
Sequenom, Inc. v Ariosa Diagnostics, Inc. [2019] FCA 1011
ORDERS
DATE OF ORDER: |
THE COURT ORDERS THAT:
1. Within 21 days of the date of these orders, the applicant file and serve minutes of orders and submissions (limited to 3 pages) to give effect to these reasons, on costs and for the further conduct of these proceedings.
2. Within 14 days of receipt of such minutes and submissions, the respondents file and serve responding minutes of orders and submissions (limited to 3 pages).
3. The interim confidentiality order made in Order 1 of the orders dated 9 June 2017 be extended until further order, save that nothing therein applies to the Court’s reasons for judgment.
4. Liberty to apply.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
[Redacted version]
BEACH J:
1 The applicant (Sequenom) is the patentee of Australian Patent No 727919 entitled “Non-invasive prenatal diagnosis” (the Patent). The Patent claims a priority date of 4 March 1997. It was filed on 4 March 1998 and granted on 19 April 2001. The invention claimed in the Patent provides a method for detecting the presence of a nucleic acid of fetal origin in a maternal serum or plasma sample. The Patent relates to prenatal diagnosis by detecting fetal nucleic acids in such non-cellular components of a maternal blood sample.
2 Sequenom seeks monetary and declaratory relief against the respondents for infringement of claims 1, 2, 3, 5, 6, 9, 13, 14, 22, 23, 25 and 26 of the Patent (the relevant claims).
3 The first respondent (Ariosa) is a US company that conducts and licenses others to conduct a non-invasive prenatal diagnosis test, marketed under the name “Harmony” (the Harmony Test).
4 The second respondent (Sonic) is an Australian pathology company which has been licensed by Ariosa to promote and supply in Australia Harmony Test results, and which through its subsidiary Sullivan Nicolaides Pty Ltd has used the Harmony Test in Australia.
5 The third respondent (Clinical) is an Australian pathology company which has been licensed by Ariosa to promote and supply in Australia Harmony Test results, and which has used the Harmony Test in Australia.
6 The respondents have cross-claimed seeking revocation of the relevant claims of the Patent on various grounds being that:
(a) the relevant claims are not for a manner of manufacture;
(b) the claimed invention lacks an inventive step;
(c) the relevant claims are not useful;
(d) the relevant claims are not fairly based;
(e) there is a lack of sufficiency; and
(f) the claimed invention was obtained by false suggestion or misrepresentation.
7 For most purposes I will describe the respondents collectively as Ariosa except where I need to distinguish between them for the purposes of the infringement case.
8 The present trial was held on issues of liability only. In summary, Sequenom has succeeded on its infringement claims save as to its case concerning the infringement of claim 26. Ariosa has failed to establish invalidity save as to claim 26 which is invalid for lack of fair basis.
9 For convenience I have divided my reasons into the following sections:
(a) Glossary of some terms – [10] to [84];
(b) Common general knowledge – [85] to [193];
(c) The Patent – [194] to [307];
(d) The experts – [308] to [338];
(e) Invalidity – General – [339] to [341];
(f) Manner of Manufacture – [342] to [528];
(g) Inventive Step – [529] to [797];
(h) Utility – [798] to [826];
(i) Sufficiency – [827] to [1038];
(j) Internal fair basis – [1039] to [1076];
(k) False suggestion – [1077] to [1160];
(l) Infringement – [1161] to [1476];
(m) Conclusion – [1477].
GLOSSARY OF SOME TERMS
10 Before discussing the science and then the Patent, it is useful to set out a glossary of some of the key terms. This glossary has been drawn from the expert evidence with some modifications to ensure its utility to both the invalidity case and the infringement case.
11 5' end and 3' end: In a single strand of DNA or RNA, the chemical convention of naming carbon atoms in the nucleotide sugar-ring means that there will be a 5'-end, which frequently contains a phosphate group attached to the 5' carbon of the ribose ring, and a 3'-end, which typically is unmodified from the ribose -OH substituent. In a DNA double helix, the strands run in opposite directions to permit base pairing between them, which is essential for replication or transcription of the encoded information.
12 Allele: A form of a gene. Usually one of two alleles, each inherited from a different parent.
13 Allosome: One of the sex chromosomes, X or Y.
14 Amniocentesis: A prenatal test involving the insertion of a needle through the mother’s abdomen into the uterus to withdraw amniotic fluid containing fetal cells shed by the fetus into the amniotic fluid.
15 Aneuploidy: The presence of an abnormal number of chromosomes in a cell, for example, 47 chromosomes instead of the expected and normal 46 (23 pairs).
16 Anucleated: A cell without a nucleus.
17 Array Binding Region: A section that is ultimately designed to bind to the microarray used in the detection step.
18 Assay Pair: For the purposes of the detection step, the Harmony Test notionally “pairs up” the target loci into assay pairs (for the Non-Polymorphic Assay) and SNP allele pairs (for the Polymorphic Assay). As depicted in the confidential Product and Process Description (PPD) which I have annexed to my reasons, [Redacted]
19 Autosomal chromosome: A chromosome that is not an allosome (a sex chromosome). Autosomes appear in 22 pairs whose members have the same form (but differ from other pairs in a diploid cell), whereas members of an allosome pair determine sex and may differ.
20 Biotinylation: The process of covalently attaching biotin to, for example, DNA. Biotinylation is rapid, specific and is unlikely to perturb the natural function of DNA due to the small size of biotin. Biotin binds to streptavidin with an extremely high affinity, fast on-rate, and high specificity and these interactions are exploited in many areas of biotechnology to isolate biotinylated molecules of interest.
21 Buffy coat: The fragment of un-clotted blood after centrifugation (spinning down) that contains most of the white blood cells and platelets.
22 Cell free DNA (cfDNA): Non-cellular DNA (i.e. DNA that is outside a cell).
23 Cell free fetal DNA (cffDNA): Non-cellular DNA from a fetus.
24 Chorionic Villus Sampling (CVS): A prenatal procedure involving the insertion of a catheter or needle through the abdomen or cervix into the placental portion of the uterus to take a sample of the chorionic villi (containing placental cells) surrounding the sac which protects the fetus.
25 Cordocentesis: Also known as percutaneous umbilical blood sampling or fetal blood sampling. A prenatal procedure performed after 18 weeks involving the insertion of a needle into the umbilical cord or intra hepatic vein under ultrasound guidance to withdraw fetal blood to detect conditions associated with fetal blood circulation including isoimmunisation, haemoglobinopathy, anaemia, thrombocytopenia and coagulation-factor abnormalities.
26 DANSR: Digital analysis of selected regions, a process of analysing sequences from assays targeted against selected genomic regions.
27 Denaturation: A process where proteins or nucleic acids such as DNA or RNA lose their structure. In the case of DNA, denaturation results in the disruption of the complementary base pair bonding (i.e. A – T and C – G) and separation of the double stranded helix into two single strands.
28 Discriminating Region: A section that is designed to allow the Harmony Test [Redacted]
29 Down syndrome: An aneuploidy of chromosome 21, with three chromosome 21s present instead of the normal two, typically associated with physical growth delays, characteristic facial features and mild to moderate intellectual disability. Down syndrome also includes cases in which the extra copy of all or part of the chromosome 21 is not a separate, third chromosome but is translocated or joined to another chromosome.
30 Edwards syndrome: Edwards syndrome is a chromosomal abnormality caused by the presence of all, or part of, an extra chromosome 18.
31 Electrophoresis: A method that separates fragments of DNA based on their size which can be stained to enable visual detection.
32 Erythrocytes: Red blood cells.
33 Fetal blood sampling: See Cordocentesis.
34 Fetal Fraction: The proportion of DNA in a sample deriving from the fetus (as opposed to the mother).
35 Fibrinogen: A blood clotting agent.
36 FISH or fluorescence in situ hybridisation: This technique is more targeted than conventional karyotyping. It utilises DNA probes which bind to specific and selected regions of interest on particular chromosomes (e.g. chromosome 21), which are then fluorescently labelled to reveal, with the aid of a fluorescent microscope, the presence, absence, relative positioning and / or copy number of the specifically targeted DNA segments.
37 FORTE: The Harmony Test’s bioinformatics algorithm (Fetal-Fraction Optimized Risk of Trisomy Evaluation) used to calculate risk scores for aneuploidy and determine fetal fraction and fetal sex.
38 Genotype: The genes in an individual’s DNA. Now this definition is very broad. In clinical practice, it would be more usual to use the term genotype to refer to the molecular genetic characteristics of an individual at a specific locus.
39 Heterozygous: A diploid organism is heterozygous at a gene locus when its cells contain two different alleles of a gene. The cell or organism is called a heterozygote specifically for the allele in question, and therefore, heterozygosity refers to a specific genotype.
40 h-index: The h-index is an author-level metric that attempts to measure both the productivity and citation impact of the publications of a scientist or scholar. The index is based on the number of the scientist’s most cited papers and the number of citations that these have received in other publications.
41 Homozygous: Where an individual has two copies of the same allele.
42 Informative locus: A polymorphic locus where the fetus has a different pair of alleles compared to its mother.
43 Karyotyping: The conventional form of chromosomal analysis, which is used to detect numerical and / or structural chromosome abnormalities in a sample of cells which have first been grown in culture. The method, broadly speaking, involves the use of various dyes which selectively stains the chromosomes in the sample.
44 LCR: Ligase chain reaction.
45 Leukocytes: White blood cells.
46 Ligation: The joining of two DNA strands. But in the context of the ligation chain reaction, ligation is the covalent joining of two adjacent nucleotides that are perfectly hybridized to the complementary target DNA.
47 Ligation Product: The oligonucleotides bind to their complementary sequences in the sample. Where all the three adjacent oligonucleotides bind, they are then ligated using the enzyme ligase to produce (partly) double stranded molecules called ligation products. DNA ligase enzymes are commonly used in molecular biology to join strands of DNA together by catalysing the formation of a bond between the 3' end of one DNA strand and the 5' end of an adjacent DNA strand. In the Harmony Test, the three oligonucleotides only join to form a ligation product if they are adjacent to one another (i.e. their binding sites on the nucleic acid of interest are next to each other).
48 Locus (plural loci): A targeted region of the genome being usually a defined or mapped position of the genome.
49 Microarray: Microarrays are used to detect and quantify specifically-targeted sequences of DNA and are also known as “gene chips”. A microarray is typically a glass slide onto which single stranded DNA molecules are fixed in an orderly manner at specific locations called spots (or features). A microarray may contain thousands of spots and each spot may contain a few million copies of identical DNA molecules that uniquely correspond to sequences of interest. Researchers know the identity and position of each short fragment in the microarray.
50 Minor Source Fraction: In the Harmony Test, the assumption is that the “minor source” of cfDNA in the sample of maternal and fetal cfDNA is of fetal origin and that the minor source fraction is an estimation of the fetal fraction.
51 NASBA: Nucleic acid sequence based amplification.
52 NIFTY Trials: The National Institute of Child Health and Human Development Fetal Cell Isolation Study (NIFTY) was a prospective, multicentre clinical project to develop non-invasive methods of prenatal diagnosis. The initial objective was to assess the utility of fetal cells in the peripheral blood of pregnant women to detect fetal chromosome abnormalities.
53 NIPD: Non-invasive prenatal diagnosis.
54 Non-Polymorphic Assay: The set of non-polymorphic oligonucleotides introduced into the sample of cfDNA in the Harmony Test.
55 OLA: Oligonucleotide ligation assay.
56 Oligo Junction: The junction of two adjacent oligonucleotides.
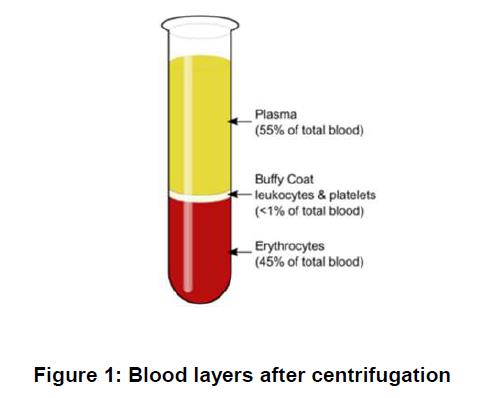
57 Oligonucleotide (oligo): See the definition for “probe”.
58 PCR: The polymerase chain reaction.
59 Percutaneous Umbilical Blood Sampling: See Cordocentesis.
60 Phenotype: The set of observable characteristics of an individual usually resulting from the interaction of its genotype with the environment, although not necessarily; for example, short long bones is the phenotype associated with the FGFR3 Gly380Arg (achondroplasia, common dwarfism).
61 Placenta: The organ comprising cells of fetal origin connecting a developing fetus to the uterine wall. It allows nutrient uptake; provides thermo-regulation to the fetus, waste elimination and gas exchange via the mother’s circulation; protects against internal infection; and produces hormones to support pregnancy.
62 Plasma: The supernatant or non-cellular liquid component of blood broadly comprising fibrinogen (a coagulation factor), salts, glucose, amino acids, vitamins, urea, other proteins and fats. It is obtained after centrifugation of blood collected into a test tube typically containing anticoagulant to prevent clotting.
63 Polymorphic Assay: The assay in the Harmony Test that is used to determine the relative proportion or dosage of markers on the selected target chromosomes. Its purpose is to estimate the minor source fraction in the sample.
64 Polymorphic locus: A locus that includes a known single nucleotide polymorphism (SNP).
65 Polymorphism: The presence of two or more different forms of a gene or sequence in an individual or population. But in clinical practice the term is usually reserved for neutral, common variants, usually present at greater than 1 to 2% of the population.
66 Pre-eclampsia: A disorder of pregnancy characterised by high blood pressure and a large amount of protein in the urine. The disorder usually occurs in the third trimester of pregnancy and worsens over time.
67 Primer: A short nucleic acid sequence that serves as a starting point for DNA synthesis. It is required for DNA replication because the enzymes that catalyse this process, DNA polymerase, can only add new nucleotides to an existing strand of DNA.
68 Probe: A probe, sequence specific probe or oligonucleotide probe is a fragment of single stranded DNA or RNA designed to bind to and target a particular complementary sequence of DNA or RNA in a given sample.
69 qPCR: Quantitative PCR.
70 qfPCR: Quantitative fluorescent PCR.
71 Read Out Cassette: For the purpose of further analysis, [Redacted]
72 Real time PCR (RT-PCR): This can also be known as quantitative PCR (qPCR). Real time PCR refers to a form of the reaction where the data is collected in real time (i.e. “live”) compared to a traditional or standard reaction where the data is collected at the end. The distinction is important as it means the data can be used for quantification. The method of detection may be, but does not need to be, by fluorescent probe. The use of specific fluorescent probes provides sequence-specific detection of DNA fragments with a complementary sequence to the probe.
73 Restriction enzyme: An enzyme that cuts DNA at or near specific recognition nucleotide sequences known as restriction sites.
74 RFLP: Restriction fragment length polymorphism.
75 Serum: The supernatant obtained after a blood sample has been allowed to clot.
76 SNP: Single-Nucleotide Polymorphisms or SNPs are variations in a single nucleotide at a specific position in genomes that are present within a population. For example, 60% of individuals in a population may have a gene with the sequence CGA at a given position in the genome and the remaining 40% of the population may have the sequence CGC at the same position. There are millions of SNPs present in the human genome.
77 SNP Allele Pair: For the purposes of the detection step, the Harmony Test notionally “pairs up” the target loci into assay pairs (for the Non-Polymorphic Assay) and SNP allele pairs (for the Polymorphic Assay). [Redacted]
78 STR: Short tandem repeat.
79 Thrombocytes: Platelets (fragments of large cells crucial to normal blood clotting).
80 Triple screen test: A prenatal test that measures the amounts of three substances in a pregnant woman’s blood: alpha-fetoprotein (AFP), human chorionic gonadotropin (hCG), and estriol (uE3). Similar tests using two markers are also known as serum screening tests.
81 Trisomy 18: See Edwards Syndrome.
82 Trisomy 21: See Down Syndrome.
83 Venepuncture: The act of obtaining access to veins (e.g. using a needle), typically from the arm, for the purpose of, for example, drawing a blood sample.
84 VNTR: Variable number of tandem repeats.
COMMON GENERAL KNOWLEDGE
85 The common general knowledge in the fields of fetal medicine and molecular genetics as at 4 March 1997 (the priority date) included the following information, which I have drawn from a statement agreed to by the parties save that I have added further detail concerning the PCR process and also further detail on Sanger sequencing which should be uncontroversial. I have also deleted some of the more basic diagrams concerning DNA.
(a) Blood
86 Blood cells make up approximately 45% of adult blood. These cells include oxygen carrying erythrocytes (red blood cells), immune cells called leukocytes (white blood cells) and thrombocytes (platelets). In the case of pregnant women, it was also known that there were fetal cells present in the mother’s blood. Plasma, which makes up the remaining 55% of the blood, is a straw coloured fluid which contains water, blood plasma proteins (including clotting factors), minerals and dissolved nutrients (such as glucose, amino acids, and fatty acids), and waste products (such as urea and lactic acid). Serum is the name given to plasma which has had the clotting factors removed.
87 Whole blood can be separated by centrifugation into three layers: (a) the upper plasma layer; (b) the “buffy coat” layer which contains the leukocytes and thrombocytes; and (c) the lower layer which contains the erythrocytes.
(b) Prenatal development
88 Following fertilisation of an ovum (egg) by a spermatozoon in a fallopian tube, the resulting single cell zygote travels down the fallopian tube and divides to form a blastocyst. Approximately 5 days after fertilisation, the blastocyst, which consists of trophoblast cells and embryonic cells, reaches the uterus and becomes embedded in the endometrium (lining) of the uterus. The trophoblast cells, which surround the embryonic cells, proliferate and embed further into the uterine lining, eventually forming the placenta. The blastocyst becomes fully implanted approximately 7 to 12 days after fertilisation.
The Placenta
89 The placenta is a composite structure made up of maternal tissues as well as those derived from the fetus. Fetal blood vessels extend to the placenta via the umbilical cord and branch into many chorionic villi, providing a large surface area for the exchange of materials between fetal and maternal blood across a layer of tissue called the placental membrane. A variety of materials, including nutrients and oxygen, are exchanged between the maternal circulatory system and the fetus via chorionic villi in the placenta and the umbilical cord. Other materials passing from the fetus or placenta into the maternal blood circulation include fetal blood cells, proteins and hormones which form the basis of the Rh disease test and the biochemical screens of maternal serum for Down syndrome discussed below. Likewise, waste materials are removed from the fetus to the maternal circulation.
Pre-eclampsia
90 Pre-eclampsia is a pregnancy disorder which affects about 6% of pregnancies, and is characterised by high blood pressure and elevated protein levels in the maternal urine. Pre-eclampsia generally occurs 24 to 26 weeks after fertilisation and often increases in severity until birth. Untreated, pre-eclampsia may lead to eclampsia (convulsions), bleeding in the mother’s brain and death of the mother. The early forms of pre-eclampsia are often associated with fetal growth restriction due to placental dysfunction.
(c) The human genome
91 The human genome represents the complete set of inherited instructions encoded in DNA in a human cell.
92 The human haploid nuclear genome consists of approximately 3 billion base pairs of DNA, organised into 23 chromosomes. Each chromosome carries a set of genes. A chromosome is a DNA-protein complex. In typical diploid individuals, that is, individuals with a balanced pairing of chromosomes, this is organised into a total of 46 chromosomes. The 46 chromosomes in a normal human somatic cell are made up of 22 pairs of homologous autosomes (non-sex chromosomes) and two sex chromosomes (XX for typical females and XY for typical males). One set of 23 chromosomes (comprising 22 autosomes and 1 allosome) is maternally-inherited and one set of 23 chromosomes is paternally-inherited.
93 DNA is a double helix molecule comprised of complementary strands of nucleotides sometimes referred to as the “building blocks” of DNA. Each nucleotide is composed of:
(a) one of four nucleotides, which are often referred to simply as “bases”, being cytosine (C), guanine (G), adenine (A) or thymine (T);
(b) a sugar called deoxyribose; and
(c) a phosphate group.
94 Nucleotides on the same DNA strand are joined in tandem by covalent bonds between the 5' position of the sugar on one nucleotide and the 3' position of the sugar on the phosphate on the next nucleotide. This forms a polynucleotide chain running from the 5' (or “five prime”) end to the 3' (or “three prime”) end. The two DNA strands, which run reverse-parallel to each other, 5'-3' and 3'-5', pair with complementary bases (A to T, C to G) to form “base pairs”.
95 Chromosomal DNA is replicated in the human cell nucleus. For this to occur the DNA-protein complexes must be disassembled and the DNA strands temporarily separated in the region of DNA being replicated. DNA helicases catalyse enzyme-dependent separation of the complementary strands from the site of previously bound initiator proteins, allowing DNA polymerase enzyme activity to synthesise two new strands using free deoxynucleotides (dNTPs). With the incorporation of each deoxynucleotide into the growing DNA strand, a pyrophosphate (two phosphate groups linked together) is released. Each strand of the original DNA molecule acts as a template for the production of a complementary strand in order to form two copies of the original DNA molecule.
96 Genes are functional units of DNA in the genome that code for particular proteins and non-coding RNAs. Different versions of a gene, for example, caused by variants such as single or multiple base changes, may be referred to as “alleles”. Where an individual has two copies of the same allele, that is, the same allele at a particular locus on each chromosome within one pair, they are said to be “homozygous” with respect to that allele. Where the alleles are different, i.e. there are different alleles at a particular locus on each chromosome within one pair, the individual is said to be “heterozygous” with respect to that allele.
(d) Genetic disorders
97 Genetic disorders are caused by changes to the genome. They may be caused by a range of mechanisms such as deletions, duplications, rearrangements and chemical modifications affecting anything from a single base to a whole chromosome or even the whole genome. Those caused by a defect in only one gene, for example, cystic fibrosis are classed as “single-gene disorders”. Other disorders may be caused by one of a number of genes. Although the spectrum is a continuum, disorders involving larger regions such as partial or whole chromosomes, for example Down syndrome, are generally referred to as “chromosomal” whilst those involving changes at the nucleotide level are generally classed as “molecular”. Genetic disorders may be inherited or arise “de novo” in the germ cell or developing embryo.
98 Prior to the priority date, prenatal testing was available for some single gene disorders, such as sickle cell anaemia, thalassemia and cystic fibrosis.
99 The cystic fibrosis gene was identified in 1989 and by the late 1990s several hundred mutations were recognized and this number has increased since that time. Prenatal diagnosis of cystic fibrosis was well-developed by the priority date and involved extracting fetal DNA from samples obtained using amniocentesis or CVS, and then analysing the extracted fetal DNA using methods such as targeted PCR.
100 Single-gene disorders can be inherited in an autosomal dominant fashion, which means that the disorder is expressed even if the person is heterozygous for the mutant allele, or in an autosomal recessive fashion, meaning that the disorder will only be expressed if the person is homozygous for mutant alleles. Disorders can also be inherited in an X-linked fashion in which a male will be more likely to have the disease phenotype as they only have one copy of the X chromosome.
Rhesus Disease
101 Rh (Rhesus) factor (also known as the Rh D antigen) is a protein found on the surface of red blood cells in so-called Rh positive individuals. Rh negative individuals lack this protein. Lack of this protein in Rh negative individuals is caused by a deletion or mutations of the gene (RhD) that encodes it in both copies of chromosome 1 which carries the RhD gene. If one copy of chromosome 1 contains the RhD gene and one does not, the individual still expresses the Rh factor and is considered Rh positive.
102 Rh disease can cause haemolytic disease of the newborn and fetus. This typically arises in second or subsequent pregnancies when a Rh negative mother is carrying a Rh positive fetus. In other words, the child inherits from its mother a copy of chromosome 1 in which the RhD gene is deleted or mutated and a copy of chromosome 1 from the father in which the RhD gene is present and functional. Since the child possesses one functioning copy of the RhD gene, the child produces Rh factor, and is thus referred to as Rh positive.
103 When an Rh negative mother carries a Rh positive fetus, the fetus expresses the Rh factor on its red blood cells. During pregnancy and birth the mother may be exposed to fetal red blood cells expressing Rh factor. The mother mounts an immune response to Rh factor, which it identifies as foreign, and thus her immune system becomes sensitised to Rh factor. A Rh negative mother sensitised to Rh factor may mount a more robust immune response destroying the red blood cells of a Rh positive fetus in subsequent pregnancies.
104 By the priority date it was routine to give all pregnant mothers a blood test to determine their Rh status. The general approach to treatment was to treat all mothers identified as Rh negative with anti-Rh factor antibodies (so called prophylactic anti-D), ensuring that any Rh positive fetal red blood cells are masked before an immune response can be raised against them by the mother’s immune system, hence preventing issues with subsequent Rh positive pregnancies. This was, however, an inefficient approach as it inevitably involved treating Rh negative mothers carrying Rh negative fetuses, who did not need the treatment. There was, therefore, a desire to develop a way to screen for the Rh status of the fetus noninvasively. This would allow prophylactic anti-D to be given only to the Rh negative women who needed it, that is, those carrying a Rh positive fetus.
Haemoglobinopathies
105 Haemoglobinopathies are genetic disorders in which the haemoglobin molecules in an affected individual’s red blood cells are abnormal. Well-known examples of haemoglobinopathies are sickle cell anaemia and alpha- and beta-thalassemia. Alpha-thalassemia is considered a lethal disease, often leading to fetal death in the third trimester with maternal hydrops (“mirror”) syndrome also commonly present. Sickle cell and beta-thalassemia can be treated but patients suffer from many symptoms and often need life-long repeated blood transfusions. Parents being confronted with a diagnosis of these fetal diseases may elect for termination.
106 Work had been carried out before the priority date to characterise the beta-globin gene and various mutations which were shown to lead to certain blood disorders.
107 The point mutations occurring in the case of beta-thalassemia may be different between the father and mother.
Sex-linked disorders
108 A number of diseases are known to be caused by a defective gene on the X chromosome, e.g. haemophilia. In many cases, these diseases primarily affect male fetuses because female fetuses will have another, non-defective, copy of the X chromosome. Work was on-going at the priority date to try to find ways to identify fetuses with possible sex-linked disorders. There was, therefore, a desire to develop a way to identify the sex of the fetus quickly, accurately and as early as possible in the first trimester. Further, in cases of congenital adrenal hyperplasia, which is not an X-linked condition, treatment with dexamethasone is needed to prevent virilisation in girls. This medication can be stopped once the fetus is known to be male. In severe X-linked diseases for which the parents may wish to elect termination, having a diagnosis as early as possible in the first trimester is valuable. DNA testing for the disease in such cases, which often took a week or longer, would only be done after the sex determination and could be omitted if the fetus was known to be female. Gender determination by ultrasound only becomes reliable for establishing the sex of a fetus from 18 weeks onwards.
109 As at the priority date, the sex of a male fetus would be apparent if a particular molecular test involved the PCR amplification of sequences known to be from the Y chromosome. In other words, if you could detect by PCR a Y-specific fragment, it was most likely that the fetus was male. This required sequence information for the Y chromosome to be known to allow for the design of primers to result in the Y specific PCR products. Further, the loci would need to be known to be unique to the Y chromosome. As at the priority date, there were Y-specific sequences published in the literature which could be used as PCR primers.
Aneuploidies
110 The presence of a variation in the number of chromosomes from the usual complement, that is, 46 chromosomes is referred to as aneuploidy. The absence of a single chromosome from a usual pair is referred to as monosomy, and the presence of an additional copy of a single chromosome to a usual pair is referred to as trisomy.
111 Aneuploidies in autosomal and sex chromosomes are responsible for a number of genetic conditions, due to abnormal dosage of genes, including Down syndrome (trisomy of chromosome 21), Edwards syndrome (trisomy of chromosome 18), Patau syndrome (trisomy of chromosome 13), Turner syndrome (full or partial monosomy of X), Klinefelter syndrome (XXY), XYY syndrome, XXYY syndrome and Triple X syndrome.
112 The most common viable autosomal trisomies are trisomies of chromosomes 21, 18 and 13. Trisomy 13 and trisomy 18 often result in miscarriage, stillbirth or, in the case of viable births, neonatal death. Trisomy 21 is not usually life-threatening but can result in significant physical and mental disability. Fetuses with aneuploidies of multiple chromosomes are unlikely to survive past the early stages of pregnancy.
113 The additional chromosome found in cases of trisomy may be paternally or maternally inherited. In trisomies 13, 18 and 21, the extra copy of the relevant chromosome is inherited from the mother in the majority of cases, that is, over 91% maternal in trisomy 13, around 95% maternal in trisomy 18 and around 90% maternal in trisomy 21.
(e) Molecular genetic diagnosis
114 Molecular genetic clinical diagnosis first became possible in the 1980s following the discovery of genetic markers, that is, known sites of variation between individuals within a population located on a particular chromosome, otherwise known as genetic polymorphisms.
115 The process of working out which allele (marker) a person had at a particular position or set of positions on a chromosome, that is, determining the genetic make-up of the alleles at the relevant loci on an individual’s chromosomes was called genotyping.
116 At the priority date the vast majority of genotyping methods depended on either detecting differences by length of genetic variation, sequence, position, conformation, or methylation. Detection methods included fluorescent labels, biotinylated probes, intercalating dyes and radiolabels.
117 There were numerous methods but predominant among them were restriction fragment length polymorphism (RFLP) analysis, short tandem repeats (STR), variable number of tandem repeats (VNTR), single strand conformation polymorphism (SSCP), denaturing gradient gel electrophoresis (DGGE), allele specific oligonucleotide (ASO), amplification refractory mutation system (ARMS), oligonucleotide ligation assay (OLA), pulsed field gel electrophoresis (PFGE), southern analysis, fluorescence in situ hybridisation (FISH), Sanger sequencing, pyrosequencing and reverse blots. Some of these methods are explained in more detail below.
118 Genetic markers used to detect human DNA per se could be as simple as detecting any one of the many repeat element families in the human genome e.g. Alu repeats. An alternative approach would be to test for the presence of a single copy gene. A more refined method of detection would rely on demonstration of differences based on polymorphism such as RFLPs detected either by genomic Southern blotting, or by PCR. Other markers commonly used at the time included STRs, VNTRs and other genetic variants.
119 Markers used to quantify DNA include the markers referred to in the preceding paragraph, along with determination of single or multiple copy sequences relative to a standard.
120 In 1997 thousands of markers that could be used for human molecular genetic testing were known. Commonly used markers included STRs, VNTRs, RFLPs and markers of structural variants. Of these variants, those used for human molecular screening/diagnosis would detect or be closely linked to the mutations underlying the specific genetic disease. Such markers would ideally be validated to document sensitivity, specificity, positive and negative predictive values. A wider number of markers may be required for diagnostic testing compared to screening.
Restriction Fragment Length Polymorphisms
121 Polymorphic loci at which base changes were known to affect restriction sites were known as restriction fragment length polymorphisms or RFLPs as I have referred to.
122 Certain naturally occurring bacterial enzymes called restriction endonucleases (or restriction enzymes) were known to cut double stranded DNA at specific points defined by the presence of a particular DNA sequence (a restriction site). Human DNA was known to contain numerous restriction sites. It was also observed after subjecting human DNA to bacterial restriction enzymes that not all individuals possess the same set of restriction sites within their DNA. If the DNA sequence at a particular locus differed from the restriction site by a single base, the relevant restriction enzyme would not cut the DNA at that site. This meant that if a section of two people’s DNA was subjected to restriction enzyme digestion the resulting fragments of DNA would either be of the same lengths (if all of the restriction sites were the same) or different lengths (if one or more of the restriction sites was absent, for example). The different fragment profiles could be observed by size separation of the products of restriction enzyme digestion by gel electrophoresis followed by a means of visualising the DNA.
Short tandem repeats
123 Short tandem repeats (STRs), also known as “microsatellites”, are sections of DNA made up of repeated short sequences, typically 2 to 4 base pairs long although they can be longer. It was found that different individuals often possessed different numbers of these repeated short sequences within a particular STR locus.
124 An individual’s STR genotype at a particular locus could be determined by creating primers for the constant (i.e. non-repetitive) DNA sequences on either side of a given STR locus and amplifying the resulting DNA fragment by PCR. Differing numbers of repeated sequences within the STR in different individuals would result in different length fragments being amplified, and the fragment lengths could be differentiated by gel electrophoresis followed by visualisation using a DNA stain or label.
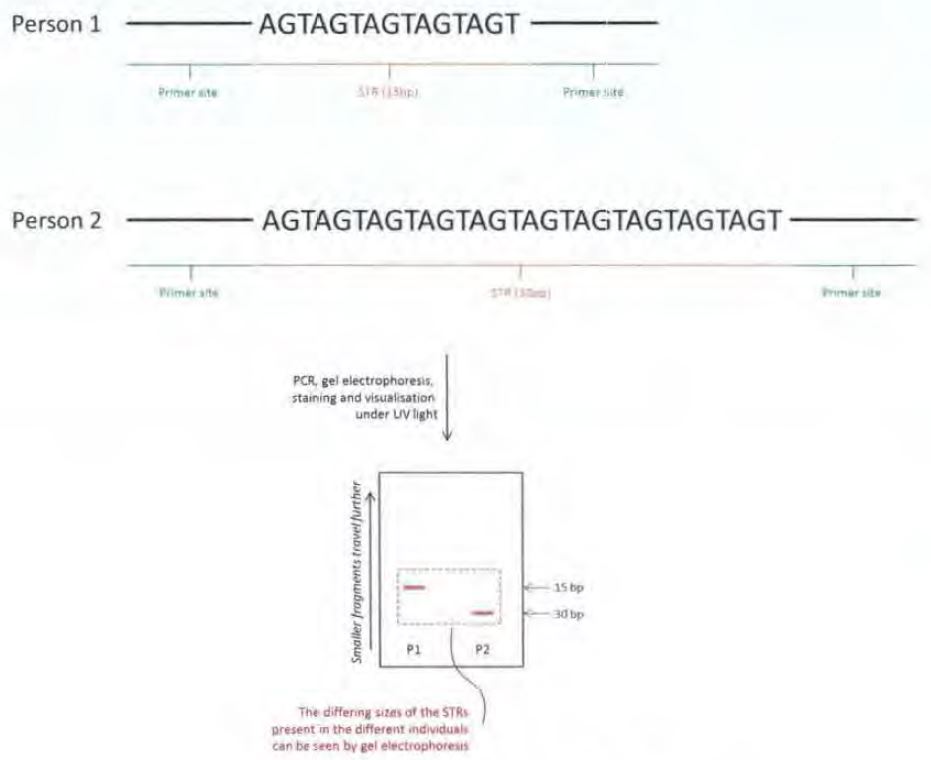
Figure 3: STRs – simplified example
125 The diagram above shows only one allele from each person, just to simplify the explanation. In reality, each person would have two copies of the STR locus, one on each chromosome, which could be the same or different as the allele drawn out above.
126 The gels produced from STR amplification could require careful analysis as the repetitive nature of STRs could lead to problems during PCR amplification and “slippage” of the DNA strands resulting in DNA fragments being produced with extra or missing copies of the short repeat units. This gave rise to extra bands in the gel known as “stutter bands” or “shadow bands” which could require careful analysis, especially when genotyping individuals with similar allele sizes, for example n base pairs and n+2 base pairs for a dinucleotide repeat marker.
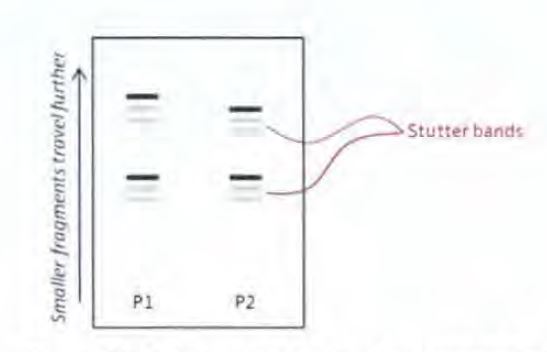
Figure 4: Schematic of stutter bands on a gel
127 STRs were used throughout the 1990s along with RFLPs as markers for linkage analysis and gene mapping. The greater heterozygosity, that is, the number of possible alleles at a given locus, of STRs, compared with RFLPs, increased the informativity of linkage analysis and assisted in the mapping of disease-causing genes. Once a disorder had been sufficiently localised to a particular part of a chromosome, that is, a defined genetic region, it was then feasible to sequence that small region to look for a gene and mutations.
Alu Repeats
128 Alu repeats are short repetitive sequences of DNA that are found throughout the human genome. They are the most common repeat sequences, and are present in tens of thousands of copies on every human chromosome. Alu repeats are generally not unique to a particular individual.
129 Amplification of Alu repeats or inter-Alu repeats may be used to establish whether human DNA is present in a sample. Primers that could be used to amplify Alu sequences had been reported by the priority date; see, for example, Nelson DL et al, “Alu polymerase chain reaction: a method for rapid isolation of human-specific sequences from complex DNA sources” (1989) 86(17) Proc. Nat. Acad. Sci. USA 6686-6690 (Nelson 1989).
Linkage analysis
130 Linkage analysis is based on the principle that the closer together one locus (e.g. a gene) is to another locus (e.g. a RFLP or STR marker) on a chromosome, the more likely it is that the gene and the marker will be passed on together to the offspring. Thus the two loci are said to be “linked”.
131 By analysing the pattern of markers present in the genomes of various members of a family in which at least one person was affected by a genetically inherited disorder, it was sometimes found that the affected (or carrier) relative(s) in a family had a consistently different marker pattern in a particular region of the chromosome to those who were not affected by the disorder. In that situation it could therefore be inferred that the gene causing the disorder was likely to be found close to those markers. By repeating this type of analysis with other families the conclusions could be refined, allowing the approximate location of the disorder-causing gene on the chromosome to be identified, that is, mapped.
132 Linkage analysis could also be used in some cases to provide a diagnosis of a genetically inherited disorder, for example if a family had a history of a genetic disorder and wished to know if another member was likely to be affected by that disorder. A good example is Huntington’s disease, the symptoms of which typically only develop when a person is in their 40s, which was diagnosed by linkage analysis in the 1980s and 1990s. Linkage analysis had to be carried out for each family separately.
133 The process required genotyping as many family members as possible in relation to a panel of markers such as RFLPs or STRs which were known to be linked with the gene thought to cause the disorder in question. The way in which the various markers were inherited down the family line could then be assessed to determine whether the presence of a particular pattern of markers (haplotype) was associated with individuals affected by the disorder in that particular family. In cases where this was possible, the relative in question could be genotyped in respect of those markers to ascertain whether or not they were likely to be affected by the disorder too.
Direct mutation analysis
134 From the late 1980s onwards people started to identify the genes which were responsible for causing genetically inherited disorders. An early example was the cystic fibrosis gene which was first identified in 1989.
135 As new methods and new technology developed through the 1990s, considerable progress was made in identifying the genes responsible for various disorders. Once the relevant genes were isolated their sequences could be determined and, in some cases, particular mutations could be identified as being responsible for causing the disorder in question by comparing the DNA sequences of affected and unaffected individuals at the relevant locus.
136 The availability of more genetic information about disorder-causing genes through the 1990s meant that this was a time of expansion in molecular genetic diagnosis, particularly in the second half of the decade and through the 2000s. Rather than having to carry out linkage analysis, with all the work on family member genotyping and the complexity which that involved, it became possible to diagnose some conditions by genotyping the individual in question at the relevant locus to determine whether a disorder-causing mutation was present. This was simplest in cases where only a single gene was responsible for the disease (known as a “single gene disease”) and where the disease was caused by a dominant allele, that is, if the individual is found to have the disease-causing gene even in only one of the relevant pair of chromosomes they can still be diagnosed with the disorder. For recessive conditions, it was common practice to check the phase, that is, check the parental origin of the mutation(s).
137 But some mutations proved very hard to characterise. For example, a large deletion in a gene on one copy of the relevant chromosome might not be seen if the patient's other copy of the gene was normal. In that case, the normal gene would be picked up but because the assay was qualitative, the fact that the other copy was missing could go unobserved. In 1997, direct mutation analysis was conducted on a much smaller scale compared to what is done now.
138 By the priority date the Human Genome Project was underway with the aim of mapping and sequencing all of the genes making up the human genome. However, the project had not been completed. It would take several more years to be finished with the full genome being published in 2003.
(f) Molecular techniques
139 By the priority date, there was a range of molecular techniques and technologies available. Some of these included the following:
(a) Gel electrophoresis.
(b) Polymerase chain reaction (PCR), including:
(i) Nested PCR; and
(ii) Quantitative PCR techniques.
(c) Sanger sequencing.
(d) Branched DNA methods.
(e) Ligase chain reaction.
Gel electrophoresis
140 Gel electrophoresis was and is used to separate molecules like DNA based on their size (molecular weight). When carried out following PCR or simple restriction enzyme digestion, the reaction products would be mixed with a suitable buffer containing a dye to increase the density of the material in the sample so that it could be seen during loading into a “well” on a gel made of agarose or polyacrylamide. An electric field would then be applied to the gel, which would cause the DNA, which is negatively charged, to migrate through the gel. The gel acts like a sieve, with smaller DNA molecules moving through it more quickly than larger DNA molecules. The gel would then be stained, typically with ethidium bromide, which binds to DNA, and then visualised with UV light. The stained molecules travel a particular distance from the starting point corresponding to the size of the molecule, so different PCR amplification products or fragments generated by restriction endonucleases will form separate bands on the gel.
PCR techniques
141 PCR is a standard molecular biology technique that involves the amplification of specific sequences of DNA using repeated cycles of denaturation, primer annealing and extension, resulting in theory at least in exponential accumulation of DNA fragments. The basic technique is illustrated by the diagram below:
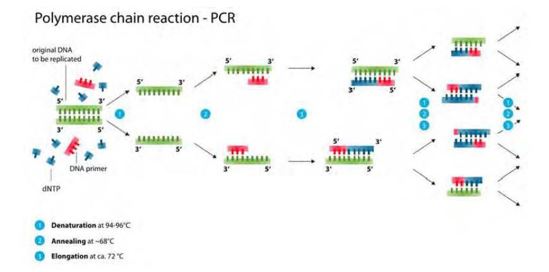
Figure 5: PCR steps: (1) denaturation; (2) annealing; (3) elongation. Template DNA shown in green; primers shown in red; new DNA strand shown in blue
142 DNA primers must be designed to bind to opposite DNA strands and flank the target of interest and initiate synthesis of a new DNA strand complementary to the target sequence of each template strand. These primer pairs are commonly referred to as the forward and reverse primers.
143 PCR requires the following components:
(a) A DNA template which is the gene or DNA sequence to be amplified.
(b) Primers, which are short single stranded pieces of DNA (i.e. an oligonucleotide) that have been synthesised to be complementary to a section of DNA. Two primers flank the gene or DNA fragment (in opposite orientation) to be amplified.
(c) DNA polymerase, being an enzyme that recognises primers and mediates the replication of the DNA template between the two primers starting from the 5' end to the 3' end by adding a complementary base to the growing strand. The most commonly used of these enzymes was Taq DNA polymerase which was derived from the heat-resistant bacteria Thermus aquaticus. As it is heat resistant it is suitable for use in PCR, which relies on thermal cycling.
(d) Nucleotides (dNTPs or deoxynucleotide triphosphates), being single units of the bases A, T, G, and C or in other words the “building blocks” for new DNA.
144 In broad terms, PCR involves the following steps:
(a) The first step is denaturation where the sample DNA is heated to break the weak bonds between the nucleotides of each strand so that the DNA separates into two separate strands.
(b) The second step is annealing. PCR does not copy all of the DNA in the sample but only a very specific gene or DNA sequence targeted by the primers. Two primers are used: one for each of the separated complementary single DNA strands. In this step, the primers bind, or anneal, to their complementary sequences on the DNA strands, marking the beginning of the sequence to be copied in the following step. Annealing occurs at lower temperatures than denaturation. And in this respect a balance needs to be achieved between a higher temperature which achieves greater specificity / lower mismatches, and the risk of denaturing the primers at those higher temperatures which prevents binding.
(c) The third step is extension where the reaction tube is heated to facilitate the extension of the nucleotide chain. From the start of the regions marked by the primers, nucleotides in the solution are added to the annealed primers by DNA polymerase to create a new strand of DNA complementary to each of the single DNA template strands. After this step, two identical copies of the original target DNA sequence are made. The extension must continue past the position of the primer on the complementary strand, so that in the next PCR round that new copy serves as a template for further amplification.
(d) The above steps are then repeated about 30 to 35 times to produce a sufficient amount of DNA copies of the original DNA target sequence. Too many amplification cycles beyond this leads to a building up of PCR “artefacts”, which create extra bands and impede analysis.
145 Let me expand further on the question of primers. PCR relies on the design of primers that will amplify a sequence of interest. As at the priority date, a number of issues needed to be considered in primer design, which sometimes required multiple rounds of optimisation, including limited sequence information and manual primer design.
146 The Gene Mutation Database and Human Genome Database were used at the priority date to design primers for use in DNA amplification methods for human molecular genetic testing. At the priority date the Gene Mutation Database and Human Genome Database had large collections of DNA sequences collated from the literature. In some circumstances these were of variable quality and reliability. Therefore, it was wise to validate any chosen marker. Both of these databases were searchable for specific DNA loci and the specific sequences could be downloaded and employed for primer design.
147 For human molecular genetic screening / diagnosis, the Gene Mutation Database had almost 500 specific disorders listed with known mutations. However, this constituted a very small percentage of total single gene disorders. For accurate human diagnostics of the majority of single gene disorders, there would be considerably more reliance on personal communication between research and clinical groups.
148 As I have indicated, as at the priority date, standard practice was to run 30 to 35 cycles of amplification, that is, to the end-point of the reaction. Running more than 40 cycles was known to result in a much higher chance of amplifying something other than the target. This can happen when the primers bind to a sequence of DNA that is very similar to the target or where there is contamination, that is, when some foreign DNA gets into the sample and is targeted by the primers. The risk of contamination affecting the results of a PCR assay is particularly high when the target sequence occurs in low levels in the sample or when there is no separation of the pre- and post-PCR laboratory areas or where appropriate equipment is not used, such as filter tips. Standard PCR is a non-quantitative technique.
Nested PCR
149 Nested PCR is a technique used to improve the specificity of the PCR reaction, that is, so that ideally only target sequences are amplified. It is particularly useful for detecting low levels of target sequence in a given DNA sample. In this method, the DNA in the sample is first amplified using “outer” primers which bind to the target sequence and possibly other non-target sequences in the sample. The sequences or “amplicons” amplified in this first round of PCR are subjected to a further PCR reaction involving one (hemi-nested) or two different “inner” primers having sequences which bind to sequences within the target amplicon.
Quantification of genetic material
150 Methods of quantification of genetic material were available at the priority date and included semi quantitative methods including densitometry scanning of areas under the peak (AUP) for Southern blots or bands on a gel. All of these could have been compared to internal controls. Other methods included PCR based methods consisting of endpoint PCR assays that sometimes used fluorescent markers. These methods also employed AUP measurements.
151 More accurate quantitative detection was achieved by quantitative PCR that measured changes in the exponential phase of PCR, again relative to a control.
152 The methods described above had a defined dynamic range and the quantification was relative.
Quantitative PCR
153 Quantitative PCR (“qPCR”), is a broad term that is used to refer to PCR methods which enable the products of a conventional PCR reaction to be quantified. By the priority date, amplification of sequences that were the targets of qPCR could be detected using a number of methods including agarose gels (a form of gel electrophoresis), fluorescent labelling of PCR products and detection with laser-induced fluorescence using capillary electrophoresis or acrylamide gels and plate capture and sandwich probe hybridisation.
Real-time quantitative PCR
154 One type of qPCR method that was available by the priority date was real-time quantitative PCR (“real-time qPCR”). Real-time qPCR involved a PCR reaction during which accumulation of the amplified PCR product was monitored by measuring a signal created by either fluorescent dyes or fluorescent probes in the reaction sample to generate an amplification curve. That is, real time qPCR is a development of standard (or end-stage) PCR, and uses fluorescent reporter molecules to monitor the amounts of PCR product present after each PCR cycle. This allows the generation of a growth curve and enables the quantification of DNA in the exponential phase by determining the number of amplification cycles necessary to achieve a specified fluorescence level.
Quantitative fluorescent PCR
155 Another qPCR method available at the priority date was quantitative fluorescent PCR (qfPCR), which was a method which required the products of the PCR reaction to be detected and quantified using a post-reaction “end point” technique, namely electrophoresis. As a result, qfPCR methods were understood to be more prone to errors in quantitation compared to real-time qPCR methods, as the latter quantified the PCR products in the same vessel as that used to amplify the DNA.
Sanger sequencing
156 Sanger sequencing is a method of direct DNA sequencing developed by Fred Sanger in about 1977. It was originally called “DNA sequencing” but is now called Sanger sequencing to distinguish it from the new generation of sequencing technologies.
157 The principle behind Sanger sequencing is similar to PCR except only one primer is used. Accordingly, amplification is arithmetic not exponential. The primer is a mixture of nucleotides and di-deoxynucleotides, which act as chain terminators during amplification, such that when a di-deoxynucleotide analogue is incorporated into the growing DNA strand, it prevents further extension from occurring. It is chance whether a normal or di-deoxy analogue is incorporated into the DNA extension product. But the ratio of normal to di-deoxy analogues is calculated to allow mainly normal nucleotides to be incorporated. The result is a series of extension products of various lengths depending on when a di-deoxy analogue is incorporated. This reaction is cycled 25 times to increase the number of extension products and to ensure that the reaction mixture contains extension products terminating at every base of the original sequence. Extension products are then separated according to size by gel electrophoresis, with each product differing in size by 1 base. The order of the separated extension products, terminating in a particular di-deoxy analogue, provides the sequence of the original sample DNA, which is demonstrated in Figure 6.
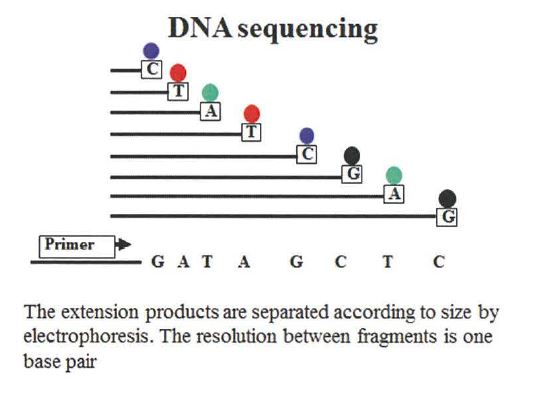
Figure 6
158 To enable detection, each di-deoxy analogue is labelled. When performing manual sequencing, usually each analogue was radiolabelled and detected on the gel after electrophoresis by autoradiography. When using automated Sanger sequencing instruments, the analogues could be labelled with one of four fluorescent colours (one for each base), such that the sequence of a sample could be determined based on the fluorescence of each analogue.
159 There were limitations with Sanger sequencing including that it could only sequence up to about 600 to 800 base pair fragments. If the DNA fragment was longer it was necessary to sequence multiple overlapping sequences.
160 Once a DNA fragment had been sequenced it was necessary to compare it to a reference sequence to identify any variations. Once sequence variations have been identified one would either be able to identify the significance of it based on previous experience or previous publications, or one could search for that mutation in a gene specific database to review what was known about the significance of that variant. Reference sequences could be sourced from the literature, gene specific databases or standard sequences.
Branched DNA methods
161 Branched DNA methods involve the use of capture probes and amplification of the signal through label probes.
Ligase Chain Reaction
162 The ligase chain reaction (“LCR”) is depicted in the Figure below.
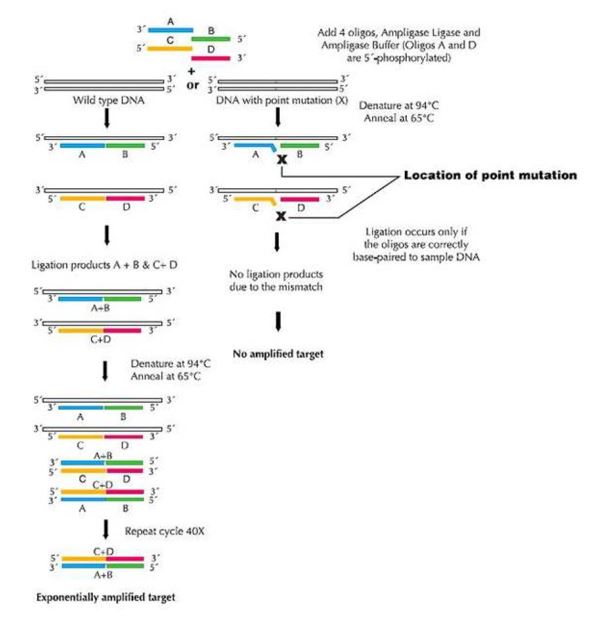
Figure 7: Ligase Chain Reaction
163 LCR is a technique that can be used in some cases to discriminate between DNA sequences differing in only a single base pair of nucleotides, such as a single base mutation.
164 LCR typically involves the use of two complementary pairs of single stranded DNA probes or oligonucleotides, each pair designed to bind to adjacent regions on both the sense and the antisense strands of a target DNA sequence. The junction of the two adjacent oligonucleotides (“Oligo Junction”) in each pair is usually positioned so that the nucleotide at the end of one of the oligonucleotides coincides with the point mutation. If, and only if, the nucleotide in the oligonucleotide at the Oligo Junction is complementary to the nucleotide at the same position in the target sequence can an enzyme called DNA ligase join the two oligonucleotides. The process can also be used to amplify a region without a single base mutation.
165 If the two adjacent oligonucleotides are able to join or ligate, the joined oligonucleotides can then themselves be used as templates for further rounds of ligation mediated amplification, which, like PCR, exponentially amplifies the paired oligonucleotides containing the target DNA sequence.
166 If the adjacent oligonucleotides are not able to join, because the nucleotide at the Oligo Junction is not complementary to the nucleotide at that position in the target sequence or because the target sequences are not present, amplification will not occur.
(g) Prenatal testing
Triple Screen
167 It was known that at around 16 weeks’ gestation in the second trimester of pregnancy, three substances, namely, alpha-fetoprotein (AFP), free beta hCG and estriol, were often present in different amounts in maternal serum in pregnancies where the fetus was affected by Down syndrome and pregnancies where it was not. This led to the development of what became known as the “triple test”. Instead of involving an invasive test, measurement of the levels of these markers could be achieved using a simple blood sample from the mother at around 16 weeks’ gestation. By considering both the risk associated with the mother’s age and the risk calculated by reference to the mother’s AFP level, a more accurate estimate of the overall likelihood that the mother was carrying a fetus affected by Down syndrome could be obtained. Determining AFP levels in maternal serum was also much cheaper than invasive testing and was not associated with a risk of miscarriage.
168 The triple test was the standard serum screening test for Down syndrome for a long time and was eventually offered to all pregnant women regardless of age in many countries. Although the precise figures vary, as a general rule, the triple test led to the identification, after invasive testing, of about 50% of cases of Down syndrome in the tested population. In other words, of the women taking the test who are actually carrying fetuses affected by Down syndrome about half of them will be identified by the triple test.
169 By the priority date work had also been carried out to find new markers which could be used at 10 to 12 weeks’ gestation as there was a desire to enable diagnosis of Down syndrome within the first trimester of pregnancy, leading to the discovery in the early 1990s that free beta hCG could be used in combination with pregnancy associated plasma protein A (PAPPA) to screen reliably for Down syndrome during the first trimester of pregnancy using maternal serum. It had also been discovered that fetuses affected by Down syndrome could be identified using ultrasound scanning in what was known as the nuchal translucency test. By the priority date researchers had started to consider whether combining the nuchal translucency test with serum markers might give even more accurate results.
170 Consequently, in addition to the use of maternal age to assess the risk that a pregnant woman was carrying a fetus with Down syndrome, it being known for decades that the likelihood of a woman conceiving a fetus affected by Down syndrome increases as the woman gets older, biochemical screening of maternal serum was used to identify women who were at higher risk of carrying an aneuploidy fetus and only those women were referred for invasive testing.
Cytogenetic techniques
171 Cytogenetic techniques were available at the priority date to analyse the number and structure of chromosomes in fetal cells, which had been extracted from the pregnant woman’s amniotic fluid, by amniocentesis, or placenta, by chorionic villus sampling (CVS). Each of these techniques carried with it a risk to the fetus.
172 Amniocentesis involves the collection of amniotic fluid, which contains fetal cells, using a needle which is inserted through the abdomen and uterus into the amniotic sac under ultrasound guidance. It is feasible from 15 weeks.
173 CVS is a technique for sampling cells that are likely to have the same karyotype as the fetus although in some cases they will differ, for example where there is a confined placental mosaicism. The sample is collected from the chorionic villus of the placenta either using a catheter inserted through the vagina or using a needle inserted through the abdomen.
174 Once the fetal cells had been isolated and cultured they could be analysed by cytogenetic techniques to determine the number and structure of chromosomes. These techniques included the following methods.
175 One method was karyotyping. The “karyotype” of an individual is the number and appearance of the chromosomes in the nucleus of the cell. Traditional karyotyping involves staining dividing (metaphase) chromosomes with a dye to allow them to be visualised under a microscope. This allowed fetuses possessing an abnormal number of chromosomes (such as an extra copy of chromosome 21 in Down syndrome) or chromosomes with abnormal structures to be diagnosed.
176 Another method was fluorescent in situ hybridisation (FISH). FISH uses a fluorescently labelled DNA probe which is designed to bind to portions of a gene of interest. The presence of trisomy 21 may be detected by the presence of three fluorescent spots in the fetal cell, rather than the expected two. The FISH technique hybridised fluorescent probes to specific sequences within a chromosome. Probes used in FISH were large and usually isolated sections of chromosomes, as opposed to small sequences used in other molecular work. Fluorescence microscopy could then be used to detect where on the chromosomes the fluorescent probe has bound. FISH was and is used to detect chromosome translocations, aneuploidies and chromosome identification.
177 Whilst these approaches were accurate and reliable, the main drawback with amniocentesis and CVS was that both procedures were invasive and were understood to increase the risk of the mother suffering a miscarriage. At the priority date the risk was believed to be around 1% for amniocentesis and 2% for CVS.
(h) Y chromosome markers & sex determination
178 The sex of a fetus determined by cytogenetics or detection of Y chromosome markers in kits was taken into account in the assessment of sex-linked genetic disorders. For example, if a pregnant woman was a known carrier of haemophilia and was having a male child, then the male child would have haemophilia.
179 As at the priority date, the sex of a male fetus would be apparent if a particular molecular test involved the PCR amplification of sequences known to be from the Y chromosome. In other words, if you could detect by PCR a Y-specific fragment, it was clear that the fetus was male. This required sequence information for the Y chromosome to be known to allow for the design of primers to result in the Y specific PCR products. Further, the loci would need to be known to be unique to the Y chromosome. As at the priority date, there were Y specific sequences published in the literature, which could be used as PCR primers.
(i) Fetal cells in maternal blood
180 The idea that fetal cells might be present in the mother’s blood had been first proposed in 1969. The possibility of being able to access whole fetal cells by taking a maternal blood sample was of great interest to those looking for a non-invasive way of obtaining information about the fetus because it would allow analysis of the fetal genome to be carried out without the need for invasive testing and the problems associated with it. All that would be needed was a blood sample from the mother’s arm. As a result, a substantial amount of work was carried out in this area through the 1980s and the 1990s.
181 The approaches and techniques that had been used for non-invasive prenatal testing using fetal cells both isolated and in a background of maternal cells before the priority date are described in a paper published in 1994 by JL Simpson and S Elias entitled “Isolating Fetal Cells in Maternal Circulation for Prenatal Diagnosis” 14(13) Prenatal Diagnosis 1229-1242.
182 By the priority date it was known that several different types of fetal cells were present in maternal blood during pregnancy, and that these fetal cells had the potential to be used for prenatal testing. These included:
(a) haematopoietic stem cells which, in the fetus, go on to make red and white blood cells;
(b) nucleated erythrocytes, which are a type of red blood cell which is typically only found in fetuses and very young children, not in adults whose red blood cells do not contain a nucleus or, therefore, chromosomes;
(c) lymphocytes and granulocytes which are types of white blood cell; and
(d) trophoblasts which are fetal placental cells which invade the tissue of the mother’s uterine wall causing changes in its vascular structure and formation of the placenta.
183 I note that in 1996, C Danae Steele and others (Steele CD et al, “Prenatal Diagnosis Using Fetal Cells Isolated From Maternal Peripheral Blood: A Review” (1996) 39(4) (December) Clinical Obstetrics & Gynecology 801-813) discussed relevant different cell types in the following terms:
TROPHOBLASTS
The possible cell types that can be isolated from maternal blood and used for prenatal diagnosis include trophoblasts, lymphocytes, and erythroblasts. Trophoblasts were the first cells to be identified in the maternal circulation because of their large size, but there are several reasons that they are not ideal for prenatal diagnosis. First, very small numbers of these cells are present in maternal blood during the first trimester, when it would be ideal to perform prenatal diagnosis. Secondly, any trophoblasts that are released into the maternal blood quickly become trapped in the lung and only rarely remain in the peripheral circulation. Thirdly, syncytiotrophoblasts [placenta] do not always share the same chromosome complement as the fetus and may be either multinucleated or mosaic. For these reasons, most investigators have concluded that trophoblasts are not the ideal cells to use for prenatal diagnosis.
LYMPHOCYTES
Fetal lymphocytes are an attractive potential source of cells for prenatal diagnosis. They express the HLA class of antigens so that if the mother and father are HLA incompatible, cells may be sorted by flow cytometry and enriched for paternal antigen expressing cells. However, this approach requires paternal HLA typing and specific antisera, and is impossible when paternal and maternal HLA antigens are shared. This approach is further limited because fetal production of lymphocytes does not begin until the second trimester, and even if lymphocytes can be isolated, it is not clear that they respond to the mitogens used to produce the metaphases needed for karyotyping. Tharapel et al flow-sorted cells for HLA differences between mother and fetus but could locate only maternal cells after metaphase had been induced.
Another potential problem is that in some cases fetal lymphocytes can persist in the maternal circulation for many years after a pregnancy…
NUCLEATED ERYTHROCYTES
Fetal nucleated erythrocytes appear to be best suited for this approach to prenatal diagnosis for several reasons. First, nucleated erythrocytes are rare in the adult circulation (except in clinical circumstances of increased hematopoesis such as pregnancy), but common in the fetus, especially in early gestation when hematopoesis occurs mainly in the yolk sac. Secondly, nucleated erythrocytes express several unique antigens such as the transferrin receptor, which make it possible to use automated cell-sorting techniques to enrich samples of maternal blood for fetal cells. Thirdly, these cells produce unique fetal hemoglobin chains such as zeta and gamma, which have the potential to be used as markers to identify these fetal cells. Lastly, erythrocytes are known to have a short life-span and are unlikely to persist from one pregnancy to the next.
Recent studies suggest that most nucleated erythrocytes that occur in first trimester maternal blood samples are maternal in origin. Enrichment of nucleated erythrocytes in general will enhance the concentration of maternal cells as well as fetal. Therefore, a unique marker for fetal nucleated erythrocytes continues to be sought.
184 All of these cell types were known to be present in all three trimesters of pregnancy.
185 From at least the late 1980s there was research being done by a number of groups on processes of recognition and enrichment of fetal cells. The aims of this research included investigating the use of fetal cells isolated from maternal blood to provide a non-invasive means of diagnosis of Down syndrome, to determine the blood type of the fetus and enable the pregnancy to be managed accordingly, to provide a way to determine the sex of the fetus (which would be useful when trying to identify fetuses with possible sex-linked disorders), and to diagnose single gene disorders.
186 However, isolating fetal cells was not easy because they were known to occur only rarely in maternal blood and were vastly outnumbered by maternal cells in the sample. Consequently, as well as work on new or improved methods and equipment for isolating fetal cells, research was also being carried out into methods for enriching the proportion of fetal cells in a sample to try to overcome these problems. Various techniques were tried including flow sorting, which relied, for example, on finding antibodies which bound to particular antigens on fetal cells, and magnetic sorting. These methods had some success in improving rates of fetal cell isolation during the course of the 1990s. However, by the priority date fetal cells could not reliably be identified from every maternal blood sample.
187 Another important issue was verifying that the cells which had been isolated were actually fetal before any analysis was carried out. Fetal cell detection had been approached using various methods. Early studies used fluorescence activated cell sorting, selecting cells identified as fetal by antibodies directed at paternal HLA type (human leukocyte antigen type). Flow cytometry was also used.
188 Flow cytometry is the use of light scattering and absorption spectra to measure various intrinsic and extrinsic properties of whole cells suspended in a solution. The solution is passed through a narrow tube past a series of laser beams arranged at different angles relative to the direction of flow, allowing only a single cell to pass at once. The passage of the cell interrupts the laser beams, and this is detected by an array of receivers arranged opposite to the beam generators. This allows each individual cell to be detected as a unique signal event, allowing the diagnostician to record many thousands of such events and draw conclusions about the properties of a large population of cells.
189 The size and internal granularity of cells can be inferred from the scattering of light from different angles. Fluorescently-labelled probes which bind to cell surface markers may also be used. Using this approach, the signals produced by flow cytometry can also be used to sort populations of cells, for example to isolate a specific sub-population of interest.
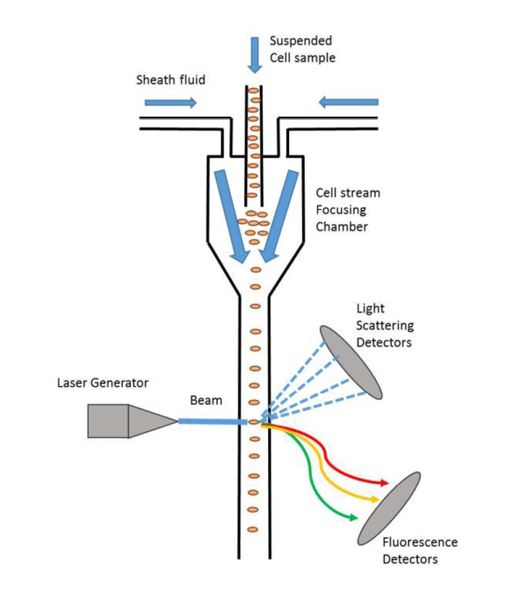
Figure 8: Illustration of the basic principles of flow cytometry
190 Fetal cell detection had also been approached using PCR with Y-chromosome specific primers. Where the reaction successfully amplified the Y-specific target sequence, it could be concluded that the cells in question were fetal because that DNA could not have originated from the mother who obviously would not possess the targeted part of the Y chromosome.
191 Another approach to detect “rare-event” cells such as fetal cells in maternal blood which was used by the priority date was via staining methods, such as FISH. This technique relied on different fluorescent probes specific for the X and Y chromosome.
192 By the priority date research had emerged suggesting that in some cases fetal cells could survive in a woman’s body for many years after the birth of the baby. This caused concern that some of the fetal cells being analysed from a given sample might not be from the current pregnancy and, therefore, could not be assumed to give reliable information on the fetal genome.
193 In spite of these problems, research into fetal cells in maternal plasma was still ongoing at the priority date given the significant clinical impact that would be felt if it was possible to overcome these problems to provide a means of allowing analysis of the fetal genome without the need for invasive testing. Some groups continue to do research on whole fetal cells in maternal circulation today.
THE PATENT
194 The Patent claims a priority date of 4 March 1997 from GB Patent No. 9704444 entitled “Non-Invasive Prenatal Diagnosis”. Non-invasive prenatal diagnosis (NIPD) was the conventional term in use at the priority date to describe techniques such as non-invasive fetal cell detection methods.
195 The Patent provides that the invention described and claimed therein relates to prenatal detection methods using non-invasive techniques, and in particular to prenatal diagnosis by detecting fetal nucleic acids in serum or plasma from a maternal blood sample; this may be treated as or part of the relevant field.
196 As I have said, whole blood can be separated by centrifugation into three layers being the upper plasma layer, the “buffy coat” layer which contains the leukocytes and thrombocytes, and the lower layer containing the erythrocytes, with serum being the plasma which has had the clotting factors removed.
197 The person skilled in the art to whom the Patent is addressed is a team comprising a person with experience in fetal medicine, and in particular, an interest in prenatal screening and diagnosis, and a person with experience in standard molecular genetics techniques including desirably but not necessarily some practical experience of the techniques involved in laboratory-based genetic analysis of patient samples in a clinical context.
(a) Professor Lo’s discovery
198 As I have indicated, as at the priority date there were various screening and diagnostic tests available to detect fetal conditions. These included tests that used cellular fetal DNA obtained through the invasive techniques of amniocentesis, CVS and cordocentesis. Research was also being conducted on the potential use of DNA from whole fetal cells, which were known to circulate in the blood of a pregnant woman, and which were able to be distinguished from maternal cells using techniques such as PCR with primers directed to the Y chromosome or the RhD gene.
199 At some point before March 1997, Professor Lo discovered using standard and routine techniques that fetal DNA could not only be detected from fetal cells in the blood of pregnant women, but also that cffDNA could be detected in the plasma and serum of pregnant women. In Professor Lo’s first experiment, he discovered that DNA from the Y chromosome could be detected in the plasma of pregnant women bearing male fetuses.
200 The Patent suggests some potential applications for Professor Lo’s discovery. It states that it may be particularly useful for sex determination, which depends on detecting the presence of a Y chromosome. The Patent states that the discovery can also be used to diagnose other conditions by detecting any paternally-inherited sequences which are not possessed by the mother. This approach depends on detecting the presence of cell-free DNA using standard techniques that cannot originate from the mother, and therefore must be fetal, and which may be causative of a disease phenotype in the fetus.
201 The Patent states that Professor Lo’s discovery can be applied to screening for chromosomal aneuploidies. The two suggested approaches set out on page 5 of the Patent rely on quantitation of the fetal DNA. I note at this point that Ariosa submits that these quantitative approaches were speculative and could not have been implemented as at March 1997.
202 Let me now discuss the Patent in more detail.
(b) Known prenatal testing methods
203 Page 1 of the Patent describes the prenatal “screening” methods used before the priority date of the claimed invention, including CVS and amniocentesis, being techniques requiring careful handling and which present a degree of risk to the mother and to the pregnancy, and the use of biochemical markers and fetal cells in maternal blood to predict abnormalities in the fetus and possible complications in pregnancy.
204 As I have said, amniocentesis involved the collection of amniotic fluid, which contained fetal cells, using a needle which was inserted through the abdomen into the uterus into the amniotic sac under ultrasound guidance and was feasible from 15 weeks gestation.
205 As I have also said, CVS was a technique for sampling cells that were likely to have the same karyotype as the fetus, although in some cases they would differ, for example, where there was a confined placental mosaicism. The sample was collected from the chorionic villus of the placenta either using a catheter inserted through the vagina or using a needle inserted through the abdomen.
206 And as I have also said, once the fetal cells had been isolated and cultured they could be analysed by cytogenetic techniques to determine the number and structure of chromosomes. These include the methods of karyotyping and FISH. The “karyotype” of an individual is the number and appearance of the chromosomes in the nucleus of the cell. Traditional karyotyping involves staining dividing (metaphase) chromosomes with a dye to allow them to be visualised under a microscope. This allowed fetuses possessing an abnormal number of chromosomes, such as an extra copy of chromosome 21 in Down syndrome, or chromosomes with abnormal structures to be diagnosed. FISH uses a fluorescently labelled DNA probe which is designed to bind to portions of a gene of interest. The presence of trisomy 21 may be detected by the presence of three fluorescent spots in the fetal cell, rather than the expected two, as depicted in the following figure:
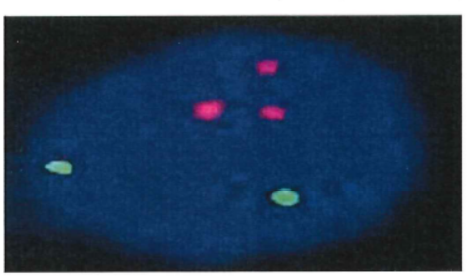
207 As I have also said, at the priority date the triple screening test involved the detection and measurement of three markers that were found to be associated with Down syndrome. The test was generally able to identify or detect 50% of cases of Down syndrome. By the priority date, work had also been carried out to find new markers which could be used at 10-12 weeks’ gestation as there was a desire to enable diagnosis of Down syndrome within the first trimester of pregnancy, leading to the discovery in the early 1990s that free beta hCG could be used in combination with PAPPA to screen reliably for Down syndrome during the first trimester of pregnancy using maternal serum. But such screening tests were also associated with less than 100% detection rates as well as false positives.
(c) NIPD using fetal cells
208 With respect to the use of foetal cells in maternal blood for non-invasive prenatal diagnosis, the Patent explains at page 1, lines 20 to 27 that the method avoids the risks associated with the conventional invasive techniques referred to above but involves time-consuming and expensive fetal cell enrichment and isolation techniques.
209 The idea that fetal cells might be present in the mother’s blood had been first proposed in 1969. The possibility of being able to access whole fetal cells by taking a maternal blood sample was of great interest to those looking for a non-invasive way of obtaining information about the fetus because it would allow analysis of the fetal genome to be carried out without the need for invasive testing and the problems associated with it. Indeed, all that would be needed was a blood sample from the mother’s arm.
210 As a result, a substantial amount of work was carried out in this area through the 1980s and the 1990s. During the course of this research, the cell-free portion of the maternal blood (i.e. serum or plasma) was treated as a laboratory waste product and was therefore routinely discarded, as the focus was on the examination of DNA found in fetal cells.
211 The approaches and techniques that had been used for non-invasive prenatal testing using fetal cells both isolated and in a background of maternal cells before the priority date were described in Simpson JL and Elias S, “Isolating Fetal Cells in Maternal Circulation for Prenatal Diagnosis” (1994) 14(13) Prenatal Diagnosis 1229-1242. That paper notes that prior to the priority date it was known that:
(a) an approach of identifying trisomic pregnancies by isolating and analysing fetal cells from peripheral maternal blood “genuinely seems practical”;
(b) the cell type that had led to the greatest success was the fetal nucleated red cell (erythroblast);
(c) fetal cells in maternal blood were very rare;
(d) the frequency of fetal cells in maternal blood clearly increases with gestation;
(e) analysis of fetal cells in maternal blood had been used to “detect” Mendelian traits such as those relating to beta thalassemia;
(f) PCR might be useful for diagnosis of conditions wherein one can seek a normal allele present in a heterozygous father mated to a mother homozygous for an autosomal recessive trait. For example, the mother might have haemoglobin SS and the father haemoglobin AS. If blood from the homozygous mother showed the normal paternal allele (A) when subjected to PCR, the fetus can be deduced to be heterozygous (AS);
(g) fetal sex could be readily detected using non-invasive fetal cell techniques, as illustrated by various previously reported methods with differing detection rates and methods wherein presumptively isolated fetal cells were subjected to PCR to detect the presence or absence of a Y DNA sequence;
(h) RhD status and aneuploidy could also be detected with varying levels of accuracy; and
(i) a number of clinical and biological questions and problems needed to be answered or resolved in order for the technique to be offered as an alternative to conventional invasive and non-invasive methods of prenatal cytogenetic diagnosis.
212 In spite of the problems associated with non-invasive fetal cell detection methods, research into fetal cells in maternal blood was still ongoing at the priority date, given the significant clinical impact that would be felt if it was possible to overcome the relevant problems to provide a means of allowing analysis of the fetal genome without the need for invasive testing.
(d) The invention described and claimed in the Patent
213 At, page 1, line 28 to page 2, line 2, the Patent states that as at the priority date there had recently been interest in the use of plasma or serum derived DNA for molecular diagnosis and that it had been demonstrated that tumour DNA could be detected by PCR in the plasma and serum of some patients.
214 At page 2, lines 5 to 19 the Patent explains that:
It has now been discovered that foetal DNA is detectable in maternal serum or plasma samples. This is a surprising and unexpected finding; maternal plasma is the very material that is routinely discarded by investigators studying non-invasive prenatal diagnosis using foetal cells in maternal blood. The detection rate is much higher using serum or plasma than using nucleated blood cell DNA extracted from a comparable volume of whole blood, suggesting that there is enrichment of foetal DNA in maternal plasma and serum. In fact, the concentration of foetal DNA in maternal plasma expressed as a % of total DNA has been measured as from 0.39% (the lowest concentration measured in early pregnancy), to as high as 11.4% (in late pregnancy), compared to ratios of generally around 0.001% and up to only 0.025% for cellular fractions (Hamada et al 1993). It is important that foetal DNA is found in maternal plasma as well as serum because this indicates that the DNA is not an artefact of the clotting process.
(Emphasis added.)
215 The inventors named in the Patent, Dr Dennis Lo and Dr James Wainscoat had discovered the presence and utility of cffDNA. The invention described and claimed in the Patent is the practical application of this discovery by a new method of detection of fetal DNA, which unlike previously disclosed cellular methods, involved human mediated discrimination between cell-free maternal and fetal DNA in an artificially prepared plasma or serum sample extracted from a pregnant female.
216 The invention was said to take advantage of a previously unknown or unsuspected property of an artificially produced serum or plasma sample extracted and isolated from a pregnant female. It is an artificial detection method deriving from and applying the inventors’ discovery of the presence of cffDNA in the maternal circulation from both the mother and developing fetus. The inventors observed that the “biological basis by which foetal DNA is liberated into the maternal plasma remain[ed] to be elucidated” (at page 33, lines 1 to 10 of the Patent).
217 The Patent states that the invention “provides a method for prenatal diagnosis” (page 2, line 23) and explains that the term “prenatal diagnosis” as used in the Patent covers (page 2, line 24):
(a) determination of any maternal or fetal condition or characteristic which is related to either the fetal DNA itself or to the quantity or quality of the fetal DNA in the maternal plasma;
(b) sex determination;
(c) detection of fetal abnormalities which may be for example chromosomal aneuploidies or simple mutations, including, for example, RhD status and haemoglobinopathies (page 4, lines 5 to 22 of the Patent); and
(d) detection and monitoring of pregnancy associated conditions such as pre-eclampsia which result in higher or lower than normal amounts of fetal DNA being present in the maternal serum or plasma.
218 At the priority date, prenatal screening tests were commonly described as having a detection rate, being the proportion of affected individuals with positive screening results.
(e) Molecular genetic techniques
219 The artificial derivation and preparation of the relevant plasma and serum samples is described in the Patent as including “a nucleic acid extraction process” and also optionally a “sequence-based enrichment method … to specifically enrich for foetal nucleic acid sequences” (page 3, lines 5 to 23).
220 Enrichment (or conversely depletion) techniques generally involve enriching one population relative to another such that both populations are still detectable. The Patent does not confine such enrichment methods to techniques such as PCR.
221 The Patent explains (page 3, lines 24 to 28) that amplification of fetal DNA sequences in the sample is normally carried out and that “standard” nucleic acid amplification systems can be used including PCR, LCR, nucleic acid sequence based amplification (NASBA), branched DNA methods and so on. However, PCR is stated to be the preferred amplification system.
(f) PCR
222 PCR is a standard molecular biology technique that involves the amplification of specific sequences of DNA using repeat cycles of denaturation, primer annealing and extension, resulting in exponential accumulation of DNA fragments. I have described this earlier.
223 A PCR reaction requires a number of synthetic ingredients, including short single stranded DNA primers designed to bind to the target sequence(s) and no other sequence(s), excess nucleotides, and DNA polymerase, being the enzyme that catalyses the extension of the nucleotide chain.
224 As I have depicted in the earlier section on common general knowledge, DNA synthesised in the first PCR cycle has the 5’ end comprising the synthetic primer sequence (shown in red) and a variable 3’ end (the first blue strands in the figure previously depicted). When the “blue” strands are denatured, the “parental” strand will re-hybridize to one primer, such that a product with a variable 3’ end will continue to be synthesised during subsequent cycles of PCR. However, only one copy of each of the products with a variable 3’ end will accumulate with each PCR cycle.
225 The second PCR cycle first generates discrete (synthetic) products with the 5’ end of one primer and the 3’ end of the other primer. Each strand of this discrete product is complementary to one of the two primers and thus acts as a template in subsequent cycles. It is these synthetic sequences that are the intended products of the PCR reaction and form the basis for any further analysis (e.g. sequencing, gel electrophoresis).
(g) LCR
226 As I have described earlier, LCR typically involves the use of two complementary pairs of single stranded typically unlabelled DNA probes or oligonucleotides, each pair designed to bind to adjacent regions on both the sense and the antisense strands of a target DNA sequence. The junction of the two adjacent oligonucleotides (Oligo Junction) in each pair is usually positioned so that the nucleotide at the end of one of the oligonucleotides coincides with the point mutation. If, and only if, the nucleotide in the oligonucleotide at the Oligo Junction is complementary to the nucleotide at the same position in the target sequence can an enzyme called DNA ligase join the two oligonucleotides. The process can also be used to amplify a region without a single base mutation. If the adjacent oligonucleotides are able to join or ligate, the joined oligonucleotides can then themselves be used as templates for further rounds of ligation mediated amplification, which, like PCR, exponentially amplifies the paired oligonucleotides containing the target DNA sequence. If the adjacent oligonucleotides are not able to join, because the nucleotide at the Oligo Junction is not complementary to the nucleotide at that position in the target sequence or because the target sequences are not present, amplification will not occur.
(h) Branched DNA methods
227 Branched DNA methods involved the use of unlabelled “capture probes” and amplification of the signal through “label probes”. Whilst the label probes contain a sequence complementary to the target sequence, detection of such sequences is achieved indirectly using synthetic sequences that are not the same as, nor complementary to, any genomic or other sequences that were originally contained in the original sample. In such assays, as depicted below, the presence and/or quantity of the target sequence was indirectly inferred or detected by way of the light emitted by a target-probe complex comprising branched DNA amplifier molecules:
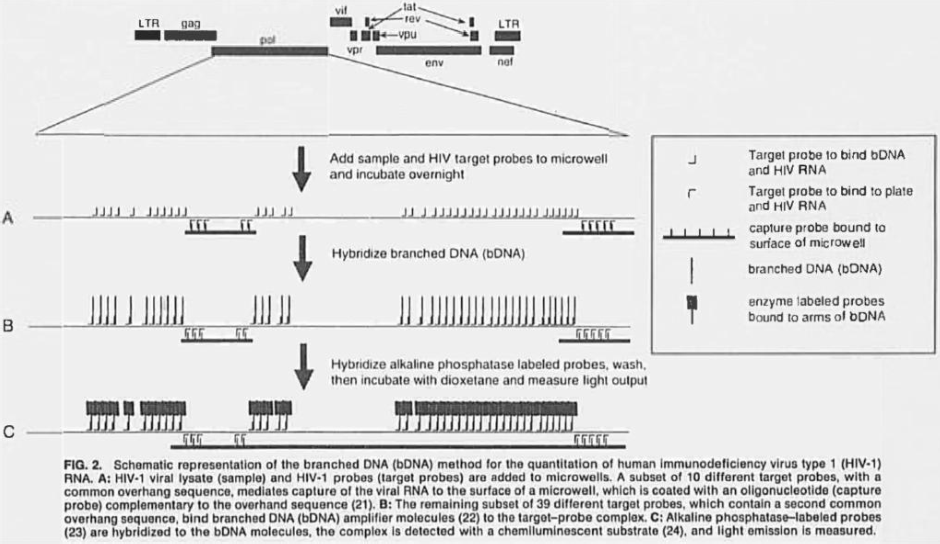
(i) Maternal and fetal conditions and characteristics
228 The Patent states (page 3, line 29 to page 4, line 4) that the method according to the invention may be particularly useful for sex determination. It explains that this application may be carried out by simply detecting the presence of a Y chromosome. In this context, no mention is made of controls for cffDNA or validation or optimisation required for a commercial clinical sex determination test. Rather, the Patent demonstrates that using only 10µL of plasma or serum, a detection rate of 80% for plasma and 70% for serum can be obtained and that this is high enough to be useful (page 9, line 11).
229 The Patent explains (page 4, lines 5 to 8) that “the method of the invention can be applied to the detection of any paternally-inherited sequences which are not possessed by the mother”.
230 Examples of the above application are stated in the Patent to include the following.
231 First, fetal rhesus D status determination in rhesus negative mothers is given as an example. I have already touched upon this to some extent, but let me re-iterate the following.
232 As explained at page 15, lines 9 to 11 of the Patent, the rhesus blood group system is involved in haemolytic disease of the newborn, transfusion reactions and autoimmune haemolytic anaemia.
233 As I have said, the Rh factor (also known as the Rh D antigen) is a protein found on the surface of red blood cells in so-called Rh positive individuals. Rh negative individuals lack this protein. Lack of this protein in Rh negative individuals is caused by a deletion or mutations of the RhD gene that encodes it in both copies of chromosome 1. If one copy of chromosome 1 contains the RhD gene and one does not, the individual still expresses the Rh factor and is considered Rh positive. I refer to my earlier discussion in the common general knowledge section.
234 Second, haemoglobinopathies, by detecting a paternally inherited mutation in the beta-globin gene not possessed by the mother, is given as an example.
235 As I have already said, haemoglobinopathies are genetic disorders in which the haemoglobin molecules in an affected individual’s red blood cells are abnormal. Well-known examples of haemoglobinopathies are sickle cell anaemia and alpha- and beta-thalassemia. Alpha-thalassemia is considered a lethal disease, often leading to fetal death in the third trimester with maternal hydrops (“mirror”) syndrome also commonly present. Sickle cell and beta-thalassemia can be treated but patients suffer from many symptoms and often need life-long repeated blood transfusions. Parents being confronted with a diagnosis of these fetal diseases may elect for termination.
236 Work had been carried out before the priority date to characterise the beta-globin gene and various mutations which were shown to lead to certain blood disorders. The use of PCR for these genes to determine the risk of a fetus being affected by such diseases was well known by the priority date arising from research into fetal cells in maternal circulation.
237 The point mutations occurring in the case of beta-thalassemia may be different between the father and mother. This meant that, using PCR primers, one could distinguish between fetal DNA, which should have paternally inherited markers, and remaining maternal DNA, which would not. As explained in the Patent (page 4, lines 19 to 22):
Provided that the father and mother carry different mutations, the paternal mutation can be used as an amplification target on maternal plasma and serum, so as to assess the risk that the foetus may be affected.
238 Third, reference was made to other paternally-inherited DNA polymorphisms or mutations on a Y or non-Y chromosome.
239 The Patent explains at page 4, line 30 to page 5, line 2 that this particular application will require the prior genotyping of the father and mother using a panel of polymorphic markers and then an allele for detection will be chosen which is present in the father, but is absent in the mother. The Patent explains at page 4, lines 27 to 30 that this type of analysis can be used to ascertain the presence of fetal nucleic acid in a particular maternal or serum sample, prior to diagnostic analysis such as sex determination.
240 Genetic markers commonly known and used at the priority date included polymorphic loci at which base changes were known to affect restriction sites, that is, RFLPs and STRs or microsatellites being sections of DNA made up of repeated short sequences.
241 Let me continue with my discussion of the Patent more generally. At page 5, lines 3 to 6, the Patent explains that the “plasma or serum based non-invasive diagnosis method according to the invention can be applied to screening for Down syndrome and other chromosomal aneuploidies” and gives two possible ways in which this might be done. As I have indicated, aneuploidies in autosomal and sex chromosomes are responsible for a number of genetic conditions, due to abnormal dosage of genes, including Down syndrome (trisomy of chromosome 21), Edwards syndrome (trisomy of chromosome 18), Patau syndrome (trisomy of chromosome 13), Turner syndrome (full or partial monosomy of X), Klinefelter syndrome (XXY), XYY syndrome, XXYY syndrome and Triple X syndrome.
242 Broadly, the first method involves the quantitative detection of fetal nucleic acid to screen women with higher cffDNA levels compared to normal pregnancies and the second method involves “the quantitation of foetal DNA markers on different chromosomes” (emphasis added). In this context, the Patent refers to the recent development of quantitative PCR techniques, such as real-time quantitative PCR (real-time qPCR) described in Heid CA et al, “Real time Quantitative PCR” (1996) 6(10) Genome Research 986-994 (Heid et al 1996).
243 As I have already indicated, quantitative PCR (qPCR) is a broad term that is used to refer to PCR methods which enable the products of a conventional PCR reaction to be quantified. By the priority date, amplification of sequences that were the targets of qPCR could be detected using a number of methods including agarose gels, a form of gel electrophoresis discussed above, fluorescent labelling of PCR products and detection with laser-induced fluorescence using capillary electrophoresis or acrylamide gels and plate capture and sandwich probe hybridisation.
244 As I have already said, another qPCR method available at the priority date was quantitative fluorescent PCR (qfPCR), which was a method which required the products of the PCR reaction to be detected and quantified using a post-reaction “end point” technique, namely, electrophoresis. As a result, qfPCR methods were understood to be more prone to errors in quantification compared to real-time qPCR methods, as the latter quantified the PCR products in the same vessel as that used to amplify the DNA.
245 Heid et al 1996 was received on 3 June 1996 and published later that year. The real-time qPCR method described in Heid et al 1996 measured PCR product accumulation through a hybridisation probe called the “TaqMan Probe”, which is labelled with two different fluorescent dyes. One dye is a “reporter” dye and the other is a “quenching” dye. When the TaqMan Probe is intact, the “quenching” dye “quenches” or absorbs the fluorescence emission of the reporter dye, such that a fluorescence signal is not generated. However, when the TaqMan probe is cut or cleaved during amplification of a target sequence, the reporter dye’s emission is no longer quenched, resulting in a detectable increase of the reporter dye fluorescence emission spectra. In such an assay, the presence and/or quantity of a target nucleic acid sequence is indirectly detected or inferred from the emission spectra levels detected by an instrument capable of measuring fluorescence in real time generated by a fluorophore that is no longer attached to the target sequence (e.g. ABI Prism 7700 Sequence Detector) (see, for example, the Patent at page 11, lines 8 to 13).
246 The background to Heid et al 1996 explains that:
Several detection systems are used for quantitative PCR and RT-PCR analysis: (1) agarose gels, (2) fluorescent labelling of PCR products and detection with laser-induced fluorescence using capillary electrophoresis (Fasco et al. 1995; Williams et al. 1996) or acrylamide gels, and (3) plate capture and sandwich probe hybridization (Mulder et al. 1994).
247 Unlike other quantitative PCR methods, real-time qPCR does not require post PCR handling, preventing potential PCR product carry-over contamination and resulting in much faster and higher throughput assays.
248 The concentration of DNA in a real-time qPCR reaction sample is determined by calculating the threshold cycle value (CT) for a sample, which is the PCR cycle number at which the concentration of DNA in a reaction sample (typically measured by the amount of fluorescence) reaches a threshold, which is placed in the exponential phase of amplification. CT values are calculated from amplification plots, such as the plot depicted below. As a consequence of the exponential nature of PCR, one cycle represents approximately a doubling in template concentration. At the priority date, qPCR was suitable for detecting a 2-fold difference.
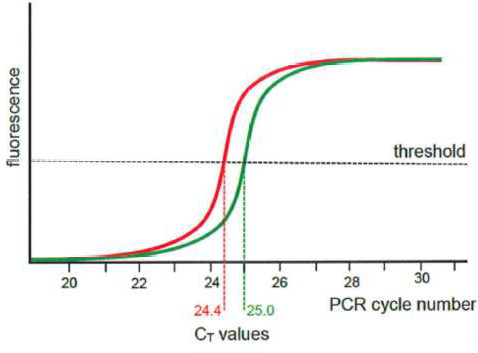
249 The Patent describes another application (page 5, line 27 to page 6, line 2) of the accurate quantitation of fetal nucleic acid levels in the maternal serum or plasma, namely, the molecular monitoring of placental pathologies such as pre-eclampsia, in which the concentration of fetal DNA is said to be elevated.
250 As I have indicated, pre-eclampsia is a pregnancy disorder which affects about 6% of pregnancies, and is characterised by high blood pressure and elevated protein levels in the maternal urine. Pre-eclampsia generally occurs 24 to 26 weeks after fertilisation and often increases in severity until birth. Untreated, pre-eclampsia may lead to eclampsia (convulsions), bleeding in the mother’s brain and death of the mother. Especially the early forms of pre-eclampsia are often associated with fetal growth restriction due to placental dysfunction.
251 The Patent says (page 6, lines 3 to 6) that it is also anticipated that it will be possible to incorporate the nucleic acid-based diagnosis methods described in the Patent into existing prenatal screening programmes and that “[s]ex determination has successfully been performed on pregnancies from 7 to 40 weeks of gestation”.
252 From page 6, line 20 to page 33, the Patent illustrates the invention with reference to five Examples, but “which do not in any way limit the scope of the invention”.
(j) Example 1 – Analysis of fetal DNA for sex determination
253 Example 1 concerns the analysis of cffDNA for sex determination and describes a project wherein 5 to 10ml of maternal peripheral blood was collected from 43 pregnant women with gestational ages from 12 to 40 weeks (at page 9, lines 1 to 2). Some samples were collected from pregnant women prior to amniocentesis and others from pregnant women recruited just before delivery. Control samples were also taken from 10 non-pregnant female subjects. The extracted blood was processed as follows (page 7 of the Patent):
Sample preparation
Maternal blood samples were processed between 1 to 3 hours following venesection. Blood samples were centrifuged at 3000g and plasma and serum were carefully removed from the EDTA-containing and plain tubes, respectively, and transferred into plain polypropylene tubes. Great care was taken to ensure that the buffy coat or the blood clot was undisturbed when plasma or serum samples, respectively, were removed. Following removal of the plasma samples, the red cell pellet and buffy coat were saved for DNA extraction using a Nucleon DNA extraction kit (Scotlabs, Strathclyde, Scotland, U.K.) The plasma and serum samples were then subjected to a second centrifugation at 3000g and the recentrifuged plasma and serum samples were collected into fresh polypropylene tubes. The samples were stored at -20°C until further processing.
DNA extraction from plasma and serum samples
Plasma and serum samples were processed for PCR using a modification of the method of Emanuel and Pestka (1993). In brief, 200 µl of plasma or serum was put into a 0.5ml eppendorf tube. The sample was then heated at 99°C for 5 minutes on a heat block. The heated sample was then centrifuged at maximum speed using a microcentrifuge. The clear supernatant was then collected and 10 µl was used for PCR.
254 At page 8, the Patent explains that the detection of a Y-specific fetal sequence from maternal plasma and serum was carried out using primers designed to amplify a single copy Y sequence designated DYS14. Serum and plasma were subjected to sixty cycles of PCR and the PCR products were analysed by agarose gel electrophoresis and ethidium bromide staining.
255 Gel electrophoresis, which I have referred to earlier, was used to separate molecules like DNA based on their size (molecular weight). Let me re-iterate some aspects and then expand on what I have previously said. When carried out following PCR or simple restriction enzyme digestion, the reaction products would be mixed with a suitable buffer containing a dye to increase the density of the material in the sample so that it could be seen during loading into a well on a gel made of agarose or polyacrylamide. An electric field would then be applied to the gel, which would cause the DNA, which is negatively charged, to migrate through the gel. The gel acts like a sieve, with smaller DNA molecules moving through it more quickly than larger DNA molecules. The gel would then be stained, typically with ethidium bromide, which binds to DNA, and then visualised with UV light. The stained molecules travel a particular distance from the starting point corresponding to the size of the molecule. Accordingly, different PCR amplification products or fragments generated by restriction endonucleases will form separate bands on the gel.
256 The bands appearing on a gel may be further analysed by “southern blotting”, such that the bands of interest are transferred from the gel to a solid membrane and the membrane is then probed with a specific detector molecule that can be used to visualise its target. This process can be depicted as follows:
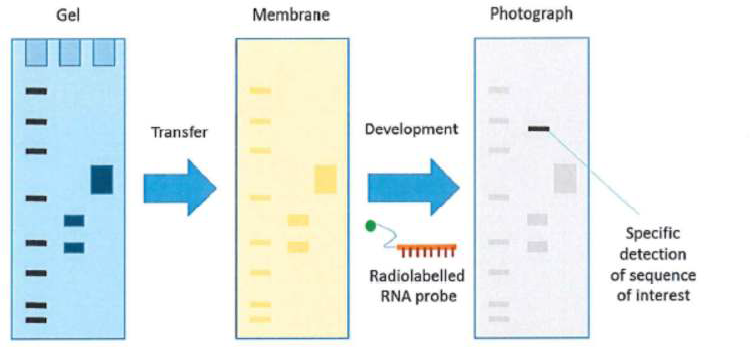
257 At page 8, lines 25 to 29, the Patent explains that PCR was able to be used to detect Y sequences in up to a 100,000 fold dilution of male genomic DNA to female genomic DNA; this is approximately equivalent to detecting Y sequences in a single male cell amidst maternal DNA.
258 Of the 30 women bearing male fetuses, Y-positive signals were detected in 24 plasma samples and 21 serum samples when 10 µl of the respective samples were used for PCR, compared to 5 out of 30 for nucleated blood cell DNA samples. None of the 13 women bearing female fetuses resulted in a positive Y signal. On the basis of these results, the Patent explained that the accuracy of the technique was high enough to be useful and could be improved further to 100% or close to 100% by, for example, using a larger volume of serum or plasma.
(k) Example 2 – Quantitative analysis of fetal DNA in maternal serum in aneuploid pregnancies
259 The background to Example 2 refers inter-alia to Bianchi DW et al, “PCR quantitation of fetal cells in maternal blood in normal and aneuploid pregnancies” (1997) 61(4) American Journal of Human Genetics 822-829, which it says demonstrated that there is increased fetal nucleated cell number in maternal circulation when the fetus is suffering from a chromosomal aneuploidy.
260 In this example, the inventors used 400 to 800µL of plasma/serum samples derived from the blood of pregnant women, extracted DNA from those samples and subjected the extracted DNA to real-time qPCR using the methods described in Heid et al 1996 (discussed above) and an SRY TaqMan system consisting of amplification primers for the SRY region on the Y chromosome and a dual-labelled fluorescent TaqMan probe. The primer/probe combinations were designed using the Primer Express software and Sequence data for the SRY gene were obtained from the GenBank Sequence database (page 11, lines 19 to 21 of the Patent).
261 Blood samples from pregnant women undergoing prenatal testing were collected prior to any invasive procedure. The fetal karyotype was confirmed by cytogenic analysis of amniotic fluid or chorionic villus samples. One of the experts before me, Professor Fisk, an expert called by Sequenom, infers from these facts that such women were in their first or early second trimester gestation. Professor Hyett, an expert called by Ariosa, infers that the samples could have been taken from 11 weeks to 24 weeks gestation.
262 5 to 10µL of extracted serum DNA was used for amplification (page 12, line 2 of the Patent). The formula used by the inventors to calculate the concentration of the target sequences “expressed in copies per ml” is described at page 12, lines 19 to 29 of the Patent.
263 The inventors also guarded against contamination by using aerosol-resistant pipette tips and separate preparation areas for different reactions, being techniques commonly applied to reduce the risk of contamination. The Patent explains at page 13, lines 7 to 10 that the 7700 Sequence Detector offered an extra level of protection by obviating the need to reopen the reaction tubes following the completion of the amplification reactions, thus minimising the possibility of carryover contamination. In addition, the TaqMan assay also included a further level of anti-contamination measure in the form of pre-amplification treatment using uracil N-glycosylase, which destroyed uracil containing PCR products (i.e. RNA).
264 Apparently, the real-time qPCR method of Example 2 was sensitive enough to detect the DNA equivalent from a single target cell in a mixture or at the 1 in 107 level.
265 The results of Example 2 demonstrated that fetal DNA concentration was statistically significantly higher in aneuploid than control (i.e. normal) pregnancies. Although there was no statement that the control pregnancies and aneuploid pregnancies were matched for gestational age, as there was in Example 4 of the Patent, the null hypothesis being that the concentration of fetal DNA is the same between the two groups, was rejected. Further, the general concentrations in the two comparator groups were different, indicating to Professor Fisk and Professor Lovett, another expert called by Sequenom, that at the priority date fetal DNA quantitation had the potential to be used as a new screening marker for fetal chromosomal aneuploidies.
(l) Example 3 – Non-invasive prenatal determination of fetal RhD status from plasma of RhD negative pregnant women
266 The method of Example 3 builds on the demonstration in 1991 that RhD negative individuals lack the RhD gene and the cloning of the human RhD gene in 1992. As noted at page 15, lines 23 to 25 of the Patent, prenatal determination of fetal RhD status had been performed using PCR based techniques on amniotic fluid samples by Bennett PR et al “Prenatal determination of fetal RhD type by DNA amplification” (1993) 329(9) New England Journal of Medicine 607-610 (Bennett et al 1993).
267 Bennett et al 1993 employed a PCR assay depicted in the figure below to detect the RhD gene in fetal DNA extracted from fetal cells, noting that the presence or absence of the RhD gene in the genome determines the genetic basis of the polymorphisms associated with Rh positivity and Rh negativity.
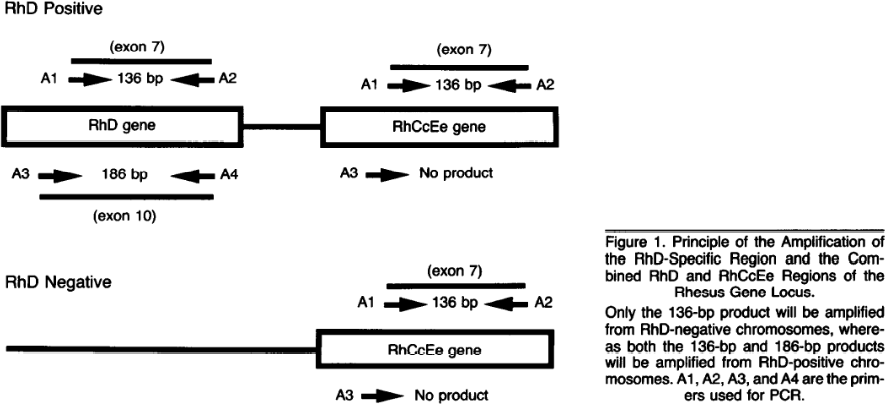
268 The authors of Bennett 1993 concluded that:
Although more studies are needed to confirm the high sensitivity and specificity of this method, the ability to determine the Rh status of the fetus early in pregnancy without invading the fetomaternal circulation should represent a major advance in the management of Rh alloimmunization. This technique may also be appropriate for the diagnosis of RhD status in embryos before implantation and in fetal cells in the maternal circulation.
(Citations omitted).
269 The Patent also refers to Lo Y et al, “Prenatal determination of fetal RhD status by analysis of peripheral maternal blood of rhesus negative mothers” 341 Lancet 1147-1148 (Lo et al 1993), where the possibility of using fetal cells in maternal blood for the determination of fetal RhD status was investigated. By using nested PCR, Lo et al 1993 were able to detect 8 out of 10 RhD positive fetuses. Three false positive results were also recorded within the 21 test cases. The authors noted that the system had not reached the precision required for routine clinical use and that the diagnostic accuracy could be further improved by combining the technique with fetal cell enrichment.
270 Nested PCR, which I have referred to earlier, is a technique used to improve the specificity of the PCR reaction so that ideally only target sequences are amplified. It is particularly useful for detecting low levels of target sequence in a given DNA sample. In this method, the DNA in the sample is first amplified using “outer” primers which bind to the target sequence and possibly other non-target sequences in the sample. The sequences or “amplicons” amplified in this first round of PCR are subjected to a further PCR reaction involving one (hemi-nested) or two different “inner” primers having sequences which bind to sequences within the target amplicon.
271 The inventors noted at page 15, lines 28 to 30 that the main problem with the fetal cell RhD determination system was that it was not sufficiently reliable without fetal cell enrichment or isolation procedures, which were tedious and expensive to perform.
272 In the project the subject of Example 3, blood samples were taken from 21 serologically RhD negative pregnant women, including some who were in the second trimester of pregnancy just prior to amniocentesis and others who were in the third trimester just before delivery (page 16, lines 13 to 17). No blood or DNA samples were taken from fathers. 800µL of plasma was used for cffDNA analysis (page 17, line 5), which was performed as described in Example 2 with modifications relating to the primer/probe sets. The Example 3 method also included a non-fetal specific beta-globin TaqMan system, which acted as a control for the presence and amplifiability of cell free DNA in the plasma samples (page 19, lines 1 to 3).
273 A RhD positive result was inferred for an amplification reaction in which the fluorescence intensity rose above a predetermined threshold value recommended by Heid et al 1996 of 10 standard deviations above the mean base-line fluorescence from cycles 1 to 15 (page 18, lines 1 to 10). The absence of a signal from a sample indicated the absence of an RhD gene and therefore the presence of the RhD negative phenotype. Other than the beta-globin TaqMan system, the Patent does not at this point discuss controls for the presence of cffDNA.
274 Example 3 demonstrated complete correlation between the fetal RhD genotype predicted from the maternal plasma real-time qPCR analysis and the result obtained from genotyping the amniotic fluid and serological testing of the cord blood, as depicted in the table below:

275 With respect to Example 3, the inventors concluded that the study:
… demonstrated the feasibility of performing non-invasive foetal RhD genotyping from maternal plasma. This represents the first description of single gene diagnosis from maternal plasma. Our results indicate that this form of genotyping is highly accurate and can potentially be used for clinical diagnosis. This high accuracy is probably the result of the high concentration of foetal DNA in maternal plasma.
276 The inventors also qualified the findings of Example 3 by noting that “[i]t is likely that for robust clinical diagnosis, multiple [RhD] primer sets are preferred. The TaqMan chemistry can easily accommodate the inclusion of multiple primer/probe sets”.
(m) Example 4 – Elevation of fetal DNA concentration in maternal serum in pre-eclamptic pregnancies
277 In this Example, the inventors used a real-time qPCR assay to show that the concentration of fetal DNA in the serum of women suffering from pre-eclampsia was elevated compared to normal pregnancies. As with other Examples, Y chromosomal sequences from male fetuses were used as a fetal marker and a proxy for the total level (concentration) of fetal DNA.
278 In the study, 400 to 800µL of serum/plasma samples were taken from women with pre-eclampsia and normal or “control” pregnancies. The gestation age of the pre-eclamptic and control subjects were matched (page 21, lines 21 to 22). Professor Hyett gave evidence that pre-eclampsia is typically diagnosed after 20 weeks gestation, and that 70% of cases of pre-eclampsia occur after 36 or 37 weeks gestation. Pre-eclampsia was defined as recorded in the Patent at page 21, lines 16 to 18 as a sustained rise in diastolic blood pressure to 90 mmHg or higher from previously lower values, with new and sustained proteinuria in the absence of urinary tract infection.
279 Real-time qPCR was performed as described in Example 2. The inventors of the Patent found that fetal DNA concentration was higher in pre-eclamptic than control pregnancies (p < 0.0001), indicating that fetal DNA concentration measurement in maternal plasma may be used as a new marker for pre-eclampsia (page 22, lines 20 to 24) and “open[ing] up research into the potential application of fetal DNA quantitation to predict the occurrence of pre-eclampsia, prior to the development of clinical signs such as hypertension and proteinuria” (page 23, lines 1 to 3).
(n) Example 5 – Quantitative analysis of fetal DNA in maternal plasma and serum
280 The introduction to Example 5 notes that to demonstrate that clinical applications for the non-invasive prenatal diagnosis of certain genetic disorders using cffDNA are possible, a number of important issues need to be resolved (page 23, lines 24 to 30). First, fetal DNA in maternal plasma and serum needs to be shown to be present in sufficient quantities for reliable molecular diagnosis to be carried out. Second, data on the variation of fetal DNA in maternal plasma and serum with regard to gestation age is required to determine the applicability of this technology to early prenatal diagnosis.
281 The genetic disorders being referred to are disorders caused by changes to the genome, such as deletions, duplications, rearrangements and chemical modifications affecting anything from a single base to a whole chromosome or even the whole genome. Those caused by a defect in only one gene (for example cystic fibrosis) are classed as single-gene disorders.
282 Example 5 addressed both of these issues using a real-time qPCR assay for measuring the copy numbers of fetal DNA molecules in maternal plasma and serum.
283 In particular, DNA was extracted from 400 to 800µL of plasma/serum and real-time qPCR analysis was performed on 40 to 80µL (page 30, lines 23 to 25) of extracted DNA as described in Example 2, using the SRY TaqMan system and the beta-globin TaqMan system described in the previous Examples (page 25, lines 20 to 23).
284 At page 27, lines 4 to 7, the Patent explains that “the concentration of beta-globin sequences in maternal plasma and serum samples was used as a measure of the total amount of extracted DNA, i.e. maternal and foetal DNA extracted from plasma and serum samples” (emphasis in original).
285 By examining the level of beta-globin sequences, the inventors concluded that:
(a) serum contained more DNA than plasma, with a mean concentration of serum DNA 14.6 times that of plasma DNA; and
(b) the total amount of plasma DNA increases as pregnancy progresses.
286 To determine the amount of fetal DNA, real-time qPCR analysis using the SRY TaqMan system was carried out (page 27, lines 23 to 25). The assay produced a positive SRY signal in each of the 27 test subjects bearing male fetuses and no signal was detected in each of the 23 test subjects bearing female fetuses (page 27, line 27), even in the women who had previously carried a male baby. Accordingly, the Patent appears to confirm its earlier statement at page 9, lines 12 to 13, namely, that accuracy of fetal sex determination can be improved by using greater sampling volume.
287 The quantitative SRY data from the 27 women bearing male subjects showed that (page 28, lines 5 to 17):
(a) the concentrations of fetal DNA in maternal plasma and serum are higher in late gestation than in early gestation;
(b) the mean concentrations of fetal DNA in maternal plasma and serum are 11.5 times and 11.9 times, respectively, higher in late gestation compared with early gestation;
(c) the absolute concentrations of fetal DNA in maternal serum and plasma were similar in individual cases;
(d) the fractional concentration of fetal DNA in early pregnancy ranges from 0.39% to 11.9% in plasma and 0.014 to 0.54% in serum; and
(e) the fractional concentration of fetal DNA in late pregnancy ranges from 2.33 to 11.4% in plasma and 0.032 to 3.97% in serum.
288 At page 29, lines 13 to 26, the Patent explains:
The most important observation in this study is the very high concentration of foetal DNA in maternal plasma and serum. Bianchi et al reported that the average number of foetal cells in maternal blood in normal pregnancies was 19 in 16 ml of maternal blood, i.e., 1.2 cells/ml during the second trimester (Bianchi et al. 1997). Therefore, the mean concentration of foetal DNA in maternal plasma and serum is 21.2 (25.4/1.2) and 23.9 (28.7/1.2) times, respectively, higher than that in the cellular fraction of maternal blood at the same gestation. The relative concentration of foetal to total plasma DNA is even higher. Thus, in early pregnancy, foetal DNA in maternal plasma constitutes a mean of 3.4% of the total plasma DNA. The respective figure in late pregnancy is 6.2%. Hamada et al reported that the frequency of foetal cells in the second trimester was 0.0035% while that in the third trimester was 0.008% (Hamada et al. 1993).
(Emphasis added.)
289 At page 31, line 27 to page 32, line 1, the Patent states that:
We envisage that foetal DNA analysis in maternal plasma and serum would be most useful in situations where the determination of foetal-derived paternally-inherited polymorphisms/mutations or genes would be helpful in clinical prenatal diagnosis.
(Emphasis added, citation omitted.)
290 The inventors also noted that the method also provides a new screening test for fetal chromosomal disorders, but with fetal cell isolation techniques still being necessary for a definitive cytogenetic diagnosis. And that for such an application, fetal DNA quantitation systems can be developed for polymorphic markers outside the Y chromosome so that quantitation can be applied to female fetuses (page 32, lines 16 to 24).
291 In this context, the Patent refers to Lo YM et al, “Two-way cell traffic between mother and fetus: biologic and clinical implications” (1996) 88(11) Blood 4390-4395. Lo et al 1996 describes a study which involved the use of PCR to detect fetal cells in maternal blood and maternal cells in fetal blood using an array of polymorphic systems from non-Y chromosomes.
292 The inventors of the Patent concluded by noting that it was “likely that foetal cell isolation and analysis of foetal DNA in maternal plasma/serum would be used as complementary techniques for non-invasive prenatal diagnosis” (page 32, lines 28 to 30).
(o) The Relevant Claims
293 Sequenom alleges that the Harmony Test used by the respondents falls within the scope of and therefore infringes claims 1, 2, 3, 5, 6, 9, 13, 14, 22, 23, 25 and 26 of the Patent (relevant claims).
294 Claim 1 provides:
A detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of foetal origin in the sample.
295 The Patent stipulates at page 38 that:
Comprises/comprising when used in this specification is taken to specify the presence of stated features, integers, steps or components but does not preclude the presence or addition of one or more other features, integers, steps, components or groups thereof.
296 Claim 2 is the method according to claim 1, “comprising amplifying the foetal nucleic acid to enable detection”.
297 Claim 3 is the method according to claim 2, “wherein the foetal nucleic acid is amplified by the polymerase chain reaction”.
298 Claim 5 is relevantly the method according to any one of claims 1 to 3, “wherein the foetal nucleic acid is detected by means of a sequence specific probe”.
299 Claim 6 is relevantly the method according to any one of the above claims, “wherein the presence of a foetal nucleic acid sequence from the Y chromosome is detected”.
300 Claim 9 is the method according to any one of claims 1 to 5, “wherein the presence of a foetal nucleic acid from a paternally-inherited non-Y chromosome is detected”.
301 Claim 13 is relevantly the method according to claim 6, “for determining the sex of the foetus”.
302 Claim 14 is relevantly the method according to claims 6 or 9, which comprises “determining the concentration of the foetal nucleic acid sequence in the maternal serum or plasma.”
303 Independent claim 22 is “a method of performing a prenatal diagnosis, which method comprises the steps of”:
(i) providing a maternal blood sample;
(ii) separating the sample into a cellular and a non-cellular fraction;
(iii) detecting the presence of a nucleic acid of foetal origin in the non-cellular fraction according to the method of any one of claims 1 to 21;
(iv) providing a diagnosis based on the presence and/or quantity and/or sequence of the foetal nucleic acid.
304 Claim 23 is the method according to claim 22, “wherein the non-cellular fraction as used in step (iii) is a plasma fraction”.
305 Independent claim 25 is:
A method of performing a prenatal diagnosis on a maternal blood sample, which method comprises removing all or substantially all nucleated and anucleated cell populations from the blood sample and subjecting the remaining fluid to a test for foetal nucleic acid indicative of a maternal or foetal condition or characteristic.
306 Independent claim 26 is:
A method of performing a prenatal diagnosis on a maternal blood sample, which method comprises obtaining a non-cellular fraction of the blood sample and performing nucleic acid analysis on the fraction.
307 I would note here that no ground of invalidity has been raised by Ariosa concerning a lack of clarity.
THE EXPERTS
308 Before proceeding further into the detail concerning the respective parties’ cases on invalidity and infringement, it is useful to say something about the experts called by the parties and their backgrounds.
(a) Sequenom’s experts
309 Professors Nicholas Fisk and Michael Lovett were both highly knowledgeable in their areas. Both are and were at the priority date very well qualified and credentialled with extensive experience and involvement in their respective fields of fetal medicine and human molecular genetics.
Professor Fisk
310 Professor Fisk obtained a Bachelor of Medicine, Bachelor of Surgery from the University of Sydney in 1980 and was awarded a PhD from University College London in 1992. In 2008, he was awarded a Master of Business Administration from Imperial College London. From 2008, Professor Fisk held management positions at the University of Queensland and since 2016 he has held a management position the University of New South Wales, being at the time of trial the Deputy Vice Chancellor (Research) at the University of New South Wales. Between 1992 and 2007, Professor Fisk was Professor of Obstetrics and Fetal Medicine at Imperial College & Queen Charlotte’s Hospital, London, where his laboratory and clinical research program achieved an international reputation for fetal diagnosis and treatment. During this period, Professor Fisk’s main research interests included human fetal mesenchymal stem cell biology, monochorionic multiple pregnancy, non-invasive prenatal diagnosis, fetal nociception, caesarean section, preterm labour, obstetric ultrasound and drug development in obstetrics. In connection with his research and work interests, Professor Fisk and his colleagues at Imperial College were awarded a number of research grants and published several papers in the scientific literature. At present, Professor Fisk’s h-index (reflecting both the number of publications and number of citations per publication during his career) is 80, with his peer-reviewed works being cited over 22,000 times.
311 Before and at the priority date, Professor Fisk was also on the editorial boards of the Journal of Obstetrics & Gynaecology (from 1992 to 2001), Fetal Diagnosis and Therapy (from 1995 to 2015) and the Journal of Maternal-Fetal Medicine (from 1995 to 2001).
312 At the priority date, Professor Fisk had training and experience in each of the areas of maternal-fetal biology, clinical practice, reproductive molecular biology and genetics including as part of his work within the Royal Postgraduate Medical School in London and Queen Charlotte’s and Chelsea Hospital. He also had experience leading and supervising a multidisciplinary collaborative team wherein certain work (e.g. routine analytical and biotechnology steps such as PCR) were delegated to technicians.
313 Now I accept that Professor Fisk’s experience involved leading and supervising multidisciplinary collaborative teams, where certain work such as routine analytical and biotechnology steps such as PCR were delegated to technicians, largely for efficiency. His affidavit stated that at the priority date, he was generally aware of molecular genetic techniques available and the scientific principles behind their application but he did not have substantial personal experience with all of the resources and techniques that were available to skilled molecular geneticists. I have kept this in mind when considering and weighing his evidence in relation to performing some molecular genetics analysis techniques. But in any event his evidence in the field of fetal medicine was of considerable assistance to me including his capacity to synthesise and simplify concepts to assist my comprehension.
Professor Lovett
314 Professor Lovett is currently the Professor and Chair in Systems Biology at the National Heart and Lung Institute at Imperial College London. He has 30 years’ experience in mammalian molecular genetics and genomics. He received a B.Sc. with Honors in Molecular Biology from the University of Edinburgh in 1977 and a PhD in Biochemistry at the Eukaryotic Molecular Genetics Group, Imperial College of Science and Technology, University of London in 1981. At the priority date, he was an associate professor of biochemistry at the University of Texas Southwestern Medical Centre in Dallas, Texas.
315 Groups led by him have developed technologies such as direct complementary deoxyribonucleic acid (cDNA) selection and targeted sequence capture, which have found wide applications in human genetics and various genome projects. Between 1999 and 2013, he was Professor of Genetics and Human Genetics Division Head at the Washington University School of Medicine in St Louis, Missouri, which at the time had one of the world’s five primary Genome centres, which were heavily involved in the Human Genome Project and responsible for sequencing the majority of the human genome. Professor Lovett is the author or co-author of over 100 scientific publications relating to mammalian molecular genetics and genomics.
316 In 1997, Professor Lovett’s research was focused on the positional cloning of genes involved in human disease using techniques such as FISH, expressed sequence tag (EST) analysis, direct cDNA selection, gel electrophoresis and blotting analysis to identify genes and map cDNAs. His particular focus was on the genetics involved in processes of hearing loss and the development of craniofacial abnormalities such as cleft lip and palette. Professor Lovett’s group was involved in numerous collaborations with groups working in other diverse fields of human disease genetics.
317 Professor Lovett demonstrated considerable knowledge of all areas covered in the expert concurrent evidence sessions. There was no doubt that Professor Lovett’s extensive practical knowledge and experience of molecular genetic techniques (both pre and post priority date) generally and in using PCR both qualitatively and quantitatively was extensive.
318 Now whilst I accept that Professor Lovett had not been involved in conducting prenatal genetic tests on samples from pregnant women, and did not have any involvement in work done on trying to develop non-invasive prenatal tests using fetal cells or cell-free fetal DNA, he had a thorough knowledge of all the available mammalian molecular genetics techniques and methods as at the priority date. Professor Lovett also had an interest in Down syndrome and prenatal testing that started whilst he was a post-doctoral fellow at the University of California. More generally, Professor Lovett is a pre-eminent geneticist who was very familiar with the genetic principles and tests of relevance to the invention claimed in the Patent and the Kazakov paper, which I will discuss later.
319 Now Ariosa contended that with the exception of a five-year period between 1987 and 1992 when he worked for Genelabs, Professor Lovett’s career has been entirely in academia. He did not have any direct experience in prenatal testing, and had not provided services for clinical diagnosis / screening. Further, Ariosa said that both as at the priority date and since, Professor Lovett has not been directly involved in any research in the field of prenatal genetics. Indeed, Professor Lovett agreed that prenatal genetics was not his specific field, although he said it was a strong interest, and that his field was human genetics in general.
320 Further, Ariosa says that to the extent that Professor Lovett now has knowledge in relation to the field of prenatal diagnosis and screening, this is knowledge learned well after the priority date from literature rather than direct, personal experience. Further, it points out that Professor Lovett also acknowledged that it would be a clinician who would also have greater knowledge of developments in the field of NIPD and problems or issues with existing products or processes used in that field than someone with his background.
321 I accept the force of Ariosa’s reservations, but they should not be over-stated. On my appraisal of the witness during the expert concurrent evidence sessions, Professor Lovett gave reliable and very cogent evidence with considerable force and expertise in the key contentious areas.
(b) Ariosa’s experts
322 Ariosa called three expert witnesses: Ms Norbury, Professor Hyett and Professor Oepkes.
Ms Norbury
323 Ms Christine Norbury is a Consultant Clinical Scientist in genetics, within the Regional Genetics Laboratories at Guy’s & St Thomas’ Hospital, London. She was awarded a BA (Hons) in Biochemistry from the University of Oxford in 1982 and a Master of Science in Clinical Biochemistry from the University of Surrey in 1984. She has been a registered Clinical Scientist since 1985 and a senior registered Clinical Scientist since 1991. As Ms Norbury explained, only a senior registered Clinical Scientist is permitted to authorise reports providing an interpretation of the results of molecular genetic analysis to clinicians. She obtained a diploma in Clinical Cytogenetics and Molecular Genetics from the Royal College of Pathologists in 1990, and has also gained fellowship of the Royal College of Pathologists.
324 Between 1987 and 2001, Ms Norbury was a Clinical Scientist within the Oxford Medical Genetics Laboratory at the Churchill Hospital, Oxford Radcliffe NHS Trust, ultimately becoming the Deputy Head of Laboratory. She was responsible for laboratory services for diagnosis or risk assessment of a range of genetic disorders. At the priority date, Ms Norbury had extensive experience in the design, implementation and performance of genetic testing for a range of genetic disorders. As Ms Norbury has explained in her affidavit evidence, at the time she joined this laboratory, molecular genetics was a very new field, the laboratory was just being established, and she was one of only two permanent staff. However, this had grown to a team of approximately 20 people by the time of her departure in 2001, illustrating the rapid growth in this area. Her role at this laboratory included “devising protocols to diagnose disorders, carrying out the necessary lab work, interpreting the results, preparing reports detailing my conclusions and authorizing the reports of the other staff”. Her role therefore included not only implementing established protocols, but also developing new protocols to enable the Oxford laboratory to provide diagnosis of conditions not previously offered. Developing new protocols required Ms Norbury “to work out, amongst other things, the tissue type to be used for the analysis, the DNA extraction process, the conditions of the PCR reaction…the approach to analysis of results” and “identifying or designing primers for use in the PCR reaction”.
325 Ms Norbury described the Oxford Medical Genetics Laboratory as “well equipped”, and in about 1996, her team first had use of a real-time PCR machine, a Roche Light Cycler. Ms Norbury describes that this was not a particularly robust piece of equipment (according to Ms Norbury the glass capillaries would often break), but had the advantage of being a closed system, reducing the risk of contamination. So, as at the priority date, Ms Norbury’s laboratory was therefore using this equipment for qualitative, rather than quantitative analysis.
326 In addition, Ms Norbury explained that whilst each of the Regional Genetic Diagnostic laboratories provided similar core services, a number of the leading molecular laboratories (which included Oxford) provided certain specialised services, and these rare or specialist interests required those laboratories to work closely with clinical and academic colleagues. During this time, Ms Norbury’s Oxford laboratory was affiliated with and collaborated with academics at Oxford University, and Ms Norbury interacted with various resident and visiting academics from around the world. Ms Norbury also explained that she and Professor Lo were in fact based at the same hospital at this time, and there was “an integrated collaboration between the laboratory – the academic and the clinical service”, so as to “facilitate this translation of academic research and clinical service delivery”. Ms Norbury also recalls attending a meeting with Professor Lo and another Oxford scientist, Ian Sargent in 1996 (in the course of working with them on a project relating to the sensitivity and feasibility of pre-implantation diagnosis of cystic fibrosis) where the scientists explained to her the work they were then undertaking relating to identifying fetal cells in maternal blood using cell specific markers.
327 After the priority date, Ms Norbury worked in a laboratory that developed a non-invasive prenatal test for sex determination based on Professor Lo’s discovery.
328 Whilst Ms Norbury was a highly talented expert in her field, her evidence before me was not completely free of problematic aspects in relation to some aspects of infringement where she seemed on occasion to adopt positions at the margin from which she had to retreat. But nevertheless I found her evidence generally to be of great assistance including the debate between her and Professor Lovett in the expert concurrent evidence sessions which fully exposed the issues that I needed to consider on qPCR techniques.
Professor Hyett
329 Professor Jonathan Hyett is an obstetrician and gynaecologist, with expertise in fetal medicine. Since 2008, he has been a Senior Staff Specialist in Obstetrics and Gynaecology and from 2010 Head of Department of High Risk Obstetrics at the Royal Prince Alfred Hospital in Sydney. This is a combined clinical and research role. Since 2009, he has also been a Clinical Professor in the Discipline of Obstetrics, Gynaecology and Neonatology at the Faculty of Medicine, University of Sydney. This is an honorary appointment, in recognition of the high quality of his academic research.
330 Professor Hyett was awarded a Bachelor of Medicine / Bachelor of Surgery from King’s College, University of London in 1991 and completed his specialist training in obstetrics and gynaecology with subspecialty training in maternal and fetal medicine between 1996 and 2003. He was awarded membership of the Royal College of Obstetricians and Gynaecologists in the UK in 1997. Professor Hyett was a lecturer in the Academic Department of Obstetrics and Gynaecology at University College London between 2000 and 2003.
331 Before the priority date (1993 to 1995), Professor Hyett had undertaken a research fellowship at King’s College, London, during which he worked on various projects relating to prenatal diagnosis. He described this fellowship as involving a strong research-based culture. His projects during this time included a project relating to determining the level of risk of the fetus having a chromosomal aneuploidy based on ultrasound imaging and other factors, as well as other projects relating to the pathophysiology of nuchal translucency which involved molecular genetics techniques and analysis. Professor Hyett also assisted with research projects being undertaken by other researchers, both within and external to King’s College. This included assisting a group based in Switzerland who were looking at the potential role of fetal cells in prenatal diagnosis, and examining whether trophoblast cells from the cervix may provide a viable, alternative source of fetal cells for analysis using FISH.
332 Between 1995 and 2000, whilst completing his specialist training, Professor Hyett undertook a number of rotations at different hospitals in the UK. Between 1995 and 1996, this included Queen Charlotte’s and Chelsea Hospital in London, where he worked with Professor Phillip Bennett and Professor Nicholas Fisk. He also spent several periods of time training in laboratories to increase his knowledge and understanding of cytogenetic and molecular genetic analysis techniques including PCR. As he explained it, in order to properly interpret and counsel patients on the results of relevant tests, it was necessary to have a thorough understanding of how the tests were performed, including potential limitations of the tests and how errors may occur in the process.
333 Now the questions asked of Professor Hyett during cross examination seemed to suggest that as at March 1997 the primary focus of his work was clinical in character, and focused on the fact that he did not have responsibility for initiating research projects. But it is plain that by that time Professor Hyett had completed a research fellowship which included research projects relating to prenatal diagnosis. He therefore had direct experience in the research work that was being undertaken in the field of prenatal diagnosis.
334 Further, after the priority date, from around 1998 Professor Hyett developed a research interest in the potential use of cffDNA for prenatal testing. In the early 2000s, Professor Hyett worked in a research group looking at the feasibility of using cffDNA in maternal plasma to screen for sex-linked gene disorders, such as X chromosome linked disorders. The same research group considered the possibility of using cffDNA to screen for chromosomal aneuploidies.
335 I found Professor Hyett to be a very helpful and reliable witness in the areas of which he had direct experience.
Professor Oepkes
336 Professor Dick Oepkes is a Professor of Obstetrics and Fetal Therapy, and Head of the Fetal Medicine Section within the Department of Obstetrics at Leiden University Medical Center in the Netherlands. Professor Oepkes was awarded a medical doctorate from the Medical School of the State University in Utrecht (Netherlands) in 1987 and completed a PhD (cum laude) in 1993 at the University of Leiden (Netherlands). From 1988 to 1993, he was a research fellow / physician-sonographer at the Fetal Medicine Unit at the Department of Obstetrics in the University Hospital at Leiden. Between 1993 and 1999, he was a Resident in Obstetrics and Gynaecology at the University of Leiden. This six year residency was required in order to become a specialist in obstetrics and gynaecology.
337 Whilst at the University of Leiden, both as a resident and research fellow, and before and at the priority date, Professor Oepkes was part of the research group led by Professor Humphrey Kanhai. Part of the research being undertaken by this group was a joint research project between the obstetrics and clinical genetics units, investigating the isolation of fetal cells from maternal blood. Although Professor Oepkes was not directly involved in this research, he was involved in counselling women and obtaining samples for the project and there was regular and ongoing informal collaboration and discussion within the broader research group in relation to this research. He was also aware of other groups undertaking research on the detection and enrichment of fetal cells from maternal blood for prenatal diagnosis, including using PCR to detect trophoblast cells by targeting the Y chromosome.
338 Professor Oepkes’ interest and involvement in research in the area of fetal medicine has continued. As he has explained in his affidavit, his current role has about a 30% research focus and he has supervised 19 PhD students in the field of obstetrics and fetal medicine. I found him to be reliable and of assistance in the evidence he gave in the area of fetal medicine.
INVALIDITY – GENERAL
339 It is convenient to the flow of my reasons to first discuss the invalidity questions and then proceed to the infringement case.
340 The issues for determination by me on validity are the following:
(a) First, whether the relevant claims are for a manner of manufacture within the meaning of s 6 of the Statute of Monopolies 1624, 21 Jac 1 c 3. Ariosa’s position is that the relevant claims are in substance to a mere discovery, namely, that cell-free fetal DNA (cffDNA) is present, and can be detected in the plasma or serum of pregnant women, and that the end result of each claim is not an artificially created state of affairs.
(b) Second, whether the claimed invention is obvious in light of a paper by Kazakov et al, published in the journal Cytology in 1995 which describes the detection of extracellular DNA in the serum of pregnant women and suggests that the source of the DNA may be fetal cells, and whether that paper could have been ascertained.
(c) Third, whether the relevant claims of the Patent are useful. Ariosa says that there are methods falling within the scope of at least some of the claims which do not fulfil the promise of the specification, namely providing a method of prenatal diagnosis.
(d) Fourth, whether the complete specification of the Patent describes the claimed invention fully, in particular whether the Patent fully describes the quantitative embodiments of the claimed invention, as set out on page 5 of the Patent.
(e) Fifth, whether the relevant claims of the Patent are fairly based on matter described in the complete specification of the Patent.
(f) Sixth, whether the claimed invention was obtained by false suggestion or misrepresentation given that it is said that methods proposed in the specification for screening for fetal chromosomal aneuploidies could not have been applied at the priority date or at the date of grant of the Patent.
341 I should say at this point that for the moment it is not necessary to deal with any contested questions concerning the construction of the relevant claims. The contested construction questions relate more to the infringement case and I will deal with these later.
MANNER OF MANUFACTURE
342 For patentability, s 18(1)(a) of the Act requires that the invention claimed in any claim be a manner of manufacture within the meaning of s 6 of the Statute of Monopolies 1624, 21 Jac 1 c 3. Further, Schedule 1 to the Act defines “invention” as “any manner of new manufacture the subject of letters patent and grant of privilege within s 6 of the Statute of Monopolies, and includes an alleged invention”.
343 It is convenient for me to set out aspects of my analysis of this ground from Meat & Livestock Australia Limited v Cargill, Inc (2018) 354 ALR 95; (2018) 129 IPR 278; [2018] FCA 51 (MLA (No 1)).
(a) D’Arcy v Myriad Genetics Inc
344 In D’Arcy v Myriad Genetics Inc (2015) 258 CLR 334 (Myriad), French CJ, Kiefel, Bell and Keane JJ (at [18]) endorsed the relevant question, citing National Research Development Corporation v Commissioner of Patents (1959) 102 CLR 252 (NRDC) at 269, as: “Is this a proper subject of letters patent according to the principles which have been developed for the application of s 6 of the Statute of Monopolies?”. Section 6 of the Statute of Monopolies declared all monopolies to be void except for:
Letters Patents and Grants of Privilege for ... the sole working or making of any manner of new Manufactures within this Realm, to the true and first Inventor and Inventors of such Manufactures, which others at the time of making such Letters Patents and Grants shall not use, so as also they be not contrary to the Law, nor mischievous to the State, by raising prices of Commodities at home, or hurt of Trade, or generally inconvenient...
345 The concept of “manner of manufacture” is to be developed on a case-by-case basis and is not susceptible to any verbal formula in lieu of the phrase “manner of manufacture”. Two necessary but not necessarily sufficient criteria for patentability of an invention are (at [28]):
1. Whether the invention as claimed is for a product made, or a process producing an outcome as a result of human action.
2. Whether the invention as claimed has economic utility.
346 The plurality observed that where the invention so far as claimed in the claim in issue falls within the existing concept of manner of manufacture, it will ordinarily be sufficient if the claimed invention satisfies those criteria. But where the claim is not within the established boundaries of what is patentable, other considerations come into play. A non-exhaustive list of these other factors was set out in the following terms (at [28]):
3. Whether patentability would be consistent with the purposes of the Act and, in particular:
3.1 whether the invention as claimed, if patentable under s 18(1)(a), could give rise to a large new field of monopoly protection with potentially negative effects on innovation;
3.2 whether the invention as claimed, if patentable under s 18(1)(a), could, because of the content of the claims, have a chilling effect on activities beyond those formally the subject of the exclusive rights granted to the patentee;
3.3 whether to accord patentability to the invention as claimed would involve the court in assessing important and conflicting public and private interests and purposes.
4. Whether to accord patentability to the invention as claimed would enhance or detract from the coherence of the law relating to inherent patentability.
5. Relevantly to Australia’s place in the international community of nations:
5.1 Australia’s obligations under international law;
5.2 the patent laws of other countries.
6. Whether to accord patentability to the class of invention as claimed would involve law-making of a kind which should be done by the legislature.
347 Factors 3, 4 and 6 were said to be of primary importance and were not mutually exclusive. Factor 5 was said to be of secondary significance. Further, it was said that such factors might also inform the “generally inconvenient” limitation in s 6 of the Statute of Monopolies. Now I would note at this point that the separate judgments of Gageler and Nettle JJ and of Gordon J did not expressly adopt what I will describe as the “other factors” approach.
348 Now various questions might be said to suggest themselves concerning the “other factors” approach. First, is this a policy-driven approach to the assessment of patentability for cases on or beyond the existing boundaries? Second, is this approach properly characterised as purposive or consequentialist or both? Third, is there a clear threshold to justify moving into such a space, and if so what? In some cases reasonable minds might differ as to whether a case is within or without existing boundaries. Fourth, has the plurality just been more transparent about the considerations to be taken into account in assessing whether new or difficult subject matter is a proper subject matter for the grant of letters patent? Fifth and further, various issues concerning the priority ranking and weighting to be given to these factors remain to be explored. For example, how are factors 3.1 and 3.2 to be ranked and weighted with factor 3.3? And what is the scope of factor 3.3? How are factors 3, 4 and 6 ranked and weighted as between themselves? How is factor 5 to be weighted with the other factors, even if it is only of secondary significance? And am I obliged to consider each and all of the factors or only some of them? For the purposes of the present case it is not necessary to answer any of these questions. That is because I do not consider that I am dealing with a new class of claim involving a significant new application of or extension to the concept of “manner of manufacture” (Commissioner of Patents v RPL Central Pty Ltd (2015) 238 FCR 27 (RPL) at [118] and [119] (per Kenny, Bennett and Nicholas JJ)). But if I am wrong, I have been able to apply this “other factors” approach without answering any of these questions, and in doing so have fortified my conclusion on patentability in any event, which is perhaps unsurprising. All of these “other factors” applied to the case before me are uni-directional. They all point to patentability. Given that conclusion, the characterisation of these other factors, their priority and their weighting is of academic interest only in the present case.
349 Now before proceeding further and for the purposes of my later analysis, it is appropriate to consider a number of topics and concepts analysed in Myriad.
What in substance is claimed?
350 All members of the Court were concerned with what in substance the claim was for when determining whether there was a manner of manufacture.
351 The claims in issue in Myriad were to an “isolated nucleic acid coding for a mutant or polymorphic BRCA1 polypeptide…”. The patentee submitted that the Court ought to treat the impugned claims as claims for a chemical compound and should not treat them as any different from any other product claims (at [27]). But the plurality in Myriad at [6] explained that “[d]espite the formulation of the claimed invention as a class of product, its substance is information embodied in arrangements of nucleotides. The information is not ‘made’ by human action. It is discerned”. This was so despite the process by which the nucleic acid sequence in Myriad was isolated involving human intervention. Further, the resulting nucleic acid sequence could not function in nature. Further, the definition of “an isolated nucleic acid” in the patent in issue in Myriad included “chemically synthesised analogs” made outside the naturally occurring environment of the nucleic acid from which the isolate was prepared. The plurality explained that it was the existence of the said information, which was the same information contained in the DNA of the person from whom the nucleic acid was isolated, which was an essential element of the invention as claimed and that the product was the medium in which that information resided (at [89]).
352 Further, Gageler and Nettle JJ considered whether the wording of the claim “properly reflects the substance of the claimed invention” (at [142]). They held that the “way in which a claim is drafted cannot, however, transcend the reality of what is in suit” (at [144]) and that “care must be taken to examine the form of claim actually made to see if it is in fact an attempt to establish a monopoly for the manufacture of a substance for a purpose for which a monopoly cannot be claimed” (at [145]). Their Honours observed that the patentee did not invent and could not claim to have invented the process of isolating nucleic acid or the process of amplifying for genetic testing the fragment comprising the BRCA1 gene (at [146]). Accordingly, it was said to be beside the point that the isolated nucleic acid was chemically, structurally and functionally different from naturally occurring DNA from which it was isolated because the patentee had not invented or claimed a new method for isolating nucleic acid (at [158]).
353 Gageler and Nettle JJ concluded that in substance the claim was (at [160]):
a claim for a monopoly over the right to apply long-established methods for the isolation and amplification of specific nucleotide fragments to the isolation and amplification of a patient’s naturally occurring BRCA1 gene, where and if it is found upon subsequent examination that the patient's BRCA1 gene happened to be afflicted by any of the specified mutations and polymorphisms
354 Accordingly it was not a valid claim of a manner of manufacture of a product (at [161]).
355 And on this aspect, Gordon J took a similar approach. Her Honour observed that the Full Federal Court’s finding that claim 1 was to a molecule which was structurally and functionally different to what occurs in nature did not take account of the words of the claim. Accordingly she observed (at [279]) that:
As a matter of substance, each of claims 1-3 focuses on the existence of one or more elements of an identified code: a code which is found in the nucleic acid isolated from a patient and which necessarily must be identical to the coding sequence in that patient. None of the asserted chemical, structural and functional differences identified by the Full Court play any part in the definition of the invention “so far as claimed” (ss 18(1)(a) and 40(2)(b)) in each of claims 1-3 or in the description (s 40(2)(a)) of the invention in the specification.
(Emphasis in original.)
356 In substance then, the Court assessed what was claimed not through the lens of the chemical properties or the chemical potentiality (or lack thereof) of the claimed product, but rather through its genetic informational content.
Not man-made – no “artificially created state of affairs”
357 As the plurality had identified that the substance of the claim was to information rather than a chemical entity, they considered that it was a claim on the boundaries of what was patentable subject matter. But their Honours also considered the “artificially created state of affairs” criterion at [91]. Their Honours observed that engaging with that criterion places the question of patentability in too narrow a frame because it distracts from the central issue of “whether an essential integer of the claims, the genetic information, takes them outside the category of that which can be ‘made’” (at [91]). Thus, the central question was, having properly construed the claims, whether the claim in substance was something that was man-made. Further, the plurality at [91] went on to observe that even if the criterion of an “artificially created state of affairs” was appropriate, “the fact of the existence of the requisite mutations or polymorphisms is a matter of chance. It is not something ‘made’. It is not ‘artificially created’”.
358 Gageler and Nettle JJ took the approach of observing (at [125]) that an “artificial state of affairs” and “economic utility” are not the only considerations relevant to the question of manner of manufacture. At [127] they identified the question as being “whether the subject matter of the claim is sufficiently artificial, or in other words different from nature, to be regarded as patentable” (my emphasis). As to the sufficient degree of the artificiality or difference, their Honours observed (at [128]) that “it is necessary that the inventive concept be seen to make a contribution to the essential difference between the product and nature”. So, the subject matter of the claim must “have about it a quality of inventiveness which distinguishes it from a mere discovery or observation of a law of nature” (at [131]). Moreover, their Honours observed that, by definition, a manner of manufacture “is an artificial thing or state of affairs which involves an element of inventiveness” (at [161]). They concluded that the presence or absence of the mutations and polymorphisms in the isolated nucleic acid was the critical discovery and was the “antithesis of a man-made artificial state of affairs” (at [162]).
359 I will address later the question of whether Gageler and Nettle JJ can be taken to have added a separate threshold requirement of inventiveness as part of the concept of “manner of manufacture”.
Economic utility
360 As to the question of economic utility, the plurality, in disposing of the idea that the isolation of the nucleic acid per se leads to an economically useful result, viz the treatment of breast and ovarian cancers, observed (at [85]):
The economic significance necessary to the patentability of an “artificially created state of affairs” in the sense used in NRDC is not demonstrated by stating that the artificially created state of affairs is a step along the way to a process or method itself claimed as an artificially created state of affairs of economic significance.
361 This rejection was reinforced by Gageler and Nettle JJ who said (at [164]):
Fifthly, it is not the isolation of nucleic acid, or even the isolation and amplification of the fragment comprising the BRCA1 gene, which leads to the “economically useful result” of treating breast and ovarian cancers. It is rather the first respondent's discovery of a naturally occurring correlation between the presence of the specified mutations and polymorphisms in such a fragment (and thus in the DNA in the cell from which the fragment is derived) and an increased probability of actual or potential malignancy.
The “other factors”
362 The plurality observed that the “purpose of the Act would not be served by according patentability to a class of claims which by their very nature lack well-defined boundaries or have negative or chilling effects on innovation” (at [29]). Their Honours found that in Myriad, the contentious claim(s) lacked well-defined boundaries because the isolated nucleic acid sequence included the products applying any process, known or unknown, to isolate the nucleic acid and thus created an unwarranted de facto monopoly on all methods of isolating nucleic acids from the BRCA1 gene. Moreover, infringement could occur without the infringer being aware of it (at [93]). Indeed, these factors would have a “chilling effect” on innovation, which effect was apparent from the face of the claim.
363 And as to maintaining to the extent feasible coherence of Australian law, the plurality pointed to Apotex Pty Ltd v Sanofi-Aventis Australia Pty Ltd (2013) 253 CLR 284 to the effect that given the established patentability of pharmaceutical products it would have been anomalous to exclude medical treatments using such products.
The application of naturally occurring phenomena to a particular use
364 A distinguishing feature of the present case that is not unimportant is that I am dealing with method claims. All judges in Myriad not only explained that they were not addressing such claims, but by implication that such claims on their face may perhaps be more readily seen as within the existing boundaries of “manner of manufacture”.
365 The plurality observed that claims to the use of the isolated nucleic acid as opposed to the product itself were not in issue in the proceeding. So at [71] they said:
A number of sections of the specification relate to methods for the use of nucleic acids in various ways and the preparation of recombinant or chemically synthesised nucleic acids and vectors. The claims in the patent relating to those matters are not in issue. Nor is there any question about the utility of the applications of isolated nucleic acids reflected in those undisputed claims.
366 And at a more general level they observed at [20]:
It is true that in Anaesthetic Supplies Pty Ltd v Rescare Ltd Lockhart J in the Full Federal Court, in a passage endorsed by Crennan and Kiefel JJ in Apotex, said:
If a process which does not produce a new substance but nevertheless results in “a new and useful effect” so that the new result is “an artificially created state of affairs” providing economic utility, it may be considered a “manner of new manufacture” within s 6 of the Statute of Monopolies.
Importantly, however, his Honour used the word “may”. Neither Lockhart J nor Crennan and Kiefel JJ should be read as holding that satisfaction of that formula would mandate a finding of inherent patentability. That is not to say that it will not suffice for a large class of cases in which there are no countervailing considerations.
(Footnotes omitted; citations omitted.)
367 Of course I accept that it is first appropriate to properly construe what in substance the process claim is to and, if necessary, consider the “other factors” identified by the plurality.
368 Further, Gageler and Nettle JJ also addressed whether claims to methods of using isolated nucleic acids may be patentable subject matter. Now at [134], their Honours observed that the essence of claim 1 was the “correlation between the incidence of cancer and the presence of the specified mutations and polymorphisms in the BRCA1 gene”, but that was not enough to make the claim a manner of manufacture. Ariosa says that it should follow that a claim to such a correlation and to the use of that correlation in a known method would also lack patentability. Now I disagree. Moreover, such a contention also ignores other aspects of their Honours’ reasoning at [137] where they distinguished a product claim from a process claim:
Of course, as NRDC implies, the application of naturally occurring phenomena to a particular use may be a manner of manufacture if it amounts to a new process or method of bringing about an artificially created state of affairs of economic significance. Even so, the inventor cannot claim to have invented the naturally occurring product as opposed to the process of application. In NRDC, the patentee could not claim to have invented, and therefore there was no suggestion of it laying claim to a monopoly over, the commonplace herbicides which were used in the course of the patentable process. Similarly, in Shell Oil Co v Commissioner of Patents, the patentee could not claim to have invented, and therefore there was no suggestion of laying claim to a monopoly over, the known compounds which were applied as part of the patentable process to a new use of plant growth regulation. So too here, in so far as the invention consists in the application of a naturally occurring phenomenon to a particular use, the inventor cannot claim to have invented the naturally occurring phenomenon as opposed to the method of use and has no claim to a monopoly over the naturally occurring phenomenon as opposed to the method of use.
(Footnotes omitted.)
369 Their Honours observed that “the application of naturally occurring phenomena to a particular use may be a manner of manufacture if it amounts to a new process or method bringing about an artificially created state of affairs of economic significance” (emphasis added). Now I accept that the word “may” is not unimportant. Further, the formalism of the claim cannot overcome its substance.
370 Now Gageler and Nettle JJ said at [147]:
It was not disputed that [the patentee] might justly lay claim to the discovery that, if an isolated fragment comprising the BRCA1 gene is found upon examination to exhibit the specified mutations and polymorphisms, their presence is or may be indicative of particular kinds of malignancy in the cell. Nor was it disputed that a process or method of using known technology to isolate a sequence of nucleic acid comprising the BRCA1 gene and examining it for the presence of the specified mutations and polymorphisms for the purpose of detecting or predicting malignancy might be patentable …
371 Of course their Honours did not accept that such process claims would necessarily be patentable. Indeed, their Honours went on to say at [147] “[b]ut, as has been observed, the discovery of a natural correlation is not patentable as such ...” and its discovery did not mean the product used in that process was patentable. It may be said that their Honours can be properly understood to be saying that the fact that process claims using the isolated nucleic acid of claims 1 to 3 were not in issue, did not mean the process claims themselves were patentable.
372 Further, I should also note that their Honours said at [152]:
In the same way here, it is one thing to say that the first respondent has invented a process which consists in isolating and examining the fragment comprising the BRCA1 gene for the presence of the specified mutations and polymorphisms as an indicium of malignancy. It is quite another and different thing to say that the first respondent, as inventor of that process, is entitled to a monopoly over the mutated BRCA1 gene, which is used merely as an ingredient in that process. The invention claimed makes no contribution to the manufacture of the substance. At best, it takes advantage of properties in the substance hitherto unknown or unsuspected. Just as there was no difference between the process and the product in Wellcome Foundation, there is no distinction between a claim to the process of isolating the BRCA1 gene for the purpose of examining it for the presence of the specified mutations and polymorphisms and the claim to the BRCA1 gene itself.
373 Further, their Honours said at [168]:
It is not disputed that a process or method of detecting the increased likelihood of certain kinds of malignancy by isolating the BRCA1 gene and examining it for the presence of any of the specified mutations and polymorphisms may be patentable subject matter as a process (subject to considerations of novelty and inventive step when compared to the prior art base). But, to repeat, claim 1 is not a claim for any such process. It is a claim for a monopoly over such isolated fragments of naturally occurring DNA as comprise the BRCA1 gene as are found upon examination to contain the (naturally occurring) specified mutations and polymorphisms.
(Emphasis added; footnote omitted.)
374 Again their Honours used the qualifier “may be”, which I will return to later. Now it may be said that their Honours’ requirement that a claim be properly construed to identify its substance and their observations that natural correlations are not patentable, answer any suggestion that their Honours would have accepted the method claims as patentable if they had been in issue. But to so say may be a rose-tinted perspective of what their Honours said.
Express exclusions in section 18
375 I should say something about s 18 of the Act.
376 Section 18(2) excludes as patentable inventions for standard patents “Human beings, and the biological processes for their generation”. Now an exclusion of that kind does not mean that everything outside that exclusion is patentable subject matter or, in particular, a manner of manufacture. This is plain from the finding in Myriad itself where the exclusion in s 18(2) was not the basis upon which Myriad was decided (at [11]). Of course, s 18(2) has no direct application to the Patent before me in any event. Section 18(3) is of no relevance.
Other matters concerning the claims in Myriad
377 First, it is apparent from each of the three sets of reasons in Myriad that although claims 1 to 3 were held not to be patentable on the basis indicated, no member of the Court gave any significant indication that claims 4 to 30 were not or did not give rise to a “manner of manufacture”. The plurality emphasised that the Court was not concerned with gene patenting generally, but only with three claims encompassing isolated nucleic acids coding for a mutant or polymorphic BRCA1 polypeptide. Their Honours found that although the claims were product claims, they were in substance claims to the genetic information embodied in, and conveyed by, the nucleic acid sequence. And as the genetic information was naturally-occurring and had not been “made” by human action, it followed that claims 1 to 3 did not define a manner of manufacture. But it is apparent that their Honours’ reasoning did not rule out patentability to the products and methods of claims 4 to 30.
378 Second, it is also apparent that the plurality considered that if claims 1 to 3 were held to be a manner of manufacture, this would have involved an extension of the categories of patentable subject matter to a new class of case. It was in that context that their Honours discussed the need to consider the potential chilling effect and “other factors” before recognising such an extension. But the “other factors” do not arise unless the claims in question require an extension of the existing concept of manner of manufacture to a new class of case or are on the border. But that is not the present case before me. The relevant claims of the Patent, directed to inventive methods and processes as I have ultimately found, are within the plain vanilla concept of manner of manufacture as outlined in NRDC and Myriad.
379 Third, as I have already indicated, Gageler and Nettle JJ noted the recognition in NRDC that the application of a naturally occurring phenomenon to a particular use may be a manner of manufacture if it amounts to a new process or method of bringing about an artificially created state of affairs of economic significance. In essence, their Honours acknowledged that a process of using known technology to isolate a sequence of nucleic acid comprising the BRCA1 gene and examining it for the presence of the specified mutations and polymorphisms to detect or predict malignancy, might be patentable (at [147]), a point that they later reiterated (at [168]). Further the words “subject to considerations of novelty and inventive step when compared to the prior art base” in [168] are not to be overlooked. They elucidate the qualification intended by the use of the word “may” in the preceding line. Their Honours implicitly recognised that a method involving the use of naturally occurring nucleic acid sequences for a particular purpose, such as a method of detection or diagnosis, may be within the established concept of a manner of manufacture and may be patentable if it satisfies the other requirements for patentability in the Act, including novelty and inventive step.
380 Fourth, Gordon J recognised the significant difference between claims 1 to 3 of the patent in suit and the remaining claims 4 to 30. Her Honour outlined the subject matter of the latter claims at [191] and later reasoned (at [256] to [258]):
What then did Myriad do? It took the idea, concept or principle that specific mutations or polymorphisms in that sequence suggest a predisposition to breast cancer and ovarian cancer and moved to carry out that idea, concept or principle, or embody it in a manner of new manufacture, in claims 4-30. The validity of those claims is not in issue.
Claim 4 may be taken as an example. In simple terms, it comprises a nucleic acid probe in which the nucleotide sequence is a portion of an isolated nucleic acid with the characteristic identified in claim 1 …
The invention in claim 4 carried into effect the idea that specifically identified mutations or polymorphisms in a sequence of the BRCA1 gene suggest a predisposition to breast cancer and ovarian cancer by testing for the presence of one or more of the specifically identified mutations or polymorphisms. That is an invention.
381 On her Honour’s reasoning, it would also appear that the invention in claims 5 to 30 was no different in principle. These included preparations and uses of polypeptides (claims 10 to 16) and various methods of diagnosis (claims 17 to 30). Now for completeness, I would note that claim 17 of the Myriad patent was expressed in the following terms:
17. A method for diagnosing a predisposition for breast and ovarian cancer in a human subject which comprises determining whether there is a germline alteration in the sequence of the BRCA 1 gene in a tissue sample of said subject compared to the nucleotide sequence set forth in SEQ.ID No:1 or a wild-tyle [sic] allelic variant thereof, said alteration indicating a predisposition to said cancer being selected from the mutations as set forth in Tables 12, 12A and 14.
382 In summary and generally speaking, the reasoning in Myriad does not assist Ariosa.
(b) Arguments against patentability
383 It may be said that in substance, there is no invention claimed, but merely the detection and discovery of naturally occurring material by known methods.
384 It may be said that the relevant claims do not expressly or in substance result in an “artificially created state of affairs” because no product is made and there is no process producing an artificial outcome as a result of human action. It may be said that an “artificially created state of affairs” requires:
(a) “physical phenomenon in which the effect, be it creation or merely alteration, may be observed”: NRDC at 276;
(b) “[a] physical effect in the sense of a concrete effect or phenomenon or manifestation or transformation ...”: Grant v Commissioner of Patents (2006) 154 FCR 62 at [32] per Heerey, Kiefel and Bennett JJ; and
(c) “something of a corporeal and substantial nature”: Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) (2007) 235 CLR 173 (Lockwood (No 2)) at [66].
385 In NRDC, after acknowledging the difficulty in delineating discovery from invention, it was said (at 264):
There may indeed be a discovery without invention – either because the discovery is of some piece of abstract information without any suggestion of practical application of it to a useful end, or because its application lies outside the realm of “manufacture”.
386 It may be said that to the extent that the method in claim 1 for example constitutes the application of Sequenom’s discovery, that application lies outside the realm of manufacture. There is no “product” or no “artificially created state of affairs” or not one which is sufficiently artificial or man-made.
387 It may be said that in the words of Gageler and Nettle JJ, the subject matter of claim 1 for example does not have “a quality of inventiveness which distinguishes it from a mere discovery or observation of a law of nature” (at [131]). That is, it is not “sufficiently artificial” and the “inventive concept” does not make a contribution to the essential difference between product and nature (at [127] and [128]).
388 Further, it may be said that sufficiency of artificiality is a concept that has also been used when considering whether a business method that uses a computer is a “manner of manufacture”. As RPL observed at [96] (per Kenny, Bennett and Nicholas JJ):
A claimed invention must be examined to ascertain whether it is in substance a scheme or plan or whether it can broadly be described as an improvement in computer technology. The basis for the analysis starts with the fact that a business method, or mere scheme, is not, per se, patentable. The fact that it is a scheme or business method does not exclude it from properly being the subject of letters patent, but it must be more than that. There must be more than an abstract idea; it must involve the creation of an artificial state of affairs where the computer is integral to the invention, rather than a mere tool in which the invention is performed. Where the claimed invention is to a computerised business method, the invention must lie in that computerisation. It is not a patentable invention simply to “put” a business method “into” a computer to implement the business method using the computer for its well-known and understood functions.
389 It may be said that as a result of Myriad, naturally occurring genetic information is not patentable per se, even when it is isolated from its natural state.
390 It is convenient at this point to deal with several aspects of the reasons of Gageler and Nettle JJ.
391 First, it may be said that Gageler and Nettle JJ have added another threshold requirement for inventiveness. By that I mean that in addition to the s 18(1)(b)(ii) requirement of inventive step there was a separate threshold requirement for inventiveness in the form of a back-door introduction of the so called “product of nature doctrine” favoured in the US. So one may point to the following passages of their Honours’ reasoning:
(a) “The question then is whether the subject matter of the claim is sufficiently artificial, or in other words different from nature, to be regarded as patentable” (at [127]);
(b) “it is necessary that the inventive concept be seen to make a contribution to the essential difference between the product and nature” (at [128]);
(c) “the subject matter of a claim have about it a quality of inventiveness which distinguishes it from a mere discovery or observation of a law of nature” (at [131]);
(d) “there are limits on the patentability of products of nature inasmuch as products of nature do not involve human intervention and therefore are lacking in the necessary quality of inventiveness to qualify as a manner of manufacture” (at [136]); and
(e) a favourable endorsement (see at [136]) of Professor Sherman’s observation that the US product of nature doctrine and the Australian test of artificially created state of affairs are the same question asked from different perspectives.
392 Now I am not at all convinced that their Honours have introduced a new threshold requirement for inventiveness. As I said in MLA (No 1), there is always a difficulty in parsing and nuancing the language of a judgment. But let me assume for the moment that this is how their Honours’ reasoning should be read. I have not applied such a new threshold for inventiveness because there are two binding authorities against me doing so. The first authority is Myriad itself. Neither the plurality nor Gordon J endorsed such an approach. The second authority is Lockwood (No 2) at [106] where the Court stated, with reference to Commissioner of Patents v Microcell (1958) 102 CLR 232 that Microcell:
stands for a narrow proposition that a Commissioner of Patents, or his or her delegate, may refuse an application for patent protection where a specification “on its face” shows the invention claimed is not a manner of new manufacture. This may arise, for example, from admissions concerning novelty. The decision in Microcell has not always been properly understood; it does not involve a separate ground of invalidity or a discrete “threshold” test.
(Footnote omitted; my emphasis.)
393 In other words, Lockwood (No 2) rejected any separate threshold test, whatever it might be said that NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1995) 183 CLR 655 at 663 and 664 stands for. Moreover, the plurality in Myriad at [12] did not refer to Philips to support any separate threshold requirement for inventiveness, but rather cited Philips in a manner consistent with how Lockwood (No 2) had described how Microcell should be read; this is consistent with the conjunction of the two references in footnote 26 to the plurality’s reasons in Myriad.
394 To complete the circle, Gageler and Nettle JJ applied Lockwood (No 2) (see [131] and footnote 144), thereby confirming my doubts as to whether their Honours intended to introduce a new threshold requirement.
395 In summary, I am bound to apply the plurality’s approach in Myriad and also Lockwood (No 2).
396 Second, reference may be made to the observations of Gageler and Nettle JJ that the discovery of a correlation between naturally occurring phenomena is not a manner of new manufacture. Now it may be accepted that an idea is not patentable per se. And it may be accepted that a “natural law” or “law of nature”, whatever that means (the concept is a contestable proposition for a philosopher of science), is not patentable per se. It may also be accepted that the discovery of an objectively ascertained statistically significant correlation between two physical phenomena is not patentable per se. But in the present case, what is sought to be patented is a method. The questions are whether that method is a manner of manufacture and involves an inventive step. Moreover, no authority requires me to exclude from the inventive step analysis, the ingenuity and very extensive research that was undertaken by Sequenom as part of determining whether the method involved an inventive step.
397 Now in the present case, and picking up some of the foregoing arguments, Ariosa submits that the invention as claimed in each relevant claim of the Patent does not involve a “manner of manufacture” for the reasons that:
(a) what is claimed in each claim is a “mere discovery” of a naturally occurring phenomenon, and not a method involving a practical application that in substance goes beyond the discovery itself, and as a result there is no “artificially created state of affairs”; and
(b) properly understood, the “end result” of each claim does not involve an artificially created state of affairs, as it results in information only.
398 Let me identify in more detail Ariosa’s submissions on the first issue, namely, that a claim would not be a claim to a “manner of manufacture” if it were in substance a claim to a discovery only.
399 In Research Affiliates LLC v Commissioner of Patents (2014) 227 FCR 378 (Research Affiliates) the Full Court (at [94], approved by the Full Court in RPL at [100]) noted that the necessary distinction is between an idea or law of nature, and its employment so as to achieve a new and useful effect.
400 Further, Ariosa says that in Myriad, the plurality of the High Court similarly identified (at [28]) two essential factors that are necessary to the characterisation of an invention as a manner of manufacture:
(a) Whether the invention as claimed is for a product made, or a process producing an outcome as a result of human action.
(b) Whether the invention as claimed has economic utility.
401 Further, Ariosa says that whilst the word “outcome” involves different language to that used in NRDC, that reference should be read with the plurality’s statement at [6] where their Honours said:
As appears from s 6 of the Statute of Monopolies, an invention is something which involves “making”. It must reside in something. It may be a product. It may be a process. It may be an outcome which can be characterised, in the language of NRDC, as an “artificially created state of affairs”. Whatever it is, it must be something brought about by human action.
(Citation omitted; emphasis added.)
402 Ariosa points out that a similar outcome to Myriad resulted in the United States in litigation on Sequenom’s US patent no. 6,258,540 (US ‘540), the corresponding US patent to the Patent (Ariosa Diagnostics Inc. Sequenom, Inc. 788 F3d 1371 (3d Cir 2015) (Ariosa (US)). The US Court held that the claims of US ‘540 were invalid, on the basis that the claims were to patent-ineligible subject matter applying the test set down by the US Supreme Court in Mayo Collaborative Services v Prometheus Laboratories, Inc (2012) 566 US 66 (Mayo) and Alice Corporation Pty Ltd v CLS Bank International (2014) 573 US 208.
403 In reaching its decision, the Court noted that the claimed methods of US ‘540 begin and end with natural phenomena, stating at 1376:
Sequenom does not contend that Drs. Lo and Wainscoat created or altered any of the genetic information encoded in the cffDNA, and it is undisputed that the location of the nucleic acids existed in nature before Drs. Lo and Wainscoat found them. The method ends with paternally inherited cffDNA, which is also a natural phenomenon. The method therefore begins and ends with a natural phenomenon. Thus, the claims are directed to matter that is naturally occurring.
404 The Court also held (at 1377) that as the claimed processes were well-understood, routine and conventional activity, they claimed patent-ineligible subject matter:
Like the patentee in Mayo, Sequenom contends that the claimed methods are patent eligible applications of a natural phenomenon, specifically a method for detecting paternally inherited cffDNA. Using methods like PCR to amplify and detect cffDNA was well-understood, routine, and conventional activity in 1997. The method at issue here amounts to a general instruction to doctors to apply routine, conventional techniques when seeking to detect cffDNA. Because the method steps were well-understood, conventional and routine, the method of detecting paternally inherited cffDNA is not new and useful. The only subject matter new and useful as of the date of the application was the discovery of the presence of cffDNA in maternal plasma or serum.
405 In June 2016, the US Supreme Court denied Sequenom’s petition for a writ of certiorari.
406 Ariosa suggested that I should follow the US and as I understood its submission seemed to suggest that Myriad was in harmony with the US position. I hardly think so, despite the comfort that Ariosa sought to draw from Gageler and Nettle JJ’s analysis.
407 Further in support of its position Ariosa made submissions concerning what it asserted was the substance of the claimed invention.
408 Now the person skilled in the art to whom the Patent is addressed is a team and the team would include a person with expertise in maternal-fetal medicine and a person with experience in standard molecular genetics techniques, including perhaps a person with some exposure to laboratory-based genetic analysis of patient samples in a clinical context.
409 Now Ariosa says that the person skilled in the art working in the field already knew that fetal DNA could provide valuable information about the fetus. They knew that they could obtain fetal DNA for analysis by a direct sampling method, such as amniocentesis or CVS.
410 Further, another known source of DNA for analysis was the fetal cells that were circulating in the mother’s blood, albeit in very low concentration.
411 Further, the cffDNA from maternal plasma or serum, once its existence was discovered, was another source of DNA, which could then be analysed using the same techniques as were being used for analysing DNA from these other known sources.
412 Further, it points out that as at the priority date, there were various screening and diagnostic tests available to detect fetal conditions. These included tests that used cellular fetal DNA obtained through the invasive techniques of amniocentesis, CVS and cordocentesis. Research was also being conducted on the potential use of DNA from whole fetal cells, which were known to circulate in the blood of a pregnant woman, and which were able to be distinguished from maternal cells using techniques such as PCR with primers directed to the Y chromosome or the Rhesus D gene.
413 Further, it points out that at some point before March 1997, Professor Lo discovered using standard, routine techniques that fetal DNA could not only be detected from fetal cells in the blood of pregnant women, but also that cffDNA could be detected in the plasma and serum of pregnant women. In Professor Lo’s first experiment, he discovered that DNA from the Y chromosome could be detected in the plasma of pregnant women bearing male fetuses.
414 Further, it points out that the Patent suggests some potential applications for Professor Lo’s discovery. It states, on page 3, that it “may be particularly useful for sex determination”, which depends on detecting the presence of a Y chromosome. On page 4, the Patent states that the discovery can also be used to diagnose other conditions by detecting “any paternally-inherited sequences which are not possessed by the mother”. This qualitative approach depends on detecting the presence of cell-free DNA (using standard techniques) that cannot originate from the mother, and therefore must be fetal, and which may be, for example, causative of a disease phenotype in the fetus.
415 Further, it points out that the Patent on page 5 then states that Professor Lo’s discovery “can be applied to screening for … chromosomal aneuploidies”. The two suggested approaches set out on page 5 of the Patent rely on quantitation of the fetal DNA.
416 Ariosa says that claim 1 of the Patent is not limited to any particular application of Professor Lo’s discovery. Claim 1 is for:
A detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of foetal origin in the sample.
417 Ariosa says that the claim is, in practical effect, a claim to the discovery that fetal nucleic acid sequences are present and can be detected in maternal plasma or serum samples. Ariosa says that as significant as Professor Lo’s discovery proved to be for prenatal diagnosis, the patentee is unable to validly claim the discovery itself, or applications of that discovery that could not be implemented at the priority date.
418 Ariosa accepts that Professor Lo’s discovery of the existence of cell-free fetal DNA circulating in the maternal bloodstream was a valuable discovery, but it says that it is uncontroversial that that discovery per se was not itself patentable. Similarly, it says that if the discovery was in fact that cffDNA was more abundant than was expected from knowledge regarding fetal cells circulating in maternal blood, again that discovery per se was not itself patentable.
419 Now Ariosa concedes that where a new scientific principle is discovered, its practical application might be patented, even if that application is straight-forward with the knowledge of that new principle. But here, Ariosa emphasises that there is a discovery of the location (or amount) of a known product (fetal DNA). But that discovery does not involve any new principle.
420 Ariosa says that as a matter of substance, the relevant claims do not involve the application of any new principle. They only involve use of the discovery for the purposes to which it is inherently adapted, since no new principle is being applied. Ariosa says that what is claimed arises directly from, and solely from, the discovery itself.
421 Ariosa says that claim 1 of the Patent does no more than claim Professor Lo’s discovery that cffDNA is detectable in maternal serum or plasma. There is no practical application of that discovery that would fall outside claim 1. The claim simply says “detect what is naturally there”, a naturally occurring phenomenon.
422 Ariosa says that there is no claim in the Patent to any new method for detecting the cffDNA once it was discovered to be present in the plasma or serum of a pregnant woman and, to use the language of the plurality in Myriad at [72], “nor could there be as no new process is disclosed”.
423 Further, Ariosa says that unlike the claims in MLA (No 1), the claims of the Patent do not claim any specific or new method of applying the detectability of cffDNA in maternal plasma/serum. The claims involve the “mere identification or discernment of the naturally occurring phenomenon”, namely, the presence of DNA, specifically cffDNA, in the plasma or serum of a pregnant woman.
424 Further, Ariosa says as to the dependent claims that they are broad examples of how to detect cffDNA in maternal plasma using well-understood, routine, and conventional activity. But this is not sufficient to produce an artificially created state of affairs.
425 Ariosa says that claim 2 refers to amplification of the fetal nucleic acid and claim 3 identifies PCR as the amplification technique to be used in the detection method of claim 1. But it says that these techniques were well-understood, routine, and conventional in 1997. Claim 5 specifies the use of a particular probe. But it says that the use of probes was also a well-known technique.
426 Ariosa says that critically, none of these claims involves anything more than the use of well-known techniques applied because of the discovery that cffDNA is present in maternal blood. Those claims only address the detection of that cffDNA. The claims set out the practical steps needed to undertake the ‘detection’ component of claim 1; that is, confirming the presence of the cffDNA that Professor Lo had discovered was present in maternal blood.
427 Further, Ariosa says that each of claims 6 and 9 specifies the chromosome from which the fetal nucleic acid to be detected is derived. Ariosa says that in effect, claims 6 and 9, taken together, cover all fetal chromosomes that are paternally-inherited and which may therefore be distinguishable from the maternal DNA if a sequence can be identified that is not possessed by the mother. So, considered together, Ariosa says that claims 6 and 9 do not effectively limit the scope of claims 1 to 3 and 5. They just identify the subsets of fetal nucleic acids which might be detected in claims 1, 2, 3 and 5.
428 Ariosa says that claim 13 is the first (and only) asserted claim that might be seen to involve a specific application of Professor Lo’s discovery. It is a claim to a method “for determining the sex of the foetus”.
429 But Ariosa says that the nature of that conclusion follows so directly from the discovery itself that in substance it involves nothing more than the discovery. Knowing that fetal DNA is present, a known probe is used to determine whether sequences from the Y chromosome are present or not. The claim therefore says one must detect a naturally occurring phenomenon, cffDNA from the Y chromosome in maternal plasma, and identify a correlation between that phenomenon and another, viz, the Y chromosome indicates a male fetus.
430 Ariosa says that claim 14 requires that the concentration of the fetal nucleic acid in the maternal serum or plasma be determined. That is, claim 14 requires detecting the presence of a fetal nucleic acid, and measuring how much is there. Ariosa says that the claim is not directed to any application of that information, and so similarly does not give rise to a manner of manufacture.
431 Ariosa says that independent claims 22 to 26 involve making a prenatal diagnosis. It accepts that whilst in form these claims might appear to involve a practical application of Professor Lo’s discovery, in substance these claims are still the “mere identification or discernment of the naturally occurring phenomenon” (in contrast with MLA (No 1) at [455]). It says that that is the case because the claims say nothing as to the method of arriving at a diagnosis, nor as to the diagnosis to be made, and hence provide no relevant limitation. They do not involve the application of any new principle, or the application of any old principle in a new way. In substance, the claims are to the discovery itself.
432 In summary, Ariosa says that the relevant claims do not, in substance, involve more than the detection (and for claim 14, the quantification) of naturally occurring material, by known methods, such that the claims are in substance claims to a mere discovery, and do not result in an artificially created state of affairs.
433 Let me turn to the other issue raised by Ariosa.
434 Ariosa says that the second manner of manufacture issue in respect of the relevant claims is that, as a matter of substance, none of the relevant claims involves as its ‘end result’ (or ‘outcome’ to use the language of the majority in Myriad) the creation of an artificially created state of affairs. Each claim instead involves as its end result, as a matter of substance, information only.
435 Sequenom relies first on the fact that the blood product is separated into different fractions, including plasma or serum, which it says is an artificial result arising from human action. For later claims it relies on the amplified material as the artificial result. But Ariosa says that the claims are not directed to a new method of fractionating blood. If they were, then the achievement of plasma or serum would be a relevant ‘end result’, involving an artificially created state of affairs. Similarly, if the claims were directed to a new method of amplifying DNA products.
436 Ariosa says that an analogous argument was rejected by the Full Court in each of Research Affiliates and RPL, where it was argued that the relevant human intervention and artificial effect arose from the requirement in the claims to use a computer.
437 Ariosa points out that those two decisions were preceded by the Full Court’s decision in Grant v Commissioner of Patents (2006) 154 FCR 62 in which the Full Court considered claims to a method relating to the protection of assets against a legal claim. The Court, after considering NRDC, said (at [29]) that a “product of a method is something in which a new and useful effect may be observed.” In respect of claims to computer programs, the Court said that courts have “looked to the application of the program to produce a practical and useful result, so that more than “intellectual information” was involved” (at [29]). The Court then concluded, in respect of the subject patent, that “the method of his patent does not produce any artificial state of affairs, in the sense of a concrete, tangible, physical, or observable effect” (at [30]) and that a “physical effect in the sense of a concrete effect or phenomenon or manifestation or transformation is required” (at [32]). The Court did not base its reasoning on any rejection of the patentability of business methods per se (at [14] and [26]), but rather it found that the requisite artificially created state of affairs did not result.
438 In Research Affiliates, the Full Court (Kenny, Bennett and Nicholas JJ) considered the patentability of claims to a computer-implemented method of generating a securities index (at [65]).
439 The Full Court reviewed NRDC, and extracted from that review the following principles (at [8], see also at [92] and [95]):
There is a “manufacture” such as might properly have been the subject of letters patent and grant of privilege under s 6 of the Statute of Monopolies whenever a process produces, either immediately or ultimately, a useful physical result in relation to a material or tangible entity (at 268, 276).
The method the subject of the relevant claim must have as its end result an artificial effect falling squarely within the true concept of what must be produced by a process if it is to be held to be patentable (at 277).
440 The Full Court (at [107]) also emphasised that in assessing whether there is a manner of manufacture the substance of the claim must be assessed:
The determination whether the claimed invention is truly “an artificially created state of affairs” in satisfaction of NRDC is made not by some mechanistic application of the criterion of artificiality or physical effect, but by an understanding of the claimed invention itself. The invention is to be understood as a matter of substance and not merely as a matter of form.
441 In Research Affiliates, the patent applicant emphasised the fact that the use of a computer involves physical effects in a computer’s memory when data is transformed and stored within a computer, which is an artificial effect (at [103] and [106]).
442 The Full Court focused on the substance of what had been invented, and said (at [115]) that there was “a useful result of the claimed process but there is no physical thing “brought into existence or so affected as the better to serve man’s purposes””.
443 Ariosa says that the same is true of the present claims. The separation of plasma or serum is incidental, as is any amplified nucleic acid material. The ‘end result’ is the information about the DNA of the fetus, which itself is no more than natural information.
444 In RPL, the Full Court (Kenny, Bennett and Nicholas JJ) considered the patentability of claims to a computer-implemented method for assessing the competency or qualifications of an individual by reference to recognised standards (referred to as recognition of prior learning, or RPL). Claim 1 involved a method of gathering evidence which involved a computer generating questions by reference to the relevant assessable criteria for a recognised qualification standard (at [36]). The Full Court referred to and applied the same requirement that there be an artificially created state of affairs, or something in which a new and useful effect may be observed ([96], [97], [100], [103], [116]), and it took the view that this was something that had to be assessed as a matter of substance, not form, and by reference to where the true ingenuity lay. The Court concluded that any ingenuity did not lie with the use of the computer, which did not involve any steps other than the normal use of a computer.
445 Ariosa points out that in Research Affiliates, the Full Court was concerned with the patentability of claims to a computer-implemented method that had as their end result a securities index. The Full Court did not accept that the index, being mere information, was an artificially created state of affairs (at [116]):
Here, that subject matter is truly the scheme, the idea, the index. As set out in the specification it may be, and in the claimed method it is, implemented in a computer, but the ingenuity of the inventors, the end result of which is the invention, is directed to the idea, which is not patentable. That method does not have an artificial effect falling squarely within the true concept of what must be produced by a process if it is to be held patentable (NRDC at 277).
446 And further at [118]:
In the context of the claim, the significance lies in the content of the data rather than any specific effect generated by the computer. The computer implementation is an essential integer of the claimed process. … However, in examining whether a claimed invention is properly the subject of letters patent, it is necessary to look not only at the integers of that claimed invention but also at the substance of that invention.
447 Further, Ariosa points out that the Full Court in RPL said of Research Affiliates (RPL at [103]):
There, the invention described in the specification was directed to the index itself and was a method of implementing a scheme which happens to use a computer to effect that implementation. There was no technical contribution to the invention or artificial effect of the invention by reason of the intervention of the inventors; the ingenuity was in the scheme not in the physical phenomenon in which the effect may be observed and which has the requisite economic utility or artificial effect.
(Citation omitted.)
448 In the present case, Ariosa says that the end result of the claimed method is information about the DNA of the fetus. This is a naturally occurring phenomenon. According to the analysis of the plurality in Myriad, that information “is not “made” by human action” (at [6]). Further, Ariosa also says that it is plain from Research Affiliates and RPL that the Full Court in each case took the view that mere information (being the index in Research Affiliates, which in fact could be seen to be the result of human intervention) is not relevantly an artificially created state of affairs.
449 Further, Ariosa says that to the extent that claims 22, 23, 25 and 26 of the Patent relate to a “method of performing a prenatal diagnosis”, it contends that this is analogous to claims to a “computer implemented method”. At [105] of Research Affiliates, the Full Court cited and quoted from the decision in Bancorp Services LLC v Sun Life Assurance Company of Canada (2012) 687 F (3d) 1266 at 1277, 1278 and emphasised the following statement:
… [s]imply adding a ‘computer aided’ limitation to a claim covering an abstract concept, without more, is insuffıcient to render the claim patent eligible …
450 That is, simply adding in the words “prenatal diagnosis” are not sufficient to alter the fact that the claims are directed to information itself, which is not an artificially created state of affairs. Therefore a product claim to that information, nor a process claim to the method of producing that information can be regarded as a manner of manufacture.
451 In MLA (No 1), I considered the issue of manner of manufacture in the context of claims to methods for identifying a trait of a bovine subject from a nucleic acid sample. I referred to the principles summarised above at [439] to [442] and [447] of MLA (No 1), and addressed the application of those principles at [455] of MLA (No 1) where I said that the “taking of a nucleic acid sample of the bovine subject” provided the requisite artificially created state of affairs. But Ariosa says that my analysis in MLA (No 1) did not look to the ‘end result’ of the method, and did not require that that end result involve an artificial effect. It says that the taking of a sample of material was not the ‘end result’ of the method. The end result of claim 1 was information, namely the identification of a trait. It says that that identification did not involve an artificially created state of affairs. It would only be subsequent steps that might be taken, acting on such information, that might result in an artificially created state of affairs (such as the breeding of a selected animal with another selected animal). Such steps were not the subject of the claim itself, and did not form part of the invention ‘as claimed’.
452 In the present case Ariosa says that each of the relevant claims of the Patent involves as its end result (if there be one in respect of claims 1, 2, 3, 5, 6, 9 and 14) information, rather than an artificially created state of affairs. Each claim requires either that the presence of a fetal nucleic acid be detected (and quantified in the case of claim 14), and for claims 22, 23, 25 and 26, that a diagnosis result. It says that it follows that none of the claims is to a manner of manufacture.
453 Finally, Ariosa says that even if the claims of the Patent were found to be processes that would satisfy the threshold requirements of manner of manufacture, the claims are still not patentable as they are not within the established boundaries of what constitutes patentable subject matter. It says that it has not been established as a matter of Australian law that a method that comprises no more than detecting (using any means) a naturally occurring phenomenon constitutes patentable subject matter. That being the case, it contends that I should consider the additional factors identified by the plurality in Myriad at [28], which include whether the claimed invention, if patentable:
(a) could give rise to a large new field of monopoly protection with potentially negative effects on innovation; and
(b) could, because of the content of the claims, have a chilling effect on activities beyond those formally the subject of the exclusive rights granted to the patentee.
454 Ariosa says that the claims of the Patent cover any method that involves the detection of cffDNA in a plasma or serum sample from a pregnant woman. Claim 26 goes even further. It purports to cover any method of prenatal diagnosis that involves “performing nucleic acid analysis” on the non-cellular fraction of a maternal blood sample. That is, the patentee seeks to secure a monopoly on any and every application of Professor Lo’s discovery.
455 Indeed, so Ariosa says, the claims of the Patent are now asserted against the Harmony Test, which implements what it describes as extremely sophisticated technology that was developed more than a decade after the priority date. It points out that Professor Lovett acknowledged the approach adopted in the Harmony Test as “state of the art” and “quite innovative”. Further, Professor Hyett noted the potential impact of the Patent, whilst in force, on the availability of other non-invasive prenatal tests using cell-free DNA, and the potential impact on research and development.
456 Ariosa says that the claimed invention gave rise to a large new field of monopoly protection with potentially negative effects on innovation, and because of its content could have had a chilling effect on activities that travel well beyond those the subject of the Patent.
(c) Analysis
457 Let me analyse each of these arguments in turn after making a few general observations.
458 As Sequenom points out, Crennan and Kiefel JJ noted in Apotex Pty Ltd v Sanofi-Aventis Australia Pty Ltd (2013) 253 CLR 284 that (at [224]):
In Australian law, the starting point is the recognition in the NRDC Case that any attempt to define the word “manufacture” or the expression “manner of manufacture”, as they occur in s 6 of the Statute of Monopolies, is bound to fail.
459 In Apotex, the High Court was considering whether claims for methods of medical treatment of the human body constituted patentable subject matter. Crennan and Kiefel JJ identified seven reasons why they concluded that Apotex’s submissions purportedly derived from NRDC had to be rejected (at [278] to [286]). Three reasons may be mentioned.
460 First, the Act contains no specific exclusion from patentability of methods of medical treatment of the human body nor can any be implied (at [279] and [280]).
461 Second, no distinction could be drawn between the subject matter of a claim for a new product suitable for therapeutic use, claimed alone or coupled with method claims and the subject matter of a claim for a hitherto unknown method of treatment using a known product (at [282]):
In each case the subject matter in respect of which a monopoly is sought effects an artificially created improvement in human health, having economic utility…
462 Third, a method claim in respect of a hitherto unknown therapeutic use of a known substance satisfies the general principle laid down in NRDC. Such a method belongs to a useful art, effects an artificially created improvement in something and can have economic utility (at [283]).
463 In approaching the claims of the Patent, what is significant is that there is no claim to the product or presence of cffDNA but rather to a method by which the discovery of the existence of cffDNA can be put to practical use. In this way the subject matter of the relevant claims can be seen to fall within the principles of NRDC and affirmed in Myriad, whilst not falling within the impermissible product claims rejected in Myriad.
464 I have already discussed in MLA (No 1) the importance of the distinction between a product claim and a method claim which applies a naturally occurring phenomenon. Nothing that Ariosa has said goes close to persuading me to revisit what I have previously said or the distinction that I have previously drawn.
465 Myriad confirms that the starting point for the resolution of the “manner of manufacture” issue is the identification of what in substance each relevant claim is for. And relevant to this inquiry is the definition of the invention, which in turn depends upon the construction of the relevant claims read in light of the specification as a whole and the relevant prior art.
466 Now the present invention as defined, for example, by claims 6 and 9, when dependent on claims 2, 3 and 5, takes advantage of a previously unknown or unsuspected property of an artificially produced serum or plasma sample extracted and isolated from a pregnant female. The invention is undoubtedly an artificial non-invasive detection method involving artificial DNA amplification methods and synthetic probes deriving from the presence of cfDNA in the maternal circulation from both the mother and developing fetus.
467 Indeed, the Patent is entitled “Non-invasive prenatal diagnosis”. The first paragraph of the Patent provides that the invention described and claimed therein “relates to prenatal detection methods using non-invasive techniques” and “[i]n particular, it relates to prenatal diagnosis by detecting foetal nucleic acids in serum or plasma from a maternal blood sample.”
468 The Patent describes (page 1) the prenatal testing methods used before the priority date of the invention, including CVS and amniocentesis, being techniques requiring careful handling and that present a degree of risk to the mother and to the pregnancy, and the use of fetal cells in maternal blood.
469 The Patent explains (page 2, lines 15 to 19) that:
It has now been discovered that foetal DNA is detectable in maternal serum or plasma samples. This is a surprising and unexpected finding; maternal plasma is the very material that is routinely discarded by investigators studying non-invasive prenatal diagnosis using foetal cells in maternal blood. The detection rate is much higher using serum or plasma than using nucleated blood cell DNA extracted from a comparable volume of whole blood, suggesting that there is enrichment of foetal DNA in maternal plasma and serum. In fact, the concentration of foetal DNA in maternal plasma expressed as a % of total DNA has been measured as from 0.39% (the lowest concentration measured in early pregnancy), to as high as 11.4% (in late pregnancy), compared to ratios of generally around 0.001% and up to only 0.025% for cellular fractions (Hamada et al 1993). It is important that foetal DNA is found in maternal plasma as well as serum because this indicates that the DNA is not an artefact of the clotting process.
470 The Patent confirms (page 2, lines 20 to 23) that the invention is not simply the presence of cffDNA in maternal serum or plasma but rather, “a detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of foetal origin in the sample”. It is explained that the claimed method thus provides a “method of prenatal diagnosis”, a term defined at page 2, line 24 to page 3, line 4.
471 Examples of techniques falling within the scope of the claimed method are given in the Patent. I have previously described these in detail.
472 As Sequenom correctly submits, the relevant claims of the Patent broadly speaking, fall into four classes.
473 The first class is a detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of fetal origin in the sample (claim 1) by amplifying the nucleic acid (claim 2) by PCR (claim 3) and / or by the use of a sequence specific probe (claim 5), wherein the presence of a fetal nucleic acid sequence from the Y chromosome is detected (claim 6) or the presence of a fetal nucleic acid from a paternally-inherited non-Y chromosome is detected (claim 9).
474 The second class is a method according to the first class for determining the sex of the fetus (claim 13).
475 The third class is a method according to the first class which further comprises determining the concentration of the fetal nucleic acid sequence in the maternal serum or plasma sample (claim 14).
476 The fourth class is a provision of a “prenatal diagnosis” following the performance of the method referred to in the first class (claims 22, 23, 25 and 26).
477 It should be well apparent that unlike Myriad, this case does not concern whether product claims to naturally occurring nucleic acid molecules per se or naturally occurring genetic information encoded by such sequences are patentable. Rather, like MLA (No 1), this case concerns method claims (MLA (No 1) at [409] and [462]). Both MLA (No 1) and the present case involve claims to methods of identifying or detecting a nucleic acid having a particular characteristic, not the nucleic acids or the information encoded by such nucleic acids per se.
478 Now the scope of the product claims at issue in Myriad was criticised by Gageler and Nettle JJ at [139] for covering situations where:
… a pathologist had no interest in looking the specified mutations and polymorphisms – indeed let it be assumed that the pathologist vehemently rejected the conclusion that the specified mutations and polymorphisms were clinically significant – and was concerned with isolating the fragment of a patient’s DNA comprising the BRCA1 gene in order only to examine it for different mutations and polymorphisms which the pathologist’s independent research had led him or her to conclude were of clinical significance? In those circumstances, the only aspect of the claimed invention of which the pathologist could be said to make any use would be the discovery of the BRCA1 gene; and as has been seen, the BRCA1 gene is not patentable as such because it is a naturally occurring phenomenon which lacks the quality of inventiveness necessary to qualify as a manner of new manufacture.
479 But I agree with Sequenom that these criticisms in relation to the scope of Myriad’s claims compared to the scope of Myriad’s invention cannot be made in this case.
480 Further, at the least the detection method of claims 6 and 9 inherently involves at least artificial steps requiring human action, whatever else may be said about the earlier claims.
481 Further, all uses of the relevant method(s) would be commensurate with the scope of the inventor’s claimed new method of prenatal detection involving the detection of cffDNA from a maternal serum or plasma sample, rather than existing methods which all focused on more problematic cellular (as distinct from non-cellular) samples of DNA. The claims cover what the inventors of the Patent invented. In other words, the inventive step or concept, that is, that paternally inherited cffDNA is detectable in maternal plasma or serum, is the subject of and is carried out in, for example, claims 6 and 9.
482 Further, in this case, a person seeking to detect cffDNA from maternal serum or plasma would necessarily be aware of that fact and would be deliberately employing the method actually invented and claimed by the named inventors. And such a method of detection would involve the use of artificial human action, such as PCR amplification (claim 3) or probes (claim 5) designed specifically to detect cffDNA.
483 Moreover, the invention is specifically defined by the integers of these claims, including “detection method”, “detection” and “performed on a maternal serum or plasma sample”.
484 In the joint expert report, the experts agreed that in this context “detect” means demonstrating the presence of non-maternal nucleic acid that is inferred to be of fetal origin. It is difficult to see how it could sensibly be said that such a method does not involve artificial human action to discriminate or distinguish between nucleic acids of maternal origin and non-maternal (i.e. fetal) origin. Moreover, without such a step, one cannot detect the presence of non-maternal (i.e. fetal) nucleic acid.
485 As Sequenom correctly submits, the substance of the above method applies and follows on from, but is different to, the identification of a natural phenomenon, namely the presence of cffDNA in maternal blood. This is made clear by, for example, page 16, lines 4 to 6 of the Patent, which explains that this important discovery offers a new approach for non-invasive prenatal diagnosis. Thus, the invention builds on, uses, practically applies and reduces to practice a discovered substance found in nature, namely, cffDNA in maternal blood, to provide a new, inventive, useful, artificial method of detection of cffDNA, and where the method is of economic significance.
486 Further, on their face claims 13 and 14 provide a practical application of the discovery of the presence of cffDNA in maternal plasma. Now Ariosa suggests that these claims do no more than identify information, which it asserts is outside the bounds of patentable subject matter. But these claims like, for example, claims 6 and 9, are on their face different to the nucleic acid sequence claims considered by the plurality in Myriad. These claims are equivalent to the detection and diagnosis claims that I considered in MLA (No 1).
487 Further, claims 22, 23, 25 and 26 are founded upon the phrase “prenatal diagnosis”. On any view they are a manner of manufacture.
488 Let me say something about inventiveness. There is no allegation that anyone performed a method as claimed in the relevant claims before the priority date of the Patent. Here the inventors named in the Patent discovered the presence and utility of cffDNA and invented a new method of detection of fetal DNA, which unlike previously disclosed methods involves the human mediated analysis of cell free DNA in the cell free component of maternal blood to distinguish between maternal and fetal DNA in an artificially isolated maternal plasma or serum sample extracted from a pregnant female.
489 Further, as was observed in NRDC at 262:
… there may be invention in the suggestion that the substance may be used to serve the new purpose; ... It is not necessary that in addition the proposed method should itself be novel or involve any inventive step.
490 Further, the Court observed in NRDC at 264 that:
But where a person finds out that a useful result may be produced by doing something which has not been done by that procedure before, his claim for a patent is not validly answered by telling him that although there was ingenuity in his discovery that the materials used in the process would produce the useful result no ingenuity was involved in showing how the discovery, once it had been made, might be applied. The fallacy lies in dividing up the process that he puts forward as his invention. It is the whole process that must be considered; he need not show more than one inventive step in the advance he has made beyond the limits of the prior art.
491 Now in my view there is no requirement that the inventive concept be seen to make a contribution to the essential difference between the product and nature. But even if I were to conclude otherwise, the inventive concept of the Patent does so. In nature, the presence of cffDNA in the maternal blood has not and cannot be detected without human action. Accordingly, unlike the claims considered in Myriad, the invention claimed adds to human knowledge and involves the suggestion of an act to be done which results in a new result, or a new process.
492 Let me turn to the question of artificially created state of affairs.
493 A claimed process that results in a new and useful effect, so that the new result is an artificially created state of affairs providing economic utility, may be considered a manner of manufacture in the vast majority of cases. Further, as I have said, a product in relation to a claimed process is simply a discernible artificially created state of affairs having economic significance.
494 In the present case the relevant claims involve human interaction and the creation of an artificially created state of affairs discernible by an observer. The artificially created state of affairs is the detection of cffDNA in the tested sample. This “product” is, by definition the result of human action and is not naturally occurring. The inventive method does not simply produce an abstract, intangible situation. It is not just the “information” encoded by the naturally occurring cffDNA itself. Moreover, production of the result requires at least:
(a) The taking of a maternal blood sample from a pregnant female.
(b) The separation of the maternal blood sample into its component parts, namely, plasma, white blood cells and red blood cells. It is obvious that such samples do not exist in the natural world. They must be artificially created by centrifuging a sample of blood to remove all cells in the presence of an anti-coagulant in the case of plasma, or without an anti-coagulant but after clotting in the case of serum.
(c) The extraction of nucleic acids from the plasma component of a maternal blood sample.
(d) A step to discriminate between maternal and cffDNA in the sample, such as, the targeting and amplification by PCR of paternally inherited sequences from the Y chromosome in the case of claim 6 and a non-Y chromosome in the case of claim 9.
495 It is difficult to see how such a process of detection and discrimination is anything but an artificial process.
496 CffDNA in a given sample is detected as a result of this process. And without human action, any cffDNA cannot be detected in maternal blood. Quite unlike the genetic information embodied in the naturally occurring nucleotides considered in Myriad or the genetic information encoded by sequences of cffDNA, the physical detection result or outcome in the present case is made by human action.
497 Further, it can hardly be said that the invention claimed in each of the claims lacks economic utility or is otherwise “essentially non-economic”. In NRDC, the High Court drew a distinction between economic subject matter and matter belonging to the field of fine arts. But the present case is nowhere near the dividing line.
498 Further, I can do no better but to again refer to the analogy of Apotex where it was accepted that a method of human treatment satisfied the requirement of economic utility. Crennan and Kiefel JJ explained (at [282] to [283]):
Fourthly, and critically, the subject matter of a claim for a new product suitable for therapeutic use, claimed alone (a product claim) or coupled with method claims (combined product/method claims), and the subject matter of a claim for a hitherto unknown method of treatment using a (known) product having prior therapeutic uses (a method claim), cannot be distinguished in terms of economics or ethics. In each case the subject matter in respect of which a monopoly is sought effects an artificially created improvement in human health, having economic utility. It could not be said that a product claim which includes a therapeutic use has an economic utility which a method or process claim for a therapeutic use does not have. It could not be contended that a patient free of psoriasis is of less value as a subject matter of inventive endeavour than a crop free of weeds. Patent monopolies are as much an appropriate reward for research into hitherto unknown therapeutic uses of (known) compounds, which uses benefit mankind, as they are for research directed to novel substances or compounds for therapeutic use in humans. It is not possible to erect a distinction between such research based on public policy considerations.
Fifthly, leaving aside, for the moment, the relevant obiter dicta in the NRDC Case, a method claim in respect of a hitherto unknown therapeutic use of a (known) substance or compound satisfies the general principle laid down in the NRDC Case. Such a method belongs to a useful art, effects an artificially created improvement in something, and can have economic utility. The economic utility of novel products and novel methods and processes in the pharmaceutical industry is underscored by s 119A of the 1990 Act and by their strict regulation in the Therapeutic Goods Act 1989 (Cth) (“the TGA”).
(Emphasis added.)
499 In the present case, the claimed method of the invention is itself of economic significance. As Sequenom points out, contrary to the nucleic acid isolate considered in Myriad, it is not merely a step along the way to another method claimed as an artificially created state of affairs of economic significance.
500 Further, the claimed method provides a significant advantage to say the least over existing fetal DNA detection methods available at the priority date within a technological field of molecular genetics and fetal medicine and produces a result, that is, the detection of cffDNA that possesses its own economic utility.
501 Let me turn to the “other factors” referred to in Myriad.
502 I held in MLA (No 1) (at [428]):
But the “other factors” do not arise unless the claims in question require an extension of the existing concept of manner of manufacture to a new class of case or are on the border.
503 In my view the claimed invention falls clearly within the concept of manner of manufacture described by the High Court in NRDC, satisfies the first two criteria identified in Myriad at [28] and is therefore patentable. But even if the substance of the invention is considered to fall outside the established boundaries of patentable subject matter, consideration of the non-exhaustive list of additional factors outlined in Myriad confirms my opinion of the invention’s patentability.
504 Now I noted in MLA (No 1) at [390] that the plurality in Myriad identified factors 3, 4 and 6 as being of primary importance and as not mutually exclusive. I noted that factor 5 was said to be of secondary significance.
505 As I say, in my view the present invention falls squarely within the established concept of manner of manufacture. This case does not concern a new class of claim and nor are Sequenom’s claims directed to a product reproducing information that is not made by human action. Accordingly, these other factors do not apply. But in any event the following observations may be made. Let me deal with factors 3, 4 and 5 in turn.
506 As to factor 3, there is no chilling effect.
507 Each of the relevant claims cover methods wherein steps are taken to “detect” the presence of a nucleic acid of fetal origin in a maternal plasma or serum sample. As explained above, “detection” requires a positive step to be taken to discriminate maternal cell-free DNA from cffDNA by, for example, amplifying, probing and targeting specific paternally inherited sequences not possessed by the pregnant female on a Y or non-Y chromosome. The boundaries of the claims are clear and are commensurate with the invention made and described in the Patent.
508 Further, these claims do not cover all applications of the patentee’s discovery that cffDNA is detectable in maternal blood. For example, Sequenom accepts that Ariosa would not be infringing such claims if it did not “detect” cffDNA by performing the Polymorphic Assay (fetal fraction), which I will explain later, or by detecting sequences from the Y chromosome. That is, Ariosa’s method would not infringe if it was confined to the non-Y Non-Polymorphic Assay, which I will also explain later when dealing with the Harmony Test.
509 Further, unlike the circumstances concerning claims 1 to 3 in Myriad, there is no risk that someone could infringe the claim without knowing it. There is no cogent evidence to support the notion that the relevant claims would have had a chilling effect on activities beyond those formally the subject of the exclusive rights granted to the patentee.
510 As to factor 4, my analysis promotes coherency of Australian law.
511 Sequenom put the attractive argument, which I accept, that for example, claims 6 and 9 would promote, rather than detract from, coherency in the application of the law by reference to the claims that I found to be patentable in MLA (No 1).
512 In MLA (No 1), I found the following method claim to involve a manner of manufacture:
A method for identifying a trait of a bovine subject from a nucleic acid sample of the bovine subject, comprising identifying in the nucleic acid sample an occurrence of at least three single nucleotide polymorphisms (SNPs) wherein the at least three SNPs are associated with the trait, and wherein the at least three SNPs occur in more than one gene [:]
[a] and wherein at least one of the SNPs corresponds to position 300 of any one of SEQ ID NOS: 19473 to 21982, or
[b] the SNP is about 500,000 or less nucleotides from position 300 of any one of SEQ ID NOS: 19473 to 21982.
513 The structure and substance of that claim resembles claims 6 and 9 of the Patent, as illustrated by Sequenom’s table that I have reproduced.
MLA claim 1 | Patent claim 6 | Patent claim 9 |
A method for identifying a trait of a bovine subject from a nucleic acid sample of the bovine subject, | A detection method performed on a maternal serum or plasma sample from a pregnant female (i.e. a method for detection of a characteristic (presence/absence of cell free-foetal DNA) of a pregnant female from a sample of the pregnant female). Claim 6 could correspondingly be expressed as a method for the purposes of prenatal detection. | A detection method performed on a maternal serum or plasma sample from a pregnant female (i.e. a method for detection of a characteristic (presence/absence of cell free-foetal DNA) of a pregnant female from a sample of the pregnant female). Claim 9 could correspondingly be expressed as a method for the purposes of prenatal detection. |
comprising identifying in the nucleic acid sample an occurrence of at least three single nucleotide polymorphisms (SNPs) wherein the at least three SNPs are associated with the trait, and wherein the at least three SNPs occur in more than one gene [:] [a] and wherein at least one of the SNPs corresponds to position 300 of any one of SEQ ID NOS: 19473 to 21982, or [b] the SNP is about 500,000 or less nucleotides from position 300 of any one of SEQ ID NOS: 19473 to 21982. | Which method comprises detecting the presence of a foetal nucleic acid from a paternally-inherited Y chromosome in the sample, wherein the foetal nucleic acid is amplified by the polymerase chain reaction and the foetal nucleic acid is detected by means of a sequence specific probe. | Which method comprises detecting the presence of a foetal nucleic acid from a paternally-inherited non-Y chromosome in the sample, wherein the foetal nucleic acid is amplified by the polymerase chain reaction and the foetal nucleic acid is detected by means of a sequence specific probe. |
514 Both claims provide a method of identification (detection) from a sample comprising nucleic acids and comprise identifying (detecting) the occurrence (presence) of nucleic acids having a particular target characteristic (association with a trait (MLA) vs. of fetal origin from a paternally inherited non-Y or Y chromosome).
515 Of the MLA claims, I said at [487]:
What is claimed are method claims. It is well accepted that method claims can use known products and apply “natural laws” to their working. A method claim cannot sensibly be characterised as a claim to information. It is a claim to inter-alia the application of information. In my view to find that the method claims in suit involve a “manner of manufacture” is to enhance rather than detract from the coherency of Australian law.
516 These observations are apposite in the present case. In view of my findings in MLA (No 1) and the similarities between the claims in issue in MLA (No 1) and the present case, to find that claims 6 and 9 relate to patentable subject matter would enhance the coherency of Australian law.
517 Further, the patentability of the relevant claims would also be consistent with the conclusion in Apotex that methods of prevention and treatment of human disease constitute patentable inventions. A new and inventive method of detecting a naturally occurring phenomenon or undertaking a form of diagnosis based thereon ought to not be distinguished from a method of preventing or treating a naturally occurring phenomenon.
518 Finally, let me say something about factor 5 and consistency with foreign law.
519 Claims equivalent to claims 6 and 9 were recently held to be patentable by Carr J in the corresponding UK proceedings between Ariosa and Sequenom (Illumina, Inc v Premaitha Health Plc [2017] EWHC 2930 (Pat) at [184] to [189]). In that case, after recognising that in deciding the question of patentability it was necessary to distinguish substance from form, Carr J concluded at [189]:
I do not accept that, properly construed, claim 1 is a claim to a discovery as such. The claims are not directed to information about the natural world, but rather to a practical process, namely a “detection method” which uses information about the natural world. Claim 1 is directed to the detection of foetal DNA in a sample of plasma or serum. Such samples do not exist in the natural world and must be artificially created. The claimed method of detection is also an artificial process which does not exist in the natural world. The claim is to a practical process of implementing a discovery, for practical applications. The actual contribution, as a matter of substance, does not fall solely within the excluded subject matter and is technical in nature.
520 But contrastingly, claims corresponding to the relevant claims were found not to be patentable in the US (Ariosa (US)). Those claims provided, by way of example (at 1373 to 1374):
1. A method for detecting a paternally inherited nucleic acid of fetal origin performed on a maternal serum or plasma sample from a pregnant female, which method comprises
amplifying a paternally inherited nucleic acid from the serum or plasma sample and
detecting the presence of a paternally inherited nucleic acid of fetal origin in the sample.
…
24. A method for detecting a paternally inherited nucleic acid on a maternal blood sample, which method comprises: removing all or substantially all nucleated and anucleated cell populations from the blood sample,
amplifying a paternally inherited nucleic acid from the remaining fluid and subjecting the amplified nucleic acid to a test for the Paternally [sic] inherited fetal nucleic acid.
25. A method for performing a prenatal diagnosis on a maternal blood sample, which method comprises
obtaining a non-cellular fraction of the blood sample
amplifying a paternally inherited nucleic acid from the non-cellular fraction
and performing nucleic acid analysis on the amplified nucleic acid to detect paternally inherited fetal nucleic acid.
521 The majority of the US Court comprising Reyna and Wallach JJ concluded that:
(a) these claims were method claims directed to matter that is naturally occurring and in particular to detecting the presence of a naturally occurring thing; and
(b) the practice of the method claims did not result in an inventive concept that transformed the natural phenomenon of cffDNA into a patentable invention.
522 In reaching the latter conclusion, the Court considered that the method or process recited by the claims of the patent in suit before it was not new and useful. But this conclusion is problematic, and is a result of the US Court’s dissection of the claims into their constituent parts, which is contrary to NRDC and Myriad.
523 Further, applying such an approach would also be inconsistent with the approach I adopted in MLA (No 1), which led to a finding of patentability in circumstances where process features of the method claim were known. In respect of the dissection of claims, in MLA (No 1) I observed at [794] albeit in the context of an inventive step assessment that:
… although MLA has asserted that each of the various steps taken by the inventors of the 253 Application were individually known and therefore the invention as claimed was obvious, I agree with Branhaven that such an approach invites error. The invention is directed to a method for inferring a bovine trait using a number of identified SNPs all of which must be associated with the trait. Even if it were accepted that each of the individual methods used by the inventors were known techniques, that would not provide a proper basis for finding that the claimed invention was obvious.
524 I also dealt with the US approach to patentability at [492] of MLA (No 1), explaining that:
The US approach accepts that a method involving the application of a “law of nature” may be patentable. Indeed, the contrary could not be seriously argued. What workable method in its application is ever free of a “law of nature”? The US debate turns more on the question of what it takes “to transform an unpatentable law of nature into a patent-eligible application of such a law” (Mayo at 72). And everyone seems to agree that you need to do more than simply state the law of nature while adding the words “apply it”. But then one is into quite unclear territory. The exposition of the test (particularly the second stage) in Mayo is too sweeping for me to work out whether I am acting consistently or inconsistently with its spirit in the conclusion that I have reached that the claims of the 253 Application (save for claim 13) are patentable.
525 Further, the observations of Linn J in the US decision perhaps better reflect the approach to patentability adopted in Australia following Myriad. Linn J reluctantly concurred with the majority. He felt he was bound by Mayo, but lamented the outcome. He also explained that the Mayo decision did not allow the Court to treat new and old methods differently in circumstances where the constituent parts of such methods were, by themselves, known or conventional. He said (at 1381):
The Supreme Court’s blanket dismissal of conventional post-solution steps leaves no room to distinguish Mayo from this case, even though here no one was amplifying and detecting paternally-inherited cffDNA using the plasma or serum of pregnant mothers. Indeed, the maternal plasma used to be “routinely discarded…
It is hard to deny that Sequenom’s invention is truly meritorious. Prior to [the invention], prenatal diagnoses required invasive methods… Dr. Mark Evans testified that “despite years of trying by multiple methods, no one was ever able to achieve acceptable success and accuracy.” In a ground breaking invention, Drs. Lo and Wainscoat discovered that there was cell-free fetal DNA in the maternal plasma. The Royal Society lauded this discovery as “a paradigm shift in non-invasive prenatal diagnosis,” and the inventors’ article describing this invention has been cited well over a thousand times… Unlike in Mayo, the ‘540 patent claims a new method that should be patent eligible. While the instructions in the claims at issue in Mayo had been widely used by doctors – they had been measuring metabolites and recalculating dosages based on toxicity / inefficacy limits for years – here, the amplification and detection of cffDNA had never before been done. The new use of the previously discarded maternal plasma to achieve such an advantageous result is deserving of patent protection…
In short, Sequenom’s invention is nothing like the invention at issue in Mayo. Sequenom “effectuate[d] a practical result and benefit not previously attained,” so its patent would traditionally have been valid… But for the sweeping language in the Supreme Court’s Mayo opinion, I see no reason in policy or statute, why this breakthrough invention should be deemed patent ineligible.”
(Citations omitted; emphasis in original).
(d) Conclusion
526 The Patent does not simply claim the discovery of cffDNA in maternal blood. Rather, it claims a new and inventive practical application of the discovery comprising a method requiring human action to detect, in an artificially created sample of maternal plasma or serum, a DNA sequence as being of fetal rather than maternal origin. And prior to the invention, no-one had worked or was working a method comprising the detection of cffDNA in plasma or serum samples extracted from pregnant females.
527 For the above reasons, the invention the subject of each of the relevant claims is a patentable invention.
528 Finally, I should note that during the course of argument I tentatively thought that claim 1, and possibly claims 2, 3 and 5, may have been problematic as manners of manufacture. But on further reflection I am satisfied that they are, although the position is obviously stronger for claims 6, 9 and following.
INVENTIVE STEP
529 Ariosa contends that claims 1 to 3, 5, 6, 9, 13, 14, 22, 23, 25 and 26 of the Patent lack an inventive step in light of the common general knowledge at the priority date considered together with Kazakov VI et al, “Extracellular DNA in the blood of pregnant women” (1995) 37(3) Cytology 232 (the Kazakov paper).
530 The applicable versions of ss 7(2) and (3), given that the application on which the Patent was granted was filed after 1 April 1991 and before 1 April 2002, are:
(2) For the purposes of this Act, an invention is to be taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed in the patent area before the priority date of the relevant claim, whether that knowledge is considered separately or together with either of the kinds of information mentioned in subsection (3), each of which must be considered separately.
(3) For the purposes of subsection (2), the kinds of information are:
(a) prior art information made publicly available in a single document or through doing a single act;
… being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant to work in the relevant art in the patent area.
531 Sequenom says that the obviousness attack fails for at least the following reasons:
(a) a person skilled in the art could not reasonably be expected to have ascertained the Kazakov paper or its abstract at the priority date;
(b) a person skilled in the art could not reasonably be expected to have treated the information in the Kazakov paper or its abstract as being relevant to work directed to solving the kind of problem said to be solved by the Patent, namely, a research project concerned with developing a new or improved product or process for use in the non-invasive prenatal diagnostic/testing field;
(c) a person skilled in the art would not have been directly led as a matter of course by the Kazakov paper or its abstract to try to detect cffDNA in a maternal plasma or serum sample; and
(d) even if a person skilled in the art tried to do so, they would not have had any reasonable expectation of detecting cffDNA in such samples.
532 In relation to the relevant legal principles, I adopt what I said in MLA (No 1) at [678] to [694] to the following effect.
533 The question is whether the claimed invention lacks an inventive step over the prior art base. An invention is taken to involve an inventive step when compared to the prior art base unless it would have been obvious to a person skilled in the relevant art in light of common general knowledge as described in Minnesota Mining and Manufacturing Co v Beiersdorf (Australia) Ltd (1980) 144 CLR 253 (Minnesota Mining) at 292 per Aickin J as it existed in the patent area (the then s 7(2) of the Act) before the priority date, whether that knowledge is considered separately or together with information of the kind described in the then s 7(3).
534 The term “obvious” means “very plain” (Aktiebolaget Hässle v Alphapharm Pty Ltd (2002) 212 CLR 411 at [34] per Gleeson CJ, Gaudron, Gummow and Hayne JJ and Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) (2007) 235 CLR 173 at [51]). The inventive element needed to sustain a patent can be small. A “scintilla of inventiveness” will be sufficient and “no smallness or simplicity will prevent a patent being good” (Meyers Taylor Pty Ltd v Vicarr Industries Ltd (1977) 137 CLR 228 at 249 per Aickin J). Relevant further content has been given to determining obviousness in The Wellcome Foundation Ltd v VR Laboratories (Aust) Pty Ltd (1981) 148 CLR 262 at 286 per Aickin J in stating:
whether the hypothetical addressee faced with the same problem would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not.
535 Further, in relation to experiments, his Honour said (at 280 and 281):
In the present case it was admitted by the respondent that the test of obviousness was an objective one, but it was argued that evidence of a subjective character was admissible. That is no doubt true in some cases because expert witnesses are often properly asked whether they found a particular invention “surprising” to them. That however does not answer the question whether evidence of the steps which the patentee took is relevant and therefore admissible. Evidence of what was in the patentee’s mind may be admissible as evidence of the state of the art, but could seldom be otherwise admissible. Evidence of what he did by way of experiment may be another matter. It might show that the experiments devised for the purpose were part of an inventive step. Alternatively it might show that the experiments were of a routine character which the uninventive worker in the field would try as a matter of course. The latter could be relevant though not decisive in every case. It may be that the perception of the true nature of the problem was the inventive step which, once taken, revealed that straightforward experiments will provide the solution. It will always be necessary to distinguish between experiments leading to an invention and subsequent experiments for checking and testing the product or process the subject of the invention. The latter would not be material to obviousness but might be material to the question of utility.
536 So, would the addressee faced with the same problem have taken, as a matter of routine, whatever steps might have led from the prior art to the invention? That question has an affinity with the Cripps question posed in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd [1970] RPC 157 and paraphrased by French CJ in AstraZeneca AB v Apotex Pty Ltd (2015) 257 CLR 356 at [15] in the following terms:
Would the notional research group at the relevant date, in all the circumstances, which include a knowledge of all the relevant prior art and of the facts of the nature and success of [the existing compound], directly be led as a matter of course to try [the claimed inventive step] in the expectation that it might well produce a useful alternative to or better drug than [the existing compound]?
537 Further, the plurality said in Aktiebolaget Hässle (at [58]) that:
The tracing of a course of action which was complex and detailed, as well as laborious, with a good deal of trial and error, with dead ends and the retracing of steps is not the taking of routine steps…
538 Further, it is erroneous to characterise as obvious the variation of all parameters or the trying of all choices until one proves successful, where the prior art did not point to it. Similarly, it is erroneous to characterise as obvious the exploration of a new technology or a promising field of experimentation, where the prior art gave no more than general guidance.
539 Further, in Aktiebolaget Hässle the plurality cited (at [74]) Judge Rich in In re O’Farrell (1988) 853 F 2d 894 at 903 who said:
[F]or many inventions that seem quite obvious, there is no absolute predictability of success until the invention is reduced to practice. There is always at least a possibility of unexpected results, that would then provide an objective basis for showing that the invention, although apparently obvious, was in law nonobvious.
540 Now impermissible hindsight should be avoided in determining whether a claimed invention lacks an inventive step. In Aktiebolaget Hässle (at [21] and [78]) the importance of avoiding hindsight was reinforced in the following terms:
[T]he warnings in the authorities against the misuse of hindsight are not to be repeated as but prefatory averments and statements of trite law. The danger of such misuse will be particularly acute where what is claimed is a new and inventive combination for the interaction of integers, some or all of which are known. It is worth repeating what was said by Lord Diplock in Technograph Printed Circuits Ltd v Mills & Rockley (Electronics) Ltd:
“Once an invention has been made it is generally possible to postulate a combination of steps by which the inventor might have arrived at the invention that he claims in his specification if he started from something that was already known. But it is only because the invention has been made and has proved successful that it is possible to postulate from what starting point and by what particular combination of steps the inventor could have arrived at his invention. It may be that taken in isolation none of the steps which it is now possible to postulate, if taken in isolation, appears to call for any inventive ingenuity. It is improbable that this reconstruction a posteriori represents the mental process by which the inventor in fact arrived at his invention, but, even if it were, inventive ingenuity lay in perceiving that the final result which it was the object of the inventor to achieve was attainable from the particular starting point and in his selection of the particular combination of steps which would lead to that result.”
… After its review of the evidence, the Full Court concluded that Astra's “development” of the formulation “was essentially an exercise in trying out various known possibilities until the correct solution emerged” (emphasis added). That view of the matter wrongly takes as the starting point the assumed result. It succumbs immediately to the seduction of hindsight. Also, the notion of trying out possibilities invites the repetition of criticisms made earlier in these reasons.
(Citations omitted.)
541 In Lockwood (No 2) (at [46]) the High Court repeated the caveat against the misuse of hindsight. Clearly, any inordinate use of hindsight is likely to conceal or gloss over all of the problems, blind alleys and choices of options encountered by the patent applicant along the path to the invention.
542 Further, it is self-evident that problems with hindsight are even further elevated where the claimed invention concerns the application of known principles.
543 Further, in Minnesota Mining & Manufacturing Co v Tyco Electronics Pty Ltd (2002) 56 IPR 248, Heerey, Emmett and Dowsett JJ said that little weight should be given to expert evidence that a claimed invention was obvious if the expert was provided with a copy of the patent or other relevant information before giving their evidence (at [45] and [46]):
The manner in which the evidence of some of the experts in the present case was bought into existence suggests that relatively little weight should be given to certain of that evidence. For example, witnesses were provided with a copy of the patent. They were either provided with a large number of other documents or found them in response to the task that was set them. That is hardly calculated to result in objective evidence as to what the hypothetical uninventive but skilled worker would have done. To give the patent to a prospective witness is tantamount to leading the witness. Further, unless the other documents were part of the common general knowledge in Australia before the priority date, they are not relevant to any question of obviousness.
Evidence by “experts” on the question of obviousness … is not always likely to be helpful: see Firebelt Pty Ltd v Brambles Australia Ltd (2002) 188 ALR 280; 54 IPR 449 at [46]. Indeed, where evidence is obtained in circumstances such as just described, the evidence is not likely to be helpful at all.
544 At the least lesser weight is to be given to the evidence of experts asserting an invention to be obvious after they have reviewed the patent or been provided with other information concerning the state of the art. Undoubtedly, there is a need for caution where an expert asserting obviousness knew both the problem and the solution.
545 Further, expert evidence as to what asserted s 7(3) information would have disclosed at the relevant time has lesser weight if an expert has undertaken an exercise of looking for the integers of the claims in the prior art and trying to find them, if necessary by combining different parts of the article without explanation of why he would have done so at its publication date in the absence of knowledge of the claims.
546 Let me say something further about common general knowledge.
547 Section 7(2) first requires consideration of what would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed before the priority date, putting to one side for the moment s 7(3) information. Common general knowledge is knowledge “generally known and accepted without question by the bulk of those who are engaged in the particular art” (British Acoustic Films Ld v Nettlefold Productions (1936) 53 RPC 221 at 250 per Luxmoore J). Information cannot be treated as part of the common general knowledge unless there is evidence of its general acceptance and assimilation by persons skilled in the art. And information does not constitute common general knowledge merely because it might be found for example in a journal, even if widely read by such persons. Further, as Luxmoore J said at 250:
In my judgment it is not sufficient to prove common general knowledge that a particular disclosure is made in an article, or series of articles, in a scientific journal, no matter how wide the circulation of that journal may be, in the absence of any evidence that the disclosure is accepted generally by those who are engaged in the art to which the disclosure relates. A piece of particular knowledge as disclosed in a scientific paper does not become common general knowledge merely because it is widely read, and still less because it is widely circulated. Such a piece of knowledge only becomes general knowledge when it is generally known and accepted without question by the bulk of those who are engaged in the particular art; in other words, when it becomes part of their common stock of knowledge relating to the art. Whatever else common general knowledge may be, it has never in my judgment included public knowledge of particular documents reports or scientific papers and the like. The knowledge of a number of individuals that a particular suggestion or particular suggestions has or have been made for the use of biasing in a particular apparatus, or a number of particular apparatus, cannot be held to be common general knowledge. It is certainly difficult to appreciate how the use of something which has in fact never been used in a particular art can ever be held to be common general knowledge in the art.
548 Further, as stated in Minnesota Mining by Aickin J (at 292), the notion of common general knowledge:
involves the use of that which is known or used by those in the relevant trade. It forms the background knowledge and experience which is available to all in the trade in considering the making of new products, or the making of improvements in old, and it must be treated as being used by an individual as a general body of knowledge.
549 So it must be knowledge that is known and available to all in the trade or at least the bulk of those who are engaged in the relevant art. Accordingly, information ascertainable by a routine literature search is not of itself taken to be common general knowledge. And patent specifications do not form part of common general knowledge without evidence that they have been absorbed into common general knowledge.
550 And as further elucidated by Jagot J in Gilead Sciences Pty Ltd v Idenix Pharmaceuticals LLC (2016) 117 IPR 252 at [216] (affirmed in Idenix Pharmaceuticals LLC v Gilead Sciences Pty Ltd (2017) 134 IPR 1 per Nicholas, Beach and Burley JJ), it is erroneous to treat a document as being part of common general knowledge simply because skilled persons could readily locate and assimilate its contents. Her Honour went on to explain (at [217]):
It may be accepted that instant recall of an article is not required. This does not mean, however, that documents found by searching for a subject-matter, rather than by some form of recall or reminder of what is already known to exist, are common general knowledge. This is so irrespective of the fact that experts in the field read widely. Further, it is not the case that mere publication and republication proves that a document and its contents have entered the common general knowledge. Nor is it the fact that a document and its contents necessarily form part of the common general knowledge merely because one expert knows or has managed to locate it and assimilate its contents. Such a document may or may not form part of the common general knowledge. The relevant inferences are to be drawn on the basis of the whole of the evidence.
551 Let me say something at this point on s 7(3) albeit briefly.
552 In addition to using common general knowledge on a stand-alone basis, common general knowledge can be aggregated with s 7(3) information. That part of the prior art base which is common general knowledge and the information referred to in s 7(3) are considered for the purpose of looking forward from the prior art base to see what the skilled person is likely to have done when faced with a particular problem. Now in a case where the problem is known and is part of the common general knowledge, the problem may be similar to that which the patentee claims to have solved with the claimed invention. But where the problem addressed by the patentee does not form part of common general knowledge, the relevant starting point is the prior art base, but not including the problem as identified in the patent specification. As was noted in AstraZeneca AB v Apotex Pty Ltd (2014) 226 FCR 324 by the plurality (at [203]):
If the problem addressed by a patent specification is itself common general knowledge, or if knowledge of the problem is s 7(3) information, then such knowledge or information will be attributed to the hypothetical person skilled in the art for the purpose of assessing obviousness. But if the problem cannot be attributed to the hypothetical person skilled in the art in either of these ways then it is not permissible to attribute a knowledge of the problem on the basis of the inventor’s “starting point” such as might be gleaned from a reading of the complete specification as a whole.
553 The purpose of the inquiry is to determine whether the invention is obvious to solve the perceived problem, looking forward from the prior art base. But of course this may not have been the patentee’s starting point.
554 Further, in AstraZeneca AB v Apotex Pty Ltd (2015) 257 CLR 356, Kiefel J summarised the effect of s 7(3) as follows (at [68] to [70]):
Before a document containing prior art information can be used along with the common general knowledge for the purposes of the s 7(2) inquiry, it is necessary that it meet the requirements of s 7(3). In Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) it was explained that prior art information which is publicly available in a single document is “ascertained” if it is discovered or found out, and “understood” means that, having discovered the information, the skilled person would have comprehended it or appreciated its meaning or import. The Court also explained that the phrase “relevant to work in the relevant art” is directed to publicly available information, not part of the common general knowledge, which the skilled person could be expected to have regarded as relevant to solving a particular problem, or meeting a long-felt want or need, as the patentee claims to have done.
Lockwood (No 2) also explains [at [127]] that, in answering the question of obviousness, the information referred to in s 7(3), like that part of the prior art base which is the common general knowledge, is considered for a particular purpose. That purpose is to look forward from the prior art base to see what the skilled person is likely to have done when faced with a problem similar to that which the patentee claims to have solved with the claimed invention. It is this aspect of the s 7(2) inquiry which assumes particular importance on these appeals.
In addressing s 7(2), it is to be borne in mind that the skilled person is an artificial construct, intended as an aid to the courts in addressing the hypothetical question of whether a person, with the same knowledge in the field and aware of the problem to which the patent was directed, would be led directly to the claimed invention. The statute’s creation of the skilled person construct for this purpose is not to be taken as an invitation to deal with the question posed by s 7(2) entirely in the abstract. Whilst the question remains one for the courts to determine, the courts do so by reference to the available evidence including that of persons who might be representative of the skilled person.
(Citations omitted.)
555 Let me say something further on the question of reasonable ascertainability.
556 As at the priority date, in order for information in addition to common general knowledge to be able to be taken into account in assessing inventive step, it was a requirement pursuant to s 7(3) (in its then terms) that a person skilled in the art could, before the priority date, be reasonably expected to have ascertained, understood, and regarded as relevant that information.
557 The word “ascertained” means “discovered or found out”. The fact that a document has been published does not mean that a person skilled in the art could be reasonably expected to have ascertained that information. The relevant question is whether the document was published in such a manner or form that it could reasonably have been expected to be found by a person skilled in the art.
558 In assessing whether a person skilled in the art could be reasonably expected to have ascertained the information, it is important to consider the characteristics of the person skilled in the art, the nature of the problem, and the information typically used by the persons skilled in the art. Section 7(3) does not assume an ascertainability by any and all skilled persons, of whatever description, of all publicly available prior art documents anywhere in the world.
559 The Full Court in Commissioner of Patents v Emperor Sports Pty Ltd (2006) 149 FCR 386 has said that there may be situations where it would not be reasonable to have an expectation that a person skilled in the relevant art would conduct a search of the patent literature. The cases which have considered the question of reasonable expectation of ascertainment, for example, JMVB Enterprises Pty Ltd v Camoflag Pty Ltd (2006) 154 FCR 348, KD Kanopy Australasia Pty Ltd v Insta Image Pty Ltd (2007) 71 IPR 615 and Delnorth Pty Ltd v Commissioner of Patents (2013) 100 IPR 175 have established a number of principles in this respect.
560 First, the mere fact that some participants in the field might undertake patent searches is not sufficient to establish a reasonable expectation of ascertainment. In Delnorth, Nicholas J refused to find that a person skilled in the art could be reasonably expected to have ascertained the relevant patents, even though there was evidence of patent searching and substantial R&D expenditure, because inter-alia (at [53]):
Mr Turner’s evidence was vague as to the nature of the patent searches undertaken by or on behalf of his company during and after the mid-1980s, and the circumstances in which they were undertaken. In particular, it said nothing about the scope of such searches including how they were targeted or how far back in time they extended.
561 Second, regard to the jurisdiction in which any prior patents, asserted to constitute s 7(3) information, were filed is important. In JMVB Enterprises Pty Ltd v Camoflag Pty Ltd, the Full Court held that it could not, on the facts of that case, be reasonably expected that a person skilled in the art in Australia would have ascertained patents filed in New Zealand (at [77]):
[T]hose contentions ignore the distinction between what might reasonably be expected of a prospective seller of caravans in New Zealand, on the one hand, and the notional person skilled in the field of campervan design in Australia. There was no evidence that any designer would actually contemplate conducting a patent search in New Zealand.
562 Third, the age of the asserted s 7(3) information is of some significance. It is conceivable that searches undertaken might extend to patents granted many years before the priority date of the patent in suit. But this may not be sufficient to establish a reasonable expectation that a person skilled in the art would search such old patents for the purpose of solving the problem to which the patent in suit was directed.
563 Fourth, it is not sufficient for a party seeking to rely upon a prior patent as s 7(3) information to establish that a person skilled in the art had some familiarity with the patent literature as at the priority date. The question is whether it could be reasonably expected that a person skilled in the art would have ascertained the specific patents which were asserted to constitute s 7(3) information. Accordingly, if it is asserted that a person skilled in the art would have ascertained a prior patent by undertaking a routine search, then unless an inference could be drawn, evidence would generally be required that the person skilled in the art may reasonably be expected to have done such a search.
564 Further, the requirement of “reasonableness” is applied to each of the “ascertained”, “understood” and “regarded as relevant” limbs of the s 7(3) test.
565 In Aspirating IP Ltd v Vision Systems Limited (2010) 88 IPR 52, it was explained at [457] to [458] that whether something could be reasonably expected requires a prediction and that in the context of s 7(3) the question is whether, having regard to the evidence, it may be reasonably predicted that the hypothetical non-inventive skilled worker faced with the relevant problem before the priority date would have ascertained, understood and regarded as relevant the particular s 7(3) information in question.
