixation is well known”. Furthermore, the letter writers remarked, formalin affects all tissues, whereas the brittle material is seen only at the surface of the polypropylene.
766 In short, they considered that the Thames study was incomplete, lacking in scientific rigor, and potentially biased.
767 Thames et al responded. They were obviously affronted by the criticism and regarded most, if not all, of it to be unjustified. They called into question the motives of their critics and took issue with much of what they had written.837 They did not deny, however, that their cleaning process was abrasive, unproven, unvalidated and had not been reported as a proper method of evaluating degradation in a polymer.
768 Moreover, the Thames analysis does not adequately explain Mr Burkley’s findings, particularly those in the 30 September 1987 memorandum. Associate Professor Guelcher considered Mr Burkley’s work much more rigorous and comprehensive, since he had directly probed the cracked outer layer and he had used multiple techniques to do so: calorimetry to measure melting temperatures, infrared spectroscopy to look, not only at the carbonyl peaks associated with polypropylene degradation but also the peak associated with the antioxidant.838
769 Having regard to Mr Burkley’s statement that Soluene treatment not only removes surface protein but also alters the oxidised polypropylene surface by removing lower molecular weight species of oxidised polypropylene with the protein, whatever the merits of the other criticisms of the Thames article by Thompson et al, the unorthodox cleaning process may well have removed lower molecular weight species of oxidised polypropylene with the protein.
770 Dr MacLean’s views echoed those expressed by Thames et al. This is perhaps unsurprising, considering that one of the people who carried out the work on his report was Dr Joshua White, a co-author of the Thames article.839 His opinion was summarised in the convenient executive summary to his report:
Although many of the applicants’ experts cited the widely reported in vivo PP degradation in the literature, careful examination of the test methods and results, however, indicates that many of these researchers have formulated their conclusions based on scientifically incomplete data and/or inadequate methods … [N]one of the studies performed on explanted mesh employed fully effective cleaning protocols leaving remnant biological matter, such as proteins and fatty acids, on the outer surface of the mesh fibers which can substantially confound the data. Furthermore, none of the authors utilize reliable quantitative methods such as molecular weight measurements of the explanted PP materials to confirm any appreciable PP degradation. Without ensuring that the adsorbed biological materials are entirely removed and without quantifying changes in molecular weight using reliable scientific techniques, any opinions that claim PROLENE is suffering from in vivo oxidative degradation are without merit.840
771 Dr MacLean contended that thorough cleaning of mesh materials conclusively proves that the cracks seen on explanted fibres are not the product of oxidation but are “associated with chemically fixed, adsorbed biological materials”. He claimed that his findings are consistent with Ethicon’s dog study “in which material degradation was conclusively ruled out and PROLENE’s ductility and compliance were maintained or improved after seven years of implantation”.
772 Exponent, the company for whom Dr MacLean works, has had a commercial relationship with Ethicon going back a number of years. Two other Exponent employees, Kevin Ong and Joshua White, were co-authors of the Thames study, which, as I have already observed was financed by Ethicon and is the only study Dr MacLean cited to deny that polypropylene meshes are subject to oxidative degradation.
773 Like Professor Iakovlev, Professor Klosterhalfen said that over years of using scanning electron microscopy he had observed in polypropylene meshes cracking and “bark-like” features which he attributed to oxidative degradation at the surface of the polymer.841 He disagreed that these were features of an organic material because, he explained, under scanning electron microscopy protein layers show a different surface morphology than do cracks in polypropylene fibres and using H&E (haematoxylin, which turns the section blue or violet, and eosin, which turns it red or pink) histology under polarised light, the birefringence of the “bark” is very distinct. He said that he had never observed such a pronounced birefringence in other proteins or soft and inflammatory tissues in the human body. Consequently, he found it hard to believe that the “bark” has a natural source.842 Professor Klosterhalfen’s view was that the presence of the “bark” or the surface cracking or degradation could stimulate the chronic inflammatory response which, in turn, could lead to the production of more scar tissue encapsulating the mesh.843 While he admitted in cross-examination that his knowledge of the effects of oxidation was derived from his reading of the literature, I do not think that this concession affects this evidence. I am satisfied that, as a very experienced pathologist with substantial expertise in the response of tissue to polypropylene mesh, he is able to distinguish organic from non-organic materials and opine about the causes and effects of the chronic inflammatory response.
774 The respondents also relied on Ethicon’s seven-year dog study to support their contention that Prolene does not undergo oxidative degradation. Dr MacLean gave evidence to this effect. He relied on the fact that no loss of molecular weight had been detected. There are problems with this evidence.
775 First, unlike the explants examined by Dr Burkley, the outer cracked surface layer of these explants was not tested. That may explain why no loss of molecular weight was detected.
776 Second, the protocol for the study844 was not followed. At the two year mark the crosshead distraction speed was increased from 5 inches per minute845 to 10 inches per minute.846 Moreover, a crosshead speed of 1 inch per minute was used for the Prolene samples and 5 inches for all other products.847 No explanation was given.
777 Third, it is not true that “material degradation was conclusively ruled out” by the study, as Dr MacLean asserted.
778 To the contrary, the study supports the conclusion that Prolene undergoes oxidative degradation in vivo.
779 Taking into account all the evidence, I am persuaded on the balance of probabilities that polypropylene, including Prolene, does undergo oxidation in vivo and that the antioxidants added to the polypropylene during the manufacturing process, do not provide permanent protection from the risk of degradation.
Did the applicants prove that oxidation of Prolene had any significant effect?
780 This question is more difficult to answer.
781 In his evidence in chief Professor Iakovlev said that the main feature of oxidative degradation of polypropylene was cracking of the polymer surface. He said that similar cracking occurs when polypropylene degrades in variable environments outside the body. He said that the material loses tensile strength and becomes brittle and that loss of tensile strength was shown in explanted polypropylene (citing the Liebert study). He also asserted that degradation leads to embrittlement of the entire surface of the mesh adding to the stiffness caused by the ingrowth of scar tissue, increasing “the mismatch between the tissues and the mesh” and hence the chance of injury to the tissues. He said that degradation invariably produces chemical products. In vitro thermal degradation of polypropylene produces an array of organic molecules and, although the conditions of thermal degradation do not match those of the body, “[i]t can be assumed that any combination of the degradation products detected during thermal or other types of degradation can be produced during degradation in the tissue”. He also said that the irregular cracked surface provides a larger area in which bacteria could shelter without being susceptible to attack from macrophages because they were too small to allow the macrophages to enter.848
782 The respondents took issue with all of these propositions. I have dealt with their arguments about the causes of the cracking above. On the other matters they submitted as follows.849
783 While Mary et al and Iakovlev, Guelcher and Bendavid reported brittleness, they did not test for brittleness; they merely inferred brittleness from the existence of cracking. While Professor Iakovlev said that embrittlement makes the mesh fibres stiffer, he is not a material scientist and has not conducted any physical testing of polypropylene or any testing for brittleness. Associate Professor Guelcher, who is a materials scientist, did not say that embrittlement makes the fibre stiffer (although he did say that it changed the surface and over time the surface effect could progress into the fibre) and he had not conducted any test to see whether an oxidised polypropylene fibre becomes stiffer either on its own or when incorporated into a mesh.850 Indeed, he was unable to attribute any clinical consequence to embrittlement. Associate Professor Guelcher, of course, is not a pathologist. Similarly, the effect of embrittlement of the mesh in the body was admittedly outside Dr Dunn’s expertise. Professor Iakovlev conceded that there was nothing in the literature he had seen to indicate that the problems with the mesh proceeded from a lack of strength, even years after implantation. He also revealed that it was a hypothesis that oxidation causes complications in patients851 and that he was unable to see how a loss of tensile strength would contribute to the complications he had seen.852
784 I note, too, that Professor Klosterhalfen testified that the difficulties with the performance of Ethicon’s meshes in vivo did not seem to have proceeded from failures under load and are not connected to the tensile strength of the mesh.853
785 In short, on the assumption that oxidative degradation occurs in vivo, the respondents submitted that the applicants’ case that it is clinically significant has not been proven.
786 Having considered all the evidence on this subject, for the reasons given above I am satisfied that it is more likely than not that Prolene does undergo oxidation in vivo when the antioxidants are depleted and that this is reflected in the surface cracking detected by Professor Kosterhalfen and Dr Iakovlev and others using scanning electron microscopy. I am also satisfied on the balance of probabilities that when this occurs it may lead to an increase in inflammation and scar tissue formation. But while the hypothesis that oxidation causes complications in patients who have been implanted with Ethicon meshes may be reasonable, I am not satisfied that it has been proven. Certainly, the evidence in this case does not establish to the requisite standard that when Prolene does oxidise in vivo it has any clinically significant effect and, in particular, that it causes a significant reduction in the tensile strength of the polymer. I accept the respondents’ submissions in this regard. The fact that the results of the Ethicon dog studies are open to question does not establish the opposite of those results.
787 The answers to the questions in dispute are as follows.
(1) The changes to pore sizes in vivo are significant.
(2) The Amid system of mesh classification is of limited utility, certainly in relation to meshes in the pelvic floor, although many of the observations made by Professor Amid remain important.
(3) The data derived from the Aachen Group’s pelvis data pool is not unreliable. In any case, it was only one resource from which the opinions of Professors Klosterhalfen and Klinge were derived.
(4) Bridging fibrosis can occur with any of the Ethicon devices and is of clinical significance.
(5) Mesh contraction is of clinical significance. It can cause complications such as mesh exposure/erosion and chronic pain, including at rest and with sexual intercourse.
(6) Since it is uncontentious that all the Ethicon devices may cause pain, including chronic pain, and that there are various mechanisms that may be responsible for it, it is unnecessary to choose between them.
(7) Prolene undergoes oxidative degradation in vivo but the evidence is not sufficient to demonstrate that the degradation is clinically significant.
PART VI: THE PERFORMANCE OF THE DEVICES
788 In Part V I discussed the problems with polypropylene mesh identified by the Aachen group. In summary, they found evidence of chronic inflammation, infection, erosion, contraction, and chronic pain after implantation of all the Ethicon devices and concluded that both Prolene and Prolene Soft were unsuitable for use in the female pelvis.
789 In this Part of the judgment I deal with the evidence from other sources regarding the performance of the devices, including the scientific literature. I was taken to a vast amount of material and an even greater amount was tendered. While I have had regard to all the articles to which I was referred, it is unnecessary to mention them all. The medical journals contained reports of studies performed on various kinds of mesh products, not all of which were made from polypropylene, let alone by Ethicon. Some of those studies involved small numbers of patients, often with short periods of follow-up. Some compared various kinds of mesh with each other. Many were single arm studies. The most reliable evidence is likely to come from a consideration of randomised controlled trials (RCTs), provided, of course, that a sufficient number of trials of “adequate quality” have been conducted.854
790 Before turning to the studies, however, I need to address some of the evidence of the epidemiologists and biostatisticians, which assists in their interpretation.
The epidemiological and statistical evidence
791 Epidemiology is the study of the occurrence, distribution and potential causes of events, including disease, over human populations. Epidemiologists may study the prevalence and incidence of a particular condition. They may also study potential risk factors. The most challenging aspect of epidemiology is determining causation.855 Biostatistics is the application of statistical principles and methods in biological research, principally, but not exclusively, in human biology and in medical contexts in particular.856
792 The evidence given by the epidemiologists and biostatisticians called by the applicants served two main purposes. The first was to assist the Court to interpret the studies on which the parties relied and to contextualise their findings. The second was to provide opinions on the safety and efficacy of surgical treatment using the Ethicon devices, including in comparison with alternative forms of surgical treatment that do not use synthetic mesh.
The qualifications of the witnesses
793 All these witnesses were well qualified to express the opinions they did.
794 Professor Hu is both an epidemiologist and physician. He described himself as “a physician-scientist-academic”, with degrees in medicine and science, and “an environmental, molecular and clinical epidemiologist” with expertise in leading multi-institutional and multi-national teams that create and conduct prospective epidemiological cohort studies to determine adverse health outcomes.857 Professor Hu was concurrently the founding dean of the School of Public Health at the University of Toronto, Professor in the School of Medicine at the same university, and Adjunct Professor in the Department of Environmental Health Sciences at the University of Michigan’s School of Public Health, holding all three positions at the time he prepared his report.
795 Associate Professor Krulewitch has been an epidemiologist for over 20 years. Previously, she worked as a nurse for over 30 years. She graduated from the University of Illinois with a Master of Science degree in 1984 and acquired a PhD in perinatal epidemiology from the University of Maryland in 1992. At the time of her first report in August 2016 she held an academic appointment in the School of Medicine at the Uniformed Services University of the Health Sciences in Bethesda, Maryland, where she teaches biostatistics to medical students and participates in research. She also has expertise in regulatory affairs which she drew on to express other opinions. In particular, from 2007 until 2012 she was employed by the FDA in the Divisions of Post-Market Surveillance and Epidemiology in the Center for Devices and Radiological Health. As a Branch Chief in the Division of Post-Market Surveillance, Associate Professor Krulewitch also oversaw the evaluation of reports of adverse events (known as “medical device reports”) including with respect surgical mesh. I discuss her evidence regarding Ethicon’s post-market surveillance in Part IX below.
796 In the Division of Epidemiology, she was the principal investigator on a number of studies of medical devices including urogynaecological mesh used in the treatment of stress urinary incontinence and pelvic organ prolapse. As Branch Chief, she led teams who conducted systematic reviews on urogynaecological mesh, which included premarket medical reviews. She participated in the writing of the FDA Safety Communications issued in 2008 and 2011 discussed in Part X below.858
797 Associate Professor Krulewitch gave unchallenged evidence that she has a thorough understanding of stress urinary incontinence, including the anatomy and physiology of the urogenital tract, and has the knowledge to collaborate with physicians and surgeons about surgical options to treat the condition. She also gave unchallenged evidence that her training in perinatal epidemiology gave her the tools to identify, evaluate, analyse and synthesise the relevant literature and to draw conclusions on the effectiveness and relative safety of different treatment options and the relationship between treatments and adverse outcomes.859 She has both designed and evaluated epidemiological studies.860
798 Professor Woodward is an eminent epidemiologist and biostatistician. At the time of his first report in August 2016 he was a Professor of Statistics and Epidemiology at The George Institute for Global Health at the University of Oxford, Adjunct Professor of Epidemiology at Johns Hopkins University in Baltimore, and Professor of Biostatistics at The George Institute for Global Health at Sydney University.861 Early in his career he worked on the design and analysis of a number of clinical trials on behalf of several pharmaceutical companies. While Professor of Medicine at New York University, based at the Mount Sinai School of Medicine where he was head of the Biostatistics Core, he helped to design and analyse clinical trials.862
799 At The George Institute, Professor Woodward served on the management committees of two large randomised controlled trials of cardiovascular disease mainly funded by a drug company. At the time of his first report, he was also, amongst other things, a senior statistician on two global collaborations of randomised controlled trials run by the Institute of certain treatments relating to the control of blood pressure and glucose as well as continuing large-scale trials in kidney disease and stroke. He is active in the practice of pooling data from multiple studies to reach overall conclusions, a practice known as meta-analysis. At the time of his first report he was an author of 518 peer-reviewed academic publications, the vast majority of which related to epidemiology or randomised clinical trials.863
800 Professor Gordon is a biostatistician. Since 1988 he has been attached to the Statistical Consulting Centre at the University of Melbourne. Since 1992 he has been its director.864 He has had substantial experience in the design and analysis of randomised controlled trials in diverse areas of medical research and has carried out many meta-analyses in order to answer research questions.865
801 The respondents submitted that, apart from providing a primer on the topic of epidemiology, the evidence of three epidemiologists and biostatisticians adduced by the applicants was of limited assistance to the Court.866 None of the experts, they argued, had experience or qualifications in pelvic surgery or had contributed to the published scientific literature in that field, and neither Professor Woodward nor Professor Hu, who were retained to give opinions about the POP devices, addressed their opinions to each of those devices (Gynemesh PS, Prolift, Prolift+M, and Prosima).867 The first proposition must be accepted. Moreover, it is true that neither Professor Hu nor Professor Woodward addressed their opinions to each of the four POP devices. Nevertheless, they both considered studies that related to those devices and other evidence that did so.
802 The respondents submitted that Professor Hu “did not even appreciate that abdominal sacrocolpopexy involved polypropylene”. He conceded as much in cross-examination.868 This proved to be a problem of recollection, however, rather than understanding. There are numerous references in his reports to the use of polypropylene, specifically Ethicon’s polypropylene mesh, in abdominal sacrocolpopexy.
803 The respondents also submitted that these witnesses did not consider the clinical significance of the pleaded complications.869 That is true, too, but only up to a point. Their conclusions were informed by studies conducted by pelvic floor surgeons in which clinical significance was no doubt taken into account.
804 The evidence of the epidemiologists and biostatisticians was of greater utility than the respondents countenanced. Not only did these witnesses assist the Court to navigate the myriad of studies upon which witnesses and the parties relied, but they also shed light on the terms and techniques used to report on them. Furthermore, they assisted the Court in the difficult task of evaluating the weight to be attached to the findings of, and conclusions reached in, those studies. I say that notwithstanding Professor Gordon’s statement that “[h]ow these findings should be weighed is really a matter for a clinical expert”.870 No such statement was made by the other biostatisticians and epidemiologists. Nor was such a proposition put to them. In any case, Professor Gordon’s statement needs to be read in context. I explain why when I deal with Professor Gordon’s evidence below.
805 It was apparent that witnesses who were pelvic floor surgeons but who lacked epidemiological or statistical expertise placed weight on study findings which were not statistically significant or which drew heavily on studies that were affected by one or more kinds of bias, without acknowledging the potential for bias. The evidence of the epidemiologists and biostatisticians also highlighted other weaknesses in the studies.
806 I therefore reject the respondents’ submission that, in the light of Professor Gordon’s statement, the Court should (invariably) prefer the evidence of pelvic surgeons to his evidence or that of the other epidemiologists and biostatisticians in assessing how the published literature should be interpreted and applied.871 Of course, there will be some matters where the evidence of the pelvic surgeons and, indeed, the evidence of other witnesses would carry greater weight. Professor Hu, for example, said that there was uncertainty about the mechanism through which mesh could directly lead to leg weakness and/or chronic pain.872 The evidence of the urogynaecologists and the pathologists, however, served to fill the gap.
Some general observations about assessing complication rates
807 As I have already noted, Dr Hinoul maintained that some of the complications were “rare”. For others he described the incidence as “low”. He did not, however, explain the basis upon which these adjectives were chosen.
808 Dr Agur included in an appendix to his report an explanation of rates drawn from advice given by the Royal College of Obstetricians and Gynaecologists (RCOG).873
Term | Number of people | Size of group/area |
Very common | 1/1 to 1/10 | One person in a family |
Common | 1/10 to 1/100 | One person in a street |
Uncommon | 1/100 to 1/1000 | One person in a village |
Rare | 1/1000 to 1/10,000 | One person in a small town |
Very rare | 1/10,000 and above | One person in a large town |
809 In the absence of anything more authoritative, this is the guide I propose to follow. When the descriptions of terms and incidence rates were put to him in cross-examination, the respondent’s pathologist, Professor Wright, said he agreed with them.874 The only other witness who was asked about them was Associate Professor Lam. He said he used different figures in his own practice. He said that he tells patients “common” is more often than 1 in 10, rare is less than 1 in 100, and uncommon is between 1 in 10 and 1 in 100.875 But he did not explain his rationale and cited no authority or professional guideline. Importantly, the range he characterised as “uncommon” was designated as “common” by the RCOG.
810 By the RCOG measure, and while the rates vary from study to study, many of the pleaded complications are not rare. In July 2011 the FDA issued a Safety Communication, the stated purpose of which was to inform health care providers and patients alike that “serious complications associated with surgical mesh for transvaginal repair of POP are not rare” (original emphasis).876
811 In his report, Associate Professor Lam said that infection and chronic pain are very uncommon following Prolift surgery.877 In cross-examination, however, he explained that the statement was based on his own experience rather than the medical literature.878
812 Withagen et al (2011), on the other hand, noted that dyspareunia rates of up to 38% had been reported after vaginal mesh placement and that de novo pain rates occurs in between 3% and 10% of cases.879 Their own study reported significant rates of pain.
813 Between September 2005 and December 2009, Withagen et al (2011) undertook a prospective observational cohort study of 374 patients who had undergone surgery for pelvic organ prolapse using Prolift at two Dutch centres specialising in pelvic organ dysfunction. Follow-up visits were planned at six weeks, six months, and 12 months. The study notably excluded women who had a compromised immune system and no simultaneous hysterectomies were performed or T-incisions made. The stated purpose of excluding these women was to reduce the risk of mesh exposure known to occur when those procedures were carried out.880 All patients received perioperative antibiotic prophylaxis.881 Even so, there were numerous complications including “rectal lesion, bladder injury, blood loss more than 500 mL, infection, hematoma, urinary retention (defined as repeated postvoid residual volume more than 100 mL measured with a bladder scanner and needing an indwelling Foley catheter or intermittent catheterization), reintervention attributable to complication, granuloma, exposure, pelvic abscess, rectovaginal or vesicovaginal fistula [referring respectively to an abnormal opening connecting the rectum and the vagina and the bladder and the vagina], pain, and dyspareunia”.882 At 12 months, 294 patients (79%) were available for follow-up. Mesh exposure occurred in 34 (12%) at six weeks, 17 (6%) at six months and 11 (4%) at 12 months. Twenty-three (68%) resolved after therapy. De novo lower abdominal or genital pain was noted in 5% of cases,883 which the authors noted was consistent with earlier reports.884 De novo dyspareunia was noted in 20 out of 78 patients (26%).885
814 These figures may well have been understated. Professor Woodward observed that Withagen et al (2011) has the dubious distinction of being the only one of the 37 studies reviewed by the Maher et al Cochrane review (2016)886 to have been assessed as having a “high risk of bias” in more than four categories: selection bias (allocation concealment), performance bias (blinding of participants and personnel); detection bias (blinding of outcome assessments), reporting bias (selective reporting), and other bias. It was unclear whether there was a risk of attrition bias (incomplete outcomes).887 No-one was blinded — not participants, not personnel, not reviewers.888
815 In their review of the English-language scientific literature published up to January 2012, de Tayrac and Sentilhes (2013) found that pain associated with mesh surgery for pelvic organ prolapse was reported at a rate of between 4% and 11% of patients.889 Associate Professor Lam agreed that that rate could not be described as “very uncommon”.890 The review covered articles which did not deal exclusively with Ethicon products but the authors concluded that the use of mesh for pelvic reconstructive surgery is associated with a risk of specific complications regardless of the surgical approach or the type of mesh.891 Furthermore, the study by Withagen et al (2011) related to Prolift and the figure for de novo pain in that study was 5%, which Associate Professor Lam conceded is not uncommon.892
816 De Tayrac and Sentilhes (2013) also found that, despite a decreased rate of anatomical recurrences with mesh, there was an increased total reoperation rate of 8.5% after transvaginal mesh surgery compared to 3.2% after surgical procedures using native tissues. Moreover, de novo apical or posterior compartment prolapse surgery was nearly twice as high after anterior compartment polypropylene mesh repair (17.7%) than after native tissue repair (9.5%).893
817 The evidence indicates that complication rates for the POP devices (used transvaginally) are significantly higher than for the SUI devices and abdominal placement of Gynemesh PS (in sacrocolpopexy).
818 In his first report Professor Korda said that the mesh erosion rate in apical repairs using the transvaginal procedures is at least 15% whereas in sacrocolpopexy it is less than 2 to 3%.894 He also said that there is a higher rate of fistulas after transvaginal mesh for vault prolapse. What is more, he stated that transvaginal mesh repair offers a poorer outcome than abdominal sacrocolpopexy with a lower anatomical success rate, lower patient satisfaction rate and shorter vaginal length.895
819 The New Zealand Accident and Compensation Corporation reported that the rate of complications related to transvaginal mesh repair was five times higher when used in prolapse repair than in urinary incontinence.896
820 In his third report Professor Korda referred to a literature review by Diwadkar et al (2009) in which the incidence of mesh erosion after sacrocolpopexy was 2.2%, but in patients who had undergone surgery for apical prolapse using vaginal mesh kits (including Prolift) the rate was 5.8%.897 Professor Korda also referred to a 2016 Cochrane review by Maher et al in which mesh erosion after transvaginal mesh repair was reported to be between 7% and 18%,898 and to the report of the European Commission’s Scientific Committee on Emerging and Newly Identified Health Risks (commonly abbreviated to SCENIHR) in December 2015, which put the figures in the range of 4% to 19%.899
821 On the other hand, Associate Professor Rosamilia gave evidence that mesh exposure rates of 9% after sacrocolpopexy have been reported in two studies.900 The first was reported by Higgs et al (2005) and related to laparoscopic sacrocolpopexy. Their results were based a median follow-up period of five and a half years.901 The second was by Nygaard et al (2013), referred to as the CARE study, which was a randomised double-blinded controlled trial relating, amongst other things, to abdominal sacrocolpopexy.902 This study had a median follow-up of seven years.
822 The 9% figure given by Associate Professor Rosamilia was not true of the CARE study. Mesh erosion was reported seven years after implantation in 23 out of 215 patients or 11%.
823 The 9% figure given by Associate Professor Rosamilia was true on the face of things for the study reported by Higgs et al (2005). There were nine cases of erosion in 103 patients. Notably all mesh erosions in that study presented at least six months after surgery, some more than three years later.903 The authors noted that in four cases the mesh had been inserted by the vaginal route and in five after laparoscopic sacrocolpopexy. Since 20 cases were performed using vaginal mesh insertion and 83 using laparoscopic mesh insertion, the mesh erosion rate was 20% (4/20) when placed vaginally and 6% (5/83) when placed laparoscopically.
824 The higher rates in these two studies might well be attributable to the length of follow-up. In their conclusions, Nygaard et al (2013) emphasised the importance of comparative trials with long-term follow-up of at least five years.904
825 To the extent that there are differences in complication rates between transvaginal and transabdominal use of mesh in prolapse surgery, they might be explained, at least in part, by the fact that the amount of mesh used in prolapse surgery using the transvaginal route is far greater than that used in incontinence surgery and transabdominal prolapse surgery. Evidence given by a number of witnesses indicated that there is a relationship between the size of the mesh and the likelihood of complications. In other words, there is a dose response to polypropylene mesh. Professor Korda, for one, stated that the larger the area of the mesh, the higher the probability of the mesh bunching, of pelvic pain, and of mesh erosion.905 In addition, the SCENIHR Opinion issued in December stated that “the available evidence suggests a higher morbidity in treating [pelvic organ prolapse] which uses a much larger amount of mesh than [the amount used to treat stress urinary incontinence].906
826 Ethicon, itself, suggested as much in a confidential paper on the use of polypropylene mesh for pelvic floor repair focussing on mesh exposure dated 21 December 2011. The paper, prepared by Christophe Vailhé, Mathew Krever and Suzanne Landgrebe under the letterhead of Ethicon Women’s Health & Urology, identified the amount of permanent synthetic material left behind as one of the most important risk factors for mesh exposure. The authors stated that “Ethicon believes that the risk of exposure will increase with the amount of mesh implanted.”907
827 Dr Hinoul was of the same opinion. The following discussion appears in the CER for Prolift+M which he signed on 25 September 2012:
Reduction of the mass and the increase in the pore size of the mesh implant foreign body are seen to alter the inflammatory response which in turn is likely to alter tissue ingrowth and the resulting properties of the reinforced tissue. As the mass of a mesh implant is reduced and the pore size is increased, the surface area exposed to the host is reduced, and the foreign body reaction to the implant is reduced.908
828 Professor Korda explained it this way:
The larger the area of the mesh the more probability is there of bunching of the mesh, pelvic pain, and mesh erosion. A total Prolift for instance is a very large area of mesh with numerous arms for placement in the various anatomical canals. There is a large individual variation in vaginal capacity and mobility hence the mesh often needs to be trimmed and tension needs to be adjusted accordingly. This is clearly more difficult to achieve with a larger surface area.
Additionally, the size and location of the arms is likely to create further complications as the arms are often placed though the obturator foramen and are attached to the coccygeus sacrospinous ligament complex thereby increasing the possibility of causing damage to the sciatic nerve, pudendal nerve or obturator nerves.
The larger the fixation arms, the more likely they are going to cause pain.909
829 Assistant Professor Margolis said that, in general, mesh complications from prolapse repair tend to be more pronounced than those arising from the use of midurethral slings and attributed this to a dose-response relationship.910
830 Professor Deprest explained that the larger the total surface of contact between the host and the implant, the “heavier” the foreign body reaction.911
831 Ostergard (2010) wrote that devices with larger surface areas are associated with higher levels of erosion through tissue. Dr Ostergard said that this was probably because the larger surface area allows more bacteria to be harboured within the interstices of the mesh and on its surface.912
832 Following their multi-centre retrospective study on the evaluation and management of complications from synthetic mesh after pelvic reconstructive surgery, Abbott et al (2014) wrote that:
[S]everal trends were identified that suggested that the synthetic mesh that is used in the application of slings for the treatment of SUI has a more predictable and less severe course of complications compared with the synthetic mesh that is used for the management of POP. For instance, those patients whose index surgery involved a sling only were significantly less likely to experience an Accordion classification severity grade 4, which is a complication that requires a return to the operating room with general anesthesia, than were those women whose index surgery involved the use of TVM. Furthermore, complications after TVM tend to be more severe, are more chronic in nature, and can be more difficult to treat. For instance, mesh erosion, pelvic pain, dyspareunia, vaginal constriction, vaginal spotting, and obstructive defecation were all significantly more common after an index surgery with TVM than 1 with sling only. 913
833 In a more recent article, Chughtai et al (2016) reported on a large observational cohort study of women who underwent transvaginal repair with mesh for pelvic organ prolapse or stress urinary incontinence between 1 January 2008 and 31 December 2012 and followed them up until 31 December 2013.914 The study identified 41,604 women. The women were split into four groups. Between 2008 and 2012, 5,070 patients underwent transvaginal prolapse repair and were implanted with mesh plus a sling for stress urinary incontinence; another 3,798 patients received mesh without a concurrent sling; 10,484 patients underwent non-mesh repair for prolapse but with a concurrent sling for stress urinary incontinence; and 22,252 patients received a sling alone. While the manufacturers were not identified, it is reasonable to assume that at least a fair number of these devices were made by Ethicon. Chughtai et al (2016) found that the combined use of POP mesh and SUI mesh sling was associated with the highest erosion and repeated intervention risk, and the SUI mesh sling alone had the lowest erosion and repeated intervention risk. They concluded that there was a dose-response relationship between the amount of mesh used and subsequent mesh erosions and other complications, and invasive repeat surgery.
834 In addition, the size and location of the arms of the mesh kits can add to the complications. Professor Korda noted that the fixation arms of the mesh are inserted through the obturator foramen where there are nerves in very close proximity to the trocar915 and said that in these circumstances the obturator nerves can be damaged.916 He also observed that often the arms are placed through the obturator foramen and attached to the coccygeus sacrospinous ligament complex which increases the possibility of damage, not just to the obturator nerves, but also to the sciatic and pudendal nerves.917
835 Other factors may deleteriously affect the outcome of mesh surgery. They include surgeon error and inexperience, as well as host or patient related factors.
836 It is common ground that both the Ethicon devices had to be placed in position with just the right amount of tension. Bunching or folding of mesh, for example, which can lead to exposure or erosion, can be caused by inappropriate placement. Associate Professor Rosamilia appeared to regard inappropriate placement as the sole cause of the phenomenon.918 But the weight of the evidence indicates that this is not so. Bunching and folding may also occur as a result of the scar tissue contracting around the mesh.919
837 It was also common ground that the Prolift procedure required significant surgical expertise. Quite apart from the need to ensure that the mesh is implanted with the right amount of tension, deep dissection is required to implant it and the surgery must be performed with great precision in a bloodless field to reduce the prospect of mesh erosion or exposure.920 The risk of mesh erosion and exposure is reportedly significantly lower in experienced hands.921 But as counsel for the respondents conceded in closing argument, the evidence shows that, even in the most experienced hands, complications, including mesh erosion and exposure, will still occur. 922 In large part this is likely to be due to the nature and construction of the devices, the so-called clean contaminated environment in which they are designed to be placed, and to patient-related factors. As Professor Korda stated, despite every effort, it is not possible to create a completely sterile environment in the vagina and bacterial colonisation would exacerbate the inflammatory reaction to the mesh.923
838 The evidence as to rates is at best merely indicative. It is impossible, however, to determine the true extent of the risks or the level of complications. As Daniel Elliott of the Mayo Clinic wrote in a review article published in July 2012, “for various reasons, the true incidence of mesh complications is unknown and, most assuredly, underestimated currently”.924 The reasons he gave were reporting variations, under-reporting, short-term reporting, patient and physician ignorance, and delayed presentation by which he meant delayed onset of symptoms.
839 It was common ground that mesh-related adverse events may occur years after vaginal mesh surgery. Two of Professor Korda’s patients, for example, in whom he had implanted TVT, suffered late erosions into the vagina, one about eight years after surgery, the other about nine years later.925 Yet, where studies were conducted before a product was released to the market, they were relatively short-term (up to 12 months) and for much of the time in question there has been a paucity of long-term data in relation to all the Ethicon devices. Consequently, most of the published studies do not account for symptoms which arise from the longer term use of the devices they review. As Dr Elliot observed, in all likelihood “there are a large number of women around the world who have yet to develop problems but given enough time will.”926
840 Further, patients who experienced late onset symptoms might not have associated them with the implant surgery. Nor might surgeons in the absence of an appropriate warning from the manufacturer. Even Professor Korda, who was regarded by Ethicon as an opinion leader and was chosen by JJM as a preceptor (or trainer) for TVT, revealed that he was unaware of a number of potential complications of implantation of TVT and TVT-O as late as 2004, and for many years thereafter.927 Dr Hinoul conceded that it was possible that, if a doctor did not associate a complaint of chronic pain with TVT, he or she could be “quite resistant” to a suggestion from the patient that her pain was associated with the product.928 That was evident in correspondence from July 2013 from an unnamed surgeon to the NSW Health Care Complaints Commission. This doctor professed to be unaware of the possibility of persistent pelvic or groin pain from TVT and referred the Commission to a review of complications of anterior TVT placement in which there was no mention of persisting pelvic or groin pain.929
841 Moreover, surgeons tend to have a limited period of contact with their patients after surgery. Professor Korda said that, generally speaking, unless a patient had problems during the first twelve months, he would not see her again.930
842 Abbott et al (2014) reported on 347 patients who attended four US tertiary referral centres for the evaluation of mesh-related complications after surgery for stress urinary incontinence and/or pelvic organ prolapse between January 2006 and December 2010.931 Abbott et al pointed out that approximately half of the women who sought treatment of a mesh-related complication at a tertiary referral centre underwent their index procedure at a different facility and noted that the same trend had been reported in other studies. They remarked that “[t]his raises the potential concern that physicians who perform these mesh procedures may not be aware of the complications their patients experience and that these providers may be responsible for future mesh-related complications with no awareness of the existing magnitude of the issue”.932
843 Dr Hinoul acknowledged in cross-examination that under-reporting of adverse patient outcomes was a significant problem.933
844 The problem of under-reporting is well illustrated by the following email exchange between Ethicon personnel in May 2010.
845 On 7 May 2010, Jonathan Meek, the Worldwide Marketing Director for Ethicon Women’s Health & Urology, wrote to various others at Ethicon, including Dr Hinoul:
Many recent data points have identified how all surgeons talk about patient outcomes being critical yet few follow them up beyond ~6 weeks and even fewer (close to none) ask practical functional outcomes questions. It leads to 2 key business questions:
• Are they not asking because they cannot effectively deal with the answer and why?
• What are the realistic surgeon motivators for them to be focused on functional outcomes[?]
The answers will unlock an insight about why surgeons use devices without data and why our brand positioning on functional outcomes have been as useful as training wheels on a walking stick…934
846 On 8 May 2010 Dr David Robinson, the Medical Director of Ethicon Women’s Health & Urology, replied:
While some surgeon investigators are interested in collecting their own patient data, for most the aim is to diagnose, operate, assess the patient postop hoping for a minimum of issues to deal with so they can move on to the next case. I do not think they ask partly because the more they ask, the more likely they will, in fact, find something that has not met the patient expectations, thus requiring more time and effort with that patient, rather than moving on to the next. To some degree, it may also be that they don't know how to deal with it if there is a problem.
I hate to be so pessimistic but I believe docs for the most part, leave it to the patient to bring up outcomes (ie, the doc is the passive recipient rather than the active inquirer) and if the patient does not bring it up, then the assumption is all is well and they can move on. 935
847 Dr Aaron Kirkemo was in furious agreement:
I concur 100%. In my practice I would constantly see my partner's patients who ostensibly “were doing great”. The minute I started running my list of questions all sorts of issues popped up. “Don't ask don’t tell” definitely applies to most surgical practices that I have seen.
848 Dr Hinoul responded that the position was the same in Europe.
849 This attitude could lead to complacency on the part of surgeons and underscores the importance of good quality studies and full disclosure by the manufacturer.
850 There are also problems with the reporting of particular complications. Professor Hu considered that the standardised questionnaires, such as the POP-Q and the urogenital distress inventory, do not assess symptoms like chronic pain or groin or leg pain in a standardised way that would allow for a quantitative analysis. Consequently, he said, there was a risk that some complications are under-reported.936 Dr Hinoul testified that the quality of life questionnaire administered to patients after surgery can mask certain adverse events.937
851 Moreover, different kinds of studies have examined the effects of the various devices and the complication rates vary (sometimes wildly) from study to study. Not all studies are of the same quality. Many of them addressed efficacy only and those that did address safety were rarely long enough, even if they were large enough, to detect the true level of complications.
852 The quality of the studies can also be affected by bias. Bias in this context includes selective choice of study population or design:938 “self-selection bias” where a patient refuses an untried treatment;939 “indication bias” because of pre-existing or unrelated conditions;940 and “reporting bias”, such as where the investigator suppresses certain information that would go against her or his favoured treatment.941 If the surgeon performing the operation also determines its success, this too might raise a question of bias.942
853 Important prognostic factors might make an outcome more or less likely, regardless of the treatment. This is another form of bias. The example Professor Woodward gave was of intensive glucose control conducted amongst relatively old people and standard glucose control conducted amongst relatively young people. In this case, he said, there will be a bias in favour of standard control because heart disease tends to affect the older population. Randomisation can balance out the characteristics of subjects in the study groups, although whether this is actually achieved will often depend on the size of the trial.943
854 The “gold standard” for finding appropriate evidence when evaluating new therapies for clinical practice is the randomised double-blinded controlled clinical trial.944 “Randomised” in this context means that each patient in the trial has a known chance, usually an equal one, of being given each treatment but the treatment to be given is unpredictable.945 It is referred to as double-blinded because neither the researchers nor the subjects know who received the treatment under investigation and who the placebo.946 Associate Professor Rosamilia, however, considered that registries are a better source of data on complications because at least some randomised controlled trials are under-powered for complications.947
855 In 1997, in an article by Dr Trisha Greenhalgh, a senior lecturer in the Unit for Evidence-Based Practice and Policy at University College London Medical School published in the British Medical Journal, wrote that even the most rigorous attempt to achieve a comparative control group will be a wasted effort if the people who assess the outcomes know the group to which the patient they are assessing was allocated. She explained by way of example that if she knew that a patient had been randomised to an active drug to lower blood pressure and not a placebo, she might be more likely to recheck a reading which was surprisingly high. She described this as an example of “performance bias”.948
856 Professor Woodward said that many people would argue that the randomised control trial is the only completely valid way to infer a causal relationship. He explained that this is because randomisation ensures that the study is not biased against the intervention (where someone is exposed to a risk simply because she is different, on average, from others who are not so exposed).949
857 Professor Gordon said that it was “widely understood and accepted that judgments about the efficacy of a medical intervention in medicine must be based on well-designed randomised controlled trials”.950 Since 1991, for example, the British Medical Journal has had a policy of not publishing trials that have not been properly randomised save in exceptional cases.951 In explaining the policy, Professors Douglas Altman and John Martin Bland, both medical statisticians, wrote:
The simplest approach to evaluating a new treatment is to compare a single group of patients given the new treatment with a group previously treated with an alternative treatment. Usually such studies compare two consecutive series of patients in the same hospital(s). This approach is seriously flawed. Problems will arise from the mixture of retrospective and prospective studies, and we can never satisfactorily eliminate possible biases due to other factors (apart from treatment) that may have changed over time. Sacks et al compared trials of the same treatments in which randomised or historical controls were used and found a consistent tendency for historically controlled trials to yield more optimistic results than randomised trials. The use of historical controls can be justified only in tightly controlled situations of relatively rare conditions, such as in evaluating treatments for advanced cancer.
The need for contemporary controls is clear, but there are difficulties. If the clinician chooses which treatment to give each patient there will probably be differences in the clinical and demographic characteristics of the patients receiving the different treatments. Much the same will happen if patients choose their own treatment or if those who agree to have a treatment are compared with refusers. Similar problems arise when the different treatment groups are at different hospitals or under different consultants. Such systematic differences, termed bias, will lead to an overestimate or underestimate of the difference between treatments. Bias can be avoided by using random allocation.952
858 The authors acknowledged that randomisation may not be possible in certain situations. But in such cases, they wrote, the goal should be to retain all the methodological features of a well-conducted randomised trial, other than randomisation.
859 Professor Gordon was asked how he would describe the quality of evidence for a new intervention for a particular condition which was not based on data derived from randomised controlled trials. He replied:
I would regard it as – as poor, and in a certain sense alarmingly poor, and that’s why the Pharmaceutical Benefits Advisory Committee, for example, asks those who want to submit new treatments to them to support that from evidence from randomised trials.953
860 He acknowledged that there were exceptions, however:
I think that when a condition is extremely rare, so if you’ve got a condition which, I don’t know, 30 people are going to get it in Australia in a year or something and it’s a serious condition, it may not be feasible then to carry out large randomised trials because, you know, you mightn’t even have a pool of available people from all around the world and, therefore, you have to do what you can with more limited information. And similarly, you know, thinking about it logically, if you – if you’re introducing a new bandaid or something and it has got a different method of opening from the previous bandaid, then I’m prepared to concede one does not need randomised trials for that, and no doubt there’s a spectrum of possible conditions and interventions here, but I don’t know. I – I think it’s – it’s baffling if you’re introducing an intervention which is going to be placed in a person’s body, a woman’s body in this case, which clearly is, you know, significant in its – in its – in its nature and potentially is going to remain there for the – for the rest of the woman’s life, it seems to me that you really should be considering the best possible evidence before you do that, and the best possible evidence comes from randomised trials. 954
(Emphasis added)
861 Associate Professor Krulewitch said that RCTs may not be feasible because of costs or other reasons. When evaluating a surgical procedure, for example, she said that it is not possible to fully blind the treatment/surgery versus the control. In these cases, her evidence was that population-based studies, retrospective case reviews, systematic literature reviews, and cross-sectional studies can be considered, although they may be rated as a lower level of evidence.955
862 Population-based studies use population averages to infer results for individuals.956
863 There are also case-control studies. A case-control study compares subjects with a particular outcome (the cases) with subjects without this outcome (the controls) and then reviews them to see what features differed between the two groups before the outcomes are ascertained. Professor Woodward pointed out, however, that this kind of study is susceptible to bias due to “recall error” because the subjects (and their carers) have a greater motivation to remember or research into the factors that may have led to their condition.957
864 In some cases it is not possible to conduct an RCT for ethical reasons. The example Professor Woodward gave was a study of the relationship between smoking and lung cancer because it is unethical to randomise people to smoke.958 An observational study may be conducted instead. In this example a group of smokers and another group of non-smokers could be recruited (limiting membership of both groups to adults with no history of cancer), followed up for many years and the incidence of lung cancer in the two groups compared. This kind of study is called a “cohort study”. Professor Woodward said that it was more prone to bias than an RCT because other factors (known as confounding factors) might explain the results as much (if not more) than cigarette consumption.
865 Regardless, Professor Woodward said that it was an essential feature of the design of any principled study to include a control group, in order to put the results in context.959 He explained that causal relationships in a study could not be reliably investigated in the absence of a control group that is “matched in all important ways”.960
866 Since all types of study design are subject to statistical error (chance), Professor Woodward said that the results of any single study, including an RCT, should be treated with caution.961
867 A sensible researcher, Professor Woodward argued, would seek to put the result of any single RCT within a general context. An “excellent” way to do this, he wrote, is by a systematic review of evidence, which identifies the studies, extracts from them “the appropriate results”, and summarises them in an overview, called a “meta-analysis”. He said that deciding which studies to include and exclude requires consideration and typically involves subjective decisions about quality.962 Dr Gordon testified that the key thing to understand about a meta-analysis is that it is a serious attempt to combine the evidence from all relevant studies of the same question. He explained that it arose out of “marked dissatisfaction with … narrative reviews of evidence where quite often senior and learned scientists were perceived to produce prejudiced and biased summaries of evidence in a selective way by citing the – the studies that pointed to a particular conclusion that they wanted to support.”963
868 In determining whether a form of treatment is beneficial or the incidence of harm resulting from the treatment, the Oxford Centre for Evidence-Based Medicine rates as optimal (level 1) systematic review of randomised trials (meta-analyses) followed by a single randomised trial (level 2). Non-randomised controlled cohort or follow-up studies (post-marketing surveillance) rank next (level 3), but for long-term harms the duration of the follow-up must be sufficient to detect them. Below these, are case series, case-control studies or historically controlled studies (level 4) and “mechanism-based reasoning” (level 5). Levels may be graded down on the basis of matters such as study quality, imprecision, inconsistency between studies, or because the absolute effect size is very small. Levels may be graded up if there is a large or very large effect size.964 Effect size refers to the size of the difference between the groups being compared.965
869 Professor Gordon insisted that in medicine judgments about the efficiency of a medical intervention must be based on well-designed randomised controlled trials. As he explained in his evidence in chief:
So the problem in general terms is that when we compare the outcomes from a study that is not randomised, any difference that we see in the outcome could be due to the different effects of the interventions used, or it could be due to features of the two groups, and, in particular, if subjects have self-selected themselves into the groups or health practitioners, surgeons or physicians, doctors have determined the allocation of subjects into the groups, then there may be features of the patients which are going to lead to particular outcomes differentially between the two groups, regardless of the treatments used. So what you’re seeing when you have non-randomised studies is an unhealthy mix of possible causes, the one that’s upfront and obvious, perhaps, the two interventions used, but many others, to a varying degree of subtlety, that – that could be there, self-selection, as I said, being an important one, health practitioner allocation being another one, and not to mention other much more basic things that occasionally are overlooked such as age and comorbidities and all sorts of things. So that’s the whole point of a randomised trial. Randomised trials were introduced precisely to get around that problem and to – to deliberately construct a situation in which the only differences between the two groups, apart from the treatment, are chance differences, literally used a randomising device, and I don’t think it’s always appreciated how powerful that – that process is. Of course, it has to be implemented well. But it – it’s an extremely powerful process because it means that you then have a pool of subjects for your trial divided into two groups by purely random process, and, therefore, they will on average be the same with respect to all the things that are relevant to the outcome, whether known or unknown. So whether they’re things that you understand are causative in the process of leading to the outcome or not. Even if you don’t know about them, they will still be guaranteed to be balanced by a randomised trial, and that’s why in general in these situations one places a lot of weight on well-designed randomised control trials.966
870 Professor Gordon testified that the history of medical interventions was littered with the publication of case series in “the early process of the intervention[s]” which found them to be “really good or promising” and which were enthusiastically received but which were followed “in the fullness of time” by “proper randomised trials” which demonstrated that the effect was nowhere near as compelling as the case series suggested. In many such cases, he added, the interventions were abandoned because they did not work.967
871 It was common ground that the most respected source of meta-analysis is the Cochrane Collaboration. The Cochrane Collaboration (named after the British epidemiologist, Archie Cochrane) describes itself as “an independent, international non-profit organisation, dedicated to making up-to-date, accurate information about the effects of healthcare readily available worldwide”.968 It produces and disseminates systematic reviews of healthcare interventions and promotes the search for evidence in the form of clinical trials and other studies of interventions.969
872 Even so, the applicants invited the Court to make findings on the percentage risk of various complications based on individual case studies or single RCTs.
873 That is an invitation I decline to accept. For all the reasons given by Professor Woodward and discussed above, this would be a dangerous course to adopt.
874 The evidence indicates that many of the studies did not follow patients for a long enough period of time to reveal how effective or safe the treatment was in the long term, a particularly critical problem in the case of a medical device designed for permanent implantation. Even in the relatively few studies that did follow patients for five years or more, as time wore on increasing numbers of patients were lost to follow-up. The longer the period of follow-up, the larger the loss.970 The quality of a study will be affected by the way it deals with this problem.971
875 In her 1997 article, Dr Greenhalgh observed that people may be lost to follow-up for any one of a number of reasons, including a suspected adverse reaction to the product (even when the patient was given the placebo), loss of patient motivation, withdrawal at the clinician’s behest (due to concurrent illness or pregnancy for example), relocation of the patient, or death of the patient.972
876 Problems created by reliance on studies where the follow-up is incomplete or compromised were well described by Dr Greenhalgh. She explained that:
Subjects who withdraw from (“drop out of”) research studies are less likely to have taken their tablets as directed, more likely to have missed their interim checkups, and more likely to have experienced side effects when taking medication, than those who do not withdraw …
Simply ignoring everyone who has withdrawn from a clinical trial will bias the results, usually in favour of the intervention. It is, therefore, standard practice to analyse the results of comparative studies on an intention to treat basis. This means that all data on patients originally allocated to the intervention arm of the study—including those who withdrew before the trial finished, those who did not take their tablets, and even those who subsequently received the control intervention for whatever reason—should be analysed along with data on the patients who followed the protocol throughout. Conversely, withdrawals from the placebo arm of the study should be analysed with those who faithfully took their placebo.973
877 For all these reasons I would put more weight on the Cochrane meta-analyses than on individual studies. That said, a meta-analysis is only as good as the studies upon which it is based. As Catto et al (2017) observed, both systematic reviews and meta-analyses depend on the quality of the trials and “can flatter to deceive if the quality of the baseline evidence is low”.974
Studies relevant to complications arising from the SUI devices
878 A number of studies were tendered on complications arising from treatments for stress urinary incontinence, including several Cochrane reviews. Below I discuss relevant outcomes from these studies.
The Nordic multicentre studies
879 In 1998 a study was published on a prospective multi-centre observational trial involving the use of Ethicon’s Prolene tape in 131 patients at six Scandinavian centres in Uppsala, Danderyd, Falun, Helsinki, NÄL and Växjö. The main objectives of this study were to assess the safety and efficacy of the TVT procedure and to obtain the surgeons’ opinion of the procedure compared to previously used techniques.
880 The patients were followed-up for one year and the results were published in the International Urogynecology Journal in an article by Ulmsten et al (1998).975
881 The authors reported that 119 out of the 131 (91%) patients were cured (according to the protocol) and another nine (7%) were significantly improved. There were only three (2%) cases which were classed as failures. The majority of patients (about 90%) were operated upon “on a day-care basis”. No defect healing and no tape rejection occurred. Three patients needed indwelling catheterisation for three days, one for more than 10 days. There were two uncomplicated haematomas and one uncomplicated bladder perforation. Having regard to these results, and notwithstanding the absence of long-term data in a device intended for permanent implantation, the authors declared that “TVT can be considered a safe and effective procedure for surgical treatment of genuine female stress incontinence”.976 At the same time, however, they also recognised that “long-term follow-up studies have to be presented before any definitive conclusions can be made”. That qualification was omitted from the rosy picture painted in the abstract.
882 The article was accompanied by an editorial comment. That editorial comment noted that this was the first prospective study of the new procedure for the treatment of stress urinary incontinence. While the editors offered some plaudits to the authors, they added:
However, many questions remain regarding the diagnostic criteria they used in selecting their patients, as well as the degree of testing performed postoperatively. Further study is needed to confirm the results found here with a very new and interesting technique in the surgical treatment of stress incontinence.977
883 Five year results were published in the International Urogynecology Journal by Nilsson et al (2001).978 This was incorrectly referred to in the applicants’ submissions as the three-year follow-up.979 The study population consisted of 90 patients who were consecutively enrolled into a prospective multicentre trial in three Nordic centres, one in Finland (Helsinki University Central Hospital), two in Sweden (Danderyds Hospital and Uppsala University Hospital).
884 These results were recorded after a median follow-up of 56 months. Eighty-five out of the initial 90 patients were fully evaluated according to the protocol.
885 Seventy-two out of the 85 patients evaluated (84.7%) were stated to be completely cured, 9 (10.6%) “significantly improved”, and 4 (4.7%) were “failures”. Patients were regarded as cured if they had a negative stress test result, a negative 24-hour pad-weighing test result (<10 g/24 h), and if the QoL (or quality of life) had improved ≥90%. To be regarded as improved the patient had to have a ≥75% improved QoL and a significant reduction in urine loss as measured by the 24-hour pad-weighing test (>50% reduction or <15 g/24 h loss). All other patients were classified as failures, even if their preoperative situation had clearly improved.980
886 Nilsson et al (2001) stated that there were few complications of surgery. There were three cases with retropubic haematomas, one patient had a bladder perforation, and in three cases intraoperative bleeding of more than 200ml occurred. In addition, four patients had initial (less than four days) post-operative voiding difficulties; seven had urinary tract infections in the first two months; and there was one case of an infection of the operating site.
887 In terms of long term complications (at five years), Nilsson et al (2001) noted that two patients had asymptomatic urogenital prolapse (grade I) and one patient had recurrent urinary tract infections, but no sign of urinary retention. The authors concluded:
The tension-free vaginal tape procedure seems to fulfil the expectations of high long-term follow-up cure rates that the short-term results have suggested. Including both subjective and objective criteria for cure or significant improvement, 95% of the TVT-operated women benefited significantly from the operation. The operation, performed under local anesthesia, was well tolerated and was associated with minimal short- and long-term postoperative morbidity. In the light of these good long-term results the TVT operation can be recommended as a routine surgical procedure for the effective treatment of female stress urinary incontinence.981
888 For several reasons these results could not reasonably be accepted as indicative of the likely outcome for TVT patients in the general population.
889 As I noted above, Ulmsten et al (1998) published one year results on 131 patients enrolled in six centres in Scandinavia, which included the three centres to which the 90 referred to in the Nilsson et al (2001) article belonged.982 While it is far from clear, it appears to be common ground that the 90 were part of the original 131 patients at the six Nordic centres.983 Associate Professor Rosamilia was unable to explain what happened to the other 41 patients.984 The applicants suggested that they were women with detrusor instability and intrinsic urethral sphincter deficiency, who had been included in the original cohort, but were now excluded. Another possible explanation is that only three of the six Nordic centres participated in the longer term follow-ups. This explanation is however unlikely to be correct. The applicants took Associate Professor Rosamilia to a report of the one year findings.985 That report indicated that the total number of patients at the three centres to which the Nilsson et al (2001) article and those which followed it related was 60, not 90, and there were 71 at the other three Nordic centres, not 41. A final possibility is that the study reported by Nilsson et al (2001) was not the same study as the one in which the original 131 patients were enrolled.
890 On the limited material to which I was taken I am unable to decide which explanation should be preferred, although I am inclined to the view that the Nilsson et al (2001) article reported on a different cohort of patients than the original study. This was the study population followed through to 17 years. It matters little, however, since it is beyond doubt that Nilsson et al (2001) excluded women with detrusor instability and intrinsic urethral sphincter deficiency. That is a matter of significance.
891 Professor Blaivas adverted to the significance of the exclusion when he was asked to comment on the study by reference to the 17 year results:
Now, so far as we can tell from this study, (1), the rate of the complication erosion is very small?---In this very high – highly selective group of patient that had a lot of exclusion criteria in the very beginning of the methods. This was the best of the best patients being operated on, by the physicians that actually designed the surgery in the first place, and I don’t believe it’s representative of what we’ve seen in clinical practice.986
892 He explained in re-examination:
And could you first of all, when you said “best of the best” what did you actually mean?---Well, there are patients who are most likely to have a successful outcome.
And where is it in the materials and methods that you get that, so as to form that opinion?---Well, firstly they could have no prior incontinence surgery, and prior incontinence surgery is a risk factor for a negative outcome. They – they had to have a positive stress test and urodynamically proven stress incontinence with no detrusor overactivity, which means there couldn’t – there can’t be a misdiagnosis, which sometimes happens and there are many people – and even myself agree that patient [sic] with detrusor overactivity are at higher risk for failure. And then they could not have intrinsic sphincter deficiency which, in this report, is diagnosed by a maximum closure pressure of less than 20 centimetres, and that has a worse prognosis for stress incontinence surgery. 987
893 I accept this evidence. It is well supported by other evidence including evidence from one of the respondents’ own witnesses. In cross-examination, Associate Professor Rosamilia accepted that the patients with detrusor instability and intrinsic sphincter deficiency who were excluded were likely to constitute the most severe cases of stress urinary incontinence and the least likely to be successfully treated.988 She had earlier testified that intrinsic sphincter deficiency is “a subset of women” whose symptoms and urodynamic findings are more severe than other patients with stress urinary incontinence. She described these women as “a severe subset.”989 As the applicants submitted, their exclusion would inevitably produce better results.990 It also follows that the results of the study would not be representative of the population as a whole which would include the most severe cases, especially since TVT was specifically indicated for patients with intrinsic sphincter deficiency. All the instructions for use for TVT (and the other SUI devices) from 8 September 2000 onwards indicated that TVT was intended for use for “treatment of… female urinary incontinence resulting from urethral hypermobility and/or intrinsic sphincter deficiency”.991
894 Associate Professor Rosamilia ultimately agreed that the Nordic studies only showed long-term efficacy for patients without both intrinsic deficiency and detrusor instability.992 She said it was not until 2008 or 2009 that data became available supporting the use of the procedure in cases of intrinsic sphincter deficiency.993
895 It should also be noted that all surgeons involved in this study were described as “experienced urogynecologists well trained in TVT surgery”.994 That circumstance, too, would be likely to guarantee better results.
896 The seven year results were published in the journal Obstetrics & Gynecology in December 2004.995 Assessment variables included a 24-hour pad weighing test, a stress test, visual analogue scale for assessing the degree of “bother”, and a questionnaire assessing the subjective perception of the women on their continence status.
897 Of the 90 women who had enrolled in the trial, 80 were available for follow-up: three had died (allegedly for reasons unconnected with the treatment), six were too disabled to be evaluated, and one was unaccounted for. Only 64 were available for objective evaluation. Both objective and subjective cure rates in the women followed up were reported to be 81.3%. Asymptomatic pelvic organ prolapse was found in five out of the 64 women (7.8%) clinically evaluated, de novo urge symptoms were reported by five out of 80 women (6.3%) interviewed, and recurrent urinary tract infections were reported by six out of the 80 (7.5%). No other long-term adverse effects of the procedure were detected. No signs were detected of tape erosion or any tissue reactions indicating rejection of the tape. The authors concluded from the absence of long-term adverse events associated with the TVT procedure and the high subjective and objective cure rates that the TVT operation was “a recommendable surgical treatment for female stress urinary incontinence.”
898 In their discussion of the results, the authors observed:
The lower the rate of those lost to follow-up, the more reliably cure rates reflect the true durability of effectiveness of treatment in a certain population. Therefore, a cure rate of more than 80% in a population such as the present one has to be regarded as reassuring. Long-term cure rates of around 70–80% have been reported for the open Burch colposuspension operation, regarded as the standard for traditional incontinence procedures.996
899 It appears, however, that the subjects lost to follow-up were ignored and the denominator used for their calculations was the number of women who were available to be examined, which skewed the results in favour of the intervention. This approach potentially overstates the benefits of the treatment since those or, at least some of those, women lost to follow-up could have been treatment failures.997 Having regard to the numbers lost to follow-up, Professor Gordon said that the study was biased because it “effectively evaluated ‘healthy survivors’ in the cohort”.
900 In 2008, the 11 year results were published in the International Urogynecology Journal.998 Assessment was once again undertaken through a 24-hour pad test, a stress test, a physical examination, and a visual analogue scale to assess the degree of “bother”. In addition, subjective continence status was assessed by patients’ global impression of improvement (PGII) as well as by condition-specific quality of life questionnaires.
901 Of the cohort of 90 women studied, 69 patients (constituting 77% of the total and 89% of those alive and capable of cooperating) were assessed. Median follow-up was 141 months after the TVT operation. Ninety percent of the women evaluated had a negative stress test and a negative pad test, being objectively cured. Subjective cure by PGII was found in 77%, with 20% regarding themselves as improved and 3% regarding the operation as a failure. No case of tape erosion or adverse tissue reaction caused by the tape was detected on vaginal examination.
902 The authors concluded:
We feel that the results of our long-term prospective follow-up of the effectiveness of the TVT operation are reassuring and that no distinct decline in cure rates occurs over a time period of 11 years. It is also encouraging to see that no late adverse effects of the polypropylene tape material was found and that erosion of the tape into adjacent tissues did not occur.
Results of the present kind should be able to achieve at any clinic where surgeons are offered systematic training and proper patient selection is undertaken.999
903 In 2013, 17 year results of the trial were published in the International Urogynecology Journal.1000 Of the cohort of 90 women, 74 could potentially be assessed (11 having died and five having such an impairment of their mental capacity that they were unable to cooperate). Of those 74 women, only 58 were evaluated: 46 in clinic; and 12 by phone.
904 In cross-examination, Associate Professor Rosamilia accepted that having only 46 out of a total of 90 patients available represented a “significant amount of patients lost to follow-up to be able to be reviewed in clinic”.1001 The applicants submitted that this cast doubt on the study’s findings in terms of long term cure since the cases lost to follow-up might have had negative outcomes and the long term cure rate might be below that reported by the authors. 1002
905 Objective cure at 17 years was seen in 42 out of the 46 women (91%) clinically examined. Women attending assessment in clinic also underwent a thorough pelvic examination, concentrating on the tape situation by careful inspection and palpation, observing signs of erosion, extrusion, fistulation, other tissue reactions and prolapse. The examinations and interviews were performed by independent doctors or nurses not involved in the early phases of the study. One woman had a “small symptom-free” exposure of the tape on the right side paraurethrally (alongside the urethra). 1003 As to exposure/extrusions, the authors made the following comment:
Age-induced atrophy of the vaginal mucosa exerts a risk of tape material extrusion into the vagina over time and can be treated or even avoided by local estrogen therapy. The single tape complication seen during the present prospective observational trial during a 17-year period was in a completely asymptomatic, continent and highly satisfied 69-year-old woman with an atrophic vaginal mucosa, who missed her 11-year follow-up visit, but had no tape exposure 7 years post-operatively. 1004
906 The authors noted that an “important observation” was that “there seem[ed] to be no shrinkage of the TVT mesh over time, as suggested by PVR [post-void residual] volumes [that is, the amount of urine left in the bladder after voiding] within normal ranges, except for two patients with concomitant diseases (Parkinson’s and grade III cystocele)”.1005
907 Nilsson et al (2013) concluded that the TVT operation was a “durable procedure”, with an efficacy lasting beyond 17 years, and with no serious long-term induced adverse effects. But since it was not a randomised controlled study, a large number of patients were lost to follow-up, and a significant population group was excluded, the conclusions are open to doubt.
908 Towards the end of their paper, Nilsson et al (2013) referred to the reported complications of the use of mesh in prolapse surgery:
The mesh complications seen in association with urogenital prolapse surgery that have alerted the FDA might not be caused by the mesh material itself. As long as a type I material is used the complications could be the result of improper training of the surgeon, resulting in an inappropriate surgical technique or choosing the wrong indication or wrong patient for the graft procedure.
The present report suggests that using a type I, macroporous, monofilament, lightweight, and soft polypropylene mesh, the risk of mesh complications even 17 years after implantation under the vaginal mucosa is negligible provided the surgery is performed by a trained and experienced surgeon.1006
909 In their concluding paragraph, they went on to say:
It is to be noted that the amount of implanted mesh with the TVT operation is small compared with the amount mostly used during urogenital prolapse surgery. This fact might make a difference to tissue performance and therefore we feel that mesh use for the treatment of urinary incontinence in the manner of a TVT operation is not associated with the problems seen in prolapse surgery. This observation could serve as a reason for a shift in the use of mesh for prolapse surgery from larger, covering meshes to smaller, tape-like suspending ones. On the other hand it might, however, be possible for larger meshes in prolapse surgery to perform as well as that in the TVT operation provided the surgical technique and the mesh material used are optimized and that the procedures are utilized with the right indications.1007
910 Those who endorsed the use of the SUI devices relied heavily on the findings of the Nordic studies, in particular the 17 year follow-up by Nilsson et al (2013). They included Ethicon in its clinical evaluations, discussed below. They also included the professional associations and expert witnesses called by the respondents in the present case. It is difficult to see how they were justified in doing so without taking into account the quality of the evidence, the size of the population, the exclusion criteria, the patients lost to follow-up, and the skill of the surgeons.
911 In cross-examination, Associate Professor Rosamilia conceded that Nilsson et al was “a poor review”1008 and, because of the number of patients lost to follow-up, it was difficult to draw reliable findings from the study.1009 These were significant concessions since this trial was one of the sources relied upon by the International Urogynecological Association for the assertion made in its July 2014 position statement, which Associate Professor Rosamilia said she had written, that studies had demonstrated long-term effectiveness.1010 The same position had been taken a few months earlier in March 2014 by the RANZCOG in its statement, with which Associate Professor Rosamilia had also been involved.1011
912 The weight of the evidence makes it tolerably clear that many, if not all, of the complications associated with incontinence surgery using polypropylene mesh, including all the SUI devices, were at least in part attributable to the mesh. Even Ethicon accepted as much. The Nilsson et al (2013) results were exceptional; the study’s findings could not be generalised to other patient and surgeon populations where the same exclusion criteria were not employed, less skilled surgeons were involved, and all other conditions were not equal. In any case the study population was a small one.
913 It will be recalled that TVT was released to the Australian market in October 1999. At the time, colposuspension was “the most popular choice for primary surgery and appeare[d] to be the most effective treatment for stress incontinence, with reported cure rates of up to 96%”.1012 The early evidence about TVT was encouraging. Reflecting on the matter in 2005, Ward and Hilton, two UK researchers, observed:
Data from three early case series, suggest objective cure rates of 84–100% up to two years. Few complications were reported. Immediate postoperative voiding difficulty was reported in 2–7%, and there was one bladder perforation and two retropubic haematomas, both treated conservatively. This evidence of efficacy led to rapid, widespread adoption of TVT before its long-term safety and effectiveness relative to existing procedures were known.1013
914 I do not understand the position to have been appreciably different in Australia.
915 Certainly no randomised controlled trials were performed on any of the SUI devices until the Ward Hilton RCT, which compared TVT surgery with colposuspension. The first results from that study were published in 2002 after only six months follow-up. This was three years after TVT was first supplied in Australia. Until that point, it was common ground that the studies were limited to preliminary and small case series with short-term follow-up. As the applicants submitted, the evidence supporting the introduction of TVT was largely confined to case series from the inventor and developer of TVT as part of the clinical trial tied to the sale of Medscand to Ethicon’s parent company, Johnson & Johnson International. The respondents argued, in effect, that the void was filled with the publication of the results from the Ward Hilton RCT.
916 This RCT was undertaken at 14 centres in the United Kingdom and the Republic of Ireland. Women with urodynamic stress incontinence unresponsive to pelvic floor muscle exercise, who had completed their family, were invited to participate. A total of 344 women were randomised, 175 to the tape arm and 169 to colposuspension.1014
917 The study was supported by Ethicon, which provided funding for the trial, materials and additional support to collaborating centres, and a grant to Dr Ward.1015 Dr Hinoul described it as “the landmark RCT that provided level 1 evidence to the medical community that TVT was set to be the new gold standard”.1016 Novarra et al (2007), in their meta-analysis of RCTs on midurethral slings, considered that it was “very well designed and performed.1017 The respondents’ urogynaecologists put great weight on its findings. Yet, while this was the first RCT comparing TVT to colposuspension, it did not provide level 1 evidence about the future of TVT.1018 Moreover, there were other issues which raise concerns about the reliability of its conclusions and the extent to which they or its findings could be generalised.
918 First, taking into account the criteria for levels of evidence set by the Oxford Centre for Evidence-Based Medicine, the fact that it was admittedly underpowered, and the number of patients lost to follow-up, Associate Professor Krulewitch described it as a low quality RCT.
919 Second, although the participants were randomly selected, neither they nor the investigators were blinded.1019 The investigators explained that it was not possible to blind either because of differences between the procedures in incision, anaesthetic, and catheterisation.1020
920 Third, there were numerous exclusions from the study. They included women with vaginal prolapse requiring treatment, women who had had previous surgery for incontinence or prolapse, women with neurologic disease, women with known bleeding diathesis or current anticoagulant therapy, women who were allergic to local anaesthetic, and women with detrusor overactivity or voiding difficulty.1021 Consequently, the findings of this study could not confidently or reliably be extended to patients with any of these conditions. Further, the generalisability of the study was limited by the fact that a substantial proportion of women in each group were on systemic hormone replacement therapy (34% in the tape group and 36% in the colposuspension group) but no analysis was undertaken based on that status.1022
921 It will be recalled that patients with detrusor overactivity were also excluded from the Nordic multicentre studies.
922 The exclusion criteria and the patients lost to follow-up potentially introduced a bias in favour of TVT.
923 The six month results of the trial were published on 13 July 2002 in the British Medical Journal.1023
924 The primary measure of cure rate was objective cure of stress incontinence based on negative stress test on urodynamic testing, combined with a negative one hour pad test. Objective cure was found in 115/175 (66%) patients in the TVT group and 97/169 (57%) in the colposuspension group (p=0.099).
925 “P” is a reference to “p-value”. It is necessary at this point to interrupt the narrative to explain the meaning and significance of the p-value for a given outcome.
926 Where the purpose of a scientific inquiry is to test a hypothesis in a comparative setting, the scientific method is to define a null hypothesis for the treatment being studied and then test it. The null hypothesis is that there is no difference between the groups being studied or no association between an exposure and an outcome (here, that there is no difference in effect between the two kinds of treatment). The p-value, as Professor Gordon explained it, is the probability (expressed as a figure between 0 and 1) of a result at least as extreme as that obtained, assuming the null hypothesis to be true.1024 In simpler terms, it is the probability that a difference as big as, or bigger than, that which was seen would have been seen if there really were no difference at all between the two groups.1025
927 A very small p-value indicates that the result obtained is very unlikely, assuming the null hypothesis to be true, and is therefore evidence against the null hypothesis.1026 Generally speaking, the p-value will be smaller when the sample sizes in the study are larger.1027 If the p-value (the chance of the observed finding) is less than 0.05 (5%), the conventional conclusion is that the null hypothesis is false. In other words, if the p-value is less than 0.05 (or the chance of that result if the treatment had no effect were less than 5%), the treatment has an effect. There is less than a 5% chance that the difference was random or due to chance.1028 If the p-value is greater than 0.05, the evidence is insufficient to refute the null hypothesis; the test is inconclusive. Put another way, an observed finding with a p-value of less than 0.05 is considered statistically significant; other findings are not.1029
928 The “power” of the study is the chance of rejecting the null hypothesis when the alternative (to the null) hypothesis is really false.1030 In other words, power is the probability that a study will find a statistically significant difference between interventions when an actual difference does exist. Low statistical power can thus interfere with the ability to identify a true difference between groups.
929 Consequently, a p-value of 0.099 indicates that at six months the difference in the objective cure rates achieved in the Ward Hilton RCT was not statistically significant.
930 Secondary outcome measures in the Ward Hilton RCT included subjective cure (measured by patient questionnaires and interviews) of incontinence and the development of voiding problems, urge symptoms and vaginal prolapse. The number of women reporting cure of stress leakage was 103 (59%) in the TVT group and 90 (53%) in the colposuspension group.1031
931 The investigators observed that the objective cure rates were lower than those previously reported, which they suspected might have been due to “the stricter definition of cure” they had used.
932 They acknowledged that the study was not adequately powered:
The number of patients recruited fell short of the target determined by a sample size calculation owing to limitations of resources and time. Although differences between the procedures for cure of stress incontinence were not shown, the numbers were not sufficient to achieve the power required to assume equivalence. Given that the objective cure for colposuspension was lower than expected from previous literature, the numbers recruited would give only 50% power to detect a 10% difference or 80% power to detect a 15% difference in cure rates. 1032
933 The applicants emphasised the inadequate power of the study, as this meant that the study did not determine a clinically significant difference between the two treatment arms.1033 It certainly affected the weight that could be attached to its findings and conclusions and the opinions of the witnesses who relied on them. When it was put to Associate Professor Rosamilia in cross-examination that the study was underpowered to detect a clinically significant difference between TVT and colposuspension as pre-set in the study protocol, she agreed but emphasised that the study was under-powered based on the authors’ own sample size calculation. She claimed that this did not mean necessarily mean it was “underpowered”.1034 She also stated that the power calculation was applicable only to the primary outcome (i.e. objective cure) but not to the secondary outcome measures.1035
934 Turning to those secondary outcome measures, Ward and Hilton reported that operative complications were more common after the TVT procedure, particularly injury to the bladder (15 patients (9%) in the TVT group and 3 patients (2%) in the colposuspension group, p=0.013), whereas colposuspension was associated with more post-operative complications and longer recovery. The authors commented that bladder injury during the TVT procedure “seems to be a relatively minor complication resulting from needle perforation”, with “no long term sequelae” observed.1036 Associate Professor Krulewitch criticised the manner of reporting this complication. She considered that the authors minimised the significance of the findings.1037
935 Retropubic haematoma, vascular injury (requiring laparotomy) and vaginal perforation only occurred with the TVT procedure but they were not statistically significant due to the small numbers. Associate Professor Krulewitch said that it was not clear whether a larger sample would show the incidence of these complications to be statistically significant.1038
936 There was also one case of tape erosion, managed by partial excision of the tape and closure of the vaginal skin. Reflecting on this, the authors observed:
In the five years since tension-free vaginal tape was first described there have been five reports of tape erosion into the urethra (Gynecare, personal communication, 2002). It is likely that this figure represents an underestimate, and long term follow up of these patients is needed to quantify the extent of this complication. 1039
937 The authors expressed the view that, in the short term, the TVT procedure was “as effective as colposuspension for the treatment of primary stress incontinence”, but noted long term follow-up was needed to assess the continuing success of the two procedures.1040
938 Two year results of the Ward Hilton RCT were published in 2004 in the American Journal of Obstetrics & Gynecology.1041
939 In their introductory remarks, the authors observed that “[e]vidence of efficacy from case series [had] led to rapid widespread adoption of TVT before its long-term safety and effectiveness relative to other procedures [were] known”.1042
940 The primary outcome measure at two years was cure of stress incontinence based on a negative one hour pad test (less than 1g change in weight). Secondary outcome measures included subjective cure and the development of voiding problems, urgency, and vaginal prolapse.
941 At 24 months, 137 women (out of the original 175) from the TVT group (79%) were followed up and 108 women (out of the original 169) from the colposuspension group (64%) were followed up. Of these, a negative 1-hour pad test was recorded in 111 (81%) patients in the TVT group and 86 (80%) in the colposuspension group.1043
942 There was an issue as to why a significantly higher number of women had dropped out of the colposuspension arm and how that matter should be addressed in analysing the results. The applicants pointed out that the patients in the colposuspension arm were described as having “milder incontinence”1044 and “less severe incontinence”.1045 They submitted that, because those undergoing colposuspension had a milder form of incontinence, they would have been easier to treat. Thus, the colposuspension cases lost to follow-up should have been treated as successes.1046 To some extent these submissions were supported by the authors themselves.
943 As to the measurement of the secondary outcomes, patients completed two questionnaires, one general (Short Form-36) and the other condition-specific (Bristol Female Lower Urinary Tract Symptoms or BFLUTS). Details are set out in tables II and III of the paper. The authors reported that 44 (25%) women in the TVT arm and 34 (20%) in the colposuspension arm reported no leakage under any circumstance after surgery. The number of women reporting “cure” of their stress leakage was 75 (43%) and 63 (37%), respectively.
944 The authors once again acknowledged that the recruitment numbers were lower than required and that the study was therefore underpowered.1047 They also acknowledged that their conclusions about cure were heavily dependent on the assumptions made about the participants who withdrew. At six months all withdrawals had been treated as failures. The authors now acknowledged that the differential dropout rate between the groups may have introduced a bias in favour of TVT. At two years a variety of assumptions were applied, three favouring TVT and only one favouring colposuspension. Nevertheless, the authors wrote:
[W]e must concur with a commentary on the 6-month article, that TVT may be better, worse, or the same as colposuspension in the cure of stress incontinence; this uncertainty remains at 2 years.1048
945 Despite this concession, the conclusion recorded in the abstract was that:
The TVT procedure appears to be as effective as colposuspension for the treatment of urodynamic stress incontinence at 2 years.1049
946 In cross-examination Associate Professor Rosamilia agreed, in effect, that, because of the numbers lost to follow-up and missing data, this conclusion was not available.1050
947 Five year results were presented in late 2006 at the 36th Annual Meeting of the International Continence Society in Christchurch, New Zealand1051 and published in the British Journal of Obstetrics and Gynaecology the following year.1052
948 The primary and secondary outcome measures were the same as the two year follow-up.
949 Ninety-eight out of the 175 women treated with TVT (56%) and 79 out of the 169 who underwent colposuspension (46.7%) returned for follow-up; 72 in the TVT group (41.1%) and 49 in the colposuspension group (28.9%) with full subjective and objective data. In cross-examination, Associate Professor Rosamilia accepted that this represented a significant loss to follow-up.1053 Ward and Hilton (2008) acknowledged that there was “presurgery withdrawal of a higher number of women in the colposuspension group who were shown to have less severe incontinence than those continuing in the trial”.1054 This was identified as a potential source of bias because it implied that the “data were not missing at random”.1055
950 A negative 1-hour pad test was recorded in 58/72 (81%) women in the TVT group and in 44/49 (90%) women in the colposuspension group. A sensitivity analysis was performed to explore the effect of different assumptions about withdrawals and missing data. No significant differences were seen between the procedures for any of these assumptions. A last observed result carried forward analysis was undertaken, in which pad test data from the last available follow-up visit were imputed for missing data. On this basis, the cure rates were 75% for TVT and 69% for colposuspension. Once again, however, the difference was not statistically significant.
951 Responses to the BFLUTS questionnaire disclosed that 38 out of 98 women (39%) in the TVT group and 36 out of 79 (46%) in the colposuspension group reported no leakage under any circumstances five years after surgery. The number of women reporting cure of stress leakage was 62 (63%) and 55 (70%) respectively. Overall, 91% of the women who had undergone TVT surgery and 90% who had undergone colposuspension regarded themselves as satisfied or very satisfied with the results of their surgery at five years.1056
952 There was a significant reduction in the number of women with cystocele in both groups five years after surgery. Enterocoele or vault/cervical prolapse and rectocele were found more commonly in the colposuspension group at five years.1057 More women in the colposuspension group (11 patients (7.5%)) underwent surgery for prolapse during the follow-up period than those in the TVT group (3 patients (1.8%)). The authors concluded that “it is likely … that colposuspension is indeed contributory to the development of vault and posterior wall prolapse, albeit in a group of women with known pelvic floor dysfunction.”1058
953 Altogether, tape-related complications were seen in six women. In the first year, one tape was “divided” for obstructed voiding, there was one suprapubic extrusion, and one vaginal erosion. Two further vaginal erosions were detected at five-year follow-up, and in addition, one woman was found to have tape within the bladder at cystoscopy after complaining of overactive bladder symptoms. There were no suture-related complications reported in the colposuspension group.
954 In their discussion of the erosions, Ward and Hilton (2008) commented:
It has been suggested that early vaginal erosion is the result of failure of vaginal skin healing rather than true erosion. In this study, there were two true vaginal erosions (only one of which was symptomatic) in the 72 women examined at 5 years.1059
955 Associate Professor Krulewitch observed that “this view discount[ed] the interaction of the tape with vaginal tissue and its effect on healing” and that “any erosion is a sign of a rejection of the tape through delayering of skin, inflammation or other autoimmune processes that prevent the proper healing of the surgical procedure”.1060
956 In any event, on the subject of erosion Ward and Hilton stressed the need for caution about relying on the figures:
Although these women suffered only minimal morbidity, it is important to be aware that synthetic sling materials have the potential to erode many years after implantation. The woman who was found to have tape within the bladder may have had an unrecognised perforation at the time of operation. It is possible that our figures represent an underestimate of tape complications and suture-related colposuspension complications as less than half the women who underwent surgery were examined at 5 years.1061
(Emphasis added)
957 The study detected no significant difference between TVT and colposuspension for the treatment of stress urinary incontinence at five years. The authors concluded that the effect of both procedures on cure of incontinence and improvement in quality of life and sexual health was maintained in the long term. They noted that vault and posterior vaginal wall prolapse were seen more commonly after colposuspension and that, although many tape-related complications may be missed operative injuries, tape erosion can occur several years after surgery.
958 Ward and Hilton recognised that the impact of the study was “somewhat limited by the high number of post-randomisation drop outs, both before and after surgery”.1062 While the authors considered it reasonable to infer that those who attended were a representative sample of the randomised group, they acknowledged that they could not be certain that the missing data was random in nature, leaving open the possibility that withdrawal or loss to follow-up was related to outcome. Thus, as before, conclusions regarding the effectiveness of the procedures drawn from the trial were said to be “heavily dependent on the handling of missing data and the assumptions made about those who withdrew”.1063
959 In cross-examination, Professor Blaivas acknowledged that the Ward Hilton RCT has “formed part of the landscape of information and discussion in the surgical community about the utility and the relative utility of these two operations”, being TVT and Burch colposuspension.1064 But he noted that the study did not ask about pain 1065 and stressed that one reason for women failing to return could be the occurrence of complications:
There are multiple papers that show that people who have severe complications often don’t go back to their treating doctor, so that’s a bit of evidence that – that’s something that I had, and that has been my experience that makes me suspect it. And so it’s – I know the difference between a speculation and a speculation armed with some evidence, and I would say it’s a speculation armed with some evidence, with some data to support it.1066
960 He also noted that:
Multiple studies have shown a prevalence of about 5% chronic pain after mesh slings, so with nearly half of the patients in the Ward Hilton (2008) series lost to follow-up at five years, [it] is not surprising that such patients might have been overlooked.1067
961 Many Cochrane reviews on incontinence treatments were tendered spanning several different topics. I refer to them elsewhere in these reasons where relevant. At this point I offer a brief overview of the more significant ones.
962 The earliest Cochrane review which looked at RCTs involving synthetic slings for the treatment of stress urinary incontinence appears to be the review by Dean et al in 2006,1068 which was updated in 2017.1069 Certainly, it is the earliest to which the parties referred.
963 Dean et al (2006) identified 22 eligible trials, 10 comparing laparoscopic (keyhole) surgery with open colposuspension and eight comparing laparoscopic colposuspension with “newer ‘self-fixing’ slings. It is unnecessary to refer to all the findings, since many of them are not relevant to the issues in the proceeding. For relevant purposes it is sufficient to note the following findings.
964 Both traditional colposuspension and the newer “self-fixing” slings had technically better results in the short term than laparoscopic colposuspension but the patients’ experience of improvement, both in the short and long term, were similar for each type of operation. Using two stitches in laparoscopic colposuspension was better than both the use of one suture and the use of mesh. The vaginal sling procedures were superior to the laparoscopic technique in that while there were no significant differences in the reported short and long-term subjective cure rates or postoperative voiding dysfunction and perioperative complications, objective cure rates at 18 months favoured slings and the duration of the surgery and hospitalisation in the case of laparoscopic colposuspension was significantly longer.
965 But the authors said that the value of the review was limited by the size and quality of the trials and the little data about long-term results.
966 The next review in sequence was Ogah et al (2009) in which, amongst other things, the authors analysed results from randomised and quasi-randomised trials comparing minimally invasive synthetic suburethral slings with traditional suburethral slings, open colposuspension, and laparoscopic procedures.1070
967 The applicants submitted that there were no trials comparing TVT with autologous fascial slings and, therefore, opinions that TVT was safer or more efficacious than autologous fascial slings were unsupported by evidence of RCTs.1071 They relied on Associate Professor Rosamilia’s evidence in cross-examination that “[t]here is no study that has compared TVT with fascial sling.”1072 But the evidence indicates that Associate Professor Rosamilia either misspoke or had something else in mind.
968 Ogah et al (2009) reviewed nine trials comparing minimally invasive synthetic suburethral slings with traditional suburethral slings.1073 Of those, at least five compared TVT with autologous fascial slings. Moreover, the meta-analysis by Novarra et al (2007), which is discussed below, reviewed three trials comparing TVT with autologous fascial slings.
969 Ogah et al (2009) concluded that in the short term minimally invasive slings were as effective as traditional suburethral slings and open, as well as laparoscopic, colposuspension, with cure rates of about 80%.1074 The authors also described these operations as “relatively safe” with shorter operating time and less post-operative voiding dysfunction and fewer symptoms of de novo urgency. At the same time, however, they described the quality of the evidence as “variable” and acknowledged that a major limitation of the review was the lack of long-term follow-up data.1075
970 Six years later, based on “low to moderate evidence”, Ford et al (2015) concluded that midurethral slings were highly effective in the short and medium term with accruing evidence of long-term effectiveness.1076
971 In their 2015 meta-analysis, Ford et al compared the results of randomised or “quasi-randomised” controlled trials where both trial arms involved an operation using a midurethral sling. Both single incision slings and other surgical procedures, such as traditional slings and colposuspension, were excluded.1077 The analysis was not limited to stress urinary incontinence but included mixed urinary incontinence. 1078
972 With the exception of a two-fold increase in the incidence of groin pain, the authors found that the transobturator tapes had fewer adverse events and that retropubic tapes had an eight-fold increase in the incidence of post-operative voiding difficulties. They also stated that the midurethral sling operations were the most extensively researched surgical treatment for SUI and had a “good safety profile”.1079 It should be noted however that the authors emphasised the need for reporting of longer-term outcome data from numerous existing trials.1080
973 Ford et al (2015) included 81 trials in their review, assessing the quality of the evidence as moderate, mainly because of the risk of bias or imprecision.1081 Moderate quality, I interpolate, meant that further research was likely to have an important impact on the confidence of the authors in the estimate of effect and could change the estimate. The authors said that there was moderate quality evidence of similar subjective cure rates for transobturator and retropubic devices in the short term (up to one year), ranging from 62% to 98% in the transobturator group and from 71% to 97% in the retropubic group. Short-term objective cure was also similar in both groups. In the long term (over five years), subjective cure rates fell, ranging from 43% to 92% in the transobturator group and from 51% to 88% in the retropubic group. The retropubic group had higher morbidity than the transobturator group, but the authors said that the overall rate of adverse events remained low. A summary of the adverse events was provided in the respondents’ submissions:
For the primary comparison, the comparison between the transobturator and retropubic route mid urethral sling, the adverse events were:
(a) major vascular injury such as retropubic haematoma or major visceral injury, for example bowel perforations (although reported by 28 of the trials, percentage reports were not included in the review);
(b) bladder perforations, with an average rate of 2.54%;
(c) postoperative voiding dysfunction, with an average rate of 5.53%;
(d) de novo urgency and urgency urinary incontinence, with an average rate of 8.35% and a rate of 8% for detrusor overactivity in both groups;
(e) vaginal tape erosion, with an average rate of 2.09%, and the one trial reporting long term vaginal tape erosion reported none;
(f) groin pain, with an average rate of 4.51%, which along with suprapubic pain was short-lasting, with most resolving within the first six months;
(g) repeat incontinence surgery, with an average rate of 2.43% within a year and 5.34% in the long term;
(h) dyspareunia, was reported in the following terms:
“In all the trials there was significant improvement in sexual function from baseline scores during the follow-up period that spanned six to 24 months. At 24-month follow-up, rates of superficial and deep dyspareunia were low, with no difference between the groups.” 1082
974 While convenient, in one important respect the above summary was incomplete. It was incomplete because it did not identify the relative incidence of several other complications reported by Ford (2015). As indicated above, the rates for a number of complications including major vascular injury, bladder/urethral perforation, postoperative voiding dysfunction and suprapubic pain were significantly lower in the transobturator group than in the retropubic group, while there was a significantly higher occurrence of groin pain in the transobturator than the retropubic group.1083
975 Summarising their main findings, the authors said this about adverse events:
Tapes passing behind the pubic bone (retropubic) seem to carry a greater risk of injuring the bladder during the operation and of women experiencing problems emptying their bladder completely after surgery. However, this operation leads to less groin pain in the short term. There is some limited evidence that this way of inserting the tape has a lower risk of requiring a repeat operation in the long term compared to tapes passing through the groin (transobturator). There is moderate quality evidence that overall reported rates of tape-related complications are low, such as erosion of the tape into the vagina at about 2% for both routes of tape insertion. The reported occurrence of problems with sexual intercourse including pain was low, and leakage of urine during intercourse … improved following insertion of these tapes. 1084
976 There were three Cochrane reviews led by Marie Lapitan of the Philippines National Institute of Health, published in 2009,1085 2012,1086 and 2016,1087 each entitled “Open retropubic colposuspension for urinary incontinence in women”. These reviews were designed to determine the effects of open retropubic colposuspension and its safety profile compared with various alternative forms of surgery for stress or mixed urinary incontinence, including midurethral sling procedures (referred to as self-fixing suburethral sling procedures) like TVT surgery.
977 The Lapitan et al (2009) review covered 46 randomised or quasi-randomised controlled trials involving 4,738 women with symptoms or urodynamic diagnoses of stress or mixed urinary incontinence that included open retropubic colposuspension surgery in at least one trial group.1088 Assessment of the methodological quality of the studies was said to be difficult in general because of insufficient details provided by the authors, particularly on random allocation concealment and blinding.1089
978 The authors concluded that:
[O]pen retropubic colposuspension is an effective treatment modality for stress urinary incontinence especially in the long term. Within the first year of treatment, the overall continence rate is approximately 85 to 90%. After five years, approximately 70% of patients can expect to be dry. Newer minimal access procedures like tension free vaginal tape look promising in comparison with open colposuspension but their long-term performance is not known and closer monitoring of its adverse event profile must be done. Laparoscopic colposuspension should allow speedier recovery but its relative safety and effectiveness is not known yet. 1090
(Emphasis added)
979 The authors observed that the sling procedures conferred similar success rates to open colposuspension but were quick to point out that the long-term adverse event profile of the sling procedure, especially with the use of TVT, was unclear. They said there was an urgent need for adequately powered trials to assess the effectiveness of open retropubic colposuspension with suburethral slings especially the self-fixing sling procedures like TVT.1091 In this and the two subsequent reviews, TVT appears to have been used as an acronym for tension-free vaginal tape in general rather than as a trade name for Ethicon’s devices.
980 Lapitan and Cody (2012) reviewed 53 trials involving 5,244 women. 1092 Amongst other things, comparisons were made between open retropubic colposuspension and a sling procedure in 20 trials of which 14 involved the TVT procedure including one three-armed study comparing Burch colposuspension, transobturator tape and TVT. The authors’ conclusions were identical to the conclusions Lapitan et al (2009) reached three years earlier, although their analysis included the review by Ogah et al (2009).
981 In their summary of the main results, Lapitan and Cody (2012) wrote:
Open retropubic colposuspension was regarded as the gold standard treatment for urinary incontinence in women before the advent of new minimally invasive procedures such as TVT. However, in countries where minimally invasive suburethral tapes are not available or the cost is prohibitive, open colposuspension remains the treatment of choice with its high success rates and acceptable levels of associated morbidity. The results of this systematic review present the evidence supporting such a view.1093
982 Notably the comparison of open colposuspension with TVT was said to be largely dependent on the Ward Hilton RCT. While Lapitan and Cody acknowledged that Ward and Hilton had demonstrated short-term advantages of TVT with less post-operative morbidity and a more rapid return to normal activities, they also noted that the trial had a high risk of performance and detection bias because neither patients nor investigators had been blinded.1094
983 Lapitan and Cody (2016) reviewed 55 trials involving 5,417 women. 1095 Following their review, they again considered that open retropubic colposuspension was an effective treatment modality for stress urinary incontinence, especially in the long term. They reported that within the first year of treatment, the overall continence rate was approximately 85% to 90%. After five years, they concluded that about 70% of women could expect to be dry. Evidence from 22 trials comparing open retropubic colposuspension with suburethral sling procedures (traditional slings, transvaginal tape or transobturator tape) found no overall significant difference in incontinence rates in all the periods of time that were evaluated: within one year of treatment, between one and five years, and at five years and more. In relation to morbidity, they wrote that, in general, the evidence did not disclose a higher morbidity or complication rate with open retropubic colposuspension compared to other surgical techniques, although pelvic organ prolapse was more common than after anterior colporrhaphy or sling procedures, while voiding problems were more common after sling procedures than after open colposuspension.1096
984 Their conclusion did not change from that reached in their 2012 review.1097
985 The Nambiar et al (2014) Cochrane review focussed on the effectiveness of mini-sling procedures in women with urodynamic clinical stress or mixed urinary incontinence. Thirty-one trials involving 3,290 women were analysed. A number of single-incision sling devices were covered, including TVT Secur. The authors found that single-incision slings resulted in higher incontinence rates compared with inside-out transobturator slings. Adverse events were significantly worse, with higher risks of vaginal mesh exposure, bladder or urethral erosion and operative blood loss. Notably, the authors concluded that TVT Secur was inferior to standard midurethral slings.1098
986 Three Cochrane reviews were led by Cathryn Glazener: “Bladder neck needle suspension for urinary incontinence in women” by Glazener and Cooper (2014); 1099 “Bladder neck needle suspension for urinary incontinence in women” by Glazener et al (2017a);1100 and “Anterior vaginal repair for urinary incontinence in women” by Glazener et al (2017b).1101
987 Glazener and Cooper (2014)1102 identified 10 trials, which included 375 women with six different types of needle suspension procedures and 489 who received alternative interventions. There was insufficient information to enable the authors to compare bladder neck needle suspension surgery with suburethral sling operations. The study was briefly raised with Associate Professor Krulewitch in cross-examination but did not rate a mention in the submissions of either party. In the circumstances nothing more needs to be said about it.
988 Glazener et al (2017a) studied the effects of bladder neck needle suspension on stress or mixed urinary incontinence in comparison with other management options. This paper appears to be a review of the same studies analysed by Glazener et al (2014).
989 The objective of the review by Glazener et al (2017b) was to determine the effects of anterior vaginal repair (anterior colporrhaphy) on urinary incontinence in comparison with other management options.1103 The selection criteria consisted of randomised or quasi-randomised trials that included anterior colporrhaphy for the treatment of urinary incontinence. Since no trials were identified which compared anterior colporrhaphy with suburethral sling procedures, this review is of no utility for present purposes.
990 Neither of the 2017 Glazener reviews was mentioned in the applicants’ submissions and both were only referenced in passing in the respondents’.1104 Nothing more need be said about them here.
The Novara and Fusco meta-analyses
991 The objective of each of these meta-analyses was to compare the efficacy, complication, and reoperation rates of midurethral tapes with other surgical treatments for female stress urinary incontinence. The authors performed a systematic review of the literature using various databases.
992 Novara et al (2007) identified 37 randomized controlled trials. They concluded that TVT outperformed Burch colposuspension and TVT and pubovaginal slings were similar in efficacy.1105 They noted, however, that the overall quality of the trials included, both in terms of methodologic and clinical parameters, was limited, which reduce the strength of the findings of any meta-analysis.
993 Novara et al (2010) was an update of Novara et al (2007). The authors stated that several randomised controlled trials had been published comparing the various surgical procedures but with “conflicting results”.1106 Of the 39 RCTs identified by the authors, patients receiving midurethral tapes were reported to have had significantly higher overall and objective cure rates than those receiving Burch colposuspension, although they also had higher rates of bladder perforation. Cure rates for patients who received midurethral tapes and pubovaginal slings were similar, although the latter had a slightly higher rate of “storage lower urinary tract symptoms” (LUTS) and a higher reoperation rate. The objective cure rates were slightly higher for retropubic tapes than for transobturator devices but subjective cure rates were similar. Patients treated with transobturator tapes had a much lower risk of bladder and vaginal perforations, haematoma, and storage LUTS. Meta-analysis demonstrated similar outcomes for TVT-O and “Monarc” (a transobturator sling system manufactured by AMS).
994 Novara et al (2010) noted, however, that the strength of their findings was limited by the heterogeneity of the outcome measures and the short length of follow-up. They observed that:
On the whole, the figures of the meta-analysis seem to support the increasing role of midurethral tapes and, specifically, of [transobturator tape] in the setting of the primary treatment of the patients with SUI. However, those data raise several concerns regarding outcome measures, reporting of complications, and follow-up. 1107
995 Notwithstanding that their meta-analysis included a significant number of RCTs with good methodological quality and generated consistent results in all the sensitivity analyses, the authors said that the variations in the outcome measures used, the length of follow-up, and the handling of cases lost to follow-up were significant concerns. They pointed out that the definitions of cure used in the various studies could significantly affect the study results and called for standardised criteria for the reporting of patient outcomes. They also pointed out that only 21 of them used validated questionnaires to evaluate subjective cure rates, the impact of stress urinary incontinence on quality of life, and sexual function, making comparisons very difficult.
996 With regard to complications, Novara et al (2010) observed that “a very limited number” of major complications were seen in the evaluated RCTs, although bowel, vascular, and nerve injuries, necrotising fasciitis, ischiorectal abscess, urethrovaginal fistulas, sepsis, and patient deaths had been reported after placement of retropubic and transobturator slings. Most of the reported complications were intraoperative ones. Only a limited number of studies providing data on the intermediate and long term functional consequences.1108 The authors said this was a matter of the utmost importance because some under-reported complications can be disabling for the affected patients while intraoperative complications like bladder injury during placement of retropubic tape had few or no implications if they were promptly recognised and treated. They noted that only two studies reported data with follow-up of five years or more, which, they observed, “clearly does not allow either the durability of efficacy or the presence of long-term morbidity and functional complications to be adequately evaluated.”1109 They also noted that in most studies patients lost to follow-up were “simply deleted from the analyses” and their outcomes assumed to be similar to the entire cohort. They observed that this approach was incorrect.
997 Novara et al updated their study and the analysis was published in 2017 with a different lead author (Ferdinando Fusco).1110 This meta-analysis was based on 28 RCTs and included over 15,000 patients. None of the applicants’ experts referred to it or was taken to it in cross-examination. Although it was tendered during Associate Professor Rosamilia’s evidence and the respondents referred to it in their written submissions, the applicants neglected to address it.
998 Fusco et al (2017) argued that their study had a number of strengths.
999 First, they claimed that it represented the most up-to-date and, except for single-incision mini-slings, the most comprehensive summary of the currently available evidence in surgical treatments of female stress urinary incontinence.
1000 Second, they claimed that their paper complied with the currently available standard of reporting of systematic review and meta-analysis.
1001 Third, they said that the review included a relatively high number of randomised controlled trials with long-term follow-up (five years or more). 1111
1002 Nevertheless, they also acknowledged that the study also had a number of limitations.
1003 First, a small, albeit “extremely low”, percentage of patients included in some of the RCTs had already received previous surgical treatments for stress urinary incontinence.
1004 Second, as was the case with the 2010 review, the evaluation of both subjective and objective outcomes was “heterogeneous” and not all the studies used validated questionnaires.
1005 Third, although the number of randomised controlled trials with long-term follow-up was relatively high, most of the studies reported only short or immediate term follow-up.
1006 Fourth, in most of the RCTs the accuracy of complication reporting was limited and did not comply with the standardised criteria.
1007 Fifth, studies comparing midurethral slings to other surgical treatments, such as bulking agent injections, were lacking.1112
1008 Notwithstanding these limitations, Fusco et al (2017) concluded that their analysis confirmed the superiority of the midurethral sling over Burch colposuspension. They said that the available literature suggested that those slings were either more effective or safer than the older surgical procedures. They also said that their analysis reconfirmed the absence of significant differences between women treated with midurethral slings and those receiving pubovaginal slings in both overall and subjective continence rates, prevalence of pelvic haematoma, vaginal erosions, and voiding LUTS but the incidence of storage LUTS was significantly lower in the midurethral sling group. Notably, while the previous meta-analysis showed higher reoperation rates for patients receiving pubovaginal slings, this one showed a similar, but not statistically significant, trend.
1009 Importantly, Fusco et al (2017) observed that, while there was a growing interest in the likelihood of chronic pain and dyspareunia after midurethral sling procedures, only a limited number of RCTs reported on long-term pain after surgery for stress urinary incontinence. One study reported “a few cases of long-term pain” at five-year follow-up with both retropubic and transobutrator transvaginal tape. One reported “scar pain” after autologous pubovaginal sling surgery.
1010 Fusco et al (2017) noted, however, that two recent studies of transobutrator transvaginal tape reported findings of 6.4% and 9% groin/inguinal pain/discomfort at seven and ten year follow-up respectively.1113 The first, by Zhang et al (2016), was a prospective study carried out in Beijing of 140 patients with stress urinary incontinence, allocated equally on a randomised basis to either Ethicon’s TVT or TVT-O, of whom 120 completed the long-term follow-up (average of 95 months). 1114 The method of randomisation was not disclosed. Zhang (2016) also reported groin/thigh pain for one TVT patient (1.72%) which Fusco et al (2017) did not mention. Fusco et al (2017) also neglected to mention the incidence of abdominal pain in both arms of the study, which was 8.62% of TVT patients and 3.23% of TVT-O patients. Indeed the total incidence of long-term pain was 10.34% for TVT patients and 8.05% for TVT-O patients. The incidence of tape exposure was 3.45% for the TVT arm and 8.05% for the TVT-O arm but, because of the small number of patients, the difference was not statistically significant. Indeed, the authors remarked that there was no significant difference in the long-term complications in the two arms.1115
1011 The second study referred to by Fusco et al (2017), which reported on a 10 year follow-up of patients after the transobturator procedure, was not tendered.
1012 Fusco et al (2017) stated that intractable suprapubic pain had previously been described following colposuspension and defined as post-colposuspension syndrome, though no reference was provided. They added that there was even less data available on the long-term prevalence of dyspareunia in patients receiving midurethral slings for stress urinary incontinence. Only a few cases had been reported in the available RCTs and the available literature (two articles were cited) seemed to suggest improvements in sexual function for sexually active patients treated with midurethral slings.
1013 The first of the two articles cited was by De Souza et al (2010), which was a prospective study comparing through standard questionnaires the effect of TVT to the Monarc sling on sexual function in women with urodynamic stress incontinence and intrinsic sphincter deficiency. Eighty-seven women were surveyed. Sexual function was reported as having improved six months after surgery and the benefit was said to have been maintained at 12 months. But the analysis at six months was of only 64 of the 87 patients (74%) who had completed all the questionnaires and at 12 months only 48 of the 64 were sexually active and had complete follow-up. Of the six-month responders only 27 (42%) underwent a TVT procedure and three of these were not followed up at 12 months.1116 The authors noted that the study had limitations, acknowledging that the total number of questionnaires analysed was small. They also disclosed that the power was calculated as a post hoc estimate. They pointed out that sexual function was not a primary outcome of their original study, which compared the effectiveness of the two devices in the treatment of intrinsic sphincter deficiency. Neither of these limitations was mentioned by Fusco et al (2017) or by Dr Hinoul who referred to the study’s conclusions in his affidavit.1117
1014 The second study on sexual function after sling surgery cited by Fusco et al (2017) was by Zyczynski et al, published in 2012.1118 Dr Hinoul referred to it, too. This involved a much larger group of women, with 597 women enrolled in a randomised trial comparing retropubic with transobturator midurethral slings.
1015 Zyczynski et al (2012) assessed sexual function with a questionnaire, reporting that two years after surgery the patients in both groups had significant, similar improvements in sexual function. Dyspareunia, incontinence during sex, and fear of incontinence during sex significantly improved after surgery. The authors concluded that midurethral sling surgery significantly improves sexual function, although they found that coexistent urge incontinence has a negative impact. They acknowledged, however, that their understanding of “a woman’s expectations and goals for sexual function” was incomplete.1119
1016 This was a retrospective observational population-based cohort study of 16,600 women in Scotland who had undergone a first, single incontinence or prolapse operation between April 1997 and March 2016.1120 The women were identified by means of a national hospital admission database. Primary outcomes were immediate postoperative complications and readmissions within five years for later postoperative complications, further incontinence surgery or further prolapse surgery.
1017 The lead author was Dr Joanne Morling of the Division of Epidemiology and Public Health at the University of Nottingham. Dr Agur and Dr Glazener were among the co-authors.
1018 For stress urinary incontinence, the authors found that in routine clinical practice mesh surgery was as effective as colposuspension (in terms of the risk of repeat incontinence surgery) but associated with a lower risk of immediate complications and subsequent prolapse surgery than colposuspension, and a similar risk of later complications and further incontinence surgery.1121
1019 Morling et al (2016) considered that their results supported the use of mesh procedures for incontinence although they added a rider that further research on longer term outcomes would be beneficial.
1020 I refer below to the findings in the prolapse group.
1021 It will be recalled that in 2004 most of the mesh kits were still in development, although Gynemesh PS had been on the market in Australia for at least six months.
1022 Maher et al (2004) set out to determine the effects of surgery in the management of pelvic organ prolapse and concluded that there was not enough evidence about the effects of different types of surgery.1122 They observed that the use of mesh to augment prolapse repair surgery had been successful in other fields such as groin hernia repair but pointed out that there were particular issues related to its use in vaginal repair. They concerned the effect on bowel, bladder and sexual function and the possibility of mesh erosion or infection. Evidence from case series highlighted some of these issues. Salvatore et al (2002) reported a mesh erosion rate of 13% after a polypropylene mesh (Prolene) overlay at vaginal repair, overactive bladder increasing from 28% to 56%, and dyspareunia increasing from 18% to 38% postoperatively.1123 Visco et al (2001) found that mesh erosion or infection rate was significantly increased when mesh was introduced vaginally rather than abdominally.1124 They studied 273 patients who underwent surgery over a seven year period between 1992 and 1999. Of those, 155 underwent an abdominal sacralcolpopexy, 88 underwent an abdominal-only sacral colpoperineopexy, and 30 underwent combined abdominal-posterior vaginal procedures. In five cases in the final category, the mesh was attached vaginally. Mersilene mesh was used in all but four patients who received Goretex. Even though Prolene was not used, the authors’ comments are of note:
Vaginal placement of mesh results in an unacceptably high rate of mesh erosion and a shorter time to erosion than any other form of vault suspension in this study. Vaginal passage of suture was also associated with an increased rate of erosion in comparison with abdominal sacral colpopexy but the rate was less than that observed with passage of vaginally fixed mesh. Therefore the use of this modification should be reserved for those patients in whom the benefit achieved by vaginal suture placement is thought to exceed the associated risk of mesh erosion.1125
1023 In her first report, Associate Professor Rosamilia noted that a 2005 publication of the International Consultation on Incontinence stated that transvaginal mesh for prolapse was not for routine use and recommended that its use be limited to clinical trials, that further research was necessary, and that techniques to reduce recurrence rates especially in the anterior wall were urgently required.1126
Studies relevant to complications arising from the POP devices
1024 Several Cochrane reviews concerning complications arising from treatments for pelvic organ prolapse were tendered. The lead author of each of those reviews was Dr Christopher Maher, a urogynaecologist from the Royal Brisbane Hospital. Professor Woodward, who was asked to look at the evidence about the POP devices only, said that the most authoritative of the Cochrane meta-analyses and the most likely to be unbiased were the most recent, published in 2016.1127 Before turning to those reviews, however, it is necessary to refer to the findings and conclusions of earlier Cochrane reviews and other relevant studies.
1025 In 2006 the French National Authority for Health (Haute Autorité de Santé) conducted an evaluation of the safety and efficacy of transvaginal mesh implants for prolapse based on a critical analysis of the literature and the opinions of some 21 experts.1128 It came to the same conclusion as Maher et al (2004):
Given the variety of types of tested implants and treated indications, the amount of follow-up observation that rarely exceeds 2 years, the absence of comparative studies with alternative techniques in the majority of cases, and the uses of imprecise and heterogeneous standards of evaluation, the data in the literature does not allow an effective evaluation on the anatomical and functional viability of implants in the treatment of genital prolapse through the vaginal approach. Some complications, several very serious, were identified. The analyzed literature does not allow an evaluation of their frequency.
According to the current state of knowledge and the consultation of experts, the use of mesh implants in genital prolapse surgeries by the vaginal route remains a matter for clinical research.1129
1026 At the time of publication of the Maher et al (2007) review, Prolift had been on the market in Australia for about two years. Maher et al concluded, based on a review of the literature in the period up to 16 April 2007, that:
[T]he evidence is not sufficient to support the use of permanent meshes or grafts at the time of vaginal repair surgery except in the context of randomised controlled clinical trials. These trials must be adequately powered to evaluate the anatomic and functional outcomes and possible adverse events. 1130
1027 They reiterated the opinion expressed in the 2004 Cochrane review that, although the use of mesh in repair surgery had been successful in other fields, such as groin hernia repair, in the area of vaginal repair of prolapse, the issues were such that evidence of an anatomical cure of the prolapse was not sufficient reason to advocate its use. The issues nominated were the effect of mesh on bowel, bladder and sexual function and the possibility of mesh erosion or infection.1131
1028 At this stage the evidence was confined to case studies which reported significant complication rates and provided cause for concern. Maher et al recommended that adequately powered randomised controlled trials should be conducted to evaluate the anatomic and functional outcomes and possible adverse events. They noted that:
The challenge in prolapse surgery is that while the prolapse itself may cause difficulties with bladder, bowel and sexual function, surgical correction may also affect these functions in unpredictable ways. Therefore, all trials need to include subjective, objective and patient determined outcomes, and the direct interaction with bladder, bowel and sexual function must be measured. The impact of interventions should also be assessed by utilising validated pelvic floor and quality of life questionnaires, morbidity and cost analysis. Ideally, long term outcomes should be reported at least at two and five years after surgery.1132
1029 In cross-examination, both Associate Professors Lam and Rosamilia agreed that the authors’ conclusion accurately reflected the state of the evidence in relation to the use of mesh augmentation for pelvic organ prolapse surgery as at 2007.1133
1030 In June 2008, following a systematic review of the published evidence on the surgical repair of anterior and/or posterior vaginal wall prolapse using mesh, which was not confined to RCTs, the UK National Institute for Health and Care Excellence (NICE) concluded that “this procedure should only be used with special arrangements for clinical governance, consent and audit or research”.1134
1031 Its report impressed upon clinicians the need to “[e]nsure that patients understand that there is uncertainty about the long term results and there is a risk of complications, including sexual dysfunction and erosion into the vagina, which would require additional procedures”.1135
1032 By 2010, the Prolift kits had been on the market for about five years. Even so, there were still no randomised controlled trials comparing results for the POP devices to any of the traditional surgical treatments for pelvic organ prolapse in women. Maher et al (2010) considered that adequately powered randomised controlled clinical trials were urgently needed.1136
1033 The authors stated that standard anterior repair was associated with more anterior compartment failures on examination than for polypropylene mesh repair “as an overlay” or armed transobturator mesh. Data relating to polypropylene mesh overlay, however, were extracted from conference abstracts without available peer review manuscripts and the authors said that they should be “interpreted with caution”.1137
1034 No differences were found in subjective outcomes, quality of life data, de novo dyspareunia, stress incontinence, re-operation rates for prolapse or incontinence. Blood loss with transobturator meshes was significantly higher than for native tissue anterior repair. Mesh erosions were reported in 10% (30/293) of anterior repairs with polypropylene mesh.1138
1035 Once again, the authors noted that the use of mesh to augment repair surgery had been successful in other fields such as groin hernia repair but, having regard to the particular issues relating to its use in vaginal repair, they considered that evidence of an improved anatomical cure of prolapse in the anterior compartment using polypropylene mesh was not sufficient reason to advocate its use. They emphasised the need for improved subjective and quality of life outcomes with reduced reoperating rates before advocating for the routine use of permanent mesh in the anterior compartment.1139 While the evidence suggested that the use of polypropylene mesh at the time of anterior vaginal wall repair might reduce the risk of recurrent cystocele on examination, improved outcomes including patient satisfaction, quality of life and reduced operations for recurrences had not yet been demonstrated.
1036 In the upper or apical compartment, they concluded that the use of mesh in open sacralcolpopexy as compared to vaginal sacrospinous colpopexy significantly improved outcomes but with increased morbidity and cost. They said that there was no evidence to suggest that the addition of any graft material at the posterior compartment repair resulted in improved outcomes. They concluded that the evidence was not sufficient to support the use of permanent meshes or grafts at the time of vaginal apical or posterior compartment repair surgery, except in the context of randomised controlled clinical trials which needed to be adequately powered to evaluate the anatomic and functional outcomes and possible adverse events.1140
1037 This was a multi-centre randomised trial the stated purpose of which was to determine the efficacy and safety of transvaginal mesh repair for prolapse of the anterior vaginal wall in comparison with anterior colporrhaphy.1141 It was the first randomised controlled trial conducted using any of the Ethicon POP devices.
1038 The results of the trial were reported by Dr Daniel Altman and others in a 2011 article titled “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” published in The New England Journal of Medicine.
1039 Altman et al (2011) observed that (before their study) “[d]espite their widespread use, none of the marketed kits [involving mesh] have been comprehensively evaluated in comparative trials”.1142 Professor Woodward described the observation as “a damning comment” on the widespread use of these kits as at the time of publication.1143 It might simply indicate, however, in the absence of any advice from the manufacturers to the contrary, that many surgeons who used them had assumed that such an evaluation had been carried out before the kits were cleared for sale.
1040 A convenient summary of the trial appeared in the applicants’ submissions, noting “P” is shorthand for “P-value”:
The study was powered to detect a 20% difference in the primary outcome measure.
189 women were assigned to the colporrhaphy group and 200 to the mesh repair group. The primary outcome measure was based on 174 women in the colporrhaphy group and 176 women in the mesh group. There was therefore missing data for 15 women in the colporrhaphy group and 24 for the mesh group. A sensitivity analysis was performed to cater for the missing data, but the authors do not state the number of missing cases analysed.
An objective and subjective cure rate of 60.8% in the mesh group and 34.5% in the colporrhaphy group was found. Complications such as bladder perforation were reported 3.5% in the mesh-repair group and 0.5% in the colporrhaphy group (P=0.07), and the respective rates of new stress urinary incontinence after surgery were 12.3% and 6.3% (P=0.05). Five patients in the mesh-repair group reported severe pelvic pain at 2 months as compared with one patient in the colporrhaphy group (P=0.22); in all except one of these patients (who was in the mesh-repair group), the pain had resolved spontaneously by the 1-year follow-up visit.
Surgical reintervention to correct mesh exposure during follow-up occurred in 3.2% of 186 patients in the mesh-repair group. The actual rate of erosion was not discussed but was later reported by Maher 2013 as being 11.5%.1144
1041 Referring only to the Altman RCT, Professor Roovers expressed the opinion that “[l]arge randomised controlled trials have shown that, as compared to native tissue surgery, vaginal mesh surgery results in better subjective and objective cure.”1145
1042 As Professor Woodward pointed out, however, a close examination of the Altman (2011) paper reveals that Professor Roovers’ opinion was unsound.1146
1043 First, the trial looked only at women who had undergone anterior compartment repair. All mesh procedures in the trial involved the use of the Prolift Anterior Pelvic Floor Repair System. Professor Roovers did not explain what basis there was for generalising the results to the repair of other compartments or, for that matter, to the use of Prolift Total, where twice the amount of mesh was used.
1044 Second, patients were followed up at two and 12 months. Professor Woodward said that a 12-month trial was an inadequate basis for drawing a reliable conclusion about the success of surgery.
1045 Third, although the sample size (389) was large in the historical context of prolapse surgery, it was moderate in the general context of multicentre RCTs and less than half of that in the PROSPECT trial (885), discussed below. With this sample size, Professor Woodward observed that the authors could not have expected to be able to draw reliable conclusions about relatively rare outcomes.
1046 Fourth, Professor Roovers’ conclusion that the trial showed benefits for “subjective and objective cure” was apparently based on the composite outcome, which was subjective assessment of prolapse plus anatomical success. In other words, it ignored safety issues. Professor Woodward said that “this hardly encompasses all aspects of subjective or objective cure”.1147 Importantly, the trial did not report on the combined endpoint of total surgery, including mesh exposure, which, he said, was “a key objective outcome in the context of mesh safety” in comparison to native tissue repair. Professor Woodward went on to say:
Altman et al (2011) says that 6 in the mesh group had surgery to correct mesh exposure, and 5 had surgery due to incontinence. For the native repair group, the only report is of a single patient that had repeat surgery for prolapse recurrence. From these numbers I compute that the relative risk for total surgery is 10.8 (95% confidence interval 1.4 to 82.5); thus the trial estimates that mesh is more than 10 times as likely to lead to surgery in the next 12 months than does native repair. Even if the 5 incontinence surgeries are ignored, the estimated risk is more than 5 times higher with mesh than native repair, although this then marginally loses statistical significance (p=0.06). These analyses undermine the conclusion that Altman has proven objective cure of mesh.1148
(Emphasis added)
1047 Professor Woodward also said that certain other findings undermined Professor Roovers’ opinion that the Altman trial proved that mesh offered superior subjective cure rates.1149 He noted that the Altman trial produced evidence which Professor Woodward described as compelling, that the risk of dyspareunia was twice as high in the mesh arm than in the native tissue arm. Altman et al (2011) also found that inguinal pain and bladder emptying difficulties during hospital stay were more common after mesh repair than after colporrhaphy (p=0.06 for pain and 0.05 for urine retention) and the respective rates of de novo stress urinary incontinence were 12.3% in the mesh arm and 6.3% in the colporrhaphy arm (p=0.05).
1048 Professor Roovers put the risk of re-operation for mesh-related complications at about 3%, based on the Altman trial and a paper by Chughtai et al (2015).1150 Given that Maher et al (2016a) reported a rate more than twice that figure (at 7%) and the PROSPECT figure was higher still (at 9% or 14%), I conclude that the Altman et al (2011) figure is likely to be an underestimate. To some extent that might be attributable, at least in part, to the precautions the surgeons took in the Altman trial to minimise complications, as Altman et al (2011) acknowledged. Further, as Professor Roovers conceded in cross-examination, his figure of 3% was based on a reintervention rate within one year.1151 Common sense suggests, and the PROSPECT results indicate, that the rate will be higher after longer follow-up.
1049 There are other reasons to question the reliability of the reported results of the Altman trial.
1050 Maher et al (2016a), which is discussed below, rated the Altman trial as having a high risk of detection bias due to lack of blinding of outcome assessments and to conflicts of interest. They pointed out that conflict of interest statements of members of the Nordic TVM Group, who were reviewers of the surgery, were not reported.1152 Professor Woodward pointed out that any conflict of interest raises the possibility that the results have been interpreted in an over-optimistic way, particularly when outcomes are not blinded.1153
1051 Altman et al (2011) disclosed the fact that the reviewers were not blinded to outcomes. They declared that one of the limitations of their study was that the postoperative assessors were aware of the treatment assignments, and conceded that it was possible that the surgeons’ beliefs about mesh kits influenced their assessments.1154
1052 Altman (2011) also disclosed that the trial had been funded in part by Ethicon. But this disclosure was diluted by the inclusion of the following statement:
The manufacturer of the mesh kit did not provide the products used in this trial and had no involvement in the study design, data collection and analysis, the writing of the manuscript, or the decision to submit the results for publication.1155
(Emphasis added)
1053 The evidence revealed that this statement was misleading.
1054 A “correction” was published in a rather inconspicuous position in the New England Journal of Medicine nearly two years later:
Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse (May 12, 2011;364:1826-36). In the Study Design subsection of Methods, the final paragraph (page 1828) should have read, “As cosponsor of the trial, the manufacturer of the mesh kit reviewed the original study protocol and a presubmission draft of the manuscript. The manufacturer did not provide the products used in the trial and had no involvement in data collection and analysis or in the decision to submit the results for publication.”1156
(Emphasis added)
1055 The sentence I have emphasised in the preceding paragraph was new. It replaced the statement in the original publication that Ethicon had “no involvement in the study design … and … the writing of the manuscript” with the statement that it had “reviewed the original study protocol and a presubmission draft of the manuscript”. It did not, however, disclose the nature of the review or the extent of Ethicon’s contribution to the final draft.
1056 It does not seem that the correction came to the attention of Maher et al (2016a). The circumstances in which it came to be made were not the subject of evidence although it is possible, if not likely, that it was prompted by Ethicon’s discovery in American litigation. Ethicon’s discovered documents in the present case, which I understand covered documents discovered in the American litigation, included a draft of Altman’s manuscript containing comments from Ethicon staff and a number of emails. 1157
1057 The evidence revealed that on 25 March 2010 Dr Altman emailed Barbara Walker of Ethicon Great Britain (Ethicon GB) asking whether there was someone in her office who could read the manuscript and “professionally revise the English text”.1158
1058 On 28 April 2010 Judi Gauld of Ethicon GB wrote to Dr Altman offering a number of “suggestions/comments” regarding changes to the manuscript.1159 One was that the mesh exposure rates probably needed more detail, including “how many were asymptomatic, required excision, treated conservatively, ongoing at 1 year, etc”. The email was sent after a meeting in Stockholm at which Dr Altman presented data from the trial. It was put to Dr Hinoul in cross-examination that it was clear from the reference to mesh exposure rates in the email that Dr Altman had initially disclosed the “gross” rates of mesh exposure and not just the cases requiring excision.1160 Dr Hinoul was reluctant to make such a concession but no other conclusion is rationally open.
1059 Yet in the pre-submission draft produced nearly three months later, there was no mention of the rates of mesh exposure and they were not reported in the published manuscript. What was reported were the rates of surgical intervention to correct mesh exposure. This is a case of selection bias. Professor Hu, who has participated in designing, studying and reporting on randomised controlled trials, considered that it should have been reported, since it was a clinically significant finding, “clearly important” to patients, and is often reported in reviews of surgery of this kind.1161 Maher et al (2013) obviously considered it important, too, because they acquired the information through “personal communication”.1162 The figure they cited was 21 out of 183, which is 11.5%. The omission of the data was favourable to Ethicon because it was more than three times higher than the figure reported in the article for surgical intervention for mesh exposure, which was 3.2%.
1060 That pre-submission draft, dated 17 July 2010 (according to the meta-data), which was obtained by the applicants and tendered in evidence, contained the following financial disclosure which had mysteriously disappeared by the time of publication:
Financial Disclosure: Dr. Altman reports having received consultant fees from Gynecare Scandinavia, Ethicon US, and Contura A/S; Dr. Falconer consultant fees from Gynecare Scandinavia. No other potential conflicts of interest relevant to this article were reported.1163
1061 The abstract also included the following statement, which was omitted from the published article:
The rate of dyspareunia was 34% in the colporrhaphy group compared to 51% in the mesh group (P=0.02).1164
1062 Since the p-value was less than 0.05, this was a statistically significant result.
1063 On 11 August 2010 Dr Altman emailed what appears to be a revised version of the manuscript1165 to a number of Ethicon employees, including Dr Hinoul, asking for their comments.1166 This version of the manuscript substituted for the statement on dyspareunia in the abstract the following statement:
Dyspareunia was present usually or always in 2/101 patients (2.0%) after colporrhaphy compared to 8/110 (7.3%) patients in the mesh group (P=0.07).
1064 The p-value for this result, which was limited to women whose dyspareunia was present “usually or always”, was greater than 0.05 and the higher incidence of dyspareunia in the mesh group lost its significance, although the dyspareunia rates originally described in the abstract (34% with colporrhaphy and 51% after mesh) could be found in the text of the article.1167
1065 In the meantime, Dr Altman was seriously thinking about joining Ethicon. Indeed, it appears that he was being courted. In another email to Dr Hinoul sent later on 11 August 2010, Dr Altman wrote:
Sorry to hear about the colleague who left the company. I would have enjoyed working with a fellow epidemiologist. Since we talked the last time, I have gotten more and more used to the thought of joining the company. I look forward to hear (sic) from you on this and other matters as soon as you have an update. 1168
1066 When he was taken to this email in cross-examination Dr Hinoul admitted that he had been speaking to Dr Altman about joining Ethicon and that Dr Altman was expressing a great deal of interest — “at a very high level” was the expression Dr Hinoul used.1169 This circumstance alone, which was presumably unknown to Maher (2013), casts a shadow over the reliability of the published manuscript.
1067 On 19 August 2010 Dr Altman sent a revised version of the manuscript to various Ethicon personnel, informing them that he had made most of the suggested changes. He advised them that tables had been adjusted according to their suggestions. And he invited them to propose additions to the manuscript while indicating “what to remove of similar word count”.1170
1068 In an email Dr Hinoul sent to Dr Altman on 20 August 2010, he proposed additional changes:
Thank you for giving us a last chance to have a look at this before submission. Of course we respect the fact that this is, and must remain, an independent study. Overall the team feels that the sexuality part of the manuscript still lacks accuracy.
The abstract, which will be the only thing most surgeons read, states: “Dyspareunia was present usually or always in 2/101 patients (2.0%) after colporraphy [sic] compared to 8/110 (7.3%) patients in the mesh group (P=0.07).” As this was not really one of the clearly predefined endpoints, we feel that reporting on the PISQ score would be more scientifically appropriate. For example: “The PISQ-12 scores, were similar for both arms of the study (35.1 (33.7-36.4) versus 35.0 (33.7-36.4) (p=0.99)”.
We feel that selecting one reported outcome somehow will be used by the mesh antagonists, whilst you may just as well have selected overall sexual satisfaction to go in the manuscript which would show a completely different impression “overall satisfaction with sexual life (‘Overall, how satisfied are you with your sexual relationship with your partner?’) with 37/92 (40%) in the colporraphy [sic] group, and 51/106 in the mesh group (48%), responding ‘usually’ or ‘always’ (P=0.37).” (This is probably reflected in the fact that the improvement in PISQ is overall larger after Prolift vs colporrhaphy as baseline PISQ scores were 33.1 versus 32.2)[.]1171
1069 It was following this email that the reference to dyspareunia in the abstract was omitted from the manuscript. Indeed, the abstract, which Dr Hinoul considered was the only part of an article surgeons usually read, contained no reference at all to dyspareunia.
1070 The text of the published article did mention dyspareunia rates but only for women experiencing it on a frequent basis (i.e., usually or always):
When we analyzed individual outcomes [in PISQ-12] that might be affected differently after the two types of interventions, pain during sexual intercourse was reported to occur “usually” or “always” by 2% of the women after colporrhaphy and by 7.3% after transvaginal mesh surgery (P = 0.07).1172
1071 This was an acknowledgment that rates of dyspareunia were higher in the mesh group but at the same time, as I have already observed, having regard to the p-value of 0.07 it was a signal that the result was not statistically significant. In the first pre-submission draft circulated to Ethicon personnel, however, the figures for dyspareunia were 46/101 (which was reported as 34%, but in fact equates to 46%) in the colporrhaphy group and 68/110 (reported as 51%, but in fact equating to 62%) in the mesh group.1173 The p-value was 0.02, signifying that the result was statistically significant. As Professor Woodward put it, “what [in the draft manuscript] was a headline result (because it was in the abstract), with a significant outcome… became an incidental result, with a non-significant outcome, in the version for public consumption”.1174
1072 Professor Hu described the communications between Ethicon and Dr Altman as “extraordinary” and said that they raised “questions with respect to undue access and influence of the industrial sponsor (Ethicon) on the work product as it developed; the lack of disclosure of this access and influence in the published manuscript; and bias in the reporting of results that resulted from the access and influence”.1175
1073 Despite these matters, Altman et al (2011) did not offer an unqualified endorsement of the use of Prolift for anterior compartment repair. Their conclusion, summarised in the abstract of the article, was that, in comparison with anterior colporrhaphy, the use of a standardised, trocar-guided mesh kit for cystocele repair resulted in higher short-term rates of successful treatment but also in higher rates of surgical complications and postoperative adverse events.1176
1074 The case for polypropylene mesh did not improve with the publication of the 2013 Cochrane review.
1075 Maher et al (2013) maintained that “evidence of an improved anatomical cure and subjective success of prolapse surgery in the anterior compartment using transvaginal polypropylene mesh remains insufficient reason to advocate its routine use”. They said that those benefits had to be weighed against the reduced blood loss, operating time, rate of de novo stress urinary incontinence and posterior and apical compartment prolapse and lower total reoperation rate after native tissue repair.1177
1076 The evidence was said to be insufficient to support the use of transvaginal permanent meshes or grafts at the time of vaginal apical or posterior compartment repair surgery except in the context of randomised controlled clinical trials, adequately powered to evaluate the anatomic and functional outcomes and possible adverse events, with blinding of reviewers and participants to minimise biases in reporting.1178
1077 Maher et al (2013) considered that the data from randomised trials were insufficient to guide practice.1179 Conclusions were drawn only in relation to the areas of surgical management of pelvic organ prolapse where at least two RCTs had been completed. Relevantly they included the following:
• Abdominal sacral colpopexy was associated with a lower rate of recurrent vault prolapse and less dyspareunia than vaginal sacrospinous colpopexy. The abdominal sacral colpopexy had a longer operating time, longer recovery time and higher cost than the vaginal surgery. Data on the subjective success rate, patient satisfaction and impact of the surgery on quality of life were too few for reliable conclusions. In single studies the sacral colpopexy had a higher objective success rate and lower reoperation rate as compared to vaginal uterosacral ligament suspension and transvaginal polypropylene mesh. Small studies compared laparoscopic sacral colpopexy to open and robotic techniques without decisive outcomes.
• The evidence suggested that the use of … polypropylene mesh at the time of anterior vaginal wall repair reduces the risk of recurrent cystocele on examination, however improved outcomes including patient satisfaction, quality of life and reduced operations for recurrences have not yet been demonstrated. Furthermore, anterior polypropylene mesh alone demonstrated an improved subjective outcome as compared to native tissue anterior repair without any difference between the groups in the rate of dyspareunia. The operating time, blood loss, rate of apical or posterior compartment prolapse and de novo stress urinary incontinence were greater in the polypropylene mesh group, which was associated with a 11.4% rate of mesh erosion and 6.8% requiring surgical reintervention.
• … There was no evidence to support the use of graft materials in the posterior compartment.
• The evidence at this stage does not support the use of transvaginal combined total, anterior or posterior mesh kits for multi-compartment prolapse. While three studies demonstrated an improved anatomical outcome after the transvaginal permanent mesh as compared to native tissue repair, no difference was found in symptoms or quality of life outcomes. The mesh exposure rate was 18%, with one half of these (9%) requiring surgical intervention. The total reoperation rate was significantly higher after the transvaginal permanent mesh at 11% compared to 3.7% following native tissue repair.1180
1078 Professor Collinet asserted that Maher et al (2013) had noted “[a] significant decrease in the rate of follow-up surgery for recurring prolapse” after mesh surgery.1181 No reference was given but Professor Gordon said that the assertion was apparently based on a table appearing on page 228 of the review (analysis 2.26) in which risk ratios were identified from six studies comparing anterior colporrhaphy with transobturator mesh (the first and largest of which was the Altman RCT.
1079 Professor Gordon pointed out, however, that, although the risk ratio favoured mesh, the results were not statistically significant using the conventional threshold since the p-value was greater than 0.05.1182 In neither of these respects was Professor Gordon’s evidence challenged or disputed and I accept it.
1080 2016 saw the publication of three Cochrane reviews of which Dr Maher was the lead author. The first encompassed review content assessed up to 6 July 2015 and was entitled “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse (Review)”.1183 I will refer to it as Maher et al (2016a). It involved a systematic review of 37 RCTs (involving 4,023 women) which compared different types of vaginal repair: transvaginal mesh; biological graft; and native tissue. The quality of the evidence was described as “very low to moderate” overall.1184 Of the 37 trials reviewed, 25 compared polypropylene permanent mesh with native tissue repair. Seventeen of these related to anterior compartment repair, the rest to permanent mesh for apical, anterior and/or posterior repair.1185 The relevant comparison was between transvaginal mesh and native tissue repair.
1081 The review identified primary and secondary outcome measures. The primary outcome measures consisted of awareness of prolapse, repeat surgery, and recurrent prolapse (defined as any stage 2 or greater vaginal prolapse on the POP-Q scale).1186 Secondary outcomes included death related to surgery; mesh exposure, injury to the bladder or bowel, and surgery for mesh exposure; disturbance of bladder and bowel function manifested, for example, in stress urinary incontinence; de novo stress urinary incontinence; bladder overactivity or urge incontinence; and de novo bladder overactivity or urge incontinence, de novo faecal incontinence; and de novo obstructed defaecation. They also included sexual function (including de novo dyspareunia) and quality of life measured by questionnaire.1187
1082 With respect to the comparison between permanent mesh and native tissue repair the review reported that awareness of prolapse at one to three years was less likely after mesh repair, based on moderate quality evidence. The authors explained that the figures suggested that, if 19% of women are aware of prolapse after native tissue repair, between 10% and 15% will be aware of prolapse after permanent mesh repair.1188 Recurrent prolapse on examination was also less likely after mesh repair. Here, the figures suggested that if 38% of women have recurrent prolapse after native tissue repair, between 11% and 20% will do so after mesh repair. But in this case the quality of the evidence was described as low.1189
1083 Rates of repeat surgery for prolapse were also lower in the mesh group, based on moderate quality evidence. There was no evidence of a difference between the groups in rates of repeat surgery for incontinence, although the quality of the evidence was said to be low. . On the other hand, moderate quality evidence demonstrated that more women in the mesh group required repeat surgery for the combined outcome of prolapse, stress incontinence, or mesh exposure. The figures reportedly suggested that, if 5% of women required repeat surgery after native tissue repair, between 7% and 18% in the permanent mesh group will do so.1190
1084 While women undergoing native tissue repair had no risk of mesh exposure, overall 134/1097 women in the transvaginal permanent mesh groups (12%) had mesh exposure. Mesh exposure was reported in 76/753 women (10%) following anterior repair only and 58/344 women (17%) after multi-compartment repair.1191 In cross-examination, Associate Professor Rosamilia agreed that, as at February 2016, this fairly represented the rates for mesh exposure overall and in particular for anterior repair only and multi-compartment repair.1192
1085 Eight per cent of women in the mesh group required repeat surgery for mesh exposure.1193
1086 Permanent mesh was associated with higher rates of de novo stress incontinence, although the evidence was described as low quality and bladder injury, based on moderate quality evidence. There was no evidence of a difference between the groups in rates of de novo dyspareunia, but the quality of the evidence was low. Effects on quality of life were said to be uncertain due to the very low quality evidence.1194
1087 The authors concluded:
While transvaginal permanent mesh is associated with lower rates of awareness of prolapse, reoperation for prolapse, and prolapse on examination than native tissue repair, it is also associated with higher rates of reoperation for prolapse, stress urinary incontinence, or mesh exposure and higher rates of bladder injury at surgery and de novo stress urinary incontinence. The risk-benefit profile means that transvaginal mesh has limited utility in primary surgery. While it is possible that in women with higher risk of recurrence the benefits may outweigh the risks, there is currently no evidence to support this position.1195
1088 Despite its strengths, Professor Woodward pointed to a number of limitations to the analyses in this review. Indeed, he said that no account was taken of time. 1196 The majority of the trials were conducted over a relatively short period (mostly one year).1197 Since most trials were no more than one year’s duration and, as Dr Hinoul acknowledged, some complications, including erosion and exposure (and their sequelae), may arise more than one year after implantation, indeed years afterwards, the results are unreliable. Moreover, they were skewed in favour of mesh.
1089 Although the review disclosed statistical evidence that anatomical outcomes are better with permanent mesh than native tissue repair, Jia et al (2008), in an article reporting on their systematic review of the literature relating to the efficacy and safety of mesh repair in surgery for anterior and/or posterior vaginal wall prolapse (which Professor Woodward described as comprehensive and Professor Hu as rigorous)1198 wrote:
One year was considered a minimum adequate period of time to assess the efficacy of prolapse repair. However, even 1-year outcomes are too early to judge whether product surgery is successful longer term. The mean time to first reoperation is reported in the literature as 12 years, and therefore, failure at 1 year should not be regarded as an adequate representation of efficacy. Prospective studies would require extended follow-up to assess meaningful mesh/graft failure.1199
1090 Jia et al (2008) also observed that it was “increasingly recognised that in prolapse surgery subjective failure is a more appropriate outcome measure of efficacy than objective failure” and that few studies reported on subjective symptoms of importance to women relating to urinary, bowel and sexual function.1200
1091 Similarly, Rudnicki et al (2016) reported on their own randomised controlled study comparing one year and three year objective and subjective cure rates and complications (relating to the use of a collagen-coated transvaginal mesh for anterior wall prolapse against a conventional anterior repair), considered that the three-year follow-up period was probably insufficient to evaluate the impact of a synthetic mesh on erosion rates and subjective outcomes.1201 They frankly acknowledged that this was a weakness of their study. Professor Woodward observed that none of the trials covered by Maher et al, nor any updates (like Rudnicki et al) and, indeed, no later studies he had seen have had longer than a three year follow-up.
1092 Professor Woodward also pointed out that several of the trials reviewed by Maher et al in the first 2016 Cochrane review did not analyse surgery following mesh exposure. As he said, this omission necessarily underestimated the risk ratio for subsequent surgery. Only seven trials reported on total surgery, consisting of surgery for recurrent prolapse, incontinence, and mesh exposure. Professor Woodward put the best estimate of the risk ratio at 2.40 (95% confidence interval: 1.51 to 3.81) which was highly significant (p = 0.0002) in favour of native tissue repair. In other words, the chance of total surgery more than doubled after mesh implantation compared to native tissue repair.1202
1093 Professor Woodward observed that the Altman RCT, which was by far the largest trial,1203 was “hugely influential in [Maher et al’s] analysis”, supplying almost half the information.1204 It was also relied upon by the respondents and their witnesses. With good reason, Professor Woodward was concerned about the potential for bias in the Altman et al (2011) study.
1094 Professor Woodward concluded that the randomised data were of insufficient quality and insufficient depth to produce a complete picture of the risks associated with the use of mesh in comparison to native tissue repair. In particular, the studies were both insufficiently powered to adequately deal with some of the key outcomes and likely to be too short to capture all the adverse outcomes.1205 “By and large”, he wrote:
[T]he RCTs have only seriously addressed the issue of recurrence of prolapse, with insufficient numbers to adequately explore the risks of subjective outcomes, and have generally underestimated the risk of repeat surgery with mesh use, relative to traditional surgery. The review has an important limitation in its lack of control for confounding, which is a reflection on the data available to the authors. In addition, the Cochrane review has no data on several outcomes [reported to arise with the use of mesh] such as infections and pain in a general sense, rather than just bladder or bowel.1206
1095 The second Cochrane review of 2016 prepared by Maher et al encompassed content assessed up to 23 August 2016. It was entitled “Surgery for women with anterior compartment prolapse (Review)”.1207 I will call it Maher et al (2016b).
1096 The objective of Maher et al (2016b) was to determine the safety and effectiveness of surgery for anterior compartment prolapse. It reviewed 33 RCTs evaluating 3,332 operations.1208 Eight trials compared traditional native tissue anterior repair with biological grafts, three with absorbable mesh, two with abdominal paravaginal repair, and 16 with permanent (polypropylene) mesh. The quality of the data was described as “generally low to moderate”. It was published after Professor Woodward’s first report but before his second. Professor Woodward did not comment on it, however, presumably because he was not asked to. Since this Cochrane review relied on many of the same studies considered by the earlier 2016 Cochrane review (with a focus on anterior compartment prolapse), Professor Woodward’s comments would apply with equal force to this one.
1097 Below I discuss the findings relating to polypropylene mesh versus native tissue repair.
1098 I begin with the primary outcomes.
1099 Awareness of prolapse (one to three years) was more likely after native tissue repair (anterior colporrhaphy) than after mesh repair, based on moderate quality evidence. According to the authors the figures suggested that if awareness of prolapse after polypropylene mesh repair occurs in 13% of women, then 18% to 30% would develop awareness of prolapse after native tissue repair.1209
1100 There was no evidence of a difference in the rate of repeat surgery for stress urinary incontinence between the two groups.
1101 Repeat surgery for prolapse, stress urinary incontinence or mesh exposure (composite outcome) was less likely after native tissue repair than after polypropylene mesh repair. The analysis suggested that, if 10% underwent subsequent surgery after polypropylene mesh repair, then 4% to 8% would require subsequent surgery after native tissue repair.1210
1102 Recurrence of anterior wall prolapse (one to three years) was more likely after native tissue repair than after polypropylene mesh repair (moderate quality evidence). The figures suggested that, if recurrent anterior wall prolapse occurred in 13% of women after polypropylene mesh repair, then 32% to 45% would have recurrence after native tissue repair. Repeat surgery for prolapse was also more likely after native tissue repair, with the authors suggesting that if 2% of patients underwent repeat surgery after mesh, then 2% to 7% would require surgery after native tissue repair.1211
1103 I turn now to the secondary outcomes.
1104 With respect to objective failure, subsequent stage 2 or greater prolapse in the posterior or apical compartment was less likely after native tissue repair than after polypropylene mesh repair. The figures suggested that, if 18% of women developed prolapse in the apical or posterior compartment on examination after polypropylene mesh repair, then 5% to 18% would develop apical or posterior compartment prolapse after native tissue repair.1212
1105 With respect to bladder function, native tissue repair was associated with a reduction in de novo urinary stress incontinence compared with mesh repair (moderate quality evidence). The figures suggested that if 10% developed de novo stress urinary incontinence after mesh repair, 5% to 10% would develop the same condition after native tissue repair. Studies provided no evidence of a difference in postoperative urge incontinence at one year or urinary voiding dysfunction at one to two years but caution was advised in interpreting the results due to low event rates and wide confidence intervals crossing the line of no effect.
1106 Intraoperative cystotomy was less likely after native tissue repair (1/416) than after polypropylene mesh repair (11/455).
1107 No data was available on bowel function.
1108 As for sexual function, there was no evidence of differences between groups in the rate of de novo dyspareunia (one to three years) on the basis of moderate quality evidence and no evidence of a postoperative difference in the rate of dyspareunia between native tissue repair and mesh repair. Nor was there evidence of a difference between the groups on the prolapse quality of life questionnaire (one year) or the pelvic floor impact questionnaire (one to two years).1213
1109 The mesh exposure rate after transvaginal polypropylene mesh was 11.3% (101/896) at one to three years. The repeat surgery rate for mesh exposure was 7.3% (56/768).
1110 Perioperative outcomes favoured native tissue repair. Operating time was shorter and blood transfusion less likely after native tissue repair. There was no difference between groups in length of hospital stay.
1111 The authors relevantly concluded that:
Native tissue repair was associated with increased awareness of prolapse and increased risk of repeat surgery for prolapse and recurrence of anterior compartment prolapse compared with polypropylene mesh repair. However, native tissue repair was associated with reduced risk of de novo SUI, reduced bladder injury, and reduced rates of repeat surgery for prolapse, stress urinary incontinence and mesh exposure (composite outcome).
Current evidence does not support the use of mesh repair compared with native tissue repair for anterior compartment prolapse owing to increased morbidity.1214
1112 Associate Professor Rosamilia agreed that as at November 2016 when this review was published, this was a fair representation of the state of the evidence relating to the use of polypropylene mesh for transvaginal repair of pelvic organ prolapse in the anterior compartment.1215
1113 The third 2016 review prepared by Maher et al was entitled “Surgery for women with apical vaginal prolapse (Review)”.1216 I refer to it as Maher et al (2016c).
1114 The objective of Maher et al (2016c) was to evaluate the safety and efficacy of any surgical intervention compared to another intervention for the management of apical vaginal prolapse. It included 30 RCTs involving 3,414 women comparing surgical procedures for apical vaginal prolapse. Evidence quality ranged from low to moderate.
1115 The authors concluded that compared to various vaginal repairs, sacralcolpopexy was associated with lower rates of awareness of prolapse, repeat surgery for prolapse, prolapse on examination, urinary stress incontinence and painful intercourse. The evidence did not support the use of transvaginal mesh compared to native tissue repairs. The evidence was imprecise, but suggested that if 18% of women are aware of prolapse after surgery without mesh, between 6% and 59% will be aware after surgery with mesh.1217
The Morling study
1116 It will be recalled that this was a very large cohort study based on hospital records which covered women who had undergone a single incontinence or prolapse procedure from 1 April 1997 to 31 March 2016. I referred above to the findings in the incontinence group.
1117 In the prolapse group, the authors found no evidence that mesh surgery for anterior or posterior compartment prolapse provided in routine clinical practice was more effective than non-mesh surgery over the longer term and, to the contrary, mesh surgery was associated with an overall increased need for repeat surgery. As for safety, they found the use of mesh in the repair of anterior and posterior compartment prolapse was associated with both increased risk of complications and lower effectiveness than the equivalent non-mesh repairs. Anterior colporrhaphy with mesh, for example, was associated with a similar risk of immediate complications but a higher risk of later complications, subsequent incontinence surgery, and subsequent prolapse surgery than anterior colporrhaphy without mesh.
1118 Professor Woodward pointed out that the risk of late complications with mesh was roughly three times the risk of late complications with native tissue repair and the risk of recurrent surgery for both anterior and posterior compartments was roughly twice as great when mesh was used than when it was not.1218
1119 For patients undergoing repair of vaginal vault prolapse, the authors of the Morling study detected no differences in outcome (complications or further surgery) after vaginal mesh or, separately, abdominal mesh surgery compared with non-mesh vaginal repair.
1120 Morling et al (2016) concluded that mesh procedures for anterior and posterior compartment prolapse could not be recommended for primary prolapse repair. They found them to have poorer overall effectiveness and substantially increased later complications compared with similar non-mesh repairs. On the other hand, they also concluded that vaginal and abdominal mesh procedures for vault prolapse repair had similar effectiveness and complications rates and that no particular vault repair procedure was clearly favoured. It should be noted that at the time of publication of the Morling study, PROSPECT (discussed below) was under way.
1121 The Morling study is significant, not least because it cast doubt over the widespread perception that the risk of recurrence of pelvic organ prolapse following native tissue procedures is high. Dr Agur said that the Morling study provided “a more accurate perception of the recurrence rate” since this was a large retrospective study and in over 18,000 colporrhaphy procedures the recurrence rate was only 3.6%.1219 Dr Agur’s figure for the number of colporrhaphy procedures was slightly out. Morling et al (2016) reported on 18,986 first, single prolapse procedures of which 1,279 involved the use of mesh which brings the number for colporrhaphy procedures down to 17,707. But the difference is hardly of any consequence. On any view of the matter this was a large study. The recurrence rate is not cited in the article. There was a lengthy appendix which was not tendered and it may have been included there. In any case, Dr Agur was not challenged about the rate or taken to task about his point. In the circumstances there is no reason why I should not accept it and I do.
1122 PROSPECT (an acronym for “Prolapse Surgery: Pragmatic Evaluation and randomised Controlled Trials”) was twice the size of the Altman RCT and the biggest randomised controlled trial conducted in the field of transvaginal mesh to date. Professor Woodward considered that it was the best designed of the trials with a sensible randomisation process and analysis plan. Furthermore, to his knowledge, none of the authors had a relevant conflict of interest.1220
1123 Conducted over the period from early January 2010 to the end of August 2013, the purpose of the trial was to compare the outcomes of primary prolapse repair using either synthetic mesh or biological grafts with the outcomes of native tissue repair. The results were reported in an article by Professor Glazener and others on behalf of the PROSPECT study group published online in The Lancet on 20 December 2016.1221 The full report of the study was published the same month by the the National Institute for Health Research under a slightly different title.1222
1124 PROSPECT encompassed two parallel group, multicentre, randomised controlled trials conducted in the United Kingdom. Thirty-five centres were involved. They consisted of secondary and tertiary referral hospitals. This meant that the trial was conducted in what Professor Woodward described as “a natural setting”, within the National Health Service, rather than in a clinical setting using specialised surgeons or centres.1223 A total of 1352 women were randomly allocated to treatments. Of them, 1348 were included in the analysis, 865 in the mesh trial (430 to standard repair alone, 435 to mesh augmentation) and 735 were in the biological graft trial (367 to standard repair alone, 368 to graft augmentation).1224 Participants, ward staff, and outcome assessors were masked to randomisation where possible; masking was obviously not possible for the surgeon. Outcomes were measured by participant-completed postal questionnaire at baseline (before surgery), and six months, one year, and two years after surgery, and in a clinic review appointment at one year.1225 The study was overseen by an independent steering committee and an independent data monitoring committee.1226
1125 In contrast to the findings of the most recent Cochrane review, this study showed that in the first two years after surgery, women derived no benefit from the use of either transvaginal synthetic mesh or biological graft to reinforce a standard anterior or posterior repair.1227 The study showed that “more than 30% of women who have prolapse surgery have a residual feeling of something coming down and more than 80% have at least one residual prolapse symptom, highlighting the poor short term outcomes of transvaginal anterior or posterior prolapse surgery with or without reinforcement”.1228 Glazener et al (2016) observed that when the PROSPECT data were added to the Cochrane review results, the summary statistics continued to favour mesh both in terms of awareness of prolapse and anatomic recurrence. Importantly, however, it highlighted a number of matters which supported the view expressed by Dr Agur that in some situations one randomised controlled trial can have greater weight than a meta-analysis of multiple trials.1229 Those matters were:
the quality of the evidence in the trials in the Cochrane meta-analysis, described as “very low to moderate, due to poor reporting of study methods, inconsistency, and imprecision in the included trials”;
heterogeneity in the trials included in the Cochrane review, since they included women who had been treated for uterine or vault prolapse;
the paucity of trials differentiating between women having primary and secondary repairs;
some trials used mesh kits rather than inlays;
variability in the inclusion criteria concerning concomitant procedures and continence surgery.
1126 In contrast, Glazener et al (2016) pointed out that PROSPECT randomly assigned a strictly defined group of women having their first repair in an anterior or posterior compartment and used only non-absorbable mesh inlays. They postulated that:
A single large trial that is free from risk of bias might be more powerful and reliable for the specific population included than a meta-analysis of many smaller trials.
1127 Glazener et al (2016) also reported no clinically important differences in either trial in individual serious adverse effects such as infection, urinary retention, dyspareunia or other pain or other non-serious adverse effects in the first or second years after surgery.1230
1128 In the mesh trial, however, 25 of the 865 women had surgery to remove part of the mesh in the first year. Eighteen (72%) were asymptomatic and 16 (64%) had exposures of less than 1 cm². One woman had total mesh removal within two weeks of surgery because of severe infection. In the second year, 17 women had surgery to remove part of the mesh. Thirteen of them (76%) were asymptomatic and 10 (59%) had exposures of less than 1 cm²). The remaining women who had a mesh complication were treated as outpatients by observation, topical treatment with oestrogen, mesh trimming, or cautery. Most mesh exposures were small or asymptomatic requiring partial removal as a day case.
1129 The cumulative number of women with a mesh complication over two years in women implanted with synthetic mesh was 51/434, which equates to 12%. Of the 51 women who had a mesh complication, 37 required surgical removal, which gives an overall rate of surgery for a mesh complication of 37/434 or 9%. These numbers related to women who had received synthetic mesh as part of their prolapse repair or a concomitant vault, uterine or continence procedure. When restricted to women who only received mesh as part of their prolapse surgery (284), the mesh complication rate in the first two years was higher (41/284 or 14%).1231
1130 Glazener et al (2016) summarised their conclusion in the abstract of the article:
Augmentation of a vaginal repair with mesh or graft material did not improve women’s outcomes in terms of effectiveness, quality of life, adverse effects, or any other outcome in the short term, but more than one in ten women had a mesh complication. Therefore, follow-up is vital to identify any longer-term potential benefits and serious adverse effects of mesh or graft reinforcement in vaginal prolapse surgery.1232
1131 Having regard to their conclusion that there was no benefit in the short term (either anatomic or symptomatic) of using synthetic mesh, the authors queried whether the potential for additional surgical procedures for exposures and extrusion in the first two years was an unnecessary risk. That query appears to have been a rhetorical one, having regard to the penultimate sentence of the article:
This additional risk suggests that in the future mesh should only be used in the context of trials aimed at identifying benefit from modifying mesh type or insertion techniques, or in defined categories of high-risk women. Long-term follow-up to assess both effectiveness and adverse effects, which is ongoing, is vital.1233
1132 The PROSPECT study did not identify the brand or brands of mesh that was implanted so that these figures should be treated with some caution. That said, they are not markedly different from figures appearing in other studies involving Ethicon’s POP devices and the respondents appear to have accepted that their products were not significantly or relevantly different from the polypropylene meshes produced by their competitors.
The prevalence of the pleaded complications
1133 As the discussion above illustrates, the evidence from the published studies was not always consistent. Nor was it presented in a form that allows for precise conclusions to be reached. The quality of the studies was variable. Some of the studies were too general and reported on all pelvic floor meshes or all meshes of a particular type. Others were limited to narrow applications, for example to the implantation of a specific device using a specific technique. Importantly, the follow-up periods varied, and were seldom long enough to provide a reliable indication of long-term risks, a matter of considerable importance for medical devices intended for permanent implantation. Often the populations studied were selective so that that the results were not readily generalisable. Some of them excluded patients who were at higher risk of achieving worse outcomes. To some extent, the expert evidence helped to discriminate between studies, but not always.
1134 For all these reasons any attempt to distil the evidence into a neat summary of incidence rates would be riddled with potential pitfalls. Mercifully, the respondents’ concession during oral argument that each of the pleaded complications is significant and their assurance that they would not take up any issue as to the precise rate of any complication makes it unnecessary to do so.1234
1135 It is nevertheless important to consider the evidence concerning the prevalence of the pleaded complications because it is relevant to the safety profile of the various Ethicon devices, which in turn is relevant to three of the applicants’ statutory claims. It is also relevant to the negligence claims because it tells us something about the magnitude of the risk the Ethicon devices posed to the safety of the applicants and the group members.
1136 No evidence was adduced about the extent of the increase in risk to which women with autoimmune disorders were and are exposed.
1137 The summary below is not intended to be an exhaustive representation of the evidence. Rather, it is a snapshot of what was revealed by it.
1138 The respondents were very critical of Professor Gordon for drawing on studies relating to non-Ethicon devices in his meta-analyses. At the same time, however, they urged the Court to put weight on the findings and conclusions of the Cochrane meta-analyses, which were not confined to their devices. In effect, therefore, if not in terms, they accepted that the conclusions of the Cochrane reviews about synthetic polypropylene mesh devices apply to their devices. That necessarily means, at least for the purposes of this case, that it is common ground that there was no material difference in the nature or rate of complications associated with the Ethicon devices and similar devices made by the respondents’ competitors, which also used polypropylene mesh or tape.1235
1139 According to the RCOG categories, the evidence indicates that the following complications are common after surgery with the SUI devices (that is, occurring in 1% to 10% of patients):
mesh exposure/extrusion/erosion;
recurrent urinary tract infections;
chronic pain;
dyspareunia;
difficulty voiding;
de novo urinary incontinence;
recurrence of stress urinary incontinence;
bladder perforations (with retropubic slings) (uncommon but not rare with transobturator slings); and
reoperation or revision surgery associated with complications.
1140 After surgery with the POP devices, the rates were higher.
1141 The evidence indicates that mesh erosion (encompassing erosion, exposure and extrusion) has been very common with transvaginal implantation and common after abdominal placement. The cure rates provided by Maher et al (2016a) show that recurrence of pelvic organ prolapse was also very common with POP devices.
1142 The studies demonstrate that the following complications were at least common:
mesh exposure/extrusion/erosion requiring surgery;
chronic pain;
dyspareunia;
de novo urinary incontinence; and
reoperation or revision surgery associated with complications.
1143 Mesh erosion is a serious adverse event and exposure is too, when surgical intervention is required.1236
1144 The reported rate of tape erosion (including extrusion and exposure) across the studies was around 2% to 5%. The true rates may well be higher, given the paucity of long-term studies and the limitations of the Nordic studies discussed above.
1145 In the Ward Hilton RCT, there were five erosions or extrusions of the mesh at the five year follow-up.1237 While the trial started out with 175 patients, only 98 were available for follow-up by the five year mark. No percentage rate was provided by the authors in their five year report, but depending on the denominator, the rate could be between 3% and 5%.
1146 Neither Ogah et al (2009) nor Ford et al (2015) discerned a significant difference between the rates of tape erosion for transobturator and retropubic slings. Ford et al (2015) reported a rate of 2.4% in the transobturator group and 2.1% in the retropubic group. Fusco et al (2017) reported a lower rate of erosion for retropubic slings (1.8%) than transobturator devices (2.8%) but noted that this was attributable mainly to the higher risk of erosion in the outside -in tapes (whereas TVT-O uses an inside-out technique). I conclude that the rate of erosion is not significantly different for TVT-O, TVT, and TVT Exact.
1147 Schimpf et al (2014) nominated an exposure rate of 2% for TVT Secur.1238 Nambiar et al (2014), however, suggested that the rate is higher for both vaginal mesh exposure and bladder/urethral erosion. They reported that, in five trials comparing TVT Secur with inside-out transobturator slings, more women in the TVT Secur groups had exposure (18/284 or 6%), than in the transobturator groups (4/278 or 1%) and the overall result was statistically significant. Only two small trials reported on mesh extrusion into the bladder or urethra. Eight women were reported to have experienced the complication, all in the single incision group.
1148 In their RCT comparing TVT Secur with TVT-O, Hinoul et al (2011) found that TVT Secur was associated with a higher incidence of mesh exposure than TVT-O. Over 12 months the rate was 7% for TVT Secur.1239
1149 I conclude that the rate of erosion for TVT Secur is likely to be higher than for the other SUI devices and is probably in the vicinity of 6% to 7%.
1150 I am unable to put a figure on the likely rate for TVT Abbrevo.
1151 In all likelihood, however, erosion is common to all the SUI devices, but most of these erosions are cases of vaginal exposure.
1152 Infection is a broad category and the evidence as to its prevalence is not settled.
1153 A number of studies reported immediate postoperative infection and urinary tract infections separately from other infections. Ward and Hilton, for example, reported symptoms of recurrent urinary tract infections, but the difference was not statistically significant: at the two- year follow-up, 10 patients with TVT (5.9%) and three with colposuspension (2.1%) had urinary tract infections.1240
1154 Fusco et al (2017) found that there was a higher incidence of urinary tract infections (10%) with retropubic slings than transobturator slings (7.9%). Ford et al (2015) commented that major complications were uncommon and unlikely to be picked up by small randomised controlled trials. They considered that a more accurate incidence could be gleaned from large national registries and databases such as MAUDE, although this method had limitations, too. Based on registry data, they reported an infection rate of 0.7% with retropubic tapes and 0.6% with transobturator tapes.1241
1155 Professor Gordon concluded that the evidence showed that the SUI devices caused fewer infections than the alternative treatments.1242 But the evidence about urinary tract infections was characterised as “equivocal”, that is, Professor Gordon could not be confident whether the incidence following surgery with the SUI devices was greater or less than with the alternative treatments. It is difficult to know what to make of these conclusions. Infections following Burch colposuspension are likely to occur intraoperatively or in the immediate post-operative period, whereas infections due to the implantation of a synthetic sling could occur at any time.
1156 Generally speaking, chronic pelvic pain is pain that has lasted for six or more months.1243 While there were reports of pain in numerous studies, it was not always clear how long the pain lasted or when it arose. Pain in different locations and of varying duration was reported in the studies.
1157 The applicants submitted that the rate of chronic pain following surgery with midurethral slings was 14%, relying on a review by Barski and Deng (2015).1244 But the figure was not supported by the source. Consideration of the authors’ methodology reveals that the figure did not represent the incidence of chronic pain. Barski and Deng (2015) reviewed retrospective cohort studies reporting on complications and calculated the percentages for each adverse event as a proportion of a pool of patients all of whom had complications. Fourteen percent of the pool of patients with complications had chronic pain. This tells us very little. It is certainly not evidence that the incidence of chronic pain after surgery with midurethral slings is 14%.
1158 In a retrospective study of 235 women conducted via postal questionnaires, Bourrat et al (2003) found that pain impairing quality of life was reported by 30% of respondents after surgery with TVT.1245 From 156 responses reporting on pain, 5.8% reported pain when walking, 9% pain with urination, and 6.4% permanent pain. Ongoing pain was particularly frequent for women with a history of incontinence surgery but was also reported in 7.7% of isolated TVT repairs. The authors did not specify the time period over which the pain was experienced, but it was likely chronic because the cohort of patients was said to have had surgery between November 1998 and December 2000, while the questionnaires were sent out in July 2001.
1159 The authors wrote that, to their knowledge, postoperative pain in such patients had not previously been studied. They were surprised by the rate of its occurrence. They recommended that a prospective study be carried out exploring the relationship between pain and TVT. I was not taken to any evidence to indicate that Ethicon ever undertook such a study.
1160 Dr Hinoul was taken to the Bourrat et al (2003) article during cross-examination. He dismissed its significance because it was retrospective, it was “only a questionnaire” (presumably patients’ accounts of their symptoms do not count for much), and the response rate was low (although I would have thought that a 67% response to a voluntary postal survey was fairly high).1246 I accept that there are limitations to the study. For a start, as the respondents pointed out, the patients were not assessed in the same way before undertaking the TVT surgery. 1247 It is significant, however, that in a PowerPoint presentation intended for internal use, Ethicon highlighted the 30% figure and said that Bourrat et al were “[p]robably the only authors who have more or less correctly assessed pain after TVT!”1248 The inference I draw is that, despite Dr Hinoul’s evidence, Ethicon understood that chronic pain was a significant complication of TVT.
1161 Blaivas et al (2015) estimated that the incidence of chronic refractory pain following sling surgery was 4.1%, where chronicity was defined as greater than six weeks.1249 Professor Blaivas said in his report that “multiple studies” had shown a prevalence of about 5% chronic pain after sling surgery, but did not identify the studies.1250
1162 Ogah et al (2009) did not report on pain in their comparison of synthetic slings with traditional slings and colposuspension.1251 They did, however, report that groin pain was more common with a transobturator approach (12%) compared with suprapubic pain in women with a retropubic sling (1.7%).1252
1163 Ford et al (2015) also reported that the rate of groin pain was higher in the transobturator group (6.4%) than in the retropubic group (1.3%), but noted that women in the retropubic group had a higher rate of suprapubic pain (2.9%) than those in the transobturator group (0.8%). They stated that both were “short-lasting”.1253 It is worth noting, however, that the duration was said to have ranged from two to 52 weeks, with a median duration of eight weeks, indicating that half of those reporting pain experienced it for over eight weeks.1254 The pain in at least some of those patients would not be characterised as “short-lasting” and may be chronic if it lasted beyond the post-operative healing period.
1164 As noted above, Zhang et al (2016),1255 whose study was cited by Fusco et al (2017), reported long-term pain after both TVT and TVT-O. At seven year follow-up, this prospective study found groin/thigh pain in four TVT-O patients (6.4%) and one TVT patient (1.72%), and abdominal pain in five TVT patients (8.62%) and two TVT-O patients (3.23%). The total incidence of long-term pain was 10.34% for TVT patients and 8.05% for TVT-O patients.1256
1165 A lower rate was reported at nine years follow-up by Karmakar et al (2017), who compared an inside-out transobturator sling (TVT-O) with an outside-in sling. The authors found that groin or thigh pain or discomfort was reported by four of the 104 patients (3.84%) in the TVT-O group.1257 A similar rate (3 to 4%) of groin pain was reported by Leval et al (2011) 12 months after implantation of TVT Abbrevo following their RCT comparing TVT-O and TVT Abbrevo.1258
1166 While Hinoul et al (2011), after another randomised controlled trial, this time comparing TVT Secur with TVT-O, reported that TVT Secur was associated with significantly less pain than TVT-O during the first two weeks postoperatively, after two weeks there was no difference in persisting pain levels.1259
1167 The evidence does not allow for a definitive conclusion to be reached on the incidence of chronic pain after implantation of an SUI device. Having regard to the findings in the various reviews and studies discussed above, however, the incidence appears to be between 1% and 10%. It is therefore common, and not rare as Dr Hinoul and others have claimed.
1168 There did not appear to be much evidence of chronic pain after native tissue repair, although Professor Korda’s training slides from 2004 on TVT stated that chronic pain had been reported in up to 12% of patients after colposuspension. 1260 Importantly, however, the weight of the evidence was that in the cases where it does arise, it is treatable. Fusco et al (2017) suggested that “[i]ntractable suprapubic pain has been previously described following colposuspension” but cited no reference. 1261 In the end, there was no reliable evidence of the rate of chronic pain after alternative forms of treatment.
1169 Like chronic pain, not many studies reliably reported on the incidence of dyspareunia. In the survey conducted by Bourrat et al (2003), the authors found that pain with intercourse was notably more frequent when associated prolapse surgery was carried out but that it occurred in 5.5% of respondents with TVT.1262
1170 In a literature review published in 2012, Shah and Badlani (2012) found that dyspareunia had been reported in 6.2% of patients after sling surgery using synthetic mesh.1263
1171 Barski and Deng (2015) stated that dyspareunia was amongst the most common complications following implantation of midurethral slings, reporting a figure of 6%.1264 As I observed previously, however, their analysis did not allow for the derivation of accurate incidence rates.
1172 Others reported on the occurrence of dyspareunia, such as Dunn et al (2014), who described the gravity of this complication, but did not report on the incidence rate in a defined or controlled population. 1265
1173 Neither party drew the Court’s attention to a study reporting on apareunia arising independently from dyspareunia. But it is unlikely that there would be any significant difference in the rates. After all, dyspareunia is likely to cause apareunia.
1174 I conclude that dyspareunia and apareunia following implantation of the SUI devices is likely to be common.
Reoperation or revision surgery
1175 There was not a great deal of evidence on the rates for reoperation and revision surgery after implantation of the SUI devices.
1176 The Ford et al (2015) Cochrane review noted that reoperation rates relating to tape insertion or postoperative voiding dysfunction drawn from registries ranged from 1.6% to 2.4% and, like the other rates drawn from the registries, this range was “largely of the same order” as those reported in the trials included in their review.1266 Nambiar et al (2014) reported that further incontinence surgery was more likely with mini-slings, including TVT Secur.1267 The evidence showed that reoperation rates were similar between retropubic and transobturator devices. Both Ogah et al (2009) and Ford et al (2015) indicated, without being able to reach a conclusion that that repeat incontinence surgery was more likely with transobturator devices than with retropubic devices. Since mesh exposure was more frequent with mini-slings, it stands to reason that revision surgery for exposure is also likely to be higher than the rates for revision surgery following implantation of midurethral or transobturator devices.
1177 I conclude that reoperation or revision surgery is likely to be common following implantation with the SUI devices.
1178 The applicants cited a number of studies, which showed that the incidence of post-operative voiding dysfunction after surgery using the SUI devices was somewhere between 2% and 8%.1268 But the evidence on the relative incidence of this complication was not consistent.
1179 Ogah et al (2009), for example, found that post-operative voiding dysfunction was less frequent after placement of midurethral slings than after traditional slings and open retropubic colposuspension,1269 while Lapitan et al (2016) concluded that voiding problems were more common after sling procedures than after open colposuspension.1270 The analysis of Lapitan et al (2016), however, involved a comparison of open retropubic colposuspension against both synthetic and traditional slings, and the results were highly influenced by one large study that reported almost no risk of developing voiding dysfunction after open colposuspension.1271 Lapitan et al (2016) stated that consistent data from other trials showed no significant different in the risk of voiding dysfunction between the two groups.
1180 Professor Korda considered that there was a significantly greater incidence of voiding problems after retropubic TVT surgery than after TVT-O because of the difficulty in achieving the right amount of tension.1272
1181 In the six month report of the Ward Hilton RCT, the authors noted that 11 patients (9%) in the TVT arm and eight (7%) in the colposuspension arm had voiding disorder.1273 One patient who had obstructed voiding required the tape to be surgically divided.
1182 Ford et al (2015), who assessed rates of postoperative voiding dysfunction in 37 trials with 6,200 participants said that the average rate across both the transobturator and the retropubic groups was 5.53% but the rates in the transobturator group were significantly lower than those in the retropubic group.1274
1183 I conclude that voiding difficulties are not uncommon after implantation with the SUI devices.
De novo or recurrent urinary incontinence
1184 In their written submissions, the applicants referred to a number of studies that showed de novo urgency or overactive bladder after sling surgery in about 8% to 10% of women.1275
1185 Ford et al (2015) reported an average rate of de novo urgency and urgency urinary incontinence of 8.3% in the short term across both transobturator and retropubic groups with no significant difference in the medium or the long term.1276
1186 Ogah et al (2009) found that de novo urgency symptoms and urgency incontinence rates were similar between synthetic suburethral slings (9%) and open retropubic colposuspension (13%). When compared with laparoscopic colposuspension and “traditional” slings (such as those using autologous fascia), synthetic slings fared better, having lower rates of de novo urgency or urge incontinence.1277
1187 Recurrence of stress incontinence was reported in one form or another by a number of studies. Usually, the efficacy rates gave a good indication of whether incontinence had returned.
1188 Ogah et al (2009) found that there was no significant difference in the efficacy of synthetic suburethral slings when compared to traditional slings, open retropubic colposuspension, and laparoscopic colposuspension, although there was conflicting evidence about the effectiveness of synthetic slings compared to laparoscopic colposuspension in the short term.1278 Lapitan et al (2016) reached a similar conclusion but, as noted, their analysis involved a comparison of open retropubic colposuspension against both synthetic and traditional slings.1279
1189 Dean et al (2017), reviewing the data on laparoscopic colposuspension, found that there were no significant differences in the reported short and long-term subjective cure rates of laparoscopic colposuspension and “self-fixing” vaginal slings, but that objective cure rates at 18 months favoured slings.1280 Rehman et al (2017), reviewing traditional suburethral slings, found that traditional slings and minimally invasive slings appeared to be equally effective in the short term.1281
1190 These outcomes were consistent with Professor Gordon’s conclusion that there was no reliable evidence to indicate that the SUI devices were more effective than the alternative treatments.1282
1191 The study by Fusco et al (2017) was the outlier. It found that patients receiving midurethral slings had significantly higher cure rates than Burch colposuspension. On the other hand, they also found that midurethral slings and pubovaginal slings had similar cure rates.1283
1192 I conclude that de novo or recurrent urinary incontinence is a common outcome of surgery using the SUI devices, but unlikely to be any more frequent than it is after traditional surgery.
Damage to surrounding organs, nerves, ligaments, tissue and blood vessels
1193 The evidence indicates that the rate of bladder perforation is lower with transobturator slings than with retropubic slings. According to Ford et al (2015), the rate was 0.6% with transobturator slings versus 4.5% for retropubic slings.1284 They reported that the average bladder perforation rate across both groups was 2.54%.1285 Major vascular injury (such as retropubic haematoma) and major visceral injury (such as bowel perforation) also occurred but significantly less often with the transobturator approach than the retropubic approach.1286
1194 Bladder perforation also appears to be more common with retropubic slings than with colposuspension.
1195 In their article on the six month follow-up of their RCT, Ward and Hilton reported that they had had bladder perforations in 15 patients (9%) in the TVT group and three (2%) following colposuspension (p=0.013). Vaginal perforations occurred in five patients (3%) in the TVT group and none in colposuspension.1287
1196 Ogah et al (2009) also found that minimally invasive synthetic suburethral slings had significantly more bladder perforations (6%) than open retropubic colposuspension (1%).1288 Lapitan et al (2016) estimated similar rates based on data comparing transvaginal tape (6.3%) with open colposuspension (0.9%).1289
1197 Ogah et al (2009) reported no statistically significant differences in the rate of bladder perforations when minimally invasive synthetic suburethral slings were compared with laparoscopic colposuspension and traditional slings.1290 In relation to both comparisons, however, the authors noted that the confidence interval was wide and they could not exclude the possibility that the data favoured traditional slings and laparoscopic colposuspension over minimally invasive synthetic slings in terms of bladder perforations.
1198 In a paper on the prevention, diagnosis and management of complications following midurethral slings, Hengel et al (2017) said that urethral injury is “relatively uncommon”, but that, if it is not recognised, leads to “a potentially devastating mesh erosion”, and that small bowel injury and perforation were rare, but serious, complications of retropubic surgery involving midurethral slings, occurring in 0.02% of cases. They also said that “[a]lthough rare, pelvic neurovascular structures, including the obturator nerve and iliac vessels, can be injured during trocar passage”.1291 They cautioned surgeons to be mindful of the risk of such an injury where there were any concerns about intra or postoperative vital signs or in the event of immediate postoperative leg weakness.
1199 Based on the RCOG classifications, I conclude that bladder and vaginal perforations are common after implantation with the retropubic SUI devices and uncommon after implantation with the transobturator SUI devices. The evidence is insufficient to enable me to reach a finding about the incidence of damage to other surrounding organs or vessels or to nerves, ligaments, tissue and blood vessels.
1200 There was not a great deal of evidence about leg weakness as a complication of any the devices.
1201 In a multicentre randomised controlled trial in the United States involving 597 women, Richter et al (2010) compared outcomes of retropubic (TVT) and transobturator (TVT-O and Monarc) devices. Twelve months after surgery, 565 (94.6%) completed the 12-month assessment. Richter et al (2010) reported that: 1292
The frequency of neurologic symptoms was … higher in the transobturator-sling group than in the retropubic-sling group (P=0.01); weakness in the upper leg was the most common neurologic symptom, occurring in 24 (60%) of those who reported neurologic symptoms.
1202 Similar outcomes were reported in the two year report of the study.1293
Offensive discharge, haemorrhage and psychiatric injury
1203 While there was evidence that complications of sling surgery could involve vaginal discharge, there was insufficient evidence to enable any finding to be made on the reported, let alone likely, incidence of this complication.
1204 Professor Deprest said that haemorrhage was a potentially serious, though uncommon, complication associated with the use of the SUI devices, which could result from hitting pelvic vessels with the needle. He observed that it could be “dramatic”, difficult to manage, and potentially lethal.1294
1205 The studies did not report on psychiatric injury as a complication of surgery with the SUI devices. No doubt long term complications, such as chronic pain, dyspareunia, and recurrent infections, could lead to psychiatric harm. The anxiety and depression experienced by some women with mesh complications were described by Dunn et al (2014).1295 There is similar evidence in the instant case. The scant evidence available from the Ward Hilton trial suggested that native tissue repair could also have a detrimental impact on mental health and their results showed that scores for emotional and social functioning, vitality and mental health were lower in the colposuspension group than the TVT group at six weeks, six months and two years.1296
Erosion, extrusion, exposure
1206 It appears that mesh exposure is the most commonly reported complication associated with the use of mesh in prolapse surgery.
1207 That was certainly the opinion of Withagen et al (2011).1297
1208 The evidence showed that exposure rates for single compartment repair have been up around 10% to 12%, and significantly higher for multi-compartment repair.
1209 Withagen et al (2011) noted that reported exposure rates varied from 0% to 25%. In his report, Associate Professor Lam stated that the incidence of vaginal mesh exposure after vaginal mesh surgery varies from 3% to 15%.1298 In cross-examination, however, he conceded that two of his three references provided higher figures and that the true rate reported in the references was between 11.5% and 20.8%.1299 He claimed to have overlooked the higher figure of 20.8%.1300 He could not explain the low figure of 3% but suggested he might have taken a figure he had read for surgical intervention for exposure of 3.5%, rounded it down to 3%, and wrongly attributed it to exposure.1301 He said that his experience with Prolift was that vaginal mesh exposure occurs in around 10% to 11% of cases but agreed that the rate increases with the passage of time.1302
1210 Maher et al (2016a) estimated an exposure rate of 12% overall after transvaginal placement of permanent mesh. After a sub-group analysis, they said that the rate of exposure following anterior repair was 10%, whereas the rate in women with multi-compartment repair was higher at 17%.1303
1211 Maher et al (2016b) reported an exposure rate of 11.3% after transvaginal placement of polypropylene mesh for anterior compartment repair.1304
1212 A very similar figure (11.5%) was obtained by Altman et al (2011) in their RCT which was exclusively concerned with Anterior Prolift.1305 As I noted above, while Altman et al (2011) did not specify the rate in their published report, the figure of 21 of 183 women (11.5%) was later communicated to and published by Maher et al (2013).1306
1213 No separate analysis for posterior compartment repair was presented by Maher et al in any of their Cochrane reviews. According to the respondents, no randomised controlled trials have been conducted to evaluate the safety and efficacy of using transvaginal mesh specifically in posterior compartment repair.1307
1214 The respondents relied on the composite analysis of mesh repair for prolapse presented by Maher et al (2016a). The competing evidence from the applicants did not present a very different figure. Professor Korda for example said that the erosion rate after posterior compartment repair with mesh was up to 10%, although he did not provide a supporting reference.1308 Nor did he elaborate on the range that could be encompassed by “up to 10%”.
1215 As noted, Maher et al (2016a) found that the exposure rate following multi-compartment repair was higher. This was consistent with other evidence that a larger amount of mesh was more likely to lead to complications such as exposure. For example, a study by Halaska et al (2012) which compared sacrospinous fixation with Prolift Total for vaginal vault prolapse found an exposure rate of 20.8% after one year.1309
1216 On any view of the matter, erosion, at least in the sense of exposure, is at least common after transvaginal implantation of any and all of the Ethicon devices.
Infection
1217 As I observed in relation to the SUI devices, infection is a broad category and its prevalence has been difficult to pin down.
1218 Based on the review by de Tayrac and Sentilhes (2013), Professor Korda said that the rate of infection after mesh surgery for prolapse repair was unknown. At the same time de Tayrac and Sentilhes (2013) observed that some studies had reported a rate of 80% bacterial mesh colonisation, 1310 which might suggest that infection could be a very common complication. De Tayrac and Sentilhes (2013) went on to say, however, that the rate of “relevant clinical infection (abscess, cellulitis, spondylodiscitis)” after mesh surgery does not seem to be more than 1%.1311 But the evidence to which they referred was low quality (level 4) and it is not clear whether the studies were designed to detect late onset infections. Only three studies were referred to. One dealt with a single case. The biggest of the three was the retrospective study by the TVM Group of 684 patients published in August 2008 in which the longest period of follow-up was six months.1312 More likely than not, the infection rates reported by de Tayrac and Sentilhes related to intraoperative and/or postoperative infection.
1219 As Professor Korda indicated, there was little evidence in published studies to identify the rate of infection. Maher et al (2016a) did not report on infection rates. In the PROSPECT trial, Glazener et al (2016) found that serious adverse events such as infection occurred with similar frequency in the mesh and non-mesh groups.1313 But the period of follow-up there was only two years. In the RCT reported by Altman (2011), while there was a higher number of urinary tract infections in the first two months after surgery, between the two month mark and one year follow-up, there was one urinary tract infection (0.5%) in the colporrhaphy group and three (1.5%) in the Prolift group.1314
1220 Consequently, while it is likely that infection is a common, or at least not uncommon, complication of mesh repair, its true incidence cannot be gleaned from published studies.
Chronic pain
1221 As with the SUI devices, insufficient evaluation has been undertaken of the POP devices to produce a reliable indicative rate for chronic pain.
1222 Altman et al (2011) reported five cases of severe pelvic pain in the mesh group at two months, but said that in all except one of these patients the pain had resolved by the one year follow-up.1315 The condition of the remaining patient at the one year follow-up was not described.
1223 De Tayrac and Sentilhes (2013) noted that the rate of polypropylene mesh-related pain reported in studies ranged between 4% and 11%, but said nothing about how long the pain lasted.1316 In their prospective observational study of 294 patients with 12 months follow-up, Withagen et al (2011) reported de novo pain in 5% of cases after transvaginal mesh repair.1317 This was one of the studies cited by de Tayrac and Sentilhes (2013).
1224 The applicants also relied on the results of a case series by Associate Professor Lam and others, which contained data on the use of Prolift and a mesh kit manufactured by American Medical Systems called Elevate. That study, conducted on patients in their tertiary centre, reported a rate of 3.9% for pain (buttock/perineal) following surgery with Prolift.1318 In cross-examination, Associate Professor Lam agreed that this accorded with his clinical experience.1319
1225 While acknowledging that the incidence of chronic pelvic pain after mesh surgery for prolapse has been poorly studied, Professor Korda gave an estimate of between zero and 30%.1320 He relied on a paper by Gyang et al (2014) which stated that pelvic pain occurred in 0 to 30% of patients following transvaginal mesh placement, based on case series. The authors of that paper relied on three sources, two of which were not in evidence.1321 The third paper was the Cochrane review by Maher et al (2007), which did not report a percentage rate for pelvic pain.1322
1226 Whatever the true rate of chronic pain for the POP devices, it does not appear to be rare, is likely to be higher than experienced by women who received only an SUI device, and that the highest rates are likely to occur following multiple-compartment repair using synthetic polypropylene mesh, including Prolene Soft.
Dyspareunia and apareunia
1227 A wide range of rates appears in the literature. In their prospective cohort study of 294 women with Prolift, Withagen (2011) reported de novo dyspareunia in 26% of patients.1323 In their literature review, Shah and Badlani (2012) noted a similarly high rate, finding that dyspareunia had been reported in up to 24.4% of patients after mesh repair for prolapse (for which they relied on a retrospective single centre study).1324 A rate of 11% after Prolift was noted in a literature review by Barski and Deng (2015), but the difficulty about using that study to derive incidence rates has been discussed above.1325
1228 A meta-analysis conducted by Ethicon itself, and referred to in CERs for Prolift, reported an average rate of 12.7% for de novo dyspareunia.1326
1229 In Associate Professor Lam’s clinical trial, 23% of women who had received Prolift implants complained of new onset pain with intercourse.1327
1230 These rates appear to be higher than those reported in some of the RCTs. Altman et al (2011) found that at one year pain during sexual intercourse was reported to occur “usually” or “always” by 7.3% of women after mesh surgery and 2% after anterior colporrhaphy,1328 but the authors did not clarify whether these figures related to de novo dyspareunia or could include pain that was also present before surgery.
1231 Halaska et al (2012) reported on a randomised controlled trial comparing sacrospinous fixation with Prolift Total used in the repair of vaginal vault prolapse.1329 At 12 months, dyspareunia was reported in 3.7% of patients with sacrospinous fixation and 8% with Prolift.
1232 Maher et al in their 2016 Cochrane reviews did not find any significant difference between permanent mesh and native tissue repair in terms of the rates of de novo dyspareunia. This was consistent with the finding of Glazener et al (2016) in the PROSPECT trial, where the authors reported that serious adverse events such as dyspareunia or other pain occurred with similar frequency in patients with and without mesh, although as I mentioned the period of follow-up was only 12 months.1330
1233 There was no evidence on incidence rates of apareunia. The focus was on dyspareunia. I was not taken to any study that reported on apareunia separately from dyspareunia. Common sense would suggest that it is unlikely that the rates are likely to be similar.
1234 I conclude that de novo dyspareunia and apareunia are likely to be common after implantation of the POP devices.
Reoperation or revision surgery
1235 Professor Korda said that the total reoperation rate after transvaginal mesh surgery for prolapse ranged from 8.5% to 22%, compared with a range of 3.2% to 9.7% for native tissue repair.1331 He relied on the literature review by de Tayrac and Sentilhes (2013). The authors of that paper noted a total reoperation rate of 8.5% based on level 1 evidence. 1332
1236 In the PROSPECT trial, 23 women (5%) in the mesh group had surgery to remove part of the mesh in the first year, and in the second year 17 women (4%) had surgery to remove part of the mesh to address complications.1333 In those two years, 5% of women with native tissue repair and 4% with mesh repair had a new prolapse operation.
1237 Maher et al (2016a) reported that more women in the mesh group required repeat surgery for the combined outcome of prolapse, stress incontinence or mesh exposure.1334 They estimated that, if 5% of women required repeat surgery after native tissue repair, then between 7% and 18% in the permanent mesh group would do so. As I have already noted, for exposure alone they found that 8% of women in the mesh group required surgery. Rates of reoperation (repeat surgery for prolapse repair), however, were lower in the mesh group than with native tissue repair.
1238 Maher et al (2016b) obtained similar results when they confined their review to anterior compartment prolapse. They found that the total rate of repeat surgery (for a combined outcome of prolapse, stress incontinence, exposure and pain) was almost twice as high after mesh (10%) than after native tissue repair (5.7%).1335 Looking only at repeat surgery for prolapse, however, the evidence suggested that, if 2% of women needed repeat surgery after mesh repair, 2% to 7% would need it after native tissue repair.
Mesh removal or excision surgery due to erosion
1239 The evidence indicates that about half of the cases of exposure following transvaginal mesh repair required excision surgery.
1240 Maher et al (2016a) reported that surgery for mesh exposure was required in 8% of women.1336 In the review on anterior compartment repair (2016b), Maher et al reported that repeat surgery for mesh exposure was required in 7.3% of women.1337 This was higher than the rate reported by Altman et al (2011) in their trial using Prolift for anterior repair. They said that surgical reintervention to correct mesh exposure occurred in 3.2% of patients with mesh repair.1338
Difficulties with voiding and defecating
1241 Maher et al (2016a) said that there was no evidence of a difference between permanent mesh and native tissue repair in the rate of voiding disorder or dysfunction.1339 Similarly, no clinically important difference in urinary retention was reported by Glazener et al (2016) in the PROSPECT trial.1340 Altman et al (2011), however, did find that bladder-emptying difficulties in the immediate post-operative period were more common after mesh repair than colporrhaphy.1341
1242 There was insufficient evidence on the prevalence of defaecation difficulties after mesh repair for prolapse. Maher et al (2016a) commented that none of the studies included in their analysis had reported on “obstructed defecation” in a format suitable for analysis.1342
1243 De novo stress urinary incontinence is also common after mesh surgery.
1244 In a PowerPoint presentation containing results obtained by Professor Lam’s centre in Sydney, a rate of 18.2% was reported for de novo stress urinary incontinence following Prolift.1343
1245 In the Altman RCT, de novo stress urinary incontinence was twice as high in the mesh group: 6.2% of patients in the colporrhaphy group and 12.3% in the Prolift Anterior group.1344 Maher et al (2016a) also reported that permanent mesh was associated with higher rates of de novo stress incontinence, but the quality of the evidence was said to be low. They estimated that, if 10% of women developed stress incontinence after native tissue repair, 10% to 17% would develop it after transvaginal mesh repair.1345 On the other hand, after 12 months, Glazener et al (2016) detected no differences in urinary outcomes between the groups.1346
1246 As Professor Roovers noted, vaginal discharge may be associated with exposure of the mesh.1347 Withagen et al (2011) reported on two cases in women with exposures as part of their prospective study on Prolift.1348
1247 While it is a complication of mesh surgery for prolapse, as Dr Hinoul accepted, 1349 there was simply not enough evidence to make a finding about its prevalence.
Recurrence of pelvic organ prolapse
1248 While recurrence of pelvic organ prolapse after native tissue repair is a given, permanent mesh repair does not prevent it. Indeed, the evidence indicates that it is common after both kinds of repair.
1249 Altman et al (2011) reported that fewer women had recurrence of prolapse after anterior mesh repair than after colporrhaphy. At one year, 60.8% of women with mesh had no prolapse (that is, 39.2% had a recurrence), compared with 34.5% after anterior colporrhaphy (65.5% had a recurrence).1350
1250 Maher et al (2016b) also found that recurrence of anterior wall prolapse was more likely after native tissue repair than after mesh repair. They estimated that if awareness of prolapse after polypropylene mesh occurs in 13% of women, then 32% to 45% would develop awareness of prolapse after native tissue repair.1351
1251 The findings reported by Cochrane review on all permanent meshes were similar. Maher et al (2016a) reported that recurrence of prolapse on examination was less likely after mesh repair, suggesting that if 38% of women had recurrent prolapse after native tissue repair, between 11% and 20% would do so after mesh repair.1352 When looking at objective failure, however, the authors said that the benefit of mesh was more pronounced when the analysis was limited to studies of anterior compartment only. On the other hand, when the analysis was restricted to studies of multi-compartment prolapse, there was no conclusive evidence of a difference between the groups. Likewise, there was no evidence of a difference in rates between the groups with grade II or greater posterior compartment prolapse.1353
1252 Halaska et al (2012) found that recurrence of prolapse was significantly higher in the native tissue group (39.4%) than in the mesh group (16.9%).1354 In the PROSPECT study, however, Glazener et al (2016) did not discern any differences between the mesh and non-mesh groups in terms of prolapse recurrence and related quality-of-life scores after one year.1355
Damage to surrounding organs, nerves, ligaments, tissue and blood vessels
1253 Injury to the bladder appears to be more common with mesh surgery than native tissue repair.
1254 Altman et al (2011) reported more bladder perforations in the mesh group (3.5%) than the colporrhaphy group (0.5%).1356 Likewise, Maher et al (2016a) reported that permanent mesh was associated with higher rates of bladder injury than native tissue repair. They estimated that if 0.5% women had bladder injury with native tissue repair, between 1% and 6% of women would have such an injury during transvaginal mesh repair.1357 The same pattern was observed when looking specifically at anterior compartment repair.1358
1255 Maher et al (2016a) referred to one trial reporting bowel injury as an outcome, but it showed no difference between mesh and non-mesh repair. De Tayrac and Sentilhes (2013) noted that bowel obstruction and ureteric complications were rare complications of mesh surgery.1359
1256 There was limited evidence on the rate and extent of haemorrhage, particularly the relative incidence as between mesh and non-mesh repair.
1257 De Tayrac and Sentilhes (2013) described massive haemorrhage as a rare but severe complication.1360
1258 Altman et al (2011) reported that the mesh group had greater intra-operative blood loss than mesh surgery (84.7ml v 35.4ml). In the mesh group, one patient had pelvic haemorrhage with blood loss in excess of 1000ml, and blood loss exceeded 500ml in four other patients.1361 This was in line with the Maher et al (2016b) review, which reported that blood transfusion was less likely after native tissue repair than after mesh repair.1362 Maher et al (2013) had explained that blood loss is significantly higher with meshes placed through a transobturator approach compared with native tissue repair.1363
1259 In the review of permanent meshes more generally, Maher et al (2016a) found that there was no evidence of a difference in the rate of blood transfusion between permanent mesh and native tissue repair.1364
Leg weakness
1260 While neither side took the Court to evidence of leg weakness manifesting after surgery with the POP devices, Dr Hinoul accepted that it was a risk.1365
1261 During oral argument the respondents submitted that Dr Hinoul had made no such admission.1366 The submission was based on the transcript of the cross-examination. My own notes suggested otherwise and the transcript was later corrected by consent after access to the audio recording. Having regard to the recording and the corrected transcript, the submission should not be accepted.
1262 As is the case with the SUI devices, however, there is insufficient evidence to come to a view as to its prevalence.
1263 The comments made above in relation to psychiatric injury associated with the use of the SUI devices apply equally to the POP devices.
Gynemesh PS used abdominally in vault repair
1264 It was common ground that the risks or potential complications are not confined to the transvaginal use of mesh. They extend to the use of non-absorbable mesh in sacrocolpopexy, which is carried out abdominally (by laparotomy or laparoscopy).1367 Moreover, the evidence the respondents themselves adduced from Professor Collinet was that the rate at which mesh specific complications occur in sacrocolpopexy is “non-negligible” and that, while the literature indicates a lower incidence of complications from sacrocolpopexy than in cases of transvaginal repair, the complications that do occur “seem more severe, and warrant more follow-ups”.1368 Associate Professor Rosamilia’s experience was that mesh exposure after sacrocolpopexy usually occurs at the vaginal vault and the exposed mesh is more difficult to access and remove than mesh exposure after transvaginal procedures which, she wrote in her report, usually occurred at the incision line along the anterior or posterior vaginal wall.1369
1265 Nevertheless, the evidence indicated that the abdominal use of Gynemesh PS in vault repair is safer than its transvaginal implantation.
1266 Professor Korda, for example, said that the anatomic outcome of abdominal sacrocolpopexy was superior to vaginal sacrospinous ligament suspension (a form of native tissue repair).1370 In his experience, postoperative prolapse symptoms, reoperation rates, dyspareunia rates, as well as length of hospital stay were similar following abdominal sacrocolpopexy and sacrospinous ligament suspension. On his understanding of the literature, complication rates were similar for the abdominal and vaginal approaches but bowel and mesh complications were more common after abdominal sacrocolpopexy. On this basis, the respondents submitted:
MR FINCH: [W]e say that Professor Korda amounts to this: the risk profile doesn’t appear to be significantly different between the native tissue repair operations and the abdominal sacrocolpopexy operations, except in the sense that he nominated possible bowel problems and possible mesh erosion problems without assigning to them a significance other than to simply note that there was a difference there. The reason for that might be obvious, of course, because what was simply involved in the operation, what product was used, but he doesn’t assign a particular level of significance to it. 1371
1267 The applicants, on the other hand, submitted that while the complication rates for Gynemesh PS, when used abdominally, were lower than for transvaginal use and for mesh kits, the nature and gravity of the complications were the same.1372 They relied on Professor Collinet’s evidence that erosion rates following sacrocolpopexy were between 5% and 10%.1373 Elsewhere in his report, however, Professor Collinet cited an erosion rate of 1% from a French database. From the same database, Professor Collinet reported the rate for chronic pain as 1.9%, but it appears that the rate reported under “persistent pain” following the abdominal approach was 1.6%, compared to 1% for the vaginal approach, although the difference was not statistically significant.1374 Professor Collinet observed:
A complete review of the literature regarding sacrocolpopexys (sic) shows a lower incidence of complications stemming from the abdominal insertion of mesh than for vaginal insertion. Nonetheless, the complications that occur after sacrocolpopexy seem more severe, and warrant more follow-ups. Although the current focus is on the use of vaginal mesh, the use of abdominal mesh, considered to be the gold standard, also has a non- negligible rate of specific prosthetic complications.1375
1268 While acknowledging that long term results were limited, Professor Roovers cited the CARE study reported by Nygaard et al (2013), which estimated that the probability of mesh erosion after sacrocolpopexy was 11% at long term follow-up (6.18 years).1376
1269 It therefore seems that mesh erosion is a common outcome of sacrocolpopexy but that in the short to medium term the rates are lower than they are after transvaginal implantation.
1270 Despite this, the respondents maintained that the safety profile of abdominal sacrocolpopexy was similar to native tissue repair.
1271 Both parties referred to a literature review by Diwadkar et al (2009), which assessed complication and reoperation rates after apical vaginal prolapse repair with mesh kits, sacrocolpopexy, and native tissue repair. 1377 Diwadkar et al (2009) found that the total complication rates for each of the groups were similar, but the reoperation rate was highest in the mesh kit group despite it having the shortest period of follow-up. The complication rates following sacrocolpopexy were summarised as follows:
The sacral colpopexy group included 5,639 patients from 52 studies, with mean follow-up of 26.5 ± 20.1 months. Thirty-nine studies addressed sacral colpopexy by laparotomy, 10 laparoscopic sacral colpopexy, and three sacrohysteropexy. This group had the highest mean total complication rate of 17.1% (range 0–52.2). Similar to the traditional vaginal surgery group, the majority of complications were managed with pharmacologic intervention (5.8%) or no intervention (5.5%). Pain (2.3%), mesh erosion (2.2%), visceral injury (1.7%), and wound complications (1.5%) were the most common complications. There were 31 cases of dehiscence in the sacral colpopexy group compared with seven and four in the traditional vaginal surgery and mesh kit groups, respectively. Pulmonary emboli and deep vein thrombosis cases were reported more commonly after sacral colpopexy.1378
1272 The applicants suggested that this article demonstrated that the abdominal use of Gynemesh PS was attended by the same complications as mesh kits, while the respondents relied on the article to suggest that the safety profiles of sacrocolpopexy and native tissue repair were not dissimilar. The respondents’ position, however, paid no regard to the potential for further mesh-related complications in the future.
1273 The CARE study by Nygaard et al (2013), upon which Professor Roovers relied, showed increasing numbers of erosions with the passage of time: 17/322 (6%) of those enrolled at the two year post-surgery mark, 23/215 enrolled in the extended trial (11%) at the seven year mark. What is more, by year seven, at least 36 of the 215 women (16.7%) required additional surgery: 11 for recurrent pelvic organ prolapse; 14 for stress urinary incontinence; and 11 for mesh complications, which I take to mean mesh-specific complications or complications unique to mesh.
1274 In its 2015 opinion on the safety of urogynaecological meshes, SCENIHR wrote:
The risks associated with sacral colpopexy are the following: vaginal mesh exposure (2–5%), de novo constipation / obstructive defecatory syndrome (10%), per-operative bladder (1%) or bowel (0.1%) injury, de novo dyspareunia (1-3%), pelvic abscess (<1%), spondilodiscitis (<0.1%) and visceral (bladder, rectum) mesh exposure (< 0.1%) (Maher et al., 2013).
…
The risk of vaginal mesh exposure is significantly increased in cases of sacrocolpopexy associated with concomitant total hysterectomy (8.6%), in comparison to 2.2% in those with previous hysterectomy (Costantini et al., 2005; Zucchi et al., 2010). Thus, if hysterectomy is required, it is recommended to perform a subtotal hysterectomy.
Even if the prevalence of complications/reintervention seems to be lower following sacral colpopexy when compared to vaginal mesh surgery (Maher et al., 2011), serious complications have been described at short- and long-term follow-up after sacral colpopexy (Nygaard et al., 2013; Arsene et al., 2014).1379
1275 The parties also referred to a multicentre retrospective study by Abbott et al (2014), which described mesh-related complications after surgery.1380 The applicants relied on this paper to show that abdominal use of mesh was attended by the same or similar complications as its transvaginal use in prolapse repair. The trouble with this article, however, was that the authors reported on patients who had undergone sacrocolpopexy with a concomitant sling procedure and those who had not, without distinguishing between the two groups. Further, as the respondents pointed out,1381 the study reported incidence rates by reference to an overall population that had experienced some form of complication already. So the erosion rate reported after sacrocolpopexy, for example, was 14 out of 25 patients (56%) who had experienced a complication.
1276 The authors observed:
Patients who were seen after TVM or sacrocolpopexies were significantly more likely to have mesh erosion and vaginal symptoms, compared with those who received a sling only (Table 3). Patients with complications after TVM had a significantly higher occurrence of pelvic pain, dyspareunia, vaginal spotting, vaginal constriction, and obstructed defecation than those after sling alone (Table 3). Compared with TVM, patients with complications after sacrocolpopexies were significantly more likely to complain of vaginal discharge but less likely to complain of dyspareunia or recurrent POP (Table 3). Voiding dysfunction was most common in those women who received a sling only (Table 3).1382
1277 The respondents also referred to the Maher et al (2016c) Cochrane review, which evaluated the safety and efficacy of surgical interventions for the management of apical vaginal prolapse.1383 The authors reviewed 30 trials comprising 3,414 women. Unfortunately it did not include a comparison of sacrocolpopexy and native tissue repair.
1278 The authors found that compared to various vaginal repairs, sacralcolpopexy was associated with lower rates of awareness of prolapse, repeat surgery for prolapse, prolapse on examination, urinary stress incontinence and painful intercourse. According to their analysis, if 7% of women were aware of prolapse after sacralcolpopexy, between 7% and 27% would likely be aware of it after vaginal procedures. Further, if 4% of women required repeat prolapse surgery after sacralcolpopexy, between 5% and 18% would require it after vaginal procedures.1384
1279 The respondents submitted that the conclusions reached by Maher et al (2016c) were consistent with Professor Korda’s opinion that abdominal sacrocolpopexy remained the “gold standard” for the management of vault prolapse. He said that compared to abdominal procedures, transvaginal mesh procedures have a lower anatomical success rate, lower patient satisfaction rate and shorter vaginal length. He also suggested that the mesh erosion after transvaginal procedures was much higher (15%) than after sacrocolpopexy (2% to 3%).1385
1280 The respondents also relied on an RCT by Maher et al (2004),1386 which compared abdominal sacrocolpopexy with vaginal sacrospinous colpopexy (the latter being a form of native tissue repair). Ninety-five women were randomly allocated, 47 to abdominal sacrocolpopexy (using Prolene mesh) and 48 to sacrospinous colpopexy. Women in both groups suffering from stress urinary incontinence underwent Burch colposuspension.
1281 Two years after surgery, the subjective and objective success rates were similar but they did not reach statistical significance. No significant differences were detected in respect of most complications. The abdominal approach was associated with a longer operating time, a slower return to activities of daily living, and a greater cost than sacrospinous colpopexy. The authors reported that preoperative dyspareunia resolved in 56% of the women in the abdominal group and 43% of the women in the vaginal group, while dyspareunia developed postoperatively in two women in the abdominal group and three women in the vaginal group (although they did not remark on statistical significance). Both procedures were said to have significantly improved the patient’s quality of life.1387
1282 A similar conclusion was reached by Morling et al (2016), following their large cohort study of patients who underwent treatment for pelvic organ prolapse between 1997 and 2016. 1388 They observed no difference in any outcomes between vaginal and abdominal mesh repair of vaginal vault prolapse compared with vaginal non-mesh repair. In particular, they noted that there was no significant difference in the rate of late procedural complications after vaginal or abdominal mesh repair for vault prolapse compared with non-mesh vaginal vault repair.1389
1283 On the other hand, as noted above, Maher et al (2013) concluded that abdominal sacrocolpopexy was associated with a lower rate of recurrent vault prolapse and less dyspareunia than vaginal sacrospinous colpopexy but with a longer operating time, longer recovery time, and higher cost.1390
The expert evidence on comparative outcomes
1284 Each of the epidemiologists retained by the applicants was asked to test two hypotheses.
1285 Professor Gordon was asked to consider whether either of the following hypotheses had been proven on the available evidence:
(1) treatment of stress urinary incontinence with the SUI devices causes equivalent or fewer complications than surgical treatment of stress urinary incontinence using the alternative treatments (the safety hypothesis); and
(2) treatment of stress urinary incontinence with the SUI devices causes an equivalent or better outcome than surgical treatment of stress urinary incontinence using the alternative treatments (the efficacy hypothesis).1391
The alternative treatments identified were open colposuspension, laparoscopic colposuspension and fascial sling repair.
1286 The same questions were put to Associate Professor Krulewitch, and questions of the same nature, limited to the POP devices, were put to Professors Hu and Woodward. None of them considered the hypotheses had been proven at any relevant time.
1287 For this purpose the applicants’ solicitors provided Professor Gordon with some 438 articles. They included randomised controlled trials, Cochrane and other reviews and meta-analyses. He excluded from his analysis non-randomised studies and studies in which the specification of randomisation was poor.1392
1288 Professor Gordon then performed his own meta-analyses of the relevant outcomes and complications, acknowledging the “practical and technical challenges” in doing so.1393 He explained, without contradiction, that it was common practice for reviews of a given research question to make use of meta-analysis.1394
1289 Before answering either question Professor Gordon made the following point:
It is important to note in this connection that “proof” is a concept that is not usually used in the summary of statistical and epidemiological evidence. This is because, no matter how convincing the evidence, from a formal point of view, there is always room for doubt. The P-value for a given outcome may be 1 in a million (i.e. 0.000001), but events of this probability do occur, so we always allow for the possibility that the null hypothesis is true, no matter what.1395
1290 Professor Gordon struck me as a scrupulously honest and thoughtful witness who endeavoured at all times to provide measured responses to the questions asked of him, both in writing and orally. Like many such witnesses, however, he made some errors. These errors became apparent in cross-examination and were readily acknowledged. Given the nature of the task he was asked to undertake, they were understandable. I will refer to the errors as they arise. I have taken them into account.
1291 With respect to the efficacy hypothesis, Professor Gordon concluded that the available evidence did not demonstrate that the SUI devices clearly cause a better (objective or subjective) outcome than the alternative treatments.1396 He considered that there was no reliable evidence to indicate that, when introduced or at the time of his report (1 September 2016), the devices were more effective for the treatment of stress urinary incontinence than the alternative treatments.
1292 Professor Gordon’s opinion was that by 1998 there was no reliable evidence on the efficacy of TVT and that, if the release of TVT onto the market should have been based on reliable evidence regarding its efficacy, then that evidence was lacking.1397 He also concluded that, except for the period between 2004 and 2006 during which there was some evidence that the SUI devices caused a better outcome for objective cure, the evidence to the end of 2015 was unconvincing regarding the efficacy of the devices in comparison with the alternative treatments. For subjective cure, Professor Gordon concluded that there was no reliable evidence favouring tape over no tape at any time during the period. Be that as it may, the applicants accepted that by 2015 the efficacy of TVT had been established.1398
1293 Professor Gordon’s opinion was that the safety hypothesis had certainly not been proven by 1998 for any relevant complication or outcome of interest.
1294 The complications he considered fell into the following categories: extrusion or erosion; infection; urinary tract infection; dyspareunia; difficulty voiding; de novo or recurrent incontinence; bladder and/or urethral injury damage; other damage; haemorrhage; haematoma; reoperation; general complications; and pain.1399 To the extent that this list differed from the pleaded complications, which were identified in the solicitors’ letter of instructions,1400 Professor Gordon explained that the omissions were due to the absence of “sufficiently useful and reliable information from the studies examined” to enable them to be formally analysed.1401
1295 Professor Gordon described as equivocal the evidence in relation to the following complications: urinary tract infection; difficulty voiding; de novo or recurrent incontinence; haemorrhage; haematoma; pain; and other damage. He wrote, “we cannot be confident that the [SUI devices] cause fewer complications than the [a]lternative [t]reatments, and nor can we be confident of the reverse; that the [SUI devices] cause more complications”; and concluded they may cause the same underlying rate of complications.1402
1296 For two complications — infections other than urinary tract infections and re-operation — Professor Gordon concluded that the SUI devices cause fewer complications than the alternative treatments.1403 He noted, however, that in the case of re-operation the evidence for consistency of the results was not substantial because the results were mostly dependent on only two studies: Ward and Hilton (2006) and Guerrero et al (2010), the latter being an RCT comparing TVT with Pelvicol (a porcine dermis sling manufactured by Bard) and autologous fascial slings.1404 In the Guerrero et al (2010) trial, a total of 201 women divided into three groups were operated on and assessed at baseline, six weeks, six months and one year.1405 The conclusions of the researchers were that Pelvicol was an inferior material for midurethral sling support and could not be recommended for the management of stress urinary incontinence in women and that TVT and autologous fascial slings yielded similar results both with regard to efficacy and adverse events at the one year follow-up.
1297 For two other complications — extrusion or erosion and bladder and/or urethral injury — the evidence from the meta-analyses suggested to Professor Gordon that the SUI devices cause more complications than the alternative treatments. In contrast to the findings concerning infection and re-operation, which were based on “relatively few studies”, he described the conclusion about bladder and/or urethral injury as “a robust and reliable finding based on several studies”. He noted that there was a tendency in the data for more erosion to be associated with longer follow-up but added that “[the] association [had] not been studied with much reliability, as more studies with longer follow-up are needed”.1406
1298 It was at this point in his report that Professor Gordon stated, “[h]ow these findings should be weighed is really a matter for a clinical expert”. I did not take this to be a concession about a weakness of his analysis of the studies but a recognition that the significance of the rates of complications was a matter for the clinical experts.
1299 His ultimate conclusion was that, for all complications, it was clearly not the case that treatment of stress urinary incontinence with the SUI devices causes equivalent or fewer complications than surgical treatment of SUI using the alternative treatments.
1300 In a second report Professor Gordon focussed on the POP devices. He presented evidence of complications drawn from 130 randomised controlled trials comparing mesh to native tissue repair for pelvic organ prolapse based on his own meta-analysis of the results of those trials.1407 He also responded to a number of questions touching on aspects of the evidence given by Dr Hinoul and several of the respondents’ expert witnesses.
1301 Professor Gordon considered data in relation to 15 complications and summarised his findings by reference to a ratio of proportions for each complication. A ratio of proportions less than one favours mesh and a ratio greater than one favours native tissue. Professor Gordon concluded that the incidence of damage to surrounding organs was 4.34 times higher for mesh than for native tissue repair and for de novo incontinence the rate was 1.35 times higher.1408 Blood loss in excess of 500mL and infection (non-urinary) were unique to the mesh arms of the trials. The risk level was estimated at 5.22 times higher for mesh in the former case and 3.02 times higher in the latter, although these findings were not statistically significant. The smallest ratio of proportions was for reoperation for prolapse. The rate of proportions was 0.46, indicating that the risk of reoperation for prolapse was lower for mesh than for native tissue repair. The results were summarised in the following table:
1302 The last two outcoes — sexual function and psychiatric injury — were based on a comparison of means. For the PISQ (the Pelvic Organ Prolapse/Incontinence Sexual Questionnaire, which assesses sexual function) and the PFIQ (the Pelvic Floor Impact Questionnaire, which assesses the impact of pelvic floor function on quality of life), the result favoured native tissue in each case.
1303 Since not all the p-values were less than 0.05, however, not all these findings are statistically significant.
1304 The major problem with Professor Gordon’s evidence was with his meta-analysis of extrusion or erosion rates (which included exposure).
1305 When he came to consider this complication, Professor Gordon observed that it was “quite unusual to attempt to make comparisons” between treatments for adverse outcomes or complications which can only occur when one of the two treatments is used. In order to shed more light on this, he said that he considered all the randomised controlled trials covered by the 438 documents he was given for the preparation of his first report but, in view of the large volume of material, he took a random sample of the relevant studies. RCTs were examined for the use of TVT, TVT-O (also referred to by Professor Gordon as TOT) or TVT Secur in any arm.1409 From this exercise he derived 255 studies, which were randomly ordered, and then searched using search terms which included erosion, extrusion, exposure and other key words. Papers and studies without reference to the search terms were excluded and the first 40 studies with a reference to the complication category “extrusion or erosion” were examined closely. Those studies had one, two or three tape treatment arms (most had two, one had three). In total there were 74 independent tape treatment arms in the 40 studies. Professor Gordon concluded that the average erosion/extrusion rate was 2.4% and the median 1%. 75% of the treatment arms had percentages lower than 4.2% and 25% had percentages greater than 4.2%. The minimum percentage was 0%, recorded in 33 (45%) of the 74 treatment arms. The maximum percentage was 15.4% from a small study arm (2/13).
1306 Professor Gordon said that there was a slight tendency for longer follow-up to be associated with a higher percentage of extrusion or erosion. Since the majority of studies had short periods of follow-up and the tape implants are a permanent intervention, he considered that the association between the rate of erosion and the length of follow-up had not yet been adequately studied.1410
1307 In cross-examination some of this evidence unravelled. It became apparent that the analysis included slings produced by manufacturers other than Ethicon.1411 That is unsurprising given the limited instructions he received. Overnight he prepared a document containing two tables, one entitled “Data from 72 arms using polypropylene mesh material in a tape implant (from 40 studies)” containing figures for erosion or extrusion regardless of manufacturer and another entitled “Data from 50 arms restricted to TVT, TVT-O, TVT-S (from 33 studies)”. He concluded that the average percentage of erosion/extrusion in the former group was 2.5% and the median 1.1%, the lower quartile 0% and the upper quartile 4.3%, meaning that 75% of the treatment arms had percentages less than 4.3% and 25% had percentages greater than 4.3%. The maximum percentage was 15.4% from a small study arm (2/13). The minimum percentage was 0% recorded in 31 (43%) of the 72 treatment arms. He concluded that the average percentage of erosion/extrusion in the latter group was 2% and the median 0.4%, the lower quartile 0% and the upper quartile 2.9%, meaning that 75% of the treatment arms had percentages less than 2.9% and 25% percentages greater than 2.9%. The minimum percentage was 0%, recorded in 25 (50%) of the treatment arms and the maximum 15.4%, from the same small study arm (2/13).
1308 While his report referred to 74 arms, in the preparation of these tables Professor Gordon realised that two of the studies included in his report did not use a polypropylene tape but a biological graft.1412 Cross-examination revealed a similar error in the small study which produced the largest figure in which the treatment was erroneously listed as TVT when in fact Gortex was used.1413
1309 In a supplementary report Professor Gordon limited his analysis to trials using Ethicon products1414 He identified 47 arms using such products. The average percentage of erosion/extrusion was calculated as 1.9% and the median was 0.8%.
1310 Unfortunately, the supplementary report also contained errors. Professor Gordon had misreported the erosion rate in a study by Bianchi-Ferraro et al (2013).1415 That study compared TVT-O with TVT Secur. In a table setting out the erosion/extrusion rates from various studies, Professor Gordon had stated that the study reported erosion/exposure rates of 3/56 (5.4%) for the TVT-O group and 5/66 (7.6%) for the TVT Secur group.1416 But the rates of exposure reported in the paper were 1/56 (2.7%) and 2/66 (3%) respectively.1417 Indeed, as the respondents pointed out, the percentage reported by the authors themselves appears to have been miscalculated, as 1/56 is actually 1.8%.1418
1311 The errors identified in Professor Gordon’s analysis mean that the rates of erosion/extrusion he gave are unreliable. In view of the other available evidence on complication rates, however, the respondents’ attack on that evidence, though justified, is of little significance.
1312 Associate Professor Krulewitch’s evidence was that none of the SUI devices had been shown to be safer than the alternative treatments, either at the time TVT was launched in 1998 or at any time thereafter. She noted that the epidemiological evidence showed that new complications have occurred with the SUI devices that are not associated with the alternative treatments, including mesh erosion and exposure.1419 Like Professor Gordon, she also concluded that by 1998 neither the efficacy hypothesis (that treatment of stress urinary incontinence with the SUI devices causes a better outcome than surgical treatment of stress urinary incontinence using the alternative treatments) nor the safety hypothesis (that treatment of stress urinary incontinence with the SUI devices causes equivalent or fewer complications than surgical treatment of stress urinary incontinence using the alternative treatments) had been proven. Indeed, in her opinion neither hypothesis has ever been proven.1420
1313 Professor Hu came to the same conclusion with respect to the POP devices, save that the relevant year was 2003 rather than 1998.1421 He was not challenged about these opinions. In cross-examination his attention was drawn to his conclusion about the position as at 2008. In his first report he stated that by 2008 the efficacy hypothesis had not been proven. Although he said at that point that his conclusion was “based on the systematic review by Jia et al (2008), as well as additional studies published in 2007 and 2008 [all discussed immediately before his conclusion]”,1422 the cross-examiner put to him that he had not set out any reasons for his conclusion. Professor Hu then took up the implied invitation to expand upon the explanation given in his report. He said:
Well, first of all, again, systematic reviews and meta-analysis are so important because of the heterogeneous nature of the results that are reported looking across the universe of studies that relate to the topic at hand. So unless actually these studies were included in a new systematic review, it would be difficult to say what the overall body of evidence quantitatively show. Second of all, as pointed out in Jia et al, it has already been recognised that subjective failure is a more appropriate outcome measure of efficacy than objective failure and these studies, save one, did not actually report results of subjective symptoms and which means that, you know, the – this more important outcome, in fact, has not been assessed. Then, as I pointed out, these are only 12 month or at most 24 month studies and in order, in my view, to really demonstrate efficacy you need longer term outcomes, and none of these studies really have longer term outcomes, so I think that, basically, went into my thinking on how to answer the question. 1423
1314 By 2016 Professor Hu considered that the overall complication rate for mesh was higher than for native tissue repair. In re-examination he testified:
[I]f we set aside the question of whether the – the complications are mesh specific or not, I think the conclusion reached by Maher still applies, which is that the mesh is associated with higher rates of reoperation for the combination of prolapse, stress urinary incontinence, or mesh exposure, and higher rates of bladder injury at surgery and de novo stress urinary incontinence. Those remain true, and in my view reflect a – what I would term as a higher complication rate overall with regards to mesh. 1424
1315 Professor Woodward’s opinion was that, apart from some evidence in relation to anterior mesh repairs, both hypotheses remain unproven.
1316 In his first report he wrote:
Taking all the evidence together, I conclude that there are insufficient data, both in terms of quality and quantity, to conclude that use of transvaginal mesh is as safe, or safer, than native tissue repair. Considering that much of the data I have reviewed are recent, use of mesh in the past must have occurred when the evidence base, on which to make a conclusion about the safety of mesh, compared to native tissue repair, was considerably weaker. Even when restricting to short-term outcomes, accepting all data as valid and including the most recent data available, I only found evidence for an advantage of mesh on anatomical outcomes, whilst there are key outcomes that have a worse prognosis when mesh is used.1425
1317 He adhered to this opinion in his second report.1426
1318 Notwithstanding their failure to cross-examine Professor Woodward or to call any epidemiologist or biostatistician to rebut the conclusions of the applicants’ four witnesses, the respondents heralded the Cochrane reviews as the more reliable reference point while, at the same time, choosing to overlook some of their uncomfortable conclusions about the safety and efficacy of polypropylene mesh in the treatment of pelvic organ prolapse.
1319 The respondents referred to the organisation’s representation on its website that the information it produces is “free from commercial sponsorship and other conflicts of interest”.1427 In effect, they were inviting the Court to put less weight on the witnesses’ evidence than on the findings and conclusions of the Cochrane reviews because the opinions of the applicants’ witnesses were not free from commercial conflicts or other conflicts of interest. But no such proposition was put to any of them in cross-examination and the inference is not open on the evidence.
1320 I accept that the Cochrane reviews are an important reference point and an important source of information. As Associate Professor Krulewitch observed in cross-examination, however, while she limited her analysis for the most part to the Ethicon devices,1428 the studies analysed in the Cochrane reviews were not confined to Ethicon’s devices. What is more, the authors of the Cochrane reviews were not available for cross-examination. In any case, the Cochrane reviews were not addressing the same questions as the applicants’ witnesses. In all the circumstances, with the exception of Professor Gordon’s evidence about erosion rates, there is no reason why the evidence they gave should not be regarded as probative of those matters.
1321 Besides, the Cochrane reviews on the POP devices support the applicants’ case.
1322 With respect to the comparative safety and efficacy of the SUI devices and alternative treatments, the respondents submitted that five Cochrane reviews — Ogah et al (2009), Dean et al (2010) and (2017), and Rehman et al (2011) and (2017) — considered a significant number of studies.1429 They argued that these reviews concluded that the SUI devices were superior in outcome or that there was no statistically significant difference in outcomes or there was insufficient evidence to reach a conclusion. Consequently, they submitted that the applicants had not established that the alternative treatments were equally or not materially less safe and effective.
1323 Even if this were true, it has no bearing on the position that obtained at the time TVT, TVT-O and TVT Secur were launched. As at 1999, when TVT was launched, only short-term, single arm studies were available. There were no comparative studies. The first RCT comparing TVT with any surgical alternative was the Ward Hilton study. Its first results were not published until 2002, a year after Mrs Sanders’s implant surgery. At least until that time, it had not been shown that TVT had equal or fewer complications or greater or equal efficacy than alternative surgical treatments for stress urinary incontinence. In any case, in 2002 only the six month results were available which was far too early to determine relative safety and efficacy.
1324 The objective of the Rehman et al reviews was to assess the effects of traditional suburethral sling procedures. Rehman et al concluded, both in 2011 and 2017, that there was insufficient information on which to judge whether traditional sling operations were better or worse than any other treatments. In both reviews they noted that long-term results were awaited and there were few trials of high quality comparing slings with other forms of surgery and only one which compared them with non-surgical treatment.
1325 The Rehman et al reviews concentrated on traditional sling procedures, rather than TVT, which was described as a modification of the suburethral sling procedure, or Prolene. The authors noted that TVT had been considered in a separate Cochrane review, citing Ogah et al (2009).
1326 It will be recalled that Ogah et al (2009) reviewed findings from trials comparing minimally invasive synthetic suburethral sling operations with traditional slings, with open colposuspension, and with laparoscopic colposuspension.
1327 With respect to the first comparison, Ogah et al (2009) did conclude that minimally invasive synthetic suburethral sling operations appeared to be as effective as traditional suburethral sling operations but the confidence intervals were wide and were compatible with traditional slings being either 13% worse or 6% better.
1328 As for adverse events, there were no statistically significant differences in bladder perforations but here, too, the confidence intervals were wide and the authors were unable to exclude the possibility that there was a big difference in favour of traditional slings. Nor were there any statistically significant differences in erosions (none was reported in any women in two trials), but the length of follow-up was short.
1329 Quality of life was assessed in six studies, not all of which used validated questionnaires, and the data were reported in different ways, so that meta-analysis was impossible. Nonetheless, the authors concluded that quality of life was equally improved by both procedures.
1330 With respect to the second comparison — synthetic suburethral slings v open retropubic colposuspension — the evidence suggested that the synthetic slings offered comparable efficacy and lower morbidity, with shorter operation times and hospital stays and fewer cases of perioperative complications or postoperative voiding dysfunction. But there was a lack of long term data and the quality of most of the trials was described as “variable”. In the short term, quality of life scores showed significantly greater improvement after minimally invasive synthetic suburethral slings. The authors concluded that, in view of their comparable efficacy and lower morbidity, the synthetic suburethral sling operations could be considered as a first line treatment for stress urinary incontinence in women instead of open retropubic colposuspension. But they added a note of caution. They remarked that “there is little information about the complication rates of tapes in the long term, or how to treat women who have had a failed tape procedure”.
1331 I note that there were significantly more bladder perforations with the synthetic slings and all the perforations in the synthetic arm occurred with TVT. Quality of life was assessed in only two of the nine trials, a feature the authors considered to be a major limitation.
1332 With respect to the third comparison — synthetic suburethral slings v laparoscopic colposuspension — there was conflicting information on cure and improvement rate, some studies favouring one procedure, others favouring the alternative. The authors stated that “[w]omen who had minimally invasive synthetic suburethral sling operations had significantly less de novo urgency and urgency urinary incontinence and shorter operating time, hospital stay and time to return to daily activities”. Both procedures led to improvement in quality of life after surgery, but with no significant differences between the groups.
1333 The authors said that there was a need for more robustly designed good quality and adequately powered randomised controlled trials with standardised objective and validated subjective outcomes, long term follow-up, and adequate reporting of adverse effects.
1334 The objective of the Dean et al reviews was to determine the effects of laparoscopic colposuspension for urinary incontinence in women. To this end the authors tested a number of different hypotheses. Hypothesis 6 involved a comparison between laparoscopic colposuspension and procedures using suburethral synthetic slings, including TVT and SupraPubic ARC, referred to in the reports as “self-fixing” sling procedures.1430
1335 Only one of five studies assessing quality of life reported a significant difference between the two procedures. That study favoured TVT, but the follow-up period was only one year.1431 Objective cure rates varied across the studies but overall laparoscopic colposuspension procedures had statistically significant lower objective cure rates.1432 Subjective cure rates were near equal in all studies comparing laparoscopy using sutures with vaginal slings including those with longer follow-up. TVT was reported to have similar subjective cure rates to laparoscopic colposuspension. The authors noted that, although there was “still a trend in favour of the sling procedures”, more studies reporting longer-term data were needed “as unanticipated and even anticipated complications may arise from these newer procedures”.1433 The conclusion, highlighted by the respondents during cross-examination of Associate Professor Krulewitch, was that “when laparoscopic colposuspension is compared with newer self-fixing sling procedures, it appears in the short term the sling procedures offer greater benefits of minimal access techniques with similar if not better cure rates”. Nevertheless, the authors observed that the value of the review was limited by the size and quality of the trials and the paucity of long-term data.
1336 Having regard to all this evidence, I find that neither in October 1999, when TVT was first supplied in Australia, nor at any time thereafter were any of the SUI devices proven to be safer or more effective in the long-term than the alternative treatments. I also find that neither in July 2003 when Gynemesh PS was first supplied in Australia nor at any time thereafter were any of the POP devices proven to be safer or more effective in the long-term than the alternative treatments.
PART VII: THE REGULATORY FRAMEWORK
1337 In this Part of the judgment I discuss the regulatory system which applies to medical devices, the essential principles or requirements to which manufacturers and suppliers must conform, and the relevant guidelines and international standards designed to assist them to do so. I also introduce the expert evidence about the respondents’ compliance with those requirements before recounting the history of the development of the Ethicon devices. That is because the development of medical devices takes place within a regulatory framework. Moreover, in recounting that history, I examine Ethicon’s evaluation of the safety and efficacy of the particular devices and an understanding of the regulatory context is useful.
1338 The evidence on these questions was given by four experts in the area, all of it adduced by the applicants. The respondents indicated an intention to lead evidence from JJM’s Director of Regulatory Affairs, Rebecca Gaudin, and an affidavit from her was filed. Without explanation, however, her affidavit was not read. Similarly, the respondents obtained a report from Elaine Duncan, a biomedical engineer and regulatory consultant, but elected not to tender it.
1339 Consequently, the applicants’ evidence went unanswered. Instead, the respondents contended that the evidence was “of peripheral relevance”,1434 “of limited assistance”,1435 or “not relevant” altogether.1436
1340 While some of the evidence may have been of peripheral relevance, this description certainly did not apply to the bulk of it and the evidence cannot be marginalised in this way.
1341 The evidence is relevant to a number of the causes of action.
1342 Non-compliance with regulatory requirements and standards is a relevant consideration in the determination of whether the devices were defective for the purpose of s 75AD of the Trade Practices Act and its ACL counterpart. As I explain later in these reasons, a product is defective if its safety is less than persons generally are entitled to expect. One factor that will have a bearing on this question is whether the product satisfied relevant regulatory requirements and standards.
1343 Evidence that the respondents failed to conform to the requirements necessary to obtain and retain European certification, when it was through European certification that Australian regulatory approval was secured, is also relevant to the negligence claims, particularly the first and second claims which relate to the respondents’ pre-market and post-market evaluation of the Ethicon devices. Moreover, in their defence, the respondents relied on the fact that they had obtained regulatory approval in Australia in order to deny that they owed the applicants and group members a duty of care, or, in the alternative, that they had discharged that duty. The evidence of the regulatory experts went to the heart of this claim and to the rebuttal of this aspect of the defence.
1344 In the absence of contradictory evidence, the evidence of the applicants’ experts may more readily be accepted. Further, it may be inferred that the evidence Ms Gaudin and Ms Duncan would have given, at least when put to the test in cross-examination, would not have assisted the respondents: see Jones v Dunkel (1959) 101 CLR 298 at 308 (Kitto J), 312 (Menzies J) and 320–321 (Windeyer J).
1345 Evidence on the operation of the Australian regulatory regime was contained in a report by Dr Derrick Beech. Dr Beech was well placed to give this evidence. From 1985 to 1996 he was the director of the Therapeutic Devices Branch of the Australian regulatory body, the Therapeutic Goods Administration. During that time he developed and directed medical device regulation in Australia. He described himself as the architect of the medical device aspects of the Therapeutic Goods Act 1989 (Cth) (TG Act). Since he left the TGA, Dr Beech has been self-employed as a medical device regulatory affairs consultant, specialising, amongst other things, in global regulatory affairs and quality systems. He was a founding member of the Medical Device Global Harmonisation Task Force. Dr Beech was not required for cross-examination and his evidence was unchallenged. I accept it.
1346 The applicants also led evidence from three overseas regulatory experts: Dr Bryan Allman, Ms Anne Holland, and Dr Peggy Pence.
1347 Dr Allman and Ms Holland gave evidence about the regulatory processes in Europe, the path that Ethicon followed to get each device on the market, and the extent to which the respondents conformed to the regulatory requirements. They also gave evidence about the regulatory processes in the United States and gave context to some of the American evidence upon which both sides relied.
1348 Dr Allman is a clinical biochemist from the United Kingdom with extensive relevant experience in regulatory affairs. Amongst his employers was Boston Scientific, one of the respondents’ competitors. Dr Allman worked for Boston Scientific for nearly six years (from June 1998 to January 2004) as its Vice-President Quality Assurance, Clinical and Regulatory Affairs. Before moving to Boston Scientific, Dr Allman worked for nearly a decade for Abbott Laboratories, an American pharmaceutical company. There, he was employed in various regulatory roles at a management level. Since leaving Boston Scientific, amongst other things Dr Allman has worked as an independent consultant on clinical, quality assurance, and regulatory matters, providing advice and support to medical device companies or companies considering entering the medical device sector.1437 Over a 30 year career in the medical device industry his responsibilities included achieving or remedying compliance with the European medical devices directives (discussed below), implementing processes for post-market surveillance, managing relationships with notified bodies (described below), and advising on European clinical evaluation and investigation requirements. He also has extensive experience in the ways in which manufacturers achieve or fail to achieve compliance with those requirements.1438
1349 Ms Holland is a biomedical engineer and quality assurance consultant from the United States, with some 30 years’ experience in regulatory compliance, quality systems development, auditing, and regulatory affairs in the medical device industry. She began her career in the mid-1980s in Colorado, working as a project engineer for a medical device manufacturer. From 1993 until 1999, she was Senior Manufacturing Engineer, Quality Assurance Manager, and Senior Quality Assurance Engineer for Sulzer Carbomedics, a medical device manufacturer in Austin, Texas.1439
1350 For the last 20 years Ms Holland has been running her own consultancy business. In that capacity, she has focussed exclusively on medical devices and has had extensive experience with long-term and permanent implantable devices. She consults with medical device manufacturers and their supply chains to develop and implement compliant solutions. She has completed over 100 supply chain/internal audits to US and international standards. Her expertise includes risk management for medical devices, design controls, quality system development, design verification and validation, process validation, internal and supplier chain auditing, and handling investigations into corrective and protective actions and regulatory remediation efforts. She has established and deployed quality management systems (QMS) for over 25 companies. She regularly analyses the design control and risk management processes and documentation of medical device manufacturers to identify their strengths and weaknesses and to evaluate whether the documentation complies with industry standards and practices. She also regularly examines their quality processes to determine operational effectiveness and compliance with regulations applying in the United States and the European Union.1440
1351 Ms Holland is also familiar with the Australian regulatory system. She gave unchallenged evidence that, “[d]ue to the similar nature of categorisation and compliance requirements for medical devices throughout the U.S., EU, and Australia, an individual with experience in the regulation or approval of medical devices in the U.S. could reasonably transfer that knowledge and experience to encompass medical devices in the EU and Australia”.
1352 Dr Pence’s evidence was directed to the requirements for medical device labelling and the extent to which the respondents complied with them. Dr Pence is a scientist with over 43 years of relevant experience. Her undergraduate degree was in microbiology and she has a PhD in toxicology with a pharmacology minor from Indiana University. During the course of her long career she has worked in a range of different capacities for a number of pharmaceutical and bioscience companies. Most relevantly, since 1995 she has been the president and chief executive officer of a company she founded to provide expert advice to industry about regulatory, nonclinical and clinical development matters, and strategic planning of product development. In that capacity she has not only provided the advice, but she has also liaised on behalf of companies with the US Food and Drug Administration (FDA) over regulatory matters, prepared regulatory submissions, and critically reviewed submissions prepared by others. She has conducted training programs in regulatory affairs. She holds US Regulatory Affairs Certification and is an instructor for candidates for Regulatory Affairs Certification. She is also a Fellow of the Regulatory Affairs Professionals Society for which she was selected by her peers based on her experience and leadership in, and contributions to, the regulatory profession.1441
1353 Although Dr Allman, Ms Holland, and Dr Pence were all required for cross-examination, none of them was shaken in his or her opinions. In these circumstances and, in the absence of any evidence to the contrary and, having regard to their qualifications and experience, unless otherwise indicated I accept their evidence.
The respondents’ arguments on the utility of the regulatory expert evidence
1354 The respondents argued that:
An expert’s subjective opinions as to asserted inadequacies in the Respondents’ dealings with the regulators are not relevant in circumstances where there is no evidence of any perceived inadequacy from the regulators. In particular, the Applicants do not seek to impugn the regulators’ actions and, in any event, it is difficult to understand how this Court could be invited to accept the opinions of the Applicants’ regulatory experts in preference to the regulators’ position.1442
1355 The argument is disingenuous.
1356 First, as the evidence of the applicants’ experts disclosed, the regulatory system largely depends on the manufacturers being frank with the regulators. As I explain below and as the respondents well knew, the regulators do not conduct the kind of scrutiny of the conduct of manufacturers that the applicants’ experts conducted in this case.
1357 Second, there was in fact documentary evidence that the regulators, including but not confined to the TGA, “perceived inadequacy”. Although in some respects this evidence was not as detailed or extensive as the evidence given by the applicants’ witnesses, it was critical of the respondents’ conduct.
1358 Third, the fact that the applicants “do not seek to impugn the regulators’ actions” is beside the point. The case is concerned with the conduct of the respondents, not the regulators.
1359 The respondents also argued that the evidence of the three overseas experts was of little assistance because the applicants did not plead that or how the foreign regulatory environment informs the respondents’ obligations. In addition, they pointed to an unsolicited remark made by Dr Allman at one point in the cross-examination that he did not know much about the current Australian system.
1360 These arguments must be rejected as well.
1361 First, the relationship between the European and the Australian regulatory regimes was not a matter that needed to be pleaded. It was a matter for evidence. As r 16.02(1)(d) of the Federal Court Rules makes clear, a pleading must state the material facts on which a party relies that are necessary to give the opposing party fair notice of the case it has to meet, but not the evidence by which those facts are to be proved.
1362 Second, since the unchallenged evidence of Dr Beech was that the Australian system is very similar to the European system and that Australia accepts European certification, Dr Allman’s admission is of no consequence.
1363 Each of Dr Allman, Ms Holland and Dr Pence was well-qualified in the field. Each had extensive relevant experience and an in-depth understanding of the regulatory requirements. Each presented as a conscientious and careful witness exhibiting no hint of partisanship. Each easily withstood cross-examination. The respondents’ submissions largely ignored their evidence. Neither in writing nor orally did they engage with it. Indeed, their submissions did not even mention the witnesses’ criticisms of their conduct. Instead, as I observed earlier, they tried to marginalise the evidence. 1443 This strategy might have been a convenient way to sideline evidence that was potentially very damaging. But it was a risky one. And it did not succeed.
Regulation of medical devices in Australia
1364 It is not possible to get a new drug onto the market without at least a seven-year safety study.1444 Surprising though this may seem, the position with respect to implantable medical devices, however, including those designed for permanent implantation, is very different. As Professor Roovers admitted, the regulatory process for taking a device to market does not ensure that the product is safe and efficacious for use in humans.1445 Furthermore, in contrast with the formalised approach to the development of new drugs, the process of evaluation of innovations in surgery has been “unregulated, unstructured, and variable”.1446
1365 Each of the devices in this proceeding is subject to the Australian regulatory regime for therapeutic goods, specifically to those aspects of the regime which apply to medical devices. This regime is administered by the TGA, a division of the Commonwealth Department of Health.
1366 The Therapeutic Goods Act relevantly prevents a medical device from being imported into or supplied in Australia unless it is included on the Australian Register of Therapeutic Goods: TG Act, Pt 4–11 Div 3.
1367 Each of the Ethicon devices is a medical device within the meaning of the TG Act. Medical device is defined in s 41BD to include:
(a) any instrument, apparatus, appliance, material or other article (whether used alone or in combination, and including the software necessary for its proper application) intended, by the person under whose name it is or is to be supplied, to be used for human beings for the purpose of one or more of the following:
(i) diagnosis, prevention, monitoring, treatment or alleviation of disease;
(ii) diagnosis, monitoring, treatment, alleviation of or compensation for an injury or disability;
(iii) investigation, replacement or modification of the anatomy or of a physiological process;
(iv) control of conception;
and that does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but that may be assisted in its function by such means[.]
1368 At all material times JJM was the Australian sponsor of the Ethicon devices. A “sponsor” includes a person responsible for importing the goods into Australia or arranging for another person to manufacture the goods for supply in Australia (or elsewhere): TG Act, s 3.
1369 A “manufacturer” of a medical device is relevantly defined in s 41BG of the TG Act to include a person who is responsible for “the design, production, packaging and labelling of the device before it is supplied under the person’s name, whether or not it is the person or another person acting on the person’s behalf who carries out those operations”.
1370 The current regime for medical devices under the TG Act was introduced in 2002 with the insertion of a new Chapter 4 (Medical Devices) and the commencement of the Therapeutic Goods (Medical Devices) Regulations 2002 (Cth) (Medical Devices Regulations or MDR). By s 41CA, the TG Act, as amended, provided for requirements for medical devices to be included in the regulations. Those requirements were to be known as the “essential principles”. The essential principles can be found in Sch 1 of the Medical Devices Regulations.
1371 The purpose of these changes, which came into effect on 4 October 2002, was to harmonise Australia’s requirements for quality, safety and performance with the recommendations of the Global Harmonisation Task Force on medical devices, which are based on the requirements of the European Community: Explanatory Memorandum to the Therapeutic Goods Amendment (Medical Devices) Bill 2002. As the EM went on to explain:
The new devices regulatory system has several key features. It provides for specified criteria for safety and performance, (the ‘essential principles’), with which devices must conform; increased use of internationally recognised standards for devices as a means of demonstrating that a device conforms with the essential principles; a risk based classification of medical devices; conformity assessment procedures to ensure devices meet the essential principles for safety and performance; and increased emphasis on post-market activities.
1372 This regime applied to each of the products when it was first supplied in Australia except for TVT, which was first supplied in 1999. The registration certificates for each of the other devices, beginning with TVT-O, incorporated the conditions in ss 41FN and 41FO of the TG Act. The conditions imposed by s 41FN apply automatically. Those covered by s 41FO are imposed by the Departmental Secretary in the Secretary’s discretion.
1373 As the EM explained, provision was made for a five-year transitional period to enable sponsors and manufacturers of medical devices already registered on the ARTG to ensure that their products complied with the new regime under Chapter 4.
1374 The classification system for medical devices, set out in Sch 2 of the Medical Devices Regulations, is based on the classification system used in the European Union. There are four classes: class I, class IIa, class IIb, class III and AIMD (active implantable medical devices). Class I is the classification associated with the lowest risk, with each higher number denoting higher levels of risk. The manufacturer of a medical device is responsible for determining its classification using a set of classification rules based on the manufacturer’s intended use of the device, including the location of the device in the body; the level of risk to patients, users and other persons; the degree of invasiveness in the body; the duration of use; and whether or not the device is powered.
Application for inclusion of a medical device on the ARTG
1375 An inclusion on the ARTG is for a “kind” of medical device: TG Act, s 41BE. This means that a single entry may cover a range of products rather than one individual device. Products classed below class III can be included on another product’s ARTG registration if they are taken to be “the same kind” of device as defined by s 41BE of the Act. All that requires, however, is that the device has the same manufacturer, the same sponsor as a device already included on the register, the same device nomenclature system code, the same medical device classification, and is “the same in relation to such other characteristics as the regulations prescribe ...”. This last feature is of no consequence. There is only one characteristic prescribed by the regulations. That is “the unique product identifier given to the device by its manufacturer to identify the device and any variants”: MDR, reg 1.6. Dr Beech explained that this means that “[a] wide and varying range of medical devices that have these same parameters can be supplied under the same ARTG entry without notification to [the] TGA”.1447
1376 The inclusion of a kind of medical device in the ARTG is subject to certain conditions. Those conditions include the requirement to have available at all times while the inclusion in the Register has effect sufficient information to substantiate compliance with the essential principles or procedures in place to ensure that such information can be obtained from the manufacturer within 20 working days: TG Act, s 41FN(3); MDR, reg 5.6.
1377 TVT-O, TVT Abbrevo, and TVT Exact were all included in the same ARTG registration as TVT on the basis that they were the same kind of device. Similarly, the Prolift+M and Prosima devices were included in the same entry as Prolift. Gynemesh PS had a separate entry, as did TVT Secur, with its unique classification as a class III device.
1378 In order to secure inclusion in the ARTG for these devices (save for TVT) JJM, as the Australian sponsor, had to make an application under s 41FC of the TG Act, certifying the following relevant matters set out in s 41FD:
(1) that the device is correctly classified according to the medical device classifications;
(2) that it complies with the essential principles established under the TG Act and set out in Sch 1 of the MDR;
(3) that appropriate “conformity assessment procedures” or other comparable procedures have been applied to devices of that kind;
(4) that either the sponsor has sufficient information to substantiate compliance with (b) and (c) above, or that the sponsor has procedures in place, including a written agreement with the manufacturer, to ensure that such information can be obtained from the manufacturer within the period specified in the regulations; and
(5) that particular advertising requirements have been complied with.
1379 There is no requirement that the information referred to in (4) above be produced to the TGA. The sponsor is required, however, to be in a position to produce it on request (such as where the TGA selects an application for auditing under s 41FH of the Act) and, if it does not have the evidence itself, to obtain it from the manufacturer. Audits are required for class III medical devices which have not already been assessed under the Agreement on Mutual Recognition in relation to Conformity Assessment, Certificates and Markings between Australia and the European Community (EC Mutual Recognition Agreement) or the Agreement on Mutual Recognition in relation to Conformity Assessment, Certificates and Markings between Australia and the European Free Trade Association: TG Act, s 41FH; MDR, reg 5.3. For an audit, the sponsor must provide (and the TGA will review and assess) information and documents such as the product’s CE certificate, Design Examination Certificate, the manufacturer’s Declaration of Conformity, labelling and other instructions for use, risk management and clinical evaluation reports (discussed below).
1380 The essential principles in the MDR include the following general principles set out in Pt 1 of Sch 1:
1 Use of medical devices not to compromise health and safety
A medical device is to be designed and produced in a way that ensures that:
(a) the device will not compromise the clinical condition or safety of a patient, or the safety and health of the user or any other person, when the device is used on a patient under the conditions and for the purposes for which the device was intended and, if applicable, by a user with appropriate technical knowledge, experience, education or training; and
(b) any risks associated with the use of the device are:
(i) acceptable risks when weighed against the intended benefit to the patient; and
(ii) compatible with a high level of protection of health and safety.
2 Design and construction of medical devices to conform with safety principles
(1) The solutions adopted by the manufacturer for the design and construction of a medical device must conform with safety principles, having regard to the generally acknowledged state of the art.
(2) Without limiting subclause (1), in selecting appropriate solutions for the design and construction of a medical device so as to minimise any risks associated with the use of the device, the manufacturer must:
(a) first, identify hazards and associated risks arising from the use of the device for its intended purpose, and foreseeable misuse of the device; and
(b) second, eliminate, or reduce, these risks as far as possible by adopting a policy of inherently safe design and construction; and
(c) third, if appropriate, ensure that adequate protection measures are taken, including alarms if necessary, in relation to any risks that cannot be eliminated; and
(d) fourth, inform users of any residual risks that may arise due to any shortcomings of the protection measures adopted.
3 Medical devices to be suitable for intended purpose
A medical device must:
(a) perform in the way intended by the manufacturer; and
(b) be designed, produced and packaged in a way that ensures that it is suitable for one or more of the purposes mentioned in the definition of medical device in subsection 41BD(1) of the Act.
4 Long-term safety
A medical device must be designed and produced in a way that ensures that if:
(a) the device is used within the period, indicated by the manufacturer, in which the device can be safely used; and
(b) the device is not subjected to stresses that are outside the stresses that can occur during normal conditions of use; and
(c) the device is regularly maintained and calibrated in accordance with the manufacturer’s instructions;
the characteristics and performances mentioned in clauses 1, 2 and 3 are not adversely affected.
5 Medical devices not to be adversely affected by transport or storage
A medical device must be designed, produced and packed in a way that ensures that the characteristics and performance of the device when it is being used for its intended purpose will not be adversely affected during transport and storage that is carried out taking account of the instructions and information provided by the manufacturer.
6 Benefits of medical devices to outweigh any side effects
The benefits to be gained from the use of a medical device for the performance intended by the manufacturer must outweigh any undesirable side effects arising from its use.
1381 Part 2 contains the essential principles about design and construction.
1382 Importantly, Pt 2 cl 7.1 provides:
7.1 Choice of materials
In ensuring that the requirements of Part I are met in relation to a particular device, particular attention must be given to:
(a) the chemical and physical properties of the materials used in the device; and
(b) the compatibility between those materials and biological tissues, cells and body fluids;
having regard to the intended purpose of the device.
1383 The other relevant principles are contained in Pt 9 (construction and environmental properties); Pt 13 (information to be provided with medical devices), and Pt 14 (clinical evidence).
1384 Clause 9.2 provides that a medical device must be designed and produced in such a way as to ensure that, as far as practicable, certain risks are removed or minimised. They include the risk of injury arising from the physical features of the device and any risks associated with reasonably foreseeable environmental conditions.
1385 Clause 13.1(1) stipulates that the certain information must be provided with a medical device including:
(c) information explaining how to use the device safely; having regard to the training and knowledge of potential users of the device.
1386 Clause 13.1(4) requires that the format, content and location of the information must be appropriate for the device and its intended purpose. Pt 13.3 contains a table listing “particular requirements” which must be provided with a medical device. Item 5 of the table reads:
Any warnings, restrictions, or precautions that should be taken, in relation to use of the device
1387 Clause 13.4 provides that instructions for the use of a medical device must be provided with the device, save in certain irrelevant circumstances. Amongst information to be provided with medical devices are:
2 The intended purpose of the device, the intended user of the device, and the kind of patient on whom the device is intended to be used (if this information is not obvious)
…
4 Information about the intended performance of the device and any undesirable side effects caused by use of the device
5 Any contra-indications, warnings, restrictions, or precautions that may apply in relation to use of the device
…
10 If applicable, an indication that the device is intended to be used only for clinical or performance investigations before being supplied
…
19 For an implantable medical device — information about any risks associated with its implantation
…
28 Information about … any particular training or qualifications required by the user of the device
1388 Clause 14 states that:
Every medical device requires clinical evidence, appropriate for the use and classification of the device, demonstrating that the device complies with the applicable provisions of the essential principles.
The requirements for medical devices cleared for sale in Europe
1389 The EC Mutual Recognition Agreement came into force on 1 January 1999. It committed Australia to recognising conformity assessment results, like testing and certification, performed by the European Union’s designated conformity assessment bodies. It covered a wide range of goods including medical devices. The underlying rationale was the elimination of “duplicative testing and recertification”.1448
1390 According to Dr Beech’s unchallenged evidence, the procedure for applying for registration of a medical device in Australia where CE marking had already been obtained is (and I infer from his evidence at all relevant times was) as follows.
1391 Apart from class III medical devices, at the time Ethicon’s devices were entered onto the ARTG, products that had received a “CE” marking were accepted without the TGA conducting any independent assessment of their safety or efficacy and without the need for the manufacturer to demonstrate that independent pre-market testing of their safety and efficacy had been carried out.1449 “CE” is an acronym for Conformité Européenne (meaning “European Conformity”). All the sponsor needed to do in order to have the devices listed on the ARTG was to produce the CE certificates.1450 The sole documentary requirement for all but class III medical devices to be included in the ARTG is the production of the CE certificate. Once an application is made for inclusion of such a device on the ARTG, the TGA checks that these accord with the CE certificate and then notifies the sponsor that the device has been entered on the ARTG.1451 Only in the case of class III medical devices is the safety and efficacy of a medical device independently assessed by the TGA before the device is listed on the ARTG. For class III devices, the TGA requires in addition to the CE certificate, a Design Examination certificate and an audit file, consisting of a Declaration of Conformity, labelling and instructions for use, risk management report and a clinical evidence report. In Dr Beech’s experience, the TGA does not obtain information from any source other than the manufacturer (through the sponsor).1452
1392 The presence of the CE mark on a medical device constitutes a representation that the device conforms to the requirements of the particular European directive which applied to that type of product at the time of certification and thereafter and is an indication to the world at large that it may lawfully be sold in all member states of the European Union. In effect, a CE mark, evidenced by a CE certificate, is a declaration by a manufacturer that its product conforms to the requirements of that directive. For medical devices, the applicable directive is the European Council Directive 93/42/EEC issued on 14 June 1993 and amended from time to time thereafter (the European Directive).1453 Dr Allman testified that, although various changes have been made to the European Directive, most notably in 2007, throughout the period with which this case is concerned, for all practical purposes the approach mandated by the European Directive to be followed when applying for certification has not materially changed, save that since the 2007 amendments commenced notified bodies have been required to exercise greater rigour in their audits.1454
1393 Notably, the declaration of conformity is described in the European Directive as “the procedure whereby the manufacturer who fulfils the obligations imposed by § 1 ensures and declares that the products concerned meet the provisions of the European Directive which apply to them”: European Directive, Annex II § 2. The manufacturer is required to ensure “application of the quality [assurance] system approved for the design, manufacture and final inspection of the products concerned” and is subject to audit in accordance with other sections of the European Directive: European Directive, Annex II § 1.1455
1394 From time to time the European Commission has published guidelines or guidance documents to assist manufacturers and other interested parties, including regulators, to comply with the terms of the European Directive. They are periodically revised.
1395 Each of these documents carries a notation in or to the same effect as the notation that appears, for example, in MEDDEV 2.7.1, to which I will come shortly:
The present Guidelines are part of a set of Guidelines relating to questions of application of EC-Directives on medical devices. They are legally not binding. The Guidelines have been carefully drafted through a process of intensive consultation of the various interest (sic) parties (competent authorities, Commission services, industries, other interested parties) during which intermediate drafts were circulated and comments were taken up in the document. Therefore, this document reflects positions taken by representatives of interest (sic) parties in the medical devices sector.1456
1396 Although “legally not binding”, the guidance documents are considered authoritative since they are the result of intensive consultation between the European Commission, competent authorities (that is, the national regulatory authorities), industry, and other interested parties.1457 Ethicon’s internal documents referred to them and it is reasonable to infer that it considered it should apply them. Certainly, in the absence of good reason to the contrary, such as national legislation which required a different course, any reasonably prudent manufacturer would follow them. It follows that a failure to comply with these guidelines could be evidence of negligence.
1397 The manufacturer must have information to demonstrate compliance with the “essential requirements” listed in the European Directive and discussed below, but the TGA does not require that information to be produced before including a device bearing the CE mark in the ARTG. The TGA requires that a medical device comply with all applicable standards as published by national and international standards bodies, although the evidence indicated that it does not require proof of compliance before listing the device on the ARTG if the device carries the CE mark.1458
1398 Each of the Ethicon devices carries a CE mark. The mark was placed on TVT in 1997 (the evidence does not reveal the precise date but it is likely to have been in about November or late October), TVT-O in December 2003, TVT Secur on 4 May 2006, TVT Exact in June 2010, TVT Abbrevo in September 2010, Gynemesh PS on 20 March 2003, Prolift on 2 March 2005, Prolift+M on 18 March 2008, and Prosima on 12 April 2007.1459
1399 TVT was listed on the ARTG on 21 July 1998, TVT Secur on 18 October 2006, Gynemesh PS on 26 May 2003, and Prolift on 30 March 2005
1400 With the exception of TVT Secur, each of the Ethicon devices was classified as a class IIb device as each was a surgically invasive and implantable medical devices intended for long-term use: see MDR, Sch 2 cl 3.4(2). TVT Secur was classified as a class III device because of the Vicryl and PDS fleece which were designed to be wholly absorbed by the patient’s body. Under Sch 2 cl 3.4(4)(c), surgically invasive and/or implantable devices that are “to be wholly, or mostly, absorbed by a patient’s body” are to be classified as Class III.
1401 Prolift+M also contained a large amount of absorbable material (Monocryl). Yet it was classified as class IIb, although the hernia mesh UltraPro, which was also made with Monocryl, was classified as a class III device.1460 An email chain from March 2008 between Ethicon and its notified body, the British Standards Institute (BSI) shows that the classification issue for Prolift+M was discussed, including as to whether UltraPro mesh should be downgraded to Class IIb.1461 In this exchange, the representative from BSI told Ethicon that it agreed with the IIb classification for Prolift+M, and confirmed that Ethicon did not have to wait for BSI’s approval to release the product as long as it has been approved by Ethicon’s quality management system. In reference to the classification of UltraPro, the representative stated:
[W]hen considering the “wholly or mainly” argument please take into account both the absorbed volume and weight of the device to be on the safe side. So if the device is wholly or mainly absorbed by weight or by volume then class III classification is most probable and if it's not i.e. the device by volume and by weight is “partially” absorbed then it could be considered as class IIb.
(Original emphasis)
1402 In Dr Allman’s opinion, the classification of Prolift+M as class IIb was inconsistent with the classification of other similar Ethicon mesh implants, such as UltraPro and TVT Secur, and was arguably incorrect.1462 It is unnecessary to consider whether it was incorrect, however, as this is an aspect of Ethicon’s conduct about which the applicants made no complaint.
1403 When a manufacturer of a medical device makes a declaration of conformity with the European Directive, it must ensure that the device has complied with each of the “essential requirements” contained in Annex 1 to the Directive: European Directive, articles 3 and 17.
1404 The essential requirements are a set of 14 requirements, set out in Annex 1 to the European Directive, designed to ensure that the benefits of the device outweigh the risks. They are the basis for, and are substantially identical to, the essential principles contained in Sch 1 of the Medical Devices Regulations.
1405 Dr Beech explained:
A manufacturer is required to state how they comply with each requirement and where the evidence for compliance is located. The bases for claiming that the device is safe and efficacious are the manufacturer’s risk management report and the manufacturer’s clinical evidence report plus compliance with Standards. It is a requirement that a medical device comply with all applicable Standards as published by National and International Standards bodies. In particular, EN (European Norm) Standards and ISO (International Standards Organisation) Standards. Evidence of compliance with Standards forms the basis of compliance with many of the Essential Requirements.1463
1406 Dr Beech described the clinical evidence report as “a key element in the work to be performed to ensure compliance with the Essential Requirements”.1464 He said that “it must contain a comprehensive literature review and details of the outcomes of clinical studies on the device itself, which provide a reasoned, clinically-valid basis to confirm that the benefit of the device exceeds the risk, as assessed by a clinician who is expert in the field”.
1407 Provided that the medical device complies with the essential requirements, as assessed by the notified body, and the quality management system is certified to ISO 13485, the notified body will issue CE notification.
1408 Notwithstanding the involvement of the notified bodies, in substance the system is self-regulating. As Dr Beech emphasised, the obligation to establish and maintain compliance with the regulatory requirements rests exclusively with the manufacturer, as does the obligation to take all possible steps to ensure that its medical devices are safe and efficacious. Notified bodies may review clinical data as part of their compliance audit. For class II products, verification (including clinical data) is conducted on a sampling basis. Only for class III products is notified body verification a requirement. Dr Allman gave unchallenged evidence that “notified body verification of clinical data is not exhaustive, and not necessarily thorough”.1465
1409 For relevant purposes the general requirements can be summarised as follows:
(1) The devices must be designed and manufactured in such a way that, when used under the conditions and for the intended purposes, they will not compromise the health or safety of patients; any risks which may be associated with their intended use must be “acceptable” “when weighed against the benefits to the patient”, and “compatible with a high level of protection of health and safety”: European Directive, Annex I § 1;
(2) the design and construction of the medical device must conform to safety principles, “taking account of the generally acknowledged state of the art”, paying particular attention to the compatibility between the materials used and biological tissues, cells and body fluids: European Directive, Annex I § 2 and §7;
(3) in selecting the most appropriate solutions, the manufacturer must apply the following principles in the following order:
(a) eliminate or reduce risks as far as possible;
(b) where appropriate take adequate protection measures in relation to risks that cannot be eliminated;
(c) inform users of residual risks: European Directive, Annex I § 2;
(4) the device must achieve the performances intended by the manufacturer: European Directive, Annex I § 3;
(5) the characteristics and performances mentioned above must not be adversely affected “to such a degree that the clinical conditions and safety of the patients are compromised during the lifetime of the device when the device is subjected to the stresses which can occur during normal conditions of use”: European Directive, Annex I § 4;
(6) the devices must be designed, manufactured and packed in such a way so that transport and storage will not adversely affect their characteristics and performances during their intended use: European Directive, Annex I § 5;
(7) any undesirable side-effect must constitute an acceptable risk when weighed against the intended performance: European Directive, Annex I § 6; and
(8) (added in 2007) demonstration of conformity with the essential requirements must include a clinical evaluation in accordance with Annex X: European Directive, Annex I § 6a.
1410 Additional requirements concerning design and construction relevantly include:
(1) requirements regarding the chemical, physical and biological properties of the device: European Directive, Annex I § 7;
(2) requirements for the elimination and control of infection and microbial contamination: European Directive, Annex I § 8; and
(3) requirements as to the information that must be supplied with the device and on its label, including instructions for use: European Directive, Annex I § 13.
1411 For the manufacturer to apply a CE mark to a device, it must maintain a technical file consisting of information sufficient to demonstrate the device’s compliance with the European Directive, including the essential requirements. The technical file is continuously updated but at any particular time, as Dr Allman put it during cross-examination, it should be “the manufacturer’s main demonstration of how they’ve complied to (sic) the Directive for a particular product”.1466 The technical file normally contains a risk management report and a clinical evaluation report (also referred to as a “clinical expert” or “clinical evidence” report) (CER). These reports are required under §§ 1.1B, 2.3.6 and 2.3.7 of Annex X of the Directive (Clinical Evaluation).
1412 The CER must contain a comprehensive literature review and particulars of the outcomes of clinical studies on the device, which, in Dr Beech’s words, “provide a reasoned, clinically-valid basis to confirm that the benefit of the device exceeds the risk, as assessed by a clinician who is expert in the field”.1467 Under § 1.1c of Annex X, the clinical evaluation report must be actively updated with data obtained from post-market surveillance.
1413 At all relevant times § 1.1 of Annex X to the European Directive required that, as a general rule, confirmation of conformity with the characteristics and performances referred to in §§ 1 and 3 of Annex X under normal conditions of use, the evaluation of undesirable side-effects, and the acceptability of the benefit/risk ratio must be based on clinical data. It also required that the evaluation of that data follow “a defined and methodologically sound procedure” based on any one of the following three approaches:
(1) a critical evaluation of the relevant scientific literature relating to the safety, performance, design characteristics and intended purpose of the device where:
(a) the device is demonstrated to be equivalent to the device to which the data relates; and
(b) the data adequately demonstrate compliance with the relevant essential requirements; or
(2) a critical evaluation of the results of all clinical investigations made; or
(3) a critical evaluation of the combination of (1) and (2).
1414 In the case of implantable devices and devices in class III, however, clinical investigations were mandatory unless it was “duly justified to rely on existing clinical data”. The clinical evaluation and its outcome had to be documented and/or fully referenced in the technical file for the device. The clinical evaluation and its outcome also had to be actively updated with data obtained from post-market surveillance and, where clinical follow-up was not deemed necessary that, too, had to be “duly justified” and documented. Where demonstration of conformity with essential requirements based on clinical data was not deemed appropriate, “adequate justification” for excluding it had to be provided “based on risk management output and under consideration of the specifics of the device/body interaction, the clinical performances intended and the claims of the manufacturer”: European Directive, Annex X, §§ 1.1b–1.1d.
1415 For the Ethicon devices, clinical investigations were not always undertaken. Ethicon chose to adopt the first approach for all the devices in question (referred to in evidence as “the literature route”). When clinical investigations were not undertaken, however, the decision to rely on existing clinical data was not always “duly justified”. When clinical investigations were undertaken, Ethicon rarely waited for them to be completed before applying the CE mark and did not critically evaluate them. The clinical evaluation reports for all the Ethicon devices revealed a distinct lack of critical evaluation of the studies on which they relied and the literature to which they referred. Some of them disclosed no evaluation at all. In addition, in some instances the literature to which the data related and upon which the reports relied concerned devices that were not demonstrated to be equivalent to the device in question.
1416 In April 2003 the European Commission published guidelines on the evaluation of clinical data subtitled (known as MEDDEV 2.7.1). It was common ground, however, that the document set out principles that had been applied or concepts that had been deployed since 1998.1468
1417 MEDDEV 2.7.1 laid down in some detail the proper approach to the kind of literature review contemplated by the European Directive and what such a review entailed.1469
1418 First, it stipulated that “due regard” needed to be paid to the extent to which the published data are relevant and applicable to the relevant characteristics of the device in question and the medical procedure for which the device is intended.
1419 Second, it stated that the review should be undertaken by a person or persons “suitably qualified in the relevant field, knowledgeable in the ‘state of the art’ and able to demonstrate objectivity”.
1420 Third, it set out the course to be followed for any clinical evaluation which took the form of a review of the relevant scientific literature. It began with a description of the methodology. It provided that a protocol should be written for the identification, selection, collation and review of relevant studies based on recognised practice for systematic review of literature. It stated that the objective of the review should be clearly defined and the types of studies that are relevant to the objective specified, taking into account the already well-established knowledge of the device. It provided that data should be taken from recognised scientific publications but stated that unpublished data should also be taken into account in order to avoid publication bias.
1421 It also specified that the review should state the sources of data and the extent of the searches that had been undertaken to retrieve the data; the rationale for the selection of the published literature; the reasons for believing that all relevant references, both favourable and unfavourable, have been identified; and the criteria for exclusion of particular references together with a justification for the exclusion.1470 It even offered examples of potential data sources: medical and paramedical databases; foreign language literature; what was referred to as “grey literature” (theses, internal reports, non-peer review journals, the internet, and industry files); references listed in primary sources; other published sources known to experts in the field obtained by personal communication; and raw data from published trials obtained from personal communication.
1422 To be equivalent, the devices should be similar with respect to all three parameters: clinical, technical, and biological with special attention to the performance, principles of operation and materials. Paragraph 4.3.1(i)(d) of MEDDEV 2.7.1 provided that the manufacturer must be able to demonstrate “equivalence” in all the following ways:
Clinical
-used for the same clinical condition or purpose;
-used at the same site in the body;
-used in similar population (including age, anatomy, physiology);
-have similar critical performance according to expected clinical effect for specified intended use.
Technical:
-used under similar conditions of use;
-have similar specifications and properties eg tensile strength, viscosity, surface characteristics
-be of similar design;
-use similar deployment methods (if relevant);
-have similar principles of operation.
Biological:
-use same materials in contact with the same human tissues or bodily fluids[.]1471
1423 If differences are identified, the significance of the differences to the safety and performance of the devices must be described. Where, for example, the new device has a new principle of operation from the device or devices the subject of the published studies, the clinical benefit of the new device has to be generated by data resulting from specifically designed clinical investigations. Otherwise, the two devices cannot be considered equivalent.
1424 The manufacturer must also be able to demonstrate the adequacy of the data in addressing the aspects of conformity set out in the objective.
1425 MEDDEV 2.7.1 proceeded to identify the way in which the clinical data should be assessed. It stipulated that the literature review should make clear the significance that is attached to particular references based on various factors including:
the relevance of the author’s background and expertise in relation to the particular device and/or medical procedure involved;
whether the author’s conclusions are substantiated by the available data;
whether the literature reflects the current medical practice and the generally acknowledged “state of the art” technologies;
whether references are taken from recognised scientific publications and whether or not they have been reported in peer review journals;
the extent to which the published literature is the outcome of a study/studies which have followed scientific principles in relation to design, for example, in having demonstrable and appropriate endpoints, inclusion and exclusion criteria, an appropriate and validated number of patients submitted, carried out for an appropriate duration, providing evidence and analysis of all adverse incidents, deaths, exclusions, withdrawals and subjects lost follow-up and identifying an appropriate statistical plan of analysis.1472
1426 It then stated that:
Ideally, evidence should be generated from a clinical trial (controlled if appropriate), properly designed cohort/case controlled study, well documented case histories or sequential reports conducted by appropriate experienced experts, whether in relation to the device itself or an equivalent device. If unpublished data is being included in the assessment, the literature review will need to weigh the significance that is attached to each report.1473
1427 It also specified the material that should not be taken into account:
The evidence should not consist of:
• isolated case reports;
• random experience;
• reports lacking sufficient detail to permit scientific evaluation (including lack of accepted and validated statistical design if this is relevant to the design of the intended study);
• unsubstantiated opinions.
1428 Moreover, MEDDEV 2.7.1 emphasised the need for “a critical evaluation of the literature” and explained what that would entail:
This critical evaluation should:
• be written by a person suitably qualified in the relevant field, knowledgeable in the “state of the art” and able to demonstrate objectivity;
• contain a short description of the medical device, its intended functions, description of the intended purpose and application of use;
• contain an analysis of all the available data considered, both favourable and unfavourable;
• establish the extent to which the literature relates to the specific characteristics and features of the device being assessed, taking due account of the exten[t] of similarity between the device(s) covered by the literature and the device under assessment;
• demonstrate that those aspects of the use of the device, including performance, addressed in the clinical part of the risk analysis are met as claimed by the manufacturer, and that the device fulfils its intended purpose as a medical device;
• analyse the identified hazards, the associated risks and the appropriate safety measures of patients, medical staff and third parties involved in the study/studies, for example by reference to the manufacturer’s risk analysis (see also ISO14155-2);
• contain a risk analysis relevant to the device design, materials and procedures involved, taking into account any adverse events, results of post-market surveillance studies, modifications and recalls (if known) (see also ISO14155-2);
• contain a description of the methods of weighting different papers and the statistical methods of analysis employed taking into account the assessment methods, the type and duration of study and the heterogeneity of the population included within the study. Particular attention should be given in circumstances where there are repeated publications on the same group of patients by the same authors in order to avoid overweighting the experience;
• include an analysis of the market experience of the same or similar devices, including the results of post-marketing studies, post-market surveillance and short- and long-term adverse events;
• contain a list of publications appropriately cross-referenced in the evaluation;
• if the clinical data relates to an equivalent device, contain a statement that equivalence with all the relevant characteristics has been demonstrated;
• include a conclusion with a justification, including an assessment of any probable benefit to health from the use of the device as intended by the manufacturer, against probable risks of injury or illness from such use taking account of the “state of the art”. If applicable, the findings should be compared with other studies covering the same field of application. These studies may involve other modalities, including alternative medical devices, medical therapy, surgery or other accepted health care methods provided they employ methods which are generally accepted as being common practice. The conclusions should make clear how the objectives of the literature review have been met and identify any gaps in the evidence necessary to cover all relevant aspects of safety and performance.
Note 1: conclusions should be relevant in the field of use, indications, contra-indications and instructions for use intended by the manufacturer
Note 2: the critical evaluation should be signed and dated by the author1474
(Emphasis added)
1429 MEDDEV 2.7.1 also stipulates at [4.3.2] that the manufacturer’s report of the literature review should be written in a format that enables the notified body to:
(1) determine that the manufacturer’s conclusions are valid;
(2) be satisfied that the data, taken together with the available pre-clinical data, is sufficient to demonstrate compliance with the essential requirements covering safety and performance of the device in question under normal conditions of use;
(3) identify gaps in the demonstration of compliance or equivalence that need to be addressed through specifically-designed clinical investigations; and
(4) determine that the claims made in the device labelling (which includes the instructions for use) are substantiated by the clinical data taken together with the pre-clinical data.1475
1430 The European Directive (93/42/EEC) was relevantly amended, effective 21 March 2010, by European Council Directive 2007/47/EC (the 2007 Directive) which clarified, and to some extent extended, requirements for clinical evaluation.1476 A requirement was added in Annex 1 that conformity with the essential requirements must include a clinical evaluation in accordance with Annex X. Although Annex X continued to provide that “as a general rule, confirmation with the requirements concerning the characteristics and performance… must be based on clinical data”, clinical evaluation became a requirement for all medical devices.
1431 The 2007 Directive included “clinical data” in the essential requirements (see § 6a of Annex I) and introduced a definition of “clinical data”. Article 1(2)(k) provided:
‘clinical data’ means the safety and/or performance information that is generated from the use of a device. Clinical data are sourced from:
- clinical investigation(s) of the device concerned; or
- clinical investigation(s) or other studies reported in the scientific literature, of a similar device for which equivalence to the device in question can be demonstrated; or
- published and/or unpublished reports on other clinical experience of either the device in question or a similar device for which equivalence to the device in question can be demonstrated;
1432 The 2007 Directive clarified that clinical evaluation and its outcome must be documented as part of the technical documentation of the device. It added the requirement for clinical evaluation to be actively updated with data obtained from post-market surveillance and, if post-market surveillance plans did not include “clinical follow-up”, a requirement to justify and document the absence of clinical follow-up. It also added a requirement for implantable and Class III devices that “clinical investigations shall be performed unless it is duly justified to rely on existing clinical data”.
1433 Guidelines on post-market clinical follow-up were published in May 2004 (known as MEDDEV 2.12.2).1477 They were intended to guide manufacturers and notified bodies on how to carry out Post-Market Clinical Follow-Up (PMCF) in order to fulfil the post-market surveillance obligations contained in the Directive.1478 They stipulated that all PMCF should be planned but they explained that the plan could take different forms. It could involve extended follow-up of patients enrolled in pre-market trials. In addition or in the alternative it could consist of a prospective study of a representative subset of patients after the device is placed on the market. It could also take the form of open registries. This plan would need to take into account, as MEDDEV 2.12.2 made clear:
results of the clinical investigation including adverse events identified;
the average life expectancy of the device;
the claims for the device made by the manufacturer;
performances for which equivalence is claimed; and
any new information.
1434 MEDDEV 2.12.2 stressed that whenever PMCF is carried out it must always be performed for the use of the product within its intended indications, according to the instructions for use.
1435 MEDDEV 2.12.2 proposed a “triage approach” and offered general advice for the evaluation of products under different circumstances:
PMCF | Product specificities | Required actions |
No PMCF | Products for which the medium/long term clinical performance and safety is already known from previous use of the device, or from fully transferable experience with equivalent devices (except **) | All received complaints and adverse events data shall be systematically reviewed, and all product related adverse events such as those described in Annex II 3.1 of the MDD must be notified to the relevant Competent Authority (ies). This includes all sources of information known by the manufacturer, including published literature. Monitoring of postmarket performance should take into account relevant data publicly available with similar devices especially when the CE marking was based on equivalence. |
PMCF | Always considered for devices where identification of possible emerging risks and the evaluation of long term safety and performance are critical (**)Products quoted as “equivalent” devices where reference product is subjected to PMCF | Same as above. ➢ Plus Post-Market Clinical Follow-up (PMCF) in the form of follow up of all or a justifiable subset of patients already enrolled in pre-marketing Clinical Investigations; or on specific sub-groups and/or prospective study or registry of a sample of products. A formal protocol should describe the duration of PMCF; identify patient population and data to be collected. (NOTE: The manufacturer must justify the design, nature, and duration of post-marketing follow-up, having regard to any published standards) PMCF report to be provided to the relevant NB for review and to competent authority if requested. |
1436 MEDDEV 2.12.2 was revised in January 2012,1479 amongst other things, to emphasise the critical importance to the long-term safety of medical devices of post-market surveillance.
1437 The revised MEDDEV 2.12.2 noted that, although clinical evidence is an essential element of the pre-market conformity assessment process, there may be limitations to the then available clinical data. It postulated that those limitations may be due to the duration of the pre-market clinical investigations, the number of subjects and investigators involved in the investigation, the relative heterogeneity of subjects and investigators and/or the controlled setting in which the investigations took place in contrast to the full range of clinical conditions encountered in general medical practice. It also observed that the extent of the data that can be gathered in the pre-market phase does not necessarily enable a manufacturer to detect rare complications or problems that only become apparent after widespread or long-term use. It described “an appropriate post-market surveillance plan” as “key” to the identification and investigation of residual risks associated with the use of the devices in the market. It stated that residual risks should be investigated and assessed in the post-market phase through systematic PCMF studies. It emphasised that clinical data derived from post-market surveillance and PMCF are not intended to replace the need for pre-market data to demonstrate conformity with the legislative requirements. Rather, they are “critical” “to ensure the long-term safety and performance of devices” after they are placed on the market.
1438 MEDDEV 2.7.1 (the guidelines on clinical evaluation issued in April 2003) was revised in December 2009 to incorporate changes introduced by the 2007 Directive “and in the light of experience”.1480 Like the 2007 Directive, it applied as of 21 March 2010.
1439 Dr Allman said that the purpose of the changes to clinical evaluation introduced by the 2007 Directive was “to ensure consistent application of pre-existing concepts and requirements”. Dr Allman explained that “[t]his reflected a view at the time that manufacturers underestimated the requirement for rigorous clinical evaluation, and clinical data, and that notified bodies had been neither consistent nor rigorous, in verifying that manufacturers had interpreted the requirements appropriately”.1481 Although, by consent, this evidence was limited to proof of the witness’s understanding, rather than the truth of the asserted facts, it was unchallenged and uncontested. Having regard to his background and experience, Dr Allman’s understanding carries some weight.
1440 The “key goal” of clinical evaluation, according to the Global Harmonization Task Force (GHTF) publication “Clinical Evidence – Key Definitions and Concepts” dated May 2007, is to determine whether any risks associated with the use of a device are acceptable when weighed against the benefits to the patient and are compatible with a high level of protection of health and safety.1482 Ethicon was mindful of the first consideration but appear to have overlooked or paid insufficient attention to the second.
1441 Dr Allman explained the significance of clinical evaluation in the following way:
Effective clinical evaluation is key to ensuring safety and performance of devices—particularly for implantable devices and new types of devices for which the determination to use (or not use) clinical data from existing (equivalent) devices as the basis for CE marking is critical. The determination to use (or not use) clinical data from existing devices, and therefore the decision to conduct (or not conduct) clinical investigations, is also an area in which manufacturers may underestimate the scientific and regulatory requirements to establish safety and performance of their devices. Performing pre-CE marking clinical investigations is also often perceived as a ‘delay’ to making devices available—not just for commercial reasons, but also because of a belief that the new device offers real clinical benefits. In my experience, it is an area in which judgements can be poor.1483
1442 Clinical evaluation reports were prepared for all the Ethicon devices, although not with the regularity Dr Hinoul suggested.1484 None of those reports contained a description of the kind referred to in MEDDEV 2.7.1 and the evidence from the regulatory experts was that they were wanting in numerous other respects. I deal with these matters in Part IX.
1443 In the United States, § 510(k) of the Federal Food, Drug, and Cosmetic Act (Title 21 of the US Code) (now § 360(e)) required device manufacturers to notify the FDA of their intention to market a medical device to enable the FDA to determine whether the device is substantially equivalent to a device already on the market, commonly referred to as a “predicate” device. A premarket notification of this kind is often referred to as a “510(k) application”. The FDA does not approve the device but the determination by the FDA of substantial equivalence entitles a manufacturer to sell the device in the United States. For this reason, a device that enters the market following the § 510(k) route is often referred to as having been cleared by the FDA. In this context, to be substantially equivalent to a predicate device, a manufacturer merely needs to satisfy the FDA that the new device is at least as safe and effective as a predicate device.1485
1444 The concept of “equivalence”, which allows a manufacturer to rely on clinical data from either the literature or clinical investigations relating to a similar device “for which equivalence to the device in question can be demonstrated”, was expressly introduced into the 2007 European Directive. But the evidence was that the concept of equivalence had “always” existed, since manufacturers who took the “literature route” had always relied on clinical data from older devices to demonstrate the performance and safety of a new device.1486 He pointed out, however, that the European requirements with respect to clinical evaluation using clinical data from “equivalent” devices can be more demanding than their US counterparts.1487
1445 This evidence was supported by MEDDEV 2.7.1, which in its first iteration (from April 2003), defined “equivalence” in the context of clinical data, as follows:
To be equivalent, the devices should have similarity with regard to the clinical, technical and biological parameters with special attention to the performance, principles of operation and materials; or if there are differences identified, an assessment and demonstration of the significance these might have on safety and performance must be set out.
1446 MEDDEV 2.7.1 included the following example:
For example we can consider the case where the device under consideration and the device referred to in the published studies do not have the same principles of operation ie the new device has a new principle of operation. Since a new mechanism of action does not necessarily result in a new clinical benefit, demonstration of the clinical benefit of the new device has to be generated by data resulting from a specifically designed clinical investigation since the 2 devices cannot be considered equivalent.
1447 As I mentioned earlier, MEDDEV 2.7.1 was relevantly revised in 2009. The 2009 version included a new definition of “equivalence”:
The devices should have the same intended use and will need to be compared with respect to their technical and biological characteristics. These characteristics should be similar to such an extent that there would be no clinically significant difference in the performance and safety of the device. The intended use relates to the clinical condition being treated, the severity and stage of disease, the site of application to/in the body and the patient population; the technical characteristics relate to the design, specifications, physiochemical properties including energy intensity, deployment methods, critical performance requirements, principles of operation and conditions of use; and biological characteristics relate to biocompatibility of materials in contact with the same body fluids/tissues. …1488
(Original emphasis)
1448 It is common ground that the respondents relied only on the “literature route” to support the CE marking for each of the Ethicon devices, although in their defence they denied the allegation that they did not conduct any pre-CE marking clinical investigation studies of the devices. That said, Dr Allman’s evidence was to the effect that the evaluation of the data did not follow a defined and methodologically sound procedure; there was no critical evaluation of the literature; equivalence was never demonstrated; and the data relied upon did not adequately demonstrate compliance with the relevant essential requirements. Many, if not all, of Dr Allman’s opinions were supported both by other regulatory experts and regulatory authorities.
1449 The objectives of clinical investigation are twofold: first, to verify that, under normal conditions of use, the performance of the devices conform to those referred to in § 3 of Annex 1, and second, to determine any undesirable side-effects under those conditions and assess whether they constitute risks when weighed against the intended performance of the device: European Directive, Annex X § 2.1.
1450 The methods for carrying out the investigations are described in §§ 2.3.1–2.3.7 of Annex X. In summary they require;
(1) an appropriate plan of investigation reflecting the latest scientific and technical knowledge and defined in such a way as to confirm or refute the manufacturer’s claims for the device;
(2) an adequate number of observations to guarantee the scientific validity of the conclusions;
(3) the use of investigative procedures appropriate to the device under examination;
(4) the performance of the investigations in circumstances similar to the normal conditions of use of the device;
(5) the performance of the investigations under the responsibility of a medical practitioner or other authorised qualified person in an appropriate environment and with access to the technical and clinical data;
(6) examination of all the appropriate features, including those involving the safety and performances of the device, and its effect on patients; and
(7) full recording of all serious adverse events and immediate notification to all competent authorities of the Member States in which the clinical investigation is being performed.
1451 Section 2.3.7 stipulates that the written report on the clinical investigation must contain a critical evaluation of all the data collected during the investigation.
The notified body and conformity assessments
1452 Under the terms of the Directive, it is the manufacturer of the device who must determine if the available clinical data are sufficient to justify CE marking or if clinical investigations are necessary before the mark is applied. It is the manufacturer who decides whether a declaration of conformity should be made.1489 Before a manufacturer can apply a CE mark to a device, however, it must appoint a “notified body” to carry out a conformity assessment: European Directive, article 16. Notified bodies are usually commercial for-profit organisations, although they must be authorised by a competent authority (usually a government authority) to assess the compliance by a manufacturer with a particular European Directive. Ethicon appointed BSI as its notified body.
1453 According to the unchallenged evidence of Dr Beech, the primary considerations applied by manufacturers in deciding which organisation to appoint as a notified body are speed of service, responsiveness, cost, ease of processing documents, and reputation. He said that “[m]anufacturers are anxious to get through the regulatory system as quickly and easily as possible to achieve revenue to recompense development costs”.1490
1454 The conformity assessment can be carried out in different ways (see European Directive, article 11), but the evidence was that in most cases manufacturers will choose the full quality assurance option (see European Directive, Annex II), in which the notified body assesses whether the manufacturer’s quality management system complies with the requirements of the Directive.1491 This was the option chosen by Ethicon for each of its devices.1492
1455 In a quality system approval, MEDDEV 2.7.1 provides that the role of the notified body is to assess the manufacturer’s procedure for clinical data evaluation. That may include a review of examples of such evaluations (in which case the notified body is supposed to pay special attention to the relevance of the data and whether the criteria referred to in MEDDEV 2.7.1 have been applied to the literature or the clinical investigations, as the case may be). But a notified body would not consider whether a conclusion in a clinical evaluation report is justified by the literature cited in the report.1493 It is only in a “design/type examination” that the notified body assesses the clinical data assembled by the manufacturer, the manufacturer’s evaluation, and the validity of the conclusions.1494 Where the manufacturer chooses the quality assurance option, as the respondents did, generally the notified body carries out a review of documentation including a sample of technical files created for different products. Dr Allman said that in a quality system approval process, the notified body does not review the technical file for the device, does not conduct testing, and does not undertake a clinical review of the products or their supporting documentation. Nor does the notified body approve medical devices or make a determination that a device is safe and effective.1495 Its role is limited to auditing the manufacturer’s procedures.
1456 On the basis of its assessment, a notified body can issue a CE certificate. Once this certificate is issued, a manufacturer may issue a Declaration of Conformity and apply a CE mark to the devices included in the scope of the quality management system certified by the notified body.
1457 Where a manufacturer relies on the literature route, claiming that a device being evaluated for CE marking is equivalent to a predicate device, Dr Allman testified that he would expect a notified body to be concerned about whether the subject device was indeed equivalent.1496 Yet, he said that he had seen no evidence that BSI’s auditors had applied their minds to the question of equivalence and none was put to him in cross-examination.1497
1458 For class III devices, the “Full Quality Assurance” option for conformity assessment additionally requires the manufacturer to submit a design dossier (essentially the technical file) to the notified body for it to review in order to assess whether the manufacturer has determined that the device meets the essential requirements in the Directive. On the basis of that assessment, the notified body will or will not issue a “Design Examination certificate” and a CE Certificate. Armed with these certificates, the manufacturer may apply CE marking to a class III device.1498 These certificates, along with an audit file, must be produced to the TGA in order for the device to be included on the ARTG.1499
Post-market surveillance and testing
1459 It is a requirement of both the European Directive and the Medical Devices Regulations that the manufacturer of a medical device monitors the clinical performance of the device in the marketplace, undertakes design review and risk management exercises to optimise its safety and efficacy, investigates adverse events, makes every effort to resolve reported adverse events, keeps detailed records, and determines corrective or preventative action.
1460 Adverse events can come to the attention of the TGA, the manufacturer, or the sponsor. The source might be a user, a clinician, a distributor, or a regulatory authority in any country in which the device is supplied. The important thing, emphasised by all the regulatory experts, is, as Dr Beech put it, that the manufacturer is obliged to pro-actively seek feedback on safety and efficacy and monitor the literature for relevant information. Section 1.1c of the European Directive requires that “the clinical evaluation and its documentation be actively updated with data obtained from the post-market surveillance”, unless post-market clinical follow-up is not deemed necessary in which case “this must be duly justified and documented”. In Australia all adverse events that might cause death or serious injury must be reported to the TGA through the Australian sponsor (here, JJM). The manufacturer is required to periodically collate and assess all post-market information and act on the finding of its own post-market surveillance report.
1461 As part of the conformity assessment procedures, manufacturers are required by the Directive to establish and keep up to date a systematic procedure to review experience in the post-production phase and implement appropriate means to apply any necessary corrective action. Since the 2007 Directive, that has expressly involved clinical evaluation.1500 Dr Allman said that, after the publication of MEDDEV 2.12.2 in May 2004, a manufacturer of implantable devices would reasonably be expected to have:
• effective procedures for systematic review of information received by the manufacturer (complaint and adverse event reports) relating to the device;
• effective procedures for systematic review of the scientific literature pertaining to the devices and similar devices;
• effective procedures for the active collection of information relevant to devices—including, where necessary, PMCF studies; and
• considered the need for PMCF studies, documented the rationale for the decision, and to have implemented such studies if necessary.1501
1462 Since the European Directive was amended, effective from 21 March 2010, post-market surveillance has also required trend reporting and the inclusion in the quality management system of a procedure describing post-market surveillance activities.1502
1463 Manufacturers were also required to report adverse events to the regulatory authorities.
1464 Under § 3.1 of Annex II to the European Directive, a manufacturer must include in its application for assessment of its quality system with a notified body an undertaking that it will notify the competent authorities immediately of the following types of incidents:
(i) any malfunction or deterioration in the characteristics and/or performance of a device, as well as any inadequacy in the instructions for use which might lead to or might have led to the death of a patient or user or to a serious deterioration in his state of health;
(ii) any technical or medical reason connected with the characteristics or performance of a device leading for the reasons referred to in subparagraph (i) to systematic recall of devices of the same type by the manufacturer.
1465 Notified bodies have a limited role to play in post-market surveillance; they merely audit the manufacturer’s procedures. They have no role to play in post-market testing of the safety and effectiveness of devices.1503 The competent authorities are responsible for monitoring adverse events to the extent that they are reported to them by the manufacturer and other sources such as healthcare professionals who use the device.1504
1466 Under s 41FN(3)(d) of the TG Act, one of the conditions that applies automatically to the inclusion of a medical device on the register is that the sponsor of the device must give information about adverse events as defined in ss 41MP and 41MPA to the TGA within the time specified by the regulations (see MDR, reg 5.7).
1467 Sections 41MP(2) and 41MPA(2), which are identical, set out the information that must be provided to the TGA regarding adverse events. That information consists of the following:
(a) information relating to:
(i) any malfunction or deterioration in the characteristics or performance of the kind of device; or
(ii) any inadequacy in the design, production, labelling, instructions for use or advertising materials of the kind of device; or
(iii) any use in accordance with, or contrary to, the use intended by the manufacturer of the kind of device;
that might lead, or might have led, to the death of a patient or a user of the device, or to a serious deterioration in his or her state of health;
(b) information relating to any technical or medical reason for a malfunction or deterioration of a kind referred to in subparagraph (a)(i) that has led the manufacturer to take steps to recall devices of that kind that have been distributed;
(c) information that indicates that a device of that kind does not comply with the essential principles;
(d) information that indicates that a certificate or other document (other than a certificate or other document issued by the Secretary under this Act) used for the purpose of an application under subsection 41FC(1) to signify:
(i) compliance with the essential principles; or
(ii) the application of relevant conformity assessment procedures to a device of that kind or the application of requirements, comparable to those procedures, to a device of that kind;
has been restricted, suspended, revoked or is no longer in effect.
1468 It is a criminal offence, punishable by imprisonment for 12 months or 1,000 penalty units or both) for a sponsor of a medical device who knows that particular information is information of a kind mentioned in subs (2) to fail to provide the information to the TGA within the specified time: s 41MP(1).
1469 Regulation 5.7 prescribes the period in which the information about adverse events must be given under s 41FN(3)(d). In short, the period ranges from 48 hours to 10 days, depending on the gravity of the potential consequences of the events.
1470 Dr Beech explained that the TGA will investigate reports of adverse events and ask for information from the sponsor about the reasons for the event and the manner in which it was resolved. The TGA may then decide to undertake its own post-market review of a device or procedure utilising a device, if it becomes aware that it had systemic issues jeopardising the safety or efficacy of the device. Dr Beech referred to the investigation into urogynaecological meshes commenced by the TGA in 2008, which included a clinical review in 2010 and a series of published statements in October 2012, August 2014, and August 2016. Since Dr Beech’s report, further statements were published by the TGA in December 2017, January 2018 and March 2019.
1471 The TGA’s statement of 2 August 2016 included an observation that the number of adverse events arising from the use of urogynaecological mesh was most likely under-reported.1505 In answer to the question why and how this could occur, Dr Beech said that accurate reporting of adverse events to the TGA depends on awareness of the adverse events and cooperation in reporting from several sources, including patients, manufacturers, sponsors and clinicians. He observed that patients do not always report problems to their surgeons and surgeons may not believe that the problems patients experienced are caused by the device. Besides, medical practitioners are not obliged to report adverse events and many do not. Dr Beech also said that under-reporting may also result from insufficient communication between manufacturers, sponsors and users as well as general ignorance of reporting mechanisms and requirements.
1472 Dr Beech explained that, because of the problem with under-reporting, manufacturers are expected to actively seek out information about the post-market performance of a device by requesting users to provide reports or by conducting detailed reviews through clinical experts or user facilities.1506
1473 Dr Beech said that the TGA would rarely carry out independent tests on medical devices, except on some consumer items such as condoms, gloves, or syringes, for which it has appropriate testing facilities. In his experience, the TGA will instead require the manufacturer of the device to investigate and report on adverse events and, although the TGA has the power to remove a device from the ARTG, in most cases a medical device will only be removed from the market by voluntary withdrawal by the manufacturer, in order to minimise reputational damage.1507
1474 The TGA has a range of powers including the power to cancel or suspend an entry on the ARTG and the power to impose conditions on the inclusion of certain kinds of devices on the register (see TG Act, s 41FP), which, if breached, can lead to suspension or cancellation.
1475 The TGA may suspend the registration for a kind of device for a specified period if it is satisfied that there is a potential risk of death, serious illness or serious injury should those devices continue to be included in the register and where it is likely that, within the period of suspension, the sponsor will be able to take action necessary to remedy the risk if the devices were to continue to be included on the Register: TG Act, s 41GA. If the suspension is not revoked before the end of the period specified by the Act (at most 6 months), the registration must be cancelled: TG Act, s 41GK. Automatic cancellation is also required under this section if the conformity assessment certificate applying to that kind of device is revoked. The TGA may decide to immediately cancel a registration for various reasons, including if it is satisfied that there is a potential risk of death, serious illness or serious injury should those devices continue to be included in the register, or if the sponsor or manufacturer requests the cancellation in writing: TG Act, s 41GL.
Overview of the evidence from the regulatory experts on the respondents’ compliance with regulatory requirements
1476 Dr Hinoul deposed that the purpose of Ethicon’s clinical evaluation reports was “to provide a systematic review and assessment of the potential risks and benefits of a device through an analysis of company sponsored studies, all relevant and available clinical data, a compilation of scientific literature relative (sic) to the safety and performance of the device (or its regulatory equivalent), and PMS [post-market surveillance] data”.1508 The evidence demonstrated that this purpose was rarely, if ever, fully realised.
1477 Dr Hinoul also deposed that:
The completion of Clinical Evaluation Reports satisfies Ethicon’s mandate to adhere to regulatory guidelines.1509
1478 To say the least, Dr Hinoul’s opinion reflected an imperfect understanding of Ethicon’s obligations.
1479 Below I summarise the evidence of Dr Allman, Ms Holland, and Dr Pence and outline their assessments of the respondents’ conduct. I will return to, and expand upon, some of this evidence later as and when it is relevant. For the most part, this evidence is concerned with the conduct of Ethicon Sàrl and Ethicon Inc. as the manufacturers of the devices. JJM was the sponsor of the Ethicon devices within the meaning of the Therapeutic Goods Act. It was not a manufacturer except, as we shall see, for the purposes of claims under the Trade Practices Act. I have referred above to the responsibilities of a sponsor of a medical device and will say something more about them later.
1480 Dr Allman conducted an extensive review of the respondents’ technical files. As a result of that review, he concluded that, although Ethicon had procedures in place that were intended to meet regulatory requirements for obtaining CE marking for each of the devices, it did not in fact have adequate clinical evidence and therefore did not have sufficient justification to affix a CE mark to any of them. He also concluded that Ethicon did not comply with the steps that a reasonable manufacturer would undertake to ensure that it was appropriate for the devices to maintain their CE marking once released onto the market.1510
1481 Dr Allman considered that the clinical evaluation of the devices, both before and after CE marking, was poor or inadequate for the following reasons.
1482 First, the evaluation process did not specify the criteria used to assess acceptability of the clinical data; did not reflect good practice; and did not meet “a reasonable interpretation” of European regulatory requirements.1511 He was particularly critical of the earlier CERs (pre-2010), describing them as “very deficient”, containing “superficial descriptions of some sort of equivalence [with predicate devices] that wasn’t a realistic equivalence”, “using data or just descriptions of publications in one or two sentences, and using that as a basis for asserting the new device met safety and performance requirements from the essential requirements” although the reports included insufficient detail to justify those conclusions.1512
1483 Second, there was an over-reliance on published clinical data and a corresponding failure to conduct pre-CE marking clinical studies of each of the devices. This was a matter of some importance since the devices were intended for innovative procedures.1513
1484 Third, and related to the first two criticisms, the conclusions in the CERs as to safety and performance were based on a comparison of a new device to Ethicon’s older devices, in circumstances where the new device had design changes that were intended to change clinical performance. Dr Allman considered this approach unjustifiable. Indeed, he described the claims of equivalence as “spurious” in the circumstances.1514 It will be recalled that para 4.1(i)(d) of MEDDEV 2.7.1 provided that the manufacturer must be able to demonstrate “equivalence” in all of a number of respects, including use at the same site in the body, use of similar deployment methods (where relevant), have similar principles of operation, and use the same materials in contact with the same human tissues and bodily fluids.
1485 Gynemesh PS was Prolene Soft by another name. Prolene Soft was designed for use in abdominal and inguinal hernia repairs. Gynemesh PS was for use in a different site in the body and, in that respect, had a different clinical parameter. There was also an obvious difference in biological parameters since the human tissues and bodily fluids in the use environment were different.
1486 Furthermore, TVT and TVT-O used different deployment methods from predicate devices. Prolift and Prosima were mesh kits, unlike Gynemesh, with special techniques for deployment. Dr Allman pointed out that the need for similarities in deployment method, indicates that a new device, even one using an existing implant, which has a new method or tools for deployment, at a minimum calls for clinical investigation to determine the impact on clinical performance of the implantation process.1515
1487 Dr Allman said:
[It is] [i]rrational to compare your new device to the old device. And it, I think, can confuse people and they start over-interpreting against a background of not wanting to do clinical trials, because that’s a big factor in bringing a new device to the market. It introduces time and it costs money, and I’m not saying companies are morally bad, or anything, but just the way project teams developing new devices work, they tend to be looking very keenly towards routes that don’t involve having to do a one-year clinical trial or investigation, to use the European jargon. And what I’m saying here in my second report is that, actually, there’s a more overriding consideration which is that of logic, that what you are doing is assuming that new device and older device have the same clinical importance. So that when you design the new device to have different clinical performance or better safety, that, to me, precludes the idea of using equivalence of new to old.1516
1488 In his opinion, Ethicon should have conducted “proper clinical investigations” (involving clinical trials designed to test the performance and safety of the new device), that is to say, studies addressing the essential requirements of the European Directive, not studies established by doctors to answer the questions in which they are interested.1517 After referring to versions of the MEDDEV 2.7.1 from April 2003 and December 2009, Dr Allman explained:
The concept of equivalence for clinical evaluation described in European guidance rests on equivalence of clinical performance—which is logically obvious. The criteria of equivalence described in the guidance are the factors to be considered in establishing probable clinical equivalence.
It is clear that if a new device is a design modification to an existing device that is intended to change or improve clinical performance, then clinical investigation of the new device is necessary.
If a new device is a design modification to an existing device which is not intended to change or improve clinical performance, then it is necessary to establish that the design changes do not change clinical performance using non-clinical studies plus knowledge of the factors which impact clinical performance. Otherwise, and especially for implantable devices, clinical investigation is necessary. Such clinical investigation might reasonably be restricted to the particular aspects of safety and performance affected by design changes.
If a new device is not a design change to an existing device, and equivalence can be demonstrated to other devices with known and acceptable safety and performance, clinical investigation may not be necessary. Demonstration of equivalence will though be dependent upon knowledge of the design features which impact clinical performance—which is more likely for well established types of device. It is less likely for innovative devices (new types of device), and for implantable devices for which the medical device directive (Directive 93/42/EEC as amended by Directive 2007/47/EC) specifically requires clinical investigation unless this can be ‘duly justified’. European guidance specifically includes ‘use of similar deployment method’ as a criterion of equivalence. This indicates that a new device using an existing implant, with a new method (or tools) for deployment, would require clinical investigation to assess the impact of the implantation process on clinical performance (at a minimum).1518
1489 Finally, Dr Allman criticised the respondents’ early CERs for failing to identify the essential requirements and for failing to state that the device met those requirements. His evidence was that a CER should set out what data is needed to answer the question of whether the device met the essential requirements and whether that data can be obtained through a literature review or through the manufacturer’s own clinical investigations.1519
1490 Dr Allman also challenged the evidence given by Dr Hinoul about Ethicon’s conduct in preparing and relying upon CERs. He correctly observed that Dr Hinoul largely “restate[d] conclusions from Ethicon CERs (or other documents) without addressing the soundness (and potential ‘non-soundness’) of those conclusions”.1520 He expressed the opinion that Dr Hinoul failed to indicate an understanding of the obligations placed on manufacturers to generate clinical evidence for their devices prior to CE marking.1521
1491 In cross-examination, it was put to Dr Allman that there was nothing in the European Directive that required Ethicon to carry out clinical investigations before applying for CE marking for each of the devices — that the manufacturer could always choose to take “the literature route”. Dr Allman replied that the literature route was a difficult option in the case of innovative devices like the Ethicon devices, because (by definition) there would be no equivalent to an innovative device and taking the literature route requires a suitable equivalent device from which available data can be used. He emphasised that, although it is up to the manufacturer to decide which clinical data route to take, the manufacturer’s decision has to be rational.1522
1492 In addition, Dr Allman observed that the early CERs did not set out plans for post-market follow-up studies to obtain clinical evidence after the products had achieved CE marking. He pointed out that studies of this kind should be conducted, according to “authoritative European guidance”,1523 where, as here:
(1) “possible emerging risks have been identified and the evaluation of long term safety is essential”, such as in 2008 after the publication of an FDA Public Health Notice;1524 and
(2) it is unclear whether complications are always reported.
1493 Dr Allman acknowledged that Ethicon’s evaluation processes improved in 2010 and came closer to complying with the European regulatory requirements. But he remained of the opinion that they were still flawed, including because Ethicon continued to rely entirely on published data from studies designed for diverse purposes and which related to the type of procedure rather than the device itself, instead of studies that were designed to establish the safety and performance of each of the particular devices.
1494 Dr Allman analysed the clinical evaluation reports that were contained in the technical files for each of the devices and concluded that none of the devices was clinically evaluated to the standard necessary to satisfy European regulatory requirements and therefore to justify CE marking.
1495 In cross-examination, the respondents challenged many of Dr Allman’s conclusions, particularly his criticisms of Ethicon’s reliance in its CERs on clinical data from predicate devices on the basis that Ethicon’s notified body (BSI) could have reached the same conclusions as Ethicon on the same material had it seen fit to do so.1525 They suggested that these were matters “about which reasonable minds might differ”.1526 Dr Allman disagreed.1527 He pointed out that the process he had undertaken of going through the CERs in detail for each device was different from what one would expect of a notified body, both in terms of the time spent in the process and the process itself. Dr Allman explained that he was tasked with reviewing the files related to each device to determine whether Ethicon was justified in applying a CE mark to it. In contrast, as a notified body, BSI was looking at Ethicon’s processes in the context of certifying its quality systems and would only be looking at certain individual devices on a sampling basis.1528
1496 Ms Holland also reviewed Ethicon’s files, including its design history and technical files, as well as submissions it made to the FDA, and assessed Ethicon’s quality assurance and risk management processes against the applicable standards and guidance documents. She applied the same method of analysis as she applies in the course of her business.1529 She expressed the view that the standards that apply to quality management systems are “industry norms” and that implementation was not optional. She pointed out that her work with medical device companies involves the application and adherence to those standards.1530
1497 Ms Holland’s evidence encompassed a timeframe from the development of TVT, first marketed in the EU in 1997, to the discontinuation and last sale in Australia of several of the devices in 2012.1531
1498 In oral evidence in chief Ms Holland expanded upon some of the matters addressed in her reports and clarified some points.
1499 In her first report Ms Holland reviewed data concerning all nine devices, but selected five of the nine devices for comprehensive analysis. Those devices were: Gynemesh PS; Prolift; Prolift+M; TVT; and TVT Secur. 1532 Her rationale for excluding the other products was this:
Although data from all products were reviewed for this report, GYNECARE PROSIMA was not selected for comprehensive analysis due to the fact that it was developed using the GYNECARE GYNEMESH PS mesh, and was introduced into the market after the GYNECARE PROLIFT and GYNECARE PROLIFT + M. All TVT products were manufactured from nonabsorbable PROLENE undyed or blue mesh. The TVT-O utilised an “inside out” surgical technique, however, was not detailed herein as the TVT SECUR also utilised the transobturator approach. The TVT-ABBREVO and TVT EXACT were both later entries to the tape market established with a pattern similar to the TVT SECUR. The TVT EXACT used a retropubic approach similar to the TVT, and the TVT-ABBREVO is considered a mini-TVT-O with a placement loop.1533
1500 Ms Holland said that during the course of her review “a clear pattern emerged” regarding Ethicon’s quality management processes that was applicable both to the five representative devices and to the devices not selected for detailed examination.1534
1501 Ms Holland was highly critical of Ethicon’s approach to quality assurance and risk management. She found that Ethicon did not adhere at any time to EU or US industry requirements and regulatory standards in any of the following areas: design validation; evaluation of complaints; management responsibility or risk management. She said that during the development of both the SUI and the POP devices, no overarching, cohesive risk management system was in place.1535 She described Ethicon’s design validation process as flawed because it did not adequately represent the population of surgeons using the devices and feedback from surgeons was not fully evaluated or implemented.1536
1502 Ms Holland said that complaints were not fully evaluated, “trended”, acted upon, or properly fed back into the risk management process. She said that action was not taken to achieve planned results and maintain the effectiveness of the quality management system, nor was it evident that risks had been reduced as far as reasonably practicable or that risk control options had been considered. What is more, she concluded, Ethicon did not fully consider whether a medical device was suitable for its intended use and in its intended use environment.1537
1503 Although Ethicon developed a number of procedures for risk management, Ms Holland said that risk management was conducted in a piecemeal fashion by relying on past risk management assessments, rather than looking at each new system as a whole. Moreover, Ethicon’s procedures required that only the risks with the highest severity ratings (severe or critical) should be subject to risk reduction and systemic analysis. Such a process, Ms Holland explained, eliminates the need for a systemic analysis of risk using standard tools such as a Failure Mode Effects Analysis (FMEA), without which it is impossible to look at the performance of the system as a whole. She said that this was a violation of ISO 14971, the standard for the application of risk management to medical devices, published by the International Standards Organisation. “Failure mode” refers to the way(s) or mode(s) in which something (here a medical device) might fail under conditions of intended use and foreseeable misuse. Failures are any errors or defects, whether actual or potential, which affect the customer. “Effects analysis” is the analysis of the consequences of those failures.
1504 FMEA encompasses the identification of the potential causes of failure, an estimate of their severity and potential frequency, and the potential for the failures to be detected. She said that a manufacturer has a duty to mitigate every identified risk as far as is reasonably practicable and that that means reconsidering the design of the product so as to eliminate any potential risks “to the fullest extent feasible”. If risk mitigation is not possible through product design, Ms Holland stated that “a manufacturer must attempt to minimise the risk by incorporating protective measures”. Consistent with the order in which they appear in the European Directive and the MDR, Ms Holland said that warnings and training are the least effective means of minimising risks and should only be used as a last resort.1538
1505 Ms Holland identified ISO 14971 as the primary standard in the medical device industry defining how to perform risk management. ISO 14971 replaced its predecessor, EN 1441, in 2000.1539 The standard lays down a methodical approach to risk management involving the establishment of a risk management plan and procedure, risk analysis, evaluation, reporting, and reviewing.1540 Ms Holland said that ISO 14971 outlines the essential steps for performing risk management. 1541 Each step is required to be documented in a risk management file.1542
1506 Ms Holland reviewed Ethicon’s internal documents that formed the basis of its quality management system. These policies and procedures changed over time and Ms Holland explained that it was a challenge to determine the dates that certain governing documents were used, due to a lack of complete revision histories and multiple document management systems.1543
1507 Ms Holland acknowledged that Ethicon developed and implemented a quality management system intended to be compliant with the relevant industry and regulatory standards. But she concluded that “implementation of the system became inconsistent and in some instances ineffective”.1544 On the basis of her detailed review of the representative devices, she identified five areas where Ethicon’s systems “became incohesive and were not deployed effectively”:
• Individual examination of the intended use of each medical device and the associated impact;
• Implementation of design controls specifically with respect to design validation;
• Deployment of risk management activities such that actions were required to mitigate potential serious risks;
• Oversight by and commitment of management to effective risk management processes; and
• Analysis of and actions upon customer feedback including but not limited to complaint data. 1545
1508 It was put to Ms Holland in cross-examination that incomplete documents, miscalculations or errors may not necessarily indicate that a quality management system is non-compliant with industry standards. She was asked whether it might be acceptable under the standards for a manufacturer to fail to follow a certain methodology with precision but in substance address its mind to the essential questions posed by that methodology. Ms Holland rejected this proposition. She said that it was insufficient for a manufacturer to claim partial compliance or to suggest issues were adequately considered but not documented. When pressed on whether during the course of undertaking an audit she would raise minor errors, Ms Holland acknowledged that mistakes do occur but that the appropriate course was to record such errors. She said that sometimes errors can be isolated, but where a number of errors are identified it may suggest non-conformity. She explained that where consistent patterns occur or whole sections of a quality management system are not adhered to, it can indicate a major non-conformity with the standards.1546
1509 In 2005 Ethicon undertook a significant revision of its policies and procedures.1547 As a result, Ethicon adopted different approaches to risk analysis depending upon whether the product was a “legacy” device. A legacy device was defined as a device released under a risk management program preceding version/revision 7 of the procedure for device design risk management known as “PR602-003” (version 7 being the 2005 revision) or a medical device which has not undergone the “compliant risk management activities” required by ISO 14971.1548 Ms Holland explained that this meant that Ethicon was required to address known hazards, remediate them or change the labelling to address the severity, duration, and frequency of the harm. TVT, Gynemesh PS, and Prolift were deemed to be legacy devices.1549
1510 Ms Holland described the risk remediation for TVT as “fundamentally flawed” because Ethicon assumed that risk management was the same for various devices regardless of differences in design or surgical approach. Further, Ms Holland observed that the risk management reports for TVT and TVT-O included post-market surveillance data from mechanically-cut and laser-cut mesh without separating out the adverse event rates and risk profiles of the different products.1550
1511 In addition, Ms Holland reported that Ethicon did not address several of the top 11 known hazards identified in the 2002 or 2006 complaint analysis. She observed that discussion notes made by the team in charge of reviewing complaints for the SUI devices recorded “no associated harm” against a number of complaints including broken, frayed or kinked mesh, post-operative pain attributed to nerve damage, and vaginal erosion/extrusion.1551 Where Ethicon determined the residual risk to be “moderate”, it considered a risk benefit analysis was not required. Only those risks it deemed “serious” were subjected to such an analysis.
1512 Ms Holland found that similar problems arose with respect to the Gynemesh PS risk management report. The overall residual risk (ORR) was determined to be “high” but no design Failure Mode and Effect Analysis (dFMEA) was conducted because the ORR was considered acceptable because Ethicon considered that the benefits outweighed the risk.1552
1513 Ms Holland explained that design controls for medical devices are not merely a regulatory requirement but a documented method of ensuring that what the manufacturer thinks it is developing is what it wanted to develop in the first place and that what comes off the production line is what the customer needs and desires.1553 She said that some of the first and essential questions that have to be answered during the planning phase are:
• Who will use the device? Is special training or special skills required?
• What is the intended use?
• What are the risks associated with its use or misuse?
• What kind of environment will it be used in?
• What materials can be used?
1514 As for the “non-legacy devices”, Ms Holland said that it did not appear that Ethicon’s risk management activities with respect to TVT Secur complied with industry standards or, indeed, Ethicon’s own standards, and it was not evident that Ethicon adequately assessed all known harms associated with the device. 1554 Ms Holland considered that the FMEA conducted for Prolift+M were more thorough than those conducted for Prolift. Nevertheless, none of the 49 potential failures was thought to require any action or mitigation to reduce the risk to as low as practicable. Twenty-three of them were defined as not associated with a hazard, although severity ratings had been assigned. Only the most severe categories of risk (9 or 10) were included in the risk management reports.
1515 Furthermore, Ms Holland considered that Ethicon had not responded effectively to most complaints. She said that “in essence, only complaints where related products are returned to Ethicon, confirmed not to meet specification, and recur (such as packaging and labelling issues) appear to be resolved in a reasonable timeframe”. In a case where the product was not returned for inspection, Ethicon’s approach was that the complaint could not be confirmed, and the problem remained unresolved, often for years. While several attempts might have been made to have the device returned, “[t]he real concern”, in Ms Holland’s opinion, was that Ethicon did not have “an active retrieval system or team to go to the field and investigate the troublesome report”.1555
1516 Even when a product was returned and the complaint was “confirmed”, no corrective and preventive action was taken unless there was also “an increasing volume of ‘noise’ from the field”.1556 After reviewing data and reports of over 5,000 complaints, Ms Holland determined that the confirmation rate was less than 1% of the total. She did not see any evidence that Ethicon had made proactive efforts to confirm a complaint if the product was not returned.
1517 Where a batch number was provided, the complaint history for only the specific batch of products was reviewed. This was not a method Ms Holland had previously encountered and she was critical of it. She said that this approach systematically limits the investigation to the one batch of product which might not be representative of the entire population. Yet the purpose of trending is to look at changes over time in the performance of a system by item (such as complaint category or complaint responsiveness).1557 She was also critical of Ethicon’s trending process because data points exceeding upper limits appeared to have been excluded from the analysis without associated action.1558 In other words, spikes in the trend were discounted or considered anomalies although a full investigation had not been carried out. Yet, she said, “any assignable/special cause of variation needs to be fully investigated and acted upon before the ‘out of control’ point(s) can be removed from consideration”.1559
1518 Only complaints relating to the most critical level of harm (9 or 10) were routinely analysed and fed back into the risk management process. Consequently, many complaints concerning harm of low or moderate severity but of frequent occurrence were not analysed in detail.1560
1519 Moreover, complaints categorised as “medical-related” were not addressed at all. Those complaints included lower extremity pain, urinary retention, vaginal extrusion, perforation, erosion, bleeding, infection, vascular injury, and post-operative complications 1561 Complaints about material fraying were not adequately evaluated, investigated or reported, 1562 although device fraying during implantation is an indication that the device could also fray in vivo and would not therefore meet device performance specifications.1563 Nor were complaints about erosion and pain.1564 And no action was taken on complaints considered liable to result in “significant harm” if they were deemed not to have had an effect on safety and efficacy.1565
1520 Ms Holland concluded that Ethicon’s complaint system was self-limiting with the result that complaints were under-reported to the regulatory authorities.
1521 A stark example of a complaint that should have been, but was not, reported is a complaint of erosion relating to TVT discussed by Ms Holland in her second report. The complaint came from a surgeon concerning a patient who was implanted with TVT in 2001. Two years after the initial TVT procedure, a second procedure was required due to the patient experiencing a recurrence of incontinence. During a pre-operative consultation for the second procedure, the surgeon visualised and felt tape in the vagina, finding that the tape from the initial operation had eroded into the vagina. In the second operation he removed part of the tape. Ethicon did not report this complaint on the basis that “no serious injury occurred”. Ms Holland said this event should have been reported as a malfunction. She explained that a serious injury does not have to occur in order for an event to be reportable. In any case, she noted that the adverse event occurred in 2003, by which time Ethicon had received complaints of erosion that were reported as serious injuries because intervention was taken. As a result, Ms Holland considered that by 2003 all erosion events were required to be reported under FDA reporting requirements.1566
1522 Ms Holland said that the problem was exacerbated by large backlogs in the complaint and “CAPA closures”. CAPA is an acronym for “corrective and preventative action”. The purpose of a CAPA is to identify the root cause of an actual or potential failure to conform to the requirements of the European Directive and identify action that could be taken to address the issue.1567 In 2011, for example, 22% of CAPAs were open for more than a year and the complaint backlog had grown to 2,500 files.1568
1523 Ms Holland also noted that Ethicon’s system allowed the complaint analyst to determine when to consult with management and/or the company’s medical director on clinical and medical issues and regulatory reporting decisions. In her experience, however, decisions of this kind are not made by the complaint analyst but by a complaint review board that includes people from quality assessment, regulatory affairs, clinical, reliability and quality systems.1569
1524 Ms Holland was firmly of the opinion that pre-clinical testing should have been performed on both the SUI and the POP devices before they were offered for sale. She said that, “regardless of the regulatory requirements”, relying on the history of similar devices in order to get a device on the market was “necessary but not sufficient”. She said that it should not have been assumed that mesh would respond the same way in the pelvic floor as it does in the abdominal wall. In fact, as we have seen, there is evidence to indicate that it does not. Yet Ms Holland said that Ethicon did not consider in its risk assessments the impact polypropylene mesh has in different anatomical locations. In particular, it did not consider whether there were different biomechanical properties and different microbiological flora with higher risk of infection and inflammatory response.1570 She said that any assessor would realise that, for a new intended use in a totally new environment of the body, additional testing should have been performed.1571
1525 During cross-examination Ms Holland was asked whether she was able to identify any standard that sets out the circumstances in which reliance on the prior history of similar devices was necessary but not sufficient. Ms Holland pointed to the obligation in ISO 14971 for manufacturers to appropriately consider the intended use of a device.
1526 ISO 14971, for example, stated that the manufacturer should make judgments relating to the safety of a medical device in order to determine its “probable suitability” for placement on the market “for its intended use/intended purpose”.1572
1527 Ms Holland testified that it was necessary to consider what a reasonable manufacturer would do,1573 no doubt because she had been asked by the applicants’ lawyers to do so. The applicants relied on her evidence in support of their claims in negligence in which they had to prove that the respondents’ conduct did not conform to the standard of care reasonably expected of a manufacturer or supplier in the respondents’ position. Ms Holland explained that the fact that a device could pass through regulatory processes was not necessarily evidence of what a reasonable manufacturer would do to make sure its product is safe and no-one was harmed. Ms Holland observed that “good ethical sound scientific judgment” is required, regardless of whether “you can get through the regulatory process”.1574
1528 Ms Holland acknowledged that Ethicon’s risk management procedures improved over time. Still, she concluded that “the execution of their risk management processes was vastly ineffective and did not meet the intent of ISO 14971 or industry practice”.1575 Ms Holland identified the following aspects of Ethicon’s conduct as “the most critical examples”:
• Risk analyses were not systematically performed. Risk analyses from previous product generations were heavily relied upon, not taking into account the differences in intended uses, product design, or surgical procedures;
• Hazards and harms were not properly identified or consistently scored. Ethicon’s idiosyncratic FMEA methodology allowed assigning severities for failure modes without defining a hazard or harm. Furthermore the severity scoring was inconsistent across FMEAs. A thorough approach to hazard analysis was not used, as hazards such as mesh tearing were evaluated as having no harms; and
• Risk reduction was limited to only the most severe risks. No evidence was found that mitigation methods were evaluated and used for risks classified as ALARP [meaning, “as low as reasonably practicable”].
1529 She concluded, based on her experience, that “Ethicon did not appropriately execute their risk management processes to enable identification and mitigation of risks, and therefore prevent or reduce harm to patients”.1576
1530 Ms Holland testified that the FMEA was undertaken “quite traditionally”, except in three important respects.1577
1531 One was that in all their analyses of all the Ethicon devices she saw no evidence that action had ever been taken. She described this as “extremely abnormal”. Indeed, she was amazed by it. She appears to have attributed Ethicon’s inaction to the fact that Ethicon set the Risk Priority Number (RPN) so high (at 294), which would require a severity of 7 or above, an occurrence of 7 or above, and a detection of 6, before action was required. The RPN is derived from multiplying the severity, occurrence and detection ratings for each line item and is used as the basis for determining the risk category.1578
1532 Another was that Ethicon determined that there was no harm associated with a hazard if it only affected less than 5% of women in whom the devices were implanted. She said that she had never seen that approach adopted in 32 years of use. She considered that this was another way in which Ethicon underestimated the frequency of occurrence of harm.1579
1533 The third was that, because the threshold was so high, Ethicon did not ever implement control mechanisms for the hazards or harms.1580
1534 Dr Pence’s evidence concerned Ethicon’s compliance with its responsibilities as a manufacturer of medical devices in relation to the development and provision of instructions for use (IFU).
1535 Dr Pence provided one report dated 18 May 2017. In the course of preparing her report she examined internal Ethicon documents, conducted her own searches and reviewed applicable industry standards.1581
1536 Her oral evidence in chief canvassed evidence already addressed in that report. She easily withstood cross-examination and it ultimately had little effect on the evidence she gave, save to clarify some points. I found her a particularly impressive witness.
1537 Dr Pence addressed five major issues:
(1) the information that ought to be included by a reasonable manufacturer in an IFU accompanying a medical device;
(2) the steps a reasonable manufacturer would take in order to obtain the information necessary to include in an IFU;
(3) the sources of information to which a reasonable manufacturer of a medical device in Ethicon’s position should have had regard in deciding on the content of the IFU for each device;
(4) the information that ought to have been included in the IFUs that accompanied the devices having regard to information that was known or knowable; and
(5) whether adverse reactions that are commonly known by clinicians are required to be included in IFUs.
1538 Dr Pence’s assessment was based on the globally-recognised industry standard for the development and provision of IFUs for medical devices set by the GHTF.1582
1539 She explained that the purpose of an IFU is to provide the physician with the information necessary to use the device safely, to ensure that the device performs as intended, and to assist the physician to make an informed decision about using the device in a particular patient, taking account of the physician’s training and knowledge.1583 She described it as the primary tool for communication between the manufacturer and the clinician about the particular device.1584 She stated that it should be consistent with available clinical data and that all the hazards and other clinically relevant information should be appropriately identified.1585
1540 In assessing Ethicon’s compliance with regulatory requirements for the content of IFUs, Dr Pence concluded that certain information required to be included in the IFUs of all devices was not provided. Following a detailed assessment of the information that was known and knowable to Ethicon when it was preparing the IFUs for each device, but without the benefit of Dr Hinoul’s concessions, Dr Pence concluded that there were significant deficiencies with respect to the information provided about potential complications, adverse reactions, and warnings. She found that a significant and extensive amount of information regarding the safety of the devices was omitted from the various IFUs. The implication of her evidence was that Ethicon’s failure to include the required information in the IFUs was contrary to the essential principles/requirements.
