
FEDERAL COURT OF AUSTRALIA
F. Hoffman-La Roche AG v Sandoz Pty Ltd [2018] FCA 874
ORDERS
First Applicant ROCHE PRODUCTS PTY LTD ACN 000 132 865 Second Applicant | ||
AND: | SANDOZ PTY LTD ACN 075 449 553 Respondent | |
DATE OF ORDER: | 12 June 2018 |
UPON THE FIRST APPLICANT UNDERTAKING TO THE COURT BY ITS COUNSEL TO:
1. submit to such order (if any) as the Court considers to be just for the payment of compensation, to be assessed by the Court or as it may direct, to any person, whether or not a party, adversely affected by the operation of these orders or any continuation (with or without variation) thereof; and
2. pay the compensation referred to in 1 above to the person or persons there referred to.
THE COURT ORDERS THAT:
1. Until 11 August 2019, the final determination of these proceedings or further order, the Respondent, whether by itself, its servants, agents or otherwise howsoever, be restrained from infringing the Asserted Patent Claims and each of them, including, without the licence of the First Applicant:
(a) supplying for use;
(b) offering for supply or sale;
(c) supplying;
(d) selling;
rituximab 500mg/mL concentrated injection vial or rituximab 100mg/10mL concentrated injection vial (together, the Sandoz Products).
2. The Respondent forthwith notify the Department of Health (Director of the PBS Price Changes Section, Pricing and Policy Branch of the Technology Assessment and Access Division) and the Minister for Health:
(a) of the granting of the interlocutory injunction set out above, and of its terms; and that
(b) for the purposes of seeking listing of the Sandoz Products on the PBS, the Respondent is no longer able to continue to provide the assurance of supply it has given, until further notice by the Respondent to the Department of Health.
3. If the respondent proposes to give further notice to the Department of Health pursuant to order 2(b) above, the Respondent shall give seven (7) days’ notice in writing to the Applicants of its intention to do so.
4. The costs of, and incidental to, the applicants’ interlocutory application for interim injunctive relief be the Applicants’ costs in the cause.
5. The parties are to confer and supply a draft short minutes of order to the Court within seven (7) days setting out the pre-trial timetable steps to bring the claim for final relief to trial with expedition.
6. Leave be granted to the Applicants to apply to the Court for the extension of Order 1, prior to 11 August 2019, having regard to the circumstances prevailing at that time.
7. These proceedings be listed for a case management hearing at 9:30am on 26 June 2018.
THE COURT NOTES THAT:
1. In this Order, Asserted Patent Claims means the claims of the patents that the First Applicant asserts, on an interlocutory basis, the Respondent threatens to infringe, being:
(a) Claims 18 and 21 of Australian Patent No. 2008207357;
(b) Claim 2 of Australian Patent No. 761844;
(c) Claim 35 of Australian Patent No. 2005211669; and
(d) Claim 3 of Australian Patent No. 2007242919.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
[1] | |
[2] | |
[10] | |
[10] | |
[15] | |
[21] | |
[23] | |
[30] | |
[30] | |
[31] | |
[59] | |
[67] | |
[67] | |
[75] | |
[81] | |
[86] | |
[89] | |
[89] | |
[93] | |
[94] | |
[106] | |
[153] | |
[154] | |
[154] | |
[207] | |
[229] |
BURLEY J:
1 In the present interlocutory application the originator of a biologic therapy used for the treatment of various cancers and rheumatoid arthritis seeks urgent orders to restrain a competitor from launching a biosimilar medicine on the basis that the biosimilar will infringe some of the claims of four asserted patents. The respondent contends that the asserted claims are invalid for want of inventive step and that the balance of convenience and justice lies against the grant of the injunction sought. For the reasons set out below I grant an interlocutory injunction, but in a more limited form than that sought by the applicants.
1.1 The parties and the broad issues
2 Rituximab is a biologic therapy prescribed in Australia to treat a number of immunology conditions including lymphoma, chronic lymphocytic leukaemia (CLL) and rheumatoid arthritis (RA). The first applicant, F. Hoffman-La Roche AG (FHLR) is a company incorporated in Switzerland. It is the registered proprietor of a number of patents relating to certain methods of use of rituximab in the treatment of a number of specified medical conditions. The second applicant, Roche Products Australia Pty Ltd (Roche Products) is the importer and supplier of products in Australia that are branded MABTHERA and that have rituximab as their active ingredient. Unless otherwise indicated, the applicants are referred to collectively below as Roche.
3 The respondent, Sandoz Pty Ltd (Sandoz), is a wholly owned subsidiary of Novartis Australia Pty Ltd. The ultimate owner of both is Novartis AG which is based in Switzerland. Sandoz has obtained a listing on the Australian Register of Therapeutic Goods (ARTG) pursuant to the National Health Act 1953 (Cth) (NH Act) for a product called RIXIMYO which is indicated for:
(1) The following forms of Non-Hodgkins Lymphoma (NHL):
(a) Previously untreated Stage III/IV follicular B-cell NHL;
(b) Relapsed or refractory low grade or follicular B-cell NHL;
(c) Diffuse large B-cell NHL (DLBCL) in combination with chemotherapy;
(2) CLL in combination with chemotherapy;
(3) RA in combination with methotrexate; and
(4) Granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in combination with glucocorticoids for the induction of remission.
4 These ARTG listings match those of MABTHERA.
5 Sandoz has applied to have RIXIMYO listed on the Pharmaceutical Benefits Scheme (PBS). On March 2018, a number of positive recommendations were made for the RIXIMYO products. The evidence suggests that it is likely that, absent an injunction, Sandoz’s products will be listed on 1 August 2018.
6 Roche is concerned that the effect of the PBS listing will be to cause a sequence of irreversible and harmful consequences to it. It now seeks interlocutory orders to restrain Sandoz from infringing the following claims (asserted claims) of the patents listed below (asserted patents):
(1) Australian Patent No. 2008207357 entitled “Combination therapies for B-cell lymphomas comprising administration of anti-CD20 antibody” (NHL Patent). The priority date of the NHL Patent is 11 August 1998 and it is due to expire on 11 August 2019. For the purposes of the interlocutory application, claims 18 and 21 are asserted to be infringed;
(2) Australian Patent No. 761844 entitled “Treatment of hematologic malignancies associated with circulating tumor cells using chimeric anti-CD20 antibody” (CLL Patent). The priority date of the CLL Patent is 9 November 1998 and it is due to expire on 9 November 2019. For the purposes of the interlocutory application, claim 2 is asserted to be infringed;
(3) Australian Patent No. 2005211669 entitled “Treatment of intermediate- and high-grade non-Hodgkins lymphoma with anti-CD20 antibody” (DLBCL Patent). The priority date of the DLBCL Patent is 11 August 1999 and it is due to expire on 2 August 2020. For the purposes of the interlocutory application, claim 35 is asserted to be infringed; and
(4) Australian Patent No. 2007242919 entitled “Therapy of autoimmune disease in a patient with an inadequate response to a TNF-alpha inhibitor” (RA Patent). The priority date of the RA Patent is 9 April 2003 and it is due to expire on 6 April 2024. For the purposes of the interlocutory application, claim 3 is asserted to be infringed.
7 Each of the claims is for a method of treating particular medical conditions using rituximab and I refer to these collectively as the “patented indications”.
8 Roche seeks an order to restrain the infringement of the asserted claims by the supply for use, importation, making, supplying, selling or keeping rituximab 500mg/50mL concentrated injection vial or rituximab 100mg/10mL concentrated injection vial which it defines to be the “Sandoz Products”. It also seeks an order compelling Sandoz to notify the Department of Health and the Minister for Health of the grant of any interlocutory injunction and inform them that it is no longer able to provide the assurance of supply that it has given (in support of the PBS application) until further notice.
9 Sandoz accepts that Roche has a prima facie case of infringement but contends that it has established a strong case that the asserted claims are invalid for lack of inventive step. It contends that the balance of convenience lies against interlocutory orders. Alternatively, it submits that the relief as sought by Roche is too broad and that if any injunction is granted, it should be narrower in scope and restricted to reflect the cascading expiry dates of each patent.
1.2 The pleaded case for interlocutory relief
10 In its points of claim for interlocutory relief (Points of Claim) Roche identifies the allegedly infringing products as being Sandoz’s two RIXIMYO products that have been registered on the ARTG. Roche alleges that Sandoz threatens to do the following in respect of RIXIMYO: supply it for use; import or make it; offer to make it; supply it; sell it; use it; and keep it for any of these purposes. Roche pleads that RIXIMYO is not a staple commercial product, that it contains a chimeric anti-CD20 antibody comprising human constant regions containing rituximab, and that RIXIMYO is a medicament that was and is manufactured for use in treating the patented indications.
11 Roche pleads that by reason of these matters the supply by Sandoz of RIXIMYO will infringe the asserted claims pursuant to ss 117(1) and 117(2)(b) of the Patents Act 1990 (Cth) (Act).
12 Section 117(1) provides that if the use of a product by a person would infringe a patent, the supply of that product by one person to another is an infringement of the patent by the supplier unless the supplier is the patentee or licensee of the patent. Section 117(2)(b) provides that the reference in subsection (1) to the use of a product by a person is a reference to any use of the product, where the product is not a staple commercial product, and the supplier had reason to believe that the person would put it to that use.
13 Roche relies upon Sandoz’s RIXIMYO Product Information sheets (PIs) which provide instructions for use in accordance with the patented indications. Paragraph [18] of Sandoz’s Defence in the substantive proceedings admits that Sandoz has reason to believe that medical practitioners will administer RIXIMYO to patients in Australia in accordance with the instructions given in the RIXIMYO PIs as approved from time to time and in accordance with any instructions that may be given in connection with the supply of RIXIMYO.
14 In its Further Amended Points of Defence and Cross-Claim on Interlocutory Relief (Defence and Cross-Claim), Sandoz states that it will not contest that it has reason to believe that medical practitioners will administer RIXIMYO to patients in Australia in the method of treatment claimed in each of the asserted claims; that its supply of RIXIMYO would amount to exploitation of each of the asserted claims; or that it threatens to authorise medical practitioners to use the method claimed in each of the asserted claims. In short, subject only to its invalidity cross-claim, for the purposes of the interlocutory application Sandoz does not dispute that it proposes to infringe the asserted claims by the supply of RIXIMYO.
15 Sandoz alleges that the invention so far as claimed in each of the asserted claims is not patentable within s 18(1)(b)(ii) of the Patents Act 1990 (Cth) (the Act) in that it does not involve an inventive step when compared with the prior art base as it existed before the relevant priority dates. Sandoz pleads that the claims lack an inventive step having regard to the common general knowledge of the person skilled in the art alone. In the alternative, it pleads that the invention so claimed would have been obvious having regard to information contained in documents to which the person skilled in the art may have regard pursuant to s 7(3) of the Act, whether alone or in combination.
16 For the NHL patent, Sandoz relies on the following prior art documents (although it makes no submission in the present application in relation to the relevance of McNeil, in (4) below):
(1) Czuczman et al, “IDEC-C2B8 and CHOP Chemoimmunotherapy of Low-Grade Lymphoma”, Blood, 86 (10 Supp. 1): 55a (1995) (Czuczman);
(2) Maloney et al., “IDEC-C2B8 (Rituximab) Anti-CD20 Monoclonal Antibody Therapy in Patients with Relapsed Low-Grade Non-Hodgkin’s Lymphoma”, Blood, 90(6): 2188-2195 (1997) (Maloney 1);
(3) Maloney et al, “IDEC-C2B8: Results of a Phase I Multiple-Dose Trial in Patients with Relapsed Non-Hodgkin’s Lymphoma”, J. Clinical Oncology, 15(10): 3166 – 3247 (1997) (Maloney 2);
(4) McNeil, “Non-Hodgkin’s Lymphoma Trials in Elderly Look Beyond CHOP”, J Nat. Cancer Inst., 90(4):266-7 (1988) (McNeil); and
(5) A combination of two or more of the documents described in paragraphs (1) to (4) above, being documents that the person skilled in the art in the patent area could be reasonably expected to have combined.
17 In its Defence and Cross-Claim, Sandoz also pleads that claim 21 of the NHL patent does not comply with s 40(3) of the Act, however, it did not press that allegation at the interlocutory hearing.
18 For the CLL patent, to which the pre-2001 version of s 7(3) applies, Sandoz relies on the following prior art documents:
(1) Maloney 1;
(2) Maloney 2; and
(3) Maloney 1 in combination with Maloney 2, being documents that are so related that the person skilled in the art would treat them as a single source.
19 For the DLBCL patent, Sandoz relies on the following prior art documents:
(1) McNeil;
(2) Link et al., “Phase II Pilot Study of the Safety and Efficacy of Rituximab in Combination with CHOP Chemotherapy in Patients with Previously Untreated Intermediate- or High-grade NHL”, American Society of Clinical Oncology, Program/Proceedings, 34th Annual Meeting (1998) (Link); and
(3) Coiffier B, “MabThera in Aggressive Lymphoma: An Update on its Efficacy and Toxicity”, Annals of Oncology, 10(Supp. 3): 213 (1999);
(4) A combination of two or more of the documents described in paragraphs (1) to (3) above, being documents that the person skilled in the art in the patent area could be reasonably expected to have combined.
20 In relation to the RA patent, Sandoz relies on the following prior art documents:
(1) Edwards JCW et al., “Efficacy and Safety of Rituximab, a B-Cell Targeted Chimeric Monoclonal Antibody: A Randomized, Placebo-Controlled Trial in Patients with Rheumatoid Arthritis”, Arthritis & Rheumatism, 46(9) S197 (2002) (Edwards 2), whether read alone or in combination with Edwards JCW and Cambridge G, “Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes”, Rheumatology, 40: 205-211 (2001) (Edwards 1);
(2) Patel DD, “B Cell-Ablative Therapy for the Treatment of Autoimmune Diseases”, Arthritis & Rheumatism, 46(8) 1984-5 (2002) (Patel);
(3) De Vita S et al., “Efficacy of Selective B Cell Blockade in the Treatment of Rheumatoid Arthritis”, Arthritis & Rheumatism, 46(8) 2029-33 (2002) (de Vita);
(4) Genentech Inc, Press Release entitled “Preliminary Positive Data from Investigational Randomized Phase II Trial Demonstrates Rituxan as a Potential Treatment for Rheumatoid Arthritis” (28 October 2002) (Genentech press release);
(5) Tuscano JM, “Successful Treatment of Infliximab-Refractory Rheumatoid Arthritis with Rituximab”, Arthritis & Rheumatism, 46(12) 3420 (2002) (Tuscano); and
(6) A combination of two or more of the documents described in paragraphs (1) to (5) above, being documents that the person skilled in the art in the patent area could be reasonably expected to have combined.
21 Roche has called the following witnesses: Mr Svend Petersen, who is the General Manager and Managing Director of Roche Products. He has sworn two affidavits, which relate to balance of convenience; Ms Carlene Todd, who is the Director of Market Access and Public Policy of Roche Products. She has affirmed two affidavits, which relate to balance of convenience; Dr John Seymour, who is a haemotologist and the Director of the Integrated Haematology Department at Peter MacCallum Cancer Centre and Royal Melbourne Hospital (RMH). He has affirmed one affidavit, which relates to the validity of the NHL, CLL and DLBCL patents and balance of convenience; Professor Eric Morand, who is a rheumatologist and the Head of the School of Clinical Sciences and Director of Rheumatology at Monash Health. He has affirmed one affidavit, which relates to the validity of the RA Patent and balance of convenience; Mr Kent Garret, who is the Director of Pharmacy at Austin Health, which comprises the Austin Hospital, Heidelberg Repatriation Hospital and Royal Talbot Rehabilitation Centre. He has affirmed one affidavit, which relates to balance of convenience; Mr Brett Rowland, who is a current legal practitioner and Special Counsel of Spruson & Ferguson Lawyers Pty Limited, Roche’s instructing solicitors. He has sworn two affidavits in support of Roche’s application, which relate to balance of convenience; Mr Jude D’Silva, who is the Business Unit Director – Established Products Roche Pharmaceuticals at Roche Products. He has affirmed one affidavit, which relates to balance of convenience.