566 Further, the Full Court in Commissioner of Patents v Emperor Sports Pty Ltd (2006) 149 FCR 386 in considering the “ascertained” limb of s 7(3), noted at [31]:
Section 7(3) does not assume an ascertainability by any and all skilled persons, of whatever description, of all publicly available prior art documents anywhere in the world. Nor does it assume that the skilled person has found the document in question, so that the only question is whether he or she has understood it and regarded it as relevant. Such a construction ignores the elements of expectation and reasonableness, as applied to the particular skilled person.
(Emphasis added.)
567 In Emperor Sports, the Full Court concluded that the asserted prior art documents in that case, being seven United States patents, were not part of the prior art base as extended by s 7(3). This was because the relevant skilled person could not reasonably have been expected to have ascertained the patents. In that case, the person skilled in the art was a Rugby League or Australian Rules Football coach, referee, umpire or administrator. The Full Court held at [35]:
Simply stated, we think it self-evident that it could not be reasonably expected that a Rugby League or Australian Rules coach, referee, umpire or administrator would conduct a search in the United States Patent Office. Such an expectation would be fanciful rather than reasonable.
568 The Full Court in Emperor Sports held that “ascertained” in s 7(3) is used in the sense of “[t]o find out or learn for a certainty by experiment, examination, or investigation; to make sure of, to get to know” (at [29]). The Court observed that this interpretation was consistent with the word “find” as suggested by the IPAC Committee in its recommendation for what became s 7(3). The IPAC Committee used the expression “reasonably have been expected to find, understand and regard as relevant” (at [30]). The Court noted that the Commissioner accepted that the change from “find” to “ascertained” appeared to be no more than a matter of drafting stylistic preference (at [30]).
569 The issue of “reasonable ascertainment” was also addressed by Branson J in EI Dupont de Nemours & Co v Imperial Chemical Industries plc (2002) 54 IPR 304 . In that case, Branson J’s conclusion as to whether an alleged s 7(3) disclosure defined as the “Lubrizol patent” could reasonably have been ascertained at [108] was that:
… although it is possible to identify the Lubrizol patent by using appropriate search terms in a search of relevant electronic data bases, the applicant has failed to establish that the patent was reasonably ascertainable in the sense required by s 7(3) of the Act. I accept the submission of the respondent that the search exercise of which the applicant called evidence was likely to have been influenced by knowledge of the result sought to be achieved, and not such as would reasonably be expected to be undertaken by the hypothetical skilled worker.
570 So a mere “possibility” of an alleged s 7(3) document being ascertained or treated as relevant is not sufficient for the purposes of s 7(3). And the relevant question is to be posed in terms of “would find” rather than “could find”.
571 Finally, the meaning of the phrase “regarded as relevant” was addressed in Lockwood (No 2) in the following terms (at [152] to [153]):
Given the history, context, purpose and specific words of limitation in s 7(3), all of which were addressed by this Court in Firebelt, the phrase “relevant to work in the relevant art” should not be construed as meaning relevant to any work in the relevant art, including work irrelevant to the particular problem or long-felt want or need, in respect of which the invention constitutes an advance in the art. The phrase can only be construed as being directed to prior disclosures, that is publicly available information (not part of common general knowledge) which a person skilled in the relevant art could be expected to have regarded as relevant to solving a particular problem or meeting a long-felt want or need as the patentee claims to have done. Otherwise the words of limitation in the last forty words of s 7(3) would have no role to play. Any piece of public information in the relevant art would be included, as is the case with the much broader and quite different formulation in the cognate provisions in the United Kingdom, which do not depend on the standard of a skilled person’s opinion of the relevance of the information.
The question of what a person skilled in the relevant art would regard as relevant, when faced with the same problem as the patentee, is to be determined on the evidence. The starting point is the subject matter of the invention to be considered together with evidence in respect of prior art, common general knowledge, the way in which the invention is an advance in the art, and any related matters. It should be mentioned that the starting point is not necessarily the inventive step as claimed, or even agreed between parties, because the evidence, particularly in respect of a combination of integers, may support a different inventive step.
(Emphasis added; citations omitted.)
(a) The Kazakov paper – Introduction
572 The English translation of the Kazakov paper produced by Newtype Communications, Inc on 22 November 2011 is as follows, with the abstract in bold.
EXTRACELLULAR DNA IN THE BLOOD OF PREGNANT WOMEN
V. I. Kazakov, V. M. Bozhkov, V. A. Linde, M. A. Repina, V. M. Mikhaylov
Institute of Cytology, Russian Academy of Sciences, and Medical Academy of Postgraduate Physician Education, St. Peterburg
The level of extracellular DNA increases in the blood of women during pregnancy. By means of PCR, the full-size Alu repeats were observed among extracellular blood DNA repeats of pregnant women. Furthermore, with Tc65 type primer the PCR method allowed to observe in the blood DNA fragments flanked by inverted Alu repeats (inter Alu repeats). The presence of such a type of inter Alu repeats was estimated in the blood of women being in the first trimester of pregnancy only, but was not estimated among blood DNA fragments of women of the last trimester of pregnancy. It is discussed which types of cells may serve as a source of extracellular blood DNA (either trophoblast cells, lymphocytes or decidual cells), the significance of such DNA for pregnancy being appreciated.
It has been shown that extracellular DNA is contained in the blood of humans and animals (Stroun et al., 1977; Fedorov, Yaneva, 1982; Vladimirov et al., 1992). An increase in the content of extracellular DNA in the blood of humans has been described during pathological processes taking place in various types of tissue of the body, especially during certain inflammatory processes of the gastrointestinal tract, during tumor and infectious diseases of viral etiology, and during disseminated lupus (Anker et al., 1975; Leon et al., 1977; Shapiro et al., 1983; Stroun et al., 1987; Goto et al., 1991).
The molecular mass of the extracellular DNA of the blood is on average between 1.106 to 15.106 Da (Stroun et al., 1977; Fedorov, Yaneva, 1982). According to EM microscopy data, the extracellular DNA of healthy persons is double-strand and linear (Dennin, 1979). According to Vasyukhin et al. (1991), human extracellular DNA contains unique sequences. It has also been shown that the extracellular DNA of the blood during disseminated lupus has sequences capable of forming Z-DNA (Van Helder, 1985).
It is believed that the high-molecular component of extracellular DNA in the blood comes from living cells (Stroun et al., 1977). It has been demonstrated in vitro that certain types of cells, especially lymphocytes, excrete extracellular DNA into their surroundings (Rogers et al., 1972; Rogers, 1976; Stroun et al., 1977; Fedorov, Yaneva, 1982). Furthermore, the blood also contains low-molecular DNA, corresponding to nucleosomes in its mobility. In the blood of rats, its content increases after total X-ray exposure (Belokhvostov et al., 1987; Vladimirov et al., 1992; Tishchenko et al., 1993). According to Tishchenko et al. (1993), the low-molecular DNA in the blood of rats after total irradiation is enriched in GC sequences. It has been conjectured that the low-molecular DNA in the blood is a product of intensified extrachromosomal synthesis of ring DNA, detected in many types of cells of humans and mammals (Vladimirova et al, 1992). Our attention was drawn to the proposition that the rise in the level of extracellular low-molecular DNA in the blood of rats after irradiation is the consequence of increased activity of Ca/Mg-endonuclease in the cells (Tishchenko et al., 1993). This proposition is in good agreement with data on the intensification of the process of apoptosis in the cells of various tissues of the body after irradiation (Khanson, Komar, 1985).
It is clear from the above that analysis of the extracellular DNA in the blood of humans and animals is of both theoretical and practical interest. It is not ruled out that the nucleotide composition of the extracellular DNA of blood is not so random as not to reflect the peculiarities of the processes of differentiation and cell death taking place in various tissues at each particular moment in the life of the organism. All of this dictated our choice of the blood of pregnant women as the object of our research. According to available data, cellular proliferation, differentiation, and cell death occur in the uterus during pregnancy (Fedorova, Kalashnikova, 1986; Mikhaylov et al., 1989, 1992a, 1992b; Mikhaylov, 1993). It was anticipated that these processes exert an influence on the specifics of the nucleotide composition of the extracellular DNA in the blood of pregnant women.
Material and method
We studied the blood sera of men, nonpregnant women, and women in the first and third trimester of pregnancy and those with late toxicosis of pregnancy. The blood was taken by syringe from the cubital vein under sterile conditions, placed in a centrifuge test tube, and left at room temperature until clotted. Immediately after the formation of a thrombus, it was removed from the walls of the test tube and centrifuged at 400 g for 10 min. The serum was centrifuged yet again at 2000 g for 10 min at 4° C. The serum obtained in this way was kept at -60° C. The use of serum instead of plasma for the analysis of the DNA of the blood can be justified if one observes the conditions for formation of a thrombus at room temperature and immediate removal of the serum from the thrombus (Leon et al., 1977; Shapiro et al., 1983). Such was done in the present investigation.
After this the serum was treated twice with phenol, mixtures of phenol and chloroform (1:1), chloroform, and isoamyl alcohol (24:1) and precipitated with ethanol at -20° C. The DNA preparations were analyzed in 1% agarose gel, and then used as the matrix in a polymerase chain reaction (PCR).
The PCR was carried out using thermophilic DNA polymerase from Thermus thermophilus. We performed 30-35 amplification cycles. The amplified fragments were analyzed in 8% PAAG. The annealing temperature of the primers was 55° C for primers B1 and C2 and 60° C for primer Tc65, the concentration of Mg2+ ions being 2-5 mM. The primers for the PCR were synthesized by us with the solid-phase triether phosphite method in the β-cyanoethyl modification on the Gene assembler instrument from Pharmacia. The sequence of the Tc65 primer was taken from literature sources (Nelson et al., 1989), the sequences of the other primers were found with the help of the Oligo program (Microsoft) for the consensus sequence of Alu repeats of the MF family and designated as B1 and C2 by us.
When the PCR was carried out with the pair of primers B1 and C2, an Alu repeat with a length of 239 base pairs was amplified as a result of the reaction. When only one primer Tc65 was used in the PCR, DNA fragments were amplified that were flanked by two Alu repeats with their terminal 3’ regions facing each other (inter-Alu repeats).
The sequences of the primers are:
Results and discussion
It was not our goal to study in detail the changes in the concentrations of DNA in the blood serum of pregnant women. From the data of authors using radioimmunological methods of determination (Leon et al., 1977; Shapiro et al., 1983), the concentration of DNA in the blood sera of healthy donors varied from 0 to 0.1 mcg/ml; according to authors using biochemical methods (Anker et al., 1975; Stroun et al., 1987), from 0.1 to 0.6 mcg/ml. Our data on the concentration of DNA in the blood of men and nonpregnant women corresponded to the literature data.
According to our data findings, during pregnancy there is an increase first of all in the concentration of low-molecular DNA, the increase being most pronounced during gestosis. The size of such DNA is from 150 to 2500 base pairs. There is a belief that the increase in concentration of low-molecular extracellular DNA in the blood is a consequence of increased synthesis of extrachromosomal DNA (Vladimirov et al., 1992). However, in evaluating the data presented, one must consider that a substantial number of endometrial cells perish during pregnancy, especially at the end of pregnancy and in connection with gestosis (Kottsova et al., 1989; Mikhaylov et al., 1992a). According to our data, the death of decidual cells is attended by a gradual loss of nuclear DNA and fragmentation of peripheral parts of the nuclei and cytoplasm and, apparently, this occurs by apoptosis (Mikhaylov et al., 1992a, 1992b; Mikhaylov, 1993). One of the major mechanisms of cell death of the apoptosis type is internucleosomal degradation of DNA due to activation of Ca2+ endonuclease (Wyllie, 1980). This does not rule out the possibility of extrachromosomal copying of DNA as the source of the extracellular DNA in the blood. Regardless of whether intensified synthesis of extrachromosomal DNA or apoptotic cell death is the source of the extracellular DNA in the blood, the presence of Alu repeats in it testifies to its nuclear origin (Fig. 1; see insert VIII).
Worthy of attention is the presence of inter-Alu repeats among the DNA of the blood serum of pregnant women. It is known that approximately a third of the Alu repeats of humans belongs to the inverted repeats, and the mean distance between inverted pairs is approximately 50 t.p.n. (Jelinek, Schmid, 1982). The bulk of such inverted repeats apparently contain a unique internal fragment. There are several kinds of inter-Alu repeats. With the help of the Tc65 primer, we detected inter-Alu repeats in the blood of women only in the first trimester of pregnancy (Fig. 2). When the PCR was carried out with the Tc65 primer, only those DNA fragments are amplified in which the spacer DNA is flanked by Alu repeats mutually facing each other by their 3’ regions, which makes these fragments even more unique. Thus, if the presence in the blood of pregnant women of an increased quantity of low-molecular DNA fragments is a sign of increased cell death occurring in the uterus in all phases of pregnancy, then the presence of inter-Alu repeats indicates the functioning of other mechanisms for such sequences getting into the serum. It is important that inter-Alu repeats have been detected only in the blood of women in the first trimester of pregnancy. This fact most likely reflects the difference in content of the cellular processes that are characteristic of the early and late stages of pregnancy. While at the end of pregnancy there is observed a death of decidual cells, which explains increase in concentration of low-molecular DNA in the blood; in the initial period of pregnancy when implantation and placentation of the embryo in the uterus occurs, there is decidualization of the endometrium and growth of the embryonal vesicle, i.e., a process of differentiation of the cells of the uterine mucous membrane and cells of the trophoblast.
It is likely that, in the early stages of pregnancy, an excretion of inter-Alu repeats from the cells occurs. Which type of cells can excrete inter-Alu repeats is a matter of great interest. This question also confronts investigators when discussing the sources of high-molecular serum DNA (see the survey: Fedorov, Yaneva, 1982). Excretion of DNA in vitro has been well characterized for lymphocytes (Fedorov, Yaneva, 1982). It is assumed that the lymphocytes might be the source of such DNA during tumor diseases. In the early stages of pregnancy, substantial lymphocyte infiltration is also observed in the uterine mucous membrane.
Thus, in the early stages of pregnancy in humans, cells of the fetus (trophoblasts) and the mother (cells of the endometrium and lymphocytes) may excrete DNA. In view of the above, and also considering the transposonic and recombinogenic nature of the Alu repeats (Tomilin, 1992), it can be conjectured that the inter-Alu repeats discovered by us in the blood serum of pregnant women may play some kind of regulatory role in the early stages of pregnancy. The cloning and sequencing of these fragments is of particular interest. What has been said does not rule out the presence of other inter-Alu repeats in the blood of pregnant women, which can be identified by means of other primers and may have their own features of distribution in the blood in the course of pregnancy.
573 I have set out the Kazakov paper in full as it is short and it is necessary to go through some of its science in some detail.
574 Let me make some introductory remarks before getting into the detail.
575 The Kazakov paper is an article published in the Russian journal Tsitologiia entitled “Extracellular DNA of pregnant women blood”. It was published in 1995 by the Russian Academy of Sciences. There is no evidence that it was peer reviewed.
576 The aims of the Kazakov paper were stated to be to test the hypothesis that there was extracellular DNA in the blood of pregnant women and to ascertain whether the level of such DNA changed during pregnancy.
577 In my view it was not an aim of the paper to test whether cell free DNA from a fetus was detectable in the circulation of pregnant women and the paper does not suggest that any such DNA was detectable. The Kazakov paper merely speculates that trophoblast cells, maternal lymphocytes or maternal decidual cells may serve as a source of extracellular DNA.
578 The experiments reported in the Kazakov paper in light of its stated objectives involved using PCR primers to amplify DNA extracted from the blood serum of:
(a) men;
(b) non-pregnant women; and
(c) pregnant women.
579 The following sets of primers were used:
(a) one pair (known as B1 and C2) was used to amplify “Alu repeats”; and
(b) a single primer (Tc65) was used to amplify “inter-Alu repeats”.
580 The results of the experiments are described with reference to two figures. Unfortunately, the version of the Kazakov paper that was provided to me was of poor quality such that the figure images are unclear.
581 Figure 1 shows a polyacrylamide gel (following electrophoresis) containing the amplification products obtained using primers B1 and C2 on serum samples from a male, non-pregnant female, a pregnant female in the first trimester, a pregnant female in the third trimester and a pregnant female with pre-eclampsia. But I would note that the method performed was not quantitative, no replicates were carried out and no statistical analysis was performed on the data.
582 Figure 2 purports to show the PCR amplification products obtained using the Tc65 primers from blood serum samples from 8 pregnant women in the first trimester of pregnancy. But I would note the following matters. First, there is no data from PCR reactions performed on samples taken later during pregnancy, even though the paper’s purported key finding is the increase in the level of extracellular DNA during pregnancy. Second, there is no data from non-pregnant women or men. Third, the reactions depicted in the gel in Figure 2 were very inefficient or did not work, as no “smear” of DNA, which would be expected following digestion with the Tc65 primer, can be observed. Fourth, no positive or negative controls were used.
583 Now based on Figure 1, the authors conclude that “during pregnancy there is an increase first of all in the concentration of low-molecular DNA, the increase being most pronounced during gestosis”. But I agree with Sequenom that this would not appear to be a reasonable conclusion based on the Figure. The authors further appear to suggest that the purported elevation in the level of low molecular weight DNA in the serum of pregnant women and those with pre-eclampsia, may have originated from dead maternal endometrial cells.
584 Based on Figure 2, the authors purport to conclude that:
... [i]t is important that inter-Alu repeats have been detected only in the blood of women in the first trimester of pregnancy. This fact most likely reflects the difference in content of the cellular processes that are characteristic of the early and late stages of pregnancy.
585 But again the conclusion is problematic to say the least and would be understood as such by the person skilled in the art. Indeed, Ms Norbury concedes that there is “little supporting data or relevant control information” in Kazakov and specifically notes that “there are no controls used in Figure 2, and it is very unclear. I therefore do not think you can draw any conclusions from Figure 2”.
586 Now Professor Oepkes has stated that he “would not have found the introduction or methods sections particularly relevant from a prenatal diagnosis and screening perspective” and that he instead would have “taken more interest in the results and discussion section”. But it is problematic to accept that, absent hindsight, any properly trained scientist would be willing to accept the results and conclusion of a scientific publication whilst disregarding the method by which those results were obtained.
587 Let me separately say something about the Kazakov abstract at this point.
588 The Kazakov abstract first states that “extracellular” “full-size Alu repeats” were observed in the blood of pregnant women and that such DNA “increases in the blood of women during pregnancy”.
589 Relevantly, at the priority date, a person skilled in the art would have been generally aware that “Alu repeats” were repeating DNA sequences found ubiquitously throughout the human genome that were rarely unique to a particular individual and did not differ by gender or physiological conditions such as pregnancy.
590 The abstract then explains that different nucleic acid sequences, namely, “inter Alu repeats” were observed in “the blood of women being in the first trimester of pregnancy only, but was not estimated among blood DNA fragments of women of the last trimester of pregnancy.”
591 Finally, the abstract states that “[i]t is discussed which types of cells may serve as a source of extracellular blood DNA (either trophoblast cells, lymphocytes, or decidual cells), the significance of such DNA for pregnancy being appreciated.”
592 But the Kazakov abstract does not contain any hypothesis or conclusion that the difference between the DNA results (i.e. the presence of inter-Alu repeats) obtained from women in their first trimester as opposed to their third trimester of pregnancy may be related to the biology of the development of pregnancy.
593 In my view, which I will elaborate on later, the Kazakov abstract draws conclusions that were inconsistent with established knowledge in the prenatal diagnosis field at the priority date, and displays little internal consistency.
594 Before proceeding further, let me say something about the person skilled in the art or in this case the relevant skilled team as I have previously identified.
595 First, the skilled team would have an interest in non-invasive prenatal testing, because of the risks presented by invasive testing. In particular, they would have an interest in developing ways to test, non-invasively, for the sex of the fetus and for rhesus D status, as well as conditions such as thalassemia.
596 Second, the skilled team would be aware that trophoblasts formed part of the placenta, and that fetal cells (including trophoblasts) and proteins were present in maternal blood. Further they would be aware of the work to develop non-invasive prenatal testing methods using fetal cells from maternal blood, including the various techniques used in such testing including the successful use of Y chromosome and RhD gene primers and PCR, as well as the problems associated with isolating fetal cells from maternal blood.
597 Third, as I have previously indicated, the skilled team would include a member who was familiar with molecular genetics analytical techniques, including the design and selection of primers including primers capable of detecting sequences from the Y chromosome, the use of PCR methods, and an awareness of the low levels at which DNA can be detected using PCR methods. Further, I am prepared to accept that the skilled team includes a person with some interest in fetal medicine including prenatal screening and diagnosis.
(b) Whether the hypothetical skilled addressee could reasonably be expected to find the Kazakov paper
598 “[A]scertained” in the context of s 7(3) means ‘discovered’ or ‘found out’. And a document could be ascertained if it was published in such a manner or form that it could reasonably have been expected to be found by a person skilled in the art. The meaning of “understood” in s 7(3) is that having discovered the information, the addressee would have comprehended it or appreciated its meaning.
599 The expectation as to what the skilled addressee “could” ascertain is a reasonable expectation, not a fanciful one. And it is to be assessed in light of the characteristics of the relevant hypothetical skilled person and the particular problem faced, the overcoming of which is said to involve an inventive step. The inquiry is hypothetical. In terms of expert evidence, the evidence which is relevant concerns the steps that the experts consider they would have taken if they had been faced with the particular problem.
600 Now Ariosa submits that the Kazakov paper satisfies the requirements of s 7(3) of the Act, being a published paper that as at the priority date the hypothetical person skilled in the art could reasonably have been expected to have ascertained, understood and regarded as relevant.
601 In summary, Ariosa says the following:
(a) The Kazakov paper was published and indexed on leading literature databases (Medline and Embase) with an English title and abstract before the priority date.
(b) For the purposes of considering s 7(3), the particular problem faced by the hypothetical skilled person should be characterised as a research project on non-invasive prenatal testing.
(c) The hypothetical skilled person undertaking such a hypothetical project would undertake a literature search, including using literature databases such as Embase and Medline.
(d) The exercise of conducting a literature search is an iterative and to some extent an individual process. Different search terms would be used iteratively and additively to produce a list of search results of a manageable size for closer review.
(e) Whilst the experts expressed differing views as to the precise search terms they could have used, ultimately Professor Fisk agreed that the terms proposed by Professor Hyett and the combinations used by Ms Pettit-Young, a research librarian I refer to below, were not unreasonable.
(f) A number of the combinations of search terms proposed by Professor Hyett and used by Ms Pettit-Young resulted in the Kazakov paper appearing in search results of a manageable volume.
(g) The hypothetical skilled address would have considered the title and abstract of the Kazakov paper of interest to the project and could have obtained a translation of the paper without unreasonable difficulty if desired.
602 Ariosa points out that although the Kazakov paper was published in the Russian language journal Tsitologiia (or Cytology) in 1995, the paper as published included an English title “Extracellular DNA of pregnant women blood”, a reference list in English, and an English language abstract extracted above.
603 Ariosa says that the English language title and abstract of Kazakov were indexed in, and searchable on, the Medline and Embase databases by the priority date. It also says that the Medline and/or Embase databases were relevant databases used by the person skilled in the art as at the priority date.
604 In elaboration, Ariosa has made the following submissions.
605 The evidence of Professor Hyett is that, if he were undertaking a research project on non-invasive prenatal testing, he would have conducted literature searches on such databases, using keyword and mesh heading searches over the previous 2 to 3 (or potentially 5) years. This would have been an iterative process and involved using different search terms or combinations of terms depending on the list of results produced, which would include the title and abstract of the article.
606 Professor Fisk agreed that the literature search process would be an iterative one. Professor Fisk also accepted that “[t]here are many ways you could do this search”, and that he would “use Boolean terms which are usually ‘and’ or ‘or’”. In the case of Professor Lovett, his affidavit evidence is consistent with an iterative search strategy.
607 In his affidavit, Professor Hyett explains that he was asked to identify search terms which he considered to be relevant to a project on non-invasive prenatal diagnosis as at the priority date. The potential search terms that he identified included “prenatal diagnosis”, “pregnancy/pregnant”, “fetal cells”, “blood”, “DNA”, “PCR”, “FISH”, “trisomy 21”, “trophoblast”, and “erythroblast”. Professor Hyett described that he would have tried combinations of two terms together, and depending on the number and nature of those results, would then have narrowed the results by adding further search terms.
608 Ariosa says that notably, Professor Hyett identified these search terms “before [he] was aware of or introduced to the Kazakov paper”. This is in contrast to Professors Fisk and Lovett, who both had detailed prior knowledge of the Kazakov paper and its contents (including the abstract) from their involvement in litigation in the US (in the case of Professor Fisk) and in the UK (in the case of Professor Lovett), prior to designing and undertaking the searches described in their affidavits.
609 Ms Pettit-Young, who is a research librarian, performed searches on the Ovid Medline and Embase databases using combinations of the search terms identified by Professor Hyett. The Kazakov paper appeared in 13 of the searches performed by Ms Pettit-Young, being searches using combinations of the search terms provided to her.
610 Further, Ariosa points out that there were key areas of dispute between the experts in relation to the potential search terms, being the reasonableness or necessity of the search terms “prenatal diagnosis” and “trophoblast”.
611 Professor Fisk’s starting point was that he would have used “prenatal diagnosis”, “antenatal diagnosis” or “fetal diagnosis” in a search, and then would have narrowed this by adding further terms such as “non-invasive”, “circulating fetal cells” or “fetal cells in maternal blood”. However, Professor Fisk accepted that it would not be unreasonable, as an alternative to “prenatal diagnosis”, “antenatal diagnosis” or “fetal diagnosis” to use another search strategy using the term “pregnancy” or “pregnant” and then adding terms such as “PCR”, “DNA” and “maternal blood”.
612 Professor Lovett also accepted that “blood”, “pregnant”, “DNA”, “PCR” and “trophoblasts” in various combinations were not unreasonable search terms.
613 Professor Fisk’s other primary criticism of the search terms proposed by Professor Hyett appeared to be the inclusion of the search term “trophoblast”. Professor Fisk initially said that he would not use this search term, but would instead use “circulating fetal cells” or “fetal cells in maternal blood”. Similarly, Professor Lovett’s evidence was that he did not think he “would ever go to trophoblast” because it was “terribly specific”.
614 But Ariosa says that this evidence of Sequenom’s experts should not be accepted, for the following reasons.
615 First, it was well-known in the field of prenatal diagnosis at the priority date that trophoblast cells existed in maternal blood, and had been the subject of significant research as a potential source of fetal DNA for non-invasive prenatal diagnosis. Now Professor Fisk suggested that “trophoblast” would not be a reasonable search term because there were difficulties associated with isolating and identifying trophoblasts in maternal blood, due to their apparent low frequency and the belief that trophoblasts tended to become trapped in the maternal lung. But Ariosa says that in Simpson JL and Elias S, “Isolating Fetal Cells in Maternal Circulation for Prenatal Diagnosis” (1994) 14(13) Prenatal Diagnosis 1229-1242 at p 1230, the authors stated:
Trophoblasts are the most obvious candidate cell, given their intimate relationship with the uterus. … The attractiveness of trophoblasts as candidate cells has led many to continue to search for ways to recover such cells.
616 Further, numerous articles identified by Professor Fisk’s own search included the term “trophoblast” in the title or abstract, thus according to Ariosa underscoring the significant ongoing work relating to trophoblasts at the priority date.
617 Further, Ariosa says that Professor Hyett explained that his inclusion of “trophoblast” as a search term was based on his knowledge and experience at the priority date in relation to research being done on these cells, even though they were known to be rare:
PROF HYETT: … I have always recognised that there were three cell types that you could potentially use for non-invasive prenatal diagnosis. Trophoblast was one of those. Lymphocytes were another. Nucleated red cells were another. And around this time, there were markers being developed that made it easier to identify trophoblast cells based on the HLA typing system. So there was certainly discussion around this time around reproducing some methodologies to actually try and look at trophoblasts rather than nucleated red blood cell.
MR SHAVIN: Yes. But you accept that it was known at this time that it was very hard to use trophoblasts because they were very rare?
PROF HYETT: I accept that, but as I say, there were some markers that had been developed which my chairman at the time, Professor [Rodeck], was aware of, and he suggested to me that they would be an interesting area for research in non-invasive prenatal diagnosis.
618 In his affidavit, Professor Hyett also gave evidence of his direct involvement in research regarding trophoblast cells before the priority date.
619 Ariosa points out that Professor Oepkes was also of the view that “trophoblast” was a reasonable search term, given the work on prenatal diagnosis using fetal cells in maternal blood which was being conducted before the priority date:
…I think that the term trophoblast definitely is something that in the field of trying to identify fetal cells in the maternal circulation would be a major search term that was together with the haematopoietic precursor cells – that was the type of cell we were looking for with the groups interested in that field, to identify ways for prenatal non-invasive diagnosis.
620 Further, Ariosa says that even Professor Fisk acknowledged that the presence of trophoblast cells in maternal blood, and research regarding trophoblast cells, was well-known at the priority date:
PROF FISK: … this was known from Schmorl 1893 – in fact, one of the few foreign-language papers that’s ever cited in the literature – when he, a German pathologist, discovered – and this is well-known from the post-mortem of women dying of eclampsia – that he could find trophoblast cells in their lung.
MR CORDINER: So they’re well-known to anybody in the area of prenatal diagnosis, one would have thought, who’s looking at, as you say, non-invasive methods for testing prenatal diagnosis.
PROF FISK: Yes. That was a classic paper that indicated there is some unidirectional traffic – cell traffic.
621 Now in order to capture such research, Professor Fisk said he would have used alternative terms “fetal cells in maternal blood” or “circulating fetal cells”. But he accepted that an alternative strategy would be to use the specific cell types in a Boolean search such as “fetal cells or trophoblast or erythrocytes”:
PROF FISK: Yes. Trouble is, if you use “fetal cells” on its own, you will get fetal liver cells, fetal skin cells, all sorts of things…
MR CORDINER: Yes
PROF FISK: … particularly in relation to invasive fetal – invasive prenatal diagnosis and maybe some fetal pathology.
MR CORDINER: So in terms of a search, though, in order to ensure that you’re not missing potentially interesting work, one might search for fetal cells or trophoblast or erythrocytes or any of the fetal cells which might be of potential use. Is that a fair or reasonable search…
PROF FISK: Yes, I did…
MR CORDINER: … string?
PROF FISK: … this search in good faith as my best attempt and trying to focus on the key issues, which is why I chose more broadly the terms “circulating fetal cells” or “fetal cells in maternal blood”, which would encompass them all.
…
MR CORDINER: I’m just saying is that reasonable: that somebody would use a search term like “trophoblast” without also having to use – that is, requiring “fetal cells” to also be there in the journal?
PROF FISK: There are a whole range of search strategies you could use …
622 Given the above, Ariosa says that it is clear that “trophoblast” would be a reasonable term to include in a literature search for a project on non-invasive prenatal testing.
623 And as to Professor Fisk’s insistence on including one of the search terms “prenatal diagnosis”, “antenatal diagnosis” or “fetal diagnosis”, Ariosa says that as Professor Oepkes explained, a researcher would not want to restrict themselves to a particular field of inquiry, as they would thereby exclude ideas from related fields.
624 Further, Ariosa points out that searches on the Embase and Medline databases with a date range of between 1995 and 1997:
(a) using “blood + trophoblast*”, “blood + pregnan* + trophoblast*” and “blood + pregnan* + DNA + trophoblast*”, would have included the Kazakov paper in the search results, amongst a total number of between 13 and 228 results, depending on the particular search string and database used; and
(b) using “blood + pregnan* + DNA”, “blood + pregnan* + PCR” and “blood + pregnan* + DNA + PCR”, would have returned the Kazakov paper as a result, amongst a total of between 53 and 211 articles, depending on the particular search string and database used.
625 And indeed Professor Hyett said that he would have reviewed results of this magnitude, explaining that it was often the case that a meta-analysis might start with more than a thousand articles before being reduced to a selected set.
626 Further, Ariosa says that this volume of results is within the range that Professor Fisk could be expected to have reviewed. Professor Fisk accepted that a list of approximately 200 results would be a manageable list to review for the purposes of a research project. Professor Fisk also acknowledged that he would regularly peruse up to 100 articles per week, either in full or by reviewing the abstracts of those papers, from the journals listed in his affidavit evidence.
627 Accordingly, in light of the evidence of both Professors Fisk and Hyett, Ariosa says that it is not unreasonable to expect that the hypothetical skilled addressee would have reviewed the results generated from the search term combinations, inter-alia:
(a) “pregnan* + PCR” (571 results on Medline); or
(b) “pregnan* + DNA” (1036 results on Medline).
Analysis
628 In my view, finding the Kazakov abstract would have at least involved conducting a literature search using peculiar search terms. Further, finding the Kazakov paper would have first required selecting the Kazakov abstract from the results of a literature search that had somehow returned the Kazakov abstract, sourcing the original Kazakov paper from an unspecified library and then having the paper translated from Russian into English.
629 Now Ariosa’s experts speculated in the joint expert report that the Kazakov abstract and/or paper may have been found through word of mouth, “submission for review, assistance in publication, reports in current awareness studies…personalised electronic newspapers detailing new research findings or through structured literature review”. But the Kazakov abstract and/or paper was not identified by any of these postulated methods.
630 Further, none of the experts were aware of the Kazakov abstract or the Kazakov paper before the priority date. Indeed, none of the experts had encountered the Kazakov paper until Ms Norbury and Professor Oepkes heard mention of it at conferences nearly 20 years after the priority date.
631 Professor Hyett was given a copy of the paper by Ariosa’s lawyers after commencement of this proceeding, although a few years before this proceeding he had “heard through discussions with [his] colleagues and at various conferences, that an Eastern European-based group had written a paper on cffDNA in maternal blood in the mid-1990s”.
632 Ms Norbury first heard of the paper at a presentation by Professor Nicolaides at the Royal Society in 2016 when it was mentioned but otherwise did not see a copy of the paper until the UK proceedings when given a copy by Herbert Smith Freehills LLP.
633 Professor Fisk first became aware of the paper in the US proceedings in April 2013.
634 Professor Lovett had not heard of it or seen a copy until he was provided with a copy of the paper in the UK proceedings.
635 Professor Oepkes was first given a copy by Herbert Smith Freehills LLP for the UK proceedings but believes he had heard of it at conferences over the last few years.
636 Further, as I have said, the Kazakov paper was written in Russian and would need to have been translated.
637 Now Ariosa points out that although the Kazakov paper was published with an English language abstract, Professor Hyett gave evidence that he had on occasion obtained translations of papers he considered may be of interest, and he would have obtained a translation of the Kazakov paper. His evidence was that this was not an overly difficult undertaking.
638 Further, there was evidence that Professor Oepkes previously had papers of interest translated, and Ms Norbury also agreed that she would have had articles translated if she was interested in the abstract of the article:
… I - there certainly wouldn’t have been a problem getting things translated if you got told of an abstract. There were lots of colleagues around who would be able to do that, and obviously a lot of Polish people speak Russian.
639 But Professor Oepkes had never in a career spanning over 30 years cited or even read a paper originally published in Russian. Professor Oepkes also agreed that in 1997 he would have experienced significant difficulty in picking up a foreign language paper via a literature search.
640 Now Professor Hyett stated that he “would have had to arrange for it to be translated from Russian to English”. But the evidence is that Professor Hyett had only on a small number of occasions arranged for other (unspecified) foreign-language articles to be translated. And he had not previously arranged a translation of a paper from Russian. More generally, in my view there was little evidence that a skilled person would have actually had the resources or the inclination to do so.
641 Let me broaden out the discussion.
642 Each of the expert witnesses gave evidence of the usual professional practices of the person skilled in the art at the priority date, including evidence as to how they personally kept up to date with developments in their fields at the priority date.
643 Each of the experts was interested in developments both within Australia and internationally, and kept their knowledge up to date by consulting various resources including, inter-alia, research journals published internationally.
644 Each of the experts identified the journals, authors and institutions they monitored regularly at the priority date.
645 Professor Lovett regularly consulted Genome Research, Physiological Genomics, Human Genetics, Charles J Epstein et al, Inborn Errors of Development: The Molecular Basis of Clinical Disorders of Morphogenesis (Oxford University Press), American Journal of Human Genetics, European Journal of Human Genetics, Genomics, Human Molecular Genetics, Nature Genetics, Nucleic Acids Research, Nature Biotechnology, Nature, Nature Methods, Nature Protocols, PLoS ONE, PLoS Genetics, Nature Communications, Science, Lancet, New England Journal of Medicine, Proceedings of the National Academy of Sciences, BioTechniques and Cell.
646 Professor Fisk attended the ICQCH library weekly to review entire new issues of American Journal Obstetrics & Gynaecology, ANZ Journal of Obstetrics & Gynaecology, Archives of Disease in Childhood (Fetal and Neonatal Edition), British Journal of Obstetrics & Gynaecology, British Medical Journal, Early Human Development, European Journal Obstetrics and Gynaecology, Fetal Diagnosis and Therapy, Journal of Maternal-Fetal Medicine, Journal of Obstetrics and Gynaecology, Journal of Perinatal Medicine, New England Journal of Medicine, Obstetrics & Gynaecology, Prenatal Diagnosis, Lancet, Ultrasound in Obstetrics & Gynaecology. Professor Fisk noted that this would sometimes result in him perusing up to 100 papers a week and that he would diligently read all articles relevant to his research field. Professor Fisk also reviewed the Table of Contents for relevant content in Archives of Disease in Childhood, Baillière’s Clinical Obstetrics and Gynaecology, Contemporary Reviews in Obstetrics Gynaecology, Fetal and Maternal Medicine Review, Human Reproduction, Human Reproduction Update, Journal of Clinical Ultrasound, Nature Medicine, Obstetric & Gynecological Survey, Pediatric Research and Pediatrics.
647 Professor Hyett attended the Royal Society of Medicine Library in London on a weekly basis and regularly reviewed the indexes of Prenatal Diagnosis, Ultrasound in Obstetrics and Gynaecology, British Journal of Obstetrics & Gynaecology, American Journal of Obstetrics and Gynecology, British Medical Journal, Lancet, New England Journal of Medicine and Obstetrics and Gynaecology.
648 Ms Norbury regularly attended journal club meetings and read journals such as Human Molecular Genetics, Nucleic Acid Research, Nature Genetics, Journal of Medical Genetics and Journal of the American Society of Human Genetics.
649 Professor Oepkes routinely reviewed Prenatal Diagnosis, American Journal of Obstetrics and Gynecology, British Journal of Obstetrics and Gynecology, Fetal Diagnosis and Therapy and Ultrasound in Obstetrics and Gynecology each time a new volume or issue was published. Professor Oepkes also reviewed journals that were more laboratory oriented, such as Clinical Chemistry.
650 But Tsitologiia or “Cytology” was not a journal reviewed by any of the expert witnesses.
651 Now a hypothetical person skilled in the art, if asked to undertake a research project on non-invasive prenatal testing, could have conducted an electronic search of the available scientific literature using Ovid Medline, Embase and/or PubMed.
652 A person skilled in the art would normally have conducted such electronic searches themselves. Literature searching was and is an iterative process which requires technical knowledge of the search area, including iteratively to devise search terms. It would have been unusual at the priority date for experienced researchers to outsource literature searches to librarians.
653 As Sequenom points out, the usual practice of a person skilled in the art undertaking literature searches at the priority date would have been to employ an iterative process involving the use of a series of appropriate keywords in combination, followed by a logical and rational selection process. Professor Hyett described the relevant “iterative” process as follows:
So I think it’s apparent that we all have different strategies for searching literature... Certainly from my perspective, I think that this is often an iterative process, whereby you use some raw search terms to search with and will often add other terms. And, yes, you will look at the volume of the work that you’ve identified, and at some point you will make a decision that you’re going to look at that list in a bit more detail, looking at the titles, potentially looking at the abstracts.
654 But I agree with Sequenom that the only truly “iterative” search strategies in evidence did not return the Kazakov abstract.
655 Now the relevant inquiry for the purposes of the hypothetical search concerns the hypothetical research project on non-invasive prenatal testing. Accordingly, it is clear that the broadest iteration of an effective literature search in the field of prenatal diagnosis at the priority date would have involved the use of at least the terms “prenatal”, “diagnosis” or both, so that as a starting point, all literature in the prenatal diagnosis field would be found. Searches outside the field are likely to have involved innovative thinking, rather than the approach of the hypothetical non-inventive person skilled in the art. As explained by Professor Oepkes:
By starting a literature search by typing in a broad term like “prenatal diagnosis”, that in itself really limits you to your own little field and – and immediately discards a lot of potentially interesting scientific work done from adjacent fields that – that often inspire us. Especially the – our – our innovative ways of – of thinking and – and doing research often requires connecting to – to other fields, in medical, even non-medical fields.
656 Professor Hyett suggested a range of search terms that he would have used if conducting searches in the context of the relevant project. And indeed the first term identified by Professor Hyett was “prenatal diagnosis”. But Professor Hyett did not give evidence of any combination of search terms he would have employed at the priority date if undertaking the relevant project nor did he perform or instruct another person to perform an “iterative” search. Professor Hyett did not give evidence establishing that he would or even could have found the Kazakov paper by the searches he says he would have performed.
657 Rather, Ariosa relies on the evidence of Ms Pettit-Young who stated that she was instructed by Ariosa’s lawyers to advise whether the Kazakov paper and Lo YM et al, “Presence of fetal DNA in maternal plasma and serum” 350(9076) Lancet 485-487 (Lo 1997) and/or their abstracts, were indexed and searchable (in English) on Ovid Medline and/or Embase as at or before both 4 March 1997 and 4 March 1998 and to perform the combination of keyword searches on Ovid Medline and Embase for the period of 1 January 1995 to 4 March 1997 set out in a list in her affidavit.
658 But the search strategy described by Ms Pettit-Young does not incorporate all of the keywords identified by Professor Hyett, and appears to include without explanation only six of the 15 terms listed. Of these six terms, Professor Fisk gave evidence that it would not have been useful to employ at least four of them if undertaking broad research in the field, and that many of the terms proposed by Professor Hyett would be expansively broad if used in isolation or additively, and likely to return hundreds if not thousands of mostly irrelevant results.
659 I agree with Sequenom that the result lists returned by Ms Pettit-Young’s searches support Professor Fisk’s concerns. Moreover, the keywords and the combinations in which they were searched were not identified, devised or designed by any of the expert witnesses. Professor Hyett confirmed that he did not speak to Ms Pettit-Young, and did not suggest to Ariosa’s lawyers that any of the search terms he identified in his affidavit evidence should be excluded from any search strategy.
660 Further, Professor Hyett did not suggest that any hypothetical search strategy should include the term “trophoblast” but exclude the terms “fetal cells” and “erythroblasts”. He conceded that there was in fact no logical reason to include the term “trophoblast” to the exclusion of the broader class of fetal cells unless one was specifically trying to focus on the body of literature relating to trophoblasts, which were known by this time to be very difficult to work with. I note that a search containing the terms “trophoblasts” or “erythroblasts” or “lymphocytes” was not performed by any of Ariosa’s witnesses.
661 Further, Ms Norbury and Professor Oepkes also confirmed that they did not advise Ariosa’s lawyers to exclude from any hypothetical search as at March 1997 the search terms “maternal serum screening”, “combined first trimester screening”, “nuchal translucency”, “ultrasound”, “fetal cells”, “trisomy 21”, “FISH” or “erythroblast” as listed in Professor Hyett’s affidavit evidence.
662 Now Ms Norbury said during cross-examination that she had conducted a literature search of her own volition after becoming aware of the evidence of the Kazakov paper and stated that Kazakov did come up as result number 561 out of 1400. However, she provided no evidence as to when she conducted the search, or what methodology she applied. And in any event it would seem that Ms Norbury’s search was conducted after the Kazakov abstract and the Kazakov paper were drawn to her attention in 2016.
663 Further, I agree with Sequenom that there is little to support the suggestion that the idiosyncratic and hindsight affected search strategy relied upon by Ariosa is a strategy that could have been reasonably employed by a person skilled in the art at the priority date undertaking the relevant project without knowledge of the claimed invention.
664 Indeed, it is unclear on what basis Ariosa’s lawyers developed the instructions and search strategy that Ms Pettit-Young was told to carry out.
665 I tend to agree with Sequenom that the narrow date range of 1 January 1995 to 4 March 1997 which Ms Pettit-Young was instructed to apply to her searches from the outset was not consistent with the usual practice of skilled addressees as at the priority date.
666 Further, given that the search strategy employed by Ms Pettit-Young was not designed by a person skilled in the art as at the priority date but was designed by Ariosa’s lawyers in the context of this proceeding and included knowledge of the title, abstract and publication date of Kazakov, the search exercise appears to have been influenced by hindsight knowledge of the result sought to be achieved.
667 Indeed, the table in Ms Pettit-Young’s affidavit demonstrates that the only circumstances in which the Kazakov abstract could have been found via literature searching was where one searched either specifically for the article, or only used search terms that were known to be included in the Kazakov abstract or title.
668 So, although Ms Pettit-Young’s evidence demonstrates that the Kazakov abstract and bibliographic information could have been returned by a literature search at the priority date in that this information had been recorded in electronic databases by this time, it fails to demonstrate that it would have been reasonable to expect a person skilled in the art to have found it.
669 In my view, the evidence of Professors Fisk and Lovett reflects the approach expected to have been taken by the skilled addressee and establishes that a person skilled in the art undertaking the relevant project could not reasonably be expected to perform a literature search in the field of prenatal diagnosis at the priority date using terms that could have identified the Kazakov abstract.
670 Professor Fisk noted that in the context of the relevant project, a search using the term “pregnancy” as an initial term in an iterative search instead of “prenatal diagnosis” would not be useful as it would retrieve results relating to the diagnosis of adults in pregnancy. Similarly, using a broad term such as “fetal cells” in isolation would return results on fetal cells that were not relevant to the relevant project, such as papers involving invasive techniques.
671 Professor Fisk maintained that as at the priority date:
I would have thought that practitioners skilled in the art would have embraced the mainstream terms in the field: prenatal, fetal or antenatal diagnosis. These were the names of the societies, of the organisations, of the meetings, of the whole field.
672 Further, Professor Fisk also pointed out that the searches carried out by Ms Pettit-Young, whilst achieving what appeared to be a manageable number of results in some cases, had in those cases, lost significant specificity. Accordingly, the searches carried out by Ms Pettit-Young would fail to retrieve results in the prenatal diagnosis field that might otherwise have been relevant to the relevant project, whilst also retrieving irrelevant results.
673 Further, Professor Fisk did not consider that it would be useful to limit any search by use of the word “trophoblast”, including because by the priority date this was already considered an unlikely candidate for prenatal diagnosis, and because this term was too narrow. Contrastingly, using “fetal cells” or “fetal cells in maternal blood” properly ensured that all potential candidate cell types would be captured by the search strategy.
674 I must say that I have found Professor Fisk’s evidence to be cogent and compelling.
675 Professor Lovett similarly viewed the search strategy undertaken by Ms Pettit-Young as contrived. It was his view that had he been undertaking the search at the priority date, he would have certainly included within the strategy the term “prenatal diagnosis”. Now although this was a search term given to Ms Pettit-Young, it was only used in combination with other words, and did not return the Kazakov paper in any such instance. In particular, he noted that some of the terms and combinations of terms used by Ms Pettit-Young were “terribly specific” (in relation to inclusion of the term “trophoblasts”) and noted that no “sane scientist” would include the terms “DNA” and “PCR” in the same search. Overall, Professor Lovett considered that the search strategy employed by Ms Pettit-Young was not one that a person skilled in the art could have reasonably conducted at the priority date.
676 Moreover, as Professors Fisk and Lovett explained in the joint expert report, the Kazakov paper was not identified by any of the methods then postulated by Ariosa’s witnesses and considered that it was:
… extremely unlikely that Kazakov 1995 would have been retrieved or prioritised by literature search methods in the field of prenatal diagnosis, as supported by the sample searches in each affidavit and Pertl & Bianchi’s systematic literature review in 2001. Similarly, it was not mentioned at the state of the art National Institutes of Health conference in 2003. Even if it were directly pointed out, Kazakov would not have been prioritised – it was published in an obscure, non-English language journal by unknown authors and the abstract does not point towards pre-natal diagnosis or fetal DNA.
677 Generally, the evidence demonstrates that a person skilled in the art at the priority date would have used a number of criteria to identify an article from a literature search result list. This assessment would have included an analysis of:
(a) the title of the paper;
(b) the journal in which the paper was published;
(c) the authors;
(d) the language of the article; and
(e) potentially a review of its abstract.
678 But having regard to these criteria, no person skilled in the art at the priority date would reasonably have been expected to identify and select the Kazakov paper from a literature search result list relating to the broad field of prenatal diagnosis, let alone to the relevant project. As Professor Lovett bluntly explained:
But I think we also have to consider – and this may sound elitist. And it isn’t meant to. I apologise if it does. But it’s just a fact of life. If you have a search in front of you, and if it has got beyond a couple of hundred, you have to filter it. And you go down that and you say, “Cytology? I’ve never heard of it.” You say, “Kazakov? I’ve never heard of him. It’s written in Russian?” Now, I mean, the credibility is…credible science by and large – and there’s few exceptions to this – in biological sciences is published in English. So there’s another thing that takes you away from it. And – and, you know, that’s not even thinking about reading the abstract yet.
679 Let me finally at this point elaborate on one matter that I have previously touched on.
680 Professors Fisk, Hyett and Oepkes (as fetal medicine experts) regularly conducted literature searches in the prenatal diagnosis field at the priority date. Professor Lovett and Ms Norbury similarly maintained their knowledge in the field of genetics. After the priority date, all experts had been involved in research and/or testing specifically involving cffDNA.
681 But as noted by Professor Fisk, to his knowledge, no one in the prenatal diagnosis field retrieved the Kazakov abstract or the Kazakov paper by the priority date, and he did not learn about the Kazakov abstract or the Kazakov paper from anyone in the field.
682 Professor Hyett also noted that he did not find the Kazakov abstract in any literature search he performed, despite having published in the field, and as a consequence having undertaken literature reviews, since the publication of the Kazakov abstract. In fact, the Kazakov paper was not cited in any of the published prenatal diagnosis literature until 2013.
683 Further, Professor Oepkes noted that despite having conducted careful and systematic reviews of the literature in the prenatal diagnosis field over the last 25 years, he had never identified the Kazakov abstract or the Kazakov paper.
684 Ms Norbury also agreed that she did not hear of the Kazakov paper or abstract until 22 years after it was published, despite:
(a) having worked in the prenatal diagnosis field since the early 1990s; and
(b) being involved before and at the priority date in the setting up of diagnostic services for various disorders which required “lots of searches and information being reported back and shared between colleagues”.
685 In summary, none of these experts had come across the Kazakov paper or its abstract at the priority date or even in the course of their extensive relevant post-priority date research.
686 Further, other well-known researchers in the prenatal diagnostics field similarly did not find the Kazakov paper. For example, Dr Diana Bianchi and Dr Barbara Pertl (Pertl B and Bianchi D, “Fetal DNA in Maternal Plasma: Emerging Clinical Applications” (2001) 98(3) Obstetrics and Gynecology 483-490) conducted a comprehensive literature search spanning thirty years, which encompassed the publication date of the Kazakov paper, in an effort to discover all articles describing the detection of fetal DNA in maternal plasma. Dr Bianchi and Dr Pertl did not find the Kazakov abstract or the Kazakov paper.
687 This all confirms my view that neither the Kazakov abstract nor the Kazakov paper fall within s 7(3).
688 Now Ariosa has put forward possible reasons why the Kazakov paper or abstract were not identified in, for example, Pertl B and Bianchi DW, “Fetal DNA in Maternal Plasma: Emerging Clinical Applications” (2001) 98(3) Obstetrics & Gynecology 483-490. But I do not need to descend further into the detail.
689 Ariosa also says that it is not determinative of the question to be addressed by me under s 7(3) that none of the experts had in fact identified Kazakov before the priority date. The relevant inquiry is whether the hypothetical skilled addressee could reasonably have been expected to have ascertained the Kazakov paper. Generally, I agree. Of course the fact that none of the experts had found the Kazakov paper is not determinative, but this fact is not irrelevant either. And it is an attractive jury point. At the least this fact confirms my otherwise reached conclusion that neither the Kazakov abstract nor the Kazakov paper were reasonably ascertainable.
(c) Whether the hypothetical skilled addressee could reasonably have been expected to consider the Kazakov paper as relevant
690 Ariosa points out that Ms Norbury, Professor Oepkes and Professor Hyett explain that the title of the Kazakov paper contains language that is directly relevant to non-invasive prenatal diagnosis or testing. They also point to the reference to trophoblasts, which were known to be fetal at the priority date, in the abstract to the Kazakov paper. Their evidence was that based on the title and abstract they would have found the suggestion in the Kazakov paper, that the serum of pregnant women contains extracellular DNA that may be from trophoblasts and therefore of fetal origin, very interesting and relevant to a research project on non-invasive prenatal testing.
691 For example, Professor Hyett stated:
In my opinion, the description in the Abstract is significant from a prenatal testing perspective, as it suggests that pregnancy-related extracellular DNA can be detected in maternal blood.
692 In cross-examination, Professor Oepkes stated:
At that time, our group and others – other groups, including friends in Amsterdam were working hard on trying to detect these foetal cells in maternal circulation for non-invasive prenatal diagnoses with the aim to make – make our diagnosis safer and we concentrated very, very much on, what you call, enrichment of these cells because we all agree that the cells were never trophoblast or nucleated. Red cells were extremely rare in a tube of maternal blood.
We tried to enrich the sample by increasing its various cell sorting techniques – the cells – because we wanted to look at their DNA and wanted to be sure it’s foetal. And then this paper suggests that there is DNA outside of these cells in maternal plasma which is the plasma or serum for – for this type of research was discarded. We were not interested in that part. There were serum markers in terms of hormones for prenatal diagnosis. They were using that in this research field. We did not care about the plasma or serum and the suggestion of this paper, both in the title and the abstract that DNA is present outside of the cell in the plasma or serum and that some of that DNA may come from trophoblast cells, therefore, it could be foetal or – or pregnancy DNA. That, it’s, I think, the key and – and essential interest of this - this paper, the suggestion, not the proof or not – not showing that that it’s there or quantifying it, just a suggestion that outside of the cells in this plasma or serum, there is DNA floating around that could be of origin from the pregnancy and, therefore – or for people of interest in this field could mean that this could be used for diagnostic purposes, yes, would like to leave it to them.
693 Similarly, Ms Norbury stated that she would have been attracted and interested to see that there was extracellular DNA in the plasma or serum as she had an interest in sources of DNA for molecular genetic testing.
694 Contrastingly, Professor Fisk’s position was that he would not have considered the Kazakov abstract to be interesting, due to the journal of publication, the authors of the article and the language of the article. Similarly, as I have said earlier, Professor Lovett suggested that he would have excluded the Kazakov paper or abstract for further review, based on the fact that he did not recognise the journal it was published in and because it was published in Russian.
695 But Ms Norbury said that this is a “closed way of approaching development”. Moreover, Ariosa says that this way of approaching development is not one that should be taken to be representative of the hypothetical addressee, faced with a research project on non-invasive prenatal testing at the priority date. As Ariosa pointed out, Professor Oepkes explained:
… any language papers that would come up from searches would be potentially of interest, and if needed we have had papers translated from Chinese, Japanese – all sorts of languages. It’s not a restriction by itself that it has to be in English.
Analysis
696 In my view, even if a person skilled in the art had obtained a translation of the Kazakov paper, the person skilled in the art would have realised that the paper does not contain any disclosure, express or inherent, relevant to the development of prenatal diagnosis using non-invasive techniques and thus it would not be regarded as relevant for the purposes of s 7(3).
697 First, the Kazakov paper, as originally published in Tsitologiia, included an English translation of the title that reads “[e]xtracellular DNA in pregnant women blood”. But I agree with Sequenom that this title does not resonate with research in the area of prenatal diagnosis and does not disclose the contents of the article in a way which suggests any relevance to the relevant project.
698 Second, Tsitologiia was an obscure journal at the priority date and even today. A skilled addressee was less likely to be drawn to articles that were published in obscure and unknown journals or from unknown authors or institutions, and would have generally discounted articles from low-quality journals.
699 Third, at the priority date, significant, reputable, high-impact and/or high-quality work, including from non-English speaking authors, would usually be published in full in English. A person skilled in the art would have considered that an article not published in English was unlikely to be relevant or useful.
700 Fourth, a person skilled in the art would not have regarded the Kazakov paper, as an article published in Russian by unknown authors, as relevant to the relevant project.
701 Fifth, the abstract states that “[i]t is discussed which types of cells may serve as a source of extracellular blood DNA (either trophoblasts, lymphocytes, or decidual cells), the significance of such DNA for pregnancy being appreciated”. But this sentence does no more than explain that the paper discusses which types of cells may serve as a source of extracellular blood DNA and lists various different cell types, including fetal cells and maternal cells. It does not equate to a hypothesis that the difference between DNA results obtained from women in their first trimester as opposed to their third trimester of pregnancy “may be related to the biology of the development of pregnancy”. And even if such a hypothesis was contained in the abstract, that would not render the abstract (or the Kazakov paper) relevant to the relevant project, as the abstract does not provide or refer to any evidence to suggest that the detected extracellular DNA could be of fetal origin.
702 Sixth, the Kazakov abstract draws conclusions that were inconsistent with established knowledge in the prenatal diagnosis field at the priority date, and displays internal inconsistency. For example, the abstract confusingly states in the first sentence that “[t]he level of extracellular DNA increases in the blood of women during pregnancy” and then later that “a type of inter Alu repeats was estimated in the blood of women being in the first trimester of pregnancy only”.
703 Further, Professor Hyett’s view that the abstract suggests “the difference between the results obtained from women in their first trimester as opposed to their third trimester of pregnancy may be related to the biology of the development of pregnancy” is problematic. In any event, Professor Hyett conceded that there are parts of the abstract that he would not fully understand and that he would have taken the abstract to a colleague with more extensive laboratory experience before taking any steps in relation to the Kazakov abstract or the Kazakov paper.
704 Further, Kazakov’s alleged finding that certain generic DNA sequences are uniformly detected in the first trimester of pregnancy but at no other time would not suggest to a person skilled in the art that Kazakov amplified extracellular DNA of fetal origin. A person skilled in the art knew that the number of fetal origin cells in the maternal blood, the only plausible source of fetal DNA known at the priority date, increased during pregnancy. A person skilled in the art would have strongly expected that cffDNA levels would increase throughout the pregnancy and be most easily detected in the third trimester. Further, there is no reliable evidence in the paper to suggest that the DNA detected in the Kazakov paper was pregnancy related. As described by Professor Lovett, “there’s no data whatsoever to support the assertion that it’s fetal in origin”.
705 Seventh, the paper demonstrates poor experimental methods and procedures and is littered with unsubstantiated speculations. Ms Norbury agreed that the experiments performed were strange and did not make any sense.
706 Eighth, the paper does not relate to or discuss any potential applications of the detection of cell free Alu sequences or inter Alu sequences in the field of non-invasive prenatal diagnosis.
707 Ninth, the paper does not provide any evidence that cffDNA is present in the maternal circulation, let alone in any detectable quantities.
708 Tenth, it was not an aim of Kazakov et al to ascertain whether cffDNA was present in the blood of pregnant women, although Professor Hyett appears to have suggested otherwise.
709 Eleventh, Kazakov et al do not hypothesise that some of the cfDNA was of fetal origin.
710 Generally, the evidence establishes that the Kazakov paper lacks credibility, cogency and consistency which are important determinants of a paper’s relevance to a person skilled in the art. Professor Hyett said that “the quality of science is important in part of an equation for you determining what you would do next.” Professor Oepkes agreed that the quality of a paper is taken into account in the assessment of a piece of literature as to its relevance.
711 I agree with Sequenom that given the numerous deficiencies, the skilled person undertaking the relevant project would have dismissed the paper, assuming they somehow found it, as being relevant in the sense that I have referred to.
(d) Whether the claimed invention was obvious in light of the common general knowledge and the Kazakov paper or abstract
712 Let me begin with Ariosa’s contentions.
713 Ariosa contends that the relevant claims of the Patent are obvious when considered in light of the common general knowledge and the Kazakov paper or abstract at the priority date.
714 It says that Kazakov suggests both in the abstract and the full paper that the source of the extracellular DNA detected in the serum of pregnant women may be trophoblast (i.e. fetal) cells. The skilled addressee with experience in fetal medicine and an interest in the field of prenatal diagnosis at the priority date would have found the idea that there may be cell-free (extracellular) DNA of fetal origin in the maternal serum of a pregnant female interesting. They would also not have considered it unlikely since he or she may know or otherwise will learn (from the Kazakov paper but not the abstract) that cell-free DNA exists generally in the serum of adults and increases in certain pathological processes such as during infections and cancer. He or she would have appreciated that if this idea were correct, it may provide a source of fetal DNA for use in prenatal testing, such as for determining fetal sex, and prenatal diagnosis of rhesus D and other single gene disorders. In this regard there had been work for some number of years on the use of fetal cells in maternal blood. Fetal cells in maternal circulation were at very low concentration. Therefore, PCR was being used to identify Y chromosomes and RhD genes in DNA extracted from whole blood with some success. Nested PCR with Y chromosome markers was able to work on low concentrations of fetal cells to maternal cells. There were, however, many disappointments and the desire for a better alternative to invasive testing was still very strong.
715 Ariosa says that the person skilled in the art would have been motivated to investigate whether the maternal serum did contain cell free DNA of fetal origin. To do so he or she would try the very simple experiment of searching for Y chromosome markers in plasma or serum. He or she would have expected this to be able to be done easily, using known, standard and routine methods, which had been used to analyse DNA extracted from whole fetal cells, such as a PCR method using primers for sequences from the Y-chromosome or rhesus D gene. He or she would have run multiple samples with controls if need be.
716 Ariosa says that the evidence of Ms Norbury establishes that the skilled addressee with experience in molecular genetics-based laboratory techniques at the priority date would have used well-known PCR primers for Y-chromosome DNA to test the suggestion in the Kazakov paper. And if that DNA was present, this would have revealed that fetal DNA was present in the maternal serum. She considered such an experiment to have been simple to carry out. It would not have required any additional resources, with samples being readily available, or primer design. As Ms Norbury explained, it would not have cost anything to run “because it’s an assay that we have up and running all the time” and they “had a bank of samples already in there”.
717 Ariosa says that the person skilled in the art would in fact get the result in the Patent because cffDNA was more prevalent than expected. He or she could also have used or would use next any markers that are not maternally inherited, for example, RhD gene sequences in an RhD negative mother. This would provide a determination for sex and other characteristics of the fetus within the meaning of diagnosis for claims 22 and following.
718 Ariosa said that Professor Lovett’s position was that if the Kazakov paper was “an agreed publication”, whilst he would expect the amount of extracellular DNA from trophoblasts would be “vanishingly small”, nevertheless he would understand there to be “tiny amounts of the fetal cells in the circulation” and that he would understand the extracellular DNA to come from apoptosis of those circulating cells.
719 Professor Lovett then said that he would have thought he could detect that extracellular DNA by looking for Alu repeats because every one of those cells contributes 1 million targets. But Ariosa says that that cannot be right because it is not possible to discern fetal from maternal extracellular DNA by looking for Alu repeats. Nevertheless, so Ariosa says, Professor Lovett did not say that he could not have detected SRY sequences of this vanishingly small amount of extracellular fetal DNA. Ariosa says that he could not have disputed that given the accepted sensitivity of nested PCR with SRY primers this could have been detected.
720 Generally, Ariosa says that with the Kazakov paper in hand, the skilled addressee would have expected to be able to detect fetal DNA through PCR techniques, such as nested PCR of SRY sequences.
721 Now I would note at this point that the Kazakov paper was the subject of a number of criticisms by Professors Fisk and Lovett, which I will elaborate on later. And based on these perceived shortcomings, they suggest that they would have dismissed it entirely, or at most, sought to repeat the experiments described in the Kazakov paper. But Ariosa says in response that whilst it may be accepted that there are criticisms that can be made of the Kazakov paper, the evidence of Sequenom’s experts is neither realistic nor credible, in circumstances where the potential clinical implications of an alternative source of fetal DNA for prenatal testing were significant, and highly sought-after.
722 Further, Ariosa submits that particular care should be taken in relation to the views expressed by Professors Lovett and Fisk. It says that Professor Lovett was not involved in the field at the priority date and appears to have added to his assessment of the Kazakov paper knowledge beyond that which would have been possessed by the person skilled in the art at the priority date. But Ariosa had to accept his criticisms of the experimental methodology of the Kazakov paper.
723 But Ariosa said that Professor Lovett seemed to be so overwhelmed by those defects as to be unable to attribute any significance to other aspects of the paper which would have been interesting and informative to the skilled person. For example, the other witnesses appeared to read much of the Kazakov paper with the interest of somebody reading a review paper. A good example, it says, is the information in the first paragraph.
It has been shown that extracellular DNA is contained in the blood of humans and animals … An increase in the content of extracellular DNA in the blood of humans has been described during pathological processes taking place in various types of tissue of the body.
724 Ariosa says that such information was not known by Ms Norbury, Professor Oepkes or Professor Hyett. It says that that kind of information would have stimulated a level of enthusiasm for the Kazakov paper’s ideas which Professor Lovett did not share by reason of his learning and understanding beyond the prenatal testing field. Ariosa says that Ms Norbury in contrast said “it was such a – a profound finding, for me, to know that there was DNA in the extracellular fluid, because of the ramifications of being able to do further analysis”.
725 Further, Professor Lovett gave the following evidence:
MR RYAN: Now, Professor, of course, you weren’t in the prenatal diagnosis field in 1997, were you?
PROF LOVETT: No.
MR RYAN: No. And so your attempt to tell the court what you would have done had you seen this paper is, of course, an exercise in putting yourself into a hypothetical situation, isn’t it?
PROF LOVETT: It’s for – everybody here – they didn’t see it in 1997.
MR RYAN: But you more so than the others because you weren’t even working in the field?
PROF LOVETT: No, because, actually, I was in a much bigger field and, quite clearly, I had some knowledge base which most people here don’t have.
MR RYAN: Yes?
PROF LOVETT: So I knew what Alu was, I know what inter-Alu is and I knew about extra-chromosomal DNA, so – and I – you know, as I mentioned earlier on, you know, once you know that there is antibodies to DNA in SLE and things like that, you quickly realise there’s DNA in blood.
726 Ariosa says that he plainly meant extracellular DNA in blood (or more accurately, in serum) because he had already said that he had “known [that] for decades”. Against that background he did not regard the paper as giving him anything useful at all.
727 Professor Fisk said in reference to Ms Norbury:
… her gestalt moment was that there was DNA present in serum, nothing to do with pregnancy. I’ve just had a quick look at the Kazakov paper. I mean, four of the – there are four references to DNA in serum in the 1970s. So here’s a paper that is at least a decade and a half after that observation – it’s apparently reasonably well known.
728 But Ariosa says that although it is not altogether clear whether Professor Fisk himself knew that fact, he was seemingly quite unimpressed by the paper making the point.
729 Generally, according to Ariosa, the overly critical analysis of any information provided by the paper infected the evidence of both Professor Lovett and Professor Fisk.
730 Further, Ariosa says that Professors Lovett and Fisk were inflexible in their openness to any new information. Each of them said he would pay no attention to papers not published in English. But Ariosa says that it is difficult to reconcile that approach with the notional approach of the person skilled in the art under the present patent system which contemplates a world-wide prior art base.
731 Further, Professor Fisk said:
Now, it’s very easy to say we will just do one or two quick experiments and we will answer this and come up with an earth-shattering discovery. The number of blind alleys that there are, the number of junior investigators who come with good ideas that won’t work because they haven’t read the literature thoroughly, or don’t appreciate the technical or logistic problems. Our job as mentors was to guide young investigators, to nurture and encourage their careers, just in the same way that the three of us here who are in the maternal fetal medicine field have had our own careers nurtured, by avoiding us going down blind alleys that aren’t going to go anywhere and trying to point us in areas that are likely to be very productive.
732 But Ariosa says that the difficulty with this approach is that it is not representative of the ordinary person skilled in the art. All that was being proposed was, indeed, one or two quick experiments and Professor Fisk’s reluctance to contemplate performance of them demonstrates a lack of interest in pursuing new ideas suggested by a published paper, which approach should not be attributed to the hypothetical person skilled in the art.
733 Ariosa says that the person skilled in the art armed with the Kazakov paper would have been motivated to investigate, using routine, standard techniques (being the same techniques that were being used to investigate fetal cells as a source of fetal DNA), the credible suggestion that the extracellular DNA detected in the serum of pregnant women included extracellular DNA of fetal origin. The Kazakov paper describes the amplification of Alu and inter-Alu repeats from the serum of pregnant women. The evidence of Sequenom’s experts is that such sequences were repetitive sequences found throughout the human genome. Ariosa says that it may be inferred that authors of the Kazakov paper likely did amplify fetal DNA.
734 Further, Ariosa says that the person skilled in the art would not have tried to repeat the approach in the Kazakov paper, as suggested by Professors Lovett and Fisk, but would have undertaken a simple PCR assay using a primer that was specific to the fetus, such as a primer directed to the Y chromosome or another paternally inherited sequence. That would necessarily involve performing claims 1 to 3, 5, 6, 9, 13, 14, 22, 23, 25 and 26, and would have confirmed the presence of cffDNA.
Analysis
735 Before proceeding further I should say something by way of background.
736 Before the priority date, research in the prenatal diagnosis field was focused on the use of fetal cells (as distinct from non-cellular components) extracted from peripheral maternal blood samples to test for various prenatal conditions. It was thought that fetal cells from maternal blood could offer an alternative and advantageous source of cell specimens to those obtained by invasive techniques such as amniocentesis, CVS and percutaneous umbilical cord sampling.
737 As at the priority date it was known that fetal cells in maternal blood were extraordinarily rare. And the rarity of fetal cells in maternal blood led to difficulties detecting and enriching fetal cells amidst the overwhelming presence of maternal cells.
738 As at the priority date researchers in the prenatal diagnosis field were working to overcome the difficulties with using fetal cells by investigating various cell purification methods. This included the identification of cell types with markers that were unique to fetuses and thereby could be used to distinguish fetal cells from maternal cells.
739 Now even if a person skilled in the art could reasonably have been expected to ascertain, understand and regard as relevant the information contained in the Kazakov abstract or the Kazakov paper, they would not have been directly led as a matter of routine to try to detect fetal DNA in the serum or plasma of pregnant women as a method of prenatal diagnosis in the reasonable expectation that it might well produce a useful result, namely, that cell-free DNA of fetal origin would be detected.
740 I agree with Sequenom that it is implausible that a person skilled in the art would have explored or designed any experiment in the field of non-invasive prenatal diagnosis based on the disclosures in the Kazakov paper, let alone the Kazakov abstract.
741 If a skilled person did anything with the Kazakov abstract, given its deficiencies, they would have, as a matter of course, sought to obtain a translation of the Kazakov paper. But having obtained a translation of the Kazakov paper, notwithstanding my view that a person skilled in the art could not reasonably be expected to have done so, a skilled person would be moved to dismiss the information in it as poor science and speculation.
742 Generally, it seems clear on the evidence that Kazakov’s methods do not reflect a proper scientific method. For example as Sequenom points out, despite purporting to undertake a quantitative analysis, no steps were taken by the authors to ensure that equal proportions of starting material had been placed into the starting reactions. Further, although seeking to draw broad-ranging conclusions about biological mechanisms, no technical replicates (or replicates of any nature) were performed and no controls were incorporated into the experiments giving rise to the results in Figure 2. This renders it impossible for the authors to determine with any certainty whether differences between test subjects were due to natural variation driven by biological factors. These failures meant that the authors were unable to carry out any analysis as to whether the results were statistically significant.
743 More generally, the preponderance of the expert evidence before me establishes that the conclusions drawn by the authors are unsubstantiated and problematic to say the least.
744 The following points can be made concerning Figure 1.
745 First, all of the experts agreed that Figure 1 is not credible. As articulated by Ms Norbury, the experiments giving rise to Figure 1 “don’t really demonstrate that there’s anything going on”.
746 Second, in Figure 1, a band appeared in all of the lanes containing DNA from human samples including pregnant women, men and non-pregnant women. This does not support the conclusion of the authors that “during pregnancy there is an increase first of all in the concentration of low-molecular DNA, the increase being most pronounced during gestosis”.
747 Third, the authors rely on a visual inspection of a gel in Figure 1 to draw the conclusion set out in the preceding point. But the method used in the Kazakov paper does not enable quantitative analysis.
748 Fourth, even if there were differences in intensities of the bands in Figure 1, this would not provide a basis on which the conclusions in relation to Figure 1 could legitimately be drawn, as the differences are too small to be reliably detected by eye. It is accepted that standard PCR is a non-quantitative technique.
749 The following points can be made concerning Figure 2.
750 First, the data in Figure 2 is of very poor quality and would not have been accepted in a reputable peer reviewed journal. Professor Lovett described Figure 2 as “rubbish” and “laughable”.
751 Second, Figure 2 purports to show the results of a PCR reaction carried out on human DNA using the Tc65 primer. This primer was known at the priority date to produce a smear of human DNA on a gel. However, no such smear can be seen in Figure 2. This result is unexpected, and the only conclusion a person skilled in the art could draw in relation to Figure 2 is that the reaction was very inefficient or had not worked. As Professor Lovett noted, “scientific method says throw it away”.
752 Third, because no positive or negative controls were used, a person skilled in the art would be unable to gain any insight into why the characteristic smear of DNA digested with Tc65 primer had not been produced in Figure 2. Due to the absence of controls, and the additional inadequacies of scientific procedure discussed earlier, it would be unrealistic for a person skilled in the art to make any meaningful interpretation of the data.
753 Fourth, Figure 2 allegedly shows the results of PCR reactions performed on women during the first trimester of pregnancy. No data is presented from PCR reactions carried out later in pregnancy, and there is no data from PCR reactions carried out on non-pregnant women or men. In these circumstances, a person skilled in the art would not have accepted the conclusion that inter-Alu repeats were not detectable in the blood of women later in pregnancy without seeing the data for those women, particularly given that this was purported to be the key result of this part of the paper and it was known that Alu repeats were ubiquitous, rarely unique to a particular individual, and did not differ by gender or physiological conditions such as pregnancy.
754 Along with the lack of controls, it would seem that the lack of data from non-pregnant women and men severely limits what a person skilled in the art could reasonably conclude from Figure 2.
755 Let me make some other points, which Sequenom submitted and I accept.
756 First, given that Figure 1 shows that Alu repeats were detectable in the blood of all human patients, pregnant or otherwise, the observation that inter-Alu repeats are only detectable in the blood of women in the first trimester of pregnancy lacks credibility and is clearly contradictory. As Professor Lovett articulated, this claim “beggars belief”. Similarly, Professor Fisk noted that this conclusion defies understanding and logic. A person skilled in the art at the priority date would have known that inter-Alu repeats should be detected in all samples in which Alu repeats are detected (i.e. all human samples).
757 Second, it was known at the priority date that the level of fetal cells in maternal blood, like many other physiological variables, increased over the term of a pregnancy, rather than decreased.
758 Third, the authors of the Kazakov paper speculate that the source of the “extracellular DNA” could have been from trophoblast cells (fetal origin), or lymphocytes or decidual cells (non-fetal origin). The paper does not, as Ms Norbury suggests, hypothesise that extracellular DNA in maternal circulation could have come from the mother or the fetus or both. At the priority date, trophoblast cells were known as cells coming from the placenta that were difficult to isolate in the maternal circulation including because they often became trapped in the maternal lung. As a result, at the priority date, there were several reasons why trophoblasts were not considered to be ideal for prenatal diagnosis, including because the volume of trophoblast cells within maternal circulation was according to Professor Hyett “very, very low, very difficult to detect”. The reference in the Kazakov paper to lymphocytes was similarly to maternal lymphocytes.
759 Fourth, given what the person skilled in the art knew at the time in relation to fetal-maternal traffic, placental mass, fetal cell numbers and the breakdown of DNA, and the fact that the Kazakov paper looked for commonplace DNA sequences expected to be found in all humans, the fact that the sequences were allegedly not found in the third trimester of pregnancy would point away from a fetal origin for the “extracellular DNA”.
760 Fifth, it would not have been clear to a person skilled in the art how a woman’s DNA profile could vary so absolutely between the first and third trimesters. Professor Hyett speculated that the “extracellular DNA” may come from a cell only present in early pregnancy, but he failed to identify a cell type that met this criterion. As explained by Professor Fisk, no such cell was or is known to exist.
761 Relatedly, during cross-examination Professor Hyett postulated a mechanism to explain the speculation in the Kazakov paper that extracellular DNA was present in the third (but not the first) trimester of pregnancy:
But an alternative to that would be that in the first trimester of pregnancy when you have a process where the placenta is very much in a development phase and it’s embedding and implanting and invading into the maternal compartment that it may be that process of developmental biology which actually exposes maternal circulation to a load of placental DNA at that point in time. That’s an alternative hypothesis.
762 But this alternative theory was post priority date hindsight conjecture on Professor Hyett’s part. And as Professor Fisk explained, Professor Hyett’s speculation is not supported by the biological mechanisms underlying the development of the placenta:
So the modelling that takes place when the placenta develops, not just first implants, is a joint maternal fetal process … The trophoblast invades some of these vessels to assist in that process, but decidual cells, which are of the mother, and trophoblast will be involved in this process. This is broadly what we refer to as trophoblast invasion. Now, from memory, this occurs in two phases. There is one phase broadly at the end of the first trimester. But there is another, a second wave trophoblast invasion which is probably the more substantial that happens in the second trimester.
So this is best manifest biophysically by a commonly used test in pregnancy to determine the resistance in the uterine arteries because women normally have high resistance in their uterine arteries, but when they’re pregnant they have a … blow resistance waveform, and that’s usually not manifest till 18, 20, even 24 weeks. So that’s when the second wave of trophoblast invasion is going on. So if it’s related to the process of trophoblast invasion and – and the remodelling of the maternal blood vessels in the decidua, I would posit that it’s a process that goes well into the second trimester.
(Emphasis added.)
763 In my view there is no data or other evidence to support Professor Hyett’s speculation. Rather, as pointed out by Sequenom, the following may be noted. As explained by Professor Fisk, it “makes sense” that the process of trophoblast invasion is “very active in the last part of mid-pregnancy, where, if this postulation is correct, you would expect more DNA to be released”. Further, it is more likely that the process leading to cell free DNA in the maternal extracellular fluid is apoptosis and cell death, and, given that cell death is greater in the second, and even greater in the third trimester than in the first trimester, it does not follow that there is a specific process that would lead to DNA release from trophoblasts only in first trimester, but not during the general growth and development of the placenta. Further, Professor Hyett agreed that he would have known at the priority date that the level of fetal cells would increase throughout pregnancy. Further, Professor Hyett acknowledged that the experiment involving the Tc65 primer, which gave rise to the conclusion that the DNA was only present in the first trimester, was not conducted on any women other than women in the first trimester. Further, consistently with Professor Hyett’s comments, Professor Fisk noted that trophoblast cells are “tiny earlier in gestation and huge in relative mass later in gestation”. Further, Professor Fisk is not aware of any cell type that is present in early pregnancy but not late pregnancy, and Professor Hyett also has not identified a cell type with these characteristics, despite postulating that such a cell type or “molecular marker” might exist. Finally, Professor Hyett noted that this was an “alternative hypothesis” to which he “would not know the answer”. And in order to determine if the hypothesis was correct he would need to perform a study.
764 In my view, the Kazakov paper does not teach the person skilled in the art anything about prenatal diagnosis or fetal nucleic acids in the serum or plasma of pregnant females. More specifically, it does not teach or provide the idea that cffDNA could be detected in maternal blood.
765 Now Ms Norbury claimed that the paper and in particular its references would have taught her at 1997 that DNA was present in extracellular-fluid. But the presence of extra chromosomal DNA in blood is something that has been known since the 1970s.
766 Now as I have indicated, Ariosa has relied on the evidence of Professor Hyett and Ms Norbury, who stated that the Kazakov paper appears to show that the level of extracellular DNA increases in the blood of women during pregnancy and demonstrates the presence of “pregnancy-specific” extracellular DNA in maternal serum. But they reached these conclusions notwithstanding that they accepted that the conclusions in the paper were not supported by experiments undertaken with proper scientific method. Moreover, these conclusions by Professor Hyett and Ms Norbury are problematic in any event.
767 The first conclusion cannot be drawn from Figure 1. This figure involved a qualitative, not quantitative analysis, and is directly inconsistent with Kazakov’s asserted findings in Figure 2. Indeed, Ms Norbury conceded that Figure 1 and Figure 2 do not back up anything. And she agreed that the methodology giving rise to Figure 1 was the wrong technique, but puzzlingly went on to state that “the [e]xperimental data is not especially relevant to my line of thought”.
768 As I have already said, at most the Kazakov paper demonstrates that extracellular DNA is present in men, non-pregnant women and pregnant women. But there is no evidence in the Kazakov paper to suggest that the DNA detected by the authors was pregnancy related, let alone fetal.
769 Further, Professor Hyett’s review of the article was also problematic.
770 Professor Hyett noted correctly that “there are a number of points in this paper where you can be very critical about the science that’s represented”. But these errors in the scientific technique would apparently not have deterred Professor Hyett from taking the paper to a molecular geneticist colleague, on the basis that the weak experimental argument would have merited further investigation given that there was an important statement being made.
771 Further and in any event, it is likely that if Professor Hyett took the paper to a person skilled in the art with the experience of Professor Lovett, such a molecular geneticist would have dismissed the paper in terms of the quality of the technique and the conclusions. Moreover, if Professor Hyett had taken the paper to a person skilled in the art with the experience of Ms Norbury, such a molecular geneticist would have simply observed that extracellular DNA was present in human blood. But this was a fact known since the 1970s. Further, the steps taken by a molecular geneticist like Ms Norbury would have depended on how widely she canvassed and who she spoke to and the peculiar knowledge of those persons.
772 Further, in my view the explanation provided by both Professor Hyett and Ms Norbury of what they understood from the Kazakov paper is hindsight affected. Further and in any event, at its highest, Professor Hyett’s evidence is that he would simply have had a “conversation about this paper, about the methodology of the paper: “Is this reasonable? Could we reproduce this? How would we work off this?””. But I agree with Sequenom that a person skilled in the art would not have regarded the speculations in the Kazakov paper to be reasonable and would not have worked off the paper.
773 Further, Professor Oepkes seemed to suggest that he would not have understood the body of the Kazakov paper and would have ignored two critical sections of the paper. But he did seem to suggest that he would have done something with the Kazakov paper. But this is inconsistent with his view that papers “lacking” data or exhibiting “poor science” “would” be “discard[ed]”.
774 Further, Ms Norbury adopted the position that she would not need to obtain a translation of the Kazakov paper. She said that in 1997, and based on only the unsubstantiated speculation in the Kazakov abstract, she would have tested a sample of maternal plasma or serum using a primer directed to the Y sequence. But as Sequenom points out, this contradicts the reason Ms Norbury gave as to why the Kazakov paper was interesting to her as at 1997. Ms Norbury stated that the “reminder” or “revelation” in the Kazakov paper that there is DNA in the extracellular fluid and the “five or six” references to this in the introduction was “essentially the main stimulant to thinking it’s worth pursuing”. She maintained that it was these references that sparked her interest that there is DNA in the serum or plasma. But these motivating references are contained only in the introduction section of the Kazakov paper, not the abstract. Therefore they would not have been accessible without a translation. When confronted with this inconsistency, Ms Norbury conceded that in the absence of reviewing the Kazakov paper she would have had “less information to go on” and that it would have been “less likely” that she would have “taken it forward”. But in any event, in the circumstances where Ms Norbury would have taken it forward, her next steps would have depended on who she spoke to and the peculiar knowledge of those persons. As Ms Norbury explained:
So if I discussed it with, say, Patricia Boyd over in the maternity unit and she said, “Yes. I know that there’s extracellular DNA in the plasma”, then that would have linked the two together, but you just don’t know whether those two conversations would ever have happened. They may have done, they may not have done.
775 As Sequenom correctly characterised the matter, it was Ms Norbury’s speculative evidence that she might have taken the abstract and spoken to a colleague and that this conversation might have given her the same insight as was in the introduction to the Kazakov paper. That is, that there is cell free DNA in extracellular fluid. At its highest, Ms Norbury’s evidence is that the Kazakov abstract merited a lot more conversation. But even if this scenario had played out, it would not have advanced Ms Norbury’s knowledge beyond what had been known in the field since the 1970s.
776 In my view, the evidence relied upon by Ariosa in relation to what a person skilled in the art would have understood from the Kazakov paper is inconsistent with how a person skilled in the art would have reasonably read a scientific publication at the priority date.
777 As I say I have much preferred the evidence of Professors Fisk and Lovett on these matters, which demonstrates that a person skilled in the art at the priority date would not have been motivated to do anything with the Kazakov paper and consequently would not have been led by the Kazakov paper to the claimed invention. As I have already said, in light of the numerous flaws and contradictory information in the Kazakov paper, the most likely outcome of a review of the Kazakov paper would have been to discard it.
778 Finally, if the person skilled in the art had been motivated to do anything at all with the Kazakov paper, it would have been to repeat the experiments described in the Kazakov paper using a properly designed experimental methodology. In order to properly test the Kazakov paper’s hypothesis, the person skilled in the art would repeat the PCR amplification conducted to generate Figure 1 using quantitative PCR on samples from multiple patients, whilst also running multiple control samples, so that any statistical differences between the concentrations of DNA in the blood could have been determined. A person skilled in the art would not have been able to predict what the result of this experiment would be. And insofar as any elevation in the amount of cell-free DNA was identified in the blood of pregnant women and those with pre-eclampsia, a person skilled in the art at the priority date would likely have considered a possible explanation to be that the DNA was derived from dead endometrial cells. The experiment would not have been informative as to the source of the cell-free DNA that was amplified.
779 A person skilled in the art would have also repeated the PCR amplification using Tc65 primers on blood samples from women in the early and late stages of pregnancy, as well as non-pregnant women. Appropriate positive and negative controls would have been included. This experiment would have confirmed that inter-Alu repeats are present in the blood of women at all stages of pregnancy, as well as in the blood of non-pregnant women and men.
780 Further, a person skilled in the art would not have been led to confirm or investigate any alleged hypothesis contained within the Kazakov paper by performing the experiments in the Kazakov paper using a primer directed to the Y chromosome. Ariosa’s conjecture is the product of hindsight analysis.
781 Let me deal with another matter. As I have indicated, a reasonable expectation of success is relevant to the obviousness inquiry.
782 Now even if a person skilled in the art performed an experiment on maternal plasma or serum using Y chromosome sequence markers, the person skilled in the art would have no reasonable expectations of being able to detect cffDNA. In my view, and assuming that the person skilled in the art did not reject the Kazakov paper or abstract out of hand, such a person would be left with no more than a research project.
783 Ms Norbury says that she would have wanted to check the idea in the Kazakov paper that some of the extracellular DNA in the serum of pregnant women might be of fetal origin. But, Ms Norbury agreed that she would not have been optimistic of the result, that it would in fact have been “surprising that there was going to be DNA there at all”, and that it would be surprising that any DNA in the extracellular fluid had not been degraded.
784 Now although Ms Norbury represented that it would have been simple to check if there was any Y material, and that this could have been done as a first run, a research project Ms Norbury was involved in in 2005 demonstrated the opposite. Ms Norbury in response sought to suggest that she would have performed a range of dilutions to check for false negatives and that “sometimes actually the more dilute samples work better than the more concentrated ones, because not only have you got a different amount of DNA there, you’ve got a different amount of contamination there”. But such an analysis imports an expectation of detecting cell-free fetal DNA in plasma acquired by Ms Norbury via her reading of the post priority date Lo 1997 paper. Ms Norbury was not aware at 1997 that there was any cell-free DNA in plasma. As Sequenom points out, if she had run experiments of the type described in the relevant 2005 poster and obtained only the 19 failed gels, she would not have recognised these as fails. Rather they would have been consistent with her lack of knowledge of cell free DNA in plasma and her expectation of not being able to detect cffDNA in maternal plasma or serum samples. It is only with the post priority knowledge of cffDNA that she would have been able to determine that the gels failed.
785 Professor Hyett stated that the Kazakov paper merited further investigation and that he would have discussed with his lab-based colleagues whether it was possible to perform the PCR step using a primer that was specific to the fetus, such as a primer directed to the Y chromosome. But he did not give direct evidence as to whether he would have had any expectation of detecting fetal DNA. Indeed, as he stated, “the whole basis of a scientific experiment is that you are testing a hypothesis to which you do not know the result”. Further, he noted that it would have been possible that one did the experiment but actually did not demonstrate any fetal DNA during that experiment.
786 Further, Professor Oepkes indicated that he would have expected his lab-based colleagues to be able to “check” whether maternal serum did actually contain DNA of fetal origin “quite easily”, that is, the laboratory technique itself was straightforward, but also did not state his expectation of actually detecting fetal DNA.
787 More generally, each of Ariosa’s experts appeared to concede that if the Kazakov paper had caused a researcher to test for Y material in maternal plasma or serum, this step would have involved lateral thinking.
788 Even more generally, in my view a person skilled in the art at the priority date would have had no reasonable expectation of successfully detecting fetal nucleic acid in the non-cellular part of maternal blood, even if they had tried to conduct an experiment of the type proposed by Ms Norbury after reading the Kazakov abstract or paper.
789 First, the knowledge in the prenatal diagnosis field at the priority date taught away from any such expectation. Fetal cells from whole maternal blood were the focal point in the field of non-invasive prenatal diagnosis at the priority date. Researchers were attempting to develop non-invasive fetal nucleic acid detection methods based on recovering fetal cells from maternal blood, but were facing an array of difficulties, including that these fetal cells were extraordinarily rare, and that there was a limited understanding of the interaction between the fetal and maternal circulations. And in their attempts to detect fetal cells, it was routine for researchers to discard the cell-free portion of the blood. For example, in an invited editorial about Dr Lo’s cffDNA based non-invasive prenatal diagnosis methods, Bianchi DW, “Fetal DNA in Maternal Plasma: The Plot Thickens and the Placental Barrier Thins” (1998) 62(4) American Journal of Human Genetics 763-764, Dr Bianchi stated “[i]n the past, the plasma has been discarded” (at 763). Dr Bianchi also explained in Bianchi DW et al, “Large Amounts of Cell-free Fetal DNA Are Present in Amniotic Fluid” (2001) 47(10) Clinical Chemistry 1867-1869 that: “Before 1997, we routinely discarded the plasma layer of the density gradient, unaware of the fact that it could contain nucleic acids” (at 1867).
790 Second, fetal origin cells were considered the only plausible source of fetal DNA at the priority date. But fetal cells were extraordinarily rare, with the reported ratio of fetal cells to maternal cells ranging from about 1 in 5 million to 1 in 100 million. In Professor Oepkes’ own words:
… isolating fetal cells was not easy because they were known to occur only rarely in maternal blood and were vastly outnumbered by maternal cells in the sample (I used to say that the chance of finding a fetal cell was ‘one in a million’, though detailed research on this suggested that they were even less frequent than that).
791 Third, a person skilled in the art at the priority date would not have expected fetal cells in maternal blood to release detectable levels of fetal DNA into maternal circulation. In relation to trophoblast cells, it was known that these cells expressed a cell surface marker involved in fetal protection from the maternal immune response. Therefore, the person skilled in the art would have expected that fetal trophoblasts were “immuno-privileged” and were therefore protected from destruction by the maternal immune system and hence would not be damaged so as to release DNA. As Sequenom points out, this understanding was supported by the fact that it was known that fetal cells had been detected in maternal blood decades after the birth of the fetus, suggesting that fetal cells were able to persist in maternal blood without being destroyed by the mother’s immune system. Trophoblasts were also challenging to work with within the context of non-invasive prenatal diagnosis.
792 Fourth, even if the person skilled in the art had believed that detectable fetal DNA was released into maternal plasma, they would have expected such DNA to be degraded very rapidly due to the presence of deoxyribonucleases.
793 In summary, it seems to me that the evidence demonstrates that at the priority date, a person skilled in the art would not have had a reasonable expectation of developing a successful method to detect cell free fetal nucleic acid using maternal serum or plasma. It was only after the contribution of Drs Lo and Wainscoat that a person skilled in the art had any reasonable expectation that cffDNA could be successfully detected in plasma and serum, and that this could be used for prenatal diagnosis.
(e) Conclusion on obviousness
794 Accordingly, Ariosa’s obviousness attack fails.
795 First, a person skilled in the art could not reasonably have been expected to have ascertained the Kazakov paper or indeed abstract at the priority date.
796 Second, a person skilled in the art could not reasonably have been expected to have treated the information in the Kazakov paper or abstract as being relevant to work related to the development of a new or improved non-invasive prenatal diagnosis technique.
797 Third, a person skilled in the art would not have been directly led as a matter of course by the Kazakov paper or abstract to try to detect cffDNA in a maternal plasma or serum sample. And even if a person skilled in the art tried to do so, they would not have had any reasonable expectation of success.
UTILITY
798 Section 18(1)(c) of the Act requires that an invention so far as claimed in any claim be useful, with the onus of showing otherwise resting on Ariosa.
799 Three questions may be posed. What has been promised (as gleaned from the specification) for the invention as delineated by the relevant claim? Is the promise useful? Has the promise been met? In this context the specification (including the claims) must be construed from the perspective of a skilled person, but not in a way that such a person would appreciate would lead to unworkability when by construction it could be given a more limited but workable meaning.
800 There is no need for the specification to provide support to demonstrate that a promise of the claims was satisfied (SNF (Australia) Pty Ltd v Ciba Speciality Chemicals Water Treatments Ltd (2011) 92 IPR 46; [2011] FCA 452 at [296] per Kenny J). And as Jagot J accepted in Apotex Pty Ltd v AstraZeneca AB (No 4) (2013) 100 IPR 285; [2013] FCA 162 at [352], “[u]ltimately, an asserted lack of utility must be established by appropriate evidence, not by mere speculation that the invention will not work or meet the promise set out in the specification”.
801 Further a lack of commercial viability or commercial practicality does not establish lack of utility. This point was made in Lane Fox v Kensington and Knightsbridge Electric Lighting Co Ltd [1892] 3 Ch 424 at 431 per Lindley LJ, cited by the High Court in Advanced Building Systems Pty Ltd v Ramset Fasteners (Aust) Pty Ltd (1998) 194 CLR 171 at [24], with that Court stating:
It is no objection to the validity of a patent granted under the Act that it is commercially impracticable; its utility depends on whether, by following the teaching of the complete specification, the result claimed is produced.
802 Further, when construing the specification, it is important to consider the nature of any promises. Unless the specification would be understood to convey a clear assertion that the invention will achieve a particular outcome across the breadth of the claims, inutility will not be demonstrated by showing that in some cases that outcome may not be achieved. A claim may have utility even though the promised advantage is not achieved in all cases. In Rescare Ltd v Anaesthetic Supplies Pty Ltd (1992) 111 ALR 205; [1992] FCA 811, the claimed method of treatment utilised a nose-piece apparatus applied to patients suffering from snoring sickness so as to maintain air pressure sufficient to keep the nasal passages open. It was argued that the claim was not useful as not all patients would achieve a beneficial result. Gummow J said (at 232):
One looks at the claim to see whether there is a failure to fulfil that promise. It is not necessary to show utility that the promise be fulfilled in every case. On the evidence, the claimed invention plainly is of considerable practical utility in the treatment of substantial numbers of persons who are “patients” within the meaning of claim 1.
803 Further, there are limits on any broad principle, if there be one, that everything within the scope of a claim must be useful. Lehane J in Aktiebolaget Hässle v Alphapharm Pty Ltd (1999) 44 IPR 593; [1999] FCA 628 at [227] observed in respect of such a broad principle that “[a] degree of caution, however, is required”. If qualifications and expedients necessary to make the invention work are left to the reader to supply, that does not necessarily equate with inutility.
804 Further, to identify by the process of construction what the patentee intended to do by his or her invention, one must construe the whole of the specification.
805 As Gummow J in Rehm Pty Ltd v Websters Security Systems (International) Pty Ltd (1988) 81 ALR 79 at 84 and 96 to 97 describes this construction question, it is a search for any “promise of the invention”. He adopted the formulation of Lindley LJ in Fawcett v Homan (1896) 13 RPC 398 at 405 that the basic principle is that “[i]f an invention does what it is intended by the Patentee to do, and the end attained is itself useful, the invention is a useful invention”. So having identified the promise of the invention (at 97):
It is in the light of this that one has to consider the question of utility, and the question is whether in the sense of patent law the device is useless for that purpose.
806 Now Ariosa contends that claims 1 to 3, 5, 9 and 14 are invalid on the ground that the claimed inventions are not useful. But before getting into Ariosa’s arguments, I should note that for the moment I do not need to get into the detail of the construction question that arises on the infringement case in terms of the meaning of the phrase “a prenatal diagnosis” in claims 22, 23, 25 and 26.
807 Ariosa says that the Patent sets out a clear promise, which is that the claimed invention “provides a method for prenatal diagnosis” (page 2 lines 20 to 23). There the Patent states:
This invention provides a detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of foetal origin in the sample. The invention thus provides a method for prenatal diagnosis.
(Emphasis added.)
808 The said promise is said to be consistent with the title of the Patent, “Non-invasive prenatal diagnosis”.
809 Further, the Patent provides the following definition of “prenatal diagnosis” (page 2 lines 24 to page 3 line 4):
The term “prenatal diagnosis” as used herein covers determination of any maternal or foetal condition or characteristic which is related to either the foetal DNA itself or to the quantity or quality of the foetal DNA in the maternal serum or plasma. Included are sex determination, and detection of foetal abnormalities which may be for example chromosomal aneuploidies or simple mutations. Also included is detection and monitoring of pregnancy-associated conditions such as pre-eclampsia which result in higher or lower than normal amounts of foetal DNA being present in the maternal serum or plasma. The nucleic: acid detected in the method according to the invention may be of a type other than DNA e.g. mRNA.
810 Ariosa says that a “maternal or foetal condition or characteristic” is therefore “related to” but distinct from “the foetal DNA itself or to the quantity or quality of the foetal DNA in the maternal serum or plasma”. That is, a “maternal or foetal condition or characteristic” is not simply the genotype of the fetus, but is rather the expression of that genotype, or its link to the “quantity or quality of the foetal DNA in the maternal serum or plasma”. Further, Ariosa says that no practical application of Dr Lo’s discovery is disclosed in the Patent, other than in the context of providing prenatal diagnosis as broadly defined.
811 But Ariosa says that claims 1 to 3, 5, 9 and 14 are not limited in the scope of their application to use in a method for prenatal diagnosis. To the extent that those claims include within their scope the mere detection of the presence of a fetal nucleic acid, in the manner specified by each relevant claim, and where that detection is not part of a method of providing a prenatal diagnosis, the claim includes within its scope subject matter that does not meet the said promise.
812 Ariosa says that this would be the case, for example, where a nucleic acid of fetal origin is detected which does not provide any information about a condition or characteristic of the fetus or a pregnant woman. As explained by Ms Norbury, the detection of a SNP “merely identifies the existence of a single base pair difference between the fetus and the mother. Unless the SNP that is identified is known to cause a pathogenic mutation, or is an identified linked marker, the identification of a single SNP cannot be relied on to make a diagnosis in relation to any characteristic or condition of either the mother or the fetus”.
813 Ariosa also says that the more complex example provided by the Polymorphic Assay of the Harmony Test, I will explain this all later, also illustrates the broad scope of claims 1 to 3, 5, 9 and 14. Sequenom relies on the Polymorphic Assay as itself giving rise to an infringement of each of those claims, yet, so Ariosa says, the Polymorphic Assay does not provide a prenatal diagnosis and nor does it provide a screening test within the broader concept of the field of prenatal diagnosis. Ariosa says that at most it identifies that a fetus has a particular DNA sequence at a particular locus. But that sequence is not itself the cause of any particular characteristic or condition of the fetus or mother. But Professor Lovett did say under cross-examination:
MR RYAN: Professor Lovett, just one question about this. I might come back to this later, I think, but while we’re here, it’s true, isn’t it, that none of these SNP markers in the polymorphic assay is manifested in any particular condition?
PROF LOVETT: Well, as we will come back to, I’m sure, later, I would argue that knowing a SNP is knowing a potential condition, and that may sound terribly contrived, but it’s not. We know that between two individuals, there’s a SNP every kb or so – kilobase. Some of those are going to be private. Some of them are going to be shared. But of those, the vast majority are what’s called variance of unknown significance. So we don’t know if they cause anything. So you can go around and judiciously choose SNPs that have never been associated with anything on the face of this earth and then 10 years from now it comes along and you’ve found, “Oh my goodness. I’ve just found an Alzheimer’s susceptibility.” So that’s my caveat. A SNP can be a predictor of a condition.
814 Further, Ariosa says that Professors Fisk and Lovett agreed in the joint expert report that the Polymorphic Assay of the Harmony Test “does not directly detect a maternal or fetal condition”. But there was then a suggestion by Professors Fisk and Lovett that an exception to this would be the detection of twins. This was said to be on the basis that “one would detect two different profiles of informative polymorphic loci”. But Ariosa says that there is no evidence to suggest that the Harmony Test could be used in this manner, and it is not in fact used in that way. The Harmony Test can be performed on twin pregnancies, but only if it is known the mother is carrying twins before the test is performed. For example, the undated Sonic brochure titled “Non-Invasive Prenatal Testing: Information for medical practitioners” states “the accuracy of the test for detecting trisomies 21, 18 and 13 appears to be very good, provided the laboratory is advised that it is a twin pregnancy”. Further, the internal Ariosa “Quick Reference Guide – Frequently Asked Questions” for training purposes dated February 2016, rev 1.0 state that it is “critical to obtain accurate information regarding the number of fetuses in the pregnancy”, and explains that the algorithm is modified in the case of twins. Ariosa says that apart from the unsupported and entirely speculative assertions of Professors Lovett and Fisk in the joint expert report, there is no other evidence which even suggests that the Polymorphic Assay could be used to detect a twin pregnancy. Ms Norbury’s evidence was that “it would just be impossible to analyse the data” from the Harmony Test, unless it was known in advance that the pregnancy was a twin pregnancy. Indeed, Ms Norbury stated that if the Harmony Test was run on an unknown twin pregnancy:
…all I think you might see if – with the statistical power is that the agreement between all the markers was not as tight as you might normally see but to start thinking, well, we’ve got 10 per cent from twin A and 10 per cent from twin B and then we’re going to have this perfect theoretical adjustment of ratios, I think, is highly unlikely because we – we’re not going to have two twins that are exactly the same because there’s always some variation between twins in terms of their position and often one is the, sort of, dominant twin to the other one in terms of size.
815 Generally, I would reject Ariosa’s lack of utility arguments.
816 First, the Patent does not promise or convey a clear assertion that every detection result provides or can be used to provide a “prenatal diagnosis”. Rather, it states that the invention provides a method of prenatal diagnosis. And this is how the alleged promise would be understood by the person skilled in the art in the context of the Patent as a whole.
817 Further, Professor Fisk noted that the Patent is “particularly directed to a method of prenatal diagnosis by detecting cell-free fetal nucleic acids in the non-cellular component of a maternal blood sample”. And he explained that:
…Reading the Priority Document and the Patent as a whole, it is clear that both describe the same invention, namely, a method comprising the detection of fetal nucleic acids (e.g. DNA or RNA) in serum or plasma from a maternal blood sample which can be used for non-invasive “prenatal diagnosis” (as that term is defined in the Priority Document and the Patent).
818 Professor Lovett explained that:
It is clear to me from the above disclosures (and a reading of the Priority Document as a whole) that the invention described in the Patent is a method comprising the detection of foetal cell free DNA in serum or plasma prepared from blood extracted from a pregnant female.
819 Further, and as Sequenom points out, the Patent explains that the invention “relates” to non-invasive detection methods and particularly prenatal diagnosis by detecting fetal nucleic acids in serum or plasma from a maternal blood sample (page 1 lines 3 to 6). It explains that the “invention provides a detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of foetal origin in the sample” (page 2 lines 20 to 23). It explains that the “method according to the invention can be applied to the detection of any paternally-inherited sequences which are not possessed by the mother and which may be for example genes which confer a disease phenotype in the foetus” (emphasis added) (page 4 lines 5 to 8). It explains that the detection of non-Y sequences “can also be used to ascertain the presence of foetal nucleic acid in a particular maternal plasma or serum sample, prior to diagnostic analysis such as sex determination” (page 4 lines 27 to 30). It says in relation to Example 5 that the “high concentration of foetal DNA in maternal plasma and serum has allowed us to reliably detect the presence of foetal genetic material” (page 30 lines 18 to 20). It also says in this context that the named inventors “envisage[d] that foetal DNA analysis in maternal plasma and serum would be most useful in situations where the determination of foetal-derived paternally-inherited polymorphisms/mutations or genes would be helpful in clinical prenatal diagnosis (Lo et al. 1994)” (page 31 line 27 to page 32 line 1).
820 Further and in any event, the phrase “prenatal diagnosis” is broadly and inclusively defined (page 2) as covering the determination of any maternal or fetal condition or characteristic which is related to either the fetal DNA itself or to the quantity or quality of the fetal DNA in the maternal serum or plasma, including the detection of simple mutations. And in the context of this definition, the Patent also says that “[t]he nucleic acid detected in the method according to the invention may be of a type other than DNA e.g. mRNA” (page 3).
821 Further, the purpose of the relevant detection method will dictate the sequence sought to be detected or quantified by the person skilled in the art. And the aim of any cffDNA detection method would be formulated prior to the conduct of the assay. For example, if the person skilled in the art wanted to determine a single gene disorder, the person skilled in the art would target a sequence associated with the disorder. Contrastingly, if the person skilled in the art wanted to confirm or quantify the presence of cffDNA in a sample, for example, as a control in a broader prenatal diagnosis method or to determine fetal fraction, the person skilled in the art could target one or more SNPs, regardless of the SNP’s association with a genetic disorder. Detection and the purpose for detection underpin the method claimed and meet the Patent’s identified promise.
822 Generally, in my view the Patent’s identified promise is fulfilled by each of the detection claims, which encompass methods for sex determination by the detection or quantification of sequences from the Y chromosome and methods for the determination of RhD status via the detection of sequences from a non-Y chromosome.
823 Second, and as Sequenom points out, even if the Patent did promise that every detection result provides or can be used to provide a prenatal diagnosis, such a promise is fulfilled to some extent by each of the detection claims.
824 The detection or quantification of a SNP can be used:
(a) to determine a fetal characteristic related to cffDNA, namely, the fetus’ genotype at the detected SNP;
(b) as a preliminary step to check for the presence of cffDNA as part of a method of determining the sex of a fetus;
(c) to determine a genetic disorder that is linked with a particular genetic disorder, where the SNP is already known to cause a pathogenic mutation or is an identified linked marker; and
(d) to determine the fetal fraction as part of a broader prenatal diagnosis method.
825 Indeed, an example of the potential utility of detecting polymorphisms not linked to a particular genetic disorder is provided in Pertl B et al, “Detection of male and female fetal DNA in maternal plasma by multiplex fluorescent polymerase chain reaction amplification of short tandem repeats” (2000) 106(1) Human Genetics 45-49. The purpose of that paper was to develop an assay for the detection of circulating fetal DNA in maternal plasma using nine different polymorphic STR loci. The authors explained that their approach may have implications for non-invasive prenatal diagnosis and that compared with other fetal DNA detection systems that use fetus-derived Y sequences to detect only male fetal DNA in maternal plasma, their proposed technique can be applied to both female and male fetuses (p 45). It was stated that using PCR amplification of nine different, highly polymorphic STRs, they could demonstrate that fetal DNA can be detected by the presence of paternally-inherited fetal-specific alleles in maternal plasma samples (p 46). It was concluded that “[t]he present report extends the clinical utility of maternal plasma DNA amplifications and makes a prospective analysis of risk pregnancy complications feasible independent of fetal gender” (p 48). Now this was after the priority date, but it is none the worse for that. This potential existed as at the priority date.
826 Ariosa has not discharged the onus of showing the requisite inutility.
SUFFICIENCY
827 Let me set out some legal principles.
828 At the relevant time, s 40(2)(a) of the Act provided:
(2) A complete specification must:
(a) describe the invention fully, including the best method known to the applicant of performing the invention; and …
829 Generally speaking, only one embodiment within each claim need be enabled for sufficiency purposes. An invention is sufficiently disclosed if a skilled person could make a single embodiment of the invention which falls within the scope of the claims. The following test for sufficiency was formulated in Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [25]:
The question is, will the disclosure enable the addressee of the specification to produce something within each claim without new inventions or additions or prolonged study of matters presenting initial difficulty?
(Citation omitted.)
830 In Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (2004) 217 CLR 274 (Lockwood (No 1)), the High Court said at [60]:
For the purposes of s 40(2)(a), it is not necessary for the inventor to disclose all the alternative means; it is enough that there is disclosure in the sense of enabling the addressee of the specification to produce something within each claim without new inventions or additions or prolonged study of matters presenting additional difficulty.
(Citation omitted.)
831 In SNF (Australia) Pty Ltd v Ciba Speciality Chemicals Water Treatments Ltd (2011) 92 IPR 46 at [234], Kenny J explained:
A specification is not insufficient merely because some experiment of a routine character (as distinct from prolonged study of matters presenting initial difficulty) is necessary in the particular case … Nor is a specification insufficient because it fails to give detailed instructions as to matters which a “practical person … would naturally settle, and expect to have to settle … himself”, provided he “would find no difficulty in so doing”.
(Citations omitted).
832 Further, the plurality in AstraZeneca AB v Apotex Pty Ltd (2014) 226 FCR 324 observed (at [205]):
… although the complete specification must describe the invention fully, including the best method known to the inventor of performing the invention, it does not follow that the inventor must explain how he or she arrived at the invention. It is the invention itself that must be fully described, not the route that was travelled by the inventor to arrive at it. Again, the way in which the inventor came to the invention described and claimed may also have an evidentiary significance. However, an invention may be the result of chance or luck as much as long experiment and the question whether an invention, or an alleged invention, involves an inventive step is an objective one.
(Citation omitted.)
833 Further, where a claim in effect claims alternative methods, the specification must describe how to perform the invention for each such method, not just one method within the claim. A patentee should not be permitted to avoid a finding of lack of sufficiency by drafting a claim to cover two or more alternative methods and then only providing a sufficient description as to one. Generally speaking I accept what was said in Tramanco Pty Ltd v BPW Transpec Pty Ltd (2014) 105 IPR 18 at [206] to [208] by Nicholas J (referred to by me in GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Apotex Pty Ltd (2016) 119 IPR 1 at [693] (GSK v Apotex)) at [207]:
In Kimberly-Clark and Doric the High Court was concerned with various product claims each of which consisted of a number of features all of which were essential (in the sense of non-optional) integers of the claim. However, there are some types of claim that may need to be approached slightly differently including, in particular, claims to methods for producing one or more specified results. For example, a claim for a method of producing one or more of outcomes A, B or C might be infringed if the alleged infringer uses the method to produce outcome A, but not outcome B or C. Whether there is infringement in such a case will depend upon (inter alia) the proper construction of the claim and, in particular, whether it requires the use of the method to produce only one or more of outcomes A, B or C, as opposed to all three of them. Assuming the former construction (in the present case it appears to have been assumed by both parties at the trial and on appeal that claim 1 is to be construed in this way), it would seem to me to be wrong in principle to hold that the description of the invention is sufficient if the specification enables the use of the method to achieve outcome A, but not outcomes B or C. It would be inconsistent with the purposes of the Act to confer a monopoly on a patentee for a method of producing any of outcomes A, B or C, if the patentee’s disclosure only enabled the use of the method to produce some of those outcomes.
834 Further, in Apotex Pty Ltd v Warner-Lambert Company LLC (No 2) (2016) 122 IPR 17 at [244] (Apotex (No 2)), Nicholas J reinforced his observation that the Kimberly-Clark test for sufficiency may not be able to be applied to all forms of claim and said (at [245] to [247]):
Tramanco was not concerned with methods of medical treatment, but with a method for determining one or more parameters relating to the performance of the suspension system of a vehicle. In the present case the method claims, as I have construed them, are to methods of treating pain, or different types of pain, in all kinds of mammals including, in particular, humans.
As we are reminded from time to time, judgments are not to be construed as if they were statutes, and it is always necessary to read and understand them in context. Application of the test of insufficiency propounded by the High Court in Kimberly-Clark based upon a test originally formulated outside the context of methods of treatment claims cannot be sensibly applied to a case such as this. In my respectful opinion, it would be a nonsense to say that the invention was sufficiently described if it enabled the skilled addressee to perform the method of the invention on rats (or other mammals) but did not enable it to be performed on humans.
In my view, s 40(2)(a) requires that the Patent in this case enable the skilled addressee to perform the claimed invention in relation to humans without new inventions or additions or prolonged study of matters presenting initial difficulty. It is only by requiring this degree of enablement that the patentee could be sensibly understood to have given consideration for the grant of a patent which the skilled addressee would understand to be essentially directed to the treatment of pain in humans.
835 Now it is important to note that the claim under consideration in Tramanco at [207] was in form and in substance a claim to a method for producing alternative results or outcomes. And it is apparent that his Honour regarded the claim in Apotex (No 2) in similar terms.
836 Now Ariosa says that it is relevant to consider the context in which the High Court in Kimberly-Clark made the statement at [25]. I agree. A principal issue before the High Court in Kimberly-Clark was the Full Court’s identification of the scope of the specification. It found that the Full Court had erred by excluding the claims from their consideration of what was described by the specification. A further error was that the Full Court did not approach the issue of full description by considering the knowledge of a skilled addressee. Ariosa says that the test in [25] was stated by the High Court in the context of this second issue, namely, whether the addressee of the specification, being a person skilled in the art, would be capable of producing something within each claim at the priority date of the claim or at least by the date of filing of the patent. The Full Court’s error had involved approaching that issue by reference to someone who knew nothing of the prior art.
837 Ariosa says that the High Court’s focus in this widely quoted test was therefore on the practical requirement of s 40(2)(a), enabling the production of something within the claim without new inventions, additions, or prolonged study, and the skills of the relevant person in respect of whom that practical requirement is to be assessed.
838 Ariosa says that in the context of addressing the first error, as to the scope of the specification, the High Court considered what amounted to “describing”, as distinct from “defining”, the invention as used in s 40(2)(b). In doing so the High Court considered the range of meanings that the word “invention” can have, and explained that the relevant meaning of “invention” for determining sufficiency is “the embodiment which is described, and around which the claims are drawn” (Kimberly-Clark at [21]). The High Court then said that this understanding of the invention was “important when assessing, for the purposes of s 40(2), the reaction of the relevant skilled addressee” (Kimberly-Clark at [21])).
839 Ariosa submits that it can be seen from the above discussion that there is a degree of tension between the expressed concept of a singular ‘embodiment’, around which the claims are drawn, and the recognition by the High Court in the test quoted above that the full description requirement is to be met in respect of each claim.
840 Ariosa says, by reference to the Full Court’s decision in Tramanco, that:
(a) a specification may relevantly disclose more than one ‘invention’ for the purposes of s 40(2)(a), involving more than one relevant ‘embodiment’;
(b) as a matter of substance, rather than merely form, a claim may be to more than one ‘invention’, each such invention relating to a different embodiment described in the specification; and
(c) where that occurs, the full description requirement in s 40(2)(a) must be satisfied for each ‘invention’, properly understood.
841 Ariosa says that it also follows that if a claim to an embodiment comprising a relevant ‘invention’ were not fully described and hence not a valid claim, then the subject matter of that claim cannot be transformed into patentable subject matter by merging it into a claim covering an additional separate embodiment, being an embodiment that gives rise to a separate ‘invention’ for which there is a full description.
842 Ariosa points out that in Tramanco, the Full Court considered the issue of sufficiency of claims to a method of measuring and logging certain performance characteristics of a vehicle’s suspension system.
843 Claim 1 provided as follows (Tramanco at [3]):
A method for logging the performance of a vehicle suspension system including the steps of measuring the dynamic effect of an impulsive load with an electronic weighing system, wherein the electronic weighing system is mounted onboard the vehicle, and determining one or more parameters selected from the group consisting of the dampening ratio of the suspension, the oscillation frequency of the suspension and the impact loading of the vehicle.
844 Of those three parameters, the issue was with the determination of the ‘impact loading of the vehicle’. The trial judge construed that integer as relating to the impact loading of the vehicle on the road, which Tramanco (the patentee) conceded was not enabled.
845 Ariosa says, however, that the Full Court found that the correct construction was that the claim integer referred to the impact loading on the suspension of the vehicle. The sufficiency attack failed on that construction because the Full Court found that the evidence did not demonstrate that the skilled person could not ‘determine’ that integer.
846 Ariosa says that, importantly, Nicholas J said that the patentee’s further concession that claim 1 was not fully described if the patentee was wrong about the construction of claim 1 was a concession ‘properly made’ (Tramanco at [208]). That is, there was a concession that claim 1 would lack sufficiency if only two of the three alternative parameters were enabled.
847 Nicholas J explained his reasoning (at [207]):
In Kimberly-Clark and Doric the High Court was concerned with various product claims each of which consisted of a number of features all of which were essential (in the sense of non-optional) integers of the claim. However, there are some types of claim that may need to be approached slightly differently including, in particular, claims to methods for producing one or more specified results. For example, a claim for a method of producing one or more of outcomes A, B or C might be infringed if the alleged infringer uses the method to produce outcome A, but not outcome B or C. Whether there is infringement in such a case will depend upon (inter alia) the proper construction of the claim and, in particular, whether it requires the use of the method to produce only one or more of outcomes A, B or C, as opposed to all three of them. Assuming the former construction (in the present case it appears to have been assumed by both parties at the trial and on appeal that claim 1 is to be construed in this way), it would seem to me to be wrong in principle to hold that the description of the invention is sufficient if the specification enables the use of the method to achieve outcome A, but not outcomes B or C. It would be inconsistent with the purposes of the Act to confer a monopoly on a patentee for a method of producing any of outcomes A, B or C, if the patentee’s disclosure only enabled the use of the method to produce some of those outcomes.
848 Further, Ariosa says that in Apotex (No 2) the patentee claimed a method for treating pain comprising administering a therapeutically effective amount of a specified group of compounds “to a mammal in need of said treatment” (Apotex (No 2) at [100]).
849 The Tramanco issue arose because the specification provided details for determining an appropriate dose to administer to a rat, but not for other mammals, including humans.
850 The patentee’s primary submission was that enabling the treatment of rats satisfied the ‘test’ in Kimberly-Clark. Nicholas J rejected that submission, saying that the patent was also, indeed primarily, directed to the treatment of humans (Apotex (No 2) at [241] to [243]).
851 His Honour then referred to the statement of principle from Tramanco referred to above, and said that for the reasons given there “it may not be possible to apply the Kimberly-Clark test of sufficiency to every manner of claim” (Apotex (No 2) at [244]). Nicholas J considered that it was necessary to consider whether there was enablement of a method of treating humans, and concluded that the evidence showed that the claims were so enabled.
852 The Full Court rejected Apotex’s appeal against that finding (Warner-Lambert Co LLC v Apotex Pty Ltd (No 2) (2018) 355 ALR 44 at [135] (Warner-Lambert)).
853 Now I would note that in MLA (No 1), I considered sufficiency, including the Tramanco issue. I distinguished the reasoning in Tramanco on the basis that the method of claim 1 of the patent in suit was a “claim to a general method that may be employed using any combination of features within the scope of the claim”, whereas the claim in Tramanco was “in substance a claim to a method for producing alternative results or outcomes”, as was the claim in Apotex (No 2) (MLA (No 1) at [906]). No doubt was cast upon the reasoning of the Full Court in Tramanco, but I recognised the need to identify, as a matter of substance, whether the claim was directed to a single method of general scope, or whether it was directed to more than one such method. If the latter, then each method would need to be enabled.
854 And as I stated, in my view the peculiar claims at issue in Tramanco were to a method for logging the performance of a vehicle suspension and determining one or more parameters selected from the group consisting of A, B, and C (i.e. the claim included methods for determining only one parameter out of A, B, C, combinations of two parameters A+B, A+C, B+C and all three parameters A+B+C).
855 But leaving aside special classes of claims to methods producing one or more specified optional outcomes, it is well settled that only one embodiment within each claim need be enabled for sufficiency purposes. An invention is sufficiently disclosed if a skilled person could make a single embodiment of the invention which falls within the scope of the claims (see also GSK v Apotex at [688] to [690] and MLA (No 1) at [885]).
856 And the enabled embodiment must be a common sense example of the invention in substance disclosed and claimed. For example, where a claim of a patent may theoretically encompass alleviating the pain of rats but a person skilled in the art would not for a moment understand the patent to be directed to the medical treatment of rats experiencing pain (but rather for the treatment of humans), the test in Kimberly-Clark will not be satisfied by simply demonstrating that the invention could be used to treat rats as distinct from humans (Apotex (No 2) at [241] to [247]).
857 Let me make some other observations.
858 There is no requirement for the patentee to provide an explanation of how the invention works.
859 Further, in Gilead Sciences Pty Ltd v Idenix Pharmaceuticals LLC (2016) 117 IPR 252 Jagot J provided a useful summary of the principles relevant to a sufficiency analysis, and in doing so accepted that in this context, the person skilled in the art (at [438]):
(a) is a person with reasonable skill and knowledge in the art, possessed of common sense and the common general knowledge pertaining to the art, who reads the specification to understand how to carry the invention into effect;
(b) is not constrained by funding or the time they can address to a problem provided that the time and funding is not a result of the need for new inventions or additions or prolonged study of matters presenting initial difficulty;
(c) may have a capacity for original research;
(d) may be well-versed in the literature and be able to carry out searches and read specifications as they become available, noting that the literature, specifications and the searches must form part of the common general knowledge in order to be available to the skilled addressee;
(e) has the advantage over the addressee for obviousness of having the invention in view in that they are trying to carry out the invention and achieve success, not searching for a solution in ignorance of it; and
(f) is taken to be honestly and intelligently trying to make the invention work.
860 In order for a challenge to the validity of a patent based upon alleged insufficiency of description to be successful, it must be shown that the person skilled in the art would be required to undertake work requiring new inventions or additions or prolonged study of matters presenting initial difficulty. In other words, it must be demonstrated that the work required to be undertaken by the skilled addressee is something other than routine. But steps can be “difficult yet routine” (Gilead at [438]).
861 On appeal, in Idenix Pharmaceuticals LLC v Gilead Sciences Pty Ltd (2017) 134 IPR 1 Nicholas, Beach and Burley JJ accepted (at [230]) that to perform a method within a claim a person skilled in the art might need to apply their will and their resources, and the work might be complex, time consuming and expensive. But this did not necessarily entail that the steps required were not routine for the notional skilled person or team.
862 Further, s 40(2)(a) does not require that the description in the Patent is sufficient to enable widespread commercial or clinical application of the invention.
863 As was held by the Full Court in Warner-Lambert (at [131]):
… s 40(2)(a) is concerned with matters of description, not matters of proof… it is not to the point that, in clinical practice, a clinician would want proof of safety and efficacy beyond the description of the invention in the specification itself. This is not the province or function of the requirement in s 40(2)(a).
864 The decision in Warner-Lambert concerned a method of treating pain, or different types of pain. The respondent asserted that the specification did not describe any safe and effective doses, and dosage regimes, which the person skilled in the art could utilise for the purpose of treating a human patient experiencing pain. That is, before the method could be carried out, it would have been necessary to undertake further research and study to obtain an understanding of the safety and toxicity profile of the drug. The Full Court approved (at [126], [128] and [130]) the following observations of Nicholas J in Apotex (No 2). First, the work undertaken to prove the safety and efficacy of a compound for regulatory and marketing approval purposes is not necessarily relevant or material to the determination of whether the requirements of s 40(2)(a) have been met in a given case. Second, the issue is not whether the patentee could have supplied the person skilled in the art with more information than it did, but whether it has supplied enough information. Third, there is a difference between whether a person skilled in the art would choose to perform the invention and whether the person skilled in the art could perform the invention based on the description in the specification. And in this context the Full Court recognised the distinction between what a person skilled in the art would do on the basis of the description in the specification, and what a person skilled in the art could do. The Full Court observed that enablement under s 40(2)(a) is concerned with the “could” not the “would”.
865 Finally, at this point I should also note one other matter. Ariosa seeks to rely on post priority date evidence, such as scientific research, to establish a lack of sufficiency. I agree that post priority date evidence may have some relevance.
866 In Gilead, Jagot J looked at the issue of sufficiency which required consideration of whether the Idenix patent enabled the skilled person to make or synthesise any compound falling within claim 7 and dependent claims. In this regard, Idenix’s own attempts to make a compound within claim 7 of the Idenix patent, including because of the level of expertise possessed by the Idenix team, was considered to be relevant. Her Honour held (at [506] and [509]):
I consider that it is apparent that Idenix attempted to synthesise a compound within claim 7 as a priority from July 2002. According to Idenix’s own documents it failed to do so between that time and 2005. As discussed, I do not accept the notion that the Idenix team, assisted by persons such as Dr Coe, Professor Fleet and Dr Barker, did not possess all of the common general knowledge relevant to the synthesis. From (and no doubt before) 27 June 2003 [the date that the patent was filed] they possessed all the information in the Idenix patent. With the benefit of the Idenix patent and the common general knowledge (and, in my view, knowledge that exceeded the common general knowledge given the involvement of Dr Storer, Dr Coe and Professor Fleet, as well as the attendance by Idenix chemists at a specialist fluorination conference), Idenix’s documents record consistent failures in all attempts to make such a compound.
…
Also as noted, if it is suggested that the work is irrelevant because the Idenix team probably possessed greater expertise than the notional skilled addressee (that is, the common general knowledge and additional knowledge), I consider that suggestion would be inconsistent with the approach in Lockwood No 2 at [119]. That is to say, if a skilled team which possesses all of the common general knowledge and additional knowledge (even inventiveness) cannot synthesise a compound within claim 7 then the notional skilled addressee possessing only the common general knowledge may be inferred even more readily not to have been able to do so.
(a) The sufficiency attack and some science
867 Ariosa said that the “invention” as claimed in each of the relevant claims other than claim 13 is not fully described.
868 First, it is said that the relevant claims include within their scope methods directed to or that rely on: (a) detecting the presence of paternally-inherited sequences in maternal serum or plasma which are not possessed by the mother; and (b) quantitating the level of fetal nucleic acid in maternal serum or plasma either as a total level, to be correlatively assessed, or by quantitating markers on two different chromosomes for comparison.
869 Second, it is said that the specification does not enable the person skilled in the art to perform the invention insofar as it relies on quantitating the level of fetal nucleic acid in maternal serum or plasma.
870 Third, it is said that the specification of the Patent does not disclose how the aneuploidy detection methods contemplated in paragraphs (a) and (b) on page 5, lines 5 to 26 of the Patent could be carried out, reliably or at all, for female fetuses using the claimed method of detection of nucleic acid of fetal origin.
871 In essence, Ariosa asserts that the “invention” described in the Patent involves two distinct embodiments.
872 A first embodiment involves the performance of a diagnosis by reference to the quality of the fetal DNA, specifically whether the maternal serum or plasma includes any targeted sequences not possessed by the mother.
873 A second embodiment involves the performance of a diagnosis by reference to the quantity of the fetal DNA, either as to the total levels of cffDNA, or the relative quantity of fetal markers on specific chromosomes.
874 Further, Ariosa seeks to divide the asserted second embodiment into:
(a) one directed to a correlation between total fetal DNA levels and some other condition; and
(b) another directed to measuring the level of fetal DNA markers on different chromosomes.
875 Ariosa’s principal submission is to then assert that each of the above three embodiments represents a different invention, each of which needs to be fully described to satisfy s 40(2)(a). And in this respect Ariosa says that the quantitative embodiments are not enabled by the disclosure in the Patent.
876 Contrastingly, Ariosa does not suggest that a person skilled in the art could not perform the qualitative methods of, for example, detecting cffDNA from a Y or non-Y chromosome (claims 1 to 13) or diagnosing fetal sex or RhD status (claims 22, 23, 25 and 26) without new inventions or additions or prolonged study of matters presenting initial difficulty.
877 I would say now that in my view, Ariosa’s approach is an attempt to circumvent the authorities of Kimberly-Clark and Lockwood (No 1) and to impose a requirement that the claims be enabled across their full breadth. Now although such a standard may apply to patents the subject of the amendments introduced by item 8 of Schedule 1 of the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth), it does not apply to the Patent.
878 Moreover, a similar attack to the one now made by Ariosa was raised and rejected by me in both GSK v Apotex and MLA (No 1).
879 In GSK v Apotex, I relevantly explained at [691] to [695]:
The submission of the respondents that the statement of principle in Kimberly-Clark should be understood as being limited to claims to a “single product with a particular feature” is in my view contrary to Kimberly-Clark and Lockwood (No 1). Those cases contain no recognition of such a limitation. The enablement of a single embodiment within each claim is sufficient for the purposes of s 40(2)(a).
Apotex has contended that Kimberly-Clark and Lockwood (No 1) each only concerned a simple product. It accepted that in such cases it may be sufficient for the patent to contain “such instructions as will enable all those to whom the specification is addressed to produce something within each claim”. However, it is said that the High Court did not consider the present question, where the claims encompass multiple formulations of the pharmaceutical composition, including as to the amount of paracetamol in each layer, the type and amount of matrix-forming polymer and as to the in vitro dissolution profile of the formulation when tested in accordance with the claimed dissolution apparatus at certain time intervals. It is said that each of these features is claimed across a broad range, so that the claims encompass multiple formulations.
In this context, Apotex has made reference to Tramanco Pty Ltd (ACN 010 101 872) v BPW Transpec Pty Ltd (ACN 006 645 272) (2014) 105 IPR 18; [2014] FCAFC 23 (Tramanco). But the claims considered in Tramanco are different. The claims in Tramanco were to a method for logging the performance of a vehicle suspension and determining one or more parameters selected from the group consisting of A, B and C. The claim included methods for determining only one parameter out of A, B, C, combinations of two parameters A+B, A+C, B+C and all three parameters A+B+C. Contrastingly, the claims of the Patent are to ranges in a dissolution profile. They are not options of different parameters. The observations of Nicholas J at [207] as to the need to enable each of the options of parameters included within the claims in Tramanco are inapplicable. Claim 1 of the Patent relevantly refers to a dissolution profile, albeit that it is expressed in terms of ranges over the three time intervals.
The Patent provides sufficient information to enable a skilled formulator to produce a formulation with a profile which satisfied regulatory acceptance targets for replicating the profile of Formulation C (and Formulation D) without new inventions or additions or prolonged study of matters presenting additional difficulty.
Professors Davies and Tucker each accepted that the Patent provided enough information to allow them to make a Formulation C (and D) having the percentage (approximately) of paracetamol released at 15, 60, 120 and 180 minutes as Formulation C (and D) as specified in table 2 (and 3) of the Patent. Formulations C and D each fall within claims 1, 2 and 3 of the Patent. Accordingly, for that reason alone, the Patent satisfies the test for sufficiency.
880 In MLA (No 1) I also held (at [890], [893] and [906] respectively):
Further, MLA contends that where (as here) a claim in effect claims alternative methods, the specification must describe how to perform the invention for each such method, not just one method within the claim. It contends that a patentee should not be permitted to avoid a finding of lack of sufficiency by drafting a claim to cover two or more alternative methods and then only providing a sufficient description as to one. In support of this contention it cites Tramanco Pty Ltd v BPW Transpec Pty Ltd (2014) 105 IPR 18; [2014] FCAFC 23 (Tramanco) at [206] to [208], per Nicholas J (referred to by me in GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Apotex Pty Ltd (2016) 119 IPR 1; [2016] FCA 608 at [693])…
MLA says that in the present case, claim 1 specifies one method that requires one of the at least three SNPs to be a specified SNP, and another method that requires one of the at least three SNPs to be within 500,000 nucleotides of a specified SNP. Accordingly, MLA says that even if limb (a) is sufficiently described, which in any event it does not accept, that cannot provide an answer to limb (b) of claim 1. It says that its contention should apply, by parity of reasoning, to different traits. That is, the claims describe a different method for each trait to be identified or inferred. Therefore each method must be sufficiently described for each such trait.
… Now as a preliminary matter, I would observe that the observations in Tramanco at [207] and Apotex (No 2) at [244] have little application to the present case. The claim under consideration in Tramanco at [207] was in form and in substance a claim to a method for producing alternative results or outcomes. It is apparent that his Honour regarded the claim in Apotex (No 2) in similar terms. But claim 1 of the 253 Application is not a claim to such a method. It is not a claim to a method for producing alternative results or outcomes. Rather it is a claim to a general method that may be employed using any combination of features within the scope of the claim.
(Emphasis added.)
881 Let me now elaborate in more detail on Ariosa’s arguments, which are not without some merit at least on the science in relation to using quantitative methods.
The asserted embodiments said to be in substance described
882 Ariosa contends that determining as a matter of substance what invention (or inventions) are described in the Patent involves ascertaining what the Patent describes as a relevant method or methods of applying the discovery by Professor Lo that cffDNA exists in the blood of a pregnant woman, rather than merely the discovery per se.
883 Ariosa says that taking an approach most favourable to the patentee in this regard, the person skilled in the art could first determine what is the broadest categorisation of a method (or methods) that is described in the Patent which involves the application of Professor Lo’s discovery.
884 The second sentence of the Patent says that the invention in particular relates to “prenatal diagnosis by detecting foetal nucleic acids in serum or plasma from a maternal blood sample”. And on page 2 (lines 24 to 27) it says that the term “prenatal diagnosis” as used in the Patent covers the determination of any maternal or fetal condition or characteristic which is related to the quantity of the fetal DNA, or the quality of the fetal DNA.
885 Ariosa says that it should first be noted that there is a qualitative aspect that is shared by each of these quantitative and qualitative analyses in that the broader ‘qualitative’ aspect is identifying that the cell-free DNA is fetal, rather than maternal.
886 Ariosa says that the application of the discovery insofar as it involves assessing the specifically qualitative aspect of the fetal DNA involves the “detection of any paternally-inherited sequences which are not possessed by the mother and which may be for example genes which confer a disease phenotype in the foetus” (page 4, lines 6 to 7). The Patent gives a description of how that qualitative application might be assessed from page 3, line 29 to page 5, line 2. Those examples include:
(a) determining the sex of the fetus;
(b) determining the rhesus D status of the fetus in a rhesus D negative mother;
(c) determining whether the fetus carries a mutation not possessed by the mother that is known to cause a haemoglobinopathy; and
(d) determining whether the fetus carries some other paternally-inherited DNA polymorphism or mutation that is linked to a particular disease.
887 Ariosa then says that the application of the discovery insofar as it involves a quantitative assessment of the fetal DNA is described in the Patent from page 5, line 3 to page 6, line 2. The Patent asserts that the total amount of cffDNA circulating in maternal plasma is higher when:
(a) the fetus has a chromosomal aneuploidy; or
(b) the mother has a placental pathology, such as pre-eclampsia.
888 Further, the Patent asserts that a chromosomal aneuploidy can be diagnosed by measuring the quantity of fetal DNA derived from a relevant chromosome such as chromosome 21, and determining whether the quantity of that fetal DNA is greater than the quantity of fetal DNA from a reference chromosome (page 5, paragraph (b)).
889 Ariosa says that it follows from these passages that the broadest conception of a relevant invention described in the specification of the Patent involves two distinct embodiments. A first embodiment applies Professor Lo’s discovery by performing a diagnosis by reference to the quality of the fetal DNA, specifically where the maternal serum or plasma includes targeted sequences not possessed by the mother. A second embodiment applies Professor Lo’s discovery by performing a diagnosis by reference to the quantity of the fetal DNA, either as to the total levels of cffDNA, or the relative quantity of fetal markers on specific chromosomes.
890 And as to the second embodiment, and as I have previously indicated, Ariosa says that it can be seen that the two approaches encompassed by that ‘embodiment’ are conceptually quite distinct. On the one hand, there is a method directed to an asserted correlation between total fetal DNA levels and a condition. On the other hand, there is a method directed to measuring the level of “fetal DNA markers on different chromosomes” i.e. measuring the level of fetal markers on one chromosome, and comparing it with the level of fetal DNA markers on a reference chromosome.
891 And again, Ariosa submits that each of these approaches is a different embodiment, representing a different invention resulting from the application of Professor Lo’s discovery.
892 Moreover, Ariosa says that whilst these two quantitative approaches may be collectively grouped together as involving ‘prenatal diagnosis’ based on quantification, that level of abstraction is too high.
893 As I say, I have a problem with Ariosa’s division. But I will return to this later. Let me delve further into the detail and some of the science.
Whether the correlative quantitation embodiment (paragraph (a) on page 5 of the Patent) is fully described
894 Ariosa points out that paragraph (a) on page 5 of the Patent asserts that the total level of cffDNA is higher in pregnancies where the fetus has a chromosomal aneuploidy. The assertion is also made at page 5, lines 27 to 29 that the level of cffDNA is also higher in pregnant women suffering from pre-eclampsia.
895 Contrastingly, Ariosa says that GB patent application no 9704444 entitled “Non-Invasive Prenatal Diagnosis” filed on 4 March 1997 (the priority document) was more circumspect than the specification of the Patent. In particular:
(a) Page 5, lines 12 to 15 of the Patent states that “it has also been demonstrated that the level of foetal DNA in maternal plasma and serum is higher in pregnancies where the foetus has a chromosomal aneuploidy than in normal pregnancies”. This can be contrasted with the corresponding statement in the priority document, which stated “it may be expected that the level of foetal DNA…”.
(b) Page 5, lines 16 to 18 of the Patent states: “Quantitative detection of foetal nucleic acid in the maternal plasma or serum e.g. a quantitative PCR assay, can be used to screen pregnant women for chromosomal aneuploidies”. Again, and in contrast, in the priority document the words instead read “could be used to screen…”.
(c) Page 5, line 27 to page 6, line 2 of the Patent states “Another application of the accurate quantitation of foetal nucleic acid levels in the maternal serum or plasma is in the molecular monitoring of certain placental pathologies, such as pre-eclampsia. The concentration of foetal DNA in maternal serum and plasma is elevated in pre-eclampsia. This is probably due to the placental damage which occurs”. In contrast, the corresponding paragraph in the priority document reads “Another potential application of the accurate quantitation… It is likely that placental damage in pre-eclampsia may result in alterations in foetal DNA concentration in material [sic] serum and plasma”.
(d) Examples 2 to 5 were not included in the priority document. They were introduced at the time the PCT application no PCT/GB98/00690 was filed on 4 March 1998.
896 Let me expand on the following submissions of Ariosa concerning the question of elevated cffDNA in aneuploidy pregnancies.
897 Example 2 of the Patent (beginning at page 9, line 15) relates to the quantitative analysis of cffDNA in maternal serum in aneuploid pregnancies. The results are presented in Figure 1, and based on these results, the Patent concludes at page 14, lines 18 to 19 that “[i]n this study we demonstrate that the concentration of foetal DNA in maternal serum is elevated in aneuploid pregnancies”.
898 But according to Ariosa’s experts there are significant difficulties with the results presented for Example 2, and they do not demonstrate what the Patent says they do.
899 The experiment described in Example 2 involves a very small sample size. The precise numbers are not provided in the Patent, but Professor Hyett estimated a total of approximately 16 samples, comprising a “relatively small” number of controls (8) being compared to a “relatively small” number of aneuploidy cases. In Professor Hyett’s experience, in order to validate the outcome purportedly demonstrated in this example, it would be necessary to replicate the experiment with at least 30 aneuploid samples, which, in the case of Down syndrome, would require screening of approximately 15,000 women.
900 In addition to the small sample size, there are other issues with the design of the study and the lack of information provided in the Patent. The Patent does not specify the gestational age at which the samples reported in Example 2 were taken. It merely says that the samples were collected “prior to any invasive procedure” and that the fetal karyotype was confirmed by amniocentesis or CVS (page 10, lines 7 to 10). This suggested that the gestational age of the subjects in Example 2 could have been anywhere from 11 to 24 weeks.
901 Ariosa says that this wide range is particularly significant when considered in light of Example 5 and Figure 4 of the Patent. Figure 4 shows a high degree of variability in the levels of cffDNA both between individual women, and at different stages of gestation, including in the period between 11 and 24 weeks. This is made clear, for example, by the variable scale on the Y-axis. This variability had the potential to skew the results.
902 According to Ariosa, because of this variability, it would be important that the data from such a study was tightly controlled and matched for gestation as between the control and aneuploid groups. Example 2 does not refer to the samples being matched for gestation in contrast to Example 4. Professor Fisk accepted that it would be optimal in such an experiment to control for gestational age.
903 Ariosa also notes that all of the aneuploid pregnancies in Figure 1 fall within the same concentration range as the control group in Figure 2. Now Professor Lovett attempted to resist this proposition, on the basis of his assumption that the samples in Example 4 and Figure 2 were of later gestational age than those in Example 2 and Figure 1. Professor Hyett’s evidence was that pre-eclampsia is a condition that is diagnosed after 20 weeks’ gestation, with 70% of cases being diagnosed after 36 or 37 weeks. Thus according to Ariosa, on the evidence there is nevertheless a potential overlap in gestational age between Figures 1 and 2, casting considerable doubt over the results.
904 Ariosa notes that the results in Figure 1 show that the mean concentration for the aneuploid group is higher than the mean concentration of the control group, and that a p-value is reported in the Patent that would be considered statistically significant. However, Ariosa says that insufficient data is provided to demonstrate a correlation between elevated cffDNA and aneuploidy and to support the asserted p-value, for the following reasons.
905 There is significant overlap between the control and aneuploidy groups as shown in Figure 1. As Professor Hyett explained:
PROF HYETT: So in the context of providing information about the efficacy of a screening test, one would normally provide detail about the sensitivity and specificity. And ideally, if that were then done in a broader population, you would have knowledge of the prevalence of disease in the population so you could then also infer what the positive and negative predictive values would be. The data that – presented in figure 1 show that the comparator here is between a relatively small number of controls and a relatively small number of cases for Down syndrome. If you look at the figure which is on, I think, page - - -
HIS HONOUR: It’s 1.044.
PROF HYETT: Yes, you can see that there’s actually significant overlap between the two .....
HIS HONOUR: Yes.
PROF HYETT: You can – I think there are eight cases in each group if you tease the circles out. Some of them are double circles in the aneuploidy group.
HIS HONOUR: Yes, they look a bit thicker .....
PROF HYETT: if you were to concentrate on the Aneuploidy Common column you could imagine if you wanted to pick up all of those aneuploidy cases you would have to set a very low threshold in order to have 100 per cent sensitivity, and you can see that by doing that about 60 per cent of your controls are then going to be identified as being in the high-risk group, so that’s not a particularly effective screening paradigm. If you look at this the other way, you can see that if you wanted to have very, very high specificity, so, in other words, not identify any controls as being high-risk, then you would only pick up about 50 per cent of the aneuploidy pregnancies in that circumstance. Now, it’s, I think, very difficult to make those statements on the basis of this number of pregnancies. Typically, if you wanted to do a study that made some statement about sensitivity or specificity of a test, a ballpark figure would be that you would use 30 cases in that series.
In – within the experiment that’s described, there’s actually no data about what the mean and standard deviation of these two distributions are, although there is a statement that the – it was statistically significantly different. But from my perspective, the – the data are not presented in a way that actually identifies this as a screening tool. It identifies that there is a difference in level of cell-free fetal DNA in the aneuploidy population compared to controls.
906 Professor Oepkes was of a similar view, but also explained how in the case of existing screening tests which involved some but not too much overlap, the data would be used to calculate a likelihood ratio. However, in this case:
So if you have these two overlapping samples and distributions, then there could also be a likelihood ratio coming from this particular measurement to further improve your – the accuracy of your screening. And then the problem here is that the overlap is so significant that this likelihood ratio coming from this experiment would be very small and the additional value in a screening program where we already had some markers would be hardly significant, or at least very weak.
And then, on top of that – and I think that that is a major drawback of this idea – is that it was only applicable in male foetuses, making these two things together at that date for people in the field a totally uninteresting proposition to try and use this or explore it further.
907 In relation to the p-value derived using the Mann-Whitney U parametric test, Professor Fisk acknowledged that it was important not to give too much strength to the individual p-values. Professor Hyett’s evidence was that there is an absence of data provided in the Patent in relation to this value and there is no data in relation to the mean or the standard deviation, the latter being necessary to determine the breadth of the two population distributions. As Professor Hyett explained:
MR SHAVIN: Now, at the foot of the page, Professor, you accepted that the results in figure 1 show that the mean concentration is elevated in aneuploid pregnancies in the study. And this is reported to be statistically significant. And you accept that, don’t you?
PROF HYETT: That statement is made. Yes.
MR SHAVIN: Yes. And you accept that statement is true, that is, that there is a statement that shows statistical significance of the patent.
PROF HYETT: Yes. When I look at figure 1 and I look at the Mann-Whitney value that’s quote[d], I’m quite surprised that it is that significant based on that data. Now, that’s a personal observation. I don’t have the absolute values of each of those data points available to me. I don’t have the means available to me. I don’t have the standard deviations available to me. But when I look at that data, I’m surprised that the P value is as listed.
908 Professor Fisk attempted to overcome this criticism on the basis that the Mann-Whitney test was used for non-normally distributed data, and that means and standard deviations were relevant when dealing with normally distributed data on a bell curve. But Professor Hyett explained that for the purposes of assessing value as a screening test, the p-value was not sufficient:
MR SHAVIN: And do you agree with his description at a high level of the Mann- Whitney test?
PROF HYETT: Yes. If you were to use this from the perspective of screening, what you would likely do is actually take the log of these data, which typically would normalise the distribution, that then allows you to develop the likelihood ratios for comparison between normal and abnormal populations. So that would be the next step that you would do to actually convert this into data that you could then use to define its value for screening.
MR SHAVIN: Professor Fisk.
PROF FISK: Could I just ask. So, Professor Hyett, I understood that a log transformation, of course, still requires it to be normally distributed. My understanding is that most screening data were reported in multiples of the medians because of this point.
PROF HYETT: But I think you will find if you took the log value of these data, yes, you would next then see whether you have normalised the distribution at that point. And I disagree with you. I think there’s a number of screening markers where that has been done and be shown that they actually do normalise at that point. And the fact that they’re expressed of multiples of the median at that point is precisely because they fit around that style of curve.
909 Professor Lovett accepted that the p-value and the results in Figure 1 do not “demonstrate that the concentration of foetal DNA in maternal serum is elevated in aneuploid pregnancies” (page 14, lines 18 to 19 of the Patent). The reported p-value merely suggests that there is a difference in the distributions of the control and aneuploid groups that statistically is calculated as being unlikely to have arisen by chance:
MR RYAN: I was just going to ask Professor Lovett, having in mind what has just been said about the P value, how does that P value assist one to say whether or not it has been demonstrated that the level of foetal DNA in maternal plasma and serum is higher in pregnancies where the foetus has a chromosomal aneuploidy than in normal pregnancies?
PROF LOVETT: I’m sorry. Can you restate the question. Sorry.
MR RYAN: The assertion on the patent at page 5 in paragraph (a) - - -
PROF LOVETT: Yes.
MR RYAN: - - - says that, at about line 12 – says that:
It has been also demonstrated that the level of foetal DNA in maternal plasma and serum is higher in pregnancies where the foetus has a chromosomal aneuploidy than in normal pregnancies.
How does the P value assist or otherwise in one’s assessment of that assertion?
PROF LOVETT: The P value is giving me – as Professor Fisk articulated, it is giving me a quantitative measurement of the degree of similarity or difference of the two distributions, of the two rank orders.
MR RYAN: But the statement on paragraph 5 that we’ve just been looking at is unqualified, isn’t it? it’s an assertion of a fact.
PROF LOVETT: Yes. I mean, they could have put the P value in there.
MR RYAN: Yes. And you wouldn’t say, would you, that figure 1 would support such an absolute statement as we saw on page 5?
PROF LOVETT: No. I actually think it does support it. I think they’ve done a quantitative detection and the distributions are different.
MR RYAN: Now, as a matter of fact, do you accept that it is not accurate to say that the level of fetal DNA is higher in aneuploid as compared with normal pregnancies?
PROF LOVETT: For the limited number of samples that they’ve used, then there is a different distribution between them. Are you asking me for every single one, is it higher? No.
910 Both Ms Norbury and Professor Oepkes also emphasised that the method described in Example 2, which involved quantitation of the Y chromosome, could only be implemented in relation to male fetuses. Ariosa submits that this is a further significant limitation on the potential of this method to be used for screening for chromosomal aneuploidies.
911 In any event, so Ariosa says, despite the suggestion in the Patent that the level of cffDNA might be used to screen for chromosomal aneuploidy, this suggestion has not been able to be put into practice in the 20 plus years since the priority date, despite considerable effort.
912 Even if the method could be used, Professor Hyett gave evidence that:
… I do not agree that the method described in paragraph (a) could be used, as at the Relevant Date or today (either as a diagnostic test or as a screening test), without significant further research.
913 Further, Ms Norbury referred to a review article by Gerovassili A et al, “Free fetal DNA in maternal circulation: a potential prognostic marker for chromosomal abnormalities?” (2007) 27(2) Prenatal Diagnosis 104-110. This paper identifies that after numerous studies there is conflicting evidence in relation to purported elevations of cffDNA in maternal serum or plasma of aneuploid pregnancies. It identifies that for trisomy 21, some studies reported increases, whilst others reported no significant difference; for trisomy 13 some studies reported a lower cffDNA level; and for trisomy 18, some studies reported no significant differences. A possible explanation for these widely varying results was suggested as being the small sample sizes used in the previous studies, and the wide range of DNA levels at each particular gestational age. Given the limitations of previous studies, the aim of the study reported in the Gerovassili paper was to “examine in a prospective study, possible quantitative differences in maternal plasma concentration of fetal and total cfDNA in a large number of chromosomally abnormal pregnancies”. In contrast to the experiment reported in Example 2 (and in Example 4) of the Patent, the paper also described that additional data collected and incorporated into the analysis included gestational age, maternal age, storage time, crown rump length, maternal weight and body mass index and smoking status, which were included as potential factors that could influence the level of cffDNA. Notably, the study focused on first-trimester pregnancies.
914 The discussion in the paper was as follows and the conclusion speaks for itself in supporting Ariosa’s thesis:
DISCUSSION
Since 1997, when fetal cfDNA in maternal circulation was discovered, a new field of research has emerged, driven by its potential clinical applications for non-invasive prenatal diagnosis. Quantitative aberrations of fetal cfDNA in maternal plasma were noticed in chromosomal aneuploidies, but conflicting results were reported among different studies. This is thought to be because of variables that might affect the level of circulating fetal cfDNA and the relatively small numbers of subjects studied.
One of the variables could be related to the timing of the plasma sampling. Previous studies have concentrated on second-trimester pregnancies by which time the invasive procedure (CVS) may have been carried out. This study concentrated on first-trimester pregnancies and the advantage is that the samples were obtained concurrently, just prior to the CVS, therefore giving the test prognostic value that can be employed prior to, or in place of any invasive procedure.
We found inverse correlation between total cfDNA circulating maternal plasma and NT measurement of the fetus (r = -0.15, p = 0.001), which, however, explains only 2.5% of the variability in cfDNA levels (R2 = 0.025). To our knowledge, this is the first study that correlated NT with cfDNA levels.
Our results suggest that there are no differences in total cfDNA levels between 264 controls and 72 Down syndrome pregnancies (median of 341.3 and 345.3 GE/mL, respectively) and contradict previously reported results (median of 36 153 and 5832 GE/mL, respectively) (Spencer et al., 2003). Previous studies used serum and archived samples while in this study plasma was separated within 4 h of venepuncture and DNA was extracted from plasma within 2 months. Total cfDNA levels increase after 6 h of venepuncture when samples are left standing (Angert et al., 2003). This could explain the difference in the results.
Fetal cfDNA levels did not differ significantly between controls and trisomy 21 samples in this study, in agreement with previous results (Ohashi et al., 2001; Hromadnikova et al., 2002; Spencer et al., 2003). Other studies however, (Lo et al., 1999; Zhong et al., 2000; Lee et al., 2002) show a two-fold increase in trisomy 21 samples compared to controls. Again, the difference may be related to the age of the samples; we utilized fresh plasma samples as opposed to archived ones. Also, the number of cases analysed in this study is larger and the average gestation age is lower compared to the other studies. In first-trimester pregnancies, fetal cfDNA levels in the maternal circulation are very low.
Similarly, fetal cfDNA levels did not differ significantly between controls and trisomy 18 in agreement with all previously reported studies (Zhong et al., 2000; Ohashi et al., 2001; Wataganara et al., 2003). We did not find any significant differences in the levels of fetal cfDNA between controls and trisomy 13 fetuses, which differ from other publications (Zhong et al., 2000; Wataganara et al., 2003). However, we believe that the number of samples studied in total (four in our study, three in Zhong’s, and five in Wataganara’s) is too small to draw any conclusions. No relation was found in the levels of total cfDNA between normal and Turner’s syndrome bearing pregnancies and any of the triploidy cases. Furthermore, we found a considerable degree of overlap between the values observed in trisomic and control fetuses.
In this study, we did find a small inverse correlation between fetal cfDNA (DYS14) and maternal weight (r = -0. 15, p =0.02) and more importantly with the BMI of the subjects (r = -0.17, p = 0.01) agreeing partly with previous findings. It was previously discussed that maternal weight is inversely correlated with the levels of free fetal DNA in the second-trimester pregnancies (r = -0.26, p = 0.07) but not in the first trimester (Wataganara et al., 2004). It is thought that since the levels of fetal cfDNA increase with GA, the effects are more pronounced in the second trimester, which could explain a lesser correlation in the first-trimester samples in this study.
A trend towards inverse correlation was also found between the levels of fetal cfDNA (DYSI4) and storage time. However, our results did not reveal important changes due to storage time in general (small r value), similar to the results of Lee et al., (2002). Sozzi et al., (2005) showed that prolonged storage of both isolated DNA and whole plasma samples led to substantial DNA degradation. We did not notice any storage effect on our samples as on average, the analysis was done within 2 months of their collection.
It has been reported that smoking during pregnancy is associated with increased chromosomal instability in amniocytes (De la Chica et al., 2005). However, in our study, no significant difference was found between smoking and non-smoking groups and the fetal and total cfDNA.
In conclusion, we present here the largest study performed to date on prospectively collected samples on the quantification of fetal and total cfDNA in maternal circulation. Our results suggest that quantification of fetal cfDNA in first-trimester pregnancies is not an ideal prognostic marker for chromosomal abnormalities with the methods currently used. While the results may not be applicable to the broader population, this diagnostic test however, would normally be applied to pregnancies at higher risk of chromosomal abnormalities.
915 Further, Professor Oepkes reviewed 13 post-priority date papers considering the suggestion that cffDNA is elevated in aneuploidy pregnancies. He summarised that the papers showed conflicting results as to whether such elevation occurs in fact, and concluded that:
I agree with most authors of these studies that several drawbacks of quantification of cell free fetal DNA as a test for fetal aneuploidy, mainly the large overlap with cell free DNA levels in normal pregnancies, the non-specific association with various pregnancy complications, and the restriction to male fetuses make it unlikely that this method would be of any use clinically. In my opinion evaluation of levels of cell free fetal or total DNA could not be used as a marker to screen for aneuploidy in clinical practice.
916 In responding to this evidence, Professor Lovett relied on four of the 13 papers cited by Professor Oepkes, and concluded on this basis that:
Having reviewed these papers and Professor Oepkes’ comments on them, I consider that it is clear that many researchers (e.g. Zhong 2000, Lee 2002, Spencer 2003, Farina 2003) have conducted well-designed studies and confirmed Dr. Lo’s comments in the Patent (and in Lo 1999) that the level of cell free foetal DNA is higher in aneuploid pregnancies compared to normal pregnancies. However, the significant overlap between the populations does not make this fact a particularly attractive screening marker. I do not consider that anyone has conclusively disproven that the total level of cell-free DNA is higher in aneuploid pregnancies compared to normal pregnancies.
(Citations omitted.)
917 But I agree with Ariosa that the papers relied upon by Professor Lovett for this statement are selective. On Professor Lovett’s analysis, the majority of the studies reported in these papers did not support his hypothesis, given that Professor Lovett only referred to four out of the 13 papers. Further, the Lee T et al, “Down syndrome and cellfree fetal DNA in archived maternal serum” (2002) 187(5) American Journal of Obstetrics and Gynecology 1217-21 (Lee 2002) and Farina A et al, “Evaluation of Cell-free Fetal DNA as a Second-Trimester Maternal Serum Marker of Down Syndrome Pregnancy” (2003) 49(2) Clinical Chemistry 239-242 (Farina 2003) papers are in effect the same study. The Farina paper also said that any use of such a method would have to “await the discovery of a reliable gender-independent fetal DNA marker that can be assayed by real-time PCR”. Further, the results of the Spencer K et al, “Increased total cell-free DNA in the serum of pregnant women carrying a fetus affected by trisomy 21” (2003) 23(7) Prenatal Diagnosis 580-583 (Spencer 2003) study were that the median concentration of cffDNA was not significantly higher in trisomy 21 cases.
918 In summary, according to Ariosa the preponderance of the evidence is that the suggestion that the level of cffDNA can be used as a screening test for aneuploidy pregnancies has been widely investigated over a period of more than 20 years, and has not been supported by any conclusive study, nor implemented in practice.
919 Now I am prepared to accept such a proposition on the preponderance of the science, but I do not think this carries Ariosa far enough to establish a lack of sufficiency as I will later explain.
920 Let me now deal with another aspect of Ariosa’s case concerning elevated cffDNA in pre-eclamptic pregnancies (see page 5 line 27) which can be related back to paragraph (a) on page 5.
921 Example 4 of the Patent relates to the quantitative analysis of cffDNA in maternal serum in pre-eclamptic pregnancies. The results are in Figure 2. The Patent concludes in relation to the experiment described in Example 4 and Figure 2 (at page 22, lines 20 to 22):
Our data indicate that the concentration of foetal DNA is higher in pre-eclamptic compared with non-pre-eclamptic pregnancies. These results indicate that foetal DNA concentration measurement in maternal plasma may be used as a new marker for pre-eclampsia.
922 However, the Patent then goes on to state (at page 22, line 30 to page 23, line 3):
Our discovery also opens up research into the potential application of foetal DNA quantitation to predict the occurrence of pre-eclampsia, prior to the development of clinical signs such as hypertension and proteinuria.
923 According to Ariosa, Example 4 and the accompanying Figure 2 suffer from many of the same difficulties identified above with respect to Example 2 and Figure 1. This includes that the method described in Example 4, which utilises the Y chromosome, will only be applicable to male fetuses.
924 But in contrast to Example 2, the description of the method of Example 4 indicates that the “pre-eclamptic and control subjects were matched for gestational age” (page 21, lines 21 to 22). But the Patent does not specify the manner in which the subjects were matched, and whether this was at a group level (i.e. all subjects fell within a particular range of gestational age, and if so, what that range was) or on an individual level (i.e. each pre-eclamptic subject was matched with a control subject of the same gestational age). Professor Fisk appeared to suggest that it was the latter, when he stated that “each case with pre-eclampsia is gestation-age-matched for a control case”.
925 Further, there is no information provided as to the gestational age of subjects, other than that the subject women had pre-eclampsia according to the criteria set out in the Patent, and the control subjects were not on medication and had no hypertension or proteinuria. Now pre-eclampsia is a condition that is diagnosed after 20 weeks’ gestation, with the majority of cases being diagnosed after 36 or 37 weeks. As a result, the potential gestational age of the subjects covers a broad range, comprising the second half of pregnancy.
926 Professor Hyett’s evidence was that even though the subjects in this example may have been matched in some way for gestational age, the lack of information about gestational age is significant. In particular, the Patent describes Example 5 (and Figure 4) as showing that “foetal DNA concentration increased as pregnancy progressed” and that “[w]omen recruited late in pregnancy had higher foetal DNA concentrations in their plasma and serum” (page 31, lines 13 to 17). The wide variation in the level of fetal DNA in the latter half of pregnancy, both within a patient and as between patients, is evident from Figure 4. For example, in the relation to Case S-7, at approximately 32 weeks, the concentration of SRY was approximately 80.9 copies/ml, but at approximately 38 weeks, this concentration had increased to 406.5 copies/ml. The concentration of case S-7 at 38 weeks can be compared to the concentration of case S-3 at approximately the same period, of only 39.8 copies/ml. But Professor Fisk appeared to assume that Example 4 is likely to be skewed to later gestation.
927 Similarly to Example 2, the Patent reports that the mean concentration of cffDNA of the pre-eclamptic group is higher than the control group, and that a p-value was reported that would typically be considered statistically significant. However, as with the aneuploidy example, no data in relation to the absolute values of the concentrations for each subject, the means or the standard deviation are provided.
928 Further, Figure 2 also features a significant overlap between the pre-eclamptic and control groups, which Ariosa’s experts identified as important in interpreting the purported results. This was explained by Professor Hyett as follows:
MR SHAVIN: And then he says, building from that, the results indicate that foetal DNA concentration measurement in maternal plasma may be used as a new marker. And you accept that’s talking about a possibility for the future.
PROF HYETT: Correct.
MR SHAVIN: So here you have - - -
HIS HONOUR: And a possibility of an additional marker, in other words, added to something else. So could be an incremental - - -
PROF HYETT: Yes. I mean, my – if I can unpack this, as well, the scenario that we were talking about with non-invasive prenatal diagnosis, from my perspective, around 1995, was that he had spent 10 or 15 years looking at foetal cells, which was taking us down an alley that did not appear to be giving us any progress in the context of defining that this was going to be useful for non-invasive prenatal diagnosis. Therefore, seeking alternatives was very relevant. If you take the concept of looking for biomarkers for pre-eclampsia at the end of pregnancy from a clinical perspective, I think there was less importance about defining biomarkers like this in that circumstance, because there was good progress being made using other sets of markers, first, to make – to define the disease and, secondly, to define severity of disease.
MR SHAVIN: Yes. This is a statement that’s made at page 22 of [the] patent, where he’s simply saying, “I’ve looked at this data, and this might be a useful marker.” He’s saying no more nor less than that, isn’t he?
PROF HYETT: Yes.
MR SHAVIN: And there’s nothing here in this patent that suggests that that is something that would be a complete waste of time?
PROF HYETT: The spread of the data – if we go to figure 2 for one moment. So bear in mind that pre-eclampsia is a condition that kills pregnant women, and therefore misdiagnosis potentially is quite significant. So here you can see that if you wanted to have 100 per cent detection of pre-eclampsia in these series of observations, you would actually include all of the control cases as well. So from my perspective, that means you have a test that has 100 per cent sensitivity and zero per cent specificity. I would suggest that’s not a very useful marker.
MR SHAVIN: I’m suggesting to you that nowhere in this patent does the patentee suggest that the proposition on lines 22 and 23 of the specification provide a diagnostic tool with a 100 per cent outcome. That’s simply not what he says, is it, Professor?
PROF HYETT: Well, you asked me to interpret the data – or I was asked in the expert report, together with my colleagues, to identify whether I felt this was going to be a useful tool or not and whether the data supported the statement made in the – in the patent. I – when I look at the data and the spread of the data, I see no value in this for this purpose.
929 Professor Oepkes appeared to agree in the joint expert report that based on Figure 2, the total level of fetal DNA in maternal plasma may be used as a new marker for pre-eclampsia, but with the caveat that based on that data considerable further work would need to be conducted to assess its clinical utility.
930 Now in the context of cffDNA varying with gestation age and the apparent lack of control for this variable in examples other than Example 4, Professor Fisk sought to overcome the deficiency by stating that “the data are the data”. But Professor Fisk accepted that the paper by Hahn S et al, “Both Maternal and Fetal Cell-Free DNA in Plasma Fluctuate” (2001) 945(1) Annals of the New York Academy of Sciences 141-144 showed that the amount of cell-free DNA observed in pregnant women changes dramatically over time, including over a period of several days. And he also accepted that it was necessary to control for such variation as otherwise the results could be impacted, and may in fact be of no value. The conclusion in Hahn was expressed as:
Our studies have indicated that the quantities of circulatory DNA varied considerably in normal healthy individuals. This feature is not solely attributable to cellular contaminants not adequately removed from the plasma fraction as considerable fold variations were also evident in a repeat experiment in which two different clearance strategies were used. It is possible that our data should be reevaluated in a more descriptive manner as used in other clinical chemical settings. This issue is being addressed currently. The most important issue of our work, however, is that, where we have been able to assay the amount of fetal DNA, we have determined fold fluctuations of a similar level to the elevations previously recorded in pregnancies with aneuploid fetuses. These results, hence, indicate that care should be taken when using 2-fold elevations in free fetal DNA levels as being indicative of a fetal aneuploidy.
931 Now I should note that Professor Fisk relied upon Martin A et al, “Can the quantity of cell-free fetal DNA predict preeclampsia: a systematic review” (2014) 34(7) Prenatal Diagnosis 685-91, which purported to find a statistically significant increase in cffDNA among women who subsequently developed pre-eclampsia. But he accepted that this conclusion was subject to many caveats that needed to be fulfilled. In relation to a more recent paper, Rolnik DL et al, “Maternal plasma cell-free DNA in the prediction of pre-eclampsia” (2015) 45(1) Ultrasound in Obstetrics & Gynecology 106-111, Professor Fisk appeared to agree that even 17 years after the priority date, it remained unknown whether there was a correlation between the level of cffDNA and pre-eclampsia. The conclusion of Rolnik, referring also in part to the Martin paper, was that measurements of total cfDNA and fetal fraction in maternal plasma at 11 to 13 weeks and 20 to 24 weeks were not predictive of pre-eclampsia.
932 Ariosa has submitted that like the aneuploidy example, there is therefore no evidence of any implementation of the method described in Example 4. Rather, the evidence is that the asserted correlation remains unproven, and no such test has been used or exists, despite considerable efforts having been made. Again, I accept what it says in terms of the relevant science, but it may not be sufficient to show a lack of sufficiency.
Whether the quantitative comparison of chromosomal markers embodiment (paragraph (b) on page 5 of the Patent) is fully described
933 Let me deal with another type of quantitative embodiment raised by Ariosa.
934 Paragraph (b) on page 5 of the Patent suggests that a second quantitative approach to screening for chromosomal aneuploidies might be to quantitate fetal DNA markers on different chromosomes.
935 According to Ariosa, and as explained by Ms Norbury, this entirely theoretical approach is based on the assumption that if a fetus possesses an extra copy of a chromosome, cffDNA from that fetal chromosome would be present in maternal plasma or serum in a higher amount than cffDNA from a fetal reference chromosome. The suggested approach requires the skilled addressee to identify informative fetal DNA markers on two different chromosomes, one being the chromosome of interest, such as chromosome 21, and the other being a reference chromosome. It then requires the skilled addressee to determine the relative amount of the fetal markers on the target chromosome and the fetal marker on the reference chromosome present in a maternal sample.
936 Now all of the experts accepted that this approach could not have been implemented to detect maternally-inherited chromosomal aneuploidies, as in this case the additional maternally-inherited chromosome would be the same as one of the mother’s chromosomes, and would be indistinguishable from the background maternal cfDNA in the sample using the PCR technology available at the priority date.
937 But Professors Lovett and Fisk maintained that this approach could have been implemented at the priority date for paternally inherited aneuploidies using, for example, paternally inherited markers on chromosome 21 and markers on a reference chromosome. In doing so, Professors Lovett and Fisk relied on technical papers describing technologies that they said could be used.
938 Professors Lovett and Fisk relied on a series of papers including Mansfield ES, “Diagnosis of Down syndrome and other aneuploidies using quantitative polymerase chain reaction and small tandem repeat polymorphisms” (1993) 2(1) Human Molecular Genetics 43-50 (Mansfield 1993), Pertl B et al, “Rapid molecular method for prenatal detection of Down’s syndrome” 343(8907) Lancet 1197-1198 (Pertl 1994), Pertl B et al, “Rapid detection of trisomies 21 and 18 and sexing by quantitative fluorescent multiplex PCR” (1996) 98(1) Human Genetics 55-59 (Pertl 1996), Pertl B et al, “Quantitative fluorescence polymerase chain reaction for the rapid prenatal detection of common aneuploidies and fetal sex” (1997) 177(4) American Journal of Obstetrics and Gynecology 899-906 (Pertl 1997), and Pertl B et al, “Detection of male and female fetal DNA in maternal plasma by multiplex fluorescent polymerase chain reaction amplification of short tandem repeats” 106(1) Human Genetics 45-49 (Pertl 2000) regarding quantitative fluorescent PCR (qfPCR) to assert that suitable markers, being STRs, were available at the priority date which could be used to discriminate paternally-inherited fetal DNA from maternal DNA. But Professor Lovett described qfPCR as a substandard technique, and appeared to accept that it could not have been used to detect a 2:1 difference in dosage of two fetal STRs against a background of maternal DNA. Let me elaborate.
939 Professor Lovett explained that the Pertl papers describe a different methodology of qPCR to real time qPCR. These papers describe qfPCR, which measures the level of fluorescence and therefore the amount of DNA from a target sequence at the end of the assay, rather than during its course. He explained that it was not as accurate as real time qPCR and was “semi-quantitative”. As noted above, he also described qfPCR as “substandard” compared to the method in Heid et al 1996, and accepted that it was not sufficiently sensitive to detect an exact 2:1 dosage difference, noting that “it’s probably good with maybe … two-fold. But it’s not as accurate as measuring in that logarithmic range”.
940 Nevertheless, Professor Lovett says that the Pertl papers, particularly Pertl 2000 “validates the teaching of the patent. The patent says go forth and look for these things, and you will be able to discriminate them. And here, Pertl has done that …”. Professor Fisk also relies on the Mansfield and Pertl papers to demonstrate the ability to measure a 2:1 difference in the amounts of two markers using qfPCR.
941 But I agree with Ariosa that there are several points which should be emphasised in relation to these papers, which may have been glossed over.
942 All of the Pertl papers published before the priority date relate to experiments conducted using qfPCR to examine pure samples of fetal DNA obtained, for example, from invasive procedures such as amniocentesis and CVS. As noted by Ms Norbury, these papers “are a good example of the extensive research that was required over a period of several years to attempt to validate the methodology proposed by Mansfield in 1993, and its potential application to prenatal diagnosis. This is not routine work, and it was not until Pertl 1997 was submitted for publication in late 1996 that the authors were prepared to conclude that the diagnostic value of qPCR techniques had been established”. Professor Oepkes also described the Pertl (and Adinolfi) group as working on the development of qfPCR for rapid testing of “pure fetal material taken from amniocentesis or chorionic villus samples”.
943 Moreover, even in these experiments which were using pure fetal DNA, the relative levels of fetal chromosome markers were still difficult to compare. For example, in Figure 2 in Pertl B et al, “Rapid detection of trisomies 21 and 18 and sexing by quantitative fluorescent multiplex PCR” (1996) 98(1) Human Genetics 55-9 (previously defined as Pertl 1996) at 58, the fetus is reported as displaying a diallelic trisomic pattern for the D21S1414 marker on chromosome 21, as well as for both the MBP Locus A and MPP Locus B on chromosome 18. But it is apparent from the electrophoretogram, that the two peaks for each of those markers is not at a ratio of 2:1 as would be expected. As pointed out by Ms Norbury, in the first line of the Table 3 of Pertl 1996 (at 57), trisomy 21 fetal samples are reported as having relative peak areas for diallelic samples from 1.68 to 2.37, which is a “quite wide range”. This is contrary to Professor Fisk’s claim that the dosages reported in Pertl are “quite precise”. Consistent with Ms Norbury, Professor Oepkes noted that the studies reported in the Pertl papers were “retrospective studies on samples that beforehand were known to come from normal or aneuploid pregnancies. They were small in number, and even there, their technique was not 100 per cent correct in all cases”.
944 The results in the pre-priority date Pertl papers also clearly demonstrate that different markers from different chromosomes, or even the markers from the same chromosome, amplify with significantly different efficiencies. Professor Lovett appeared to agree, pointing to “inherent differences in the PCR ability”, and the “variability in the PCR ability of different primer pairs … So you would – if you were doing a panel of these things, you would throw those least-sensitive markers away”.
945 Ms Norbury also pointed to the variability seen in the results in Pertl B et al, “Detection of male and female fetal DNA in maternal plasma by multiplex fluorescent polymerase chain reaction amplification of short tandem repeats” (2000) 106(1) Human Genetics 45-9 (previously defined as Pertl 2000).
946 It is worth setting out the following extracts from Pertl 2000:
Results
Dilution experiments were performed to determine the sensitivity of the fluorescent PCR assay. Male DNA was diluted in female DNA (50%, 25%, 10%, 5%, 2.5%, 1%, 0.5%, 0.1%, 0.05%, 0.01%), leading to a fetal-specific allele detection sensitivity of 1% overall (range 0.01–2.5%). As estimated by these dilution experiments, the most sensitive markers for the detection of fetal-specific alleles were D21S11, D21S1411, D21S1412, and AMXY; the least sensitive markers were D13S631 and D13S634.
Twelve mother/child pairs were studied. Analysis of maternal plasma samples collected before delivery revealed the presence of fetal-specific alleles in all cases. An example of the alleles demonstrated in maternal plasma is shown in Fig. 1. The arrow indicates the paternally inherited fetal allele.
All samples were informative in at least four of nine different STR markers (range 4–8). Fetal-specific alleles were detected with a mean of 4.7 STR markers per sample (range 1–6) (Table 1).
Fig. 1 Electrophoretogram of the amplification products from a maternal plasma sample using two short tandem repeat (STR) markers (D21S1411 and D21S1412). The marker D21S1412 shows two major polymerase chain reaction (PCR) products corresponding to two maternal alleles and including one maternally inherited fetal allele. A third, smaller “paternal” allele is indicated by the arrow, corresponding to the fetal allele that is inherited by the father. The marker D21S1411 shows a homozygous pattern with one major PCR product corresponding to the maternal alleles and including one maternally inherited fetal allele. The arrow indicates the paternally inherited fetal allele.
All experiments were done in triplicate. In some cases, fetal alleles, which were missed in the first experiment, could be detected in repeat experiments and vice versa. This phenomenon is expressed in Table 1 as “variably amplified” fetal alleles. Including the “variably amplified” alleles, paternally inherited fetal alleles were detected in 84% of informative short tandem repeats and missed in 16% (Table 1).
The rate of informativeness and the sensitivity of detection of fetal alleles were different using different markers (Table 2). The markers D21S11 and D21S1411 were the most informative and sensitive markers. Fetal alleles were amplified in ten of ten informative pairs using D21S11 and in eight of nine informative pairs using D21S1411. This was in contrast to D18S535, which was informative in 11 of 12 pairs, although fetal alleles were detected in only 4 of 11 informative cases despite the highly polymorphic nature of the marker.
Using the marker AMXY, a Y-specific sequence was detected in all plasma samples from pregnant women carrying male fetuses. No correlation was observed between the presence of contractions of the mother and the rate of detection of fetal alleles in the maternal plasma sample.
Discussion
Our results demonstrate the feasibility of detection of male and female fetal DNA in maternal plasma with the use of highly polymorphic STR markers and fluorescent multiplex PCR.
In the present study, we used PCR amplification of nine STRs to detect fetal-specific alleles in maternal plasma samples. Dilution analyses were performed to assess the sensitivity of the different STRs (i.e., the ability to detect fetal DNA representing only a small percentage of total plasma DNA). The sensitivity of PCR amplification of different STRs was estimated to be 0.01–2.5%. Hence, the described technique has a lower sensitivity for detecting fetal DNA in maternal plasma than the SRY system described by Lo et al. (1998, 1999). This lower level of sensitivity may be due to the nonselective nature of PCR amplification of STRs in that both the target (i.e., the fetus’) and the background (i.e., the mother’s) sequences are amplified together (Lo et al. 1996). Under these conditions, the excess of the background sequences could out-compete the rare target sequences for amplification. However, because of the high concentration of fetal DNA present in maternal plasma (Lo et al. 1998), our proposed technique was sensitive enough to detect fetal-specific alleles in all mother/child pairs studied.
The mean informativity of the nine STRs used in this study was 65% (range 44–89%). Fetal-specific alleles were amplified in 84% when all informative STR markers were used. The markers D21S11 and D21S1411 were the most informative and sensitive markers. PCR amplification of these two markers together with the marker AMXY should be the most useful combination for large- scale clinical application.
We intentionally performed this study using samples obtained at term to maximize the amount of fetal DNA present. Future studies need to be performed earlier in gestation at a time when noninvasive prenatal diagnostic information may be more clinically useful.
Moreover, the potential of quantitative analysis of fetal DNA in maternal plasma/serum using fluorescent PCR has still to be investigated. It should be possible to quantify the amount of fetal DNA that corresponds to the paternal allele using an appropriate internal standard.
The results of this study show that we can now confirm the presence of fetal DNA in maternal plasma independent of gender. The ability to detect male and female DNA in maternal plasma samples represents a major advantage over other DNA detection systems that use fetus-derived Y sequences. Although more studies are needed to determine the sensitivity and specificity of this method at various gestational ages, multiplex fluorescent PCR amplification of STRs may be an important technique for a wide range of clinical applications. Published reports to date include the measurement of fetal DNA in maternal plasma as a marker for pre-term delivery (Leung et al. 1998) and pre-eclampsia (Holzgreve and Hahn 1999; Lo et al. 1999). Furthermore, there may be a quantitative difference in the concentration of fetal DNA in maternal plasma between normal and aneuploid pregnancies (Bianchi et al. 1997). In these studies, amplification of Y chromosomal sequences was used to quantify fetal DNA and to monitor pregnancy complications in women carrying male fetuses. The present report extends the clinical utility of maternal plasma DNA amplifications and makes a prospective analysis of risk pregnancy complications feasible independent of fetal gender.
(Tables omitted.)
947 Pertl 2000, which was published three years after the priority date, reported on a study undertaken on maternal serum containing a mixture of fetal and maternal serum. Using a panel of STRs, the authors were able to detect cffDNA at the D21S1412 allele and the D21S1411 allele, each of which were on chromosome 21.
948 Now there were complexities in interpreting results from qfPCR when using STRs as markers.
949 But in addition to these complexities which arise when simply attempting to detect the fetal allele in a mixture of maternal and fetal cfDNA (not quantify it), the discrepancy in the size of the peaks for the maternal alleles in Figure 1 of Pertl 2000 demonstrated the different efficiencies with which different markers amplify in a PCR assay.
950 Professor Lovett characterised Pertl 2000 as a “proof of concept paper”. Now this is not without its significance given that the paper was published several years after the priority date, and does not make any attempt at quantifying the fetal or maternal alleles. It was also not attempting to detect an aneuploidy pregnancy. The sole purpose of the paper was to prove that cffDNA from chromosome 21 could be detected using a non-Y chromosome marker in a maternal serum sample, i.e. a heterogeneous mixture of fetal and maternal cfDNA in which the fetal DNA represented only a very small percentage of the total DNA.
951 Further, the authors of Pertl 2000 specifically refer to the difficulties associated with detecting the low concentration of cffDNA in the maternal sample using qfPCR, stating that they intentionally used samples collected late in gestation which had a higher cffDNA concentration:
We intentionally performed this study using samples obtained at term to maximize the amount of fetal DNA present. Future studies need to be performed earlier in gestation at a time when noninvasive prenatal diagnostic information may be more clinically useful.
Moreover, the potential of quantitative analysis of fetal DNA in maternal plasma/serum using fluorescent PCR has still to be investigated. It should be possible to quantify the amount of fetal DNA that corresponds to the paternal allele using an appropriate internal standard.
952 This discussion in Pertl 2000 also demonstrates that even three years after the priority date, it was not possible to accurately or reliably quantify the low amount of cffDNA in a maternal sample (even using a sample from late gestation) using quantitative PCR, which would be necessary in order to detect a 2:1 dosage difference between two fetal chromosomes in a paternally-inherited aneuploidy assuming that such a detection rate is possible. Indeed, Pertl 2000 represented a demonstration of the qualitative detection of the fetal allele, not an example of the quantitation of the fetal allele.
953 As Ms Norbury explained, the method described in Pertl 2000 was purely qualitative, insofar as it demonstrated the presence of the fetal allele, rather than quantitating the amount of the fetal allele.
954 Further, in relation to the results presented in Pertl 2000, Professor Lovett accepted that there were different efficiencies in the amplification of the two alleles represented in Figure 1 of Pertl 2000, but suggested that one could deal with this by “optimising primers” in terms of “length, perhaps GC content” and by multiplexing; but notably, the assay reported in Pertl 2000 was multiplexed. He also recognised that:
The – genome is not a level playing field. So there are certain places that are maybe polymorphic when looked at – a population-based study, and then you actually go in there and you’ve got poorly hybridising primers or you just have technical problems in terms of getting those guys to amplify. And the solution to it is to add depth. So what you do is you add lots of redundancy, in terms of your markers. So if you run this through – and the way to do this would have been to take all these markers and run it through loads of normal DNA samples. And you look at the noise and the variability and you say “These guys are – a bit problematical. I’m going to add in markers around them.”, And that way you add with the depth, more certainty about your ability to detect a particular piece of DNA.
955 Professor Fisk said:
The question becomes, really, a technical one about the reliability of comparison of various markers and we’ve addressed together some of these difficulties, but one would have done these working up the tests with adjusting for these things or selecting the primer so one broadly got comparable doses, otherwise the assay wouldn’t be able to be done.
956 But Ms Norbury explained the following matters. The number of markers available at the priority date was just 5% of what is now known and was “still quite limited compared to what it is now”. Further, the information now available relating to sequence variation was not available in 1997 to assist in primer design/optimisation. Further, if a PCR assay was targeting a specific marker at a particular location on a chromosome, then the primer had to be “target specific”, which meant the ability to optimise the primer design was restricted by the “inherent … nature of the genomic sequence” surrounding the target marker. Further, once a primer had been designed, it was necessary to test it and work out its efficiency. Further, the “combination of alleles” had to be considered. One reason is because “you get this concept of allele dropout where people have two very different size alleles, that the shorter one essentially takes over the reaction and the … longer one just drops out”. Consistent with this, it was reported in Pertl 1996 that the authors had to carry out two separate assays, one with primers for the D21S11 marker and the other with primers for D21S1414 because the primers could not be used together.
957 Further, the markers used in all of the work by the Pertl group including after the priority date were STRs. And Professor Lovett said that these were the types of markers he would use to implement the method of the Patent. But Professor Lovett conceded that you could not use STRs in real-time qPCR, which Professor Lovett says was the only PCR technology available at the priority date which could be used to detect a 2:1 difference in chromosome dosage as the qfPCR technology was “substandard”:
MR RYAN: Now – well, can you actually use STRs in real-time PCR?
PROF LOVETT: Well, that would be tough because you couldn’t do it in the conventional sense, but you could run it down a gel, right, at – at specific time points. It – it can’t be done, right, in the conventional sense because you’re picking up the background maternal material. Right? You would have to do this, initially at least, probably with fluorescence or something like that.
958 Professor Lovett therefore had to accept that he would need to use RFLPs or SNPs as markers for use in real time qPCR. But 1997 was “just the dawning era of single-nucleotide polymorphisms”. There “had been a couple of papers describing SNPs but the sudden take-off of that didn’t happen until afterwards”. Further, because RFLPs (and SNPs) are biallelic only, they are much less likely to be informative:
PROF LOVETT: So I would do first STRs, then I would move to RFLPs, in 1997.
MS NORBURY: Okay. So at 1997, the main markers, as we discussed on day 1, were the STRs. And if you will recall, that was because they were so informative, around eighty per cent, whereas the – there was less – less ..... information about single nucleotides, and they were – were far less informative, at, you know, less than – definitely less than 30 per cent. So – so you would have problems, then, actually finding informative markers. So it would be a combination of having – having a marker and then being able to detect the difference.
MR SHAVIN: Professor Lovett, would you like to comment on that?
PROF LOVETT: I – I agree that the – the informativeness of an RFLP or the limited number of SNPs that were available then would be a hurdle; but numbers overcome this. You just do a bunch of them. You know? So you – you do essentially what Pertl did: you take nine markers and you run through. And you run through with a bunch of RFLPs. You find one that is informative. Right?
MR SHAVIN: Yes.
PROF LOVETT: So you can actually see that the fetus has got that marker. And then you build PCR primers around it.
MR SHAVIN: Yes.
MS NORBURY: I think you would need – I mean, you would do a lot more than nine. You would be doing – having to do hundreds.
MR SHAVIN: But doing each one would be routine. There would just be a bit of work involved.
PROF LOVETT: Mmm.
MS NORBURY: There would be quite a bit of work involved in terms of – of optimising the PCR, because we – I think we – we went through the concepts of variable efficiency and possibility of allele dropout and – and SNPs in the – the primer site as well; all of that information, and the position of the markers, was much less well established in 1997.
MR SHAVIN: Yes. But in the bespoke work that you were doing at around that time, that was part of the routine work you did, was it not?
MS NORBURY: To design efficient PCR reactions?
MR SHAVIN: Yes.
MS NORBURY: Well, I think I made it clear that we were doing qualitative analysis, not quantitative analysis. We weren’t doing any quantitative analysis in 1997. So no, that wasn’t routine work.
959 Professor Lovett accepted that because of the need to use SNPs (or RFLPs) in real time qPCR, the level of informativeness of each marker was significantly reduced. Therefore in the context of screening for Down syndrome you would use “hundreds and hundreds of markers or an awful lot of them so that you – you increase your changes of informativity”. But he did not identify any such markers. Professor Lovett agreed with Ms Norbury that selecting an optimal marker would have to be done on a case by case basis because “different individuals will have different alleles”, and “you couldn’t predict without prior genotyping and knowing what the parent alleles are”. I tend to agree with Ariosa that Professor Lovett’s proposition that as at 1997 a person skilled in the art could have simply gone and found “hundreds of markers” so that you could “run this thing” and “run it on every DNA you can put your hands on” to “get nice, clean read outs of the alleles you know are there” had its problematic aspects.
960 Further, in order to deal with the lack of sensitivity of qfPCR, Professors Lovett and Fisk relied on another series of papers, being Heid et al 1996, Timothy M Woudenberg and J Stevens, “Quantitative PCR by Real Time Detection” (Proceedings SPIE 2680, Ultrasensitive Biochemical Diagnostics, 1 April 1996) (Woudenberg 1996) and Gibson UEM et al, “A Novel Method for Real Time Quantitative RT-PCR” (1996) 6(10) Genome Research 995-1001 (Gibson 1996) regarding real time quantitative PCR (real time qPCR), which they say was sufficiently sensitive to detect a 2:1 dosage difference as at the priority date. But Professors Lovett and Fisk accept that the STRs used in the qfPCR papers would not work for real time qPCR. And they provided no other examples of markers available at the priority date which could have been used with real time qPCR to discriminate between paternally-inherited fetal DNA, although they speculated that SNPs and RFLPs could be used. I also accept that the evidence establishes that a significant amount of work would be required to identify such markers, including prior genotyping. Further, even if such markers could be identified, real time qPCR would not have been sufficiently sensitive in 1997 to measure a 2:1 difference in the level of cffDNA from two different fetal chromosomes let alone reliably and consistently, given its presence at low dosages in a mixture of maternal DNA.
961 I agree with Ariosa that a review of the papers on which Professors Lovett and Fisk relied establish that, at best, implementing the method proposed in paragraph (b) on page 5 of the Patent would have constituted a significant research project. And indeed history shows that it was not until 10 years after the priority date, and the advent of new technology in the form of digital PCR and massive parallel sequencing, that the method was in fact implemented.
962 As I say, Professor Lovett relied on Heid et al 1996, Woudenberg 1996, and Gibson 1996, which were published in the year before the priority date, to establish the commercial availability of qPCR and its ability to detect a 2:1 difference in the levels of two genetic signals. All three papers were early reports on what was as at the priority date a very recently developed technology.
963 Professor Lovett explained real time qPCR as follows:
And the cleverness of that is that you have a tiny little piece of DNA which has a quencher and a fluorescence dye on each end. And it hybridises to your target. And you also have PCR primers. And so off goes the polymerase along the DNA. And this assay became known as TaqMan, after Pacman. Right? So as it goes along, as it finds that fluorescence and – and quenched nucleotide, it starts to eat it. And it releases the fluorescence away from the quencher. So for the first time, your detector sees that fluorescence. And it – what that says is the polymerase got to that point already.
So very, very accurate. And you can see the number of fluorescence molecules going up. And, as Professor Fisk said, at something like 10 standard deviations above the – above the base, you see – you’re in full logarithmic range. If you started with a smaller amount of material, then that’s – that happens later. If you started with a lot of material, then your primers bind better, because there’s more target, and you get up to that quicker. So with adequate controls in there to normalise everything, you can very accurately tell the level of something that’s in there.
964 A number of co-authors of these papers relating to real time qPCR were from Applied BioSystems, a company that produced the ABI Prism 7700 Sequence Detector, one of the first machines to enable real time qPCR. The papers were an early attempt to validate real time qPCR and the equipment produced by Applied BioSystems. As noted by Professor Lovett, as at the priority date, the technology was state of the art, and in its first year. Indeed, Professor Lovett did not himself acquire a real time qPCR machine until 1997, and whilst he had heard of equipment such as the 7700 Detector and the LightCycler, as at the priority date he had not personally used them. Professor Fisk had never undertaken qPCR himself. In contrast, Ms Norbury’s laboratory did have a LightCycler at the priority date, although they were not using it for quantitative analysis and it was not the most robust piece of engineering.
965 Heid CA et al, “Real time Quantitative PCR” (1996) 6(10) Genome Research 986-94 (previously defined as Heid et al 1996) is referenced in paragraph (b) on page 5 of the Patent, where it is said to describe a recent development that might facilitate the approach suggested in paragraph (b).
966 In Figure 1B, Heid et al 1996 reports “amplification plots of serially (1:2) diluted human genomic samples amplified with β-actin primers”. For each amplification plot, “CT values are calculated by determining the point at which the fluorescence exceeds a threshold limit (usually 10 times the standard deviation of the base line)”. Plotting the CT value against the log of input of genomic DNA generates the graph shown in Figure 1C of Heid et al 1996. The linear fit of this data can provide a model against which “relative copy numbers can be determined for an unknown sample”, by calculating the CT value of the sample and then solving for the x-value, being the relative DNA copy numbers.
967 In the Patent, the inventors used the same approach as described in Heid et al 1996 to quantify the amount of cffDNA amplified from the Y chromosome using an SRY primer and, in a separate assay, the total amount (both fetal and maternal) of cell-free DNA from the beta-globin sequence.
968 Table 1 of Heid et al 1996 reports the results of an experiment designed to establish the reproducibility of sample preparation. The authors of Heid et al 1996 explain that:
Sample Preparation Validation
Several parameters influence the efficiency of PCR amplification: magnesium and salt concentrations, reaction conditions (i.e. time and temperature), PCR target size and composition, primer sequences, and sample purity.
969 As Ms Norbury noted, the authors of Heid et al 1996 used ten replicates of a sample and measured the CT of each sample. They found that the highest CT difference between the samples was 0.85. Because of the exponential nature of PCR (i.e. each cycle results in a doubling of the amount of target DNA sequence), a CT value of 1.0 represents a 2-fold difference in the amount of target sequences. Therefore, even between replicate samples of the same material, amplified using the same primer, the variation observed in the amount quantified due to the factors identified was approaching 2:1.
970 Professor Lovett therefore pointed to Timothy M Woudenberg and J Stevens, “Quantitative PCR by Real Time Detection” (Proceedings SPIE 2680, Ultrasensitive Biochemical Diagnostics, 1 April 1996) (referred to earlier as Woudenberg 1996) as being a better example of the ability of real time qPCR to detect a 2:1 difference. Professor Lovett described the experiment in Woudenberg 1996 as follows:
… what you’re doing here is you’re following the cycles of the PCR and you’re seeing fluorescence coming along and it’s getting higher and higher for a particular product, and then it plateaus at the end. And what these guys are doing here is they’ve got this fancy machine which is the first prototype, and they’re showing that you can see, very reproductively, a twofold difference in all their standards.
971 As noted by Professor Lovett, the authors ran through “loads and loads of replicate measurements” and found that the reproducibility of replicate samples was “more than sufficient to discern factors of two in starting copy”. However, as with Heid et al 1996, the work reported in Woudenberg 1996 was assessing the ability to discern a 2:1 difference between serial dilutions of the same starting material, which was amplified in the same reaction conditions, using the same primers. That starting material was a sample of human male DNA, i.e. it was a homogenous sample. As noted by Professor Lovett, “I know full well that you can’t use a quantitative PCR machine to measure mixtures” such as mixtures of maternal and fetal DNA. But Professor Lovett did consider that a comparison could be performed across multiple markers:
MR RYAN: Yes. And if we just move through this hypothetical now. You’ve got your markers – you’re undertaking a real-time PCR assay – you’ve got to undertake separate assays in respect of each marker. Is that correct?
PROF LOVETT: Yes.
MR RYAN: Yes. So when you’re dealing, as you are here, with a mixture of maternal and foetal cell-free DNA, you have the difficulty, don’t you that, you don’t know whether you started off with the same amount of foetal DNA in each sample?
PROF LOVETT: You’re doing this all relative to two markers or more on the same sample. It doesn’t matters that samples vary between each other because you’re measuring everything relative within that one.
MR RYAN: Well, now, let’s see – you see – let’s assume you see a difference between the two assays. How do you tell whether that difference is because of different amounts of efficiency of the assays, different amounts of starting materials or difference in dosage. How do you ascertain that?
PROF LOVETT: Well, you can do essentially what was done in the patent which is to have an internal control and you would definitely run that anyway and you would probably put in multiple internal controls which are going to PCR up everything – foetus and maternal.
972 Woudenberg 1996 also noted that whilst the technique could be used with “exceedingly low copy number”, its “reproducibility [was] limited by molecular shot noise”. Professor Lovett was not sure what that term meant, but Ms Norbury explained that it was “noise on the baseline” or “amplification artefacts”.
973 Woudenberg 1996 discussed the possibility of running a multiplex PCR, and the authors emphasised that “this does not present a problem as long as the two reactions are close in initial copy and efficiency”, as well as the need for two probes to hybridise with “perfect complimentarity [sic]”. Further, the multiplex contemplated by Woudenberg appears to have been “time-multiplexed”, i.e. multiple PCR reactions were conducted at the same time across a sample block as depicted in Figure 1 of Woudenberg 1996.
974 According to Ariosa, taken together, Heid et al 1996 and Woudenberg 1996 demonstrate that as at the priority date, real time qPCR was a very new technology. And to the extent that it had the sensitivity to distinguish a 2:1 difference in the amount of genomic material, at best it was only in the case of:
(a) a homogenous sample of genomic DNA, i.e. not DNA from two different sources, and particularly not a mixture of DNA in which the target DNA was only a very small fraction of the total DNA; and
(b) a comparison between serial dilutions of the same genetic sequence, which is achieved using the same primer (i.e. a primer for the β actin gene) to amplify that sequence in increasingly dilute samples, under precisely the same reaction conditions.
975 I agree with Ariosa’s contentions.
976 Further, Professor Lovett accepted that the comparison contemplated in paragraph (b) on page 5 of the Patent would need to be undertaken by running and comparing the results of two separate assays if attempting to use real time qPCR. But where real time qPCR or indeed any qPCR technique is performed in separate assays, then all of the parameters identified in Heid et al 1996 come into play, including the size of each sequence being targeted for amplification, the design of the primers needed to target those sequences, potential contamination, different reaction conditions (even where conditions are carefully controlled) and the different amounts of starting material (i.e. the amount of cffDNA from the targeted chromosome) in each assay. As Ariosa submitted, the issue of having different amounts of starting material in each assay is particularly difficult to control in samples of maternal plasma and serum, which contain a non-homogenous and non-uniform mixture of fetal and maternal cfDNA, where the fetal cfDNA is present in a low concentration. Professor Fisk appeared to acknowledge that the non-homogenous nature of the maternal plasma/serum sample can result in replicate samples having different amounts of cffDNA.
977 According to Ariosa, given these factors, there is just no way of knowing whether any difference observed between two assays is partly or wholly attributable to different efficiencies between the assays, different amounts of starting materials or an actual difference in dosage. I tend to agree.
978 Now Professor Lovett suggested that a person skilled in the art could “do essentially what was done in the patent which is to have an internal control and you would definitely run that anyway and you would probably put in multiple internal controls which are going to PCR up everything – foetus and maternal”. But it is unclear what Professor Lovett was referring to in terms of the internal control.
979 Further, Ariosa says that at best, the Patent discusses the use of a calibration curve, which was run in parallel and in duplicate with each analysis. As Ms Norbury states, “we’ve established that the efficiency of different reactions varies and … in the patent, I don’t think there’s any example where they run a multiplex. They’re always running two separate assays”. But there is no suggestion in the Patent that markers for either the SRY or beta-globin gene could be included in the same real-time qPCR reaction.
980 Further, Ms Norbury gave evidence that in order to implement the comparison described at page 5, paragraph (b) of the Patent, in addition to certain other requirements:
A real-time PCR experiment would have been set up in which the polymorphic marker alleles of interest can be identified against the background of the maternal alleles. The two markers would be analysed in parallel. The theory would be that a 2:1 ratio would be observed in an aneuploid pregnancy whereas a normal pregnancy would be expected to result in a 1:1 ratio of the two alleles, in both cases against a large background of maternal DNA.
However I do not think that it would have been possible to get a real-time PCR assay to provide a reliable comparison of the concentrations of the two fetal chromosome markers in 1997 (assuming that all the steps I have identified above could have been successfully carried out) such that the assay could have been successfully used to detect an aneuploidy pregnancy. There would, for example, be too many variables to take account of and balance as part of the experiment, as each case might have a different combination of markers and alleles, each with their own sensitivity (the relative differences in sensitivity might be similar to the imprecision of the dosage change under investigation) - it would be necessary to run a number of replicates and I would not expect to achieve a robust dosage measurement in every case.
981 Moreover, in Gibson UEM et al, “A Novel Method for Real Time Quantitative RT-PCR” (1996) 6(10) Genome Research 995-1001 (previously referred to as Gibson 1996) the authors acknowledged that whilst it was theoretically possible to simultaneously amplify two RNA markers in the same reaction tube, there were numerous practical difficulties with doing so, which is why the authors ran separate reactions for the two different markers one of which was an internal control of a known amount of RNA.
982 Now Example 5 of the Patent does describe the use of real time qPCR to quantitate the amount of cffDNA from the Y chromosome (using the SRY primer) and the total cfDNA (by targeting a sequence from the beta-globin gene). Two separate assays were conducted, the first using the SRY TaqMan system, and the second using the beta-globin TaqMan system. The concentrations of the SRY gene and the beta-globin gene were determined from these two assays and then compared. Now the inventors did not appear to control for the factors described above, but nor did they need to. The purpose of Example 5 was to establish a basic protocol for estimating the level of cffDNA present in maternal serum relative to total cfDNA. This comparison was not concerned with accurately measuring the relative dosage of two sequences in order to diagnose an aneuploidy. It was also not concerned with trying to quantitate the relative dosage of two target fetal sequences that both represent only a very small percentage of the total DNA. And as Ms Norbury emphasised:
So when you’re looking at low levels, you don’t have the sensitivity to discriminate those differences in quantity.
983 Further, despite Professor Fisk’s evidence of the apparent ease of applying real time qPCR to the quantitation of fetal DNA as at the priority date, a group associated with Professor Fisk experienced a number of challenges in the application of real time qPCR to the detection and quantification of cffDNA, well after the priority date. This work is reported in Birch L et al, “Accurate and Robust Quantification of Circulating Fetal and Total DNA in Maternal Plasma from 5 to 41 Weeks of Gestation” (2005) 51(2) Clinical Chemistry 312-20 (Birch 2005), of which Professor Fisk is a co-author, as well as in a poster presentation in 2004. Professor Fisk agreed that as identified by the authors of the paper and the poster, “fetal DNA is at very low level in samples”. He also agreed with the concern expressed by the authors arising from the “non-homogenous distribution of the DNA” such that “[i]n one sample … you might have a certain amount of fetal DNA, but because it’s at such low levels, the next sample might have a different relative quantification”. Professor Fisk confirmed that to attempt to address these concerns, the authors pushed the PCR cycles beyond 40 cycles being the number of cycles recommended by the manufacturer. But the greater the number of PCR cycles required to detect amplification of fetal DNA, the greater the chance of amplifying something other than the target.
984 Further, the work described in Birch 2005 was not attempting to use real time qPCR to quantitate and compare the relative levels of two different fetal markers from two different chromosomes. The authors were attempting to implement a similar method as described in Example 5 of the Patent, namely measuring the amount of cffDNA based on the SRY gene, and the total cfDNA based on the “the housekeeping gene GAPDH” (at 313). The authors also note that “both targets were detected independently in singleplex reactions to prevent outcompetition of the SRY amplification” (at 313), which is the same as in Example 5 of the Patent in which the SRY and beta-globin genes were quantified in separate real-time qPCR reactions.
985 Finally, I note that in Birch 2005 the following was also said:
To investigate another potential route, we carried out STR analysis on extracted plasma to detect fetal DNA in the maternal samples by identification of paternally inherited alleles. For most loci, either one or two majority peaks should be seen, corresponding to homozygous or heterozygous maternal targets at each locus. When fetal DNA is present in detectable quantities, an additional minority peak corresponding to the paternally inherited fetal allele may also be seen, if distinguishable by size from the two maternally derived targets. This approach could also be used in detection of expanded repeat mutations. In practice, however, we found that the minority fetal DNA was detectable only when present at high concentrations, as determined by use of artificial mixtures of known amounts of female and male genomic DNA (Fig 4). The 112-bp amelogenin male-specific amplicon was detectable only in the 2:1 mixture of female to male DNA, whereas the female 106-bp amplicon was clearly seen in all samples. Informed by the results from the artificial mixtures, we tested third-trimester plasma samples because earlier samples did not have sufficient fetal DNA to allow detection by STR analysis. The samples analyzed ranged from 29 to 41 weeks of gestation, but only a single weak 112-bp amplicon was detected, in the male 41-week sample shown (Fig 4).
(Figures omitted.)
986 Let me draw together some conclusions in terms of making a quantitative comparison of chromosomal markers.
987 In my view, it is apparent from the evidence, including the technical papers relied on by Professors Fisk and Lovett that at best the implementation of the approach suggested in paragraph (b) on page 5 of the Patent would have involved an extensive amount of work in the nature of a research project. Further, even with a significant amount of work, it is doubtful that the method could have been implemented as at 1997.
988 First, all of the experts agree that as at 1997, this approach could not be used in the case of maternally inherited aneuploidies, which account for 95% of Down syndrome cases. The clear evidence of Professors Oepkes and Hyett was that any approach which excludes maternally inherited aneuploidies does not provide a screening test and would not be useful.
989 Second, to detect Down syndrome, it would be necessary to identify alleles on both a reference chromosome and chromosome 21 that were informative as between the mother and the fetus. This would require prior genotyping of the mother and father, and in many cases, an informative allele may not be found. This is particularly the case when looking for informative RFLPs or SNPs, which are biallelic and inherently less informative than STRs. In each case the mother must be homozygous (e.g. AA) for a particular SNP, and the fetus either homozygous for a different SNP or heterozygous (i.e. BB or AB). Moreover, neither Professor Lovett nor Professor Fisk identified any particular RFLPs or SNPs that were known and could have been used in real time qPCR as at the priority date. Professor Lovett conceded that “one of those optimisations is to optimise the markers you choose. So you run them and you run them and you run them until you get very used to the artefacts. And you throw away things that repeatedly give you artefacts”.
990 Third, it would be necessary to design primers targeting each of these identified genetic markers that amplified with the same or comparable efficiency. Now whilst Professor Lovett made a number of assertions regarding running “hundreds of markers” to achieve this optimisation, the practical and technical difficulties associated with this are demonstrated by the Pertl papers. These papers demonstrated that the authors struggled to optimise a small number of STR markers which, for example, resulted in different sized fluorescent peaks for markers that were theoretically present in the same concentration in the sample. Further, the ability to optimise primer design was restricted by the inherent nature of the genomic sequence surrounding the target marker.
991 Fourth, there would need to be enough cffDNA present in the maternal plasma or serum sample to allow detection of any cffDNA.
992 Fifth, cffDNA exists against a very high background of maternal cell-free DNA, where cffDNA is anywhere between 1 to 2% and 10% of the total cell-free DNA. Even if the level of cffDNA were as high as 10% of the total cell-free DNA present in a sample of maternal plasma or serum, the additional paternally inherited allele will represent only 5% of the total cell-free DNA. Using the substandard, semi-quantitative qPCR methodology described in the Pertl papers, it would not have been possible to resolve this difference.
993 Sixth, real time qPCR was very new technology at the priority date, and there is no evidence that it had been used to undertake the quantitative comparison contemplated in paragraph (b) on page 5 of the Patent. The evidence showed that it had been validated to detect a 2:1 difference in the amount of DNA present in increasingly dilute samples of the same DNA sequence, coming from the same starting material, and using the same primer in the same reaction conditions. This appears to have been the limit of the technology as at the priority date. Further, the experts agreed on the variability in efficiency of PCR assays, and that the comparison contemplated in paragraph (b) on page 5 would require separate real time qPCR assays to be run. But given factors such as the low concentration of cffDNA in maternal plasma/serum, the high concentration of maternal cell-free DNA in maternal plasma/serum, the non-homogenous nature of the mixture of fetal and maternal cell-free DNA, the variability in the amount of starting material i.e. the amount of cffDNA from assay to assay, the difficulty of designing multiple primers with comparable efficiency and the inherent limitations of the real time qPCR technology at the priority date, it was problematic to use real time qPCR in the way suggested by paragraph (b) on page 5 of the Patent.
994 Further, as Professor Lovett noted:
The patent is 1997, right. PCR in 1997 is a very different thing than PCR in the 2010s. … the stuff in the patent in 1997 is state of the art PCR.
995 Seventh, Professor Lovett also suggested that as at the priority date it would have been possible to detect XYY aneuploidy, although accepting that the analysis required for each pregnancy would be expensive and time consuming. But this is in contrast with the evidence of Ariosa’s experts in the joint expert report. This approach still depends on finding multiple informative markers on another reference chromosome and comparing the amount of the Y markers to the paternally inherited alleles on the reference chromosome. Professor Lovett agreed that the only relevant difference between detection of XYY and detection of a paternally inherited aneuploidy is that you are only “removing one problem … that is the problem of finding an informative marker on the problem chromosome”. But this suggestion otherwise suffers from the same problems identified earlier.
996 Indeed, Pertl 2000 noted that the sequences the authors were amplifying in the reported experiments had a “lower sensitivity for detecting fetal DNA in maternal plasma than the SRY system described by Lo et al”. The reason for this is suggested to be (at 48):
This lower level of sensitivity may be due to the nonselective nature of PCR amplification of STRs in that both the target (i.e., the fetus’) and the background (i.e., the mother’s) sequences are amplified together (Lo et al. 1996). Under these conditions, the excess of the background sequences could out-compete the rare target sequences for amplification.
997 Accordingly, any attempt to implement the approach suggested in paragraph (b) on page 5 of the Patent to detect the XYY aneuploidy would suffer from the additional difficulty that amplification of Y chromosome markers would be substantially more efficient than the amplification of markers from a non-Y paternally inherited reference chromosome, making any comparison even more difficult.
998 Professor Hyett further noted that the XYY aneuploidy accounts for about 0.5 per cent of all forms of aneuploidy that he sees as a clinician. Professor Fisk explained that sex chromosome aneuploidies can have less clinical significance than other aneuploidies, and that there could be people sitting in the court room with XYY who would not know that they had it.
999 Eighth, the difficulties associated with implementing the approach suggested in paragraph (b) of page 5 of the Patent are also reflected in the fact that a method of using cffDNA to screen for aneuploidy was not developed until 2007. As Ms Norbury explains:
The difficulty of applying the idea presented in the Patent, that is the detection of cell-free fetal DNA in maternal serum, to identify a fetal aneuploidy is reflected in the fact that it was not until 2007 that Professor Lo’s group reported that they had developed a methodology.
1000 Professor Oepkes also explained:
… I think that the PCR was ultimately not used to detect or predict or screen for trisomies using maternal plasma DNA. The clinical application had to wait for completely different techniques like the next generation sequencing more than 10 years later …
1001 Further, Professor Lo’s paper Lo YM et al, “Digital PCR for the molecular detection of fetal chromosomal aneuploidy” (2007) 104(32) Proceedings of the National Academy of Sciences of the United States of America 13116-21 included the following statements in its introduction:
The discovery of cell-free fetal DNA in maternal plasma in 1997 offered new possibilities for non-invasive prenatal diagnosis. This method has been readily applied to sex-linked and certain single-gene disorders, but its use for fetal chromosomal aneuploidies has been a challenge. First, fetal nucleic acids coexist in maternal plasma with a high background of maternal nucleic acids that can often interfere with analysis. Second, fetal nucleic acids circulate in maternal plasma in a cell-free form, making it difficult to derive chromosome dosage information.
(Citations omitted.)
1002 Further, in the early 2000s, Professor Hyett, together with other leading researchers in the field, had given much consideration as to how to screen for chromosomal aneuploidies using the approach suggested in paragraph (b) on page 5 of the Patent. But they formed the view that it was not possible or feasible to implement this method at that time.
1003 Indeed, Professor Hyett gave evidence that:
… In my view, there was no technology available at this time that could accurately define the quantities of DNA with a degree of resolution that would allow this type of diagnosis.
1004 In cross-examination, Professor Hyett also emphasised that:
If you look at the Pertl paper, which looks at a series of nine different STRs, when they used all of those STRs together, they had an 85 per cent diagnostic – rate of diagnostic accuracy, so they didn’t have a 100 per cent rate of diagnostic accuracy. So that, again, limits the value of that tool in clinical practice.
It means that you would have to inform your patient that you have a test available to them but 15 per cent of the time you would have the wrong answer. And that then only applies to 10 per cent of the population of Down foetuses. I think, again, when we looked at the Pertl paper and we looked at this issue of dosage, we saw how variable dosage can be when you look – it’s expressed in that format in terms of quantitation. And, again, I think that that makes it highly unlikely that this will be a useful clinical tool in this setting.
Summary and claims
1005 In summary, Ariosa says that a person skilled in the art could not as at the filing date have performed a method of prenatal diagnosis based on the quantity of cffDNA in a sample of maternal plasma or serum. Ariosa says that the approach suggested in paragraph (a) on page 5 of the Patent, and in Examples 2 and 4, does not work, as the asserted correlation between elevated levels of cffDNA and aneuploidy (or pre-eclampsia) has not, to this day, been proven, and this method has never been able to be put into practice. Further, Ariosa says that the approach suggested in paragraph (b) on page 5 of the Patent was not feasible with the technologies and techniques then available. So, Ariosa says that despite suggesting these two approaches, the disclosure in the Patent did not enable the skilled addressee to practise such a method without new inventions or additions or prolonged study of matters presenting initial difficulty.
1006 In terms of the claims, in summary Ariosa has contended the following.
1007 Claims 1 to 3 and 5 all relate to a detection method, with claims 2, 3 and 5 specifying particular laboratory techniques to be used as part of the detection method. Each of those claims contemplates use of the said method as part of a diagnosis resulting from a qualitative embodiment, as well as from the quantitative embodiment or embodiments. Insofar as those claims encompass the quantitative embodiments, they are not fully described, for the reasons discussed above.
1008 Claims 6 and 9 specify, respectively, that the methods of detection involve either a nucleic acid sequence from the Y-chromosome or from a paternally-inherited non-Y-chromosome. Each of those claims encompasses within its scope both the qualitative and quantitative embodiments discussed above. For claim 6, this is reinforced by the presence of claim 13, which limits the method of claim 6 to a qualitative embodiment, by specifying that the detection of a fetal nucleic acid sequence from the Y-chromosome is for the purpose of determining the sex of the fetus.
1009 Claim 14 expressly encompasses the first of the quantitative embodiments discussed above, involving the correlation of the fetal nucleic acid concentration to a medical condition. But even if claim 14 were to be construed as encompassing both quantitative embodiments, for the reasons discussed above, there is not a full description of either embodiment.
1010 Claim 22 encompasses both the qualitative and quantitative embodiments. This is reinforced by the drafting of the claim, which in subparagraph (iv) expressly refers to providing a diagnosis based on the presence, sequence or quantity of the fetal nucleic acid. Claim 23 is dependent on claim 22, and does not include any relevant limitation. Insofar as claims 22 and 23 encompass the quantitative embodiment(s), they lack full description.
1011 Claims 25 and 26 encompass both the qualitative and quantitative embodiments, and accordingly lack full description.
(b) Conclusions
1012 I would reject Ariosa’s lack of sufficiency argument, notwithstanding that it has made some very cogent arguments on the science. I have set out the evidence on the science and my views about it just in case I am wrong on the law and my conclusions in construing the Patent.
1013 In my view there is only one invention around which the claims are drawn. This is the method of detecting cffDNA in an artificially created plasma or serum sample isolated from blood extracted from a pregnant female. The claims are drawn around this invention to include particular applications, ranging from detection of Y sequences, detection of sequences for a particular purpose and quantification of the detected sequences. The examples are expressly stated to be examples of the one invention.
1014 The relevant claims cover in substance and in form a method producing one outcome, namely, detection of a particular specified sequence, for example Y (claim 6) and non-Y (claim 9). Further, claims 1 to 3, 5, 6, 9 and 14 do not require a diagnosis to be made on the basis of a process of relative or absolute quantitation of cffDNA.
1015 Further, the patent enables a person skilled in the art to detect cffDNA on Y and non-Y chromosomes for the purposes of, for example, sex and RhD status determination.
1016 In my view the Patent provides sufficient information to enable a skilled person to produce something within each of the relevant claims without new inventions or additions, or prolonged study of matters presenting additional difficulty.
1017 Now Ariosa’s lack of sufficiency attack has significantly focused on the question of whether a person skilled in the art could detect or diagnose aneuploidy and/or pre-eclampsia at the priority date. But the aneuploidy and pre-eclampsia examples are only two of a wide range of uses for the invention described and claimed in the Patent. By way of illustration, in the definition of prenatal diagnosis on page 2 of the Patent “aneuploidy detection” is listed as a subset of an example comprising the detection of fetal abnormalities and pre-eclampsia is the last mentioned example in the definition.
1018 Further, the aneuploidy and pre-eclampsia examples of the Patent are described in very qualified terms as potential future applications requiring further investigation and development. Let me elaborate.
1019 The Patent describes two possible ways the invention might be applied to screening for Down syndrome and other chromosomal aneuploidies at page 5, the first the subject of Example 2 and a second not discussed in any further detail in the Patent.
1020 At page 6, lines 3 to 5, the Patent states that it is anticipated that it will be possible to incorporate the nucleic acid-based diagnosis methods described therein into existing prenatal screening programmes.
1021 The Patent states at page 14, lines 18 to 25 that the results of Example 2 indicate that fetal DNA quantitation has the potential to be used as a new screening marker for fetal chromosomal aneuploidies, that a “large scale population-based study could be carried out to develop cut-off values for screening purposes”, and that it “would also be useful to investigate the correlation of foetal DNA concentration with other biochemical markers for maternal serum biochemical screening”.
1022 At page 22, line 20 to page 23, line 3, the Patent states that the results of Example 2 indicate that fetal DNA concentration measurement in maternal plasma “may be used as a new marker for pre-eclampsia” and that “[f]urther research will be required to investigate whether the level of foetal DNA is related to the severity of pre-eclampsia. Our discovery also opens up research into the potential application of foetal DNA quantitation to predict the occurrence of pre-eclampsia, prior to the development of clinical signs such as hypertension and proteinuria”.
1023 At page 33, lines 4 to 6, the Patent states that it is also likely that increased amounts of fetal DNA may be found in conditions associated with placental damage, such as pre-eclampsia. It then states that “[t]he real time quantitative PCR system described here offers a powerful tool to study these unexplored pathophysiologic aspects of foetal DNA in maternal plasma and may improve our understanding of the fetomaternal relationship” (page 33, lines 6 to 10).
1024 Contrastingly, the following statements are made in the Patent regarding sex determination (relevant claims excluding claim 9) and RhD status (relevant claims excluding claim 6):
(a) the method according to the invention may be “particularly useful” for sex determination, “it is demonstrated herein that using only 10µl of plasma or serum a detection rate of 80% for plasma and 70% for serum can be achieved” (page 3, line 29 to page 4, line 4);
(b) “Sex determination has successfully been performed on pregnancies from 7 to 40 weeks of gestation” (page 6, line 5);
(c) “In [Example 3], we have demonstrated the feasibility of performing non-invasive foetal RhD genotyping from maternal plasma. This represents the first description of single gene diagnosis from maternal plasma” (page 19, lines 6 to 9);
(d) “The high concentration of foetal DNA in maternal plasma and serum has allowed us to reliably detect the presence of foetal genetic material” (page 30, lines 18 to 20); and
(e) “we were able to detect foetal SRY sequences as early as the 7th week of gestation” (page 31, lines 10 to 11).
1025 I agree with Sequenom that even if Ariosa can establish that its asserted quantitative diagnosis embodiments were not feasible at the priority date, and I must say that Ariosa is strong on the science on this aspect, it does not follow that any of the relevant claims (as opposed to the specific aneuploidy claims 16 and 19 that are not asserted) lack sufficiency. A person skilled in the art could perform something falling within the scope of each of the relevant claims in any event.
1026 Let me now deal with each of the claims in turn.
Claims 1, 6 and 9
1027 It would have been straightforward for a person skilled in the art to perform such methods at the priority date using the information disclosed at, for example, page 1, line 1 to page 5, line 2 of the Patent, along with the common general knowledge. The Patent not only informs the person skilled in the art that cffDNA is present in detectable amounts in the maternal plasma and serum of a pregnant female, but also presents a method by which this DNA may be detected, namely PCR.
1028 Further, an example of a method falling within claim 1 would be the positive identification of a sequence from the Y chromosome in maternal plasma or serum, as such a sequence obviously could only be of fetal origin.
1029 Further, each of the Examples in the Patent discloses information that would have enabled a person skilled in the art at the priority date to perform the method described in these claims.
1030 Further, it would have been straightforward at the priority date to detect fetal DNA by testing for the presence of a sequence specific to the Y chromosome. These assays were well established by 1997.
1031 Further, the method of the invention could readily be used to determine RhD status by detecting cffDNA from the RhD gene located on chromosome 1. Shortly after the publication of the Patent, researchers were describing the analysis of fetal DNA extracted from maternal plasma for RhD status detection as being advantageous given it was rapid, robust and easy to perform.
1032 Further, the experts agreed that in addition to implementing the “obvious Example (a) on page 4 of fetal RhD in Rh negative mothers” involving the detection of cffDNA on chromosome 1 they could have implemented a method to detect the presence of a paternally inherited polymorphism or mutation present on a Y or a non-Y chromosome (claims 1, 6 and 9). However, this agreement was qualified in some respects.
Claims 2, 3 and 5
1033 Amplification (claim 2), PCR (claim 3) and the use of a “sequence specific probe” (claim 5) was standard at the priority date.
Claim 14
1034 Claim 14 refers to “determining the concentration of foetal nucleic acid sequence in the maternal serum or plasma”. An example of this method is described in Example 5 of the Patent.
1035 Clearly, in Example 5 real-time qPCR was used to perform a method of claim 14 involving the quantification of a sequence from the Y chromosome and the quantification of a sequence present in both the mother and the fetus (beta-globin). And a person skilled in the art at the priority date could have replicated the study described in Example 5. As Sequenom points out, this single application alone is sufficient to enable claim 14.
Claims 22, 23, 25 and 26
1036 Ariosa does not allege that claim 13 (determining the sex of a fetus) is invalid pursuant to s 40(2)(a). But determining the sex of a fetus is plainly a kind of prenatal diagnosis (claims 22, 23, 25 and 26) and the detection of Y sequences is indicative of fetal sex, being a fetal characteristic. Accordingly, something within each of the relevant claims has been enabled. Indeed, multiple methods falling within those claims have been enabled including diagnosis of RhD status.
1037 Finally, much has been made by Ariosa of the Tramanco point discussed by Nicholas J. But in my view it does not assist Ariosa. Each of the method claims that I am considering are in form and in substance not expressed as having variable parameters selected from say A, B and C such that the claimed method permitted one parameter out of A, B or C, combinations of two parameters A+B, A+C, B+C, or all three parameters A+B+C. Each of the method claims that I am considering do not stipulate that optionality. Each relevant claim is stipulated as one method.
1038 In summary, Ariosa has not established a lack of sufficiency.
INTERNAL FAIR BASIS
1039 Section 40(3) requires a real and reasonably clear disclosure in the body of the specification of what is claimed. The language of s 40(3) points to a comparison between the claims and what is described in the body of the specification only. As stated by Barwick CJ in Olin Corporation v Super Cartridge Co Pty Ltd (1977) 180 CLR 236 at 240:
the question is a narrow one, namely whether the claim to the product being new, useful and inventive, that is to say, the claim as expressed, travels beyond the matter disclosed in the specification.
1040 Section 40(3) does not use the word “invention”, but it requires that the claims “be fairly based on the matter in it that discusses the ‘invention’”; Lockwood (No 1) at [53]. This is the embodiment(s) which is described and around which the claims are drawn, but it does not mean the inventive step taken by the inventor or the advance in the art made by the inventor. Lockwood (No 1) described the relevant test in the following terms (at [68] and [69]):
Erroneous principles. The comparison which s 40(3) calls for is not analogous to that between a claim and an alleged anticipation or infringement. It is wrong to employ “an over meticulous verbal analysis”. It is wrong to seek to isolate in the body of the specification “essential integers” or “essential features” of an alleged invention and to ask whether they correspond with the essential integers of the claim in question.
“Real and reasonably clear disclosure”. Section 40(3) requires, in Fullagar J’s words, “a real and reasonably clear disclosure”. But those words, when used in connection with s 40(3), do not limit disclosures to preferred embodiments.
“The circumstance that something is a requirement for the best method of performing an invention does not make it necessarily a requirement for all claims; likewise, the circumstance that material is part of the description of the invention does not mean that it must be included as an integer of each claim. Rather, the question is whether there is a real and reasonably clear disclosure in the body of the specification of what is then claimed, so that the alleged invention as claimed is broadly, that is to say in a general sense, described in the body of the specification.”
Fullagar J’s phrase serves the function of compelling attention to the construction of the specification as a whole, putting aside particular parts which, although in isolation they might appear to point against the “real” disclosure, are in truth only loose or stray remarks.
(Footnotes omitted, save that the italicised phrase is drawn from Rehm Pty Ltd v Websters Security Systems (International) Pty Ltd (1988) 81 ALR 79 at 95 per Gummow J.)
1041 So, one should not use an “over meticulous verbal analysis”. Further, the focus is not on an identity of language between the claims and the disclosure in the body of the specification. Rather, one is looking for a generalised disclosure in the body that provides support for the claims in substance. Moreover, it is inappropriate to isolate in the body of the specification essential integers or features of an alleged invention and to ask whether they correspond with the essential integers of the claim.
1042 Now fair basis can be established by a comparison with the consistory clause(s). But even if a claim is based on and mirrors the form of the consistory clause(s), it will not be fairly based if other parts of the specification show that the invention is narrower than the consistory clause(s). What has been described as a “coincidence of language” between a claim and part of the body of a specification does not per se establish fair basing if that part of the language of the specification does not reflect the description of the invention in the light of the specification as a whole.
1043 Further, it is appropriate to state that the complete specification is not to be read in the abstract, but is to be construed in the light of common general knowledge and the relevant art before the priority date.
1044 Further, where a feature included in a claim is a limiting feature, there is no need for it to be the subject of an explicit disclosure in the body of the specification, if the subject matter of the claim falls within the scope of what is more broadly described in the specification. As explained in DSI Australia (Holdings) Pty Ltd v Garford Pty Ltd (2013) 100 IPR 19; [2013] FCA 132 at [240] per Yates J:
… the inquiry as to fair basis is directed to the question of claim width: see, for example, Olin Corporation at CLR 240 ... A claim may be fairly based for the purposes of s 40(3) of the Act where it adds a feature to a combination otherwise described in the specification and, by that addition, limits the described invention, as a matter of definition, to a more restrictive form than that to which the patentee might otherwise be entitled. In short, a claim may be fairly based for the purposes of s 40(3) of the Act even when all the characteristics by which the invention is defined in the claim are not described in the body of the specification itself, provided those characteristics are truly limiting ones in the sense that I have described.
1045 Relatedly, the claims need not be restricted to precise embodiments described in the specification. As Gummow J said in Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479; [1994] FCA 936 at 497:
it is no objection to any particular claim that it claims a monopoly for less than every feature described in the body of the specification. It cannot be the case that, for example, a claim is restricted to the precise embodiment which is depicted in the body of the specification.
1046 Ariosa says that the invention disclosed in the specification of the Patent is at its broadest a method of prenatal diagnosis involving detecting cffDNA. It follows that each of claims 1 to 3, 5, 6, 9 and 14 is not fairly based as none of those claims is limited in scope to use in a method of providing a prenatal diagnosis. They are claims to detection methods at large involving detecting cffDNA.
1047 Ariosa says that there are cases where the detection of a nucleic acid of fetal origin does not provide any information about a condition or characteristic of the fetus or a pregnant woman.
1048 Further, Ariosa says that insofar as the Patent discloses that a diagnosis may be undertaken by reference to detected cffDNA, the substance of what is disclosed as an invention does not extend beyond the identification of a DNA sequence that is possessed by the fetus but not by the mother and the use of a primer or probe to target that sequence in order to detect the presence of the identified fetal nucleic acid.
1049 Ariosa says that such disclosure underpins, and is expressly described in, each of the examples in the Patent, including the quantitative examples. Each of the qualitative techniques described on pages 4 to 5 of the Patent, and which form the basis of the experiments reported in two of the Examples, depends on detecting the presence of cfDNA that cannot originate from the mother, and therefore must be fetal. Further, the quantitative technique described at paragraph (a) of page 5 of the Patent is exemplified in one of the Examples of the Patent using a Y chromosome primer, and plainly cannot be possessed by the mother. And the quantitative technique described at paragraph (b) of page 5 of the Patent is said to involve “foetal DNA markers on different chromosomes”; the experts agree in the joint expert report that “fetal DNA markers” means “[p]olymorphisms present in the fetus and not in the mother”.
1050 But Ariosa says that claims 1 to 3, 5, 9, 14, and 22 to 26 are not limited to such an approach. They are so broad that they include within their scope the detection of fetal nucleic acid even if that nucleic acid is possessed by the mother (as well as the fetus). Those claims therefore lack fair basis.
1051 Separately, Ariosa says that claim 26 is a claim that includes within its scope analysis of any nucleic acid, including maternal nucleic acid (cell-free DNA), which extends beyond what is disclosed as an invention in the Patent. Whilst there is a definition of “prenatal diagnosis” on page 2 at lines 24 to 27, it is an inclusive definition and accordingly does not limit the scope of the claims, particularly when it is the only claim that excludes an express limitation to the nucleic acid being of fetal origin. Accordingly it says that claim 26 is not fairly based.
1052 I would reject Ariosa’s attack save as to claim 26.
1053 The Patent as a whole describes an invention comprising the detection of cffDNA in an artificially produced plasma or serum sample prepared from blood extracted from a pregnant female. Let me set out some of the relevant parts of the specification that make this point good.
1054 At page 2, lines 5 to 19 the Patent explains that:
It has now been discovered that foetal DNA is detectable in maternal serum or plasma samples. This is a surprising and unexpected finding; maternal plasma is the very material that is routinely discarded by investigators studying non-invasive prenatal diagnosis using foetal cells in maternal blood. The detection rate is much higher using serum or plasma than using nucleated blood cell DNA extracted from a comparable volume of whole blood, suggesting that there is enrichment of foetal DNA in maternal plasma and serum. In fact, the concentration of foetal DNA in maternal plasma expressed as a % of total DNA has been measured as from 0.39% (the lowest concentration measured in early pregnancy), to as high as 11.4% (in late pregnancy), compared to ratios of generally around 0.001% and up to only 0.025% for cellular fractions (Hamada et al 1993). It is important that foetal DNA is found in maternal plasma as well as serum because this indicates that the DNA is not an artefact of the clotting process.
(Emphasis added.)
1055 At page 2, lines 20 to 24, the Patent confirms that the invention is not the discovery of the presence of cffDNA in maternal serum or plasma but rather, “a detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of foetal origin in the sample”.
1056 At page 4, lines 5 to 9, the Patent further explains that “[t]he method according to the invention can be applied to the detection of any paternally inherited sequences that are not possessed by the mother and which may be for example genes which confer a disease phenotype in the foetus”. That is, the invention may be applied to the detection of Y chromosome sequences present in male fetus as well as the detection of paternally-inherited non-Y chromosome sequences possessed by both male and female fetuses.
1057 The example application (c) on page 4 of the Patent expressly refers to paternally-inherited DNA polymorphisms or mutations present on either a Y or non-Y chromosome and the ascertainment of the presence of such sequences “prior to diagnostic analysis such as sex determination”. “Polymorphisms” are not limited to well-known polymorphic markers used in gene mapping and linkage analysis.
1058 At page 31, line 27 to page 32, line 1, in discussing example 5, the Patent explains that the named inventors “envisage[d] that foetal DNA analysis in maternal plasma and serum would be most useful in situations where the determination of foetal-derived paternally-inherited polymorphisms/mutations or genes would be helpful in clinical prenatal diagnosis (Lo et al. 1994)”.
1059 As noted above, page 32 of the Patent also refers to the potential development of polymorphic markers outside the Y chromosome so that the method can be applied to female fetuses.
1060 Generally, these statements properly describe the invention. And contrary to Ariosa’s assertions, the foregoing general disclosure provides support for the relevant claims of the Patent. The broadest claim, claim 1, is commensurate with, and does not travel beyond the matters identified.
1061 Now Ariosa seeks to distil from the Examples described in the Patent:
(a) first, an implied requirement that the sequence detected be known in advance (e.g. by way of prior genotypic or phenotypic analysis) to not be possessed by the pregnant female and asserts that there is no real and reasonably clear disclosure of a detection method that does not satisfy such a requirement; and
(b) second, a requirement that the invention be used as part of a method of providing a prenatal diagnosis.
1062 The first allegation is made against all relevant claims and the second allegation is confined to claims 1 to 3, 5, 9 and 14. Further, Ariosa asserts that to the extent that the invention as claimed in claim 26 is for any “nucleic acid analysis”, there is no real and reasonably clear disclosure of such an invention in the specification of the Patent.
1063 Now as Sequenom characterises it, Ariosa’s first assertion of the requirement for alleged prior knowledge is intended to result in a squeeze between Sequenom’s infringement allegations and the validity of the relevant claims. In particular, Ariosa asserts that to the extent that the claims cover methods like the Harmony Test that do not involve the alleged prior knowledge requirement, such methods are not fairly based on the matter described in the specification.
1064 But the broad description of the invention described earlier does not refer to the alleged prior knowledge requirement. Further, in response to the question in the joint expert report “[d]oes the Patent require that the “nucleic acid of foetal origin” be known in advance to be of foetal origin?”, the experts all said:
No and the Examples in the Patent are consistent with this.
GN wishes to note that defining a nucleic acid as being of fetal origin is always an inference and could be a false positive such as arising from a vanishing twin or from confined placental mosaicism.
1065 Further, it is well apparent that whilst some of the Examples of the Patent involve the detection of Y sequences known in advance of the test to not be possessed by the pregnant female or through prior genotypic or phenotypic analysis, it being easier for skilled persons to detect cffDNA using such a step, the invention described and claimed in the Patent is not so limited.
1066 Further, the invention described in the body of the Patent covers methods wherein steps are taken to discriminate or distinguish between maternal and fetal DNA, such that it may be inferred or discerned that the sequence is of fetal origin. But the Examples “do not in any way limit the scope of the invention” (page 6 lines 16 and 17 of the Patent).
1067 Moreover, I agree with Sequenom that Ariosa’s complaint regarding the alleged prior knowledge requirement is really one of enablement.
1068 For example, Professor Oepkes has said that:
Detecting a nucleic acid of fetal origin in a serum or plasma sample from a pregnant female if I did not have access to the maternal and paternal genomes (in other words, if I was looking for a sequence which I did not already know was possessed by the father and not possessed by the mother) is a problem which is not solved by the text of the Priority Document. As I have discussed above, the approach which the Priority Document discloses involves looking in maternal serum/plasma for sequences which are already known to be paternally inherited and not possessed by the mother. If I did not know this, or could not determine it (by reference to maternal and paternal genetic material) before starting my search for the sequence in question, then I would be stuck. Without that knowledge, identifying the presence of a particular sequence in maternal serum or plasma would not tell me anything about the fetus.
1069 Further, Professor Hyett has explained that “in order for [the RhD] example to work it would first be necessary to know that the mother is rhesus-negative”.
1070 Further, Ms Norbury has said that:
(a) “…The different approach taken by the Harmony Test is not disclosed by or taught by the Patent. Further, in 1997, the Harmony Test’s approach would not have been thought possible, nor in fact would it have been possible with the technology available at the time”; and
(b) “…Further, for any given marker, the paternally inherited allele will represent half the amount of DNA present. For example, if the level of cell-free fetal DNA is as high as 10%, the paternally inherited DNA will represent only 5%. Even if the paternally inherited allele is known (and assuming it is different to the maternally inherited allele), detecting this amount of DNA above the baseline signal is likely to be difficult. Where the paternally inherited allele is not known, it is likely to be very difficult and very often impossible to detect the paternally inherited allele above the baseline of signal”.
1071 But the specification did not need to describe in any greater detail than the detail included in a claim to satisfy s 40(3). It is also not necessary for a patentee to describe or enable every possible means of producing a particular claimed result. There only has to be the enablement of a single embodiment falling within the scope of each claim. In the present case, both the specification and the claims define the invention in terms of a method of detection of cffDNA in a plasma or serum sample prepared from blood extracted from a pregnant female. And the skilled addressee would not read the Patent as being confined to the methods used in the Examples.
1072 Let me now deal with Ariosa’s alleged prenatal diagnosis requirement. Whilst the provision of a “prenatal diagnosis” is stated to be the primary application or field of the invention, there is a general disclosure in the body of a detection method not tied to the diagnosis of a fetal or maternal condition. In particular, as Sequenom points out, the Patent states that:
(a) “[t]his invention relates to prenatal detection methods using non-invasive techniques” (page 1 lines 1 to 3);
(b) in particular, the invention relates to prenatal diagnosis “by detecting foetal nucleic acids in serum or plasma from a maternal blood sample” (page 1 lines 2 to 6);
(c) “[t]his invention provides a detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of foetal origin in the sample” (page 2 lines 20 to 24);
(d) “[t]he method according to the invention can be applied to the detection of any paternally-inherited sequences which are not possessed by the mother and which may be for example genes which confer a disease phenotype in the foetus” (page 4 lines 5 to 8);
(e) the detection of paternally-inherited DNA polymorphisms or mutations present on either a Y or a non-Y chromosome “can also be used to ascertain the presence of foetal nucleic acid in a particular maternal plasma or serum sample, prior to diagnostic analysis such as sex determination” (page 4 lines 27 to 30); and
(f) under example 5, the named inventors “envisage[d] that foetal DNA analysis in maternal plasma and serum would be most useful in situations where the determination of foetal-derived paternally-inherited polymorphisms/mutations or genes would be helpful in clinical prenatal diagnosis” (page 31 line 27 to page 32 line 1).
1073 Further and in any event, the phrase “prenatal diagnosis” is broadly and inclusively defined on page 2 of the Patent as covering the determination of any maternal or fetal condition or characteristic which is related to either the fetal DNA itself or to the quantity or quality of the fetal DNA in the maternal serum or plasma, including the detection of simple mutations. In the context of this definition, the Patent also says at page 3 that “[t]he nucleic acid detected in the method according to the invention may be of a type other than DNA e.g. mRNA”. These references provide support for claims 1 to 3, 5, 9 and 14 of the Patent.
1074 Finally, let me deal with the claim 26 question. Claim 26 of the Patent provides:
A method of performing a prenatal diagnosis on a maternal blood sample, which method comprises obtaining a non-cellular fraction of the blood sample and performing nucleic acid analysis on the fraction.
1075 Sequenom says that the term “prenatal diagnosis” is defined in the body of the specification as covering the “determination of any maternal or fetal condition or characteristic which is related to either the fetal DNA itself or to the quantity or quality of the fetal DNA in the maternal serum or plasma” (page 2 lines 24 to 27). Therefore, the term “prenatal diagnosis” as used in the specification and claim 26 of the Patent inherently requires the detection of fetal DNA. As such, claim 26 does not travel beyond the detection method invention described in the body of the specification. I disagree. The definition of “prenatal diagnosis” is not exhaustive. Accordingly the claim is invalid for want of fair basis.
1076 Accordingly, Ariosa’s fair basis attacks fail save as to claim 26.
FALSE SUGGESTION
1077 Section 138 (3)(d) of the Act provides that the Court may revoke a patent either wholly or so far as it relates to a claim, on the ground that the patent was obtained by false suggestion.
1078 In summary, it is necessary to demonstrate, first, that a suggestion in the application was in fact false and, second, that the suggestion materially contributed to the Commissioner’s decision to grant the patent.
1079 Section 138 explicitly contemplates revocation claim by claim (Apotex Pty Ltd (formerly GenRx Pty Ltd) v Sanofi-Aventis (2008) 78 IPR 485 at [125] per Gyles J (Sanofi-Aventis)). So, as explained by Emmett J in ICI Chemicals & Polymers Ltd v Lubrizol Corp Inc (1999) 45 IPR 577 at [179]:
I do not consider that a representation or suggestion which was not material to the grant of particular claims, could constitute a basis for revocation of those claims, even if the representation or suggestion may have been material to the grant of other claims.
1080 In Prestige Group (Australia) Pty Ltd v Dart Industries Inc (1990) 26 FCR 197 (Prestige) both Gummow J and Lockhart J discussed the expression “false suggestion or representation” as used in s 100(1)(k) of the Patents Act 1952 (Cth). They identified two instances of conduct that could fall within that provision, namely, first, statements in the patent itself and, second, conduct of the patentee during the process of application for grant. Lockhart J said that the rationale for this first class of case is (at 199):
…that it is not generally necessary that the inventor make statements of this kind to have the grant of a valid patent. But if he asserts an inventive merit of his invention and promises a particularly beneficial or useful result, this may persuade members of the public into believing his claims are valid and act on the faith of that by, for example, becoming a licensee or by not using the alleged invention.
1081 Whitford J also said in Intalite International NV v Cellular Ceilings Ltd. (No. 2) [1987] RPC 537 at 547:
…The mere fact that misleading statements may be found in the body of a patent specification – they not infrequently are, for the enthusiasm of those responsible for the compilation of these documents often leads to an over-egging of the pudding – is not sufficient to establish invalidity. What has to be established is a misrepresentation of such materiality that the Crown was misled into granting a patent.
1082 A claim may be said to have been obtained by false suggestion if the suggestion can be inferred to be so material as to have actually misled or deceived the Crown into making the grant of the claim. But it is not sufficient that the representations were merely likely to mislead or deceive the Commissioner. In Gilead Sciences Pty Ltd v Idenix Pharmaceuticals LLC (2016) 117 IPR 252 at [683] Jagot J said at [683]:
it is clear that the question is whether it can be inferred that the Commissioner was actually misled by the representations and not whether the representations were merely likely to mislead or deceive the Commissioner.
1083 The alleged suggestion must have been a material inducing factor. As Lockhart J said in Prestige (at 279 to 280), the issue is:
whether the conduct constituting the false suggestion or representation materially contributed to the Commissioner’s decision to grant the patent even if other circumstances or causes also played a part in the making of that decision. It is sufficient if the conduct is a material inducing factor which led to the grant. It goes too far to say that the false suggestion or representation must be material in the sense that without it the patent would not have proceeded to grant.
1084 Further, as explained by Emmett, Weinberg and Bennett JJ in Ranbaxy Australia Pty Ltd v Warner-Lambert Co LLC (2008) 77 IPR 449 (Ranbaxy) at [137] to [138]:
In the absence of an allegation of fraud, which involves an examination of the state of mind of the patent applicant, it is not sufficient to make out the ground of false suggestion or misrepresentation to prove simply that a false or misleading statement was made and nothing else. That is to say, even if a suggestion or representation is shown to be false or misleading, that, of itself, is not sufficient reason to draw an inference that the suggestion or representation contributed to the decision to grant the patent.
In the present case, there was no explicit evidence to the effect that, if there had been no assertion that the effectiveness of the relevant compounds was surprising and unexpected, the enantiomer patent would not have been granted. While inferences can be drawn, in the absence of any evidence concerning the commissioner’s decision making process, the inferences must be reasonably cogent.
1085 But it is not necessary to prove that “but for” the representation, the patent would not have proceeded to grant. Nor is it necessary to prove a deliberate intent to deceive.
1086 Further, as explained by Kenny J in Foster’s Australia Ltd v Cash’s (Australia) Pty Ltd (2013) 219 FCR 529 (at [107]):
In order to establish that the false suggestion or misrepresentation was material, it is unnecessary to show that, without it, the Patents would not have proceeded to grant. Rather, whether or not a false suggestion or misrepresentation is a material inducing factor depends on the circumstances of the case. The inquiry is an objective one as to whether or not it is objectively likely that the false suggestion or misrepresentation materially contributed to the decision to grant the Patents.
1087 The alleged misrepresentations which Ariosa maintains in this case are with respect to the state of matters as they existed at the time of and prior to the priority date. Therefore, their correctness is to be assessed at the time that the relevant statements were made.
1088 Factors relevant to an assessment of the objective materiality of a statement contained in a patent or in correspondence with the Commissioner include the following.
1089 First, is the statement significant to the invention or an essential feature of any asserted claims? If so, it is more likely to be material.
1090 Second, does the statement relate to a result claimed to be produced by the invention or to a useful purpose to which the result produced by the invention may be applied? Statements in the latter category are unlikely to be material, provided that there are other purposes for which the result is useful. On this issue, Lockhart J in Prestige cited (at 200) the following section of Blanco White TA, Patents for Inventions and the Protection of Industrial Designs (5th ed, Stevens & Sons, 1983) at 4-405:
It is not easy to distinguish between the sort of failure to fulfil a promise of results made in the specification that will amount to lack of utility and the sort that merely amounts to a false representation and accordingly will invalidate only if the patent has been “obtained” upon it. The distinction has been phrased as one between a promise of results and a mere wrong statement of the purposes for which that which is attained can be used; also as one between a promise of results and a “mere puff”, or between a false representation of the attributes of the product claimed and an accurate representation as to its attributes coupled with an expression of an “over-sanguine and erroneous view of its character”. The drawing of this distinction in particular cases is by no means easy, unless some result to be attained is set out in the claim; if this is so, it will normally negative any implication that some different result, set out in the body of the specification, is to be attained by the invention.
1091 Third, does the statement closely relate to the invention described and claimed in the Patent?
1092 Fourth, was the statement made to support a claim to novelty or to overcome another ground of invalidity? A statement in this category is more likely to be material.
1093 Fifth, has the Commissioner chosen to give evidence? As said in Ranbaxy at [83]:
Bearing in mind that the grant of a patent is a right in rem, the commissioner could be expected to take a position if a misrepresentation did in fact play a part in the decision to grant a patent and it is a relevant factor that the commissioner chooses not to give evidence. In the absence of such evidence, it is for the court to make a finding, based on the evidence before it. In the absence of explicit evidence that the commissioner, or the commissioner’s delegate, was in fact misled, it may nevertheless be inferred that a representation in fact contributed to the decision to grant a patent, if the representation was objectively likely to contribute to such a decision and the patent was in fact granted.
(Citations omitted.)
1094 Sixth, does the statement simply constitute a genuinely held opinion? As said by Bennett J in Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth (2011) 119 IPR 194 (Sigma) at [113]:
…a statement in a patent that a step was impossible does not amount to a false suggestion simply because it can later be demonstrated that it was not impossible. A statement that a course of inquiry or experimentation was fruitless does not amount to a false suggestion just because it can later be shown that the course of inquiry or experimentation would bear fruit. If the specification stated that it was impossible to formulate venlafaxine hydrochloride using hydrogel technology, the fact that it was later shown to be possible does not demonstrate false suggestion. There is no suggestion that the statement was false to the knowledge of the inventors or of Wyeth prior to grant.
1095 Seventh, does the statement constitute an opinion based upon reasonable scientific logic and data upon which reasonable minds may differ? As observed in Sanofi-Aventis at [124] and Arrow Pharmaceuticals Ltd v Merck & Co Inc (2004) 213 ALR 182 at [120], false suggestion:
…is a difficult ground to establish where the statement that is attacked purports to be a conclusion drawn from a published article. The patent office and others with an interest are all able to read the article and form independent judgments. Establishment of the ground would at least require a conclusion that the representation was deliberately false and intended to mislead.
1096 In Apotex Pty Ltd v Les Laboratories Servier [2013] FCA 1426, Rares J (at [201]) observed that:
…The essence of each of a false suggestion or misrepresentation by a patentee in the patent is a promise that the invention will produce a particular result or results for which he or she seeks the protection of a limited monopoly. As Parker J explained, this promise can be distinguishable from incorrect assertions by the patentee of one or more useful purposes to which the result obtained by invention can be applied. In other words, if the invention actually produces the substantive result or results asserted by the patentee in the specification, the mere fact that one or more additional purposes, embodiments or examples is wrong, ordinarily, will not amount to a false suggestion. It is only if what is asserted is of such a nature that the Court finds that the Commissioner has been deceived by it, as a material inducing factor, into making the grant, that the discretion to revoke will arise.
1097 The ground of false suggestion is to be assessed objectively.
1098 Now Sequenom has relied on a statement by Bennett J in Sigma at [113] to support a submission that the test was a subjective one. But her Honour did not there address the above authorities, or directly address the issue of whether the test was objective or subjective. Her Honour’s observations were in relation to a representation that achieving a particular formulation was “impossible”. The trial judge had found that that was not a literal representation, and Bennett J’s comments on appeal should be understood in that light. Her Honour’s final observation as to the patentee’s knowledge would also have been relevant to the “fraud” component of s 138(3)(d), had the trial judge been wrong about the nature of the representation that was made.
1099 It is possible that a false suggestion or misrepresentation is made implicitly rather than explicitly in the specification. Ariosa’s case relies upon false suggestions or misrepresentations arising, expressly and by inference, from the face of the specification. This is not a case where Ariosa seeks to rely upon other correspondence or statements made by the patentee to the Patent Office during prosecution.
1100 Further, Nicholas J in Apotex (No 2) held (at [151]) that, whilst a person seeking revocation on this ground “must establish that the Commissioner was deceived or misled in a manner that caused or contributed to the grant of the patent” it is not necessary to “establish that the patent would not have been granted were it not for the false suggestion or misrepresentation relied upon”. His Honour adopted the test enunciated by Lockhart J in Prestige (at 201, with whom Gummow J agreed at 218):
…namely, whether the conduct constituting the false suggestion or representation materially contributed to the Commissioner’s decision to grant the patent even if other circumstances or causes also played a part in the making of that decision. It is sufficient if the conduct is a material inducing factor which led to the grant. It goes too far to say that the false suggestion or representation must be material in the sense that without it the patent would not have proceeded to grant.
1101 This test was also accepted by the Full Court in Ranbaxy at [82].
1102 Further, the materiality of a false suggestion or misrepresentation to the grant of a patent can therefore be inferred from the circumstances, including the nature of the suggestion or statement made. As Heerey J held in WM Wrigley Jr Co v Cadbury Schweppes Pty Ltd (2005) 66 IPR 298 (at [125]):
If a representation was made which was objectively likely to contribute to a decision to grant a patent, and a patent is granted, it may be inferred that the representation in fact contributed to that decision…
(a) Ariosa’s submissions
1103 Ariosa originally asserted that the applicant for the Patent falsely represented:
(a) first, that the method according to the invention can be successfully used to determine the sex for female fetuses (alleged gender representation);
(b) second, that it had been demonstrated including by way of Figure 1 in the Patent that the level of fetal DNA in maternal plasma and serum is higher in pregnancies where the fetus has a chromosomal aneuploidy than in normal pregnancies (alleged level representation); and
(c) third, that techniques and technology were available prior to the grant of the Patent to screen for or detect chromosomal aneuploidies such as Down Syndrome in male and female fetuses using the method according to the invention, including by the methods referred to in paragraphs (a) and (b) on page 5 of the Patent (alleged aneuploidy representation).
1104 But the alleged gender representation has now been abandoned. Ariosa’s articulation of the false representations is contained in the particulars of invalidity. It is said that by statements in the specification of the Patent, the applicants for the Patent represented to the Commissioner of Patents prior to the grant of the Patent that:
(i) [abandoned]
(ii) ‘it has been demonstrated’ that the level of foetal DNA in maternal plasma and serum is higher in pregnancies where the foetus has a chromosomal aneuploidy than in normal pregnancies (see page 5 lines 12 to 15, page 14 lines 18-20 and page 32 lines 16-19);
(iii) the results reported in Figure 1 in the Patent demonstrated the alleged relationship between an increased level of foetal DNA and a foetal chromosomal aneuploidy (see page 14, lines 11 to 23);
(iv) techniques and technology were available prior to the grant of the Patent to screen for or detect chromosomal aneuploidies such as Down’s Syndrome using the method according to the invention, including in relation to female foetuses, including by:
(A) quantitative detection of foetal nucleic acid in the maternal serum plasma or serum e.g. a quantitative PCR assay as referred to in paragraph (a) on page 5 of the specification; and
(B) quantitation of foetal DNA markers on different chromosomes using accurate PCR techniques, such as real time quantitative PCR, as referred to in paragraph (b) on page 5 of the specification (see page 5, lines 3 to 26).
1105 In support of these assertions, Ariosa has submitted the following.
1106 As part of the introduction to Example 2 titled “Quantitative analysis of foetal DNA in maternal serum in aneuploid pregnancies”, the Patent sets out a real time quantitative PCR study and concludes, by reference to Figure 1, that the study “demonstrate[s] that the concentration of foetal DNA in maternal serum is elevated in aneuploid pregnancies” (page 14 lines 18 to 19).
1107 But Ariosa says that the best evidence is that there is in fact no difference between the concentration of cffDNA in aneuploid and normal pregnancies.
1108 The Patent then states that the results of the study indicate that “foetal DNA quantitation has the potential to be used as a new screening marker for foetal chromosomal aneuploidies” (page 14 lines 19 to 21). Indeed, the Patent states more definitively at page 5 in paragraph (a) that “Quantitative detection of foetal nucleic acid in the maternal plasma or serum e.g. a quantitative PCR assay, can be used to screen pregnant women for [fetal] chromosomal aneuploidies” (emphasis added). There are significant difficulties with the results presented for Example 2, and Ms Norbury, Professor Oepkes, and Professor Hyett are of the view that the data in the Patent does not support “the potential to use total levels of fetal DNA in maternal plasma as a screening tool for fetal chromosomal aneuploidies”.
1109 Ariosa says that the quantitative PCR method described in the Patent at page 5 paragraph (a) (and exemplified in Example 2) in fact could not be used (and did not have the “potential” to be used) as a screening test for fetal chromosomal aneuploidies at the priority date or date of grant (19 April 2001) or even today.
1110 Finally, the Patent states that the method described in the Patent at page 5 paragraph (b) could be applied to screening for Down Syndrome and other chromosomal aneuploidies. That method involved “quantitation of foetal DNA markers on different chromosomes” (page 5 lines 19 to 20). However, the statement that such a method could be used, both at the priority date and the date of grant, was incorrect.
1111 In relation to the question of materiality to grant, Ariosa says that the Patent claims that the invention described and claimed in the Patent “provides a method for prenatal diagnosis” and that “it will be possible to incorporate the nucleic acid-based diagnosis methods described herein into existing prenatal screening programmes” (page 2 line 23 and page 6 lines 3 to 5).
1112 The statements identified above underpin, and provide the basis for, the claims that rely on the concentration of cffDNA.
1113 The representation is also as to a practical benefit of the preceding claims, going back to claim 1.
1114 Ariosa points out that in Ranbaxy, the relevant false representation was one that exaggerated the benefit of the invention, as distinct from constituting the sole benefit. In that case the patentee represented that one of the two enantiomers had been found to be ten times as effective as the other, when it was only twice as effective.
1115 Ariosa says that by making the statements in the Patent in suit, the patentee was telling the Commissioner that such methods were, at the priority date and the date of grant, workable. It was an assertion of the benefit of the disclosed invention that was intended to assist in persuading the Commissioner and members of the public that the invention as claimed in each of the asserted claims was valid. Ariosa says that it is, moreover, highly unlikely that the Commissioner would have allowed the patent to grant with such a false suggestion within it, even though the test only requires that the conduct be a material inducement to grant.
1116 Accordingly, it says that claims which encompass the methods identified on page 5 paragraphs (a) and (b) of the Patent (and Examples 2 and 4) should be revoked.
(b) Analysis
1117 In summary, in my view the alleged representations do not properly reflect the disclosure in the Patent as a whole and what the person skilled in the art would understand the statements regarding the above matters to mean in the context of the Patent as a whole. Further, each of the above representations were not false as at the time they were made, that is, the filing date of the Patent. Moreover, there is nothing in the evidence to suggest that the Commissioner’s decision to grant the relevant claims was influenced in any way by the alleged representations.
1118 Let me deal with the representations in the following order:
(a) the alleged level representation; and
(b) the alleged aneuploidy representation.
1119 I will discuss the question of materiality later.
Alleged level representation
1120 The Patent states that it has “been demonstrated that the level of foetal DNA in maternal plasma and serum is higher in pregnancies where the foetus has a chromosomal aneuploidy than in normal pregnancies” (page 5 lines 12 to 15). This statement is made in the context of the Patent’s discussion of two possible ways which might be used to screen for Down syndrome and other chromosomal aneuploidies and, in particular, relates to the specific disclosure in Example 2 of the Patent. And this is consistent with the understanding of the person skilled in the art of the aneuploidy applications described in the Patent as a whole.
1121 Professor Lovett explained that the alleged level representation is confirmed by Example 2, “wherein the authors of the Patent properly conclude, based on the statistically significant results of a study reported in that example, that “[f]oetal DNA concentration was higher in aneuploid than control pregnancies””.
1122 Further, Professor Hyett agreed that the results of the Example 2 study were statistically significant and demonstrate that the mean level of cffDNA was higher in aneuploid pregnancies compared to normal pregnancies, expressly stating:
In my opinion, the results in Figure 1 show that the mean concentration is elevated in aneuploid pregnancies in this study and this is reported to be statistically significant.
1123 Professor Hyett further agreed that the general concentrations of the two comparator groups (control vs aneuploid) were different.
1124 Further, Professor Oepkes similarly noted that the statistics provided in the Patent suggest a statistically significant difference between the aneuploid and control groups studied in Example 2 of the Patent. Having said that, at the same time he appears to suggest that a difference between the two groups as a whole is not supported by the data provided.
1125 Professor Fisk explained that the data in Figure 1 enables the null hypothesis to be rejected. Figure 1 demonstrates that the concentration of fetal DNA in maternal circulation is not the same between the two populations. The data in Figure 1 is statistically significant.
1126 The alleged level representation is made in the context of and with reference to the results produced by Example 2. That is, the Patent simply states that the named inventors had conducted a study the results of which demonstrated that the level of cffDNA is higher in aneuploid pregnancies compared to normal pregnancies. For example, the Patent explains that “[i]n this study we demonstrate that the concentration of foetal DNA in maternal serum is elevated in aneuploid pregnancies. These results indicate…” (emphasis added) (page 14, lines 18 to 19).
1127 The results of the study, on which the statement is based, are reproduced in Figure 1 of the Patent. Professor Lovett agreed that the data in Figure 1 supports the statement on page 5 paragraph (a) in the Patent.
1128 Now although Example 2 of the Patent does not specifically address whether the control and aneuploid samples were matched for gestational age, Professor Lovett considered that this assumption could be appropriately made.
1129 Further, the inventors are clearly describing overall differences between aneuploid and normal pregnancy groups and not stating, as an absolute fact, that cffDNA is always higher in every aneuploid pregnancy. This is apparent from the method and results of the Example 2 study, which themselves are reproduced in the Patent. As Ms Norbury explains:
…The Patent also states that the fetal DNA concentration was higher in aneuploid than control pregnancies (Mann-Whitney U Test, p=0.006). The Mann-Whitney U Test is a nonparametric test that can be used to test whether two independent groups have come from the same population (or group). In this case, I understand that the test has been used to consider the likelihood that the samples are from the aneuploid or control pregnancies groups. Although the p value of 0.006 suggests that the results from the aneuploidy and control pregnancies are distinguishable, I would not have relied on the results reported in Figure 1 as a basis to diagnose an aneuploidy pregnancy given the overlap in the data points.
1130 Moreover, the existence of “overlap” between the two populations is a feature of all screening tests. It does not detract from the legitimacy and correctness of the alleged level representation, which is simply that, it had been demonstrated that the level of cffDNA was higher in aneuploid pregnancies compared to normal pregnancies.
1131 Further, there is no suggestion that the Example 2 study was not actually performed or that the authors of the Patent did not draw the conclusions referred to above in light of the results of the study that were known at the relevant date.
1132 Further, in a study received for publication on 16 June 1999 entitled “Increased Fetal DNA Concentrations in the Plasma of Pregnant Women Carrying Fetuses with Trisomy 21” (1999) 45(10) Clinical Chemistry 1747-1751 (Lo 1999), Dr Lo et al. stated, consistently with the Patent, that:
(a) “[t]he development of quantitative assays for circulating fetal DNA has provided powerful tools for studying the variations in fetal DNA concentrations in different physiological (11, 12) and pathological (13, 14) conditions” (at 1747);
(b) “[t]he main limitation of quantitative analysis of Y-chromosomal sequences in maternal plasma is that this approach can only be used in pregnancies involving male fetuses. The extension of this type of study to pregnancies involving female fetuses would require the development of quantitative assays for autosomal or X-chromosomal polymorphisms. Qualitative versions of these systems have already been developed (18), and it would be possible to convert these assays into a real-time quantitative format” (at 1750);
(c) “[o]ur data suggest that circulating fetal DNA may potentially be used as a marker for fetal trisomy 21” (at 1750); and
(d) “[a]bnormally high concentrations of circulating fetal DNA are found in a proportion of women carrying with trisomy 21” (at 1747).
1133 Now Professor Oepkes reviewed a series of post priority date papers relating to the issue of whether the relative or absolute concentrations of cffDNA in the maternal circulation is higher in aneuploidy pregnancies. But at their highest, the papers reviewed by Professor Oepkes show conflicting results. They do not and cannot prove that the alleged level representation was false at the time it was made. At the filing date, the only results available to the researchers (and the person skilled in the art) were those outlined in the Patent. As noted above, there is no suggestion that the results reported in the Patent are themselves false or misleading. And the experts agree that the results show a statistically significant difference in the level of cffDNA in aneuploid pregnancies compared to normal pregnancies. In summary it has not been established that the alleged level representation was false at the time it was made or even today.
Alleged aneuploidy representation
1134 The Patent describes the potential application of the invention to the detection of aneuploidy in qualified terms. It states that the plasma or serum-based non-invasive prenatal diagnosis method according to the invention can be applied to screening for Down syndrome and other chromosomal aneuploidies and gives two possible ways in which this might be done (page 5 lines 1 to 5).
1135 The potential of the method of the invention to be applied to screening for aneuploidy is not an essential feature of the invention described and claimed in the relevant claims of the Patent (cf claim 19 which is not asserted).
1136 The Patent neither suggests nor promises when read in context that techniques and technology were immediately available to screen for or detect chromosomal aneuploidies. The Patent simply describes possible ways in which the invention might be applied to such conditions, noting that systems such as those described in Lo et al., “Two-Way Cell Traffic Between Mother and Fetus: Biologic and Clinical Implications” (1996) 88(11) Blood 4390-4395 had been developed and may be usefully applied in connection with the development of such a method.
1137 And so the Patent explains (page 32 lines 21 to 24) with reference to Example 5 that:
…foetal DNA quantitation systems can be developed for polymorphic markers outside the Y chromosome so that quantitation can be applied to female foetuses. Autosomal polymorphic systems which may be used for this purpose have already been described (Lo et al. 1996).
(Emphasis added.)
1138 Professor Lovett confirmed that he would have appreciated that the method could potentially be used for women carrying female fetuses by using PCR primers that amplify paternally-inherited non-Y chromosome sequences not possessed by the pregnant mother, such as paternally-inherited polymorphisms or mutations.
1139 Professor Fisk similarly explained:
I agree that the example provided in the Patent for the first method of aneuploidy detection is limited to the detection of male aneuploid pregnancies (because the example involved the quantification of sequences from the Y chromosome). However, the Patent also provides information (e.g. in Example 3) regarding the detection of non-Y chromosome sequences, which could, in my view, be applied to the detection of aneuploid female fetuses or aneuploid pregnancies overall independent of gender. For example, the Patent explains at page 32, lines 21 to 24 that:
Foetal DNA quantitation systems can be developed for polymorphic markers outside the Y chromosome so that quantitation can be applied to female foetuses. Autosomal polymorphic systems which may be used for this purpose have already been described (Lo et al. 1996).
1140 Further, in relation to Example 2 of the Patent, the only Example directly relating to aneuploidy detection, the inventors concluded that their results “indicate that foetal DNA quantitation has the potential to be used as a new screening marker for foetal chromosomal aneuploidies” (page 14 lines 18 to 25). They also recognised that “a large scale population-based study could be carried out to develop cutoff values for screening purposes”.
1141 Further, Professor Fisk confirmed that Example 2 and Figure 1 of the Patent demonstrate that “the absolute concentration of fetal Y chromosome DNA in maternal serum is elevated in aneuploid pregnancies and that fetal DNA quantitation therefore has the potential to be used as a new screening marker for fetal chromosomal aneuploidies”.
1142 He also agreed with the statements in the Patent that the data reported in connection with Example 2 indicate that “foetal DNA quantitation had the potential to be used as a new screening marker for foetal chromosomal aneuploidies”. He said that whilst:
it would be impossible to use cffDNA levels clinically to “diagnose” positively (in the narrow sense) aneuploidy, I agree with the statements in the Patent that the data reported in connection with Example 2 indicate that “foetal DNA quantitation had the potential to be used as a new screening marker for foetal chromosomal aneuploidies.” (p. 14, lines 19-21).
1143 He also considered that the Patent provided sufficient information to justify such an application.
1144 Further, as Sequenom points out, the fact that numerous research teams investigated the Patent’s suggestions regarding the levels of cffDNA in aneuploid pregnancies in some sense supports the legitimacy of the Patent’s claims regarding the potential for fetal DNA quantitation to be used as a new screening marker for fetal chromosomal aneuploidies.
1145 Professor Oepkes identified 13 studies where researchers in the prenatal diagnosis field evidently held the view that there was potential, and investigated this potential. Professor Oepkes accepted that the groups that undertook this work were reputable researchers with good standing and intelligence.
1146 In particular, Gerovassili A et al, “Free fetal DNA in maternal circulation: a potential prognostic marker for chromosomal abnormalities?” (2007) 27 Prenatal Diagnosis 104-110, published 10 years after the priority date, featured a table summarising the studies that had been conducted as at 2007. The paper demonstrated that 5 of the 11 studies showed a significant elevation in levels of fetal DNA in maternal circulation between aneuploid and control groups. However, 5 of the 11 studies showed a decrease in levels of cffDNA, although the magnitude of these decreases was generally modest compared to the reported increases. As noted by Professor Fisk, the Gerovassili paper concluded that fetal cfDNA is not an ideal prognostic marker for chromosomal abnormalities in first trimester pregnancies. So, the implication from this paper is that even 10 years after the priority date, the potential for the quantitation of fetal DNA as a marker for aneuploidy had not been ruled out. Further, the Patent did not promise that cffDNA was an ideal marker for aneuploidy screening.
1147 Further, Professor Lovett seemed convinced that fetal DNA quantitation had the potential to be used as a marker for fetal chromosomal aneuploidies, and that it was a reasonable proposition for the patentee to make that assertion based on the data in Figure 1 of the Patent.
1148 Further, the alleged aneuploidy representation and alleged level representation were both supported by statistically significant results (Example 2) and a rational scientific explanation. With respect to the latter, the Patent explained (page 32 lines 16 to 23) that:
Bianchi et al recently reported that foetal cells in maternal blood were increased in aneuploid pregnancies (Bianchi et al. 1997) and it has been demonstrated (Example 2) that the foetal DNA concentration in maternal plasma and serum is also elevated in these pregnancies. This provides a new screening test for foetal chromosomal disorders. For this application, foetal DNA quantitation systems can be developed for polymorphic markers outside the Y chromosome so that quantitation can be applied to female foetuses.
1149 In summary, the alleged representations, if made, were not false at the relevant time. Let me then turn to the question of materiality.
Not material to grant
1150 There is no evidence to suggest that the alleged representations, even if false, actually misled the Commissioner and contributed to the grant of any of the relevant claims.
1151 Ariosa has not established that it is objectively likely that the alleged representations materially contributed to the Commissioner’s decision to grant those claims.
1152 First, the alleged representations cannot be said to be significant to the invention or an essential feature of any of the relevant claims. The alleged statements do not closely relate to the invention described and claimed in those claims.
1153 Second, the alleged representations relate to potential applications of the invention. It is clear from the specification that the patentee contemplated that the invention would be applied more broadly than merely the detection of aneuploidy, being the subject of only one dependent claim, claim 19, which is not asserted nor challenged. This is illustrated by the following matters as Sequenom submitted.
1154 The definition of prenatal diagnosis at page 2 line 24 to page 3, line 4 of the Patent encompasses the determination of any maternal or fetal condition or characteristic which is related to the fetal DNA itself or to the quantity or quality of the fetal DNA in the maternal plasma or serum. Chromosomal abnormalities are listed as just one example of the fetal abnormalities that may be determined using the invention; other examples provided are sex determination and simple mutations. Similarly, pre-eclampsia is listed within this definition as one example of a pregnancy-associated condition that can be detected or monitored using the invention.
1155 Further, of the five examples in the Patent, only Example 2 directly relates to the detection of aneuploid pregnancies, and this example promises only the potential of a new screening marker, and notes that a large scale population-based study could be carried out to render the method suitable for widespread application of the method.
1156 Further, page 4 of the Patent sets out various examples of how the invention can be applied, none of which related to aneuploidy detection. Those examples include the determination of fetal rhesus D status, haemoglobinopathies, and the detection of paternally-inherited DNA polymorphisms or mutations present on either a Y or non-Y chromosome.
1157 The Patent enables persons skilled in the art to work a number of useful methods falling within the scope of each relevant claim.
1158 Third, the alleged statements were not introduced to overcome any novelty or inventive step objections, nor are they referable to any other ground of invalidity raised against the relevant claims.
1159 Fourth, the Commissioner has chosen to not give any evidence in this proceeding. Sequenom wrote to the Commissioner and received a response indicating that the Commissioner did not want to exercise any right under Rule 34.23 of the Federal Court Rules 2011 (Cth).
1160 In summary, Ariosa’s false suggestion assertions are not made out.
INFRINGMENT
1161 Sequenom alleges that the Harmony non-invasive prenatal test (Harmony Test) falls within the scope of claims 1 to 3, 5, 6, 9, 13, 14, 22, 23, 25 and 26 of the Patent. It therefore alleges that the performance in Australia of the Harmony Test by Sonic and Clinical infringes those claims. It also alleges infringement by the performance of the Harmony Test by Ariosa itself in the United States on samples collected in Australia by Sonic and Clinical. The respondents deny infringement.
1162 The Harmony Test analyses the cell free DNA present in the plasma of pregnant women to provide an estimate of the risk that the fetus is carrying an autosomal aneuploidy, namely one of trisomy 13, trisomy 18 and trisomy 21. In addition, if requested by the patient, the Harmony Test provides an estimate of the risk that the fetus is carrying a sex chromosome aneuploidy such as X, XXX, XXY, XYY and XXYY. Patients can also opt to be provided with a determination of the sex of the fetus. The detailed way in which the Harmony Test works is set out in the confidential Product and Process Description (PPD), which as I have already said is a schedule to my reasons.
1163 The Harmony Test uses sophisticated and complex technologies and approaches to undertake the analysis of cell free DNA and to provide risk assessments for each patient based on that analysis. It incorporates:
(a) the Digital Analysis of Selected Regions (DANSR) assay, which enables highly multiplexed sequencing of selected loci from specific chromosomes of interest; and
(b) the FORTE (fetal-fraction optimised risk of trisomy evaluation) algorithm, a proprietary algorithm that uses the results from the DANSR assay to distinguish between high-probability and low-probability results.
1164 The Harmony Test was developed some 15 years after the priority date. If I might say so it is a very impressive piece of science. And it relies on some technologies and approaches that were not foreshadowed by the Patent. These include the digital analysis of selected regions of the genome using digital technology that was not available as at 1997, together with sequence information derived from SNP databases that were developed several years after the priority date.
1165 Before proceeding further, let me describe the Harmony Test in some detail. For this purpose what follows for the most part is a description of the non-confidential elements thereof.
(a) The elements of the Harmony Test
1166 For simplicity, the description of the Harmony Test that I am about to describe reflects the processing of samples relating to singleton pregnancies only. But similar elements apply for the processing of samples relating to twin pregnancies, egg donor pregnancies and egg donor twin pregnancies, all of which can be analysed by the Harmony Test.
1167 The Harmony Test is a non-invasive prenatal screening test that uses cfDNA in the blood of a pregnant woman to estimate the risk that the fetus suffers from certain fetal genetic disorders. In some instances, the Harmony Test also assesses the likely chromosomal gender of the fetus.
1168 Scores are calculated and given to reflect an estimate of the risk of the fetus carrying the following fetal genetic disorders (autosomal aneuploidies) and are reported, save in some circumstances, to the patient in every completed Harmony Test:
(a) trisomy 13 (Patau syndrome);
(b) trisomy 18 (Edwards syndrome); and
(c) trisomy 21 (Down syndrome).
1169 In addition, the likely chromosomal gender of the fetus and scores reflecting an estimate of the risk of the fetus carrying the following fetal genetic disorders (sex chromosome aneuploidies) are reported, save in some circumstances, to the patient only on the patient’s request:
(a) monosomy X (Turner syndrome);
(b) trisomy X (Triple X syndrome);
(c) trisomy XXY (Klinefelter syndrome);
(d) trisomy XYY (XYY syndrome); and
(e) aneuploidy XXYY (XXYY syndrome).
1170 Since about 25 August 2015, patients have been able to opt for a report covering, in addition to autosomal aneuploidies, either or both of the following:
(a) likely chromosomal gender; and/or
(b) one of the following sex chromosome aneuploidy options:
(i) all of the sex chromosome aneuploidies set out above; or
(ii) monosomy X only.
1171 From the time that Sonic and Clinical have promoted the Harmony Test in Australia patients were able to opt for a report covering, in addition to autosomal aneuploidies, either of the following:
(a) likely chromosomal gender; or
(b) likely chromosomal gender and all of the sex chromosome aneuploidies.
1172 The Harmony Test can be summarised as consisting of the following key stages:
(a) DNA isolation.
(b) The DANSR assay.
(c) Microarray analysis.
(d) FORTE.
1173 Before proceeding further, let me go over again some concepts.
1174 During pregnancy, maternal blood contains both cfDNA originating from the fetus (cffDNA) and cfDNA originating from the mother. The cffDNA is typically approximately 10% of the total amount of cfDNA in a pregnant woman’s blood. However, this percentage varies at different stages of a pregnancy and between individuals.
1175 Approximately 50% of the cffDNA derives from the maternal genome and approximately 50% derives from the paternal genome. This is because in a euploid pregnancy, meaning a pregnancy in which the fetus’ genome is made up of 23 pairs of chromosomes, one chromosome of each pair is maternally-derived while the other is paternally-derived.
1176 The cffDNA present in maternal blood is double-stranded and is found in fragmented form, not as complete chromosomes. The average size of a cffDNA fragment is approximately 200 base pairs.
1177 A collected blood sample first undergoes a centrifugation protocol. This separates the blood into three components: (a) the buffy coat; (b) erythrocytes (red blood cells); and (c) plasma, being the fluid part of the blood, which includes minerals, salts, hormones and proteins. The cffDNA and maternal cfDNA are both contained within the plasma component. The plasma component is then separated from the buffy coat and erythrocytes. The cfDNA is then extracted from the plasma component.
1178 The Harmony Test uses a targeted approach. That is, it targets cfDNA fragments whether of fetal or maternal origin from specific chromosomes, rather than all chromosomes.
1179 The Harmony Test is comprised of a so-called Non-Polymorphic Assay and a so-called Polymorphic Assay.
1180 The Non-Polymorphic Assay interrogates loci on particular chromosomes of interest. The loci interrogated are located on several chromosomes, including chromosomes 13, 18 and 21, and the X and Y chromosomes. The Non-Polymorphic Assay relates to an analysis of whether these chromosomes are relatively over-represented and thus the risk that the fetus may suffer from one or more of the genetic disorders listed above and the likely chromosomal gender of the fetus.
1181 The Polymorphic Assay interrogates hundreds of SNPs within loci on particular chromosomes. The Polymorphic Assay relates to the estimation of the fraction of total cfDNA that is treated as cffDNA (the fetal fraction). If the fetal fraction is reported as less than 4% then the sample is rejected and no results are reported to the patient.
1182 Let me say something further on both of these types of assays.
1183 The Non-Polymorphic Assay involves an analysis of sequences from the Y chromosome for the purpose of sex determination and the detection of Y chromosome aneuploidies. The Non-Polymorphic Assay involves looking for sequences that are of fetal origin because the Y chromosome is not possessed by pregnant women. On the basis of the detection of nucleic acid from the Y chromosome, the Harmony Test determines and diagnoses fetal sex and various aneuploidies, both being fetal conditions or characteristics.
1184 Leaving aside the “Y chromosome” aspect of the Non-Polymorphic Assay (Non-Y Non-Poly Assay), the Non-Polymorphic Assay measures the relative intensities of DANSR assays corresponding to non-polymorphic loci to estimate the relative concentration of each of chromosomes inter-alia 13, 18 and 21.
1185 Combined with results from the Polymorphic Assay, the Harmony Test is able to detect very small increases in the total number of chromosome 21, 13 and 18 sequences and ascribe the increase to the fetus being a “high risk” for aneuploidy. In so doing, the Non-Y Non-Poly Assay does not distinguish between maternal and fetal sequences.
1186 The Polymorphic Assay involves looking for sequences of fetal origin, i.e. minor fractions at loci, arising from the presence of paternally inherited fetal sequences not possessed by the pregnant mother. Given the extremely large number of alleles assessed with the Polymorphic Assay, it would be implausible that any individual Harmony Test would not detect a paternally-inherited fetal nucleic acid from a non-Y chromosome.
1187 The Polymorphic Assay examines [Redacted] loci on chromosomes 1 to 12 (non-Y chromosomes) unlikely to be aneuploid in order to identify “informative loci” from which the fraction of fetal DNA in the sample can be determined. A locus is considered informative if a major fraction and a minor fraction are identified. It is an assumption of the Harmony Test that the so-called “minor fraction” is the fetal fraction. The Polymorphic Assay involves the detection and quantification of SNP alleles at a large number of loci in order to identify these informative loci from which the fraction of fetal DNA in the sample can be determined. The SNP alleles are chosen to increase the likelihood that they differ between mother and fetus (i.e. are informative). And as I say, the Polymorphic Assay relates to the estimation of the fraction of the total cfDNA that derives from the minor source, as opposed to the major source. The Polymorphic Assay assumes that the “minor source” is of fetal origin and that the minor source fraction is an estimation of the fraction of the total cfDNA that is cffDNA. This assumption is robust.
1188 Further, the Harmony IVD Kit Instructions, rev 3.0, describe informative loci as loci where the maternal genotype is homozygous for one allele and the fetus has inherited a different allele. The Instructions state that the “Harmony Test estimates the fraction of the cfDNA sample that originated with the fetus…The Harmony Test requires samples to have fetal fraction values of 4% or greater in order to provide a result”.
1189 In summary, the Polymorphic Assay determines the relative concentration of cffDNA in a maternal plasma sample (the fetal fraction) and partially genotypes the fetus at a large number of polymorphic informative loci, being variations of usually unknown significance. The partial genotype of the fetus is a fetal characteristic, albeit likely a benign variant or a variant of unknown significance. As Professor Fisk explained:
…it does involve genotyping, of course, be it single or multiple markers, but here it’s up to [Redacted] markers. If you think of this as a genotype, let’s take it in criminal terms. So I would like to infer an analogy from the Golden State Killer case recently. It was actually in the news last week because a person who did the sleuthing has revealed herself. So if somebody – say a pregnancy was a result of a crime, rape, for instance, and there was no sperm sample at the time, there was no identity for the father, you could determine with a shortlist with a high degree of accuracy the paternal genotype based on the results of the polymorphic assay.
1190 In terms of carrying out the Non-Polymorphic Assay and Polymorphic Assay the following may be noted.
1191 First, the isolated cfDNA is denatured, meaning that the double stranded cfDNA splits into single stranded cfDNA.
1192 Second, each targeted locus is interrogated using a so-called “triplet”, which comprises three individual oligonucleotides. The three oligonucleotides within a “triplet” are termed the “Left”, “Mid” and “Right” oligonucleotides respectively.
1193 Left oligonucleotides are designed such that they each contain a universal primer binding site at their 5' end, and a sequence designed to hybridise to the locus in question. Mid oligonucleotides each contain a sequence designed to hybridise to the locus in question but do not contain universal primer binding sites. Right oligonucleotides are designed such that they each contain a sequence designed to hybridise to the locus in question, followed by a universal primer binding site at the 3' end. The universal primer binding site contained in Right oligonucleotides is different from the universal primer binding site contained in Left oligonucleotides. [Redacted]
1194 The 3 oligonucleotides within a “triplet” bind adjacent to one another at the targeted locus or SNP. As a result, double-stranded cfDNA-oligonucleotide complexes are formed in solution.
1195 In both the Polymorphic Assay and Non-Polymorphic Assay, oligonucleotides bind to both the cffDNA and maternal cfDNA in the sample.
1196 Third, if all three of the oligonucleotides from a particular “triplet” bind to their corresponding locus, those three oligonucleotides are joined together to form a single continuous oligonucleotide (the ligation product). Ligation products therefore contain two universal priming sites – a Left universal priming site and a Right universal priming site. All the ligation products have the same Left universal priming site and the same Right universal priming site, but the Left universal priming sites are different from the Right universal priming sites. Ligation products are formed in proportion to the relative numbers of corresponding loci in the sample after the initial steps of the DANSR assay.
1197 Fourth, ligation products are then amplified using PCR that makes use of universal PCR primers. The universal PCR primers bind to the universal primer binding sites that are contained within the ligation products. The ligation products and their corresponding PCR products are collectively termed the DANSR products.
1198 Fifth, after amplification, the number of copies of the DANSR products for each “triplet” will be in proportion to the relative number of the different ligation products initially formed, assuming no amplification errors or efficiency issues. Therefore, the number of copies of the DANSR products for each “triplet” will be in proportion to the relative numbers of corresponding loci in the sample after the initial steps of the DANSR assay.
1199 Both the Non-Polymorphic and Polymorphic Assays involve the same underlying biochemical strategy. The principles of the design of each of the oligonucleotides are largely the same regardless of whether it is for use in the Polymorphic or Non-Polymorphic Assays. The technical steps involved in the Polymorphic and Non-Polymorphic Assays are substantially similar. Let me elaborate further on what I have said above.
1200 Broadly speaking:
(a) the oligonucleotides or probes bind to target sequences on one strand of the cfDNA in the sample;
(b) the above oligonucleotides comprise three smaller sequentially adjacent oligonucleotides, probes or “triplets”, as depicted below:
[Image redacted]
1201 Each component of the triplet contains a sequence complementary to the target sequence. Together, [Redacted]:
(a) if, and only if, the three oligonucleotides bind to a target sequence, the triplet sequences join together to form a ligation product (akin to LCR);
(b) anything not bound is washed away and removed from the sample;
(c) the ligation products are then separated from their corresponding target sequences, that is, the original maternal and cffDNA in the sample, and the maternal cfDNA and cffDNA deriving from the original sample is then discarded;
(d) the ligation products containing sequences complementary to the original fetal and maternal cfDNA as well as synthetic sequences are amplified by PCR to produce products termed by the PPD as DANSR products. After amplification, the number of copies of the DANSR products for each triplet will be in proportion to the number of different ligation products initially formed on the assumption that there is no amplification errors or efficiency issues, such that the number of copies of DANSR products for each triplet will be in proportion to the relative numbers of corresponding loci in the original sample. This process is depicted by Figure 3 of the PPD, extracted for convenience, below:
[Image redacted]
(e) [Redacted]
(f) [Redacted]
(g) [Redacted]
(h) [Redacted]
1202 In the case of the Non-Polymorphic Assay, each locus within an assay pair has been assigned the same assay binding region. So, hybridisation is competitive between the loci within each assay pair.
1203 In the case of the Polymorphic Assay, each allele within a SNP allele pair has been assigned the same array binding region. So, hybridisation is competitive between each allele in a SNP allele pair.
1204 Calculation of the fetal fraction relies on the Harmony Test identifying informative SNPs, being SNPs which differ between the mother and the fetus. The primary type of informative allele is described in the PPD as arising when the mother is homozygous for a particular allele and the fetus is heterozygous for the allele. When a locus is identified as having a major and minor fraction (i.e. being informative), the fetal fraction can be calculated as being twice the amount of the minor fraction divided by the total of the major fraction and the minor fraction, since the fetal DNA consists both of paternally inherited and maternally inherited DNA which are present in equivalent amounts.
1205 Professor Fisk notes that the Harmony IVD Kit Instructions state that data from the identified informative loci are used to estimate the fetal fraction of a given sample:
…FORTE next evaluates the relative intensities of DANSR assays corresponding to the two alleles of each polymorphic locus to estimate the allele frequency of the polymorphic locus in each sample. FORTE then identifies loci that are informative for estimating fetal fraction in each sample (i.e., loci where the maternal genotype is homozygous for one allele and the fetus has inherited a different allele), and uses the allele frequencies of these informative loci to estimate the fraction of fetal DNA in each sample.
(Emphasis added by Professor Fisk.)
And at pp 24 to 25:
The Harmony Test estimates the fraction of the cfDNA sample that originated from the fetus and reports the estimate as the Sample QC Metric FetalFraction. The Harmony Test requires samples to have fetal fraction values of 4% or greater in order to provide a result…
…The ability of the Harmony Test to detect minor source cfDNA at fractions from 4% was illustrated in the same set of male:female mixtures by comparing the Harmony Test Fetal Fraction metric to FPYN, an independent measure of fetal fraction which uses chromosome Y – specific loci (Figure 3).
(Emphasis added by Professor Fisk.)
1206 Let me now say something about the microarray analysis.
1207 Custom microarrays are used to carry out the relative quantification needed for the analyses described above. On these custom microarrays, the DANSR products are used to estimate the amounts of corresponding targeted loci.
1208 Relative quantification on the custom microarrays occurs via the binding of fluorescent labels.
1209 Labelled microarrays are then imaged to allow analysis by the FORTE computer algorithm, which I will describe in a moment.
1210 Each patient sample is analysed on a separate microarray, and the Harmony Test is designed so that 96 patient samples are analysed simultaneously.
1211 As I have said, FORTE is the proprietary Harmony Test computer algorithm. FORTE is used to estimate the fetal fraction. FORTE does not estimate the amount of cffDNA contained within a particular volume of the sample at any stage. FORTE incorporates the estimate of fetal fraction into the risk assessments described below. Information relating to the estimate of the fetal fraction is also reported to the patient in the final test result. FORTE also incorporates other factors such as maternal age and gestational age into such risk assessments. FORTE is then used, save where the sample fails a series of quality control criteria including where the fetal fraction is estimated as less than 4%, to estimate risk scores for the autosomal aneuploidies and, if requested by the patient, sex chromosome aneuploidies. FORTE is also used, save where the sample fails a series of quality control criteria including where the fetal fraction is estimated as less than 4%, to estimate likely fetal chromosomal gender, if requested by the patient.
1212 Where a sample is not rejected, estimated risk scores are provided to a patient for each autosomal aneuploidy separately. Further, individual estimated risk scores for sex chromosome aneuploidies and the likely chromosomal gender of the fetus are provided to a patient where these have been estimated in accordance with the patient’s request.
1213 Each risk score and indication of likely chromosomal gender reported to a patient reflects only a probability estimation based on the sample provided by that patient.
(b) General
1214 Sonic and Clinical have used the Harmony Test in Australia; in the case of Sonic, it has been used since September 2015, and in the case of Clinical it has been used since March 2016.
1215 Each of Sonic and Clinical have been licensed by Ariosa to use the Harmony Test in Australia.
1216 But prior to Sonic and Clinical being licensed by Ariosa to use the Harmony Test in Australia, each operated a send out model whereby they collected blood samples from pregnant women in Australia and sent those samples to Ariosa in the US. Ariosa used the Harmony Test to produce results which were provided to Sonic and Clinical in Australia which they forwarded to healthcare professionals in Australia. The periods during which each sent out samples to Ariosa in the US (the send out periods) were:
(a) in the case of Sonic, June 2015 to September 2015 and January 2017 to March 2017; and
(b) in the case of Clinical, December 2014 to March 2016.
1217 Sonic and Clinical have each promoted and supplied Harmony Test results in Australia produced by their own and Ariosa’s use of the Harmony Test in the United States.
1218 Ariosa authorised Sonic and Clinical to engage in the conduct that I have just described.
1219 Now it is clear that the Harmony Test is a detection method performed on maternal plasma samples comprising the detection of paternally-inherited fetal DNA from Y and non-Y sequences. It is also clear that the method is used to determine the sex of fetuses, XYY aneuploidy status, and fetal genotypes. Sequenom says that the Harmony Test falls within each of the relevant claims. Contrastingly, the respondents deny infringement primarily by adopting various constructions of several integers that I will elaborate on later. But at this point let me briefly discuss some legal principles.
1220 Section 13 of the Act provides that the Patent gave Sequenom the exclusive rights, during the term of the Patent, to “exploit” the invention as defined in the relevant claims and to authorise another person to exploit the invention.
1221 Schedule 1 to the Act states that where the invention is a method or process, “exploit” in relation to an invention includes use of the method or process or to make, hire, sell or otherwise dispose of a product resulting from such use, offer to make, sell, hire or otherwise dispose of such a product, use or import it, or keep it for the purpose of doing any of those things.
1222 Accordingly, a person will be taken to have infringed a method claim of a patent if that person:
(a) within Australia uses the invention; or
(b) exploits, in Australia, a product resulting from the use, anywhere, of the method or process; or
(c) authorises or procures from anywhere an infringing act referred to in (a) or (b).
1223 The construction of a claim determines whether an alleged infringing product or method falls within the scope of the invention as defined in that claim, on both textual and substantive bases. Accordingly, it is a question of whether the alleged infringing method or product is covered by the language of the claim. Therefore it is irrelevant whether an infringing variant might constitute an inventive improvement over the invention disclosed in the patent. As recognised by O’Loughlin J in Winner v Morey Haigh & Associates (Australasia) Pty Ltd (1996) 33 IPR 215 at 223 quoting Blanco White TA, Patents for Inventions and the Protection of Industrial Designs (5th ed, Stevens, 1983) p 45 [3-004]:
…If what the defendant has done does fall within the claim, it will infringe no matter what additional inventive features it contains. [I]nfringement cannot be avoided by the “super-adding of ingenuity to robbery”.
1224 That is, an infringing product can be very clever, the result of major research effort, and can have required significant ingenuity, which I might say are suitable descriptions of the Harmony Test, but nevertheless fall within the scope of the claims of the patent in suit; see also Regeneron Pharmaceuticals Inc v Genentech [2012] EWHC 657 (Pat) per Floyd J at [92] to [93] and [208], upheld in Regeneron Pharmaceuticals Inc Genentech Inc; Bayer Pharma AG v Genentech Inc [2013] EWCA Civ 93 at [53] and [172] per Kitchin LJ.
1225 In terms of the infringement issues that I now need to decide, I would note that there has been a narrowing of the issues.
1226 Ariosa does not press the argument that the Harmony Test does not detect nucleic acid of fetal origin on the basis that the detection is of the Read Out Cassettes. But it maintains the construction point that the nucleic acid detected must be known or inferred in advance to be of fetal origin and that this does not occur in the Harmony Test other than for the Non-Polymorphic Assay for the Y chromosome sequences.
1227 Ariosa does not press the argument that the Harmony Test amplifies the DANSR product rather than the nucleic acid of fetal origin or that the Read Out Cassettes comprise a “probe”. That is, subject to claim 1 being found to be infringed it accepts that there is amplification for the purposes of claim 2 and the use of a probe in claim 5.
1228 Ariosa does not take the point that the Polymorphic Assay is not a relevant detection by reason of the matters raised by Ms Norbury in the joint expert report, except to the extent these matters are relevant to the issue of whether the Harmony Test provides “a prenatal diagnosis” or “a diagnosis”.
1229 Ariosa agrees that the issue in dispute in relation to claim 14 is an issue of construction in relation to the term “concentration” only, and does not contend that any additional non-infringement argument arises if I were to adopt Sequenom’s construction, as I do, that “concentration” in claim 14 also includes relative concentration and is not limited to absolute concentration.
1230 Further, Ariosa does not contend that when reporting fetal sex the Harmony Test is not a diagnostic test.
1231 Finally, it is not in doubt that in relation to all relevant claims, the Harmony Test is performed on plasma extracted from maternal blood samples.
1232 Accordingly, the following matters remain in issue:
(a) In relation to claim 1, whether detection for the purposes of claim 1 requires that the nucleic acid of fetal origin be known in advance to be fetal.
(b) In relation to claim 14, whether “concentration” as used in claim 14 means relative and/or absolute concentration.
(c) In relation to claims 22 to 26, whether the Harmony Test XYY detection method and genotyping in the Non-Polymorphic Assay is a prenatal diagnosis for the purposes of claims 22 to 26.
(d) In relation to claim 25, whether the Harmony Test includes a test for fetal nucleic acid indicative of a maternal or fetal condition or characteristic.
1233 In this respect Sequenom says that in relation to claims 1, 2, 3, 5 and 6, in the course of the Non-Polymorphic Assay of the Harmony Test the presence of a fetal nucleic acid sequence from the Y chromosome is detected by means of a sequence specific probe and amplified by enrichment and by PCR.
1234 Further, it says that when requested by the pregnant female’s clinician, the Harmony Test reports the gender of the fetus (claims 6, 13, 22, 23, 25 and 26) and the risk that the fetus is carrying a sex chromosome aneuploidy, such as XYY (claims 22, 23, 25 and 26).
1235 Further, it says that the Non-Polymorphic Assay is relevantly performed for determining the sex of the fetus (claims 6 and 13).
1236 Further, it says that in the course of the Polymorphic Assay of the Harmony Test, paternally-inherited fetal nucleic acid from a non-Y chromosome is detected by means of a sequence specific probe and amplified by enrichment and PCR (claims 1, 2, 3, 5 and 9).
1237 Further, it says that in the course of the Polymorphic Assay of the Harmony Test, the presence of a fetal nucleic acid from a paternally-inherited non-Y chromosome is detected (claim 9) and the concentration of the sequence in the maternal plasma is determined (claim 14).
1238 Further, it says that the Polymorphic Assay detects paternally-inherited mutations and the genotype of the fetus and is thus a test for fetal nucleic acid indicative of a fetal characteristic (claim 25) and a “diagnosis” (claims 22, 23 and 26).
1239 Further, it says that the Harmony Test:
(a) is a “method of performing a prenatal diagnosis” as that term is defined and understood in the context of the Patent (claims 22, 23, 25 and 26);
(b) provides a diagnosis based on the presence, quantity and / or sequence of the fetal nucleic acid (claims 22 and 23); and
(c) comprises a test for fetal nucleic acid indicative of a fetal condition or characteristic (claim 25).
1240 Before proceeding further I would note that although I have found claim 26 to be invalid, it is convenient to leave claim 26 in my discussion just in case I turn out to be wrong.
(c) Construction
1241 There are a number of construction questions involving the claim integers that are in dispute between the parties. The relevant claim integers are:
(a) “detecting the presence of a nucleic acid of foetal origin” in claim 1;
(b) “determining the concentration of the foetal nucleic acid sequence in the maternal serum or plasma” in claim 14; and
(c) “a prenatal diagnosis” in claims 22 to 26.
1242 Let me first say something concerning the legal principles, which are not in doubt.
Legal principles
1243 A patent should be construed through the eyes of the hypothetical person (or team) who is likely to have a practical interest in the subject matter and may often work in the art in which the invention is connected.
1244 The following principles distilled from what I said in MLA (No 1) at [213] to [221] are of relevance in the present case.
1245 The principles governing the construction of patent specifications, including claims, are well established. A claim is construed from the perspective of a person skilled in the relevant art as to how such a person would have understood the patentee to be using the words of the claim in the context of the specification as a whole. Further, a claim is to be construed in the light of the common general knowledge including the art before the priority date.
1246 A measure of common sense should be used. And ordinary words should be given their ordinary meaning unless a person skilled in the art would give them a technical meaning or the specification ascribes a special meaning.
1247 In terms of how the body of the specification may be used in construing a claim, the claim should be construed in the context of the specification as a whole even if there is no apparent ambiguity in the claim. Nevertheless, it is not legitimate to narrow or expand the boundaries of the monopoly as fixed by the words of a claim by adding to these words glosses drawn from other parts of the specification. More particularly, if a claim is clear and unambiguous, to say that it is to be read in the context of the specification as a whole does not justify it being varied or made obscure by statements found in other parts of the specification.
1248 Now the specification may stipulate the problem in the art before the priority date and the objects of the invention that are designed to address or ameliorate this. Accordingly, the specified objects may be useful in construing a claim in context. Nevertheless, the specified objects are not controlling in terms of construing a claim; glosses cannot be drawn from the objects.
1249 A claim should be given a purposive construction. Words should be read in their proper context and a too technical or narrow construction should be avoided. Further, the integers of a claim should not be considered individually and in isolation. Further, a construction according to which the invention will work is to be preferred to one in which it may not. But to give a claim a purposive construction “does not involve extending or going beyond the definition of the technical matter for which the patentee seeks protection in the claims” (Sachtler GmbH and Co KG (formerly Sachtler AG) v RE Miller Pty Ltd (2005) 221 ALR 373; [2005] FCA 788 at [42] per Bennett J). To apply a purposive construction does not justify extending the patentee’s monopoly to the ideas disclosed in the specification. I also adopt what was said in Artcraft Urban Group Pty Ltd v Streetworx Pty Ltd (2016) 245 FCR 485 at [72] to [78] per Greenwood J (agreed to by Rares J at [142], [145] and [146]). Further, I would also refer to Lord Hoffmann's observations in Kirin-Amgen Inc v Hoechst Marion Roussel Ltd (2004) 64 IPR 444; [2004] UKHL 46 at [27] to [34] concerning a purposive approach to construction.
1250 As I have said, a claim is to be construed from the perspective of how a person skilled in the art would have understood the patentee to be using the words, informed by the notional skilled addressee’s relevant general knowledge and what has been disclosed in the specification. But to consider such a perspective does not entail that the Court necessarily requires expert evidence to assist on construction. If it is clear that the claims are to be read according to their ordinary meaning with no special meaning given to any word or phrase, if the science or technical issues are easily comprehensible and if, more generally, the Court does not require expert assistance in understanding the context of the claims, then expert evidence on construction may not only be unnecessary, but unhelpful and distracting. The nature and complexity of the patent in suit and the issues raised will determine the utility or necessity for expert evidence on construction. In the present case, I have to some extent been assisted on questions of construction by the expert evidence adduced by the parties, but the significance and weight of such evidence should not be over-stated. After all, the proper construction of a claim is ultimately a question of law for me, albeit that I must adopt the perspective that I have just described.
1251 In terms of the skilled addressee, one is using a hypothetical construct. The following principles are applicable:
(a) First, to identify the characteristics of the skilled addressee, the field to which the invention relates must be identified.
(b) Second, the skilled addressee is taken to be a person of ordinary skill (as opposed to a leading expert) in that field and equipped with the relevant common general knowledge including the art before the priority date.
(c) Third, the qualifications and experience of the skilled addressee will depend on the particular case, having regard to the nature of the invention and the relevant industry. Formal qualifications are not essential. Practical skill and experience in the field may suffice. A patent specification is addressed to those having a practical interest in the subject matter of the invention; such persons are those with practical knowledge and experience of the kind of work in which the invention is intended to be used.
(d) Fourth, the hypothetical person skilled in the art may possess an amalgam of attributes drawn from a team of persons whose combined skills, even if disparate, would normally be employed in interpreting and carrying into effect instructions such as those contained in the specification.
(e) Fifth, as the skilled addressee comes to a reading of the specification with the common general knowledge of persons skilled in the relevant art, they read it knowing that its purpose is to describe and demarcate an invention. But the person skilled in the art is not particularly imaginative or inventive.
(f) Sixth, the skilled addressee does not come to reading the specification seeking failure.
1252 As I have said, the legal construct may not be a single person but may be a team of persons whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those contained in the relevant instrument.
1253 As I have said, a patent application should be construed through the eyes of the hypothetical person (or team of persons) who is likely to have a practical interest in the subject matter of the invention and may often work in the art with which the invention is connected (KD Kanopy Australasia Pty Ltd v Insta Image Pty Ltd (2007) 71 IPR 615; [2007] FCA 481 at [16] per Kiefel J (as she then was) and GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Apotex Pty Ltd (2016) 119 IPR 1; [2016] FCA 608 at [277(c)]).
1254 A patent specification is addressed to those likely to have a practical interest in the subject matter of the invention. The skilled addressee is a person acquainted with the general elements and conditions of the industry who has practical knowledge and experience of the kind of work in which the invention is intended to be used.
1255 The skilled addressee comes to a reading of the specification with the common general knowledge of persons skilled in the relevant art, and they read it in a common sense and practical manner knowing that its purpose is to describe and demarcate an invention and not to be a textbook in mathematics or genetics or a shopping list of chemicals or hardware. The person skilled in the art does not come to read the specification seeking failure.
1256 The person skilled in the art is not a reference to a specific person but is a legal construct. The notional person is not an avatar for expert witnesses whose testimony is accepted by the Court. It is a pale shadow of a real person – a tool of analysis. However, it is a legal construct drawn by reference to the available evidence.
1257 The legal construct may not be a single person but may be a team of persons “whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those which are contained in the document to be construed”. This is particularly so where the art is one having a highly developed technology, in which case the hypothetical skilled addressee may be a team whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those which are contained in the document to be construed.
1258 In the present case the Patent is addressed essentially to a multi-disciplinary team. In terms of the characteristics of the person skilled in the art, again I refer to what I have previously said.
“Detecting the presence of a nucleic acid of foetal origin” (claims 1, 2, 3, 5, 6 and 9)
1259 The detection method of claim 1 comprises “detecting the presence of a nucleic acid of foetal origin in the sample”.
1260 Ariosa contends that, as a matter of construction, the method of claim 1 involves the “detection” of nucleic acid sequences that are known or inferred in advance to be of fetal origin and not possessed by the mother.
1261 It says that the context provided by the Patent shows that “a nucleic acid of foetal origin” denotes that the nucleic acid is a particular nucleic acid, with a known characteristic and is being targeted for detection, i.e. the person looking for that nucleic acid has prior knowledge that enables them to target and detect a sequence on that target.
1262 It says that all of the broad methods and specific Examples described in the Patent show such an approach being taken. In each case, the relevant nucleic acid sequence that is targeted for detection can be assumed (with a high degree of certainty) in advance to be fetal in origin, if found. The only unknown is whether the targeted nucleic acid will be detected in the relevant serum or plasma sample.
1263 Further, it says that each of the qualitative techniques as described on pages 4 to 5 of the Patent depends on detecting the presence of cell free DNA that cannot originate from the mother, and therefore must be fetal. Each of these approaches requires access to information about the parental genomes in advance of performing the claimed detection method in order to distinguish between the fetal and maternal components of cell free DNA. Sex determination involves knowing in advance that the mother does not possess a Y chromosome. This is also the basis of the experiment reported in Example 1. Paragraph (a) on page 4 involves knowing that the mother was Rhesus D negative and so would not have the Rhesus D gene, as also described in Example 3 of the Patent. Paragraph (b) on page 4 involves knowing that the father and mother carry different mutations, and using the paternal mutation as an amplification target on maternal plasma and serum, so as to assess the risk that the fetus may be affected by a haemoglobinopathy. Paragraph (c) on pages 4 to 5 requires prior genotyping of the father and mother to find mutations or polymorphisms present in the father but not in the mother.
1264 Further, it says that the quantitative technique described at paragraph (a) of page 5 of the Patent is exemplified in Example 2 of the Patent using a Y chromosome primer which is known in advance to interrogate nucleic acid sequences that could only belong to the fetus, are plainly paternal, and not of the pregnant female.
1265 Further, it says that the quantitative technique described at paragraph (b) of page 5 of the Patent is said to involve “the quantitation of foetal DNA markers on different chromosomes”, which necessarily requires prior knowledge of these relevant markers. And it points out that Ms Norbury said that the implementation of this approach would require “prior genotyping and knowing what the parent alleles are”.
1266 Further, Ariosa points to the experts’ response to question 16 in their joint expert report. They were asked “Does the Patent require that the “nucleic acid of foetal origin” is known in advance to be of foetal origin” and answered “No and the Examples in the Patent are consistent with this”. But Ariosa says that the Examples only include experiments in which it was assumed that what was being targeted was a fetal nucleic acid sequence, because it was a sequence from the SRY gene, or a sequence from the rhesus D gene when the mother was rhesus negative. Ariosa says that the expert’s answer “No” is explained because one “infers” rather than “knows” that the targeted sequence will be of fetal origin if found. It points out that Professor Oepkes observed:
PROF OEPKES: I – I agree with what Ms Norbury said before on these issues. So for 16, require that the nucleic acid is of fetal origin, to know that in advance – I think it’s – it’s quite clear for some examples that what is essential is that in advance it should be known that the – the target that is looked for is not present in – in the mother, like the Y gene or the – the rhesus D gene, if you want to use this – the test described in – in the patent.
HIS HONOUR: Yes.
PROF OEPKES: It’s generally known that women don’t have a Y gene and that if you find it in a woman that is pregnant, then it is very likely coming from her fetus, because it’s – it’s highly unlikely that it comes from somewhere else. You – you don’t need to test the father for the presence of a Y gene of the father. But it is inferred that – that you know the fetal origin, because you know you are looking for something that is not present in the mother, and because she’s pregnant, it has to be from the fetus, unless there’s a tumour producing it.
…
PROF OEPKES: That – that is sort of the language ..... so is it known? You – you – I think you have to know it. But you would need to test for it.
HIS HONOUR: So you say as long as you target something which is non-maternal – you then infer from that, if you find it, that it’s fetal.
PROF OEPKES: If it – if it is a pregnant woman’s blood, then that is the most logical conclusion, unless you have a contamination or some weird tumour-like process .....
1267 Ariosa says that when Professor Oepkes said that “you would need to test for it”, that meant you would need to test for it to “know” as a matter of fact the nucleic acid was not maternal.
1268 Ariosa points out that Ms Norbury further commented that it will not be possible to interpret the results without prior knowledge of the relevant markers:
MS NORBURY: Yes. So this is again around it being an inference, and actually in order to actually perform the test itself – if you like, the wet chemistry – you don’t need to know, apart from if you’re – we’ve talked at length before about knowing if you’ve got informative markers or not. But if you do them and not know they’re informative, you can’t interpret some of the outcomes without knowing some further information. But as a sort of practicality, you wouldn’t necessarily need to have the information upfront and, therefore, this is a sort of scenario that often occurs in clinical diagnosis that you might have to see if you could interpret the results with the information you did have, and then pursue further information if needed.
…
1269 Ariosa also says that the claims also provide relevant context. Claims 2 and 3 require the amplification of “the foetal nucleic acid” and claim 4 requires use of a “foetal sequence specific oligonucleotide primer”. Ariosa says that those claims reinforce that the fetal nucleic acid must be identified in advance. The skilled person needs to know the boundaries of what has been claimed, and therefore whether what they are intending to amplify, and use a primer in respect of, is a fetal nucleic acid or not.
1270 Let me analyse Ariosa’s arguments, although I would say now that I reject its “known in advance” argument.
1271 Claim 1 and its dependent claims claim a “detection method” which method comprises “detecting the presence of a nucleic acid of foetal origin”.
1272 It seems to me that “detect” is not a term of art. The Oxford Dictionary of English broadly defines the term as, inter-alia, “[t]o uncover, lay bare, expose, display (something covered up or hidden)” and “[t]o discover, find out, ascertain the presence, existence, or fact of (something apt to elude observation)”. This ordinary English definition is consistent with the use of the term in the body of the Patent, that is, that its analysis methods can be used to ascertain the presence of fetal nucleic acid in maternal plasma and serum, and its non-exhaustive, exemplary cffDNA methods.
1273 Further, in the joint expert report, the experts agreed that consistently with the foregoing, in the context of the Patent as a whole “detecting the presence of a nucleic acid of foetal origin” in claim 1 of the Patent simply means demonstrating the presence of non-maternal nucleic acid that is inferred to be of fetal origin. Most of the experts also appeared to agree that the Patent does not require that the “nucleic acid of foetal origin” is known in advance to be of fetal origin.
1274 Further, in the context of the phrase “detecting the presence of a nucleic acid of foetal origin”, detection requires discriminating or distinguishing between nucleic acids of fetal origin on the one hand, and nucleic acids of maternal origin on the other. Without such a step, one cannot detect or identify cell-free fetal DNA, as opposed to simply extracellular DNA from an unknown source, in the sample.
1275 But claim 1 and its dependent claims contain no limitations as to:
(a) the necessity of a prior genotyping step or “knowing in advance” that the targeted sequence is likely to be fetal; or
(b) false positive or false negative rates for clinical application.
1276 Claims 1, 6 and 9 do not require any prior genotypic or phenotypic analysis. They simply require specified (e.g. from Y and / or non-Y chromosomes) fetal DNA sequences to be distinguished and therefore “detected”.
1277 Although the respondents contend that the claims require the person skilled in the art to target sequences that they would know or infer in advance of performing the detection assay would not be possessed by the pregnant female, e.g. through the prior genotyping of the parents or the assessment of the mother’s phenotype (e.g. in the case of RhD detection), as I have said most of the experts agreed that the Patent does not require the “nucleic acid of foetal origin” to be known in advance to be of fetal origin. And as the experts noted, most of the Examples in the Patent are consistent with this.
1278 Further, at page 4, the Patent gives RhD detection as the first example of a method of the detection of paternally-inherited sequences which are not possessed by the mother. However, such methods are not described as involving a prior analysis of the father’s genotype or the genotyping of the mother. In this example, a partial paternal genotype is deduced from a partial fetal genotype.
1279 Further, Professors Fisk and Lovett observed that the identification of a nucleic acid of fetal origin can happen at any time e.g. without a priori knowledge of a paternal genotype.
1280 Let me address the next question of construction.
“Determining the concentration” (claim 14)
1281 Ariosa points out that the detection method of claim 14 of the Patent comprises “determining the concentration of the foetal nucleic acid sequence in the maternal serum or plasma”. It says that “concentration” refers to absolute concentration.
1282 It says that as initially agreed by all of the experts in the joint expert report:
Concentration is an amount per unit volume. The experimental data that are reported most commonly use copies per ml. These are derived by measuring relative concentrations compared to a standard.
1283 Ariosa says that the ordinary meaning of “concentration” is an amount per unit volume, and that in the Patent the experimental data that are reported most commonly use copies per ml.
1284 But I would note at this point that Professors Fisk and Lovett asserted by way of supplementary statements to the joint expert report that “concentration” includes both absolute concentration and relative concentration.
1285 Professor Fisk said:
1. In considering the Joint Expert Report and my Affidavit in preparation for the hearing, I realised that in the answer to Q19, I omitted additionally to specify the second meaning of “concentration” referred in the Patent, namely “relative” or “fractional concentration”, as I had addressed in my first affidavit at [76] and [109]-[111].
2. I…seek leave to vary my response to Q19 by adding the following:
“NF notes that concentration can also be defined in relative terms as an amount proportionate to another amount. Experimental data are reported in the Patent as the fractional concentration of fetal DNA relative to total DNA.”
1286 Professor Lovett said:
1. In preparing for the hearing my attention was drawn to the response to Q19 in the Joint Report. In seeking to explain what I had meant by the second sentence I realised that I had not clearly identified the contrast between absolute concentrations, which are accurately described in the first sentence, and relative concentrations, to which we referred in the second sentence.
2. The meaning I had intended to convey in the second sentence was:
By referring to “measuring relative concentrations compared to a standard” I intended a reference to the measurement of a reporter (eg beta globin) that is present in both the maternal and fetal genomes versus the measurement of a separate reporter (eg SRY) that is only present in a fraction of the sample.
3. …On reflection, I think the ambiguity arose by the use of the words “these are derived”. I intended “these” to refer to concentration, but it is clear they could be misunderstood as referring to “copies per ml”, which was not my intention.
1287 Now Ariosa has invited me to draw an inference that this construction only arose with an eye to infringement, but I am not prepared to draw such an inference.
1288 Let me return to Ariosa’s arguments.
1289 Ariosa says that the context provided by the specification itself, and by the claims, makes clear that “concentration” is being used in its ordinary meaning. It says that Ms Norbury commented: “concentration is amount [per] volume, and in all of the examples in the patent they talk about copies per mil”. Further, it says that as Professor Hyett also explained:
PROF HYETT: So I – I would agree that concentration is defined with some units – it’s an amount per mil – and that if you divide one concentration by another in that process, you actually abolish the units.
HIS HONOUR: Yes. Yes.
PROF HYETT: It’s then a fraction. So they’re for me conceptually different…
1290 Contrastingly, the construction asserted by Sequenom is that it can also mean “fractional concentration” or “relative concentration”, and therefore that it includes within its meaning a ratio. But Ariosa says that claim 14 refers to the “concentration of the foetal nucleic acid sequence in the maternal serum or plasma”. It says that no comparator is referred to, which is to be contrasted with claim 20 which refers to “fractional concentration” and includes a comparator (“total DNA”). It says that the reference to the concentration in the serum is also consistent with concentration being a measure of something in a volume.
1291 Ariosa also points out that Professor Lovett agreed that there is a relevant distinction between claim 14 and claim 20:
PROF LOVETT: Yes, and it could be either, all right? He’s using – they use it so interchangeably in here. It could be either, and he doesn’t say “absolute concentration”, or he doesn’t even say, you know, what unitage. So it could be either.
MR RYAN: Do the words in 14 “concentration of fetal nucleic acid sequence in the serum or plasma” – are those later words, “in the serum or plasma”, cause you to modify your ambivalence?
PROF LOVETT: No, because it then says, “is, by quantitative PCR”, and I know that quantitative PCR measures relative concentrations.
MR RYAN: But you agree, plainly, that when he wants to underline the fact that he’s talking about fractional concentration in claim 20, he does say.
PROF LOVETT: I agree that he makes the distinction there, yes; that’s absolutely true.
1292 Claim 14 is dependent on claims 6 to 12. Claims 6 to 9 refer to a specific sequence from the Y chromosome being detected, and claims 9 to 12 similarly refer to a similar sequence from a non-Y chromosome being detected, and the specific example of the RhD gene is given. Ariosa says that none of those claims suggests any comparator with which the concentration of the specified sequence is to be compared.
1293 Ariosa says that there are, however, specific references to the significance of the concentration of fetal nucleic acid. At paragraph (a) on page 5 the Patent asserts that there is a correlation between the concentration of fetal DNA and aneuploidy, which is expanded upon in Example 2 of the Patent. At the bottom of page 5, an alternative correlation is asserted, this time with pre-eclampsia. Ariosa says that the Patent therefore makes clear that it is the concentration of fetal nucleic acid that is of significance.
1294 Conversely, it says that there is no example or description in the Patent of any prenatal diagnosis of any kind that arises from determining the “fractional concentration” of fetal nucleic acid. The only description of fractional concentration is in the context of Example 5 of the Patent, and there the description is given solely for the purpose of demonstrating that the amount of cffDNA that is actually present in maternal plasma or serum was found to be significantly higher than would be predicted by reference to the concentration of whole fetal cells. The higher amount meant that a relatively small volume of plasma or serum could be analysed.
1295 Moreover, Ariosa says that claims 18 and 19, which are dependent on claim 14 (via claim 17) specifically pick up the asserted correlation between concentration and each of aneuploidy and pre-eclampsia. Claim 16, which refers to a comparison of “the level of foetal DNA” in the serum or plasma is also consistent with “concentration” having its ordinary meaning.
1296 Further, Ariosa says that the Patent also reports the results of the Examples by reference to a serial dilution curve, and expresses the amount of cffDNA or total cell free DNA (i.e. fetal and maternal) in a defined volume (i.e. units of copies / ml). An equation is also provided for determining concentration expressed in copies / ml. This is referred to in the Patent as being the “absolute concentration” of cffDNA or of total cell free DNA (i.e. fetal and maternal) (page 12 line 19).
1297 I do not accept Ariosa’s contentions concerning the construction of “concentration”.
1298 In my view the term “concentration” in the context of the Patent refers to the absolute or relative concentration of the fetal nucleic acid sequence in the maternal plasma. I reject Ariosa’s contention that “concentration” in claim 14 is limited to absolute concentration.
1299 I accept Professor Fisk’s explanation that in the field of molecular biology concentration can be expressed as an absolute percentage of mass in a given volume or as a fraction or percentage of another relevant parameter (i.e. relative concentration). And as I have set out earlier, in a supplementary statement to the joint expert report, Professor Fisk confirmed this view and noted that experimental data are reported in the Patent as the fractional concentration of fetal DNA relative to total DNA.
1300 Professor Lovett also considered that the term “concentration” as used in claim 14 covers the determination of the absolute or relative concentration of the fetal nucleic acid sequence in the maternal plasma. And as I have also set out above with reference to an answer that he gave in the joint expert report, Professor Lovett later explained:
By referring to “measuring relative concentrations compared to a standard” I intended a reference to the measurement of a reporter (e.g. beta globin) that is present in both the maternal and fetal genomes versus the measurement of a separate reporter (e.g. SRY) that is only present in a fraction of the sample.
1301 In my view claim 14 does not include any limitation to absolute concentrations, and covers both absolute and relative or fractional concentrations. Both concepts are described and exemplified in the Patent.
1302 For example, the first mention of “concentration” in the Patent relates to “relative” concentration (page 2 line 12):
In fact, the concentration of foetal DNA in maternal plasma expressed as a % of total DNA has been measured as from 0.39% (the lowest concentration measured in early pregnancy), to as high as 11.4% (in late pregnancy), compared to ratios of generally around 0.001% and up to only 0.025% for cellular fractions (Hamada et al 1993).
1303 Further, Example 5 refers to both absolute and relative concentrations and provides methods to calculate both figures.
1304 It is well apparent that the Patent uses the term “concentration” to refer to both absolute and relative concentrations. But the Patent does not always use the term relative or fractional when referring to relative concentration.
1305 Further, Ariosa can draw no comfort from claim 20. The fact that claim 14 includes both relative and absolute concentration is not inconsistent with claim 20, which relevantly depends on broader claims, including claim 14, and provides:
The method according to any one of claims 1 to 19, wherein the sample contains foetal DNA at a fractional concentration of total DNA of at least about 0.14%, without subjecting it to a foetal DNA enrichment step.
1306 So claim 20 narrows relative concentration in terms of percentage, which is not inconsistent with claim 14 referring to both relative and absolute concentration. Indeed, if “concentration” in claim 14 only meant absolute concentration then there may be a tension with its inclusion in claim 20. The better reading is that claim 14 refers to both types and that claim 14 is narrowed by claim 20 stipulating a percentage when relative concentration is in play.
1307 Let me now turn to the other integer in dispute.
“Prenatal diagnosis” (claims 22, 23, 25 and 26)
1308 Claims 22, 23, 25 and 26 each relate to “a method of performing a prenatal diagnosis” and claim 22 further involves the step of “providing a diagnosis”. Ariosa says that the term “a prenatal diagnosis” or “a diagnosis” here means a diagnostic test, not a screening test. Ariosa says that that is not to say that the term “prenatal diagnosis” cannot cover both as an umbrella term, but the Patent does not do so when using the term “a prenatal diagnosis”.
1309 Let me begin by discussing Ariosa’s contention as to how “prenatal diagnosis” is understood by the skilled addressee.
1310 Ariosa says that as explained by Ms Norbury “prenatal diagnostic tests are those which enable a clinical diagnosis and decision”. As explained by Professor Hyett “[a] diagnostic test is one that allows a clinician to rely on the result to determine that a patient has a particular condition”. Professor Hyett also explains that, in the context of fetal medicine, the consequences that may arise from a diagnosis are significant, including a decision in relation to termination of pregnancy. As a result, diagnostic tests in this field must produce highly accurate and reliable results. As further explained by Professor Oepkes, prenatal diagnostic tests include amniocentesis and CVS, which are considered to be very accurate and reliable, but are invasive and increase the risk of the mother suffering a miscarriage.
1311 Ariosa says that prenatal diagnostic tests may be contrasted to prenatal screening tests. As explained by Ms Norbury, a prenatal screening test involves gathering information that allows a risk assessment to be made, and from which further tests may be ordered (if required) to allow a diagnosis to be made. Results from a screening test could not be relied upon by a clinician to make a clinical decision, such as to terminate a pregnancy. That is, screening tests results are not clinically actionable. Professor Hyett described that a screening test provides an assessment of the risk that a person has a condition. Where a disease has a low prevalence in the population and the diagnostic test for that disease carries a relatively high risk, a screening test can be used to identify a sub-group of “high risk” patients, for whom specific diagnosis is more worthwhile, and the screening process also allows patients to be identified as “low risk” and reassured that further testing is less likely to be fruitful. Screening tests typically reduce the number of invasive tests that need to be conducted. Professor Oepkes was of a similar view. He explained in the context of Down syndrome that the aim of screening methods is to identify more accurately those women at particular risk of carrying a fetus affected by Down syndrome, so that they can be offered invasive testing.
1312 Ariosa says that Professor Hyett further explained that diagnosis and screening can lie on a spectrum and the criteria for each may depend on the context in which it is delivered and the consequences of an incorrect diagnosis:
PROF HYETT: I think that defining screening and diagnosis, really, they lie on a spectrum, because a diagnostic test in an ideal world has 100 per cent sensitivity and 100 per cent specificity and, therefore, also has 100 per cent positive predictive value despite what the prevalence may be. The reality is throughout medicine there are very few tools that meet those absolute levels and, as a consequence, there is some trade-off for the standard you have of diagnosis. Now, if you’re making a diagnosis in the context of them potentially having a conversation about termination of pregnancy, one wants a very high standard where you approach those limits as close as you can. If you were having a different conversation with a patient about whether they should take an anti-hypertensive for their high blood pressure as an adult, you may have a different standard for diagnosis.
1313 Further, Ariosa says that Professor Fisk broadly agreed with the above descriptions of diagnosis and screening, but explained that these concepts exist along a spectrum or continuum. He said:
So, your Honour, looking for consensus here, I think I’ve heard that screening and prenatal diagnosis, broadly, can be an umbrella term; that screening and diagnosis are like a spectrum – I think the word “continuum” was used, but I think we would agree on the principles that there are things which separate the two. The context is very important here: both the context of positive and negative results versus intermediate results…The patent uses the term in an umbrella term. In fact, at times it uses it somewhat interchangeably.
1314 Ariosa says that he further explained that for determining fetal sex and RhD status, non-invasive prenatal testing (NIPT) had developed to a point where it was sufficiently certain that such tests could be considered diagnostic. Professor Fisk also considered that the broad field of fetal medicine includes two sub-areas of prenatal diagnosis and fetal therapy.
1315 Ariosa says that Ms Norbury explained that the term “prenatal diagnosis” is sometimes loosely used as an “umbrella term” for any prenatal risk assessment, and Professor Hyett gave a similar explanation in relation to uses of the term “prenatal diagnosis” in the context of a description of an area of fetal medicine. But Ariosa says that “prenatal diagnosis” as a field of endeavour, which plainly encapsulates screening, is very different to “a prenatal diagnosis” of a particular human subject as contemplated by claims 22 and following of the Patent.
1316 Let me next turn to Ariosa’s contentions as to how “prenatal diagnosis” is used in the Patent.
1317 Ariosa says that the first paragraph of the Patent describes how the invention “relates to prenatal detection methods using non-invasive techniques”. The Patent then describes how, “[i]n particular, [the invention] relates to prenatal diagnosis by detecting foetal nucleic acids in serum or plasma from a maternal blood sample”.
1318 The third paragraph of the Patent then describes prior art screening tests for Down Syndrome and distinguishes those with the “use of foetal cells in maternal blood for non-invasive prenatal diagnosis (Simpson and Elias 1993) [which] avoids the risks associated with conventional invasive techniques”. The conventional invasive techniques are clearly the prenatal diagnostic tests available then and now.
1319 Ariosa says that whilst the paper, Simpson JL and Elias S, “Isolating Fetal Cells From Maternal Blood: Advances in Prenatal Diagnosis Through Molecular Technology” (1993) 270(19) JAMA 2357-2361 (Simpson and Elias 1993) cannot be used to construe the Patent (unless that paper is taken to be incorporated by reference), it is worth noting that in Simpson and Elias 1993, the authors isolated nucleated red blood cells and conducted FISH with chromosome-specific probes which allowed them to detect trisomy 21 and trisomy 18, in the same way that CVS and amniocentesis diagnosed those fetal aneuploidies. The authors there also distinguished between prenatal diagnostic tests and screening. They observed that “isolation of fetal cells from maternal blood for prenatal diagnosis may be accomplished in the first trimester as well as in the second trimester”, but further considered that:
Ideally, the sensitivity, specificity, positive predictive value, and negative predictive value will prove sufficient for this noninvasive approach to serve as the definitive diagnostic test for fetal aneuploidy. Even if this stringent requirement is not met, we believe that this method could still play an important role in prenatal screening for fetal aneuploidy, either as an independent test or as an added benefit in combination with other tests…
1320 Ariosa then says that on page 2 of the Patent (lines 20 to 23) it is explained that “[t]his invention provides a detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of foetal origin in the sample” (consistent with claim 1 of the Patent). The Patent goes on to say that the invention “thus provides a method for prenatal diagnosis”. Notably, according to Ariosa, the Patent here does not say “a prenatal diagnosis”. Again, Ariosa says that the distinction is important because whilst prenatal diagnosis as a field may encompass both screening tests and prenatal diagnostic tests, it contends that a prenatal diagnosis or a prenatal diagnosis test differs from a screening test.
1321 Further, Ariosa says that from line 24 on pages 2 to 3, the Patent goes on to say that:
The term “prenatal diagnosis” as used herein covers determination of any maternal or foetal condition or characteristic which is related to either the foetal DNA itself or to the quantity or quality of the foetal DNA in the maternal serum or plasma. Included are sex determination, and detection of foetal abnormalities which may be for example chromosomal aneuploidies or simple mutations. Also included is detection and monitoring of pregnancy-associated conditions such as pre-eclampsia which result in higher or lower than normal amounts of foetal DNA being present in the maternal serum or plasma. The nucleic acid detected in the method according to the invention may be of a type other than DNA e.g. mRNA.
1322 Ariosa says that this passage is not defining “a prenatal diagnosis” but instead is defining “prenatal diagnosis” in general as an umbrella term to contemplate determination (diagnosis) and detection (screening) of maternal or foetal conditions or characteristics. But the Patent then distinguishes between a diagnosis (or determination) and screening (or detection or quantification) of a fetal or maternal condition. For example, pages 3 to 4 line 29ff then explains that methods can be used for “sex determination” where finding a Y chromosome will be determinative of the fetus being male.
1323 Ariosa says that the methods described on page 4 of the Patent, which relate to detecting a specific fetal nucleic acid, all relate to methods that would be determinative as to whether the fetus is rhesus D positive, has a relevant haemoglobinopathy, or has the specific DNA polymorphism or mutation that is associated with a disease. At page 4 lines 29 to 30 the Patent contemplates that that the presence of a relevant fetal nucleic acid may be capable of being used for “diagnostic analysis”. In particular, paragraph (c) on page 4 describes sex determination as a “diagnostic analysis”, rather than a screening test making clear that “determination” should be equated with diagnosis (see page 4, lines 29 to 30 of the Patent).
1324 Ariosa says that language contrasts with the introduction on page 5 (at line 4) of the Patent to the use of a method that can be applied to “screening”. The sentence used there is instructive as to the patentee’s use of that terminology as it refers to the “plasma or serum non-invasive prenatal diagnosis method according to the invention” which can be “applied to screening”. That is, the term “diagnosis” is being used in this context as an umbrella term, but the patentee is being more precise as to the application of that method, namely in a screening method.
1325 Further, on page 6, lines 3 to 6, the Patent explains how the “nucleic acid-based diagnosis methods” described in the Patent may be incorporated into prenatal screening programmes. It says that what is meant by “nucleic acid-based diagnosis methods” is unclear and does not appear to assist in construing the term “a prenatal diagnosis” in the claims.
1326 Ariosa says that the next mention of “diagnosis” in the Patent is in Example 2 on page 9 from line 15 which distinguishes between prenatal screening and diagnosis of fetal chromosomal aneuploidies. The Patent notes that invasive methods such amniocentesis (a known diagnostic test) are not suitable for screening (because of the risks associated with them). Lines 27 to 29 on page 9 revisit Simpson and Elias 1993 noting that fetal nucleated cells in maternal circulation offer a new source of material for non-invasive diagnosis. Simpson and Elias 1993 concerned the possibility of diagnosis by FISH of nucleated fetal cells, as is done for amniocentesis.
1327 Example 3 on page 16 lines 3 to 6 of the Patent concludes, in the context of RhD status determination, that the discovery of fetal DNA in maternal plasma and serum “offers a new approach for non-invasive prenatal diagnosis”. Example 3 then goes on to explain at page 19, lines 6 to 12 how non-invasive fetal RhD genotyping from maternal plasma provides a “single gene diagnosis from maternal plasma” and that the highly accurate results can “potentially be used for clinical diagnosis” but that multiple primer sets would be preferred to provide “robust clinical diagnosis”. Ariosa says that here the Patent is describing a diagnostic test rather than screening.
1328 The next reference to prenatal diagnosis is made in Example 5 at page 23 line 24ff and likely used (but not clearly), so Ariosa says, in the umbrella sense because, at page 24 line 10 to 13, the Patent explains that they “show that the reliable detection of foetal DNA is achievable and therefore useful for the non-invasive prenatal diagnosis of selected genetic disorders”.
1329 Further, in the discussion of Example 5, at page 31 line 25ff, the Patent describes how fetal DNA analysis in maternal plasma and serum “would be most useful in situations where the determination of foetal-derived paternally-inherited polymorphisms/mutations or genes would be helpful in clinical prenatal diagnosis”. According to Ariosa what follows then is a list of examples of diagnostic rather than screening tests. Examples include foetal sex determination for the prenatal diagnosis of sex-linked disorders, foetal rhesus D status determination in sensitized rhesus negative pregnant women (Lo et al. 1993), autosomal dominant disorders in which the father carries the mutation and autosomal recessive genetic disorders in which the father and mother carry different mutations (Lo et al. 1994), certain hemoglobinopathies (Camaschella et al. 1990) and cystic fibrosis. Ariosa says that may be distinguished from the next paragraph at page 32 line 16ff where a new screening test for aneuploid pregnancies is discussed and compared.
1330 Further, Ariosa says that the experts acknowledged in the joint expert report, that “[i]n other parts of the Patent the usually discrete terms screening and diagnosis are used broadly and interchangeably, and often do not meet accepted criteria”. Ariosa says that the accepted criteria the experts appear to be referring to were the criteria for a screening test versus a diagnostic test. But according to Ariosa, the Patent does distinguish between the general field of prenatal diagnosis, which may include screening, and a particular diagnostic test, which is not a screening test.
1331 Further, Ariosa says that Professor Fisk accepted that the phrase “diagnostic test” was “also used in a narrower sense to cover more accurate invasive tests”. Nevertheless, he went on to explain that the terms “method of performing a prenatal diagnosis” and “providing a diagnosis” in the claims and specification of the Patent had a broader meaning of including screening tests, but that was based on his understanding that the phrase “prenatal diagnosis” was used broadly to cover diagnostic tests and screening tests. But Ariosa says that that is the field of prenatal diagnosis, not the conducting of a prenatal diagnosis.
1332 In my view, it is worth setting out what he said in more detail (at [112] to [114]):
As is apparent from my discussion of the field of fetal medicine above, at the priority date, the phrase “prenatal diagnosis” was generally used broadly to cover methods designed to investigate the risk that a fetus has a birth defect or fetal characteristic (such as fetal sexing).
While the phrase “diagnostic test” was also used in a narrower sense to cover more accurate invasive tests like amniocentesis and CVS, it is my view, based on a reading of the Patent as a whole and in view of my knowledge outlined above, that in the claims and specification of the Patent, the phrase “method of performing a prenatal diagnosis” or “providing a diagnosis”, has the broader meaning referred to in paragraph 112 above (and includes, for example, determining the risk that a fetus possesses a particular characteristic or condition so that, for example, a medical or clinical decision may be made to progress with the pregnancy or perform further tests).
I consider that at least the following matters support my opinion:
(a) First, the Patent uses the word “screening” to cover invasive “diagnostic” techniques such as amniocentesis and chorionic villus sampling (see, for example, page 1, lines 7 to 11, and page 5, lines 5 to 16);
(b) Second, the Patent expressly teaches that the “non-invasive diagnosis method according to the invention can be applied to screening for Down’s Syndrome and other chromosomal aneuploidies” (page 5, lines 3 to 6) (my emphasis);
(c) Third , the Patent also teaches that “it is anticipated that it will be possible to incorporate the nucleic acid-based diagnosis methods described herein into existing prenatal screening programmes” (page 6, lines 3 to 5) (my emphasis);
(d) Fourth, at the priority date, as is apparent from my discussion at paragraphs 24 to 35 above, research was being undertaken as to methods involving cellular fetal DNA obtained from maternal blood samples. The cellular fetal DNA research work is described in the Patent as using fetal cells for non-invasive prenatal diagnosis. The cellular fetal DNA work was commonly referred to as methods of non-invasive prenatal diagnosis in the literature, even though, at the priority date, the technique did not have a clinical application and it was being explored whether the test could be used as a screening method or as a definitive diagnostic test;
(e) Fifth, the Patent's definition of “prenatal diagnosis” includes the “detection” of aneuploidy, in circumstances where the Patent refers to screening for aneuploidy; and
(f) Sixth, the Patent states that the invention is useful even if a detection rate of less than 100% is achieved (see, for example, page 23, lines 15 – 24).
(Original emphasis.)
1333 Finally, Ariosa says that the language used in claims 22, 25 and 26 does not refer to “prenatal diagnosis method” in the broad, but rather to a method of performing “a prenatal diagnosis”, and to “providing a diagnosis”. That language, read in the overall context of the Patent, has the narrower meaning of diagnostic test, not a screening test. It also contrasts with the language used in claims 18 and 19 which refer to “detection of” pre-eclampsia and aneuploidy in the broader sense.
1334 Generally speaking I do not accept Ariosa’s arguments.
1335 The Patent explains (page 2 line 24) that, in the context of the Patent, “prenatal diagnosis” covers:
…determination of any maternal or foetal condition or characteristic which is related to either the foetal DNA itself or to the quantity or quality of the foetal DNA in the maternal serum or plasma. Included are sex determination, and detection of foetal abnormalities which may be for example chromosomal aneuploidies or simple mutations. Also included is detection and monitoring of pregnancy-associated conditions such as pre-eclampsia which result in higher or lower than normal amounts of foetal DNA being present in the maternal serum or plasma. The nucleic acid detected in the method according to the invention may be of a type other than DNA e.g. mRNA.
1336 Professor Fisk’s evidence is that at the priority date the phrase “prenatal diagnosis” was generally used broadly to cover methods designed to investigate the risk a fetus had a birth defect or fetal characteristic.
1337 Further, Professors Fisk and Lovett gave evidence that in the context of the Patent the term “prenatal diagnosis” is used in its broader “umbrella” sense and encompasses methods of determining the likelihood or risk that a fetus has a particular fetal condition or characteristic. That is, it includes “screening” techniques as well as accurate “diagnostic” techniques, being terms used interchangeably in the Patent as well as by the respondents’ expert witnesses and other persons skilled in the art at the priority date.
1338 As explained by Professor Fisk:
The Patent uses the term in an umbrella term. In fact, at times it uses it somewhat interchangeably. It refers to non-invasive prenatal diagnosis of fetal cells in maternal blood. For aneuploidy that’s not likely to be diagnostic in the pure sense. It also refers to screening tests of amniocentesis and CVS, which we would cluster definitely in the diagnostic end.
1339 Further, Professor Hyett stated that, in the context of the Patent, the term covers a range of possibilities and is used in a broad sense:
MR SHAVIN: Yes. And what I’m suggesting to you is that the language that has been used in the patent is indicating to you something that is broader than you might, independently of the patent, have understood by the word “diagnosis”.
PROF HYETT: It’s certainly broader than I would use to define screening or diagnosis, and personally, I find it on occasion confused.
1340 Now although “screening” and “diagnosis” may be distinguished in some contexts outside the Patent, in the particular context of the Patent the terms were used broadly and interchangeably, although some examples in the Patent lend themselves more to diagnosis and others more towards screening.
1341 Further Professor Fisk considered that “screening” tests are encompassed by the phrase “method of prenatal diagnosis”.
1342 Further and more generally, I agree with Sequenom that the definition and the use of the term “diagnosis” generally in the Patent is inconsistent with the various problematic narrower definitions of the term “diagnosis” advanced by the respondents and their experts, namely:
(a) “highly accurate” invasive tests upon which:
(i) a clinician can rely to determine that a patient has a particular condition; or
(ii) a “clinical decision” can be made to offer termination of pregnancy; or
(iii) a fetus can be “defined” as having a normal chromosomal content or having aneuploidy;
(b) the “final” test preceding clinical “treatment”, “intervention” or “advice”; or
(c) the “gold standard” test for a particular condition.
1343 Further, I note that in this respect, the Patent relevantly:
(a) indicates that detection rates of 70 to 80% are high enough to be useful;
(b) notes that the methods of the invention in Example 3 can potentially be used for clinical diagnosis; and
(c) notes that the real time quantitative detection methods of the invention in Example 5 are an attractive candidate for large scale clinical application.
1344 Further, as Professor Fisk explained, both “screening” and “diagnostic” tests involve the making of assumptions and are associated with degrees of certainty, confidence levels and errors. Professor Hyett similarly acknowledged that narrowly defined diagnostic tests can involve errors and assumptions.
1345 Further, screening and diagnostic tests, even if narrowly defined, can lie on a spectrum. Further, there can also be overlap between “screening” and “diagnostic” tests and the line between them can be blurred. As explained by Professor Hyett:
I think that defining screening and diagnosis, really, they lie on a spectrum, because a diagnostic test in an ideal world has 100 per cent sensitivity and 100 per cent specificity and, therefore, also has 100 per cent positive predictive value despite what the prevalence may be. The reality is throughout medicine there are very few tools that meet those absolute levels and, as a consequence, there is some trade off for the standard you have of diagnosis. Now, if you’re making a diagnosis in the context of them potentially having a conversation about termination of pregnancy, one wants a very high standard where you approach those limits as close as you can. If you were having a different conversation with a patient about whether they should take an anti-hypertensive for their high blood pressure as an adult, you may have a different standard for diagnosis.
1346 As Sequenom points out, the overlap can be illustrated by the language used by persons skilled in the art before the priority date. For example, Dr Bianchi said of non-invasive fetal cell detection techniques, “[a]ny prenatal diagnostic screening test relying exclusively on placental material may give a falsely abnormal fetal result in some cases (emphasis added)” (Bianchi DW, “Prenatal Diagnosis by analysis of fetal cells in maternal blood” (1995) 127(6) Journal of Pediatrics 847-856.
1347 And even seemingly accurate diagnoses can be erroneous. Indeed, as recognised by Ms Norbury “[t]he reports that I prepared described results, set out an interpretation of those results, explained why I had made the particular diagnosis and included calculations of my confidence in the diagnosis” and “[a]llelic drop out is also another source of inaccuracies and / or failure in genetic diagnosis…It can be difficult to recognise when allelic drop out occurs, as the PCR appears successful, with the results suggesting that the individual tested is homozygous for that allele. This can result in a misdiagnosis, loss of the ability to differentiate between individuals, and false assumptions about parentage or genetic diversity”.
1348 Further, even if screening tests are not used to make a clinical decision to terminate a pregnancy, they are used to make clinical decisions or to give advice as to the necessity of further tests.
1349 Further, the Patent’s definition is consistent with the way persons skilled in the art used the term at the priority date, namely, to describe the field of techniques used to obtain information regarding whether a fetus has a risk of having a particular condition or characteristic. For example, Professor Hyett explained that he “presented lectures on prenatal diagnosis” when working at University College Hospital London between 1998 and 1999 and UCL between 2000 and 2003 which “were focused on the tests available at that time, such as ultrasound, second trimester serum screening and combined first trimester screening, CVS and amniocentesis, as well as some of the issues associated with screening for chromosomal aneuploidies such as trisomy 18 and 21…These lectures also covered other genetic conditions such as cystic fibrosis, sickle cell anaemia and thalassemia, but in less detail than other topics”. And he explained that throughout his career, he had “published widely in the area of prenatal diagnosis, particularly in relation to the use of ultrasound to screen for chromosomal aneuploidies and identify other abnormalities, the underlying pathophysiology of increased nuchal translucency, identification and use of cell free fetal DNA isolated in maternal circulation, as well as in relation to pre-eclampsia and Rhesus D”.
1350 Further, Professor Fisk noted that the term “prenatal diagnosis” was used broadly in, for example, the titles of the journals Fetal Diagnosis and Therapy and Prenatal Diagnosis, being journals that covered “screening” and “diagnostic” methods.
1351 Further, as Sequenom points out, Professor Oepkes similarly recognised that cffDNA techniques have developed after the priority date to “diagnose” fetal chromosomal aneuploidy, even though all of the experts agree that such modern techniques cannot be said to diagnose aneuploidy in any narrower sense of that term.
1352 Finally, Ariosa made reference to some evidence given by Professor Fisk in the US in 2013, but I do not think that this evidence takes Ariosa anywhere. Professor Fisk could not recall what he meant. Further, as he said, there can be accuracy issues with both types of test. If it needs to be said, I do not consider that this other evidence at all impacted upon Professor Fisk’s reliability.
1353 Moreover, various disclosures in the Patent support Sequenom’s position.
1354 First, the Patent uses the word “screening” to cover invasive “diagnostic” techniques such as amniocentesis and CVS. Given that amniocentesis and CVS were diagnostic tests at the priority date, it cannot be said that the patentee uses “screening” and “diagnosis” carefully or precisely to distinguish between such tests.
1355 Second, the Patent’s definition of “prenatal diagnosis” is broad enough to cover the determination and detection of simple mutations such as haemoglobinopathies, in circumstances where the Patent explains (page 4, lines 17 to 23) that the invention can be used to assess the risk that the fetus may be affected by haemoglobinopathies.
1356 Third, the Patent teaches that the non-invasive diagnosis method according to the invention can be applied to screening for Down syndrome and other chromosomal aneuploidies (page 5 line 5).
1357 Fourth, the Patent also notes that “[i]t is anticipated that it will be possible to incorporate the nucleic acid-based diagnosis methods described herein into existing prenatal screening programmes” (page 6 line 3).
1358 Fifth, the Patent’s definition of “prenatal diagnosis” includes the detection of aneuploidy, in circumstances where the Patent refers to screening for aneuploidy.
1359 Sixth, the Patent states that the invention is useful even if a detection rate of 70 to 80% is achieved (page 23 lines 20 to 24).
1360 Seventh, the Patent notes that the cffDNA methods described therein provide a new screening test for fetal chromosomal disorders (page 32 line 20).
1361 Eighth, the Patent’s disclosure recognises that cffDNA would likely provide a mechanism of screening for chromosomal aneuploidy, rather than being a diagnostic tool. Accordingly, the Patent’s disclosure is inconsistent with a narrow definition of “diagnosis”.
(d) Conclusions in respect of the relevant claims
1362 Let me now summarise my conclusions in respect of the relevant claims.
Claim 1 – detecting the presence of a nucleic acid of fetal origin
1363 As I have indicated, the Polymorphic Assay relies on the interrogation of hundreds of loci on chromosomes 1 to 12, in order to identify informative SNPs. But prior to performing that assay, it is not known or inferred in advance which loci will be informative.
1364 As I have indicated, Ariosa says that there is a difference in approach between the Polymorphic Assay and the approach described in the Patent. It says that the techniques described in the Patent involve first identifying a nucleic acid sequence which ought not be found in the mother’s genome, then seeking to detect that sequence in maternal serum or plasma. Thus, it says that the techniques described in the Patent require that it be known or inferred in advance that, if the sequence is found in the maternal serum or plasma, it will be fetal in origin (subject to rare events that might affect the result). Put another way, the claimed invention relies on detecting a fetal nucleic acid sequence that is known (or inferred) in advance to be derived from the father and not the mother.
1365 In contrast, so Ariosa says, the Polymorphic Assay of the Harmony Test interrogates hundreds of SNPs without any prior knowledge or inference available that any particular one of the hundreds of SNPs will be informative, i.e., that the mother and fetus will have different alleles for that SNP. If a SNP is informative, the Harmony Test can distinguish the maternal and fetal allele on the microarray, and uses this information to estimate the fetal fraction.
1366 Let me deal with a related point that excited some interest during the trial. Ms Norbury cited the prospect of false positives, from for example, de novo mutations, as a reason why it is not known in the Harmony Test that the minor fraction is fetal (i.e. paternally-inherited) or that even the Y chromosome is paternally inherited.
1367 But in my view, it seems to me that there was a very small prospect of false positive results.
1368 Both the Non-Polymorphic and Polymorphic Assays of the Harmony Test operate on the principle of detecting the presence of paternally-inherited nucleic acid of fetal origin from a Y and non-Y chromosome, respectively. It is a fundamental assumption of the Non-Polymorphic Test that the Y chromosome is paternally inherited, and of the Polymorphic Assay that the minor fraction is fetal (i.e. paternally inherited).
1369 Professors Fisk and Lovett considered that the Polymorphic Assay involves detecting the presence of a nucleic acid of fetal origin. The informative loci identified have a major and minor (fetal) fraction. Professor Lovett considered it implausible that any individual Harmony Test would not detect a paternally inherited fetal nucleic acid.
1370 I agree with Sequenom that the probability that even one of the “minor source” alleles detected in the Polymorphic Assay results from a de novo mutation rather than being paternally-inherited is vanishingly small. As Professors Fisk and Lovett explained in relation to whether there were any circumstances in which the informative loci would not be the result of a paternally inherited fetal sequence:
Yes, de novo mutations can result in informative events where the apparent fetal event is not paternally inherited. These are extremely rare and occur at a rate of approximately 1 in 100 million per SNP per generation. We note that the Harmony Test employs over 600 SNPs and even allowing for informativity, the odds of multiple such events are extremely small. In any case this would not alter the validity or accuracy of the test.
1371 Further, the prospect of a de novo mutation or any other sources of potential false positives like, for example, the “stochastic PCR errors” identified by Ms Norbury, is largely ignored in the PPD. The Harmony Test marketing material quotes a false positive rate of 0.006%.
1372 Further, the Harmony Test involves the parallel interrogation of hundreds of SNPs. Therefore, even in the highly unlikely event that a particular SNP was due to a de novo mutation, in the large majority of cases the SNPs specific to the fetus will have come from the father. Professor Lovett puts the chance that even one fetal SNP is due to a de novo mutation at just 1 in 100 million. Professor Fisk also put it into perspective. He said that assuming 100,000 tests per year, one would expect one single aberrant SNP result out of the 600 in one person every 10 years, on the fetal side.
1373 Professors Lovett and Fisk also note that the odds of multiple SNP mutations are extremely small and would not alter the validity or accuracy of the Harmony Test.
1374 I agree with Sequenom’s experts that the exceptions cited by Ms Norbury as to the risk of falsely inferring that the minor source SNP at an informative locus is a paternally-inherited fetal nucleic acid (e.g. de novo mutations and maternal mutations) are so vanishingly small that the paternally inherited cffDNA inference is “is so solid statistically that it’s an extremely good inference”. At a statistical level, it is almost a certainty and “extremely more likely than not”.
1375 Further, it had been reported in the “Harmony Prenatal Test – Choose the NIPT that you can trust for every patient” by Ariosa dated 2016 (the Fetal Fraction Brochure) that the Harmony Test’s quantification of fetal cfDNA is accurate and reproducible:

1376 Now all of the above is very interesting, but as I say I have rejected Ariosa’s “known in advance” construction. Therefore infringement is established.
1377 But let me make some other points nevertheless.
1378 The respondents concede that the detection of Y sequences as part of the Harmony Test’s Non-Polymorphic Assay involves the detection of paternally inherited, fetal sequences within the meaning of claims 1 and 6.
1379 In the case of the Y in the Non-Polymorphic Assay, the Harmony Test operates by using the principle of looking for sequences that are known to be of fetal origin. Accordingly, even on the respondents’ “known in advance” argument, the Non-Polymorphic Assay detects nucleic acid known to be of fetal origin. Obviously the detection of Y chromosome sequences as part of the Harmony Test’s Non-Polymorphic Assay involves the targeting of sequences known to be paternally inherited, fetal sequences, as the Y chromosome is not possessed by pregnant women.
1380 Again as to all of this, Ms Norbury raised a vanishingly small prospect of a false result, this time of the Y chromosome coming from the mother, to suggest that even in the case of a Y chromosome, it could never be known (without genotyping the father) that it came from the father. But she had to agree that it was a fundamental assumption of the Harmony Test that the Y comes from the father.
1381 Further, and as to the Polymorphic Assay, even if Ariosa was correct on the construction question, the Harmony Test using the Polymorphic Assay would still infringe.
1382 Professors Fisk and Lovett said that the Polymorphic Assay of the Harmony Test operates on the principle of looking for sequences that are known to be of fetal origin. They acknowledged that individual polymorphic markers in the Harmony Test are not a priori known to be of fetal origin. But as a group, the panel of polymorphic markers used in the test is so comprehensive as to virtually guarantee that a substantial number will be minor fraction sequences and thus known to be of fetal origin. But Ms Norbury disagreed that the Polymorphic Assay operates using this principle. She stated that the Polymorphic Assay looked for polymorphisms that were found in the general population. And they were not known to be of fetal origin. On her evidence, the Polymorphic Assay was used to determine the minor source fraction that was inferred but not known to be fetal.
1383 But it is a truism that assumptions and inferences are part of all scientific methods. Everything is essentially a deduction and inference. It is trite to observe that in the context of the Patent, defining a nucleic acid as being a paternally inherited fetal sequence is always an inference.
1384 Further, in providing an overview of fetal development, Professor Hyett described the following assumptions inherent in CVS, a test he regarded as diagnostic:
…CVS collects chorionic villi, and therefore uses a placental tissue to define the fetal karyotype. This makes the assumption that the fetus and the placenta will always have the same karyotype. There are some circumstances where this is not the case.
1385 Professor Hyett also noted that the beta thalassemia Example:
…assumes that it is known that the father has a different beta-globin gene mutation to the mother. I understand this example to suggest that, for example in the case of PCR, a primer for this known paternal mutation would be used, and if this paternal mutation was present in the serum or plasma sample, it could be assumed that the DNA detected was fetal, and that therefore the fetus had inherited the paternal mutation. However, as at the Relevant Date, I would have understood that it would not be possible to use this method to identify whether the fetus has inherited the maternal mutation, since it would not be possible in that circumstance to distinguish the maternal and fetal mutations in the serum/plasma sample. Further, in circumstances where the mother and father had the same mutation within the gene, it would not be possible to distinguish between the maternal and fetal DNA or determine whether the fetus had inherited the mutated gene from either the mother or the father.
1386 Similarly, at the priority date linkage analysis involved assumptions that the target gene of interest had been or was likely to have been inherited with the adjacent or linked genetic markers (i.e. RFLP markers) detected by the analysis. The markers were used to trace the presence of a nearby allele indirectly. And determination that a fetus was male following the detection of a Y signal involved an assumption that the Y chromosome did not come from, for example, a mosaicism in the mother or a transplanted organ.
1387 Further and in any event, the accuracy of the Harmony Test’s methods means that it is problematic to treat the identification of informative paternally inherited (fetal) markers in the Polymorphic Assay as a mere assumption.
1388 And indeed the identification of fetal sequences in the Harmony Test is not treated in ambiguous terms in the respondents’ own instructional and marketing material. Let me give the following examples as correctly submitted by Sequenom.
1389 The Harmony IVD Kit Instructions (emphasis added):
(a) state that “FORTE then identifies loci that are informative for estimating fetal fraction in each sample”;
(b) explain that informative loci are defined as loci “where the material genotype is homozygous for one allele, and the fetus has inherited a different allele”; and
(c) refer to the Harmony Test’s ability to “detect” minor source cfDNA at fractions from 4% (i.e. fetal DNA).
1390 Sonic’s presentation on the Harmony Test explains that the fetal fraction is the percentage cfDNA “from the fetus” and that “the fetal fraction MUST be measured. Accurately.” (emphasis added).
1391 Further, Mr Andrew Sparks and Mr Eric Wang, the signatories of the PPD, in an article published in 2012 that I have referred to earlier titled “Noninvasive prenatal detection and selective analysis of cell-free DNA obtained from maternal blood: evaluation for trisomy 21 and trisomy 18” explained that:
[i]nformative polymorphic loci were defined as loci where fetal alleles differ from maternal alleles. Because DANSR exhibits allele specificities >99%, informative loci were readily identified when the fetal allele proportion of a locus was measured to be between 1-20%. A maximum likelihood estimate using the binomial distribution was employed to determine the most likely fetal fraction based upon measurements from several informative loci.
1392 Further, the Fetal Fraction Brochure explains that the DANSR and FORTE core technologies of the Harmony Prenatal Test “give you confidence by measuring fetal fraction accurately and reproducibly; [r]eporting fetal fraction to provide a confident result; [i]ncorporating fetal fraction into results to accurately distinguish high and low probability results (even at low fetal fraction)” and defines fetal fraction as “the amount of fetal cell-free DNA circulating in the mother’s blood compared to the total cell-free DNA”. The Fetal Fraction Brochure similarly states that the Harmony Test “provides better quality control as compared to other tests by accurately quantifying fetal cfDNA”.
1393 Further, an Ariosa brochure titled “Harmony Prenatal Test: Clear Answers to Questions that Matter” dated 2016 states that the Harmony Test incorporates “precise fetal DNA measurements”.
1394 Further, an undated presentation on the Harmony Test titled “Considerations in Selecting NIPT: Choosing the Right Test for your Practice” confirms that the Harmony Test “[a]ccurately measures fetal DNA amount using SNPs”.
1395 Further, an Ariosa brochure titled “The Harmony IVD Kit: Bringing the leading, global NIPT brand to your laboratory” dated 2016 explains that “[s]ingle nucleotide polymorphisms (SNP) technology is also employed to distinguish and quantify fetal fraction” (emphasis added).
1396 In my view the Harmony Test’s approach to detecting fetal DNA falls within the scope of the plain words of the detection claims of the Patent.
1397 Moreover, I agree with Sequenom that the respondents’ attempt to deny infringement on the basis of alleged assumptions and inferences in the Harmony Test method is artificial and at odds with the views of the experts (other than Ms Norbury), the respondents’ own published material and the purposive meaning given to the term “detect” by persons skilled in the art.
1398 Further, if it needs to be said in terms of the integers of claim 1, it is apparent from the description in the PPD that the Harmony Test is performed on the plasma component of a blood sample taken from a pregnant female.
1399 The Harmony IVD Kit Instructions state at p 2:
The Harmony Test is intended for use in analysis of cfDNA samples isolated from plasma from pregnant women…The Harmony Test requires cfDNA that has been isolated using a commercially available cfDNA extraction kit from approximately 4mL of plasma…
1400 Further, the Harmony Brochure states that “Harmony™ involves testing millions of short fragments of cfDNA in maternal plasma”.
Claim 2 – …comprising amplifying the fetal nucleic acid
1401 Ariosa accepts that there is amplification in the Harmony Test for the purposes of claim 2. Accordingly, infringement is established.
Claim 3 – …wherein the fetal nucleic acid is amplified by PCR
1402 The confidential PPD describes the use of PCR. Page 3 of the Harmony IVD Kit Instructions refers to “[t]hermal cycling of the 96 UPCR [universal polymerase chain reaction] reactions” which Professor Fisk says is a reference to amplification via PCR. Accordingly this integer is satisfied for claim 3. Accordingly, infringement is established.
Claim 5 – …by means of a sequence specific probe
1403 Ariosa accepts that the Harmony Test uses a probe for the purposes of claim 5. Accordingly, infringement is established.
Claim 6 – …presence of a fetal nucleic acid sequence from the Y chromosome is detected
1404 The evidence of both Professors Fisk and Lovett is that the Harmony Test Non-Polymorphic Assay involves the detection of loci on the Y chromosome. The Non-Polymorphic Assay involves looking for sequences that are of fetal origin as the Y chromosome is not possessed by pregnant women.
1405 In response to the question, “[d]oes the interrogation of loci on the Y chromosome in the Non-Polymorphic Assay of the Harmony Test detect the presence of a nucleic acid of foetal origin in a maternal plasma sample within the meaning of claim 1 of the Patent?”, and the question, “[d]oes the interrogation of loci on the Y chromosome in the Non-Polymorphic Assay of the Harmony Test detect the presence of a foetal nucleic acid sequence from the Y chromosome within the meaning of claim 6 of the Patent?”, Professor Fisk, Professor Lovett and Ms Norbury all answered “Yes”, with Ms Norbury “noting” the “possibility” of a false positive. A Harmony Test document by Ariosa dated 2016 records that the Harmony Test’s fetal sex microarray is 99.8% accurate.
1406 The presence of a fetal nucleic acid from the Y chromosome is detected as part of the Harmony Test. For example, in the PPD, chromosome Y is listed as a target chromosome within the Non-Polymorphic Assay, which is always run simultaneously with the Polymorphic Assay.
1407 It is of course an assumption of the Harmony Test that the Y chromosome comes from the father.
1408 The Harmony IVD Kit Instructions at p 23 states:
The Harmony Test was used to determine fetal sex in 787 of the 791 specimens described above. These specimens included 395 pregnancies where clinical genetic testing outcomes indicated no male fetus was present (absence of Y chromosome), and 392 pregnancies in which a male fetus was present (presence of Y chromosome).
1409 Accordingly, infringement is established.
Claim 9 – …presence of a fetal nucleic acid from a paternally-inherited non Y chromosome is detected
1410 The Polymorphic Assay identifies major and minor fractions of informative loci. The minor fraction is paternally inherited fetal DNA. Professor Lovett considers it implausible that any individual Harmony Test would not detect a paternally inherited (or fetal) nucleic acid.
1411 Accordingly, infringement is established.
Claim 13 – …for determining the sex of the fetus
1412 Claim 13 is a method for determining the sex of the fetus. It is dependent on claim 6, which provides for the detection of the presence of a fetal nucleic acid sequence from the Y chromosome.
1413 Now Ariosa says that the Harmony Test does not definitively “determine” fetal sex. It says that the Harmony Test does not make a determination of the likelihood that a fetus is female that is based on detecting the presence of a fetal nucleic acid sequence from the Y chromosome. Rather, it determines the likelihood of a female fetus based on the absence of the detection of a sequence from the Y chromosome as stated in an example Harmony Test Report in evidence:
Fetal Sex test quantifies the Y chromosome. A “female” result indicates absence of Y chromosome and a “male” result indicatives presence of Y chromosome…
1414 But Sequenom says that at the priority date, in the context of NIPD, “determination of fetal sex” simply meant detecting the presence of a Y DNA sequence and covered methods having varied levels of specificity (i.e. false negative rates) and sensitivity (false positive rates). That is, the claims cover the determination of the likelihood of a particular fetal condition or characteristic, such as gender.
1415 Further, in responding to Ms Norbury’s suggestion that Example 1 of the Patent does not determine fetal sex but rather “male status”, Professor Fisk explained that:
I consider such a distinction to be completely artificial for the purposes of considering the disclosure in the Patent. The Patent conventionally and understandably equates the presence or absence of a signal from a sequence on the Y chromosome with gender determination. The Patent does not, for example, delve into very rare discrepancies between genotype and phenotype in genetic males, such as testicular feminisation or 5-alpha reductase deficiency. In my view, Example 1 clearly determined what is understood in the Field by fetal sex, albeit with a degree of confidence of less than 100%.
1416 Consistently with Professor Fisk’s views, the Patent states that “sex determination” may be simply carried out by detecting the presence of a Y chromosome. In this context, no mention is made of the necessity to, for example, have a control for non-Y chromosome cffDNA to have additional confidence that a fetus where a Y sequence is not detected would be female.
1417 Further, in terms of determination, as Professor Hyett and his co-authors noted in Hyett JA et al “Reduction in diagnostic and therapeutic interventions by non-invasive determination of fetal sex in early pregnancy” (2005) 25(12) Prenatal Diagnosis 1111-1116:
Lo et al. (1997) demonstrated the presence of ffDNA in the plasma of pregnant women carrying male foetuses and that this was potentially a useful source of material for genetic prenatal diagnosis (Lo, 2000). Several groups have reported their ability to determine fetal sex by positively identifying male DNA sequences (to the SRY gene) in maternal plasma (Table 1) (Lo et al., 1997, 1998; Smid et al., 1999; Zhong et al., 2000, 2001; Al-Yatama et al., 2001; Costa et al., 2001; Honda et al., 2001, 2002; Nelson et al., 2001; Rijnders et al., 2001, 2003; Sekizawa et al., 2001; Farina et al., 2002; Hromadnikova et al., 2002; Guibert et al., 2003). Early studies using conventional PCR techniques had lower sensitivity and specificity than those more recent series that have used real-time quantitative PCR…
(Emphasis added.)
1418 In the same article, Professor Hyett relevantly noted that the “disadvantage of this technique, which uses primers to identify the male SRY gene, is that the diagnosis of a female fetus is based on a negative test result, and therefore carries the risk of falsely being negative” (emphasis added).
1419 Further, even if determination imports an extremely high degree of accuracy, such a standard is satisfied by the Harmony Test. The Harmony IVD Kit Instructions themselves state (at page 4) that the Harmony Test can be used to “determine fetal sex”. It is not suggested that further invasive tests are needed to “confirm” this determination or diagnosis, and the respondents concede that the Harmony Test sex option is a prenatal diagnosis.
1420 An example Harmony Test result in evidence similarly explains that the Harmony Test’s “Fetal Sex” test “…quantifies the Y chromosome. A “female” result indicates absence of Y chromosome and a “male” result indicates presence of Y chromosome” and has an accuracy of over 99% for male or female sex.
1421 The results report the fetus to be either a “male” or “female”. They do not, for example, state that there is a “low risk” that the fetus is male.
1422 In the joint expert report, all of the experts agreed that the Non-Polymorphic Assay of the Harmony Test is performed for determining the sex of the fetus within the meaning of claim 13 of the Patent.
1423 Accordingly, in my view the respondents’ assertions that the Harmony Test does not definitively “determine” fetal sex for the purposes of claim 13, but rather determines the “likely chromosomal gender” is artificial and does not place the Harmony Test method outside the scope of the claims. Infringement is established.
Claim 14 – …determining the concentration of the fetal nucleic acid sequence in the maternal serum or plasma
1424 Claim 14 of the Patent provides:
The method according to any one of claims 6 to 12, which comprises determining the concentration of the foetal nucleic acid sequence in the maternal serum or plasma.
1425 As discussed above, Ariosa’s argument is that the term “concentration” in claim 14 is a reference to an amount per unit volume, not a ratio or relative concentration. But I have already rejected this argument.
1426 As to infringement, the Harmony IVD Kit Instructions use the term “concentration” to refer to relative concentration. And Ms Norbury agreed that concentration was used in a relative manner in the following extract from the Instructions:
A targeted amplification process termed Digital Analysis of Selected Regions (DANSR) is used to simultaneously amplify from each of the 96 DNA specimens UPCR products corresponding to approximately 6800 ~75bp long genomic intervals including approximately 1200 non-polymorphic loci on each of chromosomes 4, 13, 18, 21, and X; approximately 100 non-polymorphic loci on chromosome Y; and approximately 700 single nucleotide polymorphism (SNP)-containing polymorphic loci on chromosomes 1-12, in proportion to the concentrations of their cognate loci and alleles in the original DNA specimens.
1427 The Polymorphic Assay involves the detection and quantification of SNP alleles at a large number of loci (on chromosomes 1-12). The quantity and thus concentration of each allele at an informative locus in the Polymorphic Assay is estimated by detection of the different light intensities generated at each of two different wavelengths. The allele generating the lesser signal is necessarily the paternally inherited allele of fetal origin. By calculating or very accurately “estimating” fetal fraction using the procedure outlined in the PPD, the Harmony Test includes the step of determining the concentration of fetal nucleic acid sequence in the maternal serum or plasma.
1428 Accordingly, the Harmony Test infringes claim 14.
Claims 22, 23, 25 and 26
1429 Claims 22, 25 and 26 each refer to “a method of performing a prenatal diagnosis” and/or “providing a diagnosis”.
1430 As I have indicated, Ariosa says that the term “prenatal diagnosis” is characterised as a type of test which provides highly accurate and reliable results on which a clinician and a patient can base a clinical decision, that is a decision in relation to treatment or intervention. But it says that other than sex determination (which is only provided if requested by the patient), the Harmony Test is a screening test. It is a screening test which provides a risk assessment that the fetus carries certain genetic disorders and, if the patient requests it, a risk assessment of sex chromosomal aneuploidy. If a patient is assessed to be high risk by the Harmony Test, the patient will be referred for further testing using invasive techniques. No clinical decisions are made based on the results of the Harmony Test.
1431 Now Ariosa concedes that the Harmony Test fetal sex option is a prenatal diagnosis. Accordingly, the issue which remains is whether the Harmony Test’s detection of XYY and genotyping in the Polymorphic Assay is a prenatal diagnosis. And as I have said, it is.
1432 Further, let me make the more general point that in the context of the Patent, “diagnosis” encompasses prenatal risk assessments.
1433 The Harmony Test accurately determines, for example, fetal sex and detects fetal chromosomal aneuploidy, being explicit examples of the method of prenatal diagnosis defined in the Patent. The Harmony Test also detects and determines paternally inherited mutations and genotypes the fetus. The Harmony Test is therefore a “method of prenatal diagnosis” within the meaning of the relevant claims. By providing a determination of fetal gender (i.e. a male or female result – not a “risk score” as suggested by Ms Norbury), a fetal fraction, and of a “high” or “low” risk of aneuploidy, the Harmony Test also “provides a diagnosis” within the meaning of the claims. As noted above, Ariosa concedes that the Harmony Test’s determination of fetal sex constitutes a “diagnosis” within the meaning of the claims.
1434 If the Harmony Test reports that there is a high risk that the fetus has XYY aneuploidy, confirmatory invasive testing may be offered to the mother. If, on the other hand, the Harmony Test reports that there is a low risk of XYY aneuploidy (and other conditions), no further testing would typically be performed. Thus, “clinical decisions” are made on the basis of the Harmony Test’s XYY results.
1435 The Harmony Test materials give a sensitivity of 100% and a specificity of 99.7% for sex chromosome aneuploidies.
1436 Further, in the case of sex chromosome aneuploidies, the chance of a major handicap is small. Hence the consequence of a detection of XYY aneuploidy is not in the realm of a high risk trisomy 21, 18 or 13 result, which must be confirmed by amniocentesis or CVS before termination may be offered. Thus, the Harmony Test’s XYY detection method provides a highly accurate and reliable result which is at the diagnostic end of the spectrum, even on the narrow definition of diagnosis.
1437 Let me deal with another matter concerning the expression “a nucleic acid”. Ariosa also contends that claims 22, 23 and 25 require a diagnosis to be made on the basis of “a” (singular) nucleic acid and that this does not occur with the Harmony Test, which provides risks assessments based on complex calculations performed using information relating to a large number of different nucleic acid sequences.
1438 More particularly, Ariosa says that claim 22 refers to detecting the presence of “a nucleic acid of foetal origin”, and then providing a diagnosis based on “the presence and/or quantity and/or sequence of the foetal nucleic acid”. Read in context in the Patent, that process requires a diagnostic conclusion to be based on an analysis of a single nucleic acid. But it says that that does not occur with the Harmony Test, which provides risks assessments based on complex calculations performed using information relating to a large number (thousands) of different nucleic acid sequences. Further, it says that claim 23 is dependent on claim 22, and has the same requirement.
1439 I would reject what I would describe as a technical argument.
1440 The “nucleic acid” detected in the Harmony Test’s sex determination and XYY aneuploidy detection methods may properly be identified as fetal nucleic acid from the Y chromosome comprising a number of different discrete sequences from that nucleic acid. The “nucleic acid” detected as part of the Harmony Test’s Polymorphic Assay may properly be identified as fetal nucleic acid from a non-Y chromosome.
1441 Further, claims 22 and 23 do not in any event require a diagnosis to be made solely on the basis of the fetal nucleic acid. Claim 25 similarly only requires the nucleic acid to be indicative of a fetal condition or characteristic.
1442 Let me now deal with another issue concerning the integer “separating the sample into a cellular and non-cellular (plasma) fraction” (claims 22 and 23).
1443 The PPD at [4.1] provides:
A collected blood sample firstly undergoes a centrifugation protocol. This separates the blood into three components: (1) the buffy coat; (2) erythrocytes (red blood cells); and (3) plasma (the fluid part of the blood, which contains, among other things, minerals, salts, hormones and proteins). Foetal cfDNA and Maternal cfDNA are both contained within the plasma component.
1444 Plasma is a non-cellular fraction of the blood sample.
1445 Accordingly, the Harmony Test infringes claims 22 and 23.
1446 As to claim 25, Ariosa says that it refers to testing for “foetal nucleic acid indicative of a maternal or foetal condition or characteristic”, but that does not occur with the Harmony Test. Ariosa says that there is no fetal nucleic acid (or even fetal nucleic acids) that are of themselves indicative of any maternal or fetal condition or characteristic that are tested for in the Harmony Test. Ariosa says that the process used is a different one, involving a complex test which utilises data aggregated from a large number of loci on various chromosomes together with data from other sources to assess the probability as to whether the fetus is likely to have an aneuploidy. But I reject such a technical argument. Further, XYY aneuploidy and fetal sex are fetal “conditions” or “characteristics” within the meaning of claim 25 and thus the Non-Polymorphic Assay of the Harmony Test infringes claim 25. Further, the Polymorphic Assay determines the relative concentration of cffDNA in a maternal plasma sample and partially genotypes the fetus at a large number of informative polymorphic loci by detecting paternally inherited SNP mutations. The partial genotype of the fetus is also a fetal characteristic within the broad language of claim 25.
1447 I do not need to say anything further about claim 26, save to say that the Harmony Test falls within its scope if claim 26 was otherwise valid.
Conclusions
1448 In summary, the Harmony Test falls within the scope of each of the relevant claims as correctly submitted by Sequenom.
1449 First, the Harmony Test is performed on plasma extracted from maternal blood samples (all relevant claims).
1450 Second, the Harmony Test is a detection method (claims 1, 2, 3, 5, 6, 9 and 22).
1451 Third, the Harmony Tests reports the gender of the fetus (claims 6, 13, 22, 23, 25 and 26) and the risk that the fetus is suffering from aneuploidy such as XYY (claims 22, 23, 25 and 26).
1452 Fourth, the Harmony Test detects paternally inherited mutations and determines the partial genotype of the fetus by detecting paternally inherited mutations (claims 22, 23, 25 and 26).
1453 Fifth, the Harmony Test is a “method of performing a prenatal diagnosis” (claims 22, 23, 25 and 26). It provides a diagnosis based on the presence, quantity and sequence of the fetal nucleic acid (claims 22 and 23) and it comprises a test for fetal nucleic acid indicative of a fetal condition or characteristic (claim 25).
1454 Sixth, in the course of the Non-Polymorphic Assay of the Harmony Test, the presence of a fetal nucleic acid sequence from the Y chromosome is detected by means of a sequence specific probe and amplified by enrichment and PCR (claims 1, 2, 3, 5 and 6).
1455 Seventh, the Non-Polymorphic Assay is relevantly performed for determining the sex of the fetus (claim 13).
1456 Eighth, in the course of the Polymorphic Assay of the Harmony Test, nucleic acid of fetal origin from a paternally inherited non-Y chromosome is detected by means of a sequence specific probe and amplified by enrichment and PCR (claims 1, 2, 3, 5 and 9).
1457 Ninth, in the course of the Polymorphic Assay of the Harmony Test, the presence of a fetal nucleic acid from a paternally inherited non-Y chromosome is detected (claim 9) and the concentration of the sequence in the maternal plasma is determined (claim 14).
(e) Infringement – legal characterisation and other matters
1458 In my view Ariosa has infringed the relevant claims (other than claim 26) of the Patent as pleaded in the second further amended statement of claim.
1459 Further, Sonic and Clinical by their use of the Harmony Test in Australia have each infringed the relevant claims (other than claim 26) of the Patent. Sonic and Clinical have each also infringed Sequenom’s exclusive rights by promoting and supplying Harmony Test results to medical practitioners during the send out periods. Ariosa has also authorised that conduct.
1460 Further, the Harmony Test results constitute a “product” resulting from the use of the method of Sequenom’s claimed invention and the promotion and supply of such results infringes the exclusive rights in that claimed invention granted to Sequenom.
1461 Now in relation to the send out periods the respondents say that the importation into Australia of patient reports generated by the performance of the Harmony Test in the USA on samples collected in Australia did not constitute an infringement of the Patent.
1462 As explained by Mr Rubano there are two ways in which the Harmony Test may be provided, either under the “Test Send Out” (TSO) model or the Ariosa cell-free System (AcfS) model. Under the TSO model, Sonic between June 2015 and October 2015 and Clinical between December 2014 and March 2016 collected samples of blood from pregnant women in Australia, and sent those to Ariosa in San Jose, California. Ariosa performed the Harmony Test in its laboratory in San Jose and generated a report for each Harmony Test performed. An original copy of the Harmony report in PDF format was automatically saved on Ariosa’s server in San Jose. The Harmony report was made available by Ariosa via an online secure document storage and sharing system known as ShareFile provided by a third party, Citrix; this is a distributed server system in which physical or virtual servers are located at various locations around the world and the data stored on one server is replicated on all other servers in the system. A copy of the Harmony report could then be downloaded by representatives of Sonic and Clinical from the ShareFile server. Since 2 February 2016, Ariosa has operated its own file sharing system, the Harmony customer portal, and has progressively migrated its TSO customers from ShareFile to the Harmony customer portal. From 25 January 2017, Sullivan Nicolaides was migrated to this portal. Reports are made available in a similar way to the ShareFile system and the Harmony customer portal is hosted on Ariosa’s server located in San Jose. Once the Harmony report was downloaded by a clinic, it was the responsibility of the clinic to provide a copy of the report to the requesting doctors and patients.
1463 As I say, the respondents reject Sequenom’s assertion that the performance of the Harmony Test in the USA on samples collected in Australia by Sonic and Clinical infringes the Patent.
1464 Moreover, they reject Sequenom’s characterisation of the “product” of the Harmony Test.
1465 Section 13(1) of the Act provides that “a patent gives the patentee the exclusive rights, during the term of the patent, to exploit the invention” and to authorise others to do so. The definition of “exploit” is found in the Dictionary in Schedule 1 of the Act, which provides:
“exploit”, in relation to an invention, includes:
(a) where the invention is a product--make, hire, sell or otherwise dispose of the product, offer to make, sell, hire or otherwise dispose of it, use or import it, or keep it for the purpose of doing any of those things; or
(b) where the invention is a method or process--use the method or process or do any act mentioned in paragraph (a) in respect of a product resulting from such use.
1466 In Apotex Pty Ltd v Warner-Lambert Company LLC (No 2) (2016) 122 IPR 17, Nicholas J held at [296] to [298] that:
The definition of “exploit” makes no reference to the patent area. As I have said, the express territorial limitation upon the patentee’s exclusive rights is found in ss 12 and 13. In my respectful view, there is therefore no reason to read down the words of either para (a) or para (b) of the definition of “exploit” to found any territorial limitation. This is because the Act expressly provides that a patent only has effect in the patent area: see also s 70 of the Patents Act 1952 (Cth).
Paragraph (b) of the definition of “exploit” refers to the doing of an act referred to in para (a) which includes to make or import a product. The patentee’s exclusive rights are infringed (subject to available defences) if another person does any such act within the patent area. The fact that the patented method is performed outside the patent area does not avoid infringement of a method claim (including a Swiss claim) if the product imported and sold in Australia was made using the patented method because “the acts of importation and sale occur within the patent area”. The relevant act of infringement is not the use of the method outside the patent area but the exploitation (by importation and sale) in Australia of a product made using the patented method.
In my respectful opinion, contrary to the approach taken by Lindgren J, the relevant territorial limitation is reflected in the language of ss 12 and 13(3) and there is therefore no justification for importing words of territorial limitation into the definition of “exploit”. It follows that I take a somewhat different approach to the construction of the definition of “exploit” to that taken by Lindgren J in Alphapharm, though I do not think the difference has any impact on whether or not Apotex threatens to infringe the Swiss claims in this case.
1467 This finding was considered and upheld by the Full Court in Warner-Lambert Co LLC v Apotex Pty Ltd (No 2) (2018) 355 ALR 44 at [156] to [164] and [167] to [168], having also considered Lindgren J’s approach in Alphapharm Pty Ltd v H Lundbeck A/S (2008) 76 IPR 618 at [691] to [694].
1468 The respondents deny that the Harmony Test results or reports are properly described as “products” of the claimed methods, for the purpose of the definition of “exploit”. They say that whilst a report may have been generated as a result of the performance of the Harmony Test, that report merely records information. They say that in theory, the information in the report could have been communicated to a doctor or patient in Australia by telephone. They say that the fundamental nature of information is not confined by its method of conveyance. Consistently with this, they say that to the extent that the Harmony Test or any aspect of it falls within the scope of the claims of the Patent, the Harmony Test results or reports are not a “product” within the meaning of the definition of “exploit” in the Act.
1469 Indeed they go so far as to say that there is nothing in the Act to suggest that the importation of a “product” can extend to the importation of information, and that there is nothing in Northern Territory v Collins (2008) 235 CLR 619 that would support such an approach.
1470 In summary, the respondents say that the Harmony Test results or reports are not a “product” that can be imported (or kept or sold) within the meaning of the definition of “exploit” in the Act. It is instead information that could be brought within Australia by any form of communication, or by a person receiving that communication directly overseas and themselves travelling to Australia whilst retaining that information in their memory.
1471 But in my view the respondents’ arguments lack any bounce.
1472 The term “product” is not defined in the Act. Accordingly and in context, the term is to be given its ordinary meaning, namely as covering anything resulting from the patented method that can be commercially exploited. Such an ordinary meaning was adopted in Northern Territory v Collins, where the Court was concerned with the interpretation of the word “product” in the context of s 117 of the Act; a term occurring in several places of the same statute should usually be similarly construed.
1473 Further, it has been said that “[i]n relation to a process, the product is the state of affairs in which an effect may be observed” (Cancer Voices Australia v Myriad Genetics Inc (2013) 99 IPR 567 per Nicholas J at [88]) and that “vendible product” should be “understood as covering every end produced or artificially created state of affairs which is of utility in practical affairs and whose significance thus is economic” (CCOM Pty Ltd v Jiejing Pty Ltd (1994) 51 FCR 260 at 291 per Spender, Gummow and Heerey JJ, referring to NRDC at 276 to 277. Now I accept that these observations were made in a different context and with different qualifications. Nevertheless they provide a relevant analogy.
1474 Moreover, interpreting the definition of “product” broadly limits the undesirable possibility that a potential infringer could utilise a patented process overseas and then import the resulting product into Australia, and so circumvent the monopoly granted by the method patent.
1475 In my view, the reality is that the Harmony Test is carried out for the purpose of obtaining the results. The results are clearly commercially valuable; patients pay in the order of $400 to obtain them. The Harmony Test results including being recorded in electronic or paper form are the product of a process for the purposes of the definition of “exploit”. Moreover, by supplying the Harmony Test results to clinicians in Australia, Sonic and Clinical infringed the relevant claims during the send out period.
1476 Finally, I would note that it is not now in dispute that Ariosa authorised the relevant conduct of Sonic and Clinical that I have referred to above and that it is a joint tortfeasor with them, assuming relevant infringement is established.
CONCLUSION
1477 Sequenom has succeeded on its infringement case save for its case on infringement concerning claim 26. The respondents have failed on their invalidity case save that they have established that claim 26 is invalid for lack of fair basis. I will direct that the parties bring in minutes of orders to reflect these reasons and for the further steps to be taken in this matter. I will also hear the parties on any question of costs.
I certify that the preceding one thousand, four hundred and seventy-seven (1477) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Beach. |
Associate:
SCHEDULE
PRODUCT AND PROCESS DESCRIPTION (Confidential)
[Redacted]
VID 611 of 2016 | |
SONIC HEALTHCARE LIMITED (ACN 004 196 909) | |
Third Cross-Claimant: | CLINICAL LABORATORIES PTY LTD (ACN 006 823 089) |