1541 Dr Pence examined early clinical evaluations of TVT and its prototype, internal Ethicon documents, and documents from the FDA’s Manufacturer and User Facility Device Experience (MAUDE) database to assess whether the IFUs for the SUI devices conformed to regulatory standards. She concluded in effect that they did not, noting numerous omissions. MAUDE, I interpolate, is a database which houses medical device reports submitted to the FDA.
1542 Dr Pence undertook a similar analysis of documents concerning the POP devices and came to a similar conclusion. She examined early clinical evaluations of Prolift and Prosima, internal Ethicon documents, reviews by authoritative bodies such as the the French National Authority for Health, and documents from the MAUDE database. Again, she concluded that information was missing from the IFUs in relation to potential complications, adverse reactions and warnings.
1543 I discuss the detail of this evidence below in Part XI.
1544 During cross-examination Dr Pence rejected the proposition that the standards governing the content of IFUs allow scope for a manufacturer to exercise judgment regarding whether to disclose particular risks. She explained there was some scope for the manufacturer to exercise judgment concerning the amount and type of information required to be disclosed in relation to a risk, but not whether to disclose the risk itself. 1586 In re-examination she reiterated that all residual risks, including any foreseeable and expected risks, are required to be disclosed in IFUs as adverse reactions, warnings, precautions or contraindications. 1587
1545 The respondents’ position was that there was no requirement under the standards to include in the IFU adverse reactions that are commonly understood by clinicians. Dr Pence was firmly of the view that this position was incorrect. To the contrary, in order to comply with the requirements for the content of IFUs, Dr Pence stated that the manufacturer should convey all safety information necessary for safe and effective use of the device and for making patient management decisions in a complete, accurate, balanced, and objective manner.1588 She was adamant that all the hazards and other clinically relevant information should be identified appropriately, regardless of the assumed knowledge of the user.1589
1546 Indeed, Dr Pence said that a reasonable manufacturer cannot assume that all clinicians who use a medical device are familiar with the potential adverse reactions. She stated that the manufacturer is responsible for making disclosures in the IFUs because the manufacturer has access to information that either is not available to, or is not routinely accessible by, clinicians.1590 In cross-examination, Dr Pence noted that there was a huge disparity between the resources available to clinicians and manufacturers. She considered, in effect, that it was unreasonable to expect clinicians to have the same knowledge. She pointed out that, unlike clinicians, manufacturers have entire departments dedicated to evaluating relevant literature, running clinical studies, gathering information from the MAUDE database and assessing that information.1591
1547 It was put to Dr Pence during cross-examination that the identity of the user is important in considering what is to be included in an IFU.1592 Dr Pence agreed this could be an important factor, but she maintained that the identity of the user did not relieve a manufacturer of its responsibility to comply with applicable standards. She stated that the requirement to include all relevant information in an IFU took into account the fact that users can have differing levels of skill and training.1593
PART VIII: DEVELOPMENT AND PRE-MARKET EVALUATION OF THE DEVICES
1548 The respondents’ submissions on this issue devoted a considerable amount of space to the studies of Prolene sutures and hernia mesh, beginning in the 1960s. The apparent rationale for this approach was that the material used to manufacture the Ethicon devices was the same material that had been used first in the sutures and then in the hernia mesh.1594
1549 The utility of this approach is questionable. As I observed in Part V, there are very significant differences between the sutures and the devices and between the environment of, and the mechanical forces at work in, the female pelvis and the abdomen.
1550 In any case, the applicants did not take issue with the respondents’ pre-market evaluation of either Prolene sutures or hernia mesh.
1551 For these reasons it is unnecessary to deal with the respondents’ lengthy submissions on the studies of Prolene sutures and hernia mesh. The point was neatly put by Ms Holland in her second report when referring to the deficiencies of Ethicon’s risk assessments:
[T]here is no disagreement that any clinical experience with PROLENE sutures and mesh [should] be considered when evaluating future devices of the same material. However, the evaluation should not have solely relied on prior history of similar devices for safety and effectiveness due to the new indication and use in pelvic floor repair versus the abdominal wall (hernia). What was not considered in these risk assessments was the impact of polypropylene mesh on anatomical location with respect to differing biomechanical properties (erosion/adhesion risk), microbiological flora (infection risk), as well as inflammatory response.1595
1552 Professor Klinge, who, having regard to the close relationship he and Professor Klosterhalfen enjoyed with Ethicon at the critical times, gave evidence that the Ethicon companies did not conduct their own studies on mesh for specific use in the pelvic floor, and failed to define the physiological forces at work in the pelvis and incorporate this research into the development of the Ethicon devices.1596 No objection was taken to the evidence and it was both unchallenged and uncontradicted.
1553 Otherwise, the respondents relied on Ethicon’s clinical evaluation reports and the fact that the devices had been cleared for sale. As I have already observed, they did not advert to, let alone engage with, the evidence of the applicants’ experts about the shortcomings of those evaluations. In his affidavit, Dr Hinoul purported to summarise the CERs, at times offering an endorsement of the approach and/or their conclusions.
The SUI devices
TVT
1554 As I explained in Part III above, TVT emerged from the work of Professor Ulmsten and his team.
1555 Dr Axel Arnaud, the then Medical Director for Ethicon France, learned of Professor Ulmsten’s technique in September 1995. He visited the professor in Sweden on 20 November 1995 and spent the day with him. Professor Ulmsten explained the rationale for the technique and arranged for Dr Arnaud to attend four procedures. Dr Arnaud was impressed by what he saw and by Professor Ulmsten. In a document he wrote some five years later, entitled “The history of TVT”, he recorded his preliminary conclusions:
1. The rationale for this technique was very good. It was not just based on a brilliant idea, but really the result of many years of serious scientific research.
2. Contrary to what the video tape was showing, this procedure was a true mini- invasive procedure, and in particular, it could be performed under local anesthesia in about half an hour. Furthermore, it looked quite easy to perform and was very patient friendly.
3. Its efficiency to cure USI [urinary stress incontinence], at least for the short term could hardly be challenged by anyone. This was simply demonstrated during the procedure by asking the patient to cough before and after the mesh placement.
4. The long term efficiency was not substantiated by clinical results but the fact that a mesh was used backed a reasonable anticipation of excellent long term results.1597
1556 He added that “the key questions and concerns were about the safety of [the] procedure”, noting that “it was broadly admitted that the use of any mesh through the vaginal route was associated with a high rate of complications such as rejection/infection and urethral erosion”.
1557 Dr Arnaud arranged for several Ethicon colleagues and physicians to visit Professor Ulmsten. All were said to be very sceptical about the procedure before going to Uppsala. Afterwards, however, according to Dr Arnaud, “the general opinion was that the technique was excellent and in any case worth further efforts although none could explain how it worked”. Dr Arnaud said that he became convinced of the potential for the procedure to have “a great future”. He reported the results of the physicians’ visit to the head of Ethicon European Marketing, Jacques Dumont, who “usually trusted” him and Mr Dumont was convinced with “very little of effort (sic)”. Mr Dumont asked Dr Arnaud to bring the Ulmsten procedure to the attention of the appropriate people in Somerville, New Jersey, where Ethicon Inc. was based. After a two to three hour meeting with members of Ethicon’s Research and Development team, during which Dr Arnaud advocated in favour of Ethicon capitalising on Professor Ulmsten’s work, Mr Dumont “received the green light for a deal” with Medscand Medical AB (Medscand), a small Swedish company, which had applied for a patent for the Ulmsten “slingplasty”.
1558 From August 1996 until February 1997 Dr Arnaud worked on securing a deal with Medscand. The deal was finalised on 13 February 1997. That day the respondents’ parent company, Johnson & Johnson International (JJI), signed a license and supply agreement with Medscand under which Medscand granted JJI an exclusive worldwide licence to Professor Ulmsten’s intra-vaginal slingplasty device for treating stress urinary incontinence, including any improvements, and all patents and “know-how” (cl 3.1). The device incorporated Ethicon’s Prolene mesh, covered by a removable plastic sheet, swaged on two metallic needles and a handle, which can be attached to the needles. Medscand was required to supply the devices to JJI in a “finished” form (cl 4.1(c)). Medscand, with JJI’s assistance, was required to conduct the studies necessary to obtain regulatory approval in Europe (cl 3.4).1598
1559 On 15 November 1999 Ethicon entered into an asset purchase agreement with Medscand by which it acquired all rights to the slingplasty business, including the right to manufacture, distribute and sell the products of the business, and ownership of all fixtures and equipment, books and records, and all proprietary rights.1599
1560 After the license and supply agreement had been signed in February 1997, a small European team was assembled to prepare for the market launch. It was this team that came up with the name “TVT”. Apart from coming up with the TVT name, in a matter of months the following tasks were undertaken: an artist was engaged to create the illustrations explaining the concept and the steps of the procedure; a Finnish company was contacted to shoot a professional videotape illustrating the procedure; the clinical strategy was designed, including a randomised controlled trial comparing TVT to open Burch colposuspension; an experts’ meeting was organised for 27 June 1997 in Dublin involving 11 experts from all over Europe; and a symposium was organised for the IUGA meeting in Amsterdam on 31 July the same year.
1561 A number of in vitro cytotoxicity studies were also conducted to assess the cytotoxic potential of the components of the Ulmsten device.1600 Thomas Barbolt, a research fellow in the employ of Ethicon reported in August 1997 that, after an evaluation of all the test results, the needle, PE needle guard, heat-shrink tubing, and PE sheath were considered to be non-cytotoxic as was the “raw material PP mesh”. On the face of things, the latter conclusion is troubling in the face of the statement which preceded it:
Overall, there is some evidence to suggest that the PP mesh from the sterile Ulmsten device may have cytotoxic potential.
1562 The applicants, however, made nothing of it and Dr Barbolt appears to have taken the view that, given the number of procedures in which the device had been implanted (around 1000) including over 200 documented cases, the in vitro observations did not translate into any adverse patient outcomes of clinical significance. He inferred from the available data that the Ulmsten device had “fewer complications in terms of tissue reaction than other comparable devices”. It does not seem, however, that he took into account the potential for adverse tissue reactions in the long-term arising from the use of Prolene mesh or the precautions taken by the surgeons in the cases he had in mind.
1563 A literature review was also undertaken on the biocompatibility of Prolene sutures and Prolene mesh implants. This uncritical review, completed on 1 October 1997, considered scientific articles published from 1962 to 1997 but did not outline the protocol or the research method, and did not state whether, and, if so, which or why some research findings or articles were not taken into account. The author, Dr Brigitte Hellhammer, then Manager of Scientific Information and Documentation at Ethicon, summarised the results as follows:
[E]xperimental and clinical studies demonstrate the relative inertness and biocompatibility of Prolene sutures and implants. Throughout the studies reviewed, tissue reaction to these materials is described as acute inflammatory gradually subsiding to a mild degree, with minimal foreign body reaction over extended periods of implantation. Within few weeks, Prolene mesh is fully incorporated into connective tissue. It demonstrates a low incidence of seroma formation and a good tolerability in the case of infection. There is no clinical evidence that Prolene mesh would impair wound healing or elicit any unfavourable effects on cells and tissues. These observations have been made in a variety of tissues.1601
1564 The pre-market phase ended with the presentation of the technique and the product by Laura Angelini to the European Marketing Meeting in Vienna later in 1997.1602
1565 The following year the Handbook of Biomaterials Properties was published. In chapter 1, Professor Williams wrote that there is “no such thing as an inert biomaterial”.1603 The same year an article by Klinge et al was published on the results of studies on the effect of implantation in the abdominal wall of a number of dogs of Marlex, a monofilament polypropylene mesh, and Prolene Soft hernia mesh, made by Ethicon. The study was conducted over three and six months. The authors reported that the mesh shrank considerably over time as “a consequence of the physiological wound contraction, initially by dehydration of soft tissue and later by maturation and cross linking of the collagen fibres” but that a reduction in the amount of polypropylene reduced the extent of the inflammatory response and the corresponding fibrosis.1604
1566 In 1999, in their chapter in the textbook Incisional Hernia, Professors Klinge and Klosterhalfen confirmed that surgical meshes are not inert, even years after implantation, and lead to chronic irritation of the recipient tissues, and recommended that the use of Prolene meshes be avoided.1605
1567 By October 1999, when TVT was first supplied in Australia, no randomised controlled study had been undertaken, but some early results were available from Nordic studies, which are discussed below, as well as a Taiwanese study. There were five Nordic studies in all but four of them appear to relate to the same cohort of patients.
1568 The first of the Nordic studies involved 75 women with genuine stress incontinence who were followed up for two years after surgery at Uppsala University, employing the Ulmsten method and using a sling made from Prolene mesh. Results were published in 1996 in the International Urogynecology Journal. 1606
1569 Ulmsten et al (1996) reported no intra- or postoperative complications. Sixty-three patients (84%) were said to have been completely cured during the two-year follow-up period. Six patients (8%) were said to have significantly improved in that they did not lose urine apart from an occasional leakage during severe cold, for example. In the remaining six patients in the cohort of 75 (8%) no improvement was seen. These failures were obvious at the first postoperative check-up after two months. All but five patients were able to void properly directly after surgery. All 75 were released from hospital the same day as the surgery or the following day without catheterization. Mean sick leave was 10 days and mean operation time 22 minutes. No defect healing or rejection of the sling reportedly occurred.
1570 A comparison of these results with the earlier studies showed a higher cure rate with the modified technique, which, according to Ulmsten et al (1996), suggested an improvement in the surgical technique. The number of patients who were reported as cured was higher and the mean operation time was less than that in the previous study, which the authors also considered indicative of an improvement in surgical technique. The authors were particularly pleased with the absence of defect healing and sling rejections which they attributed on the probabilities to the properties of Prolene “possibly being better accepted by the tissues in which it was implanted than Mersilene or Gore-tex”. They also described “the strong adhesive forces created around the present sling” which, in contrast to those used previously, was said to have prevented sliding.
1571 Ulmsten et al (1996) were very encouraged by the results. But they added a note of caution:
Even if the results so far are in accordance with those reported here, and by the same token the preliminary results from an ongoing Scandinavian multicenter study encompassing 500 patients seem to confirm these, we must bear in mind that long-term results are necessary before the ultimate place of a new surgical method can be established. Unfortunately, few surgical methods for the cure of stress incontinence have been exposed to prospective long-term follow-up studies. Until such an evaluation has been done the IVS plasty can only be characterized as a promising new technique that should be further evaluated in larger series of prospective studies over a longer period. As indicated above, such studies are in progress.1607
(Emphasis added)
1572 Ulmsten et al (1996) were also acutely aware that the results may have been influenced by the fact that the operations were carried out by experienced gynaecologists who had been involved in the development of the procedure.1608
1573 Yet Ethicon did not wait for the long-term results or for more widespread use before releasing TVT to the market. Dr Hinoul conceded as much in cross-examination .1609
1574 TVT was released in Europe in 1997,1610 before the one year results of the Nordic multi-centre study had been published, and well before any long term results were available. At that point in time Ulmsten et al (1996) was the only published article on the use of transvaginal tape using Prolene mesh.1611
1575 It was cleared for sale in the United States in January 1998.
1576 In its § 510(k) notification, Ethicon contended that TVT was substantially equivalent to the ProteGen sling. Dr Hinoul deposed that:
Boston Scientific’s ProteGen Sling (Vesica Sling Kit) was already cleared for use. The ProteGen Sling was already cleared for use, and after a thorough analysis of its similarities and differences with the TVT Device, also served as evidence that TVT PROLENE® Mesh would be safe...1612
1577 If Dr Hinoul intended to represent that Ethicon carried out a thorough analysis of the similarities and differences between the two products, I cannot agree. The technical file, which he referenced, does contain a table which compared the two products. What it does not contain, however, is any assessment and demonstration of the significance these might have on safety and performance. If ProteGen were indeed substantially equivalent to TVT, then one might also reasonably have expected that the literature review in the clinical evaluation report would refer to, and critically analyse, the literature on ProteGen. Yet it did not.
1578 What is more, Dr Hinoul went on to say that ProteGen was recalled in 1999 after many patients experienced complications, though he did not say what these complications were. I was not taken to any evidence to indicate that the experience with ProteGen triggered a review by Ethicon of TVT, as one might have expected of a reasonably prudent manufacturer. Indeed, a note on the TVT technical file dated 26 July 2000 from the quality system and compliance supervisor, Agnes Sifferlen, stated that the original risk analysis written by Medscand was still valid.1613
1579 Despite relying on ProteGen for the purpose of regulatory clearance in the United States, in his affidavit Dr Hinoul was quick to point to some of the differences between the devices.1614
1580 Dr Allman’s opinion, which was not challenged, was that the two devices did not meet the European definition of equivalence.1615 He pointed out that the devices were very different. First, they were made from different materials. ProteGen was made from woven polyester, not knitted polypropylene, and incorporated an absorbable component (bovine collagen). Second, the devices used different implantation methods and implantation tools. Third, the two devices used different fixation methods. TVT relied on fixation to tissue by friction followed by tissue ingrowth and ProteGen by suture anchored to bone. As Dr Allman observed, it is logically difficult to sustain an argument that the safety and performance of ProteGen could be used to define the safety and performance of TVT.
1581 The consideration for the agreement with Medscand was the payment by JJI to Medscand of “milestone” payments. The first payment (of US$400,000) was due on 28 February 1998, unless “the Clinical Trials as specified in Exhibit C have not been completed” by then, in which case the payment was not due until the trials were completed. Exhibit C was not in evidence.1616 The applicants submitted, however, without contradiction, that the reference to “Clinical Trials” was in all likelihood a reference to the clinical trials mentioned by Medscand’s medical director, Dr Margareta Eriksson, in her clinical report of 17 October 1997.1617 According to the description in Dr Eriksson’s report, this was one trial conducted in six medical centres in Scandinavia. The trial was “designed as an open, non-randomized, prospective, multi-center study”. The “primary end points” were “effectiveness of the procedure, operative and post-operative complications, as well as patient feedback”. The patients were monitored during the procedure and followed up at two, six, and 12 months after surgery. It appears from Dr Eriksson’s description of the materials and methods that this is the Nordic study to which Ulmsten et al referred. It involved 131 patients at six Scandinavian centres.
1582 In 1998 one year results from this study were published in the International Urogynecology Journal.1618 The stated object of the study was to determine “how easy, effective and, above all, safe the procedure could be in ‘ordinary’ gynecologic units”. Ulmsten et al (1998) reported that 119 (91%) of the patients were cured and another 9 (7%) were significantly improved. There were 3 (2%) failures within three days of the procedure. One bladder perforation occurred and one wound infection, the latter in a patient with vaginal wall atrophy. One patient developed a retropubic haematoma the size of a hen’s egg which was reported to have spontaneously vanished after an ultrasound. No tape rejections were reported.
1583 Once again, the results were encouraging. The authors were particularly encouraged by the low complication rate in “less experienced hands”. They concluded that TVT could be considered “a safe and effective procedure for the surgical treatment of genuine female stress incontinence”, that it could be carried out in a standardised way under local anaesthetic on a day-care basis, and that the cure rate seemed to be as good as that reported with traditional surgical procedures. But they again sounded a note of caution that “long term follow up studies have to be presented before any definitive conclusions can be made”.
1584 When the report was published in the International Urogynecology Journal, it attracted editorial comment. The authors were complimented for going into community hospitals to see if the results of the earlier study could be reproduced by the average practising gynaecologist and for using a validated quality of life assessment. But the editor(s) were apparently concerned that the results would give rise to complacency in the medical community, pointing out that:
[M]any questions remain regarding the diagnostic criteria they used in selecting their patients, as well as the degree of testing performed postoperatively. Further study is needed to confirm the results found here with a very new and interesting technique in the surgical treatment of stress incontinence.
1585 In his affidavit Dr Hinoul did not refer to a pre-launch clinical evaluation report on TVT and in their submissions the respondents do not identify one. They begin their submissions with a report issued in 2000, after TVT had been on the market in Europe for three years.
1586 The only report in evidence on TVT included in the TVT technical file that predates CE marking is the one signed by Medscand’s Dr Eriksson in October 1997.1619 This report is entitled “Scandinavian Multicenter Study of the Tension Free Vaginal Tape Procedure”. It described the clinical investigations that had been undertaken to date and simply adopted the conclusions of the first report of the Nordic study. No other report appears to have been prepared either by Medscand or Ethicon before TVT was cleared for sale. Having reviewed its technical file for TVT, Dr Allman noted that the clinical data used to support CE marking of the TVT device seemed to consist of short summaries of published data from a few centres and was heavily biased to data from the centre in which the technique was first introduced and TVT was developed. He also commented that it was not clear whether all the data were generated with the same version of the TVT device. He concluded, without contradiction, that Ethicon had undertaken no clinical investigations of its own.1620
1587 The first clinical evaluation report to which Dr Hinoul referred in his affidavit was on Prolene mesh, written by Dr James Browning on 16 March 1998. 1621 It was prepared for a different purpose and is discussed below. It would not have satisfied the requirements of the European Directive, not least because TVT is a different device.
1588 Dr Hinoul then referred to a risk/benefit analysis by Dr Isenberg dated 15 June 2000, but that was after the device had received CE marking and after it had been released for sale in the United States and Australia.1622
1589 The European Directive, which prescribed the steps to be followed by a manufacturer in order to affix CE marking,1623 relevantly defined “manufacturer” in Article 1(f) in the following way:
“[M]anufacturer” means the natural or legal person with responsibility for the design, manufacture, packaging and labelling of a device before it is placed on the market under his own name, regardless of whether these operations are carried out by that person himself or on his behalf by a third party.
The obligations of this Directive to be met by manufacturers also apply to the natural or legal person who assembles, packages, processes, fully refurbishes and/or labels one or more ready-made products and/or assigns to them their intended purpose as a device with a view to their being placed on the market under his own name…
1590 Dr Allman’s unchallenged evidence was that the effect of this definition is that the manufacturer of a device might not be the person who designed or made it. The person who supplies the device under his own name or mark is the manufacturer who carries all the obligations of the manufacturer set out in the Directive. He said that such an entity is often referred to as the “legal manufacturer” to distinguish the entity with regulatory responsibilities from the maker of the device. He added that this is clear from the terms of the Directive itself but is also emphasised by the European Commission in other documents.1624
1591 It follows that, although at the time the CE mark was applied to TVT, JJI had not yet acquired the assets of Medscand (merely operating under its exclusive licence to sell the Medscand product under the Ethicon trademark), Ethicon was the manufacturer of TVT for the purposes of the Directive and carried the obligations imposed on the manufacturer by the Directive, including the obligation to ensure that the essential requirements were met.
1592 Importantly, the Nordic multicentre study upon which Dr Eriksson relied involved a small, carefully selected group of patients. Those with significant vaginal prolapse, who had undergone previous surgery for stress urinary incontinence or prolapse, and those with voiding difficulties were excluded. Sixty percent of the post-menopausal women were on hormone replacement (oestrogen) therapy. Dr Hinoul testified that oestrogen would probably have a beneficial effect on wound healing and so reduce the extent of complications, particularly erosions and infection.1625 Furthermore, all patients were seen by experienced urogynaecologists. As Ms Holland observed, the results would not necessarily be reproducible in the wider population.1626
1593 The results of later Nordic studies were equally encouraging. Once again, however, post-menopausal patients were taking systemic or local oestrogen therapy. The procedure was invariably carried out under local anaesthetic so as to ensure that the tape was adjusted to suit “the patient’s individual tissue requirements”.1627
1594 No studies were undertaken before TVT was cleared for sale to assess its safety or efficacy across a broader population.
1595 None of these matters was disclosed in the instructions for use issued with TVT. Neither did the IFU stipulate that the procedure should be carried out under local anaesthetic or point out the risks to the patient if it were not. Nor did they mention the exclusion of women with previous incontinence or prolapse surgery. They did not recommend the use of oestrogen therapy for postmenopausal patients or point out that such women who had participated in the study had been given oestrogen therapy. And they did not require training in TVT procedures or recommend that the surgery only be carried out by experienced urogynaecologists.
1596 Moreover, at this stage there was clearly a question mark over the long-term outcomes (as to both the safety and efficacy) of TVT. Yet no indication was given in the instructions for use that long-term data were not yet available and that a randomised controlled trial was under way but not complete.
1597 The extent of Ethicon’s own evaluation of the device was very limited, as Dr Arnaud indicated in his history of TVT written in July 2000:
3. Evaluation (January-April 1996)
The next step I considered was about confirming my personal positive impression. I planned to ask some key experts to further visit Pr. Ulmsten in order to get their feed-back. I wanted to invite two Gynecologists from each of the big four countries for a workshop/visit in order to get their feed-back. So, I asked support from my European colleagues, in particular in order to identify the appropriate Physicians in their countries. I received some names and tried to get them together in Uppsala. For practical reasons, this ended up not to be possible in a reasonable time frame. Then, I opted for multiple sessions and again asked for support from my European counterpart. James Browning started the process by visiting Ulmsten with Drs. J. Bibby and P. Hilton (UK). Then, together with S. Salvati we organized the other visits in order to bring there (sic) Drs. R. Nappi, A. Ferrari, G. Vittori (Italy), Drs. R. Villet and B. Jacquetin (France) and Drs. Fisher and Hoffmeister (Germany). After completion of this process, I had to sum up the feed-back we received from our experts. Basically, each of them was very skeptical about the procedure prior coming to Uppsala. But after the visit, the general opinion was that the technique was excellent and in any case worth further efforts although none could explain how it worked. This was because the underlying concept was clearly challenging all the current concepts about the pathophysiology of USI [urinary stress incontinence].
Nevertheless, this convinced me the Ulmsten procedure could have a great future.1628
1598 The position was not advanced by the additional results from a Taiwanese study.
1599 This was a prospective, non-randomised study of 70 women with stress urinary incontinence who were treated using the TVT procedure. The results were discussed in an article by Drs Wang and Lo published in the Journal of Reproductive Medicine in May 1998. 1629 The respondents placed no reliance on it either contemporaneously or in this proceeding and Professor Krulewitch was the only witness to refer to it. It found that “successful correction” of stress urinary incontinence had been achieved in 84% of the patients, a result that was comparable to more extensive surgical procedures for the same condition. There were 26 adverse events: 11 patients (16%) suffered blood loss in excess of 200ml necessitating catherisation and vaginal tamponade (the insertion of a tampon); three patients had bladder perforations; and 12 patients (17%) had postoperative voiding difficulties requiring a further procedure. The authors declared that these complications compared favourably with complications from traditional surgical procedures. But the follow-up period was short, ranging from three to 18 months. The sample size was small. And, like the Nordic studies, there was no comparator arm.
1600 Indeed, it was common ground that there had been no comparative, let alone randomised controlled trials, assessing the safety and efficacy of TVT at the time of its launch in Australia in 1999. The first randomised controlled trial had not even begun and its six-month results were not published until July 2002.1630 The Ward Hilton RCT, which was a multicentre randomised comparative trial conducted in 14 centres in the United Kingdom and Ireland, is discussed above in Part VI.
1601 Dr Allman’s opinion was that the clinical evaluation conducted by Ethicon was not sufficient to justify CE marking. He said that Ethicon should have conducted “Ethicon-controlled” clinical investigations before CE marking was obtained or instituted post-market clinical follow-up studies at the time of CE marking.1631
1602 In his affidavit Dr Hinoul asserted that the TVT device has been the subject of numerous risk assessments including those by the original manufacturer, Medscand.1632 As Ms Holland pointed out, however, those risk assessments were manifestly inadequate.
1603 Ms Holland explained that design controls and risk management are integrally linked:
The design of the device is determined by the User Needs and technical feasibility of fulfilling those needs. Risk analysis is the iterative process of determining if the risk posed by the design embodies the crucial and basic concepts of patient safety and whether the risks associated with use of the device outweigh the ultimate benefit to the user. The purpose of risk management is to protect people from physical injury or damage to health. Both the design control and risk management processes begin with the specific device and its intended use. The design process must consider the hazards related to the use and reasonably foreseeable misuse of the device and accessories.1633
1604 She pointed out that risk planning is an essential starting point for defining risk management activities. Manufacturers are “required to responsibly define their criteria for determining that a risk is acceptable within the risk management plan and ensure that a process is in place to apply and assess measures to control the risk”. After risk control measures are applied, the medical benefit of the device must outweigh the residual risk. The object is to minimise all risks.1634
1605 Ms Holland reviewed the initial design documents prepared by Medscand and Ethicon and could not find any “User Needs, Design Inputs, Design Outputs or Design Reviews” before TVT was marketed.
1606 Ms Holland concluded that Ethicon had not complied with the requirements for quality management systems for the design of a medical device.1635 Her review of the design history and technical files from 1999 and 2000 disclosed that critical documentation regarding the TVT system design and associated processes were missing.1636 In particular, while Ethicon was required to have design requirement documents, design review and design verification records, and a dFMEA, no such documents were on the design history file.1637 Only one risk related document could be found. That was an application Failure Mode and Effect Analysis (aFMEA).
1607 Ethicon relied on the results of audits it conducted at Medscand to determine compliance with QMS requirements for TVT, but both BSI audits found “noncorformances”. The 1996 audit showed that there were noncorformances relating to device specification, the DHF, document control, and complaint handling. A follow-up audit in 1998 found nine new noncorformances. If any action had been taken to correct the noncorformances found in 1996, it was not documented. A remediation of the design history file was undertaken by Ethicon to generate missing design control documents in order to fulfil regulatory requirements and prevent future problems related to “change control”, but the new documents — a Device Design Safety Assessment (DDSA) and a retrospective dFMEA — were created between 2000 and 2002, respectively three and five years after the CE mark was applied to TVT and two and four years after it was first sold in Australia.1638
1608 The only hazards associated with TVT which were identified in the DDSA were infection and shipping damage. At the very least, as Ms Holland observed, this raised questions about the thoroughness of the assessment. Ms Holland reserved her strongest criticism, however, for the retrospective dFMEA, which she characterised as “not credible” for the following reasons:
(1) Neither industry practice nor internal procedures were utilised to perform the analyses.
(2) Severity and probability risk classes were not systematically applied and mitigation efforts were not commensurate with risk.
(3) Information conflicted with design requirements, manufacturing processes or post-market data. For example, certain risks, like “wrong mesh composition” and “mesh not cuttable” were classified as “not imaginable”. In the case of the former, Ms Holland pointed out that the material specification for Prolene mesh required the identity of the material to be checked against the raw material. Consequently, as she observed, the hazard was not only imagined but a mitigation strategy had been devised. She concluded that it was likely that the dFMEA was conducted in a vacuum, by the incorrect team, or merely as a “check a box” exercise.
(4) Known hazards such as degradation were omitted.
(5) The class of risk was reduced from “high” to “low” or “impossible” without requiring any mitigation efforts.
(6) No safety measures appear to have been added.
(7) Exceptionally, all risks were found to be acceptable even when extreme harm was known to occur, as in the case of erosion and mesh removal.
(8) No hazards resulted in a risk priority number or class that required any action to be taken, which, in Ms Holland’s experience was “very rare”.1639
1609 Furthermore, the dFMEA identified over-tensioning of the tape as a hazard classified according to severity/frequency as “long term/critical and probable”. Yet, risks associated with tensioning of the tape were not defined, although it was known that over-tensioning could lead to urinary retention. The dFMEA effectively dismissed it, stating that urinary retention is caused by the user. While “removal due to failure” was identified as a long term and critical hazard, the dFMEA stated that removal would not generate other hazards, ignoring the risks arising from revision surgery.1640
1610 In addition, the dFMEA did not consider risks associated with partial removal or hazards that might arise from removal required for other reasons, such as erosion or chronic pain.
1611 Moreover, although design controls were effective in 1996, including the requirement for design validation, Ms Holland said that review of the design history file and available technical files did not indicate that design validation had been conducted before TVT was put on the market, and she found no evidence that Ethicon had verified that TVT met the needs of users in the anticipated use environment.1641
1612 A DDSA re-evaluation conducted by Ethicon in 2002 noted that the risk assessment completed in July 2000 failed to mention 11 hazards including: vaginal extrusion; erosion/urethral; perforation by mesh; infection; vaginal incision; urethral tear; broken mesh; torn mesh; and kinked or twisted mesh.1642 These were erroneously described as “potential new hazards”. Various assumptions were made about what hazards need to be included in any evaluation. Lower extremity pain and post-operative complications, for example, were considered “symptoms or more general classifications of existing hazards” and post-operative complication complaints, which included pain, were said to be “typically caused by procedural events and not related to device function”. For the reasons given in Parts IV and VI, this was not true.
TVT-O
1613 According to Dr Hinoul,1643 TVT-O was created to enable surgeons to avoid the retropubic space and thereby reduce the risk to bladder, bowel and vascular structures of post-operative voiding dysfunction and blood loss. Certainly the TVT-O procedure involved traversing a route located away from the bladder, the urethra, the pelvic viscera, and vessels.1644 He said surgeons wanted a device that maximised safety and involved minimal passage through tissues. The focus of the respondents’ concerns, however, was in keeping ahead of their competitors. Safety was not irrelevant, but improved safety was only a means to a commercial end.
1614 In a memorandum issued on 1 May 2002 Dr Arnaud wrote “[w]e need a next generation TVT in order to move ahead from the competitors’ me-too products that recently reached the market place”.1645 In his account of the history of TVT-O,1646 Dr Arnaud wrote that, after five years of “monopolistic position on the market”, the Gynecare business, established by Ethicon in conjunction with its acquisition of TVT, was faced with two difficulties. The first was “the massive entrance of competitors”. The second was the proposal by Dr Delorme to modify the procedure, modified by Professor Mellier who went on to work for American Medical Systems, one of the respondents’ chief rivals. AMS developed the Monarc transobturator sling and, in Dr Arnaud’s words, sales of TVT “started to slow down (scil) before really diving”. Dr Arnaud attributed the decline in sales to the “obvious” fact that the transobturator approach was better than the retropubic. This was a major problem for Ethicon. Dr Arnaud described its position at this time as “a critical phase of its existence” with TVT responsible for 65% of sales.
1615 Daniel Smith, a member of the Research and Development team at Ethicon Inc. and the leader for the TVT-O project, later confirmed that the project was “created in response to competition in France using [transobturator] devices and rapidly stealing our TVT retropubic sales at an alarming rate”.1647
1616 Dr Arnaud worked assiduously to avert what he saw as a looming commercial disaster. In 2002 he began to cultivate a Belgian surgeon and Professor of Urology at the University of Liège, Jean de Leval. Professor de Leval was offering what Dr Arnaud described as “a clever modification of the transobturator approach”.1648 Dr Arnaud succeeded in persuading Professor de Leval away from AMS. In October 2002, in his pitch to the company for a “deal” with Professor de Leval, Dr Arnaud described “[t]he trans-obturator technique” as “a major threat” to the “TVT business since surgeons who have had the chance to compare it to the TVT usually prefer the trans-obturator route thanks to its better safety”. In his summary, Dr Arnaud listed the following additional considerations:
• The announced entry of AMS on this market is likely to dramatically increase the damage to our TVT business.
• The trans-obturator technique could also become a major source of growth for us providing we are bringing a better technique/product than our competitors.
• Pr de Leval has developed an innovative “in-out” surgical technique which is very likely to be more attractive to surgeons than the Porges and AMS supported “outside-in” techniques.
• Pr de Leval has also developed the ad-hoc device to perform the technique.
• The product includes a TVT-like tape and sheath, works perfectly, is nice, and basically ready to be marketed in its current version.
• European marketing has expressed a strong wish to have such a product in its portfolio.
• A provisional patent application has been filed in the US
• We could get an exclusive worldwide license with a very reasonable amount of cash.1649
(Original emphasis)
1617 Ethicon set out to enter into an exclusive arrangement with the University of Liège to license Professor de Leval’s technology. In an appropriation request dated 24 January 2003, Howard Zauberman, Ethicon’s Worldwide Vice President, New Business Development, said that the rationale was to drive sales of TVT in the wake of competition from “me-too” products and the rapid adoption of trans-obturator tapes.1650 He was concerned that more competitors would enter the market. He postulated that AMS and another competitor could launch obturator-specific products in early 2003.1651 Consequently, he stressed that “speed to market [was] critical” in the face of the following risk:
We predict a continued risk to our TVT franchise if we do not introduce a competitive obturator approach. If we were not to introduce a product with such a surgical approach, the lost profit (cash flow) to TVT™ would have a present value (PV) of $8.0MM.1652
1618 Although commercial considerations were the driving force, I accept that Ethicon was not oblivious to, or uninterested in, safety questions.
1619 In the same memorandum, Dr Arnaud had reflected on ways to “improve the safety of TVT”. One of the matters he raised was “[p]otential long term complications due to the mesh”. He did not identify those complications, however, merely observing that:
Some Physicians are concerned by potential complications which may occur in the long term due to the mesh. A solution may be the development of a long term absorbable TVT.1653
1620 Nonetheless, TVT-O as it came to be known offered some safety benefits. The technique avoided the retropubic space and the potential for injury to the organs and vessels in that space, such as bladder perforation, that were posed by TVT. In the appropriation request, Mr Zauberman wrote that transobturator vaginal tape (TOVT) “fulfils a basic surgical principle of moving needles away from important anatomical structures” and that “TOVT offers the advantage of lower bladder perforation rate”. He noted that Professor de Leval had reported no perforations for TOVT in 70 patients compared to 8% for “standard TVT”. He attributed the success of the TOVT to the reduction in the risk of bladder perforation and ease of use. He claimed that “the obturator hole approach does not compromise efficacy while reducing the complication rate”.
1621 The reduction in the incidence of bladder perforations also appears to have been Dr Arnaud’s main safety concern. In cross-examination Dr Hinoul conceded that bladder perforation was “a fairly frequent occurrence”. He said that, depending on the skill of the surgeon it ranged from between 1 and 5 percent.1654 This was an underestimate. Dr Arnaud put the rate at “around 10%”.1655 In his clinical evaluation report on TVT-O, Dr Martin Weisberg, then Medical Director of Gynecare, said that bladder perforation occurred “in around 3.8% of cases overall”, but “can be as high as 19%” in women who have had prior surgeries.1656 He characterised it as the “most frequent complication” of midurethral sling procedures.
1622 The Delorme technique used by Ethicon’s competitors was an “outside in” approach1657 in which the guide enters through an incision lateral to the labia majora (the larger, outer folds of the vulva), passes through the muscles and obturator fascia (the connective tissue covering the pelvic surface of the internal obturator muscle) exiting periurethrally (around the urethra) in the same area as TVT and, after the mesh is fastened to the guide, pulled through the incision in the thigh.1658 Professor de Leval’s modification, which was incorporated into TVT-O, and which Ethicon claimed to be an improvement,1659 involved an “inside out” approach. That means that the sling is inserted through a small vaginal incision midway under the urethra, passes laterally through the obturator foramen, and exits through the upper legs. This is known as a “hammock” shaped sling orientation. The mesh has to be trimmed on either end once it has been properly placed.
1623 On 25 November 2002, in a memorandum on Professor de Leval’s technique, Dr Arnaud proposed a “clinical strategy”, which was essentially focussed on marketing support, and based on the assumption that no clinical trials would be needed to obtain regulatory approval either in Europe or the United States.1660
1624 The first step in the strategy was a clinical report of Professor de Leval’s early experience. While he noted that the follow-up would be short, Dr Arnaud said that “the primary purpose [was] to demonstrate the feasibility of the procedure and excellent short term results”.
1625 The second step was a Belgian multicentre prospective study to begin at the earliest in the first quarter of 2003 sponsored by Professor de Leval and involving five Belgian investigators whom Professor de Leval knew personally.
1626 The third step was a Gynecare sponsored worldwide prospective study using Ethicon products to test the device as marketed by Gynecare and to start as soon as the Gynecare product became available.
1627 On the assumption that Dr Arnuad’s proposal was accepted, there was no evidence indicating that either step 2 or step 3 was undertaken.
1628 On 3 December 2002 there was a meeting of clinical and regulatory staff to discuss the strategy for the TVT-O project, then referred to within Ethicon as “Operation Mulberry”. Dr Arnaud and Ethicon’s then Medical Director, Dr Martin Weisberg, were in attendance. The meeting discussed the existing data and, with respect to CE marking, minutes of the meeting circulated by email two days later record:
* Marty feels that based on the clinical evidence, CE marking without additional studies is a viable and acceptable route
* Additional inventor cases collected until CE marking should be used to strengthen launch data. With this data, marketing would not be able to make specific claims
* If claims are desired (likely to be the case in the longer term), 2 options exist (depending on desired claim). (i) clinical trial proof e.g. lower bladder perforation, likely to be a large-ish trial with longer follow up. This information can be leveraged from Project Haydn. (ii) observational follow-up study and surgeons using their knowledge of anatomy will be able to judge this claim in light of observational follow up.1661
The next step was for Dr Weisberg to “write justification for CE marking without additional clinical data based on [Dr Arnaud’s] report”.
1629 Dr Arnaud also wrote that “[t]he safety can be somewhat demonstrated by Pr de Leval experience which involves over 70 patients” but he added that “it would be wise to get clinical experience from other investigators before launch” and that it was necessary to check the safety in the hands of others.1662
1630 Early in 2003 Ethicon became aware that a number of patients of Professor de Leval were experiencing leg pain with his obturator device. In March Dr Weisberg sent an email to Dr Arnaud to seek answers to a number of questions concerning the incidence of leg pain. Dr Arnaud responded as follows. The incidence of leg pain was 20% to 30%, but that included “minor discomfort as well as pain”. The pain was usually bilateral, “with a unilateral predominance”. The pain was usually “muscular or ligament, never nerve pain”. It occurs in the upper thigh and could be radiating from the hip, noting that the procedure requires the patient to be positioned in hyperflexion on the table. Its severity is “variable”. It usually lasts less than 48 hours. In “very rare cases” that may extend to a few days but “never after a week.” There is no motor weakness, although sometimes the pain interferes with walking. Professor de Leval suggested treatment with non-steroidal anti-inflammatory drugs. Dr Weisberg noted this kind of pain had not been reported in the literature with the “outside-in” approach but said informal discussions suggested that it was common.1663
1631 I note that, notwithstanding Dr Arnaud’s emphatic exclusion of nerve pain, years later, in the context of a discussion of the literature relating to TVT-O, Dr Hinoul observed that thigh pain experienced after transobturator procedures might be induced by the presence of a foreign body reaction in the proximity of the peripheral obturator nerve branches.1664
1632 Despite this information, on 9 April 2003 the Ethicon team working on Operation Mulberry agreed to proceed on the basis that a clinical trial would not be required.1665
1633 For the purpose of demonstrating to “authorities” and the medical community that Professor de Leval’s method was safe and efficacious, Ethicon engaged a firm of clinical research specialists, MedAlliance, to review the professor’s work. The plan backfired. MedAlliance was concerned about a number of aspects of Professor de Leval’s clinical trial. One particular concern was the number of adverse events and the fact that at least one patient had experienced “a serious adverse event in which there [was] a risk it might be classified potential severe adverse device effect”: a thrombophlebitis (an inflammation of the wall of a vein with associated thrombosis or blood clotting) and later an abscess of the thigh. Another was that the trial had been started without complying with Belgian regulations which required, amongst other things, an assurance from the manufacturer of the device that it complied with all applicable essential requirements except for those that would be verified by the clinical investigation. MedAlliance concluded that Professor de Leval was unable to comply with the relevant regulations, which it attributed to his lack of technical knowledge, legal infrastructure, and human, organisational, material and financial resources.1666
1634 Dr Arnaud counselled Professor de Leval to stop the study. In an email of 25 May 2003, he proposed instead two clinical studies but said that they “should be as ‘light’ and inexpensive as possible”.1667
1635 On 28 May 2003 the Gynecare Research and Development team decided that the first de Leval clinical study would be published but without Ethicon approval and would be used “for marketing purposes only” and the second de Leval clinical study, which was intended to support the release of TVT-O, “will stop as a clinical trial”. An unplanned third de Leval clinical study would be prepared using MedAlliance to ensure compliance using an alternative attachment method and those results would be used to launch the Ethicon device, although follow-up data would not be available for submission of the US application for regulatory approval. Planning for a fourth clinical study, to be sponsored by Ethicon, was still under way and would be used “for post-launch marketing literature, surgeon training and pre-enrolment of preceptors”. It was expected to start in October 2003.1668
1636 Professor de Leval submitted a draft of an article on his first study to Ethicon in May 2003. On 8 May 2003 Janice Burns, a senior marketing manager for Gynecare, circulated comments on the article to Ethicon colleagues that she, Laura Angelini (Ethicon’s Vice President, Global Strategic marketing) and Steve Bell (a director of marketing with Ethicon in Europe) had prepared. Amongst those colleagues were Drs Arnaud and Weisberg.
1637 The draft article began with a description of retropubic TVT and noted, to the chagrin of Ethicon’s marketing team, that:
The use of retro-pubic TVT has been associated with various and relatively frequent per- and post-operative complications, including bladder perforation, temporary or persistent retention, pain, urinary infection, and de novo instability. Other rare but severe — and possibly underestimated — complications have been reported with this approach. Indeed, the blind passage of the needle in the retro-pubic space can result in injuries to other organs than the bladder, in particular the urethra, vessels, nerves and bowel.1669
1638 For good reason, the applicants submitted that the draft article should have alerted Ethicon to a number of matters that should have been (but were not) considered for further evaluation before it launched TVT-O:
First, of the patient population Prof. de Leval had operated on, only 69.2% suffered from SUI, with the remaining patients suffering pelvic organ prolapse that was associated with symptomatic or potential SUI. The impact on (scil) that patient population, insofar as results were proposed to be extrapolated to a broader patient population, ought to have been considered;
Second, post-operative assessment was done at 1 month. The follow up period was noticeably short, prompting Prof. de Leval to note that long term follow up was required in order to assess longer term efficacy of the product;
Third, 2.8% of patients required immediate tape release due to urinary retention issue;
Fourth, Prof. de Leval identified a number of issues associated with the TVT “Classic”: there were “various and relatively frequent pre and post-operative complications” associated with the procedure including bladder perforation (in 0–23% of patients), temporary or persistent retention (1.5%–12.9%), pain, urinary infection and de novo instability. Other rare but severe — and possibly underestimated — complications were noted. These included vascular injuries, bowel injuries and deaths. Prof. de Leval attributed injury to the urethra, vessels, nerves and bowel to the blind passage of the needle through the retropubic space in the TVT “Classic” procedure. Rather than see this as a prompt for further evaluation, Ethicon sought to alter this part of the draft report, on the basis that it presented the TVT “Classic” too negatively, and sought to suggest that these concerns related to ‘certain groups’ of patients, emphasising the need to “expand on the strengths of TVT”;
Fifth, Prof. de Leval described his technique as involving alignment of the tape with the assistance of Babcock forceps, used to grasp the tape — Ethicon considered this “not good practice with monofilament tapes” and sought to refer instead to the instructions for placement of the TVT “Classic”;
Sixth, in 15.9% of cases, the patient complained directly of thigh pain following the procedure which lasted up to 2 days, and 1.9% of patients complained of severe pain that persisted for a week;
Seventh, one patient developed an abscess requiring drainage, following which Prof. de Leval adopted a practice of prophylactically administering a powerful antibiotic in all patients undergoing the procedure — this was a practice never recommended in any IFU for the TVT-O;
Eighth, 8.4% of patients in Prof. de Leval’s series experienced de novo urgency by 1 month follow up. 1670
1639 In the covering email Ms Burns identified the following “key points”:
• The paper describes TVT (scil) too negatively and we need to protect our base business.
• Could we expand on the key benefits of the GYNECARE TVT mesh?
• Can we expand more on the complication of urethral perforation of outside in?
• There is concern about describing the technique in too much detail prior to the launch date
• Can we describe the technique being needleless (scil)? 1671
1640 Notes taken on 4 August 2003 by Ethicon’s Daniel Smith referred to a decision by the “Gynecare board” “to take the risk of not requiring a clinical trial”.1672 No heed was taken of the observation by Dr Arnaud in his November 2002 memorandum that the safety of the inside-out obturator approach needed to be tested in the hands of other investigators.
1641 By 5 December 2003 a biocompatibility report was available.1673 It noted that no tests were carried out on the handles or the tape since they were made using the same materials and processes that were used to make the TVT device. The two wire guides and the “atraumatic winged guide” were not tested either, apparently because they were made from electropolished 304 stainless steel, which is an FDA approved material for medical devices. The only components that were tested were the plastic tube and Prolene mesh attachment. Results of testing those components for cytotoxicity, sensitization, intracutaneous reactivity, acute systemic toxicity, and “EO residual analysis” were considered “acceptable”.
1642 The clinical evaluation report signed by Dr Weisberg on 16 December 2003, which supported the launch of TVT-O, purported to review published clinical trials and oral presentations on the outside-in transobturator approach.1674 These were no more than potted summaries of the data derived from the document. Contrary to the requirements of the European Directive, there was no critical evaluation of the available scientific literature.
1643 The literature review in Dr Weisberg’s report consisted of two journal articles and four oral presentations, all involving the “outside-in” technique. As Dr Allman observed, the report did not describe the approach taken to identify the publications relied upon. It did not comply with MEDDEV 2.7.1 published in April 2003,1675 in that it did not state:
the extent of the searches of databases or other sources of information;
the rationale for the selection of the cited published literature;
the reason for believing that all relevant references, both favourable and unfavourable, had been identified; or
the criteria for exclusion of particular references together with a justification for exclusion.
1644 The only material dealing with the “inside-out” technique upon which Dr Weisberg relied to support his conclusion that the technique was “safe and effective” in the treatment of stress urinary incontinence in women was unpublished data from two studies by Professor de Leval.
1645 Dr Weisberg noted that at the time of preparation of his report Professor de Leval had performed 159 procedures using his “inside-out” transobturator approach using TVT tape. He said that 138 of them had been compared to 134 using the TVT technique. He said there were no complications from haemorrhage or secondary haematoma formation in the transobturator group but four cases in the traditional TVT group, nine bladder perforations in the traditional TVT group and none in the transobturator group, one vaginal erosion in each group, and no urethral erosions in either. The most frequent complaint was reportedly post-operative thigh pain. That was said to have occurred in 19 (26%) cases but to have been of “transient (24–48 hours)” duration and managed with non-steroidal anti-inflammatory medications. One patient in the transobturator group developed phlebitis with a secondary abscess on the tenth day. The source of this information was not disclosed. Presumably it was conveyed directly or indirectly by Professor de Leval. Dr Weisberg noted that the report had not yet been published and the study had not been completed. 1676
1646 It is likely, though not certain, that this was the study MedAlliance had criticised, Dr Arnaud had urged Professor de Leval to abandon, and the results of which were intended to be used only for marketing purposes.
1647 Dr Weisberg also referred to an additional single centre study of women implanted with TVT by the inside-out transobturator approach. The study was performed by Professor de Leval and was reportedly designed to evaluate intraoperative device performance and “secondarily” to collect intraoperative adverse events and early safety and effectiveness data. The parameters of the study, however, were not described and, at the time of the report, performance data was available for only 21 patients and then only after two weeks’ post-operative follow-up. The only adverse event reported by the investigator was thigh pain in six of those patients (29%), treated with non-steroidal anti-inflammatory drugs, which was said to have resolved after two to seven days of treatment.
1648 It is likely that this was the first of the two “light” and inexpensive studies that Dr Arnaud had suggested be undertaken after the first study was aborted.
1649 The report made no reference to the MedAlliance findings nor to the position taken to the de Leval studies by Ethicon’s own Research and Development team.
1650 Based on the literature review, communications with Professor de Leval, and the findings of “the project’s due diligence team” (which he did not disclose), Dr Weisberg concluded that:
(1) There was “clear clinical evidence” that the outside-in transobturator approach for suburethral sling placement is a safe and effective surgical technique.
(2) The fact that the transobturator “inside-out” approach results in the identical placement of the tape as in the “outside-in” procedure and, on the basis of the unpublished data from Prof de Leval, the efficacy of the two procedures were equivalent.
(3) Urethral injury was much less likely to occur with the “inside-out” approach because the passage of the guide and tape in the area of the urethra is performed under direct visualisation. For the same reason, “logically” the “inside out” technique will allow positioning of the tape at the mid urethra to be more accurate.
(4) The “inside-out” transobturator technique seems more precise than previous techniques, appears easy to perform, is reproducible, may not require cystoscopy, and is likely to result in fewer complications.
(5) The results in terms of curing incontinence seem to be equal to the techniques described previously in the short-term. 1677
1651 For these reasons Dr Weisberg said he was “confident” that “inside-out” transobturator approach was a “safe and effective” treatment for women with stress urinary incontinence and that additional clinical studies were not necessary “at this time”.
1652 In Dr Allman’s opinion this argument was not sufficient to support CE marking and the report did not meet the requirements for clinical evaluation required by the European Directive.1678 Amongst other things, he noted that the CER described the reported “assumed” advantages and disadvantages of the obturator approach and described and discussed published clinical trials and oral presentations relating to the “outside-in” technique. He pointed out, however, that the approach to identifying the publications was not described and that the CER relied on unpublished data from the surgeon who first described the inside-out technique based on a single centre study when only two weeks of follow-up data were available from just 21 patients.
1653 Ethicon had intended to draw on published literature derived from a surgeon-sponsored study “beginning immediately following the required Ethics Committee and Belgian Competent Authority approvals”. Once again, however, marketing imperatives were paramount. In an email circulated to Board members on 9 June 2003, Sean O’Bryan, Ethicon Inc.’s Senior Project Manager, Regulatory Affairs, wrote that “[f]or Marketing NEEDS, it was also agreed that an ETHICON sponsored study will be initiated with US and EU sites once we have a finished device”. But he did not contemplate that the study would necessarily be completed by the time the product was launched. Rather, he wrote that “[e]very effort will be made to start the study prior to launch so that valuable data can be used for marketing at the time of launch”.1679 In effect, Ethicon assumed that the study results would be favourable.
1654 TVT-O was launched worldwide on 22 December 2003 and, as I noted earlier, it was first supplied in Australia in March 2004.1680 In February 2004, Daniel Smith boasted that the project was completed in less than nine months, which he described as “[a] new record for Gynecare”, and submitted a proposal to Human Resources that an award be granted to the project team. In the submission he explained, amongst other things, that:
The original project plan was estimated to be 24 months, which was honed to 18 months with a break through goal of 12 months. upon chartering and increased market pressure eroding the TVT base business the board issue a revised edict of <9 months or Y/E 2003. Unaware of how this goal would or even could be achieved, the team set out to do what was thought to be an impossible task regardless of recourses (sic) or money.1681
1655 It is difficult to resist the conclusion that the new goal could only have been reached by cutting corners, including by compromising on safety evaluation.
TVT Secur
1656 TVT Secur, it will be recalled was a single-incision sling. It proved to be a triumph of hope over experience.
1657 At the time of the development of TVT Secur, Ethicon was said to be under pressure from as many as 12 competitors. Ethicon’s boast was that TVT Secur would allow the business to differentiate itself from its competitors based on safety, inventory (one code for both obturator and retropubic approaches), and ease of use.1682
1658 TVT Secur was described in Ethicon’s launch plan as “a game changer” for the incontinence market because it added to the clinical evidence obtained from TVT “the uniqueness of a new technology that removes the need to exit incisions in the pelvis to fine tune the tension on the sling”.1683
1659 Despite this optimistic picture, TVT Secur was not a success, certainly not in Australia, nor apparently in some other countries. Early feedback from surgeons here was not encouraging although Ethicon personnel expressed confidence that better outcomes would be achieved as experience increased.1684
1660 The initial concept for TVT Secur apparently came from Professor Ulmsten in 2003.1685 It was conceived as a less invasive device. The concept was evaluated in independent market research conducted with 150 surgeons in the European Union and the United States. According to a summary of that research, the concept of a needleless approach was assumed to result in fewer complications. One third of those surveyed (50) could detect no drawbacks. But the other 100 identified as “the key disadvantages” insufficient information and the need to see (long-term) data proving the product’s efficacy and safety, especially concerning “its holding power, the material used and the means by which the mesh is held in place”.1686
1661 Development began in 2004. The rationale for its development was the need to stay ahead of the competition:
In US and Europe the sub urethral sling market continues to be highly competitive with an estimated over 25 competitors Worldwide. This number includes Global brands as well as local manufacturers. Annual sales of the GYNECARE TVT brand (TVT & TVT O) in the direct markets is estimated to reach $~100MM by end of 2004 with a profitability of ~91%. This high profitability being key to the GYNECARE business as no other product within the pipeline can demonstrate such a profitability, which could assist to replace profit for this critical platform. Prior to the launch of TVT Obturator such markets as France had lost up to 30% of the TVT business to competitor obturator approaches. The launch of TVT Obturator in January 04 has been highly accepted by the surgical community as well as providing a strong market differentiation with an ‘Inside Out’ approach. It expected to contribute approximately 23% of the total TVT sales in the direct five markets in 2005.
GYNECARE continues to be market leader in the sub urethral sling market with an estimated market share in US at 46% and 59% European D4. It remains strongly recognized that GYNECARE developed this market and coupled with the skills, competencies and capabilities within the organization such market dominance can be sustained. However, product innovation and advancement is required in order to stay ahead of the competition.
To maintain market leadership in the increasingly competitive surgical SUI market. Market and IP intelligence confirms competitor development plans for ‘mini type’ TVT devices…
1662 Janice Burns, who prepared the document, stated that “[t]he main benefits of the concept relate to the safety of the needleless approach, which is assumed to result in less risk to patients and fewer complications”. Market research conducted in October 2004 revealed, amongst other things, that a third of the 150 surgeons surveyed could detect no drawbacks with the product but that the rest were concerned about the dearth of information and wanted to see long-term data proving that the product was safe and effective.1687
1663 Three pre-clinical studies on ewes and ewe cadavers were conducted in 2004 and 2005. The 2004 cadaver study was designed to assess the fixation force of TVT Secur once implanted. The purpose of the cadaver study carried out in 2005 was to reconfirm the initial holding power of the end of the implant after certain design and manufacturing changes.1688 Various cadaver laboratory studies were conducted but all were focussed on the fixation capacity of the device.1689
1664 Ms Holland said that the specification for TVT Secur was based on TVT. She observed that it was reasonable from an engineering perspective to ensure that the tape stayed in place without sutures. She pointed out, however, that when the device was first supplied in Australia, the specification for the pull-out force was lowered from 500 grams, the minimum fixation force considered sufficient for the duration of the cadaver studies, to 164 grams — a reduction of about 67%.
1665 As Ms Holland observed, this change, which was approved by management, was “apparently conducted in order to achieve the marketing need to continue to use the seven-year historical data from PROLENE for the TVT SECUR”.1690 In a memorandum to Daniel Smith, the project leader, entitled “Mechanical Cut vs. Laser Cut Mesh Rationale”, Allison London-Brown, from Gynecare’s marketing department, wrote:
Marketing Need: Keep relationship with PROLENE mesh and launch a new technology.
In May 2005, the GYNECARE Management met to discuss the recommendation to approve the use of LCM for the TVT SECUR project … The team … stated the use of a “physiological range” maximum, to define the upper limit of force placed on the mesh by the urethra and muscles during coughing/sneezing. The maximum force of<=l64g was the derived by (sic) the fact that at 164 grams the current TVT mesh becomes permanently deformed. It was agreed that we could not have obtained 7 years of great clinical results if the mesh had in fact permanently elongated.1691
1666 Ms Holland considered that the comparison with TVT Secur, which utilises different materials and has a different size, shape factor, and surgical approach, was not a valid method to determine a specification. Indeed, Ms London-Brown had noted that “TVT SECUR is a new product/technique” and, apart from the common use of Prolene mesh, there was “little relationship to the 7-year database.” Ms Holland said that in her experience problems emerge over time when the role of marketing moves from determining the user requirements to establishing the engineering specifications. She added that “specifications are intended to be specific for each design and intended use rather than as a promotional tool”.
1667 A dFMEA was performed once, in November 2005.1692 It classified risks based on examination of each component of the device, the function it performed, the potential failure mode, the potential effect/hazard of failure, the class of fault (whether the failure mode in and of itself leads to the hazard or potential effect or whether something else needs to happen), the nature of the harm, the severity of the harm, the potential cause, the likelihood of a potential failure mode occurring as a result of the identified cause (dubbed “occurrence”), at what stage of the design cycle the potential cause of failure can be detected (“detection”), RPN, and risk category/further action.
1668 Risks were classified either as “Broadly Acceptable (BA)”, defined in Ethicon’s Operating Procedure for Failure Modes and Effect Analyses as an area of risk accepted by the user community as “part of doing business”, or “As Low as Reasonably Practicable (ALARP), defined as an area of risk that is tolerated by the user community but where it would be desirable to reduce the potential for risk if a “practical and cost effective” solution were available.
1669 No identified risk was said to require a risk benefit analysis, defined as an area of risk that is “tolerated by the user community based on the morbidity or mortality of alternative therapies not using the medical device [under review]”.1693
1670 In every single case the occurrence rating was 4, denoting that there was a moderate likelihood (<1 in 1000) of the potential failure mode occurring as a result of the identified cause.
1671 Ms Holland reviewed the documents relating to dFMEA and concluded that it did not fully comply with either Ethicon’s own procedural requirements or industry standards. She explained:
None of the 39 potential failure modes determined to be in the as low as reasonably practicable region (“ALARP”) had any further actions taken to reduce the associated risk, nor was there any indication that options were considered. OP650-011 [Ethicon’s operating procedure for its design Failure Modes and Effect Analyses] states the “team should discuss actions that could be taken to further eliminate the potential cause, reduce the frequency with which it could occur and/or improve the detection”. Three possible corrective actions are provided including improving the current controls, redesign of the medical device element, or improve the reliability program. Industry guidance states that “any risk should be reduced to the lowest level practicable”, bearing in mind the benefits of accepting the risk and the practicability of further reduction. Ethicon did not do this. At the time the TVT SECUR was being designed, practicability included two components: technical practicability and economic practicability. However, the important concept that appears to have been overlooked is that “major risks should normally be reduced even at considerable cost.” In other words, patients should not be put at risk because the cost of the hazard reduction impacts a company’s profitability. In fact, the concept of economic consideration in risk reduction has since been removed from the current version of ISO 14971 to comply with the Essential Requirements, which is necessary to obtain approval to sell a device in the EU and CE marking.1694
Moreover, Ms Holland observed that, in 41 out of the 65 identified potential failure modes defined in the dFMEA, harms were not determined. Instead, they were described as “N/A”. That was so even in cases where the potential effect was “breach of aseptic technique” rendering the device unusable. I gather that Ethicon assumed that surgeons would work this out for themselves. But Ms Holland, presumably acting on the assumption that some might still use the device, observed that in such a case an infection could occur. She said that, if harms were appropriately defined, then the outcome of the analysis may well have been different and the associated overall residual risk may have increased. Indeed, if all harms had been included, the ORR may have exceeded 29, which, under Ethicon’s own procedures, would have required risk benefit analyses.
1672 Although it was designated a class III product,1695 no clinical trials of TVT Secur were undertaken before the device was first supplied in Australia in April 2007. Ethicon decided that a clinical study was unnecessary before marketing it.1696
1673 Before launch, however, Ethicon prepared a “Design Validation Protocol” for a study the stated purpose of which was:
[T]o provide objective evidence that the GYNECARE TVT SECUR SYSTEM (including devices, packaging, and labeling) satisfies defined user needs and intended uses within actual or simulated-use environments in a manner consistent with the Instructions for Use (IFU) for GYNECARE TVT SECUR SYSTEM and the OR staff's and surgeons usual techniques.1697
1674 Surgeons of differing levels of experience, who had previously trained on TVT products, were to be recruited to evaluate both the device and its packaging, including the labelling and instructions for use.1698
1675 According to the Design Validation Report recording the outcomes of the study,1699 it appears that 13 surgeons participated, performing 16 placements1700 in unembalmed female cadavers.1701 Twelve of the 13 were classed as experienced (in implanting TVT) and one inexperienced. Eight were urogynaecologists, two urologists, and three gynaecologists.1702 While the report concluded that the “device conforms to user needs”, it noted that, because of “multiple issues raised within the study framework”, a follow-up design validation study would be performed.
1676 A second design validation study was then conducted. The outcomes were reported on 10 February 2006.1703 Six surgeons, five of whom were classed as experienced (but with no previous experience of the device) successfully performed the placements in cadaver models, reportedly demonstrating “both the adequacy of the training materials/method and the ability to successfully use the TVT S device and place the implant”. The cadaver models, however, were torsos only, which meant that patient positioning could not be considered. The surgeons were trained using the IFU, a video of the procedures carried out on cadavers (both “U” and “Hammock”), and by demonstration.1704
1677 According to the Dr Weisberg, who signed the first clinical evaluation report on the device, the studies “confirm[ed] adequate holding ability in sheep and human cadavers”.1705
1678 Ms Holland was critical of the design validation. She wrote:
In my opinion, the design validation did not represent the simulated use conditions and led to false confidence in the performance of the device in the hands of inexperienced users without the benefit of one-on-one training by the design team.1706
1679 Her opinion was not challenged in cross-examination. The facts bear it out.
1680 Ms Holland was also critical of the risk management assessment of TVT Secur. She observed that review of the aFMEA documents on TVT Secur showed that all the identified harms were mitigated by training. But all the IFU said on the question of training was that the device should be used only by physicians trained in the surgical treatment of stress urinary incontinence. No specific surgical technique or training requirements for the device itself were provided.1707
1681 None of Ms Holland’s criticisms was addressed in the respondents’ submissions.
1682 A pre-market CER was signed on 2 December 2005 by Dr Weisberg, then Senior Medical Director of Ethicon.1708 It relied on the first design validation study and also on the sheep cadaver testing concerning the processes of implantation and affixation.
1683 Dr Weisberg said that TVT Secur addressed certain clinical needs: the reduction of complications from TVT procedures; the use of a less invasive procedure than existing procedures; and the ability of the same device for the retropubic and transobturator approaches. The complications he highlighted were bladder perforation, which he said was the most frequent complication of all retropubic midurethral tension-free support procedures according to the literature (at a rate of 3.8 to 19 percent); perforation of large vessels and intestinal viscera, also reported with the retropubic approach; and thigh pain.
1684 The list of potential complications in the report mirrored the list of adverse reactions in the IFU:
• Punctures or lacerations or injury to vessels, nerves, bladder, urethra, or bowel may occur during instrument passage and may require surgical repair.
• Transitory local irritation at the wound site and a transitory foreign body response may occur. This response could result in extrusion, erosion, fistula formation or inflammation.
• As with all foreign bodies and surgical implants, PROLENE mesh and absorbable materials may potentiate or exacerbate an existing infection.
• Over-correction, i.e., too much tension applied to the tape, may cause temporary or permanent lower urinary tract obstruction.
• Under-correction or incorrect placement may result in incomplete or no relief from urinary incontinence.1709
1685 The applicants tendered an unsigned CER dated 23 August 2005.1710 It appears to be a draft of the December 2005 CER. Its author or authors were never identified although it was prepared with the intention that it be signed by Dr Weisberg, as his name appears on the front page. A different and more detailed list of potential complications was included in this version. The unsigned 23 August 2005 CER identified the following anticipated events as potential complications:
• Punctures or lacerations of vessels, nerves, bladder, urethra or bowel
• Inflammation or infection at the surgical site
• Urinary tract infection
• Abnormal postoperative bleeding
• Dysuria
• Pain
• Hematuria
• Leg pain
• De novo detrusor instability or urgency
• Intra-operative damage to the mesh Irritation at the wound site
• Foreign body response
• Mesh exposure,
• Mesh erosion
• Fistula formation
• Urethral obstruction due to over-correction resulting in urinary retention
• Hematoma
• Venous thrombosis
• Abscess formation
• Reaction to anesthesia
• Death1711
1686 No evidence was adduced to account for the omission of the various complications listed in the 23 August 2005 draft.
1687 Despite the fact that Ethicon had concluded that a second study was necessary because of the multiple issues raised during the first, in the December 2005 CER Dr Weisberg declared that TVT Secur was safe and effective and that additional clinical studies were not necessary before it was released to the market. His conclusion was said to be based on two matters. The first was that mesh in TVT Secur was “the same as the currently marketed TVT mesh products, in terms of indications for use, material, construction and key dimensions”. The second was the risk assessment.1712
1688 It is true that the indications of use were the same as the currently marketed TVT mesh products but there were significant differences between the devices in material, construction, and dimensions. Supplementary submissions filed by the respondents on 20 June 2018 acknowledged as much.1713 It will be recalled that, in contrast to TVT and TVT-O, TVT Secur was made using not only Prolene but also Vicryl and poly-p-dioxanone (PDS), that the device consisted of a piece of Prolene tape sandwiched between layers of absorbable fleece made from Vicryl and PDS, and that it required a single-incision. For these reasons, the first basis for the conclusion as to safety and efficacy was unsound. Moreover, while the “key dimensions” to which Dr Weisberg was referring were “the width and thickness of the ‘working’ section”, he offered no reason as to why the full dimensions did not need to be taken into account. TVT Secur was significantly shorter than TVT and TVT-O.
1689 The risk assessment upon which Dr Weisberg purported to rely was not identified in the references contained in the endnotes to the CER. There is a “biocompatibility risk assessment” which was written by Dr Holste, but it is dated 3 February 2006 and therefore post-dates the CER.1714 The only risk analysis to which the respondents referred in their submissions also post-dates the CER. It is dated 3 March 2006 and is signed by Dr Robinson. In contrast to the risks Dr Hinoul conceded could arise with all the SUI devices, this assessment identified only two risks: the incidence of urinary retention; and the risk of premature removal of the protective inserter blade cover.1715 With respect to the former, urinary retention, Dr Robinson classified the risk as “moderate” but concluded that the incidence would be the same as for TVT and TVT-O because of similarities in the “tape and placement”. He relied on reports of retention after TVT and TVT-O surgery reported to Ethicon (1989 in over 900,000 procedures or 0.2%), not clinical studies, and failed to make allowance for the problem of under-reporting. In the absence of any analysis of the potential effects of the differences, this reasoning is also unsound. With respect to the latter, the risk of premature removal of the protective inserter blade cover, Dr Robinson said it was not high, but did not explain why, and claimed that it was comparable to other surgical risks posed by knives or scalpels. He concluded:
In general terms, based upon the above analysis, the anticipated medical benefits of the TVT Secur device outweigh the individual residual risks of the items discussed above.1716
1690 No explanation was given for the narrow focus of the risk assessment.
1691 Since TVT Secur has only a single-incision application, Dr Hinoul said in a post-market CER that it was not equivalent to TVT and TVT-O.
1692 Dr Robinson’s approach was also at odds with the opinion expressed by Ms London-Brown referred to above and Ms Holland’s expert opinion that the data from TVT and TVT-O “could not appropriately be leveraged to accept the risks associated with [TVT Secur]”.1717
1693 A second CER on TVT Secur was signed by both Dr Weisberg and Dr Robinson on 28 February 2006.1718 It added some later test results on elongation of laser cut Prolene and some other references, including a reference to Dr Holste’s biocompatibility risk assessment. Otherwise it was substantially the same as the December 2005 version.
1694 Both CERs asserted that reduction or elimination of bladder perforation, perforation of the large vessels and intestinal viscera (experienced with TVT), and thigh pain (experienced with TVT-O) were addressed by TVT Secur. Neither explained how.
1695 Like its predecessor, the 2006 CER also declared that TVT Secur was a safe device, effective for treatment of stress urinary incontinence, and that additional clinical studies were unnecessary before the product was released to the market. Dr Allman said that these conclusions were unjustified and the report itself was insufficient to support CE marking.1719 He said it was illogical to rely on safety and performance data from predicate devices when TVT Secur was designed to be different from, and an improvement on, those devices. He also pointed out that the regulatory requirement for clinical data is to confirm conformity with the essential requirements under normal conditions of use and to evaluate side-effects but that the report did not do this. It referred to no clinical data, “just broad statements about ‘clinical history’”. Indeed, he said that no clinical data showing the device under normal conditions of use appear to have been gathered. Dr Allman also said the report did not constitute clinical evaluation as required by the European Directive and MEDDEV 2.7.1. Dr Allman’s opinion on this point was not challenged. For all the above reasons, it should be accepted.
1696 It will be recalled that Ethicon conducted market research in 2004 which revealed concerns about the dearth of information about TVT Secur and the lack of long-term data to demonstrate its safety and efficacy. On 31 March 2006, when the Ethicon Women’s Health and Urology team (including Dr Robinson, Dan Smith, Ms London-Brown and Janice Burns) met in Singapore with “select surgeons” to introduce them to the new device. One of those surgeons was Dr Frazer. An email summarising the meeting said Dr Frazer expressed “significant interest in having EWH&U establish an RCT site in [Australia]”. Another, Professor Haylen, indicated that he would not want to be approached until data was available.1720
1697 According to Ethicon’s Australian “Launch Plan and strategy” document for TVT Secur, the incontinence market was under pressure from as many as 12 different competitors and “[t]he longer that we are unable to launch SECUR, the more that these pressure (sic) will exist and effect (sic) our business in the longer term”.1721 The document claimed that Ethicon was “committed to change the Incontinence playing field by offering a device that is safer due to its ability to be positioned without driving into the pelvic cavity or through the Obturator Membrane”. It went on to say:
This claim will be unique to the market allowing us to;
Protect existing business and gross profit especially from BSX [Boston Scientific]
Increase share from competitors especially AMS
Grow the market via strategies to attract customers not currently performing Incontinence e.g Obstetricians1722
1698 Improved safety was a laudable objective. Moreover, there was a rational basis to it. In the absence of clinical comparative studies, however, it is difficult to see how the claim was justifiable.
1699 In an email to colleagues sent on 25 October 2007, Dr Aran Maree (Ethicon’s Medical Director) reiterated his concerns about the high failure rates across multiple centres seen with TVT Secur in comparison to TVT-O. He expressed concern that TVT Secur may have been launched without enough clinical data to justify the roll-out.1723
1700 In these circumstances, I conclude that in all likelihood the failure to recommend or conduct clinical studies, let alone a randomised controlled trial, before the device was launched, whether overseas or in Australia, was attributable to marketing considerations.
TVT Exact
1701 No studies or clinical trials of TVT Exact were undertaken before market launch in Australia in July 2010. By May 2011 Ethicon was fielding concerns that the tape could cause retention and that, because the trocar is smaller in diameter than the TVT trocar, shrinkage of the mesh was more significant.1724
1702 Ethicon’s marketing strategy for TVT Exact relied heavily on TVT. Key claims, according to an undated Ethicon PowerPoint presentation by Zeb Viana, were that the new device:
• Improved ergonomics and tactile feedback for enhanced control
• Uses same trajectory and proven mesh as TVT Classic
• Requires less force during needle passage1725
1703 The pre-market CER, which was signed by Dr Kirkemo on 28 May 2010,1726 was based on a review of the clinical literature published after 1 January 2006 and before September 2009. The review was confined to 88 publications discussing the safety and efficacy of TVT and TVT-O. Literature dealing with any other sling was excluded. The publications reviewed were said to have been drawn from the PubMed database, a medical database containing journal articles and online books maintained by the US National Library of Medicine.
1704 Under the heading “Complications”, Dr Kirkemo acknowledged that:
Most of the complications caused by the suburethral tapes could be attributed to the biomechanical properties of the mesh used and to other factors such as tissue healing, infection and the surgical technique used.1727
1705 He noted that “major complications” associated with tapes had been reported in the literature, including bladder injury, urethral erosion, tape tension, bowel injury, foreign body reactions (causing, among other things, late onset erosions), urethral diverticulum (a condition in which a pocket, sac or pouch forms in the urethra); vascular injuries, nerve injuries, sexual function, hematoma, urinary retention, haemorrhage, vaginal injury, thigh abscess, and fistula. He then proceeded to summarise the reports of a number of studies (mostly case studies) where those risks were discussed.
1706 This discussion was followed by: a section on “TVT in special cases” (obese women, elderly women, and women who underwent vaginal delivery after TVT); a review of comparative studies of TVT and TVT-O, noting the nature and rates of complications; and a reference to studies examining risk factors for determining the success of treatment.
1707 The CER referred to the findings of the Ogah et al (2009) Cochrane review and the entire review appears to have been appended to the CER, along with a copy of the report of the TOMUS study by Richter et al (2010).
1708 In each case, the CER did little more than offer brief summaries of the selected articles. While adverse events reported in those studies were described, there was little to no evaluation of their significance or any discussion as to whether (and, if so, how) the adverse events should be addressed, such as through changes to the design of the product or to the content of the IFU. On the face of the report, it is not apparent why these complications were considered acceptable.
1709 Despite the reported complications, the conclusion was the standard one, namely, that the data revealed in the literature were “sufficient to demonstrate compliance with the essential requirements covering safety and performance” “under normal conditions of use” and no additional clinical data were required.1728 It is not readily apparent why Dr Kirkemo reached this conclusion, particularly when he had quoted a passage from the Ogah et al (2009) Cochrane review in which it was noted that “[m]ost of the trials had short term follow up and the quality of the evidence was variable”1729 and the TOMUS study only measured outcomes up to 12 months.
1710 The literature review was followed by a review of Ethicon’s complaints data and “a systematic review” of the MAUDE database.1730 If any conclusions were drawn from this review, they were not included in the report.
1711 It will be recalled that the European Directive required that clinical investigation” be performed for implantable devices “unless it is duly justified to rely on existing clinical data”: European Directive, Annex X § 1.1a. The methods of clinical investigations are set out in § 2.3 of Annex X, where it states that:
2.3.1. Clinical investigations must be performed on the basis of an appropriate plan of investigation reflecting the latest scientific and technical knowledge and defined in such a way as to confirm or refute the manufacturer's claims for the device; these investigations must include an adequate number of observations to guarantee the scientific validity of the conclusions.
2.3.2. The procedures used to perform the investigations must be appropriate to the device under examination.
2.3.3. Clinical investigations must be performed in circumstances similar to the normal conditions of use of the device.
2.3.4. All the appropriate features, including those involving the safety and performances of the device, and its effect on patients must be examined.
1712 Dr Allman said that reliance on the TVT data was unjustified since the procedure used with TVT Exact was different in that it involved one less cystoscopy. He added that a conclusion that review of safety and efficacy data for TVT was sufficient to assess TVT Exact would require a more critical assessment of the differences between the devices and the impact, both good and bad, on clinical use.1731 As for the conclusion based on the literature review, Dr Allman correctly observed that:
This statement simply follows the summary and review of the literature review. There is no argument to support the conclusion and there are no criteria to support the assertion that the EXACT device meets the essential requirements. Furthermore, the conclusion depends upon an assumption of equivalence between the EXACT device and the devices used in the studies reported. The EXACT device is designed to be an improvement over those devices and uses a different implantation procedure. It is not necessarily equivalent and equivalence was not demonstrated.1732
1713 As the applicants submitted, although the report identified a number of complications with TVT and TVT-O, it did not include any consideration of the extent to which those complications were likely to arise with TVT Exact. Furthermore, as the applicants also submitted, the report assumed that the complications were acceptable and that no additional clinical data was required without indicating the basis for the assumptions or why they were reasonable. Indeed, the conclusion following the literature review did not reveal what factors were taken into account to determine compliance with the essential requirements of safety and performance.1733
1714 The review of complaints and adverse events did not include complaints generated during clinical trials or in feedback as part of a product registry. The complaint rates recorded from Ethicon’s own records were exceedingly low in comparison with the incidence of the various complications recorded in the medical literature. The rate for erosions for example was 0.00008%. The applicants argued that the differences in the incidence of complications disclosed in the medical literature and the rates in Ethicon’s complaints data should have, but apparently did not, trigger concern about the adequacy of Ethicon’s complaint reporting system.1734 There is force in that argument.
TVT Abbrevo
1715 It will be recalled that TVT Abbrevo is an obturator sling like TVT-O, but the tape is significantly shorter than the tape used in TVT and TVT-O and the instruments are different.
1716 The target market for TVT Abbrevo consisted of gynaecologists, urogynaecologists and female urologists who wanted to minimise the amount of mesh in the sling, which Ethicon recognised would reduce the risk of “unintended consequences”.1735 According to Mr Viano’s presentation, it would deliver “less post-operative pain”.1736 Reduction in pain was attributed to “82% less mesh in active adductor muscles” and “32% less mesh overall”.1737
1717 A biocompatibility assessment completed by Dr Sandy Savidge on 15 March 2010 stated that the device consisted of materials with “acceptable biocompatibility test results for this application, or have been used in predicate devices that have a combination of clinical history of safe use as well as acceptable biocompatibility testing”. Based on the clinical history and the biocompatibility testing, Dr Savidge concluded that TVT Abbrevo did not represent a significant biocompatibility risk for patients.1738
1718 The technical file includes a synopsis of preclinical data prepared by Dr Hinoul “in support of TVT Abbrevo’s equivalence to TVT-O”.1739 The report is undated. Dr Hinoul examined the similarities and the differences between the two devices. Notwithstanding the significant differences in the amount of mesh used in the two devices, he concluded that “substantial equivalence” in efficacy could be demonstrated based on the following matters:
1. the long clinical history of the GYNECARE TVT mesh tapes
2. the proven equivalence in clinical efficacy demonstrated in robust meta-analyses comparing GYNECARE TVT with GYNECARE TVT Obturator System
3. the fact that the GYNECARE TVT ABBREVO mesh is identical to the GYNECARE TVT Obturator mesh ensuring an identical foreign body response for both short term and long term tissue integration
4. the preclinical evidence that TVT ABBREVO consistently perforated the critical structures providing fixation (the obturator externus muscle, the obturator internus muscle and the obturator membrane) along the same consistent pathway as the TVT Obturator System
5. the findings from initial pull out studies demonstrating that the TVT ABBREVO could not be pulled out of the obturator tissues at supra-physiological forces, predetermined at 164 grams
6. the almost identical procedural steps to perform the TVT ABBREVO in comparison to the well established and externally validated TVT Obturator System technique1740
1719 The pre-market CER for TVT Abbrevo, signed on 17 August 2010 by Dr Hinoul,1741 identified the rationale for the device under the heading “Claims”:
To reduce the amount of mesh left behind in the body, to reduce the pain possibly caused by the tape’s presence in the adductor muscles and to further improve the ergonomics of the original procedure.1742
1720 Those claims were intended to be met by modifications to TVT-O which were said to lead to a reduction in tissue trauma, and a reduction in the total length of mesh left behind in the body (to a total of 12 cm).
1721 The report also claimed that the efficacy of TVT Abbrevo was supported by “preclinical and clinical evidence for both the foreign body response to the mesh graft placed in the body as well as short-term and long-term fixation of the graft”.1743
1722 The preclinical data consisted of cadaveric anatomic studies comparing TVT Abbrevo to TVT-O, demonstrating equivalent placement and initial fixation. Long-term fixation was expected to be similar since the two devices used the same mesh and passed through the same critical structures.
1723 The report also stated that the clinical evaluation was based on an assessment made through a combination of:
• A compilation of relevant scientific literature that is currently available, as well as a written report containing a critical evaluation of this compilation.
• The results of clinical investigations relevant to the device’s prototype.1744
1724 The report largely relied on data relating to TVT and TVT-O. The long-term data was summarised as follows:
Long-term data for the predicate device (TVT-Obturator System) has shown that treatment using the TVT-O sling offers durable results with objective success reported in 88.4% after 3 years (Waltregny, 2008) and 82.4% after 4 years (Liapis, 2010). For the TVT procedure, using the same mesh material, 11 year data have been published, reporting a success rate of 90% (Nilsson, 2008).1745
1725 It also relied on findings from an unpublished single-centre study conducted by Professor de Leval at the University of Liège in Belgium which was said to have demonstrated “similar efficacy for the prototype of the GYNECARE TVT ABBREVO when compared to TVT-Obturator System after one year of follow-up”. At the time, the study was unpublished but had apparently been accepted for publication by the International Journal of Urogynecology and Pelvic Floor Dysfunction, subject to some undisclosed revisions. A draft of the manuscript was annexed to the report.1746
1726 The Belgian study was a randomised, single-centre, single-blinded, prospective trial. According to the draft manuscript by Professor de Leval, Alexandre Thomas and Dr David Waltregny annexed to the CER, 87 women suffering from stress urinary incontinence were recruited in the “original” (TVT-O) procedure and 88 women in the “modified” (TVT Abbrevo) procedure. All procedures were performed by Professor de Leval. Of the total, 170 women were followed up at one year, with cure rates of 91.7% for TVT-O and 90.7% for TVT Abbrevo (p=0.824). The incidence and intensity of groin pain was reportedly higher in the original procedure group at day 0 (p=0.003) and day 1 (p=0.011), but not afterwards. One year after their procedures three to four percent of patients in each cohort still complained of groin pain. The authors concluded that the “modified” inside-out transobturator tape procedure was as efficient and safe as TVT-O with less immediate postoperative groin pain.1747
1727 At the same time, however, de Leval et al acknowledged that it was “obvious”, that their “data originating from a single-center, single-surgeon, randomized study should be repeated in a multi-center multi-surgeon context for external validation”.1748 In these circumstances, it should have been obvious to the respondents that it was premature to put the device on the market before the data was validated in this way.
1728 In the CER Dr Hinoul reviewed available literature from “2004 through early 2010”, including the Ogah et al (2009) Cochrane review, and concluded as follows:
The above literature review, taken together with the available pre-clinical data and the clinical data regarding the prototype of the GYNECARE TVT ABBREVO, are sufficient to demonstrate compliance with the essential requirements covering safety and performance of the GYNECARE TVT ABBREVO™ under normal conditions of use. No additional clinical data is required.1749
1729 He did not, however, offer any reasons to support his conclusions. He did not address the significance of the reported complications (associated with the use of TVT, TVT-O and TVT Secur) or explain how the data in relation to those devices could support the use of the new device. Nor did he explain why no additional clinical data were required in the face of the acknowledgment by Professor de Leval and his fellow authors that the results of the Belgian study needed to be validated by a multi-centre, multi-surgeon study.
1730 Ethicon conducted a “biocompatibility risk assessment” of TVT Abbrevo.1750 As part of this assessment, however, no additional testing on the mesh was performed. Instead, the assessment drew on the “long history of safe clinical use” of Prolene mesh in the suture and the other SUI devices.
1731 Once again, Dr Allman considered that the conclusion of the CER was unjustified by the clinical evidence presented and the report did not satisfy the European regulatory requirements for clinical evaluation.1751 He explained that:
It is based on a claim of equivalence to a device for which the ABBREVO is intended to be an improvement, a study with an early version of the device, and a meta-analysis of surgical approaches to stress urinary incontinence. Normal conditions of use would not usually be interpreted as use by the surgeon who first described the procedure and who developed the device. No criteria are given to justify claims of conformance to the essential requirements. The report does not meet the objectives stated in the report and does not meet the requirements of MEDDEV 2.7.1...1752
1732 Dr Allman said that Ethicon should have conducted clinical investigations of TVT Abbrevo, with clearly defined acceptance criteria before CE marking or considered post-market clinical follow-up studies.
1733 None of these opinions was challenged in cross-examination. They should be accepted.
1734 By October 2010, when TVT Abbrevo was launched in Australia, no further clinical trials of the device had been conducted.
The POP devices
1735 The POP devices were developed against the background of opinion that traditional methods of treating uterine prolapse were unsatisfactory, that there was a high incidence of recurrence, and patients complained of sexual, bladder and bowel dysfunction after anterior or posterior repair.1753 Some surgeons were experimenting with the transvaginal use of polypropylene mesh, including Prolene, in prolapse repair.1754
1736 On 16 May 1997 Ethicon held a meeting in London with three surgeons, during which each recounted their experiences with the use of mesh for repair of pelvic floor disorders. The consensus of the meeting, according to Ethicon’s Dr James Browning, was that: the concept of mesh repair for vaginal or uterovaginal surgery was valid, but that it should be tested in a wider European and US “arena”; a comprehensive publication database should be compiled; pre-clinical work should focus on the mechanism of repair and the time needed for support; and observational clinical work could be undertaken using currently available Prolene mesh. Support was expressed for a strategy involving early marketing of a Prolene mesh and the simultaneous development of a “2nd generation prolapse mesh”. Importantly, Dr Browning reported that the meeting noted that “Prolene was the best of the currently available meshes and was felt to be adequate if not ideal to start clinical trials right now”. 1755
1737 Less than 10 months later, on 16 March 1998, and although no clinical trials had begun, let alone finished, Dr Browning signed a clinical evidence report pronouncing the use of Prolene for the tension-free repair of uterovaginal prolapse to be “safe and efficacious”. This conclusion was said to be based on “large-scale informal clinical use over the previous decade” and a review of the clinical literature.1756 The report was five pages long excluding references. But for the double-spacing and the generous gaps between paragraphs it would have been much shorter. It included no critical discussion of the literature. The only studies that were mentioned (four in number) had significant limitations. They and the other problems with this CER are discussed below.
1738 On 1 May 1998 Ethicon amended the technical file for Prolene mesh to include an indication for uterovaginal prolapse.1757
1739 In June 1998 Ethicon launched Gynemesh, a Prolene mesh for repair of anterior prolapse. According to an internal company discussion paper dated August 1998 written by its then Product Development Manager, Rowan Norrie, it was recognised at the time that Prolene was “far from the ideal material for this indication”.1758 Despite this, the discussion paper records, the decision was taken to launch the product for the following reasons:
• To raise awareness of the possibility of using a mesh for prolapse repair;
• To gain entry into this growing market before competitors;
• To spend time seeking out key surgeons as product champions and
• To allow time to carry out further market research into what the ideal product for this indication might be.
1740 In the short period it had been on the market, the discussion paper noted there were a number of concerns had been raised about the product, chief among which were:
• Fear of rejection and problems with removing mesh at a later date;
• Fear of infection;
• Concern about mesh eroding into bladder or rectum;
• Concern about the stiffness of the mesh and the risk of the mesh protruding through the vagina.
1741 The issues of rejection and mesh removal, however, were not considered in the Gynemesh CER.
1742 The parameters of “a successful outcome” were defined in the discussion paper as follows:
• Reduced rate of recurrence
• Immediate reduction of bulge
• Improved bowel and bladder function
• Less dyspareunia
• Reduced hospital stay
• Reduced time in theatre
• Ease of use
• Minimum risk of complications such as rejection
• No increase in the infection rate
1743 On 6 April 2000 Ethicon Inc. lodged with the FDA a pre-market § 510(k) notification of its intent to market “Prolene Soft (Polypropylene) Nonabsorbable Synthetic Surgical Mesh”1759 for use in “the repair of hernia or other fascial defects that require the addition of a reinforcing or bridging material to obtain the desired surgical result”.1760 The premise for the notification was twofold: that it was a modification of Prolene and was substantially equivalent to Prolene mesh in material, composition, and indications; and that it was also substantially equivalent to Mersilene mesh in its indications.1761 Mersilene, it will be recalled, was made of polyester, not polypropylene.
1744 No additional biocompatibility tests were performed.1762 Additional preclinical testing was deemed unnecessary on the basis that preclinical performance had been established for Prolene and Mersilene meshes and Prolene Soft mesh has the same indications statement.1763
1745 On 23 May 2000 the FDA confirmed in writing that it had determined that Prolene Soft was substantially equivalent for the indications for use and cleared the device for marketing “subject to the general controls provisions of the [Federal Food, Drug, and Cosmetic] Act”, which it pointed out included “requirements for annual registration, listing of devices, good manufacturing practice, labelling, and prohibitions against misbranding and adulteration”.1764
Gynemesh PS
1746 On 22 June 2001 Ethicon held a meeting of its Scientific Advisory Panel to discuss the use of mesh for pelvic floor repair. The minutes of the meeting reveal that porosity was identified as critical for tissue ingrowth and contraction of tissue needed to be avoided in order to avoid erosion.1765 The consensus of the meeting was that erosion was a risk, possibly a response to infection, typically seen by three months, usually six to 12 months, but could present late (three years was nominated). Erosion into the vagina was described as “not a good situation”, erosion into the bladder, urethra or rectum as “a very bad situation”. Both the volume and the type of material were said to play a role.
1747 Amongst the “hurdles” was the recognition that “vaginal wall characteristics [were] not well understood”. The Panel requested that a clinical study be conducted including short-term three or six month safety assessment for complications with a minimum one year primary effectiveness endpoint with cystocele and rectocele separately evaluated.1766
1748 Ethicon also commissioned market research to understand current surgical practice regarding mesh usage and to “gain perceptions about a new mesh product concept”. Thirty surgeons were surveyed. All were based in the United States. Twenty-seven were urogynaecologists and three urologists.1767 The top five desired attributes were: functional strength in vivo; erosion resistance; infection resistance; promotion of tissue-in-growth; and the ability to be sutured/use fixation.1768 The two attributes chosen by most respondents were infection and erosion resistance. The author of the report, dated 15 August 2001, proceeded to explain:
• Infection with meshes is a common concern for surgeons because they are implanting meshes in people who are particularly sensitive to infections and there is concern that the mesh would have to be removed if this occurs.
• There is concern over erosion through the anterior wall of the vagina and one respondent mentioned that the urethra and sacrum are both sensitive structures and any erosion in those areas would be ‘catastrophic’. Another commented that she avoids using mesh whenever possible due to the risk of erosion. Many have seen cases of erosion with meshes (particularly Gore-Tex). And some have had to explant a mesh for this reason.
• In response to question 6 the benefits of low risk of erosion and infection were given as related to specific mesh products and not generically to all currently available mesh products. Therefore, low risk of infection and erosion is not a benefit that is currently present with mesh and this is what respondents want to see in any new mesh materials.1769
1749 The Gynemesh PS § 510(k) notification proceeded on the basis that the device was identical in material and composition to Prolene Soft and Prolene and, although it was made from a different polymer, substantially equivalent to Mersilene. The purpose of the notification was to enable a modification to be made of the indication for use from “the repair of hernia or other fascial defects that require the addition of a reinforcing or bridging material to obtain the desired surgical result” to “a reinforcement of fascial structures of the pelvic floor in the vaginal wall prolapse when surgical treatment is intended either as mechanical support or bridging material for the fascial defect”.1770 The contraindications and adverse reactions were the same as for Prolene Soft.1771 No additional biocompatibility tests were carried out.1772
1750 The clinical studies relied on as “performance data” in support of the revised indication relied on meshes other than Prolene Soft, that is meshes of a different composition from Gynemesh PS, and save in one case involved very small patient populations (ranging from 12 to 57 patients).1773
1751 Ms Holland gave unchallenged evidence that in the technical file for Gynemesh PS no consideration was apparently given to the change in indication (from treatment for hernias in the abdominal wall to repair of pelvic organ prolapses). At the very least, Ms Holland said, the new indication should have been addressed in the risk evaluation. In fact, the new indication was not “addressed as significant” in any of the submission files, despite the vast differences between Prolene sutures, hernia mesh, and Gynemesh PS.
1752 Ms Holland considered that, if there had been a focus on indication and anatomical location, rather than an assumption made that all human tissue reacts in the same way, then a controlled study would have been performed to test the claims of equivalence. Such a study was carried out by Fan et al (2014).1774 This was an animal study involving 40 rabbits, 20 of which were implanted with polypropylene mesh and 20 with porcine-derived, cross-linked urinary bladder matrix (cUBM) materials. An erosion reaction of 67% occurred when the polypropylene mesh was used vaginally and there was no sign of erosion when it was used abdominally. Further, the polypropylene mesh showed a stronger chronic inflammation response than the cUBM. Ms Holland said that a review of biocompatibility studies found that there was a lack of information in support of the Prolene mesh products.
1753 The pre-market clinical evaluation report on Gynemesh PS was signed by Dr Weisberg on 20 September 2002. Excluding the title page, table of contents, and the endnotes listing the references, it is three pages long. The discussion of the “clinical evidence” and “potential complications” appears to have been cut and pasted from the Gynemesh CER discussed above. The differences between the two in these respects are inconsequential.
1754 The description in the 2002 Gynemesh PS CER of the physical characteristics of the device included the assertion that Gynemesh PS “affords excellent strength, durability and surgical adaptability with sufficient porosity for necessary tissue ingrowth”.1775 No reasons were given and no clinical data were cited.
1755 The report’s conclusion was that Gynemesh PS “appears to be safe and efficacious on the basis of large scale use of predicate devices over the last decade and a review of the clinical literature”.1776 Despite the endorsement of this conclusion that appeared in his affidavit, Dr Hinoul conceded in cross-examination that “the report and the material to which it referred did not represent a sufficient and adequate basis on which to come to a conclusion that the use of the mesh [for] the indication identified was safe and efficacious”.1777
1756 The differences between Gynemesh PS and the predicate devices were not examined.
1757 No reference was made to any clinical tests or to the studies of Prolene Soft hernia mesh. The only studies to which reference was made related to other meshes (Marlex, Prolene, and Vicryl). They are the same studies relied upon to support the conclusions in the Gynemesh CER. Here, as there, no critical analysis was undertaken of any of the publications mentioned.
1758 The Marlex study was the subject of an article by Dr Thomas Julian, published in 1996,1778 six years before the CER. It appears to be the only one of the referenced articles that found its way into evidence in this proceeding. Professor Hu said it was his understanding that, before 2003, this was the only prospective clinical trial that formally compared the outcome of pelvic organ prolapse repair using polypropylene mesh with native tissue repair.1779 He was not challenged about this in cross-examination. In this trial, twenty-four patients with severe recurrent anterior vaginal wall prolapse were recruited to a trial and divided equally into two groups. There is no suggestion that this was a randomised controlled trial. In the absence of randomisation, Professor Gordon pointed out that differences in recurrence rates, for example, could not be attributed to the different treatments used.1780 Three of the 12 in the mesh group (25%) had mesh-related complications, all noted within six months of surgery. The applicants submitted that these were all erosions but on close inspection of the article that appears not to be the case. Contrary to an assertion made in the CER, however, which indicated that there were no erosions, there was in fact one case of erosion, which was preceded by granulation1781 and one case of exposure which caused “a lightning-like sensation” on the penis of the patient’s sexual partner caused by two tiny perforating mesh fibres.1782 Neither of these findings was mentioned in the CER. Hispareunia was not included as a potential complication. Importantly, Julian recommended against the use of the mesh as a primary procedure for anterior repairs, reserving it for patients who have had two or more “reparative failures”.1783 This recommendation was not mentioned in the CER either.
1759 The Vicryl study was by Drs Lobel and Sand. The results of the study were apparently presented to an annual meeting of the International Urogynecological Association held in Amsterdam in 1997. An abstract was tendered in evidence1784 but no peer-reviewed journal article. The abstract revealed that this was a two-armed study but not a randomised controlled trial. The selection and exclusion criteria are obscure. Only 39 patients received the mesh. The non-mesh arm was larger (44 patients) and had a “significantly higher grade of preoperative cystocele”. Professor Gordon said that “such a difference at baseline makes the interpretation of any mesh/non-mesh comparison invalid”.1785 The authors concluded that the use of that mesh in anterior colporrhaphy was safe and appeared to decrease the recurrence of central cystocele. There were apparently operative and postoperative complications but the authors did not disclose what they were. Rather, they stated that none of them could be attributed to mesh placement. No explanation for this conclusion was given. The follow-up period was variable, ranging from six to 42 months. Neither the number lost to follow-up nor the arm to which those lost belonged was disclosed.
1760 Be that as it may, unlike Gynemesh and Gynemesh PS, Vicryl was an absorbable polyglactin 910 knitted mesh. Its relevance to Gynemesh is obscure and it was not explored in the CER.
1761 The CER referred to only two studies of the use of Prolene. The first was Foote et al (1997). It was said to describe the use of Prolene in primary posterior vaginal repairs. The only reference to outcome is a statement that “[a] 93% cure rate at 6 months was reported in 29 women”. Complications, if any, were not mentioned. Two references are cited in the report to the Foote study. One is a “personal communication awaiting publication”. Professor Gordon expressed the opinion that evidence of this kind, not available for scrutiny, should be regarded as having “low quality” and “cannot really support a conclusion”.1786 The only report of the study in evidence was that which Professor Gordon found. It is an abstract from papers published by the International Urogynecology Journal in 1998. As Professor Gordon observed, the abstract reveals that the study involved 16, not 29 women.1787 The reported cure rate at six months’ follow-up was 87% (13/16). Short term complications occurred in three patients (urinary tract infections in two and vaginal infection in one). At six months two women had “suture/mesh protrusion”, which was reported as having been “satisfactorily trimmed” in the outpatient clinic.
1762 Professor Gordon concluded that the references to the Foote article in the CER were in fact to an article footnoted by Foote et al (1997) in the abstract.1788 This was a study by Mahendran et al (1996).1789 That study involved 29 women who were implanted with Prolene mesh where a cure rate of 27/29 (93%) is reported. Notably, however, this was a study of women who underwent laparoscopic sacrocolpopexy.
1763 Professor Gordon was not challenged about his conclusion and, in the absence of any evidence to suggest he was mistaken, I accept it. It appears that Dr Browning made an error in the Gynemesh CER which was transposed into the Gynemesh PS CER.
1764 The Cervigini study used Prolene Soft, but the size of the cohort was also very small. It involved 35 patients. The period of follow-up is not apparent from the CER. Nor are the details of the study.
1765 Notwithstanding their limitations (none of which are discussed in the CER) by reference to these studies and “other literature”, which Dr Weisberg did not identify, the conclusion of the CER was that “the use of polypropylene mesh for vaginal prolapse surgery is safe and efficacious”. Professor Gordon insisted that judgments about the efficacy of a medical intervention in medicine must be based on well-designed randomised controlled trials. Since Dr Weisberg did not cite such evidence, Professor Gordon said that Dr Weisberg’s conclusion regarding efficacy was not well-founded.1790
1766 Ms Holland was also critical of the use to which these studies were put. She said that such a conclusion could not be justified on the basis of sporadic studies with small sample sizes using different material.1791
1767 The nature and extent of the literature search was not disclosed. The CER did not refer to any of the research undertaken by Professors Klosterhalfen or Klinge although it included work on Prolene Soft itself. With good reason, Professor Gordon described Professor Weisberg’s use of evidence as selective and consequently of limited value.
1768 Only three potential complications were identified: infection; edge extrusion; and fistula/erosion. This is the entire text of the section on potential complications.
In placing polypropylene mesh beneath the vaginal skin and close to the lower urinary tract, pouch of Douglas and rectum, the following complications should be considered:
(a) Infection
As this is foreign body material, and passing through a contaminated field, this is a theoretical risk. In the TVT series, PROLENE mesh placed through the anterior vaginal wall was associated with zero infection/rejection in larger series, the rate appears to be below 1%.
(b) Edge Extrusion
This was described by Foote in 7 out of 29 patients: the mesh was trimmed in the outpatient clinic and the problem resolved. Cervigni reports this in 1 out of 35 cases.
(c) Fistula/Erosions
Vaginal erosions are a well known complication associated with the use of synthetic materials for pelvic organ prolapse repairs. In none of the above series is this complication described. Experience with GYNECARE TVT Tension-Free Support for Incontinence has indicated few reported incidences of this complication.1792
1769 The reference given to “the TVT series” in paragraph (a) above was to Cervigni’s article on the use of Prolene Soft in the repair of anterior vaginal prolapse. This, too, must be an error. No reference is given for the statement in paragraph (c) that experience with TVT indicated few reported “incidences” of fistulas or erosion.
1770 In all the circumstances, I conclude that Dr Weisberg signed this CER without independently reviewing the literature upon which it was based.
1771 In his affidavit Dr Hinoul said the report considered potential complications, including infection, edge extrusion, and fistula/erosion. As Dr Allman observed, the complications were described, but they were not considered. 1793 As Dr Allman also observed, the report contained no discussion of the rationale for accepting the risk of these complications apart from a statement that in the publication describing edge extrusion (reporting on a study of 29 patients), the mesh was trimmed and the problem resolved. Neither was there a discussion about whether it was necessary to evaluate the rate or severity of these complications with respect to Gynemesh PS.1794
1772 Norwas any attention paid in the Gynemesh CER to the difference between the amount of mesh necessary to prevent a recurrence of prolapse and that necessary to prevent a recurrence of stress urinary incontinence, despite the fact that three years earlier in the book published in the wake of the 1998 Suvretta meeting Professors Klinge and Klosterhalfen had emphasised that tissue reaction is dose dependent.1795 Nor did the CER refer to Professor Klosterhalfen’s report, prepared for Ethicon’s use, sent to Ethicon in September 2000 one of the key findings of which was that Prolene Soft was “not a large pore mesh”.
1773 In its early work, the TVM Group used a standard Prolene mesh, undertaking median longitudinal colpotomies on the anterior and posterior vaginal walls. After an analysis of the first 100 procedures revealed an erosion rate of 17.5%, however, the TVM Group switched to Prolene Soft (Gynemesh PS) and made various adjustments to the technique, including eliminating the colpotomy. The erosion rate fell to 2.7%.1796 The Group attributed the reduction in erosions to the absence of scarring opposite the mesh. The prosthesis used by the TVM Group resembled what became Prolift Total but, unlike the Ethicon devices, the TVM prosthesis was designed to be cut, as required, if only a single compartment repair was necessary.1797 The work of the TVM Group is therefore relevant to both Gynemesh PS (which is cut to shape by the treating surgeon) and Prolift (which is supplied with a pre-cut mesh). Yet, the 2002 CER on Gynemesh PS did not refer to any of the findings of the TVM Group or include any advice about a colpotomy.
1774 Dr Arnaud reviewed Dr Weisberg’s draft report.1798 In an email to Dr Weisberg on 13 October 2002, apart from some typographical errors, he described it as “perfect for me”. He had only one qualification. He was concerned about what Dr Weisberg had said about “Potential complications/Fistulas&Erosions”. He wrote:
This is a problem which arises rather commonly in practice even with polypropylene and it might be wise to be more elusive on this.1799
1775 On comparison of the signed report with the draft, it appears that, after this email, the first sentence was inserted at the beginning of the brief discussion under the heading “Fistula/Erosion”:
Vaginal erosions are a well known complication associated with the use of synthetic materials for pelvic organ prolapse repairs.
1776 Dr Allman was critical of the CER, finding it to be of poor quality.1800 He said that not only did it fail to advert to the essential requirements of the European Directive but it also failed to conform to them. He noted in particular that it failed to describe the method used to identify the relevant scientific literature and in these circumstances it was impossible to determine if all the relevant literature had been identified. Having regard to the age of the articles (from five to 10 years before the CER), however, he considered it unlikely. The report cited only eight unique references, one of which was a “communication” without giving the details necessary to identify it.
1777 Dr Allman also pointed out that there was no analysis to justify the relevance of the results drawn from the four publications that were briefly discussed in the report to the specific properties of, and specific indications, for Gynemesh PS (which included abdominal as well as transvaginal placement) in normal conditions of use. Consequently, his opinion was that the conclusion that its use appeared to be safe and efficacious for pelvic floor repair was not justified and the CER was insufficient to justify CE marking of Gynemesh PS.1801
1778 During cross-examination of Dr Allman the respondents drew attention to a letter signifying that “the” technical file for Gynemesh PS was sent to BSI on 20 March 2003.1802 Since the letter did not refer to the CER, however, or even to the “complete” technical file, I cannot be confident that the CER was included, although it would be surprising if it was not. Even so, there is no evidence of the review undertaken by BSI. Neither the fact that BSI was sent the file nor, assuming it received it, the absence of evidence that it did not find fault with the CER assists the respondents, not least because the uncontradicted evidence is that the role of the notified body is limited to auditing the quality management systems. While it might examine a CER to see whether it conforms to the essential requirements, the evidence indicates that that is not commonly done. Having regard to the conclusions BSI reached following its review of Ethicon’s technical files in 2012, discussed in Part IX below — that for various reasons Ethicon’s CERs did not meet the essential requirements — it is most unlikely that it was done in this case.1803
1779 In his summary of the biocompatibility risk assessment for Gynemesh PS in a 2002 memorandum Thomas Barbolt of Ethicon’s Research and Development team wrote:
[T]he preclinical study results and the extensive clinical experience with current PROLENE mesh, and natural and blue PROLENE suture demonstrate that this polypropylene base material, with or without copper phthalocyanine blue pigment, is intrinsically safe and without significant adverse effects for patients. It is considered that GyneMesh PROLENE Soft Mesh manufactured with PROLENE Soft Mesh will result in the same level of safety demonstrated by the currently marketed products, and no further preclinical testing is necessary.1804
1780 Dr Allman observed that it makes little sense to describe a material used as an implantable device as “intrinsically safe”.1805 As he said, and the expert biomaterials evidence unequivocally shows, whether or not an implantable material is safe depends in part on where in the body it is used and what it is used for. A material used safely for one purpose in one location is not necessarily safe when used in other locations or for another purpose. I have already discussed the differences between the Ethicon devices, Prolene sutures, and hernia mesh.
1781 The applicants submitted that a clinical study should have been undertaken before Gynemesh PS was taken to market. Ms Holland testified that any assessor would realise that, for a new intended use in a totally new environment in the body, additional testing should have been performed.1806 In her first report she was critical of the DDSA, performed on 13 December 2002, which ignored this. She wrote:
The DDSA relied entirely on the PROLENE Soft Mesh utilised for hernia repair including, but not limited to biocompatibility and biostability data. Of special interest within the DDSA is the fact that although the GYNEMESH PS was for an entirely new intended use, no special training of the intended user was indicated; in fact it indicated that the IFU was sufficient for identification of the critical steps in setting up and operating the device for all intended users.1807
(Original emphasis)
1782 Ms Holland pointed out that seven potential hazards for the new mesh were identified in the DDSA and all were given risk levels of III or below, requiring no further actions, and accompanied by the comment that a clinical study design would assess parameters. But no clinical study was submitted with the technical file at the time of the request for CE marking.1808
1783 In fact a clinical study was begun before the CER was signed but the respondents did not wait for the results before applying for CE marking. The Gynemesh PS technical file includes a notation that a clinical study was in progress but was being “conducted as a post-launch activity”.1809 No reason was offered in evidence to explain the decision to launch the product before the results of the study were available.
1784 Despite the concerns expressed about erosion and the recognition of erosion as a potential adverse reaction, erosion did not appear in the original Gynemesh PS IFU. I note this IFU was not in evidence, presumably because it was not supplied with the devices in Australia. Mention of erosion was added in 2003. Its omission from the original IFU is vexing. In a file note dated 21 February 2003 Dr Weisberg expressed the view that this represented a minor change and did not invalidate the completed design validations performed for the original US submission. He said that it was commonly understood by clinicians that erosion is a potential adverse event.1810 Whatever the true position may have been, it was obviously considered a necessary inclusion. Otherwise the amendment would not have been made. In any case, Ms Holland remarked and I accept, particularly in the absence of expert evidence to the contrary, that:
The belief that residual risks should not be mitigated in some way, at the very least by the labeling, is contrary to the intent of ISO 14971 and industry practice.1811
Prolift
1785 On 28 February 2003 Professor Jacquetin filed an application in the US patents office to patent the TVM method and apparatus for treating pelvic organ prolapses in female patients described in the abstract in the following way:
An anterior implant adapted to treat central and lateral cystoceles present in a female patient includes laterally extending stabilizing straps for supporting the implant between the patient's bladder and vagina independently of the patient's arcus tendineous fascia pelvis. Rectocele and hysterocele repairs can be carried out using a single posterior implant which, like the anterior implant, is provided with laterally extending stabilizing straps for supporting the implant between the patient’s rectum and vagina.1812
1786 Ethicon planned to capitalise on the clinical data from the planned TVM clinical study.1813 It established its own project, which became known as Project D’art, the objective of which was “to provide a collection of single-patient use tools and implants for use in the repair of vaginal vault prolapse and anterior and posterior vaginal defects”, enabling transvaginal access and fixation of the implants to “appropriate pelvic floor structures”.1814 The goal was to provide a pre-cut mesh insert together with tools for a standardised set of pelvic floor repair procedures.1815 The intended users were to be urogynaecologists, gynaecologists, and urologists with experience in pelvic floor repair to whom training would be available.1816
1787 A draft product description document dated 30 July 2003 identified the following requirements under the heading “biocompatibility”: that the mesh material must cause an acceptable foreign body reaction to facilitate connective tissue infiltration and incorporation by surrounding tissue, and that it must be biocompatible with the host tissues. It also stipulated that “labelling and Instructions for Use shall contain sufficient information to ensure safe and proper use of the product”.1817
1788 It also contained the following so-called risk analysis:
Similar risk of erosion to products and surgeries currently marketed for pelvic floor repair.
Similar risk of infection to products and surgeries currently marketed for pelvic floor repair.
Similar recurrence rate to existing repairs.1818
1789 It is impossible to understand how these conclusions were possible at such an early stage of development.
1790 The plan was to start both European and US studies, it seems, by September 2003; to make a submission to the regulator for “approval” to market the device by January 2004 on the basis that it was substantially equivalent to another Ethicon product; to secure CE marking in August 2004, and to launch the device by September 2004, a mere two months after the six month data became available.1819
1791 At this stage, based on her own experience and knowledge of the literature, Associate Professor Rosamilia conceded in cross-examination that there was insufficient evidence to support the use of permanent meshes or grafts at the time of vaginal repair surgery, except in the context of adequately powered randomised controlled clinical trials.1820
1792 As Ethicon continued to work on Project D’art, patients were recruited to two clinical trials of the TVM technique sponsored by Ethicon. One was a non-randomised non-controlled observational study conducted at eight sites in France (the French study). Professor Michel Cosson, a member of the TVM Group, was the principal investigator. The objective was to demonstrate the “usability” of Prolene Soft for anterior, posterior and vault prolapse repair using the TVM technique. The plan was to recruit 90 female patients in order to obtain an “evaluable” group of 82 patients. Ninety patients were enrolled and all 90 underwent surgical correction of their prolapse with TVM.1821
1793 The study protocol for the French study included the following selection criteria:
1. Patient was a candidate for anterior and posterior surgical repair of the pelvic floor. Required to have a symptomatic prolapse of at least International Continence Society (ICS) Stage Ill as defined in Appendix A of the study protocol (Appendix 16.1.1).
2. Patient was at least 21 years of age.
3. Patient did not wish to have more children.
4. Previous or simultaneous hysterectomy. Uterus not retained.
5. Absence of uncontrolled diabetes.
6. No coagulation abnormality.1822
1794 The protocol also stipulated that the trial would be deemed a failure unless the rate of prolapse after the procedure (with an upper confidence level fixed at 90%) was less than 20%.
1795 The first patient was screened on 23 January 2004 and the last some two years later.1823
1796 Another clinical study was started in the United States (the US study), again under Ethicon’s sponsorship. Like the French study it was a single arm study and it used Gynemesh PS. It was conducted at three sites in the US and, despite the fact that it began before Prolift was launched anywhere in the world, it was described as a “post-marketing study”, presumably of Gynemesh PS. The first patient was screened on 7 May 2004 and the last on 12 December 2005. The principal investigators were David Robinson (later replaced by Elizabeth Babin), Dennis Miller, and Vincent Lucente. Dr Robinson was an Ethicon employee, Dr Lucente was a consultant on a retainer to Ethicon. According to the 12-month report for the study, the original plan was to recruit 90 patients “in order to obtain an evaluable group of 82 patients”, but only 85 women signed up.1824
1797 Its selection criteria differed in three respects from the selection criteria for the French study and consequently covered a broader group of women. First, the candidates were required to have a minimum of stage II prolapse, rather than stage III. Second, there was no requirement for previous or simultaneous hysterectomy.
1798 Even before the TVM studies began, the TVM Group was very concerned about the phenomenon of mesh contraction.
1799 On 5 February 2004, an email from Professor Cosson to Scott Ciarrocca and Professor Jaccquetin, copied to Dr Arnaud and Ophélie Berthier, advised that mesh shrinkage was a major concern.1825
1800 On 10 May 2004 Ms Berthier, marketing manager at Gynecare France, circulated an email to various Ethicon personnel, including Zenobia Walji, Gene Kammerer, and Dr Arnaud advising them that the main concern of Professors Jacquetin and Cosson was “the shrinkage of the mesh which may lead to pain, dyspareunia”. She stated that Ethicon would need to address this issue “when thinking about a next generation mesh”.1826
1801 At the same time, however, Ethicon was worried about potential competition. For this reason it was concerned to accelerate the development of its POP devices. Noting that the Gynecare product was to be marketed the following year, Dr Kammerer wrote in another internal email the same day:
[I]t seems our competition is ahead of us in this area. We need to think about how we can fast forward this project, get more support from both Gynecare and Ethicon as well as quickly optimize the construction.1827
(Emphasis added)
1802 The first paper by the TVM Group describing the TVM technique was published in the Journal de Gynécologie Obstétrique et Biologie de la Reproduction in around September 2004.1828 The authors were cautious. It had only begun its multi-centre prospective study. It had plans to evaluate the clinical feasibility of the technique, its efficacy, and its complications according to the protocol the Group had devised, at 12 months, three years, and five years. The authors of the paper (Debodinance et al) observed that “[t]he literature review is sparse and manifestly lacking in serious, methodologically sound studies to validate the materials and techniques used”. Once again, emphasis was placed on the problem of mesh contraction, which the authors noted was ignored in the literature they reviewed. At this stage the Group concluded:
The TVM technique is the fruit of serious reflexion about the complete treatment of genital prolapse. It uses a synthetic material chosen after several tests. This technique should be reserved for Grade 3 and 4 prolapses, and is possible as a first-line treatment. The procedure takes no longer than 120 minutes, and is feasible for any surgeon interested in pelvic floor surgery and accustomed to vaginal surgery, following a brief training period. The progress of the TVM group will result in a long-term study.1829
1803 In October 2004 Professor Jacquetin prepared a PowerPoint presentation on “Failures and complications of vaginal pelvic floor reconstructive surgery: is TVM (trans vaginal mesh) the solution?”1830 The problem, according to Professor Jacquetin was that pelvic surgeons prefer native tissue repair to synthetic mesh and that they fear complications of synthetics, particularly the high vaginal erosion rate. Professor Jacquetin said that, in view of the long term success of abdominal hernia repair using tension-free synthetic mesh and “the good tolerance of TVT in incontinence surgery” (citing an erosion rate of less than 1% though the source was not mentioned), “it becomes tempting to succumb to the spell of the prosthetic repair”. He remained cautious, however, adding (without alteration):
But you have to be very carefull because << the ideal synthetic material has yet to be developped … >>1831
1804 After reviewing the literature which recorded the use of various brands of synthetic mesh by surgeons to treat (mostly anterior) prolapse in women, Professor Jacquetin’s “temporary conclusion” was that more randomised controlled trials were needed with “very strict” protocols “including erosion, extrusion, stiffness, shrinkage evaluation and … long term results”. He predicted that mesh repair was “probably the future” of vaginal pelvic floor repair, but said that “the ideal mesh” (synthetic or biological) was yet to be discovered. After explaining the work of the TVM Group, Professor Jacquetin stressed the need for randomised controlled trials and he limited the indications to “1 – very large prolapse: grade IV” and “2 – recurrent prolapse” and queried “3 – No simultaneous hysterectomy”, later indicating that avoiding hysterectomy was one way to reduce the incidence of erosions. Professor Jacquetin’s ultimate conclusions at this time, as reflected in these slides were that:
implantation of mesh by the vaginal route was not validated;
vaginal surgery is very “empirical” and there were no long or even short term results, only the good impression of the promoting surgeon;
at this point in time the development of transvaginal mesh for the treatment of pelvic organ prolapse was similar to the beginning of the road with TVT in 1996; and
more RCTs were required with very strict protocols, including erosion, extrusion, stiffness, shrinkage evaluation and long term results.
1805 Professor Jacquetin expressed the view that mesh repair was “probably the future”, but was not presently justified. As he put it: “Yes but not yet!”
1806 Despite these cautionary notes, Ethicon pressed ahead with its plans. By 1 November 2004, it had a “Draft Global Launch Plan” for its product, Prolift, which was to be the culmination of the work of the TVM Group.1832
1807 The same month an article by the TVM Group appeared in the Journal de Gynécologie Obstétrique et Biologie de la Reproduction. The article repeated much of what appeared in the September article and incorporated the views expressed by Professor Jacquetin in his PowerPoint presentation. 1833
1808 Importantly, the TVM Group identified at this early stage the need to remain alert to a number of matters.
1809 The first was porosity. An estimated threshold pore size of 75 µm was said to be required to obtain adequate tissue integration. “In the absence of pores with an adequate size”, the authors stated, “the prosthesis will be encapsulated instead of being integrated into the surrounding tissue”. They said that encapsulation is responsible for a high rejection rate.
1810 The second was retraction (contraction).1834 Retraction was said to be “impossible to forecast” and “highly variable”, dependent in part on “both the knitting and the inflammatory reaction”, and apparently “enhanced with one-thread materials”. It was described as a “much more worrying phenomenon” which had not been reported in earlier studies. Its symptomatic manifestation was said to be dyspareunia.
1811 The third was erosion. The authors said that erosion is seen with rigid, low-porosity, thick meshes. They described it as “deleterious for the organs” and noted that it could occur many years after implantation. They claimed, however, that primary or secondary erosions of the vagina healed easily with local treatment, or following simple scissor excision of the revealed lesion.
1812 The fourth was chronic infection. While acute infection was said to be “very rare”, chronic infection was said to be “the real problem” associated with mesh placement. This was because bacterial cells adhered to the mesh surface and encapsulated it, forming a “biofilm” (defined as “an assembly of bacterial colonies fixed upon a support and locked up into an encapsulating matrix”).
1813 The authors concluded as follows:
The TVM technique results from thoughtful reasoning aimed at developing complete surgical repair of genital prolapse. The technique is based on the use of a carefully selected and tested synthetic material. This technique should be reserved to the management of grade 3 and 4 prolapse, possibly as first-line treatment. The intervention lasts no more than 120 minutes and can be performed, following a short training period, by any surgeon interested in pelvic floor surgery and with experience in the vaginal route. The reasoning followed by the TVM group led to the initiation of a study that will analyze the long-term results.1835
1814 A design validation protocol for Prolift was completed on 9 December 2004. According to the protocol, the study had two purposes. The first was:
…to provide objective evidence that the ANTERIOR, POSTERIOR and TOTAL GYNECARE PROLIFT PELVIC FLOOR REPAIR SYSTEMS (including the devices, packaging, and labeling) satisfy defined user needs and intended uses within actual or simulated-use environments in a manner consistent with the Instructions for Use (IFU).1836
1815 This was to be accomplished “utilizing a simulated work environment for product dispensing and a human cadaver model for product evaluation”. The protocol stated that the study recognised “the expected variation in user experience, technique, and ability”.
1816 The design validation report, dated 7 February 2005, recorded that eight doctors (two gynaecologists, four urogynaecologists, and one urologist, all but one with more than two years’ experience) participated in the evaluations.
1817 A DDSA was also conducted.1837 It referred to what Ms Holland described as “the regulatory blunder”, stating that there were “no known predicate/similar devices”.1838 Ms Holland considered that the analysis was flawed in several respects.1839
1818 First, it stated that the design validation and IFU supported the fact that the critical steps in setting up and operating the device could be performed adequately by all intended users. At the same time, however, it recognised that training was needed before the device could be used safely and effectively and, although it apparently developed a professional education program, there was no evidence to indicate that such a program was deployed to all users.
1819 Ms Holland said that it was “accepted industry practice” for manufacturers “to include typical users with variation in user experience, technique and ability”. Indeed, the sample base for the study was to include both relatively inexperienced and experienced users. Yet, Ms Holland observed that only one inexperienced user was included in the design validation and that was not representative of the worldwide user group.1840
1820 Second, use of the final design and inclusion of lot variation are expected during manufacturing and should be included in validation activities but here only one batch of product, manufactured expressly for design validation, was used. Furthermore, during the design validation, lot and batch numbers for the six packaging samples showed that no inspection was performed on the mesh. Ms Holland concluded that there was therefore no objective evidence to demonstrate that the packaging samples were representative of the final design and met the protocol requirements.1841
1821 Third, although the validation protocol stated that users would be given training on the Prolift components and surgery, the instructions for use did not require training before use and put the onus on the surgeon to contact a company sales representative to arrange for training.
1822 Fourth, the intent of design validation is to obtain user feedback and address it. Yet, here, no action was apparently taken to address any user feedback regarding the packaging and all surgeon feedback relating to the product was ignored. She acknowledged that one statement was added to the surgical technique, which related to removing the cannula before reinserting the guide, but since (she understood) that the surgical technique was not routinely sent with the product she considered “the addition was all for naught”.
1823 Fifth, while the design validation report included a statement that no additional follow-up was necessary, no reasons were given. Ms Holland said that in her experience it was necessary to consider each user comment based on the risk associated with the feedback and to document the results of the analysis in the report.1842
1824 Sixth, hazards identified in the DDSA relied on the Prolift IFU which omitted many of the warnings, precautions, contraindications and adverse reactions necessary for safe and effective use.1843
1825 A dFMEA was also carried out because the DDSA showed hazards with ratings of IV or V indicating the potential for a “severe” or “critical” hazard. 1844 Ms Holland said that, when each component was evaluated for potential failures, a number of breakdowns in the process became apparent:
• None of the 50 potential failures required further action;
• 34 out of 50 potential failure modes relied on the control method of “Cadaveric User Evaluations” or design validation which were shown … to represent actual use conditions; and
• 11 out of the 50 potential failure modes, or 22%, do not have harms associated with them. It is not possible to assess a severity level if a harm is not present.1845
1826 The instances of severe harm included “incorrect body shapes”, where the implant was too small for the patient causing improper support and creating a hazard that excessive tension would be used in repair,1846 and “incorrect strap configuration”, where the strap was too wide and incompatible with the guide system giving rise to the risk of excessive trauma. In each case the remedy proposed was cadaver studies and user valuations and, in the latter case, also design calculations.
1827 Ms Holland observed a similar pattern in her review of the aFMEA.1847 Ms Holland said that “[i]n principle, the aFMEA should be a mechanism to help establish the contents of the IFU to allow safe and effective use of the device for all users”.1848 Yet, as she observed, many of the contradictions, warnings, and precautions did not appear in the IFU, which was the control method upon which the aFMEA relied for 98% of the failure modes identified.
1828 The aFMEA identified several hazards where the potential harm was classed as severe but in respect of which no further action was considered necessary. They included:
(1) placement of the proximal cannula (anterior, presumably indicating a risk confined to Prolift Anterior), which could lead to damaged organs, partial paralysis, infection, promotion of erosion, and recurrence;
(2) placement of the distal cannula (anterior), which could lead to damaged organs;
(3) partial paralysis, infection and recurrence;
(4) placement of the posterior cannula, which could lead to damaged organs, partial paralysis, infection, and recurrence;
(5) adjustment of the implant, which could lead to repeat surgery, bunching of mesh, erosion, recurrence, and damage to organs; and
(6) positioning of the implant, which could lead to recurrence, tissue trauma, and erosion.1849
1829 In each case the potential cause was described as “lack of training” and the control method was “IFU”, noting that the IFU recommended “professional education training”.1850
1830 Ethicon’s pre-market CER for Prolift was signed by Dr Charlotte Owens on 14 January 2005.1851 Oddly enough, it made no reference to any of the publications of the TVM Group. Nor did it refer to either of the clinical studies proposed by Ethicon when Project D’art was conceived, both of which were well short of completion. Dr Allman observed that the Prolift CER was very similar to the 2002 CER for Gynemesh PS, relying on most of the same clinical evidence.1852 The applicants described it as a “cut and paste” job.1853
1831 This is all that appeared under the heading “Potential Complications”:
An eighty-eight subject series of patients with vaginal placement of GYNECARE GYNEMESH PS for treatment of pelvic organ prolapse was reviewed to evaluate three potential complications associated with mesh placement trans-vaginally.
Infection
As this is a foreign body, and passing through a contaminated field, infection is a theoretical risk. In the series referenced there were no infections reported.
Mesh Exposure
In the series referenced above, investigators reported 8 subjects with mesh exposures (9.1%). These mesh exposures were handled by office intervention with 1 exception.
Fistula
Vaginal erosions are a well known complication associated with the use of synthetic for pelvic organ prolapse repairs. In the series referenced there were no fistulas reported.
Hematoma
A potential risk of infection is associated with hematoma formation in the presence of synthetic material. The GYNECARE GYNEMESH clinical evaluation did not include any reported infections associated with hematoma adjacent to mesh.
Contraction
Tissue contraction has been reported when mesh is used for tissue support in hernia repair. The reports are rare and causative factors are not understood. There were no instances of tissue contraction reported in the GYNECARE GYNEMESH clinical evaluation.1854
(Original underlining)
1832 As the study was unpublished, and not annexed to the CER, it was not and could not be independently evaluated.1855 The form or stage of prolapse the subject of the report, qualifications and experience of treating surgeons, and the characteristics of the patient population were all unspecified. The report was also silent about the length of follow-up and the number of patients lost to follow-up. It is unclear from the report whether the review of the 88 patients was evaluating only the first three potential complications (infection, mesh exposure, and fistula) or all five. The references to haematoma and contraction appear to be additions to the report but it is not apparent when those additions were made or by whom. The description under the heading “contraction” minimised the significance of the subject, despite the advice Ethicon had received about its importance from Professor Jacquetin and the concerns expressed by the TVM Group. Dr Owens did not explain why other potential complications were not evaluated. Neither did she address the important differences between Gynemesh PS and the Prolift kits, let alone what effect they might have on patient safety. Nor was any attention apparently paid to the performance of Gynemesh PS in the two and a half years since the Gynemesh PS CER was finalised.
1833 Dr Owens declared Prolift “safe and efficacious”, “based on large-scale use of polypropylene over the last decade and a review of clinical literature and clinical trials as described”.1856
1834 It is not clear what she meant by “clinical trials as described”, since she only referred to one in her report: the case series of 88 patients treated with Gynemesh PS.
1835 Dr Allman considered that her conclusion was not justified, as “[n]o clinical evidence was presented to demonstrate the safety and performance of the mesh under the normal conditions of use of the complete PROLIFT device”.1857 I agree. As Dr Allman observed, Dr Owen had regard to only one component of the device: the mesh. Risks associated with the transvaginal method of placement or the tools provided with the Prolift kit were ignored.
1836 Professor Gordon said that a series of cases or follow-up studies of patients who have received one of the treatments for a particular indication do “not [provide] an appropriate context for sound scientific comparison of treatments” and that it was “widely understood and accepted that judgements about efficacy of a medical intervention in medicine must be based on well-designed randomised controlled trials”.1858 In this context, “random” means that each patient has a known, usually equal, chance of being given the same treatment but the treatment she is given cannot be.1859
1837 The Prolift CER report did not contain any search criteria. The nature and extent of the literature search were not identified. The report contained a heading “7. References” but the section was not populated; no references were listed. The footnotes referred to the studies to which I mentioned above and listed several others but the references for the other citations were obscure, denoted merely by “REF” or “Ref” followed by an apparently random number. The final footnote was also impenetrable: “ETHICON data on file Protocol 2002-001”.
1838 On the face of things, the review of the clinical literature was negligible. It was also superficial.
1839 The report cited four articles. One was the article by Foote et al (1997) supposedly reporting a 93% cure rate at six months in 29 women who had undergone primary posterior vaginal repairs using Prolene mesh. Another was the article by Julian (1996) on the efficacy of Marlex mesh in anterior repair. Both these articles, it will be recalled, which relied upon in the 2002 CER for Gynemesh PS. As I pointed out above, not only did the articles not relate to Gynemesh PS but they had numerous other shortcomings.
1840 The report also mentioned the article by Nilsson et al (2004), reporting on the seven year follow-up of the Nordic TVT study, but only in support of the proposition that using polypropylene mesh as a midurethral sling in a tension-free manner showed “a high clinical success rate” and “a low complication rate”.
1841 A number of difficulties have already been noted about the Nordic trials, including this one.
1842 Besides, given this was a report about treating stress urinary incontinence with a tape made from Prolene, its relevance to the efficacy of Prolift, or even Gynemesh PS, for the treatment of pelvic organ prolapse is obscure. Nor, it seems to me, could the results of this study have had any bearing on the safety of Prolift, particularly since the quantity of mesh and the fixation methods used in Prolift repair were so different from those used in TVT.
1843 The other article cited in the Prolift CER was by Dr Cheryl Iglesia and others, published in 1997.1860 This was a literature review concerning the use of mesh in gynaecologic surgery. It related to the use of synthetic mesh implants in sacrocolpopexies (15), where the method of application was transabdominal, not transvaginal, as suburethral slings (21) or as pelvic slings (5), and in which concern was expressed about the mesh-related complications.1861 Dr Owens neglected to mention, let alone evaluate the significance of, the observation by Iglesia et al (1997) that “[i]t is likely that published reports underestimate the rate of graft-related complications because follow-up was limited in most of these studies”. Although the Prolift CER mentioned the revision and removal rate in sacrocolpopexies of 2.7%, it neglected to mention the reported erosion rate of 9%. The report also failed to mention the increased amount of mesh used in Prolift, particularly Prolift Total, let alone consider the effect that might have on the potential for complications.
1844 Otherwise the report cited a “communication” by Cervigni reporting “the safe use of PROLENE mesh in the repair of anterior prolapse in 35 patients”, which was also mentioned in the 2002 Gynemesh PS CER.1862
1845 The failure to refer to any of the published work of the TVM Group is baffling, although, as Professor Korda pointed out, the results obtained by the TVM Group were “short-term results in the hands of skilled and experienced surgeons” and “could not be extrapolated to safety and efficacy in the hands of ordinary skilled gynaecologists, urogynaecologists and urologists”.1863
1846 Dr Owens’ consideration of the question of contraction was lamentable.
1847 Whatever the position might have been with hernia mesh, and whatever might have been said about contraction in the Gynemesh PS CER, having regard to what Ethicon had been told by Professors Jacquetin and Cosson, the assertion about tissue contraction with Gynemesh was misleading.
1848 Professor Gordon described the Prolift CER as “incoherent” and said that it suffered from the same problems as the CER on Gynemesh PS signed by Dr Weisberg. Noting that the conclusion for safety and efficacy was based on case series, rather than randomised controlled trials, he said that it “falls considerably short of established standards of scientific evidence” and the evidence upon which Dr Owens relied did not support her conclusion.1864
1849 Dr Hinoul conceded in cross-examination that Dr Owens’ report was inadequate to justify the product’s safety as a stand-alone document but steadfastly, and for no good reason, refused to concede that the statement about the absence of reports of tissue contraction in the Gynemesh clinical evaluation was misleading.1865 Moreover, his responses to the apparent inconsistency between the reported concerns of Professors Jacquetin and Cosson and the assertion of Dr Owens are troubling.
1850 Dr Hinoul said he assumed that Dr Owens was referring to the clinical data she had available to her at that point. If that is so, then as I said, the evaluation was manifestly inadequate. He also said that “the document on itself is an insufficient one” but added that “shrinkage, pain and dyspareunia are issues that can occur with all pelvic floor repairs”, presumably in an attempt to minimise the significance of the findings that so concerned Professors Jacquetin and Cosson. When Mr Bannon SC pressed him in cross-examination to acknowledge that the assertion about contraction in the Prolift CER was misleading he offered the following disturbing response:
Well, it is an internal document, that is – I take that back. It is there for regulatory purposes. It – it is not – it could be better.1866
1851 The reluctant acknowledgment that the report “could be better” was, to say the least, an understatement.
1852 In his affidavit Dr Hinoul contended that “[t]his risk assessment corresponds to the risk/benefit analyses I have performed”.1867 In the light of the numerous problems with Dr Owens’ assessment, if Dr Hinoul’s contention is an accurate representation of the nature of his risk/benefit analyses, it only serves to undermine those analyses.
1853 Three months after the Prolift CER was signed, in an abstract presented to an IUGA conference in Paris, Professors Jacquetin and Cosson, and other members of the TVM Group presented their three-month findings on their prospective study of 264 patients implanted with Gynemesh PS used as Prolift was designed to be used. The incidence of contraction noted in the abstract (referred to as “mesh shrinkage”) ranged from 7.69% to 35.9%.1868
1854 In contrast to the Prolift CER, the TVM Group conducted a much more comprehensive review of the literature and pronounced the results “relatively disappointing”. In their article published in November 2004 to which I referred above,1869 two months before Dr Owens signed the CER, Berrocal et al (2004) wrote that, although a number of articles dealt with the use of mesh in the surgical management of vaginal prolapse, those articles “usually describe[ed] non-validated techniques and materials and frequently present[ed] retrospective results with follow-up durations rarely exceeding 6 months”. They pointed out that most of the studies focused on cystocele (anterior prolapse) and few dealt with the posterior area and, while erosion was often reported, all neglected to mention contraction.
1855 Considering that the TVM Group was convened by Dr Arnaud, it is inconceivable that Ethicon would not have had a copy of this article at the time Dr Owens signed off on the Prolift CER.
1856 Prolift was first supplied in Europe in January 2005 and approved for sale in the United States in March 2005. Prolift was first supplied in Australia in June that year. Contrary to the opinion of the TVM Group, however, the IFU for Prolift did not recommend that the technique or the product be reserved for women suffering from grade 3 or 4 prolapses. Nor did it refer to the risk of dyspareunia as a result of contraction.
1857 As Ethicon frankly admitted in its “Prolift+M Clinical Strategy” finalised on 6 September 2007, Prolift was launched without clinical evidence.1870 This is consistent with Dr Allman’s view that the information disclosed in the 2005 CER for Prolift was inadequate to justify CE marking.1871 In December 2011 the American College of Obstetricians and Gynecologists and the American Urogynecologic Society observed that “[m]esh kits for repair of [pelvic organ prolapse] were first marketed to urologists and gynecologists as a way to improve success rates for POP repairs with native tissue, but without well-designed trials to establish the safety and efficacy of these devices”.1872
1858 Collinet et al (2005) published an article to the International Urogynaecology Journal on risk factors for and management of mesh exposure in transvaginal mesh repair in 2005. Their concluding paragraphs sounded a strong note of caution, which Ethicon did not heed. First, they were adamant that no hysterectomy should be carried out at the same time and that the number and extent of colpotomies needed to insert the mesh needed to be limited in order to enhance the prospect of healing. Second, they thought that experimental studies, clinical trials, and long-term follow-up were necessary.1873
1859 In September 2005, nine months after Prolift was taken to the European market and three months after it was first supplied in Australia, the TVM Group reported to the International Continence Society (ICS) on a multicentre retrospective study of the TVM technique in 96 women under 50 which had begun in November 2002. Its report was presented to the Court in a conference abstract.1874 In it the TVM Group described Prolift as “an interesting improvement in organ prolapse surgery” in the short to medium term. It warned, however, that the durability of anatomical results should be assessed over a longer period and observed that the medium-term outcome was disappointing. It expressed concerns about the high rate of granuloma formation (thick scarring with nodules of inflammation indicating inflammation of the scar without erosion or exposure), vaginal erosion (at 6.5%) and the impairment it would cause to “young patients” (those under 50), and the high rate of recurrent organ prolapse (8.3%) after a relatively short period of time (the mean was 3.6 months).
1860 The 12-month results from the French study were not published until 27 June 2006 (about six and a half months before Mrs Gill’s implant surgery).1875 As the following extract from the report indicates, the study was a failure according to the pre-defined success criterion. The upper confidence interval (CI) was 26.6%, not less than 20%, which was the specified criterion for success. The authors nonetheless expressed the view that TVM had “an invaluable role” in treating patients with vaginal prolapse “in terms of reasonable success rates and a lower rate of recurrence/re-operation compared to other published studies”. They concluded:
The primary effectiveness variable was recurrence of prolapse at 12 months post-procedure (failure of procedure), with failure being defined as a prolapse of ICS Stage II or more or a surgical re-intervention. The results show a failure rate at 12 months of 18.4% with a 90% Cl of 11.9–26.6. Thus the study did not meet the pre-defined criteria of a failure rate of less than 20% (upper limit of 90% Cl). Of the 16 failures there was only one patient with a prolapse of ICS Stage III; fifteen patients had prolapse of ICS Stage II. In 10 of the Stage II patients, the leading edge of the prolapse was inside the introitus.
The secondary effectiveness parameters show a failure rate at 6 months of 12.6% (90% Cl: 7.3, 20.1). Other secondary effectiveness parameters show a reduction in the number of patients reporting sexual activity limited by prolapse at 12 months compared with baseline (29 [32.2%] vs 6 [6.9%]). The incidence of dyspareunia in those patients who were sexually active was 4/61 (7%) at baseline, 8/42 (20%) at 6 months and 3/40 (8%) at 12 months (Tables 14.2.8.2 and 14.2.4.1). One patient (3004) was reported as having dyspareunia at baseline but was not sexually active due to prolapse. The 3 cases of dyspareunia at 12 months were all new onset. Dyspareunia was resolved at 12 months in the 5 patients who reported the condition at baseline.
1861 The authors considered that both the mesh exposure rate (10%) and complication rates were low and noted significant improvements in quality of life scores.
1862 After three years, however, the results were worse.1876 Anatomical failure rates, which had risen from 11.6% after 6 months to 17.4% after one year, sat at 20% after three years, which the authors acknowledged indicated “that the primary endpoint (upper confidence interval<20%) was not met”. The overall incidence of mesh exposure had risen from 10% to 14.4% (13/90, nine of whom had undergone a concurrent hysterectomy) and “the incidence of mesh erosion requiring surgery was 8/13 (8/90 = 8.9%)”. Considering that only 85 patients were available for follow-up these percentages are likely understated. Of the 61 sexually active women at baseline, 25 (41%) had ceased sexual activity by three years. The incidence of de novo dyspareunia was 5/57 (8.8%). Of the total number of sexually active patients at three years 6/39 (15.4%) reported dyspareunia. Increased vaginal wall stiffness was observed in 11 patients (12.6%) and was always associated with mesh contraction which the authors of the article (Jacquetin et al) attributed to the increased fibrosis responsible for the loss of elasticity of the vaginal walls.1877
1863 The 12-month results from the US study became available one day after those of the French study, on 28 June 2006.1878 The primary objective, according to the report was correction of the prolapse and was evaluated by determining the proportion of patients in which correction was achieved (to stage 0 or 1 on the ICS scale). This rating scale is known as the POP-Q scoring system. As the report explained, this is a standard system to describe pelvic organ prolapse in women. The patient is examined and the positions of a number of predefined reference points are measured in centimetres above or proximal to the hymen (negative numbers) or below or distal to the hymen (positive number), with the plane of the hymen being defined as zero. All measurements are recorded in whole numbers. Prolapse is classified under this system from 0 to IV.
1864 Failure of correction was defined as either stage II or higher or a surgical re-intervention to repair a recurrence of vaginal prolapse. Other criteria for evaluation were: recurrence of vaginal prolapse in the area treated with mesh; vaginal prolapse occurring in an area not treated with Gynemesh PS; peri-operative complications; patient tolerance of the mesh; post-operative complications and quality of life. The criterion for success was similar to the French study, namely that the upper confidence level did not exceed 20%. Like the French study, the study would be deemed a failure if it did not show that the prolapse (recurrence) rate was less than 20%. Unlike the French study, however, the results showed a failure rate of 12% at 12 months with a 90% confidence interval of 6.7–19.6. Since the upper figure was less than 20%, this study was not a failure. The other results were also reportedly rosier, although mesh exposure levels, urinary incontinence, and voiding difficulties were still high, though classed as “low”:
During the 12-month follow-up period adverse events were reported for 56 (65.9%) patients. The most commonly reported events were urinary incontinence (17 [20.0%] patients), mesh exposure (12 [14.1%] patients) and void dysfunction (9 [10.6%] patients). Other adverse events were each reported in less than 10% of patients. Eleven (12.9%) patients experienced adverse events that were considered to have a causal relationship to the investigational device. 1879
1865 Amongst the “other adverse events” under 10%, were seven patients (8.2%) who experienced urinary tract infections, five (5.9%) vagina atrophy, one whose ureter was severed, and one who suffered a haemorrhage.
1866 The conclusion was that:
[T]he study demonstrates the invaluable role of TVM in treating patients with vaginal prolapse in terms of success rates and a lower rate of recurrence/re-operation compared to other published studies. Significant improvements in QOL, particularly with respect to the PSI, were observed and activities of daily living QOL scores also improved over the 12-month study period. Furthermore, complication rates were low. The safety data also demonstrate a predictable safety profile with a favourable benefit/risk ratio for patients requiring treatment for vaginal prolapse.1880
1867 Further assessments were scheduled three years and five years after implantation.1881
1868 Various changes were made to the protocol during the currency of the study. Five patients withdrew, one between baseline and six months, one between six months and one year, and three following the 12 month assessment.1882
1869 Ms Holland observed that, since ease and consistency of implantation were seen as both an objective and key to the success of Prolift, the DDSA (as revised on 12 October 2004) established that special training of the intended user was necessary and that a professional education program had been developed.1883 She noted that the Prolift IFU, however, merely stated that training on the use of the Prolift system was “recommended and available” (emphasis added) and invited surgeons to contact their company sales representatives to arrange for that training. Similarly, she noted that, although Prolift incorporated entirely new instruments, form and fit factors, as well as a new surgical technique, the surgical technique was not included with each device, master box of devices, or otherwise provided, such as by an enclosed instructional disc or web link. Rather, the IFU referred readers to the recommended surgical technique guide for further information on the pelvic floor procedure and directed them to contact their company sales representative to obtain the guide. Ms Holland commented:
In my experience with both implantable and non-implantable devices that may have complex installation and/or user interface processes, passive training such as Ethicon’s approach is not sufficient to obtain the desired level of reliable training outcomes. Although surgeons are generally knowledgeable in their field, specifics relating to use of the device improve the probability of success. I believe making it necessary for the physician or his/her staff to actively search out the corporate sales representative, obtain the surgical technique, and go through the nuances of the surgical technique prior to use, is in error and the training often does not occur or does not occur to the degree needed to achieve consistent positive results. This practice does not seem to be isolated to PROLIFT, but rather is the norm for Ethicon and may contribute to easily preventable problems and harm to patients.1884
1870 This evidence was not the subject of cross-examination. I accept it.
1871 DDSA “Revision C” for Prolift, dated 25 February 2005, characterised the risk of erosion as 4 (severe) but the probability of occurrence as 3 (<1 in 10,000 or 0.01%).1885 As Dr Allman observed, however, the rate of erosion reported in the TVM studies was much higher.1886 The enormous discrepancy between complication rates, including for erosion, derived from reports or assumed in Ethicon’s risk evaluations on the one hand and complication rates reported in numerous studies on the other was not the subject of comment or analysis in any of the clinical evaluation reports.
1872 Professor Collinet said that by the time Prolift was released to the market, “the problem of mesh exposure [had] been practically resolved due to very low incidence and satisfactory treatment techniques”.1887 The weight of evidence was to the contrary. As Professor Korda observed in March 2017, it is still a problem. That was his own experience and, as he pointed out, it was also supported by the opinion of the Scientific Committee on Emerging and Newly Identified Health Risks, published in December 2015,1888 which reported that mesh exposure was the most frequently reported complication with rates ranging from 4 to 19%. Professor Korda said that he did not regard an exposure rate of between 4 and 19% as low, nor had it been his experience that treatment was easy or satisfactory.1889
Prolift+M
1873 Even before Prolift was launched in Australia and notwithstanding the way in which the subject was dealt with in the Prolift CER, concern about the problem of scar contraction around the mesh prompted Ethicon to explore alternatives.
1874 I referred to an email from Gene Kammerer sent in April 2005 to Thomas Barbolt and others at Ethicon in which he said that “scar contracture translates into procedural complications”.
1875 The heading to the email (“ULTRAPRO vs PROLENE Soft Mesh”) suggests that Mr Kammerer was considering UltraPro, which was a polypropylene mesh used in hernia repair and contained an absorbable component (Moncryl), as an alternative to Prolene Soft which had been used in Prolift. As I have noted above, UltraPro was launched in 2002. Professor Klinge described it as the first successor to Vypro, which was also a “lightweight”, multifilament polypropylene hernia mesh with an absorbable component (polyglactin 910).1890 It had pores of between 3mm and 5mm. Vypro was launched in 1998.1891 In an Ethicon internal document entitled “Rationale for using Prolene/Monocryl in pelvic floor repair”, dated 18 August 2006, the following statements appeared:
In the last 10 years the usage of lightweight meshes implanted in hernia surgery showed several benefits. The lightweight and large porous concept decreased inflammation, foreign body reaction and patient complaints about pain by increasing compliance, patient comfort and quality of life after hernia repair. The strength of the lightweight meshes is less than in heavy meshes but still more than double the natural abdominal wall strength and due to the larger pores, the compliance is much more physiologic than in heavy meshes. In the last years, more than 1 million meshes were implanted per year, showing that modern hernia repair is not imaginable without meshes.
In the 90s several companies developed new mesh systems to increase stability and comfort and decrease the amount of foreign body material to the patient. By adding an absorbable component like Monocryl to Prolene meshes (e.g. Ultrapro), the mesh gets stiff and comfortable in handling for the surgeon and when the Monocryl part is absorbed even lighter and softer for the patient. These partly absorbable meshes have been studied in preclinical trials and used in hernia surgery successfully for many years now.
…
Lightweight meshes like Ultra Pro used in Hernia surgery proved to:
... cause less inflammation traditional meshes
... induce less fibrosis small porous meshes
... generate less foreign body reactions than heavyweight meshes
... get better integrated to host tissue than small porous heavy meshes
... lower patient complaints compared to heavyweight meshes
... provoke less pain compared to traditional heavyweight meshes
... improve patients quality of life
... keep almost physiological abdominal wall mobility
... have an outstanding biocompatibility
... facilitate an excellent handling
Without any negative side effects compared to traditional heavyweight meshes!1892
1876 At a meeting in Hamburg on 2 June 2006, attended by Professors Cosson, Klosterhalfen, Deprest, Jacquetin, and Dr Lucente, together with various Ethicon employees, attendees were invited to identify “the unmet needs” associated with pelvic mesh. Those “unmet needs” included no shrinkage, no long term contraction, reduced fibrosis, no folding, no vaginal distortion, elasticity simulating physiology, no chronic pain, reduced erosion and less inflammatory response. The meeting also discussed the complaint that contraction leads to dyspareunia and decreased sexual function.1893 All these matters were given a priority of 10 points. The inference I draw from this is that they were of the highest priority. Professor Klosterhalfen told the meeting that fibrosis is responsible for complications in mesh usage and that even after 20 years the tissue still reacts to the mesh. He also told the meeting that every individual reacts differently to the mesh, that tension on the mesh causes changes in pore size which in turn cause changes in elasticity, that mesh can cause nerve damage as the mesh bears down on the nerve, and there is no such thing as an inert material.
1877 Another expert meeting was held on 23 February 2007 with several of the same participants in which similar issues were discussed.1894 At the meeting Professor Cosson queried whether polypropylene was the best material as “fractures are observed in pp after time” and because of the problem of shrinkage.
1878 At around this time Ethicon was extolling the benefits of UltraPro. In a PowerPoint presentation by Ophélie Berthier, UltraPro was described as a partially absorbable, monofilament, large pore, pliable, vaginally-compliant mesh with little memory. She stated that in general surgery for abdominal hernia repair UltraPro had been proven to cause less inflammation, induce less fibrosis, have better integration to host tissues, maintain the mobility of the abdominal wall, have an outstanding biocompatibility, and improve patients’ quality of life.1895
1879 In the meantime, Ethicon had begun work on a new project which it called “Project Lightning”, designed to address some of the unmet needs identified at the June 2006 Hamburg meeting.1896 The project led to the development of Prolift+M. The key difference between Prolift and Prolift+M was the addition of the absorbable Monocryl component. In a PowerPoint presentation dated 26 September 2008 Ethicon claimed that the addition of Monocryl “yield[ed] a greater distance between pores and resist[ed] the ability of bridging fibrosis”.1897 The source for its assertion was a paper on hernia repair.
1880 On 6 September 2007 Ethicon outlined its clinical strategy for Prolift+M. In its introductory remarks the document referred to the risks of exposure and contraction:
Mesh exposure is a common complication which can be managed by excision and closure. Mesh retraction (“shrinkage”) is less common but it is considered more serious. It can cause vaginal anatomic distortion, which may eventually have a negative impact on sexual function. Its treatment is difficult. Additionally, the scar plate that forms with in-growth of tissue into the mesh can cause stiffness of the vagina that further impacts sexual function in a negative manner.1898
1881 The strategy document explained that Ethicon was exploring the use of “a lighter weight alternative mesh for POP repair” in order to minimise these complications and that that mesh “would serve to replace” the Gynemesh PS in Prolift. UltraPro was identified as a suitable candidate and it was rebadged Gynemesh M within Ethicon Women’s Health.
1882 UltraPro, however, was then indicated for tissue reinforcement and long lasting stabilisation of fascial structures of the abdominal wall. According to the strategy document, apparently prepared by the Research and Development team, some gynaecologists had used UltraPro for pelvic organ prolapse repair but the information gathered was “limited due to: the small number of cases, the mesh was placed from an abdominal rather than vaginal approach, [and] the size of mesh used was small relative to the current intended use”. 1899 Acknowledging that the Prolift system was launched without clinical evidence, the authors declared that the strategy for Prolift +M was to be different. It was “deemed appropriate that early data [be] available in the early stages of the commercial launch”. Ethicon proposed a prospective single arm study at approximately 10 sites in the US and the European Union. It decided against “an appropriately powered comparative study” of Gynemesh PS and Gynemesh M because it assumed that Gynemesh M would bring about the benefits it was intended to achieve.1900 The primary objective of the proposed study was to evaluate the anatomical success of the Prolift+M system in women with symptomatic POP-Q stage III or IV, requiring surgical correction of pelvic organ prolapse. Secondary objectives included evaluation of reported outcomes, length of procedure, length of hospital stay, post-operative pain, return to normal activities, and peri-and post-operative complications. Follow-up was to be at three months, one year, and three years. An interim analysis of the data was to be completed on approximately 60 patients to provide early results to support the early launch phase. Dr Allman observed, correctly in my view, that the clinical strategy for Prolift+M reflected Ethicon’s commercial needs and “did not reflect the need for clinical data to support CE marking of the device, or an analysis of the type of data to support CE marking”.1901
1883 As it happened, however, no clinical studies of Prolift+M were carried out before the device was placed on the market.1902 Prolift+M was cleared for supply on the basis of Ethicon’s claim, which proved to be false, that it was equivalent to other Ethicon devices that were already on the market.
1884 In his affidavit Dr Hinoul said that, at the time Prolift+M was launched, Ethicon had more than a decade’s experience with Prolene mesh in incontinence surgery and many years of data were available on Gynemesh PS and Prosima. He added that the delivery tools, anatomical locations and indications for Prolift and Prolift+M were the same and that there was preclinical data on the mesh and a significant amount of clinical data on a very similar technique. He also said that Ethicon had conducted a specific study on Prolift+M and had data available to demonstrate its safety and efficacy. 1903
1885 There are a number of difficulties with these assertions.
1886 First, they failed to acknowledge the problems with Prolift which Prolift+M was intended to address.
1887 Second, on 6 January 2009, after the device had been cleared for sale in Europe and the United States, the Worldwide Marketing Director for Ethicon Women’s Health & Urology, Jonathan Meek, told his colleagues that Prolift+M “will enter the market with little clinical evidence, a hypersensitive market, and an aspiration positioning”.1904
1888 Third, the specific study on Prolift+M to which Dr Hinoul referred, according to the reference he cited in support of his statement, was the subject of the article by Milani et al, of which Dr Hinoul was a co-author. In 2009, when Prolift+M was launched in Australia, only limited three month results were available.
1889 An unpublished paper by Drs Hinoul, Kirkemo and Robinson, Judi Gauld and Colin Urquhart was presented to the 34th annual meeting of the International Urogynecological Association in June 2009.1905 It only reported on “the interim analysis” of 65 of the 125 patients who were enrolled in the study. Even then the mesh exposure rate is recorded as 6.2%. The manuscript was rejected for publication after it came in for criticism from the peer reviewers.
1890 There was no follow-up at six months. The 12 month results were not published until January 2011.1906 By this time the exposure rate had risen to 10.2%.
1891 It is impossible to understand how three months’ worth of data, no matter how encouraging they might have seemed, could be said to be data demonstrating the safety and efficacy of a medical device designed to remain in the body permanently. Dr Hinoul’s statement that Ethicon had data available to it at the time Prolift+M was launched to demonstrate its safety and efficacy cannot be accepted.
1892 Once again, Ms Holland was critical of Ethicon for failing in its design validation to “properly simulate user groups and use conditions”.1907 She said that the design validation report shows that all six surgeons who participated in the validation were experienced, US-based, Prolift users.1908 She noted that variation in experience, geography, or materials were not incorporated into the design validation process in accordance with industry practice “to enable an understanding of user to user variation”.1909
1893 Ms Holland observed that, in order to demonstrate that the mesh provides sufficient strength to support pelvic structures and to satisfy a request for additional burst strength data, the development team originally tried to determine an acceptable burst strength design requirement. Edward Jacobs, Ethicon’s Principal Design Engineer, Worldwide Quality Engineering, emailed Dr Arnaud asking him whether he agreed that any mesh that was strong enough for use in incisional hernia repair would be strong enough for pelvic floor repair. After receiving his affirmative response,1910 the design input was changed from a measurement of burst strength to needing to demonstrate sufficient strength for use in incisional hernia repair. Ms Holland was implicitly critical of this too. She said she could find no evidence of data to support Dr Arnaud’s opinion.1911
1894 Inexplicably, the dFMEA did not refer to several potential hazards covered by the comparable analyses for Gynemesh PS and Prolift. They included “tissue reaction”, “haemorrhage/bleeding”, “treatment not successful – recurrence” and “extended surgery”.1912 Yet, Ms Holland’s evidence was that severity rankings across products for the same intended use should remain consistent.
1895 The pre-market CER for Prolift+M was signed by Dr Robinson on 5 February 2008.1913 The report stated that it aimed to meet the following requirements:
a) Confirming that the market acceptance of GYNECARE PROLIFT System includes acceptance that the product meets the claims for use
b) The modifications made to the mesh for the GYNECARE PROLIFT+M System are shown to be acceptable, that the modified system will continue to meet claims.
1896 Dr Allman criticised these aims, noting that they did not meet the requirement for clinical evaluation as stated in the guidelines on medical devices relating to the evaluation of clinical data (MEDDEV 2.7.1) “to establish conformity of the device with the pertinent essential requirements of the Directive as they relate to safety and performance, and to demonstrate that the device performs as intended by the manufacturer”.1914
1897 The literature review referred to papers on the use of synthetic materials in prolapse repair and three papers on the use of Prolift. The review of these papers is discussed below, as the same papers were reviewed in the pre-market CER for Prosima, which pre-dated the Prolift+M CER.
1898 The Prolift+M CER acknowledged that no clinical investigations had been conducted on the use of Prolift, before considering the modifications to Prolift to produce the Prolift+M system. Dr Robinson referred to a study by Cobb et al (2006) analysing heavy weight (Marlex: 95 g/cm2), mid-weight (Prolene Soft: 45 g/cm2) and light weight (UltraPro: 28 g/cm2) polypropylene mesh in a porcine ventral hernia model.1915 The objective was to assess burst strength and stiffness of these meshes five month post-implantation. A secondary aim was to evaluate the percent of mesh shrinkage of the different weight meshes.
1899 Cobb et al (2006) reported that all implanted meshes showed increased stiffness of the abdominal wall with reduced height and diminished curvature at maximum abdominal distention. The extent of stiffness increased with mesh weight and decreased with larger pore size. Importantly, there was an “intense inflammatory response to the mesh resulting in dense, unorganized scar plate formation”. The entire mesh was encapsulated by connective tissue, forming a rigid scar plate. This was said to contribute to stiffness of the abdominal wall as well as shrinkage of the mesh.1916 Further, after mesh incorporation, there was reportedly no difference in the degree of shrinkage of the mesh regardless of weight.1917 The authors acknowledged that some studies had shown a correlation between lightweight mesh and reduced amount of shrinkage, but noted that their data did not reflect this.
1900 They concluded:
This study demonstrates that the implantation of a macroporous, LW polypropylene mesh results in less restriction of abdominal wall compliance while providing more than adequate strength for repair of ventral hernias. The lighter weight polypropylene mesh with an absorbable component adapted following implantation with a reduction in both burst strength and stiffness, becoming more physiological in regards to abdominal wall compliance. Continued, prospective evaluation of various polypropylene mesh formulations in the clinical setting will allow for a more thorough assessment of the role of these bioprosthetics in the repair of ventral hernias.1918
1901 Dr Robinson adverted to the finding about increasing abdominal wall compliance with lightweight mesh (UltraPro), but did not mention the findings about shrinkage.
1902 Instead, he relied on papers by Professors Klinge and Klosterhalfen to note that reduced mass and increased pore size resulted in a reduced foreign body reaction. An example of this move towards lowering the mass of implanted mesh, he said, was the decision to switch to Gynemesh PS in Prolift instead of Prolene (used in the SUI devices).
1903 By reference to “internally conducted animal studies”, Dr Robinson stated that Gynemesh M was biocompatible and elicited a minimal foreign body reaction. But he went on to say:
Although several benefits are anticipated with the use of a lower mass mesh implant, the benefits are expected to be subtle and likely difficult to demonstrate in a clinical setting as can be the case with evolutionary improvements. These differences include the potential for a reduction in the rates of mesh exposure and mesh contraction. Of the two, mesh exposure is more common but it is usually considered as a benign complication that can be addressed medically or in some cases with excision and closure. Mesh retraction (“shrinkage”) is a less common complication but considered more significant. Retraction may be associated with vaginal anatomic distortion and the possibility of negatively affecting sexual life or increasing the risk of postoperative pain. Treatment of mesh contraction is difficult, and may require significant effort in attempting to surgically release the vaginal distortion.1919
Despite this, Dr Robinson’s concluded that the data were sufficient to demonstrate compliance with the essential requirements covering safety and performance of Prolift+M under normal conditions of use, and no clinical data was required.
1904 The Prolift+M CER included a discussion of the rationale for the (presumed) advantages of Gynemesh M. They were that implantation of polypropylene mesh is associated with an increase in problems associated with foreign material implants: increasing use of materials with reduced implant mass and increased pore size; that the strength of currently available meshes exceeded their physiological characteristics; and that, based on the experience with UltraPro in hernia repairs Gynemesh M would provide the necessary strength and long lasting stabilisation of fascial structures in vaginal wall prolapse. As Dr Allman observed, the possible difficulties in handling lower mass implants during surgical insertion were described but suggested to be countered by the incorporation of absorbable filaments in Gynemesh M. He pointed out that this was “a plausible argument to support the design rationale for the new device but it was not an argument that supported the conclusion of the report”. It was common ground that equivalence with Prolift was assumed.1920 Dr Allman was critical of the Prolift+M CER for assuming that Prolift+M was an equivalent device to Prolift, which was made from Prolene Soft whilst at the same time asserting that Prolift+M, which used Gynemesh M, was designed to be an improvement on Prolift. He said they were not equivalent devices.1921 In these circumstances, he said there was no justification for using the literature route to certification.1922 Further, the report also assumed that UltraPro was equivalent to Prolift+M because they were made of the same material, although UltraPro had not previously been used in the female pelvis to repair pelvic organ prolapses, without explaining why it was appropriate to treat the two devices as equivalent.1923
1905 It was put to Dr Allman in cross-examination that the statement made in the first sentence of the passage from the CER extracted in the penultimate paragraph indicated that “the writer [was] plainly alive to the possible utility of a clinical trial” but discounted it for the reasons given in that passage. Dr Allman did not accept the explanation. He pointed out that it was necessary to conduct large studies to determine subtle differences in benefits and, more importantly, that clinical trials are necessary to evaluate safety issues.1924
1906 It was also put to Dr Allman in cross-examination that, since the CER stated that the indications for use and the training were essentially the same as for Prolift, Prolift+M was UltraPro by another name, and there was literature concerning UltraPro, the report did justify why a clinical study was unnecessary, Dr Allman rejected the proposition. He pointed out, in effect, that, since UltraPro was indicated for tissue reinforcement and long-lasting stabilisation of fascial structures of the abdominal wall and Prolift+M was indicated for tissue reinforcement and long-lasting stabilisation of fascial structures of the pelvic floor in vaginal wall prolapse, in the absence of any explanation as to why the comparison was apposite, there was a gap in the reasoning leading to the conclusion.1925
1907 BSI praised the “clinical equivalency matrix” in the CER, describing it as excellent.1926 Dr Allman was baffled by the glowing review. Whether or not the matrix was excellent, however, does not address the question at hand. The matrix was a process, not an outcome. Nor did its contents demonstrate equivalence.
1908 The Prolift+M CER also included a review of complaint data for Prolift and UltraPro. Over the period 2005 to early 2007, 113 complaints were said to have been reported for Prolift, of which 62 were classified as serious (with one death being reported, which apparently resulted from uncontrolled bleeding). Mesh exposure was the most commonly reported serious event with 21 reports. There were also 14 cases of operative injuries, four haemorrhages, 5 cases of urinary retention, and six cases of “significant pain”. No comment was made about whether or why these reports were acceptable or about the impact of this review on Prolift+M.1927
1909 As for UltraPro, apparently 30 complaints were recorded by Ethicon over the period 2004 to mid-2007, some of which were said to be inapplicable (as they relating to labelling and packaging). A small number related to mesh tear (9), infection (4), seroma formation (3), pain (1) and material reaction (1). A search of the MAUDE database showed 17 entries, which fell broadly within the same categories as the complaints recorded by Ethicon. Dr Robinson indicated that the reported number of problems was low and in any event these problems had been considered in the development of Prolift+M.1928
1910 The overall conclusion, which was the so-called “risk/benefit analysis”, was as follows:
The primary location for the risk/benefit solution is found in the Risk Management Report. GYNECARE PROLIFT +M Pelvic Floor Repair System is a modification to a currently available system and is intended to be an additional rather than a replacement product. The modification is focused on a change in the mesh material, from non-absorbable PROLENE Mesh to a partially absorbable mesh that includes a reduced mass of PROLENE Mesh in the construct, adding polyglecaprone tibers (GYENCARE GYNEMESH M). The newer material has been used successfully for a number of years in hernia repair products (as ULTRAPRO Mesh). The newer mesh, as is the mesh currently used in the GYNECARE PRO LIFT System, is classified as Type I, monofilament macroporous mesh. Both components, the absorbable and non-absorbable, are well tolerated by the body and the composite mesh has been shown, in bench and modeling studies, to provide adequate strength for pelvic organ prolapse support. The conclusion is the change represents a low risk.
Benefit from the use of a partially absorbable, low mass mesh is mostly theoretical. While reduction in the mass of implant material is desirable; the benefit from this reduction most likely cannot be definitively demonstrated. It is assumed that a reduction in the mass of implanted material will lead to improvements; these may include reduced tissue stiffening and contracture and a resultant reduced awareness of the mesh implant by both the patient and the examining physician. These improvements in tum may reduce patient discomfort, especially during intercourse. The reduced tissue response to the mesh may also lower the rate of mesh exposure events. It is felt, though, that these changes will be subtle and therefore not demonstrable in a clinical study. However, because the risks associated with this modification are low, the risk/benefit ratio is acceptable.1929
(Emphasis added)
1911 Dr Allman drew attention to the conclusion that the benefit of this mesh was mostly theoretical, and could not be definitively demonstrated. In his view, given the design differences between Prolift and Prolift+M, a more rigorous argument, with clinical data, was needed to conclude that the risk/benefit ratio was acceptable.1930 Dr Allman found that the report did not meet the requirements of the European Directive or MEDDEV 2.7.1, that its conclusions were not justified by the data presented, and that it was insufficient to justify CE marking. 1931
Prosima
1912 Prosima was developed as “Project Mint”. According to a May 2006 PowerPoint presentation prepared by Ethicon Women’s Health & Urology,1932 it was designed to meet the dissatisfaction of physicians with current prolapse procedures, physicians who thought there were drawbacks to suture plication and wanted to limit failures/recurrences and stay current with the rapid pace of change, were looking for easier ways to perform procedures with good outcomes, who wanted a safer procedure with fewer complications and less dissection, but who were concerned about erosion and other complications of mesh use. “Mint” was said to have the potential to be positioned as a procedure requiring less surgical skill than Prolift, creating an opportunity for Ethicon to expand its “physician base”, and for patients whose prolapse was not severe enough to warrant Prolift, creating an opportunity to expand the patient base. This “positioning”, the document proclaimed, would lead to better outcomes and fewer recurrences and would be easier and faster than the available alternatives.
1913 The clinical strategy for Prosima stated:
Whilst clinical data are not required for regulatory approvals (510k and CE mark) for the PROSIMA system, it is considered necessary to generate evidence regarding performance of this new system to support the commercial launch. A multi-centre, prospective, single-arm study will be conducted in women with Stage II or greater POP. In addition to measuring anatomic success (St 0-1), specific patient reported outcomes will be assessed at 6 and 12 months post-procedure. Complications will be reported throughout the study. The data will be analysed once all six-month data are available with the primary analysis to be completed with 12-month follow-up data.1933
(Emphasis added)
1914 As Dr Allman pointed out,1934 the strategy reflected a misunderstanding of European regulatory requirements. Clinical data were necessary to comply with the requirements for CE marking.
1915 A clinical evaluation report for Prosima dated 19 March 2007 was signed by Dr Robinson, who had taken over from Dr Weisberg as Medical Director of Ethicon Women’s Health and Urology.1935 It described Prosima as “a pre-shaped mesh cut from GYNECARE GYNEMESH PS role stock”. It noted that the Prosima performed a similar function to Prolift in that both systems involved the implantation of pre-cut Gynemesh PS to provide tissue reinforcement and long-lasting stabilisation of fascial structures of the pelvic floor and the placement of stabilisation straps (six for Prolift Total and four for the Prosima equivalent). It also noted that there were a number of differences. The similarities and differences were set out in the following table (without alteration):
Feature | GYNECARE PROLIFT System | GYNECARE PROSIMA System |
Mesh Implant | Pre-cut GYENECARE GYNEMESH PS | Pre-cut GYENECARE GYNEMESH PS |
Implantation Approach, Anterior Repair | Trocar passage per Obturator foramen with vaginal dissection | Vaginal |
Approach, Posterior Repair | Transgluteal trocar passage with vaginal dissection | Vaginal |
Anterior Mesh | Placed between bladder and vagina | Placed between bladder and vagina |
Posterior Mesh | Placed between rectum and vagina | Placed between rectum and vagina |
Anterior Fixation Straps (on each side) | Two on each side passed through the arcus tendineus fascia pelvi via the obturator foramin | One on each side, to paravaginal space against obturator internis fascia |
Posterior Fixation Straps (on each side) | One, passed through buttock and ischiorectal fossa and through the middle part of sacrospinous ligament | One, passed through tunnels to lay on the sacrospinous ligament |
Initial Fixation | Friction fixation of straps | Vaginal Support Device |
Permanent fixation | Tissue ingrowth into mesh | Tissue ingrowth into mesh |
1916 While acknowledging the “identifiable differences” between the two systems, Dr Robinson observed that they had many common characteristics, including intended use, approach and “reinforcement implant material”. In these circumstances he expressed the opinion that “the clinical acceptance of [Prolift] serves as an initial indicator of safety and effectiveness of [Prosima]” and he relied on literature relating to Prolift. It will be recalled that Prolift had been on the market at this point for two years.
1917 Later in the report, after a high level comparison of Prosima with Prolift, Dr Robinson observed that Prosima “appear[ed] to have some reduced risk to injury at implant insertion”, but went on to note:
The reliance on the VSD for initial stabilization along with 1) the reduced size of the implant 2) the reduced number of fixation straps and 3) the requirement for 2-piece insertion in all cases may limit the product's effectiveness in the most severe prolapse cases (Stage IV according to the ICS classification). Currently, clinical evidence for the use of the product in these severe cases is not available, and for this reason the product, cannot at this time, be indicated for these cases.1936
1918 He then proceeded to discuss the literature review undertaken earlier that month. He indicated that PubMed was the sole search engine used, and that the search encompassed only English publications from the previous three years, clinical trials, meta-analyses, controlled clinical trials, and reviews. Search terms were “pelvic organ prolapse”, “anterior colporrhaphy”, “posterior colporrhaphy”, and “prolapse repair mesh”. Notably no meta-analyses were produced in response nor any controlled clinical trials. After excluding articles deemed from their titles to be irrelevant or duplicates, the final list included eight papers describing clinical trials and 16 review papers but it appears that the review was limited to six papers in the former category and 11 in the latter (a total of 17) on the basis that they were “readily available”. It appears from the list of references that only seven or eight of these were cited. The remaining three were not identified. Additional literature collected during the development of previous products in the field of pelvic organ prolapse was also said to have been “consulted”. Some six of the 15 references pre-dated the three-year search period. The last of these is the Maher et al (2004) Cochrane review. Dr Allman was critical of the literature review for not following a systematic evaluation.1937
1919 The only article apparently relating to Gynemesh PS that was discussed in the report related to the predicted outcomes of Prolene Soft in hernia models. There was no discussion, however, of any potential differences in outcome for implantation of the mesh in the pelvic floor rather than the abdominal wall and hence the suitability of Gynemesh PS for pelvic floor repairs.
1920 Dr Robinson did refer to a study by Milani et al (2005) of 63 women, who underwent anterior and posterior repair using Prolene mesh.1938 Notably, women were excluded if they had not completed their family, were diabetic, or had a recognised or suspected immuno-suppression. These exclusions were not mentioned in the CER, however. After a mean follow-up of 17 months, the authors reported good anatomical outcomes with a success rate of 94% but a high rate of morbidity. They expressed concerns about a number of complications, including dyspareunia (which increased by 20% in the anterior repair group and by 63% in the posterior repair group) and erosion (in 13% of the anterior group and 6.5% of the posterior group). The authors concluded that the use of Prolene mesh “should be abandoned”.
1921 Dr Robinson adverted to only some of the study findings, and misreported them in parts. He wrote:
Milani concluded that good anatomical results where (sic) achieved when anterior and posterior vaginal prolapse was repaired with PROLENE mesh, but reported a high rate of morbidity. He concluded that PROLENE Mesh should not be used to reinforce these repairs. The two main concerns were erosion of the material through the vaginal wall and de novo dyspareunia. Mesh erosion through the vaginal wall was observed in 6.5% of the cases occurring after a mean time of 14 months. The author felt the reported change in sexual function among women treated in this series was the most clinically relevant finding. He did note that in an earlier series of patients who underwent similar posterior repair without the use of mesh reinforcement also reported a high rate of de novo dyspareunia after surgery.1939
1922 Three articles relating to Prolift were discussed or mentioned.
1923 One was the article by Fatton et al (2007) reporting on the early (three-month) results from a retrospective multicentre French study of 110 patients with stage III or IV pelvic organ prolapse conducted by the TVM Group.1940 A summary of the outcomes, apparently taken from the abstract, appeared in the Prosima CER, together with the following statement:
The authors concluded that the procedure and implants were safe, with early indication of effectiveness.
(Emphasis added)
1924 Noting that the follow-up was very short for a device installed for permanent use and the absence of a comparative arm, Professor Gordon disagreed with Dr Robinson’s statement.1941 Professor Hu observed that the report made no mention of whether patient symptoms or satisfaction outcomes were assessed.1942
1925 In fact, Dr Robinson’s characterisation of the authors’ conclusion was misleading, if not false. The conclusion of the authors that appears in the abstract was more guarded:
According to the peri-operative and immediate post-operative results, Prolift™ repair seems to be a safe technique to correct pelvic organ prolapse. Anatomical and functional results must be assessed with a long-term follow-up to confirm the effectiveness and safety of the procedure.1943
(Emphasis added)
1926 The conclusion appearing at the end of the article was that the findings of the study “suggest” that the transvaginal use of mesh using the Prolift technique is safe. Fatton et al (2007) stated that longer follow-up was necessary to confirm the effectiveness of the procedure and prospective evaluation of functional outcome was necessary in order to support the widespread use of the technique.
1927 In the body of the article Fatton et al (2007) observed that various issues needed to be addressed in future studies, including a prospective randomised trial comparing anatomical and functional outcomes of mesh reinforcement and site-specific fascial repair alone. They added that future studies should also include longer follow-up to assess procedure efficacy and to prove a low rate of long-term complications.
1928 The second article relating to Prolift, which was mentioned in the Prosima CER, was by Daniel Altman and Christian Falconer for the Nordic TVM Group, published in February 2007.1944 It was concerned with perioperative morbidity associated with transvaginal mesh repair of pelvic organ prolapse. Altman and Falconer (2007) explained in the article that their focus was on “immediate morbidity caused by the surgical technique rather than mid-to long-term complications such as rejection, erosion, and infections, typically ascribed to the biomaterials themselves”. They pointed out that prospective comparative studies were necessary to determine whether the transvaginal mesh procedures were beneficial compared to traditional suture techniques. As Professor Hu noted, because the reported outcomes were confined to perioperative morbidity, the study was of no utility in understanding long-term complications such as mesh erosions.1945
1929 This article related to the findings of a larger study than that reported by Fatton et al (2007) (involving 248 women in 25 centres in contrast to the 110 patients in the study by the TVM Group). The evidence was classified as level III but it was not discussed in the CER, let alone analysed. The Prosima CER did not mention that serious complications occurred in 11 patients (4.4%), all but one involving visceral injury. Indeed, all that was said about this paper was that it described perioperative and postoperative management of problems associated with Prolift.
1930 Altman and Falconer (2007) observed that much of the current knowledge of in vivo characteristics of biomaterials in pelvic floor surgery derived from research on midurethral sling procedures like TVT. They emphasised the importance of considering the different anatomical conditions associated with pelvic floor surgery, a point overlooked in Ethicon’s CERs that relied on articles relating to midurethral slings. They wrote:
Compared with suburethral tapes, biomaterials used at pelvic organ prolapse repair increase the biomaterial load considerably because of the increased size of the mesh. This may increase the risk for adverse tissue reactions and biomaterial-associated complications. Although the polypropylene compound used for TVT and trans- vaginal mesh is identical, other characteristics, such as elasticity and pore size, differ. One should, therefore, not assume that the biomaterial properties are the same for the two procedures, and results from incontinence surgery may not be directly applicable to pelvic organ prolapse surgery.1946
1931 They concluded that caution was advisable until long-term prospective safety studies describing biocompatibility were available. If this article was worthy of mention, one would have thought that so too was the conclusion.
1932 The third article relating to Prolift, which was mentioned in the Prosima CER, was by Collinet et al (2006) on the TVM technique, mesh exposure management, and risk factors.1947
1933 Collinet et al (2006) was a report on a retrospective case series of 277 patients two months after surgery. Mesh exposure occurred in 34 of the women (12.3%), 25 of whom (9%) required surgery in which part of the mesh was removed. The focus of this paper was on mesh exposure and associated risk factors, identified as concomitant hysterectomy (OR=5.17 [p= 0.001]) and inverted T colpotomy (a form of vaginal incision surgery, OR=6.06 [p=0.01]). No data was presented on patient symptoms or satisfaction outcomes, such as urinary urgency or dyspareunia.
1934 The CER noted only that the article identified factors that appeared to influence the occurrence of mesh exposure and, despite its obvious relevance, did not refer to its conclusion:
Nowadays, based on these data, we can only advise that caution be exercised when carrying out this new surgical procedure. 1948
1935 In the final section of the literature review, Dr Robinson referred to the conclusion of Maher et al (2004) that there was a lack of sufficient evidence about the effects of different surgeries to treat pelvic organ prolapse and insufficient evidence about the most common types of prolapse surgery. Consequently, Dr Robinson inferred that, to rely on the literature evidence for a particular type of pelvic floor surgical intervention, would not appear possible. His conclusion is therefore surprising. It reads:
Although inconclusive, there is literature that suggests the use of reinforcement synthetic mesh (specifically Type 1, macroporous mesh) has a place in providing added support to weakened pelvic floor tissue. Further, that a standardized approach to pelvic floor repair, like the standardized approaches that have been developed to treat stress urinary incontinence, appear to provide a benefit to patients and allow for a better understanding of the treatments. One such system, the GYNECARE PROLIFT Pelvic Floor Repair System has reported results and surgeon acceptance consistent with a product that meets a need for the management of this difficult and prevalent condition. The subject of this review, the GYNECARE PROSIMA Pelvic Floor Repair System, shares a number of characteristics with the GYNECARE PROLIFT System. The GYNECARE PROSIMA System is expected to meet the product indication, to provide tissue reinforcement and long-lasting stabilization of fascial structures of the pelvic floor. The performance expectation of the system is further supported with a clinical series conducted at two centers.1949
1936 Dr Robinson proceeded to discuss the clinical investigation conducted by the “the surgeon who developed the surgical procedure”, presumably Dr Marcus Carey. The analysis was said to be based on a draft manuscript of a study involving 95 patients. The manuscript was not tendered but the study appears to be the prospective observational study, which reported initial results on the use of a Prosima-like device, later published by Professor Carey and others in 2008 in the British Journal of Obstetrics and Gynaecology (the Carey study).1950 According to Dr Robinson, “the results of this study represent evidence of the safety and effectiveness performance of the [Prosima] system”, although the procedure and design of the mesh used in Prosima had “slightly” changed since the study had been conducted.1951
1937 The Carey study was a single arm non-randomised study. The women involved had prolapse assessed as POP-Q stage II or more and underwent surgery with Gynemesh PS (cut to reflect the device later marketed as Prosima) and a vaginal support device. They were followed up at six and 12 months. As Professor Gordon observed,1952 it is not clear how many patients were evaluated at the 12 month mark. Objective success rate was 92% at six months and 85% at 12 months. Subjective success rate was 91% at six months and 87% at 12 months. There were four (4.2%) mesh exposures, two of which required surgery. The number of women reporting sexual dysfunction decreased from 58% pre-operatively to 23% at 12 months. Quality of life scores were said to have significantly improved at 12 months compared with baseline.
1938 Although no criteria were identified in the CER for determining what level of risk was acceptable, Dr Robinson stated that the findings of the Carey study were acceptable and “agree[d] with the expectations derived from the literature review”. He did not acknowledge, however, the short period of follow-up or the possibility of an increase in erosion rates over time. The authors of the published article were cautiously optimistic. Their final words were:
This study reports encouraging outcomes with the surgery described. Further clinical studies, including comparative studies, are required to establish the role of this surgery.1953
1939 Since the manuscript was not in evidence, I am unable to tell whether these words were added after the peer review process or appeared in the manuscript.
1940 Be that as it may, the respondents were prepared to affix the CE mark and release the Prosima device to the market before the additional clinical studies Carey et al said were necessary to establish the safety and efficacy of the device had been conducted and notwithstanding the conclusion of the Maher et al Cochrane review.
1941 Dr Hinoul deposed that the results of the clinical investigation represented evidence of the safety and effectiveness of Prosima.1954
1942 Professor Gordon disagreed. He said that the evidence available at the time of the release of each of Gynemesh PS, Prolift and Prosima was weak; reliance was placed on case series and studies with short-term follow-up, and there were no results from randomised controlled trials.1955
1943 That opinion would have come as no surprise to Ethicon. The minutes of a meeting held on 31 May 2007 (a little after three weeks since Prosima had been released in Europe), attended by Ophelie Berthier, Axel Arnaud, Judi Gauld, and Dhinagar Subramanian, relevantly record that:
A single observational study with 12 month follow-up is unlikely to be enough to satisfy the needs for clinical and HE&R [Health Economics and Reimbursement] data.1956
1944 They recommended that future RCTs be “considered”.
1945 The next part of the Prosima CER dealt with complaint/adverse events pertaining to Gynemesh PS and Prolift from the time of their original release to January 2007. This consisted of a summary of the numbers and types of complaints over the period derived from Ethicon’s records and the MAUDE database. As already noted, the numbers were low. But in this report as in all others there was no acknowledgment or consideration of the unreliability of complaint data.
1946 The overall conclusion was that the literature review and the clinical data together provided evidence that:
[Prosima] … will provide tissue reinforcement and long-lasting stabilization of fascial structures of the pelvic floor, maintaining the vaginal canal during the healing period. Currently, evidence is lacking for the use of this product in very severe (ICS Stage IV) prolapse.1957
1947 Noting Dr Robinson’s reference to the lack of evidence for using the product to treat patients with stage IV prolapse, Dr Hinoul drew attention to the warning in the Prosima IFU to that effect and to the corresponding statement that the use of Prosima in such patients was “not recommended”. 1958
1948 Apart from complications relating to the Prosima technique, no potential complications were identified. Dr Robinson said that he consulted the final drafts of the dFMEA and the aFMEA to assess the risk/benefit profile of the Prosima system. But he did not disclose the outcomes. He also referred to the Risk Management Report. Once again, however, he did not disclose its conclusions. Nor did he undertake a risk/benefit analysis himself. In the section entitled “Risk?Benefit Analysis” he stated:
Because, as Maher described in the Cochrane Review (Ref. 14), there is insufficient evidence on the effects of different types of surgery for pelvic organ prolapse, defining a standard for acceptable performance is difficult.1959
1949 Although the report included a table comparing Prolift with Prosima it did not include a comparison with procedures, such as native tissue repair, that do not involve the use of polypropylene mesh.
1950 Moreover, as Professor Hu noted, the report is devoid of “any kind of summary discussion or conclusions regarding the safety of the mesh product”.1960
1951 Despite citing the Maher et al (2004) Cochrane review,1961 Dr Robinson did not refer to the following discussion it contained on mesh augmentation in prolapse surgery:
The use of mesh to augment repair surgery has been successful in other fields such as groin hernia repair (Scott 2004). However, particular issues related to its use in vaginal repair concern the effect on bowel, bladder and sexual function and the possibility of mesh erosion or infection. Evidence from case series suggest possible concerns. Salvatore et al reported functional outcomes after a polypropylene mesh overlay at vaginal repair including a mesh erosion rate of 13%, overactive bladder increasing from 28% to 56% and dyspareunia increasing from 18% to 38% postoperatively (Salvatore 2002). Visco et al suggested that the mesh erosion or infection rate was increased four-fold when mesh was introduced vaginally as compared to the abdominal route in the management of pelvic organ prolapse (Visco 2001).
The evidence supporting the use of polyglactin mesh repair for anterior vaginal wall prolapse came from two small trials with conflicting results and is not sufficient to support the use of permanent meshes or grafts at the time of vaginal repair surgery except in the context of randomised controlled clinical trials. These trials must be adequately powered to evaluate the anatomic and functional outcomes and possible adverse events.1962
1952 Professor Hu observed that most of the studies to which Dr Robinson referred only reported short-term results (two years or less), were restricted to interventions in one compartment, and/or were lacking the collection of data on outcomes such as dyspareunia or chronic pain. He also noted that the CER did not appear to contain any conclusion regarding the safety and efficacy of the Prosima device in particular or polypropylene mesh in general.1963
1953 The minutes of a meeting Ethicon appears to have convened in London on 26 April 2008, over a year after the CER was signed, noted that the bottom line was that Prosima needed to “meet the challenges of being non-complex, safe and effective”. According to the minutes, the meeting considered data from the investigators’ case series, most of which reported on six month follow-up. While the results were said to be promising, the follow-up period was admittedly “not long” and the number of patients followed up by each investigator was “small”. At six months, the failure rate was 26.8%. Nevertheless, in the concluding remarks the minutes noted that “[a]ll present agreed that the product was safe”. Upon what basis this view was reached is not apparent from the evidence.
1954 One of the attendees, identified as “HZ” (likely Halina Zyczynski, an investigator), was said to have commented on a “6 or 7-fold increase in mesh exposure for smokers – CARE study in US”. It was noted that patient smoking details should be collected at the 12 month visit. Although the applicants make no complaint about it, no warning or advice to this effect was included in the IFUs in evidence for any of the Ethicon devices at any time.
1955 In an email dated 11 April 2008 to David Robinson, Judi Gauld and others of Ethicon, Dr Carey had proposed giving context at the Investigator Meeting “to better frame the clinical data”.1964 An internal Ethicon discussion followed, including an email from Judi Gauld (then Manager, Clinical Research and Development) which stated:
We did state in the protocol that the study would be considered a success if the failure rate had an upper 95% CI of less than 20% at 12 months. We have already failed that.
I am concerned here that this looks like a good bit of spin going on, and due to his commercial interest, this is not going to come over as objective as perhaps it should.
PART IX: POST-MARKET EVALUATION OF THE DEVICES
Introduction
1956 There is no dispute that the respondents were obliged to undertake post-market surveillance and evaluation of the safety and efficacy of the Ethicon devices. The regulatory requirements and guidelines are discussed above in Part VII. They included monitoring the clinical performance of the devices in the marketplace; undertaking design review and risk management exercises to optimise the safety and efficacy of the devices; monitoring and investigating adverse events; reporting them to competent authorities (generally referred to as “vigilance reporting”); and deciding on corrective or preventative actions.1965 They also required taking pro-active steps to obtain feedback on safety and efficacy, monitoring the literature, and collating and assessing all the information.
1957 Dr Allman explained that there are three categories of post-market surveillance:
• Reactive monitoring of the information received by a manufacturer—complaints, adverse events, publications. Logically, this cannot be used to determine either efficacy or effectiveness. It can (and must) be used as a monitoring tool that might provide an alert to a quality problem with the device, or an alert to a design problem associated with new or expanding use of the device. The reliability and usefulness of reactive monitoring of complaints/adverse events depends on the manufacturer’s complaint reporting process and the willingness of users (or others) to report complaints or events.
• Post-market studies set-up to assess the device in real-world use. Most of these studies are ‘registries’ in which investigators record patient information and outcomes for review and analysis. Investigators use the devices as part of their normal practice. The advantage of a registry, compared to monitoring complaints and adverse events, is that it ensures consistent reporting of information. Such studies can be viewed as establishing effectiveness. In European terms, these would be described as post-market clinical follow-up studies.
• Pre-market studies extended into the post-market phase, or additional clinical investigations to assess a particular performance/safety issue or question. In European terms, these would also be described as post-market clinical follow-up studies. Long-term follow-up of patients recruited into pre-CE marking studies (for example) can be used to assess longer-term safety and performance, with CE marking legitimately based on shorter-term follow-up—though not ‘preliminary’ results from the investigation. Follow-up, and decision, points need to be pre-defined in clinical study protocols, etc. If additional clinical investigations are intended or implemented to answer questions of safety or performance, the decision to proceed to, or continue, CE marking requires justification.1966
1958 Dr Hinoul described Ethicon’s post-marketing activities in his affidavit. In sum, he referred to the gathering of post-market surveillance data identifying risks through trends in complaints data, analyses of relevant scientific literature, complaints and adverse events data, MAUDE and data from the UK Medicines and Healthcare products Regulatory Agency (MHRA), medical device reporting, and medical device vigilance reporting. I referred earlier in these reasons to the MAUDE database, the FDA’s adverse events database. Dr Allman said it was not clear what Dr Hinoul meant by MHRA data (and Dr Hinoul did not explain what he meant in his oral evidence), since the MHRA does not have a publicly accessible adverse events database.1967 Medical device reporting and medical device vigilance reporting are mandatory for medical device manufacturers. The terms are synonymous. Their purpose is to notify regulators of adverse events. The former is the US term, the latter the European.1968
1959 As Dr Allman observed, this was a description of “a basic form of post-market surveillance relying purely on the information reported to Ethicon”.1969 Dr Allman went on to point out that:
Dr Hinoul does not explain, nor justify, the decision to conduct only reactive post-market surveillance and not to conduct post-market clinical follow-up studies as required by authoritative European regulatory guidance. He does not address the well-known deficiencies of reactive surveillance—the probability of underreporting of complaints and adverse events, and problems of publication bias/delay in the scientific literature, for example. He does not address the difficulties with post-market surveillance when there are no ‘baseline’ criteria for safety and performance determined by clinical investigation or evaluation.1970
1960 Dr Allman’s opinion was that Ethicon displayed a poor understanding of the European regulatory requirements for clinical evaluation and post-market surveillance. He emphasised that these requirements were and are more demanding than the corresponding requirements in the United States.1971
1961 Based on the materials provided to her, Associate Professor Krulewitch concluded that there was no evidence that the respondents had conducted any post-market studies on the SUI devices and that the respondents had no systematic plan to monitor potential new complications and late complications of those devices.1972 That is not to say that she endorsed Ethicon’s post-market evaluation of the POP devices, rather the applicants did not seek her opinion on the question.
1962 Associate Professor Krulewitch’s evidence was consistent with the views of the regulatory experts. Although Associate Professor Krulewitch appears to have been primarily retained because of her expertise as an epidemiologist, it will be recalled that she was previously employed by the FDA’s Center for Devices and Radiological Health in the Division of Post-Market Surveillance and, while working at the FDA, she oversaw the evaluation of reports of adverse events related to surgical mesh. She was therefore well-placed to give the evidence.
1963 The applicants argued that the post-market evaluation of all the Ethicon devices was inadequate, and in some cases totally lacking, and failed to conform to the regulatory requirements.1973 Their submissions on this question were detailed and the response to them largely superficial.
1964 The respondents relied principally, and in some cases exclusively, on the clinical evaluation reports. Each of these reports included a description of the device in question and its intended application together with reviews of the literature and of complaints made and adverse events reported to the respondents and to one or more of the regulators. The conclusion drawn in each of them was that the data derived from the literature review, taken together with any available pre-clinical data, were sufficient to demonstrate compliance with the essential requirements covering safety and performance of the device in question under normal conditions of use. Each of them pronounced that no additional clinical data was required.
1965 The respondents made three points.
1966 First, they submitted (apparently on the basis of Dr Hinoul’s evidence) that CERs were prepared every three years and that they analysed company sponsored studies, relevant and available clinical data, and scientific literature relative to the safety and performance of the devices.1974 They argued, by reference to the CERs alone, that it was “plain” that “Ethicon conducted regular, rigorous and detailed investigations on its products”.1975
1967 Second, to the extent that the applicants relied on the evidence of the regulatory experts, as I observed above, they dismissed it as irrelevant or of limited assistance and did not engage with the substance of it.
1968 Third, they submitted that, as part of preparing the CERs, the respondents regularly reviewed complaints and adverse events. They did not attempt to defend the review. Rather, they submitted that, if the review was said to be inadequate, then it was up to the applicants to prove how a different process would have produced a different result.
1969 I deal with the CERs in detail below. It is sufficient at this juncture to make the following observations.
1970 CERs were indeed produced, but not as often as the respondents submitted or at the intervals they asserted. For the most part, these reports could scarcely be described as evaluations at all. In short, Ethicon’s post-market clinical evaluations were neither regular nor rigorous and, not until 2012 at the earliest, could they be described as detailed. Furthermore, after Ethicon outsourced the preparation of the CERs in about early 2010, it is difficult to see how it could be said that they regularly reviewed complaints and adverse events as “part of the preparation” of those reports.
1971 As I observed in Part VII above, the regulatory evidence cannot be dismissed or side-lined. Amongst other things, that evidence demonstrates that the CERs were deficient in numerous respects, review of complaints and adverse events was unsatisfactory, and the conclusions drawn from them largely unjustified. The reports were criticised by Dr Allman and, in some cases, also by the TGA and Ethicon’s notified body, BSI, and found to be wanting. But Dr Hinoul did not answer those criticisms and the respondents failed to address them either in their written submissions or in oral argument.
1972 In the circumstances, unless otherwise indicated, I accept that those criticisms were well-founded.
Monitoring of complaints made to the respondents
1973 The respondents received numerous complaints about the Ethicon devices.
1974 In their written submissions the applicants referred to an email sent on 15 December 2005 by Carolyn Brennan, a Worldwide Customer Quality Project Manager at Ethicon, to Dr Robinson, Ethicon’s Medical Director. The email concerned TVT only.1976 It attached two files. The first was an Excel spreadsheet of year to date (November 2005) data.1977 It disclosed that 227 complaints had been received including 143 categorised as “patient events”, the remaining 84 as device-related. The patient events included: de novo detrusor instability (1), hernia (1), incisional bleeding (1), migration of tape after implant (1), nerve damage (2), haematoma formation (3), vascular injury (3), allergic reaction (4), intra-operative complication (7), infection (11), post-procedural incontinence (11), pain (13), urinary retention (13), vaginal/bladder/urethral erosions (19), bowel/bladder/vaginal/urethral perforation (22), and post-operative complications not otherwise described (31). The largest number of “device-related” complaints was mesh fraying (18). No information was included in the document about the dates of implantation and no particulars were provided.1978
1975 The second data set was contained in a 762 page document entitled “All TVT: 01-01-97 through 11-15-2005”.1979
1976 I was also taken to an email chain from February and March 2006 in which Kevin Mahar, US Vice-President, Sales and Marketing, for Ethicon Women’s Health & Urology, flagged a proposal to recommend that the board “have some formal review of complications and the communication processes from time to time (not just specific to TVT, but all products)”.1980 The obvious inference and the inference I draw is that there was no system of formal review at this time.
1977 The email chain included an email from Dr Robinson in which he said he thought that it would be helpful to have “a discussion [about] how to create and use complication data across all the product line (sic) and also how awareness of those complications should be used in periodically reviewing our IFUs [instructions for use] and Warnings and Precautions”.1981 If Ethicon had been attentive to its post-market obligations, such a discussion would have taken place many years earlier and appropriate action would have been taken.
Reports to regulatory authorities of adverse events
1978 Dr Pence reviewed the complaints recorded in the MAUDE database for TVT, TVT-O and the POP devices. Her analysis showed that the five most commonly reported adverse events for TVT over the period from 1999 to 2010 were: urinary problems; erosion; pain; bleeding; and sexual dysfunction.1982 The five most commonly reported events for TVT-O from 2004 to 2011 were the same, save that “bleeding” was replaced by “infection”.1983 The most commonly reported events for Ethicon’s POP devices combined over the period 2002–2012 were erosion/exposure, pain, and dyspareunia.1984
1979 The applicants submitted that, because there was under-reporting of events in practice, the incidence of events derived from the MAUDE database was unreliable. The significance of the review undertaken by Dr Pence, they argued, was that it demonstrated that, if Ethicon had effectively monitored the MAUDE database, it would have encountered “some clear signposts as to issues upon which effective post-market evaluation” of the products should have been focused.1985
1980 The applicants further submitted that Ethicon had a practice of dismissing (and determining not to report) complaints on the basis that they were the subject of adequate warnings in the IFUs, even though that was not the case.1986 Ethicon would then proceed to feed back into its clinical evaluation process low adverse event rates as evidence of the safety of its products.1987
1981 The evidence supports all these submissions.
1982 Dr Pence gave evidence about Ethicon’s reporting of adverse events to the FDA. In the United States, manufacturers of medical devices are required to report to the FDA all post-market events where the device had or may have caused or contributed to a death or serious injury, or had malfunctioned, and that device (or a similar device marketed by the manufacturer) would be likely to cause or contribute to a death or serious injury if the malfunction were to recur. “Serious injury” is defined as an injury or illness that was life threatening; resulted in permanent impairment of a body function or permanent damage to a body structure; or necessitated medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure. A “malfunction” is defined as a failure of the device to meet its performance specifications or otherwise perform as intended. Performance specifications include all claims made in the labelling for the device. Malfunctions of long-term implants are reportable if the malfunction is likely to cause or contribute to death or serious injury if it recurs.1988
1983 As I indicated above, these reports to the FDA are termed “medical device reports”. As noted above, since August 1996 medical device reports received from manufacturers have been recorded in MAUDE, together with voluntary reports (for example, from physicians and consumers, included since June 1993), distributor reports (since 1993), and user facility reports (since 1991).1989
1984 Dr Pence provided 29 examples of complaints received by Ethicon in relation to its SUI devices which, in her opinion, were reportable but were not submitted to the FDA as medical device reports.1990 There was a range of complaints which Dr Pence grouped into three broad categories: erosions/extrusions; organ perforations; and other product malfunctions, such as fraying and unravelling.1991 Dr Pence pointed out that Ethicon had reported erosions/extrusions and organ perforations on numerous occasions in the past. She could not see why events of this kind were not always reported. She considered that many of Ethicon’s explanations were disingenuous and led to under-reporting of adverse events. Dr Pence also gave evidence that a reasonably prudent manufacturer would have performed due diligence to follow-up some of these events to determine if there were any longer-term sequelae.1992
1985 It is unnecessary to refer to each report discussed by Dr Pence. A few examples will suffice.1993
1986 On 30 January 2002, Ethicon was notified of a patient presenting with a “minimal asymptomatic vaginal extrusion”. Ethicon did not report the event on the basis that there was no death, serious injury or device malfunction. Dr Pence said that it was speculative for Ethicon to conclude that the extrusion would not worsen or require further treatment. She noted that a serious injury need not have taken place for an adverse event to be reportable; it is sufficient if the device would be likely to cause serious injury.
1987 Another notification about a vaginal extrusion was received on 21 August 2002. Ethicon did not report the event because it considered it likely that the event was caused by an episode of post-operative coughing. Dr Pence described Ethicon’s response as disingenuous, given that it knew that extrusion was an adverse reaction associated with tape implantation. In any event, she said, if post-operative coughing were the cause of an extrusion, then the IFU should have been amended to identify this possibility.
1988 Dr Pence made a similar point about other occasions where erosion, extrusion or exposure was attributed to something other than the Ethicon device. Where the adverse event was attributed to the use of corticosteroids, for example, Dr Pence said that the IFU should have been amended to include use of corticosteroids as a risk factor. Similarly, where Ethicon declined to report exposures on the basis that they were influenced by such factors as “inadequately estrogenized vaginal mucosa, trauma, improper closure and other things”, Dr Pence said that, if that were the case, then these matters should have been disclosed in the IFUs.
1989 At least two instances of organ perforation were not reported on the basis that they resulted from surgeon practice or error. Dr Pence criticised this reasoning, as the relevant regulation does not exempt from reporting requirements events caused by user error (the rationale being that these events may still alert the FDA of the need for improved labelling to prevent future injuries).
1990 Dr Pence undertook a similar exercise in respect of the complaints received for the POP devices. She reviewed 116 reports that were determined to be non-reportable, and provided a number of examples of events that she considered should have been reported.1994
1991 In April 2008, for instance, Ethicon received a report from a doctor about three patients with “significant dyspareunia” after implantation of Prolift.1995 In the report the doctor noted:
I have had significant problems with the Prolift kit in my patients … I have 3 current patients with dyspareunia, 2 additional ones that I have taken back to the OR for excision of mesh. I have currently discontinued use of Prolift due to these concerns.
1992 Ethicon determined not to report the event on the basis that there was “no evidence to suggest that the device itself caused any impairment or damage to body function or body structure”; “[p]ost-operative complications can occur as a result of multiple factors, including patient characteristics, nature of treatment rendered, and various extraneous factors”; and “[t]here is no indication of medical or surgical intervention.”1996 Dr Pence observed that the final statement was false, as the doctor had noted two cases that required surgery for mesh excision.
1993 In April 2006 Ethicon received information about a patient who had a urethral extrusion three to four months after surgery with Prolift Anterior.1997 Ethicon decided not to report the event because the mesh was trimmed in the doctor’s office. It also described the event as “not reportable” because the “event occurred post- procedure and no actual device malfunction is cited or indicated”; there was “no evidence to suggest that the device itself caused any impairment or damage to body function or body structure”; and because “[p]ost-operative complications can occur as a result of multiple factors, including patient characteristics, nature of treatment rendered, and various extraneous factors”. Dr Pence suggested that this appeared to be a standard comment in relation to exposures. She was critical of Ethicon for not reporting the event, particularly when the French and US studies of the TVM technique had reported exposure rates of 10% and 14.1% respectively.1998
1994 In July 2008 Ethicon received a complaint regarding a patient undergoing a “disability evaluation” after surgery using a Prolift Total device.1999 The patient was reported to have a “constellation of symptoms including urinary incontinence, frequency, and some bowel complaints.” The case was determined to be “a third party litigation”. Ethicon’s rationale for not reporting this event was:
No actual device malfunction is cited. There is no evidence to suggest that the device itself caused any permanent impairment or damage to a body function or body structure. There is no indication of medical or surgical intervention. Pelvic floor with Prolift is not intended to treat or prevent urinary incontinence. Every pelvic floor repair procedure is associated with a risk of post-operative incontinence resulting from the change in the anatomical relationships of the pelvic organs and tissues. Urinary incontinece (sic), urgency and ‘bowel problems’ could very well be existing symptoms associated with anterior and posterior pelvic organ prolapse.2000
1995 Dr Pence criticised this rationale. She said that, if the manufacturer becomes aware of information reasonably suggesting that its device may have caused or contributed to a serious injury, it is required to report that information to the FDA. She said that the contribution of the device to the events reported could not be ruled out. She pointed out that, since Ethicon knew that the US study on the TVM technique had reported that 20% of the study subjects experienced urinary incontinence, it was reasonable to consider that Prolift may have contributed to the reported events.2001
1996 Similarly, on numerous occasions Ethicon determined that adverse events were not reportable to the UK regulator, the MHRA.2002
1997 On at least one occasion, the MHRA advised Ethicon that its decision not to report an event (of a patient experiencing mesh exposure, recurrent incontinence and dyspareunia after TVT surgery) was not compliant with MEDDEV 12.12.1, the guidelines on post-market clinical follow-up.2003 The MHRA advised that “expected and foreseeable side effects are only not reportable if the adverse incident is clinically acceptable in terms of the individual patient benefit”.2004 In response, Ethicon determined to write to the MHRA reiterating its position that the event (and others like it) should be characterised as non-reportable.2005 An invitation to contact the MHRA “to try and understand their perspective” was not apparently taken up.2006
1998 The applicants also pointed to a number of other occasions where Ethicon determined that an event was not reportable when, according to the applicants, it should have been reported to the MHRA. One such instance related to the following complaint forwarded for consideration to Dr Hinoul on 18 November 2015 (without alteration):
TVT has caused pain infection discomfort affected my life greatly. TVT fitted by an urologist in 2005. It worked for stress incontinence. Months after the op I had pains all over my body and was told I have fibromyalgia. In this last year 2015 I have had increased UTI and vaginal discharge, unable to have sex as its too painful and I bleed. I have now discovered the mesh has come through my vaginal wall. I'm in pain and a bloated abdomen.2007
1999 Dr Hinoul took the view that no report needed to be made since exposure was warned about in the IFU so the patient should have been counselled about it, fibromyalgia was not a consequence of the tape placement, and, in any event, “technically” the event was “clinically acceptable in terms of the individual patient benefit”.2008 He did not explain why the event was clinically acceptable.
2000 A couple of weeks later, Dr Hinoul was asked to consider the following complaint arising from implant surgery that took place in May 2013. The complaint was in the following terms:
When i awoke from the operation i complained about a pain in my buttock of which i got told it was normal. I was discharged the day after the operation. Shortly after i had electric pains shooting down my leg and is now burning in the right buttock and back and inside thigh. I have a cutting pain in my pelvis/groin area like something is cutting my vagina. sex in some positions is so painful i have gone celibate. The pain is getting worse were i am a recluse and a prisoner in my own home. I cannot sit on my right hand side and can hardly bend my right hip as of the electric pains shooting down my leg. I had a private scan and showed partial erosion, loop on the right hand side and the right arm is out of place.2009
2001 In his response to the request, Dr Hinoul wrote that the complications were “within what can be expected for these procedures”, relying on a number of statements that were only added to the TVT IFU in October 2015 and determined that the event was not reportable on the basis of those statements. When this was pointed out to him during cross-examination, he acknowledged the difficulty, but claimed that he “could have just as well justified it with the earlier IFU”.2010 He argued that the following statement in the earlier IFU could have captured it:
Punctures or lacerations of vessels, nerves, bladder or bowel may occur during needle passage and may require surgical repair.2011
2002 I regard this evidence as disingenuous since it was not apparent from the information Dr Hinoul was given what it was that had caused the patient’s pain.
2003 On another occasion, Dr Hinoul considered that a complaint of chronic pain did not need to be reported as “pain is a potential post-operative complication related to any kind of continence procedure”.2012 As will be seen, that opinion was not shared by the regulatory authorities.
2004 I accept the applicants’ submission that Ethicon’s approach to complaints reporting limited the capacity of the regulatory authorities to adequately determine the safety of the devices.2013
2005 The respondents’ internal documents showed that they recognised that inadequate reporting was an issue. A JJM presentation entitled “Project Blackbelt December 2012” identified the issue and the following as priority matters that needed to be addressed:
1. Non conformance that compromises patient safety
2. …
3. Non conformance that compromises obligations to TGA
…2014
2006 JJM’s failure to adequately report complaints, and thereby conform to regulatory requirements, was drawn to its attention in an email sent on 9 November 2012 from Dr Meng Chen, then Associate Medical Director of Worldwide Customer Quality, to Dr Glen Mason, Director of Medical Affairs at JJM. The issue arose in respect of a complaint with which JJM had not dealt for well over a year.2015 When asked for follow-up, Dr Mason replied that “no further information [was] forthcoming” and that the file should be closed. Dr Chen replied:
According to the US federal Food, Drug and Cosmetic Act and health laws/regulations in other countries and regions, “closing a complaint file containing a serious adverse event with the level of information as currently available in this particular file” (as I understand your advice), is simply out-of compliance.
The company process (PR-00000118, product complaint handling) requires that we conduct diligent product quality investigation as well as clinical investigation for each case containing serious adverse event and death. The outcome of the investigation is a conclusion on whether and how our product caused or contributed to the reported adverse event. The conclusion has to be justified with the evidence (quality and clinical) we obtain during a complaint investigation. The conclusion and the investigation process have to be reported to the US FDA (for all cases with SAEs worldwide) and to local country or regional health authorities (if occurred outside US) following strict timelines. We, here at the WCQ, conduct investigations occurring in the US. We rely on our affiliates such as staff in your own organization to conduct direct, timely and in depth investigation for events occurred in other countries and regions…
…The local affiliates are expected to document the timeline of the investigation attempts and the respondents’ detailed response. These responses may range from a thorough answer to the questionnaire or we will be unable to provide anything on legal grounds or no response and no explanation. In this case, “no information is forthcoming” from a JNJ employee simply does NOT meet the requirements of the US and the international laws and regulations. The company process requires the exact event reporter's response and the time of the response to our request for investigation be documented in the complaint file and be reported to the US FDA and international health authorities. A hypothetical example is “Dr. Smith has written to us on MM/DD/YYYY, No information is forthcoming considering patient X privacy” (scil). Other documented reason may be “we have made requests to Dr. Smith on these dates via email and fax, the email and fax requests have been forwarded to the Ethicon WCQ and have been attached to the complaint file”.
Again, using this case as an example, we only know that a hematoma formed and it led to a drastic drop in hemoglobin level in the patient. But we do not know what injury caused the extravasation, whether it occurred during the pelvic tissue dissection or during the insertion of the Prosima Inserter; whether the patient had an anatomical deviation of a pelvic vessel, or the operating physician was new to Prosima System and forced an Inserter onto a vessel. We owe it to the patient, the surgeon, the regulators and ourselves to find out, to document and to report our findings in detail and accurately! 2016
2007 Dr Chen’s commitment to investigating all complaints of serious adverse events was certainly not reflected in the approach to reporting taken by Dr Hinoul.
2008 Nine audit reports from BSI were in evidence. The first was dated 3 June 2008. It was prepared by Karl Reese. Mr Reese reviewed the technical documentation for Gynemesh PS, Prolift+M, Prosima, and other items which are not relevant to this case.2017 The last, dated 14 May 2014, by Laurel Macomber, was irrelevant since it was not concerned with the documentation for any of the Ethicon devices.2018
2009 In June 2008 BSI found serious deficiencies in post-market surveillance with respect to Gynemesh PS, Prolift+M and Prosima.2019 Although Ethicon devised a Corrective Action Plan (CAP) to address them which included a commitment to update “all Ethicon legacy technical files” by the end of the first quarter of 2009. As I explained in Part VII, “legacy” device was a term used by Ethicon to describe devices released under a risk management program pre-dating version seven of Ethicon’s Company Procedure for Medical Device Risk Management Plan, known as PR602-003, which was issued on 31 January 2004.2020 In this version of PR602-003, Ethicon’s risk management process was overhauled in order to comply with ISO 14971, published in 2000.2021
2010 In June 2009, BSI’s findings were raised to major non-conformity status after Ethicon’s CAP was rejected because it contained “insufficient information for Post Market surveillance feeding into Risk and Clinical, as well as insufficient details on implementation dates”.2022
2011 By 2010, Prolift and TVT Exact were included in the BSI assessment. In 2011, additional minor non-conformities were identified, which included the failure of the risk management files and the CERs to meet requirements.2023 Importantly, with respect to all files, BSI noted that the risk management report only appeared to have been completed on the basis of complaints, only addressed design/application risks, and did not consider process risks or other post-market surveillance activities.2024 BSI required a CAP as a precondition for continued certification.2025
2012 BSI also found that none of the CERs was consistent with the requirements of MEDDEV 2.7.1 for: clear description of search methodology; clear exclusion data; citation of weighting methods; details of specific analysis of each cited paper; demonstration of equivalence; clear link to post-market surveillance; and because there was insufficient justification for the lack of post-market follow-up.2026
2013 In an audit in September 2012, BSI discovered that none of the risk management files had been finalised in accordance with the previous CAP; few of the CERs had been updated as required by another CAP; and the updated implementation plans indicated that completion may take three years, well-beyond the original or extended timelines.2027
2014 Having regard to the failure to implement corrective action in accordance with the CAP and the result of an audit of the technical files for Prolift and Gynemesh PS, the minor non-conformities were elevated to major non-conformities.
2015 The auditors identified four major non-conformities, including the failure of the risk management files and the CERs to meet the requirements of the European Directive.
2016 None of the risk management reports had been finalised and “very few” CERs had been updated in accordance with the CAP to enable the CERs to include deficiencies that could have an impact on the risk management reports. No files had both an updated CER and a risk management report.2028 Only four of 119 technical files included CERs which conformed to the new procedures. The auditors considered that these non-conformities were indicative of a “breakdown in the management system to effectively control the activities for which they [were] intended”.
2017 Updated implementation plans for the CAP indicated that completion would not be finalised until the end of the third quarter of 2015.2029
2018 The significant findings of the audits are discussed in more detail below in relation to the devices covered by the files BSI examined. It should be noted, consistent with Dr Allman’s evidence discussed in the earlier Part of this judgment on Ethicon’s pre-market evaluations, that the auditors made it clear that their assessment was based on sampling. Consequently, as was pointed out in some of the BSI reports, “nonconformities may exist which have not been identified”.2030 In these circumstances and having regard to BSI’s findings, it is reasonable to infer that the non-conformities were not confined to the audited files and were more likely than not equally applicable to Ethicon’s evaluations and risk assessments for the other Ethicon devices.
The SUI devices
TVT
2019 The parties disagreed about the identity of the first post-market CER on TVT. The applicants submitted that it was the report signed by Drs Weisberg and Robinson on 7 March 2006,2031 some nine years after the device was approved for sale in Europe and nearly seven years after it had been on the market in Australia. I will come to this report in due course.
2020 The respondents, on the other hand, suggested that it was a report dated 15 June 2000,2032 shortly after the launch of TVT in the United States and Australia. The applicants, for some obscure reason, characterised it as a CER supporting the launch of TVT.2033
2021 The report dated 15 June 2000 was entitled “Benefit–Side Effect Analysis for TVT Device”. It was apparently signed by Dr Richard Isenberg, the then Director of Medical Affairs, Gynecare Products Division. I was not taken to any evidence indicating the circumstances in which it was prepared. It does not purport to be a CER but the copy tendered by the applicants is preceded by a page, presumably taken from the TVT technical file, which is entitled “Clinical Expert Report” and states that the CERs will be updated in order to comply with the company’s procedure for evaluation of clinical data for CE marking.
2022 In terms of post-market evaluation, this report offered little detail. Excluding the reference list, it was two pages long. It did not include any real review of the literature. Moreover, it did not mention whether, and if so how many and what kind of, adverse events associated with the use of TVT had been reported to the respondents.2034 It failed to mention the extent of any complaints or any medical device or vigilance reports.
2023 Three kinds of benefit were identified:
(1) “Primary Benefit”: alleviation of the symptoms of stress urinary incontinence and restoration of continence. Dr Isenberg referred in this context to the results of the Nordic trials.
(2) “Secondary Benefits: Direct”: obviation of the risks of general anaesthesia, since the procedure could be carried out under local anaesthetic. In addition, because the patient was conscious, she could cooperate during the procedure, that is, “she [could] provoke leakage at different stages of the operation”, apparently increasing the likelihood that continence would be restored and that voiding would not be impaired by the repair. The procedure was also said to minimise costs and loss of workplace productivity “because it was generally performed in an outpatient setting”.
(3) “Secondary Benefits: Derived”: a number of co-morbidities were said to be reduced, such as urinary tract infections, urinary frequency, vulvar maceration, and falls. Further, TVT was expected to improve quality of life indicators such as social engagement, self-image and satisfaction. No studies were cited in support.
2024 The report stated that “risk analysis” had identified a number of potential complications and side effects associated with the use of TVT, although it did not indicate how or by whom the analysis had been carried out. They were:
Bladder penetration, urethral penetration, bleeding from pelvic floor/Space of Retzius [the retropubic space], lateral vascular injury, damage to nerves, bowel perforation, bowel obstruction, urinary retention, urinary infection, detrusor instability, mesh rejection.2035
2025 It added that worldwide experience showed that such events are rarely reported, although it did not indicate what incidence rate was considered “rare”. Clinical trial complication rates based on the reports of the Nordic trials were also said to be low, once again without specifying any numbers or criterion, and limited to urinary retention, “simple” bladder perforation, and retropubic haematomas.
2026 It neglected to mention that not all of these potential complications and side-effects were included in the TVT instructions for use and it does not appear the report prompted a review of the IFU.
2027 The CER, signed by Drs Weinberg and Robinson on 7 March 2006 covered both TVT and TVT-O and was limited to assessing the impact on safety and efficacy, if any, of switching from mechanically cut to laser cut mesh, which was said to provide smoother edges and was intended to address the issue of fraying.2036
2028 The report claimed that the switch was made in response to customer needs, explaining:
Customers expressed a desire for a TVT mesh with smoother edges rather than edges with the ends of individual fibers exposed. Customer feedback also indicated that there was some dissatisfaction with the potential fraying effect of mechanically cut mesh.
2029 Tests showed that on average the mechanically cut mesh lost approximately twice the number of particles as the laser cut mesh. In an email to Sungyoon Rha of Ethicon Inc. sent on 12 June 2006, Gene Kammerer said that according to new French standards there was a significant difference in particle loss between TVT and its competitors with TVT losing “[a]pproximately 10 fold more … at 8% of the strip falling off”. The respondents’ position, however, was that the fraying was of no clinical significance in that there was no clinical data demonstrating a difference in either efficacy or safety between mechanically cut and laser cut Prolene mesh used in its TVT devices.2037
2030 This report did not address the other complaints that had already been received in relation to TVT, did not review the literature (published or unpublished) generated since the previous CER, and did not contain a risk/benefit analysis for the product as a whole. As such, this report could hardly be characterised as post-market evaluation of TVT — or, for that matter, TVT-O.
2031 The failure to attend to these matters as required or to prepare regular CERs came to the attention of BSI. The first BSI report in evidence is dated 3 June 2008,2038 a decade after TVT had received regulatory clearance in Europe. Indeed, there was no evidence about any earlier audits. The absence of this evidence was unexplained. In the 2008 report, which detailed the conclusions reached from a two-day review of “Technical Documentation” relating to a number of devices, including some of the POP devices, BSI stated that in some cases “legacy” technical files “remain[ed] deficient in reference to standards and some are deficient in critical areas such as Risk Management, Clinical Data, and Post Market Surveillance”. It also noted that “legacy” technical files referred to “obsolete standards”, something apparently also picked up in a previous audit.
2032 During a technical assessment visit conducted on 17 and 18 June 2009, BSI reviewed the technical documentation for Mersilene Tape, TVT-O and TVT.2039 On 18 June 2009 Sheryl Bagalio, Manager of Regulatory Affairs for Ethicon, emailed Drs Robinson and Kirkemo asking: “When was last CER for TVT?”2040 Dr Robinson was unable to provide a clear answer. He told her he had a copy of a TVT-O CER but no other CERs for any of the other slings apart from “one done long ago for a disposable TVT that never happened”. Ms Bagalio thanked him for the clarification, adding an explanation for her inquiry:
We're getting dinged with a major non-conformity for our Risk Analysis which includes Post Market Surveillance and Clnical (sic) Expert Report – they have been reviewing and I wanted to make sure we had the most updated CER.
2033 In the report prepared following its review of Ethicon’s files, BSI identified a major non-conformity in the following terms:
Legacy Files:
Legacy File Corrective Action Plan remains outstanding and deficient from previous Technical Audit…
Background:
Legacy file CAP was rejected on July 22, 2008 due to insufficient information for Post Market Surveillance feeding into Risk and Clinical, as well as insufficient details on implementation dates. An updated CAP response was received the day prior to this follow-up Technical Audit. “Estimated Completion Table” was missing 2 or 3 legacy files requiring updating…2041
2034 Whereas a minor non-conformity, according to BSI, indicated a single identified lapse, a major non-conformity signified a breakdown in the ability of the management system to effectively control the processes for which the system was intended.2042 A major non-conformity was a serious matter. It placed in jeopardy the validity of certification.2043 It was therefore necessary to investigate the underlying cause of any non-conformity to determine what corrective action was required.
2035 It was more than a year before a CER was forthcoming.
2036 At some stage, probably in late February 2010, Ethicon engaged a firm called “The Marketing Connexion” to prepare CERs for TVT and TVT-O.2044 By 18 March 2010, however, it seems that the process had not yet begun. In Dr Allman’s experience it was uncommon to engage an independent person to prepare CERs,2045 The CERs certainly improved as a result — but only up to a point. For the first time, they identified the search strategy employed for the literature review, the scope of the review, the databases searched, and the selection criteria.
2037 On 26 July 2010 Dr Robinson was advised that the CERs for TVT and TVT-O were “nearly complete”.2046
2038 Dr Robinson signed the CER for TVT on 2 August 2010.2047 This was about a decade after the June 2000 report, 11 years after the device was first supplied in Australia, and 13 years after it first acquired CE marking in Europe. Moreover, this was despite the 2007 amendment to the European Directive which made it clear that clinical evaluations must be actively updated with post-market data.2048
2039 Although the European Directive required that CERs be signed by their authors, the extent of Dr Robinson’s input into this report is unclear.
2040 This 2010 CER was purportedly based on an assessment of the risks and benefits associated with the use of the TVT device through a compilation of relevant scientific literature and “a critical evaluation” of that compilation.2049 The applicants correctly submitted that the CER:
relied on a literature search strategy which was not disclosed in such a way as to enable the search to be replicated (and therefore its reliability to be tested);
referred to, but did not analyse, the impact of the lack of long term data on the effect of the device;
referred to, but did not properly analyse, the impact of patient factors on complication rates and failed to consider the absence from the IFU of a warning about them;
contained “a cursory literature review conclusion statement” (that the data demonstrated compliance with the essential requirements covering safety and performance of the device and no additional data was required) with no supportive reasoning;
included a review of complaints and adverse events which should have “triggered awareness on the part of Ethicon of the under-reporting of adverse events by reference to complication rates in the medical literature”; and
recognised that severe harm could be caused by a range of complications yet did not contain any analysis of the manner in which these complications could be treated.2050
2041 The literature review was said to have been conducted by “several independent researchers” under the direction of, and in cooperation with, the Ethicon Women’s Health and Urology World-Wide Director of Medical Affairs.2051 It includes the following reflection:
In brief, the literature thoroughly recognizes the evolution and advancement in surgical apparatus/application and potentially superior outcomes delivered by mid urethral slings. Development of less traumatic routes for sling insertion and well regarded techniques are documented. While these advancements are not without their own set of issues, such as: discussion over retropubic vs. transobturator approach, surgeon technique differences, paucity of long term results, little standardization in patient assessment, and absence of globally agreed upon definition of “cure” (therefore success) or a clear set of relevant complications to avoid /prepare for; from a purely technical stand point, mid urethral slings have advanced surgical practice beyond colposuspension procedures.2052
2042 But the report does not proceed to consider the significance of the issues it identified to the ultimate issue or to the risk profile of TVT.
2043 A number of complications mentioned in the literature were discussed. Of these, the 2010 CER stated:
Most of the complications caused by the suburethral tapes could be attributed to the biomechanical properties of the mesh used and to other factors such as tissue healing, infection and the surgical technique used.2053
2044 A number of specific complications mentioned in the literature were then discussed in turn, mostly by reference to individual case reports. They were: bladder injury; urethral erosion; tape tension; bowel injury; foreign body reactions; urethral diverticulum; vascular injuries; nerve injuries (leading to pain); sexual function; urinary retention; vaginal injury; haemorrhage; and pelvic-cutaneous fistulas. The examination of these cases and the other literature to which the report referred was said to provide “insights into the rate of complications” and their management. It is difficult to see how an individual case report could provide any insight into the rate of complications. At most, it might include the author’s opinion on the subject or a reference to someone else’s research or opinion.
2045 The implications of the findings reported in these reports, however, were not explored.
2046 The discussion of bladder injury, which the evidence in this case indicated was a common complication of TVT, referred to only two case reports.
2047 The first concerned a patient who had “postoperative complications” five years after implantation which the CER said had been attributed to perforation by the tape of the bladder muscle. The complications were not identified. The CER stated that it was assumed that “this complication” (presumably perforation of the bladder muscle) had occurred either because of “the clinical history of the patient” (who had apparently undergone a hysterectomy in the past and had a history of fibrosis surrounding a mammary implant and Sjögren syndrome, an autoimmune condition) or to “a technical defect by the surgeon during the TVT procedure”.
2048 This raised obvious questions about whether TVT was contraindicated in certain cases and whether the IFU included such contraindications or appropriate warnings to guard against a similar complication arising in other cases. But if they occurred to the author, they were not discussed in the CER. Nor was there any discussion of the reasonableness of the assumptions.
2049 The case report was the work of a number of French physicians or surgeons. It was published in the Journal de Gynecologie Obstetrique et Biologie de la Reproduction in 2007.2054 The publication is in French. Unhelpfully no English translation was tendered. But the abstract appears in both French and English, as does the title. Reference to the publication itself reveals that the discussion in the 2010 CER was misleading. The title of the article is “Érosion vésicale tardive après pose de TVT®” (Bladder erosion few years after TVT® procedure). The English translation of the abstract reads as follows:
The surgical treatment of the stress urinary incontinence mainly use tension free vaginal tape done through the Retzius [retropubic] space: a case of bladder erosion occurring 5 years after a TVT® (Gynecare·Ethicon, USA) procedure is reported. First clinical signs occur 2 years after the operation and the removal of the tape (included in the muscle of the bladder) by vaginal and sus-pubic route was necessary 5 years later because the quality of life was dramatically altered. This complication could be related to the patient (past hysterectomy, history of fibrosis surrounding a mammary implant, Gougerot-Sjögren disease) or to the surgeon with a technical defect during the TVT procedure. The late complications of TVT procedure should be recorded in a national register.
2050 In the introduction to the article, the authors pointed out, that 10 years since the new surgical techniques for treating urinary incontinence in women have been used in France, late complications were beginning to be described: erosions of surrounding tissues (rectum, bladder, ureter, and vagina with scarring), late infections often after exposure of the material, and prosthetic retractions (contraction) causing disabling pain.
2051 In the case in question, the patient presented within two years of TVT surgery with urinary retention justifying removal of a section of the tape. On that occasion, cystoscopy was normal. About three and a half years after the original surgery, however, the patient complained of progressive onset and slowly increasing pelvic pain along the trajectory of the tape, worse on defecation and during sexual intercourse, and complications associated with a shortened bladder: dysuria, pollakiuria (daytime urinary frequency), and urge incontinence. Her symptoms worsened and a year later a decision was made to excise the tape. After the literature was reviewed, several hypotheses were raised to explain the phenomenon, including the foreign body reaction to the prosthesis. Noting that it was conceivable that individual reactions to foreign materials are variable, the authors wrote that certain patients scar more easily than others. They considered that this was particularly plausible with their patient given her medical and surgical history and having regard to the late onset of her symptoms and their inevitable progressive character.
2052 If Dr Robinson, as author or, at least signatory, of the 2010 TVT CER, had read even the abstract, he should have been alert to these matters and considered whether the IFU should be amended to make it clear that erosions could occur years after TVT surgery and that doctors and patients were aware of the dramatic alteration in quality of life that could ensue. It is not apparent from the CER that he gave these matters any consideration.
2053 In the 2010 CER, urethral erosion was said to have been reported as a rare complication of synthetic slings. No reference was given. Three case reports were then summarised. One of the cases concerned a patient who complained of pain on urinating after TVT surgery. The tape was found to have caused bladder erosion. The study was reported in the Dutch Journal of Medicine in 2007.2055 The authors of the report, Heesakkers and Vierhout, made the following pertinent observation in the abstract:
Since a few years the surgical treatment of stress incontinence consists more and more of the placement of a tension-free tape. Because of the minimally invasive technology that is relatively simple, the number of surgeries for stress incontinence has tripled in 4 years time. Literature claims large success percentages and low complication rates, but these figures cannot always be reached in daily practice. Physicians must therefore be careful in selecting patients suitable for placing tension-free mid-urethral tapes and realistic results should be stated.
2054 This observation was not included in the 2010 CER and no reference was made in the IFUs to the need for care in the selection of patients.
2055 Two case reports relating to late onset erosions were discussed under the heading “[f]oreign body reactions”.2056 In one of them bladder stones had formed in two patients which were attributed to the continuous contact of the polypropylene mesh with urine. A 3 x 2cm calculus was found in the left bladder wall of the first patient after she presented with urgency, dysuria, recurrent urinary tract infections and intermittent haematuria 16 months after TVT surgery. The second patient presented with similar symptoms 26 months after her TVT surgery. She was found to have a 4.6 x 4.5 cm calculus in the neck of her bladder.
2056 It was suggested that the arms of tapes inadvertently placed close to the mucosa, which can be missed on cystoscopy, may predispose patients to the formation of bladder stones because of pressure necrosis of the mucosa and gradual penetration of the tape into the bladder.2057
2057 In the abstract of the article reporting on these cases, the authors wrote that “[h]igh clinical suspicion of bladder complications is necessary when evaluating patients with urinary symptoms after a TVT operation”.2058 This note of caution did not find its way into the 2010 CER, although it would surely warrant consideration of the sufficiency of the warnings contained in the IFU.
2058 A commentary appeared after the article. The observations made in it were relevant but appear to have been overlooked by Dr Robinson, if not also the person who prepared the report. The author of the commentary, Massimo Porena of the Department of Urology at the University of Perugia in Italy, wrote that “[i]n recent years a mass of data has been generated on the safety and efficacy of MUS [midurethral slings] but few reports have dealt with the true incidence of complications and even fewer with the treatment of complications”. He said that one of the major obstacles was the lack of national or worldwide registers which means that investigators lack “the denominator to calculate the true incidence of complications”. He noted that published complication rates tended to under-report major complications. He said that physicians needed to be aware that the “so-called” mini-invasive techniques can have complications which can impact negatively on quality of life. He observed that bladder injury, reported to have an incidence of 24% with TVT, was not always recognised during initial cystoscopy and can become the source of late complications such as chronic pain, urinary tract infection and bladder stones, requiring further surgery.
2059 Bowel injuries were said to have been reported as a complication of the TVT procedure only recently. According to the 2010 CER, they typically involved displacement of the mesh into the abdominal cavity resulting in perforation of the bowel.
2060 Three case studies were discussed. In the first, the patient underwent exploratory laparotomy four days after implantation because of nausea, vomiting, abdominal distention, and bowel contents passing through a TVT exit site. The device was found to have perforated the small bowel and re-entered the retropubic space causing a mechanical obstruction. The tape was cut and removed in its entirety and three centimetres of small bowel were resected. Bowel perforation was attributed to the entry of the TVT introducer into the peritoneal cavity.2059
2061 In the second, a patient presented with symptoms of bowel obstruction three years after TVT surgery. The symptoms included abdominal pain, distention, nausea, and vomiting. Exploratory surgery revealed an adhesive band containing a piece of TVT mesh obstructing the distal terminal ileum and attaching it to the right pelvic sidewall. Remnants of TVT mesh were identified in both the ileum and pelvic sidewall. The tape was excised. It was noted that after the intraperitoneal placement of the TVT tape adhesions were formed between the segment of tape, the bowel, and the pelvic sidewall.2060
2062 The third case study involved a woman who underwent TVT surgery at the same time as a colpoclesis (a form of native tissue repair of pelvic organ prolapse). Surgeons noted a 4cm bulge over the site of the left TVT incision that appeared to be a hematoma with overlying mild erythema (redness of the skin or mucous membranes). The erythema was noted to have expanded two days after surgery. On the third day after surgery, the patient was reported to have developed “bilious vomiting” with slight abdominal distention and diminished bowel sounds. An exploratory laparotomy reported an inguinal hernia displaced medially with loops of small bowel in the hernia sac. Although the TVT sling was properly positioned, one loop of bowel was seen to have been perforated by the sling mesh. The mesh was trimmed and a bowel resection was performed to correct the injury.2061
2063 While instances of each of these complications were identified, there was no discussion of their significance. In particular, there was no discussion about why these risks were acceptable, whether they should (or even could) be addressed through design changes or the provision of suitable warnings in the IFUs. Although the IFU was mentioned in the 2010 CER, no consideration was given to the sufficiency of the warnings or the description of the adverse reactions in the light of the information gleaned from the literature review.
2064 There was no attempt to synthesise the discussion and provide a reliable risk/benefit profile.
2065 As part of the complication of “foreign body reactions”, the possibility of late onset erosion was expressly recognised. But there was no proposal for incorporating a warning about late onset erosions in the IFUs or any consideration of whether the then current wording was sufficient to encompass it. The CER stated:
Synthetic mesh material can cause tissue reactions and erosion into nearby structures like the urethra, the bladder, or the vagina. As the non-absorbable polypropylene material of the TVT tape is thought to stay intact over time, but there certainly are changes in the aging tissues in which the tape is implanted, there has been a fear of seeing late-onset erosion problem.2062
2066 This was particularly problematic as at this time the TVT IFU warned of erosion and extrusion as complications that “could” result from a “transitory foreign body response” which “may occur”.
2067 The final sub-heading in the complications section was “Other studies” which identified a range of complications, including one report of a patient who complained of severe pain in the immediate postoperative period. It appears that the tape was then removed.
2068 The conclusion of the review was that:
The above data, taken together with any available pre-clinical data, are sufficient to demonstrate compliance with the essential requirements covering safety and performance of the Gynecare TVT* Tension-free Vaginal Tape/ Tension-free Vaginal Tape Accessory Abdominal Guide under normal conditions of use. No additional clinical data is required.2063
2069 Dr Allman’s opinion was that the conclusion was not justified by the evidence presented, that the report did not comply with European regulatory requirements, and was insufficient to support continued CE marking.2064 The basis for the opinion seems to be that the report described literature which could have included publications describing the use of different devices and that material could not be used to draw a conclusion about a specific device without addressing the equivalence of the different devices. Dr Allman seems to have believed that the references to “TVT” in the report were references to transvaginal tapes. In this respect Dr Allman appears to have been mistaken. That is not to say, however, that the conclusion was in fact justified by the evidence presented.
2070 The 2010 CER cited the Ogah et al (2009) Cochrane review, together with its conclusion that “minimally invasive synthetic suburethral sling operations appeared to be as effective as traditional suburethral slings… but with shorter operating time and less post-operative voiding dysfunction and de novo urgency symptoms”.2065 The CER also cited the opinion of Ogah et al (2009) that the quality of evidence for most of the trials was moderate. But the CER did not refer to the authors’ observations that “very few trials reported outcomes after one year, and the long term efficacy and adverse effects have yet to be determined”.2066 That said, as I have already observed, the CER did recognise that there was a “paucity of long term results”. On the other hand, it did not explain how, in these circumstances, the conclusion about safety and efficacy was available. No critical evaluation of any of the articles referred to in the review was undertaken and there was no discussion of the strength and weaknesses of the various studies.
2071 The complaint/adverse event review in the 2010 CER is in four parts. Part I consists of a review of internal complaints, which I take to mean complaints made to Ethicon. Part 2 deals with complaints registered on the MAUDE database. Part 3 compares the Ethicon record of complaints with the information on the MAUDE database. Part 4 is entitled “product recall” and notes that as at the date of the report there had been no recalls of TVT.
2072 This review has a number of curious features.
2073 First, the internal complaint review refers to complaints for the period 2006 and 2007. It is a mystery why these two years were selected. The risk management report covers the same two years. That report is undated. Perhaps it was completed in 2008. Even so, there is no obvious reason why the complaints made in the period from 2008 to 2010 were excluded. Furthermore, given that this was the first TVT CER since the device had been on the market or at least (if I am wrong about the 2000 report signed by Dr Isenberg) the first in a decade, there is no apparent reason why complaints over the entire period were not covered.
2074 Second, complaints for those years are summarised (only by type and number) and totalled (335), then compared to the corresponding volume of devices distributed for the same period (357,197) to produce an overall complaint rate of 0.00053%.
2075 A “Harm/Hazards Summary Table” follows, taken from the Risk Management Report for TVT and TVT-O. The table includes a list of certain kinds of harm, classifies each harm in the list by number, and assigns a numerical rating for the frequency of its occurrence. The table also contains a brief description of the “hazards” responsible for the harm. “Nerve Damage/Pain” is listed as a “harm” but, in contrast to what appears in the risk management report and for no apparent reason, the qualification “major” is omitted.
2076 The following harms were ranked as 10: blood loss; erosion into the urinary tract; vaginal exposure; infection; and internal organ damage. A ranking of 10 denoted that the impact of the harm was “catastrophic”. The other kinds of harm (extended surgery, fistula formation, nerve damage/pain, unintended tissue reaction, and tissue damage) were classified as 9 (“almost catastrophic”). The difference between “catastrophic” and “almost catastrophic” according to Ethicon’s FMEA Severity Ranking Scale was that in the latter case the user is forewarned that medical device failure is occurring; in the former the user is not.2067
2077 The risks of blood loss and erosion into the urinary tract were classed as remote (≤ 1/50,000). All other harm except for tissue damage was classed as unlikely (≤ 1/10,000). Tissue damage, astonishingly, is classed as extremely remote (≤ 1/100,000).
2078 The overall residual level was assessed as moderate.
2079 The frequency rates from which this assessment was partly derived bear no relationship to the figures appearing in the medical literature, including the studies described in the literature review section of the 2010 CER. They revealed exposure rates, for example, of between 0.7% and 2.6%. As Dr Allman observed, this alone throws into question the reliability of the conclusion about the overall residual risk level.2068
2080 The CER stated that it was a policy and procedural requirement of Ethicon that, when the overall residual risk is moderate, a review be undertaken of complaints about comparable devices listed on the MAUDE database. Such a review was reportedly undertaken. The CER asserted that the review did not indicate any harms/hazards in addition to those “that were indicated by review of the labeling and the internal complaint database”. No consideration was given, however, to whether that information might affect the frequency ratings given in the harm/hazards table. Nor did the review generate a revision of the IFU to include harms/hazards appearing on the database but omitted from the IFU. Two obvious examples of harm missing from the IFU at the time were “unintended tissue reaction” and “nerve damage/major pain”.
2081 Third, all this information and the resulting conclusions were derived from the complaints data and failed to take into account data from clinical trials. Dr Allman was critical of this approach. It is an approach that is of next to no utility as it is calculated to gravely underestimate the extent of the risks. The respondents did not submit that it was justified or defensible — whether because of regulatory requirements, guidelines, international standards or otherwise.
2082 Fourth, it is difficult to believe that by 2010 Ethicon truly regarded the complication rates to be this low, since they were completely out of step with the complication rates reported in the literature. The applicants submitted that Ethicon should have known that adverse events were under-reported.2069 In fact, of course, it did. Dr Hinoul testified during cross-examination that, in contrast to prospective clinical trials which “try to get all the data comprehensively”, “[w]hen you look at spontaneous reporting from patients and doctors to authorities and to companies — indeed we realise fully well that there is significant under-reporting”.2070
2083 The August 2010 CER was followed by a CER on the TVT Family of Products, signed by Dr Hinoul on 3 May 2013.2071 It covered all of the SUI devices except TVT Secur (on the basis that TVT Secur was a single-incision sling and “therefore not similar to others”). The decision to produce a single CER for the midurethral slings was at odds with an agreement reached by Dr Robinson and Michael Thomas of The Marketing Connexion that “functionally and, more specifically, insertion wise these three [TVT, TVT-O, and TVT Secur] are differentiated enough to require stand alone CER’s”.2072
2084 The May 2013 CER was drafted by a regulatory affairs consultant, Dr Jill Bunyan, who provided the first draft to Ethicon on 30 March 2013. The email attaching the first draft raised a number of queries. Some were identified as “original issues [that] remain[ed] to be addressed”:
• I am not 100% sure which are the trademark names to use for the TVT report. The file lists the following. Obviously there is the general ‘family’ name; Secur is excluded; Abbrevo, Exact and Obturator products are clear but there are three others. Which is TVT ‘classic’?
• Please provide a full list of product codes
• NB do I need to note that the Secur range is excluded?
• Currently the Technical file for TVT-Exact is missing (may be part of the general TVT one?), as is the IFU for TVT-Abbrevo (in ENGLISH), and the previous CER for TVT-Exact.2073
2085 It also raised some new queries:
Are midurethral and suburethral slings the same? The terms seem to be used interchangeably?
Is TOT or TMUS the correct term for transobturator tapes in general, or does TOT imply the outside-in approach specifically?
2086 These queries clearly indicate that, irrespective of Dr Bunyan’s expertise in regulatory affairs, her knowledge of SUI devices and of Ethicon’s products in particular was incomplete. It is also apparent from her email that she had been asked to prepare the draft without being provided with all necessary documents, including the technical file for TVT Exact, MAUDE and complaints data, and that she had not been informed of the literature search strategy.
2087 At the same time, multiple risk management reports required updating. Notwithstanding the differences between the various devices, Ethicon employees decided to produce a single report.2074
2088 Dr Hinoul signed the CER on 3 May 2013.2075 Excluding tables and appendices, it was about 200 pages long. On 11 July 2013, Lee Hackman (Staff Quality Engineer, Ethicon Surgical Care) sent some “recommended verbiage” to Dr Hinoul for inclusion in the report.2076 This concerned changes to the section on “benefit versus risk analysis” to accord with risk management procedures. Apparently, a new revision of the risk management procedure was in the works but had not yet been approved, so the then extant version remained operative. The outcome either way was that Ethicon stated that the overall residual risk was acceptable.
2089 The applicants roundly criticised this exercise:
Thus, the key section of the report that ought to have been the subject of careful consideration simply adopted a pro forma approach, presumably to tick regulatory boxes. Despite informing the Court (at [304 of his affidavit] that he had “issued” the 16 July 2013 Clinical Expert Report, and summarising its findings (at [304] – [309]), Dr Hinoul failed to disclose that the reason that report was signed was to incorporate prepared ‘verbiage’ as Ethicon’s Risk Benefit Analysis. This too tells against the credibility of the evidence Dr Hinoul has given in relation to Ethicon’s post-market clinical surveillance.2077
2090 It is not clear, however, why this amendment was said to be so problematic. While the email from Mr Scanova recommended the insertion of “recommended verbiage”, it also emphasised that the risk assessment summaries previously prepared were not affected. Dr Hinoul then signed a revised CER on 16 July 2013,2078 incorporating the relevant amendments, but otherwise in a substantially similar form to the May 2013 CER.
2091 The conclusion of the CER was as follows:
The literature review data, taken together with previously available clinical and pre- clinical data, are sufficient to demonstrate State of Art compliance with the essential requirements covering safety and performance of the GYNECARE TVTTM Family of products under normal conditions of use. No additional clinical data is required. No previously unidentified harms or hazards were identified in this review.
No information was provided from either the literature or PMS data to indicate that any new performance issues are occurring in the clinical setting that were previously unknown.
After manual review and analysis, no new hazards, increased risk or unexpected adverse events were noted. The clinical evidence provides support that GYNECARE TVTTM meets the Essential Requirements of the Medical Device Directive 2007/47/EC.2079
2092 The July 2013 CER was a seemingly comprehensive document containing references to a large number of studies. The respondents praised the literature review contained in the report (which was copied from the May 2013 CER).2080 They argued that it was extensive, pointing out that the table reviewing the relevant literature spanned 21 pages and the list of excluded articles excluded (and the reasons for exclusion) 55.
2093 Based on a review of 152 papers, this report concluded that there were “no unexpected changes in the safety and performance of all GYNECARE TVT products”, and further that:
There were no safety concerns or adverse events found in the literature related to product design or manufacturing defect. The complications and risks reported in the literature are consistent with known risks and complications described in the device labelling.
The above data, taken together with previously available clinical and pre-clinical data, are sufficient to demonstrate State of the Art compliance with the essential requirements covering safety and performance of GYNECARE TVT TM under normal conditions of use. No additional clinical data is required. No previously unidentified harms or hazards were identified in this review.2081
2094 Dr Allman said that this conclusion was not justified. He explained that, while “general comments about the ‘state of the art’ (outcomes and complications with this type of device)” might be justified, “conclusions about the compliance of a specific device to the essential requirements are not possible without specifying the criteria applied to the reported data to determine compliance”.2082
2095 The report concluded that TVT met the essential requirements of the European Directive. Dr Allman said that this conclusion was similarly unjustified, because “justifying conclusions on compliance to the essential requirements requires criteria” (and the criteria were not identified). Dr Allman was also critical of the observation in the report that no information had been provided to indicate that there were any “new performance issues” occurring in a clinical setting that were “previously unknown”. He said that this does not mean that the rates of known issues are acceptable.
2096 The July 2013 CER also came under criticism from the TGA.2083 Before I deal with this criticism, however, I should provide some context.
2097 On 25 September 2013, the TGA asked JJM for certain information.2084 As part of that request, the TGA required, amongst other things: a checklist summarising how each of the devices conformed to the applicable essential principles (or the essential requirements of the European Directive); post-market data, including the description and rate of adverse events as well as a risk analysis and corrective actions taken; risk assessment documentation; and the clinical evidence used to establish conformity. Notably, the TGA also sought clarification of Ethicon’s post-marketing vigilance plans, noting that post-marketing surveillance was passive, relying on complaints made directly to JJM.2085
2098 JJM replied on 7 November 2013, purporting to provide the information sought, enclosing a number of documents, including the IFUs for the devices, the July 2013 CER for the TVT Family of Products, post-market complaints data, and two risk management reports — a “legacy” report for TVT and TVT-O and a report on TVT “laser cut mesh”.2086
2099 The tables containing the checklist of essential requirements ran to nearly 200 pages. References were provided to Ethicon documents such as Quality System Certificate, Finished Goods Specification, IFUs, Risk Management Reports and Clinical Expert Reports. Little effort was made, however, to explain how these documents demonstrated compliance with the essential requirements.
2100 The table containing the post-market data was difficult to interpret.2087 The adverse events were not individually identified, merely described obscurely as “post procedure complication” and “inter-operative event”. Complaint rates were presented as consolidated percentages. The total complaint rate in Australia for TVT, for example, was recorded as 0.07% and for TVT-O as 0.10%. It was not clear on the face of the table whether these complaints were serious in nature.
2101 The TGA undertook an assessment of the material provided, advising JJM of the outcome on 28 April 2014.2088 The TGA clinical assessor concluded that the risk analysis documentation was not adequate to perform a full evaluation. Some of the deficiencies of the legacy RMR2089 give the flavour of the evaluator’s concerns:
• The summary table of harms and hazards contains a numerical rating system for harm severity and frequency that has not been defined in the report. Therefore, this cannot be evaluated.
• It is unclear how each of the harms listed in the report have been identified and how data from the published literature and post-marketing experience has been used to inform this report.
• The table in Appendix B lists complaints titles and total numbers recorded. It is not clear over what time period this data was collected, what devices were involved in each complaint case and from where the data was collected. Furthermore, no case analysis is presented.
• Appendix B also lists the sales information for each device, however it is not clear which countries this data refers to. It appears that this data is out of date, with no results presented later than 2007.2090
2102 The TGA clinical assessor was also critical of the July 2013 CER.
2103 The assessor said that the presentation, analysis and synthesis of clinical data contained “major deficits”. They included:
• Inability to reproduce the results from the literature search due to:
- Lack of detail regarding the search process.
- Inconsistent application of inclusion and exclusion criteria.
- Unclear reasoning to establish the final list of studies “included”.
- Inconsistency between the results presented in the literature search and the - number of studies presented in the literature critical analysis.
- Lack of current published literature, with no studies published after 2012.
• Lack of adequate critical analysis of the studies identified from the literature search.
• Major selection bias in the Literature Critical Analysis, with only a small number of author selected studies included for discussion. It is unclear why the authors have not discussed the total body of literature identified in the literature search. This indicates major selection bias in the data discussed. Therefore, the evaluator cannot have confidence that the authors report is a balanced and evidenced based document.
• Multiple sub-divisions of clinical data sets presented in various separate places across the dossier. Some subdivisions are inconsistent and overly complicated.
• Inadequate synthesis of data, with no discussion of the total body of literature leading to a clear, evidenced based, logical conclusion.
• Lack of discussion regarding safety, with excessive pages of data presented in Appendix IV that has not been analysed by the author.
The authors have not presented their synthesis, discussion and evidenced based conclusions of the total body of data identified in the literature search. Therefore, the evaluation process is limited and the TGA cannot have confidence in the brief generalised conclusion documented in Section 6.5 of the report.2091
(Original emphasis)
2104 The assessor noted that, while numerous sources of data were relied upon, the July 2013 CER simply set out “multiple isolated sets of information” without presenting an “overall synthesis and analysis of this information”.2092
2105 A number of these criticisms, such as the inability to reproduce the results of the literature search, the lack of adequate critical analysis, and selection bias in the literature review, could be levelled at the other Ethicon CERs. As we shall see, they were also made by the TGA of the 2013 Gynemesh PS CER.
2106 After reviewing the TVT post-market surveillance complaints data from January 2010 to January 2013, the assessor observed that there appeared to be a “massive increase” in the erosion rates from 2012 (3.6%) to 2013 (6.4%) and commented:
It is possible that the rates of erosion may be increasing despite a reduction in demand due to the amount of time required to develop these types of longer-term complications. It is unclear if this rate will continue to increase over time, as the rate of erosion in the longer term (i.e. longer than 2 years) is largely unknown. The author has not addressed this major increase in rates of erosion or considered the reasons for this.2093
2107 The assessor also observed that there was a large increase in the rates of other major complications, despite the reduction in demand. These complications included bladder perforation, post-operative complications, urinary retention, and vaginal exposure.
2108 With respect to TVT Exact, the assessor similarly noted a large increase over the same period in erosions (into the urinary tract) from 1 in 2011 to 72 in 2012 and, although 12 was the recorded number for 2013, the 2013 figures only captured fewer than 12 weeks of data. The assessor was similarly critical of the report’s failure to address this increase in erosion rates or to consider the reasons for it.2094
2109 A similar trend was picked up in the post-market surveillance data for TVT-O. Again, despite a reduction in demand for the device, cases of erosion rose from 63 in 2011 to 1159 in 2012 and 147 in less than three months in 2013. Yet, the major increase in erosion rates was not addressed and the reasons for it were not considered.2095
2110 The assessor noted that the author of the CER had acknowledged that post-operative mesh exposure/erosion was the most common and significant adverse event, yet failed to discuss the finding, its potential causes, or risk mitigation strategies.
2111 The assessor observed that the post-market vigilance plan for the various TVT devices covered by the CER involved “passive post-marketing surveillance in the form of complaints made directly to the Sponsor”, that post-marketing studies or registries did not appear to have been conducted, and no planned studies were documented.
2112 The TGA concluded:
The information provided by the Sponsor [JJM] has raised a number of concerns, most importantly related to the incidence of adverse events including erosion. At this stage, there has not been sufficient analysis and discussion presented by the Sponsor, especially regarding safety. Therefore, based on the information provided, the evaluator cannot have confidence in the Sponsor's conclusion that the benefit of using the GYNECARE TVTTM Family of products outweighs the associated risks. This can be amended if the Sponsor could provide some additional information to clarify the issues raised in this evaluation.2096
2113 A month earlier, on 25 March 2014, the Canadian regulator, Health Canada had written to Johnson & Johnson Medical Products (JJMP), seeking material about the Ethicon devices, after information had come to its attention that the devices might not meet the safety and effectiveness requirements of the Medical Devices Regulations (SOR/98-282) (Can).2097
2114 The letter, written by the Director of the Medical Devices Bureau, stated that Health Canada had received “numerous” adverse event reports related to various surgical meshes of the type listed on Ethicon’s licence. It pointed out that clinical evidence had demonstrated that the use of surgical mesh for the treatment of stress urinary incontinence could lead to a number of adverse events “including but not limited to: acute or chronic pain in the groin, thigh, pelvic and/or abdominal area; mesh extrusion, exposure or erosion; infection; voiding dysfunction; dyspareunia; neuromuscular damage; organ perforation; recurrence of incontinence; bleeding or hemorrhage; vaginal scarring and/or tightening; and mesh contraction and shrinkage.” It also noted that one or more surgical procedures could be required to treat these complications and that some may not always be completely corrected. Ethicon was asked to provide a copy of the labelling demonstrating that it contained up-to-date information on the potential complications including but not limited to the above-mentioned adverse events. In the event that any had been omitted, Ethicon was directed to revise its labelling to include them. A similar direction was made with respect to the POP devices. Ethicon’s response to the labelling request is dealt with below in Parts X and XI.
2115 In addition, Ethicon was directed by Health Canada to provide “a comprehensive systematic review of the device-specific short term and long term clinical safety and effectiveness for [the] device in comparison to traditional surgery methods that do not use mesh.”
2116 As the applicants observed, this was something which had not been included in any formal evaluation of the devices before they were launched on the market.2098 The request was reasonably prescriptive. JJMP was specifically asked for:
a) A table providing a side-by-side comparison of the short term and long term rates specific for your device versus traditional surgery for the following: each of the clinical performance measures (objective success rates and quality of life); individual adverse event types (including items listed in question 2); and the overall adverse event rate.
b) A concise discussion of the results and conclusions for the evidence in (a) supporting the safety, effectiveness and risk-benefit profile of your device.2099
2117 JJMP was also asked to “[e]nsure that references are properly cited, appropriate statistical methods are employed, and the methods and criteria used for data search and selection are clearly specified in your response” and, if clinical data were not provided for each model under its licence, to “provide sufficient objective evidence to support that the data are representative of the different models”.
2118 JJMP was given a month to respond and informed that, if a response was not forthcoming within that time, the licences could be suspended. Despite the tight timetable, it appears that Dr Weisberg did not learn of the request until 2 April 2014 and no action was taken to comply with it until 11 days before the due date.2100
2119 Apparently Ethicon outsourced this project to an external consultant, Dr Caroline Charles of SciLink Medical Writing.2101
2120 On 13 April 2014, Liza Ovington, Director, Strategic Medical Operations, Johnson & Johnson, forwarded Health Canada’s request to Dr Charles and two other external consultants. It is clear from the covering email that Ms Ovington was under the mistaken impression that Health Canada’s request was limited to Gynemesh PS and Artisyn Y (a different mesh product sold by Ethicon). She wrote:
[P]lease see attached the request regarding Artisyn Y Mesh and Gynemesh PS from Health Canada – obviously we cannot undertake a true systematic review as requested but agreed to take our most recent CER data for each device and try to extract and organize the relevant info as much as possible in the format they describe within the attachment …
Realize that this is not exactly what they are requesting but the best we can do in the timeframe provided …
2121 The applicants submitted that this email amounted to “an admission on Ethicon’s part that the information it had in its CERs for the devices was not a comprehensive, systematic review of the device- specific short term and long term clinical safety and effectiveness of the implants”. “Had Ethicon been complying with the Essential Requirements”, they argued, “it should have been in a position readily to provide Health Canada with the information it sought”.2102 I reject the submission. The statement that “a true systematic review as requested” could not be conducted must be read with the observation in the second sentence of the email, which reflected on the time constraints. Fairly read, it is simply a statement that the requested review could not be undertaken within the time afforded by Health Canada.
2122 In its response, Ethicon provided “comprehensive tables” detailing device-specific short and long term clinical and safety effectiveness in comparison with non-mesh surgical methods. The information was said to have been drawn from the CERs for the various devices. Ethicon referred to a large number of studies and then set out a concise discussion of the results, as requested by Health Canada.2103 As part of its discussion on long term results, Ethicon wrote:
Safety:
According to available clinical data, TVT seemed to be similar or better than traditional surgery for the majority of the long-term adverse event endpoints reported. The only differences related to mesh specific adverse events are mesh exposure or – on rare occasions – erosion. The literature reports that these specific adverse events associated with the mesh are well characterized and manageable in the majority of cases. Vault and posterior vaginal wall prolapse are seen more commonly after colposuspension. In the references cited, there were a few adverse events that were reported at a higher rate for TVT than for traditional surgery which included recurrent urinary tract infection at 24 months, dysuria (60 months) and urinary retention. These results were reported from studies that were single center and may have had a small sample size. However, when looking at a larger body of evidence as summarized in the Cochrane Database of Systematic Reviews 2009, other symptoms of obstruction such as urgency, urgency incontinence or voiding dysfunction seems to favor mid-urethral slings over colposuspension.
Effectiveness:
According to available clinical data, TVT seemed to be equivalent or better than traditional surgery for the majority of the long-term objective success rate endpoints reported. In addition, TVT seemed to be equivalent or better than traditional surgery for all subjective success rate endpoints reported.2104
2123 As the applicants submitted, Ethicon’s position on single centre studies in this context is at odds with its reliance on single centre studies where the results appeared favourable to its devices.2105
2124 The last CER for the TVT Family of Products which was tendered in evidence was approved by Dr Hinoul in 2015.2106 The report was 931 pages long. No date appears on the face of the report, but in their written submissions the respondents gave the date as 19 January 2015. Presumably this was based on the meta-data. But the report refers to publications later than January 2015.2107 Further, Dr Hinoul said in his affidavit that he had approved the report in 2015, and he included an assessment of complaints and device vigilance reports to 30 June 2015,2108 as well as a literature search which finished on 14 September 2015.2109
2125 This report was a vast improvement on its predecessors. Unlike the previous CERs, it included a critical analysis of equivalent and/or non-equivalent devices, a detailed literature search and review protocol. Moreover, it graded the relevant documents by a variety of different measures, including the quality of the evidence, and exposed the criteria used in the literature appraisal. It was followed by extensive tables extracting data from each article or abstract considered to be relevant (323 references, with 60 of those supporting more than one SUI device).2110 It then set out the relevant data from the studies individually, although with little evident synthesis.2111 The literature review was followed by a consideration of post-market complaints data.
2126 The report acknowledged a “significant” increase in complaints over the years. Dr Hinoul attributed this to the increase to litigation and, in particular, to “lawyer advertising”.2112 I will return to this matter shortly.
2127 The respondents relied on the conclusion of the 2015 CER, which they extracted in full in their submissions.2113 They did not refer in their submissions to the preceding section containing the “benefits versus risks analysis”.2114
2128 In analysing the risk benefit profile of the devices, Dr Hinoul drew a distinction between the retropubic slings (TVT and TVT Exact) and the transobturator slings (TVT-O and TVT Abbrevo). Long-term complications associated with both categories of devices were said to be tape erosion, urinary retention, de novo urgency and/or detrusor instability.2115 Yet no consideration was apparently given to the absence of any indication in the IFUs that these complications could occur in the long-term.
2129 When discussing the studies under each category, long-term complications were barely mentioned. For example, complications of the retropubic slings were largely characterised as intra-operative or post-operative. Post-operative complications included post-operative voiding difficulties, urinary tract infections, pain, nocturia, de novo urgency/urge incontinence, dyspareunia, and erosion/extrusion requiring further surgery. But it was not clear how late they could arise, how long they could last, and how severe they might be. The same was true for the suburethral slings (TVT-O and TVT Abbrevo), where the post-operative complications identified were pain/pain, urinary tract infections, voiding difficulty and urinary retention, urethral irritation, de novo urge incontinence, tape erosion/extrusion. As Dr Allman pointed out, only two studies were available for TVT Exact but there was no consideration of whether further studies should be conducted and, if not, why not.
2130 Having considered the literature, Dr Hinoul stated:
Importantly, within the context of complications reported by the studies in this literature review, no reports were identified which raised new safety concerns, i.e. complications not already identified within the context of the respective device RAS documents. The clinical literature reviewed as part of this CER therefore does not change the existing safety profile of the subject devices.2116
2131 The reference to “RAS documents” was to two Risk Assessment Summaries (RAS), one for the retropubic devices, the other for the transobturator devices. Dr Hinoul directed the reader to those documents for further information.
2132 The RAS for TVT and TVT Exact bore a release date of 31 July 2013.2117 The document grouped together these two devices due to their “similarity and use in a retropubic procedural approach”. It purported to summarise the residual risks of the devices based on the severity and probability of harms associated with their use. It was not intended to provide “a determination of acceptability of the risks”. Rather, it relied on other Ethicon documents such as risk management reports, clinical evaluation reports and Failure Mode Effect Analyses (FMEAs) to summarise the risks.
2133 The summary of harms was presented in a “Harms – Hazards Table”, which categorised the risks by reference to severity, frequency and a “harm risk level”.2118 Three types of “harm risk levels” were identified in the document (BA, ALAP, and RBA) but these terms were not defined. Instead, the document stated that the definitions were contained in a further document cited as revision 21 of PR602-003.2119 I have already noted that PR602-003 was Ethicon’s Company Procedure for Medical Device Risk Management.
2134 As I noted earlier in relation to other risk documents produced by Ethicon, “BA” was an acronym for Broadly Acceptable:
Harms or Hazards at this risk level will conform to the requirements defined in published standards and have been reduced as far as possible by the efforts of teams in product design and process development. Harms and Hazards are reduced as far as possible, using the generally acknowledged, “State of the Art” design, labeling, manufacturing and controls technology.
2135 “ALAP” was an acronym for “As Low As Possible”:
Harms or Hazards at this risk level will require documentation from the project team substantiating that the risk is reduced as far as possible, using the generally acknowledged, “State of the Art” design, labeling, manufacturing and controls technology.
2136 “RBA” was an acronym for “Risk Benefit Analysis”:
Harms at this level will require coverage of the specific Harm in the Risk Benefit Analysis portion of the Risk Management Report…
2137 The RAS stated that none of the harms had a risk level of RBA, that is to say that none of them had a risk level frequency high enough to justify coverage in the Risk Management Report. Nor were any harms categorised as BA.2120
2138 The following were identified as ALAP: blood-borne pathogen transmission; blood loss; delayed wound healing; exposure; failure of treatment; fistula formation; infection; inflammatory or unintended tissue reaction; internal organ damage; nerve damage/pain; skeletal/cartilage damage; and soft tissue damage.
2139 For each of these risks, three things were identified: primary hazards contributing to the harm; actions taken to reduce the risk as far as possible; and methods for notifying the user of the residual risk.
2140 Save for the first complication (blood-borne pathogen transmission), the “actions taken to reduce [the] risk as far as possible” were the same for each of the risks:
Known implantable material (TVT PROLENETM Mesh) was selected to reduce risks associated with the use of these products in this application. Further, surgeons using these products must be familiar with SUI procedures and techniques. Additionally, studies were performed with surgeons during product development, in order to evaluate the risks inherent in the procedure, and to refine the technique for application of the mesh so that risks were minimized. Design Validation was also performed to validate that the device could successfully be used as intended.
2141 I interpolate that no consideration appears to have been given to establishing registries or conducting post-market clinical follow-up studies in order to reducing the risks to a minimum.
2142 This was followed by an identical statement for “methods for notifying the user of this residual risk”:
This risk has been communicated to users via the IFU.
2143 Notably, under “nerve damage/pain”, the following contributing hazards were identified:
(a) Improper dissection technique, (b) Incorrect patient position, (c) Residual foreign body, (d) Device Passage causes puncture/laceration, and (e) Removal of implant/re-do procedure.
(Emphasis added)
2144 As I explain below in Part XI, I do not see where or how the potential for pain caused by a residual foreign body response, which the evidence shows could be late in onset and endure for the long term, was conveyed by the IFUs for the SUI devices in July 2013.
2145 The RAS for TVT-O and TVT Abbrevo, which also bore a release date of 31 July 2013, took a similar approach.2121 It adverted to the “similarity [of the devices] and [their] use in an obturator procedural approach”.2122 A harm/hazards table was used to present risks, followed by a summary in narrative form. Again, none of the risks was categorised as RBA (requiring risk-benefit analysis in a risk management report). The same list of risks as the retropubic devices was addressed under the category of ALAP, with the same content under “actions taken to reduce [the] risk as far as possible” and “methods for notifying the user of this residual risk”. Based on the views stated in the RAS, the risk management report concluded that the overall residual risk (ORR) was acceptable.2123
2146 Returning to the 2015 CER, Dr Hinoul considered post-marketing surveillance data for two periods: 1 January 2010 to 30 January 2013; and 1 January 2013 to 30 June 2015. In his affidavit, Dr Hinoul emphasised the low “overall complaint rates” I n these periods.2124 Between 1 January 2010 and 30 January 2013, for example, the overall complaint rates were said to be 1.3% for TVT, 0.3% for TVT Exact, 0.6% for TVT-O, and 0.1% for TVT Abbrevo. Dr Hinoul did not explore whether these figures could be reliable in view of the higher numbers reported in clinical studies and the known issue of under-reporting in practice.
2147 In this CER, Dr Hinoul observed that no new adverse events had been reported in the period January 2010 to January 2013, but there was a “potential safety signal noticed regarding the overall complaint rate”.2125 This issue had apparently been addressed in two other documents: PQI 13-060 and CAPA-002538 Analysis. These documents were also said to be applicable to the latter period, as to which the CER noted that the “majority of reported events during the annual PMSR period are related to on-going Pelvic Floor Flat Mesh and TVT Sling litigations”.2126
2148 The first document, PQI 13-060, was a “Product Quality Issues” assessment form with an “origination date” of 8 May 2013.2127 It was completed by Sathya Iyer, PMS Manager, on 20 December 2013 and approved by Korey Phillips, PMS Director, on 27 December 2013. It included a section on “Health Hazard Evaluation” signed by Dr Hinoul on 10 January 2014.
2149 This document described the “PQI issue under review” as “an increased trend of litigation alleging injuries related to Pelvic Floor Repair Systems and Gynecare Tension free Vaginal Tape (TVT) family of products”. The document stated that all complaints received were recorded, but it had become necessary to segregate complaints by reference, among other things, to “commercial vs. litigation complaints”.2128 Dr Hinoul adverted to this in the CER. He stated that Ethicon had evaluated external data on lawyer advertising to determine if the litigation complaint increases could be related to an increase in lawyer advertising and concluded that there was a strong correlation between the increase in lawyer advertising and the increase in lawsuit-related product complaints.
2150 PQI 13-060 suggested that appropriate risk management systems were in place, and a continuous review of the information received found “no triggers that signal[led] … any quality or design issues with the products”.2129 At the same time, it estimated that since 2011, “the TVT family of products and Pelvic floor repair kits [had] experienced between four to eight fold increases in reported events with a corresponding increasing event rate recorded over that same period”. Most of these were said to relate to erosion/exposure.
2151 Setting aside the increase in “litigation-related complaints”, the “commercial complaint rates [were] flat”, which was said to be “a true representation of the product performances”. Further, the flat rate of commercial complaints was said to indicate that the increase in overall complaint rates was “not a manufacturing issue”.2130
2152 It is difficult to understand the relevance or utility of analysing complaint data to assess the impact of lawyer advertising. It was not suggested that the complaints were unfounded or vexatious. In any case, erosion or exposure, for example, can hardly be fabricated. Lawyer advertising is likely to have heightened awareness of the potential connection between the devices and certain complications. It might also have coincided with a spike in late-onset complications. In any case, as I have observed, Ethicon knew that there was a significant problem of under-reporting. In any event, there is no apparent reason for distinguishing between “commercial complaint rates”, whatever that means, and “litigation-related complaints” in an evaluation of the safety profile of a medical device.
2153 A search of the MAUDE database was not conducted. The explanation given in the report was that there were “no Non-Ethicon equivalent devices identified for GYNECARE TVT Family”.2131 This is surprising, given that the 2013 CER listed numerous “equivalent devices” made by 10 different manufacturers.2132 One of those devices was Boston Scientific’s Advantage Fit, which Professor Korda testified was “essentially” the same as TVT and which he said he was using.2133 In the 2013 CER the search of the MAUDE database generated 765 reports about Advantage/Advantage Fit.
2154 Ethicon determined that no corrective action was required because the increase in complaints was associated with an increase in the trend of litigation alleging injuries.2134 This might well be an explanation for the failure to take corrective action. For the reasons given above, however, it was not a sound justification for inaction.
2155 In the 2015 CER, Dr Hinoul stated that the complaints received were “generally consistent with the type of events that would be expected with treatment of stress urinary incontinence and the events listed in the GYNECARE TVT Information for Use (IFU)”. He said that Ethicon performed “monthly complaint reviews with cross functional groups for potential safety signals” but that “[n]o trends or potential quality issues were raised through this process”.2135
2156 The concluding paragraph of the risk–benefit analysis, which was adopted in large part in the overall conclusion of the report, used more guarded language than the earlier CERs:
The overall data continue to support the proven safety and effectiveness of GYNECARE TVT, GYNECARE TVT Exact, GYNECARE TVT-O and GYNECARE TVT ABBREVO, when used as intended. Based on the current state of the art, field performance data, and critical literature analysis related to the product, the medical benefits of the four subject devices are concluded to outweigh the risks associated with the devices. Furthermore, it is concluded that the devices meet the Essential Requirements of the MDD for state of the art for performance and safety. This review confirms that the evidence demonstrates an acceptable benefit-risk profile for these products when placed in appropriately selected patients by experienced surgeons. Adverse events associated with slings such as GYNECARE TVT and GYNECARE TVT-O are well characterized and are manageable in the majority of cases. Importantly, within the context of this review, no reports were identified within the literature which raised new safety concerns, i.e. complications not already identified and/or documented within the context of the respective subject device Risk Assessment Summaries. The report does, however, point to a significant increase in lawsuit-related product complaints. An analysis revealed a strong correlation between the increase in lawyer advertising and the increase in lawsuit-related product complaints. Whilst Ethicon acknowledges that in a small, yet significant subgroup of patients, complications are to be considered serious, it would be incorrect to conclude that the high litigation-related complaint rates poses a challenge to the large body of evidence in the literature of these products and their endorsement by the majority of independent and objective societies and physicians. Nonetheless, Ethicon remains diligent in collecting and reporting adverse events and complaints, regardless of their source or underlying support, including litigation-based reports which are predominately communicated to the company through legal complaints which are simply allegations often unaccompanied by medical records.2136
(Emphasis added)
2157 This CER was not analysed in the applicants’ submissions in any detail. It was overlooked entirely in their principal submissions and only touched upon in supplementary submissions.2137
2158 Dr Allman described it as “a good attempt” at addressing the European regulatory requirements.2138 He noted that the purpose of the evaluation was clearly stated, literature search criteria were well described, the extensive published literature was summarised at great length, and post-market surveillance data (complaint and adverse event reports) were described and discussed. Nevertheless, for the following reasons, he concluded that the report suffered from “the same major flaws” as the earlier CERs:
Conclusions simply follow a description of the literature, without a clear definition of the criteria used to reach the conclusion. Where criteria are implied, this is in the form of statements of generality: for example ‘complaints are generally consistent with the type of events that would be expected with the treatment of stress urinary incontinence and the events listed in the GYNECARE TVT Information for Use’ and ‘data are therefore generally supportive of the safety profile of TVT procedures, including GYNECARE TVT’. Mention of a potential complication, or harm, continues to be considered adequate warning of a risk. Increases in numbers of complaint, and adverse event, reports are attributed to litigation with an implication that these are not substantiated—but without discussion of any attempt by Ethicon to obtain more information from the complainant. Despite the large number of publications reviewed for the complete TVT Family, evidence for individual devices is limited. Only two studies evaluated safety of TVT-Exact in the treatment of stress urinary continence; one involving 76 patients followed for twelve months, the other involving 13 patients followed for three months. Discussion (description) of the literature relating to TVT does not clearly distinguish between abdominal and transvaginal placement of the Implant.2139
TVT-O
2159 The first post-market clinical evaluation report for TVT-O was signed by Dr Robinson on 6 August 2010.2140 This was nearly seven years since regulatory clearance had been obtained in the United States and Europe, and more than six years since the device was released on the Australian market. Once again, the delay is unexplained and inexcusable.
2160 The report was drafted by The Marketing Connexion, the business which had prepared a draft of the August 2010 CER for TVT.2141 Presumably for this reason, both CERs adopted a similar structure and used similar language. In his report, Dr Allman stated that he would not discuss the TVT-O CER in detail as he had already discussed the August 2010 TVT CER and both were “very similar” and indeed “sections [were] identical”.2142
2161 Once again, the search strategy employed to identify the literature was difficult to scrutinise and impossible to replicate. The discussion of complications reported in the literature focused on individual case studies, without any attempt at synthesis, and did not examine potential treatment options. The complaint review was also inadequate because it did not acknowledge the issue of under-reporting. This was particularly problematic as this CER was finalised two years after the 2008 notification issued by the FDA, which raised concerns about risks attending the use of SUI devices. Given those circumstances, the applicants submitted that Ethicon should have been particularly “alert and analytical” in its adverse event reporting for the SUI devices.2143
2162 There was no other individual CER for TVT-O showing post-market analysis. That assessment appeared in the 2013 and 2015 CERs for the TVT Family of Products discussed above.
2163 The 2015 CER cited a retrospective comparative study by Shaw et al (2015) reporting on 225 patients (125 with TVT Abbrevo and 100 with TVT-O).2144 The objective of the study was to determine whether decreasing the burden of mesh in the groin would decrease groin pain. At six months, the authors found that 1% of the women with TVT Abbrevo reporting groin pain compared to 9.6% with TVT-O and concluded that the use of TVT Abbrevo reduces post-operative groin pain compared with the full length TVT-O with no reduction in efficiency. The authors began their discussion with the following observation:
Groin pain following a transobturator (TO) midurethral sling is extremely frustrating to patients and difficult to treat. It may be found in 16 % of patients at 2 months and in 3 % at 1 year.
2164 They later wrote:
Theories of the origin of groin pain after TVT-Obturator sling include direct obturator nerve damage or indirect nerve compression and a myofascial syndrome arising from muscle hypertonia secondary to excessive tension or incorrect placement of the tape. Regardless of the precise mechanism, obturator neuropathy after TVT-Obturator sling placement is a difficult problem to manage, for which there is no consensus. Conservative oral and injection analgesic measures have been advocated. Where these fail, vaginal excision or lysis of mesh material may improve pain in two-thirds of patients. It is not yet clear whether excision of the sling belly alone is adequate to relieve the pain versus groin dissection and removal of the offending arm.
2165 These concerns did not rate a mention in the CER.
2166 Four of the six studies evaluated for the purpose of the CER reported that TVT Abbrevo was associated with less post-operative pain than TVT-O.
2167 This evidence did not prompt any consideration of whether TVT-O should be removed from the market. Since groin pain was “a recognized complication within the RAS”, the information reported by Shaw et al (2015) was dismissed. As it did “not represent new information”, Dr Hinoul wrote that the incidence of groin pain in the study did not undermine the safety profile of TVT-O.2145 No consideration was apparently given to whether the incidence of the complication might affect its safety profile.
TVT Secur
2168 It will be recalled that TVT Secur was first supplied in Australia in April 2007. It did not take long before the respondents became aware of problems surgeons were experiencing with the device. The evidence indicated that similar problems had been encountered in Europe before the launch of TVT Secur in Australia.
2169 On 12 September 2007 Anne Capplis, JJM’s Quality Assurance Manager, reported to Dr Aran Maree, JJM’s Medical Director for Australia and New Zealand, that one surgeon had had problems in 10 out of 18 cases (56%) involving TVT Secur.2146
2170 On 24 October 2007, noting that TVT Secur had not yet been launched in New Zealand, Dr Maree briefed JJM and Ethicon personnel about concerns he had “regarding the high ‘failure’ rates across multiple centres” JJM was seeing with TVT Secur compared to TVT-O.2147 He identified four matters of concern:
(1) This product may have been launched as a substitute for TVT-O without enough clinical data to justify such a roll-out.
(2) TVT Secur (scil) may rather be a product with a niche application in some patients for whom TVT-O is a less attractive option, after full disclosure of the higher failure rates observed with this product.
(3) The original (and current?) training program may not result in competency in device insertion or result in clinical efficacy. There appear to be “tricks” to insertion of the product and removal of the inserters which prevent dislodging the device in the process etc.
(4) As a company we need to ensure that we protect the good name of J&J reputation and avoid such issues going forward elsewhere with this product and with other products.2148
2171 On 31 October 2007 representatives of JJM discussed with representatives of Ethicon Inc. the Australian complaint data and patient outcomes with TVT Secur.2149 They also discussed “overseas affiliate experience”. According to the minutes of that meeting, anecdotal evidence indicated that “a very specific technique” was required to ensure “potential optimal efficacy”. In particular, greater tension that was previously thought necessary had to be applied on insertion and the technique used during withdrawal of the inserter had to be modified to avoid inadvertent dislodgment of the mesh. Neither aspect was included in the initial TVT Secur procedural training in Australia. Moreover:
Due to initial usage technique issues and sub-optimal performance, it was understood that some countries may have ceased marketing the product (eg Norway), and others such as Germany were experiencing market difficulties.2150
2172 In view of the local complaint history, declining surgeon preference for the product locally, the apparent technical difficulties of the procedure, the need for significant retraining, and the uncertainty of the outcome, the meeting proceeded to consider what benefits TVT Secur had over the alternative TVT product. Two were identified: a reduction in post-implantation thigh pain experienced by some patients implanted with TVT-O and the reduced potential for bowel rupture. Neither was considered significant. The meeting concluded that there was “little significant surgical benefit in using SECUR over other existing procedures” and came to an in-principle agreement to withdraw the product from the local market “until further high quality objective data [became] available confirming optimal technique and enhancing product efficacy, or second-generation product [became] available which addresses current issues”.2151
2173 In an email to Catherine Beath from the Regulatory Team in the United States on 2 November 2007, Dr Maree reported on the outcome of the meeting.2152 He also advised Ms Beath that about 20 Australian surgeons had been trained in the use of TVT Secur and that those with “significant patient cohorts” had indicated their concerns that the product had failed six weeks after implantation. These problems were not unique to Australia. Amongst other things, Dr Maree told Ms Beath that in a telephone conference he had discussed with Dr Robinson and Joe Megan, Marketing Director Asia-Pacific for Ethicon Women’s Health & Urology the experiences elsewhere:
It would appear that some surgeons are claiming to have a satisfactory success rate with a modified technique, but that, at this time, we cannot be sure that this is the universal experience. We also discussed the fact that there were three Investigator-initiated Study Randomised Controlled Trials underway, that there had been a meeting in Paris in May of this year to discuss issues surgeons were experiencing with this product. It is my understanding that some suggestions had come out in the form of (i) increased tension required with this mesh with “pillowing of peri-urethral tissues required”, (which is quite the opposite of TVT-O recommendations), as well as (ii) new tips and tricks to avoid dislodging the device when removing the inserters and (iii) new tips for minimal dissection when introducing the product. We also discussed the fact that at this time some or all of these suggested changes may not be incorporated into the Instruction For Use or technical training manual.2153
2174 The failure rates in Australia were undeniably high at approximately 13 of about 20 cases (65%) for one surgeon, approximately six out of about 20+ cases (roughly 20%) for another, and “lots of early failures” — “8 at least and still counting” from about 20 cases (at least 40%) for a third.2154 These failures were clearly unexpected. The Australian surgeons with significant patient cohorts were described in the TVT Secur “Launch Plan and strategy” document as JJM’s “Alpha Preceptors”.2155 Dr Maree noted that Professor Frazer had performed about 700 TVT cases over the years and Professor Carey had trained with Dr Lucente in the United States and, despite using his modified technique, was seeing no substantial improvement in success rates. “In short”, wrote Dr Maree in his email to Ms Beath, “some of our customers are indicating their concern and a loss of confidence in this product”.2156
2175 Dr Maree also told Ms Beath that, as JJM was in possession of “new information which is relevant to the safe & effective use of this product”, it was obliged under the Australian regulations to bring this to the attention of the regulator. He said there was “a very high likelihood” of a call from the regulator asking for an explanation of the reported patient adverse events. He said that JJM considered the most appropriate “customer-focussed and Credo-aligned position” was to withdraw TVT Secur from the market “until we are confident that a modified technique, appropriately documented and tested by way of clinical study, can be taught to our surgeons and will lead to optimal patient outcomes with this product”.2157
2176 Two weeks later, on 15 November 2007, Dr Maree provided Ethicon with a draft letter to Australian doctors offering them further training and informing them of JJM’s intention to restrict further supply to surgeons who had completed the training.2158 A letter was apparently settled with the TGA on 26 February 2008 and circulated to all user surgeons in March.2159 The letter reported that, in the 12 months since TVT Secur had been launched, during which approximately 200 devices had been sold, JJM had received reports of 24 patients with “recurrent/persistent post-procedural incontinence”. JJM agreed with the TGA to halt the sale of the devices until they had come up with a plan to address the problems. 2160
2177 In the meantime, on 9 January 2008, Dr Maree reported to his US colleagues on his meeting with TGA and foreshadowed once again the possibility of a safety alert. He also advised that Dr Garcia, a materials scientist and head of the TGA Laboratories, had asked him how the use of the term “tension-free tape” could be justified when they were training surgeons on appropriate “tensioning” of the device.2161
2178 An undated PowerPoint presentation on the Australian experience, apparently from an Ethicon entity, most likely Ethicon Inc., noted concerns from JJM about anecdotal reports of high post-procedure incontinence about six weeks after implantation with TVT Secur and speculated that it might be linked to “proctor training”, a similar issue identified in Germany in February 2007. Interestingly, the document also recorded that the mean failure rate for any TVT device was about 15 to 20%, “so physicians only ‘complain’ when this rate [is] exceeded”. It recommended against a global safety alert, saying “complaint data indicates no issue and is consistent with that anticipated during development”. It recommended that in Australia physicians be retrained and the product be “allocated” to those who had undergone the retraining. 2162
2179 In March and April 2008 audit questionnaires were issued by JJM to doctors in Australia who had used TVT Secur.
2180 In his response, Professor Frazer had nothing positive to say. He expressed no interest in the additional training and said he was unable to provide an objective opinion because his experience had been “so negative”. He volunteered, however, to look at “any good quality medium term (6 months+) data the company may have”.2163 A fellow of Professor Frazer, who had performed about 20 TVT Secur procedures, emphatically rejected the opportunity for retraining, despite the promise of “new improved techniques”.2164 A gynaecologist, whose preceptor was Professor Frazer, told JJM that there was no ideal Australian TVT Secur patient and expressed no interest in retraining in the new techniques 2165 Another surgeon, who was a TVT Secur preceptor and had undertaken 15 to 20 TVT Secur cases, said there was a need to collect sufficient data to evaluate the cause of the significantly high failure rates of early cases done by most surgeons. He asked for “patient information about the risk-benefit evaluation of TVTS vs other established products”.2166 Yet another surgeon reported a 100% failure rate: four failures out of four procedures.2167
2181 In a status report on 1 May 2008, Dr Maree noted that no surgeons had asked to be newly trained in the use of TVT Secur and that, although consultations were still under way with surgeons to determine the level of interest in formal retraining, JJM had no plans to conduct any further training in the use of the product.
2182 On 4 May 2008, in an email to a colleague arising out of correspondence with the TGA regarding orthopedic implants, Dr Maree remarked that “there are disturbingly recurrent themes here between TVT-Secur and [certain of Johnson & Johnson’s hip and knee joint replacement implants]”.2168 Dr Maree went on to say:
I am confident that action we are taking now with (i) ongoing collaboration with Operations to elevate overall Quality standards, (ii) my ongoing communication with all Marketing Directors and product managers and (iii) the New Product Introduction process, which will incorporate a pre-launch risk assessment, training agenda and a post-launch performance review should go a long way to bringing us to where we need to be to fulfil our Credo responsibilities.2169
2183 On 5 June 2008, in an email to Mark Yale, Director of Worldwide Risk Management for Ethicon, Inc., Dr Maree wrote that he understood that no product had been shipped since the beginning of the year (later confirmed by Mr Budden) “as demand has completely fallen away for this product in Australia in line with local KOL [key opinion leader] opinion”.2170
2184 By 11 June 2008, no surgeon had indicated an interest in JJM’s offer of additional training and JJM had decided not to embark upon further training. A total of 73 units were still on hold, there had been no sales of TVT Secur that year, and none of the devices had been shipped since the beginning of the year.2171
2185 Notes taken of an Ethicon meeting on 18 June 2008 with Dr Carl Nilsson, described as a key opinion leader in Europe, reveal that the complication rate in Germany was “huge” and that training was “so poor in so many countries” (original emphasis). Dr Nilsson stressed the need for clinical data. He told the meeting that there was no documentation to indicate that the mini-sling was either safer than TVT or equally effective. He also told the meeting that a “mini-sling hammock will never work”. He said that work was being done to block the marketing in Finland of the Mini-Arc and the AMS mini-sling, for lack of clinical data.2172
2186 In his affidavit Dr Hinoul minimised the extent of the problem. He intimated that only three Australian surgeons had an issue with the device and that they had reported efficacy rates with TVT Secur that were lower than expected.2173 Although he alluded to problems in Germany, he barely discussed them. He said that Dr Lucente’s reported experience with TVT Secur in 108 patients showed how results improve with experience. He failed to mention, however, that, even with “revised Lucente training”, Ethicon had noted that the success rate in Australia at least was below 50%.2174
2187 Dr Hinoul referred to a Quality Board Review of TVT Secur, referencing a PowerPoint presentation, which mentioned both the Australian and the German experiences but which regarded them as anomalous. It seems that the review was based exclusively on complaints data over the period from September 2006 to October 2007.2175 It concluded that a global safety alert was not required because complaint data indicated “no issue” and was “consistent with that anticipated during development”. Dr Allman said that complaint analysis is not a reliable tool for excluding a more widespread but localised occurrence of the same problem elsewhere.2176 He considered that the situation should have been treated as a Field Safety Corrective Action requiring notification to European regulatory authorities and a Field Safety Notice should have been issued to users of TVT Secur to inform them of the “correct” use of the device and the changes in training materials.2177
2188 The first post-market CER for TVT Secur was not finalised until late October 2010,2178 more than four years after it had been cleared for sale in Australia and two and a half years after the respondents had stopped selling it here. Michael Thomas from the Marketing Connexion emailed a draft to Dr Robinson on 28 October 2010.2179 Mr Thomas submitted the draft for Dr Robinson to review and looked forward to his comments and recommendations for finalising the report. But Dr Robinson made no comments or recommendations. It is not clear whether he even reviewed it, for Mr Thomas’s email attaching “the CER” was forwarded to Neal Brunner “to distribute as needed”.2180 Dr Robinson signed the draft without making any alterations on 1 November 2010.
2189 The 2010 TVT Secur CER was based on a review of selected literature, complaints and adverse events reported to Ethicon from 2007 until August 2010, and data generated from MAUDE during the period from January 2005 to August 2010.
2190 Although the Australian, Norwegian or German experience of the device had been the subject of at least one discussion Dr Robinson had had with Dr Maree, there was no mention of it in the CER. I can only assume that the information was not conveyed to the Marketing Connexion, to which the drafting of the report had been outsourced.
2191 The report noted that the body of published literature on mini-slings, including TVT Secur, was limited because of “the relatively short period” during which the device had been in clinical use. Of the 65 articles identified and selected for in-depth examination, only 27 were considered relevant and only 10 were summarised in the CER. Of the 10, only four involved TVT Secur. The report noted that, although many of the studies that were cited concluded that TVT Secur and other mini-slings were safe and effective, several indicated that there was a learning curve that needed to be taken into account. It compared the cure rates reported in the various studies, observing that most of them achieved objective cure rates of 80% or higher but the follow-up period was only 12 months. In contrast, a relatively small study of 41 patients based on a median follow-up of 30.2 months reported an objective cure rate of only 40%, signalling the need for larger, longer term studies to address the question of long-term efficacy. Studies comparing TVT Secur with TVT and TVT-O were said to be “very limited”. Indeed, only one offered direct comparative research. As Dr Allman observed, “this highlights the paucity of clinical evidence used to support initial CE marking of SECUR”.2181
2192 Notwithstanding the acknowledged limitations and the experience with TVT Secur in Australia and Germany, the conclusion drawn from the literature review was in accordance with the template:
The above data, taken together with any available pre-clinical data, are sufficient to demonstrate compliance with the essential requirements covering safety and performance of GYNECARE TVT Secur* System under normal conditions of use. No additional clinical data is required.
2193 The report concluded that the risk for the device was “moderate”. This led to a further review of the MAUDE database of TVT data which, according to the CER, did not indicate any harms or hazards additional to those revealed from the review of the Ethicon complaints database. As Dr Allman pointed out, however, “it is not just the type of event that is important in assessing risk (and risk-benefit); severity and frequency are also critical to determine risk-benefit”.2182
2194 Dr Allman said that the conclusion based on the literature review was not justified by the evidence presented. He said that to reach “justified conclusions” and to meet the essential requirements, it was necessary to identify criteria for acceptable safety and performance as well as for intended safety and performance. Yet, no such criteria were identified in the CER.
2195 On 29 May 2012, an internal report prepared by Ethicon Inc. stated that TVT Secur was associated with inferior subjective and objective cure rates at one year and higher reoperation rates in comparison to TVT and TVT-O. Nevertheless, Ethicon had concluded that the use of TVT Secur was “an acceptable choice of therapy for a carefully selected patient population when implanted by experienced surgeon (sic)”.2183
2196 Another post-market CER for TVT Secur was signed by Dr Hinoul on 18 September 2013,2184 following what appears to be a draft dated 9 August 2013.2185 None of the later CERs for the TVT “family” contained an evaluation of TVT Secur.
2197 The September 2013 CER purportedly reviewed relevant scientific literature published between January 2010 and February 2013. A total of forty-eight articles were evaluated. The evaluation was said to reveal no significant changes in the safety and performance of the device. No safety concerns or adverse events related to product design or manufacturing defects were said to be found in the literature and the complications and risks reported in the literature were said to be consistent with the known risks and complications described in the product labelling. The conclusion derived from the literature review was that:
The above data, taken together with previously available clinical and pre-clinical data, are sufficient to demonstrate State of the Art compliance with the essential requirements covering safety and performance of GYNECARE TVT SECUR under normal conditions of use. No additional clinical data is required. No previously unidentified harms or hazards were identified in this review.2186
2198 The CER also reviewed complaints reported to Ethicon between January 2010 and April 2013. The most common complaints related to mesh exposure/erosion. The figures for erosion, “urethral or otherwise unspecified”, rose from 12 in 2010 to 524 in 2012 and 346 in 2013.
2199 As Dr Allman pointed out that the overall complaint rate for TVT Secur (at 4.86% of the total units distributed) was much higher than that reported in the 2010 CER.2187 As in the CERs for the other SUI devices, the increase in complaints was attributed “primarily” to an increase in litigation since late 2011. Consequently, further risk reduction was said to be unnecessary and the current risk within acceptable levels. But the opinion was not supported by any reasons or analysis.
2200 A search of the FDA’s MAUDE database over the period from January 2010 to January 2013 was conducted to identify serious adverse events involving equivalent devices. The CER recorded the number of reports per device each year but contained no discussion of them. As Dr Allman observed, all three equivalent devices showed a dramatic increase in reported adverse events in 2012.2188
2201 Dr Allman said that the CER did not meet European regulatory requirements and was insufficient to justify CE marking. He pointed out that no criteria were identified to justify the statement about compliance with the essential requirements. He said that, in the absence of a baseline for the safety and performance of the device, the different nature and timing of the studies, and the different devices involved, “it is not justified to state that safety and performance is unchanged”.2189 He also remarked that it was difficult to reconcile the statement on the inferiority of performance of the device made in the 29 May 2012 document mentioned above with the conclusions in CER about “State of the Art compliance with the essential requirements”.2190
2202 These devices were covered in the CERs relating to the TVT Family of Products. Three additional studies relating to TVT Abbrevo were cited in the 2015 CER on the TVT Family of Products. The largest of these was the Shaw et al (2015) study mentioned above.2191
2203 It will be recalled that Dr Allman’s evidence was that Ethicon should have conducted clinical investigations of TVT Exact and TVT Abbrevo, with clearly defined acceptance criteria, before CE marking these devices, or considered post-market clinical follow-up studies.2192 But I was not taken to any evidence to indicate that post-market clinical follow-up studies were considered, let alone undertaken.
The POP devices
Gynemesh PS
2204 In his affidavit, Dr Hinoul suggested that the respondents had first undertaken post-market surveillance of Gynemesh PS in 2009, which was seven years after the product had been released.2193 He was referring to a review he had undertaken of clinical studies from 2005 to 2008 in which he had concluded that Gynemesh PS and Prolift had both been shown to be “very effective, safe and with a positive benefit to risk profile”. As the applicants submitted,2194 this evidence was misleading. Dr Hinoul’s review was of the literature on Prolift only, not Gynemesh PS.2195 That said, the Prolift literature was hardly irrelevant, since the mesh used in Prolift was the same mesh used in Gynemesh PS.
2205 The review to which Dr Hinoul referred was conducted with Dr Vanja Sikirica of the Health, Economics and Reimbursement division of Ethicon Inc. A “non-discussed” “poster presentation” of the results was given at the 34th IUGA meeting held in Lake Como in June 2009.2196
2206 Dr Hinoul’s evidence about the conclusions of the review was not only inaccurate in that they were confined to Prolift but they were also overstated. The conclusions as reported in the abstract (the poster presentation), which Dr Hinoul cited in support of his evidence, were that the evidence for Prolift repair was “growing”; prospective, randomised, and appropriately powered trials are needed to understand longer-term outcomes, but that “current peer-reviewed data shows that Prolift is an effective pelvic floor repair device with limited complications and high patient satisfaction”. The follow-up period of the studies was short. Furthermore, for no apparent good reason the authors excluded from their review meta-analyses. Dr Hinoul’s evidence about what the review had concluded about the safety of Gynemesh PS and Prolift were certainly at odds with the conclusions of the Maher et al Cochrane reviews. It will be recalled that Maher et al (2007) concluded that the evidence was not sufficient to support the use of permanent meshes in vaginal repair surgery except in the context of randomised controlled trials.2197
2207 Furthermore, the figures reported in the abstract of the article do not suggest that Dr Hinoul’s evidence was justified by the review either. The overall recurrence rate was 9.3%; exposure rates were 6.9%; mesh excision/resection was 6.4%; and the reported dyspareunia rate was 6.7%. In the absence of long-term results, it is difficult to understand how the device could be declared to be “effective with limited complications”, let alone “very effective, safe and with a positive benefit to risk profile”.
2208 There were other problems with the review.
2209 In cross-examination it emerged that the manuscript of an article fleshing out the details of the review, which was submitted to the International Urogynecology Journal after the Lake Como meeting,2198 was rejected for publication after all three peer reviewers questioned its validity. One of the peer reviewers cited two studies in which “poor outcomes” had been reported but which were not included in the review although they had been presented to the Lake Como meeting which Dr Hinoul attended.2199 The second reviewer also considered the review was incomplete and referred to two other studies that were not included. That reviewer had “serious concerns” as well about conflicts of interest, given the relationship between the authors and Ethicon which stood to benefit financially from the publication. The third reviewer drew attention to reliance on studies with very short periods of follow-up, writing:
Pooled analysis of outcome data associated with Prolift system.
The data should be re-analyzed only to include reasonable follow up (at least 6 months - which is still too short). Inclusion of 2 months data is not acceptable and severely impacts the integrity of the outcomes reporting. Similarly abstract data only should be excluded. The authors emphasize the improvement on subjective symptoms of the study populations but fail to note the severity of symptoms in the complications groups. These patients, albeit the minority, when assessed, uniformly have abysmal PRO data. This fact is needs (sic) to be mentioned as this is a real concern.2200
2210 Despite Dr Hinoul’s evidence, in their submissions the respondents relied only on the clinical evaluation reports.
2211 Based on those reports, and without referring to any independent evidence, including Dr Allman’s, the respondents submitted that it was “plain that Ethicon conducted regular, rigorous and detailed investigations on Gynemesh PS”. They relied on the following matters:
(a) 6 May 2010 – “Clinical Expert Report Gynecare Gynemesh PS”. This CER referred to more than 50 items of literature.
(b) 26 April 2013 – “Clinical Evaluation Report Gynecare Gynemesh PS”. This CER conducted an extensive literature review, specifically noted a series of Ethicon sponsored studies, considered the post market surveillance data, and concluded:
(i) “No new and/or unacceptable risks have been identified in the literature review covering January 2010 to January 2013”;
(ii) “The literature review data taken together with previously available clinical and pre-clinical data, are sufficient to demonstrate State of Art compliance with the essential requirements covering safety and performance of [Gynemesh PS] under normal conditions of use … No previously unidentified harms or hazards were identified in this review”;
(iii) “No new information was provided from either the literature or PMS data to indicate that any new performance issues are occurring in the clinical setting that were previously unknown”; and
(iv) “After manual review and analysis, no new hazards, increased risk or unexpected adverse events were noted”.
(c) 3 May 2013 – “Clinical Evaluation Report Gynecare Gynemesh PS”. This CER was in substantially the same terms as the one dated 26 April 2013.
(d) 19 November 2014 – “Clinical Evaluation Report Prolene Mesh Family of Products [including Prolene and Prolene Soft]”. This CER conducted a detailed literature review, considered the post-market surveillance data, and concluded that the “PROLENE Mesh Family of Products has a history of safe and effective use. The evaluation of the literature data and complaints of the last 3 years confirms that, when used for the intended indications, the benefits of the product outweigh the possible risks. No additional harms or hazards were identified in the course of this review”.
(e) 15 May 2015 – “Clinical Evaluation Report [Gynemesh PS]”. Significantly, this CER considered 27 articles that reported on the outcomes of 1,123 patients treated with Gynemesh PS, approximately 566 patients treated with equivalent devices, and 39 patients treated with a non-equivalent device. The CER concluded that:
“GYNECARE GYNEMESH PS meets state of art design requirements, and is safe and effective when used clinically as intended…
The evaluation of the clinical literature data – from January 2010 through February 17, 2015, and analysis of PMS data from January 1, 2010 through January 31, 2015 confirms that when used as intended, the benefits to the patient outweigh the possible risks associated with the use of the device … No new or increased harms or hazards were identified during the course of this review …
The Post-market Surveillance Data (PMS) provides further evidence for the benefits of use outweighing the risks. The review of Ethicon’s complaint database during this CER reporting period did not identify any unknown adverse events or any potential concerns for medium/long term safety or clinical performance”.2201
2212 Contrary to the respondents’ submission, these reports were far from regular. For a start, the first post-market CER was produced in 2010, seven years after the product was launched and nearly eight years since the pre-market CER was signed on 20 September 2002. Dr Allman thought that there might have been a report dated 17 November 2004 because of a reference in the technical file but no report with this date was tendered in evidence and the respondents did not advert to one.
2213 Neither could these CERs be said to be rigorous, although they were certainly more rigorous than the original pre-market CER.
2214 I begin with the first post-market CER signed by Dr Robinson on 6 May 2010.2202 It was neither detailed nor rigorous. Excluding the cover sheet and index pages, it was 21 pages long.
2215 The literature review in the May 2010 Gynemesh PS CER was based on 238 IUGA abstracts; 10 “relative articles” on safety and complication rates (presumably articles which compared the relative risks of complications from different surgical procedures) drawn from the PubMed search engine during the period from 2000 to 2009; one Cochrane review, and six documents produced by NICE.
2216 The “overall findings” were these.2203 The overall success rates for anterior/vagina wall prolapse surgery using synthetic grafts were said to be higher than the success rates of traditional techniques, with “gross success” greater than 86% compared to 72% respectively “after a variable, but short-term follow-up”. Mean reported erosion rates using synthetic grafts in the anterior compartment were said to range from 2.6% to 6.8%, from 0 to 12.1% in the posterior compartments and from 4.7% to 14.4% in the middle compartments. Other complications were said to be rare. The most frequently reported related to bladder perforations, which were said to range from 1.8% to 3.8% of cases in those studies reporting on it.
2217 Professor Hu observed that there were inconsistencies in the report.2204 While the “overall findings” included the mean reported erosion rates listed above, elsewhere in the report the author had noted that “Erosion rates range from 3.2 to 19.3% depending on the compartment receiving the graft reinforcement and the particular mesh employed”.2205 Further, the author stated that “(h)emorrhagic incidences and de novo dyspareunia appear to be less than in techniques involving traditional vaginal repairs or abdominal sacralcolpopexies”. Yet, as Professor Hu also pointed out:
[E]lsewhere in the report on page 12 a study by Lowman et al. (2008) is acknowledged that found a de novo dyspareunia rate after Prolift mesh surgery of almost 17%; and a study by Blandon et al., (2009) is acknowledged that found that 10 of 22 patients (45%) who received mesh for prolapse reported dyspareunia. Mention of these rates was then followed by a review of similar rates of de novo dyspareunia among case series of patients receiving traditional repair surgery for pelvic organ prolapse (i.e., Native tissue repair), but, again, it is impossible to compare such rates and come to any conclusion regarding whether mesh v. Native tissue repair is associated with higher rates of dyspareunia or other such complications without conducting a rigorous randomized controlled trial.
2218 The “general conclusion” was this:
Scientific evidence demonstrating a low recurrence rate of prolapse after novel needle suspension techniques with mesh is accumulating. This improved outcome compared to traditional repairs to treat urogenital prolapse is partially offset by an additional morbidity related to the use of mesh grafts. This is almost solely related to mesh erosion. Chronic or life threatening complications are considered rare. Hemorrhagic incidences and de novo dyspareunia appear to be less than in techniques involving traditional vaginal repairs or abdominal sacrocolpopexies.
2219 The author dismissed the concerns of clinicians and the FDA, the findings of the Cochrane reviews and NICE, writing:
In summary, notwithstanding the attention clinicians, the U.S. FDA, the Cochrane Database of Systematic Reviews and NICE have given to the question of complications associated with synthetic mesh used in pelvic organ prolapse surgery, the complication rate is considered “rare” and for in (sic) instances can be minimized through specialized training (Urogynecology) and meticulous surgical technique. Furthermore, when complications are encountered thorough awareness and immediate action also lessens severity. Additionally, based on the experiences cited by numerous researchers, the more serious complications (sans mesh erosion) occur no more frequently with mesh than in the traditional prolapse treatment techniques. And, finally, mesh use is associated with higher prolapse cure success rates, at least in the follow-up periods that are currently available.
2220 No citations were given for any of these statements. The figures cited were given without reference to a source, not only in these summaries but often also in the so-called “Detailed Findings”.
2221 No consideration was given to the cases of erosions which could not be “easily managed”. Nor was any evaluation conducted of the strengths and weaknesses of the various studies that were purportedly reviewed. No attempt was made, for example, to weigh the value to be accorded to any particular study to take into account such matters as potential sources of bias, selection and exclusion criteria, or patients lost to follow-up, or to identify whether, and if so, which studies related to polypropylene mesh and which considered Gynemesh PS or Prolift. Furthermore, although the report referred to the Maher et al (2010) Cochrane review, which reviewed 40 randomised controlled trials evaluating 3,773 women, it neglected to point out that Maher et al (2010):
(1) observed that data relating to polypropylene mesh were extracted from conference abstracts without any peer reviewed manuscripts and should be interpreted with caution;2206
(2) found that there was not enough evidence on most types of common prolapse surgery or the use of mesh or grafts in vaginal prolapse surgery;
(3) observed that evidence was lacking of benefit to women, including symptoms and quality of life improvement, for the use of grafts over native tissue repairs;
(4) said that “no data exist on efficacy or otherwise of polypropylene mesh in the posterior vaginal compartment”;2207
(5) found that, while the data from three trials comparing anterior vaginal repair using polypropylene mesh with native tissue anterior colporrhaphy showed that the former produced superior objective outcomes, considered that the trials were too small to draw conclusions about subjective outcomes;2208 and
(6) noted that mesh erosions were reported in 10.3% (30/292) of women who had a polypropylene mesh.2209
2222 It is impossible to understand how a number of the conclusions in the CER had been reached, at least in relation to mesh repair in all compartments. For the most part, each of them proved to be wrong. In particular, although it is true that the FDA did refer to complications as “rare” in a 2008 alert, in an “update” it issued in 2011, a year after this CER was signed, it was at pains to emphasise that the complications associated with the use of synthetic mesh for pelvic organ prolapse surgery were “not rare”.
2223 Then there are references to complaints. The report includes a summary of 49 complaints made to Ethicon in the period from February 2007 until January 2010. No reason is given for confining the period in this way when the device had been on the market for five years before this period began and there is no discussion of the severity of the complaints or their sequelae. The report also includes the result of a MAUDE search over the period from January 2000 to March 2010 where 125 reports of injury are noted.
2224 Finally, the 2010 CER includes a risk/benefit analysis said to have been “in-depth” which culminated in an overall residual risk score of 46 and an overall residual risk level of “high”. The report notes that the procedures and practices consistent with regulatory guidelines and company policy “indicate[d] the need for a complete Risk/Benefit analysis”. Indeed, the procedures required such an analysis if the overall residual risk score was 29 or more.2210 The process was described in revision 15 of PR602-003.2211 It was summarised by Dr Allman in his second report as “a more analytical process”, referring to risk-benefit ratios and a risk-benefit analysis worksheet, and, for a device with a high level of residual risk, requiring approval at Vice-Presidential level.2212
2225 The CER concluded that “as a result of this process and a thorough review of all other pertinent information …” the overall residual risk is “considered acceptable in view of well documented benefits/patient outcomes”. As we shall see, this conclusion was based on a false premise. The conclusion that the overall residual risk was acceptable was not reached as a result of such a process; no complete risk/benefit analysis was conducted. Ethicon failed to comply with its own protocol.
2226 Dr Allman considered that the report’s conclusion was unjustified. He observed that there was some analysis of publications describing the experience of Gynemesh PS but that the literature review addressed mesh grafts and gynaecological and urological applications in general, despite recognising that efficacy and safety differ depending on the types of mesh used.2213
2227 Dr Allman noted that the CER described concerns over the “emergence of a new morbidity”. Yet it did not mention post-market clinical follow-up, although clinical follow-up had been required since 2007 unless not undertaking it was justified. He also pointed out that the fourth revision of MEDDEV 2.12.2 issued in May 2004 provided that post-market clinical follow-up should always be considered where identification of possible emerging risks and the evaluation of long term safety are critical. He said that Ethicon should have considered the need for studies to assess Gynemesh PS in normal conditions of use, particularly in the light of the deficiencies in the original CER and the apparent importance of the surgical technique in minimising complication rates.2214
2228 Like the 2010 TVT and TVT-O CERs, the 2010 CER for Gynemesh PS was outsourced to The Marketing Connexion as part of the remediation project. The draft CER was emailed to Dr Robinson at 3.08pm on the day it was signed.2215 As the applicants submitted, it is reasonable to infer that his review was cursory. A comparison of the draft with the final document bearing Dr Robinson’s signature indicates that he made no changes although the draft was sent together with a summary of “key notes and items for particular attention” which Dr Robinson was asked to review first.2216 The notes revealed, amongst other things, that:
(1) The draftsperson undertook to re-write and re-present the report in a timely manner if, upon review, Ethicon’s Medical Affairs team determined that material information had been omitted or misrepresented. Given the time frame, such a review could not have been undertaken.
(2) The primary source material for the literature review was a report about Prolift prepared by Drs Hinoul, Robinson, and Kirkemo. The Marketing Connexion supplemented that material with the results from the 44 abstracts from the 2009 IUGA meeting relating to mesh kits. This incorporated the data from the abstracts that Dr Hinoul did not include in his article. One consequence was that the erosion rates for Prolift and the AMS analogue devices (Perigee and Apogee) were changed from 3.2%–13.6% to 3.2%–19.3%.
(3) Bladder perforation was discussed as a complication, although it had been omitted from the Prolift review.
(4) A complete risk/benefit analysis was required. The Marketing Connexion was not in a position to carry it out. Furthermore:
As the prior CER published in the current series (Morcellation Devices) didn’t require a risk/benefit analysis (based on Ethicon policies and procedures) it’s not entirely clear if the Harms/Hazards Analysis together with the complaint data (internal and external), and literature review meet the standard for a valid risk/benefit analysis for the Gynemesh CER. The concluding statement in Section F currently in this draft CER suggests that it does. However, this is simply a place holder until Dr Robinson has made a final determination.
i. Without the prior CER for Gynemesh, the original ORR Score and ORR Level are not known and, therefore, no comparative conclusion can be drawn at this time regarding a possible change and its implications.2217
2229 In his reply to the email to which the draft CER was attached, Dr Robinson noted that “the CER will serve as the Risk Benefit Analysis”.2218 There is force in the applicants’ submission that Ethicon’s preparedness to adopt an outsourced “place holder” statement as its Risk Benefit Analysis makes plain that its process of undertaking post-market clinical evaluation of Gynemesh PS was tokenistic.
2230 On 10 May 2010 Michael Thomas of The Marketing Connexion emailed Dr Robinson thanking him for the feedback and explaining that he did not respond immediately since no changes were requested. He then wrote:
On to the next one … Prolift I think is next. We should be able to use the same Lit Review, don’t you think?
2231 Dr Robinson agreed. Mr Thomas replied later that day by return email advising that if “all technical input on Prolift” is complete then that CER should be ready by 21 May.
2232 On 20 June 2012 Ethicon informed its notified body, BSI, that it would be pursuing an indication for use change for Gynemesh PS to limit mesh placement to the abdominal approach. On 27 July 2012 BSI contacted Ethicon.2219 James Newman, BSI’s “scheme manager”, advised that BSI would review the change at the next audit and would be looking to see that appropriate documents were updated to reflect this change. In addition, Mr Newman informed Ethicon’s representatives, Lucy Paterson and Laura Vellucci, that, on account of the variation to the indication for use, the CER should be revised and that any changes to the contraindications, warnings and precautions, and adverse reactions should be reflected in a revised risk management report. He also told them that the report should consider as well risks associated with off-label use (transvaginal placement) because of the possibility that the device could be used according to its original (broader) indication for use. Mr Newman said that the post-market surveillance plan should be revised, too, to reflect the narrower indication for use and that there had to be clinical data to support use of the product abdominally.2220
2233 Nearly five months passed before Ethicon embarked upon the process of preparing a new CER.2221 A deadline of 21 January 2013 was set.2222 The preparation of this report also seems to have been outsourced. The 21 January deadline was not met. In the meantime substantial amendments were proposed for the Gynemesh PS IFU.2223
2234 A first draft of the CER was submitted by Dr Jill Bunyan, an independent regulatory affairs consultant.2224 She informed Ethicon of a number of difficulties she was having. Amongst other things she wrote (without alteration):
I am slightly unclear for which procedures mentioned in the papers are those that Gynemesh PS is currently recommended/promoted for use in the EU and it may be that we can alter table 2 to separate out ‘recommended uses’ from ‘other uses / previous uses’. Much of the negative/unfavourable data may therefore be for uses for which Ethicon has already stopped selling the product. The downside of course is that a review of each paper would have to be redone to determine which surgical route was utilised.
As a brief note, there were only 5 RCTs located within the 2010-2012 timeframe. Only one of them used the product via the abdominal route and the results are combined with those for the vaginal route so no comparison is possible. I have seen in the filenote of the BSi conversation in July which was provided today, that ‘supporting clinical data for the use of mesh in POP repair’ would be needed. I do not feel that we can fully support this based on the data available. Hopefully the inclusion of the various clinical guidelines will go some way to resolving this issue.
As indicated previously it would also be helpful to put in a brief paragraph on Ethicon’s response to e.g. the FDA warning letters and other NICE guidance, especially if additional studies, surveillance or educational programs have been initiated?
NB at present I have included mesh kits (ProLift, Prosima) which include Gynemesh as being the same device. Given the difference in side effect profile, is this appropriate?
2235 Dr Bunyan also sought clarification of complaints data and sales figures, since the information she was given from the complaints file were “surprisingly different” from those presented in the risk management report.
2236 As the applicants submitted, Dr Bunyan’s email should have caused Ethicon to consider whether the device had a risk/benefit profile sufficient to enable it to remain on the market even with its revised indication.2225 Her suggestion that an explanation of Ethicon’s response to the FDA warning letters was needed should have prompted Ethicon to disclose to her the post-market surveillance orders that the FDA had made in respect of Ethicon’s products. Arguably, the anomalies in Ethicon’s complaints data should have led to a review of the way in which Ethicon was handling complaints. But none of these steps appears to have been taken.
2237 On 22 January 2013, Dr Meng Chen brought to Dr Hinoul’s attention two ongoing, independent, long-term (5 year) multi-centre studies using Gynemesh PS (one in the UK and one in China). Both were apparently large studies. She also informed him that the Chinese study was “headed by the top medical school in the nation” and had sites “all over the country” and that an adverse event incidence report from the UK study had been sent to the UK regulator, the MHRA. She told Dr Hinoul that Ethicon “should have large studies like these two on our radar.2226
2238 Two days later, Dr Hinoul asked Lisa Ovington to pass on to Dr Bunyan the risk benefit profile sent to the MHRA earlier that year. He did not mention the two studies Dr Chen had told him about the resulting adverse event report provided to MHRA.2227
2239 Amongst other things, the applicants drew attention to the following matters that were included in the draft report but omitted from the final version signed by Dr Hinoul:
(1) a statement that no previous CERs had been prepared with the exception of ongoing simple literature reviews, the last of which was in May 2010; and
(2) a statement that few RCTs had been conducted comparing efficacy and/or safety of different techniques.2228
2240 The applicants also pointed out that a statement in the CER about complications in a study by Tijdink et al (2011)2229 was amended to include a statement emphasising the success of mesh surgery.2230 Furthermore, they noted that Dr Hinoul took issue with the statement made in the draft report that, although Diwadkar et al (2009)2231 determined that the total complication rate was higher for sacrocolpopexy (17.1%) than vaginal mesh kits (14.5%), vaginal mesh kits induced more severe complications requiring surgical correction.2232 Dr Hinoul commented:
NO that is NOT the conclusion of DIWADKAR. The findings of this meta-analysis demonstrate that total complication rates appear to be similar for traditional vaginal surgeries, sacral colpopexies, and vaginal mesh kits for the treatment of apical prolapse.
2241 The reference to total complication rates appeared in the first paragraph of the conclusion. But it was a small part of it. The following sentence began with the use of the telling conjunction “however”. The conclusion in full reads as follows. It demonstrates that Dr Hinoul misrepresented it:
The findings of this meta-analysis demonstrate that total complication rates appear to be similar for traditional vaginal surgeries, sacral colpopexies, and vaginal mesh kits for the treatment of apical prolapse. However, despite having the shortest follow-up period out of the three groups, the reoperation rate for complications and total reoperation rate (including complications and prolapse recurrences) was highest in the vaginal mesh kit group. Most of these reoperations are necessitated by fistulae and mesh erosions. These complications are difficult to prevent, affect quality of life, and often are not managed medically. Although visceral injury and mesh erosion also led to reoperations in the sacral colpopexy and traditional vaginal surgery groups, the majority of all complications in these groups was managed pharmacologically. An additional key finding was that, despite the longer follow-up and greater number of participants, there was a lower total complication rate in the traditional vaginal surgery group compared with sacral colpopexy. The relatively higher rate of visceral injuries and wound complications in the sacral colpopexy group is likely attributed to the abdominal approach.2233
(Emphasis added)
2242 Diwadkar et al (2009) also noted that there were no clinical trials or other comparative studies to date that compared the three main approaches to repairing the apical compartment in women undergoing surgery for pelvic organ prolapse. They said that, while sacrocolpopexy was considered by some “the gold standard” apical suspension procedure, since it had a relatively low rate of reoperation for prolapse recurrence, “this was at the expense of a high complication rate”. In contrast, traditional vaginal procedures had a higher reoperation rate for prolapse recurrences but fewer complications requiring surgical intervention. They continued:
Most importantly, our results suggest that, despite the lowest reoperation rate for prolapse recurrence, vaginal mesh kits have the highest rate of complications that require surgical intervention, which, on balance, results in the highest rate of total reoperation after apical suspension for pelvic organ prolapse. This raises the concern that the risks of these newer procedures may be greater than their benefits. One can speculate that more recurrences and complications may be diagnosed with time, given the relatively shorter mean follow-up period in the mesh kit group.