22 Sandoz has called the following witnesses: Professor Henry Prince AM, who is a haematologist and the Director of the Centre for Blood Cell Therapies at the Peter MacCallum Cancer Centre and the Director of Molecular Oncology and Cancer Immunology at Epworth Healthcare. He has affirmed one affidavit, which relates to the validity of the NHL, CLL and DLBCL patents and balance of convenience; Professor Russell Buchanan, who is a clinical rheumatologist and Associate Professor of Medicine at the University of Melbourne and the Director of the Rheumatology Unit at Austin Health. He has affirmed one affidavit, which relates to the validity of the RA patent and balance of convenience; Dr David Liew, who is a Consultant Rheumatologist and Clinical Pharmacology Fellow at the Rheumatology Unit at Austin Health. He has affirmed one affidavit, which relates mainly to balance of convenience; Dr Chen Au Peh, who is a Consultant Renal Physician at the Royal Adelaide Hospital. He has affirmed one affidavit, which relates mainly to balance of convenience; Mr Glenn Samwell, who is the Head of BioPharma Australia at Sandoz. He has affirmed one affidavit, which relates to balance of convenience; Mr Matthew Swinn, who is a current legal practitioner and Partner of King & Wood Mallesons, Sandoz’s instructing solicitors. He has sworn one affidavit, which relates to balance of convenience.
2. THE LAW APPLICABLE TO INTERLOCUTORY RELIEF
23 The principles concerning the grant of interim injunctive relief are not controversial. When considering an application for an interlocutory injunction, the Court must address itself to two main inquiries, namely whether the applicant for relief has established a prima facie case in the sense explained in Beecham Group Ltd v Bristol Laboratories Pty Ltd [1968] HCA 1; 118 CLR 618 at 622-623, and whether the balance of convenience and justice favours the grant of an injunction or the refusal of that relief.
24 The requirement of a “prima facie case” does not mean that the applicant for relief must show that it is more probable than not that it will succeed at trial. It is sufficient if the applicant shows a sufficient likelihood of success to justify, in the circumstances, the preservation of the status quo pending trial. How strong that probability needs to be depends upon the nature of the rights that are being asserted and the practical consequences likely to flow from the order that is sought; Australian Broadcasting Corporation v O’Neill [2006] HCA 46; 227 CLR 57 (Gummow and Hayne JJ) at [65].
25 In Samsung Electronics Co Ltd v Apple Inc [2011] FCAFC 146; 217 FCR 238 the Full Court said:
[60] At [19] (p 68) in O’Neill, Gleeson CJ and Crennan J said:
As Doyle CJ said in the last-mentioned case, in all applications for an interlocutory injunction, a court will ask whether the plaintiff has shown that there is a serious question to be tried as to the plaintiff's entitlement to relief, has shown that the plaintiff is likely to suffer injury for which damages will not be an adequate remedy, and has shown that the balance of convenience favours the granting of an injunction. These are the organising principles, to be applied having regard to the nature and circumstances of the case, under which issues of justice and convenience are addressed. We agree with the explanation of these organising principles in the reasons of Gummow and Hayne JJ. (See [65]–[72], and their reiteration that the doctrine of the Court established in Beecham Group Ltd v Bristol Laboratories Pty Ltd (1968) 118 CLR 618 should be followed. See also Firth Industries Ltd v Polyglas Engineering Pty Ltd (1975) 132 CLR 489 at 492 per Stephen J; Winthrop Investments Ltd v Winns Ltd [1975] 2 NSWLR 666 at 708 per Mahoney JA; World Series Cricket Pty Ltd v Parish (1977) 16 ALR 181 at 186 per Bowen CJ.)
[61] The requirement that, in order to obtain an interlocutory injunction, the plaintiff must demonstrate that, if no injunction is granted, he or she will suffer irreparable injury for which damages will not be adequate compensation (the second requirement specified by Mason ACJ in Castlemaine Tooheys at p 153) was not mentioned in Beecham. Nor was it referred to by Gummow and Hayne JJ in O’Neill. Nonetheless, Gleeson CJ and Crennan J included that requirement in their articulation of the relevant “organising principles” (at [19] (p 68) in O’Neill). They also agreed with the explanation of those principles given by Gummow and Hayne JJ at [65]–[72] (pp 81–84) in the same case. One way of reconciling the views of Gleeson CJ and Crennan J with those of Gummow and Hayne JJ on this point is to treat “irreparable harm” as one of the matters which would ordinarily need to be addressed in the Court’s consideration of the balance of convenience and justice rather than as a distinct and antecedent consideration. This has been the approach taken by some judges (eg Ashley J in AB Hassle v Pharmacia (Australia) Pty Ltd (1995) 33 IPR 63 at 76–77; Gordon J in Marley New Zealand Ltd v Icon Plastics Pty Ltd [2007] FCA 851 at [3]; Kenny J in Medrad Inc v Alpine Pty Ltd (2009) 82 IPR 101 at [38] (p 109); and Yates J in Instyle Contract Textiles Pty Ltd v Good Environmental Choice Services Pty Ltd (No 2) [2010] FCA 38 at [55]–[64]).
[62] The assessment of harm to the plaintiff, if there is no injunction, and the assessment of prejudice or harm to the defendant, if an injunction is granted, is at the heart of the basket of discretionary considerations which must be addressed and weighed as part of the Court’s consideration of the balance of convenience and justice. The question of whether damages will be an adequate remedy for the alleged infringement of the plaintiff’s rights will always need to be considered when the Court has an application for interlocutory injunctive relief before it. It may or may not be determinative in any given case. That question involves an assessment by the Court as to whether the plaintiff would, in all material respects, be in as good a position if he were confined to his damages remedy, as he would be in if an injunction were granted (see the discussion of this aspect in Spry, The Principles of Equitable Remedies (8th edn, 2010) at pp 383–389; at pp 397–399; and at pp 457–462).
[63] The interaction between the Court’s assessment of the likely harm to the plaintiff, if no injunction is granted, and its assessment of the adequacy of damages as a remedy, will always be an important factor in the Court’s determination of where the balance of convenience and justice lies. To elevate these matters into a separate and antecedent inquiry as part of a requirement in every case that the plaintiff establish “irreparable injury” is, in our judgment, to adopt too rigid an approach. These matters are best left to be considered as part of the Court’s assessment of the balance of convenience and justice even though they will inevitably fall to be considered in most cases and will almost always be important considerations to be taken into account.
[64] Gleeson CJ also observed in Lenah Game Meats (at [18] (p 219)), that, where there is little or no room for argument about the legal basis of the applicant’s claimed private right, the court will be more easily persuaded at an interlocutory stage that a prima facie case has been established. The court will then move on to consider discretionary considerations, including the balance of convenience and justice. But, as his Honour also observed at [18] (p 219):
The extent to which it is necessary, or appropriate, to examine the legal merits of a plaintiff’s claim for final relief, in determining whether to grant an interlocutory injunction, will depend upon the circumstances of the case. There is no inflexible rule.
[65] The resolution of the question of where the balance of convenience and justice lies requires the Court to exercise a discretion.
[66] In exercising that discretion, the Court is required to assess and compare the prejudice and hardship likely to be suffered by the defendant, third persons and the public generally if an injunction is granted, with that which is likely to be suffered by the plaintiff if no injunction is granted. In determining this question, the Court must make an assessment of the likelihood that the final relief (if granted) will adequately compensate the plaintiff for the continuing breaches which will have occurred between the date of the interlocutory hearing and the date when final relief might be expected to be granted.
[67] As Sundberg J observed in Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth (2009) 81 IPR 339 at [15] (p 342), when considering whether to grant an interlocutory injunction, the issue of whether the plaintiff has made out a prima facie case and whether the balance of convenience and justice favours the grant of an injunction are related inquiries. The question of whether there is a serious question or a prima facie case should not be considered in isolation from the balance of convenience. The apparent strength of the parties’ substantive cases will often be an important consideration to be weighed in the balance: Tidy Tea Ltd v Unilever Australia Ltd (1995) 32 IPR 405 at [416] per Burchett J; Aktiebolaget Hassle v Biochemie Australia Pty Ltd (2003) 57 IPR 1 at [31] (p 10) per Sackville J; Hexal Australia Pty Ltd v Roche Therapeutics Inc (2005) 66 IPR 325 at [18] (p 329) per Stone J; and Castlemaine Tooheys at 154 per Mason ACJ.
26 The point set out in the passage at [67] is of particular relevance in the present application. Sandoz, whilst accepting that its proposed conduct will prima facie fall within the scope of the claims, contends that it has a sufficiently strong case of invalidity so as to weigh materially in Sandoz’s favour on the question of balance of convenience. That approach has led to the filing of a significant body of evidence going to this subject, with each side relying on the opinions of experienced and highly skilled medical practitioners to support their respective positions.
27 In this context it is apposite to note that a countervailing argument for invalidity must be considered with some care. It is not sufficient to balance the scales by establishing a triable revocation case on the cross-claim. If that is as far as it goes then, assuming (as here) that the applicant for relief has shown a triable issue on infringement, the conclusion would remain that the applicant has a triable question. As Jessup J said in Interpharma Pty Ltd v Commissioner of Patents [2008] FCA 1498; 79 IPR 261 (Interpharma):
[17] … as a matter of analysis, unless the case for invalidity is sufficiently strong (at the provisional level) to qualify the conclusion that, overall, the applicant has a serious question, or a probability of success, the court should move to consider the adequacy of damages, the balance of convenience and other discretionary matters. It is the applicant’s title to interlocutory relief which is under consideration, and the bottom-line question, as it were, is whether the applicant has a serious question, or a probability of success, not whether the respondent does in relation to some point of defence raised or foreshadowed.
28 This passage has been adopted as correct by a number of single judges of this Court; Janssen Sciences Ireland UC v Alphapharm Pty Ltd [2017] FCA 1399 (Yates J) at [96]; Merck Sharp & Dohme Corp v Apotex Pty Ltd [2012] FCA 928; 97 IPR 414 (Merck) (Jagot J) at [5] and; Organic Marketing Australia Pty Ltd v Woolworths Ltd [2011] FCA 279 at [60] (Katzmann J).
29 Faced with the same issue, Jagot J in Merck observed (at [6]-[7]) that the assessment of the strength of Apotex’s case for invalidity (an inventive step challenge) relied heavily on expert evidence. Were Apotex’s evidence to be the only evidence available, the assessment would have been straightforward. But as might be expected, Merck had also filed expert evidence, addressing the invalidity case. None of this evidence was, or could be, tested by cross-examination. All was prepared by apparently well-qualified experts and, on its face, appeared to be rational and persuasive. Yet the evidence of the experts would lead to different conclusions. Her Honour accordingly asked the parties for assistance in determining whether (and if so, how) she could prefer the evidence of one expert over another on a rational basis when there was no lack of persuasive force apparent from the face of the expert reports and none of the evidence had been tested. She proceeded to consider the evidence of the experts against the metric of whether there was a rational basis upon which the evidence of one could be preferred over another.
30 Before turning to apply the considerations relevant to the grant or refusal of the interlocutory orders sought, it is necessary to set out some background matters which have been addressed in considerable detail in expert and lay evidence. In many instances the experts disagree on matters of fact and opinion. The present hearing is not the place to make findings of fact, or to resolve disputes of opinion and I do not purport to do so here. This section addresses some matters of background that may facilitate a better understanding of the issues in dispute.
31 The immune system normally functions to protect the body from infections. There are various components and many different types of cells that make up the immune system. They include B-cells (or B lymphocytes) and T-cells (or T lymphocytes), which are sub-types of white blood cells.
32 One function of B-cells is to produce antibodies as part of an immune response. Antibodies recognise, target and bind to specific proteins on the surface of foreign bodies (antigens) such as bacteria, viruses and some cancer cells, and cause cell destruction. T-cells are (inter alia) responsible for providing ‘help’ to B-cells and induce the release of chemicals called cytokines (predominantly by macrophages). Cytokines are small proteins that act as signals between different cells upon release. The release of cytokines can induce, exacerbate, or perpetuate inflammation. Tumour necrosis factor (TNFα) and interleukins are examples of cytokines, of which there are at least 30.
33 As stated above, rituximab is a biologic therapy. Broadly, biologic therapies (also referred to as “biological medicines” or “biologics”) are large, complex molecules derived from a biological source, such as bacterium, yeast or blood. They are different to synthetic small molecule medicines in terms of production processes, the complexity of chemical structure, purity and immunogenicity (which is discussed further below). The manufacture of a biologic involves extensive research and development to select the most appropriate cell lines to be modified so that they can be made to reproduce in a manufacturing context.
34 In the case of rituximab, it is a monoclonal antibody. Monoclonal antibodies can be made in a laboratory by injecting mice with antigens from a human cell, which is then harvested, fused with cancerous B-cells (producing a hybridoma), and humanized to avoid triggering an immune response upon administration to human patients.
35 Rituximab binds to the CD20 antigen, which is a protein molecule that can appear on the surface of B-cells. Once bound, rituximab coats the surface of the cell and triggers the body’s immune system to destroy the cell. The CD20 antigen is not present on immature or developing B-cells. As a consequence, rituximab can be used to target mature B-cells, whilst still enabling immature B-cells to develop and replenish the supply of B-cells following treatment. CD20 is also present on almost all types of B-cells, which makes it a good target for treatment of diseases with a pathogenesis involving B-cells.
36 B-cells are thought to play an integral role in the pathogenesis of particular types of leukaemias and lymphomas, and RA. As a consequence, rituximab has been found to be effective in the treatment of these diseases, each of which are discussed further below.
37 Lymphoma is a general term given to cancers that develop in the lymphatic system due to a malignant change to B-cells and T-cells. The lymphatic system is a network of lymph vessels that branch out into the tissues of the body.
38 Lymphoma is not a single disease but a diverse group of diseases. There are several different classification systems used to classify lymphomas, including the World Health Organisation (WHO) Lymphoma Classification System, International Working Formulation and Revised European American Lymphoma Classification. The WHO Lymphoma Classification System recognises 43 different classifications (or sub-types) of lymphoma. Five of these sub-types are classified as Hodgkin’s Lymphoma, characterised by the presence of Hodgkin’s or Reed Sternberg cells. The residual 38 sub-types are classified as NHL.
39 Lymphomas can also be characterised by the speed in which they grow. Low-grade, or indolent, lymphomas grow slowly, cause fewer detectable symptoms and are generally incurable. They typically tend to grow back or relapse within a few years after the first treatment and have a relentless progression of the disease with subsequent episodes of therapy providing a diminishing benefit. Intermediate and high grade, or aggressive, lymphomas grow quickly, cause severe symptoms and (at least today) are generally curable in at least a proportion of patients (although the expert haematologists disagreed as to the proportion that were curable at the priority dates). They do not demonstrate the same course of relapse and remission as low grade lymphomas.
40 The majority of NHL lymphomas are B-cell Lymphomas, accounting for approximately 80% of diagnosed cases in Australia each year. B-cell Lymphomas include DLBCL, Follicular Lymphoma (FL) and Small Lymphocytic Lymphoma (SLL), amongst others. These are discussed further below.
41 DLBCL is the most common aggressive sub-type of NHL and is generally very responsive to treatment and (at least today) curable. It accounts for approximately 30-40% of all NHL. It is characterised by large malignant B-cells that may be observed to be diffuse throughout a biopsy.
42 FL is the most common type of indolent lymphoma. FL makes up about 70-80% of all indolent lymphomas and about 20-30% of all cases of NHL. FL is usually well-controlled with treatment but like most indolent lymphomas is not commonly curable. It is characterised by tumor cells that appear in a circular or clump-like pattern, which replace the normal structure of a lymph node.
43 SLL is a less common type of indolent lymphoma. The structure and infiltrating pattern of SLL cells are different to those of FL cells, and SL cells tend to express antigens that are not commonly seen on B-cells.
44 Bulky disease is a term used to describe the presence of sites where there is a large amount of lymphoma and the patient has large tumours above a certain threshold maximum tumour diameter. Bulky disease can occur with many different types of lymphoma, including FL and DLBCL. However, the adverse prognostic implications of bulky disease are most profound in DLBCL.
45 Leukaemias manifest primarily in the blood. Different types of leukaemias are named after the cells that are affected and how quickly they grow.
46 CLL is a slow growing leukaemia of mature B-cells and the most common type of leukaemia in the Western world. It is the leukaemic counterpart of SLL. CLL and SLL cells are morphologically indistinguishable. When the cells are found predominantly in the circulating blood and bone marrow, the cancer presents as CLL. When lymphoma cells are found predominantly in the lymphatic system, the cancer presents as SLL.
47 At the priority dates, CLL cells were known to have a number of features which overlap with the features of SLL cells. However, according to Dr Seymour (the expert haematologist for Roche), before these dates SLL and CLL were considered distinct diseases, and it is only more recently that there has been an understanding that they are related entities. This appears to be an area of dispute.
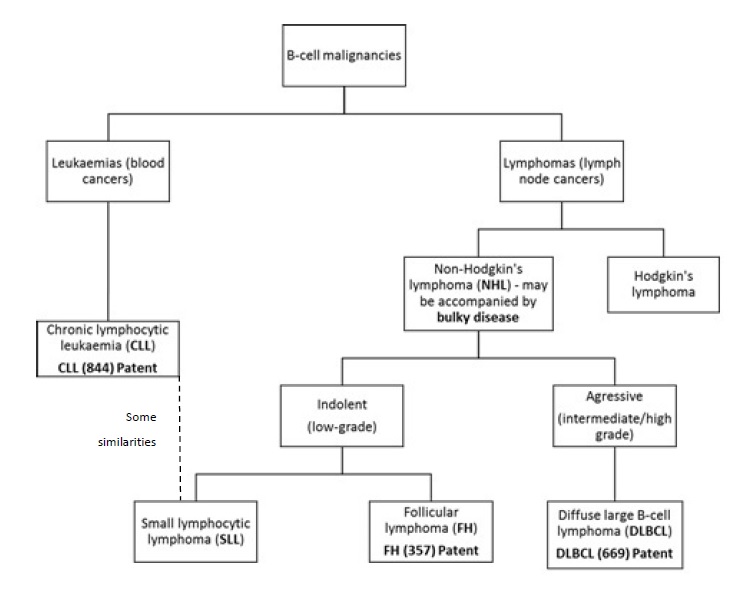
48 The flowchart below demonstrates the relationship between the diseases described above. This chart was provided by Roche during the course of the hearing. It has been slightly modified to accommodate a difference of opinion between the experts as to what grade of lymphoma DLBCL represents.
49 RA is an autoimmune disease in which the immune system inappropriately attacks joint tissue, causing painful chronic inflammation and irreversible destruction of cartilage, tendons and bones. Untreated, RA causes progressive, irreversible and erosive joint damage, which ultimately leads to the destruction of the joint itself. This leads to permanent disability and loss of productivity, and is a recognised cause of premature death.
50 The aetiology of RA remains unknown and it is currently incurable. B-cells play an integral role in the disease pathogenesis of RA, although the exact nature of this role is still unknown today. Whether this was commonly known before the priority dates is a matter of contention between the experts.
51 Classes of drugs used in the treatment of RA include conventional synthetic disease-modifying anti-rheumatic drugs (CS-DMARDS) and biological disease-modifying anti-rheumatic drugs (bDMARDS).
52 CS-DMARDS reduce damage to joints by controlling the inflammatory process in the joints and act by suppressing the body’s immune system. One particular CS-DMARD is methotrexate. Methotrexate is (and was at the priority date) the standard first-line treatment for RA. At the priority date, methotrexate was prescribed as a monotherapy or in combination with other approved treatments. Approximately a third of patients respond very well to monotherapy methotrexate and another third respond well to methotrexate in combination with another CS-DMARD. Combination therapies were generally considered more therapeutically effective than methotrexate alone.
53 bDMARDS are a newer class of medicines for the treatment of RA that target various naturally occurring substances in the immune system involved in generating inflammation. The use of bDMARDs in the treatment of RA was first presented in the late 1990s. They are generally classified as tumor necrosis factor inhibitors (TNFα-inhibitors) and non-TNF inhibitors. It appears that only TNFα-inhibitors were available in Australia before the priority date.
54 As stated above, TNFα is a type of cytokine, which induces, exacerbates or perpetuates inflammation. TNFα-inhibitors block the inflammatory effects of TNFα. Inflixmab, the first TNFα inhibitor, was approved and listed on the ARTG in August 2000 and received PBS listing in November 2003. Etanercept, the second TNFα inhibitor, was approved and listed on the ARTG in March 2003 and received PBS listing in August 2003.
55 A biosimilar medicine is a biologic that has been approved by the Therapeutic Goods Administration as similar to another biologic which has already been approved for marketing (the reference biologic). There are particular differences between biosimilars and generic small molecule drugs. For example, in contrast to the active pharmaceutical ingredients in generic small molecule drug products, the active component of biosimilars are much larger more complex molecules that have the potential to vary in many ways from the reference biologic. As a consequence, they are subject to a different set of regulatory considerations.
56 Further, a particular risk for biologics (both reference biologics and biosimilars) is that they can provoke an immune response in patients (referred to as immunogenicity). This can have a variety of consequences for the patient including the need to administer higher or more frequent doses to maintain the therapeutic effect; adverse side effects; or ineffective therapy. It can be difficult to predict which patients will have such a response. Clinicians are also concerned that immunogenicity may be heightened by multiple switches between biologics. There is no dispute that there are well publicised and common concerns amongst hospital directors of pharmacy and clinicians about immunogenicity in relation to biosimilars.
57 Under a Strategic Agreement between the Commonwealth Government and Medicines Australia Limited, a series of changes to the PBS regime will soon be introduced. These changes will enable the Pharmaceutical Benefits Advisory Committee (PBAC), which inter alia recommends new medicines for listing on the PBS, to make particular recommendations on a case by case basis when considering a PBS listing of a biosimilar product. These include recommending:
(1) a different prescribing process for biosimilars and reference biologics (such as MABTHERA) through allowing a lower level of authority (meaning the administrative arrangements that apply prior to prescribing certain PBS medicines) to the biosimilar than exists for the reference biologic at the point of introduction of the biosimilar, which may be at commencement of therapy or continuation of therapy (or both); and
(2) for treatment of naïve patients only, the prescribing of the new biosimilar compared with the reference biologic is the preferred choice for these patients, which may be further reinforced in the prescribing software.
58 These are referred to as the “biosimilar uptake drivers”. Roche emphasises that it is currently uncertain how these proposed biosimilar uptake drivers will operate in practice and the extent of their impact. However, it expects them to accelerate the adoption of biosimilars. Sandoz disputes that the drivers will have a material impact on uptake.
59 MABTHERA has been registered on the ARTG since 6 October 1998. It is approved for the indications set out in paragraph 3 above.
60 MABTHERA is a concentrate solution for an intravenous (IV) infusion or subcutaneous injection. Most patients receive an IV infusion either as an inpatient or (most commonly) an outpatient of a hospital or clinic. MABTHERA was first listed on the PBS on 1 February 1999 for the indication of relapsed refractory FL. The scope of the PBS listing has now been expanded to include treatment of out-patients with:
(1) Untreated/relapsed lymphoid cancers in combination with chemotherapy, limited to the number of cycles recommended by the standard guidelines; and
(2) RA where the patient fails a 6 month course of traditional disease-modifying drugs (DMARDS), such as methotrexate, and has poorly controlled disease (being 20 or more affected joints, 4 or more large affected joints and raised erythrocyte sedimentation rate (ESR) or C reactive protein (CRP) levels). Once the patient has received an initial approval, the patient needs to achieve a 50% improvement in joint count and 20% improvement in ESR or CRP levels, documented every 6 months, to remain on the PBS subsidised agents.
61 Sales are only subsidised by the Commonwealth under the PBS scheme where the patient is an outpatient. No subsidies are provided for treatment of inpatients.
62 There is no dispute that rituximab generally and MABTHERA in particular is a high cost medication. It may be dispensed under three different PBS reimbursement programs; highly specialised drugs, efficient funding of chemotherapy arrangements, and the general PBS schedule. The evidence of Ms Todd indicates, using an average Australian NHL patient as an example, that for a public hospital, the dispensed price for MABTHERA would be $2,641.56 per treatment. If subsidised under the PBS, the patient would make a co-payment of $39.50 (or $6.40 for a concessional patient) and there is no cost to the hospital. If the treatment is not for the PBS listed indications listed in paragraph 60 above, the full cost of the treatment is borne by either the hospital or the patient unless the patient qualifies for a compassionate program.
63 The evidence indicates that MABTHERA may be prescribed “off-label”, that is, for indications other than those for which it is registered on the ARTG. By comparing the PBS indications against the ARTG registrations and the patented indications one may see that some off-label use is subsidised by the PBS (because it extends to any treatment of an outpatient with any type of untreated or relapsed lymphoid cancer in combination with chemotherapy). In contrast, other types of off-label use will not be subsidised by the PBS (for example, the treatment of inpatients and where MABTHERA is used to treat lymphoid cancers as a monotherapy, rather than in combination with chemotherapy). Moreover, some on-label use is also not subsidised by the PBS. This is particularly the case for treatment of RA, as only a limited subset of RA patients will be entitled to PBS subsidies.
64 As explained in more detail below, the asserted claims extend to off-label and on-label uses and PBS and non-PBS subsidised uses. However, there also remains a proportion of both off-label and on-label uses that do not fall within the scope of the asserted claims.
65 Sales of MABTHERA are made by Roche Products to public and private hospitals, wholesalers and compounding or mixing houses.
66 Supply of pharmacy products to public hospitals in Australia is the subject of competitive tenders. Tender submissions are called for 3 to 6 months before the existing tender expires. They can also be called upon a change in market conditions, including the arrival of a cheaper equivalent product on the market. Ms Matthews gives evidence that, due to the concerns about immunogenicity and switching, there is a strong desire on the part of clinicians and the hospital administration of the RMH to have consistency for patients following a switch from the reference biologic to a biosimilar.
67 According to the “Field of the Invention” the NHL patent relates to the use of anti-CD20 antibodies or fragments thereof in the treatment of B-cell lymphomas, particularly the use of such antibodies and fragments in combined therapeutic regimens.
68 The “Background of the Invention” states that the use of the CD20 antigen as diagnostic and/or therapeutic agents for B-cell lymphoma has previously been reported. It goes on to say:
Previous reported therapies involving anti-CD20 antibodies have involved the administration of a therapeutic anti-CD20 antibody either alone or in conjunction with a second radiolabelled anti-CD20 antibody, or a chemotherapeutic agent.
In fact, the Food and Drug Administration has approved the therapeutic use of one such anti CD20 antibody, Rituxan®, for use in relapsed and previously treated low-grade non-Hodgkin’s lymphoma (NHL). Also the use of Rituxan® in combination with a radiolabelled murine anti-CD20 antibody has been suggested for the treatment of B-cell lymphoma.
However, while anti-CD20 antibodies and, in particular, Rituxan® (US.; in Britain, MabThera®; in general Rituximab), have been reported to be effective for treatment of B-cell lymphomas, such as non-Hodgkin’s lymphoma, the treated patients are often subject to disease relapse. Therefore, it would be beneficial if more effective treatment regimens could be developed.
More specifically, it would be advantageous if anti-CD20 antibodies had a beneficial effect in combination with other lymphoma treatments, and if new combined therapeutic regimens could be developed to lessen the likelihood or frequency of relapse.
69 The patent then provides a “Summary of the Invention” followed by a “Detailed Description of the Invention”.
70 The Detailed Description commences by referring to combined therapeutic regimens for the treatment of B-cell lymphomas. Those include a method for treating relapsed B-cell lymphoma, where a patient having prior treatment has relapsed and is administered an effective amount of a chimeric anti-CD20 antibody (including, rituximab). The prior treatments include bone marrow or stem cell transplantation, radiotherapy and chemotherapy. The previous chemotherapy may be selected from a wide group of chemotherapeutic agents and combination regimens, including CHOP, ICE, Mitazantrone, Cytarabine and a number of other listed forms. The Detailed Description also refers to methods for treating a subject having B-cell lymphoma where the subject is refractory for other therapeutic treatments, including those listed above.
71 The NHL patent provides that the combined therapeutic regimens disclosed can be performed whereby said therapies are given simultaneously, that is, the anti-CD20 antibody is administered concurrently or within the same timeframe (i.e., the therapies are going on concurrently, but the agents are not administered precisely at the same time).
72 The relevant claims in the NHL patent are claims (which is dependent on claim 16) and 21:
16. A method for treating low grade or follicular non-Hodgkin's lymphoma in a human patient comprising administering to the patient rituximab in combination with chemotherapy, wherein the chemotherapy is cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or cyclophosphamide, vincristine, and prednisone (CVP), and wherein a therapeutically effective amount of rituximab is administered to the patient simultaneously with said chemotherapy.
…
18. The method of claim 16 wherein the chemotherapy is CVP.
…
21. A method for treating low grade B-cell non-Hodgkin's lymphoma in a human patient comprising treating the patient with CVP therapy followed by administering to the patient rituximab maintenance therapy provided for 2 years, wherein rituximab is administered at a dose of 375 mg/m2.
73 Claim 18 is accordingly for a method of treating either low grade or follicular NHL involving the simultaneous administration of rituximab with a particular type of chemotherapy, being cyclophosphamide, vincristine, and prednisonc (CVP).
74 Claim 21 is a method for treating only low grade B-cell NHL by treating the patient with CVP therapy followed by administering rituximab “maintenance therapy” for 2 years with rituximab administered at 375mg/m2.
75 The “Field of the Invention” identifies that the CLL patent is directed to the treatment of haematologic malignancies associated with high numbers of circulating tumor cells by the administration of a therapeutically effective amount of chimeric or humanized antibody that binds to the B-cell surface antigen Bp35 (CD20).
76 The “Background of the Invention” then restates, in broadly the same form, much of the detail in the Background quoted above in the NHL patent.
77 The “Brief Description of the Invention” commences by stating that the inventors have “developed a novel treatment for hematologic malignancies characterized by a high number of tumour cells in the blood involving the administration of effective therapeutically effective amount of an anti-CD20 antibody”. A specific object of the invention is to treat B-prolymphocytic leukemia (B-PLL) or CLL comprising administration of a therapeutically effective amount of RITUXAN® (that is, MABTHERA).
78 In the “Detailed Description of the Invention” the patent states:
The invention involves the discovery that hematologic malignancies and, in particular, those characterized by high numbers of tumor cells in the blood may be effectively treated by the administration of a therapeutic-CD20 antibody. These malignancies include, in particular, CLL, B-PLL and transformed non-Hodgkin’s lymphoma.
This discovery is surprising notwithstanding the reported great success of RITUXAN® for the treatment of relapsed and previously treated low-grade non-Hodgkin’s lymphoma. In particular, this discovery is surprising given the very high numbers of tumor cells observed in such patients and also given the fact that such malignant cells, e.g., CLL cells, typically do not express the CD20 antigen at the high densities which is characteristic of some B-cell lymphomas, such as relapsed and previously-treated low-grade non-Hodgkin’s lymphomas. Consequently, it could not have been reasonably predicted that CD20 antigen would constitute an appropriate target for therapeutic antibody therapy of such malignancies.
79 Roche asserts the infringement of claim 2 of the CLL patent, which is dependent on claim 1. The two claims provide:
1. A method of treating chronic lymphcytic leukemia (CLL) in a human patient by administering 500-1500 mg/m2 of rituximab to the patient.
2. The method of claim 1, wherein said rituximab is administered in combination with chemotherapy.
80 Accordingly, the claimed invention may be summarised to be a method of treating CLL by administering 500-1500mg/m2 of rituximab to a patient in combination with chemotherapy.
81 The DLBCL patent is entitled “Treatment of intermediate- and high-grade non-Hodgkins lymphoma with anti-CD20 antibody”. The “Field of the Invention” concerns methods of treating DLBCL with anti-CD20 monoclonal antibodies and fragments thereof.
82 The “Background of the Invention” commences with the following statements:
Non-Hodgkin’s lymphoma is characterised by the malignant growth of B lymphocytes. According to the American Cancer Society, an estimated 54,000 new cases will be diagnosed, 65% of which will be classified as intermediate- or high-grade lymphoma. Patients diagnosed with intermediate-grade lymphyoma have an average survival rate of 2-5 years, and patients diagnosed with high-grade lymphoma survive an average of 6 months to 2 years after diagnosis.
Intermediate- and high-grade lymphomas are much more aggressive at the time of diagnosis than are low-grade lymphomas, where patients may survive an average of 5-7 years with conventional therapies. Intermediate- and high-grade lymphomas are often characterized by large extranodal bulky tumors and a large number of circulating cancer cells, which often infiltrate the bone marrow of the patient.
83 The Background goes on to observe that conventional therapies have included chemotherapy and radiation, possibly accompanied by bone marrow or stem cell transplantation if a donor is available. While patients often respond to conventional therapies, they usually relapse within several months. A relatively new approach has been to treat patients with a monoclonal antibody directed to a protein on the surface of cancerous B-cells.
84 The “Summary of Invention” states that the present invention concerns the use of anti-CD20 antibodies for the treatment of DLBCL. The inventors are said surprisingly to have found that rituximab, already approved for the treatment of low-grade follicular NHL, is effective to treat DLBCL in combination with chemotherapy in patients who have relapsed from or are refractory to chemotherapy.
85 Claim 35 is presently advanced by Roche. It is:
A method for treating a patient with diffuse large cell lymphoma accompanied by bulky disease, comprising administering to the patient a therapeutically effective amount of unlabeled Rituximab and CHOP chemotherapy, wherein the unlabeled Rituximab is administered on Day 1 of each chemotherapy cycle and the CHOP is administered on Day 1 of each chemotherapy cycle.
86 The RA patent is entitled “Therapy of autoimmune disease in a patient with an inadequate response to a TNF-alpha inhibitor”. The “Field of the Invention” states that the invention concerns therapy with antagonists which bind to B-cell surface markers, such as CD20. In particular, the invention concerns the use of such antagonists to treat autoimmune disease in a mammal who experiences an inadequate response to a TNFα-inhibitor.
87 Claim 3 of the RA patent is asserted by Roche. It is dependent on claims 1 and 2. Claims 1 – 3 are as follows:
1. A method of treating rheumatoid arthritis in a human patient who experiences an inadequate response to a TNFα-inhibitor, comprising administering to the patient an antibody that binds to CD20, wherein the antibody is administered as two intravenous does [sic] of 1000mg.
2. The method of claim 1, wherein the antibody comprises rituximab.
3. The method of claim 1 or claim 2, wherein the patient is further treated with concomitant methotrexate (MTX).
88 When read together, having regard to the dependencies, claim 3 is for a method of treating RA in a patient who experiences an inadequate response to a TNFα-inhibitor, comprising administering rituximab in 2 x intravenous doses of 1000mg, wherein the patient is further treated with concomitant methotrexate.
5. CONSIDERATION OF THE QUESTION OF ARGUABLE CASE
89 Sandoz challenges the validity of each of the patents on the basis that they lack an inventive step. As noted in Samsung at [67], the apparent strength of the parties’ substantive cases will often be an important consideration to be weighed in the balance of whether or not to grant interlocutory injunctive relief. Sandoz accepts, for the purposes of the present application, that its proposed conduct will infringe the asserted claims. That concession yields the result that, subject only to the invalidity challenge, Roche has established the strongest of arguable cases. The critical question then becomes; what is the strength of Sandoz’s invalidity challenge?
90 A great deal of expert evidence has been filed by both sides concerning the cross-claim. The expert opinion evidence reflects numerous disagreements including; as to what the person skilled in the art knew at the priority dates; what research work they would undertake; and what expectation of success there would have been in relation to that work. Sandoz does not ask that the Court try to resolve the evidentiary conflicts reflected in the affidavit materials. Indeed it is not appropriate to do so. It is not the function of the Court to conduct a preliminary trial of the action or, in general, to resolve conflict between the parties’ evidence and grant or refuse the application on the basis of such findings; Warner-Lambert Co LLC v Apotex Pty Ltd [2014] FCAFC 59; 311 ALR 632 (Full Court) (Warner-Lambert) at [72], [91].
91 Nevertheless, Sandoz urges that the Court can conclude that there is a “very serious challenge” to the validity of the patents and that this weighs heavily against the grant of the orders sought. In this connection, Sandoz states that it does not invite the Court to make findings in respect of contested facts and opinions but it nevertheless asks the Court to conclude that it has a strong prima facie case.
92 In response, Roche accepts for present purposes that the case advanced by Sandoz on the cross-claim is arguable, but no more. It submits that the overwhelming strength of its infringement case is not weakened by the existence of a merely arguable cross-claim and that this is a relevant factor in the consideration of the application.
5.2 The Person skilled in the art
93 It appears to be common ground between the parties that the personal skilled in the art for the cancer patents is a haematological malignancy specialist with both clinical and research experience, and the person skilled in the art for the RA patent is a rheumatology specialist with clinical and research experience. There is no dispute that the expert witnesses who have given evidence are suitably qualified.
5.3 The relevant law on inventive step
94 Section 18(1)(b)(ii) of the Act relevantly provides that an invention is a patentable invention for the purposes of a standard patent if the invention, so far as claimed in any claim, when compared to the prior art base as it existed before the priority date, involves an inventive step.
95 Section 7(2) of the Act provides:
For the purposes of this Act, an invention is to be taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed in the patent area before the priority date of the relevant claim, whether that information is considered separately or together with the information mention in s 7(3).
96 Section 7(3) has been amended over the years and care must be taken to ensure that the correct form is used for each patent. The form that is applicable to the challenge to the CLL patent is that which applied immediately before amendments were made in 2001 to the Act, and is as follows:
For the purposes of subsection (2), the kinds of information are:
(a) prior art information made publicly available in a single document or through doing a single act; and
(b) prior art information made publicly available in 2 or more related documents, or through doing 2 or more related acts, if the relationship between the documents or acts is such that a person skilled in the relevant art in the patent area would treat them as a single source of that information;
being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant to work in the relevant art in the patent area.
97 The form of section 7(3) applicable to the consideration of the validity of the NHL, DLBCL and RA patents is that which immediately followed the 2001 amendments:
The information for the purposes of subsection (2) is:
(a) any single piece of prior art information; or
(b) a combination of any 2 or more pieces of prior art information;
being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood, regarded as relevant and, in the case of information mentioned in paragraph (b), combined as mentioned in that paragraph.
98 Plainly enough, the onus to establish lack of inventive step rests upon the party challenging validity; s 7(2).
99 In AstraZeneca AB v Apotex Pty Ltd [2015] HCA 30; 257 CLR 356 (AstraZeneca) French CJ said (footnotes omitted):
[15] Relevant content was given to the term "obvious" by Aickin J in Wellcome Foundation Ltd, posing as the test:
"whether the hypothetical addressee faced with the same problem would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not."
The idea of steps taken "as a matter of routine" did not, as was pointed out in AB Hässle, include "a course of action which was complex and detailed, as well as laborious, with a good deal of trial and error, with dead ends and the retracing of steps". The question posed in AB Hässle was whether, in relation to a particular patent, putative experiments, leading from the relevant prior art base to the invention as claimed, are part of the inventive step claimed or are "of a routine character" to be tried "as a matter of course". That way of approaching the matter was said to have an affinity with the question posed by Graham J in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd. The question, stripped of references specific to the case before Graham J, can be framed as follows:
"Would the notional research group at the priority date, in all the circumstances, which include a knowledge of all the relevant prior art and of the facts of the nature and success of [the existing compound], directly be led as a matter of course to try [the claimed inventive step] in the expectation that it might well produce a useful alternative to or better drug than [the existing compound]?"
That question does not import, as a criterion of obviousness, that the inventive step claimed would be perceived by the hypothetical addressee as "worth a try" or "obvious to try". As was said in AB Hässle, the adoption of a criterion of validity expressed in those terms begs the question presented by the statute.
100 See also Kiefel J at [66] and [67].
101 Before a document containing prior art information can be used along with common general knowledge for the purposes of the s 7(2) inquiry, it is necessary that it meet the requirements of s 7(3). For the pre-2001 version of s 7(3) it has been held that prior art information which is publicly available in a document is “ascertained” if it is discovered or found out. “Understood” means having discovered the information, the skilled person would have comprehended it or appreciated its meaning or import. The words “relevant to work in the relevant art”, in the context of the pre-2001 version of s 7(3), are directed to publicly available information not part of the common general knowledge, which the skilled person could be expected to have regarded as solving a particular problem or meeting a long-felt want or need; AstraZeneca per Kiefel J at [68], citing Lockwood Security Products v Doric Pty Ltd [No 2] [2007] HCA 21; 235 CLR 173 (Lockwood No 2) at [132].
102 In AstraZeneca, Keifel J said (footnotes omitted, emphasis original):
[69] Lockwood [No 2] also explains that, in answering the question of obviousness, the information referred to in s 7(3), like that part of the prior art base which is the common general knowledge, is considered for a particular purpose. That purpose is to look forward from the prior art base to see what the skilled person is likely to have done when faced with a problem similar to that which the patentee claims to have solved with the claimed invention. It is this aspect of the s 7(2) enquiry which assumes particular importance on these appeals.
103 The test posits a looking forward from the prior art base, without the benefit of hindsight.
104 In Lockwood No 2, the High Court said:
[51] In Alphapharm, this court reiterated that “obvious” means “very plain”, as stated by the English Court of Appeal in General Tire & Rubber Co v Firestone Tyre and Rubber Co Ltd . The majority in Alphapharm also confirmed that the question of whether an invention is obvious is a question of fact, that is, it is what was once a “jury question”. Broadly speaking, the question is not a question of what is obvious to a court. As well as being a question of fact, the question of determining whether a patent involves an inventive step is also “one of degree and often it is by no means easy”, because ingenuity is relative, depending as it does on relevant states of common general knowledge. This difficulty is further complicated now by the need, in some circumstances, to consider s 7(3) information as well as common general knowledge.
105 The emphasis that the High Court placed on obviousness being a “jury question” is of importance in the present case. Where an inventive step challenge involves competing issues of fact and opinion it will often be difficult for a party to establish any more than that a case is arguable. It is deceptively simple to assert in submissions that a simple invention as reflected in a claim which consists of two integers is obvious. However, in each case the question must be considered against the background of the common general knowledge of the invention as claimed. Where these matters are complicated by conflicting evidence and a priority date that is more than 15 years in the past, it may be undesirable for a Court to find at the interlocutory stage that the inventive step challenge is any more than “arguable”.
5.4 Consideration of the invalidity case
106 I commence my observations by repeating that I do not here make any findings of fact or conclusions in relation to the various arguments articulated. The task at hand is to determine as best one can, whether the cross-claim advanced may, as Sandoz urges, be described as “strongly arguable”, or whether, as Roche urges, it is simply “arguable”; and feed my preliminary view on this subject into the calculus for deciding whether or not to grant the relief sought.
107 Sandoz advances its challenge to the three cancer patents (the NHL patent, the CLL patent and the DLBCL patent) on the basis of the evidence of Professor Prince, who gave a lengthy affidavit in which he responded to a series of questions from the solicitors representing Sandoz. He first provides a background to the haematological conditions that he treats on a regular basis, being “NHL, FL, CLL and DLBCL”. He then identifies the treatments for these conditions that were available at the priority dates and then describes rituximab, how it works, the way it is prescribed and how it is dispensed and administered.
108 Next, Professor Prince considers:
the extent to which, as at the Relevant Dates, I would have considered it to be a matter of routine to investigate the use of rituximab in treating different types of lymphoma and leukaemia if I had been a member of a team seeking to investigate new treatments for lymphoma and leukaemia.
109 In answering this question Professor Prince identifies a significant number of factual matters that he considers haematologists would have known concerning the way that the relevant conditions were treated as at the relevant priority dates.
110 Having regard to those matters, Professor Prince expresses his opinion that the likelihood that he and his Australian colleagues would have wished to investigate the uses for rituximab in treating patients with a wide range of B-cell lymphomas and related leukemia conditions is “very high”. Once rituximab was available, investigations into the various applications for it and the optimal dosing and combined treatment regimens would have occurred, not only as a matter of routine, but in his opinion as an urgent priority in the expectation that it may well produce new treatment options for patients.
111 Professor Prince then turns to answer the question posed by the solicitors (set out at [108] above) in relation to the treatment of each of these conditions. He then considers each piece of prior art in the context of his knowledge at the priority dates and then turns, for the first time, to consider each patent and each relevant claim in the context of his knowledge.
112 Before turning to the more particular matters to which Professor Prince refers, it is necessary to record that his evidence was answered by Dr Seymour, also a haematologist. Dr Seymour takes issue with some of the factual matters to which Professor Prince refers.
113 One of the more important points of difference is that Dr Seymour strongly disagrees that it was clear “at the outset” that rituximab had the potential to be useful in the treatment of a variety of lymphomas and leukaemias. Dr Seymour considers that the efficacy of rituximab across a broad range of lymphomas and leukaemias only appears to be obvious in hindsight. He accepts that at the priority dates there was a theoretical rationale for believing that it may have the potential to be effective in some of the different types of NHL, which may have warranted evaluation. However he, and he believes other physicians, did not expect that rituximab would be effective in multiple different types of B-cell lymphomas and leukaemias. There was no precedent for any such therapeutic agent to have such broad efficacy, and no other agent has achieved such broad efficacy since. Dr Seymour expresses the view that he still finds rituximab’s broad spectrum of activity to be “remarkable”. He states that he could not have predicted it and does not believe that it could have been predicted by other physicians.
114 Roche submits that this point of difference goes to a critical aspect of the inventive step challenge and applies to the whole of the inventive step cross-claim. It submits that the evidence of Professor Prince consistently addresses the wrong legal issue. Specifically, it contends that he does not address whether the inventions as claimed are obvious in the sense required by the modified Cripps Question (quoted by French CJ in AstraZeneca at [15]), but instead gives evidence that (to paraphrase) it would be worthwhile “to undertake further investigation of options for treatment in the expectation that these may well produce new treatment options”. Statements in the evidence that Professor Prince may have, at the priority date, considered it desirable, worthwhile, or interesting to investigate various options for treatment do not, Roche submits, address the legal test and simply indicate that Professor Prince would have engaged in largescale research programs with rituximab.
115 I now turn to the more specific evidence that Professor Prince gives in relation to the NHL patent.
116 Professor Prince observes that rituximab had already been approved by the FDA for the treatment of patients with relapsed or refractory low-grade or follicular B-cell NHL as at the priority dates. He states that it would have been clear to him that it would be “logical and ethical” to further investigate treatment paradigms using rituximab as a first-line treatment in combination with CHOP or CVP. The typical first-line treatment as at the priority date was CHOP or CVP chemotherapy and so the most logical and ethical investigation would have been therapy of a combination of rituximab with one or other of these forms of chemotherapy.
117 Furthermore, Professor Prince states that it was reported in the literature at the priority dates that rituximab showed synergy with chemotherapy treatments, for the patients who could tolerate it, and he would have investigated this synergy by administering combination therapies of rituximab with CHOP and CVP respectively in the expectation that it may well lead to better outcomes than the administration of one or other of these alone. His view was that, for the sake of patient and outpatient-clinic convenience, he would have administered both rituximab and the chemotherapy on the same day at the start of a chemotherapy cycle. These matters concern the contents of claim 18 of the NHL patent.
118 Next, Professor Prince considers that the most straightforward approach would have been to trial rituximab in the dose of 375mg/m2, because this was the FDA approved dose for treatment of patients with relapsed or refractory low grade or follicular B-cell NHL.
119 Furthermore, he observes that at the priority dates it was known that low-grade NHL was considered to be incurable and that it was common for patients to relapse within 18 months to 2 years of first-line treatment. Professor Prince considers that maintenance therapy during this relapse period was considered appropriate both at the relevant dates and now. He draws an analogy with interferon-α, another immunological therapy for cancer, which was being used in combination with chemotherapy as a maintenance therapy for NHL at the relevant date. Professor Prince concludes that he would have been interested in investigating the role of rituximab as a maintenance treatment during the typical relapse period of 18 months to 2 years with the expectation that it may well prolong a patient’s remission period. These matters address the contents of claim 21.
120 After providing this evidence, Professor Prince was provided with the prior art documents, including Czuczman, Maloney 1 and Maloney 2. It is not necessary to describe the prior art in detail. Broadly, they report studies that investigated non-simultaneous administration of rituximab and chemotherapy (in particular CHOP) in low grade or follicular lymphoma. Czuczman reports a synergy with chemotherapeutic agents and non-overlapping toxicities. It is common ground between the experts that the prior art documents did not disclose simultaneous administration of rituximab and the chemotherapy agent together; the use of CVP (as opposed to CHOP); or the use of rituximab in maintenance therapy. The experts disagree about whether it would have been obvious to try a treatment regime with those integers, with the relevant expectation of success.
121 In particular, Dr Seymour:
(1) Disputes any assertion that it was well-established and widely appreciated at the priority dates that rituximab shared synergy with chemotherapy treatments. He notes that the only studies investigating synergy prior to Czuczman were in vitro studies. Nevertheless, he acknowledges that Czuczman was promising and that the combination of rituximab and CHOP had greater effectiveness than expected.
(2) Opines that he would not have expected CVP to have the same “remarkable” effect as CHOP. In his view, CVP combined with rituximab was potentially less effective than CHOP combined with rituximab, because CVP only had one of the putative sensitizing agents alleged in earlier articles to have a sensitizing effect with rituximab, not two.
(3) Would not have chosen to administer rituximab and chemotherapy simultaneously for a number of reasons, including the fact that the earlier articles report an advantage in administering rituximab before chemotherapy so as to exploit the sensitisation phenomenon, and his concern that the immunosuppression caused by simultaneous treatment might impair the efficiency of rituximab.
(4) Strongly disagrees with the suggestion that the use of rituximab in maintenance therapy was obvious at the priority dates for a number of reasons. He gives evidence that maintenance using further chemotherapy had proved ineffective; that the net benefit of interferon-α maintenance was debated and it was not widely accepted that it actually provided an overall benefit to patients due to the toxicity associated with prolonged administration; in his view, maintenance therapy with rituximab would entail prolonged B-cell depletion and the impact of this was not known, particularly in NHL patients who were known to be somewhat immunosuppressed from the outset; and at that time there was a possible concern that maintenance therapy could lead to the development of more resistant disease. Further, there is no mention in the prior art of using rituximab for maintenance therapy, although Maloney 1 recommends the investigation of using it for that purpose. Czuczman describes treatment lasting 18 weeks, not 2 years.
122 When one looks at claim 18 of the NHL patent, one sees that it appears to involve four fairly simple integers; (1) the simultaneous administration of (2) rituximab, with (3) CVP, to (4) treat low grade or follicular NHL. The evidence indicates that there is a dispute as to whether the person skilled in the art would have taken steps to combine these integers. Certainly it appears possible that such a person, motivated by the new and exciting rituximab treatment, may have done so. But whether it was obvious to do so will require the resolution of a number of matters in debate, some of which I have touched upon above. Unlike other grounds of invalidity, the question of whether a claimed invention involves an inventive step is nuanced, fact rich and involves balancing questions of fact and degree, akin to a jury question (Lockwood No 2 at [51]). Having regard to the whole of the evidence, including the above, I am unable to conclude that Sandoz’s challenge to claim 18 is any more than arguable.
123 The same conclusion applies in relation to claim 21 of the NHL patent. There are further complexities given the additional feature of administering rituximab as maintenance therapy for two years.
124 It should also be noted that Sandoz submits that Czuczman, Maloney 1 and Maloney 2, with the common general knowledge, deprive claims 18 and 21 of inventive step both individually and in combination. However, Professor Prince does not give evidence about the effect of the combination of these prior art documents, so there remains a question about whether this would occur. In any event, such combination would not appear to resolve the differences of opinion between the experts outlined above.
125 I turn now to consider the CLL patent.
126 Professor Prince gives evidence that he and other haematologists would have been highly motivated to investigate the application of rituximab in CLL.
127 With the possible exception of a study by the MD Anderson Cancer Centre in Texas, which I discuss in more detail below, at the priority date there were no reports of the use of rituximab in the treatment of CLL. However, Professor Prince gives the opinion that at the priority date it would have been a matter of routine to investigate the potential use of rituximab in CLL in the expectation that it may well result in a new treatment therapy. He notes that CLL is the most common type of leukaemia and as such it would have been an obvious group of patients to include in any investigation into the application of rituximab. He notes that the standard treatment for patients with CLL was chemotherapy and that CLL was considered to be incurable. He would have started by investigating the application of rituximab as a monotherapy for patients who were refractory to the standard chemotherapy, as those patients had no treatment options available to them. However, he states that the investigation would also have included the application of rituximab in combination with chemotherapy for the reasons discussed above in relation to the NHL patent. Professor Prince says that at the time, it was known that CLL cells “were relatively fragile compared with other lymphoma cells” and that there was a theory that rituximab and chemotherapy were likely to have an increased synergistic effect in these more fragile CLL cells.
128 As to dosage amounts, Professor Prince notes that at the priority date, he was aware that a clinical trial was being conducted by the MD Anderson Cancer Centre in Texas with rituximab for CLL in low doses. He also considers that haematologists were beginning to recognise that there was a compartmentalised effect when treating CLL with lymphocytes in the circulating blood stream which could affect the dosage regimen for treatment of CLL in comparison to NHL. In his view, one theory was that there is an absorption effect where a therapy such as rituximab was being ‘sponged up’ by circulating cells and therefore not having impact in other comparts such as lymph nodes and bone marrow. There was also a theory that CLL cells expressed less CD20 antigen. According to Professor Prince, the preliminary data of the trial was presented at the Australian Society of Haematology and the International Lymphoma Workshop at the International Conference on Malignant Lymphoma. However, the work was not published until after the priority date. Professor Prince contends that the above-mentioned information would have encouraged him to investigate whether doses higher than 375mg/m2 would be effective and well tolerated.
129 Further, Professor Prince states that if it became apparent that there was a lower response rate for patients with CLL at the dose of 375mg/m2, with no clinical data to support a maximum dosage range and evidence of good tolerability, it would have been a routine step to increase the dose and investigate the patient’s response.
130 Professor Prince also considered that the disclosures in Maloney 1 and Maloney 2 supported the conclusions outlined above. In particular, he notes that Maloney 1 and 2 reported investigating rituximab to treat patients, including patients with SLL, which (as outlined above) is the leukaemic equivalent of SLL. Maloney 1 also suggests investigating rituximab in combination with chemotherapy and in other B-cell histologies, which he understands to encompass CLL. Further, Maloney 1 discloses that an earlier Phase I trial of single doses up to 500mg/m2 showed clinical response with no dose-limiting toxicity. Professor Prince considered that this meant that a dose of at least 500mg/m2 was feasible.
131 Dr Seymour disagrees with a number of aspects of Professor Princes’ evidence. Having regard to the integers of claim 2 of the CLL patent, Dr Seymour takes issue with Professor Prince’s statement that it would have been a matter of routine to administer rituximab in combination with chemotherapy to treat CLL. He considers that it was not established that rituximab could be administered safely to CLL patients as a monotherapy and the administration of it in combination with chemotherapy would have yielded further adverse events. At the priority date he was aware of concerning toxicity being demonstrated in a patient having even modest elevations of malignant lymphocytes in the blood and CLL patients typically have very high lymphocyte counts in the blood. Further, he disputes that the work by the MD Anderson Cancer Centre was publicly known prior to its publication in November 1998, after the priority date. Dr Seymour considers that Maloney 1 explicitly excluded patients with CLL (characterized by the presence of greater than 5000 lymphocytes per microgram). He also notes that Maloney 1 describes increased adverse events and lower efficacy for SLL patients compared with those having FL. In his opinion the similarities between CLL and SLL were sufficient to give rise to toxicity concerns that would have discouraged him from the use of rituximab to treat CLL.
132 Dr Seymour also notes that neither Maloney 1 nor 2 disclose a dose range of 500 – 1500mg/m2.
133 In its submissions Sandoz correctly identifies that the concerns that Dr Seymour raises about toxicity and absence of efficacy of rituximab in CLL demonstrate that there is an evidentiary conflict between the experts, which are matters for resolution at trial. It submits that the prima facie position established by Sandoz demonstrates that there is a “very serious” question over the validity of claim 2 of the CLL patent. However, as I note in paragraph [122] above, the variables that arise are significant. The evidence is not sufficient for me to conclude other than that the case advanced is arguable.
134 It should also be noted that Sandoz submits that Maloney 1 and Maloney 2, with the common general knowledge, deprive claim 2 of the CLL Patent of inventive step both individually and in combination. However, Professor Prince does not give evidence about whether these articles would be treated as a single source and, if so, the effect of doing so. Therefore, there remains a question about whether this would occur. In any event, such combination would not appear to resolve the differences of opinion between the experts outlined above.
135 I now turn to consider the lack of inventive step case insofar as it concerns the DLBCL patent.
136 Professor Prince gives evidence that, as at the priority date, Australian haematologists would have been highly motivated to investigate the application of rituximab to patients presenting with intermediate-grade to high-grade lymphomas, the most common of which is DLBCL. He gives the opinion that a significant number of patients with such lymphomas were being cured with first-line chemotherapy treatments, but that patients who did relapse with DLBCL were left with what is referred to as “salvage treatment options”, which involved multi-agent chemotherapy drugs not previously used with the patient and (in the case of younger patients) additional very high doses of chemotherapy supported by a stem cell transplant. Approximately 60% of such patients were non-responsive and relapsed again. Professor Prince says that as at the priority date “anecdotal case studies” (by which he means case studies presented at conferences and seminars that he and other Australian haematologists attended) and clinical trial information on the efficacy of rituximab in treating DLBCL, including with bulky disease, were being discussed at conferences and seminars that he attended. He notes that DLBCL is an aggressive lymphoma which requires urgent treatment and patients who were refractory to the standard treatment had no other treatment options available to them. Link had also reported positive findings for treating DLBCL with rituximab.
137 Professor Prince also states that it was known at the priority date that 20% of patients with DLBCL presented with bulky disease and chemotherapy was known to reduce the size of tumors associated with bulky disease. As outlined above, in Professor Prince’s opinion there was a synergistic effect between rituximab and chemotherapy, including CHOP and the administration of both rituximab and CHOP on day 1 of treatment was logically the most desirable option for the convenience of patients and smooth operation of the outpatient clinic at which treatment was administered.
138 Accordingly, he considers that Australian haematologists with access to rituximab would have investigated the use of treatment of DLBCL, with and without bulky disease, and with and without chemotherapy (and, in particular, CHOP) in the expectation that it may well result in a new treatment for patients.
139 Dr Seymour takes issue with a number of the factual matters to which Professor Prince refers. For instance, whilst he knew rituximab to be effective in the treatment of low grade lymphoma, he did not automatically expect that it would be effective in other types of lymphoma such as DLBCL and preliminary results of rituximab monotherapy in DLBCL were not particularly promising. He does not recall the anecdotal case studies in respect of the efficacy of rituximab in treating DLBCL accompanied by bulky disease. He also disagrees that there was a known synergistic effect between rituximab and chemotherapy for the reasons outlined in paragraph [121(1)] above. Furthermore, whilst he accepts that there is a convenience in administering both rituximab and CHOP on day 1, he has reservations, which he explains on a number of bases, about whether dosing CHOP and rituximab on the same day would have been safe or efficacious.
140 Sandoz submits that there is an evidentiary contest to be resolved at trial, and that for present purposes the Court should find that there is a “serious challenge” to the validity of claim 35. Again, I consider that the evidence does not permit any provisional conclusion as to the strength of the validity case beyond the observation that it is arguable.
141 I now turn to consider the inventive step challenge insofar as it concerns the RA patent.
142 Professor Buchanan responds to the following question posed by the solicitors acting for Sandoz:
discuss the extent to which, at the relevant date, if I was working as a part of a research team seeking to investigate new and better treatment strategies for RA, I would have considered investigating the use of writ rituximab in new treatments for RA.
143 For present purposes it is not necessary to address the detail of Professor Buchanan’s analysis. It suffices to refer to his evidence given having regard to the elements of claim 3 of the RA patent, noting that prior to addressing these elements, and prior to being provided with a copy of the RA patent, Professor Buchanan had set out his understanding of the relevant common general knowledge and prior art documents.
144 In his affidavit Professor Buchanan considers that the following was relevantly known to him and other rheumatologists as at the priority date:
(1) RA was conventionally treated with methotrexate as a standard first-line treatment, either alone or with one or more DMARDs;
(2) combination therapies were common practice in treating patients with RA, particularly patients with an inadequate response to a monotherapy;
(3) TNFα-inhibitors were also well-established treatments for RA;
(4) TNFα-inhibitors as at the priority date included etanercept, infliximab and adalimumab; and
(5) there were patients who were non-responsive or refractory to DMARDs and/or TNFα-inhibitors.
145 Professor Buchanan states that Edwards 1 and Edwards 2 reported investigating rituximab to treat patients with RA in several contexts. Edwards 2 describes a study treating RA in patients by administering rituximab as 2 x intravenous doses of 1000mg to 4 patient groups, including 1 group with rituximab alone (group B) and another with rituximab in combination with methotrexate (group D). It reports that the interim analysis of the study after 24 weeks is that the strongest results were for group D.
146 Edwards 1 reports that an open study of B-lymphocyte depletion in subjects with refractory RA (refractory to methotrexate and other DMARDs) was undertaken. It reports that all patients received rituximab as 4 x IV infusions over 3 hours on days 2, 8, 15 and 22 of doses of 300mg on day 2 and 600mg on the balance of the days. Oral prednisolone was also administered and cyclophosphamide as an IV infusion on days 4 and 17.
147 According to Professor Buchanan, Edwards 1 reports that all patients showed rapid improvement in synovitis (inflammation of the synovial membrane), which is one of the symptoms of RA.
148 Professor Buchanan attended the conference where Edwards 2 was presented and he read the abstract at the time of its publication. He considers it likely that he read Edwards 1 at or about the time of its publication. In his opinion, claims 1 to 3 of the RA patent summarise the progressive developments as at the priority date in the field of rheumatology with respect to the treatment of patients with RA who were refractory to DMARDs and bDMARDs. In his view, the claims do not record what he would consider to be a “eureka moment” of invention. Edwards 2 reports the success of initial studies of a combination treatment of rituximab with methotrexate where rituximab was administered at the dosage identified in claim 1. The patient group most suitable for such a combination therapy is those who were refractory to TNFα- therapy and he would have had every expectation that the results reported in Edwards 2 would also be achieved in such patients.
149 Professor Morand answers the evidence of Professor Buchanan and takes issue with a number of the factual matters and conclusions expressed by him. Without being exhaustive, Professor Morand first, would qualify the statements made by Professor Buchanan to the effect that TNFα-inhibitors were established treatments for RA and that they were known to have included etanercept, infliximab and adalimumab. He accepts that infiliximab had been approved for use on the ARTG by 2 August 2000 and that etancercept was approved by 18 March 2003 (the priority date of the RA patent being 9 April 2003), but his view is that none were in widespread use in Australia until August 2003, when etanercept was listed on the PBS. Notwithstanding these matters, Professor Morand does accept that he was aware from the literature available before the priority date that these drugs had been used to treat RA patients in the United States and Europe with considerable success. He was also aware at that time that there were some people who did not respond to TNFα-inhibitors.
150 Secondly, Professor Morand notes that Edwards 2 does not disclose that any patients had previously been administered TNFα-inhibitors. He considers that it is unlikely that they were. He notes that the dosage administered was 2 x 1000mg, giving a total of 2000mg, but before the priority date that dosage was not widely known. He considers that if any dosage of rituximab was known as at that date, it was the lymphoma dosage of 4 x 375mg/m2, which was based on the calculated body surface area of a patient and is quite different to the fixed dosage. In his view, before April 2003 it was not (and is still not) possible to predict whether a patient would respond to treatment with a TNFα-inhibitor. He also considered that those who failed to adequately respond were considered to be particularly difficult to treat. Although Professor Morand may have hoped that rituximab (with or without methotrexate) would provide a treatment option for this patient group, he would not have had such an expectation because there was no data on which to base it. Professor Morand did not regard any of the prior art documents to teach him otherwise.
151 Sandoz submits that the conflicts reflected in the evidence of Professor Morand demonstrate that it has a “powerful prima facie case” that claim 3 of the RA patent lacks an inventive step. Again, the level of disagreements between the experts on the variables that arise in the consideration of inventive step are such that, on my provisional estimate, the lack of inventive step case is arguable, but no more.
152 It should again be noted that Sandoz submits that the various prior art documents, with the common general knowledge, deprives claim 3 of inventive step both individually and in combination. However, Professor Buchanan does not give evidence about the effects of the combination of these prior art documents (other than Edwards 1 and Edwards 2), so there remains a question about whether this would occur. Again, such combination would not appear to resolve the differences of opinion between the experts outlined above.
5.5 Conclusion in relation to arguable case
153 As matters stand, Roche has established that it has a strong arguable case that each of its patents will be infringed if Sandoz commences to supply RIXIMYO in accordance with the PIs that has been approved. Having regard to the conflicting evidence going to a number of aspects of the case on lack of inventive step, in my view it is apparent that the invalidity case is arguable, but it is not, for present purposes (and I emphasise, on a provisional view), possible for me to conclude that it is strongly arguable, as Sandoz urges. Put another way, the evidence of the experts on both sides appears to be rational and persuasive and, having regard to the differences between them, I can see no rational basis for concluding other than that the lack of inventive step is arguable.
6.1 Consideration of the arguments
154 Roche submits that the following factors weigh in favour of the grant of an interlocutory injunction.
155 First, it is the owner of the patents, the infringement of which is not in dispute. Whilst there is, as Roche accepts, an arguable case on Sandoz’s cross-claim for invalidity based on an absence of an inventive step, this is not sufficient to displace the conclusion that the balance lies in Roche’s favour. For the reasons set out in section 5 above, I agree.
156 Secondly, MABTHERA has been on the market since 1999. Roche has a well-established market position in relation to the patented indications. The status quo is that the only rituximab product on the Australian market is MABTHERA. The launch of RIXIMYO in the face of Roche’s patent rights will adversely affect the status quo and severely affect the business of FHLR and Roche Products.
157 Thirdly, Roche contends that any loss that it suffers as a result of the launch of a generic rituximab will not be able to be adequately calculated or quantified by an award of damages. In this regard it is necessary to distinguish between the causes of any loss that Roche contends it will suffer and the difficulties that Roche contends will be involved in quantifying that loss.
158 The categories of loss that it asserts it will suffer are:
(1) a 16% price drop mandated by the PBS (there is also a separate 14.5% price drop, but this will occur regardless of Sandoz’s actions);
(2) additional decreases in price caused by (i) competition with Sandoz and possibly at least one other generic; and (ii) subsequent additional decreases in price by reason of mandatory price disclosure obligations under the PBS;
(3) loss of market share; and
(4) loss to goodwill as a result of (i) any partial increases in price upon the grant of a final injunction; and (ii) the possibility of terminating staff, which may also result in loss of market presence in Australia.
159 The factors that Roche contends are relevant to the irreparable nature of the harm and difficulties in assessing the loss are said to be:
(1) that the 16% price drop is unlikely to be reversed and Roche will not be able to increase its prices to what they were before Sandoz’s entry;
(2) as a result of the likely entry of at least one second generic rituximab (discussed further below), it will be difficult to assess each generic’s liability for the loss;
(3) future losses may need to be assessed, which would require hypothesising in relation to future pricing, market share and so forth; and
(4) the losses to goodwill referred to in paragraph [158(4)] above are unquantifiable.
160 The evidence of Mr Petersen is that (in approximate terms as at 2017); 47% of sales of MABTHERA are for FL, worth $68 million; 7% of sales are for CLL, worth about $9.8 million; 29% of sales are for DLBCL, worth about $42 million; and 9% of sales are for RA, worth about $12.5 million. Approximately 8% of sales are for uses that are not within those indications.
161 Mr Samwell’s evidence is that Sandoz’s launch of RIXIMYO is contingent on it obtaining PBS listing, but upon launch it will be made available for all patients. For PBS subsidised indications, it is likely that the biosimilar product would be “A” flagged with the result that even where a doctor prescribes rituximab by reference to the brand name MABTHERA (rather than the active ingredient rituximab) a hospital would be entitled to dispense the biosimilar product on the basis that it is considered therapeutically interchangeable with MABTHERA, unless the doctor ticks a box on the prescription that indicates that it must not be substituted.
162 Mr Petersen estimates that Roche will lose a substantial share of the rituximab market and that Roche’s loss is likely to be in the order of tens of millions of dollars. He provides an analysis of likely loss of market share by reference to the United Kingdom (UK) experience, and claims that such loss would likely be exacerbated by the operation of the “biosimilar uptake drivers” implemented as a matter of government policy. In the case of RIXIMYO, the PBAC made a number of positive recommendations designed to encourage the prescribing of that product by clinicians. Given the sales volumes to which I have referred above, I accept that the loss of revenue to Roche in the first 12 months will be very significant. Even if one notes that the NHL and CLL patents will expire in the latter half of 2019, it is not unrealistic to expect that sales in respect of all patented indications in the next 12 months alone would be very substantial.
163 The listing of RIXIMYO on the PBS will cause a mandatory 16% price reduction of the approved price of MABTHERA, which will be increased to 25% on 1 October 2018. It is not necessary in the present case to address the detail of the arrangements that lead to the 16% price drop. The details of the scheme pursuant to which this would take place have been set out in detail in the first affidavit of Ms Todd and are not in dispute. In short form, upon the introduction of a biosimilar brand the MABTHERA products would move from PBS “Formulary F1” to “Formulary F2”, which would prompt the price reduction. The evidence of Mr Petersen and Ms Todd is that, whilst the Minister has a discretion to reverse a product price reduction, in their experience this has never occurred. I accept that if the present injunction is refused, but at trial a final injunction is granted, the evidence indicates that the prospect that Roche would be in a position to restore its prices to their present level is remote.
164 Roche next submits that it will be faced with competition not only from Sandoz, but also from sales of at least one additional biosimilar. This will have a confounding effect on the calculation of damages, because it will lead to Roche competing with two or more biosimilar products in the market for rituximab.
165 In this context Roche draws attention to the fact that Sandoz is not the only company with a rituximab product approved and listed on the ARTG. On 16 April 2018, Celltrion Healthcare Australia Pty Ltd (Celltrion) registered two products, RITEMVIA and TRUXIMA on the ARTG in respect of the same indications as MABTHERA. The agenda for the July 2018 PBAC meeting includes an entry seeking to include the TRUXIMA brands of rituximab on the PBS for all indications for which the reference biologic (that is, MABTHERA) is listed. Ms Todd believes that if Roche is unsuccessful in restraining Sandoz, then the July PBAC meeting will consider and approve the listing of TRUXIMA, which she expects will take place on 1 December 2018, with the consequence that a second biosimilar product would be “A” flagged for prescription. The solicitors for Roche, Spruson and Ferguson, have written to Celltrion, informing it of the patents, and seeking details as to its proposals, but the evidence discloses no substantive response.
166 The evidence indicates that Celltrion rituximab products have been released on the market in the UK, and that they compete there with Roche’s UK MABTHERA products. Mr Petersen gives evidence that in April 2017 TRUXIMA received UK regulatory approval for all of the indications for which MABTHERA is approved in the UK. In June 2017, Sandoz International launched a further biosimilar version of rituximab under the name RIXATHON, which was approved for the same indications. Mr Petersen gives evidence that the price of the biosimilar products was in the order of 40% less than that of MABTHERA and that, upon the introduction of those products to the market, the use and market share of MABTHERA decreased significantly.
167 Sandoz points out that the regulatory regime in the UK is different to that in Australia, and that accordingly the history of events there is not relevant to what is likely to happen in Australia. That is likely to be so in many respects, but in my view two matters are able to be drawn from the evidence concerning the UK experience. First, that it is more than idle speculation to consider that one or other of the Celltrion products will be imminently ready for launch in Australia. Indeed, the evidence suggests that it is likely. Secondly, the market experience in the UK indicates a price drop of about 40%. In the present case Mr Samwell has given evidence that the introduction of biosimilar products competing with reference biological products will not cause prices to drop in the way that the market has seen for small molecule drugs, where reductions of up to 80% have been experienced. However, Sandoz supplies no information as to the pricing that it expects to adopt if it is permitted to launch its product. No doubt that is for sound commercial reasons, but the result is that the Court has no metric by which to gauge the likely price drop other than, perhaps, information from the experience in the UK. Mr Petersen gives evidence that in his view the price drop will be similar if not greater than that experienced in the UK. This is particularly as a result of the further mandatory price disclosure mechanism, which in turn will lead to additional price drops.
168 The mandatory price reduction mechanism arises under the NH Act by operation of what is euphemistically called a “simplified price disclosure” mechanism, which applies to manufacturers of medicines listed in the F2 formulary. Under the NH Act the sponsors of drugs listed on the F2 formulary must disclose to the Department of Health’s Price Disclosure Data Administrator certain data in relation to the products sold (other than those sales to public hospitals). This data is then used to determine the “weighted average disclosed price” (WADP) of the listed items. The calculation takes into account the average PBS approved ex-manufacturer prices of all relevant brands, the disclosed net revenue generated by those brands and the volumes sold. If the WADP is at least 10% less than the approved ex-manufacturer price (AEMP) in a given period, then the AEMP will be adjusted downwardly, thereby causing a further reduction in price for the listed products. In this equation, the greater the market share of the generic products, and the greater the rebates or discounts given in respect of those products, then the greater the reduction in the WADP and therefore the greater the reduction in the AEMP.
169 Mr Petersen gives evidence that the future price disclosure obligations for MABTHERA and other rituximab products in respect of all discounted sales to community pharmacies and private hospitals will trigger corresponding price reductions from the second year of sales onwards. The first reduction would not occur until October 2019 and then would occur every 6 months after that for 3 years. As a consequence, the first reduction would occur after the earliest patent has expired, however, Roche notes that the reduction would occur sooner than it otherwise would have if Sandoz were injuncted.
170 As a consequence of competition from Sandoz and also Celltrion, Mr Petersen expects that Roche will be obliged to reduce the price of MABTHERA substantially. This, Roche submits, will produce a spiralling effect on prices after the first 12 months because of the operation of the simplified price disclosure mechanism.
171 Mr Petersen gives evidence that it is not technically or commercially feasible for Roche Products to set different prices for units of MABTHERA depending on whether they are used for inpatient treatment or dispensed for outpatient treatment. This is because Roche Products is not able to identify the particular end patients ultimately receiving the products, especially where MABTHERA is sold to a compounding house which subsequently distributes the drug. As a consequence, whilst the mandatory price reductions only concern that proportion of sales to which a PBS subsidy applies, Roche will apply the same price to all of its MABTHERA products, regardless of their end use.
172 Although, as Sandoz submits, these pricing consequences to Roche are a matter of commercial choice, I accept that as a matter of practical implementation this will be a consequence of any mandatory price drop.
173 Roche points to a number of other side effects of the price reductions which it considers would flow from the introduction of RIXIMYO into the Australian market. One is that if interlocutory relief is refused and a final injunction is ultimately granted such that RIXIMYO products were required to be withdrawn from the market, it would be necessary to raise the prices of the MABTHERA product to some extent (although it alleges that it would not be able to return to former prices). This would inevitably result (according to Mr Petersen) in the loss of some of the goodwill amongst Roche’s customers and also give rise to risks for the reputation of Roche Products in Australia generally and, consequently, the business of FLHR. Sandoz counters this point by noting that the majority of rituximab customers are hospitals (although there is still at least a small proportion of patients who pay for rituximab themselves). Hospitals, being more sophisticated customers, would understand the nature of the pharmaceutical business. Sandoz therefore contends that any damage to goodwill would be diminished. Further, Sandoz submits that Roche would not raise its prices again in any event because at least two if not three of the patents will have expired and Petersen indicates that the same price will be charged for Mabthera across the board.
174 Mr Petersen expects that, given the drop in revenue (noted in paragraph [162] above), within 12 months following the launch of the RIXIMYO products, Roche would be likely to be required to terminate some staff. He notes that there is presently no marketing staff dedicated specifically to MABTHERA but that it is likely that the salesforce allocated to other products would be terminated and this would result in a reduction of Roche Products’ presence in the Australian marketplace and have a consequential impact on the goodwill enjoyed by Roche.
175 Fourthly, Roche contends that Sandoz has been aware since at least December 2017 of the patents and of the intention of Roche to assert its rights. It submits that Sandoz has taken steps to prepare for the launch of RIXIMYO in full knowledge of the possible consequences of an interlocutory injunction being granted, and so, in weighing the balance of convenience, substantial weight should be placed in favour of the patentee given that the respondent “deliberately took the applicants’ innovation, knowing of the claim that this was an invention entitled to protection”: Tidy Tea Ltd v Unilever Australia Ltd [1995] FCA 1439; 32 IPR 405 at [415]. As Roche noted in opening, the method of treatments patents have been on foot in the absence of the compound patent since 2013 and all but one were amended, with no oppositions thereto.
176 Finally, Roche also points to the fact that there has been no allegation of delay on Roche’s part in bringing its action against Sandoz.
177 Sandoz answers the Roche claims that damages would not be an adequate remedy in two parts. First, it submits that the difficulties that Roche contends would confront the calculation of its loss are overblown and are not as complicated as Roche claims. Secondly, it submits that the harm that it and the public will suffer if at final hearing a final injunction is refused and a claim is made on the undertaking as to damages, is far harder to calculate and would result in greater irreparable harm.
178 As to Roche’s claims of irreparable harm, Sandoz first submits that it will undertake to keep records of sales and use of MABTHERA, including of the relevant indication for which RIXIMYO is prescribed. Mr Samwell gives evidence that the administration of rituximab takes place predominantly in a small number of hospitals and infusion clinics and that it would not be an onerous task to track the administration in each of these outlets. Mr Samwell gives evidence that more than 50% of the revenue for public non-compounding hospitals is attributable to ten hospitals and almost 60% of the revenue generated by rituximab for private non-compounding hospitals is attributable to less than 10 hospitals. If Sandoz is permitted to launch, it could enter into agreements with its customers requiring records to be kept and to be available for inspection by Sandoz from time to time to ensure that it is possible to trace sales for patented- and non-patented indications. Furthermore, the evidence of Ms Matthews (the Director of pharmacy, at RMH is that at present, records are kept at RMH in the ordinary course of dispensing with respect to what brand was administered to which patient and for what indication. This includes records of off-label use, for which applications must be made on a case by case basis.
179 In response, Mr Petersen states that the administration of rituximab takes place at approximately 270 hospitals and oncology infusing clinics in Australia, which, it submits, presents a far more complicated picture of record keeping than Sandoz suggests would apply.
180 Sandoz next submits that the assessment of harm suffered by Roche should not be assumed to be complicated by other competitors in the market. The evidence of Mr Samwell quotes a 2009 report by the US Federal Trade Commission, which estimated that the cost of developing a new biosimilar is between US$100 – US$200 million and takes between 8 – 10 years. By contrast, the development of small molecule generic drugs cost between US$1 –US$5 million and take between 3 – 5 years. This means, Mr Samwell explains, that there is less likely to be a large number of generics competing with biosimilar products and less likely to be radical price reductions. Even so, Mr Samwell gives evidence that he is aware that not only Celltrion, but also Pfizer is likely to seek to launch a biosimilar rituximab in Australia and that both have been involved in actions in other jurisdictions in relation to Roche’s rituximab patents.
181 Further, Sandoz submits that the uptake of a new biosimilar medicine is relatively slow compared with small molecule compounds and that Roche’s anxiety as to the likely market share and loss of sales that it will experience is unfounded. In this regard the expert evidence indicates that not only is it necessary to satisfy clinicians as to the safety and efficacy of a biosimilar relative to the reference product, but also there is a tendency on the part of practitioners to be reluctant to switch treatment for an individual patient from the reference product to a biosimilar. Professor Prince gives evidence that at present Epworth Healthcare is supplied MABTHERA by a compounding pharmacy. If the decision were to be made at a hospital level to administer a biosimilar rituximab, and he was satisfied with efficacy data that he has been presented to support the decision, he would be comfortable with transitioning (that is, switching) patients to the biosimilar. Professor Buchanan is more cautious. He gives evidence that he would be reluctant to switch patients already being treated with MABTHERA to a biosimilar, but he would be comfortable to use a biosimilar on patients who had not previously been treated with another form of rituximab (referred to as “naïve” patients). The cause for the caution arises (and it is not in dispute between the experts) arises at least in part because of the concerns about immunogenicity described in paragraph [56] above. That is, because biologic therapies are complex medications produced using a biological source, such as cells or bacteria. There can be variations and modifications between different batches from the same biologic supplier. A biosimilar is intended to be an equivalent for the reference biologic, but it cannot be identical to the reference products because the biosimilar is unlikely to have the same cell line and it is impossible to reproduce identically every step in the manufacturing process. The consequence is that some clinicians are cautious about prescribing biosimilars to patients who are already receiving the reference medicine.
182 In this connection Sandoz submits that it is not to be concluded that the introduction of a first biosimilar rituximab would have swift market penetration, despite the government policy of biosimilar uptake drivers. It submits that the direct evidence of caution from clinicians suggests a more sedate progression than might be expected with small molecule generic products. The consequence of all of this, Sandoz contends, is that for a biosimilar medicine there will be fewer market entrants and less market volatility.
183 As to Celltrion’s imminent launch, Sandoz contends that Roche can only speculate that Celltrion will launch in December 2018, being the earliest possible date.
184 In my view the evidence does not permit the conclusion that there will not be other generic competitors in the market in the short term. To the contrary, the evidence indicates that it is at least likely (absent an interlocutory injunction) that the Celltrion TRUXIMA product will be launched in Australia after 1 December 2018. That event is likely immediately to introduce some confounding effects on the assessment of loss suffered by Roche.
185 In further answer, Sandoz submits that the assessment of a claim for compensation by Sandoz under the usual undertaking as to damages is likely to be more complex, time-consuming and costly than required if damages were sought by Roche at the conclusion of any proceedings. Sandoz submits that three cases that are yet to be determined in this Court establish this point: Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth (Federal Court of Australia proceeding no. VID 195 of 2009 and other related proceedings – NSD 596 of 2009 and NSD 1124 of 2009) (Venlafaxine Proceedings); Apotex Pty Ltd v Sanofi-Aventis & Ors (Federal Court of Australia proceeding no. NSD 1639 of 2007 and other related proceedings – NSD 1311 of 2008 and NSD 1408 of 2008) (Clopidogrel Proceedings); Watson Pharma Pty Ltd v AstraZeneca AB (Federal Court of Australia proceeding no. NSD 2342 of 2011 and other related proceedings NSD 208 of 2012 and NSD 673 of 2011) (Rosuvastatin Proceedings).
186 Sandoz relies on the evidence of Mr Swinn, who is the solicitor for Sandoz in the present proceedings and for Sigma in the Venlafaxine Proceedings. He notes that in each case one or more generic pharmaceutical companies were restrained by interlocutory injunction from supplying their product(s). In each, the generics subsequently established that a final injunction should not be granted, because the patent was revoked. Proceedings are now on foot in each case whereby persons who claim to have been adversely affected by the grant of the interlocutory injunction seek compensation from the patentees pursuant to the usual undertaking as to damages. In each case the parties initially included at least the companies restrained and the Commonwealth of Australia (which subsidised the cost of the originator product through the PBS), although in the Clopidogrel and Rosuvastatin proceedings the generics settled, leaving the Commonwealth as the only remaining claimant. In the Venlafaxine Proceedings suppliers to the generics have also claimed.
187 Mr Swinn gives evidence that in the Venlafaxine Proceedings, the pleadings reveal a very significant number of issues to be in contest concerning the hypothetical position that the various claimants would have been in, had they not been restrained by the interlocutory injunction (that is, the “counterfactual” position). The variables in issue include whether each generic claimant would have launched its generic product at all; if so, when it would do so; whether each generic would have confined its sales to the non-PBS market or sought listing on the PBS; if so, when each generic product would have been listed on the PBS; the total size of the market for the generic products in each month of the claim period; the prices at which the generic products would have been sold by each market participant each month during the claim period; the portion of the market that would have been captured by each generic; and whether the originator would have launched its own generic or authorised a third-party to do so or taken other measures to protect its market. According to Mr Swinn, the econometric evidence going to loss proposed a total of 22 alternative counterfactual scenarios, depending on the possible factual outcomes determined by the Court. This, Mr Swinn says, has led to approximately 4,500 pages of affidavit material going to factual issues and issues requiring expert opinion evidence.
188 Mr Swinn records that the Colpidogrel compensation claim was heard over 6 weeks in 2017 and that judgement is reserved; that the Venlafaxine claim is listed to be heard over 6 weeks in June and July 2018; and that the Rosuvastatin proceedings are yet to be set down.
189 Sandoz contrasts the time and complexity of these proceedings with claims for damages made by patentees who ultimately succeed in establishing infringement and points to Bayer Pharma Aktiengesellschaft v Generic Health Pty Ltd [2017] FCA 250; 124 IPR 23 as an example of a case where such a claim was advanced, heard over 8 days and determined in short measure.
190 Sandoz submits that in the event that any injunction is discharged at final hearing, any analysis of loss that it suffers will be entirely hypothetical and will require consideration of counterfactual situations of varying types and complexity, such as those that have dogged the cases referred to above, including the likely impact of the possible entry of TRUXIMA or another generic rituximab and the entry into the market of Roche’s own GAZYVA product, which does not contain rituximab but has therapeutic relativities to MABTHERA. GAZYVA is presently listed on the PBS for CLL and an application has been made for GAZYVA to be listed on the PBS for untreated FL. Furthermore, the market for biosimilar products is relatively new, which will make it much harder to determine what might have happened, the impact of the biosimilar uptake drivers (if any) and the benefits of any first mover advantages that may accrue.
191 Sandoz raises the following further points which it contends play in its favour and against the grant of the interim injunction.
192 It submits that the launch of RIXIMYO will yield public benefits including:
(1) a reduced cost to the Commonwealth of PBS approved uses of rituximab;
(2) a reduction of substantial costs presently absorbed by hospitals and patients for “non-approved” uses of rituximab and thereby improved patient access to a life-saving drug; and
(3) (by reason of the reduced cost) the fostering of new research into potential new indications for rituximab.
193 It submits that the consequence should be that a public interest factor weighs against the grant of the interim relief sought.
194 It is perhaps axiomatic that a discounted starting price for rituximab would reduce the cost to the Commonwealth of any subsidy under the PBS. That submission has diminished force for Sandoz because of an absence of evidence (as I have mentioned) as to what discount it would give for RIXIMYO. The UK experience suggests about 40%. However, one might properly be wary of profit-making commercial parties invoking the public interest in support of their private ends. In every case concerning the threatened launch of a generic product to upset a patented monopoly there is a contingent public interest in a price drop. But this is dependent upon a finding that the monopoly is not valid, or that the generic product is non-infringing. Until that finding is made it is to be doubted that it is intrinsically in the public interest for a pre-emptive launch to take place only by reason of an asserted drop in price. These observations address submissions (1) and (3) above.
195 In pointing to a public interest in reducing costs absorbed by hospitals and patients for “non-approved” uses of rituximab (submission (2) above), Sandoz raises a more cogent point. It is necessary to explain the factual background to this submission in a little more detail.
196 As outlined in section 3.3 above, there exist significant limitations as to when a prescription for rituximab will be subsidised under the PBS. This is particularly the case for the treatment of RA. According to Professor Buchanan, approximately 80% of patients meet the criteria for continuation. He personally has 4 current private patients who have self-funded rituximab treatments for RA as they do not meet the PBS criteria for subsidised treatment. This is at a cost of $18,000 to $20,000 per person on an annual basis.
197 Furthermore, a clinician may prescribe rituximab “off-label” which means that the drug has not been approved and is therefore not registered on the ARTG for the prescribed indication. Such use is not always subsidised under the PBS. The consequence is (absent ad hoc subsidies given either by the government, private health insurance or the manufacturer such as through Roche’s compassionate assistance program) that either the hospital or the patient will have to pay the full price for the drug.
198 Professor Prince gives evidence that he is aware of a significant number of off-label uses for rituximab in haematology or other immunological fields. In the field of rheumatology, Professor Buchanan gives evidence that he is aware of rituximab being widely prescribed for the treatment of auto immune diseases other than TGA approved indications. Dr Liew gives evidence of a retrospective review of patients receiving rituximab off-label at Westfield Hospital in New South Wales between July 2002 and January 2006. He reports that the study shows a growing use of such off-label indications, resulting in a cost to the hospital of $160,907 in a six-month period when, at the time, a 100mg vial of rituximab cost $943 and a 500mg vial cost $2357. Dr Liew refers to other articles published in 2010 and 2013 which report an increasing trend of off-label use. However, in this connection it is also relevant to note the evidence of Ms Todd to the effect that the PBAC conducted a review of off-label use of rituximab and found that while there was a broad unmet clinical need, the quantity and quality of available evidence for uncommon indications was limited and that rigorous assessment of cost-effectiveness for uncommon indications would be impractical due to small patient numbers and inadequate supportive evidence. As a consequence, a decision was made not to list rituximab for broader indications.
199 The evidence of off-label use is of significance, because it directs attention to the fact that care should be taken to ensure that the scope of any interlocutory injunction granted should not, absent good reason, extend beyond the scope of the monopoly reflected in the claimed invention.
200 Next, Sandoz submits that RIXIMYO is a very important product for it. Mr Samwell gives evidence that Sandoz has the opportunity to be the first mover in the market for a biosimilar rituximab product in Australia. This will give it a significant commercial advantage over the suppliers of other biosimilar rituximab products because it will:
(1) trigger tender opportunities for Sandoz with hospital and healthcare groups;
(2) enable it to be the first biosimilar to start to try to win, as Mr Caine QC colourfully put it, the “hearts and minds” of clinicians by demonstrating its efficacy and overcoming their reluctance to use a biosimilar or switch from MABTHERA; and
(3) give it leverage in respect of future biosimilar medicine launches.
201 The affidavit evidence of Mr Samwell expresses the opinion that the period of access of the first generic to the market is of more importance for the biosimilar market than in the case of small molecule generics, because there is a lengthy period of “conversion” in which biosimilar sponsors must invest effort in presenting the safety and efficacy case to key clinicians. He says that biosimilars represent a new paradigm in drug development and, in his experience, clinicians and other healthcare professionals are often not aware of the science behind the manufacture and regulation of both reference biologics and their biosimilars. The time in which Sandoz would be the only biosimilar is an invaluable commercial advantage that, if restrained, it would lose.
202 As I have noted, Mr Samwell gives evidence that he is aware of two potential competitors, one being Celltrion and the other being Pfizer. Sandoz contends that the period of exclusivity (by virtue of being the first biosimilar to market) will confer a benefit, even if it is confined to 4 months. The advantage is compounded by the fact that once a clinician’s trust is gained, there is a reluctance to switch patients again. This in turn makes it more difficult for subsequent biosimilar brands to establish market share. Mr Samwell draws attention to the introduction of three other biosimilar products in Australia to support his contention of the advantages gained by a first mover; filgrastim (a haematology therapy for stem cell transplants and chemotherapy induced neutropenia), etanercept (a treatment for RA) and epoetin lambda (a treatment of anaemia and chronic kidney failure).
203 In the case of filgrastim the first biosimilar mover was Pfizer, who within the first 12 months apparently secured in the region of 20% of the market share. In the case of etanercept the first mover achieved just 4.5% share. For epoetin lambda, the first mover was Sandoz, which obtained an estimated 40% of the market by volume.
204 It might be said that the evidence that Mr Samwell relies upon is unconvincing in the context of the present proceeding. First, unlike the experience with filgrastim, it appears highly unlikely that Sandoz will have the benefit of an 11 month period during which it is the “first mover”. Secondly, the data in support of the proposition that Sandoz was in a favourable position by reason of being the first mover for epoetin lambda is not at all clear and suggests that factors other than being the first mover were at play. Thirdly, the etanercept data might cause one to doubt that there is any advantage at all. What the contrasting results in the three examples advanced suggest is that the question of any benefit at all being accrued by a first mover is significantly affected by the nature of the drug in issue, the marketing or other efforts taken by the first mover and, no doubt, a number of other variables such as the countermeasures taken by the originator. The present information suggests that it is not really possible to know what advantage a first mover may have. Logic suggests that typically there will be some advantage, but the variables make it difficult to assess. This observation no doubt is to the benefit of Sandoz in the calculus of convenience, because it represents a factor that is difficult to quantify or predict.
205 In response to the first mover issue, Roche submits that the Celltrion products would likely be available within 4 months of the launch of RIXIMYO. They submit that the evidence of Mr Garrett (a representative of a potential purchaser of biosimilar rituximab who works at Austin Health), Mr Petersen and Mr D’Silva (from Roche) indicates that hospitals are likely to wait until the Celltrion products become available before making a decision to stock a biosimilar and, if so, which one.
206 Celltrion’s products were not registered on the ARTG until after Sandoz had filed and served its balance of convenience evidence. As a consequence, there is no evidence from Sandoz which addresses these contentions. However, Sandoz submits that Roche’s evidence is largely speculative and is at odds with the clinical evidence as to the urgent need for less expensive rituximab. It also notes that Sandoz has already been approached by public authorities, private hospitals and healthcare providers to participate in tenders for rituximab (for supply in the second half of 2018). On the other hand, Mr Samwell’s own evidence is that there are a number of private and public hospital tenders for which Sandoz may not be in a position to participate in during 2018.
207 Roche has established a prima facie case for infringement of the asserted claims. Sandoz has by its evidence demonstrated that it has an arguable case for challenging the validity of those claims, however, for the reasons set out in section 5 above, the evidence does not permit a provisional view that rises above that level. This yields the conclusion that Roche has established a probability of success in the sense contemplated by Jessup J in Interpharma identified in paragraph [27] above.
208 It is important to note that the prima facie case on infringement is based on admissions made by Sandoz with the result that the case advanced by Roche pursuant to s 117 of the Act is sufficiently established for present purposes. Tied up in the admission is the acceptance that by supplying RIXIMYO, Sandoz will be supplying to a person who it has reason to believe will use the product for the patented indications. Naturally each supply cannot be for each indication, but the admission made does not distinguish between patented indications. Each of Sandoz’s ARTG registrations for RIXIMYO is in respect of all of the patented indications. That is to say, the prima facie case established by Roche applies equally in respect of all of the patented indications.
209 In the present case the expiry of the asserted patents is relevant. The NHL patent expires on 11 August 2019, the CLL patent expires on 9 November 2019, the DLBCL patent expires on 2 August 2020 and the RA patent expires on 6 April 2024. It is unlikely that the proceedings will be concluded before the expiry of the NHL or CLL patents. In its statement of claim, Roche alleges infringement of over 70 claims in respect of 5 patents. The cross-claim was only filed in April 2018 and a timetable for the filing of evidence and the trial has not yet been proposed. In the normal course of such cases there is also likely to be an appeal.
210 This timing indicates that the outcome of the interlocutory application is likely to have the practical effect of determining the final question of injunctive relief at least for the asserted NHL and CLL claims, which enhances the need to have regard to the strength of the parties’ case when assessing the risk of doing an injustice by the grant or refusal of the interlocutory orders sought; Warner-Lambert at [70]. For the reasons that I have identified in section 5 above, this factor lies in favour of Roche.
211 Further, Roche has the benefit of a longstanding monopoly for each of the patented indications. This is not to be confused with the monopoly that Roche once had in respect of the use of rituximab per se. The patent for rituximab alone expired on 12 November 2013. The patented indications represent a narrower monopoly based on use. Nevertheless, the status quo is that this monopoly has been in place for many years and that the supply of rituximab for the patented indications represents a substantial and important area of commerce for Roche. The fact that the patentee’s trade in its product is old and established, and that the proposed trade in the putative infringer’s product is new is a matter to be given particular weight in some cases; Warner-Lambert at [96] and [98]. I regard it to be relevant here.
212 The introduction of RIXIMYO will automatically produce a 16% price drop by moving MABTHERA from F1 to F2 in the PBS formulary. I accept the evidence of Mr Petersen that Roche will find it necessary to drop the price of rituximab generally as a result of a price drop, regardless of the product’s end use.
213 I have no doubt that Sandoz will reduce the price of RIXIMYO further than the mandatory price drop. This is because it will be necessary for it to attract customers away from Roche. Sandoz adduces no evidence of the likely extent of the price reduction, but the evidence of the experience in the UK suggests that it may be in the order of 40%, or perhaps more. This will have a substantial effect on the revenue to Roche and in my view is likely in the first 12 months to lead to a reduction in staff that will have an impact on Roche’s business more generally.
214 In addition, whilst there was some debate as to the timing, it does seem to me to be likely that Celltrion will (absent restraint) launch TRUXIMA in Australia upon its listing on the PBS, which is likely to be on 1 December 2018. I have no doubt that this will lead to further downward movement of prices for rituximab products. After a year of the first biosimilar launch, the mandatory price disclosure obligations will arise which will bring into effect yet further price reductions.
215 The evidence of Mr Samwell also indicates that there is a real prospect that a further biosimilar, manufactured by Pfizer, may well enter the market at some point.
216 These factors indicate that there are likely to be significant complexities in the calculation of any damages that Sandoz is obliged to pay Roche, in the event that no interim injunction is granted and Roche establishes infringement at trial.
217 Sandoz seeks to counter the effects of these matters by undertaking to keep good records of sales, and entering into contractual terms with customers requiring them to keep records of sales. However, three matters give rise to concerns that this proposal sounds better than it is likely to be in reality.
218 First, the evidence indicates that sales of rituximab are likely to be to several different types of customer. One is public hospitals, who would acquire a suitable cheaper and efficacious biosimilar rituximab after a competitive tender process, which may take several months to complete. Rituximab will also be sold to private hospitals, community pharmacies and mixing or compounding houses who compound rituximab and then supply it to hospitals and pharmacies.
219 Ultimately, it is the clinician who, after deciding upon a treatment option, generates a prescription which will be submitted to the hospital pharmacy. The evidence indicates that generally the prescription identifies the clinical name (rituximab) not the brand name of the drug supplied, and the biosimilar will be dispensed unless the clinician specifically directs otherwise. Often rituximab will be administered in conjunction with other treatments, such as chemotherapy, but it is not apparent what form of record is made of the overall treatment regime and past treatment regimes.
220 This chain of supply indicates that there are significant complexities attached to the keeping of records. Sandoz does not appear to have a contractual relationship with either the clinicians or the patients. There is no suggestion that it will have any means of access to the records of either. Ms Matthews gives evidence that RMH keeps records of dispensing in respect of particular indications, but it is not apparent that they keep records of, for example, the other drugs that are co-administered with rituximab, at what time the particular drugs are administered or whether the patient has been resistant to other forms of treatment.
221 Secondly, unlike in many such cases, every sale by Sandoz of RIXIMYO will not necessarily be an infringing use. Accordingly, even assuming that there is no second or third biosimilar in the market, a sale of RIXIMYO cannot be assumed to be a sale lost to Roche. At present that would be the case in respect of certain off-label uses. This gives rise to a significant and perhaps unusual complication. In the case of claim 21 of the NHL patent, the claimed method will be infringed (in loose terms) only where rituximab is administered for 2 years at a dose of 375mg/m2 following CVP therapy. For claim 2 of the CLL patent, rituximab must be administered in a particular dose in combination with chemotherapy. For claim 35 of the DLBCL patent the rituximab must be administered on day 1 of a chemotherapy cycle together with CHOP being administered on the same day. The position is yet further complicated for claim 1 of the RA patent, where the method is infringed if rituximab is administered to a patient who has experienced an inadequate response to a TNFα-inhibitor and the patient is further treated with concomitant methotrexate. I am not satisfied on the evidence available to me that adequate records will be taken of those particular uses such that an accurate assessment of damage to Roche can be made.
222 Thirdly, in the event that Celltrion’s TRUXIMAB enters the market, the complexities of record keeping are likely to be exacerbated.
223 On the other hand, on the assumption that an interim injunction is granted but Sandoz succeeds in invalidating the asserted patents, I also accept that Sandoz will face significant complexities in establishing its claim on the undertaking as to damages. There is force in its submission that there are several unknown factors in the calculation of such damages that will require the consideration of competing hypothetical “counterfactuals” along the lines of those that have dogged the Venlafaxine and other similar cases. One is that it is unknown how the market will respond to the introduction of a biosimilar in this market. Indeed, biosimilar products are generally new to the Australian market and it may be difficult to model, on a hypothetical basis, what could have happened if the launch were permitted. Another is that it is unpredictable how, in a hypothetical market, the various competing suppliers of biosimilars would have behaved, and what countermeasures Roche might take to respond to mitigate the effects of the competition, and how effective those measures might be. Sandoz also submits that the approaching expiry dates of the NHL and CLL patents in particular tilt the balance of convenience in its favour, because it means that by the time the proceedings are concluded all damages incurred by Roche in respect of those patents will have been in the past, and will be crystallised, whereas the calculation of compensation for Sandoz on the undertaking for damages will be hypothetical. I consider that this final point is most appropriately addressed by consideration of the form of order made, which I discuss further below.
224 Furthermore, I accept that absent restraint Sandoz is likely to be the first mover in the rituximab biosimilar market. However, on the basis of the evidence presently available it does not appear that this is likely to be a significant factor. TRUXIMAB appears likely to be launched within months of the proposed August 2018 launch of RIXIMYO. The penetration of a new biosimilar rituximab is likely to be relatively slow.
225 I accept that third parties will be affected by the grant of an interlocutory injunction. The mandatory price drop will not take place; with the consequence that hospitals and patients who self-fund their rituximab treatment will have to continue to pay more and the Commonwealth will not have the benefit of reduced public expenditure following the 16% price drop. Furthermore, by not having a price drop it is possible that more research may be conducted into the use of rituximab which could be for the benefit of other patients. However, there is evidence of a compassionate compensation scheme by Roche and often ad hoc subsidies for other uses. Furthermore, as I have said in [194] above, it is to be doubted that significant weight should be placed on public interest arguments where the alleged infringer has failed to dislodge the prima facie case of patent infringement.
226 In considering the balance of convenience, I am also conscious that Sandoz has been well aware at least since it obtained its ARTG listing for RIXIMYO that Roche intended to assert its patent rights. Indeed, I infer that it is likely that Sandoz has been aware of this likelihood from well before that date. This is a matter weighing slightly in favour of the grant of relief, because Sandoz must be considered to have been alert to the possible consequences of its actions.
227 Roche has asserted that it will also suffer reputational harm in the event that a final injunction is granted, but an interlocutory injunction is not, because it will attempt to raise the price of MABTHERA towards the level that it enjoyed in the halcyon days of its monopoly. The market will look askance at this act, and its reputation will suffer. I give this consideration little weight, given the nature and sophistication of the market.
228 Having regard to the matters that I have identified in the consideration of the parties’ arguments (section 6.1), and weighing the balance of the matters that I have considered in this section, in my view the balance favours the preservation of the status quo and the rights protected by the asserted claims in respect of which a serious question to be tried as to the entitlement to final injunctive relief has been established. The matters raised by Sandoz, particularly in respect of the difficulties in calculating its loss should final relief not be granted, certainly make the balance more finely tuned. But overall the balance of the case and justice lies in favour of the granting of interlocutory injunctive relief.
229 There is a contest as to the appropriate form of orders to be made. Sandoz submits (in the alternative to its primary submission that no interlocutory orders should be granted) that any interlocutory injunction should be tailored to take into account the fact the NHL patent will expire on 11 August 2019 and the CLL patent on the 9 November 2019. It submits that it is most unlikely that any final relief would cover these patents, because by the final determination of the proceedings, at least these two are likely to have expired. Roche submits that the admission of infringement made by Sandoz for the purpose of the interlocutory application includes all of the patented indications. It submits that a form of order against the supply of RIXIMYO generally is appropriate in circumstances where there is reason to believe that it will be put to the infringing use, even if efforts are made to supply it for non-infringing uses. It submits that in the present case there is no evidence of efforts made by Sandoz to supply for non-infringing use. It submits that even after the expiry of the NHL and CLL patents, the on-patent use would be widespread and substantial – about 46% of the market – and there is no suggestion that Sandoz would seek to exclude patented indications from its ARTG registration and PIs.
230 There is some complexity associated with the grant of an injunction where the alleged infringement arises pursuant to ss 117(1) and 117(2)(b) of the Act. The effect of these provisions is that if the supplier had reason to believe that the supplied product would be put to the infringing use, then the supply for any use of the product, being a non-staple commercial product, is an infringement, even if the use for which it is actually supplied is a non-infringing use; Warner Lambert at [26]. That is not easy to apply literally where a product is supplied in large quantities for use by a large number of consumers where the first supplier in the relevant supply chain has reason to believe that some but not all of the consumers to whom the product might ultimately be supplied will put it to an infringing use. In AstraZeneca v Apotex [2014] FCAFC 99; 226 FCR 324 (AstraZeneca FFC) the Full Court postulated at [444] (in the context of a claim for final relief) that it may be undesirable to impose a blanket restraint upon a supplier who has reason to believe that only some consumers, perhaps a very small minority, may put the product that is or may be supplied to them to an infringing use.
231 The present interlocutory application has not been conducted on the basis that Sandoz has reason to believe that only a small minority of consumers may put RIXIMYO to an infringing use. To the contrary, the evidence suggests that at present all but about 8% of the use is likely to fall within the patented indications. Sandoz has accepted that RIXIMYO will be supplied for an infringing use, but submits that as each patent expires, the proportion of threatened infringement will diminish.
232 On the basis of the current market figures, after 9 November 2019 (when the NHL and CLL patents have expired) over 50% of uses that Roche supplies for would be for indications that are not the subject of the asserted claims. Needless to say, after the expiry of the patents the calculus as to the balance of convenience and justice will shift. Roche’s prima facie case in respect of threatened infringement in respect of those patented indications evaporates, and the case becomes one focussed on the balance of convenience as it applies to the residual indications. At that point it is possible that there could be significant non-infringing use.
233 The parties have approached the present interim application on the basis that all of the four asserted patents are in play. The draft orders proposed by Roche seek blanket interim relief in relation to all uses of the Sandoz products. I am not satisfied that it is appropriate to permit such orders to extend beyond the expiry date of the NHL patent, which event could materially alter the landscape of the balance of convenience and justice. Nor do I consider it productive to speculate as to the circumstances that may prevail as at the date. No doubt the parties will work to ensure that a hearing has been or will imminently be conducted by then. With these factors in mind I will make the orders set out below, which will cease, absent further order or absent determination, on 11 August 2019.
234 The interlocutory orders (upon the giving of the usual undertaking as to damages) will be that:
1. Until 11 August 2019, the final determination of these proceedings or further order, the Respondent, whether by itself, its servants, agents or otherwise howsoever, be restrained from infringing the Asserted Patent Claims and each of them, including, without the licence of the First Applicant:
a) supplying for use;
b) offering for supply or sale;
c) supplying;
d) selling;
rituximab 500mg/mL concentrated injection vial or rituximab 100mg/10mL concentrated injection vial (together, the Sandoz Products).
2. The Respondent forthwith notify the Department of Health (Director of the PBS Price Changes Section, Pricing and Policy Branch of the Technology Assessment and Access Division) and the Minister for Health:
a) of the granting of the interlocutory injunction set out above, of its terms; and that
b) for the purposes of seeking listing of the Sandoz Products on the PBS, the Respondent is no longer able to continue to provide the assurance of supply it has given, until further notice by the Respondent to the Department of Health.
3. If the Respondent proposes to give further notice to the Department of Health pursuant to order 2(b) above, the Respondent shall give seven (7) days’ notice in writing to the Applicants of its intention to do so.
4. The costs of, and incidental to, the Applicants’ interlocutory application for interim injunctive relief be the Applicants’ costs in the cause.
235 Leave will be granted to Roche to apply to the Court prior to 11 August 2019 for a continuation of order 1 having regard to the circumstances prevailing at that time.
236 Directions will also be made that the parties confer and propose short minutes of order for the expeditious conduct of the proceedings and supply draft short minutes to my Associate within 7 days. A case management conference in the proceeding will be conducted on a convenient date shortly thereafter.
I certify that the preceding two hundred and thirty six (236) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Burley. |
Associate:

