
Mylan Health Pty Ltd (formerly BGP Products Pty Ltd) v Sun Pharma ANZ Pty Ltd (formerly Ranbaxy Australia Pty Ltd) [2019] FCA 28
ORDERS
(FORMERLY BGP PRODUCTS PTY LTD) First Applicant BGP PRODUCTS OPERATIONS GMBH Second Applicant | ||
AND: | (FORMERLY RANBAXY AUSTRALIA PTY LTD) First Respondent ALKERMES PHARMA IRELAND LIMITED Second Respondent | |
AND BETWEEN: | (FORMERLY RANBAXY AUSTRALIA PTY LTD) Cross-Claimant | |
AND: | (FORMERLY BGP PRODUCTS PTY LTD) First Cross-Respondent BGP PRODUCTS OPERATIONS GMBH Second Cross-Respondent ALKERMES PHARMA IRELAND LIMITED Third Cross-Respondent | |
DATE OF ORDER: |
THE COURT ORDERS THAT:
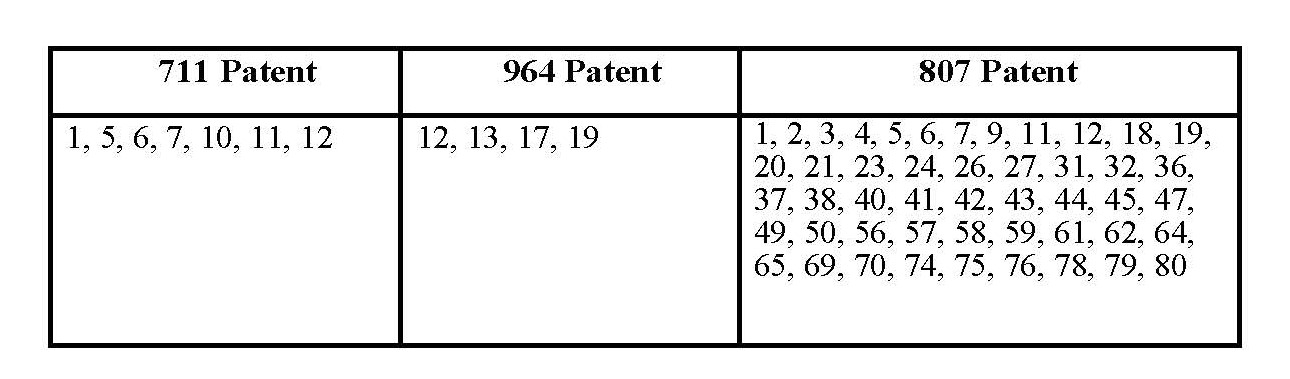
1. Claims 1, 5, 6, 7, 10, 11 and 12 of Australian Patent No. 2006313711 be revoked.
2. Claims 12 and 13 of Australian Patent No. 731964 be revoked.
3. Claims 1, 2, 3, 4, 5, 6, 7, 9, 11, 12, 18, 19, 20, 21, 23, 24, ,26, 27, 31, 32, 36, 37, 38, 40, 41, 42, 43, 44, 45, 47, 49, 50, 56, 57, 58, 59, 61, 62, 64, 65, 69, 70, 74, 75, 76, 78, 79, 80 of Australian Patent No. 2003301807 be revoked.
4. The second further amended originating application be dismissed.
5. The Confidential Schedule to the attached reasons for judgment not be published or disclosed to any person apart from the solicitors and counsel for the parties without the agreement of the first respondent or the leave of the Court.
6. Within 14 days, the parties file and serve (by way of exchange) brief written submissions (limited to 3 pages in length) on questions of costs.
7. Within 21 days, the parties file and serve (by way of exchange) brief written submissions in reply (limited to 2 pages in length) on questions of costs.
8. Order 5 made on 4 May 2016 be discharged.
9. Upon the applicants, by their counsel, undertaking to the Court during the period of the stay:
A. to prosecute any appeal expeditiously; and
B. forthwith to serve on the Commissioner of Patents copies of these orders pursuant to s 140 of the Patents Act 1990 (Cth) with a request that particulars of Orders 1, 2 and 3 (“the Revocation Orders”) be registered in accordance with s 187 of the Patents Act 1990 (Cth),
the Revocation Orders be stayed:
(a) initially for a period of 21 days from today; and
(b) if an appeal from any of the Revocation Orders is lodged within that period, until the final determination of that appeal or further order.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
NICHOLAS J:
Introduction
1 In this proceeding, the applicants seek injunctive relief restraining the first respondent from infringing certain claims of three patents. The three patents (“the Patents”) are:
Australian Patent No. 2006313711, a method of treatment patent titled “Use of fenofibrate or a derivative thereof for preventing diabetic retinopathy” (“the 711 Patent”). The asserted priority date for the 711 Patent is 10 November 2005. The second applicant owns, and the first applicant is the exclusive licensee of, the 711 Patent.
Australian Patent No. 731964, a formulation patent titled “Pharmaceutical composition of fenofibrate with high biological availability and method for preparing same” (“the 964 Patent”). The asserted priority date for the 964 Patent is 17 January 1997. The second applicant owns, and the first applicant is the exclusive licensee of, the 964 Patent.
Australian Patent No. 2003301807, a formulation patent titled “Nanoparticulate fibrate formulations” (“the 807 Patent”). The asserted priority date for the 807 Patent is 24 May 2002. The second applicant and the second respondent co-own the 807 Patent.
2 The first respondent proposes to market in Australia a number of fenofibrate products that were entered in the Australian Register of Therapeutic Goods (“ARTG”) on about 29 February 2016, specifically 45 mg and 145 mg fenofibrate film coated tablets presented in a bottle or in a blister pack under the brand names FENOFIBRATE RBX, FENOFIBRATE SUN and FENOFIBRATE RAN. These products are collectively called “the Ranbaxy Products” in the pleadings and evidence and I will adopt that nomenclature in these reasons. The second respondent is a co-owner of the 807 Patent but has not taken any active role in the proceeding. For ease of reference, I will refer to the first respondent as the respondent.
3 The respondent denies that it will infringe any of the claims upon which it is sued (the relevant claims) by importing into Australia or supplying in Australia, any of the Ranbaxy Products. It also challenges the validity of the relevant claims and seeks orders for their revocation on various grounds. It does not dispute any of the asserted priority dates.
4 The relevant claims are:
5 An order was previously made that the question of infringement of the 807 Patent insofar as it requires certain experimental evidence to be adduced be deferred until after the determination of all other issues of liability in the proceeding. That experimental evidence relates to the following integers:
(a) whether the composition exhibits bioequivalence upon administration to a human subject in a fed state as compared to a fasted state, by reference to AUC and Cmax, which is a requirement of claims 1, 2, 3, 40, 41 and 42 (and their dependent claims);
(b) whether the difference in AUC of the composition administered in a fed versus fasted state is less than about 35% or lower, which is a requirement of claim 7 (and its dependent claims); and
(c) whether the composition redisperses in a biorelevant media, which is a requirement of claims 1, 2, 3, 40, 41 and 42 (and their dependent claims).
6 The respondent has made a number of admissions relevant to the applicants’ infringement case concerning the manufacturing process and specifications relevant to the production of the Ranbaxy Products. These admissions form the basis of the brief description of the respondent’s manufacturing process that is set out in the Confidential Schedule that forms part of these reasons.
The witnesses
7 There was a considerable body of evidence adduced by both parties. Most of the oral evidence was given in concurrent sessions. The only exceptions to this were Professor Elam and Mr Butler who were cross-examined separately.
8 The witnesses called by the applicants were:
Professor Michael Stephen Roberts
9 Professor Roberts is a NHMRC Senior Principal Research Fellow, a Professor of Therapeutics and Pharmaceutical Science at the University of South Australia and a Professor of Clinical Pharmacology and Therapeutics at the University of Queensland. He is also the Director of the Therapeutics Research Centre at the Princess Alexandra Hospital in Brisbane. Professor Roberts has been a Professor since 1989 and a NHMRC Research Fellow since 1994.
10 After obtaining a Bachelor of Pharmacy in 1970, Professor Roberts practiced as a pharmacist and completed a Master of Science and Doctor of Philosophy at the University of Sydney. He then worked as a lecturer and academic at the University of Tasmania and the University of Otago.
11 Between 1978 and 1984, he worked on a project at the University of Tasmania to develop a slow release oral dosage form of aspirin which could be used in the prevention of stroke and heart attacks. From 1986 to 1989, as Head of the Department of Pharmacy at the University of Otago, Professor Roberts conducted various research projects for pharmaceutical companies, including overseeing research studies relating to the examination of bioavailability and bioequivalence of oral dosage in human volunteers. He has also been involved in the creation, development and testing of fast release oral dosage forms.
12 Professor Roberts is a named co-inventor of several patents. He has written many peer-reviewed articles, including in relation to oral delivery, pharmacokinetics, pharmacodynamics, toxicology, and drug formulation.
Professor Clive Allan Prestidge
13 Professor Prestidge is a Professor of Pharmaceutical Science at the University of South Australia. He has degrees in Chemistry and Pharmaceutical Science and has worked in a variety of academic and research roles since completing his PhD in 1990. Since 1994, Professor Prestidge has led a research group at the University of South Australia whose primary focus is nano-structured drug delivery systems and interfacial science applied to pharmaceutical formulation and delivery.
14 Professor Prestidge’s research projects before May 2002 involved using nanomaterials, either as a drug or excipient. This included research on particle-based suspensions and colloidal (nano-particle-based) drug delivery, and use of laser diffraction and laser light scattering measurement techniques.
15 Professor Prestidge has published widely in relation to nanostructured pharmaceutical delivery systems, pharmaceutical characterisation, biointerfacial processes and lipid particle based drug delivery. He is a named inventor of several patents.
Professor Richard Charles O’Brien
16 Professor O’Brien is an endocrinologist and the clinical Dean of Medicine of the Austin Clinical School at the University of Melbourne. He has a special interest in lipid disorders and diabetes, and describes himself as a clinical lipidologist. He is Director of the Lipid Services at Austin Health. His degrees include a Doctor of Philosophy from the Faculty of Medicine at the University of Melbourne awarded in 1992 for a thesis entitled “Diabetic nephropathy: early pathophysiology and therapeutic intervention.” He has held numerous clinical positions since 1982 and various academic positions at Monash University and the University of Melbourne before becoming clinical Dean of Medicine in 2007.
17 Professor O’Brien was the Head, and later the Director, of the Diabetes Section of Monash Medical Centre (1992 to 2004), the Director of Lipid Services and Deputy-Director of Diabetes at Southern Health (2005 to 2007), the Chair of the Lipid Management in Diabetes Guidelines Committee of the NHMRC (2010 to 2012) and the Chair of the Expert Committee on Hyperlipidaemia for the Australian Diabetes Society (since 2010). He has authored or co-authored articles and texts concerning the management of dyslipidaemia and treatment of patients with type 2 diabetes.
18 In addition to his academic and clinical duties, Professor O’Brien has been a member of advisory boards associated with a number of pharmaceutical companies, including, since 2007, the first applicant (then known as BGP Products Pty Ltd) in relation to the interpretation of trial data and educational material relating to fenofibrate.
Feras Karem
19 Mr Feras Karem is the Managing Director of Pharmacy 4 Less Pty Ltd which operates 45 pharmacy stores around Australia. Mr Karem holds a Bachelor of Pharmacy and a Master of Business Administration. Mr Karem gave evidence in respect of the practice of filling prescriptions and dispensing medicine to patients. Mr Karem was not cross-examined.
Professor Ronald Paul Mitchell
20 Professor Mitchell is the Professor of Clinical Ophthalmology & Eye Health at the University of Sydney and an ophthalmologist at the Westmead Hospital where he is the director of its Ophthalmology Department. His clinical practice involves management of age-related macular degeneration, diabetic retinopathy and other vascular retinopathies. Professor Mitchell has practiced as an ophthalmologist for over 25 years.
21 Professor Mitchell obtained a Bachelor of Medicine and Bachelor of Surgery (with Honours) from the University of New South Wales in 1975 and a Doctor of Medicine from the University of New South Wales in 1981. His post-doctoral program involved investigation into diabetic retinopathy and was conducted in conjunction with another ophthalmologist, Dr Paul Beaumont.
22 Since 1978, Professor Mitchell has held a number of academic and leadership positions in the field of ophthalmology, including membership of the Retinopathy Sub-Committee of the Australian Diabetes Society (1985 to 2004), a board member and, later chairman, of the Ophthalmic Research Institute of Australia (1993 to 2001) and chairman of the Information and Data Sub-Committee of the Visual Impairment Prevention Program for NSW Health (1999 to 2002). He has authored or co-authored over 1,000 publications, including in respect of the pathology, management and treatment of diabetic retinopathy.
23 Professor Mitchell was the principal author of the Clinical Practice Guidelines for the Management of Diabetic Retinopathy published by the National Health and Medical Research Council in June 1997 (the 1997 NHMRC Guidelines) and the subsequent version of those guidelines published in 2008. Professor Mitchell also assisted in the development of the protocol for the “Fenofibrate Intervention and Event Lowering in Diabetes Study (“the Field Study”) and in the conduct of that study as an investigator. He later co-authored an article reporting on this study entitled “Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (Field Study): a randomised controlled trial” that was published in The Lancet in November 2007.
24 The witnesses called by the respondent were:
Dr Paul Ernest Beaumont AM
25 Dr Beaumont is a consultant ophthalmologist at the Retinal & Vitreous Centre in Sydney. He obtained a Bachelor of Medicine and Bachelor of Surgery from the University of New South Wales in 1967.
26 During the period from approximately 1970 to 1974, Dr Beaumont worked as an Honorary Ophthalmology Registrar, and subsequently, an Ophthalmology Registrar and a Senior Ophthalmology Registrar, at the Prince of Wales Hospital in Randwick. While at the Prince of Wales Hospital, he trained under the late Professor Frederick (Fred) Hollows AC and carried out research into the causes and treatment of diabetic retinopathy. Before moving to private practice in 1976, Dr Beaumont worked under Professor Hollows as an Honorary Neuro-Ophthalmologist at the Prince Henry Hospital and Prince of Wales Hospital.
27 Since 1976, he has been engaged in private practice as an ophthalmologist specialising in medical retinal disease and, in particular, the treatment of diabetic retinopathy, retinal venous occlusion and macular degeneration. Dr Beaumont estimates that, as at November 2005, approximately 40% of the patients he treated had diabetic retinopathy.
28 Dr Beaumont later served as chairman of the NSW branch of the Royal Australian and New Zealand College of Ophthalmologist (2001 to 2003), chairman and director of the Macular Degeneration Foundation (2001 to 2013), vice-president of the Diabetic Association of New South Wales (1974 to 1975) and chairman and director of the Eye and Vision Research Institute (1978 to 2011). Dr Beaumont has authored or co-authored several publications in peer-reviewed academic journals.
29 In 2013, Dr Beaumont received the award of Member of the Order of Australia “for significant service to medicine, particularly in the field of ophthalmology”.
Christopher Butler
30 Mr Christopher Butler is the Office Manager of Internet Archive in San Francisco, California. Mr Butler was called by the respondent and gave evidence in relation to the working of the Internet Archive Wayback Machine (“the Wayback Machine”).
Leilani Epifanio Au
31 Ms Au is a Deputy Managing Editor at MIMS Australia Pty Limited (MIMS Australia) and has been in this role since around October 2016. Ms Au holds a Bachelor of Science (Pharmacy) degree and became a registered pharmacist in the Philippines in 1997. Ms Au gave evidence about works published by MIMS Australia or its predecessors. Ms Au was not cross-examined.
Ngaire Pettit-Young
32 Ms Pettit-Young is the Managing Director of Information First Pty Ltd. Ms Pettit-Young holds a Bachelor of Arts and a Graduate Diploma of Information Management and has worked as a research and reference librarian for over 20 years. Ms Pettit-Young was not cross-examined.
Ben Teeger
33 Mr Teeger is a solicitor employed by Ashurst Australia and gave evidence in relation to searches he conducted using the Wayback Machine for historical versions of the Accord Website. Mr Teeger was not cross-examined.
Professor John Norman Carter AO
34 Professor Carter is a consultant endocrinologist with particular expertise in the diagnosis and management of diabetes. He is a Clinical Professor of Medicine at Sydney Medical School, University of Sydney and Consultant Emeritus (Endocrinology) for the New South Wales Ministry of Health’s Northern Sydney Local Health District Network.
35 Professor Carter was awarded his first degree in medicine (BSc (Med)) from the University of Sydney in 1967 followed by degrees (MB, BS) from the University of Sydney in 1969. Between 1973 and 1975 he was a Clinical Fellow in Endocrinology and a National Health and Medical Research Council (“NHMRC”) Research Fellow at the Garvan Institute at St Vincent’s Hospital in Sydney.
36 After completing a postgraduate fellowship in Canada, Professor Carter returned to Australia in 1977 and was appointed inaugural Director of the Endocrine Unit at the Repatriation General Hospital based in Concord where he worked until 1983. Between 1983 and 2013 he was a Senior Visiting Medical Officer (“VMO”) (Endocrinology) at Hornsby Ku-ring-gai Hospital and the Sydney Adventist Hospital.
37 Professor Carter has been a member of the Australian Diabetes Society since 1974, an Honorary Life Member since 2009, and has also served as a Councillor and President of that organisation. Since 1993 he has also been a member of the Endocrine Society of Australia which is a non-profit association of scientists and clinicians who conduct research or practice in the field of endocrinology.
38 Between 1992 and 1994 Professor Carter was a member of the NHMRC’s Expert Panel in Diabetes. Between 1992 and 1993 he was also Chairman of the National Action Plan Management Group responsible for finalising the National Action Plan for the prevention and control for non-insulin dependent diabetes in Australia (“NAP”). From 1993 to 1997, he was Chairman of the Diabetes National Action Plan Implementation Committee (DNAPIC), which was responsible for implementing recommendations contained in the NAP.
39 In 1996, the Commonwealth Government established the Ministerial Advisory Committee on Diabetes (MACOD), which reported to the then Commonwealth Minister for Health and Family Services, the Honourable Dr Michael Wooldridge MP. Professor Carter was appointed as the Chairman of the MACOD by the Minister, a position he held from 1996 to 2000.
40 In the course of his duties as Chairman of the MACOD, Professor Carter commissioned the “National Diabetes Strategy and Implementation Plan” (NDSIP). The purpose of the NDSIP was to guide the allocation of government funding relating to improvements in diabetes care and prevention, as well as to suggest structural and functional reorganisation to ensure equitable access to effective, efficient and economically viable diabetes services for all Australians. In 1998, the Commonwealth Government established the National Diabetes Taskforce (NDT) to oversee the implementation of the NDSIP. Professor Carter was a member of the NDT from 1998 to 2002 and its successor, National Diabetes Strategy Group (NDSG). From 2002 to 2006, he was a member of the NDSG.
41 In 2000, Professor Carter received the award of Officer of the Order of Australia “for service to medicine, particularly through research and policy development on diabetes, and through endocrinology”.
Dr Louisa King
42 Dr King is the Principal of and a Patent and Design Searcher at King Search Pty Limited. Dr King holds a Bachelor of Science (organic chemistry) degree and was awarded a Doctor of Philopshy on 1992. Dr King’s doctoral research was in the field of organic chemistry. Dr King was not cross-examined.
Associate Professor David Alexander Vodden Morton
43 Mr Morton is an Associate Professor of Pharmaceutical Science at Monash University. He obtained a Bachelor of Science (Chemistry) in 1986 from the University of Bristol and a PhD in Chemistry from the same university in 1990. Mr Morton immigrated to Australia in 2007.
44 Before May 2002, Mr Morton worked at the United Kingdom Atomic Energy Authority, the Centre for Drug Formulation Studies (CDFS) at the University of Bath and Ventura Ltd. As part of his responsibilities at CDFS and Ventura Ltd, Mr Morton worked on projects involving formulations and the application of particle size to drug delivery systems for pharmaceutical companies.
Dr Desmond Berry Williams
45 Dr Williams is a Senior Lecturer in the School of Pharmacy and Medical Sciences at the University of South Australia. He graduated with a Bachelor of Pharmacy from the South Australian Institute of Technology in 1975 and was awarded a Master of Science in 1981 and Doctor of Philosophy in 1983 from the University of Kansas.
46 In 1983, Dr Williams joined the Research and Development Division of F H Faulding & Co Ltd (“Faulding”) as a Development Pharmacist based in Adelaide. He was promoted to the position of Clinical Research Manager of the Research and Development division of Faulding in 1985. At Faulding, Dr Williams worked on improving bioavailability of new and existing pharmacological compounds and controlled release dosage formulations. In 1996, Dr Williams took up a position as Group Manager of Research and Development at Sigma Pharmaceuticals Pty Ltd where he worked on new formulations of existing drug products in various dosage forms, including oral (tablets and capsules), parenteral products, lotions and creams.
47 Dr Williams joined the University of South Australia as a Senior Lecturer in 1999. During his tenure at the University of South Australia, a focus of Dr Williams’ research was on biopharmaceutics and pharmacokinetics, drug product formulation design and product assessment.
Professor Marshall Elam III
48 Professor Elam III is the Chief of the Division of Clinical Pharmacology in the Departments of Pharmacology, Preventative Medicine and Medicine (Cardiovascular Diseases) at the University of Tennessee Health Science Centre in Memphis, Tennessee.
49 Professor Elam has a Bachelor of Science (Biology) cum laude, a Doctor of Philosophy (Pharmacology) and a Doctor of Medicine. Professor Elam has been licensed to practice medicine in the State of Tennessee since 1979 and in 1985 was licensed to practice as a specialist in the field of cardiology.
Andrew Rankine
50 Dr Rankine is a partner at Ashurst Australia. Dr Rankine was not cross-examined.
The 711 Patent
The common general knowledge
51 The matters referred to in this section of my reasons form part of the common general knowledge in the field of the invention described in the 711 Patent as at 10 November 2005.
Diabetes mellitus
52 Diabetes mellitus (“diabetes”) is an endocrine (or hormonal) disorder characterised by an absolute or relative deficiency of insulin. Insulin is a hormone produced in the endocrine part of the pancreas, which is responsible for regulating glucose metabolism in the body.
53 Diabetes is a common disorder in Australia, with more than 7% of the population over 25 years of age suffering from it. The incidence of diabetes increases with age and, in Australia, over 20% of people aged over 75 suffer from it. The condition remains undiagnosed in a large proportion of people who suffer from diabetes in Australia.
54 Patients with diabetes are at risk of developing a wide variety of acute and long-term complications including macrovascular complications, such as coronary artery disease (leading to heart attack), cerebrovascular disease (leading to strokes), peripheral vascular disease (leading to infection or gangrene), microvascular complications, including diabetic retinopathy (eye disease), diabetic nephropathy (kidney disease) and diabetic neuropathy (nerve damage).
Type 1 Diabetes
55 Type 1 diabetes is the less common of the two forms of diabetes, accounting for around 10% to 15% of cases in Western countries such as Australia. Type 1 diabetes is most often diagnosed in children and young adults (although it can present at any stage of life). In patients affected by type 1 diabetes, there is a progressive destruction of the cells (known as “islet cells”) in the pancreas that are responsible for insulin synthesis and release. Generally speaking, by the time that insulin deficiency becomes clinically apparent, the islet cells in patients affected by type 1 diabetes have typically lost at least 90% of their insulin-producing capacity. Type 1 diabetes is an autoimmune condition.
56 People affected by type 1 diabetes may present with a range of symptoms and signs, including fatigue, increased thirst (polydipsia), increased urine production (polyuria), weight loss, increased appetite and dehydration. In some cases, people affected by type 1 diabetes may develop life-threatening acute complications, such as the metabolic disturbance known as diabetic ketoacidosis.
Type 2 Diabetes
57 Type 2 diabetes is the more common form of diabetes in Western countries, including Australia, and accounts for around 85% of cases in Western countries. Patients affected by type 2 diabetes suffer from insulin resistance, whereby insulin is less efficient in transporting glucose from the blood stream into cells.
58 In order to exert its normal biological actions, insulin must initially attach to specific insulin receptors on surface membranes of cells. In people affected by insulin resistance (including people with type 2 diabetes), there is typically a reduction in the number of insulin receptors and, in addition, a reduction in the attraction (or affinity) between the insulin and its receptors.
59 In the initial stages of type 2 diabetes, the body responds to insulin resistance by increasing the production of insulin from the islet cells in the pancreas, leading to an increase in the levels of insulin circulating in the blood (hyperinsulinaemia). In this early stage of type 2 diabetes, blood glucose levels typically remain within the normal range, but as the disease progresses, due to both insulin resistance becoming more severe and the islet cells becoming less able to maintain increased levels of insulin production, blood glucose levels rise. Over the longer term, insulin production typically declines to such an extent that insulin deficiency develops and insulin therapy is required to normalise blood glucose levels.
Dyslipidaemia
60 Dyslipidaemia is a condition caused by abnormal levels of the following lipids (fats) in the body:
(a) cholesterol, which is insoluble in blood and exists as one of the following complexes in the body:
(i) very low density lipoprotein cholesterol (“VLDL-C”);
(ii) low density lipoprotein cholesterol (“LDL-C”);
(iii) high density lipoprotein cholesterol (“HDL-C”); and
(b) triglycerides.
61 VLDL-C is the form in which cholesterol is secreted from the liver, where most cholesterol is made. As VLDL-C enters the bloodstream it is converted into LDL-C, which transports the cholesterol around the body. HDL-C is often referred to as the “good” cholesterol, because it transports cholesterol present in the body back to the liver, to remove it from circulation. A patient's total cholesterol levels refer to the sum of their LDL-C, VLDL-C and HDL-C levels (ie. the sum of all lipoproteins that are complexed with cholesterol). Triglycerides are another type of lipoprotein principally used for energy. A large proportion of dietary fat is transported around the body in the form of triglycerides.
62 A patient is described as having dyslipidaemia (abnormal lipid levels) or hyperlipidaemia (elevated lipid levels) if they have:
(a) elevated LDL-C levels;
(b) elevated triglyceride levels; and/or
(c) low HDL-C levels.
63 Since around the mid-1990s a class of drugs called statins have been the primary drug treatment for patients with elevated total cholesterol and LDL-C levels. For patients with elevated triglyceride levels, and/or low HDL-C levels, another class of drugs called fibrates were a primary drug treatment. Professor Carter estimated that as many as 25% of his patients were being treated with fenofibrates for lipid control as at November 2015 which I am satisfied would be fairly typical for that time. One of these, fenofibrate, became available in Australia in late 2004. Lipidil, in which fenofibrate is the active ingredient, was first described in the 29th Edition of the MIMS Annual dated June 2005 (“MIMS”). According to MIMS at p 2-181:
During clinical trials with fenofibrate, total cholesterol was reduced by 20 to 25%, triglycerides by 40 to 50%; and HDL cholesterol was increased by 10 to 30%.
Diabetic retinopathy
64 Retinopathy is “disease of the retina” whether or not it is associated with diabetes. Diabetic retinopathy is a long-term complication associated with diabetes that affects a patient's eyes. It is a complication related to increasing changes to the blood vessels in the retina.
65 The first stage of the development of retinopathy in a patient is called non-proliferative retinopathy. This is characterised by lesions in the form of microaneurysms (macular swelling across the course of a retinal capillary), and then haemorrhages (which may indicate blood vessel leakage), in the retina. Both of these types of lesions occur in the capillary bed of the retina. Other lesions characteristic of retinopathy include:
(a) hard exudates (lipid material deposited in the retina from the blood stream);
(b) cotton wool spots (small areas of infarcted retina, which is retina that has lost its blood supply); and
(c) vessel calibre changes (particularly involving the veins, which become dilated).
These early signs of retinopathy generally do not affect vision unless the macula is affected, especially where the non-proliferative retinopathy involves only a few of the lesions characteristic of retinopathy.
66 Non-proliferative retinopathy can progress to proliferative retinopathy, which is vision threatening. In this stage of retinopathy, new blood vessels develop from the retinal veins or from vessels at the optic disc. These have a propensity to bleed (haemorrhage) into the vitreous of the eye. Following haemorrhage, fibrous tissue can develop at the site of the new vessels, which can then lead to traction on the retina and retinal detachment. The vitreous haemorrhage can itself black out vision and lead to severe vision loss.
67 Another condition called macular oedema can occur at any stage of non-proliferative or proliferative retinopathy. This is caused when capillaries (small blood vessels) in the retina close to the macula become occluded (closed or non-perfused), such that there is reduced flow through these capillaries. In response, adjacent capillaries start to leak fluid (oedema) into the retina. The macula is structured so that all of the supporting cells are aligned to give a maximum light signal to the cones at the base of the macula, which means that it can accumulate fluid easily, leading to swelling and damage to the macula.
68 Because of its importance in providing sharp vision, the macula is critical to vision function. Macular oedema is the most frequent cause of vision loss in people with diabetic retinopathy. However, oedema can also occur at other parts of the retina, away from the macula. This generally does not need to be treated unless it threatens the macula.
69 In addition to the damage caused by swelling as a result of oedema or macular oedema, the leakage of fluid from the bloodstream into the retina can result in the formation of small deposits of lipid and other materials in the retina. These small deposits are called ‘hard exudates’. Hard exudates can form at any stage of diabetic retinopathy, as a result of fluid leakage from the retinal capillary bed. Hard exudates tend to occur in the outer part of the retina and, in most patients, close to or involving the macula. Hard exudate deposition is damaging when it occurs at the macula because it can cause irreversible structural changes to the macula within a period of months. If present for a long period, it will result in permanent vision loss.
Risk factors for diabetic retinopathy
70 The longer a patient has had diabetes, the more likely it is that diabetic retinopathy will develop, particularly if the patient’s blood glucose levels have been persistently elevated. Persistently elevated (poorly controlled) blood glucose levels, also known as hyperglycaemia, is the primary risk factor for diabetic retinopathy.
Detection and treatment of diabetic retinopathy
71 The person conducting the diabetic retinopathy screening will be looking for signs of the early stages of retinopathy, typically microaneurysms or haemorrhages, which look like red dots in the retina. It is the pattern and symmetry of these signs in both eyes that indicates the presence of diabetic retinopathy, because such signs in isolation in one eye could result from, for example, elevated blood pressure or other conditions.
72 In general, screening for diabetic retinopathy should be conducted for all people with diabetes at least every two years. Once retinopathy is detected in a patient, the patient needs to be reviewed, generally by an ophthalmologist, on a regular basis to assess the progress of the condition, including whether there are any signs of the development of vision threatening retinopathy, usually proliferative retinopathy or macular oedema, which would require treatment. Where the diabetic retinopathy in a patient has progressed to proliferative retinopathy or macular oedema, treatment is required. As at November 2005, the main treatment options for preventing the progression of vision-threatening diabetic retinopathy, once it was detected in a patient, were the following:
(a) Laser treatment: Laser treatment kills off areas of ischemia (loss of capillary bed, which is a network of capillaries) in the retina, and areas where the blood supply has been compromised. The laser treatment causes the new blood vessels to regress and stop bleeding, which is the main complication of proliferative retinopathy.
(b) Pan-retinal photocoagulation: This is widespread laser treatment of the peripheral retina, used for proliferative retinopathy. In this condition, the peripheral retina becomes ischemic (loss of capillary bed). Pan-retinal photocoagulation to these ischemic areas leads to regression of the new blood vessels, reducing the risk of bleeding.
(c) Steroid injection: Injection of steroids into the eye can reduce the swelling associated with macular oedema. However, this treatment option has a number of side effects and, in 2005, was used much less frequently than laser treatment.
(d) Vitrectomy: Proliferative diabetic retinopathy can cause traction on the retina or the macula, which may lead to retinal detachment and blindness. Vitrectomy is a surgical operation whereby the vitreous of the eye is removed from the patient's eye, and any tractional bands are cut and/or removed.
73 These four treatment options are intended to reduce the rate of progression of diabetic retinopathy or macular oedema, and to reduce progressive visual loss, in patients with proliferative retinopathy or macular oedema. As a result of the findings from the “Early Treatment Diabetic Retinopathy Study” (“the ETDRS”), it was recommended that laser treatment around the macula be used to clear hard exudates threatening or beginning to involve the macula. Laser treatment does not have a direct effect on hard exudates. Rather, the laser treatment reduces fluid leakage as a result of oedema or macular oedema and thus stops the further deposition of hard exudates. The body can then naturally clear the existing hard exudates from the macula.
The specification
74 The specification is said to relate to the use of fenofibrate as a derivative thereof for the treatment of retinopathy.
75 The specification states at page 1 line 7 to page 2 line 5:
Diabetes is a disorder in which the body is unable to metabolize carbohydrates (e.g., food starches, sugars, cellulose) properly. The disease is characterized by excessive amounts of sugar in the blood (hyperglycemia) and urine, inadequate production and/or utilization of insulin, and by thirst, hunger, and loss of weight. Diabetes affects about 2% of the population. Of these 10-15% are insulin dependant (type 1) diabetics and the remainder are non-insulin dependant (type 2) diabetics.
Diabetic retinopathy represents one of the most debilitating microvascular complications of diabetes. Diabetic retinopathy is a specific microvascular complication of both type 1 and type 2 diabetes. After 20 years of diabetes nearly all patients with type 1 diabetes and over 60% of patients with type 2 diabetes have some degree of retinopathy. Additionally, retinopathy develops earlier and is more severe in diabetics with elevated systolic blood pressure levels. On average, a careful eye examination reveals mild retinal abnormalities about seven years after the onset of diabetes, but the damage that threatens vision usually does not occur until much later. It can lead to blindness in its final stage. It is the second leading cause of acquired blindness in developed countries, after macular degeneration of the aged. The risk of a diabetic patient becoming blind is estimated to be 25 times greater than that of the general population. At present there is no preventive or curative pharmacological treatment for this complication. The only treatment is laser retinal photocoagulation or vitrectomy in the most severe cases.
Diabetic retinopathy is a progressive diabetic complication. It advances from a stage referred to as “simple” or initial (background retinopathy) to a final stage referred to as “proliferative retinopathy” in which there is formation of fragile retinal neovessels, leading to severe hemorrhages, sometimes with detachment of the retina, and to loss of vision. The microvascular lesions in simple retinopathy are characterized by microaneurysms, small petechial hemorrhages, exudates and venous dilations. This simple retinopathy form can remain clinically silent for a long period of time. At this simple retinopathy stage cellular and structural deterioration of the retinal capillary can be observed in the postmortem examinations of retinas from diabetic patients, compared to the retinas from normal subjects of comparable age. If proliferative retinopathy is left untreated, about half of those who have it will become blind within five years, compared to just 5% of those who receive treatment.
76 There follows a discussion in relation to the treatment of diabetic retinopathy including laser treatment of pre-proliferative retinopathy, severe pre-proliferative or proliferative retinopathy.
77 The specification states at page 2 lines 25-33:
Preventing the development or progression of diabetic retinopathy has the potential to save vision at a relatively low cost compared to the costs associated with a loss of vision. Thus, it is an object of the present invention to provide further means which contribute to the prevention of the development or progression of diabetic retinopathy.
The present invention is based on the discovery that patients taking fenofibrate or a derivative thereof need fewer treatment [sic] by retinal laser therapy than placebo-allocated patients. The results obtained from a large clinical trial demonstrate the favourable effect of fenofibrate in the prevention of retinopathy.
78 Then follows a consistory statement that mirrors the language of claim 1. A number of terms are then defined. “Prevention” is defined as “preventing the development or progression of diabetic retinopathy.” The specification then states at page 3 lines 3-5:
According to the present invention, “diabetic retinopathy” is defined as severe non proliferative grade of diabetic retinopathy, proliferative grades of diabetic retinopathy, macular edema and hard exsudates [sic].
79 The expert evidence established that “non-proliferative” and “proliferative” stages of diabetic retinopathy are mutually exclusive in the sense that a patient cannot have both non-proliferative and proliferative retinopathy at the same time. In the circumstances, I consider that the skilled addressee would understand the definition of diabetic retinopathy to encompass any one or more of the various conditions referred to in the definition in a patient suffering from diabetes. This is how I interpret the definition of diabetic retinopathy as used in the claims.
80 The specification then refers to the association between type 2 diabetes and high cardiovascular risk and the fact that very few trials of lipid-lowering therapy have focused on type 2 diabetes. There is then a discussion of two previous studies trialling the use of fibrate therapy. The specification states at page 8 lines 8-11:
Both studies were limited to people with prior myocardial infarction (MI) and have reported reductions in major cardiovascular events among participants with low HDL and high triglyceride (TG) at baseline, which were greater than those seen with use of the same fibrate among those without dyslipidaemia.
81 There is then a reference to a third study which showed reduced progression of coronary artery disease in type 2 diabetes compared with placebo.
82 The specification then refers to what is known as the Field Study. The specification states at page 8 lines 21-29:
As fibrates are known to correct the typical dyslipidaemia of diabetes, their role in cardiovascular risk reduction in diabetes may be especially important. A study called Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) has been carried out which study is a multicentre, double-blind, placebo-controlled trail evaluating the effects on coronary morbidity and mortality of longterm treatment with fenofibrate to elevate high-density lipoprotein (HDL) cholesterol levels and lower triglyceride (TG) levels in patients with type 2 diabetes and total blood cholesterol between 3 and 6.5 mmol/L (115 and 250 mg/dL) at study entry.
83 The specification then proceeds to discuss the significance of elevated low-density lipoprotein (“LDL”) cholesterol and triglycerides (“TG”) levels in patients with type 2 diabetes. The specification states at page 9 lines 3-35:
Blood total cholesterol levels are not substantially different between patients with type 2 diabetes and those of nondiabetic populations of similar age and sex. However, evaluation of other lipoprotein fractions shows that those with diabetes more often have a below-average HDL cholesterol level and elevation of TG levels in the blood which together confer an independent additionnal [sic] risk of CHD. Furthermore, although low-density lipoprotein (LDL) cholesterol levels are not substantially raised, the HDL particle is often smaller and denser than in similar nondiabetic populations, which is considered to be a more atherogenic state. An increased number of LDL particles, as seen in diabetes, is reflected in an elevated level of plasma apolipoprotein B, a more powerful predictor of risk for cardiovascular events than either total cholesterol or LDL cholesterol.
The strength of cholesterol-CHD relationship is very similar for those with type 2 diabetes as for nondiabetics, although at a higher background rate of CHD. Evidence from Helsinki Heart Study, which tested long-term fibrate (gemfibrozil) use in hypercholesterolaemic men and women without prior coronary disease, showed a significant reduction in coronary events, with the reduction among the small numbers of people with diabetes not being separately significant but appearing somewhat greater. The reductions in events observed were greater than would have been expected on the basis of lowering of LDL cholesterol alone. So, whether substantially increasing low HDL cholesterol levels and reducing elevated triglyceride levels independently reduces cardiovascular events and mortality and should be a specific target for therapy remains less well agreed.
For patients with type 2 diabetes and its typical dyslipidaemia, many physicians believe that fibrates are the logical first choice of drug treatment. The fibrates have been in clinical use for a long time, being well tolerated and with few short-term side -effects. Fenofibrate has been widely used and marketed for more than 20 years and is an effective agent for reducing plasma triglyceride and raising HDL cholesterol. Although the effects on lipid fractions may vary with the population under study, a fall of 15 % or more in total cholesterol, mediated through a reduction in LDL cholesterol, is often seen with long-term use. In parallel, HDL cholesterol elevation of 10-15 % is common, together with large reductions in plasma triglycerides of 30-40 %. In addition, a reduction in plasma fibrinogen of about 15 % has been observed.
84 This is followed by a detailed discussion of the Field Study which is said to have been designed to provide the first properly randomized evidence as to whether fenofibrate confers a benefit on clinical cardiovascular events in persons with type 2 diabetes. The principal study outcome was said to be the combined incidence of first nonfatal MI or CHD death among all randomized patients. Various secondary outcomes are then described including the effect of fenofibrate on major cardiovascular events and mortality. There follows a brief discussion of what are referred to as tertiary outcomes which are said to include at page 11 lines 12-15:
… the effects of treatment on development of vascular and neuropathic amputations, nonfatal cancers, the progression of renal disease, laser treatment for diabetic retinopathy, hospitalization for angina pectoris, and numbers and duration of all hospital admissions.
85 The specification includes a table (Table 1) describing the baseline characteristics of the 6051 subjects who participated in the study.
86 The specification then refers to the results for what is described as the tertiary outcome related to the laser treatment for diabetic retinopathy. These are said to show that more placebo-allocated patients (253 or 5.2%) than fenofibrate-allocated patients (178 or 3.6%) required one or more laser treatments for retinopathy. The specification states at page 14 lines 23-27:
These results provide the first evidence of the favourable effect of fenofibrate on the need for retinal laser therapy. As the patients taking fenofibrate had fewer treatment by retinal laser therapy than the placebo-allocated patients, the prevention and treatment of retinopathy by fenofibrate has been clearly demonstrated.
The claims
87 The relevant claims of the 711 Patent are for:
1. Use of fenofibrate or a derivative thereof for the manufacture of a medicament for the prevention and/or treatment of retinopathy, in particular diabetic retinopathy.
…
5. Use according to any of claims 1 to 4, wherein said medicament contains 200 mg, 160 mg, 145 mg or 130 mg of fenofibrate or a derivative thereof.
6. Use according to any of claims 1 to 5, wherein said medicament is an oral formulation of fenofibrate or a derivative thereof.
7. A method for the prevention and/or treatment of retinopathy, the method comprising administration of fenofibrate or a derivative thereof to a patient in need thereof.
…
10. The method according to any one of claims 7 to 9 wherein said method further comprises administration of a statin.
11. The method according to any one of claims 7 to 10 wherein 200 mg, 160 mg, 145 mg or 130 mg of fenofibrate or a derivative thereof is administered.
12. The method according to any one of claims 7 to 11 wherein the fenofibrate or derivative thereof is administered in an oral formulation.
88 Claims 1, 5 and 6 are a Swiss-style claim.
89 Claims 5 and 6 of the 711 Patent are dependent on claim 1. It is not disputed by the respondent that if claim 1 is infringed, claims 5 and 6 will also be infringed.
90 Claims 7 and 10-12 are method of treatment claims. It is not disputed by the respondent that if claim 7 is infringed, each of claims 10-12 will also be infringed.
Infringement
91 The applicants contend that the respondent threatens to directly infringe claims 1, 5 and 6 of the 711 Patent (the Swiss-style claims), and to indirectly infringe claims 7 and 10-12 of the 711 Patent (the method of treatment claims) pursuant to s 117(1) when read with s 117(2)(b) of the Patents Act 1990 (Cth) (“the Act”). The applicants do not rely on s 117(2)(c) of the Act for the 711 Patent.
Swiss-style claims
92 Swiss-style claims were considered by Yates J in Otsuka Pharmaceutical Co Ltd v Generic Health Pty Ltd (No 4) (2015) 113 IPR 191 (“Otsuka”) at [100]-[121]. As his Honour explained, such claims are appropriately characterised as claims to a method or process for the manufacture of a known medicament where the novelty of the claim arises from the therapeutic use to which the medicament is applied.
93 It is useful to first consider the respondent’s submission that a Swiss-style claim cannot be infringed by importation and supply in the patent area of a medicament that is manufactured outside the patent area. The submission is based on a legal argument as to the proper interpretation of the word exploit as defined for the purposes of the Act. I do not accept the submission for the reasons stated in the judgment in Apotex Pty Ltd v Warner-Lambert Company LLC (No 2) (2016) 122 IPR 17 (“Apotex No 2”) at [296] – [298] and, on appeal, Warner-Lambert Company LLC v Apotex Pty Limited (No 2) (2018) 129 IPR 205 at [167] – [168] (“Warner-Lambert No 2”).
94 However, it does not follow that the applicants have established that the Swiss-style claims will be infringed. In order to decide whether such a finding is justified, it is necessary to say a little more about the requirements of a Swiss-style claim.
95 In Warner Lambert Company LLC v Generics (UK) Ltd [2018] UKSC 56, the United Kingdom Supreme Court accepted that a Swiss-style claim for a second medical use of a drug (pregablin) was “purpose limited” in the sense that the monopoly claimed is a monopoly for the preparation or manufacture of a medicament for the designated purpose, namely, the treatment of a particular medical condition.
96 One of the principal issues that was considered by the Supreme Court (even though it was strictly not necessary to decide the point for the purpose of disposing of the appeal) was the proper construction of Swiss-style claims, especially those involving second medical uses. At the hearing of the appeal before the Supreme Court, the parties conceded that, as a matter of construction, a Swiss-style claim required a “mental element” in the sense that the manufacturer must have manufactured the relevant medicament with the intention that it would be used to treat the medical condition designated in the claim. The judgments given make clear that all members of the Supreme Court considered that this concession was rightly made. The crux of the debate before their Lordships was whether the mental element involved an objective or a subjective intention. The majority held that, properly construed, a Swiss-style claim requires that the manufacturer who makes the relevant medicament do so with the objective intention that it be used to treat the medical condition designated in the claim.
97 Lord Sumption referred to the distinction between subjective intention and objective intention which he described as “legally fundamental”. His Lordship said at [72]:
…Subjective intention is a state of mind, ascertained as a matter of fact. A person may subjectively intend X if, for whatever reason, he deliberately does an act which is liable to bring X about, desiring it to happen. The degree of probability of X occurring may be relevant to the question whether it should be inferred as a fact that such a desire existed, but that is a question of proof and not of principle. Objective intention by comparison is not so much a matter of fact as an artificial construct for attributing legal responsibility. A person is taken to intend the ordinary and natural consequences of his acts. He objectively intends those consequences if they were foreseeable to a reasonable person, whether or not they were actually foreseen by him. Policy considerations may determine the degree of probability with which the consequence must be foreseeable if legal responsibility is to be attributed on that basis.
98 For reasons which will become apparent, it is not necessary to decide in the present case whether the Swiss-style claims are concerned with the objective or the subjective intention of the manufacturer. However, it seems to me that the reasons given by those of their Lordships who favoured the “objective intention” are particularly persuasive, especially in so far as they highlight the difficulties that may be involved in ascertaining the subjective intention of a manufacturer, an intention that may or may not be carried into effect, and about which distributors, pharmacists, medical practitioners and patients often will have little or no knowledge.
99 The applicants submitted that a Swiss-style claim will be infringed if the manufacturer has prepared the relevant medicament knowing that it is suitable for use in the treatment of the condition designated in the claim. On their analysis, the manufacturer’s intention is irrelevant to the question of infringement. In support of their submission the applicants referred to the following passage in the judgment of Yates J in Otsuka at [172]:
For the purpose of determining infringement of a Swiss type claim, does it matter that the alleged infringer does not actually advertise or promote the medicament specifically for the therapeutic use defined in the claim? I do not think it necessarily does. The question is whether, objectively ascertained, the medicament that results from the claimed method or process is one that has the therapeutic use defined in the claim. The question is not really about how the alleged infringer markets its product, although, plainly, its conduct in that regard may well assist in determining, objectively, whether the accused product has the claimed therapeutic use.
100 The principal point that was considered by Yates J in Otsuka concerned the question of whether a Swiss-style claim should be characterised as a process or method claim (a matter of importance given the statutory definition of exploit) rather than the true scope of the purpose limiting requirement of a Swiss-style claim. In fact, it does not appear from my reading of his Honour’s judgment that any party addressed the latter issue. The particular observation relied upon by the applicants (in the last sentence of the quoted paragraph) while directed to the question of suitability for a particular therapeutic use, does not indicate that a Swiss-style claim imposes no requirement as to the purpose or intention of the manufacturer. In my view that aspect of Swiss-style claims was not a matter that was addressed by his Honour.
101 Suitability for use cannot be determinative of the question of infringement of a Swiss-style claim. If it were, it would render a person liable for infringement who manufactured a medicament for the purpose of using it to treat an indication which it had been used to treat before the priority date of the Swiss-style claim merely because the medicament might also be used for the purpose of treating a second indication that provided the novelty-conferring subject matter of the claim. If the applicants’ submission is correct, there would be an infringement even if the manufacturer took steps to ensure that the product was not used to treat the designated condition, and had no reason to believe that it would be so used. I do not think there is any doubt that this would be an absurd result, and contrary to the policy behind modern patent legislation.
102 The crucial question is whether the manufacturer has made (or will make) the relevant medicament with the intention that it be used in the treatment of the designated condition. In my view, the answer to that question is to be ascertained objectively in light of all the relevant facts and circumstances including, in particular, the approved product information (“PI”) for the product, any product labelling, and the nature, size and other pertinent characteristics of the market into which the product is to be sold.
103 Of course, it is for the applicants to establish that the manufacturer has made (or will make) the relevant medicament with the objective intention that it be used to treat the condition designated in the Swiss-style claims. The fact that it may be reasonably foreseeable or even likely that a substantial portion of the product manufactured will be used to treat that condition is certainly not determinative at least not where the product is also used extensively in the treatment of other non-designated conditions.
104 The Ranbaxy Products were originally approved for the same indications as the applicants’ fenofibrate product marketed under the name Lipidil. In particular, the Ranbaxy Products were indicated “for the reduction in the progression of diabetic retinopathy in patients with type 2 diabetes and existing diabetic retinopathy”. This was recorded in the PI for the Ranbaxy Products, as was supporting clinical trial information for that indication. However, the PI for the Ranbaxy Products has since been amended to remove any reference to diabetic retinopathy. According to the amended PI, fenofibrate is indicated as an adjunct to diet in the treatment of hypercholesterolaemia, various types of dyslipidaemia, and dyslipidaemia associated with type 2 diabetes.
105 The applicants referred to the following three matters in support of their infringement case:
(a) the amended PI for the Ranbaxy Products asserts that the Ranbaxy Products are bioequivalent to Lipidil;
(b) the PI for Lipidil expressly states that Lipidl is indicated for the reduction in the progression of diabetic retinopathy and includes details of the FIELD and ACCORD trials relevant to that indication; and
(c) the PI for the Ranbaxy Products does not assert that the Ranbaxy Products are not indicated for diabetic retinopathy or that it is somehow not relevantly bioequivalent for that purpose.
106 The applicants submitted that these matters demonstrated that there is a sound medical basis for considering that the Ranbaxy Products are suitable to be administered for the therapeutic use designated in each of the Swiss-style claims. Although I accept this submission, for the reasons I have stated it is not to the point, and does not provide a sufficient basis for finding that any of the Swiss-style claims have been, or will be, infringed by the respondent. Further, the three matters to which the applicants referred do not indicate that the Ranbaxy Products have been, or will be, made for the purpose of being used for the prevention or treatment of diabetic retinopathy. In my view the evidence does not support a finding that the Ranbaxy products will be manufactured with the intention that they be used for the prevention or treatment of diabetic retinopathy. The infringement case based on the Swiss-style claims must fail.
Method of treatment claims
107 Section 117 of the Act provides:
117 Infringement by supply of products
(1) If the use of a product by a person would infringe a patent, the supply of that product by one person to another is an infringement of the patent by the supplier unless the supplier is the patentee or licensee of the patent.
(2) A reference in subsection (1) to the use of a product by a person is a reference to:
(a) if the product is capable of only one reasonable use, having regard to its nature or design—that use; or
(b) if the product is not a staple commercial product—any use of the product, if the supplier had reason to believe that the person would put it to that use; or
(c) in any case—the use of the product in accordance with any instructions for the use of the product, or any inducement to use the product, given to the person by the supplier or contained in an advertisement published by or with the authority of the supplier.
108 It is not necessary for a patentee to succeed under s 117 that he or she identify any particular person or persons who the supplier has reason to believe will use the product in question in an infringing manner: AstraZeneca AB v Apotex Pty Ltd (2014) 226 FCR 324 (“AstraZeneca FC”) at [433].
109 Section 117(2)(b) proceeds on the basis that a product (not being a staple commercial product), may be capable of various uses, some of which would infringe the claims of the patent in suit, and others that would not. The respondent did not contend that fenofibrate is a staple commercial product for the purposes of s 117(2)(b).
110 In Apotex Pty Ltd v Sanofi-Aventis Australia Pty Ltd (2013) 253 CLR 284 Crennan and Kiefel JJ construed the method of treatment claim in the patent in suit in that case as limited to use of the relevant method for a specific purpose. Their Honours said at [289] that “[c]laim 1 is recognisably a claim limited to the specific purpose of preventing and treating psoriasis.” Hayne J adopted an analysis similar to that of the Full Court in Apotex Pty Ltd v Sanofi-Aventis Australia Pty Ltd (No 2) (2012) 204 FCR 494, where Keane CJ said at [37]:
The claim is for a method of treatment of a human ailment. That method necessarily presupposes a deliberate exercise of diagnosis and prescription by a medical practitioner. The treatment or prevention of psoriasis by the use of the claimed method presupposes a diagnosis of psoriasis and the consequent prescription of the application of leflunomide. That this is so is hardly controversial: as is apparent from all the potential constructions of the patent advanced by the parties, the claim in the patent involves the application of leflunomide by a medical practitioner.
111 The applicants accept that claim 7 and its dependent claims are properly construed as claims that require the deliberate exercise of diagnosis and prescription by a medical practitioner (based on sound medical evidence) of fenofibrate (or a derivative thereof) for the purpose of preventing or treating diabetic retinopathy.
112 The applicants submitted that if the respondent had reason to believe that a medical practitioner would, based on sound medical evidence, diagnose and prescribe the Ranbaxy Products for the purpose of preventing or treating diabetic retinopathy then any supply of the Ranbaxy Products by the respondent is an infringement of the relevant method of treatment claims.
113 In support of its contention that the respondent has the necessary reason to believe, the applicants relied upon the same three matters to which I previously referred in the context of the Swiss-style claims.
114 The evidence does not permit me to draw any precise conclusions as to the extent to which fenofibrate is currently prescribed by medical practitioners in order to treat diabetic retinopathy. The applicants submitted that about 50% of the time that fenofibrate is currently prescribed, it is prescribed solely for the purpose of treating diabetic retinopathy. The evidence relied upon by the applicants in support of that submission is mostly anecdotal and not very persuasive. In any event, the submission indicates that, on the applicants’ own case, in about 50% of the cases in which fenofibrate is prescribed, it is prescribed either to manage the patient’s lipid levels or to manage lipid levels and to prevent or treat diabetic retinopathy.
115 I think it likely that fenofibrate is mostly prescribed for the purpose of managing a patient’s lipid levels in circumstances where the medical practitioner either wishes merely to manage lipid levels or both manage lipid levels and reduce the development or the progression of diabetic retinopathy. On any view, a large proportion of the fenofibrate that is currently prescribed is for what should be considered non-infringing use. This includes cases in which fenofibrate is used specifically for the purpose of reducing levels of triglycerides, increasing levels of HDL-C or, more generally, to control lipid levels in patients who will not tolerate a statin.
116 Medical practitioners do not generally identify on a prescription for fenofibrate the indication for which the drug has been prescribed. Practitioners instead keep patient notes recording their diagnosis and (at least implicitly) their reasons for making the prescription. Most medical practitioners generally do not tick the "brand substitution not permitted" box on the prescription. If ticked, in most cases, that would prevent the pharmacist from offering a patient the Ranbaxy Products if the prescription had been written for fenofibrate. If the “brand substitution not permitted” box has not been ticked, regardless of whether the brand name is used on the prescription, the pharmacist will typically offer the patient a generic product if it is available, which most patients will accept, particularly if it results in cost savings.
117 The applicants submitted that if the Ranbaxy Products were launched, medical practitioners could be expected to permit the supply of (by not ticking the “brand substitution not permitted” box) the Ranbaxy Products for the prevention or treatment of diabetic retinopathy, regardless of whether “fenofibrate” or “Lipidil” is specified on the prescription. As to the content of the PI for the Ranbaxy Products and the absence of any reference to diabetic retinopathy, the applicants submitted:
Medical practitioners do not typically read the PI for a generic product, but assume that the indications for the generic will be identical to the original drug unless any difference is specifically drawn to their attention.
In any event, if they read the PI for the Ranbaxy Products, it sets out the information relevant to the utility of the drug for diabetic retinopathy, in that it says it is bioequivalent to Lipidil, which is commonly known to be indicated for diabetic retinopathy and does not state any reason why the Ranbaxy Products cannot be used to prevent or treat that condition.
Independent third-party materials directed to medical practitioners, such as the RACGP Diabetes Guidelines, refer to the TGA having approved the use of fenofibrate (not specifically Lipidil) for the treatment of diabetic retinopathy.
118 I accept this submission which is in my view supported by unchallenged evidence. Further, the matters referred to by the applicants in their submissions are matters that are likely to be known to the respondents at the time of supply of the fenofibrate to wholesalers or pharmacists. In those circumstances, I am satisfied that the respondent has reason to believe that a significant portion of the Ranbaxy Products that it proposes to supply will be used in a manner which would infringe claims 7, 10, 11 and 12 of the 711 Patent.
119 Accordingly, I am satisfied that the respondent threatens to indirectly infringe claims 7 and 10-12 (the method of treatment claims) by supplying the Ranbaxy Products in the patent area in the circumstances referred to in s 117(1) when read with s 117(2)(b) of the Act.
Validity
120 The validity issues that arise in relation to the 711 Patent are as follows:
(a) Whether the relevant claims lack novelty having regard to each of the following documents:
(i) The ACCORD Eye Study Protocol: Version January 2004 (“the Accord Protocol”); and
(ii) European Patent Application EP D 482 498 A (“the Squibb Patent”) when read with the Physicians’ Desk Reference, 59th Edition 2005 at pages 523-525 and 1325-1328 (“the Desk Reference”).
(b) Whether the relevant claims lack novelty having regard to the following acts:
(i) The prescribing by medical practitioners, dispensing and administering to patients with type 2 diabetes and dyslipidaemia oral formulations containing fenofibrate (including Lipidil 67 mg capsules and 160 mg tablets) to prevent or treat the symptoms and complications of diabetes including diabetic retinopathy;
(ii) The prescribing by medical practitioners, dispensing and administering to patients with type 2 diabetes of an oral formulation containing 160mg of fenofibrate to prevent or treat the symptoms and complications of diabetes including diabetic retinopathy in the course of the Accord Study;
(iii) The prescribing by medical practitioners, dispensing and administering to patients with type 2 diabetes of an oral formulation containing 200mg of fenofibrate to prevent or treat the symptoms and complications of diabetes including diabetic retinopathy in the course of the Field Study.
(c) Whether the invention, as claimed, lacks any inventive step:
(i) in light of the common general knowledge;
(ii) in light of the common general knowledge together with the information made publicly available in the documents referred to in (a) above?
(iii) in light of the common general knowledge together with any of the information made publicly available through any one of the acts referred to in (b) above.
121 The respondent carries the legal burden of establishing that the relevant information was made “publicly available” before the 10 November 2005 priority date. As Crennan J explained in JMVB Enterprises Pty Ltd v Camoflag Pty Ltd (2005) 67 IPR 68 (“JMVB”) at [53]-[54]:
[53] Prior art will only be relevant to the consideration of novelty or inventive step if it was “made publicly available” before the priority date of the claim in question. The respondent bears the onus of proof with respect to establishing that the prior art information was “publicly available” at the relevant priority date: s 7(1) Patents Act.
[54] The requisite degree of publication will be met if the prior publication or act communicates the information to any one member of the public in a manner which left that person free, in law and in equity, to make use of the information: Humpherson v Syer [1887] 4 RPC 407; Stanway Oyster Cylinders Pty Ltd v Marks (1996) 66 FCR 577 at 581; 144 ALR 627 at 630–1; 35 IPR 71 at 74–5 …
See also Insta Image Pty Ltd v KD Kanopy Australasia Pty Ltd (2008) 78 IPR 20 at [124].
Novelty
122 The respondent contended that the relevant claims were not novel in light of one or more publications or prior acts that made information publicly available before the priority date: see s 18(1)(b)(i) and s 7(1) of the Act.
Documentary Disclosures
123 When considering whether a documentary disclosure constitutes an anticipation it is necessary to consider the document as at the date of its publication and in light of the common general knowledge as it stood at that time. This proposition is in my view supported by a preponderance of authority including the decisions in General Tire & Rubber Co v Firestone Tyre & Rubber Co Ltd [1972] RPC 457 at 485, JMVB at [55] and Bradken Resources Pty Ltd v Lynx Engineering Consultants Pty Ltd (2012) 210 FCR 21 at [219].
124 Other relevant principles were discussed by the Full Court in AstraZeneca FC. With regard to sufficiency of disclosure, the plurality said at [299]-[302]:
[299] … In Meyers Taylor Pty Ltd v Vicarr Industries Ltd (1977) 137 CLR 228 Aickin J (at 235) said:
The basic test for anticipation or want of novelty is the same as that for infringement and generally one can properly ask oneself whether the alleged anticipation would, if the patent were valid, constitute an infringement.
[300] But it is important to note that the reverse infringement test is not applied by simply asking whether something within the prior art document would, if carried out after the grant of the patent, infringe the invention as claimed. In Flour Oxidizing Company Ltd v Carr & Company Ltd (1908) 25 RPC 428, Parker J (at 457) observed:
… where the question is solely a question of prior publication, it is not, in my opinion, enough to prove that an apparatus described in an earlier Specification could have been used to produce this or that result. It must also be shown that the Specification contains clear and unmistakable directions so to use it.
[301] These observations are the wellspring of a long line of cases that recognise that, in order for a prior art document to be anticipatory, there must be (to adopt the language in General Tire) a clear description of, or clear instructions to do or make, something that would infringe the patentee’s claim if carried out after the grant of the patentee’s patent. In Bristol-Myers Squibb Company v FH Faulding & Company Ltd (2000) 97 FCR 524 (Bristol-Myers), Black CJ and Lehane J reviewed the relevant authorities and concluded (at [67]):
What all of those authorities contemplate, in our view, is that a prior publication, if it is to destroy novelty, must give a direction or make a recommendation or suggestion which will result, if the skilled reader follows it, in the claimed invention. A direction, recommendation or suggestion may often, of course, be implicit in what is described and commonly the only question may be whether the publication describes with sufficient clarity the claimed invention or, in the case of a combination, each integer of it. But in this case medical practitioners hardly needed to be told that it was possible to infuse a particular dose of taxol over three hours, or how to do it. Nor, equally obviously, is that the point of the claims. The claims of the earlier of the petty patents are for a method for administration of taxol to a patient suffering from cancer; the claims of the later one are for a method of treating cancer. In each case the method involves a particular regimen for the infusion of taxol. The context was that great difficulties had been encountered in using taxol, despite its known anti-carcinogenic properties, in the treatment of cancer, because of the drug’s side effects. Each of the trials reported in the articles referred to was an investigation directed towards finding a solution of the difficulties: directed, particularly, to ascertaining safe dosage levels. But, though methods falling within the claims of the patents were used in each trial, none of the reports can be said to teach (a word which in this context encompasses direct, recommend and suggest) that which the petty patents claim.
[302] Sufficiency of disclosure is a cardinal anterior requirement in the analysis of whether a prior art document anticipates a claimed invention. It is only after the stage of assessing the sufficiency of disclosure — which involves a determination about whether a prior document has “planted the flag” as opposed to having provided merely “a signpost, however clear, upon the road” or, perhaps, something less — that the notion of reverse infringement comes into play as the final and resolving step of the required analysis. It is not the first step of the required analysis; nor is it the only step.
125 I should also refer to the Full Court’s decision of Merck & Co Inc v Arrow Pharmaceuticals Ltd (2006) 154 FCR 31 (“Merck”) in the patentee’s appeal against the decision of the primary judge (Gyles J) (see Arrow Pharmaceuticals Ltd v Merck & Co Inc (2004) 63 IPR 85). Claim 3 of the patent in suit in that case claimed a method of treatment involving the oral administration of a drug “according to a continuous schedule having a dosage interval which is once weekly.” Gyles J found that this claim was anticipated by the publication of an article in Lunar News (relevant parts of which appear at [25] of his Honour’s reasons) which included the following statement:
An intermittent treatment program (for example, once per week, or one week every three months), with higher oral dosing, needs to be tested.
126 Gyles J found (at [104]-[105]) that the Lunar News article was an anticipation of claim 3 because the article disclosed a continuous schedule of oral administration having a dosage interval which was once weekly. His Honour said at [107]:
It is submitted for Merck that the decision of the Full Court in Faulding establishes that to suggest that something needs to be tested is not to anticipate that which is suggested. Black CJ and Lehane J said (at FCR 548–9 [67]; ALR 461; IPR 576):
What all those authorities contemplate, in our view, is that a prior publication, if it is to destroy novelty, must give a direction or make a recommendation or suggestion which will result, if the skilled reader follows it, in the claimed invention.
That statement of principle was based upon a review of certain of the authorities. Those authorities stand for the proposition that the claimed invention must be disclosed as such and not simply as a possibility. If the Lunar News article had said “in view of these problems a continuous dosing schedule with various intervals greater than one day should be tested” it would not anticipate claim 3, even though a weekly dosage interval would be both technically and practically contemplated by that suggestion. On the contrary, here, the disclosure is quite precise and accords with the gist of the claimed invention. I do not accept the submission on behalf of Merck that the passage in question from Faulding adds an additional requirement for anticipation, namely that the publication should recommend the use of the invention as disclosed. That is not what the passage from Faulding says and it does not follow from the authorities analysed in that judgment. The essential difference in the treatment of the prior publications in Faulding lay in the view that one publication pointed clearly to one solution which was the invention rather than other publications which did not so point. That was a factual rather than a legal judgment and cannot be translated to the present circumstances.
127 On appeal, the Full Court rejected the patentee’s challenge to Gyles J’s findings on novelty. The Full Court referred to [107]-[108] of Gyles J’s reasons with apparent approval, clearly endorsing (at [112]) his Honour’s reasoning in relation to the Lunar News article. As to the need for further testing or clinical trials, the Full Court in Merck referred at [104]-[107] to a number of authorities which distinguished between steps leading to the discovery of the invention and subsequent checking and testing and remarked at [109]-[110]:
[109] Merck’s argument sought to rely by way of analogy on a detailed examination of the various publications which the Full Court in Faulding held were or were not anticipations. But, as the primary judge in the present case pointed out, these were questions of fact. We do not read Faulding as support for any proposition of law that, in the case of pharmaceutical patents, no publication can amount to an anticipation unless clinical trials have been actually conducted.
[110] In any event, Faulding at [67] speaks of a requirement that an alleged anticipation “teach”, in the sense of “direct, recommend or suggest”, that which is claimed. So a characterisation of an alleged anticipation as suggestion is not necessarily fatal to a novelty argument. Nor is it necessary that a reader, or even all readers, agree with the suggestion. Thus the fact that a Merck medical witness said that he would not have followed the Lunar News article for fear of side effects, does not of itself mean that disclosure was insufficient.
128 As the Full Court makes clear in Merck at [110], it is not necessary that a reader agrees with the teaching. Nor is it an answer to say that the method disclosed is one that the skilled addressee would be unwilling to perform without first undertaking clinical trials. What is essential is that the prior disclosure conveys a direction, recommendation or suggestion which, if it were to be followed by the skilled addressee, would result in the claimed invention.
The Accord Eye Study
129 The ACCORD Eye Study was a sub-study conducted within a larger study known as the ACCORD trial. The ACCORD trial was funded by the National Heart, Lung and Blood Institute which is a division of the United States’ National Institute of Health (“NIH”), the nation’s medical research agency.
130 The ACCORD trial was designed to evaluate the effects on major cardiovascular disease events of:
intensive glycaemia control (that is, intensive control of blood glucose levels) (“the glycaemia trial”);
fenofibrate treatment (one 160 mg tablet once daily) to increase HDLcholesterol levels and lower triglyceride levels, in combination with simvastatin (an average of about 22 mg) to reduce total cholesterol and LDL-cholesterol levels (“the lipid trial”); and
intensive blood pressure control (“the blood pressure trial”).
131 All of the 10,251 patients who participated in the ACCORD trial took part in the glycaemia trial. The participants who were assigned to the treatment arm of the glycaemia trial received intensive control of blood glucose levels. The participants assigned to the control arm of the glycaemia trial received less intensive, conventional therapy for control of blood glucose levels.
132 Of the 10,251 patients involved in the glycaemia trial, 5,518 patients were also involved in the lipid trial (in which those in the treatment arm received fenofibrate in combination with a statin, while those in the control arm received a statin only) and 4,725 patients were also involved in the blood pressure trial (in which those in the treatment arm received intensive control of blood pressure, while those in the control arm received less intensive, conventional therapy for the control of blood pressure).
133 The primary outcome measure for the ACCORD trial was the first occurrence of a major cardiovascular event, specifically non-fatal myocardial infarction (that is, a non-fatal heart attack), non-fatal stroke, or cardiovascular death. Secondary outcome measures included treatment differences in other cardiovascular outcomes, total mortality, microvascular outcomes, health-related quality of life, and cost-effectiveness. The analyses of the results of the ACCORD trial were published in the New England Journal of Medicine in June 2008 (in relation to the glycaemia trial) and in April 2010 (in relation to the lipid trial and blood pressure trial).
134 In addition to the glycaemia, lipid and blood pressure trials, the ACCORD trial also involved a number of ancillary studies. These included an eye sub-study known as the ACCORD Eye Study (“the Eye Study”).
135 The Protocol for the Eye Study (“the Protocol”) is dated 30 January 2004. The Protocol was made available to investigators in the ACCORD trial before November 2005. Professor Elam, who was a Principal Investigator for the ACCORD trial, was also involved in the design and conduct of the Eye Study as a member of the ACCORD Eye Study Group. Professor Elam, in his capacity as a Principal Investigator for the ACCORD trial, and as a member of the ACCORD Eye Study Group, read the Protocol a number of times, and was familiar with its contents.
136 Professor Elam said that he provided the Protocol to the clinical coordinator and other investigators at the Memphis Veterans Administration Hospital. He said that he also discussed the Protocol with his fellow investigators and that a study coordinator working under his supervision discussed the information in the protocol with patients with type 2 diabetes who were participants in the ACCORD trial and who were potentially eligible for the Eye Study.
137 Professor Elam said he also discussed the Protocol with other physicians at conferences and meetings of professional societies and organisations before November 2005 to raise broader awareness of the Eye Study among physicians and patients, in the hope of having more patients referred for assessment.
138 Professor Elam said that he could not recall any restriction on the publication or communication of the Protocol. Nor could he recall being asked to agree to or give any undertaking to keep the Protocol confidential or any suggestion or request that the information in it be kept confidential. His evidence was that it was his understanding that he was not under any restriction that prevented him from disclosing the Protocol or any of its contents to other people, including people who were not involved in the study.
139 Professor Elam said that in his experience, there was a general practice of marking documents relating to clinical trials “confidential” where it was intended that those documents be kept confidential, but that this was not done for the Protocol. He said he was also aware that in the case of trials funded by NIH, such as the ACCORD trial, there was a policy that information about those trials, including trial protocols, be made freely accessible.
140 It was suggested to Professor Elam in cross-examination that, although the Eye Study may have been funded by NIH, the drugs used in the trial were supplied free of charge by their manufacturers. Although it was not put to him directly, the suggestion seemed to be that it was unlikely that these companies would be making drugs available free of charge for use in the Eye Study without confidentiality agreements in place. I do not think this follows at all, particularly in circumstances where the Eye Study was exploring the effectiveness of drugs that were already widely prescribed. I also note that the article by Chew et al (of which Professor Elam was a co-author) published in the New England Journal of Medicine and reporting the results of the Eye Study noted that, although the study drugs were donated by the manufacturers, they “… did not participate in the study design or conduct, data accrual or analysis, or manuscript preparation.”
141 The applicants relied on evidence given by Professor O’Brien that protocols for clinical trials are typically considered confidential and only made available to investigators, ethics committee, regulatory bodies, and funding bodies in that context, and that investigators were typically required to sign confidentiality agreements relating to their participation in clinical trials. They also relied on evidence from Professor Mitchell who said he did not consider it likely that an ophthalmologist not involved in the ACCORD trial would have had access to the Protocol. However, this evidence, which was expressed at a very high level of generality, failed to engage with the much more direct evidence given by Professor Elam. I do not find evidence given by Professor O’Brien or Professor Mitchell on this point persuasive.
142 Professor Elam’s cross-examination confirmed, in my view, that although he had no specific recollection of discussions at the time, he did not believe, either now or prior to November 2005, that there were any confidentiality restrictions communicated to him and that any such restriction would have been, he believed, contrary to the NIH policy at the time.
143 It was suggested to Professor Elam in cross-examination that the NIH policy document dated 21 September 2016 entitled “NIH Policy on the Dissemination of NIH – Funded Clinical Trial Information” (which did not take effect until 18 July 2017) was inconsistent with his recollection of NIH policy at the time of the Eye Study. I do not think it is. There is nothing in that document to suggest that the NIH policy prior to the new policy coming into effect was anything other than as described by Professor Elam.
144 Professor Elam’s evidence, which I accept, satisfies me that the investigators to whom the Protocol was provided were not the subject of any express or implied confidentiality obligation that prevented them from disclosing the Protocol or any of its contents to other people and that they were not the subject of any obligation, in law or in equity, that prevented them from doing so.
145 According to Professor Elam, the administrators of the ACCORD trial maintained a website at www.accordtrial.org where information concerning the ACCORD trial was publicly accessible prior to November 2005. In addition, the administrators of the Eye Study maintained a website at www.accordanc.org where information concerning the Eye Study was publicly accessible prior to November 2005. He and his fellow investigators at the Memphis Veterans Administration Hospital, as well as his colleagues involved with the steering committee of the ACCORD trial and in the ACCORD Eye Study Group, accessed these websites for the purposes of reviewing and sharing information concerning the ACCORD trial and the Eye Study, including the Protocol. He also recalled that there was a secure part of those websites in which specific patient information was stored, which he (and other investigators) could access using login details and a password provided for that purpose.
146 This evidence, which was not challenged in cross-examination, plainly suggests that the Protocol was publicly available from the relevant website and was accessed by Professor Elam and other investigators before November 2005. It is consistent with the administrators electing not to impose any confidentiality obligations preventing or restricting access to the Protocol.
147 The respondent also relied on evidence given by Mr Ben Teeger who accessed historical versions of the ACCORD website via the Internet Archive Wayback Machine (“the Wayback Machine”). Mr Teeger’s evidence was that he searched for www.accordanc.org/eyeinfo.cfm using the Wayback Machine and accessed a version of the site which had previously been archived on 14 April 2004. The archived webpage he accessed, as captured by him, is reproduced below:
148 Mr Teeger’s evidence was that although he could click on and access the documents or locations identified in the bottom four hyperlinks via the archived version of the webpage, when he clicked on the first of those hyperlinks, which I infer was a link to the Protocol, he was unable to download that document. Instead, a message appeared that the Wayback Machine “doesn’t have that page archived.”
149 In his evidence, Mr Butler, the Office Manager of the Internet Archive, explained that this particular page may not have been archived for a range of reasons, including that the web crawler’s parameters may have been set in such a way to stop it from capturing all links, that the algorithm governing the web crawler’s behaviour may prioritise capturing images over links or particular pages over other pages, or that the document was password protected.
150 The applicants invited me to infer that the Protocol was not archived by the Wayback Machine on 14 April 2004 because it was password protected. This was one of the possible explanations given by Mr Butler.
151 The five hyperlinks (including “Download ACCORD-Eye Protocol”) appeared below the heading “ACCORD-EYE Sub Study Public Information”. In my view this strongly suggests that the five files or locations to which the five hyperlinks pointed were accessible by the public as at 14 April 2004. This is consistent with Professor Elam’s evidence, which I also accept, that the Protocol was publicly available via the ACCORD trial website before the priority date.
152 In the circumstances, I find that the information in the Protocol was made publicly available prior to November 2005 to Professor Elam and other investigators and to the public at large via the website www.accordanc.org/eyeinfo.cfm.
153 On page 1 the Protocol states:
5,800 dyslipidemic ACCORD participants (HDL < 40 mg/dl) will be randomly assigned in a double masked fashion to either a placebo or fenofibrate 160 mg daily for reduction of triglyceride levels and increase in high-density lipoprotein cholesterol levels, after low-density lipoprotein cholesterol has been lowered with statin therapy (simvastatin 20 mg daily) to target LDL levels of approximately 100 mg/dl or lower.
154 On page 3, the Protocol states that “A study designed to evaluate the effects of treatment on diabetic retinopathy within the ACCORD trial is described here.” On page 4, the document explains:
Diabetic retinopathy (DR) is an important complication of type 2 diabetes mellitus…. Clinically significant macular edema and proliferative retinopathy are major causes of vision loss, even to the point of legal blindness. …
Many patients in the older type 2 diabetes population studied in ACCORD have both DR and CVD and DR has been suggested to be a risk factor for CVD. For these reasons it is important to better delineate the relationship between DR and CVD and the relationship between their responses to control of glycemia and other risk factors.
155 On page 5, the Protocol states that:
The ETDRS study has shown a relationship between progression to high risk proliferative DR over 5 years and baseline serum triglycerides in the age group 50-69. Progression was 23% higher in those with serum triglycerides > 190 mg/dl versus those whose serum triglycerides were normal, after adjustment for 11 significant covariates. It might be noted parenthetically that in the type 1 diabetes DCCT Trial, although reduction in A1C levels appeared to be the major mechanism for the decrease in retinopathy produced by intensive glycemic management, the latter treatment also produced a significant decrease in serum triglyceride levels over the 6.5 years of follow-up. It is therefore reasonable to hypothesize that fibrate therapy which decreases serum triglycerides will reduce the risk of DR.
(footnotes omitted, emphasis added).
156 On page 6, the Protocol identifies the second aim of the study as being:
In type 2 diabetic patients whose low density lipoprotein cholesterol levels have been reduced appropriately by statin therapy, will the addition of fibrate therapy, to reduce triglyceride levels and raise high density lipoprotein cholesterol levels, decrease the risk of DR?
157 On page 7, the Protocol identifies the second primary hypothesis for the study as being:
In middle aged or older people with type 2 diabetes at high risk for having a CVD event: … In the context of good glycemic control, a therapeutic strategy that uses a fibrate to lower triglyceride levels and raise HDL cholesterol levels in patients already receiving a statin drug from the treatment of LDL cholesterol levels, will reduce the rate of development or progression of DR compared to a strategy that only uses a statin drug for treatment of LDL cholesterol levels.
158 The respondent submitted that the statements, in the context of a protocol describing the administration of a 160 mg fenofibrate tablet daily to patients with type 2 diabetes, recommend or suggest the use of fenofibrate for the purpose of preventing or treating diabetic retinopathy. In expressing its submission in these terms, the respondent was drawing on the language used by Black CJ and Lehane J in Bristol‑Myers Squibb Co v F H Faulding & Co Ltd (2000) 97 FCR 524 (“Bristol-Myers”) at [67] which is reproduced by the plurality in AstraZeneca FC at [301].
159 The applicants accepted that although the Protocol conveys a suggestion that fibrate therapy (in particular a 160 mg tablet taken daily), may reduce the risk of diabetic retinopathy in patients suffering from type 2 diabetes, it was not sufficient to amount to an anticipation. The crux of the applicants’ submission was that the Protocol did no more than articulate a hypothesis which may or may not be correct.
160 In support of their submission the applicants referred to the evidence of Professor O’Brien and Professor Mitchell who said that on a reading of the Protocol as a whole, they would not have been directed to use fenofibrate in the prevention or treatment of retinopathy. This is because, as I understood their evidence, neither regarded the hypothesis articulated in the Protocol as likely to produce to a positive result. Professor O’Brien said that he would not have been confident that lipid lowering therapy could reduce the risk of microvascular complications such as diabetic retinopathy. He said that he would not have prescribed fenofibrate for that purpose even if he was aware of the Protocol prior to November 2005 and would have awaited the outcome of the Eye Study before considering the use of fenofibrate therapy in patients with diabetic retinopathy. Professor Mitchell’s evidence was to a similar effect.
161 I think this evidence is answered by the Full Court’s observation in Bristol-Myers at [72]:
Prudent practitioners might well take the view that they would prefer to await the final outcome of the trials, both as to efficacy and as to safety, before rushing to embrace the proposed method. But, in our view, there can be no serious doubt that the abstract teaches the [invention].
162 In my view the Protocol suggested to the skilled addressee who read it prior to November 2005 that fenofibrate could be used in daily doses of 160 mg for the prevention and treatment of diabetic retinopathy. The fact that Professor O’Brien and Professor Mitchell would not have acted on this suggestion, preferring instead to await the outcome of clinical trials, is no answer to the proposition that the Protocol discloses the precise method of treatment that was later claimed. Nor is it an answer to say that the disclosure was made in the context of a proposed clinical trial aimed at testing a hypothesis.
163 It is not necessary in this case to decide whether the disclosure would be novelty defeating if the relevant hypothesis could be characterised as lacking any scientific support. However, in my view the evidence establishes that there was, at the very least, a sound scientific basis to believe that the hypothesis could well be proven correct. The observational data reported in the ETDRS Report No 22 by Chew EY, et al, published in 1996 in the Archives of Ophthalmology (“ETDRS 22”) indicated that elevated lipid levels were associated with an increased risk of retinal hard exudate formation, and that by lowering lipid levels, and with them the risk of retinal hard exudate formation, the risk of developing diabetic retinopathy in patients who did not already suffer from it, or the risk of the condition worsening in those who did, may well be reduced. This is the same process of reasoning that led Professor Carter to prescribe fibrates, including fenofibrate, prior to November 2005 for use by patients with type 2 diabetes who were at risk of developing diabetic retinopathy or who, if already suffering from that condition, were at risk of having it progress further.
164 I am satisfied that the Protocol clearly discloses a method of administering fenofibrate in a daily dose of 160 mg to patients suffering from type 2 diabetes who were already taking a statin in the expectation that this would reduce the risk of development or progression of diabetic retinopathy in those patients beyond what it would be were they to have continued to take a statin alone. Use of fibrate in accordance with this method would clearly infringe each of the method of treatment claims which are therefore invalid for lack of novelty. Further, since the novelty of the Swiss-style claims depends upon the use described being new, those claims are also invalid for lack of novelty in light of the use it is proposed in the Protocol for fenofibrate in 160 mg dosages.
The Squibb Patent when read with the Desk Reference
165 The Squibb Patent, published some 13 years before the priority date, described “a method for preventing diabetes and diabetic complications in mammalian species by administering a cholesterol lowering drug”. The Squibb Patent states at page 2 lines 1-26:
The present invention relates to a method for preventing diabetes and diabetic complications in mammalian species by administering a cholesterol lowering drug, such as an HMG CoA reductase inhibitor, such as pravastatin, alone or in combination with an ACE inhibitor, such as captopril, zofenopril, fosinopril, enalapril, ceronapril or lisinopril.
In accordance with the present invention, a method is provided for preventing diabetes or preventing onset of or reducing risk of complications normally associated with diabetes in mammalian species, wherein a therapeutically effective amount of a cholesterol lowering drug alone or in combination with an angiotensin converting enzyme inhibitor, is administered systemically, such as orally or parenterally to a diabetic patient.
The term “complications normally associated with diabetes” includes atherosclerosis (leading to myocardial infarction and cerebral ischemia), peripheral artery disease, nephropathy, retinopathy and neuropathy.
The term “diabetes” or “diabetes mellitus” as employed herein includes Type I diabetes and Type II diabetes.
It is theorized that low density lipoproteins (LDL-C) may be responsible for nephrotoxicity. Use of the cholesterol lowering drug in diabetics, in accordance with the present invention, will cause reduction in LDL-C and therefore lower the incidence of nephrotoxicity in such patients.
…
Cholesterol lowering drugs or drugs which are inhibitors of cholesterol biosynthesis which may be used in the method of the invention include HMG CoA reductase inhibitors, squalene synthetase inhibitors, fibric acid derivatives, bile acid sequestrants, probucol, niacin, niacin derivatives and the like.
166 After discussing the use of HMG CoA reductase inhibitors and squalene synthetase inhibitors, the Squibb Patent states at page 7 lines 40-42:
The other serum cholesterol lowering drugs when present will be employed in dosages normally employed as indicated in the Physician’s Desk Reference, for each of such agents such as in an amount within the range of from about 2 mg to about 7500 mg and preferably from about 2 mg to about 4000 mg.
167 Further down page 7 it is said at lines 53-55:
The compositions described above may be administered in the dosage forms as described above in single or divided doses of one to four times daily. It may be advisable to start a patient on a low dose combination and work up gradually to a high dose combination.
168 The “complications normally associated with diabetes” as set out on page 2, lines 10-12 of the Squibb Patent include retinopathy. The cholesterol lowering drugs which may be administered that are specified at page 2, lines 24-26 include fibric acid derivatives. Fenofibrate is a fibric acid derivative and Example 7 refers to tablets containing 250 mg of fenofibrate.
169 Example 7, when read in the context of the Squibb Patent as a whole as at the date of its publication, provides a clear and unmistakeable direction to use the fenofibrate taken orally in tablet form as a treatment for the complications of diabetes include retinopathy. In my view claims 1, 6, 7 and 12 of the 711 Patent are anticipated by the Squibb Patent.
170 The Squibb Patent does not provide any clear disclosure of the use of fenofibrate with statins (also known as HMG CoA reductase inhibitors) as referred to in claim 10 of the 711 Patent. Nor does it provide any disclosure of the invention defined in claims 5 and 11 which specify various doses all of which are different from the 250 mg doses specified in Example 7 of the Squibb Patent.
171 With regard to claims 5 and 11 of the 711 Patent, the respondent submitted that they were anticipated by the Squibb Patent when read with the Desk Reference. The principal difficulty with this submission is that the edition of the Physician’s Desk Reference that is in evidence was not published until some 13 years after the date of publication of the Squibb Patent. In my view it is not open to the respondent to treat the Squibb Patent and the edition of the Physician’s Desk Reference in evidence as related documents for the purposes of s 7(1)(b) of the Act.
172 Accordingly, claims 5 and 11 of the 711 Patent are not shown to have been anticipated by the Squibb Patent.
Public Acts
173 A prior use of the claimed invention will not constitute an anticipation if it does not make available the information necessary to enable the person skilled in the art to perform the invention publicly available: Jupiters Ltd v Neurizon Pty Ltd (2005) 65 IPR 86 at [125]-[147]. How much information must be made publicly available to amount to an enabling disclosure will depend on the facts of the case and what additional information could be gleaned by the person skilled in the art from the information made publicly available without undue burden.
174 Professor Carter's evidence was that, as part of the multifactorial approach to the management of diabetes patients, he would first introduce a number of non-pharmacological interventions, including exercise and diet modification. If this did not appropriately address the patient's blood glucose levels, blood pressure and blood lipid levels, Professor Carter would commence pharmacologic treatments. For patients with elevated blood glucose levels (hyperglycaemia), this would include the prescription of one or more oral hypoglycaemics, such as metformin, a sulphonylurea, or insulin injections. For patients with elevated blood pressure, this would include the prescription of one or more antihypertensive agents, such as an angiotensin converting enzyme (“ACE”) inhibitor, an angiotensin II receptor blocker (“ARB”) or, if these were not effective, a diuretic, a calcium channel blocker, and/or a beta-blocker.
175 For patients with elevated lipid levels, Professor Carter would first introduce fish oil supplements, followed by statin therapy. Fibrate therapy would only be instituted where the other interventions did not achieve Professor Carter’s desired lipid levels, or where the patient was intolerant to statins. Professor Carter’s evidence was that, while it could not be sure, he believed that, at the priority date, around 25% of his lipid-lowering prescriptions would have been for fibrate therapy.
176 By treating the abnormalities such as hyperglycaemia, hypertension, hyperlipidaemia, Professor Carter was aiming to achieve certain goals directly referable to the condition – for example, for hyperlipidaemia, he aimed to achieve certain levels of total cholesterol, LDL cholesterol, HDL cholesterol and triglycerides. He would prescribe drugs to achieve those goals, with the intention and expectation that, by treating these goals, including, fenofibrate, the incidence of all of the long-term complications of diabetes would be reduced.
177 Professor Carter’s evidence was that it would be rare for fenofibrate to be prescribed on its own to a patient in the context of seeking to address the patient's elevated lipid levels and, apart from statin-intolerant patients, he would have only prescribed fibrate therapy if a patient had persistently elevated lipid levels after the commencement of statin therapy.
178 If the reverse infringement test were to be applied for the purpose of determining whether Professor Carter’s actions when prescribing fenofibrate as a treatment for diabetic retinopathy was novelty defeating, then it would follow that at least some of the claims of the 711 Patent would be invalid. This is because Professor Carter was prescribing fenofibrate (either with or without a statin) in the expectation that it would treat the complications of diabetes including diabetic retinopathy. The fact that he was prescribing a number of drugs as part of a multi-factorial approach with a view to reducing the risk of such complications, including the development or progression of diabetic retinopathy, would not place his actions outside the scope of the claims.
179 The difficulty for the respondent is that the Act requires that for a prior act to deprive a claimed invention of novelty, it must be one that makes “prior art information” publicly available which only then may form part of the prior art base referred to in s 18(1)(b) of the Act. This is why the prior secret use of an invention that does not make any information relating to it publicly available cannot be an anticipation even though the prior use meets the requirements of the reverse infringement test.
180 In the present case, for the novelty challenge based on Professor Carter’s prior acts to succeed, there would need to be some evidence to show that these prior acts made the relevant information publicly available by, for example, him having informed a patient that he was prescribing the patient fenofibrate to treat complications of diabetes including retinopathy. Professor Carter’s evidence does not show that he ever made any disclosure to this effect to any patient to whom he prescribed fenofibrate. Accordingly, the challenge to the validity of the claims of 711 Patent based on Professor Carter’s prior acts fails.
181 The acts of investigators administering fenofibrate during the course of the Eye Study were also relied upon by the respondent as novelty defeating. This aspect of the novelty case, which received little attention in closing submissions, appears to have assumed that the Eye Study was an open label study which enabled the investigators to know whether or not they were administering fenofibrate. However, this would not be possible in a double blind study in which neither the investigator nor the participant would know whether fenofibrate or placebo was being administered. The Protocol states at page 3 that the participants would be randomly assigned in “a double masked fashion to either placebo or fenofibrate” which clearly indicates that the Eye Study was double blinded.
182 The Field Study was also a double blind study. What I have just said in relation to the Eye Study applies to the Field Study as well.
183 The respondent’s novelty case based on prior public use fails.
Inventive Step
184 Section 18(1)(b)(ii) of the Act provides that an invention is a patentable invention for the purposes of a standard patent if the invention, so far as claimed in any claim, involves an inventive step when compared with the prior art base as it existed before the priority date of that claim. At the relevant times subs 7(2) and (3) of the Act provided:
(2) For the purposes of this Act, an invention is to be taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed in the patent area before the priority date of the relevant claim, whether that knowledge is considered separately or together with the information mentioned in subsection (3).
(3) The information for the purposes of subsection (2) is:
(a) any single piece of prior art information; or
(b) a combination of any 2 or more pieces of prior art information;
being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood, regarded as relevant and, in the case of information mentioned in paragraph (b), combined as mentioned in that paragraph.
185 The expression “prior art information” is relevantly defined to mean information that is part of the prior art base for the purposes of subs 7(3) in relation to deciding whether an invention does or does not involve an inventive step. At the relevant times the expression “prior art base” was defined in para (a) of the definition to mean information that is made publicly available whether in or out of the patent area. The term “patent area” is relevantly defined in Sch 1 to mean Australia.
186 The operation of s 7 was considered by the Full Court in AstraZeneca FC and by the High Court in AstraZeneca AB v Apotex Pty Ltd (2015) 257 CLR 356 (AstraZeneca HC). In the High Court, French J said at [18]:
[18] The text of s 7(2) required, in unambiguous language, that “the onus to establish the absence of an inventive step rests upon the party challenging validity”. It was, as Jessup J correctly observed in the Full Court, a deeming provision. Section 7(2) would defeat a claim for want of inventive step unless one of the alternative conditions set out in s 7(2), read with s 7(3), was satisfied. Those conditions involved the following elements:
1. An hypothetical person skilled in the relevant art.
2. The person being, therefore, notionally possessed of the common general knowledge as it existed in the relevant area before the priority date of the impugned claim.
3. The invention being obvious to that person in the light of the common general knowledge.
4. Alternatively, that person being provided with prior art information made publicly available in a single document or through doing a single act, or made publicly available in two or more related documents or through doing two or more related acts if the relationship between them satisfied the requirement of s 7(3)(b).
5. That prior art information, as defined by s 7(3), being information that the person could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant to work in the relevant art in the patent area (the relevance requirement). “Ascertained”, in this context, means “discovered or found out”. “Understood” means that, having discovered the information, the person would have “comprehended it” or “appreciated its meaning or import”.
6. The invention being obvious to the person in the light of the common general knowledge considered together with either of the classes of prior art information defined in s 7(3).
The judicial determination whether want of inventive step is established pursuant to s 7 is mediated through the legal construct of the hypothetical person skilled in the relevant art. The construct is of a kind well-known to the law and used for setting parameters for evaluative judgments. It is a tool of analysis and is given statutory recognition, for that limited purpose, in s 7.
(footnotes omitted)
187 Kiefel J (as her Honour then was) said at [68]:
[68] Before a document containing prior art information can be used along with the common general knowledge for the purposes of the s 7(2) inquiry, it is necessary that it meet the requirements of s 7(3). In Lockwood Security Products Pty Ltd v Doric Products Pty Ltd [No 2] it was explained that prior art information which is publicly available in a single document is “ascertained” if it is discovered or found out, and “understood” means that, having discovered the information, the skilled person would have comprehended it or appreciated its meaning or import. The Court also explained that the phrase “relevant to work in the relevant art” is directed to publicly available information, not part of the common general knowledge, which the skilled person could be expected to have regarded as relevant to solving a particular problem, or meeting a long-felt want or need, as the patentee claims to have done.
[69] Lockwood [No 2] also explains that, in answering the question of obviousness, the information referred to in s 7(3), like that part of the prior art base which is the common general knowledge, is considered for a particular purpose. That purpose is to look forward from the prior art base to see what the skilled person is likely to have done when faced with a problem similar to that which the patentee claims to have solved with the claimed invention. It is this aspect of the s 7(2) inquiry which assumes particular importance on these appeals.
[70] In addressing s 7(2), it is to be borne in mind that the skilled person is an artificial construct, intended as an aid to the courts in addressing the hypothetical question of whether a person, with the same knowledge in the field and aware of the problem to which the patent was directed, would be led directly to the claimed invention. The statute’s creation of the skilled person construct for this purpose is not to be taken as an invitation to deal with the question posed by s 7(2) entirely in the abstract. Whilst the question remains one for the courts to determine, the courts do so by reference to the available evidence including that of persons who might be representative of the skilled person.
(footnotes omitted)
188 Section 7(2) of the Act uses the word “obvious” in the course of describing what must be established before an invention can be held not to involve an inventive step. Something may be “obvious” in light of the common general knowledge, or the common general knowledge coupled with relevant s 7(3) information, if it is “plain or open to the eye or mind, something which is perfectly evident to the person thinking on the subject” (Olin Mathieson Chemical Corp v Biorex Laboratories Ltd [1970] RPC 157 at 188) or something which “would at once occur to anyone acquainted with the subject and desirous of accomplishing the end” (Vickers, Sons & Co Ltd v Siddell (1890) 15 App Cas 496).
189 An invention may also be obvious in light of the common general knowledge if the person skilled in the art faced with the same problem as the inventor would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not (Wellcome Foundation Ltd v VR Laboratories (Aust) Pty Ltd (1981) 148 CLR 262 (“Wellcome”) at 286 per Aickin J) if the person skilled in the art would be directly led as a matter of course to take such steps in the expectation that doing so might well produce a useful or better alternative to the prior art (Aktiebolaget Hassle v Alphapharm Pty Ltd (2002) 212 CLR 411 (“Alphapharm”) at [50]-[53]). However, a claimed invention is not obvious merely because the person skilled in the art would consider that it was “worthwhile to try”.
190 The plurality in Alphapharm referred at [76] with approval to the following observations of Judge Rich in In re O’Farrell (1988) 853 F 2d 894 at 903:
The admonition that ‘obvious to try’ is not the standard under §103 has been directed mainly at two kinds of error. In some cases, what would have been ‘obvious to try’ would have been to vary all parameters or try each of numerous possible choices until one possibly arrived at a successful result, where the prior art gave either no indication of which parameters were critical or no direction as to which of many possible choices is likely to be successful … In others, what was ‘obvious to try’ was to explore a new technology or general approach that seemed to be a promising field of experimentation, where the prior art gave only general guidance as to the particular form of the claimed invention or how to achieve it.
191 Judge Rich’s reference to numerous or many possible choices and the exploration of a new technology where the prior art provided only “general guidance” reflects some of the difficulties posed by the “obvious to try” standard also rejected by the High Court.
192 That said, it should not be thought that the Cripps question, as reformulated by the High Court, does not allow for the possibility that the person skilled in the art might be directly led, as a matter of course, to try more than one alternative. There will be some cases in which the person skilled in the art will be directly led, as a matter of course, to try a number of different alternatives in the expectation that each may well produce a useful alternative.
193 There are four other matters of importance in relation to the question of obviousness:
The Court must be wary of the misuse of hindsight or ex post facto analysis in deciding whether a claimed invention lacks any inventive step. There are many authoritative warnings to this effect including those of Aicken J in 3M at 293-294 and Wellcome at 286, and the plurality in Alphapharm at [21].
Whether an invention is obvious is a question of fact or, as it is sometimes described, a “jury question”. However, the question is not whether the claimed invention is obvious to the Court, but whether it would be obvious to the person skilled in the art who is taken to be equipped with the relevant common general knowledge as it stood at the priority date together with any relevant s 7(3) information but who lacks any capacity for inventiveness.
The amount of inventiveness required to sustain a patent for a claimed invention is quite small. A “scintilla” of inventiveness is all that is required: Alphapharm at [195].
Merely because one pathway to an invention is shown to be obvious, does not mean that another such pathway might not also be obvious. Two or more pathways may be obvious, even though some of them might be more obvious than others.
194 Adapting the modified Cripps question to the circumstances of this case, the question is whether:
Would the notional team at the relevant date, in all the circumstances, which include a knowledge of all the relevant prior art, directly be led as a matter of course to try fenofibrate (whether alone or in combination with a statin) as a medicament for use in the prevention or treatment of diabetic retinopathy in the expectation that it might well produce a useful or better alternative to other therapies used for that purpose?
195 When postulating the modified Cripps question solely in the context of s 7(2) of the Act (ie. in the absence of any relevant s 7(3) information) the prior art referred to must be confined to prior art information that forms part of the common general knowledge in the field at the relevant date. To the extent it is shown that there is any additional information of the kind referred to in s 7(3) then the notional research group will also be taken to know that information.
The Notional Team
196 The applicants submitted that for the method of treatment claims the 711 Patent is addressed to a clinical team including a non-inventive endocrinologist and ophthalmologist. As part of this clinical team, the endocrinologist would have primary responsibility for the selection of any systemic medication for administration to patients, acting on the advice of a consulting ophthalmologist, who would defer to the endocrinologist in relation to matters of systemic treatment. The ophthalmologist would have primary responsibility for the selection of any surgical or non-systemic intervention for use in patients, acting on the advice of a consulting endocrinologist with regard to the addition of any systemic medication to the treatment regime.
197 The respondent submitted that the notional team would include an endocrinologist and a consulting ophthalmologist though I did not understand them to accept that the team members responsibilities would be divided up in the manner suggested by the applicants. Presumably this was because, with an eye to the evidence of Dr Beaumont, they would not wish to see the consulting ophthalmologist assuming primary responsibility for the selection of medications. However, nothing much turns on this point because I do not regard Dr Beaumont as a particularly good proxy for a consulting ophthalmologist in the notional team. The applicants characterised him as something of a maverick in his field. To the extent they are to be understood as suggesting that Dr Beaumont was a particularly creative and independently minded thinker then I am inclined to agree. He strikes me as the antithesis of the non-inventive worker in the field.
Use of lipid lowering therapy to treat diabetic retinopathy
198 There is a significant difference in opinion between the experts in relation to the use of lipid lowering therapy to treat diabetic retinopathy.
199 Professor Mitchell’s evidence was that, as at 10 November 2005, there were no medical therapies available to reduce the risk of a patient developing diabetic retinopathy or its rate of progression other than by addressing a patient's blood glucose and blood pressure levels. His evidence was that studies of the lipid-lowering agent clofibrate reported some effect on hard exudates in patients with diabetic retinopathy, and that no consistent beneficial effect in visual acuity on the underlying causes of diabetic retinopathy (capillary bed closure or non-perfusion in the retina) was demonstrated.
200 Professor Mitchell did not consider that any form of lipid-lowering therapy could address the underlying causes of diabetic retinopathy until after 10 November 2005 when fenofibrate was demonstrated to do so. He says that as a result of the outcomes of the FIELD and ACCORD studies, if screening of his patients reveal signs of either non-proliferative or proliferative retinopathy, or macular oedema, he recommends the use of fenofibrate. But it was not until after he became aware of the outcomes of those studies that he considered or expected that the use of fenofibrate would have a beneficial effect in reducing the risk of the development or progression of diabetic retinopathy.
201 Professor Carter’s evidence was to a slightly different effect. He considered that, as at 10 November 2005, poor lipid control was known to be associated with microvascular complications associated with diabetes including diabetic retinopathy. In this regard, he pointed to the ETDRS 22 which stated at page 1084:
Although our data are observational, they suggest that a reduction of elevated serum lipid levels may help prevent vision loss associated with retinal hard exudate. Preservation of vision may be an additional motivating factor for lowering serum lipid levels in persons with diabetes in whom they are elevated.
202 Professor Carter’s evidence (which I accept) was that he prescribed fibrates (including fenofibrate) to patients to reduce the risk of these patients developing both macrovascular and microvascular complications of diabetes including diabetic retinopathy. He considered that the risk of his patients developing retinal hard exudate could be reduced by the use of lipid lowering agents including fenofibrate. Similarly, he considered these same that lipid lowering agents could be used to slow the progression of retinal hard exudates in patients already affected by diabetic retinopathy.
203 Dr Beaumont’s evidence was that he was prescribing fenofibrate before 10 November 2005 for the treatment of diabetic retinopathy. He says that in the 1970’s, he was prescribing various pharmacological agents to treat patients suffering from diabetic retinopathy. These treatments included insulin (to control blood glucose levels), antihyperalgesic (to reduce blood pressure) and other drugs, including clofibrate, to lower patients lipid levels. With respect to clofibrate and other lipid-lowering drugs, Dr Beaumont’s evidence was that these were prescribed by him not for the purpose of improving visual acuity in patients whose retinas were already damaged by the formation of hard exudates, but to slow or prevent their formation in the first place.
204 Dr Beaumont’s evidence was that he understood, well before 10 November 2005, that if hard exudates form in the foveal region of the macula, central vision will be compromised due to the irreversible damage to photoreceptor cells (the retinal cells responsible for vision) at the site of the hard exudates. He says that he understood that the elevated serum lipids (including cholesterol and triglycerides) was a risk factor for the development and progression of diabetic retinopathy because it contributed to the deposition of retinal hard exudates at or close to the macula. In Dr Beaumont’s view, use of lipid-lowering agents, including fibrates, could reduce the risk of the development and progression of diabetic retinopathy. As Dr Beaumont explained in his affidavit evidence:
Well before November 2005, I understood that there is not a simple or linear relationship between blood lipid levels and the development of hard exudates and visual impairment in patients suffering from diabetic retinopathy. Rather, I understood that the extent of hard exudates (i.e., fatty deposits) present in the retina of a patient affected by diabetic retinopathy reflected an interplay between several factors, including the lipid levels in a patient's blood circulation, the degree of leakage from retinal blood vessels (by which fatty material moves into the retinal tissues to form hard exudates) and the rate at which fatty material is transported within, and cleared from, the retina after hard exudates have formed. In this way, I understood before November 2005 that, while elevated blood lipid levels contributed to the risk of hard exudates forming in patients with diabetic retinopathy, other factors also influenced hard exudate formation.
205 Dr Beaumont and the late Professor Fred Hollows wrote a paper published in the Australian Journal of Ophthalmology in 1975 entitled “Treatment of Diabetic Retinopathy” (“the Beaumont paper”) in which they described diabetic retinopathy as a “multifunctional disease” that in most cases requires a combined and often complex therapeutic approach. The pharmacological management of hyperlipidaemia and hypertension in situations where diet, exercise and weight loss fail was briefly discussed in this paper, though the authors did not specifically refer to fibrates or any other pharmacological agent in this context.
206 Another paper written by Dr Beaumont and Professor Hollows published in Lancet in 1972 entitled “Classification of Diabetic Retinopathy with Therapeutic Implications” proposed a fourfold classification of diabetic retinopathy one of which is described as “diabetic lipid retinopathy”. This paper referred to the characteristic feature of this class of retinopathy (at pages 420-421) as “the progressive accumulation of hard exudates … or the presence of large areas of hard exudates in the retina.” The recommended treatment for this class of retinopathy is said by the authors (at page 421) to consist of weight reduction and clofibrate administration.
207 The paper by Dr Beaumont and Professor Hollows in the Lancet attracted some criticism in correspondence that was published in the Lancet shortly after the paper first appeared. Relevantly for present purposes the authors of the correspondence, Dr Kohner and two of her colleagues, state:
Some of the comments about treatment are unacceptable, because they are not supported by adequate evidence. In particular, there is no real evidence to show that phenformin, ethylestrenol, or treatment of hyperlipidaemia or hypertension has any beneficial effect on established diabetic retinopathy.
and:
Diabetic retinopathy presents such a varied picture that its understanding and description are difficult. It is important to distinguish hypothesis from proven facts when it comes to statements about pathogenesis and especially about treatment.
208 A reply by Dr Beaumont and Professor Hollows was also published in the same issue as Dr Kohner’s letter. It does refer to evidence of the efficiency of treatment with phenformin and ethylestrenol but says nothing about clofibrate.
Common general knowledge
209 It is necessary to say more concerning some of the more contentious aspects of what the respondent said was common general knowledge as at November 2005.
210 The notional team will possess the common general knowledge in the relevant field in so far as it is relevant to the subject matter of the Patents. This will include the background knowledge and experience available to all those persons engaged in the relevant field within the patent area including publications to which they would refer as a matter of course: Minnesota Mining and Manufacturing Company v Beiersdorf (Australia) Ltd (1980) 144 CLR 253 (“3M”) at 292 per Aickin J, ICI Chemicals & Polymers Ltd v Lubrizol Corporation Inc (1999) 45 IPR 577 at [112] per Emmett J.
211 The High Court emphasised in Alphapharm at [31] that information cannot be treated as part of the common general knowledge in the absence of evidence of its general acceptance and assimilation by persons skilled in the art.
The NHMRC Guidelines
212 In June 1997, the NHMRC released its Clinical Practice Guidelines for the Management of Diabetic Retinopathy which refer to risk factors for diabetic retinopathy. As previously mentioned, Professor Mitchell was a principal author of this document.
213 The 1997 NHMRC Guidelines include (at page xvi) a “Collective summary of guidelines for the management of diabetic retinopathy by level of evidence”. The only guideline shown in this summary as supported by level 1 evidence (being the highest quality evidence on which a recommendation could be based) was the following:
Strict glycaemic control is the most important modifiable factor in preventing development of retinopathy. Efforts to achieve strict glycaemic control should be undertaken in all diabetic patients, while taking care to avoid severe recurrent hypoglycaemia
214 In the section titled “Overall summary of key points in management of diabetic retinopathy”, the 1997 NHMRC Guidelines state:
Apart from intensive glycaemic control and laser treatment, no other therapies to date have been shown to influence the development or progression of retinopathy in large multicentre randomised controlled trials.
215 This remained the position as at 10 November 2005.
Blood pressure
216 At the time the 1997 NHMRC Guidelines were published, there was inconsistent evidence regarding the impact of high blood pressure, also known as hypertension, on diabetic retinopathy. For example, the 1997 NHMRC Guidelines state on page 17:
In the WESDR, a 10-mmHg increase in the mean systolic blood pressure for the last two visits, was found to increase the incidence of retinopathy, after controlling for other risk variables, in younger-onset, but not in older-onset diabetes. However, other studies have failed to implicate hypertension as an independent risk factor for retinopathy.
(footnotes omitted)
217 However, by November 2005, blood pressure levels were understood to be a significant risk factor for diabetic retinopathy. The results of a large study called the UK Prospective Diabetes Study (UKPDS) showed that improving blood pressure reduced the risk of both macrovascular and microvascular complications, including diabetic retinopathy in diabetic patients. The study also showed that even patients without elevated blood pressure levels may reduce their risk of diabetic retinopathy by reducing their blood pressure. Those results were not published until after the publication of the 1997 NHMRC Guidelines.
Blood lipid levels
218 The 1997 NHMRC Guidelines recommended that practitioners should “[a]im to adequately manage ... serum lipids in all people with diabetes (level III evidence).” The reference to “level III evidence” reflects the fact that there was a lower level of evidence to support the recommendation than for the recommendation concerning glycaemic control (for which there was “level I evidence”).
219 The 1997 NHMRC Guidelines included, at page 12, section 1.4 entitled “Risk Factors”, and a table of the following “Key Points”:
Key Points
• All people with diabetes are at risk of developing retinopathy.
• The known duration of diabetes is the principal determinant of the presence of diabetic retinopathy (level I evidence).
• Poor glycaemic control is the most critical risk factor for the development and progression of diabetic retinopathy (level I evidence).
• The combined effects of diabetes duration and glycaemic control on the development and progression of retinopathy appear similar in both IDDM and NIDDM cases.
• Elevated serum total cholesterol is associated with an increased risk of retinal hard exudates, and an independent increased risk of visual impairment.
• Less consistent evidence implicates hypertension and pregnancy in the development and progression of diabetic retinopathy among IDDM patients.
• Early nephropathy measured as microalbuminuria is a marker for progression to PDR.
• It is not possible to avoid mild hypoglycaemic episodes when attempting strict glycaemic control, but it is important to avoid severe hypoglycaemic episodes that could be potentially life threatening. The DCCT showed that the risk of severe hypoglycaemia increases as the HbA1c falls
Guidelines
1. The most important factor preventing the development of retinopathy, in both IDDM and NIDDM, is strict glycaemic control (level I evidence).
2. Efforts to achieve strict glycaemic control should be undertaken in all patients with diabetes, while taking care to avoid severe recurrent hypoglycaemia.
3. Aim to adequately manage elevated blood pressure and serum lipids in all people with diabetes (level III evidence).
Under the sub-heading “Hyperlipidaemia” on page 17 the 1997 NHMRC Guidelines state:
A recent report of observational data from the ETDRS found that patients with elevated total serum cholesterol levels were twice as likely to have retinal hard exudates as patients with normal levels. They were also more likely to develop hard exudate during the study. Increasing hard exudate deposition appeared to be independently associated with an increased risk of visual impairment.
The footnote to the last sentence refers to the ETDRS 22, a report of an observational study from the larger clinical study ETDRS. The ETDRS did not investigate the effect of lipid-lowering agents on retinal hard exudates, because lipid-lowering agents were not part of the study design. Accordingly, ETDRS 22 did not demonstrate any effect of the use of lipid-lowering agents on retinal hard exudates. However, ETDRS 22 did suggest that there was an association between dyslipidaemia and diabetic retinopathy. The objective of ETDRS 22 was to evaluate the relationship between serum lip levels, retinal hard exudates, and visual acuity in patients with diabetic retinopathy. The conclusions drawn, as set out in the abstract, were as follows:
These data demonstrate that elevated serum lipid levels are associated with an increased risk of retinal hard exudate in persons with diabetic retinopathy. Although retinal hard exudate usually accompanies diabetic macular edema, increasing amounts of exudate appear to be independently associated with an increased risk of visual impairment. Lowering elevated serum lipid levels has been shown to decrease the risk of cardiovascular morbidity. The observational data from the Early Treatment Diabetic Retinopathy Study suggest that lipid lowering may also decrease the risk of hard exudate formation and associated vision loss in patients with diabetic retinopathy. Preservation of vision may be an additional motivating factor for lowering serum lipid levels in persons with diabetic retinopathy and elevated serum lipid levels.
220 The 1997 NHMRC Guidelines also discuss studies from the 1960s investigating the effect of a lipid-lowering fibrate called clofibrate on patients with diabetic retinopathy. As reported on page 46 of the 1997 NHMRC Guidelines, these studies did not show “any convincing benefit”.
221 It was common general knowledge by 10 November 2005 that blood lipid levels were a significant risk factor for macrovascular complications like cardiovascular disease and that it was necessary to control blood lipids in diabetes patients to address their risk of developing macrovascular complications. It was not common general knowledge that lipid lowering agents had a beneficial effect on the underlying causes of diabetic retinopathy. However, ETDRS 22, which was common general knowledge as at 10 November 2005, had noted that elevated serum levels were associated with an increased risk of hard exudates formulation and associated vision loss in patients with diabetic retinopathy and that preservation of vision may provide an additional reason for seeking to lower serum lipid levels in patients suffering from diabetic retinopathy who had elevated serum levels. Professor O’Brien accepted, and I find, that the results presented in ETDRS 22 were statistically significant and clinically important.
222 It was also common general knowledge as at 10 November 2005 that fenofibrate was an effective cholesterol lowering drug that was approved for use in Australia for the treatment of hypercholesterolemia. Fenofibrate was also believed to be more effective at lowering cholesterol than other fibrates (eg. gemfibrozil) available at that time. However, as at 10 November 2005, fibrates, including fenofibrate, were not prescribed for the treatment of hypercholesterolemia if target lipid levels could be achieved by the use of a statin unless there was some other reason why a patient’s elevated lipid levels could not be treated with those drugs (eg. because of side effects).
223 It was also common general knowledge as at 10 November 2005 that fibrates including fenofibrate were particularly effective in circumstances where the patient had elevated triglycerides, low HDL cholesterol, and normal to slightly elevated LDL cholesterol. Elevated triglycerides coupled with reduced levels of HDL cholesterol were a hallmark of what was (and is) sometimes called diabetic dyslipidaemia.
224 On the basis of Professor Carter’s and Professor O’Brien’s evidence, I infer that between about 15% to 25% of their patients were being treated with fibrates, most of whom were also being treated with a statin. I also infer that by 10 November 2005, most of these patients were being prescribed fenofibrate.
225 Professor Carter prescribed fenofibrate (and statins) as part of his multifactorial approach to the treatment of diabetes for the purpose of addressing various complications including retinopathy. Professor O’Brien prescribed fenofibrate for the purpose of addressing macrovascular complications including cardiovascular disease.
226 There was a debate between the parties as to whether or not the 1997 NHMRC Guidelines formed part of the common general knowledge. The evidence indicated that the NHMRC Guidelines, while reflecting what might be described as the standard of care to be provided to patients suffering from, or at risk of developing, diabetic retinopathy, were more directed at general practitioners many of whom, it was said, did not read such guidelines. However, what I think emerges from the evidence in relation to the NHMRC Guidelines is that, while the document itself may not have been shown to satisfy the requirements necessary to constitute common general knowledge, it constitutes a useful record of information at least some of which formed part of the common general knowledge. In this regard, I am satisfied that, consistent with the evidence of endocrinologists, it was common general knowledge as at 10 November 2005 that “[e]levated serum total cholesterol is associated with an increased risk of retinal hard exudates” even if the document itself was not.
227 I am satisfied that ETDRS 22 was common general knowledge. Each of the ophthalmologists, Professor Mitchell and Dr Beaumont, who gave evidence were very familiar with ETDRS 22 before 10 November 2005. I am satisfied that ETDRS 22 would have formed part of consulting ophthalmologist’s background knowledge as at 10 November 2005. So I am satisfied that ETDRS 22 would form part of the common general knowledge that is to be attributed to the notional team. If I am wrong about that, then I would hold that ETDRS 22 was s 7(3) information that would have been ascertained, understood, and regarded as relevant by the notional skilled team as at 10 November 2005.
228 However, as the expert evidence also made clear, ETDRS 22 did not, and was not designed to, determine whether lipid lowering agents could be used to reduce the risk of the development or progression of hard exudates or diabetic retinopathy more generally. Nor did it demonstrate a causative link between blood lipid levels and the development or progression of hard exudates.
Inventive Step Analysis
229 For some years prior to 10 November 2005, lipid lowering therapy was the standard of care for patients with diabetes who had elevated serum lipid levels. Of the pharmacological treatments that were in use at that date, statins (such as atorvastatin) were the most commonly prescribed treatments. However, a considerable number of patients were prescribed fibrates, including fenofibrate, most often for use by patients who failed to achieve their target lipid profiles despite the use of a statin, particularly where their triglycerides levels remained elevated or where HDL levels remained below their target levels, as well as in patients who were unable to tolerate a statin due to side effects.
230 ETDRS 22 pointed to an association between lipid levels and diabetic retinopathy. It provided support for the hypothesis that the use of a lipid lowering agent for the treatment of hyperlipidemia, may well prevent or slow the development or progression of retinal hard exudates. This was the same hypothesis that encouraged Professor Carter to prescribe fibrates, including fenofibrate, prior to 10 November 2005 as part of his multifactorial approach to the treatment of patients suffering from, or at risk of developing, diabetic retinopathy. Although Professor Carter is not someone who I would consider uninventive, his motivation for using fenofibrate as part of his multifactorial approach and for the purpose of preventing or slowing the development of hard exudates in the retina, was founded on a hypothesis for which ETDRS 22 provided direct support.
231 In my view the following four matters that are of particular importance to the inventive step analysis:
The ETDRS 22 was merely an observational study that was not designed to evaluate the effectiveness of any relevant pharmacological treatment on hard exudates or diabetic retinopathy more generally. But it still provided clinically important evidence pointing to an association between lipid levels and hard exudates.
Statins were the preferred, and most widely prescribed, lipid lowering drugs as at 10 November 2005. They had been shown (by the CARDS study) not only to reduce lipids, but also to reduce the risk of cardiovascular events even in subjects whose LDL count was at or below recommended levels.
As at 10 November 2005, fenofibrate was a well-known lipid lowering agent that was particularly well suited for use in the treatment of patients with elevated triglycerides and low HDL levels. It had a known safety profile, and was already being widely prescribed as a treatment for macrovascular complications in patients suffering from diabetes.
There were no pharmacological treatments that had been shown to prevent or slow the development of hard exudates or diabetic retinopathy more generally.
232 The respondent submitted that the modified Cripps question should be answered in the affirmative. In essence, the respondent’s case was that, in the circumstances, ETDRS 22 would have directly led the notional team to try fenofibrate as a treatment to prevent or slow the development of hard exudates. The suggestion, which was at least implicit in the respondent’s submission, was that ETDRS 22 provided strong support for the hypothesis that the lipid lowering effects of fenofibrate might well prevent or slow the development of hard exudates that were a characteristic of diabetic retinopathy.
233 The applicants submitted that the modified Cripps question should be answered in the negative for a number of reasons. The applicants submitted that although ETDRS 22 reported an association between elevated total cholesterol levels, LDL-C levels and triglyceride levels with the development of obvious retinal hard exudates in patients enrolled in the ETDRS over the course of the study, the notional team would discount the association with triglyceride levels because:
(a) This association was inconsistent, in that triglyceride levels were not associated with the presence of obvious retinal hard exudates in patients enrolled in the ETDRS at the commencement of the study;
(b) The notional team would understand from ETDRS 22 that the blood lipid levels of the patients enrolled in the study were only collected at the commencement of the study, and the associations reported in ETDRS 22 were only corrected (ie. adjusted) using blood glucose levels collected at the commencement of the study; and
(c) The notional team would not know whether the development of retinal hard exudates in patients over the course of the study was related to other factors that occurred during the study period, such as poor glycaemic control.
234 According to the applicants’ submission, the notional team would not consider that the association between lipid values collected at the commencement of the study and the development of retinal hard exudates over the course of the study were reliable. It was submitted that the notional team would (at most) focus on associations between hard exudates, total cholesterol and LDL-C levels, because those lipid fractions were known to fluctuate less with poor glycaemic control than triglycerides. This submission was essentially directed to the quality of the methodology used in the ETDRS 22 study and, in particular, its ability to reliably detect an association between retinal hard exudates and elevated levels of serum triglycerides against which fibrates were known to be particularly effective.
235 The applicants further submitted that even if the notional team had considered it worthwhile to try to reduce serum lipid levels in order to prevent or slow the development of hard exudates in light of ETDRS 22:
(a) the notional team would have been directly led to the use of statin therapy, and not fibrate therapy, for the treatment of diabetic retinopathy;
(b) the notional team, based on the common general knowledge as at 10 November 2005, would not have the requisite expectation of success that the administration of a statin would have a beneficial effect in the prevention or treatment of diabetic retinopathy; and
(c) if the notional team would have been directly led to the use of fibrate therapy, the expectation of success would be even lower than for statins.
236 I will start with the first of the applicants’ submissions. There was oral evidence given by Professor O’Brien and Professor Carter that demonstrates that the authors of ETDRS 22 adjusted for certain variables, a procedure that was no doubt necessary given the data that they had, but which was less than perfect, particularly if they were seeking to determine whether there is an association between elevated levels of triglycerides and retinal hard exudates that is independent of blood glucose levels. The difficulty stems from the fact that patients with high triglyceride levels generally have high blood glucose levels, which are known to be causally related to diabetic retinopathy. This may make it difficult to identify the existence of an independent association between triglycerides levels and hard exudates in circumstances where blood lipid levels were only measured at the commencement of the study.
237 It is fair to say that Professor Carter acknowledged that there were difficulties with the ETDRS 22 methodology. Nevertheless, as he explained in his oral evidence:
Well, we still can’t get away from the fact that this is a significant association between high triglycerides at baseline and the development of hard exudates during the study. It’s nothing more than that. It’s an association, and I would suggest that this is just one of a number of pieces of evidence that makes the association between lipids and hard exudates even stronger when it’s looked at overall, not just the ETDRS.
238 Later in his oral evidence Professor Carter referred to some of the other evidence which he alluded to in that answer. These included observational studies by Duncan, Cullen and Ireland dating back to the 1960’s and early 1970’s using clofibrate, a small pilot study by Gordon et al in 1991 using a statin, and a study by Sen et al in 2001 suggesting that there was a need for further investigation of the use of the statins in the prevention of diabetic retinopathy. Having referred to these studies Professor Carter said:
So what I am leading up to is that there is a whole body of evidence, none of which could I say is fantastic data but when you put it all together it presents a fairly compelling case that there is a significant association between lipids and retinopathy, and a strong suggestion that – that if you can intervene and do something about hard exudates you can improve the outcome for the patient, particularly with respect with visual acuity. Now, all of this is on top of a theoretical basis.
239 It is apparent that Professor Carter’s thinking was very much informed not only by ETDRS 22, but also by various articles none of which was suggested to form part of the common general knowledge, a point that was understandably seized upon by the applicants in their submissions. However, that is not a basis for rejecting Professor Carter’s view that ETDRS 22 showed a significant association between high triglycerides at baseline and the development of hard exudates.
240 The theoretical basis for Professor Carter’s reasoning was simple – if retinal hard exudates are lipids which have leaked into the macular, could a beneficial effect be achieved by reducing lipid levels in the patient’s blood? This was, as I understood Professor Carter’s evidence, a question that had occurred to him many years before ETDRS 22 was published. The publication of ETDRS 22 raised the same question, particularly with respect to elevated triglyceride levels.
241 ETDRS 22 showed a relationship between progressive high risk proliferative diabetic retinopathy over five years and baseline serum triglycerides in the age group 50-69, which is no doubt an important age group in the context of type 2 diabetes and diabetic retinopathy. It provided a reasonable basis to hypothesize that a reduction in elevated serum triglyceride levels achieved through the use of fenofibrate would reduce the risk of diabetic retinopathy at least in this age group.
242 While the methodology of ETDRS 22 was less than perfect, I think the significance of any methodological shortcomings have been overstated by the applicants. Professor Carter and Dr Beaumont clearly thought the observations were highly credible. Professor O’Brien also agreed that ETDRS 22 was clinically important and was apparently content to make reference to ETDRS 22 in the 1997 NHMRC Guidelines (of which he was the principal author) in support of the proposition that “[e]levated serum total cholesterol is associated with an increased risk of retinal hard exudates” without mentioning any of the methodological shortcomings that the applicants have fixed upon.
243 Further, Professor Mitchell understood, based on ETDRS 22, that it would be reasonable to think that fenofibrate “… could have a benefit for the eye in people with diabetic retinopathy” and that practitioners prescribing fenofibrate for the purpose of lowering lipids, could quite reasonably do so in the belief that this may also be helping to preserve the patient’s vision. This was certainly the view of Professor Carter and Dr Beaumont.
244 This brings me to the balance of applicants’ submissions on this topic.
245 The early studies referred to by Professor Carter focused on fibrates and were conducted before the development of statins. However, by 10 November 2005, which was after the publication of the CARDS study, it seems unlikely that Professor Carter would have considered the body of research to which he referred as a starting point for a clinical trial that investigated the effects of fibrates on hard exudates given that statins were known to be more effective at reducing lipids than fibrates. That does not necessarily mean that it would not have been worthwhile to conduct such a trial using fibrates. But it does suggest to me that the notional team might not have gone down that pathway without first investigating the use of statins in the treatment of hard exudates and diabetic retinopathy more generally or, at least, using both a fibrate and a statin in combination as Professor Carter often did.
246 It was submitted by the applicants that the notional team would have understood as at 10 November 2005 that there was a concern about interactions between statins and fibrates which would have deterred them from using them either in combination with statins or alone (because statins were the standard of care in diabetic patients). The affidavit evidence of Professor Mitchell and Professor O’Brien that was relied on in relation to this submission does not support it. Further, the NHMRC’s “National Evidence Based Guidelines for the Management of Type 2 Diabetes Mellitus – Part 7 Lipid Control in Diabetics” dated 16 September 2004 (another set of NHMRC guidelines which Professor O’Brien helped to prepare) noted at p 8:
Treatment with a statin and fibrate should be considered in people with moderate to marked elevation of both LDL cholesterol and triglycerides. Because of the increased risk of myositis, the person should be fully informed and carefully monitored.
247 The risk of myositis (inflammation of the muscles) was managed through proper monitoring. It did not deter either Professor Carter or Professor O’Brien from prescribing the combination therapy where appropriate. As to the use of fibrate therapy alone, there were of course some patients who were intolerant of statins for whom statin therapy was the first line of treatment for elevated cholesterol levels.
248 In my opinion, the notional skilled team would have been directly led by ETDRS 22 to try fenofibrate (because of its effectiveness in reducing elevated triglyceride levels and increasing HDL cholesterol levels) in combination with statins (because of their effectiveness in reducing elevated LDL cholesterol levels) in the expectation that this might well prevent or slow the development or progression of retinal hard exudates.
249 I should add that Professor O’Brien said in his oral evidence that, in his assessment, the prospect of fenofibrate proving effective in the treatment of diabetic retinopathy was “no better than fifty-fifty”. That statement seems to imply that there was no support at all in ETDRS 22 for the hypothesis that the use of a lipid-lowering agent in patients with elevated serum lipid levels would prevent or slow the development or progression of retinal hard exudates. For the reasons I have given I do not think this is correct. Nor do I think the use of a percentage based analysis is useful in this case especially when it is expressed in terms of “no better than fifty-fifty”. The modified Cripps question may receive an affirmative answer even if the hypothetical person (or team) skilled in the art has prospects of success that are less than fifty-fifty. Whether or not it does so must depend on the circumstances of each case.
250 I am satisfied that claims 1 and 7 of the 711 Patent are invalid for lack of inventive step. Given that MIMS Annual (Twenty-Ninth Edition, June 2005) indicates that fenofibrate (as Lipidil) is made available as (inter alia) a 160 mg tablet, it is clear that all of the other dependent claims on which the respondent is sued are also invalid for lack of inventive step.
Manner of Manufacture
251 The respondent submitted that it was apparent from the face of the 711 Patent that fenofibrate was well-known and used before 10 November 2005 for the treatment of diabetic patients with dyslipidaemia and that the majority of such patients develop diabetic retinopathy. It submitted that the alleged invention is said to arise from the “discovery” that patients treated with fenofibrate needed fewer treatments by laser for diabetic retinopathy than patients treated with a placebo.
252 In support of its submission, the respondent relied upon the following passage from the judgment of Gageler and Nettle JJ in D'Arcy v Myriad Genetics Inc (2015) 258 CLR 334 (“Myriad”) who, after referring to Commissioner of Patents v Microcell Ltd (1958) 102 CLR 232, NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1995) 183 CLR 655 and Advanced Building Systems Pty Ltd v Ramset Fasteners (Aust) Pty Ltd (1998) 194 CLR 171 said at [131]:
Notwithstanding that Microcell did not establish a discrete “threshold” test, each of those decisions is consistent with the requirement, essential to the concept of a “manner of manufacture”, that the subject matter of a claim have about it a quality of inventiveness which distinguishes it from a mere discovery or observation of a law of nature. That requirement is separate and distinct from the other requirements of inventive step and novelty. As Brennan, Deane and Toohey JJ stated in Philips [at 663-664], an alleged invention will:
“remain unsatisfied if it is apparent on the face of the relevant specification that the subject matter of the claim is, by reason of absence of the necessary quality of inventiveness, not a manner of new manufacture for the purposes of the Statute of Monopolies. That does not mean that the threshold requirement of ‘an alleged invention’ corresponds with or renders otiose the more specific requirements of novelty and inventive step (when compared with the prior art base) contained in s 18(1)(b). It simply means that, if it is apparent on the face of the specification that the quality of inventiveness necessary for there to be a proper subject of letters patent under the Statute of Monopolies is absent, one need go no further.”
(footnotes omitted)
253 In Myriad, the Court was concerned with the inherent patentability of a claim to an isolated DNA sequence that mirrored naturally occurring genetic information. It was in that context that Gageler and Nettle JJ observed that a claim to a mere discovery or observation of a law of nature could not be patentable subject matter. The High Court in Myriad did not base its decision on the “threshold” test which requires an examination of whether the claimed invention is new and inventive on the “face of the specification”.
254 Every valid claim to a new method of treatment using an existing therapeutic agent involves a discovery or observation of a natural phenomenon arising from the new use of the therapeutic agent, distinct from a mere discovery or observation of a law of nature. The type of “mere discovery or observation of a law of nature” which Gageler and Nettle JJ were directing their observations to in Myriad was bare genetic information. The debate over whether claims for a new method of use is a manner of manufacture has been answered in the affirmative by the High Court in Apotex Pty Ltd v Sanofi-Aventis Australia Pty Ltd (2013) 253 CLR 284.
Injunctive Relief
255 The parties made submissions in relation to the question of injunctive relief against the possibility that I would find the method of treatment basis valid and likely to be infringement by the respondent by the supply of the Ranbaxy Products.
256 The respondent contended that if the method of treatment claims were found to be valid and likely to be infringed then, as a matter of discretion, no injunctive relief should be granted. As the Full Court explained in AstraZeneca FC at [444]:
It may be undesirable to impose a blanket restraint upon a supplier who has reason to believe that only some consumers, perhaps a very small minority, may put the product that is or may be supplied to them to an infringing use. This is because the effect of such an injunction may be to deny a supplier access to a market, and consumers’ access to a product, in circumstances where the supplier could have no reason to believe that the majority of consumers would put the product to an infringing use. It seems to us that, all other things being equal, the more difficult it is for the patentee to establish that there is a likelihood of widespread infringing use, the more difficult it should be for the patentee to obtain injunctive relief in the broad terms restraining any supply of the relevant product. In the present case, even if AstraZeneca established that the generic parties had reason to believe that some consumers would engage in infringing use, the likely scale of that activity, were it to occur, was not shown to be such as would justify the grant of the wide injunction that AstraZeneca sought. Given our conclusion in relation to the validity of claims of the 051 or low dose patent, it is not necessary for us to consider what other injunctive relief, if any, might have been appropriate in lieu of that sought by AstraZeneca.
257 The question whether a blanket injunction restraining any supply of a product should be granted based on s 117(2)(b) of the Act will often be problematic in cases involving claims for new methods for treating medical conditions that utilise therapeutic agents that were known and used before the priority date to treat other medical conditions. The fact that the new method of treatment involves using a therapeutic agent that is widely used for non-infringing purposes (ie to treat conditions not designated in the relevant claim) provides a powerful argument in favour of refusing such an injunction particularly if there is no evidence to suggest (as in this case) that the alleged infringer will induce or encourage any direct infringement of the method of treatment except by the mere act of supplying the therapeutic agent used to perform the method of treatment.
258 The respondent offered to give to the Court the following undertaking in support of its contention that the more broadly framed injunctive relief sought by the applicants restraining the supply of the Ranbaxy Products should be withheld on discretionary grounds:
Until the expiry or revocation of Australian Patent No. 2006313711:
1. The Ranbaxy Products will only be promoted and marketed in Australia for their TGA-approved indications (that is, “as an adjunct to diet in the treatment of hypercholesterolaemia; types II, III, IV and V dyslipidaemia; and dyslipidaemia associated with type 2 diabetes”);
2. The Ranbaxy Products will not be promoted and marketed in Australia for the treatment or prevention of diabetic retinopathy;
3. The respondent will notify, by writing, all Australian pharmacists that the Ranbaxy Products are not to be dispensed for the treatment or prevention of diabetic retinopathy; and
4. The respondent will notify, in writing, all Australian prescribers that the applicants’ fenofibrate products are not to be prescribed for the treatment or prevention of diabetic retinopathy and, if a prescriber wishes to prescribe fenofibrate for the treatment or prevention of diabetic retinopathy, they should prescribe LIPIDIL® by brand name and indicate on the prescription that “brand substitution is not permitted”.
259 Given the conclusions I have reached in relation to the validity of the method of treatment claims, it is not necessary for me to decide whether or not the applicants should be awarded the quia timet injunctive relief they seek. However, in light of the undertakings proffered by the respondent and the extensive use of fenofibrate as a lipid-lowering agent, I would have refused the injunctive relief sought by the applicants. I would not have required the respondent to give all of the proffered undertakings as a condition of such a refusal. In particular, I am not satisfied that it would be either necessary or desirable to require the respondent to write to medical practitioners as proposed in the fourth of the proposed undertakings. It is open to the applicants to write to medical practitioners suggesting that they prescribe Lipidil for the prevention or treatment of diabetic retinopathy should the applicants choose to do so. Had the relevant claims been found valid, it would have been open to the applicants to advise medical practitioners that by failing to act in accordance with that suggestion they may be liable for authorising a patent infringement. Of course, I accept that the applicants may have strong commercial reasons for not wishing to engage in such correspondence.
The 964 Patent
The Specification
260 The claimed priority date of the 964 Patent is 17 January 2007. The respondent did not contend for any different priority.
261 The 964 Patent states at page 1 lines 5-37:
The present invention relates to a novel pharmaceutical composition having high bioavailability through improved dissolution, and a method for preparing it. The invention more particularly relates to a pharmaceutical composition for administration by oral route, containing an active ingredient of poor aqueous solubility.
Numerous active ingredients suffer from the disadvantage of being poorly soluble in an aqueous medium, thus having an insufficient dissolution profile and, consequently, poor bioavailability within the organism, following oral administration. The therapeutic dose required to be administered must thus be increased in order to obviate this disadvantage. This particularly applies to numerous hypolipemiant active ingredients, such as those belonging to the fibrate family.
Fenofibrate is a well-known hypolipemiant from the family of fibrates, which is commercially available in various doses (100 and 300 mg for example Secalip®) but in a form leading to poor bioavailability of the active ingredient. Indeed, due to it [sic] poor hydrosolubility, fenofibrate is poorly absorbed in the digestive tract and consequently its bioavailability is incomplete, irregular and often varies from one person to another.
To improve the dissolution profile of fenofibrate and its bioavailability, thereby reducing the dose requiring to be administered, it would be useful to increase its dissolution so that it could attain a level close to 100%.
Moreover, for patient comfort, it is advantageous to seek a dosage form that only requires the medicament to be taken once daily while giving the same effect as one administered several times daily.
262 This is followed by a discussion of prior art at page 2 lines 1-33:
EP-A-0330532 discloses a method for improving bioavailability of fenofibrate. This patent describes the effect of co-micronizing fenofibrate with a surfactant, for example sodium laurylsulfate in order to improve fenofibrate solubility and thereby increase its bioavailability. This patent teaches that co-micronizing fenofibrate with a solid surfactant improves fenofibrate bioavailability to a much greater extent than the improvement that would be obtained either by adding a surfactant, or through solely micronizing the fenofibrate, or, yet again, through intimately mixing the fenofibrate and surfactant, micronized separately. The dissolution method employed is the conventional rotating blade technique (European Pharmacopoeia) : product dissolution kinetics are measured in a fixed volume of the dissolution medium, agitated by means of a standardized device; a test was also carried out with an alternative technique to the European Pharmacopoeia, using the continuous-flow cell method.
The process of EP-A-0330532 leads to a new dosage form in which the active ingredient, co-micronized with a solid surfactant, has improved fenofibrate dissolution, and thus increased bioavailability, which makes it possible, for a given level of effectiveness, to decrease the daily dose of the medicament: respective 67 mg and 200 mg instead of 100 mg and 300 mg.
However, the preparation method in that patent is not completely satisfactory inasmuch as it does not lead to complete bioavailability of the active ingredient, and suffers from several disadvantages. The technique of co-micronizing fenofibrate with a solid surfactant does, it is true, improve dissolution of the active ingredient, but this dissolution remains, however, incomplete.
263 The 964 Patent then describes a need and the inventor’s discovery at page 2 line 34 to page 3 line 12 as follows:
There is thus a need to improve fenofibrate bioavailability in order to attain, over very short periods of time, a level close to 100% (or, in any case, better than the following limits: 10% in 5 minutes, 20% in 10 minutes, 50% in 20 minutes and 75% in 30 minutes in a medium consisting of 1200 ml water to which 2% Polysorbate 80 is added, or of 1000 ml of water to which 0.025M sodium lauryl sulfate sodium [sic] is added, with a blade rotation speed of 75 rpm), and this even when dissolution media having a low surfactant content are used.
Applicant has found that, surprisingly, it is possible to resolve this problem by a new method for preparing a pharmaceutical composition by spraying a suspension of the active ingredient onto an inert hydrosoluble carrier. The present invention also relates to pharmaceutical compositions thus prepared.
264 This is followed by further discussion of prior art, a number of consistory statements, and more detailed description with reference to two graphs. The consistory statements that appear at page 5 line 1 to page 6 line 4 are as follows:
Thus, the present invention provides an immediate-release fenofibrate composition comprising:
(a) an inert hydrosoluble carrier covered with at least one layer containing a fenofibrate active ingredient in a micronized form having a size less than μm, a hydrophilic polymer and, optionally, a surfactant; said hydrophilic polymer making up at least 20% by weight of (a); and
(b) optionally one or several outer phase(s) or layer(s).
In one embodiment, a surfactant is present with the active ingredient and the hydrophilic polymer.
The invention also provides a composition comprising fenofibrate having a dissolution of at least 10% in 5 minutes, 20% in 10 minutes, 50% in 20 minutes and 75% in 30 minutes, as measured using the rotating blade method at 75 rpm according to the European Pharmacopoeia, in a dissolution medium constituted by water with 2% by weight polysorbate 80 or in a dissolution medium constituted by water with 0.025M sodium lauryl sulfate.
A method for preparing a pharmaceutical composition is also provided, comprising the steps of:
(a) preparing a fenofibrate suspension in micronized form with a particle size below 20 μm, in a solution of 25 hydrophilic polymer and, optionally surfactant;
(b) applying the suspension from step (a) to an inert hydrosoluble carrier;
(c) optionally, coating granules thus obtained with one or several phase(s) or layer(s).
Step (b) is preferably carried out in a fluidized-bed granulator.
The method can comprise a step in which products obtained from step (b) or (c) are compressed, with or without additional excipients.
The invention also provides a suspension of fenofibrate in micronized form having a size less than 10 μm, in a solution of hydrophilic polymer and, optionally, surfactant.
265 The specification states at page 8 lines 16-25:
The compositions according to the invention comprise, in general, based on the total composition weight excluding the outer phase or layer, an inert hydrosoluble carrier making up from 10 to 80% by weight, preferably 20 to 50% by weight, the fenofibrate representing from 5 to 50% by weight, preferably from 20 to 45% by weight, the hydrophilic polymer representing from 20 to 60% by weight, preferably 25 to 45% by weight, the surfactant making up from o to 10% by weight, preferably 0.1 to 3% by weight.
266 Importantly, this observation assumes the presence in compositions of the invention of an inert hydrosoluble carrier and hydrophilic polymer in both non-preferred and preferred weight ratios.
267 The description of the method of the invention includes at page 9 lines 12-27:
The composition according to the invention is prepared by a novel process comprising spraying a suspension of the active ingredient in a micronized form in a solution of a hydrophilic polymer and, optionally, a surfactant, onto the inert cores.
When a surfactant is present, the active ingredient can be co-micronized with the surfactant. One will then use with advantage the teachings of EP-A-0330532.
The method according to the invention consists in using the fluidized bed granulation principle, but with specific starting materials, in order to arrive at an improved dissolution profile and thus, at elevated bioavailability. In particular, the invention employs a suspension of the micronized active ingredient in a solution of a hydrophylic polymer and, optionally, a surfactant.
268 The specification goes on to say that compositions can be prepared by other methods at page 10 lines 13-15:
The compositions according to the invention can also be prepared by other methods, for example by spraying a solution of the micronized active ingredient onto the hydrosoluble inert carrier.
269 But while other methods may be used, the example given is consistent with the view that the resulting composition will include both fenofibrate in micronized form and an inert hydrosoluble carrier.
270 The specification explains why the patent applicant believes the composition of the invention has an improved dissolution profile at page 11 lines 15-21:
The invention also covers this novel suspension. Without wishing to be tied down to a specific theory, applicant believes that this novel method, through the use of a micronized active ingredient suspension in a hydrophilic polymer solution, enabled a novel composition to be obtained in which the active ingredient is in a non-re-agglomerated form.
271 So the theory behind the invention, at least as explained in this passage, also assumes that compositions of the invention will include a micronized fenofibrate suspension in a hydrophilic polymer solution.
272 The first graph (Fig 1) relates to a comparative study of the dissolution profile of a composition of the invention compared to that of Lipanthyl® 200M. The second (Fig 2) is a graph illustrating a comparative study of the dissolution profile of a composition of the invention and that of pharmaceutical products commercially available on the German market.
273 There are four examples given illustrating the invention “without limiting it”.
The Claims
274 Claim 1 of the 964 Patent (upon which the respondent is not sued) and claims 12, 13, 17 and 19 (on which it is sued) of the 964 Patent claim are as follows:
1. An immediate-release fenofibrate composition comprising:
(a) an inert hydrosoluble carrier covered with at least one layer containing fenofibrate in a micronized form having a size less than 20 μm, a hydrophilic polymer and, optionally, a surfactant; said hydrophilic polymer making up at least 20% by weight of (a); and
(b) optionally one or several outer phase(s) or layer(s).
…
12. A composition comprising fenofibrate having a dissolution of at least 10% in 5 minutes, 20% in 10 minutes, 50% in 20 minutes and 75% in 30 minutes, as measured using the rotating blade method at 75 rpm according to the European Pharmacopoeia, in a dissolution medium constituted by water with 2% by weight polysorbate 80 or with 0.025M sodium lauryl sulfate.
13. The composition according to any one of the preceding claims, in the form of a tablet.
…
17 A suspension of fenofibrate in micronized form having a size less than 20 μm, in a solution of hydrophilic polymer and, optionally, surfactant.
…
19 The suspension of fenofibrate according to claim 17 or 18, in which the hydrophilic polymer concentration is from 5 to 40% by weight, preferably from 10 to 25% by weight.
The Issues
275 The parties identified five issues in relation to the 964 Patent one of which fell away during closing submissions. That leaves the following issues arising with respect to the 964 Patent:
Issue 1: Are claims 12 and 13 fairly based on the matter described in the specification?
Issue 2: Does claim 12 (and dependant claim 13) on its proper construction, require the use of a dissolution medium of 900 mL for determining the dissolution profile?
Issue 3: Does the respondent threaten to infringe claims 12 and 13?
Issue 4: Does the respondent threaten to infringe claims 17 and 19?
276 In closing submissions the respondent accepted that the outcome of Issue 3 is solely dependent upon the outcome of Issue 2. Issue 4 issues raises for consideration the applicability of the English Court of Appeal’s decision in Beecham Group Ltd v Bristol Laboratories [1978] RPC 153 (“Beecham”) in an infringement proceeding taken under the Act.
Issue 1 – Are claims 12 and 13 fairly based on the matter described in the specification?
277 Section 40(3) of the Act, as it stood prior to its amendment by the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth), states that a claim must be (inter alia) “fairly based on the matter described in the specification”.
278 In Lockwood Security Products Pty Limited v Doric Products Pty Ltd (No 1) (2004) 217 CLR 274 (“Lockwood No 1”) the High Court (Gleeson CJ, McHugh, Gummow, Hayne and Heydon JJ) referred with approval to a passage in the judgment of Barwick CJ in Olin Corporation v Super Cartridge Co Pty Ltd (1977) 180 CLR 236 (and also approved Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [15]) in which the Chief Justice said at 240:
The question whether the claim is fairly based is not to be resolved … by considering whether a monopoly in the product would be an undue reward for the disclosure. Rather, the question is a narrow one, namely whether the claim to the product being new, useful, and inventive, that is to say, the claim as expressed, travels beyond the matter disclosed in the specification.
279 The High Court in Lockwood No 1 held that that s 40(3) requires a “real and reasonably clear disclosure” of what is claimed. But it also made clear that the question whether there is such a disclosure is not answered solely by reference to the description of preferred embodiments. The Court said at [68]-[99]:
[68] Erroneous principles. The comparison which s 40(3) calls for is not analogous to that between a claim and an alleged anticipation or infringement. It is wrong to employ “an over meticulous verbal analysis”. It is wrong to seek to isolate in the body of the specification “essential integers” or “essential features” of an alleged invention and to ask whether they correspond with the essential integers of the claim in question [CCOM Pty Ltd v Jiejing Pty Ltd (1994) 51 FCR 260 at 281, per Spender, Gummow and Heerey JJ].
[69] “Real and reasonably clear disclosure”. Section 40(3) requires, in Fullagar J’s words, “a real and reasonably clear disclosure” [Société des Usines Chimiques Rhône-Poulenc v Commissioner of Patents (1958) 100 CLR 5 at 11]. But those words, when used in connection with s 40(3), do not limit disclosures to preferred embodiments.
“The circumstance that something is a requirement for the best method of performing an invention does not make it necessarily a requirement for all claims; likewise, the circumstance that material is part of the description of the invention does not mean that it must be included as an integer of each claim. Rather, the question is whether there is a real and reasonably clear disclosure in the body of the specification of what is then claimed, so that the alleged invention as claimed is broadly, that is to say in a general sense, described in the body of the specification.” [Rehm Pty Ltd v Websters Security Systems (International) Pty Ltd (1988) 81 ALR 79 at 95, per Gummow J]
Fullagar J’s phrase serves the function of compelling attention to the construction of the specification as a whole, putting aside particular parts which, although in isolation they might appear to point against the “real” disclosure, are in truth only loose or stray remarks.
(some citations omitted)
280 Lockwood No 1 is also authority for the proposition that a consistory clause may provide a fair basis for a claim which mirrors its language but not if there are other matters disclosed in the specification which show that the invention is narrower than the consistory clause suggests. The High Court said at [99]:
… the correct position is that a claim based on what has been cast in the form of a consistory clause is not fairly based if other parts of the matter in the specification show that the invention is narrower than that consistory clause. The inquiry is into what the body of the specification read as a whole discloses as the invention [Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 612-613]. An assertion by the inventor in a consistory clause of that of which the invention consists does not compel the conclusion by the court that the claims are fairly based nor is the assertion determinative of the identity of the invention. The consistory clause is to be considered by the court with the rest of the specification.
281 In the case of the 964 Patent, the invention described in the body of the specification has a number of aspects.
282 One aspect of the invention described is an immediate release fenofibrate composition containing (inter alia) fenofibrate in a micronized form having a size less than 20 μm. I will refer to this as the immediate release formulation. The immediate release formulation is the subject of independent claim 1.
283 Another aspect of the invention described is a method of preparing the formulation of the invention. This method comprises a number of steps the first of which is the preparation of fenofibrate in micronized form with a particle size below 20 μm suspended in a solution of hydrophilic polymer and, optionally, a surfactant. This is the method claimed in independent claim 14. The form and size of the fenofibrate as used in the method corresponds with the form and size of the fenofibrate referred to in the immediate release formulation.
284 A further aspect of the invention described is the suspension that is prepared as the first step of the method. This is the suspension claimed in independent claim 17.
285 The respondent does not suggest that any of the three “aspects of the invention” to which I have referred is not the subject of a real and reasonably clear disclosure in the specification.
286 The question is whether the specification also contains a real and reasonably clear disclosure of an invention consisting of “[a] composition comprising fenofibrate” with a particular dissolution profile as claimed in independent claim 12 that need not contain fenofibrate in micronized form.
287 In my view, when read as a whole, the specification discloses an invention comprising a fenofibrate composition that includes fenofibrate in micronized form having a size less than 20 μm, an inert hydrosoluble carrier and a hydrophilic polymer. It does not disclose an invention (or an aspect of an invention) comprising any composition of fenofibrate that has the improved dissolution profile.
288 The passage at page 2 line 34 to page 3 line 6 of the specification does not describe the invention, but the problem which is said to have been solved. The problem is said to have been solved by making the product most broadly described at page 5 lines 1-10 using the method described at page 5 lines 21-34. The dissolution profile specified at page 5 lines 13-20 is not a statement of the invention but a statement of an advantage of the invention. To the extent that the consistory statement at page 5 lines 13-20 of the specification may suggest otherwise, I regard it as inconsistent with what is disclosed elsewhere in the document including at page 8 lines 16-25, page 9 lines 12-27 (when read with page 10 lines 13-15), page 11 lines 15-21, and, I would add, other parts of the consistory clause.
289 The formulations used in the Examples include particles of micronized fenofibrate that are less than 20 μm in size, and an inert hydrosoluble carrier and a hydrophilic polymer. That does not itself imply that the invention as described is limited to a product, or a method of making a product, in which these particular ingredients are not present. The specification makes clear that the invention described is not limited by the Examples. But the description of the composition of the formulations used in the Examples form part of the matter disclosed and must be taken into account when seeking to characterise the invention described in the specification when read as a whole.
290 It follows that I accept the respondent’s contention that claims 12 and 13 are not fairly based on the matter described in the specification.
Issues 2 and 3 – Does claim 12 (and dependant claim 13) on its proper construction, require the use of a dissolution medium of 900 mL for determining the dissolution profile?
291 This issue arises out of the absence of any specified volume for the dissolution medium in claim 12. The applicants rely on experimental evidence using a dissolution medium of 1,000 mL to which 0.025M of sodium lauryl sulfate was added.
292 The applicants contended claim 12 does not prescribe any specific dissolution medium volume and that, properly construed, it leaves a choice of dissolution medium volume open to the person conducting the dissolution test. They submitted that it would not have been appropriate for the inventors of the 964 Patent to specify a volume of dissolution medium, as the volume that should be used for a certain fenofibrate composition depends on the amount of fenofibrate in the composition being tested.
293 The applicants also submitted that if, contrary to their primary submission, claim 12 is to be construed as requiring the use of a specific volume, then that volume is to be determined by reference to the specification, which describes at page 3 the attainment of the desired dissolution rate in “a medium consisting of 1200 ml water to which 2% Polysorbate 80 is added, or of 1000 ml of water to which 0.025M sodium lauryl sulfate sodium [sic] is added”. They submitted that because there is no reference to 900 mL in the specification, there is no proper basis upon which it could be considered an integer of claim 12.
294 The respondent relied on the absence of a specified volume of dissolution medium in claim 12 and the consistory statement for that claim at page 5, lines 13-20. It submitted that the evidence of Dr Williams and Professor Roberts showed that, in the absence of any contrary indication in claim 12, then the standard volume for dissolution testing in accordance with the European Pharmacopoeia, ie. 900 mL, is to be used. The respondent submitted that if that is correct, it would not be open to find that there had been infringement of claim 12 because there is no evidence as to the dissolution rate of the Ranbaxy Products in a 900 mL dissolution medium.
295 Claim 12 recites that the dissolution profile should be measured using the “rotating blade method … according to the European Pharmacopoeia”. The “rotating blade method” described in the European Pharmacopoeia uses the “paddle apparatus” (which has rotating blades). This is a standard form of testing. It is not in dispute that the skilled addressee would understand the reference to the “rotating blade method” to be a reference to the paddle apparatus. That apparatus is described in the European Pharmacopoeia as using a cylindrical vessel with a “nominal capacity of 1000 ml”. However, the description in the “Method” section of the European Pharmacopoeia for the paddle apparatus makes clear that the dissolution volume is as “prescribed” for the test being performed.
296 Relevantly, the skilled addressee would understand:
over the course of a dissolution test, the concentration of drug in the dissolution medium volume increases as the drug dissolves;
the extent and rate of dissolution of a drug will slow down as the concentration of the drug in the dissolution medium approaches saturation; and
a drug will not continue to dissolve in a dissolution medium once the concentration of the drug in the medium has reached saturation.
297 In light of these matters, the skilled addressee would select a volume of dissolution medium that is sufficient to ensure that the dissolution rate of the drug will not be significantly slowed down as the dissolution test proceeds. All that is required is that the dissolution medium volume used is “in excess of the solubility for [the] compound” being tested.
298 It was common ground between the experts that the typical volume used for the purposes of a dissolution test conducted using the rotating blade method according to the European Pharmacopoeia is 900 mL. However, in the absence of any express indication in the claim as to what volume should be used, I am satisfied that the skilled addressee would look to the specification for the purposes of determining the appropriate volume. The passage at page 2 line 34 to page 3 line 6 is significant in that it specifies two possible alternatives, one involving a medium consisting of 1200 mL of water to which 2% Polysorbate 80 is added, or 1000 mL of water to which 0.025M of sodium lauryl sulfate is added. The choice of a medium that includes either Polysorbate 80 or sodium lauryl sulfate is reflected in claim 12 which makes clear that the dissolution medium may be constituted in either of these ways. I think the skilled addressee seeking to ascertain whether a composition was within the scope of claim 12, would look to the specification for guidance as to what volume of dissolution medium to use which he or she would find in the passage to which I have referred.
299 It follows that I do not accept the respondent’s contention that claim 12 (and dependant claim 13) requires the use of a dissolution medium of 900 mL.
Issue 4 – Does the respondent threaten to infringe claims 17 and 19?
300 It is common ground that the Ranbaxy Products are not, and do not include, any product within the scope of claims 17 or 19 although products within the scope of those claims will be used outside the patent area in the course of their manufacture. The applicants argued that claims 17 and 19 will be infringed by the importation and sale of the Ranbaxy Products in the patent area is principally based on some observations made by Russell LJ when delivering the judgment of the Court of Appeal in Beecham at 200-201. The question that arises in the present case is whether his Lordship’s reasoning on this topic is applicable to infringement proceedings taken under the Act.
The Saccharin doctrine
301 The so-called Saccharin doctrine is named after the case of Saccharin Corporation Ltd v Anglo-Continental Chemical Works Ltd (1900) 17 RPC 307 in which a patented process was found by Buckley J to be infringed by importation of a product made by use of the patented process overseas. At the time that the judgment in Saccharin was handed down, under the relevant UK legislation a patent was infringed if another person either “directly or indirectly” made use of or put into practice the claimed invention. Buckley J said at 318-319:
I had better deal first with the contention that has been raised by the Defendants to the following effect. The article which was imported and sold, they say, was saccharin. The Plaintiffs’ process is one for the manufacture of ortho-toluene-sulpho-chloride, and that product is not saccharin. From that they argue that they have not imported or sold in infringement of the Patent. Now the facts are that, after the ortho-toluene-sulpho-chloride has been manufactured, you have for the production of saccharin first to change the chloride for an amide group and then to oxidise into saccharin and water. The article imported and sold, therefore, is produced by certain subsequent chemical operations from the article produced under the patented process. The article imported and sold is not ortho-toluene-sulpho-chloride, but ortho-toluene-sulpho-chloride is contained in saccharin. The Defendants say that the importation of that article is not an infringement. Now the grant in Letters Patent is a grant to a Patentee to make, use, exercise, and vend the invention, to have and enjoy the whole profit and advantage by reason of the invention; and to the end that he may have and enjoy the sole use and exercise and the full benefit of the invention all others are precluded from either directly or indirectly, making use of or putting in practice the said invention, or any part of the same, or in anywise imitating the same.
After reviewing the relevant authorities, his Lordship continued at 319:
If the patented process were the last stage in the production of the article sold, the importation and sale of the product would, in my opinion, plainly be an infringement. Does it make it any the less an infringement that the article produced and sold is manufactured by the use of the patented process which is subjected to certain other processes? In my opinion it does not. By the sale of saccharin, in the course of the production of which the patented process is used, the Patentee is deprived of some part of the whole profit and advantage of the invention, and the importer is indirectly making use of the invention. In my judgment, therefore, this contention fails.
302 The Saccharin doctrine was subsequently applied by the English Court of Appeal to the importation of a product made by the use overseas of a product patented in the United Kingdom. In Beecham, the judgment of the Court of Appeal delivered by Russell LJ, concluded that as the production overseas by the defendants of a finished product (a chemical) made use of two patented products and a patented process of the plaintiff, the supply of that chemical in the United Kingdom infringed the product patents. It is apparent that the conclusion reached by the Court of Appeal was founded on the proposition that the monopolies granted to the plaintiff pursuant to its patents prohibited the defendant from directly or indirectly making, using or putting into practice any of the patented inventions.
303 In the House of Lords, Lord Diplock, who delivered the principal speech, while approving of the ultimate disposition of the case by the Court of Appeal, made it clear that the House of Lords was not to be taken as either approving or rejecting the decision of the Court of Appeal in relation to the relevant product claims.
304 It is important to note that the case was decided under the provisions of the Patents Act 1949 (UK). The scope of a patentee’s rights under that Act was still determined by the terms of the actual grant (as interpreted by the courts) which expressly prohibited the use of the relevant invention whether directly or indirectly without the consent of the patentee.
The Patents Act 1977 (UK)
305 The position in the United Kingdom must now be considered in light of s 60(1)(c) of the Patents Act 1977 (UK) which provides that, where the patented invention is a process, it is an infringement to dispose of, offer to dispose of, or use or import, any product obtained “directly” by means of that process or to keep any such product whether for disposal or otherwise. The Court of Appeal in Pioneer Electronics Capital Inc v Warner Music Manufacturing Europe GmbH [1997] RPC 757 considered the impact of s 60(1)(c) on the Saccharin doctrine and held, according to the headnote to the report, that:
By confining infringement to products 'directly obtained', section 60(1)(c) would appear to have altered the previous English law that there was infringement where an imported product had been obtained directly or indirectly by means of a patented process provided always that the use of the patented process had been substantial.
Nourse LJ (with whom Leggatt LJ and Schiemann LJ agreed) said at 765 that “[A]n enactment confined to products directly obtained would appear to have altered the law in regard to products indirectly obtained.”
306 It appears that since the 1977 Act took effect, the Saccharin doctrine is no longer the law in the UK. Hence, questions that might have previously been resolved by reference to the Saccharin doctrine must now be resolved by reference to the relevant provisions of the 1977 Act.
The Patents Act 1952 (Cth)
307 In Re Application of Eli Lilly & Co [1982] 1 NSWLR 526, Wootten J considered an application by Eli Lilly for an extension of the term of its patent for monensin granted under the Patents Act 1952 (Cth). The product that Eli Lilly in fact marketed and sold in Australia was not monensin but monensin sodium. Wootten J applied the Court of Appeal’s judgment in Beecham to find that the sale of a product that involved the use of the claimed substance abroad would involve an infringement of the patented substance under the 1952 Act. His Honour observed at 533:
In the present case the importation of monensin sodium into Australia would have involved the use abroad of both a substance and a process claimed in the Australian patent. On the authority of the Court of Appeal in the former and of the House of Lords in the latter, the sale of the imported monensin sodium in Australia would have involved a breach of the plaintiff’s patent.
308 Neither party referred me to any other Australian decision applying Beecham in a situation involving the use outside the patent area of a patented product to manufacture another non-patented product that is subsequently imported.
Canada
309 I was referred by the applicants to Canadian authorities which indicated that the Saccharin doctrine still applies in situations involving the importation of non-patented products made overseas using a patented process or, although this is perhaps less well settled, a patented product.
310 In Pfizer Canada Inc and Warner-Lambert Company, LLC v Minister of Health and Ranbaxy Laboratories Limited 2007 FC 898 it was held that the Saccharin doctrine was not confined to process patents and should be extended to product patents. Snider J of the Federal Court of Canada observed at [80]:
[I]t seems to me that the focus of any analysis must be on whether the inventor has been deprived, even in part or even indirectly, of the full enjoyment of the invention. Under this approach, I see no reason why I should necessarily limit the application of the doctrine to process claims. Cannot, in certain circumstances, the use of a product offshore result in a loss of a patentee's advantage?
At paragraph [88] her Honour concluded that under Canadian law, the Saccharin doctrine was not confined to the importation of non-patented products the making of which would have infringed process claims.
The Patents Act 1990 (Cth)
311 Section 13 of the Act confers on the patentee the exclusive right to exploit the invention the subject of the patent throughout the patent area. Section 13 provides:
13 Exclusive rights given by patent
(1) Subject to this Act, a patent gives the patentee the exclusive rights, during the term of the patent, to exploit the invention and to authorise another person to exploit the invention.
(2) The exclusive rights are personal property and are capable of assignment and of devolution by law.
(3) A patent has effect throughout the patent area.
The definition of “patent area” includes Australia and the other areas referred to in s 12. The term “exploit” is defined in Schedule 1 to mean:
exploit, in relation to an invention, includes:
(a) where the invention is a product–make, hire, sell or otherwise dispose of the product, offer to make, sell, hire or otherwise dispose of it, use or import it, or keep it for the purpose of doing any of those things; or
(b) where the invention is a method or process–use the method or process or do any act mentioned in paragraph (a) in respect of a product resulting from such use.
312 The definition of exploit is elaborate. It is expressed to be inclusive but is otherwise quite explicit in defining what will constitute exploitation of a product claim and a method (or process) claim. As I explained in Apotex (No 2) at [297]:
Paragraph (b) of the definition of “exploit” refers to the doing of an act referred to in para (a) which includes to make or import a product. The patentee’s exclusive rights are infringed (subject to available defences) if another person does any such act within the patent area. The fact that the patented method is performed outside the patent area does not avoid infringement of a method claim (including a Swiss claim) if the product imported and sold in Australia was made using the patented method because “the acts of importation and sale occur within the patent area.” The relevant act of infringement is not the use of the method outside the patent area but the exploitation (by importation and sale) in Australia of a product made using the patented method.
The Full Court agreed with this analysis: Warner-Lambert (No 2) at [167].
313 However, the definition of exploit says nothing (except by its silence) about a situation in which a patented product is used outside the patent area to make another product that a person subsequently imports into the patent area.
314 The applicants submitted that if it is an infringement of a method claim to import into the patent area a product made outside the patent area by the use of a patented method then it creates an incongruity in the law if it is not also an infringement to use a patented product outside the patent area to produce another product that is subsequently imported. They also submitted that a patentee would be deprived of the full enjoyment of its exclusive rights if the patented product could be used outside the patent area to produce a patented product that is an important part of another product subsequently imported and sold in Australia.
315 I do not accept these submissions.
316 What the applicants’ call an incongruity in the law is the outcome of a legislative scheme that distinguishes product claims from method claims, and that defines the concept of exploitation for product claims differently from the way in which it is defined for method claims. There is no reason to think that the differences are attributable to anything other than a deliberate drafting choice aimed at defining the exclusive right so as not to prevent a person from importing non-patented goods that have been made outside the patent area using a patented product. The Patents Act 1977 (UK), which was the first UK Patents Act to define what constitutes infringement, took the same approach albeit in the context of a different legislative scheme.
317 While it is true that the definition of exploit is inclusive, it would be surprising if this was intended to provide a gateway for the recognition of a right to obtain relief for a type of infringement that may fairly be described as indirect, but without providing any express limitation or qualification in respect of that right: cf. Wilderman v Berk (1925) 42 RPC 79 (a case in which Tomlin J expressed the view that the patented process or product used overseas had to play an important part in the manufacture of the imported article).
318 As to the argument that a patentee would be deprived of the full enjoyment of its exclusive right if the Saccharin doctrine was held not to be law in Australia, it is in my opinion circular. The exclusive right is defined not by the terms of the actual grant but by s 12 (which draws on the definition of exploit) and so the scope of the exclusive right enjoyed by the patentee must be determined by reference to that and other relevant provisions of the Act.
319 It follows that the respondent does not threaten to infringe claims 17 or 19.
The 807 Patent
The Specification
320 The claimed priority date of the 807 Patent is 24 May 2002. The respondent did not contend for any different priority.
321 The 807 Patent is directed to a hypothetical skilled team (“the notional team”) which would include a pharmaceutical scientist with expertise in particle engineering and a research pharmacist with expertise in pharmacology equipped with the common general knowledge in those fields as at 24 May 2002.
322 The 807 Patent describes the field of the invention at page 1 lines 5-7:
The present invention relates to a nanoparticulate composition comprising a fibrate, preferably fenofibrate or a salt thereof. The nanoparticulate fibrate, preferably fenofibrate, particles have an effective average particle size of less than about 2000 nm.
323 Nanoparticulate compositions and prior art methods of making them are discussed in a section of the specification headed “Background Regarding Nanoparticulate Compositions”. The specification states at page 1 lines 10-19:
Nanoparticulate compositions, first described in U.S. Patent No. 5,145,684 (“the ‘684 patent”), are particles consisting of a poorly soluble therapeutic or diagnostic agent having adsorbed onto the surface thereof a non-crosslinked surface stabilizer. The ‘684 patent does not describe nanoparticulate compositions of a fibrate.
Methods of making nanoparticulate compositions are described in, for example, U.S. Patent Nos. 5,518,187 and 5,862,999, both for “Method of Grinding Pharmaceutical Substances;” U.S. Patent No. 5, 718,388, for “Continuous Method of Grinding Pharmaceutical Substances” and U.S. Patent No. 5,510,118 for “Process of Preparing Therapeutic Compositions Containing Nanoparticles.”
This is followed by a lengthy discussion of other US patents in which nanoparticulate compositions are described.
324 Fenofibrate is described in the section of the specification headed “Background Regarding Fenofibrate”. The compound is described at page 4 lines 16-20 as follows:
The compositions of the invention comprise a fibrate, preferably fenofibrate. Fenofibrate, also known as 2-[4-(4-chlorobenzoyl) phenoxy]-2-methyl-propanoic acid, 1-methylethyl ester, is a lipid regulating agent. The compound is insoluble in water …
325 This is also followed by a discussion of other US patents in which fenofibrate is described. The patents referred to include US Patents 6,074,670 and 6,277,405 both for “Fenofibrate Pharmaceutical Compositions Having High Bioavailability and Method for Preparing It”, US Patent 6,074,670 which refers to “immediate-release fenofibrate composition comprising micronized fenofibrate and at least one inert hydrosoluble carrier”, US Patent 4,739,101 describing a process for making fenofibrate, and US Patent 6,277,405 directed to micronized fenofibrate compositions having a specified dissolution profile.
326 The specification also refers to two International Applications, WO 01/80828 for “Improved Water-Insoluble Drug Particle Process” (“the 828 Patent”) and WO 02/24193 for “Stabilised Fibrate Microparticles” (“the 193 Patent”). Both of these publications are said to describe a process for making small particle compositions of poorly water soluble drugs. Both documents, which it may be inferred were publicly available before the priority date of the 807 Patent, are specifically incorporated by reference (see page 42B lines 28-30).
327 After referring to the process described in the 828 Patent and the 193 Patent, the specification next states at page 5 lines 12-21:
The process requires preparing an admixture of a drug and one or more surface active agents, followed by heating the drug admixture to at or above the melting point of the poorly water soluble drug. The heated suspension is then homogenized. The use of such a heating process is undesirable, as heating a drug to its melting point destroys the crystalline structure of the drug. Upon cooling, a drug may be amorphous or recrystallize in a different isoform, thereby producing a composition which is physically and structurally different from that desired. Such a “different” composition may have different pharmacological properties. This is significant as U.S. Food and Drug Administration (USFDA) approval of a drug substance requires that the drug substance be stable and produced in a repeatable process.
328 This is followed by a further reference to another prior art publication describing compositions of fibrate and vitamin E TGPS (a water soluble derivative of vitamin E) comprising particles the diameters of which are within defined ranges in which the mean diameter of about 100 nm to about 900 nm, with 50% of the particles of the composition below the range 350 nm to 750 nm (“D50”) and 99% below the range 500 nm to 900 nm (“D99”). The specification then states at page 6 lines 1-2 that this publication “… does not teach that the described compositions show minimal or no variability when administered in fed as compared to fasted conditions.”
329 The specification includes a description of a number of advantages that are said to arise using formulations of the fibrate composition to the invention. Compositions of the invention are said to significantly increase the bioavailability of fenofibrate which can enable use of a smaller solid dosage size. Compositions of the invention are also said to have an improved pharmacokinetic profile that is not substantially affected by the fed or fasted state of a human to whom such a composition is administered. This is a reference to what is referred to as the “food effect” whereby the taking of a poorly soluble drug with food (ie. in a fed state) may enhance it bioavailability compared to taking the drug without food (ie. in a fasted state).
330 The specification asserts that the invention encompasses a fibrate, preferably fenofibrate, composition in which administration of the composition to a subject in a fasted state is bioequivalent to the administration of the composition to a subject in a fed state. The specification adopts the following measure of bioequivalence at page 16 lines 26-30:
“Bioequivalency” is established by a 90% Confidence Interval (CI) of between 0.80 and 1.25 for both Cmax and AUC under USFDA regulatory guidelines, or a 90% CI for AUC of between 0.80 to 1.25 and a 90% CI for Cmax of between 0.70 to 1.43 under the European EMEA regulatory guidelines.
331 The specification also refers to the redispersibility of the fibrate compositions. This feature is explained at page 18 lines 13-26:
An additional feature of the fibrate, preferably fenofibrate, compositions of the invention is that the compositions redisperse such that the effective average particle size of the redispersed fibrate particles is less than about 2 microns. This is significant, as if upon administration the nanoparticulate fibrate compositions of the invention did not redisperse to a substantially nanoparticulate particle size, then the dosage form may lose the benefits afforded by formulating the fibrate into a nanoparticulate particle size.
This is because nanoparticulate active agent compositions benefit from the small particle size of the active agent; if the active agent does not redisperse into the small particle sizes upon administration, then "clumps" or agglomerated active agent particles are formed, owing to the extremely high surface free energy of the nanoparticulate system and the thermodynamic driving force to achieve an overall reduction in free energy. With the formation of such agglomerated particles, the bioavailability of the dosage form may fall well below that observed with the liquid dispersion form of the nanoparticulate active agent.
332 The specification refers at page 6 lines 3-10 to clinical studies demonstrating the association between cholesterol and the development of atherosclerosis. The specification then states at page 6 lines 11-24:
Fenofibric acid, the active metabolite of fenofibrate, produces reductions in total cholesterol, LDL cholesterol, apo-lipoprotein B, total triglycerides, and triglyceride rich lipoprotein … in treated patients. …
Because fibrates, including fenofibrate, are so insoluble in water, significant bioavailability can be problematic. In addition, conventional fibrate, including fenofibrate, formulations exhibit dramatically different effects depending upon the fed or fasted state of the patient. Finally, conventional fibrate, including fenofibrate, formulations require relatively large doses to achieve the desired therapeutic effects. There is a need in the art for nanoparticulate fibrate formulations which overcome these and other problems associated with prior conventional microcrystalline fibrate formulations. The present invention satisfies these needs.
333 The specification includes a number of definitions. According to the specification at page 6A lines 10-12, the word “comprise” (and its variants) “is not intended to exclude other additives, components, integers or steps.” At page 12 lines 3-11 there is a rather curious definition of stable, which states:
As used herein with reference to stable fibrate, preferably fenofibrate, particles, “stable” includes, but is not limited to, one or more of the following parameters: (1) that the fibrate particles do not appreciably flocculate or agglomerate due to interparticle attractive forces, or otherwise significantly increase in particle size over time; (2) that the physical structure of the fibrate, preferably fenofibrate, particles is not altered over time, such as by conversion from an amorphous phase to crystalline phase; (3) that the fibrate, preferably fenofibrate, particles are chemically stable; and/or (4) where the fibrate has not been subject to a heating step at or above the melting point of the fibrate in the preparation of the nanopartic1es of the invention.
334 The specification includes under the heading “Summary of the Invention” a number of consistory statements. The first consistory statement at page 8a lines 1-16 mirrors the language of claim 1. This is then followed by five more consistory statements that mirror the language of claims 2, 3, 40, 41 and 42. The first three consistory statements (mirroring claims 1, 2 and 3) refer to compositions (ie. products) and the other three (mirroring claims 40, 41 and 42) refer to methods of treatment.
335 The consistory statements are followed by a detailed description of the invention including by reference to two graphs (Fig 1 and Fig 2).
336 The specification states at page 10 line 16 to page 11 line 24:
The present invention is directed to nanoparticulate compositions comprising a fibrate, preferably fenofibrate. The compositions comprise a fibrate, preferably fenofibrate, and preferably at least one surface stabilizer adsorbed on the surface of the drug. The nanoparticulate fibrate, preferably fenofibrate, particles have an effective average particle size of less than about 2000 run.
As taught in the '684 patent, and as exemplified in the examples below, not every combination of surface stabilizer and active agent will result in a stable nanoparticulate composition. It was surprisingly discovered that stable, nanoparticulate fibrate, preferably fenofibrate, formulations can be made.
Advantages of the nanoparticulate fibrate, preferably fenofibrate, formulations of the invention as compared to conventional non-nanoparticulate formulations of a fibrate, particularly a fenofibrate such as TRICOR® (tablet or capsule microcrystalline fenofibrate formulations), include, but are not limited to: (1) smaller tablet or other solid dosage form size; (2) smaller doses of drug required to obtain the same pharmacological effect; (3) increased bioavailability; (4) substantially similar pharmacokinetic profiles of the nanoparticulate fibrate, preferably fenofibrate, compositions when administered in the fed versus the fasted state; (5) improved pharmacokinetic profiles; (6) bioequivalency of the nanoparticulate fibrate, preferably fenofibrate, compositions when administered in the fed versus the fasted state; (7) an increased rate of dissolution for the nanoparticulate fibrate, preferably fenofibrate, compositions; (8) bioadhesive fibrate, preferably fenofibrate, compositions; and (9) the nanoparticulate fibrate, preferably fenofibrate, compositions can be used in conjunction with other active agents useful in treating dyslipidemia, hyperlipidemia, hypercholesterolemia, cardiovascular disorders, or related conditions.
The present invention also includes nanoparticulate fibrate, preferably fenofibrate, compositions together with one or more non-toxic physiologically acceptable carriers, adjuvants, or vehicles, collectively referred to as carriers. The compositions can be formulated for parenteral injection (e.g., intravenous, intramuscular, or subcutaneous), oral administration in solid, liquid, or aerosol form, vaginal, nasal, rectal, ocular, local (powders, ointments or drops), buccal, intracisternal, intraperitoneal, or topical administration, and the like.
A preferred dosage form of the invention is a solid dosage form, although any pharmaceutically acceptable dosage form can be utilized. Exemplary solid dosage forms include, but are not limited to, tablets, capsules, sachets, lozenges, powders, pills, or granules, and the solid dosage form can be, for example, a fast melt dosage form, controlled release dosage form, lyophilized dosage form, delayed release dosage form, extended release dosage form, pulsatile release dosage form, mixed immediate release and controlled release dosage form, or a combination thereof. A solid dose tablet formulation is preferred.
337 The specification also states at page 24 lines 2-15:
The invention provides compositions comprising fibrate, preferably fenofibrate, particles and at least one surface stabilizer. The surface stabilizers preferably are adsorbed on, or associated with, the surface of the fibrate, preferably fenofibrate, particles. Surface stabilizers especially useful herein preferably physically adhere on, or associate with, the surface of the nanoparticulate fibrate particles but do not chemically react with the fibrate particles or itself. Individually adsorbed molecules of the surface stabilizer are essentially free of intermolecular cross-linkages.
338 At page 26 lines 1-10 the specification states:
The choice of a surface stabilizer for a fibrate is non-trivial and required extensive experimentation to realize a desirable formulation. Accordingly, the present invention is directed to the surprising discovery that nanoparticulate fibrate, preferably fenofibrate, compositions can be made.
Combinations of more than one surface stabilizer can be used in the invention. Useful surface stabilizers which can be employed in the invention include, but are not limited to, known organic and inorganic pharmaceutical excipients. Such excipients include various polymers, low molecular weight oligomers, natural products, and surfactants. Surface stabilizers include nonionic, anionic, cationic, ionic, and zwitterionic surfactants.
339 This passage is followed by a lengthy list of surface stablizers that can be used to perform the invention. The surface stablizers referred to include hydroxypropyl methylcellulose (also known as hypromellose or HPMC), sodium lauryl sulfate, gelatin, commercially available Tweens (eg Tween 20 and Tween 80) and many more. At page 27 lines 15-16 the specification also states that “[i]f desirable, the nanoparticulate fibrate, preferable fenofibrate, compositions of the invention can be formulated to be phospholipid-free.”
340 The specification later states at page 30 line 22 to page 31 line 13:
In one embodiment of the invention, the preferred one or more surface stabilizers of the invention is any suitable surface stabilizer as described below, with the exclusion of PEG-derivatized vitamin E, which is a non-ionic compound. In another embodiment of the invention, the preferred one or more surface stabilizers of the invention is any suitable surface stabilizer as described below, with the exclusion of phospholipids. Finally, in another embodiment of the invention, the preferred one or more surface stabilizers of the invention is any substance which is categorized by the USFDA as GRAS (“Generally Recognized As Safe”).
Preferred surface stabilizers of the invention include, but are not limited to, hypromellose, docusate sodium (DOSS), Plasdone® S630 (random copolymer of vinyl pyrrolidone and vinyl acetate in a 60:40 ratio), hydroxypropyl cellulose SL (HPC-SL), sodium lauryl sulfate (SLS), and combinations thereof. Particularly preferred combinations of surface stabilizers include, but are not limited to, hypromellose and DOSS; Plasdone® S630 and DOSS; HPC-SL and DOSS; and hypromellose, DOSS, and SLS.
The surface stabilizers are commercially available and/or can be prepared by techniques known in the art. Most of these surface stabilizers are known phannaceutical LO excipients and are described in detail in the Handbook of Pharmaceutical Excipients, published jointly by the American Pharmaceutical Association and The Pharmaceutical Society of Great Britain (The Pharmaceutical Press, 2000), specifically incorporated by reference.
341 The specification also includes a description of other pharmaceutical excipients that may be used as binders, fillers, lubricants, sweeteners and flavourings. It is said that such excipients are known in the art.
342 The description of excipients is followed by a description of the “nanoparticulate particle size”. At page 32 line 24 to page 33 line 24 the specification states:
The compositions of the invention contain nanoparticulate fibrate particles, preferably nanoparticulate fenofibrate particles, which have an effective average particle size of less than about 2000 nm (i.e., 2 microns), less than about 1900 nm, less than about 1800 nm, less than about 1700 nm, less than about 1600 nm, less than about 1500 nm, less than about 1400 nm, less than about 1300 nm, less than about 1200 nm, less than about 1100 nm, less than about 1000 nm, less than about 900 nm, less than about 800 nm, less than about 700 nm, less than about 600 nm, less than about 500 nm, less than about 400 run, less than about 300 nm, less than about 250 nm, less than about 200 nm, less than about 150 nm, less than about 100 nm, less than about 75 nm, or less than about 50 nm, as measured by light-scattering methods, microscopy, or other appropriate methods.
By “an effective average particle size of less than about 2000 nm” it is meant that at least 50% of the fibrate, preferably fenofibrate, particles have a particle size of less than the effective average, by weight, i.e., less than about 2000 nm, 1900 nm, 1800 nm, etc., when measured by the above-noted techniques. Preferably, at least about 70%, about 90%, or about 95% of the fibrate, preferably fenofibrate, particles have a particle size of less than the effective average, i.e., less than about 2000 nm, 1900 nm, 1800 nm, 1700 nm, etc.
In one embodiment of the invention, at least 99% of the fibrate particles (“D99”) have a particle size less than about 500 nm, less than about 450 nm, less than about 400 nm, less than about 350 nm, less than about 300 nm, less than about 250 nm, less than 15 about 200 run, less than about 150 nm, or less than about 100 nm. In another embodiment of the invention, at least 50% of the fibrate particles (“D50”) have a particle size less than about 350 nm, less than about 300 nm, less than about 250 nm, less than about 200 nm, less than about 150 nm, less than about 100 nm, or less than about 75 nm. In yet another embodiment of the invention, the mean particle size of the fibrate composition is less than 20 about 100 nm, less than about 75 nm, or less than about 50 nm.
In the present invention, the value for D50 of a nanoparticulate fibrate, preferably fenofibrate, composition is the particle size below which 50% of the fibrate particles fall, by weight. Similarly, D90 is the particle size below which 90% of the fibrate particles fall, by weight.
343 This is followed by a description of what are said to be several “exemplary nanoparticulate fenofibrate tablet formulations” and “exemplary embodiments of the invention”. I do not propose to set these out but it is worth nothing that the exemplary embodiments include descriptions of the invention in which it is said that the fenofibrate particles have an effective average particle size of less than about 2000 nm associated with a surface stabilizer that is not a phospholipid.
344 The description of the exemplary embodiments is followed by a section of the specification that describes methods for making the nanofibrate compositions. The specification states at page 36D lines 2-4 that “[t]he nanoparticulate fibrate, preferably fenofibrate, compositions can be made using, for example, milling, homogenization, or precipitation techniques. Exemplary methods of making nanoparticulate compositions are described in the '684 patent.” This statement is followed by numerous references to prior art describing methods of making nanoparticulate compositions.
345 The specification also includes a description of milling, microprecipitation and homogenization methods used to prepare the nanoparticulate fibrate compositions. These three methods are described at page 37 line 20 to page 39 line 5 as follows:
1. Milling to Obtain Nanoparticulate Fibrate Dispersions
Milling a fibrate, preferably fenofibrate, to obtain a nanoparticulate dispersion comprises dispersing the fibrate particles in a liquid dispersion medium in which the fibrate is poorly soluble, followed by applying mechanical means in the presence of grinding media to reduce the particle size of the fibrate to the desired effective average particle size. The dispersion medimn can be, for example, water, safflower oil, ethanol, t-butanol, glycerin, polyethylene glycol (PEG), hexane, or glycol. A preferred dispersion medium is water.
The fibrate, preferably fenofibrate, particles can be reduced in size in the presence of at least one surface stabilizer. Alternatively, the fibrate particles can be contacted with one or more surface stabilizers after attrition. Other compounds, such as a diluent, can be added to the fibrate/surface stabilizer composition during the size reduction process. Dispersions can be manufactured continuously or in a batch mode.
In one embodiment of the invention, a mixture of a fibrate and one or more surface stabilizers is heated during the milling process. If a polymeric surface stabilizer is utilized, the temperature is raised to above the cloud point of the polymeric surface stabilizer but below the actual or depressed melting point of the fibrate. The utilization of heat may be important for scale up of the milling process, as it can aid in the solubilization of the one or more active agents.
2. Precipitation to Obtain Nanoparticulate Fibrate Compositions
Another method of forming the desired nanoparticulate fibrate, preferably fenofibrate, composition is by microprecipitation. This is a method of preparing stable dispersions of poorly soluble active agents in the presence of one or more surface stabilizers and one or more colloid stability enhancing surface active agents free of any trace toxic solvents or solubilized heavy metal impurities. Such a method comprises, for example: (1) dissolving a fibrate in a suitable solvent; (2) adding the formulation from step (1) to a solution comprising at least one surface stabilizer; and (3) precipitating the formulation from step (2) using an appropriate non-solvent. The method can be followed by removal of any formed salt, if present, by dialysis or diafiltration and concentration of the dispersion by conventional means.
3. Homogenization to Obtain Nanoparticulate Fibrate Compositions
Exemplary homogenization methods of preparing active agent nanoparticulate compositions are described in U.S. Patent No. 5,510,118, for “Process of Preparing Therapeutic Compositions Containing Nanoparticles.” Such a method comprises dispersing particles of a fibrate, preferably fenofibrate, in a liquid dispersion medium, followed by subjecting the dispersion to homogenization to reduce the particle size of the fibrate to the desired effective average particle size. The fibrate particles can be reduced in size in the presence of at least one surface stabilizer. Alternatively, the fibrate particles can be contacted with one or more surface stabilizers either before or after attrition. Other compounds, such as a diluent, can be added to the fenofibrate/surface stabilizer composition either before, during, or after the size reduction process. Dispersions can be manufactured continuously or in a batch mode.
346 The specification refers at page 42B page 57 to eight examples given to illustrate the invention. The information presented includes a description of various formulations including details of particle sizes, redispersibility and a study of the food effect. The results presented are said at page 52 lines 11-14 to show that in one of the examples tested (Example 5) the pharmacokinetic profile of the fibrate was not affected by the fed or fasted state of a subject ingesting the composition (ie. no food effect). At page 58 lines 8-18 it is also asserted that, when compared to the conventional microcrystalline form of fenofibrate 160 mg dosage form “the nanoparticulate fenofibrate dosage form of the invention exhibit dramatically improved rates of dissolution.”
The Claims
347 The 807 Patent has 80 claims. Most of the construction issues that arise in relation to the 807 Patent concern the meaning of claims 1 and 40 but there are a number of other claims that raise issues of their own.
348 The relevant claims include the following:
1. A stable fenofibrate composition for oral administration comprising:
(a) particles of fenofibrate having a D50 particle size of less than about 500 nm, and
(b) at least one surface stabilizer,
wherein:
(i) the composition exhibits bioequivalence upon administration to a human subject in a fed state as compared to administration to a human subject in a fasted state; where bioequivalency is established by:
(a) a 90% Confidence Interval for AUC which is between 80% and 125%, and
(b) a 90% Confidence Interval for Cmax, which is between 80% and 125%;
(ii) the composition redisperses in a biorelevant media; and
(iii) the composition is phospholipid-free.
2. A stable fenofibrate composition for oral administration comprising:
(a) particles of fenofibrate having a mean particle size of less than about 500nm, and
(b) at least one surface stabilizer, wherein the surface stabilizer is not selected from the group consisting of sorbitan esters, polyoxyethylene castor oil derivatives, polyoxyethylene sorbitan fatty acid esters and polyoxyethylene stearates,
wherein:
(i) the composition exhibits bioequivalence upon administration to a human subject in a fed state as compared to administration to a human subject in a fasted state; where bioequivalency is established by:
(a) a 90% Confidence Interval for AUC which is between 80% and 125%, and
(b) a 90% Confidence Interval for Cmax, which is between 80% and 125%;
(ii) the composition redisperses in a biorelevant media;
(iii) the composition is phospholipid-free.
3. A stable fenofibrate composition for oral administration comprising:
(a) particles of fenofibrate having a D90 particle size of less than about 700 nm, and
(b) at least one surface stabilizer,
wherein:
(i) the composition exhibits bioequivalence upon administration to a human subject in a fed state as compared to administration to a human subject in a fasted state; where bioequivalency is established by:
(a) a 90% Confidence Interval for AUC which is between 80% and 125%, and
(b) a 90% Confidence Interval for Cmax, which is between 80% and 125%;
(iii) the composition redisperses in a biorelevant media; and
(iv) the composition is phospholipid-free.
4. The composition of claim 1, 2, or 3, wherein the particles of fenofibrate are in a dosage form for oral administration which is a tablet or capsule of about 145 mg of fenofibrate.
5. The composition of claim 1,2 or 3, which is bioequivalent to a micronized fenofibrate oral solid dosage form of 54 mg, 160 mg, or 200 mg.
6. The composition of claim 5, which is a single daily dose.
7. The composition of any one of claims 1 to 6, wherein the difference in AUC of the fenofibrate composition, when administered to a human subject in the fed versus the fasted state, is selected from the group consisting of less than about 35%, less than about 30%, less than about 25%, less than about 20%, less than about 15%, less than about 10%, less than about 5%, and less than about 3%.
…
11. The composition of any one of claims 1 to 10, wherein the fenofibrate is present in an amount selected from the group consisting of:
(a) about 50 to about 500 g of fenofibrate per kg of composition;
(b) about 100 to about 300 g of fenofibrate per kg of composition;
(c) about 200 to about 225 g of fenofibrate per kg of composition; and
(d) about 119 to about 224 g of fenofibrate per kg of composition.
12. The composition of anyone of claims 1 to 11 comprising a dosage of about 145 mg of fenofibrate, wherein:
(a) the dosage is therapeutically effective; and
(b) the composition is bioequivalent to a 160 mg micronized fenofibate tablet or 200 mg micronized fenofibate capsule, wherein bioequivalency, when administered to a human, is established by a 90% Confidence Interval of between 0.80 and 1.25 for both Cmax and AUC.
…
20. The composition of any one of claims 1 to 19, wherein the particles of fenofibrate have a particle size in which the D99 is less than about 500 nm.
21. The composition of any one of claims 1 to 20, wherein the particles of fenofibrate have a particle size in which the D50 is less than about 350 nm.
…
23 The composition of any one of claims 1 to 22 wherein:
(a) the composition is formulated for administration selected from the group consisting of oral, pulmonary, rectal, opthalmic, colonic, parenteral, intracisternal, intravaginal, intraperitoneal, local, buccal, nasal, and topical administration.
(b) the composition is formulated into a dosage form selected from the group consisting of liquid dispersions, oral suspensions, gels, aerosols, ointments, creams, tablets, lyophilized formulations, and capsules;
(c) the composition is formulated into a dosage form selected from the group consisting of controlled release formulations, fast melt formulations, delayed release formulations, extended release formulations, pulsatile release formulations, and mixed immediate release and controlled release formulations; or
(d) any combination thereof.
24 The composition of any one of claims 1 to 23 further comprising one or more pharmaceutically acceptable excipients, carriers, or a combination thereof.
…
26. The composition of anyone of claims 1 to 25, wherein within about 10 minutes at least about 40%, at least about 50%, at least about 60%, at least about 70%, or at least 80%, of the composition is dissolved, wherein dissolution is measured in a discriminating aqueous media comprising sodium lauryl sulfate at 0.025 M, and wherein the rotating blade method (European Pharmacopoeia) is used to measure dissolution.
27. The composition of any one of claims 1 to 26, wherein within about 20 minutes at least about 70%, at least about 80%, at least about 90%, or at least about 100%, of the composition is dissolved, wherein dissolution is measured in a discriminating aqueous media comprising sodium lauryl sulfate at 0.025 M, and wherein the rotating blade method (European Pharmacopoeia) is used to measure dissolution.
…
31. The composition of any one of claims 1 to 30, wherein the mean particle size of the particles of fenofibrate is selected from the group consisting of less than about 400 nm, less than about 300 run, less than about 250 nm, less than about 200 nm, less than about 150 run, less than about 100 nm, less than about 75 run, and less than about 50 nm.
32. The composition of any one of claims 1 to 31, wherein the D90 particle size of the particles of fenofibrate is selected from the group consisting of less than about 400 nm, less than about 300 nm, less than about 250 nm, less than about 200 nm, less than about 150 nm, less than about 100 nm, less than about 75 nm, and less than about 50 nm.
…
40. A method of treating a condition selected from the group consisting of hypercholesterolemia, hypertriglyceridemia, coronary heart disease, cardiovascular disorders, peripheral vascular disease, symptomatic carotid artery disease, mixed dyslipidemia, and increased risk of pancreatitis comprising administering to a subject an effective amount of a composition, wherein:
(a) the composition comprises particles of fenofibrate having a D50 particle size of less than about 500 and at least one surface stabilizer;
(b) the fenofibrate particles present in the composition redisperse in a biorelevant media; and
(c) administration of the composition to a human subject in a fasted state is bioequivalent to administration of the composition to a human subject in a fed state, wherein bioequivalency of the composition is established by:
(i) a 90% Confidence Interval for AUC which is between 0.80 and 1.25; and
(ii) a 90% Confidence Interval for Cmax, which is between 0.80 and 1.25.
…
43. The method of any one of claims 40 to 42, wherein the composition is bioequivalent to a micronized fenofibrate oral solid dosage form of 54 mg, 160 mg, or 200 mg.
…
50. The method of any one of claims 40 to 49, wherein the composition comprises a dosage of about 48 mg of fenofibrate, wherein:
(a) the dosage is therapeutically effective; and
(b) the composition is bioequivalent to a 160 mg micronized fenofibate [sic] tablet or 200 mg micronized fenofibate [sic] capsule, wherein bioequivalency, when administered to a human, is established by a 90% Confidence Interval of between 0.80 and 1.25 for both Cmax and AUC.
…
64. The method of anyone of claims 40 to 63, wherein within about 10 minutes at least about 40%, at least about 50%, at least about 60%, at least about 70%, or at least 80%, of the composition is dissolved, wherein dissolution is measured in a discriminating aqueous media comprising sodium lauryl sulfate at 0.025 M, and wherein the rotating blade method (European Pharmacopoeia) is used to measure dissolution.
65. The method of anyone of claims 40 to 64, wherein within about 20 minutes at least about 70%, at least about 80%, at least about 90%, or at least about 100%, of the composition is dissolved, wherein dissolution is measured in a discriminating aqueous media comprising sodium lauryl sulfate at 0.025 M, and wherein the rotating blade method (European Pharmacopoeia) is used to measure dissolution.
The Issues
349 The parties identified a large number of issues in relation to the 807 Patent.
Construction
(1) What is the meaning of “stable” in claims 1 to 3?
(2) Is the “composition” in each of the asserted product claims:
(a) A finished dosage form having each of the features (a), (b), (i), (ii), and (iii) identified in claim 1? Or
(b) The fenofibrate nanoparticulate of (a) and (b) and having each of the features (i), (ii), and (iii) identified in claim 1?
(3) Do claims 26, 27, 64 and 65 require a dissolution medium volume of 900 mL or 1000 ml when 0.025 M sodium lauryl sulphate is used?
(4) Is the “composition” in claims 40 to 42:
(a) A finished dosage form having each of the features (a), (b) and (c) identified in those claims? or
(b) The fenofibrate nanoparticulate of (a) and having each of the features (b) and (c) identified in those claims?
Infringement
(5) Composition: Does the Ranbaxy Product “include” a “stable composition” as the latter expression is defined at sub-paragraph 21(a) of the Second Further Amended Statement of Claim (the Stable Composition) [namely includes a stable fenofibrate composition for oral administration comprising of fenofibrate and at least one surface stabiliser (the stable composition), wherein the stable composition:
(a) contains at least one primary surface stabilizer;
(b) contains at least one secondary surface stabilizer;
(c) does not contain phospholipids; and
(d) redisperses in a biorelevant media].
(6) Particle Size:
(a) For claims 1 and 40: do the Ranbaxy Products contain fenofibrate in the form of particles having a D50 particle size of less than 500 nm?
(b) For claims 19, 21, 57 and 59: do the Ranbaxy Products contain fenofibrate in the form of particles having a D50 particle size of less than 400 nm, 350 nm, 300 nm, 250 nm or less than 200 nm?
(c) For claims 2 and 41: do the Ranbaxy Products contain fenofibrate in the form of particles having a mean particle size of less than 500 nm?
(d) For claims 31 and 70: do the Ranbaxy Products contain fenofibrate in the form of particles having a mean particle size of less than 400 nm, 300 nm, 250 nm or less than 200 nm?
(e) For claims 3 and 42: do the Ranbaxy Products contain fenofibrate in the form of particles having a 090 particle size of less than 700 nm?
(f) For claims 32 and 69: do the Ranbaxy Products contain fenofibrate in the form of particles having a 090 particle size of less than 400 nm, 300 nm, 250 nm or less than 200 nm?
(7) Stable: for claims 1 to 3 and dependent claims is the Stable Composition stable within the meaning of claims 1 to 3?
(8) Phospholipidfree: For claims 1 to 3 is the Stable Composition phospholipid free?
(9) Bioequivalence: claims 5, 12, 43 and 50:
(a) Do:
(i) the admission at Notice of Dispute [34] (Volume 1, Tab 14); and
(ii) the Ranbaxy Products PI; and/or
(iii) the results recorded in the Lipidil Clinical Study Reports (Roberts #2, [7.2]) and in the Synopsis (Roberts #3, [3])
establish on the balance of probabilities that the Ranbaxy Products are bioequivalent to a 160 mg micronized fenofibrate tablet or a 200 mg micronized fenofibrate capsule for the purposes of claims 12 and 49?
(10) Claim 26, 27, 64 and 65 issues: In light of the answer to 4(d) above, does the admission as to the dissolution profile of the Ranbaxy Products in 1000 ml of dissolution media (0.025 M SLS) establish infringement of claims 26, 27, 64 and 65?
Validity
(11) Is the term “stable” in claims l to 3 (and their dependent asserted claims) clear?
(12) Does the 807 Patent disclose an invention on the face of the specification, in light of the incorporation of WO 01/80828 on page 5?
(13) Does the invention as claimed in each of the asserted claims of the 807 Patent lack an inventive step in light of the common general knowledge taken together with the information in the 704 Patent?
(14) Does the disclosure in the 807 Patent enable the addressee of the specification to produce something within each of the asserted claims without new inventions or additions or prolonged study of matters presenting initial difficulty?
(15) Are the asserted method claims of the 807 Patent fairly based?
(16) Do the asserted method claims of the 807 Patent lack utility?
Issues 1 and 11
350 The respondent’s primary submission is that claim 1, and in each of the other asserted product claims of the 807 Patent, lack clarity by reason of the uncertain meaning of the term “stable” as used in those claims and is invalid on that basis. The respondent submitted that if, contrary to its primary submission, the Court finds that the term “stable” is capable of being given a clear meaning in each of the asserted product claims, then the Court should find that the term “stable” simply requires that the finished composition retain its physical and chemical properties to remain suitable for use until the time of its administration. The applicants’ submission was essentially the same as the respondent’s alternative submission.
351 Section 40(3) of the Act, as it relevantly applies, requires that the claims be clear. A claim is not unclear merely because more than one potential construction is open. As the High Court has said, “any purely verbal or grammatical question that can be resolved according to ordinary rules for the construction of written documents, does not, once it has been resolved, leave uncertain the ambit of the monopoly claimed”: Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610. A lack of precise definition in claims is acceptable, so long as they provide a workable standard suitable to the intended use: 3M at 274.
352 I previously referred to what I called the curious definition of “stable” at page 12 lines 3-11 as it applies to references to stable fibrate, preferably fenofibrate, particles. The word “stable” is not used in claim 1 to describe fibrate, or fenofibrate, particles but to a “stable fenofibrate composition”. As I will shortly explain, in my opinion claim 1 refers to a stable fenofibrate composition for oral administration which requires that the final dosage form be stable. On that basis, I do not think the definition referred to at page 12 lines 3-11 applies to the term “stable fenofibrate composition” as used in claim 1. In my opinion claim 1 (and each of the other asserted product claims) requires that the composition retain its essential physical and chemical properties for a reasonable period of time calculated from the date of manufacture.
Issue 2
353 The dispute between the parties is whether the “composition” of claim 1 is, as the applicants contend, the stabilised fenofibrate composition described in paragraphs (a) and (b) of claim 1 or, as the respondent contends, the final dosage form in which the fenofibrate and surface stabilizer reside.
354 The applicants submitted that the stable fenofibrate composition for oral administration referred to in claim 1 is a composition that has the physical characteristics specified in paragraphs (a) and (b) and paragraphs (ii) and (iii) of claim 1 and which, upon administration, will exhibit the pharmacological properties referred to in paragraph (i) of claim 1.
355 On this interpretation of claim 1, the size of the particles of fenofibrate referred to in paragraph (a) is not the size of the particles in the form in which they are ultimately administered but the size of such particles at an intermediate stage in the production of the composition in the form in which it is administered to human subjects.
356 According to the applicants’ submission, the requirement that the composition be “for oral administration” means that the composition must be capable of being formulated to be given orally and does not limit the composition to a formulation that is in a state that is ready to be taken orally.
357 The expert evidence relied upon by the applicants in support of their construction of claim 1 included that given by Professor Prestidge. His evidence was that in pharmaceutical science, the word “composition” generally refers to the final dosage form. However, there were two things that led Professor Prestidge to think that the claim was not directed to a final dosage form but to a composition comprising fenofibrate nanoparticles and surface stabilizer. First, he considered that the formulation of fenofibrate nanoparticles and stabilizer is the key point of the patent. Secondly, Professor Prestidge considers that the practical difficulties involved in measuring particle size in a final dosage form (especially a tablet) provides a further reason for interpreting claim 1 in this way.
358 Professor Morton on the other hand saw the presence of the words “for oral administration” and the need for the composition to exhibit the bioequivalence referred to in paragraph (i) as indicating that the claim was directed to a composition that is in finished dosage form.
359 In his closing submissions Mr Caine QC submitted on behalf of the applicants that claim 1 would cover an intermediate product, comprising a formulation of fenofibrate and surface stabilizer that may be used in the preparation of a composition suitable for oral administration (eg. a tablet, capsule or oral suspension) and that it would be an infringement to keep the intermediate product for the purposes of sale.
360 One obvious difficulty with that interpretation of the claim is that it would not be possible to determine whether the intermediate product infringed without knowing what form it ultimately took because only then could it be determined whether the composition exhibited the pharmacological properties referred to in paragraph (i) of the claim. It is not disputed by the applicants that the pharmacological properties, or at least those referred to in paragraph (i), may be affected by decisions made in formulating the finished product including as to dosage form (eg. tablet, capsule or oral suspension) and excipients.
361 The word “comprising” is, as I have mentioned, not intended to exclude other additives or components, and so claim 1 cannot be read as if it required that the intermediate product include only fenofibrate and surface stabilizer. Hence, the claim is capable of being construed as applying to a composition in finished dosage form suitable for oral administration such as a tablet or capsule. On the applicants’ construction, claim 1 would therefore cover both an intermediate product and products in finished dosage form.
362 Ultimately, the construction of the claims is a matter for the Court to determine. In my opinion the compositions referred to in claim 1 are compositions in finished dosage form (eg. a tablet, capsule or oral suspension) suitable for oral administration.
363 My reasons for preferring this construction of the claim are as follows.
364 First, although the crux of the invention is the stabilized nanoparticulate that is described in the body of the specification, the utility of the invention depends on the pharmacological properties of the compound in the human subjects to whom it is administered.
365 Secondly, the language of the claim is not apt to describe an intermediate product that may not be suitable for oral administration. The claim does not refer to a composition that is capable of being put into a form that may be given orally, but to a formulation for oral administration. This conveys, in my view, that the composition is suitable for oral administration not that it is capable of being formulated so that it is suitable for oral administration.
366 Thirdly, the claim uses the word composition on four occasions. Each time it does so it is referring to the same composition. I do not accept that it may be understood as referring to a composition consisting of an intermediate product comprising only fenofibrate particles and stabilizer and a different composition in finalised dosage form that is used to assess bioequivalence in human subjects. The claim is in my opinion referring to the same composition on each occasion it uses that word.
Issue 3
367 I previously addressed this same issue in the context of the 964 Patent. What I said then applies to claims 26, 27, 64 and 65 of the 807 Patent. These claims require a dissolution medium of 1000 mL when 0.025M sodium lauryl sulfate is used.
368 In the case of the 807 Patent the skilled addressee would look to the specification for guidance which is found at page 57 lines 12-20 and which shows the use of 1000 mL of aqueous medium containing sodium lauryl sulfate at 0.025M.
Issue 4
369 In my opinion claims 40, 41 and 42 use the word “composition” to describe a finished dosage form. The applicants contended that the word “composition” should be construed consistently with claim 1. I agree. It seems to me that what is being referred to in these claims, while structured slightly differently to claim 1, it is a composition in finished dosage form that is suitable for administration to a person suffering from one or more of the designated medical conditions.
Infringement
Background
370 The Court previously ordered that the following questions be determined separately and subsequently to all other issues of liability in the proceeding, namely, whether the Ranbaxy Products, or any of them, possess:
(a) the feature identified in the paragraph marked (i) in each of claims 1, 2 and 3 of the 807 Patent (and the corresponding feature in each claim of the 807 Patent that is dependent, directly or indirectly, on claim 1, 2 and/or 3);
(b) the feature identified in the paragraph marked (ii) in each of claims 1, 2 and 3 of the 807 Patent (and the corresponding feature in each claim of the 807 Patent that is dependent, directly or indirectly, on claim 1, 2 and/or 3);
(c) the feature identified in claim 7 of the 807 Patent (and the corresponding feature in each claim of the 807 Patent that is dependent, directly or indirectly, on claim 7);
(d) the feature identified in the paragraph marked (b) in each of claims 40, 41 and 42 of the 807 Patent (and the corresponding feature in each claim of the 807 Patent that is dependent, directly or indirectly, on claim 40, 41 and/or 42);
(e) the feature identified in the paragraph marked (c) in each of claims 40, 41 and 42 of the 807 Patent (and the corresponding feature in each claim of the 807 Patent that is dependent, directly or indirectly, on claim 40, 41 and/or 42); or
(f) the feature identified in claim 45 of the 807 Patent (and the corresponding feature in each claim that is dependent, directly or indirectly, on claim 45 of the 807 Patent).
371 Paragraph (a) of the order covers the bioequivalence requirement in claims 1-3 (and their dependant claims), para (b) the redispersion requirement in those claims, and para (c) the additional bioequivalence requirement under claim 7 (and its dependent claims). Paragraphs (d)-(f) of the order relate to those same requirements in so far as they appear in claims 40, 41 and 42 (and their dependent claims).
Issues 5 and 7: Stable Composition
372 The respondent did not dispute that the Ranbaxy Products are stable fenofibrate compositions for oral administration within the meaning of the relevant claims if “stable” is found to mean that the finished composition retains the physical and chemical properties that make it suitable for such use at least up to the time of its administration. Given my finding in relation to the meaning of the term “stable” as used in the relevant claims, it follows that this requirement of claims 1, 2 and 3 (and their dependant clams) is satisfied. Given my interpretation of those claims, it is not necessary to determine whether the intermediate fenofibrate compositon used in the manufacture of the Ranbaxy Products is stable in the relevant sense.
Issue 6: Particle size
373 The respondent contended that the evidence does not established that the particles in the Ranbaxy Products have a D50 particle size of less than about 500 nm, or that it satisfies the other particle size requirements specified in the relevant claims.
374 There is no dispute as to the size of the fenofibrate particles in the intermediate product produced during the first stage of the respondent’s manufacturing process. The D50 particle size of these particles is set out in the Confidential Schedule. The question is whether the fenofibrate particles in the Ranbaxy Product in its finished form are within the D50 particle size limit specified in claims 1 and 40 after the intermediate product has been used to manufacture the finished tablets.
375 The respondent relied upon the evidence of Professor Morton who said that, when a tablet is compressed, aggregates are formed. He said that in the present case the fenofibrate particles in the tablets may be much larger than is permitted by the relevant claims. In Professor Morton’s opinion, the processing of the intermediate product will cause the fenofibrate particles to become highly agglomerated. Dr Williams gave evidence to a similar effect.
376 The applicants’ submitted that it is the respondent’s intention (which it says may be inferred from its manufacturing protocols) that the initial size of the fenofibrate particles be maintained in the finished dosage form. They relied upon the evidence of Professor Prestidge who said:
Looking at the way this has been designed with excipients – a number of excipients that, once they come in contact with water, do all they can to drive disintegration, dispersion – you would – and the fact that we've seen some evidence here to say that the combination […] acts very well at retaining [the particle size] – gives us some confidence that there is a good dispersion here.
I believe that they've gone in there at [the initial particle size]. I believe each of the steps has been specifically designed to keep in that form, particularly upon redispersion into water in a dosage form, and there's nothing I've seen that suggests they should be significantly changed from their original size […]
The “combination” referred to by Professor Prestidge is the combination of the surface stabilizers used in the manufacture of the intermediate product.
377 The applicants also relied upon the evidence of Professor Roberts who said:
… the Ranbaxy Product [are] well-designed to maximise the impact of the appropriately formulated fenofibrate particles (that is, of an appropriate size) to achieve redispersion of the fenofibrate particles, as well as to achieve rapid dissolution and absorption. These behaviours in the tablet are best achieved by choosing excipients and manufacturing processes to enable the rapid release (as well as redispersion and subsequent dissolution) of fenofibrate from the tablets.
378 I am satisfied, on the balance of probabilities, that the fenofibrate particles in the Ranbaxy Products manufactured in accordance with the respondent’s manufacturing protocols will have, in their finished form, the D50 particle size set out in the Confidential Schedule. I therefore find that the Ranbaxy Products satisfy the particle size requirements of claim 1 and 40 of the 807 Patent. However, I think it becomes a matter of mere conjecture as to whether the smaller particle size requirements of other claims are satisfied and I am not persuaded that any more specific finding with respect to particle size is justified given the generality of the evidence relied upon by the applicants.
Issue 8: Phospholipid free
379 In closing submissions the applicants accepted that if claims 1, 2 and 3 (and their dependant claims) are to fenofibrate compositions in finished dosage form, then the requirement that composition be phospholipid-free is not satisfied. On that basis, I am satisfied that none of those claims is infringed by the manufacture or supply of the Ranbaxy Products.
Issue 9: Bioequivalence requirement of claims 5, 12, 43 and 50
380 The respondent did not dispute that the Ranbaxy Products are bioequivalent to a 160 mg micronized fenofibrate tablet or a 200 mg micronized tablet or a 200 mg micronized fenofibrate capsule for the purposes of claims 5 and 43. However, it did dispute that the Ranbaxy Products are bioequivalent within the meaning of claims 12 and 50. Unlike claims 5 and 43, claims 12 and 50 require that bioequivalency be established by a 90% Confidence Interval of between 0.80 and 1.25 for both Cmax and AUC.
381 In support of their infringement case based on claims 12 and 50, the applicants relied upon what were referred to as the Fournier Study and the Abbott Study. In essence, the applicants submitted that the PI for the Ranbaxy products, when taken with the Fournier Study and the Abbott Study, show:
the 145 mg Ranbaxy Products have been demonstrated to be relevantly bioequivalent to 145 mg Lipidil tablets;
the 145 mg Lipidil tablets have been demonstrated to be relevantly bioequivalent to 160 mg Lipidil tablets;
therefore, the 145 mg Ranbaxy Products have been demonstrated to be relevantly bioequivalent to 160 mg Lipidil tablets.
382 Dr Williams criticised this process of reasoning on the basis that it was not appropriate to draw inferences that depended upon combining the results obtained from different studies (ie the Fournier Study and the Abbott Study) that had been conducted by different investigators, at different times, in relation to different populations, and under different conditions. I accept Dr Williams’ evidence on this issue. I am not persuaded that the specific bioequivalence requirement in claims 12 and 50 is satisfied.
Issue 10: Dissolution Profile
383 The respondent contended that the evidence does not established that the Ranbaxy Products have the claimed dissolution profile specified in the relevant claims. This contention was based on the related contention that the dissolution profile must be determined using a 900 mL dissolution medium. The respondent did not dispute that if the applicants’ construction of the relevant claims was accepted then this requirement of the relevant claims was satisfied.
Issue 12
384 The question here is whether the 807 Patent discloses an invention on the face of the specification in light of the incorporation of WO 01-80828, a patent specification entitled “Improved water-insoluble drug particle process” published on 1 November 2001. In its written submissions the respondent said “… that this issue will follow the inventive step issue” and that it would not address the matter separately.
385 It is not apparent to me how the inventive step issue has any bearing on the applicants’ contention that the 807 Patent does not claim a manner of manufacture. In the circumstances, I treat this issue as having been abandoned by the respondent.
Issue 13
386 The question here is whether each of the asserted claims of the 807 Patent lacks an inventive step in light of the common general knowledge when considered with the information disclosed in the 704 Patent.
387 In answering this question it is necessary to make some findings in relation to the common general knowledge.
388 There were numerous matters that the applicants accepted formed part of the common general knowledge. These included the following matters which I am satisfied were common general knowledge as at 24 May 2002:
(a) For drugs administered orally in a solid dosage form to reach their site of action in the body, the drug must be released from the dosage form (disintegration), enter solution in the gastrointestinal (GI) tract (dissolution) and be absorbed across the walls of the GI tract, where it enters into systemic circulation and is distributed to tissues and organs, including the site of action.
(b) As the drug is released from the dosage form and absorbed from the GI tract, the concentration of the drug in the blood begins to rise. Once the drug has entered circulation, it may be delivered to organs from which it may be metabolised and/or excreted. The amount of drug in the blood over time reflects the balance between the rate of absorption and the rate of elimination or excretion.
(c) When the rate of absorption and of elimination are equal, the concentration of drug in the blood reaches a peak. This peak is called Cmax.
(d) The area under the curve (AUC) of a concentration-time profile reflects the total amount of the administered drug that reaches the systemic blood circulation.
(e) AUC can be used to quantify a drug’s “oral bioavailability” (that is, the fraction of the drug that reaches the systemic blood circulation in its active form).
(f) Bioavailability may be measured as “absolute bioavailability” (the amount of drug that reaches the systemic blood circulation when administered orally, compared with the amount of drug that reaches the systemic blood circulation when administered intravenously) or relative bioavailability (comparing the bioavailability of two different oral dosage forms).
(g) In developing solid oral dosage forms, it is frequently an objective to maximise oral bioavailability.
(h) To maximise oral bioavailability, it is necessary to consider a drug’s physicochemical properties, as well as physiological factors which influence drug absorption from the GI tract.
(i) For poorly soluble drugs, the rate at which a drug enters solution (its “dissolution rate”) is typically the rate-limiting step in the process of absorption (and thus, bioavailability).
(j) The Biopharmaceutics Classification Scheme (“BCS”), which was introduced in 1995, provides a basis for making predictions concerning the likely rate and extent of drug absorption following oral administration. It provides, inter alia, that:
Class I drugs (with high solubility and high permeability) exhibit rapid dissolution, and the rate of drug absorption may be primarily influenced by the rate of gastric emptying.
Class II drugs have low solubility and high permeability. They are poorly soluble, but once in solution, are rapidly absorbed. For these drugs, dissolution is the rate limiting step.
(k) Particle size could be reduced using “comminution” or “non-comminution” methods.
(l) Comminution methods included fluid energy (air jet) mills which produce drug particles from 0.5 to 20 µm; ball milling, which can produce very small drug particles of 200 nm or less; media milling which produces particles from 50 nm to over 1 µm; and high pressure homogenisation which produces particle sizes from 50 nm to over 1 µm.
(m) In non-comminution methods, particle size is achieved by nucleation and growth of a precipitate until it reaches the desired size range. Non-comminution methods include precipitation and crystallisation, spray drying and freeze drying.
(n) Surface stabilizers are commonly used in methods of particle size reduction.
(o) When drug particles are reduced in size, the resulting small particles will generally demonstrate an increased tendency to clump or aggregate, to form larger units. This may be called agglomeration or flocculation. This occurs because reducing particle size increases the free energy of the system, and particles behave to reduce the energy.
(p) Particle aggregation is undesirable, since it will reduce the total surface area of the particles, and thereby reduce the dissolution rate.
(q) Particle aggregation can be prevented by adding materials which accumulate at the surface and impede particle aggregation (“surface stabilizers”). These surface stabilizers reduce the adhesion between small drug particles and provide an energy barrier to particle aggregation, either by providing a physical barrier to particle aggregation (steric hindrance), or by impeding particle aggregation by electrostatic or related forces.
(r) Relatively large polymeric molecules such as HPMC can be used for steric hindrance.
(s) Surfactants assist dissolution.
(t) For new formulations of drugs in clinical use, full clinical safety and efficacy trials will not be required if a new formulation is shown to be “bioequivalent” to an existing formulation with marketing approval.
(u) Fibrates are typically BCS Class II drugs. That is, they have low solubility in aqueous fluids and dissolution is the rate limiting step for absorption after oral administration.
389 There are other matters that the respondent contends but that the applicants do not accept to be common general knowledge as at 24 May 2002. These are:
(a) The more rapid and complete the drug’s dissolution in the aqueous fluids of the GI tract and absorption from the GI tract, the less likely it is that food will impact drug absorption. Accordingly:
For BCS Class I drugs, food typically has little effect on drug absorption.
For BCS Class II drugs, the presence of food may result in an increase in drug absorption (a positive food effect). (There are a number of reasons for this. In particular, high fat meals may increase absorption, because fatty substances within the GI tract aid in the dissolution of poorly water soluble drugs.)
(b) The dissolution rate can be quantified by the “Noyes-Whitney” equation, which indicates that the rate at which a drug administered in a solid form will dissolve in the fluids in the GI tract after oral administration depends on factors which include the drug’s aqueous solubility and the total surface area.
(c) All else being equal, a drug’s dissolution rate increases as aqueous solubility and surface area increase. Smaller drug particles generally dissolve faster than larger particles.
(d) Accordingly, drug particle size has important implications for bioavailability.
(e) As the twentieth edition of the leading textbook, Remington, “The Science and Practice of Pharmacy” (published in 2000) explains, there is:
… a direct relationship between the surface area of a drug and its dissolution rate. Because the surface area increases with decreasing particle size, higher dissolution rates may be achieved through reduction of the particle size … the mere increase in the surface area of the drug does not always guarantee an equivalent increase in dissolution rate. Rather, it is the increase in the effective surface area or the area exposed to the dissolution medium, and not the absolute surface area, that is directly proportional to the dissolution rate.
(f) Particle size reduction was widely used before May 2002 to increase dissolution rates.
(g) Surfactants and surface active agents (such as the cationic surfactant sodium lauryl sulfate) can be used to impede particle aggregation by electrostatic and related repulsion forces. In this context, “surface active agents” are examples of “surface stabilizers”.
(h) The identification of a suitable surface stabilizer, or suitable combination of surface stabilizers, requires routine trial and error testing, because absolute prediction of a surface stabilizer’s performance in a given system is very rarely achievable. Candidate surface stabilizers can be identified through a literature review then tested for suitability.
390 In their closing submissions the applicants did not dispute the correctness of the propositions set out in paragraphs (a), (b), (f) or (g). I am satisfied that these matters were common general knowledge as at 24 May 2002. As to paragraph (d), I do not think there is any doubt that it was common general knowledge as at that date that “drug particle size has important implications for bioavailability”. Paragraphs (c) and (e), were the subject of some submissions by the applicants which I will now consider.
391 With regard to paragraphs (c) and (e), the applicants relied on the evidence of Professor Prestidge and Dr Roberts in support of the following propositions:
(i) It is difficult to predict whether reducing the particle size of a drug will lead to an increased dissolution rate or removing the food effect due to the complex role of food in the dissolution of a drug.
(ii) For example, food can affect the gastric emptying of the drug particles and the dissolved drug which are relevant to the drug reaching the intestines where most drugs are normally absorbed.
(iii) Food can also affect the drug transit through the rest of the gastrointestinal tract and also affect the stability of the drug in the gastrointestinal tract and its metabolic transformation of drugs in the gastrointestinal wall and in the liver.
(iv) In addition, different food components (for example fat, protein and carbohydrates) and the effect of food on different drugs can vary between the drugs.
(v) Accordingly, without sufficient data, there is no proper basis for an expectation about the extent of the food effect for a fenofibrate formulation.
(vi) Increasing the dissolution rate of a drug will not necessarily lead to a reduction in the food effect.
(vii) Reducing the particle size of a drug to increase in surface area per unit weight of the drug, all else being equal, will not necessarily lead to the drug dissolving faster than larger particles and an improved bioavailability.
392 In his affidavit evidence, Professor Prestidge said:
• The Noyes-Whitney equation is used to describe the rate of dissolution for a drug. It can be inferred by the Noyes-Whitney equation that the rate of dissolution for a drug can be modified by altering the surface area of the drug, ie changing the particle size of the drug. In particular, decreasing the particle size of a drug to maximise its surface area will generally increase the dissolution rate of the drug.
• Generally, if the dissolution rate of a drug increases, the bioavailability of the drug will increase. However, this will depend on the particular drug and the reason for the low bioavailability.
• Furthermore, in practice, the dissolution rate and food effect of a drug particle, regardless of its size, may be affected by the type of surface active substance that is used, as different surface active substances can have a different role at the drug particle interface. For example, the surface active substance could reduce surface wettability and dissolution (ie interaction of the drug particle with the water). There are also a range of other excipients used in pharmaceutical formulations which have an effect on drug dissolution and bioavailability. Therefore, while I understand the Noyes-Whitney equation to represent the general principle that reducing the particle size of a drug increases the dissolution rate, which may lead to a reduced food effect, in practice, it is not this straightforward because bioavailability may be affected by the stabiliser and other excipients used to formulate the drug.
• In addition, food can impact the way a drug interacts with the body through several mechanisms, such as causing a delay in gastric emptying, stimulating bile flow, changing gastrointestinal pH, alternating luminal metabolism, and interactions of the drug with the food itself. Food can also increase blood flow to the liver and cause changes in first pass extraction, which leads to differences in bioavailability between the fed and fasted state. It is thus difficult to predict whether reducing the particle size of a drug will lead to an increased dissolution rate or removing the food effect due to the complex role of food in the dissolution of a drug.
393 I accept this evidence in so far as it suggests that there are a number of factors that come into play when assessing whether reductions in drug particle sizes will increase dissolution rates and the food effect. However, it is important to note that it is expressed at a high level of generality and is not directed specifically to drugs with low solubility and high permeability (ie. BCS Class II drugs). I do not think Professor Prestidge’s evidence, and evidence of Dr Roberts to like effect, is inconsistent with the general proposition that, at least in the case of low solubility and high permeability drugs, reducing the particle size is likely to increase bioavailability and reduce the food effect. As Dr Roberts himself said “… it is generally understood in the pharmaceutical community that as the dissolution rate of a drug increases, the food effect of the drug decreases.”
394 I am satisfied that a person skilled in the art as at 24 May 2002 would understand that, as a general proposition, higher dissolution rates may be achieved through the reduction of particle size. The skilled addressee would understand that whether a reduction in particle size of a drug would lead to an increased dissolution rate or removal of the food effect can depend upon the role played by food in the dissolution of a drug. However, generally speaking, all else being equal, smaller drug particles would be more likely to dissolve faster than larger drug particles. Further, the more rapid and complete the drug dissolution, the less likely that there will be a food effect. I think Professor Prestidge confirmed as much in his oral evidence when he said:
… if you’re exploring a nanoparticle option as a – as a formulation strategy for something like fenofibrate, you would be trying to produce a stable formulation with the smallest possible size, because we know size correlates with surface area, correlates with dissolution and hence absorption in the body, so that’s the whole reasoning for doing this.
Inventive Step Analysis
395 I have previously set out relevant principles of law relating to obviousness.
396 The respondent contends that all of the claims of the 807 Patent on which they are sued are invalid for lack of inventive step. It advanced that case on the basis that the claimed invention in each case would have been obvious to the notional team in light of the common general knowledge as it stood as at 24 May 2002 or, alternatively, the common general knowledge as at that date together with the information contained in the 704 Patent.
397 The applicants submitted that the notional team seeking to develop an improved fenofibrate formulation would be faced with a large number of choices and could reasonably pursue many different formulation approaches without the required expectation of success. In particular, the applicants submitted that choices would need to be made by the notional team in relation to particle size, surface stabilizer, concentration of surface stabilizer and the concentration of fenofibrate. The task would be complex and detailed, and involve a good deal of trial and error, with dead ends and retracing of steps and significant uncertainty of outcome. It would not be a matter of routine nor would it be an exercise that could have been embarked upon with the required expectation of success. All this, it was submitted, precluded a finding that the invention was obvious.
398 In general terms, the invention is a composition of fenofibrate for oral administration that has improved bioavailability, an improved pharmacokinetic profile, and a therapeutic effect which is not substantially affected by the fed or fasted state of a human to whom the drug is administered. The invention also encompasses a method of treatment that involves orally administering the composition to a person suffering one or more medical conditions which fenofibrate is used to treat.
399 It is clear that the invention described in the 807 Patent is not one in which any inventive step resides in the inventors’ perception of the problem to which their experimental work was then directed. Page 6 lines 15-24 of the specification contains an acknowledgement that the bioavailability of fenofibrate, a drug that is insoluble in water, had different effects when administered orally depending on whether it was administered in a fed or fasted condition. Although the specification refers also to “other problems” (eg. the size and number of the conventional oral dosages) these are directly related to low bioavailability of the drug when orally administered in its conventional (ie. micronized) form. I am satisfied that the various problems which the 807 Patent is directed were well-known and common general knowledge as at 24 May 2002. In particular, it was common general knowledge, as at that date, that fenofibrate was a poorly soluble drug the therapeutic effect of which depended, when taken in oral dosage form, on whether the patient was fed or fasted at the time of administration (“the food effect”).
400 It is also clear that the invention described in the 807 Patent is not one in which any inventive step resides in the use of the formulations of the invention in a method of treatment. The whole purpose of preparing a pharmaceutical composition with a specified pharmacokinetic profile in humans is to enable it to be used as a method of treatment.
401 If any of the relevant claims involves an inventive step, it will reside not in the inventors’ perception of the problems associated with the oral administration of fenofibrate, but in the inventors’ solution to those problems represented by the particular formulations claimed.
402 The respondent submitted that the claimed invention was obvious because, as at 24 May 2004, the notional team, seeking to solve the problem to which the 807 Patent is directed, would have been directly led, as a matter of course, to develop a pharmaceutical formulation consisting of very small particles of fenofibrate stabilized by HPMC and SLS in an aqueous dispersion suitable for use in the preparation of a stable composition suitable for oral administration the bioavailability of which did not depend on the food effect. It contended that the notational team would have followed this development pathway in the expectation that the resulting composition may well eliminate the food effect.
403 The respondent’s contention that the claimed invention was obvious was largely based upon the common general knowledge and also the oral evidence of Professor Roberts. In particular, the respondent placed considerable reliance on oral evidence given by Professor Roberts (mostly found at transcript p 793-799). The respondent’s inventive step analysis can be summarised as follows.
404 First, the problem to be addressed was common general knowledge. The notional team would know of the need for a new fenofibrate formulation suitable for oral administration that eliminated the food effect.
405 Secondly, the notional team would understand that the best way to go about improving bioavailability and eliminating the food effect, would be to produce very small particles of fenofibrate for use in the new formulation. This is because, generally speaking, the smaller the particle size the more rapid and complete the dissolution of the drug.
406 Thirdly, the notional team would also understand that it could produce very small fenofibrate particles of 200 nm or less using a variety of well-known techniques including ball milling, media milling, high pressure homogenization or a combination of these techniques.
407 Fourthly, the notional team would understand that, because very small fenofibrate particles would have a tendency to agglomerate, it would be necessary to use surface stabilizers in order to prevent or reduce any such agglomeration.
408 Fifthly, the notional team would try HPMC and SLS, both of which were commonly used in the formulation of pharmaceuticals to prevent or reduce particle agglomeration and to assist in dissolution.
409 Sixthly, determining the concentration of surface stabilizer to fenofibrate in the new formulation would be straightforward. The notional team would use the critical micelle concentration (“CMC”) as a guide for determining how much HPMC and SLS was required in the new formulation to achieve and maintain the desired particle size.
410 By following this development pathway, the notional team would more than likely produce stable fenofibrate particles of about 200 nm or less in size that were suitable for use in the preparation of a new fenofibrate formulation with high bioavailability and no food effect.
411 I have previously set out my findings as to the common general knowledge. As to Professor Roberts’ evidence, I can say that I found it of considerable assistance in understanding how the notional team would have proceeded if seeking to address the problem to which the 807 Patent is directed as at 24 May 2002.
412 Professor Roberts accepted that if he was seeking to reduce or eliminate the food effect, he would seek to reduce the size of the fenofibrate particles and that this would lead him to identify a preferable surfactant or surface stabilizer. He also accepted that SLS and HPMC, either alone or in combination, would be leading candidates that he would try if he were seeking to reduce the particle size of fenofibrate. He said that, based on his 2002 knowledge, he “would give them a go” meaning, as I understood his evidence, that he considered such a combination would be worth trying.
413 Professor Roberts knew that HPMC could be used as a surface stabilizer, that it was very effective at coating particles so that they did not agglomerate, and that it also had a steric effect (ie. providing a steric barrier against the interparticle forces that would otherwise cause the particles to aggregate). He also knew that SLS (a negatively charged surface active agent) would assist with the dissolution of the fenofibrate in the GI tract. And he also knew that the mechanism of action of the HPMC and SLS would have a complimentary effect.
414 Professor Roberts also dealt in his oral evidence with the issue of stabilizer concentration. His evidence showed that it would be quite a simple process to establish a suitable ratio of stabilizer to fenofibrate by using the CMC to establish the maximum concentration of stabilizer that should be used. As he explained, once the CMC is exceeded, there will be little if any additional effect. In the course of his answers he made it clear that the relevant CMC could be established by a range of methods that were common general knowledge well before 2002.
415 Professor Roberts made clear in his evidence that he could not be certain that a fenofibrate formulation that used HPMC and SLS would work. After indicating that the 807 Patent showed that such a combination could be used to produce stabilized fenofibrate particles of about 300 nm in size, he was asked the following questions:
MR MURRAY: Yes. Are you able to express an opinion as to your level of optimism putting the 807 aside?
PROF ROBERTS: It’s a gamble. I mean, a whole – all sorts of developments are gambles. Sometimes they work; sometimes they don’t. I think I would be very – what’s the word? – foolhardy to suggest to you that I would be sure it would work.
MR MURRAY: Thanks. Do you think the logic – the rationale for that approach of changing the surface agent using, for example, SLS and HPMC, has a found rationale as a matter of chemistry value [sic]?
PROF ROBERTS: I might have used that, but others may not agree with me.
MR MURRAY: I’m only asking about you .....
HIS HONOUR: Yes, but it’s – I think he’s asking whether you would accept that it’s a logical way to proceed.
PROF ROBERTS: I think it is.
HIS HONOUR: Back in 2002.
MR MURRAY: Yes.
PROF ROBERTS: I think it is.
416 The applicants placed emphasis on Professor Roberts’ use of the word “gamble”. However, my impression of this evidence as a whole was that he would have had a reasonable expectation (but could not be certain) that a formulation that used HPMC and SLS to stabilize nanoparticles of fenofibrate would work, ie., they could be combined to produce a stable formulation of fenofibrate particles that were less than 300 nm in size and small enough to eliminate the food effect.
417 Professor Roberts’ evidence satisfies me that as at 24 May 2002, if the notional team had been presented with the problem addressed in the 807 patent, it is likely that it would have sought to produce a new fenofibrate formulation using very small fenofibrate particles (which Professor Prestidge described as the “nanoparticle option”) and a combination of SLS and HPMC to stabilize the fenofibrate particles and to assist their dissolution in the GI tract.
418 From that point the work of the notional team in optimizing the fenofibrate formulation to maximise bioavailability by reducing the fenofibrate particle size, and increasing the stability of the formulation, would be completely routine. This work would involve straightforward experimentation aimed at optimizing the formulation by varying the ratio of fenofibrate, HPMC and SLS in order to minimise particle size and maximise the stability of the formulation. None of this work would require the exercise of any inventive ingenuity on the part of the notional team or necessitate the use of any information that was not common general knowledge as at 24 May 2002.
419 The development pathway I have just outlined was acknowledged by Professor Roberts to be logical. It is a development pathway that I am satisfied the notional team would have been likely to follow as at 24 May 2002 in the expectation that it may well produce a fenofibrate formulation that substantially eliminated, or at least substantially reduced, the food effect.
420 I will now consider those features of the formulations of the invention as defined in the claims upon which the applicants placed reliance in resisting the respondent’s contention that the claimed invention was obvious.
421 The applicants emphasised that the formulations of the invention must be stable. I am satisfied that this requirement would be satisfied by the optimized formulation produced by the notional team. Professor Roberts’ evidence explains how SLS and HPMC would be used to stabilize the fenofibrate particles.
422 The applicants also emphasised that the formulations of the invention must contain fenofibrate particles less than about 500 nm in size. But the evidence satisfies me that particle sizes as small as 100 nm or less could be achieved using high pressure homogenization and that these sizes could be maintained using SLS and HPMC to impede particle aggregation. These particle size reduction and stabilization techniques were well-known to pharmaceutical formulators as at 24 May 2002.
423 The applicants emphasised that the formulations of the invention must meet the specified bioequivalence requirements. I think it is implicit in the teaching of the 807 Patent that the bioequivalence requirement will most likely be satisfied using a formulation comprising stable fenofibrate particles of 500 nm or less in size. The smaller the particle size, the larger the surface area of the particles, which leads to a more rapid and complete dissolution in the GI tract. The dissolution process will also be assisted by the presence of the surfactant SLS. Rapid and complete dissolution of the fenofibrate particles in the GI tract is likely to eliminate the food effect.
424 The applicants also emphasised the requirement that the formulations of the invention must meet the specified redispersion criteria. However, it is difficult to see what, if anything, this requirement adds to the bioequivalence requirement. If the fenofibrate particles did not disperse in the GI tract (which the “biorelevant media” referred to in the claims is intended to replicate) then the formulation could not reasonably be expected to exhibit the bioequivalence required by the claims. In my view, the redispersion requirement adds nothing of substance to the other requirements of the relevant claims.
425 The applicants also relied on the requirement in some of the claims that the formulation be phospholipid free. I do not think that the notional team would be drawn to phospholipids as offering a suitable stabilizer alone or in combination with other compounds. HPMC and SLS would have been perceived to be much more suitable stabilizers and were obvious candidates whereas phospholipids were not. In any event, if the formulations of the invention otherwise lack an inventive step, they are not rendered inventive by the addition of the requirement that they be phospholipid free.
426 The dependent claims of the 807 Patent introduce additional requirements beyond those found in the independent claims. These additional requirements relate to the form of the oral dosage (eg. tablet or capsule), its composition (eg. amount of fenofibrate present), its pharmacokinetics (eg. dissolution profile), its therapeutic effect (eg. blood level concentrations of the relevant metabolite, fenofibric acid) or the use to which it is put (eg. as an adjunctive therapy).
427 I did not understand the applicants to suggest that any of the dependent claims upon which the respondent was sued could be valid if it was accepted that the independent claims upon which they were dependent were invalid for lack of inventive step. Just in case I am mistaken in my understanding, I should state that I am satisfied that none of the dependent claims involve any inventive step. The reduction in tablet size, the reduction in the amount of fenofibrate present, and differences in the dissolution profiles are all the function of the size of the fenofibrate particles of 500 nm or less in size in the relevant pharmaceutical compositions.
The 704 Patent
428 Given my previous conclusion that the claims of the 807 Patent were obvious in light of the common general knowledge as at 24 May 2002, it is unnecessary for me to consider the respondent’s alternative case based upon the common general knowledge and the 704 Patent. However, given the attention the 704 Patent received at the trial, I will briefly explain why I do not think that the 704 is of any assistance to the respondent’s obviousness case.
429 The 704 Patent (US 2002/0012704) entitled “Water-Insoluble Drug Particle Process” was published on 31 January 2002. It was not disputed by the applicants that the 704 Patent constitutes s 7(3) information that, as at 24 May 2002, a person skilled in the art could reasonably be expected to have ascertained, understood and regarded as relevant to the development task.
430 The 704 Patent states that fenofibrate is a poorly water soluble compound used to reduce triglycerides levels in hypertriglyceridemic patients and plasma cholesterol and LDL-cholesterol in hypercholesterolemic and mixed dyslipidemic patients.
431 Broadly stated, the 704 Patent describes the process of making small particles of fenofibrate that involves the following five steps:
(a) mixing at high shear a mixture of fenofibrate and one or more surface active substances in an aqueous carrier above the melting point of the fenofibrate to form a heated suspension containing the fenofibrate (Step 1);
(b) homogenizing the suspension to form a heated homogenate containing the fenofibrate (Step 2);
(c) cooling the homogenate to form a transiently stable cooled homogenate containing the fenofibrate (Step 3);
(d) applying a particle stabilizing energetic process to form a cooled dispersion of small particles containing the fenofibrate (Step 4); and
(e) optionally drying the cooled dispersion to form dried small particles containing the fenofibrate (Step 5).
432 The 704 Patent contains a more detailed description of the process which includes a number of definitions of terms used throughout the specification. These include definitions of “small particles” and “transiently stable”. These definitions are important to an understanding of the five step process described in the specification and the particular formulations subsequently described.
433 The specification also includes a discussion of surface active substances that might be used in performing the process. It includes an extensive list of surface active stabilizers that are said to be useful to the invention. Phospholipid surface active substances including Phospholipon 100H and Phospholipid 90H are described as preferred, with the phospholipid known as Lipoid E80 (“E80”) being most preferred.
434 The specification describes how a suspension is prepared by mixing and heating milled particles of fenofibrate and one or more surface active substances in an aqueous carrier. The suspension is subjected to an “energetic process” (eg. high shear mixing or microfluidization) into a heated homogenate. The heated homogenate is then cooled to form a cooled homogenate that is “transiently stable”. The inventors state that it was generally found that cooled homogenate with transiently stable particles of about .3 µm (ie. 300 nm) in size could be achieved.
435 The specification also includes a description of six different cooling methods that may be applied to the heated homogenate. One of these methods, which involved fast cooling in an isothermally cooled 4°C water bath, was said to “produce cooled homogenates that maintained small particles containing … fenofibrate to a greater degree than those produced by slower cooling methods.” This was said to be especially true when E80 was used as the phospholipid substance.
436 The smallest particles sizes recorded (.26 µm and .54 µm) relate to the cooled homogenate. These particles are described as only “transiently stable”. However, the particle sizes ultimately achieved were closer to 1.0 µm in size (.87 µm or more).
437 The various examples referred to in the 704 Patent include Example 15 which is as follows:
A mixture of 225 parts of Lipoid E80 as the surface active substance, 750 parts of fenofibrate, 375 parts of sorbitol, and 750 parts of sucrose is homogeneously dispersed in 6000 parts of 10 mM pH 8.0+/-0.2 aqueous phosphate buffer using a ProScientific 400 high shear mixer at 2,000 to 3,600 rpm at ambient temperature for 30 minutes, and then heated to 95° C., 15° C. above the melting point of the drug, during continuous high shear mixing at 2,500 to 4,000 rpm. The heated suspension is then recirculatively homogenized for 10 batch volume cycles or passes using a Microfluidizer M110Y operated at 3,400 to 3,600 psig while maintained at 85° C. to 99° C. to form a heated homogenate containing the drug. After 10 passes, the heated homogenate is cooled by passage through a heat exchanger cooled by chilled water at 5° C. to 10° C. and the transiently stable cooled homogenate is further homogenized for 10 to 20 batch volume cycles or passes using a Microfluidics M110 EH homogenizer operated at 18,000 psig (peak) while maintained at 4 ° C. to 13° C. The resulting cooled dispersion comprising small particles containing fenofibrate of size less than 1.0 micron in diameter is then dried by freezing to about 40° C. and lyophilization under vacuum to produce dried small particles containing fenofibrate.
438 It is unnecessary to venture into this description in detail. The particles of fenofibrate that were ultimately produced in Example 15, which were said to be less than 1.0 µm in size, were well above the particle sizes referred to in the claims of the 807 Patent.
439 Example 20 relates to the food effect of a fenofibrate formulation prepared in accordance with Example 15. The specification states that the formulation was administered to eight human subjects in a cross-over study that measured bioavailability of a single 160 mg oral dose in both fed and fasted conditions. The conclusion drawn from the data obtained (based on AUC ratios measuring patients’ plasma levels of the drug over time) was that the bioavailability increased by less than 8% when the dose was taken in a fed condition compared to a fasted condition.
440 The applicants relied on evidence given by Professor Prestidge that the content of the 704 Patent would not lead him to expect that he could produce fenofibrate particles having a particle size of less than 0.50 μm (500 nm) that are stable. His evidence was that the skilled addressee would understand from reading the 704 Patent that the authors of that document had already attempted to optimise the fenofibrate composition disclosed in that document and that they had “done quite a wide range of … detailed formulation”. He also observed that:
They’ve explored different phospholipids; they’ve explored different phospholipid to drug ratios; they’ve explored a number of other variables in terms of the cooling method, so there’s – there’s quite a large amount of optimisation gone on in this process” and he understands that the authors of the 704 Patent have “then come out with this – what presumably is their – is their lead formulations.
441 The evidence relied on by the applicants also included the following paragraph in Professor Prestidge’s affidavit responding to evidence given by Professor Morton:
… Associate Professor Morton states that, from using the 704 Method, “I would expect to achieve a fenofibrate particle size in the order of 300 nm with a limited number of homogenization passes (in the manner described in paragraph 92 of the 704 Patent)” and that “a fenofibrate average particle size as small as 50 nm could be achieved by increasing the number of homogenization passes used in the manufacturing process (in the manner also explained in paragraph 92 of the 704 Patent)”. I agree that the 704 Patent provides a basis for an expectation that particle sizes of 0.30 μm (300 nm) and 0.05 μm (50 nm) are achievable with repeated homogenisation passes. However, the content of the 704 Patent does not lead me to expect that I could produce fenofibrate particles having a particle size of less than 0.50 μm (500 nm) that are stable. Indeed, I refer to my statements in … above regarding the fact that the particles in a cooled homogenate in step (c) of the 704 Method are transiently stable. Specifically, I refer to … above, where I discuss the cooled homogenates referred to in paragraph [0092] of the 704 Patent with particle sizes of 0.3 μm (300 nm) and 0.05 μm (50 nm), which I understand are transiently stable, and thus not physically stable. With repeated emphasis in the 704 Patent that the particles in the cooled homogenates are only transiently stable, I do not consider there to be any basis, having read the 704 Patent, for an expectation to be able to prepare stable particles having a particle size less than 0.50 μm (500 nm).
442 Professor Roberts gave evidence to the same general effect. He said that he did not see any proper basis to expect that particles of less than .5 µm (500 nm) produced using the method described in the 704 Patent would be stable. He said that he would not expect that he would be able to achieve stable fenofibrate particles with a D50 particle size of less than .5 µm (500 nm) following that method.
443 In my opinion the evidence does not establish that a skilled team endeavouring to apply the teachings of the 704 Patent with a view to producing a fenofibrate composition containing small particles of fenofibrate, could produce a stable formulation of particles of about .5 µm (500 nm) or less in size using a phospholipid as the surface stabilizer.
444 The expert witnesses who gave evidence in relation to the suitability of various surface active agents agreed that phospholipids, including E80, were not particularly good surface stabilizing agents. In the circumstances, it is necessary to be very cautious before concluding the notional team would be able to obtain fenofibrate particle sizes of 500 nm or less by using a phospholipid as a surface stabilizer even if it was minded to use such a compound for that purpose.
445 The 704 Patent is significant in that it confirms that by reducing the size of fenofibrate particles in a fenofibrate composition suitable for oral administration, it may be possible to eliminate or substantially eliminate the food effect. However, in this respect, I do not think it would tell the notional team anything more than it would already deduce from the common general knowledge.
Issue 14
446 The respondent contended that if any of the claims of the 807 Patent involve an inventive step, then the specification does not comply with the requirements of s 40(2)(a) of the Act in that it does not describe the invention fully. Given my findings in relation to inventive step, it is not necessary for me to consider the applicants’ submission on this issue in any detail although there are a number of observations that I will make in relation to it.
447 The respondent’s insufficiency challenge was predicated on the assumption that the working of the invention required something more than mere routine optimisation of a fenofibrate formulation intended for oral administration which would necessarily be beyond the assumed capacity of the non-inventive team equipped only with the common general knowledge. In those circumstances, so the respondent submitted, it would follow that the invention described in the specification was not one that could be worked without invention or prolonged study of matters presenting initial difficulty: see Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [25]. It is sufficient to say that I do not think this follows. The specification contains a detailed description of preferred embodiments and how they may be prepared. None of the expert witnesses suggested that the specification contained insufficient information with which the non-inventive team, armed only with the common general knowledge, could (at least not without an impermissible degree of difficulty) manufacture one or more formulations within the scope of each of the relevant claims.
Issue 15
448 The respondent contended that each of the methods of treatment claims was not fairly based on the matter described in the specification. These claims were said by the respondent to be analogous to those that were held invalid by the Full Court in Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth (2011) 119 IPR 194 on the basis that the specification contained what was tantamount to a disclaimer in the specification of methods that are in fact within the scope of the relevant claims.
449 The respondent’s fair basis challenge focussed on what was said to be the absence of certain requirements (or limitations) with respect to the formulations referred to in the method claims including any requirement that the fenofibrate particles be “stable” and any requirement that the composition be “phospholipid free”.
450 I do not accept the respondent’s submission with respect to either of the absent requirements. Of course, the fact that these are expressed to be requirements of the product claims does not mean that they must also be made requirements of the method claims. As to stability, the respondent’s submission assumes that the method claims would encompass the use of a formulation that was not stable. I do not think that is correct. The composition described in the method claims includes one or more stabilisers and must have a particular pharmacokinetic effect. I do not accept that the method claims can be sensibly understood (and would not be so interpreted by the skilled addressee) as encompassing a composition that was unstable to the point that the pharmacokinetic effect could not be achieved assuming it was administered within a reasonable period.
451 As to what is said to be a disclaimer of phospholipids as a stabiliser, the specification makes clear (at page 27 lines 15 to 16) that these may (but need not) be phospholipid free.
452 Similar submissions were made by the respondent in relation to the absence of any exclusion in the relevant claims of particular stabilisers (S630 and DOSS) and manufacturing techniques (involving a heating step) which were said to have been disclaimed in the specification but are not excluded by the relevant claims. I do not think that the discussion of these matters in the specification amounts to a disclaimer of these stabilisers or manufacturing techniques.
Issue 16
453 The respondent also contended that each of the method claims was not useful and was invalid on that basis. This contention was said by the respondent to be pressed only if the particle size integer in each of the relevant claims was construed as referring to primary particle size without regard to aggregation or agglomeration.
454 The submissions made in support of the respondent’s contention that the method claims were not useful assumed that the specification promised that the relevant compositions and methods of the invention were stable while also recognising that various stabilizers (D630 and DOSS) that were not excluded by the relevant claims would, if used, produce an unstable composition.
455 I do not think the specification promises that the compositions referred to in the method claims are stable. The promises made by the patentee are set out at page 6 lines 16 to 23. Those promises relate to the matters of food effect and dosage size.
456 It follows that, the challenge to the validity of the method claims on the ground that they are not useful must fail regardless of how the particle size integer in each of the method claims is construed.
Disposition
457 All of the claims of the 711 Patent upon which the respondent is sued are invalid and should be revoked.
458 Claims 12 and 13 of the 964 Patent are also invalid and should be revoked. While the respondent did not challenge the validity of claims 17 or 19 of the 964 Patent, I am not satisfied that the respondent threatens to infringe either of those claims.
459 All of the claims of the 807 Patent upon which the respondent is sued are invalid and should be revoked.
460 The further amended originating application will be dismissed.
461 The parties will be given the opportunity to make submissions in writing in relation to costs.
462 There will be orders accordingly.
ADDENDUM
463 After publication of the above reasons the respondent informed me that, contrary to what appears in [458], it did challenge the validity of claim 17 of the 964 Patent on the ground that it lacked fair basis.
464 I have reviewed the respondent’s closing submissions and accept that what it says is correct and that [458] contains an error in so far as it states that the respondent did not challenge the validity of claim 17 of the 964 Patent. The following paragraphs of these reasons deal with the respondent’s challenge to the validity of claim 17 of the 964 Patent.
465 The consistory statement states at page 6 lines 1-4 that “[t]he invention also provides a suspension of fenofibrate in micronized form having a size less than 10 μm, in a solution of hydrophilic polymer and, optionally, surfactant.” Since claim 17 refers to a fenofibrate composition in micronized form having a size less than 20 µm, that statement cannot itself provide a fair basis for claim 17.
466 However, the consistory statement at page 5 lines 23-25 describes a fenofibrate suspension (in the context of describing the relevant method) that is essentially the same as that referred to in claim 17. The question is whether there is anything disclosed in the specification to indicate that this aspect of the invention is narrower than that broadly described at page 5 lines 23-25.
467 The respondent submitted that the specification as a whole makes clear that the invention is narrower than what is identified in the consistory statement in that the suspension of fenofibrate of the invention is limited to a suspension that contains a hydrophilic polymer from 20% to 45% by weight. In support of this submission the respondent relied on the discussion of the prior art at page 3 line 7 to page 4 line 32 and the statement at page 8 lines 6-25 which is discussed at [265]-[266] above.
468 I do not regard the discussion of the prior art as limiting the invention. That discussion concerns different active ingredients and manufacturing techniques to those that are within the scope of the invention. It is not inconsistent with the broad statement at page 5 lines 23-25.
469 When the specification is read as a whole, including the consistory statement, it describes (a) an immediate release fenofibrate composition containing fenofibrate in micronized form (with a particle size of less than 20 µm), an inert hydrosoluble carrier, and a hydrophilic polymer that must make up at least 20% of the total composition by weight (page 5 lines 1-12), (b) a method of manufacturing such a composition (page 5 lines 23-34) and, finally, (c) a suspension of fenofibrate produced as the first step in that method (page 5 lines 23-25).
470 Although the method described is a method for producing the immediate release fenofibrate composition described at page 5 lines 1-8, and page 8 lines 16-25, I do not interpret the description as excluding the use of hydrophilic polymer that is less than 20% by weight in the fenofibrate suspension described at page 5 lines 23-25 or page 6 lines 1-4.
471 The fenofibrate suspension described at page 5 lines 23-25 and page 6 lines 1-4 does not include any inert hydrosoluble carrier. In that respect it would be inapt to say that the fenofibrate suspension must contain hydrophilic polymer in any one or more of the ratios specified at page 5 lines 7-8 and page 8 lines 21-23 because, unlike the compositions described in those passages, the fenofibrate suspension described does not contain any inert hydrophilic carrier.
472 It is true, as the respondent submitted, that claim 19 specifies a polymer concentration of 5% to 40% by weight for the suspension the subject of claims 17 and 18. But the concentration ratio specified in claim 19 is not the same as the ratios referred to at page 5 lines 7-8 or page 8 lines 21-23. Each of those is a ratio of polymer to the total composition (excluding any outer layers) whereas claim 19, properly construed, refers to a ratio of polymer to fenofibrate and any surfactant that makes up the suspension.
473 The respondent’s challenge to the validity of claim 17 fails.
I certify that the preceding four hundred and seventy-three (473) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Nicholas. |
Associate:
CONFIDENTIAL SCHEDULE