2243 Dr Hinoul well knew of these findings. Soon after the study was published he circulated a copy to colleagues at Ethicon observing that “[t]his paper really answers the question on mesh safety in an up to date and exhaustive manner”.2234
2244 The draft 2013 CER included a statement that the most significant of the adverse events associated with Gynemesh PS was mesh exposure, which could result in serious consequences for the patient. Following this statement Dr Bunyan noted:
The company has therefore taken the step of limiting the product indications to placement via the abdominal route only (laparotomy or laparoscopic approach) in order to minimise the risk of this occurring. This is in compliance with the recommendations presented in a number of recent UK/EU Guidelines.2235
2245 While this conclusion was a logical one, it provoked an agitated response from Dr Hinoul and it was omitted from the final report. Dr Hinoul’s response reads as follows:
Absolutely NOT!!!!!!!!!!!!!!!!!!! This was a commercial decision not based on safety or performance.2236
2246 In cross-examination Dr Hinoul conceded that the account of the reason given by Dr Bunyan in the draft report would not have come from someone outside the company but refused to concede that it was likely that it had come from someone within the company. These two positions are impossible to reconcile and reflect adversely on Dr Hinoul’s credit.
2247 The draft also contained the following paragraph:
The risk management file is available in the technical document for each product and is reviewed under a separate internal procedure at Ethicon. It has been advised that although overall residual risk level remains high for this product, that the Overall Residual Risk is considered acceptable for the now proposed limited indications.2237
(Emphasis added)
2248 The emphasised phrase was deleted from the draft. 2238 Once again, Dr Hinoul’s response was an agitated one. It reads simply “NO!!!!!!!” In cross-examination, Dr Hinoul accepted that the limitation had probably been given by someone within Ethicon. He was reluctant to admit that it was he who deleted it from the draft.2239 If it was not Dr Hinoul who deleted the phrase, then, having regard to the response from Dr Bunyan, it was done at his direction. The response from Dr Bunyan was:
Happy to delete, but this is what the RMR seems to report?
2249 The final version of the CER, which was approved by Dr Hinoul on 26 April 2013, did not include the limitation.2240 Nevertheless, a new sentence was added which goes some way toward mitigating the omission. It reads:
The benefits of using the product as recommended therefore continue to justify the associated risks.
2250 In the section entitled “Critical Analysis of Complaints & Adverse Events”, Dr Bunyan had written that there were 473 product-related complaints on the database and that the most common related to post-operative mesh exposure. 2241 The draft version had included the conclusion that “mesh exposure … remains the most significant adverse event reported”.2242 But this was omitted from the final version. Dr Hinoul’s comment in the draft was to ask whether it should be noted that “the increase in reports is related to mesh litigation”. As I have already observed, even if this were true, the relevance of such an observation is obscure. Mesh exposure is an objective sign, it cannot be feigned.
2251 There is force in the applicants’ submission that Dr Hinoul’s interventions had ensured that “any comment that exposed the real risks associated with the device was made anodyne”.
2252 The 26 April 2013 CER signed by Dr Hinoul noted that the indication for use for Gynemesh PS had been altered to limit mesh placement to the abdominal approach and relied on the statement in the FDA’s 2011 Safety Communication2243 that transabdominal mesh repair appeared to have a lower rate of mesh complications, than transvaginal POP surgery. But in four different places the report emphasised that the revised indication had been made for commercial reasons.2244 The FDA Safety Communication also warned of serious risks associated with transvaginal use but the CER failed to mention these aspects of the document despite references elsewhere to articles relating to transvaginal POP surgery and the advice Ethicon had been given by BSI in June 2012 to consider risks associated with off label use (transvaginal placement). Nor did it advert to the § 522 orders the FDA had imposed on Ethicon in 2012 (discussed below in Part X) which appear to have been the impetus for the change of indication and with which it had not complied. Furthermore, the CER failed to mention negative expert feedback.
2253 The conclusion of the report was that Gynemesh PS complies with the essential requirements covering safety and performance under normal conditions of use and that no additional clinical data were required.
2254 Dr Allman said that the conclusion was only partly justified by the evidence presented. As he explained, since one of the essential requirements is that devices must achieve the performances intended by the manufacturer, a review of the literature demonstrating that there were no unexpected safety or performance issues cannot be used to justify compliance with the essential requirements without first identifying how the device was intended to perform.2245 Dr Allman pointed out that the data upon which the 2013 CER relied derived from the use of mesh for transvaginal repair when by this stage Gynemesh PS was indicated only for abdominal use. He concluded that the conclusions of the CER regarding compliance with the essential requirements were not justified and that the CER did not meet European regulatory requirements and was insufficient to justify CE marking.2246
2255 Dr Allman’s opinion on this point must be accepted. Before the publication of the long-term outcomes of the CARE study by Nygaard et al (2013), the evidence is that little was known about long-term durability, complications, and pelvic floor symptoms after abdominal sacrocolpopexy. In that publication, Nygaard et al (2013) observed that “[t]he few studies assessing outcomes beyond two years [were] limited by small sample sizes, inconsistent outcome assessment, potentially biased examiners, and non-standardized follow-up”.2247
2256 Professor Hu observed that a notable omission from the 2013 CER was any acknowledgment of the consequences of mesh exposure, erosion or extrusion. He referred to the observations in the paper by Tijdink et el (2011) that mesh excision to treat complications after surgery for either pelvic organ prolapse or stress urinary incontinence is successful in the majority of cases but “with a substantial risk of serious complication”, that excision to treat the complications is “challenging and often requires extensive dissection and careful operating technique”, and that “serious complications may be associated with more extensive or complex surgery”. Professor Hu expressed the opinion that it was necessary to include in the device labelling “the significant risk of complicated surgery to treat mesh exposure, erosion or extrusion which, in turn, occurs in a substantial (10% or more) of mesh repairs”.2248 He also considered that pelvic pain, dyspareunia and “other impacts on quality of life” should arguably be added to the adverse reactions section of the IFUs.
2257 Professor Hu also disagreed with Dr Hinoul’s statements that no additional clinical data were required and that the benefits of using the product as recommended continue to justify the associated risk. He explained that “the handful of randomized controlled clinical trials that were available up to January 2013, the most recent date of literature review associated with this clinical report, [constituted] too sparse a database from which to draw final conclusions regarding the safety and efficacy of the product”. Professor Hu said that Dr Hinoul’s statements were at odds with the opinion statement of the American College of Obstetricians and Gynecologists published by the American Urogynecologic Society in December 2011, to which Dr Hinoul did not refer, citing the following summary and the conclusions, which were consistent with the conclusions of the Maher et al Cochrane reviews:
With the use of a composite of anatomic success, patient-oriented improvement and satisfaction, and total reoperation rates, success rates of native tissue repairs may be higher than previously thought. Based on available data, transvaginally placed mesh may improve the anatomic support of the anterior compartment compared with native tissue repairs; however, there are insufficient data on the use of mesh for the posterior or apical compartments. The risk/benefit ratio for mesh-augmented vaginal repairs must balance improved anatomic support of the anterior vaginal wall against the cost of the devices and increased complications such as mesh erosion, exposure, or extrusion; pelvic pain; groin pain and dyspareunia.2249
(Emphasis added)
2258 The conclusions of the opinion statement were that continued audit and review of outcomes were required, together with the development of a registry for surveillance for all current and future vaginal mesh implants and that rigorous randomised trials should ideally be conducted comparing the effectiveness of synthetic mesh and native tissue repair with long-term follow-up.2250
2259 Professor Hu noted that Dr Hinoul had referred to a review of synthetic mesh in the surgical repair of pelvic organ prolapse by Keys et al (2012) but omitted to mention its conclusions. Professor Hu reported that Keys et al (2012) (the full citation did not appear in the CER or in Professor Hu’s report and the article itself was not tendered) concluded that:
Mesh-based techniques show better anatomic results than traditional repair of anterior POP, but subjective outcomes are equivalent. Further research and Level I evidence are required before mesh-based repair of POP can be standardized.
2260 Professor Hu pointed out that the finding that subjective outcomes were equivalent contradicts the statement in the CER that the benefits of using the product as recommended continue to justify the associated risks.2251
2261 The 2013 CER also purported to evaluate complaint data and adverse event reports over the period from 2010 to 2012. As Dr Allman observed, while it noted that the rate of complaints received by Ethicon was 0.71% in terms of total units distributed, there was no discussion of the dramatic increase in the numbers of adverse events reported to the FDA over the same period: from 24 in 2010 and 52 in 2011 to 395 in 2012, the overwhelming majority of which were related to mesh erosion.2252 Further, while the report estimated the frequency of permanent or long-term impairment from Prolift to be 1/100 to 1/1000, it did not explain why a 1/100 risk of permanent or long-term impairment was considered acceptable. Nor did it explain why additional studies were not considered necessary to establish whether the true risk was 1/100 or 1/1000. 2253
2262 On 3 May 2013 Dr Hinoul signed another copy of the CER. The respondents did not account for this. Changes were made to the “benefits versus risk analysis” section, first to note that the risk analysis had identified a number of high risk categories and to include a discussion on those categories. The categories were erosion, internal organ damage and nerve damage and/or pain. The conclusions, however, did not change. The analysis was superficial, relied on a limited selection of material, and tended to downplay the seriousness of these complications. Once again, no reference was made to the conclusions of the FDA about these matters.
2263 By this time, Ethicon had received 3,241 product-related complaints.
2264 The 3 May 2013 CER also came in for trenchant criticism from the Australian regulator, the TGA, which I discuss below.
2265 The last of the Gynemesh PS clinical evaluation reports is dated 15 May 2015.2254 It was also signed by Dr Hinoul. Its stated objective was to update the previous CER of 3 May 2013 “per the new revision of PR-0000277 Rev 9, Franchise Procedure for Evaluation of Clinical Data for CE-Marking”. Its purpose was “to determine the State of the Art of the device, including whether additional knowledge has become available, pertinent to recognized or emerging hazards and whether the acceptability of these risks, should any be identified, changes the benefit-risk profile of the device and that use of the device, including safety and performance continues to meet its claims”. An additional purpose was to provide “a comprehensive analysis” of available and relevant data, including clinical and safety data relating to Gynemesh PS or devices said to be equivalent to Gynemesh PS.
2266 According to the report, some 27 articles were included in the review, 20 of them reporting on 1,123 patients treated with Gynemesh PS, approximately 566 patients treated in six studies with allegedly equivalent devices identified as Prolene mesh, Prolene Soft mesh, and Surgipro mesh (a product of Covidien), and 39 patients treated in two studies with Marlex (made by Bard), an allegedly non-equivalent device.
2267 Dr Hinoul’s overall conclusion from the literature review was that “these articles provide supporting evidence regarding the acceptable safety and performance profile of [Gynemesh PS] when the device is used as indicated”.2255
2268 The period covered by the review was January 2010 to February 2015. As Professor Hu pointed out, the CER ignored, without explanation, key publications that appeared in the literature in that period, including the Cochrane review by Maher et al (2013) and several prospective randomised controlled trials involving Gynemesh PS or “equivalent” devices and which compared the devices with native tissue repair. Professor Hu cited eight such studies as examples. Those examples included Altman et al (2011) and de Tayrac (2013). In these circumstances, the review could scarcely be described as comprehensive. With good reason, in the light of the omissions and the associated risks underscored in the various studies, Professor Hu disagreed with the overall conclusion.2256
2269 Dr Allman did not comment on these omissions, presumably because he was not briefed with the literature to which the CERs referred or asked to consider it. He said that the 2015 CER was “a good attempt to address European regulatory requirements for clinical evidence” but at the same time identified “several serious flaws” in it.
2270 First, he observed that, although the device was described as “one of the most studied meshes on the market” and was said to have been “safely and effectively used for treatment of disorders”, there was “extensive discussion of device equivalence and justification for the use of data generated with Prolene Mesh and Prolene Soft Mesh used in hernia repair”.2257
2271 Second, Prolene Soft mesh was chosen for the Gynemesh PS device because its characteristics were said to be different from Prolene mesh.
2272 Third, most of the clinical studies reporting experience with Gynemesh PS addressed transvaginal placement. Indeed, 912 of the 1,123 patients mentioned by Dr Hinoul (81%) were treated using a transvaginal or paravaginal approach.
2273 Fourth, and “most importantly”, the report claimed that the device conformed to the essential requirements and that the risks were acceptable without explaining or justifying those conclusions.
2274 The CER asserted that post-market surveillance follow-up was unnecessary because: “there are no unanswered questions of long-term safety and performance; no previously unknown risks identified in the literature or other data sources for similar marketed devices: and sufficient history to omit further verification of safety and performance of device when exposed to a larger and more varied population of clinical users”, and “the medium/long-term safety and clinical performance of the device(s) have been demonstrated and are known”. Similarly, it asserted that PMCF clinical studies were unnecessary “due to the low, well characterized risk of the products demonstrated in [Ethicon’s] risk documents, the long-term reactive data available on the device(s) and the clinical information provided by literature reviews through the CERs”.2258
2275 Dr Allman said that, given the limited data described in the CER for abdominal placement of the device, this was a weak argument and it did not support Dr Hinoul’s conclusion regarding the safety and performance of Gynemesh PS.2259
2276 Dr Allman also commented on the erosion rates mentioned in the CER that had been derived from the reports to Ethicon (0.02% and 0.01%), contrasting them the rates reported in the TVM studies. As Dr Allman observed, this suggested that “erosion events” were under-reported and Ethicon should have treated the information accordingly. Dr Hinoul had remarked that the rates were “distinctly different from rates that [he] review[ed] as a medical director in clinical trials”2260 but did not take the matter any further.
2277 The 2015 CER for Gynemesh PS did not refer to the paper by Nygaard et al (2013) reporting on the long-term outcomes of abdominal sacrocolpopexy revealed by the CARE trial, published in the Journal of the American Medical Association in 2013.2261 As I mentioned in Part VI, the CARE trial was a multi-centre, RCT in women without stress urinary incontinence undergoing abdominal sacrocolpopexy for pelvic organ prolapse.
2278 The object of the trial was to determine whether adding a prophylactic anti-incontinence procedure (Burch urethropexy) affected the incidence of de novo stress urinary incontinence, a common event after surgery for pelvic organ prolapse. At two years, the incidence of stress urinary incontinence was 32% (47/147) after urethropexy as against 45.2% (70/155) after no urethropexy (P= .03). To understand the balance between positive and negative outcomes and the effect of a Burch urethropexy over time, women in the CARE trial who had completed their final two-year visit were invited to participate in an extended CARE study. The primary objectives of the extended study were to compare the long-term anatomic success rates, stress continence rates, overall pelvic floor symptoms, and pelvic floor-specific quality of life, and to describe mesh-related adverse events in women undergoing abdominal sacrocolpopexy who were or were not randomised to undergo urethropexy.2262
2279 Nygaard et al (2013) chose what they described as “a clinically relevant definition of anatomic failure”. By five years, nearly a third of women in the study met their definition of failure, although 95% had no repeat treatment for pelvic organ prolapse. The authors argued that, “despite progressive loss of anatomic support, abdominal sacrocolpopexy generally provides relief of POP symptoms”. They acknowledged, however, that although the low reoperation rate for prolapse recurrence could imply that women found the treatment adequate, for older women “other health and social concerns may assume primacy over vaginal bulge symptoms”.
2280 Nygaard et al (2013) expressed surprise at the magnitude of treatment failure rates. They suggested that the lower success rate might in part be explained by “the rigor of their data collection, with unbiased outcome assessment and use of validated outcome measures, or by nonstandardized aspects of surgical technique”. They joined the chorus of voices calling for comparative effectiveness trials with long-term follow-up (of at least five years).
2281 Nygaard et al (2013) extracted “three key points” from the data.2263
2282 First, abdominal sacrocolpopexy is less effective than desired. There was no consensus on defining cure after POP surgery and depending on definition, two year cure rates for abdominal sacrocolpopexy range from 19% (perfect anatomic support) to 97% (no retreatment for POP).
2283 Second, no “clinically significant trade-offs” could be identified in undertaking surgery to treat stress urinary incontinence at the time of abdominal surgery for pelvic organ prolapse.
2284 Third, complications related to synthetic mesh continue to occur over time. The authors emphasised that “[l]ong-term follow-up was mandatory to understand the long-term patient burden associated with surgical materials and devices”.
2285 In their concluding remarks they wrote:
Placing synthetic mesh transabdominally to treat POP requires balancing a need for greater effectiveness with the probability for more complications. As evidenced by our results, patients who have received procedures in which mesh is placed trans-abdominally have problems that become apparent long after the time of surgery.
We anticipate that continued research in mesh development will lead to new materials and applications with fewer adverse events, but our data highlight the importance of careful long-term evaluation of new devices…
Based on our results, women considering abdominal sacrocolpopexy should be counseled that this procedure effectively provides relief from POP symptoms; however, the anatomic support deteriorates over time. Adding an anti-incontinence procedure for women continent preoperatively decreases, but does not eliminate, the risk of de novo SUI. Surgical counseling about the ongoing risk of mesh-related events even for abdominal sacrocolpopexy is critical. Women should be aware that symptoms such as vaginal bleeding, discharge, and pain may be due to mesh erosion and should seek help accordingly.2264
2286 Nygaard et al (2013) did not mention which brands of mesh were used in the procedures. It is surprising all the same that there was no reference to it in the CER, given the limited indication for Gynemesh PS and when Professor Lam described it in cross-examination as “the only large randomised trial [that] looked at sacrocolpopexy”.2265 It is inconceivable that Dr Hinoul was unaware of it. The Journal of the American Medical Association was not an obscure publication. Moreover, the copy of the article tendered in evidence was an Ethicon document and it discloses on its face that it was downloaded from the journal’s website by Ethicon’s Dr Chen only two days after it was published.
2287 The trial was important for a number of reasons. It was publicly-funded. Its findings were generalisable in that it involved surgery conducted by 21 surgeons at seven sites. Although the two-year anatomic failure and reoperation rates were said to be consistent with those reported in “a large body of literature”, the five-year failure rates were higher than those cited “in the few smaller longer-term studies”. The study’s strengths were said to include its randomised design, including the blinding of both participants and outcome assessors to randomisation assignment, the five-year or longer follow-up, the multicentre nature of the study, and the use of validated measures to assess both anatomic and symptomatic outcomes. In addition, all validated outcomes were assessed by trained study personnel and not by the surgeons.
2288 In the circumstances, the omission of a reference to, let alone a discussion or analysis of, the CARE trial was at least careless. It also suggests that BSI’s concern about selection bias in literature reviews was still a problem two years after it raised the issue with Ethicon.
Prolift
2289 Although Prolift was launched in 2005, five years passed before the first post-market clinical evaluation report was produced and another three years before the second. The respondents offered no explanation for the delays.
2290 Professor Collinet reported that the TVM Group continued its research after the commercial release of Prolift, particularly research into the incidence of mesh contraction. He said that mesh contraction was a persistent problem, along with the possibility of de novo dyspareunia. He proclaimed, however, that the problem of mesh exposure “ha[d] been practically resolved, due to very low incidence and satisfactory treatment techniques”.2266 This conclusion is at odds with the preponderance of the evidence.
2291 On 24 May 2006 a review was completed of the complaints received during the first year of Prolift.2267 The complaints included post-operative complications (which were not particularised), 10 cases of organ injury, and four cases of erosion. The complaints were categorised, totalled, and compared to the number of devices sold (50 out of 25,480, with a frequency of 0.20%). The review was conducted by Jeffrey Everett, an Ethicon design quality engineer. Mr Everett reported that “no new Hazards or Failure Modes were identified”; all frequencies, occurrence and severity [were] within risk tolerance of the Risk Analysis for [the] product”; and no change to that analysis was necessary. But the review did not mention the severity of any of the reported complications and did not acknowledge the problem of under-reporting.
2292 A second post-launch complaint review was conducted. Its results were reported by Mr Everett on 28 March 2007 and a revised version was presented on 14 June 2007.2268 In less than a year the number of complaints of post-operative complications, organ injury, and erosion had more than doubled. There were also “new” complaints. They included vaginal exposure, vascular injury, nerve damage, haematoma, “orientation” of the implant, and allergic reaction. Once again, however, Mr Everett wrote that no new hazards or failure modes had been identified, and all frequencies, occurrence, and severity were within acceptable risk tolerance for the product, without defining or explaining what an acceptable risk tolerance was and why the level of risk was acceptable.
2293 On 5 December 2007, shortly before Dr Hinoul joined Ethicon, an article of which he was the lead author was submitted to The Journal of Minimally Invasive Gynecology. The article was published in the journal’s September/October 2008 issue.2269 Hinoul et al (2008) reported the findings of a case study of patients designed to estimate the anatomical and functional outcome of Prolift Anterior for the repair of anterior vaginal wall prolapse. It was said to be the first published prospective study in the PubMed database evaluating Prolift Anterior that reported on both anatomic and functional outcomes.
2294 Fifty-one patients met the inclusion criteria. Patients with predominant middle or posterior compartment prolapse were excluded. Forty-eight underwent treatment with Prolift between May 2005 and October 2006 in a hospital in Genk, Belgium. All procedures were performed by Dr Hinoul who was reported to have had “extensive experience with prolapse surgery using mesh”. Patients were assessed clinically at two, six, 12, and 24 months after surgery. The median follow-up period was 14 months. Six patients were lost to follow-up.
2295 Anterior colporrhaphy was performed instead of Prolift surgery where concomitant hysterectomy was also indicated or where the patient had a history of endometrial or breast cancer. All patients received vaginal oestrogen therapy for at least four weeks before surgery. It is reasonable to infer that these were precautionary measures. In cross-examination, when asked why he chose to administer oestrogen, Dr Hinoul replied that it “felt more comfortable operating on these ladies when the vaginal tissue was in optimum for healing”, that it meant that they would be “well-epithelised” at the time of surgery.2270 Yet none one of these precautions was recommended in the IFUs for any of the Ethicon devices.
2296 Objective cure was realised in 46 out of 48 (95.8%) patients. Subjective cure was achieved in 40 out of 42 (95.2%) patients. Notwithstanding the precautions Dr Hinoul took, however, there were significant complications. Postoperatively, 5 out of 48 (10.4%) patients developed an erosion, described in the article as “the most feared complication of vaginal mesh techniques” and surgical intervention was required for two of them (4.3%). Urgency symptoms persisted in 3 out of 21 patients (14%). De novo urgency and frequency developed in only 1 patient (2%). De novo stress incontinence, however, developed in 4 out of 30 (13%). In 9 out of 29 (31%) of sexually active patients, dyspareunia due to the prolapse was present before surgery and disappeared after surgery; 3 out of 20 (15%) reported de novo dyspareunia.
2297 One of the patients requiring surgical reintervention had insulin-dependent diabetes. That prompted a change in approach. The authors explained:
Considering that poor wound healing is a well-established complication in diabetic patients, subsequent to this case, we refrained from offering mesh procedures to patients with insulin-dependent diabetes.2271
2298 Had insulin-dependent diabetes been included in the IFU as a contraindication or been referred to in a suitably crafted warning, the need for a second operation for insulin-dependent diabetic patients might have been obviated. Yet none of the IFUs for the Ethicon devices indicated that mesh repair was contraindicated or should be avoided in such cases, not even after Dr Hinoul began working for Ethicon.
2299 Hinoul et al (2008) observed that their rate of shrinkage was lower than that which was reported by Fatton et al (2007) (2% as against 17%). They speculated that in taking care to stretch the mesh slightly to achieve a flat placement they might have protected against shrinkage. They went on to point out, however, that their erosion rate was higher than the rate described by Fatton et al (2007) (4.7% as against 10.4%). They again speculated that the care taken to stretch the material might have facilitated development of erosion. No other potential cause seems to have occurred to them and no investigation was apparently instigated. One factor that might have influenced the different results was that Fatton et al (2007) were reporting findings at three months whereas Hinoul et al (2008) were reporting findings at up to two years. Surgeon experience or the “learning curve”, however, was effectively excluded. Hinoul et al (2008) said that in this study erosions occurred during the first and last procedures “suggesting that its occurrence did not depend on the surgeon’s experience”.
2300 In an attempt to understand the incidence of what they called “bothersome persistent postoperative pain”, Hinoul et al (2008) noted its occurrence “in some cases” following transobturator anterior vaginal mesh procedures, such as implantation of Prolift Anterior, but said that it was unclear to the authors what proportion of these cases was specifically related to the use of mesh. Since two patients who complained of persisting postoperative pain had also undergone a concomitant TVT-O procedure, the authors queried whether the pain might have been attributable to the increased load of polypropylene in the obturator space which could have caused additional scarring and subsequent irritation of peripheral obturator nerve branches or traction on the muscles.
2301 Hinoul et al (2008) concluded that Prolift Anterior “seems to provide a valuable surgical alternative” in that it had favourable anatomic and functional outcomes. But they proceeded to offer a note of caution:
The postoperative complication rate is 14.6%; 6.3% of patients required reintervention because of mesh-related complications. Patients deserve extensive counseling of what is known and not known about available new surgical techniques. The results of this study help to improve the quality of this counseling. Comparative studies will show whether these innovative techniques are the first choice of treatment in patients with anterior vaginal wall prolapse. As long as the results of such studies have not been published, prudence continues to be warranted because longer-term follow up of the evaluated surgical technique may show adverse effects that have not yet revealed themselves.
2302 The first post-market CER on Prolift was signed by Dr Robinson on 2 July 2010.2272
2303 The overall findings were identical to those made in the Gynemesh PS CER some two months earlier. So, too, was the general conclusion. In fact, save for substituting the name “Prolift” for “Gynemesh PS” in the conclusion to the literature review, the entire literature review in the Prolift CER is merely a copy of the Gynemesh PS CER. Consequently, the criticisms of the Gynemesh PS CER are apply equally to the Prolift CER.
2304 The complaints data, however, were quite different. The rate of complaints to Ethicon about Prolift were considerably higher than the rate of complaints about Gynemesh PS (424 in the case of Prolift, of which 186 were associated with harm, compared to 49 for Gynemesh PS, of which 47 were associated with harm) and the reports to MAUDE were nearly twice as high (233 for Prolift compared to 124 for Gynemesh PS). Yet, no consideration appears to have been given to the significance of the differences.2273
2305 The 2010 Prolift CER acknowledged that the safety and effectiveness of the Prolift system compared to conventional surgical repair for pelvic organ prolapse had not been demonstrated in randomised controlled clinical trials but stated that “randomized, controlled clinical evaluations” were under way. It also stated that in the United States “substantial equivalence of [Prolift] to synthetic mesh with the same indication had been demonstrated through benchtop and cadaveric testing and that information on the clinical performance of mesh for pelvic floor repair was available in the published literature”.2274 It referred to the publication in 2008 of the results of the French and the US TVM studies but barely mentioned their findings or the significance of them. It certainly did not analyse or evaluate them. Nor did it mention that the French TVM study failed its pre-defined criterion for success. Hinoul et al was cited, as it had been in the 2010 Gynemesh PS CER but only to note (without more) the erosion (now called exposure) and reintervention rates. Apparently it did not occur to the author of either report to consider the significance of the precautionary measures taken in that study or to reflect on either the note of caution or the adequacy of the IFUs.
2306 The risk/benefit analysis in the 2010 Prolift CER was based only on complaints data and did not apparently take into account the outcomes reported in the scientific literature. As I have already observed and Dr Hinoul admitted, complaints data are notoriously unreliable. Even so, the overall residual risk score was given at 68 and the overall residual risk level characterised as “high”. At the same time, the risk was considered acceptable “in view of well documented benefits/patient outcomes”. In the absence of a randomised controlled trial and in the light of the statements made in the Cochrane reviews and the failure to evaluate the studies referred to in the report, I struggle to see how that conclusion was open.
2307 Dr Allman was critical of the 2010 CER for various reasons. He said it suffered from the same deficiencies as the 2010 Gynemesh CER. He went on to say:
Although the PROLIFT report does contain data from two observational studies using pre-cut surgical mesh of the same type, and ‘similar’ shape, to that provided in the PROLIFT system, surgeons were not supplied with the complete PROLIFT system and were free to choose their own implantation instruments. Those studies have not been published (at least no references to publication have been provided) so it is not possible to independently determine how reliable those studies are, nor how relevant they are to use of the complete PROLIFT system. There are no clinical data available demonstrating the safety and performance of the PROLIFT device system in its normal conditions of use. The conclusion of the 2 July 2010 Clinical Expert Report, that available data are ‘sufficient to demonstrate compliance with the safety and performance of GYNECARE PROLlFT Floor Repair Systems under normal conditions of use’, is not justified on the basis of the evidence presented. The report does not meet the requirements of MEDDEV 2.7.1 Rev 3 December 2009 and does not meet regulatory requirements for clinical evaluation of EC Directive 93/42/EEC.2275
2308 The 2010 CER was also criticised by Ethicon’s notified body, BSI, in reports dated 25–27 September 2012,2276 5 October 20122277 and 14 May 2014. 2278 Like Dr Allman, BSI considered it was not consistent with the requirements of the European Directive or MEDDEV 2.7.1. The requirements in question were those concerning the characteristics and performances under the normal conditions of use of the device, and the evaluation of the side effects and of the acceptability of the benefit/risk ratio based on clinical data. It noted that this must include either a critical evaluation of the relevant currently available scientific literature relating to the safety, performance, design characteristics and intended purpose of the device or a critical evaluation of the results of all clinical investigations made.
2309 BSI pointed out that there was no clear description of the search terms, the exclusion criteria were unclear, weighting methods and details of the analysis of each paper were not cited, and insufficient evidence was provided to substantiate the report’s conclusion. What is more, it was also critical of the risk management report.
2310 BSI found that the risk management report for Prolift had been completed in 2010 but had not been updated to the new procedure and was deficient in the following additional respects:
It was based on complaints only, even though an aFMEA, dFMEAs, and CER were completed, contrary to ISO 14971 and despite purporting to fully comply with that Standard.
It did not does not address consideration of process risks.
It did not update the aFMEA and dFMEA with post-market surveillance information and did not include many of the harms identified in the “RMR/CER”.2279
2311 Eighteen months later, the position had not been rectified.2280
2312 In the period leading up to the signing of the 2010 CER, Ethicon’s Medical Affairs department became aware of a number of serious complications that had arisen with the use of Prolift.
2313 On 6 May 2010, for example, Drs Hinoul and Arnaud were notified by email that a patient had died during a Prolift procedure, apparently due to blood loss following injury to “a big vessel”.2281 The email disclosed that the implanting surgeon had been trained nearly three years earlier in one of the Prolift training centres by Dr Debodinance and had regularly used Prolift since, without problems. The Regulatory Affairs and Legal Departments were informed but neither saw “any elements that is (sic) likely to engage [Ethicon’s] responsibility as manufacturer”. Even so, the author of the email, Séverine Lacourt, Director, Ethicon Women’s Health & Urology, wrote “we might guess that the injury was done with the ancillary, so a report of the event is possible”. She sought advice as to how “the situation” might be handled. Dr Hinoul forwarded the email to Dr Robinson, adding that he presumed no action on his part was necessary since Regulatory Affairs and Legal had been informed. Dr Robinson said that Worldwide Customer Quality should be notified, but that was it. It does not seem to have occurred to anyone to consider amending the Prolift IFU to warn of the risk of haemorrhage.
2314 The following month, Ethicon’s Medical Affairs Department discussed the findings of a randomised controlled trial. This was a double-blinded multicentre RCT comparing traditional vaginal prolapse surgery without mesh with vaginal surgery with mesh (Prolift) in women with stages 2–4 prolapses (the Iglesia study).2282 At the time the results were apparently recorded in an abstract, but they were later the subject of an article published in the journal of The American College of Obstetricians and Gynecologists, Obstetrics & Gynecology.2283
2315 Sixty-five women were recruited to this RCT from January 2007 to August 2009. At this point the study was halted due to the high vaginal mesh erosion rate (15.6%) at three months with no difference in overall objective or subjective cure rates, calling into question the value of using synthetic polypropylene mesh for vaginal prolapse repairs. There can be no doubt that Dr Robinson was aware of these findings because he was copied into at least two emails about them: one from Scott Jones (Product Director, Urogynecology & Reconstructive Pelvic Surgery, Ethicon Women’s Health & Urology); and the other from Dr Hinoul.2284 Yet there was no reference to the Iglesia study in the 2010 CER. Dr Hinoul and a number of members of the TVM Group complained that, for various reasons, “level 1 evidence” could not be derived from the study. It is unnecessary to reproduce their reasons. It is sufficient to note that Professor Gordon’s evidence was that the evidence generated by the Iglesia study was “still useful evidence about efficacy”, since it was derived from a randomised controlled trial, and “a good deal more useful than data from case series upon which [Ethicon] relied”.2285
2316 In June 2010, Dr Chen notified Drs Hinoul and Robinson of eight cases of “serious complications associated with the pelvic floor repair meshes”, which included the following six, highlighted by the applicants in their submissions:
(1) a patient who died following an anterior Prolift procedure and pelvic floor surgical trauma with anti-coagulants likely as a result of post-operative bleeding and pelvic hematoma;
(2) another patient undergoing an anterior repair suffered a venous hematoma during an anterior repair with Prolift and was in ICU;
(3) a patient who died of pulmonary embolism three days after a Prolift+M Total repair;
(4) a 67 year old “otherwise healthy” patient who suffered a pulmonary embolism and died on the same day she received a Prolift Total and TVT implant;
(5) a 66 year old patient died as a result of blood loss that began during dissection for a Prolift procedure;
(6) 66 year old patient died on the operating table, apparently from vessel injury, during a Prolift procedure.2286
2317 The complaint investigation reports show that in all but one case the Ethicon device was exonerated. That may account for the reference to only one death in the CER. In the remaining case, the device was originally exonerated but was later said to have caused or contributed to the event. Yet, a decision was made that the incident need not be reported on the ground that it did not occur in “EEA” or Switzerland. This was an Australian death. In another case the surgeon was blamed for his or her (presumed) failure to perform a risk assessment and administer thromboembolism prophylaxis or post-operative anticoagulants. While the conclusions might well have been justified, Ethicon appears to have given no consideration to whether deficiencies in the IFUs or the TVM technique or both might have contributed to any of the adverse outcomes.
2318 No further CER was undertaken for Prolift before it was “decommercialised”.
2319 After Ethicon ceased supply of Prolift in August 2012, a further CER for the device was approved by Dr Hinoul on 26 April 2013.2287 It was longer than the earlier CERs. In addition to referencing the French and US TVM studies,2288 it summarised the findings of Altman et al (2011) and Withagen et al (2011), both of which, it will be recalled, were Ethicon-sponsored RCTs.
2320 The 2013 Prolift CER also contained an updated literature review, which relied on a meta-analysis by Maher et al (2009), an internal meta-analysis by Ethicon in 2012, and a systematic literature review by Bot-Robin et al.2289 The Maher et al (2009) review is discussed in Part VI and was also referenced in the 2010 Prolift CER.
2321 According to the 2013 Prolift CER, the internal Ethicon meta-analysis reviewed 27 “Level 1 and 2 studies” undertaken during 2012 and involving 3,194 patients implanted with Prolift. It found that the average success rate for Prolift was 84% (across a broad range between 38% and 97%).2290 It reported on five complications. At six months, the rate of mesh exposure was 7.6% (in a range of 0 to 16.7%). Bladder lesions were reported in 1.9% of patients, rectal lesions in 0.3%, and vascular lesions (capturing haematomas, blood transfusions, or re-intervention for haemorrhage) in 2.5%. De novo dyspareunia was reported in 12.7% of patients, but the potential range was wide — between 5% and 25%. As Dr Allman observed, no comment is made on the wide range of success rates.2291
2322 The Bot-Robin et al (2012) literature review was in evidence.2292 The authors concluded that the use of mesh to treat cystocele through the vaginal route gave a better anatomical outcome compared with traditional surgery, although they raised a caution in relation to their findings (“We must remain careful: our review does not represent a meta-analysis”).2293 They also concluded that the rate of complications, “especially” de novo dyspareunia, remained equivalent between the two techniques. In the section on their findings, the authors reported that 5.6% of patients with mesh had chronic pelvic pain as found in non-randomised studies, with three cases of chronic pain after surgery with mesh reported in randomised trials.2294 The authors considered that the main complication of prosthetic surgery by vaginal route was mesh “exposition” (presumably exposure), with its frequency varying between 0 and 35.7%. Dr Hinoul cited this finding and drew on the following discussion from the paper:
Our increasing knowledge of this particular risk leads to a standardization of surgical technique: no systematic hysterectomy, no inverted-T colpotomy when dissecting cystocoele, infiltration, dissection and positioning of the mesh while keeping the fascia on the vaginal wall, absence of colpectomy, meticulous check-up so there is no transfixion of prosthetic arms in lateral vaginal cul-de-sac. Surgical experience is also linked to the complication rate, with a learning curve evoked by Dwyer. Moreover, management of this complication seems simple enough for most authors. It should not be considered as a major complication.
2323 In his conclusion on the literature review, Dr Hinoul stated that, in their meta-analysis, Diwadkar et al (2009) had “demonstrated that traditional (native tissue) vaginal repairs, vaginal mesh repairs and sacrocolpopexy all have inherent risks with similar total complication rates”, and purportedly extracted Diwadkar’s findings in a table. I have referred above to the flaws in this analysis.
2324 The section on post-market surveillance was also updated, including complaints for Prolift, Prolift+M and Prosima during the period June 2011 to June 2012. Dr Hinoul acknowledged that the complaint rates had increased, but attributed the increased to an increase in litigation.2295 This approach is problematic for reasons given above in relation to the 2015 TVT and Gynemesh PS CERs.
2325 Dr Allman was also critical of this approach, observing that it was not clear what this was intended to convey. He said that adverse event reports have to be investigated irrespective of their source, and “it should not be assumed that reports from one source are less credible than from other sources or assumed to be caused by over reporting”.2296
2326 The discussion on risk benefit analysis identified three “high risk categories”.
2327 The first was mesh exposure.
2328 Dr Hinoul relied on a meta-analysis by Abed et al (2011), who reported an erosion rate of 10.3% after vaginal prolapse repairs using graft materials,2297 and a retrospective cohort study by Tijdink et al (2011), which reported that after insertion of Prolift 0.6% of patients between 2005 and 2009 at the hospital in which the authors worked required excision for severe mesh-related complications and 11% for minor complications.2298 After citing these two studies, Dr Hinoul estimated that “[t]his incidence, in combination with the findings of the GYNECARE PROLIFT™ literature review justifies an estimated frequency of permanent or long-term impairment to be 1/100 to 1/1000”.2299 Presumably, he meant to write 1/1000 (i.e. 0.1%) to 1/100 (i.e. 1%). It is not clear how he could have reached that that view. Nor is there any apparent reason why these two studies should have been selected as the basis for the risk calculation to the exclusion of all the other studies were mentioned in the literature that was purportedly reviewed and summarised in the tables to the report showing significantly higher rates of exposure. They included Withagen et al (2011) in the exposure rate was noted to be 14/83 (16.9%) and Bot-Robin et al (2012) where the highest percentage in the RCTs they reviewed was 35%, according to the CER. The Maher et al (2013) Cochrane review, it will be recalled, found a mesh exposure rate of 18%, with 9% requiring surgical intervention.
2329 The second high risk category was internal organ damage. Dr Hinoul stated that the incidence of internal organ damage, particularly the bowel and bladder, was 0.3% to 1.9% respectively in an analysis of 3,194 patients implanted with Prolift (this appears to come from the internal Ethicon meta-analysis cited above). Another study on Prolift+M mentioned by Dr Hinoul, but not identified by title or author, apparently found damage to the bladder in 2.3% of patients. Without explanation, Dr Hinoul concluded that, together, these studies justified “a 1% overall frequency assignation to this complication”. He claimed that internal organ damage during vaginal surgery was not life-threatening and that the majority of events were easily managed during the index surgery. He acknowledged that they could become fatal if they led to sepsis but said that to date no such reports had been made.
2330 The third category was nerve damage and/or pain. Dr Hinoul reported a number of different rates and varying severity levels, without little to no consideration of the significance of the complication:
In a review of the GYNECARE PROLIFT™ literature nine studies reported on the issue of dyspareunia and on average was reported in 12.7% of patients. Dyspareunia was described in 70 studies at an incidence rate of 9.1% in a meta-analysis reported by Abed et al 2011. The prospective GYNECARE PROLIFT™ +M study (#300-06-007) describes pelvic pain (including pelvic discomfort and vaginal pain) in 6.3% of patients. In 3.1% of patients de novo dyspareunia was also identified over a 36-month period. These dyspareunia rates are comparable with rates observed in traditional pelvic organ prolapse repairs reported by Lowman et al 2008. The injuries are often mild and transient. In rare instances they can lead to permanent damage.2300
2331 It is not apparent how Dr Hinoul reached the opinion that injuries were “often” mild and transient and in “rare instances” could lead to permanent damage, nor why rare instances of permanent damage were acceptable.
2332 As part of the conclusion of his risk-benefit analysis, Dr Hinoul expressed the view that the adverse events associated with transvaginal mesh kits were well characterised and manageable in the majority of cases. Further:
The anticipated medical benefits are considered to outweigh the overall or individual residual risks associated with this device. GYNECARE PROLIFT™ has demonstrated beneficial impact on anatomical and functional outcomes, in combination with its overall comparable complication rates in respect of traditional pelvic organ prolapse repairs (native vaginal tissue repairs (colporrhaphies), sacrospinous ligament fixations and abdominal sacrocolpopexies) and the manageability of the mesh specific complications support the conclusion that GYNECARE PROLIFT™ Pelvic Floor Repair System is a state of the art treatment option for certain patients suffering from pelvic organ prolapse.2301
(Emphasis added)
2333 As described above, Dr Hinoul’s consideration of the complications focused on the incidence rates for Prolift, with little discussion of complication rates for alternative treatments. It is not then clear how he could arrive at the emphasised conclusion about the comparable rate of complications as between Prolift and traditional surgeries.
2334 The overall conclusion of the 2013 Prolift CER reads as follows:
There is a substantial and accumulating body of clinical evidence on the performance of Ethicon, Inc.’s transvaginal mesh including GYNECARE PROLIFT™ Pelvic Floor Repair System. This evidence includes data from randomized clinical trials (RCTs) and non-RCTs. Despite the limitations inherent in looking across multiple trials (slight definitional differences, varying time points and patient populations etc.), this review has confirmed that the evidence demonstrates an acceptable benefit-risk profile for these products. Adverse events associated with transvaginal mesh kits are well characterized and are manageable in the majority of cases. No new adverse events have been identified since the initial launch of Ethicon, Inc’s mesh based solutions and the available 3 and 5 year longer term follow up data are not indicative of an increasing adverse event rate. Ethicon Inc. does acknowledge that in a small, yet significant subgroup of patients, these complications are to be considered serious.2302
2335 While Dr Allman described this CER as “well written and thorough”, he did not consider that its conclusion was justified.2303 He noted that there was no definition of the performance and safety criteria defining acceptability for the device. He said that merely because the performance of the device had not changed did not mean that the safety and performance were adequate and met the essential requirements. He concluded that the report did not satisfy the MEDDEV 2.7.1 and did not meet the requirements of the European Directive.2304
2336 Further, Dr Allman pointed out that, if the final sentence in the overall conclusion was intended to convey the idea that serious adverse events in a small, yet significant group of patients were considered acceptable, the criteria upon which this conclusion was reached were not identified. He said that, absent a definition of serious complications, statements to the effect that risks are acceptable are illogical.2305
2337 BSI, too, found that this CER, like the 2010 Prolift CER, did not satisfy the terms of the European Directive and was inconsistent with MEDDEV 2.7.1 for the same reasons and also because there was insufficient information on state of the art and safety/performance relative to alternative forms of treatment.2306
2338 Dr Allman concluded that each of the Prolift CERs was insufficient to justify CE marking and none of them complied with the European regulatory requirements and guidelines for clinical evaluation.2307
Prolift+M
2339 It will be recalled that Prolift+M received regulatory clearance in Europe on 18 March 2008 and was launched in Australia in December 2009. The first post-market CER, however, was not signed until 25 September 2012,2308 a few months after Ethicon had decided to stop selling and manufacturing it. 25 September 2012 was the day BSI was due to begin an audit in which the “full technical files” were expected to be reviewed.2309 The applicants submitted that it might be inferred that the report was completed under time pressure2310 and not as part of a considered post-market evaluation exercise. They pointed to the fact that Ethicon’s Regulatory Affairs department contacted Dr Hinoul the day before asking for the report but that circumstance alone is insufficient to warrant the drawing of that inference.
2340 On 3 July 2010, Dr Hinoul signed a clinical evaluation report on Gynemesh M, the material from which Prolift+M was manufactured, but which was not supplied in Australia (and is no longer manufactured).2311 The applicants did not refer to it either in written submissions or oral argument although the respondents relied upon it.2312 The respondents highlighted three aspects of the report: the overall complaint rate drawn from a review of complaints made between February 2007 and January 2010 (0.0010%); clinical data for Prolift+M; and the conclusion of the Risk/Benefit Analysis that “the overall risk associated with Gynemesh M is considered acceptable in view of benefits/patient outcomes, based on the literature review, clinical data and complaint reviews detailed in the CER”.2313
2341 I have already referred to the difficulties of drawing conclusions from complaint data.
2342 I note that on 25 June 2010 — a little over a week before the report was signed — Dr Hinoul had been informed by email that the first round of adverse event listings at 24 months follow-up would be circulated in two weeks’ time.2314 I infer that Dr Hinoul did not consider it necessary or desirable to wait for the results before signing off on the Gynemesh M CER.
2343 I will deal with the Risk/Benefit Analysis next before coming to the clinical data for Prolift+M.
2344 The Risk/Benefit Analysis largely replicated verbatim what appeared in the CER for Gynemesh PS, indicating that it was cut and pasted from that report, despite the differences in the two products. It is sufficient to quote the last section where the only difference between the two CERs is emphasised in the note to the table:
The analysis outcomes for Gynecare Gynemesh PS are as follows:
GYNECARE Gynemesh PS | |
Overall Residual Risk Score | 46 |
Overall Residual Risk Level | High |
According to the procedures and practices consistent with regulatory guidelines and company policy, the above scores and assessments indicate the need for a complete Risk/Benefit analysis. As a result of this process and a thorough review of all other pertinent information, including: a detailed clinical literature review as provided in Section D of this report, clinical data on Prolift+M which uses the same mesh as provided in section C and the complaint reviews (internal and MAUDE Database) as provided in Section F, the overall residual risk associated with Gynecare Gynemesh is considered acceptable in view of well documented benefits/patient outcomes.2315
2345 No reason is given to explain the relevance of risk rates for Gynemesh PS to the risk rates for Prolift+M when Prolift+M was made of a different mesh. There were no sections C, D or F in this CER.
2346 On the basis of what appears in this CER and the history concerning the production of the contemporaneous Gynemesh PS CER, I am not satisfied that a complete or proper risk/benefit analysis was conducted on Gynemesh M or Prolift+M.
2347 The clinical data consisted of the findings of a prospective multicentre cohort study of Prolift+M, funded by Ethicon at 11 international sites, three in the United States and eight in Europe. This was a single arm study. Despite the absence of a recommendation in the IFU to restrict use of the device to such women, eligibility for the study was confined to women with symptomatic pelvic organ prolapse at ICS POP-Q stage III or IV, who were suitable for surgical repair. One hundred and twenty-eight women consented to participating in the study. Surgery was completed in 127 cases. It was abandoned in one case after a cystotomy. One hundred and twenty-four patients were available for clinical follow-up at one year. An article on the one year outcomes was published in the American Journal of Obstetrics and Gynecology later that year by Milani et al (2011).2316
2348 Notwithstanding the concerns previously expressed about concurrent hysterectomies by the TVM Group, amongst others, the protocol for the study indicated that perineal repair, vaginal hysterectomy and/or mid urethral sling procedures for incontinence could be performed concurrently. A synopsis of the study including 12 month results was included as an appendix to the 2010 Gynemesh M CER. The results were said to suggest adequate anatomic support, consistent with the original Prolift mesh, “demonstrate high global patient’s impression and functional improvements (sic)”. The authors said that there were no apparent safety concerns arising from the change from Gynemesh PS to Gynemesh M. Mesh exposure rate was 10.2%. Only one of 49 patients who had reported sexual activity at 12 months (2%) complained of de novo dyspareunia. The low rate of de novo dyspareunia together with the absence of clinically relevant mesh shrinkage was said to be encouraging. It was noted that longer-term evaluation was continuing.
2349 An article reporting on the three-month results of this study was rejected for publication by the same journal after it received unfavourable appraisals from the peer reviewers.2317 The applicants were critical of Dr Hinoul for not referring to their comments in his affidavit under the mistaken impression that they pertained to the article that was in fact accepted for publication.2318 Nevertheless, the comments of the peer reviewers are instructive, as they highlight the lengths to which Ethicon was prepared to go to promote its products.
2350 The manuscript of the article stated that the lifetime prevalence of pelvic organ prolapse was 30%, which the first reviewer said was overstated, supported by reference to figures which the first reviewer said had been misquoted. Reviewer 1 noted that the introduction was too long and unfocused and failed to explain why a “lighter weight” mesh was being introduced. Reviewer 1 wrote: “If the authors are not honest about the problems with the prior mesh, this case series seems illogical”. Reviewer 1 also pointed out that, although the stated aim of the study was to assess whether the new mesh characteristics would provide the same strong anatomical support as Gynemesh PS, the assessment could not be made in the absence of a control group.
2351 The second reviewer was concerned about bias. Amongst numerous other matters the reviewer was critical of the authors for attributing recurrence of prolapse in one patient to a coughing episode. Reviewer 2 also made the following comment:
For this study I think you should just report the data and give a straightforward discussion of it. You are trying a little too hard to counter the “anti-mesh” argument that is appearing in the literature. If the data support the use of this product then the data will speak for themselves over time.
2352 Both reviewers 2and 3 considered the publication at three months after surgery was too early to report results.
2353 Like reviewer 1, reviewer 3 considered the study was limited by the fact that it was a single arm study with no control group. Reviewer 1 also remarked that the low rates of mesh erosion and dyspareunia were encouraging (at that stage erosion had occurred in six patients (5%) and there was no de novo dyspareunia), but added that “no definitive conclusions [could] be drawn except that this newer mesh might be an improvement”. This comment supports Dr Allman’s opinion that Prolift+M should have been subjected to a clinical trial before CE marking was applied.
2354 A redraft was circulated by Dr Hinoul and further changes debated among the authors. At this stage, the 12 month data were available. Both Drs Milani and Rogers said that they were hoping for a lower rate of mesh exposure (the results were comparable to the experience with Prolift). Dr Hinoul suggested, however, that a lower rate was not expected. This is unlikely having regard to the reasons behind the development of Prolift+M. There is no doubt that Drs Milani and Rogers were disappointed about the erosion rates. Dr Milani raised the obvious question whether mesh exposure was not “as much” related to weight and other factors might play a role, too. This was a matter which should have been carefully evaluated in the risk/benefit analysis of Prolift+M. But it was not. Nor was it discussed in the 2012 CER although the abstract of the published article was reproduced there. To the extent that the results of this study had a bearing on the safety and efficacy of Gynemesh M, it should also have been considered in any risk/benefit analysis of Gynemesh M. But it appears that it was not taken into account in that either.
2355 On 1 June 2010, Dr Hinoul received a PowerPoint presentation for a Pelvic Floor Platform Management Team meeting. In it, Ethicon acknowledged that the current mesh kits had “important shortcomings”.2319 It also acknowledged that both Prolift and Prolift+M were “evaluated unfavorably on many dimensions of ‘ease of use’, even by those physicians who report a preference for the brand”. Notably, on the question of “the ability to place the mesh (in proper place and flat)”, Ethicon noted that, despite a general perception that the quality of the mesh was “high”, the “mesh arms often get tangled, particularly with the 6-arm total repair”. It stated that EWHU was in the process of developing a “next generation” mesh kit, Neo, to improve upon those shortcomings. The “key learnings” from market research included that:
(1) perfect anatomical repair is not required; the hallmark of treatment success was patient satisfaction; and
(2) complications are primarily linked to mesh (usually too much mesh) and surgeon technique or user error (poor dissection planes, too much tension on mesh).2320
2356 None of these matters appears to have been taken into account in the risk analysis.
2357 On 11 April 2012 Professor Deprest provided Ethicon with a draft IUGA abstract on a study he was conducting on graft complications of mesh implants in sheep models.2321 Twenty ewes were implanted with Gynemesh M.2322 The following findings were made at necroscopy:
vaginal mesh insertion leads to an increased risk of graft related complications, mainly contraction but also exposure;
all 50x50mm vaginal meshes showed “remarkable contraction” and 80% exhibited folding (this was an average of 100% in the five ewes who were sacrificed at 60 days and 60%; (3/5) of those who were sacrificed at 90 days).
there was exposure in three of the 10 (30%) vaginal explants (two after 60 days, one after 90);
the abdominal explants showed less contraction, only one exhibited folding, and mesh was exposed in none of them;
reduction in the dimensions of the mesh to 35x35mm in a second group of 10 ewes dramatically reduced the incidence of folding in five of them after 60 days in the vaginal mesh to 20% and there were no exposures (the 90 day period at which the remaining five were to be sacrificed and the mesh explanted had not yet elapsed).
2358 The authors concluded that “[i]t seems that vaginal complications are better studied in sheep”. The results, which suggested that the addition of Monocryl did not ameliorate the incidence of mesh-related complications, caused them to wonder whether size or weight was responsible.
2359 In an email the next day to Peter Meier and Boris Batke, copied to Dr Holste, Dr Hinoul described the abstract as “unfortunate”, perhaps because, as we shall soon see, at this time Ethicon was struggling with its response to a § 522 notice from the FDA. Dr Hinoul continued:
To me the problem is the folding when placed vaginally, which occurs intra-operatively (that is why the small mesh does not show it to the same degree). These folds now will cause inappropriate ingrowth interpreted as shrinkage. To me the data speak to the fact that the model is inappropriate to study vaginal mesh placement. I would be interested to see the thickness of the vaginal epithelium.2323
2360 Dr Holste sent a short and sharp reply:
Until now the sheep is the most accepted model for intra-vaginal implantations. Vaginal epithelium morphology is comparable to humans.
2361 On 17 April 2012, Dr Hinoul’s displeasure was conveyed in an email to Professor Deprest by Susanne Landgrebe, one of Ethicon’s Research and Development managers.2324 She told Professor Deprest that Dr Hinoul was “not completely happy about the wording as people could misunderstand” and asked whether it was “still possible to do minor changes”. But it was too late. The abstract had already been submitted. Ms Landgrebe later apologised to Professor Deprest “for this little panic” at Ethicon. She explained that “the medical and marketing people”, in particular, were under pressure “due to the FDA discussion”. Although she had previously told Professor Deprest that Dr Hinoul would call him, she now said that he would not but that “[h]e would like to see in the conclusion that it is clearly stated that this study was done for model development”.2325
2362 Dr Hinoul deposed that on 27 June 2012, around the time of “decommercialisation” of all the vaginal mesh POP devices, he prepared a document entitled “Benefit Risk Profile of Transvaginal Mesh Products Used for the Treatment of Pelvic Organ Prolapse”.2326 He does not say why it was created but the inference is open that it was prepared to support the public explanation given by the respondents at the time of “decommercialisation”, discussed in the next Part of this judgment. The summary reads:
There is a substantial and accumulating body of clinical evidence on the performance of Ethicon, Inc.’s transvaginal mesh: GYNEMESH® PS and mesh kits: GYNECARE PROSIMA™ Pelvic Floor Repair System, GYNECARE PROLIFT® Pelvic Floor Repair System and GYNECARE PROLIFT + M™ Pelvic Floor Repair System. This evidence includes data from multiple randomized clinical trials (RCTs) and non-RCTs. Despite the limitations inherent in looking across multiple trials (slight definitional differences, varying time points and patient populations etc.), Ethicon, Inc’s review by medical affairs has confirmed that the evidence demonstrates an acceptable benefit-risk profile for these products when placed in appropriately selected patients by experienced surgeons. Adverse events associated with transvaginal mesh kits are well characterized and are manageable in the majority of cases. No new adverse events have been identified since the initial launch of Ethicon, Inc’s mesh based solutions and the available 3 and 5 year longer term follow up data are not indicative of an increasing adverse event rate. Ethicon Inc. does acknowledge that in a small, yet significant subgroup of patients, these complications are to be considered serious, as discussed in the Adverse Event section below.
2363 As Dr Allman pointed out that, 2327 if the acknowledgment of serious complications relates to the high risk categories of harm described, for example, in the 26 April 2013 Prolift CER, this includes acceptance of a 1% prospect of permanent or long-term impairment. Why this was considered an acceptable risk was never explained.
2364 In his affidavit Dr Hinoul deposed that the risk benefit assessment was “predominantly based” on four company-sponsored clinical trials studying Gynemesh PS, Gynecare Prosima and Prolift+M.2328 None of these was a randomised controlled trial. All were follow-up studies of patients who had undergone surgical procedures using the nominated treatments. No comparisons were made. For these reasons Professor Gordon’s unchallenged evidence was that company-sponsored trials “do not provide sound scientific evidence about efficacy and do not support Dr Hinoul’s conclusions”.2329
2365 Dr Hinoul also deposed that he relied on three company-sponsored investigator-initiated randomised controlled trials by Carey et al (2009), Altman et al (2011), and Withagen et al (2011) comparing transvaginal placement of Gynemesh PS and Prolift with native tissue repairs. Each of these studies has its own problems which I discuss elsewhere in these reasons. Putting those matters to one side for the moment, it should be noted, as Professor Hu did, that “Altman et al (2011) found that mesh was associated with a statistically significant higher risk of longer-lasting surgery, intraoperative hemorrhage and blood loss, stress urinary incontinence as well as new stress urinary incontinence, vaginal wound revision, and (as apparent from the originally drafted manuscript v. the manuscript published), dyspareunia”, 2330 findings to which Dr Hinoul neglected to refer in this report. Professor Hu pointed out, too, that the Withagen et al (2011) study2331 also found statistically-significant higher rates of some complications in comparison with native tissue repair (longer operation times, haematoma, and temporary urinary retention) and recorded an exposure rate in the mesh group of 16.9%.
2366 In the adverse events section of the report Dr Hinoul asserted:
To be able to put the benefit-risk profile related to Ethicon Inc.’s mesh based POP repair solutions in perspective, it is essential to recognize that all procedures to treat pelvic organ prolapse can be associated with similar complications. A meta-analysis by Diwadkar et al. demonstrated that traditional (native tissue) vaginal repairs, vaginal mesh repairs and sacrocolpopexy all have their own inherent risks that differ in number and severity, however, they all have similar total complication rates (Table 6.3).2332
(Original emphasis)
2367 Professor Hu described Dr Hinoul’s characterisation of the findings of the Diwadkar et al (2009) meta-analysis as disingenuous.2333 I agree. I referred to this study and to its conclusion earlier in Part VI. In the present context, Professor Hu highlighted the conclusion and the following findings.
The vaginal mesh kit group included 3,425 patients from 24 studies, with a mean follow-up of 17.1 +/- 13.8 months. The mean total complication rate for this group was 14.5% (range 0 -23.1). In contrast to the traditional vaginal surgery and sacral colpopexy groups, the majority of complications in this group required surgical intervention under general anesthesia (Dindo grade IIIb) … Although fistulae were reported rarely (0.2%, range 0 -4.2), the rate was highest for this group. Although pain-related complications were common in the sacral colpopexy group, dyspareunia rates were highest in the mesh kit group (2.2%, range 0-23.1) … the total reoperation rate was the highest (8.5%, range 0 -30.0) because of a higher rate of reoperations for complications such as mesh erosion.2334
2368 Professor Hu criticised the Benefit-Risk Profile on a number of different bases. First, he said it apparently fails to account for the “different lines of evidence pointing to higher rates of non-mesh-specific complications associated with mesh surgery [in comparison with] Native tissue repair”. Second, he said that it failed to “rigorously discuss and account for the types and rates of mesh-specific complications”. Third, he said it failed to properly describe the results of “a key published systematic review of the literature, which, in fact, concluded that vaginal mesh is associated with higher rates of complications requiring reoperation and total reoperations”, which appears to be a reference to Diwadkar et al (2009).2335
2369 The CER was signed the day Dr Hinoul swore that he had prepared it. It did not take the form of any other risk analysis undertaken at Ethicon. It made no reference to the essential requirements, the European Directive or the guidance documents, or to the ISO standards. In these circumstances the inference the applicants invited me to draw2336 — that it had been created for the purpose of supporting the communication to the market about the reason for “decommercialisation” — should be drawn. In other words, it was not a genuine risk assessment or post-market evaluation.
2370 As I observed at the beginning of this discussion, the first post-market CER for Prolift+M was signed by Dr Hinoul some three months later, on 25 September 2012.2337
2371 For the purposes of the literature review six databases were searched: Cochrane, Google Scholar, Medline, PubMed, NICE, and Scopus. Six articles relating to Prolift+M were identified, four relating to prospective studies, two to pre-clinical (histological and biomechanical) data. As Dr Allman observed, the abstracts of the articles were simply copied verbatim into the report before the conclusion is stated.2338
2372 The conclusion was formulaic: that the aforementioned data on Prolift+M, taken together with the available data on Prolift and UltraPro are sufficient to demonstrate compliance with the essential requirements covering safety and performance of Prolift+M under normal conditions of use. This is followed by the self-serving statement that “[c]linical data collection over a medium term follow up was … adequately reported upon”. No reasoning process accompanied either assertion.
2373 As Dr Allman observed, there was no analysis of the data or the literature. Without analysis, he said, and a definition of the standard applied to determine safety and effectiveness, the conclusion is not justified and the report does not meet the regulatory requirements set by MEDDEV 2.7.1 and the European Directive.2339
2374 There were other problems with this CER.
2375 First, it relied on Prolift data when Prolift+M was designed to improve on Prolift by reducing the complication rates associated with it. This was acknowledged in the background discussion included in the CER. Any genuine evaluation of Prolift+M would have considered whether this goal had been realised. This CER did not. For a start, the Deprest sheep study was not even mentioned, although it went directly to this question.
2376 Second, the stated purpose of the CER was to “determine whether additional knowledge has become available pertinent to recognized or emerging hazards and whether the acceptability of these risks, should any be identified, change the benefit-risk profile of the device”.2340 Yet, it did not refer to the FDA alerts or the § 522 notice.
2377 Third, the two pre-clinical studies were conducted on rabbits, despite the advice of Dr Batke as to the superior suitability of the sheep models.
2378 Fourth, in the second and most recent of the two studies, published in 2012,2341 involving 24 rabbits, 12 of which were implanted with Prolift+M and 12 with Prolift, the vaginal extrusion rate was reported to be “at least 50%, coinciding with a minimum of 20% of contraction”. The authors said that there were no measurable effects of adding polyglecaprone (Monocryl) in abdominal explants. They concluded that:
The addition of polyglecaprone fibers [Monocryl] did not compromise the biomechanical properties nor did it prevent vaginal extrusion and contraction. The latter as well as some other limitations preclude the rabbit vagina to be a suitable model for biomechanical testing.
2379 This cried out for comment. Yet none was offered.
2380 Fifth, as the applicants submitted, the CER relied heavily on Ethicon sponsored studies without considering the potential for bias. Ethicon employees were among the authors of two of the four articles reporting prospective data, the abstracts of which were reproduced in the CER. Dr Hinoul, himself, was one of them.
2381 The first one mentioned was by Milani et al (2011) referred to above. The article acknowledged the contribution made by members of the Research and Development team at Ethicon Women’s Health & Urology to the study design, previously the subject of peer review criticism. What it does not disclose is that not only was Dr Hinoul the operating surgeon but Dr Hinoul actively recruited women to the study. This disclosure was made in Dr Hinoul’s affidavit (at [732]) but only after the article had been discussed twice before. Neither disclosure was made in the CER.
2382 It will be recalled that Milani et al (2011) reported one year outcomes from a single arm study. The article picked up on some of the matters raised by the peer reviewers of the manuscript which reported the three-month outcomes, acknowledging the potential for bias:
The major drawback of this study is the lack of a control group, for example, with conventional POP surgery. Cohort studies are exposed to selection bias and confounding. In this study, no attempt was undertaken to avoid the surgeons’ potential bias during follow-up visits or to capture the patients’ characteristics that were screened before entry into the trial. Ideally, these should have been performed by an investigator blinded to the procedure. Future preferably randomized, controlled studies are necessary to generate evidence on a risk/benefit analysis of different mesh versus conventional repairs.
2383 The absence of a control group means that the results could not be reliably compared with the use of traditional surgery (to determine the respective merits both in terms of benefits and risks and to see whether any additional risks that might be created by the use of mesh were worth taking). Professor Woodward observed that:
Any single arm (one treatment group) study is prone to bias because blinding (as recommended in RCTs) is meaningless in this situation, and thus who is recruited, and how their data are assessed, might be manipulated – knowingly or otherwise – by the investigator. For instance, “difficult” subjects may not be recruited and a subject may be coerced into giving a subjective response that supports the investigator’s hypothesis. It would have been reassuring to have seen data to negate this worry in this extreme situation of the sponsor’s representative having input to recruitment.2342
2384 The second abstract, relating to another Ethicon-sponsored study, was presented to the IUGA and AUGS annual meetings in 2012. The lead author was Dr Lucente. Dr Hinoul was again the second-named author. This abstract included results from the same study reported by Milani et al (2011) but at three years. The manuscript, however, was still under preparation.2343 The Maher et al (2010) Cochrane review observed that data extracted from conference abstracts where no peer-reviewed manuscripts are available should be treated with caution.2344 No such treatment was accorded to the abstract in the Prolift+M CER.
2385 In addition, while only three patients did not return for clinical assessment at one year, at the three year mark 19 (15%) were lost to follow-up. As the number of patients lost to follow-up could have affected the interpretation of the findings, this should have been taken into account. Usually, as I understood the evidence, patients lost to follow-up are treated as failures. It is not apparent from the abstract that they were treated that way in this case. The three-year outcomes showed that the mesh exposure rate had risen from 10.2% to 14.8%, which indicates that the exposure rate was calculated by reference to the total number of patients who underwent implant surgery and not by reference to the total number of patients who presented for follow-up three years hence. If the rate had been calculated by reference to the latter, then the rate would have been 17%, not 14.8% as reported. Professor Woodward made this point in his second report. He wrote:
Dr Hinoul says there were 19 mesh exposures at 3 years, and claims this is a 14.8% rate. However, this is a misleading. He has got 14.8% as the ratio of 19 to 128, because 128 women started the study. However, only 109 were followed-up to 3 years, so the denominator should have been smaller, which would have given a lower rate. What he has done is to assume that every woman that was lost to follow-up was free of mesh exposure at 3 years, which is not necessarily true. It would not be appropriate to take 109 as the denominator either, because this would over-estimate risk, especially in the early part of the study. Survival analysis should have been used.2345
2386 Either way, the increase in the exposure rate was worthy of comment. The de novo dyspareunia rate had also risen from 2% to 9%. Yet, the authors of the abstract, stated that no major safety concerns were identified. Why were these matters not of concern? On the face of things, the conclusion was not consistent with the results.2346
2387 The third abstract was by Khandwala and Jaychandran (2011). While the abstract does not disclose this, the article, which was tendered in evidence,2347 reveals that Dr Khandwala was a consultant to Ethicon.
2388 This abstract also reported on results from a single arm study of Prolift+M. The authors concluded that “Prolift+M surgery is safe and effective with minimal postoperative morbidities”. But the period of follow-up was very short. The article, but not the abstract, stated that the primary objective of the study was to assess improvement in anatomy and function six months after Prolift+M surgery. Even at the six-month mark, however, 29 of the 167 patients enrolled (17.4%) did not present for review. It is not apparent how that circumstance was taken into account in the reporting of the data. Moreover, the authors noted that, although the incidence of mesh exposure in the series (3.6%) was lower than that quoted in the literature (literature to which the Prolift+M 2012 CER did not refer), their study had only a six-month follow-up and mesh exposures have been detected “up to 12 months postoperatively”, citing three-year old data from the TVM Group. In view of the short period of follow-up, it is difficult to see how the conclusion in the abstract could be so emphatic. The conclusion of the article, however, is more circumspect. It reads:
This study suggests that Prolift+M mesh surgery is a safe and effective procedure for the management of vaginal wall prolapse. However, this is only a 6-month follow-up and our longer term planned follow-up at 12, 24, 36, and 60 months would help us better understand its role. Eventually, a randomized clinical trial between the Prolift+M and traditional surgery would identify its true role in the management of genital organ prolapse.
2389 Dr Hinoul discussed the article in his affidavit (at [735]). He referred there to the note of caution, apparently recognising that longer follow-up was necessary. In any genuine clinical evaluation it is reasonable to expect that he would have been equally cautious. He also asserted (at [633]) that it demonstrated that Prolift+M was not superior to Prolift. Professor Woodward described this conclusion as “extremely dubious” owing to the limited evidence and uncontrolled analyses.2348
2390 The “complaint/adverse event review” in the CER summarised safety data collected in the Ethicon sponsored trial, complaints made to the company from launch to August 2012, and complications registered in the MAUDE database from January 2007 to September 2012. No reference was made to complications registered in the MHRA database. Dr Hinoul stated that it was Ethicon’s belief that the data drawn from the company-sponsored trial “provides the most in depth understanding of the safety profile of the Prolift+M™ device to date”.2349 This underscores the fact that Ethicon well understood the problem of under-reporting and the unreliability of reported complaints as a measure of the extent of complications.
2391 The cumulative rate and severity of adverse events associated with Prolift+M repair drawn from the trial were included in a table in the CER. They were not insignificant. The applicants described the data as alarming. Notably, the number of adverse events increased at each stage of review and at the three year review 65.6% of patients were found to have experienced at least one adverse event, 15.2% had complications which interfered with their usual activities, and 5.8% were incapacitated by them, unable to work or carry out their usual activities.
Table 3: Cumulative rate and severity of adverse events following Prolift+M repair:
N=128 | 0-3 months | 0-12 months | 0-24 months | 0-36 months |
Number of patients reporting at least one AE | 45 (35.2%) | 69 (53.9%) | 80 (62.5%) | 84 (65.6%) |
Total number of AEs | 68 | 109 | 128 | 138 |
Mild | 52 (76.5%) | 83 (76.1%) | 101 (78.9%) | 109 (79.0%) |
Moderate | 12 (17.6%) | 20 (18.3%) | 21 (16.4%) | 21 (15.2%) |
Severe | 4 (5.9%) | 6 (5.5%) | 6 (4.7%) | 8 (5.8%) |
2392 “Mild” was defined as “[a]wareness of a sign or symptom that does not interfere with the subject’s usual activity or is transient, resolved without treatment and with no sequelae”, “moderate” as “[i]nterferes with the subject’s usual activity”, and “severe” as “[i]ncapacitating with inability to work or perform usual activities”.
2393 The Prolift+M 2012 CER also included a table comparing complication rates from traditional vaginal repair, sacrocolpopexy, and mesh kits, purportedly to put the risk/benefit profile of Ethicon’s POP devices in perspective.2350
Table 8: Complication rate meta-analysis comparing traditional vaginal (native tissue) repairs with sacral colpopexy mesh repairs and transvaginal mesh kit repairs:
Traditional vaginal repair | Sacral colpopexy | Mesh kits | |
Number of studies | 48 | 52 | 24 |
Total Complication rate | 15.3 | 17.1 | 14.5 |
Mesh exposure/ infection | 0.5 | 2.2 | 5.8 |
Cystotomy | 0.4 | 1.0 | 0.7 |
Ureteral injury | 0.3 | 0.2 | 0.1 |
Bowel injury | 0.4 | 0.5 | 0.3 |
Bleeding complication | 2.8 | 1.6 | 1.1 |
Wound complications | 0.5 | 1.5 | 0.2 |
PE/ DVT | 0.1 | 0.3 | 0 |
Dyspareunia | 1.5 | 1.5 | 2.2 |
Total reoperation rate | 5.8 | 7.1 | 8.5 |
Reoperation for prolapse recurrence | 3.9 | 2.3 | 1.3 |
Total reoperation rate | 5.8 | 7.1 | 8.5 |
Reoperation for prolapse recurrence | 3.9 | 2.3 | 1.3 |
2394 The CER emphasised that all procedures to treat pelvic organ prolapse can be associated with similar complications. It drew on a meta-analysis by Diwadkar et al (2009) to prove the point. As Dr Hinoul pointed out, the meta-analysis covered articles related to mesh and mesh kits manufactured by various companies, not just Ethicon. He failed to point out, however, that the article and therefore the rates extracted from it related to complication and reoperation rates after apical vaginal prolapse repair. Only clinical trials and observational studies addressing apical prolapse repair and associated recurrence or complication rates were included in the analysis. In any case, the search was confined to the period from January 2005 to December 2007, which necessarily meant that it excluded Prolift+M, since Prolift+M did not come into the market until 2008. For the same reason long-term complications that can arise in mesh surgery but not, or are less likely to occur, in traditional surgery will not have been taken into account. Any impartial assessment would have taken these considerations into account and referred to in the CER. They were not mentioned in this CER.
2395 What is more, the table in the CER included a figure of 0.5% for mesh erosion in the traditional surgery repair column, as, indeed, did the Diwadkar et al (2009) article. But Diwadkar et al (2009) attributed that to concomitant procedures using midurethral slings made of mesh, which tends to undercut the notion that there are similar complications with all three procedures.2351 The CER did not refer to the confounding factor, noted by Diwadkar et al (2009), of concomitant procedures or any of the other limitations they mentioned in their article. Diwadkar et al counselled that their comparisons should be treated with caution.
There are several reasons that comparisons among the three surgical approaches should be interpreted with caution. First, very few randomized trials comparing these techniques are currently available. Of the 105 studies included in our analysis, only 4% represent clinical trials. Most of the studies included are retrospective case series of a single procedure without a comparison group. As such, the type of analysis we could perform is limited, and formal meta-analytic techniques could not be performed. Second, studies addressing traditional vaginal procedures and sacral colpopexy were likely better quality studies compared with studies on vaginal mesh kits, given the longer follow-up periods and larger sample sizes. The lowest rate of prolapse recurrence seen in the mesh kit group may be because the addition of mesh improves objective anatomic cure, but it also may be a reflection of the follow-up period of 17.1 months, which is approximately half the follow-up period of the traditional procedures group. Future trials with longer follow-up ultimately will determine whether recurrence rates increase or remain unchanged. Third, given the variability of follow-up times among studies, time-to-event analyses would be ideal. However, because the timing of adverse events and reoperations often was not reported, these could not be performed. Finally, the number of patients enrolled and the number of patients lost to follow-up was not always indicated in every study.2352
2396 Furthermore, while the data from Diwadkar et al (2009) were extracted, no reference was made to the authors’ conclusion. The conclusion postulated in the CER was that all procedures to treat pelvic organ prolapse can be associated with similar complications. This was misleading. The conclusion reached by Diwadkar et al (2009) was quite different:
In summary, there are no clinical trials or other comparative studies to date that compare these three main approaches to repairing the apical compartment in women undergoing surgery for pelvic organ prolapse. In this meta-analysis, we attempted to summarize the available observational studies to provide some guidelines on the relative complication and reoperation rates of these approaches. Sacral colpopexy is considered by some the gold standard apical suspension procedure. In support of this, sacral colpopexy had a relatively low rate of reoperation for prolapse recurrence. However, this was at the expense of a high complication rate. Traditional vaginal procedures, in contrast, had a higher rate of reoperation for prolapse recurrence but fewer complications that required surgical intervention. Most importantly, our results suggest that, despite the lowest reoperation rate for prolapse recurrence, vaginal mesh kits have the highest rate of complications that require surgical intervention, which, on balance, results in the highest rate of total reoperation after apical suspension for pelvic organ prolapse. This raises the concern that the risks of these newer procedures may be greater than their benefits. One can speculate that more recurrences and complications may be diagnosed with time, given the relatively shorter mean follow-up period in the mesh kit group. On the other hand, this may reflect the “learning curve” of this recently adopted new technology. More long-term studies on vaginal mesh kits and clinical trials that directly compare these surgical techniques are needed to support these findings definitively.2353
(Emphasis added)
2397 Before calculating the estimated frequency of complications, the risk/benefit analysis in the 2012 Prolift+M CER identified several “high risk categories”: mesh exposure, internal organ damage, nerve damage and/or pelvic and vaginal pain (dyspareunia), soft tissue damage, and deep venous thrombosis.
2398 The risk of internal organ damage was said to be 1%, based on an unidentified Prolift analysis of 3,194 patients which reportedly found bowel and bladder injuries in 0.3% and 1.9% of cases. That was said to correspond to the reports from the Prolift+M study of 0 (bowel) and 2.3% bladder injuries. But internal organ damage in vaginal surgery was not considered life-threatening in the risk analysis unless sepsis occurred.
2399 The rate of pelvic pain in the Prolift+M study was 6.3% and of de novo dyspareunia 3.1%. The average rate of dyspareunia in nine unidentified studies of Prolift was said to be 12.7% and in the meta-analysis by Abed et al (2011) 9%. The CER stated that these rates corresponded to rates observed in traditional pelvic organ prolapse repairs and the injuries were often mild and transient, although in rare cases they will lead to permanent damage. “Rare” was not defined. Nevertheless, the CER went on to refer to a rate of 0.6% of Prolift patients requiring excision for “severe mesh-related complications” drawn from Tijdink et al (2011) and to give an estimated frequency of permanent damage as “maximally 1/100 to 1/1000” allegedly based on “the data in the literature” and the three-year outcomes of the Prolift+M study.
2400 Deep vein thrombosis was said to be the most common non-surgical complication after major pelvic surgery and one which was potentially fatal when it leads to pulmonary thrombo-embolism. But it is not relevant in this case so nothing further need be said about it here.
2401 The conclusion of the analysis was as follows:
The PROLIFT +M™ Pelvic Floor Repair System is a mesh kit used to treat pelvic organ prolapse. Review by medical affairs has confirmed that the evidence demonstrates an acceptable benefit-risk profile for these products when placed in appropriately selected patients by experienced surgeons. Adverse events associated with transvaginal mesh kits are well characterized and are manageable in the majority of cases. No new adverse events have been identified since the product’s initial launch.
The anticipated medical benefits outweigh the Overall or Individual Residual Risk(s) associated with this device. The Prolift +M™’s proven beneficial impact on anatomical and functional outcomes in combination with its comparable overall complication rates in relation to traditional pelvic organ prolapse repairs (including sacrospinous ligament fixations and abdominal sacrocolpopexies) and the manageability of the mesh specific complications support the Prolift+M™ Pelvic Floor Repair System as a state of the art treatment option for certain patients suffering from pelvic organ prolapse.
2402 The Prolift+M study was limited to patients with grade 3 or higher prolapses, although the IFU did not specify that it should be used only in such cases. I fail to see how the risk benefit profile was justifiable without taking this into account. Nor is it apparent that similar results would necessarily be achieved in less severe cases of prolapse.
2403 There is force in the applicants’ submission that the conclusion was “inadequate” because there was no significant analysis of the issues. In the absence of a definition of “acceptable”, the reference to “an acceptable benefit risk profile” is meaningless. The CER relied on use of the product in “appropriately selected patients” by “experienced surgeons” when the surgical population to whom the device was marketed and sold was broader than that and it did not consider whether the IFU should be amended to limit the use accordingly.2354
2404 The next Prolift+M CER in evidence was one approved by Dr Hinoul and dated 26 April 2013, the same date on which Dr Hinoul approved the final CER for Prolift.2355 This was some months after Ethicon had stopped supply of the devices in Australia.
2405 The CER for Prolift+M adopted the same structure as the 2013 Prolift CER and relied on some of the same data. Further, despite the addition of more detail in relation to the reviewed data, the CER’s conclusions were an almost verbatim copy of what appeared in the September 2012 Prolift+M CER.
2406 While a lengthy tabulated summary of the literature was presented, the analysis of the literature was brief. For example, while 20 studies relating to Prolift+M were purportedly considered, the “critical analysis” of that literature appeared in three paragraphs:
In the period since the product was placed on the market in 2008, 20 clinical studies have been published relating to GYNECARE PROLIFT +M™, nine of which represent primary clinical data derived from the Ethicon study #300-07-006 data set. The remaining level 2b studies including those reported by Khandwala et al 2011, by Milani et al 2012 and those by Bhatia et al used comparable primary outcome measures (POP-Q) and assessment time points (12 months).
Overall, the performance and safety data in these independent data sets is comparable with that reported in study #300-07-006. In one series of publications by Bhatia et al 2010 the use of GYNECARE PROLIFT +M was compared directly with GYNECARE PROLIFT. No significant differences were observed in the primary outcome measure (anatomic success) or in other measures at 1 year.
Overall, the literature reports conclude that GYNECARE PROLIFT +M™ provides for a safe and effective means of managing pelvic organ prolapse with minimal post-operative morbidities.2356
2407 A prospective study on Prolift+M (“#300-07-006”) was described, which appears to be the same study relied on in the July 2010 Gynemesh M CER, discussed above. One year results were reported by Milani et al (2011) and three year results by Lucente et al (2012).
2408 The approach to analysis of the post-market surveillance data was the same as in the other CERs from this period, with litigation being blamed for the increase in complaint rates.
2409 As part of the risk benefit analysis, three high risk categories were discussed. They were the same as those in the April 2013 Prolift CER (exposure, internal organ damage, and nerve damage and/or pain). The same data were relied on, too, but with the addition of the “prospective GYNECARE PROLIFT+M study (#300-07-006)”. The estimations of frequency of harm were equally implausible.
2410 For example, the exposure rate of 10.3% reported by Abed et al (2011) was cited followed by a rate of 10.2% taken from the prospective GYNECARE PROLIFT+M study (#300-07-006) with 4/15 (i.e. 26.7%) of those requiring in-patient surgical intervention in the first 12 months. Dr Hinoul then referred again to the finding of Tijdink et al (2011) that 0.6% of patients after Prolift required excision for severe mesh-related complications and 11% for minor complications. Based on these data, he stated again that the frequency of permanent or long-term impairment was “1/100 to 1/1000”.
2411 The section on internal organ damage was a copy of the same section of the 2013 Prolift CER, and the section on nerve damage and/or pain relied on the same data. Once again, no attempt was made to explain why the complication was acceptable.
2412 Dr Hinoul’s conclusion on the risk-benefit analysis was the same for Prolift+M as for Prolift. The overall conclusions of the two CERs were also the same.
2413 Given the similarities between the two reports, Dr Allman’s opinions about these two CERs were similar.2357 He said that the conclusion of the 2013 Prolift+M CER was not justified and that the report did not meet the requirements of MEDDEV 2.7.1 or the requirements for clinical evaluation in the European Directive. Accordingly, he considered that the CER did not justify CE marking for the device. Since this report was prepared after Ethicon decided to stop distribution and supply of the device, this meant that there was no adequate basis for applying a CE marking for the entire period in which the device was distributed.2358
2414 An update to the 2013 Prolift+M CER carried the electronic signatures of Dr Hinoul, E Valerio (Director of RA), and M Day (Director Med Ops). It bore a release date of 6 July 2015.2359 The respondents submitted that this CER “confirmed that the evidence demonstrates an acceptable benefit-risk profile for the device under normal conditions of use, that no new types of adverse events had been identified since the initial launch of Prolift+M and that the available longer term follow-up data did not indicate an increasing adverse event rate”.2360
2415 This updated CER was more detailed than the earlier reports and adopted a similar structure to the other CERs prepared around this time (see the discussion of the 2015 CERs for the TVT Family of Products and Gynemesh PS above). It contained an updated literature review and assessment of complaints data, but shared some of the same flaws as its predecessors.
2416 Importantly, if the release date is taken as the date of signature, then the report was executed around three years after Ethicon had stopped supplying the product and almost three months after the device had been removed from the ARTG.2361 In these circumstances, a detailed analysis of the report is not required. It suffices to say that I have taken it into account and found nothing in it to justify any different view about the adequacy of Ethicon’s post-market evaluation of Prolift+M or the safety and efficacy of the device.
Prosima
2417 There was an unexplained hiatus of five years between the launch of Prosima and the production of a post-market clinical evaluation report.
2418 The first post-market CER for Prosima was signed by Dr Hinoul in September 2012.2362 Two versions of this report were tendered. Both were signed by Dr Hinoul. The first version bears the date 2 September 2012;2363 the second 25 September 20122364. The first version is not paginated and is plainly a draft. The last page contains the heading “References” but includes no list of references. The risk benefit analysis begins with a reference to Prolift but goes on to discuss bench-top testing (omitted from the 25 September 2012 version), includes a different harms/hazards summary table from that which appears in the later version, a different discussion of high risk categories, and a different risk benefit conclusion. In essence, the risk benefit analysis relates to a different product — Artisyn Y-shaped mesh, a synthetic mesh used to treat vaginal vault prolapse in sacrocolpopexy. No explanation was given for this. Dr Hinoul was not cross-examined about the two versions. In their submissions the applicants criticised the 25 September 2012 report, which it accepted was the first post-market CER, for incorporating a “risk benefit conclusion” that related to Artisan, pointing to the conclusion in the first version of the report. But the conclusions in the two versions are quite different, except for a single sentence: the anticipated medical benefits outweigh the Overall or Individual Residual Risk(s) associated with this device”. In the circumstances this criticism is unjustified.
2419 Dr Hinoul circulated another draft of the report to Laura Vellucci, a member of the regulatory department in Ethicon Women’s Health & Urology, and others on 20 September 2012, asking them whether they had any suggestions.2365 Karl Reese of BSI responded with a number of “suggested additions and edits”, including a number derived from the BSI audit findings, noting “the requirement to align with EN ISO 14155 and MEDDEV 2.7.1”. Dr Hinoul sought and received approval for the inclusion of the following statement “on state of the art”:
The Prosima™’s proven beneficial impact on anatomical and functional outcomes in combination with its comparable overall complication rates in relation to native tissue repairs and the manageability of the mesh specific complications support the Prosima™ Pelvic Floor Repair System as a state of the art treatment option for certain patients suffering from pelvic organ prolapse.
2420 It is difficult to understand the basis on which approval was given. The use of the term “state of the art” to describe Prosima was surely premature, even at this stage. Furthermore, there is an apparent inconsistency between referring to Prosima as “state of the art”, when it was made from Gynemesh PS, which Ethicon itself considered inferior to Prolift+M, and when Ethicon referred to Prolift+M as “the next generation” mesh.
2421 The 2012 Prosima CER followed the format of the Prolift+M report of the same date. The literature review began with a description of the device, described the search strategy, and identified publications said to be pertinent to Prosima and Prolift.2366 It then set out verbatim the abstracts of the six articles generated by the search that related to Prolift. As with the Prolift+M report there was no discussion, let alone analysis of any of the data reported in any of these articles. The risk analysis section seems to have been cut and pasted from the Prolift+M report (or vice versa). The only differences appear to be the inclusion of Prosima-specific information and the deletion of data relating to Prolift+M. It is difficult to resist the conclusion the applicants invited me to draw that the report was prepared in haste and under the pressure of the impending audit.
2422 Two of the abstracts were taken from articles reporting on Ethicon-funded studies: Sayer et al (2012)2367 and Zyczynski et al (2010).2368 Both concerned a prospective cohort study of Prosima conducted at 11 sites in the United States, the United Kingdom, Germany, and Australia. Sayer et al (2012) reported medium term outcomes (24– 34 months), Zyczynski et al (2010) 12-month outcomes. Sayer et al (2012) reported that the primary anatomic success, defined as POP-Q 0-1, was 69.1%, but in 84.5% of cases the leading edge of the vagina was above the hymen. Pelvic symptoms and sexual function were said to have improved significantly from baseline. Zyczynski et al (2010) declared that the overall success rate was 86.9%, although overall objective anatomic success (stage 0 or I) at 12 months was 76.9% and for who had an isolated anterior repair it was 79.3%.
2423 Ethicon employees also contributed to the presentation of the data in the articles. Dr Hinoul was a co-author of Sayer et al (2012). Dr Robinson and Vanka Sikirica were co-authors with Dr Carey of Zyczynski et al (2010). Judi Gauld was a co-author of both. Some of the other authors (Drs Sayer, Van Drie, Slack and Jones) all had consultancy positions with Ethicon. These conflicts of interest were disclosed in the article but not in the CER.
2424 I refer below to some of the limitations of the Prosima study and the reliability of the conclusions drawn in the Sayer et al (2012) article. At this point I note that the CER stated that the rate of mesh exposures reported by Sayer et al was 9.1%. That reflects what the figure in the abstract. As table 1 in the article shows, however, the true rate was at least 10% because the 11 patients in whom the complication arose were 11 of 110 who consented to follow-up at or after two years, not 11 of the 126 patients in whom the device was implanted. I say “at least” 10% because it appears from the text of the article that there were recurrent exposures in two of these cases. Other reported complications included worsening of, or de novo, stress urinary incontinence in 6 patients, dyspareunia in 10 sexually active patients, overactive bladder symptoms in five patients, and further prolapse surgery for four patients. Dyspareunia was not mentioned in the abstract. The article, but not the abstract, also disclosed that the exposed mesh was partially excised in eight patients, two of whom had second excisions. The percentages given for all of these complications were understated because, like the exposures, they were calculated using a denominator of 126, not 110. Consequently, the rate of worsening or de novo stress urinary incontinence was 6% not 5% as reported. Overactive bladder symptoms was 5% not 4.1%, and further prolapse surgery 4% not 3.3%. Forty-nine patients were sexually active and, of these 10 (21%) reported dyspareunia at baseline and reported improvement at follow-up. It is not clear from the article whether there were any cases of de novo dyspareunia.
2425 The Zyczynski et al (2010) abstract did not mention the mesh exposure rate. The journal article from which it was drawn, which was tendered by the applicants, discloses that mesh exposure occurred in 12 of the 136 patients (9%), eight (67%) of whom required excision surgery and four exposures were ongoing at the 1 year mark. The authors acknowledged the following limitations of the study: the lack of a comparative group, randomisation, and blinding. They also acknowledged that their findings of functional and anatomic improvement were preliminary with respect to the durable relief of prolapse symptoms. Moreover, they noted that, owing to limitations in sample size, the study could not contribute to the debate about whether mesh-augmented repairs should be used in primary repairs or reserved for secondary repairs.2369 The limitations recognised by the authors were not mentioned in the CER.
2426 Hong et al (2011) was a single case report of complications that had occurred during surgery for stage II uterine prolapse using Prosima and in the post-operative period.2370 The complications were serious. They were described in the abstract as “internal pudendal artery injury and a massive presacral hematoma that formed after surgery”. For a week after surgery, the patient reportedly experienced difficulty micturating (urinating) and difficulty defecating due to presacral haematoma compression. It took 71 days for the haematoma to resolve.
2427 The Carey et al (2008) abstract related to a prospective observational study, conducted in two tertiary referral urogynaecology practices, of 95 women with POP-Q stage II or more pelvic organ prolapse who underwent surgery with what became Prosima.2371 The conclusion recorded in the abstract was that ‘vaginal surgery using mesh and a VSD is an effective procedure for pelvic organ prolapse” with reported “objective success” at six and 12 months after surgery of 92 and 85% respectively and comparable figures for subjective success. Two patients experienced mesh exposure (4%), two of whom required surgical excision of the eroded mesh. Results were described in the article, though not the abstract, as “encouraging”. But both the abstract and the article noted that further clinical studies were required to establish the role of this surgery.
2428 The final abstract extracted but not discussed in the 2012 Prosima CER was also by Carey and others. The article from which it was drawn was published online on 7 July 2009.2372 This was a prospective randomised controlled trial designed to compare vaginal repair augmented by mesh with traditional colporrhaphy for the treatment of pelvic organ prolapse. It was conducted at a tertiary teaching hospital. One hundred and thirty-nine women with stage II or greater prolapse requiring both anterior and posterior compartment repair were recruited to the study. Sixty-nine were allocated to the mesh group and 70 to the traditional colporrhaphy group.
2429 For subjects attending the 12-month review, success in the mesh group was 81.0% (51 of 63 subjects) compared with 65.6% (40/61) in the no mesh group, but the differences were not statistically significant (p = 0.07). A high level of satisfaction with surgery and improvements in symptoms and quality-of-life data were observed at 12 months in both groups compared to baseline. Once again, however, the outcomes between the two groups were not significantly different. Vaginal mesh exposure occurred in four women in the mesh group (5.6%). De novo dyspareunia was reported at 12 months. by five of 30 (16.7%) sexually active women in the mesh group and five of 33 (15.2%) in the no mesh group Carey et al (2009) concluded that “[i]n this study, vaginal surgery augmented by mesh did not result in significantly less recurrent prolapse than traditional colporrhaphy 12 months after surgery”. That conclusion was reported in the abstract and in the 2012 Prosima CER. It raised the obvious question, however, given the added complication of vagina mesh exposure in the mesh group and the possibility of other mesh-related complications in the longer term, on what basis could it be said that the benefits of mesh-augmented surgery with Prosima outweighed the risks? Yet, the question was left begging in the CER.
2430 In order to mitigate the significance of the adverse events, Dr Hinoul compared 12-month adverse events associated with non-mesh repairs taken from three investigator-initiated randomised controlled trials: Carey et al (2009), to which I have just referred, Withagen et al (2011), and Altman et al (2011) with the results of adverse events associated with the use of Prosima based on safety data collected in the Ethicon-sponsored studies discussed by Sayer et al (2012) and Zcyzynski et al (2010), just as he had done in the Prolift+M CER. A number of observations should be made about this exercise.
2431 First, it assumes that mesh-related complications all arise within the first 12 months of surgery when Dr Hinoul well knew that they could occur much later.
2432 Second, it assumes that the study populations were comparable.
2433 Third, it overlooks the potential for bias in all the studies.
2434 In this CER, as in all others, no apparent, or at least transparent, attempt was made to determine whether the authors’ conclusions were substantiated by the available data and no account appears to have been taken of the duration of the studies and the heterogeneity of the population, contrary to MEDDEV 2.7.1. Although MEDDEV 2.7.1 expressly referred to the need to assess both short and long-term adverse events an,d although all these devices were intended for permanent implantation, the 2012 Prosima CER contained no acknowledgment of the limitations that might well be attached to the conclusions of the authors because of the short periods of follow-up. Professor Woodward observed that none of the Prosima studies to which Dr Hinoul referred in his affidavit, the longest of which was Sayer et al (2012) (at two to three years), was large or of long duration.2373
2435 Once again, there was no adequate explanation of the parameters for considering what an acceptable risk was and no reasons were given. Why did the author consider the reported rates of exposure and other complications acceptable? Why was the device safe if it carried the risk of those complications? The risk/benefit section was the same as that which appeared in the 2012 Prolift+M CER. In each case it is apparent that a cut and paste exercise was undertaken. On the basis of what appears in this CER and the history concerning the corresponding section of the Gynemesh PS CER, I am not satisfied that a complete or proper risk/benefit analysis was conducted.
2436 The conclusion of the 2012 Prosima CER was as follows:
Polypropylene-based materials are the most commonly used synthetics for pelvic organ prolapse repair. The above data on Prosima™, taken with the available longstanding data on Gynemesh PS (the mesh used in Prosima™), are sufficient to demonstrate compliance with the essential requirements covering safety and performance of PROSIMA™ under normal conditions of use. Clinical data collection over a medium term follow up was sponsored by the company and adequately reported upon.2374
2437 As with the other CERs, Dr Allman considered that the report’s conclusion was not justified as no criteria were defined to assess conformity with the essential requirements. He concluded that the report was insufficient to justify CE marking and did not meet the requirements under the European Directive or MEDDEV 2.7.1.2375
2438 Following its audit in September 2012, BSI, too, had found that the 2012 Prosima CER did not satisfy the requirements of the European Directive. Its reasons were the failure to state the inclusion and exclusion criteria and the absence of a discussion of the reason individual citations had been excluded.2376
2439 BSI also identified deficiencies in the draft risk management report for Prosima:
six high risks were identified but the contribution to the risk posed by the device (rather than the procedure) was not made clear;
there was no justification in the risk/benefit analysis for the conclusion that the benefits of the device outweighed the risks;
like the risk/benefit analysis in the CER, it did not identify whether any information on residual risks or methods to mitigate residual risks had been conveyed to users;
process risks were not included in the review of the overall residual risk. 2377
2440 These deficiencies had not been remedied more than 18 months later, in May 2014, when another BSI audit took place.2378
2441 The last Prosima CER in evidence was dated 2 May 2013. It also carried Dr Hinoul’s signature.2379 As Dr Allman observed, this was an update of the 25 September 2012 CER.
2442 The 2013 Prosima CER reviewed studies relating to a total of 496 women of whom 121 (fewer than a quarter) were followed for two years. It recognised that there were limitations in most of the studies. It stated that the main drawbacks were the absence of a control group or a comparative treatment, the short periods of follow-up (mostly one year), and the small numbers of women treated. It acknowledged that longer term follow-up in more women was required in order to assess the long term safety and effectiveness of the device.2380
2443 This CER reported a substantial increase in complaints, all of which were attributed to an increase in litigation which in turn was attributed to the FDA alert and consequently discounted. Apparently on this basis, the rates of complaint for all the mesh kits were described as “acceptable”. Excluding the cases the subject of litigation, the rate of erosion was deemed “appropriate”.2381 I struggle to understand how it was legitimate to exclude cases the subject of litigation. I also struggle to understand why the rates of erosion, with or without litigation, could be described as “acceptable”. Once again, no criteria for determining acceptability were identified.
2444 Dr Allman noted that the conclusions of the report were substantially the same as its predecessor and said they have the same flaws. Neither met the requirements of MEDDEV 2.7.1 or the requirements for clinical evaluation contained in the European Directive.2382
PART X: REMOVAL OF SOME OF THE ETHICON DEVICES FROM THE MARKET AND THE ARTG
2445 None of the POP devices is presently for sale in Australia, the United States or Europe. Neither is TVT Secur. These devices have been removed from the ARTG, though it is possible they may still be in circulation, since no recall notices were ever issued.
2446 Ethicon’s position was that the decision to stop selling these products (and, indeed, to stop manufacturing them) had nothing to do with any lack of safety or efficacy. In a document entitled “Background Information GYNECARE Pelvic Floor Repair Products and GYNECARE TVT Secur”, the origin and purpose of which was not the subject of evidence, Ethicon maintained that it continued to have confidence in the safety and effectiveness of these products and the decision to discontinue selling them was unrelated to safety or efficacy.2383
2447 In this Part of the judgment I consider the background to these decisions.
2448 The story of the decision to no longer sell the above-mentioned devices probably begins with the publication of an alert to doctors issued by the FDA on 20 October 2008 and updated the following day.2384
2449 The alert, entitled “FDA Public Health Notification: Serious Complications Associated with Transvaginal Placement of Surgical Mesh in Repair of Pelvic Organ Prolapse and Stress Urinary Incontinence”, stated:
This is to alert you to complications associated with transvaginal placement of surgical mesh to treat Pelvic Organ Prolapse (POP) and Stress Urinary Incontinence (SUI). Although rare, these complications can have serious consequences. Following is information regarding the adverse events that have been reported to the FDA and recommendations to reduce the risks.
Nature of the Problem
Over the last three years, FDA has received over 1,000 reports from nine surgical mesh manufacturers of complications that were associated with surgical mesh devices used to repair POP and SUI. These mesh devices are usually placed transvaginally utilizing tools for minimally invasive placement.
The most frequent complications included erosion through the vaginal epithelium, infection, pain, urinary problems, and recurrence of prolapse and/or incontinence. There were also reports of bowel, bladder, and blood vessel perforation during insertion. In some cases, vaginal scarring and mesh erosion led to a significant decrease in patient quality of life due to discomfort and pain, including dyspareunia.
Treatment of the various types of complications included additional surgical procedures (some of them to remove the mesh), IV therapy, blood transfusions, and drainage of hematomas or abscesses.
Specific characteristics of patients at increased risk for complications have not been determined. Contributing factors may include the overall health of the patient, the mesh material, the size and shape of the mesh, the surgical technique used, concomitant procedures undertaken (e.g. hysterectomy), and possibly estrogen status.
Recommendations
Physicians should:
• Obtain specialized training for each mesh placement technique, and be aware of its risks.
• Be vigilant for potential adverse events from the mesh, especially erosion and infection.
• Watch for complications associated with the tools used in transvaginal placement, especially bowel, bladder and blood vessel perforations.
• Inform patients that implantation of surgical mesh is permanent, and that some complications associated with the implanted mesh may require additional surgery that may or may not correct the complication.
• Inform patients about the potential for serious complications and their effect on quality of life, including pain during sexual intercourse, scarring, and narrowing of the vaginal wall (in POP repair).
• Provide patients with a written copy of the patient labeling from the surgical mesh manufacturer, if available
…
2450 Readers were also directed to the FDA website for further information, while hospitals and other user facilities were reminded of their reporting obligations and encouraged to report adverse events relating to surgical mesh that do not meet the requirements for mandatory reporting.
2451 The FDA’s concerns did not dissipate after the publication of this alert. Indeed, in an internal PowerPoint presentation produced in March 2011, Ethicon noted that there had been an increase in the FDA’s concerns.2385 The author of the document reflected on what the FDA’s growing concerns meant for the company. The author raised the possibilities of a redefinition of the premarket clearance requirements, changes to labelling, and clinical trials. Still, another PowerPoint presentation dated 11 May 2011, the work of Ethicon Women’s Health & Urology, described the urogynaecological business as “highly profitable”. Despite the FDA’s 2008 alert, sales and income had grown considerably since 2005. Projected revenue from sales of POP devices in the United States was expected to rise by $50,000,000 in the period from 2010 to 2018 and from sales of SUI devices by nearly $40,000,000.2386
2452 On 13 July 2011, the FDA issued an “update” to its 2008 alert. 2387 Its stated purpose was to alert health care providers and patients alike, both actual and prospective, that the problem was more serious than the 2008 report might have suggested:
The FDA is issuing this update to inform you that serious complications associated with surgical mesh for transvaginal repair of POP are not rare. This is a change from what the FDA previously reported on Oct 20, 2008. Furthermore, it is not clear that transvaginal POP repair with mesh is more effective than traditional non-mesh repair in all patients with POP and it may expose patients to greater risk …
(Original emphasis)
2453 The FDA advised that there had been a significant increase in the reports of adverse events it had received for urogynaecological mesh devices — from “over 1,000” in the period 2005 to 2007 to an additional 2,874 in the following three years to 31 December 2010. 1503 related to POP devices and 1371 to SUI devices.
2454 According to the alert, the most frequent of the reported complications for POP devices included mesh erosion through the vagina (also called exposure, extrusion or protrusion), pain, infection, bleeding, dyspareunia, organ perforation, and urinary problems. There were also reports of recurrent prolapse, neuro-muscular problems, vaginal scarring/shrinkage, and emotional problems. Many of these complications required additional intervention, including medical or surgical treatment, and hospitalisation.
2455 The FDA declared that, in order to better understand the use of these devices, it had conducted a systematic review of the published literature from 1996 to 2011 to evaluate their safety and effectiveness. It announced that the review showed that transvaginal repair of pelvic organ prolapse with mesh “does not improve symptomatic results or quality of life over traditional non-mesh repair”. It noted in particular that mesh used for this purpose introduced risks not present in traditional surgery for the repair of pelvic organ prolapse; mesh placed abdominally appears to result in lower rates of mesh complications compared to transvaginal placement; there was no evidence that transvaginal repair with mesh to support the top of the vagina (apical repair) or the back wall of the vagina (posterior repair) provides any added benefit compared to traditional surgery without mesh; and the anatomic benefit that may result from using mesh for anterior repair may not produce better symptomatic results. It went on to report:
The FDA’s literature review found that erosion of mesh through the vagina is the most common and consistently reported mesh-related complication from transvaginal POP surgeries using mesh. Mesh erosion can require multiple surgeries to repair and can be debilitating for some women. In some cases, even multiple surgeries will not resolve the complication.
Mesh contraction (shrinkage) is a previously unidentified risk of transvaginal POP repair with mesh that has been reported in the published scientific literature and in adverse event reports to the FDA since the Oct. 20, 2008FDA (sic) Public Health Notification. Reports in the literature associate mesh contraction with vaginal shortening, vaginal tightening and vaginal pain.
Both mesh erosion and mesh contraction may lead to severe pelvic pain, painful sexual intercourse or an inability to engage in sexual intercourse. Also, men may experience irritation and pain to the penis during sexual intercourse when the mesh is exposed in mesh erosion.
The complications associated with the use of surgical mesh for POP repair have not been linked to a single brand of mesh.
(Original emphasis)
2456 The FDA reiterated the recommendations it had made in the 2008 notification but added the following recommendations for health care providers:
• Choose mesh surgery only after weighing the risks and benefits of surgery with mesh versus all surgical and non-surgical alternatives.
• Consider these factors before placing surgical mesh:
• Surgical mesh is a permanent implant that may make future surgical repair more challenging.
• A mesh procedure may put the patient at risk for requiring additional surgery or for the development of new complications.
• Removal of mesh due to mesh complications may involve multiple surgeries and significantly impair the patient’s quality of life. Complete removal of mesh may not be possible and may not result in complete resolution of complications, including pain.
• Mesh placed abdominally for POP repair may result in lower rates of mesh complications compared to transvaginal POP surgery with mesh.
• Inform the patient about the benefits and risks of non-surgical options, non-mesh surgery, surgical mesh placed abdominally and the likely success of these alternatives compared to transvaginal surgery with mesh.
• Notify the patient if mesh will be used in her POP surgery and provide the patient with information about the specific product used.
• Ensure that the patient understands the postoperative risks and complications of mesh surgery as well as limited long-term outcomes data.
2457 It also made a number of recommendations to patients including as to the questions they should ask their surgeons. The notification concluded with a plea to health care providers to promptly report adverse events and guidance as to the information to be imparted.2388
2458 The FDA alert attracted a good deal of media attention in the United States.2389
2459 The FDA produced a white paper at around the same time which set out its position in greater detail. 2390 From its review of the scientific literature, the white paper identified the following safety concerns with transvaginal mesh in POP repair:
(1) Patients who undergo POP repair with mesh are subject to mesh-related complications not experienced by patients undergoing traditional surgery without mesh.
(2) Adverse events associated with transvaginally placed mesh can be “life-altering” for some women. “Sequelae (e.g. pain) may continue despite mesh removal”.
(3) Mesh-associated complications are not rare. The most common is vaginal mesh erosion with 10% of women experiencing erosion within 12 months of surgery (based on data from 110 studies including 11,785 women).
(4) There were increasing reports of mesh contraction, causing vaginal shortening, tightening and/or vaginal pain in association with transvaginal POP repair with mesh.
2460 The FDA noted that abdominal pelvic organ prolapse surgery (sacrocolpopexy) appears to result in lower rates of “mesh complications” in comparison to mesh surgery for the same condition performed transvaginally, with the median rate of vaginal mesh erosion reported at 4% within 23 months of surgery.
2461 The FDA also concluded from the literature review that, while transvaginal pelvic organ prolapse surgery with mesh often restores anatomy, it has not been shown to improve clinical benefit over traditional non-mesh repair.
2462 Based on a review of adverse events reported to the FDA and of the scientific literature, the FDA described its concerns about the safety and effectiveness of the use of surgical mesh for transvaginal repair of pelvic organ prolapse as “serious”.
2463 Summarising its key findings, the FDA stated that:
Based on evaluation of adverse event reports and assessment of the scientific literature, the FDA has NOT seen conclusive evidence that using transvaginally placed mesh in POP repair improves clinical outcomes any more than traditional POP repair that does not use mesh, and it may expose patients to greater risk.
2464 The FDA flagged that it was considering upgrading the risk classification for transvaginal POP mesh from class II to class III, which would require manufacturers to submit premarket approval applications, including relevant clinical data for the devices; clinical studies to address the risks and benefits of mesh to treat both pelvic organ prolapse and stress urinary incontinence; and expanding post-market monitoring of device performance. The FDA also announced that it was convening a meeting of the Obstetrics-Gynecology Devices Panel to discuss and make recommendations regarding the safety and efficacy of transvaginal mesh surgery for both pelvic organ prolapse and stress urinary incontinence.
2465 Dr Hinoul’s evidence was to the effect that Ethicon was untroubled by the FDA’s latest alert. His explanation was that Ethicon did not consider that the alert contained new information about the risks and potential complications patients could suffer as a result of Prolift. In fact, Dr Hinoul added, Ethicon considered that it was complying with the FDA’s recommendations even before the alert was issued and that it was doing so because of the information in the IFUs and professional education materials.2391
2466 It is difficult to believe that Ethicon was untroubled by this alert. Ethicon is unlikely to have welcomed it. It could not have been good for business. At the very least, the prospect of upgrading the risk classification for the POP devices alone would surely have concerned Ethicon, not least because of the additional costs this would have entailed.
2467 The FDA’s Obstetrics and Gynecology Devices Panel met in September.2392 The Panel concluded that vaginal mesh for pelvic organ prolapse repair poses risks that are unique to mesh, such as vaginal erosion, and that the clinical benefit of mesh compared to surgical repair of pelvic organ prolapse without mesh was “questionable”. Consequently, the FDA decided that additional studies were needed to fully evaluate the risk/benefit profile of POP devices and it recommended that these devices be reclassified from the current class II to class III. The FDA also recommended post-market studies to address the safety and effectiveness of the current mini-slings, based on the Panel’s view that they were not well understood and that premarket evaluation of new mini-slings should be supported by clinical studies.2393 This recommendation would undoubtedly have troubled Ethicon because, if it were to be implemented, it would have been burdensome. As will be seen, it was a burden Ethicon was not prepared to assume.
2468 By mid-November 2011, JJM noted that sales were down and, following the FDA update, that surgeons were taking a more conservative approach to mesh usage, either moving away from Ethicon products or not using mesh at all.2394 In cross-examination, Dr Hinoul admitted to having a recollection of Ethicon coming to the same conclusion and that the reason for the change in the approach of surgeons was “safety concerns”.2395 He also conceded that the contraction of the market reflected “a concern amongst surgeons and perhaps patients as well as to the safety of the POP mesh products” as a result of the FDA communications.2396
2469 On 3 January 2012, the FDA issued notices to Ethicon under § 522 of the Federal Food, Drug and Cosmetic Act. Section 522 gives the FDA the power to require a manufacturer to conduct post-market surveillance of any class II or III medical device in certain specified circumstances. Those circumstances relevantly include where the failure of the device would be reasonably likely to have serious adverse health consequences or where the device is intended to be implanted in the human body for more than a year.
2470 On 3 January 2012, the FDA informed Ethicon that each of Gynemesh PS, Prolift, Prolift+M, Prosima and TVT Secur was subject to post-market surveillance under § 522 because they were class II devices the failure of which “would be reasonably likely to cause mesh erosion (i.e. organ perforation), severe pain, and fistula formation, which would meet the definition of ‘serious adverse health consequences’ at 21 C.F.R. § 822.3(j)” and also because the devices were intended to be implanted in the body for more than one year. It reminded Ethicon that the effect of an order for post-market surveillance was that the manufacturer was bound to submit a plan to conduct the surveillance which the FDA would assess in order to determine whether it would result in the collection of useful data on unforeseen adverse events or other information necessary to protect public health. The notices indicated that failure to discharge the obligations imposed would result in regulatory action and that that might include revoking regulatory clearance or taking steps to prevent Ethicon from continuing to supply the devices.2397
2471 The FDA ordered Ethicon to submit a plan addressing a number of questions. Those questions included: the rates associated with various adverse events at several intervals over a three-year period post-implantation; quality of life of, and recurrence rates of prolapse, for patients over the same periods; rates of adverse events and quality of life following “resurgery” within 36 months of initial prolapse surgery; and comparative rates of effectiveness to non-mesh surgery. To address the questions, the FDA recommended that Ethicon conduct a randomised clinical trial or a prospective cohort study over three years comparing its devices to controls, such as transvaginal urogynaecological surgery without mesh. The FDA also made recommendations as to the design of the study. Finally, it advised Ethicon that it was considering reclassifying urogynaecological surgical mesh used for the transvaginal repair of pelvic organ prolapse to class III, in which case clinical data would be necessary to support a premarket approval application.
2472 Ethicon had no inclination to conduct either of the studies recommended in by the FDA. The following day, Laura Vellucci wrote to Brian Kanerviko, Worldwide Director of Regulatory Affairs for Ethicon Women’s Health & Urology, and Catherine Beath, Vice-President of Regulatory Affairs, to whom Mr Kanerviko reported, suggesting that the company might not have to perform the study if the products were withdrawn from the market.2398 The reply is not in evidence.
2473 Mr Kanerviko tried in vain to persuade the FDA to let Ethicon avoid the comprehensive studies the FDA had proposed. His assessment of the data held by Ethicon was that it was very unlikely to satisfy the FDA’s requirements. In a document prepared on 18 January 2012 he wrote that the Prosima studies did not have “head to head comparison” and they only had data for 29 months; the comparative data for native tissue repair as against Prolift+M was “weak”; there were two studies for Prolift with 12 months of data; and one of them related to the anterior compartment only; and there was no data for Gynemesh PS.2399 Dr Kanerviko forwarded the email to Dr Hinoul seeking his views. If Dr Hinoul replied to the email, his reply was not tendered in evidence.
2474 On 1 February 2012 Ethicon submitted “study plans” for Prolift and Prolift+M, Gynemesh PS, Prosima, and TVT Secur.2400 None of these “study plans”, however, included a proposal for a new randomised controlled trial or, indeed, any new study. Rather, Ethicon sought to rely on existing data or sponsored studies that were still in progress.2401 In view of the premise for the § 522 orders — that existing studies did not provide sufficient evidence of the safety or relative advantage of the devices in question over alternative procedures — it was unsurprising that, in early April 2012, the FDA informed Ethicon that it did not accept its proposals.2402
2475 The deficiencies the FDA identified in its correspondence included the following matters.
2476 In the case of Prolift, the data from the two published articles upon which Ethicon relied were insufficient to satisfy the requirements of the order. Those articles related to the Withagen and Altman randomised controlled trials. Amongst other things, the FDA pointed out that:
(1) neither article provided safety and effectiveness outcomes at the intervals the FDA required (six, 12, 18, 24 and 36 months) — the Withagen data was collected at six and 12 months, the Altman data at two and 12 months;
(2) the Withagen trial involved a select patient population; it included only patients with recurrent prolapse;
(3) Altman et al compared the use of mesh to traditional anterior colporrhaphy rather than to transvaginal native tissue repair;
(4) neither article provided data on quality of life measures at 18, 24 and 36 months or comparative data at those intervals for women who had not undergone surgery transvaginally and for women who had been treated without mesh; and
(5) neither article provided data on the rates of adverse events and quality of life after “resurgery”.2403
2477 For both Prolift and Prolift+M, the FDA was concerned that, while Ethicon had provided data on the number of operations using each device, it had failed to differentiate between compartments (that is to say, it had failed to indicate whether these were anterior, apical or posterior repairs or a combination thereof). In the case of Prolift+M, although Ethicon had provided rates from a single arm prospective study which had study visits at 3, 12, 24, and 36 months, Ethicon did not specify the number of patients evaluated at each visit. Moreover, Ethicon only presented rates of adverse events at six months and then did not make it clear how that data had been obtained. Similarly, Ethicon had given the FDA data on rates and severity of mesh exposure at six months purportedly drawn from the same single arm study without making clear how the data was obtained. Furthermore, Ethicon informed the FDA that all but two (17/19) cases of exposure were considered mild, yet 14 of the 19 patients had required surgical intervention to manage the exposure. In these circumstances the FDA asked for Ethicon’s “definitions for measuring severity of mesh exposure”. The FDA also pointed out that it had sought information about recurrent prolapse in its § 522 order but the rates of recurrent prolapse at the intervals sought were not provided. While Ethicon had indicated that it planned to address some of the FDA’s questions with the randomised controlled trial that was under way, the FDA had a number of concerns about Ethicon’s proposal that these questions would be addressed by that RCT. They included concerns:
(1) about the absence of a detailed statistical analysis plan on how the non-inferiority hypothesis for adverse events, revision/re-surgery and effectiveness at 36 months was to be tested;
(2) that the RCT was not adequately powered to compare anatomic outcomes by the treated compartment;
(3) that the longest period of follow-up in the proposed protocol for the RCT was 24 months, not 36 which the FDA required, and the study visit schedule did not include an 18-month visit;
(4) about the exclusion from the proposed study for no apparent reason of women under 45; and
(5) that the RCT was being conducted entirely outside the United States (in the Netherlands), which gave rise to a concern about whether the findings could be generalised to the US population.2404
2478 In cross-examination, Dr Hinoul conceded that in theory different environmental factors could affect whether findings relating to one population group were applicable “worldwide”.2405
2479 Similar problems presented themselves with the Prosima study plan.2406 In that case the FDA was concerned, amongst other things, that:
(1) Ethicon provided follow-up data for only 29 months, when it had sought 36 months’ data;
(2) data on recurrent prolapse was not provided;
(3) although intraoperative visceral injuries and visceral adhesions are among the most common adverse events seen in medical device reports, they were not included in Ethicon’s list of adverse events;
(4) Ethicon’s study plan for addressing certain questions proposed pooling control arm data from ongoing RCTs without describing how the data could be pooled;
(5) the plan was neither a randomised clinical trial nor a prospective cohort study that compared Prosima to a concurrent control through the three years of follow-up the FDA required, which the FDA believed “could yield potential bias” and Ethicon provided no information regarding potential biases or how they would be addressed;
(6) each arm of the study had a different period of follow-up;
(7) there was no plan to evaluate “the comparability of the two arms”; and
(8) the quality of life data presented to enable a comparison to be made between the outcomes from Prosima and native tissue repair were deficient in that the data was derived from validated questionnaires in three RCTS and they were different from those proposed for use in the Prosima study.
2480 In its post-market surveillance plan for Gynemesh PS,2407 Ethicon proposed using data collected for the Prosima study on the basis that they had the same intended use and were made of the same material. But the FDA did not consider that the products were comparable. The FDA said that there were “clinical implications raised by the fact that PROSIMA is a mesh kit with a pre-configured mesh, and GYNEMESH PS is a stand alone, sheet mesh”. Those implications were said to include the following matters:
(1) the inclusion of the vaginal support device and balloon in the Prosima kit results in a “fixation-free system”, whereas Gynemesh PS had to be fixed to the tissue;
(2) Prosima has a defined surgical technique and step-by-step instructions for surgeons, whereas Gynemesh PS comes without such instructions and surgeons are left to tailor the mesh to their own preferences which results in “significant variability in procedure”;
(3) Prosima includes instruments to facilitate placement of the mesh while Gynemesh PS does not, once again a variation in procedure;
(4) the quantity and surface area of mesh used when placing Gynemesh PS will vary significantly in comparison to the pre-configured mesh in the Prosima kit; and
(5) the need to trim Gynemesh PS for placement in the respective anatomical compartments may result in edges of cut mesh that are rougher than the laser cut edges of Prosima and this may lead to additional tissue trauma.
2481 Finally, the FDA disapproved of Ethicon’s post-market surveillance study plan for TVT Secur on the ground that it was unlikely to result in the collection of useful data to address the questions raised by the § 522 order.2408 Ethicon’s proposal was to answer the questions through a meta-analysis of four publications which compared TVT Secur to multi-incision retropubic or transobturator mesh slings. But the meta-analysis did not include a discussion of the proportion of all women undergoing stress urinary incontinence surgery who were exposed to the device and the types of surgical procedures performed. The outcome and adverse event data in the studies reviewed in these publications were collected at different intervals and not at all the intervals required by the FDA. The occurrence of neuromuscular problems was not addressed in any of the four studies. And, while all four studies reported data on the need for re-surgery for stress urinary incontinence, none of them collected data on adverse events and quality of life measures after re-surgery.
2482 The FDA sought responses within 30 days including, if Ethicon wished to submit them, amended study plans. It was prepared, however, to extend time on request, subject to approval, as long as Ethicon justified the need for the extension and provided a reasonable estimate of when the requested information would be given.2409
2483 At this point Ethicon abandoned the negotiations.
2484 After consultation with senior management, on 2 May 2012 Mr Kanerviko wrote to the FDA to inform it that Ethicon would “stop commercializing” (that is to say, stop selling) TVT Secur and had no intention of resuming sales in the future. He asked for up to 120 days to do so in order to notify customers and give them sufficient time to select alternative treatment options for their patients. In the meantime, he advised, Ethicon would “discontinue or revise, as appropriate, all marketing materials” and notify its customers. He also asked the FDA to place a hold on the § 522 order.2410
2485 Letters in like terms were sent on 9 May 2012 seeking a similar indulgence in relation to the § 522 orders for the POP devices, informing the FDA that Ethicon would stop the sale of Prosima, Prolift and Prolift+M within 120 days and had no intention of resuming sales in the future. In the meantime, he advised, Ethicon would “discontinue or revise, as appropriate, all marketing materials” and notify its customers.2411
2486 In each of these letters, Mr Kanerviko maintained that Ethicon believed the devices were safe and effective options for women with stress urinary incontinence or pelvic organ prolapse, as the case may be, and claimed that the decision in each case had been reached “after considering the commercial viability of the product in the United States in light of the complexities of the clinical study requirements, the significant adverse publicity and the litigation environment”. He added that “[t]he size and competitiveness of the market place and the availability of other treatment options for women were also factors”. Dr Hinoul defended this position in cross-examination. He maintained that the decision was made for commercial reasons and denied that safety issues had anything to do with it.2412
2487 The same day Mr Kanerviko advised the FDA that Ethicon intended to “update” the product labelling for Gynemesh PS to limit the indication for use to sacrocolpopexy only.2413
2488 Precisely two months later, on 9 July 2012, the FDA agreed to Ethicon’s request to conditionally suspend the § 522 orders.2414
2489 Documents that one might have expected to have been prepared in the context of decisions made in response to the FDA’s § 522 orders, such as board papers, were not produced. Nor was a minute of the decisions. Dr Hinoul testified that he had not seen any such document. He told the Court that legal advice was taken about the decision to decommercialise the products but, following a call from the applicants for the respondents to produce a copy of the advice, the Court was informed that no such advice could be found.2415
2490 The applicants were at great pains to demonstrate that safety concerns played a part in Ethicon’s decision to withdraw these devices from the market. They argued that statements to the contrary by Ethicon personnel, including Dr Hinoul, were, in effect, dishonest. They contended that the devices were withdrawn from the market because they were not sufficiently safe to remain on sale. In substance, there were three foundations to their argument. First, they pointed to the decline in the market for transvaginal POP mesh products following the FDA’s announcements in 2011 and contended that this reflected safety concerns in the medical community. Second, they contended that Ethicon’s failure to conduct the post-market surveillance ordered by the FDA or to negotiate with the FDA about modifications to its § 522 orders reflects the company’s view that the data that would be collected in these processes would not prove that the devices were safe. They argued that Ethicon’s response to the § 522 orders amounted to an admission that its devices were not safe. Third, they contended that Ethicon’s decision to withdraw the devices from the market rather than do as the FDA required, reflects its desire to avoid regulatory scrutiny.2416
2491 The first point is well-made and probably also the third. But the second is not so obvious. At no time did Ethicon acknowledge that any of its devices caused an unacceptable level of harm to the women in whom they were implanted. In all likelihood the cost to Ethicon of complying with the FDA’s § 522 orders would have been substantial and the benefits for the business difficult to gauge. The applicants did not dispute that the market for the POP devices was declining. The climate did not suggest that there were prospects of a revival. As I read the evidence, Ethicon would not have withdrawn the devices had the FDA agreed to what it proposed in its initial responses to the § 522 orders but it was unwilling to invest in the studies the FDA required of it. I accept that interest in the POP devices waned after the FDA notification. It is likely that the decline in interest was attributable to concerns of surgeons and patients about the safety issues associated with them. To this extent, safety was a factor in the decision to withdraw those devices from sale, but only indirectly. I am not persuaded that Dr Hinoul’s evidence that the decision was taken for commercial reasons was false. That said, it is fair to conclude that, more likely than not, Ethicon realised it could not satisfy either the market or the FDA that, with respect to the transvaginal use of Gynemesh PS, the mesh kits, or TVT Secur, the benefits outweighed the risks.
The decisions made by JJM and the TGA
2492 The applicants similarly claimed that safety concerns were relevant to Ethicon’s decision to place a hold on supply of TVT Secur in Australia in 2008 and to remove Gynemesh PS from sale in this country in 2017.
2493 As I have already said, TVT Secur is no longer sold in Australia, the United States or Europe. Manufacture has ceased and its Australian registration has been cancelled. That much is common ground. The parties initially disagreed about the date it was taken off the market, but it is now agreed that a hold was put on supply of the device in Australia in March 2008 and that ARTG registration was cancelled in June 2012.2417 The parties continued to disagree, however, about the reasons why it was taken off the market.
2494 The respondents relied on evidence given by Dr Hinoul that the decision was made for commercial reasons and not for reasons related to the safety or efficacy of the device.2418
2495 The applicants relied on the evidence that the hold on supply was placed after consultations with “user surgeons” about concerns over the performance of the device. This evidence shows that there was a temporal connection. Without more it does not denote a causal relationship. That said, the contemporaneous documents leave little room for doubt that the decision to put a hold on the supply of TVT Secur in Australia was not taken merely for commercial reasons, unless commercial reasons include an assessment that the product was unlikely to be profitable because of the problems experienced with the performance of the device.
2496 The applicants also referred to clinical data sets from a report of a symposium held by the IUGA2419 and submitted that it showed that TVT Secur had “lower success rates than with the other TVT products, post-surgery leaking, bladder perforation, de novo urgency, persistent urge, tape exposure, groin pain and worsening incontinence”.2420
2497 This document suggests that complication rates were not of concern, except for recurrence or worsening of stress urinary incontinence, de novo urge incontinence and voiding dysfunction. The focus of the document was on the objective cure rates from the available data and the theory that the more cases a surgeon performed the higher the objective cure rate would be.
2498 On the material to which I was taken, I am not satisfied that the safety of TVT Secur was a significant issue for the respondents. But efficacy certainly was. Obviously a product with a poor performance record was unlikely to sell well. The feedback from surgeons would not have been encouraging, to say the least. So I accept that the decision to put a hold on the supply of TVT Secur was made in part for commercial reasons but the driving factor was the high reported failure rate. Clearly, Dr Maree and his team were not satisfied that as things stood marketing and selling the device was not justifiable.
2499 According to the minutes of the meeting of JJM executives on 1 November 2007, referred to above, there were three reasons for its in-principle agreement to withdraw the device from the Australian market,. They were: “patient outcome, company ethics, and commercial considerations”.2421 Those minutes indicate that poor performance outcomes were the primary concern.
2500 On 31 May 2012 JJM informed the TGA that Ethicon had decided to discontinue TVT Secur and all the POP devices except for Gynemesh PS.
2501 On 29 August 2012 JJM informed the TGA that Ethicon was revising the indications for use for Gynemesh PS to restrict placement of the mesh “transabdominally” for prolapse repair.
2502 Within two months the TGA began a review of all urogynaecological mesh devices included in the ARTG.2422 It is unnecessary for present purposes to delve into the detail of that review.
2503 That month the TGA asked JJM for copies of all training materials used or provided by JJM or the manufacturer2423 and of the IFUs for the various devices.2424 In its reply to the request for training materials, JJM advised that clinicians wanting training on Prosima, Prolift or TVT are provided with materials in advance of the preceptor delivering the “practical” session and attached the training materials. Although training had been provided on Gynemesh PS at the time of the product launch, JJM advised that training was no longer provided.2425
2504 On 25 September 2013, the TGA directed JJM to provide it with a list of products supplied under each ARTG entry and a checklist summarising conformity with each of the essential principles, requiring that “[e]vidence of compliance to each applicable principle must refer to documents, reports etc…”.2426 The TGA also required other material including information about the number of devices distributed in Australia;, details of adverse events the subject of complaints in Australia or overseas; risk analysis on the reported adverse events; and corrective action taken by the manufacturer resulting from the analysis of complaints and adverse events. In addition, the TGA sought copies of “the clinical evidence used to establish conformity with Australian Essential Principle 14” and copies of any unpublished clinical data on the various devices. Essential principle 14 stipulated that:
Every medical device requires clinical evidence, appropriate for the use and classification of the device, demonstrating that the device complies with the applicable provisions of the essential principles.
2505 JJM responded to this request on 7 November 2013.2427 The response included a table listing the essential requirements, indicating whether they applied to the devices in question, which standards applied (international, national or internal), and whether waiver was justified. It did not indicate, however, whether the relevant requirements were satisfied. If the other information sought by the TGA was supplied, it did not find its way into evidence.
2506 On 8 January 2014 the TGA asked Ethicon for details of the § 522 post-market surveillance study plans the FDA had required of it.2428 In its response, dated 18 February 2014 and signed by Stacy Kluesner, Ethicon advised that the FDA had granted Ethicon permission to suspend the post-marketing surveillance study of each of the relevant devices. The reason in each case was Ethicon’s decision to discontinue marketing the device, not just in the United States but in all global markets.2429
2507 In the case of Gynemesh PS, Ms Kluesner said that the decision was based on Ethicon’s determination to discontinue the marketing of the device in the United States for transvaginal repair of pelvic organ prolapse and the FDA’s hold on the surveillance study was conditional, amongst other things, on Ethicon submitting “an add-to-file” to the registration to modify the indication for use to remove any references to transvaginal repair and updating all website and promotional materials to reflect the narrower indication “for abdominal use only”.2430 Ms Kluesner also informed the TGA that Ethicon had ceased manufacturing TVT Secur, Prolift, Prolift+M, and Prosima in December 2012.2431
2508 In March 2014 the RANZCOG and the Urogynaecological Association of Australia (UGSA) published a joint position statement in support of the continued use of midurethral slings to treat female stress urinary incontinence.2432 The salient parts of that statement read:
Mid-urethral slings … have been shown to be as effective as more invasive traditional surgery with major advantages of shorter operating and admission times, and a quicker return to normal activities, together with lower rates of complications. This has resulted in MUS becoming the operation of choice in Europe, the United Kingdom, Australasia and the USA for treatment of SUI.
The USA Food and Drug Administration (FDA) released a white paper and safety communications regarding safety and effectiveness of transvaginal placement of surgical mesh specifically for pelvic organ prolapse. A prolapse is where some of the pelvic organs bulge downwards giving rise to symptoms of an uncomfortable vaginal lump. Media attention on this totally distinct and separate issue of mesh use in women has the potential to cause unnecessary confusion and fear in women considering MUS for treatment of stress urinary incontinence. Both RANZCOG and UGSA wish to strongly emphasise that the US FDA publications clearly state that MUS were not the subject of their safety communication.
There is robust evidence to support the use of MUS from over 2,000 publications making this treatment the most extensively reviewed and evaluated procedure for female stress urinary incontinence now in use. These scientific publications studied all types of patients, including those with co-morbidities such as prolapse, obesity and other types of bladder dysfunction. It is, however, acknowledged that any operation can cause complications and for MUS, these include bleeding, damage to the bladder and voiding difficulties. Nevertheless, the results of a recent large multi-centre trial have again confirmed the excellent outcomes and low risks of complications to be expected after treatment with MUS. Additionally, long term effectiveness has been demonstrated in studies following patients for up to 17 years. In Australia, it has been the operation of choice to treat for female SUI since 2004. RANZCOG and UGSA support the use of monofilament polypropylene mid-urethral sling for surgical treatment of female stress urinary incontinence.
2509 An almost identical position statement was issued by the IUGA in July 2014, citing the same references. Associate Professor Rosamilia identified herself as the author.2433
2510 While there are certainly differences between midurethral slings and transvaginal mesh, these endorsements by the professional associations are troubling.
2511 First, the paper cited in support of the statement about the relative effectiveness and safety of midurethral slings was by Cody et al (2003).2434 But the paper does not seem to support the statement.
2512 Cody et al (2003) noted that data was limited and that there was no data from randomised controlled trials beyond two years from surgery so that “long-term effects are therefore currently not known reliably”. Moreover, the authors’ conclusions were far more circumspect than the position statement suggested. They wrote:
The long-term performance of TVT in terms of both continence and unanticipated adverse effects is not known reliably at the moment. Despite relatively few robust comparative data, it appears that in the short to medium term TVT’s effectiveness approaches that of alternative procedures currently available, and is of lower cost. As TVT is a less invasive procedure, it is possible that some women who would currently be managed non-surgically will be considered eligible for TVT. Increased adoption of TVT will require additional surgeons proficient in the technique. It is likely that some of the higher rates of complications, e.g. bladder perforation, reported for TVT are associated with a ‘learning curve’. Appropriate training will therefore be needed for surgeons new to the operation, in respect of both the technical aspects of the procedure and the choice of women suitable for the operation. Further research suggestions include unbiased assessments of longer term performance from follow-up of controlled trials or population-based registries; more data from methodologically sound RCTs using standard outcome measures; a surveillance system to detect longer term complications, if any, associated with the use of tape; and rigorous evaluation before extending the use of TVT to women who are currently managed non-surgically.
2513 Second, the position statement cited Nilsson et al (2013) in support of its representation as to the long-term efficacy having been demonstrated in studies following patients for up to 17 years.2435 The difficulties with relying on that study and its conclusions are discussed above in Part VI.
2514 Third, description in the RANZCOG statement of symptoms from the use of midurethral slings ignores the most serious mesh-specific symptoms and the potential for chronic pain.
2515 Professor Blaivas issued a blistering criticism of the RANZCOG statement.2436 He said it “totally whitewashe[d] the entire mesh controversy” by enthusiastically and uncritically endorsing the use of slings as both more effective and safer than native tissue alternatives, downplayed the complications, and did not even mention the two most serious complications: mesh erosion and chronic pain. He said it also failed to acknowledge the poor quality of the literature with respect to complications, the overall poor follow-up, and the lack of any meaningful outcome measures or even methodology to assess complications. He claimed that the references had been “cherry pick[ed]” to support the views of the authors.
2516 On 28 April 2014 the TGA’s clinical assessor reported on the material submitted by JJM regarding the remaining SUI devices. The assessment was critical of the risk assessment documentation and various aspects of Dr Hinoul’s CER on the TVT Family of Products dated 16 July 2013. These criticisms are discussed above. Many of the same criticisms were also levelled at his CER on Gynemesh PS.2437
2517 On 1 July 2014 the TGA’s clinical assessor determined that the clinical data for Gynemesh PS submitted by JJM did not meet the requirements of the Medical Devices Regulations, in particular essential principles 1, 2, 6, and 14.2438
2518 It appears from the TGA’s Clinical Evidence Assessment2439 that JJM had furnished the TGA with a copy of Dr Hinoul’s 3 May 2013 Gynemesh CER, a report on Gynemesh PS dated 16 May 2013 entitled “Product Risk Management Report”, post-marketing data from January 2010 to January 2013, and the most recent Gynemesh PS IFU. Notwithstanding its title, the second of these documents was not a risk management report. Rather, it was a brief summary of the methods used to evaluate the risks and the conclusions that were drawn. The TGA noted that no risk management report had been provided and therefore “no information regarding the application of routine and additional risk reduction activities”. The TGA also observed that no list had been submitted of “identified clinical risks linked to appropriate risk reduction activities”.
2519 The TGA levelled the following criticisms at the 3 May 2013 Gynemesh PS CER.
2520 First, it was unclear from the CER or the so-called risk management report how the high risk categories were determined but the risks discussed in the CER included exposure internal organ damage and nerve damage and/or pain. Yet:
The discussion surrounding each risk refers to literature that has not been specified (or provided in the references to the CER). For example, the discussion regarding internal organ damage states: “Damage to internal organs, in particular the bowel and bladder, were encountered at an incidence of 0.3% and 1.9% respectively in the analysis of 3194 patients, in whom GYNECARE PROLIFT™ was used and reported in the published literature.” No further details regarding this published literature have been provided, such as the date of this publication or a reference.
2521 Second, the discussion regarding exposure rates quoted figures of over 10% while the final conclusion is that the literature review justifies a frequency of permanent or long-term impairment to be 1/100 to 1/1000 and “[n]o evidence for justification” is offered to support the conclusion.
2522 Third, the majority of studies cited in the report to support the frequency rates of the high risk categories do not reflect the surgical approach indicated for the device.
2523 Fourth, multiple risks were excluded from the discussion, “for example, erosion (this should be specifically discussed and not assumed to be classed as exposure only) blood loss, infection, voiding dysfunction and dyspareunia”.
2524 Fifth, the risk analysis documentation supplied by JJM was insufficient to perform an evaluation or to meet the legislative requirements for essential principles 1 and 6.
2525 Sixth, no formal literature search protocol was provided, that is one “with a clear aim, inclusion and exclusion criteria, plus the planned search terms and search steps”. The assessor observed that “[t]hese criteria must be determined prior to the execution of the search to reduce selection bias and ensure that the methods used to complete the literature search do not change as the search progresses”. The assessor noted that the search report referred to “irrelevant references”, apparently identified in an earlier process before the inclusion and exclusion criteria were applied, but said that it was unclear how that process had been undertaken. The assessor said that the details of the literature search indicate that articles were included or excluded from the search on a case by case basis and no clear lists of “exact inclusion and exclusion criteria” were provided.
2526 Seventh, the literature search report contained “a number of major deficits”; the information was insufficient to meet “the legislative requirements for Essential Principle 14 regarding the literature protocol and literature search” and the deficits had “negatively impacted the quality of the clinical data submitted”. The major deficits identified by the TGA included the following items:
• The search report lists a number of searches in various databases. It is unclear how the final list of articles was achieved, how each study corresponds to the individual searches performed and at what inclusion and exclusion criteria were applied at each stage of the search process.
• A list of the included and excluded articles has been provided. It is unclear which searches were used to identify each article.
• It is not possible for an evaluator to replicate the search protocol and report submitted by the Sponsor due to lack of information provided. The application of inclusion and exclusion criteria is not consistent and the final list of studies included in the literature review is difficult to relate back to the search protocol/report.
2527 Eighth, the clinical data evaluation was also deficient. The 2013 Gynemesh PS CER summarised 50 papers in a table but there was no critical analysis of the data in the summary and copies of only five papers were supplied. The section which followed the table, entitled “Literature Critical Analysis”, discussed only a selection of the 50 listed papers. Consequently, the assessor wrote, the analysis showed selection bias and does not represent the total body of literature identified in the report. Moreover, the critical analysis of the selected papers was limited and the section included multiple references that were not identified in the literature search.
2528 Ninth, despite revising the indications for Gynemesh PS in June 2012 to allow for abdominal placement only, the evidence presented in the analysis of the literature focuses on transvaginal placement. It was unclear from either the evidence presented or the discussion in the Literature Critical Analysis that the placement of mesh via an abdominal approach was in fact superior to the transvaginal approach.
2529 Tenth, the rates of erosion presented in the clinical data were high and there was insufficient discussion about acceptable rates of exposure, potential risk groups, and appropriate risk reduction activities. Although the “Literature Critical Analysis” briefly identified a number of important technique-related factors that may increase the risk of mesh exposure, including concurrent hysterectomy, incision type, and depth of tissue dissection, “[t]his data has not been discussed in the risk analysis and links to the application of additional risk reduction activities (including training materials) [have] not been made”.
2530 The TGA concluded that Dr Hinoul’s presentation, analysis, and synthesis of clinical data contained major deficits. Those deficits included:
• Inability to reproduce the results from the literature search due to:
• Lack of detail regarding the search process.
• Inconsistent application of inclusion and exclusion criteria.
• Unclear reasoning to establish the final list of studies “included”.
• Insufficient evidence relevant to the (scil) current indication of the device.
• Lack of adequate critical analysis of the studies identified from the literature search.
• Major selection bias in the Literature Critical Analysis, with only a small number of author selected studies included for discussion. It is unclear why the authors have not discussed the total body of literature identified in the literature search. Therefore, the evaluator cannot have confidence that the authors report is a balanced and evidenced based document representing the total body of relevant literature.
• Inadequate synthesis of data, with no discussion of the total body of literature leading to a clear, evidenced based, logical conclusion.
• Lack of discussion regarding clinical safety.
2531 In the absence of sufficient evidence regarding the limited indication for Gynemesh PS, the TGA could not understand why Dr Hinoul concluded that no additional clinical data were required. In the light of the inadequate analysis of the total body of literature, selection bias in the critical analysis, and the deficits in the literature search protocol and report, the TGA described the evaluation process as “limited”. Based on the information provided, it concluded that the literature requirement for essential principle 14 had not been demonstrated.
2532 The TGA then examined the post-market data contained in the CER and additional data submitted by JJM in February and March 2014 in response to a request from the TGA. The assessor said that the data presented by Ethicon did not allow for the interpretation of adverse event/complaint rates per year and in the context of yearly distribution data and that it was unclear whether the complaints and adverse events referred to separate clinical cases. Nevertheless, the TGA observed that the rate of erosion/exposure based on the data presented was over 2% and it was unclear why risk assessment had not been conducted for this risk, considering the high number of erosion cases compared to other adverse events. The TGA also observed that the data presented by JJM showed “a clear and concerning upward trend in reports of erosion”, yet the trend had not been identified or discussed in the report. Rather, the author (Dr Hinoul) concluded — without “justification, evidence or discussion” — that the current risks were “within acceptable levels” and that “further risk reduction” was unnecessary.
2533 Next, the TGA considered the post-marketing device vigilance plan, which it characterised as involving “the collection of passive post-marketing surveillance” of complaints made directly to JJM. Based on the lack of evidence regarding the new indication for Gynemesh PS and the clear increase in erosion rates associated with it, the TGA said that it was reasonable to conclude that additional device vigilance was required.
2534 The TGA said that the risk reduction plan proposed “routine risk reduction activities only”, consisting of IFUs. No additional activities, such as training materials, were discussed in the CER or risk analysis and none was apparently proposed.
2535 The TGA then referred to the statements in the CER about the absence in the literature of new and/or unacceptable risks and that “[t]he benefits of using the product as recommended … continue to justify the associated risks”. The TGA noted that many studies presented high rates of erosion (up to 35.7%) but said that the CER had not provided an adequate discussion of this risk and justification for accepting such a high incidence. The TGA said the risk-benefit evaluation could not be supported and that JJM had submitted insufficient evidence to support their claims regarding safety and efficacy and to satisfy essential principle 6.
2536 On 20 August 2014 the TGA issued a media release announcing the completion of its review of urogynaecological surgical mesh implants. 2440
2537 In its media release the TGA advised that it had undertaken a literature search of published materials since 2009 and had found the overall quality of the literature to be poor. It concluded that there was “an absence of evidence to support the overall effectiveness of these meshes as a class of products”, while noting that the literature identified “the known adverse outcomes associated with their use”. It declared that the evidence to support the use of these meshes for transvaginal pelvic organ prolapse repair, especially posterior repair, was “not well-established”, but that their use in the treatment of stress urinary incontinence and abdominal pelvic organ prolapse repair was adequately supported by the evidence.
2538 The TGA indicated its intention to reassess the clinical evidence for each individual mesh implant to determine whether it complies with the essential principles.
2539 On 17 October 2014 Patrick O’Meley, a biomedical engineer in the Device Vigilance and Monitoring Department of the TGA’s Office of Product Review, advised Suzanne Winter of JJM that the TGA had completed its assessments of JJM’s submissions on the various devices and enclosed copies of the clinical evidence assessments. He advised that action was urgent. He drew particular attention to the deficiencies in the IFUs and the training materials for the various devices.2441 I discuss the detail in the next Part of these reasons. It is sufficient to note at this point, that, as a result of this letter, substantial amendments were made to the IFUs for those Ethicon devices that remained on the ARTG. The products were recalled for “Product Correction” in the meantime.2442
2540 On 13 July 2017, Pamela Carter from the TGA wrote to JJM advising that a decision had been made to impose a new condition of inclusion of the entry in the ARTG on all the SUI devices remaining on the ARTG that the following addition be made to the instructions for use and labelling for the devices:2443
There have been spontaneous device-vigilance reports of chronic severe pain following use of mesh for treatment of stress urinary incontinence (SUI) via the retropubic and transobturator approaches. Patients should be informed of this possibility when making a decision about surgical treatment for SUI. As is usually the case with spontaneous device-vigilance reports, it is not possible to estimate the frequency of this adverse event. Data from clinical trials show that the main short-term complication with retropubic mesh is bladder perforation at the time of operation; and that the main short term complication with transobturator mesh is groin pain.
2541 In the reasons for the decision, Ms Carter noted that “medical device adverse events reports suggest that, compared to retropubic mesh, transobturator mesh might be more commonly associated with rare, chronic, intractable, severe, debilitating, life-changing pain”. She was concerned that the risk of chronic pain had not been sufficiently addressed so as to adequately inform patients of this risk.
2542 Within four months, however, she had decided that this condition ought to be varied.
2543 On 11 December 2017 Ms Carter wrote to JJM requiring instead of the above that as a condition of inclusion of entry in the Register that the IFUs clearly indicate in the warnings and precautions and/or adverse reactions sections that the use of the devices may result in bladder perforation, severe chronic pain, and groin pain.2444 Noting that the current reference in the adverse events section of the IFUs included “acute and/or chronic pain”, Ms Carter said that she considered it important that patients are informed that a possible adverse reaction may be “severe” chronic pain. Similarly, she wrote, the risk of groin pain should be included in the IFU “so that this information can be included in the counselling provided to the patient prior to surgery in order to ensure that the patient is fully informed prior to consenting to the procedure”.2445
2544 In order to consider whether to seek a review of this decision and, if not, to “liaise with the manufacturer to determine how a change to the IFU can be made globally and the timeline for such a change”, JJM decided to place a hold on supply of all TVT products in Australia effective 17 January 2018.2446 In its advice to customers JJM continued to maintain it is “fully confident” in the safety and efficacy of these devices.2447
2545 In the meantime, on 28 November 2017, the TGA announced that it had decided to remove all the other mesh products used as single-incision mini-slings from the ARTG on the ground that there was a lack of adequate scientific evidence to enable it to be satisfied that the risks to patients associated with their use for the treatment of stress urinary incontinence were outweighed by their benefits. Cancellation notices were issued to the Australian sponsors. Cancellation took effect on 4 January 2018.2448
2546 As at 17 January 2018, the ARTG entry for TVT (99193), which also covered TVT-O, TVT Exact and TVT Abbrevo, included four “specific conditions”:
a) The person in relation to whom the kind of medical device is included in the ARTG (the sponsor) must provide to the Therapeutic Goods Administration, Department of Health (the TGA) consecutive annual reports. Reports should be for the period 1 July to 30 June. The first report must be provided by no later than 1 October 2016 and cover the period 1 July 2015 to 30 June 2016. Subsequent reports are to be provided on 1 October of each year for the duration of the device’s inclusion on the ARTG. The annual report must include records of all complaints and adverse events relating to problems with the use of the device that have been received by the manufacturer and/or sponsor over the year.
Note: The reports must clearly identify each device of the kind (e.g. by the unique product identifier, model name or catalogue number) to which the complaints and adverse events relate.
This condition applies for the entire period that the ARTG entry remains current.
b) The sponsor also, as required under s 41FN (3)(d), must report to the TGA all adverse events that occur in Australia. However, reporting exemption rules in the Australian Regulatory Guidance for Medical Devices do not apply to this ARTG entry. These adverse event reports should be submitted in accordance with the timeframes specified in the Therapeutic Goods (Medical Devices) Regulations (2002) (Regulation 5.7). Adverse event rates in each report should include confirmed and unconfirmed events and be based on the event description not whether the event has been confirmed or the cause has been identified.
c) This medical device ARTG inclusion is limited to some medical devices of the kind. These devices of the kind are medical devices identified by the manufacturer as:
a. Gynecare TVT W/Abdominal (810041A)
b. Gynecare TVT Device (810041B)
c. Gynecare TVT Obturator (810081)
d. TVT Exact Retropubic System (TVTRL)
e. TVT ABBREVO (TVTOML)
Other devices of the kind must not be supplied under this ARTG entry in Australia until and unless evidence of the compliance of those devices with the essential principles is provided and accepted by the TGA.
Note: If a sponsor requires a variation to the ARTG entry to include details of the medical devices that are to be imported, supplied or exported under an entry, the sponsor will need to submit a Device Change Request to the TGA.
All these conditions apply for the entire period that the ARTG entry remains current.
d) It will be a condition of inclusion of the Entry in the Register that the instructions for use documents (IFU) clearly indicate in the ‘warnings and precautions’ and/or ‘adverse reactions’ sections that the use of these devices may result in:
• Bladder perforation ·
• Severe chronic pain ·
• Groin pain2449
2547 By another letter dated 13 July 2017, Ms Carter notified JJM that she had decided to impose a new condition of inclusion of entry in the ARTG relating solely to Gynemesh PS.2450 The new condition entailed adding to the instructions for use and labelling for Gynemesh PS the following precaution, effective 11 August 2017:
This device is not intended for any pelvic organ prolapse repair via a transvaginal approach.
2548 Ms Carter advised that the current indications for use did not clearly articulate this.
2549 The reasons given for the decision were that, since the earlier review, new evidence had been published on the safety and performance of urogynaecological mesh devices. Ms Carter cited examples including the Maher et al (2016) Cochrane review, the PROSPECT study by Glazener et al (2017), and the study reported by Morling et al (2017). After summarising the findings from a number of studies and noting that the surgical techniques and patient populations were “materially similar” to those in Australia, Ms Carter wrote that the reported post-operative complications and adverse effects were relevant to Australia. She noted that the Maher et al (2016) Cochrane review of 25 randomised trials revealed that 12% of patients experienced considerable pain and other problems associated with mesh exposure when mesh is used for pelvic organ prolapse repair via a transvaginal approach. She stated:
Given the clinical evidence and findings of the Studies, in particular the large number of women covered by them, I consider that, because of the extent of post-operative complications and adverse events, there is now an issue as to whether the risk of use of the Device, and the benefits of its use, are acceptable when used to treat pelvic organ prolapse via a transvaginal approach.2451
2550 JJM was not disposed to accede to the TGA’s request.
2551 On 26 July 2017, following meetings with key decision makers, including regulatory and marketing personnel, Heather Paterson, a sales and marketing manager at JJM, advised her colleagues by email that “[a]pproaching from a strictly commercial point of view, it was a pretty simple decision to discontinue [the device]”. She noted that sales had significantly declined in recent years and that the demand forecast for the ensuing quarter was for only 20 units. Ms Paterson said she was not aware of the changes that would have to be made to the IFU or labelling but, no matter how small they might be, it was “not worth it from a commercial perspective”.2452
2552 Rebecca Gaudin, who had been copied into the email, wrote back two days later to advise that the cost of bringing the IFUs into alignment with the TGA’s requirements would be USD 65,000.2453
2553 On 8 August 2017 Ms Gaudin advised the TGA that JJM had decided to discontinue Gynemesh PS.2454 At the same time, however, she maintained that JJM had continuing confidence in its safety and efficacy and informed the TGA that JJM considered the amendment was unnecessary and unjustified. She said that its decision was based on a review of the cost of implementing the TGA’s requirements and the company’s conclusion that the costs of making the change were more than the total value of yearly sales of the product in 2016. She indicated that JJM would advise its customers that “this is not a product safety issue” and, for this reason, they “do not need to take any action with patients who have already received one of [the] products”.
2554 The applicants referred in their submissions to a number of internal JJM documents which were produced in preparation for the public announcement of the decision in which the same sentiments were expressed, emphasising the commercial nature of the decision.2455
2555 On 17 August 2017 the TGA advised JJM that it had reviewed its proposed action and confirmed that it would treat the discontinuation of Gynemesh PS as a withdrawal and not a recall.2456 “Withdrawal”, it explained, means “a sponsor’s removal from supply or use of therapeutic goods for reasons not related to their quality, safety or efficacy”. The TGA also approved the proposal to write to customers in the terms indicated by Ms Gaudin. On 21 August 2017, it notified JJM that a decision had been made to cancel the entry of Gynemesh PS on the ARTG and that the cancellation would be effective from the following day.2457 It is common ground that cancellation was in fact effected on 22 August 2017.2458
2556 The applicants maintained that safety factors influenced JJM’s decision. As they put it in their written submissions, despite the emphasis in internal documents on the decision being unrelated to safety, “the fact remains that the TGA’s imposition of a condition on the registration of the product” was entirely related to safety concerns and it was the imposition of the condition which resulted in the removal of the product from the market.2459
2557 The applicants also submitted that, as none of the key decision makers at JJM who determined that Gynemesh PS should be withdrawn from the Australian market gave evidence in the trial as to the basis for their decision, it should be inferred that their evidence would not have assisted the respondents’ case.2460
2558 I reject the applicants’ submissions. As to the latter, there was no need to call any of these people. The documents the applicants themselves tendered established the basis for the decision. It appears from Ms Paterson’s email that Ms Gaudin was one of the key decision makers, as might reasonably be expected, since she was the Director of Regulatory Affairs for JJM. As to the former, all the applicants can show is a temporal connection between the TGA’s decision and the JJM decision. True it is that the TGA’s decision was made for safety reasons but the preponderance of the evidence shows that JJM’s decision was unrelated to the safety issues that troubled the TGA. By endorsing JJM’s proposal to inform patients that safety considerations did not feature in its decision, the TGA accepted the respondents’ position. Moreover, the speed with which the decision was taken suggests that the decision had been under consideration for some time before the TGA imposed the new condition.
2559 On 28 November 2017 the TGA decided to remove from the ARTG all transvaginal mesh products used in the treatment of pelvic organ prolapse. In its media release issued the same day the TGA explained that the decision was taken following its review of the most recent published international studies and an examination of the clinical evidence for each such product included in the ARTG and supplied in Australia. It concluded that “the benefits of using transvaginal mesh products in the treatment of pelvic organ prolapse do not outweigh the risks these products pose to patients”.2461
PART XI: INFORMATION PROVIDED BY THE RESPONDENTS ABOUT THE ETHICON DEVICES
2560 In this Part, I discuss the evidence regarding the warnings and other information about the Ethicon devices provided by the respondents in their instructions for use, and marketing and professional training materials. I also assess the adequacy of the warnings and other information in the light of the evidence outlined earlier in these reasons about the risks posed by the devices.
2561 In Australia, a medical device must be sold accompanied by a document setting out the instructions for its use, unless the device is of a specified class and can safely be used for its intended purpose without instructions.
2562 With the exception of the original instructions for use for TVT, which were prepared by Medscand, IFUs for each of the Ethicon devices were prepared by Ethicon Inc. and distributed across the world, including Australia, in the boxes in which the devices were sold.2462
2563 Dr Pence described the IFU as “an essential component of medical devices”, “the cornerstone of risk management”, and “the primary tool for communication between the manufacturer and the clinician about the particular device”.2463 She explained that the purpose of the IFU is to provide the physician with the information necessary to use the device safely, to ensure that the device performs as intended, and to assist the physician to make an informed decision about using the device in a particular patient, taking account of his or her training and knowledge.2464 She said that the IFU should be consistent with available clinical data and that it should “appropriately” identify all the hazards and other clinically relevant information.2465 She said that the manufacturer should ensure that it provides “all safety information necessary for safe and effective device use and for making decisions about implant usage for a particular patient”.2466 She stated that, in determining its contents, the manufacturer should have regard, amongst other things, to the guidelines relating to medical devices published from time to time, by the Global Harmonization Task Force; published scientific literature (both clinical and preclinical), internal studies, adverse event reports for the device and for similar devices; adverse events data bases, and information gleaned from clinical experience data, meetings with experts, the clinical experience of its consultants, and medical and scientific conferences.2467 She said that the manufacturer has the responsibility to ensure that the IFU always accurately reflects current knowledge.2468
2564 Dr Pence largely based her assessment of Ethicon’s IFUs on the GHTF guidelines. These guidelines (often referred to as guidance documents) were devised for the purpose of providing non-binding guidance to regulatory authorities for use in the regulation of medical devices but were intended for use not only by regulators but also by industry and conformity assessment bodies.2469 The GHTF was an international body consisting of regulatory authorities and the medical device industry that was established in 1992 to address the need for harmonisation in the development of medical devices in order to enhance patient safety and increase access to safe, effective and clinically beneficial medical technologies around the world.2470 In 2012 it was disbanded and its work was taken over by the International Medical Device Regulators Forum. The legislative framework for the regulation of medical devices in Australia adopts the philosophies of the GHTF.2471
2565 Dr Pence was not asked to, and did not undertake, a systematic review of the published literature.2472 She relied principally on the information she derived from Ethicon’s internal documents and the GHTF document entitled “Label and Instructions for Use for Medical Devices”, first published in 2000 (under the title “Labelling for Medical Devices”) and revised in 2005 and 2011.2473 That document listed particulars that should be included on labels and in instructions for use. The 2000 version provided that the labelling should include, amongst other things, “any warnings and/or precautions”, “the performance intended by the manufacturer and any undesirable side-effects”, and, for implantable devices, “information regarding any particular risks in connection with its implantation”.2474 These recommendations were adopted in Australia and incorporated into the Medical Device Regulations in Sch 1 cl 13.4(3), items 3, 4, and 19.
2566 In the 2011 iteration, the GHTF guidelines on labelling and instructions for use stated, amongst other things, that an IFU should include “any residual risks, contraindications and any expected and foreseeable side-effects, including information to be conveyed to the patient in this regard”.2475 Since 2005, the GHTF has recommended that any residual risks identified by the manufacturer in its risk analysis be “reflected” as contraindications or warnings.2476 “Risk” is defined in another GHTF publication (“Essential Principles of Safety and Performance of Medical Devices”, dated 20 May 2005), sourced from a 1999 International Standard, as the “combination of the probability of occurrence of harm and the severity of that harm”.2477 The GHTF stipulated that information contained in an IFU should be consistent with the available clinical data, and all the hazards and other clinically relevant information should be identified appropriately.2478
2567 In conformity with these guidelines, Dr Pence stated that IFUs should include, amongst other things, the following:
(1) the intended use or purpose of the device including the intended device user;
(2) the performance of the device as intended by the manufacturer;
(3) any residual risks, contraindications, and any expected and foreseeable side-effects, including information to be conveyed to the patient in that regard;
(4) any requirements for special facilities or special training, or particular qualifications of the device user;
(5) information that allows the user and/or patient to be informed of any warnings, precautions, measure to be taken, and limitations of use regarding the device, including, where appropriate;
(a) warnings, precautions and/or measures to be taken in the event of malfunction of the device or changes in its performance that may affect safety;
(b) for implantable devices, information regarding any particular risks in connection with its implantation; and
(6) where the manufacturer has included clinical investigations as part of pre-market conformity assessment to demonstrate adherence to the essential principles set out in the GHTF guidelines, the investigation, outcome data and clinical safety information, or a reference to where that information might be found (this being a new requirement introduced in the 2011 version of the guidelines).2479
2568 Dr Pence also referred to the opinion of the National Research Council, an American not-for-profit organisation, that it was important to record in medical device labelling both the harmful effects that can occur as a result of using a device and the incidence of harmful risks. She said that, in communicating risk, harmful effects should not be minimised, and the incidence of those effect should be communicated if known.2480
2569 Dr Pence said that information about the severity of the residual risks and whether they can be remedied must also be included. She emphasised that information about the duration (“permanency” and “chronicity”) and the severity of the risks was “all important” since it enables the patient to make an informed decision about whether to agree to implantation with the device and the clinician to assess whether the device is suitable for the patient.2481
2570 Dr Pence explained that the “adverse reactions” section of the IFU should include all adverse events or undesirable side effects that are “expected and foreseeable” including any relevant information that must be conveyed to the patient. She also stated that this section should include, where appropriate, a direction to the reader to see other sections of the IFU for additional information that is important for the physician and patient to know in respect of the potential safety concerns associated with the use of the device.2482 In cross-examination it was put to Dr Pence that the Australian regulations (presumably the Medical Device Regulations) do not refer to “expected and foreseeable” side effects but to “undesirable” side effects.2483 The probable reason for this is that the Medical Device Regulations came into force in 2002 before the change in language in the GHTF guidelines from “undesirable” to “expected and foreseeable”. Dr Pence replied that there was no “difference in substance” between the two.2484 Even if there were some discernible difference, it is difficult to see how that would assist the respondents’ case.
2571 In her report, Dr Pence said that the warnings section should describe any serious complication, such as one that may result in persistent or significant incapacity or have the potential to substantially disrupt a patient’s ability to conduct normal life functions and any complication that might require medical or surgical intervention to preclude impairment of a body function or permanent damage to a body structure.2485 In addition, Dr Pence said that the warnings section should refer to other clinically significant complications, such as those that occur frequently and which could lead to a potentially serious outcome, and describe the actions to be taken to reduce their likelihood or severity and the means of monitoring or managing them.2486
2572 Dr Hinoul deposed that, although updates were made to the IFUs for each of the Ethicon devices over time to insert additional language and particular complications, the original IFU for each device included all complications specific to the device of which surgeons might not otherwise have been aware.2487 Dr Pence firmly disagreed. She stated that these comments are contradicted by the concerns raised by the regulatory authorities and by her own evaluation of the instructions for use to which I will come shortly. The concerns of the regulatory authorities led to extensive revisions to the instructions for use in 2009 and 2015 and I will come to them in due course.
2573 Dr Pence also disagreed with the notion, advanced by Dr Hinoul and which lay at the heart of the respondents’ defence, that adverse reactions which are commonly understood by clinicians need not be listed in an IFU. In her report she wrote:
A reasonable manufacturer cannot assume that all clinicians who may use a medical device are familiar with the potential adverse reactions. Additionally, incidence of certain adverse reactions have been shown to be greater with mesh implantation as compared to traditional non-mesh repair, and this information is important for the clinician in regards to making decisions about whether mesh implantation is suitable for a patient. Labeling, in this context the Instructions for Use, is the cornerstone of risk management.2488
2574 Dr Pence proceeded to explain that not all complaints received by manufacturers are available to clinicians. Indeed, many of them are not even passed on to the applicable regulatory authorities, although since 1984 US manufacturers of medical devices (like Ethicon Inc.), have been required to report to the FDA all post-marketing device-related deaths, serious injuries, and certain malfunctions. Since August 1996, these reports have been recorded in a publicly available database known as the Manufacturer and User Facility Device Experience Database (MAUDE database).2489 Dr Pence illustrated her point with some examples drawn from Ethicon’s records which, in her opinion, should have been but were not reported to the FDA.
2575 Nor, Dr Pence observed, was “complaint trending information” available to clinicians. She pointed out that, even commonly understood adverse reactions may occur with greater frequency and/or severity with the use of a medical device. She said that this information was important to enable clinicians to decide whether the device is suitable for a particular patient and for making decisions about the patient’s management. She also observed that unpublished studies and reports of unpublished tests (such as biocompatibility and animal tests) are not available to clinicians.2490 She remarked that, even where clinical studies sponsored or supported by the manufacturer are published, there may be a lengthy delay between the information reaching the manufacturer and the publication of the study findings during which the manufacturer is aware of adverse reactions but the only clinicians who are aware of them are those who participated in the study.
2576 In any case, Dr Pence said that selective reporting of the results of clinical trial and under-reporting of adverse events in peer-reviewed publications have been “well-documented”. She cited an article by Daniel Hartung and others, published in the Annals of Internal Medicine in 2014, which reported significant discrepancies between the reporting of serious adverse events in peer-reviewed publications and reports to ClinicalTrials.gov, the single largest publicly accessible trial registry in the United States.2491
2577 In her oral testimony, Dr Pence expanded upon this evidence.2492 She said that one cannot assume that every clinician understands all the risks or the extent of the risks. She pointed out that some of the clinicians were generalists. Moreover, she added, the company has “the most information” about its own products and groups of people dedicated to evaluating the scientific and medical literature about them”. Those resources, she observed, are beyond those of most clinicians.
2578 Indeed, it is for these reasons, Dr Pence explained in her report, that the regulatory model for medical devices fixes the manufacturer with the primary responsibility for “assuring conformity with all regulatory requirements”, including those relating to instructions for use.2493 In order to comply with the applicable requirements for the content of an IFU, she said that it is the manufacturer’s responsibility “to convey all safety information necessary for safe and effective use of the medical device and for making patient management decisions in a complete, accurate, balanced, and objective manner”.2494
2579 As one might have expected, Dr Pence agreed in cross-examination that the medical literature is available to surgeons to enable them to decide whether or not to recommend any of the devices to their patients. She added the qualification, however, “if they have time to look at it”. She observed that most surgeons do not have the time or the resources to look at all the literature and, in any event, the quality of the literature is variable. She continued:
The – even the FDA in its literature review cited that the literature – there’s very little long-term – long-term data available and they had a concern in their evaluation in 2011 that even the randomised controlled trials had not looked at safety appropriately. So, in addition, I mentioned earlier about the underreporting of adverse events, so with regard to the very many issues that exist and potential bias as well and literature and some of the literature, that that is not the appropriate tool for surgeons to get their information. It is a resource, but not the place where they should get their information. Their information should be in the instructions for use.2495
2580 Once again, Dr Pence emphasised the vast discrepancy in resources and information held by manufacturers on the one hand and surgeons on the other.2496 And, while she acknowledged that there were other sources of information available to surgeons, she did not resile from her central point that the manufacturers will know more about the risks associated with the use of their products than surgeons and that they are obliged to disclose what they know in the instructions for use.
2581 In her report, Dr Pence referred to an FDA publication from September 1993 entitled “Human Factors Principles for Medical Device Labeling”, said to be “just as applicable to supplementary information and labels on medical devices as they are to the instructional booklets that accompany devices”.2497 This was essentially a guide to the preparation of labelling for medical devices, including instructions for use. The authors noted that the National Research Council identified a number of “risk communication content areas that are especially important to medical device labeling”. They were: harmful effects that can occur as a result of using the device, the incidence of harmful effects, and environmental factors that influence device use. The authors pointed out that these matters take into account many factors including the conditions under which a device is used and the type and incidence of harmful effects associated with device use. They offered the following “tips on risk communication”:
• Do not minimize harmful effects associated with device use.
• Include incidence of harmful effects if known.
• Provide remedies for harmful effects or refer to appropriate authority.
• Describe environmental factors that influence operators and devices.2498
2582 The position taken by Dr Pence was not only supported by the regulatory documents and the actions taken by the regulators but also by some of Ethicon’s own material. For example, a Gynecare PowerPoint presentation on TVT-O, marked for “internal use only”, referred to a report by Bourrat et al (2003) in an article (published in the French journal Progrès en Urologie) of a 30% incidence of pain in women with stress urinary incontinence (159 out of 235), which impaired their quality of life.2499 The presentation described Bourrat et al as “[p]robably the only authors who have more or less correctly assessed pain after TVT!”.2500 In cross-examination Dr Hinoul said that the article would not have come to his attention at the time because he was still in clinical practice.2501
2583 The same Gynecare document also noted the following pertinent comments of Professor de Leval:
[I]t must be acknowledged that the description of pain in the literature is extremely poor. Is it transient? Do authors report on immediate post-op. Pain? Or simply on more lasting pain symptoms? Thus, the information from the literature must be cautiously analyzed.
2584 Dr Hinoul agreed that the statement of “adverse reactions” in each Ethicon IFU represented Ethicon’s authorised statement as to the adverse reactions to its implants. He also agreed that a doctor would be entitled to expect that the company supplying the product would have access to more knowledge about its product than others in the medical community.2502
2585 Dr Pence identified a number of problems with the IFUs based on an extensive review of the respondents’ files and reports on the Ethicon devices available in the FDA’s MAUDE database. Her focus was on what she described as “the safety information missing” from the IFUs. A good deal of her time was spent on deducing from the documents what potential complications were known to the respondents at various times and those of which they should have known. That course was no doubt undertaken because of the respondents’ pleading. If she had had the benefit of Dr Hinoul’s admissions about the information known to the respondents since the first of the Ethicon devices was launched on the market in Europe in 1997, much of that time and effort would have been unnecessary.
2586 Twenty-five IFUs for the SUI devices were admitted into evidence. The IFUs were in use in Australia from 1999 to at least 2015. Below is an overview of those IFUs.
2587 The early IFUs accompanying TVT are an important starting point for the discussion of all the IFUs since they were used as templates for those supplied with the other SUI devices and some of the contents also found their way into the IFUs supplied with the POP devices.
TVT
2588 In October 1999, when TVT was first supplied in Australia, it was accompanied by an IFU prepared by Medscand, the company that developed the device and with whom Ethicon had entered into a licence and supply agreement. Although Ethicon acquired TVT in November 1999,2503 it did not update the accompanying IFU until 8 September 2000.2504 Nor did it advise the market of any shortcomings in the original Medscand IFU.
2589 Between 1999 and 2015, there were nine different versions of the IFU for TVT. Only five are relevant, as four of the versions did not contain material changes bearing on the facts in issue in this proceeding.
2590 The introductory information in the Medscand IFU read as follows:
Please read all information carefully.
Failure to properly follow instructions may lead to injury and result in improper functioning of the device.
Important:
This leaflet is designed to provide instructions for use of the TVT (Tension-free Vaginal Tape) single use device, reusable introducer and reusable rigid catheter guide. It is not a reference to T.V.T surgical technique for correcting USI (Urinary Stress Incontinence).
The device should be used only by properly trained, qualified medical personnel.
These instructions are recommended for general use of the device. Variations in use may occur in specific procedures due to individual technique and patient anatomy.2505
(Original emphasis)
2591 In the first TVT IFU drafted by Ethicon, in use from 8 September 2000, an amendment was made to emphasise that “[t]he device should only be used by physicians trained in the surgical treatment of Stress Urinary Incontinence”. In the 22 December 2003 IFU, this sentence was further amended to add “and specifically in implanting the TVT device”.2506
2592 The original Medscand IFU stated that TVT is made of knitted polypropylene filaments identical in composition to the Prolene sutures. It continued with the following description of the strength and elasticity of the material:
This material has been reported to be minimally reactive and to retain its strength for a very long time in clinical use. The mesh has high burst strength (approximately 14kg/cm2) and tensile strength.
Prolene® mesh is knitted by a process which interlinks each fiber junction and which provides for elasticity in both directions. The fiber junctions are not subject to the same work fatigue exhibited by more rigid metallic meshes. This bi-directional elastic property allows adaptation to various stresses encountered in the body.2507
2593 In the 8 September 2000 IFU, this section was changed in two material respects: to limit the description of the “material” to Prolene sutures (by substituting “Prolene” for “[t]his material”) and to remove the references to the mesh having high burst and tensile strength. In the 29 November 2010 IFU the section was amended again by deleting the final sentence.
2594 The respondents adduced no evidence to explain these changes. Nor was the subject explored in cross-examination of Dr Hinoul. The most that can be said about them in the circumstances is that they were implicit acknowledgments that the earlier description was misleading, if not false, in these particular respects.
2595 The Medscand IFU also included an “Actions” section which included a statement that animal studies showed that implantation of Prolene mesh elicits “a minimal inflammatory reaction in tissues, which is transient …”
2596 The composition of Prolene has not altered since the 1960s. Yet, when TVT was launched in Australia in 1999, the section on adverse reactions was sparse. It simply read:
Adverse reactions associated with the use of this device include transitory local irritation at the wound site and a transitory foreign body response. Like all foreign bodies Prolene® mesh may potentiate an existing infection. The plastic sheath initially covering the Prolene tape is designed to minimise the risk of contamination and should be removed at the end of the procedure.
2597 As I discuss below, the references “a minimal inflammatory reaction in tissues”, to transitory local irritation at the wound site, and to the possibility of a transitory foreign body response and its consequences were misleading.
2598 These representations, or statements to similar effect, were included in all TVT IFUs until 7 October 2015 when amendments were made after intervention by the Canadian and Australian regulators.2508 Those amendments are discussed below.
2599 Four adverse reactions were included in the 8 September 2000 TVT IFU. These warnings, which featured in many other later IFUs for the SUI devices, were:
• Punctures or lacerations of vessels, nerves, bladder or bowel may occur during needle passage and may require surgical repair.
• Transitory local irritation at the wound site and a transitory foreign body response may occur. This response could result in extrusion, erosion, fistula formation and inflammation.
• As with all foreign bodies, PROLENE mesh may potentiate an existing infection. The plastic sheath initially covering the PROLENE mesh is designed to minimize the risk of contamination.
• Over correction i.e. too much tension applied to the tape, may cause temporary or permanent lower urinary tract obstruction.2509
2600 The following statement was also included:
Prolene® (polypropylene) mesh in contaminated areas should be used with the understanding that subsequent infection may require removal of the material.2510
2601 Yet, in the Surgeon’s Resource Monograph, published in 2001, which carried the Gynecare logo, the following complications were described as “specific complications that may be encountered when utilizing the GYNECARE TVT system”: vaginal bleeding; retropubic haematoma and pain; vaginal perforation during surgery; bladder perforations; inability to void after procedure; injured urethra; urethral erosion; mesh protrusion (or defective healing); vascular injuries; bowel perforations; de novo urge and possibility of post-operative obstruction; infection of the mesh; urinary tract infection; and device failure.2511
2602 This monograph included expert opinions on the use of TVT gathered from a 17-surgeon panel.2512 In his introduction, Dr Carl G Klutke observed, amongst other things that:
Every surgical procedure has unique risks and complications. An operation that is new and different needs to have a safety record. So far, the total published rate of complications using the GYNECARE TVT device has been minimal. Precise adherence to the procedure described by Ulmsten et al minimizes complications, but deviation from the technique or inexperience with it may lead to severe complications.
2603 The IFU for TVT at that time did not suggest that severe complications could arise from the use of TVT, whether as a result of deviation from the technique or inexperience or otherwise. Nor did it mention several of the “specific complications” mentioned in the Surgeon’s Resource Monograph. And the monograph was not supplied with the device.
TVT-O
2604 Seven versions of the IFU were supplied with TVT-O over the years, but only three of them included relevant revisions.
2605 The text in the original version of the TVT-O IFU, used from 7 January 2004, was very similar to the text used in the IFU supplied with TVT at the time. The only significant difference was in the warnings and precautions section. For some undisclosed reason, unlike the TVT IFU, the TVT-O IFU did not include the warning that using mesh in contaminated areas carries the risk of subsequent infection which may require removal of the material. It also contained two statements that were not in the TVT IFU:
• Transient leg pain lasting 24-48 hours may occur and can usually be managed with mild analgesics.
…
• Prophylactic antibiotics can be administered according to the surgeon’s usual practice.2513
2606 During July 2003 there was much discussion in the marketing team and with Drs Weisberg and Arnaud about whether a warning should be given in the IFU about the risk of leg pain. Initially, Daniel Smith of Gynecare R&D considered no warning should be given. He revised his position after hearing from Dr Weisberg, who, on 23 July 2003, proposed a warning in these terms:
Transient (up to 48 hours) leg or groin pain has been noted in some patients. This usually resolves with mild analgesics for non-steroidal anti-inflammatory medications.2514
2607 In an email sent on 29 July 2003, Brian Luscombe, Ethicon Inc.’s US Product Director, Female Pelvic Medicine & Reconstructive Surgery, expressed his concerns about the high incidence of transient leg pain reported in “15–25%” of Professor de Leval’s patients. Since this was not an apparent complication of the AMS Monarc device, he wrote that he thought it was necessary to include a warning about it, but expressed concern that it would put Ethicon at a competitive disadvantage to AMS.2515
2608 On 8 August 2003, Zenobia Walji, then Director, Marketing for Gynecare, counselled, in an email, of the necessity to thoroughly understand the cause and effect of leg pain and to see if it could be avoided by changing the technique and “fine tuning” the instructions for use. If not, she said that design changes needed to be considered. If this were an inherent risk of obturator techniques, she added, “then our focus would be not to ‘improve competitiveness’, but rather inform physicians of these inherent risks”. Dr Arnaud’s response was to minimise the significance of the problem and, inferentially, to counsel against providing a warning. He wrote that Professor de Leval did “not consider post-op pain as a relevant clinical issue with his procedure and I do not think we should challenge that at this point (i.e. accept it is “truly abnormal pain which can be called a complication”).2516
2609 As with the TVT IFU, significant changes were made in the last iteration of the TVT-O IFU in evidence, supplied from 22 September 2015,2517 in response to correspondence from the Canadian and Australian regulators. These changes were made to the adverse reactions and the warnings and precautions sections and were identical to the changes to the IFU for TVT.
TVT Secur
2610 This device was only on the market in Australia for less than a year (between April 2007 and March 2008). The text used in the warnings and precautions sections of the IFU was not materially different from that used in the IFUs for TVT and TVT-O at the time, except in two respects.
2611 First, the introductory information section did not include any recommendation that the device be used only by physicians specifically trained in implanting it, although such a recommendation was included both in the 22 December 2003 TVT IFU and the 7 January 2004 TVT-O IFU. The omission is surprising and it was unexplained.
2612 Second, the adverse reactions section contained the following additional bullet point which is not found in any of the IFUs for the other SUI devices:
Under-correction or incorrect placement may result in incomplete or no relief from urinary incontinence.
2613 The 2005 CER for TVT Secur listed the following possible complications as anticipated events, although many of them were not included in the IFU:
Punctures or lacerations of vessels, nerves, bladder, urethra or bowel;
Inflammation or infection at the surgical site
Urinary tract infection
Abnormal postoperative bleeding
Dysuria
Pain
Haematuria
Pain
De novo detrusor instability or urgency
Intra-operative damage to the mesh
Irritation at the wound site
Foreign body response
Mesh exposure
Mesh erosion
Fistula formation
Urethral obstruction due to over-correction resulting in urinary retention
Hematoma
Venous thrombosis
Abscess formation
Reaction to anaesthesia
Death 2518
TVT Exact
2614 It will be recalled that TVT Exact was introduced to the Australian market in July 2010.
2615 Over time, five versions of the IFU were supplied with the device. The third version, in use from 23 October 2013 until 26 November 2014, overlapped for a couple of months with the fourth version, which was in use from 12 August 2014 until 9 September 2015. As with TVT-O, only three included any relevant changes.
2616 The only significant difference between the first IFU introduced with TVT Exact and that which was supplied with TVT and TVT-O was the following additional sentence included in the introductory information section:
This is not a technique manual nor a substitute for appropriate training and experience in surgical technique for correcting Stress Urinary Incontinence.2519
2617 This sentence was also not included in the IFU for the TVT Abbrevo.
TVT Abbrevo
2618 TVT Abbrevo was introduced to the Australian market in October 2010.
2619 Three versions of the TVT Abbrevo IFU were in evidence, two of which are relevant, with the latest mirroring the changes made to the IFUs of the other SUI devices in response to the regulators’ criticisms. The first version was in use from 10 September 2010 until 27 November 2014, the second from 1 July until 15 September 2015, and the third, from 24 September 2015.
2620 The IFUs for TVT Abbrevo contained one warning which was not included in any of the IFUs for the other SUI devices:
Users familiar with other SUI devices and procedures should carefully follow the instructions for this device.2520
2621 Otherwise, the IFUs for TVT Abbrevo contained the same text as the IFUs for the other SUI devices, including the two additional warnings about transient leg pain and prophylactic antibiotics that were included in each version of the TVT-O IFU. As with the TVT-O IFUs, the warnings and precautions section did not include the warning contained in the TVT IFU that using mesh in contaminated areas can result in subsequent infection which may require removal of the material.
2622 Dr Pence considered the IFUs of the SUI devices as a group, treating them as “the TVT family of products”. That was a reasonable approach considering the similarities in the various IFUs, although, as must be apparent from the above analysis, I am mindful of the differences where they occur. Based on the potential complications that she deduced the respondents knew or ought to have known, Dr Pence reported that the following complications were missing from the IFUs until 2015, at which time some, but not all, were added:
• Pain, including leg, thigh, or gluteal pain and chronic pain
• Infection (Only “may potentiate an existing infection” was included in IFU.)
• Seroma
• Adhesion formation
• Hematoma
• Hemorrhage (Note that the Warnings and Precautions, however, include that “[r]etropubic bleeding may occur postoperatively” and that if bleeding should occur, “the patient is instructed to contact the surgeon immediately”.)
• Vaginal bleeding
• Urinary problems, including:
• Voiding dysfunction
• De novo detrusor instability or urgency, transient or persistent (Note that in the IFUs in-use from 12/22/2003, the Warnings and Precautions included that “de novo detrusor instability may occur following the TVT procedure” or other TVT family of products procedures.)
• Urinary retention
• Urinary tract infection
• Dysuria (Note that the Warnings and Precautions include that if dysuria should occur, “the patient is instructed to contact the surgeon immediately”.)
• Worsening or recurrence of incontinence/device failure
• De novo dyspareunia
• Pain experienced by partner during intercourse, impaired physical relationship
• Chronic foreign body reaction (Note that the IFUs indicated “a transitory foreign body response may occur” until the 2015 revisions. While “transitory” has been removed from the 2015 IFUs, these IFUs still fail to advise the surgeon of the chronicity of the foreign body reaction.)
• Delayed/defective healing
• Complications requiring re-surgery, mesh removal, tape adjustment, partial excision
• Urethral injury (TVT and TVT EXACT)
• Vaginal perforation
• Death (Note that the numbers of deaths in the MAUDE database were previously assessed for TVT and TVT-O as part of the expert evidence I provided in vaginal mesh litigation in the United States. Fifteen (15) deaths were reported associated with TVT implantation in the MAUDE database from 1998 to 2010; six deaths were reported in the MAUDE database associated with the TVT-O through 2011.).2521
2623 Dr Pence observed that chronic foreign body reaction, delayed/defective healing, and urinary tract infections were never included as warnings in an IFU. Neither was dysuria, although Dr Pence pointed out that the warnings and precautions section included a warning that if dysuria did occur, “the patient is instructed to contact the surgeon immediately”.
2624 Dr Pence deduced other adverse reactions that were known by, or knowable to, Ethicon were “missing” from the IFUs either before 2015 and/or were never included in any of the IFUs. She provided the following list, with the dates by which they were known, or knowable, in brackets; each of the complications about which there was never a warning was emphasised in bold:
• Recurrent urinary tract infections (2001)
• Hematuria (2001–2005)
• Vaginal discharge (2002)
• Vaginal irritation, irritation at wound site (2002)
• Shrinkage, due to contraction and scarring (2002)
• De novo urge incontinence (2002)
• Worsening of pre-existing urge incontinence (2002)
• Abscess (2005)
• Venous thrombosis (2005)
• Pulmonary embolism (08/2003-01/2006)
• Loss of motor function/numbness (08/2003-01/2006)
• Vaginal scarring, distortion (2006)
(Original emphasis)
2625 Dr Pence expressed the opinion that a reasonable manufacturer of medical devices in the position of Ethicon should have warned of these risks.2522 Not all of the above-listed potential complications, however, found their way into the applicants’ pleadings. The applicants included only chronic pain, for example, and not pain simpliciter or leg pain, and did not mention death. Nor, for example, did they include seroma, adhesion formation, haematoma, or hispareunia.
2626 Dr Pence’s opinion, which largely conforms to, and is consistent with, the regulatory requirements and/or guidelines was that:
Serious complications such as those that may result in a persistent or significant incapacity or have the potential to substantially disrupt a patient’s ability to conduct normal life functions should be described in the Warnings section of the labeling, in addition to those that may require medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure. Other clinically significant complications, e.g., those that occur frequently and have implications for patient management or may lead to a potentially serious outcome, also should be included in the Warnings section. As applicable, the description of a complication in the Warnings section also should include actions to be taken to reduce the likelihood or severity of the event and how to monitor for or manage the event.2523
2627 Based on her analysis of the information Ethicon knew or should have known and her expertise as to the information that should be included in IFUs, Dr Pence concluded that the IFUs for all the SUI devices were, and remained, incomplete and warnings in the following terms or to the following effect should have been given:
• Risk of bladder perforation is increased in patients with a history of prior incontinence surgery; risk of other organ injury is increased in patients with a history of prior surgery(ies).
• Mesh extrusion or erosion may occur and is a persistent or lifelong risk; some will require surgical correction, and multiple surgeries may be necessary.
While the IFU included erosion and extrusion as adverse reactions, there was no warning that such risk was lifelong or that patients could have multiple erosions requiring multiple surgeries, yet this was known.
• There is the potential risk that a patient may experience chronic, unresolvable pain.
• Nerve damage may result in loss of lower extremity motor function or numbness.
• Erosion through the vaginal mucosa may cause irritation to the patient’s intimate partner.
• Risk of erosion or mesh exposure, or defective healing, is increased by intra-operative injury, excessive sling tension, inadequate suturing, premature resumption of intercourse, infection, previous surgery, vaginal atrophy or vaginal injury.
• De novo dyspareunia may occur and be persistent.
• The TVT is intended to be a permanent implant, but complications (e.g., foreign body reaction, infection, and erosion) may require implant removal; complete removal may not be possible, and morbidity associated with explant may be significant. (Note: The 2015 revised IFUs included the following statement under Adverse Reactions: PROLENE Mesh is a permanent implant that integrates into the tissue. In cases in which the PROLENE Mesh needs to be removed in part or whole, significant dissection may be required.)
• Chronic pain may result from foreign body reaction and/or scarring and contraction.
• The extent of contraction or shrinkage is related to the intensity of the foreign body reaction; excessive foreign body reaction results in a massive scar plate and, thus, more shrinkage. There are differences among individual patients regarding the extent of foreign body reaction, i.e., there are “high and low responders”.
• “The formation of scar tissue throughout the mesh causes a contraction within the tissue. Since the mesh is compressible along its length it can be acted on by the tissue.” This is the cause for shrinkage, not that the mesh itself shrinks.”…[S]hrinkage rate is influenced by many parameters as the degree of fibrotic reaction is dependent on the mesh material/weave/width etc.” The “rule of thumb” regarding the amount of shrinkage occurring with the TVT mesh is 30%.
• TVT mesh has been reported to fray, narrow, curl or deform with tension which may lead to erosion, exposure, pain for the patient, and discomfort for the patient’s intimate partner during intercourse. Particles released on fraying have been reported to migrate through the vaginal wall and cause pain and pain during intercourse.
• The polypropylene used in the TVT mesh gave rise to local sarcomas in a study in which it was implanted in laboratory rats.2524
2628 I accept Dr Pence’s evidence that at the time of her report the IFUs for all the SUI devices were deficient. Having regard to the other evidence in this case, and Dr Hinoul’s testimony in particular, which indicated the extent of the respondents’ knowledge, however, I would place no limitation on the times the warnings should have been given. They should have been provided with the devices from the time each of them was made available for sale.
2629 Fourteen IFUs for the POP devices were tendered. Below is an overview of those IFUs during the period from first supply to the end of the trial.
Gynemesh PS
2630 A total of six Gynemesh PS IFUs were in evidence. They were supplied at different times in Australia, with multiple versions being in use over the same periods. The earliest Gynemesh PS IFU was dated 30 March 2003. The third version, in use from 18 December 2008 until 30 November 2010 overlapped with the fourth version, in use from 8 December 2008 until 14 April 2014, the fourth version crossed over with the fifth which was in use from 16 March 2013,2525 and the fifth version crossed over with the sixth which was in use from 3 April 2015.2526 Substantial changes were only made in the final two versions, which were in use until supply was halted in August 2017.
2631 The changes made in the 16 March 2013 IFU mirrored the changes made to the other POP devices in 2009 following requests from the FDA. The 2015 amendments were made in response to correspondence between Ethicon, the TGA and Health Canada about the adequacy of product labelling for the TVT products.
2632 A cautionary introduction was inserted in the 16 March 2013 IFU and its content remained the same in the 3 April 2015 IFU. The text was similar to that used in the Prolift IFUs, advising the user to read all information carefully, warning that a failure to follow instructions properly might lead to device failure and injury. The introduction also included the following two sentences, neither of which appeared in the Prolift IFUs:
GYNECARE GYNEMESH™ is intended for use only by physicians who are trained in the surgical procedures and techniques required for pelvic floor reconstruction (including abdominal sacrocolposuspension/sacrocolpopexy) and the implantation of synthetic meshes. The physician is advised to consult the medical literature regarding techniques, complications, and hazards associated with the intended procedures.
2633 The 30 March 2003 Gynemesh PS IFU2527 mentioned only one contraindication: that the surgeon should be aware that when the mesh material is used in “infants, children, pregnant women or women planning future pregnancies” that the product will not stretch significantly as the patient grows. This section was amended in the 16 March 2013 IFU2528 so as to advise that the product should not be used in such patients, and to include the same three contraindications that had been added to this section in the Prolift IFU four years earlier in 2009 (see below).
2634 The 30 March 2003 Gynemesh PS IFU adverse reactions section read as follows:
Potential adverse reactions are those typically associated with surgically implantable materials, including infection potentiation, inflammation, adhesion formation, fistula formation, erosion, and extrusion. 2529
2635 In the 31 March 2006 Gynemesh PS IFU,2530 the words “and scarring that results in implant contraction” were added, but they disappeared from the 16 March 2013 version. In the 2013 IFU a number of adverse reactions were added. The additions included pain, pelvic pain, and pain during intercourse, but qualified by the phrase “typically associated with pelvic organ prolapse repair procedures”. Other adverse reactions added were haematoma, urinary incontinence, urinary retention or obstruction, voiding dysfunction, wound dehiscence, nerve damage, recurrent prolapse, and mesh exposure and extrusion through the vaginal epithelium.
2636 In the 3 April 2015 Gynemesh PS IFU, significant changes were made to this section in line with changes made to the IFUs for the SUI devices around that time.
Prolift
2637 Prolift received regulatory approval in Australia on 30 March 2005 and was first supplied two months later in June 2005. Four versions of the IFU were in evidence. Since two of those versions did not contain any relevant changes, it is only necessary to refer to the contents of the first Prolift IFU issued on 11 January 20052531 and the amendments effected in the version in use from 1 October 2009.2532 The most significant amendments were made to the contraindications, adverse reactions, and warnings and precautions sections. A new section was also added, entitled regarding clinical performance. These changes were made after an exchange of correspondence between the FDA and Ethicon regarding alleged deficiencies in product labelling for both Prolift and Prolift+M.
2638 The relevant changes are discussed below when I examine the adequacy of the warnings about the pleaded complications.
Prolift+M
2639 There were two different versions of the IFU which were consecutively supplied with Prolift+M. It was common ground that the differences were not material. The first Prolift+M IFU was dated 12 December 2008.
2640 The relevant contents of the Prolift+M IFU were identical to the 1 October 2009 Prolift IFU, except for the clinical performance section, which contained only an abridged version of the section of the Prolift IFU, discussed below.
Prosima
2641 As with Prolift+M, two versions of the IFU for Prosima were in evidence but there was no material difference between them. The first Prosima IFU was dated 19 June 2007.2533
2642 For the most part, the relevant parts of the Prosima IFU were identical to the comparable parts of the 11 January 2005 Prolift IFU. In some sections, however, the Prosima IFU incorporated only some of the amendments that were incorporated into the 1 October 2009 Prolift IFU.
2643 Additions were made to the Prosima IFU which were not included in the IFUs for any of the other POP devices. One such addition was made to the indications section. It related to the support systems that were unique to Prosima. It read:
The Systems provide maintenance of the vaginal canal during the period of healing following surgical repair of vaginal wall prolapse, while supporting the position of the Mesh Implants.2534
2644 Finally, although the Prosima IFUs contained the same performance section as in the Prolift IFUs, there was no “clinical performance” section and no reference to trials which had been undertaken or were under way.
Dr Pence’s assessment
2645 Based on her analysis of the information known by, or knowable to, Ethicon and her expertise with respect to the information that should be included in IFUs, Dr Pence concluded that the warnings in the POP devices were incomplete. She considered that failing to include them meant that surgeons were denied the full scope of the safety information necessary to weigh the potential risks and benefits of implanting the device and to fully inform patients of the potential risks to get their consent. In her opinion, a reasonable manufacturer would have warned surgeons (and hence, patients) of those risks.2535
2646 In the case of Gynemesh PS, she stated that, from the time of the first IFU (in 30 March 2003), or from the time during which the second IFU was in use (from 31 March 2006), Ethicon knew, or should have known, of a number of potential adverse reactions although they were not mentioned in an IFU until either 2013 or 2015, or were never included. 2536
2647 Complications that were not included until 2013 were: pain; haematoma; voiding dysfunction; de novo stress urinary incontinence; urinary retention; de novo dyspareunia; prolapse recurrence/device failure; wound dehiscence; and nerve damage.
2648 Complications that were not included until 2015 were: chronic pain, infection (previously only “infection potentiation”); seroma; haemorrhage; de novo urge incontinence; vaginal discharge; de novo dyspareunia which may not resolve; vaginal tightening or shortening; foreign body reaction; complications requiring one or more surgeries, mesh revision or removal; neuromuscular problems; internal organ damage, perforations; and death.
2649 Complications that were never included were: cellulitis; abscess; urinary tract infection; sexual dysfunction due to vaginal constriction or perineal band; decrease in sexual activity; palpable mesh in vagina; chronic foreign body reaction or chronic inflammation; granuloma; and defaecatory dysfunction.
2650 For the Prolift, Prolift+M and Prosima devices, based on her review of its records, Dr Pence determined that Ethicon knew or should have known of the potential for the following adverse reactions at the time of the first IFUs, although they were not included in any version: chronic pain; infection (only infection potentiation was included); cellulitis; abscess; seroma; haemorrhage (although Dr Pence noted that in the warnings and precautions section of the Prosima IFU, there was a warning that bleeding may occur postoperatively and that the patient should be advised to contact the surgeon immediately if unusual pain, bleeding, or other problems occur); urinary tract infection; vaginal discharge; vaginal tightening, shortening; sexual dysfunction due to vaginal constriction or perineal band; decrease in sexual activity; palpable mesh in vagina; chronic inflammation (only inflammation was mentioned); granuloma; complications requiring one or more surgeries, mesh revision or removal (other than surgery to repair punctures or lacerations during implantation); defaecatory dysfunction; and death.2537
2651 Dr Pence found that the following adverse events were not included in the Prolift IFU until 2009, and were never included in the Prosima IFU, although each was included in the Prolift+M IFU: haematoma; voiding dysfunction; de novo stress urinary incontinence; urinary retention; prolapse recurrence/device failure; and wound dehiscence.2538
2652 Dr Pence observed that the references to pain differed from device to device. The original Prolift IFU in use from January 2005 stated only that “transient leg pain may occur and can usually be managed with mild analgesics”. This was included in the IFU for Prolift+M but not the IFU for Prosima. The Prolift+M and Prosima IFUs also included “pelvic pain and pain with intercourse” but only as potential adverse reactions “typically associated with pelvic organ prolapse repair procedures”. The same statement was added to the Prolift IFU in 2009 together with a general warning about pain as a potential adverse reaction “typically associated with surgery employing implantable materials of this type” which was also included in the Prolift+M IFU. None of the IFUs, however, mentioned chronic pain or pain that might not resolve in time or with treatment.
2653 Dr Pence also identified a number of additional complications associated with each of the POP devices which her review of the documents suggested may not have been known until a later date or for which the precise date was unknown. They were: implant rejection; unintended tissue reaction; and dermal/fascia tissue damage (reported as early as 2005 or 2007); pain with defaecation (reported in 2010); and emotional problems (reported to the FDA based on a review of the MAUDE database from 2008 to 2010).2539
2654 Dr Pence also reviewed the warnings and precautions section of each of the IFUs. She concluded that they, too, were incomplete. She said that warnings in the terms indicated below or to the same effect should have been included in all IFUs either from the date of the first IFU of each product, or as otherwise indicated in parentheses:
• Mesh extrusion or erosion may occur and is a persistent or lifelong risk; some will require surgical correction, and multiple surgeries may be necessary.
• There is the potential risk that a patient may experience chronic, unresolvable pain (by 2006-2007).
• Chronic pain and vaginal tightening and shortening may result from foreign body reaction and/or scarring and contraction.
• The extent of contraction or shrinkage is related to the intensity of the foreign body reaction; excessive foreign body reaction results in a massive scar plate and, thus, more shrinkage. There are differences among individual patients regarding the extent of foreign body reaction, i.e., there are “high and low responders.”
• “The formation of scar tissue throughout the mesh causes a contraction within the tissue. Since the mesh is compressible along its length it can be acted on by the tissue.” This is the cause for shrinkage, not that the mesh itself shrinks. “…[S]hrinkage rate is influenced by many parameters as the degree of fibrotic reaction is dependent on the mesh material/weave/width etc.”
• Sexual dysfunction and resultant decrease in sexual activity may result from mesh contraction and vaginal constriction.
• De novo dyspareunia may occur and be persistent.
• Development of new pelvic symptoms and urinary complications after pelvic floor repair with synthetic mesh can adversely affect quality of life and in some patients may be life-altering. Patients should be counseled carefully about the potential risks versus the benefits of mesh implantation prior to surgery.
• Transvaginal repair with mesh may or may not provide anatomic benefit compared to traditional, non-mesh pelvic organ prolapse repair; anatomic benefit afforded by traditional repair may not result in better symptomatic results (as early as 2005).
• Complications have been shown to be higher with mesh placement compared to traditional non-mesh repair (as early as 2005-2006).
• Implantation of vaginal mesh is permanent. Some complications associated with implanted mesh may require one or more additional surgeries that may or may not correct the complication (by 2004-2006).
• For complications requiring removal of the Mesh Implant (e.g., erosion or exposure, foreign body reaction, inflammation, infection, and fistula formation), complete removal may not be possible, and morbidity associated with explants may be significant.
• The polypropylene used in the TVT mesh gave rise to local sarcomas in a study in which it was implanted in laboratory rats.2540
2655 I accept Dr Pence’s evidence that the IFUs for all the POP devices were, and remained, incomplete at the time of her report. Once again, however, having regard to the other evidence in this case and Dr Hinoul’s testimony in particular, I would place no limitation on the times the warnings should have been given.
Intervention by the regulators
2656 On 31 May 2007 the FDA wrote to Ethicon in response to Ethicon’s § 510(k) pre-market submission for both the Prolift and Prolift+M systems outlining a number of concerns.2541 In a follow-up letter in December 2007, the FDA identified deficiencies in the draft revisions of the IFUs for both devices and sought additions to the IFUs.2542 The FDA informed Ethicon that its draft IFUs did not adequately address issues of “usability and potential adverse events”.
2657 For the Prolift IFU, the FDA asked Ethicon to include under the heading “Clinical Performance Data”:
(1) a statement that, as at December 2007, no prospective, controlled clinical trials had been conducted to evaluate the safety and effectiveness of Prolift as mechanical support or bridging material of the fascial defect in repair of vaginal wall prolapse;
(2) a statement that limited data were available from a prospective, non-randomised, non-controlled observational study using Gynemesh, a surgical mesh made of the same non-absorbable polypropylene as Prolift, which appears to be a reference to the French TVM study;
(3) a statement that, although the mesh used in that study was provided in pre-cut configurations, the insertion tools provided in the Prolift kit were not available;
(4) the inclusion criteria, primary and secondary endpoints;
(5) a description of the study population; and
(6) a summary of the clinical data from both the French and the US studies of the TVM technique.
2658 For both Prolift and Prolift+M IFUs and based on the data from the two TVM studies, the FDA asked Ethicon to make a number of other changes, including by giving textual prominence to all indications, contraindications, warnings, precautions, and adverse reactions, and by adding:
(1) immediately above the “indications” statement, the statement that:
[t]he safety and effectiveness of synthetic mesh or film support in transvaginal surgical procedures to treat pelvic organ prolapse have not been demonstrated in prospective, randomized clinical trials”;
(2) the statement that “[p]hysicians should be trained in use of surgical mesh for treatment of pelvic organ prolapse and in management of complications resulting from such procedures” following the statement “CAUTION: Federal (USA) law restricts this device to sale by or on the order of a physician”;
(3) a statement that the device is contraindicated for implantation in pregnant women and in areas of active and latent infection;
(4) as a contraindication, implantation into pregnant women and implantation into areas with active and latent infection;
(5) the following items to the adverse events section:
(a) haematoma;
(b) urinary incontinence;
(c) urinary retention/obstruction;
(d) void dysfunction;
(e) pain;
(f) infection;
(g) adhesions;
(h) wound dehiscence;
(i) nerve damage;
(j) recurrent prolapse;
(k) contracture; and
(l) procedure failure; and
(6) a warning that surgeons perform cystoscopy to confirm bladder integrity or to detect bladder perforation.
2659 Furthermore, the FDA directed Ethicon to move to the forefront of the IFUs, the contraindications, warnings, precautions, adverse reactions, performance, sterility, disposal, and storage sections. Previously these sections had followed a section on the recommended surgical technique. It also directed Ethicon to highlight the indications, contraindications, warnings, precautions, and adverse reactions by writing them in “prominent text as compared to the rest of the instructions for use”.
2660 The FDA also told Ethicon to develop a patient brochure, to be provided when counselling a patient regarding options for treating pelvic organ prolapse, and told Ethicon what it wanted included in the brochure. In particular, it specified the following matters for inclusion:
(1) explanation of pelvic organ prolapse, including anatomical issues, causes and symptoms;
(2) the range of treatment options, both non-surgical and surgical, including both native tissue and mesh repair;
(3) the risks and benefits of the various treatment options;
(4) a reference to the MAUDE database website;
(5) a statement to the effect that the safety and effectiveness of mesh for vaginal repair have not been proven in randomised, controlled clinical trials; and
(6) instructions for post-operative care.
2661 The FDA noted in the letter that Ethicon had not responded to its request for data to support the statement in the IFUs for Prolift and Prolift+M that “the bi-directional elastic property allows adaption to various stresses encountered in the body”. In the circumstances, it asked Ethicon to remove the statement from its labelling and to submit a revised IFU for review.
2662 Various other matters were also canvassed in the letter.
2663 The FDA directed Ethicon that it was not permitted to market the devices until adequate information had been provided and the FDA had given the company the go-ahead to do so.
2664 Bryan Lisa, Regulatory Affairs Manager at Ethicon, replied to the FDA in February 2008, stating that it had updated the IFU for the Prolift and Prolift+M products to address the FDA’s concerns, at the same time pushing back on some of the FDA’s proposals.2543 Most, but not all, of the FDA’s requirements were met, and Ethicon’s changes were included in the original IFU supplied in Australia with Prolift+M from 12 December 2008, but they were not included in the updated version of the IFU for Prolift until 1 October 2009.
2665 A notable omission from the Prolift IFU was the FDA’s request for the inclusion of a statement about the safety and effectiveness of synthetic mesh or film support in transvaginal surgical procedures to treat pelvic organ prolapse not having been demonstrated in prospective, randomised clinical trials. No such statement appeared anywhere in the IFU. It transpires that Ethicon submitted to the FDA that such a statement was inaccurate and should not be included. In a letter to the FDA in February 2008. Ethicon wrote:
A review of available literature identified several published studies that were randomized, controlled evaluations of transvaginal mesh intended to treat pelvic organ prolapse (Attachment III). Based on this information, we feel that the statement, as written, is not accurate. We also believe that the above statement has a potential impact to payors (sic) in the US and other countries. Therefore, in order to take the least burdensome approach with the labeling, we propose the following statement, which addresses how this device has been cleared to market in the US:
“In the US, substantial equivalence of GYNECARE PROLIFT Pelvic Floor Repair Systems to synthetic mesh with the same indication has been demonstrated through benchtop and pre-clinical testing.”
The statement also is applicable to the GYNECARE PROLIFT+M IFU.2544
(Original emphasis)
2666 Ethicon also argued against including references to two particular studies (published in 2006 and 2007) “when the body of literature available is constantly changing”. It contended that the relevant risk information was captured in the warnings, precautions, and adverse events sections of the IFU and that “a least burdensome approach is to direct surgeons to obtain further information on transvaginal mesh literature through our sales representatives, who can provide lists of available literature that have been screened for fair balance through our internal copy review process”. The FDA appears to have accepted Ethicon’s argument at face value and condoned the inclusion, instead, of the following statement before the indications section in the IFUs for both products:
Additional information on the clinical performance of mesh for pelvic floor repair is available in published literature. Contact your company sales representative for assistance.2545
2667 The FDA’s requirement that Ethicon stipulate that, as at December 2007, no prospective controlled clinical trials had been conducted to evaluate the safety and effectiveness of Prolift for its indicated purpose did not appear under the heading “clinical performance data”, as the FDA had proposed. Rather, it was included in the introductory information.
2668 As I mentioned in Part X above, on 25 March 2014, the Medical Devices Bureau of the Therapeutic Products Directorate of Health Canada, wrote to Johnson & Johnson Medical Products (JJMP), about the SUI and POP devices.2546 In the letter, the Director of the Bureau, Cindy Evans, observed that Health Canada had received “numerous adverse event reports” related to various surgical meshes of the type listed on the company’s licence. At the time, of the Ethicon devices, the licence appears to have covered TVT, TVT-O, TVT Exact, TVT Abbrevo, and Gynemesh PS (transabdominal indication only).2547
2669 The letter stated that information had recently been brought to the attention of Health Canada that the devices listed on the company’s licence might not meet the safety and efficacy requirements of the relevant regulations and sought answers to various questions designed to enable the Minister to determine that question: see Medical Devices Regulations (SOR/98-282) (Can), s 39. The relevant provisions are similar to those contained in the European Directive and the Australian Medical Devices Regulations. Section 10 of the Canadian Regulations, for example, requires that a medical device be designed and manufactured to be safe and, to this end, requires that a manufacturer takes, in particular, reasonable measures to:
(a) identify the risks inherent in the device;
(b) if the risks can be eliminated, eliminate them;
(c) if the risks cannot be eliminated,
(i) reduce the risks to the extent possible,
(ii) provide for protection appropriate to those risks, including the provision of alarms; and
(iii) provide, with the device, information relative to the risks that remain; and
(d) minimize the hazard from potential failures during the projected useful life of the device.
2670 Noting that clinical evidence had demonstrated that the use of surgical mesh for the treatment of stress urinary incontinence has the potential to lead to certain adverse events, Health Canada asked JJMP for a copy of the labelling for the devices demonstrating that it contained up-to-date information on the potential complications including but not limited to those adverse events and, in the event that any had been omitted, directed JJM to revise the labelling to include them. Those adverse events included “acute or chronic pain in the groin, thigh, pelvic and/or abdominal area; mesh extrusion, exposure or erosion; infection; voiding dysfunction; dyspareunia; neuromuscular damage; organ perforation; recurrence of incontinence; bleeding or hemorrhage; vaginal scarring and/or tightening; and mesh contraction and shrinkage”. “In addition”, the letter noted, “one or more revision surgeries may be necessary to treat these complications, while some complications may not always be completely corrected”.2548 As I read the letter, the direction to amend the labelling was intended to capture this information as well.
2671 On 16 April 2014, Stacy Kluesner, Ethicon’s Regulatory Affairs Manager, forwarded to the medical writer a “first draft response” to Health Canada addressing only the adverse events in the current labelling and the changes Ethicon proposed to make.2549 Notably, she proposed adding to the adverse reactions in the IFUs only the following:
“pain”, rather than “acute or chronic pain in the groin, thigh, pelvic and/or abdominal area”, noting that “it encompasses both acute and chronic pain and does not restrict pain to only certain areas”;
voiding dysfunction;
dyspareunia; and
recurrence of incontinence.2550
2672 Ms Kluesner queried whether nerve damage should be included in the IFU to be consistent with the POP devices. She said that Ethicon believed that “vaginal scarring and/or tightening” and “mesh contraction and shrinkage” were not applicable to TVT but floated the idea of including the former anyway in order to placate the regulator.
2673 Ethicon’s response to Health Canada was sent on 20 May 2014.2551
2674 Ethicon purported to have undertaken a comprehensive review of its product labelling. In its response to Health Canada, it provided comparative tables between its labelling at the time and the changes proposed by Health Canada. The following table (extracted in full) compared Health Canada’s list of adverse events to the information in the instructions for use supplied with the relevant SUI devices:2552
TABLE 2: Comparison of Health Canada’s Adverse Events to TVT Labelling. | ||
Health Canada Request | Current TVT Labelling | Action to Be Taken |
acute or chronic pain in the groin, thigh, pelvic and/or abdominal area | Not included | Add under the “ADVERSE REACTION” section: Acute and/or chronic pain will be added under the “ADVERSE REACTION” section. Please also see Ethicon’s response to “neuromuscular damage” below. |
mesh extrusion, exposure or erosion; | Similar language is currently included: ADVERSE REACTION Transitory local irritation at the wound site and a transitory foreign body response may occur. This response could result in the extrusion, erosion, fistula formation and inflammation. | Modify the “ADVERSE REACTION” Section as follows: Mesh extrusion, exposure, or erosion into the vagina or other structures or organs. As with any implant, a foreign body response may occur. This response could result in extrusion, erosion, exposure, fistula formation and/or inflammation. |
Infection | Currently included in the instructions for use as follows: WARNINGS AND PRECAUTIONS Do not use on GYNECARE TVT procedure for patients who have a urinary tract infection. PROLENE Mesh in contaminated areas should be used with the understanding that subsequent infection may require removal of the material. ADVERSE REACTION As with all foreign bodies, PROLENE Mesh may potentiate an existing infection. | Modify the “ADVERSE REACTION” Section as follows: As with all surgical procedures, there is a risk of infection. As with all foreign bodies, PROLENE Mesh may potentiate an existing infection. Modify the WARNINGS AND PRECAUTIONS Section as follows: Do not use GYNECARE TVT in a patient who has a urinary tract infection. |
voiding dysfunction | Not included | Add under the “ADVERSE REACTION” section: • Voiding dysfunction |
Dyspareunia | Not included | Add under the “ADVERSE REACTION” section: • Pain with intercourse which in some patients may not resolve. |
neuromuscular damage | Information is currently included in the instructions for use as follows: ADVERSE REACTION • Punctures or lacerations of vessels, nerves, bladder or bowel may occur during needle passage and may require surgical repair. | Add under the “ADVERSE REACTION” section: • Neuromuscular problems, including acute and/or chronic pain in the groin, thigh, leg, pelvic and/or abdominal area |
organ perforation | Information is currently included in the instructions for use regarding the risk of organ perforation during the procedure as follows: WARNINGS AND PRECAUTIONS or ADVERSE REACTION • Cystoscopy should be performed to confirm bladder integrity or recognize a bladder perforation. • Punctures or lacerations of vessels, nerves, bladder or bowel may occur during needle passage and may require surgical repair. | Add under the “ADVERSE REACTION” section: • Punctures or lacerations of vessels, nerves, structures or organs, including the bladder, urethra or bowel, may occur and may require surgical repair. |
recurrence of incontinence | Not included | Add under the “ADVERSE REACTION” section: • Recurrence of incontinence |
bleeding or hemorrhage | Similar information is currently included in the instructions for use regarding the risk as follows: WARNINGS AND PRECAUTIONS Bleeding associated with the procedure is included under the “Warnings” section as follows: • Retropubic bleeding may occur post-operatively. • Or Bleeding may occur post-operatively • Should dysuria, bleeding or other problems occur, the patient is instructed to contact the surgeon immediately. | Add under the “ADVERSE REACTION” section: • Bleeding including hemorrhage, or hematoma. |
vaginal scarring and/or tightening | Not included | None - Ethicon believes that this adverse event is not applicable to TVT. The only vaginal scarring would be that which is consistent with any small incision. |
mesh contraction and shrinkage | Not included | Add under the “ADVERSE REACTION” section: • Excessive contraction or shrinkage of the tissue surrounding the mesh Justification for different language: The mesh does not contract or shrink. |
one or more revision surgeries may be necessary to treat these complications, while some complications may not always be completely corrected. | Not included | Add under the “ADVERSE REACTION” section: • One or more revision surgeries may be necessary to treat these adverse reactions. • PROLENE mesh is a permanent implant which integrates into the tissue. In cases where the PROLENE mesh needs to be removed in part or whole, significant dissection may be required. |
2675 The following comparative table was provided in relation to Gynemesh PS:2553
TABLE 10: Comparison of Health Canada’s Adverse Events to Gynemesh PS Labelling. | ||
Health Canada Request | Current Gynemesh PS Labelling | Action to Be Taken |
mesh extrusion, exposure or erosion; | Included under the “ADVERSE REACTION” section. | Modify the “ADVERSE REACTION” Section as follows: • Mesh extrusion, exposure, or erosion into the vagina or other structures or organs. • As with any implant, a foreign body response may occur. This response could result in extrusion, erosion, exposure, fistula formation and/or inflammation. |
acute or chronic pain | Similar information is currently included in the instructions for use. “Pain,” “pelvic pain” and “pain with intercourse” are included under the “ADVERSE REACTION section of the instructions for use. | Add under the “ADVERSE REACTION” section: • Acute and/or chronic pain Please also see Ethicon’s response to “neuromuscular damage” below. |
Dyspareunia | Similar information is currently included in the instructions for use under the “ADVERSE REACTION” section as “pain with intercourse,” which has the same meaning as dyspareunia. | Add under the “ADVERSE REACTION” section: • Pain with intercourse which in some patients may not resolve. |
mesh contracture and its sequelae (including vaginal tightening and/or shortening) | Similar information is currently included in the instructions for use under the “ADVERSE REACTION” section as “contracture.” | Add under the “ADVERSE REACTION” section: • Excessive contraction or shrinkage of the tissue surrounding the mesh, vaginal scarring, tightening and/or shortening. Justification for different language: The mesh does not contract or shrink. |
vaginal scarring | Similar information is currently included in the instructions for use under the “ADVERSE REACTION” section as “scarring.” | Will be added under “ADVERSE REACTION” Section. See above row for wording. |
Infection | Similar information is currently included in the instructions for use. Risk of infection associated with its use in contaminated areas is described under “PRECAUTIONS.” “Infection potentiation” is included under the “ADVERSE REACTION” section. | Modify the “ADVERSE REACTION” Section as follows: • As with all surgical procedures, there is a risk of infection. As with all foreign bodies, PROLENE Soft Mesh may potentiate an existing infection. |
organ perforation | Similar information is currently included in the instructions for use regarding the risk of organ perforation during the procedure as follows: WARNINGS • A digital rectal examination may be performed to detect possible rectal perforation. • Cystoscopy may be performed to confirm bladder integrity or to detect possible bladder or ureteral perforation. | Add under the “ADVERSE REACTION” section: • Punctures or lacerations of vessels, nerves, structures or organs, including the bladder, urethra or bowel, may occur and may require surgical repair. |
neuromuscular damage | Similar information is currently included in the instructions for use under the “ADVERSE REACTION” section as “nerve damage.” | Add under the “ADVERSE REACTION” section: • Neuromuscular problems, including acute and/or chronic pain in the groin, thigh, leg, pelvic and/or abdominal area |
bleeding or hemorrhage | Similar information is currently included in the instructions for use under the “ADVERSE REACTION” section as “Hematoma.” | Add under the “ADVERSE REACTION” section: Bleeding including hemorrhage, or hematoma. |
recurrence of prolapse | Similar information is currently included in the instructions for use under the “ADVERSE REACTION” section as “recurrent prolapse.” | None |
defecatory dysfunction | Not included | None. Ethicon has performed a detailed analysis and review regarding this adverse event in association with Gynemesh PS as a result of this inquiry. We have reviewed our complaints from January 2007 until January 2013. During this time, there was only one complaint in which a defecatory problem was alleged. With this one complaint, the patient had multiple devices implanted in addition to Gynemesh PS. This complaint was inclusive and no detailed medical information was provided. In addition, Ethicon’s Clinical Evaluation Report (CER) was reviewed for to see if this adverse event had been reported in the literature for Gynemesh PS. This report captured published literature on Gynemesh PS from January 2010 through January 2013. There was no information within the CER that suggested defecatory dysfunction was an adverse event associated with Gynemesh PS. |
voiding dysfunction | Included in the instructions for use under the “ADVERSE REACTION” section. | None |
other urinary problems | Included in the instructions for use under the “ADVERSE REACTION” section as “urinary incontinence, urinary retention or obstruction.” | Modify the “ADVERSE REACTION” Section as follows: “urinary incontinence, urge incontinence, urinary frequency, urinary retention or obstruction.” |
one or more revision surgeries may be necessary to treat these complications, while some complications may not always be completely corrected. | Not included | Add under the “ADVERSE REACTION” section: • One or more revision surgeries may be necessary to treat these adverse reactions. • PROLENE Soft mesh is a permanent implant which integrates into the tissue. In cases where the PROLENE Soft mesh needs to be removed in part or whole, significant dissection may be required. |
2676 IFUs were provided to the TGA during a review of urogynaecological mesh devices which began in October 2012. In a clinical evidence assessment dated 28 April 2014, the TGA noted that several adverse events, such as haemorrhage, haematoma, urinary retention and urinary tract infection were not mentioned in the IFUs and that “important adverse events… including de novo detrusor instability” were also missing and leg pain was not included in the TVT-O IFU.2554
2677 On 17 October 2014 the TGA asked JJM to make substantial amendments to its IFUs. This request was made to JJM as the Australian sponsor. It was limited to Gynemesh PS and some SUI devices because they were the only devices remaining on the market the time. The request sought the following revisions of the IFUs:
1) The risk of erosion associated with this type of device must be clearly discussed in a prominent section.
a. Currently, this adverse event is part of a large paragraph of adverse events. The nature of this particular adverse event is such that it should be discussed separately to enhance the safety of the device. It is appropriate to phrase this in general terms such as, “urogynaecological polypropylene mesh devices have been associated with cases of erosion”.
b. This specific section on the risk of erosion should include a comment regarding patient selection, including factors that may increase the likelihood of complications and adverse events including erosion for this type of device in particular patients such as:
1. obesity,
2. smoking,
3. age,
4. use of transvaginal approach, and
5. any other risk factors identified in the field experience and clinical literature (some of which are identified in the attached reports).
2) A complete description must be included in the IFU of all known complications for this type of device including:
a. ongoing pain (specifically noting that ongoing pain may not be resolved on explant)
b. other complications identified in the clinical literature, risk assessment documentation, and post-market data that have been submitted to the TGA in relation to each device. Please see the attached clinical evaluation reports for further details.
Where risks identified in the materials submitted for this review have not been included in the IFU, compelling justification for this decision must be provided to the TGA.2555
2678 The TGA additionally requested that, as soon as possible, the surgical guides and other information and training materials be changed to address these issues.
2679 The TGA informed JJM that the deficiencies it had identified were considered to be breaches of compliance with one or more of the essential principles set out in Sch 1 of the Medical Devices Regulations.
2680 In November 2014, JJM replied to the TGA request, proposing the same changes to its labelling as were provided to Health Canada.2556 The TGA approved the proposal and ordered that “distribution of the new IFU should now be subject of Recall for Product Correction”.2557 JJM obliged and issued an urgent notice of recall for product correction on 3 September 2015.2558
2681 In July 2017, the TGA wrote to JJM notifying the company that a new condition would be imposed on the SUI devices requiring further warnings to be added to each product’s IFU2559 and, on 17 January 2018, a hold was put on the supply of each of the TVT devices in Australia (except for TVT Secur which had already been withdrawn from the market), after the TGA included the following condition on their registration:
It will be a condition of inclusion of the Entry in the Register that the instructions for use documents (IFU) clearly indicate in the “warnings and precautions” and/or “adverse reactions” sections that the use of these devices may result in:
• Bladder perforation
• Severe chronic pain
• Groin pain.2560
2682 Surgeon training in the new TVT technique was not introduced, it seems, until after TVT had been on the market for some time. In recognition of the risk of severe complications with inexperience or deviation from the Ulmsten technique, a summit meeting was held in June 2000 with the object of producing a resource monograph. The Surgeon’s Resource Monograph which was published the following year distilled the experience of a number of “proctors” of the TVT system for the purpose of training surgeons “in a safe, reproducible and standard way”.2561 Although the monograph carried the Gynecare logo, it also contained a warning that the views expressed in it do not necessarily represent the views of Ethicon and in closing submissions Mr Finch SC told the Court that it was not produced by the respondents.2562
2683 The monograph described the surgical procedure in some detail. As the respondents submitted,2563 it also addressed the panel’s experiences with patient selection and surgical procedure as well as a number of complications, providing opinions as to their likely causes and recommendations as to how to treat them and avoid them. The complications went beyond those disclosed by the IFU issued with the TVT system in 1999. They included urethral erosion, mesh protrusion (or defective healing), vascular injuries, bowel perforations, de novo urge incontinence, infection of the mesh, and urinary tract infection. The monograph also included tabulations of informal and published data on device efficacy and complications.
2684 A second monograph was produced in 2002 directed to surgery involving TVT using an abdominal retropubic approach, acknowledging the collective experiences of a number of American physicians who participated in “post-marketing clinical evaluation” of the TVT system.2564 It, too, provided instructions about the procedure and advice on the use of the tools. And it included a discussion of the history of TVT, a review of the clinical data derived from the medical literature, and a report of a very recent multi-centre study of 75 patients with a description of intraoperative and post-operative complications.
2685 There is no evidence, however, of the distribution of these monographs and importantly, no evidence to indicate that they made their way to Australia.2565
2686 A monograph of the same type was produced in 2007 about Prolift.2566 Dr Hinoul deposed that this monograph was distributed at “professional education events, at medical conferences and through sales representatives”.2567 But at which events or in which countries he did not say.
2687 JJM offered a professional education program in Australia from the early 2000s. As the Prolift IFU advised that training in the use of the device was “recommended and available” and encouraged users to contact the company sales representative to arrange it, Dr Hinoul deposed that the program was designed to supplement the information contained in the IFUs.2568 The program deployed “preceptors”. A preceptor was a person identified by JJM as a “key opinion leader” and specialist pelvic floor surgeon with experience in treating women with stress urinary incontinence and pelvic organ prolapse. In around 2005 and 2006 JJM identified surgeons who could either be trained or who could train other surgeons on using the devices.2569
2688 In around 2008 the professional education program became known as “GUARD”, standing for “Gynaecology and Urology – Applying Recent Developments”. The GUARD program was split into three stages: 1) preceptor training; 2) proctorship; and 3) ongoing support.2570 Stage one, “preceptor training”, involved spending a day with an experienced surgeon who would provide “hands-on” training, including observation and assistance with two to three procedures. During this stage, participants were provided with materials made up of procedural DVDs, IFUs, surgical technique or procedure guides, and published clinical papers. Further material was provided to participants at various intervals prior to and following this practical training stage. Stage two was “proctorship”, where the preceptor would attend the participant’s hospital and supervise the first three cases performed by the participant. It was supposed to take place within 6 to 8 weeks after the preceptor training stage. During stage three, the preceptor surgeon would provide remote support for the participant via telephone or email.2571
2689 A list of training materials for TVT, TVT-O, TVT Abbrevo, Prolift and Prosima appeared in JJM’s response to the TGA request for information dated 22 November 2012.2572 The author of the response was Susanne Winter, Regulatory Affairs Associate for JJM. Ms Winter told the TGA that similar training was provided for Gynemesh PS at the time the product was launched but was no longer provided.
2690 Neither Surgeon’s Resource Monograph appeared in the list of training materials for TVT. No clinical papers were supplied with the Prolift training material, although participants in the training program were provided with a Prolift brochure. Brochures on Prosima and TVT Abbrevo were also supplied to participants attending training in the use of those devices.2573 The training materials mentioned in Ms Winter’s letter were not tendered, although some of them, such as the IFUs, were already in evidence and it is possible that the Prolift and Prosima brochures were too. I was informed by counsel for the respondents, however, during closing argument that, of that material, only the IFUs included any warnings.2574
2691 Evidence was given by several urogynaecologists about the JJM training program. Professors Korda and Collinet, as well as Dr Hinoul, described their experience as preceptors, while Dr Chughtai spoke briefly about his experience attending training sessions. The regulatory experts also gave their opinions on the adequacy of the respondents’ training in meeting the regulatory requirements.
2692 The training was problematic, as JJM itself recognised, at least with respect to Prolift and TVT-O.2575 Jan Jackson, a JJM employee, had been complaining for months about a lack of available doctors to provide training on Prolift or TVT-O before a discussion was scheduled in late February 2006. It appears that the problem affected regional Victoria, including Bendigo, Mildura, and Wangaratta, where the “bigger users” were situated. But it is not clear whether the problem was confined to those localities. In an email sent to Jonathan Meek on 23 February 2006, Ms Jackson reported that training had been an issue with TVT-O since June 2005. From the tenor of Ms Jackson’s email it would appear that, like Mr Meek, she was part of the JJM marketing team. One of her concerns was that she did not have surgeons from the second half of 2005 “to grow [her] business in TVT O in the first half of 2006”.
2693 Regarding training for Prolift, Ms Jackson reported:
We do not have anyone in Victoria that has adopted Prolift in Victoria to the point that they could train anyone new. I think our launch in June having six surgeons all getting a fairly weak exposure to Prolift left Vic with no one being confident enough to do the procedure let alone teach the procedure. Anna Rosamilia from the ones present at the launch has done probably six cases but is still struggling with how to do the procedure with ease so she is still a long way off being able to teach. We offered her a couple of Monday dates from Judith Goh who could come to Melbourne but she could not manage the Monday to get a list. We need to talk about the possibility of having someone here to ensure she can do the procedure with ease.
2694 Associate Professor Rosamilia explained in cross-examination that she found the Prolift procedure to be quite difficult2576 with the particular difficulty being the “external skin incisions with blind trocar passage” which “added an extra risk in terms of potential for bleeding or injury to the bladder or bowel”.2577 She also commented that it was very hard to get the mesh to sit flat without folding, which was a problem she saw with complications that arose later where the mesh had been implanted by a “very inexperienced user”.2578 She agreed that the difficulties she identified made it hard for all gynaecologists and urologists to reproduce the Prolift procedure safely and effectively. She said that it was her view that Prolift surgery should have been restricted for use in specialist centres where surgeons experienced in pelvic floor surgery could perform the operation, although she had not expressed that view in 2005.2579
2695 Professor Korda was engaged by JJM in Australia as a preceptor for TVT and TVT-O from 2001 onwards. He described the preceptoring process as a way of mentoring other surgeons by teaching them how to perform operations.2580
2696 Professor Korda explained that, when he was a preceptor, he would be sent to a hospital to teach someone the procedure and two or three cases would be selected for this purpose. He said that the evidence available today shows that in order to gain the necessary competence surgeons should be performing at least 20 before performing the operations.2581 But it has always been up to the particular surgeon to decide his or her own competence before beginning to perform these operations independently.2582
2697 Professor Korda said that transvaginal mesh implant surgery to treat prolapse is a difficult and complex surgery that should only be performed by experienced surgeons who have received training and, preferably, accreditation by the institution where they conduct the operations.2583 He said that the message preceptors gave for mesh surgery for prolapse repair “was focused on the ease of surgery and the good anatomical results” and the risks were either not mentioned or underestimated.2584 The inference is open that the preceptors were used, whether wittingly or otherwise, as vehicles to promote the respondents’ products.
2698 In cross-examination Professor Korda spoke candidly about his own experience as a preceptor for TVT:
KORDA: [W]e now know what ideal preceptorship should be and I can tell you that that was not ideal preceptorship …
FINCH SC: We can forgive you for not knowing in 2001 what we now know[.]
KORDA: Exactly. What was involved in those days is that you – I mean, I can tell you how I was taught how to do it. I mean, I went to a preceptorship run by Dr Bernard Haylen at the Mater Hospital and I watched him do one case, and assisted him with another and then he assisted me with a third and that was my preceptorship and from then on I was doing these procedures. Mind you, by that stage I was very experienced pelvic surgeon who did a lot of colposuspension so the area where the TVT was inserted was well known to me. So I could see the potential problems and dangers. And to a large extent these sort of preceptorships were the same. I went to Adelaide to teach John Svigos on how to do this procedure. The manufacturer paid for my airfare, my hotel accommodation and took me to a very nice restaurant, we had a very nice bottle of Grange and I enjoyed my preceptorship as a preceptor very much.2585
2699 Between 2006 and 2012 Professor Collinet was engaged by Ethicon to take part in and lead training sessions on the technique for implanting Prolift in various countries in Europe as well as in Japan and South Africa.2586 Generally speaking that involved Professor Collinet demonstrating the technique in one case or assisting the surgeon.2587
2700 Professor Chughtai gave evidence about attending training sessions offered by Ethicon in relation to the POP devices, which he described as either a half or full day of lectures followed by a cadaver lab to trial the product. He stated further that:
Every training event that I had attended did speak about the role of appropriate tensioning and dissection. Both these parameters did vary among the proctors teaching the courses. This would highlight the difficulty in achieving a reproducible and standardized technique. Attempts were also made to demonstrate the appropriate dissection and tensioning on cadavers. It must be noted that cadaver dissection and tensioning is quite different and not necessarily applicable on patients, thus not necessarily reducing the learning curve.
…
These training sessions were inadequate to take a novice, competent urologist or gynaecologist to performing mesh implant surgery. Although, if these surgeons (course attendants) were all experts in native tissue repairs and had familiarity with pelvic anatomy from a transvaginal approach, even then these sessions would not suffice to provide adequate training alone. Mesh implants require precision in both tensioning and position of implant, further they also require a full thickness dissection to allow for safe placement, Thus a full or half day course would not be sufficient to train even an expert surgeon without previous mesh implant experience.2588
2701 Ms Holland’s opinion was that the respondents’ approach to training for Prolift was “not sufficient to obtain the desired level of reliable training outcomes”. Nor, she considered, did it reflect the critical training program that was planned by the design team and defined in the Device Design Safety Assessment for Prolift as part of the device’s risk analysis process.2589
2702 Dr Allman said that it was not clear from the evidence before him that the respondents did what was necessary to make sure their training programs worked and that training was provided to everybody who needed the training.2590 Ms Jackson’s email indicates that it is unlikely that they did so, certainly for Prolift and TVT-O.
2703 In its 2014 media release following its review of urogynaecological surgical mesh implants in Australia, the TGA identified “inadequate training/experience for implanting surgeons as a factor in increasing the risk of complications”.2591
2704 Ms Holland wrote in her first report:
In my experience with both implantable and non-implantable devices that may have complex installation and/or user interface processes, passive training such as Ethicon’s approach is not sufficient to obtain the desired level of reliable training outcomes. Although surgeons are generally knowledgeable in their field, specifics relating to use of the device improve the probability of success. I believe making it necessary for the physician or his/her staff to actively search out the corporate sales representative, obtain the surgical technique, and go through the nuances of the surgical technique prior to use is in error and the training often does not occur or does not occur to the degree needed to achieve consistent positive results.2592
2705 On the whole of the evidence and in the absence of evidence to indicate that Ms Jackson’s experience was isolated, I infer that the respondents did not provide training to all surgeons who used the devices or take reasonable steps to ensure that the training they did provide adequately equipped those who received it with the skills and information to conduct the operations with minimal risk of injury.
2706 The devices were promoted in various ways, including through the activities of sales representatives, training of surgeons, presentations to users and potential users, and product brochures. To some extent they were also promoted through the publication of articles relating to Ethicon-sponsored studies. In this section of the judgment I examine the principal methods of marketing as revealed by the evidence.
2707 This evidence is important, not least because, for the purpose of determining whether the Ethicon devices are defective, it is a mandatory consideration.
2708 Ethicon classified surgeons into four types: 2593 “Community Practitioner”; “Practice Driven”; “Technical Innovator”; and “Clinical Innovator”.2594 In a document entitled “OB/GYN space has clear segmentation requiring targeted tactics”, Ethicon offered the following descriptions:
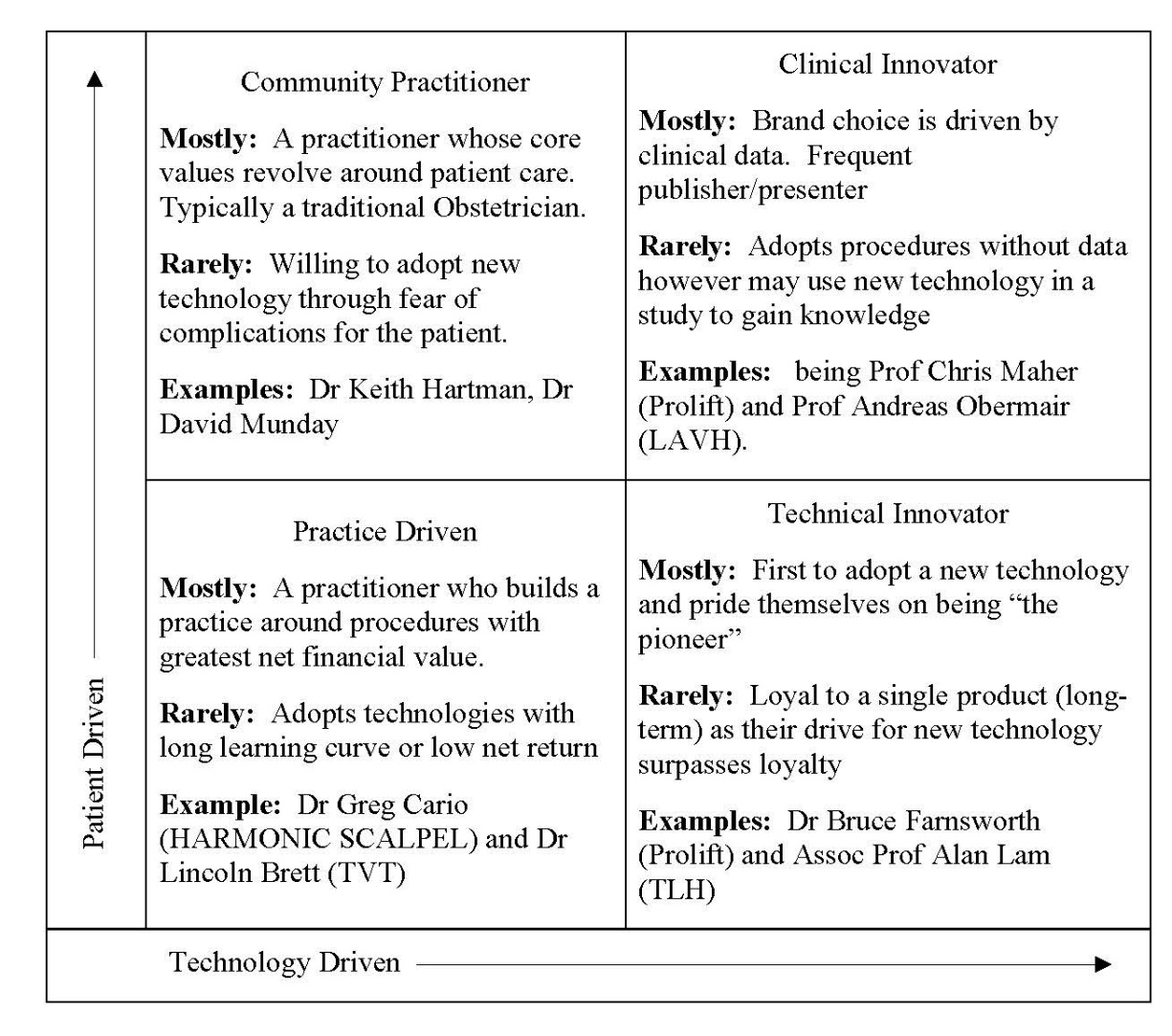
2709 The second page of this document sets out the tactical execution of the strategy proposed for each type with a cascading “influence flow” as follows:
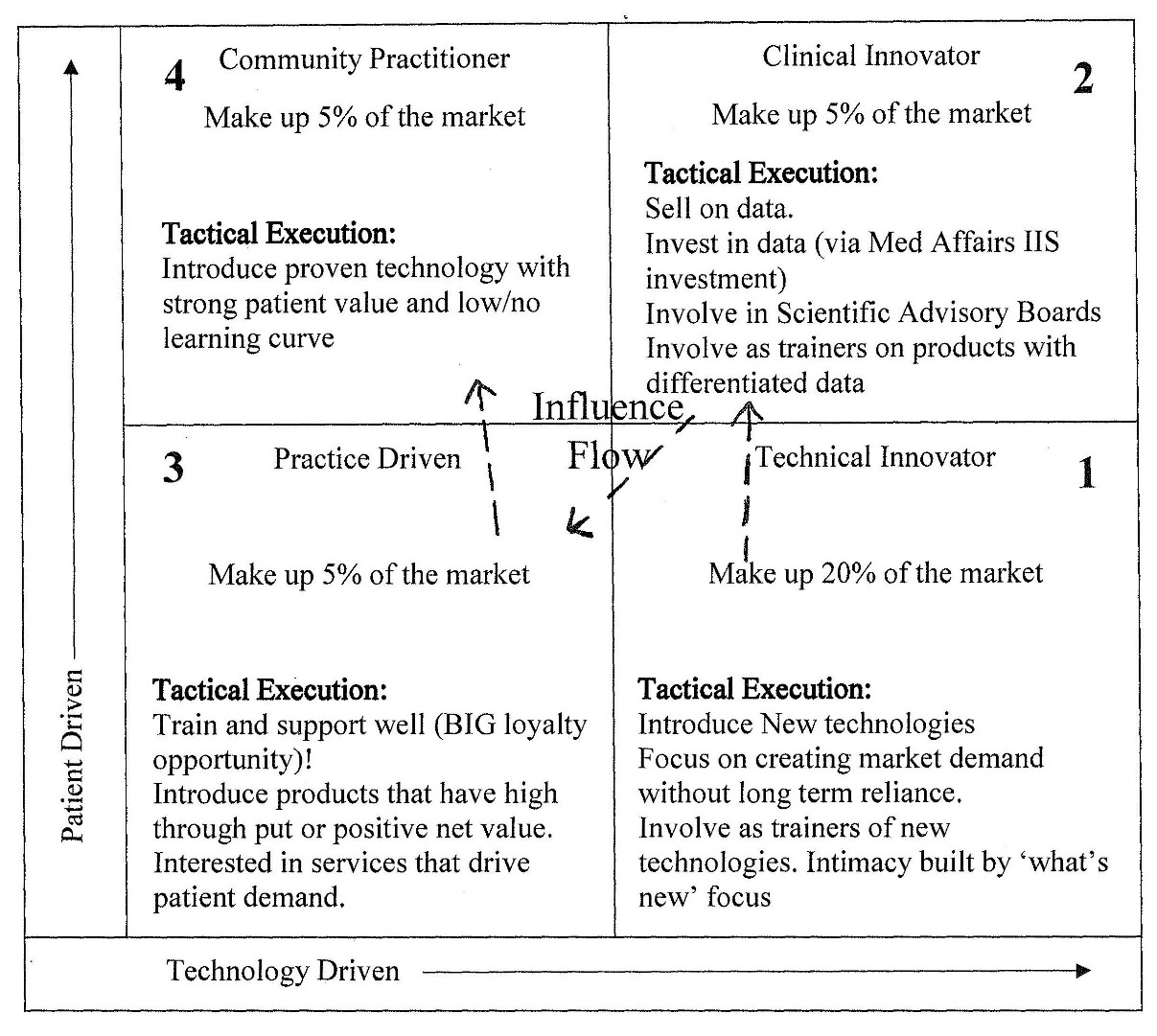
2710 The respondents made a special play for the “Practice Driven Physician”: the practitioner who was focussed on profitability and efficiency, attracted by the idea of performing multiple procedures in shorter time for higher rewards. They considered that this kind of practitioner was representative of a large portion of “the physician population”. An internal Ethicon marketing document entitled “The Practice Driven Physician” 2595 contained the following portrait of their primary target:
Practice Driven – Physician Portrait
…
• Large portion of the physician population, driven by the increasing dynamics of current HC environment
• More likely to refer highly complex cases out
• Reimbursement savvy
• Calculates – higher risk/higher reward
• Less specialized – Even distribution of patients being treated for all conditions
• More likely an early to mid career physician
• More likely to adopt new products which reduce site of care to receive increased reimbursement
• More likely to “market” their practice
• Sometimes seen as the solo practitioner
• Sees it as a business where they are “practicing medicine”
• Less inclined to attend multiple conferences
• Least likely to be doing robotic procedures
• More likely to spend a higher percentage of their time in a private hospital (EMEA)
• Less prevalent outside the US, as most physicians are employed by national healthcare systems
• More likely to push new product through the procurement process
(Emphasis added)
2711 The reference to the lower inclination to attend multiple conferences is interesting in view of the respondents’ submission that gaps in the information they provide could be filled by information derived from other sources including attendances at conferences.
2712 No evidence was called to explain this document but it appears to be an outline for an advertisement or a series of advertisements. It is undated but from its content it is reasonable to assume that it was written and conceived in about 2007, when Prosima was launched on the US and European markets.
2713 It begins with a list of props:
Photo of expensive car
Photo of European ski trip
Boating magazine
Shirt and tie with lab coat
(sign on back – “Surgery is the Cha-ching thing”)
2714 A “script outline” or multiple outlines follow:
Physician in office at desk-
Takes call from friend/colleague
• Just got back from a week in St. Moritz- fabulous ski conditions, beautiful resort.
• Yeh – I picked up the Lamborghini on Friday– an amazing machine.
• And I’m finally going to invest in that sailboat this summer so we can cruise to the Caribbean.
• My practice is now at the level of efficiency where I can take some time away to really enjoy life.
• Gotta run, my next patient will be here shortly and I'm getting a call from the hospital I need to take ...
2715 The next script outline involves the physician receiving a call from the “OR Nurse” (presumably the nurse in the operating room):
There’s an opportunity to add one more GYNECARE TVT to the surgical schedule tomorrow? Sure. You know I can do a TVT-O in eight minutes. That makes 4 TVTs and an LSH [Laparoscopic Supracervical Hysterectomy] before lunch. That works for me. Gives me time to do 3 THERMACHOICE procedures in my office before I leave for the health fair where I’m giving a talk to 50 women about pelvic health conditions and treatments. The last 2 community outreach programs brought me a dozen new patients, so be prepared for a heavy surgical schedule in the next few months.
(Original emphasis)
2716 Another has the physician receiving a call from an Ethicon representative who offers him training Prosima:
• Yes, I’d like to set up time to learn that new prolapse procedure– what did you call it “PROSIMA”? I’ve been referring out a number of cases for prolapse repair lately and you know I didn’t want to take on that PROLIFT procedure. It didn’t work for my practice. But I’d like to provide treatment for my patients with less severe prolapse who are symptomatic.
• How’s the reimbursement for that procedure? Good, good. You know I use your products because I can do more procedures in less time with better reimbursement.
• I can only do the training over the weekend, though. I’ll be in the OR for 5 procedures Friday morning and a full office schedule in the afternoon, including 3 ablations. Then I’m speaking to a group of patients at the health fair.
• By the way, do you have that permission form to link my new web site to your patient site- PelvicHealthSolutions? I’ve heard my patients like that they can assess their symptoms on your site and come in to me with a list of questions. I like my patients well-informed because it makes my time with them more efficient. And it usually leads to better outcomes. And you know I pride myself on my reputation as a good surgeon. My patients talk – and they send their family and friends to me. That’s important to me.
• We make a great team!
(Original emphasis)
2717 Professor Korda gave evidence that he had been visited over the years by many company representatives who tried to convince him to use mesh for prolapse surgery. He was given DVDs and pamphlets outlining its benefits. He was invited to numerous meetings run by drug companies where visiting “opinion leaders”, paid by the manufacturers (including the respondents) gave lectures, played DVDs, and often demonstrated live surgery on operative techniques. He said that, mesh erosion aside, in none of the lectures, DVDs, or demonstrations did they speak of the complications and they did not refer to the impact of the mesh surgery on the patient’s quality of life. Although mesh erosion was mentioned, he said that it was “played down as a small price to pay for the ‘anatomical success’” and “it was implied that the mesh erosions were easy to manage”. He said that no-one raised the risk that the mesh-induced pelvic pain was incurable, the difficulties of surgical management of mesh contraction and mesh excision, the hazards of mesh excision, or the requirement for numerous operations. He added that it was never suggested at any of these meetings that inserting the mesh required further surgical training. He said that, as a result, gynaecologists were led to believe that “the new mesh techniques had been studied in sufficient patients to realistically determine the efficacy and adverse events and that both the technique and the device [had] been approved by the FDA”.2596 This evidence was not challenged in cross-examination and no evidence was led to contradict it.
2718 The applicants tendered a number of the respondents’ product brochures. The evidence does not indicate whether these were the only product brochures published by the respondents about the Ethicon devices and it may be doubted that they were. In the absence of evidence to the contrary, it is reasonable to infer that they were at least representative.
2719 A brochure for “the TVT family of products”, entitled “Precision Knowledge Total confidence”, which was directed to surgeons, included in its description of the benefits of TVT:
> Excellent host tissue incorporation
…
> No late onset adverse events
- No tape erosion
- No tissue reactions2597
2720 The reference given for “excellent host tissue incorporation” was to the article by Professor Amid on the classification of biomaterials published in 1997. It will be recalled that Professor Amid was considering hernia meshes only.
2721 The reference given for “no late onset adverse events” was to Nilsson et al (2008), the 11-year follow-up of the Nordic trial of TVT. This was a true reflection of the reported outcome of the trial, which, it will be remembered involved a carefully screened group of patients. As the examples in Part IX demonstrate, however, it was not a true reflection of the information in Ethicon’s possession. To Ethicon’s knowledge there were late onset adverse events with TVT and there were also instances of tape erosions and tissue reactions. It follows that the representations in the brochure might have been literally true when read with the reference, but they were misleading.
2722 Wendy Bartlett of Johnson & Johnson NZ wrote to three JJM employees on 14 February 2000 to advise that urologists had been “dogged by erosions and patient rejection of the slings”.2598 The minutes of a meeting of Ethicon’s Scientific Advisory Panel on Pelvic Floor Repair held in Chicago on 22 June 2001 recorded a consensus that “erosion is a risk”, that it was “typically seen” by three months, usually by 6–12 months and that it could present three years later.2599
2723 In July 2002 the six month findings of the Ward Hilton RCT were published in the British Medical Journal. That report included the following statement:
Erosion of inorganic material into the vagina, bladder, and urethra has been reported after several sling procedures and has resulted in the withdrawal of one device. In our study there was one case of tape erosion. This was managed by partial excision of the tape and closure of the vaginal skin. In the five years since tension-free vaginal tape was first described there have been five reports of tape erosion into the urethra (Gynecare, personal communication, 2002). It is likely that this figure represents an underestimate, and long term follow up of these patients is needed to quantify the extent of this complication. 2600
2724 The following month, Ethicon received a draft of a report by members of the Institute of Applied Health Sciences in the University of Aberdeen entitled “Systematic review of the clinical effectiveness and cost-effectiveness of tension-free vaginal tape (TVT) for treatment of urinary stress incontinence”.2601 The research was commissioned by the National Health Service R&D Health Technology Assessment Programme on behalf of NICE. The final version was published in August 2003.2602 The authors, Cody et al (2003), stated that “in the longer-term, the main concern is complications associated with the use of the tape, particularly erosion into the vagina or urinary tract”. They noted that “current evidence suggests that these occur only rarely” but they were careful to add that it was too soon to judge this reliably. Table 19 of their report (extracted below) listed numerous late complications of TVT taken from case series reports:
Complication | No. of studies for which relevant data are reported (no. of women studied) | Median rate (%) (IQR) | Mean (%) |
Sling infection | 3 (225) | 1.0 (0) | 0 |
UTI | 19 (2601) | 6.7 (3.1 to 7.9) | 5.5 |
Defective healing | 19 (2974) | 0 | 0.5 |
Thrombosis | 3 (1795) | 0.5 (0.3 to 0.6) | 0.2 |
Postoperative pain | 4 (375) | 3.0 (0.9 to 5.3) | 2.3 |
Voiding difficulty | 5 (380) | 1 (0 to 4.4) | 8.3 |
New urge symptoms/detrusor instability | 27 (1644) | 4.4 (1.1 to 7.2) | 6.6 |
Voiding dysfunction | 7 (402) | 0 (0 to 0.4) | 0 |
New/recurrent prolapse | 3 (190) | 0 (0.0 to 1.1) | 1.0 |
Dyspareunia | 3 (206) | 0 | 0 |
Pain | 3 (152) | 4.4 (2.2 to 4.4) | 2.6 |
Tape rejection | 24 (2895) | 0 | 0 |
Tape erosion | 3 (278) | 1 (0.5 to 1.8) | 1.1 |
Recurrent UTI | 3 (191) | 1.1 (0.6 to 1.5) | 1.0 |
Readmission | 19 (4377) | 2.4 (1.4 to 5.0) | 2.4 |
Dysuria | 4 (348) | 7.9 (7.1 to 8.5) | 7.5 |
Urinary retention | 14 (731) | 4.4 (0.4 to 9.1) | 6.2 |
Infection | 12 (2427) | 0 (0 to 0.9) | 0.7 |
Reoperation (for incontinence) | 8 (3196) | 1.5 (1.3 to 2.4) | 1.4 |
2725 In July 2004 Ethicon GB received a report from a surgeon of a patient with TVT eroding into her bladder 16 months after the device had been implanted and seeking advice as to the best way to repair it.2603
2726 A search of the literature about TVT conducted by Ethicon’s Risk Manager in March 2005 in response to an inquiry from a law firm referred to one case of tape erosion, three cases of tape expulsion, one case of tape migration, four cases of bladder erosion, two cases of urethral erosion, 17 cases of vaginal erosion, and three unspecified cases of erosion.2604
2727 The report of the five-year follow-up of the Ward Hilton RCT, published on 26 October 2007, referred to three cases of erosion into the vagina (one at eight months and two at five years), and one case of erosion into the bladder after five years, requiring cystotomy and tape removal. Commenting on these outcomes, the authors wrote:
Erosion of synthetic sling material into the vagina, bladder and urethra has been described following the TVT and related procedures. It has been suggested that early vaginal erosion is the result of failure of vaginal skin healing rather than true erosion. In this study, there were two true vaginal erosions (only one of which was symptomatic) in the 72 women examined at 5 years. Although these women suffered only minimal morbidity, it is important to be aware that synthetic sling materials have the potential to erode many years after implantation. The woman who was found to have tape within the bladder may have had an unrecognised perforation at the time of operation. It is possible that our figures represent an underestimate of tape complications and suture-related colposuspension complications as less than half the women who underwent surgery were examined at 5 years.2605
2728 Professor Ward, the trial coordinator and lead author of the article, was supported by a grant from Ethicon. There could be no doubt that Ethicon knew of these results at least by the time the article was published.
2729 On 24 August 2007, Ethicon was advised by the FDA that a search of the MAUDE database for the period of August 2004 to August 2007 had returned “a significant number of reported adverse events” including 334 reports for TVT of a range of adverse events including mesh erosion and extrusion.2606
2730 As the applicants submitted, these brochures told “a story of simple fixes for [stress urinary incontinence] that would restore the patient to a life free from social embarrassment and inhibition”.2607 All the SUI devices were promoted by representations to this effect which tended to minimise the significance of any accompanying warning.
2731 An early brochure for TVT published in 2001 entitled “GYNECARE TVT Tension-Free Support for Incontinence: Information for Patients” begins with the following description of stress urinary incontinence:
Incontinence is not a fatal condition, but it can significantly interfere with social, professional and personal activities with deterioration in the quality of life and sometimes social isolation. It is therefore important and worthwhile to have treatment.2608
2732 It proceeded to outline various treatment options, and compared “[c]onventional surgery” to the “new technique” and “exciting new concept” of the TVT device. It stated that compared to conventional surgery, which requires four to six days in hospital and some discomfort from the abdominal wound, the TVT procedure “avoids the pain and long hospital stay of the more major operations” as it is “a minimally invasive procedure that has little post-operative pain associated with it so the patient leaves the hospital on the same day or the following day after the operation”. The following statement was made about complications:
Complications can occur and include bleeding or bladder injury when the needles are passed, difficulty emptying the bladder, urgency and urinary infection. So far, there have been no significant long-term side effects.
2733 There followed the same series of questions and answers covering various complications and returning to normal activities.
2734 To the question “Will I have pain after the operation?”, this answer was given:
Some mild pain may occur over twenty-four to forty-eight hours after surgery. This may be controlled by simple pain relief.
2735 To the question of “Does the mesh remain in place forever?”, the answer was:
Yes, evidence from long-term follow-up studies show that the tape is inert and remains in place to support the urethra.
2736 Both answers were misleading, the first because it did not admit of the possibility of severe and chronic pain, the second because the tape is not biologically inert.
2737 In cross-examination Dr Hinoul was asked if it was misleading to give this response when Ethicon knew that the mesh was not biologically inert. He replied that he thought the answer referred to the most common definition of inert, that it does not degrade and remains the way it has been inserted.2609 I doubt that this was an honest answer, but, even if it was, the response to the question in the brochure was misleading, not least because it ignored the risk that the tape could migrate.
2738 The brochure mentioned the risk of mesh exposure but not erosion, and minimised its significance, stating that “[it] is treated by antibiotics and closure of the wound”. There was also a warning that “[a] few patients have temporary difficulty” with emptying their bladders.
2739 A 2004 brochure promoting TVT, entitled “Stress urinary incontinence”, assured the reader that she need not live with the problem anymore.2610 It included a similar question and answer section. The question and answer relating to pain from the 2001 brochure are reproduced verbatim. In addition to the warning about urinating difficulty following surgery there was a statement to the effect that, to obtain relief in the event that the problem persists, the tape may need to be loosened or divided. Unlike the 2001 brochure, there was no reference to the risk of erosion or exposure or to the treatment of this complication by antibiotics and closure of the wound. Instead, the final question in this section is “What are the risks?” which was answered as follows:
All medical procedures have risks. Although rare, complications include difficulty urinating, injury to blood vessels of the pelvic sidewall and abdominal wall that may cause excessive bleeding, and bladder and bowel injury. For a complete description of risks, see the adverse events section of the attached product information.2611
2740 On the following page, four testimonials appeared, attributed to women who had been implanted with TVT, in which they extolled the dramatic improvements to their lives after the operations. The indications, contraindications, warnings and precautions and adverse reactions sections from the TVT and TVT-O IFUs were also extracted, but in much smaller print. The only adverse reactions section read as follows:
• Punctures or lacerations of vessels, nerves, bladder, urethra or bowel may occur during needle passage and may require surgical repair.
• Transitory local irritation at the wound site and a transitory foreign body response may occur. This response could result in extrusion, erosion, fistula formation or inflammation.
• As with all foreign bodies, PROLENE mesh may potentiate an existing infection. The plastic sheaths initially covering the PROLENE mesh are designed to minimize the risk of contamination.
• Over correction, i.e. too much tension applied to the tape, may cause temporary or permanent lower urinary tract obstruction.
2741 Not only was no suggestion made that long-term complications could arise but the impression created by these representations is that, with the exception of lower urinary tract obstruction, any complications would be short-lived.
2742 A 2010 brochure on the TVT family of products,2612 which was revised and updated in 2013,2613 contained a section on available treatments but, in contrast to some of the earlier brochures, did not mention any alternative form of surgical treatment or the use of bulking agents. It is true that the list did not purport to be exhaustive, but the description could lead the reader to think that “the TVT System” was the only surgical option.
2743 Similar to the 2004 brochure discussed above, to the question “What are the risks?”, the following answer was given:
All medical procedures present risks. Although rare, complications include difficulty urinating, injury to blood vessels of the pelvic sidewall and abdominal wall, and bladder and bowel injury. For a complete description of risks, see the adverse events section of the attached product information.
2744 The adverse events section of the attached product information was brief. It read:
• Punctures or lacerations or injury to vessels, nerves, bladder, urethra, or bowel may occur during instrument passage and may require surgical repair.
• Improper placement of the TVT device may result in incomplete or no relief from urinary incontinence or may cause urinary tract obstruction.
2745 This was misleading. It represented that adverse reactions only occur because of intra-operative error on the part of the surgeon. It neglected to mention mesh erosion, exposure, or contraction and their potential repercussions. Nor was there any acknowledgment of the possibility of long-term complications.
2746 The only relevant difference in the updated 2013 version was the addition of a third bullet point in the adverse events section of the attached product information:
• Transitory local irritation at the wound site and a transitory foreign body response may occur. The response could result in extrusion, erosion, fistula formation and inflammation.
2747 I discuss the deficiencies in representations of this kind (which were commonly made in the IFUs) below.
2748 The brochures in evidence directed at surgeons emphasised the benefits of the SUI devices over those manufactured by the respondents’ competitors. They were more muted in tone than the patient brochures but they paid little attention to the potential risks.
2749 A 2009 surgeon brochure on the TVT family of products, entitled “Make DATA and SAFETY YOUR CHOICE”2614 compared Ethicon’s midurethral synthetic tapes to American Medical Systems’ counterparts, boasting of lower rates of bladder and urethral perforations with TVT than AMS’ “Sparc”, no urethral, ureter or bladder perforations with TVT-O compared to Monarc, and a lower mean operative time for TVT Secur compared to AMS’ “MiniArc” with comparable blood loss. It did not mention any other risks.
2750 A 2003 surgeon brochure introducing TVT-O entitled “GYNECARE is Turning the Obturator Approach ‘Inside-Out’”2615 stated that it was “Designed to optimize safety” by “avoiding the retropubic space”, with a reduced potential for urethral and bladder injury. It claimed that the TVT-O approach maintains “key critical success factors” of TVT which it referred to as “the Gold Standard with Long-Term Demonstrated Safety and Efficacy”. It reproduced the contraindications, warnings and precautions, and adverse reactions sections of the TVT-O IFU, with all of their limitations. As the applicants noted, it did not indicate that any particular training is necessary or, for that matter, desirable.2616
2751 A brochure promoting TVT Secur was entitled “No bigger than your palm. No less than a revolution”.2617 It described the procedure as less complicated and less invasive, avoiding problematic spaces and organs, involving less dissection, less anaesthesia, less postoperative pain, and reduced risk of leg pain. For the claim that the procedure caused less post-operative pain, it stated that the “device and insertion approach avoids major nerve bundles, reducing the potential risk of leg pain”. It referred to the fact that studies of TVT Secur were under way, including a post-market evaluation of 50 patients and a prospective, multicentre study of 300 patients. It also included a section called “From a Name You Trust” which represented that TVT had the following complication rates:
• Low incidence of serious reported complications (<0.03%)
• Low retention rate (<3%)
• No reported urethral erosions in multiple clinical studies of 50+ patients
2752 The source for the first dot point was “Data on file”.
2753 The same brochure did not include a reference to any potential risks associated with implantation of the device except for a reproduction of the adverse reactions section of the IFU in barely legible subscript at the foot of a page headed “Security Like Never Before”.
2754 A 2006 brochure on TVT Secur did not include a reference to any potential risks associated with implantation of the device except for an unsourced representation that it produces “less postoperative pain” and, like the previous one I mentioned, a reproduction of the adverse reactions section of the IFU in barely legible subscript at the foot of a page headed “Security Like Never Before”.
2755 A brochure promoting TVT Exact alongside TVT referred to TVT Exact as “built upon 17 years of [TVT] Retropubic success”.2618 Another brochure comparing TVT Abbrevo with TVT-O stated that TVT-O was “now backed by 5 years of follow-up data” but that TVT Abbrevo was “[a]s effective as [the TVT-O] now supported by 3 year follow-up data” and that “35% fewer patients experienced groin pain immediately after surgery”.2619
2756 Many of these brochures used the same reassuring line: “Choose proven outcomes surgeons can trust”.
2757 TVT Exact was marketed to surgeons as a product “built upon 12 years of retropubic success” with the “same dependable design” but with an improved ergonomic handle which will “extend the control of your hand”,2620 although, as the applicants submitted, the benefits of the accompanying tools were not supported by any data and there was no pre-market clinical trial of the device.2621
2758 Similarly, TVT Abbrevo was promoted based on the “reliable results” after three years of the TVT-O. One brochure advertising this device represented that TVT Abbrevo gives the surgeon the confidence of “delivering efficacy” and “achieving excellent results with less mesh”.2622 The source given for this claim was also “Data on file” and reports relating to TVT-O, such as the De Leval study. The De Leval study was also the source cited for the statement that a randomised controlled trial of a modified obturator procedure with a shorter mesh, similar to TVT Abbrevo showed overall improvement comparable to TVT-O at one year and significantly less post-operative groin pain. No mention was made, however, of the risk of erosion or other complications. There was no hint of the possibility of any long-term detriments. Surgeons were merely referred to the instructions for use for “complete product details”.
2759 A leaflet promoting Gynemesh PS represented that the product was “[a]n inert synthetic mesh” for cystocele, rectocele and vault repairs, “engineered to provide the combination of qualities you need for pelvic support”, 2623 “conforms to the anatomy lies flat”. It was said to be made from the same material as Prolene suture and therefore to have low tissue reactivity. It was also said to have a low rate of infection in comparison to natural materials, not to harbour bacteria, and to allow for macrophage penetration and tissue in-growth for rapid healing. Those representations were misleading as was the title “Finally, lightweight, permanent support … by design!”
2760 This leaflet was produced in 2002, three years after the publication of the textbook Incisional Hernia in which Professors Klinge and Klosterhalfen had pointed out that surgical meshes are not inert and lead to chronic irritation of the host tissue.2624 In 1998, Professor Williams wrote that “it is now recognized that no material is totally inert in the body”; even very stable materials will interact to some extent with tissues. He said that there was in fact no such thing as an inert biomaterial.2625
2761 Moreover, Gynemesh PS was not designed or engineered for pelvic support. As I have previously observed, Gynemesh PS was Prolene Soft Mesh, manufactured for abdominal hernia repairs, by another name. There was no sound foundation for the representations that Gynemesh PS conforms to the anatomy of the pelvic floor, that it lies flat, or that it did not harbour bacteria.
2762 The only reference to the possibility of complications appeared in an extract from the IFU, which suffered from the same problems as the IFU.
Patient brochures
2763 A number of patient brochures on the POP devices were in evidence. Many carried the title “Get the Facts, Be Informed, Make YOUR Best Decision”.2626
2764 A 2005 Prolift brochure informed patients that the mesh was “a soft synthetic material specially designed for placement through the vagina to support pelvic organs that have ‘dropped out’ of their normal position (prolapsed)”.2627 But as Mark Yale, Director of Worldwide Risk Management for Ethicon Inc., candidly remarked in his email of 8 May 2008:
This mesh was not “specifically designed” for Prolift application, we pulled a mesh out of our existing bag of tricks. This statement is unsupportable from (scil) a design history standpoint.2628
2765 It also represented that Prolift was different from “other surgical alternatives” in the following respects:
[The Prolift operation] can be completed in less than half the time of traditional surgery. Patients may experience less pain, quicker recovery and go home the next day.
It allows for the restoration of sexual function by restoring normal vaginal anatomy.
Using this new surgical procedure there is often no need to perform a hysterectomy if the uterus itself is not diseased.
2766 This description suggested that patients would be significantly advantaged if they opted for Prolift over traditional surgery. Without warning of the prospect that patients might also experience a great deal more pain than with traditional surgery, that the pain could be chronic and resistant to treatment, this representation was misleading. Yet no such warning was given. To the contrary, as with the SUI devices, risks were minimised and users were reassured.
2767 In answer to the question “What are the risks?” the same brochure stated:
All surgical procedures present some risks. Although rare, complications associated with the procedure include injury to blood vessels of the pelvis, nerve damage, difficulty urinating, bladder and bowel injury. There is also a small risk of the mesh material becoming exposed into the vaginal canal.2629
2768 This account creates the impression that the nominated risks, with the exception of the risk of mesh exposure, are risks that could occur with traditional pelvic surgery and that, apart from the risk of mesh exposure, there are no other risks associated with Prolift. Even then, the brochure does not indicate that exposure has any potential repercussions. The risk of erosion into the bladder, the urethra or the bowel was not mentioned, although it did appear in an extract from the Prolift IFU, which appeared on the following page.
2769 The extract reproduced the contraindications, warnings and precautions, and adverse reactions in the IFU. It went some way towards correcting the misleading impression given by the answer. On the other hand, the extract, like the IFU itself, understated the risks, not least because it neglected to refer to the risk of chronic pain and the difficulties associated with removal of the device, should removal become necessary. It was also misleading because the only reference to pain was to “transient leg pain”.
2770 It will be recalled that in their letter in December 2007 the FDA asked Ethicon to develop a patient brochure to be provided when patients are counselled about options for treating pelvic organ prolapse and indicated what matters should be addressed in that brochure.2630 Those matters were:
an explanation of pelvic organ prolapse, including anatomical issues, causes and symptoms;
alternative treatment options;
risks and benefits of the various options, “including a reference to the FDA MAUDE database website”;
a statement to the effect that that safety and effectiveness of mesh for vaginal repair have not been proven in randomised, controlled clinical trials; and
instructions for post-operative care.
2771 In his response to the FDA letter, Mr Lisa advised that Ethicon had “provided the patient brochure with updated language in the ‘What are the Risks?’ section (Attachment IV)” and said that the same language would appear in the Prolift+M brochure and educational material.2631 Attachment IV was entitled “Proposed Patient Brochure” .2632 The first page contained two proposals and a brochure was included in the attachment. The second was that indications, contraindications, warnings, precautions, and adverse events would be updated “with the agreed upon labelling”.
2772 This was the first proposal:
PROPOSED “WHAT ARE THE RISKS?" SECTION (Page 13)
All surgical procedures present risks. Complications associated with the procedure include injury to blood vessels of the pelvis, nerve damage, difficulty urinating, pain, scarring, bladder and bowel injury. There is also a risk of the mesh material becoming exposed into the vaginal canal.
2773 As is apparent, this was no different from the description included in the earlier brochure.
2774 This information was provided after a conference call between Ethicon and FDA representatives on 22 January 2008.2633 I am at a loss to understand how or why (if it be the case) the FDA was comfortable with it.
2775 Another 2008 Prolift brochure entitled “stop coping. start living.” also purported to set out the facts.2634 It repeated a good deal of the material appearing in the earlier brochure including the prospect of less pain, a speedier recovery, and the restoration of normal anatomy. The answer to the question about the risks, however, was more expansive. It added the following representations:
Mesh exposure can be associated with pain during intercourse for the patient and her partner. Exposure may require treatment, such as vaginal medication or removal of the exposed mesh.
Synthetic mesh is a permanent medical device implant. Therefore, you should carefully discuss the decision to have surgery with your doctor and understand the benefits and risks of mesh implant surgery before deciding how to treat your condition.
2776 In very small print, a statement on the second last page of the brochure stated that Prolift and Prolift+M should not be used in the presence of active or latent infections or cancers of the vagina, cervix, or uterus, contraindications not mentioned in the earlier Prolift brochure or in the Prolift IFU until 1 October 2009.
2777 Once again, however, no reference was made to the risk of chronic pain. Nor did the brochure advert to the potential difficulties associated with the removal of exposed mesh and the complications that might ensue. It did not mention the risks of erosion into organs or the potential consequences. In the absence of an acknowledgment of these potential problems, the brochure suggested that, should surgical removal of exposed mesh be required, there would be no difficulties or complications.
Surgeon brochures
2778 A 2005 Prolift brochure promoted Prolift as an innovative, standardised system designed to enhance the surgical technique. 2635 It boasted that Prolene Soft was a mesh that “does not potentiate infection”, has a large pore size which “fosters proper tissue incorporation”, and is “[l]ightweight, soft, and supple”. It proclaimed the virtues of Gynemesh PS based on the claim made in a presentation by Dr Robinson that in an 88 patient, multicentre prospective study (over one year) it had an 84% success rate and only one patient required operating room intervention. This is likely to be the same study to which Dr Owens referred in the Prolift pre-market CER discussed in Part VIII. The reason for the intervention was not disclosed and the brochure neglected to mention the risks posed by, or contraindications for, Gynemesh PS, let alone the Prolift device.
2779 The respondents acknowledged that no surgeon brochures or presentations regarding Prolift contained relevant warnings and the IFU was the only material provided for surgeons that included relevant warnings.2636
2780 A 2010 surgeon brochure for Prolift+M promoted the device as suitable for all surgeons of varying skills by the following message which appeared on the cover page:2637

2781 The weight of evidence adduced in this case did not support the notion that the Prolift+M was a pelvic floor repair system that surgeons of any skill level could master.
2782 The applicants referred to three surgeon brochures promoting Prosima. The first, entitled “The first fixationless mesh system that maintains anatomical position”,2638 boasted that Prosima “Demonstrates anatomical and functional SUCCESS” “at 2+ Years”. The boast about anatomical success was said to be based on the results of “patient subset followed at 2 years” but the size of the cohort was not mentioned. The boast about functional success cited “Clinical Study Report, Protocol Number 300-06-005 … Data on file, Ethicon…”.
2783 With some justification, the applicants criticised Ethicon for failing to include a reference in this brochure to the randomised controlled trial conducted by the inventor of Prosima, Dr Marcus Carey, which demonstrated no better result using mesh than native tissue colporrhaphy.2639 The RCT was a conducted in Melbourne. 139 women were recruited. Its stated objective was to compare vaginal repair augmented by mesh with traditional colporrhaphy for the treatment of pelvic organ prolapse. The primary outcome measure was “the absence of POP-Q stage ≥2 prolapse at 12 months”. The results were published in the British Journal of Obstetrics and Gynaecology in July 2009. According to that report, the results showed no statistically significant difference in the outcomes for each group at 12 months.
2784 The “Clinical Study Report” mentioned in the leaflet was not tendered in evidence. Certainly neither side took the Court to it. But the study and its results were reported in an article by Sayer et al (2011) published in the International Urogynecology Journal.2640 The unpublished one year report dated 28 October 2009 was also tendered by the applicants2641, as was “Final Version 1” of the study protocol dated 9 March 2007.2642 It is this report that carries the reference number 300-06-005 that appears in the brochure.
2785 According to the protocol, the study was a prospective, multi-centre, single arm design and subjects were to be assessed at various intervals up to 12 months. It was not a randomised controlled trial. Before enrolling any study subjects, each investigator was required to attend cadaver laboratory training using Prosima and then complete the procedure on device run-in subjects under the supervision, and to the satisfaction, of a clinical observer. Conditions were placed on entry into the study. Amongst other things, they included that the women had symptomatic pelvic organ prolapse of POP-Q stage I or II, suitable for surgical repair and that they had no coagulation disorder and no medical condition or psychiatric illness which, in the opinion of the investigator, could affect their ability to complete the study visits under the protocol. Overall success was to be measured by achievement of a POP-Q score of stage ≤1, without further re-intervention for POP.
2786 According to the one-year report, 136 patients who underwent the procedure were included in the full analysis. Only 126 of these patients attended the follow-up visit at six months and the six-month study results across all 11 study sites demonstrated a failure in 33 patients (26.8%).2643 Among the complications that were reported, mesh exposure occurred in 12 patients (8.0%), eight of which had resolved after partial mesh excision and four of which had not resolved at one year, and de novo urge and stress urinary incontinence were each reported by 4% of the study subjects.
2787 The median length of follow-up reported in 2011 was 29 months (with a range of 24 to 34 months). Sayer et al (2011) said the primary anatomic success (as defined by the protocol) was achieved in 69.1% of cases. The rate of mesh exposure was 9.1%, 5% reported stress urinary incontinence, and 3.3% required further prolapse surgery. The conclusion, highlighted in the abstract, was that “[t]hese results indicate this non-anchored mesh repair is a safe and effective treatment for women with symptomatic vaginal prolapse in the medium term”.
2788 But several matters should be noted about the study, which were not disclosed in Ethicon’s leaflet.
2789 First, it was attended by a number of not insignificant complications.
2790 Second, there was no control group, a feature of the study that even the authors considered to be a “major drawback”.
2791 Third, the study was not large.
2792 Fourth, 19% of the original patients in the study (26 of 136 women) were not assessed after 12 months because they did not agree to return thereafter. Seven refused to return, seven could not be contacted, five had previously withdrawn, and “logistical reasons” were given for the last seven. Although patients lost to follow-up are often treated as failures, the article does not report that the 19% of patients who did not return after 12 months were treated in this way.
2793 Fifth, notwithstanding what was said in the article and in the brochure, one of the authors, Judith Gauld of Ethicon GB, noted in an email to Dr Robinson and Jonathon Meek on 12 April 2008, that Prosima had failed its primary objective in the clinical trial.2644 Ms Gauld wrote:
We did state in the protocol that the study would be considered a success if the failure rate had an upper 95% CI of less than 20% at 12 months. We have already failed that.2645
2794 Sixth, the brochure did not mention the authors’ conflicts of interest, although they were well known to Ethicon and had been disclosed in the article. Dr Hinoul and Ms Gauld disclosed their employment with Ethicon. All the other authors disclosed that they had “consultancy positions with Ethicon”. It is most unlikely that most readers of the brochure would know of this relationship.
2795 An April 2010 Prosima brochure targeted at surgeons also made various claims about the success of the products based in part on a two-year single arm study result, once again without disclosing either the limitations of the study or the adverse events that were reported by it.2646 That brochure posed the question: “What could a truly tension-free repair mean for you and your patients?” It claimed that “Truly tension free repair with GYNECARE PROSIMA™ minimizes potential complications”. The “proof” offered in support of the claim was eliminating needle passes while reducing the amount of foreign material left behind. But the evidence did not indicate that Ethicon had conducted any study which would justify the claim.
2796 Another Prosima brochure tendered by the applicants was entitled “Symptomatic Moderate Prolapse” and featured on the cover a couple engaged in ballroom dancing.2647 It also declared Prosima to be an anatomical and functional success. It contained statements such as “GYNECARE PROSIMA Pelvic Floor Repair System has the CLINICAL PROOF”, relying on the results from the same clinical study which Ms Gauld acknowledged had been a failure according to the standard set by the study protocol.
2797 Each of the Prosima brochures directed the reader to the IFUs “for complete contraindications, warnings, precautions, and adverse reactions” (emphasis added). I accept the applicants’ submission that this was a clear message from the manufacturer and supplier that surgeons need look no further than the IFUs for information on those matters.2648
The use of key opinion leaders
2798 The respondents’ marketing strategy also involved the deployment of leading medical specialists as “key opinion leaders” to “contribute to discussions on new product development, pipeline management and prioritization for product development”.2649 While no criticism could be levelled at a company for consulting experts for these purposes, the consultancy agreements in evidence required that the services be provided exclusively to Ethicon, unless Ethicon gave its consent to the contrary. Further, one such agreement, prepared for Professor Deprest by the marketing department of Ethicon Women’s Health & Urology, bound him, amongst other things, not to make any representation relating to respondents’ products or its clinical outcomes, “unless such representations [had] been reviewed and approved in advance by [Ethicon]”.2650 At least for the period of the agreement, such a clause is likely to have silenced potential critics. Moreover, as the applicants submitted, this kind of arrangement invested surgeons, regarded as leading lights in their fields of expertise, in the development of Ethicon’s products.2651
2799 Key opinion leaders were also invited to join Ethicon’s “Innovation Council”. Another consultancy agreement prepared by the marketing department of Ethicon Women’s Health & Urology for Professor Deprest engaged him to participate in Ethicon-sponsored speaker programs, advisory board meetings concerning the company’s products or other topics designated by the company, “Peer-to-Peer meetings, 1–1 monthly meetings with R&D, Medical Affairs and WW Marketing, and product evaluations”. It was also an exclusive agreement and it contained the same clause as the agreement mentioned above prohibiting representations about the company’s products or its clinical outcomes without Ethicon’s prior approval.2652
2800 As well as patient brochures, the respondents also published information about the Ethicon devices on websites called “www.controlsuddenurineloss.com” and “www.gynecare.com”. The URLs for both websites were printed on some brochures, from as early as 2001.2653
2801 The applicants tendered what they said was copies of webpages from “www.gynecare.com” (the Gynecare website) as it appeared in January 2005, in February 2005, and in March 2006.2654 The content is similar to of the content of the brochures but the warnings are easier to read. The following adverse reactions were recorded on one of the pages:
• Punctures or lacerations of vessels, nerves, bladder or bowel may occur during needle passage and may require surgical repair.
• Transitory local irritation at the wound site and a transitory foreign body response may occur. This response could result in extrusion, erosion, fistula formation and inflammation.
• As with all foreign bodies, PROLENE mesh may potentiate an existing infection. The plastic sheath initially covering the PROLENE mesh is designed to minimize the risk of contamination.
• Over correction i.e. too much tension applied to the tape, may cause temporary or permanent lower urinary tract obstruction.2655
2802 The February 2005 version included a testimonial purportedly from a patient entitled “My Secret – Fear of Embarrassment”. It told the story of a thirty-five year old woman called “Jody” who suffered from stress urinary incontinence, the humiliation of which led her to obtain help. It recounts an incident where Jody had wet herself through her clothes at an amusement park with her young daughter, then researched treatment options online where she discovered the TVT device. Jody then contacted Dr Cheryl Iglesia who is said to have told her that the TVT treatment would “restore her body’s ability to control urine loss by placing a ‘sling’ or a mesh tape beneath the urethra”. The story concluded with the following:
Not long ago, Jody had the procedure using TVT and returned home the same day. She couldn’t believe such a simple procedure could have such a huge impact on her life.
The treatment has changed Jody’s life in every way. She is back to her active self, balancing the demands of family, work and life – without SUI. Today she enjoys spending time on the beach, playing with her kids, and taking long walks. She is also looking forward to buying a new treadmill to replace the one she threw out when she had SUI!
Jody says that taking the time to talk to a doctor and undergo the treatment was the best decision she has made.2656
2803 The website also contained a link where the reader could search for a doctor that offered TVT surgery. As the applicants submitted, in addition to the patient brochures, this was an illustration of how the respondents communicated directly with patients, without a doctor as an intermediary.2657
2804 The document depicting the Gynecare website pages shows that these were generated using the “Wayback Machine” website. Evidence of this kind has been ruled inadmissible in a number of cases in this Court: see, for example, Rodney Jane Racing Pty Ltd v Monster Energy Company [2019] FCA 923 at [179] and the cases referred to there. In the absence of evidence to establish that the webpages formed part of the records of the business conducted by the persons responsible for the Wayback Machine, the evidence is hearsay: Voxson Pty Ltd v Telstra Corporation Limited (No 10) [2018] FCA 376 at [36]–[37] (Perram J). In each of those cases objection had been taken to the evidence. In the present case, however, no objection was taken. As I pointed out in Fair Work Ombudsman v Grouped Property Services Pty Ltd [2016] FCA 1034; (2016) 152 ALD 209 at [226]:
But is not for the judge to raise for herself and then determine questions of admissibility: Harrington-Smith v Western Australia (No 7) (2003) 130 FCR 424 at [13] (Lindgren J). When the Evidence Act 1995 (Cth) speaks of evidence not being admissible, “not admissible” means “not admissible over objection”: Commissioner of Taxation v SNF (Australia) Pty Ltd (2011) 193 FCR 149 at [26] (Ryan, Jessup and Perram JJ), following Seltsam Pty Ltd v McGuinness (2000) 49 NSWLR 262 at [149] (Spigelman CJ). Certainly in civil proceedings, inadmissible evidence admitted without objection can be used “as proof to the extent of whatever rational persuasive power it may have”: Jones v Sutherland Shire Council [1979] 2 NSWLR 206 at 219 (Samuels JA), quoting CT McCormick, Law of Evidence (1st ed, 1954, West Publishing Co) at [54]. See, too, Harrington-Smith at [13].
The use of preceptors and the preceptor training program
2805 Ethicon’s preceptor training program was a key part of its marketing strategy. Professional education was a strategy for ensuring that surgeons used the Ethicon devices in preference to other alternatives. That is apparent from an undated JJM marketing presentation entitled “Strategies for Growth: OTW (One to Win)”, which listed professional education as one of the three key strategies. The other two were “Demand Generation” and “Relationships”.2658 The matters referred to in this document also explain the respondents’ reluctance to share, through the IFUs and the brochures, what they knew about the risks and limitations of the devices with the user community.
2806 Under the heading “Demand Generation”, this document listed:
• <3% of ‘potential patients’ present for surgery – We have to create calls for action
• 75% of the OBS community are looking to get out within 10yrs – Incontinence and Thermachoice must be seen as the obvious choice for their practices
• Few Drs are commercially skilled – We have to help customers to attract referrals
• SECUR and PROLIFT – Use Prof Ed to ensure surgeons adopt
(Original emphasis)
2807 The three points then appeared under the heading “Prof Ed”:
• Create Preceptorship network to provide a structured training process
• The Preceptorship team to be our local ‘faculty’
• The Faculty will drive local round-table discussions / communities of practice
2808 The close relationship between Ethicon’s commercial interests and professional education is also apparent from a PowerPoint presentation dated 1 June 2010 in which a roadmap for the growth of the pelvic floor market appears. 2659 The first dot point under the heading “Leadership in Kit Market” reads:
Drive Pelvic Floor Growth through Professional Education with a Niche Focus2660
2809 The close relationship between training and marketing is illustrated by the fact that Ms Jackson reported the training issues she had experienced to Jonathan Meek from the marketing department.2661
The adequacy of the respondents’ warnings and other information it provided
2810 The applicants alleged that there were numerous deficiencies in the warnings and other information provided by the respondents about the safety and efficacy of the Ethicon devices to patients and surgeons. Taken collectively, if not individually, they argued, these matters demonstrate that the warnings were inadequate.2662
2811 The question of the adequacy of the warnings and other information provided by the respondents about the devices is relevant to several of the causes of action. For the reasons given below, the applicants’ allegation is made out and their argument should be accepted.
Adequacy of warnings and other information provided about the SUI devices
2812 It will be recalled that the first IFU drafted by Ethicon was the 8 September 2000 TVT IFU. Professor Blaivas was critical of this IFU because it:
minimised the material properties of polypropylene by stating that it “elicits a minimum to slight inflammatory response, which is transient,” that there is a “deposition of a thin fibrous layer of tissue,” that the “mesh remains soft and pliable,” that “normal wound healing is not noticeably impaired,” and that the mesh is “not subject to degradation or weakening by the action of tissue enzymes;”
trivialised the unique complications associated with mesh devices by stating “potential adverse reactions are those typically associated with surgically implantable materials”;
incorrectly stated that a “transitory local irritation at the wound site and a transitory foreign body may occur;”
failed to address the following risks: late onset adverse events; the recurrent nature of many erosions; the recurrence of stress urinary incontinence (worse than original condition) if complications develop; permanent vaginal scarring and distortion; chronic pain; sexual impairment; and dyspareunia; the management of these complications; the need for multiple operations; the failure of surgery to alleviate the problems; or the difficulty of, and risks involved in, removing the device. 2663
2813 All these criticisms were justified. Moreover, they were equally applicable to the subsequent IFUs in which the same representations were replicated and the same omissions were made.
Chronic inflammatory reaction of the tissues surrounding or attached to the tape
2814 No warning was given about the risk of a chronic inflammatory reaction at any point in time. All the IFUs for the SUI devices referred to a “transitory local irritation at the wound site and a transitory foreign body response”. All but the Medscand TVT IFU stated that both these responses “may occur”.
2815 The statement that a transitory foreign body response may occur was false for two reasons.
2816 First, a foreign body response was not a possibility; it was a certainty. As Dr Hinoul acknowledged, a foreign body response will always occur. Indeed, the devices were designed with the intention that they adhere to the tissues as a result of the foreign body response.2664 Yet, as at the time of the trial, the IFUs for those of the Ethicon devices that remained on the market continued to state that a “foreign body reaction may occur”.
2817 Second as Dr Hinoul also acknowledged, the response is not transitory but permanent.
2818 Under cross-examination Dr Hinoul said that the reference to “a transitory foreign body response” derived from a request made by the FDA.2665 In his affidavit he referred to the FDA writing to Ethicon in 1973 asking them to include in the IFU for the Prolene suture a statement that “transitory local inflammatory reactions have been reported” and then again in 1988 approving a change to the labelling warning of a “minimal, transient acute inflammatory reaction”.2666 This explanation merely illustrates the inattention in Ethicon’s approach to safety to the differences between its sutures and the knitted mesh. In no way does it justify the inclusion of such a warning in the IFUs for the SUI devices.
2819 Consequently, despite what the respondents were telling consumers in their IFUs and patient brochures, it was common ground that the foreign body or inflammatory response to polypropylene implants, including the devices in question, is not “transient” or “transitory” but chronic and permanent. As the respondents put it in final submissions, it lasts as long as the foreign object remains in the body.2667
2820 The warning about “transitory local irritation” appeared in all IFUs for the SUI devices that are in evidence. This was also false.
2821 On 2 June 2006, if not before, Professor Klosterhalfen explained to an Ethicon expert meeting in Hamburg on meshes for pelvic floor repair, that the foreign body reaction was a “chronic wound”. In 2007, an article by Professor Klinge and others was published which referred to “the obligate foreign body reaction” in non-absorbable synthetic meshes in the treatment of both genital prolapse and stress urinary incontinence and described a “chronic inflammation at the interface …”.2668
2822 In 2015, all of the IFUs for the SUI devices remaining on the market were updated so that the warning read:
Transitory local irritation at the wound site may occur.
As with any implant, a foreign body response may occur. This response could result in extrusion, erosion, fistula formation and/or inflammation.
2823 As can be seen, the adjective “transitory” continued to qualify the duration of the local irritation at the wound site but was removed from the description of the foreign body response. Nevertheless, the auxiliary verb “may” was still used to describe the occurrence of both the foreign body response and the local irritation. For this reason the new representation was also misleading.
2824 In addition to the inadequate and misleading warnings regarding local irritation and the foreign body response, all IFUs for the SUI devices referred to animal studies that purportedly showed that Prolene mesh elicited a minimal inflammatory reaction in tissues that was transient.
2825 The Medscand TVT IFU stated:
Performance
Animal studies show that implantation of Prolene® mesh elicits a minimal inflammatory reaction in tissues, which is transient and is followed by the deposition of a thin fibrous layer of tissue which can grow through the interstices of the mesh, thus incorporating the mesh to adjacent tissue.2669
2826 This section appeared in the same, or very similar form, in the subsequent TVT IFUs, all TVT-O IFUs, as well as the TVT Secur IFU.
2827 On the assumption that the statement in the IFUs accurately reflected what the animal studies showed, it was nonetheless misleading.
2828 First, no information was given about the duration of the animal studies. Professor Klinge observed that animal studies are mainly done by testing the material in the abdominal wall without contamination and without subjecting the animals to mechanical strain. Consequently, he said, the precise conditions of the preclinical testing should have been identified.2670
2829 Second, PA Consulting, the firm, it will be recalled, engaged by Johnson & Johnson to investigate mesh erosion in pelvic floor repair, reported that mesh erosion is difficult to model in pre-clinical studies and there was a “lack of a definitive animal model for in-vivo design validation”. PA Consulting went on to make the following points.
• The situation with animal models is confusing; whilst there are claims in the literature for successful animal models as a predictor of product behaviour and performance, these have not been reproduced when adopted by J&J
• The differences in animal anatomy to human anatomy make a model difficult. There are challenges relating to anatomical structures and organization; and histological relevance
• The consensus in J&J is that the animal models are not yet good enough and the organization is endeavouring to develop a new animal model
• This is challenging; study size is an issue (to detect low failure rates) and there are many influencing factors. The follow up period is also significant
• Primates perhaps represent the best model, but are very expensive to use in this way and there may be regulatory limitations to consider
• Sheep are perhaps the next best in terms of vaginal anatomy, but again are costly to utilize2671
(Emphasis added)
2830 Third, the evidence established that the inflammatory reaction elicited in human tissues is not transient. Although immediate or acute post-operative inflammation due to implantation may resolve within days or weeks, the inflammatory foreign body reaction around the implants persists indefinitely.2672 It is, indeed, chronic, not transient.
2831 This section did not undergo any significant changes from the original version in the Medscand IFU until the removal of the word “transient” in the 29 November 2010 TVT IFU as follows:
Animal studies show that implantation of PROLENE Mesh elicits a minimal inflammatory reaction in tissues and stimulates the deposition of a thin fibrous layer of tissue that can grow through the interstices of the mesh, thus incorporating the mesh into adjacent tissue …2673
2832 The 10 September 2010 TVT Abbrevo IFU and its subsequent versions and the 4 May 2010 TVT Exact IFU and its subsequent versions contained sections in the same or very similar form as the revised version above. It appears, however, that the approach to the removal of the qualifier “transient” was not uniform. The TVT-O IFUs dated between 2010 and 2015 retained the reference to a transient inflammatory reaction despite it not appearing in the other IFUs for the other SUI devices during this period.
2833 In any case, there is an intense, not minimal or slight, host defence response immediately after implantation.
2834 In cross-examination Associate Professor Lam agreed that the warning about the inflammatory reaction being minimal to slight in intensity and transient in duration did not accurately represent the nature of the chronic foreign body reaction and, in particular, that it did not indicate that the chronic foreign body reaction may result in “clinically manifest complications” such as erosions, late erosions, an increasing incidence of erosions with time, contractures or pain.2674
2835 The reference to “the deposition of a thin fibrous layer of tissue which can grow through the interstices of the mesh, thus incorporating the mesh into adjacent tissue” was also misleading. This is because in some cases, as the evidence of Professors Klosterhalfen and Klinge demonstrated, as the pores diminish in size or collapse under strain, the plastic deformation of the geometry of the mesh can lead to bridging fibrosis and even encasement of the mesh in a rigid scar plate.
Erosion
2836 The respondents did not alert Australian users to the risk of erosion until, at the earliest, 8 September 2000, after TVT had been on the market for nearly a year.
2837 The initial IFU for TVT prepared by Medscand and adopted by Ethicon omitted any mention of the risk of erosion. The 8 September 2000 TVT IFU incorporated a reference to extrusion and erosion, but only as a potential consequence of “a transitory foreign body response”.
2838 By referring to complications as a consequence of a “transitory foreign response”, the respondents conveyed the erroneous notion that the listed consequences of the foreign body response could only occur soon after implantation.
2839 The evidence did not reveal why the respondents saw fit to tie those consequences to the so-called transitory foreign body response. As the applicants pointed out, the IFU for Gynemesh PS did not do this. Having regard to Dr Hinoul’s concessions as to the state of Ethicon’s knowledge at the time each of the devices was launched, the difference in the Gynemesh PS IFU cannot be attributed to a change in understanding or the acquisition of new information.
2840 Other material prepared by the respondents did nothing to clarify the matter and it, too, minimised the significance of the risks and their potential consequences. An early brochure for TVT, for example, published in 2001mentioned the risk of mesh exposure, but not extrusion or erosion:
10. Are there any side effects from the mesh?
Occasionally a portion of the mesh may be exposed but this is uncommon. It is treated by antibiotics and closure of the wound.2675
2841 On any view of the matter, this was an understatement of the potential side effects of mesh implantation in general and of the potential consequences of mesh exposure in particular. Indeed, it trivialised the adverse events that the respondents knew could arise from mesh implantation.
2842 In 2015, following the intervention of the TGA, the respondents revised the warnings about erosion. The 7 October 2015 TVT IFU, the 22 September 2015 TVT-O IFU, the 24 September 2015 TVT Abbrevo IFU and the 18 September 2015 TVT Exact IFU all removed the qualification that the foreign body response was transitory and separated the reference to transitory local irritation from the reference to a foreign body response. “Exposure” was also added as a potential consequence of the foreign body response. As amended, and with the amendments highlighted, the warnings read:
• Transitory local irritation at the wound site and a transitory foreign body response may occur.
• As with any implant, a foreign body response may occur. This response could result in extrusion, erosion, exposure, fistula formation, and/or inflammation.
(Emphasis added)
2843 The following statement was also listed as an adverse reaction:
• Mesh extrusion, exposure or erosion into the vagina or other structures or organs.
2844 None of the IFUs, however, expressly warned of the risk of late onset complications, including late onset exposure, extrusion, or erosion. There is no good reason why such a warning was not included.
2845 Professor Blaivas said that the failure to include a warning about “late erosions, chronic inflammation and changes to the physical characteristics of the mesh sling … contributed significantly to [a] widespread lack of knowledge about the potential for late erosions due to properties of the sling itself”.2676
2846 The IFUs for the SUI devices should have made it clear that any and all of these adverse events could occur at any time after implantation.
2847 Further, although risks of erosion and extrusion were mentioned, the likelihood of these complications occurring was not addressed, nor was their possible severity or duration. No information was provided about the potential consequences of erosion, exposure or extrusion for the patient.
Infection
2848 There is always a possibility of clinical infection with a synthetic prosthesis or graft.2677 Yet, it was not until 2015 that any of the IFUs for the SUI devices warned that the devices could cause an infection.
2849 When TVT was launched in Australia in 1999 the Medscand IFU contained two references to infection.
2850 The first appeared under the heading “Adverse Reactions”. It warned of the possibility that the mesh could “potentiate an existing infection”.
2851 The other reference to infection appeared under the heading “Warnings and Precautions”. It read:
Prolene® (polypropylene) mesh in contaminated areas should be used with the understanding that subsequent infection may require removal of the material.
2852 In the 8 September 2000 TVT IFU, revised the first reference to read:
• As with all foreign bodies, PROLENE mesh may potentiate an existing infection. The plastic sheath initially covering the PROLENE mesh is designed to minimize the risk of contamination.2678
2853 The same, or very similar, wording was used in the IFUs for TVT-O, TVT Secur, TVT Abbrevo and TVT Exact until 2015.
2854 As Professor Klinge observed, the statements in the IFUs that the mesh could “potentiate an existing infection” and that the plastic, removable sheath around the sling is designed to minimise the risk of contamination were both inadequate and misleading.2679
2855 They were inadequate because the IFUs did not warn of the risk that the devices could cause infection, either at the time of implantation or more worryingly, weeks, months or years later. “Potentiate” is defined in the Macquarie Dictionary as “to increase the effect or potency of, especially by working in conjunction with”. They were misleading because they insinuated that the risk was confined to women who had an infection at the time of surgery.
2856 The second statement in the Medscand IFU that Prolene mesh in contaminated areas should be used with the understanding that subsequent infection may require removal of the material was also misleading because it did not indicate that the mesh could cause an infection. Moreover, any transvaginal operation involves using mesh in a clean-contaminated area.2680 It is therefore difficult to understand why this warning drew no connection between the implantation of the mesh and the risk of developing an infection.
2857 A statement to similar effect was included in all subsequent IFUs for TVT, the 16 December 2005 TVT Secur IFU and all IFUs for TVT Exact. No such warning or similar warning appeared in any of the IFUs for TVT-O or TVT Abbrevo.
2858 The 10 September 2010 TVT Abbrevo IFU included a new warning that reusing the device or portions of it “may create a risk of product degradation and cross-contamination, which may lead to infection or transmission of bloodborne pathogens to patients and users”. Similarly worded warnings were included in the 12 August 2014 TVT Exact IFU, the 9 December 2014 TVT IFU and the 15 December 2014 TVT-O IFU. No explanation was given by the respondents for the four-year delay in amending the other IFUs. In any case, this additional warning did not capture all potential causes of infection.
2859 All IFUs for the SUI devices stated that the relevant procedure should not be used for patients who have a urinary tract infection. But they did not warn that the devices could cause a urinary tract or other infection. None included information concerning the likelihood, severity or duration of infections.
2860 In the 2015 versions of the IFUs for the SUI devices, the respondents broadened the scope for infection, adding a warning in these terms:
As with all surgical procedures, there is a risk of infection.
2861 The statement minimised the significance of the risk in three ways: first, by intimating that the risk of infection was no greater than with any surgical procedure; second, by implying that implantation of mesh could not cause infection; and third, by failing to mention the risk of late-onset infections. Professor Klinge deposed that the mesh used in all the Ethicon devices is susceptible to an increased risk of secondary, mesh-related infections as a result of the bacteria that has adhered to the mesh during implantation and as it is passed through and implanted into the clean-contaminated environment.2681 I referred to the evidence given by Professors Korda and Deprest on this subject in Part IV of these reasons which was to similar effect.
2862 In July 2000, in his history of TVT, Dr Arnaud acknowledged that as early as 1995, when Professor Ulmsten was still developing the first TVT device and surgery, infection was a known complication. Dr Arnaud wrote that “it was broadly admitted that the use of any mesh through the vaginal route was associated with a high rate of complications, such as rejection/infection and urethral erosion”.2682 It will also be recalled that in September 2003 Drs Arnaud and Robinson were telling Ethicon personnel that there was a “high risk of infection” for pelvic meshes and that “the vaginal approach is a rather unique situation in surgery” because the mesh is “placed through a septic cavity”.2683 Contrary to what the warning suggests, there was not merely an ordinary intra-operative risk of infection arising from the surgery itself, but there was also a superadded risk because the mesh was a foreign body implanted in a clean-contaminated area and because the interstices of the mesh encouraged bacterial colonisation. Unlike intra-operative risks, this risk was a perennial one. It could eventuate at any time on or after implantation. From the outset, the IFUs should have warned of this risk.
Chronic pain
2863 No warning about chronic pain appeared in any of the IFUs for the SUI devices until after this proceeding commenced. It did not appear in an IFU until the revisions in 2015 made at the behest of the TGA, by which time TVT had been on the market for over 16 years.
2864 Until 2015, the IFUs for TVT, TVT Exact and TVT Secur did not mention pain at all.
2865 The 7 January 2004 TVT-O IFU contained one reference to pain:
Transient leg pain lasting 24-48 hours may occur and can usually be managed with mild analgesics.2684
2866 This warning was repeated in subsequent IFUs for TVT-O. It also appeared in the 10 September 2010 TVT Abbrevo IFU and later iterations of its IFUs. It goes without saying that a reference to a risk of transient pain is not a warning of the risk of chronic pain. Indeed, the statement that there is a risk of transient leg pain without also mentioning the risk of chronic pain suggests that pain will only be transient.
2867 Having regard to the potential consequences of chronic pain and the difficulties in treating it, this was a serious omission.
2868 Brochures for patients and surgeons did nothing to fill the gap caused by the absence of a warning in the IFUs. The early brochure for TVT published in 2001, entitled “GYNECARE TVT Tension-Free Support for Incontinence: Information for Patients”, is a good example. As noted above, the brochure included a question and answer format for common issues a patient might wish to know about. To the question “Will I have pain after the operation?”, the answer is given:
Some mild pain may occur over twenty-four to forty-eight hours after surgery. This may be controlled by simple pain relief.2685
2869 Not only was the risk of chronic pain not disclosed, despite the evidence that Ethicon knew about it before the product was launched, but, as with the TVT-O and Abbrevo IFUs, the clear inference was that there was no possibility of chronic pain.
2870 In cross-examination Dr Hinoul agreed that the respondents would have no reason to expect that a surgeon would deliver a different message about the device than the information given in the brochure, including as to the risk of pain.2686 He also agreed that an ordinary, reasonable patient reading the brochure would expect not to suffer long-term pain as a complication, but added that “the vast majority of patients” would not suffer this complication.2687
2871 To the extent that any warning missing from the IFUs might have been conveyed as part of surgeon training, the only training materials in evidence were the PowerPoint slides prepared by Professor Korda, which did not include a warning about chronic pain.2688 To the contrary, they suggested that chronic pain would not be a complication of TVT. Under the heading “Pain”, the slides stated, among other things, “Although chronic pain has been reported in up to 12% of patients following colposuspension and 10% after needle suspension, it has not been previously reported with TVT”.2689
2872 In 2015 a large number of potential adverse reactions were added to the IFUs for all SUI devices, save for TVT Secur because it was no longer on the market. In the adverse reactions section, the following warnings were added to the 18 September 2015 TVT Exact IFU, the 22 September 2015 TVT-O IFU, the 24 September 2015 TVT Abbrevo IFU and the 7 October 2015 TVT IFU:
• Acute and/or chronic pain.
• …
• Neuromuscular problems, including acute and/or chronic pain in the groin, thigh, leg, pelvic and/or abdominal area may occur.
2873 While the term “chronic pain” now appeared, there was no information regarding the likelihood of it occurring, how severe it might be, how long it could last, or how difficult it could be to treat.
2874 As the applicants submitted, a reference to pain or chronic pain which did not capture the potential level of pain, the difficulty of treating it, or its potentially life-altering consequences was “plainly inadequate”.2690
2875 The failure to adequately warn about the risk of chronic pain was a fundamental inadequacy in the IFUs accompanying the SUI devices.
Dyspareunia and apareunia
2876 The risk of a device causing dyspareunia or apareunia was not mentioned in any IFU for the SUI devices until 2015.
2877 While dyspareunia and apareunia are risks associated with other surgical treatments for stress urinary incontinence, in the light of the way the devices were marketed and in view of the essential principles, they should have been included in the IFUs. Otherwise, surgeons and patients alike might think that the risks arising in other kinds of surgery would or might be avoided by the use of an SUI device.
2878 Once again, the brochures did nothing to compensate for the lack of warning in the IFUs. In contrast, some brochures downplayed the risks and suggested that patients would return to normal sexual function within several weeks of their procedures. A 2004 patient brochure for TVT and TVT-O includes a series of answers to questions that a patient might expect to ask regarding the procedure and devices. It stated:
When can I have sexual intercourse?
Usually 4-6 weeks following the procedure or after your first visit back to see your doctor.2691
2879 It was not until 2015 that a warning was given about the risks of developing dyspareunia and apareunia. The following adverse reaction was added to the 7 October 2015 TVT IFU, the 22 September 2015 TVT-O IFU, the 24 September 2015 TVT Abbrevo IFU and the 18 September 2015 TVT Exact IFU:
• Pain with intercourse which in some patients may not resolve.
Heightened inflammatory response in patients with autoimmune disorders
2880 No warning of the possibility of a heightened inflammatory response was included in the IFUs for any of the SUI devices until 2015 and no warning appeared in the IFU for TVT Secur at any time.
2881 In the Medscand IFU the only patients for whom TVT was said to be contraindicated were pregnant patients, patients on anti-coagulation therapy, and patients with urinary tract infections. It was not until the 8 September 2000 TVT IFU, after TVT had been on the market in Australia for at least a year, that patients with future growth potential, including women with plans for future pregnancy, were added. No other contraindications were included in any iteration of the TVT IFU.
2882 The first reference to a warning suggestive of the possibility that some patients are more susceptible to complications than others appeared in the 18 September 2015 IFU for TVT Exact. This IFU introduced a new section under the heading “Patient Factors” the full text of which was as follows:
Physicians should use their surgical experience and judgment to determine if PROLENE Mesh is appropriate for certain patients. Patient-specific factors may impair wound healing, which may increase the likelihood of adverse reactions. 2692
2883 This statement was also included in the 22 September 2015 TVT-O IFU, the 24 September 2015 TVT Abbrevo IFU and the 7 October 2015 TVT IFU.
2884 This warning was not sufficient to alert the reader of the potential effects of implantation of any of the devices on a patient with an autoimmune disorder or whose immune system was compromised. No direction or assistance was afforded the surgeon about which factors might have the stated effect. The evidence was that “relative contradictions” to the use of biomaterials include poorly controlled diabetes, morbid obesity, heavy smoking, a history of previous pelvic radiation, severe urogenital atrophy, concurrent use of systemic steroids, and patients with autoimmune conditions.2693 This kind of information should have been included in the IFUs.
2885 At the very least, the IFUs should have noted that the devices had only been trialled on a particular patient cohort and identified those classes of patients who were excluded from the pre-market studies of TVT and their follow-ups. In addition, users should have been notified that there had been no long-term follow-up from any of the studies and that in the absence of long-term follow-up it was not possible to identify other patients for whom the device might be contraindicated but that, having regard to the unpredictability of the foreign body response in a particular patient, the device should not be used in any patient with an autoimmune disorder or whose immune system was suppressed.
2886 The respondents argued that no complaint could be made because all the IFUs indicated that a foreign body response is a potential adverse reaction and, to the extent that it is suggested that they do not indicate that the inflammatory response can be affected by autoimmune disorders, this was a matter that could reasonably be expected to be within the knowledge of specialist surgeons.
2887 I reject both arguments.
2888 First, the mere reference to a foreign body response tells one nothing about the particular susceptibility of certain classes of patients.
2889 Second, for reasons given in the following chapter, even if this were a matter of which specialist surgeons should have been aware, it should have been included in the IFUs. Professor Klinge, who it will be recalled was a consultant to Ethicon, deposed that the IFUs for all the devices should state that they should not be used in a patient with a compromised immune response.2694 No objection was taken to the inclusion of this statement in his affidavit and he was not challenged on the point during cross-examination.
Reoperation and revision surgery
2890 As noted above, all IFUs in evidence for TVT, TVT Secur and TVT Exact included a statement that using mesh in contaminated areas could result in subsequent infection which might require removal of the material. No warning to this effect was ever given for TVT-O or TVT Abbrevo.
2891 In addition to not adequately warning of the risk of the infection as discussed above, this statement did not sufficiently address the prospect of reoperation or revision surgery.
2892 First, in the absence of mentioning other reasons for reoperation, the warnings implied that infection was the only potential cause of the need to remove the material.
2893 Second, there was no warning of the difficulty of removing the mesh, the fact that some portions of the mesh might never be able to be removed, or of the potential consequences of leaving mesh behind.
2894 The 8 September 2000 TVT IFU added another warning:
Punctures or lacerations of vessels, nerves, bladder or bowel may occur during needle passage and may require surgical repair.2695
2895 Statements to similar effect appeared in all the other IFUs for all the SUI devices. This warning also did not sufficiently cover the risk of the need for reoperation or revision surgery. The inclusion of the term “during needle passage” confined the warning to incidents during surgery. It suggested reoperation might be required where punctures or lacerations were caused by surgical instruments, but not where it was required to address complications caused by the device.
2896 This warning was amended in 2015. Instead, the following statement appeared in the 2015 revisions to all IFUs for the SUI devices still on the market, with changes marked:
Punctures or lacerations of vessels, nerves, structures or organs, including the bladder, urethra or bowel may occur during needle passage and may require surgical repair.
(Emphasis added)
2897 This statement was misleading when read in the context of the IFU as a whole and in circumstances in which users were not informed that the IFU had changed. Without identifying that this kind of damage could arise after surgery, including long after surgery, one might reasonably infer that the reference related only to damage during surgery.
2898 The 2015 revisions also included the following statement towards the end of the list of “Adverse Reactions”:
• One or more revision surgeries may be necessary to treat these adverse reactions.
2899 This was the first time a warning was given of the possibility that further surgeries might be required to treat complications caused by implantation of the devices. Once again, however, the warning was deficient. There was no reference to the fact that revision surgeries might be incapable of successfully treating some or all of the complications caused by the mesh.
The difficulty or impossibility of removing mesh
2900 No warning of the difficulty or impossibility of removal was included in an IFU for any SUI device until 2015.
2901 In 2015, the following statement was added to the IFUs for all SUI devices remaining on the market:
PROLENE Mesh is a permanent implant that integrates into the tissue. In cases in which the PROLENE Mesh needs to be removed in part or whole, significant dissection may be required.
2902 The applicants submitted that this update “inched toward” an acknowledgement of the risk, but pointed out that there was still no explanation as to how mesh removal surgery should be performed, or any acknowledgement that in some cases it may not be possible to remove the device completely depending the period of time for which it had been implanted. The warning was unquestionably inadequate.2696
2903 There was no evidence that the respondents offered any advice or training on mesh removal techniques, nor of any warnings given during training as to the difficulties of mesh removal.2697 To the extent that the evidence touches on this matter, it suggested that they did not. As late as 2011 they were fielding enquiries about what to do. The applicants referred to an email chain from 2011 discussing a query from an English surgeon.2698 The query was described as follows:
[The surgeon] has recently operated on a patient who had chronic leg pain after placing in a tape. He has removed the vaginal section of the tape but is unsure about how to go about removing the tape from the leg muscles. Do we have any data/recommendations on this? Or is there any surgeon in the UK experienced in this type of procedure?
2904 The query eventually found its way to Dr Hinoul. In his response, Dr Hinoul acknowledged that Ethicon had not conducted clinical trials evaluating treatment measures for patients with thigh pain, but had “anecdotal reports as well as information from the literature regarding the potential cause and management of this condition”.2699 He then went on to discuss the potential causes of thigh pain and strategies for its management, mentioning as one of the options “localized excision of mesh”.2700
2905 At no time did the IFUs include a warning that the removal of the mesh might not alleviate the patient’s symptoms, advise of the difficulties associated with removal, or indicate that complete removal might not be achievable.
Difficulties with voiding
2906 The Medscand TVT IFU did not warn that the device may cause difficulties with voiding. In the 8 September 2000 TVT IFU, Ethicon added the following warning:
• Over correction i.e. too much tension applied to the tape, may cause temporary or permanent lower urinary tract obstruction.
2907 The same warning appeared in all other IFUs in evidence for all of the SUI devices.
2908 The applicants rightly submitted this warning was inadequate. It suggested that over-correction or tensioning may be the result of surgeon technique and did not acknowledge the inherent potential for the mesh to over correct due to contraction.2701
2909 The applicants criticised Ethicon for failing to include a warning about the potential for excessive contraction or shrinkage of the tissue surrounding the implant. The applicants submitted that excessive contraction or shrinkage ought to be the subject of a warning as it can lead to complications such as urinary tract obstruction, retention, irritation of the urethra and de novo urge incontinence. This was particularly problematic in circumstances where a warning to this effect was included in the IFUs for the same devices in Canada after intervention from the Canadian regulator.2702
2910 The product brochures did not provide appropriate warning of this complication either. In some cases, they downplayed the risks. A 2004 brochure for TVT and TVT-O for patients includes a series of answers to questions that a patient might expect to ask regarding the procedure and devices. It states:
Will I have difficulty emptying my bladder?
Some patients may experience temporary difficulty urinating following surgery and may require catheterisation back in the recovery ward. If urinating difficulty persists the tape may have to be loosened or even divided to relieve the problem.2703
2911 In 2015, a large number of potential adverse reactions was added to the IFUs for the SUI devices that were still on the market. Relevantly, these included “[v]oiding dysfunction” and “[u]rinary retention”.
De novo and recurrent urinary incontinence
2912 The TVT Medscand IFU did not warn of the risk of de novo and recurrent urinary incontinence.
2913 The 8 September 2000 TVT IFU included the following statement under the heading “Warnings and Precautions”:
The patient should be counseled that future pregnancies may negate the effects of the surgical procedure and the patient may again become incontinent.
2914 This same warning was included in all other IFUs for all of the SUI devices. No mention was made of the fact that patients might experience recurrence of incontinence in the absence of further pregnancies or for other reasons. Nor was there any mention of the prospect of de novo urinary incontinence.
2915 The 22 December 2003 TVT IFU included a new warning:
As with other incontinence procedures, de novo detrusor instability may occur following the TVT procedure. To minimize this risk, make sure to place the tape tension-free in the mid-urethral position.
2916 A warning to similar effect was incorporated into all IFUs for the other SUI devices.
2917 The adverse reactions section of the 16 December 2005 TVT Secur IFU contained the following additional warning which was not found in any of the IFUs for the other SUI devices:
Under-correction or incorrect placement may result in incomplete or no relief from urinary incontinence.
2918 The 2015 revisions to the IFUs for all the SUI devices remaining on the market included the following under the heading “Adverse Reactions”:
• Recurrence of incontinence
• Urge incontinence
• Urinary frequency
Offensive discharge
2919 No adequate warning was ever provided of the risk of offensive vaginal discharge.
2920 Up until the 2015 revisions, the prospect of a device causing any kind of vaginal discharge was not mentioned in any IFU for any SUI device, let alone the risk that the discharge could be offensive.
2921 In 2015 the following was added to the list of “Other Adverse Reactions” in each of the IFUs for the SUI devices remaining on the market:
• Atypical vaginal discharge
2922 The Macquarie Dictionary defines “atypical” as “not typical; not conforming to the type; irregular; abnormal”. The word is neutral. Its use in the IFUs suggested the discharge might be out of the ordinary, but it did not suggest that the discharge could be offensive.
Damage to surrounding organs, nerves, ligaments, tissue and blood vessels
2923 The risk of damage to surrounding organs, nerves, ligaments, tissue and blood vessels was not mentioned in the Medscand TVT IFU.
2924 As discussed above, the 8 September 2000 TVT IFU introduced a warning about “punctures or lacerations of vessels, nerves, bladder or bowel may occur during needle passage … ”.
2925 This statement was misleading because it implied that injury of this kind could only occur during surgery when mesh migration, exposure, extrusion and erosion could also cause damage of that kind to vessels, nerves, bladder or bowel. It was also misleading because it neglected to mention damage to the urethra.
2926 As already noted, the statement was amended in 2015 to substitute “bladder or bowel” for “structures or organs, including the bladder, urethra or bowel” and to remove the phrase “during needle passage”. For the reasons already given, the warning remained deficient.
Haemorrhage
2927 No adequate warning was given about the risk of haemorrhage in any IFU for any SUI device until 2015.
2928 The Medscand TVT IFU made no reference to the possibility of bleeding, let alone haemorrhage. The 8 September 2000 TVT IFU included under the heading “Warnings and Precautions”:
Retropubic bleeding may occur postoperatively. Observe for any symptoms or signs before releasing the patient from hospital.
2929 Statements to similar effect were included in all IFUs for SUI devices from then on. Warning of post-operative bleeding does not fully encompass the risk of haemorrhage.
2930 In 2015 the following was listed as an adverse reaction in all IFUs for the SUI devices remaining on the market:
• Bleeding including hemorrhage, or hematoma.
Leg weakness
2931 No warning of any kind was ever given in any IFU for an SUI device of the risk of developing leg weakness.
Psychiatric injury
2932 No warning of any kind was ever given in any IFU for an SUI device of the risk of psychiatric injury.
The absence of any or any adequate clinical or other evaluation of the risks
2933 The absence or limitations of clinical studies and the shortcomings of Ethicon’s own clinical evaluations were not disclosed in any of the IFUs for the SUI devices and no mention was made of the rates or incidence of complications.
2934 In some IFUs, the respondents adverted to a lack of clinical information regarding the use of the devices in the event of pregnancy, but did not warn of the overall lack of evaluation of the risks of the devices or the limitations of the evaluations that had been undertaken.
2935 The 22 December 2003 TVT IFU stated:
Since no clinical experience is available with vaginal delivery following the TVT procedure, in case of pregnancy delivery via cesarian section is recommended.
2936 Statements to similar effect were included in all IFUs for SUI devices published after 2003.
2937 The 7 January 2004 TVT-O IFU stated:
Since no clinical information is available about pregnancy following sub-urethral sling procedure with the GYNECARE TVT Obturator System, the patient should be counseled that future pregnancies may negate the effects of the surgical procedure and the patient may again become incontinent.
2938 Statements to similar effect were included in the 16 December 2005 TVT Secur IFU and all IFUs for TVT Abbrevo.
2939 Neither representation was sufficient to alert surgeons or patients to the absence of, or limitations in, the evaluations of the risks of the devices.
Adequacy of warnings and other information provided about the POP devices
2940 The applicants submitted that the starting point in reviewing the IFUs for the POP devices was to recognise that (with the possible exception of Gynemesh PS when used abdominally) no warning would have been adequate to permit surgery to be performed to implant one of the POP devices in any patient save in extremely exceptional circumstances.2704 The applicants argued that it might be possible for a patient to consent to surgery with a POP device where the patient had severe prolapse, her primary surgery had failed and she was willing to have surgery as part of a clinical research project.2705 If they were to be launched, the POP devices should have been indicated for such limited use.2706
2941 The applicants submitted that their approach was consistent with the TGA’s decision to remove all mesh implants for transvaginal POP repair surgery from the ARTG in November 20172707 and with the evidence given by Professor Deprest that the POP devices produce an unacceptable rate of adverse events such as exposure and contracture.2708
2942 Associate Professor Lam agreed that, as at 2004, the evidence was insufficient to support the use of permanent meshes in vaginal prolapse repair surgery except in the context of randomised controlled clinical trials. He also agreed that the manufacturer should have alerted potential users of its products to the state of the evidence.2709 Moreover, he accepted that the position was no different in 20072710 or, in the case of posterior vaginal repair, in 2013.2711 Associate Professor Lam also agreed that by 2016 the risk/benefit profile was such that transvaginal mesh had limited utility in primary surgery and that there was no evidence that the benefits of mesh surgery outweigh the risks. 2712 No such information was included in any of the IFUs or product brochures and the respondents did not point to any evidence to indicate that they conveyed the information to potential users in some other way.
2943 The applicants also criticised the fact that Gynemesh PS was indicated and promoted for prolapse repair without limitation as to use, so that it might be used both transvaginally and abdominally.2713 The 16 March 2013 Gynemesh PS stated:
GYNECARE GYNEMESH™ is indicated for use as a bridging material for apical vaginal and uterine prolapse where surgical treatment (laparotomy or laparoscopic approach) is warranted.
2944 As the applicants submitted, this updated warning was inadequate. In July 2017, the TGA wrote to JJM stating that this instruction did not “clearly articulate that the Device [was] not intended for use for pelvic organ prolapse repair via a transvaginal approach”.2714 It imposed a condition that the following precaution be added to the IFU for the device:
This device is not intended for any pelvic organ prolapse repair via a transvaginal approach.
2945 It is difficult to see why that was not made clear at the time of the change of indication in 2013.
2946 Under the heading “Performance”, the 20 March 2003 Gynemesh PS IFU represented that animal studies showed that:
[I]mplantation of PROLENE mesh elicits a minimum to slight inflammatory reaction, which is transient and is followed by the deposition of a thin fibrous layer of tissue which can grow through the interstices of the mesh, thus incorporating the mesh into adjacent tissue…2715
2947 The same or similar statements appeared in all IFUs in evidence for all the POP devices. As with the IFUs for the SUI devices, all made the false representation that the inflammatory reaction to Prolene was transient. These statements were also misleading because they failed to alert readers to the possibility that the inflammatory reaction could be excessive and they failed to identify those patients most at risk.
2948 None of the Prolift, Prolift+M or Prosima IFUs mentioned a foreign body reaction or chronic inflammation, only inflammation.
Erosion
2949 The original adverse reactions section of the 30 March 2003 Gynemesh PS IFU read as follows:
Potential adverse reactions are those typically associated with surgically implantable materials, including infection potentiation, inflammation, adhesion formation, fistula formation, erosion, and extrusion.
2950 In the subsequent version, being the 31 March 2006 Gynemesh PS IFU, this warning was slightly expanded, with changes marked as follows:
Potential adverse reactions are those typically associated with surgically implantable materials, including … adhesion formation, fistula formation, erosion, and extrusion and scarring that results in implant contraction.
2951 This version of the warning appeared in all subsequent Gynemesh PS IFUs until 2013. It appeared in the 11 January 2005 Prolift IFU and its later version until 1 October 2009. It was also included in all Prosima IFUs until the product was discontinued.
2952 Both versions above omitted any reference to the risk of developing late onset erosion, which exists for as long as the mesh is implanted. They did not acknowledge that such a complication may not resolve, even with treatment.
2953 In the 1 October 2009 Prolift IFU, this section was amended to remove the reference to “scarring resulting in implant correction” and replace it with the generic terms “contracture” and “scarring”. The following passage highlights the changes:
Potential adverse reactions are those typically associated with surgically surgery employing implantable materials of this type, including … adhesion formation, fistula formation contracture, scarring, and mesh exposure, erosion, or extrusion and scarring that results in implant contraction.
(Emphasis added)
2954 This remained the warning until Prolift was discontinued. This was also the version of the warning used in all Prolift+M IFUs from 12 December 2008 until the product was discontinued.
2955 The 16 March 2013 Gynemesh PS IFU changed the warning as follows:
Potential adverse reactions are those typically associated with surgery employing implantable materials of this type… adhesion formation, fistula formation, contracture, scarring, and mesh exposure, erosion, or extrusion, e.g., through vaginal epithelium.
(Emphasis added)
2956 The 3 April 2015 Gynemesh PS IFU again revised the warning:
Potential adverse reactions are those typically associated with surgery employing implantable materials of this type… adhesion formation, fistula formation, contracture, scarring, and mesh extrusion, exposure, or erosion , e.g., through vaginal epithelium into the vagina or other structures or organs.
(Emphasis added)
2957 It also went on to add for the first time:
As with any implant, a foreign body response may occur which could result in extrusion, erosion, exposure, fistula formation and/or inflammation.
Infection
2958 The original adverse reactions section of the 30 March 2003 Gynemesh PS IFU read:
Potential adverse reactions are those typically associated with surgically implantable materials, including infection potentiation…
2959 The risk of “infection potentiation” was included in all subsequent Gynemesh PS IFUs and all other IFUs for POP devices in evidence.
2960 The 30 March 2003 Gynemesh PS IFU also included the following warning:
The use of GYNECARE GYNEMESH PS in contaminated wounds should be used with the understanding that subsequent infection may require additional surgical procedures such as removal of the mesh.
2961 Warnings to the same or similar effect were included in all later Gynemesh PS IFUs, as well as all those for Prosima and Prolift+M. Curiously, a general warning that infection may require removal of the mesh was not included in any Prolift IFU. Instead, the 1 October 2009 Prolift IFU and those issued later included the following warning:
GYNECARE GYNEMESH™ PS Mesh must not be used following planned intra-operative or accidental opening of the gastrointestinal tract. Use in these cases may result in contamination of the mesh, which may lead to infection that may require removal of the mesh.
2962 A very similar warning appeared in the 12 December 2008 Prolift+M IFU, as well as the 16 March 2013 Gynemesh PS IFU, in addition to the more general warning.
2963 The 3 April 2015 Gynemesh PS IFU was the first and only IFU for a POP device that warned of a risk of infection, rather than the risk of “potentiating” an existing infection. It stated:
As with all surgical procedures, there is a risk of infection. As with all foreign bodies, GYNECARE GYNEMESH™ may potentiate an existing infection.
2964 This warning remained deficient. It was also misleading. It implied that the risk of infection could only arise intra-operatively or post-operatively. It did not advert to the risk that infection could occur long after surgery.
2965 For the Prolift, Prolift+M and Prosima devices, Dr Pence observed that a warning of the risk of infection was never included (only infection potentiation) and that none of the IFUs in evidence ever warned of the risk of developing urinary tract infections.2716
Chronic pain
2966 The 3 April 2015 Gynemesh PS IFU included a long list of adverse reactions that had previously not appeared in any IFU for a POP device. Among them was “acute and/or chronic pain”. It also stated:
Neuromuscular problems, including acute and/or chronic pain in the groin, thigh, leg, pelvic and/or abdominal area may occur.
2967 No IFUs for Prolift, Prolift+M and Prosima ever warned of the risk of chronic pain and where they did refer to pain the references were not only inadequate, they were also misleading.
2968 The first reference to the word “pain” in any IFU for a POP device did not occur until the 11 January 2005 Prolift IFU, which stated:
Transient leg pain may occur and can usually be managed with mild analgesics.
2969 The same warning was included in all subsequent Prolift IFUs and all IFUs for Prolift+M.
2970 The 19 June 2007 Prosima IFU included the statement:
Potential adverse reactions are those typically associated with pelvic organ prolapse repair procedures, including pain with intercourse and pelvic pain. These may be self-resolving over time.
2971 A similar warning appeared in the 12 December 2008 Prolift+M IFU and its subsequent revision in 2011. It was also included in the 1 October 2009 Prolift IFU and its subsequent revision. It did not appear in the Gynemesh PS IFU until 16 March 2013, despite two revisions to the IFUs occurring in 2008. The respondents provided no explanation for the six year delay in putting this warning in the Gynemesh PS IFU.
2972 As the applicants submitted, the clear inference to be drawn from this warning was that a patient would not experience pain that was materially different from that which could occur after native tissue procedures.2717 This was misleading because the evidence demonstrated that adverse outcomes associated with mesh surgery could be worse, indeed much worse, than after native tissue repair.
Dyspareunia and apareunia
2973 No warning was given of the risk of developing dyspareunia or apareunia until 2007 when “pain with intercourse” was included as an adverse reaction in the 19 June 2007 Prosima IFU:
Potential adverse reactions are those typically associated with pelvic organ prolapse repair procedures, including pain with intercourse and pelvic pain. These may be self-resolving over time.
2974 Warnings to similar effect were later included in the 12 December 2008 Prolift+M IFU, the 1 October 2009 Prolift IFU and the 16 March 2013 Gynemesh PS IFU.
Heightened inflammatory response in patients with autoimmune disorders
2975 At no time did any of the IFUs for the POP devices list as a contraindication the presence of an autoimmune disorder or long-term use of immune-suppressants. Nor did any advise that the chronic inflammatory response to implantation could be affected by the presence of autoimmune, connective tissue and like disorders which affect the immune response to healing. The following statement was included among the precautions in the 12 December 2008 Prolift+M IFU:
In patients with compromised immune systems or other conditions that would compromise healing the risks and benefits should be carefully weighed.
2976 The same statement was listed in the precautions of the 1 October 2009 Prolift IFU and a statement to very similar effect in the 16 March 2013 Gynemesh PS IFU. No explanation was given as to why doctors and patients had not been notified sooner.
2977 The respondents submitted that, at least from those dates, the applicants’ claim that there had been a failure to warn of the risk to immunocompromised patients was unsustainable. I reject the submission. A doctor is obliged to weigh the risks and benefits of all treatments for all patients. Apart from drawing attention to such conditions, this precaution told the reader nothing.
2978 Other material provided by the respondents did not rectify the omissions from the IFUs. An undated Prolift patient brochure posed the question “Is GYNECARE PROLIFT (PFRS) right for me?” The answer it offered was this:
Pelvic floor repair procedures with GYNECARE PROLIFT (PFRS) are appropriate for almost all patients, including overweight patients, elderly patients, and even those who have undergone previous operations for pelvic organ prolapse or stress incontinence. As with any surgery of this kind, this procedure should not be performed on pregnant women, infants or children. It should also not be considered by women who plan a future pregnancy. Only a complete physical examination and consultation with your physician can determine which procedure is right for you. 2718
(Emphasis added)
2979 It is difficult to see any reasonable basis for the representation that Prolift was “appropriate for almost all patients”. The failure to refer to the risks posed to patients with autoimmune disorders is a notable omission.
2980 In the 3 April 2015 Gynemesh PS IFU, a new section was inserted entitled “patient factors” identical in its terms to the “patient factors” section introduced in the TVT IFUs later that year. It will be recalled that physicians were advised to use their surgical experience and judgment to determine if the product was appropriate for the patient and told that “patient-specific facts may impair wound healing which may increase the likelihood of adverse reactions”.
2981 On the other hand, all IFUs for the POP devices stated that “normal wound healing is not noticeably impaired”. As Professor Klinge observed, that statement is not always true. Erosions occur and an erosion is not a sign of “normal wound healing”. Moreover, he added, having regard to the intense foreign body reaction that occurs at the mesh-tissue interface, “the chronic inflammatory response leads to chronic wound healing issues in many patients”.2719 Without any warning that a noticeable impairment in normal wound healing is not invariably the case and indicating the kinds of patients for whom it might not hold true, such as immunosuppressed women, this statement was also misleading.
2982 As they did with respect to the IFUs for the SUI devices, the respondents argued that no warning was required because these were matters which it could reasonably be expected were within the knowledge of specialist surgeons. But the evidence indicated otherwise.
2983 In his third report, Professor Korda wrote that “patients with vaginal atrophy, who smoke or use immunosuppressant medications should not undergo mesh implant surgery” and that contradictions of this kind should be included in the manufacturer’s instructions for use.2720 In examination in chief, Professor Korda was asked why patients with these attributes should not undergo mesh implant surgery. He replied:
It actually came out of a study that was performed by the European mesh group that looked at this about five or six years ago and identified patients at high risk. What they found is that patients at high risk were those who have immunosuppressed conditions, for instance, rheumatoid arthritis or any other type of immune disease and/or smoking is associated with a lowered immune response, and, obviously, vaginal atrophy is another condition that doesn’t allow you to dissect the vagina as thickly as we saw on that lady in the video. But this is information that was not around until the inquiry.2721
2984 When asked what he meant by “not around”, Professor Korda replied that obstetricians, gynaecologists, and urologists did not know “that people who were immunosuppressed shouldn’t have mesh”.2722 As I noted in Gill (No 1), he was not challenged about this evidence in cross-examination.
2985 Although Professor Korda did not identify the study he had in mind, it is likely to be the study by the TVM Group, reported by Elmér et al and published online on 19 April 2012.2723
2986 In her affidavit, Dr Robyn Leake, who treated Mrs Gill for some of the complications of her Prolift surgery, deposed that “an autoimmune disorder, psoriasis, might have predisposed her to some complications due to idiosyncratic scarring of tissue around the Prolift mesh implant”.2724 She did not indicate, however, when she became aware of this possibility. Since she was not required for cross-examination, the respondents did not put to her that she was aware of it at the time.
Reoperation and revision surgery
2987 A general warning of the risk of requiring reoperation or revision surgery was not given in any IFU for a POP device until 2015, by which point Gynemesh PS was the only device left on the market.
2988 In the 3 April 2015 Gynemesh IFU, the following warning was added:
• These adverse reactions may require surgical treatment.
• As with any surgery, one or more revision surgeries may be necessary to treat these complications.
2989 As Dr Pence observed, however, the IFUs for Prolift, Prolift+M and Prosima did not advert to the prospect of multiple operations, mesh revision or removal (other than surgery to repair punctures or lacerations during implantation or in response to infection).2725
2990 In the light of Dr Hinoul’s concessions and based on the evidence as a whole, the IFU’s should have advised, from the outset, of the possibility that additional surgery could be required and that further surgery may not cure or correct a complication.
The difficulty or impossibility of removing a device
2991 As discussed above, a precaution in the following terms was included in the IFUs for Prosima and Prolift+M from the outset, and added in the 1 October 2009 Prolift IFU and the 16 March 2013 Gynemesh PS IFU, that “[if] the Mesh Implant is used in contaminated areas it must only be with the understanding that subsequent infection may require its removal”. This statement was misleading in that it indicated that removal would only be required in the case of infection and it was deficient in that it did not advert to the difficulties of removal or to the fact that removal might not alleviate pain. Moreover, none of the IFUs provided any instruction on the method of removal.
2992 The first allusion to potential difficulties with mesh removal did not appear in any IFU until 3 April 2015 when they were mentioned for the first time in the Gynemesh PS IFU. By then, however, all the other POP devices were no longer sold or manufactured.
2993 From 2015 the Gynemesh PS IFU said:
GYNECARE GYNEMESH™ is a permanent implant that integrates into the tissue. In cases in which the GYNECARE GYNEMESH™ needs to be removed in part or whole, significant dissection may be required.2726
2994 As the applicants submitted, this statement did not properly convey the trauma associated with, and the potential impossibility of, mesh removal. Further, it did not state that mesh removal may not resolve the complication sought to be treated, such as with some patients with chronic pain.2727
Difficulties with voiding
2995 The risk of the devices causing difficulties with voiding was not included in an IFU for POP device until 2007.
2996 The first mention of this complication was in the 19 June 2007 Prosima IFU, which stated:
Dissection for pelvic floor repair procedures has the potential to impair normal voiding for a variable length of time.
2997 The same warning appeared in the 12 December 2008 Prolift+M IFU, the 1 October 2009 Prolift IFU and the 16 March 2013 Gynemesh PS IFU. The respondents provided no explanation for the delay.
2998 The 12 December 2008 Prolift+M IFU added “voiding dysfunction” to the list of potential adverse reactions. It was later added to the 1 October 2009 Prolift IFU and the 16 March 2013 Gynemesh PS IFU. The risk of “voiding dysfunction” was never listed as an adverse event in any Prosima IFU.
2999 No mention was made of the risk of developing difficulties with defaecation in any IFU for any POP device in evidence.2728
De novo and recurrent urinary incontinence
3000 The 12 December 2008 Prolift+M IFU stated:
Prolapse repair may unmask pre-existing incontinence conditions.
3001 This warning was later included in the 1 October 2009 Prolift IFU and the 16 March 2013 Gynemesh PS IFU.
3002 The 12 December 2008 Prolift+M IFU also listed “urinary incontinence” among a number of potential adverse reactions. The same term appeared in the 1 October 2009 Prolift IFU and the 16 March 2013 Gynemesh PS IFU.
Offensive discharge
3003 No adequate warning was ever given about the risk of offensive discharge in any IFU for a POP device.
3004 The 3 April 2015 Gynemesh PS IFU only referred to “atypical vaginal discharge”.
3005 The first mention of recurrent prolapse appeared in 2008.
3006 The 12 December 2008 Prolift+M IFU stated:
Potential adverse reactions are those typically associated with surgery employing implantable materials of this type, including… recurrent prolapse…
3007 This same warning appeared in the 1 October 2009 Prolift IFU and the 16 March 2013 Gynemesh PS IFU. But no such warning was ever included in a Prosima IFU.
Damage to surrounding organs, nerves, ligaments, tissue and blood vessels
3008 The first warning relevant to this complication appeared in the 11 January 2005 Prolift IFU, which stated:
Punctures or lacerations of vessels, nerves, bladder, urethra or bowel may occur during GYNECARE PROLIFT Guide passage and may require surgical repair.
3009 A very similar warning was included in all Prosima IFUs and all Prolift+M IFUs.
3010 A warning to similar effect was not included in a Gynemesh PS IFU until 2015. The warning with the 3 April 2015 Gynemesh PS IFU read:
Punctures or lacerations of vessels, nerves, structures or organs, including the bladder, urethra or bowel, may occur and may require surgical repair.
3011 The 11 January 2005 Prolift IFU also provided the following instruction:
The GYNECARE PROLIFT Pelvic Floor Repair Systems should be used with care to avoid damage to vessels, nerves, bladder and bowel. Attention to patient anatomy and correct use of the device will minimize risks.
3012 An instruction was included in all Prosima IFUs and all Prolift+M IFUs to similar effect. They also mentioned the possibility of perforation of the vaginal wall. For example, the 19 June2007 Prosima IFU said:
Use the GYNECARE PROSIMA Systems with care, and with attention to patient anatomy, to avoid damage to vessels, nerves, bladder, bowel, and vaginal wall perforation. Correct use of the GYNECARE PROSIMA System components will minimize risks.
3013 The 1 October 2009 Prolift IFU added “nerve damage” and “vaginal wall perforation” to the list of adverse reactions.
3014 No IFU for Gynemesh PS contained a warning about damage to surrounding organs, nerves, ligaments, tissue and blood vessels until 2013. At that point, “nerve damage” was added to the list of adverse reactions. The 16 March 2013 Gynemesh PS IFU also stated:
Use GYNECARE GYNEMESH™ with care, and with attention to patient anatomy and to proper dissection technique, to avoid damage to vessels, nerves, bladder, bowel, and vaginal wall.
Haemorrhage
3015 No warning of the possibility of haemorrhage was given in any IFU for a POP device until 2015. The 3 April 2015 Gynemesh PS IFU listed “bleeding including hemorrhage, or hematoma” among the potential adverse reactions.
Leg weakness
3016 No warning of any kind was ever given in any IFU for a POP device of the risk of developing leg weakness.
Psychiatric injury
3017 No warning of any kind was ever given in any IFU for a POP device of the risk of psychiatric injury.
The absence of any or any adequate clinical or other evaluation of the risks
3018 No IFU for Gynemesh PS ever addressed the absence of any or any adequate evaluation of the risks of the device.
3019 Both IFUs for Prosima merely stated:
Use of the GYNECARE PROSIMA System has not been fully evaluated in patients with Stage IV pelvic organ prolapse. Therefore its use in these patients is not recommended.
3020 The 1 October 2009 Prolift IFU made the following comment in the introductory section:
The safety and effectiveness of the GYNECARE PROLIFT™ Systems compared to conventional surgical repair for pelvic organ prolapse have not been demonstrated in randomized controlled clinical trials. In the United States, substantial equivalence of the GYNECARE PROLIFT™ Systems to synthetic mesh with the same indication has been demonstrated through benchtop and cadaveric testing. Information on the clinical performance of mesh for pelvic floor repair is available in published literature. Contact your company sales representative for assistance.
3021 A similar statement was included in the 12 December 2008 Prolift+M IFU.
3022 In his closing argument lead counsel for the respondents drew attention to this passage in the Prolift IFU.2729 It seems to me, however, that it provides little assistance to the respondents’ case. The first sentence was only included because the FDA required it and, even then and for no apparent reason, it was disconnected from the clinical performance section of the IFU, contrary to the FDA’s suggested template. The respondents acknowledged that the second sentence was meaningless to an Australian surgeon.2730 The third was little more than a statement of the obvious. Moreover, it did not even inform the reader that the literature included information about Gynemesh PS or where the most useful information could be found. Finally, the company sales representative would be unlikely to assist the surgeon to navigate the literature in an objective way.
3023 A new section on clinical performance was also inserted in the 1 October 2009 Prolift IFU in response to the FDA direction. Most of the information the FDA had required was included but it was not set out as clearly as it appeared in the FDA letter. The IFU disclosed that randomised controlled clinical evaluations of the device were under way, but that no data was yet available. It stated that the only available data came from two early observational studies of a pre-cut mesh in a similar shape to Prolift that were initiated in 2004. It also mentioned inclusion criteria as the FDA had required.2731
3024 In the “Clinical Performance” section of the IFU, the US and French TVM studies are then described with a purported summary of the results including rates of adverse events, as follows:
Study populations available for follow-up at 12 months were 83 patients in the US and 87 patients in France with a median patient age of 62 and 66.5, respectively.
The 12-month postoperative study results were as follows (US, France): proportion of subjects with ICS Stage II or greater (12.0%, 18.4%), met pre-defined criteria of upper limit of 90% CI less than 20% (yes, no), Prolapse Symptom Index (PSI) mean (6.6, 3.1), Mean QOL score (0.7, 0.4).
Adverse events, expressed as percentages, were as follows (US, France): hematoma (3.5, 4.5), abscess (0, 1.1), urinary tract infection within 6 weeks post-procedure (8.2, 16.9), mesh exposure (14.1, 10.0), surgical intervention for mesh exposure (7.1, 5.6), vesico-vaginal fistula (1.2, 1.1), recto vaginal fistula (1.0, 0), moderate/severe vaginal retraction (3.6, 12.6).2732
3025 Ethicon’s explanation for the modified format was “to save space”, since the information was to be translated into 28 languages.2733 While that might be true, the results were not presented as clearly as they appeared in the FDA’s suggested template. It is very easy to see in the FDA’s template that the French study did not meet the pre-defined criteria of correction of prolapse of less than 20%. It is not so obvious in the Ethicon revision.
3026 The summary also omitted any reference to dyspareunia, despite the fact that at 12 months the French study reported de novo dyspareunia in three cases (8% of the patients studied).2734
3027 This section of the IFU concluded with the suggestion that the user contact his or her sales representative for information about more recent data specific to Prolift that could become available in the published literature.
3028 The “Clinical Performance” section in the IFUs for Prolift+M contained only the first two sentences of the same summary in the Prolift IFU. In other words, it informed the reader that randomised controlled clinical evaluations of Prolift (not Prolift+M) were under way and that preliminary data was available from two early observational studies of pre-cut Gynemesh PS. But it did not inform the reader of the results or refer the reader to a publication in which the results could be found. And it did not explain how studies on Prolift were relevant to the clinical performance of Prolift+M.
Conclusion
3029 Dr Hinoul deposed that all the complications “specific to” the Ethicon devices were covered in the initial IFUs.2735 While he acknowledged that changes had been made to the IFUs from time to time, he said that these changes only incorporated risks that were common to all types of pelvic floor surgery for incontinence and/or prolapse. He did not countenance the notion that the IFUs were deficient in any respect or that the respondents’ approach was inappropriate, even in hindsight.
3030 I reject this evidence.
3031 The obligation to inform users of the risks in the use of the devices was far more extensive than Dr Hinoul acknowledged and the respondents consistently failed to discharge it. As Dr Allman put it:
Informing users of a risk requires informing users of the probability and severity of harm, and how that risk was established, not just informing users of a possible harm.2736
3032 None of the warnings or other information provided by the respondents satisfied this requirement.
3033 The respondents did provide warnings about some risks but the warnings they gave did not extend to all known risks or to all the pleaded complications. No information was provided about the probability of the risks and next to no information about their severity. Nor did the respondents disclose the manner in which the risks were established.
3034 The applicants rightly complained that, at all relevant times, the warnings and other information provided by the respondents about all the Ethicon devices were deficient.
3035 The significance of these deficiencies is discussed in Parts XIII and XIV of this judgment.
PART XII: PRELIMINARY ISSUES RAISED BY THE APPLICANTS’ CLAIMS
3036 Before dealing with the various causes of action, it is convenient to deal with some questions of both law and fact raised against the applicants.
3037 There was a festering issue about whether the three applicants were capable of representing all group members since they received only three of the Ethicon devices. The respondents argued that it was necessary to have an applicant to “present” a particular product to the Court before the Court could make adverse findings about it.2737 I rejected a similar argument in Philipsen v American Medical Systems LLC (No 2) [2018] FCA 1580. For similar reasons, the argument should be rejected here, too.
3038 I observed in Philipsen at [21] that Pt IVA of the Federal Court of Australia Act envisages that, as long as certain conditions are satisfied, a person may bring a claim on behalf of others who have different claims. In Ethicon Sàrl v Gill (2018) 264 FCR 394 at [50] the Full Court held that “the regime [in Pt IVA] expressly contemplates and provides for the individuality of claims within group proceedings”. In Philipsen I held that the applicant, who was allegedly implanted with a medical device used to treat pelvic organ prolapse, could bring a representative action, not only on behalf of women who had been implanted with devices manufactured by the respondents for use in treating that condition, but also on behalf of women who had been implanted with devices manufactured by the respondents for use in treating stress urinary incontinence.
3039 Section 33C of the FCA Act deals with the commencement of representative proceedings. It states that:
(1) Subject to this Part, where:
(a) 7 or more persons have claims against the same person; and
(b) the claims of all those persons are in respect of, or arise out of, the same, similar or related circumstances; and
(c) the claims of all those persons give rise to a substantial common issue of law or fact;
a proceeding may be commenced by one or more of those persons as representing some or all of them.
(2) A representative proceeding may be commenced:
(a) whether or not the relief sought:
(i) is, or includes, equitable relief; or
(ii) consists of, or includes, damages; or
(iii) includes claims for damages that would require individual assessment; or
(iv) is the same for each person represented; and
(b) whether or not the proceeding:
(i) is concerned with separate contracts or transactions between the respondent in the proceeding and individual group members; or
(ii) involves separate acts or omissions of the respondent done or omitted to be done in relation to individual group members.
3040 As Lee J observed in Dillon v RBS Group (Australia) Pty Limited [2017] FCA 896 at [43], the term “claim” is to be given a wide meaning; it “need not be based on the same conduct, and may arise out of quite disparate transactions”. It is clear from the terms of s 33C(1) that, provided the conditions set out in paragraphs (a)-(c) are made out, one or more persons can represent all members of the group.
3041 In Dillon Lee J remarked at [44]:
The ‘claims’ of all persons referred to in this ‘gateway’ provision are only required to be in respect of, or arise out of, similar or related circumstances and give rise to one substantial common issue of law or fact. It necessarily follows that the claims of the applicants (who represent the group) and group members (represented persons) can be quite different.
(Emphasis added)
3042 Section 33C should be read with s 33D, which deals with the standing of a person to bring proceedings on behalf of others.
3043 Section 33D(1) provides that a person who falls within the description in s 33C(1)(a) and who has a sufficient interest to commence a proceeding on his or her own behalf against another person has a sufficient interest to commence a representative proceeding against that other person on behalf of other persons referred to in that paragraph. A person who has commenced a representative proceeding but who ceases to have a claim against the respondent retains a sufficient interest to continue the proceeding: FCA Act, s 33D(2).
3044 “Sufficient interest” is not defined in the Act. In Finance Sector Union of Australia v Commonwealth Bank of Australia (1999) 94 FCR 179 at [18] the Full Court (Wilcox, Ryan and Madgwick JJ) rejected the notion that the expression invoked a special interest test similar to that applied to private suits for injunctions in aid of public rights. Rather, their Honours held that, if a provision allows “any person” to bring an application for a contravention, any person has a sufficient interest to commence a proceeding on his or her own behalf. Support for the Full Court’s interpretation may be found in the Explanatory Memorandum to the Federal Court of Australia Amendment Bill 1991 (Cth), through which Pt IVA was introduced into the FCA Act. It said of s 33D (at [13]):
This section provides that a person who has standing to bring an individual claim has standing to bring a representative proceeding on behalf of other persons under section 33C. The section also enables the representative party to continue to represent the group members even though he or she no longer has a claim against the respondent, for example, through settlement of his or her individual claim.
(Emphasis added)
3045 The purpose of this provision is to overcome the common law rule in relation to standing which would prevent one person from suing another on behalf of a third person: Symington v Hoechst Schering Agrevo Pty Ltd (1997) 78 FCR 164 at 167 (Wilcox J), cited with approval by French J in Revian v Dasford Holdings Pty Ltd [2002] FCA 1119 at [13]. Its effect is that, provided an applicant has standing to sue the respondents on her own behalf, she can bring a representative proceeding on behalf of others whose claims concern or arise out of the same, similar or related circumstances and give rise to a substantial common issue of law or fact.
3046 The only other condition that must be satisfied relates to the commencement of a claim. Section 33H requires that the application commencing a representative proceeding or a document filed in support of it must describe or otherwise identify the group members to which the proceeding relates and specify the nature of the claims made on their behalf, the relief claimed, and the common questions of law of fact. As the Full Court observed in Ethicon at [26], no suggestion was made that the claim was not validly commenced.
3047 These conditions are not merely minimum requirements, they also mark “the outer limit of the connection between the group members”: Timbercorp Finance Pty Ltd (in liq) v Collins (2016) 259 CLR 212 at [107] (Gordon J). Timbercorp concerned a case involving the equivalent regime in Pt 4A of the Supreme Court Act 1986 (Vic).
3048 It is common ground that seven or more women have claims against the respondents. The claims of the three applicants arise out of the same, similar or related circumstances because they all involve women who were supplied by JJM with a medical device made by a related company (either Ethicon Sàrl or Ethicon Inc.) from treated polypropylene knitted according to Ethicon’s design for use in pelvic surgery and who complain that, as a result of being implanted with such a device, they suffered complications of a like kind. Although there are some issues which only affect one or other of the applicants, or subsets of the group members and others which affect individual group members only, those claims give rise to substantial common issues of law and fact. As I pointed out in Philipsen at [31], “substantial” in this context merely means “real or of substance”. In the present case, one substantial common issue of fact, for example, is whether the pleaded complications could be caused by any or all of the devices and, if so, whether the respondents provided any or sufficient information, to alert consumers of the risk. There were also a number of common substantial issues of law. One of those issues was how s 75AC of the Trade Practices Act should be construed, which feeds into a mixed question of law and fact, namely, whether each of the devices had a defect or safety defect within the meaning of the TPA and the Australian Consumer Law respectively. The fact that not all of the Ethicon devices were supplied to the applicants does not mean that they were ineligible to represent those women who had been supplied with the other devices.
3049 It follows that all the relevant conditions were satisfied and the applicants are capable of representing all group members.
3050 If the respondents were suggesting that, although the matter passed through the s 33C gateway, the Court should decline to make findings about the other devices, then an application should have been made under s 33N(1) to declass the proceeding on the basis that, as constituted, it did not provide “an efficient and effective means of dealing with the claims of group members” or that it was “otherwise inappropriate that the claims be pursued by means of a representative proceeding”. But no such application was made.
The effect of the amendments to the group definition
3051 This question was answered by the judgment in Ethicon at [51]–[52]. In short, the amendments to the group definition take effect from the date the amendments were made.
3052 There were multiple amendments to the group definition over the course of this proceeding. For present purposes, it suffices to outline the following procedural history.
3053 The proceeding was commenced by the filing on 15 October 2012 of an Originating Application and Statement of Claim. At that time, the group was defined as “persons … who had surgery performed on them in Australia” to implant “one or more” of Prolift Total, Prolift Anterior or Prolift Posterior.
3054 On 29 January 2013, by the filing of an Amended Originating Application and Statement of Claim pursuant to an order made on 18 December 2012, the group was expanded to include women who had been implanted with Gynemesh PS, Prolift+M, Prosima, TVT, TVT-O, TVT Secur, and TVT Abbrevo. The group members were also now defined by reference to the following criteria:
(1) they had surgery performed on them in Australia to implant one or more of the devices specified;
(2) they were supplied with one or more of the devices for specified purposes; and
(3) they have suffered from one or more defined complications attributed to the devices and/or the consequences of surgical removal of the devices.
3055 0n 8 April 2016, by the filing of a Third Further Amended Originating Application and Statement of Claim pursuant to an order made on 6 April 2016, women who had been implanted with TVT Exact were added to the group.
3056 Mrs Gill was covered by the group definition in the original Statement of Claim filed in October 2012, while Mrs Dawson and Mrs Sanders were included in the group definition in the Amended Statement of Claim filed in January 2013.
Identifying the common questions
3057 Another bone of contention involved the identification of the common questions.
3058 The applicants specified common questions in the various iterations of the originating application. In time the list not only grew, it also became unnecessarily prolix and controversial. Unfortunately, although the parties were urged to reach agreement, agreement was not forthcoming.
3059 I was informed by the respondents that the title “Agreed Statement of Common Questions” appearing on a document filed by the applicants on 13 April 2017 and incorporated into the Fourth Further Amended Originating Application was a misnomer. Unhelpfully, the respondents offered no alternative version. They submitted that the definition of the common questions should be left until after the publication of this judgment. This is undesirable. The common questions should be defined, and if necessary, redefined as early as possible. The parties’ lack of agreement was appropriately deprecated in Gill v Ethicon Sàrl (No 3) [2019] FCA 587; (2019) 369 ALR 175 at [10]–[12] and the remarks made there need not be repeated here.
3060 Be that as it may, the applicants ultimately acquiesced in the respondents’ proposal. In the circumstances, the correct form of the common questions (and the answers to them) will be determined at a later date at the time final orders are made.
Allegedly fatal gaps in the evidence
3061 The respondents argued that there was insufficient evidence on the efficacy, incidence, and severity of complications associated with TVT Secur, TVT Abbrevo, Prolift+M, and Prosima to support the applicants’ claims and therefore to allow the Court to make findings on the issues with respect to these devices.
3062 I reject the argument.
3063 It is true that most of the studies, in particular those reporting randomised controlled trials, related to TVT, TVT-O, Prolift and Gynemesh PS. But there was ample evidence to support the applicants’ allegation that all of the devices posed safety risks and had the potential to cause all the pleaded complications. It was that evidence that made Dr Hinoul’s concession to this effect inevitable. Then there is the respondents’ concession in oral argument that, with the exception of the increased risks for patients with autoimmune disorders which was yet to be added to the pleading, each of the pleaded complications was clinically significant, not only in terms of severity but also incidence.2738
3064 The evidence demonstrates that complications occurred with all the Ethicon devices, including TVT Secur, TVT Abbrevo, Prolift+M, and Prosima. The complications were similar in nature to the complications reported with the other Ethicon devices, although the gravity and incidence of complications could differ from device to device. Professor Klosterhalfen’s data pool, for example, contained explants from all devices that were removed for complications.2739 I have already observed that the data pool was not apt to provide incidence rates. It is notable, however, that he found all to be harmful.
3065 This is an unsurprising result, given the common use of Prolene and Prolene Soft or Gynemesh PS and the results of the mechanical testing conducted on Ethicon’s meshes, discussed in Part V. It will be recalled that the mechanical testing showed that the pore sizes of Prolene, Gynemesh PS, and UltraPro (used in Prolift+M) collapsed with the application of mechanical force within the expected physiologic range and consistent with the forces the meshes would be expected to experience during implantation and in vivo.
3066 There was also evidence from published studies and Ethicon's own documents. These documents are discussed elsewhere in the judgment. The limitations of the published studies and Ethicon's CERs, for example, have also been addressed.
3067 In this section, I summarise the evidence about reported outcomes with respect to the four devices in question.
TVT Abbrevo
3068 The evidence disclosed that TVT Abbrevo was similar in efficacy to TVT-O. Groin and/or thigh pain was common after implantation (over 1%), albeit lower than that associated with TVT-O.
3069 It will be recalled that the pre-market CER for TVT Abbrevo, discussed in Part VIII, annexed a single centre study by de Leval et al (2011).2740 This was a randomised trial comparing TVT-O with a modified device that was eventually branded as TVT Abbrevo. In total, 175 patients (87 women in the TVT-O group and 88 in the modified device group) were enrolled. Patients were followed up at various points up to the one year mark, with 170 women presenting for final follow-up. While some patients reported severe groin pain up to one day after the operation (more in the TVT-O group), there were no reports of severe pain by 12 months. However, 3% to 4% of patients in each group still reported groin pain (albeit not severe, as defined by the protocol) at 12 months.2741
3070 In the 2013 and 2015 CERs on the TVT Family of Products discussed in Part IX, Dr Hinoul referred to a study by Waltregny et al (2012), which compared TVT-O with a prototype of TVT Abbrevo. This was a three-year follow-up report of the de Leval (2011) study.2742 At three years, one patient in the TVT Abbrevo group (1.3%) and three patients in the TVT-O group (4.1%) reported thigh pain. Between one and three year visits, one tape had to be cut in the Abbrevo group because of lower urinary tract obstruction.
3071 The de Leval (2011) and Waltregny et al (2012) studies were included in the Ford (2015) Cochrane review assessing the clinical effects of midurethral sling operations.
3072 The 2013 CER also discussed a study by Tommaselli et al (2012), which compared TVT-O with TVT Abbrevo.2743 An abstract of this study was in evidence.2744 Similar cure and complication rates were reported for both groups. Ethicon’s post-market surveillance data reported in the CER included 25 reports of erosion (“urethral or otherwise unspecified”) and five reports of vaginal exposure following the use of TVT Abbrevo in 2012 and 11 reports in 2013.2745 They also included one case of haematoma formation, one case of mesh migration, one case or post-operative lower extremity pain, one case of urethral damage, one case of vaginal perforation, and one of vaginal exposure in 2011 and two cases of unspecified “post-operative complication”, two of post-operative groin pain, and one of post-operative infection and one of post-procedural incontinence in 2012.
3073 The PMS data in the 2015 CER2746 showed a marked increase in complaints in the 18 months since the period surveyed in the 2013 CER, mostly due to a dramatic increase in complaints of erosion, from 36 cases in 2011-2013 to 882 in 2013-15. The CER also included a table of “complaints by patient consequences” for the same period 2013-2015 period. This disclosed 788 cases of pain, 782 “surgical procedure[s]”; 778 erosions, 306 “other”; 191 infections, 101 cases of “bleeding”, 92 of “scar tissue”, 63 of “neurodeficit/dysfunction”, 44 of incontinence, 34 of organ perforation, 34 of extrusion, 12 of urinary retention, 10 fistulas, nine of urinary tract infections, eight adhesions, five of urinary frequency, four of vaginal discharge, two of urgency, and sundry other matters.
3074 Since complications are under-reported, it is likely that the complaints data represent a tiny fraction of the actual incidence of complications.
TVT Secur
3075 Contrary to the respondents' submissions, the safety and efficacy of TVT Secur was considered by a number of studies that were admitted into evidence. The preponderance of these studies demonstrated that TVT Secur was less effective than any of the other slings. This was consistent with the respondents' own documents and their decision to withdraw the device from the Australian market a short time after its launch. Adverse events included excessive blood loss; bladder, urethral and vaginal perforation; persistent urinary incontinence; de novo urgency; urinary retention, exposure/erosion; dyspareunia; infection; and the need for repeat surgery. There was evidence that potentially more women with single-incision slings had exposure but fewer women had pain when compared with transobturator slings.
3076 The Cochrane review by Nambiar et al (2014) on single-incision sling operations found that TVT Secur had consistently been shown to provide poorer control of incontinence than standard midurethral slings and higher rates of side effects.2747
3077 The authors found the following statistically significant differences when single-incision slings were compared with retropubic slings:
more women had persistent urinary incontinence after single-incision sling surgery;
objective measures and quality of life outcomes favoured retropubic slings; and
de novo urgency was more common in the single-incision sling group.2748
3078 A number of complications were also reported. These included major vascular injury (reported in one study), bladder or urethral perforation (reported in four trials, but said to be uncommon), difficulty voiding (said to affect less than 4% of women), infection and dyspareunia (reported in one study), mesh exposure and extrusion.
3079 When compared with transobturator slings, women were three times more likely to need repeat incontinence surgery after single-incision slings than after transobturator slings. The results were worse from trials evaluating TVT Secur.2749
3080 In addition, more women who received single-incision slings had tape exposure and the result was statistically significant, favouring inside-out transobturator slings. Pain, both acute and chronic, however, occurred with both types of slings but was more common after transobturator slings than after single-incision slings.
3081 Similar conclusions were reached by Abdel-Fattah et al (2011).2750 Their meta-analysis covered the period from 1996 to 2011. They reviewed evidence of the efficacy and safety of single-incision slings compared with standard midurethral slings. Three studies evaluating TVT Secur were included in their analysis. They found that single-incision slings were associated with significantly lower patient-reported and objective cure rates at six to 12 months compared with standard midurethral slings. Repeat incontinence surgery and de novo urgency incontinence were significantly higher in the single-incision sling group. Consistently with the other studies, however, patients in the single-incision sling group experienced less post-operative pain than those who received the standard midurethral slings.
3082 Another meta-analysis relevant to the occurrence of adverse events after the use of TVT Secur was by Schimpf et al (2014). That paper reported adverse events by reference to a number of categories including retropubic, transobturator, and mini-sling (such as TVT Secur). The authors' review included several studies evaluating TVT Secur.2751
3083 Schimpf et al (2014) reported the following incidence rates for mini-slings:
blood loss greater than 200ml occurred in 1.1% of patients (compared to 0.22% for obturator and 1.5% for retropubic);
dyspareunia was experienced by 0.74% of patients (compared to 0% for retropubic and 0.16% for obturator);
exposure rate was 2% (compared to 1.4% for retropubic and 2.2% for obturator);
1.4% of patients had to return to the operating room for erosion (compared to 1.9% for retropubic and 2.7% for obturator);
0.31% of patients had wound infection (compared to 0.75% for retropubic and 0.74% for obturator);
urinary tract infections occurred in 3.6% of patients (compared to 11% in the retropubic group and 4.3% in the obturator group);
bowel injury occurred in 0.74% of patients (compared to 0.34% in the retropubic group and 0% in the obturator group);
overactive bladder/urgency was reported in 5.4% of patients (compared to 6.9% for retropubic and 5.3% for obturator);
retention lasting more than six weeks post-operatively occurred in 3.3% of patients (compared to 2.7% in retropubic and 2.4% in obturator), with 1.9% returning to the operating room for urinary retention (compared to 1.2% with retropubic and 1.1% with obturator);
groin pain was reported in 0.62% of patients (compared to 1.5% for retropubic and 6.5% for obturator);
leg pain was reported in 1.6% of patients (compared to 0.62% with retropubic and 16% with obturator);
bladder perforation occurred in 0.85% of cases (compared to 3.6% with retropubic and 0.7% with obturator);
urethral perforation occurred in 2.7% of cases (compared to 0.41% with retropubic and 0.2% with obturator); and
vaginal perforation occurred in 1.3% of cases (compared to 0.73% with retropubic and 2.8% with obturator).
3084 In their discussion, the authors noted:
Review of the evidence showed that both objective and subjective cure outcomes were improved with use of a full-length sling compared to a minisling.
…
With respect to a comparison of AEs, the route (retropubic vs obturator) of the traditional full-length MUS is an important consideration (Table 3). For example, minislings have similar rates of postoperative OAB symptoms (5.4%) compared with obturator slings (5.3%), but somewhat lower rates than retropubic slings (6.9%). Exposure of the sling postoperatively is similar with either obturator slings (2.2%) or minislings (2.0%), but retropubic slings have somewhat lower rates than either (1.4%). Dyspareunia is rare with any type of sling, but is somewhat more common with a minisling (0.99%) than either a retropubic (<0.001%) or obturator (0.16%) sling. Minislings have the highest rate of urethral perforation (2.7% vs <1% for either retropubic or obturator), but the lowest rate of groin pain (0.62%) when compared to either route of MUS (1.5% for retropubic, 6.5% for obturator). Metaanalyses of the AE data failed to show a significant difference for OAB symptoms after surgery or return to the operating room for retention.
In summary, for women considering minislings or traditional full-length MUS, we recommend traditional full-length MUS to maximize cure rates (Table 4).2752
3085 In addition to these reviews, a number of studies were in evidence, some of which were RCTs considered by the reviews conducted by Nambiar et al (2014) and Schimpf et al (2014).
3086 The lead author of one of those studies was Dr Hinoul. Hinoul et al (2011) reported on a randomised controlled trial comparing TVT-O (98 patients) and TVT Secur (96 patients).2753 One year follow-up results were reported for 75 out of 96 patients in the Secur group and 85 out of 98 patients in the TVT-O group. In the Secur group, seven exposures were noted at the incision site, including three at six weeks, three at six months, and one at 12 months. Patients in the Secur group experienced significantly less pain in the first two weeks after surgery. Both subjective and objective cures appeared to be significantly better with TVT-O. The authors therefore described TVT Secur as “less efficacious” than TVT-O. In their conclusion, the authors observed:
The study failed to confirm the anticipated lower perioperative morbidity rate of TVT Secur since we noted increased blood loss, higher tape exposure and bladder injury rates, and a more common need for surgical re-intervention. Considering possible economic benefits or patient preferences, the role of TVT Secur remains to be studied.2754
3087 Following another RCT comparing TVT Secur with TVT-O, Bianchi-Ferraro et al (2014) found that the difference in cure rates at 24 months (which were higher with TVT-O) was not statistically significant.2755 Over that period, tape exposure occurred in 5.3% of patients with TVT-O and 7.5% of patients with TVT Secur. Consistently with the other studies, pain was reported by more women in the TVT-O group. Thigh pain was reported by 15/56 women (26.7%) with TVT, compared to one woman out of 66 (1.5%) with TVT Secur. Other adverse events in both groups included urinary retention, infection, de novo urgency, and surgical revision for stress incontinence.
3088 Finally, an RCT by Tommaselli et al (2013) compared TVT-O and TVT Secur after 36 months. The authors did not detect statistically significant differences in either the objective or subjective cure rates. They concluded that TVT Secur was not inferior to TVT-O in terms of cure rate and caused less post-operative pain, but warned that the risk of severe bleeding could not be ruled out with TVT Secur.2756
3089 The clinical effectiveness of TVT Secur was also analysed in an Ethicon-sponsored study by Tincello et al (2011), who looked at registry data on women who had undergone surgery with TVT, TVT-O and TVT Secur. After one year the authors concluded that the objective and subjective efficacy of TVT Secur was similar to that of TVT, but lower than that of TVT-O.2757
3090 Some of these studies were cited in the 2010 and 2013 post-market CERs that were prepared for TVT Secur. Those CERs also included post-market surveillance data on TVT Secur. The September 2013 CER showed that in 2012 there were 524 complaints made to Ethicon of erosion (urethral or otherwise unspecified) and 346 just for the period from January to April 2013.2758
3091 Further, the 2013 and 2015 CERs on the TVT Family of Products, which did not purport to evaluate TVT Secur, also referred to a number of studies on TVT Secur. Many of these were reviewed by Nambiar et al (2014) in their Cochrane review. Examples include studies by Masata et al (2012), comparing TVT-O and TVT Secur, which appears to have reported significantly lower subjective and objective cure rates with TVT Secur, and Hota et al (2012), who found that TVT Secur was 3.5 times more likely to result in a positive cough stress test (signifying failure) at one year follow-up than TVT-O.2759
3092 The evidence was consistent with the RANZCOG position statement published in May 2017, which concluded that the evidence available on single-incision slings had not then demonstrated equivalence to traditional midurethral slings. The College recommended that surgery with single-incision slings should be performed only within the context of a properly conducted clinical trial or where arrangements are in place for clinical governance such as a long-term prospective audit.2760
TVT Exact
3093 The respondents did not claim that there was a gap in the evidence on TVT Exact. For completeness, however, I make the following observations.
3094 Few studies concerning TVT Exact were tendered. Nevertheless, the mesh in TVT Exact is the same width and length as TVT, and it is implanted using a retropubic approach like TVT. The only difference appears to be that the trocar with sheath accompanying TVT Exact is thinner than the TVT trocar and one less cystoscopy is recommended.
3095 Perhaps for this reason, too, few studies were conducted on TVT Exact. This is consistent with the view Dr Hinoul took in his affidavit:
The studies discussed above dealing with retropubic mid-urethral slings are relevant to the TVT-Exact. This is due to the fact that the TVT-Exact utilises the same mesh as the TVT and is implanted in the same manner … [T]here has been little literature published that specifically studies the TVT-Exact…2761
3096 It is therefore likely that outcomes with TVT Exact were similar to those with TVT. This view is supported by the conclusions of a retrospective study by Thubert et al (2016) reporting on 12 month outcomes in patients with TVT and TVT Exact.2762 The authors reported that both success rates and the prevalence of complications following surgery with both devices were similar. The prevalence of bladder injury was unchanged with TVT Exact compared to TVT. One difference of note appeared to be that patients with TVT Exact had less immediate post-operative pain.
3097 There was a (non-blinded) randomised controlled trial evaluating TVT Exact, but the comparator was a transobturator device called SLING-IUFT so the outcomes are difficult to contextualise. The study was conducted by Dr Rosita Aniuliene in Lithuania and reported by Aniuliene et al (2015). 2763 This was a prospective study in which 76 patients were allocated to the TVT Exact group and 78 patients allocated to SLING-IUFT. Twelve month outcomes were reported, showing objective cure in 94.5% of patients with TVT Exact but only 61.2% with SLING-IUFT. The TVT Exact group, however, experienced haematoma (3.9%) and bladder perforation (1.3%) whereas these complications did not occur with SLING-IUFT. Post-operative urinary retention was significantly higher in the TVT Exact group (19.7%) than in the SLING-IUFT group (1.3%), although post-operative groin pain was more significant in the SLING-IUFT group (6.4%) than TVT Exact (1.3%).
3098 The 2015 CER on the TVT Family of Products disclosed that Ethicon had received 87 complaints of erosion (urinary tract) with TVT Exact from 2010 to January 2013, with 72 of these being made in 2012.2764
Prolift+M
3099 In its 2011 safety communication, the FDA stated that serious complications associated with surgical mesh for transvaginal repair of pelvic organ prolapse were “not rare”.2765 There is no reason to think that Prolift+M fell outside the scope of that warning.
3100 While there were no randomised controlled trials on this device, there was evidence from cohort studies evaluating the efficacy and safety of Prolift+M. There was also post-market surveillance data in Ethicon’s CERs, as well as insights from Prof Klosterhalfen’s data pool. Due to the absence of RCTs, it was difficult to assess whether Prolift+M was similar in efficacy to the other POP devices. But there was ample evidence of common complications. Mesh exposure rates appeared to be similar to Prolift, in the order of 10%, often requiring excision. There were a number of reports of de novo dyspareunia following implantation of Prolift+M. In addition, de novo incontinence (stress or urge) appeared to be very common.
3101 Milani et al (2011) conducted a multi-centre cohort study evaluating outcomes in 128 women, with follow-up at three months and one year after prolapse repair surgery with Prolift+M.2766 Anatomic success at one year was found in 77.4% of women. De novo dyspareunia was reported by 2% (1 out 49 sexually active women). Five women (3.9%) reported pelvic pain, seven (5.5%) had voiding dysfunction, and 17 (13.3%) had worsening or de novo stress urinary incontinence. Bladder perforation occurred in three patients (2.3%). Finally, 13 patients (10.2%) had a mesh exposure in the first year, seven of whom required partial mesh excision; the remaining six were treated with the use of topical oestrogen.
3102 Three year follow-up results were presented at a conference in Brisbane.2767 Anatomic success was 75.9%. Mesh exposure was observed in 19 patients (14.8%). Of these, 15 (were resolved following partial mesh excision surgery. None of the patients reportedly had de novo pelvic pain, but three (2.8%) had pain when the mesh was palpated during pelvic examination. De novo dyspareunia was observed in three out of 33 sexually active patients (9%).
3103 Khandwala and Jayachandran (2011) reported on the safety and efficacy of Prolift+M in 167 women.2768 Follow-up was short at six months. Six patients (3.6%) had mesh exposure, with one requiring excision. One patient (3.5%) had novo dyspareunia and one complained of generalised vaginal and groin pain. In addition, 8.7% experienced de novo urge urinary incontinence, 0.7% experienced voiding dysfunction, 2.2% had recurrent urinary tract infections, and 2.2% had faecal incontinence.
3104 In 2013 Khandwala reported on a separate prospective cohort study of 157 patients, who were implanted with Prolift+M, 134 of whom (85%) returned for follow-up at 12 months. “Pure” anatomic success based on POP-Q lower than stage II was reported in 94% of women. Three out of the 134 women followed up (2%) had mesh exposure. De novo stress incontinence was reported by 11 subjects (8.2%) and 15 (11.2%) reported de novo urge urinary incontinence. Recurrent urinary infection was noted in three subjects (2.2%). De novo dyspareunia was reported in three out of 50 (6%) of the sexually active women.2769
3105 Evidence of complications was also seen in the 2013 CER for Prolift+M, which reported the complaints received by Ethicon in the period from the launch of the device in 2008 to January 2013. A total of 246 complaints were made of erosion into the urinary tract, 26 cases of non-specific erosion, a further 36 cases of vaginal exposure, and an assorted number of other complaints. Pain was apparently reported only by four patients, and urinary tract perforation/tissue damage by five.2770 The analysis in the 2015 Prolift+M CER was confined to complaints received in the same reporting period covered by the 2013 CER.
Prosima
3106 Prosima was made using the same mesh as Prolift and is similar in other respects. In its CERs on Prosima Ethicon relied on the literature relating to Prolift. The evidence did not suggest that the performance of Prosima was sufficiently different from the other POP devices for it to fall outside the scope of the FDA’s warning or the conclusions drawn by the biomaterials experts.
3107 A number of studies on Prosima are discussed below and also in the pre and post-market evaluation sections of this judgment. They show, among other things, that the incidence of the following adverse events was common: mesh exposure (often requiring excision), infection, de novo incontinence, and the need for repeat surgery.
3108 Carey et al (2008) studied 95 women who underwent surgery in centres in Melbourne, Australia and Cambridge in the United Kingdom with a device that was the prototype for Prosima.2771 Eighty women were followed up at 12 months. The authors reported an objective cure rate of 85% and subjective cure rate of 87%. There were no major intraoperative complications, but there was one rectal perforation. There were four exposures, two of which required excision surgery. Two women experienced stress incontinence and one had obstructive voiding.
3109 Zyczynski et al (2010) reported on a prospective cohort study conducted at 11 sites in the US, UK, Germany, and Australia.2772 The study enrolled 136 women, with 130 followed up at one year. It was fully sponsored by Ethicon.2773
3110 On examination, 76.9% of women were POP-Q stages 0/1, representing anatomic success, but in 86.9% of women the leading edge of the vaginal compartment was above the hymen. Mesh exposure occurred in 12 patients (8%). Of those, eight were resolved with partial excision, but four were ongoing at one year. De novo urge and stress incontinence symptoms were each reported by 4% of women. There were two (1.3%) reports of urinary tract infection and four reports (2.7%) of incision site infection. Three patients (2.2%) underwent reintervention for prolapse, which included one previously untreated posterior compartment prolapse and one sacrocolpopexy.
3111 Around the same time, a report was published by Sayer et al (2011) on medium term outcomes following surgical repair with Prosima. This was also an Ethicon sponsored study.2774 One hundred and thirty six women received the surgery, and 110 were followed up after a median time of 29 months.2775 The proportion of women with POP-Q 0/1, denoting anatomic success, was 69.1%, but the leading edge of the vagina was above the hymen in 84.5%. Mesh exposure occurred in 11 (9.1%) women. The mesh was partially excised in eight patients, two of whom required second excisions. Four women reported de novo dyspareunia. Six patients (5%) reported worsening or de novo symptoms of stress urinary incontinence, five of whom underwent a subsequent midurethral sling placement. Five (4.1%) reported overactive bladder symptoms. Four (3.3%) required further surgery for prolapse.
3112 In the 2013 CER on Prosima, Dr Hinoul cited a number of other studies on Prosima. Chuang et al (2012) was one. In that study the mesh exposure rate after surgery with Prosima was 6%.2776 D’Afiero (2012) compared Prolift and Prosima, finding that de novo dyspareunia rates were higher with Prolift.2777 Krofta et al (2011) reported peri-operative complications in the form of one bladder perforation and one severe bleeding, and a mesh exposure rate of 4.16% at three months.2778 In 2012 there were 100 complaints of bladder erosion.2779
PART XIII: THE APPLICANTS’ STATUTORY CLAIMS
3113 The statutory claims are made under the Trade Practices Act and the Competition and Consumer Act. The CCA commenced on 1 January 2011. In the CCA, consumer protections, which were previously in the body of the TPA, were moved into a schedule (now Sch 2), entitled the Australian Consumer Law.
3114 The principal amending act — the Trade Practices Amendment (Australian Consumer Law) Act (No. 2) 2010 (Cth), which also commenced on 1 January 2011 — saves the operation of the TPA as in force immediately before the commencement of the ACL. Item 6 of Sch 7 of this Act provides as follows:
6 Acts or omissions that occurred before commencement
(1) The Trade Practices Act 1974 as in force immediately before the commencement of this item continues to apply, after that commencement, in relation to acts or omissions that occurred before that commencement.
(2) Without limiting subitem (1), action may be taken, under or in relation to Part VC or VI of that Act as so in force, in relation to those acts or omissions.
3115 Save for the limitation of actions provisions discussed in Part XVII below, it is common ground that at all material times the relevant provisions of the TPA were in the form in which they appear, or were not materially different from, the form they took as at 30 August 2010, and that the differences in wording that appear in the comparable provisions of the CCA are inconsequential.2780
To whom are the obligations owed?
3116 With the exception of the cause of action for defective goods under s 75AD of the TPA and s 138 of the ACL, which accrues to “individuals”, the liability of the manufacturers under the pleaded causes of action is to the “consumer”. In the absence of any contrary intention, “consumer” is defined for relevant purposes in s 4B of the TPA and the CCA and s 3 of the ACL. It is common ground that the applicants and the group members fall within those definitions. In any event, the applicants pleaded that they and the group members were “consumers” in relation to the Ethicon devices and the making of such an allegation gives rise to a statutory presumption that they were consumers, which applies unless the contrary is proved: see TPA s 4B(3), ACL s 3(10). The contrary was not proved.
Upon whom are the obligations imposed?
3117 With the exception of the misleading or deceptive conduct claim, the relevant statutory obligations are imposed upon “manufacturers”.
3118 For the purposes of Pt V Div 2A of the TPA (which includes ss 74B and 74D) and of Pt VA (which includes ss 75AC and 75AD) and the equivalent provisions of the ACL, “manufacturer” has an extended definition. It covers not only a corporation that actually manufactured goods but also:
a corporation that holds itself out to the public as the manufacturer (where the actual manufacturer does not have a place of business in Australia);
a corporation that imports the goods into Australia;
a corporation that uses its brand name in relation to the goods; and
a corporation that permits another person to promote the goods as goods manufactured by the corporation: TPA, ss 74A and 75AB; ACL, s 7.
3119 “Manufactured” is defined to include “grown, extracted, produced, processed and assembled”: TPA, s 74A and 75AA. “Manufactured” is not defined in the ACL. But “manufacturer” is defined in s 7 of the ACL to include “a person who grows, extracts, produces, processes or assembles goods”. Section 18A of the Acts Interpretation Act 1901 (Cth) relevantly provides that in any Act, where a word is given a particular meaning, other parts of speech have corresponding meanings. It follows that in the ACL “manufactured” includes “grown, extracted, produced, processes or assembled”.
3120 “Corporation” is defined in s 4 of both Acts to mean certain types of bodies corporate, including a foreign corporation, a trading corporation formed within Australia, as well as the holding company of any such body corporate.
3121 At all relevant times Ethicon Sàrl and Ethicon Inc. were “manufacturers”, at least because they manufactured the Ethicon devices and used their brand name in relation to them, and JJM was a “manufacturer” because it imported them into Australia. It is beyond doubt that proceedings may be brought against a foreign manufacturer and a local deemed manufacturer for the same contravention(s): Leeks v FXC Corporation (2002) 118 FCR 299 at [13] (Finn J).
How far do the obligations extend?
3122 There is a presumption that “a statute is to be construed as limited in its operation to the territory or the nationals of the State which enacts it”: Meyer Heine Pty Ltd v China Navigation Co Ltd (1966) 115 CLR 10 at 43 (Windeyer J); see, too, Kitto J at 23 and Menzies J at 38. Whether the TPA and the CCA are to be interpreted otherwise is a matter of construction.
3123 Section 5 of the TPA extends the operation of Pt V other than Div 1AA (and therefore ss 52, 74B and 74D), but not Pt VA (which includes s 75AD), to conduct that takes place outside Australia in certain circumstances. Subject to what I say below, the only relevant difference in the comparable provision of the CCA (also s 5) is that the equivalent of s 75AD (s 138 of the ACL) is not excluded. In other words, s 138 also has extraterritorial operation. The only potentially relevant circumstance in this case is where the corporation is incorporated or “carrying on business within Australia”: TPA, para 5(1)(g); CCA, para 5(1)(g).
Do the obligations extend to overseas corporations without a place of business in Australia?
3124 The respondents submitted that, as the two Ethicon respondents are incorporated overseas and neither has a place of business in Australia, the statutory causes of action do not apply to them.
3125 I reject the submission.
3126 There is a general presumption that the TPA applies to “conduct within Australia or of Australian nationals”. Section 5(1) extends the territorial operation but only to a limited extent: Bright v Femcare Ltd [2000] FCA 742; (2000) 175 ALR 50 (Lehane J) at [77]–[78]. Similarly, in Bray v F Hoffman-La Roche Ltd (2002) 118 FCR 1 at [50]–[51] Merkel J said that:
[Section] 5 of the TPA is to be accounted for only on the basis that the Act as a whole, including s 5 itself, has been framed on the assumption that when conduct is made a contravention of the Act it is only conduct in Australia that is meant unless the conditions set out in s 5 apply.
…
[U]nless expressly provided otherwise, the legislature intended that the Act is only to apply to extra-territorial conduct in the circumstances and subject to the conditions laid down in s 5.
(Emphasis added)
3127 In other words, “the conduct prescribed by Pt V of the TPA will be taken to be conduct within Australia unless s 5(1) applies”: Worldplay Services Pty Ltd v Australian Competition and Consumer Commission (2005) 143 FCR 345 at [18] per Ryan and Kiefel JJ, Tamberlin J agreeing at [28]–[43]. In Bright at [78] Lehane J observed:
Conduct, then, gives rise to a liability under, for example, s 52, if two conditions are met: first, it is engaged in within Australia (by a corporation) or outside Australia (by a body referred to in s 5(1)); secondly, it is conduct in trade or commerce ([which includes] trade or commerce between Australia and a place outside Australia).
3128 The same is true of all the relevant provisions of the TPA and the ACL.
3129 In the present case each of those conditions was met. This case is not concerned with the extra-territorial operation of the Act. The statutory claims are concerned with conduct relating to the supply of goods. As to misleading or deceptive conduct, see Australian Competition and Consumer Commission v Valve Corporation (No 3) [2016] FCA 196; (2016) 337 ALR 647 (Edelman J) (ACCC v Valve Corporation (No 3)) at [177]. Supply of the Ethicon devices took place in Australia because the devices were received in Australia by JJM, an Australian company, delivered to Australian hospitals and doctors, and implanted in women in Australia. The allegedly misleading conduct occurred in Australia because it is based on the information provided to, and received by, Australian consumers in Australia: see Valve Corporation v Australian Competition and Consumer Commission (2017) 258 FCR 190 at [104]–[105], [116], [134]. Where the relevant conduct occurs in Australia, in Trade Practices Commission v Australia Meat Holdings Pty Ltd [1988] FCA 338; (1998) 83 ALR 299 at 356 Wilcox J described it as “a misuse of language” to speak of the statute being given an extra-territorial effect.
3130 Each of the sections of the TPA and the ACL alleged to have been contravened applies to conduct by corporations “in trade or commerce”. “Trade or commerce” is defined in s 4 of both Acts to mean “trade or commerce within Australia or between Australia and places outside Australia”. The respondents denied in their pleading that the conduct of Ethicon Sàrl and Ethicon Inc. was “in trade or commerce”. Having regard to the statutory definition, this denial is baffling. The phrase “trade or commerce between Australia and places outside Australia” plainly includes the export of foreign-made goods to Australia.
3131 Accordingly, I find that the relevant conduct of Ethicon Sàrl and Ethicon Inc. was in trade or commerce.
Were Ethicon Sàrl and Ethicon Inc. carrying on business in Australia?
3132 In any case, I am satisfied that at all relevant times both Ethicon Sàrl and Ethicon Inc. were carrying on business in Australia.
3133 The expression “carries on business” is not defined in either Act. Its meaning can vary according to context: Luckins v Highway Motel (Carnarvon) Pty Ltd (1975) 133 CLR 164 at 178 (Gibbs J). In Hope v Bathurst City Council (1980) 144 CLR 1 at 8–9 Mason J (with whom Gibbs, Stephen and Aickin JJ agreed) held that in its ordinary or popular meaning “carrying on business” means the undertaking of a commercial enterprise as a going concern, that is to say, “activities engaged in for the purpose of profit on a continuous and repetitive basis”.
3134 In ACCC v Valve Corporation (No 3) at [195] Edelman J observed that there was little direct authority on the meaning of the expression in the context in which it is used in the TPA. His Honour pointed out that, where courts have considered the meaning of the same expression, they have applied the ordinary meaning, and proceeded to follow suit, citing Bray (at [59]–[60]).
3135 In Gebo Investments (Labuan) Ltd v Signatory Investments Pty Ltd [2005] NSWSC 544; (2005) 190 FLR 209; (2005) 54 ACSR 111 at [38] Barrett J pointed out that “under the general law, carrying on business generally involves conducting some form of commercial enterprise, systematically and regularly with a view to profit”.
3136 In Luckins at 178 Gibbs J held that the expression “would usually connote, at least; the doing of a succession of acts designed to advance some enterprise of the company pursued with a view to pecuniary gain”. In Smith (on behalf of National Parks and Wildlife Service) v Capewell (1979) 142 CLR 509 at 517–519, Gibbs J reiterated that “carry on business”, in its ordinary meaning, “signifies a course of conduct involving the performance of a succession of acts, and not simply the effecting of one solitary transaction”. In Bray at [62], in observations noted with evident approval by Edelman J in Valve Corporation (No 3) at [197], Merkel J said that “carrying on business will usually involve ‘a series or repetition of acts’”, citing Thiel v Federal Commissioner of Taxation (1990) 171 CLR 338 at 350 (Dawson J).
3137 In Luckins, the statutory expression was “carrying on business within the State”. The context was the application of the Companies Act 1961 (WA) to a Victorian company operating a tourist bus business, which included long distance tours to Western Australia, but which was not incorporated in Western Australia and which had never had an office or place of business in Western Australia. The question was whether the company was carrying on business in Western Australia at the time it executed a debenture over assets of the company. If so, for the debenture to be enforceable, it had to be registered under the Western Australian Act. The evidence was that none of the tours ever started or finished in Western Australia; it was rare for anyone to join a tour there; and anyone who did always left the tour in another State or Territory. Nevertheless, the majority of the Court (Barwick CJ dissenting) held that the company was carrying on business in Western Australia because it despatched busloads of passengers through Western Australia and in the course of so doing entered into commercial transactions with various people in various parts of the State, not just on isolated occasions but over a period of time.
3138 In Gebo the question was whether a foreign company was carrying on business in Australia so as to give the Court jurisdiction under the Corporations Act to wind it up.
3139 As the decisions in both those cases make clear, a company may be carrying on business in a particular geographical area although most of its business is conducted elsewhere: Gebo at [39].
3140 In Norcast S.ár.L v Bradken Limited (No 2) (2013) 219 FCR 14, a bid-rigging case brought under the cartel provisions of the CCA, Gordon J considered the meaning of “carrying on business” in the Corporations Act 2001 (Cth). Her Honour cited Gebo with approval at [255]:
At general law, carrying on a business generally involves conducting some form of commercial enterprise, systematically and regularly with a view to profit: Gebo Investments at [38]. It is unnecessary to restate the “usual elements” of a finding of carrying on business in Australia. It is, however, necessary to point out that a company may be found to carry on business in Australia even though the bulk of its activities are conducted elsewhere (Gebo Investments at [38]–[41]) and that it conducts its activities in Australia by reason of its control over or connection with an Australian company: Adams v Cape Industries Plc [1990] Ch 433 at 530 and Bray v F Hoffman-La Roche Ltd (2002) 118 FCR 1 at [60]–[63].
(Emphasis added)
3141 An appeal from this judgment was dismissed: Bradken Limited v Norcast S.ár.L (2013) 219 FCR 101.
3142 Similarly, in Anchorage Capital Partners Pty Limited v ACPA Pty Ltd [2018] FCAFC 6 at [99], the Full Court acknowledged that a company may be found to carry on business in Australia even if it does not maintain an office in Australia or the bulk of its business is carried on outside Australia, but noted that each case will depend on its own facts.
3143 In Vautin v BY Winddown, Inc [2016] FCA 632, Rares J granted leave to a consumer to sue a foreign manufacturer for a contravention of the Australian Consumer Law because the facts gave rise to a prima facie case that the foreign company carried on business in Australia by selling the products that it manufactured to an Australian company as its exclusive dealer for the purposes of sale to persons in Australia.
3144 Here, the evidence establishes that the two Ethicon companies were engaged in a systematic course of conduct in Australia. This is not a case involving a small number of isolated transactions. They were selling their products in Australia over a number of years through a related company and promoting them jointly with that company. Supplying goods on a regular basis to an Australian company for the purpose of sale to Australian consumers is “carrying on business” in Australia. The respondents admitted in their defence that both companies supplied their goods to JJM for sale in Australia throughout the period covered by the statement of claim. In the ordinary course, the seller would profit from such an enterprise. In the absence of evidence to the contrary, I infer, based on the principle in Blatch v Archer (1774) 1 Cowp 63; [1774] 98 ER 969, that both Ethicon Inc. and Ethicon Sàrl derived profit from the sales.
3145 Further, Ethicon Inc. and JJM were in regular contact about the Ethicon devices and Ethicon Inc. handled Australian complaints about all of them. Ethicon Inc. also engaged Marcus Carey, the Australian surgeon, working in Australia, to provide consulting services to it in relation to the testing and commercialisation of what became Prosima.2781 In relation to TVT Secur, JJM’s Medical Director, Dr Aran Maree, confirmed on 5 November 2007 in an email sent, amongst others, to Ethicon Inc. (including Mark Yale, Ethicon Inc.’s Director, Worldwide Risk Management and Dr David Robinson, its Worldwide Medical Director) that there was to be “no unilateral discussion by the local subsidiary with the Australian competent authority regarding any field actions without ETHUS QA/RAIRisk Management input and sign off”.2782 It is unlikely that any different approach was adopted in relation to the other Ethicon devices.
3146 While, on the evidence, most of the dealings appear to be between Ethicon Inc. and JJM, there is evidence that Ethicon Sàrl was directly supplying JJM. On 15 November 2000 Christine Curtis of Ethicon Inc. asked logistics managers and purchasing managers to “begin placing new purchase orders” for TVT directly with Ethicon Sàrl. The evidence also reveals that the following day Joanne Pacheco of JJM forwarded Ms Curtis’s email to Vicky Jansen, apparently from Johnson & Johnson International, copied to Isabelle Miloda whose email address suggests she was with Ethicon Sàrl, seeking urgent advice as to where she should place her purchase orders.2783 There is no reason to think that JJM did not do as requested. I was also taken to an email chain commencing 5 September 2006 with a request from Henry Vaotangi of the purchasing department at JJM to Christine Theoret of Ethicon Sàrl for confirmation of his understanding that he could send to Ethicon Sàrl JJM’s purchasing order for samples of TVT Secur. The chain includes an email from Irena Stoïmenova of Johnson & Johnson International, which begins with the statement that a quantity had already been sent to Australia, and another from Ms Stoïmenova to two of her colleagues at Ethicon Sàrl asking for stock to be shipped to JJM.2784 This was said to be illustrative of a course of conduct and the respondents did not submit otherwise.2785
3147 For completeness, I note that until relatively recently, s 5(3) of the TPA/CCA precluded a person who makes a claim under s 82 from relying at a hearing on conduct to which ss 5(1) or 5(2) extended without the written consent of the Minister. The Minister was obliged to give consent unless it was not in Australia’s national interest to do so or the law of the foreign country required or specifically authorised the conduct: TPA, s 5(5).
3148 The respondents did not plead that the written consent of the Minister had not been obtained and the point was not raised in their submissions. The applicants contended that the effect of the 2017 amendments is that the requirement for Ministerial consent has been removed and the respondents did not take issue with their contention. It is true that the requirement has been removed. Subsections (3)–(5) of s 5 were inserted by s 8 of the Trade Practices Revision Act 1986 (Cth). By Sch 14 Pt 1 of the Competition and Consumer Amendment (Competition Policy Review) Act 2017 (Cth), they were repealed.
3149 But these changes were not retrospective. Schedule 14 commenced on 28 October 2017. Part 1 cl 2 provides that the repeal of subsections (3) and (4) applies “in relation to hearings commencing on or after the commencement of this item”. The hearing in this case started more than three months before the changes came into effect.
3150 Nevertheless, the weight of authority supports the conclusion that Ministerial consent is not necessary before a proceeding begins and is only required before relief in the nature of damages is granted: Auskay International Manufacturing & Trade Pty Ltd v Qantas Airways Ltd [2008] FCA 1458; (2008) 251 ALR 166 at [57] (Tracey J) and the cases referred to there. In the circumstances, I will defer making an award of damages or compensation with respect to the statutory counts against either Ethicon corporation until or unless Ministerial consent is obtained. The parties should agree upon a suitable period for the completion of that process.
3151 The pleading is convoluted and, for that reason, not always easy to follow.
3152 With respect to the SUI devices, which the applicants called “the Tape Implants”, the applicants pleaded their case in this way.
61 By reason of:
(a) the fact that:
(i) the Tape Implants were designed and manufactured by the Respondents for the Tape Purpose;
(ii) the Tape Purpose was known to the Respondents, as pleaded at paragraph 41 above;
(iii) the Respondents marketed, promoted and supplied the Tape Implants as reasonably fit for the Tape Purpose, as pleaded at paragraph 42 above;
(iv) the purposes for which the Tape Implants were commonly supplied and the purpose for which one or more of the Tape Implants were acquired by each of the Tape Sub-Group Members was for the Tape Purpose, as pleaded at paragraph 43 above; and
(v) the purposes for which the Tape Implants were commonly supplied and acquired and the purpose for which one or more of the Tape Implants were acquired by each of the Tape Sub-Group Members, being the Tape Purpose, was known to the Respondents, as pleaded at paragraph 44 above;
(b) the matters pleaded in paragraphs 45, 46, 47 and, or alternatively, 48 above; and, or alternatively
(c) the fact that none of the packaging of the Tape Implants, their Instructions For Use, nor any other source of information disseminated by the Respondents, or any of them, to Tape Sub-Group Members, Treating Hospitals or Treating Doctors gave any (or any sufficient) warning, advice or information as to some or all of the Tape Warning Matters
the safety of the Tape Implants was not such as persons generally were entitled to expect and the Tape Implants had a defect for the purposes of sections 75AC(1) and 75AD(1) of the TPA and, or alternatively, a safety defect for the purposes of sections 9 and 138 of Schedule 2 of the CCA.
62 By reason of the matters pleaded at paragraph 61(a) to (c) above, the Tape Implants were not reasonably fit for the Tape Purpose, within the meaning of section 74B of the TPA and section 55 of Schedule 2 of the CCA.
63 By reason of the matters pleaded at paragraph 61(a) to (c) above, the Tape Implants acquired by each of the Tape Sub-Group Members were not of merchantable quality within the meaning of section 74D(3) of the TPA, or acceptable quality within the meaning of section 54 of Schedule 2 of the CCA.
3153 The “Tape Purpose” and the purpose for which the SUI devices were commonly acquired and were acquired by each of the members of the sub-group is described at [40] in the following way:
The Tape Implants were designed and manufactured to:
(a) be implanted in women for the safe and effective surgical treatment of pure or predominant SUI;
(b) provide urethral support safely and effectively in patients; and
(c) alleviate involuntary urine leakage from SUI.
Sub-paragraphs (b) and (c) really add nothing to sub-paragraph (a).
3154 To support the allegation, the applicants relied on the product brochures and instructions for use in relation to the various devices.
3155 Thus, the applicants’ primary case is that the SUI devices were defective, not reasonably fit for the purpose for which they were acquired, and not of merchantable or acceptable quality because they were purportedly designed, manufactured, marketed, promoted and supplied as safe and effective for the surgical treatment of stress urinary incontinence but were not fit for that purpose and were not as safe as persons generally were entitled to expect.
3156 The alternative case is that the SUI devices were defective, not reasonably fit for the purpose for which they were acquired, and not of merchantable quality because neither the packaging of the devices nor the instructions for use gave any or any sufficient warning, advice or information as to some or all of a number of matters. Stripped of the cumbersome cross-referencing and repetition in the pleading, those matters are:
(1) that there was a risk that the SUI devices would cause the pleaded complications;
(2) that treatment of the complications was difficult or impossible, carried with it an added risk of new or aggravated complications, and/or could require surgery to remove them in whole or in part and removal was difficult;
(3) that alternative acceptable treatments were available which did not have the same risks or greater risks and were as safe or “not materially less safe” for the purpose for which the devices were acquired as surgery for stress urinary incontinence using the SUI devices;
(4) that before the SUI devices were released in Australia, none of the respondents had undertaken any or any adequate evaluation of the risks, including long-term risks, and effectiveness, including in the long term; and
(5) that the chronic inflammatory response to the SUI devices “may be affected by conditions which affect the immune response and healing, including autoimmune and connective tissue disorders”. 2786
3157 The applicants alleged that the risk of the pleaded complications arose from, or was increased by, the following matters or circumstances:
(1) the material from which the devices were made includes polypropylene;
(2) the design of the devices (including pore size, filament structure and weave, tensile strength, elasticity, density, and porosity) promotes bacterial colonisation; aggravates any inflammatory response; increases the risk of degradation of the devices; renders them incompatible with the anatomic location in which they are implanted;
(3) the material and design of the devices meant that the inflammatory response to the devices could become chronic and/or cause them to become totally infected and render them susceptible to scar plate formation, contraction, shrinkage, fraying, roping, curling, bunching, banding, deforming, collapsing, elongating, oxidative degradation, “not being inert”; and/or bridging fibrosis;
(4) the anatomical structures, tissues and locations in which they were implanted, passed through, attached to “or brought into proximity” with (including the vagina, “a clean contaminated environment that cannot be completely sterilised”); and/or
(5) the technique by which they were designed to be implanted (reference was made in this context only to TVT-O and TVT Abbrevo which are inserted through the obturator membrane).2787
3158 The applicants went on to plead that the members of the “Tape Sub-Group” (those members of the class who received one of the SUI devices) suffered loss and damage by reason of the fact that the safety of the “Tape Implants” (the SUI devices) was not such as persons generally were entitled to expect, not reasonably fit for the Tape Purpose, and/or not of merchantable quality. This was inelegant in that each of the class members must prove that her loss and damage was caused by the deficiencies in the device which she received.
3159 The claim with respect to the POP devices (referred to in the pleadings as “the Mesh Implants”) takes the same form and the allegations are almost identical.
3160 Thus, although the questions raised by the various statutory counts are different, with the exception of the misleading or deceptive goods claim, the applicants’ case is essentially the same. For this reason both parties took the view that the applicants’ success or failure on the defective goods case would spell success or failure on the fitness for purpose cases.2788 That may be so, but the converse does not necessarily follow. In the present case, however, having regard to the common position taken by the parties, it is unnecessary to consider this possibility with respect to any of the Ethicon devices.
3161 At all relevant times s 75AD of the TPA (cf. s 138 of the ACL) provided that:
If:
(a) a corporations, in trade or commerce, supplies goods manufactured by it; and
(b) they have a defect; and
(c) because of the defect an individual suffers injuries;
then:
(d) the corporation is liable to compensate the individual for the amount of the individual’s loss suffered as a result of the injuries; and
(e) the individual may recover that amount by action against the corporation; and
(f) if the individual dies because of the injuries – a law of a State or Territory about liability in respect of the death of individuals applies as if:
(i) the action were an action under the law of the State or Territory for damages in respect of the injuries; and
(ii) the defect were the corporation’s wrongful act, neglect or default.
3162 If two or more corporations are liable for the same loss, the corporations are jointly and severally liable: TPA, s 75AM.
3163 Since it is common ground that the respondents were corporations who, in trade or commerce, supplied the Ethicon devices and, for the purposes of the Act, were all manufacturers, the only remaining issues are whether the devices have a defect and, if so, whether the applicants’ injuries were caused by it.
3164 “Defect” was defined in s 75AC of the TPA (cf. s 9 of the ACL).
3165 Goods have a defect “if their safety is not such as persons generally are entitled to expect”: TPA, s 75AC(1). In assessing safety, s 75AC(2) requires that “all relevant circumstances” be taken into account including:
(a) the manner in which, and the purposes for which, they have been marketed; and
(b) their packaging; and
(c) the use of any mark in relation to them; and
(d) any instructions for, or warnings with respect to, doing, or refraining from doing, anything with or in relation to them; and
(e) what might reasonably be expected to be done with or in relation to them; and
(f) the time when they were supplied by their manufacturer.
3166 There is no material difference between s 75AD of the TPA and s 138 of the ACL or between s 75AC of the TPA and s 9 of the ACL. Apart from the substitution of “safety defect” for “defect”, the provisions are identical.
3167 It is common ground that the relevant time at which the assessment is to be made is the time the goods were supplied by the manufacturer. This picks up paragraph (f). But the phrase “the time when they were supplied by the manufacturer” is ambiguous. Does it mean the time when they were supplied to the applicant (here, the time when the devices were implanted in the group members by their surgeons)? Does it mean the time when they were supplied to the surgeons or the hospitals from which they acquired them? Does it mean the time when they were supplied to the market, that is, the time when the goods were put into circulation by the manufacturer? Since the time of supply to an individual is beyond the control of the manufacturer, it seems likely that the relevant time is the time of supply to the market. That construction is supported by the Explanatory Memorandum to the Trade Practices Amendment Bill 1992 which inserted s 75AD into the Act. It stated (at [20]):
The final specified factor is the time at which the goods were supplied … The critical time is when the alleged defective good which caused the loss was put into circulation by its manufacturer. Goods which met community expectations at that time are not defective at a later time because the safety expectations of the community have increased.
3168 It is also the approach taken in the English authorities, when interpreting a provision similar to s 75AC(2): see, e.g. Wilkes v DePuy International Ltd [2016] EWHC 3096 (QB); 153 BMLR 91; [2017] 3 All ER 589; [2018] 2 WLR 531 (Hickinbottom J) at [79].
3169 I therefore conclude that the time when the goods were supplied refers to the time when the goods were put into circulation by the manufacturer.
3170 As is perhaps obvious, the question of whether the level of safety of the goods in question is less than persons generally are entitled to expect imports what was described in the Explanatory Memorandum as an objective standard. In other words, the answer depends on what the public at large is entitled to expect, not on the expectations of the applicants: Carey-Hazell v Getz Bros &Co (Aust) Pty Ltd [2004] FCA 853; (2004) ATPR ¶42-014 at [186] (Carey-Hazell (2004)); Merck Sharp & Dohme (Australia) Pty Ltd v Peterson (2011) 196 FCR 145 at [191]. Burton J observed in A v National Blood Authority [2001] 3 All ER 289 at [31] in relation to the virtually identical definition of “defect” in the comparable English legislation:
[T]he court decides what the public is entitled to expect… [s]uch objectively assessed legitimate expectation may accord with actual expectation; but it may be more than the public actually expects, thus imposing a higher standard of safety, or it may be less than the public actually expects. Alternatively, the public may have no actual expectation – e.g., in relation to a new product…
3171 Those expectations, whatever they may be, are informed by all relevant circumstances. They include, but are not confined to, those specified by s 75AC(2).
3172 Since the question of whether there is a defect requires consideration of the way goods are marketed and instructions or warnings that have been given with respect to their use, the expectations persons generally are entitled to have about the goods will be affected by what the manufacturer has said about them. Thus, even if a product presents certain risks, that product may well not have a defect if the manufacturer gives appropriate warnings about those risks, defines appropriate limitations on the indications for use, and does not promise more in terms of safety than the product can deliver. The applicants conceded as much.2789 The instructions or warnings mentioned in s 75AC(2)(d) relate to the doing or refraining from doing something with, or in relation to, the goods. In the present case that would encompass instructions or warnings about use in relation to pregnant women or women of child-bearing years, for example, or about implantation in certain other kinds of patients. It would ordinarily not encompass warnings or advice about potential adverse effects. Nevertheless, it was common ground and properly so, consistent with the judgments in Peterson v Merck Sharpe & Dohme (Aust) Pty Ltd (2010) 184 FCR 1 (Jessup J) and on appeal in Merck (together, the Vioxx case), that information or warnings given by a manufacturer about the potential adverse effects of the use of a product are also relevant since they could affect the level of safety that persons generally are entitled to expect.
3173 Naturally enough, the defect must exist in the particular goods that are alleged to have caused injury to the individual: Peterson at [912]. Consequently, the applicants must prove that the particular Ethicon devices had such a defect.
3174 It has been clear at least since the Vioxx case that a product may be defective even if the defect is one which only affects some people. In endorsing the primary judge’s decision that Vioxx had a defect within the meaning of s 75AC, the Full Court in Merck at [201] described the defect in this way:
The defect was one which affected some people, not all. The defect was that in some people, by a mechanism not known and the subject of no hypothesis, it increased the risk of [myocardial infarction] and provided no information, advice or warning as to this effect [.]
3175 It also follows from Merck that the risk need not be high, although the extent of the risk posed by the product which will render a product defective may vary from case to case. The relevant risk in that case was very low (0.5% risk of myocardial infarction) but the potential consequences were grave, indeed fatal. The applicants also referred to Medtel Pty Ltd v Courtney (2003) 130 FCR 182 in this context. But that case did not involve a claim under s 75AD.
3176 As Andrews J observed in Gee v DePuy International Limited [2018] EWHC 1208 (QB) at [97]:
One may think it self-evident that if a product fails to meet the objective safety standard set out in [the Act] … the defect is whatever it is about that product (its state, or condition, or the risks to health and safety … that it poses) that leads the Court to conclude that it fails to meet the safety standard.
3177 An inference that goods have a defect may not be made only because safer goods of the same kind were supplied after the time of supply by the manufacturer (s 75AC(3)) or, where the goods complied with a mandatory Commonwealth standard, at the time of supply the standard was not the safest possible standard having regard to the latest scientific or technical knowledge (s 75AC(4)). As the Explanatory Memorandum recognised at [26], however, these circumstances may still be relevant factors to take into account in determining the safety of the goods. Indeed, the respondents accepted that current information about the safety of the Ethicon devices could be taken into account.2790
3178 The defect or defects need not be identified with any particular level of precision: Batchelder & Anor v Holden Ltd [2009] VSC 29 at [14] (Beach J). Nor is there a ne(ed to prove the mechanism by which the defect occurred or could have occurred: Merck at [200]–[201]; see, too, Ide v ATB Sales Ltd [2008] EWCA Civ 424; [2008] PIQR P251; [2008] All ER (D) 374 (Apr) where Thomas LJ (with whom Dyson and Ward LLJ agreed) held at [19] that, in a defective products claim under the Consumer Protection Act 1987 (UK) (UK Consumer Protection Act), it is unnecessary to ascertain the cause of the defect. The only relevant causation question is whether the defect caused the damage in any particular case.
3179 Apart from the matters listed in s 75AC(2), the respondents contended that the following additional matters need to be taken into account: the knowledge of the medical practitioners inserting the devices; the knowledge of the general community about permanent prosthetics, and the state of scientific knowledge, which changed over time and is still developing.2791
3180 The respondents do not argue that the knowledge of “persons generally” about any of the Ethicon devices or of their risks was extensive or detailed. Rather, they submitted that “there should be no suggestion that the general community (and, in particular, those having a device inserted), does not understand that permanent prosthetic implants are intended to be permanent”.2792 That proposition may be accepted, but it does not assist the respondents. If anything, the community in general (the only cohort with which the section is concerned) is surely entitled to have higher expectations about the level of safety attaching to permanent prosthetic implants than they might have about temporary prosthetics, prosthetics expected to wear out over time (like knee or hip prostheses), or about some medicines or drugs that are only intended to be taken for a limited period and which have no serious side-effects.
3181 Section 75AD appeared in Pt VA of the TPA. Part VA was inserted into the Act in 1992. It was based on the 1985 European Council Directive 85/374/EEC (the 1985 Directive on Product Liability).2793 Indeed, it was very closely modelled on the comparable provisions of that Directive.2794 As the Full Court observed in Merck at [187], the 1992 amendments introduced a strict product liability regime, that is to say, it enabled a consumer to recover compensation from the manufacturer for loss or damage without having to prove that the manufacturer was negligent. That means that, generally speaking, the focus is upon the condition or state of the product rather than the acts and omissions of the producers.
3182 It is readily apparent, then, that, subject to the manufacturer raising and making out a defence, the section applies regardless of whether the manufacturer was negligent and even if the manufacturer had no knowledge of the defect. As the applicants submitted, the policy of the section is that, if a manufacturer cannot meet the reasonable expectations of the community (as to the safety of the goods), then it should either cease manufacture or provide such information concerning the attendant risks as would alter those expectations.2795
3183 The Explanatory Memorandum referred at [15] to the variety of matters that might constitute defects within the meaning of s 75AC:
[T]here are a number of different types of potential defects. Design defects relate to matters such as the form, structure and composition of the goods. Manufacturing defects are those related to matters such as the process of construction and assembly. Instructional defects are those caused by incorrect or inadequate warnings and instructions. All these categories of “defect” fall within the meaning ascribed to defect in section 75AC.
3184 The Explanatory Memorandum noted at [18] that instructions and warnings are particularly crucial in relation to goods the manufacturer knows to be potentially hazardous, “as it is through these sources that the manufacturer can detail the nature and extent of the potential hazard and provide adequate instructions to assist consumers in avoiding that hazard”. It mentioned that “the general presentation of the product can influence consumer expectations by exaggerating safety aspects or minimising reference to possible risks”.
3185 Thus, if a product is marketed in such a way as to exaggerate its safety or minimise its potential risks, the expectations of “persons generally” are likely to be raised. They may be led to believe that the product is safer than it actually is. In effect, the section holds the manufacturer to the truth of its promotional material. If a product does not live up to the manufacturer’s claims about its safety, it is likely have a defect.
3186 Furthermore, what might reasonably be expected to be done with or in relation to the goods (that is the use to which the goods might reasonably be put) is wide enough to include potential misuse: see s 75AC(2)(e). Certainly, this is what Parliament contemplated, as appears from the Explanatory Memorandum at [19]:
[I]n some cases a manufacturer will be under an obligation to warn consumers of the potential consequences of misuse which could be anticipated by the manufacturer. This may in certain circumstances go beyond merely stating that a certain course of action should not be adopted and require the manufacturer to detail the specific consequences of such misuse (ie, to detail the type of injury or damage which may be suffered). If the loss does result partially from misuse, the manufacturer will be able to reduce the amount of compensation payable to reflect that part of the damage caused by contributory acts by the injured person (see section 75AN below), but this does not relieve the manufacturer of the obligation to warn.
3187 Thus the effect of the statutory definition of “defect” is that a product might be defective because it carries certain risks against which no warning could adequately protect the user, because it carries risks against which suitable warnings could adequately protect the user but those warnings were not provided, or merely because the information provided with the product was deficient.
3188 In Carey-Hazell (2004), a case about an allegedly defective prosthetic mitral valve, Kiefel J (as her Honour then was) observed at [199] that without a warning or instruction the use of a product might be unsafe and a warning is necessary to remove “some inherent dangerous quality”. In Peterson, Jessup J held at [915]–[918] that the safety of the drug was less than persons generally were entitled to expect because the consumption of the drug had the potential to increase the risk of myocardial infarction in circumstances which included the absence of any relevant information or warning from the manufacturer.
3189 This is the approach taken by the High Court of England and Wales where the statutory scheme is very similar. The relevant statute is the UK Consumer Protection Act, which implemented the 1985 Directive on Product Liability in the United Kingdom. It is sufficient for present purposes to note that that Act provides a remedy in damages against the producer of a product where a consumer has suffered damage caused wholly or partly by a defect in the product and that the definition of “defect” is very similar to the definition of defect in s 75AC of the TPA. For the purposes of Pt 1 of the UK Consumer Protection Act, which deals with product liability, s 3(1) provides that there is a defect in a product “if the safety of the product is not such as persons generally are entitled to expect” and, for those purposes, “safety” in relation to a product includes safety with respect to products comprised in the product and safety in the context of risks of damage to property as well as of death or personal injury. In determining for the purposes of subs (1) what persons generally are entitled to expect, s 3(2) provides that all the circumstances must be taken into account, including:
(a) the manner in which, and purposes for which, the product has been marketed, its get-up, the use of any mark in relation to the product and any instructions for, or warnings with respect to, doing or refraining from doing anything with or in relation to the product;
(b) what might reasonably be expected to be done with or in relation to the product; and
(c) the time when the product was supplied by its producer to another[.]
3190 In Abouzaid v Mothercare (UK) Ltd [2000] All ER (D) 2436; [2000] EWCA Civ 348, a 12 year old boy was injured when he was hit in the eye by a metal buckle after an elastic strap from a sleeping bag sprung from his grasp while he was trying to fasten the straps together. The trial judge found that the risk or propensity of elastic to spring back was a safety defect and the appeal was dismissed. Pill LJ (with whom Wright J agreed) said at [27]:
The risk is in losing control of an elastic strap at a time when it is stretched and eyes are in the line of recoil. The product was defective because it was supplied with a design which permitted the risk to arise and without giving a warning that the user should not so position himself that the risk arose. Members of the public were entitled to expect better from the appellants. A factor in that expectation is the vulnerability of the eye and the serious consequences which may follow from a blunt injury to the eye. Expectations would be different if the worst which could occur was an impact of elastic on the hand. It is not necessary for the Court to determine precisely what more should have been done. It is clear that more could have been done, for example a non-elasticated method of attachment or instructions to fasten the straps from behind the seat unit, together with a warning.
3191 Importantly, the law does not require that goods be “absolutely free from risk”: Merck at [191]. As the Explanatory Memorandum to the Trade Practices Amendment Bill stated at [21]:
[I]n addition to the factors specified in subsection 75AC(2), the court must take all relevant circumstances into account in determining the safety of goods. Safety expectations may also depend on matters such as the nature of the product and community knowledge of that product. For example, there are a number of known negative side effects associated with certain pharmaceuticals and vaccines. It is also generally accepted and known that these side effects cannot be avoided. Such products are known to confer substantial benefits which flow to the wider community at large. The small statistical chance of injury associated with them does not of itself mean that they are “defective”.
3192 Indeed, as Burton J said in A v National Blood Authority at [31], there will be some harmful characteristics about which no complaint could be made, such as the propensity of knives, guns and poisons to cause the kind of harm they were designed or intended to cause. In other words, a product which is generally known by its very nature and the way it has been presented (for example, in its instructions for use, labelling, and marketing) to cause personal injury or damage will not be defective on that account. In A v National Blood Authority Burton J proceeded to cite with approval the following explanation given by Professor Howells in The Law of Product Liability at [1.19]:
The emphasis on the autonomy of the individual and his free choice to expose himself to risks has generally relieved the producer of … liability. However this free choice must be an informed choice and so there has been a need to define which types of system damage users can be expected to be aware of from their general life experience (i.e., that knives can be sharp) and those that they have to be warned about (i.e., risks associated with drinking and smoking).
3193 His Honour suggested that “drugs with advertised side-effects” may fall into the latter category.
3194 By implication, the Full Court in Merck considered that s 75AC did not contemplate that a product which carried “a small statistical chance of injury” is, for that reason alone, at least, a defective product. Whether goods have a defect within the meaning of the Act depends on all relevant circumstances, including, amongst other things, the nature and extent of the warnings given by the manufacturer concerning the risk or risks. In Merck at [191], [196], and [201] the Full Court upheld the primary judge’s finding that Vioxx had a defect because the consumption of the drug (designed to treat arthritis) increased the risk of myocardial infarction in only 0.5% of cases in the absence of such a warning.
3195 Incorrect or misleading warnings might also make a product defective for this purpose.
3196 As the applicants acknowledged, a finding that particular goods have a safety defect does not, without more, signify that the goods should never have been on the market or that they should be removed from the market.2796
3197 The respondents submitted that the applicants’ case was that the Ethicon devices were defective for one or more of the following three reasons: first, because they had the pleaded complications; second, because of the availability of alternative forms of treatment which were equally or not materially less safe and effective options; and third, because the respondents failed to warn of “inadequate clinical evaluation” or the other matters complained of in the pleadings.2797 The respondents’ written submissions rested on these assumptions. They argued that none of these matters, considered alone, rendered the devices defective. That is not, however, my understanding of the applicants’ case. As I understood their case, these matters, along with others, such as the fact that neither condition is life threatening, that surgery is elective, and that alternative forms of treatment are available, were all circumstances to be taken into account in determining whether the devices were defective.2798
3198 In broad terms, the applicants’ case was that each of the Ethicon devices did not have the level of safety persons generally were entitled to expect (and were therefore defective) because of the risks they posed and having regard to the information the respondents supplied about them, including in the IFUs and the promotional material. They argued that persons generally were entitled to expect the devices to be as safe as first-line elective surgical treatment of all cases of prolapse repair or surgical treatment of stress urinary incontinence, as the case may be, in patients of all ages against a background where native tissue repair surgery was available with, at worst, comparable efficacy.2799 The applicants also submitted that, given the availability of native tissue repairs, persons generally were entitled to expect that a product for use in first-line elective surgery to treat either pelvic organ prolapse or stress urinary incontinence would not expose patients to a range of additional or increased risks including irremediable chronic pain.2800
3199 In relation to the POP devices, the applicants submitted that the rate and severity of the risks were such that those devices were inherently unsafe and no warning could have saved them from being defective. At one part of their submissions, they characterised the defect as “the risks of adverse events”.2801 More particularly, they alleged that the risks and (potential) complications of each implant were and are that:
(a) the mesh used in each of the [Ethicon devices] would result in a tissue reaction caused by a foreign body response in the form of a scar;
(b) the tissue reaction mesh response of some women might be stronger than in others but there was no way to predict the extent of tissue reaction in an individual patient;
(c) the scar tissue resulting from use of the mesh would apply a contracting force to the mesh;
(d) contraction of the mesh tissue construct could cause pain;
(e) the scar tissue caused by the foreign body response to the mesh could cause pain;
(f) complications of use of the [Ethicon devices] could include acute and chronic pain which could be difficult to treat;
(g) chronic pain resulting from the [Ethicon devices] could be very damaging and debilitating, require multiple operations which might not be successful, and such further operations could themselves cause more scarring and more pain;
(h) complications from the [Ethicon devices] could result in recurrence of the original condition sought to be treated namely prolapse or stress urinary incontinence as the case may be;
(i) pain resulting from use of the [Ethicon devices] would be nerve related;
(j) a complication from use of the [Ethicon devices] could be mesh erosion or extrusion into the vaginal canal or into another organ, which could cause pain and be difficult to treat;
(k) mesh extrusion or exposure from use of the [Ethicon devices] could occur many years after implantation;
(l) pain as a result of use of the [Ethicon devices] could arise many years after the date of implantation;
(m) two or three years follow up after implantation of the [Ethicon devices] without mesh erosion does not guarantee a future free of erosion;
(n) two or three years follow up after implantation of the [Ethicon devices] without pain does not guarantee a future free of pain;
(o) a woman who received one of the [Ethicon devices] was at a lifelong risk of erosion;
(p) a woman who received one of the [Ethicon devices] was at a lifelong risk of pain;
(q) a complication of the [Ethicon devices] could include dyspareunia and apareunia;
(r) other complications of the [Ethicon devices] could include difficulty voiding, difficulty defecating, offensive discharge, de novo urge or stress incontinence, damage to surrounding organs, ligaments, tissues and blood vessels;
(s) the [Ethicon devices] could be difficult or impossible to remove safely or without complications, any removal process carried with it a risk of new or aggravated surgical complications and might require one or more surgical procedures to attempt to remove the Implants, and removal in whole or in part might not fully alleviate the relevant complication.
3200 In substance, there was no dispute about the existence of these risks or complications. As I have already noted, Dr Hinoul conceded as much in cross-examination.
3201 The applicants submitted that the warnings provided by the respondents were insufficient to guard against these risks. In particular, they argued that the instructions for use provided with the SUI devices were deficient in at least the following respects:
(1) They failed to adequately warn of the risk of pain, including acute and/or chronic pain; pain with intercourse which in some patients may not resolve; and neuromuscular problems including acute and/or chronic pain in the groin, thigh, leg, pelvic and/or abdominal area.
(2) They did not warn of the likelihood of the mesh contracting and shrinking.
(3) They should have “decoupled” extrusions, erosions and fistula formation from transitory local irritation.
(4) They advised that “[a] transitory foreign body response may occur” when there was no doubt a foreign body response would occur and it would be permanent.
(5) They failed to provide adequate warnings regarding the so-called tape removal complications, including that one or more revision surgeries may be required and that, given the nature of the mesh as a permanent implant that integrates into the tissue, significant dissection may be required.
(6) They failed to provide any or any adequate warnings regarding the increased exposure to risk faced by certain patient groups, such as patients on anticoagulant medications, smokers, obese patients or patients with autoimmune conditions.
(7) They failed to provide any or any adequate information about the comparative risks of tape surgery relative to alternative treatments.
3202 To the extent that the safety concerns associated with the devices could have been overcome by appropriate warnings in the IFUs, the applicants submitted that the instructions, information, and warnings were deficient in numerous respects.
3203 Both sides relied on evidence of the performance of the devices in comparison with the alternative forms of surgical treatment of stress urinary incontinence and pelvic organ prolapse, effectively submitting that this comparative evidence is a relevant circumstance for the purpose of determining whether the Ethicon devices were defective under s 75AC.
3204 The applicants contended that the availability of native tissue repair was also a relevant circumstance. They argued that contextual matters relevant to the safety persons generally are entitled to expect include the facts that neither stress incontinence nor prolapse is life-threatening, surgery for both is elective, and native tissue repair is available and is at least of comparable efficacy. Given the availability of native tissue repair, the applicants contended, persons generally are and were entitled to expect that a product for use in first-line elective surgery for a non-life threatening condition would not expose the patient to new, additional or greater risks of harm than if they were to undergo treatment that did not involve the use of one of the Ethicon devices.2802
3205 The respondents argued, among other things, that it was not possible to make “absolute generalisations” about the safety and efficacy of medical devices because of differences between patients, surgeons and the devices themselves. In relation to the various treatment options, they submitted that it was not appropriate to view them as true alternatives, as the appropriate treatment will depend on individual patient factors and surgeon preference. As such, “comparisons between tailored native tissue repairs and synthetic mesh kits, are problematic”.2803
3206 It is one thing to say that factors specific to the patient and treating surgeon would have a role to play in an individual case. That proposition is uncontroversial. It is another to suggest that, where different treatment options are available for the alleviation of a condition, such as stress incontinence or pelvic organ prolapse, their outcomes cannot and should not be compared. After all, that is the very objective of randomised controlled trials in this field.
3207 The matters to which the applicants drew attention are relevant. It was not necessary for the applicants to establish “absolute generalisations” about the safety and efficacy of the various treatment options. Neither was it necessary for them to establish in relation to each pleaded complication that the severity was worse, and/or that the incidence rate was higher, when an Ethicon device was used than when it was not. The availability of alternative treatment options and other contextual matters are, however, relevant circumstances to be taken into account in deciding whether the devices were defective.
3208 The respondents contended that the evidence of comparative safety was also relevant to the issue of the adequacy of the warnings. They submitted that the presentation of risks to patients requires a comparative exercise, so that a doctor would tell patients about the safety and efficacy of the various options available to them. They noted that many of the pleaded complications were not specific to the devices, but could arise after any surgical procedure (such as damage to surrounding organs, nerves, ligaments, tissue and blood vessels, haemorrhage, and infection) or after any pelvic floor surgery (such as dyspareunia, difficulty voiding or defecating, offensive discharge and leg weakness). They submitted that, if a complication could arise from any form of pelvic floor surgery, it was reasonable for the manufacturer to expect that treating surgeons knew about it and it did not need to be included in the IFUs.2804
3209 It is true that several of the complications could arise with any form of pelvic floor surgery. And it may be accepted that doctors acting reasonably would consider the risks and benefits of all available treatments for a particular patient. It may also be accepted that, before recommending a particular course of treatment, doctors would discuss with their patients the risks and benefits of that treatment. For the reasons discussed below, however, that does not mean that the respondents were not required to warn of the risks to which patients were exposed, including those that could arise with other forms of pelvic surgery, and that a device would not be defective in the absence of such a warning. I do not accept that, if a complication that can be caused by any of the Ethicon devices as well as by other forms of pelvic floor surgery, it was reasonable to omit any mention of it in the IFU.
The relevance of the physician/surgeon intermediary
3210 The respondents submitted that the extent of safety persons generally are entitled to expect necessarily depends, at least in part, on the direct source of the information provided to the ultimate user of the product in question.2805 They pointed out that the Ethicon devices were not supplied directly to the person for whom they were designed but through medical specialists. They drew attention to an observation made by the Full Court in Merck at [191], quoting from the Explanatory Memorandum, that “the role which intermediaries may play in the supply of goods may also need to be taken into account” and in which the Court noted that prescription pharmaceuticals are supplied to the consumer by pharmacists only on the prescription of a qualified medical practitioner. The respondents argued that, a physician has a duty to “remain abreast of product characteristics and to decide which facts should be told to the patient” (citing Carey-Hazell (2004) at [219] per Kiefel J) and that medical practitioners should be presumed to be competent. They contended that the knowledge of the intermediary (here, the treating physician/surgeon) includes the following:
(1) the fact that any surgery carries risks of complications and a risk of failure (in a general sense);
(2) the fact that different treatment options will necessarily carry different risk profiles and cure rates;
(3) any specific complications associated with the various treatment options (including the implants as well as any other treatments offered by the treating surgeon) for stress urinary incontinence and pelvic organ prolapse as reported in the medical literature;
(4) the rate of any of the complications associated with the various treatment options (including the implants as well as any other treatments offered by the treating surgeon) for the respective conditions as reported in the medical literature;
(5) the risk of reoperation associated with the various treatment options (including the implants as well as any other treatments offered by the treating surgeon), as reported in the medical literature;
(6) the rate of the need for reoperation associated with the various treatment options (including the implants as well as any other treatments offered by the treating surgeon), as reported in the medical literature; and
(7) the severity of the risks associated with each of the matters outlined above.
3211 The respondents cross-examined Dr Agur and Assistant Professor Chughtai about their surgical training and, in Assistant Professor Chughtai’s case, training offered by the manufacturer in relation to POP devices specifically.2806 They established that Dr Agur received training in around 2003 to 2005 about the risk of erosion in relation to the SUI devices and pain as a risk of all pelvic surgery and that Assistant Professor Chughtai became aware at the time of his training in 2010 to 2012 of the risk of mesh erosion or mesh extrusion when mesh is used abdominally or vaginally.2807 They pointed to evidence indicating that, in spite of the absence from the IFUs for Gynemesh PS of a reference to the risk of dyspareunia, Dr Jeannette Lim, who implanted the device in Mrs Dawson, had warned her that she was at risk of dyspareunia.2808 They also pointed to evidence from Associate Professor Lam that he was aware that Prolift could cause dyspareunia. They also claimed that surgeons could be expected to know that there was uncertainty about long-term results.2809
3212 Against this context, the respondents contended, in effect, that omissions from the IFUs of references to complications like dyspareunia, which can arise from other forms of pelvic surgery, were immaterial because these were risks that had to be considered by the surgeon regardless of the procedure selected.2810
3213 It may be accepted that it is relevant that the Ethicon devices are supplied in the first instance to doctors and hospitals and not directly to patients. In Wilkes v DePuy at [108] Hickinbottom J said that it was a relevant circumstance that a learned intermediary had chosen a particular prosthesis for a particular patient and had available to him “his general professional knowledge” and “the specific IFU including warnings”. In Gee v DePuy at [169] Andrews J agreed that “the existence of a learned intermediary and the information and warnings provided to that intermediary are plainly relevant circumstances”, adding that the weight to be given to those circumstances will vary from case to case.
3214 It may also be accepted that, before a medical device is implanted, the surgeon would be obliged to disclose to the patient a material risk inherent in the proposed treatment unless the surgeon reasonably considered disclosure of the risk would cause damage to the patient: Rogers v Whitaker (1992) 175 CLR 479 at 490. I interpolate that in Rogers v Whitaker at 489–490 the High Court held that a risk is material if, in the circumstances of the particular case, a reasonable person in the patients’ position would be likely to attach significance to it or the surgeon is or should reasonably be aware that she would.
3215 Furthermore, it may be accepted that treating surgeons would be aware of the risks of pelvic surgery and that it is unlikely that the IFU would be the sole source of information for most, if not all, surgeons. And I am prepared to assume that some pelvic surgeons would have been aware of many, if not most, of the risks associated with the implantation of the various Ethicon devices as a result of their own experience or research.
3216 Nevertheless, to the extent that the respondents argue that manufacturers are excused from liability with respect to risks or complications that should be known to doctors or which they are able to discover for themselves, that argument should be rejected.
3217 First, the object of the TPA is “[the enhancement of] the welfare of Australians through … fair trading and provision for consumer protection”: TPA, s 2. It has been described as “a fundamental piece of remedial and protective legislation”, which is “to be construed so as ‘to give the fullest relief which the fair meaning of its language will allow’”: Marks v GIO Australia Holdings Limited (1998) 196 CLR 494 at [99] (Gummow J). In the same case at [124], Kirby J noted that the Act provides “important remedial protection for consumers” and remarked that attempts to divert the Court from a construction which would frustrate and defeat its objectives should be resisted.
3218 The Act does not excuse a manufacturer from liability with respect to defects that are unknown to consumers but should or might be known to doctors. To the contrary, consistent with the protective purpose of the legislative scheme, the Act envisages that the manufacturers will fully inform the “learned intermediaries”. The significance of the position of the so-called learned intermediaries was explained in the Explanatory Memorandum at [24]:
The role which intermediaries may play in the supply of goods may also need to be taken into account. For example, prescription pharmaceuticals are supplied to the consumer by a qualified pharmacist and only on the prescription of a qualified medical practitioner. Due to the complex nature and effects of these products, complete instructions and warnings may not be provided to the consumer by the manufacturer. However, detailed product information is provided to doctors and pharmacists by the manufacturer so these learned intermediaries are sufficiently informed to be able to decide whether or not it is appropriate to dispense pharmaceuticals to particular consumers. This factor will be relevant in determining whether a pharmaceutical is defective, particularly where a claim of a defect in information provided is made.
(Emphasis added)
3219 It was taken up again at [50]:
As noted above in relation to matters relevant to determining whether goods are defective, due to the complex nature of pharmaceuticals, detailed product information is provided to the qualified intermediaries rather than directly to the consumer. The information is provided with the expectation that it will be used to properly inform the consumer about the product as the doctor or pharmacist sees fit. A product cannot be considered to be defective if it acts in an injurious or damaging manner due to the failure of the intermediary to properly inform the consumer, provided that the proper information is provided by the manufacturer to the intermediary.
(Emphasis added)
3220 This point was not lost on the Full Court in Merck. Indeed, the Court cited this passage from the Explanatory Memorandum at [191] of its judgment. The respondents emphasised the first three sentences of the Explanatory Memorandum at [24].2811 But the point that was being made was that the members of the public might not expect the manufacturer to provide them directly with complete instructions and warnings, not that the manufacturer has no or some limited obligation to provide this information in the first place. In a particular case, the manufacturer’s obligation might be discharged by providing the information to the learned intermediary. Nothing in the Explanatory Memorandum, however, or in the judgment in Merck would support a contention that the manufacturer need not provide the so-called learned intermediaries with detailed information about its products and associated risks because they ought to know from their studies or their reading what the risks are or because that information is available to them from other sources, such as medical journals.
3221 Second, the respondents’ reliance on Carey-Hazell (2004) was misconceived. In the extract cited by the respondents, Kiefel J was dealing with a defence raised by the manufacturer to the consumer’s case in negligence. What her Honour said in that extract had nothing to do with a claim under s 75AD of the TPA. Her Honour was explaining the approach to a manufacturer’s duty of care in the United States which has recognised a “learned intermediary” defence. There is no such defence in Australia. The respondents took her Honour’s remarks out of context. This is the full context:
The effect of a “learned intermediary” upon a manufacturer's duty has been the subject of considerable case law in the United States. In Sterling Drug, Inc. v Cornish 370 F.2d 82 (8th Cir 1966) the term was used in relation to a physician who acted as a liaison between a patient and drug manufacturer. The content of the manufacturer's duty was to warn prescribing physicians. The doctrine has since been extended to medical device cases: Phelps v Sherwood Medical Industries 836 F.2d 296 (7th Cir 1987) (which involved the manufacturer of a heart catheter) and Brooks v Medtronic, Inc. 750 F.2d 1227 (4th Cir 1984) (a cardiac pacemaker). It has been explained that the reason for describing the duty in this way is because it is a physician's duty to remain abreast of product characteristics and to decide which facts should be told to the patient. Once adequate warnings are given to the physician, the choice of treatment and the duty to disclose properly fall upon the doctor …
(Emphasis added)
3222 Third, in A v National Blood Authority Burton J did not consider that the knowledge of the doctors and surgeons of a risk of contamination of the products in question was determinative of the public’s safety expectations. In that case it was an agreed fact that, for two decades before blood products were supplied, the medical profession and blood producers had known that a certain percentage of donated blood in the UK (thought to be between 1 and 3%) was infected with hepatitis C (then known as hepatitis non-A, non-B). The claimants argued that this knowledge was irrelevant to the legitimate expectation of the public. The defendants, on the other hand, argued that, since the risk was known to those who mattered — the medical practitioners through whom the blood was supplied — it was relevant to that question, although it was “not seriously argued … that there was any public understanding or acceptance of the infection of transfused blood by [h]epatitis C” (at [55]). Noting that doctors and surgeons knew of the risk but did not tell their patients (except on rare occasions when asked), Burton J found that “it was certainly … not known and accepted by society that there was such a risk”.
3223 In short, the fact that the goods in question are supplied through a surgeon does not excuse the manufacturer from its obligation to warn of the risks attendant upon the use of its products, even where those risks might or should be known to the surgeon. It merely affects the way the obligation is discharged. As the learned intermediary doctrine is applied in the United States and Canada, a manufacturer who provides a suitable warning to the doctor need not also provide that warning to the patient. If such a warning is given to the doctor, the manufacturer has a complete defence. In Gross v Gynecare, 2016 WL 1192556 (N.J. Super. Ct. App. Div. 2016), a case about Prolift, the Appellate Division of the Superior Court of New Jersey explained at [35]:
The learned intermediary doctrine imposes a duty on a manufacturer to warn physicians of the risks involved with its product, thereby placing the physician in the role of intermediary between manufacturer and patient. See Ehlis v. Shire Richwood, Inc., 367 F.3d 1013, 1016 (8th Cir. 2004) (North Dakota law); McElhaney v. Eli Lilly & Co., 575 F. Supp. 228, 231 (D.S.D.), aff'd, 739 F.2d 340 (8th Cir. 1984). Thus, the law considers a manufacturer's warning to a physician to be a warning to the patient, and a manufacturer “need not communicate directly with all ultimate users” of its product. In re Norplant Contraceptive Prods. Liab. Litig. 215 F. Supp. 2d 795, 803 (E.D. Tex. 2002), aff'd sub nom. White v. Wyeth Labs., 69 F. App’x 658 (5th Cir. 2003).
The court observed at [36] that the purpose of the doctrine is to enable patients to make informed and intelligent decisions about whether to undergo a recommended treatment by balancing the risks against the benefits. The court went on to explain that, once there is a finding that the warning by the manufacturer to the physician is inadequate, the defence “simply drops away”. Nothing in the doctrine excuses the manufacturer from liability because information is available to the physician from other sources.
3224 Fourth, while I accept, as I have said, that it is a relevant consideration that the goods are supplied through a learned intermediary, I do not accept that the safety persons generally are entitled to expect from the goods depends on whether the user of the goods deals directly with the manufacturer or through a learned intermediary. Nor do I accept that there is support for that proposition in the judgment of the Full Court in Merck.
3225 In Merck the Full Court did say at [191] that:
The role which intermediaries may play in the supply of goods may also need to be taken into account. For example, prescription pharmaceuticals are supplied to the consumer by a qualified pharmacist and only on the prescription of a qualified medical practitioner. Due to the complex nature and effects of these products, complete instructions and warnings may not be provided to the consumer by the manufacturer.
(Emphasis added)
3226 The respondents drew attention to this passage, overlooking the words “to the consumer” and the passage that followed:
However, detailed product information is provided to doctors and pharmacists by the manufacturer so these learned intermediaries are sufficiently informed to be able to decide whether or not it is appropriate to dispense pharmaceuticals to particular consumers. This factor will be relevant in determining whether a pharmaceutical is defective, particularly where a claim of a defect in information provided is made.
3227 Neither the Full Court’s judgment in Merck nor Kiefel J’s judgment in Carey-Hazell (2004) provides support for the notion that the respondents were not obliged to provide detailed information and warnings about the pleaded complications because doctors should be presumed to have knowledge of them or access to publications from which that knowledge could be derived.
3228 In any case, the evidence indicated that at least some of the effects on the human body of polypropylene mesh, including the Ethicon devices, were not well-known to the medical or surgical community during the relevant period and that some of the information was not available in the journals commonly read by the cohort. Assistant Professor Chughtai, for one, said that it would be unlikely that a reasonably competent gynaecologist or urologist would be aware of the scientific literature on the inflammatory response to mesh.2812
3229 Collinet et al (2006) observed that the published data about the management of mesh exposure was sparse.2813 Based on the material before the Court, little changed in the following decade.
3230 Moalli et al (2008) acknowledged that the quality of the host tissue and the technique of sling placement were well-known to most surgeons as potential contributors to “sling complications”. But they recognised that there were “knowledge deficits”, the greatest of which, they said, was in the area of material properties. In this area, the authors wrote, surgeons are “completely dependent on the mesh information supplied by a representative of the vendor”.2814 They went on to observe that:
Even more problematic is that many of the representatives have little knowledge of biomechanical factors that may be relevant and tend to focus on aspects of the sling which facilitate the operation for the surgeon. This practice may not be the best approach and may impact clinical outcomes.
3231 An inquiry made of JJM by an Australian surgeon as recently as 2013 underscores the gap in the knowledge of the user community and the importance of providing information in the IFU. The inquiry concerned the possible relationship of a patient’s pain, in particular persisting groin pain, to her TVT surgery.2815 The inquiry was forwarded in an email to Dr Hinoul and others, alongside a letter from the doctor setting out the details of the case.2816 The letter had been written in response to a complaint lodged with the NSW Health Care Complaints Commission. It disclosed the doctor’s resistance to accepting the possibility of groin pain as a consequence of TVT surgery and purported to annex a “copy of a relatively recent review of the complications of Anterior TVT in which there [was] no mention of persisting pelvic or groin pain”.2817
3232 Professor Korda said that the literature does not reflect the devastation caused by some of the mesh complications.2818 He went on to explain:
The literature merely states for example that the mesh erosion rate is for instance 10 per cent after mesh implant surgery. It does not state that those who suffer erosion are often sexually active young women who are unable to have intercourse because of a repulsive vaginal discharge, bleeding and chronic pain on sitting, standing or movement and that this may not be easily corrected.
It is my view that the published literature does not adequately convey the pain and suffering of the patients who have had mesh complications.
The published literature does not mention the fact that the placement of mesh is permanent and the patient has to live with the mesh for the rest of her life. Nor does the literature allude to the fact that when patients are counselled for the mesh placement they should be advised that if they have mesh surgery and complications develop there may not be a cure for their problem.
3233 In any case, no matter how learned the intermediary may be, it is highly unlikely, if not inconceivable, that the intermediary would know as much about the product as the manufacturer itself. In cross-examination, Dr Hinoul conceded that Ethicon had spent hundreds of thousands of dollars on reports, studies and consultancies relating to the foreign body reaction and had access to a body of knowledge on the subject which far surpasses the knowledge of gynaecological surgeons, regardless of their experience.2819 He also conceded that surgeons are entitled to think that the manufacturer is likely to have more knowledge about its own product than members of the medical community.2820
3234 Professor Blaivas, a very experienced urologist, observed that the mesh manufacturers provided no guidance as to exactly where within the thickness of the vaginal wall the mesh should be placed and experimental studies do not address the issue.2821 This is a matter of concern since placing mesh too close to the vaginal epithelium or the lower urinary tract or in the vaginal wall can cause the mesh to erode.2822
3235 Professor Korda, another very experienced urogynaecologist, gave evidence that polypropylene mesh implants carried a risk of certain complications that were unknown to pelvic surgeons versed in native tissue repair. He said that “complications such as mesh erosion, mesh contracture, bunching, severe chronic pelvic pain, severe dyspareunia and pain on movement, sitting, standing were not seen before the introduction of mesh surgery”.2823
It is my view that the literature does not reflect the devastation that some of the complications of mesh surgery result in. The literature merely states for example that the mesh erosion rate is for instance 10 per cent after mesh implant surgery. It does not state that those who suffer erosion are often sexually active young women who are unable to have intercourse because of a repulsive vaginal discharge, bleeding and chronic pain on sitting, standing or movement and that this may not be easily corrected.
It is my view that the published literature does not adequately convey the pain and suffering of the patients who have had mesh complications.
The published literature does not mention the fact that the placement of mesh is permanent and the patient has to live with the mesh for the rest of her life. Nor does the literature allude to the fact that when patients are counselled for the mesh placement they should be advised that if they have mesh surgery and complications develop there may not be a cure for their problem.2824
3236 That evidence alone is an answer to the respondents’ submission that “there is no suggestion that there was a relevant knowledge gap between the [r]espondents and the general surgical community”.2825 True it is that this evidence did not refer specifically to implantation of Prolene or Gynemesh PS or the Ethicon devices but there could be no doubt that Professor Korda had Ethicon’s products in mind.
3237 The effect of Professor Korda’s evidence was that he did not appreciate the extent of the potential harm from the use of transvaginal mesh in prolapse repair until around the time of the publication of the Altman article in 2011, which was more than four years after Mrs Gill’s implant surgery and about two years after Mrs Dawson’s. It was at that time, he wrote in his first report, that he began seeing patients who had been implanted with mesh by other gynaecologists and were seeking treatment for the complications. Not all implanting surgeons would see a similar cohort of patients. He said that:
These patients had such alarming problems and poor quality of life that I felt that I could not inflict these problems on any of the patients that came to me for treatment of prolapse. I felt that inserting mesh caused too much harm in exchange for very little extra benefit.
In my view, it is preferable to have a prolapse recurrence than the complications I was seeing.2826
3238 Professor Deprest reported that, for each novel product, he and other consultants were typically trained by an expert in the specifics of individual polypropylene kits for transvaginal anterior, posterior and combined repairs. Nevertheless, it seems, this proctorship did not prepare him for all potential complications, for he wrote that:
As the experience grew we, like others, came across a number of complications, in essence mainly graft exposures, and to a lesser extent chronic pain, either associated with mesh folding yet at times without a direct anatomical substrate.2827
3239 Dr Agur included in an appendix to his first report a narrative explaining the warnings he provided patients over the years. At no time did he refer in that narrative to the risk of chronic pain. Nor did he refer to the potential for additional surgery to remove the mesh, the difficulties that could present, and the complications that might ensue.
3240 Notwithstanding his own practice of warning of an increased risk of complications in patients with certain conditions, Dr Agur considered that the failure of mesh manufacturers to identify whether mesh would be indicated or contraindicated in certain patients “directly resulted in the blanket use of mesh in a one-size-fits-all approach”.2828
3241 The respondents tendered a PowerPoint presentation used by Associate Professor Lam when advising patients about prolapse surgery.2829 In submissions they pointed out that he was operating at the same time as Mrs Gill received her Prolift implant and drew attention to information he conveyed to patients that was not included in the IFUs for the POP devices.2830 For example, risks said to be specific to mesh/graft repairs were:
Mesh erosion – 1 to 10%
Mesh extrusion
Mesh contracture leading to painful scars
Mesh infection2831
3242 Notably, however, no distinction was drawn between the pain, inflammation, dyspareunia, or painful scars that might arise with prolapse surgery in general and mesh surgery in particular. No mention was made of the risk of chronic pain after mesh surgery, the potential for multiple operations to treat erosions, or difficulties associated with removal of the mesh. Further, while Associate Professor Lam testified that this information was provided to patients consenting to Prolift, he also confirmed that the presentation was current.2832 It does not necessarily follow that this was the information he conveyed to patients in 2006–7, when the advice was given to Mrs Gill or in 2009 when Mrs Dawson was being considered for a mesh implant.
3243 The presentation also included an almost verbatim copy of the representations made about Gynemesh PS in the IFUs for the POP devices including that it elicits a minimum transient to slight inflammatory reaction. When asked about whether he would inform patients about a continuing foreign body reaction, he said that he would provide an explanation “if the patient was to ask that specific question”.2833 In that event, he said he would tell the patient what he told the Court, namely that:
that an inflammatory reaction is important as part of a healing process, first and foremost. Without that inflammatory normal process of healing, the wound would break down. So in the initial six weeks to eight weeks, that is a normal post-surgical healing process. As we leave behind an implant, a foreign body, the patient’s tissues will react to that, and that reaction is likely to be 10 different from patient to patient, and also from time to time, from the stage when it was first put in to subsequent times and the longer the patient is followed up. The analogy is much the same as a dental implant, or a heart valve implant, or a knee or a hip. There has got to be some degree of foreign body reaction to the presence of that tissue which is foreign to our system. Clinically, that means in daily living, most of the time we’re able to and have clearly benefitted from the prosthetic materials that are used so commonly in medical care, in surgical care, because such a reaction does not harm the patient. Sometimes there are suboptimal outcomes, and that’s where we need to actually follow up the patients and have a sort of a regular feedback to understand that.2834
3244 Professor Korda was taken in cross-examination to a PowerPoint presentation he had given in 2004 as a preceptor for TVT.2835 In re-examination he testified that the slides reflected his understanding of the complications of the devices at the time of the presentation and “until recently”. 2836 The slides did not refer, and were not intended to refer, to a number of admitted or otherwise established complications. In particular, they did not refer to dyspareunia, complications associated with the removal of the tape, tape erosion due to mechanical movements of the body, complications due to irritation of the urethra by the tape, fibrotic tissue surrounding the tape, contraction of the tape by fibrotic tissue, fibrosis due to chronic inflammation, voiding dysfunction or other complications due to subsequent tensioning of the tape by reason of contraction of the tape by scar tissue, or chronic pain. Nor did they refer to the particular risks polypropylene mesh posed for a woman with a compromised immune system.
3245 In those slides, he said this about pain:
• Although chronic pain has been reported in up to 12% of patients following colposuspension and 10% after needle suspension, it has not been previously reported with TVT
• Leg pain has been observed after the transobturator approach
• Severe pain has been reported after IVS 14%2837
3246 The evidence established that at all material times the respondents knew that there was a risk of chronic pain following implantation with all of their devices. It appears from Professor Korda’s slides and his evidence in this case that for a good deal of that time he did not.
3247 Professor Korda also testified that, since he was both a user of the devices and a preceptor, teaching surgeons and registrars in training how to perform the operations to implant them, he expected that Johnson & Johnson would have informed him of any complications of which it was or became aware.2838
3248 As the applicants submitted, it is absurd to think that average gynaecologists and urologists, acting reasonably, would attend every conference at which the POP and SUI devices were discussed or read every article in every journal in which their complications were canvassed.2839 This is particularly unlikely in the case of the busy surgeon who was a key target of Ethicon’s marketing. Furthermore, as Dr Hinoul candidly observed in his email to Dr Altman about his manuscript, most doctors only read the abstracts of articles.2840 It will be recalled that the reference to dyspareunia in the draft of the abstract to the Altman article was deliberately omitted after intervention from Ethicon.
3249 I accept the evidence of Assistant Professor Chughtai that an average, reasonably competent gynaecologist would expect the manufacturer to provide her or him with information about the use, surgical technique, risks of adverse events, and studies supporting safety and efficacy.2841
3250 Fifth, I do not accept as a general proposition that surgeons could be expected to know that there is uncertainty about the long-term consequences of implantation of polypropylene mesh. Moreover, I doubt very much whether most surgeons would have any appreciation of the path to regulatory clearance for medical devices. Certainly, as Mr Finch SC acknowledged in closing argument, no evidence was led as to what surgeons knew about how the regulatory process for medical devices worked.2842 Absent information to the contrary from the manufacturer or supplier, they might well have assumed that the devices had been subjected to long-term trials before they were cleared for sale and that if that were not so the IFU and other product information would have made that clear.
3251 Sixth, the European Directive and the Medical Devices Regulations both require manufacturers to inform users of the residual risks. Moreover, it is evident from the actions of the TGA that, consistent with the evidence of the regulatory experts, its expectation was that risks about which doctors might or should be aware should nonetheless be disclosed in the IFUs for the various devices. The general public is entitled to expect nothing less.
3252 The respondents also submitted that the applicants’ failure to call surgeons with whom the applicants or any other patient decided to have “the index surgery” was “particularly significant”. They contended that, without evidence from them, the Court could not be satisfied that the information provided by the respondents or the warnings they gave were inadequate.2843 This argument must be rejected, too. Whether the safety of goods is not such that persons generally are entitled to expect does not depend on the knowledge of individual surgeons.
What might reasonably be expected to be done with or in relation to the devices?
3253 This consideration admits of no range of possibilities. The devices were designed to be implanted permanently in the female pelvis. That was what might reasonably be expected to be done with or in relation to the devices.
3254 Yet, the respondents submitted in oral argument that “what might reasonably be expected to be done with or in relation to a product includes what [a learned intermediary] might reasonably be expected to say [to the patient] about the product”.2844
3255 This submission must be rejected.
3256 First, it distorts the language of para 75AC(2)(e), which is directed to the use to which the devices might reasonably be expected to be put. The subparagraph is not concerned with what might be said about the devices. Second, the construction it advances does not accord with the purpose of the paragraph. To the extent that this is not obvious from the words used, it is clear from the Explanatory Memorandum at [19]:
A further factor which must be taken into account is the use to which the product could reasonably be expected to be put [paragraph 75AC(2)(e)]. This “use” includes all reasonably expected secondary uses and likely potential misuse. Thus in some cases a manufacturer will be under an obligation to warn consumers of the potential consequences of misuse which could be anticipated by the manufacturer. This may in certain circumstances go beyond merely stating that a certain course of action should not be adopted and require the manufacturer to detail the specific consequences of such misuse (ie, to detail the type of injury or damage which may be suffered). If the loss does result partially from misuse, the manufacturer will be able to reduce the amount of compensation payable to reflect that part of the damage caused by contributory acts by the injured person (see section 75AN below), but this does not relieve the manufacturer of the obligation to warn.
3257 As the applicants submitted, the Explanatory Memorandum not only treats the paragraph as referring to physical acts, it also indicates that the purpose of the provision is to deal with matters that are likely to require the manufacturer to take action to protect the safety of users.2845
3258 As the earlier discussion illustrates, the section is not intended to relieve the manufacturer of liability for known risks to safety merely because a third party intermediary might be aware of them and might reasonably be expected to inform the recipient about them. To the contrary, it is intended to protect the safety of recipients by ensuring that the manufacturer of goods conveys to them, whether directly or indirectly through a learned intermediary, all the information necessary to properly inform them of the risks pose by the devices, whether through use or misuse.
The manner in which and the purposes for which the devices were marketed
3259 To the extent that a product falls short of the manufacturer’s safety claims, it is likely that it would fall below the level of safety persons generally are entitled to expect. As the Explanatory Memorandum stated at [17], “consumers are entitled to expect a high degree of safety from goods which are marketed in a manner depicting simplicity and safety”. In effect, the legislature holds the manufacturer to the truth of its claims.
3260 The respondents’ promotional material was designed to encourage surgeons to recommend the devices to patients and patients to accept those recommendations. The devices were marketed as safe and effective to treat either stress urinary incontinence or pelvic organ prolapse. As I noted previously, risks were minimised and some not mentioned at all. Notwithstanding the way the respondents put their case, a number of their product brochures told surgeons, in effect, that they could and should rely on the IFUs for “complete” contraindications, warnings, precautions, and adverse reactions.
3261 Dr Hinoul deposed that the respondents were “not in direct contact with the patient”.2846 That evidence must be rejected. The respondents produced two kinds of product brochures: brochures directed to patients and brochures targeted at surgeons. Both kinds were integral to Ethicon’s marketing strategy. Dr Hinoul testified that Ethicon recognised the potential value of advertising the devices directly to patients by way of patient brochures.2847
3262 The respondents argued, in effect, that the brochures were of no consequence. They submitted that the relevant audience for many of the marketing brochures were medical professionals with expertise in pelvic floor surgery and with access to a range of information as to treatment options and the efficacy and risks of those options.2848 They asserted that brochures were only provided to patients once a treating surgeon had formed the view that surgery was appropriate and suitable for the patient, and as part of the provision of information generally concerning treatment options and the potential complications associated with surgical intervention.2849 They cited the evidence of Associate Professor Lam that he provides his patients with information brochures as part of an “extensive decision making and informed consent process” regarding the appropriate treatment options for a patient with SUI or pelvic organ prolapse.
3263 I accept that the surgeon brochures were directed to medical professionals with expertise in pelvic floor surgery and access to other information. Many of them might well be sceptical of the manufacturer’s claims. But others might be comforted by them.
3264 Any woman who read the patient brochures would be astonished to learn that she could develop chronic pain or that complications could arise years after she was implanted with the device. Doctors and patients alike were led to believe that complications were rare and, if they arose, they would be temporary, inconsequential, and/or easily treatable. The reality was different. While the evidence does not reveal complication rates for any of the SUI devices as high as those reported for the POP devices and, while certain complications were reportedly rare, many were not. Furthermore some complications could be permanent, disabling, and resistant to treatment.
3265 The respondent pointed to the absence of evidence that any of the applicants or their pelvic surgeons relied on any of the brochures, that any treating surgeon or patient was provided with them, or that any surgeons provided such materials to their patients.2850 In these circumstances, they invited the Court to infer that evidence of the influence of these marketing materials on patients or treating surgeons would not have assisted the applicants.2851
3266 With respect to the issue at hand, I reject the invitation.
3267 First, as the applicants’ argued, the very fact that brochures were produced by JJM meant that it could “safely be inferred that this material was deployed on the Australian market”.2852 There is no reasonable prospect that the brochures were left in storage in the offices or warehouses of JJM and not distributed to their intended audience. Besides, Associate Professor Lam’s evidence indicates otherwise. In the absence of any evidence to the contrary, it is reasonable to infer that brochures produced in conjunction with the supply of the devices were supplied around the time the devices came on the market or shortly thereafter.2853 After all, all evidence is to be weighed according to the proof which it was in the power of one side to have produced and the other to have contradicted. That is the inference I draw.
3268 Second, since the way that goods are marketed is a matter the Court must take into account in determining whether they have a defect, the absence of evidence that the applicants or their surgeons relied on the brochures is beside the point.
The use of any mark in relation to the devices
3269 It will be recalled that the relevant circumstances affecting the determination of the extent of the safety persons generally are entitled to expect include the use of any mark in relation to the goods. “Mark” is not defined in the Act and there is no apparent reason why it should be limited in its meaning. Here, all the devices carried the CE mark.
3270 It will also be recalled that the manufacturer of a medical device is obliged to ensure that it complies with the essential requirements contained in the European Directive. Dr Hinoul deposed that CE marking “proves” that the devices meet those requirements. Having regard to the procedure for affixing the CE mark, that evidence must be rejected.2854 All that CE marking proves is that the manufacturer asserts that a device meets those requirements. Nevertheless, the presence of the CE mark is a representation by the manufacturer that the device complies with those requirements. It is also a representation that the steps required by the Directive to affix the mark have been undertaken. Consequently, persons generally are entitled to expect that devices carrying that mark meet the essential requirements, are safe and effective for their indicated use, that risks that cannot be eliminated are reduced as far as possible through design and construction, that appropriate protection measures are taken in relation to those that cannot be eliminated, and that users are informed of “any residual risks”.
3271 Assistant Professor Chughtai gave unchallenged evidence that most gynaecologists and urologists depend on the regulator to ensure the safety of a device before “widespread adoption”.2855 He continued:
Thus, most general urologists and gynaecologists are not likely to look into the method of approval or to primarily evaluate the medical literature before proceeding with a novel device. They may count on word of mouth and device representatives, likely most of the generalists would assume a device is safe once it is readily available on the market.
3272 In short, it is reasonable for the public to expect that a medical device which carries CE marking and has been cleared for sale meets the regulatory requirements and standards. If it does not, it is unlikely to have the safety persons generally are entitled to expect. An isolated episode of non-compliance or an insignificant infraction may not matter. Repeated and systematic non-compliance, however, certainly does.
Instructions and warnings: some general observations
3273 Paragraph 75AC(2)(d) of the TPA speaks of “instructions for, or warnings with respect to, doing, or refraining from doing, anything with or in relation to them”. In its terms it does not capture warnings or information about risks which might arise even if the manufacturer’s advice is followed to the letter. Plainly, however, disclosure or non-disclosure of any risks will affect the safety expectations persons generally are entitled to have. As was pointed out in the Explanatory Memorandum to the Trade Practices Amendment Bill 1992 (Cth) at [18]:
In relation to goods which are known by the manufacturer to be potentially hazardous, instructions and warnings are particularly crucial, as it is through these sources that the manufacturer can detail the nature and extent of the potential hazard and provide adequate instructions to assist consumers in avoiding that hazard. Similarly, the general presentation of the product can influence consumer expectations by exaggerating safety aspects or minimising reference to possible risks.
3274 Dr Hinoul deposed that all the complications “specific to the mesh device” were covered in the initial instructions for use.2856 He also deposed that he had “been involved in the writing of IFUs, for example, for the TVT Abbrevo device, to ensure that it warns the pelvic floor surgeons of risks specific to the device, given the general knowledge in field”.2857 While he acknowledged that changes had been made to the IFUs from time to time, he said that these changes only incorporated risks that were common to all types of pelvic floor surgery for incontinence and/or prolapse. He did not countenance the notion that the IFUs were deficient in any respect or that Ethicon’s approach was inappropriate — even in hindsight.
3275 I accept that Dr Hinoul was involved in the writing of a number of IFUs. Otherwise I reject this evidence.
3276 First, the first IFU for TVT (the Medscand IFU) omitted any mention of the risk of erosion, exposure or contraction of the mesh. Although it is true that polypropylene sutures may erode and there are reports of erosion with autologous slings, the risks of erosion, exposure and contraction were admittedly “mesh-related” complications. Furthermore, the first reference to the “implant contraction” in an IFU for an Ethicon device did not appear until the 11 January 2005 Prolift IFU. None of the IFUs for the SUI devices ever referred to it.
3277 Second, infection was also a “mesh-related” complication. As Dr Weisberg pointed out in his CER for Gynemesh PS, since polypropylene mesh is passed through a contaminated field, infection is a potential complication. The IFUs for the SUI devices warned against using the devices in women with urinary tract infections and advised in terms to the effect that “as with all foreign bodies Prolene mesh may potentiate an existing infection”. But until 2015 none of them warned that the mesh could actually cause infection.
3278 Third, the first allusion to potential difficulties with mesh removal did not appear in any IFU until 3 April 2015 when they were mentioned for the first time in the Gynemesh PS IFU. By then, however, all the other POP devices were no longer sold or manufactured.
3279 Fourth, a number of other complications associated with the implantation of the Ethicon devices and known to the respondents at all relevant times, were not included in the IFUs until 2013, some not until 2015. Some were never included.
3280 Fifth, several statements made in the IFUs were taken from the IFUs for Prolene or Prolene Soft sutures. Some of them, like those directed to the use of mesh in infants and children which appeared in the IFUs for all the POP devices, were inappropriate. Some, to Ethicon’s knowledge, were false. Some were misleading. All reflected a lack of care in the formulation and revision of the documents.
3281 One glaring example is provided by the statements concerning the foreign body response.
3282 The statement appearing in all the IFUs for the SUI devices until September or October 2015 that a transitory foreign body response may occur was false for two reasons.
3283 In the first place, a foreign body response was not a possibility; it was a certainty. As Dr Hinoul acknowledged in cross-examination, a foreign body response will always occur.2858 Indeed, the devices were designed with the intention that they adhere to the tissues as a result of the foreign body response. Yet, the most recent IFUs in evidence for those of the Ethicon devices that remained on the market at the time of the trial continued to state that a foreign body reaction may occur.
3284 In the second place, as Dr Hinoul also acknowledged, the response is not transitory but permanent.
3285 Dr Hinoul denied that the statement was misleading, claiming that the reader would understand the reference to “foreign body response” to be a reference to an abnormal or unintended foreign body response.2859 I am far from convinced that this is so. In any case, even if some or, indeed, all readers were to interpret the statement in this way, it is unquestionably misleading because it was liable to lead the reader into the belief that there is no possibility of a permanent or long-lasting abnormal foreign body response. The overwhelming weight of evidence demonstrates that such a belief would be false.
3286 Dr Meng Chen, Ethicon’s Associate Medical Director of Worldwide Customer Quality, was under the impression that the suggestion Ethicon was making was that the complications were transitory. In an email to Bryan Lisa of Ethicon US sent on 29 January 2009, Dr Chen wrote:
Pardon me again, from what I see each day, these patient experiences are not “transitory” at all.2860
3287 Yet it was not until 2015 that the error was rectified and then only at the instigation of a regulator.
3288 The IFUs for all the SUI devices that remained on the market at the time of the trial continued to warn of “transitory local irritation at the wound site”, although as early as 2 June 2006, if not before, Professor Klosterhalfen, in an address to an Ethicon expert meeting in Hamburg on meshes for pelvic floor repair, stated that the foreign body reaction was a “chronic wound”.2861 In 2007 an article by Professor Klinge and others was published which referred to “the obligate foreign body reaction” in nonabsorbable synthetic meshes in the treatment of both genital prolapse and stress urinary incontinence and described a “chronic inflammation at the interface…”.2862
3289 Under cross-examination Dr Hinoul said that the reference to “a transitory foreign body response” derived from a request made by the FDA in respect of Prolene.2863 In his affidavit he referred to the FDA writing to Ethicon in 1973 asking them to include in the IFU for the Prolene suture a statement that “transitory local inflammatory reactions have been reported” and then again in 1988 approving a change to the labelling to warn of a “minimal, transient acute inflammatory reaction”.2864 This explanation merely illustrates the inattention in Ethicon’s approach to the safety of its products to the differences between its sutures and the mesh.
3290 Professor Klinge pointed to other statements in the IFUs about the inflammatory response to the mesh and other representations about its qualities which were inaccurate, incorrect or otherwise misleading.2865 To the extent that these statements are reproduced in the product brochures, the product brochures are also misleading.
3291 First, all the IFUs contain an assertion to the effect that animal studies show that implantation of the mesh elicits a minimum to slight inflammatory reaction which is transient and is followed by the deposition of a thin fibrous layer of tissue which can grow through the interstices of the mesh, thus incorporating the mesh into adjacent tissue.
3292 Professor Klinge explained that animal studies are mainly done by testing the material in the abdominal wall without contamination and without subjecting the animals to mechanical strain. Consequently, he said, the precise conditions of the preclinical testing should have been identified. In any case, as Professor Klinger stated, there is an intense (not minimum to slight) host defence response immediately after implantation.
3293 Moreover, the inflammatory reaction elicited in the tissues is not transient. Although immediate or acute post-operative inflammation due to implantation may resolve within days or weeks, the inflammatory foreign body reaction around the implants can persist for years.
3294 Associate Professor Lam agreed that the warning about the inflammatory reaction being minimal to slight in intensity and transient in duration did not accurately represent the nature of the chronic persistent foreign body reaction that occurs and, in particular, that it did not indicate that the chronic foreign body reaction may result in complications such as erosions, late erosions, an increasing incidence of erosions with time, contractures or pain.2866
3295 Having been told that a minimal and transient inflammatory response to the mesh could occur, persons generally are entitled to expect no more severe or enduring a reaction.
3296 Second, the representation in the IFUs for the POP devices that “[t]he mesh remains soft and pliable” is misleading, too, given the evidence about contraction and the development of a solid mass in the area of the implanted mesh. It is at least, as Professor Klinge observed, incomplete.2867
3297 Third, the IFUs for the POP devices state that “normal wound healing is not noticeably impaired”. But that is by no means invariably so. Erosions may occur and, as Professor Klinge noted, they are not a sign of normal wound healing.2868 Professor Klinge also pointed out that, having regard to the fact that the intense foreign body reaction that occurs at the mesh-tissue interface, “the chronic inflammatory response leads to chronic wound healing issues in many patients”. For these reasons, the unqualified statement that “normal wound healing is not noticeably impaired” was also misleading.
3298 Fourth, the IFUs for all the Ethicon devices include a representation that the mesh is not subject to degradation or weakening by the action of tissue enzymes. Yet, as I discussed in Part XI, Prolene is subject to oxidative degradation, although the clinical significance of the degradation remains controversial.
3299 Fifth, the description in the IFUs for all the devices except for Prolift+M includes the sentence: “This material, when used as a suture, has been reported to be non-reactive …”. As Professor Klinge remarked, “there is no justification for comparing the tissue reaction to a suture to the tissue reaction to mesh”.2869 A representation of this kind can lull the reader into a false sense of security.
3300 Sixth, the description of the devices in the IFUs for TVT, TVT-O, TVT Secur, Gynemesh PS, Prolift, and Prosima also includes the representation that the mesh has “elasticity in both directions” or a “bi-directional elastic property [which] allows adaptation to various stresses encountered in the body”. Professor Klinge said that there is no scientific data to justify these statements and they are incorrect since they fail to take into account the anisotropic behaviour of pelvic tissues or the biomechanics of the pelvis. Neither Prolene nor Gynemesh PS was built to meet the specific elasticity requirements that are needed for application in the pelvis.2870 Professor Klinge deposed that:
In my opinion, mesh with elasticity in two directions does not account for the anisotropic behaviour of pelvic tissues or the biomechanics of the pelvis. In general, Ethicon's claim of "bidirectional elasticity" begs the questions: "Why is it important to have elasticity in two directions and not all directions?"; "How much elasticity is there in each direction, and why is that important?"; "Is the same amount of elasticity required in the arms as in the central portion of the mesh?"; "If so, how much, and if not, why not?". Only by understanding what questions to ask, and answering those questions, could one attempt to design a pelvic floor mesh that will adapt to the stresses, and this has not occurred. In my opinion, Ethicon never established that any bidirectional elasticity in the Mesh Implants to allow adaptation to various stresses in the pelvis. Furthermore, the design of the Prolift uses arms for fixation, comparable to ligaments intended to withstand uniaxial forces, but not biaxial stress as in a flat mesh area.2871
3301 The development of a rigid scar/mesh compound or scar plate that occurs with bridging fibrosis causes a loss of elasticity.2872 The study conducted by Professors Mühl and Klinge showed that under minimal strain or tension TVT slings failed to return to their original or near-original geometric shape and design. Rather, they became permanently elongated.2873 Besides, whatever elastic properties the Ethicon devices possessed, they were not adapted to the stresses in the female pelvis.
3302 Seventh, the IFUs for Gynemesh PS, Prolift and Prosima stated that “[t]he mesh affords excellent strength, durability, and surgical adaptability …”. Professor Klinge deposed that the words “excellent strength” indicate that the mesh is optimised for the physiological requirements of the pelvic floor.2874 The evidence is to the contrary.
3303 Eighth, those IFUs also represent that Gynemesh PS has “sufficient porosity for necessary tissue ingrowth”. Yet, deformation and collapse of the pores causes chronic inflammation and bridging fibrosis and inhibits growth through the pores of fatty tissue. As Professor Klinge put it, there is tissue ingrowth but it is not the type of ingrowth that avoids complications. For this reason the representation is misleading.2875
3304 Finally, it is misleading to describe Gynemesh PS as a mesh “…knitted into a unique design that results in a mesh that is approximately 50% more flexible than standard PROLENE mesh”. It is misleading because it is capable of causing the reader to erroneously think that Gynemesh PS was designed for the specific purpose of prolapse repair in women when it is in fact a mesh designed for use in the abdominal wall rebadged for pelvic use as Gynemesh PS. Professor Klinge stated in his affidavit:
Although flat mesh PS may have some advantages under mechanical strain both have pore collapse, both have roping, scar shrinkage and mesh fraying and both result in inelastic shrinking mesh/scar ropes. Neither is a specific construction to meet the specific elasticity requirements that are needed for the application in the pelvis. Both the Prolene and PS mesh were originally used in the repair of abdominal wall as a tension free procedures. They are now used in a procedure with tension for the repair of SUI and POP. Ethicon has provided specific developments for the intra-abdominal placement, for the groin (open or laparoscopically), but has provided no distinct textile construction for the pelvic floor. Therefore the statement that PS mesh affords 50 percent more flexibility is misleading because this flexibility has not been designed for the specific application in the female pelvis.
3305 It was not until 2015 that the IFUs contained a warning about the prospect of further surgery. Statements to the following effect, or in similar terms, were added to the IFUs for the SUI devices:
• One or more revision surgeries may be necessary to treat these adverse reactions.
• PROLENE Mesh is a permanent implant that integrates into the tissue. In cases in which the PROLENE Mesh needs to be removed in part or whole, significant dissection may be required.
3306 At the same time the respondents added to the adverse reactions for Gynemesh PS a warning in the same terms as the last bullet point, substituting Gynemesh PS for Prolene Mesh.
3307 A precaution was included in some IFUs for TVT, TVT Secur, TVT Exact, Gynemesh PS, Prosima and Prolift+M to the effect that if the implant is used in contaminated areas it must only be with the understanding that subsequent infection may require its removal. This statement was misleading in that it indicated that removal would only be required in the case of infection and it was deficient in that it did not advert to the difficulties of removal or to the fact that removal might not alleviate pain. Moreover, none of the IFUs provided any instruction on the method of removal.
3308 The obligation to inform users of the risks in the use of the devices was far more extensive than Dr Hinoul acknowledged and Ethicon consistently failed to discharge it. As Dr Allman put it:
Informing users of a risk requires informing users of the probability and severity of harm, and how that risk was established, not just informing users of a possible harm.2876
3309 The IFUs did not satisfy this requirement.
3310 The respondents treated the omissions from, and misrepresentations in, the IFUs as matters of no or little moment. Dr Hinoul deposed that the IFU is “not intended to be a comprehensive guide for surgical treatment because its intended audience consists of pelvic floor surgeons who are trained to treat the conditions and who must use their training, experience and education in conjunction with the procedural steps detailed in the IFU”.2877 The respondents pointed to company-sponsored professional training and literature and company presentations at medical and scientific conferences. They also pointed to information accumulated from the surgeon’s medical education and training, data from studies discussed in the medical literature, information from conferences, and their own clinical experience. They submitted that:
(1) as the potential user of the devices is the treating surgeon and not the patient, they did not have any control over whether, and if so the manner in which, the contents of the IFU would be conveyed to each patient;
(2) the IFUs use short-form expressions such as “erosion”, “extrusion”, “fistula” and “inflammation” for conditions that should be well-understood by any competent and properly trained surgeon;
(3) if a complication associated with the devices was not included in the IFUs but was a complication associated with all surgical interventions for SUI and POP (such as dyspareunia) then the omission is immaterial, as the surgeon was obliged to have regard to the risk regardless of which procedure was selected;
(4) each IFU contained “a precaution” to the effect that users should be familiar with surgical procedures and techniques used for treating stress urinary incontinence (in the case of the SUI devices) and pelvic floor repair and non-absorbable or synthetic meshes (in the case of the POP devices);
(5) the IFUs were not intended to be the sole point of reference for a surgeon undertaking the procedure; and
(6) none of the pelvic surgeons who gave evidence suggested that he or she relied solely or even primarily on the IFU for information as to the risks associated with the implants.2878
3311 I accept that the IFUs will not be the only source of information for surgeons. I also recognise that the IFUs were drafted for pelvic surgeons familiar with pelvic floor surgery. Moreover, I accept that none of the relevant witnesses gave evidence that he or she relied solely on the IFUs for information as to the risks associated with the devices. But that does not mean that surgeons are not entitled to rely on the IFUs or to depend on the manufacturer for accurate information about the risks posed by the devices and the precautions that should be taken to guard against or minimise them. Indeed, unless they had reason to know that an IFU was deficient in these respects, they might well consider that there was no need to look beyond it.
3312 The fact that surgeons can draw on their knowledge of the causes of erosion, extrusion, fistula, and inflammation does not necessarily mean that they are in a position to give “appropriate advice” to the patient about the use of particular medical devices. Nor does the fact that some of the complications associated with the devices can also arise with native tissue repair excuse the respondents from including them in the IFUs, as the respondents submitted.2879 Certainly, this is not the position contemplated by the European Directive, the TG Act or the Medical Devices Regulations, as I pointed out in Part VII of these reasons. Neither is it the position taken by the TGA, as the evidence demonstrated. I refer in particular to the advice provided to JJM by the TGA in October 2014, discussed in Parts X and XI.
3313 I reject the proposition that it is unreasonable to expect manufacturers to include in an IFU warnings about matters of which they were aware because the surgeons should also have been aware of them or could have discovered them by other means or from other sources. Dr Pence pointed out the fallacy of this argument. A manufacturer who fails to draw attention in an IFU to risks associated with the use or implantation of its medical devices endangers the safety of patients because it leaves to chance the prospect that a particular risk or contraindication of which the manufacturer is aware but the doctor is unaware (either as to its incidence or extent) will be made known to the patient. Such an approach does not serve the statutory purpose.
3314 In any case, the respondents encouraged surgeons and patients to rely on the IFUs as an exhaustive statement of the risks associated with the devices. Marketing brochures tendered by the applicants directed both surgeons and patients to go to the instructions for use for “complete product information” and to the adverse events section for “a complete description of risks”. A Prosima brochure, for example, which bore the logos of both Ethicon Women’s Health & Urology and JJM, suggested that they need not look beyond the IFUs. It stated:
For complete contraindications, warnings, precautions, and adverse reactions, see Instructions for Use.2880
3315 Furthermore, during cross-examination Dr Hinoul either volunteered or conceded that:
the statement of adverse reactions in an IFU represents the manufacturer’s official or authorised statement of a product’s adverse reactions;2881
while journal articles might come to conflicting conclusions about the safety of a drug or device or procedure, a surgeon must take seriously a statement by the manufacturer or supplier about the risks of the product;2882
a surgeon is entitled to think that the manufacturer is likely to have more knowledge about its own product than members of the medical community;2883 and
for these reasons it was important that all risks associated with implantation of the Ethicon devices had to be included in the IFUs.2884
3316 If a risk arises because of the material from which the device is constructed, the way it is constructed, the way in which some people might react to it, or because of the technique used to implant it, then, it seems to me, regardless of whether the risk might also arise from other forms of pelvic floor surgery, persons generally are entitled to expect it to be included in the IFU. In any case, the uncontradicted evidence of Dr Pence was that all risks which cannot be eliminated by changes to a product’s design or composition or modifications to the surgical procedure or both must be the subject of appropriate warnings in the IFU or product labelling. The evidence from the TGA’s investigation of the Ethicon devices appears to bear this out.
3317 The fact that some doctors were or may have been aware of risks not mentioned in the IFUs and may or may not have warned some patients of those risks would not mean that the devices were not defective. Whether or not a device has a defect is determined by reference to the reasonable expectations of persons generally. In my opinion, persons generally are entitled to expect that, if the manufacturer of a medical device is aware, or ought reasonably to be aware, of a risk to patient safety arising from the implantation of its device, then it would disclose that risk in the instructions for use supplied with the device and in brochures, leaflets, and other material it produces about the device.
3318 While the respondents effectively argued that it was unnecessary to warn about complications where those complications would have been known to doctors or because doctors had access to such information through other channels, for the following reasons the more likely explanation is that the respondents did not consider it was in their commercial interests to be full and frank with the public about the risks associated with their products.
3319 Senior members of Ethicon’s marketing team were involved in discussions about the contents of the IFUs and what should be reported to the regulatory authorities. They were also involved in decisions about the content of presentations at medical conferences. In these circumstances, the conclusion is irresistible that decisions about the disclosure of information about the safety of products were influenced by marketing considerations.
3320 The following emails are illustrative of the nature and extent of their influence.
3321 On 30 July 1998, nine days after the TGA had cleared TVT for sale in Australia but more than a year before the device was on the Australian market, the marketing team circulated a summary of “key points” from the US marketing research study on TVT. Some of the matters highlighted in this document were that:
exposure to vaginal bacteria was thought to increase the likelihood of infection and physicians suggested that the interstices in the tape might promote the growth of bacteria;
the mesh might be more likely to promote infection than a single strand of suture because of the interstices in the mesh;
physicians want to be reassured with study data that rates of infection associated with TVT are low and that rates of erosion are either very low or non-existent;
physicians want to see long-term efficacy combined with a low incidence of complications;
physicians are very interested in the incidence of complications, specifically infection, osteomyelitis, bladder perforation, vessel perforation and urethral erosion;
physicians are concerned about causing urinary retention [perhaps by over-correcting the incontinence, a matter raised in internal Ethicon correspondence in February 2000],2885 osteomyelitis, erosions or infections as a result of using synthetic slings; and
most physicians do not perform stress incontinence surgery very often so that becoming proficient at any new stress incontinence surgical technique is “challenging”.2886
3322 None of these matters was disclosed in the Medscand IFU for TVT. The respondents did not alert Australian users or recipients to the risk of erosion until, at the earliest, 8 September 2000, after the device had been on the market for nearly a year. Nor did they warn that the mesh used in any of the Ethicon devices could cause infection. In the October 2015 version of the IFUs for the SUI devices then on the market, which remains current, the respondents inserted the statement that “as with all surgical procedures, there is a risk of infection”. This statement minimised the significance of the risk since the risk was not one merely associated with surgical procedures; there was a superadded risk because the mesh was a foreign body implanted in a clean contaminated area and there was a risk of bacterial colonisation. The risk of bladder or vessel perforation was not mentioned in the Medscand IFU. The first mention of a risk of urinary retention or a puncture or laceration of the urethra in TVT surgery did not appear until the 2015 version of the TVT IFU. The limitations of clinical studies were not disclosed in any of the IFUs and no mention was made of the rates or incidence of complications.
3323 In October 2000, following a visit to Ethicon by a urology professor and active user of TVT in which he explained a technique to explant a Prolene mesh tape, Zenobia Walji and Laura Angelini, both members of the marketing team, expressed concern about disseminating the information outside the business. Ms Angelini wrote (without alteration):
Theoretically, I can envisage no need for TVT explant. And I agree with Zenobia that if we, in any way, publish such an information, we start giving the reason to believe that explant of the TVT may be needed in some circumstances. Frankly, I do not want to dig my own grave ... !
There are too many people (competitors and other docs) who could be interested in spreading out misinformation or misusing our information about TVT. In my opinion, we must be very careful in avoiding “overinformation”.2887
3324 Yet, it was common ground that in some cases there could be a need to explant TVT and that explanting any device is difficult. Even so, no reference to removal of TVT, whether in part or in whole, appeared in the IFUs for any of the SUI devices until the last iteration in 2015. A 2001 TVT brochure purportedly setting out “information for patients” posed the question: “Are there any side effects from the mesh?” The answer that was given avoided any mention of the need for explantation. It was: “Occasionally a portion of tape may be exposed but this is uncommon [and] is treated by antibiotics and closure of the wound”.
3325 On 29 July 2003 Brian Luscombe, a marketing administrator, expressed concern about the complication of “transient leg pain” with “Mulberry” (which became TVT-O) and which he said had been experienced by 15 to 25% of Dr Leval’s patients. 2888 His concern did not relate to patient safety or comfort. Rather, it related to the absence of similar reports with Monarc, the AMS transobturator tape, and of a warning about it in the Monarc IFU. He wrote:
I am troubled by this apparent complication of MULBERRY that does not appear to be an issue with MONARC. Namely, that patient[s] of De Leval’s have had transient leg pain in 15-25% of patients. While this may not be a big deal, I do not believe there are reports of this with MONARC. This is concerning, especially now that we feel it necessary to include as a Warning and Precaution - AMS has no such Warning and Precaution and they will use this against us when we enter the market.
I am not sure that a sufficient explanation has been given as to why this complication would be unique to MULBERRY. One possible explanation may be in the size of our Helical Passer (which is larger than MONARC's) and/or in the position on the leg where we penetrate the skin and soft tissues (DeLeval's exit points on the leg are more lateral than MONARC's entry point, which is within the crease of the leg, not 2 cm outside of it. If they are not seeing this then we will have a competitive challenge convincing MONARC users to switch to MULBERRY as well as retaining the physicians we train.
3326 On 11 January 2005 Dr Arnaud proposed an addition to the IFU for Prolift in these terms:
WARNING: Early clinical experience has shown that the use of mesh through a vaginal approach can occasionally/uncommonly lead to complications such as vaginal erosion and retraction which can result in an anatomical distortion of the vaginal cavity that can interfere with sexual intercourse. Clinical data suggest the risk of such a complication is increased in case of associated hysterectomy. This must be taken in consideration when the procedure is planned in a sexually active woman.2889
3327 Ophélie Berthier, the Marketing Manager, suggested that the reference to “vaginal erosion” be replaced by “mesh exposure” and “retraction” by “tissue incorporation or tissue contraction”.2890 Why, I ask rhetorically, was this a matter for a marketing manager?
3328 In fact, Dr Arnaud’s proposal was not taken up. No such warning was ever included in the IFU for Prolift or any of the other Ethicon devices. No explanation for this course was offered in evidence.
3329 On 24 March 2005 Laura Angelini sent an email to Kimberley Hunsicker, Worldwide Manager, Gynecare Clinical Affairs, about a prospective presentation by Dr David Robinson to the AAGL (presumably the American Association of Gynecologic Laparoscopists).2891 The substance of the communique is set out below as it appears in the email. The lower case entries were made by Ms Hunsicker. Ms Angelini’s contribution appears in upper case. “Vince” is Dr Vincent Lucente.
As we discussed, the content was to be video oriented YES with early complications. I WOULD LIKE THAT WE SPIN IT MORE ON THE SAFETY ASPECT RATHER THAN COMPLICATIONS. CAN WE INCLUDE THAT IN THE TITLE AS WELL? I have attached a draft for our internal review only. Vince felt it was unethical to exclude dyspareunia data, I ACCEPT THAT WE NEED TO REPORT THE CASE OF DYSPAREUNIA BECAUSE I AGREE THAT IT WOULD BE UNETHICAL NOT TO MENTION SINCE WE KNOW ABOUT IT. HOWEVER THE WAY IT IS PRESENTED IN THE ABSTRACT IS GOING TO KILL US. WE CANNOT REALLY SAY ANYTHING ON DYSPAREUNIA (AND CERTAINLY CANNOT MENTION ANY PERCENTAGE, IF WE HAVE NOT REALLY ANALYSED THE DATA AND ASKED THE QUESTION. SO I AM NOT IN FAVOR (AND I KNOW JACQUETIN WOULD ALSO NOT BE) OF PUTTING THIS IN THE TABLE. MAYBE MENTION SEPARATELY AND CLEARLY SAY THAT THIS IS ONE REPORT BUT ANALYSIS HASN'T BEEN DONE YET. ALSO MISSING IS THE INFO ON LENGTH OF FOLLOW UP. IF WE SAY EARLY COMPLICATIONS PEOPLE WOULD LIKE TO KNOW AT HOW MANY MONTHS.
(Emphasis added, original use of upper case)
3330 Although omitting dyspareunia data was considered unethical, Kimberley Hunsicker, responded:
I will remove the dyspareunia and if it comes up we can speak to it. The reason it is low is because it is only self reported as an adverse event and not solicited in a questionnaire. This is the reason for my clarifying comments in the abstract. To my knowledge, we will not have “solicited” information (questionnaires) on dyspareunia at the end of the trial. Therefore, there will no scientific analysis specific to dyspareunia other that which is reported as an adverse event.
3331 In February 2006 Scott Ciarrocca (at that stage Manager, Electro/Mechanical Platform, Ethicon Product Development) sought Professor Jacquetin’s views about two proposed additions to the adverse reactions section of the Prolift IFU.2892 The first was:
Dissection for Prolift and any similar procedure has the potential to impair normal voiding for variable length of time.
The second was:
Pain with intercourse and pelvic pain have been reported infrequently. Both tend to be mild and are most often self-resolving.
3332 Voiding dysfunction and dyspareunia did not appear in the Prolift IFU until October 2009, more than three and a half years later.
3333 When he was informed that Professor Jacquetin was of the opinion that any proposal to modify the IFU should be considered in a broader context in which all aspects of the IFU were reviewed, he told the “team” that he was “not enthusiastic” about taking such a course and asked for their thoughts. The only reply he appears to have received came from a member of the marketing team, Giselle Bonet. Unsurprisingly, she agreed with him.
3334 On 3 April 2006 Mr Ciarocca was informed by John Clay, manager of regulatory affairs, in an email copied to a number of other Ethicon personnel including Giselle Bonet, that it had been decided to include the following addition to the adverse reactions section:
Dissection for pelvic floor repair procedures has the potential to impair normal voiding for a variable length of time.2893
3335 This was a revision of the earlier proposal made in February 2006. It removed the reference to Prolift. This had the effect of making it appear as if impairment of normal voiding was a potential adverse reaction to any pelvic floor procedure, and that it was not related to the design or construction of Prolift or the technique of implantation. Doubtless, this was also its purpose. No explanation for the revision was offered in evidence.
3336 Mr Clay advised that it was proposed to add to the warnings and precautions section a warning not to use Prolift in patients who are on anticoagulant therapy.2894 This was said to be taken from the “TVT-O insert”. He also advised that it was decided not to include the statement about pain with intercourse and pelvic pain. Once again, no explanation was given.
3337 It is difficult to avoid the conclusion that marketing considerations prevailed to the potential detriment of patient safety.
3338 Mr Clay had earlier queried whether the warning about patients on anticoagulants, which was given in relation to TVT-O, should be expressed in the same way for Prolift or should be made “more general to pelvic floor repair”. Dr Robinson’s opinion was sought. He asked: “When you say make it general to pelvic floor, does that mean extend to the Gynemesh PS labelling?” He said he thought that would be “equally appropriate”.
3339 Once again, however, there was pushback. Mr Ciarocca noted that no such warning had been included in relation to Gynemesh PS but suggested limiting the statement to Prolift, saying “GYNEMESH can theoretically be used to do a small repair that does not present major risk to bleeding complications – but PROLIFT will always be pretty invasive”. Dr Robinson’s reply was emphatic: “You wouldn’t want to do either on a patient on an anticoagulant”.
3340 It took more than three years before changes were included in the Prolift IFU and there is no evidence to indicate that the market was informed of those changes any earlier. When the amended IFU was issued, the warning about patients on anticoagulant therapy had been diluted. Instead of advising that Prolift not be used on patients who were on anticoagulation therapy, which was the original proposal, the warning, included for the first time in the IFU issued on 1 October 2009, read:
Patients on anticoagulation agents undergoing surgery using the GYNECARE PROLIFTTM System must have their anticoagulation therapy carefully managed.
3341 The same warning was given for Gynemesh PS but not until 16 March 2013 — nearly seven years after the exchange between Mr Ciarrocca and Dr Robinson.
3342 A warning in the same terms was included in the IFU for Prolift+M on 12 December 2008.
3343 In the light of Dr Robinson’s advice to Mr Ciarrocca, the warning was manifestly inadequate and potentially very dangerous.
3344 On 10 December 2008, in an internal email arising out of a complaint, Dr Meng Chen noted the emphasis placed by the FDA on the importance of informed pre-operative consent, and observed that “[o]ne of the paths for a better pre-operative consent is to provide an updated IFU to the operating physicians that reflecting the current knowledge of the manufacturer's (sic) on the potential adverse reactions”.2895 She suggested that the IFUs for all SUI devices be updated “particularly in the area of ‘Potential Adverse Reactions’”. Yet, the first update to the adverse reactions section for the SUI devices was not introduced until October 2015.
3345 Persons generally are entitled to expect manufacturers of an implantable medical device, particularly one designed to remain in the body permanently, to err on the side of caution. Certainly that is how a reasonably prudent manufacturer would behave. It is what the 1985 Directive on Product Liability required. But it was not how the respondents behaved.
3346 Although it may not have been possible to predict which patients would have an adverse reaction, as Dr Hinoul claimed2896, the respondents either knew or ought to have known that certain classes of patient were at higher risk of developing adverse reactions than others. In some instances they provided a warning of sorts about them, but in other instances they did not.
The time the devices were supplied by the manufacturer
3347 In determining the extent of safety of goods, one of the relevant circumstances to be considered is the time the goods were supplied by the manufacturer. The significance of the time the goods were supplied by the manufacturer is that goods which met community expectations at that time are not defective if the safety expectations of the community are higher at a later time: see Explanatory Memorandum at [20].
3348 The respondents submitted that, if the state of scientific knowledge at the time of supply does not permit identification of the defect, then persons generally are not entitled to expect a product to have “characteristics” “which are not able realistically to be considered, identified, or implemented by a manufacturer having regard to the state of scientific knowledge during the time of supply of the [product]”.2897 They submitted that in determining whether any of the devices had a defect, the Court had to consider what was foreseeable at the relevant time. They contended that the presence of the so-called state of the art defence in s 75AK(1)(c) (referred to in Europe as the “development risks” defence) does not detract from the argument.2898
3349 There are a number of difficulties with the respondents’ arguments.
3350 First, it is an arid one.
3351 Although the respondents submitted that the state of scientific knowledge has changed since the devices were first designed, developed and manufactured and, indeed, is still developing, the pleaded complications were known to the respondents at all relevant times. Moreover, as Dr Ostergard observed in an article cited by Professor Klinge, “an abundance of information was available to mesh manufacturers before the FDA cleared most meshes.”2899
3352 The respondents submitted that, if the Court were to find in favour of the applicants on the dispute concerning the pore size sufficient to minimise complications, then the Court could only find that the respondents knew or could have known about it after 2007 “after Dr Mühl and his team’s introduction of ‘effective porosity’”.2900 They pointed to the evidence given by Professor Klinge that the publication of the article by Mühl et al that year “introduced to the world the idea of effective porosity” and that the knowledge was still developing.2901
3353 As the applicants submitted, however, the importance of maintaining an open pore structure was not a recent development. It was discussed at the first Suvretta meeting in 1994 in the context of hernia meshes, before any of the Ethicon meshes had been developed.2902 At that time surgeons undertaking hernia repairs in the abdominal wall well understood that mesh should only be used in a tension-free environment.2903
3354 Second, the respondents accepted that scientific knowledge acquired after the time of first supply can be taken into account to determine whether the safety of the device was not such as persons generally were entitled to expect. That was an appropriate concession consistent with the purpose of the TPA. It is supported by the recent decision of the High Court of Justice of England and Wales in Gee v DePuy.
3355 Gee v DePuy was a representative action brought against a manufacturer of hip implants under the UK Consumer Protection Act. Andrews J observed at [84]:
It was common ground that the level of safety that the public is entitled to expect must be evaluated at the time when the product is first put on the market by the producer, though strictly speaking, that time is one of the circumstances which the Court must take into account. However, in determining whether the product met that level of safety, the Court is entitled to have regard to everything now known about it that is relevant to that enquiry, irrespective of whether that information was available at the time it was put on the market or has come to light subsequently. That is obviously the correct approach, otherwise a claimant would never be able to establish that a product, whose lack of safety only comes to light one or two years after it was first marketed, was defective at the time of its initial circulation.
3356 Third, if the respondents were right there would be no need for s 75AK(1)(c).
3357 Section 75AK(1)(c) (cf. ACL s 142(c)) provides that it is a defence to an action under s 75AD that “the state of scientific or technical knowledge at the time when they were supplied by their actual manufacturer was not such as to enable [the] defect to be discovered”.
3358 The clear intention of Parliament was to limit the relevance of any deficiency in the state of scientific or technical knowledge at the time of supply to that which was unable to be discovered and to impose an onus on the manufacturer to prove the deficiency. The onus imposed by s 75AK(1)(c) is a legal onus, not merely an evidentiary one as the respondents submitted in closing argument. The applicant bears no onus in this regard. For these reasons I reject the respondents’ argument (unsupported by authority) that the presence of the defence in s 75AK(1)(c) is immaterial.
3359 Similar arguments were rejected by the Court of Appeal of England and Wales in Abouzaid at [25]–[26] (Pill LJ), [40]–[47] (Chadwick LJ), [53] (Wright J) and by Burton J in A v National Blood Authority at [64]. In Abouzaid Chadwick LJ at [43] said that, whether the state of scientific and technical knowledge at the time of supply was such that a producer of products of the relevant kind might have been expected to discover the defect was relevant to the statutory defence under s 4(1)(e) of the UK Consumer Protection Act (the equivalent of s 75AK(1)(c)) but it had nothing to do with the question of the degree of safety persons generally are entitled to expect.
What level of safety are persons generally entitled to expect?
3360 Since goods have a defect within the meaning of s 75AC if their safety is not such as persons generally are entitled to expect, it is first necessary to determine what that level of safety is.
3361 In Peterson at [917] Jessup J held that persons generally are entitled to expect that, to the extent that a drug is known or believed to have side-effects, or to carry the potential for side-effects (especially serious ones), then the supplier of the drug will provide medical practitioners “in whatever terms, and by whatever means, are appropriate” “with information or warnings sufficient to permit a balanced, cautious and informed judgment to be made”. This finding was not disturbed on appeal.
3362 There is no good reason why the standard should be any lower for a medical device intended for permanent implantation in the human body. Indeed, in the present case the respondents conceded in oral argument that persons generally are entitled to expect that the manufacturer would warn of complications of which it was or ought to have been aware. While there was a qualification to the concession in that it applied only to “clinically significant complications”, that was immaterial in this case because all were either admittedly or proven to be of that order. To the extent that the information they conveyed is said to be deficient, however, the respondents argued that they did not need to provide it, since manufacturers could reasonably expect that the deficit would be filled by the treating surgeon.2904 I have dealt with this argument above. Even if the argument had any foundation, it is beside the point. Section 75AC is not concerned with the reasonable expectations of manufacturers but with the reasonable expectations of the public at large.
3363 The level of safety persons generally are entitled to expect, however, is fact-specific. What holds true for one product or one class of product does not necessarily hold true for another.
3364 While the general public might well recognise that drugs may have side-effects and may be familiar with the side-effects of some of them, I do not believe that, but for the publicity associated with this litigation, it could be said that “persons generally” are likely to know that implantation of a medical device of the kinds in question could have side-effects. Nor do I think that persons generally would appreciate that, unlike a new drug, long-term safety studies are not routinely undertaken for new implantable medical devices.
3365 Nevertheless, the question is not what persons generally know but what they are entitled to expect. The applicants submitted that Peterson is distinguishable because there is no evidence here that implantable medical devices are generally understood to have at least some adverse side-effects.2905 It is true that there is no such evidence, but that is not the end of the matter.
3366 It has been held that “safety is inherently and necessarily a relative concept”, that no “medicinal product” (whether a drug or a medical device) can be absolutely safe, and that it cannot therefore reasonably be expected that such a product will ever be risk-free: Wilkes v DePuy at [65]; Gee v DePuy at [110]. Both these cases dealt with hip prostheses. But the statement is also true of pelvic prostheses. Factors such as the variability of vaginal anatomy and tissue thickness in patients, other factors that affect wound healing or the body’s immune response to the implantation of a foreign body, differences in training, experience, skill and ability of surgeons, and (in the case of the POP devices) the level of precision necessary to place the mesh at a depth so as to avoid erosion all mean that complications cannot always be prevented.2906
3367 That said, Assistant Professor Chughtai gave unchallenged evidence that a reasonably competent gynaecologist or urologist would expect that any mesh implant would have minimal and self-limiting inflammation.2907
3368 The respondents did not deny that there are complications associated with the use of all the Ethicon devices. Rather, they emphasised the evidence that all surgical interventions to treat the relevant conditions have potential adverse outcomes.2908 Of that there can be no doubt. I accept that persons generally would reasonably expect that pelvic floor surgery is not without risk. But to the extent that there are risks associated with the devices or their implantation, regardless of whether they may also be associated with other forms of pelvic floor surgery, persons generally are entitled to expect that the manufacturer would alert them to those risks, in the first instance by including them in the instructions for use supplied with the various devices, and would not provide them or their doctors with misleading information.
3369 As the applicants submitted, the safety of the devices which persons generally were (and are) entitled to expect is affected by their purpose (the surgical treatment of stress urinary incontinence or pelvic organ prolapse as the case may be), the use to which they were to be put (as a permanent implant in the female pelvis), by the facts that neither condition is life-threatening, that treatment (including surgery) is elective, and that even if the surgical option is chosen there are well-established and well-accepted alternative forms of surgical treatment that do not involve the use of mesh.2909 As Dr Elliott put it in his 2012 review article on transvaginal mesh kits for prolapse repair, “[i]t is one thing to have a high complication rate when dealing with life or death issues without suitable alternatives — in this situation a complication, even a severe one, may be acceptable when a patient has no other choice and his or her life is at stake”; it is another where the patient has a choice and her life is not in danger: Dr Elliott noted that some complications of mesh surgery are not rare, but accepted the possibility that others were. “But”, he asked rhetorically, “what is the acceptable percentage rate of life-altering, irreversible, preventable complications when treating a quality-of-life problem especially when equally successful treatment alternatives exist that do not have these problems?”.2910 This was a question Ethicon failed to address in their clinical evaluations of any of the Ethicon devices.
3370 What persons generally are entitled to expect of medical devices is also affected by what the manufacturer says or does not say about them.
3371 Professor Korda, whose evidence was largely confined to the POP devices, gave some evidence about the kind of information he would have expected manufacturers to provide:
I would have most certainly expected the manufacturer of mesh implants to provide information about the lack of inertness of the material, the severity and duration of the inflammatory response, the short term and long term complications, the material risks, the severity and chronicity of complications and the indications and contraindications for the use of mesh implants prior to my using of mesh for prolapse surgery.2911
3372 Professor Korda also said that he would have expected that the information that was provided would be up-to-date, accurate, and a balanced and objective representation of relevant literature, and that it would include information about the quality of the studies, the safety and efficacy of the procedures, and the length of follow-up.
3373 These are reasonable expectations which persons generally would share.
3374 As the applicants submitted, because the level of safety set by the statute depends in part on the way in which and the purposes for which goods are marketed and the instructions and warnings given in relation to them, where a medical device is supplied for the purpose of treating a condition without specifying a degree of severity of the condition which would justify its use and unless the indication for the device is limited for use in a particular kind or kinds of patient and not contraindicated for certain patients, then the level of safety must be taken to be the same for all patients.2912
3375 Until late 2015, in their IFUs and their marketing material, including their patient and surgeon brochures, the respondents represented that their meshes would elicit a minimum to slight inflammatory reaction, that the fibrotic reaction would be uniform, that the mesh would remain soft and pliable, and that normal wound healing would not be appreciably impaired. Few contraindications were identified. Persons generally were entitled to expect that, unless the IFUs indicated that these effects could not necessarily be expected in certain specified classes of women, then they were applicable to all.
3376 At the very least, therefore, I would hold that where a medical device carries risks when used as intended, persons generally are entitled to expect that the manufacturer would provide medical practitioners with the information and warnings necessary to enable them to make a balanced, cautious, and informed judgment about whether to recommend implantation with one or other of the devices to their patients and to enable patients to make a balanced, cautions, and informed decision about whether to consent to such a procedure. That includes information about known and foreseeable risks. It includes information about those patients who are particularly at risk. It includes information about the true state of affairs, including the real risks of implantation with the device, the extent of the risks in the short and the long-term, and the limitations, if any, of the available data. Persons generally are entitled to expect that manufacturers are not selective about which risks or other information to disclose. The same holds true of direct communications to the public, such as in patient brochures and on the internet. Unless a manufacturer provides frank warnings about the risks associated with the use of its products, medical devices included, persons generally are entitled to expect that the product does not carry those risks. It is no answer to say that the risk is rare, since the rarer the risk, the greater the chance that neither the patient nor the doctor will know about it. Nor is it an answer to say that other products or other procedures carry the same or similar risks.
3377 Ideally, no product intended for permanent implantation in the body should be on the market until it has been subject to properly designed randomised controlled trials. No doubt to the surprise of many, there is no such legislative requirement. Be that as it may, persons generally are entitled to expect that commercial considerations are not prioritised over patient safety and that a device that bears a CE marking would not be available for sale in Australia, unless it satisfied the requirements for CE marking. A medical device intended for permanent implantation, such as the Ethicon devices, which has not been the subject of adequately powered clinical trials in a large group of patients and where long-term data are not available before it is released to the market, lacks the safety persons generally are entitled to expect, unless it is sold accompanied by advice alerting users to the lack of data or limitations of the available data and candid information as to risks and contraindications.
Was the safety of each device less than persons generally are entitled to expect?
3378 All the SUI devices have common features and points of similarity. So do the POP devices. Many of these features are shared by all the Ethicon devices. But there are also some differences. The respondents pointed to the following matters:
(1) differences in implantation technique between the retropubic devices (TVT and TVT Exact) and the transobturator devices (TVT-O and TVT Abbrevo); TVT-Secur could be implanted using either of these approaches;
(2) differences in the quantities of mesh used in the various devices;
(3) differences in the types of mesh: the SUI devices were made of Prolene; Gynemesh PS, Prolift and Prosima were made of Prolene Soft; Prolift+M was made of Gynemesh M (Prolene Soft to which Monocryl had been added);
(4) unlike Prolift, Prolift+M, and Prosima, Gynemesh PS was cut and shaped by the surgeon;
(5) the fact that Gynemesh PS could be implanted abdominally (by open or laparoscopic surgery) or transvaginally; and
(6) Prosima had a different shape than the other POP devices and used a vaginal support device and a balloon, which were not used in any of the other mesh kits.2913
3379 I accept the respondents’ argument2914 that, because of these differences (or some of them), evidence as to cure rates, both objective and subjective; complication rates; and (up to a point) the type and severity of complications arising from a procedure using one device is not necessarily applicable to a procedure using a different device.
3380 The applicants’ approach was to consider the devices as a group (by indication) in the first instance and then examine the differences.2915 This seems reasonable to me. Indeed, it was essentially the same approach taken by the respondents. As the respondents submitted in closing argument, it is necessary to consider the significance of the differences.2916
3381 I begin with the SUI devices.
The SUI devices
3382 All the SUI devices were made from Prolene. The evidence from Professors Klosterhalfen and Klinge discussed in Part V, which I have accepted, was that Prolene was and is a small pore mesh, which increases the risk of injury to the patient and unnecessarily compromises patient safety, leading to such complications as infection, excessive scarring through and around the mesh, erosion, chronic pain, and dyspareunia.
3383 On the other hand, the evidence is that the TVT procedure is less invasive than colposuspension or any of the older sling procedures, is often if not usually performed under local or regional anaesthetic, and is followed by a shorter period of hospitalisation.2917 The respondents argued that it was now “the gold standard” for the surgical treatment of stress urinary incontinence for women. There was some support for this accolade in the literature, including in a review article by Ashley Cox and others from the University of Toronto published in 2013.2918
3384 The respondents pointed to the absence of evidence from Professor Blaivas, Assistant Professor Margolis and Dr Agur that any of the SUI devices had a defect within the meaning of s 75AC of the TPA to support their contention that the applicants had not made out their case. 2919 That is a matter of no moment.
3385 Before the passage of the Evidence Act 1995 (Cth), evidence of that nature would arguably have been inadmissible: see, for example, RW Miller & Co Pty Ltd v Krupp (Aust) Pty Ltd (1991) 34 NSWLR 129 at 130–131 (Giles J). Section 80 of the Evidence Act abolishes the so-called ultimate issue rule, relevantly providing that evidence of an opinion is not inadmissible only because it is about an ultimate issue. But it is difficult to see how the opinion of an expert as to whether the safety of a product fails to meet the standard “persons generally are entitled to expect” would carry any particular weight. That question is quintessentially “a jury question”, that is, one for the trier of fact. Satisfaction of the statutory test falls to be considered on the basis of all the evidence taking into account all relevant circumstances, including the factors referred to in the section.
3386 Contrary to the respondents’ argument, neither is it a matter of any consequence that Professor Blaivas did not advocate for the removal of TVT from the market. Indeed, had he done so, the respondents might well have objected to the evidence. Whether TVT should be removed from the market is beyond the scope of this inquiry.
3387 The respondents also submitted that the expert evidence from both parties indicated that the midurethral sling is safe.2920
3388 It is true that a number of the expert witnesses, including Professors Korda and Dr Agur who were called by the applicants, continue to offer a midurethral sling as a primary treatment option for women with stress urinary incontinence. It is also true that various professional organisations have described the midurethral sling as the “operation of choice” or the “gold standard” for the treatment of stress urinary incontinence. Furthermore, as the respondents pointed out, in the 2015 Cochrane review on midurethral sling operations for stress urinary incontinence in women (Ford et al (2015)) midurethral sling operations were said to “have a good safety profile”.
3389 Nevertheless, this evidence is not decisive.
3390 First, the evidence did not address the statutory test. None of the experts was invited to do so. There is no reason to think that those who did endorse Ethicon’s devices, whether expressly or by implication, including the professional organisations, applied their minds to the statutory test or took into account all the relevant factors that bear upon satisfaction of the test.
3391 Second, the evidence is at odds with the opinions of Professors Klosterhalfen and Klinge, which was to the opposite effect.
3392 Third, not all the evidence regarding the continued use of midurethral slings by the applicants’ experts was focussed on the devices in question and it was not pitched as high as the respondents submitted.
3393 Professor Korda did say that he continues to use TVT-O, but he testified that he now only uses a retropubic midurethral tape for women with intrinsic sphincter deficiency and then only when the pressure in the urethra is low and “the only operation that will cure it is a proper kinking or obstruction of the urethra”, which can be better achieved with a retropubic transvaginal tape. The retropubic midurethral device he uses is not made by Ethicon, however. It is a Boston Scientific product called “Advantage Fit”, although he did add that the product was “essentially” the same as TVT.2921
3394 Assistant Professor Chughtai testified that he offered polypropylene midurethral slings as a primary treatment for women with stress urinary incontinence, but he was not asked and did not volunteer which slings he used.2922
3395 Dr Agur considered that there was probably still a place for retropubic tapes in certain indications where colposuspensions could not be performed. His view, however, was that such surgery should only be performed by experienced accredited surgeons and for patients who fully understand all the available alternatives and associated risks.2923 On the other hand, he considered that transobturator slings, both multiple and single-incision, should be restricted to use in research. He explained:
The recent Cochrane review [Ford et al (2015)] has shown a significantly higher need for repeat continence surgery raising concerns on efficacy. Its advantages of less blood loss and less operation time are not clinically important. The lower risk of intraoperative bladder injury does not appear to be advantageous in the long term. However, the significantly higher risk for chronic groin and thigh pain is a serious disadvantage. Therefore, if complete surgical removal is very difficult (and probably impossible), compared to retropubic tape implants, such serious adverse events cannot be addressed and can result in lifelong disability. Taking the above risks into consideration, and as there is hardly any absolute indication for the multiple- or single-incision transobturator procedures, I believe they should be performed only after permission from an ethics committee e.g. within research context.2924
3396 Professor Deprest said that he now used a single-incision or mini-sling called an Altis sling, manufactured by another company.2925 That was Professor Roovers’ evidence, too.2926 It is reasonable to infer that they did so because they were concerned for safety reasons to see that patients were implanted with as little mesh as possible.
3397 Altis is made by Coloplast AS. The evidence is that that device was cancelled from the ARTG on 28 November 2017, effective 4 January 2018.2927 The reason given by the TGA was that:
[T]here is a lack of adequate scientific evidence before the TGA for it to be satisfied that the risks to patients associated with the use of mesh products as single incision mini-slings for the treatment of stress urinary incontinence are outweighed by their benefits.
3398 In the cancellation notice the TGA emphasised that “mini-slings are different devices to mid-urethral slings”.
3399 The Royal Australian and New Zealand College of Obstetricians and Gynaecologists also distinguished between midurethral and single-incision slings. In its most recent position statement on the subject, issued in May 2017, RANZCOG did recommend midurethral slings, but only in “routine cases”, describing it as “highly effective in the short and medium term for treatment of urinary stress incontinence” and carrying “less risk than most other available major continence surgeries”.2928 It claimed that “a large number of studies” have shown that midurethral sling surgery to be “highly effective” and to have “improve[d] women’s quality of life overall”. It said that there was “robust evidence” to support “the traditional MUS” from over 2,000 publications and referred to it as “the operation of choice” for female stress urinary incontinence in Australia and New Zealand. At the same time, however, far from endorsing the single-incision sling, RANZCOG recommended that:
Single incision Slings (SIS) should be performed within the context of a properly conducted clinical trial or where arrangements are in place for clinical governance such as long-term prospective audit.2929
3400 The College also referred in this Statement to a number of complications associated with the implantation of synthetic slings, emphasising the importance of obtaining informed patient consent:
Complications must be discussed with women considering surgery including the different complications associated with each MUS route. Discussion must include bleeding, damage to the bladder and urethra, bowel and major vessel perforation. Voiding difficulties which may require catheterisation, loosening or even division of the sling at a later stage which may result in recurrent SUI. De novo urge incontinence or worsening of pre existing (sic) over active bladder symptoms can occur. Sling insertion can cause pain and dyspareunia and with the TOR [transobturator route], groin pain can occur. This is usually short lived but may become intractable. In some women these long term adverse outcomes have had severe effects on everyday activities and their quality of life. The mesh is a permanent material that can result in mesh exposure and infection which may occur soon after surgery or many years later. This can result in the need for mesh removal which may be difficult, may have complications and may not completely resolve chronic pain or other adverse symptoms.
3401 It concluded with the observation that surgeons performing midurethral sling surgery need to be aware of the risks and benefits of each approach and appropriately trained to perform the operations and manage the possible complications.
3402 In its letter to JJM of 13 July 2017, the TGA referred to the RANZCOG Statement, noting a number of its comments, in explaining the reasons for imposing as a condition for entry on the ARTG the inclusion in the instructions for use for TVT, TVT-O, TVT Exact, and TVT Abbrevo of a reference to spontaneous device vigilance reports of chronic severe pain following use of mesh for the treatment of stress urinary incontinence via the retropubic or transobturator route.2930
3403 Fourth, in their 2015 Cochrane review, Ford et al did not give an unqualified endorsement of the midurethral sling, let alone of all the SUI devices. In the paragraph immediately following their reference to the safety profile of midurethral sling operations they wrote:
A salient point illustrated throughout this review is the need for reporting of longer-term outcome data from the numerous existing trials. This would substantially increase the evidence base and provide clarification regarding uncertainties about long-term effectiveness and adverse event profile.2931
3404 Later, under the heading “Limitations of the review”, they emphasised the point again:
Most of our results are based on moderate quality evidence. Most trials did not describe their methods clearly, thus leading to some degree of uncertainty in the findings. At present there are only a limited number of randomised controlled trials (these produce the most reliable results) that have published data beyond five years after surgery. This means that evidence about how effective and safe these procedures are in the longer term lags behind the evidence for them in the short and medium term (up to five years). We encourage researchers to publish longer-term data to help increase the reliability of longer-term results in this area.2932
3405 The respondents submitted that not all the pleaded complications will make a device defective for the purpose of s 75AC. They contended that it is the incidence of clinically significant complications, not the incidence of complications per se that matters.2933 I accept that this is so. But in the light of the concession in closing argument that each of the pleaded complications is clinically significant,2934 it is difficult to know where this takes them. The example they gave was the risk of bladder perforation that arises with the retropubic SUI devices, like TVT, although the respondents did not argue that they were not obliged to warn of the risk.2935 They pointed to Professor Korda’s evidence in cross-examination that perforation of the bladder causes negligible injury:
What does one expect next to happen to the perforation? Do you need to surgery correct it with a suture?---No. I mean, the actual trocar with which you perforate the bladder is only about .7 – it’s about seven millimetres, roughly, width, and if you put a hole in the bladder that size, it heals by itself, if it’s a clean perforation, by just keeping the bladder empty. The more modern retropubic tapes have an even smaller trocar and both Johnson & Johnson and (scil) the one I use, which is by Boston, is a much finer needle. So perforation of the bladder under those circumstances literally causes negligible injury.2936
3406 But what happens if the perforation is not clean? And what about the position with the earlier retropubic tapes? In Part IX I referred to a case study that was mentioned in the 2010 TVT CER in which the authors suggested that inadvertent placement of the arms of the tapes close to the mucosa can be missed on cystoscopy and can predispose patients to the formation of stones in the bladder. I also referred to a commentary on the article in which the observation was made that injury to the bladder is not always recognised during initial cystoscopy and can be the cause of late complications, including chronic pain, urinary tract infection, and bladder stones, and require further surgery.
3407 In any case, Professor Korda did not consider perforation of the bladder to be an insignificant complication. Indeed, he described it as a major problem with the retropubic TVT. He said that it caused “significant incidence of injury to the bladder”.2937 Besides, the rationale for the obturator devices was that they avoided bladder perforations. If bladder perforations were an insignificant complication, why bother developing the obturator devices? The market would surely have been small, if not negligible.
3408 The respondents also argued that the applicants had not established that inadequate clinical evaluations had been undertaken with respect to the Ethicon devices. They referred to “[t]he extensive pre-market testing and continued studies”. They emphasised that the Ford et al (2015) Cochrane review described the midurethral sling operations as “the most extensively researched surgical treatment for stress urinary incontinence in women”. They also submitted that Professor Blaivas had conceded that “TVT was the most well studied operation”.2938
3409 This argument is problematic, too.
3410 First, the applicants did establish that Ethicon’s clinical evaluations of the devices were inadequate. I discussed the deficiencies in the clinical evaluations in Parts VIII and IX. Further, the pre-market testing of the devices was not extensive. As I mentioned in Part VIII, there were limitations affecting the use to which the Nordic studies could be put and no studies were undertaken before TVT was cleared for sale to assess its safety or efficacy across a broader population. TVT-O was rushed to market with limited data. These and other matters are discussed in more detail below in the context of the first and second negligence claims.
3411 Second, Ford et al (2015) did not consider whether the studies were adequate for present purposes, that is to say adequate to demonstrate that the devices reached the level of safety persons generally were entitled to expect, having regard to the mandatory considerations in the TPA. What is more, they expressed concern about the paucity of data relating to long-term risks.
3412 Third, Professor Blaivas did not concede that surgery using any of the Ethicon devices was “the most well studied operation”. Indeed, he did not mention TVT. Like Ford et al, he was referring to midurethral sling operations in general.2939 He did agree that midurethral sling surgery was the least invasive surgical option to treat urinary incontinence and its effectiveness was well-documented in the short and medium term.2940 He also agreed that there was increasing evidence of its long term effectiveness, although he stressed that none of the surgical procedures does very well in the long term and, like the alternatives, the symptomatic improvement “drops off in time”.2941 But he did not endorse any of the SUI devices. He did not say, for example, that TVT had a good safety profile. To the contrary, his opinion was that TVT carried significant long-term safety risks and that insufficient warnings had been given about those risks.2942 His position, he explained, was that information about potential complications or safety risks associated with a device, whether rare or not, needed to be passed on to doctors and patients. Professor Blaivas said that, had it not been for the worldwide litigation about pelvic mesh, even he would not have known about all the risks.2943 He considered, in effect, that the endorsements of TVT by RANZCOG and others were ill-informed.2944
3413 Notwithstanding the support for the SUI devices from some of the medical experts and the professional medical groups, I am persuaded that at all relevant times none of them met the level of safety persons generally were entitled to expect. I have reached this conclusion because of the biomaterials evidence about the problems with Prolene mesh, the deficiencies in the information the manufacturers supplied with the devices, the way in which the devices were marketed and promoted, and the shortcomings of the clinical evaluations.
3414 As to the first matter, the evidence established that, following implantation, contraction of the mesh/tissue construct occurs with the Prolene mesh used in the SUI devices and that, under minimal strain, its pores will deform and collapse, increasing the risk of injury to patients in whom it is implanted. In a not insignificant number of patients, the precise number or percentage of whom is unknown, this may cause significant and serious complications, including the pleaded complications. Some women are particularly susceptible. Yet, the respondents gave no or no sufficient warning of these complications.
3415 As to the second and fourth, the essential requirements contained in Annex I to the European Directive required an assessment of whether residual risks related to the use of the devices, which could not be “designed out”, were acceptable when weighed against the benefits to the patients and compatible with a high level of protection of health and safety. They also required manufacturers to inform users of the residual risks. Ethicon declared that its devices conformed to the essential requirements. In the absence of such a declaration, the devices would not have obtained CE marking. The evidence demonstrates that the declarations were not justified.
3416 As to the third, the devices were marketed and promoted as both safe and effective — as effective, if not more effective, than other incontinence procedures but with fewer risks. The evidence establishes that there was no sound foundation for these representations. At least in some categories of risk, such as chronic pain, whether in the pelvis or referred from the pelvis, the risk was higher and the consequences, if the risk came home, more extreme. Promotional material minimised the risks and exaggerated the benefits.
3417 The manufacturers did provide warnings about some risks but the warnings they gave did not extend to all the known risks. The applicants rightly complained that the warnings about the various complications were deficient with respect to the following matters: pain; extrusions, erosions and fistula formation; the description of the nature of the foreign body response; tape removal complications; patient-specific factors; comparative risks; and mesh contraction.2945
3418 Further, many of the representations the respondents made about the devices were misleading.
3419 First, the respondents repeatedly told users that transitory local irritation at the wound site and a transitory foreign body response may occur. As I have already explained, these representations were false. The foreign body response is neither transitory nor merely possible. It is chronic and inevitable. Moreover, the IFUs were deficient in that they failed to indicate that implantation of the devices would and were intended to generate a chronic foreign body reaction, that the foreign body reaction would cause chronic inflammation of the tissues at the interface of the mesh and the tissues, that the extent of the reaction and the inflammatory response in an individual patient was unpredictable, and that, depending on the nature and extent of the inflammatory response, significant complications could occur.
3420 The adjective “transitory” continues to qualify the duration of the local irritation at the wound site but was removed in 2015 from the description of the foreign body response. But the auxiliary verb “may” continues to be used to describe the occurrence of the foreign body response. For this reason the current representation is also misleading.
3421 Second, the IFUs for all the SUI devices, with the exception of the Medscand IFU for TVT, which made no mention of erosion, extrusion or exposure, drew a causal connection between a “transitory foreign body response” and “extrusion, erosion, fistula formation, and inflammation”. They stated that:
Transitory local irritation at the wound site and a transitory foreign body response may occur. This response could result in extrusion, erosion, fistula formation and inflammation.
3422 By coupling the phrase “transitory foreign body response” with the complications of “extrusion, erosion, fistula formation and inflammation”, the respondents conveyed the erroneous notion that these complications could only occur soon after implantation.
3423 In 2015, following the intervention of the TGA, Ethicon revised the warning to remove the adjective “transitory” from the foreign body response, to separate transitory local irritation at the wound site from the foreign body response, and to add mesh extrusion, exposure and erosion as separate entries. It did not, however, specifically warn of the risk of late extrusion, exposure or erosion. There is no good reason why such a warning was not included. The IFUs for the SUI devices should have made it clear that any and all of these adverse events could occur at any time after implantation.
3424 Third, many of the IFUs referred to animal studies that purportedly showed that Prolene mesh elicits a minimal to slight inflammatory reaction in tissues, which is transient. Assuming this is what the animal studies showed, these representations were misleading, as Sandy Savidge, one of Ethicon’s in-house scientists, told Dr Kirkemo in September 2009.2946 He reiterated the point in another email the following month.2947 Professor Klinge said that it was incorrect to describe the inflammatory reaction as transient. He explained that, although immediate or acute post-operative inflammation due to the implantation may resolve within days or weeks of implantation, the inflammatory foreign body reaction around the implants persists for years.2948 It is, indeed, chronic, not transient.
3425 Fourth, until the last iteration of the IFUs, issued on and from 22 September 2015, there was no warning about the risk of chronic pain or dyspareunia.
3426 It is true, as the respondents argued, that pain is a complication of any surgery. Yet, the evidence indicates that chronic pain of the kind reported in connection with the Ethicon devices is almost unheard of in traditional forms of surgery for the treatment of stress urinary incontinence.2949
3427 The risk of chronic pain with the SUI devices might well have been uncommon, if not rare, but it is a significant risk and the consequences are potentially grave. As I observed earlier, the need to warn is more urgent with respect to risks that may be rare. Since there is less chance that a medical practitioner will be aware of them, they may not associate a patient’s complaint with the device, and may compromise her treatment. Speaking of “persistent groin pain” after TVT-O implantation, which they considered “extremely rare”, Hazewinkel, Hinoul and Roovers wrote in 2009 that there was a paucity of literature on how to diagnose and manage it.2950
3428 Moreover, the tenor of the respondents’ marketing material was that surgery involving the devices carried the risk of fewer complications. In these circumstances, the failure to refer to risks which were common to other kinds of pelvic surgery could have caused users to mistakenly believe that at least some of those risks could be avoided by using the SUI devices and the TVT procedure.
3429 I therefore accept the applicants’ submission that the respondents’ failure to warn about the potential for chronic pain, dyspareunia, or localised pain associated with the implantation of the particular devices was a significant omission.2951
3430 Fifth, all the IFUs for the SUI devices stated that the TVT procedure should not be used for patients who have a urinary tract infection and warned that Prolene mesh could “potentiate” an existing infection. Yet, the meshes used in all the Ethicon can also cause infection.
3431 Further, the IFUs for the SUI devices with the exception of those for TVT-O and TVT Abbrevo advised that Prolene mesh in contaminated areas should be used with the understanding that subsequent infection might require its removal. There would appear to be no good reason why such a warning should have been omitted from the IFUs for TVT-O and TVT Abbrevo. In any case, even this advice had the potential to mislead because it did not make it clear that the mesh itself could cause an infection.
3432 The risk of infection from implantation of the SUI devices was not the subject of a warning until the September 2015 IFUs, after TVT had been on the market for nearly two decades. Even then, the warning was inadequate. The warning that was given was to the effect that the risk of infection was no different than the risk of infection that arises with all surgical procedures. That warning minimised the extent of the risk since it did not capture the risk of infection that arises with Prolene mesh well after the patient recovers from surgery. Indeed, it obscured that information.
3433 It will be recalled that in September 2003 Drs Arnaud and Robinson were telling Ethicon personnel that there was a high risk of infection for pelvic meshes and that “the vaginal approach is a rather unique situation in surgery” because the mesh is “placed through a septic cavity”.2952 This information should have been incorporated in the IFUs from the outset. Its omission exposed patients to an unnecessary risk of significant injury.
3434 Sixth, at no time did the instructions for use include a warning that the removal of the mesh might not alleviate the patient’s symptoms, advise of the difficulties associated with removal, or indicate that complete removal might not be achievable.
3435 Seventh, the warning given about damage to surrounding organs, nerves, ligaments, tissue and blood vessels did not change until 2015. It was that “punctures or lacerations of vessels, nerves, bladder or bowel may occur during needle passage …”. This was misleading in that it implied that the punctures or lacerations could only occur during surgery. Yet migration, exposure or extrusion and erosion could also cause damage of that kind to vessels, nerves, bladder or bowel. It was also misleading because it could be read as overlooking the potential for damage to the urethra. It was amended in 2015 to substitute for “bladder or bowel” “structures or organs, including the bladder, urethra or bowel” and to remove the phrase “during needle passage”. In my opinion, however, this representation was misleading, too, when read in the context of the IFU as a whole and in circumstances in which users were not informed that the IFU had changed. That is because, without identifying that this kind of damage could arise after surgery, including long after surgery, one might reasonably infer that the reference related only to damage during surgery.
3436 Eight, not until the last iteration of the IFUs in 2015 was there a warning about dyspareunia. While dyspareunia is a risk associated with other surgical treatments for stress urinary incontinence, in the light of the way the devices were marketed and in view of the essential requirements, it should have been included in the IFUs. The current warning that “[p]ain with intercourse which in some patients may not resolve” is reasonable.
3437 Ninth, the IFUs warned only of the possibility of retropubic bleeding postoperatively. Not until 2015 did they expressly warn of the risk of haemorrhage (an escape of blood from a ruptured blood vessel).
3438 Tenth, the IFUs failed to warn of the risk of recurrent urinary incontinence until 2015.
3439 Eleventh, the IFUs did warn that applying too much tension to the tape could cause lower urinary tract obstruction, either temporarily or permanently. Lower urinary tract obstruction would cause difficulty voiding. As I pointed out in Part XI, both Health Canada and the TGA considered that a specific warning of the risk of voiding dysfunction should have been given.
3440 There was not much evidence to support the applicants’ complaint about the failure to warn about “leg weakness”. Dr Hinoul was asked about leg weakness in the context of POP mesh but not in relation to the SUI devices, although he suggested that leg weakness could follow any surgery in the “pelvic organ prolapse space”.2953 There was some evidence of a higher incidence of neurologic symptoms, such as leg weakness following surgery with transobturator devices, like TVT-O, when compared with retropubic devices, like TVT: Richter et al (2010) reporting on 12 month results2954 and Albo et al (2012) reporting on 24 month results.2955
3441 Further, there was evidence that the obturator devices could cause “leg pain”. There was no warning about leg pain until 2015. All the IFUs for the obturator devices — TVT-O and TVT Abbrevo — are deficient in this respect until September 2015 when the “adverse reactions” section was amended to include pain and, more specifically “neuromuscular problems, including acute and/or chronic pain in the groin, thigh, leg, pelvic and/or abdominal area”. This was an extraordinary omission in the light of what Ethicon learned early in 2003, before CE marking was obtained, and Dr Arnaud’s email to Dr Weinberg that the leg pain experienced by a number of Professor de Leval’s patients, who had received the obturator device using the inside-out technique, had not been reported in the literature.2956 Be that as it may, the applicants did not plead that any of the IFUs were deficient because of the absence of a specific reference to leg pain, so I shall say no more about it.
3442 While Dr Hinoul was reluctant to acknowledge any deficiencies in the IFUs, the failure by the respondents to argue with the TGA about its recommendations suggests that they accepted that the warnings should have been given or, at least, that it was reasonable to provide the warnings.
3443 In 2015 Ethicon also amended its IFUs for all the SUI devices to include in the list of adverse reactions:
• One or more revision surgeries may be necessary to treat these Adverse Reactions.
• PROLENE Mesh is a permanent implant that integrates into the tissue. In cases in which the PROLENE Mesh needs to be removed in part or whole, significant dissection may be required.
3444 Although from this point on, all the IFUs contained a detailed list of instructions for the implantation of the devices, there were still no instructions about the manner in which any revision surgery was to be performed. Assistant Professor Margolis said that he had to teach himself. In his report he wrote that he had to develop his skills and expertise in partial removal of mesh systems on his own through direct patient contact, communication with other physicians and extensive research in the peer reviewed literature since none of the IFUs for TVT or TVT-O adequately addressed the management of complications.2957 He also said that:
Once the transvaginal mesh implant is scarred into place, it is impossible to remove all of the mesh material. It is irreversible. If the mesh is removed within 2 weeks of implantation, which is rarely the case, much of the foreign body can be removed because it has not been scarred into place by the previously described scarification process.
Once set in by the fibroblastic process, the mesh can only be partially removed. The mesh becomes so densely adherent and cemented to adjacent tissues as to make its complete extraction impossible. Even if the mesh is removed, the scar tissue that results from mesh implantation, mesh chronic foreign body reaction, and mesh explant surgery is permanent and irreversible …
3445 I found Assistant Professor Margolis to be an unsatisfactory witness. His credibility was badly damaged in cross-examination. He was given to overstatement. He quoted selectively from references and, at times, wrongly. When challenged, he obstinately stood his ground. He criticised others for “cherry picking” articles that only supported their opinions, but he was caught out doing something similar himself.2958 For these reasons, where his opinions were called into question, I have placed no weight on them unless they were supported by other evidence.
3446 On this subject, however, he was not challenged, and this aspect of his evidence is not at odds with the evidence of other witnesses. It was also supported by the Hazewinkel et al (2009) article of which Dr Hinoul was a co-author.2959 Speaking of TVT-O, Hazewinkel et al warned that “the ultimate option” of removing the tape “should not be postponed too long as scarring around the tape progresses over time and makes removal more difficult and nerve damage may become irreversible over time”.
3447 Dr Agur, although garrulous, was much more measured than Assistant Professor Margolis and, despite the occasional overstatement, emerged from cross-examination with his credibility intact. He said that surgery for complete tape removal is generally reserved for patients suffering chronic pain on account of the implant and the surgery is complex, highly challenging, and associated with a high risk of injury. I did not understand any of this evidence to be controversial.2960
3448 For the first time, in 2015 Ethicon included a warning suggestive of the prospect that some patients are more susceptible than others to complications. The warning was in the following terms:
Physicians should use their surgical experience and judgment to determine if PROLENE mesh is appropriate for certain patients. Patient specific factors may impair wound-healing, which may increase the likelihood of adverse reactions.
3449 Even then Ethicon gave no indication as to what “patient specific factors” it had in mind.
3450 The evidence was that “relative contradictions” to the use of biomaterials include poorly controlled diabetes, morbid obesity, heavy smoking, a history of previous pelvic radiation, severe urogenital atrophy, concurrent use of systemic steroids, and patients with autoimmune conditions.2961
3451 None of the IFUs for the SUI devices gave surgeons any information about the comparative risks of surgical treatment of SUI and native tissue repair. As the applicants submitted, information about comparative risks is information which would enable a patient to make an informed decision about whether to undergo surgery involving the implantation of any of the SUI devices.2962 Assistant Professor Chughtai said that it would be expected that the IFU would include a comparison to the existing “gold standard”.2963 Still, I am not persuaded that the expectations of the general public concerning the safety of a particular medical device include that the manufacturer would supply them with information as to its relative safety.
3452 At no point in time did Ethicon inform users of the potential for mesh contraction or shrinkage of the tissues surrounding Prolene, although it accepted that it was a risk associated with the SUI devices. Dr Hinoul conceded that excessive contraction or shrinkage of the tissues surrounding the mesh can occur following implantation of TVT.2964 Ethicon agreed to Health Canada’s request to include it in the adverse events in the IFUs for the SUI devices.2965 There is no good reason why it was omitted from the IFUs accompanying the devices sold in Australia. As I concluded earlier, mesh contraction or shrinkage of the surrounding tissues has clinical significance. Dr Hinoul conceded that it could lead to tightening of the mesh tissue construct which, in turn could lead to urine retention, irritation of the urethra, and de novo urge incontinence.2966 It was mentioned in all the IFUs for Prolift, Prolift+M, and Prosima, and the second and subsequent IFUs for Gynemesh PS. Its omission from the IFUs for the SUI devices had a deleterious effect on the safety persons generally were entitled to expect. In the absence of this information, persons generally were entitled to think that this was not an adverse event that could arise with SUI devices.
3453 In my opinion, the differences between the various devices are immaterial for present purposes. While the differences in implantation technique between the retropubic and the transobturator devices exposed patients to different risks or different levels of risk, in each case the risks to which patients were exposed by the use of Prolene mesh and the added risks associated with the particular implantation technique were sufficient, in the absence of suitable warnings and in the light of the representations the respondents made about the risks and safety in general, to render the SUI devices defective within the meaning of the TPA. There was no difference in the quantity of mesh used for TVT, TVT-O and TVT Exact. TVT Exact and TVT used the same retropubic technique. TVT Abbrevo used less mesh than TVT and the rates of complications were reportedly lower than with TVT and TVT-O, but the risks were not insignificant and the warnings were still deficient. The retropubic devices exposed patients to a higher risk of bladder perforation but the transobturator devices exposed patients to a higher risk of leg pain.
3454 It will be recalled that TVT Secur was also made from Prolene, albeit a smaller quantity than that used in TVT and TVT-O and sandwiched between layers of absorbable fleece made from polyglactin 910 and poly-p-dioxanone. But the Nambiar et al Cochrane review published in 2014, which reviewed several single-incision slings including TVT Secur, concluded that TVT Secur was “considerably inferior to retropubic and inside-out transobturator slings”. It described TVT-Secur as “a specific type of mini-sling that has consistently been shown to provide poorer control of incontinence, along with higher rates of side effects, compared with standard mini-urethral slings”. 2967
3455 The results of the 31 trials involving over 3,000 women reviewed in the Nambiar et al (2014) meta-analysis demonstrated that women were more likely to remain incontinent after surgery with single-incision slings than with retropubic slings like TVT (roughly twice as likely) and that, although the duration of the operation was slightly shorter for single-incision slings, there was a higher risk of de novo urgency. Four of the five studies in the comparison included TVT Secur. Single-incision slings also resulted in similarly higher incontinence rates compared to inside-out transobturator slings and the adverse event profile for the former was “significantly worse”, with higher rates of mesh exposure, bladder/urethral erosion and operative blood loss. Although postoperative pain was less common with single-incision slings and rates of long-term pain or discomfort were marginally lower, Nambiar et al (2014) said that the clinical significance of those differences was questionable. Once again, most of the findings were derived from trials involving TVT Secur.
3456 I referred earlier to the paper by Hinoul et al (2011) reporting on the results of a randomised trial comparing TVT Secur and TVT-O, which concluded that TVT Secur was “less efficacious” than TVT-O and was associated with more bleeding problems, more postoperative urinary tract infections, more common mesh exposure, more bladder lesions and a more frequent need for surgical re-intervention.2968
3457 A little over a year later, on 29 May 2012, Ethicon privately acknowledged that, after at least six randomised controlled trials with one year follow-up, TVT Secur was “associated with inferior patient-reported and objective cure rates at 1 year, and higher reoperation rates when compared to standard mid-urethral slings” like TVT and TVT-O.2969
Conclusion in respect of the SUI devices
3458 Having regard to the nature and extent of the risks associated with all the devices, the deficiencies of the respondents’ warnings and the other information they provided, the repeated failure to comply with the requirements for CE marking, and the way in which the devices were marketed, at no relevant time was the safety of any of the SUI devices such as persons generally were entitled to expect. Accordingly, each SUI device had a “defect” or “a safety defect” within the meaning of the TPA and the ACL respectively.
The POP devices
3459 Notwithstanding the differences between the various devices, the each of Gynemesh PS, Prolift and Prosima was made of Prolene Soft and had an identical pore configuration.
3460 The Prolift arms and the additional load of Prolift Total more probably than not created a higher risk of complications than the other devices but, to the extent that all the POP devices were made from Prolene Soft, all carried the risks generated by the use of the materials, including the risk of the pleaded complications.
3461 The respondents referred to a number of allegedly distinguishing features of Prolift+M. But in closing submissions, their lead counsel said that the only significant difference was the addition of Monocryl.2970 He conceded that any other structural or knit differences, while of interest, were immaterial.2971
3462 In fact, the addition of Monocryl did not make any significant difference. Although Prolift+M was partially absorbable owing to the addition of Monocryl which, as the applicants put it, notionally increased the sizes of the pores, the evidence about effective porosity indicates that in vivo Prolift+M behaved in the same way as the other devices. Had suitable clinical studies been conducted before the device was taken to market, that would have been evident.
3463 While Gynemesh PS could be used abdominally as well as transvaginally, both approaches were attended by potential complications. The use of Gynemesh PS in abdominal sacrocolpopexy appears to result in fewer complications, at least in some respects, than when used transvaginally (erosion rates in particular appear to be significantly lower). But its indication was not limited to abdominal use until March 2013 at the earliest. Even so, the level of complications from abdominal use was not insignificant. After March 2013, the warnings contained in the IFU continued to be deficient. For a start, the March 2013 IFU did not make it clear that the device should not be implanted transvaginally. It will be recalled that the TGA later required as a condition of continuing registration that the IFU expressly state that “[the] device is not intended for any pelvic organ prolapse repair via a transvaginal approach”.2972
3464 The information provided in the IFUs for all the POP devices was deficient in numerous respects from the first iteration to the last. Those deficiencies are discussed in Part XI and again below in the context of the misleading or deceptive conduct and negligence claims. It is sufficient to note at this point that the deficiencies included the occurrence and duration of the foreign body reaction and, in no particular order, the risks of a chronic inflammatory reaction of the tissues surrounding the mesh; chronic pain; late onset erosions, extrusion, and exposure; infection; dyspareunia and apareunia; defaecating difficulties; the potential consequences of erosion, extrusion, and exposure, including difficulties associated with removal of the mesh; the potential for multiple operations to treat complications; and the potential for a heightened inflammatory response in patients with autoimmune disorders or with other vulnerabilities. For the reasons given above and below, many of the representations made in the IFUs and the respondents’ promotional material were misleading.
3465 While amendments were made to the IFUs from time to time, they did not adequately capture either the true nature and extent of the adverse events that could arise from implantation with the devices or the categories of patients for whom surgery using those devices was contraindicated.
3466 The POP devices were promoted as safe and effective treatments for pelvic organ prolapse with benefits that native tissue repair could not offer. The Prolift patient brochure, for example, is a case in point. 2973 Not only did it differentiate between Prolift and traditional surgery in a positive way, it minimised the risks associated with Prolift surgery. While the brochure informed patients that they could experience less pain, it did so only in the context of intra-operative and (early) post-operative pain. It provided no indication of the risk of chronic pain or severe pain months or years after implantation. While it informed patients that the procedure allowed for the restoration of sexual function, it did not warn them of the risk of dyspareunia or apareunia let alone chronic dyspareunia or apareunia. While it mentioned the risk of the mesh becoming exposed in the vaginal canal, it said nothing about the potential consequences of mesh exposure. Nor did it refer to the risk of erosion into vessels or organs and the potential consequences if such an event were to occur.
3467 Moreover, while the brochure informed readers that there were some risks, readers were assured that risks associated with the procedure were “rare” and that there was only “a small risk” of mesh exposure into the vaginal canal. Because of the limited available data and the problem of under-reporting, and with no indication of what was meant by rare or small, this was deceptive. It was liable to give patients a false sense of security. It is true that the relevant parts of the IFU were included in the brochure, but there was nothing in them to suggest that there was a risk of long-term complications such as chronic pain, and for the reasons discussed in Part XI, the instructions for use were deficient in numerous other respects.
3468 For the reasons already canvassed, it is no answer to assert, as the respondents did, that all surgery carries risk and that some of the pleaded complications can arise with native tissue repair too. Nor does it matter that “the objective evidence” (whatever that may be) may be inconclusive as to the extent of safety of the POP devices.
3469 With the exception of Prosima, the POP devices were sold without discrimination as to the severity of the prolapse or the age or health of the patient, and regardless of whether the patient had undergone native tissue repair, which had failed, or the patient was at high risk of recurrence.2974 Prosima was indicated as suitable for women with stage 2 and 3 prolapses. Yet, as the applicants put it in their closing submissions, there was “a virtual consensus” among the urogynaecologists called as experts in this case that all the POP devices should not be used for primary prolapse surgery in ordinary practice, with the possible exception of severe cases.2975
3470 There was a wealth of evidence to indicate that the POP devices were never safe for general use in the wide range of patients for whom they were indicated and promoted.
3471 In 2006 the French National Health Authority concluded that the use of mesh implants for genital prolapse surgery by the vaginal route remained a matter for clinical research.2976
3472 In 2004 and 2007 the Maher et al Cochrane reviews concluded that they were only suitable for use in randomised controlled trials. That conclusion never changed over time in the case of posterior and apical repair. By 2016 Maher et al concluded that, owing to significant morbidity, the evidence did not support its use as a first-line intervention for anterior compartment repair.2977
3473 In cross-examination, Associate Professor Lam generally agreed with Maher et al, based on his own experience.2978 He also agreed that a manufacturer of transvaginal mesh should inform potential users that the evidence was insufficient to support the use of mesh at the time of vaginal repair surgery except in the context of randomised controlled trials.2979
3474 Professor Roovers was invited to agree as well, but declined to take up the invitation. That said, he did not disagree either.2980 Moreover, he said that there was very limited evidence on the safety and efficacy of any mesh implant until the Altman paper, which was limited to anterior repair, in 2011 — six years after the launch of Prolift.2981 The problems with the Altman paper are discussed above in Part VI.
3475 In its opinion on the safety of surgical meshes used in urogynaecological surgery, published in December 2015, the European Commission’s Scientific Committee on Emerging and Newly Identified Health Risks wrote that, based on the available scientific evidence, “[t]he implantation of any mesh for the treatment of [pelvic organ prolapse] via the vaginal route should only be considered in complex cases, in particular, after failed primary repair surgery” and should only be used when other surgical procedures have already failed or are expected to fail.2982 It identified the following factors as influences upon surgical outcomes:
material properties (biocompatibility, tissue integration, long-term stability, and mechanical performance over time which includes flexibility, elasticity, aging and resistance to deformation);
product design (such as the physical characteristics of the mesh, including the pore size);
the amount of mesh used;
the route of implantation (transvaginal or transabdominal);
patient characteristics, such as age, obesity, and smoking;
associated procedures, such as hysterectomy; and
surgeon experience.2983
3476 In substance, this was the opinion of most of the respondents’ witnesses.
3477 By the time of the trial none of the urogynaecologists called by the applicants and only one called by the respondents would countenance the use of polypropylene mesh for surgical repair of pelvic organ prolapse in a primary procedure.
3478 The outlier, Professor Collinet, said that at the time of the commercial release of Prolift, the problem of mesh exposure had been “practically resolved”, “due to very low incidence and satisfactory treatment techniques”.2984 Dr Agur said that he was surprised to read the statement, noting that the problem persists to this day and is a perennial issue as long as polypropylene mesh continues to be implanted underneath the skin of the vagina.2985
3479 Be that as it may, Professor Collinet also testified that the TVM Group considered that, in order to minimise the risk of complications, it was necessary to limit the use of Prolift to “very large prolapse grade IV” and recurrent prolapse. Yet, the respondents never took this course.2986
3480 Professor Korda observed that:
The principle of using grafts in reconstructive surgery is to reinforce existing tissue. The material must be safe, biologically compatible, and must provide both anatomic and functional results, especially in pelvic floor surgery. The ideal material should be chemically and physically inert, non-carcinogenic, mechanically strong while remaining flexible, non-allergenic, non-inflammatory, and non-modifiable by body tissue. It must be sterile, convenient to use and affordable, with minimal risk of subsequent infection or rejection.
Currently, no graft has all these properties. Moreover, in pelvic organ prolapse surgery, the optimal implant should restore normal anatomy and function to the vagina and the surrounding pelvic organs and have longer longevity than autologous tissue. Once implanted, it should not result in adhesion formation on the visceral surfaces.2987
3481 Professor Korda said that the safety and efficacy of mesh implant surgery has still not been demonstrated.2988 He said that no acceptable qualitative or quantitative measures of outcome have been demonstrated to convince him of the safety of that surgery for pelvic organ prolapse repair and no randomised controlled trial has documented long term efficacy and safety.
3482 His opinion was supported by the findings of a review of the scientific literature conducted by Drs Mickey Karram and Christopher Maher published in the International Urogynecology Journal in 2013.2989 The review was extensive, covering PubMed, Medline, Cochrane library and the Cochrane database published up to January 2012. The later Cochrane reviews confirmed the position.
3483 Apart from Gynemesh PS cut into strips for apical repair, Professor Korda would only countenance the use of the POP devices for anterior repair and then only in special circumstances, in older patients, when the indications are “very strong”, the patients have been told that there is no data to determine the products’ long term efficacy and ultimate complications, they have been warned of the significant material risks they are likely to encounter and they accept those risks. He added that it was generally more acceptable to have recurrence of prolapse than the complications he had seen from mesh implant surgery. 2990
3484 He observed (correctly) that the indications for mesh use were never properly identified by the respondents.2991
3485 Under cross-examination Professor Deprest told the Court that his view was that the complication rate for mesh in the surgical repair of pelvic organ prolapses was “unacceptable”.2992 Like Associate Professor Rosamilia, he did not distinguish between meshes of different manufacturers or between Ethicon meshes.
3486 Associate Professor Rosamilia supported the use of midurethral slings but only considered that the benefits of transvaginal mesh “potentially outweighed the risks in women with severe stage prolapse or recurrent prolapse”.2993
3487 Professor Deprest used mesh kits from the outset, it seems, but only for those patients he considered were at the highest risk of recurrence should they undergo native tissue repair.2994 Together with Professor Roovers and others, he is an author of the Consensus Statement of the European Urogynaecologic Association published in 2017 which recommended limiting the use of synthetic mesh for pelvic organ prolapse to “complex cases with recurrent prolapse” and “restricted to those surgeons with appropriate training who are working in multidisciplinary referral centres”.2995 The unstated, but implicit, reason is that the risks outweigh the benefits in all other cases.
3488 Professor Roovers considered that vaginal mesh surgery was still in “its development phase”.2996 He said that:
Concerning the time of introduction of vaginal implants, Dr Blaivas states that "none of these products had demonstrated efficacy and safety to an acceptable level, at the time of introduction”. It is important to realize that it will never be feasible to do this. Implantation in animal models has limited predictive value for surgery in human beings, so there will always be a first “experiment” in human beings. The debate is how long one should wait before there is wide implementation of innovative surgical procedures, and how much data in small safety studies are needed first.2997
3489 He claimed that this was not the responsibility of industry. He argued that the professional societies should provide “recommendations for this”, and that it was their responsibility to protect patients against the “too rapid implementation of products that have not been adequately evaluated”. But this evidence was contradicted by the evidence of the regulatory experts and is at odds with the statutory scheme and, as it will be seen, the respondents’ obligations at common law.
3490 Associate Professor Lam expressed his agreement with Professor Korda’s view that mesh implants could only be justified in the anterior compartment and then only in the following limited circumstances:
where there is little other choice of treatment and the symptoms of prolapse are so grave that they intrude on the quality of the patient’s life; or
where recurrence rates are predictably so high that the use of mesh may justify running the risk of complications;
where there has been a large recurrent prolapse, or where perineal 3/4D ultrasound demonstrates avulsion of the puborectalis muscle “where a better outcome has been demonstrated with the use of mesh, provided that patients accept the high incidence of complications”.2998
3491 The opening remarks of Professor Korda’s first report, which neatly summarised his own opinion, fairly reflects the position which emerged from the evidence as a whole:
I have been viewing with great interest the evolving evidence in the use of mesh for prolapse surgery since 2001 at which time it was suggested that mesh insertion gives superior results in prolapse repair.
Over the years during my attendances at various conferences of the International Continence Society and International Urogynaecological Association as well as specific lectures and presentations, I have been observing the reports of complications associated with mesh insertion such as damage to pelvic nerves, rectal perforation, bladder perforation, mesh erosion and dyspareunia. I have been waiting for the development of clinical evidence which would support and justify safe mesh insertion in prolapse surgery. To date, in my opinion, this is yet to be presented.2999
3492 While Ethicon had hoped that Prolift+M would lead to a reduction in some of the complications, particularly erosion and contraction, its hope was not realised. To the contrary, even the early figures for Prolift+M showed higher rates of erosion than for Prolift.
3493 Professor Deprest was very much in favour of restricting the use of mesh in prolapse repair. He wrote in his report:
I am completely in line with how Dr Agur wants to safeguard the patient as well as the use of mesh, by restricting their use. This can indeed be within trials, but their safe use in experienced hands and subsets of patients should be clinically possible. Dr Agur does acknowledge at some stage a certain need for mesh surgery (Grade III-IV or recurrence), which could certainly be defendable practice; if possible within a trial, that would be even better.3000
3494 Despite Ethicon’s repeated unqualified representations to the regulators that all the Ethicon devices were safe and efficacious and despite the indications in the IFUs, not even Dr Hinoul could justify the unrestricted use of the POP devices. The Benefit/Risk Profile of the POP devices he signed on 27 June 2012 stated:
Despite the limitations inherent in looking across multiple trials (slight definitional differences, varying time points and patient populations etc.), Ethicon, Inc's review by medical affairs has confirmed that the evidence demonstrates an acceptable benefit-risk profile for these products when placed in appropriately selected patients by experienced surgeons. 3001
(Emphasis added)
3495 Finally, for the reasons given in Parts VIII and IX, none of the POP devices satisfied all the requirements for CE marking at any relevant time.
Conclusion in respect of the POP devices
3496 The weight of evidence leads inexorably to the conclusion that at all relevant times the safety of all the POP devices was below the level that persons generally were entitled to expect. Notwithstanding the differences in the various devices, each of them exposed women to significant risks of injury against which inadequate warnings were given and in respect of which misleading representations were made. The respondents were not candid with the public about the risks of, and contradictions for, use of the devices or the limitations of the available data. The respondents represented that the benefits of using the POP devices outweighed the risks for women with any level of prolapse when the evidence did not support that. None of the POP devices was the subject of an adequately powered clinical trial, before it was released to market. The respondents represented that the devices met the essential requirements for CE marking when the material upon which they relied to affix and maintain the CE mark was insufficient to satisfy those requirements.
3497 At its highest, the evidence supported what Dr Daniel Elliott postulated in his 2012 review article:
Possibly, in some highly experienced hands, mesh kits are successful and possibly indicated in some highly selected, well-informed patients. But not all hands qualify as ‘highly experienced’, not all patients qualify as ‘highly select’, and not all patients are fully informed. In the overwhelming majority of patients, mesh kits are not indicated, they do not provide any proven benefit, and they needlessly increase the complication rate.3002
3498 On balance, I am persuaded that the mesh kits were only ever suitable for use in the context of a clinical trial and then only with appropriate warnings about the nature and extent of the potential complications. Even if they could be said to have been suitable for use in treatment of severe cases of pelvic organ prolapse or where native tissue repair had failed or where there was a high risk of recurrence or in highly experienced hands and for a highly select group of patients, they were sold without such limitations.
3499 At all relevant times, Gynemesh PS was also defective within the meaning of s 75AD of the TPA and s 138 of the ACL because its implantation exposed women to the risk of significant injury, the warnings given by the respondents were insufficient to protect users from those risks, and the information supplied with and about the device was liable to lull users into a false sense of security. The limitation made to the indication for use, which only came into effect on 16 March 2013, might have lessened the complication rate but it did not raise the level of safety to that which persons generally were entitled to expect, because the information accompanying the sale of the device was still insufficient to put users on notice of the true nature and extent of the risks.
3500 It follows that at all material times each of the POP devices had a “defect” within the meaning of the TPA and a “safety defect” within the meaning of the ACL. Provided that the applicants can prove that they suffered injuries because of the defect and none of the defences succeeds, they are entitled to relief, including compensation.
The defence under s 75AK(1)(c)
3501 Section 75AK of the TPA provides for a number of defences. The respondents pleaded one. That is the defence under s 75AK(1)(c). Section 75AK(1)(c) provided that:
In a liability action, it is a defence if it is established that:
…
(c) [T]he state of scientific or technical knowledge at the time when [the goods in question] were supplied by their actual manufacturer was not such as to enable that defect to be discovered.
3502 “Liability action” is defined in s 75AA to mean an action under s 75AD, 75AE, 75AF or 75AG. Thus, for present purposes, the defence is confined to the appellants’ case under s 75AD.
3503 The defence is maintained in the ACL in s 142(c). It is in identical terms save for the inconsequential insertion of “safety” before “defect”, the substitution of “defective goods action” for “liability action” and the deletion of “actual”.
3504 As Professor Harland observed in an article published in the Sydney Law Review, the defence is a very narrow one.3003 It is not enough for a manufacturer to prove that it could not reasonably be expected to have discovered the defect; the defect must be one that no-one could have discovered at the time it supplied the goods. Moreover, the relevant time is the time the goods containing the defect were supplied, not the time when goods of that kind were first supplied. If these matters were not clear from the text, they are clear from the Explanatory Memorandum at [55]–[56], cited by the Full Court in Merck at [204] and by the respondents in their submissions, which stated:
It is the objective state of scientific and technical knowledge, not the subjective knowledge of the individual manufacturer, which is to be taken into account. It is only if the defect could not have been discovered by anybody that the manufacturer will be able to succeed. A manufacturer must expect that there may be further scientific or technical advances during the period of testing and production. The manufacturer should therefore satisfy itself that there have been no further technical advances which affect the safety of the goods before putting them into circulation.
Similarly, a manufacturer must keep up to date with advances in knowledge after it first puts a product into circulation to ensure that new information is taken into account in the manufacture of subsequent goods, as new information may expose defects in goods. The crucial time is therefore when the alleged defective good which caused the injury was supplied by the manufacturer, not the time at which the manufacturer first supplied goods of that type.
(Emphasis added)
3505 In the Vioxx case, both the primary judge and the Full Court proceeded on the basis that this was the way the defence should be construed. The respondents in the present case did not argue otherwise.
3506 The respondents’ submissions were weak. They merely emphasised the development of scientific learning. While they could show that with the passage of time there was an increase in knowledge about certain matters, they did not prove that the knowledge could not have been acquired at an earlier point in time. In cross-examination, for example, Professor Klinge was asked when it became apparent to him that what might be significant about the structure of Prolene Soft was the effective pore size influenced by the existence of the diagonal braces. Although when pressed for a date he said 2007, his immediate response was illuminating:
When – when we have been asked to look to the tissue response of the – the Prolift mesh, because then we – we look to what happens within the pores of the Prolene mesh, the Prolift mesh, and then it became clear that you had all these pores are filled by scar tissue as well, and then it became evident that the reason for this are these crossing fibres that are reducing the pore size.3004
3507 There is no reason to believe that, had he been asked to look at the tissue response to Prolift before 2007, he would not have reached the same conclusion. In re-examination Professor Klinge testified that, before 2007, they had not been asked to do that and they were only asked to do so by US lawyers because of “the upcoming problems with the pelvic floor meshes in the field”.3005
3508 It was evident from Professor Klinge’s affidavit that, even before TVT was conceived, the respondents were on notice of the necessity to design a mesh to suit the biomechanical demands of the specific part of the body in which it was to be implanted. That was the subject of Professor Klinge’s presentation at the first Suvretta meeting in 1994. I referred in Part V to Professor Williams’ remark in his chapter on “General Concepts of Biocompatibility” in the Handbook of Biomaterial Properties published in 1998. It bears repeating in this context:
To recognize the very effective performance of a material under one set of conditions but then to assume that the same material can perform equally well under entirely different circumstances is inherently dangerous since it takes into account neither the variations one might expect to see in the host response from site to site nor the fact that what is appropriate for one situation may not be appropriate for another.3006
3509 This danger should have been obvious to the respondents, not least because it accords with common sense. In their eagerness to take their products to market, however, little or no attention was given to this basic principle.
3510 Animal studies conducted in 2006 showed that vaginal implantation of dermal collagen graft materials may cause a different tissue response than that which is generated by abdominal implantation.3007 There is no reason to believe that similar studies could not have been conducted well before CE marking of TVT.
3511 The respondents submitted that it was not until 2007 that it could be established that the Amid classification was no longer appropriate for pelvic meshes and that a pore size of greater than 1,000 microns is necessary to minimise the risk of complications.3008 This submission must be rejected for the reasons given in Part V above.
3512 In any case, it is only necessary to pull at the mesh with both hands to see that the pore sizes reduce under strain and Amid had referred to the problem of the interstices in 1997, before the launch of the first of the Ethicon devices.
3513 Furthermore, it is difficult to see how the defence was justified when the respondents were admittedly aware of all the risks before any of the devices were supplied.
3514 It follows that the respondents have not made out their defence.
Conclusion
3515 At all material times Ethicon Sàrl manufactured and supplied all the SUI devices, Prolift, Polist+M, and Prosima when those devices had a defect within the meaning of s 75AC of the TPA. Consequently, if any applicant and/or group member suffered injury because of the defect in the device she received, Ethicon Sàrl is liable to compensate her for the amount of the loss and damage she sustained as a result of that injury, unless the action is statute-barred and the limitation period is not extended.
3516 At all material times Ethicon Inc. manufactured and supplied Gynemesh PS when it had a defect within the meaning of s 75AC of the TPA. Accordingly, Ethicon Inc. is liable to compensate any applicant and/or group member who suffered an injury because of the defect for the amount of the loss and damage she sustained as a result of that injury.
3517 JJM was the deemed manufacturer and supplier of all the devices and is jointly and severally liable with Ethicon Sàrl and Ethicon Inc.
Unfitness for purpose and unmerchantable quality
3518 There is no material difference between the applicants’ s 74B case and their s 74D case. For that reason, I propose to deal with them together.
The legal framework: unfitness for purpose
3519 Section 74B provided as follows:
(1) Where:
(a) a corporation, in trade or commerce, supplies goods manufactured by the corporation to another person who acquires the goods for re-supply;
(b) a person (whether or not the person who acquired the goods from the corporation) supplies the goods (otherwise than by way of sale by auction) to a consumer;
(c) the goods are acquired by the consumer for a particular purpose that was, expressly or by implication, made known to the corporation, either directly, or through the person from whom the consumer acquired the goods or a person by whom any antecedent negotiations in connexion with the acquisition of the goods were conducted;
(d) the goods are not reasonably fit for that purpose, whether or not that is a purpose for which such goods are commonly supplied; and
(e) the consumer or a person who acquires the goods from, or derives title to the goods through or under, the consumer suffers loss or damage by reason that the goods are not reasonably fit for that purpose;
the corporation is liable to compensate the consumer or that other person for the loss or damage and the consumer or that other person may recover the amount of the compensation by action against the corporation in a court of competent jurisdiction.
(2) Subsection (1) does not apply:
(a) if the goods are not reasonably fit for the purpose referred to in that subsection by reason of:
(i) an act or default of any person (not being the corporation or a servant or agent of the corporation); or
(ii) a cause independent of human control;
occurring after the goods have left the control of the corporation; or
(b) where the circumstances show that the consumer did not rely, or that it was unreasonable for the consumer to rely, on the skill or judgment of the corporation.
3520 There is no dispute about ss 74B(1)(a) and (b). It is agreed that JJM is the (deemed) manufacturer, that JJM supplied the devices to others for re-supply, and that a person supplied one or other of the devices to the applicants. Here, that person was the surgeon who implanted the device. There is no evidence as to who it was who acquired the devices but a supplier for the purposes of s 74B(1)(b) need not be the person who acquired the goods from the manufacturer: Merck at [170].
3521 It follows that each of the applicants must prove:
(1) the particular purpose for which the device in question was acquired;
(2) that that particular purpose was conveyed expressly or by implication to the manufacturer, if not directly then, through the person from whom she acquired the device (here, the treating surgeon);
(3) that the device was not reasonably fit for the relevant purpose; and
(4) that she suffered injury “by that reason”.
3522 As to the first and second issues, the Full Court held in Merck at [171] that:
The hinge on which s 74B turns is the purpose for which the consumer acquired the goods, as that purpose (expressly or implicitly) is made known by the consumer, in this case, to the person from whom the consumer acquired the goods, namely the pharmacist. Identification of purpose in s 74B(1)(c) is necessarily a subjective matter: Rasell v Cavalier Marketing (Australia) Pty Ltd [1991] 2 Qd R 323 at 330.
3523 Nevertheless, it has been held that in some cases the manufacturer may be presumed to know the consumer’s purpose: see Carey-Hazell (2004) at [212]. In Merck the Full Court (at [171]) held that, by having a prescription filled by a pharmacist, Mr Peterson implicitly made known that Vioxx was being acquired for the purpose of use as a medication for treatment for arthritic pain without gastrointestinal side-effects.
3524 This is a similar case. Where goods are commonly acquired for a particular purpose, however, it seems to me, notwithstanding what was said in Carey-Hazell (2004) and Merck, that the intention of the Act was that such cases would be covered by s 74D rather than s 74B. I cannot see why it was necessary for the applicants to sue for breaches of both ss 74B and 74D.
3525 As to the third issue, the test is objective (Merck at [174]): whether the particular device was as fit for its purpose as a reasonable consumer in the position of the applicant would expect: Graham Barclay Oysters Pty Ltd v Ryan (2000) 102 FCR 307 at [533]–[536] (Lindgren J, Lee J agreeing at [69]). Although there was an appeal to the High Court, the Full Court’s decisions on the TPA counts were not disturbed: Graham Barclay Oysters Pty Ltd v Ryan (2002) 211 CLR 540 (Graham Barclay Oysters (HC)).
3526 It is not necessary for the applicants to prove that they relied on the skill or judgment of the manufacturer; the onus is on the manufacturer to prove non-reliance: Graham Barclay Oysters at [515] (Lindgren J, Lee J agreeing at [69]). That is a legal onus, not merely an evidential one. In effect, there is a statutory presumption that an applicant relies on the skill or judgment of the manufacturer that the goods are fit for the purpose for which they are acquired. In the present case, since the proposition was not put to any of the witnesses, let alone proved, it should be taken that the applicants relied on Ethicon’s skill or judgment.
3527 Section 74B(1) does not apply if, amongst other things, the circumstances show that the consumer did not rely, or it was unreasonable for the consumer to rely, on the skill or judgment of the corporation: s 74B(2) (ACL, s 55(3)). The respondents raised this as a defence in their pleading, but the defence was abandoned in closing submissions.3009
The legal framework: unmerchantable quality
3528 The applicants allege that each of the devices was not of merchantable quality within the meaning of s 74D of the TPA. At all material times s 74D provided:
74D Actions in respect of goods of unmerchantable quality
(1) Where:
(a) a corporation, in trade or commerce, supplies goods manufactured by the corporation to another person who acquires the goods for re-supply;
(b) a person (whether or not the person who acquired the goods from the corporation) supplies the goods (otherwise than by way of sale by auction) to a consumer;
(c) the goods are not of merchantable quality; and
(d) the consumer or a person who acquires the goods from, or derives title to the goods through or under, the consumer suffers loss or damage by reason that the goods are not of merchantable quality;
the corporation in liable to compensate the consumer or that other person for the loss or damage and the consumer or that other person may recover the amount of the compensation by action against the corporation in a court of competent jurisdiction.
(2) Subsection (1) does not apply:
(a) if the goods are not of merchantable quality by reason of:
(i) an act or default of any person (not being the corporation or a servant or agent of the corporation; or
(ii) a cause independent of human control;
occurring after the goods have left the control of the corporation;
(b) as regards defects specifically drawn to the consumer’s attention before the making of the contract for the supply of the goods to the consumer: or
(c) if the consumer examines the goods before that contract is made, as regards defects that the examination ought to reveal.
(3) Goods of any kind are of merchantable quality within the meaning of this section if they are as fit for the purpose or purposes for which goods of that kind are commonly bought as it is reasonable to expect having regard to:
(a) any description applied to the goods by the corporation;
(b) the price received by the corporation for the goods (if relevant); an
(c) all the other relevant circumstances.
3529 The equivalent provision in the Australian Consumer Law is s 54, which is not materially different. It merely substitutes “acceptable” for “merchantable”.
3530 This cause of action has seven elements:
(1) the goods were supplied by a corporation to another person;
(2) the supply was in trade or commerce;
(3) the corporation was the manufacturer of the goods;
(4) the person who acquired them did so for the purpose of re-supply;
(5) there was a supply to a consumer otherwise than by sale by auction;
(6) the goods were not of merchantable quality (in that they were not as fit for the purpose or purposes for which goods of that kind are commonly bought as it is reasonable to expect having regard to any description applied to the goods by the corporation, the price received by the corporation, and all other relevant circumstances); and
(7) the consumer suffered loss or damage “by reason” that the goods were not of merchantable quality.
3531 Only the last two elements are in issue.
3532 Two questions presently arise for consideration.
3533 First, at all material times were the relevant Ethicon devices as fit for the purpose for which they were commonly bought as it is reasonable to expect having regard to the description applied to them by the manufacturer and any other relevant circumstances? Second, if not, did any of the applicants suffer loss or damage “by reason” of that circumstance? Only the first raises a common issue.
3534 So what is the correct approach to the first question?
3535 First, the purpose for which the goods are bought must be determined. The relevant inquiry is into the purpose or purposes for which goods of the relevant kind are commonly bought as is reasonable to expect, having regard to the manufacturer’s description, the price received for the goods, and all other relevant circumstances.
3536 Second, the relevant expectations are those of a reasonable consumer in the position of the actual consumer: Graham Barclay Oysters at [533]–[534] (Lindgren J), Lee J agreeing at [69], Kiefel J at [611]; Courtney v Medtel Pty Ltd (2003) 126 FCR 219 at [216] (Sackville J).
3537 Third, it would be wrong to measure the reasonable expectations of the hypothetical reasonable consumer against the specialist technical knowledge of the manufacturer: Graham Barclay Oysters at [536].
3538 Fourth, the question is to be answered on the basis of what it was objectively reasonable to expect at the time of supply to the consumer: Medtel at [64] (Branson J, Jacobson J agreeing at [81]; see also Moore J at [43]). The answer will always depend on the circumstances. As Branson J explained at [64]:
The test contained in s 74D(3) is a test that requires the making of a comparison. It calls for the fitness for purpose of the goods in question to be measured against what it was objectively reasonable to expect, in terms of fitness for purpose, in all the relevant circumstances. Those circumstances include the description applied to the goods by the manufacturer and the price received by the manufacturer for the goods. What it is objectively reasonable to expect in terms of fitness for purpose of goods of one description may be quite different from what it would be reasonable to expect of goods of another description. What it would be reasonable to expect in terms of fitness for purpose of an inexpensive product might be quite different from what it would be reasonable to expect of an expensive product of the same kind.
3539 Fifth, goods may be not of merchantable quality within the meaning of the section if the evidence shows that there is a risk they may fail; it is unnecessary to prove actual failure: Medtel at [72]–[74].
3540 Sixth, the assessment is to be undertaken in the light of all the information about the goods available at the time of trial: Medtel at [70], [81]. Consequently, after-acquired information may be taken into account for this purpose.
3541 The underlying legislative policy, as Lindgren J explained in Graham Barclay Oysters at [538], is to require manufacturers to meet the reasonable expectations of consumers as to the fitness of their goods for their purpose or purposes. His Honour continued:
Consistently with that policy, if the manufacturer knows that it cannot be sure to meet those expectations, it must cease manufacturing, or, if possible, ensure that the consumer has agreed to bear the risk (perhaps by an appropriate warning with the result that the consumer’s otherwise reasonable expectations are made unreasonable).
3542 At a broad level, pelvic mesh devices are acquired for the purpose of treating either stress urinary incontinence or pelvic organ prolapse. More particularly, it is reasonable to infer from the way in which they were promoted that they were acquired for the purpose of treating one of those conditions more effectively or at least as effectively as other surgical interventions and with fewer risks to safety.
3543 It might not be reasonable to expect that a product of this kind would be free of risk. Nevertheless, it is certainly reasonable to expect that, whatever risks attend the use of such products, the manufacturer would disclose them so that the consumers could make an informed decision as to whether or not to consent to their implantation. In Ryan v Great Lakes Council (1999) 102 LGERA 307, a case in which consumers of oysters who contracted hepatitis A after consuming infected oysters grown in polluted waters, for example, Wilcox J rejected an argument that the oysters were as fit for human consumption as it was reasonable to expect because it was impossible for a grower to guarantee the absence of a virus. His Honour held at [374] that the issue posed by s 74D(3) was not whether it was possible for the grower to ensure that the oysters were free of viruses, but whether a purchaser would act reasonably in expecting them to be. He said that one of the relevant circumstances affecting the question as to what was reasonable to expect was the absence of a warning by the oyster growers of the possibility of a virus in the oysters. This aspect of the decision was not disturbed on appeal: Graham Barclay Oysters at [527]–[538] (Lindgren J, with whose conclusion and reasons Lee J and Kiefel J agreed at [69] and [611] respectively). The appeal to the High Court was confined to the claim in negligence. In the present case, I am satisfied that consumers of implantable medical devices would act reasonably in expecting that the devices would not carry risks if those risks were not disclosed by the respondents to the hospitals to which they were supplied and the people to whom they were promoted, including medical professionals and patients.
Conclusion
3544 It was common ground that if the applicants made out their case under s 75AD then the causes of action under s 74B and s 74D must also succeed.3010 In these circumstances I need say nothing more. Only the question of causation remains to be determined. I will come to that in due course.
Misleading or deceptive conduct
What must be proved?
3545 Section 52(1) of the TPA prohibited a corporation in trade or commerce from engaging in conduct that is misleading or deceptive or likely to mislead or deceive. Section 18(1) of the ACL, which replaced it, prohibits a person in trade or commerce from engaging in that conduct. The only difference is that s 18 captures individuals as well as corporations. The difference is of no consequence in this case.
3546 The relevant principles are well established.
3547 First, the conduct with which the sections are concerned is conduct which leads or is likely to lead a person or persons into error: Rhone-Poulenc Agrochimie SA v UIM Chemical Services Pty Ltd (1986) 12 FCR 477 at 490 (Bowen CJ). The words “likely to mislead or deceive” serve no other purpose than to emphasise that it is not necessary to show that anyone was actually misled or deceived by the corporation’s conduct.
3548 Second, the section is not merely concerned with representations. “Engaging in conduct” means doing or refusing to do an act and “refusing to do an act” includes “refraining (otherwise than inadvertently) from doing that act”: TPA, s 4(2); CCA, s 4(2). Thus a corporation may fall foul of the section by failing to comment or disclose certain information, including by failing to mention a qualification to an absolute statement (see, for example, Rhone-Poulenc at 489–90). There need not be a recognised duty of disclosure under the common law or in equity (Rhone-Poulenc at 490 per Bowen CJ; at 508 per Jackson J); silence “may simply be the element in all the circumstances of a case which renders the conduct in question misleading or deceptive”: Commonwealth Bank of Australia v Mehta (1991) 23 NSWLR 84 at 88 (Samuels JA), approved in Demagogue Pty Ltd v Ramensky (1993) 39 FCR 31 at 40 (Gummow J, with whom Black CJ and Cooper J agreed). As Gummow J put it in Demagogue at 41, consistently with the natural meaning of the terms used in s 52, the question is whether in the light of all relevant circumstances constituted by acts, omissions, statements or silence, the conduct is or is likely to be misleading or deceptive. Thus, a vendor’s silence in circumstances where there is no duty to speak at common law might be misleading or deceptive where it induces, or is likely to induce, the purchaser into error: Rhone-Poulenc at 508 (Jackson J).
3549 In Demagogue at 41 Gummow J expressed his agreement with French J in Kimberley NZI Finance Ltd v Torero Pty Ltd [1989] FCA 400 that unless the circumstances gave rise to a reasonable expectation that if some relevant fact existed it would be disclosed, it is difficult to see how mere silence could support the inference that the fact does not exist. Black CJ observed at 32 that:
Silence is to be assessed as a circumstance like any other. To say this is certainly not to impose any general duty of disclosure; the question is simply whether, having regard to all the relevant circumstances, there has been conduct that is misleading or deceptive or that is likely to mislead or deceive. To speak of “mere silence” or of a duty of disclosure can divert attention from that primary question. Although “mere silence” is a convenient way of describing some fact situations, there is in truth no such thing as “mere silence” because the significance of silence always falls to be considered in the context in which it occurs. That context may or may not include facts giving rise to a reasonable expectation, in the circumstances of the case, that if particular matters exist they will be disclosed.
3550 In Johnson Tiles Pty Limited v Esso Australia Limited (2000) 104 FCR 564 at [70] French J disagreed with the Chief Justice’s statement that silence is “to be assessed as a circumstance like any other”. He emphasised what was apparent from the Chief Justice’s reasons, namely that conduct is to be assessed by reference to context or circumstances, so that if a corporation fails to disclose a fact which in the circumstances of the case would reasonably be expected not to exist, then non-disclosure may convey the misleading impression that it does.
3551 Third, a representation may be literally true, but in the circumstances misleading. To paraphrase an example given by Stephen J in Hornsby Building Information Centre Pty Ltd v Sydney Building Information Centre Ltd (1978) 140 CLR 216 at 227, it could be misleading or deceptive to advertise a play as starring Hugh Jackman when the role is played by an unknown actor who bears the same name as the film star. A half-truth is also apt to mislead: Fraser v NRMA Holdings Ltd (1994) 52 FCR 1 at (Gummow J). Similarly, a failure to qualify an absolute statement may render the statement misleading, as may a failure to reveal changes or developments since the making of the statement: Rhone-Poulenc at 490 (Bowen CJ).
3552 Fourth, where, as here, conduct is directed, not to particular individuals, but to a section of the public or a class of persons, the question of whether the conduct in question is misleading or deceptive or likely to mislead or deceive is to be answered by reference to the effect of the conduct on the ordinary (Parkdale Custom Built Furniture Pty Ltd v Puxu Pty Ltd (1982) 149 CLR 191 at 210 per Mason J) or reasonable (Parkdale at 199 per Gibbs CJ) person. To that end, it is necessary to identify the relevant class of persons to whom the conduct is directed and “to isolate by some criterion a representative member of the class”: see Campomar Sociedad, Limitada v Nike International Limited (2000) 202 CLR 45 at [103]. There may, however, be more than one relevant class (as in Primary Health Care Ltd v Commonwealth of Australia [2017] FCAFC 174). This is such a case. The conduct in the present case was directed both to medical practitioners, principally gynaecologists and urogynaecologists, and to women suffering from stress urinary incontinence and/or pelvic organ prolapse.
3553 The class includes (or classes include) the experienced and the inexperienced, the gullible and the astute, the intelligent and the less intelligent, the informed and the uninformed, but the section is concerned with the effect of the conduct on reasonable members of the class and not on those who fail to take reasonable care of their own interests: Parkdale at 199 (Gibbs CJ).
3554 Fifth, conduct is likely to mislead or deceive if there is a real and not remote chance or possibility that the reasonable member of the class, or a not insignificant number of people in the relevant class, would be misled or deceived. See, for example, Global Sportsman Pty Ltd v Mirror Newspapers Ltd (1984) 2 FCR 82 at 87.
3555 Sixth, the question is to be determined objectively, in context, and having regard to the relevant surrounding facts and circumstances: Taco Company of Australia Inc v Taco Bell Pty Ltd [1982] FCA 136; (1982) 42 ALR 177 at 199, 202; Parkdale at 199 (Gibbs CJ).
3556 Seventh, conduct can be misleading or deceptive regardless of the intention of the alleged wrongdoer; the section is concerned with the effects or consequences (actual or likely) of the conduct: Hornsby Building Information Centre at 228 (Stephen J), 232 (Jacobs J), 234 (Murphy J). Nor does it matter whether the wrongdoer took reasonable care. No question of fault is involved. See Parkdale at 197. That said, where an intention to deceive is established, a court may more readily infer that the intention will probably be effective: Campomar at [33].
3557 In Johnson Tiles French J considered whether the facts which are not disclosed have to be known to the party failing to make disclosure. His Honour noted the general principle at [66]–[67] and referred to what the Full Court said in Fraser v NRMA Holdings Ltd (1995) 55 FCR 452 at 465–6:
[F]or the purposes of s 52, if by reason of what was said and what was left unsaid the conduct of the corporation is misleading and deceptive or likely to mislead or deceive, a contravention would occur even if the corporation through its directors and officers did not have knowledge of the undisclosed facts which rendered the conduct in breach of s 52. A contravention of s 52 may occur without knowledge or fault on the part of the corporation, and notwithstanding the exercise of reasonable care: Parkdale Custom Built Furniture Pty Ltd v Puxu Pty Ltd at 197.
3558 At this point his Honour said that “[t]his is not to say that knowledge may not be a relevant consideration” and “in a case where disclosure would reasonably be expected of a fact if that fact were known to the corporation”, failure to disclose may convey the implication that the fact is not known.
3559 In Fraser at 468, the Full Court stated that:
Where the contravention of s 52 alleged involves a failure to make a full and fair disclosure of information, the applicant carries the onus of establishing how or in what manner that which was said involved error or how that which was left unsaid had the potential to mislead or deceive. Errors and omissions to have that potential must be relevant to the topic about which it is said that the respondents' conduct is likely to mislead or deceive … The need to make full and fair disclosure must be tempered by the need to present a document that is intelligible to reasonable members of the class to whom it is directed, and is likely to assist rather than to confuse.
3560 Further, the Full Court held at 467 that:
Whilst s 52 does not by its terms impose an independent duty of disclosure which would require a corporation or its directors to give any particular information …, where information for that purpose is promulgated, unless the information given constitutes a full and fair disclosure of all facts which are material to enable … a properly informed decision [to be made], the combination of what is said and what is left unsaid may, depending on the full circumstances, be likely to mislead or deceive ...
What is the conduct complained of?
3561 That is not an easy question to answer, certainly not on the basis of the applicants’ convoluted pleading which tended to obscure rather than to elucidate the conduct. The pleading was of a type described by Jackson J in Mio Art Pty Ltd v Macequest Pty Ltd [2013] QSC 211; (2013) 95 ACSR 583 at [63]:
[W]here a pleading alleges a lengthy historical account of facts that occurred over an extensive period of a commercial relationship, then particular specific causes of action are pleaded on the basis that the reader is invited to find the relevant material facts for any cause of action in all that has gone before, the price for the death of that hero, brevity, is not paid in the valuable coin of precision. Instead, the reader is invited on a would-be treasure hunt, with the unlikely satisfaction that after looking in every nook and cranny, and trying every combination possible, there will be an Archimedian “Eureka” moment.
3562 Misleading or deceptive conduct was pleaded for the first time in the Fourth Further Amended Statement of Claim, which was filed by leave on 10 July 2017. The amendment was foreshadowed at a case management hearing on 29 May 2017. It appears to have been an afterthought following a lightbulb moment during the preparation for the hearing which was due to start a little more than a month later. I was informed by the applicants’ lead counsel that it did “nothing other than [tack] onto what’s already there in the failure to warn case”.3011
3563 The allegation is made in relation to both the POP and the SUI devices and the pleading is in the same form. It is sufficient therefore for present purposes to refer to the pleading in relation to the POP devices, since it appears first. It relevantly reads:
39A. Further and in the alternative, the matters pleaded in paragraphs 5, 15, 16, 17, 18, 19, 20, 23, 23A, 23B, 23BA and 23C, 31A, 32, 32A, 35A, 36 and 36C are repeated.
39B By reason of the matters pleaded at paragraph 39A, each of the Respondents engaged in conduct that was misleading or deceptive or likely to mislead or deceive in contravention of section 52 of the TPA and section 18 of Schedule 2 of the CCA.
3564 Paragraph 5 described the role of JJM as importer, supplier, and promoter of the devices in Australia. Paragraph 15 described the treating hospitals and doctors as recipients of representations made by JJM and/or one or other or both of the Ethicon respondents. It did not, however, particularise any representations. Paragraphs 16 and 17 were allegations that the marketing, promotion, and supply of the devices by the Ethicon parties and JJM on their behalf was “in trade and commerce in Australia”. All of these matters were relevant to whether there was a contravention of s 52 of the TPA or s 18 of the ACL but they did not define the character of the contravening conduct.
3565 Paragraph 18 defined the purpose of the POP implants. In paragraph 19 the applicants alleges that the purpose was known to the respondents. These are allegations of fact that are material to other causes of action but not this one. It is not until paragraph 20 that we first acquire a glimpse of the impugned conduct, albeit in the context of the unfitness for purpose and unmerchantable quality cases. There, the applicants alleged that the respondents promoted, distributed and supplied the POP devices as medical devices that were reasonably fit for “the Mesh Purpose” (that is, for use in pelvic surgery for the treatment of pelvic organ prolapse, to restore pelvic anatomy and function, and “thereby alleviate the symptoms [of pelvic organ prolapse]”. The particulars refer to extracts from brochures and IFUs for each of the POP devices.
3566 Paragraph 23 described the risks and complications of the POP devices, and 23A the difficulties and risks associated with removal surgery. Paragraph 23B dealt with native tissue repair as an available and safer alternative.
3567 Paragraph 23BA alleged that before the POP devices were released, supplied, distributed, marketed or promoted in Australia none of the respondents undertook any or any adequate evaluation of their risks.
3568 Paragraph 23C alleged that the respondents failed to give any or any sufficient information or warning of the risks or of their failure to evaluate them adequately or at all before taking the POP devices to market.
3569 The remaining paragraphs made similar allegations in the context of the claim in negligence.
3570 But what is it about this conduct that makes it misleading or deceptive within the meaning of the statute? What, if any representations were made, whether expressly, implicitly or by silence that the applicants contend were liable to cause them to fall into error?
3571 Unsurprisingly, the respondents had trouble coming to grips with the way the applicants put their claim.
3572 In chapter 2 of their written submissions, which were filed at about the same time the applicants filed theirs, they described the pleaded claim as obscure and incoherent. They went looking for representations in the pleading but had trouble finding them. At the time the order was made for the filing of the Fourth Amended Statement of Claim, I also fixed a timetable for the respondents to request further and better particulars of this claim and for the applicants to reply but if such a request was made and a reply forthcoming that correspondence was not tendered.
3573 It is trite that a pleading should state the pleader’s case with sufficient clarity to enable the pleader’s opponent to understand the case the opponent is called upon to meet: Gould v Mount Oxide Mines Ltd (in liq) (1916) 22 CLR 490 at 517. This pleading barely gets there.
3574 What is more, r 16.42 of the Federal Court Rules requires that a party who pleads fraud, misrepresentation, unconscionable conduct, breach of trust, wilful default or undue influence must state in the pleading particulars of the facts on which the party relies. The claim under s 52 of the TPA and s 18 of the ACL involves a plea of misrepresentation. For the purposes of such a claim the pleader is required to identify what it is alleged that the impugned statements conveyed to their intended audience: Forrest v Australian Securities and Investments Commission (2012) 247 CLR 486 at [26]. This pleading does not do that.
3575 This is not, however, a judgment on the pleading.
3576 In their opening submissions, the appellants did clarify their position somewhat. They submitted that the respondents’ conduct in marketing the devices and continuing to market them without proper disclosure or warning as to their complications and the gravity of them was misleading or deceptive or likely to mislead or deceive. They submitted (correctly) that the cause of action for misleading or deceptive conduct is not limited to the making of specific representations. Where, as here, a course of conduct is alleged that was misleading, by reference to the marketing and promotion of the devices by means of brochures, through IFUs and otherwise, the whole of the conduct is to be considered, citing Campbell v Backoffice Investments Pty Limited (2009) 238 CLR 304 at [102]. As McHugh J said in Butcher v Lachlan Elder Realty Pty Ltd (2004) 218 CLR 592 at [109], in a passage cited with approval by the plurality in Campbell at [102]:
The question whether conduct is misleading or deceptive or is likely to mislead or deceive is a question of fact. In determining whether a contravention of s 52 has occurred, the task of the court is to examine the relevant course of conduct as a whole. It is determined by reference to the alleged conduct in the light of the relevant surrounding facts and circumstances. It is an objective question that the court must determine for itself. It invites error to look at isolated parts of the corporation’s conduct. The effect of any relevant statements or actions or any silence or inaction occurring in the context of a single course of conduct must be deduced from the whole course of conduct. Thus, where the alleged contravention of s 52 relates primarily to a document, the effect of the document must be examined in the context of the evidence as a whole. The court is not confined to examining the document in isolation. It must have regard to all the conduct of the corporation in relation to the document including the preparation and distribution of the document and any statement, action, silence or inaction in connection with the document.
3577 The applicants did, however, rely on “numerous specific statements” about the devices. They pointed to the following statements the respondents made about the POP devices, all of which they argued, were misleading or deceptive or likely to mislead or deceive:
the mesh is non-reactive;
the mesh elicits a minimum to slight inflammatory reaction;
any inflammatory reaction is transient;
the mesh remains soft and pliable and is not subject to degradation;
normal wound healing is not impaired;
POP mesh implant surgery can be completed in half the time of traditional surgery;
POP mesh implant patients experience less pain;
the POP devices restore sexual function;
the POP devices provide long lasting stabilisation of fascial structures of the pelvic floor in vaginal wall prolapse;
they will correct prolapse defects and restore normal support; and
the surgical procedure for implanting the POP devices simplifies the repairing process.
3578 The applicants also relied on similar statements made about the SUI devices. They, too, were said to have been made from a non-reactive material and to elicit a minimal inflammatory reaction. Once again, the inflammatory reaction was said to be “transient”. The material from which it was made was said to cause transitory local irritation and a transitory foreign body response and not subject to degradation and providing for elasticity in both directions. The implant surgery was described as quick and simple. In addition, they submitted, the respondents made statements to the following effect:
the SUI tape device is for treatment of stress urinary incompetence (without further elaboration);
the SUI tape device affords long term clinical efficacy and safety;
long-term clinical efficacy and safety studies have been carried out; and
multiple clinical studies have proven the safety of the SUI tape devices.
3579 They did not at this point in their submissions, however, specifically address in the context of this cause of action what was misleading about these statements nor did they identify where these statements were made. While some of the sources are readily identifiable (the IFUs and the brochures), it was not clear whether these were the sole sources upon which the applicants relied.
3580 The applicants’ closing written submissions essentially repeated what they had said in their opening. They argued that the respondents’ conduct in marketing (and continuing to market) the devices without proper disclosure or warning about their potential complications and the gravity of them was misleading or deceptive or likely to mislead or deceive within the meaning of s 52 of the TPA and s 18 of the ACL.3012 They also argued that the respondents’ failure to provide appropriate warnings of the risks of the devices and to adequately assess the safety of the devices before they were launched also constituted misleading conduct.3013 In closing submissions they referred to the same conduct and statements without identifying specifically where the conduct occurred or the statements were made. In the result, I was left in the unhappy position of having to trawl through the material to assess them for myself. To say the least, this is unsatisfactory. Nevertheless, the evidence was not hard to find.
Was the conduct complained of misleading or deceptive?
3581 At all relevant times, the information in the IFUs and the brochures the respondents provided to patients and surgeons about the devices omitted warnings of the pleaded complications and the limits to the evaluation of the devices. The respondents also failed to provide information about the gravity of the risks and, with few exceptions, about how they could be mitigated. What is more, that same information made inaccurate and, at times, false representations about the devices. The question is whether this conduct considered as a whole was misleading or likely to mislead. I believe it was.
3582 In the Vioxx case, it was the combination of the manufacturer failing to provide relevant information whilst continuing to allow inaccurate information to circulate unchecked that constituted misleading conduct. In Peterson, Jessup J held it was misleading or deceptive for the manufacturer to omit warnings from product information about the results of a randomised control trial which showed the risk of myocardial infarction in patients taking Vioxx was higher than the product information disclosed. The product information had said that myocardial infarction had been observed in less than 0.1% of patients taking Vioxx in osteoarthritis studies. However, trial results from mid-2000 contained a figure of 0.5% based on patient-years. As a result, Jessup J held that, from the point that those results were available until the product information was amended in November 2001, the manufacturer had engaged in misleading conduct by failing to give practitioners advice about the results whilst unamended product information was still in circulation. His Honour said at [904] that “the misleading conduct was constituted by the failure to draw to the attention of doctors and other health care professionals the cardiovascular risk message that emerged from [the trial]”. This finding remained undisturbed on appeal.
3583 I identified the deficiencies in the respondents’ warnings and other information at length in Part XI of these reasons and will assume familiarity with that discussion in order to keep repetition to a minimum. I also commented on the misleading nature of the information provided by the respondents in the context of the applicants’ defective goods claim.
3584 It is apparent from that discussion that the respondents neglected to warn of certain risks, gave incomplete warnings about other risks, and made inaccurate or false representations about a variety of matters. The evidence established that the respondents engaged in a course of conduct in which they exaggerated the benefits of the devices and minimised the risks. This course of conduct was pursued from the time the first Ethicon device was supplied in Australia, if not earlier, and continued for years. It was curbed, and then only to some extent, by the intervention of the regulators.
3585 The following examples will suffice for present purposes. Taken in context, both individually and collectively, they support the applicants’ misleading and deceptive conduct claim.
3586 First, in every IFU for all the Ethicon devices the respondents falsely represented that the mesh elicited a minimal to slight inflammatory reaction which was transient and asserted that a foreign body reaction may occur, when they well knew that it would invariably occur. These representations were doubtless calculated to lull the reader into a false sense of security.
3587 Scott Jones (Product Director, Pelvic Floor Repair of Ethicon US), for example, pointed out in an email on 12 November 2008 that “permanent heavyweight meshes cause a persistent inflammatory reaction at the mesh-tissue interface for months to years after implantation”.3014
3588 In an email sent on 29 September 2009 by Katrin Elbert of Ethicon US reported that Sandy Savage, also of Ethicon US, told her that the reference to “transient” in the IFUs was misleading.3015
3589 In their report on three-month results of their Prolift+M study in March 2009, Dr Hinoul and his collaborators from Ethicon stated that “the mesh induces an acute and chronic foreign body reaction, which can lead to both exposure and shrinkage”.3016 They wrote at the time that their understanding of these reactions to the mesh as it is integrated into the host tissue led to “design modifications to mitigate [those] responses”.
3590 As I have already noted, Dr Hinoul denied that the statements in the IFUs were misleading, but I do not accept his denial.
3591 Second, the respondents did not warn of the risk of chronic pain until 2015. Instead, where references were made to the duration of pain, the IFUs and brochures for various devices stated that it was “transient”. I have referred to Professor Korda’s evidence about the information disseminated at meetings run by the manufacturers in which no mention was made of the risk of incurable mesh-induced pelvic pain and other matters, including the difficulties of surgical management of mesh excision. Some brochures claimed the pain could be managed by “simple pain relief”, suggesting that pain would be short-term.3017 The omission of a reference to the risk of chronic pain coupled with references to the risk of transient pain was likely to lead the reader to the erroneous belief that there was no prospect of chronic pain.
3592 Third, while the risk of erosion was the subject of a warning in all but one of the IFUs, the respondents never warned of the risk of late onset erosions. By coupling the warning about erosions with the erroneous statement that the foreign body response would be transitory, the respondents implied that if there were to be an erosion, it would occur soon after implantation. A not insignificant number of the relevant class might well also conclude, as Dr Meng Chen apprehended, 3018 that the potential complications were also transitory, particularly in the absence of any reference to the risk of long-term complications.
3593 Fourth, all IFUs downplayed the risk of infection and many implied that the only risk was that implantation could promote or enhance a pre-existing infection. The statement that a pre-existing infection could be potentiated was in the nature of a half-truth. It disguised the fact, well-known to the respondents, that the use of polypropylene mesh in the pelvic cavity could cause an infection.
3594 None of the IFUs for the SUI devices ever warned that Prolene mesh could cause an infection. The most recent IFUs (from October 2015) suggest that the risk of infection is related to the surgical procedure and is no different from the risk of infection with any operation. No indication is given of the risk of late infections caused by bacterial colonisation of the mesh or in connection with erosion, exposure or extrusion of the mesh.
3595 The IFUs for the POP devices warned that the mesh might have to be removed in the event of an infection if the mesh was used in contaminated wounds. The first indication, however, that the mesh itself could become contaminated did not appear until December 2008 with the publication of the 2008 Prolift+M IFU. Even then, the warning was limited to circumstances in which the gastrointestinal tract was opened.
3596 Fifth, until 2007 for the POP devices and 2015 for the SUI devices, the respondents did not warn of the risk of dyspareunia or apareunia. Instead, brochures for various devices claimed that patients could usually resume sexual activity within four to six weeks after surgery without any indication that it could be painful or too painful to tolerate. While the risk of dyspareunia may be no greater with the devices than in native tissue repair, the brochures implied that any pain with intercourse would be confined to the immediate post-operative period. That was capable of leading a not insignificant number of patients, if not also some doctors, into error.
3597 Sixth, the respondents never warned of the increased risk of a heightened inflammatory response in immunosuppressed patients or advised that implantation with the devices was contraindicated in such cases. Indeed, a brochure claimed that Prolift was “appropriate for almost all patients”.3019 Moreover, such patients were not identified in the contraindications sections of the IFUs when other kinds of patients were, both patients and surgeons might reasonably think, contrary to the fact, that the devices were appropriate for them and that they were at no added risk.
3598 Seventh, although they were aware of the possibility, the respondents did not warn until 2015 that revision surgery or multiple surgical procedures might be necessary or about the potential effects of that surgery. Nor did they warn of the difficulties of removing the mesh.
3599 Eighth, the respondents did not inform consumers of the limitations of clinical evaluations of the devices or the shortcomings of any available studies. Instead, they boasted that the devices had proven clinical safety and efficacy. Once again, the combination of the omissions and the positive assertions was likely to mislead a not insignificant number of doctors and patients.
3600 Ninth, the statement in the Prolift brochure that the mesh was “specially designed for placement through the vagina to support [prolapsed pelvic organs]”3020 was false. Gynemesh PS was not specially designed for transvaginal placement in prolapse repair. Similarly, the statement in the Gynemesh PS leaflet that Gynemesh PS was engineered for pelvic support was also false.3021 It was Prolene Soft by another name: a mesh designed or engineered for abdominal placement to reinforce the abdominal wall.
3601 This kind of conduct is reminiscent of the conduct with which Australian Competition and Consumer Commission v Reckitt Benckiser (Australia) Pty Ltd (No 4) [2015] FCA 1408 (ACCC v Reckitt Benckiser (No 4)) was concerned and which Edelman J declared to be misleading or deceptive in contravention of s 18 of the ACL. In that case, Reckitt Benckiser marketed and sold a range of analgaesics under the brand name Nurofen, labelling them “Nurofen Migraine Pain”, “Nurofen Tension Headache”, “Nurofen Period Pain”, and “Nurofen Back Pain”. In so doing Reckitt Benckiser represented that each of the four products in the range was specifically formulated to treat a particular kind of pain although, amongst other things, each of the products in the range contained the same active ingredient and was made to the same formula. While the declarations were made following admissions by the company, his Honour observed in his penalty judgment that “the admissions might be characterised as close to bowing to the inevitable”: Australian Competition and Consumer Commission v Reckitt Benckiser (Australia) Pty Ltd (No 7) [2016] FCA 424 at [69].
3602 Tenth, the repeated representations made by the respondents that the meshes used in the Ethicon devices were inert were false, too, since no biomaterial is inert.
3603 Professor Korda’s unchallenged evidence about the promotional activities of the respondents and the professional training they offered demonstrated that these misrepresentations were not confined to the IFUs and the brochures. Professor Korda’s testimony about his ignorance of some of the pleaded complications indicates that the respondents did not even share their knowledge about undisclosed risks with the preceptors of their devices and indicates that he and other surgeons are likely to have been misled by the respondents’ conduct.3022
3604 A woman contemplating surgery to implant any of these devices would reasonably have assumed that the information provided by the manufacturer would include warnings of all the risks that could arise with the use of the devices, particularly risks of which the manufacturer was aware, and that the information would be accurate, reliable, and not apt to mislead. To the extent that the respondents did not warn of the pleaded complications, it was reasonable for patients to infer that those risks did not exist. To the extent that the respondents made false representations as to the safety and efficacy of their devices, it was reasonable for patients to believe them and rely upon them.
3605 A surgeon might well make similar assumptions. Of course, medical practitioners have access to more resources and greater knowledge about the devices and the scientific context in which the information was provided. But a medical practitioner reading the product information would reasonably have assumed that what was said about their adverse side-effects represented what the manufacturer knew about them: Peterson at [903].
3606 The evidence establishes that these would have been erroneous assumptions.
3607 Having regard to all the relevant circumstances, I am satisfied that, at all material times the Ethicon devices were or have been on the market, the respondents engaged in a course of conduct which was misleading or deceptive or likely to mislead or deceive a not insignificant number of patients and surgeons as to both their safety and efficacy.
PART XIV: THE APPLICANTS’ CLAIMS IN NEGLIGENCE
3608 Part VA of the TPA does not operate as a code for product liability compensation claims. It is clear from s 75AR of the Act that Pt VA is not intended to exclude or limit the concurrent operation of any law in force in a state or territory, including the common law, and is not to be taken to limit, restrict or otherwise affect any right or remedy a person would have if Pt VA had not been enacted.
The elements of the cause of action
3609 In order to make out a cause of action in negligence, an applicant must prove that a respondent owed her a duty of care, that it breached that duty by failing to take reasonable care for her health or safety, and that any such breach caused her damage.
3610 The damage to Mrs Sanders and Mrs Gill occurred in Western Australia, the damage to Mrs Dawson in Victoria. It is common ground that those circumstances affect the law that applies to all substantive questions, including questions of the kind and amount of damages that may be recovered and, in the cases of Mrs Sanders and Mrs Gill, whether the actions are statute-barred: John Pfeiffer Pty Ltd v Rogerson (2000) 203 CLR 503 at [100].
3611 The result is that the modifications to the common law effected by the State legislation discussed below apply where relevant. This legislation is picked up and applied as surrogate Commonwealth law by operation of s 79 of the Judiciary Act 1903 (Cth): Okwume v Commonwealth [2016] FCA 1252 at [272] (Charlesworth J); Rizeq v Western Australia (2017) 262 CLR 1 at [63], [96]–[97] (Bell, Gageler, Keane, Nettle and Gordon JJ).
3612 Claims in negligence that may be pursued by members of the group the applicants represent will also be subject to statutory modifications in the states and territories in which the torts affecting them occurred. I will deal with the modifications affecting the applicants in each context in which they arise for consideration. It is sufficient to note at this point that the Civil Liability Act 2002 (WA) (CLA WA)) came into effect on 1 January 2003.3023 To the extent provided for by that Act, it applies to claims for damages for harm that arises out of an incident happening on or after 1 December 2003: CLA (WA), s 5A(3). Consequently, it applies to Mrs Gill but not Mrs Sanders. The relevant amendments made to the Wrongs Act 1958 (Vic) (Wrongs Act (Vic)) were made by the Wrongs and Other Acts (Law of Negligence) Act 2003 (Vic), which commenced on 3 December 2003. They apply to Mrs Dawson’s case.
The allegations
3613 The allegations made against the manufacturers, Ethicon Sàrl and Ethicon Inc., are essentially the same with respect to all the products. No material distinction was made between the SUI and the POP devices.
3614 In substance, the case as pleaded was that the manufacturers breached their duty of care by:
(1) designing, manufacturing, promoting, and supplying the Ethicon devices which had all the problems identified by the biomaterials experts and carried the risk of the pleaded complications;
(2) failing to conduct any or any adequate evaluation of the safety and effectiveness of the Ethicon devices before they were released in Australia;
(3) failing to conduct any or any adequate evaluation of the safety and effectiveness of the Ethicon devices after they were released in Australia; and
(4) failing to inform the women in the respective sub-classes, their treating doctors or hospitals of the characteristics of the devices and the risks identified in the pleading.
3615 Item (1) above was ultimately not pressed.3024 Notwithstanding the way in which the case was pleaded, the applicants’ case against the respondents was that each of the Ethicon devices could cause a number of potentially serious complications, yet the manufacturers failed to:
(1) undertake any adequate pre-market clinical or other evaluation of their safety and efficacy of; and/or
(2) undertake any adequate post-market clinical or other evaluation; and/or
(3) provide certain information about the risks associated with the use of the devices or the deficiencies in the evaluations of them.
3616 The applicants’ case against the supplier, JJM, was similar save to the extent indicated below.
3617 In their closing written submissions, the applicants summarised their negligence case in the following way.
3618 First, they argued that the respondents were negligent in failing to undertake sufficient steps to evaluate the safety of the devices before they were launched. Had such steps been undertaken, they contended, the devices would not have been launched. Alternatively, the devices would only have been released accompanied by full and frank warnings about the risks associated with their use and, to the extent that the available information was limited, by disclosures of the shortcomings of that information.
3619 Second, they argued that the respondents were negligent in failing to undertake sufficient steps to monitor and evaluate the devices after they had been launched. They contended that proper steps would have seen the withdrawal of the devices at an early date or the provision of the appropriate information and warnings.
3620 Third, they argued that the respondents were negligent in failing to warn of the risks of the devices of which they were (or ought to have been) aware and of the fact that they had not undertaken sufficient steps to evaluate their safety before launch.
3621 Moreover, they contended that:
Far from undertaking such warnings, the Respondents suppressed the extent of risks, suppressed notification to authorities of adverse events, ignored data, proactively sought to have relevant data excluded from published papers or presented in a way that obscured the significance of data and sought and obtained regulatory approval on a basis by obscuring from auditors relevant information which would have likely resulted in a failure to obtain or maintain such approval.3025
3622 Had it not been for the respondent’s failure to take any one of these precautions, the applicants claimed that they would not have been injured.
3623 Similar allegations were made against JJM. The only significant difference, whether by accident or design, was that no allegation was made in the applicants’ pleadings about any deficiency in JJM’s post-market evaluation of the SUI devices.
The manufacturers’ duty of care
3624 Despite views to the contrary expressed by some of the respondents’ witnesses, the fact that medical practitioners owe their patients a duty of care does not detract from the existence of a duty of care on the part of the manufacturer or supplier. Indeed, notwithstanding what appeared in their defence, the respondents accepted that they owed a duty to exercise reasonable care to avoid injury to consumers.3026
3625 As Ipp JA noted in Amaca Pty Ltd (Under NSW External Administration) v A B and P Constructions Pty Ltd [2007] NSWCA 220; (2007) Aust Torts Reports ¶81–910; (2007) 5 DDCR 543 at [94], the scope or content of the duty owed by a manufacturer to a consumer has been settled since the beginning of the modern law of negligence when Lord Atkin held in Donoghue v Stevenson [1932] AC 562 at 599 that a manufacturer has a duty to take reasonable care to avoid injury to the consumer of its products. In Graham Barclay Oysters (HC) at [106] McHugh J, drawing on Lord Atkin’s formulation of the duty, said:
The duty of care owed by a manufacturer or producer to a consumer is a duty to take reasonable care to avoid injury to the consumer. To formulate the duty in more specific terms invites error because it is likely to mix a question of law (whether a duty existed) with a question of fact (whether a breach occurred).
3626 Reasonable care includes “responsibility for ensuring that appropriate and necessary information about the product is communicated to persons who will use or consume [it] and who it can be foreseen may suffer loss or damage”: Peterson at [782].
3627 In the context of the present case, the manufacturers had a duty to take reasonable care in the design, testing, evaluation, supply, and marketing of the devices. That duty extended to providing accurate information about the performance and safety of the devices, including warnings about potential complications and contraindications. The duty was not confined to the period before the devices were made or placed on the market, it was a continuing obligation to evaluate their safety and keep abreast of information about the nature and extent of potential complications and to convey that information to users of the devices. Thus, as the Court of Appeal of England and Wales observed in Wright v Dunlop Rubber Co Ltd (1972) 13 KIR 255 at 272:
If the manufacturer discovers that the product is unsafe, or has reason to believe that it may be unsafe, his duty may be to cease forthwith to manufacture or supply the product in its unsafe form. It may be that in some circumstances the duty would be fulfilled by less drastic action: by, for example, giving proper warning to persons to whom the product is supplied of the relevant facts, as known or suspected, giving rise to the actual or potential risk. Factors which would be relevant would be the gravity of the consequences if the risk should become a reality, and the gravity of the consequences which would arise from the withdrawal of the product.
3628 JJM is deemed to be a manufacturer for the purposes of the statutory claims but at common law it is a supplier.
3629 Trindade and Cane observed in The Law of Torts in Australia (Oxford University Press, 5th edition, 2012) at 635 that the principle in Donoghue v Stevenson “covers everyone in the chain of manufacture, distribution and supply”. But a vendor of goods does not, by reason of that circumstance alone, owe a duty of care to the purchaser or end-user/consumer: Laundess v Laundess (1994) Aust Torts Reports ¶81-316; (1994) 20 MVR 156, McPherson’s Ltd v Eaton (2005) 65 NSWLR 187. In Laundess (at 61,874; 160) Mahoney JA, with whom Meagher JA and Powell JA agreed, said that there must be “something more” than a mere relationship of vendor and purchaser and (at 61,876; 161) that whether or not a duty of care will be imposed upon a vendor of goods must depend on the nature of the goods, the risk involved, and the circumstances of the case. In Eaton, Ipp JA, with whom both Mason P and Hodgson JA agreed, observed at [62] that the authorities supported this position. His Honour referred to a number of them and identified the presence in each of them of something more than the mere vendor-purchaser relationship. The additional factors his Honour identified from the case law included:
actual knowledge of the relevant risk: Clarke v Army and Navy Co-operative Society Ltd [1903] 1 KB 155, Watson v Buckely, Osborne Garrett & Co Ltd [1940] 1 All ER 174, Cuckow v Polyester Reinforced Products Pty Ltd (1970) 19 FLR 122;
knowledge that should put the vendor on inquiry: Fisher v Harrods Ltd [1966] 1 Lloyd’s Rep 500;
the presence of an obvious danger: Burfitt v Kille [1939] 2 KB 743; and
where, in the case of the sale of a second-hand car, the defect would be discovered by a competent mechanic: Andrews v Hopkinson [1957] 1 QB 229; or
where the distributor was also the manufacturer or producer, as in Thompson v Johnson and Johnson Pty Ltd [1991] 2 VR 449, Graham Barclay Oysters, and Dovuro v Pty Ltd v Wilkins (2003) 215 CLR 317.
3630 Eaton concerned the duty of care owed by a non-manufacturing distributor of goods to an employee of a purchaser of those goods. McPhersons was a hardware retail outlet which sold, amongst many other things, millboard. One of its customers was Mr Eaton’s employer. Mr Eaton died from mesothelioma, a tumor of the lining of the lung caused by inhaling asbestos dust to which he was allegedly exposed when he was working with millboard acquired from McPhersons. His widow brought the action and was the first respondent to the appeal. McPhersons had no knowledge of the dangers of asbestos and the dangers are not apparent from the material itself.
3631 In Eaton at [5] Mason P emphasised that it was necessary to consider whether there is “a broadly stated duty to take reasonable care to avoid foreseeable risk of injury in which the reasonableness of the distributor’s conduct is a factual issue dependent on the circumstances” or there is “a more confined duty expressed as having a particular content or scope”. The President said at [17] that the distributor’s duty of care is confined by what the distributor knows or has reason to know.
3632 Ipp JA accepted that the fact that the finding of the trial judge that McPhersons ought to have known of the dangers of asbestos was capable of constituting the “something more” at [91]. His Honour said at [93] that “ought to know” refers to “the knowledge that a person, acting reasonably in all the circumstances of the case, should know”. At [95]–[96], his Honour stated that whether or not that person ought to know of the risk depends largely on whether it is reasonably foreseeable to a reasonable vendor in the position of that person that the relevant risk might materialise.
3633 His Honour concluded that the trial judge erred in law by proceeding on the basis that the mere vendor-purchaser relationship gave rise to a duty of care and also in the making of the finding that McPhersons ought to have known of the dangers of asbestos. His Honour observed at [100] that the trial judge had asked himself the right question (whether a retailer in the position of McPhersons ought reasonably to have known that such exposure constituted a risk of injury) but that he failed to conduct “that specific inquiry” or apply “that standard”. His Honour explained at [101]:
In conducting the requisite inquiry, the judge was required to have regard to the fact that McPherson’s was a retailer (and not a manufacturer); that it sold a vast number of products made up of a possibly unknowable number of constituents; and that products containing asbestos comprised less than one per cent of the products that it sold. McPherson’s did not have actual knowledge of the dangers of asbestos. Thus, the judge needed to determine the practical difficulties (even with the resources available to it) that faced McPherson’s in learning about and keeping track of the dangers connected with each one of its products it sold. His Honour needed to determine whether, in this context, there was any fact that should have led McPherson’s to know, not only that the inhalation of asbestos was dangerous, but that asbestos fibres, in the quantities likely to be released when millboard was cut, might be dangerous. This required proof of actual facts from which a reasonable inference might be drawn, not merely the exercise of some moral or other judgment based on a world view of the duties of retailers generally. In addition, regard had to be had to the reasonableness, in the existing circumstances, of McPherson’s explanation that it relied on the manufacturers to inform it if there were any dangers in the use of the products they sold.
3634 At [104] Ipp JA went on to say that, before a finding could be made that McPhersons should have investigated the possible dangers of asbestos, there had to be evidence and a consideration of facts from which it could be said that it had reason to know that the asbestos in the millboard was possibly dangerous. At [106] his Honour set out a number of examples of facts that might support a finding of reasonable foreseeability. They included that “evidence that the dangers of asbestos were generally known to hardware retailers as a group, or published in articles that a retailer such as McPherson’s, acting diligently, could be expected to read, or that there was public discussion of such dangers (not confined to particular specialist groups), or that McPherson’s held itself out in a way that induced others to rely on it to ensure that the products it sold would contain warnings as to the safe way to work with asbestos and to cut the millboard”.
3635 The present case is very different from Eaton.
3636 JJM was not a mere vendor or distributor of Ethicon’s products. It was a member of the same corporate group as Ethicon Sàrl and Ethicon Inc., with ready access to all their expertise and information. It published material promoting the Ethicon devices. It was also the sponsor of the Ethicon devices within the meaning of the Therapeutic Goods Act. Because of its close relationship with the manufacturers and especially because of its role as sponsor of their products, JJM had reason to know of the various risks. If it did not know, it ought to have known of the risks of which Ethicon was aware, including the pleaded complications, as well as the limitations in the clinical evaluations conducted by Ethicon.
3637 Since the Therapeutic Goods Amendment (Medical Devices) Act 2002 (Cth) commenced and the Medical Devices Regulations came into operation, the sponsor of a medical device has been required, amongst other things, to:
have available throughout the time a device is included on the ARTG:
(a) sufficient information to substantiate compliance with the essential principles or procedures in place, including a written agreement with the manufacturer, to ensure that such information can be obtained from the manufacturer within 20 working days: TG Act, s 41FN(3)(a); MDR, reg 5.6;
(b) sufficient information to substantiate that the conformity assessment procedures or comparable requirements have been applied: TG Act, s 41FN(3)(b);
certify with respect to a medical device the subject of an application for inclusion on the ARTG that:
(c) devices of that kind comply with the essential principles: TG Act, s 41FD(d);
(d) it has available sufficient information to substantiate compliance with the essential principles or have procedures in place to ensure that such information can be provided from the manufacturer to the TGA within 20 working days: TG Act, s 41FD(e); MDR, reg 5.2;
(e) appropriate conformity assessment procedures have been applied: TG Act, s 41FD(f);
(f) the manufacturer has appropriate conformity assessment evidence for the device: TG Act, s 41FD(g);
(g) that the applicable provisions of the Therapeutic Goods Advertising Code and any other requirements in relation to advertising (which include prohibitions against misleading advertising) have been complied with: TG Act, s 41FD(h); and
(h) the conformity assessment evidence remains valid while the device is supplied in Australia.3027
apply to include the medical devices in the ARTG, including certifying that the information provided is complete and correct;
upon request:
(i) provide documentation relating to the medical device to the TGA: TG Act s 41FN(1)(b);
(j) allow a person authorised by the TGA to enter and inspect any premises, including outside Australia, where the devices are manufactured or located and to conduct tests or require tests to be conducted on the devices: TG Act s 41FN(1)(a);
notify the TGA of certain incidents and performance issues within a specified time: TG Act s 41FN(3)(d); MDR reg 10.4; and
give the manufacturer information relevant to its obligations under the conformity assessment or comparable procedures and whether the devices comply with the essential principles: TG Act, s 41FN(e).
3638 When checking to see that there is clinical evidence, the sponsor is directed to look for a section in the technical dossier that is clearly labelled “Clinical Evidence” and which includes, amongst other things:
identification of the essential principles relevant to the specific design of the device;
clinical data or justification as to why none are required; and
a clinical evaluation report containing a comprehensive analysis of the clinical data relevant to the device, written by a clinical expert competent in the appropriate field and able to give an objective assessment of the clinical data.3028
3639 It is the responsibility of both sponsors and manufacturers to ensure that the current regulatory requirements are “fully” met.3029
3640 As I mentioned in Parts VII and VIII these obligations did not apply to TVT until 2007, but they applied to all the other Ethicon devices throughout the relevant period. It is most unlikely that JJM would not have had access to the TVT technical file at all material times.
3641 In addition, by publishing and providing product brochures, which purported to include warnings of the risks posed by the devices, JJM held itself as having the relevant knowledge.
3642 In any case, whatever the true state of affairs, no point was taken against the applicants that the knowledge of one respondent was not also the knowledge of the other. Besides, as I mentioned at the outset of these reasons, neither during the trial nor in their submissions did the respondents draw any distinction between the positions of the manufacturers and JJM with respect to any of the causes of action.
3643 Having regard to all these matters, I find that, with one qualification, JJM’s duty of care was co-extensive with the duty of care owed by the manufacturers. All three respondents were obliged to exercise reasonable care in the supply and marketing of the devices.
3644 The exception relates to the allegation that JJM had a duty to undertake clinical evaluations of the Ethicon devices. I am not persuaded, either as a matter of law or fact, that the scope of JJM’s duty extended that far. Rather, JJM’s duty of care was to take reasonable steps to ensure that the information they conveyed about the devices was accurate, sufficient to alert both medical practitioners and prospective patients about the true risks associated with the use of the devices, and was not apt to cause them to form an erroneous impression about their safety and efficacy, either absolutely or in comparison with alternative forms of treatment. Reasonable care would also have included informing doctors and prospective patients of any shortcomings of the clinical evidence or evaluations. The applicants did not establish that its obligations went any further.
3645 Despite the breadth of their defence, in their written submissions the respondents’ indicated that the dispute was confined to three areas:
(1) the relevant standard of care and whether it is to be informed by “the concept of the ‘learned intermediary’”;
(2) the alleged failure to warn, the content of any duty to warn, and whether any failure had “a causally relevant effect”; and
(3) the causal relationship between the loss and damage to the applicants and the respondents’ alleged failure to conduct any or any adequate pre-market evaluation of the devices.3030
3646 It is well established that the standard of care is determined by what a reasonable person in the position of the respondent or respondents would do in response to the reasonably foreseeable risk: Graham Barclay Oysters (HC) at [192] (Gummow and Hayne JJ). The response will be affected, amongst other things, by the nature of the product, the gravity of the risk and the severity of the consequences should the risk eventuate. In the case of an inherently dangerous product or a product designed for human consumption or implantation, particularly permanent implantation, the level of caution required of a reasonable manufacturer (and of a supplier in the position of JJM) will necessarily be high.
3647 The respondents submitted that the standard of care must depend, in part, on the intermediaries between the entity who owes the duty and the persons to whom the duty is owed. In the present case they were said to be the treating surgeons with whom the patients agreed to have the implant surgery. The respondents argued that such individuals obtain the relevant information about the patient’s circumstances, have specialist training, skills and experience which enable them to assess the available information about the risks associated with the device and to communicate them to the patient. They argued that surgeons are uniquely qualified to assess a patient’s circumstances against the benefits and risks associated with the devices and communicate them to the patient, are capable of selecting and recommending products based on an assessment of their own surgical skills and preferences, and are the people with whom the patient ultimately decides to have the surgery.
3648 All these matters are true, but they can have no bearing on the first or second negligence claim. As for the third, they do not absolve the manufacturer from informing prospective patients, whether directly or indirectly, of the nature and extent of the risks posed to them by implantation of the devices and doing so clearly, completely and honestly. As the plurality observed in McLean v Tedman (1984) 155 CLR 306 at [8], “[t]he standard of care expected of the reasonable man requires him to take him to take account of the possibility of inadvertent and negligent conduct on the part of others”. In the present context, this means that the respondents were expected to take into account the possibility that the medical practitioner might fail to inform the patient of the relevant risks unless they were specifically drawn to their attention by the respondents.
3649 Earlier I referred to some of the US and Australian authorities, albeit in a different context. The approach of the Supreme Court of Canada is not materially different, as the judgment of that court in Hollis v Dow Corning Ltd [1995] 4 SCR 634 illustrates.
3650 Hollis was a negligence case about a post-surgical rupture of a breast implant. The plaintiff, Ms Hollis, was not warned by the implanting surgeon of the risks of post-surgical complications or of the possibility that the implants might rupture inside her body. The implanting surgeon received little warning from the manufacturer (Dow) about the possibility of a rupture although Dow had been aware for years that implant ruptures could cause adverse reactions in the body arising from loose gel. The only reference to rupture in the warnings issued by Dow before Ms Hollis’s implant surgery suggested it could only occur with “abnormal squeezing or trauma”. Ms Hollis recovered damages from Dow on the basis of negligent manufacture. That finding was overturned on appeal but the Court of Appeal dismissed the appeal on the ground that Dow had failed to provide an adequate warning of the risks of rupture. The sole issue before the Supreme Court was whether the Court of Appeal erred in finding Dow liable to Ms Hollis for failing to issue an adequate warning of the risk of a post-surgical implant rupture. On the question of the role of the “learned intermediary”, La Forest J who delivered the judgment of the majority (and with whom on this point the minority agreed) said at [27] that, in general, the duty to warn is owed directly by the manufacturer to the consumer although in exceptional circumstances it may be satisfied by warning a “learned intermediary”. He referred to its roots in the US case law and its application to highly technical products and to circumstances where the consumer primarily relies on the judgment of the intermediary. He continued at [28]–[29]:
In such cases, a warning to the ultimate consumer may not be necessary and the manufacturer may satisfy its duty to warn the ultimate consumer by warning the learned intermediary of the risks inherent in the use of the product …
The rule operates to discharge the manufacturer’s duty not to the learned intermediary, but to the ultimate consumer, who has a right to full and current information about any risks inherent in the ordinary use of the product. Thus, the rule presumes that the intermediary is “learned”, that is to say, fully apprised of the risks associated with the use of the product. Accordingly, the manufacturer can only be said to have discharged its duty to the consumer when the intermediary’s knowledge approximates that of the manufacturer. To allow manufacturers to claim the benefit of the rule where they have not fully warned the physician would undermine the policy rationale for the duty to warn, which is to ensure that the consumer is fully informed of all risks. Since the manufacturer is in the best position to know the risks attendant upon the use of its product and is also in the best position to ensure that the product is safe for normal use, the primary duty to give a clear, complete, and current warning must fall on its shoulders.
(Emphasis added)
3651 In Hollis the Court held that the duty was satisfied by warning the implanting surgeon, noting that neither the implant nor its packaging was placed in the hands of the ultimate consumer and that Dow, in contrast to the respondents in the present case, had never communicated its warnings directly to patients. In these circumstances, La Forest J concluded at [31], a manufacturer in Dow’s position could discharge its duty to the ultimate consumer “by giving the treating surgeon clear, complete and current information concerning any general and specific risks that arise from the ordinary use of the product”.
3652 The way in which information is to be conveyed will obviously be affected by the characteristics of the audience or readership. Indeed, the Global Harmonization Task Force recommended in 2005 guidelines entitled Labelling for Medical Devices that “instructions for use should be written in terms readily understood by the intended user and, where appropriate, supplemented with drawings and diagrams”.3031 But I do not accept the respondents’ contention that the standard of care expected of the manufacturer, or the supplier for that matter, is affected by the fact that the devices are supplied through surgeons or following consultation with surgeons.
3653 The general principles for determining whether a defendant was negligent were set out by Mason J in Wyong Shire Council v Shirt (1980) 146 CLR 40 at 47−48:
In deciding whether there has been a breach of the duty of care the tribunal of fact must first ask itself whether a reasonable man in the defendant’s position would have foreseen that his conduct involved a risk of injury to the plaintiff or to a class of persons including the plaintiff. If the answer be in the affirmative, it is then for the tribunal of fact to determine what a reasonable man would do by way of response to the risk. The perception of the reasonable man’s response calls for a consideration of the magnitude of the risk and the degree of the probability of its occurrence, along with the expense, difficulty and inconvenience of taking alleviating action and any other conflicting responsibilities which the defendant may have. It is only when these matters are balanced out that the tribunal of fact can confidently assert what is the standard of response to be ascribed to the reasonable man placed in the defendant’s position.
3654 In Peterson at [800] Jessup J said that, although Wyong Shire Council v Shirt was not a product liability case, these principles were of universal application. Subject to any statutory modifications, I respectfully agree. I also agree with his Honour’s comment at [801] that it is “a distraction to attempt to equate the position of the respondents with that of a surgeon advising a patient about the risks involved in undertaking a particular procedure”.
3655 Adapting the remarks of Jessup J to the facts of the present case, the respondents were not in a one-on-one relationship with the consumers of the Ethicon devices. They were manufacturers and a supplier of mass-produced products, packaged and labelled by them without discrimination as between those consumers. Any decisions they made about supplying the devices, or about providing a warning, would have had to be made generally, although a suitably crafted warning might well have discriminated between them. They did not pretend to understand the circumstances of particular patients. To the contrary, by relying on the “learned intermediary” concept, they implicitly disclaimed any such understanding.
3656 A manufacturer breaches its duty of care if, by exercising reasonable care, it should have foreseen and avoided the consumer’s loss: Dovuro at [30] (McHugh J). To determine whether or not the duty has been breached it is necessary to consider what a reasonable person in the position of the respondents would have done if confronted by a foreseeable risk. The inquiry is a prospective one. It cannot be confined by the circumstances in which the applicants were injured: Vairy v Wyong Shire Council (2005) 223 CLR 422 at [125]–[129] (Hayne J); Adeels Palace Pty Ltd v Moubarak (2009) 239 CLR 420 at [31] (French CJ, Gummow, Hayne, Heydon and Crennan JJ).
3657 While there are slight differences in the wording of the various statutes in force in the different state and territory jurisdictions, none of the respondents will be liable for any harm caused by a failure to take precautions against a risk of harm unless:
(1) the risk is foreseeable in the sense that it is a risk of which it knew or ought to have known;
(2) the risk was not insignificant; and
(3) in the circumstances, a reasonable person in its position would have taken those precautions.
See, for example: CLA (WA), s 5B(1); Wrongs Act (Vic), s 48(1); and Civil Liability Act 2002 (NSW), s 5B(1) (CLA (NSW)).
3658 These provisions were enacted as part of a suite of legislative reforms across the country following the publication of the report produced in September 2002 by the Panel of Eminent Persons chaired by the Hon David Ipp AO QC, which undertook a comprehensive review of the law of negligence and which is commonly referred to as “the Ipp Report”.
3659 Whether or not a risk is “not insignificant” for this purpose turns on the prospect, not the severity of the harm: Garzo v Liverpool/Campbelltown Christian School [2011] NSWSC 292 (Garling J) at [104]–[105]. According to the Ipp Report at [7.15], the phrase “not insignificant” was not intended to be a synonym for “significant”. Rather, it was intended to indicate “a risk that is of a higher probability than is indicated by the phrase ‘not far-fetched or fanciful’ but not so high as might be indicated by a phrase such as ‘a substantial risk’”. The Panel explained that the selection of the double negative was a deliberate one. They considered that “significant” was apt to indicate a higher degree of probability than they intended.
3660 The risk addressed by these sections is the risk that materialised in the particular case: Garzo v Liverpool/Campbelltown Christian School [2012] NSWCA 151 at [7] (Basten JA), at [22] (Meagher JA); Southern Colour (Vic) Pty Ltd v Parr [2017] VSCA 301 (Santamaria, Ashley and Kaye JJA) at [55]; Coles Supermarkets Australia Pty Ltd v Bridge [2018] NSWCA 183 at [20]–[22] (Leeming and Payne JJA). “[A]n unduly narrow formulation of risk”, such as one which “obscures the true source of potential injury” or which “too narrowly focusses on the particular hazard which caused the injury” or which “fails to capture part of the plaintiff’s case” is to be avoided: Southern Colour at [55]; Coles Supermarkets at [22].
3661 In determining whether a reasonable person would have taken precautions against a risk of harm, a court must take into account, amongst other relevant things:
(1) the probability that the harm would occur if care were not taken;
(2) the likely seriousness of the harm;
(3) the burden of taking precautions to avoid the risk of harm;
(4) the social utility of the activity that creates the risk.
See: CLA (WA), s 5B(2); Wrongs Act (Vic), s 48(2); CLA (NSW), s 5B(2); Civil Liability Act 2004 (Qld), s 9(2); Civil Liability Act 1936 (SA), s 32(2); Civil Liability Act 2002 (Tas), s 11(2); Civil Law (Wrongs) Act 2002 (ACT), s 43(2).
3662 Despite differences in the language between the statutory provisions and the common law principles as expounded in Wyong Shire Council v Shirt, it has been held that s 5B(2) of the CLA (NSW), which is similar, and in some cases identical, to the provisions in the Acts listed above, substantially restates those principles: Waverley Council v Ferreira [2005] NSWCA 418; (2005) Aust Torts Reports ¶81–818 at [45] (Ipp JA, with whom Spigelman CJ and Tobias J agreed); Council of the City of Greater Taree v Wells [2010] NSWCA 147; (2010) Aust Torts Reports ¶82-063; (2010) 174 LGERA 208 at [55] (Beazley JA, with whom McColl and Basten JJA agreed). Further, in Waverley Council v Ferreira at [27] Ipp JA said the common law requirement to identify what a reasonable person in the defendant’s position would do in response to a reasonably foreseeably risk (for which proposition his Honour cited Graham Barclay Oysters (HC)), was “consistent with” s 5B(1) of the CLA (NSW). Drawing on what was said at [7.5]–[7.8] of the Ipp Report, his Honour explained at [47] that the rationale for the enactment of s 5B(2) was to ensure “that courts would focus more directly on the issue ‘whether it would be reasonable to require precautions to be taken against a particular risk’” and “avoid conflation of the concept of foreseeability of risk with the conclusion that a reasonable person would have taken precautions against it”. At [51] he observed that “s 5B(2) provides a framework for deciding what precautions the reasonable person would have taken to avoid the harm and involves weighing the factors set out in ss 5B(2)(a) and (b) against those in ss 5B(2)(c) and (d) (subject, of course, to each being applicable in the particular circumstances of the case)”.
3663 At all relevant times, for the purpose of determining whether the risk was “not insignificant”, subs 48(3) of the Wrongs Act (Vic) provided that:
(a) insignificant risks include but are not limited to risks that are far-fetched or fanciful; and
(b) risks that are not insignificant are all risks other than insignificant risks and include, but are not limited to, significant risks.
3664 Section 49 of the Wrongs Act (Vic) also provided:
In a proceeding relating to liability for negligence—
(a) the burden of taking precautions to avoid a risk of harm includes the burden of taking precautions to avoid similar risks of harm for which the person may be responsible; and
(b) the fact that a risk of harm could have been avoided by doing something in a different way does not of itself give rise to or affect liability for the way in which the thing was done; and
(c) the subsequent taking of action that would (had the action been taken earlier) have avoided a risk of harm does not of itself give rise to or affect liability in respect of the risk and does not of itself constitute an admission of liability in connection with the risk.
3665 I note that s 50 of the Wrongs Act (Vic) states, in effect, that where a defendant has a duty to give a warning or other information to a plaintiff in respect of a risk or other matter, the duty is discharged if the defendant takes reasonable care in giving the warning or other information.
3666 The CLA (WA) does not contain a provision like s 49 of the Wrongs Act (Vic), but s 5C of the CLA (NSW) is in identical terms. As the applicants observed, however, this provision reflects the position at common law: Vairy v Wyong Shire Council at [7] (Gleeson CJ and Kirby J); Derrick v Cheung [2001] HCA 48; (2000) 181 ALR 301; (2000) 33 MVR 393 at [13] (Gleeson CJ, Gaudron, Kirby, Hayne and Callinan JJ); Mobbs v Kain [2009] NSWCA 301; (2009) Aust Torts Reports ¶82–037; (2009) 54 MVR 179 at [92] (McColl JA, with whom Giles and MacFarlan JJA agreed).
3667 As McHugh J explained in Dovuro at [38] (Gummow and Heydon JJ agreeing at [62] and [177] respectively):
A defendant is not negligent merely because it fails to take an alternative course of conduct that would have eliminated the risk of damage. The plaintiff must show that the defendant was not acting reasonably in failing to take that course. If inaction is a course reasonably open to the defendant, the plaintiff fails to prove negligence even if there were alternatives open to the defendant that would have eliminated the risk.
3668 Thus, the high level questions here are:
(1) Would a reasonable manufacturer and/or supplier in the respondents’ position have foreseen that its conduct involved the alleged risks of injury to the applicants or to a class of persons including the applicants?
(2) Would the exercise of reasonable care on the part of a manufacturer and/or supplier in the respondents’ position have involved taking the precautions the applicants allege were not taken in this case?
(3) Did the respondents fail to take those precautions?
(4) If so, was their conduct unreasonable?
3669 In the present case the first question only arises in relation to one particular aspect of the failure to warn case, because in all other respects the respondents admitted that they knew of the relevant risks. As the applicants submitted, there is no question that the risks were not insignificant. Indeed, the respondents conceded they were significant. Furthermore, for the most part the consequences to class members if those risks materialised were potentially grave. The dispute concerns the precautions that should have been taken, whether they were not in fact taken, and if so, whether the respondents acted unreasonably in failing to take them.
3670 A manufacturer’s duty of care to a consumer is not discharged merely because it complied with the relevant regulatory requirements: Peterson at [792]–[795]; approved by the Full Court in Merck at [162]. Moreover, while the actions and inactions of the regulatory authorities and the views expressed by the professional associations may be relevant to the reasonableness of the respondents’ conduct, they are not decisive. As the Court of Appeal of the Supreme Court of Victoria observed in analogous circumstances in Thompson v Johnson and Johnson at 494:
[T]o accept that proposition would permit the respondents to abrogate the duty of reasonable care owed by them. It is not the response of such a body which determines whether a person in the position of the respondents is or is not negligent. That is for the courts to decide. However, it is a relevant fact to be taken into account when determining whether reasonable care has been exercised.
3671 Much will depend on the circumstances of the case. In the present case, the products in question received CE marking on the basis of Ethicon’s representations. The evidence of the regulatory experts that the respondents did not conform to the regulatory requirements and standards diminishes the significance of regulatory clearance. Furthermore, since it is reasonable to assume that a prudent manufacturer would comply with the regulatory requirements and standards before obtaining CE marking or offering the devices for sale, evidence of non-compliance is indicative of a failure to take reasonable care for the safety of the applicants and group members.
Did the respondents breach their duty of care to the applicants?
3672 The correct approach, then, is to proceed on the basis that each of the respondents owed each applicant a duty to take reasonable care, identify the reasonably foreseeable risk, consider what was a reasonable response to that risk, and then decide whether the respondents failed to respond in that way.
Was the risk of injury foreseeable?
3673 The relevant risk for this purpose is the risk of injury of the same kind as that which the applicants sustained, not the risk of the particular damage or its extent: Chapman v Hearse (1961) 106 CLR 112 at 120–121; Commonwealth v McLean (1996) 41 NSWLR 389 at 403 (Handley and Beazley JJA).
3674 Foreseeability is well established. At all relevant times the respondents either knew or should have known of the risk of injury of the kind the applicants sustained.
What was a reasonable response to the risk?
3675 The evidence established that implantation of all the Ethicon devices could cause serious injury and that most of the pleaded complications are not rare. Indeed, many are common or at least not uncommon.
3676 As Jessup J observed in Peterson at [803], the object of a reasonable person confronted with the existence or prospect of a risk of which he or she knew or ought to have known would naturally be “to eliminate or to minimise the scope for the risk coming home, or at least to warn the consumer, or to provide information or advice in the nature of a warning, so that he or she might weigh the size and nature of the risk, or the prospect of a risk, against the benefit of [using the product], and make his or her own decision”.
3677 The allegations as pressed were that the reasonable response to the risk in the present case was to conduct a proper evaluation of the safety and efficacy of the devices both before and after they were taken to market and if, following such evaluations the respondents were justified in selling them, to inform consumers of the various risks which could arise with implantation of the devices and the limitations of the available data. There was no dispute that such evaluations should have been undertaken; the question was whether the applicants had proved that they had not been. There was, however, a dispute about the extent of the information the respondents were obliged to provide.
3678 Inconvenience might well have been a factor in the respondents’ response to the risks of the Ethicon devices, but no cogent evidence was adduced to suggest that the costs of taking any of the precautions the applicants alleged should have been taken to reduce the risks were prohibitive or outweighed the interests of patient safety or that there was any difficulty in conducting the appropriate evaluations. Nor was it suggested that the respondents had any conflicting responsibilities.
3679 Ethicon did redesign a number of the devices. TVT-O, TVT Secur, TVT Abbrevo, Prolift+M and Prosima were all redesigns of existing devices. At least one of the reasons for doing so was to reduce risks posed by earlier products. But the devices were not withdrawn from the market when the new and purportedly improved versions were made available for sale and not all known or knowable risks were identified or the subject of warnings (or suitable warnings) sufficient to minimise those risks.
First alleged breach: inadequate pre-market evaluation
Overview of the arguments and some general findings
3680 The applicants’ argued that, for a variety of reasons set out in detail below, the pre-market evaluations of the Ethicon devices evinced a want of reasonable care for the safety of the women for whose benefit they were intended and promoted. In particular, they claimed that the pre-market evaluations were deficient because:
(1) the respondents “prioritised” commercial considerations over safety;3032
(2) their risk analyses and design validation studies were wanting or “poorly considered”;3033
(3) they failed to consider treatment options for potential complications of which they were aware, such as chronic pain;3034
(4) their engagement with the regulatory requirements for clinical evaluation, especially their approach to clinical evaluation reports, was perfunctory;3035 and
(5) they did not satisfy the European Directive in numerous respects.3036
3681 In substance I accept these arguments. I explain why below. Before doing so, however, I need to address the respondents’ arguments.
3682 The respondents argued that the applicants had failed to make out their case because the evidence indicated that Ethicon had conducted extensive studies, including long-term in vivo studies on Prolene sutures.3037 They also submitted that the applicants’ case should fail because they did not adduce evidence from a regulator that registration would have been withheld had the respondents disclosed certain facts about the testing process conducted for each of the devices.3038
3683 Both these arguments must be rejected.
3684 First, this case is not concerned with the effects of implantation of Prolene sutures. It is concerned with the effects of Prolene mesh and Prolene Soft mesh. For the reasons given above in Part V, it is largely beside the point that Ethicon conducted long-term studies on the effects of implantation of Prolene sutures. Many of the complications that arise with the implantation of Prolene and Prolene Soft arise from the structure of the mesh — the distribution of the pores, the effective pores size under load and after tissue integration, and the pore geometry. Other factors, such as the quantity of the material used in the meshes, are also relevant.
3685 Similarly, Ethicon’s work on hernia meshes is of limited utility. That work was conducted in the context of meshes designed to reinforce the abdominal wall. The evidence did not suggest that any attention had been given in that work to implanting meshes in the female pelvis where both the environment and the mechanical forces are different.
3686 As Ms Holland said, while it was appropriate for Ethicon to incorporate the extensive knowledge it had gained from its use of Prolene sutures and hernia mesh when designing and developing the Ethicon devices, it was not appropriate to rely on existing data without conducting a biological re-evaluation when the new device has a different manufacturing process, a different manufacturing location, different design and a different intended use. That follows from the basic principles of materials science discussed in Part V above.
3687 Second, the applicants’ case did not require evidence from the regulator. The uncontested evidence about the regulatory system from several experts in the field demonstrated that the scheme governing medical devices was largely self-regulating. The applicants are entitled to succeed if they prove on the balance of probabilities that the evidence upon which Ethicon relied to support CE marking was insufficient to justify it.
3688 It will be recalled that medical devices may not be sold in Australia unless they are included on the ARTG. In the present case, the respondents relied on CE marking to sell the Ethicon devices in Australia. As I observed earlier, under the terms of the relevant European Directive it is the manufacturer of a device who must determine if the available clinical data are sufficient to justify the application of a CE mark or if clinical investigations are necessary before the mark is applied. In order to apply the CE mark to a device, the manufacturer is obliged to maintain a technical file consisting of information sufficient to demonstrate that the device complies with the European Directive, including the essential requirements. The manufacturer must also have a clinical evidence or evaluation report and to actively update it with data obtained from post-market surveillance. In the case of medical devices like the Ethicon devices, the manufacturer is required to conduct clinical investigations unless taking the literature route is duly justified. The clinical evidence report must contain a comprehensive literature review and particulars of the outcomes of clinical studies that provide a reasoned, clinically-valid basis to confirm that the benefits of the device exceed the risks. The comprehensive literature review must include a critical evaluation of the relevant scientific literature or the results of all clinical investigations or both.
3689 No reasonably prudent manufacturer, who elected to take the literature route, would affix a CE mark to a device without conducting such a review, without undertaking a thorough risk analysis, and without providing a reasoned, clinically-valid basis to confirm that the benefits of the device exceed the risks. A reasonably prudent manufacturer of medical devices would adhere to the regulatory requirements both in letter and in spirit. A reasonably prudent manufacturer of medical devices would also follow the regulatory guidelines. Where, on a proper analysis of the manufacturer’s responsibilities, clinical investigations (trials) are required, then a reasonable manufacturer in the position of the respondents would undertake those investigations and would not affix the CE mark, let alone take the device to market, before the results of the trials are known and analysed. In the case of a device intended for permanent implantation, the trials should be of sufficient duration to take into account late onset complications.
3690 The respondents also argued that the applicants could only succeed on this claim if they established that withholding or withdrawing the devices from the market was the only course reasonably open to the respondents, citing Peterson at [810]. That argument must also be rejected.
3691 In Peterson at [809]–[810] Jessup J said that the “kernel” of the applicant’s case was that Merck should not have made rofecoxib available for incorporation into Vioxx until the completion of any relevant research investigations, clinical trial or observational studies. Rofecoxib, I should explain, was a non-steroidal anti-inflammatory drug developed by Merck’s scientists to be used in the treatment of arthritis and was the active ingredient of Vioxx. His Honour accepted that Merck ought to have recognised that it might ultimately be found that the consumption of Vioxx caused or contributed to various cardiovascular events, fully described at [58] of his reasons. His Honour went on to say, however, that “[i]n the state of the science as it existed until September 2004” (when Vioxx was withdrawn from the market because of the occurrence of adverse cardiovascular events in a major clinical trial), he was “not persuaded that the only course reasonably open to the respondents was to have withdrawn Vioxx from the market altogether; or that the only course reasonably open to Merck was to have made rofecoxib unavailable to MSDA [Merck Sharpe & Dohme Australia Pty Ltd]”. MSDA, I interpolate, was a subsidiary of Merck and the manufacturer, distributor and seller of Vioxx in Australia. His Honour proceeded to explain why:
Rofecoxib was not a drug which, as a direct and inevitable consequence, caused CVT events in all cases. Indeed, the occurrence of CVT events in the Vioxx arm of the VIGOR trial (including those which presumably were not caused or contributed to by Vioxx) was only 1.67 such events for every 100 patient-years. As against this, I must recognise that Vioxx was a drug which worked a lot of good: for many people (including the applicant) it brought relief from the pain and discomfort associated with arthritis without gastrointestinal side-effects. As a prescription drug, it required the judgment of a medical practitioner before any person could consume it. For rofecoxib to have been withdrawn upon nothing more than the signal of potential risk generated by VIGOR would have been to deny a great many arthritis sufferers a means of securing comfort which they may freely have chosen, even if fully informed. It is notorious that many drugs carry side-effects, yet resort is had to them as the result of the balancing by patients of the risks and benefits to be derived, informed and assisted by the professional judgments of their medical practitioners.
3692 Consequently, his Honour held at [811]–[812] that in the circumstances as they existed until September 2004, Merck’s duty of care as manufacturer and supplier of rofecoxib could be (and was) discharged by the provision to MSDA of information necessary to enable MSDA to formulate and disseminate adequate instructions and warnings about the possibility of the drug causing or contributing to the cardiovascular events.
3693 With respect, his Honour may have put the test too high. The question is not whether withholding or withdrawing the product was the only course reasonably open to the manufacturer, unless of course this was the way Peterson was argued. The question here at least is whether it was unreasonable for the respondents to release the devices for sale in Australia without having undertaken the steps the applicants alleged they ought to have taken. I accept, however, that the answer might be no different. If the devices could confer substantial benefits for a large number of women and the benefits outweighed the risks, then it might not have been unreasonable to release them for sale, provided that the respondents disclosed the known and foreseeable risks and such other information of which the respondents were or should have been aware that would have enabled the women for whom the products were designed and marketed to make an informed decision about whether to receive them. In that event, however, unless they were accompanied by such information, it would have been unreasonable for the respondents to release them for sale.
3694 The respondents submitted that, for four reasons, evidence of non-compliance with specific regulatory requirements alone could not be determinative of whether the respondents discharged their duty of care.3039
3695 The first reason was that the applicants did not plead “that the regulatory environment informs the respondents’ obligations”, and if so, how non-compliance bears on the pleaded allegations of breach.
3696 The second was that some of the regulatory requirements are subjective, requiring an element of judgment from the manufacturer. The respondents argued that the evidence of regulatory experts of “their subjective view of what the regulations require” and of whether Ethicon’s processes met those requirements was of no assistance, citing Caltex Refineries (Qld) Pty Limited v Stavar (2009) 75 NSWLR 649 at [44] and [123] per Allsop P but without explaining its significance.
3697 The third was that any identified non-compliance is irrelevant.
3698 The fourth was that “the regulator’s oversight of the goods is of some moment, because what was done is a matter of record, as is the CE marking and inclusion on the ARTG of each of the implants”.
3699 I accept that evidence of non-compliance with some regulatory requirements may be irrelevant. I also accept that evidence of non-compliance with regulatory requirements alone may not be determinative. But I do not consider that the alleged deficiency in the pleading is of any consequence. No objection was taken to the evidence on that account and the respondents had ample opportunity to meet the applicants’ case. Neither do I accept that the opinion evidence of regulatory experts is of no assistance.
3700 I am unable to see the relevance of the remarks of Allsop P in Stavar. The evidence of the regulatory experts in the present case was undoubtedly of assistance.
3701 Moreover, the instances of non-compliance with regulatory requirements are not isolated and they are far from irrelevant.
3702 The difficulty with the respondents’ reliance on CE marking is that it was Ethicon, not the regulator, who affixed the mark and it was CE marking which led to the inclusion of the devices on the ARTG.
3703 The expert evidence canvassed the precautions that are required of a reasonable manufacturer before CE certification is permissible.
3704 Dr Allman said that the manufacturers must follow and comply with the terms of the European Directive and its revisions.3040 He said that doing so might well exceed what would be necessary for a notified body to issue a CE certificate, especially for class II medical devices where the notified body verifies compliance of individual devices on a sampling basis only.3041 While the so-called literature route is an option in an appropriate case, if a new device is a design modification to an existing device intended to change or improve clinical performance, then a clinical investigation of the new device is necessary.
3705 Ms Holland, a senior quality assurance specialist, explained the obligations of manufacturers with respect to managing risks. She said that manufacturers are required to identify risks and they have a duty to mitigate every one of those risks as far as is reasonable practicable. That means reconsidering the design of the product so as to eliminate any potential risks “to the fullest extent feasible” and “if risk mitigation cannot occur through product design, a manufacturer must attempt to minimise the risk by incorporating protective measures”. She said that warnings and training are the least effective means of minimising risk and should only be used as a last option.3042 Where changes are made to a product, she said that each change should be reviewed to determine if the change could affect the risk assessment for the product. She said that, used properly, “the risk management process will create and maintain a robust product design as it will help ensure that the product that is on the market is safe, performs as intended, and that known or knowable risks will be identified, and warned about or mitigated”.3043
3706 As I explained in Part VIII, Ethicon had no cohesive risk management system and its design validation process was flawed. It failed to conform to the essential requirements. Amongst other things, it failed to address all known hazards. It failed to eliminate or reduce risks as far as possible. And it failed to inform users of all residual risks.
3707 Ms Holland noted that dyspareunia was not addressed in the risk management documents for any of the SUI devices and that revision surgery was only mentioned in the context of the prospect that the mesh might need to be removed in the event of subsequent infection.3044 The difficulty of removing the mesh was not mentioned at all in risk management documents.3045 Alone of the POP devices, only pre-market evaluation documents on Prosima mentioned risks associated with secondary surgery and then only in the context of “loss of balloon functionality”.3046
3708 Ms Holland’s uncontradicted evidence was that Ethicon’s risk management process for the SUI devices was focussed on establishing evidence that risk management activities had been undertaken and was intended to meet its interpretation of the regulations rather than to prevent harm to women. She concluded that the process was not effective in identifying, evaluating, or warning of the risks.3047Although Ms Holland noted that the process became “more mature” over time,3048 based on her examination of the Ethicon files, she described the risk management system as “broken”.3049 She formed the opinion that:
Ethicon’s company culture allowed a minimalistic approach to be taken to risk management activities and did not require that actions were actually taken to minimise risks to the greatest degree “practicable”. My review did not see any redesign or mitigation actions resulting from the risk management process for tape or mesh products. In my experience this practice is not acceptable nor is it industry practice since many of the management teams with which I have worked appear to be more risk adverse and their performance is often based upon field problems related to products for which they are responsible.3050
3709 As Ms Holland noted, internal audits of Ethicon’s risk management process conducted in 2010 found that the risk assessment process did not follow the procedure set out in its risk management plan; there was a lack of the required additional approvers for high risk items; a lack of consistency in applying severity rankings to harms, and a lack of training for those performing risk management activities; MAUDE and other databases for safety-related risks were not analysed for safety-related risks; and foreseeable hazards were not addressed under both normal and “fault” conditions.3051
3710 The pre-market clinical evaluation reports for each device were largely perfunctory. None included a comprehensive literature review or a truly critical evaluation of the relevant scientific literature or of the results of clinical investigations. Insufficient attention was given to the essential requirements. The literature route was invariably taken as the quicker and easier option when it was unjustified. Further, Dr Allman’s evidence, which I accept, was that:
The conclusions of CERs used to justify initial CE marking of the Implants relied on equivalence to other devices rather than clinical investigation of the new device. Equivalence was not justified. The Implants were either innovative devices or design changes to an existing device which was innovative. Design changes were intended to impact clinical performance. The CERs did not provide logical justification for conclusions regarding safety and performance, and for claims of compliance to the essential requirements. 3052
3711 Even if the manufacturer could have satisfied the regulatory requirements by relying on the scientific literature, the decisions to take the Ethicon devices to market without clinical trials designed to test their performance and safety in the user population for which they were intended or to do so before the results of clinical trials were known, let alone evaluated, showed a want of reasonable care for the safety of the women for whom they were indicated.
3712 Ms Holland’s evidence, which I also accept, was that:
[P]reclinical testing should have been performed on both the tape and mesh prior to marketing the devices for a new indication for use. Regardless of the regulatory requirements to place devices on the market, reliance on the prior history of similar devices for safety and effectiveness is necessary, but not sufficient. Use of mesh in a pelvic floor repair versus the abdominal wall (hernia) should not be assumed to respond in similar fashion. What was not considered in these risk assessments was the impact of polypropylene mesh on anatomical location with respect to differing biomechanical properties (erosion/adhesion risk), microbiological flora (infection risk), as well as inflammatory response. 3053
3713 Nearly seven years after TVT was first supplied in Australia and nearly three years after the Australian launch of Gynemesh PS, Davila et al (2006) observed in their article following the 2005 IUGA Grafts Roundtable:
It is clear that a significant amount of work is necessary in the preclinical testing of materials. This includes biocompatibility studies as well as identification of animal models for fascial repair. It is recognized that more site-specific tests be (sic) developed to determine the safety and efficacy of graft materials. This is particularly important in translational research as related to the similarities and differences in the pathophysiology and treatment of abdominal wall hernias as related to genital prolapse. Because many animal studies utilize the abdominal wall hernia model to evaluate a specific graft material, their value in predicting usefulness in vaginal prolapse repair is unclear. In the interim, animal models for vaginal prolapse and correction of such should be searched for and identified. … An important aspect of graft utilization should also be the identification of those situations where grafts should not be used. The specific clinical scenarios are currently not well-recognized and are open for discussion.3054
(Original emphasis)
3714 Even later, in an internal Ethicon draft report on biomechanical considerations for pelvic floor design, written in February 2011, the authors (identified as Jurgen Trzewik and Christophe Vailhe) referred to the importance of animal models in pelvic floor research. At the same time, however, they remarked that it was “of course” necessary to know what is happening in the human female. They candidly acknowledged that “[t]he development of knowledge to understand the mechanics of pelvic floor disorders is imperative; yet, we are only just beginning to determine the necessary criteria on which to base design for pelvic floor implants”.3055
3715 These observations, made 14 years after the release for sale of TVT, 11 years after the release of Gynemesh PS and six years after the launch of Prolift, are a damning indictment of the approach taken by the respondents to the devices in question.
The SUI devices
3716 The applicants argued that Ethicon’s pre-market evaluation of the SUI devices was wanting in the following respects.
3717 First, the design, development and pre-market evaluation of the SUI devices was undertaken without Ethicon taking “any substantive action” to assess their risk/benefit profile.
3718 The applicants submitted that Ethicon knew or should have known that it could not assume that a mesh used for hernia repair could be rebadged for use in the pelvic floor. They further submitted that Ethicon knew or should have known that the results obtained in a study of the surgical treatment of a small cohort of carefully selected patients by a group of highly skilled surgeons would not be comparable to the results achieved by less expert and less experienced clinicians in relation to a broader patient population. They also argued that Ethicon’s pre-market evaluation of each of the devices was deficient in that it did not undertake targeted in vivo or in vitro studies to determine the impact of using Prolene mesh transvaginally or carry out a single clinical investigation into the question of how the complications it knew were associated with the devices could be treated.3056
3719 Second, Ethicon prioritised commercial considerations over patient safety.3057
3720 Third, the SUI devices were launched on the market before their safety and efficacy had been established, particularly in the long-term, at best relying on small case series including those conducted by the inventor and developer of TVT, Professor Ulmsten.
3721 Even the respondents’ expert witnesses acknowledged that this situation obtained until 2002 when the initial results of the Ward Hilton randomised controlled trial were published. Yet, the information Ethicon supplied to the public about the devices, in particular in the IFUs, did not refer to the absence of robust evidence or long-term evidence of safety and efficacy.3058
3722 Fourth, the clinical evaluations of all the SUI devices failed to conform to the regulatory requirements which would entitle a manufacturer to affix the CE mark to a medical device.3059 Quite apart from the evidence of Dr Allman to this effect, the applicants submitted that the respondents’ failure to adequately warn of the potential risks associated with the use of the devices “in and of itself” did not conform to the essential requirements under the European Directive. In the result, the SUI devices should never have received a CE marking and consequently should never have been entered onto the ARTG.3060
3723 The respondents, on the other hand, maintained that all the devices had been subject to extensive testing. They repeatedly referred to the statement in the Ford et al (2015) Cochrane review that “mid-urethral sling operations have been the most extensively researched surgical treatment for stress urinary incontinence in women and have a good safety profile” and a statement attributed to Dr Blaivas in cross-examination that TVT is the most well studied operation.3061 They also relied on the CERs and Dr Hinoul’s summaries of them.
3724 Quite apart from conflating the first two negligence claims, this aspect of the respondents’ submissions failed to grapple with the applicants’ arguments or the evidence marshalled in support of them.
3725 The applicants’ submissions on the inadequacy of the pre-market evaluation of the Ethicon devices should be accepted. They fairly capture the effect of the evidence, which I discussed in greater detail in Part VIII and which I summarise below.
TVT
3726 In October 1999, when TVT was first supplied in Australia, Ethicon had undertaken no pre-market evaluation and there were no studies reporting long-term results in the user population. The clinical evaluation report prepared for Medscand was a woefully inadequate basis upon which to rely. Dr Hinoul conceded as much. As I noted in Part VI, the Nordic studies involved a select population group, a circumstance Dr Eriksson, the author of the report, neglected to mention and, I infer, take into account. Before TVT was cleared for sale, Ethicon undertook no clinical investigations of its own to assess its safety or efficacy across a wider population although it was indicated and marketed as suitable for most women with stress urinary incontinence. In the circumstances, I accept Dr Allman’s opinion that, given the paucity of clinical data to support CE marking of the device, before applying the CE mark Ethicon should have conducted clinical studies of its own with clearly defined acceptance criteria. 3062 In my opinion, its failure to do so demonstrated a want of reasonable care. A reasonably prudent manufacturer would not have acted in this way.
3727 Further, based on Ms Holland’s uncontradicted evidence, I find that before TVT was put on the market: there was no overarching, cohesive risk management system in place; the necessary design documentation had not been created; no design validation had been conducted; and Ethicon had not verified that TVT met the needs of users in its anticipated use environment.
3728 On the basis of Ms Holland’s evidence, I also find that the only risk analysis undertaken before TVT was launched was recorded in an application Failure Mode and Effect Analysis and that Ethicon did not undertake any Device Design Safety Assessment, as it should have, before the device was placed on the market.3063 The only aFMEA in the TVT design history file was dated 24 June 1999.3064 This was the fifth revision of the original which was prepared on 22 September 1998. It only examined ways in which steps required by the procedure would fail. Every single identified risk was declared acceptable. The document offered no explanation as to why this was so and the respondents called no evidence about it. Nor did they offer any justification for the lack of any other form of risk analysis or for the absence of a DDSA.
3729 Many of the failure modes itemised in the aFMEA, such as penetration of the bladder, urethral wall, blood vessels and nerves, were not mentioned in the Medscand IFU although they were included in the 8 September 2000 IFU. Most of the pleaded complications, however, were not captured, and therefore not assessed, by the aFMEA. While “too hard tension” was identified as a failure mode and its effects included urinary retention, urinary retention was not listed in the IFU as an adverse event until 2015 and, although the 8 September 2000 IFU warned that excessive tension applied to the tape could cause lower urinary tract obstruction, no such warning appeared before then.
TVT-O
3730 TVT-O was taken to market with unseemly haste. Although Dr Hinoul denied it, there was abundant evidence that the product was rushed to market. I accept that at the time the product was launched the respondents believed TVT-O to be safe. Having regard to the limited extent of the investigations that had been carried out into transobturator devices in general and the inside-out approach in particular, this was little more than a leap of faith. It is clear from the evidence that the development of TVT-O was driven by concerns about loss of income due to pressure from competitors and that matters of safety were subsidiary considerations which yielded to commercial imperatives.
3731 Dr Arnaud recognised that the safety of TVT-O needed to be tested in the hands of surgeons other than Professor de Leval, yet Ethicon was content to launch the device without that experience. This was not only unwise, it was also imprudent. Moreover, the existing studies were of such short duration that they offered no insight into the potential for long-term complications. As I observed earlier, the CER upon which Ethicon relied to launch the device did not comply with the requirements of the European Directive in several respects, including by failing to undertake a critical evaluation of the scientific literature. It also failed to acknowledge the shortcomings of the de Leval studies. To say the least, the assertion made in the CER that there was “clear clinical evidence” that the outside-in transobturator approach was a safe and effective surgical technique was an overstatement. I accept Dr Allman’s opinion that the reasons given in the CER to support the assertion and the conclusion that no additional studies were presently required were insufficient to support CE marking.3065
3732 In my opinion a reasonably prudent manufacturer would have carried out a critical evaluation of the scientific literature and undertaken additional studies to evaluate the risks and benefits of the device in the hands of a wide cohort of users on a wide group or groups of patients over such a period of time as would have provided some insight into the nature and extent of the potential long term risks. Ethicon’s failure to do so evinced a lack of reasonable care. It did not conform to the regulatory requirement for clinical data to confirm conformity with the essential requirements under normal conditions of use.
3733 Writing in 2005, a year after TVT-O was first supplied in Australia, Ward and Hilton observed that the TVT procedure had been extensively investigated but “among the wealth of literature there [were] few randomised trials or large cohort studies”. They acknowledged many of the existing studies were of poor quality with small patient numbers and non-standard outcome measures. They added:
Evidence of the safety and efficacy of the newer procedures, such as transobturator tapes, is gradually emerging, but not before the widespread adoption of these techniques for first-line treatment of stress incontinence.3066
3734 Ethicon did consider whether to include in the IFU for TVT-O a warning about the risk of leg pain, said to be transient, suffered by 15 to 25% of patients in Professor de Leval’s study. This was apparent on the face of internal Ethicon email correspondence.3067 As noted above, in an email dated 29 July 2003, Brian Luscombe, the US Product Director for Gynecare, expressed concern about it since it was not apparently reported with Monarc, the AMS obturator tape, and was not the subject of a warning in its IFU. The concern, however, did not relate to the welfare of patients but to the prospect that AMS would use it against Ethicon when it entered the market. Zenobia Walji, Director of Marketing for Gynecare, counselled a principled approach. She considered it was necessary to thoroughly understand the “cause-effect” of the complication. She wondered whether the risk could be avoided by changing technique alone and “fine tuning the IFU”. If neither course resolved the matter, she asked whether there was a design change that could avoid the complication and, if so, should the change be made now or when the “next gen product” is developed. She also wondered whether there was a way to be sure that the Monarc procedure also avoided the complication. She said that if it were an inherent risk of the obturator techniques then Ethicon’s focus would not be to “improve competitiveness” but to inform physicians of the risk. Dr Arnaud’s response was rather dismissive. In an email dated 14 August 2003, he wrote:
1. We need to differenciate (sic) basic post-op pain from truly abnormal pain which can be called a complication.
Any surgical procedure gives some kind of post-op pain because of the surgical trauma. This will occur with TVT as well as with every obturator procedure. Any procedure consisting in passing a needle through tissues will generate pain on or close to the needle route. In the case of Mulberry [a reference to TVT-O], the needle being passed in the muscle of the thigh, it is not surprising to have some kind of pain at this level.
2. Most of the time, post-operative pain is considered so common that it is not even reported in publications. Since Jean de Leval wanted to monitor every single parameters (sic) concerning the procedure he has designed, he noticed and reported in his paper a 20% rate of post-op pain. This includes minor discomfort as well as pain that disappears within 48 h with minor antalgic drugs.
3. Two patients had a painful post-op course that lasted about a week. According to Jean, the pain seemed to be of articular origin and it is very likely that it was a hip pain due to the position on the table.
Jean de Leval does not consider post-op pain as a relevant clinical issue with his procedure and I do not think we should challenge that at this point.3068
(Original emphasis)
3735 No investigation was conducted. In its initial CER for TVT-O Ethicon assumed that the data collected about TVT would apply to TVT-O.3069 For the reasons given by Dr Allman, that assumption should not have been made. It was contrary to the regulatory guidelines and below the standard of care one would reasonably expect of a manufacturer of a medical device intended for permanent implantation.
TVT Secur
3736 The pre-market evaluation of TVT Secur was also below the standard of care required of a manufacturer of a medical device intended for permanent implantation. The decision to release the device onto the market was at best premature.
3737 In view of the fact that TVT Secur was a new product and involved a new technique, it was imprudent of Ethicon to uncritically rely, as it did, on the TVT database to support the launch of the device. As Dr Hinoul observed, TVT Secur was not equivalent to TVT or TVT-O. The design validation studies were insufficient to support its launch, since they did not match the conditions in which the device would be used. As Ms Holland’s evidence established, contrary to industry guidance Ethicon failed to reduce the risks to the lowest level practicable.
3738 Although the risk management assessment concluded that all the identified harms were mitigated by training, the IFU failed to identify the training that was required. The risk benefit analysis identified only two risks: urinary retention and the possibility of premature removal of the protective inserter blade cover. None of the admitted complications was mentioned, let alone weighed in the balance. In any case, the report paid scant, if any, attention to the differences between TVT Secur and its predecessors on rates of urinary retention. Similarly, the pre-market CER did not address all relevant risks. Nor did the CER consider the potential impact of the differences in material, construction, and dimensions between TVT Secur and TVT.
3739 Dr Hinoul maintained that a clinical evaluation of the risks and benefits of the device had been carried out despite the fact that all the risks he admitted were known to Ethicon at the time were not addressed, let alone considered. This was the subject of cross-examination and his testimony was evasive and unpersuasive:
[Y]ou agree that an evaluation of the risks and benefits of the product which did not address or even mention certain known risks would be an inadequate evaluation. You would agree with that, wouldn’t you?---I think we referred to the predicate device and the predicate device’s performance and if there are reasons to believe that the performance will be equivalent, that is what you have to satisfy the regulators with.
HER HONOUR: Dr Hinoul, can you not answer the question or do you not want to answer the question? Would you put the question again, please, Mr Bannon.
MR BANNON: Do you agree that an evaluation of the risks and benefits of the product which did not address or even mention certain known risks would be an inadequate evaluation?---I – I do not think you have to address all the risks if you refer to the appropriate equivalent device of which you know the risks. So it would imply – the document would refer to the risk profile of the equivalent product.
How would an investigator know that you had evaluated all the known risks by the process you mentioned?---And I think that is exactly why they have audits and they come and they question us and we have to be able to answer those questions.
…
How would a regulator or a notified body know whether you had evaluated known risks if you didn’t list them?---Because in our complaint reporting we do list complaints and issues and we have an instruction for use that lists the adverse events.3070
3740 The testimony was also disingenuous, not least because the instructions for use did not list all the potential adverse events.
3741 Once again, and for the reasons he gave, I accept Dr Allman’s evidence that the opinions expressed in the CER that TVT Secur was a safe device for treatment of stress urinary incontinence and that additional studies were unnecessary before the device was taken to market were unjustified.3071 This is a clear case in which clinical studies should have been undertaken. Without them, Ethicon could not properly evaluate the potential risks posed by implantation of the device against the potential benefits.
TVT Exact
3742 For the reasons given in Part VIII, Ethicon’s failure to undertake studies or clinical trials of TVT Exact before it was supplied in Australia was not duly justified, as required by the European Directive. Furthermore, the failure of the pre-launch CER to consider the extent to which complications associated with TVT and TVT-O were likely to arise with TVT Exact and to assume, without justification, that those complications were acceptable also fell short of the essential requirements and evinced a want of care on the part of the manufacturer. No reasonably prudent manufacturer would take a medical device of this kind to market without complying with the essential requirements.
TVT Abbrevo
3743 TVT Abbrevo was designed as an improvement on TVT-O. Ethicon’s objective was to reduce adverse effects, particularly pain, by reducing the amount of mesh. As I observed in Part VIII above, the pre-launch CER largely relied on data relating to TVT and TVT-O. While that data was obviously relevant, it was insufficient. It was reasonable to hypothesise that a reduction in mesh would lead to a reduction in complications, but a reasonably prudent manufacturer would test the hypothesis in a real-world environment. The CER referred to the findings from Professor de Leval’s single-centre study, which compared TVT Abbrevo with TVT-O, after one year’s follow-up. As Professor de Leval, himself, recognised, those findings needed to be validated externally in a multi-centre multi-surgeon context. A reasonably prudent manufacturer would have heeded this advice. Ethicon did not do so before launching the device in Australia. Furthermore, for the reasons given by Dr Allman, the conclusions of the CER to support CE marking were not justified by the clinical evidence presented and the report did not satisfy the European regulatory requirements.3072 In these circumstances, Ethicon’s conduct fell short of what was required of such a manufacturer.
The POP devices
3744 The applicants submitted that, like the SUI devices, Ethicon’s pre-market evaluation of the POP devices suffered as a result of its “desire to push product to market whilst undertaking the minimum amount of pre-market clinical study”.3073 They submitted, in effect, that impelled by these commercial imperatives, Ethicon relied on the hernia meshes as predicate devices, which they submitted “over-rode rational assessment of the risks inherent in permanently implanting polypropylene mesh via a blind, transvaginal, passage”.3074 Moreover, they submitted, Ethicon failed to consider treatment options for the complications that it knew could, and in some women would, arise through the use of the devices or the impact these devices could have on some women who experienced some of these known complications, such as chronic pain. They argued that despite the call for randomised clinical trials by, amongst others, the surgeons Ethicon had recruited to design Prolift, it was “rarely willing to do more than the bare minimum to study its devices prior to launching them on the medical community”.3075 None of the POP devices, they contended, was supported by clinical data sufficient to justify CE marking at any time before it was released to the market.3076
3745 There was a wealth of evidence to support these arguments.
Gynemesh PS
3746 The pre-market evaluation for Gynemesh PS was woeful. Despite the acknowledgment by its Scientific Advisory Panel in June 2001 that the characteristics of the vaginal wall were not well-understood and that erosions could present years after implantation and despite its request for a clinical study separately evaluating risks associated with treatment for anterior and posterior prolapse, Dr Weisberg signed the CER declaring the product to be safe and efficacious without the benefit of such a study. The CER was perfunctory. The literature review was minimal and uncritical. As I mentioned in the discussion of this report in Part VIII of these reasons, reference was made to studies on Marlex, Prolene and Vicryl. By the time the report was signed, six years had elapsed since the publication of the Marlex study. Furthermore, its findings were misrepresented. The two studies on Prolene concerned very small cohorts of women. The Vicryl study was five years old and its relevance obscure.
3747 Given the admissions about the state of Ethicon’s knowledge about the potential risks, the discussion of potential complications was manifestly inadequate. It failed to address all foreseeable risks and the significance of the three risks that were mentioned was minimised. Nor did it refer to the findings of the TVM Group or the advice it had received from Professor Klosterhalfen. Despite the available evidence that the tissue reaction is dose dependent, no consideration was given to the potential for differences in the incidence of complications because of the difference in the quantity of mesh that would be required to treat stress urinary incontinence on the one hand and pelvic organ prolapse on the other.
3748 For the reasons given by Dr Allman, discussed in Part VIII, the CER was insufficient to justify CE marking. Indeed, Dr Hinoul conceded that the report and the material to which it referred were an insufficient basis for the report’s conclusion that the use of Gynemesh PS for the purposes for which it was indicated was safe and efficacious.
3749 In all the circumstances, I accept the applicants’ submission that a clinical study should have been undertaken before the device was taken to market. The prudent course would have been to undertake a randomised controlled trial. At the very least, Ethicon ought to have waited for the clinical study they had begun to conclude and for its results to be analysed. The decisions to obtain CE marking and to proceed to sell the device were precipitous and opportunistic. They were inconsistent with the actions of a reasonably prudent manufacturer.
Prolift
3750 For all the reasons given in Part VIII, the pre-market evaluation of Prolift was not conducted with the level of care required of a manufacturer of an implantable medical device.
3751 Put shortly, the decision to proceed with the certification and sale of Prolift was equally rash. The inventors of the TVM technique were much more cautious than the respondents. They considered that transvaginal implantation of mesh was “not validated” at the very time the respondents was planning to launch Prolift. They proposed that long term studies be carried out first. They also considered that the technique should be reserved for the surgical treatment of grades 3 and 4 prolapse. The respondents, on the other hand, did not wait for long term studies before taking Prolift to market and did not limit its indication to surgery for grades III or IV prolapse.
3752 The respondents ignored the advice given to them by Professor Jacquetin that implantation of mesh by the vaginal route had not been validated, that randomised controlled trials were necessary, that the indications for transvaginal mesh be limited to grade IV and recurrent prolapse, and that simultaneous hysterectomy should be avoided.
3753 For the reasons given by Ms Holland, the design validation for Prolift was inadequate.
3754 The CER signed by Dr Owens suffered from many of the same deficiencies as the Gynemesh PS CER signed by Dr Weisberg. The likely reason is that, as Dr Allman observed, it largely repeated the Gynemesh PS CER without specifically addressing the safety and performance of the Prolift system. It, too, was manifestly inadequate. As Dr Allman explained, it did not conform to the requirements of the European Directive. It did not justify CE marking.
3755 The pre-market evaluation conducted in relation to Prolift fell well below the standard of care expected of a reasonably prudent manufacturer of a device of this kind.
Prolift+M
3756 The same is true of Prolift+M.
3757 Before Prolift+M was first taken to market, no studies were undertaken to examine whether the results apparently achieved with UltraPro in general surgery for abdominal hernia repair could be replicated in the pelvic floor. A reasonably prudent manufacturer would have conducted such studies. The respondents realised that studies were necessary but declared the device safe and effective before the results of the studies were known.
3758 When Prolift+M was launched in Australia, only three-month results were available from a single arm study limited to women with symptomatic POP-Q grade III or IV prolapse. This data was insufficient to demonstrate the safety and efficacy of the device for its intended use, since the device was designed as a permanent implant and indicated for use for women with pelvic organ prolapse regardless of the POP-Q stage it had reached. For the reasons given by Ms Holland, the design validation did not simulate user groups or use conditions. For the reasons given by Dr Allman, the CER was insufficient to justify CE marking.3077
Prosima
3759 For all the reasons discussed in Part VIII, the pre-market evaluation of Prosima was inadequate and the CER, signed by Dr Robinson, was insufficient to justify CE marking. The only clinical study on Prosima conducted before it was launched was the single arm observational study by the inventor, Dr Carey. At the time it was made available for sale, there was insufficient evidence to conclude that Prosima was safe or effective for the purpose for which it was intended and for use by surgeons who did not possess the surgical skills necessary to safely implant Prolift.
3760 Ethicon had been warned against releasing Prosima but defied those warnings. On 9 February 2009, Scott Jones, the Product Director, Pelvic Floor Repair, for Ethicon Women’s Health & Urology reported in an email to Jonathan Meek that every surgeon to whom he spoke at a recent summit said that the company was “making a huge mistake by commercializing Prosima”. Amongst the areas of concern he highlighted were:
--We shouldn’t focus on applying mesh to a stage 2 defect because the future of these repairs is unknown
--We shouldn’t be training less skilled surgeons or surgeons with lower surgical volumes especially in light of the FDA PHN [Public Health Notice].
…
--Plication has a roughly 30% rate of recurrence, and PROSIMA has a roughly 30% failure rate
…
--Customers feel like we are making the same mistakes that we made with SECUR, namely rushing to market without all the answers.3078
3761 Dr Allman concluded that the pre-market CER for Prosima did not comply with European regulatory requirements for clinical evaluation and was insufficient to justify CE marking. I accept his opinion that, given the lack of clinical data to support CE marking of Prosima, Ethicon should have conducted clinical investigations of the device, with clearly defined acceptance criteria, before taking it to market.3079
Was the pre-market evaluation of any of the Ethicon devices insufficient to discharge the manufacturers’ duty of care?
3762 The pre-market evaluation of all Ethicon devices was insufficient to discharge Ethicon’s duty of care. All evinced a want of reasonable care. None met the essential requirements of the applicable European Directive. By failing to consider the impact of the potential risks on the quality of life of the women in whom they were implanted and the issues surrounding treatment of complications, Ethicon failed to properly evaluate whether the risks were acceptable when weighed against the benefits to patients and were compatible with a high level of protection of health and safety. All their clinical evaluation reports were deficient in this respect. They were also deficient in numerous other respects, discussed at length in Part VIII. None of them complied with the regulatory requirements and guidelines. For example, the search, inclusion and/or exclusion criteria were opaque. There was obvious selection bias. Devices were treated as equivalent to other devices when they did not meet the criteria for equivalence. There was little to no critical analysis of the literature. Limitations of the studies reported in the literature, for instance, such as the level of evidence, the number of patients, the materials and methods deployed, or the length of follow-up, were not discussed and rarely acknowledged. None of the CERs contained a comprehensive literature review. Reliance on the literature was often, if not invariably, unjustified and certainly not “duly justified”. Indeed, the conclusions in all the CERs suffered from a paucity of reasoning.
3763 Ethicon also failed to take sufficient account of how the Ethicon devices might fare when used as intended, that is, by a broad range of surgeons, of varying skills and experience, in a diverse patient population. Instead, it placed considerable reliance on studies performed by highly experienced surgeons, often designers of the devices, and failed to properly account for the restrictions placed on the study populations by those surgeons. And it routinely ignored the observations made by the surgeons about the need for additional data. Although the European Directive stipulated that clinical investigations should be performed unless it was “duly justified to rely on existing clinical data”, Ethicon often chose not to conduct clinical studies until after a device was taken to market. When it embarked on pre-market clinical studies, it rarely waited for the results before making the devices available for sale. Moreover, when clinical investigations were not undertaken or the results were not yet available, the clinical expert reports on which it relied to secure CE marking, did not “duly” justify reliance on existing data.
3764 The preponderance of evidence supports the applicants’ case that all the devices were introduced with unseemly haste and without due attention to the safety of the women for whom they were indicated and to whom, both indirectly and directly, they were promoted.
3765 Professor Gordon considered that, in the absence of long term follow-up studies, none of the POP devices could be classified as “safe”. Although he was asked to provide opinions on the evidence relied upon to support the POP devices only, his remarks are also applicable to the SUI devices. He wrote:
The [POP devices] entail the insertion of a synthetic mesh into a woman’s body. The intention of the treatment is for it to remain permanently. There can be complications arising from the mesh itself, such as extrusion and erosion, and when this occurs re-operations may be required. It is possible – and there is some evidence for this, which I discuss in my response to Question 6 of the letter of instruction – that the rate of extrusion and/or erosion may increase with time since the operation. Given all this, it seems to me that an appropriate judgement of “safety” was not feasible during the early stages of the development of mesh, prior to its release onto the market, since at that time no long term follow-up studies had been conducted. 3080
3766 Consequently, Professor Gordon disagreed with Dr Hinoul’s opinion that the studies allowed a conclusion to be drawn that the various POP devices were safe and efficacious for the treatment of pelvic organ prolapse before the devices was released to market. He said that the available evidence was weak, observing that, since the results of randomised controlled trials were not yet available, series of cases or follow-up studies over a short term were relied upon. Noting the preference of the Pharmaceutical Benefits Advisory Committee (the independent body appointed by the Australian Government to recommend new medicines for listing on the Pharmaceutical Benefits Scheme) for data derived from randomised trials, he continued:
This evidence falls short of the usual standards of evidence for the introduction of a new intervention for a particular condition. These standards require randomised trials, meta-analysis if possible, and for a permanently installed physical material, should not rely on only short-term follow-up, in my opinion. 3081
3767 Professor Hu was asked whether the trials or studies conducted or relied upon by Ethicon for the purpose of introducing the POP devices to the market had the capacity properly to identify and investigate the potential for new complications. He said that the trial and seven case series relied upon for this purpose had very little capacity to properly identify and investigate the potential for new complications. He noted that there was only a single prospective trial, which was small in size (with only 12 subjects per group) and had major limitations. There were seven case series which had no controls for comparison. Neither the prospective trial nor the retrospective case series reported data on patient satisfaction, sexual function, or other indices of quality, or followed patients beyond two years. Professor Hu described this as “a sparse data base” and one which lacked adequate years of follow-up to enable conclusions to be drawn about the safety of the product. In fact, he said that the sparse information that was available was “highly suggestive of mesh-specific complications that cast doubt on the statement in the company’s § 510(k) document that mesh products ‘have an established history of safe clinical use as implantable materials.’”3082
3768 This evidence was not the subject of cross-examination.
3769 In cross-examination Associate Professor Rosamilia agreed that there was widespread adoption of transvaginal mesh kits with no robust clinical trials before regulatory approval and application.3083
3770 In an article published in October 2010, Professor Roovers, who, it will be recalled, was a witness for the respondents, observed that the technology was “offered on the market, even though the evaluation process [had] not yet been completed” and was offered to gynaecologists, without taking into account their volume of work and without the technical skills required for prolapse surgery and the management of complications.3084
3771 In a later article, published in July 2016, Professor Roovers wrote:
The patient is the one who has the most to lose in this debate. The patient is also the one for whom it is most difficult to determine what to believe and what not to believe. Indeed, there are many women who have suffered from an overly rapid introduction of a new surgical technique that was not yet ready to implement in daily clinical practice.3085
3772 In his evidence in chief, he made the following observation:
With very limited evidence, and with very limited knowledge about the surgical principles, vaginal mesh surgery was widely implemented in a very short period. There may have been good reasons for that, of which the high risk on recurrence following native tissue repair is probably the most important. However, I believe that patients have suffered from the over-rushed introduction of vaginal mesh surgery.3086
3773 In cross-examination, he said that there was very limited evidence about the safety and efficacy of the POP devices in humans before they were released to the market.3087 He laid the blame on the medical profession. Given the legal obligations of the manufacturers, however, they were also responsible. Indeed, since the timing of the introduction of the devices to the market and the information they supplied to users was entirely in their hands, they were at least equally, if not primarily, to blame.
3774 As was the case with the SUI devices, at all material times Ethicon was driven by commercial considerations. Staying ahead of the competition was their pre-eminent concern. Safety was a secondary consideration.
Second alleged breach: inadequate post-market evaluation
3775 It was uncontroversial that a manufacturer’s duty of care does not cease once the devices are released to the market. Rather, it was common ground that Ethicon had a continuing duty to monitor the performance of the devices. This was also a regulatory requirement.
3776 The applicants submitted that Ethicon failed to undertake post-market evaluation of the devices adequately and, in certain respects, at all.3088 They identified three primary areas where deficiencies in this process were apparent. These were:
(1) Ethicon’s post-market clinical follow-up on the devices, both in terms of clinical studies and ongoing review of the scientific literature;
(2) Ethicon’s complaint handling and event reporting to regulatory authorities; and
(3) Ethicon’s responses to enquiries raised by regulators.
3777 Had sufficient evaluation been undertaken, they contended, it would quickly have become apparent that the adverse events, the impact of those events on patients, and the warnings accompanying the devices were “all unacceptable”.3089
3778 I accept these submissions.
3779 Annex X of the European Directive stated that “the clinical evaluation and its documentation must be actively updated with data obtained from the post-market surveillance”. Dr Beech explained that this means that the manufacturer is obliged to be proactive in seeking feedback on the clinical performance of a device.3090 Evidence to similar effect was given by the other regulatory experts. I referred in Part IX to Dr Hinoul’s account of the post-market evaluation undertaken by Ethicon. It was correctly characterised by Dr Allman as reactive, rather than proactive. It was, in effect, a minimalistic approach to post-market evaluation and fell well below what was expected of a reasonably prudent manufacturer. As Dr Allman explained:
Post-market surveillance conducted by Ethicon relied on passive monitoring of complaint and adverse event reports, assessment of the published literature, and Ethicon sponsored studies. Given the nature of the Implants, the risks identified as part of risk management processes, and the absence of clinical investigation pre-CE marking to establish safety and performance of the Implants, passive post-market surveillance was insufficient. Ethicon should have conducted post-market clinical follow-up studies. Ethicon sponsored studies were conducted primarily for commercial (marketing) purposes.3091
3780 The fourth revision of the guidelines on post-market clinical follow-up (MEDDEV 2.12.2), which was issued in May 2004, referred to manufacturers’ obligations to carry out systematic reviews of post-production experience (called “post-market surveillance” and often referred to by the acronym PMS).3092 Implementation of a programme for post-market surveillance was stated to be key to identifying and investigating risks associated with the use of medical devices placed on the market. Manufacturers were advised to have a defined strategy in place for each of their products. Post Market Clinical Follow-Up was considered to be an important part of this process. Dr Allman said:
A manufacturer, of implantable devices, and after publication of MEDDEV 2.12.2 (i.e. from 2004), would reasonably be expected to have:
• effective procedures for systematic review of information received by the manufacturer (complaint and adverse event reports) relating to the device;
• effective procedures for systematic review of the scientific literature pertaining to the devices and similar devices;
• effective procedures for the active collection of information relevant to devices—including, where necessary, PMCF studies; and
• considered the need for PMCF studies, documented the rationale for the decision, and to have implemented such studies if necessary.3093
3781 Dr Allman explained that adequate post-market surveillance would have required post-market clinical follow-up studies to assess safety and performance of the devices outside the rigorously controlled environment of a clinical study.3094
3782 Ms Holland said that “pro-active activities” such as periodic literature reviews, reviews of field actions, and customer feedback requests would be reasonable actions for a manufacturer to take when monitoring an implantable mesh device upon commercialisation. She also said, however, that, when an increase in adverse events or device malfunctions is seen, additional device testing or other applicable surveillance should be conducted.3095 She explained that the post-market evaluation performed by a manufacturer should be in direct proportion to the risks associated with the device.3096
3783 Dr Pence’s evidence was to similar effect. She said that a manufacturer should perform clinical evaluation routinely once a device is on the market to determine if there is new information relating to safety and performance obtained from clinical experience.3097
3784 For the reasons discussed at length in Part IX and summarised below, the post-market evaluation of all the Ethicon devices was deficient. It fell well below the level of care required of a reasonably prudent manufacturer.
3785 First, post-market clinical evaluation was haphazard and for years the requirement for clinical evaluation reports was overlooked, if not ignored.
3786 Second, as Dr Allman noted, the BSI audits indicated that Ethicon’s procedures for risk management and clinical evaluation did not conform to the European regulatory requirements (he described them as “deficient versus European requirements”) — from the time the Ethicon devices were first supplied until 2012 when some of the devices were withdrawn from the market. Indeed, the BSI audits indicated that there had been a breakdown in the ability of the risk management system to effectively control risk. Furthermore, Ethicon was slow to respond to the findings of the audits. Based on the contents of the BSI audit report following the September 2012 audit, Dr Allman testified that Ethicon had a “very serious problem” with its quality systems which needed to be addressed.3098 Ms Holland’s evidence was to like effect.
3787 Third, no proper consideration was given to post-market clinical follow-up of the necessary kind, despite the requirement to do it or justify not doing it, included in the European Directive and the indication in the MEDDEV 2.12.2 that post-market clinical follow-up should always be considered where identification of possible emerging risks and the evaluation of long term safety are critical.
3788 Professor Krulewitch, whose evidence was confined to the SUI devices, said that, based on the respondents’ records provided to her by the applicants’ lawyers, there was no evidence that the respondents had conducted any post-market studies on those devices. She said that, in the light of the many unknown or unanticipated outcomes, post-market studies would have been very important for the respondents to evaluate new complications as they arose. Yet, there was “no systematic plan” to monitor potential new complications or late complications.3099 I appreciate that Professor Krulewitch was not briefed with all the relevant evidence on these matters, including the CERs, but her evidence on this question is supported by other evidence, including the CERs themselves.
3789 Dr Hinoul’s response to Professor Krulewitch’s evidence about Ethicon’s failure to conduct post-market clinical follow-up was that Ethicon “continually provided funding and/or product for surgeons to use in postmarket studies”.3100 As Dr Allman pointed out, this confuses the nature of company sponsored studies, which are often conducted for marketing purposes, and proposed by the investigators, with the studies deemed necessary by the manufacturer of a device to examine specific aspects of device safety and performance in the post-marketing phase of the life cycle of the device. His view, which I accept and which was well supported by other evidence, was that Ethicon had no coherent plan for generating post-market clinical evidence.3101
3790 Dr Allman’s opinion, which I accept in the absence of any expert evidence to the contrary, was that, absent due justification for not undertaking them, post-market clinical follow-up studies were required to assess safety and performance of all the devices “outside the rigorously controlled environment of a clinical study” — in other words under normal conditions of use.3102 At the very least, the “passive post-market surveillance of the sort Ethicon did conduct” could have been compared to the baseline established by the follow-up studies.3103
3791 Fourth, there was no system of formal review of complaints before March 2006.
3792 Fifth, as Dr Pence’s evidence disclosed, Ethicon tended to minimise the significance of complaints and to avoid responsibility for adverse events.
3793 Sixth, Ethicon failed to take heed of the likelihood that adverse events were under-reported and failed to comply with their reporting obligations, a course of conduct which was liable to mislead both the regulators and the notified body (BSI) about the number and extent of the complaints and therefore the safety of their devices. Its investigation of the sharp increase in complaints following the FDA notification in 2011 was manifestly inadequate. The apparent correlation between an increase in complaints and an increase in lawyer advertising or litigation was not a sufficient basis for its failure to take corrective action.
3794 Finally, post-market clinical evaluation reports were not routinely or regularly prepared until 2010, by which time several of the Ethicon devices had been on the market for years. Although the quality of the reports improved, as Dr Allman observed, following the BSI audit findings, they were still inadequate. Moreover, as Dr Allman also observed, the improvements in the reports could not compensate for the lack of suitable evidence from studies specifically intended to examine the safety and performance of the devices against pre-determined criteria.
3795 Like the pre-market CERS, the post-market CERs were deficient in numerous respects, discussed at length in Part IX. They drew upon clinical data which included non-equivalent devices. They failed to clearly state the criteria used to found their claims that the devices satisfied the essential requirements. To the extent that they referred to the scientific literature, their references were selective and often omitted advice given by the authors of the articles about the use that should be made of their findings. They rarely engaged in what could pass for a critical, let alone objective, analysis of the literature and, as the TGA pointed out with respect to the July 2013 CER on the TVT Family of Products3104 and the May 2013 CER on Gynemesh PS,3105 there was major selection bias in the so-called critical analysis. Where the literature review indicated that the IFU might require attention, no review of the IFU was suggested, let alone undertaken. The conclusions, particularly with regard to safety, were not justified. They relied on the number of complaint/adverse event reports Ethicon had received when, as Dr Allman remarked, though useful to monitor safety and performance, complaint and adverse event data are unsuitable to establish safety and performance or benefit over risk. Dr Allman explained in re-examination:
[Y]ou have no idea what proportion of bad things that happened get reported. That depends on people’s willingness to report and your own complaint handling processes. So it’s useful to see if complaint rates go up because that’s telling you something has happened, but you have no idea what the underlying adverse event rate is. That was particularly important, I think, for Ethicon devices with erosion, because that would not necessarily be reported by users. It’s mentioned in the instructions for use. It’s not described properly as a risk, so incidence and severity, but if people think it’s a normal expectation, they wouldn’t report it.3106
3796 Moreover, since it was well aware of the problem of under-reporting, Ethicon’s reliance on complaints data to demonstrate a low incidence of adverse events in comparison to sales of the devices was disingenuous.
The SUI devices
3797 Turning to the evaluations of the individual devices, no or at least no adequate post-market clinical evaluation was conducted on TVT until 2010, when the first post-market CER was produced, although by then the device had been on the market in Australia for more than a decade. On any view of the matter, this omission fell below the relevant standard of care.
3798 For the reasons given in Part IX, if the 2000 and 2006 CERs for TVT purported to be post-market clinical evaluations, they, too, were inadequate. The 2010 CER was also deficient in significant respects. Any thorough review of the literature should have prompted Ethicon to consider whether the IFU required amendment to include additional warnings, adverse events, and/or contraindications. Any consideration should have focussed on patient safety and paid close attention to the regulatory requirements, standards, and guidelines.
3799 A 2008 report from BSI noted deficiencies in relation to “legacy files”, which would have included TVT and TVT-O, in critical areas such as risk management, clinical data, and post-market surveillance.3107
3800 In 2011, BSI again found deficiencies in the risk management reports the company audited, which included TVT Exact, and found that none of the CERs was consistent with the requirements of MEDDEV 2.7.1 in numerous respects.3108 More likely than not it would also have found deficiencies in the files for the other SUI devices had they also been the subject of an audit.
3801 Despite the length of the July 2013 CER for the TVT Family of Products, the conclusion that TVT met the essential requirements was unjustified and the report did not meet European or Australian regulatory standards. It will be recalled that the TGA’s assessor was critical of the evaluation in numerous respects including irreproducibility of search results; major selection bias; inadequate synthesis of data; a lack of discussion about safety; a lack of critical analysis of the selected studies; and a failure to address the apparent increase over the previous three years in erosion rates, described as “massive”, or to consider the reasons for it.3109
3802 Although the 2015 CER was a vast improvement on its predecessors, it was still deficient for the reasons given by Dr Allman and set out in Part IX.
3803 The failure to produce a post-market clinical evaluation report on TVT-O until 2010, more than six years after the device was first supplied in Australia, was below the standard of care reasonably required of a manufacturer of medical devices. When one was finally produced in August 2010, it suffered from numerous deficiencies discussed in Part IX. Later surveillance was covered by the 2013 CER for the TVT Family of Products, the deficiencies of which I have already canvassed.
3804 For the reasons given above in Part IX and discussed by Dr Allman in his reports, neither the 2010 CER nor the 2013 CER met the regulatory requirements for clinical evaluation or justified continued CE marking of the device.
3805 For the reasons given in Part IX and discussed in the context of the CERs for the TVT Family of Products, the post-market surveillance on TVT Abbrevo and Exact was also wanting.
3806 Similarly, the failure to produce a post-market clinical evaluation report on TVT Secur until more than three years had elapsed since it had first been supplied in Australia was indicative of an inattention to consumer safety.
3807 It seems that no post-market surveillance was conducted on TVT Secur after September 2013. Certainly, no report was produced. The CERs for the TVT “family” of products did not include TVT Secur.
The POP devices
3808 Despite regulatory requirements, Ethicon did not conduct a clinical evaluation of the safety and efficacy of Gynemesh PS until 2010, by which time it had been on the market in Australia for seven years.
3809 No explanation was proffered for the failure to undertake any formal post-market clinical evaluation over this long period. Dr Allman said that the lapse of time was “too long to be considered good practice”, particularly in the light of the concerns of clinicians and regulators which were mentioned in the 2010 report.3110 There was a further gap of three years before the next one appeared.
3810 There were also delays in the production of the CERs for the other POP devices. It was five years after its release to market before the first post-market CER for Prolift was produced and another three years before the second. Prolift+M was launched in Australia in December 2009. It took until September 2012 for Ethicon to complete a post-market CER. Similarly, it was five years after Prosima was launched until a post-market CER was completed. This was hardly the behaviour of a manufacturer proactively monitoring the performance of its devices.
3811 Then there is the matter of the nature and conduct of the evaluations.
3812 Apart from the general deficiencies discussed above, there was no justification for excluding from the 2010 CER for Gynemesh PS reference to, let alone consideration of, complaints to Ethicon made before February 2007 or of a discussion of the severity of the matters to which they related or their outcomes.
3813 Although the residual risk level was considered to be high, the overall residual risk was deemed “acceptable in view of well documented benefits/patient outcomes”. For the reasons given in Part IX, there was no genuine risk analysis and the designation of the overall residual risk as acceptable was unsound. Reliance on the IFUs was not good enough when, for the reasons indicated in Part XI and in this Part, information in the IFUs was both deficient and, in significant respects, misleading.
3814 External stakeholders expressed public concern about the dearth of clinical evidence available about the POP devices and the risks associated with their use.
3815 In 2005, the year Prolift was launched and after Gynemesh PS had been on the market in the United States for three years, the IUGA Grafts Roundtable noted that although industry and current clinical trends suggested that graft materials could be used in all cases of reconstructive pelvic surgery, there was no evidence-based medicine to support the expansion of the use of graft materials in pelvic reconstructive surgery or the use of such materials for all forms of surgery.3111
3816 Six years after the launch of Prolift, Withagen et al (2011) noted that, despite the widespread application of vaginal meshes in recent years, there was “still little scientific evidence on the pros and cons, as well as the risk factors for postoperative problems”.3112
3817 As I mentioned in Part V, the position of the IUGA Grafts Roundtable was that the Amid classification was not applicable to pelvic grafts and that rat models used to evaluate the use of grafts for hernia repair techniques were not useful in assessing the likelihood of graft infection in the pelvis. In their article recording the conclusions of the Roundtable, Davila et al (2006) wrote that perhaps the most troubling aspect of the use of grafts in reconstructive surgery related to the development of what they described as “healing abnormalities”, such as erosion, rejection, and infection, the aetiology of which they considered was poorly understood. They pointed out that the available laboratory testing and animal models used in the research of biomaterials did not adequately reflect the same environmental challenges that present in the female pelvis over a lifetime. They also noted that, although surgeons were becoming skilled at graft implantation, expertise needed to be developed in the management of complications, including explantation of grafts.3113
3818 As the manufacturer of such materials, Ethicon was obliged to address these matters. A reasonable and prudent manufacturer would do no less. As I have already discussed, there is no doubt that it would have been alive to them. It held meetings with experts at which similar concerns were raised. Professor Deprest, a co-author of the Davila et al (2006) article, attended one of them in Hamburg on 2 June 2006, the month after the publication of the article. Professor Klosterhalfen also attended, along with other surgeons and Ethicon personnel.3114 The minutes record that highlights from the presentations included the following observations:
there was “a huge need” for more research and data in biomechanics of pelvic organ prolapse and pelvic floor repair using meshes:
shrinkage is not controlled by “softness” of mesh (in other words the move from Prolene to Prolene Soft did not control mesh contraction);
chronic pain, though not a frequent complication, was the complication of most concern to surgeons;
vaginal pain after implantation is feared because there is no real treatment option;
fibrosis was responsible for complications in mesh usage;
every individual reacts differently to a mesh;
tension on the mesh changes the sizes of the pores;
meshes can cause nerve damage due to mechanical irritation; and
there is no inert material.
3819 The highest priorities in terms of unmet clinical needs were:
eliminating shrinkage and long-term contraction (noting that severe contraction causes dyspareunia and reduces sexual function);
reducing fibrosis;
eliminating vaginal distortion, maintaining a “normal” vaginal wall and “normal” sexual function; and
eliminating chronic pain.3115
3820 Ethicon’s inaction and its dismissive approach to the concerns of clinicians, the FDA, NICE, and the Cochrane reviews were unwarranted and inconsistent with what might reasonably have been expected of a reasonably prudent manufacturer. Ethicon’s response to the increase in adverse event reports to the FDA was also dismissive and inconsistent with its duty of care. It did not conduct a proper investigation into the reasons for the increase. Rather, it assumed in its favour that it was artificial. As Dr Allman put it, Ethicon’s response was “to maintain that it was not real … on the basis of their own (flawed) assessment of the risks”.3116 His evidence, which I accept, was that the FDA’s 2008 Public Health Notice should have:
prompted a re-evaluation of the safety of the devices under the applicable European regulations,
led to consideration and initiation of post-market clinical follow-up studies.3117
3821 Moreover, as discussed above, when Ethicon presented a post-market surveillance plan to the FDA for Gynemesh PS suggesting that data collected for Prosima could be used, the FDA categorically rejected the plan.3118
3822 Similarly, as late as 2014 the TGA concluded that the clinical data for Gynemesh PS submitted by JJM did not satisfy the requirements of the Medical Devices Regulations and, in particular, essential principles 1, 2, 6, and 14. Essential principle 1 relevantly required that a medical device will not compromise the clinical condition or safety of a patient and that the risks associated with its use are acceptable when weighed against the intended benefit and compatible with a high level of protection of health and safety. Essential principle 2 relevantly required that hazards and associated risks be identified, then eliminated or reduced as far as possible and that users be informed of any residual risks. Essential principle 6 required that the intended performance benefits outweigh any undesirable side effects. Essential principle 14 required clinical evidence demonstrating compliance with the applicable provisions of the essential principles.
3823 Like Dr Allman and Ms Holland, the TGA was critical of Ethicon’s approach to risk management.3119 Like Dr Allman, the TGA levelled a number of criticisms at the 3 May 2013 Gynemesh PS CER. Given that this report was a significant improvement on previous CERs, it is likely that the TGA would have been critical of those, too, had it reviewed them.
3824 The positions taken by the FDA and the TGA lend support to the opinions of the regulatory experts.
3825 The CERs for the other POP devices were also inadequate. The deficiencies in those reports are covered extensively in Part IX and do not need to be repeated here.
Was the post-market evaluation insufficient to discharge the manufacturers’ duty of care?
3826 No post-market evaluation of any Ethicon device complied with the regulatory requirements. Evaluation was not undertaken actively or regularly. Neither was it appropriately updated. Post-market surveillance did not include clinical follow-up studies and the failure to do so was not documented or justified. Ethicon did not have adequate clinical evidence to support continued CE marking of any the Ethicon devices.
3827 I find that Ethicon supplied and marketed the Ethicon devices without conducting sufficient post-market evaluations. The evaluations that were undertaken were haphazard, often perfunctory, and passive. For all the reasons canvassed above, they did not conform to the standard of care expected of a reasonable person in the position of Ethicon in response to the foreseeable risks of injury and having regard to the gravity of the potential consequences.
Third alleged breach: failing to provide adequate warnings and other relevant information
3828 It will be recalled that the applicants alleged that the respondents failed to provide any or any adequate information, advice or warnings about the pleaded complications and the absence of any or any adequate clinical or other evaluation of the risks.
3829 This claim raised four main issues: first, on what basis did a duty to warn arise; second, did reasonable care require warnings about the pleaded complications and/or the other matters; third, to whom should the warnings have been directed; and fourth, was the information supplied by the respondents sufficient to discharge their duty.
The nature and purpose of the duty to warn
3830 In their written submissions, the applicants equated the duty of a medical device manufacturer and supplier to warn the user of the risks associated with their products with the “comparable” duty of a medical practitioner to warn a patient of the risks of medical treatment, outlined in Rogers v Whitaker.3120
3831 In Rogers v Whitaker at 490, the High Court held that a doctor has a duty to warn a patient of a material risk inherent in the proposed treatment. It defined a material risk in the following way:
[A] risk is material if, in the circumstances of the particular case, a reasonable person in the patient’s position, if warned of the risk, would be likely to attach significance to it or if the medical practitioner is or should reasonably be aware that the particular patient, if warned of the risk would be likely to attach significance to it.
3832 The effect of the applicants’ position, if correct, is that the manufacturer has a duty to warn only of risks associated with the use of its device to which a reasonable person in the position of the consumer (here the surgeon and the patient) would be likely to attach significance.
3833 In the Vioxx case, however, Jessup J went further. In Peterson at [786] his Honour distinguished Rogers v Whitaker, holding that the manufacturer’s duty is not confined to warning of “material” risks. He pointed out that no such restriction can be found in the formulation of the duty in Donoghue v Stevenson; the duty is to warn of reasonably foreseeable risks. Neither side submitted that his Honour was wrong to do so and no appeal was brought from this aspect of his Honour’s decision.
3834 In closing oral argument, the applicants acknowledged that their position did not align with what Jessup J held in Peterson. Initially, they said the Jessup J “may have been too generous”.3121 Eventually, however, they adopted his Honour’s approach.3122 They were right to do so.
3835 The duty of a manufacturer or supplier of a medical device is not, and the duty of the respondents was not, “comparable” to the duty owed by a medical practitioner to a patient. In Peterson at [785] Jessup J explained that the obligation to warn of material risks has a particular relevance to the doctor-patient relationship. As his Honour observed, the correct starting point is not Rogers v Whitaker, but Donoghue v Stevenson.
Did reasonable care require warnings about the pleaded complications and/or the other matters?
3836 The short answer to this question is that it did.
3837 At all material times the relevant risks were not only foreseeable but known. They were also risks which patients and doctors would consider were at least “not insignificant”. I did not understand the respondents to suggest otherwise.
3838 The respondents submitted that the users of the devices were trained medical professionals who should be familiar with surgical procedures and techniques involving pelvic floor repair and non-absorbable or synthetic meshes and pointed to the instructions to this effect in the IFUs for all the devices. Such a person, they argued, would appreciate the risks of using the devices. Besides, they added, JJM offered a professional education program and surgeons who required training and guidance on the relevant procedures for implanting the devices could enrol in that program if necessary. They also argued that, in any event, to the extent that the IFUs were said to be deficient or confusing, it was reasonable for the respondents to expect that medical practitioners, acting reasonably, would seek guidance or clarification should they not understand a statement in the IFU. In other words, the respondents’ argument was that they were not obliged to mention risks of which they were aware because the users of the devices (the physicians or surgeons) either knew of them already or had at their disposal other sources from which they could have acquired the information. I have already rejected that argument in the context of the defective goods case. For the following reasons, it must be rejected here, too.
3839 As Jessup J remarked in Peterson at [796], “the duty to warn the physician seems clearly to be based on the silent proposition that, absent warnings, the physician may know little about the characteristics of a product alleged to be harmful”. It is not to be assumed that the physician or surgeon has other potential sources of information. As his Honour observed, the assumption is that the medical professional is best equipped to receive the warning which might well be technical and unintelligible to the layperson, to factor it into the decision-making process, and to communicate it in an intelligible way to the patient.
3840 It is notable that since the commencement of the Medical Device Regulations on 4 October 2002, there has been an unqualified statutory obligation to provide certain information with any medical device. This information includes:
Any warnings, restrictions, or precautions that should be taken, in relation to use of the device.
3841 The Regulations also required that the instructions for use include, amongst other things:
Any contra-indications, warnings, restrictions, or precautions that may apply in relation to use of the device.
…
For an implantable medical device—information about any risks associated with its implantation.
3842 The applicants did not argue that these requirements gave rise to a private right to sue for breach of statutory duty and it is not therefore necessary to decide whether they do. Nevertheless, a breach of those requirements can provide evidence of negligence: see O’Connor v SP Bray Ltd (1937) 56 CLR 464 at 477 (Dixon J).
3843 The notion that the medical literature was sufficient to alert the medical profession of the true nature and extent of the risks associated with implantation of mesh was rebuffed by a number of witnesses. I referred to some of this evidence above when dealing with the same point in the context of the defective goods claim.
3844 It is no answer to the applicants’ claim to point to the fact that professional education was offered and surgeons who elected to undergo it could have asked questions. As I have already stated, the respondents had an obligation to warn of all reasonably foreseeable risks regardless of what they might presume to think surgeons already knew or could learn from their own research or upon inquiry or after further education. As the applicants argued, the existence of the duty of a medical practitioner to warn or provide information does not obviate the duty of care owed by a manufacturer; the duties are at least co-extensive. One reason for this is the likelihood that the manufacturer would “overvalue a product and underemphasise its risk”: Hollis at [46].
3845 In Peterson, Jessup J held at [836] that, if an appropriate warning had been provided to medical practitioners, the manufacturer did not have additional obligations to warn the patient or consumer. This was on the basis that, once a medical practitioner had formed a professional judgment, providing a separate additional warning to the patient would be unnecessary and somewhat confusing. There is some merit in this view. This case, however, unlike Peterson, involves a manufacturer and supplier that directly communicated with patients through brochures and a website. The respondents pursued a marketing strategy that targeted both surgeons and patients. Having embarked upon that course, the respondents were bound to take reasonable care to ensure that the information they imparted was complete and sufficient to warn their target market of all reasonably foreseeable risks of harm.
3846 In Hollis at [20]–[23] La Forest J described the manufacturer’s duty to warn in this way
[A] manufacturer of a product has a duty in tort to warn consumers of dangers it knows or ought to know are inherent in the product’s use. This duty to warn is a continuing duty, requiring manufacturers to warn not only of dangers known at the time of sale, but also of dangers discovered after the product has been sold and delivered … All warnings must be reasonably communicated, and must clearly describe any specific dangers that arise from the ordinary use of the product …
The duty to warn serves to correct the knowledge imbalance between manufacturers and consumers by alerting consumers to any dangers and allowing them to make informed decisions concerning the safe use of the product.
The nature and scope of this duty varies with the level of danger entailed by the ordinary use of the product. Where significant dangers are entailed by the ordinary use of the product, it will rarely be sufficient for manufacturers to give general warnings concerning those dangers; the warnings must be sufficiently detailed to give the consumer a full indication of each of the specific dangers arising from the use of the product
In the case of medical products … the standard of care to be met by manufacturers in ensuring that consumers are properly warned is necessarily high. Medical products are often designed for bodily ingestion or implantation, and the risks created by their improper use are obviously substantial … Given the intimate relationship between medical products and the consumer’s body, and the resulting risk created to the consumer, there will almost always be a heavy onus on manufacturers of medical products to provide clear, complete and current information concerning the dangers inherent in the ordinary use of their product.
3847 His Honour proceeded to refer to the vulnerable position of consumers who depend on the manufacturer and the doctor to provide enough information to enable them to make an informed decision about their treatment and the “enormous informational advantage” that pharmaceutical companies enjoy over consumers. At [26] he said:
In light of the enormous informational advantage enjoyed by medical manufacturers over consumers, it is reasonable and just to require manufacturers, under the law of tort, to make clear, complete and current informational disclosure to consumers concerning the risks inherent in the ordinary use of their products. A high standard for disclosure protects public health by promoting the right to bodily integrity, increasing consumer choice and facilitating a more meaningful doctor-patient relationship. At the same time, it cannot be said that requiring manufacturers to be forthright about the risks inherent in the use of their product imposes an onerous burden on the manufacturers. As Robins J.A. explained in [Buchan v. Ortho Pharmaceutical (Canada) Ltd. (1986), 12 O.A.C. 361] at p. 381, “drug manufacturers are in a position to escape all liability by the simple expedient of providing a clear and forthright warning of the dangers inherent in the use of their products of which they know or ought to know”.
3848 I respectfully agree.
To whom should the warnings and other information have been provided?
3849 Warnings should have been provided (both with the devices and in the promotional material) to prospective users, including the applicants, group members, their treating surgeons or physicians, and the hospitals to which the devices were distributed.
Did reasonable care require warnings about all the pleaded complications and/or the other matters?
3850 I find that a reasonably prudent manufacturer and supplier in the position of the respondents would have warned prospective users of all but one of the pleaded complications. The exception concerns the risk of psychiatric injury. There is no evidence to indicate that there was a risk of psychiatric injury independently of an antecedent physical injury. On the available material, I am not satisfied that a reasonably prudent manufacturer and supplier in the position of the respondents would have warned of the risk of psychiatric injury.
3851 The evidence established that not only was each of the risks of the remaining pleaded complications reasonably foreseeable, but in all but one case they were also admittedly known to Ethicon at the time the devices were first marketed. These risks were significant for the most part and certainly not insignificant and the consequences could be serious. These considerations alone would have required more comprehensive warnings.
3852 A reasonably prudent manufacturer and supplier in the position of the respondents would also have provided guidance about how to manage the potential risks arising from implantation with all of their meshes, regardless of what they might surmise physicians knew, should have known, or could have learned from other sources or by other means. They would also have provided information about the extent of the risks or at least directed readers to where that information could readily be obtained and, if the evidence was insufficient to enable such an assessment to be made, because, for example, the long-term effects had not been adequately studied or studied at all, then they should have alerted users.
3853 These conclusions are supported by the regulatory evidence in particular. That evidence, discussed in Part XI, established that the warnings and information provided by the respondents fell well short of what was reasonably required.
3854 In their supplementary submissions filed on 14 June 2018, the respondents’ argued that an implanting surgeon would appreciate the possible implications for a patient presenting with an autoimmune disorder. Even if this were true, for the reasons given above, a reasonably prudent manufacturer or supplier in the position of the respondents would still issue a warning about it. The consequences of not doing so could be dire. As it happens, however, the respondents’ argument was not supported by reference to any evidence and was in fact contradicted by the evidence.
3855 Professor Korda was a preceptor for TVT. He did not issue such a warning and he professed to have no knowledge of the relevant risk. Moreover, his uncontested evidence, which I accept, was that the risk was generally unknown to the surgical community until 2012 — around the time or after Prolift, Prolift+M, and Prosima had been withdrawn from sale and removed from the ARTG.3123 Some support for Professor Korda’s evidence comes from the absence from Associate Professor Lam’s PowerPoint presentation of any reference to the position of women with autoimmune disorders, let alone any warning that the risks for them were any different than for other women.3124
3856 The evidence about the respondents’ training program, such as it was, did not suggest that this issue was covered.
3857 Dr Pence noted that Ethicon “had information that certain patient populations were more likely to experience negative outcomes” as a result of the use of the POP devices.3125 Her evidence was that that information should have been included in the IFUs for those devices. She referred to a presentation by Dr Hinoul to the FDA Obstetrics and Gynecology Medical Devices Advisory Committee in September 2011 in which he advised the Committee that certain patient populations were at higher risk of complications from vaginal meshes for pelvic organ prolapse repair and proposed that predisposing risk factors be included in the labelling. Dr Hinoul told the Committee:
One of the most important questions we need to ask ourselves is also why these adverse events are occurring. And the risk factors for mesh exposures are becoming more and more apparent. Several studies published this year show that hysterectomy, patient age, smoking, diabetes, and surgeon experience predispose patients to mesh exposure. Patient selection and risk factors, appropriately stated in the device's labeling, as well as the surgeon's training, are therefore part of our proposal.3126
(Emphasis added)
3858 The risk, however, was not confined to the POP devices. Moreover, well before 2011 Ethicon was or ought to have been aware of these matters and alerted users to them.
3859 Assistant Professor Chughtai said that “most general urologists and gynaecologists are not likely to look into the method of approval or to primarily evaluate the medical literature before proceeding with a novel device” and it would be “unlikely” that a reasonably competent gynaecologist or urologist would be aware of the scientific literature on the inflammatory response to mesh.3127 Indeed, when he responded to a question raised by the applicants’ solicitors as to what surgical and patient factors were likely to influence whether mesh implant surgery is likely to be safe and efficacious, he did not mention autoimmune disorders and like conditions.3128
3860 Assistant Professor Margolis said that surgeons do not routinely read scientific literature dealing with, amongst other things, the duration of the inflammatory response to biomaterials. He said that they rely on the manufacturer to provide that information.3129
3861 I find that a reasonable manufacturer and supplier in the position of the respondents would have warned of the heightened risk of adverse reactions for patients with autoimmune disorders or whose immune systems were suppressed for other reasons. Indeed, as I have already indicated, I am persuaded by the evidence that the devices were contraindicated in such cases and a warning to this effect should have been given in the IFUs and the promotional material, including the surgeon and patient brochures. The consequences of not doing so were potentially serious. A woman in this position would attach significance to such information.
3862 As with all the other pleaded complications except for psychiatric injury, the warnings should have been provided from the time each of the Ethicon devices was first supplied.
3863 Ethicon’s failure to adequately conduct clinical testing of the various devices in the user population before they were made available for sale and over a sufficient period to enable their suitability for permanent implantation to be properly evaluated meant that there were shortcomings in the respondents’ knowledge of the extent of the risks and the incidence of the potential complications. A reasonable and prudent manufacturer and supplier in the position of the respondents would have included this information in the IFUs and the other material furnished by the respondents to medical practitioners and patients. As Dr Allman put it, informing users of a risk means providing them with information, not merely about possible harm but also about the probability and severity of harm, and how that risk was established.3130
3864 I also accept the applicants’ submission that doctors and hospitals should have been notified of any change of significance made to an IFU, especially a change to the adverse events section.3131 No reasonably prudent manufacturer or supplier in the respondents’ position would do otherwise.
3865 JJM issued such a notification on 3 September 2015 in relation to Gynemesh PS and the SUI devices that were still on the market, although it seems likely that it was only because the TGA required it.3132 The pro forma document,3133 which was tendered by the applicants during cross-examination of Dr Hinoul, was addressed to the Chief Executive Officer and marked for the attention of the Nursing Unit Manager of the Operating Theatre and the Chief of Surgery. To ensure that the information was brought to the attention of the relevant personnel, JJM stipulated that the notice be distributed to all users of the products and be “visibly posted in [the] facility for awareness” and required written confirmation that it had been distributed to all staff within the facility who used the products.
3866 There is no evidence to indicate that notifications such as this were given at any earlier time and it appears to be common ground that they were not.
3867 The respondents contended that it was not open to the applicants to make this submission because it should have been put to Australian pelvic surgeons that updates to an IFU should be brought to their attention and it was not. They argued:
In this regard, as has been made clear by the Respondents on a number of occasions, any contention that the implanting surgeons would have done anything different suffers from the critical evidentiary gap in the Applicants’ case. This is another instance where the Applicants failed to put forward any direct evidence from a relevant implanting surgeon involved in the decision to proceed to surgery with an Ethicon product.3134
3868 I reject this contention. These considerations, if valid, go to the question of causation in an individual case, not breach.
3869 The applicants submitted that in cross-examination Dr Hinoul conceded that it would be important to inform users of such a change.3135
3870 The respondents denied that this was the effect of Dr Hinoul’s evidence. They argued that his evidence was that a change to an IFU should be brought to the attention of users if the change would be construed as a very different IFU with new adverse events but that this was not the case with respect to the IFUs for the Ethicon devices.3136 This was the evidence:
And may we take it you would agree that, if there was a change to the adverse reaction in the IFU, it would be important to bring it to the attention of the users of the IFU?---If it would indeed have – be construed as a very different IFU with new adverse events, yes. Again, as I’ve stated from the beginning we felt that the original IFU covered the – the adverse events appropriately.3137
3871 Dr Hinoul’s answer is ambiguous. It is possible to construe it in the way the respondents sought to. On the other hand, it is also possible that Dr Hinoul was initially obfuscating but then acquiesced in the cross-examiner’s proposition before gratuitously reiterating the respondents’ position that there was nothing wrong with the original IFU. That was certainly the way I took his answer at the time.
3872 In any case, while not all of the IFUs may have added “new adverse events”, many of them did. Regardless, there would be little point in making changes to instructions for use unless the changes were brought to the attention of physicians and surgeons. No reasonably prudent manufacturer or supplier would make such a change without also taking all reasonable steps to notify the user population. Otherwise the safety of patients would be unnecessarily imperilled.
3873 A reasonably prudent manufacturer or supplier would also have recalled the product brochures at the same time as the IFUs in order to make the necessary changes.
3874 I am not persuaded, however, that the respondents were obliged to inform users of the availability of alternative treatments, a matter also raised in the applicants’ pleading. No submissions were directed to this question and the expert evidence did not go this far. Nor am I persuaded that the respondents were required to provide information comparing the safety and efficacy of the devices with alternative treatments. They were not entitled, however, to misinform or mislead users about these matters.
Was the information supplied by the respondents insufficient to discharge their duty of care?
3875 On 19 December 2008, three months after the FDA alert, in an email to colleagues in the regulatory and quality departments, Ethicon’s Dr Meng Chen drew attention to a complaint from a TVT patient who felt she had not consented to an operation which could result in the post-surgical complications she had developed. Dr Cheng observed that the FDA had stressed the importance of pre-operative consent. She pointed out that:
One of the paths for a better pre-operative consent is to provide an updated IFU to the operating physicians that reflect[s] the current knowledge of the manufacturers (scil) on the potential adverse reactions.3138
3876 This was not a course the respondents ever took.
3877 Dr Hinoul was taken to this email in cross-examination.3139 He said he discussed the matter with Dr Cheng at the time, reviewed the IFUs, and felt that they were “adequate”. I cannot agree.
3878 I discussed the deficiencies in the respondents’ warnings and other information at length earlier in these reasons. As is apparent from that discussion, the information about the potential risks in the IFUs and promotional material for all devices was below the standard required of a reasonably prudent manufacturer or supplier in the position of the respondents. It fell well short of capturing all known, let alone reasonably foreseeable risks, and was liable to mislead the reader about the safety and efficacy of the various devices. From the first of the IFUs to the last, the warnings they contained and the information they conveyed were insufficient to discharge the respondents’ duty of care.
Conclusion
3879 Each of the respondents was negligent. The risks were known, not insignificant, and on Ethicon’s own admission, serious harm could ensue if they eventuated. A far more cautious approach was warranted than the respondents took.
3880 There is no reason to believe that the burden of taking the relevant precautions was so great as to render the respondents’ conduct reasonable in the circumstances.
3881 There was no evidence to suggest that it would have been oppressive for the respondents to include appropriate warnings or to conduct and complete suitable clinical studies to assess their efficacy and safety. Nor was there any evidence to suggest that it would have been unreasonable for them to do what the regulatory experts said was necessary to satisfy the regulatory requirements and to conduct themselves in the way reasonably expected of a manufacturer or supplier in their position.
3882 No submissions were made about the social utility of the devices. Naturally I accept that there is utility in medical innovation and that there was utility in developing medical devices that would provide more effective long-term relief from stress urinary incontinence and pelvic organ prolapse than traditional surgical procedures offered. But this does not excuse or justify the respondents’ conduct, particularly when alternative established methods of treatment were available which did not expose patients to the additional risks created by the use of the devices.
3883 As the manufacturer of TVT, Ethicon Sàrl breached its duty of care to Mrs Sanders by failing to conduct adequate clinical evaluations of the device before her implant surgery on 12 March 2001. It also breached its duty of care to Mrs Gill by failing to conduct an adequate evaluation of Prolift, more particularly, Prolift Total, before her implant surgery on 12 January 2007. Equally, the company breached its duty of care to both these applicants by failing to issue adequate warnings about the risks to which they were exposed if they were to undergo surgery with the relevant device and about the shortcomings of their evaluations. As the manufacturer of Gynemesh PS, Ethicon Inc. breached its duty of care to Mrs Dawson by failing to conduct an adequate evaluation of its safety before her implant surgery on 8 May 2009 and by failing to provide adequate warnings of the risks to which she was exposed if she were to agree to implantation with the device and of the shortcomings of their evaluations.
3884 JJM also breached its duty of care to the applicants. Like Ethicon Sàrl and Ethicon Inc., it failed to provide adequate warnings of the risks of the pleaded complications and of the limitations of the clinical evaluations.
PART XV: THE APPLICANTS’ CIRCUMSTANCES
Background and medical history
3885 The first applicant, Kathryn Gill, was born on 4 December 1970.3140 She was just 36 when she was implanted with Prolift Total on 12 January 2007.
3886 She met her husband, Steven, in Melbourne in 1997.3141 They lived together for about 4 years before marrying in 2001. They moved to Perth in 2002 when Mr Gill took up a position as Executive Officer of the Western Australian Rock Lobster Council. She and her husband have two children, both boys, born on 14 April 2002 and 3 February 2004.3142
3887 Mrs Gill suffered from gestational diabetes during the course of both of her pregnancies and post-natal depression following the birth of each child.3143 She also experienced what she described as “feelings of sadness and lethargy” around the time her father died in 1999 for which she received counselling and took anti-depressants for about a year after which she felt much better.3144
3888 She has had psoriasis, an autoimmune skin condition, since she was a child. From time to time it affected the skin on her elbows, knees, scalp and stomach but in about 2003 she was diagnosed with psoriatic arthritis, which causes inflammation of the joints and which affected her fingers, hips, knees and ankles. From time to time her knees and ankles get sore but then settle down.3145
3889 From time to time Mrs Gill has also experienced neck pain, which she attributes to tension, and pain in other parts of her body that cause her to clench her jaw and stiffen her neck and for which she has received chiropractic treatment.3146
3890 Nevertheless, before 2004, when her second child was born, Mrs Gill considered that she had always been fit and healthy. She led a fairly active life.3147 She quit smoking in 2000 and described herself as “conservationist/environmentalist/natural therapies/healthy living person”.3148
3891 She began sailing at the age of 10.3149 Whilst at university she played in the rollerblade hockey team for Monash University competing in the men’s league and was a member of a rollerblading demonstration team providing entertainment at sporting functions.3150 In 1992 Mrs Gill injured her back playing rollerblade hockey when she was tackled into a wall. She could not play sport for a few weeks. Thereafter she has suffered from intermittent lower back pain for which she was treated with physiotherapy and remedial massage. The pain consists of cramps down the back of her hamstrings on both sides.3151
3892 On weekends she competitively sailed dinghies (referred to as 125 sailing dinghies) and won several State championships and two Australian championships. She stopped sailing competitively at age 27 to take up yachting/cruising with her husband.3152 She regularly swam, snorkelled, and jogged.3153 She enjoyed camping in the bush regularly, including driving a 4WD to remote locations to go fishing.3154 She also tried to maintain a vegetable garden.3155
3893 After the birth of her second child, Mrs Gill attended fitness classes which incorporated Pilates, body balance and a lot of walking and gentle jogging. She was also part of a very active mothers’ group that met once or twice a week and also exercised together. She visited her family in Melbourne as much as she could, taking advantage of the free travel for her baby, and joined her husband on many of his work trips both within Western Australia and across the country.3156
3894 Before she underwent prolapse surgery in 2007 she was responsible for most of the household chores. She did all the household cooking and cleaning as well as the grocery shopping and the laundry for the family, although her husband “regularly did his own laundry” too.3157
3895 As we shall soon see, her work was also very active.
Education and employment history
3896 Mrs Gill attended Monash University, graduating with a Bachelor of Science, majoring in Environmental Science and English Literature, and then Deakin University where she obtained a Graduate Diploma of Environmental Management.3158 From 2000 to 2002, before the birth of her first child, she worked full-time for SeaNet as a Fisheries Extension Officer with OceanWatch Australia, NSW.3159
3897 Whilst working with Coastcare and Eco-Logic in the late 1990s she was required to take students snorkelling in the ocean, canoeing in estuaries, and accompany them on bushwalks. During the years she worked for SeaNet she regularly went out on commercial fishing boats and assisted with such tasks as hauling nets, sorting fish, and stowing ropes and pots, work she described as physically demanding. She prided herself on her ability to keep up with the bigger, more experienced men on the crew.3160
3898 After moving to Western Australia in early 2002 she spent a couple of years out of the workforce bearing and raising her children.3161 She returned to the workforce on a casual basis in 2004 as a lecturer at the Challenger TAFE or Institute in the employ of the Fremantle Maritime Training Centre (FMTC), lecturing in subjects “pertaining to” the Diploma of Maritime Science and the Certificate II in Marine Tourism.3162
3899 Following the birth of her second child in 2004, Mrs Gill noticed a bulge at the introitus.3163 She consulted a GP, who advised her to continue with her post-partum exercises.3164 One morning in late February 2004, however, she had a bright blood loss in “a small gush”, which worried her and made her feel uncomfortable, so she presented to Glengarry Hospital.3165
3900 Sometime in 2004, Mrs Gill consulted the obstetrician who managed her second pregnancy, Dr Richard Tai, who told her she had a vaginal prolapse but assured her that it was not significant.3166
3901 In about the middle of the year she developed pneumonia and was ill for around 10 weeks. During that time she coughed repeatedly which strained her abdominal muscles. She felt that the coughing was making her prolapse worse and she thought the bulge in her vagina was growing. She said that she felt it pushing out with every cough. She did not feel pain, however, only a general discomfort. At the same time she was regularly constipated and also experienced pain with intercourse. The prolapse prevented her from using tampons. In addition, when she need to pass urine she felt that she had to go urgently. At times she did not make it to the toilet in time and wet herself. She said she was unable to run to the toilet because, if she tried, she could feel the prolapse coming out.3167
3902 These symptoms persisted after she recovered from pneumonia but she was determined to work. In about December 2004 she applied for work with the FMTC (now part of TAFE) and was offered a casual position as a lecturer teaching marine biology.3168 Mostly, she said, she had set classes which were on Tuesdays. Now and again she also covered for other staff members when they were sick or on leave. The most she ever worked, however, was three days a week.3169
3903 Mrs Gill said that the main issue posed by the prolapse was related to her inability to use tampons during menstrual cycles. In the warmer months she took her marine biology students snorkelling once or twice a week and this became awkward if she was menstruating. She said she would often wear a pad under her wetsuit and just hoped that no blood leaked out. She considered menstrual cups to contain the flow whilst snorkelling but because of the prolapse she could not keep a cup in place. She said she was very self-conscious of her menstrual issues but they were manageable and she was determined to continue to work.3170
3904 On 1 February 2005 she saw her GP, Dr Dianne Prior, about her prolapse and was referred to Dr Jay Natalwala, an obstetrician and gynaecologist.3171 Dr Natalwala saw her on 7 February 2005.3172 He considered that she had “quite an impressive Grade II–III cystourethrocoele” with the vagina seen just beyond the introitus, and “a Grade II–III rectocoele with poor perineal support.”3173 According to his letter to Dr Prior, Mrs Gill was complaining of a lump in her vagina, worse with exercise, and she told Dr Natalwala that she could not use tampons and was unable to have intercourse.3174 Stress incontinence was not a major problem but she reported urinary frequency, urinary urgency and severe urge incontinence to Dr Natalwala. According to the letter, she was “wet almost on a daily basis” and her bowels were “always constipated with a perineal bulge”. Dr Natalwala arranged for her to see a pelvic floor physiotherapist but suggested that she would require “some sort of operative intervention”.3175
3905 Mrs Gill saw the physiotherapist on 25 February 2005. She complained of a sensation of vaginal prolapse particularly after gym exercises. She also reported some bladder urgency since the birth of her last child and would leak small amounts of urine approximately once per day. She was given a floor exercise program, and advised to cease doing multiple sets of sit-ups which could aggravate her prolapse and to avoid high impact aerobic exercise, such as running or skipping, in favour of low impact aerobic exercises, such as cycling and swimming.3176
3906 On 10 March 2005 Mrs Gill returned to see Dr Natalwala, this time with her husband. According to Dr Natalwala’s notes and his letter to Dr Prior at this consultation they had a long discussion about treatment options and he recommended against the use of mesh, offering her vaginal repair with a possible paravaginal repair and culdoplasty instead.3177 In his letter to Dr Prior he wrote:
I have seen Kathryn today with her husband Stephen and I have discussed the situation of her prolapse in great detail including why this may have happened, where the damage is and it is not just fascial damage but also partial nerve damage which inevitably goes hand in hand with this. There is also a family history component of this in terms of collagen and elastin problems.
I have discussed with her that the mesh is probably not for her. She is too young for us to use this at this stage. I have offered her a vaginal repair with a possible paravaginal repair and a culdoplasty on my list in May. I have fully discussed with her the operation, the outcome and recovery from this including the risks associated with this.3178
3907 Mrs Gill claimed to have no recollection of this or, indeed, of anything discussed at this appointment.3179 There is no reason, however, why Dr Natalwala’s account should not be accepted, having regard to his contemporaneous records.
3908 On 15 April 2005 Mrs Gill rang Dr Natalwala’s rooms to cancel her operation scheduled for 12 May 2005. According to the note of the phone call, Mrs Gill said she would return in a year’s time to discuss the matter further.3180 She never did.3181
3909 Mrs Gill candidly disclosed in her affidavit that the sexual relationship she enjoyed with her husband had never returned to normal after the birth of their second child which she attributed to fatigue from looking after two small children and then the prolapse. But, she added, she remained keen to have sex when the opportunity arose.3182
3910 Around the middle of 2006, Mrs Gill’s bowel symptoms deteriorated and she needed to insert her fingers into her vagina and push her vaginal wall backwards to defaecate.3183 Mrs Gill said that her sexual relationship with her husband significantly deteriorated at this time. She lost her libido and withdrew from her husband. She explained:3184
It got to the point where I did not want to have sex at all, and I withdrew from Steven in that respect. I believed that because of my prolapse there were parts inside of me that could potentially get pushed out by having sex. I feared it would not be pretty, and that it would actually be quite horrible. I felt that I could not guarantee that I would not defaecate during sex. I was also worried that my bladder would get pushed, and that this would result in my wetting myself. I felt self-conscious that I smelt like urine. I was constantly trying to clean and wash myself.
3911 Mrs Gill consulted a psychologist, Debra Roberts, for sexual health counselling aimed to help her to learn to live with her symptoms. After several months she recommended that Mrs Gill seek a physical solution.3185
3912 In September 2006, Mrs Gill saw another GP, Dr Sobha Eranki. Dr Eranki referred her to Dr Nicolas Tsokos, a gynaecologist.3186 Mrs Gill did not end up seeing Dr Tsokos and does not remember why.
3913 But Dr Eranki also referred Mrs Gill to another gynaecologist, Dr Vincent Chapple.3187 On 23 October 2006 Mrs Gill presented to Dr Chapple complaining of urge incontinence, urgency, and frequency, a vaginal lump, and difficulty defecating. 3188 In his notes of the consultation Dr Chapple recorded a history of sexual problems with reduced libido, a vacuous [enlarged, very open and loose] vagina, a “problem with work”, and an inability to run.3189 In his letter to the GP he added that she had “unwanted fertility”.3190 He diagnosed a large cystourethrocele, grade III uterine prolapse and a moderate rectocele with a normal anteverted uterus with no pelvic masses or tenderness. Also in his letter to the GP Dr Chapple reported:
Mrs Gill's symptomatic prolapse requires surgical treatment and nowadays with the development of prosthetic devices to augment repairs, patients have a lot of options which are quite difficult to work through. I started this process today and discussed vaginal hysterectomy with anterior posterior vaginal repair +/- a mesh prosthesis. 1 have also discussed a prolift, abdominal sacral hysteropexy, which would be regarded as a gold standard procedure in somebody who wishes to retain their fertility, which obviously this is not the case with Kathryn's unwanted fertility is part of her symptom problems. In spite of this however she is not at this time prepared to let go of her uterus which also makes contemplation of a vaginal hysterectomy as part of the repair a problem.3191
3914 He left her to think about things and arranged to see her in a few weeks’ time to “go over” the matter again and “come to some decision”.
3915 Mrs Gill saw Dr Chapple in his rooms on two further occasions, on 30 November and 6 December 2006, when they again discussed options for treatment, including the use of a Prolift implant.3192
3916 On 30 November 2006 Dr Chapple wrote to Dr Eranki:
Kathryn was reviewed again today with her partner to discuss planned surgery for symptomatic utero-vaginal prolapse. We again went over the various surgical options being; 1) vaginal hysterectomy, anterior/posterior repair + - Mesh prosthesis. 2) abdominal sacro-hysteropexy or laparoscopic sacro-hysteropexy or 3) Prolift. The last two of these procedures are designed to obviously retain the uterus. My feeling is that this really is what Kathryn is looking for as she has not entirely given up on the prospect of future pregnancies.
Due to this I would lean away from a Prolift procedure as this clearly is designed for women who have ceased their family and the manufacturers do not recommend its use if future pregnancies are planned. Having said this I did discuss with Kathryn that anatomically I am getting the best results with Prolift since its introduction although it does seem that there is a longer recovery and certainly more post-operative pain than you would see with the other procedures.
Again unfortunately no firm decision was made and Kathryn and her partner have gone away to consider things further. I will let you know what is decided in due course.3193
3917 On 6 December 2006 Ms Gill informed Dr Chapple that she wished to proceed with the mesh implant surgery. She asked whether he would be able to insert a Mirena (an intrauterine device or IUD) at the same time. He said he could.3194
3918 The clinical notes record that he again discussed the pros and cons of surgical options but Mrs Gill can no longer remember what was said. She conceded that it was possible that the information she was given about the mesh may have been given over all three consultations prior to the operation.3195
3919 Mrs Gill was admitted to the Joondalup Private Hospital in Perth on 12 January 2007 and underwent surgery the same day.3196 She was implanted with Prolift Total and a Mirena IUD was inserted for contraception and control of her menstrual cycle.3197 According to the hospital records a cystoscopy was carried out and this revealed no evidence of damage and the ureteric orifices were visible.3198
3920 She was discharged four days later, on 16 January 2007.3199
The aftermath of the prolapse surgery
3921 Mrs Gill experienced a lot of pain in her pelvic region following the surgery, which, she said, she had expected. She rested and tried to avoid coughing, as she knew that would place strain on her abdomen and pelvis. After a couple of weeks, however, in anticipation of her recovery from surgery, she began to become a little more active. But the pain did not disappear, nor did it improve.3200
3922 On 30 January 2007, two weeks after her discharge from hospital, she returned to see Dr Chapple. In her affidavit she said she told him she was in pain, he reassured her that she was still in the process of healing from the surgery, gave her a prescription, and told her he would review her six weeks after surgery.3201 His notes are consistent with this evidence but they also raise the possibility of an infection (“?vaginal wound infection”).3202 Indeed, the drugs he prescribed included Augmentin and Flagyl, both antibiotics used to treat infections, and Diflucan, an antifungal medication. His notes also record the development of “throbbing, pressure-like pain” the day before the consultation and worse sitting and standing than lying down. On examination she was “generally swollen”, most tender in the posterior region.
3923 Later Mrs Gill noticed that it was becoming painful for her to defaecate. She had pain for about half an hour after opening her bowels. She also noticed that her stools were coated with a lot of mucus. She then became sick and started to experience fevers, shivers and shakes, nausea, diarrhoea, vaginal bleeding, and cramping.3203
3924 Late in the afternoon of 20 February 2007 she telephoned Dr Chapple’s rooms. She said that she had been unwell all day, had woken with cramping, and then for the past hour had been very cold, achy, the glands in her neck were raised, it was sore to swallow, she had a headache, and her back was also very sore. The Patient Communication Form, which contained notes of the conversation, record that Mrs Gill also mentioned that her bowels were “awful – everything coated in mucous and a lot of mucous”.3204 She had had pain around her coccyx for the past week and defecation was painful and the pain lingered for half an hour afterwards. Dr Chapple advised her to see her GP and indicated he would review her separately.3205
3925 Mrs Gill saw a GP at the local medical centre. He referred her to the Emergency Department at Joondalup Private Hospital. In the referral letter he wrote that she had a high fever, sore throat and increasing lower abdominal and pelvic pain since her prolapse surgery six weeks earlier. He said that she was prostrate, complaining of worsening pelvic pain, and was very tender in the left iliac fossa. He noted the possibility that she could have a virus causing the fever but expressed concern that she might have “a pelvic collection/infection” that was responsible for her symptoms.3206
3926 Evidently, she was put on a drip, which helped to break the fever around 2.00am and blood tests were undertaken.3207 The hospital notes record that she was diagnosed with viraemia.3208
3927 Dr Chapple reviewed her on 22 February 2007.3209
3928 She was unwell. She complained of fever, pelvic pain and mucoid diarrhoea. She also had rigors, an abrupt attack of shivering and a sensation of coldness accompanied by a rapid rise in body temperature. 3210 On examination the uterus was well supported and not tender but the posterior vaginal wall was very tender and there was tenderness generally throughout the pelvis. Dr Chapple arranged for her to be admitted to the Glengarry Private Hospital the same day for intravenous antibiotic treatment. He told her she had a postsurgical infection.3211 According to his notes he wondered whether there was an erosion near the cervix on the right. The same day, in a letter to Dr Eranki, he wrote that he had reviewed the blood tests done at Joondalup Emergency Department on 20 February 2007 and now thought that “the likeliest scenario” was “infected mesh”.3212
3929 Mrs Gill remained in hospital until 28 February 2007. The hospital notes indicate that the IUD was removed and sent to pathology.3213 Mrs Gill continued to experience bowel pain, cramps and fever after she was discharged.3214
3930 She returned to work at the beginning of the semester but was unable to continue due to her symptoms.3215 She had severe stomach cramps and diarrhoea for about an hour on the morning of 2 March 2007.3216 On 7 March she was readmitted to Glengarry Private Hospital for observation.3217 A CT scan was performed which apparently showed no abnormality.3218 Dr Chapple saw her the next day and noted she had “rigor” that morning. On 9 March she underwent a colonoscopy. The gastroenterologist, Dr Mark Glaser, concluded she had pseudo-membranous pan colitis, most likely due to clostridium difficile, and prescribed the antibiotic, Flagyl.3219 Clostridium difficile was in fact isolated on microbiological examination.3220
3931 Mrs Gill was discharged from hospital on 10 March 2007. She continued to feel very tired and weak. She spent a lot of time lying down and sleeping. The pain in her pelvic region did not improve. On one occasion she and her husband attempted to have intercourse, but she found it was too painful to continue. She became increasingly concerned and anxious that “something just wasn't right”.3221
3932 On 12 March 2007 Mrs Gill telephoned Dr Chapple’s rooms inquiring about the recovery period from the implant surgery. She said that she had stopped vomiting, her diarrhoea was “ceasing” but she felt very tired and could only spend a small amount of time out of bed.3222
3933 Mrs Gill returned to see Dr Chapple on 26 March 2007 presenting with back pain. His notes record that she felt that her bowel problem had been bad over the weekend and that she had tried to have intercourse but could not manage it.3223 On examination he noted that she had no residual prolapse but that the anterior mesh had concertinaed together and was slightly tender. He tried to reassure her and indicated he would review her in 10 weeks about her periods which, according to his letter to Dr Eranki, had returned with a vengeance since the Mirena had been removed.3224
3934 Mrs Gill said that she was left with the impression that Dr Chapple was unable to explain the extent of the problems she was having and his letter to Dr Eranki is consistent with her impression.3225
3935 From then on, things did not improve. She continued to be very tired, so tired in fact that she could not even manage grocery shopping without frequent rests or assistance. On a number of occasions, her mother flew over from Melbourne to help her with general duties, such as grocery shopping and school drop-offs.3226
3936 Mrs Gill continued to suffer a lot of pain. She described the pain as being of three kinds:3227
a constant, aching pain in the region of her coccyx (which she rated at 6 out of 10 in severity);
sporadic severe pain akin to period pain with coughing or on sudden movements (of such an intensity that she could not speak and at times struggled to breathe and had to lie down to let it pass, rated at 8 or 9 out of 10); and
“terrible” pain on defaecation, fluctuating in intensity, but with a difficult bowel movement (such as hard or large stool, or diarrhoea), around 9 out of 10 in severity, and she was unable to talk or concentrate; she had to focus on breathing, and she became sweaty, pale and started shaking. The pain built up and then peaked, and lasted from one to five minutes. She needed to sit or lie very still “until the waves of pain, cramps and spasms subsided”. She compared it to the feeling of labour pains near the coccyx. She said that it felt like her coccyx was being pushed against so hard that it was going to crack.
3937 She also regularly bled, 3228 which I took to mean from the vagina and with greater frequency than could be attributed to menstruation.
3938 She spent a lot of time sleeping and just lying in bed. She found that any kind of bending aggravated her pain. Even sitting in a chair was a problem. She found the best thing for her to do was to either stand or lie down. Her husband put a mattress on the floor of the family room for her. At dinner time, she stood to eat, and afterwards lay on the mattress with her sons next to her.3229
3939 She abstained from sex apart from the one attempt, which was so painful she could not continue as she felt her vagina was not stretching and intercourse was tearing her.3230
3940 On 18 April 2007 Mrs Gill returned to see Dr Glaser, the gastroenterologist. He told Dr Franki that she had responded very well to Flagyl and the clostridium difficile organism had been eradicated. He noted that Mrs Gill told him that at the end of her 10-day course of Flagyl she felt very well, her energy levels improved, and she started exercising again, but that she had had a recurrence of pain overlying the coccyx and fatigue the previous week. Her bowel motions had increased to three or four a day but without blood or mucous. She had no fever. Dr Glaser was unable to explain the fatigue and thought that the residual variability of bowel pattern was probably explained by post-infectious irritable bowel syndrome.3231
3941 Sometime in the middle of 2007, whilst trying to insert a tampon, Mrs Gill felt “something sharp” inside her. She deposed:
I could feel something sharp inside of me. It was an uncomfortable rather than painful feeling. However, if I moved suddenly, I could feel the mesh slightly tear me, and that resulted in a sharp pain. I felt very confused after I felt the sharp mesh inside of me. I knew that something must be wrong. I did not understand what was happening to me, and I did not know whether it was my fault or not. I assumed that it must have been my fault, and that I must not have been properly following my post-operative instructions. I made that assumption because I went into the surgery believing that if I rested after the surgery I would have a good recovery and I would be fine. Because I was still having problems, I reasoned that I must have done something wrong, although I did not know what that might have been. It was very confusing, disappointing and upsetting for me.3232
3942 Mrs Gill was reviewed by Dr Chapple on 11 June 2007. This was about six months after the prolapse surgery. She told Dr Chapple she had been unable to have sexual intercourse since the operation and that about three weeks before the consultation she could feel mesh in her vagina. She complained of lower abdominal cramps with exertion. Dr Chapple’s notes indicate that on examination there was a “2mm mesh erosion” in the anterior fornix to the right. He diagnosed her with vaginismus. Professor Korda explained that vaginismus is the recurrent and persistent involuntary contraction of the perineal muscles surrounding the outer third of the vagina when vaginal penetration is attempted, whether with a penis, a finger, a tampon or a speculum. He said that it can be global, when a woman is unable to place anything inside her vagina, or situational, in which she is able to insert a tampon and can tolerate a pelvic examination but cannot have intercourse.3233
3943 Dr Chapple proposed treatment with high doses of topical vaginal oestrogen for six to eight weeks and tried to reassure Mrs Gill. In a letter to Dr Eranki he indicated that if the mesh erosion were still in evidence he would “bring her in” and excise the eroded piece of mesh.3234
3944 Mrs Gill decided to seek a second opinion. She received a referral from her GP to see Dr Jessica Yin, a urologist.3235
3945 Dr Yin saw Mrs Gill on 14 June 2007 at the Hollywood Private Hospital. Dr Yin confirmed the mesh erosion with Mrs Gill. She told her it could heal or keep eroding and that she had two options: excision surgery or a trial of oestrogen to see if it could heal spontaneously. Mrs Gill chose the oestrogen option because she was reluctant to have another operation. She arranged for a flexible cystoscopy and thorough abdominal, vaginal and rectal examinations and asked Mrs Gill to obtain a number of routine blood tests.3236
3946 On 26 July 2007 Dr Yin reviewed Mrs Gill. Her mesh erosion had not healed. Dr Yin reported to the GP that there had been “some effort at granulating over [the area of the erosion] with friable vaginal tissue” but “significant mesh” was still palpable.3237
3947 She discussed with Mrs Gill the risks of a mesh excision “particularly bleeding, infection, prolapse, over granulation, altered sensation and the low risk of fistula formation and non-healing”. Nevertheless, she recommended proceeding to excise the mesh as she believed it was “likely to speed up her healing in [that] area”. She also recommended that Mrs Gill avoid the use of tampons as the mesh was “quite prickly” and was likely to collect cotton material.3238 Mrs Gill decided to proceed with the surgery as the oestrogen trial had not worked and she felt that she had no other option.3239
3948 On 20 August 2007 Mrs Gill presented to the GP with a number of complaints including depression and psoriatic arthritis.3240
3949 On 10 September 2007 she was admitted to admitted to Hollywood Private Hospital under the care of Dr Yin, who excised a piece of mesh, measuring 2 cm by 1 cm, from her anterior vaginal wall.3241 A rectal examination revealed no evidence of erosion into the rectum but the posterior Prolift arm was felt close to the mucosa. She was discharged from hospital the following day on antibiotics.3242
3950 After an episode of rectal bleeding, she had another colonoscopy on 3 October 2007 and a random rectal biopsy was obtained. But the colonoscopy was normal. There was no abnormality found in the rectum to account for the rectal bleeding.3243
3951 Mrs Gill returned to work about six weeks after the surgery, working two to three days a week.3244 She stated that, although she was a casual employee, her employer “gratuitously” paid her “during this period”,3245 which I take to be a reference to the period after she returned to work following the excision surgery.
3952 Mrs Gill returned to see Dr Yin in October. Although she was still complaining of vaginal discomfort, her vagina was easily able to admit a speculum and Dr Yin reported that her tissues were well-healed with no evidence of ongoing mesh erosion. She noted, however, that Mrs Gill was struggling. In a letter to Dr Eranki dated 17 October 2007 she wrote:
Unfortunately Katie is going through a bit of a bad patch which she attributes to her ongoing health problems. She feels her husband has had enough of her being sick. They are currently seeking counselling which I have encouraged but hopefully as her health problems dissipate her overall energy and mood may improve.3246
3953 The records from the GP’s surgery reveal two consultations in January and February 2008 in which Mrs Gill reported trouble in the marriage. The notes for the second of these consultations indicate that Mrs Gill had a decreased libido and that the sexual relationship with her husband had been poor since the prolapse surgery.3247 When she presented at the practice on 26 March that year she was diagnosed with depression and prescribed Esipram,3248 a selective serotonin reuptake inhibitor (SSRI).
3954 During 2008 she started to receive massage therapy for her pain on the recommendation of Dr Hira Singh, whom she had seen about an iron deficiency.3249
3955 Mrs Gill returned to see Dr Yin on 14 May 2008. In her report to Dr Eranki, Dr Yin noted that Mrs Gill had been getting what amounts to dysfunctional uterine bleeding and that she remained lethargic and unwell. Dr Yin also noted that Mrs Gill was undertaking marriage counselling. It appears that she had resumed sexual intercourse because Dr Yin wrote that she had noticed dyspareunia with deep penetration. Examination revealed a further mesh erosion in the right vault and the left arm of the mesh was very tight and tender. She was “actively” bleeding. Dr Yin arranged for Mrs Gill to see Dr Robyn Leake about the dyspareunia and bleeding. Dr Yin said that she strongly suspected that the dyspareunia “has a lot to do with [the] mesh erosion and the tightness of her Prolift in general”. In the meantime she “tentatively” arranged for the excision of the mesh.3250
3956 Mrs Gill saw Dr Leake exactly a week later. She complained of pain on the left side of the pelvis since the insertion of the Prolift, which she said felt a lot like contractions and which were precipitated by bowel movements. She also reported pain with intercourse. Examination revealed four main areas of concern:
superficial pinpoint tenderness around the vestibule of the vulva (the area surrounding the opening of the vagina), which she suspected was vestibulitis;
an area of mesh erosion anterior to the cervix;
some tenderness on the left side where the arm of the Prolift closest to the cervix was located; and
the arm of the posterior Prolift at the left posterior aspect of the cervix seemed to be quite tight and tender and produced pain upon palpation.3251
3957 Dr Leake deposed that the left posterior arm of the mesh needed to be removed. She considered that partial removal of the arm would be likely to alleviate Mrs Gill’s pain. She felt that by dividing the left posterior mesh arm, the cervix and the upper vagina would move more freely. She also said that the anterior arm should be divided in order to relieve the contraction-like pain and the risk of recurrent erosions. She considered that the eroded mesh anterior to the cervix should also be removed and the vaginal tissue closed over the top of the area.3252 But she was reluctant to remove all the mesh because “it seems to have ha[d] given her a good result with regards to her prolapse”.3253
3958 Mrs Gill was very upset and depressed by the news that she would have to undergo another operation. She said that found it hard at this time to remain buoyant and positive.3254
3959 On 19 June 2008 she was readmitted to Hollywood Private Hospital.3255 The next day Dr Leake, with the assistance of Dr Yin, removed the posterior arm of the Prolift as she considered that the tension under it could be provoking the Mrs Gill’s dyspareunia and the pain she felt after defecation.3256 She also noted a small area of mesh erosion overlying the left posterior arm, which suggested “undue tension”. Dr Leake carried out a Fenton’s procedure (an operation to remove scar tissue and widen the vaginal opening) as there was some inflexibility of the perineum which could also have been contributing to the dyspareunia. Dr Leake removed the anterior right arm half way to the obturator fossa in order to reduce the tension between the cervix and the point of fixation of the mesh.3257 By allowing more movement of the cervix, Dr Leake intended to alleviate Mrs Gill’s dyspareunia and make intercourse more comfortable.3258
3960 Mrs Gill was discharged on 23 June 2008 on a course of Indomethacin for pain relief.3259 She returned to work about two weeks later, again working two to three days a week.3260
3961 On 27 June 2008 she attended Dr Leake’s rooms for post-operative review. According to Dr Leake’s report to Dr Yin she did very well postoperatively and had no problems voiding, but within three days she was back in hospital with pain and bleeding.3261 Dr Yin examined her under anaesthetic on 4 July 2008 and found a small vaginal abscess which she lanced and drained.3262 She was discharged from hospital on 6 July 2008 with oral antibiotics.3263
3962 In her affidavit Dr Leake said that it was tightness of the mesh around the cervix that caused the erosions Mrs Gill had experienced but the excessive tension was not due to incompetence on the part of Dr Chapple or faulty operating technique. I referred earlier in these reasons to Dr Leake’s opinion that it was impossible for even skilled surgeons to get the tension right in every case. She also noted that Mrs Gill had an autoimmune disorder, psoriasis, which, she said, might have predisposed Mrs Gill to some complications due to idiosyncratic scarring of tissue around the Prolift mesh implant.3264
3963 On 23 July 2008 Mrs Gill returned to see Dr Yin. She was struggling with what she described as ongoing deep-seated abdominal pain which, on questioning, appeared to be “crampy lower abdominal pain”, worse when opening her bowels. She also reported some ongoing bleeding from the vagina and she had some blood-stained discharge on examination but Dr Yin did not think she had an infection or a collection (an abscess) and queried whether she might have been having an episode of colitis.3265
3964 She was reviewed by Dr Leake in August 2008. By then her bowels had settled down and the bleeding had disappeared. They discussed contraception and menstrual control. On examination the posterior vagina was not tender and felt well healed. The right upper area adjacent to the cervix was a little tense but not tender. The introitus was markedly improved, although a little sore on the left.3266
3965 Her symptoms continued to improve and with that her quality of life and her mood also improved. In her affidavit Mrs Gill said that the constant, aching pain in the area of her coccyx had fallen from a 6 out of 10 to a 3 and, although the pain never entirely disappeared, it became more of a background ache which was easier to live with. The severe sporadic pain she had previously experienced reduced in frequency but never disappeared, flaring up with seemingly innocuous activities like twisting to get out of the car, rolling over in bed or sneezing. Her bowel movements became less painful. She attributed this to the use of daily laxatives since the operation in June 2008 which softened her motions so that she did not have to strain her abdominal muscles to evacuate her bowels. She was also able to resume intercourse with her husband without pain, albeit irregularly and tentatively. She described a short period in August 2009 when she had pain on intercourse again but it resolved. With the diminution in her pain Mrs Gill started to become more active. She resumed jogging and started to take camping trips with the family.3267 Nonetheless, she continued to be treated with anti-depressants. In February 2009 Efexor XR modified-release capsules were substituted for Lexapro.3268 Efexor XR is a serotonin-noradrenaline reuptake inhibitor (SNRI).
3966 When Mrs Gill saw Dr Leake on 9 September 2008, she told Dr Leake she was delighted with the way her vaginal comfort had improved since the procedures. She also told her that she was now able to have comfortable sex for the first time in three years.3269
3967 In her affidavit Mrs Gill said that the improvements she has experienced following the surgery on 19 June 2008 had “plateaued” and her physical condition was largely unchanged until about mid-2013 when she experienced a rather abrupt deterioration.3270
3968 In September 2008 her heavy periods were a greater problem. She had flooding for several days at a time and passed large clots. Dr Leake noted that she had a family history of fibroids and recommended she have an ultrasound of the uterus to see if she had any.3271
3969 A transabdominal and transvaginal ultrasound was performed on 3 November 2008. There was no evidence of fibroids but the myometrial echo pattern and the greater thickness in the posterior wall in comparison to the anterior wall were reportedly consistent with adenomyosis, a condition in which fragments of endometrial tissue (the mucous membrane lining the inside of the uterus) infiltrates the myometrium (the wall of the uterus).3272
3970 On 1 December 2008 Mrs Gill was readmitted to the Hollywood Private Hospital under the care of Dr Leake for insertion under general anaesthetic of a Mirena IUD to help with her heavy periods.3273 Curettings were sent for analysis and in a report dated 8 December 2008 the pathologist, Dr Anup Naran, concluded that there was no evidence of endometriosis, endometrial hyperplasia or malignancy.3274
3971 Recovery was not all plain sailing, however. In the middle of 2009 Mrs Gill said that she had “a bit of a meltdown”. She found herself crying constantly. She was advised to take a break so she flew to Victoria and stayed with her mother for a week or so, resting and sleeping a lot.3275 The meltdown may have been triggered by the death of a friend with two young children following a myocardial infarction during chemotherapy for lung cancer.3276 But her depression continued and the marriage was again strained. In late August Dr Flynn wrote another script for Effexor XR with five repeats and referred her to a clinical psychologist, Flavia Bises, whom she consulted on six occasions between September and November 2009.3277 It appears that in the meantime, the Effexor was changed to Pristiq, another SNRI, and the change was associated with significant mood swings which exacerbated Mrs Gill’s depression and interfered with her therapy. But after yet another change in medication to Cymbalta, a selective SNRI, her mood improved. Ms Bises recommended a further six sessions.3278
3972 In February 2010, however, Mrs Gill’s husband, Steven, had an accident while the family were holidaying in Victoria. He fell down a flight of stairs, suffering serious injuries. Mrs Gill had to care for him. He was left with permanent visual impairment and Mrs Gill had to drive him to and from work. He returned to full-time work after a few months, though, at which time she resumed working two to three days a week.3279
3973 After her return to Western Australia Mrs Gill returned to see Debra Roberts, the psychologist who had provided her with sexual health counselling before her prolapse surgery. Ms Roberts informed Dr Flynn on 8 November 2010 that Mrs Gill had a severe depression which was having a negative impact on her sense of self, her relationships, and her ability to cope. Ms Roberts said that she engaged well in therapy and recommended an extension of her mental health care plan for a further six sessions.3280 Two days later Mrs Gill presented to Dr Flynn, reporting a worsening of her depression, a lack of support from her workplace, fatigue, and her marriage in a perilous state.3281
3974 On 11 November 2010 Mrs Gill consulted a psychiatrist, Dr George Ataris. In a report to Dr Flynn he noted that she was dysphoric with restricted affect and flat throughout the consultation. He took a history from her which it is unnecessary to detail here. He diagnosed her with a major depressive episode, recurrent, with atypical features of hypersomnia, and recommending increasing her Cymbalta to “a supra maximal dose”, which he hoped would restore her energy.3282 She was reviewed a month later on 8 December. Dr Ataris thought she had responded to the increased dose of Cymbalta and reported that there were currently “no significant issues other than the ongoing relationship issues” which he considered “pivotal” to her recovery. Weight gain, which had been a problem for some time since the prolapse surgery, remained an issue but Dr Ataris indicated she would benefit from a graded exercise program.3283
3975 In December 2010 Mrs Gill told her GP she was working hard on “fixing things” and by March 2011 she reported feeling better after a good holiday. She was running three times a week, spending time with friends, and her work commitments had reduced. In April she informed Dr Flynn that she was feeling happier and her motivation was good but her sleep was still disturbed, she was constantly tired, and she was anxious.3284
3976 On 25 July 2011 Mrs Gill saw Dr Leake for the last time. She told her that her periods were regular and much lighter but that she had a new problem. The new problem was a period-like pain with orgasm and from constipation, which she said radiated into her back and bottom. On examination Dr Leake found that the mesh was “just palpable”, both anteriorly and posteriorly, but did not find an erosion or abnormal tension.3285
3977 In her affidavit Dr Leake stated that, with the benefit of hindsight, it was clear that all of Mrs Gill’s gynaecological symptoms for which she consulted her were secondary to the use of Prolift mesh to repair her prolapse. Dr Leake noted that after the removal of the mesh and healing of the erosions in 2008, Mrs Gill was able to return in a relatively short time to intercourse and her vestibulitis symptoms disappeared as did other symptoms of which she complained, including bowel-related symptoms. She noted, however, the continued vaginal bleeding which she suspected was due to fibroids.3286
3978 In August 2011 Mrs Gill presented to her GP in distress. She reported that she was not coping well, was eating compulsively, and following a reduction in her dose of Cymbalta, she was fighting and crying. She felt she had no support in Perth and missed her mother in Melbourne.3287
3979 The following month she had a long consultation with a different doctor at the medical centre, Dr Lewin Bedford-Brown. He noted that she had been on Cymbalta for the last year and that she thought she was getting terrible side-effects from it including hot flushes and irritability. He advised her to gradually decrease her dose of Cymbalta and wrote her a script for Effexor XR which he recommended she begin to take gradually.3288
3980 On 17 October 2011 Mrs Gill presented to a doctor at the medical centre and they discussed her mental state.3289 A week later, Dr John Terry, also a GP at the medical centre, prepared another mental health plan. He referred her to Dr Maretha Cronje, a clinical psychologist, for counselling.3290 She had six sessions, beginning on 3 November 2011, which included cognitive behavioural therapy.3291 She noted that Mr Gill was also having therapy.3292
3981 By 22 December 2011 Mrs Gill told the GP that she felt “normal”, was coping well “with lots of stuff”, and her mood and energy were good.3293
3982 In early 2012 the family moved to Geelong to be closer to Mrs Gill’s family. She hoped the move would help her to improve. She obtained a teaching position at a TAFE institute in Geelong working up to four days a week when the work was available. 3294 She began attending the Newtown Medical Centre.3295 At what appears to be the first consultation there in April 2012 she saw Dr Laura Bridges.3296 She told Dr Bridges about her saga with anti-depressants and said that she felt a little agitated on them and wondered whether she could be weaned off them. She told her that her mood was good and had been so for some time. Dr Bridges reduced the dose from 60mg to 30mg daily.3297 She also referred her for investigations of palpitations which Mrs Gill said she had felt a few times a week over the previous two months.3298
3983 In May 2012, however, Mrs Gill returned to the medical centre where she saw Dr Genevieve Seabrook. She told Dr Seabrook that she had started decreasing the Cymbalta and was feeling very dizzy and nauseous and more anxious and depressed. She said her husband was fed up. Dr Seabrook suggested switching her to an SSRI and issued her with a prescription for Luvox 50mg 1 daily. A week later she saw Dr Seabrook again. Mrs Gill said there had been a mix-up at the chemist and she had been given Lovan 20mg which she had started five days ago and that her dizziness had settled and she was much less anxious, though she still felt sad. Dr Seabrook ceased prescription of the Luvox 50mg daily and instead prescribed Lovan 20mg 1 daily and gave her the names of some psychologists she could see for marriage counselling.3299
3984 By mid-June 2012 Mrs Gill reported feeling much improved on the Lovan with no anxiety symptoms. She told Dr Seabrook that the marital issues continued and her husband had lost his job. Both she and he had seen a marriage counsellor. Dr Seabrook discussed mental health care plan issues with Mrs Gill and referred her to Jacqueline Payne, a psychologist. 3300 Mrs Gill attended for treatment on four occasions, the last in November 2012.3301
3985 By March 2013, however, she was still struggling with anxiety and depression. Dr Seabrook renewed her Lovan prescription and referred her to another psychologist, Dr Emily Hill.3302
3986 During April and May 2013 Mrs Gill attended six counselling sessions with Emily Hill, a psychologist in Dr Mackey’s employ.3303 In a letter to Dr Seabrook dated 23 April 2013 Ms Hill wrote that Mrs Gill reported symptoms consistent with a diagnosis of major depressive disorder, noting that she also felt “stressed, anxious and jittery most of the time”.3304 Mrs Gill told her that she had experienced several traumatic events throughout her life which she felt she had not adequately resolved.3305 On 23 October 2013 Dr Hill informed Dr Seabrook that Mrs Gill had responded relatively well to cognitive behavioural therapy, based on her reports at their last session on 31 May of feeling less depressed and experiencing fewer intrusive thoughts about the past.3306
3987 In the middle of 2013, things took another turn for the worst. Mrs Gill could feel something inside her again and when she explored her vagina with her fingers she could feel something sharp and her vagina felt lumpy. Once again intercourse became painful and if she orgasmed she would experience severe cramps. She also started bleeding after intercourse and her bowel movements became painful once more. What is more, she noticed that she leaked a little urine especially when she sneezed or strained and was not always able to make it to the toilet on time.3307
3988 She deposed that this deterioration in her symptoms “absolutely gutted” her.3308
3989 On 29 May 2013 Mrs Gill consulted Dr Seabrook. Dr Seabrook recorded the following history:
Needs r/f Melb uro-gynae for r/v ? vag Infection due to mesh erosion
Has felt bit unwell with vag discomfort, itch & discharge - has started Canestan Cr[eam] - improved but still some itch
Had prolapse surg 2007 with mesh - since then 4 further ops for Cx's [complications] incl prev erosion of mesh
Has spoken to Perth urologist who recommended Melb spec.3309
3990 Dr Seabrook referred her to Dr Caroline Dowling, a Melbourne urological surgeon.3310 At a consultation on 29 May 2013 Mrs Gill said that Dr Seabrook told her that other women had had similar problems as hers as a result of mesh implant surgery for pelvic prolapse and suggested that she google “mesh-implant problems”. Mrs Gill did as she suggested. She deposed that when she did so:
I found various pieces of information about mesh problems which I had never before heard of, such as that the problems associated with mesh could be permanent and also very debilitating. I also saw a story on the ABC's 7:30 Report about a lady, Julie Davis (the former lead applicant in the proceedings). This was the first time I heard there was legal action against Ethicon and Johnson & Johnson in respect of mesh implants for pelvic prolapse.3311
3991 On 5 June 2013 Mrs Gill saw Dr Dowling. In her report to Dr Seabrook written the same day, Dr Dowling recorded a history of her implantation with Prolift in 2007 and her subsequent “issues with vaginal erosions and one of the arms being quite tight” and of the three operations by Dr Yin. She noted she had been “quite well probably since 2008” until about three weeks before the consultation when she had some night sweats and when she explored digitally she felt she could feel something sharp. She also noted:
She was also experiencing some pressure on her coccyx and increasing problems with bowel dysfunction including constipation and quite a lot of pain between motions and quite regular motions at other times. She has a background of low back and crampy like labour pain which is constant and she is aware of all day but is not quite worth taking pain relief for. Her urinary symptoms do not bother her as much although she does admit to having a small amount of urinary leakage on the way to void. She is using a Poise pantiliner. She has had no urinary tract infection nor haematuria. She feels that the vagina is somewhat lumpy to palpation but is not aware of any prolapse or tissue protrusion. She finds intercourse deeply painful but otherwise tolerable. She has a Milena in situ and was wondering whether she may have actually palpated the strings. She is having some issues with quality of orgasm.3312
3992 On examination Dr Dowling noted a rough area to palpation at the introitus posteriorly and also just distal to the cervix on the anterior wall but she could not see any mesh. So she organised for Mrs Gill to have a CT scan of the abdomen and pelvis with contrast. In the meantime she placed her on some topical vaginal oestrogen, gave her some Movicol (presumably to soften the bowel motions) and referred her to a physiotherapist in Geelong.
3993 The CT scan was taken on 18 June 2013. The radiologist found a large mass measuring 5.6cm in diameter emanating from the myometrium, which he considered most likely represented a uterine fibroid. 3313
3994 Dr Dowling reviewed Mrs Gill on 16 July 2013.
3995 At this time Mrs Gill was still having problems with vaginal bleeding. She was getting bright fresh blood after intercourse and after Dr Dowling’s examination. Her husband was also aware of “something sharp with intercourse”. Dr Dowling recommended that Mrs Gill undergo an examination under anaesthesia with a view to changing her Mirena, checking for mesh exposure and, if possible, treating it at the same time.3314
3996 Mrs Gill said that she became very anxious and tense at this point. She explained:
The whole reason why I had the mesh implant surgery in the first place was to get better and not have to have further surgery, and yet over six years later, I still was not better and was facing the prospect of yet another operation. I certainly was not looking forward to further surgery, and I knew it would potentially take a few months to recover. However, I accepted that the surgery was necessary so I decided to proceed.3315
3997 On 8 August 2013 Mrs Gill was admitted to St Vincent’s Private Hospital in Melbourne. At the operation Dr Dowling found a 2cm mesh exposure lying vertically in the midline just distal to the cervix in the anterior vaginal wall which she excised. She also performed an anterior vaginal repair and a colleague, Dr Alison de Souza, changed Mrs Gill's Mirena. Rectal examination revealed bands [formed by scar tissue] from the previous mesh surgery which Dr Dowling believed could contribute to poor bowel emptying.3316 A benign cyst was removed at the same time. Post-operatively she was given topical oestrogen. The histopathological examination confirmed the presence of mesh associated with mixed inflammatory tissue and actinomyces [a species of bacteria] like organisms.3317
3998 In her letter to Dr Seabrook of 8 August 2013, Dr Dowling expressed the hope that Mrs Gill would not have to have this kind of procedure again but said that she would need ongoing observation, “given the number of times it [presumably mesh exposure] has been an issue so far”.3318
3999 Mrs Gill was discharged from hospital on 9 August 2013.3319 She deposed that she was largely bed-ridden for several days thereafter.3320 For the first time, she also experienced pain originating in her lower right pelvic area and travelling down the front of her right groin and leg, which she termed “sporadic nerve pain”.3321
4000 About six weeks later Mrs Gill tried to return to work but her capacity to work declined. She said that she became unreliable because of the pain she was experiencing.3322
4001 In September 2013 she obtained a further script for Lovan 20mg and on 29 October 2013 Dr James Carter, another GP at the Newtown Medical Centre, devised a further mental health care plan. She presented to Dr Carter with anxiety issues due to her recurrent gynaecological surgery since the prolapse repair as well as relationship problems. At this time Mrs Gill was also seeing a psychologist, Helen Handsjuk and she also had an exacerbation of her psoriasis for which she was prescribed a topical ointment.3323
4002 Although the consultations with Ms Handsjuk were described as “sex counselling”, the report from Ms Handsjuk to Dr Seabrook revealed a more complex picture. On 14 December 2013, after six consultations, Ms Handsjuk wrote:
Kathryn presented with symptoms of anxiety and depression pertaining to her sexual relationship with husband Steve with whom she has been since 1997.
Kathryn described her sexual relationship with Steve as being mutual, adventurous and enjoyable until after the birth of their second son in 2004 after which she suffered with PND followed with pain due to prolapsed uterus, bowel and bladder. A faulty mesh during the first operation, resulted in several other corrective operations and will necessitate future operations as the mesh erodes and protrudes, causing pain and discomfort. With a fear of potentially losing Steve if she continued to disclose the level of pain, Kathryn subconsciously kept the pain to herself as well as the frustration and resentment resulting from obligatory SI.
The psychological interventions of C.B.T. [cognitive behavioural therapy] and sexual counselling have gradually been addressing the fear, communication, disengagement from psychological and physical pain, and progressive new re-engagement with one another. A general plan has been discussed but will require substantially more time to implement.3324
4003 On 12 November 2013 Mrs Gill returned to see Dr Dowling. According to Dr Dowling’s report to Dr Seabrook, she had improved pain control and was managing better now her anxiety had reduced. She did, however, report intermittent pains, especially after intercourse and any heavy activity like travelling or moving suitcases. Dr Dowling wrote that they seem to be “shooting like pains which originate in the pelvic region and can go down into her legs”. She added that Mrs Gill also suffered a different “visceral” pain with a very large bowel action which she has been able to control well using Movicol daily and has only had it twice since the last consultation (daily before that). Evidently Mrs Gill also reported having more success with more frequent intercourse and was finding the physiotherapist and the counsellor very helpful.3325
4004 On examination Dr Dowling found what seemed to be a persistent residual suture just distal to the cervix on the anterior vaginal wall which she considered should be investigated.
4005 In December 2013 the physiotherapist, Celia Bolton, reported to Dr Dowling that Mrs Gill had complained of urinary urgency, urinary urge incontinence and nocturnal frequency; apareunia due to posterior vaginal wall and deep pain; constipation and difficult and incomplete defaecation with associated post-defaecation pain in the coccyx area; right groin pain; and vaginal discomfort with voluntary pelvic floor exercises “like fingers down a blackboard”. Although there had been improvement in suppressing her urge and she was now continent, the defaecatory problems persisted despite daily Movicol and defaecation retraining. She noted that her right groin pain, anal pain, and sexual activity had not improved. She referred her to a physiotherapist in Perth. I note that Ms Bolton undertook a musculoskeletal hip assessment and concluded that it (presumably the hip or the musculoskeletal system or both) did not appear to contribute to Mrs Gill’s pain.3326
4006 Early the following year Mrs Gill returned to Perth with the family. She said that she and her husband had decided that this was the best place to raise their sons and to enjoy an outdoor lifestyle.3327
4007 On 29 May 2014 Mrs Gill had an external haemorrhoid incised [cut into] and drained s after she presented with rectal bleeding with a bowel motion.3328
4008 On 4 June 2014 she presented to Dr Jill Christophers at the Seacrest Medical Centre with ongoing pain in the right groin and she was referred back to Dr Yin.3329 Dr Christophers also referred Mrs Gill for a pelvic ultrasound which revealed a 5.4cm fibroid in the posterior uterine wall.3330
4009 On the same day, Chloe Serrao, a continence and women’s health physiotherapist, reported to Dr Yin that Mrs Gill had been referred by the Victorian physiotherapist for ongoing physiotherapy management of her difficulties and pain associated with evacuation and dyspareunia. Ms Serrao diagnosed her with the following problems:
• Pelvic pain – low abdominal, coccyx pain and groin pain following evacuation 7-l0/10 burning, tearing spasm pain lasting minimum 30 mins, low grade constant pain 3/10. Significant worsening in the last few weeks
• Bowels: difficulties emptying, focusing on avoiding straining, type 2-3 stool, looser stool caused faecal incontinence, emptying up to 3-4x day, recent burst of haemorrhoids with straining, aware poor fluid intake
• Dyspareunia – onset following prolift repair, deep vaginal pain 7/10 penetration, can alter position to reduce sometimes, not attempting very often due to pain
• PFM dysfunction: Increased resting tone and pain reproduced through iliococcygeus, puborectalis and obturator internus with reproduction of pain caused with evacuation, incomplete relaxation
• Increased muscle tone through abdominals and gluteals
• Prolapse: aware of worsening lump, grade l cystocele and uterine descent at rest and on cough
• Psychosocial: anxiety, stress, worried mesh erosion again
• Urgency, UUI occasional and minimal bother to patient, RTUS small PVR 30mls but voided without strong urge.3331
4010 In her letter to Dr Yin Ms Serrao drew Dr Yin’s attention to Mrs Gill’s high anxiety that her symptoms were related to another mesh erosion.
4011 On 10 June 2014 Dr Yin referred wrote to Dr Tim Jeffery, a urogynaecologist with “expertise with Prolifts and their potential problems”. She asked him to attend a cystoscopy and examination under anaesthetic with her to see what was feasible for Mrs Gill. In the letter, Dr Yin noted that since Mrs Gill had returned to Perth she continues to complain about pelvic pain “but remains stoic about this”. She also stated that she continued to have “significant discomfort with a large bowel motion that we think is related to the compression effect of [the] posterior bands”. She told Dr Jeffrey that Mrs Gill “would be delighted to have all of her mesh removed but understands that this is an impossibility” but that she was willing to think about a possible hysterectomy if it would allow access to the vaginal vault and allow release of the posterior bands.3332
4012 To make matters worse, at about this time the manager of the FMTC informed Mrs Gill that there was no more work for her.3333 Mrs Gill looked for other work and managed to find a casual position with the Department of Education as a Relief Education Assistant (Special Needs), working at the Belridge Secondary Education Support Centre, working one day a week for the princely sum of $26 per hour. She formed the opinion that casual lecturing work would be unavailable in the future due to funding cuts to TAFE and decided to increase her skills by obtaining a Diploma of Education (secondary), to enable her to work as a secondary school teacher.3334
4013 Mrs Gill saw Dr Jeffrey on 14 August 2014.3335 Following an examination he confirmed her fears. He found a small suburethral mesh ulceration. There was very significant tenderness in the posterior vaginal wall, and the right-sided mesh arm from the posterior prolapse passing to the sacrospinous ligament on the right was extremely tense and tender. Palpation of this mesh arm produced a significant episode of pain radiating from the rectum to the pelvic side wall and down the right leg. He reported to Dr Yin that this was typical of a mesh arm too tight through the sacrospinous ligament. He also noted that Mrs Gill had “a bulky fibroid uterus” which was putting pressure on the rectum. He discussed with Mrs Gill her various surgical options. He wrote:
She understands that further division of the mesh may not change her symptoms. I discussed the role of mesh contracture, which is why her symptoms are becoming more obvious as the mesh shrinks in response to shrinking scar tissue. When she becomes post-menopausal this is going to get worse.
(Emphasis added)
He said that ideally Mrs Gill needed to consider a further vaginal repair procedure dividing all the mesh arms off the anterior and posterior Prolift. He noted that there was a risk of recto-vaginal fistula formation if the rectum were damaged during the posterior mesh manipulations. He discussed the role of hysterectomy with Mr and Mrs Gill but did not think it was indicated at that point although he thought it might well be necessary in the future.
4014 Dr Jeffrey sought a second opinion from Dr Nicolas Tsokos. Dr Tsokos examined Mrs Gill on 28 October 2014 and reported to Dr Jeffrey:
Kathryn’s problems remain as a relatively persistent right groin pain and an intermittent rectal and low abdominal pain usually associated with defecation of a large stool but perhaps most troubling is her persistent dyspareunia (scil).
On examination there is possibly a small mesh erosion mid urethrally, a recurrence of cystocele, a degree of rectocele recurrence and an acutely anteverted bulky uterus. The vagina overall seems of reasonable capacity and length but there are two very tight bands of mesh palpable rectally.
As always mesh and its complications provide a difficult issue, particularly in a relatively young woman in whom the inability to have satisfactory intercourse is creating a marital problem. I perceive that she really has three options for treatment.
One is consider using agents such as Lyrica and I have provided her with a script for this to see if there can be some resolution of her pain though I doubt that her dyspareunia (scil) will be resolved.
Given that there may be a small mesh erosion anteriorly examination under anaesthetic and excision of that mesh would seem reasonable but the question really is whether an attempt should be made to excise the posterior arms of mesh because of the attendant risk of rectal damage. Perhaps this is an issue that we could discuss.
Finally, there may be some value in removing the uterus. I don't know that this is a definite and whether one would combine it with an attempt of excision of the mesh. My own feeling would be not to attempt to combine a hysterectomy with excision of the mesh but then it is not clear in my mind whether hysterectomy would be significantly beneficial.
As I say l think it would be best if we could discuss her case. Perhaps we could arrange for EUA and initial mesh excision as a combined case.3336
4015 Lyrica, I interpolate, is an anticonvulsant indicated for the treatment of diabetic nerve pain, pain after shingles and fibromyalgia.3337
4016 In June 2015 she began full-time studies for a graduate Diploma of Education at the University of Notre Dame in Fremantle, hoping to qualify by September 2016.3338 At the same time she continued working one day a week for the Belridge Secondary Education Support Centre.3339
4017 In the meantime, in December 2015 or January 2016, Mrs Gill’s prolapse returned. She felt it was not as bad as the initial prolapse. She deposed that “what protrudes from [her] vagina is about the size of a grape”, which she pushes back but it inevitably “slowly falls back out again over the course of a day”. She expressed deep concern that she would need yet another operation.3340 In her first affidavit she deposed:
I do not want to have another operation. I know that, based on my previous operations, this will stop my life for anywhere between one to three months, depending on my level of pain and the extent to which it takes to recover. I know that I will then probably need to try to lose the weight that I will most likely gain following the surgery, provided my specialists allow me to exercise. I get anxious thinking about when would be a good time to have the surgery given that I am studying full time and working part time. Towards the end of the year, I would like to secure work as a teacher, and as a new employee, I do not want to have to ask for time off work to have further surgery. I also get clingy with my boys because I anticipate that when I have the operation I will be away from them and that I will be unable to be a good mother for them while I am recovering.3341
4018 She added that she was worried about her husband driving the boys, a task she undertakes despite the fact that his driver’s licence was reinstated in early 2014 because she “knows” his vision has not fully returned.
4019 In February 2016 Dr Jeffrey reviewed Mrs Gill. He noted that Mrs Gill was having increasing pelvic pain and discomfort with intercourse, bowel action and “generally moving around”. Clinically, however, he felt that the pain was more related to her uterus than her previous mesh. The uterus was particularly tender to move and compression of the uterus produced a lot of the pain she experiences which Mrs Gill attributed to the mesh. Dr Jeffreys could not find any evidence of mesh ulceration and examining her and touching the mesh during the procedure did not produce significant pelvic discomfort. He thought that uterine adenomyosis was probably the cause of a significant amount of her pelvic discomfort.3342
4020 Dr Jeffrey recommended that Mrs Gill consider an abdominal hysterectomy which he felt would significantly improve her symptoms. He told the GP that her uterus is adenomyotic and fibrotic and that this was unrelated to her previously placed mesh.
4021 In March 2016 Mrs Gill was required to complete a two-week full-time teaching practical as part of her Graduate Diploma. For six of the ten days she worked, she had unexplained vaginal bleeding and was in strong pain over the course of the two weeks, but because she needed to stay alert and teach, she was only able to take Panadol and Ibuprofen. At the end of each day she was exhausted and went to bed at 6pm. Her husband had to do all the cooking, shopping and caring for the children over this time. On the weekends she mostly stayed in bed on both days because she was too tired and sore to do much else. She was “incredibly irritable and anxious during these two weeks” because she had to work through pain and could not predict when the pain and bleeding would start or stop. Before the most recent onset of pelvic issues in 2013 she deposed that she aspired to securing full-time employment at the FMTC or as a teacher elsewhere, once her boys reached High School. Now she despaired of whether she would ever cope with full-time employment.3343
4022 Mrs Gill returned to see Dr Jeffrey on 16 March 2016 in the company of her husband. Dr Jeffrey wrote in his report to the GP that they had decided to go ahead with the hysterectomy and were keen to proceed with it on 7 June. He noted that Mrs Gill understood that the operation “may or may not influence her pain” and that it “may not interfere with her mesh problems”. Yet he remained of the view that many of her symptoms related to adenomyosis in the uterus rather than the mesh.3344
4023 In April 2016 Mrs Gill was referred to Dr Stas Vashevnik, a gynaecologist “re previous mesh operation for prolapse”.3345 He examined Mrs Gill on 15 April 2016. Like Dr Jeffery he could not find evidence of a mesh erosion into the vagina and he, too, felt that a lot of Mrs Gill’s pain was due to the bulky uterus and fibroid. He emphasised that this was certainly a cause of her increased bleeding and “Mirena being ineffective”. He was strongly of the opinion that a hysterectomy via a laparoscopic approach with conservation of the ovaries would relieve a lot of her pain symptoms and stop the bleeding as well. He left her to think about it.3346
4024 Four days after this examination, on 19 April 2016, Mrs Gill saw a consultant psychiatrist, Dr Patricia Jungfer, at the request of her solicitors.3347 Dr Jungfer diagnosed her with a generalised anxiety disorder with panic disorder, an adjustment disorder with depressed mood, and a chronic pain disorder. In her first report Dr Jungfer wrote:
Mrs Gill’s difficulties with respect to anxiety occur within the context of her chronic pain. The generalised anxiety disorder and her adjustment disorder arise due to her lack of ability to resolve the situation, the persistent pain and the uncertainty regarding her longer term medical health. My understanding is, based on her information, that she is unable to have the mesh removed and therefore is likely to expect long term and chronic pain. Therefore, I would argue that the anxiety she experiences will be that of a continuous state as she is always concerned and anxious regarding the possible damage that the implant will cause. I would argue that this results in a permanent impairment. The permanent impairment results in changes with regards to, in particular, her self-care, socialisation, concentration, interpersonal relations and her ability to engage in paid employment. She has a mild to moderate impairment from a psychiatric perspective as she utilises positive adaptive strategies to minimise the impact of these psychiatric symptoms.3348
4025 Dr Jungfer considered that the fact that Mrs Gill continues to experience pain and the restriction it imposes on her activities exacerbates and worsens her anxiety state. She concluded that the difficulties with respect to anxiety, the generalised anxiety state, and the panic attacks were the direct result of the complications of the implant surgery. On the basis of Mrs Gill’s account that her anxiety impairs her concentration, reduces her resilience, and increases her fatigue, she expressed the view that Mrs Gill was unable to work full-time and that, as she continues to experience pain and ongoing anxiety, her earning capacity will remain impaired. She was also of the opinion that there had been a significant reduction in Mrs Gill’s self-esteem and self-confidence and a significant strain on her marital relationship because she and her husband could not have a sexual relationship without her experiencing substantial pain.
4026 Dr Jungfer’s diagnoses were called into question by another psychiatrist, Dr Anthony Samuels, who examined Mrs Gill at the request of the respondents. I will deal with the controversy when I come to consider damages. It is sufficient at this point to observe that it was common ground that “the mesh complications” had a significantly deleterious effect on Mrs Gill’s mental state.
4027 On 20 April 2016 Mrs Gill underwent urodynamic studies to determine the flow rate of urine during urination. These studies were carried out by Associate Professor Clara Shek at the request of Dr Christophers in conjunction with a cystourethoscopy.
4028 Mrs Gill presented to Associate Professor Shek with pelvic pain and deep dyspareunia following her Prolift prolapse repair. Associate Professor Shek noted her long history of chronic constipation and obstructive defecation. In her report to Dr Christophers on 20 April 2016,3349 she wrote that the obstructive defecation had improved after the prolapse repair but there was a new onset of pain on defecation when she had a bulky and hard motion for which she was taking Movicol. She also noted mixed urinary incontinence over several years with the urge component “at a bother score of 8/10 and stress urinary incontinence at a bother score of 4.6/10”. In addition, she said that there was urinary frequency and occasional poor urinary stream. Mrs Gill reported a vaginal bulge “at a bother score of 1.8/10”, two episodes of faecal incontinence, and flatal incontinence. She also apparently told her that she was planning to have a hysterectomy for menorrhagia and that her husband had had a vasectomy.
4029 Clinical examination revealed evidence of recurrent prolapse in all three compartments, tenderness at the left vaginal vault, but no lesions or mesh in the rectum and no mesh in the vagina or urethra.
4030 Associate Professor Shek diagnosed Mrs Gill with moderate urodynamic stress incontinence and a recurrent three-compartment prolapse against a background of bilateral levator avulsion and marked hiaital ballooning. She recommended conservative treatment including pelvic floor muscle exercises, bladder retraining and dietary modification, preferably with a trained continence therapist, and consideration of a trial of oral or transdermal anticholinergics for her overactive bladder symptoms. If her stress incontinence symptoms worsened, Associate Professor Shek said she could be offered a suburethral sling but the effect of implantation of a suburethral sling on her overactive bladder symptoms were difficult to predict.
4031 Professor Korda examined Mrs Gill at the request of her solicitors on 21 April 2016 and his findings and opinions were included in his first report of 24 August 2016.3350
4032 At this stage she was still considering the options raised by Dr Tsokos.
4033 At the consultation on 21 April 2016 Mrs Gill complained to Professor Korda of urinary frequency (every hour), nocturia (the passage of urine at night), twice; stress and urge incontinence; a slow urinary stream; occasional feelings of incomplete bladder emptying, and a recurrence of her prolapse. She also complained of constipation, having to use Movicol continuously, incomplete bowel emptying with occasional faecal incontinence and complete flatal incontinence. She complained of deep dyspareunia, especially on the right side. She had severe pain in her coccyx and nausea during orgasms and constant low abdominal, right groin and lower back pain which radiates to her loins. She told Professor Korda that she felt exhausted all day.
4034 Abdominal examination revealed no obvious tenderness or masses and her abdomen was soft. The vaginal examination revealed a normal introitus, the presence of a cystocoele, a second degree uterine prolapse and a first degree posterior vaginal wall prolapse. There was tenderness in the right sacrospinous ligament and in the left vaginal vault. GH was 4.25 cm, PB 5 cm. The levator palpated to Oxford grade II to III bilaterally with bilateral defects, indicating pelvic floor weakness.3351 Rectal examination revealed no mesh erosion.
4035 Professor Korda listed her injuries and disabilities as: moderate urodynamic stress incontinence with a mean urethral closure pressure of 57 cm of water without voiding dysfunction or evidence of detrusor overactivity; recurrent three compartment prolapse against a background of bilateral evulsion of the levator muscles and a ballooning of the urogenital hiatus to 35.24 cm2; bowel dysfunction manifest by pain during constipation, incomplete bowel emptying, flatal incontinence and occasional faecal incontinence. He noted her complaints of dyspareunia and difficulties during orgasm, manifested by pain in the coccyx and nausea, and of constant lower abdominal and groin pain and lower back pain radiating into the kidneys. He expressed the view that all her current symptoms and disabilities resulted from the insertion of the Prolift device, the resultant erosion and the multiple surgical procedures required to correct and manage her symptoms, although he retreated a little from this in cross-examination. I will return to this subject in Part XVIII.
4036 She returned to see Dr Jeffery on 9 May 2016. They had a long discussion about her options. She indicated to him that she was not convinced that she wanted to proceed with an abdominal hysterectomy.3352 He arranged to see her six months hence. In the meantime she was to see a pelvic floor physiotherapist in an attempt to improve some of her symptoms.
4037 It appears that Mrs Gill lost confidence in Dr Jeffery. In cross-examination, when taken to Dr Jeffery’s report of 16 March 2016, she said that “quite a bit” of their discussion which led to her decision not to have a hysterectomy was missing. She explained:
Because I asked him if by removing my uterus will it improve my groin pain, the damaged nerve there. He said no. Will it improve pain around my coccyx with my bowel motions? He said, no, it will not impact that. Will it stop the sharp pains I get with the tearing of the movement? No. So I said, what is the purpose of the hysterectomy? He said it might stop a bit of period pain, which is the adenomyosis. Which – that pain is when you actually poke my uterus. So I was not going to have another surgery with the hope if I get poked in my tummy, it won’t hurt me.3353
4038 It was put to her in cross-examination that she may have confused the appointments, since there is a long discussion about options in the 9 May 2016 report. Mrs Gill did not deny it. She merely said that she “took a lot of paperwork in for him to read about mesh, and how actually removing the uterus can actually put more strain on the mesh and – by creating a vaginal vault”. She told the Court that it appeared that that was “the first time he had heard things like that” and she lost a lot of confidence in him and started to look for a different doctor.3354 It is more likely than not that Mrs Gill was recounting what happened during the consultation of 5 May, since she returned to see him after the March consultation and, although he intended to review her again six months after the May consultation, this was the last time she saw him.
4039 In the end, Mrs Gill elected not to go ahead with a hysterectomy.
4040 On 18 July 2016 Mrs Gill saw a colorectal surgeon, Dr Alan Meagher, at the request of her solicitors. She told him about her ongoing bowel problems and her pain. She said that, despite the multiple excisions of mesh and despite brief improvements, her pain had never settled. In particular, she told him that “the pain in her lower back where the mesh is attached continues to be an ongoing significant hassle”. Although she said that she had learned how to manage the pain better, it persisted and interfered with her daily activities. She told Dr Meagher that if she had an orgasm it set off her coccyx pain and she feels cramping pain like labour pains in her pelvis. If she became constipated she said it felt as though she is passing blunt sticks through her anus. Notwithstanding multiple examinations of her rectal area, she said that no-one had been able to find the mesh eroding through the rectum but she often still had some rectal bleeding. She also complained of a degree of flatus, faecal incontinence and urgency which she had not experienced before her prolapse surgery. She expressed great concern about the faecal urgency and incontinence given that she was about to start high school teaching.3355 There was significant scarring around her vagina.
4041 Dr Meagher carried out a vaginal and a digital rectal examination. He found marked thickening of the vaginal wall posteriorly and in his report to the solicitors he indicated that it was significantly tender. He also noted some blood discharge in the vagina. In the rectum he found some palpable thickening which he said was probably related to mesh but could just be bilateral scarring. He said that the area was abnormally thickened and tender. He found only very small haemorrhoids and there was no evidence of a fissure.
4042 In his report to the Court Dr Meagher wrote:
As an independent expert witness I don’t think there’s any real doubt that the Prolift implant did more likely than not cause physical damage to Mrs. Gill’s body. Specifically the areas of colorectal function which have been affected include the need to take Movicol to avoid large bowel movements. When Mrs. Gill has large bowel movements she suffers really severe episodes of pain.
Mrs. Gill also suffers ongoing chronic background pain which is difficult to describe – it does appear to be related to the mesh and the scarring around the mesh and at times appears centred on the coccyx.
She also suffers a degree of faecal urgency, incontinence of flatus and incontinence of liquid faeces as a result of the surgery and partly as a result of the need to take Movicol to avoid the pain. There is also a degree of faecal urgency and Mrs. Gill needs to stay near a toilet and again is concerned about the effect this may have on her work3356
4043 Dr Meagher’s unequivocal view was that if Mrs Gill had not been implanted with the Prolift, more likely than not she would avoided the physical damage to her body that led to the revision surgery and he had no doubt that the complications of the Prolift implant had had “a very substantial effect” on Mrs Gill’s life.3357
4044 At the time she signed her first affidavit on 27 July 2016 Mrs Gill gave the following description of her ongoing symptoms and disabilities:
188. I continue to experience different kinds of pain in my pelvic region.
189. I have an almost constant pelvic ache just above my pubic bone. The pain feels as though it commences from just inside my hips, and runs across my waist above my pubic bone. It is a cramp-like sensation as though my muscles are bruised, and also feels a little like period pain. I would rate the pain at about a two out of 10. Mentally, I try my best to block out the pain. The pain fatigues me and can lower my tolerance for other irritations such as a headache or loud TV, or the boys fighting. Because I am fatigued, I generally go to bed early or lie down with a heat pack on the area to give me some relief.
190. I also experience what I describe as nerve pain; the pain starts in my lower right pelvic area and travels down the front of my right groin/leg area. This is most prominent when I am sedentary or at rest (usually at night while trying to get to sleep, and while studying or working at my computer). This pain is relatively constant.
191. I experience a sharp, sudden pain, which is almost like the feeling of a tearing injury. I feel the pain in my lower pelvic region; it can be on the left or right hand side, or central. This kind of pain can occur from sneezing, by turning or moving sharply, bending to pick up groceries, pushing a shopping trolley, twisting quickly while getting out of the car, turning over in bed, from performing household chores such as vacuuming and scrubbing, and from tickling and playing with my boys. This pain also occurs when my bladder is full. If I have any pressure on my bladder for example during the night I get pain. This kind of pain stops me instantly and I have to wait for the pain to pass before I can continue. The pain can last from 10 seconds to two minutes.
192. I also experience pain in the vagina on the right hand side and at the top of the vagina with sexual intercourse. I have to move during intercourse to avoid contact with these areas.
193. I also experience a deep pain based in the coccyx area. This pain is awful; it is deep and strong and sickening. The only thing I can liken the pain around my coccyx to is labour pain, but it is right at the back and it builds to something that is all consuming; there is nothing I can do to overcome it. This pain around the coccyx region is triggered by bowel movements. For the past three or four years, I have taken laxatives to reduce straining as this triggers the pain. Every now and then I get a bit out of balance and I end up with harder motions and that sets off the pain again. So I can see that it is really closely related to my bowel motions. The pain around the coccyx region is also triggered by orgasm and so I have avoided orgasm to avoid the pain.
194. Often when I have pain in the coccyx region, I cannot focus or speak and need to lie down if possible, and concentrate on my breathing. The pain causes me to get shaky, and sometimes feel sick. I often want to cry during and after these bouts of pain. It takes a big effort to pull myself together and get on with whatever I was doing beforehand, and pretend it didn't happen. Fortunately, this kind of pain does not happen often, probably about once per month.
195. I continue to have bleeding/spotting, which usually occurs after low impact activities (I avoid high impact activities). For the last year, this has occurred on average about once or twice a week. There have been many occasions when I have gone out on the water in a boat with my family and the constant movement and bouncing around on waves has caused me to bleed. This makes me upset and anxious as I do not want to stop boating and miss out on time with my family. Bleeding/spotting can also occur from laughing a lot, dancing, having sex, doing a low impact exercise circuit .or performing housework. The bleeding/spotting can last anywhere between half a day to two days. This has been the case for about the last 12 months.
196. I experience flatulence. I sometimes have no control over wind. I also have issues with urinary leakage if I do not get to the toilet quick enough. I worry about the impact of these things on my teaching aspirations.
197. Before the mesh operation I did not experience pain in these regions, although I did experience some pain with intercourse in connection with vaginal prolapse.
198. My sexual relationship with Steven continues to suffer; we probably have intercourse about once a month on average compared to two to three times a week prior to my pelvic organ prolapse. I no longer really want to have sex, and whenever we do I find that I need to keep telling Steven whenever it hurts. I find it particularly painful on the right side of my vagina. I am also now only able to have sex in one or two positions. Intercourse is therefore always quite tentative and cumbersome. Also, as mentioned above, climaxing can lead to terrible pain. I estimate that about one in three orgasms result in awful pain, so I often deliberately avoid climaxing. I have found I also experience pain following intercourse.
199. I now have a sense of discomfort whenever my bladder is full, and I often wake several times a night to empty my bladder.
200. If I get bumped or hit in the stomach, I end up with very bad cramping pain. The pain is sharp and is felt in my lower pelvic region. Even relatively minor hits to the stomach result in severe pain. For instance, I recall on one occasion I was swimming with my boys and one of them playfully threw a Wahu beach ball at me which hit me in the stomach. I was not looking at the time so I could not brace myself and I ended up being in terrible pain.
201. The pattern of good and bad episodes of pain is not predictable. I have had periods of up to two or three weeks where I have not experienced pain. But there has always been an end to these episodes of respite and pain has returned.
202. On average, I generally have one day each week where my pain is noticeably worse than the other days. I often cannot predict when that may occur. I find those days particularly exhausting, and I often end up having to take Panadol and just lie down to wait for the pain to pass.
203. I am often extremely fatigued. To try to manage this I often go to bed very early - around 7:00pm. I generally fall asleep easily. However, even though I go to bed early I find that when I wake in the morning I am still fatigued, and this only gets worse over the course of the day.
4045 She also deposed that, ever since her implant surgery she had had “a constant sense of being worried and anxious”. She said that she gets very anxious whenever she thinks about her medical condition and the decisions she needs to make about treatment and she is afraid of suffering more complications. She worries about what her doctors will advise her to do. She continues to worry that she is in her present predicament because she did something wrong and acted contrary to medical advice.3358 She is very concerned and distressed about her children often seeing her as sick or unavailable. She worries about the impact her experiences will have on them, especially her unavailability due to illness. She feels guilty because she perceives she has hurt them. She is distressed about her “lack of functional ability” in comparison to how she once was. She continued:
206 I think I have failed as a woman in that I have not been able to fulfil all the responsibilities of a woman. I have self-critical thoughts; for example, that I have been sabotaging my sex life unconsciously and that I am too lazy and not trying hard enough to heal myself and provide for my husband and children. These thoughts always come back to the feeling that my problems are my fault.
207 The thing that scares me the most is what possibly could happen in the future. When I get a strong pain I think, “imagine if I have that constantly for the rest of my life.” And then that scares me, because I think, is that possibly what my future is? Is it just going to slowly get worse? So far, no one has been able to give me an answer and to me that is just as bad as being told that it will get worse. I feel frightened that I have a medical condition that even the best surgeons do not know how to manage and they cannot predict what will happen to me. I do not know who to go to. I do not know who to trust. I do not know who can deal with it properly. I am in a holding pattern at the moment, where I do not want anyone to touch me. These thoughts lead me to feel very anxious and overwhelm me at times.
208 My constant worrying and anxiety pervades pretty much all aspects of my life. Sometimes my anxiety causes my heart to race, and I start to shake and become tearful. These symptoms seem to occur more often in the second half of the day when I am more fatigued.
209 Since my mesh surgery I have experienced, and continue to experience, low self-esteem and self-confidence. I have also become increasingly emotional and prone to mood swings. I am also often tearful.
210 I do not believe I am much fun to be around anymore, and even though I have a good network of friends, I find myself often avoiding engaging with people. As a result, I have become more socially withdrawn.
211 I now find it difficult to relax.
212 I find myself comfort eating at times.
4046 On a bad day with pain she finds it very hard to concentrate or focus. She described the pain as “very distracting”. On these days she deposed that she is unable to make important decisions or undertake mentally demanding work and becomes quite forgetful.
4047 She said that the constant sense of anxiety she now has and the feeling of being unable to cope is “much different” from the pervasive sadness she experienced with post-natal depression.
4048 Between July and September 2016 Mrs Gill was required to complete a 10 week full-time teaching practical component. During that time she said that she suffered similar symptoms to those experienced in March 2016, including bleeding and strong pain. She took over the counter analgesics, eschewing stronger painkillers to ensure that she remained alert whilst with the students. She described this period in the following way:3359
The first four weeks of full time work absolutely wrecked me. The pain and exhaustion at times was so severe that I believed I would not be able to complete the 10 weeks and gain my qualification. At one stage I called my university supervisor at Notre Dame University, Anne Coffey, and told her I would need to withdraw. With Anne's help, an agreement was reached with my supervising teacher to reduce from full time to four days a week. When I was not actively teaching during the day I would lie down in the school’s (scil) 'sick bay' to perform my preparatory work for my next class. The reduction in days and ability to [lie] down in the sick bay allowed me to get through the practical but it was horrible.
4049 Throughout this period her husband was responsible for all the cooking, shopping and caring for the children. Mrs Gill said that she was unable to participate in any recreational activities and had limited engagement with her children as she spent the weekends recovering in order to be able to work during the week.3360
4050 In late October 2016 Dr Christopher referred her to a pain clinic and on 4 April 2017 she saw Dr Phillip Kriel, a specialist pain management physician. He noted that she continued to have significant pain in both the pelvis and the right lower abdomen which interfered with many aspects of her life and that she used both nociceptive and neuropathic descriptors for the pain, a factor her counsel submitted tended to support the evidence of Professors Klosterhalfen and Iakovlev about the entrapment of nerves in scar tissue. Nociceptive pain is defined in The New Shorter Oxford Dictionary as pain in response to, or caused by, a painful stimulus, and neuropathic pain as pain caused by disease or dysfunction of one or more peripheral nerves.
4051 Dr Kriel considered all Mrs Gill’s pain to be “consistent with the mesh”. He noted that she was currently taking two Panadol tablets when needed three times a day, anti-inflammatories (Voltaren, Naprogesic, Celcoxib or Nurofen) as needed, Diazepam if the pain was “out of control” twice a month, and Buprenorphine tablets as needed for strong pain. He made a number of recommendations for treatment, while expressing doubt that there was “much more on the medicine front” that could be improved upon. Those recommendations included Nortripyline 10-20mg two hours before bedtime to add neuropathic pain cover as well as to increase the quality of her sleep. He also expressed the opinion that she could “gain much by modest natural antiinflammatories in the form of fish oil 5g per day and curcumin 600mg per day”, that she had “a lot to gain” from hydrotherapy three to four times a week, that she could “gain much by further pain psychology through Anna Dale at Balanced Health” at first on her own, but later with her husband, and that she had the potential to gain much from a trial of neuromodulation [spinal cord stimulation].3361
4052 In early 2017 Mrs Gill felt that she was “at the end of [her] tether and needed some hope of treatment” especially for her ongoing pain. She did not think there were any surgeons in WA who could help her so she asked for a referral to Dr Thierry Vancaillie, a gynaecologist and pain specialist and clinical professor in gynaecology at the University of NSW, whom she believed to be a surgeon who specialised in the treatment of transvaginal mesh complications. Although at this time she knew that the implant was hard to remove, she was optimistic that he might be able to help her.3362
4053 She consulted Professor Vancaillie on 3 July 2017.3363
4054 When he examined her, he found, amongst other things, that the right Alcock canal (also known as the pudendal canal) was exquisitely tender and that residual mesh was palpable to a few millimetres above the canal and about one centimetre caudal from the ischial spine. He also noted that her uterus was enlarged by the fibroid but was mobile and non-tender.
4055 Professor Vancaillie briefly discussed with Mrs Gill the presence of the uterine fibroid. He considered that it did not need to be surgically treated, presumably because her uterus remained mobile and non-tender. He diagnosed her with a chronic pain syndrome (pudendal neuropathy) secondary to mesh insertion and removal surgery. He considered that further surgery would be difficult “to say the least” and would probably not be beneficial. He advocated what he described as a “conservative approach with topical as well as oral medications”.3364
4056 Professor Vancaillie informed Mrs Gill that he could feel residual mesh but that further surgery to remove the mesh was difficult and probably not beneficial because of the multiple surgeries she had had in the past. Mrs Gill was devastated by this news. Although she did not want further surgery, she was upset because it made her feel that her condition might never improve.3365
4057 Professor Vancaillie prescribed Zaldiar 37.5mg/325mg tablets, which Mrs Gill found to be the first strong pain medication that sufficiently alleviated her pain whilst not significantly affecting her cognitive ability; Allegron 10mg tablets once nightly to treat her urinary symptoms and to help her sleep; Amitriptyline + Estriol organogel AH formulation once daily; and Valium 5mg vaginal suppositories.3366
4058 On the occasion of this consultation, Mrs Gill spoke to Elizabeth Howard, an osteopath and pain educator, who shared rooms with Professor Vancaillie. Ms Howard gave her advice about strategies to reduce her pain, including healthy eating, counselling, and regular exercise. This gave her some confidence that she had options to better manage her pain in order not to let it get out of control.3367
4059 Professor Vancaillie reviewed Mrs Gill on 15 November 2017. He took a history that she had derived “good benefit” from Zaldiar but still experienced pain with sitting. He prescribed a daily low dose of Palexia SR, a slow release opioid analgaesic, with the Zaldiar for breakthrough pain. He postulated that it might give her “a reasonable quality of life”.
4060 On 16 January 2018 Mrs Gill was examined by Dr Bernadette Brown, a Urogynaecology Fellow, at the “mesh clinic” of King Edward Memorial Hospital.3368 On examination Dr Brown found a fullness palpable suprapubically which she thought was likely to be the uterine fibroid. Mrs Gill was tender in her right groin adjacent to the adductor longus tendon and on vaginal examination there was a small area of mesh exposure on the posterior wall. The distal mesh edge was palpable at the introitus and the cut edges of the mesh were exposed along that length. She also found mesh exposure centrally on the anterior vaginal wall with the proximal mesh edge exposed along its entire length. She described the fibres as “quite elongated” and said that they appear to rub on the cervix. Her notes read: “tender +++on [right] anterior wall/apex where mesh fibres are exposed”. Pain prevented a full speculum examination.3369
4061 Dr Brown noted in her report to Drs Dowling and Yin that Mrs Gill had made it plain that she would not undergo any further surgical procedure to treat her mesh complications, adding “[s]he feels that she has been experimented on and wishes to wait until sufficient skill levels have been obtained before she will proceed with full mesh excision”.
4062 Mrs Gill asked Dr Brown about treatment options for the uterine fibroid. Dr Brown considered that at that particular time a hysterectomy was not a viable option but the question could be revisited if Mrs Gill were to undergo a mesh excision in the future. They also discussed the possibility of trialling Zoladex to distinguish between vaginal bleeding from adenomyosis/fibroid from vaginal bleeding from mesh exposure. Dr Brown referred Mrs Gill to a dietician, noting that she had gained weight because the pain was preventing her from exercising. Dr Brown was suspicious that, on top of her mesh-related complications, she had interstitial cystitis (chronic pain affecting the bladder) and was concerned about the possibility of mesh exposure in the bowel and the bladder.
4063 Mrs Gill continues to have the emotional problems she described at [204]–[214] of her first affidavit. She often feels irritable and has little tolerance for minor irritants. She withdraws from social situations due to lower self-esteem and embarrassment caused by flatal, faecal, and urinary incontinence. She said she does not like the person she is anymore and often cancels social plans. In her second affidavit, she described ongoing pain and discomfort in the following way:
i. I rate my constant, deep, aching pain across my lower abdominal behind my pubic bone similar to severe period pain ranging from two out ten to six out of ten on average.
ii. I rate my “shooting” pain in my lower abdomen as six out of ten to seven out of ten. It is this type of pain that affects my ability to concentrate, articulate sentences and my memory. This is very frustrating and embarrassing for me. My concentration is so affected on some days that I cannot complete important tasks such as paying household bills as I worry I will make mistakes.
iii. When I have a full colon or large bowel motion I experience pain of nine out of ten. During these episodes I feel like I am going through a deep labour contraction near my coccyx bone. I feel shaky, pale and dizzy when this happens and I am unable to talk. I use Movicol to avoid this pain which makes my bowels difficult to control. I have accidental bowel motions which I find extremely embarrassing, especially when this happens at work. This may happen once a month. I am still trying to find the balance between enough Movicol to reduce pain and too much that I lose control of my bowels.
iv. I have a constant right groin pain that I rate on average one out of ten to three out of ten. This sometimes becomes a sudden sharp pain that shoots down my right groin to my thigh and prevents me from supporting my weight. It can take up to 10 minutes for this to pass. When this happens I struggle to breath and must support myself on someone or something close to me until it passes.
v. I suffer differing pains throughout the week depending on my schedule. By Wednesday my pain is in the foreground. I cannot ignore it, I get irritable, forgetful and struggle to get through the day. On days like this I go home and lie down straightaway.
vi. Due to my pain and exhaustion levels Steven and I have not been intimate for almost a year. This is placing a significant strain on our marriage.
4064 Mrs Gill has made adjustments to avoid aggravating the pain. In her first affidavit she offered the following examples. She tries to avoid going grocery shopping and only does so if she is feeling up to it. Otherwise she pays to get her groceries delivered. She has been having her groceries delivered on and off for since about 2011. Whenever she does the laundry, she puts the basket on a stand while hanging out washing to avoid repetitive bending. She tends to stand more than sit because she has found that standing is less likely to cause cramping. And she avoids lifting heavy items like dog food, potting mix, or pool salt, and always seeks assistance.3370
4065 In September 2017 Mrs Gill completed her Graduate Diploma in Education (Secondary) and secured a contract at Duncraig Secondary Education Centre working three days per week with special needs children.3371 She finds that she is exhausted after a day at work and tends to lie down on the lounge when she gets home.3372 She struggles to keep up the three days. She said she felt that she was paying a very high price in pain in order to work and that was affecting her family. She painted a rather grim picture in her oral evidence, saying that:
[The work] has certainly tipped the balance in my life, in that it exacerbates my pain every day I work, and I spend the days off trying to settle my pain, and I find that in the last year that I’ve been working it has been quite rare for me to have a pain-free day, which tipples over into my family life. I don’t have a great quality of life at the moment. When I’m in pain, I’m quite irritable. I have low tolerance. I don’t like who I am and I’m sure my kids don’t like who I am. But my whole aim is to get on top of my pain by Monday so I can start again so that I can work effectively. So haven’t found that balance yet and I’m not sure how to either.3373
4066 Her explanation for why she switched to special needs teaching was similarly evocative:
I am qualified to teach year 12 ATAR marine and maritime studies which is my background and my strength. When I was doing my prac at school, I found the content – I loved teaching the mainstream teaching and I loved teaching navigation skills and chart reading. However, the environment is very difficult for me with my pain issues and my bowel issues. A class of 24 to 30 students, I couldn’t leave to go to the bathroom if I needed to. It was very difficult to mask the pain, whereas I find in the special needs [classes] with four education assistants and students coming and going to the toilet, I find that I can fit in with that routine a lot more easily and I find less judgment if I smell or if anything goes wrong. In mainstream with a whole lot of 17 year old boys and girls, it’s not nice for me, whereas my students who need support, there’s no judgment. I can be who I am. So as much as it’s probably not where my career – where I planned to be, it’s what I can deal with what I have.3374
4067 Mrs Gill rations her time engaging in activities with the family.3375 Whereas it was once a joy to do so, “any sort of movement like being in a boat or going four-wheel driving” exacerbates the pain and often causes bleeding. When asked how she felt about this and how it affected her relationship with her sons, then aged 13 and 15, she replied:
Oh, it’s awful because I – I don’t want them to feel like they’re being rejected and I’m not wanting to be involved. And I feel like I’m letting them down, and sometimes they don’t quite understand that why would I choose one and not the other or why can’t I do both. So I – and I – I hate not being part of their memories. I hate not being part of their special events. So when I am home on my own knowing they’re off doing a family event, I – I hate that because I’m meant to be part of their family.3376
4068 Nevertheless, Mrs Gill began exercising again towards the end of 2015 and returned to sailing dinghies on weekends. She said that Dr Jeffrey told her she was unlikely to cause further injury from sailing dinghies.3377 The boat she sails is small, 12.5 feet, and she sails with a crew member. When she was questioned about this in cross-examination, she said that she used to be “a very, very good sailor” and had competed in the Australian titles. She said she missed sailing “dearly” and in 2015 the family agreed to support her return to competition. She sailed for up to three hours on a weekend. It seems that the competition took place in January and she needed “a lot of pain medication” to do it, but she was very happy to have done so. She told the Court that she had not sailed since.3378 Her exercise consists of gym classes once a week on average. Mrs Gill said that she is unable to do any running, jumping or high impact activities in those classes and is given alternatives by the instructor.3379
4069 She has also resumed camping with the family but whenever they go camping, she avoids lifting heavy items like water containers and rests when she is tired.3380
Background and medical history
4070 The second applicant, Diane Dawson, was born on 23 March 1959. She married Geoff Dawson in May 1978. The couple have three children, born in 1978, 1981, and 1983.
4071 The first child was delivered with the aid of forceps and an episiotomy (an incision during birth into the tissues surrounding the perineum, the opening of the vagina).3381 The third was also an instrumental delivery.3382
4072 Mrs Dawson is now a grandmother. The oldest of her seven grandchildren was 10 at the time she swore the first of her three affidavits in July 2016.3383
4073 On 1 July 1988 Mrs Dawson presented to Dr Philip Hall, an obstetrician and gynaecologist, with intermittent irregular vaginal bleeding over the previous five years and some post-coital bleeding. On 27 July 1988, Dr Hall examined her under anaesthetic, performed a D&C (dilatation and curettage), cauterised the cervix, undertook a diagnostic laparoscopy, divided some adhesions, and aspirated some fimbrial (fallopian tube) cysts.3384 The surgery was successful and the bleeding resolved.3385 When Dr Hall reviewed Mrs Dawson on 12 September 1988 he noted a dramatic change. He said that her principal symptoms were now those of premenstrual bloating with which she seemed to be able to cope satisfactorily.3386
4074 Mrs Dawson has a long history of urinary incontinence dating back to her childhood. Dr Donald Moss, a urologist, whom she consulted in April 1990, wrote in a report to Dr Hall that when she was young, she was enuretic (incontinent of urine) during dreaming, that she had annual episodes of urge incontinence while she was a schoolgirl, and that she had always had a tendency to “giggle incontinence”.3387 Dr Moss attributed these symptoms to “a degree of bladder irritability”. Following the laparoscopy and cervical diathermy, however, she experienced urge incontinence of a trickle of urine if she delayed unduly and, on occasions, lost a larger volume of urine. Bladder drill and pelvic floor exercises made no significant difference. At the time of the examination by Dr Moss, Mrs Dawson also reported some dyspareunia deep on vaginal pressure but no coital wetting. Notably, on examination there was good support of the anterior vaginal wall and no demonstrable stress incontinence.
4075 On 2 May 1990 Dr Moss performed a cystoscopy but was unable to find a specific cause for her “early neurogenic bladder”.3388 Mrs Dawson said that, after this procedure, she did not have any problems with urinary incontinence for “many years”.3389 But she reportedly had some incontinence in July 1994, according to a report from a gastroenterologist, Dr Arunasalam Ambikapathy.3390
4076 She has also had a long history of constipation dating back to at least November 1992.3391 In July 1994, when she was seen by Dr Ambikapathy, she also reported increased flatulence over the previous six months which was becoming a source of embarrassment. He considered that she did not have a serious underlying bowel disease but “a motility disorder” and suggested modifications to her diet and fluid intake, the use of a bulk forming agent, and an exercise program.3392 When he saw her again about three weeks later he reported that she had made modifications to her diet and seemed to be improving. He noted she was less flatulent and had more energy.3393 He did not see her again until February 1998 when she was referred by her GP with problems with her bowel function and some pelvic pain.3394 Dr Ambikapathy performed an upper gastrointestinal endoscopic examination on 5 February 1998 and diagnosed mild gastritis.3395 Although he found no evidence of peptic ulcer disease, he put her on a trial of Pepcidine and Prepulsid.3396 The effect of Mrs Dawson’s evidence was that her bowel function and pelvic pain resolved soon afterwards.3397
4077 Mrs Dawson suffered from depression after the birth of her second child.3398 Since then, she has had recurrent episodes of depression for which she has received intermittent treatment. The history recorded by Dr Jungfer, who assessed her at the request of her solicitors, referred to these episodes as episodes of major depression which occurred within the context of psychosocial stress.
4078 In 2001 she experienced an episode of “left tennis elbow”, from which she completely recovered and in 2005 she had an episode of bursitis in her right shoulder from which she also fully recovered.3399
4079 Mrs Dawson is a non-smoker, having quit smoking the day before her surgery in July 1988.3400
4080 Mrs Dawson attended high school for less than four years. She left at the age of 16 after completing one term in form 4 (now referred to as year 10) and worked as a cashier at several grocery stores.3401 Since 1985, however, she has been employed by the St John of Good Hospital in Ballarat in various different capacities and since 1996 she has been employed by the hospital as a purchasing officer.3402 She described this job as a sedentary role in which she works seated at a desk for about eight hours a day, five days a week.3403
4081 Mrs Dawson was first diagnosed with a prolapse in 1999. On 16 November 1999 she saw Dr Judith Fleming, an obstetrician and gynaecologist, and diagnosed a cystocoele “with primary cervical descent”. Dr Fleming noted that she had marked urgency and some stress incontinence as well as persistent intermenstrual bleeding and poor libido. She removed a large and fleshy cervical polyp, sent the polyp to histopathology, and referred Mrs Dawson to an incontinence physiotherapist.3404
4082 Mrs Dawson returned to see Dr Fleming on 12 October 2000. Mrs Dawson was certain that her prolapse was increasing and Dr Fleming agreed but considered that the deterioration was minor and encouraged her to continue her pelvic floor exercises.3405 By August 2001, however, when Dr Fleming saw her next, the prolapse had increased further, she had increasing problems of constipation, unsatisfied defaecation, and unspecified prolapse symptoms. She also reported “terrible” periods and poor libido. Dr Fleming decided that the prolapse required surgical repair and, at Mrs Dawson’s request, arrangements were made for this to be carried out together with a vaginal hysterectomy.3406
4083 On 18 September 2001, Dr Fleming performed surgery on Mrs Dawson at Ballarat Base Hospital.3407 The operative findings were as follows: “a bulky uterus, primary cervical descent to the upper third of the vagina, upper cystocele, small rectocele, deficient perineum, small rectocele, deficient perineum, small enterocele, [but] no urethrocele”. A hysterectomy and anterior and posterior colporrhaphy were performed. 3408 The surgery was complicated by a post-operative infection.3409
The aftermath of the first prolapse repair
4084 After the procedure Mrs Dawson’s heavy vaginal bleeding stopped but she started to experience occasional discomfort and dryness with sexual intercourse.3410 When she was reviewed by Dr Fleming on 29 October 2001 she was reported to be “doing very well” with marked improvement of the bladder and the bowel but she was still “quite tender for intercourse and to sit”.3411
4085 In November 2002 Mrs Dawson saw a gastroenterologist, Dr Grant Phelps with “a 12 month history of intermittent anal type bleeding, passing variable amounts of brightish blood after defecation”.3412 He carried out a colonoscopy on 7 November 2002. The colonoscopy was normal. Dr Phelps thought she had irritable bowel symptoms and that the bleeding was presumably due to haemorrhoids although haemorrhoids were “not well seen”.3413
4086 In March 2003 she returned to see Dr Fleming. She reported feeling sharpness on intercourse with deep pain and dryness. Dr Fleming prescribed Ovestin, a hormone replacement.3414
4087 Mrs Dawson was seen on 4 March 2004 by Dr Russell Dalton, obstetrician and gynaecologist. She apparently told him that she had difficulty emptying her bowel and was conscious of a vaginal bulge. She also described significant pain in her mid vagina with intercourse and on deep penetration and had “slight urgency”. In his report to the GP Dr Dalton stated that she was “quite anxious about her vaginal and pelvic pain”. On examination she had a significantly tender point at the mid vaginal posteriorly which caused pain in her bowel and she was markedly tender at the vaginal vault. He diagnosed pelvic and vault adhesions and possible rectovaginal adhesions. He recommended that she undergo an examination under anaesthesia, a laparoscopy and adhesiolysis (a surgical procedure to remove or divide adhesions in order to restore normal anatomy and function and relieve painful symptoms).3415 Mrs Dawson consented to the procedure which was performed on 26 April 2004.
4088 At laparoscopy Dr Dalton found that both ovaries appeared normal but were adherent to the vaginal vault via restricted adhesions; a loop of mid ileum was attached to the vaginal vault via a broad and dense adhesion; the bowel was attached to the right ovary; the left adnexum had some adhesions along the ovarian ligament to the vaginal vault; and there was a 3cm left-sided fimbrial cyst.3416 The adnexa, I interpolate, are tissues or structures in the body adjacent to or near another, related structure. The ovaries and fallopian tubes, for example, are adnexa of the uterus: Mosby’s Dictionary of Medicine, Nursing & Health Professions (Elsevier, 9th ed, 2012) at 44. A fimbral cyst is a small cyst at the end of the fallopian tubes.3417
4089 Dr Dalton carried out a sharp dissection to free the small bowel from the vaginal vault and divided the adhesions to free the ovaries from the vaginal vault. He also excised the left fimbral cyst. Since there was an obvious cause for her abdominal pain, he was hopeful that Mrs Dawson’s symptoms would resolve with the division of the adhesions and the evidence from Mrs Dawson was that her bowel function and abdominal pain improved after the surgery, although other evidence, discussed in Part XVIII below, indicates that the improvement in bowel function was not permanent.3418
4090 The unchallenged evidence, corroborated by Mr Dawson, is that Mrs Dawson has always been active and “on the go”. She enjoyed walking by herself and with her husband and girlfriends. She rode a bicycle for leisure and performed regular daily exercises at home, sometimes using an exercise bike. Mrs Dawson enjoyed travelling, throughout Australia and overseas. In Australia she and her husband undertook some long drives from their home in Ballarat, including two 18,000 km trips, camping at various locations along the way. In 1999 they drove across the Nullarbor Desert to Perth and up the Western Australian coast to Darwin and home through the centre of Australia. In 2007 they did the same trip in reverse order. In 2002 they drove through the middle of New South Wales up to Cairns. Mrs Dawson was also a keen gardener, regularly spending many hours a week in the garden, an activity she enjoyed and found therapeutic.3419
4091 Notwithstanding periodic decline in her libido and some occasional pain with intercourse after her hysterectomy in 2001, both Mr and Mrs Dawson described their sexual relationship before Mrs Dawson’s implant surgery in 2009 as “fairly healthy”, having intercourse on average once every one to two weeks.3420
4092 In 2008 Mrs Dawson developed mild pain in her lower back, bottom and pelvis after prolonged sitting. In early 2009, however, the pain worsened. At about the same time, her bowel function deteriorated and she needed to digitate to push back her vagina in order to defaecate. She also experienced urge incontinence daily albeit with minimal loss of urine. Nevertheless, she described herself as smelling of urine on occasions at the end of the day. She felt a lump in her vagina and a “dragging” sensation which was worse at the end of the day.3421 Otherwise, she described herself as “very healthy”; the only other issue she had was her depression.3422 She was very active and walked a lot. She and her husband walked their dogs. She socialised, sewed, was actively involved in the Country Women’s Association, and kept her house “beautiful”.3423 She was very involved with the grandchildren. She claimed to have had a good relationship with her husband, although it is clear from the other evidence that the relationship has been a rocky one over the years, and in her second affidavit Mrs Dawson disclosed that she and her husband had separated for three to four months in 2007.3424
4093 On 22 January 2009 Mrs Dawson saw her GP, Dr Terence Gibson. He noted a “feeling of prolapse”, her difficulty “holding urine” and her occasional incontinence, and her tendency to constipation. He found her coccyx tender. Dr Gibson diagnosed her with vaginal wall prolapse and referred her to Dr Jeanette Lim, a urogynaecologist.3425
4094 Mrs Dawson presented to Dr Lim on 28 January 2009. In her letter to Dr Gibson, Dr Lim described Mrs Dawson as a lovely woman with symptoms of recurrent prolapse, urge incontinence and superficial dyspareunia, the latter since her hysterectomy and vaginal repair in 2001. She thought her urge incontinence might be attributable to an excessive intake of fluids (more than three litres a day).3426
4095 Examination revealed a moderate sized cystocoele and rectocoele both descending to 1 cm above the introitus as well as “quite atrophic” vaginal epithelium. Dr Lim recommended physiotherapy, which Mrs Dawson declined because she felt she had had “enough physiotherapy” and a course of topical oestrogen, additional lubrication, and “positioning” during sexual intercourse, presumably to ease the superficial dyspareunia. She also recommended that Mrs Dawson cut her use of caffeinated beverages and restrict her fluid intake to six to eight glasses of water a day. As for the prolapse, Dr Lim discussed with Mrs Dawson three treatment options: “physiotherapy, a ring pessary, and surgery”. Mrs Dawson was said to be very keen to proceed with surgical management. In her report to Dr Gibson, Dr Lim said that she provided Mrs Dawson with information about a vaginal repair and mesh and vaginal support device. 3427
4096 At a follow-up consultation on 26 February 2009, Dr Lim conducted a urodynamic study and a cystoscopy on Mrs Dawson. The former revealed mild urodynamic stress incontinence and an overactive bladder, the latter moderate trabeculation. Dr Lim raised with Mrs Dawson the possibility of performing an anti-incontinence procedure at the time of her prolapse surgery. Mrs Dawson declined to have the additional procedure at this time, although she was aware that her pre-existing stress incontinence could worsen after a cystocoele repair. At this consultation Dr Lim obtained Mrs Dawson’s consent for an anterior and posterior repair with mesh and vaginal support device and cystoscopy.3428
The implant surgery
4097 On 8 May 2009, Dr Lim carried out an anterior and posterior repair using Gynemesh PS cut to size (10 cm x 15 cm), with arms placed to “tunnels cut to [the] pelvic sidewalls and to ischial spines posteriorly” and secured to fascia with 2.0 Monocryl. She also inserted a vaginal support device and performed a cystoscopy. The cystoscopy was normal. There were no foreign bodies or perforations.3429
4098 Given the use of Gynemesh PS, the use of a single-incision, and the deployment of the vaginal support device, Professor Roovers and Professor Rosamilia considered that the surgical procedure was similar to the Prosima technique except that the mesh was not pre-cut.3430 The surgery was performed a little over a year after the publication of the article by Carey et al (“Vaginal surgery for pelvic organ prolapse using mesh and a vaginal support device”) reporting favourable 12-month results from surgery on 95 women with POP-Q stage II or more pelvic organ prolapses using Gynemesh PS cut in a Y-shape and deploying a vaginal support device. Eighty-four of the 95 women were operated upon in Melbourne. A number of the women in the study had undergone previous surgery including hysterectomy and vaginal repair.3431 Notably, except for mesh exposure, the article did not describe the various risks of which Mrs Dawson said she had not been warned.
4099 Mrs Dawson was discharged from hospital on antibiotics on 11 May 2009.3432
The aftermath of the implant surgery
4100 She was bed-ridden for a few days following discharge and her husband “essentially waited on [her] hand and foot”. She then started becoming more mobile, but avoided lifting or prolonged standing.
4101 In the few weeks following the operation, she experienced some post-surgical pelvic pain, as she had expected. Overall, she said she felt as though she had obtained a very good result. For example, she did not have any further bladder symptoms, and in those few weeks her bowel function also returned to normal.3433
4102 On 21 May 2009 Dr Lim saw her for a post-surgical review. Mrs Dawson was concerned that she was still having some vaginal bleeding and that she felt a “dragging” in the posterior vaginal wall but she reported that her bowels were working well and that she felt well.3434
4103 Mrs Dawson attended on Dr Lim for a second post-surgical review on 3 June 2009 at which time the vaginal support device was removed. Both she and Dr Lim were delighted with the results. Dr Lim’s notes recorded that she had no pain or bleeding, that she had no bladder symptoms, that her bowel function was now normal and she was happy that she no longer needed to digitate, and that she had no symptoms of prolapse. There was no evidence of erosion or infection on examination and no rectal haematoma. Dr Lim considered she had excellent anterior, posterior and vault support. In view of the positive findings Dr Lim arranged to review Mrs Dawson six months later. She was pronounced fit to resume “normal activity”, including sexual activity, in three weeks.3435
4104 Mrs Dawson returned to work after about six weeks after she was discharged from hospital during which time she received sick pay.3436
4105 Soon after the 3 June 2009 consultation with Dr Lim, however, Mrs Dawson experienced a range of troubling symptoms. In particular, she began to feel “terrible pain inside and across [her] bottom” and “a prickle deep inside [her] bottom”, like she had “a board running across [it]”. She variously described the pain as “horrible”, “terrible”, unforgettable, “excruciating”, knife-like, far worse than any pain she had experienced before the implant surgery, and which she ranked as 10 out of 10 in severity. She also experienced pain radiating down her legs, pain deep inside her vagina and she was very tender in and around her vagina. The dragging sensation she had felt earlier became worse. Her bowel problems returned; once again she needed to digitate in order to pass a motion. She experienced pain with intercourse.3437 She said that the greatest problem she had was that she could not tolerate sitting for very long. She said that when she resumed driving, about six weeks after the operation, she sometimes had to tightly squeeze the steering wheel to try and prop herself up so as to avoid sitting on her buttocks.3438 She recalled sitting at work one day asking herself whether she would be able to cope with the pain for the rest of her life, a prospect she described as unimaginable.3439
4106 These symptoms persisted for months.3440
4107 On 18 and 22 June 2009 Mrs Dawson consulted her GP, Dr Mark Churcher, who told her that he thought the severe pain was in the region of her coccyx, arranged for x-rays of the pelvis and the coccyx, and referred her to an orthopaedic surgeon, John Nelson.3441 Mrs Dawson saw Dr Nelson on 2 July 2009. He remarked in his report to Dr Churcher that on examination Mrs Dawson was obviously uncomfortable with sitting and was tender not only over the point of her coccyx but over the entire area around it, even to light touch. He expressed the firm view that the “local surgery” (presumably a reference to the implant surgery) had had “some effect, either directly or indirectly in the coccygeal area”. He also informed Dr Churcher that he suspected that “all the ‘bits’ that were tightened” during the surgery “tend to pull on the coccyx” and she might have developed coccydinia, To exclude any “local collection” or any other problem within the bone, however, he asked Mrs Dawson to have an MRI as soon as possible.3442 The same day Dr Nelson wrote to Dr Lim, enclosing his report to Dr Churcher, and asking her whether she had had any experience of patients who, after “this type of procedure” had developed coccydinia (pain in the coccyx), directly or indirectly, and whether there was any other investigation or treatment she felt was appropriate.3443
4108 Dr Lim replied to Dr Nelson on 14 July 2009, in effect, rejecting Dr Nelson’s suspicion that the surgery was the cause of the pain. She wrote:
As [Mrs Dawson] had only a moderate sized cystocele and rectocele no fascial plication was performed and therefore no tensioning or pulling of tissues would have occurred. Instead, mesh reinforcement was placed in a tension free fashion with the body of the mesh overlying the vesico-vaginal fascia and arms of the anterior mesh to the fascia of the obturator internus muscle of the lateral pelvic side wall. The posterior mesh was once again placed in a tension free fashion with the body of the mesh lying on the recto-vaginal fascia with the arms of the mesh being placed in tunnels leading to the ischial spines. These mesh prosthesis are not secured to sacrospinous ligaments or any muscular structures and therefore would not be pulling on the coccyx or any of the pelvic musculature. Instead it relies on ingrowth of the patient’s native tissue and scar tissue into the mesh prosthesis to provide ongoing support to the pelvic organs. Similarly as there was no perineal defect no perineorrhaphy [suturing of the perineum] was required and therefore no sutures were placed in the perineal body which would cause any pulling on tissues in the lower part of the vagina.3444
4109 Dr Lim said that she was unaware of any incidence of prolapse repair of this kind causing coccydinia, especially when no fascia, muscle or perineal body plication had been undertaken and the mesh prosthesis was not fixed to any ligaments or muscle tissue. Apart from the MRI, which she endorsed, she could suggest no further investigations for the coccydinia.
4110 An MRI of the sacrococcygeal spine was performed on 18 July 2009. It revealed “no MR abnormality” that would explain Mrs Dawson’ clinical presentation.3445
4111 Mrs Dawson returned to see Dr Churcher on 23 July 2009. His notes record “coccydinia, can’t (scil) sit, pain +++”.3446 She was referred back to Dr Lim, who saw her six days later.
First mesh exposure and excision surgery
4112 Mrs Dawson was particularly troubled by her paracoccygeal pain and wanted to be re-examined. She was concerned that her dragging prolapse sensation had returned. In her report to Dr Churcher of 29 July 2009, Dr Lim reported the following examination findings:
Examination today revealed a very well supported anterior and posterior vaginal wall. There was no indication of any recurrent prolapse at all. The vault remains well supported. A detailed vaginal examination did not reveal any hard scarring or ridges in the vagina caused by the mesh but did reveal a small 2 mm. midline anterior wall mesh erosion. On vigorous coughing Diane was not able to demonstrate any stress incontinence today however she tells me that her bladder was probably not full enough for this to be evident. I also performed a per-rectal examination where I was not able to demonstrate any tenderness on palpation of the areas where the mesh arms would have been placed para-rectally to the Ischial spines. There were also no rectocele demonstrated on per-rectal examination or areas where stool may be able to be trapped. I then examined her in the area of the coccyx externally and Diane tells me that her pain is not directly on the coccyx but rather just to the right side of it and she did appear to be tender on palpation of that area and the right peri-anal area.3447
4113 Dr Lim advised Dr Churcher that the mesh erosion was currently asymptomatic and could be amenable to some oestrogen therapy and asked Mrs Dawson to resume the use of topical vaginal oestrogen. Until the pain in the coccygeal region was sorted out, however, she considered treatment for other issues, including the pre-surgical urodynamic stress incontinence and the return of the bowel symptoms, should be deferred.
4114 At this time Dr Nelson described Mrs Dawson as “somewhat desperate about [her] situation” and noted in a letter to Dr Churcher of 30 July 2009 that she “really [was] struggling because of her inability to sit”. He arranged for an examination under anaesthetic of the sacro-coccygeal joint and a local steroid injection in the hope that it might “break the [pain] cycle” for her.3448
4115 Mrs Dawson was readmitted to St John of God Hospital under Dr Nelson’s care on 7 August 2009.3449 Three days later she returned to see Dr Churcher. According to the consultation notes, she told Dr Churcher that her “broader buttock area felt better” but that the sharp area “like a grating or splinter” remained until “all recurred one day after the injection” and she was unable to sit flat on her bottom or drive.3450 Dr Churcher prescribed Endep (amitriptyline) 10mg 1 per night on the assumption that the pain was neuropathic.3451
4116 Dr Churcher referred her to Dr Stephen Tobin, a local general surgeon. In his referral letter he noted that she had “marked [right] paracoccygeal pain, present for nearly 2 years”, but “dramatically worse” since the implant surgery in May that year. He noted that the MRI was “unremarkable” and reported that Mrs Dawson was “looking for an organic cause”.3452
4117 Dr Tobin examined her the following day, 11 August 2009. He noted in his report to Dr Churcher of the same date that Mrs Dawson sat on her left side and appeared uncomfortable during the examination. Although rectal examination did not apparently generate “a lot of pain”, posterior examination and examination of the coccyx externally caused “exquisite discomfort both overlying the mid line and to the right of the midline”.3453 He recommended that she continue with Celebrex and wait six to eight weeks to see the effect of the steroid injection.3454
4118 Dr Tobin contacted Dr Lim on 20 August 2009 and suggested that she continue to see Mrs Dawson.3455
4119 On 9 September 2009, however, Mrs Dawson saw Dr Tobin again. She was “quite upset” with continuing pain in the coccygeal area and also pain in the upper legs, all of which appeared to date from the “gynaecologic surgery earlier [that] year”, which was presumably a reference to the May implant surgery.3456 He did not examine her, however, recommending that she continue in the care of the other two surgeons, Dr Lim and Dr Nelson.3457
4120 Mrs Dawson saw Dr Lim the same day. Dr Lim noted that she was upset about her continuing buttock/coccygeal pain, despite some improvement after the steroid injection. In her report to Dr Churcher, Dr Lim wrote that Mrs Dawson was complaining of a “prickly feeling” across both buttocks in a “broadband” and of more “aching all over her body, including the supra-pubic area and also her arms” and that, with sexual intercourse, she was experiencing some pain at the vault. Examination revealed a 1x6mm erosion in the mid anterior vaginal wall. Once again, she counselled conservative treatment, also recommending amitriptyline at the low dose prescribed by Dr Churcher. Noting that Mrs Dawson had ceased taking Efexor, an anti-depressant soon after her prolapse surgery, she wondered whether this could be a factor in her current symptoms. She arranged to see her again in four weeks.3458
4121 Dr Lim reviewed Mrs Dawson on 8 October 2009.3459 She reported that the very low dose amitriptyline had had no side effects and that Mrs Dawson had no pain in the right coccygeal region. Nevertheless, she noted that Mrs Dawson still thought that “something [was] not quite right” and that she sometimes felt like she was sitting on a board. She also noted that, although the pain with sexual intercourse had improved, it had not completely resolved. She said that Mrs Dawson remained keen for further investigation. She confessed to being “more and more depressed with the increased pressure at her workplace and social pressures at home as well as having to deal with the recent death of her mother-in-law. On examination, the tenderness in the vault had disappeared but the small anterior wall erosion was still evident.
4122 Dr Lim suggested that Mrs Dawson increase her dose of amitriptyline to 25mg to try to eliminate her dyspareunia altogether. In the meantime she proposed scheduling her for an examination under anaesthetic and excision of her mesh erosion with cystoscopy, with the assistance of Dr Tobin.
4123 Mrs Dawson was readmitted to St John of God Hospital on 14 October 2009. That day she was examined under anaesthetic and Dr Lim excised a 2mm x 1cm mesh erosion (significantly larger than she had diagnosed) “completely”. The cystoscopy confirmed that the bladder was uninjured.3460 Apparently, the erosion was not at the site of her pain.3461 Dr Tobin conducted a preliminary rectal examination under anaesthetic. He found “a lax pelvic floor” but no demonstrable rectal prolapse. He also performed a flexible sigmoidoscopy, that is to say he examined the sigmoid colon while Mrs Dawson was sedated, using a flexible tube inserted through the anus. He found no abnormality save for a “minor mid rectal scar”. He recommended that she continue with conservative and supportive care under Dr Lim’s guidance.3462
4124 Histopathology of the excised mesh confirmed that there had been a mesh erosion with “foreign/mesh material associated with inflammation and foreign body reaction”. Microscopy reportedly showed blood admixed with scant fragments of mesh partly surrounded by granulation tissue and foreign body giant cells. 3463
4125 A little over a week after the operation, Mrs Dawson returned to see Dr Churcher. She told him her bottom had been sore ever since and that she continued to have a “pricking” feeling in her perianal region and a sore spot on the right lateral aspect of her bottom. She could only sit forward or to the left side and expressed considerable frustration.3464
4126 Six weeks after surgery, on 2 December 2009, Dr Lim reviewed Mrs Dawson.3465 Dr Lim arranged for her to have a 3D ultrasound and MRI. An MRI of the abdomen and pelvis was carried out at the Royal Women’s Hospital on 9 February 2010.3466 In correspondence shortly thereafter, Dr Lim stated that the MRI revealed no evidence of recurrent vaginal prolapse or any mesh-related pathology.3467 According to Dr Tobin, however, the report itself apparently showed a degree of rectocoele.3468 Moreover, the radiologist who performed it, Dr Jagjeet Makhijani, whose findings and conclusions were approved by Dr Andrew Dobrotwir, found that on straining, there was abnormal descent of the bladder and vesicourethral function, consistent with a cystocoele, and urethral prolapse and abnormal indentation of the posterior vaginal wall by the rectum with abnormal rectal descent, consistent with an anterior rectocoele. He concluded, relevantly, that she had bladder and urethral prolapse associated with an anterior rectocoele.3469
4127 In the meantime, Mrs Dawson presented to Dr Churcher on 30 December 2009. On this occasion she told him she had become increasingly angry and stressed, crying more, felt tired easily, was not relating well to people and was rude, and felt like she wanted to withdraw. Her sleep was variable. She had intrusive thoughts. She was also nauseous. He noted that she had resumed Efexor 75mg two weeks earlier. He doubled the dose and added a nightly dose of 100 mg of carbamazepine, an antiepileptic drug used to treat neuropathic pain.3470 By January the following year the change in medicines had apparently improved both her mood and her sleep.3471 The chronic pain, however, persisted. In February 2010 she discussed the situation with Dr Churcher. He encouraged her to continue with the carbamazepine, to increase it to 200mg during the day and 400mg at night, and to do more exercise, stretching and yoga.3472 When she returned to see him in late April, however, she had petechiae (small red or purple spots caused by bleeding into the skin or other organ) and a tremor, both of which Dr Churcher attributed to the carbamazepine and he reduced the dose to 100mg.3473
4128 In the meantime Dr Lim referred Mrs Dawson to Dr Carey for an opinion on whether her problems were mesh-related and whether he could do anything to improve her current condition.3474 But Mrs Dawson did not follow-up on this referral because she decided against further surgery.3475
4129 Mrs Dawson was periodically reviewed thereafter by Dr Churcher. She continued to have ongoing pelvic pain. On 26 July 2010 he wondered whether she had a small central erosion through the anterior wall of the vagina near the vault and prescribed some more topical oestrogen. On review a month later, she was tender over the scar in her vagina and the left fornix and very sensitive in “all areas”, although she improved when she relaxed.3476 Although pelvic pain remained a problem, during 2012 and 2013 the notes from UFS Medical suggest that depression and anxiety were predominant.3477 When Dr Tobin reviewed Mrs Dawson on 25 August 2010 he considered that the deep seated pelvic pain and vaginal pain had settled down, apparently because she was concerned about her problems with defecation. At the same time, however, he noted that sexual activity was virtually non-existent because she felt “uncomfortable” (his expression, not necessarily hers) and in his report to Dr Churcher he also said that he understood that pelvic examination in the office was also “uncomfortable” (again, his word, not necessarily hers). Apparently he did not attempt an examination himself. After he saw her again on 5 October 2010 he thought that most of her pelvic discomfort had resolved.3478 That assessment appears to have been based on a history rather than an examination.
4130 On 7 June 2012 Dr Churcher referred Mrs Dawson to Dr James Swan, an obstetrician and gynaecologist, based in Geelong for “opinion and management regarding ongoing pelvic floor issues”. Those issues were listed in the referral letter as follows:
right inner buttock pain, increasing with a full rectum;
pelvic and low back discomfort;
a sensation of “BA” pushing to the right and needing buttock pressure to successfully pass a motion;
deep dyspareunia and slight superficial pain.3479
4131 Mrs Dawson saw Dr Swan on 27 June 2012. In his report to Dr Churcher following that examination he wrote:
As well as her persistent buttock and leg pain, she is apareunic because of severe dyspareunia, and is also experiencing both genuine stress incontinence and detrusor instability.
She also complains of mechanically poor ability to defecate and empty her rectum and often uses digital support both vaginally and anally to achieve this.
Examination revealed a slightly shortened vagina, with a tender apical vaginal vault. There was no cystocele or rectocele, but her perineum was noted to be shortened and there was no bladder neck descent.3480
4132 He advised that the situation would be “extremely difficult” to manage. He said that in the first place he was “aggressively reoestroeganising her vagina with Vagifem 1 pv nocte for three weeks and then twice weekly thereafter” and commencing her on Ditropan 5mg bd to try to reduce her detrusor instability.
4133 Dr Swan reviewed her in August 2012 when she reportedly told him she felt much better, that she was not experiencing the same pelvic pain as before, and that her leg pain had resolved. Her vaginal vault was “much less tender with minimal tenderness on the right side” and, although the left side was still tender, it was not nearly as bad as it had been. She elected to cease Ditropan after two weeks of use but was noticing less urgency and minimal stress incontinence. She still had significant difficulty initiating and completing defaecation. He suggested that she continue with regular use of Vagifem twice weekly to maintain “oestroeganisation”.3481
Second mesh exposure and excision surgery
4134 Dr Swan reviewed Mrs Dawson again on 14 March 2013. At this time she still had apareunia, was now complaining of pain with a full rectum, continued to experience difficulty initiating and completing defaecation, and her buttock pain had returned. Examination confirmed the shortened vagina and the vaginal vault was tender, especially in the midline with evidence of midline scarring at the apex and tenderness in the left fornix. The anterior vaginal wall was well supported but she had “a grade 1 rectocele predominantly involving the lower 1/3 of the posterior vaginal wall with a narrowed and inelastic introitus”.3482
4135 The next day Dr Swan referred Mrs Dawson to Professor Peter Dwyer in Melbourne.3483 In his letter to Professor Dwyer, he recounted his examination findings from the June consultation somewhat differently. Instead of a slightly shortened vagina and a tender apical vaginal vault, he wrote that the examination revealed a significantly shortened vagina with a very tender scarred vault, particularly in the midline and to the left fornix.
4136 Professor Dwyer examined Mrs Dawson on 2 September 2012. In his report to Dr Swan dated 3 September 2013, he noted that Mrs Dawson had had chronic pelvic pain since the mesh implant surgery in 2009 as well as severe dyspareunia and difficulty defaecating. He reported that Mrs Dawson was “very sensitive to vaginal examination and experienced quite severe pain with a one finger vaginal examination … mainly on the posterior and lateral vaginal walls”. On rectal examination he found “a band of very tender tissue palpable on the left side”. He also noted that she was having some leakage with coughing, bending and laughing but that this was only an occasional problem with which she was presently unconcerned.
4137 Professor Dwyer advised that the best option at this point was surgical removal of as much of the mesh as possible. He informed Mrs Dawson that there was no guarantee that removal of the mesh would cure her pain or her difficulty with defaecation, although he told her that he “would expect a 60 to 70% chance of considerable improvement”.3484
4138 Mrs Dawson said that she was greatly relieved to hear what Professor Dwyer had to say. She explained:
I had spent a number of years without having anyone being able to tell me definitively what was wrong, so to finally have a specialist identify the problem for me, and tell me how it could be fixed, was wonderful. In fact, I recall breaking down in tears because I was so relieved by what Professor Dwyer had told me and that I finally knew what the cause of my problems was.3485
4139 On 26 November 2013 Mrs Dawson saw Dr Amber Kennedy, a urogynaecology registrar at the outpatients’ clinic of the Mercy Hospital for Women in Heidelberg, Victoria, where Professor Dwyer intended to operate. She wrote to Dr Swan on 18 December 2013:
Her case was discussed with the urogynaecology fellow, Dr Kris Cvach and consultant, Dr Yik Lim and she has recently been seen by Professor Peter Dwyer in his private rooms. As you know, she has had a very difficult time since 2009 when she had an anterior and posterior vaginal mesh repair, suffering with severe chronic pelvic pain, rectal pain, dyspareunia and difficulty with defaecation. Diane has been completely apareunic for over 2 years now and the severity of the pain is significantly affecting her quality of life. She does have mild urinary stress incontinence however this does not present as her primary concern.
On examination today, Diane has a large area of palpable mesh that is not exposed but is very painful, both anteriorly and posteriorly, a very shortened vagina and narrowed introitus. As was the plan from Professor Dwyer’s rooms, I have booked her for an attempted vaginal mesh removal. Diane is aware that possible complications for this procedure include entry into the bladder or bowel requiring further surgery or procedures and that we cannot guarantee complete resolution of her pain after this is performed. She will be reviewed prior to surgery by a consultant to be further examined and to ascertain which arm or arms of the mesh are to be removed.3486
4140 The examination findings at the pre-admission assessment are also notable. Mrs Dawson was tender “all around” but especially at the apex of the vagina and in the left posterolateral aspect.3487
4141 Mrs Dawson was admitted to the Mercy Hospital on 30 January 2014.3488 Professor Dwyer operated the following day, excising the mesh arm on the left and a small (less than 5 mm) mesh at the vault.3489 Mrs Dawson was discharged from hospital on 2 February 2014.3490
4142 A pathologist, Dr Tineke Fancourt, reported that the segments sent to the laboratory showed “fragments of fibrous tissue surrounding foreign body with a small amount of adipose tissue, nerves and blood vessels”; “associated mild chronic inflammatory infiltrate consisting of lymphocytes, histiocytes and plasma cells”; but “no evidence of a significant acute inflammatory infiltrate”.3491 He concluded that there was no evidence of malignancy in relation to the vaginal mesh.
4143 Professor Dwyer reviewed Mrs Dawson on 13 March 2014, six weeks after the operation. He reported to Dr Swan that she had “generally” made a good recovery, that she told him she felt much better and that her pain had largely settled. He noted, however, that she was still having some gluteal pain which she told him she had had ever since her mesh repair in 2009. Her urinary control was “good” and her bowel function “generally good”. On examination, however, she remained “extremely tender vaginally in all areas”. Despite that finding, Professor Dwyer considered that the vagina seemed to be healing well and appeared healthy.3492
4144 Mrs Dawson was reviewed by Dr Lin Li Ow, a urogynaecology fellow at Mercy Hospital, on 29 May 2014. She reported to Dr Ow “quite severe constipation” over the previous two months and was unable to empty her bowels. Examination once again showed that the vagina was healing well. This time Mrs Dawson reportedly tolerated the examination well and did not complain of much pain. Despite the history of severe constipation, the rectal examination showed no evidence of faecal impaction and her stools were reportedly soft. Dr Ow recommended daily Movicol to ease the constipation and vaginal dilators to improve Mrs Dawson’s vaginal capacity.3493
Third mesh exposure and excision surgery
4145 On 27 November 2014 Dr Ow reviewed Mrs Dawson again, this time with Dr Lore Schierlitz, a consultant urogynaecologist. Mrs Dawson apparently told Dr Ow that she felt much better in that she was able to sit down comfortably with no issues but was totally unable to tolerate intercourse due to pain everywhere in the pelvic region. She said that the vaginal dilators made no difference. She reportedly had no prolapse symptoms, constipation, urgency or urge incontinence, but very occasional stress incontinence. Despite the reported improvement, examination was difficult because her pelvic floor was hypertense and painful. A few strands of mesh were visible on the posterior wall and a 0.5cm area of granulation tissue was found on the posterior wall 0.5cm from the introitus. Dr Ow recommended Ovestin for the erosion and physiotherapy (trigger point therapy) for areas of pain in the vagina and the pelvic floor generally.3494
4146 By March 2015, however, it was apparent that conservative treatment was not enough.
4147 She was examined by a continence physiotherapist, Heather Deane, on 3 March 2015 there was a line of tenderness from the upper third of the left posterior vaginal wall diagonally to the lower third of the right vaginal wall and there was an area of mesh exposure along this line. Palpation of the line of tenderness elicited pelvic floor contraction and pain reactions.3495
4148 When Dr Schierlitz saw Mrs Dawson again on 19 March 2015, although her bowel function had improved under the instruction of the physiotherapist, pain was still a barrier to the resumption of sexual activity. On examination the levator ani muscles were still tense and tender (“like a bruised feeling”) and two patches on the anterior vaginal wall that were tender and had underlying mesh. There was an area on the left lateral fornix where some mesh was both palpable and visible. Finally there was tender area on the left side in the posterior vaginal wall which Dr Schierlitz thought might relate to the remaining mesh.3496
4149 Dr Schierlitz discussed the treatment options with Mrs Dawson and recommended surgery. 3497
4150 Mrs Dawson was devastated by the prospect of further surgery. She explained in her first affidavit:
When Professor Dwyer had told me in September 2013 that I needed surgery, the news came as a relief because I felt the cause of my problems had finally been identified and that my problems would finally be resolved. But when Dr Schierlitz told me I needed further surgery, I did not feel any relief at all.
Instead, I was deeply frustrated, and I broke down crying. I started to believe that there was simply no light at the end of the tunnel for me. I remember saying to Dr Schierlitz, “This has wrecked my womanhood. I am scared that I may never be able to have intercourse again.” She told me, “Hopefully one day you will be able to have intercourse again. She did not sound confident.
4151 The planned mesh excision took place on 15 May 2015.3498 Mesh was excised at the anterior vaginal wall, the vault, and the left vaginal wall.3499 A cystoscopy was also performed. It showed that the bladder was intact and no mesh had eroded into the bladder. It is clear from the discharge summary, signed by Dr Schierlitz, that she was of the view that the mesh was the or at least a cause of Mrs Dawson’s pain.3500
4152 Histopathology reported the presence in the specimens of densely collagenous tissue containing birefringent and foreign material consistent with mesh and showing patchy inflammation including lymphocytes and foreign body-type giant cells forming loose aggregates around the foreign material.3501
4153 Mrs Dawson was discharged on 18 May 2015.3502 She was largely bed-ridden for a few days but slowly became increasingly mobile. She was absent from work on paid sick leave for four weeks.3503
4154 Dr Schierlitz reviewed Mrs Dawson on 28 May 2015. In her letter to Dr Swan reporting on that consultation she noted that the procedure had been “relatively uneventful” and that she had managed to remove most of the mesh that was palpable and painful on prior examination. At the consultation Mrs Dawson told Dr Schierlitz that her pain was much improved, her bowel function was normal (she had no constipation and no difficulty evacuating), she had no urgency, urge or stress incontinence, and she was voiding well three to four times a day with a good flow. She had not yet attempted sexual activity. The sutures were still present, the vaginal epithelium was healing well, and, although there was some pain with the speculum examination, there was very little on digital examination.3504
Fourth mesh exposure and excision surgery
4155 Mrs Dawson did not see Dr Schierlitz again until 9 July 2015.3505
4156 At this consultation Mrs Dawson reported that she had begun to experience pain in her bottom and back passage again shortly after the 28 May consultation. This was confounded by constipation and difficulty with defaecation and abdominal bloating. Overall, however, Dr Schierlitz considered that Mrs Dawson’s vaginal pain was still better than it had been before the mesh had been removed. On examination the introitus was pain-free, the lavatory inner muscles were less tense, and she had lost the bruised feeling. Her anterior vaginal wall was much improved. There was no palpable or visible mesh exposure and no pain on palpation. The left lateral vaginal wall was not tender and there was no palpable mesh there either. Overall, the vagina had a good vaginal length with the capacity to admit a mid-sized bivalve speculum.
4157 On the posterior vaginal wall, however, there was now an edge of palpable mesh at the mid-vaginal level and on speculum examination some strands of the mesh were visible, with mesh exposure. This area was the area of tenderness and pain that Mrs Dawson experienced when her rectum filled before defaecation. There was some bleeding from the area of mesh exposure after the examination.
4158 On rectal examination, two small haemorrhoids were noted, but there was no mesh palpable in the rectum, although pushing on the posterior vaginal wall during the examination caused a vaginal resurge pain.
4159 Dr Schierlitz decided to proceed conservatively at this stage. She recommended meticulous bowel care, increased fluid intake, and the use of Nurofen for pain relief when needed. She also advised Mrs Dawson to use Ovestin cream twice weekly. Dr Schierlitz informed Mrs Dawson, however, that if the posterior vaginal wall continued to be painful or resumption of sexual activity caused pain or discomfort to her or her husband, then further surgery may need to be considered.
4160 Mrs Dawson was devastated to learn of the prospect of further surgery. She said that recovering from each operation took a lot out of her and she felt weaker than she had been before the implant surgery in 2009.3506
4161 Unfortunately in the weeks that followed, Mrs Dawson continued to feel what she described as “a pulling pain across [her] bottom”, which made it very difficult for her to sit. By the end of each day her bottom ached and she had a dragging sensation inside her.3507
4162 At the time of the next consultation with Dr Schierlitz on 3 September 2015, Mrs Dawson was still experiencing vaginal pain although not as much pain on defecation as before, as she was not straining as much. Her pelvic floor was sore and aching. She was bloating and there was a dragging feeling in the vagina which increased with a full rectum. Bladder function, however, was now “pretty good” and bowel function was better overall. On examination Dr Schierlitz detected an area of granulation tissue (about 2.5 cm) in the mid anterior vaginal wall which also showed a protruding suture, which Dr Schierlitz trimmed. Dr Schierlitz attributed Mrs Dawson’s discomfort to a prickling sensation which improved after the suture was trimmed. On the posterior vaginal wall to the right, however, there was still an area of palpable mesh (1 x 2cm) and the fibres of the mesh were visible on speculum examination. That area was “quite tender and [had] a needle prick sensation”. The rectal mucosa was mobile over the mesh and the mesh was not palpable in the rectum but there was tenderness in the vagina with pressure on the right side during the rectal examination.3508
4163 In her report to Dr Churcher, Dr Schierlitz wrote that she had had a long discussion with Mrs Dawson to see how she would like to proceed and noted that, despite a significant improvement in the pain since the last operation, she was “still quite upset” that she was still having pain. 3509 Mrs Dawson gave the following account of her reaction to being told that she still had eroded mesh which could be treated by further surgery:
Once again, I broke down crying out of disappointment and frustration. I wondered whether my problems with the mesh were ever going to end. Given that conservative measures (such as the use of Ovestin cream) had not fixed my mesh erosion, I agreed to have further surgery. I was of the view that the mesh was the cause of my pain, so I wanted it removed from me. I would have been happy to have the whole of the mesh removed if that were possible. I wanted to feel normal as a woman and as a human being again, and I still clung to the hope that removing parts of the mesh might possibly fix my problems.3510
4164 On 30 October 2015 Mrs Dawson was readmitted to the Mercy Hospital and Dr Schierlitz excised mesh from both the anterior and the posterior vaginal wall.
4165 Just before the operation, while she was waiting to be admitted, Mrs Dawson felt exhausted, both physically and emotionally.3511
4166 At the operation, three small strands were excised from the anterior wall and three pieces from the midline of the posterior wall.3512 The indication for the surgery was “mesh exposure, pain”.3513 The mesh specimen sent to pathology was reported as showing fragments of benign squamous mucosa in which fibres of foreign material are present within the stroma (the supporting tissue) associated with mild chronic inflammation and foreign body type giant cells.3514
4167 She was discharged on 1 November, took six weeks off work on sick pay, but her symptoms did not improve following the operation.3515
4168 Dr Schierlitz reviewed Mrs Dawson next on 17 March 2016, five months after the last operation. No report was tendered from this consultation but it seems that Mrs Dawson was worse at this point. The doctor’s notes record “pain ”, which I infer means that the pain was much worse. They also record that Mrs Dawson had difficulty sitting and walking for long periods. Once again she was constipated and had to strain to defaecate but there was no blood or mucous in the bowel. Dr Schierlitz considered that there was no recurrent prolapse3516 and referred her for physiotherapy for “hypertense pelvic floor with levator co-contraction”.3517
4169 On 1 June 2016 Mrs Dawson was seen by Associate Professor Clara Shek for urodynamic testing and cystourethroscopy.3518 No mesh material was visible in the bladder or the uterus but vaginal examination was painful and levator assessment was abandoned because of pain. The lower posterior vaginal wall was scarred. Associate Professor Shek concluded that Mrs Dawson had detrusor overactivity and chronic pelvic pain following the mesh repair but found “no significant pelvic organ prolapse”.
4170 On 2 June 2016 Professor Korda examined Mrs Dawson at the request of her solicitors and reviewed the urodynamic studies conducted by Associate Professor Shek.3519
4171 Mrs Dawson told him she was experiencing vaginal pain, pain in her buttocks radiating into her legs, a prickly feeling inside her pelvis which awoke her at night, and a burning feeling when she sat and when she extended her legs. She also told him she was constipated, needed constant Metamucil (a fibre supplement made up of psyllium husks), needed to push on a bulge in her right buttock to empty her bowels, and complained of urinary stress incontinence, occasional urinary urge incontinence, flatal incontinence, and severe dyspareunia which made sexual intercourse impossible. She indicated that on 13 May 2016 she noticed a “needle” coming out of her rectum which was whitish in colour.3520
4172 Professor Korda conducted an abdominal and a pelvic examination. The abdominal examination revealed no obvious abnormalities. The results of the pelvic examination, however, were quite different. It confirmed a narrowed introitus and a contracted, narrowed vagina with a fibrosis around the left anterior and left posterior portion of the vagina. Her vagina was rigid, not pliable, and only admitted one finger. A tender remnant of the mesh was palpable on both the anterior and posterior walls. The right ischiorectal fossa was tender and she indicated that this was the spot where she had to push in order to complete defaecation.
4173 Professor Korda concluded that Mrs Dawson had detrusor overactivity and chronic pain syndrome as a result of mesh insertion. More specifically, he concluded that the Gynemesh implant resulted in dyspareunia and chronic pain syndrome. Apart from the constipation, the need to digitate, the prolapse symptoms, lower back pain and dragging pain that preceded the implant surgery, he attributed all the symptoms she described to him at the time of the consultation to the Gynemesh implant. In his report he also attributed her urinary dysfunction to the implant but in cross-examination, when reminded of the pre-existing urinary symptoms, revised his opinion, stating that the mesh repair worsened her urinary dysfunction.3521
4174 He considered that the prognosis for her chronic pain syndrome was very poor. He pointed out that chronic pain syndromes are “traditionally …very difficult to treat” and require lifelong treatment using a multidisciplinary approach with a combination of psychotherapy, physical therapy and pharmacotherapy. Based on his experience, he put the likelihood of a cure at less than 10%. He said that the detrusor overactivity would require lifelong treatment which could control but not resolve her symptoms. He said that the defaecation disorder required evaluation with anal physiology studies, a combination of behavioural therapy, neuromodulation, and perhaps surgical intervention, but the likelihood of a complete and permanent cure was low, again in the vicinity of less than 10%. He thought the opinion of a colorectal surgeon would be useful.3522
4175 Mrs Dawson was examined by a colorectal surgeon, Professor Anthony Eyers, on 28 June 2016.3523 He, too, conducted an abdominal examination and those findings were also unremarkable. On anorectal examination there was no undue perineal descent but there was noticeable asymmetry with bulging evident on the right, apparently centred posterior to the mid-lateral line.
4176 The digital rectal examination, however, revealed “a palpable area of deficiency of the pelvic floor” on the left, above the anal canal in the posterior aspect and anterior to this area an edge of palpable residual mesh. There was a similar apparent deficiency of the pelvic floor on the right through which the lateral edge of the distal sacrum and coccyx were unusually prominent and palpable immediately under the bowel wall without the usual intervening soft tissue. Both the palpable mesh and the sacro-coccygeal ridge were very tender. Professor Eyers considered all these abnormalities were consistent with her previous mesh prolapse surgery and the subsequent procedures to remove the mesh. In particular, he stated:
The end result of the use (and attempted removal), of the mesh, is that Mrs Dawson's long-standing constipation has been aggravated, rather than improved, by failure to correct her associated obstructed defaecation. Both factors were present to some extent before the pelvic repair performed by Dr Lim. It appears that the obstructed defaecation was originally caused by her rectocele. But now the situation appears to involve more than a simple rectocele, (located anteriorly), and to result from a more diffuse (albeit similar) process, as evidenced by her having to apply pressure laterally (especially on the right side) to help her to defaecate.3524
4177 He added that the insertion and later removal of the mesh had caused what he referred to as “collateral damage” to the normal tissues alongside and around the mesh. He said that this explains the abnormalities he was able to palpate in the parametrial and pararectal tissues and the irregularities he could feel in the pelvic floor. He said that these palpable abnormalities were “indicative of residual mesh and/or the secondary effects of surgery performed [in an attempt] to remove the mesh”. He also said that it was possible that there was an element of pudendal neuropathy which was contributing to her situation. Regardless, he considered that her disabilities were most likely permanent.
4178 Professor Eyers noted that Mrs Dawson continued to be troubled by pain in the inner aspect of her right thigh and her vagina and, in the absence of successful treatment, he expressed the opinion that the pain would continue to trouble her. He recommended that Mrs Dawson be referred to a pain management specialist or pain clinic.
4179 He also noted her on-going bowel issues, which he considered had worsened since the mesh surgery. He said there was no available treatment likely to correct them, so that it was likely she would continue to be troubled by them, too, and would need fibre supplements and aperients (mild laxatives).
4180 Professor Eyers was not required for cross-examination and the respondents adduced no evidence from a colorectal surgeon. None of the respondents’ witnesses commented on his opinions. There is therefore no reason why I should not accept his opinions and I do.
4181 In July 2017, after reading Professor Eyers’ report, Professor Korda concluded that Mrs Dawson’s detrusor overactivity was well-controlled with Vesicare but stated that it would have to be taken for the rest of her life and would require constant medical supervision and re-assessment. He agreed that Mrs Dawson’s defaecation disorder was originally caused by a simple rectocoele which had been aggravated by the insertion of the mesh and the later correction surgeries which damaged adjoining and contiguous organs around the rectum. He remained of the view that her chronic pain syndrome had a very poor prognosis, that the likelihood of a cure for her defaecation disorder was exceedingly low, and her prognosis extremely poor. He also expressed the view that it was more likely than not that Mrs Dawson had pudendal neuropathy because of mesh placement near the sacrospinous ligaments which are near the pudendal nerve. Like Professor Eyers, he considered that her disabilities were most likely to be permanent.3525
4182 Professor Korda also adhered to the opinion that, but for the mesh implant, it is more likely than not that Mrs Dawson “would have avoided vaginal pain, pain in her buttocks which radiates down her legs, a prickly feeling inside her pelvis, a burning feeling when she extends both legs, mesh erosions and repeated revision surgery”.3526
4183 On 8 September 2016 Mrs Dawson was seen by Dr Debiyoti Karmakar, a urogynaecology fellow, for review of her pain management.3527
4184 Dr Karmakar took a history that there had been some improvement in her pain after focused physiotherapy and that she was using vaginal dilators but that her quality of life was still quite impaired since she was unable to have intercourse and her rectal pain was debilitating at times. Mrs Dawson also reported an increase in her overactive bladder symptoms with more frequent urge incontinence.
4185 On examination with the speculum, bimanual and per rectum examinations, Dr Karmakar noted that the genital hiatus was about 4cm and tolerated the insertion of one finger but two fingers were painful. No mesh exposure was seen or felt. He found it difficult, however, to assess the vaginal walls as examination was uncomfortable even with a small speculum. The right sacrospinus ligament was tender to touch (7/10) and the pain was sharp. There was also pain on the left rectal wall (8/10), which Mrs Dawson described as a pulling sensation.
4186 Dr Karmakar prescribed Endep cream to help with the vaginal symptoms and Vesicare 5g once a day for her overactive bladder symptoms.
4187 Mrs Dawson apparently told him that she wanted to explore surgery and wished to see Dr Schierlitz as soon as possible.
4188 Dr Schierlitz reviewed Mrs Dawson again in November 2016 and February 2017.3528
4189 At the February 2017 consultation, Mrs Dawson reported a pulling pain from the left side of the vagina and referring into the rectum originating predominantly on the posterior wall. There was also a sharp pain at the introitus on the posterior fourchette (a thin tissue at the bottom of the inner folds of the vulva). The bladder function was quite good but the bowel function was complicated by constipation and difficulty evacuating. She remained inactive sexually due to the pain and the band in the mid vagina.
4190 On examination there was some tenderness on the posterior fourchette, but Dr Schierlitz thought that “the main pain originate[d] from a scar band on the left side of the vagina, about 1.5 cm from the introitus”. There was a pulling sensation referred into the rectum and the bottom and top of the vagina was mildly tender. No mesh was palpable, however, and no mesh exposure detectable. Nevertheless, after a long discussion with Mrs Dawson a decision was made to “have one more try to divide the vaginal band and see if that relieves her discomfort and help[ed] with the referred pain”.3529
4191 On 17 March 2017 Mrs Dawson returned to hospital where, under anaesthetic, vaginal scar tissue was divided and excised and a cystourethroscopy was performed.3530 The specimen sections sent to the pathologist revealed vaginal scar tissue with mild chronic inflammatory infiltrate consisting mainly of lymphocytes.3531
4192 Before the operation Mrs Dawson was in a great deal of pain. She said she was hardly able to walk or sit and “had a bulge coming outside of [her] bottom”, which she sometimes had to push in order to defaecate and which “felt like touching a balloon”.3532
4193 Professor Korda expressed the opinion that the formation of the vaginal band, scarring and the ongoing pain involving Mrs Dawson’s vagina, rectum and buttock more likely than not resulted from the mesh implant surgery performed on 8 May 2009. Further, Professor Korda opined that it was more likely than not that the surgery Mrs Dawson underwent on 17 March 2017 was necessary because of the mesh implant surgery that was performed on her on 8 May 2009. His opinion was based on the fact that the scar tissue was related to the initial mesh insertion and subsequent revision surgeries and was most unlikely to have developed from any other procedure she had had.3533 This opinion was not challenged in cross-examination.
4194 About a month after the 2017 operation, on 18 April 2017, Mrs Dawson returned to see Dr Churcher. According to his contemporaneous notes, the surgery had resolved the pulling sensation, she still had some faint “prickling”, but was very happy and had stopped taking Palexia.3534 Mrs Dawson’s evidence, which I accept, was that the Palexia had caused her to feel nauseous and did not always alleviate the pain.3535
4195 On 26 April 2017 a defecating proctogram was performed. This is an x-ray examination of the rectum taken during defaecation. During defaecation, a rectal prolapse was identified and the radiologist reported that the study was suggestive of rectal prolapse.3536 But Dr Stewart, who reviewed Mrs Dawson on 15 May 2017, considered that the defecating proctogram showed no significant internal prolapse but a degree of anismus (contraction rather than normal relaxation of pelvic floor muscles during attempted defaecation).3537 After an endoanal ultrasound and anal manometry, however, Dr Stewart said that there was no evidence of significant anismus. He said that the external sphincter was partially deficient anteriorly and she had associated low sphincter pressures.3538 But he did not explain the significance of these findings and neither side referred to them in submissions.
Current position
4196 In her first affidavit sworn in July 2016 Mrs Dawson stated that, following the surgery in October 2009, her pain did not improve and, as a result, she became increasingly angry and stressed. She said that the pain associated with sexual intercourse was worse than it had been before the excision surgery. She identified two types of pain. First, there was “entrance pain”. She said that her genitals felt “puffed up and swollen” and it was extremely painful when her husband tried to enter. Second, if he did penetrate, she felt like a knife was cutting inside her. As a result of the dyspareunia, they gave up trying. She said she was crying a lot, felt very tired, and felt that her pain was draining her of energy.3539 She continued:
I was depressed at how my life had changed so significantly. In particular, I no longer had a sexual relationship with my husband, I no longer wanted to see people, I could not sit for very long, and I could not exercise and walk like I used to. I was extremely frustrated at not being able to find a solution to my predicament; none of the doctors were able to explain to me why I was in so much pain. There did not seem to be an end in sight for me. I felt wounded thinking that this was how things were going to be for the rest of my life. I remember thinking that this was definitely not the way I wanted to live.3540
4197 Mrs Dawson added that the cessation of their sex life had affected her marriage. She said that her husband stopped cuddling her so as not to get aroused because he knew she would not have sex with him.
4198 At the time of swearing the first affidavit in late July 2016, Mrs Dawson described the following problems.
4199 She had an ache across her bottom all day every day. Regularly, she felt like she had “a prickle in [her] bottom” and, when seated, she felt a tired, burning sensation across her bottom. Sometimes, she said, she felt like the area was on fire. There was a pulling sensation from the left to the right of her bottom, running down the top of her legs beneath her buttocks and into the top of her right inner thigh. The pain gets worse over the course of the day, rising from 3 out of 10 (with 10 being the worst pain imaginable) in the mornings to 7 out of 10 in the evenings. She could only cope with work because she was able to regularly get out of the chair. Nonetheless, walking would aggravate the pain in her bottom.
4200 She had an ache in her pelvic floor, which was not constant, but appeared to occur after walking or being on her feet for long periods. At its worse she ranked it 7 out of 10.
4201 She also had pain deep inside her vagina which was constant but which fluctuated in intensity and was aggravated by walking or prolonged sitting. On occasions she also felt a sharp, stabbing type pain in her vagina.
4202 She felt a constant pulling pain in the bottom of her buttocks radiating down her legs which is often worse after walking.
4203 She also felt a constant dragging sensation inside her like a recurrent prolapse, although she has been told that she has no recurrent prolapse.
4204 Her sleep was disturbed. She was waking a couple of times a night with pain in her bottom, particularly since the operation in October 2015.
4205 Her energy levels were significantly depleted and she was often tired.
4206 She was always concerned about hurting her pelvis and fearful that if she tripped and fell and landed on her bottom the pain would be unbearable.
4207 Her genitals felt swollen and burning and she was conscious of an unpleasant odour from her vagina, particularly since the operation in October 2015.
4208 She and her husband have attempted intercourse three or four times unsuccessfully since the implant surgery. The intimacy they once shared had all but disappeared. She would avoid doing anything to arouse her husband.
4209 She continued to take Metamucil to keep her stools soft, but not every day because it caused bloating. She often felt faeces bulging inside her and, although she detests it, needed to push the bulge with her fingers against the outside of her bottom to defaecate.
4210 She felt that her pain was aggravated by heavy lifting and tried to avoid it where possible. She is also much weaker than she used to be. Consequently, the amount of work she is able to do in the garden is reduced and, although she continues to shop for groceries, she cannot carry as many bags at one time as she used to and she needs to take greater care when lifting and carrying.
4211 She had gained about 10kg in weight since her implant surgery which she attributed to her reduction in physical activity caused by her pain and other disabilities.
4212 She had also noticed significant changes in her emotional well-being since the implant surgery. She said that, in particular:
(a) I have become extremely angry and bitter as a result of the many problems I have had since my implant surgery. It is true that I have suffered from depression at various times during the course of my adult life, but I have never been angry like I am now. This is the most significant change to my psychological well-being since my implant surgery.
(b) My mood generally is terrible; I am always annoyed and irritable. I regularly have mood swings.
(c) I no longer feel like a woman; it is as though my womanhood has been taken from me. I do not feel pretty or attractive, and instead I feel like I am ugly and fat. I no longer feel good about myself at all, and my self-esteem has significantly dropped. I never felt this way before my implant surgery.
(d) I remain deeply frustrated and saddened by the fact that I still have not obtained a resolution for my ongoing pain, and by the enormous impact my pain has had, and continues to have, on my day to day life.
I believe that my ongoing issues with anger, bitterness and low self-esteem have affected the quality of my relationships with friends and family. For example:
(a) I have found myself becoming more argumentative. This permeates a lot of my interactions with others. Also, it’s not always what I say, but how I say it, that creates conflict with friends or family. I actually believe that my anger can often be detected in the tone in which I speak, and this is noticeable to others.
(b) I used to always be a sociable person; I used to have a lot of friends, and I loved spending time with them. This is no longer the case; I am now far less social. As far as I am concerned, I am not often a joy to be around, and this makes me reluctant to be social.
I find myself going through phases of sociability. Sometimes I feel okay interacting with friends, but at other times I have depressing thoughts and I start thinking that I am nothing, and that I am worthless. This causes me anxiety, which leads to me pulling back and becoming socially withdrawn.
4213 She expressed concern that, if her husband were to die before her, she would never be able to remarry because she would have nothing “sexually, or feminine” to contribute to the relationship.
4214 Mrs Dawson was seen by Dr Jungfer at the request of her solicitors on 31 May 2016. Dr Jungfer noted that Mrs Dawson became “tearful and distressed when speaking about the changes and the loss experiences in her life”, which, she said were “congruent with the subject matter discussed”.3541 In oral evidence Dr Jungfer described her as “acutely clinically depressed”.3542 Dr Jungfer’s opinion was that she had developed a major depressive illness in response to the complications associated with the mesh implant and her inability to achieve “a good symptomatic recovery” and that this major depressive illness was chronic.3543 At the time of the assessment Dr Jungfer considered that no other factor was then contributing to her mood state. She said that changes in socialisation, reduction in recreational activities and the loss of intimacy in her relationship with her husband were secondary to the complications arising from the mesh surgery. She considered that it was likely that Mrs Dawson would have had a further depressive episode regardless of whether she had undergone the mesh surgery and suffered the complications but she considered it unlikely that she would have developed persistent depressive symptoms amounting to a chronic major depressive disorder had it not been for those events.3544 Like Professor Korda, Dr Jungfer recommended that Mrs Dawson be referred to a pain management service where she expected her mental state would receive appropriate attention. She pointed out that most pain management services are multi-disciplinary and include a psychiatrist and psychologist as well as a pain management physician. She thought Mrs Dawson’s mood disorder was unlikely to recover while she continued to be in pain.
4215 These opinions were controversial. Dr Lisa Brown, who examined Mrs Dawson on behalf of the respondents about six months later, had a different opinion. She did not doubt that Mrs Dawson had been suffering from depression but she considered that the recent depressive symptoms were attributable to the fact that Mrs Dawson had stopped taking anti-depressants in 2009 after her mesh surgery.3545 I will determine the dispute later in these reasons when I come to consider the extent of Mrs Dawson’s injuries.
4216 In her second affidavit, which was sworn on 28 July 2017, Mrs Dawson confirmed that after the final operation in March 2017 she felt much better, indeed much better than she had felt since the mesh was originally inserted. But the relief was short-lived. Towards the end of May 2017 she began to feel worse. As at 28 July 2017 she reported continuing pain in the nature of a burning sensation in the top of her legs and underneath her buttocks, a needle-like sensation inside her bottom, and a burning pain in and around her vagina, but no continuing pulling sensation across the bottom.3546 She said that when she applies Ovestin as directed by Dr Schierlitz using a sweeping motion to try to stretch the vaginal tissues, she feels an intensification of the burning sensation and pain at the tops of her legs, in and around the vagina and groin.3547 Sometimes the pain woke her from her sleep.3548
4217 Her bottom and pelvis continue to ache after a day’s work. Some accommodation has been made for her. She was supplied with a gel chair and her manager arranged a trial of a “sit/stand” desk.3549 The gel chair was helpful. The sit/stand desk, however, was not because there was “no room on it”, which I took to mean insufficient room to enable her to work satisfactorily, and it was removed after a week.3550 At her request, commencing 22 August 2017, Mrs Dawson’s working hours were reduced to four days a week.3551 In her second affidavit she was pessimistic about the future and was embarrassed about having to continually ask for time off.3552 During additional evidence in chief given in November 2017, when asked how she was coping with four days a week, she said that it was a lot better but she still gets tired and sore, has difficulty sitting, and finds that “really stressful”.3553 She was unsure how she would manage work in the future.3554
4218 Her pain adversely affected her recreational pursuits as well, including gardening, quilting and taking long walks which she and her husband had previously enjoyed, and the activities to which she can take her grandchildren.3555 The pain has also affected her capacity to drive long distances and to carry out household chores.3556 She struggles with household duties, has depended on the help of her husband, who retired in 2007, and has had to employ a cleaner as well.3557
4219 Since the implant surgery Mrs Dawson has travelled overseas and in Australia but not without discomfort. She and her husband took a bus tour to Europe over 21 days in May 2010 and in 2013 or 2014 they went to China and Tibet with a friend also for 21 days. Between those two trips they travelled to the South Island of New Zealand and in 2016 to the North Island for 10 days.3558 Mrs Dawson said that she found it difficult to sit and was tired a lot and did not interact with others in the way she would have liked.3559 She felt like she had a board inside her running across her bottom but that feeling disappeared after Professor Dwyer removed the left arm of the mesh.3560
4220 She still felt sad and angry at her plight and her emotional state, the impact on her relationships (including her marital relationship), remained as she described it in her first affidavit.3561 In her oral testimony she recounted a recent occasion at work when she was in a lot of pain when someone “just pressed [her] buttons” and she “blew up at her”.3562
4221 Mr and Mrs Dawson attempted to have sexual intercourse on about 18 September 2017. On that occasion she experienced knife-like pain pushing up and down inside her vagina and pain radiating down the right leg and a burning and pulling sensation under her right buttock. The next day she spent a great deal of time on the toilet with increasing rectal mucus. As a result of the pain she vowed never to have intercourse with her husband again.3563 In oral evidence she testified that, since the operation in 2009 she had had intercourse with her husband five, possibly six times. She said that it felt like someone was cutting her inside and she would scream with pain.3564
4222 Mrs Dawson told the Court that for several years since the complications of the surgery she has “felt terrible down below”. She lamented that she “couldn’t get feelings like a woman”, she thought that she did not look any good, did not feel any good and she felt that her “womanhood had gone”.3565
4223 Mrs Dawson’s account was supported by her husband’s evidence upon which he was not cross-examined.
4224 As at 8 February 2018, the last medical record available indicates that Mrs Dawson was prescribed the following medication: Efexor-XR modified release capsules (Capsule, Modified Release) 75 mg [28] (an antidepressant); Lyrica capsules 75mg [56] (an analgesic for neuropathic pain); Movicol (single dose sachets) [30] (to soften her bowel movements); Ovestin (oestrogen) vaginal cream; Palexia SR sustained release tablets 100mg [28] (an opioid analgesic), and Vesicare tablets 5mg (equiv. solifonacin 3.8 mg) [30] (a muscarinic receptor antagonist that reduces muscle spasms of the bladder muscles) for her overactive bladder symptoms.3566
4225 In oral evidence Mrs Dawson testified that she takes Efexor and Lyrica in the morning and at night applies three ointments. In addition to the Ovestin vaginal cream, she uses xylocaine 5% and amitriptilyne 2% to ease her pain. The xylocaine is applied internally through a dilator. She also uses a TENS machine to stimulate the nerves which she places near the back of her right leg at the site of her pain and in the groin as well as on her back since “sometimes that goes through to the vagina as well”.3567 These changes were prescribed by Dr Schierlitz.3568
4226 Dr Jungfer testified in November 2017 that the complications from the mesh surgery continued to contribute to the episode of major depression she diagnosed about 17 months earlier, despite the fact that notes from consultations with her GP and a psychologist to whom she was referred by her GP rarely mention the implant surgery or its consequences or indeed pain as a contributing factor.3569 When invited to comment on the significance of the absence in the clinical notes of references to pain or the subsequent operations, Dr Jungfer gave the following reply:
It can – essentially, it can mean two things. The first is that the psychologist was choosing to focus on things that the psychologist thought that she could change or that she thought were more significant. So it doesn’t mean that Mrs Dawson didn’t report the pain, but she also – you will have clinical situations where patients will come in and, because they know they’re seeing you for depression and they feel that, you know, the depression – this is what the person wants to hear, so to speak, or by the nature of the questions, they might not mention physical symptoms or physical problems. So it could be that Mrs Dawson didn’t present a history related to that because that’s not what she thought she was there for. It might also be that at the time that she was seeing the psychologist, the pain wasn’t as significant for her as these other issues that were going on in her life. And it might also be that the psychologist’s focus was not on the pain. So there are multiple explanations.3570
4227 It was not put to Dr Jungfer in cross-examination that the absence of such references signified or even suggested that the pain and/or its sequelae did not contribute to Mrs Dawson’s depression. Nor was Mrs Dawson challenged about her own evidence that she was depressed at the significant changes in her life since the implant surgery.
Background
4228 Mrs Sanders was born in the United Kingdom on 10 August 1946, which means that she is currently 73 years of age. She was 54 when she received the TVT device.3571
4229 Mrs Sanders gave birth to two children on 20 March 1975 and 5 December 1978.3572 The first involved an attempted vacuum extraction followed by a forceps delivery.3573
4230 Before the injuries the subject of this proceeding, Mrs Sanders described herself as happy and easy going. She had no history of mental disorders and no relevant family history.3574
Education and employment history
4231 After completing high school in about 1962 Mrs Sanders studied design for a few months before taking up an apprenticeship at a hairdresser. In 1967 she opened her own salon, which she operated for about seven years. Except for some part-time employment as a store clerk in 1977, she has not been in paid employment since.3575
4232 Between 1978 and 1997, however, Mrs Sanders sewed and made craft items, such as toys, dolls, and baby items, which for many years she would take to fairs to sell.3576
4233 Mrs Sanders has a long history of urinary incontinence. She dated her symptoms back to 1993. She first experienced leakage of urine with walking, running, laughing and coughing and she also found herself having to rise at night to urinate urgently.3577 She was not inclined to speak about her symptoms because she was embarrassed by them. She did not seek assistance and managed with the use of panty liners. But the symptoms gradually worsened. By October 2000 she was unable to run, walk, or do any form of exercise without some leakage so she decided to tell her general practitioner, Dr Clare Matthews. Dr Matthews recommended pelvic floor exercises and referred her to an urologist, Dr John Taylor.3578
4234 Mrs Sanders consulted Dr Taylor on 19 October 2000. He diagnosed her with “typical stress urinary incontinence”, some urgency, and occasional urge incontinence. According to Mrs Sanders’ unchallenged evidence, he told her he would “make things better” and “fix the incontinence” and would put her on a waiting list for a TVT implant. He described the operation as “a simple in and out, day procedure”.3579
The implant surgery
4235 On 12 March 2001 Mrs Sanders was admitted to the King Edward Memorial Hospital in Perth and underwent surgery the same day. Mrs Sanders believed that Dr Taylor performed the surgery. In fact, it was carried out by Dr Sandra McNeill, a surgical registrar still undergoing her specialist training, under the supervision of Dr Tsokos.3580
4236 A TVT retropubic device was implanted under local anaesthetic.3581 The operation report indicates that, after the TVT was introduced, cystoscopy revealed a normal bladder with no perforations.3582 It also records that the TVT was adjusted with “tapping”, which means that the surgeon tapped on the patient’s abdomen to simulate an increase in intra-abdominal pressure experienced with a cough or a squeeze in order to adjust the tension of the implant.3583
The aftermath of the implant surgery
4237 Mrs Sanders remained in hospital until 14 March 2001.3584 The contemporaneous notes indicate that the surgery proceeded “without undue event” and that before Mrs Sanders was discharged she was well and voided with no problems.3585 Mrs Sanders deposed that she had had a bad reaction to the anaesthetic, however, and felt nauseous after the operation. She also said that she had a urine test and was told that she had blood in her urine.3586 But she seems to have recovered quickly. The progress notes from the post-operative check-ups at the hospital in March and May 2001 indicate that she was feeling well, with no problems (19 March), and that she was pleased with the results of the operation (31 May).3587 These records are confirmed by Mrs Sanders’ evidence. She described her recovery as “uneventful” and the results of the surgery as “brilliant”. Her urinary symptoms improved and she was not getting up at night to urinate as before. Two months after the operation she resumed sexual intercourse which was pain-free.3588
4238 Between 2001 and 2006 Mrs Sanders said that she continued to be happy with the results of the surgery and did not have any problems.3589 She mentioned that in around 2002 to 2003 she had symptoms of urinary frequency and was told by her GP that she had a bladder infection. She was prescribed some medication and the symptoms disappeared.3590
4239 Mrs Sanders’ evidence is supported by evidence given by her husband, Peter. In his affidavit, upon which he was not cross-examined, he deposed that he did not recall his wife complaining to him about being unwell before 2008.3591
Injury
4240 In 2007 Mrs Sanders started to experience discomfort urinating. She also began to urinate more frequently. At the time she believed these symptoms were related to her earlier bladder infection, so she began taking non-prescription medication for the treatment of bladder infections. She was also prescribed antibiotics and underwent a pap-smear and an ultrasound of her urinary tract. Both were reported to be normal.3592
4241 In early 2008 she began to find sexual intercourse painful, as if there was something sharp in her vagina, like a blade. Her husband also described feeling something sharp and said that intercourse was painful for him, too. Mrs Sanders discussed the matter with her husband and started to have sex less frequently, abstaining altogether from sometime in 2008 or 2009.3593 Mr Sanders’ account of this period is slightly different from his wife’s. He spoke of his own pain but did not mention that she was also in pain.3594
4242 Sometime in 2008 Mrs Sanders developed a constant dull ache in her right groin, which she maintains she still has, on both the right and left sides of the groin.3595
4243 In around 2008 to 2009 Ms Sanders started to feel “something sharp” in her vagina when sitting. She said that this pain was different from the dull ache in her groin. She described it as a sudden, stabbing pain that “felt like a blade was sticking into me”, sometimes radiating down both of her legs. At times she felt the pain while sitting down, most often while driving or using the sewing machine. She said it would give her a jolt. Although it only lasted for a matter of seconds at a time, it was a severe pain. She ranked it as 9 out of 10 in intensity. After only five minutes on the sewing machine, she would have to stand up and lie down flat in bed until the pain subsided. This pain meant that Mrs Sanders used her sewing machine less, formerly once a week, now once a month, and did not go to fairs to sell her craft items as often. She and her husband bought a pedal-free sewing machine in the hope that it might ease the pain, but it did not. She had trouble standing for long periods because of the ache in her groin.3596 The pain “really got [her] down” and she felt miserable.3597 Between 2009 and 2010 Mrs Sanders experienced the sharp pain in her vagina more frequently, causing her to stop sewing completely.
4244 In 2010 her urinary symptoms also deteriorated, to the point that when she was not taking antibiotics she had both frequency and discomfort urinating (dysuria). Mrs Sanders gave the following description of her urinary problems, which she was told at the time were caused by urinary infections:
The problems did not feel like what I had experienced with cystitis [bladder infection]. I did not have a burning sensation with urination. The pain affected my whole body. It went into my arms and my fingers. My jaw locked and it felt like I was in labour and like I needed to push something out. On occasion, I was not able to pass urine at all. I needed to sit on the toilet for up to a couple of hours trying to urinate. After one or two days on antibiotics, I could urinate normally, but I still felt unwell for a couple of days after that. It zapped all my energy and I did not feel like doing anything. After I completed a script of antibiotics, I was okay for a week or two and then the symptoms returned.3598
4245 At about this time Mrs Sanders said that she started to take painkillers regularly to help with the dull groin pain and the stabbing vaginal pain and the pain associated with what she was told were urinary infections. She said that she took six Panadol tablets throughout the day and at night two Panadol and two Advil (an over-the counter ibuprofen-based non-steroidal anti-inflammatory drug) tablets. Whenever she was unable to get an appointment to see a GP, she went to the chemist to purchase over-the-counter medication for bladder infections.3599
4246 It was at about this time that her husband said that he remembered his wife saying “I feel like something is there and something isn’t right” and that he noticed that she was taking Panadol and Advil in addition to antibiotics.3600 It was also at about his time that he noticed changes in the home. He said:
Ann and I used to joke that she was really house proud. It was around 2010 Ann stopped cooking and cleaning like she used to. She would not do the windows, mop the floors or make the beds, she would just do general tiding (sic). Anything more physical around the house I would do for her. Ann used to cook extravagant main meals. It was around this time her cooking decreased, and instead she would make meals that were easy to prepare like salads. I started to cook very simple meals during the week to help out. It became a real effort to take Ann out anywhere. At first these changes were difficult on our relationship as we could not do the things we used to. However, our relationship adapted and we just did what we had to do.3601
4247 By January 2011 the dull ache in her groin and sharp pain in her vagina had become so painful that she went to see a general practitioner, Dr Juliet Tran.3602 Dr Tan recorded the following history:
pt reports she has haematuria [blood in the urine], saw KEMH 5 yrs ago, had ring inserted , had checkup but no examination performed at the time, feels it is sticking into her when she sits down
urinary freq, pressure3603
4248 The notes record that Mrs Sanders refused specialist review but Mrs Sanders denied that she had.3604
4249 Although these notes make no mention of pain, the reason given for contact is “dysuria”, which means difficult or painful urination. Professor Korda said that dysuria is usually associated with urgency and frequency of urination if due to cystitis or urethritis and the pain is burning in nature, relieved by curing the underlying cause, although a high fluid intake usually helps.3605
4250 Plainly Dr Tan thought Mrs Sanders had a urinary infection because she prescribed antibiotics (Alprim). Her notes also record that Mrs Sanders had “ring pessary at vaginal orifice level on examination”. Dr Tan recommended that Mrs Sanders see a specialist. She told her that “it needs to be repaired as obstructing urine outflow”. It is not clear to what noun the pronoun “it” was intended to refer but Mrs Sanders has never been fitted with a ring pessary. Dr Tan referred Mrs Sanders to the outpatients’ department at King Edward Memorial Hospital. The referral reads:
She had a ring inserted 5 yrs ago as she had urine stress incontinence and damage to her muscles during labour. The ring is pressing into her when she sits and is at the level of the vaginal orifice. Please arrange treatment.3606
4251 On 28 January 2011 Mrs Sanders presented to the Emergency Centre at King Edward Memorial Hospital where she was triaged. Mr Sanders drove her there because “she was in some pain”.3607 The presenting problem repeats the error about the ring pessary, presumably taken from Dr Tan’s referral. It reads:
Had ring inserted 6 yrs ago – now painful sensation in vagina when sitd (sic) down and is at level of vaginal orifice
Recurrent UTI’s. Haematuria. Has just completed course antibiotics & has script for rpt. No urinary symptoms currently.3608
4252 Mrs Sanders was then examined by a registrar, Dr M Howlett, whom Mrs Sanders referred to as “a young doctor”.3609 The history as recorded relevantly reads:
64 y/o P2
referred by OPC triage for r/o ring pessary
Pt reports never had ring pessary
6 years ago had ? birch colposuspension w mesh
Past 6/12 – recurrent UTI [urinary tract infections]
=> No Sx [symptoms] now
– Has always been able to feel pricking pain when sat down + felt mesh
really feels well
Mild urgency for 6 yrs. Good urine flow3610
4253 On examination, Dr Howlett noted that Mrs Sanders was well but could “see + feel mesh at introitus”. Dr Howlett also noted that she had “mildly atrophic walls”. Vaginal oestrogen was prescribed and Mrs Sanders was referred to the urogynaecology outpatients’ clinic. Dr Howlett’s referral note reads:
Thank you for seeing this P2 who was referred for r/o ring pessary by OPC.
She has not got a ring pessary in situ, but rather had + now mesh protruding.
Causing her mild discomfort on sitting
mild urgency otherwise no urinary Sx [symptoms]
report 6/12 recurrent UTI [urinary tract infections].
Please r/v for trimming mesh3611
4254 On 19 May 2011, Mrs Sanders was seen at King Edward Memorial Hospital by a consultant gynaecologist, Dr Alanagh Gilbert. In her first affidavit she said that she told Dr Gilbert that she felt something itchy and sharp sticking into her, that she had recurrent bladder infections, and was no longer able to have sexual intercourse because of pain.3612
4255 The notes of the consultation read:
TVT 10 years ago
didn't feel right from the start
feels something itchy
doesn’t have stress incont
urgency & occ urge incontinence
–no hormonal therapy
–vaginal dryness
–not able to have sexual
has had recurrent UTI’s – treated by fluids
O/E small piece of mesh visible LHS
for EUA/cystoscopy/r/o vaginal tape3613
4256 As the respondents submitted, pathology results from urine testing for urinary tract infections in the period from June to October 2011 do not indicate that Mrs Sanders had urinary tract infections and on 13 October 2011 the pathologist wrote that “[m]icroscopy indicates that UTI is unlikely”.3614
4257 In her letter to Dr Tan, Dr Gilbert wrote:
I saw Ann in the Urogynaecology Clinic today. She had a TVT placed 10 years ago for urinary stress incontinence. She has never felt quite right in the vagina since then but it has become worse recently.
On examination she has an area of mesh erosion anteriorly. She does not have any stress incontinence currently and only has mild urge.
I have arranged for her to come in for an examination under anaesthesia cystoscopy and removal of vaginal mesh and we will let you know how all goes.3615
4258 In her affidavit Mrs Sanders denied that she had had the symptoms of which she complained to Dr Gilbert since her incontinence surgery. Rather, she said that she had had these symptoms “for a long time”.3616 In cross-examination she also denied that she had ever told Dr Gilbert that her vagina had never felt right since the operation in March 2001.3617
4259 Mrs Sanders deposed that she was unconcerned when she was told she needed an operation to remove the mesh because she believed she would “just go back to getting urinary leakage with sneezing, coughing and walking” and that she could cope with these symptoms. The pain she was experiencing, however, was another matter. She said that she could not cope with that. She added that she felt a sense of relief at the knowledge that she would not have to put up with the pain and constant bladder infections.3618
4260 On 28 June 2011 Mrs Sanders presented to Dr Matthews with right groin pain on sitting. She also reported that she was urinating frequently but had difficulty passing urine, and that sometimes she had a sense of urgency in having to get to the toilet before wetting herself. She said that she told Dr Matthews that sometimes her jaw locked when she strained to urinate and that it felt like she was in labour. On examination internal rotation of the right hip was reduced and she was tender on the right side of the pubic symphysis, a joint between the pubic bones in the centre of the pelvis in front of the bladder. Dr Matthews requested pathology of the midstream urine, requested x-rays of the hip and pelvis and a pelvic ultrasound.3619
4261 Mrs Sanders said in her first affidavit that Dr Matthews offered her Valium but she refused, thinking that she was being treated for depression when she wanted treatment for her pain. She wondered whether Dr Matthews believed her. She said it was “like hitting [her] head against a brick wall”.3620 But on a return visit a week later, she said that Dr Matthews prescribed some painkillers but they did not alleviate the pain.
4262 Dr Matthews’ records do not include an entry one week later. The next entry is for 14 July 2011 when Mrs Sanders presented with hip pain. I assume this was the consultation Mrs Sanders had in mind. On this occasion Dr Matthews reviewed the scans that were performed on 5 July 2011. As reported by the radiologist, the ultrasound findings were normal but the x-ray of the pelvis and right hip showed mild degenerative changes in the right hip joint with tiny marginal osteophyte formation and reduced medial joint space and no other abnormalities. Mrs Sanders was tender over the right greater trochanter, a part of the femur near its joint with the hip bone and Dr Matthews prescribed Panadol Osteo tablets.3621
4263 On 8 August 2011 Mrs Sanders was admitted to King Edward Memorial Hospital for surgery. The surgeon was Dr John Daborn, a urogynaecologist. After appropriate preparation and draping, local anaesthetic was injected around the mesh and the exposed mesh was excised with sharp dissection at the midurethra.3622 According to the operation report, Dr Daborn found “TVT eroded at mid urethra” and inflamed bladder mucosa.3623 The report indicates that the eroded tape was excised but does not state how much tape was removed. Dr Daborn wondered whether Mrs Sanders had a squamous metaplasia at the bladder trigone (the triangular region of the wall of the bladder, which lies between the openings of the two ureters and the urethra).3624 Professor Korda explained that squamous metaplasia is a “benign non-cancerous change (metaplasia) of (non-squamous) surfacing lining cells (epithelium) to a squamous morphology”. 3625
4264 Dr Daborn also took a bladder biopsy.
4265 A histopathology report was tendered and, although the findings and conclusions appear to be instructive, neither party referred to them in submissions.3626
4266 The pathologist, Dr Sukeerat Ruba, reported that the biopsy showed “acute on chronic cystitis with histological features in keeping with a cystitis glandularis”. Dr Ruba said that the vaginal mesh sections showed irregular fragments of dense fibrous tissue in which there was birefringent foreign body-type material surrounded by a multinucleated giant cell and histiocytic reaction with associated chronic inflammation. As Professor Iakovlev explained, birefringence is refractivity or brightness in polarised light.3627 Birefringence is a characteristic of polypropylene.3628
4267 On the face of things, the biopsy findings tend to confirm Mrs Sanders’ account that she had a history of cystitis and the findings concerning the mesh sections support the evidence given by Professor Klosterhalfen and others about the chronic inflammatory reaction to Prolene.
4268 Mrs Sanders said that the following day Dr Daborn explained to her that only part of the mesh could be removed and, that the mesh had eroded into her bladder, causing inflammation. He also told her she might require further surgery. Ms Sanders recalled that this was the first time she had heard the word erosion in relation to mesh. She repeated that it was never mentioned to her at the time of her incontinence surgery.3629 She said that Dr Daborn was the first doctor to tell her that the TVT had eroded and that the problems caused by the TVT were likely to require more operations and might be permanent.3630
4269 Mrs Sanders was discharged on oral antibiotics the next day.3631
The aftermath of the excision surgery
4270 Mrs Sanders said that she felt unwell after the mesh excision surgery. The symptoms she associated with the bladder infections returned. She felt pain while urinating, difficulty urinating, and urinary frequency.3632 Later in August she said that she started to experience an increasing urge to pass urine urgently. She said that she often leaked urine before making it to the bathroom and her stress urinary incontinence returned: she experienced leakage during exercise, coughing, and laughing.3633
4271 During the first six weeks after she was discharged from hospital Mr Sanders assisted her with various tasks such as shopping, heavy lifting, cleaning, and driving and he took two or three days off work to care for her immediately after her operation.3634
4272 Her urinary symptoms continued to deteriorate. She claims she now has no bladder control at all, which affects her confidence wherever she goes. She wears pads all the time and changes them multiple times a day. She also carries a change of clothes with her at all times in case she has “an accident”.3635
4273 In addition, the dull aching pain she experienced across her pelvis and groin increased, and towards the end of 2011 she felt the sharp pain in her vagina returning when she was sitting down.3636 She said, however, that the sharp pain is not as bad as it was before the mesh excision surgery on 8 August 2011. Now she feels the pain about once a week and sometimes every couple of weeks when she is sitting down and it lasts a couple of seconds before disappearing.3637
4274 On 22 September 2011 Mrs Sanders was reviewed at the urogynaecology clinic by Dr Tsokos. He reported to Dr Tan that her vagina was now “well-healed” although she still had urinary symptoms but that she was not keen on further treatment or investigation at this time.3638 Mrs Sanders deposed that he told her she could have another operation to fix her problems but that she said that she did not want to go through further surgery. In her affidavit she explained that, by then, she had lost faith in doctors being able to fix her problems.3639
4275 On 13 October 2011 Mrs Sanders saw Dr Matthews. Her notes indicate that Mrs Sanders told her that she over the past few weeks she had “urgency” and was passing small quantities of urine. They do not record any complaint of pain. Pathology was requested from a urine sample and the microscopy indicated that a urinary tract infection was unlikely.3640
4276 Over the next few months Mrs Sanders saw two other doctors who prescribed non-surgical treatments, none of which resolved her problems. One of these doctors was Dr Tim Jeffery, who examined Mrs Sanders at King Edward Memorial Hospital on about 27 October 2011. Dr Jeffrey reported that Mrs Sanders had recovered from her recent mesh removal but had now developed “some urgency type symptoms”. On examination he could not find any mesh ulceration or evidence of vaginal prolapse. He put her on Vesicare 5mg daily to try to stabilise her urgency symptoms.3641
4277 For the next few years, Ms Sanders continued to rely on antibiotics to help alleviate her urinary symptoms, but these symptoms never completely resolved. Sometimes she felt fine for a couple of months, without urinary frequency or pain, then her symptoms would return for a few weeks. In August 2012 she resumed her craft work and returned to her sewing machine “approximately every month and sometimes every couple of months”. If she sewed, however, she could not do so for longer than 30 minutes at a time and could not return until her pain subsided.3642
4278 Mrs Sanders continued to seek treatment for her urinary symptoms and continued to be prescribed antibiotics for what she believed to be bladder infections.
4279 In June 2013 she travelled to the United Kingdom for six months. She said that getting around the country was difficult and she mostly drove. She said that she had two urinary tract infections while she was there with frequency, pain on and “difficulty with” urination, and urgency.3643
4280 On 8 April 2014 Mrs Sanders consulted Dr Matthews. The reason given for the consultation as recorded in the doctor’s notes is hip pain and the history is of right groin pain for “a couple of months” and of the right leg “giving way”, then an ache in the left groin, and pain (apparently on or since the previous Wednesday, six days earlier) in the lower back radiating into both groins with pain into the right upper inner thigh. Examination was difficult because of the pain. Her lumbar spine and paravertebral muscles were tender and she had pain laterally in the hips. Internal rotation of both hips was reduced and caused pain and she had pain on lying with both hips flexed. Straight leg raising was severely limited and the doctor was unable to elicit reflexes.
4281 Dr Matthews prescribed Mobic capsules (a non-steroidal anti-inflammatory drug) 15mg 1 daily and Panadeine Forte tablets 500mg/30 mg 2 qid (four times a day) prn (as needed), and requested a CT scan of the lumbar spine and x-rays of the hips.3644 She also advised Mrs Sanders to use a heat pack and to return the next day.
4282 The next day Mrs Sanders presented to the surgery, apparently with the scans. On this occasions she was seen by Dr Peck Tang. Dr Tang reviewed the results and examined Mrs Sanders. Evidently the scans showed multilevel facet joint degeneration and mild spondylosis as well as degeneration in the right hip joint but the objective findings did not correlate with the degree of Mrs Sanders’ pain. Dr Tang thought the pain was likely to be a combination of facet joint osteoarthritis radiating into the right groin and right hip osteoarthritis. She prescribed Fentanyl patches, one every 72 hours.3645
4283 On 3 June 2014 Mrs Sanders saw Dr Matthews with pain in her right hip and groin, which woke her at night, and pain in the lumbar area on both sides. According to Dr Matthew’s notes, she told the doctor that she did not take the Fentanyl patches but she did take the Mobic and that helped a little. The history includes a report that walking stirs the pain up a lot and that the right leg gives way at times. Dr Matthews gave her another prescription for Mobic and added a prescription for Panadol Osteo tablets 665mg tid (three times a day). She also referred her to a specialist, Dr Judith Cole.3646
4284 Mrs Sanders returned to see Dr Matthews on 23 September 2014 with persistent right groin pain and right hip pain. She also told her that she had had a urinary tract infection in July and another the day before the consultation. On examination her right groin was tender. She was given a prescription for Keflex capsules 500mg 1 tid.
4285 Mrs Sanders said that she saw another GP, Dr Ajibola Oki at a different medical centre on 18 July 2014 while Dr Matthews was on holiday. She said that she told Dr Oki she had increased urinary incontinence and frequency and believed she had another bladder infection. She said that Dr Oki prescribed antibiotics and that, by the time she returned four days later, she felt better and thought the infection had gone. Mrs Sanders said that the doctor told her that the previous urine sample she had provided proved positive for blood and bacteria in very high levels and asked her to complete the course of antibiotics and to provide another urine sample. Although she said she did as she was told, she was unable to recall what happened.3647
4286 Dr Oki’s notes were tendered. They tend to support Mrs Sanders’ account. The consultation notes for 22 July, for example, record that she was clinically better, her urine had improved, and that her urine samples showed “blood/bacteria+++, mixed bacteria”. The comment by the pathologist on the pathology results, however, which accompanied the notes, was merely that a urinary tract infection could not be excluded.3648 The comment on the follow-up tests on the sample requested on 22 July, collected and reported on 28 July, was “[m]icroscopy indicates that UTI is unlikely”.3649
4287 On 23 September 2014 Mrs Sanders returned to Dr Matthews with persistent right groin pain and also right hip pain.
4288 On 4 December 2014 Mrs Sanders said that she saw Dr Matthews in relation to urinary frequency and urinary discomfort as well as pain in the groin and right hip. The reason given for contact is “inguinal hernia”, which is a defect in the abdominal wall through which either bowel or fat can protrude.3650 Presumably Mrs Sanders presented with groin pain and Dr Matthews suspected that the cause of her groin pain was an inguinal hernia. The doctor’s notes, however, make no mention of the urinary symptoms.3651 Mrs Sanders said that Dr Matthews told her she might have a hernia and referred her to see Dr Michael Levitt, a general surgeon. Mrs Sanders saw Dr Levitt on 14 January 2015. Dr Levitt reported to the GP:
Thank you for referring Ann whose right groin pain is associated with exquisite tenderness over the groin, the inguinal ligament and, most especially, over the pubic bone. Her clinical features are much more in keeping with a diagnosis of osteitis pubis [inflammation of the pubic symphysis and surrounding muscle insertion] than they are with pain originating from what is, to be honest, a barely detectable inguinal hernia.
I am arranging to evaluate the possible diagnosis of osteitis pubis by means of an MR1 scan and have introduced regular oral non-steroidal anti-inflammatory painkillers to help reduce her pain. Naturally 1 shall report back to you with news of the result of her MRI scan.3652
4289 The MRI (of the pubic symphysis and hips) was performed the next day.3653 The radiologist’s summary was that there was a small insertional tear of the left adductor longus tendon and a small anterosuperior labral tear of the right hip, and minor degenerative change in the pubic symphysis. Otherwise, he wrote, the hip joints were “grossly normal on wide field of view imaging”. The only additional finding of note was of mild right and moderate left superficial trochanteric bursal oedema (swelling/excessive watery fluid).
4290 Before making a final decision about surgery, Dr Levitt sought an opinion from Dr Scott Isbel, a physician.3654
4291 Dr Scott Isbel saw Mrs Sanders on 6 March 2015.3655 He took a history of right anterior groin, right lateral hip and right buttock pain with an irritable hip joint, worse if bumped or jarred. He noted that the recent MRI suggested no significant joint arthropathy although there was a small labral tear. On examination there was tenderness in the lower back and the right-sided facet joint, moderate tenderness in the right great trochanter, and the hip joint was irritable with flexion adduction and internal rotation.
4292 Dr Isbel thought that Mrs Sanders was likely to have low grade synovitis in the right hip joint, gluteal tendon insertion and bursitis pain, and referred pain in the lumbar facet joint. He recommended a trial of a CT-guided injection into the right hip joint and on 31 March 2015 Mrs Sanders received a cortisone injection into her right hip joint.3656
4293 On 29 July 2015 Dr Levitt saw Mrs Sanders again. In his report to Dr Matthews he wrote that the injection into the hip joint had “definitely provided her with symptomatic improvement and there really can be little doubt now that her pain is musculoskeletal in origin and that her right inguinal hernia is coincidental only”.3657
4294 In the meantime Mrs Sanders saw Dr Matthews for a flu injection on 23 April 2015 and told her that she had had five urinary tract infections since December and that Keflex works quickly. It seems that at this point Dr Matthews prescribed Keflex for prophylactic purposes.3658 The notes for the next consultation on 27 August 2015 include a history that Mrs Sanders was very well and had had no more urinary tract infections since the prophylactic Keflex was started. She therefore planned to continue with this course of treatment.3659
4295 In late April 2015, however, Mrs Sanders began to worry about the long-term effect antibiotics was having on her health. She said that she started skipping tablets to reduce her intake, but as soon as she did so her symptoms returned. She said that she tried alternatives to antibiotics, including trialling pessaries for approximately three weeks, but they did not relieve her symptoms. It is likely that these events occurred at a later point in time. Dr Matthews’ notes for the consultation on 1 December 2015 record once again that Mrs Sanders had no more urinary tract infections with daily Keflex. They then state: “discussed other options eg trial of vaginal oestrogen then after a month try stopping keflex”.3660
4296 According to the notes, at the consultation on 1 December 2015 Mrs Sanders’ only complaint of pain was about the right hip. She told Dr Matthews that the right hip pain was getting worse and that the hip injection only helped for a couple of weeks. She also reported night pain and “limping”. Dr Matthews referred her to Professor Richard Carey-Smith, an orthopaedic surgeon.3661
4297 Mrs Sanders saw Professor Carey-Smith on 21 December 2015.3662 He took the following history, which Mrs Sanders accepted in cross-examination as accurate:
[Mrs Sanders] has been limping around for 2 years with a painful right hip. This is increasingly interfering with her daily activities and limits her ability to keep up with her friends whilst walking. Sleep is not yet disturbed. Unfortunately she has ballooned putting on nearly 20kgs over the last couple of years related to decreased activity. She has had some problems with a bladder sling which causes her very severe urinary tract infections and clearly we will need to be aware of this at the time of joint replacement. Otherwise she is not in too bad a shape.
4298 His examination findings included a positive Trendelenberg gait which was obviously antalgic, poor straight leg raise on the right side, groin pain, pain on internal rotation and flexion, and a mild flexion deformity of the right hip.
4299 Professor Carey-Smith diagnosed her with central arthritis in the right hip and recommended a total hip replacement.
4300 Mrs Sanders returned to see Professor Carey-Smith on 19 February 2016 with up to date imaging. His diagnosis had not changed. Nor had his recommendation for a hip replacement for on this occasion he advised her about the risks associated with the procedure.3663 In her second affidavit Mrs Sanders said that she was “an emotional mess” when she saw Professor Carey-Smith because of what had happened with the tape implant. She said that she told him she did not know whether she wanted to go through with the operation. She claimed she had “lost all faith in medicine”. But she was reassured by Professor Carey-Smith.3664
4301 On 31 May 2016 Mrs Sanders was assessed by Dr Jungfer at the request of her solicitors.3665 Dr Jungfer reviewed Mrs Sanders’ medical records and took a history from Mrs Sanders herself, which was broadly consistent with her evidence.3666 She noted that Mrs Sanders became “tearful and emotional when speaking about the loss experiences associated with her incontinence, the change in her functional capacity and the difficulties that she experiences”. I noted that Mrs Sanders became emotionally labile during oral evidence in chief when, in response to a question from Mr Graham SC about how she saw her current lifestyle in comparison to the life she enjoyed before her continence surgery in 2001, she replied: “Well, to say I’m devastated is an understatement …”3667
4302 Dr Jungfer diagnosed her with an adjustment disorder with depressed mood. Dr Jungfer explained:
An adjustment disorder is a cluster of symptoms which reflect a psychological response to an identifiable stress. The psychological response can include emotional and behavioral symptoms. These symptoms or behaviours are required to be clinically significant and cause marked distress as well as significant impairment in social, occupational, or other important areas of functioning. For a DSM-5 diagnosis of an adjustment disorder the symptoms should not be explicable on the basis of another psychiatric condition.3668
4303 Dr Rosalie Wilcox, another psychiatrist, assessed Mrs Sanders on behalf of the respondents on November 2016.3669 She obtained a similar history to that provided to Dr Jungfer and her diagnosis was the same.3670 She considered that the adjustment disorder more likely than not occurred as a consequence of the impairment of Mrs Sanders’ urological function that had become more pronounced since the operation of 8 August 2011.
4304 Dr Wilcox noted that before the continence surgery in March 2001, Mrs Sanders had had stress incontinence, but she “provided no history to suggest that she had other symptoms such as recurrent UTIs or pain in the groin” and that “[i]t was not until some years after the surgery that she became aware of sharp pain in her vagina and she did not seek any medical advice until the pain became associated with urinary tract infections”.3671
4305 Dr Wilcox noted that an adjustment disorder is caused by a stressor. Consequently, there is an expectation that it will resolve once the stressor has been removed. On the other hand, Dr Wilcox observed, if the stressor persists, as has occurred with Mrs Sanders, the condition is less likely to resolve and she could have symptoms of varying severity on a long term basis.3672 Like Dr Jungfer, Dr Wilcox considered that the prognosis was closely tied to the status of the urological symptoms and it was likely that her mood would fluctuate in response to the severity of her symptoms.3673 Unlike Dr Jungfer, however, she did not recommend treatment. In particular, she said that there was no need for anti-depressants and, based on her attitude to medication, she did not consider Mrs Sanders would take any if it were recommended.3674
4306 Dr Wilcox administered a Beck Depression Inventory (BDI-II), a 21 question self-report inventory that measures characteristic attitudes and symptoms of depression. Dr Wilcox observed in her report that the BDI is regarded as having a high degree of reliability. Mrs Sanders achieved a score of 27, which Dr Wilcox stated suggested moderate depression. She noted that Mrs Sanders had indicated that she felt she was being punished, that she obtained less pleasure from things she used to enjoy, that it was hard to get interested in anything and she did not consider herself as worthwhile or as useful as she used to.
4307 Dr Wilcox also administered an Anxiety Inventory. Apparently Mrs Sanders achieved a score of 6, which indicated that she was not bothered by symptoms of anxiety.
4308 Once again Mrs Sanders was “teary when she spoke of the impact of the urinary incontinence on her quality of life”.3675
4309 Dr Wilcox was asked to describe the effect of Mrs Sanders’ psychiatric condition on her ability to perform domestic duties, her relationships, her confidence, her self-esteem and her ability to socialise. This was her response:
In my opinion her psychiatric condition does not impact to any extent on her ability to perform domestic duties. I note she would like to be more helpful around the home and is mainly limited in her ability to manage domestic chores due to symptoms of a physical nature. She still manages to do most chores but at a slower pace.
As a result of her psychiatric condition her level of enthusiasm is decreased. She is frustrated by her reduced capacity to manage activities and this in turn results in irritability and intolerance of others. In addition her lower mood is associated with hypersensitivity as she is overly conscious of her urinary symptoms and as a consequence she is embarrassed and ashamed and is less willing to socialise. Due to obsessive ruminations her sleep is impaired and she is more fatigued and has less capacity to enjoy activities.3676
4310 Dr Wilcox also considered, however, that it was likely that Mrs Sanders’ reduced mobility secondary to her right hip pain affected her ability to perform domestic duties and that it was also probable that her weight gain had had a negative impact on her self-image and self- esteem. Dr Wilcox noted that Mrs Sanders had acknowledged that she was not comfortable with the weight gain and had been trying to lose weight for some time.3677
4311 Following a review on 6 June 2017 in the wake of the hip replacement surgery, Dr Jungfer considered that Mrs Sanders had more anxiety symptoms than were evident on the previous assessment on 31 May 2016 and revised her diagnosis to “adjustment disorder with mixed anxiety and depressed mood”. She said that she was satisfied that Mrs Sanders meets the diagnostic criteria for this condition since she reported both depressive and anxiety symptoms.3678
4312 Dr Jungfer also revised her views about treatment. Having canvassed the matter with Mrs Sanders at the second consultation, Dr Jungfer was informed that she would not seek counselling of any kind. Mrs Sanders told her that ventilating her problems with her daughters and obtaining their feedback was beneficial and that she would not be comfortable disclosing her issues to a third party. Dr Jungfer therefore agreed with Dr Wilcox that counselling was not necessary. On the other hand, she considered that anti-depressants were reasonable in order to modify her pain but that this treatment should be initiated by a psychiatrist, having regard to Mrs Sanders’ anxiety and her perception of the negative consequences from medical intervention. She suggested as an alternative treatment from a chronic pain management service. 3679
4313 The diagnosis was not in dispute.3680 Indeed, none of Dr Jungfer’s opinions about Mrs Sanders was disputed. Although the doctor was required for cross-examination, the cross-examination was confined to her opinions concerning Mrs Gill and Mrs Dawson.
4314 On 2 June 2016 Professor Korda saw Mrs Sanders at the request of her solicitors.3681
4315 At this consultation she reported bilateral groin pain, which had increased since the excision surgery; dysuria, lower back pain, right hip pain, and urinary symptoms. In addition, she complained of constipation and faecal incontinence. She also mentioned that she had developed a limp and was awaiting a hip replacement.3682 She told Professor Korda that she had not had sexual intercourse since the revision surgery in August 2011.
4316 On examination her abdomen was generally tender and there was mild discomfort on palpation in both inguinal regions, which Professor Korda said was suggestive of the presence of bilateral inguinal hernias.3683 Mesh was easily palpable through her lower vagina to the right of the urethra, around 5mm from the vaginal opening and the mesh was covered by only a very small amount of vaginal epithelium over the sharp bristles of the mesh, which Professor Korda wrote was suggestive of “incipient erosion”. In oral evidence he was asked whether by incipient erosion he meant the beginning of erosion or something else. This was his reply:
Well, it means that the examination indicated that in the case of Mrs Sanders there was a very thin bit of tissue, almost like a little membrane only, between the mesh and the examining finger, which really would suggest that it wouldn’t be difficult for that part of the mesh to be eroded into the vagina, for instance, from friction or from further thinning of the vagina from lack of oestrogens in the menopause. That’s what I meant.3684
4317 By friction Professor Korda was referring to the movement of the vaginal skin during sexual intercourse.3685 He explained that the repetitious activity of vaginal penetration would move the skin over the area of mesh and the mesh is firm and hard and does not move against the vaginal skin.3686
4318 Contrary to Professor Blaivas’s assumption that Mrs Sanders had “mild” vaginal atrophy,3687 Professor Korda found that she had “quite marked” vaginal atrophy (cell degeneration), and tenderness under the pubic arches. But her introitus was normal and she had well supported anterior, apical and posterior vaginal compartments.3688 Her cervix looked healthy. Her uterus was normal in size, shape and configuration, and the vaginal fornices (the three vaulted spaces at the top of the vagina) were clear.
4319 Professor Korda also reviewed urodynamic investigations carried out by Associate Professor Clara Shek the day before his examination. Marked urodynamic stress incontinence was observed in the supine position and moderate urodynamic stress incontinence in the erect position.3689
4320 A 3D/4D perineal ultrasound examination revealed that the bladder had descended 9.5mm below the pubic symphysis and there was a rectocoele 20.8mm below the pubis. A suburethral sling was seen dorsal to the distal urethra in an unusual location with a sling pubis gap of 18.5 mm.3690 The posterior vaginal wall appeared thickened (an explanation for which eluded Professor Korda during cross-examination),3691 but there was no anterior compartment or posterior compartment mesh, that in the axial plane there was a left partial defect in the puborectalis muscle but no hiatal ballooning with a hiatus on Valsalva of only 24.2 cm, and the external anal sphincter was intact. 3692
4321 Professor Korda expressed the opinion that the unusual position of the sling was not due to inappropriate insertion at the initial surgery but was caused by the excision of a portion of the sling in August 2011. In oral evidence he explained that, since the TVT had been working very well without any problem for some seven or eight years, “one must assume that the original insertion was appropriate”. He continued:
When the erosion occurred, they removed segment of that tape so that the rest of it may have moved to an unusual position because the tension of that device, which normally is U-shaped, was altered so that on one side there was less tension and it moved out. I didn’t want to suggest that abnormal position was due to inappropriate surgery.3693
4322 Professor Korda also referred to Associate Professor Shek’s examination findings. She had noted that there was pain on the left side of the vagina on palpation of the levator muscles. Although she could not feel mesh erosion, she could palpate the mesh through the lower vagina to the right of urethra around 5mm cranial to the introitus. Professor Korda considered that feeling the mesh so close to the vaginal epithelium on the balance of probabilities indicated “incipient erosion” and signalled the possible need for further revision surgery.3694
4323 On cystourethroscopy, no mesh or sling material was found in the bladder or the urethra.
4324 As reported by Professor Korda, Associate Professor Shek concluded that Mrs Sanders had marked urodynamic stress incontinence with low compliance bladder and possible urethral instability.
4325 When asked to explain the meaning of a low compliance bladder, Professor Korda said:
The best way to understand a low compliance bladder is that the bladder is like a bag which sort of is in a collapsed state when it is empty, and as it fills up – and the bladder usually fills up at a rate of about 60 mils a minute, and the normal bladder capacity is 500 mils. So as it fills up, the bladder does not increase because the bladder just expands to accommodate the urine, and usually it’s only when it reaches full capacity that the patient or any individual has to go to the toilet because the bladder sends messages to the brain ..... that is normal compliance. If the bladder is in some ways thickened for some reason or it doesn’t expand properly, then the pressure in the bladder, as you fill it, linearly goes up with the filling volume. So, in other words, as you fill the bladder, the bladder pressure goes up in a linear way. And that is defined as a low compliance bladder, and such patients do have symptoms which are urinary frequency and urgency, and they are not dissimilar symptoms to urinary tract infections.3695
4326 Professor Korda listed Mrs Sanders’ then current injuries and disabilities as apareunia; urodynamic stress incontinence; low compliance bladder; urethral instability; bilateral groin pain; episodes of dysuria; lower back pain; right hip pain; pain radiating from the groin up through her body as far as her jaw, arms and fingers; constipation; and faecal incontinence.3696 I will deal with the relationship between these conditions and TVT when I come to consider damages in Part XVIII.
4327 On 1 August 2016 the hip replacement surgery was performed by Professor Carey-Smith at Osborne Park Hospital.3697 Following the surgery, Mrs Sanders felt nauseous from the anaesthetic and vomited “all the time”, but she said that she “came good after two days” and had no other issues apart from the vomiting. She was discharged from hospital on 4 August. She said that the hospital gave her some strong painkillers to take home but that she did not take them because she does not like to take any medication that makes her feel “not with it”.3698
4328 On 10 August 2016 Mrs Sanders saw her GP, Dr Matthews, to discuss pain relief following her hip operation. Dr Matthews prescribed Tapentadol (Palexia) SR tablets 50mg 1 bd (by day), an opioid analgaesic, and Tramadol Actavis capsules 50 mg, an analgaesic indicated for the treatment of moderate to severe pain, 1–2 qid prn.3699 In the end she said (and I accept) that she took Advil and Panadol for the pain, in the morning, afternoon and night, around four to six tablets a day.3700
4329 In the six weeks following her hip replacement, Mrs Sanders reported that she was able to ambulate around the shops with the aid of one elbow crutch and unaided within the home. She also reported some achiness in her right hip joint with “sustained positions” but was able to manages this well by “doing exercises throughout the day”.3701
4330 On 14 November 2016 Mrs Sanders presented to the orthopaedic clinic of Osborne Park Hospital for a post-surgery check-up.3702 She indicated that she was very happy with the outcome of the surgery. By this time her hip pain was gone and she no longer felt a grinding or burning pain in her hip socket. Nevertheless, the hip was tender over the scar and it felt like her scar was inflamed.3703 Arrangements were made for her to have a corticosteroid injection into her trochanteric bursa and surrounding tissues.3704 Mrs Sanders said that she had the injection the following day.3705
4331 At the follow-up appointment in the orthopaedic clinic on 23 January 2017 Mrs Sanders reported that she was significantly better, her trochanteric bursitis (inflammation around the capsule of the hip joint) had reduced after the corticosteroid injection, and she was now pain free.3706 In his report, Ryan Ridley, Senior Physiotherapist, noted that she was “able to do all of her activities of daily life independently”, she was sleeping at night, and was “very content with life”.3707
4332 In her second affidavit Mrs Sanders said that she was not sure what Mr Ridley meant when he wrote that she was able to do all her activities of daily life independently. Although she no longer had pain in her right hip, she stated, her husband still did and does everything for her. She explained:
Pete hoovers, mops the floors, puts the washing out, and washes the windows. I cannot stand for very long or walk long distances. For example, I cannot walk the length of a shopping centre. This has nothing to do with the hip, this relates to my lower back pain and my groin pain.3708
4333 By this time, however, she had an additional problem. She had developed pain in her left shoulder.3709 She presented to Dr Lisa O’Rourke in the Ashton Avenue Medical Centre (where Dr Matthews practised) with pain in her left shoulder, specifically in the rotator cuff which Dr O’Rourke attributed to a likely rotator cuff tear and inflammation. She took a history of two dislocations of the same shoulder in childhood and an episode of frozen shoulder two years ago.3710 Dr Rourke referred her for an x-ray and ultrasound of the shoulder which was carried out on 2 November 2016.3711 The only abnormal findings from the two investigations were seen on the ultrasound. It revealed subacromial subdeltoid bursal thickening and bunching on abduction, suggestive of bursitis. The following day Mrs Sanders received an ultrasound-guided steroid injection into the left subacromial bursa. When she saw Dr O’Rourke again on 2 December 2016, however, she apparently told the doctor that her shoulder was worse in spite of the steroid injection.3712
4334 On 13 December 2016 Mrs Sanders had an MRI of her left shoulder. The radiologist’s comment on the findings was as follows:
Mild anterolateral acromial downslope and thickening of the coraco-acromial ligament, plus mild AC joint arthropathy, resulting in impingement of the subacromial/subdeltoid bursa, which (scil) is thickened and oedamatous, and supraspinatus tendinopathy with some bursal surface fraying but no high-grade rotator cuff tear or tendon retraction scene and muscle bulk is preserved. The remainder of the rotator carpet intact.
Posterior superior labral fissuring with a probable small posterior superior labral tear.
Surrounding muscle bulk is preserved.3713
4335 Mrs Sanders was assessed by a physiotherapist on 20 December 2016. She told him that her left shoulder pain was recent (over the last two months) and that her pain was aggravated by overhead reaching and eased with rest.3714 On 2 May 2017, Dr John Hill, a consultant orthopaedic surgeon, examined Mrs Sanders at the request of her solicitors.3715 The obvious purpose of the examination was to isolate the symptoms and disabilities that were related to the osteoarthritic hip from the pain allegedly attributable to the effects of the TVT implant.
4336 Dr Hill received a history, consistent with the other evidence, that after the hip replacement Mrs Sanders had almost immediate relief from the lateral hip pain and improvement in her mobility. Mrs Sanders told him, consistent with her account to the Court, that the hip surgery did not relieve her back, pubic or medial groin pain. She reported to Dr Hill continuing vaginal pain “most of the time”, punctuated by episodes of more severe “shooting” pain. She also reported ongoing right pubic pain. She complained of increased pain with prolonged standing or walking for more than half-way around a nearby oval and told Dr Hill that her walking capacity had slowly deteriorated since 2006. Notably she also told him that the total hip replacement had not yet made a significant difference to her walking capacity and endurance, although there had been a reduction in night pain. Her sleep was disturbed most of the time, she reported, with vaginal pain. She told Dr Hill that the only medication she takes, apart from daily Keflex, was Panadol and Advil which “take the edge off the pain” and “a natural tablet to help [her] sleep”. She said that she was reluctant to take anything stronger than the Panadol and Advil because it makes her “feel like a zombie” and she cannot drive.
4337 The examination of the right hip revealed a faded posterolateral surgical scar of approximately 15cm which was not tender but she was noted to be “quite markedly tender in the central pubic area”. Straight leg raising, abduction and rotation were in the normal range and did not cause pain. On examination of the lumbar spine she was able to flex forward and reach the level of her lower shins, extension was smooth and synchronous, and her lateral flexion was not significantly restricted.
4338 Dr Hill noted in his report that the pain Mrs Sanders had suffered in her groin for many years, particularly from July 2011 to 1 August 2016, seemed to be localised to the inner aspect of her groin and pubic area rather than the more central area groin pain from hip arthritis is usually felt. Dr Hill also noted that over the two years before the consultation, Mrs Sanders had developed increasing more laterally placed hip pain and that serial x-rays disclosed an increase in the degenerative changes of her right hip joint.
4339 Dr Hill was of the opinion that Mrs Sanders had had complete relief from the more laterally situated hip pain but that the more medial groin pain in the pubic area had not changed since the hip replacement and neither had the symptoms in the back. He considered that the mild degenerative changes evident on x-rays “would certainly be a factor” in her back pain. He stated that, while the MRI showed some mild degenerative changes in her pubic symphysis, which could possibly account for her pubic pain and tenderness, it was his experience that this degree of degenerative change in the pubic symphysis is not uncommon and is frequently asymptomatic. He recommended that Mrs Sanders undergo a radionuclear bone scan of the lumbar spine and pelvis to determine any inflammatory activity in the symphysis pubis which could account for any pain in that area, and any synovitis in the lower lumbar joints which might account for the backache. He added that the bone scan should exclude a possible osteitis pubis, which could also account for pubic pain
4340 Based on Mrs Sanders’ account of her situation, Dr Hill considered that the combination of vaginal pain and low back ache was causing a significant disability. He did not expect the lower back problem to deteriorate in the future and thought that with a simple exercise regime under instruction from a physiotherapist she might well improve.
4341 A whole body bone scan was performed on 25 May 2017. It reportedly showed:
1. Low-grade increased tracer uptake involving the spine consistent with degenerative change. The SPECT images demonstrate low-grade tracer uptake responding to the right L3/4 facet joint and left L4/5 facet joint. There is mild tracer uptake consistent with degenerative change involving the inferior aspect of the right sacroiliac joint.
2. Increased activity involving the upper and lower limbs consistent with osteoarthritis.
3. No convincing evidence of loosening of the right hip prosthesis. Low moderate grade tracer uptake involving the prosthesis, most prominent at the superolateral aspect of the right acetabulum is consistent with a stress response.3716
4342 In an addendum to the report, however, the radiologist, Dr Andrew Patrikeos, indicated that he had reviewed the images for signs of osteitis pubis but no focal increased uptake is evident in relation to the os pubi and there was no convincing sclerosis adjacent to the symphysis pubis. He stated that the findings did not suggest osteitis pubis.3717
4343 No evidence was adduced to explain the significance of these findings. But in view of the comments made by Dr Hill, who was not required for cross-examination, it is reasonable to infer and I do infer that there was no evidence of inflammatory activity in the symphysis pubis to account for the pain in that area and that osteitis pubis has been excluded by the bone scan. As I read Dr Patrikeos’s report, however, he did not exclude synovitis in the lower lumbar joints which could account for Mrs Sanders’ lumbar pain, which was raised by Dr Hill as a possibility.
Current position
4344 Mrs Sanders continues to experience discomfort and pain in her groin and vagina and throughout her pelvis. She continues to take an antibiotic tablet every day, as well as two paracetamol (Panadol) and two ibuprofen (Advil) tablets every night.
4345 In her first affidavit she said that she had a constant dull ache in her pelvis, which feels like a heavy period. She distinguished it from the pain in her right hip, which arose in 2011, and which she described as “sort of a burning feeling further around my right side”. The pain was severe. She rated it as 7 out of 10. She also experienced a sharp, sudden pain in her groin when was bumped on her leg or near her pelvis, when she moved suddenly, or when walking on uneven ground. Although the pain lasts for seconds only and subsides, it is even more severe at 9 out of 10. On occasions she feels a sharp pain in her vagina when she is sitting down which she likened to a blade. This, too, lasts only a few seconds but it is equally severe. On top of this she had pain which flares up from time to time and she attributed to bladder infections. She said:
Every couple of months, the symptoms flare up and I usually take extra antibiotics until the symptoms subside. When I get urinary tract infections, I experience pain when urinating, I struggle to pass urine and feel like I need to go to the toilet frequently. It feels like I am in labour and I need to push my insides out. I get a ‘lock jaw’ from straining and numbness down my arms. I can spend a couple of hours at night on the toilet unable to pass urine until the medication kicks in. When I have a severe infection I see blood in my urine in the toilet.
4346 She was and continues to be troubled by incontinence. Once or twice a night she needs to go to the bathroom. She wears continence pads all the time and needs to change them multiple times during the day. Whenever she leaves home, she takes a spare pair of clothes in case of leakage. She leaks urine with exercise, coughing and laughing. She has no control over her bladder. Often she has a sudden urge to go to the bathroom but cannot make it in time.
4347 She is worried about having to take antibiotics in the long term and the effect it might have on her health. Her incontinence and “bladder infections” have affected her confidence. Sometimes she has to cancel plans, which upsets her.3718
4348 She has had to make adjustments to manage her symptoms. She avoids activities that will cause her pain or embarrassment. For example, she first checks where the bathrooms are before going anywhere. She often avoids social outings. She only carries out chores that do not require lifting or stretching. After ironing or vacuuming for five minutes or so, she has to sit down. While she manages to get the small chores done, it takes her a long time and her husband finishes those she cannot complete.3719
4349 She cannot go out for walks along the beach anymore. She struggles to keep up with family and friends when walking around the shops or at the farmers’ markets. Standing for long periods or walking provokes groin, back and hip pain. She stopped selling her craft at fairs in 2015.
4350 While her marriage is very strong and she and her husband enjoyed sex weekly before 2008, they have abstained from sex since 2008 because of the pain with intercourse. She often feels she is letting her husband down and holding him back from enjoying activities himself. Pain inhibits her capacity to play with her granddaughter.
4351 In her second affidavit she described her pain as follows.
I now feel the aching groin or pelvic pain on my right and left side. This heavy dull ache is in the centre of my groin very low down. If I could draw a diagram, I would draw a line very low down in my pelvis in between my legs. There is a dull ache all across this area. I experience this dull ache all the time, and it does not matter if I am walking or sitting. While I feel the pain all the time, sometimes the pain is exacerbated by walking or standing. I also experience this dull ache on the inside of my thighs on both sides. The pain feels a bit like period pain.
I continue to feel a sharp pain in my vagina. Before my surgery on 8 August 2011, the pain felt like there were big shards of glass inside my vagina. It felt as though the shards moved when I moved and they would give me a jolt. Since the surgery in August 2011, the pain has not been as bad, but I still have pain. Instead of shards of glass, it now feels like pins or needles are pressing into my vagina; it is a ’jab jab jab’ sort of pain. This pain is there all the time. It feels as though it is inside the vagina in the left and right side and not in the centre of the vagina.
The other type of pain that I experience is a really violent type of pain. This pain started around 2009. It is a sharp pain that shoots up from my vagina through my body three or four times, ‘bang bang bang’, and then stops. This pain can happen at any time. The pain takes my breath away. In June this year, for example, I was sitting in a chair holding a cup of hot coffee. I felt the pain shoot through my body and I dropped my drink; that is how bad the pain was. This shooting pain can happen every two or three days and sometimes once a week. I usually experience this pain when sitting. It is not dependent on walking or standing, and it is felt more towards the right side of the inside of my vagina. This pain is gradually getting worse, and I am feeling the pain more often. As per paragraph [72] of my First Affidavit, in 2016 I felt this pain once every week and sometimes every couple of weeks.3720
4352 Mrs Sanders said that the hip operation only relieved the pain she felt in her hip, and did not change or lessen any of her groin or lower back pain. She continues to abstain from sexual intercourse because she does not want to feel more pain in her vagina. Having regard to the groin and lower back pain as well as the urinary leakage, she said that she could not walk or exercise as she was able to before.
4353 Moreover, she continues to experience urinary symptoms. She still leaks urine with walking, coughing and laughing. She often has a sudden urge to go to the bathroom and feels that she has no control over her bladder with the result that she when the urge strikes her, she becomes incontinent.
4354 The pain has also taken a toll on her mental health. She has been both anxious and depressed and grieves for the life she once enjoyed. She deposed that she has lost her confidence and feels “absolutely worthless”.3721 Sleep is disturbed by both pain and ruminations about the mesh.3722 In oral evidence she said that her life has completely changed as a result of this pain and she is frightened about the future.3723
4355 Mr Sanders said that his wife is not the person she was before her problems arose in about 2008. He described her as “irritable when she has an infection”. He said she often remarks that she wished she had never had the operation. He confirmed her evidence that, from at least 2011, if not from 2008, they have abstained from sexual intercourse.3724
4356 The prognosis is uncertain. Professor Blaivas described it as “guarded at best”. She continues to have significant urinary symptoms. Although Associate Professor Shek found no mesh erosion on cystourethroscopy, Professor Blaivas pointed out that she had abnormal findings on pelvic examination, including levator tenderness and a “bristly” area on palpation. Like Professor Korda, he considered that the palpable bristles were a “forme fruste” (an incomplete manifestation) of another erosion and further surgery would be required to deal with it. If she wanted the dyspareunia treated, he added, that would likely require removal of all the suburethral portion of the mesh. What is more, if the groin pain were found to be due to the mesh, all the mesh would need to be removed.
4357 Causation is not a common question. It falls to be considered separately for each applicant. Before I do so, I wish to make some general observations.
The legal approach to causation
4358 The law approaches causation quite differently from science or philosophy.
4359 For a start, in a civil case the quest is not for a certain connection. Causation, like all other questions, is to be proved on the balance of probabilities. The determination of the question does not involve a process of scientific reasoning or mathematical calculation. Rather, “[i]t involves other kinds of reasoning in judgments, the correctness of which cannot be demonstrated by mathematics or ordinary logic”: Jones v Sutherland Shire Council [1979] 2 NSWLR 206 at 227 (Mahoney JA).
4360 As Mason CJ (with whom Toohey and Gaudron JJ agreed) explained in March v Stramare (E&MH) Pty Ltd (1991) 171 CLR 506 at 509:
In philosophy and science, the concept of causation has been developed in the context of explaining phenomena by reference to the relationship between conditions and occurrences. In law, on the other hand, problems of causation arise in the context of ascertaining or apportioning legal responsibility for a given occurrence. The law does not accept John Stuart Mill's definition of cause as the sum of the conditions which are jointly sufficient to produce it. Thus, at law, a person may be responsible for damage when his or her wrongful conduct is one of a number of conditions sufficient to produce that damage.
4361 Similarly, in Henville v Walker (2001) 206 CLR 459 at [97] McHugh J remarked:
The common law concept of causation recognises that conduct that infringes a legal norm may be causally connected with the sustaining of loss or damage even though other factors may have contributed to the loss or damage. Every event is the product of a number of conditions that have combined to produce the event. Some philosophers draw a distinction between a condition that is necessary only and a cause that is both necessary and sufficient to produce the event. The common law has avoided the technical controversies inherent in the logic of causation. Unlike science and philosophy, the common law is not concerned to discover universal connections between phenomena so as to enable predictions to be made. The common law concept of causation looks backward because its function is to determine whether a person should be held responsible for some past act or omission. Out of the many conditions that combine to produce loss or damage to a person, the common law is concerned with determining only whether some breach of a legal norm was so significant that, as a matter of common sense, it should be regarded as a cause of damage.
4362 At common law, in order to succeed in an action for damages for negligence, the moving party must prove that the injuries, loss and damage were caused by the negligent act(s) and/or omission(s) of the person or persons who are sued. That does not mean, however, that the negligence of the respondent(s) need be the only cause. It is enough that their acts or omissions caused or materially contributed to their injuries: March v Stramare at 514 (Mason CJ). A material contribution is one which is more than trivial or negligible: see Bonnington Castings Ltd v Wardlaw [1956] AC 613 at 621 (Lord Reid).
4363 In March v Stramare, Mason CJ said that, generally, the necessary causal connection is made out “if it appears that that the plaintiff would not have sustained his or her injuries had the defendant not been negligent”; in other words the plaintiff would not have been injured but for the negligence of the defendant. His Honour did not accept, however, that the “but for” test ever was or should become the exclusive test of causation in negligence cases (at 508). He observed at 515 that the common law tradition involved applying common sense to the facts of the particular case to determine whether the conduct of the putative wrongdoer caused a particular occurrence.
4364 In March v Stramare, the driver of a car (Mr March) was injured when, under the influence of alcohol and while driving at an excessive speed, he collided with a truck which was parked at night in the centre of a six-lane highway with its parking and hazard lights illuminated. The trial judge had found that the driver and owner of the truck was negligent but that Mr March was also negligent and apportioned 70% of the responsibility to Mr March. On appeal, Mr March’s negligence was found to be the sole effective cause of the accident and the action was dismissed. The High Court overturned the appeal judgment and restored the judgment of the trial judge in favour of Mr March.
4365 As Mason CJ explained at 518–519:
As a matter of both logic and common sense, it makes no sense to regard the negligence of … a third party as a superseding cause or novus actus interveniens when the defendant's wrongful conduct has generated the very risk of injury resulting from the negligence of … a third party and that injury occurs in the ordinary course of things. In such a situation, the defendant's negligence satisfies the “but for” test and is properly to be regarded as a cause of the consequence because there is no reason in common sense, logic or policy for refusing to so regard it.
4366 McHugh J considered (at 534) that it was preferable to use the “but for test”, which he called the “causa sine qua non test”, as the exclusive test of causation. His Honour acknowledged that there were exceptions, however, and nominated as an obvious one the case where the damage is the result of the simultaneous operation of two or more separate and independent events each of which was sufficient to cause the damage. At 536–537 he reached the same conclusion as Mason CJ. Noting that the defendants had conceded that injury to another road user was reasonably foreseeable as a result of the manner in which the truck was parked, that no challenge was made to the trial judge’s finding that the defendants were negligent or that they owed a duty of care to careless and drunken as well as careful drivers, his Honour held that the collision that occurred was the very kind of thing that was likely to happen if there was a lack of care on the part of the defendants. He said that both the damage to March and the manner in which it occurred were “fairly within the risk created by the defendants’ breach of duty”.
4367 Deane J came to the same conclusion at 520–522.
4368 Mason CJ (at 515) deprecated the approach of “commentators” and apparently favoured by McHugh J (at 534–536), which divided causation into two questions: causation in fact (determined by the application of the “but for” test) and the attribution of legal responsibility for the damage (scope of risk or liability) in which value judgments may intrude. His Honour considered that such an approach puts too much weight on the “but for test” to the exclusion of the “common sense” approach and implies that value judgment has or should have no part to play in determining causation in fact. His position, which was supported by Deane J (at 522–523) as well as Gaudron and Toohey JJ, was, as Toohey J put it at 524, that in a negligence case “causation is essentially a question of fact… into which considerations of policy and value judgments necessarily enter.” In Travel Compensation Fund v Tambree (2005) 224 CLR 627 at [81] Callinan J said that the division of causation into these two separate questions was unjustifiable. Nevertheless, this was the approach recommended in the Ipp Report and it is now enshrined in civil liability legislation across the country.
4369 The position taken in March v Stramare was reaffirmed by the High Court in a number of cases both in the context of claims for damages in negligence, for example in Roads and Traffic Authority v Royal [2008] HCA 19; (2008) 82 ALJR 870 at [32] (Gummow, Hayne and Heydon JJ; Kiefel J agreeing at [135]) and involving claims for compensation under the Trade Practices Act, for example in Wardley Australia Ltd & Anor v The State of Western Australia (1992) 175 CLR 514 at 525 (Mason CJ, Dawson, Gaudron and McHugh JJ).
4370 I will deal with the extent to which legislation has affected the common law when I come to consider the issue in relation to the three applicants. I turn now to the relevant provisions of the Trade Practices Act.
Causation under the Trade Practices Act
4371 Under s 74B(1) of the TPA a corporate manufacturer is liable, in the circumstances set out in that section, to compensate a consumer (or a person who acquires the goods from a consumer etc.) who suffers loss or damage “by reason that” the goods it supplied are not reasonably fit for the purpose for which they were acquired.
4372 Similarly, under s 74D(1) of the Act a corporate manufacturer is liable in the circumstances set out in that section to compensate a consumer or other person there described who suffers loss or damage “by reason that” the goods are not of merchantable quality.
4373 The compensation payable under each section is “for the loss or damage” (emphasis added).
4374 A reference to “loss or damage” in the Act includes “injury” (TPA, s 4K) and “injury” includes injury to the person: Pritchard v Racecage Pty Ltd (1997) 72 FCR 203 at 217 (Branson J, Spender J and Olney J agreeing at 205 and 206 respectively).
4375 Section 75AD, however, makes a corporate manufacturer liable to compensate the individual who suffered injuries “because of” a defect of its goods and to recover compensation for “the amount of the individual’s loss suffered as a result of the injuries” (emphasis added).
4376 At all material times, s 82(1) relevantly provided that, subject to subs (1AAA) (discussed below), a person who suffers loss or damage “by conduct” of another person done in contravention of a provision of certain Parts of the Act, including Pt V, may recover the amount of that loss or damage from that person or against any person involved in the contravention.
4377 This captures the applicants’ claims under ss 52, 74B and 74D which all fall within Pt V, but not s 75AD, which falls within Pt VA and is not one of the Parts mentioned in the subsection.
4378 The right to recover damages for personal injuries caused by a contravention of s 52 was removed by the Trade Practices Amendment (Personal Injuries and Death) Act 2006 (Cth): TPA, s 82(1AAA). This amendment implemented recommendation 19 of the Ipp Report. The reasons behind the recommendation appear at [5.23]–[5.34] of the Report.
4379 Subsection 82(1AAA) provided:
A person who suffers loss or damage by conduct of another person may not recover the amount of the loss or damage by an action under subsection (1) to the extent to which:
(a) the action would be based on the conduct contravening a provision of Division 1 of Part V; and
(b) the loss or damage is, or results from death or personal injury; and
the death or personal injury does not result from smoking or other use of tobacco products.
4380 The effect of subs 82(1AAA) in the present case is that any applicant or group member may not recover damages for a contravention of s 52 or its counterpart in the ACL (s 18), if she was injured after 20 April 2006, being the date the amendment commenced. Subsection 82(1AAA) was repealed with the repeal of the TPA and the enactment of the Competition and Consumer Act. But its substance was re-enacted in s 137C of the CCA. Consequently, the only applicant who might be entitled to recover damages for injury caused by the respondents’ misleading or deceptive conduct is Mrs Sanders, whose implant surgery, it will be recalled, took place in 2001.
4381 Despite the difference in language, it has been held that “by” in s 82 means “by reason of” or “as a result of”: Munchies Management Pty Limited v Belperio (1988) 58 FCR 274 at 286 (Fisher, Gummow and Lee JJ). In these circumstances, there is no reason in principle why what was decided in Wardley should not also apply to ss 74B(1) and 74D(1) and 75AD. Indeed, in Carey-Hazell (2004) Kiefel J said as much about s 75AD at [195].
4382 The approach to causation in a statutory cause of action is necessarily affected by the subject-matter, scope and purpose of the Act: Travel Compensation Fund v Tambree at [30] (Gleeson CJ), [49] (Gummow and Hayne JJ), [79] (Callinan J). In I & L Securities Pty Ltd v HTW Valuers (Brisbane) Pty Ltd (2002) 210 CLR 109 Gleeson CJ observed at [26]:
The relationship between conduct of a person that is in contravention of the statute, and loss or damage suffered, expressed in the word “by”, is one of legal responsibility. Such responsibility is vindicated by an award of damages. When a court assesses an amount of loss or damage for the purpose of making an order under s 82, it is not merely engaged in the factual, or historical, exercise of explaining, and calculating the financial consequences of, a sequence of events, of which the contravention forms part. It is attributing legal responsibility; blame. This is not done in a conceptual vacuum. It is done in order to give effect to a statute with a discernible purpose; and that purpose provides a guide as to the requirements of justice and equity in the case. Those requirements are not determined by a visceral response on the part of the judge assessing damages, but by the judge's concept of principle and of the statutory purpose.
4383 Having regard to the objects of the Trade Practices Act, s 82 is to be applied “in a way that promotes competition and fair trading and protects consumers” and a “narrow, inflexible construction of the section” is not called for: Henville v Walker at [96] (McHugh J).
4384 In Wardley at 525 the plurality (Mason CJ, Dawson, Gaudron and McHugh JJ) said that “s 82(1) should be understood as taking up the common law practical or common-sense concept of causation” discussed in March v Stramare “except in so far as the concept is modified or supplemented expressly or impliedly by the provisions of the Act”. That remains the position under the Act: see, for example, 3Meg.com Pty Ltd v TM & SM Pike Pty Ltd (2012) 43 WAR 350 at [62].
4385 Beyond the “not negligible” threshold, however, the strength of the requisite connection is immaterial: Carey-Hazell (2004) at [195] (Kiefel J). As the Full Court observed in Como Investments Pty Ltd (in liq) v Yenald Nominees Pty Ltd [1997] FCA 12 at 6; (1997) 19 ATPR ¶41–550 at 43,619 in a passage cited with approval by McHugh J in Henville v Walker at [109]):
The law does not consider cause and effect in mathematical or in philosophical terms. The law looks at what influences the actions of the parties. Acknowledging that people are often swayed by several considerations, influencing them to varying extents, the law attributes causality to a single one of those considerations, provided it had some substantial rather than negligible effect.
4386 In Como Investments, which was an action under s 52 of the TPA, the Full Court went on to observe at 8 that:
Where a representation is relevant to the decision in question, and in its nature persuasive to induce the making of that decision, it accords with legal notions of causation to hold that it has a causative effect. And where a respondent, who may be taken to know his own business, has thought it was in his interests to misrepresent the situation in a particular respect, the Court may infer that the misrepresentation was persuasive. These inferences arise from the making of the representation followed by the respondent doing the thing it was calculated to induce him to do.
All this is a matter of common sense. It has also been stated in the authorities.
4387 Absent evidence to the contrary, this is the inference a court should draw.
4388 In Henville v Walker, both Gleeson CJ at [14] and Hayne J at [163]–[164] said that it was not essential that the contravention of the statute be the sole cause of the loss or damage to satisfy the requirements of s 82(1). Rather, it was sufficient if the contravening conduct was a cause. Gaudron J at [61] observed that the common-sense approach requires no more than that the act or event in question should have materially contributed to the loss or injury and there was nothing in the Act to suggest that a different approach should be taken to a misrepresentation that constitutes a contravention of s 52(1). McHugh J said at [106]:
If the defendant's breach has “materially contributed” [citing Bonnington Castings Ltd v Wardlaw at 620] to the loss or damage suffered, it will be regarded as a cause of the loss or damage, despite other factors or conditions having played an even more significant role in producing the loss or damage. As long as the breach materially contributed to the damage, a causal connection will ordinarily exist even though the breach without more would not have brought about the damage. In exceptional cases, where an abnormal event intervenes between the breach and damage, it may be right as a matter of common sense to hold that the breach was not a cause of damage. But such cases are exceptional.
(Emphasis added)
4389 In Travel Compensation Fund v Tambree at [32] Gleeson CJ observed that, if a person acts, or fails to act, in a particular way in reliance on certain information, the loss or damage may flow directly from the act or omission and only indirectly from the making of the representation. His Honour continued:
Where the reliance involves undertaking a risk, and information is provided for the purpose of inducing such reliance, then if misleading or deceptive conduct takes the form of participating in providing false information, and the very risk against which protection is sought materialises, it is consistent with the purpose of the statute to treat the loss as resulting from the misleading conduct.
4390 In obiter dicta in Brosnan v Katke [2016] FCAFC 1; (2016) ATPR ¶42–515 at [123]–[124], however, a Full Court of this Court suggested that it might not be sufficient that the contravening conduct materially contributed to an applicant’s loss or damage. At [123] the Court said:
The two tests (“but for causation” and “material contribution”) are “different beasts”: Clements v Clements [2012] SCC 32; [2012] 2 SCR 181, 189-190 [14] (McLachlin CJ). In some areas of the law, such as the tort of deceit, the necessity (or “but for”) test for causation is relaxed in favour of a “material contribution test”. It is sometimes said that this is because of the difficulty involved in weighing the potency of different contributions to mental decision making: Reynell v Sprye (1852) 1 De GM & G 660, 708-709; (1852) 42 ER 710, 728-729 (Lord Cranworth ); Smith v Kay (1859) 7 HLC 750, 759; (1859) 11 ER 299, 303 (Lord Chelmsford LC); Arnison v Smith (1875) 41 Ch D 348, 369 (Lord Halsbury LC). But cf Mills v Mills [1938] HCA 4; (1938) 60 CLR 150, 185-186 (Dixon J); Eclairs Group Ltd and Glengary Overseas Ltd v JKX Oil & Gas plc [2015] UKSC 71 [20]-[21] (Lord Sumption), [54] (Lord Mance).
4391 The Court went on to say that, before “the material contribution approach” could be applied to s 82, it would be necessary to give close attention to the terms and context of the section, to the authorities, and to the validity of taking a different approach to mental decision making.
4392 But the Full Court did not receive full submissions on the point and did not determine the question one way or another.
4393 In fact, the High Court gave close attention to the terms and context of s 82 in Henville v Walker and I & L Securities Pty Ltd. The Full Court did not refer to either of those judgments in Brosnan or to the authorities discussed by the High Court in those cases. The result of those analyses is that the question of whether an applicant suffered loss or damage “by conduct” of a respondent in contravention of one or other of the relevant provisions of the Act devolves into a question of “whether the contravention was a cause of (in the sense of materially contributed to) the loss”: I & L Securities Pty Ltd at [62] (Gaudron, Gummow and Hayne JJ).
4394 With respect, the reference by the Full Court to Clements v Clements [2012] 2 SCR 181 is misplaced. In Clements v Clements, McLachlin CJ was referring to a material contribution to risk, not a material contribution to injury, loss or damage. Furthermore, in Evans v Queanbeyan City Council [2011] NSWCA 230; (2011) 9 DDCR 541 at [23]–[26], Allsop P recognised that the meaning of material contribution to injury may differ in some contexts in the two jurisdictions.
4395 It is well established in Australia that a material increase in the risk of harm, without more, is insufficient to enable a conclusion to be reached that there was a material contribution to the harm: see, for example, Bendix Mintex Pty Ltd v Barnes (1997) 42 NSWLR 307 at 316 (Mason P); Evans at [22], approved by the Full Court in Merck at [102]–[103]. It must be shown that it was probable that the risk came home: Bendix v Barnes at 318 (Mason P). In Merck, Mr Peterson ultimately failed because the Full Court held that he had not proved that it had. Nevertheless, as French CJ explained in Amaca Pty Ltd v Booth (2011) 346 CLR 36 at [43]:
[I]f the association between two events is shown to have a causal explanation, then the conclusion may be open, if the second event should occur, that the first event has been at least a contributing cause of that occurrence. An after-the-event inference of causal connection may be reached on the civil standard of proof, namely, balance of probabilities, notwithstanding that the statistical correlation between the first event and the second event indicated, prospectively, no more than a “mere possibility” or “real chance” that the second event would occur given the first event.
4396 In this case, the applicants submitted at one point that the relevant questions on causation in relation to the statutory claims were whether “because of the defect, an individual suffers injuries” (para 75AD(c)); “the consumer suffers loss or damage by reason that the goods are not of merchantable quality” (s 74D(1)(d)); and “the consumer suffers loss and damage by reason that the goods are not reasonably fit for that purpose” (para74B(1)(e), overlooking the causal question posed by s 82 for the purposes of the s 52 claim.3725 On the other hand, they went on to submit that “the test for causation is simply whether the [device] caused the particular injury” and “not whether the absence of any warning caused injury” (original emphasis).3726 This latter submission was said to be derived from Merck where the Full Court held that Mr Peterson’s claim must fail because he had not proved that his myocardial infarction was caused by ingestion of Vioxx. Put in this way, the submission must be rejected. It takes what the Full Court said in Merck out of context.
4397 The Full Court in Merck held at [175] that the claim under s 74B was not made out since it had not been demonstrated that Mr Peterson’s consumption of Vioxx was a necessary precondition of his heart attack. In the case of the s 75AD claim, the Full Court emphasised at [201] the need to establish that the claimant’s injuries were suffered “because of the defect” (original emphasis). The Full Court held at [201] that Mr Peterson had not demonstrated that the increased risk affected him “in the sense that the MI [myocardial infarction] he suffered was caused by (because of) his consumption of Vioxx”.
4398 The Full Court should not be taken to have articulated a test that requires a claimant merely to prove a causal connection between the use of a product and the claimant’s loss or damage. The whole discussion of causation was premised on the rule that “a plaintiff must establish as a necessary condition of recovery that he or she would not have suffered loss but for the defendant’s actionable misconduct”: Merck at [104]. Whether or not Mr Peterson would have had a heart attack had he not taken Vioxx was part of the inquiry into whether he would have had a heart attack had it not been for the manufacturer’s actionable misconduct, but it was not the whole inquiry.
4399 In this proceeding, the respondents argued that if there were a defect then it comprised the absence of suitable warnings and that the applicants needed to prove that their loss or damage was caused by the absence of the warnings.3727 The applicants, on the other hand, argued that the defect was the risk of harm posed by the goods, not the absence of a warning.3728
4400 Everything turns, then, on how the defect is properly characterised.
4401 The role of a safety warning is to alert the consumer to the risks associated with the use of the product to protect her or him from harm. In the case of a medical device, warnings as to adverse events and contraindications, and advice as to the limits of the available information serve to assist patients to make an informed decision about whether to undergo surgery with such a device. Where a medical device, when used as intended, exposes consumers to a risk of significant harm, then the device will have a defect unless it is accompanied by warnings sufficient to alert patients to that risk. In such a case, the defect is not the absence or inadequacy of the warnings, as the respondents contended, but the fact that the device has a propensity to cause harm that persons generally would not reasonably expect. By contrast, a product designed and marketed to cause harm and which functions as intended, such as a firearm or a poison, may only be defective because of the absence of a sufficient warning. The defect in such a case is the failure to warn. This is the “instructional defect” mentioned in the Explanatory Memorandum referred to above in Part XIII. The Ethicon devices were not in this class. They were not designed or intended to cause harm.
4402 This construction is consistent with the approach taken to the analogous provision in the United Kingdom. Section 2(1) of the Consumer Protection Act renders certain persons, including producers and importers, liable for damage caused wholly or in part by a defect in the product. The definition of defect is very similar to the definition in s 75AC of the TPA.
4403 In Gee v DePuy at [86], Andrews J said that the defect is defined by reference to the condition of the product — that is, the product’s failure to meet the level of safety persons generally are entitled to expect — rather than by reference to some fault or deficiency in it or the precise mechanism that caused the damage. The respondents relied on this observation, emphasising the distinction her Ladyship drew between a fault or deficiency in the product and the failure of the product to meet the requisite level of safety.3729 Andrews J went on to say at [87], however, in a passage to which the respondents did not refer, that when the lack of safety is due to a combination of circumstances or features, in which case it is the combination which renders the product defective, “the defect will still consist of whatever it is about the character, state or condition of the product that makes it unsafe”. The point her Ladyship was making at [86] was that the mere existence of a fault or deficiency in a product does not establish that the product has a defect because all relevant circumstances have to be taken into account in the determination of whether the product fails to satisfy the statutory definition of “defect”. She made that clear at [108] and also at [111].
4404 The respondents’ approach to causation is contrary to the decisions in the Vioxx case.
4405 At first instance, Jessup J found that Vioxx had a defect but dismissed Mr Peterson’s claim under s 75AD because he upheld the defence under s 75AK(1)(c) that the state of scientific knowledge at the time of supply was not such as to have enabled that defect to have been discovered. In Merck at [198], the Full Court observed that, although his Honour had not identified the defect in express terms, it was apparent that he characterised the defect as the increased risk of experiencing a myocardial infarction “absent the provision of any information, advice or warning as to this risk”. In upholding the state of the art defence, Jessup J emphasised the nature of the defect. He said in Peterson at [927]:
At the scientific or technical level as such, I would hold that the defect could not have been so discovered. The defect, of course, is the inadequate safety of the goods themselves. Vioxx was unsafe in that sense because it increased the risk of myocardial infarction … [I]t was not until September 2004 that the state of scientific knowledge was such as to enable the discovery of the fact that the consumption of Vioxx increased the risk of myocardial infarction.
4406 He returned to the point at [929] where he observed that “[t]he defect was something inherent in Vioxx as a matter of composition”. He stressed that it was that defect with which s 75AK(1)(c) was concerned. He held that it was not concerned with “the kind of contextual circumstances referred to in s 75AC(2)”, which, of course, include warnings and information.
4407 On appeal the Full Court rejected the submission by Merck that Jessup J was wrong to hold that Vioxx had a defect at [198]. In substance, at [201] it endorsed his Honour’s finding:
[T]he better view is that Vioxx had a defect within the meaning of s 75AC. The defect was one which affected some people, not all. The defect was that in some people, by a mechanism not known and the subject of no hypothesis, it increased the risk of MI and provided no information, advice or warning as to this effect: see [137]–[138] and [198] above.
4408 The Full Court then remarked at [211]–[202]:
But the conclusion that Vioxx had this “defect” presents two further statutory questions. First, the statute requires that an individual claimant suffer injuries because of the defect: s 75AD(c). As has already been demonstrated, Mr Peterson did not demonstrate that the increased risk affected him, in the sense that the MI he suffered was caused by (because of) his consumption of Vioxx… In other words, Mr Peterson’s claim fails at the first hurdle.
Moreover, even if Mr Peterson had demonstrated that the MI he suffered was caused by (because of) his consumption of Vioxx (which he did not), the next relevant statutory enquiry would be whether the defect (as it has been identified) was one which, given the state of scientific or technical knowledge at the time when the goods were supplied by MSDA, was not such as to enable that defect to be discovered.
(Original emphasis)
4409 The Full Court went on to approve the primary judge’s decision to uphold the state of the art defence and his Honour’s reasons, including what his Honour said at [929]: Merck at [205]–[208]. At no point did the Full Court hold that, for the purpose of s 75AD, it was necessary for Mr Peterson to prove that he would not have taken Vioxx if he had been warned of the risk that it could cause a heart attack.
4410 The respondents tried to distinguish the Vioxx case. They argued that medical devices are unlike prescription drugs in that “the implants involve the further input of the surgeon with whom the patient elects to have the index surgery, including his or her preferences, training and capacity.”3730 But the prescription of drugs also requires the “input” of the treating doctor, including his or her preferences, training and capacity. In my opinion, the distinction is unwarranted.
4411 It follows from the judgment in Merck that a product may have a defect within the meaning of s 75AC because they may cause injury in respect of which no or no sufficient warning, advice or information is given by the manufacturer. It also follows from Merck that the causal connection required by para 75AD(c) (that an individual suffers injuries “because of the defect”) is a connection between the relevant safety risk posed by the product and the injuries suffered by the user or consumer. In other words, an applicant must prove that the risk came home. If she does so, she is entitled to damages unless the respondents can make out a statutory defence or the claim is statute-barred.
The respondents’ general arguments
4412 The respondents submitted that all the applicants must fail, both under the statute and at common law, since there was no evidence from their treating doctors that, had the warnings the applicants say should have been, but were not, provided, they would have conducted themselves any differently.3731 The respondents further submitted that the applicants’ failure to call evidence from the implanting surgeons in each case was fatal to their claims because, in the absence of evidence from those surgeons, the Court could not be satisfied that the information given by the surgeons was inadequate.3732
4413 In any case, they submitted that the evidence suggests that the applicants may not have paid attention to their treating doctors, referring only to evidence from Mrs Gill;3733 that the treating doctors did not provide much detail;3734 and that, in Mrs Dawson’s case, the treating doctors considered that they had appropriately discussed the risks with the applicants.3735
4414 Similarly, to the extent that the applicants rely on the marketing and promotional materials as influencing the treating doctors to suggest implantation of the devices, the respondents submitted that there must be proof that, had the suggested warnings been made or the representations been different, events would have taken another course. They contended that there was “no satisfactory evidence that this is so”.3736
4415 First, for the reasons given above, these matters have no bearing on the claims under s 75AD or, having regard to the common position of the parties, those under ss 74B and 74D.
4416 Second, there is more than one negligence count. Only one is concerned with the inadequacy of the warnings. I will deal with this issue as and when it arises.
4417 Third, I reject the respondents’ submission that, since the applicants had failed to adduce evidence from the treating surgeons, the Court could not be satisfied that, if the respondents had warned of the pleaded complications, the surgeons would have passed on the information to their patients or that their patients would have chosen a different method of treatment.3737
4418 Contrary to the respondents’ opening submission, this was not the position taken by the Full Court in Black v Lipovac (by his next friend Lipovac) [1998] FCA 699; (1998) 217 ALR 365 at [151]–[152] and arguments to the same effect have been rejected in the United States and Canada.
4419 Black v Lipovac was a medical negligence case arising out of the death of an infant (Tom Lipovac) who suffered hypoxia and consequential brain damage in a seizure after his parents inserted an aminophylline suppository into his rectum that had been prescribed by his GP, Dr Black. Dr Black cross-claimed against the manufacturer, Hamilton Holdings Pty Ltd (Hamilton) alleging it was negligent for failing to warn general practitioners that the use of 100mg suppositories for an infant of Tom’s age and weight carried a real risk of a seizure. The trial judge upheld the claim against Dr Black and, although he found that Hamilton was negligent in failing to provide advice to physicians as to the possible toxic effects of aminophylline, he dismissed Dr Black’s cross-claim because he found that Hamilton’s negligence did not cause any of the harm suffered by Tom.
4420 In Black v Lipovac at [151] the Full Court merely affirmed the requirement for a respondent’s conduct to be a “real or direct or effective cause of the applicant’s loss”. It is true that the Full Court observed that the requirement would have been satisfied in that case if Dr Black had relied on some misleading or deceptive conduct by the manufacturer, but it did not say that it was only through direct evidence from the doctor that the requirement could be made out. Its observation must be read in the context of the evidence in the case and the trial judge’s findings. The trial judge found that the treating doctor did not rely on the information provided by the manufacturer (see [32], [61]). The evidence was summarised at [143] of the Full Court’s decision. That evidence included that Dr Black had extensive knowledge about the use of aminophylline, that he knew the recommended dosages for all forms of delivery, including suppositories, and that he was aware that aminophylline had toxic side effects. While in answer to some interrogatories he said he was not aware of certain other alleged risks, he said that, even if he had been aware of all those matters, he would have treated Tom “in exactly the same way”. Unsurprisingly, the Full Court said at [144] that “the inescapable conclusion” was that Hamilton’s negligence played no part in the harm suffered by Tom.
4421 There was no evidence of this nature in the present case. Nor was there evidence from which such an inference could properly be drawn. In my opinion, in the absence of evidence to the contrary, the Court may infer that, if the respondents had included warnings about the pleaded complications and the other relevant matters, the treating doctors would have passed the information onto their patients. After all, that would be consistent with the doctors’ professional obligations.
4422 In the Canadian case Hollis, which I mentioned in Part XIII in the context of the discussion of the role of the learned intermediary, the manufacturers, Dow, argued that there was no causal connection between the breach of its duty to warn and Mrs Hollis’s injury because, amongst other things, the implanting surgeon (Dr Birch) would not have warned her even if Dow had properly warned him. La Forest J, writing for the majority, observed at [55] that there was an assumption, which lay at the heart of this argument, that, to succeed in her claim against Dow, Mrs Hollis had to prove that Dr Birch would have warned her had he received a proper warning from Dow. His Honour held that the assumption was not well founded. He continued:
Ms. Hollis, it will be remembered, demonstrated that Dow had breached its duty to warn her of the risk of rupture, that she would not have undergone the medical procedure if she had been fully informed of the risks, and that she suffered injury from the rupture. Had Dr. Birch been adequately warned but had not passed on the information to Ms. Hollis, Dow would, it is true, have been absolved of liability by virtue of the learned intermediary doctrine. But I fail to see how one can reason from this that, for Dow to be liable, Ms. Hollis must now establish that Dr. Birch would have informed her if he had known. To require her to do so would be to ask her to prove a hypothetical situation relating to her doctor's conduct, one, moreover, brought about by Dow’s failure to perform its duty. While the legal and persuasive onus in a negligence case generally falls on the plaintiff, I do not see how this can require the plaintiff to prove a hypothetical situation of this kind.
4423 I respectfully agree.
4424 La Forest J pointed out at [61] that a number of courts in the United States had reached a similar conclusion. In McCue v Norwich Pharmacal Co, 453 F. 2d 1033 (1st Cir. 1972) at 1035, for example, the Court of Appeal for the First Circuit said that, having put a dangerous drug on the market without adequate warning, a defendant cannot be heard to say that the physician might have disregarded a proper one.
4425 In Buchan v Ortho Pharmaceutical (Canada) Ltd (1986) 12 OAC 361 the Ontario Court of Appeal said at 377 that, absent evidence to the contrary, it could be assumed that a doctor would not ignore a proper warning, fail to disclose a material risk, or otherwise act negligently and, even if the evidence were to indicate that the doctor was negligent, the manufacturer would not escape liability on that account if the doctor’s negligence were a foreseeable consequence of its breach of duty. The minority in Hollis did not appear to disagree with this proposition: Hollis at [81]. As to the latter proposition, see Mahony v J. Kruschich (Demolitions) Proprietary Limited (1985) 156 CLR 522.
4426 Even if the respondents were right to contend that the applicants had to prove that their doctors would have passed on the warning that should have been given, it does not follow that it was essential for them to put on affidavits from the doctors in which they said as much. That assumes that the only pathway to proof was by direct evidence. But facts may also be proved by circumstantial evidence. In other words, in the absence of direct evidence to this effect, evidence might be available from which an inference to that effect could and should be drawn.
4427 I now turn to assess the issue of causation in respect of each applicant.
Did Mrs Gill suffer injury caused by a defect in Prolift Total?
4428 While there is a dispute about the extent of the injuries Mrs Gill suffered as a result of the implantation of the Prolift Total device, which I deal with later, the respondents accept that the implantation of the device caused some of Mrs Gill’s injuries, including the four episodes of mesh exposure, the consequential surgical procedures, and some of her pelvic pain.3738 They deny, however, that the injuries were caused by a defect in the device. I have already rejected their arguments. Since the use of Prolift Total as intended carried risks of significant and serious injury, including infection, erosion and chronic pain, about which insufficient and misleading warnings were given, and Mrs Gill sustained injury of that kind, she suffered damage caused by the defect.
4429 Having regard to the common position of the parties that the outcome of the defect case determines the outcome of the Pt V Div 2A causes of action, I find that the respondents’ contraventions of ss 74B and 74D also caused her loss and damage.
4430 It follows that, subject to the respondents’ limitation defence, the respondents are liable to compensate Mrs Gill for that loss and damage. I discuss the question of which injuries are compensable when I come to assess damages.
Did Mrs Gill suffer damage caused by the respondents’ negligence?
4431 The question of causation is more difficult here, not least because there are three negligence claims.
4432 Section 5C of the CLA (WA) sets out the general principles that apply to the determination of causation. It provides as follows:
(1) A determination that the fault of a person (the tortfeasor) caused particular harm comprises the following elements—
(a) that the fault was a necessary condition of the occurrence of the harm (factual causation); and
(b) that it is appropriate for the scope for the tortfeasor’s liability to extend to the harm so caused (scope of liability).
(2) In determining in an appropriate case, in accordance with established principles, whether a fault that cannot be established as a necessary condition of the occurrence of harm should be taken to establish factual causation, the court is to consider (amongst other relevant things) —
(a) whether and why responsibility for the harm should, or should not, be imposed on the tortfeasor; and
(b) whether and why the harm should be left to lie where it fell.
(3) If it is relevant to the determination of factual causation to determine what the person who suffered harm (the injured person) would have done if the tortfeasor had not been at fault —
(a) subject to paragraph (b), the matter is to be determined by considering what the injured person would have done if the tortfeasor had not been at fault; and
(b) evidence of the injured person as to what he or she would have done if the tortfeasor had not been at fault is inadmissible.
(4) For the purpose of determining the scope of liability, the court is to consider (amongst other relevant things) whether and why responsibility for the harm should, or should not, be imposed on the tortfeasor.
4433 “Harm” is defined in s 3 to mean “harm of any kind” and relevantly includes personal injury and economic loss. “Personal injury” relevantly includes “impairment of a person’s physical or mental condition” and “disease”.
4434 The onus of proving any fact relevant to the issue of causation lies with the applicant: CLA (WA), s 5D.
4435 The determination of factual causation under para 5C(1)(a) of the CLA (WA) and its analogues in other jurisdictions merely involves the application of the “but for” test: Wallace v Kam (2013) 250 CLR 375 at [16], which dealt with in para 5D(1)(a) of the CLA (NSW). In other words, the test is whether the harm would have occurred but for the negligence of the tortfeasor. As the High Court put it in Wallace v Kam:
The determination of factual causation in accordance with s 5D(1)(a) involves nothing more or less than the application of a ‘but for’ test of causation. That is to say, a determination in accordance with s 5D(1)(a) that negligence was a necessary condition of the occurrence of harm is nothing more or less than a determination on the balance of probabilities that the harm that in fact occurred would not have occurred absent the negligence.
4436 Although negligence on the part of the tortfeasor is a necessary condition of the occurrence of harm, an applicant may succeed where there are other conditions which also contribute to the harm. In March v Stramare at 530, McHugh J said that the basis of the “but for” test of causation is that a person may be causally responsible for damage although his or her act or omission was only one of the conditions sufficient to produce the damage. That statement of the common law is not affected by s 5C(1)(a) of the CLA (WA) and its analogues. In Strong v Woolworths Ltd (2012) 246 CLR 182, another case involving the interpretation of s 5D(1)(a) of the CLA (NSW), French CJ, Gummow, Crennan and Bell JJ explained at [20]:
Under the statute, factual causation requires proof that the defendant's negligence was a necessary condition of the occurrence of the particular harm. A necessary condition is a condition that must be present for the occurrence of the harm. However, there may be more than one set of conditions necessary for the occurrence of particular harm and it follows that a defendant's negligent act or omission which is necessary to complete a set of conditions that are jointly sufficient to account for the occurrence of the harm will meet the test of factual causation within s 5D(1)(a). In such a case, the defendant's conduct may be described as contributing to the occurrence of the harm.
4437 Similarly, in Hunt & Hunt v Mitchell Morgan Pty Limited (2013) 247 CLR 613 (also involving s 5D of the CLA (NSW)), at [43]–[45] French CJ, Hayne and Kiefel JJ affirmed what Mason CJ said in March v Stramare. Their Honours said that causation is largely a question of fact to be approached by applying common-sense to the facts of the case, that courts are not constrained to find a single or effective cause, that two or more successive wrongdoers may be liable for the same damage, and that it is enough that the act or omission was a cause in the sense that it materially contributed to the loss.
The first and second negligence claims: inadequate evaluation of Prolift Total
4438 I now turn to consider the causal connection between Mrs Gill’s damage and the first two breaches: the inadequate pre and post-market evaluations of the devices. In their submissions, the applicants dealt with the causation questions together. They submitted that the question for resolution was whether, but for the respondents’ failure to properly evaluate the safety of Prolift Total, Mrs Gill would not have suffered injury or loss. They submitted, without contradiction, that if the answer to this question is yes, then there could be no issue that legal responsibility should attach to Ethicon’s conduct.3739
4439 The respondents argued that the applicants had failed to make out this case for the following reasons: first, that clinical testing is not the only regulatory pathway to market; second, if clinical tests had been conducted they would at least have vindicated the sale of the SUI devices because they became “the gold standard” treatment for stress urinary incontinence; third, the applicants did not call evidence from a regulator that registration would have been withheld if the respondents had disclosed certain facts about the testing process for the various devices; fourth, the applicants did not put to any of the respondents’ witnesses that they would not have placed the devices on the market had they known of the asserted problems with the testing regime”; and fifth, there was extensive evaluation conducted for all the devices.3740 I will deal with each of these arguments.
4440 First, while it is true that clinical testing is not the only regulatory avenue to market, a manufacturer has to satisfy the essential requirements for CE marking. It may well be the case that the only way to do so in a particular case is through clinical trials. In any case, as I explained in Part VIII of these reasons, even if the literature route is followed, the manufacturer is obliged to critically review the literature and “duly” justify reliance on existing clinical data. As with all the other devices, in the case of Prolift, that obligation was not discharged.
4441 Second, the fact that the TVT procedure became “the gold standard” treatment for stress urinary incontinence is no answer to the allegation that the negligent evaluation conducted by the respondents of Prolift Total in particular or Prolift in general, either before or after it was cleared for sale, caused damage to Mrs Gill. In the two year period since the device was cleared for sale, the evidence did not support the use of Prolift in the repair of a single compartment, let alone in multiple compartments. The Cochrane review by Maher et al (2007) concluded that the evidence was not sufficient to support the use of any permanent meshes in vaginal repair surgery for prolapse except in the context of randomised controlled clinical trials.3741
4442 Third, the absence of evidence from the regulator is beside the point. It was Ethicon that determined to apply the CE mark.
4443 Fourth, the person at Ethicon who made that decision did not give evidence. Nor did any member of Ethicon’s regulatory team. In the circumstances, I infer that any evidence any of these people could have given on this question would not have assisted the respondents’ case.
4444 Fifth, the proposition that the devices were “extensively evaluated” does not hold good for Prolift in general, let alone Prolift Total, which was the device Mrs Gill received.
4445 I have concluded that Ethicon was negligent in its evaluations and that they did not justify CE marking either before Prolift was taken to market or afterwards. In other words, they were not entitled to apply the CE mark. Further, at no time before Mrs Gill’s implant surgery was Prolift Total proven to be safe enough to go to market. Thus, even if clinical evaluations had been conducted with reasonable care and in accordance with the requirements of the European Directive, a reasonably prudent manufacturer would not have affixed the mark or taken the device to market until or unless its safety had been demonstrated in adequately powered randomised controlled trials.
4446 There is no evidence to indicate, and the respondents did not submit that, without CE marking, Prolift would have been sold in Australia. In any case, Ethicon’s conduct did not satisfy the requirements of the Australian law. If it was not sold in Australia, more probably than not it would not have been available to Mrs Gill’s surgeon and it would not have been implanted in Mrs Gill.
4447 It follows that Mrs Gill succeeds on the first and second negligence claims.
The third negligence claim: failure to provide adequate warnings and other information
4448 In order to succeed on this claim, Mrs Gill must prove that, but for the respondents’ failure to warn of the pleaded complications (other than the risk of psychiatric injury) and the inadequate evaluations, she would not have consented to implantation with the device. The respondents eschewed an argument both in this case and in Mrs Dawson’s that, if factual causation were established, it was inappropriate to extend their liability to the harm caused by their conduct.3742
4449 Like all other issues in the case, the standard of proof is the balance of probabilities. It is not sufficient that Mrs Gill demonstrate that it was possible that the respondents’ acts or omissions caused her damage. Moreover, if she would have agreed to the Prolift Total procedure even if the relevant warnings had been given, she must fail.
4450 As the CLA (WA) prohibits any consideration of the evidence of the injured person, during the trial it was common ground that Mrs Gill’s evidence on this subject must be disregarded. If the common position were correct, then the determination could only be made by drawing inferences from the available evidence including, for example, evidence of her conduct at or about the relevant time; evidence from her about how she felt about certain things; evidence of others in a position to assess her conduct and her apparent feelings or motivations; and other matters which might have influenced her: Neal v Ambulance Service (NSW) [2008] NSWCA 346; (2008) Aust Torts Reports ¶81–988 at [41] (Basten JA, with Tobias JA and Handley AJA agreeing at [1] and [66] respectively). That evidence would also include Mrs Gill’s account of why she agreed to surgery involving mesh in preference to the alternatives and the information imparted to her about the pros and cons of mesh implant surgery.
4451 But the common position was not correct.
4452 This case, like all cases in this Court, was heard in the exercise of federal jurisdiction. A State cannot enact a law which governs the exercise by a court of federal jurisdiction: Rizeq at [63]. In other words, State laws do not apply of their own force in proceedings which invoke the judicial power of the Commonwealth: Bass v Permanent Trustee Co Ltd (1999) 198 CLR 334 at [35] (Gleeson CJ, Gaudon, McHugh, Gummow, Hayne and Callinan JJ); Solomons v District Court of NSW (2002) 211 CLR 119 at [134] (Kirby J). They apply if they are picked up by a law of the Commonwealth and then they apply as federal law: Solomons at [134].
4453 As Bell, Gageler, Keane, Nettle and Gordon JJ explained in Rizeq at [63]:
The incapacity of a State Parliament to enact a law which governs the exercise of federal jurisdiction by a court, whether it be a federal court or a State court, explains the necessity for s 79 of the Judiciary Act and is the key to understanding the nature and extent of its operation. Section 79 is a law, enacted under s 51(xxxix) of the Constitution, which serves to ensure that the exercise of federal jurisdiction is effective. The section fills a gap in the law governing the actual exercise of federal jurisdiction which exists by reason of the absence of State legislative power. The section fills that gap by picking up the text of a State law governing the exercise of State jurisdiction and applying that text as a Commonwealth law to govern the manner of exercise of federal jurisdiction. The section has no broader operation.
4454 But s 79 does not operate to insert a provision of a State law into a Commonwealth legislative scheme where a Commonwealth law properly construed can be seen to have left no room for the State law: R v Gee (2003) 212 CLR 230 at [62] (McHugh and Gummow JJ), cited with approval by the Court in Bui v Director of Public Prosecutions for the Commonwealth of Australia (2012) 244 CLR 638 at [25] (French CJ, Gummow, Hayne, Kiefel and Bell JJ).
4455 Section 79 of the Judiciary Act relevantly provides in subs (1) that:
The laws of each State or Territory, including the laws relating to procedure, evidence, and the competency of witnesses, shall, except as otherwise provided by the Constitution or the laws of the Commonwealth, be binding on all Courts exercising federal jurisdiction in that State or Territory in all cases to which they are applicable.
(Emphasis added)
4456 Section 5C(3) of the CLA (WA), like its analogues in other States, is a State law relating to evidence. The effect of s 79(1) of the Judiciary Act is that the restriction on admissibility of the evidence imposed by s 5C(3) applies in this case only if the Constitution or a law of the Commonwealth does not otherwise provide. But there is a law of the Commonwealth which otherwise provides. That is the Evidence Act. Section 56 of the Evidence Act provides that:
(1) Except as otherwise provided by this Act, evidence that is relevant in a proceeding is admissible in the proceeding.
(2) Evidence that is not relevant in the proceeding is not admissible.
4457 The Evidence Act provides for numerous exceptions to the rule that relevant evidence is admissible. None of those exceptions, however, is apt to capture s 5C(3) of the CLA (WA). Certainly when I drew the parties’ attention to this issue, they did not contend that there was one. Nor did they contend that s 5C(3) of the CLA (WA) was an integral part of the legislative scheme so that the Court could not pick up the rest of the Act without also picking up that subsection: see Solomons at [134].
4458 It follows that s 5C(3) of the CLA (WA) is not picked up by the Judiciary Act.
4459 Mrs Gill gave evidence about what she would have done had she been warned of certain matters not disclosed by the respondents in circumstances I have found to be negligent. That evidence appears at [82] of her first affidavit. There is no dispute that that evidence is relevant. Based on the common position of the parties this evidence was received only to the extent that it might be relevant to Mrs Gill’s claims under the TPA. Since that position was erroneous, a conclusion it seems that the parties themselves now recognise, it is admitted without limitation.
4460 So what does the evidence tell us?
4461 Mrs Gill’s evidence was as follows. First, she identified several factors that she said were important to her decision as to whether to undergo prolapse repair surgery with mesh:
(a) I had been told that the mesh was likely to fix me forever, whereas if I did not have the mesh, but underwent the old prolapse repair procedure, then in a few years, I might have to have another operation because the prolapse was likely to recur. I had a prolapse that was uncomfortable and causing significant problems within my marriage. I was keen to have this problem fixed once and for all and the information provided to me by Dr Chapple about mesh implants suggested to me this could be done once only without any significant difficulties.
(b) I had been told that I could retain my uterus with the mesh procedure and this was vital to my identity as a woman. It seemed to me the mesh implant operation was the only option to get me back to the right shape and to allow me to keep my uterus. The other options either involved a hysterectomy or had a significant chance of the prolapse returning with all the problems I had been experiencing coming back again. Although I was not seriously planning on having more children, I was only 36, and I did not want a hysterectomy. To have the mesh meant I kept my uterus. The sort of symptoms I was having made me feel very unfeminine and to also have my uterus removed just made me think my body would feel like an empty box. And I just did not want to be an empty box. I just wanted to keep my uterus because it was an important part of me as a woman.
(c) I did not have any understanding that mesh was for old women. I had the impression that a mesh implant was for women who wanted to still be sexually active and physically active too. By this I mean I believed it was for women, like me, who wanted to do things like jogging and cycling.3743
4462 She added that she was also influenced by the amount and content of the information she was given about the risks associated with the operation. She highlighted the absence of information as to the following matters: the risk of chronic and severe pain or chronic pain with intercourse; the risk of vaginal shortening or narrowing or its potential consequences or methods of treatment; the fact that an erosion could cause severe pain and that treatment for it could require multiple operations; the fact that it might be impossible to remove the mesh to treat the complications; the possibility that the mesh might be unsuitable for her because of her psoriasis; and whether surgery to treat a recurrence of prolapse might be more difficult because of the mesh.
4463 The information provided to Mrs Gill by Dr Chapple before her implant surgery in January 2007 was consistent with the information contained in the 11 January 2005 Prolift IFU (in use until 13 December 2007) and which I have determined had significant deficiencies.
4464 I accept the respondents’ submission, that Mrs Gill was, or was at least likely to be, aware that the Prolift procedure was relatively new, that it was associated with a longer recovery and more pain in the post-operative period, that it was not recommended for women who planned to become pregnant, and that there was a risk of erosion. 3744 But the evidence does not indicate that she was told of the limited data available about Prolift Total and the absence of clinical trials, the prospect of chronic pain or other long-term adverse events, the unsuitability of the device for someone with an autoimmune disorder, the potential difficulties associated with treatment of adverse events, or other matters which would have been significant to her decision-making and which were known to the respondents but undisclosed in the 11 January 2005 Prolift IFU. While it is theoretically possible that Dr Chapple knew about all the pleaded complications despite the respondents’ negligence, there is no evidence that he did and the evidence of Professor Korda and others suggests that that is highly unlikely. Moreover, it is extremely unlikely, if not inconceivable, that Dr Chapple would have been aware of the deficiencies in the clinical evaluations of Prolift in general or Prolift Total in particular.
4465 Mrs Gill deposed that she would not have consented to the operation if she had been told of the following matters:
(1) that erosions did not occur only in a small percentage of cases or that they were often difficult to treat and might require multiple operations;
(2) the risk of erosion was in the order of 1 in 10 or more patients experiencing the problem;
(3) there was a risk of chronic and severe pain that might not be able to be treated effectively because the mesh might not be able to be removed
(4) there was a risk of chronic pain with intercourse which might not be able to be remedied because the mesh might not be able to be removed;
(5) the mesh was unsuitable for younger women like her who wished to remain sexually and physically active;
(6) the mesh was unsuitable for patients with an autoimmune disease like psoriasis;
(7) there was a risk of recurrence of the prolapse which would be more difficult to treat because of the presence of the mesh;
(8) there were risks of vaginal narrowing and shortening;
(9) there was a risk of contraction of the mesh, which could cause chronic pain and pain with intercourse, and which might be difficult to treat because the mesh might be unable to be removed; and
(10) the safety and efficacy of using mesh as a treatment for prolapse required more study and investigation.3745
4466 Whatever the true rate of erosion for transvaginal mesh kits, the overwhelming evidence from studies suggests that the risk in Mrs Gill’s case was high, given that she had a three compartment repair.
4467 Mrs Gill was not cross-examined on this evidence. No questions were put to her about whether, erosion aside, it mattered to her that the risk of the other matters was small or large, common, uncommon or rare. She was not asked, for example, whether she would have proceeded with the surgery in any event if those risks were rare.
4468 It is of course necessary to recognise the possibility that her evidence was affected by the wisdom of hindsight.
4469 Professor Kahneman wrote in Thinking, Fast and Slow (Penguin, 2012) at 202:
A general limitation of the human mind is its imperfect ability to reconstruct past states of knowledge or beliefs that have changed. Once you adopt a new view of the world (or any part of it), you immediately lose much of your ability to recall what you used to believe before your mind changed.
4470 Even where a witness’s evidence is unchallenged and uncontradicted, it is open to the court to disbelieve evidence found to be tainted by hindsight: Ellis v Wallsend District Hospital (1989) 17 NSWLR 553 at 560 (Kirby P) and 581 (Samuels JA); Towns v Cross [2001] NSWCA 129 at [22] (Davies JA, Mason P and Giles JA agreeing).
4471 The evidence may simply be implausible, as it plainly was in Odisho v Bonazzi [2014] VSCA 11. In that case the appellant alleged that her multiple pulmonary emboli were caused by the negligent failure of her gynaecologist to warn her that a drug she was prescribed could cause her to suffer a thromboembolic event. During cross-examination at the trial, the appellant told the Court that she would not have taken the test if the relevant risk was one in a trillion or there had only ever been one recorded episode (see [40]). The Court held that “the exaggerated nature” of this evidence “well justified” the decision of the primary judge to reject it. In Ellis, like Towns, a medical negligence case involving a failure to warn of possible risks of surgery, Kirby P observed at 560 that, no matter how honest a patient may try to be, self-interest and knowledge of the misfortunes that that have followed the particular treatment will necessarily colour her evidence. Samuels JA, with whom Meagher JA agreed, made a similar observation at 581. As McHugh J put it in Rosenberg v Percival (2001) 205 CLR 434 at [26], “human nature being what it is, most persons who suffer harm as the result of a medical procedure and sue for damages genuinely believe that they would not have undertaken the procedure, if they had been warned of the risk of that harm”.
4472 On the other hand, even where an injured person’s evidence is considered utterly unreliable, a court might nonetheless infer, based on the objective facts and its assessment of her general character and personality, that she would not have agreed to the treatment had she been warned: Rosenberg v Percival at [25] (McHugh J). In Chappel v Hart (1998) 195 CLR 232, in the footnote at 246, McHugh J said that the reliability of a plaintiff’s subjective evidence could only be determined by reference to objective factors, particularly the plaintiff’s attitude and conduct at or about the time when the breach of duty took place.
4473 So regardless of what Mrs Gill said, it is necessary to examine all the circumstances. Those circumstances include, but are not limited to, the matters Mrs Gill identified as important to her decision-making process.
4474 The first specialist Mrs Gill saw for her prolapse was Dr Natalwala. It will be recalled that he recommended against the use of mesh. On the evidence before the Court, the reason he was opposed to the use of mesh was her youth. I take that to be a reference to the fact that she was still of child-bearing years. His report to the GP indicates that he was cognisant of her family history of “collagen and elastin problems” but this only appears to have been relevant as an explanation for the prolapse.3746 The evidence does not suggest that he knew or considered it to be a factor militating against the use of mesh.
4475 Dr Chapple’s notes record that at the first consultation with Mrs Gill on 23 October 2006 they had a long discussion about the “pros and cons of the various procedures”.3747
4476 The notes do not record the contents of the conversation and Dr Chapple was not called to fill the gap. Mrs Gill gave evidence of receiving certain advice. It is clear that her memory of the various consultations was imperfect. That is scarcely surprising. Moreover, Mrs Gill only had cause to reflect upon the advice after the unhappy outcome of the procedure and her affidavit was taken nearly a decade later. In these circumstances her evidence must be treated with caution. Nonetheless, the evidence she gave about the advice she was given and not given was largely consistent with the contemporaneous records. It was also consistent with the information conveyed by the respondents in the IFU supplied with Prolift at the time Mrs Gill was being considered for, and consented to, surgery and the Prolift product brochures. In substance or effect, as Mrs Gill put it in her first affidavit, the advice was that the use of mesh would prevent a recurrence and that any complications were easy to fix and short-lived.3748 In the absence of any evidence to the contrary, I accept what she said. On the basis of that evidence and having regard to the contemporaneous records I make the following findings.
4477 At Mrs Gill’s first consultation with Dr Chapple in October 2006, surgical options were canvassed, some involving mesh and some not.
4478 One option they discussed was a hysterectomy, which she was dead against. The thought of a hysterectomy scared her because it was irreversible and she did not wish to “completely close the door on the possibility of a third child” particularly since the marriage was under strain and the future uncertain, but predominantly because she considered that her uterus defined her as a woman.3749
4479 Another option Dr Chapple raised with her was mesh repair.
4480 Dr Chapple told her that the mesh implant was like a grapevine being supported by a lattice; “the mesh acts like a lattice which your muscles and flesh will grow into” and he drew a diagram to illustrate this. He also told her that the implantation of mesh was a new treatment method which would permanently fix her prolapse and that no further surgery would be required. In particular, he told her that, after the mesh is implanted, her pelvic region would be “rock solid”, “nothing will be able to fall through”, and “[she would] be able to jump on a trampoline with [her] boys and nothing will come out”. Mrs Gill concluded that if she took this option she would not need another operation. She was given no information about what might happen to her if the prolapse were to recur or whether a recurrence would be more difficult to treat because of the mesh.3750
4481 At the second consultation Dr Chapple again discussed the surgical options with Mrs Gill. At that consultation he also canvassed the possibility of an abdominal or laparoscopic sacro-hysteropexy, and he may well have done so at the earlier consultation. Since he recognised that Mrs Gill not only wished to retain her uterus but also wanted to leave open the possibility of future pregnancies, he told her GP that he “would lean away from a Prolift procedure”.3751 He did so based on the manufacturer’s recommendation that it not be used if future pregnancies are planned. On the other hand, he also told Mrs Gill that he was getting the best results on Prolift.
4482 At the third consultation they had another discussion about the pros and cons of the various procedures.
4483 Notwithstanding what Dr Chapple told the GP, Mrs Gill did not remember Dr Chapple telling her that he would lean against Prolift and the evidence is insufficient to enable such a finding to be made. But she did remember being informed that if she chose the mesh option she would not be able to have further children because the mesh would not be able to grow or stretch with a baby and the baby could not be delivered vaginally. Rather, she testified that she recalled a conversation in the following terms or to the following effect:
[He said] if you fall pregnant after I’ve done your Prolift, you won’t be able to deliver because you will undo all my good work. So I said, “So could I have a Caesar?” and he said yes. So, to me, it wasn’t the end of my ability to have children. 3752
4484 Mrs Gill was also informed that, for a small percentage of women, their bodies reject the implant and “this can cause an erosion”. She did not remember whether Dr Chapple advised her of the potential complications associated with erosions or where they occurred.3753 Nor did she remember whether she was advised that erosions might lead to further surgical treatment or that they might be serious problems if they eventuated. The overall effect of her evidence, however, was that she was not told that any further surgery would be required. She was given the impression that erosions were uncommon minor complications that could be corrected easily and would be short-lived. This is consistent with the information that appeared in the Prolift brochures, which referred to a small risk of the mesh material becoming exposed into the vaginal canal and made no mention of the possibility of surgical intervention to remove the mesh, let alone the difficulties that could be associated with it and the potential consequences.
4485 Mrs Gill might well have been told by Dr Chapple that there was a longer recovery time with Prolift and more post-operative pain, consistent with what he mentioned in his report to Dr Eranki. But Mrs Gill’s evidence, which was not contradicted and which I accept, was that she was not advised that the use of mesh in prolapse repair surgery could result in permanent complications, such as chronic and severe pain, or that multiple revision operations could be required. Nor was she told that it could result in chronic pain during intercourse or that the procedure was best reserved for elderly women who are not sexually active. Since she was then only 36, if she had been given such advice, I have no doubt that she would have attached significance to it.
4486 Moreover, she was not told that the use of mesh might be unsuitable for her because of her psoriasis, which is an autoimmune disorder.3754 She was not informed of the risks of vaginal narrowing or shortening or the percentage of cases in which they develop. Neither was she informed of the risk of contraction or its consequences.
4487 As a result of the information she received from Dr Chapple, Mrs Gill was confident that the mesh option was the latest and best treatment for prolapse. She was under the impression that the old way of doing things meant the prolapse would inevitably recur and she would have to have another operation. She wanted to keep her uterus to keep open the possibility of having more children. She reasoned that if she w to fall pregnant again she could give birth by caesarean section.
4488 While there was no direct evidence about what Dr Chapple would have done had the respondents discharged their duty of care, the inference was open on the evidence that a proper warning from the manufacturer would have been passed on to Mrs Gill.
4489 Based on the contemporaneous records, Mr Finch SC submitted during closing argument that Dr Chapple was “a very competent and diligent doctor”3755 who engaged in “a careful consult process”.3756 It is difficult in these circumstances to accept that he would not have passed on information supplied by the manufacturer that could affect his patient’s decision as to whether to agree to Prolift surgery. Indeed, Mr Finch acknowledged that it was open to the Court to infer that Dr Chapple would have passed on information as to the risk of chronic pain had it been included in the IFU or had otherwise been brought to his attention by the respondents.3757
4490 On the other hand, Mr Finch submitted that if the IFU classed the risk as rare, in the absence of evidence from Dr Chapple about how he would have dealt with that information, there was a gap in the applicants’ case.3758
4491 One difficulty with this submission is that its premise was that the risk of chronic pain was rare and that in the counterfactual scenario the IFU would reflect that. In the light of the possibility of late erosions, however, and the relatively short time during which Prolift had been on the market when Dr Chapple was advising Mrs Gill, it would have been misleading to classify the risk as rare. Indeed, as I have observed, even at this point in time it is difficult to reach any reliable conclusion about the true incidence of chronic pain after Prolift surgery in particular or surgery with POP devices in general. Another problem is that, since Mrs Gill had an autoimmune disorder, she was more vulnerable than others to fall victim to that and other known risks. Regardless, a reasonably prudent doctor in Dr Chapple’s position would have been obliged to pass on information of this kind to his patient and, noting senior counsel’s characterisation of Dr Chapple and the evidentiary support for it, I consider that Dr Chapple would have done so. Having regard to his professional, legal and ethical obligations, the contrary inference is highly improbable. Indeed, there is no evidentiary foundation for it.
4492 For all these reasons, I find that it is more probable than not that, if the respondents had warned of the pleaded complications, then Dr Chapple would have communicated that information to Mrs Gill.
4493 Mrs Gill is an intelligent and thoughtful person with scientific training. She did not rush into surgery. It is apparent that she gave the matter careful consideration, giving weight to the benefits of each option against the risks that were made known to her. As Mr Finch SC acknowledged in argument, the fact that there had been several discussions over multiple consultations about the “pros and cons” of the surgery indicates that she was agonising over her decision and that she was worried about the cons.3759 Although she did not remember everything she was told, I have no doubt that, had she been warned that the surgery would expose her to a risk of chronic pain, she would have remembered. I also have no doubt that a risk of that kind would have weighed heavily upon her in the decision-making process. At her young age and in her circumstances, the prospect of chronic pain, dyspareunia and repeat surgery with no promise of recovery would have been repugnant.
4494 Further, while Mrs Gill assumed that she would not fall into the “small percentage” of women whose bodies might reject the mesh, she did so in a state of relative ignorance. She had not been informed of the full extent of the potential complications. Nor, importantly, had she been informed that she was in a high risk category because she had an autoimmune disorder: psoriasis. If the respondents had made known to her, whether directly or indirectly through Dr Chapple, that mesh surgery in general or Prolift in particular was contraindicated for women with autoimmune disorders or that she was at a higher risk of injury on that account, it is unlikely that she would have made the same assumption. What is more, in those circumstances, it is more likely than not that Dr Chapple, as a careful and diligent physician, would have recommended against the use of mesh and therefore against both Prolift surgery and sacrohysteropexy, on the assumption that that necessarily involved mesh, as the respondents submitted.3760
4495 While the onus rests throughout with Mrs Gill, it was never put to her in cross-examination that, even if she had been warned of the pleaded complications or the inadequate evaluations, she would still have opted for Prolift.3761
4496 Taking all these matters into account, I am persuaded that it is more likely than not that, but for the deficiencies in the information provided by the respondents, Mrs Gill would not have agreed to mesh surgery in general and Prolift in particular. Information about the potential for chronic pain and the increased vulnerability to injury for women with autoimmune disorders, for example, are risks to which a reasonable person in Mrs Gill’s position would be likely to attach significance and, given the surgeon’s legal and ethical obligations, information which almost certainly would have been passed on to her. Had Mrs Gill been informed of those risks, it is more likely than not she would have opted for some form of native tissue repair. Has she been apprised of the true extent of the risks of mesh surgery, she may even have resigned herself to the loss of her uterus.
4497 It follows that Mrs Gill suffered damage caused by the respondents’ negligence.
Did Mrs Dawson suffer injury caused by a defect in Gynemesh?
4498 As in Mrs Gill’s case, there is a dispute about the extent to which Mrs Dawson’s injuries can be attributed to the respondents’ product but there is no dispute that she suffered injury. Since the transvaginal implantation of Gynemesh PS carried risks of significant and serious injury, including infection, erosion and chronic refractory pain about which the respondents gave no, or no sufficient, warning, and Mrs Dawson sustained injury of that kind, she suffered injury caused by the defect. Having regard to the approach taken by all parties that the outcome of the defect case determines the outcome of the Pt VA cases, it follows that she also suffered injury caused by the respondents’ contraventions of ss 74B and 74D of the Act.
4499 Consequently, Mrs Dawson is entitled to be compensated for the loss she has suffered as a result of her injuries. I will discuss the question of which injuries are compensable when I deal with damages.
Did Mrs Dawson suffer damage caused by the respondents’ negligence?
4500 Section 51 of the Wrongs Act (Vic) contains the general principles applying to causation in a negligence case. It is based on the recommendations in the Ipp Report and is relevantly indistinguishable from s 5C of the CLA (WA) and s 5D of the CLA (NSW). As Beach JJA and McMillan AJA observed in Odisho v Bonazzi at [30] and footnote 18, s 51 is the equivalent of s 5D of the CLA (NSW). As their Honours noted, the text of the sections is identical, except in two respects. First, (like s 5C of the CLA (WA)), s 51(2) refers to “an appropriate case”’, s 5D(2) to “an exceptional case”. Second, unlike the CLA (NSW) and CLA (WA), while s 5D(3) mirrors the subjective test set out in s 5D(3), goes on to provide that “any statement made by the person after suffering the harm about what he or she would have done is inadmissible except to the extent (if any) that the statement is against his or her interest”. Consequently, the above discussion of the law applies equally here.
4501 Section 52 of the Wrongs Act (Vic) confirms the common law position that the plaintiff bears the burden of proving any relevant fact on the balance of probabilities.
The first and second negligence claims: inadequate evaluation of Gynemesh PS
4502 I found the pre and post-market evaluations of Gynemesh PS to be insufficient to justify CE marking. It will be recalled that the pre-market evaluation was undertaken in 2002 and that no post-market evaluation was conducted until after Mrs Dawson’s implant surgery. In these circumstances, the device should not have been on the market. If it had not been on the market, Mrs Dawson would not have been offered the device. Ethicon’s negligence was therefore a necessary condition of the occurrence of the relevant harm. No question arises as to the scope of liability. For these reasons I find that the inadequate evaluation of Gynemesh PS caused Mrs Dawson’s injuries.
The third negligence claim: failure to provide adequate warnings and other information
4503 The question here is whether, but for the respondents’ failure to warn of the pleaded complications and/or the deficiencies in the evaluations, Mrs Dawson would have undergone surgery with Gynemesh PS.
4504 The respondents submitted that the Court should find that Mrs Dawson was made aware of the following risks before she consented to the implant surgery in early 2009: bleeding, infection, anaesthetic risk, erosion, recurrence of prolapse, visceral injury, de novo stress incontinence, dyspareunia and an increase in vaginal discharge while the splint was in situ.3762 The submission rested on two pieces of evidence: Dr Lim’s report to Dr Gibson of 26 February 2009 in which she asserted as much3763 and a document entitled “Acknowledgement of Consent for Treatment” signed by Mrs Dawson on 26 February 2009.3764 In the latter document Mrs Dawson agreed to the procedure and acknowledged that it had been explained to her to her satisfaction and Dr Lim confirmed that she had explained to her the nature and effect of the procedure and she was of the opinion that Mrs Dawson understood the explanation. A post-it note apparently attached to the document, presumably completed by Dr Lim, lists the same risks identified in the letter to Dr Gibson and no others.
4505 Mrs Dawson acknowledged that she was informed that there were risks associated with the operation. Although she did not specifically recall what risks were identified, she accepted that Dr Lim might have advised her of the risks mentioned in the letter. She believed, however, that she was not given any further detail about those risks. “For example”, she deposed in her first affidavit, “[she] did not know what erosion was” and, if Dr Lim informed her of the risk of erosion, she did not explain what that meant. In particular, she was not informed about the incidence of erosion or how serious it could be or “what needed to be done to get rid of it.”3765 Moreover, she stated she did not recall being told and did not believe she was told of the following risks:
i. The mesh could erode into my vagina or cause pain, pain with intercourse or the need for multiple operations to alleviate the problem.
ii. If these complications arose, it may not be possible to remove the entire implant, or substantial parts of the implant, in order to alleviate the problem.
iii. How often these complications occurred. For instance, I was not told that mesh erosions occurred at least in 10% of cases.
iv. The effectiveness of the mesh in curing prolapse required further evaluation or that studies about its effectiveness were only preliminary.
v. The implant surgery could lead to me experiencing chronic and severe pain, which could significantly affect my quality of life.
vi. It might not be possible to remove all of the mesh and that I could instead be stuck with it being in my body for the rest of my life.
vii. If the prolapse recurred, it might be harder to fix because of the mesh than if no mesh had been used.
vii. There were other surgical options to treat my prolapse that did not run the risk of suffering erosions, chronic and severe pain or pain with intercourse.3766
4506 After the two consultations with Dr Lim, Mrs Dawson was left with the impression that the use of mesh was likely to give her “a good chance” of never having to undergo prolapse surgery again.
4507 I accept that Dr Lim did inform Mrs Dawson of the risks described in her letter to Dr Gibson. Furthermore, despite Mrs Dawson’s evidence, I consider that it is likely that Dr Lim did say something to her about the nature of an erosion although she may not have mentioned the word itself. In the absence of evidence to the contrary, however, and in the light of the contemporaneous records and the contents of the 18 December 2008 Gynemesh IFU (in use until to 30 November 2010), I consider that it is unlikely that Mrs Dawson was informed of the risk of chronic pain, the possibility of multiple operations to deal with erosions, and the difficulties associated with removal of the mesh, risks about which the respondents should have, but did not, warn. Taking into account all the evidence, I find it is more likely than not that she was not so informed.
4508 On the assumption that it was necessary for Mrs Dawson to prove that, if appropriate warnings had been given Dr Lim would have passed them on, on the balance of probabilities I am satisfied that she has discharged that burden. I mentioned in Part XIII the respondents’ submission that medical practitioners should be presumed to be competent. Absent evidence to the contrary, I agree. Medical practitioners should also be presumed to act in accordance with their legal and ethical obligations, unless there is evidence to indicate otherwise. There is no reason to think that Dr Lim would have overlooked her legal and ethical obligations. The risk of chronic pain, the possibility of multiple operations to deal with erosions, and the difficulties associated with removal of the mesh were all material risks of which she would have been bound to warn her patient. A reasonable person in Mrs Dawson’s position would have attached significance to them.
4509 The more difficult question is what Mrs Dawson would have done if she had been warned. The respondents argued that the Court should prefer the contemporaneous documentary evidence over Mrs Dawson’s recollection.3767 To the extent that there is any inconsistency between the two, that is the course I have taken. But the contemporaneous documents provide little, if any, assistance on this question.
4510 Unlike s 5C of the CLA (WA) and s 5D of the CLA (NSW), s 51 of the Wrongs Act (Vic), which is identical in all other respects with its interstate counterparts, does not render that evidence inadmissible. So even if I am wrong about the application of these sections in federal jurisdiction, Mrs Dawson’s evidence can be taken into account.
4511 Mrs Dawson’s evidence was as follows.
The reason I do not believe I was advised of these matters is because if I had been told about them, it would have run “alarm bells” for me and I never would have chosen to have the surgery. I was told the operation was likely to fix my prolapse once and for all. I understood that the mesh would remain in my body for the rest of my life. Dr Lim mentioned risks around the time of the surgery, but I believed that, once I had recovered from the operation, I was not going to have any further problems. If there were long-term risks or doubts about whether it was actually effective, then I would not have agreed to the surgery. I would have felt that the surgery with the mesh was experimental.3768
4512 This evidence was untouched in cross-examination. Mrs Dawson’s honesty, like that of the other applicants, was never impugned. These are relevant considerations. But they are not decisive. I still have to consider whether the evidence is reliable.
4513 Mrs Dawson seemed adamant that Dr Lim did not warn her of the risk of dyspareunia or pain during sexual intercourse. Certainly she did not recall such advice, and I have preferred Dr Lim’s evidence to hers in this respect. But her other evidence was not inconsistent with the contemporaneous evidence. Indeed, it was consistent with it. Moreover it is not incredible. To the contrary, as the applicants submitted,3769 it has the ring of truth about it.
4514 I am acutely conscious of the wisdom of hindsight, but in all these circumstances I accept Mrs Dawson’s evidence. I find that, but for the respondents’ negligence, it is more likely than not that Mrs Dawson would not have agreed to mesh surgery. If native tissue repair were an option, then I consider she would have taken that option. Professor Korda’s evidence was that native tissue repair was an option.3770 His view was that in January 2009 it would have been reasonable for Mrs Dawson to have undergone a repeat anterior vaginal repair using native tissue, native tissue posterior vaginal repair with levatorplasty, or a defect specific posterior vaginal repair, avoiding the use of mesh altogether.3771 Although he conceded that he did not know the state of her native tissue at the time she was being considered for surgery in 2009, he pointed out that Dr Lim’s notes described “some degree of prolapse”, but not an enormous amount. He said that the first native tissue repair had lasted for seven or eight years and a second could have cured the prolapse for a similar period of time.
4515 If native tissue would not have been an option, I am satisfied that she would have pursued conservative treatments.
Did Mrs Sanders suffer injury caused by a defect in TVT?
4516 There is no dispute that Mrs Sanders suffered injury as a result of the TVT implant. The respondents conceded that the following were injuries caused wholly or in part by TVT: the exposure requiring surgery on 8 August 2011 and the surgery itself and the adjustment disorder with mixed anxiety and depressed mood, though only to the extent that her reduction in mobility is causally related to the implant rather than her co-morbidities, particularly the osteoarthritis of her hip.3772 There is a dispute about the other alleged injuries which I will deal with in Part XVIII.
4517 Once again, since TVT carried a number of risks about which the respondents provided no, or no adequate, warning, it was defective and, since a number of those risks came home, Mrs Sanders has established that she suffered injury as a result of the defect.
Did Mrs Sanders suffer damage caused by the respondents' negligence?
The first and second negligence cases: inadequate evaluation of TVT
4518 The available evidence about the safety and efficacy of TVT at the time of Mrs Sanders' implant surgery was weak. It was common ground that before the Ward Hilton RCT, the studies were limited to preliminary and small case series with short-term follow-up. Ward and Hilton did not publish their initial six-monthly results until 2002, the year after Mrs Sanders' operation. Before Mrs Sanders' operation, Ethicon had not prepared a clinical evaluation report. Medscand apparently relied on the report of Dr Eriksson on the Nordic multicentre studies. It will be recalled that this was a non-randomised trial involving a select group of patients, and no studies had been undertaken to assess its safety or efficacy across a broader population. At that point, only one year results were available. No clinical investigations were conducted by Ethicon before CE marking was applied and no post-market clinical follow-up studies were undertaken until well after Mrs Sanders' operation. CE marking was not justified at the time the mark was applied or at any time before Mrs Sanders' surgery.
4519 No long-term results were available from the Nordic multicentre studies until 2001 at the earliest, when the three year findings were published. But they only captured 90 of the 131 women originally enrolled; nearly one-third (31%) of the cohort was not assessed.3773 Those who were likely to have the most severe type of stress urinary incontinence, and were therefore the hardest to treat, were excluded from the trial.3774
4520 While risk assessments were apparently conducted both by Medscand and Ethicon before Mrs Sanders’ operation, none of them was sufficient.
4521 In these circumstances, the CE mark should not have been applied to TVT before Mrs Sanders’ implant surgery in March 2001. More likely than not, had it not been for those negligent evaluations the mark would not have been applied in 1998 and TVT would not have been on the Australian market in March 2001, in which case Mrs Sanders would not have received the device and would not have suffered the damage that ensued from its implantation. It follows that the negligent evaluations caused the damage Mrs Sanders suffered as a result of its implantation.
The third negligence claim: failure to provide adequate warnings and other information
4522 According to Mrs Sanders’ unchallenged evidence, Dr Taylor did not inform her of any risks, either in respect of the surgery in general or in relation to TVT in particular. In a letter to Mrs Sanders' GP following the consultation, Dr Taylor wrote:
This patient has had no benefit from pelvic floor exercises and is anxious to have a surgical cure. Her name has been placed on the waiting list for a TVT mesh type sling.3775
4523 The letter does not mention any discussion with Mrs Sanders about the risks associated with TVT and no contemporaneous note of such a discussion was tendered. Of course, that does not necessarily mean that there was no such discussion. Dr Taylor might have had such a discussion but neglected to record it. In the absence of evidence from him, however, and the absence of any note, we only have Mrs Sanders' word.
4524 On 8 March 2001, Mrs Sanders signed a consent form for a procedure described as “Transvaginal tape”.3776 By her signature she indicated that she consented to that “operation procedure” and that the nature of it had been explained to her by Dr Giele. Immediately above her signature was a section entitled medical practitioner’s confirmation:

4525 It was common ground that the first item reads “infn” and is an abbreviation for “infection”, that the second reads “anas”, meaning “anaesthetic”, that the third is “rupture”, and the fourth “haemorrhage”.
4526 Unsurprisingly, Dr McNeill had no recollection of the operation. After reviewing the hospital records, however, she said that she did not believe that she consulted with Mrs Sanders beforehand. She explained that, at that time, the usual practice at the hospital was that the consultant would meet with the patient before surgery to obtain consent.3777
4527 Mrs Sanders said that she had never heard of the word “erosion” or the fact that erosion was a potential complication of TVT until after her second operation in 2011. In her first affidavit she stated:
I was not informed about the risk of tape erosion, how frequently it occurs or what the consequences were to me if it developed. I was not informed that there was any risk of pain with intercourse associated with the use of the tape. I was not told how erosions or painful intercourse were likely to be treated if they developed or even if they could develop. I was not told whether there was an alternative method of operating to fix my urinary problems or how the tape operation compared with any alternative. I was not told whether the long-term complications of the tape operation had been determined or whether the long-term effectiveness had been established. I assumed that the tape was going to be permanently in me from the time it was inserted. I assumed, because no one told me otherwise, that it was going to be safe to be inside me for the rest of my life.3778
4528 The respondents submitted that, since 15 years had passed between the 2001 operation and her affidavit, little weight should be given to this evidence.3779 That submission has considerable force. Mrs Sanders was reflecting on events that had taken place many years beforehand, after she had undergone great suffering, when she had had no reason to reflect on the matter until, at the earliest, she was told that her symptoms were attributable to the TVT, and when it is likely that she did not think about it until she was questioned by her lawyers.
4529 On the other hand, Mrs Sanders’ evidence is consistent with the contemporaneous documents.
4530 Neither in his clinical notes3780 nor in Dr Taylor's letter to Mrs Sanders' GP3781 is there any reference to a discussion about risks associated with the operation let alone risks of TVT in particular. I infer from the evidence of Dr O’Neill that she did not discuss the risks of the procedure with Mrs Sanders. The hospital notes indicate that Dr Giele, who was the doctor who obtained Mrs Sanders’ written consent to the procedure, only mentioned intraoperative risks. Certainly, there is no mention of erosion or exposure, whether late-onset or otherwise, or its potential consequences.
4531 Mrs Sanders’ evidence was also consistent with the first iteration of the TVT IFU published by Medscand. The only adverse consequences about which the Medscand IFU warned were transitory local irritation at the wound site, a transitory foreign body response, and the potentiation of an existing infection. In the second edition of the IFU, issued on 8 September 2000, these were repeated but with the additional misleading statement after the reference to “transitory foreign body response” that could result in extrusion, erosion, fistula formation, and inflammation. The 8 September TVT IFU also included warnings of the risk of punctures or lacerations of vessels, nerves, bladder or bowel during needle passage and the possibility of surgical repair. Furthermore, it warned that the application of excessive tension to the tape could cause temporary or permanent lower urinary tract obstruction. Amongst other things, however, it did not warn of the prospect of chronic pain, late onset erosions or that surgery might be required to treat the erosions, let alone of the potential consequences of surgical treatment.
4532 The evidence does not permit me to conclude, however, that the device inserted in Mrs Sanders was delivered to the hospital with the 8 September 2000 TVT IFU. In the absence of interrogatories, which were not administered, the applicants were not in a position to adduce evidence about this. Only the respondents could have known.
4533 Mrs Sanders’ medical records gave them all the information they needed to identify the relevant IFU.3782 Yet they called no evidence about it. The Product Traceability Label was affixed to her 2001 operation report:

4534 The same label appears on the operation count sheet, which was also tendered.3783
4535 Further, the respondents adduced no evidence about when the boxes containing the 8 September 2000 TVT IFUs were first ordered by King Edward Memorial Hospital, when they were dispatched to Australia or when they were received by the hospital.
4536 In considering the extent of the information Mrs Sanders was likely to have received, it is relevant that the respondents did not adduce evidence to support their submission that the TVT device was accompanied by the 8 September 2000 IFU. It is relevant because only they could have known and all the evidence is to be weighed according to the proof which it is in the power of one side to produce and the other to contradict: Blatch v Archer at 970. No evidence was given to explain the absence of the evidence.
4537 I infer that the evidence the respondents could have adduced about this matter would not have assisted their case.
4538 The applicants submitted that, overall, the evidence is consistent with the respondents providing information which related to efficacy but which failed to mention risks, in particular, the risk of erosion, and long-term adverse consequences.3784 They relied not only on the evidence of Mrs Sanders but also on a PowerPoint presentation on TVT prepared by JJM in which Dr Taylor appears as an example of surgeons who implanted the device.3785 The applicants argued, in effect, that this presentation should be taken into account in considering whether or not to accept her evidence that she was not warned of the risks of erosion and chronic pain, or of long-term complications of any kind.3786
4539 Like the product brochures, the PowerPoint slides emphasised the benefits of TVT and minimised the risks. TVT surgery was described as “a minimally invasive surgical alternative for the treatment of urinary stress incontinence” and the “only ‘tension-free’ suburethral sling procedure on the market with 5 years of proven efficacy, performance and safety” (original emphasis). Its clinical features and benefits were said to be:
• Can be performed under local anaesthesia allowing for intra-operative evaluation
• ‘Tension-free’ – no distortion of vaginal anatomy
• No fixation, anchors or sutures required
• Minimally invasive – only 3 small incisions required
• Procedure takes no longer than 30 minutes to perform
• Day only procedure – most women return home the same day
• Short recovery period allowing early return to daily activities
• Low incidence of post-operative voiding difficulties (3-4%)
• No cases of Prolene* tape rejection to date
• High success rate – with more than 90% of patients symptomatically cured or significantly improved at 5 year f/u3787
4540 Five studies were referred to without mention of any adverse effects.
4541 Another slide purported to list results at six month and two year follow-up of a randomised controlled trial comparing colposuspension and TVT suggesting comparable outcomes but more rapid recovery from surgery with TVT and fewer adverse outcomes. The RCT was not identified but, since it was common ground that the Ward Hilton study was the first RCT comparing TVT with colposuspension, this was presumably a reference to the Ward Hilton study. The PowerPoint presentation was undated but the metadata indicated that it was created on 19 June 2001 but amended on 20 August 2002.3788 The figures for the numbers in the two arms of the RCT mentioned in the slide correspond with the patients available for review in the Ward Hilton RCT at six months and two years, although the two year results were not published until 2004. The slide omitted to mention that there was an exposure in the vagina at six months and three additional cases in the following 18 months,3789 indicating that the assertion in the presentation that there had been no cases of Prolene tape rejection to date was false.
4542 It is unlikely that the information respondents imparted to the market before August 2002 was more fulsome, although the 8 September 2000 TVT IFU was more informative than its predecessor from Medscand. But in the absence of evidence as to the use, if any, to which the presentation was put, it is difficult to place much weight on it.
4543 In the absence of any evidence as to when the box containing the TVT device was dispatched to Australia and the capacity of the respondents to establish the date, I conclude that it is unlikely that it included the 8 September 2000 IFU.
4544 Even if the device given to Mrs Sanders had been accompanied by the 8 September 2000 IFU, that would not assist the respondents.
4545 First, there is no evidence to indicate that Dr Taylor had seen the 8 September 2000 IFU at the time he saw Mrs Sanders. It will be recalled that Dr Taylor saw Mrs Sanders once, in October 2000, the month after this iteration of the IFU had been issued.
4546 Second, as I have already mentioned, the respondents did not alert users of their devices when changes were made to the IFUs.
4547 In all the circumstances I conclude it is unlikely that Dr Taylor had seen it when he recommended TVT to her.
4548 Third, in any case, as I have found above in Part XI, all the IFUs for TVT were (and remain) deficient.
4549 Mrs Sanders did not say that she would not have had the operation had she been warned of the risk of erosion per se. I will come to this part of her evidence shortly.
4550 At the time of Mrs Sanders’ operation, TVT had been on the market in Australia for less than 18 months.
4551 Dr O’Neill recalled that there were about six to seven years’ worth of data on the efficacy of the TVT implants, that the short term efficacy appeared very good but that there were no long-term studies. She understood that there had been some randomised controlled trials but they were short-term studies. Her recollection was faulty in this respect. In fact, there had only been one randomised controlled trial with only six months of published data. She professed to have had at the time “some knowledge” that the long-term risks of TVT surgery included urinary retention, erosion, and dyspareunia. At the same time, however, she stated:
While risks or complications involved in TVT implants were known in 2001, their seriousness, including their long-term prevalence and, in part, the difficulty in treating complications if they arose was not well known so that patients, like Mrs Sanders, could not be given reliable information about these matters … I would not have been in a position to provide her with any reliable information about the long-term efficacy or risks of the TVT prior to her operation.3790
4552 I accept her evidence. There is no reason to think that any of the other medical practitioners involved in Mrs Sanders’ treatment, including Dr Taylor, would have been in any better position.
4553 So what would Mrs Sanders have done had the respondents provided the relevant information, whether directly to her or indirectly through her treating doctor(s)?
4554 Mrs Sanders’ evidence was that she would not have agreed to the procedure. Specifically, she said she would not have agreed if she had been told that:
(a) The operation had a risk of the tape eroding into the bladder or vagina or urethra, which might require numerous operations to treat or that may not be able to be permanently fixed;
(b) The operation had a risk of the tape causing pain with intercourse that might require a further operation to treat and that the operation may not be able to fix the problem;
(c) If an erosion occurred, the tape may have to be cut, which might lead to a return of my urinary problems;
(d) The tape could be difficult to remove if complications developed that required its removal;
(e) The long-term effectiveness of the tape in treating urinary incontinence had not been proved because it had only been on the market for a few years;
(f) It was unknown whether the tape was safe to be left inside me for the rest of my life.3791
4555 She was not cross-examined on this evidence. I do not doubt her honesty. But Mrs Sanders was reflecting on events that had taken place more than 15 years earlier and more than a decade before this proceeding was instituted, after she had undergone great suffering, when she had had no reason to reflect on the matter until, at the earliest, she was told that her symptoms were attributable to TVT. This does make it difficult to place much weight on what she said. Nevertheless, there was no evidence to suggest that, if informed of those matters, she would have agreed to TVT when there were alternative forms of treatment. Although those alternatives would have included surgical repair using native tissue with some risks, including the risk of pain with sexual intercourse, these were not novel forms of treatment, having been studied over a long period of time and they would not have carried a risk of erosion and its sequelae. The only evidence to indicate that Mrs Sanders was a risk-taker was the evidence that, many years earlier, she had established her own hairdressing business. That risk, however, bears no relationship to the risk in question.
4556 On balance, taking all relevant matters into account, I consider it more likely than not that Mrs Sanders would not have agreed to TVT surgery if she had been informed of the pleaded complications and the above listed matters in particular. But for the absence of a suitable warning, it is more likely than not that she would have undergone some other form of incontinence surgery which did not involve the use of polypropylene mesh.
4557 At common law, where, as here, there is a duty to inform a person of a risk of physical injury which would ordinarily suffice to avert the risk which called the duty into existence and the risk eventuates, breach of the duty is treated as having caused or materially contributed to that injury unless there is sufficient reason to the contrary: Chappel v Hart at [10] (Gaudron J). See also: Betts v Whittingslowe (1945) 71 CLR 637 at 649 (Dixon J) and Hampic Pty Ltd v Adams [1999] NSWCA 455; (2000) ATPR ¶41–737 at [37] (Mason P and Davies AJA). Here, there is no sufficient reason to the contrary.
4558 I find that the respondents’ want of care in the provision of information to prospective users of TVT was a cause of Mrs Sanders’ damage. More likely than not it contributed, if not directly, then indirectly, to Dr Taylor’s decision to opt for TVT over a traditional form of surgical treatment. That is so, even if Dr Taylor omitted to warn Mrs Sanders at all: see Medlin v State Government Insurance Commission (1995) 182 CLR 1 at [6] (Deane, Dawson and Toohey, Gaudron JJ).
Did Mrs Sanders suffer damage by the misleading conduct of the respondents?
4559 In the absence of evidence that the product brochures had come to the attention of Mrs Sanders or her treating doctors, there is no evidence to establish a causal connection between the misleading representations they contained and the damage sustained by Mrs Sanders.
4560 The two potentially operative IFUs, however, also misrepresented and minimised the nature and extent of the risks. For the reasons given above, it is more likely than not that those representations materially contributed to Mrs Sanders’ damage. Consequently, I find that Mrs Sanders suffered damage by some of the respondents’ misleading conduct.
4561 In Wardley at 526 the plurality stated that in many cases the common law measure of damages will be an appropriate guide to the measure of damages under s 82(1) of the TPA but emphasised that it is always necessary to look at the provisions of the Act to discern any relevant legislative intention. In Marks v GIO Australia at 513–514, however, McHugh, Hayne and Callinan JJ said that once the causal connection between the alleged loss or damage and the contravening conduct has been established, there is nothing in the Act to suggest that the amount that may be recovered under s 82(1) should be limited by drawing an analogy with the law of contract or tort. In the present case, neither side referred to Marks. Both proceeded on the basis that the measure of damages for this contravention was the measure of damages at common law. Consequently, that is how I will proceed.
PART XVII: THE LIMITATIONS QUESTIONS
4562 The respondents contended that the claims of Mrs Gill and Mrs Sanders are statute-barred. The determination of those questions involves the resolution of complex questions of law and fact.
The claims under the Trade Practices Act
4563 The periods of limitation are fixed by ss 74J and 75AO of the Trade Practices Act and Div 2 of Pt VIB of that Act. Which statutory limitations apply in any particular case depends on the date the contravention occurred.
Contraventions before July 2004
4564 Section 74J appeared in Pt V Div 2A (which includes ss 74B and 74D). It relevantly provided as follows:
(1) Subject to this section, an action under a provision of this Division may be commenced at any time within 3 years after the day on which the cause of action accrued.
(2) For the purposes of this section, a cause of action shall be deemed to have accrued:
(a) … on the day on which the consumer … first became aware, or ought reasonably to have become aware:
(i) in the case of an action under section 74B—that the goods were not reasonably fit for the purpose referred to in that section.
…
(iii) in the case of an action under section 74D—that the goods were not of merchantable quality.
…
(3) In an action under a provision of this Division, it is a defence if the defendant proves that the action was not commenced within 10 years after the time of the first supply to a consumer of the goods to which the action relates.
4565 Section 75AO provided that:
(1) Subject to subsection (2), person may commence a liability action at any time within 3 years after the time the person became aware, or ought reasonably to have become aware, of the alleged loss, the defect and the identity of the person who manufactured the action goods.
(2) A liability action must be commenced within 10 years of the supply by the manufacturer of the action goods.
4566 A “liability action” includes an action under s 75AD and “action goods” are “goods whose supply and defect is alleged in the action”: TPA, s 75AA.
Contraventions after July 2004
4567 In 2004, however, by the Trade Practices Amendment (Personal Injuries and Death) Act 2004 (No 2) (Cth) (2004 Amendment Act), both ss 74J and 75AO were amended to insert the following note:
Part VIB restricts awards of compensation for death or personal injury, and sets out time limits for commencing actions for compensation for death or personal injury.
4568 The amendments commenced on 13 July 2004. They applied prospectively to contraventions of Pt V Divs 1A and 2A and Pt VA that occurred after that date: 2004 Amendment Act, Sch 1 cl 11.
4569 Under Pt VIB, a court is prohibited from awarding damages for personal injury in a proceeding that relates to Pt IVA, Pt V Div 1A or 2A, or to Pt VA if the proceeding was commenced more than three years after “the date of discoverability” for the injury to which the damages would relate or after the end of “the long-stop period” for the injury: TPA, s 87F.
4570 “Plaintiff” is defined in s 87D to include the person by whom the proceeding is brought, however described, and therefore includes an “applicant”.
4571 The “date of discoverability” is relevantly defined in s 87G(1) as the first date the plaintiff knows or ought to have known each of the following:
(a) that the injury occurred;
(b) that the injury was attributable to a contravention of the Act; and
(c) that the injury was significant enough to justify bringing an action.
For these purposes, the plaintiff ought to know a fact if the plaintiff would have ascertained the fact had she taken all reasonable steps to do so before the date in question: TPA, s 87G(2). In determining what the plaintiff knew or ought to have known, the court may have regard to the plaintiff’s conduct and her statements, whether oral or written: TPA, s 87G(3).
4572 The “long-stop period” is defined in s 87H to mean 12 years after the act or omission alleged to have caused the injury or the period as extended by the court. The long-stop period cannot be extended, however, more than three years beyond the date of discoverability: TPA, s 87H(2). In considering whether to extend the period, s 87H(3) provides that the Court is required to have regard to the justice of the case and in particular to:
(a) whether the passage of time has prejudiced a fair trial;
(b) the nature and extent of the loss or damage;
(c) the nature of the defendant’s conduct alleged to have caused the death or injury; and
(d) the nature of the defendant’s conduct since the alleged act or omission.
4573 “Defendant” is a synonym for “respondent”.
4574 Consistent with the ordinary principles of statutory construction (see, for example, Project Blue Sky Inc v Australian Broadcasting Authority (1998) 194 CLR 355 at [69]), ss 74J and 75AO must be read subject to Pt VIB. The effect of the notes to these sections is that the limitation periods specified in them do not apply to actions for damages for personal injuries under those parts of the Act covered by Pt VIB; they are governed by the terms of Pt VIB alone. The notes are part of the Act: Acts Interpretation Act, s 13. In other words, in any action for damages for personal injury arising out of a contravention of Pt V of the Trade Practices Act that occurred after 13 July 2004, the time in which proceedings for compensation for loss or damage may be brought is fixed by Pt VIB and not Pt V.
4575 The purpose of these amendments was to give effect to the recommendations of the Ipp Report. One of the objectives of the review that culminated in the report was the introduction of consistent limitation periods and constraints on damages in cases of this kind: see Explanatory Memorandum to the Bill to the 2004 Amendment Act, [1.1], [1.7].
4576 So how is s 86G(1) to be interpreted? What defines the occurrence of injury? What is meant by “attributable to”? When will an injury be “significant enough” to justify the institution of proceedings?
4577 Oddly enough, there appear to be no cases on point. Certainly the parties did not draw my attention to any. The recommendations of the Ipp Report, however, picked up some of the language of the Limitation Act 1980 (UK) (UK Limitation Act). To the extent that that language is reproduced in Pt VIB, the English authorities provide guidance on its interpretation.
4578 Section 11 of the UK Limitation Act fixes a three year general limitation period for damages consisting of, or including, damages in respect of personal injuries. The time limit runs from either the date on which the cause of action accrued or “the date of knowledge (if later) of the person injured”.
4579 Section 14(1) relevantly provides that references in s 11 to a person’s date of knowledge are references to the date the person first had knowledge of the following facts: (a) that the injury in question was significant; (b) that the injury was attributable in whole or in part to the act or omission which is alleged to constitute negligence, nuisance or breach of duty; and (c) the identity of the defendant. Knowledge that any acts or omissions did or did not, as a matter of law, involve negligence, nuisance or breach of duty is irrelevant.
4580 Obviously “attributable” means capable of being attributed. Lord Reid said in Central Asbestos Co Ltd v Dodd [1973] AC 518 at 533B–533C that “attribute” has a number of cognate meanings, the essential element of which is a connection of some kind. Central Asbestos was an action in negligence. In that case the relevant connection was a causal connection between certain acts which involved or amounted to negligence.
4581 For the purposes of the section:
(a) an injury is significant if the person whose date of knowledge is in question would reasonably have considered it sufficiently serious to justify instituting proceedings for damages against a defendant who did not dispute liability and is able to satisfy a judgment (s 14(2)); and
(b) (knowledge includes knowledge which the injured person might reasonably have been expected to acquire from facts observable or ascertainable by him or from facts ascertainable by him with the help of medical or other appropriate expert advice which it is reasonable for him to seek but does not include knowledge of a fact ascertainable only with the help of expert advice as long as the person has taken all reasonable steps to obtain (and, where appropriate, act upon) that advice (s 14(3)).
4582 The test in s 14(2) was said to be partly subjective (would the plaintiff have considered the injury sufficiently serious) and partly objective (would he have been reasonable if he did not), “[t]aking that plaintiff, with that plaintiff’s intelligence, would he have been reasonable in considering the injury not sufficiently serious to justify instituting proceedings for damages”: McCafferty v Metropolitan Police District Receiver [1977] 2 All ER 756 at 775; [1977] 1 WLR 1073 at 108, followed by the Court of Appeal in Nash v Eli Lilly & Co [1993] 4 All ER 383 at 390; [1993] 1 WLR 782 at 791.
4583 In Halford v Brookes [1991] 3 All ER 559 at 573; [1991] 1 WLR 428 at 443, Lord Donaldson MR said of the question of knowledge in the context of s 14(3):
The word has to be construed in the context of the purpose of the section, which is to determine a period of time within which a plaintiff can be required to start any proceedings. In this context “knowledge” clearly does not mean “know for certain and beyond possibility of contradiction.” It does, however, mean “know with sufficient confidence to justify embarking on the preliminaries to the issue of a writ, such as submitting a claim to the proposed defendant, taking legal and other advice and collecting evidence.” Suspicion, particularly if it is vague and unsupported, will indeed not be enough, but reasonable belief will normally suffice.
4584 This approach was endorsed by the House of Lords in Haward v Fawcetts [2006] UKHL 9; [2006] 3 All ER 497; [2006] 1 WLR 682.
4585 In Nash, after referring to Lord Donaldson’s remarks in Halford, the Court of Appeal declined to define knowledge for the purposes of the UK Act, noting that “Parliament left the word to speak for itself” ([1993] 4 All ER 383 at 392; [1993] 1 WLR 782 at 792). Nevertheless, the Court went on to say:
In applying the section to the facts of these cases, we shall proceed on the basis that knowledge is a condition of mind which imports a degree of certainty and that the degree of certainty which is appropriate for this purpose is that which, for the particular plaintiff, may reasonably be regarded as sufficient to justify embarking upon the preliminaries to the making of a claim for compensation such as the taking of legal or other advice.
Whether or not a state of mind for this purpose is properly to be treated by the court as knowledge seems to us to depend, in the first place, upon the nature of the information which the plaintiff has received, the extent to which he pays attention to the information as affecting him, and his capacity to understand it. There is a second stage at which the information, when received and understood, is evaluated. It may be rejected as unbelievable. It may be regarded as unreliable or uncertain. The court must assess the intelligence of the plaintiff; consider and assess his assertions as to how he regarded such information as he had; and determine whether he had knowledge of the facts by reason of his understanding of the information. The section, it is to be emphasised, attaches consequence to the having of knowledge which depends upon information and understanding.
4586 Later, the Court stated ([1993] 4 All ER 383 at 395; [1993] 1 WLR 782 at 795):
As we have said above, whether a claimant has knowledge depends both upon the information he has received and upon what he makes of it. If it appears that a claimant, while believing that his injury is attributable to the act or omission of the defendant, realises that his belief requires expert confirmation before he acquires such a degree of certainty of belief as amounts to knowledge, then he will not have knowledge until that confirmation is obtained. Frequently, as it seems to us, it will be safe for the court to proceed upon the basis that a claimant did realise that he required confirmation if he acted in a manner consistent with that state of mind even if he is, as he may frequently be, unable to recall with any degree of precision what his state of mind was. Conclusions as to a claimant's state of mind will, we think, usually be more securely based upon inference from conduct in the known circumstances than from a claimant’s later assertion as to how he now recalls his then state of mind as between, for example, belief or knowledge. We add that we have difficulty in perceiving how in any case where a claimant has sought advice and taken proceedings, it can rightly be held that the claimant had not then had relevant knowledge.
4587 The Court concluded that knowledge for the purposes of s 14 is “a state of mind experienced by the plaintiff actually existing or which might have existed had the plaintiff, acting reasonably, acquired knowledge from the facts observable or ascertainable by him or which he could have acquired with the help of medical or other appropriate expert advice which it was reasonable for him to obtain” ([1993] 4 All ER 383 at 395; [1993] 1 WLR 782 at 796). The Court held that “attributable” in the expression “attributable to the act or omission” means capable of being attributed as a real (rather than fanciful) possibility, not probability ([1993] 4 All ER 383 at 397; [1993] 1 WLR 782 at 797). That interpretation was approved in Haward at [11].
4588 Nash was a case with certain similarities to the present. It was a group action. The first defendant was a multinational pharmaceutical company. The other defendants were mostly related companies. The defendants were involved in the manufacture of a drug named Opren, which was used in the treatment of arthritis. The plaintiffs were people to whom the drug had been prescribed on various dates over a period of time. The drug had a beneficial effect in the relief of arthritis but it also had a number of serious side-effects. The nature of the side-effects varied. The plaintiffs with whom the appeals were concerned mainly complained of photosensitivity and onycholysis (a condition affecting finger and toe nails causing them to become ridged and loose and to separate from the nail bed). Other patients suffered more serious complications, including liver and kidney failure resulting, in some cases, in death. The regulatory authority considered that the side-effects were unacceptable and withdrew the product licence.
4589 In my opinion, the approach taken by the English cases is the approach required under the analogous provisions of the Trade Practices Act.
4590 The language used in s 87G is also similar to that used in s 39 of the Limitation Act 2005 (WA), which is discussed below in relation to the common law claims. In AME Hospitals Pty Ltd v Dixon (2015) 48 WAR 139, McLure P, with whom Newnes JA agreed, applied the definition of “attributable” given in Roncevich v Repatriation Commission (2005) 222 CLR 115 (in the context of the phrase “arising out of or attributable to defence service” in s 70 of the Veterans’ Entitlements Act 1986 (Cth), holding at [33] that “attributable to” requires a causal connection in fact (as distinct from causation at law). McLure P also held at [39] that knowledge of whether or not the injury was attributable to the conduct of the respondents is a matter that requires expert medical knowledge and experience.
4591 Buss JA approached the matter in a different way. His Honour considered at [208] that the term “attributable to” should be interpreted having regard to the ordinary and natural meaning of “attribute”, which is “to ascribe to as belonging or pertaining”. His Honour therefore held that “attributable to” refers to a connection between the death or injury and the conduct of a person, which need not be the sole or dominant connection. At [210]–[211] his Honour said:
It is significant that Parliament used the term “attributable to” and not the term “caused by”. The term “attributable to” is able to accommodate a degree of uncertainty in relation to whether the person to whom the cause of action accrues is aware, or ought reasonably to have become aware, of a connection between the relevant death or injury and the conduct of a person, within s 39(3) or s 39(4), as the case may be.
In my opinion, it is apparent from the language and focus of s 39, read in the context of:
(a) the ordinary and natural meaning of the word “attribute”;
(b) the meaning of “aware” in s 39(3) and (4);
(c) the meaning of “ought reasonably to have become aware” in 39(4); and
(d) the subject matter and purpose of s 39 (namely the granting of an extension of time, in certain circumstances, to commence an action for damages, after the limitation period has expired, in respect of personal injury),
that the term “attributable to”, in s 39(3) and (4), requires that the person to whom the cause of action accrues was aware, or ought reasonably to have become aware, of a connection between the death or injury, on the one hand, and the conduct of a person, on the other, with sufficient confidence reasonably to justify, in all the circumstances, the commencement of proceedings against the proposed defendant on the relevant cause of action by the issue of a writ or other originating process.
4592 I doubt that the differences in approach are significant. In both cases the relevant connection is a causal connection. A causal connection can be made out even if the relevant conduct is not the sole cause of the death or injury. McLure P did not suggest otherwise. Nor did her Honour require certainty.
4593 Some assistance can also be obtained from the NSW authorities on the provisions of Pt 2 Div 6 and of Pt 3 Div 4 of the Limitation Act 1969 (NSW), which have a similar history, the former fixing the limitation period for personal injury cases, the latter dealing with the long-stop limitation period and extensions of it in the same kind of cases. Both sets of provisions were inserted in 2002 following the recommendations of the Ipp Report. The most significant difference is that, instead of the words “the injury was attributable to a contravention of the Act”, the NSW Act uses the words “the injury … was caused by the fault of the defendant”.
4594 In Baker-Morrison v State of New South Wales (2009) 74 NSWLR 454, Basten JA, with whom Ipp and Macfarlan JJA agreed, held that:
(1) taking all reasonable steps must, in appropriate circumstances, include obtaining medical and legal advice and information (at [37]);
(2) whether the injury is sufficiently serious to justify the bringing of an action requires both legal and medical expertise (at [41]) and no proper view could be formed on this question without information of this nature (at [42]);
(3) in most circumstances, the act of instructing a solicitor will be sufficient to show that the plaintiff has taken “all reasonable steps”; in some circumstances, there may be a question as to whether the plaintiff’s instructions were adequate or whether other limitations prevented the solicitor from taking proper steps in a timely fashion (at [58]);
(4) in context, the phrase “ought to have known” means that the person should have made an inquiry (at [59]);
(5) where the plaintiff has taken all reasonable steps to ascertain facts that depend on the advice of a professional person and has been given wrong advice, the plaintiff may not have the necessary state of mind (at [59]; see also Frizelle v Bauer [2009] NSWCA 239 at [30] (Basten JA, McColl JA agreeing at [1]).
4595 With one qualification, the Act does not state who bears the burden of proof, that is, whether the plaintiff/applicant must prove that her action was commenced within the period fixed by the statute or whether the defendant/respondent must prove that it is out of time. The qualification relates to the long-stop provision in s 74J(3) which squarely places the burden on the defendant to establish that the action was not commenced within 10 years of the date of supply.
4596 In their written submissions, the respondents contended that the onus was on the applicants, specifically Mrs Gill and Mrs Sanders to establish that their claims were within time.3792 But they provided no reasons and their contention was unsupported by authority. The applicants argued that the respondents carried the burden, but they cited no authority either
4597 Section 74J was inserted into the TPA by the Trade Practices Amendment Act 1978 (Cth). The Explanatory Memorandum to the Bill that became the Act says nothing about who bears the onus of proof.
4598 To the extent that the Explanatory Memorandum to the Trade Practices Amendment Bill 1992 deals with the question, it tends to support the applicants’ argument. The 1992 Explanatory Memorandum stated at [30]:
The Bill conforms with the EC Directive by leaving the onus of proof on the balance of probabilities on the issues going to liability (sections 75AD to 75AG) firmly with the plaintiff. In contrast to the Directive itself, in the Australian legal context there is no need to specifically provide that this is the case because Australian courts will presume this to be so in the absence of express words to the contrary. This is likewise the case under the equivalent British legislation, the Consumer Protection Act 1987. The Government intends that in applying the legislation the Australian courts will fully acquaint themselves with the emerging jurisprudence in Europe, especially on procedural and evidentiary matters.
4599 Despite what was said in the Explanatory Memorandum concerning the Government’s intention, neither side took me to any European authorities on the point.
4600 Both the Ipp Report and the Explanatory Memorandum to the Bill for the 2004 Amendment Act were silent on the question. Notwithstanding what appeared in their written submissions, however, in oral argument senior counsel for the respondents accepted that his clients carried the onus of proving that the actions were brought out of time, at least with respect to the time limitations in ss 74J(1) and 75AO(1).3793
4601 The concession was well-made. Although in the past the question was uncertain and the authorities were in conflict, in Australia it is now generally accepted that, unless the statute bars both the right and the remedy, the burden of proving that an action is statute-barred lies with the person making the allegation: Pullen v Gutteridge, Haskins & Davey Pty Ltd [1993] VR 27 at 71–76; Cigna Insurance Asia Pacific Ltd v Packer (2000) 23 WAR 159 at [36]–[52] (per Malcolm CJ, Kennedy J agreeing at [56]). As Brooking, Tadgell and Hayne JJA put it in Pullen, in which the question was extensively considered, at 74:
It is for the plaintiff to plead and prove the elements of the cause of action. If the accruing of the cause of action in time is no part of the cause of action, the plaintiff need not allege or prove it.
See also: Handford P, Limitation of Actions, The Laws of Australia, (Thomson Reuters, 4th ed, 2017) at [5.10.2540].
4602 This is consistent with the general principle of construction enunciated by Walsh JA in Currie v Dempsey (1967) 69 SR (NSW) 116 at 125:
In my opinion the [legal] burden of proof … lies on a plaintiff, if the fact alleged (whether affirmative or negative in form) is an essential element in his cause of action, eg, if its existence is a condition precedent to his right to maintain the action. The onus is on the defendant, if the allegation is not a denial of an essential ingredient in the cause of action, but is one which, if established, will constitute a good defence, that is, an ‘avoidance’ of the claim which, prima facie, the plaintiff has.
4603 There was a dispute in the present case about whether s 75AO(2) bars the right as well as the remedy. I deal with that question in the context of Mrs Sanders’ case where the issue arose. It is sufficient to note at this point that, for the reasons given below, the view I have reached is that, properly construed, in no respect does ss 75J or 75AO bar both the remedy and the right. They are not essential ingredients of the causes of action created by Pt V or Pt VA respectively. Sections 74J and 75AO create defences to those causes of action and the onus was therefore on the respondents to make them out. The essential ingredients of the statutory causes of action are set out in the antecedent sections, relevantly ss 74B, 74D and 75AD. Insofar as the defective goods cause of action is concerned, the statement in the 1992 Explanatory Memorandum that the Bill left the onus of proof on “issues going to liability” with the plaintiff coupled with the express reference to ss 75AD to 75AG, supports this construction.
4604 For similar reasons the same must be said of the limitation periods fixed under Pt VIB Div 2 of the TPA.
Kathryn Gill
4605 Since Mrs Gill did not receive Prolift Total until 12 January 2007, she is plainly caught by the provisions of Pt VIB.
4606 In their defence the respondents pleaded that Mrs Gill’s claims for compensation under the Trade Practices Act were statute-barred pursuant to s 87F of the Act because her cause of action accrued more than three years before the commencement of these proceedings.3794 They argued that the causes of action under ss 74B, 74D and s 75AD of the Act first accrued between June and August 2007 because it was in this period that her first mesh exposure was identified and because, with the information she then had, she ought reasonably to have become aware that Prolift was not reasonably fit for the purpose for which it was supplied, that it was not of merchantable quality, and that it had a defect. Alternatively, they argued that those causes of action accrued in June 2008, at the time of her second revision surgery for mesh exposure.3795
4607 As I have explained, the causes of action under sections 74B, 74D and 75AD accrue from the date of discoverability. That means that it is necessary to determine when Mrs Gill first knew or ought to have known that she had an injury, that the injury was attributable to a contravention of the Act, and that the injury was significant enough to justify bringing an action. It also means that the respondents had to establish that the last of these dates was more than three years before this proceeding was commenced.
4608 It is evident that Mrs Gill is intelligent and insightful. On the other hand, she is not medically or legally trained and a good deal of her understanding and insight has no doubt been acquired after years of reflection and counselling. Her evidence needs to be assessed in this light.
4609 The respondents argued that:
Mrs Gill knew at the time of her prolapse surgery that the purpose of the surgery was to insert a permanent device designed to correct her prolapse;
despite her evidence to the contrary, documentary evidence shows that in 2006 and 2007 she was aware of the name of the device;
during the period from June to August 2007 she knew that at least some of her symptoms were related to the device (and if not then certainly by June 2008 when the second revision operation was carried out); and
she had the capacity to determine the manufacturer of the device at any time from June 2007.3796
4610 For these reasons they contended that the action was statute-barred; it was brought more than three years from the date of discoverability for the injury to which her action for damages would relate. Since that date, at the latest, was in June 2008 and the Court was prohibited from extending the date by more than three years beyond the date of discoverability, it follows that the Court had no power to extend the period.
When did Mrs Gill first know that she had sustained an injury? When should she first have known?
4611 The first question, then, is when Mrs Gill knew or should have known that she had sustained an injury.
4612 There is, however, an antecedent question. That is when injury occurred.
4613 It was very difficult to pin down the position of either side on this question or, for that matter, on the question of what precisely constituted the injury.
4614 The starting point for present purposes must be the pleading. What did Mrs Gill allege was the injury?
4615 In the latest iteration of the statement of claim the applicants pleaded at [23P] that “by reason of the matters pleaded at paragraphs 23D to 23O above” (which attribute certain features to the mesh and describe “mesh risks”, “mesh complications” and “mesh removal complications”), Mrs Gill suffered loss and damage. That loss and damage was particularised in the following way:
(A) Personal injury including one or more of the Mesh Complications and Mesh Removal Complications including, in respect of the Gill First, Second and Third Revision Surgeries, the complications pleaded at paragraphs 23L, 23K, 23M and 23O above and psychiatric injury including depression generalised anxiety disorder, adjustment disorder with depressed mood and/or chronic pain disorder.
(B) Health care expenses.
(C) Additional out of pocket expenses.
(D) Economic loss.
(E) The need for gratuitous and in addition, or alternatively, commercial care.
(F) Non-economic loss.
(G) Additional particulars may be provided following the service of evidence.
4616 The “mesh complications” were alleged to be:
a chronic inflammatory reaction of the tissues in which the devices were implanted, attached and/or of the surrounding tissues;
erosion and/or extrusion and/or protrusion/and/or exposure of the devices through tissue in which the devices were implanted or attached and/or the surrounding tissues;
the consequential need for surgery to remove the mesh implant or part thereof;
chronic pain, dyspareunia, apareunia, difficulty voiding and/or defecating;
offensive discharge;
recurrence of pelvic organ prolapse;
de novo or recurrent urinary incontinence;
damage to surrounding organs, nerves, ligaments, tissue and/or blood vessels;
haemorrhage;
infection;
leg weakness;
reoperation or revision surgery associated with complications, including the above complications; and/or
psychiatric injury.
4617 In a statement of particulars I ordered Mrs Gill to file, the injuries were particularised as:
1. Chronic inflammatory reaction of tissues into which the Prolift Total mesh implant (Mesh Implant) was implanted, and surrounding tissues.
2. Erosion and/or extrusion and/or exposure of the Mesh Implant through the anterior vaginal wall 5cm proximal to the urethral meatus to the right of the midline.
3. Requirement for surgery on 20 September 2007 to excise the exposed portion of the Mesh Implant referred to in the preceding paragraph (First Revision Surgery).
4. Erosion and/or extrusion and/or exposure of the Mesh Implant anterior to the cervix.
5. Requirement for surgery on 20 June 2008 to:
(a) Excise the right deep anterior arm of the Mesh Implant to halfway along the obturator fossa;
(b) Excise the left posterior arm of the Mesh Implant near the cervix and uterosacral ligaments;
(c) Perform a Fenton's procedure (Second Revision Surgery).
6. Requirement for surgery on 4 July 2008 to excise and drain an anterior vaginal abscess.
7. Erosion and/or extrusion and/or exposure of the Mesh Implant on the anterior vaginal wall just distal to the cervix.
8. Requirement for surgery on 8 August 2013 to excise the exposed portion of the Mesh Implant referred to in the preceding paragraph (Third Revision Surgery).
9. Sub-urethral mesh ulceration with significant tenderness in the posterior vaginal wall and the right-sided mesh arm from the posterior prolapse as it was passing to the sacrospinous ligament.
10. Erosion and/or extrusion and/or exposure of the Mesh Implant under the urethra.
11. Recurrent anterior compartment prolapse (cystocele).
12. Recurrent posterior compartment prolapse (rectocele).
13. Recurrent uterine prolapse.
14. Development of bulky, fibroid, acutely anteverted uterus.
15. Generalised anxiety disorder (DSM-5).
16. Adjustment disorder with depressed mood (DSM-5).
4618 “Personal injury” is defined in s 4KA of the TPA in an inclusive way. Section 4KA was introduced in the 2004 Amendment Act. It states:
In this Act (except in section 68B):
Personal injury includes:
(a) pre-natal injury; or
(b) impairment of a person’s physical or mental condition; or
(c) disease;
but does not include an impairment of a person’s mental condition unless the impairment consists of a recognised psychiatric illness.
4619 Injury, itself, is not defined. The inference to be drawn is that Parliament intended that the ordinary meaning apply. “Injury” is relevantly defined in the The New Shorter Oxford English Dictionary as “hurt or loss …; harm, detriment, damage” and in the Macquarie Dictionary as “a particular instance of harm”. Consequently, the question devolves into an inquiry as to when harm first occurred and when Mrs Gill knew or ought to have known that she had been harmed or damaged by the mesh.
4620 The evidence does not specify when the reaction to the mesh became chronic. Nor is there any evidence to indicate whether, let alone when, Mrs Gill knew or, for that matter, ought to have known, that she had a chronic inflammatory reaction to the mesh. Her affidavits did not touch on the point. She was not questioned about it at the hearing, either in chief or in cross-examination. In any case, I do not consider that the chronic inflammatory reaction as formulated in the applicants’ pleading is an injury. The chronic inflammatory reaction to which the applicants were referring was the foreign body reaction. The foreign body reaction is the means by which the mesh was designed to adhere to the body’s tissues. In closing argument Mr Finch SC described it as a “mechanism, not a risk”.3797 If the chronic inflammatory reaction were an injury for relevant purposes, then every woman in whom polypropylene mesh has been implanted has been injured, regardless of whether she ever had or has any adverse effect, and should see a lawyer immediately.
4621 There is no doubt, however, that, regardless of when injury first occurred, by June 2007 Mrs Gill knew that the mesh had eroded. Dr Chapple told her so on 11 June 2007, having found on examination a 2 mm erosion in the right anterior fornix. On 14 June 2007 Dr Yin carried out a cystoscopy and found a palpable and viable mesh erosion that was significantly bigger than Dr Chapple had detected. Despite treatment with oestrogen, Dr Yin told Mrs Gill on 26 July that the mesh erosion was still there.3798 In September Mrs Gill was admitted to hospital to have the eroded portion of mesh excised. Dr Yin described the erosion as “sizeable” in a letter to Dr Eranki on 17 October 2007.3799
4622 Notwithstanding the applicants’ pleading, it is doubtful whether, without more, this erosion or exposure was a personal injury. Strictly speaking, mesh erosion is an adverse event, not an injury, although an erosion may cause injury, as occurred in this case.
4623 Mrs Gill’s evidence, however, which I accept, was that she was in a lot of pain in her pelvic region following her implant surgery which, contrary to her expectations, did not abate.
4624 I describe the symptoms Mrs Gill suffered in the months immediately following her implant surgery in detail in Part XV.
4625 In June 2007 Mrs Gill said that she could feel the mesh inside her, that whenever she moved suddenly she could feel it “slightly tear” her, and that that resulted in a sharp pain. Although she said that she was confused and did not know what was happening to her, she said “I knew that something must be wrong”.3800
4626 On 10 September 2007 Mrs Gill underwent surgery to excise that portion of the mesh which had eroded (the exposed mesh). In May 2008 a second mesh erosion was detected. Mrs Gill was then in a great deal of pain, and on 20 June 2008 she was admitted to hospital for further surgery to remove more mesh.
4627 Doing the best I can with the evidence, I find that it is more probable than not that injury occurred soon after Prolift was implanted, within a matter of weeks, perhaps a month at most.
4628 At that stage Mrs Gill did not know that she had sustained an injury. She did know that she had sustained an injury, however, when she could feel the mesh tear her, causing sharp pain. That was in about May or June 2007, in all likelihood before 11 June 2007 when she presented to Dr Chapple and told him that about three weeks earlier she could feel mesh in her vagina.
4629 I do not think that she ought to have known she had suffered an injury any earlier than that. In the early period after her surgery she was entitled to think that she was still in the process of recovery. When she complained of pain to Dr Chapple on 30 January 2007, he reassured her she was still healing from the surgery.3801 His notes read “on-going healing”, although he queried whether she might have a “vaginal wound infection”,3802 and arranged to see her six weeks later. In late February she was diagnosed with viraemia (a virus in the blood).3803 On 22 February 2007 Dr Chapple thought she might have a mesh infection and wondered whether she might have an erosion near her cervix.3804 On 7 March 2007 she was re-admitted to hospital. The next day she had a colonoscopy and was told she had colitis. According to the report, that was most likely due to a bacterial infection.3805 She was treated with Flagyl and “quickly responded”, according to Dr Chapple. When he saw her on 26 March 2007, he noted that the anterior mesh had concertinaed together, but recorded that it was only slightly tender.3806
4630 Although Mrs Gill presented with back pain, Dr Chapple offered no diagnosis, reassured her, and arranged to see her again in 10 weeks’ time. He thought so little of her presenting problem or of the examination finding that he failed to mention either of them to Mrs Gill’s GP, Dr Eranki, in his letter to her written the same day.3807 There is no evidence to indicate that he ever informed Mrs Gill that the mesh had concertinaed together. It is not apparent that he knew what this signified or presaged.
When did Mrs Gill first know or when should she have known that the injury was significant enough to justify bringing an action?
4631 It is convenient to deal with this question now, despite its place in the sequence in s 87G(1).
4632 I do not think that when she was first told she had a mesh erosion Mrs Gill knew that she had an injury significant enough to justify bringing an action. She had earlier been led to believe that an erosion was not a significant event and was told that it could be fixed with the use of oestrogen cream. When the cream proved ineffectual, she opted for surgery.
4633 On 10 September 2007, Mrs Gill underwent surgery to excise that portion of the mesh which had eroded. Even then she was optimistic that the revision surgery would cure her. It did not. Indeed, despite a six-week recovery period after the revision surgery following which she returned to work, her evidence was that her pain never really improved and she continued to have the same physical symptoms that she had before the operation.3808 Moreover, her mental state was perilous. She deposed that “psychologically” she remained in “a very dark place”.3809
4634 I find that Mrs Gill first knew or should have known that she had an injury significant enough to justify bringing an action in about late October 2007 by which time it was apparent to her that the revision surgery had been unsuccessful. Having regard to the medical advice she received, it is unreasonable to expect her to have known any earlier.
When did Mrs Gill first know that her injury was attributable to a contravention of the TPA? When should she first have known?
4635 The final question is when Mrs Gill knew or ought to have known that her injury was attributable to a contravention of the Act. The answer depends on when she knew or ought to have known that:
(1) the device with which she had been implanted was manufactured and/or imported by one or other of the respondents;
(2) the device had a defect of the kind alleged by the applicants or was for the same reason not reasonably fit for the purpose for which it was acquired; and
(3) her injury was caused by the defect or the unsuitability of the device for the relevant purpose.
4636 Her counsel argued that she did not become aware that a defective mesh implant was the physical cause of her injuries until about July 2013, when she consulted Dr Dowling. It was not until at this time, too, they contended, that she became aware that her injuries were attributable to the conduct of the respondents. They submitted that “it was not reasonable for Mrs Gill to have possessed the relevant medical and scientific knowledge regarding the defective nature of the implant so as to have acquired an awareness of the connection at an earlier time”.3810
4637 Mrs Gill said that she had no recollection of Dr Chapple using the name “Prolift” in connection with the discussions they had about surgery to repair the prolapse. She also stated that she did not become aware of the name Prolift and that Prolift was used inside her until after some of the mesh was removed.3811
4638 She said that Dr Chapple did not show her any pictures of the mesh kit or the equipment that would be used for the surgery. She said that she was provided with the 11 January 2005 Prolift IFU and some patient information brochures “to aid preparation” of the affidavit but had never before seen those documents or any other document containing pictures of the kit or equipment.3812
4639 Later in her affidavit Mrs Gill said that it was either at her first consultation with Dr Dowling on 5 June 2013 or at the next appointment on 16 July 2013 that she became aware that she had received a Prolift implant and that it was not until then that she knew she had sustained injuries related to Prolift.3813 She deposed that she asked Dr Dowling whether she was aware of other women who had similar problems to hers, that Dr Dowling replied that she was, and that Dr Dowling also informed her that Johnson & Johnson had withdrawn Prolift from sale. Mrs Gill stated that this was the first time that she became aware that she had been implanted with a device supplied by Johnson & Johnson. She added that the information Dr Dowling had given her made her think that there was a defect in the Prolift implant and that it was not her fault that she had experienced all the problems she had had since 2007.3814
4640 At the hearing Mrs Gill was asked what she meant by it was not her fault. She replied:
Up until then, whenever I presented with pain or problems, I was just told, “Wow, you’re unlucky, or gee, that’s no good or what did you do to cause that.” So it was always my fault. I was either not healing properly or I had overdone it or I hadn’t taken enough vitamins or I hadn’t rested sufficiently. So it was always me trying to fix what I had done and I was constantly, you know, being told, “Well, what did you do today? What caused your bleeding?” “Oh, I did some washing”, but never once has anyone offered the fact that maybe it was the mesh. It was always my fault.3815
4641 At the time she saw Dr Yin in June 2007 Mrs Gill said that, although she had been told by both Dr Chapple and Dr Yin that she had a mesh erosion, she did not understand that the problems she was experiencing were due to the mesh implant itself.3816
4642 Under cross-examination Mrs Gill went even further. She denied knowing that she had been implanted with mesh at the time she was seeing Dr Chapple and Dr Yin in June 2007. She said that she was told it was “a synthetic material like a lattice that would grow in … and hold you in place and you would be wonderful again”.3817 She conceded, however, that she had been told by both Dr Chapple and Dr Yin that a portion of the implant was exposed in her vagina and that when she called Dr Yin’s rooms on 1 August she had said that she wanted Dr Yin to deal with that portion of the device.
4643 I do not accept that Mrs Gill did not know that Prolift was the name of the mesh before she saw Dr Dowling and even more difficult to accept that she did not know that she had received a mesh implant in 2007.
4644 As to whether she knew she had received a mesh implant in 2007, in fact it was Dr Chapple’s rooms (not Dr Yin’s) that Mrs Gill called on 1 August 2007. The contemporaneous note of that call includes the statement “Dr Yin is going to deal with the mesh”.3818 The expression “deal with the mesh” appears in inverted commas in the notes. It is reasonable to infer from the use of the inverted commas that these were the very words Mrs Gill used. Moreover, as I have already observed, in her first affidavit Mrs Gill deposed that she could feel the mesh inside her, that it was tearing her, and that both Dr Chapple and Dr Yin told her she had a mesh erosion. She also indicated in that affidavit that at the first two appointments with Dr Chapple he had repeatedly referred to the use of mesh. For example, she said that at one of these appointments she asked Dr Chapple whether she would need to continue with her pelvic floor exercises after the operation and he replied: “No, because you will have a mesh supporting your pelvic floor”. She also stated that at the appointment on 6 December 2006 she told Dr Chapple “I want to proceed with the mesh implant surgery”.3819
4645 It follows that, notwithstanding her oral evidence to the contrary, I am satisfied that Mrs Gill knew that she had received a mesh implant in January 2007. In any case, as the respondents submitted, it would not matter whether she knew it was a mesh implant as distinct from “a synthetic material like a lattice …” or “something like flyscreen”, another expression she used in oral evidence to emphasise her point.
4646 As to whether she knew the name Prolift before she saw Dr Dowling, Dr Chapple’s contemporaneous notes of the consultation on 6 December 2006 after his discussion with her about the pros and cons of alternative treatments, including Prolift, includes the concluding statement “wants prolift”.3820 It was conceivable that he did not mention the name Prolift to her. That was the effect of Mrs Gill’s evidence in chief. In recounting his conversations with her, she used the expression “the mesh implant” instead.
4647 In cross-examination, however, Mrs Gill conceded that she remembered that Dr Chapple said that “he gets the best results with the Prolift”3821 and that “if you fall pregnant after I’ve done your Prolift, you won’t be able to deliver because you will undo all my good work”.3822 In the light of this evidence, it seems that Mrs Gill knew that mesh surgery using Prolift was one of the options Dr Chapple was proposing. In all the circumstances, including the absence of a reference to any other device, I find that Mrs Gill knew in January 2007 that she was to receive a Prolift implant.
4648 Further, the contemporaneous evidence indicates that Mrs Gill knew by July 2007, if not before, that she was in fact implanted with a Prolift implant. In cross-examination she was taken to the consent form she signed on 26 July 2007 for the first mesh excision. It contained the following statement:
I acknowledge that I have consented to admission to this facility for the following Operation/Procedure:
Ro PROLIFT EROSION (MESH EROSION).3823
“Ro” presumably means “removal of”.
4649 The description of the operation was handwritten. In the absence of any suggestion to the contrary, I conclude that the handwriting is Dr Yin’s. The only word Mrs Gill said she could read was “Prolift”. Mrs Gill did not deny being told that the device was called Prolift. Her evidence was that she did not recall knowing the term Prolift. Nor did she deny seeing the word Prolift on the form. She speculated that she “would have just looked at that as medical jargon and signed it”.3824 I found this evidence unpersuasive. I very much doubt that Mrs Gill would have signed a form that she did not understand, at least not without seeking an explanation from the person who presented it to her. It is possible, of course, that she paid no attention to the reference to Prolift and that she really did not then know that mesh was Prolift. I think that it is more likely than not, however, that she knew at that time that she had received a Prolift device, but that at the time she had no particular reason to remember the name.
4650 I therefore find that by 26 July 2007 Mrs Gill was aware that the mesh with which she had been implanted was called Prolift, although she may have forgotten the name.
4651 That is by no means the end of the matter. Mrs Gill’s knowledge, whether actual or constructive, that she suffered injury which was attributable to Prolift is one thing. The more difficult question is whether she knew or ought to have known that she had had an injury that was attributable to a contravention of the Trade Practices Act. The answer to that question turns on when she first knew or ought to have known both that:
Prolift had a defect and/or was not reasonably fit for the purpose for which it was acquired and/or was not of a merchantable quality; and
that she had suffered injury for that reason.
4652 In considering this question it is necessary to bear in mind the statutory meaning of goods with a “defect” (that the safety of the goods is not such as persons generally are entitled to expect) and the reason I found Prolift to be defective (because it could cause significant side-effects in respect of which the respondents had provided inadequate warnings and misleading information).
4653 As I mentioned earlier, Mrs Gill deposed that it was not until she saw Dr Dowling in 2013 that she first suspected that there was a defect in the Prolift mesh implant and that the problems she had experienced since 2007 were not her fault.3825 It seems that it was following that consultation that she first sought legal advice. In her first affidavit she stated:
With the deterioration of my symptoms, and based on what I had been told by Drs Seabrook and Dowling, as well as from my own searches on the internet, I started becoming increasingly anxious that I might be one of the women who had-been inserted with a defective mesh implant and that my problems might be long term. However, I still hoped that further revision surgery would finally resolve my problems.
At some stage in July 2013, I spoke with a lawyer in the Geelong office of Maurice Blackburn who was acting for my husband in relation to his accident that had occurred in 2010. I asked about mesh litigation as I had read something about it on the computer.
The solicitor provided me with paperwork to fill out and submit to Maurice Blackburn. I then became aware of the class action against the respondents, and I signed up with Maurice Blackburn and became a group member in these proceedings.
I did not know that I had sustained injuries that were related to Prolift mesh until around July 2013. While I knew that the mesh had caused erosions in 2007 and 2008, I did not make the link that the erosions and other complications I had suffered were likely to be due to a problem with the mesh until I went to Dr Seabrook in May 2013 and then saw Dr Dowling in July 2013.3826
4654 When she felt the mesh tearing her in May or June 2007, she stated:
I did not understand what was happening to me, and I did not know whether it was my fault or not. I assumed that it must have been my fault, and that I must not have been properly following my post-operative instructions. I made that assumption because I went into the surgery believing that if I rested after the surgery I would have a good recovery and I would be fine. Because I was still having problems, I reasoned that I must have done something wrong, although I did not know what that might have been. It was very confusing, disappointing and upsetting for me.3827
4655 After her surgery in 2007 she continued to worry that it was her fault that the mesh had eroded in the first place.3828
4656 Her subsequent conduct, including the time she sought legal advice, is consistent with this account. There is also some support for it in Dr Leake’s affidavit. Dr Leake deposed:
I do not recall providing any advice to Kathryn that her mesh erosions or pelvic pain were caused by a defect or fault with the Prolift. I did not provide advice about the cause of the tension or why it was responsible for her pain and erosions. I did not tell her that her problems were caused by incompetent surgery because I do not and never had formed that view. Based on my usual practice now, and throughout the time she consulted me, if I had provided such advice about the cause of her symptoms or whether there was a defect with the Prolift, I would have recorded it in my notes and included it in the letter I sent to the referring doctor. There are no records of my providing such advice. I therefore believe I did not give it to Kathryn.3829
4657 Moreover, there is no evidence that any other doctor gave her such advice before she consulted solicitors in 2013. If such advice had been given, it is reasonable to expect that there would have been some record of it. None of the contemporaneous medical records indicate that she had been told that Prolift was unsafe or that the advice provided by the respondents about its side-effects was deficient in any way.
4658 There is no evidence that Mrs Gill understood that mesh exposure signified that the mesh was defective, not reasonably fit for the purpose for which it was acquired or of unmerchantable quality. She was unaware of any recurrence of prolapse until 2016. The warning she said she had been given was that some women’s bodies reject the implant and that that can cause an erosion.3830 She said she was given the impression that erosions were uncommon minor complications that could be easily corrected and would be short-lived. I accept this evidence. It is consistent with the information provided by the respondents at the time. I cannot see how that knowledge would reasonably signify to an uninformed layperson that the device was defective, not reasonably fit for purpose, or of unmerchantable quality, let alone convey that the safety of the device was unsafe for the reasons pleaded and proved by the applicants.
4659 Mrs Gill’s evidence was that, until she learned from Dr Dowling in 2013 that other women were having problems and that Prolift had been withdrawn from sale, she did not know that there was anything wrong with Prolift. I accept that evidence. There is no evidence to indicate that she was aware at any earlier time that Prolift had a number of risks about which the respondents had not warned or had provided inadequate warning or, indeed, that there was any deficiency in any of the information they had provided about the device. It seems that, until she had the conversation with Dr Dowling, she believed that the fault lay elsewhere, probably with her. The first reference I have been able to find in the contemporaneous evidence to anything suggesting that Mrs Gill was aware that Prolift was defective is in the report of Dr Helen Handsjuk, six months after the consultation with Dr Dowling and after Mrs Gill had seen a lawyer. Dr Handsjuk’s report indicates that she took a history from Mrs Gill that “[a] faulty mesh during the first operation, resulted in several other corrective operations and will necessitate future operations as the mesh erodes and protrudes, causing pain and discomfort”.3831
4660 I conclude that Mrs Gill did not know that Prolift was defective until about June or July 2013. By that time, this proceeding had already commenced. Although she was not then an applicant, she was a group member.
4661 The next question is whether Mrs Gill should have known these things before then and, if so, when.
4662 To answer this question it is necessary to inquire into what steps Mrs Gill ought reasonably to have taken to ascertain that she had sustained an injury that was attributable to a contravention of the TPA.
4663 When Mrs Gill first saw Dr Yin, Dr Yin suspected she might have a mesh erosion but she also thought she might have an ongoing infection. The report she sent to Dr Eranki did not include an opinion. She records against a heading “Impression”:
? Mesh erosion
? Ongoing infection3832
4664 Be that as it may, Mrs Gill underwent a cystoscopy later that day which confirmed the presence of eroded mesh and in her affidavit she stated that Dr Yin told her “I can confirm you have a mesh erosion”.3833
4665 Given that the respondents had warned that Prolift could cause an erosion, the fact that she knew the mesh had eroded does not establish that she knew that her injury was attributable to a contravention of the TPA. It will be recalled that Mrs Gill’s evidence was that it was the information Dr Dowling gave her that made her think that there was “some defect” with the Prolift implant and that it was not her fault that she had experienced the problems she had had since 2007. This evidence was not challenged in cross-examination, at least not directly. It was only after her conversations with Dr Seabrook and Dr Dowling in 2013 that Mrs Gill conducted searches on the internet and obtained legal advice. It was never suggested to Mrs Gill that she should have taken any steps at an earlier time to discover whether she had been injured because of a defect with Prolift or because Prolift was not reasonably fit for purpose or was of unmerchantable quality.
4666 In these circumstances, and despite my misgivings about other aspects of her evidence, I am not satisfied that before this proceeding commenced Mrs Gill knew or ought to have known that she had been injured because of a contravention of the Trade Practices Act.
4667 It follows that the limitation defence is not made out. I find that the proceedings were not commenced more than three years after the date of discoverability, that is more than three years from the time Mrs Gill knew or ought to have known that she had been injured, that the injury was significant enough to justify bringing an action, and that the injury was attributable to a contravention of the TPA.
Ann Sanders
4668 As Mrs Sanders was implanted with TVT before the commencement of the 2004 Amendment Act, her claims are subject to the limitation provisions in Pt V of the TPA.
4669 The respondents raised a number of limitations issues against Mrs Sanders in their submissions.
4670 They argued that her claim under s 75AD was statute-barred because it was required to be commenced within 10 years of supply by the manufacturer.
4671 In respect of her claims under ss 74B and 74D, they submitted that:
the defence in s 74J(3) applied because her action was commenced 10 years after supply; and
her claim was subject to the requirement in s 74J(1) that a claim be commenced within three years of the cause of action having accrued.
4672 They also argued that her misleading and deceptive conduct claim was statute-barred because it had to be commenced within six years of the accrual of the cause of action.
4673 I deal with each of these arguments below.
4674 The respondents argued that, pursuant to ss 74J(3) and 75AO(2), Mrs Sanders’ claims under ss 74B, 74D and 75AD must fail because this proceeding was commenced more than 10 years after she was supplied with TVT.3834
4675 In the light of the Full Court’s reasoning in Ethicon at [51]–[52], in Mrs Sanders’ case, the date the proceeding relevantly commenced is the date on which women who had been implanted with TVT were added as group members. That was 29 January 2013, when the amended statement of claim was filed.
4676 For the following reasons, I reject the respondents’ argument that Mrs Sanders’ action is statute-barred under ss 74J(3) and 75AO(2).
4677 The point was not pleaded against her. Indeed, no application was ever made for leave to amend the defence to enable the respondents to rely on it. When these matters were put against them, the respondents contended that they were not obliged to plead the point. I cannot agree.
4678 It is common ground that, if ss 74J(3) and 75AO(2) bar the remedy rather than extinguish the right (the cause of action), the point had to be pleaded and, if it is not pleaded, it does not arise for consideration; it is said to have been waived: Commonwealth v Verwayen (1990) 170 CLR 394 at 456 (Dawson J). A limitation provision that bars the remedy and does not extinguish the right, as Cooper J observed in Commonwealth of Australia v Mewett (1995) 59 FCR 391 at 398, is irrelevant; it does not arise for consideration “until such time as a defendant raises the plea in bar to the remedy”.
4679 The respondents argued that, on their proper construction, both provisions are substantive and operate not merely to bar the remedy but also the right, and therefore extinguish the causes of action covered by Pt V Div 2A (which relevantly includes ss 74B and 74D), as well as Pt VA (which includes s 75AD). For this reason, they maintained this was a matter that they did not have to plead.
4680 In Carey-Hazell v Getz Bros & Co (Aust) Pty Ltd (2001) 112 FCR 336 (Carey-Hazell (2001)) at [36], however, French J held that, properly construed, s 75AO(1) merely bars the remedy and does not extinguish the right. He added that it was “also doubtful whether the ‘long-stop’ limitations in ss 74J(3) and 75AO(2) have an extinguishing effect”.
4681 In Carey-Hazell (2001) French J was dealing with an application for summary dismissal. It will be recalled that the case concerned a claim for damages for injuries alleged to have resulted from a thromboembolism associated with the implantation of a prosthetic mitral valve. The applicant claimed that the valve was not reasonably fit for its purpose within the meaning of s 74B of the Trade Practices Act and had a defect within the meaning of s 75AC of the Act, giving her a right of action under ss 74B and 75AD. The respondent to the proceeding, and the applicant for summary dismissal, was the importer and supplier of the valve. The application was brought before a defence had been filed.
4682 At [35] French J said:
The three year limitation period imposed by s 74J expressly assumes the existence of a cause of action under Div 2A of Pt V of the Act. It defines the time limited by reference to the day “on which the cause of action accrued”. Section 75AO(1) does not use that explicit terminology but like s 74J has a facultative operation providing that “a person may commence a liability action at any time within 3 years”. In each case the liability for compensation is defined in distinct provisions without reference to the time limitation — the relevant liability-creating provisions in this case being s 74B and 75AD. Neither of the limitation provisions, s 74J or 75AO, in terms makes compliance with the time limit an element of the cause of action to which it applies. The expiry of the time limit is not said to extinguish the cause of action. And the matters of awareness or constructive awareness addressed by these provisions are not logically congruent with the matters constituting the elements of the cause of action. In each case the expiry of the time limit leaves the cause of action intact.
4683 At [36] his Honour observed that in White v Eurocycle Pty Ltd (1995) 64 SASR 461 at 467–470 Duggan J, with whom King CJ and Nyland J agreed, had reached the same conclusion about the effect of s 74J and said that, not only was that conclusion correct, but it also applied to s 75AO. It was at this point that French J expressed his view about s 75AO(2). His Honour noted that the question of whether s 75AO(2) had the effect of extinguishing the cause of action did not arise in the case before him “save to illustrate … the general point that it is a matter of statutory construction”, contrasting s 75AO(2) with s 11A(3) of the UK Consumer Protection Act, which expressly extinguishes the right of action. Section 11A(3) of the UK Consumer Protection Act provides that an action for damages under Pt 1 “shall not be brought after the expiration of the period of ten years from the relevant time …; and this subsection shall operate to extinguish a right of action and shall do so whether or not that right of action had accrued, or time under [certain other provisions of the Act] had begun to run, at the end of the said period of ten years”.
4684 The respondents argued that the “passing observations” of French J about both ss 74J(3) and 75AO(2) were of no assistance because the summary dismissal application in Carey-Hazell (2001) was based on ss 74(1) and 75AO(1) and the differences in the language of ss 75AO(2) and 11A(3) of the UK Consumer Protection Act are not decisive in the construction of the Australian provision. They also relied upon Menzies v Paccar Financial Pty Limited [2016] FCA 400 which deals with s 143(2) of the ACL. Section 143 of the ACL, although expressed differently, is to the same effect as s 75AO.
4685 In holding in White v Eurocycle that the limitation period imposed by s 74J was procedural in character, Duggan J observed at 467 that:
The limitation periods for various types of action are grouped in a section of the Act separate from those which allow the rights of action. The actions are based on a variety of different circumstances which might arise between manufacturers, sellers, consumers and importers. Section 74J appears under the heading “Time for commencing actions”. The language of the limitation provisions is typical of that used in the usual statute of limitations of a general character.
4686 Like Carey-Hazell (2001), Menzies was also an application for summary dismissal. This time the application was successful. The background to the application is complicated and need not be repeated here. Various matters were pleaded. The relevant part of the claim brought by Mr and Mrs Menzies concerned an allegation about safety defects in prime movers sold by the respondents and manufactured by the first respondent. Nicholas J said at [67] that it was “apparent that s 143(2) is intended to provide a manufacturer with a “long-stop” limitation defence with the 10 year long-stop limitation period being calculated from the date upon which the manufacturer first supplies the defective goods”. His Honour said that that was “confirmed” by the Explanatory Memorandum to the Trade Practices Amendment (Australian Consumer Law) Bill (No 2) 2010 (Cth), referring to [12.47] and [12.49]. His Honour proceeded to hold at [68] that, since the proceeding was commenced 12 years after the year in which the prime movers were supplied, the Menzies’ action under s 138(1) of the ACL was not brought within the time prescribed by s 143(2) and was out of time.
4687 Menzies does not assist the respondents. Nicholas J did not consider whether s 143(2) was intended to extinguish the right or just the remedy. The Explanatory Memorandum is silent on this question. His Honour referred to no authorities and it is not apparent that he was referred to any. I note that the applicants were self-represented.
4688 The difference in the terms of the Australian and UK provisions is certainly not decisive and the remark French J made about s 75AO(2) in Carey-Hazell (2001) was both obiter and tentative. Still, there is much to be said in its favour.
4689 The principles of statutory construction are not in doubt. The respondents referred to the passage in the joint judgment of Kiefel CJ, Nettle and Gordon JJ in SZTAL v Minister for Immigration and Border Protection [2017] HCA 34; (2017) 347 ALR 405; (2017) 91 ALJR 936 at [14]:
The starting point for the ascertainment of the meaning of a statutory provision is the text of the statute whilst, at the same time, regard is had to its context and purpose. Context should be regarded at this first stage and not at some later stage and it should be regarded in its widest sense. This is not to deny the importance of the natural and ordinary meaning of a word, namely how it is ordinarily understood in discourse, to the process of construction. Considerations of context and purpose simply recognise that, understood in its statutory, historical or other context, some other meaning of a word may be suggested, and so too, if its ordinary meaning is not consistent with the statutory purpose, that meaning must be rejected.
4690 Neither subsection of s 75AO is concerned with a substantive right. They are both procedural in nature and properly characterised as limitation provisions since they impose a time limit on the commencement of proceedings to enforce rights. Read together, they limit the period during which an action may be commenced to 10 years from the time the applicant became aware, or ought reasonably to have become aware, of the alleged loss, the defect, and the identity of the manufacturer. The substantive right or cause of action is conferred by s 75AD. It is not an element of the cause of action that it be commenced within 10 years of supply.
4691 Similarly, none of the subsections of s 74J is concerned with a substantive right. They are also procedural in character. The substantive rights or causes of actions are conferred by the antecedent sections of Div 2A, including, relevantly ss 74B and 74D. If anything, the argument is stronger in relation to s 74J(3) because it provides that it is a defence to an action under Div 2A if the defendant proves that the action was not commenced within 10 years of the time of first supply. As non-commencement within 10 years of supply is characterised as a defence, it could not reasonably be said to be an element of the cause of action.
4692 The observations of Duggan J in White v Eurocycle apply equally to s 75AO(2). These are important contextual matters. The limitation period for the purpose of a defective goods claim appears in a separate section from that which allows the right of action and the heading to s 75AO, like the heading to s 74J, is “Time for commencing actions”. Moreover, the terms in which s 75AO(1) is couched are relevantly identical to s 74J(1). While s 74J(3) and s 75AO(2) are expressed differently, the language used in s 75AO(2) is redolent of the language used in many limitation provisions in statutes of limitations of a general character. In particular, it is similar to s 38(1)(c)(v) of the Limitation Act 1935 (WA) and s 5(1) of the Limitation of Actions Act 1958 (Vic), both of which use mandatory language. The former provides that certain actions “shall and may be commenced” within six years from the date on which the cause of action accrues. The latter that certain actions “shall not be brought after the expiration of six years from the date on which the cause of action accrued”. In both cases the defendant is required to plead and prove the defence. In all likelihood the differences between ss 74J(3) and 75AO(2) can be explained by the differences in origin of the different Parts of the Act. They were not the result of any deliberate decision either to extinguish rights or alter the onus of proof. Had that been the case, one might have expected an indication to that effect in the Explanatory Memorandum to the Trade Practices Amendment Bill 1992. But there is no such indication.
4693 Furthermore, had the Parliament intended to extinguish the right, it could easily have said so, as the UK Parliament did. After all, it has done so in other cases. Section 34 of the Civil Aviation (Carriers’ Liability) Act 1959 (Cth) is a case in point. It provides that “[t]he right of a person to damages under this Part is extinguished if an action is not brought by him or her for his or her benefit within two years after …”.
4694 I accept that the omission of a reference to extinguishment is not conclusive. Even so, the only material difference between subss 75AO(1) and 75AO(2) is the substitution of the imperative auxiliary verb “must” for the facultative “may”. On one view the difference is an important one. The respondents argued, in effect, that it was decisive. On its face, they submitted, subs 75AO(2) indicates a legislative intention to extinguish the right rather than bar the remedy.3835
4695 The respondents acknowledged that, generally speaking, a limitation provision that assumes an entitlement to take a proceeding (in this case by the use of the words “defective goods action”) is “indicative of a legislative intention to bar the remedy and not extinguish the right” but they argued that the context indicates otherwise in the present case.
4696 I am not persuaded by this argument.
4697 In Verwayen, the High Court treated a similarly worded provision in the Limitation of Actions Act 1958 (Vic) as barring the remedy and not extinguishing the right. In my opinion there is nothing in the terms of s 75AO or its statutory or historical context which points in the opposite direction.
4698 Verwayen involved one of the numerous actions for damages for personal injuries resulting from the 1964 collision between the Royal Australian naval vessels, HMAS Voyager and HMAS Melbourne. For policy reasons the Commonwealth did not contest liability or plead a limitation defence, although the injuries to the plaintiff occurred at the time of the collision and the Limitation of Actions Act 1958 (Vic) imposed a three year limitation period. But the policy changed in 1985 and the following year the Commonwealth successfully applied for leave to amend its defence in order to put liability in issue and to plead that the action was statute-barred. Consequently, the plaintiff filed a reply in which, amongst other things, he pleaded that the Commonwealth had waived the limitation defence and was estopped from relying on it. By a majority (Mason CJ, Brennan and McHugh JJ dissenting) the High Court held that the Commonwealth was not free to dispute liability. Deane and Dawson JJ held that the Commonwealth was estopped from doing so, while Toohey and Gaudron JJ held that it had waived its right to rely on either defence. Despite their differences, all members of the Court were of the same opinion about the nature of the statutory bar to commencing proceedings.
4699 The relevant provision in Verwayen was s 5(6). It, too, used mandatory language:
No action for damages for negligence…, where the damages claimed by the plaintiff consist of or include damages in respect of personal injuries to any person, shall be brought after the expiration of three years after the cause of action accrued.
(Emphasis added)
4700 Mason CJ observed at 405 that, although the terms of the subsection might suggest that satisfaction of the time condition goes to jurisdiction, similarly expressed limitation provisions have been held not to limit the jurisdiction but to bar the remedy and not the right, creating a defence to the action which must be pleaded. No member of the Court considered that s 5(6) should be regarded any differently. Mason CJ said at 404–405:
In this case there is the public policy that there should be finality in civil litigation. However, the Parliament has seen fit to implement this policy, not by imposing a jurisdictional restriction, but by conferring on defendants a right to plead as a defence the expiry of the relevant time period. In these circumstances and having regard to the nature of the statutory defence, I conclude that the purpose of the statute is to confer a benefit upon persons as individuals rather than to meet some public need which must be satisfied to the exclusion of the right of access of individuals to the courts. On that basis, it is possible to “contract out” of the statutory provisions, and it is equally possible to deprive them of effect by other means such as waiver. Put differently, the provisions are procedural rather than substantive in nature, which suggests that they are capable of waiver …
See also: Brennan J at 425–426, Gaudron J at 486, McHugh J at 497, Toohey J at 473–474.
4701 At 497 McHugh J described s 5 as “a true statute of limitation”, distinguishing it from a limitation period annexed by a statute to a right which it creates. “In the latter class of case”, his Honour explained, “the limitation period will generally be of the essence of the right”. A true statute of limitation is “a plea in confession and avoidance of that right and not a condition precedent to its exercise”.
4702 In Mewett at 534, citing Verwayen at 473–474, Gummow and Kirby JJ observed that:
First, a statutory bar, at least in the case of a statute of limitations in the traditional form, does not go to the jurisdiction of the court to entertain the claim but to the remedy available and hence to the defences which may be pleaded. The cause of action has not been extinguished. Absent an appropriate plea, the matter of the statutory bar does not arise for the consideration of the court. This is so at least where the limitation period is not annexed by statute to a right which it creates so as to be of the essence of that right. Secondly, in the circumstances the defendant may be estopped from pleading the statutory bar or otherwise be deemed to have waived the right to do so.
4703 The same approach was taken to s 36(1) of the Limitations of Actions Act 1936 (SA) in McKain v R W Miller & Company (South Australia) Pty Limited (1991) 174 CLR 1. That section, which the majority held was procedural in nature and barred the remedy but not the right, provided that “all actions in which the damages claimed consist of or include damages in respect of personal injuries to any person, shall be commenced within three years next after the cause of action accrued but not after”. I referred above to similarly worded provisions in the Limitation of Actions Act 1958 (Vic) and the Limitation Act 1935 (WA), which the courts in Pullen and Cigna held considered barred only the remedy and not the right.
4704 In my view ss 75AO(2) and 74J(3) operate in the same way. The provisions are not relevantly different in their terms and serve the same purpose. The limitation period in s 75AO(2) is not annexed by the TPA to the right created by s 75AD or to an incident of that right. Nor is the limitation period in s 74J(3) annexed by the TPA to the rights created by ss 74B and/or 74D. Having regard to the way in which these provisions are expressed, they are procedural in nature and are conditions of the remedies and not the rights. It does not matter that the time limits are imposed by the same statute that creates the causes of action: Australian Iron & Steel Ltd v Hoogland (1962) 108 CLR 471 at 488–489 (Windeyer J).
4705 Regardless, the point should have been raised in the defence. Trial by ambush is a thing of the past.
4706 In Verwayen at 482, Gaudron J remarked that, if a person in the course of litigation fails to plead a matter, take an available objection or pursue a particular point of law, the case proceeds on the basis that the point which could have been taken is not an issue. Her Honour added that “were it otherwise the conduct of litigation would be unmanageable”. At 483, she noted that a party who fails to take an objection that a condition attaching to the exercise of jurisdiction has not been satisfied may be precluded as a result of its participation in the proceedings from later raising the defect.
4707 Moreover, rule 16.08 of the Federal Court Rules provides that:
In a pleading subsequent to a statement of claim, a party must expressly plead a matter of fact or point of law that:
(a) raises an issue not arising out of the earlier pleading;
(b) if not expressly pleaded, might take another party by surprise if later pleaded; or
(c) the party alleges makes another party’s claim or defence not maintainable.
4708 The point the respondents raise is a point of law that they allege makes Mrs Sanders’ claim under s 75AD “not maintainable”. Moreover, it is a point of law which, if it were not expressly pleaded, might take an applicant by surprise if pleaded later as it plainly did in the present case. If the respondents intended to raise it, it had to be pleaded.
4709 In White v Overland [2001] FCA 1333; (2001) 67 ALD 731 at [4] Allsop J (as the Chief Justice then was) made the following pertinent observation. Any attempt at paraphrase would detract from its potency:
[I]n the efficient and proper conduct of civil litigation, even civil litigation hard fought between parties, it should always be recognised that in the propounding of issues for trial the parties should take steps to ensure that all relevant parties to the dispute are cognisant of what the issues are. Any practice of quietly leaving footprints in correspondence or directions hearings to be uncovered some time later in an attempt to reveal that a matter was always in issue should be discouraged firmly. Even if something has been said, where it is evident, or indeed suspected, that the other side is proceeding on the basis of a misconception or has not appreciated something, as a general rule, efficiency, common sense and an appreciation of the costs and resources (both public and private) likely to be wasted by confusion in litigation will mandate that a party through his or her representative ensure that the other is not proceeding on a misconception or that the other does appreciate something that has been said. Litigation is not a game. It is a costly and stressful, though necessary, evil. To paraphrase Roscoe Pound from “The Causes of Popular Dissatisfaction with the Administration of Justice” (1906) 29 ABA Rep 395, 404-406, the “sporting theory of justice” and any behavioural manifestation of it should be seen as a survival, or better, a relic, of the days when a lawsuit was a fight between two clans: cf Jackamara v Krakouer (1998) 195 CLR 516 at 526-527 per Gummow and Hayne JJ. Representatives do not owe duties to the other side’s client. They owe duties to their own client. But no one’s interests are advanced by litigation proceeding on assumptions which are seen or suspected to be false. This is very much the case when an issue, if it is to be propounded, might endanger the instructions of those acting for the other side. In saying this I need make no reference to the well-known responsibility of the Crown and emanations of the Crown to act at all times as model litigants beyond referring to what was said by the Full Court of this Court in Scott v Handley [1999] FCA 404 at [43] ff. I would expect no less than that which I have indicated of bitterly competitive commercial parties in the hardest fought of cases. In the long run, the only consequence of keeping issues hidden or not clearly identifying them is to disrupt the business of the court leading to the waste of valuable public resources and to lead to the incurring of unnecessary costs by the parties, costs which ultimately have to be borne by someone.
4710 To similar effect, in Nowlan v Marson Transport Pty Ltd (2001) 53 NSWLR 116 at 128 Heydon JA deprecated the conduct of litigation “as if it were a card game in which opponents never see some of each other’s cards until the last moment”. His Honour said that conduct of this kind is “out of line with modern trends”.
4711 More than that, for the reasons given by Allsop J in White v Overland, raising this point at the heel of the hunt, so to speak, and not in the defence is contrary to the terms of Pt VB of the FCA Act which require parties to conduct proceedings in the Court in a way that is consistent with “the overarching purpose of the civil practice and procedure provisions”, which include the Rules: FCA Act, s 37M. “The overarching purpose” of those provisions is defined in s 37M(1). It is to facilitate the just resolution of disputes according to law and as quickly, inexpensively and efficiently as possible. Since the Rules must be interpreted and applied in the way that best promotes the overarching purpose (FCA Act, s 37M(3)), r 16.08 should be construed so as to require a party who wishes to rely on a long-stop provision to plead it.
4712 In supplementary submissions lodged after their case had closed, extensive written submissions had been filed, and two weeks of oral argument had taken place, the respondents claimed in the alternative that the point raised by s 75AO(2) had indeed been pleaded.
4713 I reject that claim.
4714 It is more difficult than it should be to explain why that is so because of the way the respondents amended their pleadings over time. Standard practice (not to mention common sense) dictates that practitioners ensure paragraph numbers remain consistent across amended pleadings. This was not done in the present case. As a result, the substance of particular paragraphs and any amendments to them are found in different paragraph numbers across the various versions of the defence.
4715 The three applicants became parties to the proceeding on 6 April 2016, when the Third Further Amended Originating Application and Statement of Claim were filed. On 2 May 2016 the respondents filed an amended defence.
4716 In their Defence to the Third Further Amended Statement of Claim, the respondents did not raise the s 75AO(2) point against Mrs Sanders. Rather, they alleged at (then) paragraph [87]:
Further, and in answer to Mrs Sanders’ claim for compensation under the TPA and CCA (which is not admitted), the Respondents state that:
(a) Mrs Sanders’ alleged cause of action accrued more than 6 years before the commencement of these proceedings; and
(b) pursuant to section 87F of the TPA/CCA, her cause of action is statute barred.
4717 The defence was amended again on 7 July 2017 in response to the Fourth Further Amended Originating Application and Statement of Claim filed on 31 May 2017, in which the applicants added their misleading or deceptive conduct claim. Once again, however, the s 75AO(2) point was not raised. On this occasion the respondents merely added a reference to s 82 of the TPA to subparagraph [91(b)] (the corresponding subparagraph in the previous version of the defence being subparagraph [87(b)].
4718 In Gill (No 1), I allowed an application by the applicants for leave to file a Fifth Further Amended Originating Application and Statement of Claim. The effect of the amendments was to expand the group members (pursuant to s 33K of the FCA Act), to enable the applicants to seek declaratory and injunctive relief with respect to their misleading or deceptive conduct claim, and to add the allegation about the failure to provide information or warnings concerning the potential immune response to the Ethicon devices.
4719 On 12 April 2018, I ordered that the applicants file the documents by 13 April. I also ordered that within seven days thereafter the respondents file and serve any defence to the Fifth Further Amended Statement of Claim. The respondents filed their amended defence on 20 April 2018. The limitation defence pleaded against Mrs Sanders was unaltered.
4720 Before the respondents filed the Defence to the Fifth Further Amended Statement of Claim, paragraph [87] of their defence read as follows:
Further, pending receipt of further particulars of the Group Members’ claims, in answer to the allegations pleaded in paragraphs 24 to 39 and 59 to 81 of the 3FASOC 4FASOC, the Respondents state that the Group Members’ causes of action will be subject to the limitation periods prescribed by the:
(a) Limitation Act 1969 (NSW);
(b) Limitation of Actions Act 1958 (Vic);
(c) Limitation of Actions Act 1974 (QLD);
(d) Limitation Act 2005 (WA);
(e) Limitation Act 1935 (WA);
(t) Limitation Act 1985 (ACT);
(g) Limitation Act 1974 (TAS);
(h) Limitation of Actions Act 1936 (SA);
(i) Limitation Act 1981 (NT);
(j) Trade Practices Act 1974 (Cth); and
(k) Competition and Consumer Act 2010 (Cth).
4721 This paragraph was previously paragraph [83] of the Defence to the Third Further Amended Statement of Claim.
4722 In the Defence to the Fifth Further Amended Statement of Claim, the respondents inserted in subparagraphs [87(j)] and [87(k)] references to specific provisions of the two Acts. Subparagraph (j) read, with the amendment underlined:
Trade Practices Act 1974 (Cth); including ss 74J(1) and (3), 75AO(1) and (2), 82(2), 87F, 87G and 87H[.]
4723 The respondents’ supplementary submissions on the limitation questions were filed on 4 May 2018. Those submissions included the following contentions at [30]–[31]:
If the Court concludes that s 75AO(2) is a procedural provision, it has been pleaded at [87(j)] of the Defence and operates as a complete defence to all group members’ cause of action pursuant to s 75AD of Part VA of the TPA and section 138 of the ACL based on the TVT Classic. As Mrs Sanders is a group member [87(j)] applies to Mrs Sanders.
If the Court concludes it was necessary in relation to Mrs Sanders for the respondents to plead that provision not only through [87] but also in [91] of its Defence, no prejudice would now be suffered for the Defence to be so amended in light of the limitation being specifically pleaded against all group members in [87(j)] and given the respondents’ objection to Mrs Sanders’ common law claim as statute barred pursuant to s 38(1)(c)(vi) of the Limitation Act 1935 (WA)).: see submissions made by respondents dated 30 March 2016.
4724 I do not accept these submissions. By no stretch of the imagination could this be said to raise an allegation against Mrs Sanders that her claim was statute-barred by ss 74J(3), 75AO(2), or at all.
4725 First, the allegation that her claim was statute-barred was made in [91] of the defence and that paragraph did not refer to s 74J(3) or s 75AO(2) either in substance or in form. It merely alleged that her alleged cause of action accrued more than six years before the commencement of the proceeding and that her claim was statute-barred under ss 82 and 87F of the “TPA/CCA”.
4726 Second, Mrs Sanders did not fall within the chapeau of [87] of the Defence to the Fourth Further Amended Statement of Claim because she was not a group member. A “group member” is “a member of a group of persons on whose behalf a representative proceeding has been commenced”. Once Mrs Sanders became an applicant she ceased to be a “group member”. Rather, as a person who had commenced a representative proceeding, she was a “representative party”: FCA Act, s 33A.
4727 Third, in any event, a statement that the group members’ cause of action “will be subject” to limitation provisions in various statutes is not a plea that a party’s claim is defeated by a limitation period.
4728 Fourth, it is impossible to see how the respondents’ objection to Mrs Sanders’ common law claim as statute-barred pursuant to s 38(1)(c)(vi) of the Limitation Act 1935 (WA) would alleviate any prejudice to Mrs Sanders or put her on notice that the respondents were relying on s 75AO(2) of the Trade Practices Act to defeat her claim under ss 75AD or 74J(3) to defeat her claims under ss 74B and 74D.
4729 The respondents also contended that Mrs Sanders’ claims under Pt V Div 2A were barred under s 74J(1) because they were brought more than three years after the causes of action accrued and that her claim under s 75AD was barred under s 75AO(1) because it was not brought within three years after she became aware, or ought reasonably to have become aware, of the alleged loss, the defect, and the identity of the manufacturer.
4730 These matters were also not raised in the respondents’ defence. Nevertheless, the applicants proceeded on the assumption that they were. Either that or they wrongly assumed that they had to prove that the cause of action was commenced in time, despite the fact that the respondents had not raised the limitation defence in their pleading and notwithstanding their later argument to the contrary. In contrast to the position they took about s 75AO(2), they did not submit that it was not open to the respondents to argue that the action was not brought within the relevant three year period because the matter was not raised in the respondents’ defence. To the contrary, they filed submissions addressing the question. Since the case was conducted in this way, no question of trial by ambush arises and, subject to a compelling argument to the contrary, I would be disposed to grant the respondents leave to amend their defence to plead that the actions under ss 74B, 74D and 75AD were brought outside the three year periods provided for under ss 74J(1) and 75AO(1) respectively.
4731 I turn first to the claims under ss 74B and 74D (the causes of action under Pt V Div 2A).
4732 Unlike s 82, these provisions defined when a cause of action for the purposes of the section accrues.
4733 Section 74J(2) relevantly provided:
For the purposes of this section, a cause of action shall be deemed to have accrued:
(a) in the case of an action other than an action under section 74H, on the day on which the consumer or a person who acquired the goods from, or derived title to the goods through or under, the consumer first became aware, or ought reasonably to have become aware:
(i) in the case of an action under section 74B—that the goods were not reasonably fit for the purpose referred to in that section;
…
(iii) in the case of an action under section 74D—that the goods were not of merchantable quality.
4734 As I mentioned earlier, s 75AO(1) relevantly provided that a person may commence a liability action at any time within 3 years after the time the person became aware, or ought reasonably to have become aware, of the alleged loss, the defect, and the identity of the person who manufactured the defective goods. In other words, for the purpose of the defective goods claim, time did not begin to run until the applicant became aware, or ought reasonably to have become aware, of all of these matters.
4735 Mrs Sanders’ counsel argued that it was likely that all these causes of action accrued after the excision surgery in August 2011 when Mrs Sanders was informed by Dr Daborn that “the problems caused by the TVT were likely to require more operations and might be permanent”.3836 They submitted that this was when Mrs Sanders began to realise that her symptoms of pain and urinary dysfunction were caused by the device and were not merely transitory and that, therefore, the device was defective, or not reasonably fit for purpose or of merchantable quality.3837 Since these proceedings were commenced in October 2012, it follows, they argued, that her action was within time. In fact, as I have already observed, while this proceeding was commenced in October 2012, since Mrs Sanders did not become a group member until the filing of the amended statement of claim on 29 January 2013, that is the date on which Mrs Sanders’ claim is taken to have commenced.
4736 The respondents, on the other hand, argued that all these causes of action are statue-barred because Mrs Sanders was experiencing urinary symptoms in 2003 and vaginal irritation and pain in 2008 and, whichever is the relevant date, she is out of time because she was then aware that the TVT device was not reasonably fit for purpose, not of merchantable quality, and defective.
4737 I will deal first with the arguments relating to the Pt V Div 2A causes of action.
4738 I have difficulties with both sides’ arguments.
4739 The applicants ignored the question of when Mrs Sanders ought reasonably to have become aware of the relevant matters. The respondents did not address the statutory criteria of awareness of the elements of the contraventions; their submissions were directed to awareness of symptoms. The two are by no means necessarily identical.
4740 The respondents first pointed to urinary symptoms Mrs Sanders experienced during two short periods in February 2002 and October 2003. In her first affidavit Mrs Sanders deposed:
Between 2001 and 2006 I continued to be happy with the results of the surgery and did not have any problems.
…
[O]n 19 and 26 February 2002, I believe I saw Dr Naunton at lnnaloo Shoppers Village and provided urine samples after these appointments. Later on 7 and 25 October 2003, I believe I saw Dr Naunton at lnnaloo Shoppers Village and provided further urine samples. While I do not remember these specific appointments, around 2002 to 2003, I recall having one or two appointments in relation to symptoms of urinary frequency. Dr Naunton told me that I had cystitis, a bladder infection. She prescribed some medication for it and the symptoms went away.3838
4741 Contrary to the advice Mrs Sanders said she received from Dr Naunton, however, her pathology results from October and November 2003 show no evidence of infection.3839
4742 There is no evidence to indicate that Mrs Sanders was aware or ought reasonably to have been aware that these symptoms were related to her TVT implant, let alone to a defective, unsuitable or unmerchantable device.
4743 There is no mention of urinary frequency in the contemporaneous medical records before 2013. But on her own account Mrs Sanders had experienced a variety of urinary symptoms since around 1993,3840 well before the TVT device was implanted. In any case, apart from these isolated episodes, Mrs Sanders’ evidence does not suggest that she experienced any further urinary frequency until 2007. The respondents did not provide any evidence to suggest that TVT was the cause of her symptoms in 2002 or 2003 and, for the following reasons, I am not satisfied that it was.
4744 First, Mrs Sanders’ account suggests that they were not.
4745 After her recovery from the surgery she reported an improvement in her urinary symptoms.3841 She said that between 2001 and 2006 she “continued to be happy with the results of the surgery and did not have any problems”.
4746 There was a distinction between the symptoms she experienced in 2007 and those she experienced in February 2002 and October 2003. In particular, she did not report that she had discomfort when urinating in 2002 or 2003.
4747 Second, the respondents’ medical evidence denies any connection. None of the experts stated that the two discrete episodes in February 2002 and October 2003 were related to the implantation of the TVT.
4748 Third, Professor Blaivas’s opinion was that “the erosion and subsequent urinary symptoms [and] recurrent UTIs” were caused by the TVT and the revision surgery.3842 It is clear from his report that the reference to “recurrent UTIs” excludes the episodes in 2002 and 2003 and refers to what Mrs Sanders reported occurring in 2007.3843 Professor Korda considered that Mrs Sanders’ urinary symptoms were attributable to urethral instability, which he said was caused by “the complications that have arisen from the erosion of the TVT tape”.3844
4749 It is more likely than not that the urinary symptoms Mrs Sanders described having from 2007 onwards were caused by erosion of the TVT tape and its sequelae, including its partial removal. I will say more about this in Part XVIII, when I look at the extent of Mrs Sanders’ injuries in the context of damages. No evidence was adduced pinpointing when the erosion first occurred. But it is common ground that erosions can occur well after implantation, indeed many years later. Here, for the following reasons, the evidence indicates that this was a late onset erosion.
4750 Fourth, Mrs Sanders had an excellent recovery from the surgery, her urinary symptoms improved, she resumed sexual intercourse within two months and, with the exception of the two episodes of urinary frequency in February 2002 and October 2003, she experienced no pain or other problems for years. Dr Naunton, who saw her at the time, did not give evidence and the medical records pertaining to the relevant examinations were not tendered. In these circumstances, it is difficult to place much weight on Mrs Sanders’ account. A pathology report from Western Diagnostic Pathology was in evidence which showed that urine was collected on 7 October 2003 but, as the respondents submitted,3845 the pathology tests showed no evidence of infection.3846
4751 Fifth, the apparent consensus of medical opinion is that Mrs Sanders had a late onset erosion. Professor Blaivas described it as such. Professor Roovers said that it was unlikely that the exposure had been present before 2007 since Mrs Sanders had undergone several pelvic examinations since the TVT was inserted (which did not detect it). Professor Deprest said that she developed “a symptomatic tape exposure” in about 2011, apparently discounting the significance of the recurrent urinary tract infections and urinary dysfunction commencing in 2007 and the onset of dyspareunia the following year. Associate Professor Rosamilia considered that the TVT was not the cause of the recurrent UTIs beginning in 2007 and did not suggest that the exposure, diagnosed in 2011, occurred soon after the surgery or earlier than 2007.
4752 Sixth, apart from the isolated episodes of urinary frequency in February 2002 and October 2003, Mrs Sanders described no physical changes in her body following her continence surgery until 2007 when she began to experience dysuria and urinary frequency. It is this time that the expert witnesses regard as the beginning of recurrent urinary tract infections and/or symptoms of urinary dysfunction, which the applicants' witnesses attributed to the TVT.
4753 The respondents then pointed to the symptoms of irritation and pain Mrs Sanders experienced in 2008. These symptoms undoubtedly were caused by TVT.
4754 There is no evidence, however, to indicate that before 2011 Mrs Sanders was aware that the TVT device was the cause of the urinary symptoms she had experienced since her incontinence surgery or of her pelvic pain and dyspareunia. In her first affidavit she said that Dr Tan was the first doctor to tell her that the TVT was the cause of her pain.3847 But when Mrs Sanders presented to her on 28 January 2011, Dr Tan believed that a ring pessary was causing her pain and this appears to be the only occasion on which Mrs Sanders saw Dr Tan. In additional oral evidence-in-chief, however, the following exchange occurred between Mrs Sanders and her counsel:
Well, I – I explained to her that I was in pain, and she said, “Would you mind if I examined you?” So I said, “Okay.” She examined me and then she brought out a medical chart, it was quite big, of what a lady’s inside would look like and she said, “This is where whatever it is you’ve had fitted should be, and this is where it is on you,” and she pointed out that it was way, way down.
And did you understand from that explanation that the cause of the pain was as a result of what the thing – the thing that had been fitted to you?---I did then. Yes. Did she – what word did she use to describe this thing that had been fitted inside you?---She didn’t actually say a lot. She just said, “From what you’ve told me about the pain you’re in, I will refer you back to King Edwards Hospital,” which she did. 3848
4755 In these circumstances the reference to “the TVT” at [59] of the affidavit should be taken to mean “the device fitted by Dr McNeill which I now know to be TVT”. It is reasonable to infer that at this point Mrs Sanders became aware that TVT was the cause of her pain. But there is no evidence to indicate that she knew then that the urinary symptoms had anything to do with the TVT.
4756 Given the information in the IFU, the information she was given before her consent was obtained, and the passage of time since the surgery, this is neither surprising nor unreasonable. Mrs Sanders had no reason to suspect that TVT might be implicated before the conversation with Dr Tan.
4757 Dr Howlett, who examined Mrs Sanders the same day as Dr Tan, realised that she did not have a ring pessary and that what was protruding was mesh.3849
4758 The evidence indicates that Dr Gilbert was the first medical practitioner she saw since her recovery from TVT surgery who recorded that she had been implanted with TVT. This was recorded in a consultation note dated 19 May 2011.3850 Dr Gilbert’s source is unknown.
4759 Mrs Sanders’ sworn evidence was that she did not then know that she had received a TVT device. I accept her evidence. It is certainly plausible and it is consistent with the contemporaneous records. She said that Dr Taylor did not tell her; he referred merely to a sling. That evidence is certainly credible. The consent form she signed in 2001 referred to “transvaginal tape” and in October 2000 and March 2001 there was more than one transvaginal tape on the market. Dr Matthews certainly knew that Dr Taylor was proposing to implant a “TVT mesh type sling”, for that is what Dr Taylor informed her in his letter of 17 October 2000. So it is possible that the information in Dr Gilbert’s possession had been obtained from Dr Matthews. As the operation in 2001 was performed at the same hospital, more likely than not the information came from the hospital records in which there are numerous references to TVT and to which Dr Gilbert would have had ready access.
4760 Based on this evidence I conclude that by late January 2011, although she did not know, Mrs Sanders ought reasonably to have known that she had a mesh erosion and that the erosion was the cause of her pain. That was the earliest time these causes of action could have accrued. Since her claim commenced within three years of that time, Mrs Sanders’ action is not barred by either s 74J(1) or s 75AO(1).
4761 The respondents also pleaded that Mrs Sanders’ action was statute-barred under s 82.
4762 Section 82(1), which appears in Pt VI of the TPA, relevantly provided that a person who suffers loss or damage by conduct of another person that was done in contravention of a provision of Pt V of the Act may recover the amount of the loss or damage by action against that other person or against any person involved in the contravention. Part V includes the prohibition of misleading or deceptive conduct in s 52. Putting the limitation defence to one side, it will be recalled that Mrs Sanders is the only one of the three applicants who can bring a claim for damages arising out of that contravention.
4763 Section 82(2) provided that an action under subsection (1) may be commenced at any time within six years after the day on which the cause of action that relates to the conduct accrues: see also Trade Practices Amendment Act (No 1) 2001 (Cth), Sch 1 cl 21(2).3851 Since Pt VI of the Act did not define when a cause of action accrues for the purpose of an action for damages under s 82, common law principles apply.
4764 So what are those principles?
4765 A cause of action consists of “the essential [factual] ingredients in the title to the right which it is proposed to enforce”: Williams v Milotin (1957) 97 CLR 465 at 474. Consequently, in the absence of any statutory provision to the contrary, a cause of action is complete and accrues when all the facts have occurred which the moving party must prove in order to succeed: AME Hospitals at [138] (Buss JA). Knowledge of the legal implications of those facts forms no part of the cause of action. As Wilson J observed in Do Carmo v Ford Excavations Pty Ltd (1984) 154 CLR 234 at 245, a person may know all the facts that need to be proved to make out a cause of action “but for want of taking legal advice may not know that those facts give rise to a right to relief”.
4766 Although it was not apparent from the pleading, it was clear from the written submissions that the section was pleaded against the claim under s 52 of the Act for misleading or deceptive conduct. The applicants argued that the relevant conduct occurred between 19 October 2000 when Dr Taylor advised Mrs Sanders to undergo continence surgery using TVT and 12 March 2001 when the surgery was performed.3852 The cause of action under s 82(1) was not complete, however, until damage caused by the misleading conduct first occurred: Wardley at 525.
4767 Damages under s 82 are recoverable for actual loss or damage incurred, not for potential or likely damage; prospective loss or damage is not enough: Wardley at 526–527. That means that damage must be “beyond what can be regarded as negligible” (Cartledge v E Jopling & Sons Ltd [1963] AC 758 at 772 per Lord Reid), “material” or more than de minimis (Cartledge at 779 per Lord Pearce) or, as Buss JA put it in AME Hospitals at [137], it must be “measurable”. The cause of action accrues, that is time begins to run, however, regardless of whether the injured person knew of the damage and even if it was incapable of being discovered: Cartledge at 771, 773–774, 775, 777). Cartledge has been followed in numerous cases in Australia: see, for example, Hawkins v Clayton (1988) 164 CLR 539 at 587–588; Do Carmo at 241 (Wilson J); and Alcan Gove Pty Ltd v Zabic (2015) 257 CLR 1 at [20]. There is no reason why the accrual of a cause of action for the purpose of s 82 should be approached in a different way.
4768 The respondents submitted that:
Relevantly, s 82(1) provided that the limitation period applies from “the day on which the cause of action that related to the conduct accrues”. Whilst the s 52 claim is not easily understood, it appears to be brought on the basis that Mrs Sanders would not have proceeded with the TVT Implant. If that is correct, the relevant loss or damage by the conduct is the implantation of the Tape Implant itself. As Mrs Sanders had surgery to implant the TVT Implant on 12 March 2001, the 6-year limitation period for her s 82 damages claim expired on 11 March 2007 and there is no power for the Court to otherwise ‘extend’ the applicable limitation period in s 82(2).3853
4769 The difficulty with this submission is that it proceeds from a false premise. The cause of action is created by s 82, not s 52. The implantation of the TVT device is not the relevant loss or damage and there is no evidence of measurable damage having occurred at the time of implantation. In the absence of any persuasive medical evidence to indicate that the TVT caused non-negligible, material or measurable damage any earlier, the first sign of damage did not occur until Mrs Sanders began to experience recurrent symptoms of urinary dysfunction. That was in 2007. The more difficult question is when in 2007.
4770 On the balance of probabilities it was sometime in January 2007. Mrs Sanders deposed that “[a]round the beginning of 2007” she began to experience some discomfort while urinating. She also said that “[i]n early 2007”, to relieve her symptoms, she bought around five boxes of medication from the pharmacy “before it stopped working”. 3854
4771 On 1 February 2007 she attended the Innaloo Medical Centre and told Dr Naunton that she had discomfort urinating and was urinating frequently.3855 It may be inferred that she suffered damage sometime before that appointment occurred during which time she was taking the medication she had purchased from the pharmacy.
4772 Mrs Sanders was not cross-examined on when exactly her symptoms began. There was no evidence regarding precisely how long she had experienced urinary frequency or discomfort before she visited Dr Naunton on 1 February 2007. It is therefore possible that her symptoms began on 30 or 31 January 2007. On balance, however, I think it unlikely. Based on her own evidence, I consider it more likely than not that she had experienced recurrent urinary dysfunction for more than a couple of days before her consultation with Dr Naunton and that she consulted him after the over the counter medication had “stopped working”.
4773 It follows that Mrs Sanders’ claim under s 52 is statute-barred.
4774 The respondents did not address this defence in their closing submissions or in oral argument. In the circumstances, I assume it is not pressed.
4775 In any case, it is entirely without merit.
4776 Section 87F, as I have already explained, was inserted into the Trade Practices Act by the 2004 Amendment Act.
4777 Clause 11 of Sch 1 provided that the amendments “apply to contraventions of Pt IVA, of Div 1A or 2A of Pt V or of Pt VA of the [Trade Practices Act] that occur after this Schedule commences”. In Mrs Sanders’ case the contraventions of Pt V Div 2A and of Pt VA occurred more than three years before Sch 1 commenced
4778 There is no law of the Commonwealth which fixes a limitation period for actions in negligence. Here, the relevant State laws are picked up and applied by s 79 of the Judiciary Act. As I observed in Part XIV, it was common ground that, following the judgment of the High Court in John Pfeiffer, that means that the laws that applied in the places where the torts occurred are the laws that govern the limitation questions.
The relevant legislation
4779 Section 14 of the Limitation Act 2005 (WA) (the 2005 Act) provides that a common law claim for damages for personal injury cannot be commenced if three years have elapsed since the cause of action accrued. But the Act applies only to causes of action that accrue on or after 15 November 2005, being the day the Act came into operation: 2005 Act, s 4(1). Its predecessor, the Limitation Act 1935 (WA) (the 1935 Act), allowed for actions in negligence to be brought within six years “after the cause of action”: see s 38(1)(c)(vi). In contrast with the 2005 Act, however, it made no provision for extensions of time in respect of injuries of the kind suffered by the applicants.
4780 The 1935 Act was repealed by s 4(1) of the Limitation Legislation Amendment and Repeal Act 2005 (WA) (Amendment and Repeal Act) but, by s 4(2) of the Amendment and Repeal Act, subject to some exceptions which are presently irrelevant, the 1935 Act continues to apply to causes of action that accrued before the commencement of the 2005 Act. Section 4(3) of the Amendment and Repeal Act, however, provides that s 4(2) is subject to ss 6 and 7 of the 2005 Act.
4781 Section 6 is presently relevant. Section 7 is not.
4782 Section 6 applies to personal injury actions. It provides that s 55 or s 56 of the 2005 Act, whichever is relevant, applies to ascertain the time of accrual of a cause of action relating to a personal injury and if, under the relevant section, the cause of action accrues before the commencement day (being 15 November 2005), then the applicable limitation period is that which would have applied before that day, even if that period has expired. Section 56 is irrelevant. It is concerned only with asbestos related diseases.
4783 Section 55 is the relevant section. It applies to all other personal injury cases. It reads:
(1) A cause of action for damages relating to personal injury accrues when the only or earlier of such of the following events as are applicable occurs—
(a) the person becomes aware that he or she sustained a not insignificant personal injury;
(b) the first symptom, clinical sign or other manifestation of personal injury consistent with the person having sustained a not insignificant personal injury.
(2) This section does not apply to a personal injury that is attributable to the inhalation of asbestos.
4784 “Personal injury” is defined in s 3(1) to include “a disease, impairment of a person’s physical condition, and mental disability”.
4785 “Not insignificant” means not trifling or not of no consequence: Thomas Peacock & Sons Pty Ltd v Abreu [2013] WASCA 19 at [40] (Pullin, Newnes and Murphy JJA).
4786 The effect of subsections 4(2) and 4(3) of the Amendment and Repeal Act is that the question of when a cause of action for damages accrues in a claim for damages for personal injuries made after the commencement of the 2005 Act is to be determined by s 5 of the 2005 Act. If the earlier of the events described in s 6(1) of the 2005 Act accrued before 15 November 2005, when the 2005 Act commenced, then the period of limitation set by the 1935 Act continues to apply. If it does not, then the limitation period under the 2005 Act applies.
4787 The respondents bear the onus of proving that the action cannot be commenced because the limitation period has expired: 2005 Act, s 79(1).
Kathryn Gill
4788 It is indisputable that Mrs Gill’s cause of action accrued after the commencement of the 2005 Act. But there is dispute about how long after that date it accrued.
4789 The respondents submitted that Mrs Gill was first aware that she had sustained a not insignificant personal injury by the time she underwent the mesh excision surgery in September 2007, if not before. Alternatively, they submitted that she was by that time aware that she had a symptom, clinical sign or other manifestation of a personal injury consistent with a not insignificant injury.3856 If not then, they contended that she was certainly aware of one or the other by August 2008, when her second mesh exposure was diagnosed and surgically removed.3857 Either way, her cause of action had accrued more than three years before these proceedings were instituted in October 2012.3858
4790 On the other hand, the applicants argued that Mrs Gill does not require an extension of time because the proceedings had already begun by the time she became aware that her symptoms were attributable to a not insignificant personal injury. They submitted that her cause of action first accrued in June or July 2013 because only then did she know that the original erosion and the first revision surgery were “symptoms of a not insignificant personal injury”.3859
4791 Putting to one side the description of the mesh exposure and the surgery as “symptoms” of an injury, a proposition which is open to question (indications of injury would be more accurate), these matters are beside the point.
4792 The applicants contended that s 55 of the 2005 Act does not contain any element of constructive awareness. Despite the presence of the word “aware” in paragraph (a) and its absence from paragraph (b), they submitted that both paragraphs required actual awareness. At first blush, the argument was troubling. But the applicants cited the observations of the WA Court of Appeal in Thomas Peacock at [32] and [40] where the Court said:
Section 14 of the Limitation Act … gives effect to a parliamentary intention that if an action for damages for personal injury is to be brought, it must be brought within a period of three years from the time at which the plaintiff first becomes aware that they have suffered a ‘not insignificant’ injury. The provision in s 55 as to the time at which the cause of action accrues is plainly intended to avoid the prospect of the limitation period expiring before a person is aware they have an injury of a nature which would justify a claim for damages.
…
It is plainly the case that the purpose of s 55 is to avoid time running while a person is unaware that he or she has suffered a ‘not insignificant’ personal injury; that is, an injury which is not ‘trifling’ or ‘of no consequence’: Macquarie Dictionary. Whether or not an injury is of such nature must depend upon the extent of the injury itself. The effect of s 55 is that the cause of action accrues when the person first becomes aware that they have suffered such a personal injury or there is some symptom, clinical sign or other manifestation consistent with such a personal injury. The relevant criterion is awareness, or means of awareness, of the existence of a personal injury which is not insignificant.
4793 They also argued that their construction was supported by a contextual analysis of the text and the extrinsic materials.
4794 The legislative history is discussed at length by Buss JA in AME Hospitals at [135]–[152].
4795 In short, under the 1935 Act a cause of action for damages for personal injury accrued at the time of the occurrence of a not insignificant injury, regardless of whether the injured person was aware she had been injured, and there was no provision for an extension of time. These were the mischiefs which the 2005 Act was intended to rectify.
4796 The process of reform took an inordinately long time. The adequacy of the State’s limitation laws was referred to the Western Australian Law Reform Commission in June 1982 and a report published four months later culminated in legislative amendments but those amendments only dealt with asbestos-related diseases. Ten years later, in 1992, the Commission released a discussion paper examining proposals for general amendment of the 1935 Act and in 1997 published a report recommending a suite of changes. In his reasons in AME Hospitals at [148], Buss JA referred to a discussion paper entitled “Limitations Law Reform” released to the public in 2002 which outlined the Government’s position. The Government’s proposal was that actions in tort for personal injury should be subject to “some special rules”. One was the introduction of a power enabling the courts to extend the time for the commencement of proceedings in tort for personal injury for a three year period if certain conditions were met. The other was a provision dealing with the accrual of the cause of action:
[I]n the case of latent injury or disease, the cause of action in tort should be deemed to have accrued not when the injury or disease occurred (which is the present position) but when it first manifested itself in a not insignificant form. This overcomes the present anomalous potential for a claim for damages being time barred even before the victim was aware that he or she had contracted a disease.
4797 The Explanatory Notes to the Bill for the 2005 Act are not especially helpful. In relation to the clause that was ultimately enacted as s 55 of the 2005 Act, the Explanatory Notes stated:
Clause 54 is of particular significance in that it changes the common law in relation to when a cause of action accrues for damages relating to personal injury (other than a personal injury which is attributable to the inhalation of asbestos). Under the current law, a cause of action for a personal injury claim accrues when the injury or disease occurs. Clause 54 provides that a cause of action for damages relating to a personal injury to a person accrues when the only or earlier of such of the following events as are applicable occurs –
(a) the person becomes aware that he or she has sustained a not insignificant personal injury;
(b) the first symptom, clinical sign or other manifestation of personal injury consistent with the person having sustained a not insignificant personal injury.
(Original emphasis)
4798 The second reading speech was more informative. The Attorney General, the Hon JA McGinty, observed that the basis for the reforms stemmed from the 1992 discussion paper, noting that, since then, the Ipp report had also been released. More particularly he explained:
The initial limitation period for the commencement of proceedings for personal injury will be three years, not the current six years. The court will be empowered to extend time for the commencement of proceedings for three years from when the victim knew, or ought to have known, that the physical cause of his or her injury was attributable to the conduct of a person, whether a defendant or not, and the identity of that person. In the case of latent injury or disease, the cause of action is to accrue not when the injury or disease occurred, which is the present position, but when it first manifests itself in a not insignificant form.3860
(Emphasis added)
4799 In her reasons in AME Hospitals at [22], McLure P considered that para 55(1)(a) is concerned with patent injuries and para 55(1)(b) with latent injuries in non-asbestos cases.
4800 In its 1997 report entitled “Limitation And Notice Of Actions”, the Commission identified the problem of latent injury in the following way at [1.13]:
A continual concern of law reform commissions and legislatures over the last thirty years has been the problem of latent injury. In Cartledge v E Jopling & Sons Ltd in 1962, the English House of Lords held that, in cases involving diseases such as silicosis or asbestosis (both forms of pneumoconiosis), the cause of action accrues as soon as some damage results from the inhalation of silica dust or asbestos particles, even though the damage is undetectable and the plaintiff is unaware of it …
4801 Later, at [4.16], in the context of a discussion of personal injury cases, the Commission observed:
In most cases of personal injury, determining the time at which damage is suffered is straightforward. In the case of physical injuries received in a road accident, for example, it is usually clear that damage was suffered at the time of impact, the plaintiff will be immediately aware of the existence of the damage, and it is logical for the limitation period to start running from that point. However, in many cases the fact that injury has been suffered is not immediately apparent, and determining the date of accrual is very difficult. This applies in many cases of latent personal injury, and also to cases involving the infliction of disease. In cases of asbestosis, mesothelioma, silicosis and the like, the injury is caused by the inhalation of minute particles of fibre or dust, but only becomes apparent many years afterwards. To take another example, AIDS is caused by contact with contaminated blood or other bodily fluids, but some years may elapse before a person who is HIV positive begins to experience signs of illness. In such cases the law says that damage is suffered at the point of initial onset, even though at that point in time he can have no knowledge of it …The problem, then, is that in cases of latent disease or injury, because the cause of action accrues at the point of initial onset, the limitation period may well have run its course before the plaintiff is, or could reasonably be expected to be, aware of the existence of damage giving an entitlement to sue.
(Emphasis added)
4802 I accept that the purpose of para 55(1)(b) is to make provision for latent injuries or diseases, that is cases in which a not insignificant injury occurs at a time when the injured person does not know and could not reasonably be expected to know of it. In such a case paragraph (b) provides for the cause of action to accrue from the later time when there is some manifestation of a not insignificant injury. That manifestation provides the means of awareness of which the Court of Appeal spoke in Thomas Peacock.
4803 It may be that para (b) has an element of actual awareness because one of the conditions is that there must be a symptom, clinical sign or other manifestation of an injury. I do not, however, accept the applicants’ argument that time only begins to run under both paras (a) and (b) once the injured person becomes aware that the symptoms s/he first experiences are symptoms of a not insignificant injury. That construction is at odds with the text of the subsection, whether or not read in context. It would defeat the purpose of having two different starting points and of providing for the accrual of the cause of action at the earlier point in time. The premise for paragraph (b) is that the damage is manifest: the injured person has detectable indications of a not insignificant injury, not that the injured person has detected that s/he has such an injury. As the provision indicates, it is sufficient that the symptoms etc. of which she is presumably, if not necessarily, aware are consistent with her having sustained a not insignificant injury. Moreover, the applicants took the remarks of the Court of Appeal in Thomas Peacock at [40] out of context. When those remarks are read with [39], it is clear that the very argument the applicants advanced was expressly rejected by the Court of Appeal in Thomas Peacock at [40].
4804 Thus, it is not to the point that Mrs Gill may not have believed that her injury was significant until well after her first mesh erosion. The pain Mrs Gill experienced in connection with the tearing by the mesh was a symptom of a not insignificant personal injury. That symptom occurred in late May or early June 2007. The applicants accepted that the sharp pains Mrs Gill experienced in connection with the first erosion were the first manifestations of personal injury consistent with Mrs Gill having sustained a not insignificant personal injury.3861 Indeed, they conceded in oral argument, that it was not possible to describe a painful erosion as an insignificant personal injury.3862
4805 In any case, I am satisfied that Mrs Gill certainly became aware that she had sustained a not insignificant personal injury by late October 2007.
4806 It follows that the respondents have discharged their burden of proof. The period of limitation expired by 11 June 2007 or, at the very latest, by October 2007, five years before these proceedings were commenced. Mrs Gill’s action is therefore statute-barred.
Should leave be granted to extend the period of limitation?
4807 The 2005 Act enables a claimant to apply to a court for leave to commence an action for damages relating to a personal injury even though the limitation period provided for under the Act has expired: s 39(1). It also permits a court to grant leave, provided certain conditions are met. Those conditions are set out in subs 39(3) and (4), which relevantly provide as follows:
(3) On an application a court may extend the time in which the action can be commenced if the court is satisfied that, when the limitation period expired, a person to whom the cause of action accrues —
(a) was not aware of the physical cause of the … injury;
(b) was aware of the physical cause of the … injury but was not aware that the… injury was attributable to the conduct of a person; or
(c) was aware of the physical cause of the death or injury and that the death or injury was attributable to the conduct of a person but after reasonable inquiry, had been unable to establish that person’s identity.
(4) On an application the court may extend the time in which the action can be commenced up to 3 years from when a person to whom the cause of action accrues became aware, or ought reasonably to have become aware—
(a) of the physical cause of the … injury;
(b) that the … injury was attributable to the conduct of a person (whether a defendant or not); and
(c) of the identity of the person mentioned in paragraph (b).
4808 “Defendant” is defined in s 3(1) as a person against whom an action is brought or proposed to be brought. It was common ground that the reference to “person” in paragraphs (b) and (c) of each subsection is a reference to a defendant (respondent) or prospective defendant (respondent).3863
4809 The burden of proof lies with the person applying for the extension: 2005 Act, s 79(3).
4810 The respondents submitted that, as at August 2007 or by June or August 2008, Mrs Gill knew of the physical cause of her injury and knew that it was attributable to the Prolift device “and therefore” to the conduct of the respondents and had the capacity to, and ought reasonably to have become aware, of their identity. For these reasons they contended the Court had no power to extend time.3864 I disagree.
4811 “Injury” means the personal injury the subject of the application for leave under s 39(1): AME Hospitals at [23] (McLure P); [265] (Newnes JA). “Personal injury” is defined in s 3(1) of the 2005 Act to include “a disease, impairment of a person’s physical condition, and mental disability”. In this context, it refers to the fact of injury; as opposed to its manifestations, such as its signs, symptoms or other sequelae: AME Hospitals at [181] (Buss JA). “Physical cause” refers to the factual, not legal, cause and “directs attention to the bodily mechanism or circumstances by which, as a matter of fact, the … injury was occasioned or brought about”: AME Hospitals at [188] (Buss JA).
4812 “Conduct” encapsulates both an act and an omission: AME Hospitals at [39] (McLure P); [265] (Newnes JA). “Attributable to” denotes a causal connection in fact as opposed to law so that paragraph (b) refers to a causal relationship between the injury and the conduct: AME Hospitals at [33] (McLure P); [265] (Newnes JA).
4813 “Ought reasonably to have become aware” “imports the notion that the person, acting reasonably, should have taken steps to acquire knowledge of relevant facts or circumstances”: AME Hospitals at [202] (Buss JA). Nevertheless, the court does not consider how a reasonable person in the position of the person to whom the cause of action accrues (the applicant) would have acted, disregarding the applicant’s particular characteristics, qualities, and circumstances. To the contrary, while the test is objective, the question of whether the applicant ought reasonably to have become aware of the relevant facts is to be answered having regard to her characteristics, attributes and circumstances: AME Hospitals at [205] (Buss JA, with McLure P and Newnes JJ agreeing at [42] and [265] respectively).
4814 AME Hospitals relevantly concerned the awareness of the father of the respondent (a child), who was “floppy” and not breathing when she was born. She was diagnosed with hypoxic ischaemic encephalopathy, a degenerative brain disease resulting from a lack of oxygen, and later with cerebral palsy. The respondent was born in August 2001. The initial diagnosis was contained in a hospital report the parents received some twelve months later. By June 2003 the parents were also aware that she had cerebral palsy. But neither then, nor in 2006 (when they sought legal advice), were they informed of the cause, despite asking various medical practitioners about the possible causes. A medical report obtained by their lawyers in October 2006 from Professor Con Michael did not identify the cause of either the hypoxic ischaemic encephalopathy or the cerebral palsy, effectively exonerating the hospital and the obstetrician. The respondent’s parents accepted the opinion and did not take the matter further.
4815 In May 2012, however, the parents were contacted by the lawyers and told that they had received information that had caused them to review their “childbirth files” and, after having done so, they considered that the parents should consider obtaining a second opinion. After obtaining advice on the costs, the father instructed the lawyers to seek an opinion from the expert they recommended, Professor Gus Dekker. He provided a report in August 2012 in which he attributed the encephalopathy to the lack of appropriate electronic foetal monitoring during the process of delivery. In July 2013 an application was filed seeking an extension of time under s 39 to sue the obstetrician and the hospital where the respondent was born. The parties agreed that the cause of action accrued on the commencement of the 2005 Act (15 November 2005) and expired on 15 November 2011. The Master granted leave to extend the time. Leave to appeal was granted, but the Court of Appeal dismissed the appeal.
4816 McLure P (with whom Newnes JA agreed) held at [36]:
What constitutes actual awareness will depend on the nature of the fact or matter in issue. In this case, awareness of the physical cause of the respondent’s injury is a matter of inference, from primary facts, that requires expert medical knowledge and experience. That the respondent’s father may have been aware of some or all of the primary facts and of the opinion of a non-qualified person as to the physical cause of the respondent’s injury cannot satisfy the actual awareness requirement.
4817 Later in her reasons, at [41], her Honour held that:
[A] person will be aware of a matter which requires expert knowledge and experience if he is aware of an expert opinion which is reasonably capable of being accepted and capable of establishing the relevant facts (that is, that the physical cause of the injury is attributable to the conduct of an identified person).
4818 Her Honour went on to find that, for the purposes of the extension application, the respondent’s father became aware of the physical cause of the respondent’s injury only when he received Professor Dekker’s opinion in August 2012. Buss JA said at [96] that it was also at this time that the father became aware that his daughter had suffered an injury during birth which caused her condition and that the injury was attributable to the conduct of the obstetrician and/or the nursing staff.
4819 On the question whether the father ought to have known at any earlier point in time, McLure P said at [45] that reasonableness did not require further investigations upon receiving Professor Michael’s report, as he was a highly qualified and experienced specialist in the relevant field, had been briefed with all available documentation, his view was expressed in unqualified and unequivocal terms, and he was firmly of the opinion that the injuries were not attributable to the appellant’s conduct.
4820 Here, Mrs Gill was aware that the mesh caused her injury after she felt it tear her and suffered sharp pain. That was in about May or June 2007. But she was not aware that it was attributable to the conduct of a person until after she had consulted Dr Dowling in either June or July 2013. Awareness does not connote mere suspicion and awareness referred to in the section is actual, not constructive: AME Hospitals at [34] (McLure P), [197]–[198] (Buss JA), citing Harris v Commercial Minerals Ltd (1996) 186 CLR 1 at 9–10 in the context of a comparable provision of the Limitation Act 1969 (NSW). While actual awareness within s 39(3) of the 2005 Act “does not require knowledge of the relevant fact or facts with complete certainty as to its or their existence and beyond the possibility of error”, it does mean “awareness or knowledge of the relevant fact or facts with sufficient confidence reasonably to justify, in all the circumstances, the commencement of proceedings against the proposed defendant on the relevant cause of action by the issue of a writ or other originating process”: AME Hospitals at [200] (Buss JA).
4821 In other words, the question with which s 39(3) is concerned is when the applicant actually became aware, not when she ought reasonably to have been aware.
4822 Moreover, the relevant conduct for the purposes of both s 39(3) and s 39(4) is to be ascertained from the particulars of negligence and the evidence: AME Hospitals at [213]–[214] (Buss JA). This construction is consistent with the proper interpretation of similar provisions in the equivalent NSW Act: see Dedousis v Water Board (1994) 181 CLR 171. Thus, the applicants must prove that at the relevant time Mrs Gill was unaware of the acts and omissions of the respondents said to constitute their negligence. They include: the respondents’ omissions both before and after their devices were released to the market to undertake any or any adequate evaluations; and to give any, or any sufficient, information or warning of the risk of the device to cause one or more of the pleaded complications or of their inadequate evaluations. To the extent that she touched upon any these matters in her evidence in chief, save for the possibility of an erosion, her evidence was that she was ignorant of them.
4823 The respondents’ submission included a non sequitur. It does not follow from the fact that Mrs Gill knew that her injury was caused by the Prolift device that she was aware that her injury was attributable to the conduct of a person. She believed that she was somehow to blame, probably because the advice she had received from Dr Chapple was that the body could reject the implant. She was not aware that her injury was attributable to the conduct of the respondents before she spoke to Dr Dowling. Indeed, in oral argument the respondents accepted that she did not actually know that the injury was attributable to the conduct of the respondents.3865
4824 Provided that the Court is satisfied that when the limitation period expired the claimant was not aware that the injury was attributable to the conduct of a person, it does not matter that she might have been able to ascertain the identity of the respondents had she made reasonable inquiries. As Buss JA observed in AME Hospitals at [158], a court’s power to extend time under s 39(3) is enlivened if that court is satisfied that one of the conditions in para (a), (b) or (c) applies.
4825 In oral argument the respondents relied on s 39(4) where the phrase “ought reasonably to have become aware” appears,3866 but they took the phrase out of context. Section 39(4) applies only once the power to extend time is enlivened by satisfaction of the matters contained in s 39(3) and operates so as to limit the period of extension to “up to 3 years” from the time the claimant becomes aware or ought to have become aware of the matters referred to in s 39(4). As Buss JA put it in AME Hospitals at [160], s 39(4) relates to the length of any extension of time a court may grant.
4826 Section 39(4) is concerned with the state of awareness or knowledge that ought reasonably to have been acquired by the person to whom the cause of action accrues. As Buss JA explained at [205]:
The court does not assess how a reasonable person in the position of the person to whom the cause of action accrues would have acted and, in making that assessment, disregard aspects of that person's actual characteristics, qualities and circumstances. The task of the court is to determine whether the person to whom the cause of action accrues ought reasonably to have become aware of the matters enumerated in pars (a), (b) and (c) of s 39(4) by reference to what can reasonably have been expected of that person having regard to the actual subjective factors I have mentioned.
4827 Furthermore, as I noted above, the question of when the claimant became aware or ought reasonably to have become aware that the injury was attributable to the conduct of a person within the meaning of s 39(4)(b) is directed to the relevant conduct, in other words to the acts and/or omissions particularised in the claimant’s pleading: AME Hospitals at [214] (Buss JA). The matters referred to in s 39(4) are cumulative. Thus the last date that a claimant becomes aware or ought reasonably to have become aware of the relevant fact will determine the starting time for the three-year period during which the time may be extended.
4828 In the circumstances of this case, on the facts as proved, that three-year period did not commence, until after Dr Dowling informed Mrs Gill in 2013 that Prolift had been removed from the market. Before then Mrs Gill had no reason to think that the relevant acts or omissions of the respondent might have been responsible for her injuries. When she was given a reason, she promptly set about investigating her rights. By that time, however, the action was already on foot. If there were ever any doubt about whether a court could extend time under s 39 of the 2005 Act after the commencement of an action, it was removed by the judgment of the NSW Court of Appeal in Waldron v Joondalup Hospital Pty Ltd (2018) 98 NSWLR 552 at [136] (Sackville AJA, McColl AP and Meagher JA agreeing at [1] and [2] respectively).
4829 It follows that the Court has the power to extend time to enable Mrs Gill to sue. It may exercise that power in its discretion.
4830 In deciding whether to do so, s 44 of the 2005 Act requires that the Court take two matters into account:
(3) whether the delay in commencing the action, whatever the merits of the reasons for it, would unacceptably diminish the prospects of a fair trial; and
(4) whether extending the time would significantly prejudice the defendant (other than by reason only of the commencement of the proposed action).
4831 There is no reason to think that the delay would unacceptably diminish the prospects of a fair trial. The respondents adduced no evidence of prejudice and did not submit that there was any presumptive prejudice: see Brisbane South Regional Health Authority v Taylor (1996) 186 CLR 541 at 544 (Dawson J), 547–550 (Toohey and Gummow JJ), 551–553 (McHugh J). Indeed, they made no submissions about the exercise of the discretion. In oral argument when the question was raised with the respondents’ counsel, they eschewed the notion that they had been prejudiced and I was informed that, if the Court was satisfied that it had the power to grant an extension, the respondents did not dispute that it should exercise its discretion in Mrs Gill’s favour.3867
4832 I therefore find that extending the time would not significantly prejudice the respondents and that the limitation period should be extended to enable Mrs Gill to sue for damages for negligence.
Ann Sanders
Is the action statute-barred?
4833 At [90] of their Defence to the Fifth Further Amended Statement of Claim, the respondents pleaded that Mrs Sanders’ common law claim for damages accrued more than six years before this proceeding was issued and was therefore statue-barred under s 38(1)(c)(vi) of the 1935 Act. They had maintained this from their initial defence to Mrs Sanders’ claim (then at [86] of the Defence to the Third Statement of Claim).
4834 In their closing submissions, the respondents correctly submitted that under the 1935 Act the accrual of the cause of action and the running of time were not dependent on the plaintiff knowing that all of the facts comprising the cause of action had occurred: AME Hospitals at [39] (Buss JA). The limitation period began to run at the time of the first “not insignificant” injury.
4835 To that end, the respondents pointed to three not insignificant “injuries”, all of which occurred before the commencement of the 2005 Act:
(a) the surgery and implantation of TVT on 12 March 2001, since item 1 of Mrs Sanders’ particulars of injury was “chronic inflammation”;
(b) the urinary symptoms which developed after the implant surgery, for which Mrs Sanders consulted Dr Naunton in 2002 and 2003; and
(c) the injury Mrs Sanders experienced from the time of implant and which she reported on various occasions in 2011 as “prickling pain”, adding that it left her “never [feeling] quite right in the vagina since”.3868
4836 Nevertheless, they accepted that, if the Court were not to find that any of those items amounted to “a not insignificant injury”, then the 1935 Act would not apply.
4837 The respondents submitted that, if the chronic inflammatory reaction were an injury, that injury occurred on or around 12 March 2001 when the TVT device was implanted.3869 That may be true if the chronic inflammatory reaction to which the applicants were referring in their particulars was the foreign body reaction to the TVT. But, as I have explained above, I do not accept it was an injury.
4838 As for the urinary symptoms Mrs Sanders recalled having in 2002 and 2003, on the assumption that Mrs Sanders’ recollection is correct, for the reasons already given I am not persuaded that they were caused by the TVT device.
4839 The final “not insignificant injury” the respondents alleged was based on accounts attributed to Mrs Sanders in progress notes from King Edward Memorial Hospital and the Ashton Avenue practice.
4840 The first of these is a note made by Dr Howlett that Mrs Sanders “has always been able to feel prickling pain when sat down + felt mesh”.3870 Read in context, it is far from clear, however, that “always” is a reference to the period ever since implantation in 2001. This is how the note appears in the hospital records:

4841 Mrs Sanders was briefly taken to this statement in cross-examination. The only question she was asked was whether she recalled telling someone at the hospital on 28 January 2011 that she had “always been able to feel prickling pain when she sat down and that she felt mesh”. Mrs Sanders replied:
No, because when I first had it done in 2001, I was pleased, and it really worked. It worked. I was happy with it.3871
4842 It was not suggested to her that this evidence was false or that her memory was faulty.
4843 The second piece of evidence is the note made by Dr Gilbert on 19 May 2011 that Mrs Sanders “didn’t feel right from the start”.3872 In the letter she wrote to Dr Tan, Dr Gilbert wrote that “[s]he has never felt quite right in the vagina since [the TVT placement] but it has become worse recently”.3873 On one view this evidence is consistent with Dr Howlett’s note. On the other hand, it does not refer to “prickling pain” or feeling mesh since the operation.
4844 I accept, of course, that Mrs Sanders may well not have remembered what she said to the doctor but in cross-examination, Mrs Sanders was adamant that she did not give such a history.
And you gave a history of your problems to Dr Gilbert?---Yes. Okay. And did you tell Dr Gilbert that your vagina had not felt right ever since the time of the operation in March 2001?---No. No. I said the operation – the first operation was brilliant. She said – I think she said, “Would you have thought it wasn’t right from the word go?” And I – no, it worked. I was happy. I was pleased. 3874
4845 I accept her denial. She did not strike me as a dishonest person and it was not suggested that she was dishonest. Nor was it put to her that she was reconstructing the conversation. On balance, I conclude that Dr Gilbert was mistaken. In any case, what does not feeling right mean? On its own, it signifies nothing.
4846 The final piece of evidence appears in Dr Matthews’ notes for the consultation on 29 June 2011: that ever since her surgery she had “noticed feeling of mesh in vagina”.3875
4847 Mrs Sanders denied giving this history, too,3876 and Dr Matthews’ history note-taking is unreliable because she erroneously recorded that Mrs Sanders had had a “vaginal prolapse repair”. In any case, there was no reference here to any associated pain, prickling or otherwise.
4848 Even if I were to accept these accounts as accurate, it would be mere speculation to conclude that they signify that Mrs Sanders sustained a “not insignificant injury” at the time of surgery when none of the expert witnesses was asked to comment on the significance of the accounts.
4849 The applicants submitted that Mrs Sanders did not become aware that she was suffering from symptoms of a not insignificant injury until she consulted Dr Daborn in August 2011 and was told that the injuries associated with the TVT could be permanent.3877
4850 I reject this submission, too.
4851 First, neither Act requires that a personal injury have permanent consequences for it to be considered “not insignificant”. I was not taken to any authorities in support of such a proposition.
4852 Second, a cause of action does not only accrue when the injured person becomes aware that she has sustained a not insignificant personal injury. That circumstance is provided for in para 6(1)(a) of the 2005 Act. The applicants’ submission failed to have regard to the alternative foundation in paragraph (b): the occurrence of the first symptom, clinical sign or other manifestation of personal injury consistent with the injured person having sustained a not insignificant personal injury. If that event occurred before the injured person was aware that she had sustained a not insignificant personal injury, then that event would determine when the cause of action accrued.
4853 On the balance of probabilities, the first symptom or other manifestation of personal injury consistent with Mrs Sanders having sustained a not insignificant personal injury occurred either in 2007 when she began experiencing discomfort urinating and recurrent urinary frequency or in 2008 when she first experienced dyspareunia and groin pain. If any damage occurred before then, I would hold it to have been de minimis.
4854 I therefore find that Mrs Sanders’ cause of action in negligence accrued no earlier than January 2007. This means that her common law claim is caught by the 2005 Act, not the 1935 Act. Two consequences flow from this. One is that the action is statute-barred since, whether 2007 or 2008 is correct, the three-year limitation period under the 2005 Act expired well before the SUI subgroup was included in the pleading. The other is that Mrs Sanders is entitled to apply for an extension of time.
4855 The applicants submitted that, since the respondents only pleaded that the action was statute-barred under the 1935 Act, not the 2005 Act, “in addition or in the alternative, the respondents are estopped by their conduct from now seeking to rely upon any statutory limitation defence provided for by the 2005 Act”.3878
4856 I do not accept the submission. While it is true, as the applicants contended, that they were entitled to expect that this defence would be specifically pleaded, the respondents did plead that the action was brought more than six years after the cause of action accrued. The period of limitation under the 2005 Act is three years. The relevant provisions of the 2005 Act are no more onerous than the relevant provisions of the 1935 Act. I am not persuaded that the applicants have suffered any prejudice by the respondents’ reliance on the provisions of s 6 of the 2005 Act.
Should the limitation period be extended?
4857 On the assumption that, contrary to their submissions, the action was statute-barred, the applicants argued that an extension of time should be granted because:
(a) Mrs Sanders did not become aware that there was a problem with her TVT implant until 21 January 2011 when she consulted Dr Tan;
(b) it was not until August 2011, when she saw Dr Daborn, that she became aware that her injuries could be permanent; until then she believed her symptoms would be resolved by revision surgery;
(c) there is no evidence of actual or significant presumptive prejudice to the respondents; and
(d) the delay is not such as to unacceptably diminish the respondents’ entitlement to a fair trial.3879
4858 The respondents’ submissions were equally brief. They contended that the applicants had failed to show that, when the limitation period expired, Mrs Sanders was not aware of the physical cause of her symptoms, that the physical cause was attributable to the conduct of a person, or that Mrs Sanders was unable to establish the identity of the party responsible after reasonable inquiry.3880
4859 For the reasons given above, I find that the physical cause of Mrs Sanders’ personal injury was the erosion of the TVT tape. Mrs Sanders was not aware that the tape had eroded until she was told by a doctor. The first doctor to refer to an erosion was Dr Gilbert in May 2011. The first time she became aware that the tape had moved was during or soon after the conversation with Dr Tan on 28 January 2011. There is no reason why she ought reasonably to have become aware of these matters at an earlier time. In these circumstances, the Court has power to extend the time in which Mrs Sanders could bring her action. The three year period set by s 37(4) of the 2005 Act only expired after the SUI subgroup were added to the proceeding.
4860 In any case, I am satisfied that Mrs Sanders was not aware that her injury was attributable to the acts and omissions of the respondents said to have constituted negligence before she consulted her lawyers. There is no evidence to indicate when that was, but it is unlikely to have been before the erosion was detected. Before that time there was no reason for her to consult a lawyer.
4861 There is no good reason why the Court’s discretion should not be exercised in Mrs Sanders’ favour.
4862 The respondents adduced no evidence of prejudice and made no submissions on this question. In these circumstances I cannot conclude that an extension of time would cause them significant prejudice, whether actual or presumptive, and I do not consider that the delay in commencing the action would unacceptably diminish the respondents’ prospects of a fair trial.
4863 Accordingly, the limitation period should be extended to enable Mrs Sanders to prosecute her case in negligence.
PART XVIII: DAMAGES AND COMPENSATION
The negligence claims
4864 For the purpose of determining the applicable law, questions as to the kinds or heads of damage and as to the amounts that may be awarded are treated just like questions relating to the ascertainment of damages. They are substantive issues governed by the lex loci delicti (the law of the place where the wrongs occurred): John Pfeiffer at [100]. It follows that the law of Western Australia applies to the assessment of damages for the causes of action in negligence brought by Mrs Gill and Mrs Sanders and the law of Victoria applies to the claim for damages brought by Mrs Dawson. The unstated assumption is that, absent any comparable federal law, both the Wrongs Act (Vic) and CLA (WA) are picked up by s 79 of the Judiciary Act.
4865 One of the many complexities of this case, which has added to the labour of the lawyers and the Court, is the fact that damages must therefore be assessed having regard to statutory schemes that vary from jurisdiction to jurisdiction. Common law principles give way to the extent that they are modified by the legislation. That means that the amounts that may be awarded to the applicants, and in due course group members, will differ, not only as between the statutory causes of action and the negligence counts but also according to the time and place of the damage.
4866 Since the various claims are made in the alternative, the applicants are required to elect which remedy to take, in other words, which damages regime is to apply: Graham Barclay Oysters (HC) at [130] (Gummow and Hayne JJ); [331] (Callinan J). The respondents submitted that the election must be made before final orders are entered, relying on what Gummow and Hayne JJ said at [130]. Actually, their Honours said that the election must be made “no later than at the time of seeking final judgment in the action”, which is at least arguably earlier than the respondents submitted. Kirby J said at [228] that the election had to be made before steps were taken to enforce the judgment, which is later than either endpoint. Callinan J merely said (at [331]) that the election had to be made “in due course”.
4867 The applicants did not make an election at the trial or in written submissions. I therefore propose to give them leave to do so before judgment is entered and final orders made, consistent with the course taken by Hoeben J in Crump v Equine Nutrition Systems Pty Limited t/as Horsepower [2006] NSWSC 512 at [267].
4868 In their written submissions the applicants included a summary of the relevant principles that affect the assessment of the various heads of damage in a way that I did not understand to be controversial. These principles apply to all the claims in the present case unless they have been modified by statute and those modifications apply to the applicants. It is common ground that none of the statutory modifications applies to Mrs Sanders.
4869 Damages are payable at common law for pain and suffering, which includes physical pain and mental illness or anguish, loss of enjoyment of life, and loss of the amenities of life.
4870 “Loss of amenities” refers to the destruction or diminution of a faculty or skill caused by “the deprivation of the ability to participate in normal activities and thus to enjoy life to the full and to take full advantage of the opportunities that otherwise it might offer”: Teubner v Humble (1963) 108 CLR 491 at 506 (Windeyer J). That said, and despite legislative intervention in recent decades suggesting otherwise, “damages that result from personal injuries cannot ultimately be regarded as made up of completely separate components”: Teubner at 507 (Windeyer J). For convenience, I will refer to general damages as pain and suffering.
4871 The Court is required to consider the severity of the injury, the pain and suffering experienced by the claimant at the time of injury, the pain and suffering associated with the treatment or other reasonable measures taken to mitigate the effects of the injury, and any mental illness, distress, frustration, anxiety, and/or disappointment suffered as a result of the injury or as a result of limitations on the applicant’s ability to do what she would have done had she not been injured.
4872 The amount awarded must be proportionate to the injury and its consequences in the particular case; no other norm or standard is to be applied, even if derived from a consideration of amounts awarded by the courts in apparently similar or comparable cases: Planet Fisheries Pty Ltd v La Rosa (1968) 119 CLR 118 at 124–5. In Planet Fisheries their Honours went on to say:
The judgment of a Court awarding damages is not to be overborne by what other minds have judged right and proper for other situations. It may be granted that a judge who is making such an assessment will be aware of and give weight to current general ideas of fairness and moderation. But …[t]he awareness must be a product of general experience and not formed ad hoc by a process of considering particular cases and endeavouring, necessarily unsuccessfully, to allow for differences between the circumstances of those cases and the circumstances of the case in hand.
4873 In other words, as the applicants put it in their submissions, there is no tariff on damages for pain and suffering. The problem with the approach urged on the court in Planet Fisheries is that no two cases are truly alike. The amount of compensable loss depends, not only on the severity of the injury, but also on the consequences for the injured person, and the consequences of apparently similar injuries vary from one person to the next: Thatcher v Charles (1961) 104 CLR 57 at 71–72 (Windeyer J).
4874 Damages are awarded once and for all. Consequently, the assessment has to take into account both past and future pain and suffering and, if the damage is or may be permanent, then it has to be assessed for the duration of the applicant’s life.
4875 The tortfeasor or wrongdoer must take the injured person as it finds her. Consequently, the fact that one applicant may have had been more vulnerable to injury than another or may have had a peculiar susceptibility to develop complications that ensued does not assist the wrongdoer.
4876 While it is “impossible precisely to translate pain and suffering and the loss of enjoyment of life into money values”, and no amount of money will restore an applicant to her pre-injury position, that is the purpose of an award of damages, which means that an attempt must be made to assess a reasonable sum, having regard, as far as possible, to the prevailing standards of the community: O’Brien v Dundson (1965) 39 ALJR 78 at 78 (Barwick CJ, Kitto and Taylor JJ).
4877 In contrast to the position under the various statutory schemes, general damages at common law are at large; they are neither capped nor reduced in certain cases.
4878 Damages are payable to compensate for a loss of earning capacity provided that the loss is caused by reason of an injury caused by the wrongdoer’s negligent act or omission and that the diminution of earning capacity is or may be productive of financial loss: Graham v Baker (1961) 106 CLR 340 at 347 (Dixon CJ, Kitto and Taylor JJ); Medlin at 3 (Deane, Dawson, Toohey and Gaudron JJ).
4879 A discount rate of 3% is to be applied to the present value of future economic loss. The rate is an arbitrary one. It was fixed by the High Court in Todorovic v Waller (1981) 150 CLR 402, subject to any relevant statutory provisions to the contrary. The intention of the discount is to provide an allowance for inflation, future changes in wage rates generally or prices, and for tax, whether actual or notional, on income derived from investing the sum awarded. No further allowance for these matters is to be made.
4880 An injured person is also entitled to damages for the loss of pension or superannuation benefits that she would have received had she not been injured. This is treated as a loss of earnings and is therefore also subject to the 3% discount applied to future economic loss: Todorovic at 425–426 (Gibbs CJ and Wilson J).
4881 In the calculation of future economic loss a court is also required to take into account matters that might have affected the applicant’s earning capacity irrespective of the injury the subject of the proceedings. The major ones are sickness, accident, unemployment and industrial disputes. But not all contingencies are adverse, as Windeyer J pointed out in Bresatz v Przibilla (1962) 108 CLR 541 at 544 (Windeyer J). A court is also required to take into account the chance that, but for the injury, the applicant would have advanced in her career and/or increased her earnings. But contingencies are to be considered only in relation to their impact on the earning capacity of the injured person, not by reference to the workforce in general. See Wynn v NSW Insurance Ministerial Corporation (1995) 184 CLR 485 at 497 (Dawson, Toohey, Gaudron and Gummow JJ). As the plurality observed in Wynn at 497–498, the customary discount in New South Wales for the ordinary vicissitudes of life is 15%, a figure which is adjusted up or down having regard to the applicant’s circumstances.
4882 Provided they are reasonably necessary on account of the injury, an injured person is also entitled to recover as damages the value of the amounts expended in obtaining medical, surgical, hospital and like services such as physiotherapy, other forms of rehabilitation and attendant care, and the costs of medication, past and future.
Gratuitous care and/or services
4883 Damages are payable for care and/or services provided to an injured person provided that they are reasonably necessary as a result of the injury, even if they are provided voluntarily and the applicant is under no legal obligation to pay for them: Griffiths v Kerkemeyer (1977) 139 CLR 161; Van Gervan v Fenton (1992) 175 CLR 327.
4884 In Van Gervan v Fenton the majority held that only two questions need to be answered before a claimant can recover damages. The first question is: what are the services required to satisfy the claimant’s needs arising from the defendant’s wrong? The second question is: what is the value of those services? In answering the first question the majority held that it was wrong to make a discount or allowance for services that were provided before the injury, for example as part of the give-and-take of marriage (Mason CJ, Toohey and McHugh JJ at 338; Gaudron J at 349–350). As for the second question, in general, the value of the services is assessed by reference to the market cost of acquiring them because “the market cost of services is ordinarily the reasonable and objective value of the need for those services”: Griffiths v Kerkemeyer at 193 (Mason CJ); Van Gervan v Fenton at 333–334 (Mason CJ, Toohey and McHugh JJ) and 348–349 (Gaudron J).
4885 Since the purpose of damages in tort is to restore the injured person to the position she would have been had she not been injured, it is the applicant’s needs that are to be compensated. In Morgan v Gibson [1997] NSWCA 212 the insurer argued in vain that the damages awarded to Mrs Gibson, who it described as an obsessive cleaner, were excessive because it was unreasonable to require it to meet the cost of services necessary to restore her home to her high standards. Yet, as Meagher JA, with whom Mason P agreed, observed, the purpose of damages in tort is to restore the injured person to the position she would have enjoyed but for the tort. Any inquiry into the needs of an injured person “involves an investigation of the subjective requirements of the particular person, who must be taken as she is found”. It is the cost of providing the services to her based on her needs which must be reasonable. In that case, his Honour went on to say:
Part of the respondent’s lifestyle was to spend much time shopping for the purpose of finding the “best bargain”, an occupation she greatly enjoyed. The evidence before [the trial judge] was that the respondent’s husband now accompanied her on her shopping expeditions, which lasted from 10.00am to 5.00pm. The husband indicated that he believed he could perform it, unaccompanied, in a period of one and a half to two hours; the daughter in two and a half. Her Honour noted that these shopping expeditions constituted one of the respondent’s only weekly outings and allowed her to keep a modicum of control over her household. For the purposes of Griffiths v Kerkemeyer damages, her Honour allowed five hours. The appellant argued that her Honour fell into error in taking into consideration the fact that these expeditions were, in effect, of psychological benefit to the respondent. It was argued that it was not reasonably necessary for the respondent’s husband, Mr Gibson, to take his wife on the expeditions, to fulfil her need to have the shopping done. But that is not the only need that is being satisfied. In some cases, the plaintiff’s “need” for Griffiths v Kerkemeyer purposes is simply a need for the provision of food; in others, it is a need for the provision of assistance in obtaining food. Her case was clearly of the latter kind. It is not difficult to imagine that a person so proud of keeping an orderly household would suffer from being unable to contribute or assist in that area which was her domain. In this way, the shopping expeditions took on a therapeutic quality. Mr Gibson was providing a service which the respondent needed and which is properly to be viewed as in the realm of the type of services for which Griffiths v Kerkemeyer damages are awarded …
4886 Further, where a claim for domestic assistance includes services which directly benefit the injured person and services that benefit her and other members of her family, it is not necessary to disentangle the elements as long as the service in question is reasonably required by the injured person.
4887 In Morgan v Gibson, Meagher JA observed that “[i]t is nonsense to suggest that, because others may also derive a benefit, the respondent’s inability to perform the gardening she used to perform, is in some way a ‘need’ which must be discounted”. His Honour said that if the contrary were accepted, the basis of the principle would no longer be “need”, but “need” less “benefit to others”.
4888 Similarly, in White v Benjamin [2015] NSWCA 75; (2015) 70 MVR 188 at [70] Basten JA (with whom Meagher JA agreed) held that “where a service is reasonably required by the plaintiff, which is likely to cover the cleaning of the house in which she lives, the benefits to other members of the household may be disregarded”.
4889 After the Ipp Review, one might reasonably have expected the promulgation of a uniform set of rules. Indeed this was the recommendation of the Panel. It urged the incorporation of their recommendations “(in suitably drafted form) in a single statute (that might be styled the Civil Liability (Personal Injuries and Death) Act) … to be enacted in each jurisdiction”. Instead, as Professor Harold Luntz lamented in the preface to Assessment of Damages for Personal Injury and Death: General Principles (LexisNexis Butterworths, 2006), not only were different names chosen for the statutes in the various jurisdictions but the jurisdictions differed as to which of the recommendations they adopted and which they rejected. Even when they adopted the same recommendations, they often chose different words, sometimes for policy reasons but sometimes for no apparent reason other than the preference of parliamentary counsel for a particular style. Some of the statutory changes are directly contrary to recommendations of the Ipp Committee that the law not be changed.
4890 Mrs Sanders’ claims are unaffected by any of these changes. Mrs Gill’s damages in negligence are affected by the changes to the common law made in the CLA (WA) and Mrs Dawson’s by the changes made by amendments to the Wrongs Act (Vic). Their claims under the TPA are caught by changes made to that Act in 2004 and incorporated in Pt VIB.
4891 Section 87 of the TPA entitles the Court to make orders, amongst other things, directing the person who engaged in the contravening conduct to pay to the person who suffered the loss or damage the amount of that loss or damage: TPA, s 87(2)(d).
4892 Part VIB modifies the common law in certain respects but only for contraventions occurring on or after the commencement date. The modifications relevantly include the manner in which the following heads of damage are to be calculated: general damages (renamed “non-economic loss”), economic loss or loss of earning capacity, loss of superannuation benefits, gratuitous care and services, and interest.
4893 I shall refer to the effects of the relevant legislative changes in the contexts in which they arise for consideration.
Some issues of principle affecting all claims
4894 On a number of issues of principle the parties were at odds.
Calculation of life expectancy
4895 The first relates to the calculation of life expectancy in each case. The respondents calculations appear to have been based on the use of historical tables (rather than prospective tables which take into account predicted improvements in life expectancy), an approach which was rejected by the High Court in Golden Eagle International Trading Pty Ltd v Zhang (2007) 229 CLR 498. In Zhang Kirby and Hayne JJ said at [70] (Gummow, Callinan and Crennan JJ agreeing at [4]):
The Court of Appeal held … that “it is appropriate for the courts to make their estimations on the basis of the best information available: the projected tables would appear to be a more accurate assessment of future trends than the historical tables.” There is no reason to doubt that the Court of Appeal was correct in its conclusion that the projected tables published by the Australian Bureau of Statistics were more likely to give an accurate estimate of future life expectancy than the historical tables published by the Bureau. That being so, it follows that the Court of Appeal was right to conclude that, despite the then prevailing practice in the courts of New South Wales, the primary judge should have used the prospective rather than the historical tables.
Discounting damages for the vicissitudes of life
4896 The second issue concerns discounting damages for future out-of-pocket expenses and care or services by 15% to reflect the vicissitudes of life, for which the respondents contended.
4897 It is beyond doubt that damages for future economic loss are to be discounted to take into account the ordinary vicissitudes of life. But the position with respect to future out-of-pocket expenses and care is different. In contrast to the assessment of future economic loss, the awards for these heads of damage are calculated over the duration of the applicants’ life using life tables which already take into account the contingency of death: see Luntz H, Assessment of Damages for Personal Injury and Death (Butterworths, 4th ed, 2002) at [6.1.6]. In Sharman v Evans (1977) 138 CLR 563 at 588, Gibbs and Stephen JJ observed that once a probable life expectancy is determined the vicissitudes of life “enter not at all into the assessment of future hospital expenses …”.
4898 Wood J made the same observation in Dodge v Snell [2011] TASSC 19 at [452], also citing Sharman v Evans.
4899 There was no express reference in Sharman v Evans to damages for care and services. That was because the care in question was nursing care and the Court held that the plaintiff, a quadriplegic, would spend the rest of her life in hospital. Consequently future nursing care was subsumed within future hospital expenses.
4900 The parties provided no assistance on this point. Apart from Dodge v Snell, my own research reveals only one case where the matter was squarely raised. That was a recent decision of the ACT Court of Appeal. In Oliver v Roberts [2018] ACTCA 35; (2018) 85 MVR 259 the Court rejected a submission by senior counsel for the appellant that the primary judge had erred by discounting future commercial care for vicissitudes, contrary to Sharman v Evans. The Court held at [81] that the appellant’s reliance on the remarks of Gibbs and Stephen JJ was “mistaken”, reasoning as follows:
81 The appellant’s reliance on the above passage is mistaken. In Sharman at 588, Gibbs and Stephen JJ accepted that “hazards of life, including illness and disablement” should be taken into account. That approach was consistent with a general principle discussed in Jobling v Associated Dairies Ltd [1982] AC 794 (Jobling) (which concerned the assessment of damages for loss of earning capacity). At AC 814; All ER 65, Lord Keith said:
In considering how matters might have been expected to work out if there had been no accident, the “vicissitudes” principle says that it is right to take into account events, such as illness, which not uncommonly occur in the ordinary course of human life. If such events are not taken into account, the damages may be greater than are required to compensate the plaintiff for the effects of the accident, and that result would be unfair to the defendant.
82 In Malec v JC Hutton Pty Ltd (1990) 169 CLR 638 (Malec) at 645, Deane, Gaudron and McHugh JJ spoke of a reduction “to take account of the chance that factors, unconnected with the defendant’s negligence” would have necessitated a level of care similar to that required because of the defendant’s negligence.
83 Whether it is referred to as a discount for the “hazards of life” or “factors unconnected with the defendant’s negligence” (or even “vicissitudes”), there is no doubt that damages for future care (other than damages awarded by way of a buffer) should be discounted to compensate a claimant only for the effects of the accident and to avoid an unfair result to a defendant.
4901 It is beyond doubt that damages for future care, like all heads of damage, should compensate a claimant only for the effects of the compensable injury. With the greatest respect, however, I am of the opinion that the Court was plainly wrong to hold that damages for future care and assistance must be reduced for the ordinary vicissitudes of life.
4902 In Sharman v Evans Gibbs and Stephen JJ did accept that “hazards of life, including illness and disablement” should be taken into account but not with respect to future care. Their Honours were referring to damages for future loss of earning capacity, which they had distinguished from damages for hospital expenses. That is apparent when the passage is read as a whole. The reference to Jobling v Associated Dairies Ltd [1982] AC 794 is of no assistance because it concerned damages for loss of earning capacity. In Malec v J C Hutton Pty Ltd (1990) 169 CLR 638 at 645, Deane, Gaudron and McHugh JJ did say that the compensation for the care and attention the plaintiff received from his wife should be reduced “to take account of the chance that factors, unconnected with the defendant’s negligence, would have necessitated similar care and attention”. But the factors unconnected with the defendant’s negligence to which their Honours were referring were not the ordinary vicissitudes of life. That is clear from their Honours’ earlier observation in the same paragraph that damages for pain and suffering must be reduced to take into account the same factors. Damages for pain and suffering are not reduced for the vicissitudes of life. It is also clear from the facts, recited by their Honours at the beginning of their reasons. Those facts were as follows.
4903 The trial judge found that the plaintiff had a neurotic illness precipitated by brucellosis which he contracted as a result of the defendant’s negligence. But he also found that it was at least as probable as not that the neurotic condition from which the plaintiff suffered at the time of trial was not related to the brucellosis contracted 10 to 12 years earlier. This latter finding was reversed on appeal. The Full Court held that damages should be assessed on the basis that the neurotic condition suffered at the time of trial, when the plaintiff was 49, was caused by depression induced by the brucellosis. Matthews and Ambrose JJ went on to conclude, however, that because of his personality it was likely that the symptoms he experienced from a spinal condition would have rendered him unemployable by the age of 44 and that, as a result, he would have suffered a similar neurotic condition even if he had never contracted brucellosis. Consequently, they awarded damages for economic loss only to age 44. They refused Griffiths v Kerkemeyer damages for this reason. At the same time, however, the damages they awarded for pain and suffering included an amount for the future. Deane, Gaudron and McHugh JJ, with whom Brennan and Dawson JJ agreed, held at 642 that the majority erred in holding that the plaintiff was not entitled to economic loss after the age of 44, in their assessment of damages for pain and suffering, and in refusing to award damages for the care and attention provided by his wife.
4904 In short, there was a need to take into account the chance that factors, unconnected with the defendant’s negligence, would have necessitated similar care and attention, not because of the vicissitudes of life but because there was evidence that another condition from which the plaintiff suffered would (or could) have brought about a similar result.
4905 Neither Jobling nor Malec had anything to do with the point the ACT Court of Appeal was required to consider in Oliver.
Discounting damages for other reasons
4906 The third area of dispute relates to the correct approach to other contingencies and two contingencies in particular. The first is the chance that, had the applicants undergone a different form of surgery, they might have developed some of the same complications. The second is the significance of pre-existing conditions, that is, conditions which preceded the implantation of the respondents’ devices. The first is to be dealt with according to the principles in Malec, the second according to the principles in Watts v Rake (1960) 108 CLR 158 and Purkess v Crittenden (1965) 114 CLR 164.
4907 In Watts v Rake the trial judge assessed general damages on the basis that the accident in which the plaintiff was injured merely accelerated the plaintiff’s complete disablement from ankylosing spondylitis which he had at the time of the accident. The plaintiff appealed. The arguments of the defendant, who sought to uphold the assessment, were summarised by Dixon CJ at 160. According to that summary, the defendant argued that the plaintiff’s damages should be reduced to take into account the following circumstances: first, his predisposition to many or some of the arthritic and other conditions which rapidly developed as a result of the accident the subject of the proceedings; second, that part of his present condition was due to causes other than the accident; and third, that, even if there had been no accident, “eventually and prematurely [he would] have been incapacitated by the seeds of disability within him”.
4908 The High Court rejected all three contentions, upheld the plaintiff’s appeal and increased his damages.
4909 As to the first, the court held that the fact that the plaintiff was predisposed to the injury caused by the accident did not reduce his damages; the tortfeasor must take an injured person as he finds him. As Dixon CJ put it:
If the injury proves more serious in its incidents and its consequences because of the injured man’s condition, that does nothing but increase the damages the defendant must pay. To sever the remaining leg of a one-legged man or put out the eye of a one-eyed man is to do a far more serious injury than it would have been had the injured man possessed two legs or two eyes. But for the seriousness of the injury the defendant must pay.
4910 As to the second and third arguments, the Chief Justice said that there was a presumption in favour of the plaintiff for the defendant to overcome. He continued:
If the disabilities of the plaintiff can be disentangled and one or more traced to causes in which the injuries he sustained through the accident play no part, it is the defendant who should be required to do the disentangling and to exclude the operation of the accident as a contributory cause. If it be the case that at some future date the plaintiff would in any event have reached his present pitiable state, the defendant should be called upon to prove that satisfactorily and moreover to show the period at the close of which it would have occurred.
4911 Menzies J, with whom Dixon CJ and Windeyer JJ agreed, observed at 163 that the burden was on the plaintiff to prove that his present condition and incapacity for work resulted from the accident the subject of the proceedings, but “it was not … for him to disprove that his pre-accident ill health would eventually cripple and incapacitate him”. His Honour continued:
Prima facie, where a plaintiff was in apparent good health before an accident and is in bad health thereafter, the change would be regarded as a consequence of the accident and it is for the defendant to prove that there is some other explanation for it, e.g., that the plaintiff has aggravated his condition by some unreasonable act or omission. Similarly, although it is of course material to ascertain what was the pre-accident condition of the plaintiff who alleges that his post-accident ill health is due to the accident, it is for the defendant to prove that before the accident the plaintiff was in a condition that, without the accident, would have led to his post-accident state of health.
4912 In Purkess v Crittenden it was argued that Watts v Rake suggested that, where a plaintiff asserts that he had become permanently disabled as a result of the defendant’s negligence and the defendant sets up a case that, owing to a pre-existing condition, the plaintiff would have become permanently disabled within an ascertainable period, the defendant carries the onus of proving that case. In a joint judgment Barwick CJ, Kitto and Taylor JJ denied the suggestion. They distinguished between the legal burden of proof, which, they said is always stable, and the burden of adducing evidence, which may constantly shift. They said that it was with the latter (the evidentiary or evidential burden) with which Watts v Rake was concerned. Their Honours explained:
We understand that case to proceed upon the basis that where a plaintiff has, by direct or circumstantial evidence, made out a prima facie case that incapacity has resulted from the defendant's negligence, the onus of adducing evidence that his incapacity is wholly or partly the result of some pre-existing condition or that incapacity, either total or partial, would, in any event, have resulted from a preexisting condition, rests upon the defendant. In other words, in the absence of such evidence the plaintiff, if his evidence be accepted, will be entitled to succeed on the issue of damages and no issue will arise as to the existence of any pre-existing abnormality or its prospective results, or as to the relationship of any such abnormality to the disabilities of which he complains at the trial … [I]t is not enough for the defendant merely to suggest the existence of a progressive pre-existing condition in the plaintiff or a relationship between any such condition and the plaintiff's present incapacity. On the contrary it was stressed that both the pre-existing condition and its future probable effects or its actual relationship to that incapacity must be the subject of evidence (i.e. either substantive evidence in the defendant's case or evidence extracted by cross-examination in the plaintiff's case) which, if accepted, would establish with some reasonable measure of precision, what the preexisting condition was and what its future effects, both as to their nature and their future development and progress, were likely to be. That being done, it is for the plaintiff upon the whole of the evidence to satisfy the tribunal of fact of the extent of the injury caused by the defendant's negligence.
4913 It is on this basis that the applicants’ damages are to be assessed. The principle is not disturbed by s 5D of the CLA (WA) and s 52 of the Wrongs Act (Vic) which provide that the plaintiff always carries the burden of proving any fact relevant to causation. In Keith v Gal [2016] NSWCA 152; (2016) 77 MVR 45 the respondents submitted that s 5E of the CLA (NSW), the analogue of s 5D of the CLA (WA), was concerned with both the legal and the evidential burden and therefore displaced the principles in Watts v Rake and Purkess v Crittenden. Gleeson JA, with whom Meagher JA and Tobias AJA agreed, decided it was unnecessary to resolve the issue but observed at [144]:
One difficulty with the respondents’ argument is that it conflates the legal burden of proof which falls on the plaintiff, on the balance of probabilities, as s 5E makes clear, with the evidential burden which is considered at different points in time during the trial as evidence is adduced, and is different to the legal burden (which always remains on the plaintiff).
4914 The only other authority I have been able to find in which the subject was raised is DC v State of New South Wales [2016] NSWCA 198 in which it was apparently common ground that s 5E is consistent with the principles established by Watts v Rake and Purkess v Crittenden.
4915 I respectfully agree with the observation in Keith v Gal. I would not read s 5D of the CLA (WA) or s 52 of the Wrongs Act (Vic) any differently.
4916 In her Statement of Particulars filed on 21 July 2017 Mrs Gill claimed to have received the following injuries:
1 Chronic inflammatory reaction of tissues into which the Prolift Total mesh implant (Mesh Implant) was implanted, and surrounding tissues.
2 Erosion and/or extrusion and/or exposure of the Mesh Implant through the anterior vaginal wall 5cm proximal to the urethral meatus to the right of the midline.
3 Requirement for surgery on 10 September 2007 to excise the exposed portion of the Mesh Implant referred to in the preceding paragraph (First Revision Surgery).
4 Erosion and/or extrusion and/or exposure of the Mesh Implant anterior to the cervix.
5 Requirement for surgery on 20 June 2008 to:
(a) Excise the right deep anterior arm of the Mesh Implant to halfway along the obturator fossa;
(b) Excise the left posterior arm of the Mesh Implant near the cervix and uterosacral ligaments;
(c) Perform a Fenton's procedure (Second Revision Surgery).
6 Requirement for surgery on 4 July 2008 to excise and drain an anterior vaginal abscess.
7 Erosion and/or extrusion and/or exposure of the Mesh Implant on the anterior vaginal wall just distal to the cervix.
8 Requirement for surgery on 8 August 2013 to excise the exposed portion of the Mesh Implant referred to in the preceding paragraph (Third Revision Surgery).
9 Sub-urethral mesh ulceration with significant tenderness in the posterior vaginal wall and the right-sided mesh arm from the posterior prolapse as it was passing to the sacrospinous ligament.
10 Erosion and/or extrusion and/or exposure of the Mesh Implant under the urethra.
11 Recurrent anterior compartment prolapse (cystocele).
12 Recurrent posterior compartment prolapse (rectocele).
13 Recurrent uterine prolapse.
14 Development of bulky, fibroid, acutely anteverted uterus.
15 Generalised anxiety disorder (DSM-5).
16 Adjustment disorder with depressed mood (DSM-5).
4917 She also claimed to have suffered the following continuing disabilities:
1 Constant pelvic cramp-like ache or pain.
2 Constant lower abdominal pain, which radiates to her loins.
3 Right groin pain, which is sharp and burning in character.
4 Constant lower back/sacryl pain in the coccyx area, which increases with bowel motions.
5 Dyspareunia.
6 Exacerbation of pain when bending, lifting, carrying heavy objects or engaging in sexual intercourse.
7 Severe pelvic pain on defaecation.
8 Severe pain in the coccyx and nausea during orgasm.
9 Tenderness in the right sacrospinous ligament.
10 Tenderness in the vaginal vault.
11 Sensation of heaviness and pulling down pain in her groin, limiting ability to stand.
12 Urinary frequency (between 5-6 times per day and hourly) with little warning and need to go to the toilet quickly.
13 Nocturia (2-3 times per night).
14 Sleep disturbance.
15 Night sweats.
16 Stress urinary incontinence (SUI).
17 Urge urinary incontinence (UUI).
18 Slow urinary stream.
19 Occasional feelings of incomplete bladder emptying.
20 Faecal constipation.
21 Need to use laxative (Movicol or Osmolax) to maintain faecal continence.
22 Incomplete bowel emptying.
23 Flatal incontinence.
24 Faecal incontinence on a weekly or fortnightly basis.
25 Need to digitate to push prolapse back into place.
26 Bleeding or spotting from the vagina following intercourse and physical activity.
27 Chronic significant fatigue, which increases with daytime activity.
28 Inability to drive for more than 1-2 hours.
29 Inability to sit for more than 30 minutes without experiencing pain.
30 Inability to stand for more than 30 minutes without experiencing pain.
31 Need to alternate between standing and sitting.
32 When sitting, need to alternate between upright sitting and reclining.
33 Difficulty transferring on and off lounge chairs.
34 Difficulty transferring on and off bed.
35 Difficulty transferring in and out of a motor vehicle.
36 Need for modifications to pool at home to assist with transfers into and out of the pool.
37 Need to sit when dressing.
38 Inability to run.
39 Inability to walk for more than 2 km.
40 Difficulty walking on uneven surfaces.
41 Difficulty exercising.
42 Difficulty relaxing.
43 Avoidance of stairs.
44 Inability to push or pull a shopping trolley, mop or vacuum.
45 Inability to use tampons.
46 Inability to scuba dive or swim when menstruating.
47 Inability to engage in outdoor activities with her family, eg, kayaking.
48 Severely impaired ability to engage in social and leisure activities.
49 Significant diminution in ability to engage in sexual intercourse.
50 Significant deleterious impact on intimate relationship with husband, Steven.
51 Low libido.
52 Reduced ability to parent her two sons, Lachlan (DOB 14 April 2002) and Dylan (3 February 2004).
53 Social withdrawal.
54 Low self-esteem.
55 Weight gain (6 kg).
56 Irritability and general intolerance.
57 Constant anxiety.
58 Panic attacks.
Sub-Particulars
(a) Hyperventilation.
(b) Tachycardia.
(c) Palpitations.
(d) Overwhelming sense that something is going to go wrong or an impending sense of doom.
59 Tearfulness.
60 Constant rumination about medical care.
61 Avoidance of thoughts of medical condition.
62 Forgetfulness and poor memory.
63 Difficulty concentrating.
64 Difficulty finding words.
65 Emotional lability and mood swings.
66 Low mood.
67 Feelings of sadness.
68 Foreshortened sense of the future.
69 Requirement for assistance with activities of daily living.
70 Need for counselling to maintain the marital relationship.
71 Need for psychotherapy to assist with pain management.
72 Need to ingest opioid, non-opioid and neuropathic analgesics, non-steroidal anti-inflammatory drugs (NSAIDs), SSRI, tranquillisers and dietary supplements to control pain.
Sub-Particulars
(a) Valium 2mg (tranquilliser).
(b) Naproxyn (24 x 275mg) (NSAID).
(c) Celecoxib (brand name Celebrex) (NSAID).
(d) Difenac (NSAID).
(e) Oxycodin (opioid analgesic).
(f) Tramodol (opioid analgesic).
(g) Voltaren (NSAID).
(h) Panadol (non-opioid analgesic).
(i) lboprofen (NSAID).
(j) Fluoxetine (brand name Lovan).
(k) Pregabalin (brand name Lyrica) (neuropathic analgesic).
(l) Fish oil, curcumin, magnesium supplements
4918 The respondents conceded that Mrs Gill has proved that the following items of damage listed above are caused by the Prolift implant: items 2–10 of the injuries (the various exposures and the surgery to address them) and “certain” of her ongoing pelvic pain. 3881
4919 For the most part, I accept that Mrs Gill has or has had the injuries and disabilities listed in her Statement of Particulars since her prolapse surgery in January 2007. I do not accept, however, that they are all attributable to the Prolift device either in whole or in part.
4920 I consider below those items which were not admittedly related to the implant or which were disputed.
Chronic inflammatory reaction
4921 The respondents said nothing about the first particularised injury: chronic inflammatory reaction.
4922 There is no doubt that Mrs Gill experienced a chronic inflammatory reaction to the implanted mesh.
4923 In September 2007, nine months after her implant surgery and at the time of her first mesh excision, a pathologist, Dr Trevor Kyle, who examined a mesh specimen reported that on microscopy:
Much of the specimen consists of edematous [swollen] congested granulation tissue containing moderate and focally marked inflammation. This is mainly nonspecific and chronic. Small foci of subacute inflammation are also present. On the surface of this tissue is a small amount of birefractile foreign material consistent with mesh.3882
4924 Professor Klosterhalfen, who also examined tissue specimens taken from Mrs Gill, deposed that:
The specimens of Mrs Gill … reveal a typical chronic inflammatory reaction and foreign body reaction.
4925 For the reasons given in Part XVII, however, I do not regard the anticipated chronic inflammatory reaction as an injury in and of itself. Without more, none of the applicants or class members would be entitled to damages.
4926 I turn now to the defined areas of dispute.
4927 I will deal first with the recurrent prolapses (items 11, 12, and 13 of the particularised injuries).
4928 Professor Roovers testified that “we mostly do not see a recurrence of prolapse after … reintervention” in a case where mesh has been used for Prolift repair.3883 Based on his own experience and the publication of another group who quoted a 10% risk of recurrence, he said that the chance was “small”. Whatever the true figure might be, there was an undeniable risk of recurrence after surgical repair. Professor Roovers said that whether there will be a recurrence will depend on the reason for the surgery.3884 If the reason for the surgery is, as it was here, to treat an erosion and the surgery involves removal or excision of mesh, the risk will obviously be higher than if the surgery does not involve excision of mesh, and the extent of the risk would increase with multiple surgical procedures.
4929 Professor Roovers also testified that scar tissue produced from the operation to implant the mesh is generally sufficient to prevent organs descending into the vagina.3885 On further questioning, however, he described this as an hypothesis.3886 Professor Korda’s evidence, which I accept, is that the hypothesis is correct only in the short term. As he observed, if it were correct, there would be no recurrence of prolapse after native tissue repair which largely relies on the formation of fibrous tissue.3887
4930 I accept that, even if Prolift were not defective and even if the respondents had not been negligent, there was always a risk that Mrs Gill’s prolapses would recur. I also accept the evidence given by both Associate Professor Lam and Professor Korda that Mrs Gill was at high risk of developing recurrent prolapse, if for no other reason than that she had 3D ultrasound evidence of bilateral levator avulsion (tearing of the puborectalis part of the levator ani muscle) and hiatal ballooning.3888 In his first report Professor Korda stated that he has referred patients for mesh prolapse repair in clinical situations where there is ultrasound evidence of avulsion of the puborectalis muscle (a part of the levator ani muscle) that causes enlargement of the urogenital hiatus (the opening in the pelvic floor between the medial borders of the levator ani muscles on each side through which the urethra and vagina pass). He explained that the reason is that it has been shown that the risk of prolapse recurrence is much greater in patients with an enlarged urogenital hiatus due to avulsion than in patients in whom there is no avulsion of the puborectalis muscle.3889 Indeed, he observed that levator tears are the most substantial risk factor for female pelvic organ prolapse yet found.3890
4931 Be that as it may, I am satisfied that Mrs Gill is entitled to damages for the recurrent prolapses. That is because I am persuaded that the multiple excision procedures necessitated by the painful erosions caused a reduction in the support the device was designed to provide and increased the risk that eventuated. The first mesh erosion or exposure in 2007 was on the anterior vaginal wall and resulted in the excision of 2 x 1cm of mesh. The second in 2008 was in the right vault and over the left posterior arm. At operation Dr Leake removed the anterior right arm half way to the obturator fossa and the entire posterior arm. The third mesh erosion or exposure in 2013 was in the anterior vaginal wall just distal from the cervix. The prolapse(s) recurred in late 2015 or early 2016. In April 2016 Associate Professor Shek diagnosed a recurrent three-compartment prolapse.3891 Professor Korda confirmed the diagnosis when he examined Mrs Gill very soon after.3892
Development of a bulky, fibroid, acutely anterverted uterus
4932 The second dispute concerns “the development of a bulky, fibroid, acutely anteverted uterus”, which is item 14 of the applicants’ particularised injuries.
4933 With respect to this allegation, I accept the respondents’ submission. The evidence does not establish that Mrs Gill developed a “bulky, fibroid, acutely anteverted uterus” as a result of the implantation of the Prolift device.
4934 An anteverted uterus is a uterus which tilts forward at the cervix towards the abdomen, as distinct from a retroverted uterus which tilts backward at the cervix. 3893 Most women have anteverted uteri. Indeed, “anteversion” is defined in the Oxford Concise Medical Dictionary as “the forward inclination of an organ, especially the normal forward inclination of the uterus” (emphasis added).
4935 When Dr Chapple first examined Mrs Gill he described her as having “a normal anteverted uterus”.3894 Similarly, Dr Paul Roche, the radiologist who conducted the CT scan of her abdomen and pelvis eight weeks after Mrs Gill’s Prolift surgery, described her uterus as anteverted and of normal appearance.3895
4936 If the tilt is extremely severe, I understand that it can put pressure on the pelvis and cause pelvic pain. There is no evidence to which I was taken, however, to indicate that the tilt in Mrs Gill’s uterus is extremely severe or, if so, it was attributable to the respondents’ device. The reference to an “acutely anteverted bulky uterus” appears to be derived from Dr Tsokos’s report of 28 October 2014 but he did not attributed the condition to implantation with Prolift. He did not suggest that it had “developed” as a consequence of the device or the procedure. Nor did he say the tilt was extremely severe or even abnormal.
4937 Furthermore, as the respondents’ submitted, there was no evidence that Mrs Gill’s bulky uterus and intramural posterior fibroid are related to the Prolift device.3896 She has a family history of fibroids.3897 It is not in dispute that the fibroid is a cause of her excessive bleeding.3898
Chronic pelvic pain, dyspareunia, coccygeal, low back pain, severe pain on defaecation
4938 The third point of contention concerns the relationship between Mrs Gill’s Prolift surgery and her chronic pelvic pain and dyspareunia, constant coccygeal pain, low back pain, severe pain on defaecation, broadly captured in items 1–8 of the list of disabilities, and vaginal bleeding and spotting (item 26).
4939 While there was no dispute that chronic pelvic pain and dyspareunia can be caused by Prolift, there was a real dispute about the extent to which Prolift was responsible for Mrs Gill’s chronic pelvic pain and dyspareunia and her other pain symptoms.
4940 Professor Korda, who was the only expert witness to examine her, was firmly of the view that they were. He insisted that Mrs Gill’s current complaints of dyspareunia and difficulties during orgasm were different from the complaints with which she presented to Dr Chapple of reduced libido and difficulties with sexual intercourse due to a vacuous vagina.3899
4941 The respondents submitted that Mrs Gill not only has a uterine fibroid but also adenomyosis and that both are indicative of endometriosis which commonly causes chronic pelvic pain, sporadic period-like pain, painful intercourse, abnormal vaginal bleeding, constipation, and painful bowel movements.3900 This submission was largely based on evidence given by Associate Professor Lam.
4942 In his report Associate Professor Lam said that further gynaecological and colorectal opinions, followed by investigations with imaging tests such as transvaginal ultrasound, pelvic MRI, colonoscopy and laparoscopy, should have been considered in order to eliminate the possibility of non-mesh pathologies such as endometriosis or fibroids.3901 In oral evidence in chief he expressed the opinion that the persistent coccygeal, sporadic period-like pain, and pain when passing a stool were uncharacteristic of pelvic floor repair surgery.3902
4943 Associate Professor Lam noted that adenomyosis was diagnosed on ultrasound on 3 November 2008. He said that adenomyosis has been found to have a significant association with deep infiltrating endometriosis and that Mrs Gill pain symptoms were “suggestive of deep infiltrative endometriosis”. He also noted that women with fibroids may have co-existing endometriosis, citing an article in which a high correlation was apparently found in women with symptomatic leiomyomas (fibroids). He said that the Mirena IUD has been used to treat endometriosis and has been found to significantly reduce period pain, pelvic pain, and deep dyspareunia. He stated that Mirena is effective for five years and observed that Mrs Gill’s pain was under control between 2008 and 2013 following the insertion of the Mirena but recurred thereafter.
4944 In all these circumstances, Professor Lam felt that it was “highly probable” that Mrs Gill’s recurrent, chronic pain and bowel symptoms “may have been due” to the fibroid and endometriosis “rather than mesh-related exposure alone”. Associate Professor Lam conceded, however, that Mrs Gill’s coccygeal pain could be related to the use of the Prolift device because the posterior Prolift is anchored or attached to the sacrospinous ligament, which runs from the ischial spine to the tip of the sacrum, which is the coccyx, and that the pain and tenderness along the posterior vaginal wall could be related to the posterior mesh arms.3903 He said it was “highly probable that the large posterior intramural fibroid could contribute or account for her deep pelvic pain, deep dyspareunia, abdominal bloating, constipation and incomplete bowel emptying sensation, and exaggerate her prolapse symptoms”. He said that an MRI of the pelvis, colonoscopy and diagnostic laparoscopy would be appropriate and useful investigations to determine whether other pelvic pathology like endometriosis was present.3904
4945 It is important to recognise that it is apparent that Associate Professor Lam considered that mesh-related exposure was at least a cause of the recurrent chronic pain and bowel symptoms. The question he raises is whether it was the only cause.
4946 The question is a reasonable one. I have some difficulty with his opinion, however, and therefore the submissions which rested on it.
4947 First, the source for Associate Professor Lam’s statement that adenomyosis has been found to be significantly associated with deep infiltrating endometriosis was an article published in 2014 based on a retrospective study of 1618 women with a preoperative clinical and ultrasound diagnosis of endometriosis. That was an article by Di Donato et al entitled “Prevalence of adenomyosis in women undergoing surgery for endometriosis” published in the European Journal of Obstetrics & Gynecology and Reproductive Biology.3905 Adenomyosis was found in only 21.8% of the women studied (353/1618). It is evident from the study’s findings that endometriosis and adenomyosis do not always go hand in hand.
4948 Second, there is some doubt about whether Mrs Gill has or had adenomyosis.
4949 Adenomyosis is a benign condition of the uterus, defined by the presence of fragments of endometrial tissue (the mucous membrane lining the inside of the uterus) within the myometrium (the wall of the uterus).3906 Associate Professor Lam explained that it is a term used to describe the findings of islands of endometrial-like cells and glands in the deep muscle layer of the uterus. “Adeno” means gland, “myo” muscle, and “osis” growth.3907 Endometriosis is a condition resulting from the appearance of endometrial tissue outside the uterus,3908 primarily on the pelvic peritoneum, ovaries and rectovaginal septum.3909
4950 The symptoms of adenomyosis are dysmenorrhea (painful periods), dyspareunia, abnormal uterine bleeding and infertility.3910
4951 According to Di Donato et al (2014), adenomyosis is “a difficult diagnosis, often established only at pathological examination of the hysterectomy specimen”. The authors acknowledged, however, that improvements in transvaginal ultrasonography have facilitated the making of a more detailed assessment of the architecture of the uterus.
4952 Mrs Gill was diagnosed on the basis of an ultrasound. According to the report she had evidence of a myometrial echo pattern (an echo pattern in the muscular layer of the wall of the uterus) on a transabdominal and transvaginal ultrasound in November 2008 and a posterior vaginal wall thicker than the anterior wall. The radiologist, Dr Diana Davies, stated that these findings were consistent with adenomyosis and she concluded that Mrs Gill had uterine adenomyosis.3911
4953 The ultrasound was ordered for the purpose of seeing whether Mrs Gill had any fibroids and, if she did, what size they were. But Dr Davies reported that there was no evidence of fibroids. The first evidence of a fibroid appears to be from a pelvic ultrasound performed on 12 June 2014. That fibroid was reported as measuring 5.4cm.3912 Apart from the presence of the posterior uterine fibroid, the ultrasound was reportedly normal. No mention was made of the myometrial echo pattern previously reported or the differential in the anterior and posterior vaginal walls found in 2008 which Dr Davies had interpreted as adenomyosis.
4954 A later ultrasound taken on 16 March 2017, however, was reported as showing “diffuse heterogeneous echotexture of the myometrium in keeping with adenomyosis” which the radiologist, Dr Sirisha Madhala wrote, “suggested” adenomyosis. She also found a 7cm fibroid.3913
4955 Third, even if it were to be accepted that Mrs Gill more probably than not had adenomyosis, there is no evidence that Mrs Gill ever had endometriosis. In fact such evidence as there is appears to exclude it.
4956 In the article to which I referred above, Di Donato et al (2014) made an ultrasound diagnosis when ovarian endometriomas or endometrotic nodules were visualised.3914 According to the reports, neither ultrasound that Mrs Gill underwent revealed ovarian endometriosis or endometic nodules.
4957 Apart from colonoscopy, none of the investigations Associate Professor Lam suggested was apparently carried out. Neither of the 2007 colonoscopies, however, disclosed endometriosis. The colonoscopy performed on 3 October 2007, after an episode of rectal bleeding, was normal.3915 None appear to have been conducted since.
4958 The pathologist who examined the tissue fragments removed during the operation to insert the Mirena on 1 December 2008 reported that there was “no evidence of endometriosis”.3916 The endometrium was reported as normal in the pelvic ultrasound of 16 March 2017.3917 Professor Korda confirmed that there was no evidence in Mrs Gill’s history that she ever had endometriosis.3918 Unlike Associate Professor Lam, he had examined Mrs Gill and he did not accept that her symptoms of pelvic pain, bowel dysfunction, painful bowel movements or dyspareunia were due to endometriosis or, for that matter, could be explained by anything apart from the implantation of the Prolift Total.
4959 Professor Korda also observed that the mere fact that Mrs Gill had a fibroid and that fibroids and endometriosis can co-exist does not mean that Mrs Gill had or has endometriosis. He said that nothing in either her history or her clinical notes suggests the presence of endometriosis.3919 He characterised Professor Lam’s opinion (that because Mrs Gill has a fibroid she is also likely to have coexistent endometriosis) as “highly speculative” and his contention that the temporary relief experienced as a result of the Mirena IUD as “equally theoretical”. He pointed out that the Mirena IUD is effective in treating pelvic pain, dysmenorrhea and abnormal uterine bleeding in the absence of endometriosis.3920
4960 None of Mrs Gill’s treating doctors expressed the opinion that she had endometriosis. Dr Leake’s operative findings did not include endometriosis. The IUD was not inserted because she had endometriosis.
4961 I am not satisfied that Mrs Gill has ever had endometriosis. The evidence rises no higher than a suspicion.
4962 On the other hand, I cannot exclude the possibility that she has adenomyosis and, having regard to the whole of the evidence, I think it more likely than not that she does. But it is a vexed question whether it was a cause of any of Mrs Gill’s symptoms. Contrary to what was implicit, if not explicit, in the respondents’ submissions, all of Mrs Gill’s treating doctors did not agree that adenomyosis was responsible for some or all of her symptoms. In fact, as far as I can tell, only Dr Jeffrey was of that opinion and then only in 2016.
4963 As Professor Korda observed, adenomyosis was diagnosed long after the mesh had been inserted. He said that it classically causes heavy bleeding but “not much pain”, and that the pain associated with adenomyosis is usually pain during menstruation.3921
4964 Notably, when Dr Jeffrey examined Mrs Gill in August 2014, he did not mention adenomyosis. At that consultation, it will be recalled, he confirmed the presence of a small suburethral mesh ulceration and found very significant tenderness in the posterior vaginal wall and extreme tension and tenderness in the right-sided mesh arm. Palpation of this mesh arm produced a significant episode of pain radiating from the rectum to the pelvic sidewall and down the right leg. He told Mrs Gill that contraction of the mesh was responsible for her symptoms “becoming more obvious as the mesh shrinks in response to shrinking scar tissue” and said that they were only going to get worse after menopause.3922 Associate Professor Lam said that dyspareunia could be due to painful mesh contraction.3923 Professor Korda agreed.3924 In cross-examination, Associate Professor Lam accepted, in effect, that where palpation of contracted mesh reproduces the pain experienced by women on movement and during sexual intercourse, the pain is attributable to the contracted mesh.3925
4965 On bimanual assessment Dr Jeffrey noted the presence of the “bulky fibroid uterus” and said that it was putting pressure on the rectum. But he did not say that it was responsible for any of her symptoms.
4966 Mrs Gill was referred to Dr Tsokos for a second opinion. In the referral letter, Dr Jeffrey wrote that she had “terrible problems from mesh ulcerations and tension”.3926 Her symptoms at that time were “relatively persistent right groin pain and an intermittent rectal and low abdominal pain usually associated with defecation of a large stool but perhaps most troubling is her persistent dyspareunia”.3927 He referred to the bulky uterus but did not mention adenomyosis. Nor did he suggest that the fibroid or the bulky uterus was causing any of these symptoms.
4967 When Dr Jeffrey reviewed Mrs Gill in February 2016, however, he thought that the pain she was then experiencing was “more related to her uterus than her previous mesh”. On examination he found her uterus to be particularly tender to move and that compression of the uterus produced a lot of the pain she was experiencing. At that time he thought that uterine adenomyosis was probably the cause of a significant amount of her pelvic discomfort.3928
4968 In the light of Dr Jeffrey’s examination finding, I am persuaded that the predominant source of the pain at this time was the uterus and that adenomyosis was a possible, if not probable, explanation.
4969 On 15 April 2016 Dr Vashevnik noted the presence of the intramural posterior fibroid and felt that many of Mrs Gill’s pain symptoms were due to the bulky uterus and the fibroid. He was convinced that the bulky uterus and fibroid were “a cause of her increased bleeding and the ineffectiveness of the Mirena and the applicants did not argue to the contrary.3929
4970 Dr Vashevnik’s report of the same date records that he carried out a physical examination but, apart from noting the absence of mesh erosion in the vagina, his only finding is the presence of the bulky uterus with IUD in place. The history in the report provides little insight into his reasons for thinking that many of Mrs Gill’s pain symptoms were due to the bulky uterus and the fibroid.
4971 On 21 April 2016, however, when Professor Korda examined Mrs Gill, the only areas of tenderness he found in the vaginal area were in the right sacrospinous ligament and the left vaginal vault.
4972 Associate Professor Lam testified that Mrs Gill’s fibroid was “clearly quite large” and could cause back pain and urinary pressure symptoms, including frequency, urgency, and urge incontinence, and could cause or aggravate urinary stress incontinence. But he did not examine her.
4973 Associate Professor Lam also said that in patients who suffer from chronic pain, any trigger factors that might arise from these sort of conditions could either trigger pain or contribute to different manifestations of the pain of which the patient complains. 3930
4974 Professor Korda did not accept that the fibroid was significant. He said that 10 years after mesh insertion a posterior fibroid of modest size could not be causing her (pain) symptoms.3931 Indeed, he said that fibroids of the size that were ultimately found in Mrs Gill “never cause pain unless they undergo some form of degeneration which in this case was not so”.3932
4975 The most recent evidence from Professor Vancaille and Dr Brown does not suggest that the pain is due to either the fibroid or the adenomyosis.
4976 I accept that adenomyosis might explain some of Mrs Gill’s pain in 2016, when it was first diagnosed by a clinician. I also accept that the “crampy pelvic pain associated with the [breakthrough] bleeding” that she reported to Dr Vashevnik also in 2016 might also be explained by adenomyosis. In the absence of any evidence that adenomyosis was present before the ultrasound of 3 November 2008, however, I am not persuaded that it could account for any of the pain she felt before then. Furthermore, in the absence of any evidence from the treating doctors that adenomyosis accounts for any of the pain Mrs Gill experienced before 2016, I am not satisfied that it does.
4977 Mrs Gill gave unchallenged evidence that she first felt constant aching pain in her coccyx, sporadic period-like pain with coughing or sudden movement, and “terrible” pain on defaecation (after her Prolift surgery but before her first excision procedure in June 2007). She first experienced pelvic pain following the Prolift surgery.3933 She first noticed pain with defaecation in about February 2007, the month after the Prolift surgery.3934 When she and her husband attempted sexual intercourse, she felt too much pain to continue.3935 While she improved after her surgery in 2008, her symptoms returned in 2013 at a time when in all likelihood there was an incipient erosion.
4978 The improvement in vaginal comfort and resumption of sexual intercourse without pain that Mrs Gill reported to her GP in September 2008 followed the first second excision surgery and tends to support the conclusion that the mesh was responsible for the dyspareunia and pelvic pain. The evidence does indicate that Mrs Gill suffered from dyspareunia before the Prolift surgery.3936 When she suffered pneumonia in mid-2004 she said she experienced pain with intercourse and a host of other symptoms.3937 The evidence is not entirely clear as to whether she continued to have pain with intercourse after she recovered from the pneumonia. But the weight of the evidence indicates that she did not. After she recovered from pneumonia, the medical records do not contain a reference to such a complaint until after the Prolift surgery. When she saw Dr Natalwala in February 2005, she complained about a vaginal lump and, although she said she was unable to have intercourse, Dr Natalwala’s report makes no mention of pain or dyspareunia.3938 When she first presented to Dr Chapple in October 2006 her libido was reduced, but there is nothing to suggest that she had dyspareunia.3939 While she had difficulties with intercourse before she received the Prolift implant, apart from a period of time in 2004, those difficulties related to the lump caused by the prolapse.3940 Dr Leake, who last saw Mrs Gill in July 2011, was convinced that all of the gynaecological symptoms with which she presented to her were secondary to the use of the Prolift mesh. She noted the resumption of sexual intercourse in 2008 only a relatively short time after the mesh had been removed and the erosions had healed and the disappearance of various symptoms including the bowel-related symptoms. Only the persistent bleeding, she said, was possibly unrelated.3941
4979 Associate Professor Rosamilia attributed the considerable symptom relief in the five years between 2008 and 2013 to the success of the mesh excision surgery.3942 She was uncertain about whether the mesh was responsible for the exacerbation of the pelvic pain at the end of this period. While she noted that several factors could contribute to chronic pelvic pain, including adenomyosis, she did not attribute Mrs Gill’s subsequent pain to any other condition. Dr Dowling noted the presence on the CT scan of a 5.6cm uterine fibroid but did not implicate it as a cause of Mrs Gill’s symptoms.3943 When Mrs Gill was admitted for surgery in August 2013, two centimetres of mesh were exposed in the anterior wall and bands formed by scar tissue from the previous mesh surgery were apparent in the rectum. There were also histological signs of infection, which probably accounted for some of the pain. The histopathologist, Dr Peter Nguyen, reported that the anterior vaginal wall mesh was “associated with mixed inflammation and a collection of actinomyces like organisms.3944 A report for Ethicon on mesh erosions approved by Dr Peter Meier, apparently written in 2010, stated that actinomyoces have been implicated “as causative organisms associated with vaginal mesh infections”.3945 The 2015 Gynemesh PS CER referred to a case study in 2014 of a patient with actinomyces infection five years after implantation with Gynemesh PS.3946
4980 Professor Vancaille’s examination findings in July 2017 tell against a uterine explanation for Mrs Gill’s pain. He noted exquisite tenderness in the right Alcock canal (a structure in the pelvis through which the pudendal nerve passes, also called the pudendal canal). He found residual mesh palpable a few millimetres above the canal and about a centimetre caudal from the ischial spine. He recognised that the uterus was enlarged by the fibroid but he described it as “mobile and non-tender”. He did not attribute any of her pain to a uterine cause. In his letter to the GP he said that the uterine fibroid did not need to be treated. He diagnosed a “chronic pain syndrome (pudendal neuropathy) secondary to mesh insertion and removal surgery”.3947
4981 On the balance of probabilities the pain Mrs Gill experienced after her Prolift surgery in her pelvis, coccyx, her groin, her lower back, her vagina and her rectum was caused by the implantation of Prolift Total, mesh infection, mesh erosions or exposure, and the surgery undertaken to treat the exposures. While there may have been some contribution from the bulky uterus, if there was, it did not occur before 2016 and it was short-lived.
4982 Apart from the fibroid, the bulky uterus and the adenomyosis, the respondents pointed to no other alternative cause for Mrs Gill’s chronic pain. Mrs Gill did hurt her back in 1992 but the unchallenged evidence was that, before the Prolift implant, she had only had intermittent lower back pain which was treated with physiotherapy and remedial massage.3948 She said that over the previous five or six years, it occurred less than two or three times a year and usually disappeared within a few hours of stretching.3949 The pain she reported after receiving her Prolift implant, was of an altogether different character. She described pain within her vagina and rectum; pain in the right groin; “a sharp, sudden pain, which is almost like the feeling of a tearing injury felt in [her] lower pelvic region on both sides”; and pain around her coccyx.3950
4983 On the other hand, I am not satisfied that the vaginal bleeding and spotting are attributable to the implantation of Prolift. Nor am I satisfied that, with the exception of one discrete period of time, Mrs Gill’s inability to use tampons, another of the listed disabilities, can be attributed to Prolift. On Mrs Gill’s own account, the prolapse for which she was treated with Prolift prevented her from using tampons and that is what made things difficult for her when swimming and snorkelling.3951 On 26 July 2007, after her prolapse repair but before the first excision procedure, Dr Yin recommended against the use of tampons because the mesh was “quite prickly” and was “likely to collect cotton material”.3952 It follows that the inability to use tampons and any consequential interference with Mrs Gill’s enjoyment of life was related to the Prolift implant from 26 July 2007 until the time of the first mesh excision. Thereafter the evidence is silent. In the absence of evidence, I am unable to find that, if she had any inability to use tampons after the mesh was excised or at least after she recovered from the first mesh excision, it was caused by the Prolift implant.
Bowel and gastrointestinal symptoms
4984 A further point of contention concerned Mrs Gill’s bowel and gastrointestinal symptoms.
4985 The respondents referred to Mrs Gill’s long history of chronic constipation and obstructive defecation and Dr Jeffrey’s finding in August 2014 that she had “a bulky fibroid uterus which [was] putting pressure on the rectum”. They noted the reference in the report of the physiotherapist, Celia Bolton, that Mrs Gill had experienced faecal infrequency, straining and bloating even before she had given birth to her children.3953 They also referred to Dr Natalwala’s observation in 2005 that Mrs Gill had partial nerve damage in conjunction with her fascial damage.3954
4986 The only colorectal surgeon to give evidence, Dr Meagher, was of the opinion that the Prolift implant was the cause of most of Mrs Gill’s colorectal symptoms. He wrote in his report:
I don’t think there’s any real doubt that the Prolift implant did more likely than not cause physical damage to her body. Specifically the areas of colorectal function which have been affected include the need to take Movicol to avoid large bowel movements. When Mrs. Gill has large bowel movements she suffers really severe episodes of pain.
Mrs. Gill also suffers ongoing chronic background pain which is difficult to describe - it does appears to be related to the mesh and the scarring around the mesh and at times appears centred on the coccyx.
She also suffers a degree of faecal urgency, incontinence of flatus and incontinence of liquid faeces as a result of the surgery and partly as a result of the need to take Movicol to avoid the pain. There is also a degree of faecal urgency and Mrs. Gill needs to stay near a toilet and again is concerned about the effect this may have on her work.3955
4987 Dr Meagher noted that, although she strained a lot before her prolapse surgery, she had not had any pain.
4988 One premise for the opinions he expressed was that, although she had symptoms of a rectocoele (consisting of the need to push on the rear wall of her vaginal to begin defaecation), Mrs Gill did not suffer significant pain in the pelvis or coccyx or perineal areas before her Prolift surgery. Earlier in his report he had referred to her description of her pre-existing defaecatory problem in recording her colorectal medical history and observed that it sounded very much like “a straightforward rectocoele”.3956 The respondents, seized upon this observation and submitted that Dr Meagher misunderstood Mrs Gill’s presenting symptoms in 2005–6 and, for this reason, his opinion should be rejected:
As Mrs Gill’s presenting symptoms were described in February 2005 by Dr Natalwala as “quite an impressive Grade II-III cystourethrocoele” and “a Grade II-III rectocoele with poor perineal support”, Dr Meagher’s conclusion that all of her colorectal complaints can be levelled at the Mesh Implant should be rejected.
4989 They submitted that Mrs Gill presented with unique symptoms of extreme uterine prolapse and a rectocoele and those symptoms included defaecatory symptoms and constipation and were also indicative of fascial and nerve damage. Consequently, they argued, the Court could not be satisfied that her colorectal symptoms (at least in part) are the result of her Prolift implant and the subsequent surgeries.3957
4990 I reject the submission.
4991 First, Dr Meagher was referring to Mrs Gill’s colorectal history, not her prolapse history. He was not suggesting that she only had a single compartment prolapse. While her cystourethrocoele was apparently “quite impressive”, I do not understand why that made it “extreme”. No evidentiary foundation was laid for the submission that Mrs Gill’s symptoms were unique.
4992 Second, the presence of an impressive grade II-III cystourethrocoele, which affects the anterior wall of the vagina, was beside the point. As I understand the position, the cystourethrolcoele caused urinary incontinence. There is no reason to think that that had anything to do with her pre-existing or subsequent colorectal symptoms.
4993 Third, while Mrs Gill did have a long history of constipation and obstructive defaecation, the colorectal symptoms which Dr Meagher attributed to the implantation of the Prolift were not symptoms of constipation or obstructive defaecation. They were the severe pain she felt with large bowel movements necessitating the use of Movicol to soften the stools and faecal urgency, flatal incontinence, and faecal incontinence. In oral evidence Mrs Gill explained that in order to reduce the extreme pain, she takes “a lot of laxatives” every day. She said that the laxatives reduce the muscle spasms but “it’s a fine balance because if I take too much then I lose control”.3958
4994 There is no history of pain on defaecation or of faecal urgency, flatal incontinence or faecal incontinence before the Prolift surgery. Dr Meagher was careful to note a total absence of symptoms of faecal incontinence at the time Mrs Gill was seeing Dr Chapple. He recorded that Mrs Gill had only had flatal incontinence “following all she has been through with the mesh” and had never suffered any faecal urgency or incontinence of either flatus or faeces, liquid or solid, before the Prolift surgery. He also took note that before the Prolift surgery she had never had the kind of pain she experienced after it:
Mrs. Gill then explained that she never had these type of pains prior to the operation. Essentially she never thought about pain in the region until she'd had the mesh operation — she was just keen to try and avoid pushing on the back wall of her vagina to have a bowel movement but realises now that in fact a lot of women needed to do that. That symptom really did not get in the way of her quality of life.3959
4995 Professor Korda rejected the proposition put to him in cross-examination that Mrs Gill’s pre-existing constipation would be likely to be associated with pain and said that he did not think that constipation causes pain.3960 Although Dr Natalwala took a history of chronic constipation when he saw Mrs Gill in 2005, he did not record a history of pain in association with constipation. Indeed he did not mention pain in any of his reports.3961
4996 Fourth, while Dr Natalwala noted that Mrs Gill had suffered partial nerve damage with the prolapse well before the Prolift surgery, the respondents pointed to no evidence connecting Mrs Gill’s post-Prolift pain or other colorectal symptoms to that nerve damage.3962
4997 In cross-examination Dr Meagher maintained that Mrs Gill’s chronic pain, the pain on large bowel movements, the pain with orgasms, and faecal incontinence could not be attributed to a fibroid of the size described by Dr Jeffrey.3963
4998 Associate Professor Shek, who saw Mrs Gill in April 2016, observed that Mrs Gill “still strains at stool and there is incomplete bowel emptying”, no doubt symptoms of constipation and obstructive defaecation respectively.3964 Associate Professor Shek did not attribute these symptoms to her Prolift repair.
4999 Associate Professor Shek also saw Mrs Gill in June 2016 and noted that the obstructive defaecation had improved after the prolapse repair but there had been a “new onset pain on defaecation with a bulky and hard stool”.3965
5000 The simple point is that while Mrs Gill had constipation and a sensation of incomplete bowel evacuation before being implanted with Prolift, she did not experience pain with constipation or bowel movements until after Prolift implantation. Dr Kriel considered that Mrs Gill’s pain was significantly exacerbated by constipation, amongst other things. But the respondents must take Mrs Gill as it found her. Her pre-existing constipation only made the pain worse than it might otherwise have been.
5001 In my opinion it is more likely than not that the symptoms identified by Dr Meagher of pain on bowel movements and while straining to defaecate were caused by the Prolift implant and the faecal and flatal incontinence and faecal urgency by the surgery and the need to take Movicol to avoid the pain. Dr Yin felt that the pain with a large bowel movement was related to the compression effect of the posterior bands or scarring.3966 Similarly, Dr Dowling said that the presence of the bands could contribute to poor bowel emptying.3967 I was not taken to anything in the contemporaneous medical records that would undermine the assumption made by Dr Meagher, based on the history given to him by Mrs Gill, that none of the symptoms which Dr Meagher attributed to Prolift or to the use of Movicol had been present before the Prolift surgery.
5002 I therefore find that, notwithstanding Mrs Gill’s long history of obstructive defaecation, the Prolift surgery caused a multitude of other colorectal problems, including chronic pain, pain on large bowel movements, pain when straining to defaecate, pain with orgasms, and faecal and flatal incontinence.
5003 Associate Professor Lam testified that fluctuating pain with bowel movements could be associated with the Prolift repair because the posterior Prolift arms were inserted through the sacrospinous ligament.3968 He added, however, that it would tend to be more constant and, if it occurred, it would occur in the early post-surgical healing phase when fibrosis takes place. He said after that phase is complete, such pain tends to disappear or resolve spontaneously. That did not happen in this case, however, perhaps because of Mrs Gill’s chronic inflammation.
5004 The next point of contention relates to the psychiatric injuries of a generalised anxiety disorder (item 15) and an adjustment disorder with depressed mood (item 16).
5005 These were Dr Jungfer’s diagnoses. Dr Jungfer also diagnosed a chronic pain disorder, which she explained was a reference to chronic physical pain. She said that there was a component in her clinical presentation that suggested neuropathic pain, that is, abnormal activation of the pain fibres that regulate and manage pain.3969 She attributed all these conditions to the Prolift surgery and its complications.3970
5006 In her first report, Dr Jungfer expressed the opinion that Mrs Gill’s anxiety, the generalised anxiety state and panic attacks directly arose as a result of the complications following the Prolift surgery.3971 She explained that, before the surgery, Mrs Gill was known to be very organised and methodical and “tried to contain uncertainty within her environment”. Since then, however, she has suffered anxiety about her future in the light of her inability to obtain “confirmatory evidence” that no further damage will occur. The pain and the consequential restrictions on her activities exacerbate and worsen her anxiety state. While she was subjected to other stressors at the same time, Dr Jungfer considered that, unlike the others, the mesh-related stressors were unknown and uncontrollable.3972 She said that Mrs Gill’s personal history of depression and her family history of anxiety disorders made her more vulnerable to the development of psychological distress.3973
5007 The psychiatric history revealed in the affidavits of Mr and Mrs Gill and by the contemporaneous medical evidence is as follows.
5008 In 1999, following the death of her father, Mrs Gill was diagnosed with depression. She was treated with counselling and antidepressants. Recovery took about 12 months.3974 After the birth of each of her two children she was diagnosed with post-natal depression and treated again with antidepressants.3975 After the birth of her oldest child, the marriage came under strain.3976 In the lead-up to her first revision surgery on 10 September 2007, she was often distressed and teary. She was struggling physically and with the uncertainty surrounding her long-term prognosis. The marriage came under further strain at this point.3977 Four months after the first revision surgery, in January 2008, she was depressed again and reported problems with her marriage.3978 In February 2008 the GP took a history that she had a poor sexual relationship due to colitis, the prolapse surgery, and the “mesh repair problems”.3979 By May 2008 she told Dr Yin she was in marriage counselling.3980 Later that month she was “very upset and depressed” when told she needed another operation.3981
5009 For a time after the surgery in June 2008, Mrs Gill continued to struggle with depression. In September 2008 Dr Flynn’s progress notes indicate that she had been depressed for four years but they also record “multiple problems” following the Prolift repair and the revision procedures, including painful sex.3982 She was back on antidepressants and, notwithstanding her improvement after the June 2008 operation, she remained depressed and the marital strain continued.3983 In mid-June 2009 Mrs Gill deposed that she had “a bit of a meltdown” and found herself crying constantly.3984 In July 2009 she mentioned to her GP that a male friend died at 53 of lung cancer leaving behind two young children.3985 In August 2009 she was referred to a psychologist, Flavia Bises, who explored with her issues relating to her father and her marriage.3986 Throughout 2010 Ms Gill regularly consulted another psychologist, Debra Roberts. She reported experiencing hyperactivity, difficulty sleeping, hot flushes and irritability.3987 In November 2010, in a letter to Dr Flynn, Ms Roberts reported that Ms Gill was committed to gaining insight into her cognition and behaviours, had engaged well in therapy, and had developed coping skills.3988 The same month Mrs Gill presented to her GP with worsening depression and marital problems.3989 On 11 November 2010 she saw a psychiatrist, Dr Ataris. At that time she seemed to be focussed on the problems in the marriage. He diagnosed her with a major depressive episode.3990 He saw her again in December 2010 when he felt that the relationship issues were “pivotal in her recover[y] phase”.3991 By March the following year, after a holiday and a reduction in work commitments, her mood had improved significantly.3992
5010 Within five months, however, she was not coping as well, compulsively eating, “fighting and crying”, according to her GP’s notes.3993 In late September 2011 she told the GP that she felt she had been getting terrible side effects from her antidepressant (Cymbalta), including hot flushes and irritability and her medication was changed to Efexor.3994 In October 2011 Dr John Terry diagnosed a depression in partial remission with perpetuating symptoms of stress and arousal in the context of multiple biopsychosocial stressors. The biological stressors he identified were the prolapse with complications including multiple operations in an 18 month period, low libido, “sciatica”, hypersomnia, and “physical symptoms of stress and anxiety”. The psychological stressor was described as “mood low usually related to circumstances”. The social stressors were her husband’s job loss, his accident (in 2010) leading to partial blindness, (caring for) two young children, the death of her father in 1999, the separation from her family, and the marital relationship.3995 Ms Gill then embarked on counselling with Dr Maretha Cronje. At the same time her husband was in therapy.3996
5011 Anxiety and depression continued to be problems during 2012, then apparently focussed on persistent marital issues, and 2013.3997 In July 2013, however, when Dr Dowling recommended she undergo further surgery to change her IUD and treat any further mesh exposure, Mrs Gill became very anxious and tense.3998 Her anxiety increased as her symptoms deteriorated and she worried about whether she might be one of the women who had received a defective mesh implant and that her problems might be long-term.3999 She underwent further treatment with another psychologist, Helen Handsjuk, for “sex counselling – anxiety issues due to recurrent gynae surg since had mesh repair”.4000
5012 Having undertaken an extensive review of the clinical records, Dr Samuels expressed the opinion that the symptoms Mrs Gill had experienced since January 2007 were most in keeping with a persistent depressive disorder (dysthymia).4001 He also diagnosed her with a somatic symptom disorder with predominant pain of moderate severity.4002 On the critical question of causation he said that it was possible that the implant surgery and the subsequent treatment resulting from it worsened her underlying depressive and anxiety symptoms and that they were “not insignificant stressors”. Having regard to her history of chronic depression, longstanding relationship issues, and her husband’s head injury and its consequences, he said that he was unable to say that it was more likely than not that her psychiatric illness resulted from the implant surgery and subsequent treatment.4003 He considered that Mrs Gill had been suffering from a persistent depressive disorder that had been present in varying intensity at least from the birth of her first child.4004 On the other hand, he also wrote:
It is difficult to accurately weight the various stressors in terms of relative contribution to her current psychiatric and psychological illness. However, I would see her pre-existing history of depression, relationship issues, dislocation from family in Victoria in the early child rearing years, the implant surgery and complications and finally her husband’s very severe head injury as all being factors highly relevant to the development of these problems.4005
5013 Dr Jungfer disagreed with Dr Samuel’s diagnoses.
5014 The gap between the competing views did not close after cross-examination but it narrowed.
5015 While Dr Samuels would not accept that Mrs Gill had an anxiety disorder, he did accept that she had anxiety symptoms, which he considered to be a component of the depressive condition.4006 He did not dispute that she was in genuine and significant pain4007 and agreed with Dr Jungfer that “there are problems of chronic pain”.4008 Indeed, at one point in the cross-examination he expressed the view that they were largely saying the same things.4009
5016 Dr Samuels also made it clear that he was not denying that there was a connection between the Prolift and Mrs Gill’s depression and anxiety. His opinion was that the Prolift surgery and the complications post-surgery had exacerbated an underlying tendency to depression and anxiety.4010 Moreover, he readily agreed that, regardless of the nomenclature, Mrs Gill’s underlying psychiatric disorder became significantly worse as a consequence of the mesh complications.4011
5017 There is no doubt that Mrs Gill had was subjected to multiple stressors both before and after the Prolift surgery which resulted in episodic depression. Dr Jungfer considered that she had a major depressive illness precipitated by the birth of her children, a depressive reaction to the death of her father, and that the traumatic brain injury sustained by her husband had a deleterious effect on her mental health.4012
5018 In my view the depression was multifactorial. The adverse consequences of the Prolift surgery were a significant contributing factor but they were not the only cause. The marital relationship was a significant stressor. That said, although there were difficulties in the marriage well before the Prolift surgery, the evidence indicates that the relationship deteriorated considerably as a result of the complications of Prolift. That is particularly evident from the report of Ms Handsjuk of 14 December 2013.4013 Dr Jungfer concluded that the issues in the relationship became “long-term” because of the multiple gynaecological problems.4014 Mr Gill stated in his affidavit that his wife’s “pain and restrictions continue to take a heavy toll on her” and that her unhappiness and frustration about her inability to do the things she once could continue to impose a strain on their relationship.4015 If there were any doubt about the relationship between the marital problems and the mesh complications, it was put to rest by Mrs Gill’s evidence, which was not challenged in cross-examination. Asked in chief about her relationship with her husband, Mrs Gill testified:
And your relationship with your husband, could you just tell the court how that stands?- --Okay. It’s not easy. The early stages of – of this – when it was not understood for my pains and the bleeding and problems were caused by the mesh, it was largely looking at it that it was my fault or – he – he took it as a rejection, that I couldn’t be intimate with him and didn’t understand that it was sincere pain and an actual medical problem. He does now. But that put us under a lot of pressure because I felt I wasn’t being heard and not supported, and he felt that I wasn’t interested in him, and he really struggled with that and I really struggled with that. Now that he understands that it is a medical issue, he’s a lot more sympathetic. However, it’s still not easy. Neither of us are happy about it. Neither of us enjoy being in this situation. It’s not where we wanted to be, and – and it’s very hard.
Can you be intimate?---Not really, no. This – I think this year we tried once, and – yes, it’s – it usually causes me pain and he doesn’t want to hurt me, and I’m very anxious about having pain caused when it’s meant to be something that’s fun, it’s no longer. It’s – it’s a terrible stress.
5019 It is difficult to determine the correct diagnoses.
5020 Dr Jungfer disagreed with Dr Samuel’s diagnosis of dysthymia, based on Mrs Gill’s clinical presentation at the time she saw her in April 2016. She said that at that time her clinical presentation was “more substantially one of an anxiety diathesis [state] and that episodes of depression [were] reactive to various changes in her state of health”.4016
5021 Dr Jungfer gave the following reasons for diagnosing an adjustment disorder (without alteration):
I was of the opinion that at the time that I saw her that she had developed and adjustment disorder which are emotional or behavioural symptoms in response to an identified or identified stressors and that these symptoms were clinically significant by causing marked distress. The intensity of the stressor have caused significant impairment in social, occupational, or other important areas of functioning. The stress-related disturbance should, according to DSM-5, not meet the criteria of another mental disorder or be merely an exacerbation of a pre-existing mental disorder and should also not represent normal bereavement.
Adjustment disorders are considered to be a stress response syndrome. It is recognized that they vary widely in terms of their incidence within the population. The duration criteri[on] of an adjustment disorder is that the symptoms do not persist for more than six months once the consequences or stressor has been terminated.
5022 In my opinion, the diagnosis of adjustment disorder caused by the complications of Prolift is inapt. More likely than not Mrs Gill had an underlying depression that was periodically controlled by medication but which was aggravated by a number of stressors including the Prolift complications. Two years before the Prolift surgery, when she was referred to Dr Natalwala, the referral letter discloses that she was on Lexapro, an anti-depressant,4017 and in September 2006, when she was referred to Dr Chapple, the referral letter reveals that she was on Lexapro.4018 Mrs Gill would not have been prescribed Lexapro if she were not suffering depression or a mood disorder. When she consulted Dr Terry on 24 October 2011 he noted that she had a history of depression dating back to the age of 34 and she described depressive symptoms which started six to seven years earlier.4019 On the basis of this evidence I am not satisfied that she had no underlying mood disorder in the two years before the Prolift surgery. Nor do I believe that Mrs Gill’s own evidence indicates otherwise. She said in her first affidavit that she found the counselling she had received after the birth of her second child beneficial and her symptoms improved. She did not say that she became symptom-free. To the contrary, she said that her recovery was “complicated” by the development of the prolapse.4020
5023 In concluding that Mrs Gill had a somatic symptom disorder, Dr Samuels referred to episodes in Mrs Gill’s medical history which, he said, suggested that she has a tendency to somatise and to express her psychological distress through physical symptoms. The instances he cited included presentations to hospital emergency departments with palpitations and shortness of breath in October 2004 and in 2009; the admission to Joondalup Emergency Department in February 2007 with bowel symptoms only partially explained by clostridium difficile, persistent chronic pain which prevented her from working more than three days a week, and recent episodes of acute pain that have disrupted family events. He noted that the criteria for the disorder include:
disproportionate and persistent thoughts about the seriousness of one’s symptoms;
persistently high levels of anxiety about health and symptoms;
excessive time and energy devoted to these symptoms or health concerns.
5024 Dr Jungfer disagreed with the diagnosis of somatic symptom disorder based on the absence of evidence indicating that Mrs Gill’s concerns about her symptoms were excessive or that she devoted excessive time to them.4021 Moreover, she argued that Mrs Gill’s conduct in persisting with certain forms of treatment and not seeking medical treatment for relatively long periods of time were inconsistent with somatic symptom disorder.4022
5025 I accept that the palpitations and shortness of breath may well have been symptoms of anxiety, rather than of an underlying physical illness. According to Dr Samuels’ report (the hospital notes do not appear to have been tendered), she was admitted to hospital, possibly Sir Charles Gairdner Hospital, on 12 October 2004 with palpitations mostly associated with stress but more noticeable over the previous few days. There was no ventricular ectopic episode and there were no “sinister” symptoms, and a 12 lead ECG was normal.4023 There is no satisfactory evidence of similar episodes in 2009.
5026 But Dr Jungfer was right to reject the diagnosis of somatic symptom disorder. The evidence does not indicate that Mrs Gill’s concerns about the seriousness of her symptoms were or are disproportionate or that she devoted excessive time or energy to them.
5027 Based on the evidence Mrs Gill gave about her anxiety symptoms, the medical history and the reasons given by Dr Jungfer, I am persuaded, in addition to an aggravation of her underlying depression, that Mrs Gill developed a generalised anxiety disorder in response to the complications of Prolift and the revision surgery. In coming to this conclusion I recognise that there were anxiety symptoms that preceded the surgery, including (based on the history to Dr Ataris) during her second pregnancy, but they were episodic.4024 The diagnostic criteria for a generalised anxiety disorder according to the DSM-V (the Diagnostic and Statistical Manual of Mental Disorders), upon which both psychiatrists relied, include excessive anxiety and worry about a number of events or activities occurring more days than not for at least six months. It is not possible to determine when it began but I have little doubt that it was present by 2010 and had begun to develop before then but after the Prolift surgery.
5028 The final area of contention concerns the urinary symptoms.
5029 In his first report Professor Korda attributed all of Mrs Gill’s current symptoms and disabilities to the insertion of the Prolift device, the resultant erosion, and the multiple surgical procedures that were required to correct and manage her symptoms. This included the lower urinary tract symptoms of frequency, nocturia, stress incontinence, urge incontinence, a slow stream, and occasional incomplete bladder emptying. He did not explain why these symptoms could be ascribed to Prolift, its complications, or the subsequent treatment. With respect to all the urinary symptoms, his statement is difficult to understand since, at the same time, he acknowledged that before Mrs Gill was implanted with the Prolift device, she had urinary urge incontinence.4025
5030 The respondents argued that the evidence was insufficient to prove a causal connection between Mrs Gill’s urinary symptoms and the Prolift implant and subsequent operations. Since the urodynamic tests conducted by Associate Professor Shek disclosed that Mrs Gill had a stable bladder, they submitted that her urinary urgency, urge incontinence frequency and nocturia “could have been partially due to the large posterior intramural uterine fibroid putting extrinsic pressure on her bladder”.4026 The basis for the submission was a statement to the same effect by Associate Professor Lam. Associate Professor Lam said that the urodynamic finding of moderate stress incontinence should be interpreted with caution in the presence of extrinsic bladder compression from the large fibroid. He said that Professor Korda did not appear to have taken either possibility into account.4027
5031 Professor Korda answered Associate Professor Lam in his third report. He disagreed that these urinary symptoms could have been due to pressure from the fibroid on the bladder. He said that fibroids on the posterior wall of the uterus do not cause such symptoms and do not put pressure on the bladder because they are located away from the bladder, between the uterus and the sacrum. He considered that a posterior uterine fibroid could not cause compression of the bladder.4028 Professor Korda was not cross-examined on this evidence. Associate Professor Lam was not taken to it during evidence in chief. And the respondents did not advert to it in their submissions. In these circumstances, there is no good reason why I should not accept it.
5032 Nevertheless, I accept the respondents’ submission that Mrs Gill has not proved a connection between her urinary symptoms and the implantation of Prolift.
5033 In her first affidavit Mrs Gill stated that in 2004, more than two years before she received the device, that she felt a need to urinate urgently, that at times she did not make it to the toilet and “wet” herself, and that she was wet most days.4029
5034 In his report to Dr Prior of 7 February 2005, Dr Natalwala reported that “[s]tress incontinence is not a major problem but she does have urinary frequency, urinary urgency and severe urge incontinence, and is wet almost on a daily basis”.4030 Urinary frequency is defined as the need to pass urine eight or more times a day. Urinary urgency is a strong and sudden desire to void that is inappropriate. If not relieved, it can cause urge incontinence.4031 I would infer in Mrs Gill’s case that her symptoms of urinary frequency, urgency and urge incontinence were related to her vaginal prolapse.
5035 In his first report Professor Korda said that pelvic organ prolapse can affect bladder function and that prolapse-related urinary tract symptoms include, amongst other things, urinary frequency and urgency, a slow stream, and a feeling of incomplete bladder emptying.4032
5036 In cross-examination Professor Korda said that he thought the stress incontinence was worse because the urodynamic study by Associate Professor Shek showed that it was now moderate and therefore more of a problem than it had been when Dr Natalwala saw her, although Mrs Gill told Associate Professor Shek that she was not greatly bothered by it, which suggests otherwise. Professor Korda conceded, however, that it was not possible to say whether the Prolift surgery had exacerbated the condition or merely failed to stop it.4033
5037 For completeness, I should deal with another of the respondents’ submissions.4034 The respondents pointed to references in the medical reports of Mrs Gill’s treating doctors to a “tightness” in relation to the mesh arms. Dr Yin and Dr Jeffery mentioned it in their reports of 14 May 2008 and 14 August 2014. The respondents submitted that more likely than not this tightness was “attributable to Dr Chapple’s surgical technique rather than anything inherent in the Mesh Implant itself”, citing the following passage in Associate Professor Rosamilia’s second report:
The majority of transvaginal mesh repairs I have performed or assessed have not developed pain or dyspareunia; mesh exposure if it has occurred, has usually required 1 surgery for revision but sometimes more. The mesh-related cause in those women who been (sic) referred for pain and have an examination of tenderness has often been due to the arms being too tight, superficial and tense. The arms being placed too tightly occurs particularly with procedures that involved the passing of mesh arms into or through the pelvic ligaments such as the sacrospinous ligament as an anchor point and occurs more often when performed by inexperienced surgeons.
5038 The respondents then submitted that “[c]learly none of the Respondents can be held responsible for the decision by Dr Chapple to use the Mesh Implant in relation to Mrs Gill, nor for the technique adopted by Dr Chapple in relation to the Mesh Implant”.
5039 I reject the submission.
5040 The passage cited from Associate Professor Rosamilia’s report did not and could not exculpate the respondents from responsibility for the tightness of the mesh arms — far from it.
5041 First, implantation of Prolift Total was a procedure that involved the passing of mesh arms into or through the pelvic ligaments including the sacrospinous ligament as an anchor point. This was the procedure Ethicon recommended. The Prolift IFU stated that the posterior implant has two straps that are “secured in the sacrospinous ligament” either via a transgluteal or a vaginal approach.4035 Excessive tightness at the anchor points was a foreseeable consequence of implanting Prolift according to the recommended procedure. The evidence demonstrates that the respondents were aware before Mrs Gill’s surgery that placing mesh too tightly could cause constant pain.4036 The IFU warned against placing excessive tension on the mesh implant during handling.
5042 Second, while it is possible that he did so, there is no evidence that Dr Chapple did in fact place excessive tension on the implant. Mrs Gill’s counsel tendered Dr Chapple’s operation record and several other reports of his but he was not required for cross-examination.4037 If he had been, the respondents could have questioned him about his technique. One cannot infer from tightness in one mesh arm first detected more than a year after the surgery that the surgeon was responsible for it when the evidence demonstrates that scarring and mesh contraction can cause tightness.
5043 In any case, it will be recalled that Dr Leake, who was also not required for cross-examination, deposed that from the outset gynaecologists, without any defect in their surgical technique, made the mesh too tight and that achieving the correct tension was a matter of judgment, as well as skill and experience.4038
5044 Dr Leake also pointed out that the mesh can become too tight because of variations in the patients’ responses to the mesh. Moreover, she said that the pulley system used for fixing the upper arms of the Prolift, which involved a blind approach, contributed to the problem of excessive tension because it made it difficult to determine whether there would be sufficient or excessive tension after the operation was over.
5045 Dr Leake said that a competent gynaecological surgeon could not invariably achieve the requisite tension. Dr Chapple was an obstetrician and gynaecologist, not a specialist urogynaecologist. Ethicon could have but did not restrict the use of the device to specialist urogynaecologists.
5046 Third, insertion complications are more likely to arise during the intraoperative period and up to 48 hours after surgery. Where complications are clinically diagnosed between 48 hours and two months after surgery, healing or infection complications are more likely.4039 Mrs Gill’s complications began more than two weeks after surgery.
5047 Associate Professor Rosamilia cited two articles to support her opinion that patients of more experienced surgeons had fewer erosions.4040 But neither of the studies with which the articles were concerned involved Prolift. The abstract to the article by Dwyer and O’Reilly, which related to a study of Atrium polypropylene for the repair of large or recurrent anterior and posterior compartment prolapse, stated that vaginal mesh erosion was related to surgical experience.4041 The second article was by Achtari et al.4042 It concerned risk factors for mesh erosion after transvaginal surgery with Atrium and Vypro 11, like Prolift+M a composite polypropylene/polyglactin 910 mesh. Dwyer and O’Reilly were co-authors. The abstract stated that in Vypro II patients, the surgeon experienced in the technique had fewer erosions than the inexperienced surgeons and that surgeon experience was associated with mesh erosion. Associate Professor Rosamilia also neglected to mention that not all the findings of each of the studies reported were statistically significant, a circumstance she conceded in cross-examination.4043
Conclusion
5048 In summary, I am not satisfied that the anteverted bulky uterus and intramural posterior fibroid were caused, either directly or indirectly, by the Prolift implant. Nor am I satisfied that the vaginal bleeding and spotting or the urinary symptoms are related to the implant.
5049 On the other hand, I am satisfied that the implant caused chronic inflammation, chronic pelvic pain (including coccygeal, groin, vaginal, and low back pain), severe pain on defaecation and the need to take Movicol to avoid large bowel movements and the consequential faecal urgency and flatal and faecal incontinence. I am also satisfied that the complications of the Prolift implant aggravated an underlying depressive disorder and caused a generalised anxiety disorder. I find that the recurrent prolapses were caused by the excision of pieces of mesh, including the supportive arms, which were undertaken to treat the painful erosions.
Non-economic or non-pecuniary loss
5050 As the applicants submitted, the relevant provisions of the TPA, particularly ss 87L–87T, limit damages for non-economic loss in a manner very similar to the analogous provisions of the CLA (NSW) but differently from those in the CLA (WA).
5051 “Non-economic loss” is defined in the TPA as one or more of the following: pain and suffering; loss of amenities of life; loss of expectation of life; and disfigurement: TPA, s 87D. A court is prohibited from awarding personal injury damages for non-economic loss except in accordance with the terms of Division 3 of Pt VIB of the Act. The relevant modifications are these.
5052 First, a maximum sum is fixed by the legislation and periodically indexed: TPA, ss 87L-87N.
5053 Second, the maximum sum is reserved for “a most extreme case”, defined as “a case in which the plaintiff suffers non-economic loss of the gravest conceivable kind”: TPA, s 87P.
5054 Third, if the non-economic loss is at least 33% but less than 100% of a most extreme case, the court must evaluate the extent of the plaintiff’s non-economic loss as a percentage and the court must not award an amount that exceeds the applicable percentage: TPA, s 87Q.
5055 Fourth, where the non-economic loss is at least 15% but less than 33%, the amount the court may award is reduced by degrees to a percentage of the percentage according to a table appearing in s 87R of the Act: TPA, s 87R.
5056 Fifth, in the event that non-economic loss is less than 15%, the Court may not award any damages for non-economic loss: TPA, s 87S.
5057 Sixth, contrary to the position at common law, the Court may refer to authorities for the purpose of establishing the appropriate award: TPA, s 87T.
5058 Seventh, a court must not order interest on this head of damage: TPA, s 87ZA(1)(a); CCA, s 87ZA.
5059 As at September 2018, the maximum amount of damages for non-economic loss under the TPA was $350,750.
5060 Under the CLA (WA), non-economic loss is referred to as “non-pecuniary loss”. The definition of “non-pecuniary loss”, which appears in s 9(4), differs from the definition of non-economic loss in the TPA and the Wrongs Act (Vic). Section 9(4) provides that “non-pecuniary loss” means pain and suffering; loss of amenities of life; loss of enjoyment of life; curtailment of expectation of life; and bodily or mental harm. Despite the absence of a reference to loss of enjoyment of life or bodily or mental harm in the definitions of non-economic loss and the absence of disfigurement in the definition of non-pecuniary loss, it is extremely doubtful that the various parliaments intended to exclude those matters and neither side suggested that anything should be made of the differences. “Non-pecuniary loss”, like “non-economic loss”, is merely a new term for general damages.
5061 Under the CLA (WA), there is a threshold below which no damages are to be awarded and a formula for calculating the amount in certain cases. But the Act imposes no upper limit. The restrictions are contained in s 9 of the Act. Its terms are convoluted. Nevertheless, it is common ground that Mrs Gill exceeds the threshold and that the complexities do not affect the calculation of her damages.4044
5062 To date Mrs Gill has endured nearly 13 years of pain and suffering. As a (nearly) 49 year old woman, based on the 2019 medium life tables, she has a projected life expectancy of 37.75 years, which means that she faces the unenviable prospect of just under another four decades of unremitting pain.
5063 The applicants argued that, if Mrs Gill had undergone a native tissue repair instead of Prolift Total mesh implant surgery, it is more likely than not that she would have obtained relief from her prolapse symptoms, although they concede that she may have developed recurrent prolapse, stress incontinence and bowel dysfunction. On the other hand, they submitted, she would not have developed dyspareunia, difficulties with orgasm or “multifaceted” pain in the coccyx, groin, or lower back. But for the injuries and disabilities the subject of this action, they contended, she would have remained a strong, independent and outgoing woman, dedicated to her work as a marine scientist or a lecturer in marine science and leading an active family and social life, which included swimming, snorkelling, jogging, sailing and camping.4045
5064 They argued that non-economic loss should be assessed at 60% of a most extreme case for the purposes of the statutory counts and $300,000 to $350,000 at common law.4046
5065 The respondents, on the other hand, argued that non-economic loss should be assessed at 35% of a most extreme case for the purposes of the statutory counts and $150,000 in negligence.4047 The submission no doubt reflects the respondents’ position that a good deal of Mrs Gill’s pain and suffering should not be attributed to the use of Prolift, its admitted complications or the corrective surgery.
5066 I take into account Mrs Gill’s premorbid problems of psoriatic arthritis, sporadic lower back pain, and neck pain. There is no reason to suppose that she would not continue to experience symptoms of all three conditions in the future regardless of whether she had suffered harm as a result of implantation with the respondents’ device. In varying degrees they are likely to have interfered with her enjoyment of life from time to time. I also take into account her history of depression and her vulnerability to anxiety. It is likely that she would have been troubled to some extent by depression and anxiety throughout her life even if she had not been implanted with Prolift. Over a projected life span of 37.75 years, the chances are, too, that she would almost certainly have experienced a recurrence of her prolapse. Consequently, allowance must also be made for the chance that she would have been bothered by prolapse symptoms at least periodically in the future, even after native tissue repair. These symptoms are likely to have interfered with her lifestyle, inhibiting her to some degree from engaging in certain physical and social activities, just as they did in the past.
5067 Nevertheless, were it not for the Prolift surgery, it is unlikely that Mrs Gill would have experienced some of the colorectal symptoms she has suffered since the surgery. Nor would she have suffered the coccygeal, groin, vaginal and severe low back pain she has endured over the last 12 years, albeit with some respite in the period from about September 2008 to mid-2013, and from which she has no real prospect of permanent relief. Chronic pain is debilitating. Sometimes it can be overwhelming. It intrudes into every aspect of life. Mrs Gill has done her best to get on with life in spite of it but, to some extent, that has only increased her suffering.
5068 Taking into account the impact the Prolift surgery and its complications have had on her life, set out in detail in Part XV of these reasons and bearing in mind the long period of past and future suffering, but making due allowance for the other conditions to which I have referred above, I assess Mrs Gill’s non-economic loss for the purpose of the TPA claim at 45% of a most extreme case and her non-pecuniary loss at common law at $325,000. In these circumstances, ss 9 and 10 of the CLA (WA) do not operate to limit the amount payable.
5069 The applicants proposed apportioning 20% to the past. As a proportion of the total period that is too low. I would assign 25% to the past.
5070 Mrs Gill’s claim for past economic loss starts in July 2013 and finishes at the end of the 2017 calendar year. To put it in context, however, it is necessary to review the relevant history.
5071 After the birth of her children, on about 5 April 2005, Mrs Gill returned to work on a casual basis as a lecturer with the Fremantle Maritime Training Centre.4048
5072 The Prolift surgery was performed on 12 January 2007. From then until February 2007, Mrs Gill was unable to work. In February 2007 she tried to resume work but realised that she was still unable to work due to the pain associated with the mesh implant. She remained off work until July 2007.4049 Throughout that period, however, her employer paid most of her wages, although she was employed as a casual.
5073 From July to September 2007 Mrs Gill worked as a lecturer at the FMTC for about two days per week.4050 For about six weeks after the first excision surgery on 10 September 2007, Mrs Gill was unable to work but the FMTC continued to pay her salary.4051 From about November 2007 she returned to work, for about two to three days a week, and continued until her second excision surgery on 20 June 2008. She returned to work in the same capacity after about six weeks. Between July 2008 and January 2010 she continued to work about two days a week, with the exception of a period of about one week in mid-2009 when she flew to Victoria to stay with her mother.4052
5074 After her husband’s head injury in February 2010 Mrs Gill stopped work to care for him. She returned to work in mid-2010, two to three days a week.4053
5075 On or about 3 February 2012 Mr and Mrs Gill and their two children moved from Perth to Geelong, so that she could be closer to her family. Nevertheless, she remained in the employ of the FMTC, writing for between 3 and 12 hours a week. She was also employed in a teaching position at the Gordon Institute of Technical and Further Education in Geelong, working between half a day and four days a week, depending on the availability of work.4054
5076 In mid-2013 Mrs Gill’s symptoms deteriorated. In her second affidavit she said that about six weeks after her third excision surgery, which took place on 8 August 2013, she tried to return to work but her ability to work declined and she became unreliable because of the pain she was experiencing.4055 She did not explain what she meant by “unreliable” and the matter was not explored by either party in oral evidence. I infer that she meant that she could not always be counted on to turn up for work.
5077 Early the following year, when Mrs Gill and her family returned to Perth, Mrs Gill’s evidence was that the manager of the FMTC told her that he no longer had any work for her.4056 It appears from her salary history, evidently produced by the FMTC, that in 2014 she worked for the FMTC from 16 January until 5 June.4057 In July 2014 she obtained a casual position with the Department of Education as a Relief Education Assistant (Special Needs) working about one day a week at the Belridge Secondary Education Support Centre.4058 The evidence is silent about the hours she worked but in the absence of any indication to the contrary I would infer that it was at least six hours.
5078 Mrs Gill came to the view that casual lecturing positions would no longer be available because of funding cuts to TAFE. That was the reason she returned to university in June 2015 to study full-time for a Diploma of Education. While studying, Mrs Gill continued to work one day a week at the Belridge Secondary Education Support Centre, largely in an administrative role in the school office.4059 After completing her studies in September 2016, she began working with special needs children at Duncraig Secondary Education Centre, three days per week, and was still in that position at the time she gave evidence in November 2017. 4060
5079 Mrs Gill’s current position is in peril. As I mentioned above, she is struggling to maintain a three day working week. In her second affidavit she gave the following evidence which was not the subject of cross-examination:
17 In July 2017 I discussed my concerns with the Upper School Coordinator of Duncraig Secondary Education Centre, Christine Lester. I explained that I have no balance in my life and was considering a reduction in days. Although Christine was supportive of me she expressed concern that even if it cut down to two days per week it would be the same work load due to spending days outside of my two days at work to write reports and attend to other administrative tasks. I felt like she was trying to talk me out of it.
18 Christine at present is assisting me with my administrative tasks so I can take some down time at home. Although this is a great help, I feel she does not completely understand the situation that I am in and the lack of balance in my life. Due to the time it takes to recover from my days at work, I have no well days at home to engage in therapy or exercise. I spend all my time at work, in bed or on the lounge.
5080 Mrs Gill claimed $74,326.74 in past economic loss, made up as follows:4061
Period | Weeks | Net Weekly Income but for Injury | Actual Net Weekly Income | Net Weekly Loss | Amount |
01.07.2013 – 30.06.2014 | 52.1 | $466.19 | $447.90 | $18.29 | $952.91 |
01.07.2014 – 30.06.2015 | 52.1 | $466.19 | $198.52 | $267.67 | $13,945.61 |
01.07.2015 – 31.12.2015 | 26.0 | $466.19 | $280.77 | $185.43 | $4,821.18 |
01.01.2016 – 30.06.2016 | 26.0 | $1,020.82 | $280.77 | $740.05 | $19,241.30 |
01.07.2016 – 30.06.2017 | 52.1 | $1,020.82 | $568.00 | $452.82 | $23,591.92 |
01.07.2017 – 31.12.2017 | 26.0 | $1,020.82 | $568.00 | $452.82 | $11,773.32 |
Total | $74,326.24 |
5081 The information in the third column for the last three periods is derived from the annual salary for a teacher at the first salary increment under the Western Australian School Education Act (Teacher and Administrators) Agreement 2014.4062 No source was identified for the first three periods in either the Statement of Particulars filed for Mrs Gill or the applicants’ submissions. Mrs Gill did not give evidence about her probable or actual earnings and the source is obscure. In the absence of any argument to the contrary, however, I shall assume that all the figures in columns 3 and 4 of the table are correct and not in dispute, and that there is no issue about the maths either.
5082 The respondents submitted that Mrs Gill had not proved that she suffered any economic loss as a result of the Prolift complications.
5083 Certainly, on no view of the matter is Mrs Gill entitled to damages or compensation calculated in the manner advanced by her lawyers.
5084 As I observed earlier, damages are payable, not on the basis of a diminution in earning capacity, but because the diminution in earning capacity is or may be productive of economic loss. Even if I were to infer that the deterioration in Mrs Gill’s symptoms in mid-2013 resulted in a diminution of her earning capacity, there is no evidence to suggest that it was productive of economic loss.
5085 The income tax records show that Mrs Gill’s earnings actually increased from $16,782 in the financial year ending 30 June 2013 (which was below the tax threshold)4063 to $24,540 gross ($23,335 net) in the following financial year.4064 Although they fell to $10,343 in the financial year ending 30 June 2015, the evidence does not establish that the loss more probably than not arose because of her injuries.4065 To the contrary, it establishes that it was occasioned by cuts to the TAFE budget. The evidence does not indicate that Mrs Gill’s disabilities played any part in her employer’s decision not to offer her any more work.
5086 Dr Slesenger, to whose evidence I will come shortly, noted that Mrs Gill had stopped work for six months after her surgery in 2007. Although he did not identify which surgical procedure he had in mind, I infer from Mrs Gill’s evidence that it was the Prolift surgery. Dr Slesenger recorded that she returned to work on reduced hours, generally one to two days per week but was unable to maintain this level of attendance and “reduced to 1 day a week (she advised that she was generally unreliable and had to take regular time off work)”. Dr Slesenger then stated that she remained at Challenger Institute until 2015 when she left work “due to her unreliability and financial changes”.
5087 That history was admitted for the limited purpose of proving what Mrs Gill said to Dr Slesenger and it is at odds with Mrs Gill’s evidence.
5088 For the financial year ending 30 June 2016, the only evidence about her earnings is a PAYG payment summary.4066 It discloses a gross payment of $14,628. During that financial year, however, Mrs Gill was studying full-time in order to acquire an additional qualification, a path she chose to take, not because she was unable to pursue her former line of work on account of her injuries, but because she had “formed the belief … that lecturing work would be unavailable for a casual lecturer in the future due to funding cuts to TAFE”.4067
5089 Mrs Gill deposed that, before “the most recent onset of pelvic issues in 2013”, she had aspired to securing full-time employment as a teacher once her sons reached high school.4068 The younger of her two sons turned 12 on 3 February 2016. It is therefore clear that, but for her injuries, Mrs Gill would not have secured a full-time teaching position before February 2016 at the earliest, assuming that she had applied for, and was offered, a position she was prepared to take. At that point in time, however, she was still studying full-time. It was not until September 2016 that she completed her Diploma of Education. On her own evidence, she was in this position, not as a result of the injuries the subject of this proceeding, but as a result of funding cuts to TAFE. Consequently, if there is any entitlement to economic loss, it seems to me that it begins at the earliest, not in July 2013 but in October 2016.
5090 The only medical evidence going to Mrs Gill’s capacity to work was given by Dr Jungfer, Dr Samuels, and Dr Slesenger.
5091 Dr Jungfer’s opinion was that it was more likely than not that, as Mrs Gill continues to experience the pain and continues to have anxiety symptoms, which on self-report have impaired her concentration, reduced her resilience, and increased her fatigue, her earning capacity will continue to be impaired. Dr Jungfer emphasised that, having regard to Mrs Gill’s mental state, she is less resilient to stress.4069 None of this evidence was challenged in cross-examination and I accept it.
5092 When asked by the respondents for his opinion on Mrs Gill’s prognosis, Dr Samuels wrote:4070
Her prognosis is very much related to the physical symptoms she is experiencing. Whilst she is continuing to manifest these symptoms, in particular pain, it is likely that she will continue to feel depressed and anxious from time to time. It seems that after three days of working her symptoms become quite severe and this leads to a feeling of exhaustion and deterioration in her mood and anxiety state. At the present time she needs to have a day to fully recover and to take pain relief if required. If over time she is better able to manage her physical symptoms, it is likely that this would favourably impact upon her psychiatric condition.
5093 When asked what effect Mrs Gill’s psychiatric condition has on her ability to work, Dr Samuels replied that she managed her psychiatric symptoms very well but can become short and irritable with her children when she is in a lot of pain and has “physical problems” and can also feel quite exhausted and has a need to withdraw from people.
5094 It appears that Dr Samuels accepted that Mrs Gill was working to maximum capacity. When asked to identify the differences between any of his opinions and those expressed by Dr Jungfer, Dr Samuels did not nominate earning capacity.4071
5095 Dr Slesenger is a specialist occupational physician. Amongst other things, he is a Fellow of the Australasian Faculty of Occupational and Environmental Medicine and holds a Masters degree in Occupational Health and Safety from Adelaide University.4072 As at November 2017, he was working in two Victorian clinics/medical centres. Formerly he was an occupational health advisor to Ford Australia, Bosch Australia, CSL, and Worksafe Victoria. In the course of his work he visits various workplaces. Of particular relevance is the fact that he is often required to visit schools and other educational training facilities.4073
5096 Dr Slesenger was provided with some medical records, which he reviewed and discussed in his first report.4074 He also examined Mrs Gill on 15 July 2016. He observed her as she recounted her history. He said that she interacted well and gave a clear and consistent account of her injuries. Her aspect was normal and her eye contact good. She became emotional while discussing her functional limitations. He examined her abdomen but found no abnormalities. There was no tenderness on palpation and, apart from “fullness” in the left iliac fossa, there was no organomegaly (abnormal enlargement of bodily organs). He did not carry out an internal gynaecological examination. External gynaecological examination was normal.
5097 Dr Slesenger was asked by Mrs Gill’s solicitors whether it was more likely than not that Mrs Gill’s earning capacity had been affected by her injuries and disabilities and if so to what extent, in what way, and for what period of time. He was also asked whether any modifications to the workplace or work practices would be required and whether Mrs Gill would need to be retrained or redeployed. He did not answer all the questions. This was his response:
Taking Mrs Gill’s clinical history as a whole, there is evidence that there has been a reduction in her occupational activity. It is noted that whilst working at the Challenger Institute prior to her surgery, she was working 3 hours a day, 3 days a week and was preparing lessons for her further 1½ hours a day and in addition she was driving 2 hours between home and work each day. After her surgery, she reduced her work activity to 1 day per week and eventually left work due to a combination of financial changes at the Challenger Institute and her unreliability.
The clinical records indicate a significant psychological component to her presentation between 2008 and 2013, which Mrs Gill has attributed to her gynaecological disorder and associated impairment.
Since 2015, Mrs Gill has commenced re-training and is currently attending university 3 days a week for 3 hours a day, and is driving for 1 hour per day. She is also performing 6–7 hours of work per week at home at a sit-stand desk. These tasks are self-paced and I do not anticipate that this level of work can be replicated within a work environment.
5098 Dr Schlesinger was also asked whether he considered Mrs Gill was suited to full-time work as a secondary school teacher. This was his response to that question:
Mrs Gill attended a placement at St Steven’s School in 2016 where she was allocated for a 2-week period … She described difficulties due to pain, fatigue and also constant vaginal bleeding whilst attending this placement. She also noted that whilst having to prepare lessons and deliver classes, she was also required to attend the beach to pick up samples for study within the class.
With regard to Mrs Gill’s future earning capacity, it is unclear whether she will be able to complete her retraining and given her description of her current capacity, I am not optimistic that she will be able to complete all the components of her training requirements, including future placement. It may be useful to access copies of her educational records to confirm her progress through her Diploma in Education.
I also note that should she complete the retraining, it is unlikely that she will be able to attend work on a consistent and a reliable basis for more than 2 hours a day, 2 days a week, in addition to 2 hours a day preparatory work time at home (sit/stand desk). This in turn will restrict the amount of work available to her.
In addition, I note a number of other factors affecting her future employment:
• The availability of a sit-stand desk within the classroom environment (particularly if she is working in a supply teacher role.
• Travel distances between home and work (particularly in a supply teacher role).
• She may also be required to perform manual tasks (e.g. collecting samples during field trips).
5099 After he produced his report, Dr Slesenger was sent Mrs Gill’s two affidavits. He described the first affidavit as “broadly consistent” with the history Mrs Gill gave to him. He stood by the opinion he expressed in his first report but added that he did not believe that Mrs Gill would be able to maintain her current working schedule and that was “likely to alter within the foreseeable future”.
5100 In support of his opinion about the prospective limits on the amount of work that would be available to her, Dr Slesenger gave persuasive evidence about the difficulties Mrs Gill was likely to face on the open labour market:
In my experience, and based on my training, I’m aware that educational establishments require to be able to match their teaching needs, the pupils or the students, to the availability of staff. It has to be – one of the key issues is it has to be predictable. Staff have got to be available on certain times during the schedule of the working week. The [roster] that they arrange for the rooms to be available and the students to be in the right place at the right time is set – usually at least a term, if not two or three terms, in advance. In my experience it’s very difficult to be able to arrange a return to work for workers who have very tight restrictions in terms of the hours that they’re available, or the work tasks that they can do, or that the adaptation of the classroom environment has to be fixed for that particular teacher. It becomes more difficult if the teacher is working in a supply – a supply teacher, because there will be a limited number of schools that will be capable of offering that – those accommodations, usually at short notice, and usually with – usually with – without the – with difficulty understanding that the patient may be available on that day or not available on that day. What usually happens is they would go to the next person on the list. My experience of getting workers back to work with – with those restrictions is that it is usually unsuccessful, and the more restrictions there are, the less – the less likely it is that that person will be accommodated.4075
5101 Nonetheless, there are difficulties with Dr Slesenger’s evidence. In view of those difficulties, I am not satisfied that Mrs Gill’s residual capacity to work either now or in the future is limited to only two hours a day two days a week.
5102 First, contrary to Dr Slesenger’s expectations, Mrs Gill was able to attend work, albeit with considerable difficulty, for more than two hours a day two days a week for at least 14 months after she obtained her Diploma of Education in September 2016.4076
5103 Second, he noted and presumably took into account Mrs Gill’s constant vaginal bleeding during her placement, which I have not found to be related to the Prolift implant.
5104 Third, Dr Slesenger’s opinions were not informed by a functional assessment of Mrs Gill. He performed no such assessment in her home, at the university, during her teacher-training, or even in his rooms.4077 He did not identify any abnormality or any functional restriction of Mrs Gill in his examination,4078 probably because he conducted no internal examination or any functional assessment. His opinions rest entirely on the history he was given and, to the extent that they supported Mrs Gill’s complaints, the medical records.
5105 Fourth, Dr Slesenger did not visit Mrs Gill’s workplace or any educational facilities in Perth and there is no evidence to indicate that he spoke to any of Mrs Gill’s supervisors.4079
5106 Fifth, after reading Mrs Gill’s second affidavit Dr Slesenger noted that Mrs Gill had “approached her employers (sic) to reduce her working hours”.4080 This was a misreading of Mrs Gill’s affidavit. She said that she was “planning to request a reduction in days ...”, but had “not made a formal request yet …”.4081
5107 Sixth, Dr Slesenger was pessimistic about Mrs Gill’s work capacity following his assessment and his pessimism, in the short term, at least, was defeated by Mrs Gill’s resilience.
5108 Be that as it may, Western Australia is not a foreign country. It is unlikely that the needs and capacity of educational institutions in Perth would be appreciably different from the educational facilities with which Dr Slesenger was familiar. In competition with able-bodied teachers, I do not doubt that Mrs Gill would be at a considerable disadvantage on the open labour market.
5109 Further, I have found that Mrs Gill was struggling to maintain a three-day working week. As I mentioned earlier, it was never put to her that her pain was not genuine or that she had exaggerated her symptoms. The respondents’ psychiatrist, Dr Samuels, said that her pain was genuine. In these circumstances I am satisfied that her earning capacity is diminished. Having regard to her work ethic, it is virtually certain that she would have sought a full-time teaching position when both her children were in high school and, given her credentials and her determination, it is highly likely that she would have secured one. As was submitted on her behalf, work is obviously important to her self-worth.4082 It is not just a source of income; it helps to give meaning to her life. As she put it herself when asked why she continued to teach:
I’ve always been very proud of the work I do and I want to continue to be. I also need to have a house clean and need to be able to go to surgeons, so I need to be able to contribute to those costs. I need to work to earn. And, you know, for instance, things that relieve my pain like a massage, I haven’t done that for a while because I can’t afford it But equally, I — for my own self-worth, I want to be able to work so that I can contribute to my family and be part of the world, participate. I need to be somewhere.4083
5110 She also expressed the wish to be able to work in the marine and environmental field again, although she could not see how that was possible.
5111 For these reasons I expect she will remain in the workforce as long as she can. By the same token, in the absence of any improvement in her mental or physical condition, like Dr Slesenger, I do not see how her current work effort is sustainable.
5112 No evidence was given about the state of the employment market in Perth, let alone the availability of teaching positions for someone with Mrs Gill’s qualifications and experience. Even so, her work capacity is reduced by her pain. The pain was caused by the Prolift device. Since September 2016 she was qualified to work as a teacher. But for the pain she would have been able to do so full-time. Instead she can barely manage work three days a week and is unlikely to be able to continue to do so in the long term. In these circumstances, she is entitled to be compensated, but not in the way or in the amount for which her lawyers contended.
5113 In the case of a hypothetical event, like Mrs Gill taking up full-time teaching or not being able to continue to work at her current level, damages are not to be assessed on the basis of a simple mathematical calculation of the difference between the full-time earnings and the current earnings or the full-time earnings and a possible future reduced income. As Deane, Gaudron and McHugh JJ explained in Malec at 643:
The future may be predicted and the hypothetical may be conjectured. But questions as to the future or hypothetical effect of physical injury or degeneration are not commonly susceptible of scientific demonstration or proof. If the law is to take account of future or hypothetical events in assessing damages, it can only do so in terms of the degree of probability of those events occurring. The probability may be very high — 99.9 per cent — or very low — 0.1 per cent. But unless the chance is so low as to be regarded as speculative — say less than I per cent — or so high as to be practically certain — say over 99 per cent — the court will take that chance into account in assessing the damages. Where proof is necessarily unattainable, it would be unfair to treat as certain a prediction which has a 51 per cent probability of occurring, but to ignore altogether a prediction which has a 49 per cent probability of occurring. Thus, the court assesses the degree of probability that an event would have occurred, or might occur, and adjusts its award of damages to reflect the degree of probability. The adjustment may increase or decrease the amount of damages otherwise to be awarded … The approach is the same whether it is alleged that the event would have occurred before or might occur after the assessment of damages takes place.
5114 No challenge was made to the figures given by the applicants for full-time teaching positions. But they are only a starting point. The same must be said of Mrs Gill’s current earnings.
5115 I would award Mrs Gill damages for past loss of earning capacity from January 2017 to take into account the possibility that Mrs Gill might not have been able to secure a full-time teaching position immediately after she qualified and I would reduce the difference between the prospective income and the actual income by 15% to allow for what I regard as the level of uncertainty surrounding the hypothetical scenario.
5116 Accordingly, in the period from 1 January 2017 to 31 December 2017, the notional loss is $20,053.13 (85% of $452.82 x 52.1), which I would round down to $20,000.
5117 In the 2018 and 2019 calendar years the rates for prospective net weekly income increased to $1,104.25 and $1,192.18 respectively.4084 The differences between those two sums and Mrs Gill’s actual net weekly income ($568.00) at the time she gave evidence are $536.25 and $624.18 respectively. If it were to be assumed that Mrs Gill’s earning capacity in those years is the same as it was when she gave her evidence, that would produce a loss of $27,938.63 in the first year and $32,519.78 in the second, totalling $60,458.41 before any adjustment is made to reflect the chance that she would not have been employed full-time but for her compensable injuries and that she would not have been able to continue working three days a week in these periods. The chance of the former is very high to near certain. The chance of the latter is more difficult to assess.
5118 Doing the best I can on the evidence, I would allow $55,000 for these two years.
5119 In total, then, I assess past economic loss at $75,000.
5120 Neither the TPA nor the CLA (WA) requires any further adjustment.
5121 Past out-of-pocket expenses were agreed at $65,000.4085
5122 Past care and services were agreed at $100,000.4086
5123 The purpose of an award of interest is to compensate the injured person for the loss or detriment she has suffered by being kept out of her money since the loss or detriment first occurred: Batchelor v Burke (1981) 148 CLR 448 at 455 (Gibbs CJ).
5124 For the purpose of the TPA claims, interest on damages for economic loss, past out-of-pocket expenses, and past loss of superannuation benefits is payable at the 10-year benchmark bond rate (defined in s 87ZA(4)) on the day that the court determines the amount of damages: TPA, s 87ZA(2). But interest is not payable on damages for past gratuitous services or non-economic loss: s 87ZA(1). Since the evidence indicates that the vast majority of the past care and services was provided gratuitously and the agreed figure did not distinguish between that which was provided voluntarily and that which was provided commercially, I decline to order interest on that sum.
5125 The CLA (WA) does not alter the law with respect to interest on awards of damages in actions for damages in negligence for personal injuries. While s 32(2) of the Supreme Court Act 1935 (WA) precludes the Supreme Court from awarding interest on general damages, that section has no application to a proceeding in this Court. There is no such prohibition in the comparable provision of the FCA Act: s 51A. While the CLA (WA) is picked up and applied as surrogate federal law, the Supreme Court Act is not.
5126 Section 51A, which commenced on 22 November 1984, relevantly provides that:
(1) In any proceedings for the recovery of any money (including … damages …) in respect of a cause of action that arises after the commencement of this section, the Court or a Judge shall, upon application, unless good cause is shown to the contrary, either:
(a) order that there be included in the sum for which judgment is given interest at such rate as the Court or the Judge, as the case may be, thinks fit on the whole or any part of the money for the whole or any part of the period between the date when the cause of action arose and the date as of which judgment is entered; or
(b) without proceeding to calculate interest in accordance with paragraph (a), order that there be included in the sum for which judgment is given a lump sum in lieu of any such interest.
(2) Subsection (1) does not:
(a) authorize the giving of interest upon interest or of a sum in lieu of such interest;
(b) apply in relation to any debt upon which interest is payable as of right whether by virtue of an agreement or otherwise;
(c) affect the damages recoverable for the dishonour of a bill of exchange;
(d) limit the operation of any enactment or rule of law which, apart from this section, provides for the award of interest; or
(e) authorize the giving of interest, or a sum in lieu of interest, otherwise than by consent, upon any sum for which judgment is given by consent.
5127 Subsection 51A(3) prohibits the award of damages in certain circumstances, none of which is relevant to the present case. Subsection 51A(4) makes it clear, however, that the section does not preclude the award of interest on compensation where a liability has been met by the applicant, such as where the applicant has paid for out-of-pocket expenses.
5128 In the present case, interest on damages was claimed from the outset and no good cause was shown as to why interest should not be awarded on all heads of damage for past losses. Indeed, the respondents made no submissions on the subject, until after a year after I had reserved judgment when I invited the parties to agree on interest on past services.
5129 In an email from the respondents’ solicitors sent to my associate on 11 March 2019, the respondents submitted that there should be no interest on any amounts awarded for past services, whether paid or gratuitous, because no claim for interest on past services was made in the applicants’ particulars or their submissions when interest had been claimed for other heads of damage.
5130 I reject the submission. One would not ordinarily claim interest in a statement of particulars. What matters is that interest was claimed in the originating application. I do not take the applicants’ silence in either their particulars or their submissions to be an abandonment of their claim. When I was informed of the parties’ agreement on the figures of past services, the amount was not expressed to be inclusive of interest.
5131 Consequently, with one exception, interest on all heads of damage for past losses should be awarded in accordance with Practice Note GPN-INT. The exception relates to the claim for past non-economic loss, in respect of which interest is to be determined in accordance with the judgment in MBP (SA) Pty Ltd v Gogic (1991) 171 CLR 657 at 666–7. The figure settled on in Gogic was 4%. Since, the notional loss was sustained progressively, however, it is generally accepted that half the rate or 2% is to be applied: Grincelis v House (1998) 84 FCR 190 at 213 (Hill and Kiefel JJ), 192 (Foster J). The reason for the lower rate for interest on non-economic loss is that, in contrast to special damages for economic losses, those damages are determined based on the value of money at the date of assessment: see, for example, Wheeler v Page (1982) 31 SASR 1 at 6 (King CJ), which the High Court followed in Gogic. See, too, Grincelis v House (2000) 201 CLR 321 at [18] (Gleeson CJ, Gaudron, McHugh, Gummow and Hayne JJ). Interest on past gratuitous care or services is not to be treated in this way, however, and, in the absence of evidence as to commercial interest rates over the period in question, the rates referred to in the Practice Note are applicable to the awards for past economic loss, past out-of-pocket expenses, past loss of superannuation benefits, and past care and services: Grincelis at [18]–[21]. Once again, to allow for the fact that the damages represent amounts accruing over time, half the rate should apply: Grincelis at [22].
5132 There is a degree of artificiality about the applicants’ submissions on this question. They argued that future economic loss should be assessed on the basis that, although Mrs Gill was working three days a week, she is currently only fit to work for two days a week, and that this work capacity will reduce even further in the future to three hours a day, two days a week. They proposed deferring this latter reduction in earning capacity for 12 months.
5133 The starting point for their calculations was that, but for her injuries, Mrs Gill would have been working full-time from 2016 when her youngest son started high school.4087 From December 2018, they submitted, she would have qualified for a gross annual salary under the Agreement of $81,820, which equates to a net weekly income of $1,192.18. They argued that damages for future loss of earning capacity should be awarded based on the integers set out in the following table, save that a 5% discount rate applies under the Trade Practices Act: TPA, s 87Y(1)(b). Since no rate is fixed by the CLA (WA), the common law rate of 3% applies to the damages for negligence.
Period | 3% multiplier | Deferred multiplier | Net weekly income but for injury | Residual earning capacity | Net weekly loss | Total |
01.01.2018–31.12.2018 | 51.4 | N/A | $1,104.25 | 40% | $662.55 | $34,055.07 |
01.01.2019–04.12.2037 | 758.6 | 0.971 | $1,192.18 | 20% | $953.74 | $702,525.46 |
Total | $736,580.53 |
5134 Damages for loss of earnings are capped under the CLA (WA) at three times the average weekly earnings of full-time adult employees in Western Australia for the quarter ending most recently before the date of the award for which such an amount has been estimated by the Australian Statistician and is, at that date, available to the court making the award: CLA (WA), s 11. Under the TPA, the cap is twice the average weekly earnings published by the Australian Statistician for all employees (total earnings, seasonally adjusted): TPA, ss 87U and 87V. Since Mrs Gill’s weekly loss is significantly less than either cap, the applicants argued that the only discount should be 15% for the ordinary vicissitudes of life.4088
5135 The applicants’ approach is flawed. Like their approach to past economic loss, it treats as certain what is uncertain. It assumes, without evidence, that Mrs Gill would have secured a full-time teaching position in 2016 and that she would have remained in full-time employment until she was 67. It also assumes that her working hours would be reduced from three to two days a week and from three full days to two days of three hours each.
5136 The other problem with their approach is that it fails to take into account the possibility that Mrs Gill would be unable to work for periods of time in the future because of unrelated medical conditions.
5137 The respondents’ position is that Mrs Gill has not proved that she is entitled to any sum for future economic loss and that, if the Court were to determine otherwise, a “buffer” of no more than $100,000 is all that is warranted. For the reasons given above in relation to the claim for past economic loss, I reject the respondents’ submission that Mrs Gill has not made out a case for future economic loss.
5138 In State of New South Wales v Moss (2000) 54 NSWLR 536 at [71] Heydon JA described the assessment of future economic loss as an exercise in estimation of possibilities, not proof of probabilities:
The income earned before the injury is relevant, but only as an evidentiary aid in assessing damages for the loss of capacity to earn income: Paff v Speed at 566, per Windeyer J. Evaluation of the worth of a loss of capacity to earn — of a lost chance to earn — is of its nature a more imprecise inquiry than calculation of a lost income. It rests on the hypothesis — that the plaintiff will have undiminished capacity — which has been rendered false by events. It does not depend on calculating the income from a particular career which is no longer possible, but in calculating the damage to a capacity to carry on various careers. It is an exercise in estimation of possibilities, not proof of probabilities. H Luntz, Assessment of Damages for Personal Injury and Death, 3rd ed, at 91 [1.9.18], said: “it is not necessary for the plaintiff to establish the future loss with the same degree of precision as the present and past loss …The court is really being asked to estimate as best it can the future effect of the injuries from which the plaintiff has been proved to be suffering as a result of the defendant’s wrongful act”.
…
The inquiry — the process of estimation of possibilities — is thus an imprecise and indeterminate one to be carried out within very broad parameters. The trier of fact may have to form conclusions on “slender material[s]”: Callaghan v Wm C Lynch Pty Ltd (1962) 79 WN (NSW) 830; [1962] NSWR 871 at 877, per Evatt CJ, Herron J and Sugerman J.
5139 In many cases, pre-injury earnings can provide a very useful guide to determining probable post-injury earnings but that is by no means invariably so, as Gleeson CJ, Gummow, Kirby and Hayne JJ pointed out in Husher v Husher (1999) 197 CLR 138 at [8]. Their Honours referred to the example of a student who was injured before entering the workforce. A parent, like Mrs Gill, who has taken time out of the workforce to have or raise children or who chooses to work reduced hours while the children are still at school, is another case in point. The inquiry, as their Honours emphasised, is one about the likely course of future events and evidence of past events does not always provide a certain guide to the future.
5140 In Pollard v Baulderstone Hornibrook Engineering Pty Limited [2008] NSWCA 99; (2008) Aust Torts Reports ¶81-949; (2008) 172 IR 453, in which the Court of Appeal upheld an award of $120,000 by way of a buffer for future economic loss, McColl JA, with whom Mason P and Beazley JA agreed, observed at [84] that in a case where “the impact of an injury upon the economic benefit from exercising earning capacity” is uncertain, it is “appropriate to award damages for future economic loss by way of a buffer”. Her Honour continued:
In such a case, the Court still undertakes a comparison between the economic benefits the plaintiff derived from exercising earning capacity before injury and the economic benefit derived from exercising earning capacity after injury, although the difference cannot be determined otherwise than by the broad approach of a buffer: Penrith City Council v Parks [2004] NSWCA 201 (at [3] - [5]) per Giles JA; applied Kmart Australia Ltd v McCann [2004] NSWCA 283 (at [62]) per Pearlman AJA (Handley and Ipp JJA agreeing); see also Hornsby Shire Council v King [2005] NSWCA 67 (at [23]) per Ipp JA (Mason P and Brownie AJA agreeing); Leichhardt Municipal Council v Montgomery [2005] NSWCA 432 (at [33]) per Hodgson JA (McColl JA agreeing). In the latter case Mason P (at [2]) opined that “a buffer or cushion award is usually reserved to the situation where there is a smallish risk that otherwise secure employment prospects may come to an end, in consequence of the tort-related injury, at some distant time in the future”, but, with respect, the accepted wisdom appears to be that a buffer can be deployed in circumstances such as the present.
5141 In Leichhardt Municipal Council v Montgomery [2005] NSWCA 432 the Court upheld an award of $160,000 by way of a buffer for future economic loss.
5142 In all the circumstances, I agree with the respondents that a so-called buffer should be awarded, but I do not consider that the amount the respondents suggested would adequately compensate Mrs Gill for her loss.
5143 On the one hand, Mrs Gill has other physical and mental health issues that are likely to trouble her in the future as they have in the past. She is also a determined and resourceful individual with a strong work ethic. With help from a pain clinic she may be able to manage her pain better. On the other hand, her prognosis for recovery from the mesh-related problems is poor. She faces the prospect of further erosions and a lifetime of chronic pain. There is no guarantee that she will keep her present position, particularly if she cannot maintain her current output which, as I have said, seems likely. She is highly unlikely ever to be able to teach full-time, as she had intended, and more likely than not her only option will be part-time or casual work if and when the opportunities present themselves. Making due allowance for the various contingencies, I award $250,000 for future loss of earning capacity. Neither side argued that, if I were to award damages by way of a buffer, it was necessary to differentiate between damages for the purpose of the TPA claim and damages for the purpose of the common law claim.
Loss of superannuation benefits
5144 Mrs Gill claimed $10,101.06 for past loss of superannuation benefits made up as follows:
Period | Weeks | SG rate | Gross weekly income but for injury | Actual gross weekly income | Gross weekly loss | Amount |
01.07.2013 – 30.06.2014 | 52.1 | 9.25% | $528.14 | $471.01 | $57.13 | $275.32 |
01.07.2014 – 30.06.2015 | 52.1 | 9.50% | $528.14 | $198.52 | $329.62 | $1,631.45 |
01.07.2015 – 31.12.2015 | 26.0 | 9.50% | $528.14 | $280.77 | $247.38 | $611.03 |
01.01.2016 – 30.06.2016 | 26.0 | 9.50% | $1,307.82 | $280.77 | $1,027.05 | $2,536.81 |
01.07.2016 – 30.06.2017 | 52.1 | 9.50% | $1,307.82 | $627.66 | $680.16 | $3,366.45 |
01.07.2017 – 31.12.2017 | 26.0 | 9.50% | $1,307.82 | $627.66 | $680.16 | $1,680.00 |
Total | $10,101.06 |
5145 Once again, although the entitlement was contested, the figures were not disputed. Having regard to my findings concerning Mrs Gill’s past economic loss, however, the award for past loss of superannuation benefits should be limited to the period from 2017 to 2019 inclusive.
5146 For 2017, I calculate the loss at 85% of $3,366.45 or $2,861.48, which I would round down to $2,500.
5147 Figures were provided for the 2018 and 2019 calendar years, albeit in the context of the claim for future loss of superannuation benefits. In those years, the gross weekly income was $1,307.82 and $1,434.93 respectively. Assuming an actual weekly income of $627.66 in each year, the difference between gross weekly income and actual weekly income is $680.16 in 2018 and $807.27 in 2019. Over 52.1 weeks, that results in a gross figure of $35,436.34 in 2018 and $42,058.77 in 2019. Applying the superannuation guarantee rate of 9.50% to those two sums yields $3,366.45 in 2018 and $3995.58 in 2019. For 2018, I calculate the loss at 85% of $3,366.45 or $2,861.48, which I would also round down to $2,500. For 2019, I calculate the loss at 85% of $3995.58 or $3396.24, which I would round down to $3,300.
5148 It follows that I award $8,300 for past loss of superannuation.
5149 Future loss should be calculated on the sum I have awarded for future economic loss ($250,000) multiplied by the superannuation guarantee rate of 9.50%, deferred for 18 years. The appropriate discount rate is 5% in the case of the TPA claim and 3% in the case of the common law claim. While superannuation is payable on gross, not net income, I have not grossed up the $250,000 figure because, when applied to the years it covers, the annual amount is below the income tax threshold.
5150 Section 87Z of the TPA imposes a cap on damages for loss of superannuation entitlements. But the respondents did not argue that the cap would have been exceeded even if the Court were to award damages in the amount claimed by the applicants, either for Mrs Gill or Mrs Dawson. In the circumstances, nothing more need be said about it.
5151 Mrs Gill‘s evidence was that, because of her pain, she is unable to undertake many of the cleaning and maintenance tasks she performed independently before the Prolift surgery.4089 They include:
heavy cleaning, such as mopping, vacuuming and cleaning the oven;
seasonal cleaning, such as cleaning the windows, gutters, pergola and solar panels;
making the beds, shopping, laundry, gardening and other household tasks.
5152 Mrs Gill said that on her good days, after work she might do an hour’s work around the house, like empty the dishwasher, before she is “wiped out”. On a bad day, she goes to bed when she gets home from work and her husband looks after the children and brings her dinner to bed. She estimated that this happened at least two out of the three days she worked. On the days she does not work, she spends most of the day in bed recovering and then is “too bombed out on medication” when her husband returns home and relies on him to look after the children.4090
5153 Mrs Gill testified that her husband assists her for a minimum of seven hours a day.4091
5154 Ms Williams’ first report included a table which summarised the tasks formerly performed by Mrs Gill and the assistance she was receiving at the time of trial:
Table 3: Domestic Management Tasks | ||
Task | Previous Performance | Current Performance |
Light Cleaning Tasks | All tasks carried out by Katie. | Katie reported that a cleaning lady now shares the heavy cleaning tasks. Katie reported that she now has to pay for activities such as mopping or vacuum cleaning the next day as she is tired, sore and bleeding. She cannot clean the oven as she cannot bend to do this due to pain. Lifetime assistance with cleaning is required for Katie. |
Heavy Cleaning Tasks | ||
Spring Cleaning or Seasonal Tasks | Katie previously would do many of these tasks such as cleaning out the gutters and cleaning the solar panels. She was an active participant in these type of tasks with Steven. | These tasks don’t get done anymore according to Katie. Katie is now unable to clean the windows, gutters, pergola or the solar panels due to pain. Lifetime assistance with seasonal cleaning is required for Katie. |
Bed Making | Katie performed these tasks independently. | Lifetime assistance with changing the bed linen is required for Katie due to issues with functional mobility caused by pain. |
Meal Preparation and Cooking | Katie reported that she struggles with cooking due to pain. Steven is now required to assist with cooking. | |
Washing-up | Katie is assisted by her family. | |
Laundry | Katie reported that she gets assistance from Steven and her sons. Steven has built a pull down drying rack in the laundry and there is a heavy duty clothes horse. Katie’s sons fill the laundry trolley so that she does not have to lift a basket of clothes. Lifetime assistance with laundry is required for Katie. | |
Ironing | Ironing is now minimized and Katie paces herself. | |
Shopping | Katie reported that she now has difficulty with shopping due to pain. Katie reported that has recently started online shopping, but Steven does some of the family grocery shopping. Delivery on average costs $9 per week or $468 annually. Lifetime assistance with shopping is required for Katie. | |
Bed making | Bending over to make beds and change linen is painful for Katie. Lifetime assistance is required for Katie. | |
Gardening and Lawn Care | Katie reported that she used to enjoy gardening. Steven and the boys do this task as Katie is unable to dig or weed. They would like a gardener to assist as the garden is now overgrown. Lifetime assistance with gardening and lawn mowing is required for Katie. | |
Putting the bins out | Steven and the boys now do this task. Katie required lifetime assistance. | |
Pool Care | Steven does this task and Katie is unable to assist due to pain. Lifetime assistance with maintaining the pool is required for Katie. | |
Pet Care | Katie is able to manage basic pet care, but is assisted by her family. | |
5155 No order was sought or made under s 136 of the Evidence Act limiting the use to be made of this evidence or of other matters of history contained in the report. The information in the table can therefore be utilised to fill any gaps in the evidence given by Mr and Mrs Gill.
5156 The applicants submitted that the assessment of this head of damage should be made on the premise that Mrs Gill’s condition is chronic and permanent, unlikely to improve, and that, as she ages, her need for care and support is likely to increase. They also argued, with the support of Ms Williams, that it was unreasonable to expect Mr Gill to continue to provide assistance gratuitously and that the award should be made on the basis that “the care should be provided commercially”.4092
5157 Ms Williams’ opinion was as follows:
Katie will require assistance with heavy domestic tasks. Five (5) hours per week of heavy cleaning is supported. Heavy domestic tasks such as cleaning kitchens and bathrooms, mopping, sweeping and vacuuming are considered heavy domestic activities.
Katie will be dependent on commercial services for handyman tasks, gardening, lawn care and heavy domestic work.
12 additional hours per year is considered reasonable for seasonal cleaning such as cobweb removal, cleaning windows and washing curtains, etc.
Four (4) hours per month is supported for garden and lawn care. Pool care is also required.
Handyman services of one (1) hour per month are supported to complete basic maintenance such as changing tap washers and changing light bulbs.4093
5158 She also said that until the age of 60 she would require the following additional assistance:
12 hours a week support to assist with meal preparation, shopping, running errands like picking up prescriptions, and helping with domestic tasks not completed by the cleaner; and
one week a year live-in 24 hour assistance when she is in severe pain, is ill, or is recovering from surgery.4094
5159 Ms Williams considered that Mrs Gill’s support requirements were “most likely to increase with age”.4095 Accordingly, from the age of 60 she said that, rather than 12 hours a week assistance, Mrs Gill required assistance (in addition to the matters set out above), three hours a day for seven days a week (21 hours a week) and two weeks a year live-in 24 hour assistance.
5160 Ms Borthwick’s approach was very different.
5161 She made no provision for assistance for those tasks Mrs Gill did not perform before the Prolift repair. She also considered that Mrs Gill’s need for assistance would reduce once her sons left home. Moreover, her provision for assistance came to an abrupt end once Mrs Gill turns 80, on the ground that by that age it was “very likely that this level of assistance would have been required in any event due to age related physical changes”.4096
5162 Ms Borthwick considered that Mrs Gill should receive 3.5 hours a week domestic assistance, including spring cleaning and support for home delivery of her shopping currently provided at a cost of $10 per week. Once Mrs Gill’s sons become adults and are “preparing to leave home”, however, she reduced her recommendation to 2.5 hours a week. She said there was no need for support with gardening or lawn mowing “as it is reasonable that in any marriage, there is an equal share or at least some domestic responsibilities undertaken by each partner”. She said that outdoor and home maintenance tasks had always been Mr Gill’s responsibility.4097
5163 Ms Borthwick agreed with Ms Williams that Mrs Gill needs assistance with tasks that require her to lift and carry weights greater than 5kg or to work at low levels or in confined spaces as these activities result in an increase of intra-abdominal pressure which Mrs Gill indicated triggers pain. Nevertheless, she could not fathom how those activities could reasonably require 15 hours a week assistance.4098
5164 She did not agree with the provision for live-in care.4099
5165 Mrs Gill sought a sum of $947,144.37, purportedly based on Ms Williams’ assessment. That sum was calculated by reference to her recommendations but reduced by 25% “on account of the give-and-take of Mrs Gill’s relationship with her husband” and by a further 15% for vicissitudes. It was not entirely clear what was meant by the former or, for that matter, the latter. The written submissions did not identify the vicissitudes. In oral argument, however, it became clear that they were the ordinary vicissitudes of life.4100 While a reduction in vicissitudes is undoubtedly required, for the reasons given earlier a discount for the ordinary vicissitudes of life is inappropriate.
5166 The submissions made on Mrs Gill’s behalf calculated future care from 1 January 2018, a rather optimistic assessment given that oral argument was not due to start until the following month. Strictly, future assistance should be determined from the time of publication of judgment, not from 1 January 2018. Since a sum for the past was agreed, however, based on the position as at December 2017, I propose to deal with the claims for both future care and services and future out-of-pocket expenses for all three applicants, from that point.
5167 The respondents submitted that Ms Williams failed to consider the extent to which Mrs Gill’s ability to perform daily activities was affected by her pre-existing pelvic organ prolapse symptoms.4101 Since there is no evidence that Mrs Gill’s pre-existing pelvic organ prolapse symptoms affected her capacity to undertake the kinds of tasks with which she now claims she requires assistance, this is a red herring.
5168 Still, there are a number of difficulties with Ms Williams’ assessment.
5169 First, she provided no reasons for her opinion that Mrs Gill’s needs are most likely to increase with age and the opinion does not purport to be based wholly or substantially on Ms Williams’ expert knowledge. Indeed, the basis of the opinion was not disclosed. Since the opinion was not apparently based either wholly or substantially on Ms Williams’s training, study or experience it was arguably inadmissible and is entitled to little, if any, weight: see Makita (Australia) Pty Ltd v Sprowles (2001) 52 NSWLR 705 at [86]; Sydneywide Distributors Pty Ltd v Red Bull Australia Pty Ltd (2002) 234 FCR 549 at [14], [17].
5170 In reality, this was not an opinion but an assumption. It appears to have been based on the account given by Mrs Gill, which was noted earlier in the report, that she was worried her current symptoms could become worse as she ages and the mesh erodes further.
5171 Second, as the respondents submitted, there is no medical evidence to support Ms Williams’ recommendation for live-in care at any time in the future.4102
5172 Third, as the respondents also submitted, there is no evidence that, before her Prolift surgery, Mrs Gill was responsible for maintaining the pool.4103 Indeed, Mrs Gill apparently told Ms Borthwick that it had been her husband’s responsibility.4104 That would not matter if this case were governed by the common law (see Van Gervan v Fenton at 338 and 250), but it is not.
5173 Fourth, according to what she told Ms Borthwick, before Mrs Gill was injured Mr Gill was also responsible for the home maintenance, mowing the lawn, and the more strenuous work in the garden.4105
5174 On the other hand, there are also difficulties with Ms Borthwick’s assessment.
5175 First, as Ms Williams observed, Ms Borthwick failed to take into account the fact that, while Mrs Gill has the capacity to undertake and complete certain tasks, the cumulative effect of doing so has had a deleterious effect on the rest of her life.4106
5176 Second, the cessation of care at 80 was arbitrary. Ms Williams noted that Ms Borthwick had referred to no “geriatric research evidence” to support her opinion that the current level of care she assessed Mrs Gill required would be needed in any event once she turned 80. Ms Williams also referred to her knowledge of “many 80 to 90 year old women who continue to live in their homes independently with no support …”4107
5177 Third, Ms Borthwick does not appear to have taken account of the impact that Mrs Gill’s work is having on her capacity to undertake household chores.
5178 Fourth, Ms Borthwick assumed that the Gill children would either leave home at 18 and that, if they did, this would reduce Mrs Gill’s need for assistance with cleaning or, if they did not, their contribution to the household would be greater.4108
5179 Fifth, Ms Borthwick did not always inquire about the time Mrs Gill took to perform certain tasks.4109 In particular, she did not routinely inquire into “standard tasks”, such as stripping a bed, although she conceded that it would be relevant to make such an inquiry as the time it takes one person might be different from the time it takes another.4110 Similarly, she did not inquire about the time it took Mr Gill to perform all the various tasks he undertakes around the home.4111
5180 Sixth, Ms Borthwick contended that Mrs Gill’s rate for domestic cleaning was excessive and not in keeping with rates charged by local services. She recommended the rate in line with the services provided by Absolute Domestics of $33 per hour and noted that rates quoted in local publications ranged from $25 to $35. Yet, Ms Williams recommended the Absolute Domestics rate of $33 per hour.
5181 Under cross-examination Ms Borthwick retreated a little from her evidence in chief. Ms Borthwick had made no provision for assistance with maintaining Mrs Gill’s car because she understood that Mrs Gill had not cleaned her car before her Prolift surgery. In fact, Mrs Gill’s testimony was to the opposite effect. She said that she had always looked after her own car.4112 That evidence was not challenged in cross-examination and I accept it. On the assumption that Mrs Gill’s evidence would be accepted, Ms Borthwick agreed that provision should be made for the cost of car maintenance as it would involve the kinds of activities, including compression of the abdomen and squatting, with which she needed help on account of her injuries.4113 But she would only allow for cleaning once every six weeks (at a cost of $35). She said it was not reasonable for her car to be cleaned more often.4114 Why or how she came to that conclusion is obscure.
5182 Mrs Gill’s evidence disclosed other problems with Ms Borthwick’s history-taking. For example, Ms Borthwick stated that Mrs Gill lacks the energy or stamina to work fulltime.4115 Mrs Gill was adamant that it was not fatigue or stamina that prevented her from working fulltime; it was pain.4116 In cross-examination Ms Borthwick professed to understand that Mrs Gill felt that after three days’ work she could not tolerate the increase in pain and that is what she meant by stamina and claimed the Mrs Gill’s account did not “greatly” change her opinion.4117 Ms Borthwick also stated that the cleaner works for two, rather than three, hours a fortnight.4118 In cross-examination she said that this was an error in her recollection, although it made no difference to her recommendations.4119
5183 Further, Mrs Gill testified that, although her husband had always mowed the lawn, it was she who did the gardening. She said that she can no longer bend and weed or prune and now pays for a gardener to do the weeding. She said she loved gardening, derived joy from her roses, and missed gardening a great deal. She said that before her injuries she could spend up to six hours a week in the garden.4120 Once again, this evidence was not challenged. For this reason, to the extent that there are discrepancies between Mrs Gill’s evidence and the account recorded by Mrs Borthwick, I prefer Mrs Gill’s evidence. Ms Borthwick said that she understood that Mrs Gill did the gardens and weeded the garden beds but that Mr Gill “did the trees and shrubs and lawns”. She acknowledged that all these tasks were too physically demanding for her now as they would increase her intra-abdominal pressure which often triggers pain.4121 Despite this, she made no allocation for assistance with gardening.
5184 In their written submissions the respondents summarised the effect of Ms Borthwick’s evidence as modified by her concessions in the following table:
Expense | Weekly cost | Multiplier | Result |
Domestic assistance to age 52 (3.5 hours x $35) | $122.50 | 231.5 | $28,358.75 |
Domestic assistance to age 80 (2.5 hours x $35) | $87.50 | 796.6 | $69,702.50 |
Delivery for online shopping ([33 years —] to age 80) | $10.00 | 855.7 | $8,557.00 |
Commercial car washing ($35 every six weeks) | $5.83 | 855.7 | $4,988.73 |
Commercial gardening assistance (one hour a week at $35 per hour to age 80) | $35.00 | 855.7 | $29,949.50 |
Total | $141,556.48 |
5185 The respondents proposed a 15% discount for vicissitudes, which would bring the sum down to $120,323,01.4122
5186 It should also be noted that the respondents assumed that the 5% multiplier was applicable. That is true for the claims under the TPA because s 87Y provides that, unless a discount rate is prescribed by the regulations, which it is not, the discount rate is 5%. But it is not true of the negligence claims. The CLA (WA) is silent about the discount rate, which means that the common law rate of 3% applies. This means that the sum would be higher with respect to the common law claim.
5187 The determination of damages for gratuitous assistance is subject to certain conditions set out in Pt VIB Div 5 of the TPA (ss 87W and 87X) and ss 12 and 13 of the CLA (WA).
5188 By ss 87W and 87X of the TPA no award can be made for damages for “gratuitous attendant care services” (defined to include services of a domestic nature provided by one person to another for which the other person has not paid or is not liable to pay) unless the court is satisfied that:
(a) there is a reasonable need for the services;
(b) the need has arisen solely because of the personal injury to which the damages relate;
(c) the services would not be provided to the applicant but for the injury;
(d) the services are provided or are to be provided for at least six hours a week and for a period of at least six months; and
(e) before the applicant lost her capacity to provide the services, she had provided them for at least six hours a week and over a period of at least six months.
5189 In addition, a ceiling is imposed on the hourly rate for the calculation of damages.
5190 Section 87X relevantly provided that:
(1) A court must not, in a proceeding to which this Part applies, award personal injury damages for loss of the plaintiff’s capacity to provide gratuitous attendant care services to other persons, except in accordance with this section.
(2) The court must be satisfied that:
(a) prior to his or her loss of capacity to provide the services, the plaintiff had provided the services:
(i) for at least 6 hours per week; and
(ii) over a period of at least 6 months; and
(b) the other person would have been entitled, if the plaintiff had died as a result of the contravention of this Act to which the award relates, to recover damages under a law of a State or Territory for loss of the plaintiff’s services.
(3) If the plaintiff would have provided the services during a quarter for which, at the time the award was made, the amount of average weekly earnings was ascertainable, the court must not award as personal injury damages for the services:
(a) if the services would have been provided for at least 40 hours per week—an amount per week that exceeds average weekly earnings for that quarter; and
(b) if the services would have been provided for less than 40 hours per week—an amount per hour that exceeds 1/40 of average weekly earnings for that quarter.
(4) If the plaintiff:
(a) would have provided the services during a quarter for which, at the time the award was made, the amount of average weekly earnings was not ascertainable; or
(b) would have provided the services after the time the award was made;
the court must not award as personal injury damages for the services:
(c) if the services were provided for at least 40 hours per week—an amount per week that exceeds average weekly earnings for the quarter that, at the time the award was made, was the most recent quarter for which the amount of average weekly earnings was ascertainable; or
(d) if the services were provided for less than 40 hours per week—an amount per hour that exceeds 1/40 of average weekly earnings for that quarter.
5191 Section 12 of the CLA (WA) provides as follows:
(1) This section deals with the awarding of damages for gratuitous services of a domestic nature or gratuitous services relating to nursing and attendance that have been or are to be provided to the person in whose favour the award is sought by a member of the same household or family as the person.
(2) No damages are to be awarded for the services if the services would have been, or would be, provided to the person even if the person had not suffered the personal injury.
(3) If the amount of damages that could, if this subsection did not apply, be awarded under subsection (5) or (7) is Amount B or less, no damages are to be awarded for the services.
(4) In subsection (3) —
“Amount B” has the meaning given by section 13.
(5) If the services are provided or to be provided for not less than 40 hours per week, the amount of damages awarded for them is not to exceed the amount calculated on a weekly basis at the rate of —
(a) the amount estimated by the Australian Statistician as the average weekly total earnings of all employees in Western Australia for the relevant quarter; or
(b) if the Australian Statistician ceases to make the estimate referred to in paragraph (a), the weekly amount fixed by, or determined in accordance with, the regulations.
(6) In subsection (5)(a) —
“the relevant quarter” means —
(a) the quarter in which the services were provided; or
(b) if at the date of the award an estimate as referred to in subsection (5)(a) is not available to the court for that quarter or the services are yet to be provided, the most recent quarter for which such an estimate is available to the court at the date of the award.
(7) If the services are provided or to be provided for less than 40 hours per week, the amount of damages awarded for them is not to exceed the amount calculated on an hourly basis at an hourly rate that is one-fortieth of the weekly rate that would be applicable under subsection (5) if the services were provided or to be provided for not less than 40 hours per week.
5192 Section 13 defines Amount B in the following way:
(1) Amount B for the financial year ending on 30 June 2003 is $5,000.
(2) For any other financial year, Amount B is obtained by varying Amount B for the preceding financial year according to section 4.
(3) On or before each 1 July after this section commences, the Minister is to publish a notice in the Gazette specifying the amount that is Amount B for the financial year commencing on that 1 July.
(4) Publication under subsection (3) is for public information only and a failure to publish or a delay or error in publication does not affect what is Amount B for the year concerned.
5193 I was informed that as at December 2017 Amount B was $7,000.4123 It remains at $7,000.4124
5194 Although it is not entirely clear, the respondents’ submissions seem to proceed on the basis that these conditions applied to Mrs Gill, although Mrs Gill’s claim, like that of the other applicants, was for commercial assistance.4125 In their written submissions they did not concede that damages for future domestic assistance should be determined on the basis that the assistance would be provided commercially. They merely submitted that “at a maximum, any allowance for future care should be limited to the sum of $94,865.93.4126 That figure bears no resemblance to the submission that followed it. I assume it is a relic of a previous draft. It appears to be derived from adding the figures for the first four items in the table and then discounting the total by 15% and does not include the last item (commercial gardening assistance).
5195 Moreover, to the extent that Mrs Borthwick did not provide for assistance where the services would have been provided to Mrs Gill or for her benefit irrespective of whether she had been injured (such as lawn mowing and home maintenance), the respondents’ submission incorporated restrictions on damages for domestic assistance that are imposed by the legislation only where the assistance is provided gratuitously.
5196 The first question, then, is upon what premise should the damages be assessed. The second is how the assessment should be made.
5197 The approach to the assessment of damages for future care where the injured plaintiff claims the cost of commercial assistance has been the subject of much discussion in the NSW Court of Appeal over the last decade.
5198 In Miller v Galderisi [2009] NSWCA 353 the Court made it clear that in an appropriate case where past services had been provided gratuitously, damages for future domestic assistance could include commercial services. Miller concerned a claim for damages affected by the Motor Accidents Compensation Act 1999 (NSW), which imposed conditions or limitations on an injured person’s damages for gratuitous domestic services similar to those imposed by the TPA. The trial judge found that the respondent had a need for domestic assistance which had been increased by the accident but that the increased need did not reach the statutory threshold imposed by s 128 of the Motor Accidents Compensation Act. Nevertheless, he awarded damages on a commercial basis. The Court (Allsop P, Basten and MacFarlan JJA) held at [16] that the award was unjustifiable since it was made on the assumption that the respondent required commercial domestic assistance immediately and would continue to require it on that basis for the rest of his life. The Court considered the assumption to be incorrect because at the time of the hearing the assistance was being provided gratuitously. The Court observed, however, at [18] that there was no reason in principle why, if the evidence supports it, damages may not be awarded “in respect of a need for commercial domestic assistance likely to arise in the future after the availability of gratuitous assistance ceases”. It pointed out that such an award had been made in Nominal Defendant v Lane [2004] NSWCA 405.
5199 In Gordon v Truong [2014] NSWCA 97; (2014) 66 MVR 241, another case affected by the Motor Accidents Compensation Act, the trial judge did not award damages for future commercial assistance and refused damages for gratuitous assistance on the basis that the assistance being provided to the cross-appellant (Mr Truong) (by Ms Pham, one of her adult sons, and an adult son of Mr Truong) was below the statutory threshold. In declining to award damages for commercial assistance, he considered himself bound to do so by the decision in Miller. By a majority, the Court distinguished Miller and held that Mr Truong was entitled to an award of domestic assistance at commercial rates on the basis that, although past assistance had been provided gratuitously, it was not established that future assistance would be provided on the same basis (at [125]–[135]).
5200 Simpson JA, with whom Macfarlan JA agreed, said at [125] that the decision of the trial judge that he was bound by Miller was based on an unstated assumption of fact: that the services would continue to be provided, as they had been in the past, on the basis that the plaintiff (Mr Truong) was not liable to pay for them. Her Honour said at [126] that the assumption was unwarranted; the probabilities were that, in due course, both able-bodied sons would move out of Mr Truong’s home and establish their own homes. She said it could not be assumed that they would be in a position to devote their time to his care. As for Ms Pham, her Honour said at [127] that the probabilities were that she would return to her part-time employment which she gave up only because of the need to care for Mr Truong, in which case it would be necessary to replace her contribution to his care with paid services.
5201 In distinguishing Miller Simpson JA stated at [130]–[131]:
130 Notwithstanding an apparent correspondence of facts, there is a very significant point of differentiation between the facts of Miller and the facts of the present case. In Miller, the plaintiff's wife was in receipt of a carer's pension, granted for the very purpose of enabling her to provide care to the plaintiff. Although it was not given as a reason in this Court, it seems to me that, to award damages to enable the plaintiff to pay for assistance at commercial rates, while his wife continued to receive a pension for that very purpose, would amount to unjust enrichment, and be contrary to principle. In fact, it is difficult to say that the services in Miller were being rendered gratuitously. They were being paid for, although not by the plaintiff, and the plaintiff was not liable to pay for them. The implicit finding of this Court was that the assistance would continue to be rendered, and it would continue to be paid from resources other than those of the plaintiff. In the terms of s 141B, therefore, they were services for which the plaintiff would not be liable to pay. In the circumstance that the plaintiff's wife was in receipt of a carer's pension for the very purpose of providing the services it was an inevitable conclusion that the services would continue to be rendered on the basis that the plaintiff would not be liable to pay for them.
131 In this case, Ms Pham was also in receipt of a carer's pension, but well before the plaintiff's injury. That pension had nothing to do with the plaintiff's injury [it related to the care of one of her sons who was disabled]. Unlike in Miller, services rendered by Ms Pham to the plaintiff, additional to those she rendered pre-injury, were rendered gratuitously. There is not the same basis for a finding — or an assumption — that Ms Pham will continue to render the services gratuitously, once the plaintiff has funds.
5202 Basten JA, who dissented, considered (at [26]) that commercial domestic assistance should be treated as a possibility, not involving a finding on the balance of probabilities, “but an exercise in prediction in accordance with the principles established in Malec”.
5203 In Metaxoulis v McDonald’s Australia Ltd [2015] NSWCA 95, a case affected by the CLA (NSW), the threshold for gratuitous care was not reached and the trial judge refused to award damages for commercial assistance. Basten JA, with whom McColl and MacFarlan JJA agreed, remarked at [77] that the apparent reasons, were that the appellant’s wife might like some commercial assistance and surgery would relieve his chronic pain and satisfy his need for care and assistance. Basten JA said that the trial judge’s reasons could not be accepted. The appellant’s wife testified that she had obtained commercial assistance in the past and intended to so in the future. Basten JA said at [78] that this evidence supported the view that such assistance would be sought. His Honour also said that to suggest that this was merely her preference ignored the evidence given by the appellant. The appellant testified that a cleaner used to come to the house every week for two or three hours, that she only stopped coming because he could not justify the expense, and that if he had the opportunity, he would definitely employ a cleaner again. He also testified that he did not think his wife could continue doing the work he used to do because she was very tired after working full-time and spending the whole Sunday cleaning. At [81] his Honour stated:
The appellant’s written submission proposed an allowance of $26 per hour for two hours per week for the remainder of the appellant’s life expectancy, being 44.4 years. If that calculation were accepted, there must be a significant reduction for vicissitudes, both to take account of normal ageing processes and to take account of the significant likelihood that the appellant would have required domestic assistance at an earlier stage because of his existing injury. The adjustment is appropriately undertaken by way of a proportionate reduction in accordance with the principles identified in Malec v JC Hutton Pty Ltd. [The appellant’s calculation produced a figure of $49,111; a reduction of 40% would produce an allowance, in round terms, of $30,000. That is an appropriate figure in the circumstances.
5204 No reference was made to Gordon v Truong.
5205 In Sampco Pty Ltd v Wurth [2015] NSWCA 117, where the issue once again arose for consideration, Basten JA observed at [101] that the majority in Gordon v Truong “appeared to adopt an approach based on the balance of probabilities”. Notwithstanding the fact that his Honour was in the minority in Sampco, counsel in Sampco accepted that the Malec approach was correct.
5206 Basten JA noted that Sampco was not run on the basis that, due to a change in circumstances, gratuitous assistance might cease and commercial assistance might be required but on the plaintiff’s expressed preference for commercial services. Consequently, he said the case “did not involve speculation as to future events in foreseeable but unpredictable circumstances”. Since the plaintiff’s husband had been willing to provide the necessary domestic assistance in the past, his Honour, with whom Meagher JA and Adamson J agreed, proceeded to find that it was not unreasonable to expect that he would continue to do so in the future.
5207 His Honour pointed out at [104] that the only evidence led on the question was given by the plaintiff. It was that paying somebody to come in and do the heavier chores would make married life much better. His Honour said at [105] that this evidence was an insufficient basis for finding that the plaintiff’s husband would not continue to provide the assistance he had previously given or that there was any likelihood that, if an award for domestic assistance were made, it would be employed for the purpose of obtaining commercial assistance.
5208 In Australia and New Zealand Banking Group Ltd v Haq [2016] NSWCA 93 at [47] Basten JA described the case law as “ambivalent” with regard to whether a finding on the need for commercial services in the future involves “a Malec assessment of possibilities” or is to be made on the balance of probabilities.
5209 More recently, however, in Avopiling Pty Ltd v Bosevski; Avopiling Pty Ltd v The Workers Compensation Nominal Insurer (2018) 98 NSWLR 171 at [137], Payne JA, with whom both McColl and White JJA agreed, noted that the weight of authority in the NSW Court of Appeal was that “the Malec v Hutton approach” applies to the assessment of damages for future attendant care and considered that it was the correct approach as a matter of principle.
5210 I return to the present case.
5211 Like Mr Metaxoulis and unlike the position in all the other cases to which I have referred, Mrs Gill had procured commercial domestic assistance in the past.
5212 Mrs Gill’s evidence on this subject was as follows. For most of 2007 through 2009 she hired a babysitter for two hours in the afternoon on school days until her husband returned from work. The babysitter was usually paid about $40 per week to look after the two boys and usually also to cook dinner for the family and do some household chores. In 2010 and 2011 Mrs Gill hired a cleaner to perform household chores, including cleaning, once a fortnight. She paid $60 a visit. In 2016 she engaged a new cleaner to work once a fortnight for three hours at $105 a visit. According to what she told Ms Borthwick, the cleaner also changes the bed linen on these visits.4127 The only reason she did not retain a cleaner during 2012 to 2015 was that she did not feel that the family could afford one.4128 She also paid someone to weed the garden.4129
5213 This evidence was not the subject of cross-examination.
5214 Mrs Gill’s affidavit was silent as to her future intentions. So was her husband’s. And neither she nor her husband was asked about them during oral evidence. Still, it would be wrong to place too much weight on the absence of direct evidence of intention: White v Benjamin at [87] (Basten JA). In that case, past assistance had been provided to the plaintiff by her husband on a gratuitous basis. The plaintiff testified that she wanted to obtain commercial assistance rather than to continue to rely on her husband. The trial judge rejected her evidence. The Court of Appeal held that the trial judge had erred. Basten JA, with whom Meagher JA agreed, said at [88]:
What was required was consideration of the family circumstances, including the fact that Mr White was self-employed and apparently busy; that his wife was unable to do heavy cleaning and hanging out clothes; and that cleaning services are not the kind of personal domestic assistance which one spouse may prefer to obtain from another. Rather, they are services which are readily available and availed of by those who can afford them and who are otherwise engaged in remunerative employment or have a disability.
5215 In White v Benjamin the Court awarded damages for future domestic assistance on a commercial basis to cover assistance with cleaning and washing but reduced the market value by 15% “for vicissitudes”, which I took to refer to the chance that commercial assistance would not be provided. Basten JA said at [92] that, “although there is a significant chance that commercial assistance will not be obtained, a greater reduction would be self-fulfilling”. In Avopiling the Court followed White v Benjamin but reduced the market value by 25%.
5216 In the present case, Mr Gill is a marine biologist in full-time employment. The relationship between him and his wife is under significant strain. Since Mrs Gill was injured, she has employed a cleaner and at the time of trial they were paying a cleaner for three hours work a fortnight. There is no reason to think that they would not continue to do so in the future. Only financial considerations precluded them from doing so in the past and then only for a period of time.
5217 While neither Mrs nor Mr Gill gave direct evidence about her or his future intentions, Mrs Gill did give evidence that, but for her injuries, she had intended to return to full-time employment when her sons were in high school in which case she would have had less time and energy and possibly less inclination to devote to household chores.4130
5218 Still, other services continue to be provided gratuitously by Mr Gill with assistance from the two children. During the week Mr Gill is responsible for the cooking, the washing up and the preparation of school lunches. He also assists with the housework including the laundry, cooking, gardening and lawn care, putting out the bins, caring for the animals, and maintaining the pool.4131
5219 Mrs Gill said that she feels guilty that the boys are required to help with the housework and that her husband does too much and has no leisure time. She also said that this makes her feel like a failure as a wife and a burden on the family.4132
5220 Seasonal cleaning no longer gets done, which worries Mrs Gill.4133
5221 I infer from the use of commercial services in the past and Mr and Mrs Gill’s personal circumstances that Mrs Gill would continue to avail herself of commercial services in the future.
5222 In view of Mrs Gill’s decision to hire a cleaner and a gardener, the fact that both she and her husband would have been working full-time but for her injuries, her sense of guilt about the burden family members now carry, the fact that the marriage is already under strain, I infer that if she had the resources to do so Mrs Gill would also have engaged commercial services to attend the lawns, to clean the pool and carry out pool and home maintenance, to undertake kind of tasks Ms Williams captured under the title spring cleaning or seasonal tasks, such as cleaning the windows, the gutters, and the pergolas, and would do so in the future.
5223 As I accept Mrs Gill’s pain is genuine and disabling, I accept that she needs assistance with all these activities as a result of the injuries caused by the Prolift device. Ms Borthwick accepted that she required assistance with tasks that require her to lift and carry weights greater than five kilograms or to work at low levels or in confined spaces and that would justify assistance with all of these tasks.
5224 I now turn to the question of hours.
5225 It is not apparent from Ms Williams’ report why five hours assistance a week is necessary with heavy cleaning when Mrs Gill employs a cleaner to do it in three hours a fortnight.4134 Ms Williams offered no reasons for her opinion and none of the witnesses offered any insight into the size of the house or the number of rooms. In these circumstances, I would allow only three hours a fortnight. I would also allow an additional amount for spring cleaning. Ms Williams recommended 12 hours a year. Ms Borthwick allowed ½ hour a fortnight in the past and rolled up spring cleaning with other cleaning for the future. Half an hour a fortnight over a year is 13 hours annually. I propose awarding 12 hours a year in accordance with Ms Williams’ recommendation.
5226 Thirty three dollars is a reasonable hourly rate for regular cleaning but not for the spring cleaning tasks. The relevant tasks include working at heights and carry a level of risk that ordinary domestic cleaning does not. For these reasons I prefer Ms Williams’ assessment of $165 for three hours four times a year, which amounts to $55 per hour. I dare say that is why the rates Ms Williams cited were higher for these tasks than for the heavy cleaning.
5227 I accept Ms Borthwick’s evidence that that $35 is a reasonable figure for washing the car but it does not take into account vacuuming. I also regard six weekly intervals as too long. Instead, I would allow one wash and vacuum per month at $55, as Ms Williams proposed.4135
5228 In accordance with Ms Williams’ recommendation, for gardening and lawn care I would also allow four hours a month at $35 per hour. Although Ms Borthwick considered that the costs of maintaining the lawns should not be included, she did not quarrel with Ms Williams’ assignment of four hours a month or the nominated hourly rate. For the same reason, I also accept Ms Williams’ recommendation for one hour a month of handyman services at $55 per hour.
5229 Against the chance that Mrs Gill would not engage outside services for at least some of the activities, I would reduce the market value of these amounts by 5%. Furthermore, all amounts should be reduced by an additional 15% for the chance that one or more of Mrs Gill’s non-compensable conditions might have prevented her from carrying out some or all of these tasks even if she had not been injured.
5230 The claim for live-in help one or two days a year is not made out. Nor is the claim for assistance with shopping but the cost of home-delivery of groceries at $10 per week should be allowed.
5231 In summary, with respect to the commercial services, I would award the following sums over Mrs Gill’s life less 20% for vicissitudes:
Nature of assistance | Hours pw | Amount ph | Amount pw |
Heavy cleaning (3 hours per fortnight) | 1.5 | $33.00 | $49.50 |
Seasonal cleaning (12 hours a year) | 0.23 | $55.00 | $12.65 |
Car wash (1 per month at $55) | $12.69 | ||
Gardening, lawn care (4 hours per month) | 0.92 | $35.00 | $32.20 |
Handyman services (1 hour per month) | 0.23 | $55.00 | $12.65 |
Home delivery of groceries ($10 per week) | $10.00 | ||
Total weekly amount before discount for vicissitudes | $129.69 |
5232 Finally, while I am satisfied that from time to time Mrs Gill needs help with meal preparation and other tasks around the home and that she may need someone to run errands for her when she is in too much pain, I am not satisfied that she would engage outside help for these purposes. At the time of her report Ms Borthwick said that two hours assistance was required with meal preparation a week. Doing the best I can on the evidence, I would allow four hours a week at $33 per hour for meal preparation and other domestic tasks.
5233 As I said, these figures should be capitalised over Mrs Gill’s life but, bearing in mind that the past services were agreed as at December 2017, this should be on the footing that Mrs Gill’s life expectancy is 39.67 years, as it was at December 2017, not 37.75, as it is now. For ease of calculation I would round up the figure to 40 years.
5234 For the same reason, the future out-of-pocket expenses calculated over Mrs Gill’s life should also be assessed on the basis that she has a probable life expectancy of 40 years.
5235 In her Statement of Particulars Mrs Gill claimed a total of $242,066.73 in future out-of-pocket expenses, consisting of $219,802.94 in treatment expenses and $22,263.79 for equipment. In the submissions filed on her behalf, however, the total had increased by about $37,000. The primary reason for the increase appears to be the switch from a 5% discount rate in the Statement of Particulars to a 3% rate in the submissions. Since the TPA requires the use of the 5% rate, the 5% multiplier remains applicable to the award with respect to the statutory counts but the 3% rate applies to the common law claim.
5236 Without reference to authority, the applicants submitted that a 3% multiplier should be used because the 5% discount rate, provided for by s 87Y of the Trade Practices Act, only applies to economic loss.4136 The respondents made no submissions on the question but it appears from their submissions that they applied the 5% rate.
5237 Section 87Y relevantly provides that if an award of personal injury damages covered by Pt VIB applies includes any component, assessed as a lump sum, for future economic loss of any kind, the present value of the loss is to be determined by applying a discount rate of 5% unless a different percentage is prescribed by the regulations.
5238 Economic loss is not defined in the TPA. But non-economic loss is. In these circumstances any loss which does not fall within the definition of “non-economic loss” is “economic loss”. “Non-economic loss” is defined in s 87D of the TPA for the purposes of Pt VIB, which applies to personal injuries damages, to mean any one or more of the following: pain and suffering; loss of amenities of life; loss of expectation of life; and disfigurement. Since future out-of-pocket expenses do not fall into any of these categories, it is surely economic loss. Both the Explanatory Memorandum for the Trade Practices Amendment (Personal Injuries and Death) Bill 2004 and second reading speeches are unhelpful on this point.
5239 In my opinion, a 5% multiplier should be applied to the calculation of both future out-of-pocket expenses and future services with respect to the TPA claim.
5240 The claim for the costs of future treatment comprises the following items:
(a) $43,300 for further revision surgeries;4137
(b) quarterly specialist consultations at $350 a consultation
(c) monthly GP consultations at $50.00 a consultation;
(d) $10,000 for “other medical attendances (radiology and pathology)”;
(e) clinical psychologist (25 sessions per year until 2022 when Mrs Gill’s youngest son turns 18)) at $245 per session;
(f) $10,000 for a pain management program;
(g) 12 sessions per year with a psychiatrist at $255 per session;
(h) $25,000 for a nerve stimulator;
(i) quarterly chiropractic treatment at $50 a session;
(j) monthly remedial massage at $50 a session;
(k) annual occupational therapy assessments at $733;
(l) antidepressants at $30 a month;
(m) laxatives at $5 a week;
(n) Panadol/Nurofen at $5 a fortnight;
(o) Voltaren/Naprogesic at $2 a week;
(p) Pregabalin (Lyrica) at $10 a week;
(q) Oxycontin/Tramadol;
(r) Valium twice a month at $1.44 a week;
(s) $30 a month in multivitamins; and
(t) Buprenorphine at $2.50 a week.4138
5241 Except where indicated, the claim was made over Mrs Gill’s life expectancy.
5242 The first four items were said to be estimates, but the basis for making each of the estimates was not disclosed in the written submissions. Items 5 to 7 inclusive were based on Dr Jungfer’s evidence. Item 8 was based on evidence in Professor Korda’s report relating to Mrs Dawson. Otherwise no source for either the claims or the costings was identified. That was unsatisfactory. It was unreasonable to require the respondents or the Court to search through the mass of material in evidence, if any, that could support the claims.
5243 The applicants made no written submissions in support of these claims. That was unhelpful.
Revision surgeries
5244 I begin with the claim for revision surgeries.
5245 In oral argument Mr Graham SC, for the applicants, confirmed that there was no evidence of the present costs, but pointed to evidence of medical expenses for the 2013 operation to support the claim, which was admitted by consent.4139 He argued that the cost of the 2013 operation was $4,625.61 and submitted that, taking into account inflation, it would be reasonable to allow $5,000 for each prospective operation.4140
5246 In the Statement of Particulars and the written submissions, $30,000 had been claimed for revision surgeries.4141 Mr Graham contended, however, that, since Mrs Gill had had four erosions in 11 years (an average of one erosion every 2.75 years), over the rest of her life she could have 15 more erosions. Since the present value of mesh erosion surgery is $5,000, which equates to $34.90 per week ($5,000 divided by 2.75 years multiplied by 52.1 weeks), applying the 3% multiplier over 41 years (1,239.9), the cost of revision surgeries was $43,272.51. He argued that an allowance of $43,300 should be made instead of the $30,000 previously sought.
5247 There are several problems with this approach.
5248 First, it is based on the premise that all the integers in the equation are certain. The premise was not established by the evidence.
5249 I was taken to no evidence to suggest that, because Mrs Gill has had four erosions over 11 years, she is likely to have 15 erosions in the future. Indeed, there is no evidence about the possible, let alone probable, number of erosions or the rate at which they could, let alone would, occur, only that Mrs Gill is at risk of further erosions.4142
5250 Second, the figure upon which the initial calculation was made (the calculation of the cost of surgery) included $853.95 in costs which were not incurred in connection with the surgery. Those costs related to two consultations with Dr Dowling two months before the surgery and a CT scan three weeks before the surgery, when separate claims are made for the costs of future specialist consultations and imaging. If those costs were excluded from the initial total of $4,625.61, the cost of the 2013 surgery would fall to $3,771.66. The applicants calculated the present day cost of surgery by adding a figure to the 2013 cost, based on increases to the Consumer Price Index, to reflect inflation.4143 While it would be preferable to have had evidence of the current costs, adjusting the 2013 figures to take into account inflation is a reasonable approach. Applying that approach to the figure of $3,771.66 yields a sum of $4,111.10 or, say $4,100. If that figure were used instead of the $5,000 amount, the weekly figure would drop to $28.67 and the final figure to $35,547 using the 3% multiplier, not $43,272.51.
5251 Third, the 3% multiplier only applies to the common law claim and the 3% multiplier over 40 years is 1,224.2. If the 5% multiplier is used (917.5), the final figure is $26,514.02.
5252 The respondents submitted that Mrs Gill does not currently have a mesh erosion and there is a real issue as to whether any further surgery, if warranted in the future, will provide any tangible benefit and whether, in any event, Mrs Gill would agree to it. They submitted that, if any award were to be made, it should be modest and limited to a buffer of no more than $10,000.4144
5253 At the time the respondents’ submissions were filed, there was no evidence of a current mesh erosion. The most recent medical evidence, however, indicates otherwise. When she examined Mrs Gill on 16 January 2018, Dr Bernadette Brown found a small area of exposed mesh on the posterior vaginal wall and another area of mesh exposure on the anterior vaginal wall with long fibres apparently rubbing on the cervix. In her report to the GP, Dr Brown commented on Mrs Gill’s attitude to surgery:
Kathryn is quite clear that she does not wish to undergo any further surgeries for treatment of her mesh complications. She feels that she has been experimented on and wishes to wait until sufficient skill levels have been obtained before she will proceed with full mesh excision. She has agreed to an examination in theatre – I am suspicious that, on top of her mesh related pain symptoms, she has features of interstitial cystitis. We can perform injections of local anaesthetic and steroid if we identify any ulcerative changes within her bladder. It is also important to ascertain whether there are any other areas of mesh exposure or erosion (bladder or bowel)[.]4145
5254 Thus exploratory surgery was imminent in February 2018 when the report was tendered. Indeed, it has probably already taken place. In addition, despite her reluctance to undergo further surgery to treat the complications, there is a reasonable chance that she will decide she has no alternative.
5255 Doing the best I can, allowing for the virtual certainty of one operation and the chance of more, having regard to the passage of time and taking into account Mrs Gill’s relative youth but making due allowance for her reluctance to undergo further surgery, I would allow a buffer of $15,000.
Specialist consultations
5256 Since claims have been made for psychiatric, psychological, chiropractic and massage treatment, like the respondents I assume that the claim for specialist consultations relates to consultations with a gynaecologist, urogynaecologist or urologist.4146 Noting that Mrs Gill saw Dr Dowling three times in 2013, Dr Jeffrey and Dr Tsokos in 2014, Dr Vashevnik once in 2016, and Professor Vancaille once in 2017, the respondents submitted that an annual consultation was reasonable.
5257 The evidence does not support an allowance for four consultations a year. Even so, the premise for the respondents’ submission is wrong. In 2013 Mrs Gill saw Dr Dowling four times, if the day of the surgery is included.4147 In 2016, as well as Dr Vashevnik,4148 she also saw Dr Jeffrey three times on 17 February,4149 16 March,4150 and 5 May.4151 Mrs Gill saw Professor Vancaille, not once but twice in 2017, on 3 July4152 and on 15 November.4153 Two months after her last consultation with Professor Vancaille, she saw Dr Brown.4154
5258 The evidence indicates that Mrs Gill will remain under specialist care indefinitely. In the absence of an erosion or infection, one consultation a year might well suffice. But there will be other times when consultations at more frequent intervals will be necessary. In these circumstances, I would allow two a year at the amount of $350 per consultation and discount the total amount by 15%.
5259 No evidence was adduced about the present cost of urogynaecological consultations and I have struggled to find evidence of the costs of past consultations, which would provide a guide to the costs of future consultations. Defence Health records disclose Dr Dowling’s fees but only for the treatment in hospital on 8 August 2013.4155 Medicare records only disclose the benefits, not the fees themselves.
5260 In the circumstances, and in the absence of any argument from the respondents that the $350 figure nominated by the applicants was unreasonable, I take it to be common ground that $350 is a reasonable sum.
GP consultations
5261 The claim for $50 a month is excessive. I have no problem with the $50 figure. That seems entirely reasonable. The respondents submitted, however, and the applicants did not dispute, that between 2013, when Mrs Gill’s symptoms abruptly deteriorated, and December 2017, Mrs Gill saw GPs a total of 17 times. Moreover, the consultations did not just relate to the complications of Prolift. I accept the respondents’ submission that an award for bi-annual consultations with a GP for mesh-related ailments is reasonable.4156
Other medical attendances
5262 In the absence of any particulars, submissions, or up-to-date evidence, it is difficult to know what to make of the claim for $10,000 for the costs of diagnostic imaging and pathology. As the respondents submitted, in view of Mrs Gill’s documented history of mesh erosions and the use of CT scanning to diagnose the erosions, some allowance for this kind of imaging is apt.4157 The respondents would limit it to a buffer of “up to $1,000”. It seems to me, however, that this would be inadequate.
5263 For a start, Dr Brown proposed, amongst other things, that a biopsy be taken, which means that, almost certainly, there would be pathology costs. Second, in the event of any further mesh removal surgery, there would also be pathology costs. Third, the Medicare benefit alone for the CT of the abdomen and pelvis conducted on 18 June 2013 was $608.05.4158 Fourth, there might be other scans, such as ultrasounds, possibly even an MRI. Fifth, Mrs Gill’s life expectancy is nearly four decades long.
5264 I would allow $4,000 for diagnostic imaging and pathology costs.
Psychiatrist, psychologist and pain management
5265 Mrs Gill claims the costs of 25 sessions per year with a clinical psychologist until her oldest son turns 18; 12 sessions a year for life with a psychiatrist; and $10,000 for a pain management program. The claim is said to be based on Dr Jungfer’s recommendations.
5266 The respondent’ submitted, in effect, that no award should be made for the costs of psychiatric treatment because the Court could not be satisfied that Mrs Gill had discharged her burden of proving that she had suffered a psychiatric injury as a result of her Prolift implant and the subsequent operations.4159 I have already rejected this argument. It follows that an award should be made. The only remaining question is in what amount.
5267 Dr Jungfer made no recommendation, however, for psychiatric treatment, at least outside a pain management program, despite citing the current AMA rate for a short consultation with a consultant psychiatrist.4160 Dr Samuels, on the other hand, thought Mrs Gill would probably benefit from a review by a psychiatrist “particularly given her chronic pain state”. He suspected that other antidepressants might be more effective in the management of her pain.4161 He noted, however, that she was to attend a multidisciplinary pain clinic, which he suspected might well make “some suggestions along that line”.4162 He said that the cost of a review by a consultant psychiatrist would be in the region of $350 for a first assessment and, if there were to be a change in her medication, she might need to see the psychiatrist for some months after that.4163 He also observed that further surgical intervention had the potential to exacerbate her situation. He said that any decisions in that regard should be made in conjunction with her GP, pain specialist, and gynaecologists and that “ideally”, a psychiatrist and psychologist should be involved in the decision-making process.4164
5268 I am satisfied that an award should be made for some psychiatric treatment, but not of the order Mrs Gill’s lawyers sought. The evidence certainly does not support one consultation every month with a psychiatrist for the rest of Mrs Gill’s life.
5269 I would award $2,000. It is clear that changes in Mrs Gill’s pain management regime are likely to require review, if not adjustment, of any treatment she requires or may require for her depression and her physician will need to work together with her psychiatrist to ensure that she is on a safe and effective course of treatment. Moreover, as Dr Samuels said, a psychiatrist should be involved in the decision-making process around future surgical intervention.
5270 Dr Jungfer did consider that Mrs Gill would benefit from being able to see a clinical psychologist, preferably one with pain management experience. The purpose of this treatment, she explained, would be to “have someone to ventilate with, to look at approaches such as mindfulness, self-hypnosis for her chronic pain management as well as providing her with more general support’.4165 Dr Jungfer recommended 25 sessions a year “until there is some stabilisation of her clinical presentation or her children reach adulthood and are more independent”. She also recommended a pain management program and the use of Pregabalin, (Lyrica) to which I will come later.4166
5271 The premise upon which Dr Jungfer’s recommendations were made was that Mrs Gill had not had “any form of rational treatment with regards to her chronic pain”.4167 That may well have been true at the time of Dr Jungfer’s assessment, but it was not true by the time of the trial. By then she had seen been to a pain clinic, where she saw Dr Kriel, and had also seen Professor Vancaille, and Elizabeth Howard. Dr Kriel also recommended “pain psychology” as well as hydrotherapy.4168
5272 Dr Samuels considered that a multi-disciplinary pain clinic would be “the most appropriate setting to help her manage [her] symptoms”. He said it was likely that in such a facility she would be taught “cognitive behavioural strategies to manage her interpretation of pain and her physical symptoms”, how to pace herself better “in terms of work”, and would also explore “the relationship and family issues”.4169
5273 While Mrs Gill has had some assistance from pain management specialists, the evidence indicates that she has not had the benefit of the kind of treatment Drs Jungfer and Samuels recommended. For a start she has had no cognitive behavioural therapy. Mrs Gill said that she had not followed up the pain psychology or hydrotherapy because she did not feel well enough on her days off work but would like to “trial” them both.4170
5274 I would award $8,500 towards further treatment in a multidisciplinary pain clinic. I would also make an allowance for the cost of additional treatment by a psychologist to assist in the decision-making process over future surgery and to provide counselling as required over the years to deal with the psychological impact of her chronic pain. Dr Samuels said that he “probably would not be advocating a need for psychological support at the present time”, but he did not discount the possibility that she might need it in the future.4171 Although Mrs Gill has seen a number of psychologists over the years, she is yet to see one with any apparent expertise in the management of chronic pain.
5275 I would award $3,500 for the cost of additional treatment by a psychologist.
Nerve stimulator
5276 Mrs Gill claims $25,000 for a “nerve stimulator”. The basis of this claim is a recommendation made by Professor Korda about Mrs Dawson.4172 It is by no means clear what the applicants were proposing. Professor Korda referred to two types of nerve stimulation in his report: transcutaneous electrical nerve stimulation (TENS) and implantable nerve stimulation devices.
5277 In support of his costing Professor Korda cited an article from the Department of Pain Management at the renowned Cleveland Clinic Foundation: Nagy A Mekhail, Armin Aeschbach, and Michael Stanton-Hicks, “Cost Benefit Analysis of Neurostimulation for Chronic Pain” (2004) 20 Clin J Pain 462–468.4173 That report dealt only with implantable neurostimulation devices. It provided no insight into the cost of TENS machines. Based on the amount claimed on Mrs Gill’s behalf, which related to the implantable devices, I can only assume that the claim was for the cost of such a device.
5278 Mekhail et al conducted a cost-benefit analysis of the use in healthcare of spinal cord stimulation (SCS) and peripheral nerve stimulation (PNS) of patients with intractable chronic neuropathic pain. The study was based on answers to a questionnaire provided by patients who had received SCS or PNS implants at The Cleveland Clinic Pain Management Department between 1990 and 1998. The authors concluded from the reduced demand for healthcare resources that these forms of treatment, although associated with relatively high initial costs, might give rise to substantial long- term economic benefits and should be considered “a viable option” for early treatment of patients with intractable chronic neuropathic pain.
5279 It is hard to know what to make of this study. The authors’ conclusion was based on answers given by 128 of the 222 patients (58%) to whom the questionnaires were sent. Of the 94 who did not respond, 74 were lost to follow-up and 18 chose not to participate. How this affects the reliability of the study’s findings is unknown. The authors also referred to “several shortcomings” in their methodology, including the absence of a control group, record bias, and changes in status after implantation.
5280 I reject the claim.
5281 After all she has been through, I do not think that there is a remote chance that Mrs Gill would entertain the idea of another implant. Indeed, the little evidence there is on the subject indicates that she would not.
5282 Dr Kriel recommended a trial of neuromodulation, which Mrs Gill described to Ms Williams as spinal cord stimulation therapy, during his assessment of Mrs Gill on 4 April 2017, but she did not proceed with the trial, telling Ms Williams that she was afraid of trying it, given her experience with surgery.4174 In her oral evidence Mrs Gill described it as an “extreme measure”.4175 She said that she saw a second pain specialist in Professor Vancaille’s rooms in July that year who gave her much less invasive options.4176 I presume this was a reference to Elizabeth Howard, the osteopath and pain educator.
5283 There is no evidence to support the use of a TENS machine. In her first affidavit Mrs Gill gave evidence that she had tried a TENS machine that Dr Jeffrey obtained from a physiotherapist but said it worked for about a week only. None of the specialists, whom she has seen, including the two pain management specialists, recommended either an implantable nerve stimulator or a TENS machine.
Chiropractic treatment and remedial massage
5284 As the respondents submitted, there is no evidence that Mrs Gill requires regular or periodic chiropractic treatment or remedial massage as a result of her Prolift implant.4177 I therefore reject the claim. Dr Flynn noted at a consultation in February 2009 that Mrs Gill was seeing a chiropractor for head and neck treatment apparently owing to headaches.4178 In her first affidavit Mrs Gill said that she attends a chiropractor once every few months and a masseuse at the same practice about once a month on average, each at an average cost of $50 a session.4179 The chiropractor’s account statement shows that he was providing “spinal adjustments”.4180 To the extent that the massages indicate any cause for the pain for which the treatment was apparently given, the notes do not support Mrs Gill’s thesis. They identify other causes such as “dumped in surf” (27 July 2015),4181 getting “dunked” at the beach (28 July 2015) and driving to Exmouth (6 October 2015).4182
5285 Mrs Gill said that she had had neck pain from time to time but had “not been diagnosed with a medical condition associated with that pain”. She believed it was caused by tension and pain in other areas of her body that cause her to clench her jaw and stiffen her neck.4183 This is a plausible thesis but, in the absence of any expert opinion, it is not possible to conclude that she may require chiropractic or massage treatment in the future as a result of the implantation of the Prolift Total device.
Occupational therapy
5286 Ms Williams recommended that Mrs Gill undergo a two hour annual review with an occupational therapist for advice and assistance with “energy and pain management techniques and equipment to support [her] to maintain as much independence and quality of life as possible”.4184 The cost, as claimed, of this annual review, including two hours travel, was $733.
5287 Ms Borthwick said that she does not support the need for annual occupational therapy except for the catastrophically injured or “ill patients for whom the annual review of complex equipment of therapeutic input is required” and that Mrs Gill does not fall within either category. Instead, she recommended 12 hours of occupational therapy at the cost of $181 per hour including GST (a total of $2,172), for the purpose of providing education in task modification and pacing, home layout to reduce the need for bending and lifting, and the use of recommended equipment; and to identify possible modifications to the layout of the home.4185
5288 In her supplementary report, Ms Williams argued that a one off block of 12 hours of occupational therapy would have a minimal impact on Mrs Gill’s ability to manage her activities of daily living.4186
5289 I am not persuaded that annual assessments are required. I did not understand Mrs Borthwick to have recommended that the 12 hours be provided in one block and Ms Borthwick’s alternative recommendation was not challenged in cross-examination.
5290 I would award $3,000, based on Mrs Borthwick’s recommendation but with an allowance for the cost of travel.
Medication and supplements
5291 This claim as particularised included $30 per month for anti-depressants; $5 a week for laxatives; $5 a fortnight for Panadol or Nurofen; $2 a week for Voltaren or Naprogesic; $10 a week for Pregabalin; an allowance of $5,000 for Oxycontin/Tramadol; and $1.44 a week for Valium. But in closing submissions, the allowance for Oxycontin/Tramadol was not pressed.
5292 I will deal with the drugs first and then the laxatives.
5293 In her first affidavit Mrs Gill said that she did not particularly like taking medication so she tries to minimise her intake. She went on to say that she had taken a variety of anti-depressants since her surgery and was currently taking one 20mg capsule of Lovan a day, which costs approximately $30 a month. She said the costs of other anti-depressants were “much the same”. For pain, she said she generally took about four to six Panadol a week, but sometimes considerably more, depending on her level of pain. She estimated that on average she had spent about $5 a fortnight on Panadol or Nurofen since her implant surgery. She added that she also takes Voltaren or Naprogesic at times. She said that she had been prescribed stronger medication, such as Lyrica, OxyContin and Tramadol but had avoided taking them as much as possible because they make her feel tired and “out of it” and she did not want to be like that while she has young children who need care, supervision, and transportation.4187
5294 By the time she saw Professor Vancaille in July 2017 the only medication she was taking was Lovan, Panadol, and Valium.4188
5295 The claim for antidepressants at $30 a month is presumably based on Mrs Gill’s evidence. I accept that it is likely that Mrs Gill will be on some form of anti-depressant indefinitely. In all probability, however, that would have been the case, regardless of whether she had suffered complications from the implantation of Prolift. Dr Jungfer did not recommend antidepressants.
5296 In the circumstances I make no allowance for antidepressants.
5297 The respondents submitted that there is no evidence to support the claims for Pregalbin, Oxycontin or Tramadol or Buprenorphine.4189
5298 The respondents submitted that Mrs Gill had been prescribed Tramadol only once, on 30 January 2007.4190 Dr Chapple’s records show that on that day, which was 18 days after the Prolift surgery, he prescribed Tramadol hydrochloride 100mg 6 to 12 hourly as required.4191 In the absence of any submission to the contrary, I accept the respondents’ submission. The respondents also submitted that there was no record of the prescription having been dispensed.4192 That may well be so. In any case, Mrs Gill testified that she never took Tramadol.4193 Although Mrs Gill said that she had been prescribed Oxycontin in the past, I can find no record of it. The chance that either Oxycontin or Tramadol might be prescribed in the future is purely speculative. The claim for an allowance for Oxycontin or Tramadol is not made out.
5299 Mrs Gill deposed in her second affidavit that at the time she saw Dr Kriel she was taking Valium or Buprenorphine twice a month when her pain was out of control.4194 I can find no evidence about who prescribed these medications.
5300 In her second affidavit Mrs Gill said that Dr Vancaille prescribed the following medication for her:
Zaldiar 37.5mg/325mg tablets to treat her “strong pain”;
Allegron 10mg tables once at night to treat her urinary symptoms and to help her sleep; and
Amitriptyline + Estriol organogel AH formulation once daily; and
Valium 5mg vaginal suppository.4195
5301 Professor Vancaille’s records corroborate her account.
5302 Mrs Gill deposed that she had found the Zaldiar to be the first strong pain medication to provide sufficient pain relief without having a significant effect on her cognition. She said that she still needed Panadol and Voltaren but had reduced the Panadol to once a day.4196
5303 At that time, she said, she had not yet received the Valium suppository.4197
5304 In his most recent report, however, Professor Vancaille recommended that Mrs Gill take one low dose Palexia slow release tablet daily (Palexia SR 50mg sustained release tablets) and Zaldiar for breakthrough pain. He indicated he had provided her with prescriptions for both drugs. Palexia is a centrally acting synthetic analgesic indicated for moderate to severe pain.4198 Zaldiar is a combination of tramadol hydrochloride and paracetamol. Mrs Gill said that she never took Tramadol because she was concerned about the effect it might have on her ability to work and that Zaldiar was “a weaker” medication.4199 Having regard to the response Mrs Gill has had to Zaldiar, I think it is likely that she will continue to use it as breakthrough medication.
5305 The most recent evidence I can find on the cost of Palexia SR 50mg tablets is a patient history from Mrs Dawson’s pharmacy. It discloses that, as at 5 January 2017, Palexia SR 50 mg tablets cost $5.30 for a packet of 28.4200
5306 On the assumption that Palexia SR 50mg or an equivalent would be prescribed for the rest of Mrs Gill’s life, allowing $1.33 per week multiplied by 917.5 (the 5% multiplier for 40 years), that would amount to $1,220. Applying the 3% multiplier (1,224.2) yields a sum of $1,628.
5307 I can find no evidence, however, about the cost of Zaldiar.
5308 While Dr Jungfer recommended Pregabalin (Lyrica) at night to improve the quality of Mrs Gill’s sleep and, if tolerated, during the day to assist with pain control. But in the absence of any evidence about how this drug might interact with the drugs and when Professor Vancaille apparently prescribed Allegron to help Mrs Gill sleep, I am not persuaded that any allowance should be made for it.
5309 I accept that it is likely that Mrs Gill will also use non-prescription analgesics and anti-inflammatory medication, especially while she is working.
5310 The respondents submitted that Mrs Gill’s medication requirements “consequent on the Mesh Implant” are likely to change over time and are incapable of precise calculation. They submitted that any award for future medication expenses should be “by way of a modest buffer”, limited to a maximum of $10,000.
5311 There is force in the respondents’ submissions. It is certainly possible, if not probable, that Mrs Gill’s pain medication will change in the future as it has in the past.
5312 I would allow $10,000 for the cost of future medication.
5313 In her first affidavit Mrs Gill said that she takes Movicol daily to keep her stools soft and regular so as to prevent the terrible pain she experiences with passing “a difficult motion”. She said that she has been taking Movicol daily since around 2013 at a cost of approximately $20 a month. She takes one sachet a day. Each box contains 30 sachets.4201 This is presumably the evidentiary basis for the claim for $5 a week for laxatives.
5314 The respondents submitted that no allowance should be made for the cost of laxatives because the Court could not be satisfied that Mrs Gill’s bowel dysfunction and her need to take daily laxatives to maintain faecal continence and reduce pelvic pain were related to the implantation of the Prolift device.4202 Having regard to my findings on this question, I reject the submission. I am satisfied that, although Mrs Gill was constipated before her Prolift surgery, her need to take Movicol has been caused by the severe pain she experiences if she strains to defaecate. In the TPA action claim I would allow $3,440, which is approximately $4,587 ($5 x 917.5, being the 5% multiplier for 40 years) less 25% for the chance that she would have needed to take Movicol or some other laxative in any event. In the negligence actions I would allow $4,600, which is approximately 75% of $6,199.50 ($5 x 1239.9).
5315 The final item is multivitamins. While taking multivitamins may be a good idea, in the absence of expert evidence that they are reasonably required on account of the complications of the Prolift implant, I am not satisfied that the claim has been made out.
5316 A claim is also made for the cost of equipment, purportedly based on Ms Williams’ recommendations. In the table below, I set out the claim:
Item | Unit cost | Replacement period | Annual cost | Weekly cost |
Recliner lounge chair with stand-up function | $2,785.00 | 10 years | $278.00 | $5.34 |
Equipment for carers (fire blanket, fire extinguisher 2kg, hand wash) | $71.37 | Various | $128.73 | $2.47 |
Pressure cushion | $550.00 | 3 years | $183.33 | $3.52 |
Mobility scooter | $3,500.00 | 10 years | $350.00 | $6.72 |
Scooter maintenance & insurance | $385.00 | 1 year | $385.00 | $7.39 |
Car bar | $60.00 | 5 years | $12.00 | $0.23 |
Taxi allowance | $100.00 | 1 year | $100.00 | $1.92 |
Kitchen trolley | $250.00 | 10 years | $25.00 | $0.48 |
Shower chair | $99.00 | 10 years | $9.90 | $0.19 |
Long handled sponge | $25.00 | 1 year | $25.00 | $0.48 |
Easy reacher | $55.00 | 1 year | $55.00 | $1.05 |
Long shoe horn | $10.00 | 3 years | $3.33 | $0.06 |
Long handled vacuum | $299.00 | 3 years | $99.67 | $1.91 |
Toilet rail | $474.00 | 10 years | $47.40 | $0.91 |
5317 The value of the claim, as made, based on a capitalisation over Mrs Gill’s life, using the 3% tables, is $31,653.49. I reiterate that, for the purposes of the TPA claim, the 5% multiplier should be used.
5318 Ms Borthwick, on the other hand, recommended only the following items:4203
Item | Unit cost | Replacement period | Annual cost | Weekly cost |
Laundry basket and trolley | $39.00 | 5 years | $7.80 | $0.15 |
Traymobile | $165.00 | 8 years | $20.60 | $0.40 |
Long handled dustpan and broom | $28.00 | 4 years | $7.00 | $0.13 |
Easy reacher | $65.00 | 5 years | $13.00 | $0.25 |
5319 The recliner lounge chair with stand-up function was recommended to provide a comfortable seat for relaxing and pain relief with “multiple positions and stand-up to assist function”.4204 The respondents argued against it on the basis that Ms Williams had noted that Mrs Gill has two existing lounge chairs with armrests to provide leverage on rising or controlling descent.4205 That is not entirely correct. This is what Ms Williams wrote:
When transferring on and off the lounge chair Katie needs to push up on the armrests to provide leverage. She is also reliant on the arm rest to control the descent into the chair to minimize pain. This was observed during the interview. Katie reported that she is unable to use the “good lounge” at home as it is too low and soft and therefore, she cannot get out of it without assistance.
5320 Still, I reject this claim. I do not believe sufficient justification has been given. Ms Williams does not suggest, and there is no other evidence to indicate, that the use of the armrests in this way exposes Mrs Gill to a risk of injury. In the circumstances, it would be unreasonable to saddle the respondents with the cost.
5321 The justification Ms Williams gave for the equipment described in the applicants’ claim as “equipment for carers” is “occupational health and safety requirements”.”4206 But, in the absence of submissions on the question and since I am not satisfied that a claim for personal care has been made out, I am not persuaded that any allowance should be made for these items. I note that Mr Walsh made no similar recommendation in Mrs Dawson’s case.
5322 Ms Williams said that Mrs Gill was likely to require assistance with transportation “as she ages with chronic pain and physical limitation” and recommended a four wheel mobility scooter from 2031 (when Mrs Gill is 60) (together with annual maintenance and insurance) for outdoor and long-distance community mobility.4207 Ms Williams asserted that Mrs Gill will not age in the same way as her peers.4208 It is not readily apparent that she had the necessary expertise to offer on an opinion on this question. In any event, she provided no reasons to support her opinion. Moreover, there is no medical evidence to indicate that Mrs Gill will be unable to drive her car as a result of the injuries caused by the complications of Prolift at any time in the future. In the absence of such evidence, I am not satisfied that the claim for a scooter is made out. It follows that I reject the claim for the scooter and the expenses associated with it.
5323 Ms Williams also recommended an allowance of $100 a month to allow Mrs Gill to catch a taxi when she is unable to drive herself due to pain.4209 In Mrs Gill’s Statement of Particulars and the applicants’ written submissions, Mrs Gill appears to have claimed a more modest allowance of $100 a year. Ms Williams’ recommendation was based on a statement made in her report of 20 July 2017 that “there will be periods when [Mrs Gill] is unable to drive due to pain and therefore a taxi allowance is recommended”.4210 But the respondents objected to this evidence and it was not pressed. There was no other evidence to support it. I therefore reject the claim for a taxi allowance.
5324 Ms Williams recommended that a pressure cushion be acquired for use when sitting in a car, chair or mobility scooter to minimise pain and increase tolerance and provide a cushioned ride.4211 In view of the discomfort she experiences while sitting and her limited sitting tolerance due to pain, I consider the recommendation reasonable and I would allow it.
5325 Since Mrs Gill has difficulty getting in and out of her car, I also consider the claim for a car bar to assist with such transfers, to be a reasonable one.4212 There is no good reason to attribute Mrs Gill’s difficulty to anything other than her chronic pelvic pain.
5326 Mrs Gill gave evidence that lifting heavy items aggravates her pain. While her pre-existing lower back problem might contribute at times, I am satisfied that the predominant reason for it is the chronic pelvic pain. Ms Borthwick agreed with Ms Williams that Mrs Gill needs assistance with tasks that require her to lift and carry weights greater than five kilograms or to work at low levels or in confined spaces. She explained that these activities result in increased intra-abdominal pressure which triggers pain.4213 For this reason, it seems to me, both the claims for a kitchen trolley to enable items to be moved around the home without lifting and a long-handled cordless stick vacuum cleaner to allow Mrs Gill to spot clean are justified. Ms Borthwick supported the recommendation for a trolley (although she referred to it as a traymobile).4214 Ms Borthwick opposed the claim for a hand-held stick vacuum on the basis that she had recommended the engagement of a commercial domestic cleaner to vacuum the home weekly.4215 But this assumes that Mrs Gill would have no need to spot clean in between. The assumption is unrealistic. Indeed, Ms Borthwick herself, recommended a long-handled dustpan.4216
5327 The evidence would also support a claim for a laundry trolley, as Ms Borthwick recommended, but no such claim was made, presumably because Ms Williams did not provide for one or Mrs Gill already has one.
5328 According to Mrs Williams, Mrs Gill had “no reported difficulty” getting on and off the toilet.4217 She did say that she would require a rail for shower and toilet transfers in the future.4218 In the absence of medical evidence that Mrs Gill’s pelvic pain is likely to deteriorate with age, I am not satisfied that any future need is related to the injuries caused by the Prolift device. Consequently, I am not persuaded that Mrs Gill needs a toilet rail, either now or in the future, as a result of her injuries.
5329 Mrs Borthwick argued that there was no need for a shower chair “as Mrs Gill has a safe standing tolerance sufficient to complete her personal care” and has been able to shower independently since her Prolift surgery. It is not in dispute that Mrs Gill can stand for long enough to shower. But Ms Williams recommended the shower chair to allow Mrs Gill to sit to bathe “if required due to pain”. I do not consider the recommendation unreasonable.
5330 I would also allow the claim for long-handled sponges, an easy reacher, and a long shoe-horn. Each of these appliances was recommended to enable Mrs Gill to avoid bending.4219 Mrs Gill’s evidence was that she can experience a sharp, sudden pain in her lower pelvic region from bending to pick up groceries.4220 Ms Borthwick supported the claim for an easy reacher.4221 If Mrs Gill has difficulty bending and requires an easy reacher, then it is reasonable to conclude that she also requires long-handled sponges and a long shoe-horn.
5331 Ms Williams’ recommendations included replacing the sponges and the easy reacher annually, and the long shoe-horn every three years. Ms Williams did not explain why any of the items should be replaced at the intervals she recommended. Sometimes, Ms Borthwick did not dispute her recommendations. At other times, she proposed replacement after a shorter interval. Neither the applicants nor the respondents explained why one recommendation should be adopted in preference to another. Where Ms Borthwick did not dispute Ms Williams’ recommendations, I have accepted them. In the case of the kitchen trolley, Ms Williams recommended replacement after 10 years4222 while Ms Borthwick recommended replacement after eight years.4223 In this case, I took the view that, although it was to Mrs Gill’s detriment, the longer interval should be preferred since that is what was claimed. In some cases, common sense could fill the gap in the evidence. It is easy to understand, for example, that sponges can wear out after a year of repeated use. In the case of an easy reacher, however, common sense cannot assist. I have no idea how long an easy reacher is expected to last or why it might need to be replaced. In the circumstances, I consider that Ms Borthwick’s recommendation for replacement at five-year intervals4224 should be followed.
5332 In summary, I allow the allow the claims for the following items only:
a pressure cushion to be replaced at three-yearly intervals;
a car bar to be replaced at five-yearly intervals;
a kitchen trolley to be replaced at 10-yearly intervals;
a long-handled cordless stick vacuum cleaner to be replaced at three-yearly intervals;
easy reacher to be replaced every five years;
shower chair to be replaced at ten-yearly intervals;
long-handled sponges to be replaced every year; and
a long shoe horn to be replaced at three-yearly intervals.
5333 I am not satisfied that the other claims are reasonably necessary on account of Mrs Gill’s injuries.
5334 There is a difference in the costs quoted by the two occupational therapists for the easy-reacher and the trolley. Neither provides a source or seeks to justify her figure and the matter is not dealt with in submissions. In these circumstances, I propose to split the difference. In other words, the figures for the two items should be based on a unit value of $207.50 for the trolley and $60 for the easy reacher. For the other items, I allow the amounts as claimed. The amounts should be capitalised over Mrs Gill’s projected life expectancy of 40 years.
5335 I would discount all amounts, however, by 5% for the chance that Mrs Gill might have needed similar equipment at some point in time because of back pain unrelated to her mesh implant.
5336 In Mrs Dawson’s Statement of Particulars, the following injuries were attributed to the Gynemesh PS implant:
1. Chronic inflammatory reaction of tissues in and around the Gynecare Gynemesh PS mesh implant (Mesh Implant) that were implanted into the anterior and posterior vaginal compartments on 8 May 2009.
2. Erosion and/or extrusion and/or exposure of the Mesh Implant through the mid-anterior vaginal wall.
3. Requirement for surgery on 14 October 2009 to excise the exposed portion of the Mesh Implant referred to in the preceding paragraph (First Revision Surgery).
4. Pudendal neuropathy.
5. Erosion and/or extrusion and/or exposure of the Mesh Implant through the posterior vaginal wall.
6. Scarring of the vaginal vault.
7. Rectal tissue banding.
8. Detrusor instability.
9. Requirement for surgery on 31 January 2014 to excise the exposed portion of the Mesh Implant referred to in the preceding paragraph (Second Revision Surgery), specifically:
(a) left arm of the Mesh Implant in the posterior compartment; and
(b) mesh at the vaginal vault.
10. Erosion and/or extrusion and/or exposure of the Mesh Implant through the posterior vaginal wall.
11. Tissue granulation on the posterior wall near to the introitus.
12. Requirement for surgery on 15 May 2015 to excise a portion of the Mesh Implant from the anterior wall of the vagina, vaginal vault and the left lateral vaginal wall (Third Revision Surgery).
13. Erosion and/or extrusion and/or exposure of the Mesh Implant through the posterior vaginal wall.
14. Tissue granulation on the mid-anterior vaginal wall.
15. Requirement for surgery on 30 October 2015 (Fourth Revision Surgery) to:
(a) excise the exposed portion of the Mesh Implant referred to in paragraph 12, above;
(b) excise mesh from the anterior vaginal wall;
(c) divide a left lateral vaginal band using diathermy.
16. Fibrosis around the left anterior and left posterior portions of the vagina.
17. Rigid vagina.
18. Detrusor overactivity.
19. Lower urinary tract dysfunction.
20. Chronic pain syndrome.
21. Exacerbation of pre-existing defecation disorder.
22. Requirement for surgery on 17 March 2017 (Fifth Revision Surgery) to remove scar tissue from the left side of the vagina.
23. Major depressive disorder (DSM-5), chronic.
24. Adjustment Disorder with mixed emotional features (DSM-5).4225
5337 The respondents submitted that the Court could accept that the following injuries were caused by the Gynemesh PS implant:
items 2 and 3 (the exposure requiring surgery on 14 October 2009 and the surgery itself);
items 5,6,7, and 9 (the exposure requiring surgery on 31 January 2014 and the surgery itself);
items 10, 11, and 12 (the exposure requiring surgery on 30 October 2015 and the surgery itself); and
items 16 and 22 (the scarring on the left side of the vagina and the operation on 17 May 2017 to remove it).4226
5338 The respondents argued that Mrs Dawson had not proved that a number of the alleged injuries were “the result of or caused solely be the implant”. Those items were: detrusor instability (item 8); detrusor overactivity (item 18), and lower urinary tract dysfunction (item 19);4227 chronic pain syndrome (item 20); 4228 and the psychiatric disorders (items 23 and 24).4229
5339 I will deal with each of the disputed items in order. Before I do so, however, I wish to say something about the items which were not admitted and about which no submissions were put. The first is chronic inflammation (item 1).
Chronic inflammatory reaction
5340 There is little doubt that there was a chronic inflammatory reaction of the tissues in and around the Gynemesh PS implant.
5341 The histopathology report on mesh sections taken from Mrs Dawson during the surgery by Dr Wallace following the first erosion, noted the presence in the sections of foreign mesh material associated with inflammation and foreign body reaction.4230 This was five months after the device had been implanted. Six years later, another pathologist, Dr Trishe Y-M Leong, who examined sections of vaginal mesh taken from Mrs Dawson during the surgery following another erosion, noted the presence of fibres of foreign material within the stroma (connective tissue) of the vaginal mucosa associated with mild chronic inflammation and foreign body type giant cells (macrophages). She diagnosed chronic inflammation.4231
5342 Professor Iakovlev agreed with the findings of the original pathologists and said his findings were the same.4232 Professor Klosterhalfen confirmed that Mrs Dawson’s specimens reveal “a typical chronic inflammatory reaction and foreign body reaction” eight years after implantation.4233
5343 Professor Wright did not contradict these opinions.
5344 I find that Gynemesh PS caused a chronic inflammatory reaction of the tissues in and around the implant. Once again, however, I do not regard it as an injury as such.
5345 The second item which was not admitted but about which the respondents made no submissions was pudendal neuropathy.
5346 It will be recalled that Professor Eyers considered that pudendal neuropathy may have contributed to Mrs Dawson’s problems and, having read Professor Eyers’ report, Professor Korda considered it more likely than not Mrs Dawson had pudendal neuropathy, since the mesh was placed near the ligaments which are close to the pudendal nerve. Professor Eyers was not cross-examined. Professor Korda was asked no questions about this aspect of his opinion. None of the respondents’ witnesses referred to it or to Professor Eyers’ report.
5347 Pudendal neuropathy means damage, disease or dysfunction of the pudendal nerve. The pudendal nerve is the major nerve in the perineum. The motor and sensory innervation of the perineum is via the pudendal nerve. It runs through the gluteal region, the pudendal canal and the perineum.4234 Pudendal neuralgia is a painful neuropathic condition involving the dermatome of the pudendal nerve.4235 It is described as “burning vaginal or vulval (anywhere between anus and clitoris) pain associated with tenderness over the course of the pudendal nerves”.4236 Sometimes pudendal neuropathy and pudendal neuralgia are used interchangeably.4237
5348 Along with pelvic floor muscle spasm and infection, pudendal neuralgia is a common cause of chronic pelvic pain.4238 Depending on the chronicity of the neuralgia, pain will likely remain the longer the inciting mesh is present.4239
5349 Gyang et al (2014) described pudendal neuralgia in the following way:
The symptoms of pudendal neuralgia vary according to the branch that has been captured or injured. The Nantes criteria for diagnosing pudendal neuralgia include: (a) pain in the anatomical territory of the pudendal nerve; (b) pain exacerbated by sitting; (c) no waking at night because of pain; (d) no objective sensory loss on clinical examination; and (e) positive anesthetic pudendal nerve block.
5350 The Nantes criteria were referred to by the IUGA and the ICS in their joint report on terminology for female pelvic floor dysfunction published in 2009.4240
5351 Gyang et al (2014) stated that pudendal neuralgia can arise as a result of an unrecognised mechanical nerve injury typically referred to as “entrapment”, which may be caused by the pelvic floor muscles, mesh, or a suture directly injuring the nerve. They explained that the pudendal nerve travels beneath the lateral portion of the sacrospinous ligament close to the ischial spine, which is near where the posterior meshes are fixed in pelvic reconstructive surgery.
5352 Any one of these causes could account for Mrs Dawson’s chronic pelvic pain. In all probability all three are responsible.
5353 Pudendal neuralgia is associated with burning, sticking or sharp pain and is not restricted to the vestibule: Baggish M and Karram M (eds), Atlas of Pelvic Anatomy and Gynecologic Surgery (Elsevier, 4th ed, 2016) at 973. It will be recalled that Mrs Dawson described a burning sensation across her bottom, under her buttocks and down the backs of her legs, and burning pain in and around her vagina.4241 Sitting for long periods aggravates her pain.4242 The pain inside her vagina is aggravated by walking and prolonged sitting.4243 On occasions she said she could also feel “a sharp, stabbing type pain” in her vagina.4244 Moreover, since 2016 Mrs Dawson has been treated with Lyrica, which is a prescription drug indicated for the relief of neuropathic pain.4245
5354 I am persuaded by Professor Korda’s opinion. I consider that it is more likely than not that pudendal neuropathy is at least making a contribution to Mrs Dawson’s chronic pelvic pain and that more likely than not it was caused by the installation of the mesh.
5355 The other alleged injury about which the respondents made no submission was “rigid vagina”. But neither did the applicants. The only reference I can find to Mrs Dawson having a “rigid vagina” appears in Professor Korda’s first report. He does not comment on its significance or its cause. In the circumstances I am unable to form an opinion about its relationship to the Gynemesh PS implant.
5356 I turn now to the disputed items.
5357 The respondents argued that the Court could not be satisfied that any of Mrs Dawson’s urinary symptoms could be sheeted home to the implant and were more likely due to her pelvic floor tissue.4246
5358 There is no doubt that Mrs Dawson has or has had detrusor instability and detrusor overactivity causing lower urinary tract dysfunction.
5359 On 3 July 2012 Dr Swan reported that, amongst other things, Mrs Dawson had genuine stress incontinence and detrusor instability.4247 On 1 June 2016 Associate Professor Shek performed urodynamic studies on Mrs Dawson and concluded, amongst other things, that she was suffering from detrusor overactivity.4248 Dr Swan did not attribute either condition to the Gynemesh PS implant. Associate Professor Shek stated in her report that Mrs Dawson was suffering from detrusor overactivity “after” her mesh repair. She did not say that it was caused by the mesh repair.
5360 In his first report, and alone of all the medical experts, Professor Korda stated that the Gynemesh PS implant had resulted in the development of lower urinary tract dysfunction.4249Yet, as the respondents pointed out, Mrs Dawson’s lower urinary tract dysfunction predated the implantation of Gynemesh PS.4250
5361 When Mrs Dawson first saw Dr Lim in January 2009, she had urge incontinence and an excessively mobile bladder neck with a weak pelvic floor muscle tone. The urodynamics study performed the following month, two months before the Gynemesh PS was implanted, showed mild urodynamic stress incontinence and an overactive bladder. Dr Lim recommended that Mrs Dawson undergo an anti-incontinence procedure at the same time as her mesh repair, but she declined to do so.
5362 Mrs Dawson disclosed a long history of urinary incontinence in her first affidavit, which preceded her first prolapse and which has troubled her intermittently in varying degrees thereafter.
5363 In cross-examination, when Professor Korda was taken to Mrs Dawson’s account of her problems in early 2009, he revised his opinion. Since her symptoms of both stress and urge incontinence preceded the Gynemesh PS operation, he no longer held to the position that the Gynemesh implant had resulted in the development of her lower urinary tract dysfunction. Rather, he said that her urinary dysfunction had worsened. He admitted, however, that that the deterioration in urinary dysfunction was as consistent with the operation not fixing the dysfunction as it was of causing it.4251 In effect, he was unable to say whether Gynemesh PS had had any impact on Mrs Dawson’s urinary function.
5364 I find that Mrs Dawson’s detrusor instability or overactivity and lower urinary tract dysfunction were not caused by the respondents’ device and I am not satisfied that they were exacerbated by it.
5365 The next disputed item concerns the chronic pain syndrome.
5366 The respondents submitted that:
[T]o the extent “chronic pain syndrome” (Item 20) is claimed as a separate personal injury, the basis of this claim, in the evidence, is unclear. Certain of the experts use the phrase conceptually. However, no expert uses the phrase as a basis for a separate “personal injury”. To do so would have the effect of creating an injury which is separately compensable (rather than as part of the claim) in circumstances where her pain is found to be totally or partially referable to the Mesh Implant.4252
5367 The submission is misconceived. Both Associate Professor Shek and Professor Korda diagnosed Mrs Dawson with a chronic pain syndrome. Associate Professor Shek said that it occurred after her mesh repair. Professor Korda said it was caused by the Gynemesh implant. Unlike the urinary symptoms, there is no history of a chronic pain syndrome before the mesh implant. On the balance of probabilities, I find that it was caused by the implant.
5368 As I understand his report, Mrs Dawson’s symptoms of vaginal pain, pain in her buttocks which radiates down her leg, a “prickly feeling” inside her pelvis, and a burning feeling when she extends both legs are captured by this diagnosis.
5369 During cross-examination, when asked what he meant by “chronic pain syndrome”, Professor Korda replied:
Well, it’s – pain is a very difficult area to define. And when it refers to pain in the pelvis, it can be due to a variety of causes. Some gynaecological, some urological, some rectal. When a patient has continuous pain and is described as chronic pain, it is usually not caused by prolapse. Prolapse actually is not a cause of pain. Very rarely do you get pelvic pain from prolapse. So that any attempt to cure pelvic pain with a prolapse, whether it’s native tissue or mesh is not likely to cure pelvic pain. It’s usually a problem that is difficult to treat. It requires multidisciplinary approach. It requires the help of pain physicians, various other specialists, like psychiatrist, gynaecologists, urologists, etcetera. So a lot of centres have so-called pain centres which sort of manage these pains …4253
5370 In short, he used the expression to refer to chronic pain which is difficult to treat, which becomes progressively worse with surgical intervention, and requires multidisciplinary management.
5371 Mrs Dawson is entitled to damages for her chronic pain syndrome. The respondents need have no concern that I will compensate her twice for what is essentially the same injury.
5372 The third area of dispute relates to the psychiatric disorders.
5373 Mrs Dawson has a long history of depression going back at least a decade before she received the Gynemesh PS implant. In September 1999 she saw a psychiatrist, Dr Thacore, to whom she gave a history of recurrent depression going back 10 years.4254 Indeed, she had post-natal depression following the birth of her second child in 1983. Dr Thacore put her on Prozac, which she had taken in the past with good effect. Although she is recorded to have told him in early November 1999 that she had recovered, two weeks later she reportedly told Dr Fleming, a gynaecologist, that the depression was an ongoing problem.4255
5374 In February 2004, more than five years before her mesh surgery, Mrs Dawson was diagnosed with a recurrent depressive disorder. The psychiatrist who made the diagnosis, Dr Mani Rajagopalan, noted in a letter to Mrs Dawson’s then GP that she had experienced recurrent episodes of depression for over two decades, with the last episode occurring around 1997 or 1998.4256 He said the episodes lasted for about six months at a time and responded well to Prozac. Features of the current episode, which he said appeared to have begun 18 months before the consultation, included a depressed mood worse in the evening, daily crying spells, poor concentration, impaired work performance and tiredness with terminal insomnia. He said there were several psychological stresses including marital issues.
5375 In 2006, 2007, and 2008 Mrs Dawson was being treated by her GP for depression.4257 He prescribed first Lexapro, then Efexor.4258 When Dr Gibson referred her to Dr Lim, he told Dr Lim “[s]he has Efexor for controlled depression”.4259
5376 Dr Jungfer and Dr Brown agreed that Mrs Dawson suffers from a recurrent depressive disorder.4260 There can be no doubt that this preceded her mesh surgery.
5377 The respondents submitted that the evidence of the two psychiatrists established that, regardless of the correct diagnosis, Mrs Dawson was likely to suffer a psychiatric disorder even if she had not had mesh surgery.4261
5378 That is true, but it does not mean necessarily that Mrs Dawson did not suffer a psychiatric disorder as a result of the complications of Gynemesh PS.
5379 In her first affidavit Mrs Dawson said that from about October 2009, with no improvement of the pain, she became increasingly angry and stressed. She said she cried a lot and was depressed at how her life had changed so significantly. She said she was extremely frustrated at not being able to find a solution to her predicament and the inability of the doctors to explain why she was in so much pain. She said that there did not seem to be an end in sight. I referred in Part XV to her reflections on her current situation.
5380 Based on the history Mrs Dawson provided, on the assumption that she was euthymic at the time the mesh was inserted (that is, that her mental state or mood was normal) and on her assessment of Mrs Dawson’s current mental state, Dr Jungfer diagnosed Mrs Dawson with a major depressive illness precipitated by the chronic pain, loss and disability caused by the mesh surgery.4262 She did not consider that other factors were contributing to her mood state at the time of the consultation. She attributed changes in socialisation, the reduction in recreational activities and the loss of intimacy with her husband to the complications of mesh surgery.4263 In oral evidence she said that the complications following the mesh surgery were a cause of the latest episode of major depression and that these complications “continue to contribute as a causative factor”.4264
5381 Although Dr Jungfer recognised that Mrs Dawson had “a history of recurrent episodes of major depression in the context of psychological stress” and a strong family history of psychiatric illness, she considered that at the time Mrs Dawson underwent the mesh surgery, no psychosocial stressors were operative.4265 At the consultation Mrs Dawson reported irritability, “constant worry and concern”, “terminal insomnia”, depressed mood, and “feelings of tearfulness”. She told the doctor that she had abandoned her previous recreational activities, in part because of the pain but also because she has less motivation and initiative. Dr Jungfer noted that there had been a reduction in her self-esteem. Dr Jungfer said that the clinical records provided to her by the applicants’ lawyers suggested that, in the past when Mrs Dawson was depressed, she had also been irritable and short-tempered and that this was a component of her mood disorder.4266
5382 Based on the contemporaneous medical records in which there was no specific reference to “psychological sequelae specifically related to the mesh insertion and its removal, including pain and changes to her quality of life”, Dr Brown considered that it was more likely than not that Mrs Dawson experienced “expected feelings of emotional distress” relating to these issues and that “her more pronounced mood problems” were linked to marital and work-related matters. Apart from a period between September and December 2009, which she attributed to Mrs Dawson going off her anti-depressants in about May 2009, she concluded that her emotional reactions were normal. 4267 She wrote in her report:4268
Mrs Dawson's fluctuant depressive symptoms have been considered most likely to arise on the basis of her experience of ongoing psychosocial stressors, particularly given that she is a woman who has had a long-term pattern of responding to marital, family and work-related problems with recurrent mood symptoms. There is only limited documentary support for any contention that Mrs Dawson has experienced exacerbations of depression based on her experience of pelvic, vaginal and buttock-related pain apart from during the remainder of 2009. Rather, Mrs Dawson has been considered more likely to have responded with expected feelings of emotional distress to her pain, particularly given the emphasis in both the medical and counselling records on other issues being more significant in contributing to her mood complaints.4269
5383 In re-examination Dr Brown explained what she meant by “expected feelings of emotional distress”. She said:
Anyone who experiences pain, particularly if it’s prolonged or severe, will experience an understandable reaction. Although individuals vary, common reactions would be to feel angry, frustrated, sad, upset, and distressed. Individuals who experience chronic or severe pain can also experience what we describe, as psychiatrists, as being “normal depression”. We all experience depressive symptoms in at least a mild degree if a particular life stressor is a significant one, and that could include chronic pain. So in providing my ultimate opinion I decided, based on the information I had available to me, that Mrs Dawson’s descriptions of her emotional reactions fitted within what would be an understandable adjustment reaction, rather than an actual psychiatric disorder. Had she not complained of symptoms of those various types, it would be a more surprising outcome to her reported experience of pain.4270
5384 Dr Jungfer agreed with Dr Brown that the relapse of Mrs Dawson’s depression from September to December 2009 more probably than not related to the fact that she was unmedicated and also to the pain and the negative outcome from the surgical procedure, which I infer is a reference to the implantation of Gynemesh PS.4271
5385 The first record I can find of depressive symptoms since the mesh surgery appears in Dr Tobin’s notes of his consultation on 9 September 2009. Twice in seven lines of notes he recorded that Mrs Dawson was upset and in his letter to Dr Churcher he said that she was “quite upset today”.4272 He does not say what was upsetting her but in her first affidavit Mrs Dawson said that she broke down sobbing after Dr Tobin told her that he was unable to help her.4273
5386 On 8 October 2009 Dr Lim noted in her report to Dr Churcher that Mrs Dawson had confided in her that day that she had been feeling increasingly depressed “with the increased pressure at her workplace and social pressures at home as well as having to deal with the recent death of her mother-in-law”.4274 Dr Lim put her back on Efexor and Dr Churcher continued to prescribe it in the following years.4275
5387 In September 2011 Mrs Dawson apparently told Dr Churcher that she had “done [her] block at work”. He recommended counselling.4276 Three weeks later she saw him again. They discussed “recent issues at work” and he referred her to Dr Erica Echstein, a psychologist.4277 In the referral letter he noted that she had been “stressed and depressed for a variety of reasons” and that “both work related and domestic issues exist”.4278
5388 While Dr Echstein’s reports indicate that there were frank discussions about a number of stressors, including work-related issues and difficulties in her marriage, nowhere in Dr Echstein’s records is there a reference to pain or mesh-related issues and it does not seem as if Mrs Dawson made any connection between her changes in mood and the problems she was having with the mesh or her physical pain. While it is possible that Mrs Dawson’s dyspareunia and apareunia contributed to the marital difficulties she experienced after the mesh surgery, the difficulties in the marriage were longstanding and preceded the mesh surgery by at least a decade.4279
5389 In June 2012 Mrs Dawson had a lengthy consultation with Dr Churcher. She reported continuing problems with her pelvic symptoms but at the same time she said that she felt ready to stop Efexor and he took steps to wean her off it.4280
5390 In the years that followed there are several references in Dr Churcher’s notes to stress and stressors, but again there are no references that suggest that any possible mesh-related problem was one of them.
5391 Dr Brown was particularly influenced by the dearth of references in the clinical records to pain associated with the mesh implant or later operations as a cause of distress.4281 Dr Jungfer’s attention was drawn to some of these records in cross-examination. Unlike Dr Brown, she was untroubled by this. She said of the absence of a reference to pain and the numerous operations to deal with complications of the mesh:
It can – essentially, it can mean two things. The first is that the psychologist was choosing to focus on things that the psychologist thought that she could change or that she thought were more significant. So it doesn’t mean that Mrs Dawson didn’t report the pain, but she also – you will have clinical situations where patients will come in and, because they know they’re seeing you for depression and they feel that, you know, the depression – this is what the person wants to hear, so to speak, or by the nature of the questions, they might not mention physical symptoms or physical problems. So it could be that Mrs Dawson didn’t present a history related to that because that’s not what she thought she was there for. It might also be that at the time that she was seeing the psychologist, the pain wasn’t as significant for her as these other issues that were going on in her life. And it might also be that the psychologist’s focus was not on the pain. So there are multiple explanations.4282
5392 One might think that another possible explanation was that the pain was not a contributing factor, although this proposition was never put to Dr Brown.
5393 I accept that there were multiple stressors which contributed to Mrs Dawson’s depression. But I find it difficult to accept that, in someone with an underlying depression or vulnerability to depression, the additional stress associated with repeated surgery, which provides little or no relief from persistent pain, would not exacerbate her condition or precipitate another episode. Dr Jungfer testified that, when Mrs Dawson was not taking anti-depressants she appeared to be more vulnerable to an episode of major depression.4283
5394 At the same time, I also find it hard to accept, after all this time, that, if the mesh complications, actual or perceived, were making any significant contribution to her depressive symptoms, there would scarcely be a reference to them in the clinical notes of any of the clinicians who were treating her.
5395 It is, of course, entirely possible that Mrs Dawson lacked insight into the causes of her depression. I note that in her letter to Dr Churcher of 9 March 2012, Dr Echstein wrote that “whilst she is able to talk about work issues easily it is harder for her to be in touch with other factors in her life, internally or externally, that may be contributing to her mood”.4284 After a few consultations, however, Dr Echstein said that she was able to see that unhappiness in her marriage was contributing to her anxiety symptoms and overall distress. She said that it was possible that work stresses mirror the stressors in her marriage and there is displacement of anger and anxiety onto work, which makes it more difficult to manage work stresses. If the complications of her mesh surgery, actual or perceived, were amongst the “factors in her life … contributing to her mood” they did not emerge during therapy.
5396 I recognise that there are occasional references in doctors’ records to Mrs Dawson being “upset” or “quite upset” about her pain. Without more, however, they do not signify that her pain was making a material contribution to any depressive episode she was then experiencing.
5397 The applicants argued that I should prefer Dr Jungfer’s opinion to Dr Brown’s for the following reasons.
5398 First, Dr Brown’s opinion that there is an absence of documentary evidence to connect the Mrs Dawson’s depression with her pain “ignores the careful history taken by Dr Jungfer and reflected in her reports”. They refer to statements made by Dr Jungfer that there is “fairly clear documentation about the persistent problems with regard to [the Dawsons’] intimate relationship secondary to the mesh surgery” and Mr Dawson’s account in his affidavit of his emotional response to the change in their intimate relationship.
5399 Second, Dr Brown conceded that Mrs Dawson could have been suffering from an underlying psychiatric disorder even while she was ingesting antidepressants.
5400 Third, the Court would be cautious about accepting Dr Brown’s opinion when she made an error in her summary of Dr Lim’s report of 8 October 2009. The vice is said to appear in the italicised words of the following sentence:
On 8 October 2009 Mrs Dawson confided that she had been feeling more and more depressed because of increased pressure at her workplace and social pressures at home as well as having to deal with the recent death of her mother-in-law.
The applicants submitted that, by the use of “because of” instead of “with the”, which Dr Lim had used”, Dr Brown “clearly portrayed a causal relationship between Mrs Dawson’s feelings of depression and the psychosocial pressures that were referred to. They contended that “this causal relationship may or may not have accurately reflected either Dr Lim’s opinion or Ms Dawson’s.
5401 None of these matters is of any moment.
5402 As to the first matter, Dr Brown did not ignore the history in Dr Jungfer’s report. Rather, she considered the contemporaneous medical records provided a more reliable reference point for what was worrying Mrs Dawson in the past. As to the second, Dr Brown’s concession does not solve Mrs Dawson’s fundamental difficulty in establishing a material connection between the respondents’ wrongdoing and her depression. As to the third, it seems to me that Dr Brown’s singular error did not materially affect the meaning of the statement recorded by Dr Lim. Nor did it adversely affect Mrs Dawson. The consultation with Dr Lim on 8 October 2009 fell within the one period (September to December 2009) in which Dr Brown considered her experience of coccygeal pain may have been a contributing factor to the recurrence of her depression.4285
5403 In the result, I am not persuaded that Dr Jungfer is right. Apart from a few months in late 2009, when it is likely that Mrs Dawson had an episode of depression caused by her severe pain, I am not persuaded that Mrs Dawson suffered a psychiatric disorder as a result of damage caused by the respondents’ wrongdoing. As Mrs Dawson, herself, said in her evidence in chief, she has “always had the depression”.4286 With the exception of the period in 2009, the intrusion of the mesh complications does not appear to have made a significant difference. That is not to say, she has not been distressed by the mesh complications. To the contrary, I find that she has been.
5404 Mrs Dawson’s alleged disabilities were listed at exorbitant length in the Amended Statement of Particulars filed on 28 July 2017. It is unnecessary to extract the list here. It is sufficient to note that the respondents submitted that it was necessary for the Court to determine which of a number of those disabilities “flow[ed] totally or partially” from Mrs Dawson’s compensable injuries.4287
5405 The first was item 1(d): coccydynia (pain in the coccyx).
5406 The first reference in the medical evidence to coccydynia after the mesh implant surgery appears in Dr Churcher’s notes of 22 June 2009. He described “marked coccydynia on the right side …”.4288 But the evidence also indicates that Mrs Dawson had suffered from pain in her coccyx before she received the Gynemesh implant.
5407 When she was seen by Dr Gibson on 22 January 2009, she had a tender coccyx.4289 In his referral letter to Dr Mitchell written on 25 June 2009, some six weeks after the mesh surgery, Dr Churcher wrote that Mrs Dawson had a “12 – 18 months spontaneous onset coccydynia”.4290 The significance of pre-existing coccygeal pain was not addressed in the applicants’ submissions.
5408 The first doctor to connect the coccydynia to the mesh surgery was the orthopaedic surgeon, Dr John Nelson. In his letter to Dr Churcher of 2 July 2009, he noted that Mrs Dawson had “some local soreness of mild degree, basically with discomfort on sitting only” over the previous 12 to 18 months.4291 Since the mesh surgery, however, he wrote that Mrs Dawson had had “fairly constant pain and tenderness over the area”, which had been worrying her not only with local pressure, but also at rest. On examination, she was tender not only over the point of the coccyx, but over the entire area around it, even to light touch. He said there was “little doubt that her local surgery has had some effect, either directly or indirectly[,] in the coccygeal area”.
5409 Although he readily confessed that he lacked a sufficient understanding of the area to be certain, Dr Nelson said he was suspicious that all the “bits” that were tightened tended to pull on the coccyx and suggested that Mrs Dawson might have some coccydynia from that. He said it was obviously necessary to exclude any local collection or other problem within the bone and arranged for an MRI to be done for this purpose.
5410 Dr Nelson sent Dr Lim a copy of his letter to Dr Churcher and asked her whether she had had any experience of patients who, after “this type of procedure” had developed coccydynia, either directly or indirectly.4292 In her reply, Dr Lim emphatically rejected the notion that the mesh could pull on the coccyx. She explained:
After the relevant investigations she underwent an anterior and posterior vaginal repair with mesh augmentation. As she had only a moderate sized cystocele and rectocele no fascial plication was performed and therefore no tensioning or pulling of tissues would have occurred. Instead, mesh reinforcement was placed in a tension free fashion with the body of the mesh overlying the vesico-vaginal fascia and arms of the anterior mesh to the obturator internus muscle of the lateral pelvic side wall. The posterior mesh was once again placed in a tension free fashion with the body of the mesh lying on the recto-vaginal fascia with the arms of the mesh being placed in tunnels leading to the ischial spines. These mesh prosthes[e]s are not secured to sacrospinous ligaments or any muscular structures and therefore would not be pulling on the coccyx or any of the pelvic musculature. Instead it relies on ingrowth of the patient’s native tissue and scar tissue into the mesh prosthesis to provide ongoing support to the pelvic organs. Similarly as there was no perineal defect no perineorrhahphy was required and therefore no sutures were placed in the perineal body which would cause any pulling on tissues in the lower part of the vagina.4293
5411 Dr Lim also said that she was unaware of any incidents of prolapse repair of the kind she had performed causing coccydynia, particularly when no fascia, muscle or perineal body plication has been undertaken and there had been no fixation of the mesh prosthesis into any ligaments or muscle tissue.
5412 The MRI was performed on 18 July 2009. It apparently excluded the local collection or bony abnormality as a possible cause of the coccygeal pain. The radiologist reported “[n]o MR abnormality to explain [Mrs Dawson’s] clinical presentation.4294
5413 In his referral letter to Dr Tobin, dated 10 August 2009, Dr Churcher wrote that Mrs Dawson had “marked R paracoccygeal pain, present of nearly 2 years but dramatically worse since [the mesh] repair in May this year”. He said that Mrs Dawson was “looking for an organic cause” and suggested that the pain might be neuropathic in origin.4295
5414 In his report of 11 August 2009, Dr Tobin wrote:
One has to accept the patient’s view and that is that her significant pain problem dates from the time of her mesh repair. This does not mean pain is causally related to the surgery.4296
5415 Following the first mesh excision surgery on 14 October 2009, Dr Lim, who assisted in the operation, wrote that “there was no evidence of stenosis or mesh banding in the vagina to explain [Mrs Dawson’s] right coccygeal pain…” 4297
5416 That said, there is a clear history of persistent right paracoccygeal pain after the mesh surgery and, in her letter to Dr Carey of 17 February 2010, Dr Lim reported that around four weeks after the splint was removed Mrs Dawson complained of a number of symptoms including exacerbation of her pre-existing coccygeal pain.4298
5417 In his first report, Professor Korda wrote that soon after her mesh surgery Mrs Dawson had “deterioration in her constipation and developed severe pain in her buttocks and her coccyx and the pain was radiating down her leg”.4299 In cross-examination, he was taken to that statement.4300 But when in cross-examination he was taken to Mrs Dawson’s complaints of pain in the buttocks and coccyx radiating down her leg in early 2009, before her mesh surgery,4301 Professor Korda conceded that this was a similar description and that it was not possible to tell from the record whether the symptoms experienced after mesh repair had been caused or exacerbated by it.4302
5418 In her second report Associate Professor Rosamilia, however, noted that the repair procedure in October 2009 in which a piece of mesh in the anterior wall of the vagina was excised had exacerbated Mrs Dawson’s coccygeal pain and wrote:
Exacerbation of pre-existing pelvic pain following surgery is a common occurrence; it may be exacerbated due to the positioning in lithotomy required for vaginal surgery and the psychological stress associated with any surgery. The anterior wall of the vagina is not closely related to the coccyx and surgery on this site would not directly cause coccygeal pain.4303
5419 In his first report, Professor Roovers asserted that “in August 2010 Dr Tobin considered the obstructed defecation, which has been present since a young age, as the cause of coccydynia”, but Dr Tobin’s August 2010 report does not include any statement to that effect.4304
5420 Following his examination of Mrs Dawson in July 2017, Professor Eyers, the colorectal surgeon, reported that on digital rectal examination there was a palpable area of deficiency on both sides of the pelvic floor. Anterior to this area on the left was an edge of what he felt was likely to be palpable residual mesh. Professor Eyers considered that the lateral edge of the distal sacrum and coccyx on the right side were unusually prominent and palpable immediately after the bowel wall without the usual intervening soft tissue. Both palpable mesh and the sacro-coccygeal ridge were very tender. Professor Eyers expressed the view that all the abnormalities he detected on examination were “consistent with her having had previous genital prolapse surgery using mesh, and a series of subsequent procedures to remove the mesh”.4305 Indeed, he went further, stating:
I believe that the insertion and subsequent removal [of] the mesh has resulted in “collateral damage” to the normal tissues alongside and around the mesh. This would explain the abnormalities I could palpate in the parametrial and pararectal tissues, and any irregularities I could feel in the pelvic floor (the levatores ani).
Despite Mrs Dawson having had anal physiology studies … no objective measurement has been made of pudendal nerve function, and it remains a possibility that there is an element of pudendal neuropathy contributing to her situation.
5421 It will be recalled that Professor Eyers was not required for cross-examination.
5422 Taking all the evidence into account, including Mrs Dawson’s, and notwithstanding the evidence to the contrary, I am persuaded by Professor Eyers’ opinion.
5423 Mrs Dawson’s coccygeal pain was very much worse after her mesh surgery than before and worse again after the revision surgery in October. On the balance of probabilities the surgery made it worse. It is most unlikely that the marked deterioration occurred spontaneously. The MRI excluded as potential causes a local collection or a bony abnormality. Although the precise mechanism has not been identified, the inference is available on the evidence that the mesh implantation and revision surgery were responsible. Pudendal neuropathy is certainly a possible explanation. As Herron CJ observed in EMI (Australia) Limited v Bes (1970) 44 WCR 114 at 119 (appeal dismissed: (1970) 44 ALJR 360):
It is not incumbent upon the applicant, upon whom the onus rests, to produce evidence from medical witnesses which proves to demonstration that the applicant’s contention is correct. Medical science may say in individual cases that there is no possible connection between the events and the death in which case of course, if the facts stand outside an area in which common experience can be the touchstone, then the judge cannot act as if there were a connection. But if medical science is prepared to say that it is a possible view, then … the judge after examining the lay evidence may decide that it is probable. It is only when medical science denies that there is any such connection that the judge is not entitled in such a case to act on his own intuitive reasoning.
These remarks were cited with approval in Commissioner of Police v David Rea [2008] NSWCA 199 at [8] by Handley JA, with whom Allsop P and Johnson J agreed at [10] and [11] respectively. See also Fernandez v Tubemakers of Australia Ltd [1975] 2 NSWLR 190 at 197 (Glass JA); 198–200 (Mahoney JA), appeal dismissed: Tubemakers of Australia Ltd v Fernandez (1976) 10 ALR 303; (1976) 50 ALJR 720.
5424 The second disability the respondents queried was Mrs Dawson’s apareunia. They submitted, based on a suggestion by Professor Roovers, that it might be related to her “marital problems and emotional factors”.4306
5425 In his affidavit, upon which he was not cross-examined, Mr Dawson stated:
I recall our sex life dramatically changed after her [mesh] surgery. Whenever we tried to have intercourse Diane complained that it was hurting too much. I remember her saying “it feels like a knife is cutting inside of me”. This was something that I initially found very frustrating. I was angry because of what had happened to Diane, and because it appeared that sex would no longer be a part of our relationship. At times we still tried to have intercourse, but the pain was too much for Diane. As time went on I realised we just were not going to be able to have sex because of the pain it caused Diane. I ended up having to come to terms with the fact that this was the new reality for our relationship. 4307
5426 Apareunia is alleged to have been a problem, however, only after the first revision surgery in October 2009. Mrs Dawson’s evidence was that the pain she had experienced with sexual intercourse was worse after this procedure than before and it got to the point where she and her husband stopped having sex altogether.4308 After the January 2014 operation, Mrs Dawson said that she and her husband tried to have sex, perhaps a couple of times, but it was far too painful for her.4309 None of this evidence was challenged either. Mr Dawson said that, after the October 2009 operation, Mrs Dawson was unable to have intercourse because of the pain.4310 He said that they attempted to have sex a couple of times after the surgery in January 2014, but it was still too painful for his wife and, “as a result of this, [they] once again resolved that it was not possible for [them] to resume a sexual relationship”.4311
5427 The first reference to apareunia in the medical records appears to be in Dr Swan’s report of 3 July 2012.4312 He took a history from Mrs Dawson that, as well as her persistent buttock and leg pain, she was apareunic because of severe dyspareunia. On 14 March 2013 Dr Swan noted that Mrs Dawson still had apareunia.4313
5428 In her report to Dr Churcher dated 25 October 2015, following her examination of Mrs Dawson early the previous month, Dr Schierlitz wrote that Mrs Dawson was “still not sexually active as she is not (sic) very anxious about the pain and the discomfort and does not feel that she is in any position to resume sexual activity soon”.4314
5429 Mrs Dawson told Professor Eyers in 2017 that she no longer had a sex life and that she and her husband had tried sex three times since 2009 but could not continue because she was still too sore.4315
5430 Professor Korda’s opinion was that she had severe dyspareunia resulting in apareunia.4316
5431 I am satisfied that Mrs Dawson’s apareunia was caused by the worsening of her dyspareunia following the first mesh revision surgery and was more likely than not related to the surgery. Dr Roovers’ suggestion finds no support in the evidence.
5432 The third area of dispute related to Mrs Dawson’s bowel symptoms.
5433 The respondents submitted that the following symptoms were “clearly pre-existing conditions”: incomplete defaecation and difficulty defaecating experienced in the period between the mesh surgery and the first revision procedure, constipation, difficulty initiating and completing defaecation, the need to apply external pressure or vaginal digitation in order to defaecate, incomplete defecation, pain on defaecation, the need to take Metamucil between the first and second revision procedures, constipation, difficulty defecating, and the need to take Metamucil and laxatives between the second and third revision procedures.4317 The respondents argued that, at best, the Gynemesh implant “aggravated some aspects of her bowel symptoms”.
5434 This submission has substance.
5435 In her first affidavit Mrs Dawson admitted to a long history of constipation dating back to her 20s.4318 Indeed, she told Professor Eyers that she had “always been constipated” and that she has taken Metamucil daily and Nulax as required for as long as she could remember.4319 She also said that she had had three colonoscopies when she was in her 40s (in 2002, 2004, and 2008) owing to her constipation.4320
5436 In 2004 she started experiencing more difficulties emptying her bowel. Although surgery in April that year resulted in an improvement in her bowel function, the improvement was only temporary.4321 Dr Churcher told Dr Stewart in October 2004 that her problems with defecation had worsened since her hysterectomy and prolapse surgery in 2001, was now passing “ribbon [bowel motions]”, had to push on a buttock to initiate a bowel action, and felt like she constantly had a “loaded rectum”.4322 In November 2007 Dr Churcher referred her back to Dr Stewart with “2 years of deteriorating bowel habit”.4323 After seeing Mrs Dawson, Dr Stewart noted that she continued to have “difficulty with evacuation and a feeling of obstructed defecation”.4324 By early 2009, that is before the implant, her bowel function deteriorated again and she needed to digitate to produce a bowel movement.4325
5437 About two weeks after the mesh surgery, Dr Lim’s notes record that her bowels were working well and on 3 June 2009 she noted that her bowel function was now normal and she was happy she no longer needed to digitate.4326 Shortly afterwards, Mrs Dawson said that her bowel symptoms returned and she again had to digitate to pass a motion.4327
5438 In 2015, after a change of diet, Dr Schierlitz reported a significant improvement in her bowel function.4328 But by the time she saw Professor Eyers in 2017, she still had “on-going issues with her bowels”. He explained:
Before the original surgery in Ballarat [the mesh surgery] she occasionally needed to apply some pressure in the vagina to assist defaecation. (This is a common feature of a rectocele). She no longer needed to do this, but she always did need to press on her perineum to the right of her anus. She was aware of a bulge that is present in that location, and had found that pressing on the bulge assisted with defaecation. She could feel a pulling discomfort on the left side, when she was straining at stool.
While she continued on Metamucil, Mrs Dawson was also subject to episodes of constipation which Metamucil alone would not shift. She had tried the chocolate Movicol, but it did not work, either when she tried it a couple of years previously, or when she tried it again last year.4329
5439 Professor Eyers concluded that:
The end result of the use (and attempted removal), of the mesh, is that Mrs Dawson's long-standing constipation has been aggravated, rather than improved, by failure to correct her associated obstructed defaecation. Both factors were present to some extent before the pelvic repair performed by Dr Lim. It appears that the obstructed defaecation was originally caused by her rectocele. But now the situation appears to involve more than a simple rectocele, (located anteriorly), and to result from a more diffuse (albeit similar) process, as evidenced by her having to apply pressure laterally (especially on the right side) to help her to defaecate.
The mechanism of good defaecation is undoubtedly complex, and not yet fully understood. But it is understood that a mechanical bulging of the low rectum, either to the front (as in a rectocele), or laterally (as here), disturbs the normal mechanics and allows “pocketing” (or “ballooning”) of the stool as straining occurs. This, in turn, misdirects the vectors of the straining force from the line of the anal canal, and somehow makes it more difficult to pass the motion.
Hence the situation is improved when pressure is applied over the bulging, and patients discover that pressure, in the vagina (in the case of a rectocele) or over the bulging perineum (as here), helps them get their bowel motion out.4330
5440 In his first report, Professor Korda made no connection between Mrs Dawson’s defaecation problems and the mesh surgery.4331 After reading Professor Eyers report, however, Professor Korda said that he agreed with Professor Eyers that Mrs Dawson’s defaecation disorder was originally caused by a simple rectocoele but that it had been aggravated by the insertion of mesh and the subsequent corrective surgeries.4332
5441 It is arguable that Professor Eyers did not say in the passage cited above that the defaecation disorder had been aggravated by the insertion of mesh and the subsequent corrective surgeries. But that appears to be his opinion when the report is read as a whole. Presumably the respondents took it the same way, since Professor Korda was not challenged about his opinion in cross-examination and no evidence was led from any of the respondents’ witnesses to address it or any other aspect of Professor Eyers’ report. Indeed, none of the respondents’ witnesses commented on the relationship between the post-mesh defaecation problems and the mesh or the later surgeries.
5442 On balance, I find that the insertion of the mesh and the repeated surgical interventions to deal with its complications aggravated to some degree Mrs Dawson’s pre-existing defaecation disorder, most likely by damaging the pararectal tissues. To what degree, however, is difficult to gauge. In their written submissions the only change to which the applicants referred was the need to press on her perineum to the right of the anus rather than to digitate the posterior wall of her vagina.4333
5443 The respondents submitted that Mrs Dawson’s depressive symptoms and her need to take anti-depressants “reflect an underlying depressive order and are not the result of the Mesh Implant or subsequent surgeries”.4334
5444 With the exception of the episode in late 2009, for the reasons given above in relation to psychiatric disorders, I accept this submission.
5445 The final issue relates to the pelvic pain.
5446 The respondents submitted that Mrs Dawson’s pelvic pain was “clearly multi-factorial” and the Court could not be satisfied that the implant was its sole cause of her chronic pelvic pain. Their reasons were brief. They contended that Mrs Dawson had had “a long history (since at least 1988) of reporting and being treated for pelvic pain and has also had pre-existing coccydinia”.4335 Later in their submissions, they also referred to “at least a 19-year history of dyspareunia”.4336
5447 Insofar as this submission refers to the coccydynia, I have already dealt with it. There is also a history of pelvic pain and dyspareunia but the evidence indicates that it was intermittent and bore little resemblance to the pelvic pain that she has suffered since her mesh surgery.
5448 The reference to 1988 is to an operation report by Dr Phillip Hall dated 27 July 1988. Dr Hall aspirated a fimbrial cyst and divided some adhesions.4337 The report makes no mention of pain. Mrs Dawson first saw Dr Hall on 1 July 1988. At that time she gave a history of intermittent vaginal bleeding, very heavy periods associated with the passage of clots, and significant post-coital bleeding. Again, there was no mention of pain. Dr Hall found “a very significant vascular ectropion” on her cervix but described her pelvis as “otherwise normal”. A cervical ectropion is a condition in which cells from inside the cervix form a red, inflamed patch on the outside the cervix. At the operation, performed laparoscopically, the ectropion was treated with superficial diathermy.
5449 That said, it would appear that Mrs Dawson had been experiencing some pelvic pain because on 11 October 1988 Dr Hall reported that her pelvic pains had completely disappeared over recent months.4338
5450 On 4 April 1990, in a report to the GP, Dr Hall noted that she was aware of “pain and pressure feelings in the pelvis” but she also told him that she had been having significant menstrual problems with dysmenorrhea (period pain) that day.4339 On 4 April 1990 Mrs Dawson saw Dr Moss. There was no mention of pelvic pain in his report, only “some dyspareunia deep on vaginal pressure”.4340
5451 In July 1994 Mrs Dawson reported hypogastric pain before moving her bowels which was relieved by defaecation.4341 In May the following year she complained to her GP of right pelvic premenstrual and menstrual pain.4342 In February 1988 she had a gastroscopy and was found to have had some mild chronic gastritis. Mrs Dawson deposed that the procedure was performed to investigate problems with her bowel function and also pelvic pain but that both her bowel function and pelvic pain resolved after it.4343
5452 Mrs Dawson reported pain on intercourse to Dr Fleming on 18 March 2003 and to Dr Dalton on 4 March 2004.4344 She told Dr Fleming that intercourse feels sharp with deep pain and dryness. Dr Fleming put her on a course of Ovestin “to see if things settle down” and reduces the dryness. In the event of persistent tenderness at the vaginal vault, she flagged the possibility of a diagnostic laparoscopy to exclude intra-abdominal adhesions. It seems that the tenderness at the vault and the dyspareunia persisted.
5453 A year later Mrs Dawson described to Dr Dalton significant pain in the middle of her vagina with intercourse and on deep penetration. On examination Dr Dalton noted that she had “a significantly tender point at the mid vagina posteriorly which causes pain in her bowel” and marked tenderness at the vaginal vault or apex, which Professor Korda testified would have caused her deep dyspareunia.4345 Dr Dalton diagnosed her with pelvic pain and adhesions to the ovary and the vaginal vault, which he divided on 27 April 2004.4346
5454 Professor Korda explained in re-examination that a patient with adhesions would be expected to have pelvic pain and division of the adhesions could alleviate pelvic pain in certain circumstances.4347 That was obviously Dr Dalton’s objective and it appears to have been realised. When he reviewed her a month later, Mrs Dawson was well, made no complaint about pelvic pain or dyspareunia, the examination revealed nothing of significance or relevance, and Dr Dalton never saw her again.4348
5455 The medical records disclose no further complaints of pelvic pain until October 2007 when she presented to her GP with “right low pelvic pain”.4349 There is no further reference in the records to dyspareunia or pain with intercourse until late January 2009 when she first saw Dr Lim. On this occasion she reported superficial dyspareunia but no deep dyspareunia and she told Dr Lim that deep dyspareunia had not been a problem for her.4350 There was no mention of other pelvic pain. Professor Korda distinguished between the vaginal pain Mrs Dawson had when she saw Dr Dalton in March 2004, which appears to be the same complaint with which she presented to Dr Fleming the previous year, and the superficial dyspareunia with which she presented to Dr Lim nearly five years later.4351 He concluded that the dyspareunia was due to the adhesions that had formed between the bowel, the ovary and the vaginal vault and that, once they were divided (in April 2004) that dyspareunia was cured.4352
5456 The weight of the evidence supports the conclusion that, independent of the coccygeal pain, Mrs Dawson developed chronic and severe pelvic pain caused by the complications of the Gynemesh implant. Professor Korda acknowledged that Mrs Dawson had had pelvic pain before the mesh surgery. He understood from interviewing Mrs Dawson that the pain deteriorated significantly after the mesh repair and became progressively worse with the revision operations.4353 The contemporaneous records support his understanding and the opinions upon which it was based. On the balance of probabilities the significant deterioration in pelvic pain is attributable to the implantation of Gynemesh PS and the subsequent operations to deal with its complications. For a start, in November 2013 Dr Kennedy found a large area of palpable mesh which, though not exposed, was very painful, both anteriorly and posteriorly.4354 In March 2014 the physiotherapist, Heather Deane, noted that palpation of a line of tenderness from the upper third of the left posterior vaginal wall to the lower third of the right vaginal wall, where there was an area of mesh exposure, and which did not correlate or feel like trigger points, elicited pelvic floor contractions and “pain reactions”. 4355 In May 2015 Dr Schierlitz removed most, but not all, of the anterior mesh which was “palpable and painful” on the previous examination.4356 In September 2015 there was an area of mesh on the posterior vaginal wall which Dr Schierlitz said was palpable with fibres visible on speculum examination over an area which was “quite tender” and had a “needle prick sensation”.4357 In June 2016 Professor Korda found “a tender remnant of the mesh palpable on both the anterior and posterior walls.4358 Similarly, the same month Professor Eyers found palpable mesh above the anal canal which was “very tender”.4359
5457 The presence of tenderness on palpation in an area of mesh points to the likelihood of a causal relationship between the two. The correlation between the loss of the board-like sensation across her buttocks with the removal of the left arm of the mesh by Professor Dwyer on 31 January 2014 and the improvement in pelvic pain that followed strongly suggests an association between those symptoms and the mesh. So, too, does the improvement in the discomfort due to the “prickling” sensation after the trimming of the protruding suture in Dr Schierlitz’s rooms in September 2015. In addition, there is every likelihood of pain due to contraction of the tissues around the mesh, infection of the mesh and trauma to the pudendal nerve.
5458 Professor Korda conceded in cross-examination that the fact that Mrs Dawson’s vaginal pain (including dyspareunia) was worse after the mesh surgery than before is consistent with the fact that the mesh operation simply did not fix it.4360 More likely than not, however, based on all the evidence, the mesh and the revision operations materially contributed to this deterioration, too.
Conclusion
5459 In summary, I am satisfied that Mrs Dawson has suffered all the injuries particularised in her Statement of Particulars with the exception of the rigid vagina. While I accept that Mrs Dawson has a psychiatric disorder, except for a few months in late 2009, I am not satisfied on the evidence that it was caused or significantly aggravated by the implantation of Gynemesh PS or its sequelae.
5460 I am also satisfied that the Gynemesh PS implant caused or materially contributed, directly or indirectly, to the disabilities listed in the Statement of Particulars with the exception of:
detrusor instability, detrusor overactivity, and lower urinary tract dysfunction, including stress urinary incontinence, urinary urge incontinence, and difficulty urinating;
coccydynia;
dyspareunia; and
bowel problems.
5461 I am satisfied, however, that the implant directly or indirectly aggravated Mrs Dawson’s pre-existing dyspareunia and defaecation problems. I am also satisfied that more likely than not the implantation of the mesh caused Mrs Dawson to suffer much more severe pain in the region of her coccyx than she had previously experienced.
5462 Mrs Dawson’s non-economic loss should therefore be assessed on the basis that there were four mesh exposures over six years, each associated with pain and suffering, and culminating in surgical intervention which caused more pain and suffering. She had to undergo five separate operations in what proved to be largely futile attempts to alleviate her pain. There is a chance that she will require similar surgery in the future. She suffered a substantial deterioration in her coccygeal and pelvic pain and dyspareunia, as well as an aggravation of her bowel dysfunction, caused (directly or indirectly) by the use of Gynemesh PS.
5463 There no dispute that Mrs Dawson’s pain is genuine.4361 It is notable that both Dr Jungfer and Dr Brown commented in their reports that during their respective consultations Mrs Dawson was obviously in physical discomfort. Dr Jungfer said that she became progressively more uncomfortable in her seat and said that she could see Mrs Dawson her moving her buttocks and trying to relieve the pressure.4362 Dr Brown noticed that she shifted about from one side of her buttocks to another and that, after being encouraged to get up and move around as needed, she did so on a number of occasions to relieve her discomfort.4363
5464 Her enjoyment of life has been substantially diminished. She has managed to stay at work, but in discomfort, if not overt pain, most of the time, thanks to a sympathetic employer.
5465 Her symptoms began in about June 2009, which means that she has suffered now for over a decade, albeit with a period of some symptomatic relief between 2010 and 2012. As a 60 year-old Australian woman her life-expectancy based on the 2019 medium life expectancy tables is 27.41 years. The future is unlikely to be any better than the present.
5466 Further, while Mrs Dawson may not have experienced an enduring psychiatric disorder as a result of the complications of the mesh, damages should include some compensation both for her short-term aggravation and for her “expected” or understandable feelings of emotional distress. Where a physical injury has occurred, it is unnecessary for an injured plaintiff to prove that she has suffered a recognisable psychiatric injury before recovering damages for the emotional effects of the injury. Damages for mental suffering short of a psychiatric illness or disorder are routinely awarded in personal injuries cases where physical injury has also been sustained. Although there are limitations on the recovery of damages for mental harm, they only apply in cases where the sole damage is mental harm. Mrs Dawson has obviously been distressed by her plight. That was not just evident from what she said but by how she presented. During the course of recounting her problems in oral evidence, she broke into tears, just as she had at a similar point in her consultation with Dr Jungfer.
5467 Despite the problems she had had before her mesh surgery, Mrs Dawson had been very active. She socialised, sewed, cooked and baked, and enjoyed walking, both by herself and with others.4364 She also coped very well with household and outdoor tasks.4365 She very much enjoyed gardening and found it therapeutic.4366 Since the mesh surgery, all these activities have been curtailed. She used to be very sociable and had many friends with whom she enjoyed spending time. Now she feels she is often poor company and often avoids socialising. She also enjoyed her job and generally coped very well with it, too. Since her mesh surgery, work has been a struggle. Mrs Dawson took great pride in her home. She testified that she kept it beautiful.4367 Now she needs assistance. While the marriage was not exactly plain sailing before her mesh surgery, she apparently enjoyed a level of intimacy with her husband that is now missing from their relationship. Her sense of self has altered. Her self-esteem and self-confidence have greatly diminished. She does not see herself as the woman she once was.
5468 In the area of general damages or non-economic loss, the Wrongs Act (Vic) made two significant changes to the common law.
5469 First, it provided that no damages can be awarded for non-economic loss unless the applicant has suffered “significant injury”: Wrongs Act (Vic), s 28LE. “Significant injury” is elaborately defined in s 28LF, the terms of which need not be mentioned as it was common ground that Mrs Dawson suffered “significant injury” within the meaning of the Act.
5470 Second, the Act imposed a cap on damages for non-economic loss. That is to say the Act prescribes the maximum amount a court may award a claimant for that head of damage: Wrongs Act, s 28G. The amount was $371,380 when the section was inserted in the Act in 2002. But the amount is indexed at the beginning of each financial year by a formula set out in s 28H of the Wrongs Act (Vic) and the amount is published annually in the Government Gazette. By s 6 of the Wrongs Amendment Act 2015 (Vic) which commenced on 1 July 2016, the figure mentioned in the Act was increased substantially to $577,050, to bring it in line with the cap on damages for awards of damages for pain and suffering under the Workplace Injury Rehabilitation and Compensation Act 2013 (Vic): see the note to s 28HAAB of the Wrongs Act (Vic). The amendment applies to an award of damages regardless of the time the relevant act or omission giving rise to the claim occurred (Wrongs Act (Vic), s 28LACA(1)) and to any award of damages in a proceeding (such as this one) that was on foot but had not been finally determined before s 6 commenced: Wrongs Act (Vic), s 28LACA(2). The effect of indexation is that at the time of trial the maximum amount was $598,360. As at 1 July 2019 the maximum amount was $623,950.4368
5471 In contrast to the positon under the Commonwealth legislation, however, damages are not awarded in proportion to the maximum amount or reduced in certain cases. They are to be assessed as one would assess them at common law. The amount in s 28G simply operates as a ceiling.
5472 Counsel for the applicants submitted that, having regard to the nature and extent of her injuries, the awards made in other cases to which they referred in their submissions, and prevailing community standards, Mrs Dawson’s non-economic loss at common law should be assessed at between $250,000 and $300,000 and, with respect to the statutory claims, approximately 50% of a most extreme case.4369 The respondents, on the other hand, submitted that the appropriate award is “up to $200,000” and the appropriate award for the statutory claims is an amount commensurate with 30 to 32% of a most extreme case. The reasons for the difference are neatly encapsulated in chapter 15B of the respondents’ written submissions (at [82]):
As detailed above, Mrs Dawson has had a long history of psychiatric issues, bowel disorders, pelvic pain and urinary issues. She does not require mobility aids, and drives and works 4 days a week (and had been working 5 days a week until August 2017). Mrs Dawson requires some domestic assistance, but it is not significant. Given her history, Mrs Dawson was always going [to] experience a level of pain, disability, restriction of movement and recreational activities, and bowel and bladder dysfunction, regardless of her Mesh Implant. Had she undergone non-prosthetic pelvic floor repair surgery rather than Mesh Implant surgery, given the earlier native tissue repair had already failed, she would still likely have required at least 1 revision surgery in her lifetime. Mrs Dawson's psychiatric history and circumstances also indicate that she would have experienced recurrent bouts of depression, regardless of her Mesh Implant and gynaecological issues.
5473 The respondents are substantially correct. The applicants made no apparent allowance for the likelihood that Mrs Dawson would have undergone native tissue repair had she declined mesh surgery, in which case there is every chance that she would have had a recurrence of her prolapse symptoms and perhaps a further surgical repair. She is also likely to have battled depression regardless of the injuries the subject of this proceeding. She might also have suffered some dyspareunia and episodic pelvic pain and coccydynia. Moreover, given her long history of constipation, bowel and urinary dysfunction, her bowels and her bladder were likely to likely to have troubled her indefinitely.
5474 But the respondents must take Mrs Dawson as they find her. The effects of their wrongdoing have made her life much worse than it would otherwise have been. The factors the respondents highlighted are relevant to her claim for economic loss but they do not entitle them to a discount on her damages for non-economic loss. In fact, they did not argue that they did.
5475 The respondents also submitted that, if Mrs Dawson had proceeded with an abdominal or laparoscopic sacrocolpopexy, all the same risks and perhaps more would arise.4370 Both Professor Roovers and Associate Professor Rosamilia suggested as much. But Professor Korda pointed out that Mrs Dawson was never a candidate for sacrocolpopexy since sacrocolpopexy is indicated for vault prolapse and Mrs Dawson did not have a vault prolapse.4371 Neither Professor Roovers nor Associate Professor Rosamilia had an answer to this. Professor Korda’s point was not addressed in their reports and the point was not raised with them when they were called to give evidence. In any case, even if sacrocolpopexy might have had an incidental effect on her anterior prolapse, it would have done nothing for her rectal prolapse.
5476 Even so, the applicants’ assessment is excessive, not least because it proceeds on the footing that the respondents’ wrongs caused a recurrence of Mrs Dawson’s major depression which has endured and urinary dysfunction, when I have found that they failed to discharge their burden of proof in relation to all three of these matters.
5477 Taking all relevant matters into account, for the purposes of the claim in negligence I would award Mrs Dawson $225,000 for non-economic loss and for the purposes of the statutory counts I find that Mrs Dawson’ non-economic loss is 35% of a most-extreme case. I would apportion half to the past.
Past economic loss
5478 In contrast to the position at common law, the TPA imposes a cap on economic loss of twice the amount of average weekly earnings on the amount a plaintiff may recover (TPA, s 87U) and, again in contrast to the position at common law, unless another rate is prescribed by the regulations, a 5% discount rate is to be applied: TPA, s 87Y.
5479 Under the Wrongs Act (Vic), the maximum amount of damages that may be awarded for each week of the period of loss of earnings (whether past or future) is three times the amount of average weekly earnings at the date of the award, not twice as in the TPA: Wrongs Act, s 28F(2). This reflects an amendment made to the section by the Wrongs Amendment Act, which also applies to any award in a proceeding that was pending but not finally determined (s 28LACA(2)), regardless of the date of the relevant act or omission giving rise to the claim (s 28LACA(1)). The amount of average weekly earnings at the date of the award is calculated in the same way as in Western Australia, but by reference to the earnings of Victorian employees: Wrongs Act (Vic), s 28F(3).
5480 Mrs Dawson’s claim commences on 22 August 2017 when she reduced her working week from five to four days. All post-operative absences from work were covered by sick leave.4372 Until 21 August 2017 Mrs Dawson was working full-time. As at that date she earned a gross annual income of $64,905.86, which equates to $980.63 per week net.4373 The respondents overlooked this evidence in their submissions. They referred to records from the Australian Tax Office about her earnings in the 2016 financial year, but those records were not in evidence.4374
5481 Accordingly, the claim for past economic loss made on her behalf is based on the value of one day’s work. For want of any other available metric, her lawyers sought damages for past loss of earning capacity based on the assumption that she had suffered a net weekly loss of $196.13, being one-fifth of 20% of $980.63.
5482 The respondents submitted that there was no evidence establishing a connection between Mrs Dawson’s recent reduction in working hours and her Gynemesh implant.4375 They contended that her reduction in working hours “could be a personal preference”. That contention was based on an isolated statement taken (out of context) from her first affidavit that she believed her quality of life would certainly improve if she were able to reduce her working hours to part-time.4376
5483 Of course, without more, the mere fact that Mrs Dawson had reduced her working week from five to four days would not entitle her to damages for lost income. First, she had to prove that her earning capacity had been diminished by her compensable injury. Second, she had to prove that the diminution in her earning capacity had been productive of economic loss.
5484 Both sides addressed the first criterion but overlooked the second.
5485 I reject the notion that Mrs Dawson’s reduction in working hours was simply a matter of personal preference or, put another way, a voluntary decision unrelated to her compensable injury. The respondents’ submission that there is no evidence establishing a connection between the recent reduction in her working hours and her mesh implant is not correct.
5486 In her affidavits, supplemented by oral evidence, Mrs Dawson discussed her pain, its impact on her mood, and her energy levels, and resultant fatigue. She said that sitting for long periods significantly aggravates her pain. In her first affidavit she said that her ability to continue working was “very dependent on [her] ability to regularly stand up and stretch”. She said that she had found it “very hard work” to maintain her job and that she did not believe it was good for her health to continue to work full-time, considering the amount of pain she was in at the end of a day’s work. Within this context that she said that she believed her quality of life would improve if she were able to switch from full-time to part-time work.4377 In her second affidavit, sworn on 28 July 2017 she said:
23 In terms of my day to day experience at work, even though I do get up and walk around, most of my day is spent sitting and by the end of the day my bottom half is aching. I have been supplied with a gel chair and my manager is arranging to trial a second stand desk.
24 I have asked to reduce the number of days I work each fortnight, by one day, so I work a nine day fortnight as I no longer feel like I can work full time. My employer has agreed. I am unsure as to when this will start, but anticipate that it will be within the next few weeks. I feel that I will need to continue to reduce the number of days I work even after this, although I do not know when this might occur. I am embarrassed to continually have to ask for time off and am grateful that my employer is accommodating.
5487 In her third affidavit, sworn on 9 November 2017, exactly a week before she was called to give oral evidence, Mrs Dawson deposed that her employer had agreed to reduce the number of days she works each week to four days and that commenced on 22 August 2017. In oral evidence she was asked how she was coping with the reduced hours. She replied:
It’s a lot better but I still come home at night – from just sitting all day, it’s – I still get tired. So the other Tuesday night I came home and I just could have given everything up. I was just so sore and tired that I had had enough. But other than that day, it’s okay.4378
5488 The only medical evidence upon which the applicants relied is the evidence of Dr Slesenger.
5489 Dr Slesenger saw Mrs Dawson on one occasion, on 11 July 2016. His opinions were based on conclusions derived from his reading of the medical records which were supplied to him by the applicants’ lawyers and from the history he took during his consultation with Mrs Dawson. Like his examination of Mrs Gill, his examination of Mrs Dawson was of little to no utility. He inspected her pelvis, coccyx, gluteal and rectal areas, but only externally. Unsurprisingly, given the nature of her injuries, external examination revealed no abnormalities. He palpated her abdomen, although she complained of no abdominal pain, and found no tenderness. But he did not palpate her pelvis, coccyx, gluteal and rectal areas, which were the sources of her pain. That is not to say, however, that Dr Slesenger’s evidence is irrelevant or entitled to no weight. It simply means that his opinions were based on the history and the findings of other medical experts.
5490 When asked whether it was more likely than not that Mrs Dawson’s earning capacity had been affected by her injuries and disabilities, this was his response:
This is a difficult question to answer. However, I note the following:
• Mrs Dawson has had regular time off work after each surgery (each time between 4 and 6 weeks).
• There is a question as to whether the surgery has affected her career advancement and there is certainly evidence that Mrs Dawson had commenced a senior management training course, but was unable to complete this. Further information with regard to the dates of the course should be sought.
• Should she become job detached, I have concerns with regard to her capacity to gain employment in an open job market.
With regard to Mrs Dawson’s current work capacity, this is a difficult question to answer. I note that she has been able to remain in work despite repeated surgical interventions and I also note the secondary nature of her job tasks. Nevertheless, I also note her current functional limitations affecting her ability to set and her recent commencement of Lyrica and duloxetine to assist with her pain control. I am of the opinion that given her current level of impairment and disability, there is a strong possibility that she will have to reduce her working hours and I anticipate that there is likely to be reduced to 4 hours a day, 5 days a week with a 5-minute rest break per hour. 4379
5491 When asked directly whether Mrs Dawson was suited for full-time work, he replied:
On balance, given Mrs Dawson’s current level of symptoms, her functional limitations and her poor response to treatment to date, as well as the recent deterioration in her symptoms, I am of the opinion she is unlikely to be able to maintain full-time employment.
5492 Mrs Dawson’s tolerance for full-time work was not exhausted for more than a year after this assessment.
5493 Dr Slesenger did not expressly state that she was unfit for full-time work. But his evidence lends weight to the commonsense conclusion that the reduction in Mrs Dawson’s working hours was not just a voluntary decision unconnected to her compensable injury and provides support for a finding that her decision to request a reduction was not unreasonable.
5494 Since it is common ground that Mrs Dawson’s chronic pain is genuine and I have found that it was caused by the complications arising from the implantation of the respondents’ device and/or the surgery designed to treat the complications, if her wages had been reduced, then I would have awarded her damages commensurate with the extent of that reduction. It does not matter that the reduction in working hours was made at her request. It was not suggested to her, let alone proved, that her decision to request a reduction was unreasonable and the respondents did not plead or prove that going onto reduced hours amounted to a failure to mitigate her loss. See Medlin.
5495 But evidence was not led to establish that Mrs Dawson’s income had been reduced. In her third affidavit, all Mrs Dawson said was that her employer had agreed to her request for a further reduction in days worked and identified the commencement date.4380 Although over two months had passed since the request was put into effect, she did not say that her income was reduced accordingly. If that were an oversight, it was not rectified in oral evidence. Moreover, no wage slips or other wage records were tendered. No evidence was adduced from her employer. And no application was made for leave to reopen to tender her 2018 income tax return.
5496 Consequently, I have no option but to dismiss the claim for past economic loss.
5497 Since the claim for past loss of earning capacity has not been proven, it follows that there is no entitlement to damages for past loss of superannuation.
Past out-of-pocket expenses
5498 $107,585.60 was claimed for past out-of-pocket expenses. The parties agreed on $15,000.4381
Past domestic care and assistance
5499 According to Mrs Dawson, virtually all the past care and assistance was provided by Mr Dawson.
5500 Based on the reports of Mr Walsh, an amount of $109,427.45 was sought to compensate Mrs Dawson for the gratuitous domestic care and assistance she has received.4382 An additional sum of $870.74 was also sought to compensate Mrs Dawson for the sum she paid to a cleaner for two hours per fortnight at $39.40 per hour since 19 July 2017.4383
5501 But the parties agreed on a figure of $60,000.4384
Interest
5502 With respect to the statutory claims, interest should be paid on past out-of-pocket expenses in accordance with s 87ZA of the TPA. No interest is payable with respect to the statutory claims on past non-economic loss or gratuitous attendant care services. In the absence of any information as to whether the agreement reached between the parties incorporates paid services and, if so, to what extent, and any evidence as to the amounts Mrs Dawson paid the cleaner she employed from January 2017, I decline to award interest on past domestic assistance for the purpose of the claims under the TPA.
5503 The Wrongs Act, however, imposes no restrictions on the award of interest. Consequently, on the award of damages for negligence, just as in Mrs Gill’s case, I award interest on all past losses:
at 2% pa on non-economic loss; and
at half the rates prescribed by Practice Note GPN-INT on all other past losses, including the agreed amount for past assistance.
Future loss of earning capacity
5504 The amount claimed on Mrs Dawson’s behalf for future loss of earning capacity was $136,044.66, derived from a total of $160,052.54 less 15% for vicissitudes.
5505 Her claim for future loss of earning capacity was based on the following assumptions:
that she would continue to work four days a week for a year and suffer a net weekly loss of $196.13;
that thereafter she would work in accordance with Dr Slesenger’s prediction of four hours a day, five days a week, which equates to a residual work capacity of 50% and that if she did so she would suffer a net weekly loss of $490.32.
5506 None of these matters is certain or even virtually certain, although I accept that, if Mrs Dawson has not in fact suffered a loss of income by the reduction in her working hours, that situation is unlikely to continue indefinitely.
5507 Mrs Dawson deposed that, for financial reasons, she intended to keep working until age 67 and is in no position to retire any sooner.4385 No suggestion to the contrary was put to her in cross-examination and it appears to be common ground that any award for future economic loss or loss of earning capacity should be calculated to age 67.4386
5508 In her first affidavit Mrs Dawson expressed concern about her future employment for the following reasons:
(a) If, for whatever reason, I am no longer able to regularly stand up and stretch at work (approximately every thirty minutes or so) I do not believe I would be able to cope with my role because sitting for long periods significantly aggravates my pain. As far as I am concerned, my ability to keep working is very dependent on my ability to be able to regularly stand up and stretch.
(b) Although I continue to work on a full-time basis, I do not believe that doing so is good for my health considering the pain I find myself in at the end of each working day … up to a seven out of 10 on the pain scale. I have continued to work full time for financial reasons. However, I believe my quality of life would certainly improve if I was able to reduce my working hours to part-time.
(c) I know that if I do require further surgeries I will need to take between four to six weeks off work immediately after any procedure. The amount of sick leave I now have with my employer has significantly diminished as a result of the periods I have had off work recovering from my operations. I am also concerned that if I was to fall ill for other reasons I won't have much sick leave to draw on.
(d) If, for whatever reason, I was to lose my job in the future I believe I would have enormous difficulty finding alternative employment because: I would not be suited for any roles that might require me to be on my feet for long periods of time; I would not be suited for a purely sedentary role if I was unable to regularly stand up and stretch; if I was required to disclose to any prospective employers the nature of my injury and ongoing pain and restrictions I believe this would seriously compromise my prospects of securing work. 4387
5509 Of course, since the first affidavit was sworn, Mrs Dawson has been able to reduce her working hours to four days a week.
5510 The only assault on this evidence in cross-examination was to suggest to Mrs Dawson that her relationship with her employer was stable and to emphasise the ways in which her employer has accommodated her disabilities.4388 Mrs Dawson conceded that no-one at the hospital had ever suggested to her that she was not wanted there.4389
5511 As the respondents submitted, Mrs Dawson has demonstrated that she has the capacity to work in her current role despite her injuries, which suggests that “there is scope for ongoing employment”.4390 No doubt this is at least in part because she has a sympathetic employer and she has been a long, loyal, and valued employee. Before the implant surgery, for example, she won “the Valued Performance award” from among 600 workers at St John of God Hospital.4391 While Mrs Dawson is understandably concerned about the future, there is insufficient evidence to indicate that she is likely to lose her current position.
5512 But there is no certainty that she will be able to maintain her current work effort for another seven years. Some allowance should be made for the chances that she might have to retire early, lose her job, lose income for the one day a week she does not work, or suffer a greater loss because she had to reduce her working hours by more than one day a week. If she were to retire early or lose her job, as a disabled woman over 60, she would no doubt be at a considerable disadvantage on the open labour market.
5513 In all the circumstances, I would allow $50,000 as a buffer against the potential for economic loss to take into account the various vicissitudes, both positive and negative.
Loss of superannuation benefits
5514 I would award $4,750 for loss of superannuation benefits calculated at 9.5% of $50,000 deferred for 7 years. The discount rate is 5% under both s 28I of the Wrongs Act (Vic) and s 87Y of the TPA.
Future domestic assistance
5515 Mrs Dawson’s evidence was that, after the implant surgery in May 2009 and each of the operations in October 2009, January 2014, and October 2015, Mr Dawson cared for her for six weeks. She said that after the operation in May 2015 he cared for her for three weeks and after the operation in March 2017 for three weeks.4392 During these periods, he would spend most of the day with her, do all the washing, ironing, cooking, and cleaning, and most of the driving.4393
5516 At other relevant times, Mrs Dawson said that her husband had helped her with “heavy cleaning tasks”, such as mopping, sweeping, vacuuming, and cleaning the bathroom and had also helped with changing bed linen, cooking, washing up, laundry, ironing, and shopping. These tasks, she added, she would have done but for the pain she has experienced since her mesh implant surgery.4394
5517 None of this evidence was challenged in cross-examination and I accept it.
5518 In oral evidence Mrs Dawson said that her husband probably provides nine hours a week of care and assistance to her.4395 In cross-examination she described the various tasks he undertakes in more detail and adhered to her evidence that he would spend about nine hours doing them.4396 She volunteered that she and her husband had discussed the matter and calculated the time by reference to the various tasks and that she had refreshed her memory from a note she had made based on the discussion.4397 During the cross-examination Mrs Dawson asked the cross-examiner for permission to retrieve the note and then, apparently by reference to it, she told the Court that Mr Dawson spent two hours ironing, an hour taking her to buy groceries, two hours cleaning the toilets and showers, two hours cleaning, two hours gardening, half an hour carrying washing in and out, and two hours helping with meals.4398 No call was made for the production of the note and no challenge was made to the reliability of the figures.
5519 At times when the pain was worse, Mrs Dawson said that her husband would help with lighter cleaning tasks which she otherwise would have done.4399
5520 Since about the middle of July 2017, Mrs Dawson has used the services of a cleaner for two hours a fortnight to clean the two bathrooms and to vacuum and clean the floors.4400 It appears that the limit of two hours was imposed, not because it was a true reflection of Mrs Dawson’s needs, but it was all the Dawsons could afford.4401
5521 Timothy Walsh, the occupational therapist, retained to evaluate Mrs Dawson’s needs, estimated that four hours of assistance was required weekly, four hours on a quarterly basis, and three hours annually. In addition, since the garden in the Dawsons’ current house was “low maintenance with lawns in the front and rear and low maintenance gardens around the fence line”, he estimated that she would require assistance there in the order of 1.5 hours per month “with the expectation that Mr Dawson would contribute”.4402
5522 Weekly services comprised cleaning of all floors and wet areas, changing bedclothes, and ironing. Annual services involved moving furniture and cleaning underneath it and also cleaning the oven, although I would have thought that an oven would need cleaning more frequently.4403 Quarterly service involved the following activities:
cleaning windows inside and out;
deep cleaning the refrigerator and the pantry;
cleaning or wiping timber furniture;
cleaning high and low cupboards; and
dusting and wiping down all skirting boards.4404
5523 Mr Walsh’s conclusions were based on his observations of Mrs Dawson sitting, standing, and bending; information from the assessments of medical specialists; the affidavits of Mr and Mrs Dawson; and the information conveyed verbally to him by Mr and Mrs Dawson.
5524 Apart from Mrs Dawson’s oral evidence that the house had two bathrooms, the only evidence about the house came from Mrs Borthwick. She described the property as a new, level, 3-4 bedroom, open plan, single storey home covered in white tiles throughout except for the bedrooms and lounge room, which are carpeted.
5525 Section 28IA of the Wrongs Act (Vic) imposes limits on the extent to which a court may award damages for gratuitous attendant care, consistent with the recommendations of the Ipp Review. It provides that:
(1) No damages may be awarded to a claimant for gratuitous attendant care services unless the court is satisfied that—
(a) there is (or was) a reasonable need for the services to be provided; and
(b) the need has arisen (or arose) solely because of the injury to which the damages relate; and
(c) the services would not be (or would not have been) provided to the claimant but for the injury.
(2) Further, no damages may be awarded to a claimant for gratuitous attendant care services if the services are provided, or are to be provided—
(a) for less than 6 hours per week; and
(b) for less than 6 months.
5526 “Attendant care services” are defined in s 28B to mean any of the following:
(a) services of a domestic nature;
(b) services relating to nursing;
(c) services that aim to alleviate the consequences of an injury.
5527 “Gratuitous attendant care services” are defined in the same section to mean attendant care services that have been, or are to be, provided by another person to the claimant and for which the claimant has not paid and is not liable to pay.
5528 In Alcoa Portland Aluminium Pty Ltd v Victorian WorkCover Authority [2007] VSCA 210 at [27]–[43] esp at [40]–[41] (Chernov JA, Maxwell ACJ agreeing at [1] and Neave JA agreeing at [50]), the Victorian Court of Appeal held that subsection (2) should be read conjunctively so that the claimant is only denied damages with respect to the need for services of this kind if the conditions in both paragraphs are satisfied. In other words, a claimant cannot recover damages for such services if they are provided for less than six hours per week and for less than six months. The effect is to permit a claimant to recover damages if the services are provided for at least six hours per week or six months. In so doing, the Court declined to follow the approach to the comparable section of s 72(2) of the Motor Accidents Act 1988 (NSW) (which are identical to the words used in s 15(3) of the CLA (NSW)) taken by the NSW Court of Appeal in Geoghan v D’Aubert [2002] NSWCA 260; (2000) 36 MVR 542, despite the fact that the comparable sections were relevantly identical. On the other hand, the judgment in Alcoa is consistent with the position taken by the Queensland Court of Appeal in Grice v State of Queensland [2005] QCA 272.
5529 The applicants claimed that the limitations were in any event irrelevant because the assistance should be provided on a commercial basis, citing Mr Walsh’s opinion that “it would be more reasonable” for the services previously provided by Mr Dawson to be provided commercially. Mr Walsh explained:
– Mr Dawson reported he has been diagnosed with degenerative changes in both shoulders and his back due to arthritis. Therefore it would be unreasonable to expect him to continue to complete all of the routine household cleaning and gardening duties.
– Other than Mrs Dawson’s physical injuries she is diagnosed as having a ‘Major Depressive Illness’ (Dr Patricia Jungfer 24 August 2016) and reported significant stress and guilt that Mr Dawson has been required to take over her previous cleaning duties. To continue to expect that Mr Dawson should complete these duties only further compounds Mrs Dawson stress.
– Even with the recommended level of services Mr Dawson would still be required to complete or assist Mrs Dawson with duties including, shopping, gardening and driving.4405
5530 Ms Borthwick was also of the view that domestic cleaning should be provided on a commercial basis.4406
5531 Having regard to all these considerations as well as the fact that Mrs Dawson hired a cleaner, I agree that future domestic assistance should be determined on the basis that assistance of the kind nominated by Mr Walsh will be provided commercially.
5532 The applicants claimed $196,158.02 less 15% for vicissitudes. Consistently with all other future calculations, it begins on 16 December 2017. Since past care is agreed up until that time, I intend to deal with future assistance on the same basis as the parties did, namely, to assess it on the basis that the award covers the period from that date. The gross sum was calculated using a 3% multiplier, although the Statement of Particulars used a 5% multiplier. The applicants’ claim was as follows:
Type of care | Hours pw | Cost ph | Cost pw | Multiplier | Amount |
Weekly cleaning | 4 | $39.40 | $157.60 | 1,035.9 | $163,257.84 |
Quarterly cleaning (4 hours per quarter) | 0.3 | $39.40 | $11.82 | 1,035.9 | $12,244.34 |
Annual services (3 hours) | 0.06 | $39.40 | $2.36 | 1,035.9 | $2,444,72 |
Gardening (1.5 hours per month) | 0.4 | $43.94 | $17.58 | 1,035.9 | $18,211,12 |
5533 The hourly rates were based on the current provider fees under the National Disability Insurance Scheme.4407
5534 The claim was purportedly based on Mr Walsh’s opinion.
5535 Yet, in his second report Mr Walsh pointed out that he had inadvertently omitted a provision for one hour a week of shopping but no claim was made for assistance with the shopping.4408 Moreover, despite the fact that Mr Walsh assumed that Mr Dawson would continue to provide some assistance in the future with other domestic tasks such as cooking, washing, driving, and some of the gardening, the effect of the applicants’ submissions is that they did not press any claim for gratuitous assistance.
5536 There was no dispute that future assistance should be provided at commercial rates. Nor, following a concession made by Ms Borthwick in cross-examination, was there a dispute about the claim for gardening assistance; the respondents agreed that Mrs Dawson required 1.5 hours of assistance per month or 0.4 hours a week.4409 But with respect to the cleaning, based on Ms Borthwick’s report the respondents submitted that, instead of the 4.36 hours claimed, only 2.5 hours should be allowed.
5537 In my opinion Ms Borthwick’s allowance is insufficient. It does not pay enough attention to the kind of house Mrs Dawson kept before she was injured, although Mrs Borthwick noted that Mrs Dawson reported having “a strong need to keep her home very clean and tidy” and described herself as “a fastidious housekeeper”.4410 Nor does it take into account the fact that she suffers pain and consequential fatigue as a result of her work. Furthermore, it was based on the erroneous assumption that Mr Dawson provided her with approximately two hours of assistance per week that he had not previously provided.4411 In cross-examination Ms Borthwick conceded that, if I were to accept Mrs Dawson’s evidence that he provided nine hours of assistance a week (as I do), then she had underestimated the amount of assistance Mrs Dawson needs. She also considered that Mr Walsh’s assessment was “within the scope of being reasonable”. 4412
5538 Taking all these matters into account, I generally prefer Mr Walsh’s assessment. On the other hand, Mrs Dawson conceded in cross-examination that she could do the ironing. There should be some adjustment to his numbers to reflect this concession.4413
5539 In the circumstances, I would allow 3.5 hours of weekly cleaning and I would not reduce the amounts for quarterly and annual cleaning, which, if anything, strike me as underestimates.
5540 There is also a dispute about the hourly rate for cleaning. Ms Borthwick argued that the NDIS figures used by Mr Walsh were excessive and not in keeping with the assistance that was available locally. She recommended $33 per hour, which she said was the rate recommended by Absolute Domestics. Mr Walsh’s response was as follows:
At the time of my assessment Mrs Dawson advised that she was in the process of obtaining cleaning services, and advised that she had difficulty obtaining quotes at what she considered to be a reasonable price. When later I researched cleaning services within the region, I found that the costs varied and sometimes it was unclear if the service was insured. Therefore the NDIS rate was provided (NDIS cleaning services -$39.40 per hour, gardening service $43.94 per hour) given that these are registered service providers and work under all necessary compliances.
The following are the rates of reimbursement to household cleaning services (not care attendant rates) by government agencies:
Comcare rates above: HH2- Cleaning per hour $48.07 Comcare.gov.auTAC rates: Housekeeping per hour H600 = $36.18 tac.vic.gov.au
Worksafe rates: PH140 Home help services = $32.88 worksafe.vic.gov.au
Two local commercial cleaning providers were contacted, Meticulous cleaning Services and Absolute Domestics who quoted $33.50 and $37.00 per hour. It seems reasonable that the quoted hourly rate falls within the rate provided in my report.
5541 The last sentence is ambiguous. The ambiguity was not resolved by the provision of a follow-up report and Mr Walsh was not required for cross-examination. Oddly enough, no-one bothered to ask Mrs Dawson what she was paying her cleaner.
5542 In all the circumstances, I would fix a rate of $37 per hour, which is roughly the average of all these amounts.
5543 Contrary to Ms Borthwick’s opinion, for the reasons given above the need for future assistance should not be determined on the footing that it is only be awarded to age 80. Rather, the sum should be calculated over Mrs Dawson’s life expectancy. As at December 2017, when Mrs Dawson was three months shy of her 59th birthday, that was about 28 years on the current medium life expectancy tables. It was common ground that the amount should be reduced by 15% for vicissitudes.
5544 The final question concerns the discount rate.
5545 Without reference to authority, as they did with Mrs Gill, the applicants submitted that a 3% multiplier should be used because the 5% discount rate, provided for by s 28I of the Wrongs Act (Vic) and s 87Y of the Trade Practices Act, only applies to economic loss.4414 The respondents made no submissions on the question but, as in Mrs Gill’s case, it appears from their submissions that they applied the 5% rate.
5546 As I noted with respect to Mrs Gill, s 87Y relevantly provides that if an award of personal injury damages covered by Pt VIB applies includes any component, assessed as a lump sum, for future economic loss of any kind, the present value of the loss is to be determined by applying a discount rate of 5% unless a different percentage is prescribed by the regulations. Section 28I of the Wrongs Act (Vic) is similarly worded, except for the omission of the expansive phrase “of any kind”.
5547 Economic loss is not defined in either Act. But non-economic loss is. In these circumstances any loss which does not fall within the definition of “non-economic loss” is “economic loss”. “Non-economic loss” is defined in the s 28C of the Wrongs Act (Vic) for the purposes of Pt VB, which applies to personal injuries damages, to mean any one or more of the following: pain and suffering; loss of amenities of life; and loss of enjoyment of life. Section 87D of the TPA defines “non-economic loss” in the same way except that it includes disfigurement. Since future assistance does not fall into any of these categories, it must be an economic loss. While the Explanatory Memorandum is unhelpful and there is some ambiguity in the Premier’s second reading speech, it seems to me that the intention of the Parliament was to apply a 5% discount rate, not just to loss of earning capacity but to future expenses, including care and assistance. An assumption to this effect was made in McLellan v Savicky [2013] NSWSC 1756 at [20] (Harrison AJ).
5548 Section 14 of the CLA (NSW) is in the same terms as s 28I of the Wrongs Act (Vic). In his Annotated Civil Liability Act 2002 (NSW) (Thomson Reuters, 3rd ed, 2018), Dominic Villa asserts, without reference to authority, that the discount rate prescribed by s 14 of the NSW Act is not limited to loss of earning capacity but extends to all future economic loss including medical and nursing expenses. In Kelly v Culakovski [2014] VSCA 305 at [86] Neave, Beach and Kyrou JJA applied a 5% discount rate to the cost of future massage and Pilates treatments on the assumption that s 28I of the Wrongs Act (Vic) applied and that treatment costs were a form of economic loss. If that is right, then there is no reason why a 5% multiplier should not also apply to commercial services for care and assistance.
5549 In my opinion a 5% discount rate should be applied.
5550 In summary, Mrs Dawson reasonably requires assistance from a cleaner for three and a half hours’ each week, four hours each quarter for quarterly cleaning, and three hours a year for annual cleaning, and she also reasonably requires 0.4 hours assistance from a gardener each week. The amount for cleaning should be calculated at the rate of $37 per hour and the amount for the gardening at the rate given by Mr Walsh of $43.94 per hour. The total should be calculated over Mrs Dawson’s projected life expectancy (as at December 2017) of 28 years, using the 5% discount rate, but reduced by 15% for vicissitudes in accordance with the parties’ common position.
5551 The applicants claim for future treatment was captured in the following table, which appeared in their closing submissions:4415
Item | Unit cost | Weekly cost | Multiplier | Amount |
Clinical psychology: 20 sessions @ $245 per consultation | $ 4,900.00 | N/A | N/A | $4,900.00 |
Psychiatrist consultation: 13 sessions for the first year @ $380 for the initial consultation and then 12 sessions @ $255 per session | $ 3,440.00 | N/A | N/A | $3,440.00 |
4 further psychiatrist consultations per year. | $ 255.00 | $ 19.58 | 822.0 x 0.952 | $ 15,322.21 |
Pain management program | $ 12,000.00 | N/A | N/A | $ 12,000.00 |
Nerve Stimulator | $ 25,000.00 | N/A | N/A | $ 25,000.00 |
Physiotherapy, continence advisor and pads @ $60 per week | $ 60.00 | $ 60.00 | 822.0 | $ 49,320.00 |
Ongoing visits to GP | $ 50.00 | $ 3.85 | 822.0 | $ 3,164.70 |
Occupational therapist assessment | N/A | N/A | N/A | $ 510.00 |
Allowance for future excision surgery | N/A | N/A | N/A | $ 25,000.00 |
Over-the-counter pain killers | $ 5.00 | $ 5.00 | 822.0 | $ 4,110.00 |
Metamucil | $ 13.00 | $ 3.00 | 822.0 | $ 2,466.00 |
Ovestin vaginal oestrogen cream @$15.50 per month | $ 15.50 | $ 3.58 | 822.0 | $ 2,942.76 |
Lyrica (3 x 75mg caps/day @ $38.80 / pkt of 56 caps) | $ 38.80 | $ 14.55 | 822.0 | $ 11,960.10 |
Amitripyline (Endep) | $ 8.50 | $ 1.96 | 822.0 | $ 1,611.12 |
Vesicare | $ 45.99 | $ 10.61 | 822.0 | $ 8,721.42 |
Effexor | $ 37.80 | $ 3.69 | 822.0 | $ 3,033.18 |
Xylocaine | $ 38.99 | $ 15.75 | 822.0 | $ 12,946.50 |
Amitriptyline | $ 38.99 | $ 15.75 | 822.0 | $ 12,946.50 |
Electrodes for TENS machine | $ 31.99 | $ 1.50 | 822.0 | $ 1,233.00 |
Total | $ 200,627.49 |
5552 Unfortunately, no sources were given for any of the items and no submissions were made in support of them. In their submissions the respondents identified some references. Otherwise I had to trawl through the evidence in search of them. That was highly unsatisfactory.
5553 The first three items are based on Dr Jungfer’s recommendations. She recommended psychiatric treatment and psychological counselling only if Mrs Dawson did not see a psychiatrist and psychologist as part of her pain management program.
5554 The claim for a pain management program is also apparently based on a recommendation of Dr Jungfer. It was supported by Professor Korda and Professor Eyers. Professor Eyers recommended referral to a pain specialist or a pain clinic for expert management. Professor Korda considered that Mrs Dawson had severe chronic pain syndrome which would require “frequent visits to pain specialists, pain medication, psychological counselling, acupuncture, behavioural and relaxation feedback therapies …”.4416 The respondents resisted the claim, arguing instead in favour of Dr Brown’s proposal of six consultations with a pain management specialist at the rate of $325 per consultation, totalling $1,950.
5555 The claim for a pain management program is made out. Professor Korda’s evidence was that chronic pain syndrome is “a very difficult area to treat” and necessitates a multidisciplinary approach which combines psychotherapy, physical therapy and pharmacotherapy.4417 The best way to achieve this and, in the long run I would think the most economical and efficient way, is through a pain management program. Moreover, given the severity and chronicity of the problem, it is highly unlikely that six consultations with a pain management specialist would be enough.
5556 The respondents submitted that the expert evidence in support of Mrs Dawson had been “superseded by the more recent treatment records documenting an improvement in Mrs Dawson’s symptoms”.4418 No reference was given for this submission and I do not accept that there has been any significant improvement in Mrs Dawson’s symptoms which would render such a program unnecessary or unreasonable. No suggestion to that effect was put to Mrs Dawson or to any of the experts who made the recommendation. The most recent record from Mrs Dawson’s GP shows that her current medications include Lyrica capsules 75mg, an anti-convulsant, used in the management of neuropathic pain, and Palexia SR (sustained release) tablets 100mg, which is an opioid analgaesic used to relieve moderate to severe pain.4419
5557 Dr Jungfer’s evidence was that pain management programs typically cost around $12,000.4420
5558 The respondents said that no expert in pain management had given evidence on the need for, or reasonableness, of this treatment.4421 But evidence to this effect was given by three experts without objection. Moreover, none of those experts was cross-examined on his or her recommendation or as to the reasonableness of the costs.
5559 I allow the claim for a pain management program in the sum of $12,000. But I disallow the claim for independent psychiatric and psychological treatment. Dr Jungfer said that the majority of pain management services will include a psychiatrist and a psychologist.4422
5560 The respondents also argued that the claim for “nerve stimulator surgery” was not supported by expert evidence. As I mentioned above, however, Professor Korda recommended a “nerve stimulator”, which he said could cost around $25,000.4423
5561 The question of a nerve stimulator, however, was not raised in evidence with Mrs Dawson. Since the treatment apparently involves surgical implantation of another medical device (see Mekhail et al, above), I am not satisfied that Mrs Dawson would consent to it.
5562 In the circumstances, I reject the claim for a nerve stimulator.
5563 The claims for physiotherapy, continence advisor, and continence pads are apparently based on Professor Korda’s opinion that this treatment is required for Mrs Dawson’s detrusor overactivity. Since I am not satisfied that that the detrusor overactivity was caused or aggravated by the mesh implant, either directly or indirectly, these claims must be rejected.
5564 The respondents submitted that no expert specifically recommended GP consultations.4424 That may be so, but they do recommend a variety of medications which can only be obtained with a doctor’s prescription. The amount claimed is based on four consultations a year, which seems reasonable.
5565 There is no dispute about the claim for $510 for an occupational therapy assessment.4425
5566 As for the claim for further excision surgery, there is no certainty that any further surgery would be required. The respondents submitted that no sum should be awarded because there was no evidence of an erosion or exposure, Mrs Dawson is reluctant to undergo further surgery, and she has undergone some improvement. As there is a real chance of further exposures or erosions, some allowance should be made, however, for the chance of further surgery. I would allow $10,000.
5567 That brings me to the claim for medication.
5568 The first one listed is “over-the-counter analgaesics”. Mrs Dawson said that she has taken Nurofen (Ibuprofen) from time to time.4426 Ibuprofen is a nonsteroidal anti-inflammatory drug commonly used to relieve pain. I accept that she may derive a benefit from using it. The claim is based on the use of one packet a week. The evidence is insufficient to justify such a claim. Doing the best I can, I would allow $1,500.
5569 Cymbalta and Amitriptyline are both central nervous system anti-depressants.4427 The claim for Cymbalta was not included in the applicants’ closing submissions. I therefore take it that that claim was not pressed. Effexor is another anti-depressant. Since I am not satisfied that, with the exception of the period in 2009, Mrs Dawson’s depression was caused or aggravated by her chronic pain, I reject the claim for anti-depressants. I accept that, because of her history of recurrent depression, she is predisposed or vulnerable to develop further episodes of depression. But it is 10 years now since the development of the mesh complications. If her chronic pain has not caused or materially contributed to an episode of depression to date, the chance of it doing so in the future is remote.
5570 The respondents argued that no allowance should be made for the cost of Palexia, because prescriptions for both Palexia 100mg and 50mg were discontinued by Dr Churcher on 18 April 2017 and that Mrs Dawson had deposed in her second affidavit that, since her most recent operation, she did not need to take it.4428 As I mentioned earlier, however, the most recent medical records, tendered by consent after the respondents’ written submissions had been filed, show that the cessation was temporary. On 31 August 2017, about a month after the second affidavit had been sworn, she was again prescribed Palexia SR tablets 100mg [28] (1 mane).4429 On 3 October 2017, when she presented with pelvic pain, she told Dr Churcher that she takes Palexia at night because taking it in the morning made her eyes “unfocused and flutter”.4430 He then updated the previous prescription to substitute “nocte” for “mane”. Further prescriptions were issued on 21 November 2017, 12 December 2017, 18 January 2018, and 23 January 2018.4431
5571 In their closing submissions, however, the claim did not include Palexia. I therefore took it that the claim was not pressed.
5572 In her examination in chief Mrs Dawson said that she continues to take Lyrica.4432 Although the contrary was not put to her in cross-examination, the respondents submitted she should not receive damages for the cost of Lyrica as her prescription was stopped in October 2016 because she was allergic to it.4433 It is true that Dr Churcher’s clinical notes for 31 October 2016 contain an entry that reads “Allergy: Lyrica” and indicate that she became nauseous when using it.4434 But the most recent records indicate that Dr Churcher reinstated Lyrica on 3 October 2017, when Mrs Dawson presented with pelvic pain, issuing a prescription for Lyrica capsules 75mg [56] (1 mane) with five repeats.4435
5573 The claim for Lyrica 75mg should be allowed.
5574 Mrs Dawson’s evidence on the use of Metamucil was inconsistent. In her first affidavit she that she takes Metamucil on a daily basis at a cost on average of $13 a month.4436 In her first affidavit, however, she also said that she did not take it every day because it causes bloating.4437 In her second affidavit (at [29]) she said she was using Movicol, which, serves a similar purpose, at a cost of approximately $18 a month, and made no mention of Metamucil. In oral evidence in chief she said she no longer used Metamucil.4438 Instead, she said she continues to use Movicol, which she described as “great”.4439 Movicol powder for oral solution (single dose sachets) [30], “2 sachets daily prn” is listed in her GP’s records under “current medications”.4440 The respondents referred to these records for other purposes in their written submissions and tendered them during oral argument. Dr Churcher’s notes of the consultation on 1 August 2016 record that she was taking Metamucil but on the advice of her specialist she added Movicol.4441 The most recent records, dated 8 February 2018, also include Movicol powder amongst the current medications.4442
5575 I would allow $18 per month for Movicol ($4.16 per week), but the amount should be discounted in the light of Mrs Dawson’s pre-existing defaecation disorder. The respondents suggested a discount of 50%, which seems reasonable.4443
5576 Ovestin is an oestrogen cream, which Mrs Dawson said she uses twice a week to thicken her vaginal walls.4444 The respondents admitted that a claim for Ovestin is reasonable, but not the claim that was made.4445 I agree. The claim is for the cost of a tube per month. But Mrs Dawson did not say how quickly the tube was depleted or how often she purchased the product. Based on the dispensary and PBS (Pharmaceutical Benefits Scheme) records, the respondents submitted that the award be limited to two “units” a year, less a reduction to account for any inevitability of ongoing usage and the vicissitudes of life. I would award Mrs Dawson the value of two tubes a year. At $15.50 per tube, that amounts to $31.00 per year or $0.60 per week and $478 in total. No discount is justified. The respondents pointed to no evidence to support the proposition that it was inevitable that Mrs Dawson would have required Ovestin in any event.
5577 Vesicare is a urinary antispasmodic drug indicated for bladder function disorders.4446 It is common ground that Mrs Dawson uses the drug but the evidence indicates that she was put on a trial of the drug on 26 February 2009, before Gynemesh PS was implanted.4447 Since I am not satisfied that there is a connection between Mrs Dawson’s subsequent lower urinary tract dysfunction and the implant, I reject the claim.
5578 Mrs Dawson gave evidence on 16 November 2017 that she uses two new ointments, one was xylocaine 5%; the other was amitriptyline 2%. She said that she applies the xylocaine using a dilator onto “the scar – well, onto the sore parts inside of me to try and help with the pain”. She explained that the amitriptyline was intended to alleviate the pain but is used externally.4448 She said that both were prescribed by Dr Schierlitz.4449
5579 These items were not included in the Statement of Particulars and no application to amend was made to add them. Mrs Dawson did not give evidence as to their cost and I can find no evidence of it elsewhere. In the circumstances I reject the claim.
5580 The last item in the table is “electrodes for TENS machine”.
5581 In his first report Professor Korda stated that Mrs Dawson will need transcutaneous electrical nerve stimulation (TENS).4450 In oral evidence in chief Mrs Dawson revealed that she had started to use a TENS machine.4451
Finally, in addition to your treatment, you also use a TENS machine; is that right?---That’s correct.
What do you use that for?---For nerve stimulation. They’re trying to – they’re now saying maybe there’s some nerve things going on so they want to stimulate the nerves and - - -
In what region?---I have to put it down near the back of my leg, my right leg, just at the top where the pain is and in the groin. And then I put the other one on my back, because sometimes that goes through to the vagina as well.
I see?---At the front. Do you get some relief with that?---It certainly stimulates things, but I don’t, at the moment, believe I’ve got relief but I’ve only used it for two weeks at the moment.
I see. And your current urogynaecologist is Dr Schierlitz?---Correct.
S-c-h-i-e-r-l-i-t-z?---Could be. I am not too sure of the spelling, sorry.
Has she prescribed those changes to the medication and treatment?---She has, yes.
5582 But no claim was made in the Statement of Particulars for electrodes for a TENS machine. Professor Korda’s recommendation was not supported by reasons. No evidence was given by Dr Schierlitz about the purpose of the treatment and what she hoped to achieve. Thus far, at least, the evidence does not indicate that Mrs Dawson has received any benefit from the TENS machine. Furthermore, I can find no evidence to support the amount sought. In all these circumstances, the evidence is insufficient to justify the claim.
5583 Accordingly, I allow the following items only:
$12,000 for a pain management program;
GP consultations (four annually);
$510 for an occupational therapy assessment;
$10,000 for future surgery;
$1,500 for over-the-counter analgaesics;
Lyrica 75mg;
Laxatives (Movicol); and
Ovestin.
5584 I am not satisfied that Mrs Dawson has made out an entitlement for the other items in the table.
5585 I am satisfied that Mrs Dawson is likely to be troubled by chronic pain for the rest of her life. The pain management program will hopefully help her to manage the pain but it will not cure her. Consequently, with the exception of the pain management program, the occupational therapy assessment, the future surgery, and over-the-counter analgaesics, the amounts should be calculated over Mrs Dawson’s life span. Consistently with the claim for future care, they should be calculated from December 2017 over a life expectancy of 28 years in accordance with the most recent medium life expectancy tables and, for the reasons given above, a 5% discount rate should be applied.
5586 It seems to me, however, that there should be a further discount to take into account the chance, small though it may be, that the pain management program will reduce the need for some of the other treatments. For this reason I would reduce the value of the amount for the neuropathic analgaesic, Lyrica, by 15%. Having regard to the previous history of low back and pelvic pain, I would reduce by 10% the total sum for all future out-of-pocket expenses, except for future surgery, Ovestin and Movicol (where a discount has already been made), to take into account the chance that Mrs Dawson would have required similar treatment even if she had not undergone mesh surgery.
5587 The following claim was made for future equipment, purportedly based on Mr Walsh’s first report:
Item | Unit cost | Replacement | Annual cost | Weekly cost | Multiplier | Amount |
Steam cleaner | $850.00 | 5 years | $170.00 | $3.27 | 822.0 | $2,687.94 |
Light weight battery powered stick vacuum | $450.00 | 5 years | $90.00 | $1.73 | 822.0 | $1,422.06 |
Gel/foam cushion | $400.00 | 3 years | $133.33 | $2.56 | 822.0 | $2,104.32 |
Recliner chair | $4,500.00 | 7 years | $642.86 | $12.36 | 721.2 x 0.711 | $6,337.88 |
Computer chair | $700.00 | 5 years | $140.00 | $2.69 | 753.6 x 0.784 | $1,589.31 |
TOTAL | $14,141.51 |
5588 There was no dispute about the first two items.4452 Accordingly, I allow the claims for a steam cleaner and a lightweight battery-powered stick vacuum but a multiplier of 796.6 should be used, being the applicable multiplier for a life expectancy of 28 years.
5589 Mr Walsh recommended a gel/foam cushion costing $400, with replacements every three years. Ms Borthwick said that a ring cushion was required at a cost of $25.4453 Neither Mr Walsh nor Ms Borthwick explained why one was preferable to the other. On the other hand, Ms Borthwick did not give any reasons for disagreeing with any of Mr Walsh’s recommendations.
5590 Mr Walsh noted, however, that Mrs Dawson’s employer had recently provided her with a computer chair with a gel seat which had decreased the pain she experiences when sitting. He also stated:
When at home she finds it difficult to tolerate sitting in a standard chair or stool and is required to rest in a recliner where she can decrease the loading into her pelvis through a semi reclined position.
Ms Dawson reports she continues to experience increased pain from any standard seat and with driving.
From the information provided, Mr (sic) Dawson would benefit from a seating assessment from an occupational therapist to review her needs for a cushion. It is possible the pain with sitting could be decreased through an appropriate Coccyx cushions (sic) used in chairs or cars.4454
5591 Since Mrs Dawson has had relief from the gel seat on the computer chair, it is reasonable to think that a gel cushion would give her some comfort when sitting in a standard chair. The recommendation for a gel cushion, however, appears to be contingent on an occupational therapist reviewing her over three hours to determine whether she should receive one. Since Mr Walsh is an occupational therapist, I struggle to understand why he could not have carried out the assessment himself. Mrs Dawson makes no claim for the cost of an occupational therapist seating assessment (Mr Walsh costed it at $510, based on three hours at $170 per hour) and none has apparently been conducted. The effect of Mr Walsh’s evidence is that there is a chance only that she would benefit from such a cushion. In my opinion, the chances are greater than even, indeed much greater, given the comfort Mrs Dawson has derived from the gel seat on the computer chair. In the circumstances, I would allow the claim but at a unit cost of $350.
5592 The respondents opposed the other two items.
5593 Mr Walsh observed that Mrs Dawson already has “a work chair”, presumably referring to the computer chair with gel seat, and recliner and said that they would “not need to be currently funded”. It follows that the claim for an immediate sum of $4,500 is not supported by the evidence. If the claim were made out, any amount would need to be deferred until the time for replacement of the existing chair.
5594 The respondents argued there was no evidence to indicate that Mrs Dawson bought her current recliner chair because of the mesh implant and, for this reason, she has not discharged her onus of proving that the future expenses would be incurred because of the implant and its consequences. It is true that there is no evidence that Mrs Dawson bought her current recliner chair because of the implant but that does not mean that, when her current chair wears out and needs replacement, she will not need to obtain one with a suitable seat. In the absence of evidence that that would be any more expensive, however, the claim must be rejected.
5595 The respondents also argued that the case for a computer chair had not been established. They submitted that it was unclear from Mr Walsh’s report whether Mr Walsh’s recommendation was for a computer chair at home or at work. If it was for work, then, they contended, she had already been supplied with a “gel chair” and that was a cost to the employer, not to her. If it were for home, they contended that there was no evidence to suggest that her current computer chair was unsuitable for her needs. They argued that any award for “sitting aids” should be based on the cost of a ring cushion as recommended by Ms Borthwick or “at a maximum” the gel/foam cushion recommended by Mr Walsh.4455
5596 I accept the respondents’ submission. The evidence about the computer chair is ambiguous. There is no evidence about whether Mrs Dawson has a computer chair at home, let alone whether it is unsuitable.
5597 I reject the claims for a recliner and a computer chair.
5598 In summary, only the claims for the following items are made out:
$2,605 for a steam cleaner;
$1,378 for a lightweight battery-powered stick vacuum; and
a gel cushion to be replaced at three-yearly intervals, but at the unit cost of $350 rather than $400.
5599 I would discount the total amount by 10% for the chance that Mrs Dawson might have required these items regardless of her compensable injuries.
5600 I note that Ms Borthwick made recommendations for a number of other pieces of equipment, but Mrs Dawson made no claim for them.
The extent of the injuries
5601 Before assessing damages it is necessary to determine which of Mrs Sanders’ injuries and disabilities were caused, either directly or indirectly, by her TVT implant.
5602 In her Statement of Particulars, the following injuries were attributed to the TVT:
1 Chronic inflammatory reaction of tissues into and surrounding which the TVT Tape Implant (also known as TVT Classic or TVT retropubic) was implanted on 12 March 2001, including the bladder mucosa and trigone.
2 Erosion and/or extrusion and/or exposure of the Tape Implant into the vagina.
3 Erosion and/or extrusion and/or exposure of the Tape Implant into the mid-urethra.
4 In addition or in the alternative to the preceding paragraph, erosion and/or extrusion and/or exposure of the Tape Implant into the bladder.
5 Scarification of the urethra, perivaginal tissue and/or bladder.
6 Requirement for surgery on 8 August 2011 to excise the exposed portion of the Tape Implant referred to in the preceding paragraphs (Revision Surgery).
7 Squamous metaplasia of the trigone.
8 Detrusor instability.
9 Chronic or recurrent cystitis/ urinary tract infections (UTls).
10 In addition or in the alternative to the preceding paragraph:
(a) Low compliance bladder.
(b) Urethral instability.
11 Adjustment disorder with mixed anxiety and depressed mood (DSM-5 [309.28]).
5603 The applicants claim that Mrs Sanders has suffered the following disabilities as a result of one or more of those injuries:
1. From 2007 to 8 August 2011:
(a) Urinary frequency, worsening over time.
(b) Urge urinary incontinence (UUI), worsening over time.
(c) Pain and/or discomfort when urinating (dysuria), worsening over time.
(d) Labour-like pain when straining to urinate radiating up through her body to her jawlock
(e) Difficulty voiding and occasional inability to void urine.
(f) Need to have to sit on the toilet and strain for about 2 hours in order to urinate.
(g) Bilateral dull aching groin and/or pelvic pain, radiating across her pelvis and exacerbated by standing and walking.
(h) Sharp, stabbing, blade-like, vaginal pain during intercourse (up to 2008) and when sitting, worsening over time.
(i) Radiation of sharp, stabbing, blade-like vaginal pain down both legs, necessitating having to stop sitting and associated activities (eg, craft work) and needing to stand up or lie down.
(j) Dyspareunia.
(k) From 2008 or 2009 – apareunia/ need to abstain from sexual intercourse.
(l) Significant deleterious impact on intimate relationship with husband, Peter.
(m) Requirement to limit involvement in making and selling craft items.
2. Since the Revision Surgery on 8 August 2011:
(a) Strong and frequent urge to urinate (urinary frequency and UUI) and associated inability to urinate.
(b) Pain and/or discomfort when urinating (dysuria).
(c) Labour-like pain when straining to urinate radiating up through her body to her jawlock and down her arms and fingers associated with numbness in the arms.
(d) Difficulty voiding and occasional inability to void urine.
(e) Need to sit on the toilet and strain for about 2 hours in order to urinate.
(f) Stress urinary incontinence (SUI).
(g) Occasional feelings of incomplete bladder emptying.
(h) Complete lack of bladder control (enuresis).
(i) Nocturia (1–2 times per night).
(j) Need to wear continence pads on a constant basis and change up to 5 times during the day and once at night.
(k) Need to change clothing multiple times per day.
(l) Chronic bilateral dull aching groin and/or pelvic pain.
(m) Pain and/or tenderness of or near the pubic symphysis.
(n) Sharp, stabbing, blade-like vaginal pain when sitting.
(o) Sharp, sudden groin pain, made worse when bumped, by having to move suddenly or when walking on uneven surfaces.
(p) Dyspareunia.
(q) Apareunia/ need to abstain from sexual intercourse.
(r) Soreness/ discomfort /feeling of inflammation in the perianal area.
(s) Low back pain.
(t) Chronic fatigue.
(u) Feeling of being emotionally and physically drained.
(v) Worsening of the above physical symptoms as each day progresses.
(w) Abdominal ache, especially at the end of the day.
(x) Faecal constipation.
(y) Faecal incontinence.
(z) Flatus incontinence.
(aa) Need to ingest antibiotics (Keflex) on a daily basis to relieve pain and/or discomfort when urinating, or symptoms of urinary tract and/or bladder infections.
(bb) Need to ingest non-opioid analgesics non-steroidal anti-inflammatory drugs (NSAIDs) on a daily basis to control pain.
Sub-Particulars
(i) Paracetamol.
(ii) Ibuprofen.
(cc) Need for anticholinergic medication therapy for detrusor instability.
(dd) Reduce mobility
Sub-Particulars
(i) Difficulty walking or exercising.
(ii) Inability to go for long walks.
(iii) Need to rest after walking for 100m.
(iv)Avoidance of ascending or descending stairs or inclined surfaces.
(ee) Impaired ability to babysit for and interact with grandchildren, and associated distress.
(ff) Inability to run and play with grandchildren, and associated distress.
(gg) Reduction in physical activity.
(hh) Inability to sit for long periods of time and avoidance of prolonged sitting.
(ii) Social withdrawal.
(jj) Impaired ability to engage in social and leisure activities.
Sub-Particulars
(i) Reduced frequency of social outings.
(ii) Tendency to limit social activities to family members.
(iii) Need to ensure that location being visited has toilet facilities.
(iv) Need to reduce frequency of visits to 93 year old father.
(v) Need to carry a change of clothes whenever going outside the home.
(vi) Need to refrain from drinking too much while out due to fear of being incontinent.
(kk) Need to cancel appointments at short notice due to pain.
(ll) Significant deleterious impact on intimate relationship with husband, Peter.
(mm) Reduced ability to perform activities of daily living.
Sub-Particulars
(i) Difficulty performing activities that involve lifting and stretching and dependence upon husband to perform same.
(ii) Difficulty performing heavy domestic tasks (eg, sweeping, mopping, vacuuming, cleaning the toilet and bathroom) and dependence upon husband to perform same.
(iii) Need to sit or lay down after performing activities (eg, ironing) after a short period of time (a few minutes).
(iv) Delay in completing chores.
(v) Limited ability to cook and prepare meals.
(vi) Difficulty pushing a shopping trolley and need to lean on it for support when walking.
(vii) Difficulty transferring in and out of chairs, bed and shower.
(nn) Irritability.
(oo) Anger and frustration at no longer being physically fit and healthy.
(pp) Mood swings.
(qq) Sleep disturbance from pain and rumination on medical condition.
(rr) Constant worry that she will experience an episode of incontinence and that others will find the smell of urine offensive.
(ss) Embarrassment and distress about her incontinence and physical state; fear of discussing it with most other persons.
(tt) Rumination on her condition and pervasive sense of sadness, anxiety and worry about having undergone the Tape Implant surgery.
(uu) Distrust of the medical profession and despondency about medical opinions that her symptoms of urinary incontinence would be relieved by the Tape Implant surgery.
(vv) Anxiety about incontinence, pain and social withdrawal.
(ww) Chronic stress associated with physical symptoms.
(xx) Self-conscious and lowered self-esteem or sense of self-worth.
(yy) Difficulty concentrating.
(zz) Poor motivation.4456
5604 Professor Blaivas said that Mrs Sanders’ described symptoms and disabilities are common in the mesh complication patients he sees in his clinical practice and are well documented in the medical literature.4457 He said that “erosion into the lower urinary tract is a serious complication of mid-urethral slings” and that “late-onset erosion is a result of the material properties of polypropylene, including chronic inflammation, shrinkage, stiffening, and deformation, and, in her case, cannot be attributed to over-tensioning at the time of insertion”. It was his opinion that the erosion and subsequent urinary symptoms were all caused by the TVT and subsequent corrective surgery.
5605 For the most part there is no dispute about the symptoms and disabilities Mrs Sanders has experienced since her incontinence surgery or about the extent to which they have affected her life. The real dispute concerns the reason for them.
5606 The respondents submitted that only the mesh exposure requiring surgery on 8 August 2011 and the adjustment disorder with mixed anxiety and depressed mood should be attributed to the TVT implant and the latter only to the extent that the reduction in Mrs Sanders’ mobility is found to be causally related to the implant rather than to her co-morbidities.4458 The co-morbidities to which they referred were:
the right total hip replacement for osteoarthritis (though not, oddly enough, the osteoarthritis itself);
impingement of the subacromial/subdeltoid bursa of the left shoulder, supraspinatus tendinopathy and symptoms associated with a “frozen” left shoulder;
lumbar spine and multi-level facet joint degeneration, most marked at levels L3/4 and L4/5; and
obesity.4459
5607 The following alleged injuries and consequential disabilities are in dispute:
chronic inflammation;
erosion into the bladder and urethra;
squamous metaplasia of the trigone;
the urinary tract infections;
scarification of the urethra, perivaginal tissue and/or bladder;
detrusor instability;
low compliance bladder and urethral instability;
flatal incontinence;
constipation; and
chronic pain, dyspareunia and apareunia, unless they are found to have been caused by the mesh exposure or its consequences.
5608 I will deal with these matters seriatim.
5609 In the light of the histopathological evidence, there is no room for doubt that Mrs Sanders had chronic inflammation.
5610 I referred in Part XV of these reasons to the initial histopathological findings. Dr Ruba, the pathologist who examined the specimens from her excision surgery in August 2011, reported that they showed dense fibrous tissue in which there was birefringent foreign body-type material surrounded by a multinucleated giant cell and histiocytic reaction with associated chronic inflammation and foci of acute inflammation. Professor Klosterhalfen, who also examined the specimens, stated:
The specimen of Mrs Sanders reveals a typical chronic inflammatory reaction and foreign body reaction even after 10 years of implantation. The resection specimen shows a prominent scarring and fibrotic tissue reaction, forming a mesh fold. On top, there are histological findings of erosion with secondary low-grade infection. Indicator cells of the low-grade infection are smaller accumulations of polymorphonuclear and eosinophilic granulocytes at the interface between the mesh and host tissues …4460
5611 Professor Iakovlev came to a similar conclusion.4461
5612 Apart from the conclusion that a mesh fold had been formed, Professor Wright did not take issue with these opinions. It was also common ground that the findings were “characteristic of the tissue response to implanted polypropylene mesh in the female pelvis”.4462
5613 As I have said before, however, I do not regard the anticipated chronic inflammatory reaction itself to be an injury. In any case, the first evidence of chronic inflammation was in August 2011, at the time of the excision surgery.
Scarification of the urethra, perivaginal tissue and/or bladder
5614 The only references to “scarification” in the evidence come from Professor Klosterhalfen and Assistant Professor Margolis. Neither witness explained what he meant by it. “Scarification” is defined in the Oxford Concise Medical Dictionary as “the process of making a series of shallow cuts or scratches in the skin to allow a substance to penetrate the body”. Plainly this is not what either of them intended. Assistant Professor Margolis testified that patients with presumed synthetic sling complications or injuries have presented with scarification.4463 When asked in chief why removing a sling might not completely overcome voiding dysfunction he said:
Well, the sling has already – by the time it has caused voiding dysfunction, it has already caused damage to the urethra and it has also caused extensive scarification.. I can go in and remove the sling and I can even go in and remove part of the scar with it, but once tissue is damaged it will re-scar …4464
5615 In response to a leading question in cross-examination, Assistant Professor Margolis acknowledged that by “scarification” he meant the formation of scars.4465 I infer that so did Professor Klosterhalfen.
5616 I accept that TVT caused the formation of scar tissue. But the evidence does not permit me to say how extensive it was or precisely where it occurred.
Erosion into the bladder and urethra
5617 I am not satisfied that the TVT eroded into either the bladder or the urethra.
5618 In his operation report Dr Daborn wrote that “TVT eroded at mid urethra”, not into the urethra or the bladder.4466
5619 Despite Mrs Sanders’ account of her conversation with Dr Daborn, there is no good evidence, including no contemporaneous record, to indicate that the tape had eroded into the bladder. I consider that in this respect Mrs Sanders’ recollection is unlikely to be correct.
5620 An erosion into the bladder is a serious matter. Had Dr Daborn been of that opinion, one would expect to find a reference to it in the operation report. Yet, there is no reference in the operation report or in any other hospital record. The reference to the TVT erosion in the operation report precedes Dr Daborn’s summary of the findings he made on cystoscopy; it is not part of them. Without more, Mrs Sanders’ evidence is an insufficient basis for making such a finding. The pathologist’s findings from the bladder biopsy included “acute on chronic inflammation” and concluded that Mrs Sanders had cystitis. The evidence does not indicate that the mesh sections he examined were taken from the bladder. While the tape could have caused the inflammation in the bladder, I am not satisfied that an erosion of the tape into the bladder was responsible for it.
5621 Indeed, despite the particulars and Mrs Sanders’ evidence, her counsel did not submit that there was an erosion into the bladder. They argued that the tape had eroded into the urethra.4467 The evidence about this is at best equivocal. It appears to be based on an interpretation of the operation report which refers to mesh erosion at the midurethra and a diagram, presumably in Dr Daborn’s hand, which appears in the report.
5622 The applicants conceded that the report (and the diagrams accompanying it) could be interpreted as referring to the procedure to remove the exposed mesh and the references to “mid-urethra” are to the level of the erosion in the vagina.4468 Professor Blaivas said in this report that it was “unclear as to whether there was an erosion into the urethra in addition to the vaginal erosion”.4469 He was taken to this in cross-examination but, before waiting for the question, he volunteered that, having looked again at the diagram, he did not see any evidence that the tape had eroded into the urethra.4470 No other expert considered that there was a urethral erosion. The only person who could resolve the ambiguity is Dr Daborn and he did not give evidence. While it is possible that there was a urethral as well as a vaginal erosion, having regard to the evidence as a whole and in the absence of evidence from Dr Daborn, I am not satisfied on the balance of probabilities that there was an erosion into the urethra.
Squamous metaplasia of the trigone
5623 I now turn to the third item: squamous metaplasia of the trigone.
5624 It will be recalled that the trigone is a triangular ridge of tissue, like the triangular region of the wall of the bladder, that lies between the openings of the two ureters and the urethra.4471 Squamous metaplasia is a benign condition.4472 Dr Daborn queried whether Mrs Sanders had a squamous metaplasia of the trigone. He did not diagnose it. Besides, there is no evidence that it was caused by the TVT device or the mesh excision surgery. For all I know it was an incidental finding at operation. In any event, the applicants made no submissions about it.
Urinary tract infections, detrusor instability, low compliance bladder, urethral instability
5625 It seems to be common ground that all of Mrs Sanders’ urinary symptoms could have been caused by the TVT device or its sequelae. The question is whether more probably than not they were.
5626 In his report of 23 August 2016 Professor Blaivas stated that Mrs Sanders developed multiple complications from her TVT device.4473 These complications included dyspareunia and apareunia; severe groin pain and tenderness of the pubic symphysis; and, following the late onset vaginal erosion, new-onset urinary symptoms (including severe stress incontinence, worsening urgency and recurrent urinary tract infections). He said that Mrs Sanders’ description of her symptoms and disabilities was common in the mesh complication patients he sees in his clinical practice and is well documented in the medical literature. It was his opinion that the erosion and subsequent urinary symptoms were all caused by the TVT and subsequent corrective surgery.4474
5627 On balance, and with one certain qualification to which I shall come shortly, this opinion should be accepted.
5628 The respondents’ witnesses tended to discount, if not rule out, any relationship between these symptoms and the TVT and/or the excision surgery with few, if any, reasons.
5629 Professor Frazer did not express an opinion on the probable relationship, only on the possibilities. He said that it was possible that Mrs Sanders’ current problems could be ascribed to the TVT surgery but, then again, “it is at least conceivable that some of her symptoms, which she now, with some certainty, ascribes entirely to the TVT mesh, may be due to other causes”. He said that urinary tract infections, dyspareunia, troublesome pelvic pains all tend to increase with age. 4475
5630 I will come to the question of chronic pain and dyspareunia in due course. At this point, I will deal with the urinary symptoms.
5631 Professor Roovers said that most likely there was no causal relationship between the TVT implanted in 2001 and the recurrent urinary tract infections reported in 2007.4476 His evidence, however, was based on the false premise that Mrs Sanders had received a pessary for pelvic organ prolapse. In his report, he wrote that this supposed pessary “may have resulted in the exposure of the TVT because of pressure of the pessary on the vaginal epithelium above the tape”. No attempt was made in oral evidence to correct his misconception. Having regard to the false assumption on which his conclusion was premised, I give no weight to it.
5632 In her report Associate Professor Rosamilia was emphatic that the TVT was not the cause of the recurrent urinary tract infections. 4477 As Assistant Professor Margolis postulated, the reason she was so emphatic is that she appears to have taken the view that urinary tract infections can only be associated with the device if there has been an erosion into the urethra or the bladder. He said such a view would be incorrect since urinary tract infections from the TVT can be caused by partial obstruction of the urethra by the sling, sling infection, and sling erosion.4478
5633 Professor Deprest’s analysis of Mrs Sanders’ case was also brief and rather superficial. He concluded that she was treated for mixed incontinence in 2001, after which there was satisfactory improvement of the stress incontinence but moderate urge incontinence persisted. He added that she developed a symptomatic tape exposure around 10 years later, which, he said, was successfully revised.4479
5634 The true position is not so clear.
5635 Assistant Professor Margolis said that there were no other medical conditions in Mrs Sanders’ history that could possibly have caused her recurrent urinary tract infections other than TVT. While he accepted that urinary tract infections are common throughout the lives of women, he said that they tend to occur in young women who are more sexually active and in the very elderly especially those with significant prolapse. He said that there was a clear causal relationship between TVT sling procedures with urinary outlet obstruction and subsequent recurrent urinary tract infections. 4480
5636 Professor Blaivas also considered other potential causes. He acknowledged that recurrent urinary tract infections in postmenopausal women are common. He said that recurrent urinary tract infections have many causes including vaginal atrophy, pelvic organ prolapse, and urethral obstruction. Based on her medical records, he accepted that Mrs Sanders has vaginal atrophy but noted that it was described as mild. For this reason he considered that vaginal atrophy placed a minor role, if any. He excluded pelvic organ prolapse and urethral obstruction since there was no evidence of the latter and the first evidence of the former was not until recently when a grade II rectal prolapse was noted.4481
5637 Professor Roovers speculated that ageing might have resulted in vaginal atrophy and thus a thicker vaginal epithelium but pointed to no evidence of vaginal atrophy. 4482
5638 Contrary to Professor Blaivas and Assistant Professor Margolis, neither of whom examined Mrs Sanders, Professor Korda found that Mrs Sanders had marked vaginal atrophy under the pubic arches.4483 Neither Professor Blaivas nor Assistant Professor Margolis was asked to comment on this finding.
5639 Moreover, having reviewed the results of the microurine examinations conducted on 19 October 2000, 8 March 2001, 21 January 2011, 28 June 2011, 2 August 2011, 13 October 2011, 22 January 2014, 6 February 2014, and 22 July 2014, Professor Korda was unable to see any convincing evidence of documented urinary tract infections.4484 In oral evidence he pointed out that “the easiest thing in the world for a pathologist to do is grow bugs in urine, so [if] they can’t grow bugs in urine [,] [t]here is no infection”.4485
5640 Professor Korda’s opinion was that the symptoms Mrs Sanders ascribed to urinary tract infections can be explained by an alternative pathological process such as urethral instability and/or a low compliance bladder, both of which were suggested by Associate Professor Shek’s urodynamic study. He said that the aetiology of urethral instability is not fully understood but there are “theories that postulate an intrinsic primary defect in the urethra”. Professor Korda’s opinion was that the insertion of the TVT device, the later erosion, and the necessity for excision damaged the integrity of the urethra and caused urethral instability. Professor Blaivas also observed that symptoms of frequency, nocturia, urgency, and dysuria are associated with urethral instability.4486
5641 Professor Korda was persuasive. On the other hand, Dr Ruba, the pathologist who examined the specimen taken from the bladder biopsy, expressed the opinion that Mrs Sanders had “acute on chronic cystitis with histological features in keeping with a cystitis glandularis”.4487 In other words, he diagnosed an acute exacerbation of a chronic bladder infection. The bladder is part of the urinary tract. Assuming he was given a copy, Professor Korda appears to have overlooked Dr Ruba’s report. He was not asked to comment on the significance of his findings. Nor were any of the other witnesses.
5642 Professor Wright said in his first report that cystitis glandularis is a common incidental finding in a bladder biopsy and is associated with long-standing chronic cystitis and other causes of mucosal irritation.4488
5643 There is therefore evidence of chronic infection of the urinary tract.
5644 Whether or not Mrs Sanders did have recurrent urinary tract infections, I am satisfied on the balance of probabilities that the urinary symptoms of which Mrs Sanders’ complained in and from 2011 were caused by the TVT device. Professor Korda’s view, which I accept, was that the insertion of the TVT, the subsequent erosion, and the necessity for excision damaged the integrity of the urethra, causing urethral instability.4489 In oral examination he was invited to explain the mechanism. He replied:
But, anyway, the urethral instability is defined as a condition whereby the urethral pressure, which is the intrinsic pressure inside the urethra, much like if you think of the pressure inside a garden hose is low in a certain type of garden hose and high in a certain type of other rubber garden hose, so that’s the best way to look at it. When there is urethral instability, then that pressure is lower than the bladder pressure on an empty bladder … We don’t know what causes it, but there is no question that it’s caused by some interference with the nerve supply to the urethra. So if you put a tape around the urethra, that doesn’t necessarily cause damage to the urethra. But if you have an erosion of that tape and there is infection around that tape and then you have to remove the tape, then the surgery that you do to achieve this may damage the nerves to the urethra.4490
5645 Thus, irrespective of whether Mrs Sanders truly suffered from recurrent urinary tract infections, I find that more likely than not that the removal surgery caused the return of Mrs Sanders’ urinary incontinence.
5646 Associate Professor Rosamilia said that approximately 40% of patients will have recurrent stress incontinence of some degree after excision of exposed mesh after midurethral sling surgery.4491 Professor Korda agreed.4492 I infer that both accepted that the recurrent stress urinary incontinence can be attributed to the excision surgery.
5647 Professor Blaivas observed that recurrent stress incontinence is well-known with all anti-incontinence procedures. He considered, however, that the temporal relationship between the onset of worsening stress incontinence and the excision surgery provided “a compelling argument” that this was not simply a failure of the original operation but a complication of the mesh excision surgery necessitated by the erosion of the tape.4493 I agree.
5648 The more difficult question relates to the other urinary symptoms of urge incontinence and frequency.
5649 Associate Professor Rosamilia observed that correction of stress incontinence by a midurethral sling will correct urgency in about 60% of cases and that, if the support is removed by excision, urgency could increase but “recurrent cystitis and the natural history of overactive bladder” could also be causes.4494
5650 Assistant Professor Margolis was dismissive of the notion that urgency could be corrected by surgery for stress urinary incontinence.4495 But Associate Professor Rosamilia cited in support of her observation a paper published in the British Journal of Obstetrics and Gynaecology in 2011 of which she was a co-author.4496 That was a report of a prospective cohort study of 754 women who had undergone midurethral sling surgery. Those with persistent urgency or urge urinary incontinence at long term follow-up were compared with women whose symptoms had resolved. The reported results show that persistent urgency occurred in 40.3% of the cases and in 59.7% it resolved after surgery. On the other hand, the paper stated that “[w]omen who experienced resolution of their urgency were more likely to have undergone concomitant prolapse surgery”, particularly a vault suspension for apical prolapse, at the time of their midurethral sling surgery. Associate Professor Rosamilia neglected to mention this in her report.
5651 Professor Blaivas acknowledged that de novo and persistent overactive bladder symptoms are well-known after all incontinence procedures. Furthermore, he observed that Mrs Sanders had some urgency and occasional urge incontinence before her TVT surgery, but he said that it was worse later.4497 He went on to say:
Other causes of OAB symptoms include mesh erosion into the bladder or urethra, urethral obstruction, urethral diverticulum, neurogenic bladder and chronic UTI. There is no evidence that she had any of these conditions except UTI and possibly urethral erosion, but the symptoms persisted even after successful treatment with antibiotics. Cystitis glandularis and bladder inflammation may be contributing factors. It is also possible that, even if the mesh had (scil) not eroded into the lower urinary tract, it could be embedded (scil) in the wall of the bladder or urethra and that can cause OAB symptoms. Notwithstanding all of these, in my opinion, the TVT mesh is the proximate cause of her current OAB symptoms …
5652 On balance, I find that Mrs Sanders’ overactive bladder symptoms of urgency and frequency are attributable to urethral instability either caused or aggravated by the erosion of the tape and its sequelae. In view of her pre-existing symptoms, however, some allowance must be made for the chance that they would have persisted or recurred even if she had not undergone TVT surgery.
Flatal incontinence and constipation
5653 This claim is not made out.
5654 I can find no reference in the evidence to flatal incontinence and, so far as I can tell, the only reference in the evidence to constipation and faecal incontinence appears in Professor Korda’s 2016 report, but he did not attribute either to Mrs Sanders’ TVT implant. Dr Wilcox received a history that Mrs Sanders had less control of her bowel function since 2011 and that she was now terrified to leave the house if she had not opened her bowels in the morning. No submissions were made on this subject and I can find no evidence to connect any bowel problems Mrs Sanders may have to the TVT procedure, the erosion, the revision surgery, or to any other form of treatment.
Chronic pain, dyspareunia, and apareunia
5655 I turn now to the chronic pelvic pain, dyspareunia, and apareunia.
5656 In her report Associate Professor Rosamilia ruled out TVT as a cause of pelvic or hip pain. Professor Roovers said that TVT was most unlikely to have been a cause and that it was most likely that this pain was related to degenerative changes in Mrs Sanders’ pelvis (osteoarthritis).4498
5657 I am satisfied that some of Mrs Sanders’ pain was attributable to TVT. The osteoarthritis certainly confounded the picture. But only for a limited period of time. And Mrs Sanders was able to isolate the osteoarthritic pain. She distinguished between the pelvic pain she first experienced in early 2011 from the pain she said developed in 2014. The former, she explained, was closer to her groin and in the nature of an ache, similar but worse than the kind of pain she had felt since about 2008. The hip pain she experienced in 2014 was away from her groin and “a burning or grinding pain” which gradually deteriorated over the next two years.4499 She said that she had had a complete recovery from her hip pain after the hip surgery and this evidence was not discredited in cross-examination. The fact that she continues to suffer groin and pelvic pain despite the hip surgery tends to rule out osteoarthritis as a cause of her ongoing pain and makes it unlikely that all of the pain symptoms she had experienced before the surgery were attributable to osteoarthritis. In any case, the respondents’ witnesses did not explain how osteoarthritis in the hip could cause dyspareunia.
5658 Professor Blaivas noted that Mrs Sanders had no history of sexual impairment or dyspareunia before the TVT placement and the medical records suggest that these symptoms developed at the same time as the vaginal erosion and have persisted despite the excision surgery. He said that an independent medical examination would likely clarify the relationship between these symptoms and the mesh and excision procedure.4500 Professor Korda’s examination supports such a relationship.
5659 As for the other pain symptoms, Professor Blaivas said that a link between the TVT device and Mrs Sanders’s right groin pain and pubic tenderness was not clearly established in the medical records, noting questions raised about whether other potential diagnoses (hip arthritis and a small inguinal hernia) were sufficient to explain the severity of her symptoms. He said that there was a strong possibility that the pain in these areas was related to the device but an independent medical examination was necessary to determine the exact nature of her pain.4501
5660 Dr Levitt effectively ruled out the inguinal hernia as a cause of her pain. It will be recalled that he said that the response to the injection into the right hip joint meant that there was little doubt that the pain in her hip was musculoskeletal in origin and that the right inguinal hernia was “coincidental only”, by which I understood him to mean a coincidental finding on the MRI. The hernia was not even palpable.4502 An independent medical examination was undertaken. That was the examination by Professor Korda in June 2016.
5661 Following the hip replacement, the weight of the evidence favours the conclusion that the groin pain (both before and after the hip replacement surgery) and the other pelvic pain which has persisted since Mrs Sanders recovered from her hip surgery are not attributable to osteoarthritis or to the right inguinal hernia.
5662 After the hip surgery, in the light of Mrs Sanders’ account, in oral evidence, Associate Professor Rosamilia retreated a little from the position she had adopted in her report. While she still excluded the TVT as a cause of the hip pain, she conceded that it was possible that the other pain was caused by the TVT procedure but could also be due to other causes, which she did not identify.4503
5663 Dr Hill was the only orthopaedic surgeon to give evidence. He noted that Mrs Sanders’ groin pain appeared to be localised to the inner aspect of her groin and pubic area than the more central area which is usually associated with osteoarthritis of the hip. He concluded that she had had complete relief from the more laterally situated hip pain since the hip replacement but that there had been no change in the more medial groin pain in the pubic area. The bone scan performed in May 2017 did not point to osteitis pubis as a cause of the pubic pain.4504
5664 Notwithstanding his opinions about urinary tract infections on the occasions referred to, Professor Korda was not of the opinion that Mrs Sanders had never suffered an infection. To the contrary, he attributed her chronic pelvic pain to an infection of the exposed mesh.
5665 In his second report he wrote:
The erosion of the TVT device and the necessity for partial removal has resulted in the recurrence of urodynamic stress incontinence with a low compliance bladder and urethral instability.
Additionally, the prolonged period of erosion has resulted in colonisation of the mesh with bacteria which has now resulted in chronic pain syndrome which has resulted in apareunia and lower back pain, groin pain and hip pain.
It is highly probable that the easily palpable bristly portion of the remaining tape underlying the thin vaginal mucosa contributes to Ms Sanders' dyspareunia and if she was to engage in penetrative intercourse this area would erode further necessitating future surgery.
To some degree the hip pain may be related to the chronic hip condition for which she is awaiting hip replacement surgery and is unrelated to the TVT implant.4505
5666 In his third report in which he responded to the opinions of the respondents’ experts, he reiterated his view:
It is my opinion that following excision of the mesh, excision of the TVT and repairing the epithelium Mrs Sanders is most likely to have developed an infection of the exposed mesh which created an inflammatory process which resulted in the development of pain.4506
5667 That opinion is supported by the evidence of the pathologists.
5668 In oral evidence Professor Korda explained that the hip pain could be related to the TVT because pelvic pain is not uncommonly referred to the hip and it is often very difficult to differentiate between the various causes. In the light of the later evidence that the hip pain had resolved after the hip replacement, however, Professor Korda excluded TVT as a cause of the hip pain but he maintained that her other pains were almost certainly due to the TVT.4507
5669 I find that on the balance of probabilities Mrs Sanders’ dyspareunia, apareunia, and chronic pelvic pain, with the exception of the hip pain which was cured by the hip replacement, were caused by the erosion of the TVT and its sequelae.
5670 Professor Korda’s opinion was that, but for the TVT implantation, Mrs Sanders would not have developed dyspareunia, groin pain, pain radiating from her groin to her leg, or urethral instability. Nor, he said, would she have developed recurrent urinary incontinence “as the tape would not have been divided”. If she had undergone a colposuspension, he stated, rather than TVT implantation, not only would she have avoided the other symptoms but she would also have had relief from her urinary stress incontinence.4508
5671 I accept this evidence. While dyspareunia can occur after a colposuspension, Mrs Sanders’ dyspareunia (and apareunia) occurred years after the operation and coincided with her feeling “something sharp, like a blade” in her vagina. In my view, in all likelihood what she was feeling was the mesh eroding.
5672 Contrary to the respondents’ submission, it is beside the point that some of Mrs Sanders’ problems could have arisen even if she had had continence surgery without mesh.
5673 In Commonwealth v McLean, a jury had found the plaintiff’s throat cancer to have been caused by the defendant’s negligence. The Commonwealth had argued that he would have developed throat cancer regardless of its conduct. The Court of Appeal unanimously rejected the argument. Handley and Beazley JJA said at 411 (Santow JA agreeing at 412):
The issue was causation in fact of an actual event, and not causation of an hypothetical event where the court allows for the probability that the event would or would not have occurred: see Malec v J C Hutton Pty Ltd (1990) 169 CLR 638. Causation in fact of an actual event is an all or nothing issue. The plaintiff was not entitled to recover anything for his throat cancer unless he established, on the balance of probabilities, that it had been caused by the tort. Once the plaintiff succeeded on that issue the defendant was not entitled to any deduction for the chance that the plaintiff might have contracted throat cancer in any event: see Hotson v East Berkshire Health Authority [1987] AC 750 at 783.
(Emphasis added)
While the case is not on all fours with the present one, the point is a similar one. The question is whether the defendant’s wrongdoing caused the plaintiff’s injury, not whether he or she might have suffered the same or a similar injury in different circumstances.
5674 There was no dispute between the parties about the diagnosis of an adjustment disorder or as to its cause. There was no real difference of opinion between Dr Jungfer and Dr Wilcox and, although Dr Jungfer was required for cross-examination, she was asked no questions about Mrs Sanders. For these reasons I take it to be common ground that Mrs Sanders suffered an adjustment order with mixed anxiety and depression caused by the complications of the TVT. Dr Jungfer provided cogent reasons for excluding the hip pain as a cause of the psychiatric disorder. She explained:
Mrs. Sanders' adjustment disorder which with depressed and anxious mood has persisted despite the hip replacement surgery. Mrs. Sanders made a remarkable recovery from the hip replacement surgery with only a brief stay in hospital and fairly rapid independence and now does not report any persistent hip pain but still continues to have the pelvic pain and the persistent other physical symptoms and psychological difficulties. Therefore there is no evidence in my opinion that her adjustment disorder is related to the hip problem. One could hypothesize that she possibly has become more anxious since the hip surgery as it may well be that Mrs. Sanders at the time that I saw her was hoping that her physical symptoms would resolve particularly that of the pain symptoms with the hip surgery. They have not resolved and she has become more anxious in an acknowledgement that the persistent consequences of the mesh implantation will have an enduring effect upon her functioning. Overall difficulties have not changed in response to the hip replacement surgery.4509
5675 Based on Mrs Sanders’ history, Dr Jungfer also excluded her lumbar back pain as a cause of the adjustment disorder.4510 I accept these opinions.
Conclusion
5676 In summary, I find that Mrs Sanders underwent chronic inflammation and scarring of her perivaginal tissues as a result of the implantation of the TVT device. I also find that the TVT is responsible for her urethral instability, recurrent stress incontinence, urge incontinence, and urinary frequency. Furthermore, I find that her dyspareunia, apareunia, and chronic pelvic pain in the groin and pubic area, which sometimes radiates into her legs, were caused by the erosion of the TVT.
5677 But I am not satisfied that the TVT eroded into either her bladder or her urethra, that she has squamous metaplasia of the trigone, or that if she does it has anything to do with the TVT. Nor am I satisfied that there is any connection between her bowel problems and the TVT or the repair surgery.
General damages
5678 The applicants argued for an award of between $250,000 and $350,000.4511
5679 The respondents submitted that, having regard to the injuries and disabilities caused by the TVT implant, Mrs Sanders’ significant co-morbidities, especially in relation to her hip replacement, and the risk of complications in the event that a surgical procedure other than TVT had been adopted to treat her incontinence, the appropriate award for general damages or non-economic loss is in the range of $50,000 to $100,000.4512
5680 I cannot accept either submission.
5681 It seems to me that the applicants’ assessment is excessive. They referred to a number of authorities. For the reasons given by the High Court in Planet Fisheries I have found them to be of little to no assistance. The applicants’ figure appears to take no account of Mrs Sanders’ co-morbidities or her age.
5682 On the other hand, the respondents’ submissions are manifestly inadequate having regard to my conclusions.
5683 First, the osteoarthritis of the hip contributed to Mrs Sanders’ past pain but it was by no means the sole cause of it and has not contributed to her symptoms and disabilities since she recovered from her hip replacement surgery in about late 2016.
5684 Second, although there is evidence of degenerative changes in the lumbar spine, the findings from the bone scan and the CT scan, both done in 2014, suggest it is mild. Despite a facet joint injection at L4/5 on 9 June 2014, four days later Mrs Sanders presented to her GP with bilateral lumbar pain.4513 While I cannot rule out some contribution from other causes, on the balance of probabilities I consider that at least some, if not all, of Mrs Sanders’ low back pain forms part of the chronic pain syndrome she developed as a result of the infected mesh exposure.
5685 Third, the left shoulder was a problem in 2006. Since I do not consider that Mrs Sanders suffered injury as a result of the TVT implantation until 2007, that episode is irrelevant. She presented to her GP again in late October 2016 with pain in her left shoulder and the ultrasound findings suggested she had bursitis for which she received a steroid injection in early November 2016 but, according to the GP’s notes, that only made her worse.4514 Nevertheless, an MRI conducted in December 2016 reportedly showed impingement of the subacromial-subdeltoid bursa, which was thickened and swollen.4515 By 23 January 2017, however, following a corticosteroid injection, her bursitis had reduced and she was reportedly pain-free.4516 I conclude that Mrs Sanders was troubled by her shoulder for two to three months in 2016 to 2017 but I accept that the shoulder may well continue to cause her problems in the future.
5686 Fourth, there is insufficient evidence to suggest that the obesity is disabling or likely to become so. On the other hand, the effect of Mrs Sanders’ evidence is that her inactivity due to her pain caused or contributed to her weight gain. For a limited time that was partly caused by her osteoarthritis but during that time she would have been disabled in any event by her other pains.
5687 Mrs Sanders has had a range of disabling symptoms caused by TVT for over a decade now. They have interfered with every aspect of her life. Her lower back and groin pain, in particular, have inhibited her capacity to exercise, even to walk on the beach or for any distance, or to stand for any length of time. After only a short walk she is exhausted. Even sitting can cause severe pain. Her enjoyment of her craftwork is diminished and she has abandoned her market stall. She struggles to maintain the home, requiring assistance with various tasks she managed on her own and without difficulty before the onset of her pelvic pain in 2008. She now has two grandchildren, whom she loved to babysit, but, unlike her husband, she has not been able to participate in activities with them, such as playing outside or cycling, which makes her feel like a bystander in their lives.4517 She is frustrated by her reduced capacity and has become irritable and intolerant of others. She is deeply ashamed of, and embarrassed by, her urinary symptoms and disinclined to socialise. Her sleep is impaired. She ruminates about her plight and, based on what she told Ms Williams, the occupational therapist, she is awoken by pain, which means she is often tired.4518 Her self-image and self-esteem have suffered greatly.
5688 The prognosis is guarded. She had to undergo one surgical procedure to remove mesh and she may well have to undergo at least one other, since it is likely that she has had a further exposure or erosion. She can expect to continue to be troubled by these symptoms for the rest of her life, which, based on the 2019 medium life tables, is in the order of 15.97 years. Had it not been for the TVT surgery, she is likely to have been bothered by some incontinence symptoms but not by chronic refractory pain. The descriptions she gave of that pain were graphic and she appeared to be genuinely distressed when recounting in court its effect on her life. Although the reliability of some aspects of her evidence was challenged, no suggestion was made that she was fabricating or exaggerating her symptoms. In cross-examination Mrs Sanders was taken to medical records and asked to confirm that she had not complained of pain at certain times, but it was never put to her that at such times she was not then in pain.
5689 Taking all relevant matters into account, and making due allowance for the chance that she might experience symptoms in the future as a result of her non-compensable or pre-existing conditions, I would award $200,000 for general damages/non-economic loss. I apportion 35% to the past.
5690 Interest on past loss should be awarded at the rate of 2% per annum.
Economic loss
5691 No claim was made for past or future economic loss.
Past out-of-pocket expenses
5692 Past out-of-pocket expenses were agreed at $7,000.4519
5693 The amount was agreed at $120,000.4520
Interest
5694 Since Mrs Sanders is not affected by the 2004 amendments to the TPA, she is entitled to interest on all heads of damages for past losses, including non-economic loss and gratuitous services.
5695 In their written submissions the applicants claimed that the Court could not award interest on general damages because of the preclusion in s 32(2) of the Supreme Court Act 1935 (WA), on the erroneous assumption that that Act was applicable.
5696 For the reasons given above in relation to Mrs Gill, however, the Supreme Court Act does not apply and interest should be awarded on all past losses, including general damages and past gratuitous services.
5697 The rates of interest should be calculated and applied in the same way as I indicated with respect to the other applicants.
Future domestic assistance
5698 In her first affidavit Mrs Sanders deposed that, before her incontinence surgery in March 2001, she was responsible for all household chores.4521 She spent an hour a day and a couple of hours each weekend cleaning. I infer from her evidence that, with the exception of the time she was recovering from the incontinence surgery, that continued to be the case until the onset of her pain in 2008. Now, however, she needs assistance with the heavier tasks in particular. She said that she can put the washing in the machine but cannot hang it out. She said that she was unable to stretch or stand for very long because of hip, back and groin pain and that when she sits down she may experience sharp vaginal pain, the onset of which, I gather, is unpredictable.4522
5699 In her second affidavit, Mrs Sanders deposed that, after she had recovered from the hip surgery and was no longer troubled by the pain for which the surgery was indicated, but she was still unable to do a range of tasks she was able to carry out without difficulty before 2008. She explained that the effect of the hip surgery was to remove the pain in her hip; it had no effect on her other pain.4523 She told Ms Williams that pain is present when she is sitting, standing and lying down and she has no relief from changing positions.4524 Her account to Ms Williams was consistent with the account she gave in her affidavit.
5700 Mrs Sanders gave the following evidence about the assistance she currently requires:
As described in paragraph [94] of my First Affidavit, since 2008, I have been unable to do all the household chores I was responsible for. Pete does all the big household chores, he hoovers, irons, mops the floors, cleans the bathroom, puts the washing out and washes the windows. Pete often calls in from work to check if I am okay. While I can drive, Pete often drives me places and always drives me to my appointments. The time Pete spends on each of these tasks is detailed, he:
(a) vacuums and mops the floors three times per week, each time he spends approximately an hour;
(b) irons clothing two times a week, mid-week and on the weekends, this takes two hours each time;
(c) hangs out the washing three times per week, he spends approximately thirty minutes each time;
(d) washes the windows once per week and this takes him one hour;
(e) cleans the bathroom and shower two times per week and spends thirty minutes each time;
(f) prepares meals for me four times per week and spends approximately thirty minutes preparing the meals; and
(g) he drives me to all of my appointments and tests as they arise.4525
5701 In her oral evidence she elaborated on the reasons why she was unable, or struggles to, undertake these tasks herself.
5702 Mrs Sanders’ claim for future care rests largely on the report of Ms Williams, who assessed her on 18 May 2017 and reported on 20 July 2017. Ms Williams, it will be recalled, was not required for cross-examination.
5703 The premise for Ms Williams’ opinions was that Mrs Sanders will continue to live with her husband in a small townhouse in Perth, despite her own desire to return to England when her husband retires, and that her need for care will continue at about the same rate as it has since February 2016, save that it will probably increase with age.4526
5704 Ms Williams considered that Mrs Sanders required three hours assistance a week with heavy cleaning; 12 hours a year for help with seasonal tasks/spring cleaning, such as “high cleaning”, removal of cobwebs, washing windows and curtains; four hours a month to keep the garden and lawns neat and presentable; a monthly car wash and vacuum; “basic” ironing; and one hour a month for home maintenance such as hanging pictures, minor repairs, and changing tap washers. 4527
5705 In addition, Ms Williams said that assistance was required from a community support worker three hours a day, four days a week with respect to the following tasks:
meal preparation;
laundry and bed-making assistance;
shopping;
running errands such as picking up prescriptions;
transport if required; and
support to attend social and leisure “tasks”. 4528
5706 According to Ms Williams, this level of assistance is required for the rest of Mrs Sanders’ life.4529 The costings, set out in table 7 in annexure B to Ms Williams’ report, appear to reflect market rates.4530 The 12 hours of additional domestic assistance with a multiplicity of tasks were included in table 4, somewhat misleadingly referred to as “personal care”.
5707 Ms Williams’ report included a convenient table reflecting what she was told were the chores which Ms Sanders carried out before her surgery in 2011 and with which she now requires assistance:
Tasks | Pre Incident Performance | Current Performance |
Light Cleaning Tasks | All tasks carried out by Ann. | By pacing herself Ann reported that she is able to manage some light domestic activities and others are carried out by Pete. |
Heavy Cleaning Tasks | Pete is now responsible for all the heavier domestic tasks such as: sweeping, mopping, vacuuming, and cleaning the toilet, and bathroom. Ann will require lifetime assistance with heavy cleaning due to pain and fatigue issues. | |
Spring Cleaning or Seasonal Tasks | Pete is now responsible for all the seasonal cleaning tasks, and some tasks do not get done. Eg: cleaning windows, curtains and cobweb removal. Ann will require lifetime assistance with seasonal cleaning due to pain and fatigue issues. | |
Bed Making | Pete now makes the bed and changes the linen regularly as bending and reaching is too painful. Ann will require lifetime assistance with bed making. | |
Meal Preparation and Cooking | Pete heats ‘Lite n’ Easy’ meals for himself so Ann does not have to cook. Ann puts together simple meals for herself, but does not cook now as she cannot stand for sufficient time due to pain. Ann will require lifetime assistance with meal preparation. | |
Washing-Up | Pete now does washing up as Ann is unable to manage static standing at the sink due to pain. Future assistance is required. | |
Laundry | Pete hangs out the washing and brings it into the house. Ann will require lifetime assistance with laundry tasks due to pain. | |
Ironing | Ann finds this very painful and does one item at a time. Pete irons his own uniform. Ann will require lifetime assistance with ironing. | |
Shopping | Ann reported that shopping is painful and difficult even when visiting small shops. Pete assists by bringing home shopping items on a daily basis. Ann will require lifetime assistance with shopping. | |
Garden and Lawn Care | Pete and Ann shared this task equally. | Pete now does all gardening and lawn care required. Ann will require lifetime assistance with gardening and lawn care if living alone. |
Putting the bins out | Shared. | Completed by Pete. Ann will require lifetime assistance with rubbish management if living alone. |
Car Care | Ann was independent. | Ann is able to put fuel in her car, but cannot wash and clean it. A monthly car wash is recommended. |
5708 Both occupational therapists purportedly based their opinions on the amount of assistance Mrs Sanders was currently receiving. Even so, there is a massive difference between the opinions expressed by the two occupational therapists.
5709 In her report Ms Borthwick recorded that the only assistance Mr Sanders provided to his wife each week from 1 April 2016 to 29 September 2017, being the most current figures, consisted of 2 hours of meal preparation; ½ hour vacuuming, mopping, sweeping; ½ hour laundry; 0.2 hours changing linen (presumably on the bed or beds); 1 hour shopping, 0.3 hours cleaning the bathroom; and 0.25 hours spring cleaning — a total of 4.75 hours per week. She disregarded the ironing he did, ostensibly because he was ironing his own shirts; and also gardening, lawn care, and home maintenance because these were “not [Mrs Sanders’ role]”. Ms Borthwick considered that Mrs Sanders would continue to require the level of support she is currently receiving. But she reduced the amount to 1.75 hours per week once Mrs Sanders turned 80 apparently on the basis that by that age she would have required assistance with shopping and cleaning for three hours a week due to “age-related changes” regardless of whether she had suffered from the symptoms attributable to the TVT implant.4531
5710 Ms Borthwick’s account of the current assistance is at odds with the account Mrs Sanders gave. Mrs Sanders said that Ms Borthwick did not ask her questions about how much time it took her husband to assist her.4532
5711 For the most part, I prefer Ms Williams’ evidence to Ms Borthwick’s. Ms Williams pointed out some problems with Ms Borthwick’s approach.4533
5712 First, a number of Ms Borthwick’s assumptions about Mrs Sanders’ pre-injury activities were wrong. Ms Borthwick excluded the gardening on the basis that it was not Mrs Sanders’ role when in fact she and her husband had shared the gardening. Ms Borthwick failed to include ironing when Mrs Sanders had done all the ironing and when both she and her husband wore clothes that needed ironing.
5713 Second, Ms Borthwick did not make sufficient allowances for reasonable completion of the various tasks. Ms Williams said that 20 minutes is insufficient to fully clean a bathroom once a week to, let alone two to three times a week as Ms Sanders required, and that 30 minutes a week to complete the laundry tasks and a similar period to sweep, mop, and vacuum all the floors in a house are similarly significant underestimations.
5714 Third, Ms Borthwick’s reasons were inadequate. “Not required” or “not her role” was stated without an explanation as to how (or why, I would add) she came to this conclusion.
5715 Fourth, Ms Borthwick adopted an unusual approach to her assessment. She seemed to rely on observations of posture, gait, and some staged observations to gauge pain. In contrast, Ms Williams said she used standardised and well-known methods of assessments. She noted that having a person carry a 4kg weight in a laundry basket once in no way replicates the actual task. Further, she used other modes of assessment which were designed for purposes that bore no or too remote a relationship to the determination of the task at hand.
5716 Fifth, Ms Borthwick took no account of Ms Sanders’ background and insufficient account of her pain, its multiple locations and its effects or the impact of the psychological factors. Ms Williams also felt that she had failed to have regard to the medical evidence that Ms Sanders was unlikely to improve and the likelihood that, because of the complications of TVT, she would not age in the same way as her peers.4534
5717 Sixth, Ms Borthwick did not assess the type or level of pain Ms Sanders experienced during or following the tasks she was asked to perform.4535
5718 Ms Borthwick replied to some, but not all, of these criticisms in a supplementary report of her own completed the day before she was called for cross-examination.4536 The first and third remained unanswered. As for the second, Ms Borthwick explained her practice. She said that she acquired information by asking direct questions of clients about their reported limitations and indirectly through discussions about recent activities. She said she had had regard to all the material that was made available to her including medical and other evidence. As for the time it takes to do certain things, she said she asked the clients specific questions about the frequency and time taken to complete “most” tasks, specifically with regard to tasks such as transport which can vary greatly from one person to the next. She said that in determining what was reasonable and necessary she takes into account the Australian Bureau of Statistics data and her own experience as a commercial cleaner over a five year period after she had finished university.
5719 Ms Borthwick’s reply and her oral evidence lend support to many of Ms Williams’ conclusions. Both tend to confirm Ms Williams’ view that Ms Borthwick took insufficient account of Mrs Sanders’ types of pain or its multiple locations or how tired she felt after undertaking certain tasks or to the nature and effect of chronic pain and paid insufficient, if any attention, to the reasonable needs of Mrs Sanders, with her high standards, as opposed to a notional person with similar injuries who was not as fussy as Mrs Sanders about her house or her or her husband’s appearance. I note that in her principal report Ms Borthwick stated that “overall, [she had] outlined the amount of time required to complete tasks assessed as necessary for two middle aged/elderly people living in a small to modest sized dwelling”. This indicates that she addressed the wrong question.
5720 In cross-examination Ms Borthwick was taken to Mrs Sanders’ evidence about the extent of her pain and its effect on her capacity to undertake a number of tasks and she made some concessions which suggest that she is likely to have underestimated the extent of Mrs Sanders’ pain and her reasonable needs.
5721 Contrary to the respondents’ submissions, it is quite clear that Ms Williams did have regard to the evidence of Mr and Mrs Sanders.4537 The respondents also submitted that Ms Williams did not assess or take into account the impact of Mrs Sanders’ right hip pain on her functional capacity and that, in contrast, “it was evident from Ms Borthwick’s assessment … that her right hip pain caused her to limp, avoid long distance walking, running and high impact activity”. Ms Borthwick’s evidence clearly related to the evaluation of past services. In light of the parties’ agreement, it is now irrelevant. Ms Borthwick noted that the total hip replacement had “effectively addressed” the right hip pain and, when she observed Mrs Sanders walking, “she maintained a normal gait pattern, weight bearing evenly on both lower limbs”, had an apparently normal pelvic rhythm and walked at a “normal” pace. She said that her tolerance for walking remains reduced but this was due to her “visceral pain”.4538
5722 Having regard to the omissions from, and the contents of, her reply to Ms Williams’ criticisms and the concessions made in cross-examination, I conclude that Ms Borthwick underestimated the extent of Mrs Sanders’ disabilities. She paid insufficient regard to Mrs Sanders’ limited standing tolerance and the fatigue caused by the pain, to which Ms Williams was very much alive. Nor do I believe that she took sufficient, if any, account of the kind of house Mrs Sanders kept before her injuries or of the kind of person she was. In other words, I do not consider that she took any or any sufficient account of Mrs Sanders’ particular needs. As Meagher JA observed in Morgan v Gibson and as Ms Williams, but not Ms Borthwick, recognised:
An injured person’s “need” may not be the same as that of an objectively reasonable person in the shoes of the injured person and determining what that fictitious person might require. It is the cost of providing the services that the particular plaintiff needs which must be reasonable.
5723 With one qualification Mrs Sanders’ own evidence on the help she had required was not the subject of cross-examination.
5724 The qualification is that Mrs Sanders was cross-examined on a patient health questionnaire she completed on or about 1 August 2016 in which she ticked “no” against the question: “Do you currently receive any assistance at home?” Her testimony made it quite clear that she believed she was being asked about paid assistance.4539 She maintained that her husband had helped her before her hip surgery but not before 2008 at the earliest. Her evidence is supported by the affidavit of her husband who was not cross-examined at all and she gave a similar account to both occupational therapists.
5725 Mrs Sanders’ evidence about the extent of the assistance she received is not inherently incredible, particularly in the light of the evidence she gave in her first affidavit. She described herself as “house proud”, something her husband confirmed in his affidavit.4540 She said that she would spend an hour each day cleaning, including vacuuming and mopping.4541 She also used to cook what her husband described as “extravagant main meals”. In around 2010 he said she cooked less, began making meals that were easier to prepare and he would also help by cooking “very simple meals”. Ms Williams said that the time spent ironing was based on two people who are and always have been “well presented” and who choose to wear clothes that require ironing.4542
5726 Having regard to the principles outlined in Morgan v Gibson, these factors must be taken into account in the assessment of future needs.
5727 I find that Mrs Sanders did receive help in the home from her husband and that she needed his help as a result of the injuries she received as a result of the TVT implantation. I also find that the need for help of the kind currently provided by Mr Sanders is ongoing and likely to be required for the rest of Mrs Sanders’ life. The medical evidence indicates that the prospect of an improvement is highly unlikely.
5728 I accept that Mrs Sanders’ account may not be entirely reliable. The times she gave were plainly estimates. It is possible that Mrs Sanders overestimated some times. By the same token, she may have underestimated others. Ms Borthwick did not interview Mr Sanders, who one might have thought was in a better position to give evidence about the time he took to perform the various tasks.4543 The applicants submitted that Ms Borthwick’s inability to obtain information from Mr Sanders affected the reliability of her assessment. Yet in his own affidavit he gave no evidence about the time he spent assisting his wife.
5729 Even so, the question is what Mrs Sanders reasonably needs.
5730 I accept that from time to time in the past Mrs Sanders may have been unable to perform household chores because of other ailments and that her hip and shoulder pain may well have intruded. I also accept that in the future it is possible that she may have a recurrence of shoulder pain and that her hip replacement may wear out. It seems to me, therefore, that some adjustment should be made to the figures to take these contingencies into account. Old age alone, however, is an arbitrary basis for a discount. As Ms Williams observed in her supplementary report, Ms Borthwick provided no research to support her opinion. Ms Williams said that she knew many women from 80 to 90 years of age who continue to live in their homes independently with no support.4544
5731 On the other hand, I consider that Ms Williams’ assessment of additional assistance to be provided by a community support worker is excessive. I see no reason why Mrs Sanders requires support attending social and leisure tasks. Moreover, the basis upon which Ms Williams settled on three hours a day, four days a week (a total of 12 hours a week) is obscure. Doing the best I can on the evidence, I would instead allow seven hours a week. While Ms Williams made provision for one of the days to fall on a Saturday, she did not explain why. In the circumstances, I consider that the Monday to Friday rate of $49.90 per hour should be used.
5732 Ms Borthwick said that Mrs Sanders’ capacity for activity would improve if she were to address her depression and loss of motivation.4545 But this was not the evidence of the psychiatrists. While Dr Jungfer stated that people with psychiatric illness often have a reduced ability to tolerate pain and may have an increased perception of pain, she did not say that Mrs Sanders would have a greater capacity for activity if her depression were treated.4546 And Dr Wilcox said that Mrs Sanders’ psychiatric condition had no impact on her ability to perform domestic duties; her limitations in that regard were entirely due to her physical symptoms.4547 That said, there is a small chance that, with the assistance of a pain management service, Mrs Sanders may obtain some pain relief and so increase her capacity to contribute to, if not resume, some of the tasks with which she currently struggles or which are currently beyond her.
5733 Ms Williams’ opinion was that, although the services Mrs Sanders has required to date have been provided on a voluntary basis, future care should be provided on a commercial basis. As she pointed out:
Through life circumstances, such as ill health or death, or separation and divorce he may not be able to provide these services in the future and services must therefore be commercial. He continues to work and is entitled to his own leisure and relaxation time, in addition to completing his usual share of domestic tasks. It is reasonable that family members increase their role in domestic tasks or care for periods following unexpected illness or surgery, but not for a lifetime as required [for] Ms Sanders.4548
5734 Besides, Ms Williams considered the support that Mrs Sanders was currently receiving was insufficient to meet her needs.4549
5735 In my view it is unreasonable to expect Mr Sanders to continue to provide the services his wife requires and unreasonable to proceed on the assumption that Mr Sanders will always be available. What is more, as the purpose of damages is to restore the injured person to her pre-injury position (insofar as money can do that), it makes no sense to assume that her husband’s role and therefore the relationship she previously enjoyed with him should change in this way.
5736 Regardless, since Mrs Sanders’ case is governed by the common law and no statutory modifications apply, the assessment is to be made on the basis of “the standard or market cost of the services”: Griffiths v Kerkemeyer (Mason J) at 193 (Gibbs and Stephen JJ agreeing); affirmed in Van Gervan v Fenton at 331–334, 338 (Mason CJ, Toohey and McHugh JJ) where their Honours held that the compensation is awarded to the plaintiff for his or her loss and, in general, the reasonable value of that loss is the market cost of providing the services (see also Gaudron J at 349). In Van Gervan v Fenton, Mason CJ, Toohey and McHugh JJ said at 334 that in some cases where, for example, the cost of providing the services at a remote location is much greater than providing the same services in a densely populated area or where there is so little competition from other providers, it might be necessary to discount the market cost because it is unreasonable in the circumstances. Their Honours went on to observe at 335-336, however, that:
It does not seem reasonable that the defendant's liability to pay damages should be reduced at the indirect expense of the provider by invoking notions of marital or family obligation to provide the services free of charge or at less than market rates … Moreover, a plaintiff should be entitled to arrange his or her affairs in the way in which that person pleases and should not be constrained by monetary considerations from dispensing with gratuitous services and obtaining outside services if they are desired. Indeed, the relationship between the provider and the plaintiff may continue to exist in some cases only because outside help is able to be obtained … The use of the market cost criterion enables the plaintiff to be properly compensated by the award of a reasonable sum whether or not the gratuitous care provider continues to provide that care.
5737 Furthermore, as their Honours pointed out at 337, the claim is not one for special damages and so no question of mitigation by the plaintiff arises.
5738 Damages should be awarded at the market rates to compensate Mrs Sanders for her reasonable need for assistance with the following tasks for the designated periods for the rest of her life.
5739 Taking into account all of the above, I award the following in respect of future care and assistance:
Care type | Hours pw | Hourly rate | Weekly sum |
Heavy cleaning (kitchens, bathrooms, mopping, sweeping, and vacuuming) | 3 | $66 for first 2 hrs then $23 | $89 |
Other domestic tasks, including laundry, meal preparation, as well as shopping, running errands | 7 | $49.90 | $349.30 |
Spring cleaning | 1 | $55 | $55 |
Gardening and lawn maintenance (4 hours per month) | 0.92 | $45 | $41.40 |
Car wash (and vacuum) 1 per month (base cost $45) | .23 | $45 | $10.35 |
Ironing ($25 per week) | $25 | ||
Home maintenance (1 hour per month) | .23 | $56 | $12.88 |
Total | $582.93 |
5740 A 3% multiplier is to be applied in relation to the awards in both negligence and under the TPA, since the 2004 amendments do not apply to Mrs Sanders.
5741 Ms Borthwick quarrelled with Ms Williams’ allocation of one car wash per month. She said it was not reasonable that the car be cleaned more often than every six weeks. But she provided no reason for this opinion. I consider that once a month is not unreasonable. Ms Williams nominated a figure of $45 but did not identify her source. Ms Borthwick said a full clean of the outside and a vacuum of the inside could be effected at a cost of $35 but did nominate a provider who, she said, could do it at that cost.4550
5742 Ms Williams allowed 12 hours per month for home maintenance. Why she recommended this figure is obscure and, on the face of things, it seems manifestly excessive.
5743 With respect to cleaning there is no dispute that the rates at which the assistance should be calculated are the rates charged by Absolute Domestics.4551 Those rates are set out in annexure B to Ms Williams’ principal report and correspond to the amounts the applicants claim.
5744 The total amount should be reduced by 25% to take into account two matters: first, the possibility that Mrs Sanders will obtain some pain relief with the assistance of a pain management service enabling her to be more active around the home and second, the prospect that, regardless of the TVT implant, Mrs Sanders would have been unable to undertake those activities from time to time or continuing because of joint or arthritic problems in her hip, spine, and/or her shoulder. I appreciate that if Mrs Sanders had declined surgery she would continue to be troubled by urinary symptoms unless she underwent a native tissue repair and that, even with a successful native tissue repair, there is always a risk of recurrence. But there is no evidence to suggest that Mrs Sanders’ need for home help is related to her urinary symptoms.
Future out-of-pocket expenses
5745 Mrs Sanders seeks a total of $132,037.74 based on an assessment of $155,338.52 less 15% for vicissitudes. Under no circumstances can that degree of exactitude be warranted. Further, I have explained above why a discount for the vicissitudes of life is inappropriate. But a discount for the chance that some of the expenses may not be incurred is another matter.
5746 The claim made on Mrs Sanders’ behalf has three different components:
(a) therapy and interventions;
(b) equipment; and
(c) travel.
5747 Mrs Sanders claimed a total of $62,358.29 for future therapy and intervention.
5748 The first item was $20,000 for future revision surgeries. This sum is said to be an estimate but no foundation was given for the estimate. The respondents submitted that no allowance should be made, in effect, because there is no chance that Mrs Sanders would agree to further surgery. They pointed to the evidence in her first affidavit that, following the excision in August 2011, Dr Tsokos advised her that she could have further surgery but she told him she did not want further surgery.4552 She deposed that she had lost all faith in doctors being able to fix her problems.4553 Dr Tsokos’ report to Dr Tan confirms that Mrs Sanders was not keen on further treatment or investigation at the time.4554 She also deposed that she is afraid of having any more procedures for her incontinence.4555 When she saw Dr Wilcox in December 2016 she again indicated she was reluctant to undergo any further procedures.4556 But she also told Dr Wilcox that her daughters were keen for her to see a surgeon in the United Kingdom where they live. In her second affidavit she said that she wanted to see a UK doctor who her daughter had found who specialises in treating mesh complications. The respondents’ submissions also overlook Mrs Sanders’ evidence that, although she was apprehensive about undergoing hip surgery, having regard to the complications of the TVT implant, she was reassured by Professor Carey-Smith and had a good outcome from that surgery.4557
5749 Despite her misgivings, I believe that there is a real chance that Mrs Sanders will undergo surgery in the future to deal with further mesh exposure, if not also incontinence. Since there is by no means any certainty about this, the amount the applicants’ have allowed is excessive. I would allow $6,000.
5750 The next item relates to GP consultations.
5751 Mrs Sanders claims the cost of three GP consultations per year at $50 per consultation. No evidence was directed to this question. As the respondents submitted, consultations in the recent past have been for a variety of problems, including her right hip, left shoulder, and general health. As the right hip is no longer a problem, it is unlikely that she is likely to be concerned about it for at least another decade. The respondents did not take issue with the cost of the consultations but submitted that three per year were excessive and that only one should be allowed. While three consultations a year might be excessive, one is inadequate. I would allow two. There should be no deduction for the vicissitudes of life. Two $50 consultations per year equates to a weekly cost of $1.89. When multiplied by 728.4 to generate a figure for Mrs Sanders’ life, this equates to $1,376.68, which I would round down to $1,350.
5752 The third item is “pain management specialists”.
5753 Dr Jungfer initially recommended that Mrs Sanders undergo cognitive behavioural therapy.4558 She recommended an allowance of 10 sessions with a clinical psychologist and, although she did not see a role for pharmacotherapy, she felt that, if her symptoms deteriorated, she might need an anti-depressant. Following the second consultation and after Mrs Sanders had seen Dr Wilcox’s report, however, Dr Jungfer changed her mind. She agreed with Dr Wilcox that Mrs Sanders would not likely accept any recommendation for counselling since Mrs Sanders disliked discussing these matters with strangers. In the circumstances she suggested that she be seen by a pain management service. She said that an allowance of four to six consultations with the specialists to establish and stabilise the medication, then monitor and return to general practice care would be reasonable. The cost of the initial consultation she put at about $380 to $400 and thereafter from $255 to $270 per consultation and the cost of pharmacotherapy at $50 per calendar month.
5754 Mrs Sanders claims one consultation at $400 and five thereafter at $270, totalling $1,750. The respondents agreed that the claim was reasonable but argued that any award should be limited to $1,488 after the application of a 15% discount for vicissitudes of life.
5755 The discount for vicissitudes is misconceived for the reasons given earlier. Since that was the only basis upon which the respondents contested the claim, I would allow the claim in full.
5756 Mrs Sanders also claims the cost of anti-depressants at $40 per month for the rest of her life, totalling $9,105.
5757 The respondents opposed the claim based on Mrs Sanders’ own evidence that she was not interested in taking anti-depressants and the evidence of the two psychiatrists.
5758 What Mrs Sanders actually said was that, when she saw Dr Matthews on 28 June 2011, Dr Matthews offered her Valium. She said she was “really taken aback” to be offered Valium because she “did not want to be treated for depression” but for pain.4559 She reportedly told Dr Wilcox that, not only had she no desire for counselling, but that she was not interested in anti-depressants either.4560 On this basis, Dr Wilcox recommended against any treatment.4561 Dr Jungfer acknowledged that Mrs Sanders might not see a psychiatrist but, in making the recommendation for treatment at a pain management service, she noted that anti-depressants are used by pain management services and she believed that this might be more acceptable to Mrs Sanders.
5759 I think it is reasonable to make an allowance for the chance that Mrs Sanders will agree to treatment with an anti-depressant as part of a regime to manage her pain. I would allow $1,000.
5760 The next item relates to the costs associated with the daily use of Panadol and Ibuprofen, two each per day. Mrs Sanders claimed an amount of $15.99 for a packet of Panadol, which, at two per day, translates to $2.33 per week and totals $1,697.17 over the course of her projected life span. She claimed $8.99 for a packet of Ibuprofen, which, at two per day, translates to $3.14 per week and totals $2,287.17 over the same period. Together, the claims for Panadol and Ibuprofen total $3,984.34. This was a slight increase on the amounts sought in the Statement of Particulars, which were prepared five months before the submissions. The differences can be explained by the fact that a 5% discount rate was used in the Statement of Particulars but a 3% rate was used in the written submissions.
5761 The respondents addressed the claim made in the Statement of Particulars but not that which was advanced in the submissions. They agreed that the former was reasonable but argued that it should be reduced by 15% for the vicissitudes of life. They also quarrelled with the multiplier.
5762 The respondents argued that the correct multiplier was 602.8 but they are wrong. 602.8 is the 5% multiplier for 17 years. In the Statement of Particulars a multiplier of 625.0 was used. That is the 5% multiplier for 18 years. In their written submissions, Mrs Sanders’ lawyers used 728.4 which is the 3% multiplier for 18 years. The 3% multiplier is the appropriate one, since this claim is unaffected by the legislative changes to the TPA and the CLA (WA). Mrs Sanders was 71 at the time the submissions were prepared. Her life expectancy on the medium life tables was 18 years. The correct multiplier is therefore 728.4.
5763 For the reasons given above, there should be no deduction for the vicissitudes of life. But there is no certainty that Mrs Sanders will need to take this medication every day for the rest of her life. For this reason I would reduce the sum to a round figure of $3,500.
5764 Mrs Sanders also claims the cost associated with daily use of Cephalexin 250mg (Keflex) in the amount of $2,702.36. The respondents resisted the claim on the basis that there is no evidence in her treatment records or the opinions of Professor Korda or Associate Professor Rosamilia that she has a chronic infection. This ignores Dr Ruba’s report. Besides, Mrs Sanders’ evidence suggests that there are continuing signs of infection. In her recent affidavit she trialled going off Keflex in May 2017 and within three days she was “in incredible pain”.4562
5765 In any case Keflex was prescribed for prophylactic reasons. I think it reasonable to make an allowance for antibiotics on this basis. The respondents submitted that, if I were to make an award, it should be limited to a maximum of $953, based on a weekly cost of $1.86, the application of the “appropriate” multiplier, and a 15% discount for the vicissitudes of life.
5766 The figure claimed on Mrs Sanders’ behalf was based on costings provided by a registered nurse, included in annexure B to Ms Williams’ report.4563 Those costings are not disputed. According to them, the annual cost of the Keflex at two tablets per day is $193.45 and the weekly amount is $3.71, which is the amount claimed in the submissions. That is the amount I propose to allow. Once the correct multiplier is applied, that produces a sum of $2,702.36. While it is inappropriate to discount for the vicissitudes of life, I do think it is appropriate to discount for the chance that Keflex (or another antibiotic) will not be prescribed for the rest of Mrs Sanders’ life. I would allow $1,000 for this item.
5767 Mrs Sanders also claims the costs associated with the daily use of continence pads and wet wipes at a weekly cost of $17.12 and a total cost of $12,470.21, using a multiplier of 728.4. As the respondents submitted, if she had not received the TVT implant Mrs Sanders would have continued to experience urinary incontinence and required the permanent use of continence pads. If she had undergone an alternative form of surgery, it may have cured the stress incontinence but the cure may not have lasted for the rest of Mrs Sanders’ life. I accept the respondents’ submission that the amount claimed is excessive. I would allow 65% of $17.12 x 728.4 or $8,105.64, which I would round down to $8,000.
5768 The final item is for the cost of annual consultations with an occupational therapist at $733 per consultation. Ms Williams said that an allowance should be made for two hours annually and for life with an occupational therapist “to advise and assist on energy and pain management techniques and equipment”.4564 She costed this at $185 per hour plus an additional two hours in travel at $181.50 per hour including GST, totalling $733 per year and over Mrs Sanders’ projected life span $10,248.59, which corresponds to the amount sought.
5769 The respondents accepted the need for some occupational therapy but urged the Court to accept Ms Borthwick’s recommendation of 10 hours on a one-off basis for education in task modification, sourcing and providing appropriate equipment and training Mrs Sanders in the use of the recommended equipment at a cost of $1,810.
5770 I can see no reason why this item should be an annual cost. Certainly Ms Williams offered none. On the other hand, if the Sanders were to leave their present home, which is only rented, she might well need additional advice. I would allow a total of $2,500.
5771 In summary, I allow the following amounts:
$6,000 for future revision surgeries;
$1,350 for GP consultations;
$1,750 for pain management;
$1,000 for anti-depressants;
$3,500 for over-the-counter pain killers (Panadol and Ibuprofen);
$1,000 for prophylactic antibiotics;
$8,000 for continence pads and wet wipes; and
$2,500 for occupational therapy.
5772 These sums total $25,100.
5773 Mrs Sanders claims a total of $27,220.28 for the cost of future equipment. Many of the items making up the total were controversial.
5774 Mrs Sanders claim consists of the following items at the specified costs, apparently derived from Ms Williams’ report:
Item | Unit Cost | Replacement | Cost pa | Cost pw | Multiplier | Amount |
Recliner | $2,785.00 | 10 years | $278.00 | $5.34 | 728.4 | $3,889.65 |
Equipment for carers | $71.37 | Various | $128.73 | $2.47 | 728.4 | $1,799.15 |
Mobility scooter | $3,500.00 | 10 years | $350.00 | $6.72 | 728.4 | $4,894.85 |
Scooter maintenance & insurance | $385.00 | 1 year | $385.00 | $7.39 | 728.4 | $5,382.87 |
Pressure cushion | $550.00 | 3 years | $183.33 | $3.52 | 728.4 | $2,563.97 |
Car bar | $60.00 | 5 years | $12.00 | $0.23 | 728.4 | $167.53 |
Taxi allowance | $100.00 | 1 year | $100.00 | $1.92 | 728.4 | $1,398.53 |
Electric Adjustable Bed | $2,500.00 | 10 years | $250.00 | $4.80 | 728.4 | $3,496.32 |
Kitchen trolley | $250.00 | 10 years | $25.00 | $0.48 | 728.4 | $349.63 |
Shower chair | $99.00 | 10 years | $9.90 | $0.19 | 728.4 | $138.39 |
Long handled sponge | $25.00 | 1 year | $25.00 | $0.48 | 728.4 | $349.63 |
Easy reacher | $55.00 | 1 year | $55.00 | $1.05 | 728.4 | $764.82 |
Long shoe horn | $10.00 | 3 years | $3.33 | $0.06 | 728.4 | $43.70 |
Long handled vacuum | $299.00 | 3 years | $99.67 | $1.91 | 728.4 | $1,318.40 |
Toilet rail | $474.00 | 10 years | $47.40 | $0.91 | 728.4 | $662.84 |
Total | $27,220.28 |
5775 Ms Borthwick recommended the following items of equipment to enhance Mrs Sanders’ safety and independence:4565
Item | Unit Cost | Life expectancy | Annual cost |
Handy line | $38.90 | 5 years | $7.95 |
Sit stand stool | $150.00 | 10 years | $15.00 |
Mobility scooter | $3,500.00 | 10 years | Not required |
Shower stool | $8.50 | 10 years | $8.50 |
Robot vacuum | $400.00 | 5 years | $80.00 |
Scooter maintenance | $150.00 | 1 year | $150.00 |
Total | $261.45 |
5776 The first claim Mrs Sanders makes is for the cost of a recliner chair and its replacement after 10 years. The respondents correctly submitted that there was no medical evidence to support the claim. But there was medical evidence that I have accepted that Mrs Sanders suffers chronic pain as a result of the complications of her TVT implant and Ms Williams recommended the provision of a recliner lounge chair with stand-up function to provide a comfortable seat for relaxing and pain relief. She said that “multiple positions and stand-up assist function”.4566
5777 But the evidence is that Mrs Sanders already has a recliner chair.4567 As the respondents submitted, Mrs Sanders did not suggest that this chair was purchased to help alleviate her pain. Moreover, no evidence was given to indicate that her recliner was unsuitable. I think it is reasonable, based on Mrs Sanders’ symptoms and prognosis and Ms Williams’ opinion that the respondents contribute to the cost of a replacement in due course. I would allow $2,000.
5778 As with Mrs Gill and for the same reasons, I am not satisfied that the claim for “equipment for carers” is justified.
5779 It can be seen that the provision of a mobility scooter was a recommendation common to the reports of both occupational therapists.
5780 Ms Borthwick recommended the mobility scooter to enable Mrs Sanders to have “easier access to fairs and craft shows, activities that give her inspiration and joy”. But she considered it was unlikely that Mrs Sanders would use it, based on a comment Mrs Sanders made to her regarding her husband’s suggestion that she use a walking frame for a similar purpose. Mrs Sanders told her that she had declined because she did not want to appear old or disabled.4568 This was not the subject of cross-examination and Mrs Sanders did not give evidence that she would use one if supplied.
5781 I am not persuaded that there is any real chance that Mrs Sanders would use a mobility scooter. I therefore disallow the claim for a scooter, scooter maintenance, and pressure cushions (which related to the scooter).
5782 The next item was a “car bar”. Ms Williams recommended it to assist with car transfers.4569 Although she did not mention it in either of her affidavits, Mrs Sanders told Ms Williams that getting in and out of the car was particularly painful.4570 No order was sought or made limiting the use to which that evidence could be put. The effect of s 60 of the Evidence Act is that the evidence is not hearsay; it can be relied upon to support the truth of Mrs Sanders’ account: see, for example, Welsh v R (1996) 90 A Crim R 364 at 369 (Hunt CJ at CL, Newman J and Bell AJ); Bodney v Bennell (2008) 167 FCR 84 at [90]–[93] (Finn, Sundberg and Mansfield JJ). In the light of Mrs Sanders’ sworn account of her pain, the complaint she made to Ms Williams is not surprising and I accept it.
5783 In the circumstances, I am disposed to allow the claim. The amount sought is $167.73, purportedly based on the unit cost of $60 and replacement after five years. By my calculations, this should result in an award of $156.42, being $60 + ($60 x 0.863) + ($60 x 0.744). I therefore allow $156.
5784 The next item is a taxi allowance of $100 per year. The respondents submitted that Mrs Sanders can drive and therefore does not need a taxi allowance. The submission overlooked Ms Williams’ evidence as to the reason for the allowance. That was to deal with occasions when Mrs Sanders was in too much pain to drive herself.4571 Ms Williams’ recommendation was for $100 per month or $1,200 per year. Ms Sanders claimed only $100 per year. That amounts to $1.92 a week and over Mrs Sanders’ lifetime, $1,398.53. I do not consider that unreasonable. I would allow the claim and round it off at $1,399.
5785 Ms Williams also recommended an electric adjustable bed which, she said, would aid in transfers and assist with pain management and “bed mobility”.4572 Ms Sanders informed Ms Williams that moving in and out of a chair, bed, and in and out of the shower all cause pain. While Ms Borthwick made no similar recommendation, she did not argue against it.
5786 The amount of the claim was $3,496.32 based on one bed now at $2,500, and a replacement in 10 years. By my reckoning, the present value of such a claim would be $2,500 + ($2,500 x 0.744) which is $4,360. Since the amount claimed is less than that, I allow it in full. Contrary to the respondents’ submission, there should be no discount for the vicissitudes of life or Mrs Sanders’ co-morbidities. There is no evidence to indicate that they would require the purchase of an electric adjustable bed at some time in the future.
5787 Ms Williams recommended that Mrs Sanders acquire a number of aids: a kitchen or utility trolley to help her transfer items around the home without lifting; a shower chair to allow her to sit to bathe if necessary because of pain; long-handled sponges to help her wash her legs without bending; an easy reacher to eliminate the need to bend to pick up items from the floor; a long shoe horn to help her put on her shoes without bending; a cordless long-handled vacuum stick to enable her to spot clean without bending; and a toilet rail with spacer to assist her to transfer from the toilet. The total amount sought for these aids was $3,627.41.
5788 The applicants claim the cost of these items and their replacement at various intervals in accordance with the evidence in Ms Williams’ report. Rather than engage with the claim, the respondents argued that Ms Borthwick’s recommendations for a handy line, sit stand stool, shower stool, and robot vacuum should be preferred, but made no allowance for replacements, despite Ms Borthwick’s observations about the life expectancy of the items. They submitted that “Mrs Sanders’ unrelated co-morbidities including the issues relating to her right hip, left shoulder, lower back, and obesity, as well as her advancing age will inevitably impact upon her requirement for the use of aids including a kitchen/utility trolley, long-handled sponges, easy reacher, long shoe horn and toilet rail”.4573 I assume that they meant that these conditions would inevitably mean that these items would be required regardless of Mrs Sanders’ mesh-related pain.
5789 The respondents’ position is problematic for two reasons. First, Ms Borthwick did not explain why she did not support Ms Williams’ recommendations. Second, Mrs Sanders is no longer troubled by right hip pain.
5790 Nevertheless, the recommendations for long-handled sponges, an easy reacher, a long shoe horn, and a cordless vacuum cleaner were all made in order to enable Mrs Sanders to avoid bending. Yet, Mrs Sanders did not give evidence that she had any difficulty bending and she told Ms Williams that she was able to bend over. Ms Borthwick noted in her report that she bends forward, flexing towards floor level to retrieve items when required but does not sustain forward flexion for greater than a minute or two at a time. She also said that she demonstrated no difficulty “donning and doffing” footwear or reaching to the ground from a seated or standing positions, although she preferred to place her hand on support when reaching.4574 Mrs Sanders did report to both Ms Williams and Ms Borthwick that she could not squat. She told Ms Borthwick that crouching and squatting trigger increased pain in her groin. But Ms Borthwick said that in her experience anyone who has had a total hip replacement would be unable to complete a full depth squat anyway because acute flexion of the replaced joint is “not medically advised”. In all these circumstances, I am not persuaded that Mrs Sanders has made out a case for the costs of the long-handled sponges, easy reacher, long shoe horn and cordless vacuum cleaner.
5791 On the other hand, Mrs Sanders did give evidence that she had trouble lifting and that she avoids chores requiring lifting. She also told Ms Williams she could only lift very light weights and Ms Borthwick accepted as much.4575 I do not accept the respondents’ submission that Mrs Sanders’ unrelated co-morbidities would have interfered with her ability to do these tasks in any event.
5792 Taking all relevant considerations into account, I would allow $1,500 for aids.
5793 In their submissions the applicants also made a claim on Mrs Sanders’ behalf for the annual cost of upgrading from an economy class to a business class ticket for a return flight from Perth to London Heathrow presumably so that she could visit her daughters in greater comfort than an economy class seat would permit. The claim, which totals $4,695 per year and translates to $90.28, per week and, on the applicants’ calculation $65,759.95 in total, was not included in the statement of particulars.
5794 No provision for this significant expense was made in Ms Williams’ first report or in the appendix in which the various recommendations were costed. The claim was supported, however, by evidence elicited from Ms Williams in her supplementary report in response to direct questioning. Ms Borthwick considered that business class air travel was unwarranted. She said that Mrs Sanders’ standing tolerance was of concern, especially when she had to wait for long periods in airport terminals or at customs but airport assistance, which is free, would take care of this. But Ms Borthwick overlooked the evidence that Mrs Sanders also has trouble sitting.
5795 In her first affidavit, Mrs Sanders deposed that she cannot sit for long periods because it gives her a stabbing pain in her vagina, especially when sitting on a hard chair.4576 She also reportedly told Ms Williams that she was very concerned about the prolonged sitting on the long haul flight to the United Kingdom and that she was unsure whether she could tolerate the flight.
5796 Nevertheless, for the following reasons I am not persuaded that she has made out a case for the cost of an upgrade to business class.
5797 First, the question was not addressed in the medical evidence.
5798 In her first affidavit Mrs Sanders discussed travelling overseas since 2008 including to Canada and the United Kingdom. She said that she did not enjoy the Canadian trip as much as she had hoped because of the pain and that she had trouble “getting around the UK”. She did not say, however, that the long plane trip was problematic, perhaps because unlike in a car, it is possible to move around on a plane.
5799 Besides, Mrs Sanders would still have to sit for a prolonged period in business class. True it is, she could fully recline her seat in business class. But if, as she apparently told Ms Williams, she derives no relief from changing her position, the capacity to recline the seat would make no significant difference. Further, she reportedly told Ms Borthwick that, for their forthcoming trip to the UK, she and her husband had selected seats towards the rear of the aircraft with a space which allowed for better adjustment of posture and would not disturb others as she often rises to go to the toilet.
5800 In any civil proceeding in its original jurisdiction, the Court has the power to make binding declarations of right: FCA Act, s 21(1). In a representative proceeding the Court is expressly empowered to make declarations of liability: FCA Act, s 33Z(1).
5801 In their written closing submissions the applicants submitted that declarations should be made that:
(a) each of the devices had a defect;
(b) each of the devices was not of merchantable quality;
(c) each of the devices was not reasonably fit for the purpose for which it was acquired;
(d) the respondents breached their duty of care by failing to undertake adequate pre-market and post-market evaluation of the devices and also by failing to issue adequate warnings about the risks associated with their use; and
(e) the respondents engaged in misleading conduct both by providing limited warnings and in failing to provide adequate warnings in relation to those risks.
5802 The declarations sought in the originating application were of this nature.
5803 In their interlocutory application filed after the conclusion of the hearing, however, the applicants applied for leave to amend the originating application to also seek declarations that by, marketing, promoting, distributing and supplying the devices without providing “the proper warning set out in CRT.010.021.0001” Ethicon engaged in conduct that was misleading or deceptive or was likely to mislead or deceive the group members. “The proper warning” was in the following terms:
The implant is designed to and will elicit an acute inflammatory reaction followed by a chronic inflammatory response in all patients. The chronic inflammatory response will result in continuously regenerating scar tissue within and surrounding the implant for as long as the implant is in the body. The scar tissue will cause the implant to contract to some degree in all patients. It is not possible to predict the severity of the chronic inflammatory response of any individual patient. There is a risk that in some patients the severity of the chronic inflammatory response will result in adverse outcomes. That risk is not rare. It is not possible to identify in advance the patients who will have that response. At risk patients include healthy patients. The severity of a patient’s chronic inflammatory response can be affected by physical activity and mechanical loading of the pelvic floor. It may be affected by conditions which affect the immune response and healing, including autoimmune and connective tissue disorders. The mechanical forces in the pelvic floor are unknown and may influence the compatibility and function of the implant. The adverse events which may result are:
(a) by reason of excessive contraction of the implant or otherwise, severe chronic pain with potentially life altering consequences with or without psychiatric injury;
(b) damage to or entrapment of nerves in the scar tissue surrounding the implant or otherwise, resulting in severe chronic pain with potentially life altering consequences with or without psychiatric injury;
(c) by reason of excessive contraction of the implant or otherwise, de novo dyspareunia including severe chronic dyspareunia and consequently apareunia;
(d) erosion or extrusion of the implant into the vaginal canal resulting in infection of the tissue surrounding the non-exposed part of the implant which may be difficult to treat resulting in offensive vaginal discharge;
(e) erosion or extrusion of the implant into the vaginal canal resulting in pain suffered by the patient or her sexual partner or both;
(f) erosion or extrusion of the implant into surrounding organs such as the bladder, urethra or rectum with the risk of damage to those organs and pain;
(g) by reason of excessive contraction of the implant or otherwise, difficulty voiding or defecating;
(h) by reason of excessive contraction of the implant or otherwise, de novo stress or urge incontinence,
(i) infection.
Each of the risks may eventuate regardless of the skill of the surgeon. They are not rare. If any of those adverse events eventuate, they will likely persist for so long as the implant remains in the patient. Removal of the implant in whole or in part will not necessarily alleviate the patient’s condition. Removal of part of the implant can be difficult. Removal of the whole of the implant may be practically impossible.
Surgery to remove the whole or part of an implant can result in further scarring and tissue damage resulting in adverse outcomes including severe chronic pain which may not be able to be treated.
Surgery to remove the whole or part of the implant may result in recurrence of the original condition the mesh was designed to treat.
Surgery to remove the whole or part of the implant may make remedying the original condition of the patient which the implant was designed to alleviate more difficult to treat.
Successful treatment of erosion by excision will not necessarily prevent further erosion or other future adverse outcomes.
Whether the implant is or remains inert for the remainder of a patient’s life is unknown. The implant is made from polypropylene. It is known that polypropylene undergoes oxidative degradation in vivo. Degradation may affect the mechanical properties of the implant and the severity of the chronic inflammatory response to it.
Use of the implant may result in fraying and fragmentation of the implant or the release of polypropylene particles which may affect the severity of the chronic inflammatory response to the implant.
The adverse outcomes may not materialise until years after implantation and the risk of the adverse outcomes are life-long risks for so long as the implant remains in the patient.
Insufficient clinical testing of the implant has been undertaken to determine the safety of the implant and its long-term effectiveness.
The long-term safety of the implant has not been studied and is unknown.
The long-term efficacy of the implant has not been studied and is unknown.
Whether the safety and efficacy of the implant is the same as or better than non-mesh treatment alternatives has not yet been established.
5804 The Court’s discretion is very broad. In general, however, a declaration will not be made unless the question to which it relates is real, rather than hypothetical; the person raising it has a real interest in doing so; and there is a proper contradictor, that is, someone who has a true interest in opposing the declaration: Forster v Jododex Australia Pty Limited (1972) 127 CLR 421 at 437–438 (Gibbs J).
5805 It was not suggested, that these conditions were not satisfied in the present case. Rather, the respondents contended that the declarations sought by the applicants lacked utility. They accepted that declarations may be appropriate where it is established that the respondent intends to take action that will amount to a contravention of a law or an infringement of a duty or where the proceeding involves a matter of public interest and the circumstances call for the Court to mark its disapproval of the contravening conduct. They argued, however, that this was not such a case. In the event that the Court was of the view that declaratory relief were appropriate, they submitted that the form of the declarations will depend on the Court’s findings and should therefore await the Court’s determination of the common issues.4577
5806 Declaratory relief is appropriate. The proceeding unquestionably involves a matter of public interest. Proceedings brought under the TPA are concerned with the protection of the public. Declarations can mark the Court’s disapproval of the contravening conduct. Damages are merely compensatory. Declarations may have a deterrent effect, if not on the respondents, then on others in an analogous position: Australian Competition and Consumer Commission v Murray Goulburn Co-Operative Co Limited [2018] FCA 1964 at [15] (Beach J). There is no reason why, if declarations about the contravening conduct were to be made, declarations should not also be made about the negligent conduct.
5807 I see no point, however, in declarations that the various devices have a defect or are not of merchantable quality or fit for the purpose for which they are acquired. Without identifying the reasons the devices were defective, declarations in the form in which they appear in the originating application would be meaningless. Unlike the form of declaration made by Edelman J in ACCC v Reckitt Benckiser (No 4), it could scarcely be said that the proposed form in the present case was “closely tailored” to the contravening conduct. Based on my findings, however, the parties should be able to agree on an appropriate form.
5808 The applicants also sought an injunction pursuant to s 80 of the TPA or s 232 of the ACL restraining the respondents from supplying, distributing, marketing or promoting the devices without providing the proper warning. Leave to amend the originating application to seek that injunction was granted over the respondents’ opposition on 9 April 2018: see Gill v Ethicon SÁRL [2018] FCA 470.
5809 Section 80 is inapplicable. The effect of the transitional provisions in the Trade Practices Amendment (Australian Consumer Law) Act (No. 2) 2010 (Cth) is that any proceedings for an injunction under or in relation to the TPA that were commenced but not concluded before 1 January 2011 are taken, after that date, to be proceedings under s 232 of the ACL: see Sch 7 item 6(2).
5810 Section 232 relevantly provides that:
(1) A court may grant an injunction, in such terms as the court considers appropriate, if the court is satisfied that a person has engaged … in conduct that constitutes or would constitute:
(a) a contravention of a provision of Chapter 2, 3 or 4;
…
(2) The court may grant the injunction in application by the regulator or any other person.
…
(4) The power of the court to grant an injunction under subsection (1) restraining a person from engaging in conduct may be exercised:
(a) whether or not it appears to the court that the person intends to engage again, or to continue to engage, in conduct of a kind referred to in that subsection;
(b) whether or not the person has previously engaged in conduct of that kind; and
(c) whether or not there is an imminent danger of substantial damage to any other person if the person engages in conduct of that kind.
(5) Without limiting subsection (1), the court may grant an injunction under that subsection restraining a person from carrying on a business or supplying goods or services (whether or not a part of, or incidental to, the carrying on of another business):
(a) for a specified period: or
(b) except on specified terms and conditions.
…
5811 Chapter 2 includes the prohibition against engaging in misleading or deceptive conduct (s 18 of the ACL, formerly s 52 of the TPA), while chapter 3 includes the guarantees that goods are of acceptable quality (s 54, formerly 74D of the TPA) and fit for the disclosed purpose (s 55, formerly s 74B of the TPA), and also the liability to pay compensation for injuries caused by goods with a safety defect (s 138, formerly s 74AD of the TPA).
5812 It is trite to observe that the power conferred on the Court by s 232, like that of its predecessor, s 80 of the TPA, is extremely broad. Nevertheless, besides being appropriate in its terms, the proposed injunction should be enforceable, any breach should be detectable, and there must be utility in the making the order. An injunction will not be appropriate in its terms if, on its face, it operates over a range of conduct which includes conduct falling outside the relationship required by the section: ICI Australia Operations Pty Ltd v Trade Practices Commission (1992) 38 FCR 248 at 267 (Gummow J). That relationship is between the contraventions of the ACL and the respondent’s or respondents’ conduct.
5813 The respondents opposed the injunction the applicants sought. Their arguments were advanced in opposition to the applicants’ interlocutory application. Although I indicated in my reasons for judgment when granting leave to amend that I would give the respondents the opportunity to supplement those arguments were they to seek it, they never applied to do so. The respondents’ arguments, as I laid out in Gill (No 1), were as follows:
(a) the proposed form of injunction purports to usurp the role of the TGA in regulating the provision of medical devices in Australia;
(b) if relief is pressed, the Commonwealth would have been a necessary party and should have been joined at the outset;
(c) the TGA is better placed to supervise the content of warnings necessary to accompany the sale of a medical device and is already doing so;
(d) the form and content of the proposed warning is a “lawyers’ construct based on the applicants’ pleaded case” and should not be the basis on which sales of the devices are permitted;
(e) it would be futile to grant the application because the pre-conditions for the making of the order have not been established and, in the context of a detailed regulatory regime enforced by the TGA, the injunction could not be said to have “the character of enforcing present compliance or inducing future compliance”: Truth About Motorways Pty Limited v Macquarie Infrastructure Investment Management Limited (2000) 200 CLR 591 at [79]–[80] (Gummow J), adopted by the Full Court in Australian Competition and Consumer Commission v Dataline.Net.Au Pty Ltd (In liq) (2007) 161 FCR 513 at [98] (Moore, Dowsett and Greenwood JJ); and
(f) the proposed relief would not be for the benefit of the applicants or the group members, actual or prospective.
5814 None of these arguments has merit.
5815 With respect to the first two arguments, I maintain what I said my interlocutory judgment at [76]:
The contentions that the job of regulating the sale of the products should be left to the regulator and that any such order could not be made without adding the regulator as a party are spurious. The TGA does not have an exclusive role in protecting the safety of consumers. As Selway J observed in Australian Competition and Consumer Commission v 4WD Systems Pty Ltd [2003] FCA 850; (2003) 200 ALR 491; (2003) 59 IPR 435 at [217], “[t]he purpose of an appropriately drafted injunction may be merely to reinforce to the market place that the restrained behaviour is unacceptable”.
5816 As for the third argument, the TGA might well be better placed to supervise the content of warnings, but the evidence indicates that it did nothing about them until after Ethicon stopped manufacturing and selling the POP devices and TVT Secur was no longer sold in Australia. Not even the FDA alerts were sufficient to prompt an investigation before then. Besides, I have found that not even the most recent IFUs are adequate to protect women from the risks to which the SUI devices still on the ARTG expose them.
5817 As for the fourth argument, it is true that the proposed warning has been crafted by lawyers but that, of itself, is no basis for refusing relief. It is difficult to conceive of any injunction that is not the work of lawyers.
5818 Turning to the fifth argument, the only relevant precondition to the exercise of the power is that the Court is satisfied that the respondents (or one or more of them) engaged in conduct that constitutes or would constitute a contravention of a provision of Chapter 2, 3 or 4 of the ACL. Having regard to my findings on the statutory causes of action, this precondition is satisfied. The Court has the power to grant an injunction. The only question is whether it should exercise that power to make an order in the terms proposed. The quote from Truth About Motorways is selective. This is the full context of Gummow J’s observation with the substantive omissions emphasised:
In many cases, the remedy sought under s 80 for a prohibitory injunction would have the character of enforcing present compliance or inducing future compliance with the norm of conduct imposed by s 52.
5819 In any case, I do not accept that the proposed order does not have “the character of enforcing present compliance or inducing future compliance”.
5820 Finally, as I said in Gill (No 1) at [78]–[79], injunctive relief need not be of benefit to the applicants or the group members:
78 As the Hon Dyson Heydon AO observes in his commentary on the Act (published by Thomson Reuters) at [280.230], the words “any other person” mean what they say. In Phelps v Western Mining Corporation Ltd (1978) 20 ALR 183; 33 FLR 327 the Full Court of this Court rejected an argument that only persons who are affected by a contravention of Pt V of the Trade Practices Act (the consumer protection provisions now contained in the ACL) could seek relief under s 80, holding that the words were to be given an unlimited ambit. An applicant need have no direct or special interest in the proceeding. A competitor of an alleged contravener, for example, may seek injunctive relief. The High Court endorsed this position in Truth About Motorways. See, for example, at [13]–[15] (Gleeson CJ and McHugh J). In any case, the applicants have a direct and special interest in the present proceeding.
79 The enforcement of the Competition and Consumer Act through relief of this kind serves a public purpose. That purpose includes prevention and deterrence of undesirable trade practices. The public interest may justify the making of an injunction to restrain contravening conduct even in circumstances where the applicants have no personal interest and, as Toohey J observed in Trade Practices Commission v Mobil Oil Australia Ltd (1984) 4 FCR 296 at 300, even when the contravener is unlikely to engage in the offending conduct again. The power may be exercised in order “to mark the court’s disapproval” of the contravener’s conduct: ibid. Contrary to the respondents’ position, the TGA is not the sole guardian of the public interest when it comes to medical devices.
5821 It remains for me to consider whether the terms of the proposed injunction are appropriate.
5822 Having regard to my findings, the proposed injunction would not operate over a range of conduct which includes conduct falling outside the relationship required by the section. It would be enforceable. Moreover, any breach would be detectable. The respondents did not suggest otherwise. But it is too prescriptive. In addition, parts of it are inappropriate, particularly those parts which refer to safety and efficacy studies of the devices. Although they would have been appropriate when the devices were launched, I am not satisfied that they are appropriate now. Furthermore, the “proper warning” included the risk of pain to the patient’s sexual partner when no such complaint was included in the applicants’ pleading and “difficulty defaecating” when that risk was only pleaded in the case of the POP devices. With suitable modifications, however, I consider that it would be appropriate to enjoin the respondents from supplying, distributing, marketing or promoting any of the SUI devices still on the ARTG without including in the IFU for each of those devices and any promotional material relating to those devices a warning in the following terms or to the following effect:
The mesh is designed to, and will invariably elicit in all patients, an acute inflammatory reaction followed by a chronic inflammatory response. The chronic inflammatory response will result in continuously regenerating scar tissue within and surrounding the implant for as long as the implant remains in the body. The scar tissue will cause the mesh to contract to some degree in all patients. It is not possible to predict the severity of the chronic foreign body response in any individual patient. In some patients the chronic inflammatory response will have adverse effects. It is not possible to identify in advance the patients who will experience those effects, although some patients are at greater risk than others. At-risk patients include healthy patients. The severity of a patient’s chronic inflammatory response can be affected by physical activity and mechanical loading of the pelvic floor. It can also be affected by conditions which affect the immune response and healing, such as autoimmune and connective tissue disorders. The mechanical forces in the pelvic floor may influence the compatibility and function of the implant.
The adverse events which may result include:
(a) infection;
(b) erosion of the mesh into the vaginal canal resulting in infection which may be difficult to treat, cause offensive vaginal discharge and pain;
(c) erosion of the mesh into surrounding organs such as the bladder, urethra or rectum which may cause pain and damage those organs;
(d) damage to nerves in the scar tissue surrounding the implant or elsewhere;
(e) chronic pain, which may be severe;
(f) dyspareunia, which may be severe and may become chronic;
(g) apareunia;
(h) leg weakness;
(i) de novo or recurrent incontinence;
(j) difficulty voiding; and
(k) vaginal discharge.
Adverse events may occur years after implantation. The risk will endure for as long as the implant remains in the patient.
Each of these events may occur regardless of the skill of the surgeon.
While the true incidence of these complications is unknown, they are not rare.
Removal of the implant in whole or in part will not necessarily alleviate the patient’s symptoms. Removal of part of the implant can be difficult. Removal of the whole of the implant may be practically impossible. Surgery to remove the whole or part of an implant can result in further scarring and tissue damage which, in turn, may have adverse outcomes including severe chronic pain which may not be able to be satisfactorily treated. Surgery to remove the whole or part of the implant may also result in recurrence of stress urinary incontinence.
Removal of the eroded mesh will not necessarily prevent further erosions or other adverse events.
5823 Since all of the POP devices have been removed from the market and are no longer on the ARTG, I am not satisfied that there is any utility in extending the injunction to, or making a similar injunction with respect to, those devices.
5824 The parties should bring in short minutes of order giving effect to these reasons by 14 February 2020. Those orders should include both orders with respect to the disposition of the applicants’ claims and those required by s 33ZB of the FCA Act. The length of time I have allowed takes into account the length of the judgment and the impending summer holiday period. I encourage the parties, however, not to wait until the last moment but to do their best to ensure that orders can be made as soon as possible.
5825 By 20 December 2019 I require each of the applicants to notify the respondents and the Court whether she elects to receive damages in accordance with the relevant provisions of the TPA or at common law, as modified by local legislation where applicable.
5826 In the event that the parties are unable to agree on the form of the orders or all the orders, including the form of the common questions and the answers to them for the purpose of the s 33ZB orders, a timetable for the filing and exchange of submissions should be forwarded to my chambers by 17 January 2020, with a view to a hearing in the week commencing 10 February 2020.
5827 I will grant liberty to apply on two days’ notice.
I certify that the preceding five thousand, eight hundred and twenty-seven (5,827) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Katzmann. |
Associate:
SBM.010.001.0001 at 0015
AID.010.002.0001_8
CRT.500.007.0001
T6039–40 (TRA.500.076.0001 at 0003-0004)
ETH.MESH.08219161; ETH.MESH.08219150; MSC.010.170.0001
ETH.MESH.08219159; ETH.MESH.08219144
T2 (TRA.500.001.0001_2 at 0002_2)
T69 (TRA.500.001.0001_2 at 0069_2)
Collinet at 22 (EXP.020.005.0001_4 at 0022_4)
Korda 1 at 5 (EXP.010.078.0001_3 at 0006_3)
Hinoul at [16] (LAY.020.001.0001_4 at 0007_4])
Hinoul at [19(f)] (LAY.020.001.0001_4 at 0009_4)
Hinoul at [18] (LAY.020.001.0001_4 at 0007_4)
Hinoul at [19(f)] (LAY.020.001.0001_4 at 0009_4)
Hinoul at [19(g)-(i)] (LAY.020.001.0001_4 at 0009_4)
ETH.MESH.03913758
Hinoul at [19(f)] (LAY.020.001.0001_4 at 0009_4)
Hinoul at [217] (LAY.020.001.0001_4 at 0088_4)
ETH.MESH.09625725 at 5731
ETH.MESH.09625725 at 5818
Respondents’ written closing submissions (RWS) ch 7 at [12] (SBM.020.002.0108 at 0112)
RWS ch 7 at [15]–[24] (SBM.020.002.0108 at 0119)
Hinoul at [218] (LAY.020.001.0001_4 at 0088_4)
Usher F et al, “Marlex Mesh, a New Plastic Mesh for Replacing Tissue Defects” (1959) 78 AMA Arch Surg 138–145 (SHI.MESH.00033484)
Klinge at [41]-[43] (LAY.010.022.0001_2 at 0008_2)
Hinoul [226] (LAY.020.001.0001_4 at 0091_4)
Klinge at [48] (LAY.010.022.0001_2 at 0011_2); see also Hinoul at [226]–[228] (LAY.020.001.0001_4 at 0091_4)
CRT.500.007.0001; AWS ch 3 (SBM.010.005.0125_2); RWS ch 3 and 4 (SBM.020.026.0001)
T581 (TRA.500.007.0001_2 at 0073_2)
T582 (TRA.500.007.0001_2 at 0074_2)
T583 (TRA.500.007.0001_2 at 0075_2)
T582 (TRA.500.007.0001_2 at 0074_2)
Applicants’ written closing submissions (AWS) ch 3 at [6] (SBM.010.005.0125_2 at 0126_2); CRT.010.001.0001_2
SHI.MESH.00013425
Blaivas 1 at 8 (EXP.010.026.0001_3 at 0010_3); Hinoul at [48] (LAY.020.001.0001_4 at 0020_4)
Rosamilia 1 at 7 (EXP.020.026.0001_3 at 0009_3)
Hinoul at [49(b)(ii)] (LAY.020.001.0001_4 at 0021_4); Rosamilia 1 at 6 (EXP.020.026.0001_3 at 0008_3)
Blaivas 1 at 8 (EXP.010.026.0001_3 at 0010_3)
Oxford Concise Medical Dictionary (Oxford University Press, 8th ed, 2010) at 767
PUB.MESH.00001204; although see ETH.MESH.09917864 at 7866
ETH.MESH.09917864 at 7866; T592–93 (TRA.500.007.0001_2 at 0084_2)
PUB.MESH.00001204 at 1204; see also ETH.MESH.09917864 at 7866
T713 (TRA.500.008.0001_2 at 0102_2); PUB.MESH.00009395 at 9408
Chaikin D, Rosenthal J and Blaivas J, “Pubovaginal fascial sling for all types of stress urinary incontinence: Long-term analysis” (1998) 160 J Urol 1312-1316 (ETH.MESH.09917864)
Ibid at 1316 (ETH.MESH.09917864 at 7868)
T599 (TRA.500.007.0001_2 at 0091_2)
Barone W, Abramowitch S and Moalli P, “Host Response to Biomaterials for Pelvic Floor Reconstruction” in Host Response to Biomaterials (Elsevier Inc, 2015) at 386 (SHI.MESH.00044785 at 4796)
AWS Ch 3 at [16]–[17] (SBM.010.005.0125_2 at 0128); Blaivas 1 at 9–11 (EXP.010.026.0001_3 at 0011–13)
T713 (TRA.500.008.0001_2 at 0102_2)
Hinoul at [347] (LAY.020.001.0001_4 at 0134_4); Petros P, “Creating a gold standard surgical device: scientific discoveries leading to TVT and beyond” (2015) Int Urogynecol J (PUB.MESH.00001886)
Petros P, “Creating a gold standard surgical device: scientific discoveries leading to TVT and beyond” (2015) Int Urogynecol J (PUB.MESH.00001886 at 1887); see also Iglesia C, Fenner D and Brubaker L, “The use of mesh in gynecologic surgery” (1997) 8 Int Urogynecol J 105–115 (ETH.MESH.00155061)
Hinoul at [349] (LAY.020.001.0001_4 at 0135_4); Petros P and Ulmsten U, “The combined intravaginal sling and tuck operation. An ambulatory procedure for stress and urge incontinence” (1990) 69 Acta Obstet Gynecol Scand 53–59 (PUB.MESH.00000185)
Hinoul at [349] (LAY.020.001.0001_4 at 0135_4); Petros P and Ulmsten U, “The combined intravaginal sling and tuck operation. An ambulatory procedure for stress and urge incontinence” (1990) 69 Acta Obstet Gynecol Scand 53–59 (PUB.MESH.00000185)
Ulmsten U and Petros P, “Intravaginal Slingplasty (IVS): an ambulatory surgical procedure for treatment of female urinary incontinence”(1995) 29 Scand J Urol Nephrol 75–82 (ETH.MESH.01271090)
Ibid at 78 (ETH.MESH.01271090 at 1093)
Ibid at 75 (ETH.MESH.01271090 at 1091)
Ibid at 81 (ETH.MESH.01271090 at 1097)
Ulmsten L et al, “An ambulatory surgical procedure under local anesthesia for treatment of female urinary incontinence” (1996) 7 Int Urogynecol J 81–86 (ETH.MESH.19935677)
Dunn 1 at 5 (EXP.010.031.0001_3 at 0007_3)
MacLean 1 at 19 (EXP.020.042.0001_4 at 0019_4)
Dunn 1 at 5 (EXP.010.031.0001_3 at 0007_3)
Dunn 1 at 6 (EXP.010.031.0001_3 at 0008_3)
MacLean 1 at 21 (EXP.020.042.0001_4 at 0021_4)
Dunn 1 at 7 (EXP.010.031.0001_3 at 0009_3); ETH.MESH.06678663
ETH.MESH.02268619 at 8620
ETH.MESH.02268619 at 8620
ETH.MESH.12824382 at 4384
ETH.MESH.02607272 at 7688
ETH.MESH.00309254 at 9255
ETH.MESH.06001408
ETH.MESH.06001408
ETH.MESH.03535750
ETH.MESH.06001408
ETH.MESH.10984368 at 4381–2; Hinoul at [77(a)] (LAY.020.001.0001_4 at 0040_4)
Hinoul at [80] (LAY.020.001.0001_4 at 0041_4)
ETH.MESH.10984368 at 4382
ETH.MESH.10681859 at 1866
ETH.MESH.10589402
AWS ch 7 at [56] (SBM.010.005.0250_2 at 0261_2)
ETH.MESH.01189423 at 9426
AID.010.002.0001_8
AID.010.002.0001_8
AID.010.002.0001_8
Hinoul at [241(a)(ii)] (LAY.020.001.0001_4 at 0094_4)
Allman 2 at 3 (EXP.010.180.0001_3 at 0004_3)
ETH.MESH.08476210 at 6279–80
ETH.MESH.11803730
Hinoul at [232] (LAY.020.001.0001_4 at 0092_4); see also ETH.MESH.01317180 at 7209
Hinoul at [85]–[86] (LAY.020.001.0001_4 at 0042_4)
AID.010.002.0001_8
AID.010.002.0001_8
Hinoul at [87] (LAY.020.001.0001_4 at 0043_4)
ETH.MESH.02340568 at 0577; Hinoul at [87]–[88] (LAY.020.001.0001_4 at 0043_4)
ETH.MESH.01189423 at 9429–9433
ETH.MESH.00753158 at 3159
Hinoul at [89]–[90] (LAY.020.001.0001_4 at 0043_4)
ETH.MESH.01189423 at 9427
Nambiar A, Cody J and Jeffery S, “Single-incision sling operations for urinary incontinence in women” The Cochrane Collaboration (John Wiley & Sons Ltd, 2014) (SHI.MESH.00004889 at 4897)
Ibid (SHI.MESH.00004889 at 4895)
ETH.MESH.08219407
JJM.MESH.00082765 at 2766
AID.010.002.0001_8
ETH.MESH.01189423 at 9426
RWS Ch 1 at [23] (SBM.020.002.0015 at 0020)
AID.010.015.0001
Hinoul at [95(a)] (LAY.020.001.0001_4 at 0044_4)
AID.010.002.0001_8
AID.010.002.0001_8
Hinoul at [96] (LAY.020.001.0001_4 at 0045_4)
Hinoul at [97] (LAY.020.001.0001_4 at 0045_4)
AID.010.015.0001; Hinoul at [98] LAY.020.001.0001_4 at 0045_4)
Hinoul at [99] LAY.020.001.0001_4 at 0046_4)
ETH.MESH.10681773 at 1779
CRT.500.007.0001 at 0007
Deprest at [118(a)] (EXP.020.006.0001_4 at 0025_4)
Korda 1 at 8 (EXP.010.078.0001_3 at 0010_3)
Korda 1 at 9–10 (EXP.010.078.0001_3 at 0011_3–0012_3)
Korda 1 at 10 (EXP.010.078.0001_3 at 0012_3)
Deprest at [118(b)] (EXP.020.006.0001_4 at 0025_4)
CRT.500.007.0001 at 0008
Korda 1 at 13 (EXP.010.078.0001_3 at 0015_3)
Korda 1 at 15 (EXP.010.078.0001_3 at 0017_3)
Korda 1 at 13 (EXP.010.078.0001_3 at 0015_3)
Korda 1 at 17 fn 54 (EXP.010.078.0019_3)
Korda 1 at 17 fn 54 (EXP.010.078.0019_3)
Korda 1 at 17 (EXP.010.078.0019_3)
Rosamilia 1 at 15 (EXP.020.026.0001_3 at 0017_3)
Rosamilia 1 at 16 (EXP.020.026.0001_3 at 0018_3)
Korda 1 at 29 (EXP.010.078.0001_3 at 0031_3)
Chughtai 1 at 5 (EXP.010.001.0001_3 at 0006_3)
T1157–60 (TRA.500.014.0001_2 at 0030_2-0033_2); Chughtai 1 at 7 (EXP.010.001.0001_3 at 0008_3); Korda 1 at 29 (EXP.010.078.0001_3 at 0031_3)
Oxford Concise Medical Dictionary (Oxford University Press, 8th ed, 2010) at 588
Korda 1 at 28–29, 35–37 (EXP.010.078.0001_3); Chughtai 1 at 7 (EXP.010.001.0001_3 at 0008_3)
Korda 1 at 36–38 (EXP.010.078.0001_3 at 0038_3)
See: Chughtai 1 at 7 (EXP.010.001.0001_3 at 0008_3); Korda 1 at 28 (EXP.010.078.0001_3 at 0030_3)
AWS ch 4 at [14] (SBM.010.005.0136_2 at 0140_2)
Korda 1 at 27 (EXP.010.078.0001_3 at 0029_3)
Hinoul at [26] (LAY.020.001.0001_4 at 0012_4)
Hinoul at [544] (LAY.020.001.0001_4 at 0262_4)
ETH.MESH.06400509 at 0580
AID.010.004.0001_6; Hinoul at [241(a)(i)], [546] (LAY.020.001.0001_4)
Hinoul at [546] (LAY.020.001.0001_4 at 0263_4)
Statement of Facts (CRT.500.007.0001)
AID.010.002.0001_8
Hinoul at [543] (LAY.020.001.0001_4 at 0262_4)
Hinoul at [547] (LAY.020.001.0001_4 at 0236_4); see also ETH.MESH.20070090 at 0106 and ETH.MESH.15905318 at 5331
ETH.MESH.22134106 at 4109
RWS annexure 4B (SBM.020.008.0001 at 0002)
JJM.MESH.00614902; JJM.MESH.00616884
AID.010.002.0001_8
AID.010.002.0001_8
Hinoul at [110] (LAY.020.001.0001_4 at 0049_4)
Hinoul at [112] (LAY.020.001.0001_4 at 0049_4)
Hinoul at [114] (LAY.020.001.0001_4 at 0049_4)
ETH.MESH.03932906
Berrocal J et al, “Conceptual advances in the surgical management of genital prolapse” (2004) 33 J Gynecol Obstet Biol Reprod 577–587 at 577 (ETH.MESH.00659678 at 9678)
Stedman’s Medical Dictionary (Lippincott, Williams and Wilkins, 2006) cited in Haylen B et al, “An International Urogynecological Association (IUGA) International Continence Society (ICS) joint terminology and classification of the complications related directly to the insertion of prostheses (meshes, implants, tapes) & grafts in female pelvic floor surgery” (2011) 22 (3) Int Urogynecol J 1–15 at 6 (ETH.MESH.00187674 at 7677)
Haylen B et al, “An International Urogynecological Association (IUGA) International Continence Society (ICS) joint terminology and classification of the complications related directly to the insertion of prostheses (meshes, implants, tapes) & grafts in female pelvic floor surgery” (2011) 22 (3) Int Urogynecol J 1–15 at 6–7 (ETH.MESH.00187674 at 7677–7678)
See, for example, ETH.MESH.00000918
ETH.MESH.03932906
ETH.MESH.00659678 at 9680
ETH.MESH.01155610 at 5611
ETH.MESH.01155610
Hinoul at [164] (LAY.020.001.0001_4 at 0066_4); ETH.MESH.02341522
RWS annexure 4B (SBM.020.008.0001)
Fatton B et al, “Transvaginal repair of genital prolapse: preliminary results of a new tension-free vaginal mesh (Prolift™ technique) – a case series multicentric study” (2007) 18 Int Urogynecol J 743–752 (ETH.MESH.00093734 at 3735)
AWS ch 8 at [77] (SBM.010.005.0296_2 at 0316_2); Collinet at 28 (EXP.020.005.0001_4 at 0028_4); Korda 3 at 21 (EXP.010.129.0001_5 at 0022_5)
Rosamilia 1 at 23 (EXP.020.026.0001_3 at 0025_3)
Rosamilia 1 at 27 (EXP.020.026.0001_3 at 0029_3)
Fatton B et al, “Transvaginal repair of genital prolapse: preliminary results of a new tension-free vaginal mesh (Prolift™ technique) – a case series multicentric study” (2007) 18 Int Urogynecol J 743–752 (ETH.MESH.00093734 at 3742)
AID.010.002.0001_8
AID.010.002.0001_8
AID.010.002.0001_8
ETH.MESH.09746846
ETH.MESH.00743865 at 3865
Hinoul at [563] (LAY.020.001.0001_4 at 0269_4)
ETH.MESH.06288861 at 8872
ETH.MESH.06288861 at 8871
ETH.MESH.06288861 at 8871
Korda 3 at 4 [EXP.010.129.0001_5 at 0005_5)
Korda 3 at 4 [EXP.010.129.0001_5 at 0005_5)
Agur 2 at 2 (EXP.010.152.0001_3 at 0004_3)
Agur 2 at 2 (EXP.010.152.0001_3 at 0004_3)
Iglesia C et al, ‘Vaginal Mesh for Prolapse: A Randomized Controlled Trial’ (2010) 116 Obstet Gynecol 293–303 (ETH.MESH.00013799)
Fifth Further Amended Statement of Claim at [45(d)–(p)]; see also AWS ch 9 at [2] (SBM.010.005.0379_2 at 0379_2)
Fifth Further Amended Statement of Claim at [23(d)–(g)]
T3085–86 (TRA.500.039.0001_3 at 0084_3–0085_3)
T3088 (TRA.500.039.0001_3 at 0087_3)
T3088 (TRA.500.039.0001_3 at 0087_3)
T3089 (TRA.500.039.0001_3 at 0088_3)
T3089–94 (TRA.500.039.0001_3 at 0088_3–0089_3)
T3087–90; 92–93 (TRA.500.039.0001_3 at 0086_3; 0091_3–0092_3)
T3089 (TRA.500.039.0001_3 at 0088_3)
T6061 (TRA.500.076.0001 at 0025)
ETH.MESH.14489208 at 9220
T3088 (TRA.500.039.0001_3 from 0087_3)
T3372 (TRA.500.043.0001_3 at 0018_3)
Collinet 1 at 5 (EXP.020.005.0001_2 at 005_2)
Collinet 1 at 7 (EXP.020.007.0001_2 at 005_2)
EXP.020.008.0001; Roovers 1 at 2 (EXP.020.033.0001_3 at 0004_3)
Deprest 1 (EXP.020.006.0001_3)
T3916, 3920 (TRA.500.049.0001_2 at 0032_2, 0036_2)
Roovers 1 at 4 (EXP.020.033.0001_3 at 0006_3)
EXP.020.022.0001
Rosamilia 1 at 2 (EXP.020.026.0001_3 at 0005_3)
Deprest at [283] (EXP.020.006.0001_4 at 0061_4); T4733 (TRA.500.059.0001 at 0016)
Chughtai 1 at 11 (EXP.010.001.0001_3 at 0012_3)
Deprest at [286] (EXP.020.006.0001_4 at 0062_4)
Deprest at [284] (EXP.020.006.0001_4 at 0061_4)
Hinoul H, Goossens A and Roovers J, “Factors determining the adoption of innovative needle suspension techniques with mesh to treat urogenital prolapse: a conjoint analysis study” (2010) 151(2) Eur J Obstet Gynecol Reprod Biol 212–216 (ETH.MESH.03906063 at 6066)
Korda 3 at 8 (EXP.010.129.0001_5 at 0009_5)
Deprest at [284] (EXP.020.006.0001_4 at 0061_4)
Deprest at [284] (EXP.020.006.0001_4 at 0061_4)
T3239–40 (TRA.500.041.0001_3 at 0052_3–0053_3)
Korda 1 at 71 (EXP.010.078.0001_3 at 0073_3)
Klinge at [310] (LAY.010.022.0001_2 at 0111_2)
Klosterhalfen 1 at [165] (LAY.010.014.0001_6
Blaivas 1 at 14 (EXP.010.026.0001_3 at 0016_3)
ETH.MESH.03719171 at 9173
Chughtai 2 at 7–8 (EXP.010.112.0001_3 at 0008_3–0009_3); Deprest at [291] (EXP.020.006.0001_4 at 0063_3)
Collinet P et al, “Transvaginal mesh technique for pelvic organ prolapse repair: mesh exposure management and risk factors” (2006) 17 Int Urogynecol J 315–320 at 319–320 (ETH.MESH.02232574 at 2579–2580)
Korda 1 at 71 (EXP.010.078.0001_3 at 0073_3)
Korda 3 at 27 (EXP.010.129.0001_5 at 0028_5)
Deprest 1 at [285] (EXP.020.006.0001_4 at 0062_4)
Korda 3 at 27 (EXP.010.129.0001_5 at 0028_5)
Deprest at [289] (EXP.020.006.0001_4 at 0062_4)
Korda 1 at 90 (EXP.010.078.0001_3 at 0092_3)
Korda 1 at 70 (EXP.010.078.0001_3 at 0072_3)
Korda 1 at 89 (EXP.010.078.0001_3 at 0091_3)
Korda 1 at 70 (EXP.010.078.0001_3 at 0072_3); Korda 3 at 23 (EXP.010.129.0001_5 at 0024_5)
Korda 1 at 89 (EXP.010.078.0001_3 at 0091_3)
Chughtai 1 at 10 (EXP.010.001.0001_3 at 0011_3)
Chughtai 1 at 11 (EXP.010.001.0001_3 at 0012_3)
Deprest at [346] (EXP.020.006.0001_4 at 0072_4)
Korda 3 at 14 (EXP.010.129.0001_5 at 0015_5)
Korda 3 at 14–15 (EXP.010.129.0001_5 at 0015_5)
Korda 3 at 15 (EXP.010.129.0001_5 at 0016_5)
T1187 (TRA.500.014.0001_2 at 0060_2)
Ostergard D, “Polypropylene Vaginal Mesh Grafts in Gynecology” (2010) 116(4) Obstet Gynecol 962–966 (ETH.MESH.00544274 at 4276)
Korda 1 at 90 (EXP.010.078.0001_3 at 0092_3)
Korda 1 at 87 (EXP.010.078.0001_3 at 0089_3)
Korda 1 at 68 (EXP.010.078.0001_3 at 0070_3)
Korda 1 at 70 (EXP.010.078.0001_3 at 0072_3)
Blaivas 1 at 13 (EXP.010.026.0001_3 at 0015_3)
ETH.MESH.25280424 at 0468
PUB.MESH.00009294 at 9299
Korda 1 at 74 (EXP.010.078.0001_3 at 0076_3)
Korda 1 at 75 (EXP.010.078.0001_3 at 0077_3)
Chughtai 1 at 10 (EXP.010.001.0001_3 at 0011_3)
Collinet at 25 (EXP.020.005.0001_4 at 0025_4)
Deprest at [295]–[296] (EXP.020.006.0001_4 at 0064_4)
Collinet P et al, “Transvaginal mesh technique for pelvic organ prolapse repair: mesh exposure management and risk factors” (2006) 17 Int Urogynecol J 315–320 (ETH.MESH.00071591)
See, for example, Rosamilia 1 at 28 (EXP.020.026.0001_3 at 0030_3); Korda at 76 (EXP.010.078.0001_3 at 0078_3)
Rosamilia 1 at 29 (EXP.020.026.0001_4 at 0031_4)
Rosamilia 1 at 29 (EXP.020.026.0001_4 at 0031_4)
Korda 3 at 12 (EXP.010.129.0001_5 at 0013_5)
Korda 1 at 76 (EXP.010.078.0001_3 at 0078_3)
Korda 1 at 76 (EXP.010.078.0001_3 at 0078_3)
ETH.MESH.05642960
T3822 (TRA.500.048.0001_2 at 0029_3)
T3824 (TRA.500.048.0001_2 at 0031_2); T4199 (TRA.500.052.0001_2 at 0039_2)
Korda 1 at 76 (EXP.010.078.0001_3 at 0078_3)
Chughtai 1 at 11 (EXP.010.001.0001_3 at 0012_3)
Agur at 22 (EXP.010.049.0001_3 at 0023_3)
Deprest at [296] (EXP.020.006.0001_4 at 0064_4)
T3825 (TRA.500.048.0001_2 at 0032_2)
Korda 3 at 12 (EXP.010.129.0001_5 at 0012_5)
Agur 1 at 9–10; 21 (EXP.010.049.0001_3 at 0010_3-0011_3; 0022_3)
Agur 1 at 21 (EXP.010.049.0001_3 at 0022_3)
Agur 1 at 21 (EXP.010.049.0001_3 at 0022_3)
Korda 1 at 77 (EXP.010.078.0001_3 at 0079_3)
Korda 1 at 76 (EXP.010.078.0001_3 at 0078_3); T1183 (TRA.500.014.0001_2 at 0056_2)
Rosamilia at 28 (EXP.020.026.0001_3 at 0030_3)
ETH.MESH.04087047 at 7049
Deprest at [101] (EXP.020.006.0001_4 at 0021_4)
ETH.MESH.04087047
ETH.MESH.04087047 at 7050
Deprest at [100] (EXP.020.006.0001_4 at 0021_4)
Deprest at [102] (EXP.020.006.0001_4 at 0021_4)
Deprest at [104] (EXP.020.006.0001_4 at 0021_4)
Korda 1 at 68 (EXP.010.078.0001_3 at 0070_3)
Korda 1 at 71 (EXP.010.078.0001_3 at 0073_3)
T1195 (TRA.500.014.0001_2 at 0068_2)
Korda 1 at 71 (EXP.010.078.0001_3 at 0073_3)
Korda 3 at 22 (EXP.010.129.0001_5 at 0023_5)
T4431 (TRA.500.055.0001_3 at 0031_3)
Collinet at 28 (EXP.020.005.0001_4 at 0028_4)
Rosamilia 1 at 22 (EXP.020.026.0001_3 at 0024_3); Roovers 1 at 14 (EXP.020.033.0001_3 at 0016_3)
Rosamilia 1 at 20–21 (EXP.020.026.0001_3 at 0022_3)
Dwyer P and O’Reilly B, “Transvaginal repair of anterior and posterior compartment prolapse with Atrium polypropylene mesh” (2004) 111(8) BJOG 831-6 (ETH.MESH.06139479)
T4265 (TRA.500.053.0001_2 at 0024_2); Achtari C et al, “Risk factors for mesh erosion after transvaginal surgery using polypropylene (Atrium) or composite polypropylene/polyglactin 910 (Vypro II) mesh” (2005) 16(5) Int Urogynecol J Pelvic Floor Dysfunct 389–394 (ETH.MESH.03394815)
T4268 (TRA.500.053.0001_2 at 0026)
Agur 2 at 11 (EXP.010.152.0001_3 at 0012_3)
Agur 2 at 11 (EXP.010.152.0001_3 at 0012_3)
Korda 1 at 64 (EXP.010.078.0001_3 at 0066_3)
PUB.MESH.00009026 at 9043
T3775 (TRA.500.047.0001_2 at 0069_2)
T3777 (TRA.500.047.0001_2 at 0071_2)
T3798 (TRA.500.048.0001_2 at 0005_2)
Leake at [50] (LAY.010.002.0001 at 0014)
Leake at [50(d)] (LAY.010.002.0001 at 0015)
Leake at [51] (LAY.010.002.0001 at 0015)
Leake at [56] (LAY.010.002.0001 at 0016)
Gigliobianco G et al, “Biomaterials for Pelvic Floor Reconstructive Surgery: How Can We Do Better?”, (2015) BioMed Research International, Article ID 968087 (SHI.MESH.00007734 at 7749); Scientific Committee on Emerging and Newly Identified Health Risks (SCENIHR), “Opinion on The safety of surgical meshes used in urogynaecological surgery”, 3 December 2015 (SHI.MESH.00006213 at 6246)
T2731 (TRA.500.035.0001_3 at 0022_3)
ETH.MESH.22048410
ETH.MESH.22048410 at 8419
Santerre 1 at [174] (EXP.020.052.0001_4 at 0060_4)
Santerre 1 at [55]–[56] (EXP.020.052.0001_4 at 0018_4)
Santerre 1 at [50]–[51] (EXP.020.052.0001_4 at 0017_4)
Bellon J et al, “In vitro interaction of bacteria with polypropylene/ePTFE prostheses” (2001) 22(14) Biomaterials 2021–2024 (ETH.MESH.00144931)
T4569–71 (TRA.500.056.0001 at 0084)
Santerre 1 at [59] (EXP.020.052.0001_4 at 0019_4)
Nyhus L, “VI Mesh-Related Complications: Exception or the Rule?- Impact of Technique and Material” Meshes: Benefits and Risks (Springer, 2004) (SHI.MESH.00022986 at 3213)
Klinge at [54] (LAY.010.022.0001_2 at 0012_2)
ETH.MESH.05455878
Davila G, Drutz H and Deprest J, “Clinical implications of the biology of grafts: conclusions of the 2005 IUGA Grafts Roundtable” (2006) 17 Int Urogynecol J S51–S55 (ETH.MESH.01594955 at 4957)
Elmér C et al, “Risk factors for mesh complications after trocar guided transvaginal mesh kit repair of anterior vaginal wall prolapse” (2012) 31 Neurourol Urodyn 1165–1169 (ETH.MESH.08299594)
T4786 (TRA.500.059.0001 at 0069)
Korda 3 at 21 (EXP.010.129.0001_5 at 0022_5)
T1960, 1962 (TRA.500.025.0001_2 at 0018_2, 0020_2); Iakovlev 1 at 40–41 (EXP.010.036.0001_4 at 0041_4–0042_4); Deprest at [285] (EXP.020.006.0001_4 at 0062_4); see also T1752 (TRA.500.022.0001_2 at 0028_2); ETH.MESH.02157879 at 7880
T6125–26 (TRA.500.076.0001 at 0089–90)
Wright 1 at 30 (EXP.020.057.0001_4 at 0032_4)
T5017 (TRA.500.062.0001 at 0038)
Wright 1 at 7 (EXP.020.057.0001_4 at 0009_4)
Klinge at [79(c)] (LAY.010.022.0001_2 at 0018_2)
Wright 1 at 9 (EXP.020.057.0001_4 at 0011_4)
Klinge at [79(f)] (LAY.010.022.0001_2 at 0018_2)
T5003 (TRA.500.062.0001 at 0024)
T5048, 5053 (TRA.500.062.0001 at 0069, 0074)
Barone W, Abramowitch S and Moalli P, “Host Response to Biomaterials for Pelvic Floor Reconstruction” in Host Response to Biomaterials (Elsevier Inc, 2015) (SHI.MESH.00044785)
AWS ch 6 at [222] (SBM.010.005.0156_2 at 0206_2)
T4580 (TRA.500.057.0001 at 0007)
T4568 (TRA.500.056.0001 at 0083)
T4645–46 (TRA.500.057.0001 at 0072)
AWS ch 6 at [223] (SBM.010.005.0156_2 at 0206_2)
Deprest at [282] (EXP.020.006.0001_4 at 0061_4)
SHI.MESH.00045854 at 5864
T4764 (TRA.500.059.0001 at 0047)
T4774 (TRA.500.059.0001 at 0057)
Deprest et al, “The biology behind fascial defects and the use of implants in pelvic organ prolapse repair” (2006) 17 Int Urogynecol J S16–S25 (ETH.MESH.08999773)
T4799 (TRA.500.059.0001 at 0063)
T4775–77 (TRA.500.059.0001 at 0058–0060)
AWS ch 6 at [226] (SBM.010.005.0156_2 at 0208_2)
RWS ch 11 at [47] (SBM.020.002.0275 at 0292)
RWS Ch 11 at [50] (SBM.020.002.0275 at 0292)
RWS Ch 11 at [62] (SBM.020.002.0275 at 0295)
MacLean at 25 (EXP.020.042.0001_4 at 0025_4)
Williams D, “General Concepts of Biocompatibility” in Handbook of Biomaterial Properties (Springer Science+Business Media, 1998) (SHI.MESH.00014223 at 4225)
Guelcher 1 at 9 (EXP.010.066.0001_3 at 0011_3)
Klosterhalfen 2 at [11] (LAY.010.018.0001_3 at 0003_3)
Klinge at [91] (LAY.010.022.0001_2 at 0023_2)
Williams D, “On the Mechanisms of Biocompatibility” (2008) 29 Biomaterials 2941–2953 (SHI.MESH.00000759)
Ali S, Doherty P and Williams D, “The mechanisms of oxidative degradation of biomedical polymers by free radicals” (1994) 51 Journal of applied polymer science 1389 at 1390 (SHI.MESH.00022189 at 2190)
Williams D, “General Concepts of Biocompatibility” in Handbook of Biomaterial Properties (Springer Science+Business Media, 1998) (SHI.MESH.00014223 at 4226)
Klosterhalfen B et al, “Biological response to mesh” (2003) Eur Surg 35 (ETH.MESH.06894393 at 4395)
ETH.MESH.00272548
Williams D, “General Concepts of Biocompatibility” Handbook of Biomaterial Properties (Springer Science+Business Media, 1998) (SHI.MESH.00014223 at 4228)
Ibid (SHI.MESH.00014223 at 4229)
Ibid (SHI.MESH.00014223 at 4230)
Williams D, “Biodegradation of surgical polymers” (1982) 17 J Mater Sci 1233–1246 (SHI.MESH.00000001)
Klinge at [91] (LAY.010.022.0001_2 at 0023_2)
Cosson M et al, “Mechanical properties of synthetic implants used in the repair of prolapse and urinary incontinence in women: which is the ideal material?” (2003) 14 Int Urogynecol J 169–178 (ETH.MESH.00157897 at 7904)
Ulrich D et al “Human Endometrial Mesenchymal Stem Cells Modulate the Tissue Response and Mechanical Behavior of Polyamide Mesh Implants for Pelvic Organ Prolapse Repair” (2012) 20(3,4) Tissue Engineering 785 at 786 (SHI.MESH.00044580 at 4581)
Ulrich D et al “A Preclinical Evaluation of Alternative Synthetic Biomaterials for Fascial Defect Repair Using a Rat Abdominal Hernia Model” (2012) 7(11) PLOS ONE (HMESH_ETH_07250423)
T2046–47 (TRA.500.026.0001_2 at 0023_26–0024_2)
Guelcher 1 at 11 (EXP.010.066.0001_3 at 0013_3)
Klosterhalfen 1 at [48] (LAY.010.014.0001_6 at 0009_6)
T96 (TRA.500.0002.0001_2 at 0019_2)
Guelcher 1 at 11–12 (EXP.010.066.0001_3 at 0013_3-0014_3)
Dunn 1 at 15 (EXP.010.031.0001_3 at 0017_3)
Klosterhalfen 1 at [49] (LAY.010.014.0001_6 at 0009_6)
Klosterhalfen 2 at [18] (LAY.010.018.0001_3 at 0005_3)
Deprest at [260]–[262] (EXP.020.006.0001_4 at 0055_4)
Klinge at [86] (LAY.010.022.0001_2 at 0021_2)
Klosterhalfen 2 at [18] (LAY.010.018.0001_3 at 0005_3)
Klinge at [95] (LAY.010.022.0001_2 at 0029_2)
Klosterhalfen 1 at [150]–[151] (LAY.010.014.0001_6 at 0030_6)
Klosterhalfen 1 at [153] (LAY.010.014.0001_6 at 0031_6)
Klosterhalfen 2 at [18] (LAY.010.018.0001_3 at 0005_3)
Klosterhalfen 2 at [28] (LAY.010.018.0001_6 at 0009_6)
Klinge at [371] (LAY.010.022.0001_2 at 0131_2)
Guelcher 2 at 29 (EXP.010.138.0001_3 at 0030_3); see also Klinge U and Klosterhalfen, “Biomaterials – Experimental Aspects” in Incisional hernia (Springer, 1st ed, 1999) (SHI.MESH.00036749 at 6979)
See, for example, ETH.MESH.10511708
Klosterhalfen 1 at [172] (LAY.010.014.0001_6 at 0036_6)
Klosterhalfen 1 at [178]–[179] (LAY.010.014.0001_6 at 0038_6)
RWS ch 11 at [18] (SBM.020.002.0275 at 0283)
Klinge at [92(j)] (LAY.010.022.0001_2 at 0027_2)
Klosterhalfen 2 at [190] (LAY.010.018.0001_3 at 0066_3)
RWS ch 11 [23] (SBM.020.002.0275 at 0284)
Cobb W, Kercher K, Heniford B, “The Argument for Lightweight Polypropylene Mesh in Hernia Repair” (2005) 12(1) Surgical Innovation 63–69 (ETH.MESH.01424029 at 4032)
Deprest J et al “The biology behind fascial defects and the use of implants in pelvic organ prolapse repair” (2006) 17(1) Int Urogynecol J 16–25 (ETH.MESH.00074372 at 4379) (S23)
AWS ch 11 at [31] (SBM.010.030.0460 at 0468)
Elmer C et al, “Histological inflammatory response to transvaginal polypropylene mesh for pelvic reconstructive surgery” (2009) 181(3) J Urol 1189–1195 (ETH.MESH.00017362 at 7367)
T3484-3485 (TRA.500.044.0001_3 at 0035–7)
T4067 (TRA.500.050.0001 at 0090)
Klosterhalfen 2 at [44] (LAY.010.018.0001_3 at 0020_3)
Klosterhalfen 2 at [70] (LAY.010.018.0001_3 at 0028_3)
Ulrich D et al, “A Preclinical Evaluation of Alternative Synthetic Biomaterials for Fascial Defect Repair Using a Rat Abdominal Hernia Model” (2012) 7(11) PLOS ONE (HMESH_ETH_07250423)
ETH-01748 at 01756
ETH.MESH.00094046
Klosterhalfen 1 at [13] (LAY.010.014.0001_6 at 0003_6)
Klosterhalfen 1 at [17] (LAY.010.014.0001_6 at 0004_6)
Klosterhalfen 1 at [7]–[10] (LAY.010.014.0001_6 at 0002_6)
Klosterhalfen 1 at [12] (LAY.010.014.0001_6 at 0003_6)
Klosterhalfen 1 at [26] (LAY.010.014.0001_6 at 0005_6)
Klosterhalfen 1 at [14] (LAY.010.014.0001_6 at 0003_6)
T4858 (TRA.500.060.0001 at 0046)
ETH.MESH.02589032
Klinge at [32] (LAY.010.022.0001_2 at 0007_2)
Klinge at [24]–[36] (LAY.010.022.0001_2 at 0006_2-0007_2)
Klinge at [29] (LAY.010.022.0001_2 at 0006_2)
Klinge at [1]–[21] (LAY.010.022.0001_2 at 0002_2-0005_2)
Klosterhalfen 1 at [19] (LAY.010.014.0001_6 at 0004_6)
Klosterhalfen 1 at [19] (LAY.010.014.0001_6 at 0004_6)
Klinge at [22] (LAY.010.022.0001_2 at 0005_2); Klosterhalfen 1 [20]–[21] (LAY.010.014.0001_6 at 0005_6)
Klosterhalfen 1 at [22] (LAY.010.014.0001_6 at 0005_6)
Klosterhalfen 1 at [94] (LAY.010.014.0001_6 at 0019_6)
Klosterhalfen 1 at [23] (LAY.010.014.0001_6 at 0005_6)
Klosterhalfen 1 at [25] (LAY.010.014.0001_6 at 0005_6)
SBM.010.025.0001 at 0012–3
Klosterhalfen 1 at [25] (LAY.010.014.0001_6 at 0005_6)
Klinge U et al, “Modified mesh for hernia repair that is adapted to the physiology of the abdominal wall” (1998) 164(12) Eur J Surg 951–960 (ETH.MESH.14069949)
Kosterhalfen 1 at [69] (LAY.010.014.0001_6 at 0013_6)
Kosterhalfen 1 at [69] (LAY.010.014.0001_6 at 0013_6)
Kosterhalfen 1 at [70] (LAY.010.014.0001_6 at 0014_6)
HMESH_ETH_01841514
Klinge at [52] (LAY.010.022.0001_2 at 0011_2)
Klinge at [53] (LAY.010.022.0001_2 at 0012_2)
Klosterhalfen 1 at [64] (LAY.010.014.0001_6 at 0012_6)
Klinge at [54] (LAY.010.022.0001_2 at 0012_2)
Klinge at [55] (LAY.010.022.0001_2 at 0012_2)
Klinge at [59] (LAY.010.022.0001_2 at 0013_2)
Klinge at [61] (LAY.010.022.0001_2 at 0014_2)
Schumpelick V and Kingsnorth A (eds), Incisional hernia (Springer, 1st ed, 1999) (SHI.MESH.00036749)
Klinge U and Klosterhalfen, “Biomaterials – Experimental Aspects” in Incisional hernia (Springer, 1st ed, 1999) (SHI.MESH.00036749 at 6961–6962)
Klosterhalfen 1 at [74] (LAY.010.014.0001_6 at 0015_6)
Klosterhalfen 1 at [60] (LAY.010.014.0001_6 at 0011_6)
Klosterhalfen 1 at [62] (LAY.010.014.0001_6 at 0012_6)
Klosterhalfen 1 at [62] (LAY.010.014.0001_6 at 0012_6)
Klosterhalfen 1 at [85] (LAY.010.014.0001_6 at 0017_6)
Klosterhalfen 1 at [65] (LAY.010.014.0001_6 at 0012_6)
Klosterhalfen 1 at [67] (LAY.010.014.0001_6 at 0013_6)
Klosterhalfen 1 at [106] (LAY.010.014.0001_6 at 0022_6)
ETH.MESH.05465545_2
Klosterhalfen 1 at [109] (LAY.010.014.0001_6 at 0023_6)
Klosterhalfen 1 at [111] (LAY.010.014.0001_6 at 0023_6)
Klosterhalfen 1 at [120] (LAY.010.014.0001_6 at 0024_6)
Klosterhalfen 1 at [79] (LAY.010.014.0001_6 at 0016_6)
Klosterhalfen 1 at [134] (LAY.010.014.0001_6 at 0026_6)
Klosterhalfen 1 at [136] (LAY.010.014.0001_6 at 0026_6)
Klinge at [172] (LAY.010.022.0001_2 at 0065_2)
Klinge at [179] (LAY.010.022.0001_2 at 0067_2)
T2713 (TRA.500.035.0001_3 at 0004_3)
Wright 1 at 39 (EXP.020.057.0001_3 at 0041_3)
See, for example, ETH.MESH.01818382
Boulanger L et al, “Bacteriological analysis of meshes removed for complications after surgical management of urinary incontinence or pelvic organ prolapse” (2008) 19 Int Urogynecol J 827 (ETH.MESH.00273021)
Klosterhalfen 2 at [303] (LAY.010.018.0001_3 at 0114_3)
Klinge [199] (LAY.010.022.0001_2 at 0076_2)
ETH.MESH.22354125 at 4151
T2713–14 (TRA.500.035.0001_3 at 0004_3-0005_3)
The New Shorter Oxford English Dictionary (Oxford University Press, 4th ed, 1993) at 792
T2736 (TRA.500.035.0001_3 at 0027_3)
Klinge at [374] (LAY.010.022.0001_2 at 0132_2)
Klinge at [375] (LAY.010.022.0001_2 at 0132_2)
Klinge at [376] (LAY.010.022.0001_2 at 0133_2)
Klinge at [371] (LAY.010. 022.0001_2 at 0131_2)
Klinge at [373] (LAY.010. 022.0001_2 at 0132_2)
Deprest J et al “The biology behind fascial defects and the use of implants in pelvic organ prolapse repair” (2006) 17(1) Int Urogynecol J 16–25, (ETH.MESH.00074372 at 4373, 4379)
Klinge at [380] (LAY.010.022.0000 at 0134_2)
Klinge at [379] (LAY.010.022.0000 at 0134_2)
See RWS Ch 11 at [85] (SBM.020.002.0275 at 0305); SBM.020.019.0001 at 0003; T2147 (TRA.500.027.0001_2 at 0034_2)
RWS Ch 11 at [85] (SBM.020.002.0275 at 0305)
RWS Ch 11 at [57], [79] (SBM.020.002.0275 at 0294, 0302)
SBM.020.019.0001 at 0003
RWS Ch 11 at [81(c)] (SBM.020.002.0275 at 0303)
SHI.MESH.00028426_2
T2124 (TRA.500.027.0001_2 at 0011_2)
T2122 (TRA.500.027.0001_2 at 0009_2)
T2122 (TRA.500.027.0001_2 at 0009_2)
T2128 (TRA.500.027.0001_2 at 0015_2)
T2128 (TRA.500.027.0001_2 at 0015_2)
T2128(TRA.500.027.0001_2 at 0015_2)
T2128–29 (TRA.500.027.0001_2 at 0015_2–0016_2)
T2130 (TRA.500.027.0001_2 at 0017_2)
T2125 (TRA.500.027.0001_2 at 0012_2)
T2130 (TRA.500.027.0001_2 at 0017_2)
T2129 (TRA.500.027.0001_2 at 0016_2); T2132 (TRA.500.027.0001_2 at 0019_2)
T2132 (TRA.500.027.0001_2 at 0019_2)
T2127 (TRA.500.027.0001_2 at 0014_2)
T2125 (TRA.500.027.0001_2 at 0012_2)
T2126-27 (TRA.500.027.0001_2 at 0013_2-0014_2)
T2130 (TRA.500.027.0001_2 at 0017_2)
T2127/24-32 (TRA.500.027.0001_2 at 0014_2)
T2125/12 (TRA.500.027.0001_2 at 0012_2)
T2127 (TRA.500.027.0001_2 at 0014_2)
SHI.MESH.00022730
T2134 (TRA.500.027.0001_2 at 0020_2–0021_2)
T2135 (TRA.500.027.0001_2 at 0022_2)
RWS Ch 11 at [80] (SBM.020.002.0275 at 0303
T2135–36 (TRA.500.027.0001_2 at 0022_2-0023_2)
T2136 (TRA.500.027.0001_2 at 0023_2)
T2144–45 (TRA.500.027.0001_2 at 0032_2-0033_2)
SBM.010.025.0001 at 0012
RWS ch 11 at [86] (SBM.020.002.0275 at 0306)
Deprest at [342] and [346] (EXP.020.006.0001_4 at 0071_4)
Klosterhalfen 2 at [316] (LAY.010.018.0001_3 at 0118_3)
T2003 (TRA.500.025.0001_2 at 0061_2)
SHI.MESH.00022730 at 2773
Klinge U, Otto J and Mühl T, “High Structural Stability of Textile Implants Prevents Pore Collapse and Preserves Effective Porosity at Strain” (2015) Biomed Res Int 1-7 (SHI.MESH.00024765 at 4767)
Klosterhalfen 1 at [158] (LAY.010.014.0001_6 at 0033_6)
Mühl T et al “New objective measurement to characterize the porosity of textile implants” (20078) 84(1) J Biomed Mater Res B Appl Biomater 176–183 (SHI.MESH.00033721 at 3724)
Ibid (SHI.MESH.00033721 at 3724); Klinge at [146] (LAY.010.022.0001_2 at 0053_2)
T2792 (TRA.500.036.0001_2 at 0033_2)
RWS ch 11 at [83] (SBM.020.002.0275 at 0305)
T2134–35 (TRA.500.027.0001_2 at 0021_2-0022_2)
T2161 (TRA.500.027.0001_2 at 0049_2)
RWS ch 11 at [88] (SBM.020.002.0275 at 0305)
Klinge [113]–[121] (LAY.010.022.0001_2 at 0035_2-0041_2)
Klinge U et al, “Foreign body reaction to meshes used for the repair of abdominal wall hernias” (1999) 165 Eur J Surg 665–673 (ETH.MESH.01405349)
Klinge U et al “Impact of Polymer Pore Size on the Interface Scar Formation in a Rat Model” (2002) 103(2) J Surg Res 208–214 (ETH.MESH.01700542 at 0547)
Junge K et al, “Functional and morphological evaluation of different polypropylene-mesh modifications for Inguinal repair” (2002) 26 World J Surgery 1472–1480 (ETH.MESH.10463926 at 3932)
Rosch R et al, “Mesh implants in hernia repair; Inflammatory cell response in a rat model” (2003) 35(3) Eur Surg Res 161–166 (ETH.MESH.19736758)
See, for example, Klosterhalfen 1 at [176], [186] (LAY.010.014.0001_6 at 0037_6, 0039_6)
Klosterhalfen 1 at [186] (LAY.010.014.0001_6 at 0039_6)
Klosterhalfen 1 at [189] (LAY.010.014.0001_6 at 0040_6)
Klinge U, Otto J and Mühl T, “High Structural Stability of Textile Implants Prevents Pore Collapse and Preserves Effective Porosity at Strain” (2015) Biomed Res Int 1–7 (SHI.MESH.00024765)
AWS ch 11 at [57] (SBM.010.030.0460 at 0474)
RWS ch 11 at [50] (SBM.020.002.0275 at 0292)
Kosterhalfen 1 at [68] (LAY.010.014.0001_6 at 0031_6)
Klosterhalfen 2 at [170] (LAY.010.018.0001_3 at 0059_3)
Amid PK, “Classification of Biomaterials and their Related Complications in Abdominal Wall Hernia Surgery” (1997) 1 Hernia 15–21 (ETH.MESH.08536173)
Amid PK, “Classification of Biomaterials and their Related Complications in Abdominal Wall Hernia Surgery” (1997) 1 Hernia 15–21 (ETH.MESH.08536173)
Berrocal J et al, “Conceptual Advances in the Surgical Management of Genital Prolapse: The TVM technique emergence” (2004) 33 J Gynecol Obstet Biol Reprod 577–587 (ETH.MESH.00659678 at 9681)
Klosterhalfen 2 at [41] (LAY.010.018.0001_3 at 0015_3)
Klosterhalfen 2 at [108], [128] (LAY.010.018.0001_3 at 0043_3, 0049_3); Klinge at [108] (LAY.010.022.0001_2 at 0034_2)
Klosterhalfen 2 at [128] (LAY.010.018.0001_3 at 0049_3)
Klinge at [103] (LAY.010.022.0001_2 at 0032_2)
Klinge at [104] (LAY.010.022.0001_2 at 0033_2)
Klinge U and Klosterhalfen B, “Modified classification of surgical meshes for hernia repair based on the analysis of 1,000 explanted meshes (2012) 16 Hernia 251-258 (ETH.MESH.10569655)
Klosterhalfen 2 at [108], [131] (LAY.010.018.0001_3 at 0043_3, 0059_3)
RWS Ch 11 at 23-31ff (SBM.020.002.0275 at 0297-0305)
T5043–44 (TRA.500.062.0001 at 0064–0065)
Klosterhalfen 2 at [118] (LAY.010.018.0001_3 at 0045_3)
Barone W, Abramowitch S and Moalli P, “Host Response to Biomaterials for Pelvic Floor Reconstruction” in Host Response to Biomaterials (Elsevier Inc, 2015) (SHI.MESH.00044785 at 4815)
Davila G, Drutz H and Deprest J, “Clinical implications of the biology of grafts: conclusions of the 2005 IUGA Grafts Roundtable” (2006) 17 Int Urogynecol J S51-55 (ETH.MESH.01594955)
ETH.MESH.08638414
ETH.MESH.09651393 at 1396
Barone W, Abramowitch S and Moalli P, “Host Response to Biomaterials for Pelvic Floor Reconstruction” in Host Response to Biomaterials (Elsevier Inc, 2015) (SHI.MESH.00044785 at 4809)
Liang R et al, “Exploring the basic science of prolapse meshes” (2016) Oct 28(5) Curr Opin Obstet Gynecol 413–419 (SHI.MESH.00016387 at 6389)
ETH.MESH.05479695
ETH.MESH.05479695
ETH.MESH.05479695 slide 13
ETH.MESH.05479695 slide 2
ETH.MESH.05337221 at 7222
T5050 (TRA.500.062.0001 at 0071)
ETH.MESH.04038003 at 8016
T2776 (TRA.500.036.0001_3 at 0015_3)
ETH-01748 at 1755
ETH.MESH.02227337
ETH.MESH.05237872
ETH.MESH.04941016
Klosterhalfen 2 at [115] (LAY.010.018.0001_3 at 0044_3)
ETH.MESH.06400509 at 0571
Klinge at [344] (LAY.010.022.0001_2 at 0122_2); ETH.MESH.04020026 at 0037; ETH.MESH.10543450 at 3454; ETH.MESH.05239904 at 9905; ETH.MESH.05235980
ETH.MESH.05479695
ETH.MESH.02211890 at 1899
ETH.MESH.02211890 at 1899–1900
Klinge at [175] (LAY.010.022.0001_2 at 0066_2)
ETH.MESH.12832662
T2150–51 (TRA.500.027.0001_2 at 0037_2–0038_2)
T2069 (TRA.500.026.0001_2 at 0046_2); T2152 (TRA.500.027.0001_2 at 0040_2)
Klosterhalfen 1 at [158]–[160] (LAY.010.014.0001_6 at 0033_6)
Klosterhalfen 2 at [115] (LAY.010.018.0001_3 at 0044_3)
ETH.MESH.09651393
Moalli P et al, “Tensile properties of five commonly used mid-urethral slings relative to the TVT™” (2008) 19 Int Urogynecol J 655–663 (SHI.MESH.00033759)
Dietz et al, “Mechanical properties of urogynecologic implant materials” (2003) 14 Int Urogynecol J 239–243 (ETH.MESH.00015593)
Klosterhalfen 2 at [121] (LAY.010.018.0001_3 at 0047_3)
Klosterhalfen 2 at [122] (LAY.010.018.0001_3 at 0047_3)
Klinge at [155] (LAY.010.022.0001_2 at 0058_2)
Klinge at [155] (LAY.010.022.0001_2 at 0058_2)
Klinge at [159]–[162] (LAY.010.022.0001_2 at 0060_2, 0061_2)
Deprest at [276]–[277] (EXP.020.006.0001_4 at 0059_4)
Barone W, Moalli P and Abramowitch S, “Textile properties of synthetic prolapse mesh in response to uniaxial loading” (2016) 215 Am J Obstet Gynecol 326 (PUB.MESH.00006114)
Wright 1 at 48 (EXP.020.057.0001_4 at 0050_4)
Klinge at [178]–[179] (LAY.010.022.0001_2 at 0067_2)
Klosterhalfen 2 at [120]-[123] (LAY.010.018.0001_3 at 0046_3-0047_3)
Klosterhalfen 1 at [159] (LAY.010.014.0001_6 at 0033-6); Mühl T et al, “New objective measurement to characterize the porosity of textile implants” (2008) 84(1) J Biomed Mater Res B Appl Biomater 176–183 (SHI.MESH.00033721)
Klinge at [73] (LAY.010.022.0001_2 at 0016_2)
Klinge at [169] (LAY.010.022.0001_2 at 0065_2)
Klinge at [194], [172] (LAY.010.022.0001_2 at 0074_2, 0065_2)
Klosterhalfen 1 at [52] (LAY.010.014.0001_6 at 0010_6)
Klinge at [79] (LAY.010.022.0001_2 at 0017_2)
AWS ch 11 at [52] (SBM.010.005.0461_2 at 0474_2); Cobb W, Kercher K and Heniford B, “The Argument for Lightweight Polypropylene Mesh in Hernia Repair” (2005) 12(1) Surg Innov 63–69 (ETH.MESH.01424029 at 4032)
Barone W, Moalli P and Abramowitch S, “Textile properties of synthetic prolapse mesh in response to uniaxial loading” (2016) 215 Am J Obstet Gynecol 326 (PUB.MESH.00006114)
Mühl T et al, “New objective measurement to characterize the porosity of textile implants” (2008) 84(1) J Biomed Mater Res B Appl Biomater 176–183 (SHI.MESH.00033721 at 0725)
Deprest at [279] (EXP.020.006.0001_4 at 0060_4)
T4601 (TRA.500.057.0001 at 0028)
T4677 (TRA.500.058.0001 at 0017); see also T4562 (TRA.500.056.0001 at 0077)
T5046–47 (TRA.500.062.0001 at 0067-8)
T5047 (TRA.500.062.0001 at 0068)
T5048 (TRA.500.062.0001 at 0069)
T5015 (TRA.500.062.0001 at 0036)
T5047/10, 5150/10-20 (TRA.500.062.0001 at 0068, 0079)
Klinge at [136] (LAY.010.022.0001_2 at 0049_2)
Klinge U et al, “Foreign body reaction to meshes used for the repair of abdominal wall hernias” (1999) 165 Eur J Surg 665–673 (ETH.MESH.01405349 at 5354)
Ibid (ETH.MESH.01405349 at 5354)
ETH.MESH.01818382 at 8395
ETH.MESH.01425079 at 5087
ETH.MESH.02053629 at 3630
ETH.MESH.02053629 at 3632
ETH.MESH.01782867
ETH.MESH.01782867
Klinge at [293] (LAY.010.022.0001_2 at 0104_2)
Klinge at [312] (LAY.010.022.0001_2 at 0111_2)
Klinge at [303]–[304] (LAY.010.022.0001_2 at 0109_2)
Klosterhalfen 1 at [154] (LAY.010.014.0001_6 at 0031_6); AWS ch 11 at [211]-[213] (SBM.010.005.0461_2 at 0511_2)
Klosterhalfen 1 at [187] (LAY.010.014.0001_6 at 0040_6)
Chughtai 1 at 10 (EXP.010.001.0001_3 at 0011_3)
Ostergard DR, “Polypropylene Vaginal Mesh Grafts in Gynecology” (2010) 116 Obstet Gynecol 962–966 at 964 (ETH.MESH.00544274 at 4276)
Fatton B et al, “Transvaginal repair of genital prolapse: preliminary results of a new tension-free vaginal mesh (Prolift technique) – a case series multicentric study” (2007) 18 Int Urogynecol J 743–752 (ETH.MESH.00093734 at 3740)
Hinoul at [722] (LAY.020.001.0001_4 at 0333_4)
Amid PK, “Classification of biomaterials and their related complications in abdominal wall hernia surgery” (1997) 1 Hernia 15-21 (ETH.MESH.03805989 at 5996)
Klinge at [294] (LAY.010.022.0001_2 at 0106_2); Klinge U et al, “Shrinking of polypropylene mesh in vivo: an experimental study in dogs” (1998) 164 Eur J Surg 965–969 (SHI.MESH.00033744)
ETH.MESH.00659726
ETH.MESH.00821702
ETH.MESH.00821702
Cobb W, Kercher K and Heniford B, “The Argument for Lightweight Polypropylene Mesh in Hernia Repair” (2005) 12(1) Surg Innov 63–69 (ETH.MESH.01424029 at 4032)
Klinge at [295] (LAY.010.022.0001_2 at 0106_2); Cobb W et al, “Textile analysis of heavyweight, mid-weight, and lightweight polypropylene mesh in a porcine ventral hernia model” (2006) 136(1) J Surg Res 1-7 (SHI.MESH.00033731)
ETH.MESH.01818382 at 8383
Klosterhalfen 1 at [185] (LAY.010.014.0001_6 at 0039_6); ETH.MESH.01818382
ETH.MESH.01818382 at 8393
ETH.MESH.00570726 at 0727
ETH.MESH.07192412 at 2413
ETH.MESH.07192412 at 2413
Klinge at [308] (LAY.010.022.0001_2 at 0110_2)
Wright 1 at 42 (EXP.020.057.0001_4 at 0044_4)
Klinge at [313] (LAY.010.022.0001_2 at 0111_2)
Hinoul P et al, “A prospective study to evaluate the anatomic and functional outcome of a transobturator mesh kit (prolift anterior) for symptomatic cystocele repair” (2008) 15(5) J Minim Invasive Gynecol 615–620 (ETH.MESH.00016122 at 6125)
T3122–23 (TRA.500.040.0001_4 at 0024_4–0025_4)
T3484–85 (TRA.500.044.0001_3 at 0035–7)
T2732 (TRA.500.035.0001_3 at 0023_3)
T5057–58 (TRA.500.062.0001 at 0078–0079)
Klinge U et al, “Demands and properties of alloplastic implants for the treatment of stress urinary incontinence” (2007) 4(3) Expert Rev Med Devices 349–359 (ETH.MESH.13766913 at 6918)
Deprest at [287] (EXP.020.006.0001_4 at 0062_4)
Barone W, Abramowitch S and Moalli P, “Host Response to Biomaterials for Pelvic Floor Reconstruction” in Host Response to Biomaterials (Elsevier Inc, 2015) (SHI.MESH.00044785 at 4809)
Klosterhalfen 1 at [154] (LAY.010.014.0001_6 at 0031_6)
Klinge at [314] (LAY.010.022.0001_2 at 0112_2)
Klinge at [312] (LAY.010.022.0001_2 at 0111_2)
T3485 (TRA.500.044.0001_3 at 0037_3)
T3485 (TRA.500.044.0001_3 at 0037_3)
RWS Ch 11 at [111] (SBM.020.002.0275 at 0311)
Sergent F et al, “Experimental biomechanical evaluation of PP prostheses used in pelvic organ prolapse surgery” (2009) 20 Int Urogynecol J 597–604 (ETH.MESH.00116081 at 6085)
RWS ch 11 at [124] (SBM.020.002.0275 at 0315)
Svabík K et al, “Ultrasound appearances after mesh implantation—evidence of mesh contraction or folding?” (2011) 22 Int Urogynecol J 529 (ETH.MESH.01205232)
RWS ch 11 at [128] (SBM.020.002.0275 at 0317)
T4246 (TRA.500.053.0001 at 0008_2)
RWS ch 11 at [135] (SBM.020.002.0275 at 0320)
T4067 (TRA.500.050.0001 at 0090)
T4393 (TRA.500.054.0001_2 at 0098_2)
T3814 (TRA.500.048.0001_2 at 0025_2)
Deprest at [288] (EXP.020.006.0001_4 at 0062_4)
ETH.MESH.01819450
ETH.MESH.02992144
ETH.MESH.02992144
Milani A et al, “Trocar-guided mesh repair of vaginal prolapse using partially absorbable mesh: 1 year outcomes” (2011) 204(1) Am J Obstet Gynecol 74.e1–74.e8 (ETH.MESH.05110011)
RWS ch 11 at [131] (SBM.020.002.0275 at 0318); Rogowski A et al, “Mesh retraction correlates with vaginal pain and overactive bladder symptoms after anterior vaginal mesh repair” (2013) 24 Int Urogynecol J 2087–2092 (SHI.MESH.00015789)
Voyles C et al, “Emergency abdominal wall reconstruction with polypropylene mesh: short-term benefits versus long-term complications” (1981) 194(2) Ann Surg 219–223 (SHI.MESH.00047948)
Clarke K et al, “Intestine submucosa and polypropylene mesh for abdominal wall repair in dogs” (1996) 60(1) J Surg Res 107–114 (SHI.MESH.00024802 at 4807)
Feiner B and Maher C, “Vaginal Mesh Contraction: Definition, Clinical Presentation, and Management” (2010) 115 (2 Pt 1) Obstet Gynecol 325–330 (ETH.MESH.06139646)
Ibid (ETH.MESH.06139646)
T3874 (TRA.500.048.0001_2 at 0085_2)
Withagen M et al, “Risk factors for exposure, pain, and dyspareunia after tension-free vaginal mesh procedure” (2011) 118(3) Obstet Gynecol 629–636 (ETH.MESH.04929975)
Elliott D, “Con: mesh in vaginal surgery: do the risks outweigh the benefits?” (2012) 22(4) Curr Opin Urol 276–281 (SHI.MESH.00033536 at 3539)
SHI.MESH.00006213 at 6246
ETH.MESH.05643793 at 3795
ETH.MESH.00584846
Berrocal J et al, “Conceptual Advances in the Surgical Management of Genital Prolapse: The TVM technique emergence” (2004) 33 J Gynecol Obstet Biol Reprod 577–587 (ETH.MESH.00659678 at 9688)
ETH.MESH.00570726
ETH.MESH.00570726 at 0727
RWS Ch 11 at [123] (SBM.020.002.0275 at 0315)
Caquant F et al, “Safety of Trans Vaginal Mesh procedure: retrospective study of 684 patients” (2008) 34(4) J Obstet Gynaecol Res 449–456 (ETH.MESH.02588589 at 8592)
ETH.MESH.04020155
ETH.MESH.12922134; T3093 (TRA.500.039.0001_3 at 0092_3)
ETH.MESH.00870466 at 0469
ETH.MESH.02017152 at 7153, 7155
JJM.MESH.00446160 at 6169
ETH.MESH.02992144
ETH.MESH.04038003 at 8004
ETH.MESH.04038032 at 8043
Caquant F et al, “Safety of Trans Vaginal Mesh procedure: retrospective study of 684 patients” (2008) 34(4) J Obstet Gynaecol Res 449–456 (ETH.MESH.02588589 at 8593)
Ibid (ETH.MESH.02588589 at 8595)
ETH.MESH.06372555 at 2556
Rogowski A et al, “Mesh retraction correlates with vaginal pain and overactive bladder symptoms after anterior vaginal mesh repair” (2013) 24 Int Urogynecol J 2087–2092 (SHI.MESH.00015789)
Klosterhalfen 1 at [161]–[162] (LAY.010.014.0001_6 at 0034_6)
Klosterhalfen 1 at [160] (LAY.010.014.0001_6 at 0034_6)
Klosterhalfen 1 at [163] (LAY.010.014.0001_6 at 0034_6)
Klosterhalfen 1 at [160] (LAY.010.014.0001_6 at 0034_6)
ETH.MESH.02227224
Klosterhalfen 1 at [219] (LAY.010.014.0001_6 at 0047_6); ETH.MESH.05237919
Klosterhalfen 1 at [221] (LAY.010.014.0001_6 at 0047_6)
T4786 (TRA.500.059.0001 at 0069)
Dietz H et al, “Mechanical properties of urogynecologic implant materials” (2003) 14(4) Int Urogynecol J Pelvic Floor Dysfunct 239–243 (ETH.MESH.00015593 at 5596)
Wright 1 at 30 (EXP.020.057.0001_4 at 0032_4)
PUB.MESH.00003980 at 3981
Boulanger et al, “Tissue integration and tolerance to meshes used in gynecologic surgery: An experimental study” (2006) 125(1) Eur J Obstet Gynecol Reprod Biol 103–108 (ETH.MESH.01405343)
Wright 1 at 30 (EXP.020.057.0001_4 at 0032_4)
Elmer C et al, “Histological Inflammatory Response To Transvaginal Polypropylene Mesh for Pelvic Reconstructive Surgery” (2009) 181 J Urol 1189–1195 at 1192 (ETH.MESH.00017362 at 7365)
Wright 1 at 30 (EXP.020.057.0001_4 at 0032_4)
Elmer C et al, “Histological Inflammatory Response To Transvaginal Polypropylene Mesh for Pelvic Reconstructive Surgery” (2009) 181 J Urol 1189–1195 at 1192 (ETH.MESH.00017362 at 7367)
Pierce et al, “Long-term histologic response to synthetic and biologic graft materials implanted in the vagina and abdomen of a rabbit model” (2009) 200(5) Am J Obstet Gynecol 546.e1–546.e8 (ETH.MESH.07468578)
Pierce et al, “Long-term histologic response to synthetic and biologic graft materials implanted in the vagina and abdomen of a rabbit model” (2009) 200(5) Am J Obstet Gynecol 546.e1–546.e8 (ETH.MESH.07468578 at 8583)
Klosterhalfen B et al, “Influence of implantation interval on the long-term biocompatibility of surgical mesh” (2002) 89 British Journal of Surgery 1043–1048 (ETH.MESH.10511211)
Klosterhalfen 2 at [39]–[40] (LAY.010.018.0001_3 at 0014_3-0015_3)
Klosterhalfen 2 at [41]–[42] (LAY.010.018.0001_3 at 0014_3-0015_3)
Klosterhalfen 2 at [43] (LAY.010.018.0001_3 at 0016_3)
Iakovlev 2 at 23 (EXP.010.147.0001_4 at 0024_4)
Iakovlev 2 at 22 (EXP.010.147.0001_4 at 0022_4)
Deprest J et al, “The biology behind fascial defects and the use of implants in pelvic organ prolapse repair” (2006) 17(1) Int Urogynecol J 16–25 (ETH.MESH.00074372 at 4375)
T2782 (TRA.500.036.0001_3 at 0021_3)
Klinge at [87] (LAY.010.022.0001_2 at 0022_2)
Iakovlev 2 at 23–24 (EXP.010.147.0001_4 at 0024_4-0025_4)
ETH.MESH.00396836
ETH.MESH.09652185
ETH.MESH.03726920; ETH.MESH.03728739
ETH.MESH.09641320
RWS Ch 11 at [103] (SBM.020.002.0275 at 0309)
Barone W, Abramowitch S and Moalli P, “Host Response to Biomaterials for Pelvic Floor Reconstruction” in Host Response to Biomaterials (Elsevier Inc, 2015) (SHI.MESH.00044785 at 4811–4812)
Ibid (SHI.MESH.00044785 at 4818)
Klosterhalfen 1 at [164] (LAY.010.014.0001_6 at 0034_6)
Klinge at [276] (LAY.010.022.0001_2 at 0099_2)
Berrocal J et al, “Conceptual Advances in the Surgical Management of Genital Prolapse: The TVM technique emergence” (2004) 33 J Gynecol Obstet Biol Reprod 577–587 (ETH.MESH.00659678 at 9682)
Klinge at [276] (LAY.010.022.0001_2 at 0099_2)
Klinge U et al, “Demands and properties of alloplastic implants for the treatment of stress urinary incontinence” (2007) 4(3) Expert Rev Med Devices 349–359 (ETH.MESH.13766913 at 6917)
Berrocal J et al, “Conceptual advances in the surgical management of genital prolapse: the TVM technique emergence” (2004) 33 J Gynecol Obstet Biol Reprod 577–587 (ETH.MESH.00659678)
RWS Ch 11 at [156] (SBM.020.002.0275 at 0325)
Barone W, Abramowitch S and Moalli P, “Host Response to Biomaterials for Pelvic Floor Reconstruction” in Host Response to Biomaterials (Elsevier Inc, 2015) (SHI.MESH.00044785 at 4818)
Ibid (SHI.MESH.00044785 at 4819)
T5081 (TRA.500.063.0001 at 0010)
T5081 (TRA.500.063.0001 at 0010)
T4527 (TRA.500.056.0001 at 0042)
T5101 (TRA.500.063.0001 at 0003)
Barone W, Abramowitch S and Moalli P, “Host Response to Biomaterials” in Host Response to Biomaterials for Pelvic Floor Reconstruction (Elsevier Inc 2015) (SHI.MESH.00044785 at 4824)
Iakovlev 1 at 38 (EXP.010.036.0001_4 at 0039_4)
Iakovlev 1 at 38 (EXP.010.036.0001_4 at 0039_4)
T5100 (TRA.500.063.0001 at 0029)
Wright 2 at 3–4 (EXP.020.071.0001_4 at 0005_4-0006_4)
Iakovlev 2 at 23–24 (EXP.010.147.0001_4 at 0024_4–0025_4)
Iakovlev 2 at 24 (EXP.010.147.0001_4 at 0025_4)
Elliott D, “Con: mesh in vaginal surgery: do the risks outweigh the benefits?” (2012) 22(4) Curr Opin Urol 276–281 (SHI.MESH.00033536)
ETH.MESH.04038003 at 8004
Klosterhalfen 1 at 32 (LAY.010.014.0001_6 at 0032_6)
Korda 3 at 8 (EXP.010.129.0001_5 at 0009_5)
Korda 3 at 10 (EXP.010.129.0001_5 at 0011_5)
Klosterhalfen B, Junge K and Klinge U, “The lightweight and large porous mesh concept for hernia repair” (2005) 2(1) Expert Rev Med Devices 103–117 (ETH.MESH.02151903 at 1911)
Klosterhalfen 1 [154(c)], [223] (LAY.010.014.0001_6 at 0031_6, 0048_6)
T2141–42 (TRA.500.027.0001_2 at 0028_2–0029_2)
RWS ch 11 at [153] (SBM.020.002.0275 at 0325)
T2144 (TRA.500.027.0001_2 at 0031_2)
Iakovlev 2 at 103 (EXP.010.147.0001_4 at 0104_4)
RWS ch 11 at [145], [146]–[147] (SBM.020.002.0275 at 0323)
Wright at 52 (EXP.020.057.0001_4 at 0054_4)
Iakovlev 2 at 24 (EXP.010.147.0001_4 at 0025_4)
Iakovlev 2 at 102 (EXP.010.147.0001_4 at 0103_4)
Dunn 1 at 13 (EXP.010.031.0001_3 at 0015_3)
MacLean 1 at 24–25 (EXP.020.042.0001_4 at 0024_4–0025_4); Guelcher 1 at 4, 6–7 (EXP.010.066.0001 at 0006_3, 0008_3–0009_3)
Guelcher 1 at 6 (EXP.010.066.0001_3 at 0008_3)
Guelcher 1 at 6 (EXP.010.066.0001_3 at 0008_3)
Guelcher 1 at 6 (EXP.010.066.0001_3 at 0008_3)
Dunn 1 at 38 (EXP.010.031.0001_3 at 0040_3)
Guelcher 1 at 3 (EXP.010.066.0001_3 at 0006_3-0007_3); SHI.MESH.00041837
MacLean 1 at 23 (EXP.020.042.0001_4 at 0023_4); 2185 (TRA.500.028.0001_3 at 0016_3)
MacLean 1 at 24 (EXP.020.042.0001_4 at 0024_4)
Ali S, Doherty P and Williams D, “The mechanisms of oxidative degradation of biomedical polymers by free radicals” (1994) 51 Journal of applied polymer science 1389 (SHI.MESH.00022189)
Dunn 1 at 9 (EXP.010.031.0001_3 at 0011_3)
See Ali S, Doherty P and Williams D, “The mechanisms of oxidative degradation of biomedical polymers by free radicals” (1994) 51 Journal of applied polymer science 1389 at 1397 (SHI.MESH.00022189 at 2197)
Guelcher 1 at 18 (EXP.010.066.0001_3 at 0021_3); Dunn 1 at 17, 89 (EXP.010.031.0001_3 at 0019_3, 0091_3)
Dunn 1 at 17 (EXP.010.031.0001_3 at 0019_3)
Dunn 1 at 17 (EXP.010.031.0001_3 at 0019_3)
Guelcher 1 at 26 (EXP.010.066.0001_3 at 0028_3)
T4511–12 (TRA.500.056.0001 at 0026–0027)
T4644 (TRA.500.057.0001 at 0071)
T4644–45 (TRA.500.057.0001 at 0072–0073)
T4645, 4649 (TRA.500.057.0001 at 0073, 0076)
Mary C et al, “Comparison of the In Vivo Behaviour of Polyvinylidene Fluoride and Polypropylene Sutures Used in Vascular Surgery” (1998) 44(3) ASAIO J 199–206 (ETH.MESH.10630795)
Costello C et al, “Materials Characterization of Explanted Polypropylene Hernia Meshes” (2007) J Biomed Mater Res Part B: Appl Biomater 44–49 (ETH.MESH.05446083 at 6088)
Clavé A et al, “Polypropylene as a reinforcement in pelvic surgery is not inert: comparative analysis of 100 explants” (2010) 21(3) Int Urogynecol J 261–270 (ETH.MESH.01260885 at 0887)
Ibid (ETH.MESH.01260885 at 0887)
Ibid (ETH.MESH.01260885 at 0888)
AWS ch 11 at [249] (SBM.010.005.0461_2 at 0519_2)
Imel A et al, “In vivo oxidative degradation of polypropylene pelvic mesh” (2015) 73 Biomaterials 131–141 (SHI.MESH.00008045)
Iakovlev V, Guelcher S and Bendavid R, “ Degradation of polypropylene in vivo: A microscopic analysis of meshes explanted from patients” (2015) J Biomed Mater Res Part B: Appl Biomater (SHI.MESH.00005672)
Talley A et al, “Oxidation and degradation of polypropylene transvaginal mesh” (2017) 28:5 Journal of Biomaterials Science, Polymer Edition 444–458 (SHI.MESH.00029011 at 9012)
ETH.MESH.15958445
ETH.MESH.15955438
ETH.MESH.15955438 at 5440
ETH.MESH.15955438 at 5442
ETH.MESH.15955438 at 5454
ETH.MESH.15955438 at 5462–5465
ETH.MESH.15958452
ETH.MESH.15958336 at 8339
ETH.MESH.15958445 at 8446
ETH.MESH.18789422
ETH.MESH.12831391 at 1392
ETH.MESH.12831391
ETH.MESH.12831407
ETH.MESH.09888068
ETH.MESH.09888068 at 8100; ETH.MESH.09888187 at 8188
ETH.MESH.09888187
ETH.MESH.11336071 at 6080
ETH.MESH.09888144 at 8145
ETH.MESH.09888187 at 8188
Santerre at [238] (EXP.020.052.0001_4 at 0080_4)
T4706 (TRA.500.058.0001 at 0046)
T4707 (TRA.500.058.0001 at 0047)
T4708 (TRA.500.058.0001 at 0048)
T4708 (TRA.500.058.0001 at 0048)
Thames S, White J and Ong K, “The myth: in vivo degradation of polypropylene-based meshes” (2017) 28(2) Int Urogynecol J 285–297 (PUB.MESH.00004875)
T4953 (TRA.500.061.0001 at 0079)
T4708 (TRA.500.058.0001 at 0049); PUB.MESH.00004614 at 4617
PUB.MESH.00004614 at 4617
PUB.MESH.00004614 at 4618
Mary C et al, “Comparison of the In Vivo Behaviour of Polyvinylidene Fluoride and Polypropylene Sutures Used in Vascular Surgery” (1998) 44(3) ASAIO J 199–206 (ETH.MESH.10630795 at 0796)
ETH.MESH.03048268
RWS Ch 11 at [161] (SBM.020.002.0275 from 0328)
MacLean 1 at 45 (EXP.020.042.0001_4 at 0045_4); Santerre at 78 (EXP.020.052.0001_4 at 0080_4)
RWS Ch 11 at [261] (SBM.010.005.0461_2)
Guelcher 1 at 8 (EXP.010.066.0001_3 at 0010_3); Liebert T et al, “Subcutaneous implants of polypropylene filaments” (1976) 10(6) J Biomed Mater Res 939–951 (ETH.MESH.10575742)
T2194 (TRA.500.028.0001_3 at 0025_3)
Liebert T et al, “Subcutaneous implants of polypropylene filaments” (1976) 10(6) J Biomed Mater Res 939–951 (ETH.MESH.10575742 at 5753)
Guelcher 2 at 1–2 (EXP.010.138.0001_3 at 0003_3)
Thames S, White J and Ong K, “The myth: in vivo degradation of polypropylene-based meshes” (2017) 28(2) Int Urogynecol J 285–297 (PUB.MESH.00004875); T2922 (TRA.500.038.0001_3 at 0018_3)
Williams D, “Biodegradation of surgical polymers” (1982) 17 J Mater Sci 1233–1246 (SHI.MESH.00000001)
Williams D, “Mechanisms of Biodegradation of Implantable Polymers” (1992) 10(1-2) Clin Mater 9–12 (SHI.MESH.00040883)
T1794 (TRA.500.022.0001_2 at 0070_2)
Thompson M et al, “In vivo polypropylene mesh degradation is hardly a myth” (2017) 28(2) Int Urogynecol J 333–335 (SHI.MESH.00025319)
SHI.MESH.00025335
T2219 (TRA.500.028.0001_3 at 0050_3)
MacLean at 11 (EXP.020.042.0001_4 at 0011_4)
MacLean at 16 (EXP.020.042.0001_4 at 0016_4)
Klosterhalfen 2 at [205]–[206] (LAY.010.018.0001_3 at 0071_3–0072_3); T2057–8 (TRA.500.026.0001_2 at 0034_2-0035_2)
Klosterhalfen 2 at 72 (LAY.010.018.0001_3 at 0072_3)
T2045, 2057–58 (TRA.500.026.0001_2 at 0022_2, 0034_2–0035_2)
ETH.MESH.09888068 at 8078
ETH.MESH.09888068 at 8078
ETH.MESH.11336184 at 6212
ETH.MESH.11336071 at 6075, 6083
Iakovlev 1 at 23 (EXP.010.036.0001_4 at 0024_4)
RWS ch 11 at [160] (SBM.020.002.0275 at 0326)
T2226 (TRA.500.028.0001 at 0059)
T1830–31 (TRA.500.023.0001 at 0023_2–0024_2)
T1770 (TRA.500.022.0001_2 at 0046_2)
T2059 (TRA.500.026.0001_2 at 0036_2–0037_2)
Maher C et al “Surgical management for pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2013) at 4 (SHI.MESH.00004130 at 4141)
Woodward 1 at [2.2.2] (EXP.010.044.0001_3 at 0011_3)
Gordon 1 at 1 (EXP.010.096.0001_3 at 0006_3)
Hu 1 at 2 (EXP.010.023.0001_3 at 0003_3)
Krulewitch 1 at [1.5] (EXP.010.081.0001_3 at 0010_3)
Krulewitch 1 at [1.4] (EXP.010.081.0001_3 at 0009_3)
Krulewitch 1 at [1.7] (EXP.010.081.0001_3 at 0011_3)
Woodward 1 at 1 (EXP.010.044.0001_3 at 0003_3)
Woodward 1 at 4 (EXP.010.044.0001_3 at 0006_3)
Woodward 1 at 5 (EXP.010.044.0001_3 at 0007_3)
Gordon 1 at 6 (EXP.010.096.0001_3 at 0008_3)
Gordon 1 at 7–8 (EXP.010.096.0001_3 starting at 0009_3)
RWS ch 5 at [1] (SBM.020.002.0083 at 0084)
RWS ch 5 at [2], [7], [36] (SBM.020.002.0083 at 0084, 0085 and 0100); SBM.020.002.0002 at 0005_3
RWS ch 5 at [36(a)] (SBM.020.002.0083 at 0100); T2693 (TRA.500.034.0001_3 at 0036_3)
RWS ch 5 at [2] (SBM.020.002.0083 at 0084)
Gordon 1 at 83 (EXP.010.096.0001_3 at 0085_3)
SBM.020.016.0001 at 0002
Hu 1 at 5 (EXP.010.023.0001_3 at 0007_3)
EXP.010.057.0001_3
T5125–26 (TRA.500.063.0001 at 0054–0055)
T3772–73 (TRA.500.047.0001_3 at 0064_3–0065_3)
ETH.MESH.06372555
Lam at 12 (EXP.020.021.0001_4 at 0014_4)
T3863 (TRA.500.048.0001_2 at 0070_2)
Withagen M et al, “Risk Factors for Exposure, Pain, and Dyspareunia After Tension-Free Vaginal Mesh Procedure” (2011) 118 Obstet Gynecol 629–636 at 629–630 (ETH.MESH.04929975 at 9975–9976)
Ibid at 630 (ETH.MESH.04929975 at 9976)
Ibid (ETH.MESH.04929975 at 9976)
Ibid at 630–31 (ETH.MESH.04929975 at 9976–9977)
Ibid at 632 (ETH.MESH.04929975 at 9978)
Ibid at 634 (ETH.MESH.04929975 at 9980)
Ibid (ETH.MESH.04929975 at 9980)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) (SHI.MESH.00005748)
Woodward 2 at [6.7.4] (EXP.010.106.0001_3 at 0055_3)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) (SHI.MESH.00005748 at 5834)
De Tayrac R and Sentilhes L, “Complications of pelvic organ prolapse surgery and methods of prevention” (2013) 24 Int Urogynecol J 1859–1872 at 1863 (ETH.MESH.12358329 at 8333)
T3863 (TRA.500.048.0001_2 at 0070_2)
De Tayrac R and Sentilhes L, “Complications of pelvic organ prolapse surgery and methods of prevention” (2013) 24 Int Urogynecol J 1859–1872 at 1868 (ETH.MESH.12358329 at 8338)
T3864 (TRA.500.048.0001_2 at 0071_2)
De Tayrac R and Sentilhes L, “Complications of pelvic organ prolapse surgery and methods of prevention” (2013) 24 Int Urogynecol J 1859–1872 at 1861 (ETH.MESH.12358329 at 8331)
Korda 1 at 73 (EXP.010.078.0001_3 at 0075_3)
Korda 1 at 73 (EXP.010.078.0001_3 at 0075_3)
Korda 1 at 80 (EXP.010.078.0001_3 at 0082_3)
Diwadkar G et al, “Complication and Reoperation Rates After Apical Vaginal Prolapse Surgical Repair” (2009) 113(2) Obstet & Gynecol 367–373 (ETH.MESH.00020695); Korda 3 at 12 (EXP.010.129.0001_5 at 0013_5)
Korda 3 at 5 (EXP.010.129.0001_5 at 0006_5)
Korda 3 at 12 (EXP.010.129.0001_5 at 0013_5)
Rosamilia 1 at 28 (EXP.020.026.0001_4 at 0030_4)
Higgs P, Chua H and Smith A, “Long term review of laparoscopic sacrocolpopexy” (2005) 112 BJOG 1134–1138 (ETH.MESH.08516881)
Nygaard I et al, “Long-term Outcomes Following Abdominal Sacrocolpopexy for Pelvic Organ Prolapse” (2013) 309(19) JAMA 2016–2024 (ETH.MESH.09466922)
Higgs P, Chua H and Smith A, “Long term review of laparoscopic sacrocolpopexy” (2005) 112 BJOG 1134–1138 (ETH.MESH.08516881 at 6883)
Nygaard I et al, “Long-term Outcomes Following Abdominal Sacrocolpopexy for Pelvic Organ Prolapse” (2013) 309(19) JAMA 2016–2024 at 2023 (ETH.MESH.09466922 at 6929)
Korda 1 at 83 (EXP.010.078.0001_3 at 0085_3)
SHI.MESH.00006213 at 6216
ETH.MESH.04038032 at 8043
ETH.MESH.10684212 at 4216
Korda 1 at 83 (EXP.010.078.0001_3 at 0085_3)
Margolis 1 at 22 (EXP.010.041.0001_3 at 0023_3)
Deprest at [273] (EXP.020.006.0001_4 at 0059_4)
Ostergard D, “Polypropylene Vaginal Mesh Grafts in Gynecology” (2010) 116 Obstet Gynecol 962–966 at 963 (ETH.MESH.00544274 at 4275)
Abbott S et al, “Evaluation and management of complications from synthetic mesh after pelvic reconstructive surgery: a multicenter study” (2014) 210 Am J Obstet Gynecol 163.e1–163.e8 at 163.e6 (SHI.MESH.00001610 at 1615)
Chughtai B et al, “Association Between the Amount of Vaginal Mesh Used With Mesh Erosions and Repeated Surgery After Repairing Pelvic Organ Prolapse and Stress Urinary Incontinence” (2016) 152(3) JAMA Surg 257–263 at 257 (SHI.MESH.00029503 at 9503)
Korda 1 at 72 (EXP.010.078.0001_3 at 0074_3)
Korda 1 at 69 (EXP.010.078.0001_3 at 0071_3)
Korda 1 at 83 (EXP.010.078.0001_3 at 0085_3)
Rosamilia 1 at 29 (EXP.020.026.0001_4 at 0031_4)
Korda 3 at 12 (EXP.010.129.0001_5 at 0013_5); Klosterhalfen 1 at [210] (LAY.010.014.0001_6 at 0045_6)
Collinet at 6 (EXP.020.005.0001_4 at 0006_4); Korda 3 at 21 (EXP.010.129.0001_5 at 0022_5)
Korda 3 at 20 (EXP.010.129.0001_5 at 0021_5)
T6132 (TRA.500.076.0001 at 0096)
Korda 3 at 21 (EXP.010.129.0001_5 at 0022_5)
Elliott D, “Con: mesh in vaginal surgery: do the risks outweigh the benefits?” (2012) 22(4) Curr Opin Urol 276–281 at 278 (SHI.MESH.00033536 at 3538–39)
T1324–25 (TRA.500.016.0001_2 at 0006–0007_2)
Elliott D, “Con: mesh in vaginal surgery: do the risks outweigh the benefits?” (2012) 22(4) Curr Opin Urol 276–281 at 278 (SHI.MESH.00033536 at 3538–39)
T1320–21 (TRA.500.016.0001_2 at 0002_2–0003_2)
T3362 (TRA.500.043.0001_3 at 0008_3)
JJM.MESH.00082931 at 2937
T1326 (TRA.500.016.0001_2 at 0008_2)
Abbott S et al, “Evaluation and management of complications from synthetic mesh after pelvic reconstructive surgery: a multicenter study”(2014) 210 Am J Obstet Gynecol 163.e1–163.e8 at 163.e1 (SHI.MESH.00001610 at 1610)
Ibid at 163.e6 (SHI.MESH.00001610 at 1615)
T3356 (TRA.500.043.0001_3 at 0002_3)
ETH.MESH.00580971 at 0972
ETH.MESH.00580971 at 0971
T2681 (TRA.500.034.0001_3 at 0024_3)
T3395 (TRA.500.043.0001_3 at 0041_3)
Woodward 1 at [2.2.4] (EXP.010.044.0001_3 at 0012_3)
Woodward 1 at [2.4.3] (EXP.010.044.0001_3 at 0014_3–0015_3)
Woodward 1 at [2.4.3] (EXP.010.044.0001_3 at 0014_3–0015_3)
Woodward 1at [2.4.6] (EXP.010.044.0001_3 at 0016_3)
Woodward 1at [2.4.7] (EXP.010.044.0001_3 at 0017_3)
Woodward 1at [2.4.4] (EXP.010.044.0001_3 at 0015_3)
Krulewitch 1 at 12 (EXP.010.081.0001_3 at 0015_3); Woodward 1 [2.4.2] (EXP.010.044.0001_3 at 0014_3)
Altman D and Bland J, “Treatment allocation in controlled trials: why randomise?” (1999) 318 BMJ 1209 (SHI.MESH.00032771)
Krulewitch 1 at 12 (EXP.010.081.0001_3 at 0015_3)
T4257 (TRA.500.053.0001_2 at 0015_2)
Greenhalgh G, “How to read a paper: Assessing the methodological quality of published papers”(1997) 315 BMJ 305–308 at 307 (MSC.010.150.0001 at 0007)
Woodward 1 at [2.4.3] (EXP.010.044.0001_3 at 0014_3)
Gordon 2 at 6 (EXP.010.161.0001_3 at
Altman D and Bland J, “Treatment allocation in controlled trials: why randomise?” (1999) 318 BMJ 1209 (SHI.MESH.00032771)
Ibid (SHI.MESH.00032771)
T2590 (TRA.500.033.0001_3 at 0019_3)
T2590 (TRA.500.033.0001_3 at 0019_3)
Krulewitch 1 at 12 (EXP.010.081.0001_3 at 0015_3)
Woodward 1 at [2.4.12] (EXP.010.044.0001_3 at 0019_3)
Woodward 1 at [2.4.13] (EXP.010.044.0001_3 at 0020_3)
Woodward 1 at [2.4.9] (EXP.010.044.0001_3 at 0017_3–0018_3)
Woodward 1 at [2.3.1] (EXP.010.044.0001_3 at 0013_3)
Woodward 1 at [2.3.1] (EXP.010.044.0001_3 at 0013_3)
Woodward 1 at [2.4.15] (EXP.010.044.0001_3 at 0020_3)
Woodward 1 at [2.4.16] (EXP.010.044.0001_3 at 0021_3)
T2566 (TRA.500.032.0001_3 at 0085_3)
SHI.MESH.00019045
Krulewitch 1 at 23 (EXP.010.081.0001_3 at 0026_3); see also T2689 (TRA.500.034.0001_3 at 0032_3) (Hu)
T2589 (TRA.500.033.0001_3 at 0018_3)
T2589–90 (TRA.500.033.0001_3 at 0018_3–0019_3)
PUB.MESH.00011898
PUB.MESH.00011898
T4055 (TRA.500.050.0001_2 at 0074_2)
Ibid (TRA.500.050.0001_2 at 0074_2)
Greenhalgh G, “How to read a paper: Assessing the methodological quality of published papers”(1997) 315 BMJ 305–308 at 308 (MSC.010.150.0001 at 0008)
Ibid (MSC.010.150.0001 at 0008)
Catto J et al, “In Defense of Randomized Clinical Trials in Surgery: Let Us Not Forget Archie Cochrane’s Legacy” (2017) 71 Eur Urol 820 (SHI.MESH.00038743)
Ulmsten U et al, “A Multicenter Study of Tension-Free Vaginal Tape (TVT) for Surgical Treatment of Stress Urinary Incontinence” (1998) 9 Int Urogynecol J 210–213 (ETH.MESH.01769250)
Ibid at 212 (ETH.MESH.01769250 at 9252)
Ibid at 213 (ETH.MESH.01769250 at 9253)
Nilsson C et al, “Long-term Results of the Tension-Free Vaginal Tape (TVT) Procedure for Surgical Treatment of Female Stress Urinary Incontinence” (2001) (Suppl 2) Int Urogynecol J S5–S8 (ETH.MESH.00756407)
AWS ch 6 at [195] (SBM.010.005.0156_2 at 0199)
Nilsson C et al, “Long-term Results of the Tension-Free Vaginal Tape (TVT) Procedure for Surgical Treatment of Female Stress Urinary Incontinence” (2001) (Suppl 2) Int Urogynecol J S5–S8 at S6 (ETH.MESH.00756407 at 6409)
Ibid at S7 (ETH.MESH.00756407 at 6410)
Ulmsten U et al, “A Multicenter Study of Tension-Free Vaginal Tape (TVT) for Surgical Treatment of Stress Urinary Incontinence” (1998) 9 Int Urogynecol J 210–213 (ETH.MESH.01769250)
T4237–39 (TRA.500.052.0001_2 at 0077_2–0079_2)
T4237–39 (TRA.500.052.0001_2 at 0077_2–0079_2)
T4230 (TRA.500.052.0001 at 0074); see also ETH.MESH.11858511
T661 (TRA.500.008.0001_2 at 0050_2
T779 (TRA.500.009.0001_2 at 0050_2)
T4238–39 (TRA.500.052.0001_2 at 0078_2–0079_2)
T4165 (TRA.500.052.0001_2 at 0005_2)
AWS ch 12 at [149] (SBM.010.005.0529_2 at 0058_2)
ETH.MESH.05225354 at 5380
T4235 (TRA.500.052.0001_2 at 0079_2)
T4235 (TRA.500.052.0001_2 at 0079_2)
Nilsson C et al, “Long-term Results of the Tension-Free Vaginal Tape (TVT) Procedure for Surgical Treatment of Female Stress Urinary Incontinence” (2001) (Suppl 2) Int Urogynecol J S5–S8 at S6 (ETH.MESH.00756407 at 6409)
Nilsson C, Falconer C and Rezapour M, “Seven-Year Follow-up of the Tension-Free Vaginal Tape Procedure for Treatment of Urinary Incontinence” (2004) 104(6) Obstet & Gynecol 1259–1262 (ETH.MESH.19878441)
Nilsson C, Falconer C and Rezapour M, “Seven-Year Follow-up of the Tension-Free Vaginal Tape Procedure for Treatment of Urinary Incontinence” (2004) 104(6) Obstet & Gynecol 1259–1262 at 1261 (ETH.MESH.19878441 at 8443)
T2591 (TRA.500.033.0001_3 at 0020)
Nilsson C et al, “Eleven years prospective follow-up of the tension-free vaginal tape procedure for treatment of stress urinary incontinence” (2008) Int Urogynecol J (ETH.MESH.05151145)
Ibid (ETH.MESH.05151145 at 1148)
Nilsson C et al, “Seventeen years' follow-up of the tension-free vaginal tape procedure for female stress urinary incontinence” (2013) Int Urogynecol J (ETH.MESH.19876896)
T4244 (TRA.500.053.0001_2 at 0002_2)
AWS ch 12 at [150] (SBM.010.005.0529_2 at 0569_2)
Nilsson C et al, “Seventeen years' follow-up of the tension-free vaginal tape procedure for female stress urinary incontinence” (2013) Int Urogynecol J (ETH.MESH.19876896 at 6898)
Ibid (ETH.MESH.19876896 at 6899)
Ibid (ETH.MESH.19876896 at 6899)
Nilsson C et al, “Seventeen years' follow-up of the tension-free vaginal tape procedure for female stress urinary incontinence” (2013) Int Urogynecol J (ETH.MESH.19876896 at 6899)
Ibid (ETH.MESH.19876896 at 6900)
T4244 (TRA.500.053.0001_2 at 0002_2)
T4245 (TRA.500.053.0001_2 at 0003_2)
T4231–32 (TRA.500.052.0001_2 at 0071_2–0072_2); see also PUB.MESH.00001813
T4232 (TRA.500.052.0001_2 at 0072_2); see also SHI.MESH.00015461
Ward K and Hilton P, “A prospective multicenter randomized trial of tension-free vaginal tape and colposuspension for primary urodynamic stress incontinence: Two-year follow-up” (2004) 194 Am J Obstet Gynecol 324–331 at 325 (ETH.MESH.00016582 at 6583)
Ward K and Hilton P, “Minimally invasive synthetic suburethral slings: emerging complications” (2005) 7 TOG 223–232 at 223 (ETH.MESH.04087047 at 7047)
Ward K and Hilton P, “A prospective multicenter randomized trial of tension-free vaginal tape and colposuspension for primary urodynamic stress incontinence: Two-year follow-up” (2004) 194 Am J Obstet Gynecol 324–331 at 326 (ETH.MESH.00016582 at 6584)
Ibid at 324 (ETH.MESH.00016582 at 6582)
Hinoul at [286] (LAY.020.001.0001_4 at 0110_4)
ETH.MESH.01734903 at 4905
Krulewitch 2 at 14–16 (EXP.010.117.0001_3 at 0017_3–0019_3)
Ward K and Hilton P, “A prospective multicenter randomized trial of tension-free vaginal tape and colposuspension for primary urodynamic stress incontinence: Two-year follow-up” (2004) 194 Am J Obstet Gynecol 324–331 at 325 (ETH.MESH.00016582 at 6583)
Ibid (ETH.MESH.00016582 at 6583)
Ibid (ETH.MESH.00016582 at 6583)
Krulewitch 2 at 16 (EXP.010.117.0001_3 at 0019_3); T2428–29 (TRA.500.031.0001_3 at 0027_3–0028_3)
Ward K and Hilton P, “Prospective multicentre randomised trial of tension-free vaginal tape and colposuspension as primary treatment for stress incontinence” (2002) 325 BMJ 1–7 (ETH.MESH.13649139)
Gordon 1 at 12 (EXP.010.096.0001_3 at 0014_3)
Webb P and Bain C, Essential Epidemiology (Cambridge University Press, 2nd ed, 2011) at 430
Gordon 1 at 12 (EXP.010.096.0001_3 at 0014_3)
Ibid (EXP.010.096.0001_3 at 0014_3)
T2695 (TRA.500.034.0001_3 at 0038_3)
Woodward 1 at [2.1.3] (EXP.010.044.0001_3 at 0008_3); Gordon 1 at 14 (EXP.010.096.0001_3 at 0016_3)
Ibid at [2.1.5] (EXP.010.044.0001_3 at 0009_3)
Ibid at 3 (ETH.MESH.13649139 at 9141)
Ibid at 4 (ETH.MESH.13649139 at 9142)
AWS ch 12 at [136], [139]–[140] (SBM.010.005.0529_2 at 0056_2)
T4216 (TRA.500.052.0001_2 at 0056_2)
T4216 (TRA.500.052.0001_2 at 0056_2)
Ward K and Hilton P, “Prospective multicentre randomised trial of tension-free vaginal tape and colposuspension as primary treatment for stress incontinence” (2002) 325 BMJ 1–7 at 6 (ETH.MESH.13649139 at 9144)
Krulewitch 2 at 16 (EXP.010.117.0001_3 at 0019_3)
Krulewitch 2 at 16 (EXP.010.117.0001_3 at 0019_3)
Ward K and Hilton P, “Prospective multicentre randomised trial of tension-free vaginal tape and colposuspension as primary treatment for stress incontinence” (2002) 325 BMJ 1–7 at 6 (ETH.MESH.13649139 at 9144)
Ibid at 3, 6 (ETH.MESH.13649139 at 9141, 9144)
Ward K and Hilton P, “ A prospective multicenter randomized trial of tension-free vaginal tape and colposuspension for primary urodynamic stress incontinence: Two-year follow-up” (2004) 190 Am J Obstet Gynecol 324–331 (ETH.MESH.00016582)
Ibid at 325 (ETH.MESH.00016582 at 6583)
Ibid at 327 (ETH.MESH.00016582 at 6585)
Ibid at 329 (ETH.MESH.00016582 at 6587); T4212 (TRA.500.052.0001_2 at 0052_2)
Ward K and Hilton P, “Tension-free vaginal tape versus colposuspension for primary urodynamic stress incontinence: 5-year follow up” (2008) 115 BJOG 226–233 (ETH.MESH.04045078 at 5083); T4224 (TRA.500.052.0001_2 at 0064_2)
AWS ch 12 at [144] (SBM.010.005.0529_2 at 0567_2)
Ward K and Hilton P, “A prospective multicenter randomized trial of tension-free vaginal tape and colposuspension for primary urodynamic stress incontinence: Two-year follow-up” (2004) 190 Am J Obstet Gynecol 324–331 at 325, 328 (ETH.MESH.00016582 at 6583, 6586)
Ibid at 329 (ETH.MESH.00016582 at 6587)
Ibid at 324 (ETH.MESH.00016582 at 6582)
T4213 (TRA.500.052.0001_2 at 0053_2)
ETH.MESH.00994498
Ward K and Hilton P, “Tension-free vaginal tape versus colposuspension for primary urodynamic stress incontinence: 5-year follow up” (2008) 115 BJOG 226–233 (ETH.MESH.04045078)
T4223 (TRA.500.052.0001_2 at 0063_2)
Ward K and Hilton P, “Tension-free vaginal tape versus colposuspension for primary urodynamic stress incontinence: 5-year follow up” (2008) 115 BJOG 226–233 at 331 (ETH.MESH.04045078 at 5083)
Ibid at 231–232 (ETH.MESH.04045078 at 5083–84)
Ibid at 228 (ETH.MESH.04045078 at 5080)
Ibid at 231 (ETH.MESH.04045078 at 5083)
Ibid at 232 (ETH.MESH.04045078 at 5084)
Ibid (ETH.MESH.04045078 at 5084)
Krulewitch 1 at 62 (EXP.010.081.0001_3 at 0065_3)
Ward K and Hilton P, “Tension-free vaginal tape versus colposuspension for primary urodynamic stress incontinence: 5-year follow up” (2008) 115 BJOG 226–233 at 332 (ETH.MESH.04045078 at 5084)
Ibid at 229 (ETH.MESH.04045078 at 5081)
Ibid at 232 (ETH.MESH.04045078 at 5084)
T640 (TRA.500.008.0001_2 at 0029_2)
T644 (TRA.500.008.0001_2 at 0033_2)
T645 (TRA.500.008.0001_2 at 0034_2)
Blaivas 2 at 7 (EXP.010.166.0001_2 at 0009_2)
Dean N et al, “Laparoscopic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2006) (SHI.MESH.00002325)
Dean N et al, “Laparoscopic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2017) (PUB.MESH.00010784)
Ogah J, Cody J and Rogerson L, “Minimally invasive synthetic suburethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) (SHI.MESH.00003908)
AWS ch 12 at [132] (SBM.010.005.0529_2 at 0565_2)
T4210 (TRA.500.052.0001_2 at 0050_2)
Ogah J, Cody J and Rogerson L, “Minimally invasive synthetic suburethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) at 10 (SHI.MESH.00003908 at 3923)
Ibid at 2 (SHI.MESH.00003908 at 3915)
Ibid (SHI.MESH.00003908 at 3915)
Ford A et al, “Mid-urethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) at 47 (SHI.MESH.00004603 at 4654)
Ibid (SHI.MESH.00004603 at 4654)
Ibid (SHI.MESH.00004603 at 4654)
Ibid at 2 (SHI.MESH.00004603 at 4609)
Ibid (SHI.MESH.00004603 at 4609)
Ibid at 2–3 (SHI.MESH.00004603 at 4609–4610)
RWS ch 9 at [15] (SBM.020.002.0195 at 0201)
Ford A et al, “Mid-urethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) at 25–28 (SHI.MESH.00004603 at 4632–4635)
Ibid at 3 (SHI.MESH.00004603 at 4610)
Lapitan M, Cody J and Grant A, “Open retropubic colposuspension for urinary incontinence in
women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) (ETH.MESH.04558879)
Lapitan M and Cody J, “Open retropubic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2012) (SHI.MESH.00005007)
Lapitan M and Cody J, “Open retropubic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) (SHI.MESH.00005945)
Lapitan M, Cody J and Grant A, “Open retropubic colposuspension for urinary incontinence in
women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) at 1 (ETH.MESH.04558879 at 8885)
Ibid (ETH.MESH.04558879 at 8892–8893)
Ibid at 2 (ETH.MESH.04558879 at 8886)
Ibid at 24 (ETH.MESH.04558879 at 8908)
Lapitan M and Cody J, “Open retropubic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2012) (SHI.MESH.00005007)
Ibid at 24 (SHI.MESH.00005007 at 0036)
Ibid at 103 (SHI.MESH.00005007 at 5115)
Lapitan M and Cody J, “Open retropubic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 1 (SHI.MESH.00005945 at 5951)
Ibid at 2 (SHI.MESH.00005945 at 5952)
Ibid (SHI.MESH.00005945 at 5952)
Nambiar A, Cody J and Jeffery S, “Single-incision sling operations for urinary incontinence in women” The Cochrane Collaboration (John Wiley & Sons Ltd, 2014) at 2 (SHI.MESH.00004889 at 4894)
Glazener C and Cooper K, “Bladder neck needle suspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) (SHI.MESH.00005250)
Glazener C, Cooper K and Mashayekhi A, “Bladder neck needle suspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2017) (PUB.MESH.00011151).
Glazener C, Cooper K and Mashayekhi A, “Anterior vaginal repair for urinary incontinence in women
(Review)” (John Wiley & Sons Ltd, 2017) (PUB.MESH.00011228)
Glazener C and Cooper K, “Bladder neck needle suspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) (SHI.MESH.00005250)
Glazener C, Cooper K and Mashayekhi A, “Anterior vaginal repair for urinary incontinence in women
(Review)” (John Wiley & Sons Ltd, 2017) at 1 (PUB.MESH.00011228 at 1231)
RWS ch 5 [13(c)], [32]–[33] (SBM.020.002.0083 at 0089, 0098)
Novara G et al, “Tension-Free Midurethral Slings in the Treatment of Female Stress Urinary Incontinence: A Systematic Review and Meta-analysis of Randomized Controlled Trials of Effectiveness” (2007) 52 Eur Urol 663–679 at 663 (SHI.MESH.00000721 at 0721)
Novara G et al, “Updated Systematic Review and Meta-Analysis of the Comparative Data on Colposuspensions, Pubovaginal Slings, and Midurethral Tapes in the Surgical Treatment of Female Stress Urinary Incontinence” (2010) 58 Eur Urol 218–238 (ETH.MESH.03419524)
Ibid at 234 (ETH.MESH.03419524 at 9540)
Ibid at 589–590 (ETH.MESH.03419524 at 9539–9540)
Ibid at 235 (ETH.MESH.03419524 at 9541)
Fusco F et al, “Updated Systematic Review and Meta-analysis of the Comparative Data on Colposuspensions, Pubovaginal Slings, and Midurethral Tapes in the Surgical Treatment of Female Stress Urinary Incontinence” (2017) 72 Eur Urol 567–591 (PUB.MESH.00121617)
Ibid at 588–589 (PUB.MESH.00121617 at 1638–1639)
Ibid at 588–589 (PUB.MESH.00121617 at 1638–39)
Ibid at 588 (PUB.MESH.00121617 at 1638)
Zhang Z et al, “Retropubic tension-free vaginal tape and inside-out transobturator tape: a long-term randomized trial” (2016) 27 Int Urogynecol J 103–111 (SHI.MESH.00007402)
Ibid at 108 (SHI.MESH.00007402 at 7407)
De Souza A et al, “Sexual function following retropubic TVT and transobturator Monarc sling in women with intrinsic sphincter deficiency: a multicentre prospective study” (2010) Int Urogynecol J (ETH.MESH.04087987 at 7989)
Hinoul at [365] (LAY.020.001.0001_4 at 0142_4)
Zyczynski H et al, “Sexual activity and function in women more than 2 years after midurethral sling placement” (2012) 207 Am J Obstet Gynecol 421.e1–421.e6 (SHI.MESH.00001723)
Ibid at 421.e6 (SHI.MESH.00001723 at 1728)
Morling R et al, “Adverse events after first, single, mesh and non-mesh surgical procedures for stress urinary incontinence and pelvic organ prolapse in Scotland, 1997-2016: a population based cohort study” (2016) The Lancet 1–12 (SHI.MESH.00024517)
Ibid at 7 (SHI.MESH.00024517 at 4523)
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2004) at 8 (PUB.MESH.00009046 at 9055)
Ibid (PUB.MESH.00009046 at 9055); see further SHI.MESH.00014672
Ibid (PUB.MESH.00009046 at 9055); see further ETH.MESH.08089321
ETH.MESH.08089321 at 9325
Rosamilia 1 at 21 (EXP.020.026.0001_4 at 0023_4)
Woodward 1 at 25 (EXP.010.044.0001_3 at 0027_3)
SHI.MESH.00038518
SHI.MESH.00038518 at 8525
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2007) at 18 (SHI.MESH.00011315 at 1337)
Ibid at 17 (SHI.MESH.00011315 at 1336)
Ibid at 18 (SHI.MESH.00011315 at 1337)
T4140 (TRA.500.051.0001_2 at 0063_2); T4259 (TRA.500.053.0001_2 at 0017_2)
SHI.MESH.00028977 at 8977
SHI.MESH.00028977 at 8978
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2010) at 2 (ETH.MESH.01028668 at 8676)
Ibid (ETH.MESH.01028668 at 8676)
Ibid (ETH.MESH.01028668 at 8676)
Ibid at 26 (ETH.MESH.01028668 at 8700)
Ibid (ETH.MESH.01028668 at 8700)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 (ETH.MESH.07971133)
Ibid at 1827 (ETH.MESH.07971133 at 1134)
Woodward 1 at [3.1.28] (EXP.010.044.0001_3 at 0049_3)
AWS annexure 2 at [62] (SBM.010.007.0001 at 0036)
Roovers 1 at 13 (EXP.020.033.0001_3 at 0015_3)
Woodward 1 at [4.4] (EXP.010.044.0001_3 at 0069_3)
Woodward 2 at [4] (EXP.010.106.0001_3 at 0020_3)
Ibid at [5] (EXP.010.106.0001_3 at 0020_3)
Ibid at [7] (EXP.010.106.0001_3 at 0021_3)
Roovers 1 at 13 (EXP.020.033.0001_4 at 0015_4); see also ETH.MESH.22325924
T4084 (TRA.500.051.0001_2 at 0007_2)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) 40–41 (SHI.MESH.00005748 at 5790–5791)
Woodward 2 [6.2.2] (EXP.010.106.0001_3 at 0047_3)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1835 (ETH.MESH.07971133 at 1142)
Ibid at 1828 (ETH.MESH.07971133 at 1135)
SHI.MESH.00010243
ETH.MESH.01264341
ETH.MESH.04526463 at 6491
ETH.MESH.03136930
T3460 (TRA.500.044.0001_3 at 0012_3)
T2684–85 (TRA.500.034.0001_3 at 0027_3–0028_3)
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2013) at 44 (PUB.MESH.00001398 at 1449)
ETH.MESH.01205987 at 5987
ETH.MESH.01205987 at 5988
ETH.MESH.01258976
ETH.MESH.04526463 at 6465
ETH.MESH.01258976 at 8985
ETH.MESH.00578893 at 8896
T3454 (TRA.500.044.0001_3 at 0006_3)
ETH.MESH.04526463 at 6464
ETH.MESH.00578939 at 8941
ETH.MESH.07971133 at 1139
ETH.MESH.01205987 at 5997
Woodward 1 at [3.1.35] (EXP.010.044.0001_3 at 0052_3)
Hu 1 at 20–21 (EXP.010.023.0001_3 at 0022_3–0023_3)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1826 (ETH.MESH.07971133 at 1133)
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2013) at 27 (PUB.MESH.00001398 at 1432)
Ibid (PUB.MESH.00001398 at 1432)
Ibid at 29 (PUB.MESH.00001398 at 1434)
Ibid (PUB.MESH.00001398 at 1434)
Collinet at 29 (EXP.020.005.0001_4 at 0029_4)
Gordon 2 at 34 (EXP.010.161.0001_3 at 0036_3)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) (SHI.MESH.00005748)
Ibid at 1 (SHI.MESH.00005748 at 5751)
Ibid at 12 (SHI.MESH.00005748 at 5762)
Ibid at 7 (SHI.MESH.00005748 at 5757)
Ibid at 8 (SHI.MESH.00005748 at 5758)
Ibid at 15 (SHI.MESH.00005748 at 5765)
Ibid at 16 (SHI.MESH.00005748 at 5766)
Ibid at 16 (SHI.MESH.00005748 at 5766)
Ibid (SHI.MESH.00005748 at 5766)
T4263 (TRA.500.053.0001_2 at 0021_2)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 16 (SHI.MESH.00005748 at 5766)
Ibid at 18 (SHI.MESH.00005748 at 5768)
Ibid at 2 (SHI.MESH.00005748 at 5752)
Woodward 1 at [3.1.19] (EXP.010.044.0001_3 at 0044_3)
Ibid at [3.1.16] (EXP.010.044.0001_3 at 0041_3–0042_3)
Woodward 1 at [3.1.16] (EXP.010.044.0001_3 at 0041_3); Hu 2 at 19–20 (EXP.010.184.0001_3 at 0020_3–0021_3)
Jia X et al, “Efficacy and safety of using mesh or grafts in surgery for anterior and/or posterior vaginal wall prolapse: systematic review and meta-analysis”(2008) 115 BJOG 1350–1361 at 1358 (SHI.MESH.00007412 at 7420)
Ibid (SHI.MESH.00007412 at 7420)
Rudnicki M et al, “A 3–year follow-up after anterior colporrhaphy compared with collagen-coated transvaginal mesh for anterior vaginal wall prolapse: a randomised controlled trial” (2016) 123 BJOG 136–146 at 141 (SHI.MESH.00005889 at 5894)
Woodward 1 at [3.1.18] (EXP.010.044.0001_3 at 0042_3)
Woodward 1 at [3.1.5] (EXP.010.044.0001_3 at 0023_3)
Woodward 1 at [2.4.26] (EXP.010.044.0001_3 at 0026_3)
Woodward 1 at [3.1.36] (EXP.010.044.0001_3 at 0052_3)
Woodward 1 at [3.1.36] (EXP.010.044.0001_3 at 0052_3)
Maher C et al, “Surgery for women with anterior compartment prolapse (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) (SHI.MESH.00043256)
Ibid at 1 (SHI.MESH.00043256 at 3260)
Ibid at 19 (SHI.MESH.00043256 at 3278)
Ibid at 20 (SHI.MESH.00043256 at 3279)
Ibid at 19 (SHI.MESH.00043256 at 3278)
Ibid at 21 (SHI.MESH.00043256 at 3280)
Ibid at 22 (SHI.MESH.00043256 at 3281)
Ibid at 3 (SHI.MESH.00043256 at 3262)
T4264 (TRA.500.053.0001_2 at 0022_2)
Maher et al, “Surgery for women with apical vaginal prolapse (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) (PUB.MESH.00004281)
Ibid at 3 (PUB.MESH.00004281 at 4288)
Woodward 2 at [3.2] (EXP.010.106.0001_3 at 0018_3)
Agur 2 at 5 (EXP.010.152.0001_3 at 0006_3)
Woodward 2 [2.4.1] (EXP.010.106.0001_3 at 0009_3)
Glazener C et al, “Mesh, graft, or standard repair for women having primary transvaginal anterior or posterior compartment prolapse surgery: two parallel-group, multicentre, randomised, controlled trials (PROSPECT)” (2016) The Lancet 1–12 (SHI.MESH.00024529)
Glazener C et al, “Clinical effectiveness and cost-effectiveness of surgical options for the management of anterior and/or posterior vaginal wall prolapse: two randomised controlled trials within a comprehensive cohort study – results from the PROSPECT Study” (2016) 20(95) Health Technol Assess vii–452 (PUB.MESH.00005456)
Woodward 2 [2.4.1] (EXP.010.106.0001_3 at 0009_3)
Glazener C et al, “Mesh, graft, or standard repair for women having primary transvaginal anterior or posterior compartment prolapse surgery: two parallel-group, multicentre, randomised, controlled trials (PROSPECT)” (2016) The Lancet 1–12 at 6 (SHI.MESH.00024529 at 4534)
Ibid at 3 (SHI.MESH.00024529 at 4534)
Ibid at 5 (SHI.MESH.00024529 at 4533)
Ibid at 10 (SHI.MESH.00024529 at 4538)
Ibid at 10 (SHI.MESH.00024529 at 4538)
T1111 (TRA.500.013.0001_2 at 0036_2)
Glazener C et al, “Mesh, graft, or standard repair for women having primary transvaginal anterior or posterior compartment prolapse surgery: two parallel-group, multicentre, randomised, controlled trials (PROSPECT)” (2016) The Lancet 1–12 at 8 (SHI.MESH.00024529 at 4531)
Ibid at 9 (SHI.MESH.00024529 at 4537)
Ibid at 1 (SHI.MESH.00024529 at 4529)
Ibid at 11 (SHI.MESH.00024529 at 4539)
T6061 (TRA.500.076.0001 at 0025)
T6141 (TRA.500.076.0001 at 0105)
Richter H et al, “Retropubic versus Transobturator Midurethral Slings for Stress Incontinence” (2010) 362(22) N Eng J Med 2066–2076 at 2074 (ETH.MESH.09231613 at 1621)
Ward K and Hilton P, “Tension-free vaginal tape versus colposuspension for primary urodynamic stress incontinence: 5-year follow up” (2008) 115 BJOG 226–233 at 229 (ETH.MESH.04045078 at 5081)
Schimpf M et al, “Sling surgery for stress urinary incontinence in women: a systematic review and metaanalysis” (2014) 211 Am J Obstet Gynecol 71.e1–71.237 at 71.e5 (ETH.MESH.18852919)
Hinoul P et al, “A Randomized, Controlled Trial Comparing an Innovative Single Incision Sling With an Established Transobturator Sling to Treat Female Stress Urinary Incontinence” (2011) 185 The Journal of Urology 1356–1362 (ETH.MESH.13213961 at 3965)
Ward K and Hilton P, “A prospective multicenter randomized trial of tension-free vaginal tape and colposuspension for primary urodynamic stress incontinence: Two-year follow-up” (2004) 190 Am J Obstet Gynecol 324–331 at 330 (ETH.MESH.00016582 at 6588)
Ford A et al, “Mid-urethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) at 45 (SHI.MESH.00004603 at 4652)
Gordon 1 at 82 (EXP.010.096.0001_3 at 0084_3)
Gyang et al, “Managing chronic pelvic pain following reconstructive pelvic surgery with transvaginal mesh” (2014) 25 Int Urogynecol J 313–318 at 317 (SHI.MESH.00020223); PUB.MESH.00008915
Barski D and Deng D, “Management of Mesh Complications after SUI and POP Repair: Review and Analysis of the Current Literature” (2015) BioMed Res Int 1–8 (SHI.MESH.00007704)
Bourrat M et al, “Complications and medium-term functional results of TVT in stress urinary incontinence” (2003) 13 Progress in Urology 1358–1364 (SHI.MESH.00043573_2)
T3679 (TRA.500.046.0001_3 at 0046_3)
SBM.020.013.0001 at 0006
ETH.MESH.06616596
Blaivas J et al, “Safety considerations for synthetic sling surgery” (2015) 12 Nat Rev Urol 481–509 at 485 (SHI.MESH.00005684 at 5688)
Blaivas 2 at 7 (EXP.010.166.0001_2 at 0010_2)
Ogah J, Cody J and Rogerson L, “Minimally invasive synthetic suburethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) (SHI.MESH.00003908)
Ibid at 22 (SHI.MESH.00003908 at 3935)
Ford A et al, “Mid-urethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) at 2 (SHI.MESH.00004603 at 4609)
Ibid at 28 (SHI.MESH.00004603 at 4635)
Zhang Z et al, “Retropubic tension-free vaginal tape and inside-out transobturator tape: a long-term randomized trial” (2016) 27 Int Urogynecol J 103–111 (SHI.MESH.00007402)
Ibid at 107 (SHI.MESH.00007402 at 7406)
Karmakar D, Mostafa A and Abdel-Fattah M, “Long-term outcomes of transobturator tapes in women with stress urinary incontinence: E-TOT randomised controlled trial” 124 BJOG 973–981 at 976 (SHI.MESH.00040783 at 0786)
ETH.MESH.10681490 at 1528
Hinoul P et al, “A Randomized, Controlled Trial Comparing an Innovative Single Incision Sling With an Established Transobturator Sling to Treat Female Stress Urinary Incontinence”, (2011) 185 The Journal of Urology, 1356–1362 at (ETH.MESH.13213961 at 3966)
JJM.MESH.00043638
Fusco F et al, “Updated Systematic Review and Meta-analysis of the Comparative Data on Colposuspensions, Pubovaginal Slings, and Midurethral Tapes in the Surgical Treatment of Female Stress Urinary Incontinence” (2017) 72 Eur Urol 567–591 at 588 (PUB.MESH.00121617 at 1638)
Bourrat M et al, “Complications and medium-term functional results of TVT in stress urinary incontinence” (2003) 13 Progress in Urology 1358–1364 (SHI.MESH.00043573_2)
Sha H and Badlani G, “Mesh complications in female pelvic floor reconstructive surgery and their management: A systematic review” (2012) 28(2) Indian J Urol 129–153 at 138 (SHI.MESH.00039513 at 9521)
Barski D and Deng D, “Management of Mesh Complications after SUI and POP Repair: Review and Analysis of the Current Literature” (2015) BioMed Res Int 1–8 at 2 (SHI.MESH.00007704 at 7705)
Dunn G et al, “Changed Women: The Long-Term Impact of Vaginal Mesh Complications” (2014) 20(3) Female Pelvic Med Reconstr Surg 131–136 (SHI.MESH.00010187)
Ford A et al, “Mid-urethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) (ETH.MESH.24982894 at 2943)
Nambiar A, Cody J and Jeffery S, “Single-incision sling operations for urinary incontinence in women” The Cochrane Collaboration (John Wiley & Sons Ltd, 2014) at 2 (SHI.MESH.00004889 at 4894)
AWS ch 9 at [40] (SBM.010.005.0379_2 at 0393_2)
Ogah J, Cody J and Rogerson L, “Minimally invasive synthetic suburethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) at 1 (SHI.MESH.00003908 at 3914)
Lapitan M and Cody J, “Open retropubic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 2 (SHI.MESH.00005945 at 5952)
Ibid at 20 (SHI.MESH.00005945 at 5970)
T1195 (TRA.500.014.0001_2 at 0068_2)
Ward K and Hilton P, “Prospective multicentre randomised trial of tension-free vaginal tape and colposuspension as primary treatment for stress incontinence” (2002) 325 BMJ 1–7 at 4 (ETH.MESH.13649139 at 9142)
Ford A et al, “Mid-urethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) (ETH.MESH.24982894 at 2924)
AWS ch 9 at [43] (SBM.010.005.0379_2 at 0934)
Ford A et al, “Mid-urethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) at 27 (ETH.MESH.24982894 at 2925)
Ogah J, Cody J and Rogerson L, “Minimally invasive synthetic suburethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) at 17 (SHI.MESH.00003908 at 3930)
Ibid at 2 (SHI.MESH.00003908 at 3915)
Lapitan M and Cody J, “Open retropubic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 2 (SHI.MESH.00005945 at 5952)
Dean N et al, “Laparoscopic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2017) at 2 (PUB.MESH.00010784 at 0788)
Rehman H et al, “Traditional suburethral sling operations for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2017) at 2 (PUB.MESH.00011745 at 1750)
Gordon 1 at 83 (EXP.010.096.0001_3 at 0085_3)
Fusco F et al, “Updated Systematic Review and Meta-analysis of the Comparative Data on Colposuspensions, Pubovaginal Slings, and Midurethral Tapes in the Surgical Treatment of Female Stress Urinary Incontinence” (2017) 72 Eur Urol 567–591 at 567, 569–570 (PUB.MESH.00121617 at 1617, 1619–1620)
Ford A et al, “Mid-urethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) at 1 (SHI.MESH.00004603 at 4608)
Ibid at 2 (SHI.MESH.00004603 at 4609)
Ibid at 25 (SHI.MESH.00004603 at 4632)
Ward K and Hilton P, “Prospective multicentre randomised trial of tension-free vaginal tape and colposuspension as primary treatment for stress incontinence” (2002) 325 BMJ 1–7 at 3 (ETH.MESH.13649139 at 9141)
Ogah J, Cody J and Rogerson L, “Minimally invasive synthetic suburethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) at 2 (SHI.MESH.00003908 at 3915)
Lapitan M and Cody J, “Open retropubic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 20 (SHI.MESH.00005945 at 5970)
Ogah J, Cody J and Rogerson L, “Minimally invasive synthetic suburethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) at 16, 18 (SHI.MESH.00003908 at 3929, 3931)
Hengel A et al, “Prevention, diagnosis, and management of midurethral mesh sling complications” (2017) 11 Can Urol Assoc J S135–S140 at S136 (PUB.MESH.00121284 at 1285)
Richter H et al, “Retropubic versus Transobturator Midurethral Slings for Stress Incontinence” (2010) 362(22) N Eng J Med 2066–2076 at 2072 (ETH.MESH.09231613 at 1619)
Albo M et al, “Treatment Success of Retropubic and Transobturator Mid Urethral Slings at 24 Months” (2012) 188 J Urol 2281–2287 at 2284–2285 (ETH.MESH.12358173 at 8176–8177
Deprest at [101] (EXP.020.006.0001_3 at 0021_3)
Dunn G et al, “Changed Women: The Long-Term Impact of Vaginal Mesh Complications” (2014) 20(3) Female Pelvic Med Reconstr Surg 131–136 (SHI.MESH.00010187)
Ward K and Hilton P, “A prospective multicenter randomized trial of tension-free vaginal tape and colposuspension for primary urodynamic stress incontinence: Two-year follow-up” (2004) 194 Am J Obstet Gynecol 324–331 at 327 (ETH.MESH.00016582 at 6585)
Withagen M et al, “Risk Factors for Exposure, Pain, and Dyspareunia After Tension-Free Vaginal Mesh Procedure” (2011) 118 Obstet Gynecol 629–636 (ETH.MESH.04929975)
Lam at 28 (EXP.020.021.0001_4 at 0030_4)
T3857 (TRA.500.048.0001_2 at 0064_2)
T3860 (TRA.500.048.0001_2 at 0067_2)
T3859 (TRA.500.048.0001_2 at 0066_2)
T3858 (TRA.500.048.0001_2 at 0065_2)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 16 (SHI.MESH.00005748 at 5766)
Maher C et al, “Surgery for women with anterior compartment prolapse (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 21 (SHI.MESH.00043256 at 3280)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 (ETH.MESH.07971133)
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2013) at 44 (PUB.MESH.00001398 at 1449)
RWS ch 9 [146] (SBM.020.002.0195 at 0258)
Korda 1 at 89 (EXP.010.078.0001_3 at 0091_3)
Halaska M, “A multicenter, randomized, prospective, controlled study comparing sacrospinous fixation and transvaginal mesh in the treatment of posthysterectomy vaginal vault prolapse” (2012) 207 Am J Obstet Gynecol 301.e1–301.e7 at 301.e5 (ETH.MESH.08299299 at 9303)
Korda 1 at 75 (EXP.010.078.0001_3 at 0077_3)
De Tayrac R and Sentilhes L, “Complications of pelvic organ prolapse surgery and methods of prevention” (2013) 24 Int Urogynecol J 1859–1872 at 1863 (ETH.MESH.12358329 at 8333)
Caquant F et al, “Safety of Trans Vaginal Mesh procedure: Retrospective study of 684 patients”, (2008) J Obstet Gynaecol Res 449–456 (ETH.MESH.02588589)
Glazener C et al, “Mesh, graft, or standard repair for women having primary transvaginal anterior or posterior compartment prolapse surgery: two parallel-group, multicentre, randomised, controlled trials (PROSPECT)” (2016) The Lancet 1–12 at 8 (SHI.MESH.00024529 at 4536)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1835 (ETH.MESH.07971133 at 1142)
Ibid at 1833 (ETH.MESH.07971133 at 1140)
De Tayrac R and Sentilhes L, “Complications of pelvic organ prolapse surgery and methods of prevention” (2013) 24 Int Urogynecol J 1859–1872 at 1863 (ETH.MESH.12358329 at 8333)
Withagen M et al, “Risk Factors for Exposure, Pain, and Dyspareunia After Tension-Free Vaginal Mesh Procedure” (2011) 118 Obstet Gynecol 629–636 at 632 (ETH.MESH.04929975 at 9978)
PUB.MESH.00009026 at 9038
T3809 (TRA.500.048.0001_2 at 0016_2)
Korda 1 at 75 (EXP.010.078.0001_3 at 0077_3)
Gyang et al, “Managing chronic pelvic pain following reconstructive pelvic surgery with transvaginal mesh” (2014) 25 Int Urogynecol J 313–318 at 317 (SHI.MESH.00020223 at 0227)
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2007) (SHI.MESH.00011315)
Withagen M et al, “Risk Factors for Exposure, Pain, and Dyspareunia After Tension-Free Vaginal Mesh Procedure” (2011) 118 Obstet Gynecol 629–636 at 632 (ETH.MESH.04929975 at 9978)
Sha H and Badlani G, “Mesh complications in female pelvic floor reconstructive surgery and their management: A systematic review” (2012) 28(2) Indian J Urol 129–153 at 138 (SHI.MESH.00039513 at 9521)
Barski D and Deng D, “Management of Mesh Complications after SUI and POP Repair: Review and Analysis of the Current Literature” (2015) BioMed Res Int 1–8 (SHI.MESH.00007704)
ETH.MESH.10684755 at 4804
T3783 (TRA.500.047.0001_2 at 0077_2)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1832 (ETH.MESH.07971133 at 1139)
Halaska M, “A multicenter, randomized, prospective, controlled study comparing sacrospinous fixation and transvaginal mesh in the treatment of posthysterectomy vaginal vault prolapse” (2012) 207 Am J Obstet Gynecol 301.e1–301.e7 at 301.e3 (ETH.MESH.08299299 at 9301)
Glazener C et al, “Mesh, graft, or standard repair for women having primary transvaginal anterior or posterior compartment prolapse surgery: two parallel-group, multicentre, randomised, controlled trials (PROSPECT)” (2016) The Lancet 1–12 at 8–9 (SHI.MESH.00024529 at 4536–4537)
Korda 1 at 74 (EXP.010.078.0001_3 at 0076_3)
De Tayrac R and Sentilhes L, “Complications of pelvic organ prolapse surgery and methods of prevention” (2013) 24 Int Urogynecol J 1859–1872 at 1861 (ETH.MESH.12358329 at 8331)
Glazener C et al, “Mesh, graft, or standard repair for women having primary transvaginal anterior or posterior compartment prolapse surgery: two parallel-group, multicentre, randomised, controlled trials (PROSPECT)” (2016) The Lancet 1–12 at 8 (SHI.MESH.00024529 at 4536)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 16 (SHI.MESH.00005748 at 5766)
Maher C et al, “Surgery for women with anterior compartment prolapse (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 35 (SHI.MESH.00043256 at 3294)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 3 (SHI.MESH.00005748 at 5753)
Maher C et al, “Surgery for women with anterior compartment prolapse (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 21 (SHI.MESH.00043256 at 3280)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1826 (ETH.MESH.07971133 at 1133)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 17 (SHI.MESH.00005748 at 5767)
Glazener C et al, “Mesh, graft, or standard repair for women having primary transvaginal anterior or posterior compartment prolapse surgery: two parallel-group, multicentre, randomised, controlled trials (PROSPECT)” (2016) The Lancet 1–12 at 7–8 (SHI.MESH.00024529 at 4535–4536)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1834 (ETH.MESH.07971133 at 1141)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 17 (SHI.MESH.00005748 at 5767)
PUB.MESH.00009026 at 9042
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1832 (ETH.MESH.07971133 at 1139)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 17 (SHI.MESH.00005748 at 5767)
Glazener C et al, “Mesh, graft, or standard repair for women having primary transvaginal anterior or posterior compartment prolapse surgery: two parallel-group, multicentre, randomised, controlled trials (PROSPECT)” (2016) The Lancet 1–12 at 7 (SHI.MESH.00024529 at 4535)
T3946 (TRA.500.049.0001_2 at 0062_2)
Withagen M et al, “Risk Factors for Exposure, Pain, and Dyspareunia After Tension-Free Vaginal Mesh Procedure” (2011) 118 Obstet Gynecol 629–636 at 631 (ETH.MESH.04929975 at 9977)
T3148 (TRA.500.040.0001_3 at 0050_3)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1829 (ETH.MESH.07971133 at 1136)
Maher C et al, “Surgery for women with anterior compartment prolapse (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 20 (SHI.MESH.00043256 at 3279)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016)at 2 (SHI.MESH.00005748 at 5752)
Ibid at 17 (SHI.MESH.00005748 at 5767)
Halaska M, “A multicenter, randomized, prospective, controlled study comparing sacrospinous fixation and transvaginal mesh in the treatment of posthysterectomy vaginal vault prolapse” (2012) 207 Am J Obstet Gynecol 301.e1–301.e7 at 301.e5 (ETH.MESH.08299299 at 9303)
Glazener C et al, “Mesh, graft, or standard repair for women having primary transvaginal anterior or posterior compartment prolapse surgery: two parallel-group, multicentre, randomised, controlled trials (PROSPECT)” (2016) The Lancet 1–12 at 7 (SHI.MESH.00024529 at 4535)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1826 (ETH.MESH.07971133 at 1133)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 16 (SHI.MESH.00005748 at 5766)
Maher C et al, “Surgery for women with anterior compartment prolapse (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) (SHI.MESH.00043256 at 3280)
De Tayrac R and Sentilhes L, “Complications of pelvic organ prolapse surgery and methods of prevention” (2013) 24 Int Urogynecol J 1859–1872 at 1863 (ETH.MESH.12358329 at 8333)
Ibid (ETH.MESH.12358329 at 8333)
Altman D et al, “Anterior Colporrhaphy versus Transvaginal Mesh for Pelvic-Organ Prolapse” (2011) 364 N Engl J Med 1826–1836 at 1833 (ETH.MESH.07971133 at 1140)
Maher C et al, “Surgery for women with anterior compartment prolapse (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 22 (SHI.MESH.00043256 at 3281)
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2013) at 2 (PUB.MESH.00001398 at 1407)
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) at 18 (SHI.MESH.00005748 at 5768)
T3148 (TRA.500.040.0001_4 at 0050_4)
T6337 (TRA.500.078.0001 at 0085)
Collinet at 31 (EXP.020.005.0001_4 at 0031_4); Korda 3 at 3 (EXP.010.129.0001_5 at 0004_5)
Collinet at 31 (EXP.020.005.0001_4 at 0031_4)
Rosamilia 1 at 29 (EXP.020.026.0001_4 at 0031_4)
Korda 1 at 36 (EXP.010.078.0001_3 at 0038_3)
T6137 (TRA.500.076.0001 at 0101)
AWS ch 9 at [128] (SBM.010.005.0379_2 at 0415_2)
Collinet at 9 (EXP.020.005.0001_4 at 0009_4)
Collinet at 31 (EXP.020.005.0001_4 at 0031_4)
Collinet at 31 (EXP.020.005.0001_4 at 0031_4)
Nygaard I et al, “Long-term Outcomes Following Abdominal Sacrocolpopexy for Pelvic Organ Prolapse” (2013) 309(19) JAMA 2016–2024 at 2020 (ETH.MESH.09466922 at 6926)
Diwadkar G et al, “Complication and Reoperation Rates After Apical Vaginal Prolapse Surgical Repair” (2009) 113(2) Obstet & Gynecol 367–373 (ETH.MESH.00020695)
Ibid at 369–370 (ETH.MESH.00020695 at 0697–0698)
SHI.MESH.00006213 at 6250
Abbott S et al, “Evaluation and management of complications from synthetic mesh after pelvic reconstructive surgery: a multicenter study”(2014) 210 Am J Obstet Gynecol 163.e1–163.e8 at 163.e1 (SHI.MESH.00001610)
T6156 (TRA.500.077.0001 at 0009)
Abbott S et al, “Evaluation and management of complications from synthetic mesh after pelvic reconstructive surgery: a multicenter study”(2014) 210 Am J Obstet Gynecol 163.e1–163.e8 (SHI.MESH.00001610)
Maher et al, “Surgery for women with apical vaginal prolapse (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) (PUB.MESH.00004281)
Ibid at 3 (PUB.MESH.00004281 at 4288)
Korda 1 at 73 (EXP.010.078.0001_3 at 0075_3)
Maher et al, “Abdominal sacral colpopexy or vaginal sacrospinous colpopexy for vaginal vault prolapse: A prospective randomized study” (2004) 190 Am J Obstet Gynecol 20–26 (ETH.MESH.01264131)
Ibid at 25 (ETH.MESH.01264131 at 4136)
Morling R et al, “Adverse events after first, single, mesh and non-mesh surgical procedures for stress urinary incontinence and pelvic organ prolapse in Scotland, 1997-2016: a population based cohort study” (2016) The Lancet 1–12 (SHI.MESH.00024517)
Ibid at 5 (SHI.MESH.00024517 at 4522)
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2013) at 29 (PUB.MESH.00001398 at 1434)
Gordon 1 at 17 (EXP.010.096.0001_3 at 0019_3)
Gordon 1 at 33 (EXP.010.096.0001_3 at 0035_3)
Gordon 1 at 34 (EXP.010.096.0001_3 at 0036_3)
Gordon 1 at 8 (EXP.010.096.0001_3 at 0010_3)
Gordon 1 at 45–46 (EXP.010.096.0001_3 at 0047_3–0048_3)
Gordon 1 at 46 (EXP.010.096.0001_3 at 0048_3)
Gordon 1 at 46 (EXP.010.096.0001_3 at 0048_3)
T6933 (TRA.500.086.0001 at 0032)
Gordon 1 at 47 (EXP.010.096.0001_3 at 0049_3)
EXP.010.098.0001_2
Gordon 1 at 48 (EXP.010.096.0001_3 at 0050_3)
Gordon 1 at 81 (EXP.010.096.0001_3 at 0083_3)
Gordon 1 at 82 (EXP.010.096.0001_3 at 0084_3)
Guerrero K et al, “A randomised controlled trial comparing TVT™, Pelvicol™ and autologous fascial slings for the treatment of stress urinary incontinence in women” (2010) 117 BJOG 1493–1503 (ETH.MESH.00024825)
Ibid at 1495 (ETH.MESH.00024825 at 4827)
Gordon 1 at 83 (EXP.010.096.0001_3 at 0085_3)
Gordon 2 at 6 (EXP.010.161.0001_3 at 0008_3)
Gordon 2 at 7–9 (EXP.010.161.0001_3 at 0009_3–0011_3)
Gordon 1 at 50 (EXP.010.096.0001_3 at 0052_3)
Gordon 1 at 50–51 (EXP.010.096.0001_3 at 0052_3–0053_3)
T2659 (TRA.500.034.0001_3 at 0002_2) (Gordon)
T2661 (TRA.500.034.0001_3 at 0004_2)
T2663 (TRA.500.034.0001_3 at 0006_2)
EXP.010.332.0001_2
Bianchi-Ferraro A et al, “Single-incision sling compared with transobturator sling for treating stress urinary incontinence: a randomized controlled trial” (2013) 24 Int Urogynecol J 1459–1465 (ETH.MESH.16322900); see RWS ch 5 at [7] (SBM.020.002.0083 at 0085–0086)
EXP.010.332.0001_2 at 0009_2
Bianchi-Ferraro A et al, “Single-incision sling compared with transobturator sling for treating stress urinary incontinence: a randomized controlled trial” (2013) 24 Int Urogynecol J 1459–1465 at 1463 (ETH.MESH.16322900 at 2904)
RWS ch 5 at [7] (SBM.020.002.0083 at 0085–0086)
Krulewitch 1 at 45 (EXP.010.081.0001_3 at 0048_3)
Krulewitch 1 at 34, 38 (EXP.010.081.0001_3 at 0037_3, 0041_3)
Hu 1 at 9, 27 (EXP.010.023.0001_3 at 0011_3)
Hu 1 at 15 (EXP.010.023.0001_3 at 0017_3)
T2697 (TRA.500.034.0001_3 at 0040_3)
T2702 (TRA.500.034.0001_3 at 0045_3)
Woodward 1 at [4.8] (EXP.010.044.0001_3 at 0070_3)
Woodward 2 at 72 (EXP.010.106.0001_3 at 0074_3)
T2445 (TRA.500.031.0001_3 at 0044_3)
T2464 (TRA.500.031.0001_3 at 0063_3)
RWS ch 13 at [37] (SBM.020.002.0338 at 0348)
Dean N et al, “Laparoscopic colposuspension for urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2017) (PUB.MESH.00010784 at 0796)
Ibid (PUB.MESH.00010784 at 0797)
Ibid (PUB.MESH.00010784 at 0796)
Ibid (PUB.MESH.00010784 at 0800)
RWS ch 2 at [47] (SBM.020.002.0024 at 0043)
RWS ch 12 at [5] (SBM.020.002.0336 at 0337)
RWS ch 12 at [5] (SBM.020.002.0336 at 0337)
EXP.010.102.0001_3
Allman 1 at 2 (EXP.010.100.0001_4 at 0003_4)
EXP.010.071.0001_2
Holland 1 (EXP.010.069.0001_3)
EXP.010.209.0001_2
RWS ch 12 at [5] (SBM.020.002.0336 at 0337)
RWS ch 12 (SBM.020.002.0336)
T3488 (TRA.500.044.0001_3 at 0040_3)
T3921 (TRA.500.049.0001_2 at 0037_2)
McCulloch P et al, “No surgical innovation without evaluation: the IDEAL recommendations” (2009) 374 The Lancet 1105–1112 (SHI.MESH.00027879); T3928 (TRA.500.049.0001_2 at 0044_2) (Roovers)
Beech at [4.2] (EXP.010.063.0001 at 0006)
Holland 1 at 4 (EXP.010.069.0001_3 at 0008_3)
Beech at [5.1] (EXP.010.063.0001 at 0006)
Beech at [4.1] (EXP.010.063.0001 at 0006)
Beech at [4.1] (EXP.010.063.0001 at 0005)
Beech 1 at 3–4 (EXP.010.063.0001 at 0005–0006)
SHI.MESH.00019231
T1430, 1435, 1447 (TRA.500.018.0001_2 at 0031_2, 0036_2, 0049_2)
SHI.MESH.00019231 at 9263
SHI.MESH.00019871 at 9871
Allman 1 at 7 (EXP.010.100.0001_4 at 0008_4)
Beech at 5 (EXP.010.063.0001 at 0007)
AID.010.002.0001_8
Allman 1 at 26 (EXP.010.100.0001_4 at 0027_4)
ETH.MESH.22256670 at 57051
Allman 1 at 29 (EXP.010.100.0001_4 at 0030_4)
Beech 1 at 2 (EXP.010.063.0001 at 0004)
Beech 1 at 3 (EXP.010.063.0001 at 0005)
Allman 1 at 12 (EXP.010.100.0001_4 at 0013_4)
T1437 (TRA.500.018.0001_2 at 0038_2)
Beech 1 at 3 (EXP.010.063.0001 at 0005)
T1451 (TRA.500.018.0001_2 at 0052_2)
SHI.MESH.00019871 at 9873
SHI.MESH.00019871 at 9875–9877
SHI.MESH.00019871 at 9876–9877
SHI.MESH.00019871 at 9877–9878
SHI.MESH.00019871 at 9878
SHI.MESH.00019871 at 9878–9879
SHI.MESH.00019871 at 9880
Allman 1 at 8 (EXP.010.100.0001_4 at 0009_4)
SHI.MESH.00019689
SHI.MESH.00019689 at 9690
Allman 1 at 10 (EXP.010.100.0001_4 at 0011_4); SHI.MESH.00020010
SHI.MESH.00019825 at 9827
Allman 1 at 10 (EXP.010.100.0001_4 at 0011_4)
SHI.MESH.00033324 at 3330
Allman 1 at 15 (EXP.010.100.0001_4 at 0016_4)
Hinoul at [242] (LAY.020.001.0001_4 at 0095_4)
AWS ch 8 at [186] (SBM.010.005.0296_2 at 0339_2); MSC.020.015.0001
T1429 (TRA.500.018.0001_2 at 0030_2)
Altman 2 at 2 (EXP.010.180.0001_3 at 0004_3)
SHI.MESH.00019825 at 9832
Allman 1 at 6 (EXP.010.100.0001_4 at 0007_4)
Beech at 7 (EXP.010.063.0001 at 0009)
Allman 1 at 5 (EXP.010.100.0001_4 at 0006_4)
T1429 (TRA.500.018.0001_2 at 0013_2, 0030_2)
T1439 (TRA.500.018.0001_2 at 0041_2)
SHI.MESH.00019871 at 9884
Allman 1 at 4 (EXP.010.100.0001_4 at 0005_4)
T1462 (TRA.500.018.0001_2 at 0063_2)
T1462 (TRA.500.018.0001_2 at 0062_2)
Allman 1 at 6 (EXP.010.100.0001_4 at 0007_4)
Beech at 4 (EXP.010.063.0001 at 0006)
Allman 1 at 14 (EXP.010.100.0001_4 at 0015_4)
Allman 1 at 14 (EXP.010.100.0001_4 at 0015_4)
Holland 1 at 70 (EXP.010.069.0001_3 at 0075_3)
Allman 1 at 13 (EXP.010.100.0001_4 at 0014_4)
Allman 1 at 13 (EXP.010.100.0001_4 at 0003_4)
PUB.MESH.00003413
Beech at [6.5] (EXP.010.063.0001 at 0008)
Beech at [6.4] (EXP.010.063.0001 at 0008)
Hinoul at [242] (LAY.020.001.0001_4 at 0095_4)
Hinoul at [242] (LAY.020.001.0001_4 at 0095_4)
Allman 1 at 14 (EXP.010.100.0001_4 at 0015_4)
Allman 1 at 15 (EXP.010.100.0001_4 at 0016_4)
T1431–32 (TRA.500.018.0001_2 at 0032_2)
Allman 1 at 15 (EXP.010.100.0001_4 at 0016_4)
T1432 (TRA.500.018.0001_2 at 0032_2)
Allman 2 at 3 (EXP.010.180.0001_3 at 0005_3)
T1471–72 (TRA.500.018.0001_2 at 0072_2–0073_2)
T1432 (TRA.500.018.0001_2 at 0032_2)
Allman 2 at 3 (EXP.010.180.0001_3 at 0004_3)
T1433 (TRA.500.018.0001_2 at 0034_2)
Allman 2 at 6 (EXP.010.180.0001_3 at 0008_3)
Allman 2 at 6 (EXP.010.180.0001_3 at 0008_3)
T1458 (TRA.500.018.0001_2 at 0059_2)
Allman 2 at 2 (EXP.010.180.0001_3 at 0003_3)
Allman 1 at 15 (EXP.010.100.0001_4 at 0016_4)
T1459 (TRA.500.018.0001_2 at 0060_2)
T1467 (TRA.500.018.0001_2 at 0068_2)
T1467 (TRA.500.018.0001_2 at 0068_2)
T1436 (TRA.500.018.0001_2 at 0037_2)
Holland 1 at 10–11 (EXP.010.069.0001_3 at 0008_3)
Holland 1 at 12 (EXP.010.069.0001_3 at 0008_3)
Holland 1 at 9 (EXP.010.069.0001_3 at 0013_3)
Holland 1 at 9 (EXP.010.069.0001_3 at 0013_3)
Holland 1 at 10 (EXP.010.069.0001_3 at 0014_3)
Holland 1 at 9 (EXP.010.069.0001_3 at 0013_3)
Holland 1 at 44 (EXP.010.069.0001_3 at 0048_3)
Holland 1 at 16 (EXP.010.069.0001_3 at 0021_3)
Holland 1 at 16 (EXP.010.069.0001_3 at 0021_3)
Holland 1 at 42 (EXP.010.069.0001_3 at 0046_3)
Holland 1 at 16 (EXP.010.069.0001_3 at 0015_3)
SHI.MESH.00018192
Holland 1 at 16 (EXP.010.069.0001_3 at 0015_3)
SHI.MESH.00018192 at 8197
Holland 1 at 17 (EXP.010.069.0001_3 at 0016_3)
Holland 1 at 22 (EXP.010.069.0001_3 at 0026_3)
Holland 1 at 22 (EXP.010.069.0001_3 at 0026_3)
T1654–55 (TRA.500.018.0001_2 at 0006_2–0007_2)
Holland 1 at 55 (EXP.010.069.0001_3 at 0059_3)
ETH.MESH.10618716 at 8717; Holland 1 at 48 (EXP.010.069.0001_3 at 0052_3)
Holland 1 at 48 (EXP.010.069.0001_3 at 0052_3)
Holland 1 at 49 (EXP.010.069.0001_3 at 0053_3)
Holland 1 at 49 (EXP.010.069.0001_3 at 0053_3), ETH.MESH.10618757 at 8768
Holland 1 at 49 (EXP.010.069.0001_3 at 0054_3)
Holland 1 at 22 (EXP.010.069.0001_3 at 0027_3)
Holland 1 at 50–51 (EXP.010.069.0001_3 at 0054_3-0055_3)
Holland 2 at 68 (EXP.010.192.0001_3 at 0069_3)
Holland 2 at 46 (EXP.010.192.0001_3 at 0047_3)
Holland 2 at 80 (EXP.010.192.0001_3 at 0081_3)
Holland 2 at 77 (EXP.010.192.0001_3 at 0078_3)
Holland 2 at 78 (EXP.010.192.0001_3 at 0079_3)
Holland 2 at 63 (EXP.010.192.0001_3 at 0064_3)
Holland 2 at 48 (EXP.010.192.0001_3 at 0049_3)
Holland 2 at 69 (EXP.010.192.0001_3 at 0070_3)
Holland 2 at 70 (EXP.010.192.0001_3 at 0071_3)
Holland 2 at 69 (EXP.010.192.0001_3 at 0070_3)
Holland 2 at 48 (EXP.010.192.0001_3 at 0049_3)
Holland 2 at 73 (EXP.010.192.0001_3 at 0074_3)
AID.010.011.0001 at 0002
Holland 2 at 46 (EXP.010.192.0001_3 at 0047_3)
Holland 2 at 63 (EXP.010.192.0001_3 at 0064_3)
Holland 2 at 10 (EXP.010.192.0001)
T1710 (TRA.500.021.0001_2 at 0062_2)
SHI.MESH.00018192
T1675 (TRA.500.021.0001_2 at 0027_2)
T1675 (TRA.500.021.0001_2 at 0027_2)
Holland 1 at 55 (EXP.010.069.0001_3 at 0059_3)
Holland 1 at 55 (EXP.010.069.0001_3 at 0059_3)
T1610 (TRA.500.020.0001_2 at 0020_2)
T1610 (TRA.500.020.0001_2 at 0020_2)
T1610 (TRA.500.020.0001_2 at 0020_2)
T1610 (TRA.500.020.0001_2 at 0020_2)
Pence at [34] (EXP.010.200.0001_2 at 0012_2)
Pence at [40] (EXP.010.200.0001_2 at 0014_2)
Pence at [38] (EXP.010.200.0001_2 at 0013_2)
T1516 (TRA.500.019.0001_2 at 0005_2)
Pence at [43] (EXP.010.200.0001_2 at 0017_2)
T1567–68 (TRA.500.019.0001_2 at 0055_2-0056_2)
T1581 (TRA.500.019.0001_2 at 0069_2)
Pence at [221] (EXP.010.200.0001_2 at 0011_2)
Pence at [212] (EXP.010.200.0001_2 at 0009_2)
Pence at [216] (EXP.010.200.0001_2 at 0011_2)
T1578 (TRA.500.019.0001_2 at 0066_2)
T1555 (TRA.500.019.0001_2 at 0043_2)
T1556 (TRA.500.019.0001_2 at 0044_2)
RWS ch 7 at [40] (SBM.020.002.0108 at 0127)
Holland 2 at 11 (EXP.010.192.0001_3 at 0012_3)
Klinge at [205] (LAY.010.022.0001_2 at 0078_2)
ETH.MESH.03932912
ETH.MESH.09747038 at 7044
ETH.MESH.08695898; ETH.MESH.05972834; Hinoul at [73] (LAY.020.001.0001_4 at 0039_4)
ETH.MESH.07295656 at 0727
ETH.MESH.04381806 at 0809
ETH.MESH.03932912 at 0914
SH1.MESH.00014223 at 4225
Klinge U et al, “Shrinkage Polypropylene Mesh in vivo: An Experimental Study in Dogs” (1998) 164 Eur J Surg 965–969 (ETH.MESH.05960184)
Klinge U and Klosterhalfen, “Biomaterials – Experimental Aspects” in Incisional hernia (Springer, 1st ed, 1999) (SHI.MESH.00036749 at 6979)
Ulmsten U et al, “An ambulatory surgical procedure under local anesthesia for treatment of female urinary incontinence” (1996) 7(2) Int Urogynecol J Pelvic Floor Dysfunct 81–85 (ETH.MESH.19935677)
Ibid (ETH.MESH.19935677 at 5681)
Ibid (ETH.MESH.19935677 at 5681)
T3486 (TRA.500.044.0001_3 at 0038_3)
ETH.MESH.03932912 at 2914; RWS ch 7 at [62] (SBM.020.002.0108 at 0135)
Holland 2 at 3 (EXP.010.192.0001_2 at 0004_2)
Hinoul at 95 (LAY.020.001.0001_4 at 0095_4)
ETH.MESH.10984368 at 4420
Hinoul at [241] (LAY.020.001.0001_4 at 0094_4)
Allman 2 at 3 (EXP.010.180.0001_3 at 0004_3)
ETH.MESH.09747038 at 7046
ETH.MESH.11858511 at 8516–8521
Ulmsten U et al, “A Multicenter Study of Tension-Free Vaginal Tape (TVT) for Surgical Treatment of Stress Urinary Incontinence” (1998) 9 Int Urogynecol J 210-213 (ETH.MESH.01769250)
ETH.MESH.11858511 at 8516
Allman 1 at 35 (EXP.010.100.0001_4 at 0036_4)
Hinoul at [244] (LAY.020.001.0001_4 at 0096_4)
Hinoul at [245] (LAY.020.001.0001_4 at 0096_4)
SHI.MESH.00019231 at 9244
Allman 1 at 6 (EXP.010.100.0001_4 at 0007_4)
T3287–88 (TRA.500.042.0001_3 at 0020_3-0021_3)
Holland 2 at 3–7 (EXP.010.192.0001_3 at 0004_3-0008_3)
Ulmsten U et al, “A Multicenter Study of Tension-Free Vaginal Tape (TVT) for Surgical Treatment of Stress Urinary Incontinence” (1998) 9 Int Urogynecol J 210-213 at 211 (ETH.MESH.01769250 at 9251)
ETH.MESH.03932912 at 2913
Wang A and Lo T, “Tension-Free Vaginal Tape: A Minimally Invasive Solution to Stress Urinary Incontinence in Women” (1998) 43 J Reprod Med 429–434 (ETH.MESH.19935693 at 5697)
Ward K and Hilton P, “Prospective multicentre randomised trial of tension-free vaginal tape and colposuspension as primary treatment for stress incontinence” (2002) 13 BMJ 325 (ETH.MESH.13649139)
Allman 1 at 36 (EXP.010.100.0001_4 at 0037_4)
Hinoul at [240] (LAY.020.001.0001_4 at 0094_4)
Holland 1 at 39 (EXP.010.069.0001_3 at 0044_3)
Holland 1 at 42 (EXP.010.069.0001_3 at 0046_3)
Holland 1 at 25 (EXP.010.069.0001_3 at 0016_3)
Holland 1 at 45 (EXP.010.069.0001_3 at 0049_3)
Holland 1 at 25–26, 45 (EXP.010.069.0001_3 at 0039_3-0040_3, 0049_3)
Holland 1 at 26 (EXP.010.069.0001_3 at 0030_3)
Holland 1 at 46–47 (EXP.010.069.0001_3 at 0050_3-0051_3)
Holland 1 at 47 (EXP.010.069.0001_3 at 0051_3)
Holland 1 at 26 (EXP.010.069.0001_3 at 0039_3)
ETH.MESH.01317508 at 7510
Hinoul at [82] (LAY.020.001.0001_4 at 0043_4)
ETH.MESH.10681859 at 1866
ETH.MESH.03907468
ETH.MESH.03932909
ETH.MESH.00262013
ETH.MESH.03932909
ETH.MESH.03907327 at 7330
ETH.MESH.09746793
ETH.MESH.09746793 at 6794; JJM.MESH.00009139; Rosamilia 1 (EXP.020.026.0001_4 at 0011_4)
ETH.MESH.09746793 at 6794
ETH.MESH.03907468
T3598 (TRA.500.045.0001_3 at 0059_3)
T3598 (TRA.500.045.0001_3 at 0059_3)
ETH.MESH.22134684 at 4685
See Hinoul at [314] (LAY.020.001.0001_4 at 0122_4)
ETH.MESH.22134684 at 4685
See, for example, ETH.MESH.09746793
ETH.MESH.03927746 at 7750
ETH.MESH.03801821
ETH.MESH.03927746 at 7750
ETH.MESH.03919404
ETH.MESH.22011418 at 1699
ETH.MESH.00858080
ETH.MESH.03906958
ETH.MESH.03910890
ETH.MESH.00858096 at 8097
ETH.MESH.00865460
AWS ch 7 at [116] (SBM.010.005.0250_2 at 0276_2)
ETH.MESH.00865459
ETH.MESH.00858080
ETH.MESH.22011418 at 1656
ETH.MESH.22134684
SHI.MESH.00019871
ETH.MESH.22134684 at 4692
ETH.MESH.22134684 at 4693
Allman 1 at 46 (EXP.010.100.0001_4 at 0047_4)
ETH.MESH.00261584
ETH.MESH.00262013
ETH.MESH.00262013
JJM.MESH.00009139
JJM.MESH.00009139
JJM.MESH.00060246
ETH.MESH.01591931 at 1934
ETH.MESH.01592062
ETH.MESH.01591931 at 1934–5
ETH.MESH.22257253 at 7384
ETH.MESH.22257253 at 7382–7385
Holland 1 at 28 (EXP.010.069.0001_3 at 0032_3)
ETH.MESH.00858252
Holland 1 at 51 (EXP.010.069.0001_3 at 0055_3); ETH.MESH.15058647
ETH.MESH.03742864; ETH.MESH.15058647
Holland 1 at 51 (EXP.010.069.0001_3 at 0055_3)
ETH.MESH.22427253 at 7254
ETH.MESH.22257253 at 7268
ETH.MESH.01594589
ETH.MESH.01594589
ETH.MESH.01592121
ETH.MESH.01592121 at 2124
ETH.MESH.01592121 at 2127
ETH.MESH.01592121 at 2125
ETH.MESH.01594695
ETH.MESH.01594695 at 4700
ETH.MESH.03714002
Holland 1 at 30 (EXP.010.069.0001_3 at 0034_3)
Holland 1 at 29 (EXP.010.069.0001_3 at 0033_3); ETH.MESH.02340568
ETH.MESH.03714002
ETH.MESH.03714002 at 4014
ETH.MESH.03905059
ETH.MESH.03905059 at 5069–5070
ETH.MESH.03714002 at 0005
SBM.020.032.0001
ETH.MESH.04939001
ETH.MESH.06706004 at 6006
ETH.MESH.06706004 at 6006
Holland 1 at 52 (EXP.010.069.0001_3 at 0056_3)
ETH.MESH.01189423
Allman 1 at 41 (EXP.010.100.0001_4 at 0042_4)
JJM.MESH.00008731
JJM.MESH.00009139 at 9140
JJM.MESH.00009139 at 9139
ETH.MESH.00647410 at 7414
JJM.MESH.00046395
ETH.MESH.00574738
ETH.MESH.10681859
ETH.MESH.10681859 at 1867
ETH.MESH.10681859 at 1884
ETH.MESH.10681859 at 1882
ETH.MESH.10681859 at 1885
Allman 1 at 51 (EXP.010.100.0001_4 at 0052_4)
Allman 1 at 52 (EXP.010.100.0001_4 at 0053_4)
AWS ch 7 at [144(d)] (SBM.010.005.0250_2 at 0287_2)
AWS ch 7 at [145(e)] (SBM.010.005.0250_2 at 0287_2-0288_2)
ETH.MESH.00574738
ETH.MESH.00574738
ETH.MESH.00574738
ETH.MESH.22011418 at 1578
ETH.MESH.06402361 at 2640
ETH.MESH.06402361 at 2654
ETH.MESH.10681773
ETH.MESH.10681773 at 1779
ETH.MESH.10681773 at 1779
ETH.MESH.10681773 at 1775
ETH.MESH.10681773 at 1780
ETH.MESH.10681773 at 1811
ETH.MESH.10681773 at 1824
ETH.MESH.10681773 at 1823
ETH.MESH.10681773 at 1785
ETH.MESH.22011418 at 1578; RWS ch 7 at [84] (SBM.020.002.0108 at 0127)
Allman 1 at 50–51 (EXP.010.100.0001_4 at 0051-52)
Allman 1 at 50 (EXP.010.100.0001_4 at 0051)
ETH.MESH.10539999 at 540001
See, for example, de Tayrac R, Gervaise A and Fernandez H, “Cure de cystocèle voie basse par prothèse sous-vésicale libre” (2002) 31 J Gynecol Obstet Biol Reprod 597–599 (PUB.MESH.00005028); Husaunndee M et al, “Surgical treatment of genital prolapse using a new prosthetic lateral hysteropexy technique combining both vaginal and coelioscopic approaches” (2003) 32 J Gynecol Obstet Biol Reprod 314–320 (PUB.MESH.00005065_2)
ETH.MESH.12006257
ETH.MESH.16046418 at 6609
ETH.MESH.21667551 at 7700
ETH.MESH.10539999 at 400002
ETH.MESH.22354125 at 4136
ETH.MESH.22354125 at 4128
ETH.MESH.22354125 at 4141
ETH.MESH.22354125 at 4165
ETH.MESH.22354125 at 4169
ETH.MESH.22354125 at 4126
ETH.MESH.00219861 at 00220079
ETH.MESH.00219861 at 00220081
ETH.MESH.00219861 at 00220097
ETH.MESH.00219861 at 00220115
ETH.MESH.00219861 at 00220117
ETH.MESH.20070090 at 0117
ETH.MESH.20070090 at 0126, 0139, 4157 and 0142
ETH.MESH.20070090 at 0142
ETH.MESH.20070090 at 0146
Fan X et al, “Comparison of polypropylene mesh and porcine-derived cross-linked urinary bladder matrix materials implanted in rabbits for vagina and abdomen” (2014) 25(5) Int Urogynecol J 683–689 (SHI.MESH.00011669)
ETH.MESH.22007003 at 7127
ETH.MESH.22007003 at 7129
T3555 (TRA.500.045.0001_3 at 0016_3)
Julian T, “The efficacy of Marlex mesh in the repair of severe, recurrent vaginal prolapse of the anterior midvaginal wall” (1996) 176(6) Am J Obstet Gynecol 1472–1575 (ETH.MESH.04544688)
Hu 1 at 6 (EXP.010.023.0001_3 at 0008_3)
Gordon 2 at 22 (EXP.010.161.0001_3 at 0024_3)
Julian T, “The efficacy of Marlex mesh in the repair of severe, recurrent vaginal prolapse of the anterior midvaginal wall” (1996) 176(6) Am J Obstet Gynecol 1472–1575 (ETH.MESH.04544688 at 4691)
Ibid (ETH.MESH.04544688 at 4689–90)
Ibid (ETH.MESH.04544688 at 4690)
SHI.MESH.00033137 at 3148
Gordon 2 at 14 (EXP.010.161.0001_3 at 0016_3)
Gordon 2 at 15 (EXP.010.161.0001_3 at 0017_3)
Gordon 2 at 13 (EXP.010.161.0001_3 at 0015_3)
Gordon 2 at 14 (EXP.010.161.0001_3 at 0016_3)
Mahendran D et al, “Laparosopic sacrocolpopexy in the management of vaginal vault prolapse” (1996) 5(4) Gynaecological Endoscopy 217–222 (SHI.MESH.00033280)
Gordon 2 at 14 (EXP.010.161.0001_3 at 0016_3)
T1709 (TRA.500.021.0001_2 at 0061_2)
ETH.MESH.22007003 at 7129
Allman 2 at 23 (EXP.010.180.0001_3 at 0024_3)
Allman 2 at 23 (EXP.010.180.0001_3 at 0024_3)
Klinge U and Klosterhalfen B, “Biomaterials – Experimental Aspects” in Incisional hernia (Springer, 1st ed, 1999) (SHI.MESH.00036749 at 6379)
Berrocal J et al, “Conceptual advance in the surgical management of genital prolapse: The TVM technique emergence” (2004) 33 J Gynecol Obstet Biol Reprod 577–587 (ETH.MESH.00659678 at 9688)
ETH.MESH.04040841_2
ETH.MESH.03910186
ETH.MESH.03910183
Allman 2 at 22 (EXP.010.180.0001_3 at 0023_3)
Allman 1 at 21 (EXP.010.100.0001_4 at 0022_4)
T1443 (TRA.500.018.0001_2 at 0044_2)
ETH.MESH.06376830 at 6840
ETH.MESH.22401797 at 1969
Allman 2 at 23 (EXP.010.180.0001_3 at 0024_3)
T1710 (TRA.500.021.0001_2 at 0062_2)
Holland 1 at 48 (EXP.010.069.0001_3 at 0052_3)
Holland 1 at 48 (EXP.010.069.0001_3 at 0052_3)
ETH.MESH.00219861 at 9918
ETH.MESH.02225348 at 5602
Holland 1 at 32 (EXP.010.069.0001_3 at 0036_3)
SHI.MESH.00043531
ETH.MESH.02267181
ETH.MESH.15055676
ETH.MESH.15055676 at 5678
ETH.MESH.15055676 at 5679
ETH.MESH.15055676 at 5682
ETH.MESH.15055676 at 5681
ETH.MESH.02267181
T4258 (TRA.500.053.0001_2 at 0016_2)
ETH.MESH.00012009 at 2010
ETH.MESH.00012009 at 2011
ETH.MESH.00012009 at 2009
ETH.MESH.00012090
ETH.MESH.00900574
ETH.MESH.00584846
ETH.MESH.00584846
Debodinance P et al, “Changing attitudes on the surgical treatment of genital prolapse: Birth of the TVM [Tension-free Vaginal Mesh] technique” (2004) 33 J Gynecol Obstet Biol Reprod 577–588 (ETH.MESH.04040841_2)
Ibid (ETH.MESH.04040841_2)
ETH.MESH.03926416
ETH.MESH.03926416
ETH.MESH.00712432
Berrocal J et al, “Conceptual advances in the surgical management of genital prolapse: The TVM technique emergence” (2004) 33 J Gynecol Obstet Biol Reprod 577–587 (ETH.MESH.00659678)
ETH.MESH.00659678 at 9681; T4391–92 (TRA.500.054.0001_2 at 0096_2)
Berrocal J et al, “Conceptual advances in the surgical management of genital prolapse: The TVM technique emergence” (2004) 33 J Gynecol Obstet Biol Reprod 577–587 (ETH.MESH.00659678 at 9689)
ETH.MESH.06700221 at 0224
ETH.MESH.00308747 from 8787
ETH.MESH.00308747 at 8804
Holland 1 at 49–50 (EXP.010.069.0001_3 at 0053_3-0054_3)
Holland 1 at 36 (EXP.010.069.0040_3)
Holland 1 at 36 (EXP.010.069.0001_3 at 0040_3)
Holland 1 at 37 (EXP.010.069.0001_3 at 0041_3)
Holland 1 at 50 (EXP.010.069.0001_3 at 0054_3)
ETH.MESH.00308747 at 8818
Holland 1 at 50 (EXP.010.069.0001_3 at 0054_3)
ETH.MESH.15049498
Holland 1 at 50 (EXP.010.069.0001_3 at 0054_3); ETH.MESH.00754437 at 8822
Holland 1 at 50 (EXP.010.069.0001_3 at 0054_3)
ETH.MESH.20029268
ETH.MESH.20029268 from 9270
ETH.MESH.22134106
Allman 1 at 21 (EXP.010.100.0001_4 at 0022_4)
AWS ch 8 at [237] (SBM.010.005.0296_2 at 0361_2)
ETH.MESH.22134106 at 4110–4111
Allman 1 at 21 (EXP.010.100.0001_4 at 0022_4)
ETH.MESH.22134106 at 4111
Allman 1 at 21 (EXP.010.100.0001_4 at 0022_4)
Gordon 2 at 6 (EXP.010.161.0001_3 at 0008_3)
Altman D and Bland J, “Treatment allocation in controlled trials: why randomise?” (1999) 318 BMJ 1209 (SHI.MESH.00032771)
Iglesia C, Fenner D and Brubaker L, “The use of mesh in gynecologic surgery” (1997) 8(2) Int Urogynecol J Pelvic Floor Dysfunct 105–115 (ETH.MESH.00155061)
Ibid (ETH.MESH.00155061)
ETH.MESH.22134106 at 4110
Korda 3 at 4 (EXP.010.129.0001_5 at 0005_5)
Gordon 2 at 16 (EXP.010.161.0001_3 at 0018_3)
T3571 (TRA.500.045.0001_3 at 0032_3)
T3571 (TRA.500.045.0001_3 at 0032_3)
Hinoul at [559] (LAY.020.001.0001_4 at 0268_4)
SHI.MESH.00045785
Berrocal J et al, “Conceptual advances in the surgical management of genital prolapse: The TVM technique emergence” (2004) 33 J Gynecol Obstet Biol Reprod 577–587 (ETH.MESH.00659678)
ETH.MESH.02992144 at 2145
Allman 1 at 21 (EXP.010.100.0001_4 at 0022_4)
ETH.MESH.06252711 at 2714
ETH.MESH.02232575
SHI.MESH.00045780
ETH.MESH.00012009
ETH.MESH.08224081
ETH.MESH.08224086
ETH.MESH.00012090
ETH.MESH.00012090 at 2094
ETH.MESH.00012090 at 2094
ETH.MESH.00012107
ETH.MESH.00012125
ETH.MESH.01407442 at 7446
Holland 1 at 34 (EXP.010.069.0001_3 at 0038_3)
ETH.MESH.22133539_V2 at 3610_V2
Allman 2 at 27 (EXP.010.180.0001_3 at 0028_3)
Collinet at 17 (EXP.020.005.0001_4 at 0017_4)
SHI.MESH.00006213
Korda 3 at 7 (EXP.010.129.0001_5 at 0008_5)
Klinge at [48] (LAY.010.022.0001_2 at 0011_2)
Klinge at [57] (LAY.010.022.0001_2 at 0013_2)
ETH.MESH.05235980
ETH.MESH.00870466 at 0469
ETH.MESH.02017152 at 7153
ETH.MESH.00742724
ETH.MESH.05237408
ETH.MESH.00006796
ETH.MESH.02992144 at 2144
ETH.MESH.02992144 at 2144
ETH.MESH.02992144 at 2145
Allman 2 at 29 (EXP.010.180.0001_3 at 0030_3)
ETH.MESH.22256670 at 6701
Hinoul at [570] (LAY.020.001.0001_4 at 0272_4)
ETH.MESH.00035476
ETH.MESH.00094046
Milani A et al, “Trocar-guided mesh repair of vaginal prolapse using partially absorbable mesh: 1 year outcomes” (2011) 204(1) Am J Obstet Gynecol 74.e1–74.e8 (ETH.MESH.05110011)
Holland 1 at 38 (EXP.010.069.0001_3 at 0042_3)
Holland 1 at 38 (EXP.010.069.0001_3 at 0042_3)
Holland 1 at 39 (EXP.010.069.0001_3 at 0043_3)
ETH.MESH.06717951
Holland 1 at 38 (EXP.010.069.0001_3 at 0042_3)
Holland 1 at 54 (EXP.010.069.0001_3 at 0039_3)
ETH.MESH.02195768 at 5771
SHI.MESH.00019871 at 9873
Cobb W et al, “Textile analysis of heavy weight, mid-weight, and light weight polypropylene mesh in a porcine ventral hernia model” (2006) 136(1) J Surg Res 1–7 (SHI.MESH.00033731)
Ibid at 5 (SHI.MESH.00033731 at 3735)
Ibid at 6 (SHI.MESH.00033731 at 3736)
Ibid at 6–7 (SHI.MESH.00033731 at 3736–3737)
ETH.MESH.02195768 at 5777
T1477 (TRA.500.018.0001_2 at 0078_2)
Allman 1 at 27 (EXP.010.100.0001_4 at 0022_4)
T1475 (TRA.500.018.0001_2 at 0076_2)
T1491 (TRA.500.018.0001_2 at 0092_2)
T1492–93 (TRA.500.018.0001_2 at 0093–4_2)
T1491 (TRA.500.018.0001_2 at 0092_2)
ETH.MESH.06397498 at 7500
ETH.MESH.02195768 at 5777
ETH.MESH.02195768 at 5778
ETH.MESH.02195768 at 5778–5779
Allman 1 at 27 (EXP.010.100.0001_4 at 0028_4)
Allman 1 at 30 (EXP.010.100.0001_4 at 0031_4)
ETH.MESH.00743865
ETH.MESH.00753999 at 4000
Allman 2 at 32 (EXP.010.180.0001_3 at 0033_3)
ETH.MESH.03360387
ETH.MESH.03360387 at 0399
Allman 1 at 32–33 (EXP.010.100.0001_4 at 0033_4–0034_4)
Milani R et al, “Functional and anatomical outcome of anterior and posterior vaginal prolapse repair with prolene mesh” (2005) 112(1) BJOG 107–111 (ETH.MESH.00169718)
ETH.MESH.03360387 at 0393
Fatton B et al, “Transvaginal repair of genital prolapse: preliminary results of a new tension-free vaginal mesh (Prolift™ technique) - a case series multicentric study” (2007) 18(7) Int Urogynecol J Pelvic Floor Dysfunct 743–752 (ETH.MESH.00093734)
Gordon 2 at 16 (EXP.010.161.0001_3 at 0018_3)
Hu 2 at 16 (EXP.010.184.0001_3 at 0017_3)
Fatton B et al, “Transvaginal repair of genital prolapse: preliminary results of a new tension-free vaginal mesh (Prolift™ technique) - a case series multicentric study” (2007) 18(7) Int Urogynecol J Pelvic Floor Dysfunct 743–752 at 743 (ETH.MESH.00093734 at 3734)
Altman D and Falconer C, “Perioperative Morbidity Using Transvaginal Mesh in Pelvic Organ Prolapse Repair” (2007) 109(2) Obstet Gynecol 303–308 (SHI.MESH.00032757)
Hu 2 at 16 (EXP.010.184.0001_3 at 0017_3)
Altman D and Falconer C, “Perioperative Morbidity Using Transvaginal Mesh in Pelvic Organ Prolapse Repair” (2007) 109(2) Obstet Gynecol 303–308 at 307 (SHI.MESH.00032757 at 2731)
Collinet P et al, “Transvaginal mesh technique for pelvic organ prolapse repair: mesh exposure management and risk factors” (2006) 17(4) Int Urogynecol J Pelvic Floor Dysfunct 315–320 (ETH.MESH.00034322)
Ibid at 320 (ETH.MESH.00034322 at 4328)
ETH.MESH.03360387 at 0394
Carey M et al, “Vaginal surgery for pelvic organ prolapse using mesh and a vaginal support device” (2008) 115(3) BJOG 391–397 (ETH.MESH.00154197)
ETH.MESH.02010349 at 0356
Gordon 2 at 17 (EXP.010.161.0001_3 at 0019_3)
Carey M et al, “Vaginal surgery for pelvic organ prolapse using mesh and a vaginal support device” (2008) 115(3) BJOG 391–397 at 397 (ETH.MESH.00154197 at 4203)
Hinoul at [567] (LAY.020.001.0001_4 at 0271_4)
Gordon 2 at 17 (EXP.010.161.0001_3 at 0019_3)
ETH.MESH.03915197 at 5199
ETH.MESH.03360387 at 0399
Hinoul at 271 fn 673 (LAY.020.001.0001_4 at 0272_4)
ETH.MESH.03360387 at 0396
Hu 2 at 17 (EXP.010.184.0001_3 at 0018_3)
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2004) (PUB.MESH.00009046)
Ibid (PUB.MESH.00009046 at 9055)
Hu 2 at 17 (EXP.010.184.0001_3 at 0018_3)
ETH.MESH.03162936
Allman 1 at 13 [5.1] (EXP.010.100.0001_4 at 0014_4)
Allman 2 at 5 (EXP.010.180.0001_3 at 0006_3)
Allman 2 at 15 (EXP.010.180.0001_3 at 0016_3)
Allman 2 at 15 (EXP.010.180.0001_3 at 0016_3)
Allman 2 at 15 (EXP.010.180.0001_3 at 0016_3)
Allman 2 at 15 (EXP.010.180.0001_3 at 0016_3)
Allman 2 at 6 (EXP.010.180.0001_3 at 0007_3)
Krulewitch 1 at 62–63 (EXP.010.081.0001_3 at 0066_3–0066_3
AWS ch 12 at [1]–[13] (SBM.010.005.0529_2 at 0530_2-0532_2)
Hinoul at [242] (LAY.020.001.0001_4 at 0095_4)
RWS ch 7 at [90] (SBM.020.002.0108 at 0146)
ETH.MESH.09499231
ETH.MESH.09499232
ETH.MESH.09499232
ETH.MESH.09499233
ETH.MESH.00847438 at 7440
ETH.MESH.00847438 at 7439
Pence exhibit 3 pt A at table 3.1 (EXP.010.203.0001_2 at 0003_2)
Pence exhibit 3 pt A at table 3.2 (EXP.010.203.0001_2 at 0003_2)
Pence exhibit 4 pt A at table 4.1 (EXP.010.207.0001_2 at 0002_2)
AWS ch 12 at [83] (SBM.010.005.0529_2 at 0551_2)
AWS ch 12 at [5] (SBM.010.005.0529_2 at 0530_2)
AWS ch 12 at [5] (SBM.010.005.0529_2 at 0530_2)
Pence exhibit 3 pt A at 13 (EXP.010.203.0001_2 at 0014_2)
Pence at [107] (EXP.010.200.0001_2 at 0047_2)
Pence exhibit 3 pt C (EXP.010.205.0001_2)
Pence exhibit 3 pt A from 6 (EXP.010.203.0001_2 at 0007_2)
Pence exhibit 3 pt A at 15 (EXP.010.203.0001_2 at 0016_2)
Pence exhibit 3 pt A from 6 (EXP.010.203.0001_2 at 0007_2)
Pence exhibit 4 pt A at 13–14 (EXP.010.207.0001_2 at 0014_2–0015_2)
ETH.MESH.02645424 at 6926
ETH.MESH.02645424 at 6926–6928
ETH.MESH.02645424 at 5725–5726
Pence exhibit 4 pt A at 14 (EXP.010.207.0001_2 at 0015)
ETH.MESH.02645424 at 7100
ETH.MESH.02645424 at 7100–7103
Pence exhibit 4 pt A at 14 (EXP.010.207.0001_2 at 0015)
AWS ch 6 at [318] (SBM.010.005.0156_2 at 0232_2)
ETH.MESH.23862436
ETH.MESH.23862436 at 2439
ETH.MESH.23862436 at 2436
ETH.MESH.23862436 at 2436
ETH.MESH.25297340
ETH.MESH.25297340
ETH.MESH.25297757
T3236 (TRA.500.041.0001_3 at 0049_3)
T3236 (TRA.500.041.0001_3 at 0049_3)
ETH.MESH.25390742
AWS ch 6 at [320] (SBM.010.005.0156_2 at 0232_2)
JJM.MESH.00000896
JJM.MESH.00250500 at 0502
JJM.MESH.00332342
ETH.MESH.06397498
ETH.MESH.17650617
ETH.MESH.06397498; Allman 2 at 9 (EXP.010.180.0001_3 at 0010_3)
ETH.MESH.03742571; Holland 2 at 124 (EXP.010.192.0001_3 at 0125_3)
Holland 1 at 48 (EXP.010.069.0001_3 at 0052_3)
ETH.MESH.21996159
ETH.MESH.09479794
ETH.MESH.09479794 at 9800
ETH.MESH.09470000 at 9795
ETH.MESH.09479794 at 9801
ETH.MESH.06796086 at 6093
ETH.MESH.06796086 at 6088, 6094
ETH.MESH.06796086 at 6096
See, for example, ETH.MESH.17650617 at 0635
ETH.MESH.01784823
ETH.MESH.07226579; RWS ch 7 at [89] (SBM.020.002.0108 at 0144)
SBM.010.019.0001
ETH.MESH.07226579
ETH.MESH.07226579 at 6581
ETH.MESH.01784823
Hinoul at [238] (LAY.020.001.0001_4 at 0093_4), RWS ch 7 at [74] (SBM.020.002.0108 at 0127)
ETH.MESH.06397498
ETH.MESH.03666837
ETH.MESH.00602574
ETH.MESH.03666837 at 6840
Holland 2 at 22 (EXP.010.192.0001_3 at 0125_3)
ETH.MESH.09479794 at 9795; Allman 2 at 9 (EXP.010.180.0001_3 at 0010_3)
ETH.MESH.00831307; ETH.MESH.008270
T1478 (TRA.500.018.0001_2 at 0079_2)
ETH.MESH.00831307
ETH.MESH.04384126
Allman 1 at 10 (EXP.010.100.0001_4 at 0011_4)
ETH.MESH.04384126 at 4129
AWS ch 12 at [27] (SBM.010.005.0529_2 at 0535_2)
ETH.MESH.04384126 at 4135
ETH.MESH.04384126 at 4140
ETH.MESH.04384126 at 4141
Quereux F et al, “Érosion vésicale tardive après pose de TVT® (Bladder erosion few years after TVT® procedure)” (2007) 36 Journal de Gynecology Obstétrique et Biologie de la Reproduction 75–77 (SHI.MESH.00027669)
Heesakkers J and Vierhout M, “Some complications of tension-free mid-urethral tapes for the treatment of stress incontinence in women” (2007) 151 Dutch Journal of Medicine 1361–1366 (ETH.MESH.02602049_2)
ETH.MESH.04384126 at 4144
Tzortzis et al, “Bladder stone formation after a tension-free vaginal tape procedure: report on two cases” (2007) 79 Urol Int 181–183 (SHI.MESH.00003734)
Ibid (SHI.MESH.00003734 at 3736)
ETH.MESH.04384126 at 4143
ETH.MESH.04384126 at 4143
ETH.MESH.04384126 at 4143
ETH.MESH.04384126 at 4144
ETH.MESH.04384126 at 4153
Allman 1 at 38 (EXP.010.100.0001_4 at 0039_4)
ETH.MESH.04384126 at 4140
Ogah J, Cody JD and Rogerson L, “Minimally invasive synthetic suburethral sling operations for stress urinary incontinence in women” (2009) The Cochrane Collaboration (John Wiley & Sons Ltd, 2009) (SHI.MESH.00003908)
ETH.MESH.03742906 at 2935)
Allman 2 at 17 (EXP.010.180.0001_3 at 0018_3)
AWS ch 12 at [27(e)] (SBM.010.005.0529_2 at 0536_2)
T3356 (TRA.500.043.0001_3 at 0002_3)
ETH.MESH.10682109
ETH.MESH.00827096
ETH.MESH.08306308
ETH.MESH.13456803
ETH.MESH.10682109
ETH.MESH.13449236 and ETH.MESH.13449237
AWS ch 12 at [34] (SBM.010.005.0529_2 at 0538_2)
ETH.MESH.10178882
ETH.MESH.10178882 at 9082
RWS ch 7 at [90(j)–(l)] (SBM.020.002.0108 at 0149-0151)
ETH.MESH.10178882 at 9065
Allman 1 at 39 (EXP.010.100.0001_4 at 0040_4)
JJM.MESH.00581976
JJM.MESH.00205999
JJM.MESH.00581976 at 1997
SHI.MESH.00042634
SHI.MESH.00042634 at 2844
JJM.MESH.00581976
SHI.MESH.00042634 at 2850
JJM.MESH.00581976 at 1984
JJM.MESH.00581976 at 1989
JJM.MESH.00581976 at 1990
JJM.MESH.00581976 at 1992
JJM.MESH.00581976 at 1993
JJM.MESH.00581976 at 1995
JJM.MESH.00581976 at 2002
ETH.MESH.16357664
AWS ch 12 at [100] (SBM.010.005.0529_2 at 0556_2)
ETH.MESH.16357664 at 7666
ETH.MESH.17634752 at 4754
ETH.MESH.17634752 at 4754-5
AWS ch 12 at [104] (SBM.010.005.0529_2 at 0557_2)
ETH.MESH.16358754
ETH.MESH.16358754 at 8798
AWS ch 12 at [110] (SBM.010.005.0529_2 at 0559_2)
ETH.MESH.25280424
ETH.MESH.25280424 at 0959
ETH.MESH.25280424 at 0880
ETH.MESH.25280424 at 0443; Hinoul at [311] (LAY.020.001.0001_4 at 0121_4)
ETH.MESH.25280424 at 0444
ETH.MESH.25280424 at 0699
ETH.MESH.25280424 at 0914
RWS ch 7 at [90(o)] (SBM.020.002.0108 at 0153)
ETH.MESH.25280424 at 0936
ETH.MESH.25280424 at 0938
ETH.MESH.25280424 at 0941
ETH.MESH.10703794
ETH.MESH.10703794 at 3809
ETH.MESH.10679735 at 9736; Hinoul [302] (LAY.020.001.0001_4 at 0115_4)
ETH.MESH.10703794 at 3803
ETH.MESH.17618513 at 8522
ETH.MESH.17618513 at 8523
ETH.MESH.17618513 at 8516
Hinoul at [311] (LAY.020.001.0001_4 at 0121_4)
ETH.MESH.25280424 at 0941
ETH.MESH.25280424 at 0942–0943
ETH.MESH.14489208
ETH.MESH.14489208 at 9210
ETH.MESH.14489208 at 9211
ETH.MESH.14489208 at 9213
ETH.MESH.25280424 at 0932
ETH.MESH.10682109 at 2305–2306
T1196 (TRA.500.014.0001_2 at 0069_2)
HMESH_ETH_05065597
ETH.MESH.25280424 at 0941
ETH.MESH.25280424 at 0943–0944
SBM.010.034.0001
Allman 2 at 19 (EXP.010.180.0001_3 at 0020_3)
Allman 2 at 19 (EXP.010.180.0001_3 at 0020_3)
ETH.MESH.00831618
ETH.MESH.00831576
Allman 1 at 47 (EXP.010.100.0001_4 at 0048_4)
AWS ch 12 at [42] (SBM.010.005.0529_2 at 0541_2)
Shaw J, Jeppson P and Rardin C, “Decreasing transobturator sling groin pain without decreasing efficacy using TVT-Abbrevo” (2015) 26(9) Int Urogynecol 1369–1372 (ETH.MESH.25296843)
ETH.MESH.25280424 at 0836
JJM.MESH.00484570
ETH.MESH.06051583
ETH.MESH.06051583
JJM.MESH.00339111
JJM.MESH.00339111
JJM.MESH.00339111 at 9112
ETH.MESH.02154877
ETH.MESH.02154877 at 4879
ETH.MESH.02154877 at 4878
JJM.MESH.00009139 at 9140
ETH.MESH.02154877 at 4879
ETH.MESH.02154877 at 4879
ETH.MESH.00311769
ETH.MESH.04127325
JJM.MESH.00060364
ETH.MESH.04127133
ETH.MESH.08487851
JJM.MESH.00446287
JJM.MESH.00446283
JJM.MESH.00446291
JJM.MESH.00446267
JJM.MESH.00446259
ETH.MESH.06049989
ETH.MESH.06049989
ETH.MESH.04127314 at 7315
JJM.MESH.00060364
ETH.MESH.04048515
Hinoul at [319] (LAY.020.001.0001_4 at 0125)
ETH.MESH.00874445
ETH.MESH.02151813
Allman 2 at 20 (EXP.010.180.0001_3 at 0021_3)
Allman 2 at 21 (EXP.010.180.0001_3 at 0022_3)
ETH.MESH.10685642
ETH.MESH.01797353
ETH.MESH.01797353
Allman 1 at 43 (EXP.010.100.0001_4 at 0044_4)
Allman 2 at 21 (EXP.010.180.0001_3 at 0022_3)
ETH.MESH.05600916 at 0922
ETH.MESH.11335606
ETH.MESH.10607089
ETH.MESH.11335606 at 5674
Allman 1 at 44 (EXP.010.100.0001_4 at 0045_4)
Allman 1 at 44 (EXP.010.100.0001_4 at 0045_4)
Allman 1 at 44 (EXP.010.100.0001_4 at 0045_4)
Allman 2 at 21 (EXP.010.180.0001_3 at 0022_3)
ETH.MESH.25296843
Allman 1 at 51–52 (EXP.010.100.0001_4 at 0052_4-0053_4)
Hinoul at [577] (LAY.020.001.0001_4 at 0274_4)
AWS ch 12 at [170] (SBM.010.005.0529_2 at 0574_2)
ETH.MESH.02232773; ETH.MESH.05189826
ETH.MESH.05189826
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2007) at 18 (SHI.MESH.00011315 at 1337)
T3518 (TRA.500.044.0001_3 at 0070_3)
ETH.MESH.00578261 at 8262
ETH.MESH.00578261 at 8263
RWS ch 8 at [11] (SBM.020.002.0155 at 0163)
ETH.MESH.10680160
ETH.MESH.10680160 at 0167
Hu 2 at 21 (EXP.010.184.0001_3 at 0022_3)
ETH.MESH.10680160 at 0169
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2010) (ETH.MESH.01028668) at 8676
Ibid at 2 (ETH.MESH.01028668 at 8676)
Ibid at 17–18 (ETH.MESH.01028668 at 8691–8692)
Ibid at 18 (ETH.MESH.01028668 at 8692)
ETH.MESH.03664020; Allman 2 at 24 (EXP.010.180.0001_3 at 0025_3)
ETH.MESH.03664020
Allman 2 at 24 (EXP.010.180.0001_3 at 0025_3)
Allman 1 at 18 (EXP.010.100.0001_4 at 0019_4)
Allman 1 at 18 (EXP.010.100.0001_4 at 0019_4)
ETH.MESH.00830212
ETH.MESH.01796630; ETH.MESH.01796654
ETH.MESH.01796654 at 6655
ETH.MESH.00830212 at 0214
ETH.MESH.08311115
ETH.MESH.08311115
ETH.MESH.08311095
ETH.MESH.08309389
ETH.MESH.08311095
ETH.MESH.08313174
AWS ch 12 at [199] (SBM.010.005.0529_2 at 0585_2)
ETH.MESH.08309202
ETH.MESH.08309056
ETH.MESH.08301293
Tijdink M et al, “Surgical management of mesh-related complications after prior pelvic floor reconstructive surgery with mesh” (2011) 22(11) Int Urogynecol J 1395–1404 (ETH.MESH.08309018)
ETH.MESH.08301293 at 1361–1362
Diwadkar G et al, “Complication and reoperation rates after apical vaginal prolapse surgical repair: a systematic review” (2009) 113(2 Pt 1) Obstet Gynecol 367–373 (ETH.MESH.00020695)
ETH.MESH.08301293 at 1362
Diwadkar G et al, “Complication and reoperation rates after apical vaginal prolapse surgical repair: a systematic review” (2009) 113(2 Pt 1) Obstet Gynecol 367–373 at 371 (ETH.MESH.00020695 at 0699)
ETH.MESH.01238632; ETH.MESH.01238633.
ETH.MESH.08301293 at 1365
ETH.MESH.08301293 at 1366
ETH.MESH.08301293 at 1373
T3641 (TRA.500.046.0001_3 at 0010_2)
T3643 (TRA.500.046.0001_3)
ETH.MESH.10680186 at 0264
ETH.MESH.10680186 at 0261
ETH.MESH.08301293 at 1369
ETH.MESH.06372555
ETH.MESH.06372555 at 2555
Allman 1 at 18–19 (EXP.010.100.0001_4 at 0019_4–0020_4)
Allman 2 at 28 (EXP.010.180.0001_3 at 0029_3)
Nygaard I et al “Long-term outcomes following abdominal sacrocolpopexy for pelvic organ prolapse” (2013) 309 JAMA 2016–2024 (ETH.MESH.09466922)
Hu 2 at 26 (EXP.010.184.0001_3 at 0027_3)
Hu 2 at 27 (EXP.010.184.0001_3 at 0028_3); Committee on Gynecologic Practice, “Committee Opinion No. 513: Vaginal placement of synthetic mesh for pelvic organ prolapse” (2011) 118(6) Obstet Gynecol 1459–1464 at 1462 (ETH.MESH.06252711 at 2714)
Hu 2 at 27 (EXP.010.184.0001_3 at 0028_3)
Hu 2 at 27 (EXP.010.184.0001_3 at 0028_3)
Allman 1 at 19 (EXP.010.100.0001_4 at 0020_4)
Allman 2 at 25 (EXP.010.180.0001_3 at 0026_3)
ETH.MESH.22146153
ETH.MESH.22146153 at 6252
Hu 2 at 30 (EXP.010.184.0001_3 at 0031_3)
Allman 2 at 25–6 (EXP.010.180.0001_3 at 0026_3-0027_3)
ETH.MESH.22146153 at 6247–6248
Allman 2 at 26 (EXP.010.180.0001_3 at 0027_3)
Hinoul at [215] (LAY.020.001.0001_2 at 0085_2)
Nygaard I et al, “Long-term outcomes following abdominal sacrocolpopexy for pelvic organ prolapse” (2013) 309 JAMA 2016–2024 (ETH.MESH.09466922)
Ibid at 2017 (ETH.MESH.09466922 at 6923)
Ibid at 2020 (ETH.MESH.09466922 at 6926)
Ibid at 2023 (ETH.MESH.09466922 at 6929)
T3858 (TRA.500.048.0001_2 at 0065_2)
Collinet at 17 (EXP.020.005.0001_4 at 0018_4)
ETH.MESH.02059150
ETH.MESH.00365412
Hinoul P et al, “A prospective study to evaluate the anatomic and functional outcome of a transobturator mesh kit (prolift anterior) for symptomatic cystocele repair” (2008) 15(5) J Minim Invasive Gynecol 615–620 (ETH.MESH.00016122)
T3481 (TRA.500.044.0001_3 at 0033_3)
Hinoul P et al “A prospective study to evaluate the anatomic and functional outcome of a transobturator mesh kit (prolift anterior) for symptomatic cystocele repair” (2008) 15(5) J Minim Invasive Gynecol 615–620 (ETH.MESH.00016122 at 6126)
ETH.MESH.10684599
ETH.MESH.10684599; ETH.MESH.10680160
ETH.MESH.10684599
Allman 1 at 24 (EXP.010.100.0001_4 at 0025_4)
ETH.MESH.06796086
ETH.MESH.06376830 at 6840–1
ETH.MESH.17650617 at 0628
ETH.MESH.06796086 at 6095
ETH.MESH.17650617 at 0628, 0629–30
ETH.MESH.00830220 at 0221
ETH.MESH.00009682
Iglesia C et al, “Vaginal mesh for prolapse: a randomized controlled trial” (2010) 116(2 Pt 1) Obstet Gynecol 293–303 (ETH.MESH.00013799)
ETH.MESH.00009682
Gordon 2 at 19–20 (EXP.010.161.0001_3 at 0021_3-0022_3)
AWS Ch 12 at [251] (SBM.010.005.0529_2 at 0598_2)
ETH.MESH.10684755
ETH.MESH.10684755 at 4772
ETH.MESH.10684755 at 4804
ETH.MESH.10684755
Allman 2 at 28 (EXP.010.180.0001_3 at 0029_3)
Bot-Robin V et al, “Use of vaginal mesh for pelvic organ prolapse repair: a literature review” (2012) 9 Gynecol Surg 3–15 (ETH.MESH.08564071)
Ibid (ETH.MESH.08564071 at 4079)
Ibid (ETH.MESH.08564071 at 4078)
ETH.MESH.10684755 at 4805
Allman 1 at 25 (EXP.010.100.0001_4 at 0026_4)
Abed H, “Incidence and management of graft erosion, wound granulation, and dyspareunia following vaginal prolapse repair with graft materials: a systematic review (2011) 22(7) Int Urogynecol J 789–798 (ETH.MESH.06142574)
Tijdink M et al, “Surgical management of mesh-related complications after prior pelvic floor reconstructive surgery with mesh” (2011) 22(11) Int Urogynecol J 1395–1404 (ETH.MESH.08309018 at 9025)
ETH.MESH.10684755 at 4811
ETH.MESH.10684755 at 4812
ETH.MESH.10684755 at 4812
ETH.MESH.10684755 at 4812
Allman 1 at 24 (EXP.010.100.0001_4 at 0025_4)
Allman 1 at 25 (EXP.010.100.0001_4 at 0026_4)
Allman 1 at 25 (EXP.010.100.0001_4 at 0026_4)
ETH.MESH.06796086 at 6097
Allman 1 at 25 (EXP.010.100.0001_4 at 0026_4)
ETH.MESH.10684212
ETH.MESH.08314409 at 4410
AWS ch 12 at [277] (SBM.010.005.0529_2 at 0599_2)
ETH.MESH.06399653 at 9769; RWS ch 8 [51] (SBM.020.002.0155)
RWS ch 8 at [50] (SBM.020.002.0155 at 0187)
ETH.MESH.06399653
ETH.MESH.08918910
ETH.MESH.06399653 at 9780
Milani A et al, “Trocar-guided mesh repair of vaginal prolapse using partially absorbable mesh: 1 year outcomes” (2011) 204(1) Am J Obstet Gynecol 74 (ETH.MESH.05110011)
ETH.MESH.00579915
AWS ch 12 at [263] (SBM.010.005.0529_2 at 0601_2)
ETH.MESH.03029416
ETH.MESH.03029416
ETH.MESH.04943199
ETH.MESH.04943201
ETH.MESH.04943199
ETH.MESH.13488424
ETH.MESH.13488807
ETH.MESH.04545603
Allman 2 at 28 (EXP.010.180.0001_3 at 0029_3)
Hinoul at [637] (LAY.020.001.0001_4 at 0307_4)
Gordon 2 at 20 (EXP.010.161.0001_3 at 0022_3)
Hu 2 at 22 (EXP.010.184.0001_3 at 0023_3)
ETH.MESH.13639854
ETH.MESH.04545603 at 5607
Hu 2 at 22 (EXP.010.184.0001_3 at 0023_3)
Hu 2 at 22 (EXP.010.184.0001_3 at 0023_3)
Hu 2 at 23 (EXP.010.184.0001_3 at 0024_3)
AWS ch 12 at [275] (SBM.010.005.0529_2 at 0605_2)
ETH.MESH.10684212
Allman 1 at 28 (EXP.010.100.0001_4 at 0029_4)
Allman 1 at 28 (EXP.010.100.0001_4 at 0029_4)
ETH.MESH.10684212 at 4214
Ozog Y et al, “Biomechanical effects of polyglecaprone fibers in a polypropylene mesh after abdominal and rectovaginal implantation in a rabbit” (2012) 23 Int Urogynecol J 1397–1402 (PUB.MESH.00006352)
Woodward 2 at [6.7.1] (EXP.010.106.0001_3 at 0053_3)
ETH.MESH.10684212 at 4225
ETH.MESH.01028668 at 8676
Woodward 2 at [6.12.3] (EXP.010.106.0001_3)
AWS ch 12 at [278(d)(iii)] (SBM.010.005.0529_2 at 0607_2)
Khandwala S and Jayachandran C, “Transvaginal mesh surgery for pelvic organ prolapse - Prolift+M: a prospective clinical trial” (2011) 22(11) Int Urogynecol J 1405–1411 (ETH.MESH.04036947)
Woodward 2 at [6.7.3] (EXP.010.106.0001_3 at 0054_3)
ETH.MESH.10684212 at 4225
ETH.MESH.10684212 at 4233
Diwadkar G et al, “Complication and reoperation rates after apical vaginal prolapse surgical repair: a systematic review” (2009) 113(2 Pt 1) Obstet Gynecol 367–373 at 371 (ETH.MESH.00020695 at 0699)
Ibid (ETH.MESH.00020695 at 0699)
Ibid at 372 (ETH.MESH.00020695 at 0700)
AWS ch 12 at [278(h)] (SBM.010.005.0529_2 at 0609_2)
ETH.MESH.10684322
ETH.MESH.10684322 at 4363
Allman 1 at 29 (EXP.010.100.0001_4 at 0030_4)
Allman 1 at 30 (EXP.010.100.0001_4 at 0031_4)
ETH.MESH.25147304
RWS ch 8 at [49(b)] (SBM.020.002.0155 at 0186)
AID.010.002.0001_8
ETH.MESH.10685039
ETH.MESH.07228683
ETH.MESH.10685039
ETH.MESH.08314282
ETH.MESH.10685039
Sayer T et al “Medium-term clinical outcomes following surgical repair for vaginal prolapse with tension-free mesh and vaginal support device” (2012) 23(4) Int Urogynecol J 487–493 (ETH.MESH.13653791)
Zyczynski H et al, “One-year clinical outcomes after prolapse surgery with nonanchored mesh and vaginal support device” (2010) 203(6) Am J Obstet Gynecol 587 (ETH.MESH.00028001)
ETH.MESH.00028001 at 8007
ETH.MESH.03046515
Carey M et al, “Vaginal surgery for pelvic organ prolapse using mesh and a vaginal support device” (2008) 115(3) BJOG 391–397 (ETH.MESH.00154197)
Carey M et al, “Vaginal repair with mesh versus colporrhaphy for prolapse: a randomised controlled trial” (2009) 116(10) BJOG 1380–1386 (ETH.MESH.00354025)
Woodward 2 at [6.11] (EXP.010.106.0001_3 at 0060_3)
ETH.MESH.07229215
Allman 1 at 34 (EXP.010.100.0001_4 at 0035_4)
ETH.MESH.06796086 at 6097
ETH.MESH.0679608 at 096
ETH.MESH.17650617 at 0628
ETH.MESH.10685269
ETH.MESH.10685269 at 5305
ETH.MESH.10685269 at 5305–5306
Allman 1 at 34 (EXP.010.100.0001_4 at 0035_4)
ETH.MESH.05600916 at 0919
JJM.MESH.00572989
ETH.MESH.06925500
ETH.MESH.00564358; see also AWS ch 12 at [349] (SBM.010.005.0529_2 at 0628_2)
SHI.MESH.00015366 at 5367
SHI.MESH.00015366 at 5371
T3259 (TRA.500.041.0001_3 at 0049_3); Hinoul at [510(f)(ii)] (LAY.020.001.0001_4 at 0246_4)
ETH.MESH.07239245
Hinoul at [468] (LAY.020.001.0001_4 at 0220_4)
Hinoul at [469] (LAY.020.001.0001_4 at 0220_4)
Hinoul at [470]–[471] (LAY.020.001.0001_4 at 0221_4)
JJM.MESH.00195552
T3262 (TRA.500.041.0001_3 at 0076_3)
T3271 (TRA.500.042.0001_3 at 0004_3)
ETH.MESH.05602302; ETH.MESH.03730586; ETH.MESH.03730594; ETH.MESH.05598153; T3284 (TRA.500.042.0001 at 0023)
ETH.MESH.05603815 at 3819
ETH.MESH.05598199
T3304–05 (TRA.500.042.0001_3 at 0037_3–0038_3); ETH.MESH.04925516; ETH.MESH.04925627; ETH.MESH.04925553; ETH.MESH.05810364
T3304–05 (TRA.500.042.0001_3 at 0037_3-0038_3)
HMESH_ETH_02064584
HMESH_ETH_02064584 at 4587
HMESH_ETH_02064584 at 4585
T3306 (TRA.500.042.0001_3 at 0039_3)
HMESH_ETH_02064589 at 4590
HMESH_ETH_02064581
ETH.MESH.05602306
ETH.MESH.05602306 at 2308
ETH.MESH.04474313
ETH.MESH.04567154; ETH.MESH.06847156
T3331 (TRA.500.042.0001_3 at 0065_3)
ETH.MESH.04474315
ETH.MESH.06593814; ETH.MESH.06593812; ETH.MESH.06593809
T3317–18 (TRA.500.042.0001_3 at 0050_3-0051_3); T3450 (TRA.500.044.0001_3 at 0002_3)
AWS ch 12 at [392] (SBM.010.005.0529_2 at 0645_2)
AID.010.002.0001_8
Hinoul from [128] (LAY.020.001.0001_4 at 0054_4), RWS at [55] (SBM.020.001.0001_2 at 0017_2)
JJM.MESH.00060248
AWS ch 12 at [47]–[48], [400] (SBM.010.005.0529_2 at 0542_2
JJM.MESH.00339111 at 9112
JJM.MESH.00205999
JJM.MESH.00438962
JJM.MESH.00002216
SHI.MESH.00042634 at 3044
JJM.MESH.00205999
SHI.MESH.00043163
JJM.MESH.00207358
JJM.MESH.00287779
JJM.MESH.00287779
JJM.MESH.00287779
ETH.MESH.16308855
Rosamilia 1 at 13 (EXP.020.026.0001_4 at 0015_4)
Cody J et al, “Systematic review of the clinical effectiveness and cost-effectiveness of tension-free vaginal tape for treatment of urinary stress incontinence) (2003) 7(21) Health Technol Assess iii–189 (ETH.MESH.04055002)
Nilsson C et al, “Seventeen years’ follow-up of the tension-free vaginal tape procedure for female stress urinary incontinence” (2013) 24(8) Int Urogynecol J 1265–1269 (ETH.MESH.19876896)
Blaivas 1 at 20 (EXP.010.026.0001_3 at 0022_3)
AWS at [117] (SBM.010.005.0529_2 at 0561_2)
JJM.MESH.00582003 at 2018
JJM.MESH.00582003 at 2007
SHI.MESH.00015458
JJM.MESH.00581973
ETH.MESH.24257904 at 7907
JJM.MESH.00614771
MSC.010.172.0001
MSC.010.172.0001
MSC.010.172.0001 at 0006
MSC.010.172.0001 at 0009
MSC.010.168.0001
MSC.010.170.0001
JJM.MESH.00614765
JJM.MESH.00614765 at 4767
JJM.MESH.00614761
JJM.MESH.00614761
JJM.MESH.00614845
AWS ch 12 at [408]–[409] (SBM.010.005.0529_2 at 0647_2)
JJM.MESH.00614902
JJM.MESH.00616884
AID.010.002.0001_8
AWS ch 12 at [412] (SBM.010.005.0529_2 at 0648_2)
AWS ch 12 at [411] (SBM.010.005.0529_2 at 0648_2)
MSC.010.168.0001
Hinoul at [136] (LAY.020.001.0001_4 at 0057_4)
Pence at [38] (EXP.010.200.0001_2 at 0013_2–0014_2); T1516 (TRA.500.019.0001_2 at 0004_2)
Pence at [38] (EXP.010.200.0001_2 at 0013_2–0014_2)
Pence at [43] (EXP.010.200.0001_2 at 0016_2)
Pence at [60] (EXP.010.200.0001_2 at 0021_2)
Pence at [60]–[65] (EXP.010.200.0001_2 at 0021_2–0022_2)
Pence at [60] (EXP.010.200.0001_2 at 0021_2)
SHI.MESH.00025051 at 5053
Pence at [40] (EXP.010.200.0001_2 at 0014_2); T1549 (TRA.500.019.0001_2 at 0037_2)
Pence at [41], [212] (EXP.010.200.0001_2 at 0016_2, 0109_2); PUB.MESH.00002388 at 2405, 2494
Pence at [69] (EXP.010.200.0001_2 at 0023_2)
SHI.MESH.00025051; SHI.MESH.00025003; and SHI.MESH.00025013
SHI.MESH.00025051 at 5056
SHI.MESH.00025013 at 4022
Pence at [43] (EXP.010.200.0001_2 at 0016_2)
ETH.MESH.07366187 at 6194
Pence at [43], [212] (EXP.010.200.0001_2 at 0016_2, 0109_2)
AWS Ch 10 at [13] (SBM.010.005.0425_2 at 0428_2); Pence at [42] (EXP.010.200.0001_2 at 0015_2–0016_2); SHI.MESH.00025013 at 5022; T1557–1561 (TRA.500.019.0001_2 at 0045_2–0049_2)
Pence at [44], [213] (EXP.010.200.0001_2 at 0016_2, 0109_2)
T1516 (TRA.500.019.0001_2 at 0004_2)
Pence at [116], [204] (EXP.010.200.0001_2 at 0057_2; 0103_2)
PUB.MESH.00002388 at 2452
T1561 (TRA.500.019.0001_2 at 0049_2)
Pence at [118], [209] (EXP.010.200.0001_2 at 0059_2; 0108_2)
Pence at [118], [209] (EXP.010.200.0001_2 at 0059_2; 0108_2)
Hinoul at [154] (LAY.020.001.0001_4 at 0063_4)
Pence at [215] (EXP.010.200.0001_2 at 0110_2)
SBM.010.017.0001
Pence at [217]–[218] (EXP.010.200.0001_2 at 0110_2-0111_2)
Pence at [220] (EXP.010.200.0001_2 at 0111_2); Hartung D et al, “Reporting discrepancies between the ClinicalTrials.gov results database and peer-reviewed publications” (2014) 160(7) Ann Intern Med 477–483 (SHI.MESH.00038279)
T1531 (TRA.500.019.0001_2 at 0019_2)
Pence at [216] (EXP.010.200.0001_2 at 0110_2)
Pence at [221] (EXP.010.200.0001_2 at 0111_2)
T1576 (TRA.500.019.0001_2 at 0064_2)
T1578 (TRA.500.019.0001_2 at 0066_2)
SHI.MESH.00033371 at 3374
SHI.MESH.00033371 at 3382
Bourrat M et al, “Complications and medium-term functional results of TVT in stress urinary incontinence” (2003) 13 Progress in Urology 1358–1364 (SHI.MESH.00043573_2)
ETH.MESH.06616596
T3676 (TRA.500.046.0001 at 15)
T3229 (TRA.500.041.0001_3 at 0042_3)
ETH.MESH.08695898; Hinoul at [73] (LAY.020.001.0001_4 at 0040_4)
ETH.MESH.05225354
ETH.MESH.10589402 at 9403
ETH.MESH.02340306 at 0331
ETH.MESH.10589402 at 9403
ETH.MESH.17636681
ETH.MESH.05225354 at 5383
ETH.MESH.05225354 at 5382
ETH.MESH.00658177 at 8189
ETH.MESH.00658177 at 8180
ETH.MESH.02340829 at 0835
ETH.MESH.03803462 at 3463
ETH.MESH.03911390 at 1391
ETH.MESH.03911390
ETH.MESH.22128727
ETH.MESH.03905059 at 5069
ETH.MESH.05799233 at 9237
ETH.MESH.02341203 at 1210
Pence at [117] (EXP.010.200.0001_2 at 0057_2)
Pence at [117] (EXP.010.200.0001_2 at 0057_2)
Pence at [118] (EXP.010.200.0001_2 at 0059_2)
Pence at [119] (EXP.010.200.0001_2 at 0059_2)
ETH.MESH.08221376
ETH.MESH.22128963
ETH.MESH.02342194
ETH.MESH.08221376
ETH.MESH.02342194 at 2196
ETH.MESH.02342278
ETH.MESH.02341522
ETH.MESH.02001398
ETH.MESH.01700610
ETH.MESH.01700610 at 0619
Pence at [205]–[208] (EXP.010.200.0001_2 at 0103_2–0106_2)
Pence at [205] (EXP.010.200.0001_2 at 0103_2)
Pence at [205]–[208] (EXP.010.200.0001_2 at 0103_2–0106_2)
Pence at [206] (EXP.010.200.0001_2 at 0104_2)
Pence at [205]–[208] (EXP.010.200.0001_2 at 0103_2–0106_2)
Pence at [210] (EXP.010.200.0001_2 at 0108_2)
ETH.MESH.00083765
ETH.MESH.00643217
ETH.MESH.00372319 from 2664
ETH.MESH.00372319 at 2666
ETH.MESH.00372319 at 2667
ETH.MESH.16357664 at 7665
ETH.MESH.16358754
ETH.MESH.16357664 at 7665
ETH.MESH.17635133; SBM.010.005.0529_2 at 0558_2
ETH.MESH.17635139 at 5140
ETH.MESH.16358753; ETH.MESH.16358754-8834
ETH.MESH.16358754 at 8756
ETH.MESH.16358754 at 8805
JJM.MESH.00581976
JJM.MESH.00581973
ETH.MESH.24991657
ETH.MESH.24991657 at 1658
ETH.MESH.24258019
JJM.MESH.00614771
MSC.010.170.0001
ETH.MESH.00658177
T5667 (TRA.500.071.0001 at 0051)
RWS ch 7 at [65] (SBM.020.002.0108 at 0136)
ETH.MESH.01271289 at 1292
T5667 (TRA.500.071.0001 at 0051)
JJM.MESH.00446160
Hinoul at [169], [602] (LAY.020.001.0001_4 at 0067_4, 0286_4)
ETH.MESH.01271289; Hinoul at [155] (LAY.020.001.0001_4 at 0063_4);
RWS ch 10 at [11]–[12] (SBM.020.002.0264 at 0270)
JJM.MESH.00497822
RWS ch 10 at [13] (SBM.020.002.0264 at 0270-0271)
JJM.MESH.00201987
JJM.MESH.00438961 at 8965
T6195 (TRA.500.077.0001 at 0048)
ETH.MESH.02161141
T4189 (TRA.500.052.0001_2 at 0029_2)
T4189 (TRA.500.052.0001_2 at 0029_2)
T4190 (TRA.500.052.0001_2 at 0030_2)
T4190 (TRA.500.052.0001_2 at 0030_2)
T1192 (TRA.500.014.0001_2 at 0065_2)
T1193 (TRA.500.014.0001_2 at 0066_2)
T1194 (TRA.500.014.0001_2 at 0067_2)
Korda 3 at 9 (EXP.010.129.0001_5 at 0010_5); T1187–88 (TRA.500.014.0001_2 at 0060_2–0061_2)
Korda 1 at 67 (EXP.010.078.0001_3 at 0069_3)
T1192 (TRA.500.014.0001_2 at 0065_2)
Collinet at 4–5 (EXP.020.005.0001_3 at 0004_3–0005_3); T4333 (TRA.500.054.0001_2 at 0045_2)
T4333 (TRA.500.054.0001_2 at 0038_2-0039_2)
Chughtai 2 at 7 (EXP.010.112.0001_3 at 0008_3)
Holland 1 at 34 (EXP.010.069.0001_3 at 0038_3)
T1504 (TRA.500.018.0001_2 at 0106_2)
SHI.MESH.00015458 at 5459
Holland 1 at 34 (EXP.010.069.0001_3 at 0038_3)
AWS Ch 10 at [99] (SBM.010.005.0425_2 at 0448_2)
JJM.MESH.00450463
ETH.MESH.05144315 at 4317
Korda 1 at 66 (EXP.010.078.0001_3 at 0068_3)
JJM.MESH.00572771_2 at 2772_2
JJM.MESH.00041471 at 1472
ETH.MESH.00219861 at 220079
Ward K and Hilton P “Prospective multicentre randomised trial of tension-free vaginal tape and colposuspension as primary treatment for stress incontinence” (2002) 325 BMJ 67 1–7 at 6 (ETH.MESH.13649139 at 9144)
ETH.MESH.04174461 at 4514
Cody J et al, “Systematic review of the clinical effectiveness and cost-effectiveness of tension-free vaginal tape for treatment of urinary stress incontinence” (2003) 7 Health Technol Assess 21 (ETH.MESH.04055002)
ETH.MESH.03910799
ETH.MESH.00316780
Ward K and Hilton P “Tension-free vaginal tape versus colposuspension for primary urodynamic stress incontinence: 5-year follow up” (2008) 115 BJOG 226–233 at 232 (ETH.MESH.04045078 at 5084)
ETH.MESH.00083765 at 3766
AWS Ch 10 at [68] (SBM.010.005.0425_2 at 0441_2)
JJM.MESH.00498916_2 at 8917_2; see also JJM.MESH.00323665 at 3667
T3399–3400 (TRA.500.043.0001_3 at 0045_3–0046_3)
JJM.MESH.00454395_2
JJM.MESH.00454395_2 at 4401
JJM.MESH.00572746_2
JJM.MESH.00453802_2
ETH.MESH.01186068
JJM.MESH.00454404_2
AWS Ch 10 at [79] (SBM.010.005.0425_2 at 0443_2)
JJM.MESH.00573165_2
JJM.MESH.00572757_2
JJM.MESH.00572754_2
JJM.MESH.00134664_2
AWS Ch 7 at [47]–[48] (SBM.010.005.0250_2 at 0259_2)
JJM.MESH.00134659_2
JJM.MESH.00324295_2
SHI.MESH.00036749
Williams D, “General Concepts of Biocompatibility” in Handbook of Biomaterial Properties (Springer Science+Business Media, 1998) at 482–483 (SHI.MESH.00014223 at 4224)
JJM.MESH.00453820_2 at 3821_2
JJM.MESH.00453820_2
ETH.MESH.00329162
JJM.MESH.00453820_2 at 3825_2
ETH.MESH.00643217
ETH.MESH.00372319 at 2667
ETH.MESH.00372319 at 2697
ETH.MESH.00372319 at 2667
ETH-10187
JJM.MESH.00572938_2
T6196 (TRA.500.077.0001 at 0049)
JJM.MESH.00573161_2
JJM.MESH.00572736_2
Carey M et al, “Vaginal repair with mesh versus colporrhaphy for prolapse: a randomised controlled trial” (2009) 116(10) BJOG 1380–1386 (ETH.MESH.00588983)
Sayer T et al, “Medium-term clinical outcomes following surgical repair for vaginal prolapse with tension-free mesh and vaginal support device” (2011) Int Urogynecol J (ETH.MESH.13653791)
ETH.MESH.04420750
ETH.MESH.07157112
Pence at [135]–[136] (EXP.010.200.0001_2 at 0065_2)
AWS Ch 10 at [142] (SBM.010.005.0425_2 at 0457_2).
ETH.MESH.03162936
JJM.MESH.00572738_2
JJM.MESH.00573157
T5740 (TRA.500.072.0001 at 0054)
ETH.MESH.06129870
ETH.MESH.06129870 at 9874
AWS Ch 10 at [97] (SBM.010.005.0425_2 at 0448_2)
ETH.MESH.10132641 at 2650
See, for example, JJM.MESH.00498916_2; JJM.MESH.00323665_2
SHI.MESH.00039448
SHI.MESH.00039448
SHI.MESH.00039448
T302–03 (TRA.500.004.0001_3 at 0070_3–0071_3)
ETH.MESH.02142685
ETH.MESH.03029416
ETH.MESH.03029416
ETH.MESH.02161141
T5921 (TRA.500.074.0001 at 0063)
Blaivas 1 at 20 (EXP.010.026.0001_3 at 0022_3)
T3140 (TRA.500.040.0001_4 at 0042_4); T3172–73 (TRA.500.040.0001_4 at 0074_4-0075_5)
T3165 (TRA.500.040.0001_4 at 0067_4)
Hinoul at [221] (LAY.020.001.0001_4 at 0088_4)
RWS ch 11 at [18] (SBM.020.002.0275 at 0282)
Klinge U et al, “Demands and properties of alloplastic implants for the treatment of stress urinary incontinence” (2007) 4(3) Expert Rev Med Devices 349–359 at 354 (ETH.MESH.13766913 at 6918)
ETH.MESH.10589402 at 9404
Klinge 1 at [318(a)] (LAY.010.022.0001 at 0114)
ETH.MESH.02589032 at 9075
Klinge 1 at [318(a)] (LAY.010.022.0001 at 0114)
ETH.MESH.03427878 at 7882
T3819 (TRA.500.048.0001_2 at 0026_2)
JJM.MESH.00498916_2
Blaivas 2 at 6 (EXP.010.166.0001_3 at 0008_3)
Haylen, B et al, “An International Urogynecological Association (IUGA) International Continence Society (ICS) joint terminology and classification of the complications related directly to the insertion of prostheses (meshes, implants, tapes) & grafts in female pelvic floor surgery” (2011) 22 (3) Int Urogynecol J 1–15 at 6–7 (ETH.MESH.00187674 at 7677–7678)
ETH.MESH.05225354 at 5383
Klinge 1 at [290] (LAY.010.022.0001_2 at 0104_2)
T245 (TRA.500.004.0001_3 at 0013_3)
Klinge 1 at [290] (LAY.010.022.0001_2 at 0104_2)
ETH.MESH.03932912
ETH.MESH.00272548
ETH.MESH.02340829 at 0835
JJM.MESH.00498916_2
T3397 (TRA.500.043.0001_3 at 0043_3)
T3398 (TRA.500.043.0001_3 at 0044_3)
T5690 (TRA.500.072.0001 at 0004); JJM.MESH.00043638
JJM.MESH.00043638
SBM.010.011.0001 at 0022
JJM.MESH.00454395_2 at 4401_2
ETH.MESH.22129185 at 9189
Davila G, Drutz H and Deprest J, “Clinical implications of the biology of grafts: conclusions of the 2005 IUGA Grafts Roundtable” (2006) 17 Int Urogynecol J S51-S55 (ETH.MESH.01594955)
Klinge at [291] (LAY.010.022.0001_2 at 0104_2)
ETH.MESH.05225354 at 5383
AWS Ch 10 at [51] (SBM.010.030.0424 at 0437)
SBM.010.011.0001 at 0020
ETH.MESH.04056606 at 6608
ETH.MESH.04056606 at 6606
ETH.MESH.04056606 at 6607
T5933 (TRA.500.074.0001 at 0075)
AWS Ch 10 at [67] (SBM.010.005.0425_2 at 0441_2); ETH.MESH.16358754
JJM.MESH.00454395_2
AWS Ch 10 at [104] (SBM.010.005.0425_2 at 0450_2)
AWS Ch 10 at [105] (SBM.010.005.0425_2 at 0450_2)
SBM.010.005.0002_2 at 0011_2
MSC.010.168.0001
AWS Ch 10 at [104] (SBM.010.005.0425_2 at 0450_2); T4780 (TRA.500.059.0001 at 0063)
T4138–39 (TRA.500.051.0001_2 at 0061_2)
T4139 (TRA.500.051.0001_2 at 0062_2)
T4143 (TRA.500.051.0001_2 at 0066_2)
T4145–46 (TRA.500.051.0001_2 at 0068_2)
T5915 (TRA.500.074.0001 at 0057)
JJM.MESH.00614765 at 4766
ETH.MESH.02342194 at 2195
Pence at [206]–[208] (EXP.010.200.0001_2 at 0104_2–0106_2)
SBM.010.005.0002_2 at 0025_2
JJM.MESH.00453820 at 3826
Klinge at [318(e)] (LAY.010.022.0001_2 at 0114_2)
Korda 3 at 10 (EXP.010.129.0001_5 at 0011_5)
T1188 (TRA.500.014.0001_2 at 0061_2)
T1188 (TRA.500.014.0001_2 at 0061_2)
Elmér C et al, “Risk factors for mesh complications after trocar guided transvaginal mesh kit repair of anterior vaginal wall prolapse” (2012) 31 Neurourol Urodyn 1165–1169 (ETH.MESH.08299594)
Leake at [55] (LAY.010.002.0001 at 0016)
Pence at [206]–[208] (EXP.010.200.0001_2 at 0104_2–0106_2)
ETH.MESH.22128963 at 8966
AWS Ch 10 at [122(a)] (SBM.010.005.0425_2 at 0453_2)
Pence at [206]–[208] (EXP.010.200.0001_2 at 0104_2–0106_2)
T6356 (TRA.500.078.0001 at 0104)
T6357 (TRA.500.078.0001 at 0105)
ETH.MESH.02001398 at 1400
ETH.MESH.02001398 at 1400
ETH.MESH.00748451at 8505
ETH.MESH.00012009 at 2012, 2056
Hinoul at [152] (LAY.020.001.0001_4 at 0062_4)
Allman 2 at 8 (EXP.010.180.0001_3 at 0009_3)
T6360 (TRA.500.078.0001 at 0108)
T6102–03 (TRA.500.076.0001 at 0066–0067)
SHI.MESH.00028426_2
ETH.MESH.10681490 at 1528
ETH.MESH.05151121 at 1126
ETH.MESH.25280424 at 0578
ETH.MESH.10682109 at 2200, 2280
ETH.MESH.09908206
ETH.MESH.10682109 at 2296
ETH.MESH.25280424 at 0881
Nambiar A, Cody J and Jeffery S, “Single-incision sling operations for urinary incontinence in women” The Cochrane Collaboration (John Wiley & Sons Ltd, 2014) at 3 (SHI.MESH.00004889 at 4895)
Ibid at 14 (SHI.MESH.00004889 at 4906)
Ibid at 19 (SHI.MESH.00004889 at 4911)
Abdel-Fattah M et al, “Single-Incision Mini-Slings Versus Standard Midurethral Slings in Surgical Management of Female Stress Urinary Incontinence: A Meta-Analysis of Effectiveness and Complications” (2011) Eur Urol 1–13 (ETH.MESH.04559103)
Schimpf M et al, “Sling surgery for stress urinary incontinence in women: a systematic review and metaanalysis” (2014) 211 Am J Obstet Gynecol 71.e1–71.237 at 71.e5 (ETH.MESH.18852919 at 2923)
Ibid at 71.e16 (ETH.MESH.18852919 at 2934)
Hinoul P et al, “A Randomized, Controlled Trial Comparing an Innovative Single Incision Sling With an Established Transobturator Sling to Treat Female Stress Urinary Incontinence” (2011) 185 J Urol 1356–1362 (ETH.MESH.13213961)
Ibid at 1361 (ETH.MESH.13213961 at 3966)
Bianchi-Ferraro A et al, “Randomized controlled trial comparing TVT-O and TVT-S for the treatment of stress urinary incontinence: 2-year results” (2014) 25 Int Urogynecol J 1343–1348 (SHI.MESH.00001601)
Tomaselli G et al, “Tension-Free Vaginal Tape-O and-Secur for the Treatment of Stress Urinary Incontinence: A Thirty-Six-Month Follow-Up Single-Blind, Double-Arm, Randomized Study” (2013) 20 JMIG 198–204 at 203 (ETH.MESH.16322795 at 2800)
Tincello D et al, “The TVT Worldwide Observational Registry for Long-Term Data: Safety and Efficacy of Suburethral Sling Insertion Approaches for Stress Urinary Incontinence in Women” (2011) 186 J Urol 2310–2315 (ETH.MESH.08307408)
ETH.MESH.11335606 at 5675
ETH.MESH.10682109 at 2280, 2197
PUB.MESH.00009294
Hinoul at [388] (LAY.020.001.0001_4 at 0168_4)
Thubert T et al, “Bladder injury and success rates following retropubic mid-urethral sling: TVT EXACT vs. TVT” (2016) 198 Eur J Obstet Gynecol Reprod Biol 78–83 (PUB.MESH.00002261)
Aniuliene R, Aniulis P and Skaudickas D, “TVT-Exact and midurethral sling (SLING-IUFT)
operative procedures: a randomized study” (2015) 10 Open Med 311–317 (PUB.MESH.00001868)
ETH.MESH.25280424 at 0872
ETH.MESH.06372555
Milani A et al, “Trocar-guided mesh repair of vaginal prolapse using partially absorbable mesh: 1 year outcomes” (2011) 204 Obstet Gynecol 74.e1–74.e8 (ETH.MESH.05110011)
ETH.MESH.06284961
Khandwala S and Jayachandran C, “Transvaginal mesh surgery for pelvic organ prolapse-Prolift+M: a prospective clinical trial”(2011) Int Urogynecol J 1–7 (ETH.MESH.04036947)
Khandwala S, “Transvaginal Mesh Surgery for Pelvic Organ Prolapse: One-Year Outcome Analysis” (2013) 19(2) FPMRS 84–89 (ETH.MESH.08299637)
ETH.MESH.10684322 at 4366
Carey M et al, “Vaginal surgery for pelvic organ prolapse using mesh and a vaginal support device” (2008) 115 BJOG 391–397 (ETH.MESH.00154197)
Zyczynski M et al, “One-year clinical outcomes after prolapse surgery with nonanchored mesh and vaginal support device” (2010) 2013 Am J Obstet Gynecol 1.e1–1.e8 (ETH.MESH.00028001)
ETH.MESH.10685269 at 5292
ETH.MESH.10685269 at 5290
Sayer T et al, “Medium-term clinical outcomes following surgical repair for vaginal prolapse with tension-free mesh and vaginal support device” (2011) Int Urogynecol J (ETH.MESH.13653791)
ETH.MESH.10685269 at 5293
ETH.MESH.10685269 at 5293
ETH.MESH.07902732; ETH.MESH.10685269 at 5296
ETH.MESH.10685000 at 5306
T371–72 (TRA.500.005.0001_2 at 0059_2–0060_2)
ETH.MESH.09746846
ETH.MESH.01946717
JM.MESH.00041606
JJM.MESH.00127109; JJM.MESH.00127113
T6019 (TRA.500.075.0001 at 0073)
Fifth Further Amended Statement of Claim at [48]
Fifth Further Amended Statement of Claim at [45]
T6042 (TRA.500.076.0001 at 0006)
AWS ch 13 at [5] (SBM.010.005.0649_2 at 0653_2)
T6089 (TRA.500.076.0001 at 0053)
RWS ch 2 at [75(a)] (SBM.020.002.0024 at 0051–0053)
RWS ch 2 at [75(b)(ii)] (SBM.020.002.0024 at 0051)
Explanatory Memorandum for the Trade Practices Amendment Bill 1992
Harland D, “The Influence of European Law on Product Liability in Australia” (1995) 17 Syd LR 336 at 337
SBM.010.005.0002_2 at 0008_2
SBM.010.005.0002_2 at 0007_2
RWS ch 13 at [3] (SBM.020.002.0338)
AWS ch 13 at [14], [35] (SBM.010.005.0649_2 at 0656_2, 0662_2)
AWS ch 13 at [17], [36] (SBM.010.005.0649_2 at 0657_2, 0663_2)
AWS ch 13 at [19]–[20], [43] (SBM.010.005.0649_2 at 0657_2-0658_2, 0665_2)
AWS ch 13 at [12] (SBM.010.005.0649_2 at 0655_2–0656_2)
AWS ch 13 at 8 (SBM.010.005.0649_2 at 0657_2)
SBM.020.026.0001 at 0008
RWS ch 10 at 5 (SBM.020.002.0264 at 0268)
RWS ch 2 at [75] (SBM.020.002.0024 at 0051–55)
T1008-T1011 (TRA.500.012.0001_2 at 0018_2-0021_2); T1357 (TRA.500.017.0001_2 at 0019_2)
T1010 (TRA.500.012.0001_2 at 0020_2); T1358 (TRA.500.017.0001_2 at 0020_2)
T6238 (TRA.500.577.0001 at 0091)
T6304 (TRA.500.078.0001 at 0052)
RWS ch 10 at [7] (SBM.020.002.0264 at 0268-69)
RWS ch 2 at 26 (SBM.020.002.0224 at 0049)
Chughtai 2 at [4.3] (EXP.010.112.0001_3 at 0010_3)
Collinet P et al “Transvaginal mesh technique for pelvic organ prolapse repair: mesh exposure management and risk factors” (2006) 17 Int Urogynecol J 315-20 at 319 (ETH.MESH.00071591 at 1595)
Moalli, P et al “Tensile properties of five commonly used mid-urethral slings relative to TVT” (2008) 19 Int Urogynecol J 655-663 (SHI.MESH.00033759)
AWS ch 9 at [29] (SBM.010.005.0379_2 at 0390_2); JJM.MESH.00082926; T5709 (TRA.500.072.0001 at 0023)
JJM.MESH.00082931
JJM.MESH.00082931 at 0937
Korda 1 at [4.15] (EXP.010.078.0001_3 at 0089_3)
T3168–69 (TRA.500.040.0001_4 at 0070_4–0071_4)
T3229 (TRA.500.041.0001_3 at 0042_3)
Blaivas 1 at 14 (EXP.010.026.0001_2 at 0016_2)
Blaivas 1 at 14 (EXP.010.026.0001_2 at 0016_2)
Korda 3 at 23 (EXP.010.129.0001_5 at 0024_5)
Korda 1 at 87 (EXP.010.078.0001_3 at 0089_3)
RWS ch 13 at [61] (SBM.020.002.0338 at 0354)
Korda 1 at [4.14] (EXP.010.078.0001_3 at 0088_3)
Deprest at [197] (EXP.020.006.0001_3 at 0044_3)
Agur 2 at 8 (EXP.010.152.0001_3 at 0009_3)
MSC.020.025.0001
T6224–25 (TRA.500.077.0001 at 0077–0078)
MSC.020.025.0001 at 0024
T3771–72 (TRA.500.047.0001_3 at 0063_3–0064_3)
T3744 (TRA.500.047.0001_3 at 0036_3)
T3744 (TRA.500.047.0001_3 at 0036_3)
JJM.MESH.00043638; T1197 (TRA.500.014.0001_2 at 0070_2)
T1320–21 (TRA.500.016.0001_2 at 0002_2-0003_2)
JJM.MESH.00043638
T1320–21 (TRA.500.016.0001_2 at 0002_2–0003_2)
AWS ch 2 at [155] (SBM.010.005.0059_2 at 0118_2-0119_2)
ETH.MESH.00578939 at 8941
Chughtai 2 at [4.1] (EXP.010.112.0001_3 at 0010_3)
T6261 (TRA.500.078.0001 at 0009)
RWS ch 13 at [57] (SBM.020.002.0338 at 0353)
T6047 (TRA.500.076.0001 at 0011)
SBM.010.011.0001 at 0009
Hinoul at [142] (LAY.020.001.0001_4 at 0060_4)
T3416–19 (TRA.500.043.0001_3 at 0062_3-0065_3); ETH.MESH.02142685
SBM.020.025.0001 at 0004
RWS ch 10 at [21] (SBM.020.002.0264 at 0274-74)
RWS ch 10 at [20] (SBM.020.002.0264 at 0273); SBM.020.025.0001 at 0002
SBM.020.025.0001 at 0002
AWS Ch 10 at [71] (SBM.010.005.0001_2 at 0442_2)
T5716 (TRA.500.072.0001 at 0031)
Hinoul at [73] (LAY.020.001.0001_4 at 0040_4)
Chughtai 2 at 9 (EXP.010.112.0001_3 at 0010_3)
Hinoul at [152] (LAY.020.001.0001_4 at 0062_4)
Hinoul at [137] (LAY.020.001.0001_4 at 0057_4)
T3171–73 (TRA.500.040.0001_4 at 0073_4-0075_4)
T3173 (TRA.500.040.0001_4 at 0073_4)
ETH.MESH.04094863
ETH.MESH.05240032 at 0033
Klinge U et al, “Demands and properties of alloplastic implants for the treatment of stress urinary incontinence” (2007) Expert Rev Med Devices 4(3) 349–359.ETH.MESH.13766913 at 6918
T3096 (TRA.500.039.0001_3 at 0095_3)
Hinoul at [221] (LAY.020.001.0001_4 at 0088_4–0089_4)
Klinge at [318] (LAY.010.022.0001_2 at 0113_2–0115_2)
T3826–27 (TRA.500.048.0001_2 at 0033_2-0034_2)
Klinge at 114 (LAY.010.022.0001_2 at 0114_2)
Klinge at 114 (LAY.010.022.0001_2 at 0114_2)
Klinge at 114 (LAY.010.022.0001_2 at 0114_2)
Klinge at [318(j)] (LAY.010.022.0001_2 at 0115_2)
Klinge at [235] (LAY.010.022.0001_2 at 0087_2)
Klinge at [113] (LAY.010.022.0001_2 at 0035_2-0036_2)
Klinge at [188] (LAY.010.022.0001_2 at 0071_2)
Klinge at 114 (LAY.010.022.0001_2 at 0114_2)
Klinge at 114–5 (LAY.010.022.0001_2 at 0114_2-0115_2)
Allman 2 at 8 (EXP.010.180.0001_3 at 0009_3)
Hinoul at [148] (LAY.020.001.0001_4 at 0061_4)
RWS ch 10 at 5–6 (SBM.020.002.0264 at 0268–0269)
RWS ch 10 at [7] (SBM.020.002.0264 at 0268–0269)
JJM.MESH.00572736
T3145 (TRA.500.040.0001_4 at 0047_4)
T3229 (TRA.500.041.0001_3 at 0042_3)
T3229 (TRA.500.041.0001_3 at 0042_3)
T3229 (TRA.500.041.0001_3 at 0042_3)
JJM.MESH.00041471
ETH.MESH.00130934
ETH.MESH.08167452, admitted into evidence at TRA.500.080.0001 at 0041-42
ETH.MESH.03911390 at 1391
ETH.MESH.02286052
ETH.MESH.02286052
ETH.MESH.06828907
ETH.MESH.00132990
ETH.MESH.00757019
ETH.MESH.00757019
ETH.MESH.04092868
T3085 (TRA.500.039.0001_3 at 0084_3)
RWS ch 2 at 31 (SBM.020.002.0024 at 0054)
T6070 (TRA.500.076.0001 at 0034)
Ostergard D, “Degradation, infection and heat effects on polypropylene mesh for pelvic implantation: what was known and when it was known” (2011) 22(7) Int Urogynecol J 771–774 (ETH.MESH.00187693); Klinge U et al, “Demands and properties of alloplastic implants for the treatment of stress urinary incontinence” (2007) 4(3) Expert Rev Med Devices 349–359 (ETH.MESH.13766913)
RWS ch 11 at [65] (SBM.020.002.0275 at 0296–7)
RWS ch 11 at [96]–[103] (SBM.020.002.0275 at 0308–0309)
SHI.MESH.00038841 at 9025, 9028
Klinge at T2742 (TRA.500.035.0001_3 at 0033_3)
T6047 (TRA.500.076.0001 at 0011)
SBM.010.005.0001_2 at 0006_2
See, for example Korda 3 at 10 (EXP.010.129.0001_5 at 0011_5)
Chughtai 2 at 9 (EXP.010.112.0001_3 at 0010_3)
RWS ch 13 at [12]–[13], [24]-[25] (SBM.020.002.0338 at 0342, 0345)
AWS ch 13 from [14], [35] (SBM.010.005.0649_2 from 0656_2, 0663_2)
Elliot D, “Con: mesh in vaginal surgery: do the risks outweigh the benefits?” (2012) 22 Curr Opin Urol 2012 276–281 at 280 (SHI.MESH.00033536 at 3540)
Korda 3 at 23 (EXP.010.129.0001_5 at 0024_5)
SBM.010.005.0002_2 at 0004_2–0005_2
RWS ch 13 at 3–4 (SBM.020.002.0338 at 0340–0341)
RWS ch 13 at [7] (SBM.020.002.0338 at 0340)
AWS ch 13 at [13], [34] (SBM.010.005.0649_2 at 0656_2, 0662_2)
T6098 (TRA.500.076.0001 at 0062)
ETH.MESH.04174461 at 4470
Cox A, Herschorn, S and Lee L, “Surgical management of female SUI: is there a gold standard?” (2013) 10(2) Nat Rev Urol 78-89 (ETH.MESH.14421306)
RWS ch 13 at [15] (SBM.020.002.0338 at 0342-43)
RWS ch 13 [16] (SBM.020.002.0338 at 0343)
T1196 (TRA.500.014.0001_2 at 0069_2)
T1364 (TRA.500.017.0001_2 at 0026_2)
Agur 1 at 23 (EXP.010.049.0001_3 at 0024_3)
Agur 1 at 19 (EXP.010.049.0001_3 at 0020_3)
T4729 (TRA.500.059.0001 at 0012)
Roovers 1 at 7 (EXP.020.044.0001_3 at 0009_3)
MSC.010.168.0001
PUB.MESH.00009294
PUB.MESH.00009294
JJM.MESH.00614771
Ford A et al, “Mid-urethral sling operations for stress urinary incontinence in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2015) at 2 (ETH.MESH.24982894 at 2900)
Ibid at 3 (ETH.MESH.24982894 at 2901)
RWS ch 13 [19] (SBM.020.002.0338 at 0344); T6102 (TRA.500.076.0001 at 0066)
T6061, 6103 (TRA.500.076.0001 at 0025, 0067)
RWS Ch 13 [20] (SBM.020.002.0338 at 0344)
T1204–5 (TRA.500.014.0001_2 at 0078_2)
T1195 (TRA.500.014.0001_2 at 0068_2)
RWS ch 13 at [63] (SBM.020.002.0338 at 0355)
T723 (TRA.500.008.0001_2 at 0112_2)
T712 (TRA.500.008.0001_2 at 0101_2)
T712 (TRA.500.008.0001_2 at 0101_2)
T723 (TRA.500.008.0001_2 at 0112_2)
T723 (TRA.500.008.0001_2 at 0112_2)
T723–24 (TRA.500.008.0001_2 at 0112_2-0113_2)
AWS ch 10 (SBM.010.005.0001_2)
ETH.MESH.00592915
ETH.MESH.07512722
Klinge at [318] (LAY.010.022.0001_2 at 1113_2)
Blaivas at [4.7(b)] (EXP.010.026.0001_2 at 0013_2)
ETH.MESH.00277754
AWS ch 10 at [33] (SBM.010.005.0425_2 at 0435_2)
ETH.MESH.00272548
T3148 (TRA.500.040.0001_4 at 0050_4)
Richter H et al, “Retropubic versus Transobturator Midurethral Slings for Stress Incontinence” (2010) 362(22) N Eng J Med 2066–2076 at 2072 (ETH.MESH.09231613 at 1619)
Albo M et al, “Treatment Success of Retropubic and Transobturator Mid Urethral Slings at 24 Months” (2012) 188 J Urol 2281–2287 at 2284–2285 (ETH.MESH.12358173 at 8176–8177)
ETH.MESH.03919404
Margolis 1 at 17 (EXP.010.041.0001_3 at 0018_3)
T842 (TRA.500.010.0001_2 at 0049_2)
Hazewinkel M, Hinoul P and Roovers R, “Persistent groin pain following a trans-obturator sling procedure for stress urinary incontinence: a diagnostic and therapeutic challenge”, Int Urogynecol J (2009) 20:363–365 (ETH.METH.00277754)
Agur 1 at 10 (EXP.010.049.0001_3 at 0011_3)
Davila G, Drutz H and Deprest J, “Clinical implications of the biology of grafts: conclusions of the 2005 IUGA Grafts Roundtable”(2006) 17 Int Urogynecol J (2006) S51-S55 (ETH.MESH.01594955)
AWS ch 10 at [66] (SBM.010.005.0001_2 at 0441_2)
Chughtai 2 (EXP.010.112.0001_3 at 0010_3)
T3194 (TRA.500.041.0001_3 at 0007_3)
ETH.MESH.16358754 at 8758
T3194 (TRA.500.041.0001_3 at 0007_3)
SHI.MESH.00004889 at 4895
Hinoul P et al, “A Randomized, Controlled Trial Comparing an Innovative Single Incision Sling With an Established Transobturator Sling to Treat Female Stress Urinary Incontinence” (2011) 185 J Urol 1356–1362 (ETH.MESH.13213961)
ETH.MESH.05600916 at 0922
T6354 (TRA.500.078.0001 at 0102)
T6354 (TRA.500.078.0001 at 0102)
JJM.MESH.00614765 at 4765
JJM.MESH.00453820_2
AWS ch 13 at [15] (SBM.010.005.0649_2 at 0656_2-0657_2)
AWS ch 13 at [24] (SBM.010.005.0649_2 at 0659_2-0660_2)
SHI.MESH.00038518
Maher C et al, “Transvaginal mesh or grafts compared with native tissue repair for vaginal prolapse” The Cochrane Collaboration (John Wiley & Sons Ltd, 2016) (SHI.MESH.00005748 at 5779)
T4147 (TRA.500.051.0001_2 at 0070_2)
T4138–39 (TRA.500.051.0001_2 at 0061_2–0062_2)
T4046-47 (TRA.500.050.0001_2 at 0065_2-0066_2)
T4047, 4051 (TRA.500.050.0001_2 at 0065_2, 0070_2)
SHI.MESH.00006213 at 6216–7
SHI.MESH.00006213 at 6277
Collinet at 17 (EXP.020.005.0001_4 at 0017_4)
Agur 2 at 3 (EXP.010.152.0001 at 0004_3)
T4408 (TRA.500.055.0001_3 at 0009_3)
Korda 1 at 84 (EXP.010.078.0001_3 at 0086_3)
Korda 1 at 82–83 (EXP.010.078.0001 at 0084_3–0085_3)
Karram, M and Maher C, “Surgery for posterior vaginal wall prolapse” (2013) 24 Int Urogynecol J 1835–1841 (SHI.MESH.00002941)
Korda 1 at 85 (EXP.010.078.0001 at 0087_3)
Korda 1 at 86 (EXP.010.078.0001 at 0088_3)
T4778–80 (TRA.500.059.0001 at 0061–0063)
Rosamilia 1 (EXP.020.026.0001_4 at 0015_4, 0032_4)
Deprest 1 at [196] (EXP.020.006.0001_4 at 0044_4)
SHI.MESH.00044454 at 4459
Roovers 1 (EXP.020.033.0001_4 at 0016_4)
Roovers 1 (EXP.020.033.0001_4 at 0028_4)
Korda 1 at 84–85 (EXP.010.078.0001 at 0086_3-0087_3); Lam at 33 (EXP.020.021.0001_4 at 0035_4)
Korda 1 at 1 (EXP.010.078.0001_3 at 0002_3)
Deprest 1 at [339] (EXP.020.006.0001_4 at 0070_4)
SHI.MESH.00002941
SHI.MESH.00033536 at 3540
Harland D, “The Influence of European Law on Product Liability in Australia” (1995) 17 Syd LR 336 at 337
T2744 (TRA.500.035.0001_3 at 0035_3)
T2783 (TRA.500.035.0001_3 at 0022_3)
Williams D, “General Concepts of Biocompatibility” Handbook of Biomaterial Properties (Springer Science & Business Media, 1998) (SHI.MESH.00014223 at 4226)
Hilger W et al, “Histological and biomechanical evaluation of implanted graft materials in a rabbit vaginal and abdominal model” (2006) 195 AJOG 1826–1831 (ETH.MESH.02587798)
RWS ch 11 at [96] (SBM.020.002.0275 at 0307)
T6089 (TRA.500.076.0001 at 0054)
T6082 (TRA.500.076.0001 at 0046)
TRA.510.005.0001 at 0002
AWS ch 13 at [61] (SBM.010.005.0001_2 at 0669_2–0670_2)
AWS ch 13 at [62] (SBM.010.005.0001_2 at 0670_2)
ETH.MESH.00680021
ETH.MESH.07477967
ETH.MESH.00094046
JJM.MESH.00498916; JJM.MESH.00323665
ETH.MESH.04093117
JJM.MESH.00453820
JJM.MESH.00453820_2
JJM.MESH.00324295_2
T1320–21 (TRA.500.016.0001_2 at 0002_2–0003_2)
Government Gazette Western Australia (17 December 2002) at 5905
T5946–50 (TRA.500.075.0001 at 0002–0006)
AWS ch 13 at [60] (SBM.010.005.0649_2 at 0669_2)
T6042–43 (TRA.500.076.0001 at 0006–0007)
PUB.MESH.00002388 at 2414
PUB.MESH.00002388 at 2459
PUB.MESH.00002388 at 2420
SBM.020.002.0001_0067–0076
SHI.MESH.00025003 at 5009
AWS ch 7 at [65] (SBM.010.005.0250_2 at 0262_2); AWS ch 8 at [173] (SBM.010.030.0295 at 0336)
AWS ch 7 at [65] (SBM.010.005.0250_2 at 0262_2); AWS ch 8 at [8] (SBM.010.005.0296_2 at 0298_2)
AWS ch 7 at [66] (SBM.010.005.0250_2 at 0263_2); AWS ch 8 at [176] (SBM.010.005.0296_2 at 0337_2)
AWS ch 7 at [65] (SBM.010.005.0250_2 at 0262_2); AWS ch 8 at [172] (SBM.010.005.0296_2 at 0037_2)
AWS ch 7 at [159]–[162] (SBM.010.005.0250_2 at 0335_2–0339_2)
RWS ch 2 [54], [64] (SBM.020.002.0002 at 0013); RWS ch 7 at [35] (SBM.020.002.0108 at 0126)
SBM.020.011.0001 at 0006
RWS ch 2 at [49]–[54] (SBM.020.002.0024 at 0043–0044)
Allman 1 at 11 (EXP.010.100.0001_4 at 0012_4)
Allman 1 at 13 (EXP.010.100.0001_4 at 0014_4)
Holland 1 at 42 (EXP.010.069.0001_3 at 0046_3)
Holland 1 at 43 (EXP.010.069.0001_3 at 0047_3)
Holland 2 at 90 (EXP.010.192.0001_2 at 0091_2)
Holland 2 at 90–91 (EXP.010.192.0001_2 at 0091_2–0092_2)
Holland 2 at 109 (EXP.010.192.0001_2 at 0110_2)
Holland 2 at 89 (EXP.010.192.0001_2 at 0090_2)
Holland 2 at 100 (EXP.010.192.0001_2 at 0101_2)
Holland 1 at 63 (EXP.010.069.0001_3 at 0067_3)
Holland 1 at 63 (EXP.010.069.0001_3 at 0067_3)
Holland 1 at 58 (EXP.010.069.0001_3 at 0062_3)
Allman 2 at 6 (EXP.010.180.0001_3 at 0007_3)
Holland 2 at 10 (EXP.010.192.0001_2 at 0011_2)
Davila W, Drutz H and Deprest J, “Clinical implications of the biology of grafts: conclusions of the 2005 IUGA Grafts Roundtable” (2006) 17 Int Urogynecol J S51–S55 at S54 (ETH.MESH.01594955 at 4958)
ETH.MESH.04038003 at 8021
AWS ch 7 at [1]–[6] (SBM.010.005.0250_2 at 0251_2)
AWS ch 7 at [65] (SBM.010.005.0250_2 at 0262_2)
AWS ch 7 at [178] (SBM.010.005.0250_2 at 0294_2)
AWS ch 7 at [68] (SBM.010.005.0250_2 at 0263_2)
AWS ch 7 at [83] (SBM.010.005.0250_2 at 0267_2–0268_2)
RWS ch 13 at [17], [63] and [78] (SBM.020.002.0338 at 0343, 0355 and 0357)
Allman 1 at 40 (EXP.010.100.0001_4 at 0041_4)
Holland 1 at 45 (EXP.010.069.0001_3 at 0049_3)
ETH.MESH.22139600
Allman 1 at 47 (EXP.010.100.0001_4 at 0048_4)
Ward K and Hilton P, “Minimally invasive synthetic suburethral slings: emerging complications” (2005) 7 TOG 223–232 at 230 (ETH.MESH.04087047 at 7054)
ETH.MESH.03911390
ETH.MESH.03911390
ETH.MESH.22134684
T3615–16 (TRA.500.045.0001_3 at 0076_3–0077_3)
Allman 1 at 45 (EXP.010.100.0001_4 at 0046_4)
Allman 1 at 51 (EXP.010.100.0001_4 at 0052_4)
AWS ch 8 at [173] (SBM.010.005.0296_2 at 0337_2)
AWS ch 8 at [174] (SBM.010.005.0296_2 at 0337_2)
AWS ch 8 at [177] (SBM.010.005.0296_2 at 0338_2)
AWS ch 8 at [179]–[181] (SBM.010.005.0296_2 at 0338_2)
Allman 1 at 52 (EXP.010.100.0001_4 at 0031_4)
ETH.MESH.00569126
Allman 1 at 34 (EXP.010.100.0001_4 at 0035_4)
Gordon 2 at 12 (EXP.010.161.0001_3 at 0014_3)
Gordon 2 at 17 (EXP.010.161.0001_3 at 0019_3)
Hu 1 at 34 (EXP.010.023.0001_3 at 0036_3)
T4249 (TRA.500.053.0001_2)
Roovers J, “Response to article: Structured vaginal reconstructive prolapse surgery” (2010) 123 NTOG 295–298 at 297 (SHI.MESH.00043589_2)
Roovers J, “Collaboration with the mesh industry: who needs who?” (2016) 27 lnt Urogynecol J 1293–1295 (SHI.MESH.00044451 at 4452)
Roovers 1 at 14 (EXP.020.033.0001_4 at 0016_4)
T3949 (TRA.500.049.0001_2 at 0065_2)
AWS ch 12 at [3] (SBM.010.005.0529_2 at 0530_2)
AWS ch 12 at [2] (SBM.010.005.0529_2 at 0530_2)
Beech at 7 (EXP.010.063.0001 at 0008_4)
Allman 2 at 7 (EXP.010.180.0001_3 at 0008_3)
SHI.MESH.00019689
Allman 1 at 14 (EXP.010.100.0001_4 at 0015_4)
Allman 2 at 7 (EXP.010.180.0001_3 at 0008_3)
Holland 1 at 73 (EXP.010.069.0001_3 at 0077_3)
Holland 1 at 69 (EXP.010.069.0001_3 at 0073_3)
Pence at [51] (EXP.010.200.0001_2 at 0019_2)
T1502 (TRA.500.018.0001_2 at 0103_2)
Krulewitch 1 at [4.4] (EXP.010.081.0001_3 at 0065_3)
Hinoul at 106 (LAY.020.001.0001_4 at 0106_4)
Allman 2 at 7 (EXP.010.180.0001_3 at 0008_3)
Allman 2 at 7 (EXP.010.180.0001_3 at 0008_3)
Allman 2 at 7 (EXP.010.180.0001_3 at 0008_3)
JJM.MESH.00581976 at 1989
ETH.MESH.22146153
T1506 (TRA.500.018.0001_2 at 0107_2)
ETH.MESH.06397498
ETH.MESH.09479794 at 9801
JJM.MESH.00581976 at 1989
Allman 1 at 19 (EXP.010.100.0001_4 at 0020_4)
Davila G, Drutz H and Deprest J, “Clinical implications of the biology of grafts: conclusions of the 2005 IUGA Grafts Roundtable” (2006) 17 Int Urogynecol J S51-S55 (ETH.MESH.01594955)
Withagen M et al, “Risk Factors for Exposure, Pain, and Dyspareunia After Tension-Free Vaginal Mesh Procedure” (2011) 118 Obstet Gynecol 629–636 (ETH.MESH.04929975 at 9979
ETH.MESH.01594955
ETH.MESH.00870466
ETH.MESH.03908358
Allman 2 at 8 (EXP.010.180.0001_3 at 0009_3)
Allman 2 at 8 (EXP.010.180.0001_3 at 0009_3)
HMESH_ETH_02064581
JJM.MESH.00582003 at 2007
AWS ch 2 at [156]-[163] (SBM.010.005.0059_2 at 0119_2–0121_2)
T5937 (TRA.500.074.0001 at 0079)
T5957 (TRA.500.075.0001 at 0013)
AID.010.002.0001_8
MSC.020.025.0001
Pence at [175] (EXP.010.200.0001_2 at 0081_2)
PUB.MESH.00002755
Chughtai 2 at 9 (EXP.010.112.0001_3 at 0010_3)
Chughtai 2 at 7 (EXP.010.112.0001_3 at 0008_3)
Margolis 2 at 11 (EXP.010.124.0001_3 at 0012_3).
Allman 2 at 8 (EXP.010.180.0001_3 at 0009_3)
AWS ch 10 at [21] (SBM.010.005.0425_2 at 0431_2)
ETH.MESH.24258019 at 7909
ETH.MESH.24258019
SBM.020.035.0001 at 0003
T3220 (TRA.500.041.0001 at 0039)
SBM.020.035.0001 at 0002
T3220 (TRA.500.041.0001 at 0039)
ETH.MESH.04092868
T3242 (TRA.500.041.0001_3 at 0055_3)
Gill 1 at [6] (LAY.010.001.0001 at 0003)
Gill 1 at [11] (LAY.010.001.0001 at 0004)
Gill 1 at [5] (LAY.010.001.0001 at 0003)
Gill 1 at [31]–[33] (LAY.010.001.0001 at 0007–0008)
Gill 1 at [28] (LAY.010.001.0001 at 0007)
Gill 1 at [37]–[38] (LAY.010.001.0001 at 0008–0009)
Gill 1 at [39] (LAY.010.001.0001 at 0009)
Gill 1 at [17] (LAY.010.001.0001 at 0005)
Gill 1 at [17(g)], [30] (LAY.010.001.0001 at 0006-0007)
Gill 1 at [17(d)] (LAY.010.001.0001 at 0005)
Gill 1 at [17(a)] (LAY.010.001.0001 at 0005)
Gill 1 at [21]–[22] (LAY.010.001.0001 at 0006–0007)
Gill 1 at [17(d)] (LAY.010.001.0001 at 0005)
Gill 1 at [17(e)] (LAY.010.001.0001 at 0005)
Gill 1 at [17(f)] (LAY.010.001.0001 at 0006)
Gill 1 at [17(g)] (LAY.010.001.0001 at 0006)
Gill 1 at [18] (LAY.010.001.0001 at 0006)
Gill 1 at [19] (LAY.010.001.0001 at 0006)
Gill 1 at [10] (LAY.010.001.0001 at 0004)
Gill 1 at [13] (LAY.010.001.0001 at 0004)
Gill 1 at [17(c)] (LAY.010.001.0001 at 0005)
Gill 1 at [14] (LAY.010.001.0001 at 0004)
Gill 1 at [15] (LAY.010.001.0001 at 0005); GIL.MESH.00003387
GIL.MESH.00001121
GIL.MESH.00001121
GIL.MESH.00001121
Gill 1 at [40]–[41] (LAY.010.001.0001 at 0009)
Gill 1 at [43] (LAY.010.001.0001 at 0009)
Gill 1 at [44] (LAY.010.001.0001 at 0009–0010)
Gill 1 at [46] (LAY.010.001.0001 at 0010)
Gill 1 at [47] (LAY.010.001.0001 at 0010)
Gill 1 at [48] (LAY.010.001.0001 at 0010)
Gill 1 at [49] (LAY.010.001.0001 at 0010)
GIL.MESH.00000159
GIL.MESH.00000159
Gill 1 at [49] (LAY.010.001.0001 at 0010); GIL.MESH.00000159
Gill 1 at [50] (LAY.010.001.0001 at 0010–0011); GIL.MESH.00001481
Gill 1 at [51] (LAY.010.001.0001 at 0011); GIL.MESH.00000157
GIL.MESH.00000157
Gill 1 at [51] (LAY.010.001.0001 at 0011)
GIL.MESH.00000119; see also GIL.MESH.00000119_2 at 0121_2
Gill 1 at [52] (LAY.010.001.0001 at 0011)
Gill 1 at [58] (LAY.010.001.0001 at 0012)
Gill 1 at [57] (LAY.010.001.0001 at 0012)
Gill 1 at [59] (LAY.010.001.0001 at 0012)
Gill 1 at [60]-[61] (LAY.010.001.0001 at 0012)
Gill 1 at [62] (LAY.010.001.0001 at 0011)
GIL.MESH.00000156
GIL.MESH.00000144
T1310 (TRA.500.015.0001_2 at 0093_2)
GIL.MESH.00000144
GIL.MESH.00000144
GIL.MESH.00000591; GIL.MESH.00000106 at 0108
GIL.MESH.00000591
Gill 1 at [77] (LAY.010.001.0001 at 0016)
Gill 1 at [77]–[78] (LAY.010.001.0001 at 0016)
Gill 1 at [84]–[85] (LAY.010.001.0001 at 0019)
GIL.MESH.00000117
GIL.MESH.00000117
Gill 1 at [84] (LAY.010.001.0001 at 0019); GIL.MESH.00000389
Gill 1 at [86]-[87] (LAY.010.001.0001 at 0019)
Gill 1 at [88] (LAY.010.001.0001 at 0019)
GIL.MESH.00000106 at 0108
Gill 1 at [89] (LAY.010.001.0001 at 0020)
GIL.MESH.00000110
GIL.MESH.00000110
GIL.MESH.00000567
GIL.MESH.00001577
GIL.MESH.00000440; Gill 1 at [90] (LAY.010.001.0001 at 0011).
Gill 1 at [91] (LAY.010.001.0001 at 0011)
Korda 1 at 95 (EXP.010.078.0001_3 at 0097_3); GIL.MESH.00000106 at 0107
Gill 1 at [91] (LAY.010.001.0001 at 0011); GIL.MESH.00000106; GIL.MESH.00001572
GIL.MESH.00001608
GIL.MESH.00001008 at 1010
Gill 1 at [91]–[92] (LAY.010.001.0001 at 0020)
Gill 1 at [101] (LAY.010.001.0001 at 0022)
GIL.MESH.00000106 at 0107
Gill 1 at [92] (LAY.010.001.0001 at 0020); GIL.MESH.00000106
GIL.MESH.00000106
GIL.MESH.00001580
GIL.MESH.00000993
Gill 1 at [92]–[93] (LAY.010.001.0001 at 0020)
GIL.MESH.00000109
GIL.MESH.00000106 at 0106
GIL.MESH.00000141
Gill 1 at [94] (LAY.010.001.0001 at 0020)
Gill 1 at [95] (LAY.010.001.0001 at 0021)
Gill 1 at [96] (LAY.010.001.0001 at 0021)
Gill 1 at [101] (LAY.010.001.0001 at 0023)
Gill 1 at [97] (LAY.010.001.0001 at 0021)
Gill 1 at [97] (LAY.010.001.0001 at 0021)
GIL.MESH.00000540
Gill 1 at [102] (LAY.010.001.0001 at 0023)
Korda 1 at 96 (EXP.010.078.0001_3 at 0098_3)
GIL.MESH.00000140
Gill 1 at [104]–[105] (LAY.010.001.0001 at 0023)
Gill 1 at [106] (LAY.010.001.0001 at 0023-4); GIL.MESH.00000150 at 0151
GIL.MESH.00000014
GIL.MESH.00000014
Gill 1 [109]–[111] (LAY.010.001.0001 at 0024-5); GIL.MESH.00000200
GIL.MESH.00000788 at 0790
GIL.MESH.00001618
Gill 1 at [114] (LAY.010.001.0001 at 0025)
GIL.MESH.00000378
Gill 2 at [8] (LAY.010.029.0001 at 0003-4)
Gill 2 at [8] (LAY.010.029.0001 at 0003-4)
GIL.MESH.00001614
GIL.MESH.00000788 at 0791
Gill 1 at [120] (LAY.010.001.0001 at 0026); GIL.MESH.00000799
Gill 1 at [121] (LAY.010.001.0001 at 0026); GIL.MESH.00001900 to 1901
Gill 1 at [123] (LAY.010.001.0001 at 0026); GIL.MESH.00001941
Leake 1 at [17] (LAY.010.002.0001 at 0006–0007)
Leake 1 at [17]-[19] (LAY.010.002.0001 at 0007)
GIL.MESH.00000102 at 0103
Gill 1 at [125] (LAY.010.001.0001 at 0027)
Gill 1 at [126]–[127] (LAY.010.001.0001 at 0027)
Leake 1 at [22] (LAY.010.002.0001 at 0007–0008)
Leake 1 at [22] (LAY.010.002.0001 at 0008)
Leake 1 at [22] (LAY.010.002.0001 at 0008)
Gill 1 at [128] (LAY.010.001.0001 at 0027)
Gill 2 at [9] (LAY.010.029.0001 at 0004)
Gill 1 at [129] (LAY.010.001.0001 at 0027); GIL.MESH.00000101; GIL.MESH.00000309
GIL.MESH.00000022
Gill 1 [131]–[132] (LAY.010.001.0001 at 0027); GIL.MESH.00000300
Leake 1 at [55]–[56] (LAY.010.002.0001 at 0015–0016)
GIL.MESH.00000023
Leake 1 at [24] (LAY.010.002.0001 at 0008)
Gill 1 [133]–[134] (LAY.010.001.0001 at 0026–0027)
GIL.MESH.00002884
Leake 1 at [26]-[28] (LAY.010.002.0001 at 0009); GIL.MESH.00000099; GIL.MESH.00000086 at 0088
Gill 1 at [154] (LAY.010.001.0001 at 0031)
GIL.MESH.00000099; GIL.MESH.00000086 at 0088
Oxford Concise Medical Dictionary (8th ed, Oxford University Press, 2010) at 244
Gill 1 at [136] (LAY.010.001.0001 at 0028); Leake at [29] (LAY.010.002.0001 at 0010)
GIL.MESH.00000089
Gill 1 at [137] (LAY.010.001.0001 at 0028)
GIL.MESH.00002884 at 2898
Gill 1 at [138] (LAY.010.001.0001 at 0028)
GIL.MESH.00002953
Gill 1 at [139], [142] (LAY.010.001.0001 at 0028); S Gill at [43]–[44] (LAY.010.004.0001 at 0009)
GIL.MESH.00002957
GIL.MESH.00002884
GIL.MESH.00000062
GIL.MESH.00001881
GIL.MESH.00002884 at 2892–2894
Leake at [31]–[33] (LAY.010.002.0001 at 0010–0011)
Leake at [36] (LAY.010.002.0001 at 0011)
GIL.MESH.00002884 at 2891
GIL.MESH.00002884 at 2890–2891
GIL.MESH.00002884 at 2890
GIL.MESH.00002884 at 2890
GIL.MESH.00000846; GIL.MESH.00000072
GIL.MESH.00000072
GIL.MESH.00002884 at 2887
Gill 1 at [149]–[150] (LAY.010.001.0001 at 0030–0031)
Gill 1 at [151] (LAY.010.001.0001 at 0031); GIL.MESH.00002597
GIL.MESH.00002597 at 2598
GIL.MESH.00002597 at 2599-2600
GIL.MESH.00000648 at 2600-2601
GIL.MESH.00002597 at 2600–2601
GIL.MESH.00002597 at 2601
GIL.MESH.00000676
GIL.MESH.00002597 at 2601; GIL.MESH.00002624
Gill 1 at [153] (LAY.010.001.0001 at 0031); GIL.MESH.00002659
GIL.MESH.00000677
GIL.MESH.00000677
GIL.MESH.00002659
Gill 1 at [155] (LAY.010.001.0001 at 0031)
Gill 1 at [156] (LAY.010.001.0001 at 0031)
GIL.MESH.00002597 at 2602
GIL.MESH.00002627
Gill 1 at [157]–[158] (LAY.010.001.0001 at 0031-2)
GIL.MESH.00000679
GIL.MESH.00000010
GIL.MESH.00002653
Gill 1 at [163] (LAY.010.001.0001 at 0032)
GIL.MESH.00000684; GIL.MESH.00000685; GIL.MESH.00000521
GIL.MESH.00000008
GIL.MESH.00000684
Gill 1 at [174] (LAY.010.001.0001 at 0034); GIL.MESH.00000461
Gill 1 at [175] (LAY.010.001.0001 at 0034)
Gill 1 at [176] (LAY.010.001.0001 at 0034)
Gill 2 at [10] (LAY.010.029.0001 at 0004)
Gill 1 at [177] (LAY.010.001.0001 at 0034); GIL.MESH.00000657; GIL.MESH.00002597 at 2603
GIL.MESH.00002662
GIL.MESH.00000009
GIL.MESH.00000712
Gill 1 at [180] (LAY.010.001.0001 at 0035)
GIL.MESH.00003008
GIL.MESH.00002884
GIL.MESH.00000731
GIL.MESH.00000710
GIL.MESH.00000024
Gill 1 at [182] (LAY.010.001.0001 at 0035)
Gill 1 at [182]–[183] (LAY.010.001.0001 at 0035)
GIL.MESH.00000729
GIL.MESH.00000810
Korda 1 at 119 (EXP.010.078.0001_3 at 0121_3)
Gill 1 at [184] (LAY.010.001.0001 at 0035)
Gill 1 at [230] (LAY.010.001.0001 at 0043-4)
Gill 1 at [185]–[186] (LAY.010.001.0001 at 0036)
Gill 1 at [186] (LAY.010.001.0001 at 0036)
MSC.020.042.0001
Gill 1 at [225] (LAY.010.001.0001 at 0043)
MSC.020.042.0001 at 0002
GIL.MESH.00001304
GIL.MESH.00001306
Jungfer at 1 (EXP.010.004.0001_2 at 0003_2)
Jungfer at 19 (EXP.010.004.0001_2 at 0021_2)
GIL.MESH.00003130
Korda 1 at 120–124 (EXP.010.078.0001_3 at 0122_3–0126_3)
T1333 (TRA.500.015.0001_2 at 0015_2)
MSC.020.042.0001 at 0003
T5180 (TRA.500.064.0001 at 0025)
T5181 (TRA.500.064.0001 at 0026)
Meagher at 10 (EXP.010.013.0001_2 at 0012_2)
Meagher at 10 (EXP.010.013.0001_2 at 0012_2)
Meagher at 10 (EXP.010.013.0001_2 at 0014_2)
Gill 1 at [51] (LAY.010.001.0001 at 0011)
Gill 2 at [11]–[12] (LAY.010.029.0001 at 0004)
Gill 2 at [11]–[12] (LAY.010.029.0001 at 0004)
GIL.MESH.00003134
Gill 2 at [23]–[24] (LAY.010.029.0001 at 0007)
Gill 2 at [25] (LAY.010.029.0001 at 0007); GIL.MESH.00000086
GIL.MESH.00003340
Gill 2 at [25] (LAY.010.029.0001 at 0007–0008)
Gill 2 at [25] (LAY.010.029.0001 at 0007–8)
Gill 2 at [26] (LAY.010.029.0001 at 0007–0008)
GIL.MESH.00003498
GIL.MESH.00003500 at 3505
Gill 1 at [215] (LAY.010.001.0001 at 0041)
Gill 2 at [13] (LAY.010.029.0001 at 0004)
Gill 2 at [14]–[18] (LAY.010.029.0001 at 0004–0005)
T5162 (TRA.500.064.0001 at 0007)
T5163 (TRA.500.064.0001 at 0008)
T5164 (TRA.500.064.0001 at 0009)
T5164 (TRA.500.064.0001 at 0009)
Gill 1 at [216] (LAY.010.001.0001 at 0041)
T5202–03 (TRA.500.064.0001 at 0048)
Gill 1 at [218] (LAY.010.001.0001 at 0042)
Gill 1 at [219] (LAY.010.001.0001 at 0042)
Korda 1 at 124 (EXP.010.078.0001_3 at 0126_3)
DAW.MESH.00001179; DAW.MESH.00001190
Dawson 1 at [8] (LAY.010.007.0001 at 0003-0004)
Dawson 1 at [13] (LAY.010.007.0001 at 0004–0005); DAW.MESH.00001066; DAW.MESH.00001067
Dawson 1 at [13] (LAY.010.007.0001 at 0005)
DAW.MESH.00001064
DAW.MESH.00001058
DAW.MESH.00001056
Dawson 1 at [13] (LAY.010.007.0001 at 0005)
DAW.MESH.00001051
DAW.MESH.00000973 at 0975
DAW.MESH.00001051
DAW.MESH.00001050
Dawson 1 at [13] (LAY.010.007.0001 at 0005)
DAW.MESH.00001044
DAW.MESH.00001044
Dawson 1 at [13] (LAY.010.007.0001 at 0005)
Dawson 1 at [13] (LAY.010.007.0001 at 0004)
Dawson 1 at [13] (LAY.010.007.0001 at 0005)
Dawson 1 at [13] (LAY.010.007.0001 at 0005)
Dawson 1 at [7] (LAY.010.007.0001 at 0004–0005)
Dawson 1 at [9] (LAY.010.007.0001 at 0004)
Dawson 1 at [9] (LAY.010.007.0001 at 0004)
DAW.MESH.00001043
DAW.MESH.00001040
DAW.MESH.00001036
Dawson 1 at [17] (LAY.010.007.0001 at 0007)
DAW.MESH.00001035
DAW.MESH.00001024
Dawson at [18] (LAY.010.007.0001 at 0007)
DAW.MESH.00001034
DAW.MESH.00001032
DAW.MESH.00000564
DAW.MESH.00001030
DAW.MESH.00001024
DAW.MESH.00001023
T1282 (TRA.500.015.0001_2 at 0065_2)
T1283 (TRA.500.015.0001_2 at 0066_2)
Dawson 1 at [10]–[12] (LAY.010.007.0001 at 0004); G Dawson at [10] (LAY.010.008.0001 at 0004)
Dawson 1 at [14] (LAY.010.007.0001 at 0006); G Dawson at [9] (LAY.010.008.0001 at 0003)
Dawson 1 at [20]–[21] (LAY.010.007.0001 at 0007–0008)
T5211 (TRA.500.064.0001 at 0056)
T5211 (TRA.500.064.0001 at 0056)
T5212 (TRA.500.064.0001 at 0057); Dawson 2 at [6] (LAY.010.028.0001 at 0003)
DAW.MESH.00000582 at 0595; DAW.MESH.00000984
DAW.MESH.00001018
DAW.MESH.00001018
DAW.MESH.00001017
DAW.MESH.00000026
Roovers 1 at 21 (EXP.020.033.0001_3 at 0023_3); Rosamilia at [2.2] (EXP.020.031.0001_3 at 0013_3)
Carey M et al, “Vaginal surgery for pelvic organ prolapse using mesh and a vaginal support device”(2008) 115 BJOG 391–397 (ETH.MESH.00154197)
Dawson 1 at [27] (LAY.010.007.0001 at 0007–0008)
Dawson 1 at [26]–[30] (LAY.010.007.0001 at 0011)
Dawson 1 at [31] (LAY.010.007.0001 at 0012)
DAW.MESH.00000014 at 0015; DAW.MESH.00000005
Dawson 1 at [28] (LAY.010.007.0001 at 0011)
Dawson 1 at [33]–[36] (LAY.010.007.0001 at 0012–0003)
Dawson 1 at [34] (LAY.010.007.0001 at 0012)
Dawson 1 at [35] (LAY.010.007.0001 at 0012–0003)
Dawson 1 at [34] (LAY.010.007.0001 at 0012)
Dawson 1 at [36] (LAY.010.007.0001 at 0013)
DAW.MESH.00000400
DAW.MESH.00000774
DAW.MESH.00001012
DAW.MESH.00000788
DAW.MESH.00000582 at 0594
DAW.MESH.00000761
DAW.MESH.00000399
DAW.MESH.00000398
DAW.MESH.00000582 at 0594
DAW.MESH.00000582 at 0594
DAW.MESH.00000759
DAW.MESH.00000757
DAW.MESH.00000757
DAW.MESH.00000755
DAW.MESH.00000752
DAW.MESH.00000752
DAW.MESH.00000654
DAW.MESH.00000651
DAW.MESH.00000381
DAW.MESH.00000129 at 0130
DAW.MESH.00000380
DAW.MESH.00000377
DAW.MESH.00000582 at 0593
DAW.MESH.00000745
DAW.MESH.00000645
DAW.MESH.00000129 at 0130
DAW.MESH.00000738
DAW.MESH.00000645
Australian Medicines Handbook (Australian Medicines Handbook Pty Ltd, 7th ed, 2007) at 640
DAW.MESH.00000582 at 0592
DAW.MESH.00000582 at 0592
DAW.MESH.00000582 at 0591
DAW.MESH.00000129
DAW.MESH.00001724 at 1725
DAW.MESH.00000582 at 0591
DAW.MESH.00000582
DAW.MESH.00000683
DAW.MESH.00000683
DAW.MESH.00000316
DAW.MESH.00001724 at 1725–1726
DAW.MESH.00001724 at 1726
DAW.MESH.00001724
DAW.MESH.00000141
Dawson 1 at [62] (LAY.010.007.0001 at 0018)
DAW.MESH.00000113
DAW.MESH.00000055
DAW.MESH.00000232
DAW.MESH.00000248
DAW.MESH.00000232
DAW.MESH.00000821
DAW.MESH.00000100
DAW.MESH.00000099
DAW.MESH.00000707
DAW.MESH.00001259
DAW.MESH.00000084
DAW.MESH.00000084
DAW.MESH.00000090
DAW.MESH.00000090
DAW.MESH.00000189
DAW.MESH.00000818
DAW.MESH.00000189
Dawson 1 at [72]–[75] (LAY.010.007.0001 at 0019)
DAW.MESH.00000082
DAW.MESH.00000077
Dawson 1 at [79] (LAY.010.007.0001 at 0020)
Dawson 1 at [80] (LAY.010.007.0001 at 0020)
DAW.MESH.00000798
DAW.MESH.00000798
Dawson 1 at [81] (LAY.010.007.0001 at 0020–0021)
Dawson 1 at [82]–[87] (LAY.010.007.0001 at 0021)
DAW.MESH.00000147
DAW.MESH.00000269
DAW.MESH.00000815
Dawson 1 at [82]–[87] (LAY.010.007.0001 at 0021)
DAW.MESH.00000794 at 0796
DAW.MESH.00000800 at 0800
DAW.MESH.00001482
Korda 1 (EXP.010.078.0001_3)
Korda 1 at 140 (EXP.010.078.0001_3 at 0142_3)
T1291–92 (TRA.500.015.0001_2 at 0074_2-0075_2)
Korda 1 at 141-42 (EXP.010.078.0001_3 at 0143_3-0144_3)
Eyers (EXP.010.234.0001_2)
Eyers at 10–11 (EXP.010.234.0001_2 at 0012_2-0013_2)
Korda 4 at 3–4 (EXP.010.261.0001_3 at 0004_3–0005_3)
Korda 4 at 5 (EXP.010.261.0001_3 at 0006_3)
DAW.MESH.00001440 at 1450
DAW.MESH.00001403 at 1438
DAW.MESH.00001403 at 1438
DAW.MESH.00001403 at 1424
DAW.MESH.00001403 at 1436
Dawson 2 at [9]–[10] (LAY.020.028.0001 at 0004)
Korda 4 at 2 (EXP.010.261.0001_3 at 0004_3–0005_3)
DAW.MESH.00001440
DAW.MESH.00001403 at 1438
DAW.MESH.00001488
DAW.MESH.00001484
DAW.MESH.00001484 at 1485
Dawson 1 at [52]–[57] (LAY.010.007.0001 at 0015-16)
Dawson 1 at [54] (LAY.010.007.0001 at 0016)
Jungfer 2 at [7.1] (EXP.010.007.0001_2 at 0014_2)
T5425 (TRA.500.067.0001 at 0030); Jungfer 2 at 12–14 (EXP.010.007.0001_2 at 0014_2–0016_2)
Jungfer 2 at [9.8], [13.0] (EXP.010.007.0001_2 at 0016_2, 0018_2)
Jungfer 2 at 16 (EXP.010.007.0001_2 at 0018_2)
Brown at [19.91] (EXP.020.061.0001 at 0040)
Dawson 2 at [12]–[15] (LAY.010.028.0001 at 0004)
Dawson 2 at [16] (LAY.010.028.0001 at 0004)
Dawson 2 at [19] (LAY.010.028.0001 at 0005)
Dawson 2 at [24] (LAY.010.028.0001 at 0005); T5218 (TRA.500.064.0001 at 0063)
T5218, 5234 (TRA.500.064.0001 at 0063, 0079)
Dawson 3 at [7] (LAY.010.047.0001 at 0003)
Dawson 2 at [24] (LAY.010.028.0001 at 0004)
T5215, 5219 (TRA.500.064.0001 at 0060, 0064)
T5215 (TRA.500.064.0001 at 0060)
Dawson 1 at [95]-[97] (LAY.010.007.0001 at 0026); Dawson 2 at [26] (LAY.010.028.0001 at 0005-06)
G Dawson at [21] (LAY.010.008.0001 at 0006)
T5212, 5224 (TRA.500.064.0001 at 0057, 0069); Dawson 2 at [38]–[42] (LAY.010.028.0001 at 0006-07)
T5225–5226 (TRA.500.064.0001 at 0057, 0069)
T5239 (TRA.500.064.0001 at 0084)
T5239 (TRA.500.064.0001 at 0084)
Dawson 2 at [22] (LAY.010.028.0001 at 0005)
T5215 (TRA.500.064.0001 at 0060)
Dawson 3 at [4]–[6] (LAY.010.047.0001 at 0003)
T5215 (TRA.500.064.0001 at 0060)
T5240 (TRA.500.064.0001 at 0085)
DAW.MESH.00001798
T5210 (TRA.500.064.0001 at 0055)
T5210 (TRA.500.064.0001 at 0055)
T5425 (TRA.500.067.0001 at 0030)
T5419 (TRA.500.067.0001 at 0024)
Sanders 1 at [5], [39] (LAY.010.005.0001 at 0003, 0008)
Sanders 1 at [16], [18] (LAY.010.005.0001 at 0004-05)
SAN.MESH.00000463
Wilcox at [4.1]-[5.1] (EXP.020.067.0001 at 0006)
Sanders 1 at [11], [17] (LAY.010.005.0001 at 0004)
Sanders 1 at [19], [22] (LAY.010.005.0001 at 0004)
Sanders 1 at [29] (LAY.010.005.0001 at 0006)
Sanders 1 [31]–[32] (LAY.010.005.0001 at 0006)
Sanders 1 [33]–[36] (LAY.010.005.0001 at 0006-07)
SAN.MESH.00000541; McNeill at [13] (LAY.010.011.0001 at 0005)
SAN.MESH.00000541; Korda 2 at 4 (EXP.010.103.0001_4 at 0005_4)
SAN.MESH.00000541
McNeill at [17] (LAY.010.011.0001 at 0005)
SAN.MESH.00000519
McNeill at [18] (LAY.010.011.0001 at 0006); SAN.MESH.00000460
Sanders 1 at [39] (LAY.010.005.0001 at 0008)
SAN.MESH.00000195 at 0196
Sanders 1 at [40]-[42] (LAY.010.0005.0001 at 0008-09)
Sanders 1 at [44]-[45] (LAY.010.0005.0001 at 0009)
Sanders 1 at [45] (LAY.010.0005.0001 at 0009)
P Sanders at [16] (LAY.010.006.0001 at 0004)
Sanders 1 at [46]-[48] (LAY.010.005.0001 at 0009)
Sanders 1 [50]–[51] (LAY.010.005.0001 at 0010)
P Sanders at [17] (LAY.010.006.0001 at 0004)
Sanders 1 at [50] (LAY.010.005.0001 at 0010)
Sanders 1 at [52]-[53] (LAY.010.005.0001 at 0010)
Sanders 1 at [52] (LAY.010.005.0001 at 0010)
Sanders 1 at [56] (LAY.010.005.0001 at 0011)
Sanders 1 at [57] (LAY.010.005.0001 at 0011)
P Sanders at [20] (LAY.010.006.0001 at 0004)
P Sanders at [21] (LAY.010.006.0001 at 0005)
Sanders 1 at [59] (LAY.010.005.0001 at 0011–0012)
SAN.MESH.00000098 at 0099
Sanders 1 at (LAY.010.005.0001 at 0012)
Korda 2 at 18 (EXP.010.103.0001_4 at 0019_4)
SAN.MESH.00000102
P Sanders at [22] (LAY.010.006.0001 at 0005)
SAN.MESH.00000464
SAN.MESH.00000464
SAN.MESH.00000464
SAN.MESH.00000457
Sanders 1 at [61] (LAY.010.005.0001 at 0012)
SAN.MESH.00000470
SAN.MESH.00000001 at 0010; RWS ch 15C at [12] (SBM.020.002.0511 at 0515)
SAN.MESH.00000458
Sanders 1 at [61] (LAY.010.005.0001 at 0012)
T5279 (TRA.500.065.0001 at 0034)
Sanders 1 at [61] (LAY.010.005.0001 at 0012)
SAN.MESH.00000001 at 0007
Sanders 1 at [62] (LAY.010.005.0001 at 0013)
SAN.MESH.00000001 at 0009
Korda 2 at 9 (EXP.010.103.0001_4 at 0010_4)
SAN.MESH.00000078 at 0079
Korda 2 at 9 (EXP.010.103.0001_4 at 0010_4)
Korda 2 at 9 (EXP.010.103.0001_4 at 0010_4)
SAN.MESH.00000554
Iakovlev 2 at 172, 176 (EXP.010. 147.0001_4 at 0173_4, 0177_4)
T1754 (TRA.500.022.0001 at 0031)
Sanders 1 at [66] (LAY.010.005.0001 at 0013)
Sanders 1 at [66] (LAY.010.005.0001 at 0013)
SAN.MESH.00000078; SAN.MESH.00000480
Sanders 1 at [68] (LAY.010.005.0001 at 0014)
Sanders 1 at [69] (LAY.010.005.0001 at 0014)
P Sanders at [24] (LAY.010.006.0001 at 0005–0006)
Sanders 1 at [70] (LAY.010.005.0001 at 0014)
Sanders 1 at [71]-[72] (LAY.010.005.0001 at 0014–0015)
Sanders 1 at [72] (LAY.010.005.0001 at 0014–0015)
SAN.MESH.00000456
Sanders 1 at [73] (LAY.010.005.0001 at 0015)
SAN.MESH.00000001 at 0007, 0010
SAN.MESH.00000455
Sanders 1 at [76] (LAY.010.005.0001 at 0015)
Sanders 1 at [79]–[80] (LAY.010.005.0001 at 0015)
SAN.MESH.00000001 at 0006
SAN.MESH.00000001 at 0005–0006
SAN.MESH.00000001 at 0005
Sanders 1 at [83]–[84] (LAY.010.005.0001 at 0015)
SAN.MESH.00000205 at 0206-07
SAN.MESH.00000205 at 0208
T737 (TRA.500.009.0001_2 at 0008_2)
SAN.MESH.00000001 at 0004
SAN.MESH.00000041
SAN.MESH.00000043
SAN.MESH.00000163
SAN.MESH.00000186
SAN.MESH.00000247; SAN.MESH.00000187
SAN.MESH.00000028
SAN.MESH.00000001 at 0003–0004
SAN.MESH.00000001 at 0003
SAN.MESH.00000001 at 0003
SAN.MESH.00000167
SAN.MESH.00000031
SAN.MESH.00000180
Sanders 2 at [14] (LAY.010.027.0001 at 0005)
Jungfer 3 (EXP.010.010.0001_2)
SAN.MESH.00000541
T5258 (TRA.500.065.0001 at 0013)
Jungfer 6 at 3 (EXP.010.226.0001_2 at 0005_2)
Wilcox at 3 (EXP.020.067.0001 at 0003)
Wilcox at [11.1] (EXP.020.067.0001 at 0012)
Wilcox at 12 (EXP.020.067.0001 at 0014)
Wilcox at [12.11] (EXP.020.067.0001 at 0013)
Wilcox at 13 (EXP.020.067.0001 at 0015)
Wilcox at 13 (EXP.020.067.0001 at 0015)
Wilcox at 11 (EXP.020.067.0001 at 0013)
Wilcox at 13 (EXP.020.067.0001 at 0015)
Wilcox at 13 (EXP.020.067.0001 at 0015)
Jungfer 6 at 4 (EXP.010.226.0001_2 at 0006_2)
Jungfer 6 at 9 (EXP.010.226.0001_2 at 0006_2)
RWS ch 15C at [78] (SBM.020.002.0511 at 0536)
Korda 2 at 1 (EXP.010.103.0001_4 at 0002_4)
Korda 2 at 13–14 (EXP.010.103.0001_4 at 0014_4–0015_4)
Korda 2 at 13–14 (EXP.010.103.0001_4 at 0014_4–0015_4)
T1184–85 (TRA.500.014.0001_2 at 0057_2-0058_2)
T1185 (TRA.500.014.0001_2 at 0058_2)
T1185 (TRA.500.014.0001_2 at 0058_2)
Blaivas (EXP.010.026.0001_3 at 0029_3)
Korda 2 at 14 (EXP.010.103.0001_4 at 0015_4)
Korda 2 at 15 (EXP.010.103.0001_4 at 0016_4)
Korda 2 at 15 (EXP.010.103.0001_4 at 0016_4)
T1185 (TRA.500.014.0001_2 at 0058_2)
Korda 2 at 16 (EXP.010.103.0001_4 at 0017_4)
T1185 (TRA.500.014.0001_2 at 0058_2)
Korda 2 at 16 (EXP.010.103.0001_4 at 0017_4)
T1186 (TRA.500.014.0001_2 at 0059_2)
Korda 2 at 17 (EXP.010.103.0001_4 at 0017_4)
SAN.MESH.00000620
Sanders 2 at [17] (LAY.010.027.0001 at 0005)
SAN.MESH.00000762 at 0765
Sanders 2 at [18] (LAY.010.027.0001 at 0005–0006)
SAN.MESH.00000771 at 0775
SAN.MESH.00000603
Sanders 2 at [21] (LAY.010.027.0001 at 0006)
SAN.MESH.00000603
Sanders 2 at [22] (LAY.010.027.0001 at 0006)
T735 (TRA.500.009.0001_2 at 0006_2)
SAN.MESH.00000602
Sanders 2 at [23] (LAY.010.027.0001 at 0006)
SAN.MESH.00000762 at 0764
SAN.MESH.00000762 at 0765
SAN.MESH.00000771
SAN.MESH.00000770; SAN.MESH.00000762 at 0763
SAN.MESH.00000589
SAN.MESH.00000591
Hill (EXP.010.230.0001_3)
SAN.MESH.00000792
SAN.MESH.00000792
Sanders 1 at [101] (LAY.010.005.0001 at 0021)
Sanders 1 at [102] (LAY.010.005.0001 at 0021)
Sanders 2 at [26] (LAY.010.027.0001 at 0007–0008)
Sanders 2 at [46] (LAY.010.027.0001 at 0011)
Sanders 2 at [44] (LAY.010.027.0001 at 0010)
T5259–60 (TRA.500.065.0001 at 0014–0015)
P Sanders at [31] (LAY.010.006.0001 at 0007)
AWS ch 15 at [167] (SBM.010.005.0674_2 at 0731_2)
AWS ch 15 at [309] (SBM.010.005.0674_2 at 0816_2)
AWS ch 2 at [4] (SBM.010.005.0059_2 at 0060_2); T5668–69 (TRA.500.071.0001 at 0052–0053)
RWS ch 2 at [24]–[26] (SBM.020.002.0024 at 0034–0036); T6062 (TRA.500.076.0001 at 0026)
SBM.020.024.0001 at 0003–0004
RWS ch 2 at [72] (SBM.020.002.0024 at 0050)
SBM.020.002.0001 at 0003
SBM.020.002.0001 at 0011; RWS ch 13 at [57] (SBM.020.002.0338 at 0353)
Gill 1 at [51] (LAY.010.001.0001 at 0011)
See: Gill 1 at [51] (LAY.010.001.0001 at [0011); Dawson 1 at [22(g)] (LAY.010.005.0001 at 0007); and Sanders 1 at [36] (LAY.010.007.0001 at 0009)
See: Dawson 1 at [24] (LAY.010.005.0001 at 0010)
SBM.020.001.0001 at 0028
RWS ch 13 at [58] (SBM.020.002.0338 at 0353)
RWS ch 15A at [86] (SBM.020.002.0361 at 0385–0386)
AWS ch 15 at [166] (SBM.010.005.0674_2 at 0731_2)
SBM.020.011.0001
Maher C et al, “Surgical management of pelvic organ prolapse in women (Review)” The Cochrane Collaboration (John Wiley & Sons Ltd, 2007) at 18 (SHI.MESH.00011315 at 1337)
T6989 (TRA.500.087.0001 at 0013)
Gill 1 at [80] at (LAY.010.001.0001 at 0017–0018)
RWS ch 15A at [80] (SBM.020.002.0361 at 0383)
Gill 1 at [82] (LAY.010.001.0001 at 0018–0019)
GIL.MESH.00000157
GIL.MESH.00000106
Gill 1 at [81] (LAY.010.001.0001 at 0018)
Gill 1 at [65]–[66] (LAY.010.001.0001 at 0013)
Gill 1 at [67]–[68] (LAY.010.001.0001 at 0013–0014)
GIL.MESH.00001609
T5194 (TRA.500.064.0001 at 0039)
Gill 1 at [70(a)] (LAY.010.001.0001 at 0014)
Leake at [55] (LAY.010.002.0001 at 0016)
T6290 (TRA.500.078.0001 at 0038)
T6219 (TRA.500.077.0001 at 0072)
T6217 (TRA.500.077.0001 at 0070)
T6290 (TRA.500.078.0001 at 0038)
T6213 (TRA.500.077.0001 at 0066)
T6207–08 (TRA.500.077.0001 at 0060)
T6128 (TRA.500.077.0001 at 0071)
RWS ch 15B at [57] (SBM.020.002.0458 at 0471)
DAW.MESH.00001017
DAW.MESH.00000418
Dawson 1 at [24]–[25] (LAY.010.007.0001 at 0010–0011)
Dawson 1 at [24] (LAY.010.007.0001 at 0010–0011)
RWS ch 15B at [59] (SBM.020.002.0458 at 0472)
Dawson 1 at [22(i)] (LAY.010.007.0001 at 0008–0009)
AWS ch 15 at [305] (SBM.010.005.0674_2 at 0814_2)
Korda 1 at 142 (EXP.010.078.0001_3 at 0144_3)
Korda 4 at 4–5 (EXP.010.261.0001_3 at 0005_3-0006_3)
RWS ch 15C at [47] (SBM.020.002.0511 at 0524)
Nilsson C et al, “Long-term Results of the Tension-Free Vaginal Tape (TVT) Procedure for Surgical Treatment of Female Stress Urinary Incontinence” (2001) (Suppl 2) Int Urogynecol J S5–S8 (ETH.MESH.00756407); T4237 (TRA.500.052.0001_2 at 0077_2)
T4238 (TRA.500.052.0001_2 at 0078_2)
SAN.MESH.00000463
SAN.MESH.00000536
McNeill at [15] (LAY.010.011.0001 at 0005)
Sanders 1 at [37] (LAY.010.005.0001 at 0007)
RWS ch 15C at [43] (SBM.020.002.0511 at 0523)
SAN.MESH.00000468
SAN.MESH.00000463; SAN.MESH.00000194
SAN.MESH.00000203
SAN.MESH.00000543
T7069 (TRA.500.088.0001 at 0007)
JJM.MESH.00043093
T6783 (TRA.500.088.0001 at 0021)
JJM.MESH.00043093
MSC.020.069.0001
Ward K and Hilton P, “Prospective multicentre randomised trial of tension-free vaginal tape and colposuspension as primary treatment for stress incontinence” (2002) 325 BMJ 1–7 (ETH.MESH.13649139); Ward K and Hilton P, “A prospective multicenter randomized trial of tension-free vaginal tape and colposuspension for primary urodynamic stress incontinence: Two-year follow-up” (2004) 194 Am J Obstet Gynecol 324–331 (ETH.MESH.00016582); Ward K and Hilton P, “Tension-free vaginal tape versus colposuspension for primary urodynamic stress incontinence: 5-year follow up” (2008) 115 BJOG 226–233 (ETH.MESH.04045078)
O’Neill at [25]–[26] (LAY.010.011.0001 at 0007)
Sanders 1 at [38] (LAY.010.005.0001 at 0007–0008)
RWS ch 2 at [55] (SBM.020.002.0024 at 0044)
T3702–04; see especially 3703 (TRA.500.046.0001_3 at 0069_2–0071_3)
RWS ch 15A at [115] (SBM.020.002.0361 at 0395)
RWS ch 15A at [108]–[109] (SBM.020.002.0361 at 0394)
RWS ch 15A at [102]–[106] (SBM.020.002.0361 at 0392–0393)
T6423 (TRA.500.079.0001 at 0060)
Gill 1 at [109] (LAY.010.001.0001 at 0024)
GIL.MESH.00001614
Gill 1 at [102] (LAY.010.001.0001 at 0023)
Gill 1 at [88] (LAY.010.001.0001 at 0019)
GIL.MESH.00000106 at 0108
GIL.MESH.00000440
GIL.MESH.00000106
GIL.MESH.00000548
GIL.MESH.00000141
GIL.MESH.00000106; GIL.MESH.00001607
Gill 1 at [118] (LAY.010.001.0001 at 0025)
Gill 1 at [122] (LAY.010.001.0001 at 0026)
AWS ch 15 at [176] (SBM.010.005.0674_2 at 0752_2)
Gill 1 at [78] (LAY.010.001.0001 at 0016)
Gill 1 at [79] (LAY.010.001.0001 at 0017)
Gill 1 at [159]–[160] (LAY.010.001.0001 at 0032)
Gill 1 at [161] (LAY.010.001.0001 at 0032)
T5167 (TRA.500.064.0001 at 0012)
Gill 1 at [106] (LAY.010.001.0001 at 0023–0024)
T5196 (TRA.500.064.0001 at 0012)
GIL.MESH.00000106; GIL.MESH.00001572
Gill 1 at [76] (LAY.010.001.0001 at 0016)
GIL.MESH.00000106 at 0108
T5194 (TRA.500.064.0001 at 0039)
T5194 (TRA.500.064.0001 at 0039)
GIL.MESH.00000200
T5197 (TRA.500.064.0001 at 0042)
Gill 1 at [161] (LAY.010.001.0001 at 0032)
Gill 1 at [161] (LAY.010.001.0001 at 0032–0033)
Gill 1 at [102] (LAY.010.001.0001 at 0023)
Gill 1 at [116] (LAY.010.001.0001 at 0025)
Leake at [35] (LAY.010.002.0001 at 0011)
Gill 1 [70(a)] (LAY.010.001.0001 at 0014)
GIL.MESH.00000690
GIL.MESH.00000150
Gill 1 at [106] (LAY.010.001.0001 at 0023)
RWS ch 15C at [52]–[53] (SBM.020.002.0511 at 0527–0529); SBM.020.010.0001
SBM.020.010.0001 at 0008
Sanders 1 at [66] (LAY.010.005.0001 at 0013)
AWS ch 15 at [416] (SBM.010.005.0674_2 at 0084_2)
Sanders 1 at [44]–[45] (LAY.010.005.0001 at 0009)
SAN.MESH.00000439; SAN.MESH.00000438
Sanders 1 at [29]–[45] (LAY.010.005.0001 at 0006)
Sanders at [43] (LAY.010.005.0001 at 0009)
Blaivas 1 at 26 (EXP.010.026.0001_2 at 0028_2)
Blaivas 1 at 22 (EXP.010.026.0001_2 at 0024_2)
Korda 2 at 18 (EXP.010.103.0001_4 at 0019_4)
RWS ch 15C at [13] (SBM.020.002.0511 at 0515)
SAN.MESH.00000439; SAN.MESH.00000438
Sanders 1 at [59] (LAY.010.005.0001 at 0006)
T5255 (TRA.500.065.0001 at 0010)
SAN.MESH.00000464
SAN.MESH.00000470
AWS ch 15 at [404] (SBM.010.005.0674_2 at 0879_2); RWS ch 15C at [54] (SBM.020.002.0511 at 0529)
AWS ch 15 at [404] (SBM.010.005.0674_2 at 0879_2)
RWS ch 15C [54(c)] (SBM.020.002.0511 at 0529)
Sanders 1 at [46] (LAY.010.005.0001 at 0009)
Sanders 1 at [47] (LAY.010.005.0001 at 0009)
RWS ch 15A at [113] (SBM.020.002.0361 at 0395)
RWS ch 15A at [114] (SBM.020.002.0361 at 0395)
RWS ch 15A at [115] (SBM.020.002.0361 at 0395)
AWS ch 15 at [175] (SBM.010.005.0674_2 at 0751_2)
Legislative Assembly of Western Australia, Hansard, 7 April 2005, 562–566 at 564
T6650 (TRA.500.082.0001 at 0049)
T6649 (TRA.500.082.0001 at 0048)
T7041 (TRA.500.087.0001 at 0065)
RWS ch 15A at [118] (SBM.020.002.0361 at 0396)
T6998 (TRA.500.087.0001 at 0022)
T6997 (TRA.500.087.0001 at 0021)
T6993–94 (TRA.500.087.0001 at 0017–0018)
RWS ch 15C at [58]–[60] (SBM.020.002.0511 at 0531)
RWS ch 15C at [50] (SBM.020.002.0511 at 0526)
SAN.MESH.00000464
T5277 (TRA.500.065.0001 at 0032)
SAN.MESH.00000470
SAN.MESH.00000458; SAN.MESH.00000105
T5255 (TRA.500.065.0001 at 0010)
SAN.MESH.00000001 at 0007
T5275 (TRA.500.065.0001 at 0030)
AWS ch 15 at [400] (SBM.010.005.0674_2 at 0877_2)
AWS ch 15 at [402] (SBM.010.005.0674_2 at 0878_2)
AWS ch 15 at [401] (SBM.010.005.0674_2–0877_2)
RWS ch 15C at [69] (SBM.020.002.0511 at 0534)
RWS ch 15A at [86] (SBM.020.002.0361 at 0385–0386)
Wright (EXP.020.057.0001_3 at 0060_3)
T3907 (TRA.500.049.0001_2 at 0023_2)
T3907 (TRA.500.049.0001_2 at 0023_2)
T3910–11 (TRA.500.049.0001_2 at 0026_2-0027_2)
T3912 (TRA.500.049.0001_2 at 0028_2)
Korda 3 at 32 (EXP.020.129.0001_5 at 0033_5)
Korda 1 at 122 (EXP.010.078.0001_3 at 0124); Korda 3 at 29 (EXP.020.129.0001_5 at 0030_5); Lam at 27 (EXP.020.021.0001_4 at 0029_4)
Korda 1 at 2 (EXP.010.078.0001_3 at 0003_3)
Korda 1 at 16 (EXP.010.078.0001 at 0018_3)
GIL.MESH.00003130
Korda 1 at 120–124 (EXP.010.078.0001_3 at 0122_3–0126_3)
T3768 (TRA.500.047.0001_3 at 0060_3)
GIL.MESH.00001610
GIL.MESH.00000996
RWS ch 15A at [88] (SBM.020.002.0361 at 0386)
GIL.MESH.00000086 at 0088; Leake at [13] (LAY.010.002.0001 at 0005)
T6594 (TRA.500.081.0001 at 0071)
T1310 (TRA.500.015.0001_2 at 0093_2)
RWS ch 15A at [93] (SBM.020.002.0361 at 0388)
Lam at 21 (EXP.020.021.0001_4 at 0023_4)
T3763 (TRA.500.047.0001_3 at 0055_3)
T3760 (TRA.500.047.0001_2 at 0052_3)
Lam at 25–26 (EXP.020.021.0001_4 at 0028_4)
Di Donato N et al, “Prevalence of adenomyosis in women undergoing surgery for endometriosis” (2014) 181 European Journal of Obstetrics & Gynecology and Reproductive Biology 289–293 (PUB.MESH.00009074)
Ibid (PUB.MESH.00009074)
T3764 (TRA.500.047.0001_3 at 0056_3)
Oxford Concise Medical Dictionary (Oxford University Press, 8th ed, 2010) at 244
PUB.MESH.00008972
Di Donato N et al, “Prevalence of adenomyosis in women undergoing surgery for endometriosis” (2014) 181 European Journal of Obstetrics & Gynecology and Reproductive Biology 289–293 (PUB.MESH.00009074)
GIL.MESH.00000104
GILMESH.00000731
GIL.MESH.00003120
Di Donato N et al, “Prevalence of adenomyosis in women undergoing surgery for endometriosis” (2014) 181 European Journal of Obstetrics & Gynecology and Reproductive Biology 289–293 (PUB.MESH.00009074 at 9075)
GIL.MESH.00000378
GIL.MESH.00000089
GIL.MESH.00003120
Korda 3 at 29 (EXP.010.129.0001_5 at 0030_5)
Korda 3 at 31 (EXP.010.129.0001_5 at 0032_5)
Korda 3 at 31 (EXP.010.129.0001_5 at 0032_5)
Korda 3 at 34 (EXP.010.129.0001_5 at 0035_5)
GIL.MESH.00001663
Lam at 29 (EXP.020.021.0001_4 at 0031_4)
Korda 3 at 30 (EXP.010.129.0001_5 at 0030_5)
T3878-80 (TRA.500.048.0001_2 at 0085_2–0087_2)
GIL.MESH.00000728
GIL.MESH.00000810
MSC.020.042.0001
GIL.MESH.00001306
T3769 (TRA.500.047.0001_3 at 0061_3)
Korda 3 at 31 (EXP.010.129.0001_5 at 0032_5)
Korda 3 at 34 (EXP.010.129.0001_5 at 0035_5)
Gill 1 at [86]–[87] (LAY.010.001.0001 at 0019)
Gill 1 at [89] (LAY.010.001.0001 at 0019–0020)
Gill 1 at [99] (LAY.010.001.0001 at 0022)
Gill 1 at [43] (LAY.010.001.0001 at 0009)
Gill 1 at [43] (LAY.010.001.0001 at 0009)
GIL.MESH.00001625
GIL.MESH.00000106 at 0108
T1315–16 (TRA.500.015.0001_2 at 0098_2–0099_2)
Leake at [36] (LAY.010.002.0001 at 0011)
Rosamilia 2 at 7 (EXP.020.031.0001_3 at 0009_3)
GIL.MESH.00002653
GIL.MESH.00000008
ETH.MESH.00869977 at 9983
ETH.MESH.2214615 at 6209, 6184
GIL.MESH.00003340 at 3341
Gill 1 at [21] (LAY.010.001.0001 at 0006)
Gill 1 [25] (LAY.010.001.0001 at 0007)
Gill 1 [26] ((LAY.010.001.0001 at 0007)
Gill 1 at [43], [47] (LAY.010.001.0001 at 0009)
GIL.MESH.00000014
RWS ch 15A at [89]–[90] (SBM.020.002.0361 at 0386-87)
GIL.MESH.00000157
Meagher at 10 (EXP.010.013.0001_2 at 0012_2)
Meagher at 7 (EXP.010.013.0001_2 at 0009_2)
RWS ch 15A at [92] (SBM.020.002.0361 at 0387)
T5164 (TRA.500.064.0001 at 0009)
Meagher at 9 (EXP.010.013.0001_2 at 0011_2)
T1310 (TRA.500.015.0001_2 at 0093_2)
GIL.MESH.000001625
GIL.MESH.000000157
T5376 (TRA.500.066.0001 at 0051)
GIL.MESH.00003130
SHI.MESH.00039511
GIL.MESH.00001668
GIL.MESH.00000684
T3760 (TRA.500.047.0001_3 at 0052_3)
Jungfer 2 at 13 (EXP.010.007.0001_2 at 0015_2)
Jungfer 1 at 19 (EXP.010.007.0001_2 at 0021_2)
Jungfer 1 at 19 (EXP.010.007.0001_2 at 0021_2)
Jungfer 1 at 20 (EXP.010.007.0001_2 at 0021_2)
Jungfer 1 at 17 (EXP.010.007.0001_2 at 0021_2)
Gill 1 at [28] (LAY.010.001.0001 at 0007)
Gill 1 at [33], [35]–[36] (LAY.010.001.0001 at 0007)
S Gill at [12] (LAY.010.004.0001 at 0004)
S Gill at [31] (LAY.010.004.0001 at 0007)
GIL.MESH.00000788 at 0790
GIL.MESH.00000788 at 0791
GIL.MESH.00000019
Gill 1 at [124]-[125] (LAY.010.001.0001 at 0027)
GIL.MESH.00002884 at 2902
Gill 1 at [138] (LAY.010.001.0001 at 0028-29)
Gill 1 at [137] (LAY.010.001.0001 at 0028)
GIL.MESH.00002884 at 2898
GIL.MESH.00002884 at 2898
Gill 1 at [145] (LAY.010.001.0001 at 0030)
GIL.MESH.00000065
GIL.MESH.00002884 at 2895
GIL.MESH.00000062
GIL.MESH.00001881
GIL.MESH.00002884 at 2893
GIL.MESH.00002884 at 2891
GIL.MESH.00002884 at 2890
GIL.MESH.00000068
GIL.MESH.00000072
GIL.MESH.00002597 at 2601
Gill 1 at [163]–[164] (LAY.010.001.0001 at 0032); S Gill at [57] (LAY.010.004.0001 at 0011)
Gill 1 at [164] (LAY.010.001.0001 at 0032)
GIL.MESH.00002597 at 2603
Samuels at [475] (EXP.020.064.0001 at 0051)
Samuels at [491] (EXP.020.064.0001 at 0053)
Samuels at [504]–[506] (EXP.020.064.0001 at 0054)
Samuels at [515] (EXP.020.064.0001 at 0056)
Samuels at [503] (EXP.020.064.0001 at 0054)
T5452 (TRA.500.067.0001 at 0057)
T5442 (TRA.500.067.0001 at 0047)
T5442, 5451 (TRA.500.067.0001 at 0047, 0056)
T5451 (TRA.500.067.0001 at 0056)
T5452 (TRA.500.067.0001 at 0057)
T5452 (TRA.500.067.0001 at 0057)
T5404 (TRA.500.067.0001 at 0009)
GIL.MESH.00002662
Jungfer 5 at 9 (EXP.010.223.0001 at 0011)
S Gill at [70] (LAY.010.004.0001 at 0013)
Jungfer 2 at 6 (EXP.010.223.0001 at 0008)
GIL.MESH.00000160
GIL.MESH.00000569
GIL.MESH.00002884 at 2888
Gill 1 at [36] (LAY.010.001.0001 at 0009)
Jungfer 5 at 8 (EXP.010.223.0001 at 0010)
Jungfer 5 at 8 (EXP.010.223.0001 at 0010)
Samuels at [207] (EXP.020.064.0001 at 0026)
GIL.MESH.00001882; GIL.MESH.00000062
Korda 1 at 123 [6.5] (EXP.010.078.0001_3 at 0125_3)
RWS ch 15A at [90] (SBM.020.002.0361 at 0390-91)
Lam at 27 (EXP.020.021.0001_4 at 0029_4)
Korda 3 at 29 (EXP.010.129.0001_5 at 0030_5)
Gill 1 at [43] (LAY.010.001.0001 at 0009)
GIL.MESH.000001625
Korda 1 at 17 (EXP.010.078.0001_3 at 0019_3)
Korda 1 at 17 (EXP.010.078.0001_3 at 0019_3)
T1309 (TRA.015.0001_2 at 0092)
RWS ch 15A at [94] (SBM.020.002.0361 at 0388)
ETH.MESH.02341522 at 1524
ETH.MESH.01782783 at 2784
GIL.MESH.00000117
LAY.010.002.0001 at 0014-15
Haylen T et al, “An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint terminology and classification of the complications related directly to the insertion of prostheses (meshes, implants, tapes) & grafts in female pelvic floor surgery” (2011) 22 Int Urogynecol J 3–15 (ETH.MESH.00187674)
Rosamilia 1 at 20–21 (EXP.020.026.0001_4 at 0022_4-0023_4)
Dwyer P and O’Reilly B, “Transvaginal repair of anterior and posterior compartment prolapse with Atrium polypropylene mesh” (2004) 111 BJOG 831–836 (ETH.MESH.06139479 at 9479)
Achtari C et al, “Risk factors for mesh erosion after transvaginal surgery using polypropylene (Atrium) or composite polypropylene/polyglactin 910 (Vypro II) mesh” (2005) 16 Int Urogynecol J 389–394 (ETH.MESH.03394815)
T4264–65 (TRA.500.053.0001_2 at 0022_2–0023_2)
RWS ch 15A [151(b)] (SBM.020.002.0361 at 0406)
AWS ch 15 at [206] (SBM.010.005.0674_2 at 0764_2)
AWS ch 15 at [209], [211] (SBM.010.005.0674_2 at 0766_2)
RWS ch 15A at [151] (SBM.020.002.0361 at 0406)
Gill 1 at 4 [15] (LAY.010.001.0001 at 0005); GIL.MESH.00003175
Gill 1 at 21, 23 [101], [108] (LAY.010.001.0001 at 0022-23, 0024)
Gill 1 at [108] (LAY.010.001.0001 at 0024)
Gill 1 at [117] (LAY.010.001.0001 at 0025); Gill 2 at [8] (LAY.010.029.0001 at 0004)
Gill 2 at [9] (LAY.010.029.0001 at 0004); Gill 1 at [137] (LAY.010.001.0001 at 0028)
Gill 1 at [139]-[142] (LAY.010.001.0001 at 0028); S Gill at [43]-[47] (LAY.010.004.0001 at 0009)
Gill 1 at [149]-[150] (LAY.010.001.0001 at 0030-31)
Gill 2 at [10] (LAY.010.029.0001 at 0004)
Gill 1 at [182] (LAY.010.001.0001 at 0035)
GIL.MESH.00003401 at 3421–3422
Gill 1 at [180], [182] (LAY.010.001.0001 at 0035)
Gill 1 at [230] (LAY.010.000.0001 at 0043-44)
Gill 1 at 34 [183]–[184] (LAY.010.000.0001 at 0043-44) ; Gill 2 at 3 [13] (LAY.010.029.0001 at 0004); T5162 (TRA.500.064.0001 at 0007)
SBM.010.005.0674_2 at 0782_2
LAW.700.082.0001 at 0050
GIL.MESH.00003218; GIL.MESH.00003215
GIL.MESH.00003224; GIL.MESH.00003221
GIL.MESH.00003226
GIL.MESH.00003230
Gill 1 at [183] (LAY.010.001.0001 at 0035)
Gill 1 at [226] (LAY.010.001.0001 at 0035)
Jungfer 1 at 20 (EXP.010.004.0001_2 at 0022_2)
Samuels at 53 (EXP.020.064.0001 at 0055)
Samuels at 54 (EXP.020.064.0001 at 0055)
EXP.010.019.0001
T5339 (TRA.500.066.0001 at 0014)
Slesenger 1 (EXP.010.020.0001_3)
T5342–43 (TRA.500.066.0001 at 0017)
Gill 2 at [13] (LAY.010.029.0001 at 0004); T5162 (TRA.500.064.0001 at 0007)
T5344 (TRA.500.066.0001 at 0019)
T5344-45 (TRA.500.066.0001 at 0019–0020)
T5346 (TRA.500.066.0001 at 0021)
Slesenger 2 at 2 (EXP.010.323.0001 at 0004)
Gill 2 at [16] (LAY.010.029.0001 at 0005)
AWS ch 15 at [243] (SBM.010.005.0001_2 at 0780_2)
T5163 (TRA.500.064.0001 at 0008)
AWS ch 15 at [251] (SBM.010.005.0674_2 at 0784_2)
Email from Clayton Utz dated 14 February 2019
Email from Clayton Utz dated 14 February 2019
AWS ch 15 at [250] (SBM.010.005.0674_2 at 0784_2)
AWS ch 15A at [252]–[253]; [258] (SBM.010.005.0674_2 at 0786_2)
Gill 2 at [29] (LAY.010.029.0001 at 0009)
Gill 2 at [30]–[33] (LAY.010.029.0001 at 0009)
T5169 (TRA.500.061.0001 at 0014)
AWS ch 15 at [238] (SBM.010.005.0001_2 at 0775_2); Williams (Gill 2) at 2 (EXP.010.326.0001 at 0004)
Williams (Gill 1) at 21 (EXP.010.244.0001_2 at 0024_2)
Williams (Gill 1) at 22 (EXP.010.244.0001_2 at 0025_2)
Williams (Gill 1) at 22 (EXP.010.244.0001_2 at 0025_2)
Borthwick (Gill) at 38 (EXP.020.108.0001_2 at 0040_2)
Borthwick (Gill) at 32 (EXP.020.108.0001_2 at 0034_2)
Borthwick (Gill) at [7.4.14] (EXP.020.108.0001_2 at 0037_2)
Borthwick (Gill) at [7.4.18] (EXP.020.108.0001_2 at 0037_2)
T7157 (TRA.500.089.0001 at 0018)
RWS ch 15A at 66 (SBM.020.002.0361 at 0426)
RWS ch 15A at [235] (SBM.020.002.0361 at 0435)
RWS ch 15A at [235] (SBM.020.002.0361 at 0435)
Borthwick (Gill) at [5.1.2] (EXP.010.108.0001_2 at 0019_2)
Borthwick (Gill) at [5.1.2] (EXP.010.108.0001_2 at 0019_2)
Williams (Gill 2) at [4] (EXP.010.326.0001 at 0003)
Williams (Gill 2) at [10] (EXP.010.326.0001 at 0003)
T5502 (TRA.500.068.0001 at 0022)
T5493, 5495 (TRA.500.068.0001 at 0012, 0014)
T5495-96 (TRA.500.068.0001 at 0014-15)
T5500-01 (TRA.500.068.0001 at 0019-20)
T5172 (TRA.500.064.0001 at 0017)
T5510–11 (TRA.500.068.001 at 0029–0030)
Borthwick (Gill) at 20 (EXP.020.108.0001_2 at 0022_2)
Borthwick (Gill) at [7.1.35] (EXP.020.108.0001_2 at 0032_2)
T5173 (TRA.500.064.0001 at 0017)
T5508 (TRA.500.068.001 at 0029-30)
Borthwick (Gill) at [7.1.36] (EXP.020.108.0001_2 at 0032_2); T5173 (TRA.500.064.0001 at 0018)
T5512–13 (TRA.500.068.0001 at 0031-32)
T5172 (TRA.500.064.0001 at 0017)
T5512 (TRA.500.068.001 at 0031)
RWS ch 15A at [239] (SBM.020.002.0361 at 0437)
RWS ch 15A at [205] (SBM.020.002.0361 at 0423)
Government Gazette Western Australia (5 July 2019) at 2664
PAR.010.003.0001_2 at 0014_2
RWS ch 15A at [237] (SBM.020.002.0361 at 0436)
Borthwick at 18 (EXP.020.109.0001_2 at 0020_2)
Gill 1 at [245] (LAY.010.001.0001 at 0046)
T5172 (TRA.500.064.0001 at 0017)
Gill 1 at [227] (LAY.010.001.0001 at 0043)
Gill 2 at [29]–[30] (LAY.010.029.0001 at 0009)
Gill 2 at [29] (LAY.010.029.0001 at 0009)
Gill 2 at [29] (LAY.010.029.0001 at 0009)
Williams 1 (EXP.010.244.0001_2 at 0024_2)
Williams (Gill) (EXP.010.248.0001 at 0011)
AWS ch 15 at [260] (SBM.010.005.0674_2 at 0786_2)
AID.010.022.0001; T7141–44 (TRA.500.089.0001 at 0002–0005)
AWS ch 15 at 127–128 (SBM.010.005.0674_2 at 0802_2–0803_2)
GIL.MESH.00003384; GIL.MESH.00003232; MSC.010.209.0001; T7141–2 (TRA.500.089.0001 at 0002–0003)
MSC.010.209.0001 at 0004; T7141–42 (TRA.500.089.0001 at 0002-03)
AWS ch 15 at 127 (SBM.010.005.0674_2 at 0802_2–03_2)
T7143–44 (TRA.500.089.0001 at 0004–05)
MSC.010.209.0001 at 0004
RWS ch 15A at [259]–[264] (SBM.020.002.0361 at 0445–0446)
GIL.MESH.00003498
RWS ch 15A at [265] (SBM.020.002.0361 at 0445–0446)
Gill 1 at [85], [159], [172] (LAY.010.001.0001 at 0019, 0032, 0034)
GIL.MESH.00001306
MSC.020.042.0001
MSC.020.042.0001 at 0002
MSC.020.042.0001 at 0003
GIL.MESH.00003131
GIL.MESH.00003478
GIL.MESH.00003498
GIL.MESH.00003384 at 3385
RWS ch 15A at [267] (SBM.020.002.0361 at 0447)
RWS ch 15A at [268] (SBM.020.002.0361 at 0447)
GIL.MESH.00003033 at 3046
RWS ch 15A at [271]–[272] (SBM.020.002.0361 at 0448)
Jungfer 1 at [9.3] (EXP.010.004.0001_2 at 0020_2)
Samuels at [507] (EXP.020.064.0001 at 0054)
Samuels at [507] (EXP.020.064.0001 at 0054)
Samuels at [508] (EXP.020.064.0001 at 0054)
Samuels at [512] (EXP.020.064.0001 at 0054)
Jungfer 1 at [9.2] (EXP.010.004.0001_2 at 0020_2)
Jungfer 1 at [9.3]-[9.4] (EXP.010.004.0001_2 at 0020_2)
Jungfer 1 at [9.1] (EXP.010.004.0001_2 at 0019_2)
Gill 2 at [20] (LAY.010.029.0001 at 0007)
Samuels at [514] (EXP.020.064.0001 at 0056)
Gill 2 at [20]–[21] (LAY.010.029.0001 at 0007)
Samuels at [509] (EXP.020.064.0001 at 0056)
Korda 1 at 143 [7.10] (EXP.010.078.0001_3 at 0145_3)
SHI.MESH.00011478
Williams 1 (Gill) at 5 (EXP.010.244.0001_2 at 0008_2)
T5171 (TRA.500.064.0001 at 0016)
T5171–72 (TRA.500.064.0001 at 0016–0017)
RWS ch 15A [280] (SBM.020.002.0361 at 0449)
GIL.MESH.00002884 at 2900
Gill 1 at [249]–[250] (LAY.010.001.0001 at 0047)
GIL.MESH.00003439
GIL.MESH.00000766 at 0767
GIL.MESH.00003439
Gill 1 at [39] (LAY.010.001.0001 at 0009)
Williams 1 at 20 (EXP.010.244.0001_2 at 0023_2)
Borthwick (Gill) at 22, 38 (EXP.020.108.0001_2 at 0024_2, 0040_2)
Williams 2 at [27] (EXP.010.326.0001 at 0005-06)
Gill 1 at [246] (LAY.010.001.0001 at 0047)
GIL.MESH.00003131; GIL.MESH.00003340 at 3341
RWS ch 15A at [286(a)] (SBM.020.002.0361 at 0451)
RWS ch 15A at 92 (SBM.020.002.0361 at 0451)
GIL.MESH.00000106
RWS ch 15A at 92 (SBM.020.002.0361 at 0451)
T5170 (TRA.500.064.0001 at 0015)
Gill 2 at [22] (LAY.010.029.0001 at 0007)
Gill 2 at [27] (LAY.010.029.0001 at 0008); GIL.MESH.00000028 at 0043
Gill 2 at [27] (LAY.010.029.0001 at 0008)
Gill 2 at [28] (LAY.010.029.0001 at 0008)
MSC.010.181.0001
T5170 (TRA.500.064.0001 at 0015)
DAW.MESH.00001784
Gill 1 at 45 (LAY.010.001.0001 at 0046)
RWS ch 15A at 92 (SBM.020.002.0361 at 0452)
Borthwick (Gill) at 21 (EXP.020.108.0001 at 0022)
Williams 1 (Annexure B) at 6 (EXP.010.248.0001 at 0006)
RWS ch 15A at [289(a)] (SBM.020.002.0361 at 0454)
Williams 1 (Annexure B) at 10 (EXP.010.248.0001 at 0010)
Williams 1 at 14 (EXP.010.244.0001_2 at 0017_2); Williams 1 (Annexure B) at 12 (EXP.010.248.0001 at 0012)
Williams 3 at [17] (EXP.010.326.0001 at 0005)
Williams 1 (Annexure B) at 13 (EXP.010.248.0001 at 0013)
Williams 1 at 21 (EXP.010.244.0001_2 at 0024_2)
Williams 1 (Annexure B) at 12 (EXP.010.248.0001 at 0012)
Williams 1 (EXP.010.244.0001_2 at 0012_2)
Borthwick (Gill) at [7.4.14] (EXP.020.108.0001_2 at 0037_2)
Borthwick (Gill) at [6.1.2] (EXP.020.108.0001_2 at 0023_2)
Borthwick (Gill) at [7.4.17] (EXP.020.108.0001_2 at 0038_2)
Borthwick (Gill at [6.1.1) (EXP.020.108.0001_2 at.0023_2)
Williams 1 at 13 (EXP.010.244.0001_2 at 0016_2)
Williams 1 at 13 (EXP.010.244.0001_2 at 0016_2)
Williams 1 (Annexure B) at 13 (EXP.010.248.0001 at 0014)
Gill 1 at [191] (LAY.010.001.0001 at 0037)
Borthwick (Gill) at [6.1.1) (EXP.020.108.0001 at 0022)
Williams 1 (Annexure B) at 13 (EXP.010.248.0001 at 0014)
Borthwick (Gill) [6.1.1] (EXP.020.108.0001 at 0022)
Borthwick (Gill) [6.1.1] (EXP.020.108.0001 at 0022)
PAR.010.004.0001
RWS ch 15B at [63] (SBM.020.002.0458 at 0473)
RWS ch 15B at [64]–[65] (SBM.020.002.0458 at 0473)
RWS ch 15B at [67] (SBM.020.002.0458 at 0474)
RWS ch 15B at [66] (SBM.020.002.0458 at 0474)
DAW.MESH.00000377
DAW.MESH.00000815
Iakovlev 2 at 175 (EXP.010.147.0001_4 at 0176_4)
Klosterhalfen 1 at [258] (LAY.010.014.0001_6 at 0054_6)
Karram M and Maher F, Surgical Management of Pelvic Organ Prolapse (Elsevier, 2013) at 20
Gyang A et al, “Managing chronic pelvic pain following reconstructive pelvic surgery with transvaginal mesh” (2014) 25 Int Urogynecol J 313–318 (SHI.MESH.00020223)
Haylen B et al, “An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction” (2010) Int Urogynecol J 215–226 (PUB.MESH.00009152)
Gyang A et al, “Managing chronic pelvic pain following reconstructive pelvic surgery with transvaginal mesh” (2014) 25 Int Urogynecol J 313–318 (SHI.MESH.00020223); Haylen B et al, “An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction” (2010) Int Urogynecol J 215–226 (PUB.MESH.00009152)
Gyang et al, “Managing chronic pelvic pain following reconstructive pelvic surgery with transvaginal mesh” (2014) 25 Int Urogynecol J 313–318 (SHI.MESH.00020223)
Wolff G et al, “Mesh Excision: Is Total Mesh Excision Necessary” (2018) 17:34 Curr Urol Rep 1–7 (SHI.MESH.00024444)
Haylen B et al, “An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint report on the terminology for female pelvic floor dysfunction” (2010) Int Urogynecol J 215–226 (PUB.MESH.00009152)
Dawson 1 at [88(a)] (LAY.010.004.0001 at 0021-22); Dawson 2 at [10], [13], [14] (LAY.010.028.0001 at 0004)
Dawson 1 at [34], [94], [96], [97] (LAY.010.007.0001 at 0012)
Dawson 1 at [88(c)] (LAY.010.007.0001 at 0022)
Dawson 1 at [88(c)] (LAY.010.007.0001 at 0022)
Korda 3 at 34 (EXP.010.129.0001_5 at 0035_5)
RWS ch 15B at [65] (SBM.020.002.0458 at 0473)
DAW.MESH.00000316
DAW.MESH.00001482
Korda 1 at [7.5] (EXP.010.078.0001_3 at 0144_3)
RWS ch 15B at [65] (SBM.020.002.0458 at 0473)
T1292 (TRA.500.015.0001_2 at 0075_2)
RWS ch 15B at [67] (SBM.020.002.0458 at 0474)
T1295 (TRA.500.015.0001_2 at 0078_2)
DAW.MESH.00001042
DAW.MESH.00001043
DAW.MESH.00001025
DAW.MESH.00000582 at 0597, 0600
DAW.MESH.00000582
DAW.MESH.00000984
Jungfer 4 at 6 (EXP.010.220.0001 at 0008)
RWS ch 15B at [66] (SBM.020.002.0458 at 0474)
Jungfer 2 at [9.9] (EXP.010.004.0001_2 at 0016_2)
Jungfer 2 at [9.9] (EXP.010.004.0001_2 at 0016_2)
T5425 (TRA.500.067.0001 at 0030)
Jungfer 2 at [9.3] (EXP.010.007.0001_2 at 0015_2)
Jungfer 2 at [9.8] (EXP.010.007.0001_2 at 0016_2)
Brown at [19.90]–[19.91] (EXP.020.061.0001_2 at 0039–0040_2)
Brown at [19.91]–[19.92] (EXP.020.061.0001_2 at 0040_2)
Brown at [19.91]–[19.92] (EXP.020.061.0001_2 at 0040_2)
T5475 (TRA.500.067.0001 at 0080)
Jungfer 2 at 7 (EXP.010.220.0001 at 0009)
DAW.MESH.00000751
Dawson 1 at [46] (LAY.010.007.0001 at 0014)
DAW.MESH.00000651
DAW.MESH.00000129
DAW.MESH.00000582
DAW.MESH.00000582 at 0589
DAW.MESH.00000938
DAW.MESH.00001300
DAW.MESH.00000582 at 0588
Brown at [19.90] (EXP.020.061.0001_2 at 0039_2)
T5419 (TRA.500.067.0001 at 0024)
T5420 (TRA.500.067.0001 at 0025)
DAW.MESH.00000890
Brown at [19.91] (EXP.020.061.0001_2 at 0040_2)
T5211 (TRA.500.064.0001 at 0056)
RWS ch 15B at [68] (SBM.020.002.0458 at 0475)
DAW.MESH.00000582 at 0595
DAW.MESH.00000582 at 0595
DAW.MESH.00000961
DAW.MESH.00000400
DAW.MESH.00000774
DAW.MESH.00001012
DAW.MESH.00000582 at 0618
DAW.MESH.00000759
DAW.MESH.00000757
DAW.MESH.00000381
DAW.MESH.00000129
Korda 1 at 133 (EXP.010.078.0001_3 at 0135_3)
T1287 (TRA.500.015.0001_2 at 0070_2)
Korda 1 at 126 (EXP.010.078.0001_ at 0128_3)
T1287–88 (TRA.500.015.0001_2 at 0070_2)
Rosamilia 2 at 9 (EXP.020.031.0001_3 at 0011_3)
Roovers 1 at 19 (EXP.020.033.0001_4 at 0021_4); DAW.MESH.00000741
Eyers at 9 (EXP.010.234.0001_2 at 0011_2)
RWS ch 15B at 18 (SBM.020.002.0458 at 0475)
G Dawson at [18] (LAY.010.008.0001 at 0005)
Dawson 1 at [56] (LAY.010.007.0001 at 0016)
Dawson 1 at [67] (LAY.010.007.0001 at 0018)
G Dawson at [21(j)] (LAY.010.008.0001 at 0005)
G Dawson at [27] (LAY.010.008.0001 at 0005)
DAW.MESH.00000316
DAW.MESH.00000116
DAW.MESH.00000798
Eyers at 8 (EXP.010.234.0001_2 at 0011_2)
Korda 1 at 141 [7.3] (EXP.010.078.0001_3 at 0143_3)
RWS ch 15B at [68(c)] (SBM.020.002.0458 at 0475)
Dawson at [13(d)] (LAY.010.007.0001 at 0005)
Eyers at 8 (EXP.010.234.0001_2 at 0010_2)
Dawson 1 at [13(j)] (LAY.010.007.0001 at 0006)
Dawson 1 at [19] (LAY.010.007.0001 at 0007)
DAW.MESH.00000850
DAW.MESH.00000967
DAW.MESH.00000990
Dawson 1 at [20(a)] (LAY.010.007.0001 at 0007)
DAW.MESH.00000014 at 0015
Dawson 1 at [33(f)] (LAY.010.007.0001 at 0012)
DAW.MESH.00000798
Eyers at 8 (EXP.010.234.0001_2 at 0010_2)
Eyers at 10–11 (EXP.010.234.0001_2 at 0012_2-0013_2)
Korda 1 at 142 (EXP.010.078.0001_3 at 0144_2)
Korda 4 at 3 (EXP.010.261.0001_3 at 0005_3)
AWS ch 15 at 142–3 (SBM.010.005.0674_2 at 0816_2-0817_2)
RWS ch 15B at [68] (SBM.020.002.0458 at 0475)
RWS ch 15B at [69] (SBM.020.002.0458 at 0475)
RWS ch 15B at 19 (SBM.020.002.0458 at 0476)
DAW.MESH.00001066
DAW.MESH.00001063
DAW.MESH.00001059
DAW.MESH.00001058
DAW.MESH.00001051
DAW.MESH.00000973 at 0975
Dawson 1 at [13(f)] (LAY.010.007.0001 at 0005)
DAW.MESH.00001030; DAW.MESH.00000863
T1328 (TRA.500.016.0001_2 at 0010_2)
DAW.MESH.00000863; DAW.MESH.00000518
T1328 (TRA.500.016.0001_2 at 0010_2)
DAW.MESH.00000869
DAW.MESH.00000582 at 0597
DAW.MESH.00001018
T1327–28 (TRA.500.016.0001_2 at 0009_2)
T1329–30 (TRA.500.016.0001_2 at 0011_2-0012_2)
T1295–96 (TRA.500.015.0001_2 at 0078_2-0079_2)
DAW.MESH.00000113
DAW.MESH.00001259
DAW.MESH.00000082
DAW.MESH.00000798
Korda 1 at 140 (EXP.010.078.0001_3 at 0143_3)
Eyers at 9 (EXP.010.234.0001_2 at 0011_2)
T1294 (TRA.500.015.0001_2 at 0077_2)
T5471 (TRA.500.067.0001 at 0076)
Jungfer 2 at [7.1] (EXP.010.007.0001_2 at 0014_2)
Brown at [15.2] (EXP.020.061.0001_2 at 0014_2)
T5211 (TRA.500.064.0001 at 0056); Dawson 1 at [10] (LAY.010.007.0001 at 0004)
T5205, T5212 (TRA.500.064.0001 at 0050, 0057)
Dawson 1 at [12] (LAY.010.007.0001 at 0004)
T5211 (TRA.500.064.0001 at 0056)
Government Gazette of Victoria (27 June 2019) at 1249
AWS ch 15 at [318], [320] (SBM.010.005.0674_2 at 0827_2-0828_2)
RWS ch 15B at [76] (SBM.020.002.0458 at 0477)
Korda 3 at 38 (EXP.010.129.0001_5 at 0039_5)
Dawson 2 at [24] (LAY.010.028.0001 at 0005)
Dawson 2 at [25(c)] (LAY.010.028.0001 at 0005); AWS ch 15 at [347] (SBM.010.005.0674_2 at 0834_2)
RWS ch 15B at [97] (SBM.020.002.0458 at 0481)
RWS ch 15B at [93] (SBM.020.002.0458 at 0480)
RWS ch 15B at [93] (SBM.020.002.0458 at 0480)
Dawson 1 at [94] (LAY.010.007.0001 at 0025-26)
T5215 (TRA.500.064.0001 at 0060)
Slesenger 1 at 15 (EXP.010.017.0001_3 at 0017_3)
Dawson 3 at [7] (LAY.010.047.0001 at 0003)
Email from Clayton Utz dated 21 February 2019
AWS ch 15 at [322] (SBM.010.005.0674_2 at 0828_2)
AWS ch 15 at [323] (SBM.010.005.0674_2 at 0828_2)
Email from Clayton Utz dated 21 February 2019
Dawson 1 at [94] (LAY.010.007.0001 at 0025)
RWS ch 15B at [97] (SBM.020.002.0458 at 0481-82)
Dawson 1 at [94] (LAY.010.007.0001 at 0025)
T5233–34 (TRA.500.064.0001 at 0078–0079)
T5235 (TRA.500.064.0001 at 0080)
RWS ch 15B at [95] (SBM.020.002.0458 at 0481–0482)
DAW.MESH.00000999
Dawson 2 at [32]–[37] (LAY.010.028.0001 at 0006)
Dawson 2 at [38] (LAY.010.028.0001 at 0006)
Dawson 2 at [39]–[40] (LAY.010.028.0001 at 0007)
T5217 (TRA.500.064.0001 at 0062)
T5226–28 (TRA.500.064.0001 at 0071–0073)
T5228 (TRA.500.064.0001 at 0073)
T5228 (TRA.500.064.0001 at 0073)
Dawson 2 at [39]–[40] (LAY.010.028.0001 at 0007)
T5236 (TRA.500.064.0001 at 0081)
Walsh 1 at 4 (EXP.010.239.0001 at 0006)
Walsh 1 at 6 (EXP.010.239.0001 at 0006)
AWS ch 15 at 166 (SBM.010.005.0674_2 at 0840_2)
Walsh 1 at 6 (EXP.010.239.0001 at 0006)
Walsh 1 at 7-8 (EXP.010.239.0001 at 0009-10)
Borthwick (Dawson) at [7.4.6] (SBM.010.005.0674_2 at 0833_2)
Walsh 1 at 8 (EXP.010.239.0001 at 0010)
Walsh 2 at 3 (EXP.010.329.0001 at 0005)
RWS ch 15B at 32 (SBM.020.002.0458 at 0489)
Borthwick (Dawson) at [2.5.2], [5.1.3] (EXP.020.104.0001 at 0011, 0016)
Borthwick (Dawson) at [1.2.4] (EXP.020.104.0001 at 0005)
T5515 (TRA.500.068.0001 at 0034)
T5236 (TRA.500.064.0001 at 0081)
AWS ch 15 at [343] (SBM.010.005.0674_2 at 0832_2–0833_2)
AWS ch 15 at 174 (SBM.010.005.0674_2 at 0848_2)
Korda 1 at 143 (EXP.010.078.0001_3 at 0145_3)
Korda 1 at 141–143 (EXP.010.078.0001_3 at 0143_3–0144_3)
RWS [143(e)] (SBM.020.002.0458 at 0498)
MSC.010.190.0001; MSC.010.181.0001; DAW.MESH.00001798
Jungfer 1 at [10.1] (EXP.010.007.0001_2 at 0016_2)
RWS ch 15B at [143(b)] (SBM.020.002.0458 at 0497)
Jungfer 1 at [10.1] (EXP.010.007.0001_2 at 0016_2)
Korda 1 at 143 at [7.10] (EXP.010.078.0001_3 at 0145_3)
RWS ch 15B at [144] (SBM.020.002.0458 at 0498-99)
RWS ch 15B at [146] (SBM.020.002.0458 at 0500)
Dawson 1 at [98(a)] (LAY.010.007.0001 at 0027)
MSC.010.185.0001; MSC.010.186.0001
RWS ch 15B at 48 (SBM.020.002.0458 at 0505)
DAW.MESH.00001798 at 1802
DAW.MESH.00001798 at 1801
DAW.MESH.00001798 at 1799–1800
T5209 (TRA.500.064.0001 at 0054)
RWS ch 15B at 48–49 (SBM.020.002.0458 at 0505)
DAW.MESH.00001440 at 1444
DAW.MESH.00001798 at 1801
Dawson 1 at [98(d)] (LAY.010.007.0001 at 0027)
Dawson 1 at [77] (LAY.010.007.0001 at 0020)
T5219 (TRA.500.064.0001 at 0064)
T5214 (TRA.500.064.0001 at 0059)
DAW.MESH.00001440 at 1441
DAW.MESH.00001440 at 1446
DAW.MESH.00001798
RWS ch 15B at 47 (SBM.020.002.0458 at 0504)
Dawson 1 [98(b)] (LAY.010.007.0001 at 0027); Dawson 2 [28] (LAY.010.007.0001 at 0027); MSC.010.196.0001
RWS ch 15B at 47-8 (SBM.020.002.0458 at 0504–0505)
MSC.010.202.0001
DAW.MESH.00000014
T5210 (TRA.500.064.0001 at 0055)
T5210 (TRA.500.064.0001 at 0055)
Korda 1 at [7.10] (EXP.010.078.0001_3 at 0145_3)
T5210 (TRA.500.064.0001 at 0055)
RWS ch 15B at 50 (SBM.020.002.0458 at 0507)
Borthwick (Dawson) at [6.1.1] (EXP.020.104.0001 at 0020)
Walsh 1 at 7 (EXP.010.239.0001 at 0009)
RWS ch 15B at [149(c)–(j)] (SBM.020.002.0458 at 0507-08)
PAR.010.002.0001_2
Blaivas 1 at 26 (EXP.010.026.0001_2 at 0028_2)
RWS ch 15A at [78] (SBM.020.002.0511 at 0536)
RWS ch 15A at [79] (SBM.020.002.0511 at 0536)
Klosterhalfen 1 at [252] (LAY.010.014.0001_6 at 0053_6)
Iakovlev 1 at 151–152 (EXP.010.036.0001_4 at 0152_3–0153_3)
Wright at 116 (EXP.020.057.0001_4 at 0118_4)
T816 (TRA.500.010.0001_2 at 0023_2)
T823 (TRA.500.010.0001_2 at 0030_2)
T874 (TRA.500.010.0001_2 at 0081_2)
SAN.MESH.00000078; SAN.MESH.00000503
T6802–03 (TRA.500.084.0001 at 0035–0036)
T6806–07 (TRA.500.084.0001 at 0039–0040)
Blaivas 1 at 23–24 (EXP.010.026.0001_3 at 0025_3)
T732 (TRA.500.009.0001_2 at 0003_2)
Korda 2 at 9 (EXP.010.103.0001_4 at 0010_4)
Korda 2 at 9 (EXP.010.103.0001_4 at 0010_4)
Blaivas 1 at 25–26 (EXP.010.026.0001_2 at 0027_2–0028_2)
Blaivas 1 at 26 (EXP.010.026.0001_2 at 0028_2)
Frazer at 8 (EXP.020.016.0001_2 at 0008_2)
Roovers 1 at 24 (EXP.020.033.0001_4 at 0026_4)
Rosamilia 2 at [3.2] (EXP.020.031.0001_3 at 0018_3)
Margolis 2 at 17 (EXP.010.124.0001_3 at 0018_3)
Deprest at [116] (EXP.020.006.0001_4 at 0025_4)
Margolis 2 at 16–17 (EXP.010.124.0001_3 at 0018_3)
Blaivas at 27 (EXP.010.026.0001_2 at 0029_2)
Roovers at 24 (EXP.020.033.0001_4 at 0026_4)
Korda 2 at 9 (EXP.010.103.0001_4 at 0010_4)
Korda 2 at 18 (EXP.010.103.0001_4 at 0019_4)
T1187 (TRA.500.014.0001_2 at 0060_2)
Korda 2 at 19 (EXP.010.103.0001_4 at 0020_4)
SAN.MESH.00000554
Wright 1 at 105 (EXP.020.057.0001_4 at 0107_4)
Korda 2 at 19 (EXP.010.103.0001_4 at 0020_4)
T1186 (TRA.500.014.0001_2 at 0059_2)
Rosamilia 2 at 16 (EXP.020.031.0001_3 at 0018_3)
Korda 3 at 41 (EXP.010.129.0001_5 at 0042_5)
Blaivas 1 at 27 (EXP.010.026.0001_2 at 0029_2)
Rosamilia 2 at 16 (EXP.020.031.0001_3 at 0018_3)
Margolis 2 at 17-18 (EXP.010.124.0001_3 at 0018_2)
Lee J et al, “Persistence of urgency and urge urinary incontinence in women with mixed urinary symptoms after midurethral slings: a multivariate analysis” (2011) 118 BJOG 798–805 (PUB.MESH.00009174)
Blaivas 1 at 27 (EXP.010.026.0001_2 at 0029_2)
Rosamilia 2 at 16 (EXP.020.031.0001_3 at 0018_3)
Sanders 2 at [8], [13] (LAY.010.027.0001 at 0004)
Blaivas 1 at 27 (EXP.010.026.0001_2 at 0029_2)
Blaivas 1 at 27–8 (EXP.010.026.0001_2 at 0029_2–0030_2)
SAN.MESH.00000028
T4180 (TRA.500.052.0001_2 at 0020_2); T4277 (TRA.500.053.0001_2 at 0035_2)
Hill at 7 (EXP.010.230.0001_2 at 0009_2)
Korda 2 at 18 (EXP.010.103.0001_4 at 0019_4)
Korda 3 at 39–40 (EXP.010.129.0001_5 at 0040_5-0041_5)
T1152 (TRA.500.014.0001_2 at 0025_2)
Korda 2 at 19 (EXP.010.103.0001_4 at 0020_4)
Jungfer 4 at 5–6 (EXP.010.226.0001_2 at 0007_2–0008_2)
Jungfer 2 at 7 (EXP.010.226.0001_2 at 0009_2)
AWS ch 15 at [431] (SBM.010.005.0674_2 at 0891_2)
RWS ch 15A at [80] (SBM.020.002.0511 at 0536-7)
SAN.MESH.00000030; SAN.MESH.00000001 at 0005
SAN.MESH.00000762 at 0763-64; SAN.MESH.00000771; SAN.MESH.00000770
SAN.MESH.00000589
SAN.MESH.00000602
Sanders 1 at [95] (LAY.010.005.0001 at 0019)
Williams 2 at 5 (EXP.010.252.0001_2 at 0008_2)
Email from Clayton Utz dated 21 February 2019
Email from Clayton Utz dated 21 February 2019
Sanders 1 at [24] (LAY.010.005.0001 at 0005)
Sanders 1 at [94] (LAY.010.005.0001 at 0018-19)
Sanders 2 at [30] (LAY.010.027.0001 at 0008)
Williams 2 at 6 (EXP.010.252.0001_2 at 0009_2)
Sanders 2 at [36] (LAY.010.027.0001 at 0008)
AWS ch 15 at [454] (SBM.010.005.0674_2 at 0898_2)
Williams 2 (Sanders) at 14 (EXP.010.252.0001_2 at 0017_2); Williams 2 Annexure B (Sanders) at 10 (EXP.010.254.0001 at 0010)
Williams 2 (Sanders) at 14 (EXP.010.252.0001_2 at 0017)
Williams at 15 (EXP.010.252.0001_2 at 0018_2)
Williams 2 Annexure B (Sanders) at 10 (EXP.010.254.0001 at 0010)
Borthwick 1 at 21–2 (EXP.020.100.0001 at 0023–0024)
T5265 (TRA.500.065.0001 at 0020)
Williams 3 (Sanders) at 2 (EXP.010.320.0001_2 at 0004_2)
Williams 3 (Sanders) at 3 (EXP.010.320.0001_2 at 0005_2)
Williams 3 (Sanders) at 4 (EXP.010.320.0001_2 at 0006_2)
Borthwick 4 (EXP.020.112.0001)
RWS ch 15C at 34 (SBM.020.002.0511 at 0544)
Borthwick 1 at [7.4.10]–[7.4.11] (EXP.020.100.0001 at 0026)
T5296 (TRA.500.065.0001 at 0051)
P Sanders at [21] (LAY.010.006.0001 at 0005)
Sanders 1 at [94] (LAY.010.005.0001 at 0018)
Williams 3 at 2 (EXP.010.320.0001_2 at 0004_2)
T5528 (TRA.500.068.0001 at 0047)
Williams 3 at 2 (EXP.010.320.0001_2 at 0004_2)
Borthwick (Sanders) at [7.2.2] (EXP.020.100.0001 at 0023)
Jungfer 6 at 8 (EXP.010.226.0001_2 at 0010_2)
Wilcox at 13 (EXP.020.067.0001 at 0015)
Williams 3 at 2 (EXP.010.320.0001_2 at 0005_2)
Williams 2 at 12 (EXP.010.252.0001_2 at 0015_2)
Borthwick (Sanders) at [5.2.2] (EXP.020.100.0001 at 0019)
Borthwick (Sanders) at [7.4.21] (EXP.020.100.0001 at 0019)
Sanders 1 at [73] (LAY.010.005.0001 at 0015); SAN.MESH.00000106
Sanders 1 at [73] (LAY.010.005.0001 at 0015)
SAN.MESH.00000106
Sanders 1 at [99] (LAY.010.005.0001 at 0020)
Wilcox at [2.8] (EXP.020.067.0001 at 0005)
Sanders 2 at [14] (LAY.010.027.0001 at 0005)
Jungfer 3 at [1.5] (EXP.010.010.0001_2 at 0004_2)
Sanders 1 at [62] (LAY.010.005.0001 at 0013)
Wilcox at [8.1] (EXP.020.067.0001 at 0009)
Wilcox 1 at 12-13 (EXP.020.067.0001 at 0014-15)
Sanders 2 at [34] (LAY.010.027.0001 at 0009)
Williams 2 Annexure B (Sanders) at 16 (EXP.010.254.0001 at 0016)
Williams 2 at 13 (EXP.010.252.0001_2 at 0016_2)
Borthwick (Sanders) at [6.1.1] (EXP.020.100.0001 at 0020)
Williams 2 Annexure B (Sanders) at 6 (EXP.010.254.0001 at 0006)
Sanders 2 at [45] (LAY.010.027.0001 at 0011)
Borthwick at [6.1.2] (EXP.020.100.0001 at 0020)
Williams 2 Annexure B (Sanders) at 12 (EXP.010.254.0001 at 0012)
Williams 2 at 8 (EXP.010.252.0001_2 at 0011_2)
Williams 2 Annexure B (Sanders) at 13 (EXP.010.254.0001 at 0012)
Williams 2 Annexure B (Sanders) at 13 (EXP.010.254.0001 at 0008)
RWS ch 15C at [155] (SBM.020.002.0511 at 0561)
Borthwick (Sanders) at [4.4.1] (EXP.020.100.0001 at 0014)
Williams 2 at 7 (EXP.010.252.0001_2 at 0010_2); T5525 (TRA.500.068.0001 at 0044)
Sanders 1 at [105] (LAY.010.005.0001 at 0022)
SBM.020.002.0359 at 0360

