
FEDERAL COURT OF AUSTRALIA
InterPharma Pty Ltd v Hospira, Inc (No 5) [2019] FCA 960
ORDERS
INTERPHARMA PTY LTD (ACN 099 877 899) Applicant | ||
AND: | Respondent | |
AND BETWEEN: | First Cross-Claimant | |
PFIZER AUSTRALIA PTY LTD Second Cross-Claimant | ||
AND: | INTERPHARMA PTY LTD (ACN 099 877 899) Cross-Respondent | |
DATE OF ORDER: |
THE COURT ORDERS THAT:
1. Pursuant to s 136 of the Evidence Act 1995 (Cth) the use of the following documents be limited to the use stated below:
(a) The document entitled “Abbott-85499 Dexmedetomidine Clinical Study Report: A Phase III, Multicenter, Double-Blind, Randomized, Comparative Study Evaluating The Effect of Two Doses of Dexmedetomidine Versus Placebo In Adult Patients Undergoing Elective Coronary Artery Bypass Graft(s) Surgery”, be admitted and used solely as evidence of the previous representations that the study identified as DEX-95-004:
(i) was a Phase III, Multicenter, Double-Blind, Randomized, Comparative Study Evaluating The Effect of Two Doses of Dexmedetomidine Versus Placebo In Adult Patients Undergoing Elective Coronary Artery Bypass Graft(s) Surgery;
(ii) (ii) was initiated on 2 January 1996 and completed on 7 December 1997; and
(iii) was conducted in compliance with Good Clinical Practice (GCP) guidelines.
(b) The document entitled “Abbott-85499 Dexmedetomidine Clinical Study Report: A Phase II, Single-Center, Two-Part Study Evaluating the Safety, Efficacy, and Dose Titratability of Dexmedetomidine in ICU Sedation” be admitted and used solely as evidence of the previous representations that the study identified as W97-249:
(i) was a Phase II, Single-Center, Two-Part Study Evaluating the Safety, Efficacy, and Dose Titratability of Dexmedetomidine in ICU Sedation;
(ii) was initiated on 14 January 1998 and completed on 12 March 1998; and
(iii) was conducted in compliance with GCP guidelines.
(a) The document entitled “Duke University Medical Center Institutional Review Board (IRB)” and an attached document entitled “Duke University Medical Center – Informed Consent for a Research Project: A Phase III, Multicenter, Double-Blind, Randomized, Comparative Study Evaluating The Effect of Two Doses of Dexmedetomidine Versus Placebo In Adult Patients Undergoing Elective Coronary Artery Bypass Graft(s) Surgery: DEX-95-004, Version 2” (Duke Form) be admitted and used solely as evidence of the previous representations that:
(i) the patient informed consent form that was approved by the IRB for use in the DEX-95-004 study at the Duke Medical Center was the Duke Form;
(ii) the DEX-95-004 study was a Phase III, Multicenter, Double-Blind, Randomized, Comparative Study Evaluating the Effect of Two Doses of Dexmedetomidine Versus Placebo In Adult Patients Undergoing Elective Coronary Artery Bypass Graft(s) Surgery; and
(iii) the approval was given for a period of one year terminating on 11 October 1996.
(b) The document entitled “Information for Patients: A phase II, single-center, two part study evaluating the safety, efficacy and dose titratability of Dexmedetomidine in ICU sedation”, having the protocol “W97-249”, version 5 (part 1) dated 14 January 1998 (249 Form) and correspondence dated 19 January 1998 on behalf of the Board of Directors of the Academic Hospital Utrecht be admitted and used solely as evidence of the previous representations that:
(i) the patient informed consent form that was approved for use in part 1 of the W97-249 study was the 249 Form; and
(ii) the W97-249 study was a Phase II, Single-Center, Two-Part Study Evaluating the Safety, Efficacy and Dose Titratability of Dexmedetomidine in ICU Sedation.
2. Leave be granted to amend the Third Further Amended Particulars of Invalidity in accordance with these reasons (delivered on 20 June 2019) (these reasons).
3. The Proposed Fourth Further Amended Particulars of Invalidity stand as the Fourth Further Amended Particulars of Invalidity to the extent leave is granted in Order 2 above.
4. By 4:00 pm on 4 July 2019, each party file and serve proposed minutes of orders reflecting the conclusions in these reasons and short submissions (limited to 3 pages) addressing:
(a) the form of the proposed orders; and
(b) costs (other than with respect to the interlocutory application filed on 12 January 2018).
5. By 2:00 pm on 21 June 2019, each party inform the other parties and the associate to Justice Kenny whether it has any confidentiality issue with these reasons.
6. Until 2:00 pm on 21 June 2019 or further order, these reasons not be made available to or published to any person other than the parties and their legal advisors.
7. The question of costs be determined on the papers unless a party wishes to be heard on that question orally.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
KENNY J:
Introduction
1 This proceeding involves the construction, validity and, if valid, alleged infringement of a patent entitled “Use of dexmedetomidine for ICU sedation” (Patent). Hospira, Inc and Pfizer Australia Pty Ltd (together referred to as Pfizer) are respectively the registered proprietor and exclusive licensee of the Patent. The Patent is Australian Patent No 754484. Pfizer sells pharmaceutical products under the name “Precedex®”, the sole active ingredient of which is dexmedetomidine hydrochloride (a pharmaceutically acceptable salt of dexmedetomidine). These products are registered on the Australian Register of Therapeutic Goods (ARTG), with Pfizer as sponsor.
2 InterPharma Pty Ltd (InterPharma), which carries on the business of importing and distributing pharmaceutical products for sale in Australia, is the sponsor of a listing on the ARTG for a family of products, which contain dexmedetomidine hydrochloride as their sole active ingredient (Generic Products). The Generic Products are listed on the ARTG under the brand name “Dexmedetomidine Ever Pharma” and are registered “for sedation of initially intubated patients during treatment in an intensive care setting” (ICU sedation indication) and “for sedation of non-intubated patients prior to and/or during surgical and other procedures” (procedural sedation indication). These indications are the same as for Precedex®.
3 InterPharma, which initially commenced the proceeding to revoke the claims of the Patent, relied on a number of grounds of invalidity. The grounds relied on were that the invention as claimed: (a) was not a “manner of new manufacture”; (b) was not novel; (c) lacked an inventive step; (d) was not fairly based on the matter described in the specification; and (e) did not comply with s 40 of the Patents Act 1990 (Cth) (Patents Act) in that the claims were not clear and succinct.
4 In a cross-claim, Pfizer contended that InterPharma threatened to infringe each claim of the Patent by, amongst other things, offering to supply and supplying the Generic Products. InterPharma admitted that, subject to the question of validity, its proposed conduct threatened to directly infringe claim 1 and indirectly infringe claims 13, 15 and 26 of the Patent. InterPharma also admitted threatened infringement of claims 2, 14 and 27 of the Patent on the basis of a particular construction of those claims (which, for the reasons given below, I accept).
5 There was a substantive hearing of the claim and cross-claim on 3, 4, 7, 21 and 22 May 2018. On 6 August 2018, InterPharma filed an application to re-open the hearing. The hearing was resumed, by consent, on 15 November 2018, when further expert evidence was adduced.
6 InterPharma was, until the Patent expired on 31 March 2019, subject to interlocutory orders restraining it from apprehended infringement of the Patent. The focus at this stage of the proceeding was on Pfizer’s entitlement to final relief. For the reasons stated below, I have found that InterPharma threatened to infringe the Patent during its term and that InterPharma’s challenge to the validity of the Patent should be rejected.
Abbreviated references
7 In these reasons, the following abbreviations are used:
Belleville means the article by Belleville J, Ward D, Bloor B, and Maze M, “Effects of intravenous dexmedetomidine in humans: I. Sedation, ventilation, and metabolic rate”, Anesthesiology 1992, 77: 1125-1133.
Bloor means the article by Bloor B, Ward D, Belleville J, and Maze M, “Effects of intravenous dexmedetomidine in humans: II. Hemodynamic changes”, Anesthesiology 1992, 77: 1134-1142.
Belleville/Bloor means Belleville and Bloor together.
Böhrer 1990 means the article by Böhrer H, Bach A, Layer M and Werning P, “Clonidine as a sedative adjunct in intensive care”, Intensive Care Medicine 1990, 16: 265-266.
GCP Guidelines means the Guideline for Good Clinical Practice E6(R1), Current Step 4 version dated 10 June 1996, developed by the appropriate Expert Working Group of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use.
Hall 2010 means the article by Hall J, “Creating the animated intensive care unit”, Critical Care Medicine 2010, 38(10): S668-S675.
Jalonen 1997 means the article by Jalonen J, Hynynen M, Kuitunen A, Heikkila H, Perttila J, Salmenpera M, Valtonen M, Aantaa R, and Kallio A, “Dexmedetomidine as an anaesthetic adjunct in coronary artery bypass grafting”, Anesthesiology 1997, 86: 331-345.
Kress 1996 means the article by Kress J, O’Connor M, Pohlman A, Olson D, Lavoie A, Toledano A, and Hall J, “Sedation of critically ill patients during mechanical ventilation”, American Journal of Respiratory and Critical Care Medicine 1996, 153: 1012-1018.
Kress 2000 means the article by Kress J, Pohlman A, O’Connor M, and Hall J, “Daily interruption of sedative infusions in critically ill patients undergoing mechanical ventilation”, New England Journal of Medicine 2000, 342(20): 1471-1477.
Petty editorial means the editorial by Petty T, “Suspended life or extending death?”, Chest 1998, 114(2): 360-361.
Pohlman 1994 means the article by Pohlman A, Simpson K, and Hall J, “Continuous intravenous infusions of lorazepam versus midazolam for sedation during mechanical ventilator support: a prospective, randomized study”, Critical Care Medicine 1994, 22(8): 1241-1247.
Talke means the article by Talke P, Li J, Jain U, Leung J, Drasner K, Hollenberg M, and Mangano D, “Effects of perioperative dexmedetomidine infusion in patients undergoing vascular surgery”, Anesthesiology 1995, 82: 620-633.
Tuxen 1998 means the article by Tuxen D, “Sedation and paralysis in mechanical ventilation”, Clinical Pulmonary Medicine 1998, 5(5): 314-328.
1997 Review Article means the article by French C and Bellomo R, “The role of [alpha]-receptor manipulation in the prevention of renal dysfunction”, Current Opinion in Critical Care 1997, 3: 408-413.
Overview of the patent
8 The Patent claimed an earliest priority date of 1 April 1998 (priority date). The priority date is uncontested. The Patent expired on 31 March 2019 (Patent expiry date).
9 The application for the Patent was made on 31 March 1999. Issues of infringement and validity are therefore to be determined under the Patents Act, in the form in which it existed prior to, among other Acts, the Patents Amendment (Innovation Patents) Act 2000 (Cth), the Patents Amendment Act 2001 (Cth), and the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth).
The background to the invention
10 The invention the subject of the Patent relates to the use of dexmedetomidine or a pharmaceutically acceptable salt thereof (referred to in these reasons as dexmedetomidine unless specified otherwise) in intensive care unit (ICU) sedation; a method of sedating a patient in the ICU by administering dexmedetomidine; and the use of that compound in the manufacture of a medicament for ICU sedation.
11 The specification explains:
In addition to the actual sedation of a patient in the ICU, the word sedation in the ICU context also includes the treatment of conditions that affect patient comfort, such as pain and anxiety. Also, the word intensive care unit includes any setting that provides intensive care.
…
The aim of ICU sedation is to ensure that the patient is comfortable, relaxed, and tolerates uncomfortable procedures such as placement of iv-lines or other catheters, but is still arousable.
12 According to the specification:
Particularly, the present invention relates to a method of sedating a patient while in the ICU by administering dexmedetomidine … wherein dexmedetomidine is essentially the sole active agent or the sole active agent administered for this purpose.
13 The specification explains that ICU patients often receive a variety of drugs concurrently. It states:
The agents used most commonly are given to achieve patient comfort. Various drugs are administered to produce anxiolysis (benzodiazepines), amnesia (benzodiazepines), analgesia (opioids), antidepression (antidepressants/benzodiazepines), muscle relaxation, sleep (barbiturates, benzodiazepines, propofol) and anaesthesia (propofol, barbiturates, volatile anaesthetics) for unpleasant procedures. These agents are cumulatively called sedatives in the context of ICU sedation, though sedation also includes the treatment of conditions that affect patient comfort, such as pain and anxiety, and many of the drugs mentioned above are not considered sedatives outside the context of ICU sedation.
14 The specification describes the side effects associated with ICU sedatives as follows:
The presently available sedative agents are associated with such adverse effects as prolonged sedation or oversedation (propofol and especially poor metabolizers of midazolam), prolonged weaning (midazolam), respiratory depression (benzodiazepines, propofol, and opioids), hypotension (propofol bolus dosing), bradycardia, ileus or decreased gastrointestinal motility (opioids), immunosuppression (volatile anaesthetics and nitrous oxide), renal function impairment, hepatotoxicity (barbiturates), tolerance (midazolam, propofol), hyperlipidemia (propofol), increased infections (propofol), lack of orientation and cooperation (midazolam, opioids, and propofol), and potential abuse (midazolam, opioids, and propofol).
15 The specification further states that the combination of ICU sedatives (referred to as polypharmacy) “may cause adverse effects”:
For example, the agents may act synergistically, which is not predictable; the toxicity of the agents may be additive; and the pharmacokinetics of each agent may be altered in an unpredictable fashion.
16 According to the specification, “[t]he preferred level of sedation for critically ill patients has changed considerably in recent years”. It explains:
Today, most intensive care doctors in the ICU prefer their patients to be asleep but easily arousable, and the level of sedation is now tailored towards the patient’s individual requirements.
17 The specification identifies the known problems with α2-adrenoceptor agonists, particularly clonidine. It states that:
According to Tryba et al., clonidine has its limitations in sedating critically ill patients mainly because of its unpredictable hemodynamic effects, i.e., bradycardia and hypotension, so that it must be titrated for each individual patient. Long term treatment of critically ill patients with clonidine has been reported to be associated with such rebound effects as tachycardia and hypertension.
18 The specification states that “α2-agonists are not presently used by themselves in ICU sedation”. It continues as follows:
Further, α2-agonists are not generally used in ICU sedation even in conjunction with other sedative agents. Only clonidine has been evaluated for use in ICU sedation, and then only in conjunction with opioids, benzodiazepines, ketamine, and neuroleptics. Further, administration of clonidine as essentially the sole active agent or the sole active agent to a patient in the ICU to achieve sedation has not been disclosed to the best of the applicants’ knowledge.
19 Some of the characteristics of an “ideal sedative agent for a critically ill patient” are identified. According to the specification:
An ideal sedative agent for a critically ill patient should provide sedation at easily determined doses with ready arousability together with hemodynamic stabilizing effects. Further, it should be an anxiolytic and an analgesic, and should prevent nausea, vomiting, and shivering. It should not cause respiratory depression. Preferably, an ideal sedative agent should be used by itself in ICU sedation to avoid the dangers of polypharmacy.
20 The specification depicts the chemical structure of dexmedetomidine as follows:
21 The specification states that dexmedetomidine has been described in US Patent 4910214 (US 214) as “an α2-receptor agonist for general sedation/analgesia and the treatment of hypertension or anxiety”.
The invention as described in the specification
22 Under the heading, “Summary of the invention”, the specification states that it had been “unexpectedly found that dexmedetomidine … is an ideal sedative agent to be administered to a patient in the ICU to achieve patient comfort”. Regarding the invention as a method of sedating a patient in the ICU by administering dexmedetomidine, wherein that compound is essentially the sole active agent or the sole active agent, the specification states:
The method is premised on the discovery that essentially only dexmedetomidine … need[s] to be administered to a patient in the ICU to achieve sedation and patient comfort. No additional sedative agents are required.
23 The Ramsay Scale (an assessment of the level of sedation in experimental subjects) is described under the heading, “Brief description of the drawings”. The specification states that, in the Ramsay Scale, the level of wakefulness is scored on a scale of 1 to 6 (Ramsay Sedation Score) based on progressive loss of responsiveness to stimuli ranging from auditory to deep painful stimuli. Figure 2 (set out below) shows the Ramsay Sedation Score of a patient in the dexmedetomidine study described in example 3.
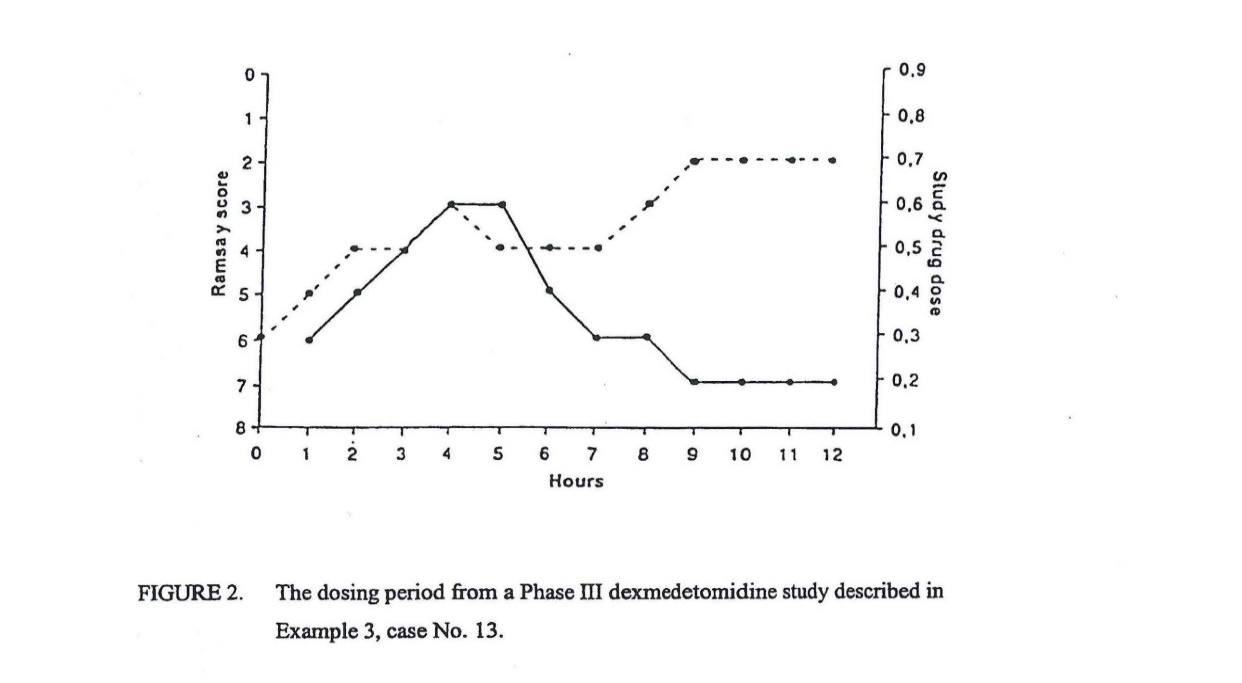
24 The dotted line signifies fluctuations in the Ramsay Sedation Score in response to dose adjustments.
25 Under the heading, “Detailed description of the invention”, the specification further explains the particular method of sedating a patient in the ICU by administering dexmedetomidine as essentially the sole active agent or the sole active agent for this purpose. It states:
Particularly, it has been found that dexmedetomidine … can be essentially the sole active agent or the sole active agent administered to a patient in the ICU in order to sedate the patient.
26 The specification goes on to describe the unique quality of the sedation in the ICU achieved by administering dexmedetomidine. It states, in particular:
Patients sedated by dexmedetomidine … are arousable and oriented, which makes the treatment of the patient easier. The patients can be awakened and they are able to respond to questions. They are aware, but not anxious, and tolerate an endotracheal tube well. Should a deeper level of sedation or more sedation be required or desired, an increase in dexmedetomidine dose smoothly transits the patient into a deeper level of sedation.
27 It is further said that:
Dexmedetomidine does not have adverse effects associated with other sedative agents, such as, respiratory depression, nausea, prolonged sedation, ileus or decreased gastrointestinal motility, or immunosuppression.
28 The specification explains that the precise amount of dexmedetomidine to be administered to achieve the desired outcome depends on a number of factors. It further states that:
The dose range of dexmedetomidine can be described as target plasma concentrations. The plasma concentration range anticipated to provide sedation in the patient population in the ICU varies between 0.1-2 ng/ml depending on the desired level of sedation and the general condition of the patient. These plasma concentrations can be achieved by intravenous administration by using a bolus dose and continuing it by a steady maintenance infusion. For example, the dose range for the bolus to achieve the forementioned plasma concentration range in a human is about 0.2-2 µg/kg, preferably about 0.5-2 µg/kg, more preferably 1.0 µg/kg, to be administered in about 10 minutes or slower, followed by a maintenance dose of about 0.1-2.0 µg/kg/h, preferably about 0.2-0.7 µg/kg/h, more preferably about 0.4-0.7 µg/kg/h.
29 The specification provides 3 examples, which exemplify the invention.
The claims
30 The independent claims of the Patent are:
Claim 1: “Use of dexmedetomidine … in the manufacture of a medicament for use in intensive care unit sedation”.
Claim 13: “A method of sedating a patient in an intensive care unit, wherein said method comprises administering dexmedetomidine … to a patient in need thereof”.
Claim 15: “A method of sedating an intensive care unit patient, comprising administering a pharmaceutical composition to the patient, wherein the pharmaceutical composition comprises an active agent and an inactive agent, wherein the active agent consists of dexmedetomidine”.
Claim 26: “Use of dexmedetomidine … in the intensive care unit sedation”.
Claim 1, dependent claims 2 to 12 and omnibus claim 38
31 The use according to claim 1 is the use of dexmedetomidine in the manufacture of a medicament for use in ICU sedation. Claim 1 is therefore a “Swiss-style” or “Swiss-type” claim in the form of “the use of [known] compound X in the manufacture of a medicament for a specified (and new) therapeutic use”: see Apotex Pty Ltd v Sanofi-Aventis Australia Pty Ltd [2013] HCA 50; 253 CLR 284 (Apotex v Sanofi-Aventis HCA) at [248].
32 In Otsuka Pharmaceutical Co Ltd v Generic Health Pty Ltd (No 4) [2015] FCA 634; 113 IPR 191 (Otsuka Pharmaceutical v Generic Health (No 4)) at [120], Yates J explained that a claim of this kind should be characterised as a method (or process) claim. Although the Swiss-style claim in that case (as here) was to a method of manufacture of a medicament, it was accepted that the novelty of the claim might arise from the new therapeutic use to which the medicament was applied: Otsuka Pharmaceutical v Generic Health (No 4) at [116]; see also Actavis UK Ltd v Merck & Co Inc [2008] EWCA Civ 444; [2009] 1 All ER 196 (Actavis UK v Merck) at [31], [49].
33 As will be seen, claims 2 to 12 are dependent Swiss-style claims, dependent on claim 1. Each of claims 2 to 12 further qualifies what is said to be the hitherto undiscovered therapeutic use of the medicament by reference to its particular administration (see [189]-[199] below). Claim 38, which is to a “use according to claim 1 substantially as hereinbefore described”, is an omnibus claim to claim 1. As stated below, I reject InterPharma’s submission that the use claimed in claims 2 to 12 and 38 can be understood as “directed to the clinician administering the product that results from the manufacture of the medicament”.
34 It is convenient to note at this point that claim 3 relates to the use of dexmedetomidine in the manufacture of a medicament for use in ICU sedation (as described in claim 1 or 2), “wherein the dexmedetomidine … is administered in an amount to achieve a plasma concentration of 0.1-2 ng/ml”; and that InterPharma made submissions about the plasma concentration integer, which are considered below.
Claims 13, 15 and 26, dependent claims 14, 16 to 25, 27 to 37 and omnibus claim 39
35 Claims 13, 15 and 26 are method (of treatment) claims. Claim 13 is for a method of sedating a patient in an ICU, wherein the method comprises administering dexmedetomidine to a patient in need of that drug. Claim 14 is a dependent claim, dependent on claim 13.
36 Claim 15 is for a method of sedating an ICU patient, comprising administering a pharmaceutical composition to the patient, amongst other things, “wherein the active agent consists of dexmedetomidine”.
37 Claim 16 is a dependent claim, dependent on claims 13, 14 or 15, wherein dexmedetomidine is administered “in an amount to achieve a plasma concentration of 0.1-2 ng/ml”. Again, I note that the significance of this latter integer is considered below.
38 Claims 17 to 25 are all dependent claims (dependent on claims 16, 17, 18, 19, 20, 21, 23 or 24). Relevant aspects of these claims are discussed below.
39 Claim 26 is an independent claim for the “[u]se of dexmedetomidine … in the intensive care unit sedation”.
40 Claim 27 is a dependent claim (dependent on claim 26), wherein dexmedetomidine is “essentially the sole active agent or the sole active agent”. Claims 28 to 37 are also dependent claims (dependent on claims 26, 27, 28, 29, 30, 31, 32, 33, 35 or 36), relevant aspects of which are further discussed below.
41 Claim 39 is a second omnibus claim, for a method according to claim 13 or 15 “substantially as hereinbefore described”.
Overview of witness evidence
Intensive care specialists
42 Three specialists in intensive care medicine gave evidence at trial. They were: Professor Russell Booth Hall; Professor Rinaldo Bellomo; and Associate Professor (Assoc Professor) Craig John French.
43 Professors Hall and Bellomo and Assoc Professor French met together on 2 May 2018, and created their Joint Expert Report (Joint Report). The Joint Report was in evidence.
44 InterPharma called Professor Hall. Since 1981, Professor Hall has been an Attending Physician, Critical Care Services, at the University Of Chicago Pritzker School Of Medicine. Amongst other things, he holds a certification in Critical Care Medicine from the American Board of Internal Medicine and is a Member of the Society for Critical Care Medicine. He is an author or co-author of very many articles published in leading medical journals.
45 Professor Hall had a specific interest in ICU sedation as at the priority date (April 1998). It may be accepted that his interest and knowledge about ICU sedation would have been significantly greater than that of the average ICU specialist at that time.
46 Professor Hall’s research interest is particularly relevant to the invention of the Patent. He was a co-author of Kress 2000, a highly influential paper leading intensive care specialists to question the previously accepted practice of deep sedation (see [537], [549] below). He was also a co-author of Kress 1996 and Pohlman 1994 (see [550] below).
47 Professor Hall is an experienced witness, who, on his own account, gives evidence in 15 to 20 cases a year, and has given evidence in United States (US) courts in two cases concerning the US equivalent of the Patent in suit.
48 Pfizer called Professor Bellomo, who has been delivering intensive care medicine to patients in Australia for well over 25 years. Professor Bellomo is the Director of Intensive Care Research at the Austin Hospital and has worked and continues to work as an intensive care specialist at that hospital and other hospitals, including the Royal Melbourne Hospital (RMH) ICU. Since 2002 he has been a Professor in the Faculty of Medicine at the University of Melbourne and since 2016 he has been a Board Member of the Australian and New Zealand College of Intensive Care Medicine.
49 Professor Bellomo came to intensive care medicine from a general physician’s background. Throughout his career, he has been engaged in clinical research in intensive care medicine, with a particular interest in the kidneys. He has an extensive list of publications, and has held editorial and reviewer positions for leading international medical journals, including US journals.
50 Pfizer also called Assoc Professor French, who has been practising in both anaesthesia and intensive care medicine in Australia since 1995. Assoc Professor French was undergoing training in both anaesthesia and intensive care medicine between 1992 and 1997. He was working as an anaesthetics registrar during 1995 and, from midway through that year, as an intensive care registrar. In 1998, he was admitted to the Fellowship of the Faculty of Intensive Care of the Australian and New Zealand College of Anaesthetists.
51 From January 1998 to 2001, Assoc Professor French worked as a consultant intensive care specialist and anaesthetist at the Western Hospital in Melbourne. In 2002, he was appointed Director of Intensive Care at the Sunshine Hospital, also in Melbourne. Since 2003, he has been the Director of Intensive Care at Western Health, a health services body that manages multiple hospitals in Melbourne. Among other things, he has been a Clinical Associate Professor at the University of Melbourne since 2011.
52 At the priority date, in April 1998, Assoc Professor French was a recently qualified specialist intensivist and anaesthetist. He considered his breadth of knowledge in intensive care and anaesthesia as at that date to be at its greatest level in his career. In subsequent years he maintained his overall general knowledge of intensive care and anaesthesia, although he gravitated towards his particular interests. Also as at the priority date, he had been involved in clinical trials for the development of new drugs within the ICU. Since 2015, he has been the chair of the Australian and New Zealand Intensive Care Society Clinical Trials Group.
53 Assoc Professor French has a particular interest in transfusion medicine in the ICU and the role of erythropoietin in clinical illness, although he had an early interest in mechanical ventilation and volatile anaesthetics. He published two papers prior to April 1998, and subsequently a large number of papers, many of them in the intensive care field. He co-authored numerous papers with Professor Bellomo. He has held editorial and reviewer roles for international, including US, medical journals.
54 As noted, Professors Hall and Bellomo and Assoc Professor French were each involved in research, including clinical trials, in intensive care medicine before April 1998. Both Professor Bellomo and Assoc Professor French specifically attested to their familiarity with the GCP Guidelines, the principles required to be followed in clinical trials, and with the investigator brochure supplied by drug companies to those designing and carrying out clinical trials. They both gave evidence about the training that was required to become an intensive care specialist in Australia. For example, Assoc Professor French had obtained an undergraduate medical degree in 1988, completed his residency in 1991, was an anaesthetic registrar from 1992 to 1995, and qualified as an intensive care specialist in 1998. Their evidence was, and it may be accepted, that the training to become an intensive care specialist called for extensive knowledge and the standard for the examinations was high. Those practising in intensive care medicine in Australia at the priority date also had another specialist qualification in medicine. Assoc Professor French and Professor Bellomo both affirmed that all intensive care specialists, regardless of their background or their additional practice, were equally skilled and equipped to administer intensive care medicine.
55 In addition to giving evidence at the trial, Professors Hall and Bellomo and Assoc Professor French each subsequently gave further evidence at the resumed hearing in November 2018.
Other witnesses
56 There were five other witnesses whose affidavits were tendered without objection and without cross-examination.
57 InterPharma adduced the affidavit evidence of Ms Carolyn Stewart. Ms Stewart is the Business and Operations Manager of the Melbourne Children’s Trials Centre at the Murdoch Children’s Research Institute. She has a Masters of Medical Science (Drug Development) from the University of New South Wales and, since the late 1980s, she has acquired extensive experience in respect of the conduct of local and international clinical trials.
58 While working for Amgen Australia Pty Ltd and its US parent company (Amgen) between 1992 and 2004, Ms Stewart was involved in drug research and development, and in clinical trials in Australia and overseas. Her responsibilities during those years included clinical trial management and conduct, writing protocols, designing and preparing participant information sheets and consent forms, and preparing study reports and publications.
59 Since working for Amgen, Ms Stewart has provided consultancy services to third parties regarding clinical trial site management and third party vendor management of clinical trials. She has also worked for the Nucleus Network Centre for Clinical Studies, where she was engaged in similar work. Ms Stewart gave evidence about the applicable legal and ethical requirements for the management and conduct of clinical drug trials and about good clinical practice.
60 InterPharma also adduced the affidavit evidence of Mr Bob van Der Kamp. Mr van Der Kamp is an Attorney at Law in the Netherlands. His evidence was confined to confirming: (1) the implementation into Dutch law of European Union (EU) Directive 91/507/EEC concerning good clinical practice for clinical trials, by a Decree on Manufacturing and Delivery of Pharmaceutical Products (Decree); and (2) that the Decree was part of Dutch law in 1998. His evidence was that the Decree became effective on 1 August 1994 and was in force until 1 July 2007. As will be seen, his evidence was relevant because one of the clinical trials on which InterPharma’s lack of novelty case relied was carried out in the Netherlands at the relevant time (see [400] below).
61 Pfizer relied on the affidavit evidence of Mr Rodney Ian Lindsay Cruise. Mr Cruise is a Patent Attorney with more than 28 years’ experience in conducting intellectual property research in the areas of patents, trademarks and designs. He was familiar with the conduct of searches on different databases of medical scientific literature and gave evidence about conducting searches of the medical literature as at the priority date. The evidence of Mr Cruise was that the most common medical scientific literature search database used as at 1 April 1998 was Medline, and that the two platforms most commonly used at that time for searching that database were PubMed and STN. His evidence was relevant to Pfizer’s response to InterPharma’s case on lack of inventive step (see [593] below).
62 At the start of the trial, Pfizer also relied on the affidavit evidence of Ms Melissa Jane Ankravs. Ms Ankravs is the Senior ICU Pharmacist and Team Leader at the RMH and is based in the ICU at the RMH. Ms Ankravs gave evidence about the role of the RMH Pharmacy and the means by which medicines were made available by the RMH Pharmacy for administration to patients in the RMH. She stated that she was involved in maintaining the stock levels in the ICU imprest at the RMH. She gave evidence about the numbers of vials of dexmedetomidine 200microg/50mL distributed to the ICU imprest and to the operating theatre imprests (located at other hospital locations outside the ICU). Her evidence was relevant to an issue that arose concerning the use of dexmedetomidine for procedural sedation, an aspect of the way InterPharma initially put its case. As this aspect of InterPharma’s case was subsequently abandoned, her evidence was not relied on in the parties’ final submissions.
63 Lastly, Pfizer relied on the affidavit evidence of Mr Michael Joseph Ryan. Mr Ryan had been the Director of Pharmacy at a number of hospitals in Melbourne between 1977 and 1994. He subsequently held the following positions: Manager of Health Information and Technology Services at the Victorian Healthcare Association (1997 to 1999); National Pharmacy Services Manager for Hospital Supplies of Australia (HSA) (1999 to 2001); and National Business Manager – Pharmacy at HSA (2001 to 2004). By 2018, he was an independent hospital and medicines management consultant, who ran his own business, PharmConsult Pty Ltd. He was also a Fellow of the Society of Hospital Pharmacists of Australia, having completed his Fellowship between 1979 and 1983.
64 It may be accepted that Mr Ryan had substantial knowledge and experience in the provision of pharmacy services within public and private hospitals, the procurement and supply of pharmaceutical products in the public health system, and in prescribing practices in hospitals. He gave evidence about the way in which medicines were dispensed to public and private hospital patients. He described tendering processes and outcomes. He gave evidence about the anticipated effect of the Generic Products entering the market, in terms of lessening restrictions related to cost-minimisation on the formulary in public hospitals. Pfizer relied on his evidence in support of an aspect of its case on infringement concerning the “essentially the sole active agent or the sole active agent” integer in claims 2, 14 and 27. It was unnecessary to refer to this aspect of Mr Ryan’s evidence in these reasons in light of InterPharma’s admission of infringement of these claims on the basis of a construction which, as will be seen, I accept. His evidence was also referred to in opening submissions in the context of the procedural sedation issue that was not ultimately pursued by InterPharma (see [62] above).
Principles of patent construction
65 In Jupiters Ltd v Neurizon Pty Ltd [2005] FCAFC 90; 65 IPR 86; 222 ALR 155 (Jupiters) at [67], a Full Court of this Court summarised the principles for the construction of patents as follows:
(i) the proper construction of a specification is a matter of law: Décor Corporation Pty Ltd v Dart Industries Inc (1988) 13 IPR 385 at 400;
(ii) a patent specification should be given a purposive, not a purely literal, construction: Flexible Steel Lacing Co v Beltreco Ltd (2000) 49 IPR 331; [2000] FCA 890 at [81] … ; and it is not to be read in the abstract but is to be construed in the light of the common general knowledge and the art before the priority date: Kimberley-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1; 177 ALR 460; 50 IPR 513; [2001] HCA 8 at [24];
(iii) the words used in a specification are to be given the meaning which the normal person skilled in the art would attach to them, having regard to his or her own general knowledge and to what is disclosed in the body of the specification: Décor Corporation Pty Ltd at 391;
(iv) while the claims are to be construed in the context of the specification as a whole, it is not legitimate to narrow or expand the boundaries of monopoly as fixed by the words of a claim by adding to those words glosses drawn from other parts of the specification, although terms in the claim which are unclear may be defined by reference to the body of the specification: Kimberley-Clark v Arico at [15]; Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610; Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 at 478; the body of a specification cannot be used to change a clear claim for one subject matter into a claim for another and different subject matter: Electric & Musical Industries Ltd v Lissen Ltd [1938] 4 All ER 221 at 224–5; (1938) 56 RPC 23 at 39;
(v) experts can give evidence on the meaning which those skilled in the art would give to technical or scientific terms and phrases and on unusual or special meanings to be given by skilled addressees to words which might otherwise bear their ordinary meaning: Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479 at 485–6 … ; the court is to place itself in the position of some person acquainted with the surrounding circumstances as to the state of the art and manufacture at the time (Kimberley-Clark v Arico at [24]); and
(vi) it is for the court, not for any witness however expert, to construe the specification; Sartas No 1 Pty Ltd, at 485–6.
66 The need to read a patent specification as a whole and in light of the common general knowledge in the art before the priority date continues to be emphasised. So too is the need to read a patent specification in a practical way and to give it a purposive construction.
This approach to construction requires the court to read the specification through the eyes of the skilled addressee with practical knowledge and experience in the field of work in which the invention was intended to be used and a proper understanding of the purpose of the invention.
See GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Generic Partners Pty Ltd [2018] FCAFC 71; 131 IPR 384 (Generic Partners) at [106].
67 As Lord Hoffmann explained in Kirin-Amgen Inc v Hoechst Marion Roussel Ltd [2004] UKHL 46; 64 IPR 444; [2005] 1 All ER 667 at [34]:
“Purposive construction” does not mean that one is extending or going beyond the definition of the technical matter for which the patentee seeks protection in the claims. The question is always what the person skilled in the art would have understood the patentee to be using the language of the claim to mean. And for this purpose, the language he has chosen is usually of critical importance. … There will be occasions upon which it will be obvious to the skilled man that the patentee must in some respect have departed from conventional use of language or included in his description of the invention some element which he did not mean to be essential. But one would not expect that to happen very often.
68 With respect to this passage, the Full Court in Generic Partners at [109]-[110] added:
It is important to note that Lord Hoffman[n] was referring here to the meaning conveyed to the skilled addressee by the language used and was not directing himself to a situation in which the skilled addressee deduced that the language of the claim, although conveying to him or her a particular meaning, could never have been intended to mean what it conveyed.
… These are situations in which the court endeavours to give effect to the skilled addressee’s understanding of the claim language in preference to a purely literal or grammatical construction, not because the skilled addressee understands that the claim contains a mistake that requires correction, but because, when read in the context of the document as a whole and the common general knowledge, the words used would convey that meaning to the skilled addressee.
Regarding the claims
69 With respect to the claims in a patent specification, as Lord Russel explained in Electric and Musical Industries, Ltd v Lissen, Ltd [1938] 4 All ER 221 at 224; (1939) 56 RPC 23 at 39:
The function of the claims is to define clearly and with precision the monopoly claimed, so that others may know the exact boundaries of the area within which they will be trespassers. Their primary object is to limit, and not to extend, the monopoly. What is not claimed is disclaimed. The claims must undoubtedly be read as part of the entire document, and not as a separate document. Nevertheless, the forbidden field must be found in the language of the claims, and not elsewhere.
See also Otsuka Pharmaceutical Co Ltd v Generic Health Pty Ltd (No 2) [2016] FCAFC 111; 120 IPR 431 (Otsuka FCAFC) at [94] (Besanko and Nicholas JJ).
70 Section 40(3) of the Patents Act requires that the claims of a patent be clear and succinct. It is an objection to the validity of a patent that “the scope of any claim of the complete specification is not sufficiently and clearly defined”: see Blanco White, Patents for inventions and the protection of industrial design (5th ed, 1983) (Blanco White) at 4-701, p 143. The need for clarity arises because, as Dixon CJ said in Martin v Scribal Pty Ltd (1954) 92 CLR 17 (Martin v Scribal) at 59, “the principles governing the definition of a monopoly operating over the public at large require a description which is not reasonably capable of misunderstanding”.
71 A court may refer to the body of the specification to identify the background of the claims and the meaning of technical terms, and also to resolve ambiguities in, or other doubts about, the claims’ construction: Austal Ships Sales Pty Ltd v Stena Rederi Aktiebolag [2008] FCAFC 121; 77 IPR 229 (Austal Ships) at [13], citing Flexible Steel Lacing Co v Beltreco Ltd [2000] FCA 890; 49 IPR 331 (Flexible Steel) at [71]-[78]; Nesbit Evans Group Australia Pty Ltd v Impro Ltd [1997] FCA 1092; 39 IPR 56 (Nesbit Evans) at 94-95. A claim is not invalid merely because it might have been better drafted nor because it is difficult to construe, “provided it can be properly and fairly construed”: Britax Childcare Pty Ltd v Infa-Secure Pty Ltd (No 4) [2015] FCA 651; 113 IPR 280 at [583] (emphasis in original); see also Flexible Steel at [80]-[81], quoting Blanco White at 4-701, and cited with approval by the Full Court in Austal Ships at [14].
72 In Martin v Scribal at 97, Taylor J observed:
The claims also must be construed without an eye on the alleged infringer’s acts. … On the other hand, it is right to construe a claim with an eye benevolent to the inventor and with a view to making the invention work — this is an application of the old doctrine ut res magis valeat quam pereat … ; and, where the language of a claim is obscure or doubtful, the doubt may sometimes be resolved by referring to words in the body of the document to explain it. This is known as the dictionary principle. …
This remains a correct approach: see Otsuka FCAFC at [95] (Besanko and Nicholas JJ).
73 In Minnesota Mining and Manufacturing Co v Beiersdorf (Australia) Ltd (1980) 144 CLR 253 (Minnesota Mining) at 274, Aickin J said that the principle with respect to clarity is that “[l]ack of precise definition in claims is not fatal to their validity so long as they provide a workable standard suitable to the intended use”. Stephen J had applied this principle in Monsanto Co v Commissioner of Patents (1974) 48 ALJR 59 (Monsanto), in holding at 61 that the use of the word “substantial” was clear in the phrase “in an amount to have any substantial effect as a cooling medium” in a claim in the patent in suit. Stephen J said, at 60, that:
The lack of clarity which is complained of results, it is said, solely from the use of the adjective “substantial” in the phrase “any substantial effect as a cooling medium”. It is said that its use makes it impossible to determine the limits of the claim to monopoly, that the specification contains no criteria of substantiality of effect and that its meaning cannot be determined by simple experiment.
… Of course what is substantial is a question of degree and is, in that sense, imprecise but in its present context it does not, I think, give rise to any ambiguity.
…
It will in each case be a question of fact and degree whether or not an injected admixture has a substantial effect as a cooling medium and this is the very sort of question which not only do courts have to answer daily, but which I believe that those skilled in the art would have little difficulty in resolving. When only small quantities of admixed fluids are involved, as will be the case when modifiers or solvents are in question, the evidence discloses that they will have no “substantial” effect as cooling media. …
There will, I think, in the present case be no difficulty in a third party ascertaining whether or not what he proposes to do falls within the ambit of the claim … .
74 The principle stated by Aickin J in Minnesota Mining has since been regularly applied in Australian courts: see, for example, Freeman v TJ and FL Pohlner Pty Ltd (1994) 30 IPR 377 at 381-382; Nesbit Evans at 94-95; Austal Ships at [14], quoting Flexible Steel at [81]. In Nesbit Evans at 95, Lindgren J, with whom Hill J agreed, said that:
… it is not unusual for a claim to define an invention partly by the use of a relative expression which necessitates exercise of judgment … and so long as the claims provide a workable standard suitable to the intended use, they will be valid … . The expressions in question must be understood in a practical, common sense manner.
75 In Flexible Steel at [81], Hely J said that:
It is permissible for an invention to be described in a way which involves matters of degree. Lack of precise definition in claims is not fatal to their validity, so long as they provide a workable standard suitable to the intended use. The consideration is whether, on any reasonable view, the claim has meaning. In determining this, the expressions in question must be understood in a practical, commonsense manner. Absurd constructions should be avoided and mere technicalities should not defeat the grant of protection.
(Citations omitted)
76 Whether or not a degree of imprecision is fatal to the validity of a claim depends on whether the claim provides a workable standard for those skilled in the art in the circumstances of the intended use. Where there is no workable standard, imprecision in a claim spells invalidity for lack of clarity, as Albany Molecular Research Inc v Alphapharm Pty Ltd [2011] FCA 120; 90 IPR 457 (Albany Molecular) illustrates. In that case, Jessup J held at [174]-[175] that the expression “substantially pure” when used in the claims in the patent in suit failed to satisfy s 40(3) of the Patents Act, because it left “the definition of the boundaries of the invention uncertain or variable”. (The lack of clarity in that case was cured by a subsequent amendment to the specification, which indicated that the claim to substantial purity was limited to compounds of at least 98% purity: Albany Molecular at [182], [186].)
Regarding the skilled addressee
77 The construction of a patent specification is the responsibility of the Court, since it is a matter of law. To construe a specification, however, the Court must place itself in the positon of the skilled addressee in Australia to whom the specification is addressed: see W R Grace & Co v Asahi Kasei Kogyo Kabushiki Kaisha (1993) 25 IPR 481 (Grace v Asahi) at 496. The words of the specification are to be given the meaning that that skilled addressee would give them in light of the common general knowledge immediately prior to the priority date and what is disclosed in the specification itself.
[S]ince documents of this nature are almost certain to contain technical material, the court must, by evidence, be put in the position of a person of the kind to whom the document is addressed, that is to say, a person skilled in the relevant art at the relevant date. If the art is one having a highly developed technology, the notional skilled reader to whom the document is addressed may not be a single person but a team, whose combined skill would normally be employed in that art in interpreting and carrying into effect instructions such as those which are contained in the document to be construed.
General Tire & Rubber Co v Firestone Tyre & Rubber Co Ltd [1972] RPC 457 (General Tire) at 485.
78 The “skilled addressee” is essentially a hypothetical construct equivalent to a person “acquainted with the surrounding circumstances as to the state of [the] art and manufacture at the time”: Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd [2001] HCA 8; 207 CLR 1 at [24], citing with approval British Dynamite Co v Krebs (1879) 13 RPC 190 at 192. Such a person is “likely to have a practical interest in the subject matter of the invention”: Catnic Components Ltd v Hill & Smith Ltd [1982] RPC 183 at 242. The skilled addressee is taken to be a person of ordinary skill in the field to which the invention relates, rather than a leading expert in the field: see Streetworx Pty Ltd v Artcraft Urban Group Pty Ltd [2014] FCA 1366; 110 IPR 82 at [67] (Beach J). As the Full Court in Jupiters at [67] said, “the words used in a specification are to be given the meaning which the normal person skilled in the art would attach to them, having regard to his or her own general knowledge and to what is disclosed in the body of the specification”.
79 The Patent relates to the field of intensive care medicine and concerns the sedation of patients in intensive care using dexmedetomidine. The skilled addressee to whom the Patent is addressed is an intensive care specialist engaged in clinical practice and in some research in the field of intensive care medicine in Australia immediately prior to the priority date. Such a person would have the requisite knowledge of the state of the “art and manufacture” at the relevant time and a practical interest in the subject matter of the invention.
80 Intensive care specialists (or intensivists) in Australia at the priority date were the specialist doctors who were responsible for the clinical care of patients in an intensive care setting in a hospital. Such specialists were typically physicians or anaesthetists with specialist intensive care training. The clinical practice of intensive care specialists in Australia at the priority date would have included sedating intensive care patients in the ICU.
81 Professor Bellomo’s evidence was (and I accept) that immediately before the priority date, intensivists in Australia generally focussed their research on “issues related to major organs of the body rather than on areas such as sedation”. Assoc Professor French gave evidence to the same effect. Assoc Professor French explained (and again I accept) that, at the priority date, sedation was thought to be “a fairly benign procedure” in that “the practice of ICU sedation was not thought to generally impact patient outcomes adversely”.
82 Although the Patent concerns the sedation of patients in the ICU using dexmedetomidine, I do not accept, as InterPharma contended, that the relevant skilled addressee was an intensive care specialist with a particular interest in research in the field of intensive care sedation. To qualify the skilled addressee in this way is to select a sub-set of intensivists, with a particular level of interest, knowledge and skill in an aspect of intensive care medicine. This is not to identify the person of ordinary skill who works in the field of the invention (intensive care medicine) in Australia, and therefore not to identify the skilled addressee. Whilst highly skilled individuals, the ordinary intensive care specialist in Australia at the priority date did not have a specific interest, let alone research interest, in the sedation of patients in intensive care.
83 It may be accepted that the skilled addressee is not a manifestation of or “avatar” for the expert witness whose testimony is accepted by the court: AstraZeneca AB v Apotex Pty Ltd [2015] HCA 30; 257 CLR 356 (AstraZeneca HCA) at [23]. The skilled addressee is a tool of analysis that is given form and content by the testimony of those witnesses. In this context, I accept that Professor Bellomo in particular was able to, and did, give relevant evidence as to how the skilled addressee would have understood the teaching, terms and expressions in the Patent and the relevant prior art. His evidence also assisted in ascertaining the knowledge that the skilled addressee would have had immediately before the priority date, including what the skilled addressee would have done as a matter of routine when faced with the problem that the invention of the Patent was intended to address.
84 The evidence of Assoc Professor French was able to, and did, assist from time to time with respect to particular matters, often in explaining or augmenting Professor Bellomo’s evidence. It is necessary to bear in mind, however, that although he had been working in the field of intensive care medicine as a registrar for some years before April 1998, Assoc Professor French did not complete his qualification as an intensive care specialist until early 1998. It will be recalled that he said, and I accept, that he had a greater breadth of knowledge of matters relevant to intensive care medicine at the time he completed his examinations for Fellowship of the Faculty of Intensive Care in 1998 than thereafter. It may be inferred from this that, as at and immediately before the priority date, his knowledge of some matters relevant to intensive care medicine would have been greater than that of the ordinary intensivist at that time. It may also be relevant to bear in mind that although Assoc Professor French had been engaged in clinical trials for the development of new drugs and had published two papers, as at April 1998, his research interest was to develop much more in the ensuing years.
85 Care also needs to be taken as regards Professor Hall’s evidence, bearing in mind that his medical training and clinical experience are in the US, including his training and experience in critical care medicine (as intensive care medicine is called in the US). For this reason, the testimony of Professor Bellomo and, subject to the qualifications already mentioned, the testimony of Assoc Professor French are likely to be of more assistance in enabling the Court to place itself in the positon of the skilled addressee in Australia than the evidence of Professor Hall. This does not mean that Professor Hall’s testimony can or should be entirely disregarded, since the evidence showed that the relevant “art and manufacture” had some important international dimensions. Rather, the fact that Professor Hall gave testimony based on knowledge and experience gained outside Australia has to be borne in mind in considering issues of evidentiary relevance, cogency and weight. It has also to be borne in mind that, in contrast to the ordinary intensive care specialist in Australia at the priority date, Professor Hall had at that date a particular research interest in sedation in critical care medicine and, in consequence, his knowledge of sedation in this field would have been more extensive than that of the ordinary intensive care specialist. It is also the case that his field of research was relevant to the invention of the Patent.
Are the claims in the Patent clear?
86 It is convenient to address the parties’ submissions on clarity at this point, since they also involve issues concerning the construction of the claims of the Patent in suit.
87 Before doing so, however, it is necessary to address InterPharma’s application for leave to amend its particulars of invalidity as regards its clarity ground in accordance with its Proposed Fourth Further Amended Particulars of Invalidity.
Leave to amend particulars of invalidity — clarity
88 When the hearing began on 3 May 2018, InterPharma was relying on its “Further Further [sic] Amended Particulars of Invalidity” filed on 12 April 2018. InterPharma subsequently prepared another document entitled “Third Further Amended Particulars of Invalidity”, which it filed on 11 May 2018. This document was created following a discussion in court on 7 May 2018, in the course of which senior counsel for Pfizer noted that InterPharma’s case on invalidity appeared to have “slimmed down somewhat” and requested that InterPharma file amended particulars of invalidity and an amended defence to cross-claim (the effect of which need not be addressed here) to reflect the case that it sought to make. The Court granted leave to InterPharma that day to amend its pleadings and particulars to this end.
89 In its closing submissions filed on 18 May 2018, however, Pfizer took issue with the form of the particulars filed by InterPharma on 11 May 2018. The matter was taken up at the commencement of the hearing of closing submissions on 21 May 2018. In response to the discussion that day, InterPharma filed another document on 24 May 2018, also entitled “Third Further Amended Particulars of Invalidity”. A Proposed Fourth Further Amended Particulars of Invalidity was filed on 25 May 2018 (incorporating the amendments made by the Third Further Amended Particulars of Invalidity filed on 24 May 2018). This document also indicated the proposed amendments for which leave was still required. Pfizer opposed a substantial part of these amendments, including InterPharma’s proposed amendments with respect to its clarity objection.
90 As indicated, when the trial began, InterPharma was relying on its Further Further Amended Particulars of Invalidity, according to which it alleged (in para 7A) that all the claims were invalid on the basis that they did not comply with s 40(3) of the Patents Act, because the limitation “for use in intensive care unit sedation” in claims 1 to 12 and 38, the limitation “a patient in an intensive care unit” in claims 13 to 25 and 39, and the limitation “in the intensive care unit sedation” in claims 26 to 37 were not clear to the skilled reader. This was said to be because it was not clear to the skilled reader:
... whether “intensive care unit sedation” referred to in claims 1 to 12, and claims 26 to 38, inclusive comprises sedation of all patients who are in an intensive care unit or only comprises sedation of a sub-set of critically ill patients who are in an intensive care unit and if so how that sub-set is defined[; and]
… whether a “patient in an intensive care unit” referred to in claims 13 to 25 inclusive includes any patient who is in an intensive care unit or only includes a sub-set of critically ill patients who are in an intensive care unit and if so how that sub-set is defined.
InterPharma retained these particulars in its Proposed Fourth Further Amended Particulars of Invalidity.
91 In opening written submissions filed before trial, InterPharma submitted as to lack of clarity that:
20. While the Patent states that an ICU encompasses “any setting that provides intensive care”, it does not further describe or elucidate the features of such a setting.
21. The Patent also states that in the ICU context, sedation includes the actual sedation of a patient and “the treatment of conditions that affect patient comfort, such as pain and anxiety”. Thus, analgesic agents such as opioids are “called sedatives in the context of ICU sedation …”.
…
23. InterPharma will contend that the claims of the Patent lack clarity because the concept of an ICU, which is a limiting feature of all of the claims of the Patent, lacked a sufficiently clear meaning before April 1998 to define a precise boundary of Pfizer’s patent monopoly. …
24. [T]he claims of the Patent are not directed to the particular qualities of the patients to whom dexmedetomidine is administered but rather the setting of that administration. The Patent covers any use of dexmedetomidine in an intensive care setting suitable to achieve sedation of a patient (or, as that term is defined in the Patent, to treat their pain or anxiety).
…
26. While intensivists agree that there are areas within hospitals that are designated as ICUs, and that these areas have certain features such as constant monitoring and availability of life support, there is no delineation between the level of care relevantly given to patients in other hospitals settings such as the operating theatre, a high care post-operative unit, high dependency unit or recovery room. …
27. The lack of clarity of what is an ICU cannot be resolved by defining a certain class of ICU patients. … Pfizer’s attempt to do so depends on excluding certain ICU patients (e.g. those not intubated) from the class for the reason that sedatives are less important for those ICU patients.
(Citations omitted)
92 In her oral opening, senior counsel for InterPharma also submitted, in relation to these limitations, that “the boundaries are not clear and it includes, on our case, the … relatively healthy compared to the critically ill who are being monitored in the ICU”.
93 Pfizer contended that InterPharma did not pursue the sub-set argument foreshadowed in its Further Further Amended Particulars of Invalidity. I reject this submission. It seems to me that InterPharma retained this argument throughout, although it also mounted other arguments (foreshadowed above) to support its fundamental contention that the limitations “for use in intensive care unit sedation”, “a patient in an intensive care unit”, and “in the intensive care unit sedation” are so unclear as to invalidate the claims in which they appear, essentially because the concept of an ICU is itself unclear.
94 InterPharma’s case in closing was not substantially different from its case in opening. In closing written submissions, InterPharma submitted:
86. The concept of an ICU lacked a sufficiently clear meaning before April 1998 to define a precise boundary of Pfizer’s patent monopoly.
87. While the Patent states that an ICU encompasses “any setting that provides intensive care”, it does not further describe or elucidate the features of such a setting.
88. As appears from the discussion earlier, the experts agreed that there were units designated as ICUs and that these were intensive care settings that shared certain features. However, intensive care could also be given outside a unit designated as an ICU. The independent claims lack clarity as there is no clear boundary to be drawn between, for example, the level of monitoring and care provided to patients in an ICU who are not on life support, and that which they received in other post-operative care units. A sensible boundary cannot be drawn by reference to the person overseeing the care: it was clear that not every intensive care setting was overseen by an ICM specialist.
89. The concept of an ICU patient cannot clarify or confine the meaning of an ICU. While the specification refers for example to “critically ill patients” and those who are “mechanically ventilated”, and provides examples of post-surgical patients who were sedated in an ICU (such as those who underwent coronary artery bypass surgery, or aortic valve replacement), there is no definition of an ICU patient in the Patent. An ICU patient is any patient who is in an intensive care setting, and the experts agree that such patients are a heterogeneous group. Further, whether a given patient is in an ICU … depends on clinical judgment, the availability of ICU beds and other care options.
90. In any event, the claims are not drawn by reference to the patients who are treated with dexmedetomidine but rather the setting of its administration. It would be impermissible to put a gloss on the claims by limiting their scope to a specific population of ICU patients described in the Patent.
91. Nor can the concept of ICU sedation itself clarify or confine the meaning of ICU. It is clear that ICU sedation could be given to any patient who is in an intensive care setting and is to be broadly construed, including the “actual sedation of a patient” and “the treatment of conditions that affect patient comfort, such as pain and anxiety”. ...
95 The sub-set argument is included at [89] of InterPharma’s closing submissions, although these submissions evidently include other arguments, foreshadowed in opening, in support of the proposition that the relevant limitations are so unclear as to invalidate the claims.
96 At a general level, this is the position that is sought to be reflected in InterPharma’s Proposed Fourth Further Amended Particulars of Invalidity. By this document, InterPharma indicates that it seeks leave to incorporate paras (f) and (g) into the particulars to para 7A. These proposed particulars allege that it is not clear to the skilled reader:
(f) … whether a patient outside a unit designated as an intensive care unit does or does not constitute a “patient in an intensive care unit”/“intensive care unit patient” referred to in claims 13 to 25 and 39 of the Patent and if so in what circumstances[; and]
(g) … whether sedation of patients outside a unit designated as an intensive care unit does or does not constitute “intensive care unit sedation” referred to in claims 1 to 12, and claims 26 to 38, and if so in what circumstances.
97 In submissions filed, with leave, on 29 May 2018, Pfizer opposed the grant of leave to permit the incorporation of paras (f) and (g), on the basis that they alleged a new basis for invalidity and that the evidence at trial had been completed. Pfizer submitted that, had these amendments been made at trial, it might have questioned witnesses and cross-examined Professor Hall on relevant matters and that it had now lost the opportunity to do so. Pfizer also submitted that the proposed amendments were unclear and ambiguous.
98 Pfizer argued that the proposed paras (f) and (g) to the particulars to para 7A would give rise to new factual issues not in issue at trial, including as to whether a patient outside a unit designated as an “ICU” did or did not constitute an “ICU patient” and whether sedation outside a unit designated as an “ICU” did or did not constitute “intensive care unit sedation”.
99 Noting that amendments to the same or similar effect had been proposed in the earlier Third Further Amended Particulars of Invalidity filed on 11 May 2018, Pfizer also emphasised that InterPharma had added, without explanation, a reference to claim 39 in para (f) of the particulars to para 7A.
100 In any event, so Pfizer contended, it was apparent from the expert evidence that the skilled addressee would have no difficulty reading and understanding each of the claims in the context of the Patent and that invalidity did not arise by virtue of failure to comply with s 40(3) of the Patents Act on the basis that the expressions “patient in an intensive care unit”, “intensive care unit patient”, “intensive care unit sedation”, and “intensive care unit” lacked clarity.
101 Save for two matters, I am not persuaded that, if leave were granted, Pfizer would have missed a relevant opportunity to meet the case delineated by paras (f) and (g) to the particulars to para 7A of InterPharma’s Proposed Fourth Further Amended Particulars of Invalidity. It should be inferred from InterPharma’s written and oral submissions in opening that, in substance, InterPharma’s clarity case at the commencement of the trial was that the use in the claims of the relevant limitations deprived the claims of the requisite clarity because the concept of an ICU was not sufficiently clear before the priority date, and intensive care could be given both within and outside a designated ICU, the concept of ICU sedation was very broad, and the lack of clarity could not be resolved by reference to a defined class of ICU patients. InterPharma’s written opening submissions made it clear that its attack on clarity included the proposition that there was no clear boundary between the level of care in an ICU and the level of care in other hospital settings, and that, in this context, it was unclear whether “intensive care unit sedation” included the sedation of patients who were not in a designated ICU. These arguments were filled out at [88] and [91] of InterPharma’s closing written submissions, but they remained the same arguments. Doubtless, the addition of paras (f) and (g) to the particulars to para 7A of the Fourth Further Amended Particulars of Invalidity will elaborate on what was earlier set out in para 7A of the Further Further Amended Particulars of Invalidity. Save in two respects, this elaboration will merely express the case that InterPharma opened and ran at trial. Pfizer was put on notice of InterPharma’s case before the commencement of the trial, when InterPharma filed its opening written submissions. It is not the case that these proposed new particulars raise factual issues that were not raised at the commencement of, or during, the trial. This is, so it seems to me, confirmed by certain of the questions addressed to the medical experts, the answers to which were included in the Joint Report, by the course of some of the concurrent evidence, and by other evidence adduced by Pfizer in chief and in cross-examination.
102 It does not seem to me, however, that leave should be given to include the words “and if so in what circumstances” in either para (f) or (g) of the particulars to para 7A. The inclusion of these words would, in my view (and substantially for the reason identified by Pfizer in connection with the proposed amendment to the particulars to para 7B), enlarge the case beyond that made at the trial, after the close of evidence and to Pfizer’s prejudice since it has lost the opportunity to adduce evidence or cross-examine Professor Hall on relevant matters. In any event, the intended significance of these words is unclear.
103 Secondly, in the absence of any explanation by InterPharma as to the late incorporation of claim 39 into para (f) of the particulars to para 7A, leave to include a reference to claim 39 should be granted only on the basis that, as it is an omnibus claim, then if InterPharma were successful in this aspect of its invalidity case, any such success should flow through to claim 39 on the same basis and no other basis.
104 Accordingly, subject to these qualifications, leave is granted to InterPharma to amend its particulars to include paras (f) and (g) to the particulars to para 7A in its Proposed Fourth Further Amended Particulars of Invalidity (hereafter referred to as the Fourth Further Amended Particulars of Invalidity).
105 There was a second basis for the alleged invalidity of the Patent’s claims on the basis of lack of clarity, as set out in para 7B of InterPharma’s Further Further Amended Particulars of Invalidity. This referred to the limitation introduced by the expression “essentially the sole active agent or the sole active agent” in claims 2 to 12, 14 to 25, 27 to 37 and 39 (although claim 15 did not include this specific limitation but an analogous limitation “an active and an inactive agent”). The particulars to para 7B of InterPharma’s Further Further Amended Particulars of Invalidity relevantly alleged that it is not clear to the skilled reader:
(c) … whether the administration of dexmedetomidine … with an anaesthetic, analgesic, anti-psychotic or a non-sedative agent in an intensive care unit constitutes its use as “essentially the sole active agent” for sedation and/or for sedating a patient.
106 By its Fourth Further Amended Particulars of Invalidity, InterPharma sought to add the words “or in what circumstances”, so that proposed para (c) of the particulars to para 7B alleged that:
(c) It is not clear to the skilled reader whether or in what circumstances the administration of dexmedetomidine … with an anaesthetic, analgesic, anti-psychotic or non-sedative agent in an intensive care unit constitutes its use as “essentially the sole active agent” for sedation and/or for sedating a patient.
(Emphasis added)
107 InterPharma also sought to delete para (b) of the particulars to para 7B of its Further Further Amended Particulars of Invalidity.
108 In its 29 May 2018 submissions Pfizer opposed the grant of leave to add the words “or in what circumstances” to para (c) of the particulars to para 7B. As Pfizer noted, InterPharma has not provided any explanation as to why it seeks to add the words “in what circumstances”. It may be that the proposed amendment is intended to align the particulars to this paragraph more closely to InterPharma’s opening written submissions, which stated:
The word “essentially” indicates that dexmedetomidine may be co-administered with another agent. If the other agent is a drug with analgesic or anxiolytic qualities then, given that the Patent defines ICU sedation to include actual sedation and the treatment of pain or anxiety, it is not clear in what circumstances involving the administration of a second agent with analgesic or anxiolytic qualities, such as an opioid or benzodiazepine, the use of dexmedetomidine would fall within these claims.
(Emphasis added)
109 Viewed in this way, it may be that the proposed amendment to para (c) of the particulars to para 7B is not intended to expand InterPharma’s case beyond its submissions in opening. The precise import of the proposed amendment is, however, uncertain. On another reading, the amendment would, as Pfizer contended, substantially expand InterPharma’s case in relation to the clarity of the “essentially the sole active agent” limitation, by advancing an alternative case to that which InterPharma ran at trial. That is, the proposed addition of “or in what circumstances” might be thought to raise the possibility that:
if contrary to InterPharma’s pleaded case, it is clear to the skilled addressee that the administration of dexmedetomidine with an anaesthetic, analgesic, anti-psychotic or a non-sedative agent in an intensive care setting does or does not constitute use of dexmedetomidine as “essentially the sole active agent” for ICU sedation, … [then] it is not clear “in what circumstances” the administration of dexmedetomidine with such an agent constitutes use of dexmedetomidine as “essentially the sole active agent” for ICU sedation.
110 This question was not put in issue in the Further Further Amended Particulars of Invalidity or in any subsequent version of InterPharma’s Particulars of Invalidity. It was not raised at trial. The proposed amendment to para (c) would in this case prejudice Pfizer since it has lost the opportunity to adduce evidence or cross-examine Professor Hall about this issue. In any event, as before, the intended significance of the proposed additional words is unclear.
111 In these circumstances, I would not grant leave to amend para (c) of the particulars to para 7B. In any event, it does not seem to me that the amendment is necessary. InterPharma will not be prevented from advancing the case it has already run with respect to this limitation at trial.
112 Pfizer did not oppose the proposed deletion of para (b) of the particulars to para 7B of InterPharma’s Further Further Amended Particulars of Invalidity. Leave will be granted to make this amendment.
113 There was a third basis for the alleged invalidity of the claims for reason of lack of clarity set out in para 8 of InterPharma’s Further Further Amended Particulars of Invalidity. Paragraph 8 asserted that claims 3 to 12 and 38 do not comply with s 40(3) because “[i]t is unclear which ‘use’ in claim 1 is the intended ‘use’ in claims 3 to 12 and 38”. Further, it was asserted that to the extent that the “use” is:
… use of dexmedetomidine to make a medicament of claim 1 it is not clear to the skilled reader how this could be construed to include a method of administering the medicament of claim 1 to a patient as part of the process for making the medicament [; and]
… the “for use” defining the purpose of the medicament in claim 1 it is not clear to the skilled reader how the method or process of manufacturing the medicament of claim 1 could be construed to include a method of administering the medicament.
114 InterPharma sought to amend para 8 by substituting a reference to “claims 2 to 12” for the references to “claims 3 to 12”. Pfizer did not oppose a grant of leave to make this amendment. Leave will be granted to make this amendment.
115 There was a fourth basis for alleged invalidity on the basis of lack of clarity set out in para 9 of the Further Further Amended Particulars of Invalidity, which was said to relate to claims 23, 24 and 25 “on the grounds that [these] claims do not comply with the requirements of section 40(3) of the Patents Act in that the claims are not clear and succinct”. By its Fourth Further Amended Particulars of Invalidity, InterPharma sought to delete para 9. Pfizer did not oppose a grant of leave to make this amendment. Leave will be granted to make this amendment.
Overview of InterPharma’s submissions as to lack of clarity
116 InterPharma submitted in closing that:
(a) none of the claims comply with s 40(3) of the Patents Act as it is not sufficiently clear from the claims when post-operative care outside a designated ICU constitutes a setting of intensive care within the meaning of those claims;
(b) claims 2, 14 and 27 (and their dependent claims and claim 39) do not comply with s 40(3) as it is not sufficiently clear from the claims what is meant by the expression “essentially the sole active agent”; and
(c) claims 2 to 12 and 38 do not comply with s 40(3) as it is not sufficiently clear from the claims how the purported Swiss-style dependent claims are intended to be construed.
117 In closing submissions InterPharma also contended that claims 3, 16, and 28 (and dependent claims 4 to 12, 17 to 25 and 29 to 36) do not comply with s 40(3) as it is not sufficiently clear from the claims what is meant by the expression administration of dexmedetomidine “in an amount to achieve a plasma concentration of 0.1-2 ng.ml”. This allegation is not to be found in the relevant Particulars of Invalidity. Pfizer did not make any submission about this omission, and I shall address it in these reasons in the manner it was argued (see [201]-[208] below). InterPharma made no submission in this regard about claim 37.
All claims: lack of clarity from use of the expressions “for use in intensive care unit sedation”; “a patient in an intensive care unit”; “an intensive care unit patient”; and “in the intensive care unit sedation”?
118 The Patent relates to the sedation of ICU patients. As already noted, InterPharma contended that all of the claims of the Patent lack clarity because the concept of an ICU, which was a limiting feature of all claims, lacked a sufficiently clear meaning before April 1998 to define the monopoly precisely.
119 Noting that the Patent states that “the word intensive care unit includes any setting that provides intensive care”, InterPharma contended that the lack of clarity arose because the Patent did not further describe the features of such a setting. Referring to the experts’ evidence that intensive care could be given outside a designated ICU, InterPharma submitted that there was no clear boundary between what was, and was not, an ICU setting. No such boundary existed, so InterPharma submitted: (a) between the level of monitoring and care for patients not on life-support in an ICU and the level of monitoring and care that such patients received in a high care post-operative unit or a high dependency unit; or (b) by reference to the overseeing specialist since not every intensive care setting was overseen by such a specialist. Although the experts were in broad agreement about the characteristics of an ICU, InterPharma submitted that “the fact that there are these other non-ICU units complicates where the edge is” and the experts were not agreed on “where the border of where they understood it to be an ICU … is”.
120 InterPharma further submitted that the concept of an “ICU patient” could not clarify the meaning of an ICU, because there was no definition of an ICU patient in the Patent, and patients in an intensive care setting were, so the experts agreed, a heterogeneous group and even more so in hospitals in regional centres than in cities. In any event, InterPharma emphasised that whether or not a given patient was in an ICU depended on clinical judgment, the availability of ICU beds and other care options.
121 InterPharma also submitted that the concept of ICU sedation could not clarify the meaning of an ICU, because: (1) ICU sedation could be administered to any patient in an intensive care setting; and (2) ICU sedation was to be broadly construed to include not only “actual sedation” but also “the treatment of conditions that affect patient comfort, such as pain and anxiety”. InterPharma noted that the Patent states that many drugs were only considered sedatives in the context of ICU sedation and that the concept of ICU sedation under the Patent extended to administering any agent to a patient who is in any ICU setting, where the agent helped to keep the patient calm and allowed them to tolerate their treatment. InterPharma further noted that the Patent claimed levels of sedation on the Ramsay Scale from 2 to 6.
122 Pfizer contended that the skilled addressee would have no difficulty in identifying “an intensive care unit”, as that expression was used in the claims and that the claims were not made doubtful or unclear by the use of the expressions “for use in intensive care unit sedation”, “a patient in an intensive care unit”, “an intensive care unit patient”, or “in the intensive care unit sedation”. I accept Pfizer’s submission in this regard, for the reasons I am about to give.
123 It may be accepted that the expression “intensive care unit” and its cognate expressions, “for use in intensive care unit sedation”, “a patient in an intensive care unit”, “an intensive care unit patient”, and “in intensive care unit sedation”, are limitations, in one or other form, of all the claims of the Patent.
“Intensive care unit”
124 As used in the Patent, the expression “intensive care unit” and its acronym “ICU” can refer to any setting that provides intensive care. The specification specifically explains that “the word intensive care unit includes any setting that provides intensive care”.
125 Professors Hall and Bellomo and Assoc Professor French had no difficulty in identifying “a setting that provides intensive care”. This is apparent from their Joint Report, where, in answer to the question, “[w]hat is a setting that provides intensive care?”, they replied that:
The ICU is a defined area within a hospital. It contains specially trained medical and nursing staff at high staff patient ratio and contains continuous patient monitoring and life support equipment.
126 Nor did the medical experts have any difficulty identifying an ICU patient. In answer to the question, “[w]ho is an ICU patient?”, their agreed response in their Joint Report was that:
Intensive care patients are those who require specialised treatments or a level of monitoring that is not able to be provided in a general hospital environment.
127 The three medical experts agreed upon the identifying characteristics of an intensive care setting, by reference to the type of patients, the type of care, the type of monitoring, the ratio of nurses or doctors to patients and the direct supervision of an exclusively rostered specialist. Their evidence was that, prior to April 1998 (and now) there were (and remain) areas within hospitals designated as ICUs, which have certain characteristics, such as specially trained medical and nursing staff at a high staff-patient ratio, continuous patient monitoring, and life support equipment.
128 Professor Bellomo’s evidence was that the term “intensive care unit” or “ICU” refers to an area in a hospital dedicated to the care of the sickest patients in the hospital; and that, in Australia, in 1998, an ICU typically had:
(a) one to one nurse to patient ratio at all times;
(b) senior doctors (including intensive care specialists) available 24 hours a day;
(c) advanced trainee doctors and residents available 24 hours a day; and
(d) continuous monitoring.
129 Professor Hall stated in evidence that he agreed with Professor Bellomo’s account of the typical characteristics of an ICU, but would add reference to the availability in an ICU of continuous intravenous infusion. Professor Bellomo and Assoc Professor French later stated their substantial agreement with Professor Hall and that continuous infusion of sedatives was unlikely to be found outside the operating theatre or the ICU. Similarly a bolus administration was almost exclusively given in an ICU. This is also consonant with the Joint Report.
130 Professor Bellomo’s evidence was that, immediately before April 1998 (and now), intensive care medicine (involving the assessment, stabilisation and ongoing care of patients who required advanced life support and/or advanced monitoring as a result of life-threatening illnesses, conditions, injuries or complications) was typically conducted in an ICU. His evidence was that an intensive care specialist was required to assess and repeatedly reassess vital functions, including breathing rate, oxygen and carbon dioxide levels, concentrations of key elements in the blood, blood pressure, heart rate, brain function, liver function and kidney function. This occurred in an ICU.
131 All three medical experts agreed that the ICU environment (or setting) can be distinguished from other hospital settings where nurse to patient ratios are lower, moment to moment patient care is not required and certain life supports, such as mechanical ventilation, are not available. In the opinions of Professor Bellomo and Assoc Professor French, immediately before the priority date (as now) a high care post-operative unit, a high care dependency unit, a coronary care unit and a step-down unit were (and are) relevantly distinguishable from an ICU because of the differences in type of patients, type of care and type of monitoring within them. Professor Hall agreed that, in his North American experience, the differences between these units was “more than language and [the] name on the door”.
132 For example, Professor Bellomo said in evidence:
My view is that the high-care post-operative area, or the high-dependency unit, as we often refer to in Australia, is a separate entity, and it is so named because it is a separate entity even though it has some features of monitoring and treatment that resemble those of the ICU.
…
The location of a high-dependency unit, geographically, may be within the structure, or the room or the floor of the ICU, or may be actually entirely physically separate, depending on hospitals, but the type of patients, and the type of care, and the type of monitoring and the ratio of nurses to – or doctors to patient is different from that of the ICU, hence a different name for it.
133 Assoc Professor French also gave evidence that:
[I]t’s not just the type of patient and the level of monitoring and the level of nursing staff, but another feature of an Intensive Care Unit in Australian practice is that it’s under the direct supervision of an exclusively rostered intensive care specialist. And, in general, the majority of stand-alone high-dependency units, coronary care units and step-down units don’t have that same overarching medical structure and leadership. So at least in the Australian context, an Intensive Care Unit is all those things that Professor Bellomo described, plus having that overarching medical leadership from an appropriately trained intensive care specialist.
134 Assoc Professor French acknowledged that hospital arrangements in regional centres may differ a little from arrangements in the cities in that, for example, in regional centres an ICU might not be under the direct supervision of an intensive care specialist, but would usually be under the direct supervision of an exclusively rostered doctor in a related discipline, such as anaesthesia. Such a doctor would, for example, “be working as a consultant anaesthetist but would also be, at times, exclusively rostered to the Intensive Care Unit”.
135 Both Professor Bellomo and Assoc Professor French agreed that, in Australia, variations in patient demand and the availability of resources at any one time might determine patient placement in some cases. Professor Bellomo’s evidence was that “the logistics, the resources, the needs, the financial burden and the patient type of operations” may result in different patient placements. He said that “[s]ometimes we have to triage. The demand on the ICU beds is greater than the availability and we have to make decisions about how best to manage the resources available”.
136 Professor Bellomo’s evidence concerning ICUs and the existence of comparable areas within a hospital, the practice of intensive care medicine, and the effect of patient demand and resource constraints was compelling. As already noted, he had well over 25 years’ experience in the practice of intensive care medicine in hospitals in Australia. This was apparent in the evidence he gave. His evidence was, moreover, supported by that of Assoc Professor French and in some material respects (as indicated above and allowing for differences in Australian and US hospital arrangements) by that of Professor Hall.
137 In substance, the evidence of the medical experts established that ICUs and intensive care settings in Australia were readily identifiable immediately prior to April 1998 by reference to their typical features, which were a one to one nurse to patient ratio, constant availability of senior doctors and advanced trainee doctors and residents, continuous monitoring, and the availability of continuous intravenous infusion. Put another way, the evidence established that as at April 1998 the ICU environment (or setting) could be readily distinguished from other hospital settings because of the differences in the type of patients, the type of care, the type of monitoring, the ratio of nurses to patients, the availability of doctors and supervision by an exclusively rostered specialist.
138 It is true, as InterPharma noted, that Assoc Professor French’s evidence was that the ICUs in hospitals in regional centres might not be under the direct supervision of an intensive care specialist, although they would be under the direct supervision of an exclusively rostered doctor in a related discipline. It does not seem to me that this diminished his evidence that an ICU was different from other areas of a hospital. Rather, this evidence showed that the same kind of arrangements were made for ICUs in regional centres and city hospitals, necessarily adjusted to take account of a difference in the availability of medical specialist staff. For like reasons, it does not seem to me that the fact that the availability of resources and patient demand at any one time might determine patient placement in or out of the ICU militated against the fact that the skilled addressee would well understand the concept of an ICU, including a setting that provides intensive care. This kind of triage reflected a different consideration, namely, how best to manage finite resources.
139 Each of Pfizer and InterPharma sought to call in aid the definition of “Intensive Care Unit” in item T1.7 of the Medicare Benefits Schedule Book (operating from 1 November 1997) (Medicare Benefits Schedule). InterPharma submitted that the definition was different from a description given by Assoc Professor French. Pfizer submitted that the definition was broadly consistent with the evidence of the medical experts. It does not seem to me that much turned on the differences between the medical experts’ evidence as to the typical identifying characteristics of an ICU and the definition in the Medicare Benefits Schedule, bearing in mind the nature and function of the Medicare Benefits Schedule. At most, the Medicare Benefits Schedule highlighted that, at the level of payment to individuals to meet hospital and medical expenses, the concept of an ICU was well-recognised.
140 Further, it does not appear to me that the experts’ disagreement about the study in Talke (discussed below) militates against the conclusion that the skilled addressee would have readily understood the concept of an “intensive care unit” or ICU immediately before the priority date in Australia.
141 The expression “intensive care unit” and its acronym “ICU” as used in the Patent would have been well-understood and its meaning would be clear to a skilled addressee immediately before the priority date in Australia. The skilled addressee in Australia as at that date would have been readily able to identify an ICU and any setting that provided intensive care.
“Patient in an intensive care unit”; “intensive care unit patient”
142 The parties did not suggest that there was any relevant difference between the expressions “patient in an intensive care unit” and “intensive care unit patient”. As noted above, the experts agreed about who were “intensive care unit patients”, being, in the words of their Joint Report, “those who require specialised treatments or a level of monitoring that is not able to be provided in a general hospital environment”. The experts agreed, moreover, that in the context of the Patent “patient in an intensive care unit” meant “[a]ny patient in an environment that provides intensive care”.
143 It may be recalled from the earlier discussion of InterPharma’s lack of clarity submissions that InterPharma contended that the concept of an ICU patient cannot clarify or confine the meaning of an ICU; there is no definition of an ICU patient in the Patent; and whether a patient became an ICU patient depended on clinical judgment and other extraneous matters. In so far as these submissions alleged a relevant lack of clarity in the expressions “patient in an intensive care unit” and “intensive care unit patient”, I would reject them for the following reasons.
144 The evidence established that in order to become an ICU patient, a patient must be admitted to an ICU in accordance with the applicable admissions policy. The evidence of Professor Bellomo and Assoc Professor French was, and I accept, that the intensive care specialist on duty would first determine whether intensive care would benefit the patient. Professor Bellomo agreed with Assoc Professor French’s evidence that the duty intensivist would:
… make an overall assessment whether the treatments that we can offer in the ICU are of benefit to the patient and those benefits outweigh the risks, or they make an assessment whether the patient’s level of disease is such they could be managed in a general ward environment.
145 When asked by the Court what united patients in the ICU environment, Professor Bellomo agreed that it was either that they had a life-threatening condition or that they were at risk of a life-threatening condition if things went wrong. This was consistent with Professor Bellomo’s evidence that immediately before the priority date ICU patients were a unique class of hospital patients, united by their need for advanced life support and/or advanced monitoring. He said that between 50% and 80% of ICU patients required intubation in order to allow for mechanical ventilation.
146 Professor Bellomo also said in evidence that the individual characteristics of ICU patients were very variable, and that such patients might arrive from various places, including emergency departments, operating theatres and other specialties within hospitals. Notwithstanding this variability, he emphasised that the care of ICU patients was complex, bearing in mind that most had unstable vital functions; required multiple medications administered in various ways; were under excessive physiological and psychological stress; and were subject to complex interacting conditions. Assoc Professor French gave evidence to similar effect. I accept their evidence in that regard. I also accept Professor Bellomo’s evidence that, while patients in an ICU might be described as heterogeneous because their individual characteristics were variable, the ICU patients at the priority date were critically ill patients, whose care was complex. All the medical experts agreed that the most severely ill patients often had multiple organ failures, while other critically ill patients might not be experiencing organ failure but be assessed to be at risk of complications or deterioration, for example, after major surgery or because of a medical condition.
147 The expressions “patient in an intensive care unit” and “intensive care unit patient” as used in the Patent would have been well-understood and their meaning would have been clear to a skilled addressee immediately before the priority date in Australia. A skilled addressee in Australia as at that date would have been readily able to identify a “patient in an intensive care unit” or an “intensive care unit patient”; and I would reject InterPharma’s submissions to the contrary.
“Intensive care unit sedation”
148 It may also be recalled from the earlier discussion of InterPharma’s lack of clarity submissions that InterPharma contended that the concept of ICU sedation could not confine the meaning of an ICU; that ICU sedation could be administered to any patient who is in an intensive care setting; and that the concept is to be broadly construed, including the “actual sedation of a patient” and “the treatment of conditions that affect patient comfort, such as pain and anxiety”. In so far as these submissions alleged a relevant lack of clarity in the expression “intensive care unit sedation”, I would reject them for the following reasons.
149 In their Joint Report, in answer to the further question, “[w]hat is ‘ICU sedation’?”, the three medical experts agreed that:
ICU sedation is the administration of medication to ICU patients in circumstances where it is necessary so that the patient can tolerate and accept intensive care treatments and environment, for example, the presence of an endotracheal tube to enable mechanical ventilation.
150 It should be observed that this statement about ICU sedation focussed on inducing patient tolerance and acceptance of intensive care treatments and environment by administering medication. The experts agreed that this was what “ICU sedation” meant in the Patent.
151 The medical experts emphasised that the concept of sedation was different to sleep. They agreed that what was meant by “sedation” differed, depending on the medical context, and that the concept of sedation in an ICU was relevantly different from the concept of sedation in other contexts.
152 Professor Hall’s evidence, which I accept, was that sedation involved bringing an ICU patient into a calmer state, along a spectrum from agitated to tranquil, tranquil to asleep. His evidence also was that a patient was brought into a calmer state, in order that the patient could accept intensive care treatment. He observed that calming a patient for this purpose did not always involve medication and might, for example, involve repositioning a mechanical device if that was making the patient uncomfortable, talking with patients and involving their family to allay the patient’s fears, and even medicating to address a “pure source of pain”. Professor Hall added that “we would go to medication that would calm them” only after going through such a checklist.
153 In order to appreciate Professor Hall’s evidence about the purpose of sedation, it is useful also to refer to his evidence that:
Within an ICU, patients have varying sedation needs. In an ICU a patient is very likely to have one or more devices in them, such as peripheral intravenous catheters (IVs), larger catheters going into a major vessel of the body to deliver medications or for monitoring (for example, arterial catheters, where the device is put in and left in the artery of the wrist or the groin) and, in the majority of cases, endotracheal tubes that pass over the tongue and into the airways to allow mechanical ventilation (or “machine breathing”).
…
[M]ost ICU patients are in a state somewhere between mild discomfort and severe pain and are anxious or agitated.
154 I interpolate here that the medical experts agreed that, as at April 1998 (and today), sedatives were (and are) normally administered in an ICU by continuous infusion, although a bolus might first be used if the rapid onset of sedation were desired. Professor Hall stated, and it was not disputed, that the amount of sedatives administered per patient is much higher in an ICU than in a general ward and that a patient being continuously infused with a sedative needs to be continuously monitored, as his or her dosage may need to be titrated.
155 Professor Hall’s evidence about the purpose of sedating an ICU patient was consistent with the evidence of Professor Bellomo and Assoc Professor French. The evidence of Professor Bellomo and Assoc Professor French was that immediately before the priority date (and today) the concept of ICU sedation involved affecting an ICU patient’s awareness or alertness to some degree to induce calm and allow the patient to accept treatment. In Professor Bellomo’s words:
“ICU sedation” … refers to the administration of a drug or drugs to an ICU patient that affect the patient’s awareness to some degree to induce calm and allow the patient to tolerate their circumstances, including necessary medical interventions such as mechanical ventilation and procedures.
Assoc Professor French deposed that:
[S]edation in an ICU or, “ICU sedation”, is the administration of medication to ICU patients in circumstances where it is necessary so that the patient can tolerate and accept intensive care, for example, the presence of an endotracheal tube to enable mechanical ventilation. ICU sedation generally affects the ICU patient’s level of alertness to some degree.
156 The three medical experts agreed that, immediately before April 1998, there was “a variable spectrum on the appropriate level of sedation depending on the patient’s circumstances and response to the drugs”. Professors Hall and Bellomo, in particular, explained this idea in similar terms. They agreed that the target level of sedation for an ICU patient would be likely to vary in the course of treatment. Professor Bellomo’s evidence was that the target for sedation was “dynamic, … related to the patient’s condition, to [the] therapeutic intent, to what you’re intending to do [to] the patient”, for example:
… whether you’re going to put lines or do interventions that would be upsetting the patient[,] whether the patient’s wakefulness might mean that the patient interacts with a breathing machine in a way that is dissynchronous … [a]nd, of course, whether the illness has begun to resolve or has resolved, therefore, justifying the step in the direction of awakening the patient with the intent to remove the ventilator.
157 Depending on the particular needs of the patient, Professor Bellomo’s evidence was that:
ICU sedation may involve drug induced calmness and tolerance, without drowsiness, or it may involve drug induced calmness and tolerance together with some level of drowsiness ranging from light drowsiness to deep “sleep”.
Assoc Professor French made it clear that this remained the case today. His evidence was that the level of sedation would vary, depending on the patient’s needs, from moderate to deep sedation, “to no sedation, and just analgesia”.
158 It is in this context that the Patent specification’s description of levels of sedation equivalent to a Ramsay Sedation Score of 2 (patient co-operative, oriented and tranquil) to 6 (asleep, no response) is to be understood. None of the claims contain a limitation as to the level of sedation.
159 The experts also understood that there was a distinction between administering a medication to address pain and/or anxiety and medicating for sedation. Professor Hall made this point in the evidence to which I have already referred in which he said that intensivists would resort to medicating to calm an ICU patient so as to accept intensive care treatment only after they had endeavoured to allay the patient’s fears through talking and family support and by administering an analgesic for pain-relief. In particular, Professor Hall said:
I think by elimination, … [if] a patient remains agitated despite those things being gone through on a checklist or addressed and then we would go to a medication that would calm them, yes.
160 The same understanding is reflected in Assoc Professor French’s observation that the level of sedation would vary, depending on the patient’s needs, from moderate to deep sedation, “to no sedation, and just analgesia”.
161 InterPharma submitted in closing submissions that:
ICU sedation under the Patent plainly extends to administering any agent to a patient who is in any ICU setting, where the agent helps to keep the patient calm and allow them to tolerate their treatment. ICU sedation must include, for example, administering an analgesic such as opioid as well as anxiolytic and anaesthetic agents such as benzodiazepines, propofol, and barbiturates.
162 This submission involves a level of confusion that was not evident in the evidence of the experts. Their evidence established that, immediately before the priority date, a drug administered for the purpose of sedating a patient in an ICU was a drug administered for the purpose of inducing calmness in the patient so as to facilitate the patient’s acceptance of intensive care treatment. Where a drug was administered in order to treat pain or anxiety without inducing such calmness and acceptance, that drug was not acting as a sedative agent. Where, for example, an analgesic drug does not have any sedative properties at the dose being administered to an ICU patient, then it is acting as an analgesic but it is not acting as a sedative agent as understood in an ICU.
163 In support of its contention that ICU sedation in the context of the Patent includes sedation or treatment of pain and anxiety, InterPharma relied on the Patent specification at p 2, lines 2-3, which states that “sedation also includes the treatment of conditions that affect patient comfort, such as pain and anxiety”.
164 As the medical experts explained, opioids such as morphine may be seen as an analgesic or a sedative, depending on the dose that is administered. The same or a similar difference, depending on dose, is manifest in other classes of drugs.
165 In answer to my question, “[i]f you were administering morphine … you wouldn’t regard that as a sedative, though, would you?”, Professor Bellomo said:
It depends on the dose that you give of morphine. So, for example, if somebody gave me maybe by continuous infusion 0.5 of a milligram, it would be unlikely that [I] would be sedated but if they gave me 10 milligrams, I would be profoundly sedated. So it’s a dose relationship.
If a high sedating dose is administered, then the morphine is being used as a sedative, whilst at a lower dose the morphine will be used as an analgesic. Professor Bellomo’s evidence was that morphine had been administered at a high sedating dose in ICUs in the 1990s and was regarded as an ICU sedative when used in this way.
166 Against this background, I accept Professor Bellomo’s evidence that the passage at p 2 of the Patent identifies that, in addition to a sedative effect, “sedation also includes treatment of conditions that affect patient comfort, such as pain and anxiety and so on” and that this passage was consistent with the experts’ agreed understanding of the meaning of “ICU sedation”, as stated in their Joint Report. Professor Bellomo’s evidence in this regard is also consistent with the subsequent statement in the specification (at p 3, lines 23-25) that “[a]n ideal sedative agent for a critically ill patient should provide sedation … . Further, it should be an anxiolytic and an analgesic … ”. Professor Bellomo was clear, however, that, in the context of the Patent, the administration of morphine for pain alone was not to be understood as the use of morphine as an ICU sedative. Having regard to the way the drug worked when administered to patients in an ICU, whether or not morphine was used as an analgesic or sedative was, to use his own words, “very much dependent on the dose”.
167 For the reasons stated, a skilled addressee in Australia immediately before the priority date would have had no difficulty understanding the expression “in the intensive care unit sedation” as used in the Patent and would have been readily able to identify whether or not a drug was administered “in the intensive care unit sedation”. I would reject InterPharma’s submissions to the contrary.
168 For the reasons stated, I reject InterPharma’s submissions that the claims lacked clarity by reason of the use of the expressions “for use in intensive care unit sedation”; “a patient in an intensive care unit”; “an intensive care unit patient”; and “in the intensive care unit sedation”. Further, it would have been clear to a skilled addressee that none of these concepts was limited by reference to any particular sub-set of patients in the ICU and that such a limitation was not required in the interests of clarity.
Claims 2, 14, 27 (and their dependent claims): lack of clarity from use of the expression “essentially the sole active agent or the sole active agent”?
169 Each of claims 2, 14 and 27 relate to the use of dexmedetomidine in ICU sedation, wherein the dexmedetomidine is “essentially the sole active agent or the sole active agent”. There are numerous dependent claims. As to claim 15, see [187] below.
170 In opening written submissions filed before trial, InterPharma’s submissions as to lack of clarity included that the meaning of “essentially the sole active agent” was not sufficiently clear before April 1998 to define the precise boundary of Pfizer’s monopoly. It was said that:
The word “essentially” indicates that dexmedetomidine may be co-administered with another agent. If the other agent is a drug with analgesic or anxiolytic qualities then, given that the Patent defines ICU sedation to include actual sedation and the treatment of pain or anxiety, it is not clear in what circumstances involving the administration of a second agent with analgesic or anxiolytic qualities, such as an opioid or benzodiazepine, the use of dexmedetomidine would fall within these claims.
171 As noted earlier, InterPharma alleged in para 7B of its Further Further Amended Particulars of Invalidity that it is not clear whether the administration of dexmedetomidine with an anaesthetic, analgesic, anti-psychotic or a non-sedative agent in an ICU constitutes its use as “essentially the sole active agent” for the purpose of ICU sedation.
172 In closing submissions, InterPharma drew attention to the fact that in their Joint Report, the medical experts agreed that the term “essentially the sole active agent” was “a difficult term to understand”, and submitted that their agreed interpretation could not assist in determining the circumstances in which the use of dexmedetomidine with another sedative or analgesic agent would be considered as the use of dexmedetomidine as “essentially the sole active agent” in the claimed method. In this context, InterPharma relied on Professor Hall’s evidence that he would have “no ability to draw any line” between a case in which the drug was used as “essentially the sole active agent” and another case where the drug was administered not as “essentially the sole active agent”, and with an anaesthetic or non-sedative agent. InterPharma also referred to the difficulty that each of Professor Bellomo and Assoc Professor French said that they had in drawing this line, and the fact that none of the experts could say where that line lay. InterPharma contended that there was no clear boundary defined by the expression “essentially the sole active agent” that could be drawn by the person skilled in the art, and that the lay understanding of “essentially” did not have any clinical significance. Senior counsel for InterPharma submitted that:
[I]n the context … where this Patent defines sedatives in an ICU far more broadly than they are understood outside the ICU ... we say this claim really has no boundary that any of the experts were able to articulate.
173 InterPharma contended in the alternative that, if the term “essentially” had a sufficiently clear meaning, then the effect of the term is that the sole agent claims permit the co-administration of dexmedetomidine with a second agent, provided that the second agent is not administered for sedation but for a different purpose such as analgesia. It submitted that this construction was consistent with the evidence from the experts that an analgesic agent is not ordinarily understood by those in the field as a “sedative agent”. For the reasons outlined below, I accept this alternative construction (see [181]-[186]).
174 I accept that, as Pfizer submitted (and contrary to the assertion in para 7B of InterPharma’s Further Further Amended Particulars of Invalidity: see [171]), none of the experts considered that the administration of dexmedetomidine with an anaesthetic or other agent administered for the purpose of ICU sedation consisted the use of dexmedetomidine as “essentially the sole active agent or the sole active agent”. This was, as Pfizer submitted, an absurd construction, which was to be avoided.
175 As will also be apparent by now, much of InterPharma’s clarity case at this point in fact depended on the alleged lack of clarity introduced by the word “essentially”. Whilst InterPharma’s Further Further Amended Particulars of Invalidity were themselves far from clear in this regard, it seems to me that this was the case that it ran at trial.
176 Pfizer submitted, and I accept, that the expression “essentially the sole active agent or the sole active agent” described the administration of dexmedetomidine by itself or “essentially” by itself for the purpose of ICU sedation. Pfizer submitted that although the use of the word “essentially” involved some imprecision, it did not involve any lack of clarity. For the reasons stated below, I agree that the expression does not lack clarity in the relevant sense.
177 Reference to the Patent confirms that this is what is intended by the expression. In discussing the background to the invention, the specification identifies that patients in an ICU may receive a variety of drugs concurrently. It further identifies that the most commonly used drugs are given for patient comfort; that such drugs are “cumulatively called sedatives in the context of ICU sedation”; and that many are not considered sedatives in any other context. The specification further states that “the combination of these agents (polypharmacy) may cause adverse effects”, and that “[p]referably, an ideal sedative agent should be used by itself in ICU sedation to avoid the dangers of polypharmacy”.
178 The Patent states that the invention relates to a method of sedating a patient in the ICU by administering dexmedetomidine. One aspect of the invention is identified as follows:
Particularly, the present invention relates to a method of sedating a patient while in the ICU by administering dexmedetomidine … wherein dexmedetomidine is essentially the sole active agent or the sole active agent administered for this purpose.
(Emphasis added)
179 There is a further description in the specification of this method of sedating a patient in an ICU by administering dexmedetomidine, wherein dexmedetomidine is essentially the sole active agent or the sole active agent, where it is said that:
The method is premised on the discovery that essentially only dexmedetomidine … need[s] to be administered to a patient in the ICU to achieve sedation and patient comfort. No additional sedative agents are required.
The specification makes it clear that this method looks to the totality of the drugs administered to an ICU patient for the purpose of ICU sedation.
180 The medical experts had no difficulty in interpreting the words “sole active agent”. In their Joint Report they agreed that “[s]ole active agent means only one agent being used”. This was confirmed in their concurrent evidence, where they agreed that “sole active agent” meant “a single agent … is being administered to calm th[e] patient”. Further, and consistently with this, Assoc Professor French said that, in his opinion, dexmedetomidine would be the “sole active agent” where it is the only agent administered to the patient for the purpose of ICU sedation. This aligned with Professor Hall’s evidence that he understood this limitation to mean that “in what is being infused, the only thing that could account for any sedative effect, whatever its quality, is dexmedetomidine”.
181 In their Joint Report, the medical experts also agreed that the words “essentially the sole active agent” mean “that the agent is being administered with another agent that has either sedative and/or analgesic properties”. Professor Bellomo’s evidence was that he understood “essentially sole active agent or sole active agent” to refer to a situation where dexmedetomidine was “essentially the only agent or the only agent administered to the patient for the purpose of achieving ICU sedation”.
182 In the course of concurrent evidence, Professor Bellomo said that whilst he agreed with Professor Hall that the expression “essentially the sole active agent” is “different and difficult because it is difficult to perceive a specific dose that would be limited in significance and impact on the sedation”. He added:
Nonetheless, I took the view that, in common conversation and lay conversation, essentially would mean that somewhere in the perception of the assessor, that about 90 per cent of the perceived sedation was thought to or believed to come from dexmedetomidine, and a little bit extra was thought to be coming from the morphine or the fentanyl. I took it to mean in a lay sense. I understand the difficulty in drawing the line, but I … assumed it to be indicative that a small amount of another drug which was doing just a little bit of assistance was the scenario [that was] referred to in the Patent.
Assoc Professor French agreed with this interpretation.
183 Professor Bellomo’s evidence was that the “line” to which Professor Hall referred was “very much dependent on the dose”:
… because even if … one mentioned a specific dose in milligrams, a specific dose would have a completely different effect in a 130-kilo man and an 82 year old 50-kilogram lady. … That’s why we said we had trouble with that argument. And that’s why we ended up describing a commonsensical understanding of that word, but not a specific or a medical understanding of that word … .
184 By way of example, Professor Bellomo explained that, in his view, dexmedetomidine would be “essentially the sole active agent” if administered to a patient for the purpose of achieving ICU sedation when administered at a maintenance dose of around 1-1.5 µg/kg/h in conjunction with a low dose of morphine, for example, at around 1-2 mg/hr. Professor Bellomo further explained that:
If you’re using two drugs like we use with morphine and midazolam, the way we used them in the 90s, very much equivalent contribution, I would not regard that as essentially the sole [active agent]. There was very much a partnership of equal. I would regard it as a sole [active agent] when one of the additional drugs would be seen to make a minimal contribution to the entire therapeutic endeavour.
185 Assoc Professor French gave evidence that, in his opinion, dexmedetomidine would be “essentially the sole active agent” when dexmedetomidine was administered as the dominant agent for the purpose of ICU sedation, where more than one drug was administered, for example, where a patient was given a low dose of morphine for pain relief along with dexmedetomidine for ICU sedation. He added that in that situation, in his view, dexmedetomidine was essentially the sole active sedative agent, acknowledging that morphine at a low dose has mild sedative effects.
186 This practical, common-sense construction, which I accept, provides a workable standard suitable to the intended use. InterPharma, supported by Professor Hall, said in effect that the use of the word “essentially” in the expression “essentially the sole active agent or the sole active agent” made it impossible to determine the boundary of the claim to monopoly. I reject this proposition. Plainly enough, as Professor Bellomo’s examples illustrate, what is “essentially” the sole active agent is a question of degree and, to that extent, a matter for judgment, but it does not seem to me to give rise to any ambiguity or other lack of clarity. Its meaning is clear, although in each case it will be a question of fact and degree as to whether dexmedetomidine is administered to a patient in an ICU as “essentially the sole active agent or the sole active agent” for the purpose of ICU sedation. Having regard to the evidence of Professor Bellomo and Assoc Professor French, understood in this practical, common sense way, it seems to me that a person skilled in the art would have little difficulty in practice in resolving the question, and in ascertaining whether what was proposed to be done fell within the ambit of the claims.
187 In this context, it may be observed that InterPharma included claim 15 in para 7B of its Further Further Amended Particulars of Invalidity, even though claim 15 included a different though verbally similar integer — “administering a pharmaceutical composition … [which] comprises an active agent and an inactive agent, wherein the active agent consists of dexmedetomidine”. In closing written submissions, InterPharma submitted that the inclusion of that integer required that dexmedetomidine be the only active agent of the pharmaceutical composition that is administered. For present purposes, this may be accepted. InterPharma also submitted that “[i]t may be noted that nothing in claim 15 precludes the administration of a further pharmaceutical composition with the claimed pharmaceutical composition. It is unclear precisely what InterPharma intended by this latter statement, and, in any event, the parties did not address it. To the extent that InterPharma challenged the validity of this claim on the basis of lack of clarity, that challenge should be rejected. It put forward nothing more to advance such a case.
188 It follows, for the reasons stated, that I reject InterPharma’s submission that there was a lack of clarity in claims 2 to 12, 14 to 25, 27 to 37 and 39 by virtue of the integer, “essentially the sole active agent or the sole active agent” (or the analogous integer in claim 15), such that the relevant claims were invalid.
Claims 2 to 12 and 38: lack of clarity as to how purported Swiss-style dependent claims are to be construed?
189 As indicated above, para 8 of InterPharma’s Fourth Further Amended Particulars of Invalidity alleged that claims 2 to 12 and 38 do not comply with the requirements of s 40(3) of the Patents Act in that the claims are not clear. In Particulars to para 8 InterPharma claimed:
(a) Claim 1 is a Swiss style claim, properly characterised as a method or process of making the claimed medicament.
(b) Claims 2 to 12 and 38 are ultimately dependent on claim 1 and claim “the use according to claim 1”.
(c) It is unclear which “use” in claim 1 is the intended “use” in claims 2 to 12 and 38.
(d) To the extent that the “use” is use of dexmedetomidine to make the medicament of claim 1 it is not clear to the skilled reader how this could be construed to include a method of administering the medicament of claim 1 to a patient as part of the process for making the medicament.
(e) To the extent that the “use” is the “for use” defining the purpose of the medicament in claim 1 it is not clear to the skilled reader how the method or process of manufacturing the medicament of claim 1 could be construed to include a method of administering the medicament.
190 In opening address at trial, senior counsel for InterPharma elaborated on this argument in the following terms:
Claim 2 can be read as a Swiss-claim in that there’s the making of the medicament and, in that medicament, [dexmedetomidine] is the sole – or essentially the sole active agent in that medicament. The difficulty is when one applies Pfizer’s construction … we have the Swiss-claim, which is a method for manufacture, but it has now turned into a method of administration addressed to the doctor.
…
… It could be construed as to the medicament and whether dexmedetomodine is in the medicament with something else, and that would be our construction on claim 2, but claim 3 doesn’t work, in that it is a direction to the manufacturer making the medicament, as well as the clinician who is administering it. … So we say that that claim is a bit unclear, because it’s a Swiss-claim tangled up with a method of administration addressed to two different people.
191 In closing written submissions, InterPharma reiterated its opening submissions, submitting that the balance of claims 4 to 12 and 38 suffered from the same default.
192 In opening address at the hearing, senior counsel for Pfizer responded that:
There seems to be some dispute between us as to how [the Swiss-style claims are] properly to be interpreted, in particular, from claims 2 onwards – or 3. InterPharma says that these are not Swiss-style claims because they are directed to a new method of administration, not use of dexmedetomidine in the manufacture of a medicament. We don’t see that … . For our purposes, first of all, we have the use in claim 1 of dexmedetomidine in the manufacture of a medicament for use in intensive care sedation. Then you have the use according to claim 1, wherein the [dexmedetomidine] or the pharmaceutically acceptable salt is essentially a sole active agent. Well … we say that’s talking about the method there.
Then the use according to 1 or 2, wherein the dexmedetomidine or pharmaceutical salt is administered in an amount to achieve a plasma concentration – we will say that’s to the method of treatment. It would be odd to try and say that that should somehow relate to the use of dexmedetomidine to make the product and it’s not the right use.
193 Senior counsel for Pfizer subsequently added that:
Ultimately, a Swiss claim is said to gain its novelty from the method of treatment and so here, when you look at the dependent claims, it’s not unusual, we would say, to ask yourself, “Well, what is that as to the method of treatment?” So, here, the first thing is – the method of treatment is sedation in the ICU. The next thing is that a method involves essentially a sole – dexmedetomidine – essentially, a sole or sole active agent. Then to achieve plasma concentrations, then – it’s all looking to the method of treatment, those additional claims, and we don’t … see why – if all of those just appeared in the first claim, why that wouldn’t still be a Swiss claim.
Pfizer reiterated this position in closing submissions.
194 I accept that, as Pfizer submitted, each of claims 2 to 12 is a dependent “Swiss-style” claim, ultimately dependent on claim 1. The dispute between the parties arose from the fact that, since claim 1 is a Swiss-style claim, it mentions of two “uses”. When claim 2 refers to the “use according to claim 1”, one might therefore ask, to which use is the claim referring. This is only a matter of semantics, however. When considered fairly, there is no real lack of clarity here.
195 The specification identifies that “an ideal sedative agent should be used by itself in ICU sedation to avoid the dangers of polypharmacy”. As senior counsel for Pfizer put it in closing submissions:
No doubt the problem arises because it’s a Swiss claim because there’s two uses identified. Your Honour then has to construe which use … we are talking about in a sensible way. And we say … to do that, you read the patent. What is the patent worried about? The patent is not worried about having two sedatives in the one medicament. That’s not what it describes as the problem. It describes the problem as people being treated with two sedatives.
196 In this context, having regard to claim 2 and each of the following dependent claims, it is clear that the relevant use is what is said to be the hitherto undiscovered therapeutic use. This is consistent with the fact that, because claim 1 is a Swiss-style claim, novelty will arise from the claimed new therapeutic use, and not from the manufacture of the medicament. It is clear that each of claims 2 to 12 further qualifies what is said to be the hitherto undiscovered therapeutic use of the medicament.
197 Senior counsel for Pfizer submitted in closing that:
It’s … said that [in] claim 2 the relevant use, if I can put it that way, wherein the dexmedetomidine or pharmaceutical salt thereof as essentially sole active agent or sole active agent, means that it’s in the making of the medicament rather than in the use for intensive care unit sedation. And we say, your Honour, when one reads not only that claim but each of the following claims, it’s plain that the limitation in each of the claims, that is, limiting claim 1, is to how the drug is administered, that is, the method of treatment, not the making of the drug itself. And that’s what is intended by these claims.
198 I accept this submission. It is equally clear, so it seems to me, that the subsequent dependent Swiss-style claims should be read in the same way. For example, I accept that, as Pfizer submitted, claim 5 is to be read as a claim for “use of dexmedetomidine in the manufacture of a medicament for use in ICU sedation”, “wherein a loading dose and a maintenance dose of dexmedetomidine are administered”. InterPharma’s submission that the “use according to claim 1” relates to the use of dexmedetomidine to make the product is contrived.
199 It follows, for the reasons stated, that I reject InterPharma’s submission that there was a lack of clarity in claims 2 to 12 and 38 by virtue of being dependent on the Swiss-style claim in claim 1, such that the relevant claims were invalid.
InterPharma’s other allegations as to lack of clarity
200 As noted earlier, InterPharma originally alleged that there was a lack of clarity in claims 23, 24 and 25. As discussed above, leave is granted to delete the relevant allegation from InterPharma’s Fourth Further Amended Particulars of Invalidity. Since the allegation is no longer pressed, it is unnecessary to consider it.
201 It should also be noted that in its written opening submissions, InterPharma submitted that claims 3, 16 and 28 (plasma concentration claims) and dependent claims 4 to 12, 17 to 25 and 29 to 36 were invalid because the words “administered in an amount to achieve a plasma concentration of 0.1-2 ng/ml” lacked clarity. InterPharma submitted:
This is because it is not clear from this expression whether it refers to an administration of dexmedetomidine in order to achieve the claimed range of plasma concentration or an administration of dexmedetomidine which inevitably achieves that range.
202 At the time these submissions were filed, however, InterPharma’s Further Further Amended Particulars of Invalidity did not include an allegation of lack of clarity on this basis.
203 InterPharma sought to add a lack of clarity allegation by inserting para 9A into its Third Further Amended Particulars of Invalidity filed on 11 May 2018 (before the parties made their written and oral closing submissions to the Court). Proposed para 9A alleged that there was a lack of clarity in claims 3 to 12, 16 to 25 and 28 to 37 on the basis that each of those claims requires that dexmedetomidine be “administered in an amount to achieve a plasma concentration of 0.1-2 ng/ml”, and it is not clear to the skilled reader:
(b) … whether this expression requires that the administration of dexmedetomidine be directed to achieving a plasma concentration in the range claimed or whether the claim requires that dexmedetomidine be administered in any amount which achieves a plasma concentration in the range claimed[; and]
(c) … when the plasma concentration is required to be achieved.
204 In written closing submissions, InterPharma made submissions in relation to the “construction” of the plasma concentration claims (see [238]-[241] below), and then stated that “[i]f InterPharma’s construction is not accepted then it is submitted that the words [of the plasma concentration claims] are inherently ambiguous and capable of bearing multiple meanings, thus do not have a sufficiently clear meaning to define a patent monopoly” (emphasis added).
205 In written closing submissions, Pfizer opposed the inclusion of proposed para 9A on the basis that it was a new argument that had not been run at trial and that Pfizer would have led evidence or cross-examined differently had the point been taken earlier.
206 Subsequently, during oral closing submissions on 21 May 2018, shortly after Pfizer took issue with the form of InterPharma’s Third Further Amended Particulars of Invalidity filed on 11 May 2018, senior counsel for InterPharma, Ms Rofe QC, confirmed that InterPharma’s point with respect to the plasma concentration claims was not one of lack of clarity, but one of construction relevant to its answer to Pfizer’s infringement case. This is apparent from the following exchange between the Court and senior counsel:
THE COURT: It’s not a lack of clarity complaint though, is it? It’s a clear enough integer. It’s a clear enough – it’s saying something which is measurable. It can be measured and everyone knows it can.
MS ROFE: Yes.
THE COURT: So what’s the problem with it?
MS ROFE: Well, it’s probably more of the infringement that it’s not done in order to achieve. Well, we say it has a meaning and it is to achieve a plasma concentration. The person administering it is administering it in order to achieve that plasma concentration and we say … that it’s not.
…
THE COURT: If I were not to accept that, is there any other difficulty with the integer?
MS ROFE: No. I think, your Honour, it is the direction to achieve the plasma concentration, which, on the evidence, is not what is done. ...
THE COURT: … I just want to understand it a little bit better. Is that a clarity point or is that something else?
MS ROFE: Your Honour, I think it’s more just a construction point relevant to infringement, although our friends are saying that it has a different construction, which we say is not clear.
(Emphasis added)
207 Consistently with that exchange, when, on 24 May 2018, InterPharma subsequently filed another document, also entitled “Third Further Amended Particulars of Invalidity” to reflect various amendments discussed during closing submissions, the previously proposed para 9A had been removed. Moreover, InterPharma’s Fourth Further Amended Particulars of Invalidity filed on 25 May 2018 did not seek leave to include para 9A or any other allegation of lack of clarity in respect of the plasma concentration claims.
208 The outcome of the discussion between the Court and senior counsel for InterPharma in the course of InterPharma’s closing submissions identified that InterPharma’s point with respect to this integer was not one of lack of clarity but one in answer to Pfizer’s infringement case. Consistently with this, InterPharma no longer included this lack of clarity allegation in the Particulars of Invalidity that it has since filed or in relation to which it seeks leave to amend. It must be inferred from this that InterPharma no longer seeks to allege lack of clarity specifically in respect of the plasma concentration claims. In any event, it seems to me that this would in substance have been a new clarity point. It is therefore unnecessary to address either this particular lack of clarity allegation or Pfizer’s opposition to raising it. Further, as will be seen, there is, in my opinion, no lack of clarity associated with the plasma concentration claims on the construction advanced by Pfizer, and which I accept.
Infringement
209 It was common ground at trial that InterPharma intended, unless enjoined, to supply the Generic Products in Australia with a product information (PI) document containing an indication for “ICU sedation”, that is, “[f]or sedation of initially intubated patients during treatment in an intensive care setting”. When the trial began on 3 May 2018, Pfizer alleged that InterPharma’s proposed conduct in respect of the Generic Products constituted a threatened infringement of each of the claims of the Patent. InterPharma denied any threatened infringement, primarily on the basis that all the claims of the Patent were invalid. For the reasons set out below, I have rejected InterPharma’s submissions with respect to invalidity.
210 Broadly speaking, as set out below, Pfizer made out its case of threatened infringement.
InterPharma’s Infringement Analysis
211 Before considering the substantive issues relevant to infringement, it is necessary to deal with an issue raised by Pfizer after the hearing in relation to a document entitled “InterPharma’s Infringement Analysis Australian Patent No. 754484” (Infringement Analysis). The Infringement Analysis was presented to the Court by senior counsel for InterPharma during closing submissions as a summary of InterPharma’s position. At its request, Pfizer was granted leave to file short submissions after the hearing concerning the Infringement Analysis. Pfizer filed written submissions dated 28 May 2018 contending, relevantly, that the Infringement Analysis “is replete with bare assertions unsupported by evidence”. Pfizer did not expressly submit, however, that the Court should disregard the Infringement Analysis.
212 InterPharma submitted, and I accept, that the Infringement Analysis was a means to direct the Court to relevant parts of InterPharma’s written closing submissions and summarised InterPharma’s position with respect to each of the claims. I accept that, as InterPharma submitted, the document was not intended to have any greater significance than InterPharma’s submissions. I have had regard to InterPharma’s Infringement Analysis on this basis only, subject to one matter.
213 It seems to me that the Infringement Analysis incorrectly stated InterPharma’s position in relation to claim 27. In written closing submissions, InterPharma admitted infringement of claim 27 (along with claims 2 and 14), if the Court took the view that the claims were sufficiently clear and were “construed to allow the co-administration of dexmedetomidine with an agent administered for analgesia (albeit it also has a sedative effect) rather than for sedation”. This is not reflected in the Infringement Analysis, which states instead that InterPharma denies infringement. There is an admission of infringement in relation to claim 28, however, which was not part of InterPharma’s closing written submissions. It appears to me, therefore, that there has been a typographical mistake in the Infringement Analysis, with the result that the relevant admission mistakenly appears in relation to claim 28, instead of claim 27. In any event, I have read the Infringement Analysis in this way, which is consistent with InterPharma’s closing written submissions and the way it ran its case at trial.
General principles and relevant matters
214 An infringement proceeding is designed to enforce the exclusive rights granted to the patentee under s 13 of the Patents Act to exploit an invention, as that term is defined in Sch 1, for the term of the patent. Pfizer instituted its cross-claim alleging threatened infringement as part of its response to InterPharma’s Patent invalidity claims. (With respect to any permanent injunctive relief that the Court might have granted, InterPharma initially sought to “carve out” the ICU sedation indication so that the Generic Products could continue to be supplied with the procedural sedation indication. It did not ultimately press this contention.)
215 The term of the Patent ended on 31 March 2019, while judgment was reserved. For this reason, the interlocutory injunction, which was the subject of Order 1 of orders made on 18 December 2017, restraining InterPharma from marketing, selling and supplying the Generic Products for use in ICU sedation, also came to an end on that date. The infringement proceeding, even if successful, would no longer support the grant of a permanent injunction. The outcome of that proceeding is, however, relevant to other issues, including costs. It therefore remains necessary to decide whether by its proposed conduct InterPharma threatened to infringe Pfizer’s rights under the Patent prior to its expiry.
216 There is no dispute about the basic test for infringement. A patent is infringed if, on a proper construction of the claim, a product possesses all the integers of that claim, without the patentee’s authorisation: see recently Davies v Lazer Safe Pty Ltd [2018] FCA 702; 132 IPR 202 at [240] (appeal dismissed: Davies v Lazer Safe Pty Ltd [2019] FCAFC 65), citing Rodi & Wienenberger AG v Henry Showell Ltd [1969] RPC 367 at 369 (Lord Upjohn); Olin Corporation v Super Cartridge Co Pty Ltd (1977) 180 CLR 236 at 246 (Gibbs J); Fresenius Medical Care Australia Pty Limited v Gambro Pty Limited [2005] FCAFC 220; 67 IPR 230; 224 ALR 168 at [49] (Wilcox, Branson and Bennett JJ).
217 As already indicated, claim 1 is a Swiss-style claim, claims 2 to 12 are dependent Swiss-style claims and claim 38 is an omnibus claim to claim 1. The parties accepted that Swiss-style claims may only be infringed directly and cannot be infringed indirectly by operation of s 117 of the Patents Act: see Otsuka Pharmaceutical v Generic Health (No 4) at [168] (Yates J). Accordingly, these claims may only be infringed directly, including by the importation, supply and offer to supply in Australia of the medicament resulting from the claimed manufacture overseas: Warner-Lambert Co LLC v Apotex Pty Ltd (No 2) [2018] FCAFC 26; 129 IPR 205 at [156]-[168] (Jagot, Yates and Burley JJ). As Yates J also said in Otsuka Pharmaceutical v Generic Health (No 4) at [169] and [172]:
This is not to say that the therapeutic use defined in [a claim] can be ignored when considering whether that claim has been, or is threatened to be, infringed. The therapeutic use defined in the claim qualifies, and thus confines, the scope of the monopoly that is claimed. …
For the purpose of determining infringement of a Swiss type claim … [t]he question is whether, objectively ascertained, the medicament that results from the claimed method or process is one that has the therapeutic use defined in the claim. The question is not really about how the alleged infringer markets its product, although, plainly, its conduct in that regard may well assist in determining, objectively, whether the accused product has the claimed therapeutic use.
218 In Mylan Health Pty Ltd v Sun Pharma ANZ Pty Ltd [2019] FCA 28; 138 IPR 402 (Mylan), Nicholas J gave further consideration to the infringement of Swiss-style claims, finding that infringement of the Swiss-style claims in the Patent in suit in that case was not established because the manufacturer’s intention in making the medicament was not established: see Mylan at [92]-[106]. His Honour held at [102]:
The crucial question is whether the manufacturer has made (or will make) the relevant medicament with the intention that it be used in the treatment of the designated condition. In my view, the answer to that question is to be ascertained objectively in light of all the relevant facts and circumstances including, in particular, the PI for the product, any product labelling, and the nature, size and other pertinent characteristics of the market into which the product is to be sold.
219 While judgment was reserved, the parties drew my attention to Mylan and his Honour’s discussion about novelty. No party relied on his Honour’s statements with respect to infringement of a Swiss-style claim. The reason for this is clear enough: in this case his Honour’s analysis, especially in [102], would have made no difference. This is because, in this case, the product information for the Generic Products unequivocally indicated a use “for sedation” of patients in “an intensive care setting”. The relevant product information is discussed further below.
220 Claims 13 to 37 and 39 (the second omnibus claim) are method claims. Pfizer’s case regarding infringement of the method claims was based on s 117 of the Patents Act. That section sets out the conditions under which a supply of a product will constitute an infringement of an indirect kind. Section 117 relevantly provided:
117 Infringement by supply of products
(1) If the use of a product by a person would infringe a patent, the supply of that product by one person to another is an infringement of the patent by the supplier unless the supplier is the patentee or licensee of the patent.
(2) A reference in subsection (1) to the use of a product by a person is a reference to:
…
(b) if the product is not a staple commercial product—any use of the product, if the supplier had reason to believe that the person would put it to that use; or
(c) in any case—the use of the product in accordance with any instructions for the use of the product, or any inducement to use the product, given to the person by the supplier or contained in an advertisement published by or with the authority of the supplier.
There was no dispute between the parties that the Generic Products are not “staple commercial products” for the purposes of s 117(2)(b) of the Patents Act.
221 Method claims may be infringed indirectly pursuant to s 117(1) by the importation, supply and offer to supply in Australia of products the use of which infringes a claim of the Patent. For Pfizer to establish indirect infringement of the method claims under s 117(1) of the Patents Act, it needed to show that InterPharma:
had “reason to believe” that a person to whom the Generic Products were supplied “would” put the Generic Products to use in accordance with a relevant method claim: see s 117(2)(b); or
provided instructions or inducements to use the Generic Products in accordance with a relevant method claim: see 117(2)(c).
222 Since 13 July 2017, InterPharma has been recorded as the sponsor of a listing on the ARTG for the Generic Products. InterPharma has a product information document approved by the TGA (Generic Products PI). The provision of product information is regulated by the Therapeutic Goods Act 1989 (Cth) (Therapeutic Goods Act).
223 As at 20 June 2017, s 23(2)(ba) of the Therapeutic Goods Act provided that an application for registration or listing of therapeutic goods on the ARTG, in the case of a restricted medicine (such as dexmedetomidine), must be accompanied by product information in the form approved under s 7D. Section 3(1) of the Therapeutic Goods Act specified that product information, in relation to therapeutic goods, “means information relating to the safe and effective use of the goods, including information regarding the usefulness and limitations of the goods”. (Section 23 has been amended subsequently, but none of these amendments concern the present discussion.)
224 Also in evidence in the proceedings were the Australian Regulatory Guidelines for Prescription Medicines, Guidance 8: Product Information, published by the TGA. They provided:
The PI is a document associated with a medicine that contains technical information relating to its safe and effective use, including:
• the characteristics of the active ingredient
• indications, contraindications and precautions
• adverse reactions that may occur
• relevant clinical trials
• dosage and storage.
The PI:
• includes scientific and technical information about the risks and benefits of the medicine to support its use for the indications for which it is approved.
• informs medical practitioners, pharmacists and other health professionals about the safe and appropriate prescribing and dispensing of the medicine.
• assists healthcare professionals to provide patient education about the medicine to support high-quality and safe clinical care.
225 The Generic Products PI refers to two “indications” for the Generic Products as follows:
INDICATIONS
ICU Sedation
For sedation of initially intubated patients during treatment in an intensive care setting. The use of DEXMEDETOMIDINE EVER PHARMA by continuous infusion in these patients should not exceed 24 hours.
Procedural Sedation
For sedation of non-intubated patients prior to and/or during surgical and other procedures.
226 Under the heading, “Dosage and administration”, the Generic Products PI further relevantly states:
NOTE: Dexmedetomidine should be administered only by persons skilled in anaesthetics or in the management of patients in the intensive care setting. Due to the known pharmacological effects, patients should be continuously monitored.
Clinically significant events of bradycardia and sinus arrest have been associated with dexmedetomidine hydrochloride administration in young, healthy volunteers with high vagal tone or with different routes of administration including rapid intravenous or bolus administration of dexmedetomidine hydrochloride.
Adults: Dexmedetomidine should be individualised and titrated to the desired clinical effect.
ICU Sedation
Initiation
For adult patients, DEXMEDETOMIDINE EVER PHARMA is generally initiated with a loading infusion of 1 (one) microgram/kg over 10 to 20 minutes, if needed. The use of DEXMEDETOMIDINE EVER PHARMA by continuous infusion in these patients should not exceed 24 hours.
For patients being converted from alternate sedative therapy a loading dose may not be required.
Maintenance of ICU sedation
Adult patients will generally require a maintenance infusion of 0.2 to 1 microgram/kg/h*. The rate of the maintenance infusion should be adjusted to achieve the desired level of sedation. As a guide, it is recommended that 0.4 microgram/kg/h should be the initial maintenance infusion. If after approximately 5 minutes sedation is not adequate, the rate of infusion can be increased in increments of 0.1 microgram/kg/h or higher. Dosages as low as 0.05 microgram/kg/h have been used in clinical studies. Patients receiving dexmedetomidine hydrochloride have been observed to be rousable and alert when stimulated. This is an expected component of dexmedetomidine sedation and should not be considered a lack of efficacy in the absence of other clinical signs and symptoms.
A dose reduction for both the loading and maintenance infusions should be considered in patients with impaired hepatic function and in patients over 65 years of age.
Dexmedetomidine hydrochloride has been continuously infused in mechanically ventilated patients prior to extubation, during extubation, and post-extubation. It is not necessary to discontinue DEXMEDETOMIDINE EVER PHARMA prior to extubation.
227 The Therapeutic Goods Act and the marketing approval granted for the Generic Products, together with the Generic Products PI, were relevant to the infringement claims. In its closing written submissions, InterPharma accepted that the Generic Products PI supplied with the Generic Products relevantly instructs, with respect to the ICU sedation indication, that:
(a) dexmedetomidine should only be administered by persons skilled in anaesthetics or in the management of patients in an intensive care setting and, due to the known pharmacological effects, patients should be continuously monitored;
(b) for adults, dexmedetomidine should be individualised and titrated to the desired clinical effect;
(c) for adult patients, dexmedetomidine “is generally initiated with a loading infusion of 1 (one) microgram/kg over 10 to 20 minutes, if needed”;
(d) the use of dexmedetomidine by continuous infusion in these patients should not exceed 24 hours; and
(e) a maintenance dose is generally required for ICU sedation.
228 InterPharma disputed, however, that the Generic Products PI “contains instructions for the use of the [Generic Products], to assist healthcare professionals in prescribing, dispensing and administering” the Generic Products. I accept that, as Pfizer contended, this assertion “is wholly without merit” in the context of the regulatory regime established by the Therapeutic Goods Act.
229 It may be accepted that specific indications in a PI for a pharmaceutical product listed on the ARTG constitute instructions to do what is indicated: see Apotex v Sanofi-Aventis HCA at [303] (Crennan and Kiefel JJ). Citing Apotex v Sanofi-Aventis HCA at [303], Pfizer submitted that the Generic Products PI was an “emphatic instruction” to recipients of Generic Products to use the Generic Products for ICU sedation in accordance with the dosage and administration stated in that PI and that s 117(2)(c) of the Patents Act was thereby engaged. I accept that submission. I further accept that, as Pfizer submitted, it can reasonably be inferred that InterPharma had “reason to believe” that a person to whom Generic Products were supplied would use the Generic Products in that manner, thereby engaging s 117(2)(b) of the Patents Act. It may, however, also be accepted that, as InterPharma submitted, whether other information in a PI constitutes a relevant instruction or inducement depends on a close consideration of the PI.
InterPharma’s admitted infringement of claims
230 At trial Pfizer submitted that InterPharma’s proposed conduct in respect of its Generic Products would infringe each of the claims of the Patent. InterPharma accepted that, to the extent that the claims were valid, its proposed conduct would infringe claims 1, 13 and 26 (the independent claims), and claims 2, 14 and 27 (the sole active agent claims) if the claims were construed as I have done. It also conceded that its conduct in respect of its Generic Products would infringe claim 15 provided the claim was valid and did not preclude administration of the Generic Products with another pharmaceutical composition.
231 In other words, it was not in dispute that, if claims 1, 13 and 26 were valid (and I have found they were: see below):
• any supply of the Generic Products by InterPharma prior to the Patent expiry date would have directly infringed claim 1, because each of the Generic Products “is a product resulting from the use of a pharmaceutically acceptable salt of dexmedetomidine in the manufacture of a medicament for use in intensive care sedation”: see Further Amended Defence to Cross-Claim filed 11 May 2018; Amended Defence to Cross-Claim filed 5 February 2018; Defence to Cross-Claim filed 20 September 2017; see also InterPharma’s closing written submissions at [134](a); and
• any supply of the Generic Products prior to the Patent expiry date would have indirectly infringed claims 13 and 26 by reason of s 117(1), together with s 117(2)(b) or s 117(2)(c) of the Patents Act, because the Generic Products PI instructs, and InterPharma has reason to believe, that a person or persons to whom it would supply the Generic Products would use the Generic Products in a method that falls within claims 13 and 26: see Further Amended Defence to Cross-Claim filed 11 May 2018; Amended Defence to Cross-Claim filed 5 February 2018; Defence to Cross-Claim filed 20 September 2017; see also InterPharma’s closing written submissions at [134](b).
232 As stated above, I have rejected InterPharma’s clarity challenge to the validity of claim 15 and, as appears below, I have rejected InterPharma’s other validity challenges. I can discern no relevant distinction as regards infringement of claim 15 and infringement of claims 13 and 26, notwithstanding the qualified nature of InterPharma’s concession with respect to claim 15.
233 As will be seen, InterPharma denied any further threatened infringement (even if the claims were valid) on the basis of its arguments about claim construction.
234 I also note at this point that, after the trial had concluded and while judgment was reserved, InterPharma applied (on 6 August 2018) for the proceeding to be re-opened on the basis that Pfizer had amended its Precedex® PI prior to trial in a manner which, so InterPharma said, was material to Pfizer’s infringement case at trial. Pfizer consented to InterPharma’s application to re-open in written submissions filed on 1 November 2018. The resumed hearing took place on 15 November 2018. The evidence and submissions referable to the resumed hearing are discussed below in relation to the parties’ submissions about the infringement of the loading dose claims.
Claims 2, 14 and 27 (essentially the sole active agent or the sole active agent)
235 InterPharma accepted in written closing submissions that, if claims 2, 14 and 27 were “sufficiently clear and are construed to allow the co-administration of dexmedetomidine with an agent administered for analgesia (albeit it also has a sedative effect) rather than for sedation”, then the supply of the Generic Products would directly infringe claim 2 (on Pfizer’s construction) and indirectly infringe claims 14 and 27. The reference to “Pfizer’s construction” of claim 2 above is a reference to Pfizer’s contention (which I have accepted) that claim 2 is properly characterised as a dependent Swiss-style claim. As stated above, I have rejected InterPharma’s clarity challenge to claims 2, 14 and 27. Further, I accepted that, on their proper construction, these claims would be satisfied where dexmedetomidine was administered to a patient for the purpose of achieving ICU sedation (at a dose to achieve that purpose) with another agent administered for a purpose other than sedation (such as analgesia) albeit there might be some sedative effect. It therefore follows from InterPharma’s admission regarding the infringement of claims 2, 14 and 27 that:
claim 2 would have been directly infringed by the exploitation of the Generic Products in Australia prior to the Patent expiry date; and
prior to the Patent expiry date, claims 14 and 27 would have been indirectly infringed by the supply of the Generic Products, by reason of s 117(1), together with s 117(2)(b) or s 117(2)(c) of the Patents Act.
Claims 3, 16 and 28 (plasma concentration)
236 Amongst other things, claims 3, 16 and 28 require that dexmedetomidine is “administered in an amount to achieve a plasma concentration of 0.1-2 ng/ml”.
237 InterPharma denied that the supply of the Generic Products would infringe claims 3, 16 and 28 on the basis that, in practice, the Generic Products would not be “administered in an amount to achieve” any particular plasma concentration as required by these claims. This proposition depended on InterPharma’s construction of these claims.
238 InterPharma contended that the plain meaning of the plasma concentration claims is that an amount of dexmedetomidine is administered to achieve (ie to target) a specific plasma concentration within the claimed range that the clinician anticipates will be suitable given the desired level of sedation and the general condition of the patient. InterPharma argued that this construction was supported by the specification of the Patent, which relevantly stated:
The dose range of dexmedetomidine can be described as target plasma concentrations. The plasma concentration range anticipated to provide sedation in the patient population in the ICU varies between 0.1-2 ng/ml depending on the desired level of sedation and the general condition of the patient. …
239 InterPharma submitted that to construe the plasma concentration claims as covering the administration of dexmedetomidine in any dose that is likely to achieve a plasma concentration anywhere in the claimed range would denude the words “to achieve” and the concept of a “target” of any meaning and would give the claims no substantial work to do. InterPharma relied on the evidence of Professor Hall that he and other clinicians “do not titrate the dose of dexmedetomidine … to achieve any particular serum level (i.e. plasma concentration)”, but instead “to achieve the … desired level of sedation”. InterPharma also referred to the evidence of Assoc Professor French and Professor Bellomo, who said that they did not routinely measure the plasma concentration in their patients and there was no standard laboratory test to do so. Professor Bellomo added that the reason for this was because clinicians titrate the dose of dexmedetomidine “to the patient’s condition … not to a particular blood level”. The evidence of Assoc Professor French and Professor Bellomo was that, even if they were told to target a certain plasma level, they would not know what infusion rate they would need to use, and would need to further investigate and test each patient. In this regard, Professor Bellomo observed that:
You would have to measure literally with high-performance liquid chromatography because there is so much variability in the disposal of the drug in the critically ill that even if you had information in normal human volunteers that if you gave a particular dose, you will get a particular level, you could not draw robust inferences about what would happen in a critically ill patient.
… You would have to measure it, which is not what we do.
240 Professor Bellomo accepted that a patient’s plasma level would be affected by their metabolism and that would differ depending on the health of the patient, including “[o]rgan dysfunction, kidney dysfunction, liver dysfunction, the severity of the illness, … other drugs that might interfere with the breakdown and metabolism and the volume of distribution of a given drug”. In closing senior counsel for InterPharma submitted that his evidence in this regard indicated that, because these factors affected the plasma concentration, there was “no correlation between the dose and the plasma concentration”. I reject this characterisation of Professor Bellomo’s evidence. As will be seen, Professor Bellomo accepted that, based on the information in the Patent, it was “highly likely” the administration of the Generic Products in the amount identified in the Generic Products PI would achieve the target plasma concentration. I would infer from this that he in fact accepted that there is a correlation between dose and plasma concentration.
241 InterPharma further submitted that it followed from the experts’ evidence that, while a dose that achieves a sedative effect in a patient (other than in a patient at the extremes) might ordinarily be expected to fall within what is the extremely wide plasma concentration range set out in the Patent, those patients would not have been administered dexmedetomidine by a clinician “in an amount to achieve” a plasma level within that range, but instead to achieve a desired level of sedation. I reject this construction of the claims for the reasons I am about to state.
242 Pfizer submitted that the plasma concentration claims relate to the dose of dexmedetomidine administered to a patient that achieves a plasma concentration within the specified range (that being the range anticipated to provide sedation in the patient population in the ICU). I accept that this is the proper construction of the plasma concentration claims.
243 This construction is, in my view, supported by both the specification of the Patent and the structure of the claims. The specification states that the dose range of dexmedetomidine “can be described as target plasma concentrations”. The specification goes on to state that “[t]hese plasma concentrations can be achieved by intravenous administration by using a bolus dose and continuing it by a steady maintenance infusion”. It also identifies the dose range (by reference to the loading dose and the maintenance dose) to achieve the plasma concentration range anticipated to provide sedation in the patient population in the ICU of between 0.1 and 2 ng/ml depending on the desired level of sedation.
244 The link between dose and plasma concentration was also reflected in the structure of the claims of the Patent. Most particularly, while claim 3 is to the administration of dexmedetomidine “in an amount to achieve a plasma concentration of 0.1-2 ng/ml”, the subsidiary claims limit the plasma concentration by reference to specific dose ranges and regimes for administering dexmedetomidine. Furthermore, the experts also agreed in the Joint Report that the expression “administered in an amount to achieve a plasma concentration of 0.1-2 ng/ml” in the context of claim 3 of the Patent meant “the amount of drug administered to a patient to achieve a plasma concentration of 0.1-2 ng/ml” (emphasis added).
245 Senior counsel for Pfizer submitted, and I accept, that a common-sense construction of the plasma concentration claims requires having regard to the fact that the claims relate to the treatment of patients by clinicians; and they are not concerned with clinical scientific research conducted in a laboratory environment. Understood in this context, it is not to the point that the skilled addressee would not, in practice, measure the plasma concentration levels of their patients nor administer dexmedetomidine with the object of achieving a particular plasma concentration. The plasma concentration claims are not claims to administration of dexmedetomidine “for the purpose of” or with the “objective” of achieving the claimed target plasma concentration. Rather, the claims relate to the amount (ie the dosage range) of dexmedetomidine administered to a patient that achieves the target plasma concentrations.
246 As discussed at [204]-[207], InterPharma indicated in passing in closing written and oral submissions that if its construction of the plasma concentration claims was not accepted, then it would allege that the claims lacked clarity. For the reasons already stated, however, InterPharma did not particularise its case in this way and it must be inferred that it did not ultimately seek to allege lack of clarity in respect of the plasma concentration claims. This is reflected in InterPharma’s Infringement Analysis, which does not refer to any lack of clarity allegation in relation to these claims (unlike other claims). In any event, I accept that the plasma concentration claims, as construed above, are sufficiently clear. Amongst other things, it is evident that the experts were able to identify the amount of dexmedetomidine administered to a patient to achieve a plasma concentration in the specified range in the context of the Generic Products PI.
247 Based on its construction of the plasma concentration claims (which I have rejected), InterPharma contended that although its Generic Products PI contains general dosage and administration instructions and information about pharmacokinetic parameters, this information does not constitute instructions to administer the Generic Products, nor did InterPharma have reason to believe the Generic Products would be administered, “in an amount to achieve a plasma concentration” within the claimed range. Accordingly, InterPharma denied that its proposed conduct would infringe the plasma concentration claims.
248 Since I do not accept InterPharma’s construction of the plasma concentration claims, this defence to Pfizer’s infringement claim falls away. The critical question, as senior counsel for Pfizer submitted, is whether or not the relevant dosage range in the Generic Products PI in fact achieves the plasma concentrations identified in the claims. The evidence indicated that it did. Professor Bellomo gave evidence that he examined the Generic Products PI and observed that, although “some variations are noted for particular circumstances”, the general dosage and administration instructions in the Generic Products PI were as follows:
(a) loading infusion of 1 µg/kg over 10 to 20 minutes; and
(b) maintenance infusion of 0.2 to 1 µg/kg/h.
His evidence was that, based on the information in the Patent, he considered it was highly likely that the administration of the Generic Products in the amount specified in the Generic Products PI would achieve a plasma concentration of 0.1-2 ng/mL in patients in the ICU. Allowing for patients “at the extremes and not represented by previously published pharmacokinetic data”, Professor Hall gave evidence substantially to the same effect. Professors Bellomo and Hall agreed that the Generic Products PI instructs clinicians to administer the Generic Products in an amount within the dosage range that would achieve a plasma concentration of 0.1-2 ng/ml in most cases. I would therefore conclude that the Generic Products PI instructs clinicians to administer the Generic Products in an amount within the dosage range that would achieve a plasma concentration of 0.1-2 ng/ml in most cases. There was no evidence to the contrary. I would infer from the experts’ evidence that InterPharma had reason to believe that the Generic Products would be used in this way.
249 For these reasons, I find that claim 3 would have been directly infringed by the exploitation of the Generic Products in Australia prior to the Patent expiry date; and that prior to the Patent expiry date, claims 16 and 28 would have been indirectly infringed by the supply of the Generic Products, by reason of s 117(1), together with s 117(2)(b) or s 117(2)(c) of the Patents Act.
Claims 4, 17 and 29 (intravenous administration)
250 Claims 4, 17 and 29 are each claims relating to the intravenous administration of dexmedetomidine. There is no dispute that the Generic Products PI instructs, and InterPharma has reason to believe, that the Generic Products are to be administered to patients intravenously. InterPharma submitted, however, that its proposed conduct would not infringe claims 4, 17 and 29 because each of these claims depend on one of the plasma concentration claims. For the reasons set out above, I have rejected InterPharma’s argument as to the plasma concentration claims. Accordingly, I find that claim 4 would have been directly infringed by the exploitation of the Generic Products in Australia prior to the Patent expiry date; and that prior to the Patent expiry date, claims 17 and 29 would have been indirectly infringed by the supply of the Generic Products, by reason of s 117(1), together with s 117(2)(b) or s 117(2)(c) of the Patents Act.
Claims 5 to 12, 18 to 25 and 30 to 37 (loading dose and maintenance dose)
251 Claims 5 to 12, 18 to 25 and 30 to 37 each relate to use for ICU sedation wherein a loading dose and maintenance dose of dexmedetomidine are administered. The focus of the dispute between the parties with respect to these claims was on the loading dose integer. This was also the focus of the re-opened trial on 15 November 2018. (The experts accepted that a reference in the evidence to “rapid intravenous or bolus administration” was a reference to a loading dose. Professor Bellomo explained that a maintenance infusion or dose is “a steady administration of the drug over a long period of time in order to maintain a certain effect”.)
252 There was no dispute that, except for the issues relating to the loading dose integer, claims 5 to 12 of the Patent would have been infringed directly by the exploitation of the Generic Products in Australia prior to the Patent expiry date; and claims 18 to 25 and 30 to 37 would have been indirectly infringed by the supply of the Generic Products by reason of s 117(1), together with s 117(2)(b) or s 117(2)(c) of the Patents Act.
253 Further to this conclusion, a matter relating to the maintenance dose integer should be noted. The Generic Products PI stated that adult patients “will generally require” a maintenance infusion (or dose) of 0.2-1 µg/kg/hr and recommended the initial maintenance infusion of 0.4 μg/kg/hr, with increments of 0.1 μg/kg/hr if sedation is not adequate at that dose. Professor Bellomo’s evidence was (and I accept) that the Generic Products PI instructs the administration of a maintenance dose within the range claimed in claims 10 to 12, 23 to 25 and 35 to 37. This was not contradicted by any other expert evidence.
The focus of the case as re-opened and heard on 15 November 2018
254 Leave was granted, by consent, to re-open the trial in this proceeding for consideration of two matters, namely:
(a) Whether or not the Product Information approved by the Therapeutic Goods Administration for the [Generic Products] Products dated 9 July 2018 (the Amended [Generic Products] PI) provides instructions to administer a loading dose of dexmedetomidine hydrochloride as claimed in claims 5-12, 18-25 and 30-39 of Australian Patent No. 754484 (the Patent); and
(b) Whether or not, in light of the Amended [Generic Products] PI, InterPharma would have reason to believe that, upon the introduction of a generic version of dexmedetomidine hydrochloride, there would be any (or alternatively any significant) use of a loading dose as claimed in claims 5-12, 18-25 and 30-39 of the Patent.
255 At the conclusion of the hearing on 22 May 2018, InterPharma’s position in relation to claims 5 to 12, 18 to 25 and 30 to 37 was that although the Generic Products PI instructed the administration of a loading dose, it had no reason to believe that the Generic Products would be used in the manner claimed in the loading dose claims. InterPharma’s submission about indirect as opposed to direct infringement of claims 5 to 12 apparently reflected its view that claims 5 to 12 were not Swiss-style claims (a view I have rejected). InterPharma’s position was apparently that (subject to its other arguments) it had infringed claims 5 to 12, 18 to 25 and 30 to 37 by reason of s 117(1) read with s 117(2)(c) of the Patents Act, but had not infringed by reason of s 117(1) read with s 117(2)(b).
256 As already stated, InterPharma filed its application to re-open on 6 August 2018, together with affidavits asserting that, without its knowledge, Pfizer had amended the Precedex® PI on 6 March 2018 (prior to trial) with the consequence that InterPharma had conducted its case under a misapprehension of fact. InterPharma asserted that it would have conducted its infringement defence differently had it known of Pfizer’s amendment to its Precedex® PI. InterPharma explained that it made its application in August 2018, since its legal representatives had only became aware in July 2018 that Pfizer had amended its Precedex® PI.
257 At the resumed hearing, InterPharma relied on the affidavit of Professor Hall affirmed on 6 September 2018, and Pfizer relied on the affidavit of Professor Bellomo affirmed on 13 November 2018 and the affidavit of Assoc Professor French affirmed on 8 November 2018. The three affidavits were tendered without objection. The experts each gave evidence in a resumed concurrent session. InterPharma also relied on written submissions filed on 19 October 2018, and Pfizer relied on written submissions dated 1 November 2018. The parties made further submissions at the resumed hearing.
258 There was no dispute that, as a practical matter, InterPharma was required to update the Generic Products PI to reflect the amendments that had been made to the Precedex® PI; and that InterPharma did in fact make corresponding amendments to its Generic Products PI. The TGA approved these amendments on 20 July 2018 (Amended Generic Products PI). Since InterPharma’s submissions depended in part on its Amended Generic Products PI, it may be useful to describe the amendments here.
259 The Amended Generic Products PI is substantially identical to the Generic Products PI, save for some formatting changes and two amendments to section 4.2 headed “Dose and method of administration”.
260 The relevant passages in the Generic Products PI originally stated:
For adult patients, DEXMEDETOMIDINE EVER PHARMA is generally initiated with a loading infusion of 1 (one) microgram/kg over 10 to 20 minutes, if needed. The use of DEXMEDETOMIDINE EVER PHARMA by continuous infusion in these patients should not exceed 24 hours.
For patients being converted from alternate sedative therapy a loading dose may not be required.
(Emphasis added)
261 The Amended Generic Products PI states:
For adult patients, DEXMEDETOMIDINE EVER PHARMA may be initiated with a loading infusion of 1 (one) microgram/kg over 10 to 20 minutes, if needed. The use of DEXMEDETOMIDINE EVER PHARMA by continuous infusion in these patients should not exceed 24 hours.
The use of loading dose of dexmedetomidine was associated with an increased rate of adverse event, including hypotension, hypertension and bradycardia, in clinical trials involving adult ICU patients.
For patients being converted from alternate sedative therapy a loading dose may not be required.
(Emphasis added)
It will be seen that the reference to dexmedetomidine being “generally” initiated with a loading dose, if needed, was removed and replaced with a statement that it “may be” initiated, if needed; and a new paragraph concerning the loading dose was incorporated into the Amended Generic Products PI.
262 In broad terms, InterPharma’s position was that, in light of the Amended Generic Products PI, it had a complete defence to alleged infringement of the loading dose claims, while Pfizer contended that the Amended Generic Products PI made no difference at all.
Instruction to administer a loading dose — s 117(2)(c)
263 As already indicated, before the re-opening, it was not in dispute that the Generic Products PI instructed the administration of a loading dose of the Generic Products. In the re-opened hearing, InterPharma contended that the Amended Generic Products PI “would not instruct the use of dexmedetomidine in accordance with any of those claims of the Patent that depend on the integer of administering a loading dose”. InterPharma contended that, having regard to the Amended Generic Products PI, there could be no infringement of the loading dose claims by reason of s 117(1) read with s 117(2)(c) of the Patents Act.
264 InterPharma submitted in written submissions that the “clear purpose” of the amendments to the Generic Products PI was to discourage the use of a loading dose and that the Amended Generic Products PI would be understood by the skilled addressee to caution against this practice. In closing submissions at the resumed hearing, counsel for InterPharma (Mr Thompson) contended that the Amended Generic Products PI:
… informs clinicians that a loading dose may be given to an ICU patient if needed and in a certain manner but does nothing to encourage such use, and, to the contrary, in referring to an increased rate of adverse events … it provides a prominent warning or caution against that use. Properly characterised, it is discouraging the use of a loading dose unless the clinician, exercising their own judgment, decides that it is necessary to do so.
…
What is submitted is that in the particular circumstances of this case, what is now contained [in the Amended Generic Products PI] falls short of constituting an instruction.
(Emphasis added)
265 InterPharma relied on the evidence of Professor Hall, who deposed that the words “is generally” in the Generic Products PI informed him and other clinicians that “as a matter of routine, dexmedetomidine should be administered by a loading dose or bolus for ICU sedation of a patient.” He stated that the replacement of “is generally” with the word “may” in the Amended Generic Products PI informed him and other clinicians “that the administration of dexmedetomidine using a loading dose or bolus is not a matter of routine”. His evidence was that the use of the words “may be” would cause him to “pause and think more about the risks of the drug versus the benefits”.
266 Professor Hall also gave evidence that the introduction of an additional paragraph regarding adverse events (the additional paragraph) in the Amended Generic Products PI “explicitly warns or cautions the clinician against the use of loading dose of dexmedetomidine” and that this instructs clinicians that “unless absolutely necessary, a loading dose or bolus should not be used”. Accordingly, his opinion was that the Amended Generic Products PI did not instruct a general recommended loading dose of dexmedetomidine for ICU sedation.
267 Pfizer submitted the Amended Generic Products PI was substantially identical to the original Generic Products PI and that it contained “instructions that a recipient may administer a loading dose of a Generic Product, if needed in a particular case”, such that s 117(2)(c) of the Patents Act was engaged. In closing submissions at the resumed hearing, senior counsel for Pfizer submitted that, for the purpose of s 117(2)(c) of the Patents Act, the Court should have regard to the Amended Generic Products PI “as it stands” and should not seek to read the Amended Generic Products PI by comparing it with the original PI. Senior counsel emphasised that the Amended Generic Products PI is not marked up with the changes made to the original Generic Products PI, and that there was no evidence before the Court that a clinician reading the Amended Generic Products PI would have in mind what was said in the previous version.
268 Having regard to the majority’s observations in AstraZeneca AB v Apotex Pty Ltd [2014] FCAFC 99; 107 IPR 177; 226 FCR 324 (AstraZeneca FCAFC) at [427] and to Jagot J’s analysis in Beadcrete Pty Ltd v Fei Yu (t/as Jewels 4 Pools) (No 2) [2013] FCA 187; 100 IPR 188 (Beadcrete) at [126], senior counsel for Pfizer submitted that it was sufficient that the Amended Generic Products PI indicated that dexmedetomidine can be administered in a loading dose to constitute an instruction for the purpose of s 117(2)(c). At the same time, senior counsel also emphasised that the Amended Generic Products PI “stretches well beyond just that, because there are so many indications that a loading dose is something that will be done, because it actually instructs you how to deal with the outcomes of [a] loading dose which might be adverse”. Pfizer relied on the evidence of Professor Bellomo and Assoc Professor French, both of whom gave significantly different evidence to Professor Hall concerning the effect of the amendments to the Generic Products PI.
269 Professor Bellomo and Assoc Professor French expressly disagreed with Professor Hall’s opinion that the Amended Generic Products PI instructs clinicians “that unless absolutely necessary, a loading dose or bolus should not be used”. Professor Bellomo deposed that the substitution of the words “may be” for “generally” instructed him that:
when the administration of dexmedetomidine is initiated, it does not need to be given in a loading dose. However, where considered appropriate in the judgement of the treating intensive care clinician, a loading dose may be given. The Amended [Generic Products] PI then provides instructions on how to administer a loading dose, should the clinician do so.
270 Neither Professor Bellomo nor Assoc Professor French regarded the additional paragraph in the Amended Generic Products PI as a “safety warning or caution” against the use of a loading dose. Professor Bellomo’s evidence was that the additional paragraph “serves to remind intensive care clinicians that there are potential adverse events that should be considered when administering a loading dose”. He explained that:
This information enables the clinician to decide whether a given patient is suitable for a loading dose and/or any steps that the clinician may need to take to counteract such adverse events should they occur (eg the administration of other medicines to increase blood pressure and/or heart rate).
271 Similarly, Assoc Professor French characterised the information in the new paragraph as “an advisory statement informing clinicians of the potential side-effects of this agent during bolus administration”. He deposed that:
As with any prescribing of drugs for patients in the ICU, I only prescribe the use of dexmedetomidine with a loading dose for ICU sedation when I consider it necessary. This paragraph does not tell me that a loading dose should not be used unless “absolutely” necessary. Instead, it informs me that if I choose to use a loading dose, I should be aware of the risks of these complications, and take them into account when deciding to administer a product. The administration of any product will always involve weighing up risks and benefits, and the potential side effects of a product will be part of this analysis. It is general practice for this type of document to include information about the potential side effects.
At the hearing, he added:
[T]he expectation would be that a reasonable clinician would take into account the clinical information provided – in the information provided … in the product statement and the clinical scenario that they were encountering, and using their knowledge and the knowledge in this document make an informed decision about what is in the best interests of the patient.
272 Professor Bellomo and Assoc Professor French both contrasted the additional paragraph with an existing statement in the Amended Generic Products PI that “[c]aution should be exercised when administering dexmedetomidine to patients with advanced heart block and/or severe ventricular dysfunction”. They both regarded that latter statement as an “explicit” warning, which differed from the additional paragraph in the Amended Generic Products PI. Professor Bellomo emphasised that the fact the additional paragraph was not in the same form as the explicit caution was indicative that it was simply “information” for clinicians to consider when making their decision about whether or not to give a loading dose, as opposed to a warning or caution against doing so.
273 While Professor Bellomo accepted that the Amended Generic Products PI did not encourage the use of a loading dose, he did not accept that it “discouraged” the use of a loading dose, as counsel for InterPharma erroneously submitted in closing submissions. Rather, both Professor Bellomo and Assoc Professor French clearly agreed that the inclusion of the additional paragraph in the Amended Generic Products PI was directed at “encouraging the correct and appropriate use” of dexmedetomidine.
274 I reject InterPharma’s submission that the Amended Generic Products PI warns against or discourages the use of a loading dose. First, the statement in section 4.2 of that document that dexmedetomidine “may be initiated with a loading infusion of 1 (one) microgram/kg over 10 to 20 minutes, if needed” expressly instructs a clinician that a loading dose of the Generic Products may be administered for initiation of ICU sedation, “if needed”. I accept that, as Professor Bellomo said, the Amended Generic Products PI indicates that there is no need for a loading dose to be given for the initiation of the administration of dexmedetomidine but that a loading dose can be given when the treating intensive care clinician judges this to be appropriate, including in light of the risk of adverse events identified in the Amended Generic Products PI. Indeed, Professor Hall accepted as much when he conceded in cross-examination that there was “still a recommendation for using a loading dose, if required” in the Amended Generic Products PI. Further, as Assoc Professor French emphasised, where a clinician considers it appropriate to use a loading dose, the additional paragraph in the Amended Generic Products PI was designed to bring the risks of adverse events to the clinician’s attention in order that they may be taken into account when deciding to administer the Generic Products. As Professor Bellomo pointed out, the Amended Generic Products PI provides specific instructions on how to administer a loading dose, should the clinician determine to do so.
275 In this regard it is significant, as Professor Bellomo said in his evidence, that the adverse events referred to in the additional paragraph were in fact described in the original Generic Products PI (ie before the amendments were made). Under the heading “Dosage and Administration”, for example, the original Generic Products PI stated that:
Clinically significant events of bradycardia and sinus arrest have been associated with dexmedetomidine hydrochloride administration in young, healthy volunteers with high vagal tone or with different routes of administration including rapid intravenous or bolus administration of dexmedetomidine hydrochloride.
(Emphasis added)
276 Further, under the heading “Precautions”, the original Generic Products PI stated that:
Hypotension, Bradycardia and Sinus Arrest
Clinical events of bradycardia and sinus arrest have been associated with dexmedetomidine administration in young, healthy volunteers with high vagal tone or with different routes of administration including rapid intravenous or bolus administration of dexmedetomidine.
…
Reports of hypotension and bradycardia have been associated with dexmedetomidine infusion. If medical intervention is required, treatment may include decreasing or stopping the infusion of dexmedetomidine increasing the rate of IV fluid administration, elevation of the lower extremities, and use of pressor agents. …
…
Transient Hypertension
Transient hypertension has been observed primarily during the loading infusion, associated with initial peripheral vasoconstrictive effects of dexmedetomidine and relatively higher plasma concentrations achieved during the loading infusion. If intervention is necessary, reduction of the loading infusion rate may be considered. Following the loading infusion, the central effects of dexmedetomidine dominate and the blood pressure usually decreases.
…
(Emphasis added)
277 The original Generic Products PI also stated that in patients over 65 years of age, “a higher incidence of bradycardia and hypotension was observed following administration of dexmedetomidine”, and recommended that “[a] dose reduction for both the loading and maintenance infusions should be considered in patients with impaired hepatic function and in patients over 65 years of age”.
278 As senior counsel for Pfizer noted, the above-mentioned passages in the original Generic Products PI (which are also in the Amended Generic Products PI) identify the matters that the clinician should take into account when deciding to give a loading dose of dexmedetomidine, and at the same time some passages outline a solution to avoid a potential adverse effect. In other words, as Professor Hall accepted in cross-examination, the Generic Products PI, in both original and amended versions, tells the clinician how to administer dexmedetomidine in a loading dose and what to do if adverse events such as hypertension, hypotension or bradycardia arise from the administration of such a loading dose.
279 The statement in the Amended Generic Products PI that “[f]or patients being converted from alternate sedative therapy a loading dose may not be required” is also significant in this context. I accept, as Pfizer submitted, that this statement undermines InterPharma’s proposition that the preceding instructions are an “explicit warning” against the use of a loading dose. To the contrary, it indicates that, at least for patients who are not being converted from alternative sedative therapy, a loading dose may be appropriate. This is consistent with Professor Bellomo’s evidence that:
[W]here a patient is not being transitioned from another sedative and the patient is severely agitated (which is often associated with high blood pressure and an elevated heart rate) the administration of dexmedetomidine in a loading dose may be both appropriate and desirable.
(Emphasis added)
280 The Amended Generic Products PI assumes there will be occasions in which the clinician will decide that a loading dose should be given, even in patients who are identified to be at risk of an adverse event. It is, in my view, clear from the evidence to which I have referred and the document itself that the Amended Generic Products PI does not discourage clinicians from giving a loading dose or warn against that use. On the contrary, the Amended Generic Products PI expressly instructs a clinician that a loading dose of the Generic Products may be administered for initiation of ICU sedation, “if needed”. This instruction is, of course, subject to the clinician’s judgment as to whether a loading dose is appropriately given. Understood in this way, the Amended Generic Products PI provides instructions (within the meaning of s 117(2)(c) of the Patents Act) for the use of the Generic Products by loading dose.
281 I accept that, as Pfizer submitted, further support for this conclusion can be found in Beadcrete, where the patent in suit claimed, among other things, glass beads of a certain size averaging between 1.5 and 4.5 mm. The respondents supplied glass beads in multiple different colours. Relevantly, the use of green and ice blue glass beads in isolation or in combination with each other (each found to have an average size that fell within the relevant claim) would have infringed the patent in suit: see at [147]. Jagot J explained (at [126]) that the instructions in the relevant respondent’s sales materials were:
… to the effect that there are “hundreds of colours and combinations”, “a magnificent range of colours and combinations”, there being “100’s of colours and combinations to choose from and a person may select “any combination of colours you wish to choose”, there being “freedom of choice with colour”. In other words, the respondents instruct that the product may be used as the customer sees fit, either with a single colour or any combination of colours from those available.
(Emphasis added)
282 Also at [126], her Honour found that:
This satisfies the requirement for instructions to use the product in both these ways; that is, by use of a single colour or a combination of colours. It follows that if one of the four colours tested by the applicants infringes the patent, infringement is established under s 117(2)(c).
(Emphasis added)
On appeal, the Full Court referred to the above passage (with other passages) with approval and said there was “no difficulty in characterizing those statements as instructing a use which infringes claim 1”: see Fei Yu (t/as Jewels 4 Pools) v Beadcrete Pty Ltd [2014] FCAFC 117; 107 IPR 517 at [83]-[85].
283 Alternatively, the Amended Generic Products PI provided an inducement to use the Generic Products as a loading dose. In AstraZeneca FCAFC at [427], the majority held that a statement in a product information document that “10, 20 and 40 mg tablets can be divided into equal halves” (emphasis added) “provided a clear inducement to consumers of its 20 mg dosage product to engage in tablet splitting” and that, if the Court had found the relevant claims valid, then it would have found the relevant party liable for infringement under s 117(1) when read with s 117(2)(c) of the Patents Act.
284 For these reasons, I find that claims 5 to 12 of the Patent would have been directly infringed by the exploitation of the Generic Products in Australia prior to the Patent expiry date; and that prior to the Patent expiry date, claims 18 to 25 and 30 to 37 would have been indirectly infringed by supply of the Generic Products, by reason of s 117(1), together with s 117(2)(c) of the Patents Act.
Reason to believe Generic Products would be administered in a loading dose — s 117(2)(b)
285 Prior to re-opening, InterPharma contended that it had no reason to believe that the Generic Products would be used in the manner contemplated by the loading dose claims. In closing written submissions it submitted that “the evidence establishes that dexmedetomidine is almost invariably given in practice only as a continuous maintenance infusion with no loading dose: the use of a loading dose of dexmedetomidine in clinical practice is rare”. In the re-opened hearing, InterPharma argued that the Amended Generic Products PI strengthened its case regarding the absence of “reason to believe”, as the amendments meant that the use of a loading dose would become even more uncommon.
286 Pfizer submitted to the contrary that InterPharma had “reason to believe” that some medical practitioners to whom Generic Products were supplied would administer a loading dose in accordance with the Amended Generic Products PI, and that this was sufficient to engage s 117(2)(b) of the Patents Act. Pfizer further contended that the evidence showed that it was likely that the use of a loading dose would increase if the Generic Products entered the market.
287 In view of the conclusion reached above with respect to the application of s 117(1) together with s 117(2)(c) of the Patents Act in the relation to the loading dose claims, it is unnecessary to decide whether s 117(1) read with s 117(2)(b) would also be engaged.
Dependent on plasma concentration claims
288 InterPharma further submitted that its proposed conduct would not infringe any of the loading dose claims (claims 5 to 12, 18 to 25 and 30 to 37) on the basis that each of these claims depend on one of the plasma concentration claims. For the reasons already given regarding infringement of the plasma concentration claims, I reject this submission.
Claims 8, 21 and 33 (duration of loading dose)
289 Finally, with respect to infringement of the various loading dose claims, InterPharma contested its infringement of the integer in claims 8, 21 and 33 that the loading dose be administered “in about 10 minutes”. InterPharma submitted that the instruction in the Generic Products PI, whether considered alone or with other information in that PI, did not constitute an instruction to administer the loading dose “in about 10 minutes” because a clinician who followed the instruction may administer the dose over a period of time which is not “about 10 minutes”. Accordingly, so InterPharma submitted, there was no instruction that would fall within claims 8, 21 and 33. I reject this submission.
290 As already stated, the Generic Products PI (in its original and amended form) under the heading “Dosage and administration” states that a loading infusion of dexmedetomidine is generally initiated over “10 to 20 minutes” if needed. This encompasses “in about 10 minutes”, which is the integer in claims 8, 21 and 33 at issue here. Further, the Generic Products PI describes two clinical trials for ICU sedation where the loading dose was given “over 10 minutes”. As Professor Bellomo observed, the Generic Products PI demonstrated that the administration of the drug in the clinical trials was efficacious, and that 10 minutes had proved an adequate period in which to administer a loading dose. Reading the Generic Products PI as a whole, I accept that a clinician would understand the Generic Products PI instructs a person to administer the Generic Products as a loading dose “in about 10 minutes”.
291 Moreover, Professor Hall gave evidence that in clinical practice if dexmedetomidine is given by a loading dose it is “usually a 10 minute bolus”. I would infer from this evidence (which was not contested) and from what appears in the Generic Products PI that InterPharma had reason to believe that in the circumstances where a loading dose was administered, it would be administered in about 10 minutes.
292 For these reasons, and having regard to the conclusions set out above regarding infringement of the loading dose claims generally, I accept that claim 8 would have been directly infringed by the exploitation of the Generic Products in Australia prior to the Patent expiry date; and that prior to the Patent expiry date, claims 21 and 33 would have been indirectly infringed by supply of the Generic Products, by reason of s 117(1), together with s 117(2)(b) or s 117(2)(c) of the Patents Act.
293 Also, for the reasons stated, in so far as InterPharma, via its Infringement Analysis, sought to make related submissions in connection with claims 9, 22 and 34 (presumably on the basis that these claims depend on one of claims 8, 21 and 33), I would reject those submissions.
Other (undisputed) matters
294 For the sake of completeness, I note that, apart from the matters discussed above, so far as each of the other integers of the loading dose claims were concerned, there was no dispute that the Generic Products PI instructs and InterPharma had reason to believe that the Generic Products:
(a) would be administered as a maintenance dose to a patient (an integer relevant to each of claims 5 to 12, 18 to 25 and 30 to 37); and
(b) would be administered to patients who are humans (an integer relevant to claims 6, 19, 31 and their dependent claims).
Claims 7, 9, 20, 22, 32 and 34 (loading dosages)
295 There was also no dispute that, if the Generic Products PI instructed the administration of a loading dose, that instruction was for a loading dose of the Generic Products of 1 µg/kg, which is the same dose claimed in claims 9, 22 and 34, and squarely falls within the range of 0.2-2 µg/kg claimed in claims 7, 20 and 32.
296 Accordingly, I accept that claims 7 and 9 of the Patent would have been directly infringed by the exploitation of the Generic Products in Australia prior to the Patent expiry date; and that prior to the Patent expiry date, claims 20, 22, 32 and 34 would have been indirectly infringed by the supply of the Generic Products, by reason of s 117(1), together with s 117(2)(b) or s 117(2)(c) of the Patents Act.
Claims 10 to 12, 23 to 25 and 35 to 37 (maintenance dosages)
297 Further, the Generic Products PI recommends the administration of a maintenance dose of 0.2-1 µg/kg/hr, and that a rate of 0.4 μg/kg/hr should be the initial maintenance infusion, with increases in increments of 0.1 μg/kg/hr if sedation is not adequate at the initial rate. Professor Bellomo’s uncontested evidence was that the Generic Products PI instructs the administration of a maintenance dose within the range claimed in claims 10 to 12, 23 to 25 and 35 to 37. Pfizer submitted (and InterPharma did not deny) that the Court should infer that InterPharma had reason to believe that a maintenance dose of the Generic Products would be administered in this infringing manner. I accept Pfizer’s submissions in this regard.
298 For these reasons, I accept that claims 10 to 12 of the Patent would have been directly infringed by the exploitation of the Generic Products in Australia prior to the Patent expiry date; and that prior to the Patent expiry date, claims 23 to 25 and 35 to 37 would have been indirectly infringed by supply of the Generic Products, by reason of s 117(1), together with s 117(2)(b) or s 117(2)(c) of the Patents Act.
Claims 38 and 39 (omnibus claims)
299 The parties scarcely addressed the construction of the omnibus claims and their submissions on infringement of the omnibus claims was slim. It is clear that Pfizer need not rely on these claims in order to establish that InterPharma’s threatened conduct would have infringed the Patent prior to the Patent expiry date. In these circumstances, it is unnecessary and inappropriate to make any further observation about the omnibus claims.
Proportionality and injunctive relief
300 Before leaving the topic of infringement, I would note one further matter, although, as will be seen, there is no need to do more.
301 In closing written submissions filed on 21 May 2018, citing AstraZeneca FCAFC at [442]-[444], InterPharma submitted that:
While it is not necessary for the patentee to identify any particular person or persons in order to successfully rely upon s 117(1) when read with s 117(2)(b) [or] (c), difficulties arise under s 117(2)(b) where there are “many users of the product only some of whom are likely to put the product to an infringing use.” Where s 117(1) is engaged in such circumstances “then some consideration of proportionality as between the extent of the infringing use that is forecast and the scope of any injunctive relief is warranted”. This is because “the effect of such an injunction may be to deny a supplier access to a market, and consumers access to a product, in circumstances where the supplier could have no reason to believe that the majority of consumers would put the product to an infringing use.”
In [AstraZeneca FCAFC] a majority of the Full Court thus concluded in obiter that:
all other things being equal, the more difficult it is for the patentee to establish that there is a likelihood of widespread infringing use, the more difficult it should be for the patentee to obtain injunctive relief in the broad terms restraining any supply of the relevant product.
(Footnotes omitted)
302 InterPharma contended that “general considerations of this kind should apply whenever the scope of any injunctive relief is being considered” under s 117(1) of the Patents Act, although it did not elaborate on the relevance of such “general considerations” in written closing submissions. Subsequently, in closing submissions at the hearing in May 2018, senior counsel for InterPharma explained that the submission was directed to the possibility that InterPharma’s proposed conduct might be found to be infringing with respect to certain categories of claims but not others, such as the loading dose claims. This aspect of InterPharma’s case was developed at the resumed hearing in November 2018 when InterPharma submitted that if the Court made a finding of infringement on the basis that InterPharma had reason to believe that some medical practitioners to whom the Generic Products would be supplied would administer a loading dose, notwithstanding the Amended Generic Products PI, then the minimal extent of any such infringement would not of itself warrant the grant of any injunctive relief. Counsel for InterPharma subsequently stated in oral closing at the resumed hearing that the issue of proportionality in respect of the infringement of the loading dose claims:
… only arises for consideration in the event that the primary claims as to the use of dexmedetomidine in an ICU as a sole active agent are held to be invalid … the independent claims would need to be invalidated … it’s in that context in circumstances where it seems to be accepted that the predominant, if not the almost invariable use, would be non-infringing today, that the question of proportionality arises.
303 Plainly enough, this issue does not arise for consideration: first, as stated below, I make no finding of invalidity and, secondly, no issue of injunctive relief remains.
Conclusion on infringement
304 For the reasons stated, Pfizer made out its case against InterPharma of threatened infringement of each of the claims of the Patent on which it relied (save for the omnibus claims, which it is unnecessary to consider).
Interpharma’s case on invalidity
305 My reasons for rejecting InterPharma’s challenge to the validity of the Patent in suit are set out below. I turn first to InterPharma’s novelty ground.
Novelty
306 Amongst other things, InterPharma submitted that the invention so far as claimed in claims 1 to 4, 13 to 17 and 26 to 29 of the Patent was not novel. Section 18(1)(b)(i) of the Patents Act provided that an invention is a patentable invention if the invention, so far as claimed in any claim, when compared with the prior art base as it existed before the priority date, is novel. Section 7(1) of the Patents Act provided that an invention is taken to be novel when compared with the prior art base unless it is not novel in the light of any one of the following kinds of information, each of which must be considered separately:
(a) prior art information (other than that mentioned in paragraph (c)) made publicly available in a single document or through doing a single act;
(b) prior art information (other than that mentioned in paragraph (c)) made publicly available in 2 or more related documents, or through doing 2 or more related acts, if the relationship between the documents or acts is such that a person skilled in the relevant art would treat them as a single source of information;
(c) prior art information contained in a single specification of the kind mentioned in subparagraph (b)(ii) of the definition of prior art base in Schedule 1.
307 Information will have been made “publicly available” if it was made available to at least one member of the public who, in that capacity, was free, in law and equity, to make use of it. It is immaterial whether or not the invention has become known to many people or a few people: Insta Image Pty Ltd v KD Kanopy Australasia Pty Ltd [2008] FCAFC 139; 239 FCR 117 at [124].
308 A prior art document must itself disclose explicitly or implicitly all of the essential features of an invention, as claimed, if it is to deprive the invention of novelty: AstraZeneca FCAFC at [350]-[352]; Samsung Electronics Company Ltd v Apple Inc [2011] FCAFC 156; 217 FCR 238 (Samsung) at [127]. As Bennett J (with whom Middleton J agreed) stated in H Lundbeck A/S v Alphapharm Pty Ltd [2009] FCAFC 70; 88 IPR 228; 177 FCR 151 (H Lundbeck) at [180], “[w]here the prior publication discloses exactly what is claimed, there is anticipation”.
309 The English Court of Appeal in General Tire at 486 stated the nature of the test for anticipation as follows:
To anticipate the patentee’s claim the prior publication must contain clear and unmistakable directions to do what the patentee claims to have invented … . A signpost, however clear, upon the road to the patentee’s invention will not suffice. The prior inventor must be clearly shown to have planted his flag at the precise destination before the patentee.
In AstraZeneca FCAFC at [302] a majority of the Full Court (with whom Jessup J relevantly agreed) remarked that:
It is only after the stage of assessing the sufficiency of disclosure — which involves a determination about whether a prior document has “planted the flag” as opposed to having provided merely “a signpost, however clear, upon the road” or, perhaps, something less — that the notion of reverse infringement comes into play as the final and resolving step of the required analysis.
310 Difficult questions can arise as to the nature of the disclosure that must be made by the prior art document to anticipate an invention of a later patentee. The discussion by Lord Westbury LC in Hill v Evans (1862) 4 De GF & J 288; 1A IPR 1 (Hill v Evans) has been frequently mentioned in this connection. In that case, Lord Westbury said at 6-7:
I apprehend that the principle is correctly thus expressed: the antecedent statement must be such that a person of ordinary knowledge of the subject would at once perceive, understand, and be able practically to apply the discovery without the necessity of making further experiments and gaining further information before the invention can be made useful. If something remains to be ascertained which is necessary for the useful application of the discovery, that affords sufficient room for another valid patent. ... [T]he question is, what is the nature and extent of the information thus acquired which is necessary to disprove the novelty of the subsequent patent? There is not, I think, any other general answer that can be given to this question than this: that the information as to the alleged invention given by the prior publication must, for the purposes of practical utility, be equal to that given by the subsequent patent. The invention must be shewn to have been before made known. Whatever, therefore, is essential to the invention must be read out of the prior publication. If specific details are necessary for the practical working and real utility of the alleged invention, they must be found substantially in the prior publication.
Apparent generality, or a proposition not true to its full extent, will not prejudice a subsequent statement which is limited and accurate, and gives a specific rule of practical application.
The reason is manifest, because much further information, and therefore much further discovery, are required before the real truth can be extricated and embodied in a form to serve the use of mankind. It is the difference between the ore and the refined and pure metal which is extracted from it.
Again, it is not, in my opinion, true in these cases to say, that knowledge, and the means of obtaining knowledge, are the same. There is a great difference between them. To carry me to the place at which I wish to arrive is very different from merely putting me on the road that leads to it. …
Upon principle, therefore, I conclude that the prior knowledge of an invention to avoid a patent must be knowledge equal to that required to be given by a specification, namely, such knowledge as will enable the public to perceive the very discovery, and to carry the invention into practical use.
311 It may be accepted that, as Pfizer submitted, a disclosure in a prior art document that merely “includes”, “encompasses” or “covers” a later specific disclosure by a subsequent patentee will not deprive an invention of novelty: see, for example, ICI Chemicals & Polymers Ltd v The Lubrizol Co Inc [2000] FCA 1349; 49 IPR 513; 106 FCR 214 at [51]; IGT (Australia) Pty Ltd v Aristocrat Technologies Australia Ltd [2008] FCAFC 131; 77 IPR 482 at [48]-[61]; Sanofi-Aventis Australia Pty Ltd v Apotex Pty Ltd (No 3) [2011] FCA 846; 196 FCR 1 (Sanofi-Aventis v Apotex (No 3)) at [180]; AstraZeneca FCAFC at [295], [296], [305]. A direction in a prior art document that is capable of being carried out in a manner that would infringe will not anticipate if it is at least as likely to be carried out in a non-infringing way: see Bristol-Myers Squibb Co v F H Faulding & Co Ltd [2000] FCA 316; 97 FCR 524 (Bristol-Myers) at [66], quoting General Tire at 486. Further, it is not permissible to supplement a prior publication by adding integers from the common general knowledge. The publication must itself disclose each and every essential feature of that invention, as claimed: see AstraZeneca FCAFC at [352], [354].
312 It can also be accepted that not every prior published description of a method falling within a claim amounts to an anticipation, as explained in Bristol-Myers. The claimed invention in that case was a process of administering taxol in the treatment of cancer. In addressing the question whether reports of a clinical trial of taxol contained clear and unmistakable directions to do what the patentee claimed to have invented, Black CJ and Lehane J stated at [67] and [69]:
[A] prior publication, if it is to destroy novelty, must give a direction or make a recommendation or suggestion which will result, if the skilled reader follows it, in the claimed invention … . The context was that great difficulties had been encountered in using taxol, despite its known anti-carcinogenic properties, in the treatment of cancer, because of the drug’s side effects. Each of the trials reported in the articles referred to was an investigation directed towards finding a solution of the difficulties: directed, particularly, to ascertaining safe dosage levels. But, though methods falling within the claims of the patents were used in each trial, none of the reports can be said to teach (a word which in this context encompasses direct, recommend and suggest) that which the petty patents claim.
…
For those reasons we respectfully disagree with the conclusion of the trial judge that each of the Phase I trial reports was an anticipation. In our view none of them was.
313 Nonetheless, their Honours held at [72] that the claimed invention was in fact anticipated by an abstract of a document reporting on an ongoing joint European-Canadian trial, that disclosed a dosage within the claims was effective, saying:
We are told that earlier studies had revealed taxol’s “activity against” certain forms of cancer; that the point of the studies then underway was to evaluate the “feasibility of shorter infusion time” and a lower dose “versus a maximum tolerated dose … ”. We are told (apparently) that what the tests have already revealed “makes even outpatient treatment with this first available representative of this new class of antitumour agents possible”. In other words, it is already known that taxol is effective against certain cancers; it is known that 175mg/m2 is a maximum tolerated dose; the purpose of the trial is to test the feasibility of a three hour infusion of that dose and a smaller dose; and the three hour infusion has already proved feasible, so that outpatient treatment has been demonstrated to be possible. Prudent practitioners might well take the view that they would prefer to await the final outcome of the trials, both as to efficacy and as to safety, before rushing to embrace the proposed method. But, in our view, there can be no serious doubt that the abstract teaches the shorter infusion period, with premedication, as a “treatment” of cancer.
As their Honours were at pains to emphasise, the disclosure in the abstract was not in the nature of an investigation as to whether the method subsequently claimed would work. Rather, it was a statement that the disclosed method had been tested and in fact worked.
314 In Merck & Co Inc v Arrow Pharmaceuticals Ltd [2006] FCAFC 91; 68 IPR 511; 154 FCR 31 (Merck v Arrow) at [104]-[112], a Full Court of this Court held that claim 3 of the patent in suit, which was a method of treatment claim relating to the oral administration of alendronate (a potent bisphosphonate compound), in weekly doses, was anticipated by an article in the Lunar News notwithstanding that it reported that “[a]n intermittent treatment program (for example, one per week, or one week every three months), with higher oral dosing, needs to be tested”: at [77]. The fact that further testing was in contemplation was not necessarily fatal to a finding of anticipation: see Merck v Arrow at [104]. After referring to Hill v Evans and C Van Der Lely NV v Bamfords Ltd (1962) 1A IPR 86; [1963] RPC 61, the Full Court stated:
Likewise in Wellcome Foundation Ltd v V R Laboratories (Aust) Pty Ltd (1981) 148 CLR 262 at 281 Aikin J, with whom Gibbs, Stephen, Mason and Wilson JJ agreed, said:
It will always be necessary to distinguish between experiments leading to an invention and subsequent experiments for checking and testing the product or process the subject of the invention. The latter would not be material to obviousness but might be material to the question of utility.
These observations are particularly relevant in the case of pharmaceutical patents like the present one where it is a matter of notoriety that prolonged testing for the purpose of regulatory approval must occur between the stage of patent application and commercial marketing.
315 In Merck v Arrow the primary judge held that the publication of the relevant article in the Lunar News deprived the invention as claimed in claim 3 of novelty because the article disclosed a continuous schedule of oral administration of alendronate having a once weekly dosage interval. The Full Court upheld this finding on appeal at [112]-[113].
316 Merck v Arrow (amongst other authorities) was referred to by Nicholas J in Mylan at [125]-[128], where his Honour made it very clear that he was not seeking to depart from previous statements of the law with respect to anticipation. The decision in Mylan relevantly turned on a factual judgment that a prior publication of a Protocol for a study known as the ACCORD Eye Study, referred to by the primary judge as the “Protocol”, disclosed that the claimed method of treatment in the patent in suit relating to the use of fenofibrate for the prevention and/or treatment of retinophaty worked. His Honour said at [162] and [164]:
In my view the Protocol suggested to the skilled addressee who read it prior to November 2005 that fenofibrate could be used in daily doses of 160 mg for the prevention and treatment of diabetic retinopathy. The fact that Professor O’Brien and Professor Mitchell would not have acted on this suggestion, preferring instead to await the outcome of clinical trials, is no answer to the proposition that the Protocol discloses the precise method of treatment that was later claimed. Nor is it an answer to say that the disclosure was made in the context of a proposed clinical trial aimed at testing a hypothesis.
…
I am satisfied that the Protocol clearly discloses a method of administering fenofibrate in a daily dose of 160 mg to patients suffering from type 2 diabetes who were already taking a statin in the expectation that this would reduce the risk of development or progression of diabetic retinopathy in those patients beyond what it would be were they to have continued to take a statin alone. Use of fibrate in accordance with this method would clearly infringe each of the method of treatment claims which are therefore invalid for lack of novelty. Further, since the novelty of the Swiss-style claims depends upon the use described being new, those claims are also invalid for lack of novelty in light of the use it is proposed in the Protocol for fenofibrate in 160 mg dosages.
Nicholas J held that, on the facts of that case, it was irrelevant that two expert medical witnesses said in evidence that they would wait for the outcome of the clinical trials and that the disclosure was made in the context of “a proposed clinical trial aimed at testing a hypothesis”: see Mylan at [162].
317 Before leaving this statement of the law, it may be noted that InterPharma referred particularly to Bristol-Myers Squibb Co v Baker Norton Pharmaceuticals Inc [1999] RPC 253 (Baker Norton) to illustrate what it said was the correct application of the applicable principles. I refer to my earlier reasons for judgment on the interlocutory injunction for a statement of the decision in that case: see InterPharma Pty Ltd v Hospira, Inc (No 3) [2017] FCA 1536 at [51]-[52]. InterPharma also placed particular reliance on the separate judgment of Beach J (agreeing in the result) in Otsuka FCAFC, where his Honour reiterated at [176] that novelty is not conferred merely by: (a) providing more information about an old use; (b) explaining the scientific theory for the mechanism which underlines a use already described in the prior art; or (c) claiming a narrower use of an old product, where that narrower use fits within the broader use for the old product already described in the prior art.
318 These principles must be applied in determining whether US 214 and/or certain patient information consent (PIC) forms deprived the invention as claimed of novelty.
Pleading issues
319 InterPharma submitted in closing written submissions that its challenge to validity on the novelty ground relied separately on the disclosures in:
(c) US 214;
(d) a PIC form from the Duke University site of clinical study no DEX-95-004 (Duke Form); and
(e) a PIC form from Part 1 of clinical study no W97-249 (249 Form).
320 I note that, prior to trial, InterPharma’s Further Further Amended Particulars of Invalidity had asserted a lack of novelty in respect of US 214 and various PIC forms including the 015 Form and the PHU 004 Form, but this was not the case pressed at trial. At the hearing on 4 May 2018, the Court was advised by Pfizer’s senior counsel (Mr Cordiner QC) that, as a result of his discussions with senior counsel for InterPharma (Ms Rofe QC), he would cross-examine Professor Hall on only two PIC forms. These forms were the Duke Form and the 249 Form. Consistently with this, Ms Rofe QC also confined her cross-examination of Professor Bellomo and Assoc Professor French to the Duke Form and the 249 Form.
321 In closing written submissions, Pfizer submitted that InterPharma should be permitted to rely only on the Duke Form and the 249 Form, since Pfizer had confined its cross-examination “as a result of InterPharma indicating that Professor Hall only need to be taken to [these] forms”. Senior counsel for InterPharma properly accepted this position on 21 May 2018 when the issue was raised in court. It must be accepted that, by the way InterPharma conducted its case at trial, it had indicated an intention to limit its case on novelty to the Duke Form and the 249 Form. Indeed, this outcome is reflected in InterPharma’s Third Further Amended Particulars of Invalidity filed on 24 May 2018.
322 When, at the hearing of closing submissions on 21 May 2018, senior counsel for Pfizer requested that InterPharma amend its particulars to reflect the case it ran at trial, including that InterPharma only relied on the form for Part 1 of clinical study no W97-249 (249 study), Ms Rofe QC said that InterPharma “would prefer to keep [Part] 2 [of the 249 study] in” because “Part 2 is substantively the same” as Part 1. Mr Cordiner QC responded that “[w]e had understood our friends not to rely on it”. His understanding was entirely consistent with InterPharma’s statement in closing written submissions that the 249 Form was from Part 1 of the 249 study. Further, in keeping with this, the form for Part 2 of the 249 study was not a focus of evidence at trial, although touched on from time to time. Nor was it a focus of InterPharma’s submissions, although Pfizer touched on it briefly in its closing submissions. In these circumstances, it does not seem to me that the form for Part 2 of the 249 study was ever accurately described as “in”, although as Mr Cordiner QC indicated, Pfizer addressed Part 2 of the 249 study briefly in closing written submissions.
323 In the absence of any express statement with respect to the form for Part 2 of the 249 study, the parties’ precise positions remain unclear. InterPharma’s Third Further Amended Particulars of Invalidity filed on 24 May 2018 relevantly stated that it relied on “the following prior art information … (v) [t]he patient information and consent forms/informed consent forms for the following clinical trials/studies of dexmedetomidine: … g. W97-249”, but it did not indicate whether this referred to the form for part 1 of the 249 study in conformity with its closing written submissions, or something more — namely, the forms for Parts 1 and 2. As will be seen, the position with respect to the next version of InterPharma’s Particulars of Invalidity went even further beyond the case it had run to that point.
324 So far as I can see, Pfizer was correct in understanding that InterPharma’s case up until 21 May 2018 was that the form for Part 1 of the 249 study, which it explicitly called the 249 Form, anticipated the invention as claimed in the Patent. InterPharma had not made any relevant allegation about the form for Part 2 of the 249 study. It ought not be permitted to extend its case at the point of closing, in the way it sought to do.
325 InterPharma’s Fourth Further Amended Particulars of Invalidity filed on 25 May 2018 again sought to amend InterPharma’s particulars with respect to its novelty case. In this document, InterPharma indicated that it sought leave to strike out particular 1(a)(v)(g), which was limited to claims 1 to 4, 13 to 17, and 26 to 29 in respect of the 249 Form, and replace it with a new para 1A, which is in the following terms:
The invention so far as claimed in each and every claim of the Patent is not a patentable invention within the meaning of s 138(3)(b) and s 18(1)(b)(i) of the Patents Act 1990 when compared with the patient information and consent forms/informed consent forms for W97-249, being prior art information published before the priority date of the claims of the Patent.
(Emphasis added)
326 InterPharma filed submissions dated 25 May 2018 in support of the proposed amendment, which relevantly submitted that proposed para 1A did not raise any new novelty ground, but aligned with the case that InterPharma had opened and to which its evidence in chief was directed.
327 Pfizer filed submissions in response on 29 May 2018 in which it submitted that proposed para 1A expanded InterPharma’s novelty case regarding the 249 Form by referring to all claims of the Patent.
328 In reply submissions filed on 30 May 2018, InterPharma submitted that it was unambiguous that its case was that all claims were anticipated by the 249 Form.
329 It will be apparent from the foregoing that, prior to the filing of the Fourth Further Amended Particulars of Invalidity on 25 May 2018, InterPharma’s pleaded novelty case with respect to the 249 Form had been that the invention so far as claimed in claims 1 to 4, 13 to 17 and 26 to 29 of the Patent was not a patentable invention within the meaning of ss 138(3)(b) and 18(1)(b)(i) of the Patents Act when compared with the prior art base as it existed before the priority date.
330 In affidavits filed before the trial commenced, Professor Bellomo and Assoc Professor French each stated that they disagreed with [150] of Professor Hall’s first affidavit. In that paragraph, Professor Hall had deposed that:
In my opinion, a study in accordance with the W97-249 PIC Form (Part 1) would do what is claimed in all of the claims of the 484 Patent, except for those claims that refer to dexmedetomidine being administered to achieve a plasma concentration of 0.1-2 nano-grams per milli-litre.
331 While each of Professor Bellomo and Assoc Professor French responded directly to this paragraph and expressed the view in their affidavits that the 249 Form did not disclose the use of dexmedetomidine for ICU sedation, neither responded directly to Professor Hall’s earlier more-wide-ranging statements at [145]-[149] of his first affidavit. I accept that, as Pfizer submitted, this was completely appropriate given the case as then pleaded by InterPharma.
332 Consistently with InterPharma’s pleaded case, in making their Joint Report the medical experts were not asked to respond to any questions concerning an alleged anticipation by the 249 Form other than with respect to claims 1, 2, 13, 14, 26 and 27 of the Patent.
333 Also consistently with InterPharma’s pleaded case, in written opening submissions dated 26 April 2018, InterPharma made submissions in support of its contention that the 249 Form anticipated claims 1, 2, 13, 14, 26 and 27 of the Patent. InterPharma concluded:
Accordingly, the 249 Form discloses and anticipates claims 1, 2, 13, 14, 15, 26, and 27 of the Patent and (on Pfizer’s apparent construction) also the balance of the claims of the Patent.
334 There was no elaboration of the reference to the “balance of the claims”, and InterPharma did not seek at this time to amend its pleadings. Pfizer submitted that it assumed, as it was entitled to do, that the reference to “the balance of the claims of the Patent” was “a reference to those claims pleaded in regard to novelty (ie claims 1 to 4, 13 to 17 and 26 to 29 and not some new case of which it had not been given notice”. I accept Pfizer’s submission. It was not for it to identify a different case from that pleaded by InterPharma because of InterPharma’s use of the oblique expression “also the balance of the claims”, particularly as it did not indicate any intention to amend its pleadings in those written submissions or, indeed, in its subsequent oral submissions.
335 As Pfizer submitted, in its written opening submissions it clearly responded to the case as pleaded against it by InterPharma. It noted at [3.1] that “InterPharma alleges that the invention so far as claimed in claims 1 to 4, 13 to 17 and 26 to 29 of the Patent is not novel”. Pfizer submitted and, in the absence of any evidence to the contrary I accept, that InterPharma took no steps at this point to indicate that this was not an accurate statement of its novelty case.
336 In oral opening submissions on 3 May 2018, Ms Rofe QC, for InterPharma, said as follows:
We’ve set out in our submissions an analysis of the disclosure from the 249 form highlighting that it discloses – and I think the experts agree – and to that extent, we might have to make the joint report confidential as well, as they talk about what’s in the patient consent forms. In relation to the 249, they agree that it discloses the administration of dexmedetomidine to ICU patients. They quali[f]y that by saying it’s for the purpose of research, but the disclosure is there. We say it’s in circumstances where – and, now, I do need to discuss some of the – the relevance of the two arms of the test and that there is no choice available in that document that does not produce something falling within the claims of the patent.
We say that that, along with the reference to the large number of patients in which it has already been tried, the large number of healthy volunteers in which it has already been tried and the fact that the company is spending the time and money to have this test, is a direction, recommendation, suggestion to do something that would infringe the claim. For example, if this document were to be found in the back of the plane seat next to one on a plane and one was a person skilled in the art looking at sedation, this would be a direction, recommendation or suggestion to do something.
There was no indication in this opening that InterPharma intended to enlarge its case on novelty in order to allege that the 249 Form anticipated all the claims of the Patent.
337 I accept that, as Pfizer submitted, InterPharma did not notify Pfizer that it was proposing to run a different case from that which it had pleaded with respect to the 249 Form until InterPharma filed and served its written closing submissions dated 18 May 2018. Those submissions at [250]-[251] signalled a different case from that which InterPharma had pleaded and opened.
338 These paragraphs read as follows:
Accordingly, there would be no way for the hypothetical PSA to conduct a study in accordance with what the 249 Form teaches that would not do precisely what is claimed in:
a. claims 1, 13, 26 and (on the basis that the PSA would as a matter of course administer the drug in a saline solution, even if the 249 Form does not clearly instruct this) claim 15 of the Patent;
b. (on Pfizer’s apparent construction of these claims) also claims 3, 16, and 28; and
c. claims 4–12, 18–25, and 29–39 of the Patent. (c.f. the Phase I trials referred to in Bristol Myers Squibb.
Further, a study in accordance with the 249 Form would necessarily involve first administering dexmedetomidine on its own. There would necessarily be some time after the maintenance dose commenced before it was determined whether the use of any rescue medication was required or not. While, consistent with the study design, rescue medications may be administered while the patient in the ICU [sic], the initial administration of dexmedetomidine in this way suffices to anticipate claims 2, 14, and 27, as the sole agent claims do not stipulate any minimum period over which dexmedetomidine must be “essentially the sole active agent or the sole active agent”.
It is plain that these paragraphs express a very different case from that opened by InterPharma.
339 InterPharma contended that Pfizer waited until evidence had closed to contend, for the first time, that InterPharma should be confined to its particularised case on the 249 Form. I reject this submission. It was for InterPharma to notify Pfizer as early as possible that it proposed to enlarge its case on novelty with respect to the 249 Form. It seems that it could have done so by the time the trial commenced.
340 Further, I accept Pfizer’s submission that had it known of an allegation of anticipation by the 249 Form of claims 5 to 12, 18 to 25 and 30 to 39, then it would have sought detailed evidence from the medical experts as to the difference of opinion “enunciated in broad terms on the issue of anticipation, included questions for the joint expert report relevant to that issue and on the basis of that evidence cross-examined Professor Hall on that issue”.
341 As Pfizer noted, InterPharma had multiple opportunities to identify how it put its case on novelty. There was no persuasive reason advanced as to why the proposed amendment was not sought to be made at an earlier stage in the proceeding.
342 In the circumstances outlined, leave to amend so as to add proposed para 1A to InterPharma’s Fourth Further Amended Particulars of Invalidity will be refused. In light of this conclusion, it is unnecessary to consider Pfizer’s alternative submissions filed on 26 November 2018.
343 Before moving to substantive matters, it is convenient to note here that in InterPharma’s written submissions on this proposed amendment filed on 25 May 2018, headed “Particulars amendment – novelty – 249 Form”, there were a number of footnotes, including footnote 2, which read as follows:
Other proposed amendments were raised in opening and were objected to by Pfizer. They concern clarity points which InterPharma says arise from the way Pfizer construes its claim. These were ventilated at trial and are not further addressed here, but are also pressed.
344 This case has proceeded on pleadings. As will be seen, InterPharma has availed itself of opportunities to amend its pleadings on a number of occasions. These reasons are directed to the issues that are raised by the pleadings in their ultimate form, and, to the extent necessary, to any request that has been made for leave to amend those pleadings. It is no part of the Court’s function in such a case as this to decide issues that are not raised or sought to be raised by the pleadings.
Admissibility of PIC forms
345 It is also convenient to address at this point an evidentiary issue concerning the admissibility of the PIC forms, which arose during closing submissions.
346 In the course of the trial, InterPharma tendered without objection an affidavit of Ms Carolyn Stewart affirmed on 13 April 2018, with its exhibits. Included in these exhibits were a number of documents that had been submitted by Pfizer’s predecessor, Abbott Laboratories Inc (Abbott), to the US Food and Drug Administration (FDA) in relation to clinical trials conducted by Abbott concerning the use of dexmedetomidine (FDA documents). These documents, which Pfizer claimed were confidential, were made available to InterPharma by Pfizer in discharging its discovery obligations. I interpolate here that, although Pfizer initially claimed that all of the documents that had been submitted to the FDA were confidential, by the end of the first day of trial senior counsel for Pfizer had advised the Court that Pfizer no longer claimed confidentiality over the contents of the PIC forms themselves.
347 Well before trial, Pfizer had admitted the authenticity of the FDA documents. It was not in dispute that the FDA documents were part of the records belonging to or kept by Abbott.
348 In closing written submissions, InterPharma submitted that the FDA documents were “plainly business records with the meaning of s 69(1) of the Evidence Act 1995 (Cth)” (Evidence Act). As a consequence, the Court asked the parties during closing submissions how s 69 of the Evidence Act operated with respect to the FDA documents in the absence of any evidence that the makers of the representations fell within the description of persons referred to in s 69(2)(a) or (b). The issue of admissibility was addressed by the parties during closing submissions and was also the subject of written submissions after the hearing.
349 Ultimately, by email dated 19 June 2018, Pfizer consented to the following documents being admitted, subject to an order pursuant to s 136 of the Evidence Act limiting the use of the documents such that:
(1) A document entitled “Abbott-85499 Dexmedetomidine Clinical Study Report: A Phase III, Multicenter, Double-Blind, Randomized, Comparative Study Evaluating The Effect of Two Doses of Dexmedetomidine Versus Placebo In Adult Patients Undergoing Elective Coronary Artery Bypass Graft(s) Surgery”, be admitted solely as evidence of the previous representations that the study identified as DEX-95-004:
(a) was a Phase III, Multicenter, Double-Blind, Randomized, Comparative Study Evaluating The Effect of Two Doses of Dexmedetomidine Versus Placebo In Adult Patients Undergoing Elective Coronary Artery Bypass Graft(s) Surgery;
(b) was initiated on 2 January 1996 and completed on 7 December 1997; and
(c) was conducted in compliance with GCP Guidelines.
(2) A document entitled “Abbott-85499 Dexmedetomidine Clinical Study Report: A Phase II, Single-Center, Two-Part Study Evaluating the Safety, Efficacy, and Dose Titratability of Dexmedetomidine in ICU Sedation” be admitted solely as evidence of the previous representations that the study identified as W97-249:
(a) was a Phase II, Single-Center, Two-Part Study Evaluating the Safety, Efficacy, and Dose Titratability of Dexmedetomidine in ICU Sedation;
(b) was initiated on 14 January 1998 and completed on 12 March 1998; and
(c) was conducted in compliance with GCP Guidelines.
(3) A document entitled “Duke University Medical Center Institutional Review Board (IRB)” and an attached document entitled “Duke University Medical Center – Informed Consent for a Research Project: A Phase III, Multicenter, Double-Blind, Randomized, Comparative Study Evaluating The Effect of Two Doses of Dexmedetomidine Versus Placebo In Adult Patients Undergoing Elective Coronary Artery Bypass Graft(s) Surgery: DEX-95-004, Version 2” be admitted solely as evidence of the previous representations that:
(a) the PIC form that was approved by the IRB for use in the DEX-95-004 study at the Duke Medical Center was the Duke Form;
(b) the DEX-95-004 study was a Phase III, Multicenter, Double-Blind, Randomized, Comparative Study Evaluating the Effect of Two Doses of Dexmedetomidine Versus Placebo In Adult Patients Undergoing Elective Coronary Artery Bypass Graft(s) Surgery; and
(c) the approval was given for a period of one year terminating on 11 October 1996.
(4) A document entitled “Information for Patients: A phase II, single-center, two part study evaluating the safety, efficacy and dose titratability of Dexmedetomidine in ICU sedation”, having the protocol “W97-249”, version 5 (part 1) dated 14 January 1998 and correspondence dated 19 January 1998 on behalf of the Board of Directors of the Academic Hospital Utrecht be admitted solely as evidence of the previous representations that:
(a) the PIC form that was approved for use in part 1 of the 249 study was the 249 Form; and
(b) the 249 study was a Phase II, Single-Center, Two-Part Study Evaluating the Safety, Efficacy and Dose Titratability of Dexmedetomidine in ICU Sedation.
350 By email dated 27 July 2018, InterPharma commented that, as it seemed to be common ground that the clinical trial documents were admissible business records, there was no reasonable justification for imposing the s 136 limitation on the use of the documents that Pfizer now seeks. I disagree. It seems to me that there is a danger that the evidentiary use of the documents might be misleading or confusing, or unfairly prejudicial to Pfizer, if admitted otherwise than for the limited uses to which Pfizer has agreed.
351 I accept that, as Pfizer submitted, in its solicitor’s email of 19 June 2018, the documents referred to in InterPharma’s submissions of 1 June 2018 do not contain the representations alleged at [12] and [14] of those submissions, namely that “the [Duke Form or 249 Form] was in fact the informed consent form that was used for [study no. Dex-95-004 at the Duke Medical Center or study number W97-249 (part 1)]”).
352 By email dated 27 July 2018, InterPharma’s solicitor claimed that Pfizer was suggesting that the PIC forms submitted to the FDA may have been other than those actually used in the clinical trials. It does not seem to me that this was the point that Pfizer sought to make. Rather, Pfizer’s position was consistent with the submissions made by its senior counsel, Mr Cordiner QC, in closing submissions on 22 May 2018 that: (a) it was for InterPharma to prove that these documents or replicas of them were provided to patients; and, if it did so, then InterPharma would establish that the documents or replicas of them were provided to patients free in law and equity to use the information in them; and (b) the business records issue (as raised by InterPharma in closing submissions) was neither here nor there because, so far as novelty was concerned, the issue was not what the author of the document knew or intended to say, but rather, assuming the document was publicly available, what were the disclosures that the document made on its face.
353 For the reasons stated, I would therefore make an order under s 136 of the Evidence Act limiting the evidentiary use of the documents referred to in [349] above in the way Pfizer proposed.
US 214
354 InterPharma contended that the invention so far as claimed in claims 1 to 4, 13 to 17 and 26 to 29 of the Patent was not a patentable invention within the meaning of ss 138(3)(b) and 18(1)(b)(i) of the Patents Act when compared with US 214. InterPharma submitted that this was because US 214 explicitly teaches, directs and suggests the use of dexmedetomidine as the sole intravenous sedative agent in mammals, including humans, in an amount to achieve the desired effect without harmful side effects. InterPharma also contended that, properly understood, the invention claimed in the Patent merely identifies the use of dexmedetomidine as a sedative in a narrower group of people that fall within the broad group claimed in US 214. For the reasons I am about to state, I reject InterPharma’s submissions.
What does US 214 disclose?
355 US 214, entitled “Optical Isomer of an Imidazole Derivative Medetomidine as an Alpha-2-Receptor Agonist”, discloses the discovery of the new chemical compound, dexmedetomidine. US 214 states that “[t]he present invention provides, as new compounds, the optically active [d-enantiomer] of medetomidine” (col 1, lines 23-25). It further states that “[t]he d-enantiomer can be expected to be of value, e.g., as a sedative-analgesic, anxiolytic or anti-hypertensive agent” (col 2, lines 27-30).
356 US 214 describes the separation of the enantiomers (col 5; example), and the properties of dexmedetomidine revealed in in vitro tests and the following in vivo animal studies:
(a) the administration of dexmedetomidine intro-peritoneally and subcutaneously to mice in a spontaneous motility and writhing test to study sedative/analgesic properties of dexmedetomidine compared to a racemic mixture;
(c) the use of dexmedetomidine in an elevated t-maze test to study anxiolytic effects of the compound in rats; and
(d) the injection of dexmedetomidine into the femoral vein of rats to study anti-hypertensive effects.
See US 214, cols 3 and 4.
357 According to US 214, the results of these animal studies showed that dexmedetomidine has an enhanced sedative/analgesic property compared to the racemic mixture (medetomidine) and other reference compounds (detomidine and clonidine) (col 3, lines 55-60). The results also showed that dexmedetomidine has an anxiolytic profile in the elevated t-maze test and “possesses clear anti-hypertensive and bradycardia effects” (col 4, lines 16-17, 61-62). US 214 further states (at col 4, lines 64 to col 5, line 8):
The d- and l-enantiomers, and their non-toxic, pharmaceutically acceptable acid addition salts or mixtures thereof may be administered parenterally, intravenously or orally. Typically, an effective amount of the compound is combined with a suitable pharmaceutical carrier. As used herein, the term “effective amount” encompasses those amounts which yield the desired activity without causing adverse side-effects. The precise amount employed in a particular situation is dependent upon numerous factors such as method of administration, type of mammal, condition for which the derivative is administered, etc. and of course the structure of the derivative.
358 I accept that, as Pfizer submitted, this discloses no more than that dexmedetomidine and the l-enantiomer ought to be given at different doses as necessary to achieve what one wants to achieve (ie the “effective amount”). There is no example of any such dose in US 214.
359 The claims of US 214 relate to dexmedetomidine. Most relevantly, claims 3 and 4 are method of treatment claims. Claim 3 is to: “[a] method of sedation/analgesia or treatment of anxiety or hypertension by administration to a subject of an effective amount of [dexmedetomidine] according to claim 1”. Claim 4 is in similar terms, referring to claim 2.
360 The focus of US 214 is on the identification and characterisation of the enantiomer of medetomidine, although the disclosure in the patent gives rise to a “very large number of possibilities” for treatments: compare AstraZeneca FCAFC at [298]. The evidence was that, after reading US 214, the person skilled in the art might, for example, consider the use of dexmedetomidine for sedation/analgesia or treatment of anxiety or hypertension. US 214 does not, however, identify which of these therapeutic outcomes (sedation/analgesia, the treatment of anxiety or the treatment of hypertension) dexmedetomidine will be suitable to treat. This is highlighted by Professor Hall’s evidence that:
If the hemodynamic effects were shown to be dominant, while the sedative effect was modest, the drug could have promise and would be more useful as an anti- hypertensive. If the sedative effect was significant and the hemodynamic changes modest and manageable, it would have potential benefit in the management of critically ill patients.
361 At best for InterPharma, the medical experts agreed in their Joint Report that the sedative properties observed in the rodent models in 1988 (reported in US 214) “are likely the same observed in 1992 in humans”. (It seems that this latter reference was to the healthy volunteer studies described in Belleville and Bloor in 1992.) The experts further agreed, however, that “[t]he effects observed in animal models [in US 214] may not be readily translated into the human population. Further research is justified”. It may be inferred from this and the experts’ subsequent evidence that the experts considered that the animal models disclosed in US 214 were a guide to a potential effect in humans, but did not disclose that the same effect would in fact be observed in human ICU patients.
362 Moreover, US 214 did not disclose that the effect in the animal models would be observed in human ICU patients at a dose that did not produce adverse effects. In concurrent session, Assoc Professor French and Professor Bellomo said:
MS ROFE: So you would say that testing in rats would not – let me – testing in animals wouldn’t give you a basis for predicting the effect in humans.
A/PROF FRENCH: No. These are animal models, so they don’t actually – we use them as a guide to having a potential effect in humans, but it is by no means certain that the same effect will be observed in humans or that the effect can be observed at a dose which doesn’t produce adverse effects.
MS ROFE: And, Professor Bellomo, you would say the same, that you cannot make a prediction from an animal model into humans?
PROF BELLOMO: Yes, I think I would. I think the experience with drugs tested in animals over the last 50 years has been that there is a very unpredictable relationship between what is found in animals and what happens in human beings.
Professor Hall agreed in substance.
363 Professor Bellomo addressed some of these considerations in his affidavit evidence. He deposed that:
While [US 214] does present animal studies with mice and rats (as well as in vitro studies), it does not provide any data on human patients or mention human patients … . I assume the “sedative analgesic effects” and “anxiolytic effects” described in animals in [US 214] refer to indicators in an animal model that dexmedetomidine may have sedative, analgesic and/or anxiolytic properties. However, the suggestion in an animal model that a drug may have these properties is not equivalent to a suggestion that the drug will be an effective sedative, analgesic or anxiolytic agent in humans, in particular, in the ICU. This is primarily because:
(a) animal models may not be good indicators of the effect of a drug in a human, and in particular ICU patients; and
(b) even if a drug is shown to have a particular property in humans (ie sedative property) it cannot be assumed that it will be an effective ICU sedative agent. For example, the drug may have unacceptable side effects at the dose required to achieve sedation.
I accept Professor Bellomo’s evidence, which was supported by the evidence of Assoc Professor French and the other evidence to which I make reference in this context. I reject InterPharma’s submission that “[t]he fact that the tests referred to in … US 214 were in animals dose not detract in any way from its teaching”. That submission is not, in my view, consistent with the evidence.
364 As Pfizer noted, the evidence of Professor Bellomo and Assoc Professor French, which I accept, made it clear that the skilled addressee would not understand that the passing reference to intravenous administration of the drug in US 214 was the same as continuous intravenous infusion, being the means by which ICU sedative agents are typically administered.
365 Further, before April 1998, there were known limitations in sedating ICU patients with clonidine, also an alpha-2 agonist and the only alpha-2 agonist available, in the sense of regulator-approved, before April 1998 for use in humans. The uncontested evidence was that the use of clonidine in ICUs before April 1998 was limited. Professor Bellomo’s affidavit evidence included that:
Most ICU patients who require ICU sedation have low blood pressure. Generally, it was not, and still is not, possible to use clonidine at a dose that has sedative effects in [the] ICU patient as the necessary dose of clonidine to achieve sedative effects will typically cause the patient’s blood pressure to drop to a dangerously low level. Accordingly, clonidine is not a suitable ICU sedative for general use and I would never consider it as better than, or a substitute for, the ICU sedative agents available as at 1 April 1998.
Further, I was not aware of any circumstances where clonidine was administered as an ICU sedative to a patient who was intubated and mechanically ventilated … . At 1 April 1998 and today I would not consider administering clonidine as an ICU sedative to a patient who is intubated and mechanically ventilated as, like most ICU patients, such patients generally have low blood pressure and the administration of clonidine at the dose required to achieve ICU sedation so that the patient can tolerate an endotracheal tube would cause the patient’s blood pressure to drop to a dangerously low level.
At 1 April 1998, I administered clonidine to ICU patients for hypertension and delirium tremens as a bolus intravenous dose. … The inability to readily administer clonidine as a continuous infusion was another reason why clonidine could not be a substitute for the primary ICU sedative agents used at that time. …
366 Assoc Professor French gave evidence to like effect. Professor Hall’s evidence was that “it was … understood, based on the experience of clonidine, that the role [of clonidine in sedation] was limited”.
367 The skilled addressee would have been aware that the concept of sedation of a patient in an ICU was relevantly different from the concept of sedation in other medical contexts. As stated above, intensive care sedation involved affecting the patient’s alertness to some degree to induce calm, in order that the patient could accept intensive care treatment. ICU sedation was known to be a complex process, bearing in mind that intensive care patients were patients requiring advanced life support and/or advanced monitoring as a result of life-threatening injuries, illness, conditions, or complications. The fact that ICU patients often have co-morbid conditions increased the risk of complications. They were very likely to have one or more devices in them and, therefore, be experiencing some degree of pain. Most were anxious or agitated. ICU sedatives were typically administered via continuous intravenous infusion so that the desired sedative state could be achieved and maintained and the dose titrated as required to respond to the ICU patient’s complex and changing needs.
368 It would have been clear to the skilled addressee that the sedative effect disclosed in US 214 may not be achievable in the difficult ICU environment at a dose safe for ICU patients, especially having regard to the experience with clonidine and the fact that US 214 disclosed “clear anti-hypertensive and bradycardia effects”: col 4, lines 61-62. Indeed, bearing in mind the known limitations in sedating ICU patients with clonidine, the skilled addressee would have understood that the “anti-hypertensive and bradycardia effects” disclosed in US 214 might make dexmedetomidine unsuitable for use in sedating ICU patients and that the matter would require further investigation.
369 Having regard to the matters referred to above, I accept the evidence of Professor Bellomo and Assoc Professor French that US 214 does not disclose the use of dexmedetomidine for ICU sedation. In his affidavit evidence, Professor Bellomo said:
As a result of reading US 214, I would not have had any expectation that dexmedetomidine would be useful as a sedative in the ICU.
US 214 does not mention the treatment of ICU patients. … [I]t cannot be assumed that a drug that is appropriate for non-ICU subjects will also be appropriate for ICU patients and that data generated from non-ICU subjects can be extrapolated to ICU patients. If a drug has been used in non-ICU subjects, additional caution, beyond that which is often exercised in other disciplines, must be exercised before that drug can be adopted in the ICU.
…
[A]lmost all ICU patients have unstable vital functions, including blood pressure and heart rate and drugs that significantly affect an ICU patient’s blood pressure and heart rate are not appropriate for ICU use. Therefore, as an ICU specialist in April 1998, I would be concerned by these characteristics of the d-enantiomer and these findings would discourage me from using dexmedetomidine in the ICU.
370 Assoc Professor French deposed that:
While US 214 suggests that dexmedetomidine may have sedative or analgesic (or anxiolytic or antihypertensive) properties in animal models, it does not provide data and inform me of whether those effects would occur in humans. It may be that it will have all, some or none of these effects in humans. It is also not possible to determine from US 214 whether dexmedetomidine in humans will have a side effect profile that would make it acceptable for use in humans, and in particular in ICU sedation. There is nothing in US 214 that suggests to me that dexmedetomidine would be suitable as an ICU sedative.
371 While US 214 opened up possibilities, it did not contain any direction, recommendation or suggestion that would result in the claimed invention if the person skilled in the art followed it. This is consistent with Professor Hall’s evidence in concurrent session that, as to US 214, “if I had looked at this document in 1988 [sic], I would have said this is a first step forward to something that might have some use”. There is no evidence to suggest that the skilled addressee could have determined from US 214 that “some use” would include ICU sedation. The skilled addressee would not discern a disclosure in US 214 of the use of dexmedetomidine to achieve ICU sedation. The further investigation required to determine whether dexmedetomidine would be suitable for use in ICU sedation would be directed to “experiments leading to an invention”: see Wellcome Foundation Ltd v VR Laboratories (Aust) Pty Ltd (1981) 148 CLR 262 at 281. They are entirely different from those contemplated, for example, in Bristol-Myers at [72].
372 Absent the disclosure of the use of dexmedetomidine to achieve ICU sedation, US 214 does not disclose the features of claims 1 to 4, 13 to 17 and 26 to 29 of the Patent: compare AstraZeneca FCAFC at [295]. I reject InterPharma’s submissions to the contrary. The notion of reverse infringement is not reached: AstraZeneca FCAFC at [302]. As indicated, US 214 may disclose to the skilled addressee that dexmedetomidine is a promising candidate for sedation/analgesia or the treatment of anxiety or the treatment of hypertension, based on the result of animal tests and findings regarding comparative potency with other substances, including clonidine. There is, however, no disclosure to the skilled addressee of the relative strength or effects of dexmedetomidine in any of these treatments and the skilled addressee would understand that, with regard to the sedative effect, it may not be achievable at a dose that is safe for patients in an ICU. For the purpose of assessing novelty, this is, in the language of General Tire, no more than a “signpost … upon the road” to the invention as claimed.
373 Further, in so far as raised by InterPharma’s pleaded case, it is apparent that US 214 does not disclose the integers of dependent claims 3, 16 or 28 of the Patent. The disclosure of a dose by reference to an “effective amount” is not a disclosure of a specific dose range to achieve the claimed plasma concentration in these claims: compare AstraZeneca FCAFC at [295].
Does the Patent merely claim a narrower use of an old product?
374 As foreshadowed above, InterPharma also contended that the claims in the Patent in suit lacked novelty because the use of dexmedetomidine as a sedative in ICU patients “falls squarely within the broader disclosure and claims of US 214”. In this regard, InterPharma relied on Beach J’s statement in Otsuka FCAFC at [176] that novelty is not conferred merely by “claiming a narrower use of an old product, where that narrower use fits within the broader use for the old product already described in the prior art”. The patent in suit in that case claimed, in so far as is presently relevant, the use of a compound effective in the treatment of cognitive impairment caused by certain forms of schizophrenia associated with the 5-HT1A receptors. The prior art (US 528) disclosed the use of that compound in the treatment of schizophrenia, including the treatment of cognitive impairment caused by schizophrenia, but not the action of the compound on 5HT1A receptors. The primary judge (Yates J) said, as to novelty:
If, at that time, aripiprazole had been used in a therapeutically effective amount to treat schizophrenia, it would inevitably have treated cognitive impairment of the kind referred to in claim 7 … .
Novelty of invention is not provided merely because information given as part of the definition of the invention in a claim is new information.
See Otsuka Pharmaceutical v Generic Health (No 4) at [320]-[321].
375 On appeal, Beach J referred to these paragraphs (and others) in the reasons for judgment of the primary judge and observed at [176]-[177]:
Like the primary judge, I agree that novelty is not conferred merely by:
(a) providing more information about an old use;
(b) explaining the scientific theory for the mechanism which underlines a use already described in the prior art; or
(c) claiming a narrower use of an old product, where that narrower use fits within the broader use for the old product already described in the prior art.
In this respect I agree with the observations in Actavis UK Ltd v Janssen Pharmaceuticals NV [2008] FSR 35 at [99] and [100] per Floyd J and Bristol-Myers Squibb Co v Baker Norton Pharmaceuticals Inc [1999] RPC 253 at [59] per Jacob J. The addition of the embryonic scientific hypothesis referring to the 5-HT1A receptor subtype is hardly the obtaining of a new technical effect or a new use. Once one accepts the significant overlap between negative and cognitive symptoms of schizophrenia, all that the patent in suit has done is to take a subset of the use and method disclosed in US 528 and to justify the narrowing based on a scientific theory as at the priority date that could hardly be said to be robust. In essence this is just a variant form of parametritis.
(Emphasis added)
376 In Actavis UK Ltd v Janssen Pharmaceuticals NV [2008] EWHC 1422; FSR 35 (Actavis UK Ltd v Janssen) at [99]-[100], Floyd J said that:
In my judgement, merely explaining the mechanism which underlies a use already described in the prior art cannot, without more, give rise to novelty. In MOBIL, the technical effects which underlay the new and old uses were different and distinct. So also in Mr Alexander’s example about disease X and disease Y. It is not the case that every discovery about the mode of action of a drug can be translated into a new purpose and claimed as such.
I think that all that is done here is to explain why the results that would be obtained with compound 84 are as good as they are. The same technical effect underlies both the old use and the new, and the new use is for the same purpose. So if, as I have held, the inevitable result of 362 is that the RSSS isomer potentiates one of the other isomers in compound 84, claim 1 is not saved by MOBIL.
(Emphasis added)
377 In Baker Norton at [59], Jacobs J said:
I think another way of putting this point is to say this is not a case of second medical use at all. The use is the same. All you have new in the patent is more information about that use. I think that is right. As Laddie J said in Evans Medical Ltd’s Patent [1988] RPC 517 at 576.
First, one must identify what the alleged invention is, that is to say what is covered by the claims of the patent, and then one must decide whether or not that invention, or any part of it, would be made inevitably by following the instruction in the prior art. If it would be, then it does not matter whether the skilled reader of the prior art would realise that he was working within the area claimed in the subsequent patent.
(Emphasis added)
378 The invention as claimed in Actavis UK Ltd v Janssen, in Baker Norton and ultimately Otsuka Pharmaceutical v Generic Health (No 4) merely explained the mechanism in an existing use or provided more information about the side effects or mechanism of an existing use. That is not this case; and it is clear that Beach J was not seeking depart from the existing authorities to state a new legal principle.
379 I reject InterPharma’s submission that in disclosing a method of sedation of ICU patients the Patent merely confined the use of an old product to a narrower class of patients. In contrast to Actavis UK Ltd v Janssen, Baker Norton and Otsuka Pharmaceutical v Generic Health (No 4), if a skilled addressee were to follow the limited information regarding the “method of sedation/analgesia” in US 214, the skilled addressee would not inevitably result in the invention as claimed in the Patent, particularly having regard to the unknown relative magnitude and quality of the sedative/analgesic, anxiolytic and anti-hypertensive properties of dexmedetomidine.
380 For the reasons stated, I would not conclude that US 214 deprives the invention as claimed of novelty, as InterPharma has submitted.
Duke Form
381 As indicated above, there was documentary evidence that the study identified as DEX-95-004 was a “Phase III, Multicenter, Double-Blind, Randomized, Comparative Study Evaluating The Effect of Two Doses of Dexmedetomidine Versus Placebo In Adult Patients Undergoing Elective Coronary Artery Bypass Graft(s) Surgery”. There was also evidence that the study was initiated on 2 January 1996 and completed on 7 December 1997; and that it was conducted in compliance with GCP Guidelines. There was evidence that the PIC form approved by the IRB for use in the study was the Duke Form. Pfizer admitted that the study was conducted in compliance with the FDA Code of Federal Regulations Title 21 Part 50, as applicable.
382 I would infer from the study completion date (7 December 1997) that the study was completed before 1 April 1998, the priority date of the Patent. I would also infer from the fact that the study was conducted in accordance with the GCP Guidelines and from the fact that the Duke Form was the PIC form approved for use in the study by the IRB, that replicas of the Duke Form were, on the balance of probabilities, supplied to the patients who participated in the study in order to obtain their consent. The information in the Duke Form was “publicly available” within the meaning of s 7(1) of the Patents Act in that it was made available to at least one member of the public who, in that capacity, was free, in law and equity, to make use of it. Professor Hall, who had been involved in clinical trial involving patient consent, deposed that a patient given such a form was “free to talk to whoever they want to talk to before they give their consent”. Ms Stewart, who was involved in conducting clinical trials in the relevant period in Australia and elsewhere, gave evidence to much the same effect. Ms Stewart was not required for cross-examination.
383 The Duke Form sought patient consent “to participate in a research study carried out by the Department of Anesthesiology at Duke University Medical Center,” Durham, North Carolina in the US. As will be seen, the Duke Form describes investigations into the administration of dexmedetomidine as an anaesthetic adjunct in surgical patients.
384 The medical experts agreed in their Joint Report that the purpose of the study was not clearly defined, although the likely outcomes of interest were outlined as follows:
Dexmedetomidine is a sedative drug which enhances the effect of anesthetic agents, reducing the amount of these drugs required for sedation and helps to keep blood pressure and heart rate under control. The potential benefits of using lower doses of anesthetics are early recovery, less shivering and decreased nausea and vomiting after surgery. In addition, dexmedetomidine has pain-killing (analgesic) effects which may increase comfort and decrease the need for other pain relieving drugs such as morphine in the early period after surgery. This may allow you to be safely removed from the mechanical ventilator earlier than would otherwise be the case.
(Emphasis added)
385 The Duke Form further informed the patient that:
You will receive a drug (midazolam) to help you to relax in the preoperative holding area. … You will not know whether you receive the study drug (dexmedetomidine) or salt water (no active medicine). … The rate of the infusion will then be adjusted to maintain a stable concentration and the infusion will continue in the intensive care unit (ICU) for 6 hours after the end of the operation.
During the operation and in the ICU, your blood pressure, pulse and heart and lung function will be very closely monitored. After surgery in the ICU, your care will be exactly the same as it would be if you were not participating in the study. You will receive routine morphine (pain medication) from your nurse as needed to treat any pain you might have. If you have nausea and/or vomiting, you will be treated with the standard anti-sickness medications through your i.v. . Your shivering will be treated with i.v. meperidine (Demerol).
(Emphasis added)
386 It will be seen from the above quoted passages that the Duke Form describes dexmedetomidine as a sedative drug which enhances the effect of anaesthetic agents during surgery, reducing the amount of anaesthetic agents required and helping to keep blood pressure and heart rate under control. In substance, the proposed study concerns the testing of dexmedetomidine as an anaesthetic adjunct, in the sense that dexmedetomidine was to be administered alongside the anaesthetic agent or agents. The evidence of Professor Bellomo and Assoc Professor French was, and I accept, that the administration of a drug as an anaesthetic adjunct only discloses the behaviour of that drug together with that anaesthetic. It may be inferred from their evidence that the use of dexmedetomidine as an anaesthetic adjunct would not establish whether or not dexmedetomidine operates as a suitable agent by itself, including for the sedation of patients in an ICU.
387 As seen above, the Duke Form disclosed that the patients would be in the ICU for 6 hours after the end of the operation. Professor Bellomo’s evidence was (and I accept) that, as indicated in the Duke Form, the only proposed benefit of the administration of dexmedetomidine after surgery was pain relief. He drew this conclusion from the statement on the face of the Duke Form that “dexmedetomidine has pain-killing (analgesic) effects which may increase comfort and decrease the need for other pain relieving drugs such as morphine in the early period after surgery”. The Duke Form did not disclose any potential benefits of dexmedetomidine in terms of sedation after surgery or in terms of ICU sedation after surgery.
388 In cross-examination Professor Bellomo agreed with senior counsel for InterPharma that the statement in the Duke Form (in the first sentence of para 3) that “[d]exmedetomidine is a sedative drug” indicated that the investigators believed this to be the case. As subsequently shown, however, this answer did not take account of the context of the whole passage, because he was taken to only part of the first sentence. In re-examination, Professor Bellomo explained that in context of the whole paragraph, the relevant sedative effect of dexmedetomidine (as stated in the first sentence of para 3) related to its effect on the anaesthetic agents and to help maintain haemodynamic stability during surgery. This was clear in his evidence in re-examination that the statement in the first sentence of para 3 of the Duke Form meant that:
[T]he sedation of dexmedetomidine is related to its effect on the anaesthetic agents to enhance them and reduce the amount of these drugs needed to achieve the sedation that they normally do achieve and to help maintain what is known as haemodynamic stability.
… It is … suggesting that it’s related to anaesthesia, because it uses the words “anaesthetic agent” in the first sentence and it uses the word “anaesthetics” in the second sentence.
389 The further potential benefits and the side effects of dexmedetomidine were described in the Duke Form in the following terms:
The potential benefits to you are earlier and more comfortable recovery after surgery, more stable heart rate and blood pressure, and reduction of pain, nausea, vomiting and shivering. On the other hand, there may not be any benefit to you. However, your participation will help to evaluate the safety and effectiveness of dexmedetomidine perioperatively in [coronary artery bypass graft] surgery and may benefit other patients in the future. The potential side effects of dexmedetomidine are excessive drowsiness (sedation), dryness of mouth, slowing of heart rate, low blood pressure and stinging in your vein. Less common side effects are dizziness and lightheadedness, headache, transient visual changes (e.g. blurred vision), nausea and vomiting, and spontaneous movements of arms and legs.
(Emphasis added)
390 Sedation is referred to in this passage not as a potential benefit, but as a potential side effect of the administration of the drug. As Pfizer observed, the investigators did not go as far as to suggest that this side effect was likely, let alone certain. In this context Assoc Professor French helpfully deposed:
The Duke PIC Form does not clearly describe the purpose of the study and sedation is only described as a potential side effect of dexmedetomidine. This does not disclose to me whether dexmedetomidine will or will not provide sedation. Further, it does not disclose that dexmedetomidine will provide ICU sedation.
Similarly, Professor Bellomo deposed that the Duke Form indicated to him that sedation was a potential side effect from using dexmedetomidine “that may or may not arise” for patients administered with dexmedetomidine in the proposed study.
391 In cross-examination, Professor Bellomo was taken by senior counsel for InterPharma to the GCP Guidelines. His attention was drawn to the following paragraphs of that document:
2.2 Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks.
…
4.8.10 Both the informed consent discussion and the written informed consent form and any other written information to be provided to subjects should include explanations of the following:
…
(h) The reasonably expected benefits. When there is no intended clinical benefit to the subject, the subject should be made aware of this.
392 Ms Rofe QC put to Professor Bellomo that:
So there is a reasonable expectation that the patients, in undergoing this study, when they’re in their intensive care unit being infused with dexmedetomidine, the ones in at least the high-dose group, would be experiencing a sedative effect?
Professor Bellomo answered: “[t]hat is what the investigators reasonably believed at that time. Correct.”
393 Professor Bellomo’s answer was a limited one; and the exchange must be considered in the context of Professor Bellomo’s evidence as a whole, including his evidence about the GCP Guidelines and his understanding of the disclosures in the Duke Form, which were consistent with his opinion that the Duke Form “does not describe a method of sedating ICU patients using dexmedetomidine or use of dexmedetomidine in ICU sedation as claimed”. His evidence at this point does not lead me to infer that the skilled addressee would have had a different understanding. It must also be borne in mind that the GCP Guidelines, at [2.2] and other points, are identifying the nature of an hypothesis being tested by the investigators in the investigation.
394 Again, it is important to emphasise that what was involved here was a clinical study to ascertain the potential benefits of the drug. The proposed study in the Duke Form was an investigation into the potential benefits of the use of dexmedetomidine as an anaesthetic adjunct during surgery and for pain relief after surgery. It was not proposed for use on its own. It must also be borne in mind that the medical experts agreed in their Joint Report, and it can be accepted, that the outcome of the study was expressed as uncertain.
395 As already stated, the Duke Form discloses the use of dexmedetomidine for the purpose of research in patients perioperatively (including in an ICU after surgery) and, while the experts agreed that it was likely that sedation would be an outcome of interest, that did not mean that sedation, being a potential side effect, was necessarily expected to occur. The Duke Form does not contain a direction, recommendation or suggestion concerning the use of dexmedetomidine for ICU sedation. It does not teach that dexmedetomidine is effective for use in ICU sedation. Indeed, it does not disclose that sedation in the ICU is a possible benefit of the study: it does no more than indicate that sedation may be a potential side effect. The Duke Form did not therefore anticipate claims 1 to 4, 13 to 17 and 26 to 29, as InterPharma alleged.
396 Further, Professor Bellomo’s evidence was that morphine and/or midazolam were likely to have been administered to patients in the study to achieve ICU sedation and, as such, he did not consider that dexmedetomidine could have been administered as essentially the sole active agent or the sole active agent. The experts also agreed in their Joint Report that agents other than dexmedetomidine were used, or potentially used, that could have resulted in ICU sedation for the patients in the proposed study. It will be recalled that the Duke Form specifically stated that:
[a]fter surgery in the ICU, your care will be exactly the same as it would be if you were not participating in the study. You will receive routine morphine (pain medication) from your nurse as needed to treat any pain you might have.
397 Accordingly, the Duke Form does not disclose the “essentially the sole active agent or the sole active agent” integer in claims 2, 3, 4, 14, 16, 17, 27, 28 and 29 of the Patent. Nor does it disclose the verbally similar integer in claim 15. As to claims 3, 16 and 28 (ie the plasma concentration claims) and claims 4, 17 and 29 as dependent thereon, the dose of dexmedetomidine given to patients is not disclosed. Further, as to claim 15, the nature of the pharmaceutical composition is not disclosed.
398 For these reasons, I reject InterPharma’s contention that the Duke Form anticipates claims 1 to 4, 13 to 17 and 26 to 29 of the Patent.
249 study
249 Form
399 As indicated above, there was documentary evidence that the 249 study was a “Phase II, Single-Centre, Two-Part Clinical Study Evaluating the Safety, Efficacy, and Dose Titratability of Dexmedetomidine in ICU Sedation”. There was also evidence that the study was initiated on 14 January 1998 and completed on 12 March 1998; and that it was conducted in compliance with GCP Guidelines. The 249 study was conducted in the Netherlands.
400 Ms Stewart’s evidence was that the GCP Guidelines were in operation in the Netherlands. Relevantly, Ms Stewart deposed that these guidelines were made pursuant to EU Directive 75/318/EEC as amended by EU Directive 91/507/EEC, which came into force in 1991. Mr van Der Kamp, an Attorney at Law in the Netherlands, attested that the good clinical practice part of EU Directive 91/507/EEC was implemented into Dutch law by the Decree on Manufacturing and Delivery of Pharmaceutical Products, which became effective on 1 August 1994 and was in force until 1 July 2007. At the time of the 249 study, applicable EU and Dutch law therefore required compliance with the GCP Guidelines. Pfizer admitted that “[a]ny conduct” of the 249 study “would have been compliant with all applicable laws.” There was also evidence that the PIC form that was approved for use in Part 1 of the 249 study was the 249 Form.
401 I would infer from the study completion date (12 March 1998) that the study was completed before the priority date of the Patent in suit. I would also infer from the fact that the study was conducted in accordance with the laws applicable in the Netherlands, including the GCP Guidelines, and from the fact that the 249 Form was the PIC form approved for use in the study, that replicas of the 249 Form (for both Parts 1 and 2 of the study) were, on the balance of probabilities, supplied to the patients who participated in the study in order to obtain their consent. This is further supported by the affidavit evidence of Ms Stewart. There was a separate consent form for each part of the study and it may be accepted that potential participants would only have been given the form for the part of the study that concerned them.
402 The information in the 249 Form, which related to Part 1 of the 249 study, was “publicly available” within the meaning of s 7(1) of the Patents Act in that it was made available to at least one member of the public who, in that capacity, was free, in law and equity, to make use of it. On the basis of her experience in the management and conduct of clinical trials, Ms Stewart’s evidence was that “prospective participants of [sic] studies must be able [to] disclose and discuss the patient information that they have been provided with their family doctor, family members, friends and anyone else so that they can make an informed decision about whether to participate in the study”. As already stated, Ms Stewart was not required for cross-examination. Her evidence was consistent with that of Professor Hall (see [382] above).
403 The medical experts agreed in their Joint Report (and it must be accepted) that the 249 Form disclosed that dexmedetomidine was a sedative drug, and that the purpose of administering dexmedetomidine in the study was to evaluate the efficacy, safety and dose titratability of dexmedetomidine in ICU sedation. They further agreed that the 249 Form disclosed “the administration of dexmedetomidine to ICU patients for the purpose of research and that sedation will be assessed.”
404 It is apparent that the study was a Phase II single-centre trial involving only 24 patients. In cross-examination, Professor Hall said, and I accept, that in the context of clinical trials of the present kind:
Phase 1 is healthy people or minimally ill people. Phase 2 is some group of patients. It might be the simplest of many … more complex groups of patients, and phase 3 would be much more large comparator trials;
As Assoc Professor French deposed, and I accept, the 249 Form indicated that the proposed study was “an early stage (phase II) clinical study”, noting that studies of this kind “generally involve a small number of patients and a single site” as was the case here.
405 The heart of the dispute between the medical experts with respect to the 249 Form was whether it disclosed that dexmedetomidine was suitable for use for ICU sedation. Professor Hall deposed that, in his opinion, “a study in accordance with the [249 Form] would do what is claimed in all of the claims of the … Patent, except for those claims that refer to dexmedetomidine being administered to achieve a plasma concertation of 0.1-2 nano-grams per milli-litre”. Professor Bellomo disagreed, on the basis that the 249 Form did not inform him that dexmedetomidine was suitable for ICU sedation. He added:
Rather, the [249] Form informs me that in a very small group of patients (twenty four) dexmedetomidine was being evaluated in a phase II trial to see if it may be worth investigating further as a potential agent to reduce the amount of other sedative and analgesic drugs that are administered to ICU patients.
Assoc Professor French was of the same opinion.
406 Bearing in mind the matters already mentioned and the following matters, it seems to me that, although the skilled addressee would understand from the 249 Form that the investigators in this study, in conformity with the GCP Guidelines, reasonably anticipated that the potential benefits justified the risks associated with a clinical trial in humans, and reasonably believed that they would achieve sedation with dexmedetomidine, nonetheless the skilled addressee would also have understood that the investigators had no certainty about these matters. Indeed, Professor Hall agreed in cross-examination that “you would not be obtaining informed consent of patients if you had certainty in either direction”.
407 The terms of the 249 Form reflected that it was indeed an exploratory study in which comparatively little was known about effects of dexmedetomidine when administered to ICU patients. For example, the 249 Form commenced by informing prospective patients who might be involved in the study that:
For a study on a new, experimental drug we seek 24 patients. The drug that will be investigated is Dexmedetomidine, a drug that will be used as a sedative on the Intensive Care Unit.
(Emphasis added)
408 Consistently with the medical experts’ agreement in their Joint Report that the 249 Form expresses uncertainty as to the possible outcomes of the study, Assoc Professor French deposed, and I accept, that the 249 Form did not tell him whether dexmedetomidine “will be an effective agent for ICU sedation”. He added that he would not have had an expectation from reading the 249 Form that dexmedetomidine would be suitable for ICU sedation and, moreover, he was unable to determine from the 249 Form whether or not dexmedetomidine was to be co-administered with other drugs.
409 Uncertainty about the therapeutic effects of dexmedetomidine when administered to ICU patients is also apparent from the comparatively slight and contingent information that the 249 Form presents about the drug. The 249 Form stated:
Dexmedetomidine is one of a group of drugs called α-adrenoceptor agonists which have anaesthetic and analgesic properties. When used during mechanical ventilation, Dexmedetomidine may mean that the use of other sedative and analgesic drugs can be reduced, and the side effects of these other drugs, such as nausea, vomiting and trembling could possibly be diminished. Dexmedetomidine may have a favourable effect on the blood pressure, and it is possible that it could relieve the pain resulting from an operation or other procedures.
(Emphasis added)
410 It is in this context that the 249 Form made it clear that:
The aim of this study is to investigate the effects and possible advantages of Dexmedetomidine during the time that you are being given mechanical ventilation on the ICU. The dosage of this drug can be adapted to ensure that you are comfortable.
411 The 249 Form emphasised the contingent nature of the advantages of being administered the drug: they were no more than possibilities, based on the data “to date”. Under the heading “Advantages and disadvantages associated with participation in this study”, the 249 Form further stated:
Even though the drug is still under research evaluation, based on data analysed upon to date you may benefit from this study, as dexmedetomidine used during ventilation may result in use of less other sedatives and pain relievers and so may also reduce the undesirable side-effects such as nausea, vomiting, shivering associated with the use of those other medications. Dexmedetomidine also appears to provide its effects on sedation and analgesia without significant ventilatory depression associated with the use of opioids and it also provides blood pressure control as well as possible reductions in pain from any surgerie [sic] completed.
The risks involved in this study are associated with the drug, the blood samples, and the other study procedures. It may also involve risks that cannot be foreseen at this moment … .
(Emphasis added)
412 In cross-examination, Professor Hall acknowledged that the 249 Form indicated no more than that the use of dexmedetomidine might result in the use of less other sedatives and other analgesics and, in this way, might reduce undesirable side effects. He also acknowledged that the 249 Form indicated merely that the use of dexmedetomidine might have an effect on sedation; and that it might or might not cause ventilatory depression and reduce pain of clinical significance to the patient from surgery. Even if the above statement that dexmedetomidine “appears to provide its effects on sedation and analgesia without significant ventilatory depression” is taken as a statement that dexmedetomidine has effects on sedation, that statement says nothing about whether that effect will necessarily be present in the ICU patients who are to be the subject of the study or, if it is, whether the effect can be observed at a dose which is safe for them. Further, as Mr Cordiner QC put it, referring to Professor Hall’s evidence, the investigators would not have known:
… the balance of any sedative effect versus any adverse effects … . You don’t know the outcomes of the study as described … . And you don’t know the extent of any advantages or disadvantages as described in this document. And … you also don’t know whether or not the drug is able to be titrated, as [it] is one of the things that is being evaluated.
413 The 249 Form stated that dexmedetomidine had been given to approximately 314 healthy volunteers and approximately 1900 patients. It reports that:
Sleepiness and a dry mouth are the most frequently occurring effects that were seen in healthy volunteers. In the patient studies, low blood pressure, and a slow heart rate were the most frequently seen undesirable effects. In addition, drowsiness, dizziness, changes in the heart rhythm, headache, transient blurred vision, or other changes in the visual acuity, nausea and vomiting were seen.
In 15 persons (6 volunteers and 9 patients), there was a transient interruption of the normal heart rhythm for a brief period of 2.4 to 20 seconds, which resolved spontaneously in 6 persons. Nine (9) persons were given medication, with a quick restoration of the normal heart rhythm, after which no further problems occurred and dexmedetomidine dosing could be continued. Of these 9 patients in 5 there was another stimulus which might have caused or contributed to the interruption of the normal heart rhythm: twice intubation, once cannulation, once the first incision and once a dose of muscle relaxant.
(Emphasis added)
Professor Hall accepted in cross-examination that the 249 Form did not state that sleepiness occurred in all healthy volunteers or patients: rather, it was frequently seen in healthy volunteers.
414 Further, while it may be accepted that the relevant patients were hospital patients, the 249 Form does not suggest that the 1900 patients were in a setting referable to an ICU or that they had characteristics of ICU patients. As to this, Professor Bellomo’s evidence in cross-examination was that:
[I]t is not possible for me to know what these 1900 patients were; whether they were a combination of different types of surgery, whether the agent was given for haemodynamic stability, or whether it was given as an adjunct to anaesthesia or both.
Assoc Professor French similarly said in cross-examination that it was not possible to say why the drug was being administered to the 1900 patients, and to what extent sedation was evaluated.
415 I would accept that, as Pfizer submitted, the skilled addressee could do no more than speculate that the 1900 patients were the subjects of elective surgery or had been in some operative setting, and dexmedetomidine might have been given as an adjunct to anaesthesia or for haemodynamic stability or perhaps for some other purpose under investigation. There is nothing to indicate that dexmedetomidine was given to these patients as a sedative in an ICU; indeed, the fact that the study was being undertaken at all is a strong indication to the contrary, as Professor Hall acknowledged.
416 It is clear from the title of the study and the 249 Form read as a whole that the study is to determine whether dexmedetomidine will be effective and safe for use as a sedative in the ICU and, if so, whether that effect will be titratable. It is within this context that the statement in the introduction of the 249 Form that dexmedetomidine “will be used as a sedative on the Intensive Care Unit” must be understood. This is because, as indicated in the study title, the administration of the drug is for the purpose of “evaluating the safety, efficacy and dose titratability of Dexmedetomidine in ICU sedation”, including whether the drug will be efficacious, safe and dose treatable at all. That is, dexmedetomidine was to be used in the study “as a sedative on the Intensive Care Unit” to ascertain whether or not it would work effectively and safely as a sedative agent in an ICU.
417 Professor Bellomo’s evidence in cross-examination helpfully elucidates the focus of the investigator’s “inquiry” in this regard in the following exchange with senior counsel for InterPharma:
MS ROFE: Now, the title there relates to dexmedetomidine in ICU sedation?
PROF BELLOMO: Yes.
MS ROFE: And in the first paragraph there, [it says]: Dexmedetomidine … a drug that will be used as a sedative in the intensive care unit. So that tells you that it has sedative properties?
PROF BELLOMO: It tells you that the investigators would use it for that purpose and so they are testing the hypothesis that, if used for that purpose, it will achieve both safety, efficacy and it will be appropriate[ly] titratable for dose, and that is the hypothesis that the investigators plan to test.
MS ROFE: So the use of the unqualified “will be used as a sedative” suggests that they consider that it does have sedative properties?
PROF BELLOMO: It does imply that the investigators at that time believed that it would act as a sedative drug, yes.
MS ROFE: And another thing that might suggest that to you is the fact that there’s two arms and no placebo. If they didn’t think there was a sedative effect, they might test it against a placebo to see if there was any difference?
PROF BELLOMO: So it’s a little bit trickier than that, because this is a two-part study, so there is a first part, which is where they’re trying to establish what is the optimal dose of the drug, so they are comparing 0.2 micrograms per kilogram per hour to 0.4 micrograms per kilogram per hour, and then you go to part 2, where they will then perform a second component of the study, and a placebo will be administered in comparison to the ideal dose of the drug as determined by the first part. So the first part, it is correct, there is no placebo; the second part, there is a placebo.
(Emphasis added)
418 Assoc Professor French gave evidence to similar effect when he said that “you hypothesise that you will get the result”. Professor Hall agreed in cross-examination that the 249 Form disclosed the possibility of the use of dexmedetomidine as an ICU sedative and that the purpose of the 249 study was to evaluate whether in fact dexmedetomidine would be an effective sedative agent in the ICU.
419 With the abovementioned considerations in mind, I conclude that the 249 Form did not anticipate claims 1 to 4, 13 to 17 and 26 to 29 of the Patent. Rather, the 249 Form recorded a proposal for a trial to evaluate a hypothesis: whether the use of dexmedetomidine in ICU sedation was safe, effective and dose titratable. The 249 Form did not disclose that it was known that dexmedetomidine was suitable for use as a sedative for patients in the ICU. I reject InterPharma’s submission that the skilled addressee “reading the 249 Form would have a reasonable expectation of seeing a sedative effect on patients in the ICU who received dexmedetomidine in the study referred to in the 249 Form”.
420 As already indicated, the skilled addressee would understand that the 249 study was an early study about the use of dexmedetomidine for ICU sedation, and that the study would not be conducted if the investigators had certainty in either direction. The skilled addressee would have known that a significant proportion of Phase II trials were unsuccessful. In this regard, Professor Bellomo deposed, and I accept as consistent with other evidence, that “[t]he end of a [P]hase II study is another common stage for the drug development to stop because it is determined that the drug is not having the anticipated therapeutic effect in the target population”.
421 Moreover, as the experts agreed in their Joint Report, immediately before 1 April 1998 (and at the time of the 249 study) most intensive care specialists would have had little, if any, knowledge of dexmedetomidine.
422 The 249 Form discloses the possibility of the use of dexmedetomidine as an ICU sedative and that a study is to be conducted to evaluate that possibility. This is an hypothesis to be tested in order to establish if it is at all well founded. This is not like the abstract in Bristol-Myers reporting on an ongoing joint European-Canadian trial and disclosing that a dosage within the claims was effective: see Bristol-Myers at [72]. Nor is it like the article in the Lunar News considered in Merck v Arrow, which disclosed a continuous schedule of oral administration having a once weekly dosage interval. The 249 Form is not in the nature of a statement that discloses that the method will work, and ought to be tested to obtain regulatory approval. The disclosure in the 249 Form is not dissimilar to the reports of a clinical trial of taxol considered in Bristol-Myer. None of the reports were said to direct, recommend or suggest the invention as claimed. Black CJ and Lehane J apparently accepted that the reports amounted to “mere speculation as to whether the method subsequently claimed would work” (to adopt the language of the respondent’s senior counsel in that case); and held that none of the reports “taught the method of the claims”: Bristol-Myers at [68].
423 Clearly enough, the 249 Form does not disclose the integers of dependent claims 2, 14, or 27 of the Patent. Nor does it disclose the verbally-similar integer in claim 15. The medical experts agreed in their Joint Report that aside from dexmedetomidine, other agents were used, or potentially used, in the study that could have resulted in ICU sedation. The 249 Form indicated that patients receiving dexmedetomidine would also be given midazolam for the purpose of achieving ICU sedation and/or morphine for analgesia, and that the potential outcome of the study is only that less sedative drugs may be needed. It was said, for example, that:
[T]he doctor will give you the following medicines if necessary, in addition to the study medication: midazolam (this is a frequently used sedative) and/or morphine (this is a frequently used analgesic). Supplementary treatment that may be necessary for your safety and comfort will be given at the discretion of the consulting doctor.
424 In this context, the evidence of Professor Bellomo is helpful. He deposed:
From the statements in the [249] Form … I consider that the investigators do not expect that dexmedetomidine will be able to replace the existing standard ICU sedatives and analgesics provided to patients, but rather, that it may decrease the amount of those standard ICU sedatives and analgesics. Accordingly, the best case scenario put forward by the investigators of this study is that dexmedetomidine may be a useful drug to complement existing ICU sedatives in use.
… it appears very unlikely to me, and I would expect would appear unlikely to others in my field, that dexmedetomidine will be administered (or could be administered) as essentially the sole active agent or the sole active agent to achieve ICU sedation.
Form for Part 2 of the 249 study
425 The above discussion has focussed on the 249 Form. As I have already stated, Pfizer was, in my view, correct in understanding that InterPharma’s case up until 21 May 2018 was that the form for Part 1 of the 249 study, which it called the 249 Form, anticipated the invention as claimed in the Patent; and that it had not made such an allegation about the form for Part 2 of the 249 study. In case I am wrong, however, I would add the following about the form for Part 2 of the 249 study.
426 As already indicated, Part 2 of the 249 study provided for a placebo and it depended on the completion of Part 1 in so far as the dose of dexmedetomidine administered to patients in Part 2 was to be determined by reference to the data in Part 1. The 249 Form and the form relating to Part 2 of the 249 study were essentially identical, although I accept that they must be read separately from one another since Part 2 was directed to a different group of patients.
427 Both Pfizer and InterPharma approached the matter on the basis that the outcome with respect to the 249 Form would be the same with respect to the form for Part 2 of the study. InterPharma did not advance any reason why there might be different outcomes. Pfizer observed, and in the absence of any contrary argument, I accept, that although the dose of dexmedetomidine administered to patients in Part 2 of the study was to be determined by reference to the data in Part 1, the Part 2 form does not disclose whether or not the dose in Part 1 will be shown to be safe and effective. Bearing this and the foregoing matters in mind, if it were necessary to decide, I would conclude that the form for Part 2 of the 249 study, like the 249 Form, discloses only a hypothesis to be tested, where the outcome of that testing was uncertain. The 249 Form and the form for Part 2 of the 249 study do not disclose that dexmedetomidine is suitable for ICU sedation.
Inevitable result?
428 InterPharma also relied on the analysis of “inevitable result” in support of its contention that the 249 Form (and it may be inferred the Duke Form) anticipated the invention as claimed. Ms Rofe QC noted in closing that she had “consistently said from the outset of this case that we have the same use with the same technical effect [in the prior art information], and in that sense, to the extent that it is inevitable the same use to treat the same disease”.
429 In closing submissions, Ms Rofe QC referred to the very helpful discussion of this analysis by Yates J in Otsuka Pharmaceutical v Generic Health (No 4), culminating at [319]-[320] and, again in this context, to Beach J’s further discussion in Otsuka FCAFC at [115], [172]-[173]. As Mr Cordiner QC observed, however, the analysis about inevitable result has to be understood:
in the context of a claim where the additional integer is narrowing in the sense that it adds an additional integer. But the inevitable use of the drug beforehand would have that feature, because all it was doing is identifying something that the therapy gave. So it’s a curious narrowing. It’s a narrowing without there being really a narrowing … .
430 With respect to the 249 Form, in written closing submissions, InterPharma submitted that:
[T]here would be no way for the hypothetical [person skilled in the art (PSA)] to conduct a study in accordance with what the 249 Form teaches that would not do precisely what is claimed in:
(a) claims 1, 13, 26 and (on the basis that the PSA would as a matter of course administer the drug in a saline solution, even if the 249 Form does not clearly instruct this) claim 15 of the Patent;
(b) (on Pfizer’s apparent construction of these claims) also claims 3, 16, and 28; and
(c) claims 4–12, 18–25, and 29–39 of the Patent (c.f. the Phase I trials referred to in Bristol Myers Squibb).
Further, a study in accordance with the 249 Form would necessarily involve first administering dexmedetomidine on its own. There would necessarily be some time after the maintenance dose commenced before it was determined whether the use of any rescue medication was required or not. While, consistent with the study design, rescue medications may be administered while the patient [is] in the ICU, the initial administration of dexmedetomidine in this way suffices to anticipate claims 2, 14, and 27, as the sole agent claims do not stipulate any minimum period over which dexmedetomidine must be “essentially the sole active agent or the sole active agent”
Further, as the study involves the administration of dexmedetomidine to ICU patients within the dose ranges claimed in the patent, on the basis that this agent is suitable as claimed for use as “essentially the sole active agent or the sole active agent” (on whichever is the true construction of the claim), then a study conducted by the hypothetical PSA in accordance with the study design would invariably involve administering dexmedetomidine as “essentially the sole active agent or the sole active agent” to at least some of the patients in the study over at least some period of the study.
431 Reference has already been made to Bristol-Myers at [67]. It is, however, also clear from this paragraph and the surrounding paragraphs that a similar argument to that advanced by InterPharma was rejected in Bristol-Myers, where Black CJ and Lehane J said at [66]-[68]:
[66] Senior counsel for the respondent relied on the decision of the Court of Appeal in General Tire & Rubber Co v Firestone Tyre and Rubber Co Ltd [1972] RPC 457, particularly two passages in the judgment (at 485):
If the earlier publication … discloses the same device as the device which the patentee by his claim, so construed, asserts that he has invented, the patentee’s claim has been anticipated, but not otherwise. In such circumstances the patentee is not the true and first inventor of the device and his claimed invention is not new … [.]
… If the prior inventor’s publication contains a clear description of, or clear instructions to do or make, something that would infringe the patentee’s claim if carried out after the grant of the patentee’s patent, the patentee’s claim will have been shown to lack the necessary novelty, that is to say, it will have been anticipated.
It is important to remember, however, that the Court also said this (at 486):
If, on the other hand, the prior publication contains a direction which is capable of being carried out in a manner which would infringe the patentee’s claim, but would be at least as likely to be carried out in a way which would not do so, the patentee’s claim will not have been anticipated, although it may fail on the ground of obviousness. To anticipate the patentee’s claim the prior publication must contain clear and unmistakable directions to do what the patentee claims to have invented … [.] A signpost, however clear, upon the road to the patentee’s invention will not suffice. The prior inventor must be clearly shown to have planted his flag at the precise destination before the patentee.
[67] What all those authorities contemplate, in our view, is that a prior publication, if it is to destroy novelty, must give a direction or make a recommendation or suggestion which will result, if the skilled reader follows it, in the claimed invention. … But in this case medical practitioners hardly needed to be told that it was possible to infuse a particular dose of taxol over three hours, or how to do it. Nor, equally obviously, is that the point of the claims. … The context was that great difficulties had been encountered in using taxol, despite its known anti-carcinogenic properties, in the treatment of cancer, because of the drug’s side effects. Each of the trials reported in the articles referred to was an investigation directed towards finding a solution of the difficulties: directed, particularly, to ascertaining safe dosage levels. But, though methods falling within the claims of the patents were used in each trial, none of the reports can be said to teach (a word which in this context encompasses direct, recommend and suggest) that which the petty patents claim.
[68] Senior counsel for the respondent acknowledged that not every prior published description of a method falling within the claims would amount to an anticipation. He accepted that a mere speculation as to whether the method subsequently claimed would work would not, of itself, destroy novelty. With somewhat greater hesitation, he accepted that a mere proposal for a trial of the method claimed might not be an anticipation. But he submitted that, in any event, the circumstance that the methods of administration described in the articles had been put into effect in the Phase I trials necessarily meant that the reports anticipated the claims. He referred, in this context, to the decision of the House of Lords in Bristol-Myers Co v Beecham Group [1974] AC 646; [1975] RPC 127. But there are several difficulties with that. Prior use, even if unwitting, of a chemical compound subsequently claimed is, as the authorities made clear, a different thing altogether: there is, of course, no question of what the use teaches. Secondly, the particular use (reported in the publications relied on here) cannot itself be relied upon as an anticipation (and it was not relied on), because it took place outside Australia; but, that being so, the fact that actual use is reported in a prior publication cannot, in principle, make any difference. The question is still, what does the prior publication teach? Each of the reports taught, no doubt, some useful things relating to the administration of taxol. But none of them taught the method of the claims.
(Emphasis added)
432 Pfizer contended, and I accept, that, having regard to Bristol-Myers at [68], the actual use of dexmedetomidine reported in the Duke Form and 249 Form was not relevant to a novelty analysis. The proposed prior use reported in the Duke Form and 249 Form cannot be relied on as an anticipation in this case because it took place outside of Australia. For the reasons already stated, the disclosure in each of the Duke Form and 249 Form was no greater than the disclosure in the reports in Bristol-Myers that the Full Court rejected as anticipating the method of treatment claims in that case.
433 In any event, the Duke Form and the 249 Form do not contain clear instructions to do or make something that would infringe the Patent. The invention as claimed in claims 13 to 17 and 26 to 29 of the Patent was limited by reference to the purpose for which dexmedetomidine is used, namely, ICU sedation, and infringement is made out by looking at the purpose, aim or object of the administration of the drug, not the effect in fact of the administration: compare Apotex v Sanofi-Aventis HCA at [289], [294]. It seems to me that the same kind of observation may also be fairly made with respect to claims 1 to 4, bearing in mind that in a Swiss-style claim, such as claim 1, the novelty arises from the therapeutic use or method of treatment. The carrying out of any supposed directions in the Duke Form or the 249 Form would not result in the deliberate administration of dexmedetomidine for the purpose, aim or object of ICU sedation. As already explained, the Duke Form describes research into the administration of dexmedetomidine as an anaesthetic adjunct, ie as an agent administered alongside general anaesthesia in surgical patients and as an agent that may decrease the need for other pain relieving drugs post-surgery. The 249 Form describes early exploratory work involving the administration of dexmedetomidine for the purpose of evaluating its safety, efficacy and dose titratability in ICU sedation. It follows that the use of dexmedetomidine as described in these two PICs would not “infringe the claims of the Patent”, since neither directed the administration of dexmedetomidine for the purpose, aim or object of ICU sedation.
434 As to the submission (see [430] above) that there would be a use of the drug on its own, to anticipate claims 2, 14 and 27, Mr Cordiner QC submitted, and I accept, that before rescue medication was used, there was no sedation, and the drug was not providing a sedative effect such as to fall within those claims. Once a rescue drug is given, then the use of dexmedetomidine was not within these claims. Further, as Mr Cordiner QC submitted, one simply does not know the results of what happened in the case of the 24 patients in the 249 study. It is difficult to apply the notion of inevitable result when the document in question (the 249 Form) described what was to happen before the trial, but not the results of the trial.
435 In any event, there are difficulties with the application of the “inevitable result” analysis in relation to inventions involving new methods of medical treatment, as Bennett and Yates JJ observed in Apotex Pty Ltd v Sanofi-Aventis Australia Pty Ltd (No 2) [2012] FCAFC 102; 96 IPR 185; 204 FCR 494 (Apotex v Sanofi-Aventis FC) at [165]: see also Actavis UK Ltd v Janssen at [90]. As the Bennett and Yates JJ said in Apotex v Sanofi-Aventis FCAFC:
There is a question whether the unyielding logic of the “inevitable result” cases can be applied uncritically in every case of alleged anticipation. If so applied, inventions claimed as new methods of medical treatment involving the administration of a known compound for a hitherto unknown and unexpected, but nevertheless useful, therapeutic use would never stand scrutiny as patentable inventions. This is because, on the logic of the “inevitable result” cases, the disclosure of a given compound for one therapeutic use must equally and inevitably disclose all therapeutic uses of the compound in susceptible recipients, notwithstanding that those uses might not have been discovered at the time of disclosure of the first use, and may never be known. Such an approach may not give true expression to the statutory test that denies novelty only in light of information that is made “publicly available”, a requirement as stated in Hill v Evans (1862) 4 De GF & J 288 at 301; 1A IPR 1 at 7 that “[t]he invention must be shewn to have been before made known” … .
(Emphasis added)
One cannot help but agree.
436 Having regard to the foregoing considerations, it does not seem to me that the 249 Form or the Duke Form anticipated the invention as claimed in the Patent in suit, even on InterPharma’s application of the “inevitable result” approach.
Grace Period — 249 Form
437 Having regard to the conclusion I have reached with respect to lack of anticipation by the 249 Form, it is unnecessary to consider Pfizer’s contention that the information within the 249 Form should be disregarded in considering novelty pursuant to s 24(1) of the Patents Act.
Manner of manufacture
Parties’ positions
438 Section 18(1)(a) of the Patents Act provides that an invention is a patentable invention, if the invention, so far as claimed in any claim, is a manner of manufacture within the meaning of s 6 of the Statute of Monopolies 1623 (Imp) (Statute of Monopolies). The term “patentable invention” is defined in the Dictionary in Sch 1 to the Patents Act as “an invention of the kind mentioned in section 18”. The term “invention” is defined as “any manner of new manufacture the subject of letters patent and grant of privilege within section 6 of the Statute of Monopolies, and includes an alleged invention”. As Dixon CJ, Kitto and Windeyer JJ made clear in National Research Development Corporation v Commissioner of Patents (1959) 102 CLR 252 (NRDC) at 269, the field of patentability is determined by ascertaining whether what is claimed is a proper subject for monopoly by a patent according to the principles that have been developed for the application of s 6 of the Statute of Monopolies.
439 In its Third Further Amended Particulars of Invalidity filed on 24 May 2018, InterPharma asserted that the invention as claimed in the Patent in suit was not a patentable invention within the meaning of s 18(1)(a) of the Patents Act in that it was not a manner of manufacture within the meaning of s 6 of the Statute of Monopolies. In its particulars, InterPharma further asserted that:
(a) The Patent expressly incorporates by reference the entirety of the US 214 (page 4, lines 25–26), which amongst other things describes the use of dexmedetomidine as an alpha-2-receptor agonist for sedation (page 4, lines 9–10), and claims a pharmaceutical composition suitable for use as a method of sedation comprising dexmedetomidine or a non-toxic pharmaceutically acceptable acid addition salt thereof in an amount sufficient to produce the desired effect in association with a pharmaceutical carrier; and a method of sedation by administration to a subject of an effective amount of such composition.
(b) Accordingly, the claims of the Patent are not patentable subject matter because on the face of the specification of the Patent the claims do not comprise a previously unknown use of a known substance, but merely a known or analogous use of a known substance.
440 As will be seen, InterPharma contended that the invention as claimed was not a “manner of new manufacture”, because it disclosed nothing but a new use of an old substance. In this regard, InterPharma relied particularly on the principles laid down by the High Court in Commissioner of Patents v Microcell Ltd (1959) 102 CLR 232 (Microcell). Citing Merck v Arrow at [66], InterPharma submitted that, by incorporating by reference US 214 in its entirety into the Patent in suit, the patentee in this case invited a comparison between what is disclosed in US 214 and what is claimed in the Patent, and that such comparison reveals an absence of a patentable invention. InterPharma submitted that the patentee’s express recognition and acknowledgement of the publication of dexmedetomidine and its uses in US 214 cannot be disregarded for the purposes of assessing whether there is a patentable invention simply because the patentee added the statement, by amendment during prosecution, that the discussion in the background “is not to be taken as an admission that any of the material referred to was published, known or part of the common general knowledge in Australia as at the priority date of any of the claims”. Senior counsel for InterPharma submitted in opening that:
[T]he patentee of the patent in suit is seeking to claim the use of dexmedetomidine in sedating a patient in an ICU. We say that is not a new use, and it’s also a use for which the known sedative properties of dexmedetomidine make it suitable in accordance with the microcell test, and a use that’s analogous to the sedation disclosed and claimed in US 214.
441 In particular, InterPharma submitted that the Patent in suit neither claimed any new substance, nor any new characteristic, use or technical effect of dexmedetomidine. In closing written submissions, InterPharma argued that US 214 discloses and claims the administration of dexmedetomidine: (a) to a human patient; (b) in an effective amount to produce the desired sedation/analgesia; (c) as the sole active agent; and (d) intravenously by continuous infusion. In support of these propositions, InterPharma referred to Professor Bellomo’s evidence in cross-examination that because the claims in US 214 are not confined to animals, he would have to assume they include “any mammal”, and that because the claims are not limited to a specific method of administration, “by natural conclusion”, they could include continuous infusion (though it is worth noting that Professor Bellomo did emphasise that the claim in this regard was broader than the specification which related to intravenous administration of the drug as an anti-hypertensive agent).
442 With the above matters in mind, InterPharma submitted that US 214 discloses that dexmedetomidine has sedative properties and that those sedative properties can be used to sedate mammals, including humans. InterPharma emphasised that the claims of US 214 are unlimited with respect to patient, type of patient (whether sick or healthy), method of administration, level of sedation or quality of sedation. InterPharma submitted that US 214 teaches that an effective amount or dose of dexmedetomidine (that is, the amount required to achieve the desired sedative activity without harmful side effects) can be determined by the skilled addressee, by a standard and non-inventive process having regard to various stated factors.
443 Having regard to these matters, InterPharma submitted that US 214 can only be construed as a direction to the skilled addressee that dexmedetomidine can be used as a safe and effective intravenous sedative in patients, including ICU patients, at doses that could be routinely worked out by the skilled addressee. In other words, so InterPharma submitted, using dexmedetomidine for the sedation of patients in an intensive care setting, including by intravenous administration, is “entirely within the contemplation of and encompassed by the US 214 claims”.
444 InterPharma submitted that even if the use of dexmedetomidine in ICU sedation was, in any sense, “new”, that use could only be said to result from the “discovery” that the drug is useful in an ICU because of its sedative/analgesic properties (or its other properties as an alpha-2 agonist) which were expressly disclosed and claimed in US 214. At most, so InterPharma said, the patentee has identified a class of people (ICU patients) in relation to which the compound claimed in US 214 has been found to be suitable, and it is the known sedative/analgesic properties of dexmedetomidine that make it suitable for such use. Accordingly, InterPharma contended that the purported new use of dexmedetomidine for ICU sedation did not constitute its use for a purpose for which the substance was not previously used due to a “hitherto unknown or unsuspected property of the material” in accordance with the Microcell test, and there was, therefore, no patentable invention.
445 That the “use” on which the US 214 disclosure and claims were based was studies limited to animals was not, so InterPharma submitted, a basis to read down its disclosure. In any event, even if it was only permissible to compare the use on animals with the ICU use, InterPharma contended that the result would be the same: US 214 discloses the use of dexmedetomidine (and its sedative/analgesic properties) as a sedative/analgesic in an animal trial, and the use of dexmedetomidine (and its sedative/analgesic properties) as a sedative/analgesic in human patients is the same as or analogous to the use in animals, and it still takes advantage of the known properties of the substance. It followed, so InterPharma argued, that the invention as claimed in the independent claims and the sole active agent claims of the Patent was not a manner of new manufacture over the invention described and claimed in US 214.
446 InterPharma submitted that the plasma concentration claims added nothing of substance to the independent claims and cannot constitute an invention. This was because these claims simply specify that dexmedetomidine is administered to achieve a plasma concentration within a certain range (which the specification states is anticipated to provide sedation in ICU patients) and that any “effective” amount of dexmedetomidine to achieve sedation of a human patient would be expected to result in a plasma concentration within the claimed range. InterPharma submitted that Pfizer had engaged in “parametritis”, by selecting a variable that has no clinical significance so as to appear to constitute a manner of new manufacture. InterPharma also contended that there can be no invention over US 214 merely in doses (or dose ranges) that are taught to be effective to achieve the sedation of a patient without harmful side effects, as US 214 teaches that such effective doses can be worked out routinely by the skilled addressee. Nor, so InterPharma said, is there an invention merely in the choice to administer a loading dose.
447 In response, Pfizer submitted that an objection to validity on the basis of manner of manufacture (as distinct from novelty and inventive step) is confined by reference only to what is disclosed as “known” on the face of the specification, without reference to common general knowledge. Pfizer contended that it was not enough that the specification simply avers to prior art to make it “known” in the relevant sense for testing lack of manner of new manufacture by reason of an “analogous use”. Rather, so Pfizer submitted, the specification must expressly or implicitly describe the prior art as well-known or common general knowledge. After referring to relevant authorities (discussed below) senior counsel for Pfizer submitted that the fundamental question is whether or not, on the face of the specification, “it is admitted that the [US] 214 patent was common general knowledge that is well-known”. The answer, so Pfizer submitted, was “no”.
448 Pfizer submitted that, on the face of the specification, it cannot be said that the Patent claims a new use of a “known” substance for which its “known” properties make it suitable (ie ICU sedation) in the sense required by the authorities. Pfizer contended that the authorities make clear that “known” in this context (ie on the face of the specification) means “an old and well-known substance” or “well-known and well-understood things”, or common general knowledge. Pfizer drew attention to the absence of any admission in the specification of the Patent that dexmedetomidine or its properties were relevantly “known” and to the fact that the Patent specifically denied that this information was known in the requisite sense.
449 In any event, Pfizer submitted that even if it were accepted (contrary to Pfizer’s case) that the information in US 214 was relevantly “known” (ie that dexmedetomidine was a “known substance” with “known properties”) it made no difference that the specification incorporated US 214 with regard to whether the invention claimed in the Patent was “new”. Pfizer contended that there were a number of reasons for this, including that, in contrast with the invention claimed in the Patent, which is directed to the specific use of dexmedetomidine for ICU sedation, US 214 gives no more than a broad general disclosure that dexmedetomidine “can be expected to be of value, e.g., as a sedative-analgesic, anxiolytic or anti-hypertensive agent”.
450 Senior counsel for Pfizer submitted, moreover, that, notwithstanding the disclosure in US 214 regarding the haemodynamic effects of dexmedetomidine and the indication in the Patent that such haemodynamic effects might be unsuitable for patients in an ICU, the Patent disclosed the surprising result that the drug can be used safely for ICU sedation. He submitted that “that is enough of an invention”.
451 Further, and in any event, Pfizer submitted that the new therapeutic use claimed in the Patent is not the same as, or analogous to, the use disclosed in US 214 (as opposed to the claims, which Pfizer said are not relevant to assessing analogous use) which is early experimental use in rodents, not humans, to investigate the properties of the newly discovered compound, dexmedetomidine. Pfizer emphasised that it was not seeking to disavow the claims of US 214 by simply focussing on the animal studies disclosed in US 214, as InterPharma had submitted. Nor was it contending that there is no patentable invention in US 214. Pfizer accepted that valid claims in patents can be made, and often by necessity are made, on the basis of animal studies. Pfizer submitted, however, that the focus should be on the prior use in US 214 because the traditional principles of “analogous use” upon which InterPharma relied did not extend to mere descriptions. In this connection, Pfizer referred to Pope Appliance Co v Spanish River Pulp and Paper Mills Ltd (1929) 46 RPC 23 (Pope Appliance), and to the discussion at 56 in that case of Morgan & Co v Windover & Co (1890) 7 RPC 131 (Morgan v Windover) and Harwood v Great Northern Railway Co (1865) 11 HLC 654. Pfizer submitted that it was not permissible to focus on the claims and disavow the limited use actually disclosed in US 214, as InterPharma had apparently done.
452 With respect to the dependent claims, Pfizer submitted that InterPharma could not rely on matters outside of the specification, which Pfizer submitted it did in suggesting that the dose claims in the Patent can be determined by a “standard and non-inventive process” and by alleged common general knowledge. The disclosure of an “effective dose” was not, so Pfizer submitted, sufficient to deprive the dependent dose claims of an invention. Further, Pfizer denied that the plasma concentration claims were an example of limiting claims by reference to a series of parameters not mentioned in the prior art.
Discussion
453 As I am about to explain, I accept, as Pfizer submitted, that the Patent did not claim anything which, on the face of the specification, was not a proper subject of letters patent according to accepted principles, and there was no absence of a manner of new manufacture.
454 It may be accepted that Microcell, NRDC and Merck v Arrow support the proposition that there will be no “manner of new manufacture” and therefore no patentable invention where all that is disclosed on the face of the specification is a new use for an old substance if its known properties make it suitable for that use. This is, of course, the proposition on which InterPharma’s case depends at this point.
455 In Microcell, the Deputy Commissioner of Patents had refused to accept Microcell’s patent application for a self-propelled rocket projector comprising a tube of synthetic resinous plastic material reinforced with mineral fibres. The application was rejected on the basis that it was not for an invention, because what was shown was no more than putting a known material to a new use in making a known article for which the material was suitable because of its known properties. The High Court effectively upheld this decision, stating at 246-247 and 249 that:
The specification in the present case does not, in our opinion, disclose a patentable invention. It seems to us to fall within a class of case to which reference is made in Terrell on Patents, 8th ed. (1934) pp. 213, 214 … . The passage reads: “As has already been stated, the Comptroller has jurisdiction, both on application and opposition, to consider whether the invention is a ‘manner of new manufacture’ within the meaning of s. 93. In the exercise of this jurisdiction, applications have invariably been refused where the invention is merely for a new use of ‘an old and well-known substance by itself, without saying more’, and in such cases it is not enough for the applicant to show that he has produced a vendible article”.
…
Many valid patents are for new uses of old things. But it is not an inventive idea for which a monopoly can be claimed to take a substance which is known and used for the making of various articles, and make out of it an article for which its known properties make it suitable, although it has not in fact been used to make that article before.
456 The Court in Microcell concluded at 250-251:
Here the specification does not on its face disclose more than a new use of a particular known product. To use Lord Buckmaster’s words, no new product is obtained, and there is no new method of manufacture suggested or an old one improved. Tubular self-propelled-rocket projectors were at the relevant time well-known articles of manufacture. Synthetic resinous plastics reinforced with mineral fibres, and in particular polyester plastics reinforced with glass or asbestos fibres, were well-known materials. These things are to be gathered from the specification itself, which contains no suggestion of novelty in relation to the article to be manufactured or the material to be used. It further appears from matter published in Australia as early as 1946 that the reinforced plastic materials referred to in the specification had been used in the manufacture of a wide variety of articles. The properties of those materials were known generally, and in particular it was well known that they possessed that combination of great strength and lightness wherein, according to the specification itself, lies their virtue for the purpose in hand. … In these circumstances we do not think it can be said, merely because it does not seem previously to have occurred to anyone to make a rocket projector out of reinforced plastic, that any inventive idea is disclosed by the specification. It follows that this appeal must be allowed.
…
We have in truth nothing but a claim for the use of a known material in the manufacture of known articles for the purpose of which its known properties make that material suitable. A claim for nothing more than that cannot be subject matter for a patent, and the position cannot be affected either by the fact that nobody thought of doing the thing before, or by the fact that, when somebody did think of doing it, it was found to be a good thing to do.
(Emphasis added)
457 The proposition that there will be no patentable invention where all that is disclosed is a new use for an old substance was further explored in NRDC (decided in the same year as Microcell). In NRDC, the High Court referred as it did in Microcell, to the words of Lord Buckmaster in Re BA’s Application (1915) 32 RPC 348 at 349, in describing an alleged invention as “nothing but a claim for a new use of an old substance” and observed at 262:
But, as the Microcell Case emphasizes, it must always be remembered how much is wrapped up in the “nothing but”. Lord Buckmaster did not use the words without explanation:— “…when once a substance is known,” he said, “its methods of production ascertained, its characteristics and its constituents well defined, you cannot patent the use of that for a purpose which was hitherto unknown”. And why? Because in the postulated state of knowledge the new purpose is no more than analogous to the purposes for which the utility of the substance is already known, and therefore your suggestion of the new purpose lacks the quality of inventiveness … . Unless invention is found in some new method of using the material or some new adaptation of it so as to serve the new purpose, no valid patent can be granted … . If, however, the new use that is proposed consists in taking advantage of a hitherto unknown or unsuspected property of the material, the situation is not that to which Lord Buckmaster’s language refers. In that case there may be invention in the suggestion that the substance may be used to serve the new purpose; and then, provided that a practical method of so using it is disclosed and that the process comes within the concept of patent law ultimately traceable to the use in the Statute of Monopolies of the words “manner of manufacture”, all the elements of a patentable invention are present: see the Microcell Case.
(Emphasis added; citations omitted)
458 Prior to Merck v Arrow, the High Court discussed the need for a manner of new manufacture to support patentability in N V Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1995) 183 CLR 655 (Phillips v Mirabella). Brennan, Deane and Toohey JJ held at 663 that the effect of the opening words of s 18(1) of the Patents Act “is that the primary or threshold requirement of a ‘patentable invention’ is that it be an ‘invention’”, which meant, so their Honours said, “‘an alleged invention’, that is to say, an ‘alleged’ ‘manner of new manufacture’ the subject of letters patent and grant of privilege within s 6 of the Statute of Monopolies’”. They continued at 663-664:
[T]hat threshold requirement of “an alleged invention” will, notwithstanding an assertion of “newness”, remain unsatisfied if it is apparent on the face of the relevant specification that the subject matter of the claim is, by reason of absence of the necessary quality of inventiveness, not a manner of new manufacture for the purposes of the Statute of Monopolies. That does not mean that the threshold requirement of “an alleged invention” corresponds with or renders otiose the more specific requirements of novelty and inventive step (when compare with the prior art base) contained in s 18(1)(b). It simply means that, if it is apparent on the face of the specification that the quality of inventiveness necessary for there to be a proper subject of letters patent under the Statute of Monopolies is absent, one need go no further.
(Emphasis added)
459 As already indicated, InterPharma placed particular reliance on Merck v Arrow in support of its no “manner of new manufacture” contentions. At issue in Merck v Arrow was the validity of a patent, with a priority date of 2 September 1999. The patent concerned a method of preventing osteoporosis, which comprised orally administering alendronate on a weekly basis. Before the priority date, it was “known” that: abnormal bone resorption was associated with osteoporosis; bisphosphonates inhibit abnormal bone resorption associated with osteoporosis; alendronate is a potent bisphosphonate; alendronate had been approved for sale and was on sale for the treatment of osteoporosis in the US since October 1995 and in Australia since October 1996. The patent claimed a method of treatment of osteoporosis with alendronate (an old, well-known, compound already approved for sale and on sale to treat osteoporosis) with the sole asserted novelty being that the drug was taken weekly. The patent incorporated numerous publications and some patents by reference into the body of the specification, including two patents (referred to in the Court’s reasons for judgment as “Strein” and “Goodship”) that disclosed weekly administration of alendronate in animals to minimise bone loss. (The relevant disclosures and preferred embodiments of the invention in one of these patents were based solely on experiments in rats: Merck v Arrow at [43]; see the discussion below.) The Court held at [66] that, by including the Strein and Goodship patents in the specification, the patentee “invited … comparison” between the disclosures of the patent in suit and those of Strein and Goodship when assessing whether, on the face of the specification, the claimed invention has the necessary element of claimed manner of new manufacture.
460 In concluding that, on the face of the specification, the claimed invention lacked this requisite element, the Court adopted and applied (at [67]-[75]) the observations of the House of Lords in Morgan v Windover (per Lord Halsbury LC at 134 and Lord Herschell at 137-138) that there is no invention in the application of well-known and well-understood things to an analogous use. The Court in Merck v Arrow at [75] concluded:
At the most, it might be said, in the words of the Lord Chancellor in [Morgan v] Windover, that Merck, by applying well-known and well-understood things to an analogous case, achieved advantages not previously thought of or practised. In the words of Lord Herschell in the same case, Merck saw or realised advantages inherent in an existing substance and practice. However this may be too generous, given Strein and Goodship. The claims in the Patent are analogous to the use of alendronate as taught in those documents. The Patent specification discloses no new substance, no new characteristic of a known substance, no new use and no new method. There is, therefore, no manner of new manufacture.
461 Also in Merck v Arrow, the Court affirmed at [63] that Microcell, NRDC and Philips v Mirabella established the following propositions:
1. The opening words of s 18(1) (“a patentable invention is an invention that”) impose a threshold requirement that the “patentable invention” be an “invention”, that is to say an “alleged” “manner of new manufacture” within s 6 of the Statute of Monopolies (Philips at 663).
2. That requirement will not be met if, on the face of the specification, the subject matter:
(a) lacks the necessary quality of inventiveness under the Statute of Monopolies (Phillips at 664);
(b) is not new (NRDC at 262, Philips at 664).
3. A new use of an old substance is not an invention if its known properties make it suitable for that use – in such a case the new purpose is “no more than analogous to the purposes for which the utility of the substance is already known” (NRDC at 262).
4. But there will be an invention if the new use consists in taking advantage of a hitherto unknown or unsuspected property of the substance (NRDC at 262).
462 It should be borne in mind, however, that it is also well-accepted that there is a relevant distinction between an unknown use of a known thing, and a use that is either known or analogous to what is known: see Merck v Arrow at [75] and Apotex v Sanofi-Aventis HCA at [235] (Crennan and Kiefel JJ). Jagot J applied this distinction in making her decision as the primary judge in Sanofi-Aventis v Apotex (No 3), when her Honour held at [241] that a reference in the patent in suit to an earlier patent disclosing the anti-inflammatory properties of leflunomide could not be equated to a disclosure on the face of the specification that leflunomide treated psoriatic arthritis or psoriasis. Her Honour explained at [242]:
The qualities of leflunomide (or its character), on the face of the patent in suit, also could not be described as “known” in the sense that term is used in this context. It is true that the patent in suit discloses leflunomide as being anti-inflammatory, but that general description cannot be said to exhaustively define the actions and thus the characteristics or qualities of leflunomide. This is consistent with the position disclosed by the objective evidence available at that time.
Jagot J’s judgment was upheld on appeal: see Apotex v Sanofi-Aventis FCAFC, especially at [195]-[196] (Bennett and Yates JJ).
463 The nature of the “manner of manufacture” objection must also be borne in mind: see, for example, Otsuka Pharmaceutical v Generic Health (No 4) at [369] (Yates J). In Dura-Post (Aust) Pty Ltd v Delnorth Pty Ltd [2009] FCAFC 81; 81 IPR 480; 177 FCR 239 at [31], Kenny and Stone JJ (with whom Perram J agreed) accepted that:
Microcell stands for “a narrow proposition that a Commissioner of Patents, or his or her delegate, may refuse an application for patent protection where a specification ‘on its face’ shows the invention claimed is not a manner of new manufacture”: see Lockwood 235 CLR 173 at [106] per Gummow, Hayne, Callinan, Heydon and Crennan JJ; Mirabella 183 CLR at 663-664 per Brennan, Deane and Toohey JJ; and Merck 154 FCR at 51-53 per Heerey, Kiefel and Dowsett JJ. As their Honours there said in Mirabella, “if it is apparent on the face of the specification that the quality of inventiveness necessary for there to be a proper subject of letters patent under the Statute of Monopolies is absent, one need go no further”.
464 This is not the same as impugning the validity of a patent on the ground of lack of novelty or of inventive step. Inquiry as to the manner of (new) manufacture is anterior, rather than subsequent: see D’Arcy v Myriad Genetics Inc [2015] HCA 35; 258 CLR 334 (D’Arcy v Myriad) at [12], [18]-[37] (French CJ, Kiefel, Bell and Keane JJ). Thus, as the Court said in Merck v Arrow at [63], an objection to patentability will be made out where it is apparent on the face of the specification that the subject matter of the claim is not a manner of new manufacture for the purposes of s 6 of the Statute of Monopolies, by reason of the absence of the necessary quality of inventiveness. In Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) [2007] HCA 21; 235 CLR 173 (Lockwood (No 2)) Gummow, Hayne, Callinan, Heydon and Crennan JJ held at [106] that Microcell stood for the proposition “that a Commissioner of Patents, or his or her delegate, may refuse an application for patent protection where a specification ‘on its face’ shows the invention claimed is not a manner of new manufacture”. Their Honours added that Microcell did “not involve a separate ground of invalidity or a discrete ‘threshold’ test”.
465 A conclusion that the subject matter of the claimed invention is not a manner of new manufacture is reached without reference to common general knowledge outside that disclosed in the specification (although matters of common general knowledge extrinsic to the specification are sometimes mentioned as being confirmatory of the conclusion reached by reference to the face of the specification). For instance, in D’Arcy v Myriad at [12], it was said by French CJ, Kiefel, Bell and Keane JJ that the definition of “invention”:
… allows for exclusion from the class of “invention”, and therefore from the class of “patentable invention” anything which is not, on the face of the specification, a proper subject of letters patent according to traditional principles. That anterior exclusion may be based upon an admission, on the face of the specification, which makes clear that the invention claimed is not novel or does not involve an inventive step.
(Citations omitted)
466 In Advanced Building Systems Pty Ltd v Ramset Fasteners (Aust) Pty Ltd [1998] HCA 19; 194 CLR 171, Brennan CJ, Gaudron, McHugh and Gummow JJ said at [39] that:
In Philips, the appellant failed in its attempt to establish that although a claimed use was nothing but a new use of an old substance this could still be a proper subject of letters patent under the 1990 Act where this character of the claimed use was apparent on the face of the specification. Rather, Brennan, Deane and Toohey JJ decided that “if it is apparent on the face of the specification that the quality of inventiveness necessary for there to be a proper subject of letters patent under the Statute of Monopolies is absent, one need go no further”. It was unnecessary to adduce evidence of the prior art base and to compare the invention claimed with the prior art base for the purposes of s 18(1)(b) if the absence of inventiveness appeared on the face of the specification. …
(Citations omitted)
467 In Bristol-Myers, Black CJ and Lehane J referred to Microcell at 250 (see [456] above) before stating at [29]:
That passage makes it quite clear that the lack of inventive step appeared on the face of the specification. It makes it clear also that the conclusion that there was no inventive step was reinforced by a consideration of material earlier published in Australia, information in which was “well-known” and “known generally”. … [T]he substance of the Court’s finding was that what was apparent on the face of the specification was reinforced by proof that particular information had passed into common general knowledge, in the relevant field, in Australia.
The majority of the Full Court followed these principles in AstraZeneca FCAFC: see [385], [392].
468 As noted above, InterPharma relied on the fact that the Patent in suit had incorporated US 214 by reference in support of its submission that the patentee had invited a comparison between what was disclosed in US 214 and what was claimed in the Patent, and that such comparison revealed an absence of a patentable invention.
469 I turn first to the Patent in suit. Under the heading, “Background of the invention”, the specification states that α2-adrenoceptor agonists “are being evaluated in general anaesthetic practice because of their sympatholytic, sedative, anaesthetic, and hemodynamic stabilizing effects”. As at the priority date of the Patent, on the evidence before the Court, I accept that the skilled addressee would have understood that the reference to “α2-adrenoceptor agonists” was essentially a reference to clonidine since no other α2-adrenoceptor agonists were commonly known in the field.
470 The specification discloses, moreover, that “Tryba et al. suggest that clonidine may be useful in ICU patients with withdrawal symptoms” and that they “briefly mention the use of clonidine for ICU sedation.” It continues:
According to Tryba et al., clonidine has its limitations in sedating critically ill patients mainly because of its unpredictable hemodynamic effects, i.e., bradycardia and hypotension, so that it must be titrated for each individual patient. Long term treatment of critically ill patients with clonidine has been reported to be associated with such rebound effects as tachycardia and hypertension.
… Only clonidine has been evaluated for use in ICU sedation, and then only in conjunction with opioids, benzodiazepines, ketamine, and neuroleptics. Further, administration of clonidine as essentially the sole active agent or the sole active agent to a patient in the ICU to achieve sedation has not been disclosed to the best of the applicants’ knowledge.
Shortly after the reference to clonidine, the specification states that dexmedetomidine is described in US 214 as “an α2-receptor agonist for general sedation/analgesia and the treatment of hypertension or anxiety.” The Patent also states that US 214 (and another US patent) discuss parenteral, intravenous, and oral ways of administration”.
471 The Patent in suit specifically provides that “[t]he US Patents discussed herein are specifically incorporated by reference in their entirety”; and that:
The discussion of the background to the invention herein is included to explain the context of the invention. This is not to be taken as an admission that any of the material referred to was published, known or part of the common general knowledge in Australia as at the priority date of any of the claims.
This was followed by the statement that it has been “unexpectedly found that dexmedetomidine … is an ideal sedative agent to be administered to a patient in the ICU to achieve patient comfort”.
472 The disclosure in US 214 is described at [355]-[373] above. As indicated at [355] above, it may be accepted that the disclosure in US 214 concerns the discovery and characterisation of the new compound, dexmedetomidine.
Not use of a “known” substance for which its “known” properties make it suitable
473 US 214 (incorporated by the Patent in suit) states that dexmedetomidine was a new compound (col 1, line 23). There is no admission in the specification of the Patent that dexmedetomidine or its properties were “known” in any relevant sense in Australia immediately before the priority date. To conclude otherwise would be to disregard the clear language of the Patent, which specifically states that nothing in the discussion of the background to the invention is to be taken as an admission that any of the referenced material (including US 214) was “known or part of the common general knowledge in Australia as at the priority date”. The specification of the Patent in suit states, moreover, that only clonidine was mentioned by Tryba et al, in discussing the usefulness of α2-agonists, where it was suggested that clonidine (and no other α2-agonists) may be useful in ICU patients with withdrawal symptoms.
474 As already noted at [440] above, InterPharma relied on the fact that the statement in the specification as to there being no admission was not in the provisional patent applications, but was added by amendment during prosecution. Mr Cordiner QC, for Pfizer, said in response:
There’s a couple of answers to that. First of all, the provisionals didn’t say that this information was common general knowledge. … The second point, even if that statement was made, it was before the grant of the patent. It’s not said that there was anything misleading about … page 4(a). The final point is, as can be seen, page 4(a) talks about common general knowledge in Australia. It’s hardly surprising that provisionals which were filed in the US didn’t include a specific statement of what was common general knowledge in Australia, and the patentee, quite properly, identified the position in Australia. And, your Honour, we don’t withdraw from the proposition that the US 214 was not common general knowledge.
475 I accept Mr Cordiner’s submissions in this regard. Critically, for present purposes, the Patent (at page 4a) relevantly states, in effect, that there is no admission that the material was “common general knowledge in Australia as at the priority date” (emphasis added), and as the provisionals were filed in the US, there is nothing untoward about a statement about common general knowledge in Australia before the grant of the Patent.
476 There is no admission on the face of the specification which makes clear that the invention claimed is not novel or does not involve an inventive step. As Mr Cordiner said, notwithstanding the disclosure in US 214 regarding the haemodynamic effects of dexmedetomidine and the indication in the Patent that such haemodynamic effects might be unsuitable for patients in an ICU, the Patent disclosed that it “has been unexpectedly found that dexmedetomidine … is an ideal sedative agent to be administered to a patient in the ICU”. Of itself, contrary to InterPharma’s submissions, the face of the specification indicates an invention.
477 Further, this is not a case in which it can be said that the specification discloses merely a new use for a known substance, for which its known properties make it suitable. I accept that, as Pfizer submitted, something (here, dexmedetomidine) is not “known” in the Microcell sense just because it has been published and referred to. Microcell and the other authorities mentioned above in this connection show that “known” in this context means “an old and well-known substance” or “well-known and well-understood things” or common general knowledge.
478 In so far as relevant, the evidence supports this outcome. The evidence established that at the priority date dexmedetomidine was not commercially available and had no established clinical use. Further, before 1 April 1998, most intensive care specialists would have had little, if any, knowledge of dexmedetomidine.
479 Furthermore, even if I were wrong about what it means to be “known” in the present context, and it were found that dexmedetomidine was, relevantly, a “known” substance with “known” properties, the incorporation of US 214 would not lead to the conclusion that the specification of the Patent in suit showed that the invention claimed is not a manner of new manufacture. As noted already, US 214 makes no more than the general disclosure that dexmedetomidine “can be expected to be of value, e.g., as a sedative-analgesic, anxiolytic or anti-hypertensive agent” (see col 2, lines 28-29). As already noted, it claims the use of dexmedetomidine for sedation/analgesia or treatment of anxiety or hypertension. It gives no more than a “broad indicative” range of therapeutic uses, which are, in any event, dependent on there being an “effective amount” of dexmedetomidine “which yield the desired activity without causing adverse side effects”: (col 5, lines 1-3); compare AstraZeneca FCAFC at [390]. In contrast, the Patent in suit is directed to the specific therapeutic use of dexmedetomidine for ICU sedation.
480 The person skilled in the art would understand, on reading the whole of the specification of US 214 and using it as the sole body of information, that the “known” “clear anti-hypertensive and bradycardia effects” reported in US 214 might make dexmedetomidine entirely unsuitable for a “new use” in ICU sedation and that further experimentation would be required. This was confirmed by the medical experts’ agreement, as stated in the Joint Report, that the properties of dexmedetomidine described in US 214 did not demonstrate that it was suitable for use as an ICU sedative. This was also consistent with the evidence of Professor Bellomo and Assoc Professor French referred to at [369]-[370] above.
481 It may also be noted that Professor Hall acknowledged that:
Based on the data in US 214, researchers were not in a position to know whether, when administered in effective amounts in humans, dexmedetomidine would have significant effects on blood pressure and heart rate.
He said elsewhere that:
The main research issue for dexmedetomidine during the 1990s was whether the drug would prove to be heavier on sedation and lighter on hemodynamic effects, or vice versa.
Not an analogous use
482 Significantly too, the new therapeutic use claimed in the Patent in suit is not the same as, or even analogous to, the use disclosed in US 214 (as opposed to the claims, which, I accept, are not relevant to an assessment of analogous use). The only use disclosed in US 214 is an experimental use in rodents, to investigate the properties of the newly discovered compound, dexmedetomidine.
483 The principles with respect to analogous use were discussed in Pope Appliance. The patent in suit in that case related to a device to be used in connection with a paper-making machine. The respondents relied on a number of prior patents as paper anticipations, although there was no evidence that the machines described in the prior patents had ever been made or proved to work successfully. The Privy Council held at 55 that no anticipation was proved.
484 Noting, however, that the judgments below were not based on anticipation, their Lordships went on to consider the submission that there was no invention, holding that the trial judge had misdirected himself, in looking to “[t]he apparent distinction between the cited prior art [patents] and Pope” and purporting to apply the doctrine of analogous use derived from Morgan v Windover. The Privy Council said at 56 that:
[I]t is clear that, in their Lordships’ opinion, the learned trial Judge misdirected himself. He arrived at the opinion that the invention was old by making a mosaic of other and prior descriptions. He also, in their Lordships’ opinion, took quite an erroneous view as to an analogous user. Analogous user is what its name denotes, something which has to do with user. He has applied the doctrine not to things used, but to things described. But as to things only described, there must either be anticipation or not. And anticipation must be judged by the canons already mentioned. Does the man attacking the problem find what he wants as a solution in the prior so-called anticipations? The distinction between anticipation by prior description and by prior user is well understood. The doctrine of analogous user only applies to cases as to things in actual use.
485 They explained (also at 56) that:
The leading case is the fishplate case, Harwood v Great Northern Railway Co ((1865) 11 HLC 654). That dealt with the question of whether there could be a good patent for a fishplate on a railway where the same fishplate had been used on a bridge. Mr Justice Blackburn … states the problem thus at page 667: “In every case arises a question of fact whether the contrivance before in use was so similar to that which the Patentee claims that there is no invention in the difference.” The contrivance, be it observed, must be a contrivance in use, not one merely described. Then there was the case of Morgan v Windover, the C-spring case ((1890) 7 RPC 131). Throughout the judgment analogous user is only applied to a known thing. In the words of Lord Halsbury, at page 134, “The application of well-known things to an analogous use is not the proper subject for a patent.”
Upon the whole matter their Lordships find that the judgements below cannot be supported. To do so would be to deprive the Patentee of the fruits of what has been found a very practical and very useful invention.
486 Pope Appliance establishes that analogous use is to be assessed by reference to things in prior use, as opposed to things that have merely been described. On this basis, it cannot be said the new therapeutic use claimed in the Patent in suit is analogous to the use disclosed in US 214, let alone the same as that use.
487 Valid claims can, of course, be made on the basis of animal studies; and Pfizer correctly emphasised that it was not seeking to disavow the claims of US 214 by focussing on the animal studies disclosed in US 214 in the context of assessing analogous use. In accepting Pfizer’s submissions with respect to analogous use, it clearly does not follow that there was no patentable invention in US 214 (contrary to InterPharma’s submission).
488 The subject of the Patent in suit in the present case is entirely different from the subject of the patent in Merck v Arrow and the synthetic resinous plastic self-propelled-rocket projector the subject of the patent in Microcell. It was clear in Merck v Arrow that alendronate and its therapeutic use inhibiting abnormal bone resorption associated with osteoporosis were “known” in the relevant sense: see [459] above. It will be recalled that alendronate had been registered on the ARTG and on sale in Australia for the treatment of osteoporosis for a number of years prior to the priority date of the patent in suit in that case. The animal studies disclosed in the specification (by reference to the Strein and Goodship patents) were to the same therapeutic use (prevention of bone loss) and the same treatment regimen (weekly administration) the subject of the patent in that case. This led the Full Court to say at [62]:
Strein teaches weekly administration of effective amounts of alendronate for the treatment of osteoporosis, an advantage being reduced GI side effects. By analogy to the treatment of osteoporosis, Goodship teaches weekly administration of alendronate in therapeutically effective amounts for treatment of prosthesis loosening and prosthesis migration. The range of dosages includes those identified in the Patent claims. All that can be said of the Patent, as against Strein, is that the former does not teach rest periods. As against Goodship, the Patent additionally teaches use of alendronate for the treatment of osteoporosis (which the Goodship application assumes) and expressly identifies the advantages of weekly, over daily, dosing.
It was evident therefore that the patent in Merck v Arrow disclosed “no new substance, no new characteristic of a known substance, no new use and no new method”: see at [75].
489 The present case, which is evidently distinguishable from Merck v Arrow, must be decided on its own facts and circumstances.
Dependent claims
490 As already stated, any issue as to the absence of manner of new manufacture must be decided by reference to matters within the specification of the Patent in suit. InterPharma impermissibly sought to rely on matters outside the specification in submitting that the dose claims in the Patent could be determined by a person skilled in the art by applying a “standard and non-inventive process”. InterPharma’s submission at this point shows that these matters were not to be found on the face of the specification. Further, it does not seem to me that the disclosure of an “effective dose” in US 214 can deprive the dependent dose claims in the Patent in suit of an invention: compare AstraZeneca FCAFC at [391].
491 A new form of administration of a known compound may constitute a manner of new manufacture: Actavis UK v Merck at [31]. Pfizer submitted that InterPharma impermissibly sought to challenge the dependent claims as lacking inventive subject matter by “filling gaps” by reference to matters that were not disclosed on the face of the specification of the Patent and suggesting that the plasma concentration claims were a form of “parameteritis”. I accept that, as Pfizer contended, this kind of analysis involves a misapplication of the limited nature of any anterior “threshold” test in Microcell: compare, Bristol-Myers at [45]. Further, the literature to which I am about to turn (see [493] below) describes a targeted plasma concentration of dexmedetomidine. It cannot therefore be said to be an example of parameteritis, that is limiting claims by reference to a series of parameters not mentioned in the prior art: see Otsuka FCAFC at [115]. As described in the Patent, targeting a particular plasma concentration is a surrogate for specifying the dose range related to the desired sedative effect to be achieved with dexmedetomidine.
Conclusion
492 For the foregoing reasons, I reject the submission that the use of dexmedetomidine claimed in the Patent was a use of an old substance where its known properties made it suitable for that use. The Patent in suit did not claim anything which, on the face of the specification, was not a proper subject of letters patent according to traditional principles.
Inventive step
493 InterPharma contended that the invention so far as claimed in any of the claims of the Patent lacked an inventive step over either:
(a) Belleville/Bloor; or
(b) Talke.
494 Section 18(1)(b)(ii) of the Patents Act provided that an invention is a patentable invention if the invention, when compared with the prior art base as it existed before the priority date, involves an inventive step.
495 Section 7, as in force at the applicable time, relevantly provided:
…
(2) For the purposes of this Act, an invention is to be taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed in the patent area before the priority date of the relevant claim, whether that knowledge is considered separately or together with either of the kinds of information mentioned in subsection (3), each of which must be considered separately.
(3) For the purposes of subsection (2), the kinds of information are:
(a) prior art information made publicly available in a single document or through doing a single act; and
(b) prior art information made publicly available in 2 or more related documents, or through doing 2 or more related acts, if the relationship between the documents or acts is such that a person skilled in the relevant art in the patent area would treat them as a single source of that information;
being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant to work in the relevant art in the patent area.
The “patent area” was Australia. The term “prior art information” was defined in Sch 1 to the Patents Act, by reference to the “prior art base”.
496 The term “prior art base” was defined in Sch 1 relevantly as:
(i) information in a document, being a document publicly available anywhere in the patent area; and
(ii) information made publicly available through doing an act anywhere in the patent area; and
(iii) where the invention is the subject of a standard patent or an application for a standard patent—information in a document publicly available outside the patent area;
…
497 Section 7(2) of the Patents Act required the party challenging validity to establish that there was no inventive step. As French CJ said in AstraZeneca HCA at [18], s 7(2) would “defeat a claim for want of inventive step unless one of the alternative conditions set out in s 7(2), read with s 7(3), was satisfied”. These conditions were said (also at [18]) to involve the following elements:
1 An hypothetical person skilled in the relevant art.
2. The person being, therefore, notionally possessed of the common general knowledge as it existed in the relevant area before the priority date of the impugned claim.
3. The invention being obvious to that person in the light of the common general knowledge.
4. Alternatively, that person being provided with prior art information made publicly available in a single document or through doing a single act, or made publicly available in two or more related documents or through doing two or more related acts if the relationship between them satisfied the requirement of s 7(3)(b).
5. That prior art information, as defined by s 7(3), being information that the person could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant to work in the relevant art in the patent area (the relevance requirement). “Ascertained”, in this context, means “discovered or found out”. “Understood” means that, having discovered the information, the person would have “comprehended it” or “appreciated its meaning or import”.
6. The invention being obvious to the person in the light of the common general knowledge considered together with either of the classes of prior art information defined in s 7(3).
(Citations omitted)
498 In Aktiebolaget Hässle v Alphapharm Pty Ltd [2002] HCA 59; 212 CLR 411 (AB Hässle) at [34], Gleeson CJ, Gaudron, Gummow and Hayne JJ cited General Tire at 497 for the proposition that “obvious” in the context of an inventive step inquiry means “very plain”, although their Honours made it clear at [36] that this statement should not be accepted at face value. Thus, they went on to say (also at [36]) that “‘obvious’ does not stand by itself in the statute to specify a ground of revocation; the reader is required to ‘have regard’ to what was ‘known or used’ on or before a particular date, and to a particular geographical area”.
499 Further, and helpfully, French CJ observed in AstraZeneca HCA at [15] with respect to obviousness that:
Relevant content was given to the term “obvious” by Aickin J in Wellcome Foundation Ltd [v VR Laboratories (Aust) Pty Ltd (1981) 148 CLR 262 at 286], posing as the test:
“whether the hypothetical addressee faced with the same problem would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not.”
The idea of steps taken “as a matter of routine” did not, as was pointed out in AB Hässle, include “a course of action which was complex and detailed, as well as laborious, with a good deal of trial and error, with dead ends and the retracing of steps”. The question posed in AB Hässle was whether, in relation to a particular patent, putative experiments, leading from the relevant prior art base to the invention as claimed, are part of the inventive step claimed or are “of a routine character” to be tried “as a matter of course”. That way of approaching the matter was said to have an affinity with the question posed by Graham J in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd [[1970] RPC 157 at 187-188]. The question, stripped of references specific to the case before Graham J, can be framed as follows:
“Would the notional research group at the relevant date, in all the circumstances, which include a knowledge of all the relevant prior art and of the facts of the nature and success of [the existing compound], directly be led as a matter of course to try [the claimed inventive step] in the expectation that it might well produce a useful alternative to or better drug than [the existing compound]?”
That question does not import, as a criterion of obviousness, that the inventive step claimed would be perceived by the hypothetical addressee as “worth a try” or “obvious to try”. As was said in AB Hässle, the adoption of a criterion of validity expressed in those terms begs the question presented by the statute.
(Some citations omitted)
500 The Full Court in Generic Health Pty Ltd v Bayer Pharma Aktiengesellschaft [2014] FCAFC 73; 222 FCR 336; 106 IPR 381 at [71] expressed a similar view, when they said:
We do not think that the plurality in [AB Hässle] were saying that the reformulated Cripps question was the test to be applied in every case. Rather, it is a formulation of the test which will be of assistance in cases, particularly those of a similar nature to [AB Hässle]. The plurality did not reject as an alternative expression of the test the question whether experiments were of a routine character to be tried as a matter of course (Wellcome Foundation Ltd v VR Laboratories (Aust) Pty Ltd (1981) 148 CLR 262, at 280-281, 286, per Aickin J). We do not think there is a divide here in terms of whether an expectation of success is relevant between a test which refers to routine steps to be tried as a matter of course and the reformulated Cripps question. It is difficult to think of a case where an expectation that an experiment might well succeed is not implicit in the characterisation of steps as routine and to be tried as a matter of course. On the other hand, we think a test formulated in terms of worthwhile to try was firmly rejected by the High Court in [AB Hässle] (see also Pfizer [Overseas Pharmaceuticals v Eli Lilly & Company (2005) 225 ALR 416] at 476, [287], per French and Lindgren JJ). The fact (if it be the fact) that the position in the United States may have shifted does not affect the binding nature of what the plurality said in [AB Hässle].
501 The relationship between obviousness and inventive step was addressed in Lockwood (No 2), where the High Court said at [52]:
[A]s a basic premise, obviousness and inventiveness are antitheses and the question is always “is the step taken over the prior art an ‘obvious step’ or ‘an inventive step’”? An inventive step is often an issue “borne out by the evidence of the experts”. There is no distinction between obviousness and a lack of inventive step. A “scintilla of invention” remains sufficient in Australian law to support the validity of a patent. In R D Werner Lockhart J stated that there must be “some difficulty overcome, some barrier crossed”. This is consonant with older authorities in the United Kingdom which recognised that some inventiveness was required to distinguish patentable advances over the prior art from advances which “any fool” could devise. It also accords with the requirement in the United States that for an invention to be “non-obvious” it must be “beyond the skill of the calling”.
(Citations omitted)
502 The concept of common general knowledge is, plainly enough, crucial, since the skilled addressee is taken to be armed with the common general knowledge for the purpose of an inquiry of the kind contemplated in s 7(2) of the Patents Act: see AstraZeneca FCAFC at [524].
503 In Minnesota Mining at 292, Aickin J said, in a well-known passage, that:
The notion of common general knowledge itself involves the use of that which is known or used by those in the relevant trade. It forms the background knowledge and experience which is available to all in the trade in considering the making of new products, or the making of improvements in old, and it must be treated as being used by an individual as a general body of knowledge.
504 In order to be properly characterised as “common general knowledge” in the relevant field, there must be evidence that information had been “assimilated” into common general knowledge: see AB Hässle at [57]. It is not permissible to attribute to the notional skilled addressee, as a starting point, knowledge of the problem addressed by the Patent unless the problem is itself common general knowledge, or if knowledge of the problem is information of a kind described in s 7(3) of the Patents Act: AstraZeneca FCAFC at [202]-[203].
505 Before a prior art document can be used for the purpose of s 7(2) of the Patents Act, it must meet the requirements of s 7(3). The prior art information referred to in s 7(3) is information which is not part of the common general knowledge: AstraZeneca HCA at [68] (Kiefel J). Such information can only be taken into account if the skilled addressee could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded it as relevant to work in the art: AstraZeneca HCA at [68] (Kiefel J), citing Lockwood (No 2) at [132], [152]. In Lockwood (No 2), the High Court said at [149]:
The exercise, of which s 7(3) is an integral part, is the exercise of determining whether “an invention” (s 7(2)) as disclosed “in any claim” (s 18(1)) “involve[s] an inventive step when compared with the prior art base” (s 7(2)). The “prior art base” for s 7(2) is enlarged by s 7(3), so as to go beyond common general knowledge and to bring into consideration “prior art information” which “could ... be reasonably expected to have [been] ascertained, understood and regarded as relevant to work in the relevant art” (s 7(3)) by “a person skilled in the relevant art” (s 7(2)). This brings to mind Lord Reid’s reference to a “diligent searcher” in Technograph Printed Circuits Ltd v Mills & Rockley (Electronics) Ltd and suggests a person skilled in the relevant art familiar with some, but not necessarily every piece of, publicly available information in the relevant art beyond common general knowledge.
(Citations omitted)
506 In Commissioner of Patents v Emperor Sports Pty Ltd [2006] FCAFC 26; 67 IPR 488; 149 FCR 386 at [31] a Full Court of this Court explained:
Section 7(3) does not assume an ascertainability by any and all skilled persons, of whatever description, of all publicly available prior art documents anywhere in the world. Nor does it assume that the skilled person has found the document in question, so that the only question is whether he or she has understood it and regarded it as relevant. Such a construction ignores the elements of expectation and reasonableness, as applied to the particular skilled person.
507 “Ascertained” in this context means “discovered or found out” and “understood” means that, having discovered the information, the person would have “comprehended it” or “appreciated its meaning or import”: AstraZeneca HCA at [18] (French CJ), [68] (Kiefel J).
508 Information is only relevant for the purpose of s 7(3) (as it stood at the relevant time) if it can be regarded as relevant to work in the relevant art. The idea of being relevant to work in the relevant art is directed to publicly available information “which the skilled person could be expected to have regarded as relevant to solving a particular problem, or meeting a long-felt want or need, as the patentee claims to have done”: AstraZeneca HCA at [68] (Kiefel J). In Lockwood (No 2) at [153], the High Court observed:
The question of what a person skilled in the relevant art would regard as relevant, when faced with the same problem as the patentee, is to be determined on the evidence. The starting point is the subject matter of the invention to be considered together with evidence in respect of prior art, common general knowledge, the way in which the invention is an advance in the art, and any related matters. It should be mentioned that the starting point is not necessarily the inventive step as claimed, or even agreed between parties, because the evidence, particularly in respect of a combination of integers, may support a different inventive step.
509 Further, as the Full Court in Ajinomoto Co Inc v NutraSweet Australia Pty Ltd [2008] FCAFC 34; 76 IPR 1; 166 FCR 530 at [61] observed:
In relation to the question of construction, Ajinomoto contends that the language in s 7(3) should be given its ordinary and natural meaning. When the section refers to work, it means real work, and not work in the abstract. Accordingly, NutraSweet was required to demonstrate that such work was actually undertaken.
The Court agreed, holding at [119] that “[t]he expression ‘relevant to work in the relevant art in [Australia]’ should, in our view, be given its ordinary and natural meaning”.
510 As to the factual environment the skilled person is notionally to be placed when asking whether he or she “could reasonably be expected” to do the things referred to, Jessup J said in AstraZeneca FCAFC at [523]:
It was submitted on [the appellants’] behalf that an important question arises with respect to the construction of s 7(3), namely, in what factual environment is the skilled person notionally to be placed when one enquires whether he or she “could reasonably be expected” to do the things referred to? That environment must, it seems to me, be limited to the common general knowledge. The subsection permits an extension to the common general knowledge only when certain conditions are satisfied. In determining whether those conditions are satisfied in a particular case, it would be circular, and contrary to the scheme of the provision, notionally to provide the skilled person with access to information which was not part of the common general knowledge.
I see no reason to disagree. This is, so it seems to me, consistent with the approach taken by the High Court in AstraZeneca HCA: see at [35]-[39] (French CJ), [59], [75]-[78] (Kiefel J), [103]-[111] (Gageler and Keane JJ), [120] (Nettle J). As Gageler and Keane JJ stated (at [113]):
The inquiry contemplated by s 7(2) is whether the invention would have been obvious to a person skilled in the art in the light of the common general knowledge alone, or “together with” a single piece of prior art information ascertained pursuant to s 7(3). What s 7(2) requires, by the plain and ordinary meaning of its language, is that where multiple pieces of prior art information are available and capable of being regarded as relevant, each must be considered, one at a time, together with the common general knowledge to answer the question whether the invention is obvious. If the invention is obvious in light of the common general knowledge plus any one of the ascertained pieces of prior art information, then the patent is invalid for want of an inventive step.
Hypothetical skilled addressee (revisited)
511 I have already discussed the concept of the hypothetical skilled addressee (see [77]-[85] above). As already stated, in this case the hypothetical skilled addressee to whom the Patent was addressed is an intensive care specialist engaged in clinical practice and some research in the field of intensive care medicine in Australia immediately prior to the priority date. Such a person would have the requisite knowledge of the state of the “art and manufacture” at the relevant time and a practical interest in the subject matter of the invention.
Common general knowledge
512 Professor Bellomo’s evidence was, and I accept, that before April 1998 in Australia all intensive care specialists were required to be knowledgeable and skilled in the administration and management of sedation for ICU patients. There was no dispute about this.
513 There was also general agreement about the nature and purpose of ICU sedation. As already indicated, the medical experts agreed in their Joint Report that:
ICU sedation is the administration of medication to ICU patients in circumstances where it is necessary so that the patient can tolerate and accept intensive care treatments and environment, for example, the presence of an endotracheal tube to enable mechanical ventilation.
514 As already explained at [154] above, the medical experts agreed that, as at April 1998 (like today), sedative agents were primarily administered intravenously by continuous infusion in an ICU or operating theatre, although a bolus might first be used if the rapid onset of sedation were desired. The amount of sedatives administered per patient is much higher in those settings (and especially in an ICU) than in a general ward. They agreed that a patient being continuously infused with a sedative needs to be continuously monitored, as his or her dosage may need to be titrated.
Alpha-2-agonists
515 All three medical experts agreed that as at April 1998 the class of drug known as alpha-2 agonists was generally known by intensive care specialists, although clonidine was the only α2-agonist available, in the sense of regulator-approved, before April 1998 for use in humans.
516 The generally known properties of α2-agonists were that they affected the sympathetic nervous system, lowered blood pressure and heart rate, and had some sedative effects. Professors Hall and Bellomo agreed that it was probably not generally known that, with drugs of this kind, heart rate and blood pressure could go up at first before going down.
517 Professor Bellomo’s evidence was, and I accept, that:
[I]t was also known that each agent within [the α2-agonist] family would have its own pharmacokinetic and pharmacodynamic profile and potentially give rise to a very different therapeutic effect when administered (eg have different effects on sedation, the sympathetic nervous system, blood pressure and heart rate). Accordingly, it was not (and is still not) possible to make reliable assumptions of the clinical effects of a new Alpha-2 agonist based on knowledge of existing Alpha-2 agonists.
518 Assoc Professor French gave evidence, which I accept, that he was aware from his anaesthesia training in the early 1990s that alpha-2 receptors were complex, and that while alpha-2 agonists as a class typically had common properties, individual members of the class might give rise to very different clinical results when administered to humans because of different pharmacodynamics and pharmacokinetic properties.
519 It may be accepted that the skilled addressee’s knowledge of α2-agonists as at the priority date typically related to clonidine; that the skilled addressee was aware that α2-agonists typically result in lowering blood pressure and/or slowing heart rate, with some sedative effect; and that α2-agonists had different pharmacodynamics and pharmacokinetic properties from one another, which meant that they might have very different clinical results when administered to humans.
520 Professor Bellomo’s evidence, which I accept, was that at the priority date clonidine was generally known to have anti-hypertensive properties and, although it typically did not have a sedative effect at the standard dose used to treat hypertension, if administered at a high dose, it could have sedative effects. His evidence was that the use of clonidine in ICUs before April 1998 was limited; and it was typically used was to treat delirium tremens in alcohol withdrawal patients because it addressed their high blood pressure and heart rate, and their agitation, in one medication. He said that, in this circumstance, clonidine was administered with at least a benzodiazepine. Professor Hall and Assoc Professor French gave evidence to the same effect.
521 Professor Bellomo also gave evidence that:
Most ICU patients who require ICU sedation have low blood pressure. Generally, it was not, and still is not, possible to use clonidine at a dose that has sedative effects in [the] ICU patient as the necessary dose of clonidine to achieve sedative effects will typically cause the patient’s blood pressure to drop to a dangerously low level. …
… At 1 April 1998 and today I would not consider administering clonidine as an ICU sedative to a patient who is intubated and mechanically ventilated as, like most ICU patients, such patients generally have low blood pressure and the administration of clonidine at the dose required to achieve ICU sedation so that the patient can tolerate an endotracheal tube would cause the patient’s blood pressure to drop to a dangerously low level.
Assoc Professor French gave evidence to like effect.
522 Professor Hall’s evidence was also that clonidine had anti-hypertensive effects, with some sedative qualities. He said that “it was also understood, based on the experience of clonidine, that the role [of α2-agonists in sedation] was limited”. Professor Hall agreed that the potentially dangerous effect on blood pressure and heart rate at the requisite dose to achieve sedative effects made it unsuitable for use in ICU sedation. In this regard, Professor Hall gave the following evidence in cross-examination:
MR CORDINER: Did you … think [clonidine] was not suitable because it had a potentially dangerous effect on blood pressure and heart rate at the dose that would be necessary to have a relevant sedative effect in the ICU?
PROF HALL: That would be one reason. Another reason, equally as important, is that it’s very long-acting once it’s given by either a bolus or a – and certainly by a continuous infusion, so you would stop the drug and still have effects for a long time.
MR CORDINER: And I think you also say at paragraph 51 that the drug was not a potent sedative, that is, clonidine was not a potent sedative?
PROF HALL: In that balance between haemodynamic effect and sedation there was often a greater relative haemodynamic effect than a sedative effect.
MR CORDINER: And clonidine had been around for a couple of decades clinically by 1998?
PROF HALL: Yes.
MR CORDINER: And so those matters were well-known in the field of ---?
PROF HALL: Yes.
MR CORDINER: ICUs?
PROF HALL: Yes.
Both Professors Hall and Bellomo also gave evidence that the sedative effects of clonidine were generally considered to be unwanted side effects of the drug.
523 The evidence of Professor Bellomo and Assoc Professor French was that at the priority date clonidine was available intravenously in Australia (although, according to Professor Hall, not in the US) and it was administered as a single dose, rather than as a continuous infusion. Their evidence disclosed that it is not desirable to administer clonidine as a continuous intravenous infusion, which is the way sedatives are generally administered in the ICU, because of its long half-life.
524 It is clear that the skilled addressee would not have considered clonidine to have been a promising drug for use in ICU sedation at the priority date.
Dexmedetomidine
525 At the priority date, dexmedetomidine was not commercially available and had no established clinical uses.
526 All three medical experts agreed that, immediately before 1 April 1998, most intensive care specialists would have had little if any knowledge of the alpha-2 agonist dexmedetomidine. Dexmedetomidine was not part of the common general knowledge as at the priority date.
527 Professor Bellomo’s evidence was that he became aware of dexmedetomidine in the course of a review of the anaesthetic literature in the preparation for the 1997 Review Article concerning the potential use of alpha-2 agonists, in renal dysfunction. This literature review disclosed a study in Jalonen 1997 on the effects of dexmedetomidine used as an anaesthetic adjunct during cardiac surgery.
528 Assoc Professor French agreed that he came across the Jalonen 1997 study and its discussion of dexmedetomidine in the course of his literature review for the 1997 Review Article. He added, however, that:
I guess my knowledge of dexmedetomidine was a little earlier than that in that when I was preparing for some specialist post-graduate exams I became aware of the broad class of alpha 2 agonist as you would, but also their role in veterinary anaesthesia as well, and it was in that context that I recall reading briefly about dexmedetomidine, but it wasn’t an area of priority learning for me because it was such a new and novel agent at that time, so I really knew very little about that drug.
529 In contrast with Professor Bellomo and Assoc Professor French, Professor Hall’s evidence was that he became aware of and specifically began following the literature concerning dexmedetomidine in the early 1990s, because he had a particular interest in ICU sedation. His evidence was that he “would have come across dexmedetomidine by virtue of it being referenced as an alpha agonist”, with known sedative properties, but that he did not believe that “it would have been known to someone without that particular interest or … who was not tracking the literature”. In cross-examination concerning his literature searches, he stated that he “agreed that many intensivists might never have heard of the medication as a sedative at that time”.
530 As already indicated, there is no evidence that information about the existence and/or properties of dexmedetomidine was part of the common general knowledge immediately before the priority date. The existence of information about the drug in the literature is not sufficient to make this common general knowledge. It is well established that information does not constitute common general knowledge merely because it might be found in a journal or by a literature search: see British Acoustic Films Ltd v Nettlefold Productions (1936) 53 RPC 221 at 250 (Luxmoore J). I accept that, as Pfizer submitted, there was no evidence in this case that the skilled addressee had dexmedetomidine and its properties “as the furniture of his or her mind at the priority date”.
Deep sedation
531 The experts agreed in their Joint Report that, as at 1 April 1998, there was “a variable spectrum on the appropriate level of sedation depending on the patient’s circumstances and response to the drugs”. As indicated at [156] above, Professors Hall and Bellomo, in particular, explained this idea in similar terms, agreeing that the target level of sedation for an ICU patient would be likely to vary in the course of treatment. Assoc Professor French substantially agreed.
532 The medical experts agreed in their Joint Report that deep sedation could be defined as a Ramsay Sedation Score of 4, 5 or 6, but they disagreed about the nature of the intensive care specialists’ opinion at the priority date about the use of deep sedation. In answer to the question, “[a]t the priority date did you perceive issues with this level of sedation?”, Professor Hall responded in the Joint Report:
Yes. Deep sedation should be restricted to a subset of patients and avoided in others.
In contrast, Professor Bellomo replied:
No. In Australia at the priority date no specific issues related to deep sedation were considered.
Assoc Professor French agreed with Professor Bellomo.
533 Professor Bellomo’s evidence was that, as at 1 April 1998, the aim of intensive care specialists, including himself, in sedating ICU patients was “to ensure their safety and comfort”, and it was thought that “the best way to achieve this for the vast majority of patients was through deep sedation”. Professor Bellomo said that at that date, deep sedation was typically achieved through the use of midazolam and propofol.
534 Professor Bellomo also gave evidence that deep sedation was not recognised as a problem before April 1998 and “[a]s at 1 April 1998, [he] did not consider that any change was required to the practice of deep sedation in the ICU”. His evidence was that it was not until the 2000s that he and other intensivists began to change their opinion about deep sedation. He said: “[s]tarting in the early 2000s, there was a cultural change in the ICU field towards a belief … that lighter sedation may be preferable to deep sedation for ICU patients”.
535 Assoc Professor French’s evidence supported that of Professor Bellomo. His evidence was that:
[A]s of now, … in Australian intensive care practice, we use, on average a lighter level of sedation and we target sedation using sedation scores. That was not our practice in 1998. Indeed, the practice in 1998 was simply to tell the nursing staff to sedate the patient with M & M, morphine and midazolam, and then they just went away and did it, which is now recognised as perhaps not being the optimal way of sedating patients. But certainly in the institutions I worked in in 1998, that was pretty much what happened.
536 Like Professor Bellomo, he said that “the goal of ICU sedation has always been the same: to keep the patient comfortable and safe”. His evidence was that:
Historically (and in 1998), on average patients were sedated to a much greater level than they are today. This was because at that time sedation was thought to be a relatively benign procedure that did not affect patient outcomes and that deep sedation was the best way to ensure that the patient remained comfortable and safe.
537 Assoc Professor French said that around the year 2000 he became aware of the Kress 2000 article, which, in his own words, was a “landmark study from the US that adopted an approach known as a ‘sedation vacation’”. His evidence was that:
The main effect of the Kress [2000] paper in Australia was to highlight that there were adverse effects associated with deep sedation in the ICU and so while it is important to ensure that the patient was calm and sedated enough to tolerate ICU procedures, deep sedation can give rise to detrimental effects.
He explained that a lighter level of sedation has become more common in the last 15 years.
538 Disagreeing with Professor Hall about intensive care specialists’ view of deep sedation, Professor Bellomo said in concurrent session:
I think many people in Australia … took a different view at that time. And the view was: why would you want to be awake through this? When the time comes, we will let you wake up. And so I think it’s important to realise there … are no randomised control trials that prove that one strategy is better than another strategy. And all the shift in the way in which we think about sedation, which Professor French has highlighted, has come through indirect evidence and thinking and reasoning and observation, but was certainly not established in Australia at that time and is still not yet convincingly proven, if that makes sense.
…
I think there have been lots of studies associating lighter sedation with better patient outcomes … and that has been influential. But, of course, association is not causation. Nonetheless, they have been influential, they have been published. They have alerted people to the potential risks of taking the view that I have described before, which is put them to sleep and wake them up when it’s time. Whether the science of medicine will prove that to be the case in due course remains uncertain, but all of these associated investigations have proven very influential.
539 In cross-examination Professor Bellomo was taken to Tuxen 1998. Referring to this article, Ms Rofe QC asked Professor Bellomo if he agreed with the statement that “[a] cooperative patient should be relaxed, comfortable and awake during the day and asleep, but easily awaken, at night.” Professor Bellomo replied:
Well, it depends on the patient, because I think, under many circumstances, this recipe for the optimal world is unsafe, so I think, in some patients, it’s very reasonable and I would think it should be applied, but in my view, in the majority of ICU patients that we were treating at the time, in the ICU, it had significant risks and may not be appropriate.
540 In any event, Professor Bellomo stated that he understood from Professor Tuxen’s statements about “uncooperative or agitated patients” that Professor Tuxen in effect agreed with him about the need for deep sedation. That is, Professor Bellomo responded that Professor Tuxen’s goal was “a cooperative patient and, if he can’t get that goal, then he goes to deep sedation. … It’s all a matter of circumstances, patient and disease”.
541 Professor Bellomo’s evidence about professional attitudes to deep sedation up to the 2000s was not undermined in cross-examination, and he did not “retreat” from the proposition that the practice of deep sedation was widespread in Australia before April 1998, as InterPharma at one point contended. Professor Bellomo made it clear that he disagreed with Professor Tuxen to the extent that Professor Tuxen expressed a contrary view.
542 Professor Tuxen’s views were not put to Assoc Professor French, and InterPharma did not call Professor Tuxen as a witness (or indicate that he was unavailable to be called). Further, there is no evidence that Professor Tuxen’s paper led intensivists in Australia to re-evaluate their approach to deep sedation or that it was itself part of such a re-evaluation
543 Professor Hall disagreed with Professor Bellomo and Assoc Professor French with respect to this issue, saying that deep sedation was recognised by him and other intensivists (presumably in the US) as a problem before April 1998, and that it was seen as preferable to be able to interact with patients in the ICU to the extent possible in each particular case. Professor Hall referred to the studies described in Kress 1996, a paper that he co-authored, amongst other studies. He noted that this paper identified a “sweet spot” of sedation, in which patients were lightly sedated, being “quiet and restful … but able to be interacted with on demand”. Professor Hall acknowledged that the problem was how to achieve this level of sedation, particularly with the sedative agents commonly used before April 1998. He said in evidence:
I believe, throughout the 90s, the concept of having lighter sedation and a patient that would come out of sedation faster was always there. And the ability to achieve it was the problem. I think there was a gap between that which you wished for and that which you could actually achieve with the tools that you had, because if you gave midazolam to a patient, it might very well accumulate and your best intentions would be thwarted and the nurses might have difficulty precisely titrating to this point … . So there’s certainly a gap between how well we could do it. But the concept that this would be the best way to treat the patient, I don’t think, was much of an a-ha moment at any point in the 90s. I think people would say that would be great if we could do it.
Professor Hall also noted that the disadvantages of propofol were well-known before 1 April 1998, in particular, that although it was much more titratable than benzodiazepines, such as midazolam, it was difficult to administer in an amount “to hit a sedation ‘sweet spot’”.
544 Professor Hall’s evidence that “throughout the 90s, the concept of having lighter sedation … was always there” was to some extent undermined in cross-examination when he was taken to his own evidence on 7 March 2012, before the US District Court in Hospira, Inc v Sandoz International GmbH. In that proceeding, Professor Hall agreed that in 1995 in the US “many patients in the [ICU] were being sedated into these states where there was no interaction with them” and that the Petty editorial in 1998 calling for lighter sedation where possible “was a voice crying out for change”. The following exchange also took place in that proceeding:
Q. And 10 years later, you wrote about that and said we still need to think about this; right?
A Or move even further, since we had made a great deal of change between ’98 and beyond.
Q. So from ’98 to 2010, some change had taken place, and you wrote in 2010 about moving even further forward; right?
A. Correct.
545 In the present proceedings, Mr Cordiner QC, for Pfizer, asked Professor Hall whether that was still correct. Professor Hall replied: “Yes. … [A]t that point, we had published a paper where we had patients out of bed, sitting in chairs and walking.”
546 The effect of Professor Hall’s evidence at this point was that he considered that the observations in the Petty editorial in August 1998 led to the change described by Professor Hall in Hall 2010. This would indicate that the “cultural change” that led intensive care specialists to prefer lighter sedation for ICU patients over deeper sedation where possible post-dated 1998 and the priority date of 1 April 1998.
547 In any event, to the extent that Professor Hall’s evidence was that he held a concern about the level of sedation of ICU patients prior to 1998, in my view, that reflect the fact that his level of knowledge and interest in ICU sedation was significantly greater than that of the skilled addressee, as Professor Hall acknowledged.
548 The uncontested evidence was that the primary goal in sedating an ICU patient was to keep the patient comfortable and safe. It was also common ground that the level of sedation for any particular patient might vary over that patient’s time within the ICU, as well as from patient to patient within the ICU (depending on the patient’s condition and the specialist’s therapeutic intent). Professor Bellomo’s reference to a “cultural change” with respect to sedation in the ICU referred to a change in overall approach to sedation in this context, as a consequence of which intensive care specialists replaced their predilection for deep sedation with a preference for lighter sedation where that was appropriate to achieve the primary goal of keeping the patient comfortable and safe. In substance, the principal point of disagreement between Professor Bellomo and Assoc Professor French on the one hand and Professor Hall on the other was about when this cultural change occurred. As seen above, Professor Bellomo and Assoc Professor French gave evidence that the change started to occur in Australia in the early 2000s, whilst Professor Hall maintained that the change was occurring in the 1990s and, particularly, before the priority date.
549 There are a number of considerations that lead me to accept that the cultural change to which Professor Bellomo referred had not occurred in Australia before 1 April 1998. Whilst all three witnesses were knowledgeable about, and experienced in, the practice of intensive care medicine in their own countries, as at the priority date, only Professor Bellomo had extensive knowledge and experience of intensive care medicine in hospitals in Australia. He was an impressive witness, whose evidence on this issue I accept. It may also be borne in mind that, as at April 1998, Assoc Professor French was a recently qualified specialist in Australia in intensive care and anaesthesia, whose evidence was (and I accept) that, as at the priority date, his breadth of knowledge was the greatest it ever was in his career. His evidence about his knowledge and experience of ICU sedation prior to April 1998 supported the evidence of Professor Bellomo. Furthermore, Assoc Professor French recalled reading the Kress 2000 paper after its publication, describing it as a landmark study. It may be inferred from this that he understood that the Kress 2000 study made a significant contribution to the generally accepted view of intensive care specialists in Australia about the desirability of prolonged deep sedation. Assoc Professor French’s evidence therefore strongly supported Professor Bellomo’s evidence.
550 In contrast with Professor Bellomo’s and Assoc Professor French’s evidence, Professor Hall’s knowledge and experience related to the practice of intensive care medicine in the US, not Australia. In addition, as already noted, Professor Hall had a particular interest in sedation in the ICU dating from the early to mid-1990s: see, for example, Kress 1996 and Pohlman 1994. It is possible that Professor Hall has placed the change at a little earlier than Professor Bellomo and Assoc Professor French because, as a researcher in the area of sedation and as a co-author of Pohlman 1994, Kress 1996 and Kress 2000, he in fact had a relevantly earlier awareness of the information conveyed in those papers and an earlier knowledge of the association between deep sedation and adverse patient outcomes than the skilled addressee would have had.
551 It should also be noted, as discussed above, that Professor Hall’s evidence that the shift away from deep sedation occurred prior to 1 April 1998 was undermined in cross-examination by reference to his earlier evidence in US District Court proceedings in 2012.
552 There is also a possibility that a shift away from deep sedation occurred earlier in hospitals in the US than in Australia, bearing in mind that the hospital systems and practice of intensive care medicine in the US and Australia differed in some ways and, indeed, the studies on which Kress 1996 and Kress 2000 were based were in the US.
553 I accept therefore that the shift away from deep sedation in intensive care medicine in Australia, based on intensivists’ appreciation that deep sedation was associated with adverse effects on patient outcomes took place, or started to take place, in ICUs in Australia in the early 2000s. It was at this time that specialists in Australia re-examined their general approach to the sedation of ICU patients, with the result that they came to view deep sedation less favourably than before and a lighter level of sedation became more prevalent.
554 I accept that the skilled addressee in Australia at the priority date considered that deep sedation was typically the best way to achieve safety and comfort for the majority of ICU patients. In this context, it was therefore important that a sedative agent used in the ICU could achieve deep sedation, as this was the most common level of sedation as at the priority date. Furthermore, at the priority date, the skilled addressee did not perceive there to be any problem with this level of sedation.
ICU sedative agents
555 It may be accepted that the effect of a drug on an ICU patient’s vital functions is particularly significant where, as is typically the case, he or she has (or is at risk of) organ failure and/or unstable vital functions (see [146] above). This was clear from Professor Bellomo’s evidence, which I accept, that ICU patients will not always respond to medications in the same way as healthy patients, because the excessive physical and psychological stress they are likely suffering can affect the way their bodies respond to drugs. He further deposed that:
[A]s almost all ICU patients have unstable vital functions the effect that a drug has on an ICU patient’s vital functions is particularly significant. If a given drug has a substantial effect on a patient’s vital functions (eg creates very low blood pressure) then that drug may not be appropriate for administration to ICU patients.
Both Professor Bellomo and Assoc Professor French also observed that, as ICU patients require multiple medications, administered through various routes, drug-to-drug interactions can occur.
556 Professor Hall’s evidence in cross-examination was consistent in some respects with the understanding of Professor Bellomo and Assoc Professor French in the above regard. In cross-examination, Professor Hall agreed that “part of the ideal sedative would be that the side effects would be completely minimal, because patients in the intensive care unit have shock and unstable blood pressures and unstable heart rates”. Also in cross-examination, Professor Hall gave evidence to similar effect in the following exchange with Mr Cordiner QC:
MR CORDINER: [I]s that the case also – just thinking about why these people are in an ICU, it’s because their vital functions are variable … and they’re at risk of organ failure or have organ failure; that’s correct, isn’t it?
PROF HALL: Yes.
MR CORDINER: Yes. So even though they’re in an environment where you’re watching them all the time, it’s best, in an ideal sedative at least, not to mess around with their haemodynamics?
PROF HALL: It would be best. So you’re balancing all the properties that we have listed here. If you manage to hit every one of them perfectly, that would be ideal. It’s a high bar.
557 I accept that particularly where a drug has a risk of substantial effect on a patient’s vital functions, then the skilled addressee would not expect that the drug would be appropriate for administration to ICU patients.
558 It was uncontested that, as at 1 April 1998, there were frequent changes in drug selection and dosage in respect of ICU patients, as intensive care specialists sought to respond to changing clinical situations. The sedation of ICU patients often involved the use of a combination of drugs. The evidence established that at the priority date dosing of existing sedatives was a matter of clinical judgment and pharmacological understanding, and it involved titrating, adjusting, combining, modifying, ceasing, and modulating all the agents virtually all the time.
559 The medical experts agreed in their Joint Report that, as at 1 April 1998, the agents commonly used for sedation included propofol, narcotics (such as morphine and fentanyl) and benzodiazepines (such as midazolam). Other classes of drugs were used less frequently, including ketamine, butyrophenones and clonidine. The medical experts also agreed that “[s]ide effects are well recognised with all ICU sedative agents. These are managed by careful administration and choice of agent.”
560 While the association of prolonged deep sedation of patients with longer term adverse clinical outcomes was not widely appreciated until after April 1998, the experts agreed that, before the priority date, there were known limitations to the drugs available for ICU sedation. An intensive care specialist with knowledge and experience of these limitations would manage these potential problems in ICU patients by careful dose administration and sometimes the use of other drugs, for example, to provide blood pressure support. As Assoc Professor French put it in cross-examination, sedation in the ICU “was a common procedure used, [and] it had recognised problems. However, … any long term complications attributable to the sedative agents were considered to be remote”. In answer to the question “[s]o it could be useful to have another sedative to be added to your armoury to deal with patients who had trouble with the existing sedatives?”, he said:
[A]s I referred to in my affidavit, I think there were problems with all sedatives as of 1 April 1988, and as of today, and therefore a new agent that offers particular advantages would be of interest.
561 The evidence of Professor Bellomo and Assoc Professor French (which I accept) was that midazolam was the sedative “agent of choice” in ICUs in Australia from the mid-1980s until the early 1990s, and that it was used in conjunction with morphine for analgesia. Further, I accept Professor Bellomo’s evidence that any association between benzodiazepines (such as midazolam) and agitation or the development of delirium was not known at the priority date.
562 The evidence of Professor Bellomo and Assoc Professor French (which I accept) was that by 1 April 1998, propofol was also being used as a sedative agent in Australian ICUs, in conjunction with a narcotic, such as morphine or fentanyl, which Assoc Professor French described as “very powerful analgesics but [which] also have at the lower doses mild sedative effects”. Propofol, which is not a benzodiazepine and belongs to another class of drugs, has profound sedative effects but minimal anxiolytic and analgesic effects. I accept Assoc Professor French’s evidence that a benefit of propofol from an intensive care specialist’s perspective was that it had a predictable context sensitive half-life. Professor Bellomo gave evidence to similar effect, that “[t]he key advantage of propofol was that it was fast acting, and typically did not accumulate in the body so patients would wake up at more predictable times and at a faster rate after the infusion was stopped.”
563 Professor Bellomo’s evidence was that propofol was first introduced as a sedative agent in Australian ICUs in the early 1990s or “a bit later”, although its use at that time was limited. Professor Bellomo said that the use of propofol became slowly more prevalent in the 1990s, but it did not become the preferred agent for ICU sedation until around the early 2000s when concerns had grown about the adverse effects of midazolam and there was greater clinical knowledge and experience about the use of propofol. This was broadly consistent with Assoc Professor French’s evidence that he did not recall using propofol in 1992 when he first started working in intensive care, that he began using it in intensive care in the mid-1990s in a limited way, and that by the time he had finished his training in 1997, “propofol was more commonly used in intensive care [for sedation], but certainly wasn’t the dominant sedative used [in the ICU]”. His evidence was that, by 1998, he was using propofol more regularly, and considered it to be a “very useful ICU sedative”, with a more favourable pharmokinetic profile than midazolam and morphine, although it was still considered expensive.
564 While propofol had known limitations, as at the priority date Assoc Professor French considered it to be a good drug and that its limitations could be managed for ICU patients. The evidence of Assoc Professor French and Professor Bellomo was, and I accept, that the primary limitation of propofol was its effect on lowering blood pressure, but this limitation was overcome by careful dose administration and use of blood pressure support where necessary. It was common ground between the experts that, at the priority date, propofol infusion syndrome was not generally known by intensive care specialists to be a problem in adults. The experts also agreed that the risk of excessive triglyceride levels through the prolonged use of propofol was managed in ICU patients by monitoring triglyceride levels and either lowering the dose or ceasing to administer the drug if these problems became unacceptable. Professor Bellomo’s opinion was, however, that this was not a common problem. Professor Hall agreed that “a clinically catastrophic event” of this kind was unlikely. Assoc Professor French added that:
[W]hile elevated triglycerides are measured in a number of patients, the clinical significance of that is uncertain and it would only be … a very small number of patients in whom you would cease the propofol because of severe hypertriglyceridemia, ie, high levels of triglycerides. The most common group of patients where I would do that would be those with acute pancreatitis, but they aren’t that common anyhow.
Both Professor Bellomo and Assoc Professor French gave evidence that, at the priority date, they considered that propofol would become the preferred agent for ICU sedation, and that they believed that others in the field would have held the same view.
565 Professor Bellomo and Assoc Professor French emphasised that the effects of propofol, like midazolam, were known at the priority date because it had by then been used extensively outside the ICU. Professor Bellomo explained that propofol (and midazolam) began being used in anaesthesia in Australia in the late 1980s and was registered in the ARTG at that time for that purpose. Assoc Professor French gave uncontested evidence that propofol was the most common intravenous induction agent used in general anaesthesia in Australian by 1995. He also said, and I accept, that “the important thing with both propofol and midazolam is that there was a substantial body of clinical use in anaesthesia that occurred prior to their translation into intensive care practice”, and “knowledge from anaesthesia with those drugs moved across with them into the intensive care environment”. His evidence in this regard was consistent with that of Professor Hall. Professor Hall explained that one of the motivations for him conducting a study involving midazolam (published in 1994) was the absence of “any trial of midazolam in [ICU] patients. It had just moved from a comfort in the perioperative anaesthesia setting” to use in an ICU.
566 I accept Professor Bellomo’s unchallenged evidence that it cannot be assumed that a drug that is appropriate for use in non-ICU patients would also be appropriate for ICU patients, or that data generated from non-ICU subjects can be extrapolated to ICU patients. The skilled addressee would have been aware of these matters. Even if a drug has been used in non-ICU subjects, the skilled addressee would exercise additional caution, beyond that which is often exercised in other clinical contexts, before adopting the drug in the ICU.
567 The evidence of the three medical experts showed that there were limited circumstances in which drugs used outside an ICU setting, such as midazolam and propofol, had been translated into the ICU setting and used as ICU sedatives without prior extensive testing in ICU patients. The evidence of Professor Bellomo and Assoc Professor French was that, in these limited circumstances, there had been extensive knowledge and clinical use of the drug outside the ICU before it was even considered for use as an ICU sedative. As already indicated, both Professor Bellomo and Associate Professor French gave evidence that propofol and midazolam were approved on the ARTG in the anaesthesia setting and there was extensive knowledge and experience with the use of those drugs in the anaesthesia setting in Australia before they were accepted as suitable drugs for ICU sedation in Australia.
568 Pfizer submitted that this evidence was not undermined by InterPharma’s attempt to argue that the title of three publications referred to in footnotes 3, 5 and 7 to Kress 1996 suggested there was research published in relation to the use of propofol and midazolam in the ICU in 1987 and later. I accept this submission, bearing in mind that none of the witnesses were taken to the papers to which those footnotes relate; there was no suggestion that the relevant research was undertaken in Australia; and, as Pfizer noted, it is not clear whether propofol and midazolam were registered for use in the relevant jurisdiction before the research was undertaken.
569 Pfizer submitted that where a drug is to be used for the first time in the ICU, greater knowledge of the drug is required than when a drug is used for the first time in non-ICU patients; and that such knowledge is obtained from extensive use in non-ICU patients. As indicated above, this was, in effect, the substance of evidence given by Professor Bellomo and Assoc Professor French in this proceeding (which I accept). I accept that it was not a part of Pfizer’s case that, for a claimed medical use to be obvious, the skilled addressee must expect the drug to achieve regulatory approval. Rather, Pfizer’s point was that, given the vulnerabilities of ICU patients generally, the skilled addressee seeking a new ICU sedative agent would not be led to use a new sedative agent in the ICU for the first time until satisfactory knowledge had been obtained from extensive use in non-ICU patients about whether or not there is a dose at which sedation can be achieved in non-ICU patients without adverse effects. The evidence indicated that this will almost always be the case.
570 Assoc Professor French made this very point clearly in cross-examination in the following exchange:
MS ROFE: But you would need to take the leap at some point to put it into that patient group. And I suggest that you would put it into the ICU patients coming from – like, the post-operative group of ICU patients rather than the septic shock or critically ill ICU patients, but you would need to put it in an ICU patient group to test?
A/PROF FRENCH: … [P]rior to putting it into any intensive care patient group to test, I would wish to see a number of studies evaluating it in less severely ill patients.
MS ROFE: So you would take a group of patients, potentially the coronary artery bypass-type patient?
A/PROF FRENCH: No, no, I would be looking at patients that don’t go to the intensive care unit. I would be looking particularly for information from the anaesthesia literature about how this drug – what properties were observed with this drug during otherwise well patients undergoing anesthesia.
Professor Bellomo gave evidence to much the same effect.
Parties’ submissions
571 InterPharma alleged that, having regard to the common general knowledge and the disclosures in Belleville/Bloor and Talke, the invention so far as claimed in all the claims of the Patent was not a patentable invention within the meaning of ss 138(3)(b) and 18(1)(b)(ii) of the Patents Act, in that the invention lacked an inventive step.
572 InterPharma submitted in closing written submissions that the skilled addressee, carrying out routine literature searches, would almost certainly have included the word “alpha agonist” in any database search. It relied on the fact that using a PubMed search employing that term disclosed Belleville. InterPharma submitted that since Belleville was followed by Bloor in the same journal, the skilled addressee could reasonably be expected to have found that paper as well.
573 Senior counsel for InterPharma augmented these submissions in her closing address. Ms Rofe QC, for InterPharma, submitted that before the priority date the hypothetical skilled addressee, knowing the problems with existing sedative agents for ICU sedation, would have had an impetus to solve the problems. Ms Rofe submitted that the fact that papers, such as Belleville/Bloor and Talke were being published in well-regarded journals such as Anesthesiology indicated that people were interested in solving a problem, being the sedation of patients in the ICU. Referring to the properties of alpha agonists that were known before April 1998, she also contended that, if the skilled addressee was looking for a new or better sedative before the priority date, then the skilled addressee would have considered alpha agonists “a logical place to start”. She submitted that “all [the experts] had an awareness of the existence of dexmedetomidine and that it fell within that class [of alpha agonists]”. She further submitted that the known properties of clonidine “didn’t forestall the entire class having the same properties”, because it was known that different alpha agonists had different properties. Referring to Professor Hall’s evidence, Ms Rofe also submitted that the fact that alpha agonists were known to lower blood pressure was not a problem with those patients who “started off with sky high blood pressure”. Relying on the evidence of Professor Hall as to the searches he conducted for this case at the request of InterPharma’s solicitors, she submitted that the skilled addressee would have commenced with a literature search, and that this search would include the term “alpha agonist”, which would have identified Belleville/Bloor. Alternatively, she said that such a search would have identified dexmedetomidine “as being worth searching”. She submitted that, if “dexmedetomidine” were the search term, then the skilled addressee would find Talke. Ms Rofe submitted that the search conducted by Professor Hall (to which he deposed in his affidavit evidence) was “very similar” to the search carried out by Professor O’Brien as described by French CJ in AstraZeneca HCA at [35].
574 InterPharma submitted in closing that the skilled addressee reading Belleville/Bloor would understand intravenous dexmedetomidine to have dose dependent sedative effects in healthy volunteers at doses that were well tolerated by the volunteers. InterPharma made it clear that it did not suggest that an intensive care specialist reading Belleville/Bloor before April 1998 would have immediately moved to using dexmedetomidine in clinical practice for ICU sedation (even assuming there was no regulatory barrier to doing so). Rather, InterPharma contended that the evidence established that, on the basis of the promising knowledge that the skilled addressee would have obtained on reading Belleville/Bloor, the skilled addressee would have had sufficient confidence to progress along the standard drug development and approval pathway and would as a matter of course have taken the standard steps along that pathway.
575 InterPharma submitted in closing written submissions that although Belleville/Bloor was only reporting on a Phase I trial in healthy volunteers, the successful trials showing a sedative effect in that population would have given the skilled addressee more confidence to proceed with further trials than could be the case, for example, with the results of Phase I trials to treat a disease. InterPharma contended that:
The obvious next step for the hypothetical PSA would have been a dose finding study in a population of patients which would have measured the agent’s sedative, ventilative, and haemodynamic effects in that population. As the drug is administered intravenously, the patients would be sick patients in a hospital. The hypothetical PSA understood that the sedative effect seen in the healthy volunteers would be seen in hospital patients including ICU patients. What remained to be determined was whether that effect would be seen at a dose acceptable to patients in progressively less healthy groups or whether the anticipated side effects would prove unmanageable. The steps that were required to get from the Belleville[/Bloor] Paper to the claimed invention were thus of a routine character to be tried as a matter of course by a hypothetical PSA with an interest in an alternative ICU sedative. It is not inventive to test a drug with established dose dependent sedative properties as a sedative in progressively sicker populations of patients along the way to testing the drug as a sedative in ICU patients.
Such tests would not have involved speculative experimentation, or any ingenuity. There is no invention in conducting tests to establish whether a drug with known dose dependent sedative properties and other side effects in humans can achieve effective sedation in a target population of unhealthy humans without those side effects becoming prohibitive. This is a paradigm example of what routine testing comprises.
576 InterPharma contended in written closing submissions that the skilled addressee would not have needed to cross any further barrier or to carry out any inventive experimentation; and that the question in the light Belleville/Bloor was whether the side effects of administering the agent in ICU patients in doses sufficient to achieve a sedative effect might prove prohibitive, which routine dosing studies in progressively sicker patient groups could establish.
577 Again, Ms Rofe QC elucidated these submissions in closing at trial. She submitted that the skilled addressee reading Belleville/Bloor would understand that intravenous dexmedetomidine had dose dependent sedative effects in healthy volunteers at doses that were well tolerated by the volunteers. She submitted that the skilled addressee would have been led by the results of Belleville/Bloor to follow the standard routine drug approval pathway leading to the claimed invention, including trials in the patient group or groups. Referring to the evidence of Assoc Professor French, Ms Rofe identified the focus of the further trials that he contemplated as investigating whether dexmedetomidine was tolerated without unmanageable side effects, contending that this would be ascertained by routine steps.
578 Ms Rofe QC submitted that the question that the experts were asked to address in their Joint Report at [8.7(b)], in the context of Talke, namely, “[w]ould you have been directly led as a matter of course to try dexmedetomidine as an agent for ICU sedation” was the wrong one. This was because, in her submission, the question “wraps the entire drug development process up into one question” when in fact there are stages, which InterPharma described as “routine steps”, leading to the claimed invention. In this context, she relied on AstraZeneca HCA at [94] and [116].
579 With respect to Talke, InterPharma submitted that the skilled addressee would understand from the title of Talke that dexmedetomidine was being infused in patients around the time of having vascular surgery, and that the authors were tracking the effects on these patients. InterPharma submitted that the skilled addressee would have regarded Talke as relevant because it disclosed an agent that could be an alternative sedative for ICU sedation. Although the skilled addressee would have understood that the primary purpose of the study was to measure the drug in the patients to find out if dexmedetomidine might reduce the risk of heart complications in post-surgical patients, InterPharma submitted that the skilled addressee would also have regard to whether dexmedetomidine might be a useful ICU sedative. InterPharma contended that the lack of clinically observable sedative effects post-operatively in the patients in the Talke study would not have discouraged the skilled addressee from carrying out further research because the skilled addressee would be aware that Talke disclosed that prior research had shown dose-dependent sedation, and would infer that the effects of the drug were reducing over time, or that the amount administered was insufficient to provide sedation. InterPharma contended that the sedative effect observed pre-operatively would have encouraged the skilled addressee; and although it was more common for ICU patients to be sedated to at least a level 4 Ramsay Sedation Score, it would have been seen as appropriate for some ICU patients to have the apparently marginally lesser level of sedation suggested by “asleep but with ready arousability”.
580 In written submissions, InterPharma took issue with the evidence of Professor Bellomo and Assoc Professor French, contending that, contrary to their evidence, the post-operative haemodynamic effects of dexmedetomidine reported in the Talke study were not substantial enough to discourage further studies. Referring to Professor Hall’s evidence, InterPharma contended that there was nothing in the reported post-operative haemodynamic data that would have led a skilled addressee to expect dexmedetomidine not to be safe to use in the ICU. Rather, InterPharma submitted that the skilled addressee with an interest in ICU sedation would have taken routine steps to the claimed invention, including doing a dosing study on the cohort the subject of Talke, or on a lower risk group of surgical patients, starting at the medium to high doses used in Talke and then looking at whether the drug was titratable upwards, to find out how many patients could be effectively sedated and whether the sedation would be adequate for a useful period of time in an ICU. Ms Rofe QC submitted that Professor Hall’s evidence led to the conclusion that, after reading Talke, there would be only routine steps leading to the claimed invention.
581 In this context, InterPharma submitted that:
The real question is whether a [skilled addressee] considering an alternative sedative for use in ICU sedation would have needed to carry out any inventive experimentation in order to administer a sedative agent disclosed in these papers on carefully selected groups of surgical patients, titrating the doses to achieve a desired sedative effect (whether “deep” sedation or otherwise) and measuring the haemodynamic and any other side effects.
582 InterPharma submitted that the answer was “no”, because the disclosures in Talke of dose-dependent sedation and the lack of prohibitive reported haemodynamic effects pointed the skilled addressee in the direction of further research that would have led to the claimed invention. All that remained to be ascertained, so InterPharma said, was whether the drug would be sufficiently safe at levels that proved effective.
583 Pfizer accepted that Belleville/Bloor ought to be treated as a single source of information for the purpose of s 7(3) of the Patents Act. Pfizer did not accept, however, that Belleville/Bloor could be ascertained, understood and regarded as relevant to work in Australia. It submitted that the skilled addressee could not have reasonably been expected to have regarded the early pre-clinical study that it disclosed as relevant to the clinical work of sedating patients in the ICU that was actually being carried out in Australia. Furthermore, it said that the skilled addressee would not even have read Belleville/Bloor because the abstract said nothing about ICU or post-operative sedation; and, in any event, Professor Hall’s far-ranging search did not establish that the skilled addressee could reasonably be expected to have ascertained Belleville/Bloor (or Talke) at the priority date. Pfizer contended that there was no analogy between Professor Hall’s search and the search by Professor O’Brien described in AstraZeneca HCA, as InterPharma had submitted.
584 Pfizer also contended that the skilled addressee would not have ascertained Talke because there was no mention in the title, the abstract or the key words of “ICU”, “post-operative sedation” or “sedation”; and the paper would not have been located by using the words “sedative” or “sedation” in a search of the PubMed database at April 1998. Pfizer also argued that the search process undertaken by Professor Hall in this regard was not that of the diligent researcher described in Lockwood (No 2) or AstraZeneca HCA. Pfizer contended, moreover, that the skilled addressee could not reasonably have been expected to have regarded Talke as relevant to the clinical work of sedating patients in the ICU because it was a small early stage study in 24 patients, no sedative effect was reported post-operatively, and it was unclear whether any of the patients were in the ICU.
585 Even if relevantly ascertained, understood and regarded as relevant, for the purposes of s 7(3) of the Patents Act, Pfizer contended that the skilled addressee reading Belleville/Bloor would not have been directly led as a matter of course to try the invention as claimed in the expectation that it might well produce a useful alternative to, or a better drug than, existing ICU sedatives. It was fanciful to suggest, so Pfizer submitted, that Belleville/Bloor would have directly led the skilled addressee as a matter of course to try dexmedetomidine with such an expectation. Pfizer further submitted that the skilled addressee was not confronted with any particular problem that was part of the common general knowledge or s 7(3) information. Pfizer emphasised that the experts agreed that nothing in Belleville/Bloor indicates that dexmedetomidine would be an appropriate agent for ICU sedation. Rather, so Pfizer contended, there were matters disclosed in Belleville/Bloor that would deter the skilled addressee from considering dexmedetomidine as an ICU sedative. Pfizer also contended that Belleville/Bloor teaches away from the integers of the dependent claims. In summary, Pfizer’s position was that Belleville/Bloor concerned an early clinical study in healthy volunteers; provides no suggestion that dexmedetomidine might be useful as an ICU sedative; and contains results that suggest dexmedetomidine might not be useful for use as an ICU sedative.
586 As regards Talke, Pfizer argued that, even if it satisfied the requirements of s 7(3), the skilled addressee reading Talke would not have been directly led as a matter of course to try the invention as claimed, in the expectation that it might well produce a useful alternative to, or a better drug than, existing ICU sedatives because, among other things, Talke does not disclose any post-operative sedation and suggests that dexmedetomidine may give rise to unpredictable effects on blood pressure and heart rate. Amongst other things, Pfizer argued that, given the results reported in Talke showing no sedative effects post-operatively, the skilled addressee would not have been led directly to try the drug as an ICU sedative.
587 Regarding the dependent claims, Pfizer argued that there was no suggestion that the skilled addressee would have been directly led to try dexmedetomidine as the sole active agent or essentially the sole active agent; Talke does not teach the administration of dexmedetomidine by a loading dose and a maintenance dose; and even the highest dose reported in Talke is outside the doses claimed in claims 11 or 12 of the Patent.
Section 7(3) — ascertained, understood and regarded as relevant
588 Assoc Professor French gave evidence that, at the priority date, he kept up to date by regularly reading anaesthetic journals, intensive care journals and general medical journals, including Anaesthesiology. He deposed:
For the journals I read, I scanned the table of contents to decide which articles would be of interest to me. I would then read the abstracts of the ones of interest and if, on reading the abstracts, the article seemed of particular relevance to my practice, I would read the whole article. I did not typically read articles directed to pre-clinical work or early clinical work, unless those articles related to mechanical ventilation or volatile anaesthetics, as I had a particular interest in these areas. The articles I read were typically reports of later stage clinical work as it was this work that was most relevant to my practice.
589 In cross examination, Assoc Professor French confirmed that, after looking at the index to a journal, he would scan the topics of editorials and the general articles, and if he found one of interest, then he “would first look at the abstract and if the abstract confirmed that it was of interest to [him], then [he] would read the article”. He also confirmed that he did not read reports of pre-clinical work or early clinical work because “they weren’t of particular interest to [him] at that time”. There was no evidence to suggest that this practice of literature review was not typical of intensive care specialists in Australia immediately before the priority date.
590 It may therefore be accepted that the skilled addressee could reasonably be expected to have scanned the topics of the articles and editorials in leading journals in the intensive care field, including Anaesthesiology, and if there was a topic of interest, then the skilled addressee would read the abstract. If the abstract confirmed that the article was of interest to him or her, then the skilled addressee would read the article.
591 Let it be assumed that the skilled addressee could reasonably be expected to have thought that the topics “Effects of intravenous dexmedetomidine in humans: I. Sedation, ventilation, and metabolic rate” (Belleville) and “Effects of intravenous dexmedetomidine in humans: II. Hemodynamic changes” (Bloor) were of potential interest such that the skilled addressee read the relevant abstract. Nonetheless it cannot be concluded that the skilled addressee would have ascertained Belleville/Bloor. This is because the abstract disclosed only early pre-clinical research in a single two-minute infusion of dexmedetomidine in 37 healthy male volunteers. Reading this, the evidence indicated that the skilled addressee would not have gone on to read Belleville/Bloor since this research would not have been thought relevant to the clinical practice of sedating ICU patients by continuous infusion. Furthermore, the key words accompanying the abstract for Belleville/Bloor did not include the terms “ICU”, “post-operative sedation” or “sedation”, the typical search terms that the skilled addressee would have used in looking for research concerning ICU sedation. It can therefore be concluded that the skilled addressee would not have ascertained Belleville/Bloor as the abstract and keywords would not have led the skilled addressee to consider the papers relevant to clinical practice.
592 Talke, which like Belleville/Bloor, was published in Anesthesiology, was in relevantly the same position in this respect as Belleville/Bloor. The title, “Effects of perioperative dexmedetomidine infusion in patients undergoing vascular surgery”, contains no reference to “ICU”, “post-operative sedation” or “sedation”. Nor is there any reference to these topics in the abstract or key words. Further, the article itself contains little more than a passing comment on sedation. Assoc Professor French deposed (and I accept) that had he been conducting a search using the words “ICU”, “post-operative sedation” or “sedation” in April 1998, he would not have expected Talke to appear in his research results.
593 Further, Mr Cruise deposed that had the skilled addressee searched via using the words “sedation”, “sedative” or any other direct variants of the word “sedation” or the search terms “ICU”, “Intensive care” or any direct variant of those terms in PubMed (or the other principal database STN Medline) as at 1 April 1998, Talke would not have been located. He also deposed that it was “highly unlikely” that Talke would appear in the results of a search if a user inputted the search term “sedative”, “sedation” and any other direct variants of the word “sedation” into the “all fields” search field in the PubMed database as at 1 April 1998. Mr Cruise was not called for cross-examination.
594 In view of the evidence referred to above, it cannot be concluded that the skilled addressee would have ascertained Talke before the priority date. It will be recalled that Professor Hall only found Talke by using the search term “dexmedetomidine”.
595 Further, besides there being no reasonable expectation as to the skilled addressee’s ascertainment of Belleville/Bloor, for the reasons set out below, the skilled addressee could not reasonably have been expected to have regarded Belleville/Bloor as relevant (within the meaning of s 7(3) of the Patents Act) to the work of intensive care sedation as it was being carried out in Australia at the priority date. This means that the skilled addressee would not have ascertained Talke by way of Belleville/Bloor and Professor Hall’s further search for dexmedetomidine, being a search that, for the reasons stated below, must be put to one side as having no bearing on the issue of ascertainment before the priority date.
596 Talke was in relevantly the same position. The skilled addressee could not reasonably have been expected to have regarded Talke as relevant to the clinical work of sedating patients in the ICU because it was an early stage study in 24 patients, directed to determining if dexmedetomidine would attenuate the rise in heart rate and blood pressure in patients during and after surgery, which would not have interested the skilled addressee. Furthermore, as discussed below, Talke reported no sedative effect of dexmedetomidine post-operatively, and it is unclear whether any of the patients were in the ICU.
597 InterPharma did not establish that work in the relevant art in Australia at the priority date was directed to the development of new ICU sedative agents (see [82] above). Rather, the evidence of Professor Bellomo and Associate Professor French (which I accept) was that before the priority date the clinical research being done in Australia was focused on issues relating to major organs of the body rather than on other areas such as sedation (see [81] above). The evidence established that, although there were known limitations to the drugs available for ICU sedation before the priority date, intensive care specialists knew how to manage these limitations. The evidence was, moreover, that as at the priority date, these specialists would have seen propofol as becoming the preferred agent for ICU sedation. At most, Assoc Professor French thought that, as at the priority date, a “new agent” would be of interest. Having regard to the evidence before me, I accept his appraisal. As already indicated, I accept that the skilled addressee could not reasonably have been expected to have regarded Belleville/Bloor as relevant to the work of intensive care sedation as carried out in Australia at the priority date.
598 As previously stated, InterPharma relied on the search process undertaken by Professor Hall in support of its submission that the skilled addressee would have commenced with a literature search, and that one of the search terms the skilled addressee would use was “alpha agonist”, which would have identified Belleville/Bloor.
599 Professor Hall deposed to the literature searches that he would have carried out if he had been involved in researching a potential intravenous agent for use in ICU sedation in March 1998 (immediately before the priority date) via the online database, PubMed. He deposed that had he been conducting a literature search using PubMed in March 1998 for intravenous agents for use in ICU sedation, he would have started by entering a range of generic search terms, including “ICU sedation”, “sedation and critical illness”, “alpha agonists and sedation”, “clonidine and sedation”, and also search terms for sedative agents that he knew could be administered intravenously.
600 I interpolate here that Professor Hall further deposed that the only intravenous sedative agent that he was aware of in March 1998 that was not already used in ICUs was dexmedetomidine, which he knew to be a selective alpha-2 agonist having sedative effects. Professor Hall explained, however, that he did not initially use “dexmedetomidine” as a search term, because this could be said to be “unfair hindsight bias”. Professor Hall stated, nonetheless, that “any researcher with an interest in ICU sedation, and indeed physicians generally, would in March 1998 be likely to be familiar with clonidine, and that it is an alpha-agonist with sedative effects” and that he therefore used the search terms “alpha agonists and sedation”.
601 Omitting the search using the term “dexmedetomidine”, the result of Professor Hall’s searches conducted between 31 January 2018 and 26 February 2018 was the identification of in excess of 6,000 papers. A search using the terms “alpha agonists and sedation” on 31 January 2018 located Belleville. (It may be useful to note that the relevant exhibit is confusing because the search terms appear to be “alpha agonist and post-operative” as opposed to “alpha agonist and sedation”, but it was clear from Professor Hall’s evidence that the reference to “and post-operative” was mistaken.) Professor Hall’s evidence was that, having conducted his searches, he identified Belleville as a paper of apparent interest.
602 Professor Hall located Bloor (and Talke) by using the term “dexmedetomidine”. I accept, however, that, given that dexmedetomidine was not part of the common general knowledge at the priority date, the search using that term can be put to one side: see further AstraZeneca FCAFC at [523] (Jessup J), quoted above at [510]. Nonetheless, it may be accepted that a reading of Belleville would have led the skilled addressee to the identification of Bloor.
603 It does not appear to me, however, that the skilled addressee would have taken the route identified by Professor Hall to locate Belleville and, in consequence, Bloor. This is because the skilled addressee’s knowledge about alpha-2 agonists at the priority date would not have led the addressee to use the search terms “alpha agonists and sedation” if carrying out a literature search to find a new ICU sedative. The evidence of Assoc Professor French (which I accept) supported the conclusion that the skilled addressee’s knowledge about alpha-2 agonists at the priority date was limited to clonidine.
604 Having regard to the evidence outlined at [519]-[524] above, clonidine was evidently not considered to be a promising drug for use in ICU sedation at the priority date and has never been approved for such use. Further, I accept that, as Pfizer submitted, the skilled addressee in Australia would have had more knowledge of and even experience in the intravenous administration of clonidine and its effects than Professor Hall because, although it was available for intravenous use in Australia, it was not so available in the US. I accept Pfizer’s submission that the skilled addressee in Australia would not have used the search term “alpha agonist and sedation” if carrying out a literature search to find a new sedative for use in the ICU, having regard to the skilled addressee’s limited knowledge of alpha-2 agonists, and the unpromising knowledge of and experience with clonidine. I therefore accept Pfizer’s submission that, having regard to the knowledge of the skilled addressee in Australia before the priority date, the skilled addressee would not have ascertained Belleville (and, in consequence, Bloor) via such a search. As Pfizer pointed out, neither Assoc Professor French nor Professor Bellomo were asked whether they would conduct a search using that term. There was no evidence to indicate that the skilled addressee would have ascertained Belleville/Bloor in any other way.
605 In any event, as indicated at [522] above, Professor Hall acknowledged that he would not have considered clonidine a promising drug for use in ICU sedation; and, unsurprisingly, his reference to clonidine as a basis for framing his literature search was challenged in cross-examination. He defended his position with the statement that “[i]t has a niche use and it’s from a class of drugs that have a potential as sedatives. … [O]ne would look at the class of alpha agonists if new ones arose that would have better balance of sedative than haemodynamic properties as sedatives”. When pressed about why the skilled addressee, who was unaware of dexmedetomidine, would think there might be some other new alpha agonist that would be more promising than clonidine, Professor Hall said:
I think that a well-trained and practising intensivist, regardless of the level of particular interest in the world of sedation, at a minimum would know that the existing agents in their entirety as a group were not perfect, not any of them solely and even in combination, and that looking down the road for new sedative agents is perfectly reasonable and could be valuable in the management of patients. And they fell into broad classes and one of those classes is alpha agonists with the sole clinically available agent only used in a limited way, but other agents in the group dependent on their balance of sedative and haemodynamic effects potentially much more useful.
606 It did not seem to me, however, that this answer took the matter any further and it did not support the conclusion that the skilled addressee would have used the term “alpha agonists” in a search of the kind Professor Hall sought to simulate, on the basis that there might be some other alpha agonist that was more promising than clonidine for use in ICU sedation.
607 Having regard to the evidence, especially that to which I have just referred, I accept Pfizer’s submission that, having regard to the knowledge of the skilled addressee in Australia before the priority date, the skilled addressee would not have used the search term “alpha agonists and sedation” in a literature search to find a new sedative agent for use on patients in the ICU and would not therefore have ascertained Belleville (and, in consequence, Bloor). In any event, if the skilled addressee had, before the priority date, undertaken literature searches of the kind described and undertaken by Professor Hall, then the skilled addressee would have found Böhrer 1990, which is listed in the results of one of his searches. I accept that, as Pfizer contended, reference to Böhrer 1990 would, as outlined below, have been likely to have dissuaded the skilled addressee from further exploring the literature by reference to a search term including “alpha agonists”. Having regard to the entirety of the evidence concerning clonidine, it does not seem to me that the skilled addressee in Australia would have been deterred only from the use of alpha-2 agonists “for protracted periods of time”, as Professor Hall claimed was true in his case.
608 As already indicated, InterPharma also submitted that Professor Hall’s search process was akin to the search undertaken by Professor O’Brien described in AstraZeneca HCA. As the reasons for judgment of Jessup J in AstraZeneca FCAFC at [522] shows, however, before making his search, Professor O’Brien was not told that there was a “new statin” that was not part of the common general knowledge. Nor was he given the prior art or patent in suit before he carried out his search. It was in the absence of such information that Professor O’Brien “undertook searches and whittled down the articles of interest by reference to their abstracts. Ultimately, he requested a full copy of each of three articles”: see AstraZeneca FCAFC at [531] (Jessup J).
609 Professor Hall was not in the same position as Professor O’Brien described in AstraZeneca HCA. Professor Hall’s pre-search familiarity with the invention claimed in the Patent can be inferred from the fact that he had twice given evidence in proceedings in the US, on both occasions concerning a foreign equivalent of the Patent in suit. Furthermore, he agreed in cross-examination that he was “intimately aware of [Belleville/Bloor and Talke], if nothing else because [of] … the evidence [he] gave in 2012 and 2016 in the US”. He deposed, furthermore, that Talke was part of the literature on dexmedomidine with which he was familiar during the 1990s and was therefore familiar with Talke well prior to beginning his searches.
610 In closing written submissions Pfizer submitted that Professor Hall was given Belleville/Bloor and Talke before carrying out his literatures searches, relying on what appears at transcript 110. It appears to me, however, that the questions that senior counsel for Pfizer put to Professor Hall were based on a misreading of the relevant parts of his affidavit (that Professor Hall had been given the papers prior to beginning his searches when in fact the affidavit did not say this at all). This misreading resulted in misleading questions and, more importantly, possibly an unintended concession that he had been given these papers by InterPharma’s solicitors prior to conducting his searches. This does not, however, detract from the admitted fact that Professor Hall was clearly familiar with Belleville/Bloor and Talke prior to undertaking the literature searches for this case.
611 Further, before the priority date, Professor Hall was aware of Böhrer 1990, which was the only study of which he was aware in which clonidine was administered as an adjunct for sedation to an ICU patient by way of continuous infusion. Böhrer 1990 reported that serious problems arose following this infusion, including cardiovascular depression, and tachycardia, hypertension, agitation and sweating following abrupt clonidine withdrawal. When asked why he failed to include Böhrer 1990 in his list of papers that were “particularly relevant” in his search of the literature, Professor Hall said that he did not include it because he “was focussed on dexmedetomidine, which was the subject of this matter”. Moreover, as Pfizer pointed out in its closing written submissions, the papers on clonidine that were selected by Professor Hall as “particularly relevant” in fact identified adverse effects when using clonidine as a transdermal patch for prophylaxis of withdrawal symptoms in children; did not relate to sedation at all; or did not suggest that dexmedetomidine would be any better than clonidine for sedative use.
612 Professor Hall had deposed that he had been asked by InterPharma’s solicitors “what literature searches [he] would have carried out if [he] was involved in researching a potential intravenous agent for use in ICU sedation in March 1998” and “to replicate the searches that [he] would have carried out, and to identify the papers that [he] would have regarded as being particularly relevant”. Professor Hall acknowledged, in cross-examination, that he knew that he needed to find Talke and, so too one might infer, Belleville. I would also infer from this, the conduct of his searches and other evidence to which I refer, that the object of Professor Hall’s research task was to identify a search path using PubMed that would find Belleville/Boor and Talke. This is supported by the further fact that Professor Hall failed to draw the attention of the court to Böhrer 1990 which, as already indicated, told against the use of clonidine as a sedative agent and would have dissuaded the skilled addressee from using the term “alpha agonist” in a literature search designed to identify new sedatives for use on ICU patients. It would follow from this that the search process described and undertaken by Professor Hall was not the research process of the diligent researcher described in Lockwood (No 2) at [149] or, indeed, AstraZeneca HCA at [15].
613 These considerations lead me to conclude that, as at the priority date, the skilled addressee could not reasonably have been expected to have ascertained Belleville/Bloor or Talke and regarded them as relevant to work in Australia. Accordingly, neither Belleville/Bloor nor Talke satisfies the requirements of s 7(3) of the Patents Act.
614 If I were wrong in these conclusions, then the question would be whether, given the common general knowledge before the priority considered with Belleville/Bloor only, the skilled addressee would have been led directly as a matter of course to try the invention as claimed in the expectation that it might well produce a useful alternative to, or a better drug than, existing ICU sedatives. The same question would also arise with respect to Talke.
615 As already noted, this is not a case where the skilled addressee is confronted with a particular problem that is part of the common general knowledge or even the s 7(3) information.
616 Belleville/Bloor related to early phase studies on the effects of dexmedetomidine on healthy volunteers. Assoc Professor French’s evidence was (and I accept) that:
[T]his is the first – this is a healthy human volunteer study. There’s a – a great deal of uncertainty remains at this point in time regarding the effects of the drug in patients, in unhealthy individuals, particularly intensive care patients.
617 The experts agreed in their Joint Report that there was nothing in Belleville/Bloor that indicated that dexmedetomidine would be an appropriate agent for ICU sedation, and that “extensive further research [was] required”. Each of them elaborated on this in evidence at trial.
618 Professor Hall’s evidence was that, reading Belleville/Bloor, he would not “as a matter of routine, use dexmedetomidine as an ICU sedative”. Professor Hall said in cross-examination that he would “aggregate that information [in Belleville/Bloor] and say this drug has potentials for this pre-operative protective effect on the patient as an adjunct to anaesthesia.” He did not mention ICU sedation in this regard.
619 Assoc Professor French explained the matter clearly in the following exchange:
THE COURT: So your real concentration in relation to unhealthy patients is on not so much sedation, but the consequences of the drug in relation to other matters, like heart rate, blood pressure?
A/PROF FRENCH: Yes. So if you – for example, the – the sedative effects may not be able to be achieved because there could be adverse effects of the drug at this point – this point in time …known or unknown which could become apparent prior to the sedative effect being achieved.
THE COURT: Another difficulty, presumably, would be the likely pharmacokinetics, if you like, the effect with the ICU ---?
A/PROF FRENCH: Well, there ---
THE COURT: --- context of other drugs?
A/PROF FRENCH: So – so yes, as – as patients become more ill, the way that they handle drugs in terms of their pharmacokinetics, how they’re distributed within the body, how the body eliminates them– metabolises and eliminates them and also, then, the interaction of that drug with the receptor and the effect that occurs – there are changes as you become more unwell.
620 Assoc Professor French made very clear in cross-examination that, given Belleville/Bloor, he would not be directly led as a matter of course to try dexmedetomidine as an agent for ICU sedation. This was consistent with his earlier affidavit evidence concerning Belleville/Bloor.
621 Professor Bellomo was not cross-examined on his evidence that Belleville/Bloor would not encourage him to conduct further research on the use of dexmedetomidine in ICU sedation.
622 There were other matters that bear on the fact that aspects of Belleville/Bloor would have deterred the skilled addressee from considering dexmedetomidine as an ICU sedative, including that:
it was an early study clearly identifying “the potential for unwanted side effects” of the intravenous use of dexmedetomidine;
the reported study environment was very different from an ICU in which patients would need much more sedative and, as Professor Hall acknowledged, as the dose of dexmedetomidine increased, so would concerns about the drug’s effects on blood pressure and heart rate;
the study provided no information about whether dexmedetomidine was suitable to be administered as a continuous infusion and little information regarding the pharmacokinetics of the drug;
although the abstract for Belleville stated that dexmedetomidine was thought not to have “appreciable ventilatory effects”, Belleville itself stated “our subjects were young and healthy; even the minimal ventilatory effects of [dexmedetomidine] seen in this study could be detrimental to more frail patients with pre-existing respiratory disease” and “[i]n older, less healthy patients who are receiving other medications, [dexmedetomidine] could still have clinically significant ventilatory effects”. While perhaps a routine note of caution, the evidence established that ICU patients have these characteristics; Professor Bellomo’s evidence was that drugs that cause ventilatory decrease can place an ICU patient in danger of respiratory complications; and Professor Hall acknowledged that caution was appropriate given that this was an early stage of testing of dexmedetomidine in healthy patients; and
Bloor reported that the administration of dexmedetomidine resulted in decreased blood pressure and heart rate. Both Professor Bellomo and Assoc Professor French expressed the opinion that this indicated that dexmedetomidine would not be useful as a sedative for use in the ICU.
623 Further, it is also relevant that Belleville/Bloor stated that:
(i) the reported increase in oxygen consumption required further investigation since the combination of increase oxygen consumption during a period of reduced ventilation and blood pressure could be detrimental;
(ii) three vagally mediated dysrhythmias were observed in the highest dexmedetomidine-treated dose group of healthy young subjects with low resting heart rates, which would be of concern; and
(iii) the marked transient reduction in cardiac output merited further study and would be undesirable in patients with impaired cardiac function. While Assoc Professor French described the authors’ hypothesis to avoid this effect by slowing down the infusion as “reasonable”, he also said that “a great deal of uncertainty remain[ed] at this point”. Professor Hall ultimately agreed that this “all just requires further investigation”.
624 Having regard to the matters to which I have referred, I would reject the proposition that, given the common general knowledge before the priority considered with Belleville/Bloor only, the skilled addressee would have been led directly as a matter of course to try the invention as claimed in the expectation that it might well produce a useful alternative to, or a better drug than, existing ICU sedatives. It is worth bearing in mind that, at the priority date, the skilled addressee would have known of the effects of propofol and midazolam. As Assoc Professor French explained:
I think the key difference, though, relates to the [fact that] propofol had been widely used in clinical anaesthesia for a number of years prior to its transitioning into intensive care. So particularly those intensivist that had an anaesthesia background had a great deal of clinical experience with the drug and understood … how it’s used and understood its adverse effects. This is now evaluating a drug in a research environment. The drug had been given – you know, only given to a relatively small number of patients in contrast to the many, many, many, many thousands of people – hundreds of thousands of people – that would have been administered propofol prior to its transition into ICU.
625 The further question is, given the common general knowledge before the priority date considered with Talke, would the skilled addressee have been led directly as a matter of course to try the invention as claimed in the expectation that it might well produce a useful alternative to, or a better drug than, existing ICU sedatives? The Joint Report indicated that Professor Bellomo and Assoc Professor French agreed they would not have been directly led as a matter of course to try dexmedetomidine as an agent for ICU sedation. Professor Hall disagreed, stating that he would have conducted a trial. None of the experts stated that they would have expected dexmedetomidine to be a useful alternative to, or better drug than, existing ICU sedatives. All the experts commented “No. Further research required.” Each of them elaborated on their opinions in evidence at trial.
626 In answer to questions from the bench, commencing “[b]y the time you had read the Talke paper, would you still be worried about the unpredictability of dex[medetomidine] as an alpha-2 agonist?”, Professor Hall said:
In this class, all we had was clonidine. And we already knew that the balance of sedation vis-à-vis haemodynamic effect was not particularly favourable, and it also accumulated. So it wasn’t offering us much over other drugs. Dexmedetomidine, in fact, was known from the animal research to be quite different in the alpha 2, alpha 1 ratio effects. As Dr Bellomo said yesterday, that at least opens the door that sedation effects could be more prominent than haemodynamic effects; but likely both are present. And after that – that’s about all I would say. I would say it’s in the right direction as ... alpha 2 agonist, but absolutely we need to – as of Talke, we need to establish the dose that truly has the sedative effect that’s desirable and watch the haemodynamics. That’s what I meant … in my affidavit by flipping it. And tachyphylaxis enters into that picture. For instance, if the sedative effect is just great for a day and then you find yourself going up, if there hasn’t been tachyphylaxis to the haemodynamic effects, then that trade-off could go awry. Absolutely. So the pattern of studying these drugs that start in the perioperative period and then move to the intensive care unit is start with the less sick ICU patients and for shorter duration in your next investigation, and then just extend that.
627 Notwithstanding Professor Hall’s last statement in this passage, I do not accept that the skilled addressee would have been led as a matter of course to try dexmedetomidine as a sedative agent in the ICU. The previous comments in this passage belie that proposition. In any event, Professor Bellomo deposed that, even if it were assumed that some of the patients in the Talke study were in the ICU post-operatively, based on that study, he would not have had any expectation that dexmedetomidine would be useful as a sedative in the ICU. Assoc Professor French was of the same opinion. I accept their evidence, bearing in mind that dexmedetomidine had no known or approved clinical use, and that its hemodynamic effects were of unknown quality and quantity, especially in relation to “the balance of sedation”.
628 Further, I accept that, as Pfizer submitted, Professor Hall’s evidence in the concurrent evidence session that he would have conducted a trial was apparently based on a combined reading of Belleville/Bloor and Talke. In this session, senior counsel for InterPharma asked Professor Hall to clarify his answer to 8.7(a) of the Joint Report, “[w]hat if anything would you have done as a consequence of reading Talke?” Professor Hall responded:
Well, I think I stated in my affidavit that nobody would take the Talke paper and then say that dexmedetomidine is now the first-line treatment for any patient in any [ICU] anywhere. And one also would be aware of the Belleville paper from Talke, because even if it eluded you somehow, it’s actually cited in Talke. So Talke tells you that dexmedetomidine has been given in a set of different doses to human volunteers, healthy, totally healthy volunteers, and it had a sedative effect after a bolus administration that lasted several hours and then went away. And they even established a starting point of that dose that fairly uniformly in human volunteers had a significant sedative effect described as sleeping but easily arousable.
629 Professor Hall subsequently corrected this by saying, in answer to a question from senior counsel for Pfizer:
Well, by reference, Talke tells you, via Belleville, that it had been previously given, but, yes, I mean, if that was interpreted, then the proper interpretation is that the normal volunteers are described in Belleville. I completely agree.
630 The following exchange at trial confirms Professor Hall’s approach:
THE COURT: As I understand [Professor Hall’s] evidence: if you read Belleville, you would see health[y] patients referred to and then you would be taken to the Talke article; is that correct?
PROF HALL: Not only that, but that it is stated in Talke that dexmedetomidine has been given to normal volunteers, reference Belleville.
MR CORDINER: Yes?
PROF HALL: So there’s a bullet point in Talke and where you could get as much information as is available is cited as one of the references, Belleville.
MR CORDINER: Belleville. And that’s where you would learn the sedative [e]ffect, after bolus administration, that lasted several hours and then went away?
PROF HALL: Yes, and if you were reading Talke with a mind to any sedative [e]ffects, that would be a standout comment in the paper.
631 The only (footnoted) reference to Belleville in Talke merely states:
In healthy volunteers, dexmedetomidine … like clonidine, has antinociceptive and sedative effects.
632 Contrary to Professor Hall’s evidence, there is no “comment” in Talke that dexmedetomidine had a sedative effect after a bolus administration that lasted several hours and then went away. Professor Hall’s evidence that he would conduct a trial in relation to ICU sedation on this basis does not establish that he would have done so based on Talke alone (having regard, of course, to common general knowledge). It is clear from his evidence that this assertion is based on a combined reading of Talke and Belleville, such a combined reading being impermissible. As Pfizer submitted, Professor Hall’s evidence in this regard was unsurprising, given that he had said that in the US proceeding he had relied on Belleville/Bloor and Talke together to say that the invention the subject of the equivalent US patent was obvious.
633 It has also to be borne in mind that Talke described the administration of dexmedetomidine for one hour pre-operatively, during surgery and 48 hours post-operatively. Talke disclosed, as all of the experts agreed in their Joint Report, that dexmedetomidine had a sedative effect in patients pre-operatively. As Assoc Professor French observed, however, Talke reported no sedative effect post-operatively, although the administration of dexmedetomidine post-operatively resulted in dose-dependent lowering of blood pressure and slowing of heart rate. Professor Bellomo and Assoc Professor French agreed that Talke would not have indicated to them that dexmedetomidine would be an appropriate agent for ICU sedation because, as noted in the Joint Report, “[n]o sedative effect was reported post-operatively with moderate hemodynamic effects”. Assoc Professor French said, moreover, that Talke would have almost given him the opposite view (that dexmedetomidine would not be suitable as an ICU sedative) in that “no sedation was observed and there were haemodynamic effects observed”. Assoc Professor French rejected InterPharma’s senior counsel’s suggestion that the skilled addressee would try a larger dose to achieve sedative effect post-operatively, observing that:
There would be significant concern that you could add – in my opinion, be a significant concern that if you increase the dose purely to try – and this is a hypothetical situation – purely to try and provide more sedation, you could potentially harm the patients by adversely affecting the haemodynamic status.
634 As already indicated Professor Bellomo and Assoc Professor French were impressive witnesses, both knowledgeable in relation to intensive care medicine in Australia, and I would accept their evidence. For the reasons given above and below, I would accept the evidence of Professor Bellomo and Assoc Professor French in preference to that of Professor Hall in so far as they disagreed with Professor Hall.
635 Amongst other things, I would not accept Professor Hall’s evidence that there was nothing in Talke to indicate that dexmedetomidine would be unsuitable for use in ICU sedation. Professor Hall accepted that Talke indicated that dexmedetomidine might have unstable haemodynamics: Talke reported that dexmedetomidine significantly (that is, by a 20% decrease) lowered patient blood pressure and slowed patient heart rates in response to the first hour of treatment and that after the first hour there were some trends between dose and haemodynamic effects. He also accepted that most patients in the dexmedetomidine group were given high doses of phenylephrine (which was given when blood pressure was dangerously low) intra-operatively, and some were also given it after surgery. He further accepted that glycopyrrolate, which was given to increase heart-rate was not given in the placebo group but was given in the dexmedetomidine group.
636 As well, Talke disclosed that high dexmedetomidine plasma levels may increase blood pressure. Senior counsel for Pfizer asked Professor Hall whether “[i]f you had to titrate [dexmedetomidine] up, as it has been described to you in Talke, would you agree there would be a risk of blood pressure or heart rate increasing [or] decreasing?”. Professor Hall answered that “[t]here’s actually both”. Further, he said:
It is most likely that you would have – if there were an adverse effect – I think it would be most likely there would be some effect on heart rate and blood pressure; it’s more or less guaranteed. Whether it was tolerable, and whether it was tolerable with additional support, or whether it was simply intolerable because it was either unpredictable or there was no simpl[e] way to reverse it would be the things to be determined. Because this is an alpha agonist, on top of all of that, caution at some dosing, it is conceivable that the blood pressure could actually rise. That would be most unusual as a sustained effect but it could, and that observation came from careful looking at clonidine where you sometimes see a small uptake in blood pressure before the more dominant hypotensive effect occurs. So I think there would always be that issue as you up-dose the drug for any purpose. If you up-dosed dexmedetomidine for the purposes of perioperative protective effects on the heart, you would have to worry about these problems and if you titrated the drug for sedative purposes, you would have to worry about these problems. You would worry about them less in a group of patients that began with lower heart rates and lower blood pressures to begin with, of course, because the change in them might be more immediately adverse, whereas patients who – it’s actually – it’s undesirable to have those effects.
637 The above evidence, and Professor Hall’s evidence that the ideal sedative would have side effects that were completely minimal because patients in the ICU have shock, unstable blood pressures and unstable heart rates, is inconsistent with his evidence that there is nothing in Talke to indicate that dexmedetomidine would be unsuitable for use as a sedative for patients in the ICU. His evidence in the latter regard should be rejected.
638 Having regard to the evidence before the Court, I accept that the skilled addressee would not have been directly led by Talke to try dexmedetomidine, being a drug that had no sedative effects post-operatively, as an ICU sedative.
639 The following considerations, amongst others, fortify this conclusion:
Talke suggested that the absence of sedative effects in post-operative patients was consistent “with recent findings of tachyphylaxis to the anesthetic effects of dexmedetomidine in rats”, a possibility that indicated that long-term administration of dexmedetomidine would have rapidly diminishing sedative effects, as a consequence of which the drug would be unsuitable for use in ICU sedation;
As agreed in the Joint Report, Talke did not describe the use of dexmedetomidine for ICU sedation. Professor Bellomo and Assoc Professor French agreed that it was not possible to determine if any of the patients discussed in Talke (in particular the aortic surgery patients) were treated in the ICU. Senior counsel for InterPharma put to Assoc Professor French, “[b]ut given … the adverse effects that a number of them seem to experience … wouldn’t a number of them have ended up in ICU?”; and Assoc Professor French responded that it was difficult to ascertain what the severity of the adverse events was and whether that would necessarily have meant that they needed to be in the intensive care unit or not. He was not taken, however, to that part of Talke under the heading, “Adverse events”, which specifically stated that “[n]one of these events was life-threatening”.
I would accept based on Professor Bellomo and Assoc Professor French’s evidence discussed at [520]-[521] above, that the skilled addressee’s knowledge about clonidine and its use as an anti-hypertensive would have deterred the skilled addressee from considering dexmedetomidine for use as an ICU sedative.
640 So far as concerns the dependent claims, there is no suggestion that the skilled addressee would have been directly led to try dexmedetomidine as the sole active agent or essentially the sole active agent. Further, Talke does not teach the administration of dexmedetomidine by a loading dose and a maintenance dose. It should also be borne in mind that even the highest dose reported in Talke (0.183 µg/kg/h) is outside the doses claimed in claims 11 or 12 of the Patent.
641 Having regard to the matters to which I have referred, I would reject the proposition that given the common general knowledge before the priority considered with Talke only, the skilled addressee would have been led directly as a matter of course to try the invention as claimed in the expectation that it might well produce a useful alternative to, or a better drug than, existing ICU sedatives.
642 For the reasons stated, it has not been shown that any of the claims of the Patent lacked an inventive step, having regard to Belleville/Bloor or Talke.
643 Before concluding the discussion of this topic, I would accept Pfizer’s submission that an annotated version of Exhibit R19 should be disregarded, for the reasons it outlined in submissions filed on 28 May 2018. I would add that I have not found it necessary to refer to the table summarising Professor Hall’s search results, which is Exhibit R19. I have instead had regard to Exhibit R4 and the evidence relevant to it.
Fair basis
644 InterPharma alleged that the Patent was invalid for the reason that the invention as claimed did not comply with s 40(3) of the Patents Act in that each of the claims were not fairly based on the matter described in the specification.
Legal principles
645 Section 40(3) of the Patents Act, as applicable to the Patent, required that the claim or claims be fairly based on the matter described in the specification. The invention as claimed must be broadly described in the body of the specification and not travel beyond the matter disclosed there.
646 With respect to this ground, the inquiry is into what the body of the specification read as a whole discloses as the invention: see Lockwood Security Products Pty Ltd v Doric Products Pty Ltd [2004] HCA 58; 217 CLR 274 (Lockwood (No 1)) at [99].
647 In Lockwood (No 1) at [68]-[69] the High Court stated:
Erroneous principles. The comparison which s 40(3) calls for is not analogous to that between a claim and an alleged anticipation or infringement. It is wrong to employ “an over meticulous verbal analysis”. It is wrong to seek to isolate in the body of the specification “essential integers” or “essential features” of an alleged invention and to ask whether they correspond with the essential integers of the claim in question.
“Real and reasonably clear disclosure”. Section 40(3) requires, in Fullagar J’s words, “a real and reasonably clear disclosure”. But those words, when used in connection with s 40(3), do not limit disclosures to preferred embodiments.
“The circumstance that something is a requirement for the best method of performing an invention does not make it necessarily a requirement for all claims; likewise, the circumstance that material is part of the description of the invention does not mean that it must be included as an integer of each claim. Rather, the question is whether there is a real and reasonably clear disclosure in the body of the specification of what is then claimed, so that the alleged invention as claimed is broadly, that is to say in a general sense, described in the body of the specification.”
Fullagar J’s phrase serves the function of compelling attention to the construction of the specification as a whole, putting aside particular parts which, although in isolation they might appear to point against the “real” disclosure, are in truth only loose or stray remarks.
(Citations omitted)
648 Section 40(3) does not concern the question of the merits of or entitlement to the claimed invention, but instead concerns the question of drafting and a single comparison of the claims with the disclosures of the specification: Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth [2011] FCAFC 132; 119 IPR 194 at [89]. However, the High Court also stated in Lockwood (No 1) at [99] that:
[T]he correct position is that a claim based on what has been cast in the form of a consistory clause is not fairly based if other parts of the matter in the specification show that the invention is narrower than that consistory clause. The inquiry is into what the body of the specification read as a whole discloses as the invention. An assertion by the inventor in a consistory clause of that of which the invention consists does not compel the conclusion by the court that the claims are fairly based nor is the assertion determinative of the identity of the invention. The consistory clause is to be considered by the court with the rest of the specification.
(Citation omitted)
Parties’ submissions
649 InterPharma submitted that the Patent was invalid for lack of fair basis because, while the claims covered the complete spectrum of sedation, there was no real and reasonably clear disclosure in the specification of the use of dexmedetomidine in ICU sedation other than where the patient is easily arousable.
650 It was common ground between the experts that the claims of the Patent encompassed any level of sedation. Senior counsel for InterPharma submitted, however, that “the true invention” described in the specification of the Patent “is the [use of] dexmedetomidine as a sedative with the easily rousable quality”. In this regard, InterPharma referred to the following passages in the specification:
“[t]he aim of ICU sedation is to ensure that the patient is comfortable, relaxed, and tolerates uncomfortable procedures such as placement of iv-lines or other catheters, but is still arousable”;
“most intensive care doctors in the ICU prefer their patients to be asleep but easily arousable”;
“[a]n ideal sedative agent for a critically ill patient should provide sedation at easily determined doses with ready arousability together with hemodynamic stabilizing effects”;
“dexmedetomidine … is an ideal agent to be administered to a patient in the ICU to achieve patient comfort”;
“[t]he quality of the sedation in the ICU achieved by administering dexmedetomidine is unique. Patients sedated by dexmedetomidine … are arousable and oriented, which makes the treatment of the patient easier”; and
“[l]ack of respiratory depression should allow dexmedetomidine to be used also for non-ventilated, critically ill patients who require sedation … yet must remain oriented and easily aroused”.
651 Moreover, InterPharma submitted that “[n]one of the [e]xamples involved a patient being deeply sedated as a result of the administration of dexmedetomidine”, but instead “involved sedation where the patient was easily rousable, alert, awake, and able to communicate”. InterPharma submitted that the Court should reject Professor Bellomo’s evidence that the level of sedation to which the specification refers includes the complete spectrum from very light to very deep on the basis that Professor Bellomo only referred to a single passage of the specification, referring to the ability to achieve a level of sedation, if required, that is “deeper” than the level of sedation that leaves the patient easily arousable. That passage, so InterPharma said, should not be read as a broad disclosure of the use of dexmedetomidine to achieve all levels of sedation, rather than the ideal level discussed throughout the specification. Senior counsel for InterPharma submitted that the reference to deeper sedation is what the High Court in Lockwood (No 1) at [69] described as a “loose or stray” remark.
652 Pfizer also relied on various passages in the specification, including the passage to which Professor Bellomo referred, in support of its submission that the claims did not travel beyond the specification. Senior counsel for Pfizer submitted that the reference to dexmedetomidine being used to smoothly transition a patient into a deeper level of sedation was “hardly a throwaway line” and, properly construed, was part of the reason why the quality of sedation achieved by administering dexmedetomidine was unique.
653 Pfizer submitted that InterPharma’s submission that no example given in the specification involved a patient being deeply sedated was incorrect, and drew attention to example 3. It also referred to Figure 2 set out at [23] above showing the Ramsay Sedation Score of a patient in the dexmedetomidine study described in example 3. Pfizer also relied on the Joint Report, and the evidence of both Professor Bellomo and Assoc Professor French as demonstrating that both the claims and the specification of the Patent describe the complete spectrum of sedation.
Analysis
654 In considering the parties’ respective submissions, it is important to consider the specification as a whole: reference may be made to [10] above and following; and to the summary of the invention of the Patent, where it is said that:
It has been unexpectedly found that dexmedetomidine … is an ideal sedative agent to be administered to a patient in the ICU to achieve patient comfort.
655 Reference may also be made to the detailed description of the invention and, in particular, to the whole of the following passage (only part of which is set out at [26] above):
The quality of the sedation in the ICU achieved by administering dexmedetomidine is unique. Patients sedated by dexmedetomidine … are arousable and oriented, which makes the treatment of the patient easier. The patients can be awakened and they are able to respond to questions. They are aware, but not anxious, and tolerate an endotracheal tube well. Should a deeper level of sedation or more sedation be required or desired, an increase in dexmedetomidine dose smoothly transits the patient into a deeper level of sedation. Dexmedetomidine does not have adverse effects associated with other sedative agents, such as, respiratory depression, nausea, prolonged sedation, ileus or decreased gastrointestinal motility, or immunosuppression. Lack of respiratory depression should allow dexmedetomidine to be used also for non-ventilated, critically ill patients who require sedation, anxiolysis, analgesia, and hemodynamic stability yet must remain oriented and easily aroused. In addition, it is water soluble and, thus, does not increase the lipid load in patients sedated for long periods of time. A predictable pharmacological response can be achieved by administering dexmedetomidine … to a patient in the ICU.
656 I accept that, when this passage is read in its entirety, the unique quality of the sedation achieved by the use of dexmedetomidine is that which is set out in the whole of the passage, rather than merely in the second, third and fourth sentences of that passage, as InterPharma sought to suggest. This means that the ability to achieve deeper sedation is, as senior counsel for Pfizer submitted, also among the reasons why sedation by administration of dexmedetomidine is said to be unique. It is therefore not the case that the reference to deeper sedation is a throwaway line, or a loose or stray remark, as senior counsel for InterPharma submitted.
657 It is well-accepted that the examples in a specification cannot be used to read down a claimed disclosure. In any event, InterPharma’s characterisation of the examples given in the specification should not be accepted. The experts agreed in their Joint Report that the level of sedation described in the Patent was Ramsay Sedation Score of 2 to 6 and that “[d]eep sedation may be defined as a Ramsay [Sedation] [S]core of 4, 5, or 6”. Example 3, case 8, to which InterPharma did not refer, detailed the administration of a loading dose of dexmedetomidine (with propofol) followed by infusions of dexmedetomidine (with propofol) and doses of morphine, where “[t]he patient’s Ramsay Sedation Score continually increased until the patient was oversedated with a score of 6”. This was deep sedation; and, as senior counsel for Pfizer submitted, there was no suggestion of easy arousability. Indeed, reference to Figure 2 (set out at [23] above) shows the Ramsay Sedation Score of another patient in the dexmedetomidine study described in example 3, ranging from 3 to 6 during the dosing period.
658 Importantly, the evidence of both Professor Bellomo and Assoc Professor French was that the specification and the claims of the Patent in suit disclosed the complete spectrum of sedation as achievable by the administration of dexmedetomidine. While Professor Bellomo agreed that a benefit of the invention as claimed was that ICU patients sedated with dexmedetomidine were asleep but easily arousable, his evidence was that the meaning of “sedation” used in the claims was the same as the meaning of “sedation” in the specification, and that “sedation” as used in the claims and specification included “the complete spectrum of sedation from very light (including where the patient is ‘easily arousable’) to very deep”.
659 Assoc Professor French’s evidence was that, notwithstanding that the specification indicated that it was an advantage of dexmedetomidine that it can provide arousable sedation, it was clear from the specification that “dexmedetomidine may be used to achieve the full gamut of sedation”, including deep sedation. He deposed that the specification made it clear to him “that dexmedetomidine has a dose dependent effect where there is a dose where patients are calm and cooperative but higher doses are available for greater levels of sedation”. Assoc Professor French said that this was also consistent with the examples in the specification, which show levels of sedation greater than arousable sedation.
660 As the foregoing discussion shows, their evidence is consistent with the Patent, read as a whole; and it should be accepted.
661 Accordingly, I accept that the claims did not travel beyond the specification; and there was a real and reasonably clear disclosure in the body of the specification of the use of dexmedetomidine for ICU sedation across the complete spectrum of sedation achievable by the administration of that agent.
Conclusion
662 For the reasons stated, InterPharma threatened to infringe the Patent in suit prior to its expiry on 31 March 2019 and InterPharma’s challenge to the validity of the Patent should be rejected.
663 The parties will be afforded an opportunity to submit minutes of orders in accordance with these reasons.
I certify that the preceding six hundred and sixty-three (663) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Kenny. |
Associate:

