
FEDERAL COURT OF AUSTRALIA
Apotex Pty Ltd v Warner-Lambert Company LLC (No 2) [2016] FCA 1238
Solicitor for the Respondent/Cross-Claimants in NSD 763 of 2013 and Applicants/Cross-Respondents in NSD 251 of 2014: | Allens Linklaters |
Counsel for the Respondent/Cross-Claimant in NSD 251 of 2014: | Ms AH Bowne SC |
Solicitor for the Respondent/Cross-Claimant in NSD 251 of 2014: | Maddocks Lawyers |
Table of Corrections | |
Para [207], last sentence be amended to also refer to “A/Prof. King” | |
25 October 2016 | Para [297], third sentence be amended to read “ the acts of importation and sale occur within the patent area.” |
4 November 2016 | Para [156], the word “based” deleted from the first sentence |
4 November 2016 | Para [174], first sentence amended to read “… that a pleading point was being taken.” |
ORDERS
APOTEX PTY LTD (ACN 096 916 148) Applicant | ||
AND: | Respondent | |
AND BETWEEN: | (and others named in the Schedule) First Cross-Claimant | |
AND: | APOTEX PTY LTD (ACN 096 916 148) Cross-Respondent | |
DATE OF ORDER: |
THE COURT ORDERS THAT:
1. The respondent and cross-claimants file and serve a draft minute of the final orders they seek in this proceeding within 7 days.
2. The proceeding stand over to a date to be fixed for the purpose of making final orders.
3. Up to and including 28 October 2016 or further order, the contents of paragraphs [275] to [281] (inclusive) and [283] of the reasons for judgment of Nicholas J dated today be kept confidential and not be made available for public inspection.
4. There be liberty to apply on 24 hours’ notice.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
ORDERS
NSD 251 of 2014 | ||
BETWEEN: | WARNER-LAMBERT COMPANY LLC First Applicant PF PRISM CV Second Applicant PFIZER IRELAND PHARMACEUTICALS (and others named in the Schedule) Third Applicant | |
AND: | GENERIC PARTNERS PTY LTD (ACN 132 833 777) Respondent | |
AND BETWEEN: | GENERIC PARTNERS PTY LTD (ACN 132 833 777) (ACN 132 833 777) Cross-Claimant | |
AND: | WARNER-LAMBERT COMPANY LLC (and others named in the Schedule) First Cross-Respondent | |
JUDGE: | Nicholas J |
DATE OF ORDER: | 21 october 2016 |
THE COURT ORDERS THAT:
1. The applicants and cross-respondents file and serve a draft minute of the final orders they seek in this proceeding within 7 days.
2. The proceeding stand over to a date to be fixed for the purpose of making final orders.
3. Up to and including 28 October 2016 or further order, the contents of paragraphs [275] to [281] (inclusive) and [283] of the reasons for judgment of Nicholas J dated today be kept confidential and not be made available for public inspection.
4. There be liberty to apply on 24 hours’ notice.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
NICHOLAS J:
1 There are two proceedings now before me. The first proceeding (“the Pfizer proceeding”) is brought by Apotex Pty Ltd (“Apotex”) against Warner-Lambert Company LLC (“Warner-Lambert”) and various related entities that form part of the Pfizer group of companies (together “Pfizer”). The second proceeding (“the GPPL proceeding”) is brought by Pfizer against Generic Partners Pty Ltd (“GPPL”). In these proceedings Pfizer seek injunctive relief against Apotex and GPPL under s 120 of the Patents Act 1990 (Cth) (“the Act”).
2 Warner-Lambert is the registered owner of Australian Standard Patent 714980 (“the Patent”) entitled “Isobutylgaba and its Derivatives for the Treatment of Pain”. The Patent was applied for on 16 July 1997 (“the application date”) with a claimed priority date of 24 July 1996 (“the priority date”). There is no dispute as to the correctness of the priority date.
3 In general terms, the Patent relates to the use of certain compounds for the treatment of pain and in the manufacture of medicaments for the treatment of pain. These include the compound known as pregabalin which is marketed and sold in Australia by Pfizer Australia Pty Ltd under the brand name Lyrica.
4 In the Pfizer proceeding, Pfizer alleges that Apotex threatens to infringe each of the 32 claims of the Patent. Apotex seeks an order for the revocation of each of the claims upon which it is sued. Apotex admits that, if claims 1-15 and 31 are valid, then it threatens to infringe them. Apotex says that it does not threaten to infringe the various Swiss claims, claims 16-30, and 32 (if that is truly a Swiss claim), because the products it is proposing to market and sell in Australia will be manufactured overseas by a third party. There is a further infringement issue peculiar to claim 32 which I also need to consider.
5 Apotex alleges that the claims of the Patent are invalid because the complete specification does not describe the invention fully, as required by s 40(2)(a) of the Act. Apotex further alleges that the claims of the Patent are invalid because the invention claimed, contrary to s 18(1)(c) of the Act, is not useful. Apotex also alleges that the Patent should be revoked because the patentee, Warner-Lambert, was not entitled to the Patent (s 138(3)(a) of the Act) and also because the Patent was obtained by false suggestion or misrepresentation (s 138(3)(d) of the Act).
6 In the GPPL proceeding, Pfizer alleges that GPPL also threatens to infringe claims 1-30 of the Patent. GPPL admits that, assuming the claims of the Patent are valid, it threatens to infringe claims 1-15 but not claims 16-30 (the Swiss claims). GPPL contends that each of the claims upon which it is sued are invalid on the same grounds that are relied upon by Apotex and it also seeks an order for the revocation of all such claims. In final submissions, GPPL adopted Apotex’s submissions in relation to both infringement and validity in respect of claims 1-30. During the course of the trial Pfizer withdrew its allegations of infringement against GPPL in respect of claims 31 and 32 and GPPL withdrew its claims of invalidity in respect of those claims.
7 The validity of the Patent is to be determined by reference to the provisions of the Act as they stood as at 24 July 1996 which is, of course, before the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth) (“the Raising the Bar Act”) took effect except for those provisions that relate to entitlement. So far as the latter issue is concerned, ss 22A and 138(4) of the Act in its current form apply in this case: see AstraZeneca AB v Apotex Pty Ltd (2014) 226 FCR 324 at [182]-[184].
8 There were 14 witnesses who made affidavits that were read at the hearing, 12 of whom were experts who contributed to one or more Joint Expert Reports and who gave oral evidence during the concurrent sessions. The other two witnesses, Ms Smith and A/Prof. Miller, made affidavits, but were not required for cross-examination. There were five “fields” that were the subject of Expert Conferences that led to five Joint Expert Reports, covering the following fields:
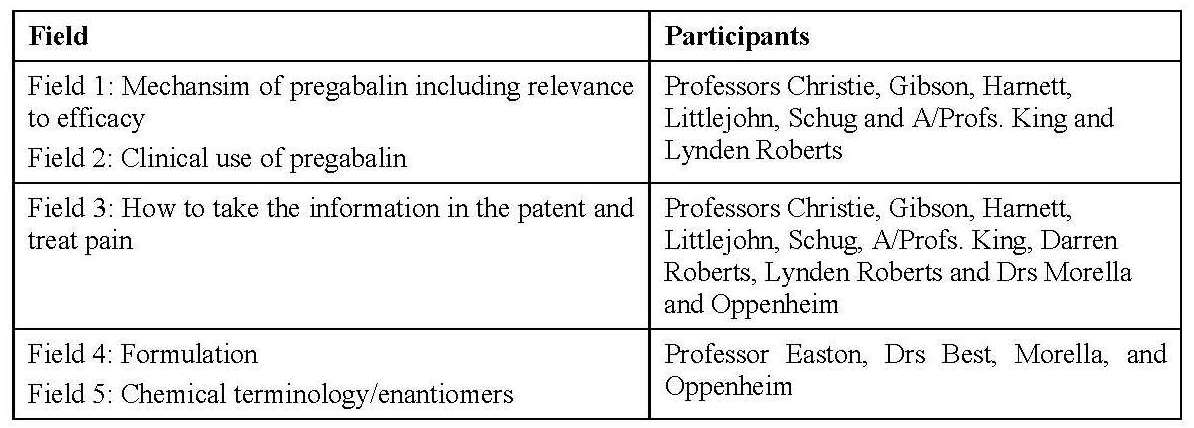
9 Further details of the witnesses and, in the case of the expert witnesses, their field of expertise, are set out below. Witnesses called by Apotex are designated (A) and those called by Pfizer are designated (P). The only witnesses separately cross-examined after the completion of the concurrent sessions were Professors Christie, Easton, Littlejohn and Schug and A/Prof. Darren Roberts.
10 My assessment of the expert witnesses was mostly very favourable. Their oral evidence given in the concurrent sessions was particularly helpful. The concurrent sessions allowed the experts to engage in a candid and spontaneous exchange of views in relation to key issues relevant to their fields of expertise. The contributions of the clinicians and their approach to the task of giving evidence was in my opinion exemplary. This is also true of Professor Christie whose evidence, along with that of Professors Gibson and Schug, was particularly informative.
11 It is necessary to make the following observation in relation to Dr Oppenheim’s affidavit evidence concerning the difficulties that would be encountered by the skilled addressee in formulating a pharmaceutical composition with which to perform the claimed methods of treatment using the Patent and the common general knowledge. In light of evidence given by Dr Oppenheim in other proceedings (to which his attention was drawn in the concurrent session) I am satisfied that these difficulties were overstated in his affidavit evidence. In final submissions Apotex placed little (if any) reliance upon this evidence and it is therefore not necessary for me to say any more about it.
12 A/Prof. King is a consultant neurologist with the Royal Melbourne Hospital where he consults with patients on a range of neurological conditions. He also holds various other positions including that of Associate Professor at the University of Melbourne’s Centre for Neurosciences. He is a member of the Australia and New Zealand Association of Neurologists, the Royal Australasian College of Physicians and the American Academy of Neurology. A/Prof. King has worked in the field of neurology in Australia for 38 years. His evidence was directed toward Fields 1, 2 and 3.
13 A/Prof. Lynden Roberts is a practising consultant rheumatologist at the Monash Medical Centre. He is also a Member of the National Steering Committee Rheumatoid Arthritis Clinical Registry. A/Prof. Roberts is also a Member of the National Gout Group Steering Committee for Australia and New Zealand and is the Continuing Professional Development Lead of the Royal Australasian College of Physicians’ Rheumatology Specialist Training Committee. A/Prof. Roberts’ evidence was directed to Fields 1, 2 and 3.
14 Professor Paul Harnett is a medical oncologist who specialises in the management of breast cancer and gynaecological cancers. Professor Harnett holds the positions of Director, the Crown Princess Mary Cancer Centre, Westmead where he is a Senior Staff Specialist in Medical Oncology; Director, Sydney West Translational Cancer Research Centre and Director, Sydney West Cancer Network, Sydney West and Nepean Blue Mountains Local Health Districts. He also holds the positions of Consultant Medical Oncologist to the New South Wales Breast Cancer Institute, Consultant Medical Oncologist Westmead Centre for Gynaecologic Cancer and Clinical Professor of the University of Sydney. Professor Harnett’s evidence was directed to Fields 1, 2 and 3.
15 Professor Stephen Gibson is a psychologist and research academic with a specialisation in pain. He is a Professor in the Department of Medicine at the University of Melbourne, the Director of Clinical Research at the National Ageing Research Institute and the Director of Research at the Caulfield Pain Management and Research Centre in Victoria. Professor Gibson has over 25 years’ experience in clinical pain research. He also works in clinical practice as a clinical psychologist at various multidisciplinary pain management clinics in Victoria. His evidence was directed to Fields 1, 2 and 3.
16 A/Prof. Darren Roberts is a clinical pharmacologist, nephrologist and clinical researcher. He is a Fellow of the Royal Australasian College of Physicians with subspecialisation in clinical pharmacology and nephrology. He has expertise in clinical trials and clinical toxicology. He has participated in multiple investigator-initiated university-based and hospital-based clinical trials in Australia, the United Kingdom and Sri Lanka. This included working as a clinical sub-investigator in various Phase 3 clinical trials. Some of the clinical trials in which he has been involved have concerned treatments for iron deficiency and auto-immune disease. A/Prof Roberts’ evidence was directed to Fields 3, 4 and 5.
17 Dr Oppenheim is a formulation chemist and principal of his own consultancy. Dr Oppenheim provides consultancy services to the therapeutic goods and food industries. His work in this area is primarily with clients that market and manufacture prescription pharmaceutical products and over-the-counter products, complementary medicines, dietary supplements, therapeutic devices and foods. He has over 40 years’ experience in this field. Dr Oppenheim’s evidence was directed to Fields 4 and 5.
18 Dr Best is an organic chemist who has worked in the field of chemistry in Australia for 26 years. He is the Managing Director of EpiChem Pty Ltd (“EpiChem”) which he founded in 2003. EpiChem supplies services in synthetic and medicinal chemistry to the drug discovery and pharmaceutical industries. EpiChem also undertakes in-house and collaborative drug discovery programs. Dr Best’s evidence was directed to Fields 4 and 5.
19 Ms Smith is a partner of Ashurst Australia, solicitors for Apotex. Her affidavit exhibited (inter alia) the product information documents published by Pfizer in relation to Lyrica and copies of correspondence between Pfizer and the United States Food and Drug Administration (“FDA”) relating to Lyrica.
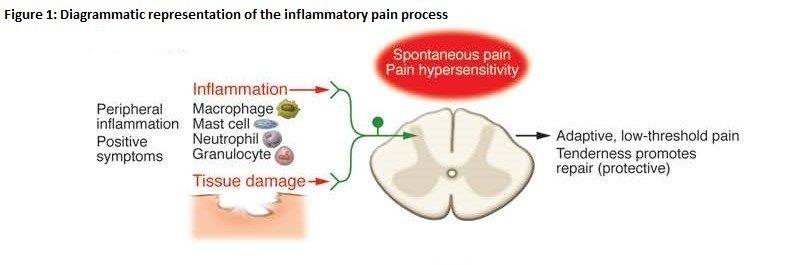
20 Professor Schug is a pain specialist and the Director of Pain Medicine at the Royal Perth Hospital. Professor Schug is also Chair of Anaesthesia at the University of Western Australia. His academic appointments have had a significant clinical component, initially in anaesthesia, intensive care and pain medicine, with increasing emphasis on pain medicine. He has almost 30 years’ experience in the field of pain medicine. His evidence was directed toward Fields 1, 2 and 3.
Professor Macdonald Christie (P)
21 Professor Christie is a Professor of Pharmacology at the University of Sydney who also has expertise in pain. His research interests include the neurobiology of pain, analgesia, and pain-relieving drugs and therapies with a focus on neuroplasticity, neuropathic pain, visceral pain, analgesia and spinal-cord injury. Professor Christie’s research work includes a number of projects aimed at identifying neural and molecular mechanisms underlying the sensitisation responsible for chronic pain states in animal models, and at developing novel peptide drugs that target those mechanisms. Professor Christie’s evidence was directed to Fields 1, 2 and 3 . He also gave some oral evidence directed to Field 5.
Professor Geoffrey Littlejohn (P)
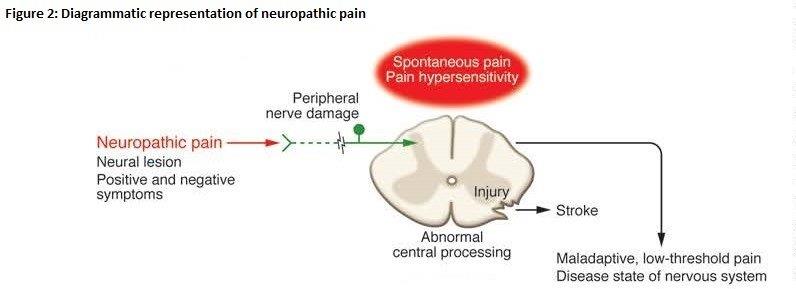
22 Professor Littlejohn is a rheumatologist. He is a Professor in the Department of Medicine at Monash University, Emeritus Director of Rheumatology at Monash Health and Adjunct Professor at Edith Cowan University, Perth. He also provides clinical and consulting services in the field of rheumatology. He has been the principal investigator in a large number of clinical trials at Monash Health using targeted pharmacological agents to treat rheumatoid arthritis, psoriatic arthritis, osteoarthritis, fibromyalgia and gout. Professor Littlejohn’s evidence was directed to Fields 1, 2 and 3.
Professor Christopher Easton (P)
23 Professor Easton is an organic chemist and a Professor at the Research School of Chemistry, Australian National University. He has been involved in various research projects undertaken in collaboration with various pharmaceutical and chemical companies. Professor Easton’s evidence directed to Fields 4 and 5.
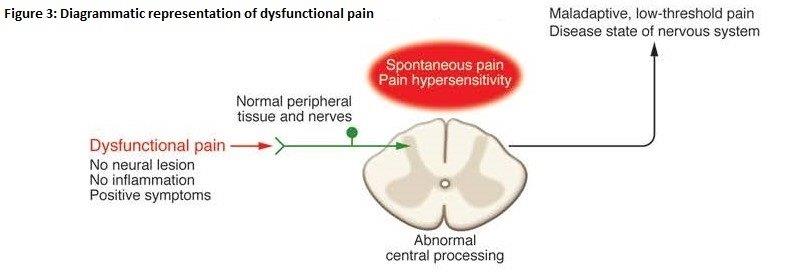
24 Dr Morella is a chemist. He has extensive experience in the fields of drug delivery and pharmaceutics and has worked as both a biotechnology scientist and consultant to industry. Dr Morella has been a member of the various advisory groups at the University of South Australia including the Medical and Pharmaceutical Biotechnology Advisory Group, the Pharmaceutical Science Curriculum Development Advisory Group, the Centre for Drug Formulation and Delivery Advisory Group, and the Pharmaceutical Sciences Advisory Committee. His evidence was directed to Fields 4 and 5.
25 A/Prof. Miller is an Associate Professor and Medical Director of the Family Medicine Research Centre (“FMRC”) at the University of Sydney. One of FMRC’s key research programs is the “Bettering the Evaluation And Care of Health” (“BEACH”) program which has been conducted since 1998 and collects data about the conduct of general practice in Australia. He provided an affidavit in which he exhibited a report (“the Beach Report”) that was relied upon by Pfizer to show that pregabalin is prescribed to treat pain associated with a wide variety of medical conditions.
26 There was no dispute between the parties as to the attributes of the skilled addressee. In the present case the subject matter of the Patent concerns the preparation and use of certain pharmaceutical compositions in the treatment of different types of pain (eg. neuropathic pain) including pain associated with different conditions (eg. cancer pain).
27 The skilled addressee is a notional person who may have an interest in using the products or methods of the invention, making the products of the invention, or making products used to carry out the methods of the invention either alone or in collaboration with others having such an interest: see Aristocrat Technologies Australia Pty Ltd v Konami Australia Pty Ltd (2015) 114 IPR 28 at [26].
28 In the present case people with a practical interest in the invention the subject of the Patent would include pharmacologists, clinicians (pain specialists and other clinicians who treat pain), pharmaceutical formulators and organic chemists. Thus, the relevant fields of expertise include pharmacology, pharmaceutical formulation, pain medicine, and organic chemistry.
29 The notional skilled addressee will possess the common general knowledge in each of these fields in so far as they are relevant to the subject matter of the Patent. In the present case it will include the background knowledge and experience available to all those persons engaged in the particular fields that I have identified including publications to which they would refer as a matter of course: see Minnesota Mining and Manufacturing Company v Beiersdorf (Australia) Ltd (1980) 144 CLR 253 at 292 per Aickin J; ICI Chemicals & Polymers Ltd v Lubrizol Corporation Inc (1999) 45 IPR 577 at [112] per Emmett J.
30 There was no issue between the parties as to the state of the common general knowledge at the priority date. Some of the relevant scientific and medical knowledge referred to in the evidence was first acquired by the experts after the priority date (particularly in relation to “central sensitisation”) and the following account of the relevant science indicates if this is so. Subject to that qualification, the matters set out in this section of my reasons (which is mostly drawn from Professor Christie’s and Professor Easton’s affidavit evidence) were matters of common general knowledge as at the priority date.
31 Pain is a complex phenomenon that reflects a blend of sensory, cognitive, emotional, and social components. It occurs in the context of fundamental biological events taking place at multiple levels of the “somatosensory system”.
32 The somatosensory system is the part of the nervous system (including the brain) that conveys sensory stimuli, such as sensations of touch, pressure, temperature, proprioception (position) and vibration, arising from the muscles, joints, internal organs, skin and fascia to the brain where it is interpreted.
33 Pain has been defined as “[a]n unpleasant sensory and emotional experience associated with actual and potential tissue damage, or described in terms of such damage” (see International Association for the Study of Pain (“IASP”), Classification of Chronic Pain, IASP Press, 2nd Edition, 1994 at p 210).
34 Descriptions of the underlying biological mechanisms associated with pain are particularly relevant in the preclinical environment where an understanding of pain mechanisms is central to the research and the development of new pain therapies and drug indications.
35 In the clinical setting there is limited availability of diagnostic tools to definitively identify the involvement or contribution of a biological mechanism in a patient presenting with pain and in this setting pain is usually categorised in other ways. Pain may be described in terms of its etiology (ie. the cause or set of causes of a disease or condition), pathology or disease-base (eg. osteoarthritis), duration (eg. acute or chronic), or by the location of the pain (eg. headache, lower back pain).
36 There are various pain mechanisms that are intimately interlinked and more than one mechanism is commonly implicated in the pain experience at any point in time.
37 Nociception is the neural process by which noxious stimuli generate neural signals, the outcome of which can be a range of autonomic responses (such as altered blood pressure and heart rate), reflex withdrawal, and the perception and experience of pain. Nociceptive pain is part of the normal response to threatened or actual physical trauma or injury, and induces a response to the noxious stimulus which is aimed at preventing or minimising injury and/or protecting or healing existing injury.
38 “Nociceptive pain” is a pain sensation initiated by a noxious stimulus and typically subsides when the noxious stimulus ceases or the threat of injury is removed. However, some diseases can generate recurrent noxious stimuli, leading to ongoing nociceptive pain (eg. osteoarthritis). The neurobiological process of nociception commences with the activation of specialised receptors called “nociceptors”.
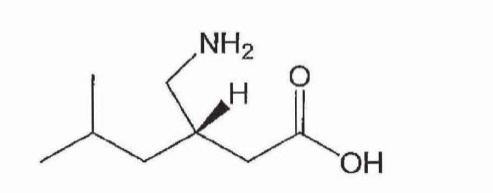
39 Neurons are electronically excitable cells that process and transmit electrical and chemical signals. Nociceptors are located on the peripheral terminals of specialised neurons called “primary sensory neurons” (or “primary afferent neurons”) that innervate peripheral tissue.
40 There are two main classes of primary sensory neurons responsive to intense noxious stimuli: medium diameter, myelinated “Aδ-fibres” and small diameter, unmyelinated “C-fibres”. Related classes of sensory neurons are the myelinated “Aβ-fibres”, responsible for the detection of low threshold, innocuous stimuli such as touch, and “Aα-fibres” which are largely responsible for proprioception.
41 Myelin is a material formed of lipid and protein that acts as an insulating sheath, which allows faster rates of signal transmission along the length of the neuron. Myelinated Aδ-fibres are responsible for the first sharp sensation of pain and reflex withdrawal. Unmyelinated C-fibres are responsible for the subsequent sensation of pain experienced over the following seconds, and provide the brain with further information about the tissue damage, including information about its location, type and severity.
42 The cell bodies of primary sensory neurons are located in the dorsal root ganglia (“DRG”) for neurons that innervate the body, and the trigeminal neural ganglion (“TNG”) for neurons that innervate the face and head. They have both a peripheral and central axonal branch that innervates their target organ/tissue and the spinal cord or brain stem, respectively.
43 Nociceptor peripheral axonal terminals are characterised by specialised, high threshold transducer receptor/ion channel complexes which respond to (or are “gated” by) intense or prolonged noxious stimuli, such as elevated temperature (eg. a burn from a hot iron), chemicals (eg. an acid burn) or mechanical forces such as pressure or shearing (eg. a hammer strike or surgical incision). Some nociceptors respond to different types of noxious stimuli while others respond only to a specific type.
44 Nociceptors convert (or “transduce”) noxious stimuli into electrical signals. When activation by a noxious stimulus causes nociceptor ion channels to open, allowing sodium and calcium cations to enter the peripheral terminal of the sensory neuron, the influx of sodium and calcium cations produce an electrical current which depolarises the terminal membrane. If the noxious stimulation produces a sufficiently strong depolarising current, specialised voltage-gated sodium channels will also open. The opening of these voltage-gated sodium channels increases the flow of sodium ions into the peripheral terminal, causing further depolarisation of the terminal membrane and resulting in the generation of “action potentials”. Action potentials are a brief fluctuation in membrane electrical potential resulting from the rapid opening and closing of voltage-gated sodium ion channels. The intensity and duration of the initiating noxious stimulus is reflected in the frequency of the action potentials.
45 Action potentials generated in the nociceptor peripheral terminals are conducted along the axon of the sensory neuron by successive firing (opening and closing) of voltage-gated sodium channels to the cell body located in the DRG and to the central terminal of the primary sensory neuron located in the dorsal horn of the spinal cord. The dorsal horn of the spinal cord is organised into distinct laminae (layers). Aδ-fibres project to lamina I and the deeper lamina II of the dorsal horn, C-fibres project more superficially to lamina I and II, and Aβ-fibres and Aα-fibres project to deep laminae III, IV and V of the dorsal horn.
46 Transfer of input from peripheral nociceptors to secondary neurons located in the dorsal horn of the spinal cord is mediated through direct synaptic contact between the central terminals of the primary (pre-synaptic) and secondary (post-synaptic) neurons. A “synapse” is the specialised junction at which action potentials are transmitted by chemical transmission from the axon terminal of one neuron (the pre-synaptic terminal) and received by the terminal of one or more target neurons (post-synaptic terminal).
47 Depolarisation of the pre-synaptic terminal by incoming action potentials leads to the opening of voltage-gated calcium channels (“VGCCs”, principally neural “N-type” VGCCs) located in the pre-synaptic membrane and triggers an influx of calcium ions into the pre-synaptic terminal. This in turn leads to the release of the excitatory neurotransmitter “glutamate”, the neuropeptides “substance P” and calcitonin gene-related peptide (“CGRP”), from vesicles located within the pre-synaptic terminal. The neurotransmitters and neuropeptides cross the synaptic cleft (the space separating the pre-synaptic and post-synaptic neurons) and bind to receptors located on the post-synaptic membrane of the secondary neuron.
48 Glutamate binds at post-synaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (“AMPA”) receptors, N-methyl-D-aspartate (“NMDA”) receptors, and kainate receptors, which are all “ionotropic” receptor ion channels. Ionotropic receptors directly gate ion channels when a ligand binds to the receptor located on the ion channel. There are also various “metabotropic” glutamate receptors. Metabotropic receptors indirectly gate ion channels.
49 Substance P binds at post-synaptic, metabotropic neurokinin-1 (“NK-1”) receptors. CGRP binds to a metabotropic calcitonin receptor-like receptor (“CRL”) and a receptor activity-modifying protein (“RAMP”) complex located on the post-synaptic terminal.
50 Secondary neurons are responsible for localising, integrating and distributing signals from peripheral neurons. The majority of sensory inputs are distributed directly from secondary neurons in the spinal cord dorsal horn to the periphery and are responsible for (inter alia) muscle reflexes. Only a small percentage (about 10%) of all sensory input is transmitted from secondary neurons to the higher regions of the brain where ultimately it may be perceived as pain. Not all nociceptive sensory input is transmitted to the brain. The central nervous system (“CNS”) contains mechanisms that can block or modify the transmission of signals before they ascend to the brain. One way this is achieved is by the mechanism called “descending modulation”.
51 During normal nociceptive transmission, binding by glutamate to AMPA ionotropic receptor ion channels located on the post-synaptic central terminal of secondary neurons leads to an influx of sodium ions, and a lesser efflux of potassium ions. This has the effect of depolarising the secondary neuron, leading to the generation of a new action potential in the secondary neuron.
52 In the basal, or normal resting state, ionotropic NMDA receptor ion channels located on post-synaptic terminals of secondary neurons are each blocked by an extracellular magnesium ion. This means that in most neurons in their normal resting state glutamate binding to the NMDA receptor does not result in significant influx of calcium ions across the post-synaptic membrane.
53 However, if depolarisation of the postsynaptic terminal is sufficiently strong (eg. in response to the influx of calcium ions and sodium ions through activation of ionotropic AMPA receptor ion channels) sodium ions and calcium ions will flow in through the NMDA receptor ion channels. This further depolarises the post-synaptic terminal.
54 “Neuroplasticity” refers to the capacity of the nervous system to adapt and reorganise itself. Increased depolarisation of the post-synaptic terminal results in increased excitability, and calcium-dependent neuroplasticity associated with activation of intracellular protein kinases, that can produce a persistent increase in postsynaptic excitability and synaptic responses in the secondary neuron.
55 The activity-dependent effects of NMDA receptor activation are a major component of the underlying mechanisms associated with the phenomenon of central sensitisation.
56 Inflammation can give rise to pain when a stimulus (usually noxious), infection, or disease results in tissue damage that triggers the body’s inflammatory response. The inflammatory response involves the initiation of a complex series of biochemical and cellular events aimed at repairing the damaged tissue.
(Source: Extracted from CJ Woolf “What is this thing called pain?” (2010) 120(11) Journal of Clinical Investigation (“Woolf (2010)”).
57 Tissue injury results in the release of numerous chemical mediators from peripheral nerve terminals, ruptured cells, and immune cells that are recruited to the site of injury. Some inflammatory mediators (eg. protons, bradykinin, adenosine triphosphate) can directly activate nociceptors, leading to spontaneous nociceptive pain. Other mediators modify the response properties of primary afferent neurons to subsequent stimuli. For example, this can occur by increasing the sensitivity of transducer receptors located in the nociceptor peripheral terminals so that they are activated by lower intensity noxious stimuli, and increasing the intensity of the response by altering the activation threshold of voltage-gated ion channels. The phenomenon of increased responsiveness and reduced nociceptive threshold in the peripheral terminal of primary sensory neurons is known as “peripheral sensitisation” and is responsible for the increased sensitivity and tenderness of injured tissue that promotes protective behaviour toward the injured site.
58 Inflammatory pain typically resolves when the underlying injury has healed. However, some inflammatory pain conditions (eg. rheumatoid arthritis) can give rise to an ongoing inflammatory response associated with chronic pain.
59 Nociceptive pain and inflammatory pain are part of the normal function of the body’s protective and healing response.
60 The IASP currently defines “neuropathic pain” as “pain caused by a lesion or disease of the somatosensory nervous system” (see Jensen et al, “Commentary: A new definition of neuropathic pain” (2011) 152 Pain at 131). However, at the priority date, the IASP defined neuropathic pain as “pain initiated or caused by a primary lesion or dysfunction in the nervous system.”: see IASP, op. cit at p 212. The deletion of the reference to “dysfunction” has led many experts in the field to treat dysfunctional pain as a pain classification different from neuropathic pain.
(Source: Extracted from Woolf (2010))
61 Neuropathic pain is considered to be a pathological condition as it provides no protective or healing benefit. It can be caused by a range of different diseases and lesions to the nervous system. Peripheral neuropathic pain results from damage to the primary sensory neurons (eg. as a result of a surgical incision), or from neuropathies such as postherpetic neuralgia, diabetic peripheral neuropathy and trigeminal neuralgia. In contrast, central neuropathic pain results from nerve damage within the spinal cord or brain and commonly arises due to spinal cord injury, brain trauma, stroke or multiple sclerosis.
62 There was some evidence given by A/Prof. Lynden Roberts to suggest that neuropathic pain will always be a chronic pain condition. Ultimately however, he accepted that this may reflect too narrow a view of neuropathic pain, which can describe both acute and chronic pain conditions.
63 Dysfunctional pain is a maladaptive condition caused by the abnormal functioning of the nervous system in the absence of any identifiable damage to the CNS, disease or condition which may account for the pain. Dysfunctional pain is an example of pain as a disease condition in itself requiring treatment in its own right. Fibromyalgia is an example of dysfunctional pain syndrome. Fibromyalgia is a maladaptive pain syndrome which manifests as diffuse pain throughout the body with widespread tenderness over discrete areas of the body, but with no identifiable precipitating pathological or physical condition.
(Source: Extracted from Woolf (2010))
64 The perception of pain is not limited to the brain’s processing of ascending signals received from the peripheral sensory apparatus (nociceptors) in response to noxious stimulation. Peripheral and central changes which affect pain thresholds and responses, referred to as “peripheral sensitisation” and “central sensitisation” respectively, can modify the way in which stimuli are detected and pain is experienced. In addition, pain perception can be modulated by regulation of the transmission of signals from the spinal cord to the brain, and also within the brain itself. This enables the brain to amplify or reduce the sensation of pain. This is a complex process that is in turn strongly influenced by psychological and social factors. The mechanism by which the brain regulates the transmission of pain signals from the spinal cord to the brain is referred to as “descending modulation”.
65 The brain can directly modulate (amplify or depress) incoming sensory signals and thus influence pain perception. The descending pain modulatory circuit takes inputs from multiple areas of the brain (including, but not limited to, the hypothalamus, the amygdala and the rostral anterior cingulate cortex), feeds them into the midbrain periaqueductal gray (“PAG”) region, which in turn sends outputs to the rostral ventromedial medulla (“RVM”). In essence, the action of descending modulation can be characterised as a complex feedback control system in which neurons within the RVM project to the spinal or medullary dorsal horns to directly or indirectly enhance or diminish nociceptive signals and thereby facilitate, as well as inhibit, pain.
66 Peripheral sensitisation refers to the reduced activation threshold and amplified responsiveness of nociceptors to stimulation of their receptive fields which occurs after initiation of the inflammatory response following tissue injury. Peripheral sensitisation is characterised by primary hyperalgesia localised at the site of injury or trauma. The term “hyperalgesia” refers to the amplification of the pain sensation following stimulation by a stimulus which is normally painful. “Primary hyperalgesia” is characterised by enhanced pain in response to heat and mechanical stimuli at the site of injury. “Secondary hyperalgesia” refers to an amplified pain response evoked outside the receptive field of the nerve or nerves directly stimulated at the point of injury.
67 Peripheral sensitisation is an adaptive process and is part of the body’s normal protective response to physical trauma or injury which induces protective behaviour toward the site of injury that promotes healing. Peripheral sensitisation typically arises during inflammation, and is fundamentally associated with increased peripheral sensitivity of the nociceptive system. There must usually be some ongoing peripheral stimulation by inflammatory mediators for it to persist.
68 At a cellular level, peripheral sensitisation involves increased nociceptor sensitivity arising from a decrease in the activation threshold of normally high-threshold nociceptors by inflammatory mediators (eg. bradykinin, prostaglandins and NGF). As a result, normally high threshold nociceptors, which normally would respond only to intense noxious stimuli, can be activated by lower intensity stimuli. Peripheral sensitisation also results in increased excitability of voltage gated ion channels at nociceptor terminals which in turn modifies the activation threshold of the ion channel, resulting in an increased number of action potentials being generated. The net result of these adaptive changes to the nociceptive system is primary hyperalgesia.
69 Central sensitisation is a phenomenon that was discovered in 1983 by a neurophysiologist named Clifford Woolf. Central sensitisation is a form of neuroplasticity leading to dynamic changes in the properties and function of neurons in the CNS, and amplified pain processing in the CNS, which ultimately manifests as pain hypersensitivity and/or spontaneous pain in the absence of sensory input. It is an important component of inflammatory, neuropathic and dysfunctional pain.
70 As at the priority date, the term central sensitisation referred to so-called “activity dependent neuronal plasticity” triggered by the increased activity evoked in secondary neurons in the spinal cord dorsal horn in response to intense, repeated or sustained noxious stimulus. Activity dependent neuronal plasticity or central sensitisation is characterised by increased sensitivity manifesting as enhanced responsiveness to noxious stimuli (hyperalgesia) and a reduction in sensory threshold such that non-noxious stimuli evoke pain (allodynia). Activity dependent central sensitisation can arise very quickly (within seconds to minutes) following intense, sustained or recurring nociceptive input.
71 An important consequence of central sensitisation is that the sensation of pain is no longer reflective of the presence, duration or intensity of noxious peripheral stimuli. The level of central sensitisation elicited in any individual subjected to a particular noxious stimulus is variable. However, in activity dependent (adaptive) central sensitisation the level of sensitisation is commonly linked to the severity and duration of the stimulus, so that the more severe and prolonged the stimulus, the greater the central sensitisation.
72 Two of the key mechanisms by which activity dependent central sensitisation arises are shown in simplified form in Figure 4. An increase in N-type VGCC activity (N-type VGCC hyperactivity) in the primary afferent synaptic transmitting terminals of Aδ-fibres and C-fibres (circled in Figure 4) amplifies signals transmitted from those fibres to the secondary neuron which transmits sensory signals to the brain, where ultimately they may be interpreted as pain. This central amplification enhances the pain response to noxious stimuli in amplitude, duration and spatial extent and causes a normally painful stimulus to be perceived as more painful. This condition is known as hyperalgesia.
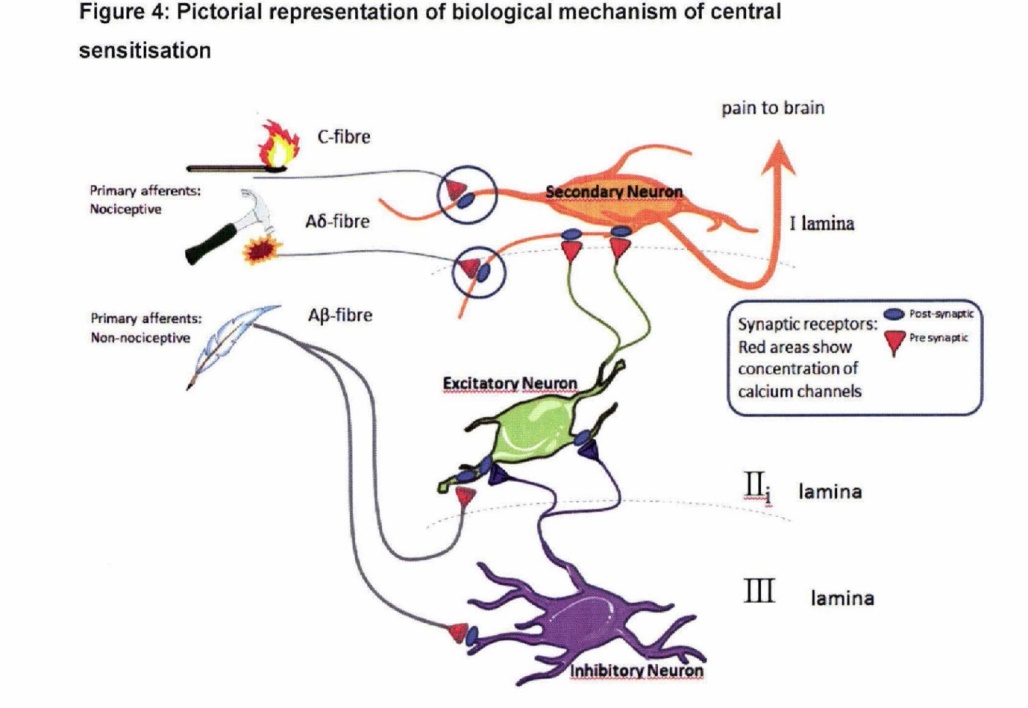
(Source: Affidavit of Professor Macdonald James Christie dated 7 November 2014)
73 As a result of central sensitisation, signals from low threshold myelinated Aβ-fibres (which are normally involved in the sensation of touch) may also be transmitted to the secondary pain transmitting neurons via excitatory neurons, as depicted in Figure 4. This transmission is usually prevented by inhibitory neurons. An increase in N-type VGCC activity in the pre-synaptic terminals of the Aβ-fibres stimulates the excitatory neurons, and at the same time down-regulates the inhibitory neurons, causing the excitatory neurons to transmit signals to the secondary neurons in the dorsal horn. An increase in N-type VGCC activity in pre-synaptic terminals of the excitatory neurons can further amplify this effect. The consequence is that a normally innocuous stimulus such as ordinary touch can give rise to stimulation of the secondary neurons, which transmit such sensory signals to the brain where it is interpreted as pain. This mechanism causes a normally non-painful stimulus to be interpreted as painful. This condition is known as allodynia.
74 Central sensitisation can be viewed as an “adaptive” phenomenon in the sense that it contributes to the normal protective and healing responses by increasing sensitivity to both noxious and non-noxious stimuli at the site of injury (primary hyperalgesia and allodynia) and the surrounding body area (secondary hyperalgesia and allodynia). While primary hyperalgesia and allodynia can be caused by peripheral sensitisation or central sensitisation, secondary hyperalgesia and allodynia are the result only of central sensitisation.
75 An example of adaptive central sensitisation is the transient central sensitisation that occurs during inflammation, which enhances the body’s protective behaviours to promote healing, but which subsides when the underlying pathological condition has resolved.
76 Central sensitisation is a complex phenomenon that is still not fully understood. Since the priority date, the body of knowledge surrounding the phenomenon of central sensitisation has expanded and it is now apparent that there are other complex pathways and mechanisms that can heighten the sensitivity of the CNS. These forms of central sensitisation can be long-lasting, or in some cases permanent, and are reflective of maladaptive central sensitisation associated with pathological pain conditions common in neuropathic and dysfunctional conditions. Maladaptive central sensitisation can occur in the absence of a noxious stimulus, or may develop after an initial stimulus has resolved.
77 Although the mechanisms involved in central sensitisation are complex, N-type VGCC hyperactivity is a common feature of central sensitisation. The reduction in hyperactivity in N-type VGCC is therefore a potential way to reduce central sensitisation.
78 Pregabalin has the chemical name (S)-3-(aminomethyl)-5-methylhexanoic acid and the following chemical structure:
79 Pregabalin was originally developed as a gamma-aminobutyric acid (“GABA”) agonist to activate GABA receptors. However, it was established after the priority date that pregabalin does not bind to GABA receptors.
80 Whilst the mechanisms of action of pregabalin were not fully understood at the priority date, it was known that pregabalin binds with high affinity to alpha-2-delta (α2δ) auxiliary protein subunits of pre-synaptic VGCCs, particularly N-type VGCCs, located on the central terminals of primary sensory neurons in the dorsal horn of the spinal cord.
81 It is now understood that by binding to α2δ subunits pregabalin acts as a “calcium channel modulator” that produces analgesic effects by the modulation of VGCCs and relieving pain associated with central sensitisation.
82 VGCCs are comprised of combinations of subunits as depicted in Figure 6. The α1 subunit spans the cellular membrane and forms a channel through which calcium ions may pass. The α1 subunit is associated with three auxiliary (accessory) subunits: the cytosolic (intracellular) β subunit; the transmembrane γ subunit; and the transmembrane α2δ subunit. The α2δ subunit stabilises the location of the α1 subunit within the membrane and also modulates its function.
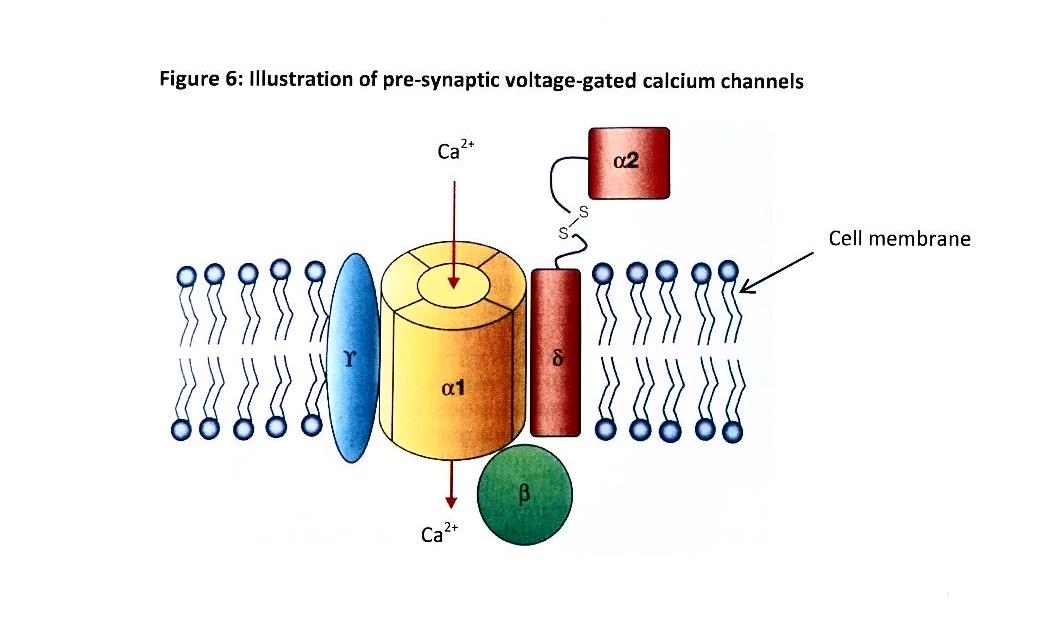
(Source: Extracted with adaptations from Wall & Melzack’s Textbook of Pain, 6th Edition, 2013 at p 496)
83 As previously explained, release of excitatory neurotransmitters from the presynaptic terminal is mediated by calcium ion influx through VGCCs. Thus, in response to cell membrane depolarisation, resulting from incoming action potentials, the channel-forming α1 subunit allows calcium ions to enter the cell. This calcium ion influx triggers the pre-synaptic release of the excitatory neurotransmitter glutamate, as well as substance P and CGRP, particularly under conditions of intense nociceptive stimulation. When glutamate is released by this mechanism, it is available to bind to post-synaptic receptors (including AMPA and NMDA receptors) and activate secondary neurons within the spinal cord dorsal horn.
84 It is now known that expression of the α2δ subunit is increased (or upregulated) in models of pain that involve central sensitisation. This increase in α2δ expression is thought to increase VGCC activity in nerve terminals, leading to hyperexcitability. Under these conditions, pregabalin may normalise VGCC activity and reduce central sensitisation by inhibiting or reducing the release of glutamate and other excitatory neurotransmitters which modulates the transmission of pain signals to the cortex. There is also some evidence to suggest that pregabalin may act in other ways that might inhibit excessive synapse formation that contributes to sensitisation, and also activate descending pain modulation which produces analgesia. It has been observed that the regions of the highest concentration of α2δ expression in the CNS (in particular the PAG region) are known to be implicated in the descending modulation of pain.
85 “Organic chemistry” is the chemistry of carbon compounds. The smallest unit of any compound is known as a molecule. A molecule is in turn made up of atoms. The complexities of organic chemistry derive from the ability of carbon atoms to form molecules in which chains or rings of carbon atoms form the “skeleton” of a three dimensional molecular structure.
86 The compounds of organic chemistry are generally compounds of carbon (“C”) and hydrogen (“H”), but may also include a range of other elements such as oxygen (“O”), nitrogen (“N”) and fluorine (“F”). These elements can be linked together by covalent bonds to form molecules. Covalent bonds result from the sharing of electrons between atoms. Organic molecules are generally made up by covalent bonds between carbon atoms, although oxygen and nitrogen atoms can also be involved in forming linking parts of molecular structures. Carbon is able to form up to four covalent bonds with other atoms. The ability to form multiple covalent bonds enables these elements to form linking parts of molecular structures.
87 A molecule can have the same chemical formula (representing the number and types of the atoms of the compound) but different structures. This means they have different chemical and physical properties. This is because the linkages in the compounds are different. Compounds which have the same chemical formula but different structures are referred to as “structural isomers”.
88 Where compounds of the same chemical formula have the same sequence of bonded atoms but differ in the orientation of atoms in space, they are referred to as “stereoisomers”. Stereoisomers can occur when the four atoms (or groups of atoms) to which a carbon is bonded in a molecule are different. In that case, there are two ways in which the atoms or groups arrange around the carbon atom. These arrangements are non-superimposable mirror images of each other in the same way that a left and right hand are non-superimposable mirror images. The carbon atom in this type of arrangement is called “chiral” (which derives from the Greek for “hand”). Each chiral carbon atom is called a “chiral centre”. Figure 3 is a diagram that represents chiral carbon atoms in compounds which are non-superimposable mirror images of each other, shown as Figure 3(a) and Figure 3(b).
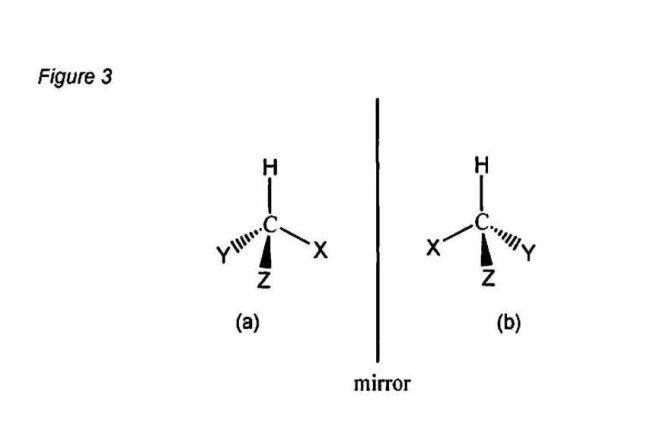
(Source: Affidavit of Professor Christopher John Easton dated 6 November 2014)
89 When representing a chiral structure, a third dimension is required to fully represent the structure. Conventionally two of the bonds are represented in the plane of the page by using plain lines (ie. the bonds with the “H” and “X” groups in Figures 3(a) and 3(b)). Bonds represented using a wedge-shaped symbol indicate that the bond projects above the plane of the page (ie. the bond to the “Z” group in Figures 3(a) and 3(b)). Bonds represented using dashes or broken lines indicate that the bond projects below the plane of the page (ie. the bond to the “Y” group in Figures 3(a) and 3(b)). In a compound that includes a single chiral centre there are only two possible stereoisomers. Both of these stereoisomers are non-superimposable mirror images of each other, as shown in Figures 3(a) and 3(b).
90 Stereoisomers which are non-superimposable mirror images are called “enantiomers”. Enantiomers have many of the same chemical and physical properties, such as chemical reactivity, melting point, boiling point, density and solubility. An exception is when polarised light is passed through a solution containing only one of the two enantiomers, the plane of polarised light is rotated either in a clockwise or anticlockwise direction. When two enantiomers have been separated from each other it is therefore possible to distinguish between them by their respective effect on the rotation of polarised light.
91 The enantiomer that causes the plane of polarised light to rotate in a clockwise direction is referred to as the “(+)” enantiomer. The enantiomer that causes anticlockwise rotation is referred to as the “(-)” enantiomer. Where a sample containing an equal mixture of two enantiomers of a particular compound is tested in this way, there will be no rotation of the plane of polarised light because the rotation caused by one enantiomer is cancelled by the rotation caused by the opposite enantiomer. An equal mixture of the two enantiomers is called a “racemic” mixture or a “racemate”. A racemic mixture is typically denoted “(±)”.
92 The usual system of assigning absolute stereochemistry about chiral centres involves designating each chiral carbon atom “(R)” (from the Latin “rectus”) or “(S)” (from the Latin “sinister”) as follows. An order of priority is assigned to the four atoms or groups bonded to the chiral carbon (based on rules that rank each of the atoms or groups bonded to the carbon). The arrangement of atoms or groups will be right-handed or left-handed depending on how the priority of the atoms or groups works out. The designation of “(R)” or “(S)” denotes the absolute stereochemistry at the chiral centre in the compound in accordance with these rules. Whether an enantiomer is (R) or (S) cannot be determined merely from knowing whether it is (+) or (-).
93 Racemic mixtures may be denoted as “(RS)” indicating that the compound contains equal amounts of the (R) and (S) enantiomers, “(±)” indicating that the compound contains equal amounts of the (+) and (-) enantiomers, or with the prefix “rae” as an abbreviation for “racemic”. If a chemical compound is described by its chemical name alone (ie. the stereochemistry is not specified), the term, by definition, includes all compounds of that chemical without being limited by particular stereochemistry.
94 When a molecule has two chiral centres, there are usually four possible stereoisomers. In the normal case, when the stereoisomers are assigned their absolute stereochemistry, the possibilities are as follows: (RR), (RS), (SR) and (SS). Among these four stereoisomers, the (RR) and (SS) isomers are non-superimposable mirror images of each other and are enantiomers in the same way that the (R) and (S) stereoisomers are enantiomers in the case of a molecule with a single chiral centre. Likewise, the (RS) and the (SR) stereoisomers are a pair of non-superimposable mirror image forms and thus they are also enantiomers. Again, the enantiomers have identical chemical and physical properties other than in relation to the rotation of polarised light and interaction with chiral systems.
95 Stereoisomers that are not enantiomers are referred to as “diastereomers”. Each diastereomer will generally have different chemical and physical properties.
THE PRINCIPLES OF CONSTRUCTION
96 There was no dispute between the parties as to the relevant principles of construction. These were summarised by the Full Court (Hill, Finn and Gyles JJ) in Jupiters Ltd v Neurizon Pty Ltd (2005) 222 ALR 155 at [67]:
There is no real dispute between the parties as to the principles of construction to be applied in this matter although there is some difference in emphasis. It suffices for present purposes to refer to the following:
(i) the proper construction of a specification is a matter of law: Décor Corporation Pty Ltd v Dart Industries Inc (1988) 13 IPR 385 at 400;
(ii) a patent specification should be given a purposive, not a purely literal, construction: Flexible Steel Lacing Co v Beltreco Ltd (2000) 49 IPR 331; [2000] FCA 890 at [81] (Flexible Steel Lacing); and it is not to be read in the abstract but is to be construed in the light of the common general knowledge and the art before the priority date: Kimberley-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1; 177 ALR 460; 50 IPR 513; [2001] HCA 8 at [24];
(iii) the words used in a specification are to be given the meaning which the normal person skilled in the art would attach to them, having regard to his or her own general knowledge and to what is disclosed in the body of the specification: Décor Corporation Pty Ltd at 391;
(iv) while the claims are to be construed in the context of the specification as a whole, it is not legitimate to narrow or expand the boundaries of monopoly as fixed by the words of a claim by adding to those words glosses drawn from other parts of the specification, although terms in the claim which are unclear may be defined by reference to the body of the specification: Kimberley-Clark v Arico at [15]; Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610; Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 at 478; the body of a specification cannot be used to change a clear claim for one subject matter into a claim for another and different subject matter: Electric & Musical Industries Ltd v Lissen Ltd [1938] 4 All ER 221 at 224–5; (1938) 56 RPC 23 at 39;
(v) experts can give evidence on the meaning which those skilled in the art would give to technical or scientific terms and phrases and on unusual or special meanings to be given by skilled addressees to words which might otherwise bear their ordinary meaning: Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479 at 485–6 (Sartas No 1 Pty Ltd); the court is to place itself in the position of some person acquainted with the surrounding circumstances as to the state of the art and manufacture at the time (Kimberley-Clark v Arico at [24]); and
(vi) it is for the court, not for any witness however expert, to construe the specification; Sartas No 1 Pty Ltd at 485–6.
97 The Patent commences with a description of the background to the invention. Relevantly, the Patent states at page 1, lines 4 to 13:
The present invention is the use of analogs of glutamic acid and gamma- aminobutyric acid (GABA) in pain therapy, as the compounds exhibit analgesic/antihyperalgesic action. Advantages of the use of the compounds includes the finding that repeated use does not lead to tolerance nor is there a cross-tolerance between morphine and the compounds.
The compounds of the invention are known agents useful in antiseizure therapy for central nervous system disorders such as epilepsy, Huntington’s chorea, cerebral ischemia, Parkinson’s disease, tardive dyskinesia, and spasticity. It has also been suggested that the compounds can be used as antidepressants, anxiolytics, and antipsychotics.
98 There is then a reference to two prior patent specifications, WO 92/09560 (“the 92 Specification”) and WP 93/23383 (“the 93 Specification”). In closing submissions Apotex accepted that these specifications were “incorporated by reference”.
99 The Patent includes a summary of the invention which relevantly states at page 1, line 17 – page 2, line 13:
The instant invention is a method of using a compound of Formula I below in the treatment of pain, especially for treatment of chronic pain disorders. Such disorders include, but are not limited to, inflammatory pain, postoperative pain, osteoarthritis pain associated with metastatic cancer, trigeminal neuralgia, acute herpetic and postherpetic neuralgia, diabetic neuropathy, causalgia, brachial plexus avulsion, occipital neuralgia, reflex sympathetic dystrophy, fibromyalgia, gout, phantom limb pain, burn pain, and other forms of neuralgic, neuropathic, and idiopathic pain syndromes.
According to a first aspect, the invention provides a method for treating pain comprising administering a therapeutically effective amount of a compound of Formula I:
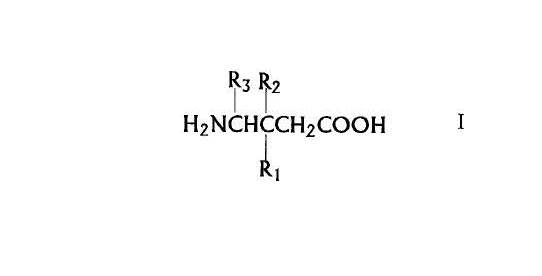
or a pharmaceutically acceptable salt diastereomer or an enantiomer thereof wherein
R1 is a straight or branched alkyl of from 1 to 6 carbon atoms, phenyl, or cycloalkyl of from 3 to 6 carbon atoms;
R2 is hydrogen or methyl; and
R3 is hydrogen, methyl, or carboxyl
to a mammal in need of said treatment.
Preferred compounds of the invention are those according to claim 1 wherein R3 and R2 are hydrogen, and R1 is -(CH2)0-2-iC4H9 as an (R), (S), or (R,S) isomer.
The more preferred compounds of the invention are (S)-3-(aminomethyl)-5- methylhexanoic acid and 3-aminomethyl-5-methyl-hexanoic acid.
A second aspect of the invention is then described which also refers to Formula I but “for the manufacture of a medicament for the treatment of pain” rather than a method of treatment.
100 It is convenient at this point to refer to the claims. The Patent contains 32 claims. Claims 1 to 15 define methods of treatment. Claim 1 is to:
A method for treating pain comprising administering a therapeutically effective amount of a compound of Formula I
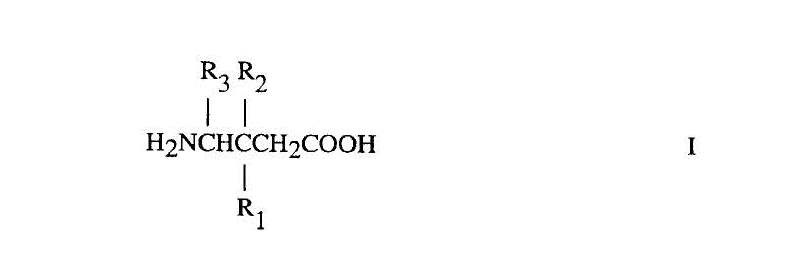
or a pharmaceutically acceptable salt, diastereomer, or enantiomer thereof wherein
R1 is a straight or branched alkyl of from 1 to 6 carbon atoms, phenyl, or cycloalkyl of from 3 to 6 carbon atoms;
R2 is hydrogen or methyl; and
R3 is hydrogen, methyl, or carboxyl to a mammal in need of said treatment.
101 There is an issue between the parties as to how the words “a method for treating pain” in claim 1 should be interpreted (“Construction Issue 1”). There is also an issue between the parties as to how the reference to “a mammal” in claim 1 and other claims should be interpreted (“Construction Issue 2”). Apotex contends that when the claims are read in the context of the Patent as a whole, the reference to a mammal means a human, and would not encompass a rat. This issue is said by the parties to be relevant to the sufficiency case.
102 Claims 2 to 15 are dependent on claim 1. It is important to note that each of the dependent claims 2 to 15 is expressed to be dependent on claim 1 rather than any other preceding claim.
103 Claims 2 and 3 narrow the class of compound claimed. Claim 2 is for:
2. A method according to Claim 1 wherein the compound administered is a compound of Formula I wherein R3 and R2 are hydrogen, and R1 is
-(CH2)0-2-i C4H9 as an (R), (S), or (R,S) isomer.
Thus, claim 2 is limited to a method of treatment that uses a compound of Formula I in which R1, R2 and R3 consist of particular molecules and in which the compound is in the form of an (R), (S) or (R,S) isomer.
104 Claim 3 is for:
3. A method according to Claim 1 wherein the compound administered is named (S)-3-(aminomethyl)-5-methylhexanoic acid and 3-aminomethyl-5-methyl-hexanoic acid.
105 The reference to “(S)-3-(aminomethyl)-5-methylhexanoic acid” is the (S)-enantiomer of “3-(aminomethyl)-5-methyl-hexanoic acid” which I shall for convenience refer to simply as “the 3-amino compound”. The presence of the (S) immediately before the reference to the 3-amino compound indicates that claim 3 is here referring to the (S)-enantiomer of the 3-amino compound. For convenience I will refer to this as “the (S) 3-amino compound” or simply as “the (S)-enantiomer”. In some parts of the Patent the 3-amino compound is also referred to as 3-Isobutylgaba and the (S)-enantiomer as S-(+)-3-Isobutylgaba or occasionally abbreviated to “(S)-(+)-IBG”. It is common ground that pregabalin, the active pharmaceutical ingredient in Lyrica, is the (S) 3-amino compound.
106 There is an issue between the parties as to how the second reference to the 3-amino compound in claim 3 should be interpreted (“Construction Issue 3”). Apotex contends that it refers to the racemate of the 3-amino compound. Pfizer contends that, read in context, it is a reference to the (S)-enantiomer, since it is the only compound in “(S)-3-(aminomethyl)-5-methylhexanoic acid and 3-(aminomethyl)-5-methyl-hexanoic acid”. Alternatively, Pfizer contends that it refers to any mixture of the (S)-enantiomer and the (R)-enantiomer including both racemic and non-racemic mixtures. Construction Issue 3 is relevant to both false suggestion and utility.
107 Claims 4 to 15 identify particular types of pain that may be treated with the relevant compounds. For example, claim 4 is to “[a] method according to Claim 1 wherein the pain treated is inflammatory pain.” The other types of pain that are specified in claims 5 to 15 are neuropathic pain; cancer pain, postoperative pain; phantom limb pain; burn pain; gout pain; osteoarthritic pain; trigeminal neuralgia pain; acute herpetic and postherpetic pain; causalgia pain; and idiopathic pain.
108 Claims 16 to 18 are Swiss claims that are for the use of the same compounds as are identified in claim 1 to 3 in a medicament for the treatment of pain. Claims 19 to 30 are for uses according to claim 16 for the same pain types that are identified in claims 5 to 15. Claims 31 and 32 resemble omnibus claims. They claim a method of treating pain and the use of a relevant compound by reference to one or more of the examples and accompanying figures as follows:
31. A method for treating pain, substantially as herein described with reference to one or more of the examples but excluding comparative examples.
32. Use of a compound of formula I or a pharmaceutically acceptable salt diastereomer or an enantiomer thereof wherein
R1 is a straight or branched alkyl of from 1 to 6 carbon atoms, phenyl, or cycloalkyl of from 3 to 6 carbon atoms;
R2 is hydrogen or methyl; and
R3 is hydrogen, methyl, or carboxyl, substantially as herein described with reference to one or more of the examples and the accompanying figures.
109 The Patent includes a detailed description of the invention. The Patent states at page 5, lines 9 to 22:
The instant invention is a method of using a compound of Formula I above as an analgesic in the treatment of pain as listed above. Pain such as inflammatory pain, neuropathic pain, cancer pain, postoperative pain, and idiopathic pain which is pain of unknown origin, for example, phantom limb pain are included especially. Neuropathic pain is caused by injury or infection of peripheral sensory nerves. It includes, but is not limited to pain from peripheral nerve trauma, herpes virus infection, diabetes mellitus, causalgia, plexus avulsion, neuroma, limb amputation, and vasculitis. Neuropathic pain is also caused by nerve damage from chronic alcoholism, human immunodeficiency virus infection, hypothyroidism, uremia, or vitamin deficiencies. Neuropathic pain includes, but is not limited to pain caused by nerve injury such as, for example, the pain diabetics suffer from.
The conditions listed above are known to be poorly treated by currently marketed analgesics such as narcotics or nonsteroidal anti-inflammatory drugs (NSAID) due to insufficient efficacy or limiting side effects.
110 The Patent also states at page 6, lines 6 to 10:
The compounds of the present invention can contain one or several asymmetric carbon atoms. The invention includes the individual diastereomers or enantiomers, and the mixtures thereof. The individual diastereomers or enantiomers may be prepared or isolated by methods already well-known in the art.
111 Some further information relevant to formation of the compounds of the invention is then provided. It was not contended by Apotex that this information was insufficient to enable the notional skilled addressee to prepare compounds of the invention. As I mentioned, Apotex accepted that the 92 Specification and the 93 Specification were “incorporated by reference” and that these provide sufficient information to enable the skilled addressee to make the (S)-enantiomer (pregabalin). Though it is questionable whether or not these documents are properly described as incorporated by reference, I am satisfied that they would be read and understood by the skilled addressee as relevant background material and that the information contained in them would enable the skilled addressee to manufacture the (S)-enantiomer (by resolution of the racemate) without undue difficulty.
112 The Patent states at page 7 lines 4 to 27:
The compounds made by the synthetic methods can be used as pharmaceutical compositions as agent in the treatment of pain when an effective amount of a compound of the Formula I, together with a pharmaceutically acceptable carrier is used. The pharmaceutical can be used in a method for treating such disorders in mammals, including human, suffering therefrom by administering to such mammals an effective amount of the compound as described above in unit dosage form.
The pharmaceutical compound, made in accordance with the present invention, can be prepared and administered in a wide variety of dosage forms by either oral or parenteral routes of administration. For example, these pharmaceutical compositions can be made in inert, pharmaceutically acceptable carriers which are either solid or liquid. Solid form preparations include powders, tablets, dispersible granules, capsules, cachets, and suppositories. Other solid and liquid form preparations could be made in accordance with known methods of the art and administered by the oral route in an appropriate formulation, or by a parenteral route such as intravenous, intramuscular, or subcutaneous injection as a liquid formulation.
The quantity of active compound in a unit dose of preparation may be varied or adjusted from 1 mg to about 300 mg/kg daily, based on an average 70-kg patient. A daily dose range of about 1 mg to about 50 mg/kg is preferred. The dosages, however, may be varied depending upon the requirement with a patient, the severity of the condition being treated, and the compound being employed. Determination of the proper dosage for particular situations is within the skill of the art.
113 There is an issue between the parties as to whether the reference to “1 mg” in the first and second sentences of this last paragraph (page 7, lines 22 to 23) should be interpreted as to mean “1 mg” or “1 mg per kilo” (ie. 1mg/kg) (“Construction Issue 4”). The issue is relevant to Apotex’s sufficiency case and, in particular, the amount of work that must be carried out by the skilled addressee to ascertain a safe and effective dose of the compounds of the invention for use as a treatment for pain.
114 The Patent describes tests performed on rats to demonstrate the comparative efficacy of various compounds in treating pain. The test results are presented in graphs that are reproduced in the six sets of Figures that are entitled as follows:
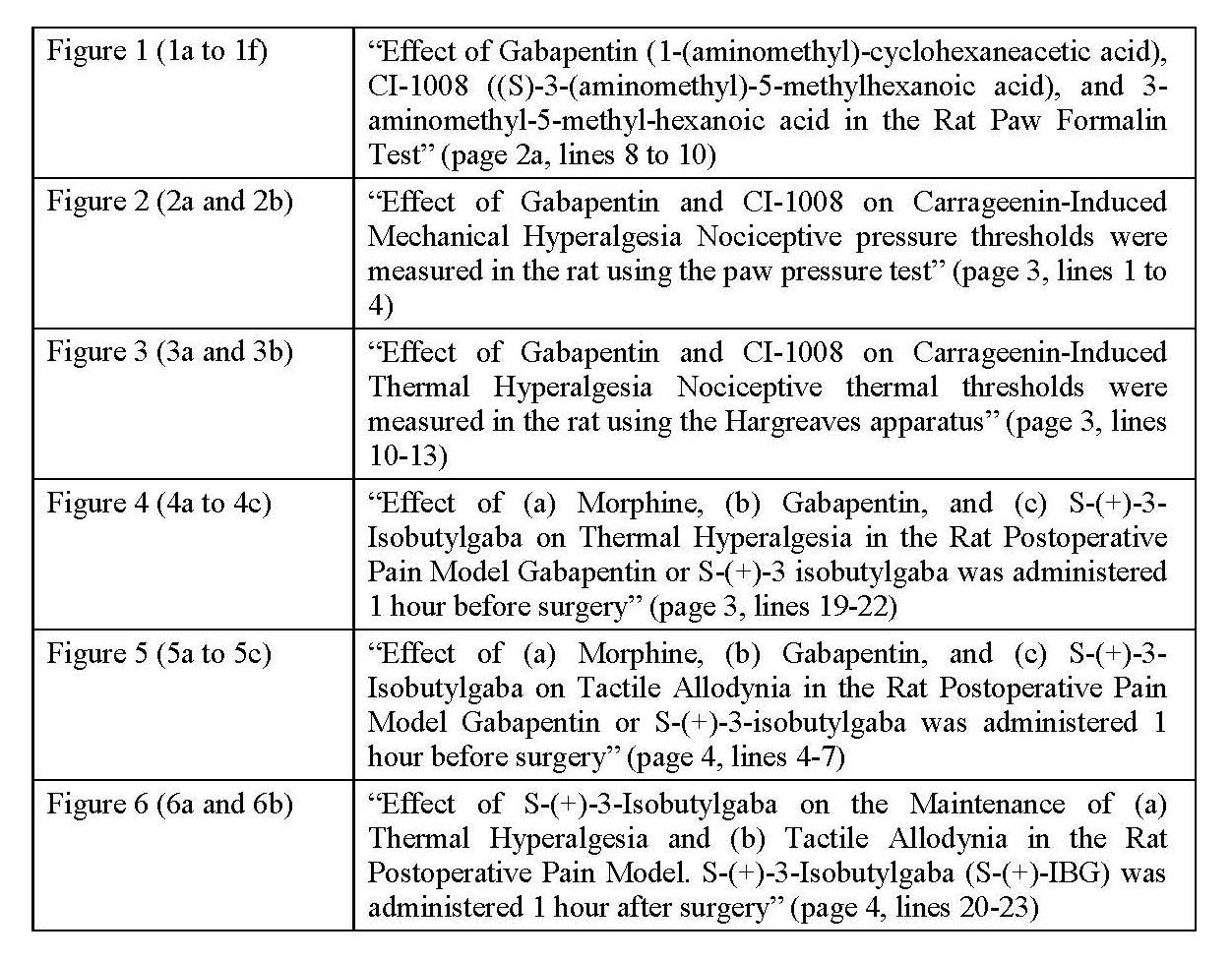
115 The reference to “CI-1008” in the Figures is a reference to the (S)-enantiomer (pregabalin). Some of the Figures (Figures 1e and 1f) also refer to “PD-144550” which is in turn referred to in the description of the Figures as “3-aminomethyl-5-methyl-hexanoic acid”, that is to say, the 3-amino compound. There is a significant issue that is relevant to the false suggestion case as to how those references should be interpreted or, more particularly, how they are likely to have been interpreted by the Commissioner when considering whether or not to grant the Patent.
116 It is common ground that PD-144550 is actually the (R)-enantiomer of the 3-amino compound. However, Apotex contends that the skilled addressee would have understood the term “PD-144550” to refer to the racemate of the 3-amino compound rather than (R)-enantiomer, and that this is how the Commissioner would have interpreted the reference to PD-144550 when considering whether or not to grant the Patent. I will return to this topic later in these reasons.
117 Figure 1 describes the results of the “Rat Paw Formalin Test”. In this test, formalin, a chemical irritant, is injected into the rat paw which triggers behavioural responses (licking or biting of the paw) which are measured over two phases, an early phase (0-10 minute period) followed by a late phase (10-45 minute period). Both gabapentin (Figure 1b) and the (S)-enantiomer (Figure 1d), administered 1 hour before the injection of formalin, demonstrated significant late phase effects. However, the (S)-enantiomer showed the strongest effect, particularly at lower doses.
118 The Patent includes some discussion of the results of the Rat Formalin Paw Test. The Patent states at page 8, lines 7 to 13:
The s.c. administration of gabapentin (10-300 mg/kg) or CI-1008 (1-100 mg/kg) 1 hour before formalin dose-dependently blocked the licking/biting behavior during the late phase of the formalin response with respective minimum effective doses (MED) of 30 and 10 mg/kg (Figure 1). However, neither of the compounds affected the early phase at any of the doses tested. Similar administration of 3-aminomethyl-5-methyl-hexanoic acid produced only a modest blockade of the late phase at 100 mg/kg.
119 Apotex contends that this last sentence would have most likely been understood by the Commissioner to refer to the racemate of the 3-amino compound.
120 Figures 2 and 3 describe a number of tests which show the effect of (inter alia) gabapentin and pregabalin on carrageenin-induced mechanical hyperalgesia (Figure 2) and thermal hyperalgesia (Figure 3). Carrageenin (or carrageenan) is an irritant which is injected into the paw that produces prolonged irritation and hyperalgesia. A device is used to apply and measure pressure applied to the paw until it is withdrawn from the device by the animal. A lower measure is indicative of mechanical hypersensitivity and hyperalgesia. The results for the (S)-enantiomer (Figure 2a) show a dose dependent increase in the pain threshold. The results for pregabalin (Figure 2b) show a similar dose-dependent response but at lower doses (1-100 mg/kg) than for gabapentin (3-300 mg/kg). Morphine, which was also tested, completely blocked hyperalgesia (also shown in Figure 2b).
121 Similar tests were performed using the carrageenan inflammatory pain model as an indicator of thermal hyperalgesia. In this test each animal was placed on a glass plate and a heat source was directed at its inflamed paw. The results show that gabapentin (Figure 3a) and the (S)-enantiomer (Figure 3b) dose-dependently reduced the time taken for the animal to remove its paw from the heat source. This is indicative of their antihyperalgesic effect.
122 Figure 4 describes the results of tests that apply a rat model of postoperative pain which involves making a standard incision of the skin, facia and muscle at a particular part of the hind paw which is then sutured. The Patent states at page 9, lines 23 to 26:
It has been suggested that this model displays some similarities to the human postoperative pain state. In the present study we have examined and compared the activities of gabapentin and S-(+)-3-isobutylgaba with morphine in this model of postoperative pain.
123 Figure 4 shows results for pre-treatments with gabapentin, the (S)-enantiomer or morphine before the surgery. Gabapentin (30 mg/kg) is shown (Figure 4b) to have a significant effect inhibiting thermal hyperalgesia 2 hours and 24 hours after surgery. The (S)-enantiomer is shown (Figure 4c) to have a much more sustained reversal of hyperalgesia than gabapentin when used at the same dose.
124 Animals with tactile allodynia will have a painful reaction to an innocuous force. Figure 5 shows the results of tests for the effect of gabapentin, pregabalin and morphine administered before surgery on the development of tactile allodynia using the same model used in Figures 3 and 4 but also using thin nylon filaments (Frey filaments) that are used to apply a force to a particular area of skin, allowing the force at which a response is elicited to be measured. The results for morphine (Figure 5a), gabapentin (Figure 5b) and the (S)-enantiomer (Figure 5c) show that pregabalin was most effective at reducing tactile allodynia, and that this effect was maintained for 3 days.
125 Figure 6 describes tests on rats for tactile allodynia and thermal hyperalgesia after surgery followed by administration of pregabalin one hour later. The results for both tactile allodynia (Figure 6a) and thermal hyperalgesia (Figure 6b) are shown. An analgesic effect was observed over a five hour period at a dosage of 6 mg/kg.
126 The Patent states at page 13, lines 10 to 16:
The results presented here show that incision of the rat plantaris muscle induces thermal hyperalgesia and tactile allodynia lasting at least 3 days. The major findings of the present study are that gabapentin and S-(+)-3-isobutylgaba are equally effective at blocking both nociceptive responses. In contrast, morphine was found to be more effective against thermal hyperalgesia than tactile allodynia. Furthermore, S-(+)-3-isobutylgaba completely blocked induction and maintenance of allodynia and hyperalgesia.
127 This passage is followed by the claims.
128 At no point does the Patent refer to any safety or efficacy testing of the compounds of the invention in humans. The tests summarised in the Patent were, as I have explained, all conducted on rats.
Construction Issue 1: “method of treating pain”
129 In its written submission Apotex submitted that there was an issue to be determined as to the meaning of the words “a method of treating pain” (claims 1 to 15 and 31) and “for the treatment of pain” (claims 16 to 30 and 32). In the present case I do not think there can be much doubt about what those words mean. The scope of the claim is limited by purpose and not by result: see Apotex Pty Ltd v Sanofi-Aventis Australia Pty Ltd (2013) 253 CLR 284 at [174], [177] and [289] per Crennan and Kiefel JJ when referring to the claim for “[a] method of preventing or treating a skin disorder which comprises administering to the recipient an effective amount of [leflunomide]”. The claim will be infringed if a person administers a therapeutically effective amount of the relevant compound to a patient in need of treatment for pain for the purpose of providing such treatment even though the treatment may not be effective in that patient.
130 This construction of the claim is supported by the expert evidence. A/Prof. King, with whom the other clinicians who gave evidence agreed, accepted that a clinician would consider themselves to be engaging in the medical treatment of pain in the event that they prescribed a drug for the purpose of providing treatment and in the expectation that the treatment would be effective. He also agreed that pain treatments do not always work, either at all or to the same extent, in every patient suffering from pain.
131 Claim 1 is not limited to a method of treating neuropathic pain or pain associated with central sensitisation. The notional skilled addressee would have understood claim 1 as defining a method of treatment for any type of pain. The language of the claim is exceedingly broad and does not limit the types of pain that may be treated with the relevant compounds. It covers all those pain types specifically referred to in dependent claims 4 to 15 and more. Nor is claim 1 limited to the use of the relevant compounds as an adjuvant (or adjunctive) therapy. Use of a relevant compound as an adjuvant therapy is within the claims, but so too is use of such a compound as the sole treatment for pain.
Construction Issue 2: “mammals”
132 Apotex contended that the scope of the claims does not extend beyond humans. However, the language of the claims is clear and unambiguous. It does not confine the methods or medicaments claimed to the treatment of humans.
Construction Issue 3: “3-(aminomethyl)-5-methyl-hexanoic acid”
133 Apotex did not challenge the validity of claim 3 on the ground that it lacked clarity. Both sides accepted that if claim 3 is ambiguous, the ambiguity could be resolved by the application of the well settled rules governing claim construction.
134 Pfizer submitted that claim 3 should be interpreted as referring to nothing more than the (S)-enantiomer. The argument advanced in support of this construction was simple: there is only one compound that is properly referred to as “(S)-3-(aminomethyl)-5-methylhexanoic acid” and “3-aminomethyl-5-methyl-hexanoic acid” and that is the (S)-enantiomer. According to Pfizer’s submission, the first term identifies a particular enantiomer of the 3-amino compound, and the second term identifies the same enantiomer (ie. the (S)-enantiomer) as it might be also be identified but without any specific reference to its stereochemistry.
135 Pfizer’s interpretation of claim 3 is not one that occurred to any of the expert witnesses who gave evidence in the case. Moreover, it suffers from a fundamental problem. Based upon a reading of the Patent as a whole, it is clear that the “3-aminomethyl-5-methyl-hexanoic acid” referred to in claim 3 is the same as that referred to in the consistory statement at page 2, line 13, the description of Figure 1 at page 2a, line 10 and the discussion of the results at page 8, lines 11 to 13. If the language used in claim 3 is given the same meaning as it is given in the body of the specification, it would mean that the inventor’s experiments comparing the effects of “(S)-3-(aminomethyl)-5-methylhexanoic acid” and “3-aminomethyl-5-methyl-hexanoic acid” as shown in the Rat Formalin Paw Test (Figures 1a-1f) involved a comparison of gabapentin and two compounds with precisely the same chemical structure and stereochemistry. That would be nonsense.
136 Further, the Patent states at page 2, lines 12 to 13 that “[t]he more preferred compounds of the invention are (S)-3-(aminomethyl)-5-methylhexanoic acid and 3-aminomethyl-5-methyl-hexanoic acid.” This would also suggest to the skilled addressee that claim 3 is to be understood as referring to more than one compound.
137 Pfizer also submitted, in the alternative, that the reference to the “3-aminomethyl-5-methyl-hexanoic acid” in claim 3 (and in other parts of the specification) should be read as a non-stereospecific term that encompasses the (S)-enantiomer (ie. pregabalin) or any mixture of the (S)-enantiomer and the (R)-enantiomer including the racemate. Pfizer primarily relied upon general scientific usage together with evidence given by Professor Easton and Dr Morella in support of this construction.
138 Apotex submitted that the skilled addressee would understand claim 3 to cover just the (S)-enantiomer and the racemate. It also submitted that if, as Pfizer submitted, the second reference to the 3-amino compound in claim 3 refers to the compound regardless of its stereochemistry, then the earlier reference to the (S)-enantiomer is redundant.
139 Professor Easton and Dr Morella were of the view that, as used in the Patent, the term “3-aminomethyl-5-methyl-hexanoic acid” does not define stereochemistry and that claim 3 should be taken to refer to the 3-amino compound in the form of the (S)-enantiomer, the (R)-enantiomer or any mixture of the two including the racemate. However, Dr Best and Dr Oppenheim understood claim 3 to refer only to the (S)-enantiomer and the racemate because when the chirality of a compound is not specified, they would usually understand this as a reference to the racemate.
140 All experts who participated in the Field 5 Expert Conference agreed that, strictly speaking, the term “3-aminomethyl-5-methyl-hexanoic acid” does not define the stereochemistry, and will include the (R)-enantiomer, the (S)-enantiomer and any mixture of the two including the racemate. However, they disagreed on the use of the term when used in the context of the Patent.
141 All experts who participated in the Field 5 Expert Conference also agreed that typically one enantiomer will have higher activity than the other enantiomer and that the activity of the racemate will be an average of the two enantiomers. However, it is apparent from their evidence that this is merely a rule of thumb.
142 Professor Christie, who did not participate in the Field 5 Expert Conference, also gave evidence that usually, but not necessarily, a reference to a compound that was not accompanied by an indication of its stereochemistry, would be understood as referring to the racemate. However, Professor Christie believed that the position is complicated in this case by the test results in Figures 1e and 1f which show that compound PD-144550 is at best about one-tenth as potent as CI-1008 (the (S)-enantiomer). This led Professor Christie to conclude that in addition to testing the (S)-enantiomer the inventor tested the (R)-enantiomer and not the racemate. Professor Easton and Dr Morella came to the same conclusion based upon their consideration of Figures 1e and 1f.
143 Dr Best and Dr Oppenheim accepted that the results shown in Figures 1e and 1f for PD-144550 were not typical of a racemate, but did not consider this as an appropriate way to go about identifying the stereochemistry of the compound tested in circumstances where the stereochemistry was not specified in the Patent. In other words, they would not use the Figures 1e and 1f test results as a basis for assigning stereochemistry to PD-144550.
144 In my view the preferable interpretation of claim 3 is that it refers to the (S)-enantiomer and the racemate of the 3-amino compound. The principal reason why I do not accept Pfizer’s alternative construction of claim 3 is that the Patent refers to “3-aminomethyl-5-methyl-hexanoic acid” as if it is a specific compound (eg. at page 8, lines 12 to 13 referring to the “administration of 3-aminomethyl-5-methyl-hexanoic acid”). For reasons previously explained that compound cannot be the (S)-enantiomer and, in the absence of any specific indication that it is the (R)-enantiomer referred to in claim 3, I think the claim is to be understood as referring to the racemate.
145 Pfizer faintly argued that the skilled addressee would have readily ascertained, as a matter of course, by consulting the 1994 Edition of “Chemical Abstracts” that PD-144550 was the (R)-enantiomer of the 3-amino compound. This would have confirmed, according to this argument, that it was the (R)-enantiomer and not the racemate that was used in the Figures 1e and 1f tests. I do not accept that submission.
146 All experts who participated in the Field 5 Expert Conference agreed that the compound known as PD-144550 was defined in the scientific literature at the priority date. However, neither Dr Best nor Dr Oppenheim believed that this information would have been readily accessible at that date.
147 Although Mr Bannon SC sought to demonstrate otherwise in cross-examination by taking Dr Best to extracts from the 1994 edition of “Chemical Abstracts” I am not persuaded that, as at the priority date, skilled addressees would have sought to access that material as a matter of course for the purpose of identifying the compound referred to in the Patent as PD-144550. It is true that if they were to have done so, they may have found the information they sought. However, this would have been more a matter of luck than anything else. Even though the 1994 edition of “Chemical Abstracts” includes a discussion of the 1994 paper by Yuen et al (discussed further below) and, in that context, makes reference to PD-144550 as the (R)-enantiomer, I very much doubt that skilled addressees would have gone to that work in the expectation that it would tell them anything at all about PD-144550.
148 Not all information in standard reference works that are widely read and consulted will be common general knowledge. Much will depend on the circumstances of the case. Information in such works might not form part of the common general knowledge if the skilled addressee would not have reason to access it in a given situation as a matter of course: see Generics (UK) Ltd v Daiichi Pharmaceutical Co Ltd [2009] RPC 23 at [25] per Jacobs LJ. Even if the 1994 Edition of “Chemical Abstracts” was readily available to the skilled addressee at the priority date I am not satisfied that he or she would have had any reason to believe that it would contain information identifying PD-144550. On that basis, I am not satisfied that the information in question was common general knowledge as at the priority date.
149 I have already set out the passage in the Patent in which the reference to “1mg” appears (see [113] above). The question is whether it is to be understood as a reference to “1mg” or “1mg/kg”. I consider that the notional skilled addressee would understand the reference to 1mg to occur in the context of a weight based dosage calculation and would interpret the reference to 1mg as a reference to 1mg/kg. This is how I think it should be interpreted.
150 Section 138(3) of the Act specifies the grounds upon which the Court may make an order revoking a patent, either wholly, or so far as it relates to a claim. These grounds include, as provided for in s 138(3)(d), “that the patent was obtained by fraud, false suggestion or misrepresentation.” The Patents Act 1952 (Cth) (“the 1952 Act”) also provided, in s 100(1)(k), for the revocation of a patent either wholly, or in so far as it relates to any claim on the ground “that the patent was obtained on a false suggestion or misrepresentation.” The latter provision mirrored s 32(1)(j) of the Patents Act 1949 (UK).
151 It is well established that, if an order for revocation is to be made under s 138(3)(d) of the Act, the false suggestion or misrepresentation relied upon must be a material factor inducing the grant of the patent. It follows that an applicant seeking revocation on this ground must establish that the Commissioner was deceived or misled in a manner that caused or contributed to the grant of the patent. However, it is not necessary for the applicant for revocation to establish that the patent would not have been granted were it not for the false suggestion or misrepresentation relied upon. Lockhart J expressed the relevant test in Prestige Group (Australia) Pty Ltd v Dart Industries Inc (1990) 26 FCR 197 in these terms (at 201):
… namely, whether the conduct constituting the false suggestion or representation materially contributed to the Commissioner's decision to grant the patent even if other circumstances or causes also played a part in the making of that decision. It is sufficient if the conduct is a material inducing factor which led to the grant. It goes too far to say that the false suggestion or representation must be material in the sense that without it the patent would not have proceeded to grant.
Gummow J agreed with Lockhart J on this point (at 218) and said that it would be sufficient that the false suggestion or misrepresentation was “a material inducing factor that led to the grant”.
152 In the present case the false suggestion or misrepresentation allegedly made to the Commissioner is said to arise out of statements made in the specification filed as part of the complete application for the Patent that were carried through into the Patent as granted. It was not submitted by Apotex that any such suggestion or representation was expressly made. Apotex’s case is that the specification filed as part of the complete application for the Patent conveyed the relevant suggestion or representation by implication.
153 The particular matters relied upon by Apotex are:
(a) The following express statements:
(i) “The more preferred compounds of the invention are (S)-3-(aminomethyl)-5-methylhexanoic acid and 3-aminomethyl-5-methylhexanoic acid.” (Patent at page 2, lines 12 to 13);
(ii) “Figure 1. Effect of Gabapentin (1-(aminomethyl)-cyclohexaneacetic acid), CI-1008, ((S)-3-(aminomethyl)-5-methylhexanoic acid), and 3-aminomethyl-5-methyl-hexanoic acid in the Rat Paw Formalin Test. Test compounds were administered s.c. 1 hour before an intraplantar injection of 50 μL formalin.” (Patent at page 2a, lines to 8 to 12);
(iii) “Effects of Gabapentin, CI-1008, and 3-Aminomethyl-5-methyl-hexanoic Acid in the Rat Formalin Paw Test” (Patent at page 7, lines 28 to 29); and
(iv) “Similar administration of 3-aminomethyl-5-methyl-hexanoic acid produced only a modest blockade of the late phase at 100 mg/kg.” (Patent at page 8, lines 11 to 13).
(b) Properly construed, each of the claims of the Patent (including claim 3) encompasses the racemate of the 3-amino compound.
154 These matters are said by Apotex to have conveyed a suggestion or representation to the effect that the racemate of the 3-amino compound had been tested for the treatment of pain as described and claimed in the Patent.
155 It is not disputed by Pfizer that if any suggestion or representation to the effect alleged by Apotex was made, then it was false. However, Pfizer says that no such suggestion or representation was made and that, even if it was, it is not shown to have been a material inducing factor that led to the grant of the Patent.
156 Whether the Commissioner (or her delegate) understood from reading the patent application that the inventor tested the racemate is largely a matter of conjecture. There was no direct evidence (leaving aside the patent application itself) to which Apotex could point that would suggest the Commissioner had this understanding. Nevertheless, I accept that on my interpretation of the Patent, the patent application did convey a false suggestion that the inventor tested the racemate and that the results of those tests were reported in Figures 1e and f. However, it does not follow that this false suggestion materially contributed to the Commissioner’s decision to grant the Patent.
157 It seems to me unlikely that the Commissioner gave any consideration to the test results except in so far as they indicated that the most preferred embodiment (the (S)-enantiomer) may be effective in the treatment of pain. After all, claims 1 and 2 cover a very wide range of other compounds that were not tested by the inventor.
158 The main focus of the inventor’s testing as reported in the patent application was on the (S)-enantiomer. The (S)-enantiomer was used in all of the reported tests. PD-144550 was used in the tests reported in Figures 1e and 1f, but not in any of the other reported tests. However, there is nothing in the evidence to suggest that the Commissioner’s decision to grant the Patent was influenced in any way by the false suggestion that PD-144550 was the racemate. From the Commissioner’s perspective, whether it was the racemate, the (R)-enantiomer or some other less preferred compound of Formula I that was tested, would seem to be of no moment. In the circumstances, I am not satisfied that the false suggestion was a material inducing factor that led to the grant of the Patent.
159 There were two other related submissions made by Pfizer.
160 Pfizer submitted that, by the application date (and, a fortiori, the date of grant), the Commissioner would have had access to more recent editions of “Chemical Abstracts” including, in particular, cumulative indexes that became available after 1996 and the related electronic database known as SciFinder and that these would have permitted the Commissioner to determine the stereochemistry of the compound referred to in the patent application as PD144550. This argument assumes that the false suggestion was material. It also assumes, wrongly in my view, that the Commissioner had some reason to look behind (so to speak) the patent application with a view to determining whether it was intended to convey the suggestion that I am satisfied it would convey to the skilled addressee.
161 Even if it was necessary for the Commissioner to consider the common general knowledge for the purpose of understanding and interpreting the patent application I do not see why the Commissioner would have any reason to consider the common general knowledge as it stood at some point after the priority date. A patent (and, a fortiori, a patent application) is to be construed in light of the common general knowledge as at the priority date. I do not accept Pfizer’s submission.
162 Pfizer also submitted that the false suggestion could not be a material inducing factor in circumstances where the (R)-enantiomer was considerably less potent than the racemate. Given my earlier findings in relation to materiality, it is not necessary for me to rule on this submission.
163 The allegation that the Patent was obtained by a false suggestion or misrepresentation is not made out.
164 Section 18(1)(c) of the Act requires that an invention be “useful”. Apotex alleges that the claimed invention is not useful in a number of respects. First, Apotex contends that pregabalin is not useful because it is not effective in the treatment of pain that does not involve neuropathic pain or central sensitisation. It alleges that central sensitisation is not clinically relevant to various pain conditions referred to in the claims and that pregabalin is therefore not useful in the treatment of such pain conditions. Second, Apotex contends that all the claims that encompass the (R)-enantiomer are invalid because the (R)-enantiomer is “inactive” or “practically inactive” and not useful as a treatment for any type of pain. Apotex accepts that it carries the onus of proof.
165 Section 18(1)(c) does not require that an invention be useful or even practicable in a commercial sense. Absent any promise in the specification to that effect, there is no requirement that an invention be commercially desirable or preferable to other products or processes in the field: see Blanco White, Patents for Inventions, 5th ed, at para 4-407. This point was made by Lindley LJ in Lane Fox v Kensington and Knightsbridge Electric Lighting Co Ltd [1892] 3 Ch 424 at 430-431 and endorsed by the High Court in Advanced Building Systems Pty Ltd v Ramset Fasteners (Aust) Pty Ltd (1998) 194 CLR 171 where the majority said at [24]:
It is no objection to the validity of a patent granted under the Act that it is commercially impracticable; its utility depends upon whether, by following the teaching of the complete specification, the result claimed is produced.
166 The claimed invention need not be simpler or cheaper to make or use, more effective, or otherwise superior, to anything already available unless the specification promises that it will be. The requirement that the claimed invention be useful merely requires that it give to the public a useful choice: British Liquid Air Co Ltd v British Oxygen Company Ltd (1908) 25 RPC 577 at 607. The basic principle, according to Gummow J in Rehm Pty Ltd v Websters Security Systems (International) Pty Ltd (1988) 81 ALR 79 at 96 quoting Lindley LJ in Fawcett v Homan (1896) 13 RPC 398 at 405, is as follows:
If an invention does what it is intended by the patentee to do, and the end attained is itself useful, the invention is a useful invention.
167 Apotex submitted that if there are products or methods within the scope of a claim that are not useful then the claim will be invalid for lack of utility. In Norton and Gregory Ltd v Jacobs (1937) 54 RPC 271 Lord Greene MR said at 276:
Now if Claim 1 be read by itself and construed in accordance with the ordinary meaning of the language used, it is apparent that the use of any reducing agent falls within it. The character of the reducing agent to be used is not defined by reference to any particular quality or any particular result. If the matter stood there, the Claim would be unquestionably bad. But it is said (and this is the substantial part of the Appellants’ argument) that the language of the Claim must be construed so as to exclude any reducing agent which a chemist of ordinary skill would know, with or without experiment, to be unsuitable in view of the result to be achieved. We are unable to accept this argument. The fact that a skilled chemist desiring to use the invention would reject certain reducing agents as being unsuitable is one thing; it is quite a different thing to say that a claim must in point of construction be cut down so as to exclude those reducing agents because a skilled chemist would not use them.
168 His Lordship’s judgment is often cited in support of the principle that a claim is bad if it is wider than what is useful or if it covers means that will not produce the desired result even if a skilled person would know which means to avoid: Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 601; WM Wrigley Jr Co v Cadbury Schweppes Pty Ltd (2005) 66 IPR 298 at [138]. However, as is also apparent from those cases, much depends upon the proper construction of the claim. Claims are not to be read through the eyes of a skilled addressee purposefully seeking to perform the claimed invention in a manner that would not provide a useful result: see Washex Machinery Corporation v Roy Burton & Co Pty Ltd (1975) 49 ALJR 12 at 19. Nor should claims be construed in a manner that the skilled addressee would appreciate would lead to unworkability if there is another construction that is equally open that avoids that result: Electric & Musical Industries Ltd v Lissen Ltd (1938) 56 RPC 23 at 39; Rehm Pty Ltd v Websters Security Systems (International) Pty Ltd (1988) 81 ALR 79 at 99.
169 It is often said that the principle that a claim is bad if it is wider than what is useful is one that must be applied with great caution. As Blanco White explains at para 4-409:
It follows from what has been said that it is often a convenient test of the utility of the invention contained in a claim to consider whether the claim includes forms of the invention which are not useful, but that this test must be applied with very great caution. The function of a claim is to delimit the monopoly given by the patent, not to give instructions for the working of the invention, and it is consequently not necessary that the claim should contain these instructions; even the body of the specification is required to contain only those instructions that the reader cannot supply for himself. It would be unreasonable to expect the claims to contain more. A distinction should accordingly be drawn between cases in which the invention claimed is not useful unless an additional feature or features be added to those claimed (the claim then being invalid), and cases where the qualifications and expedients necessary to make the article claimed work can be, and on a true construction of the claim are, left to the reader to supply for himself. Since in cases where the reader can make the thing work the courts tend wherever possible to construe claims as requiring him to do so, it is not in practice enough to ask whether the claim includes things that are not useful; it is necessary to ask also whether there is anything in the language of the claim positively pointing to some useless construction. The successful utility attacks are nearly always in cases of that sort. Examples are: where a claim specifies two alternative processes or constructions of mechanism, of which only one is useful; or the claim specifies the use of any of a group of chemical compounds, and it is not substantially true that all will work; or the claim includes a series of constructions, and only certain members of the series are useful; or more generally, the claim contains a limitation directed to a particular feature and further limitation of that same feature is needed for utility; or the feature needed for effective working is expressly made optional-as when it is added by a subsidiary claim.
It does not follow that a broad claim is invalid merely because some subsidiary claim within it lacks utility: for the invention the subject of the subsidiary claim may be intended to attain some result additional to the main object to which the broader claim is directed.
(Footnotes omitted; TA Blanco White, Patents for Inventions, 5th ed. Stevens & Sons, London, 1983)
170 A claim may have utility even if a promised advantage cannot be achieved in all cases or with the same degree of success: Rescare Ltd v Anaesthetic Supplies Pty Ltd (1992) 25 IPR 119 at 142-143 (Gummow J) and Sanofi-Aventis Australia Pty Ltd v Apotex Pty Ltd (No 3) (2011) 92 IPR 320 at [245], point 8 (Jagot J). Thus, if a claimed method of treatment for nausea was effective in a substantial proportion of patients experiencing nausea, the claim would not be invalid merely because a small proportion of patients experiencing nausea did not respond to the treatment. This is because the skilled addressee reading the specification would not expect (assuming he or she is not told otherwise) that the claimed treatment will be an effective treatment for every patient suffering nausea resulting from every conceivable condition. Even if the effectiveness of a method of treatment was established by a successful Phase 3 clinical trial, the results obtained would usually do no more than indicate a statistically significant effect across the relevant cohort, and would not imply that the method was more effective than placebo in the treatment of all trial participants.
171 Pfizer submitted that the case based upon what Apotex described as “clinically relevant” central sensitisation is not open to Apotex on the pleading. The relevant paragraph of its pleading (para 29) asserts:
The (S) enantiomer is not effective in any case, in any patient, in the treatment of pain conditions, that do not involve neuropathic pain or central sensitisation.
172 Particulars are given of twenty-two pain conditions for which the (S)-enantiomer is said not to be an effective treatment. Many are defined as a subset of a pain type: for example, “Nociceptive pain not involving central sensitisation or neuropathic pain” and “Postoperative pain caused by acute inflammatory pain arising postoperatively”. Others are expressed more broadly: for example “acute gout pain” and “septic arthritis”. The pleading does not define, or even mention, the concept of “clinical relevance”.
173 Pfizer submitted that Apotex’s inutility case is specifically pleaded and that it is that pregabalin “is not effective” in any case in the treatment of pain conditions that do not involve neuropathic pain or central sensitisation. Pfizer submitted that if a pain condition involves either neuropathic pain or central sensitisation, then the inutility case based upon the alleged lack of effectiveness in pain conditions that do not involve neuropathic pain or central sensitisation must fail. In particular, Pfizer submitted that Apotex should not be permitted to argue that the invention is not useful on the basis that pregabalin is not effective to treat pain conditions where central sensitisation is present but not to an extent that might be considered “clinically relevant”.
174 Senior Counsel for Pfizer, Mr Bannon SC, made it clear before the commencement of evidence that a pleading point was being taken. He explained Pfizer’s position to me in these terms shortly before the concurrent evidence began (T 199-200):
MR BANNON: To avoid relevance objections on various topics, looking at my learned friend’s proposed questions over the weekend, I think it’s implicit from what – follows from what I said on Friday – we rely on their pleading for inutility. We oppose a rolling implicit amendment to that.
HIS HONOUR: All right.
MR BANNON: And, in particular, the debate about inutility followed a specific requirement. They specifically plead that. Your Honour may or may not recall there was an argument about it, and there’s a judgment. But the case, we say – and the only case which has been pleaded – is that the pregabalin doesn’t work – leaving aside the R enantiomer, the [sic] pregabalin doesn’t work in various sorts of pains because those pains do not involve central sensitisation or neuropathic pain. There’s no case pleaded to say that, even if those pains do involve that, nevertheless it doesn’t work because there’s some gradation as to the extent in which they work based on clinical relevance or some other theory. We say that’s not part of the pleaded case. So questions going to that issue we say are irrelevant. But we – I – the better course is for the whole thing to proceed and - - -
HIS HONOUR: All right. And you’re not to be taken as acquiescing in a - - -
MR BANNON: Absolutely.
HIS HONOUR: - - - departure from the pleading.
MR BANNON: Yes.
175 Pfizer’s pleading point was not fully addressed until after the close of the evidence at which time it was the subject of detailed submissions.
176 Pfizer’s contention that all pain conditions involve some element of central sensitisation, even if it might be so slight as to be of no relevance to the clinician treating a patient for pain, reflects the view of Professors Christie, Schug and Littlejohn. Pfizer submitted that if, as Professors Christie, Schug and Littlejohn maintain, central sensitisation is involved in all pain states, then Apotex’s pleaded case must fail.
177 Pfizer made the following additional points.
178 First, there is no allegation that the invention lacks utility on the basis that there are particular doses or dosage regimes within the claims that would not be effective to treat the alleged categories or types of pain. Pfizer says that the case pleaded by Apotex is that pregabalin is not effective as a treatment for pain in any dose that the skilled addressee might use.
179 Secondly, Apotex does not allege that the invention lacks utility on the basis that it fails to meet any “promise” made in the specification. The pleaded case is that the claimed invention will not work at all for the particular conditions identified. In particular, there is no allegation that a particular level of efficacy or effect is required for the invention to be considered useful.
180 The first of Pfizer’s additional points is correct. Apotex does not allege that pregabalin will provide pain relief at some doses but not others. Rather, Apotex’s case is that for some pain conditions, no matter what dose is used, pregabalin will not provide any pain relief.
181 It is also true that Apotex’s case is not based upon any express or implied promise arising from the specification. But where the consistory statements and the claims describe a method of medical treatment, the skilled addressee would ordinarily understand the patentee to be saying that the claimed method is capable of providing a therapeutic benefit. If the claimed method is incapable of providing any therapeutic benefit, then the claim will be invalid on the basis that the claimed invention is not useful.
182 It is true that if Apotex’s pleading is read literally, Apotex’s case is wholly founded upon the contention that pregabalin is not effective in the treatment of any pain that does not involve the phenomenon of central sensitisation. On Pfizer’s case, if all expert witnesses were to agree that central sensitisation is involved in a particular pain condition but that it does not in fact make any actual contribution to the pain experienced by the patient, then the claimed methods must still be considered to provide a useful treatment for that particular pain condition. I do not agree that Apotex’s case is that narrow. The notion of “involvement” in this context must refer to some relevant or meaningful involvement contributing to the pain condition.
183 The first proposition the clinicians were invited to comment on during the Field 2 Expert Conference was whether “pregabalin is potentially effective in clinically relevant pain states in a mammal that involve a central sensitisation component” (emphasis added). A/Prof. Lynden Roberts and Professor Harnett are shown in the Field 2 Joint Expert Report tendered by Pfizer to disagree with this proposition on the basis that pregabalin is potentially effective only if central sensitisation is clinically relevant. Similarly, the clinicians were also invited to comment on the following proposition:
That when central sensitization is a clinically relevant component of a patient’s pain condition, that pregabalin could be effective treatment for [various types of specified pain].
184 This case was closely case-managed. If Pfizer was of the view that the case specifically addressed in those parts of the Field 2 Joint Expert Report were irrelevant to Apotex’s pleaded case then they should never have agreed to it being raised with the clinicians at the Field 2 Expert Conference without bringing the matter back to Court for directions with a view to resolving the pleading issue. In my view Pfizer was on notice of the case that Apotex proposed to make at least some time prior to the convening of the Field 2 Expert Conference and well prior to the commencement of the trial. To the extent that there has been any departure from the pleaded case, I would characterise it as both minor and technical. I am satisfied that Pfizer has not suffered any relevant prejudice given that the issue was fully explored in evidence and submissions.
185 There is no issue as to the second way in which the inutility case is put by Apotex. It alleges in the relevant paragraph of its pleading (para 35) that the (R)-enantiomer is not effective in the treatment of any pain condition.
186 In the present case claim 1 is directed to a method of treating pain in mammals including, in particular, humans. The skilled addressee would understand pain in this context in the way defined by the IASP (see [33] above). He or she would not read claim 1 as limited to pain states that involve central sensitisation. The term central sensitisation is not used anywhere in the Patent.
187 The skilled addressee would also understand claim 1 as covering pain conditions in humans that require medical treatment. The claimed invention is not directed to the treatment of minor ailments or irritations that the skilled addressee would not consider serious enough to warrant medical intervention.
188 All expert clinicians who gave evidence in this proceeding agreed that no drug will be effective in all patients. Thus, while they all agreed that pregabalin is an effective treatment for neuropathic pain, they all accepted that there will be some patients for whom it will provide no pain relief even from neuropathic pain.
189 As previously mentioned, the Patent does not convey any promise or representation as to the degree to which the claimed method of treatment will relieve pain. If the method of treatment defined in claim 1 is capable of providing some measure of pain relief to a substantial number of patients in need of treatment for pain then the claim is not invalid for lack of utility.
190 There are differences of opinion between the clinicians as to the effectiveness of pregabalin in the treatment of pain. At the Field 2 Expert Conference Professors Schug, Christie, Littlejohn and A/Prof. King agreed that “pregabalin is potentially effective in clinically relevant pain states in a mammal that involve a central sensitization component.” Professor Gibson also agreed except if pregabalin is used as the sole pain relief therapy. This was a qualification frequently raised by Professor Gibson and for convenience I shall refer to it as his “sole therapy qualification”. A/Prof. Lynden Roberts and Professor Harnett considered that pregabalin is a potentially effective treatment but only in those pain states in which central sensitisation is “clinically relevant”.
191 In their oral evidence it became clear that A/Prof. Lynden Roberts and King and Professor Harnett considered that there are some pain states in which central sensitisation is not clinically relevant in the sense that it is not making any contribution to the pain state. In those circumstances they would not prescribe pregabalin either as a sole or an adjunctive therapy.
192 By way of example, A/Prof. King, a neurologist, referred to acute pain associated with disc disease involving no neuropathic component. In his opinion, central sensitisation is not clinically relevant to such cases. A/Prof. King said the pain in these cases is usually treated successfully with bed rest, anti-inflammatories and other therapies before it becomes chronic. There would be no point in prescribing pregabalin given the availability of these therapies.
193 Another example used to illustrate this way of thinking is acute gout. This is a condition that A/Prof. Lynden Roberts and Professor Littlejohn, both of whom are rheumatologists, are often required to treat. Acute gout pain refers to pain with a rapid, intense onset, which is associated with red, hot and swollen joints. It is caused by a build-up of uric acid in the joints, which become swollen and inflamed. Chronic gout pain is experienced by patients who generally have a history of acute attacks of gout pain. The pain experienced by patients with chronic gout is due to an ongoing inflammatory process. In some rare cases of chronic gout, a patient may also experience neuropathic pain. Pain associated with gout can be extreme.
194 A/Prof. Lynden Roberts and Professor Littlejohn agreed that most gout pain is acute and will resolve relatively quickly with treatment usually over a few hours or a couple of days. A/Prof. Lynden Roberts said that in practice he has never seen signs of central sensitisation in cases of acute gout pain. He said in the concurrent evidence (T 245):
… so in people who have acute gout in the knee, for example, they present with pain in the knee. And when I examine their knee, I can move their knee and it’s very irritable. And I can see that it’s acute gout and I treat them with anti-inflammatory tablets. When I see people with another condition in the knee that’s persistent, I can find features of allodynia and hyperalgesia clinically that are very apparent clinically that would suggest that central sensitisation was the major component of their problem. And so clinically I can discriminate reasonably easily between people who have central sensitisation and people who don’t.
195 A/Prof. Lynden Roberts was cross-examined with a view to suggesting that he had no basis for believing that central sensitisation was not playing any role in the pain experienced by patients with acute gout. It is clear from his evidence that many patients he treats for acute pain conditions, including acute gout, do not show any clinical signs of central sensitisation. He therefore considers that it would be pointless to prescribe pregabalin for the treatment of acute gout pain even as an adjunctive therapy.
196 A/Prof. Lynden Roberts’ evidence on this topic may be contrasted with Professor Littlejohn’s. It was put to Professor Littlejohn that there are some pain states in which central sensitisation is not clinically relevant. I understood him to accept that proposition as correct. However, he went on to qualify that answer as follows (T 288):
I think [central sensitisation] is always clinically relevant, but whether you target that process as your key strategy or whether you target the primary process causing the pain is a clinical decision based on the context and a lot of other factors, co-morbid factors, etcetera. […] We wouldn’t use a drug to target central sensitisation for an acute attack of gout. We would treat that with high dose anti-inflammatories, but recognise the central sensitisation is playing a role in that patient’s pain and their pain and allodynia and other clinical features, but you would treat the primary cause, and that would be the same as a lot of other clinical conditions.
197 Professor Littlejohn’s position was made much clearer during cross-examination in which he gave the following evidence (T 626-627):
And […] acute gout is an example of a condition that you treat often, isn’t it?---Yes, indeed.
And it’s an example of a pain condition where central sensitisation does not manifest as the key pain that needs targeting?---I don’t know about that. I think it’s a major component of acute gout pain. It’s just that we have other ways of targeting the acute gout mechanism through high dose anti-inflammatories, which are so effective that it just turns the whole process off very quickly.
But you don’t use drugs specifically targeting central sensitisation for an acute attack of gout, do you?---No, they would take too long to work.
I see?---We – we want to work within, you know, hours to get relief.
I see. And I think you said [in] the concurrent session, you don’t use pregabalin to treat acute gout?---No.
But you would accept, I think, that a patient presenting with acute gout will be in need of treatment for their pain?---Correct.
And I think you said that acute gout is the most intense inflammation that we can see within a joint?---Correct.
And so it’s serious pain that a patient presents with acute gout?---A patient becomes sensitised to even someone walking into the room so it’s extreme pain and extreme hyperalgesia.
You don’t prescribe pregabalin in acute gout, because, in your professional opinion, it will not have any significant effect on that patient’s pain experience?---Well, I don’t know if we know that. I don’t think trials or studies have been done to establish that. It’s just that it hasn’t been required, because we can put a cortisone injection into a gouty knee and the pain will be diminished within 30 minutes so there’s no need to worry about other strategies that might be theoretically possibly [sic].
It would be possible, wouldn’t it, to administer pregabalin in combination with other treatments, wouldn’t it, in a case of acute gout?---You could certainly do that. It would be an interesting study.
And you don’t do that?---No.
And you’re not concerned that it might be beneficial to a patient to prescribe pregabalin to prevent a case of acute gout transitioning to a chronic disease?---I’m not concerned that would happen, no.
198 Professor Littlejohn went on to indicate that, in his view, central sensitisation is always present when a patient is experiencing pain but that sometimes there are other strategies that are better suited to a particular clinical situation that does not involve targeting central sensitisation. He later gave this evidence: (T 628):
But there are conditions where central sensitisation may not manifest as the key pain that needs targeting?---Yes, that is – that’s true.
And I think you agreed with me that you wouldn’t withhold a treatment from a patient you thought would provide a benefit to that patient?---No, I wouldn’t.
And so it’s your professional opinion, isn’t it, that in a patient with a condition where there’s no manifestation of central sensitisation that needs targeting, pregabalin will not provide a useful benefit to that patient?---That’s probably correct.
199 Professor Schug was asked in the concurrent evidence to comment on the proposition that pregabalin is not effective in the treatment of pain conditions that do not involve central sensitisation. His response was that all clinically relevant pain states involve some degree of central sensitisation. Professor Christie was of the same opinion. Ultimately, Professor Gibson also agreed that processes of central sensitisation occur, to a greater or lesser extent, in all pathophysiological pain states. Although Professor Gibson questioned whether all nociceptive pain causes central sensitisation, he agreed that central sensitisation will be present to some degree in all pain states warranting medical intervention. He said that the process of central sensitisation is “not like an on-off switch” and added (T 296):
[I]t can be very minor in terms of the amount of central sensitisation […] that you have through to being extensive and prolonged and severe. And so my qualification really alludes to that process. There will be cases where you, yes, have central sensitisation, because it is part of the normal response pattern of the nociceptive system to any injury or disease, but that in some cases there will be a relatively minor component of central sensitisation. In other cases there could be a relatively major component of central sensitisation.
I accept the evidence of Professor Gibson on this topic which is consistent with that of Professors Christie and Schug.
200 There are some types of pain which there may be no reason to treat with pregabalin. The pain associated with acute gout provides a very good example of this. As Professor Schug explains in his affidavit evidence, there may be various reasons why a clinician might not prescribe pregabalin to treat a particular type of pain. As he explained:
Because pain is complex and responses to treatment are variable, my experience is that a useful benefit from the use of pregabalin is obtained in only some patients. Some patients can suffer side effects like somnolence or dizziness. There are many cases of pain for which pregabalin is usually not used. For example, other analgesic options may be efficacious for particular conditions, they may be cheaper or may not have the same side effect profile. However, this does not mean that pregabalin would not be effective to treat those conditions in at least some patients, if it were administered to them. There are a range of reasons, including particularly side effect profile and cost, why pregabalin would not be prescribed in a given situation, particularly when pain is not difficult to treat and cheaper alternatives are likely to be effective.
In all cases of clinically relevant pain I assume, and I consider that most pain specialists would assume, that central sensitisation contributes to some degree which may be large or small, to the pain experience. As a result, pregabalin could be administered to any patient suffering any pain condition with the expectation that the patient may receive a benefit due to the reduction in the central sensitisation component of the pain. Whether pregabalin is the analgesic of choice in a given situation, or a benefit is actually obtained by a particular patient, are different matters for the reasons I have explained.
201 As part of the Field 2 Expert Conference, the clinicians were also asked to comment on the proposition that when central sensitisation is a clinically relevant component of a patient’s pain condition, pregabalin could be an effective treatment for various pain conditions including acute gout pain. As part of the same question they were also asked about other pain conditions although for present purposes I will focus on the one that I have mentioned. Professors Littlejohn, Schug and Christie’s collective response was as follows:
Prof Littlejohn, Prof Schug and Prof Christie agree but consider the definition of ‘clinically relevant’ central sensitization to be ambiguous because central sensitization […] might occur but not always be obvious as defined by some clinicians but is nonetheless expected to contribute to a greater or less [sic] extent to all pain types involving intense or prolonged nociceptive stimulation, or nerve injury whether or not other mechanisms (e.g. ongoing nociceptor stimulation) or pain drug targets dominate. In cases wherein other mechanisms and drug targets dominate, pregabalin could still be an effective treatment, alone or in combination with other treatments, even if the pain type is likely to respond fully or in part to other preferred treatments. Many conditions have pain contributed to by different mechanisms, particularly central sensitization.
202 Professor Gibson agreed that, leaving aside pain of entirely neuropathic origin, pregabalin could be used as an effective pain relief therapy, but not on its own. Professor Schug accepted that there would be many pain conditions for which pregabalin alone would not be an effective treatment. Mr Catterns QC, who appeared for Apotex, asked Professor Schug whether there are some pains which would be treated with pregabalin alone or in combination with other drugs. Professor Schug’s answer was as follows (T 428):
I mean as already discussed many times, pregabalin addresses not pain itself, but the central sensitisation which follows an initial painful insult, and therefore pregabalin is not qualifying as an analgesic, and I think even the patent makes clear that it is not an analgesic but an antihyperalgesic drug. And for that reason there is nearly no condition except neuropathic pain and fibromyalgia, and as a pathological pain states [sic] where the only mechanism is central sensitisation, where pregabalin on its own is effective.
203 Mr Catterns QC relied heavily on this answer as evidence that pregabalin will have no effect upon a patient’s pain where pregabalin is used as the sole treatment except in cases of neuropathic pain or fibromyalgia. However, I do not think this answer goes that far. Professor Schug, as I understood him, did not accept that when answering Mr Catterns’ question that pregabalin would have no effect on pain if used alone except in cases of neuropathic pain or fibromyalgia. The question and answer were directed to how a clinician would treat such pain conditions given that, except in cases of neuropathic pain and fibromyalgia, central sensitisation is not the only pain mechanism involved.
204 The evidence established that central sensitisation plays a role in all pain conditions but that in some cases it plays a minor role only. What this indicates is that for some pain conditions pregabalin may provide a patient with little in the way of pain relief especially when compared with other treatment options. But it does not follow that the use of pregabalin would provide such a patient with no pain relief.
205 The best example of pain that is least likely to be relieved by treatment with pregabalin is acute gout. One reason why pregabalin is not used to treat pain associated with acute gout is that there are more effective, quick-fire treatments available that resolve the underlying condition and with it the accompanying pain. Another reason is that, because acute gout is an inflammatory condition, central sensitisation will only play a minor role in the patient’s pain and, therefore, the scope for pregabalin to reduce the patient’s pain will be quite limited. But if acute gout were treated with pregabalin alone, can it be said that this would not provide a substantial number of patients with some measure of pain relief? The fact that the pain relief achieved might be quite small, because central sensitisation was contributing to the patient’s pain in a minor way, does not mean that pregabalin does not provide a useful treatment option for patients suffering from pain associated with acute gout. The fact that there may be far simpler, cheaper and more effective ways of treating pain in patients suffering from acute gout explains why clinicians do not prescribe pregabalin as a treatment for acute gout but it does not establish that pregabalin would not be useful in the treatment of that condition either alone or in combination with other therapies.
206 I am not persuaded that pregabalin would not reduce the patient’s pain to some degree in cases of acute gout. Put in slightly different terms, I am not persuaded that in cases of acute gout, treatment with pregabalin would provide no greater reduction in the patient’s pain state than would treatment with placebo. In those circumstances, Apotex’s inutility case, in so far as it is based upon the usefulness of pregablin as a treatment for acute gout or, for that matter, any other condition in which central sensitisation plays a minor role, must fail.
207 Apotex advanced the same arguments in relation to various other pain conditions including cancer pain, postoperative pain and burn pain. I do not propose to refer to the evidence in relation to each of these particular conditions because what I have said in relation to gout pain also applies to all other pain conditions in which central sensitisation plays a minor role and for which pregabalin is likely to be much less effective than other treatments. However, in deference to Professor Harnett, who like Professors Schug, Christie, Gibson, Littlejohn, A/Prof. Lynden Roberts and King, was an exemplary witness, I propose to say a little more about his evidence.
208 Professor Harnett, an oncologist, said that he would not expect pregabalin to have a therapeutic effect in treating the pain experienced by a patient with cancer if the patient’s pain condition did not involve neuropathic pain.
209 Professor Harnett said that he does not need to target central sensitisation as the mechanism causing pain in the majority of cases. I accept this evidence. It reflects the approach of a highly experienced clinician who is providing his cancer patients with the best pain relief available. There are no doubt other pain treatments (eg. analgesics) that will be of much greater benefit to patients whose pain does not have a neuropathic component. However, I do not think Professor Harnett’s evidence provides a sound basis for concluding that pregabalin would not have any therapeutic effect in the treatment of pain that does not include a neuropathic component. As with gout pain, central sensitisation is likely to play some role in cancer pain lacking any neuropathic component, even though its role might be quite minor.
210 I accept that the extent of any therapeutic effect that will be achieved using pregabalin in cases where central sensitisation plays a minor role is likely to be slight. However, in my view, it is not possible to determine in the absence of more specific evidence whether the effect is likely to be merely slight or, as Apotex contends, non-existent. The positon would be different were I able to conclude that central sensitisation does not play any role in pain that does not involve a neuropathic component. However, each of Professors Schug, Christie and Gibson, all of whom have great expertise in the mechanisms of pain, agree that this is not the case.
Effectiveness of the (R)-enantiomer
211 Apotex also contended that, with the exception of claims 3 and 18, the claims of the Patent are invalid because the (R)-enantiomer of the 3-amino compound is not effective in the treatment of pain. This contention was developed on the assumption that claims 3 and 18 do not encompass the (R)-enantiomer. Of course, if properly construed, claims 3 and 18 do encompass the (R)-enantiomer, then these claims would also be invalid if it is found that the (R)-enantiomer is not effective in the treatment of pain.
212 In support of its contention that the (R)-enantiomer is not effective in the treatment of pain, Apotex relied upon a number of scientific papers and Pfizer Australia’s Product Information document (“the Lyrica PI”) for pregabalin. I shall deal with each of these in turn.
213 The Taylor paper, by Charles Taylor et al, is entitled “Potent and stereospecific anticonvulsant activity of 3-isobutyl GABA relates to in vitro binding at a novel site labeled by tritiated gabapentin”. It was published in 1993 in Epilepsy Research, a respected, peer reviewed scientific publication.
214 By way of introduction, the authors note that in recent clinical trials, gabapentin was proven to be an effective treatment for the prevention of partial seizures (ie. as an anticonvulsant) in patients refractory to (ie. difficult to treat with) available drugs. The paper reports the results of the authors’ pre-clinical study of gabapentin and 3-Isobutlyl GABA (the 3-amino compound) relating to their use as anticonvulsants. In this study, mice were given a drug and then tested for prevention of seizures by application of electrical current at “low-intensity electroshock” and “maximal electroshock” levels. The three drugs tested were gabapentin and the enantiomers of the 3-amino compound. In the case of the (S)-enantiomer, five out of ten mice treated with a dose of 0.65mg/kg of the (S)-enantiomer were protected from low-intensity electroshocks, while three out of ten mice treated with the (R)-enantiomer were protected from low-intensity electroshock at a dose of 300 mg/kg. In the maximal electroshock test, five out of ten mice treated with the (S)-enantiomer at a dose of 20mg/kg were protected, while zero out of ten mice were protected after treatment with the (R)-enantiomer at a dose of 300mg/kg. The authors noted (at page 134):
The S(+) enantiomer was responsible for virtually all of the anticonvulsant activity previously reported […] for the racemate while the R(-) enantiomer was inactive at doses up to 100 mg/kg and only prevented low-intensity electroshock seizures in three of 10 mice following administration of 300 mg/kg; conventional maximal electroshock seizures were not prevented even with 300 mg/kg.
215 The authors also report the results of an in vitro binding study using brain tissue of rats to measure the activity or potency of the (S)-enantiomer, the (R)-enantiomer and the racemate of the 3-amino compound at inhibiting the binding of tritiated gabapentin at a receptor binding site labelled by [3H]gabapentin (an isotopic marker for gabapentin) where gabapentin was known to bind. The authors discussion of the results of this in vitro test noted that there were significant differences in potency between the (S)-enantiomer and the (R)-enantiomer, and that the (S)-enantiomer was approximately 20 times more potent than the (R)-enantiomer.
216 This paper by Po-wai Yuen et al entitled “Enantioselective Synthesis of PD144723: A Potent Stereospecific Anticonvulsant” was published in 1994 in Bioorganic & Medicinal Chemistry Letters.
217 In this paper, the authors discussed previous studies that described a high affinity gabapentin binding site, which suggested the anticonvulsant activity of gabapentin may be due to its interaction at this site. They noted that the (S)-enantiomer may also be active through its interaction with this binding site.
218 In their own study, the authors investigated the stereospecifity of the gabapentin binding site using both the (S)-enantiomer (also referred to as PD144723) and the (R)-enantiomer (also referred to as PD144550) in the brain tissue of rats to measure the extent to which these were able to displace [3H]gabapentin from the gabapentin binding site. The authors stated (at page 825):
[t]he (S)-enantiomer, PD144723, was found to be the most potent compound yet studied for displacement of [3H]gabapentin from the gabapentin binding site in vitro […] The (R)-enantiomer, PD144550, displaced [3H]gabapentin at a much higher concentration […] The comparatively poor binding of PD144550 at the gabapentin binding site compared with PD144723 illustrates the stereoselectivity of this site […]
219 The authors also reported the results of their in vivo tests using the maximal electroshock model to measure the effects of gabapentin, the (S)-enantiomer and the (R)-enantiomer in blocking maximal electroshock seizures in mice. They found that the (S)-enantiomer was most effective in blocking such seizures. They also noted that the (R)-enantiomer was inactive in blocking maximal electroshock seizures even with an intravenous dose of 300 mg/kg.
220 This paper, by Nicolas Gee et al, entitled “The Novel Anticonvulsant Drug, Gabapentin (Neurontin), Binds to the α2δ Subunit of a Calcium Channel” was published in 1996 in the respected, peer reviewed Journal of Biological Chemistry. It reports the results of another binding study using, among other biological materials, brain tissue of pigs. The authors found that (S)-enantiomer was approximately 10 times more potent at inhibiting the gabapentin binding site than the (R)-enantiomer.
221 This paper, by MJ Field et al, published in 1997 in the British Journal of Pharmacology, is entitled “Gabapentin (neurontin) and S-(+)-3-isobutylgaba represent a novel class of selective antihyperalgesic agents”. It is clear that the research work reported in this paper includes much of that reported in the Patent. For example, it includes (at page 1516):
Results
Effect of gabapentin, S-(+)-3-isobutylgaba and R-(-)-3-isobutylgaba in the rat formalin test
The s.c. administration of gabapentin (10-300 mg kg-1) and S-(+)-3-isobutylgaba (1-100 mg kg-1) 1 h before formalin dose-dependently antagonized the late phase of the formalin response with respective minimum effective doses (MED) of 30 and 10 mg kg-1 […] However, similar administration of R-(-)3-isobutylgaba (1-100 mg kg-1) only produced a modest blockade of the second phase at the highest dose of 100 mg kg-1 […] None of the compounds had any effect on the early phase of the formalin response […]
222 The authors also conducted tests for the effects of gabapentin, the (S)-enantiomer and the (R)-enantiomer using the rat carrageen-induced thermal hyperalgesia model and the mechanical hyperalgesia model. At the highest doses tested, gabapentin and the (S)-enantiomer demonstrated significant anti-hyperalgesia action in both models, while the (R)-enantiomer failed to show any such effect in either model at the highest dose tested (100 mg/kg).
223 The authors’ discussion of their research included the following (at page 1519):
The results presented here show that gabapentin and the S-(+)-isomer of 3-isobutylgaba possess antihyperalgesic actions in inflammatory pain models. Thus, both compounds dose-dependently and selectively blocked the development of the second phase of the formalin response and the maintenance of carrageenan-induced mechanical and thermal hyperalgesia.
The authors further note (at page 1520):
It has previously been shown that gabapentin binds with high affinity to the α2δ subunit of a voltage-dependent calcium channel (Gee et al., 1996). These studies further revealed that 3-isobutylgaba binds to the α2δ protein in a stereoselective manner. (S)-( + )-3-isobutylgaba was found to show a similar affinity as gabapentin, but the (R)-(-)-isomer was 10 fold weaker. The present results show that the (S)-(+)-isomer of 3-isobutylgaba displays similar antihyperalgesic potency to gabapentin in both carrageenan and formalin-induced hyperalgesia models. In contrast, (R)-(-)-3-isobutylgaba was either inactive or only weakly active at 10 fold higher doses than the (S)-(+)-isomer. Previously, a similar rank order of potency was obtained for these three compounds in experimental models of epilepsy (Taylor et al., 1993).
224 This paper by Jong Hun Jun and Tony L Yaksh is entitled “The effect of intrathecal gabapentin and 3-isobutyl γ-aminobutyric acid on the hyperalgesia observed after thermal injury in the rat” and was published in 1998 in the peer reviewed journal Anaesthesia & Analgesia.
225 In this paper the authors reported on in vivo tests they conducted aimed at measuring hyperalgesia in rats inflicted with a mild thermal injury that was treated with a spinal administration (intrathecal) of gabapentin, the (S)-enantiomer or the (R)-enantiomer. Some reversal of hyperalgesia was produced in rats treated with gabapentin and the (S)-enantiomer, but not in those treated with the (R)-enantiomer even at the highest dose (300 μg) (ie. micrograms).
226 The authors noted (at page 352):
There are now data that suggest that gabapentin may represent a class of drugs, which exert their actions through a novel mechanism. First, gabapentin binds brain tissue with high nanomolar affinity. Second, although this binding seems not to correspond with any known transmitter site, it is displaced by a structural analog S(+)-3-isobutyl GABA, but not by the stereoisomer R(-)-3-isobutyl GABA. Third, the stereoselectivity in binding is reflected in the anticonvulsant activity and now in the spinal antihyperalgesic effects. […] These convergent observations jointly suggest that gabapentin and 3-isobutyl GABA may share a common, if as yet undefined, mechanism of action.
(Citations omitted)
227 The Carlton paper, by Susan Carlton and Shengtai Zhou, is entitled “Attenuation of formalin-induced nociceptive behaviors following local peripheral injection of gabapentin”. It was published in 1998 in Pain, another respected, peer reviewed scientific journal. It reports the results of the authors’ preclinical study designed to determine if local peripheral injection of gabapentin (“GP”) or the (S)-enantiomer of the 3-amino compound could reduce formalin induced nociceptive behaviour in rats. The compositions tested included GP plus 2% formalin, in doses of 6, 60 or 600 μg, the (S)-enantiomer plus 2% formalin in doses of 300 or 600 μg, and the (R)-enantiomer plus 2% formalin in doses of 600 μg.
228 The authors reported (at page 205):
S-(+)-3-Isobutylgaba and R-(-)-3-isobutylgaba are two enantiomers which have a pharmacologic profile similar to GP but they differ significantly in potency (Taylor et al., 1993). The present study demonstrates that S-(+)-3-isobutylgaba reduces formalin-induced nociceptive behaviors with slightly less efficacy than GP. This is in contrast to Field et al. (1997 …) who reported that systemic administration of S-(+)-3-isobutylgaba was more effective than GP in reducing nociceptive behaviors in the formalin test … The S enantiomer is significantly more active than the R enantiomer in displacing [3H]GP binding and in preventing maximal electroshock seizures in mice (Taylor et al., 1993). In agreement with this, a greater efficacy of S-(+)-3-isobutylgaba compared to R(-)3-isobutylgaba has been described in other behavioural studies (Jun and Yaksh, 1998; […]; Field et al., 1997 …).
(Emphasis added)
They concluded (at page 206):
In conclusion, the present study demonstrates that local peripheral injection of GP dramatically decreases both phase 1 and phase 2 formalin-induced nociceptive behaviors. This antihyperalgesic activity could be mimicked by S-(+)-3-isobutylgaba but not by its isomer R-(-)-3-isobutylgaba. […] local anesthetic effect. Thus, in addition to its actions following systemic and intrathecal routes, local peripheral administration of GP can produce dramatic antihyperalgesic effects. GP or its analog may offer a novel therapeutic agent for topical or local treatment of pathological pain of peripheral origin.
(Emphasis added)
229 The Lyrica PI states that the active ingredient of Lyrica is pregabalin, which is said to be an analogue of the neurotransmitter gamma-aminobutyric acid (GABA) having analgesic and anticonvulsant activity. It also includes the following section describing the mechanism of action of Lyrica (pregabalin):
In vitro studies show that pregabalin binds to an auxiliary subunit (α2-δ protein) of voltage-gated calcium channels in the central nervous system, potently displacing [3H]-gabapentin. Two lines of evidence indicate that binding of pregabalin to the α2-δ site is required for analgesic and anticonvulsant activity in animal models: (1) Studies with the inactive R-enantiomer and other structural derivatives of pregabalin and (2) Studies of pregabalin in mutant mice with defective drug binding to the α2-δ protein. In addition, pregabalin reduces the release of several neurotransmitters, including glutamate, noradrenaline, and substance P. The significance of these effects for the clinical pharmacology of pregabalin is not known.
(Emphasis added)
Apotex placed great weight on the description of the (R)-enantiomer in the Lyrica PI as “the inactive enantiomer”.
230 I think the scientific papers relied upon by Apotex in support of its contention that the (R)-enantiomer is ineffective, at any dose, as a treatment for pain, must be approached with caution. All of the studies relied upon are either pre-clinical in vitro studies to estimate the relative activity of gabapentin, the (S)-enantiomer and the (R)-enantiomer at the gabapentin binding site or pre-clinical in vivo studies in rodents that have some value as predictive tools, but which may not provide a reliable basis for inferring that the (R)-enantiomer is not useful as a treatment for pain particularly if administered at a high dose. In one of those studies, as also reported in the Patent, the (R)-enantiomer was shown to have an effect at 100 mg/kg in the rat formalin test, producing a “modest blockage” in the second phase. None of gabapentin (300 mg/kg), the (S)-enantiomer (100 mg/kg) or the (R)-enantiomer (100 mg/kg) had any effect in the first phase.
231 It is significant, in my view, that none of Apotex’s witnesses proffered any opinion in their evidence-in-chief, whether based on the scientific papers to which I have referred or otherwise, that the (R)-enantiomer is not, or would not be, effective in the treatment of pain if administered at an appropriate dose. I think this is a case in which it is open to me to draw an inference to the effect that Apotex’s witnesses’ evidence on this particular point would not have assisted its case.
232 For reasons that were never explained, the experts were not asked to consider whether the (R)-enantiomer was effective to treat pain in the Expert Conferences. However, in cross-examination of Professor Christie that took place after the completion of the concurrent evidence, Mr Catterns QC took up the issue with him. Mr Catterns’ cross-examination explored, at a general level, the question whether the (R)-enantiomer was “inactive” or “practically inactive”. The relevant portion of Professor Christie’s cross-examination included the following (T 611-613):
[…] Would you agree that, in the light of the material I’ve shown you, as a practical matter, the R enantiomer is inactive?
[…]
If you mean as a practical matter in terms of utility - - -
Yes?--- - - - I would have to say that the R enantiomer is relatively inactive and would not be preferred over the S enantiomer, because the S enantiomer is more active. So that’s really the practical interpretation. When I use the term “inactive”, in fact, it encapsulates in my mind that relativity.
It’s unlikely to have an effect equivalent to the S enantiomer unless administered in doses 10, 20 or more times higher? ---Yes, that’s correct.
That is a practical limitation; would you agree? ---That’s correct, yes.
[…]
And would you agree that means it’s practically inactive? ---I’m afraid I don’t know what “practically” means in that sense.
[…]
[…] Professor, in the context of a patent for the treatment of pain - - -? ---Yes.
--- where particular dosage ranges have been given on page 7, would you agree that the R enantiomer is practically inactive?---The dosage ranges given on page 7 are, on my preferred interpretation, one milligram per kilogram to 300 milligrams per kilogram daily, and one to 50 is preferred. I’m not sure to which the one milligram per kilogram to 300 milligrams per kilogram daily – which compound that’s actually referring to. Is it referring to the more preferred compound, or is it referring to the racemate or the R enantiomer? I’m not sure about that. If it is referring to those others, then we might expect to see some activity at the upper dosage range from the R enantiomer. And I can envisage scenarios where that may be practical. It may turn out that the S enantiomer is poisonous in humans and the R enantiomer is not.
Well, we now know that the S enantiomer is not? ---Yes.
I’m now bringing ourselves forward, if I may, in time? ---Yes.
(Emphasis added)
233 It is desirable to refer again to page 7, lines 21 to 27, of the Patent at this point. It will be recalled that the Patent (as I have interpreted it) specifies a dosage range with 300 mg/kg at the upper end based upon an average 70 kg patient, and a preferred dosage range of 1 mg/kg to 50 mg/kg. However, the Patent also makes clear that these dosages may be varied depending upon (inter alia) “the compound being employed”. Thus, the Patent expressly contemplates that for what might be referred to as the less preferred compounds, it may be necessary to use a dose of greater than 300 mg/kg. I understood Professor Christie to say that at a dose of 300 mg/kg he would expect the (R)-enantiomer to exhibit some activity. His evidence to that effect was not challenged by the cross-examiner which I regard as quite significant in circumstances where no witness called by Apotex proffered a contrary opinion.
234 The Lyrica PI expressly mentions doses of pregabalin of up to 600 mg/kg per day, and A/Prof. King said that, in his experience, about 600 mg/kg per day is the maximum dose tolerated for pregabalin. However, there was evidence to indicate that some of the clinicians prescribe pregabalin at even higher doses. Professor Schug prescribes dosages of up to 900 mg/kg for some patients with spinal injuries. Apotex made no attempt to show that the (R)-enantiomer could not also be administered at dosages of between 600mg/kg and 900 mg/kg.
235 I am not persuaded that the (R)-enantiomer would be ineffective in the treatment of pain if administered at high doses. Clearly, the (R)-enantiomer is considerably less active than the (S)-enantiomer and is likely to be less active than the racemate. But in Professor Christie’s words, the (R)-enantiomer is only “relatively inactive” and that is the sense in which I would interpret the word “inactive” where used to describe the (R)-enantiomer in the Lyrica PI. Even though the (R)-enantiomer may be much less active than the (S)-enantiomer, it does not follow, and the evidence does not establish, that the (R)-enantiomer would be of no use in the treatment of pain. The fact that there may be no market for the (R)-enantiomer as a pain treatment (due to the availability of superior drugs including pregabalin and gabapentin) does not mean that the (R)-enantiomer is not useful in the relevant sense.
236 Section 40(2)(a) of the Act provides that a complete specification must “fully describe” the invention.
237 It is convenient at this point to consider the submission made by Pfizer which was said to provide a complete answer to Apotex’s insufficiency case. Pfizer submitted that the test for sufficiency to be applied in this case is that articulated by the High Court in Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 (“Kimberly-Clark”) at [25]. The High Court held that the test for sufficiency was encapsulated by the following question:
The question is, will the disclosure enable the addressee of the specification to produce something within each claim without new inventions or additions or prolonged study of matters presenting initial difficulty (59)?
The footnote refers to a statement in Blanco White, Patents for Inventions, 5th ed (1983), at para 4-502 where the learned author states:
To be proper and sufficient, the complete specification as a whole (that is, read together with the claims, and in the light of the drawings, if any) must in the first place contain such instructions as will enable all those to whom the specification is addressed to produce something within each claim “by following the directions of the specification, without any new inventions or additions of their own” and without “prolonged study of matters which present some initial difficulty.”
(Footnotes omitted)
238 In Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (2004) 217 CLR 274 (“Doric”) the High Court, citing Kimberly-Clark at [25], said at [60]:
For the purposes of s 40(2)(a), it is not necessary for the inventor to disclose all the alternative means; it is enough that there is disclosure in the sense of enabling the addressee of the specification to produce something within each claim without new inventions or additions or prolonged study of matters presenting additional [sic] difficulty.
239 Kimberly-Clark was concerned with a patent for a disposable nappy and the question was whether the specification sufficiently enabled the manufacture of a nappy having each of the essential integers of the claimed invention. The case raised two questions. The first was whether, in determining whether an invention was sufficiently described, it was permissible to have regard to what was disclosed in the claims. The second question involved a factual inquiry and a consideration of whether the evidence before the primary judge supported his Honour’s finding that the claimed invention was sufficiently described.
240 Doric was concerned with a patent for a key controlled latch assembly suitable for use in certain door locks. The High Court considered the scope of the requirement imposed by s 40(3) of the Act (as it then stood) that a claim be fairly based on the matter described in the specification. In the course of rejecting the applicability of Lord Hoffman’s reasoning in Biogen Inc v Medeva Plc [1997] RPC 1 at 45, the High Court referred at [67] to “the rule” recognised in the High Court’s construction of s 40(2)(a) in Kimberly-Clark at [25] as “an important aspect of Australian law …”.
241 As I have previously said, the language of the claims of the Patent is expressly directed to the treatment of pain in mammals generally and, contrary to a submission made by Apotex, none of the claims is confined to the treatment of pain in humans. Against that background, Pfizer submitted that the requirements of s 40(2)(a) would be satisfied in this case if the Patent contains sufficient information to enable the skilled addressee to perform the invention in relation to rats. I do not accept that submission.
242 Although the claims refer to mammals rather than humans, and the various test results reported in the Patent all relate to experiments on rats, it is clear that the Patent is primarily directed to the treatment of pain in humans. This is clear from the description of the various disorders that cause nerve damage and neuropathic pain, which are referred to in the detailed description at page 5 lines 9 to 22 of the Patent (eg. chronic alcoholism and human immunodeficiency virus infection).
243 The fact the test results presented in the Patent all relate to experiments on rats is due to the fact that these were considered useful in predicting the effect of the relevant compounds in humans. Not for a moment would the skilled addressee understand this Patent as directed to the medical treatment of rats experiencing pain.
244 For reasons explained in Tramanco Pty Ltd v BPW Transpec Pty Ltd (2014) 105 IPR 18 (“Tramanco”) it may not be possible to apply the Kimberly-Clark test of sufficiency to every manner of claim. As I explained in Tramanco at [207]:
In Kimberly-Clark and Doric the High Court was concerned with various product claims each of which consisted of a number of features all of which were essential (in the sense of non-optional) integers of the claim. However, there are some types of claim that may need to be approached slightly differently including, in particular, claims to methods for producing one or more specified results. For example, a claim for a method of producing one or more of outcomes A, B or C might be infringed if the alleged infringer uses the method to produce outcome A, but not outcome B or C. Whether there is infringement in such a case will depend upon (inter alia) the proper construction of the claim and, in particular, whether it requires the use of the method to produce only one or more of outcomes A, B or C, as opposed to all three of them. Assuming the former construction (in the present case it appears to have been assumed by both parties at the trial and on appeal that claim 1 is to be construed in this way), it would seem to me to be wrong in principle to hold that the description of the invention is sufficient if the specification enables the use of the method to achieve outcome A, but not outcomes B or C. It would be inconsistent with the purposes of the Act to confer a monopoly on a patentee for a method of producing any of outcomes A, B or C, if the patentee’s disclosure only enabled the use of the method to produce some of those outcomes.
Allsop CJ (at [53]) and Greenwood J (at [164]) agreed with these observations.
245 Tramanco was not concerned with methods of medical treatment, but with a method for determining one or more parameters relating to the performance of the suspension system of a vehicle. In the present case the method claims, as I have construed them, are to methods of treating pain, or different types of pain, in all kinds of mammals including, in particular, humans.
246 As we are reminded from time to time, judgments are not to be construed as if they were statutes, and it is always necessary to read and understand them in context. Application of the test of insufficiency propounded by the High Court in Kimberly-Clark based upon a test originally formulated outside the context of methods of treatment claims cannot be sensibly applied to a case such as this. In my respectful opinion, it would be a nonsense to say that the invention was sufficiently described if it enabled the skilled addressee to perform the method of the invention on rats (or other mammals) but did not enable it to be performed on humans.
247 In my view, s 40(2)(a) requires that the Patent in this case enable the skilled addressee to perform the claimed invention in relation to humans without new inventions or additions or prolonged study of matters presenting initial difficulty. It is only by requiring this degree of enablement that the patentee could be sensibly understood to have given consideration for the grant of a patent which the skilled addressee would understand to be essentially directed to the treatment of pain in humans.
248 Lord Parker observed in Osram Lamp Works Ltd v Pope’s Electric Lamp Co Ltd (1917) 34 RPC 369 at 391:
A Specification may therefore be considered as addressed, at any rate primarily, to the persons who would, in normal course, have to act on the directions given for the performance. These persons may be assumed to possess not only a reasonable amount of common sense, but also a competent knowledge of the art or arts which have to be called into play in carrying the patentee's directions into effect. I say “art or arts” because in carrying out the directions given by the patentee it may well be necessary to call in aid more than one art. Some of the directions contained in a Specification may have to be carried out by skilled mechanics, others by competent chemists. In such a case, the mechanic and chemist must be assumed to co-operate for the purpose in view, each making good any deficiency in the other’s technical equipment. The Specification cannot be considered insufficient merely because the mechanic without the aid of the chemist, or the chemist without the aid of the mechanic would be unable to comprehend the meaning of or to carry into effect the directions given by the patentee.
249 I have previously noted that the notional skilled addressee will possess common general knowledge in the fields of pharmacology, pharmaceutical formulation, pain medicine and organic chemistry as it stood at the application date. For this purpose the notional skilled addressee in the present case may be considered either as a notional skilled team or a notional skilled person having expertise in all these fields. This notional team or notional person must be taken to possess a complete knowledge and understanding of the common general knowledge in respect of all of these fields.
250 In essence, Apotex submitted that the Patent does not describe any safe and effective doses and dosage regimes which the skilled addressee may utilise for the purpose of treating a patient in need of treatment for pain. In support of its submission, Apotex made the following points:
The Patent does not describe any specific dose for the (S)-enantiomer or any other compound. The description of doses at page 7, lines 21 to 27 refer to a preferred dose of about 1mg/kg to about 300 mg/kg based on the average 70 kg patient, but these statements are not made in relation to any particular compound.
The Patent does not include any information in relation to solubility, bioavailability or safety. The experimental work referred to in the Patent all relate to in vivo tests on rats treated with (inter alia) the (S)-enantiomer. There is no disclosure of any pre-clinical pharmacodynamic, pharmacokinetic or toxicology tests, nor any Phase 1, 2 or 3 trials.
The fact that pregabalin is now known to be safe and efficacious is irrelevant because the sufficiency of the description is to be assessed on the basis of what is disclosed in the specification as it would be read and understood by the skilled addressee as at the application date. At that date, the compounds claimed in the Patent were new chemical entities that were not the subject of any regulatory approval and lacking any known safety and toxicity profile.
251 In his closing submissions Mr Catterns QC accepted (T 946) that the skilled addressee is provided with sufficient information to make pregabalin. He also accepted that if pregabalin was a known compound previously registered for therapeutic use in humans with a known safety and toxicity profile, then it would be within the competence of the skilled addressee to determine an appropriate dose for the treatment of a particular condition by incrementally increasing the dose until the desired therapeutic effect was achieved (ie. dose titration).
252 However, Mr Catterns submitted that, given the limited information disclosed in the Patent, the skilled addressee could not simply work within the broad dose range provided and titrate the dose up. This is because, at the application date, pregabalin was a new chemical entity and, so far as the notional skilled addressee reading the Patent would be aware, had not yet been tested in humans. He submitted that before pregabalin could be administered to a human in need of a treatment for pain, it would be necessary to undertake further research and study to obtain an understanding of the safety and toxicity profile of the drug, and this would impose an “undue burden” on the skilled addressee who was seeking to use the claimed methods of treatment.
253 The term “undue burden” is frequently used in the English authorities which have held that a patent specification must be sufficient to allow the invention to be performed over the whole scope of the claimed invention without “undue burden”: see the decision of Arnold J in Eli Lilly & Company v Janssen Alzheimer Immunotherapy [2014] RPC 1 and the authorities referred to by his Lordship at [248]-[250].
254 Mr Bannon SC submitted that Apotex’s approach to sufficiency was misconceived because under Australia law, all that was required by s 40(1)(a) was that the skilled addressee be given information sufficient to enable the invention to be performed if the skilled addressee chose to perform it. He submitted that the relevant question is not whether a clinician would choose to perform the invention, but whether he or she could perform the invention. On that basis, Mr Bannon submitted that it was not necessary for the patentee to include in the specification any information concerning the safety or efficacy of the claimed methods in order to satisfy the requirements of s 40(1)(a). He submitted that the skilled addressee is provided all the information required to perform the invention by the Patent supplemented by the common general knowledge.
255 Applying the standard of enablement postulated by the High Court in Kimberly-Clark, a claim will be invalid if the claimed invention cannot be performed “without new inventions or additions or prolonged study of matters presenting initial difficulty.” As to what constitutes “prolonged study of matters which present some initial difficulty”, the passage from Blanco White, Patents for Inventions, 5th ed (1983) at para 4-502 referred to by the High Court in Kimberly-Clark at [25] cites Valensi v British Radio Corporation [1973] 90 RPC 337 (“Valensi”) at 377. In that case the Court of Appeal reviewed the authorities and concluded that:
We think that the effect of these cases as a whole is to show that the hypothetical addressee is not a person of exceptional skill and knowledge, that he is not to be expected to exercise any invention nor any prolonged research, inquiry or experiment. He must, however, be prepared to display a reasonable degree of skill and common knowledge of the art in making trials and to correct obvious errors in the specification if a means of correcting them can readily be found.
[…]
Further, we are of the opinion that it is not only inventive steps that cannot be required of the addressee. While the addressee must be taken as a person with a will to make the instructions work, he is not to be called upon to make a prolonged study of matters which present some initial difficulty: and, in particular, if there are actual errors in the specification - if the apparatus really will not work without departing from what is described - then, unless both the existence of the error and the way to correct it can quickly be discovered by an addressee of the degree of skill and knowledge which we envisage, the description is insufficient.
256 Valensi was concerned with errors in the specification which the skilled addressee needed to understand and correct in order to make the claimed invention work. Earlier in its reasons the Court of Appeal referred to the well-known case of No-Fume Ltd v Frank Pitchford & Co Ltd (1935) 52 RPC 231. The description of the invention in that case was said to be insufficient because the specification did not disclose certain dimensions for a mechanical device. Lord Hanworth MR was satisfied that the description was sufficient because the proportions of the device could be ascertained without the need for invention. Romer LJ said at 243:
[T]he Patentee fulfils his duty if in his complete specification he describes and ascertains the nature of the invention, and the manner in which the invention is to be performed, sufficiently and fairly. It is not necessary that he should describe in his specification the manner in which the invention is to be performed, with that wealth of detail with which the specification of the manufacturer of something is usually put before the workman who is engaged to manufacture it. Specifications very frequently contain mistakes; they also have omissions. But if a man skilled in the art can easily rectify the mistakes and can readily supply the omissions, the patent will not be held to be invalid. The test to be applied for the purpose of ascertaining whether a man skilled in the art can readily correct the mistakes or readily supply the omissions, has been stated to be this: Can he rectify the mistakes and supply the omissions without the exercise of any inventive faculty? If he can, then the description of the specification is sufficient. If he cannot, the patent will be void for insufficiency.
257 Mr Bannon SC submitted that the notion of “difficulty” in this context is not concerned with how hard the work is, or how significant the burden of that work may be, but only with difficulties in working the invention that arise from some defect in the way in which the invention is described. Pfizer relied upon the following statement of McTiernan J in Samuel Taylor Pty Ltd v SA Brush Co Ltd (1950) 83 CLR 617 at 625:
In complying with the second condition i.e., in describing in what manner the invention is to be performed, the Patentee does all that is necessary, if he makes it plain to persons having reasonable skill in doing such things as have to be done in order to work the patent, what they are to do in order to perform the invention. If, as may happen, they are to do something the like of which has never been done before, he must tell them how to do it, if a reasonably competent workman would not himself see how to do it on reading the Specification…”
(Pfizer’s emphasis)
258 I do not accept that the scope of the obligation imposed by s 40(2)(a) of the Act is as narrow as Pfizer suggests. Nor do I understand McTiernan J to have held that the obligation is satisfied provided that the working of the patent does not require the skilled addressee to do something that has never been done before. There may well be things that have been done before in relation to which the patentee must still provide the skilled addressee with some directions if it is not within the ordinary skill and knowledge of a person working in the relevant field. Further, in describing the invention fully, it may be necessary for the inventor to explain by reference to his or her own experimental work what it is that he or she has discovered. In the present case I doubt that it would be possible for the inventor to fully describe the invention without doing so by reference to the animal tests referred to in the Patent that apparently provided the basis for the inventor’s discovery that the relevant compounds would be useful in the treatment of pain in humans. The results of these tests appear to lie at the heart of the inventive concept.
259 The description of the invention will not be insufficient merely because the skilled addressee is expected to apply considerable skill, effort and resources to make it work. If the steps required to be taken to work the invention are readily apparent to the notional skilled addressee, and they are standard or routine steps within the competence of the notional skilled addressee, then the test for sufficiency will be satisfied.
260 In Eli Lilly and Company v Pfizer Overseas Pharmaceuticals (2005) 64 IPR 506 Heerey J was concerned with the alleged infringement by the applicant (“Lilly”) of a claim (claim 10) to a method of treatment for impotence that required the use of a particular compound (a particular cGMP-specific PDEV inhibitor) within a much broader class of compounds which, for convenience, I shall refer to as compounds of the invention. The specification described various in vitro tests on human tissue for determining the potency of various compounds of the invention to inhibit the cGMP-specific PDEV. The specification discussed some toxicity testing of some of the relevant compounds (which were not specifically identified) in rats, dogs and mice and also reported that various “especially preferred” compounds of the invention had been administered to volunteers in both single and multiple dose studies and that one of these compounds induced erections. Again, none of the compounds administered were specifically identified.
261 Lilly alleged that claim 10 was invalid because the specification did not provide sufficient information to enable the skilled addressee to perform the claimed method. Heerey J found that a pharmacologist and a medicinal chemist with experience in drug discovery and development would be members of the notional skilled team and the medicinal chemist would be able to synthesise the relevant compound using information incorporated in the patent. His Honour said at [193]:
It would be necessary to test for oral bioavailabilty, toxicity and effectiveness, but the evidence shows that while these steps call for skill, they are essentially routine for those skilled in this area. The term routine here (and in other contexts in this case) is not used as a synonym for simple and easy. In the present case the hypothetical skilled workers at the hypothetical workbench are persons holding academic qualifications at the Ph D level together with practical experience. It would not be necessary to employ such persons unless the task they had to perform was a difficult one. Yet this does not of itself mean that the patent could not be worked without further invention.
262 His Honour’s decision on this issue was upheld on appeal: see Pfizer Overseas Pharmaceuticals v Eli Lilly and Company (2005) 225 ALR 416. French and Lindgren JJ (with whom Crennan J agreed on this issue) observed at [342]:
It was not disputed that a range of tests would have to be performed before the compound was administered to a human to determine whether it were efficacious. Lilly submitted therefore that a considerable investment in time and resources was required before any compound could be tested for efficacy in humans. Given that time and cost it submitted that forcing a skilled addressee to discriminate between even two compounds by not identifying that for which the patentee generated a result constituted a requirement that the skilled addressee undertake prolonged study of matters presenting initial difficulty within the meaning of Kimberly-Clark […].
263 French and Lindgren JJ rejected this submission and held that there was more than an adequate basis for Heerey J’s conclusion. It is significant that Heerey J’s approach to the issue of sufficiency focused on whether testing for toxicity and effectiveness of the particular compound was routine or whether it required further invention. No member of the Full Court suggested that this approach was incorrect.
264 It is necessary to make some findings in relation to the steps that must be followed to obtain regulatory approval permitting the clinical use of a new drug. Before a new drug can be registered and approved for clinical use, it must first be evaluated for safety and effectiveness. To a large extent the process is determined by practices and procedures prescribed by regulatory authorities. The usual stages of the process are as follows:
Pre-clinical studies – The new drug is tested in vitro and in animal (eg. rats, rabbits and monkeys) tests to evaluate its toxicological, pharmacodynamics and pharmacokinetic properties. The data obtained from the pre-clinical studies provides safety and toxicity information which will determine whether or not the drug is sufficiently safe for use in clinical trials.
Phase 1 studies – The drug is tested in humans for the first time, initially in very low doses in a small group of healthy volunteers. Phase 1 is mostly concerned with determining a safe dosage range and identifying any potential issue with respect to toxicity or side effects. Dose response data is examined to determine the relationship between dose and clinical response. The relationship between dose and effect (both desirable and undesirable) can be charted on a dose response curve. The dose response data is then used as the basis for dosing in later phases.
Phase 2 studies – The drug is tested on a larger group of volunteers (typically a few hundred) in order to evaluate efficacy, to further evaluate safety, and to refine the dosing regimen.
Phase 3 studies – The drug is tested on a still larger group of people (from several hundred to several thousand) to determine its efficacy in comparison to other drugs and/or placebo, to monitor for any adverse effects, and to collect any information that will allow the drug to be used safely.
Sometimes a Phase 2 study may be permitted to transition into a Phase 3 (which is referred to as a Phase 2/3 study).
265 Once these studies have been completed and a proper dosage range is determined, the drug may then be approved and registered on the Australian Register of Therapeutic Goods (“ARTG”). In most cases this will occur after the drug has been approved for clinical use by the US Federal Drug Administration (“FDA”) or the European Medicines Agency (“EMA”)
266 The steps required to take a new drug from discovery to approval are complex, time-consuming and expensive. Professor Christie’s evidence indicated that to start with a new compound (eg. pregabalin) and move through the pre-clinical stage alone may take several years and the Phase 1 and Phase 2 studies may each occupy several more years. Further, not all new drugs make it through all stages of the process; for example, some drugs are shown to be safe and effective at the Phase 2 stage but not at the Phase 3 stage.
267 The experts who participated in the Field 3 Expert Conference answered several questions relating to the issue of sufficiency in the Joint Report. Paragraph 3.6 of the report states:
3.6 How to take the information in the Patent and treat pain:
Given the information in the Patent, what steps need to be taken to treat pain in humans in clinical trials?
Given the information in the Patent, what steps need to be taken to treat pain in humans using a regulatory agency approved product?
We agree with the following brief outline of the steps that are required here, including:
1. Further animal studies for toxicology and pharmacokinetics
2. Further animal studies to identify which diseases or conditions are most promising for clinical development
3. Preformulation and formulation development
4. Phase 1 clinical trials (small size) - safety in healthy volunteers, pharmacological understanding to help with dosing.
5. Phase 2 clinical trials (trials of smaller size) - determining the dose(s) to be used and safety in people with specific disease states and effect so that the size of the phase 3 trial can be calculated
6. Phase 3 clinical trials (trials of larger size) - efficacy in the disease state compared to placebo or other standard, including safety signals
7. Registration
8. Phase 4 pharmacovigilance
The process is complex and expected to take 10 years and more than $ 1 billion per API [active pharmaceutical ingredient]. It is well known that there is an attrition rate in taking new drugs all the way through the development process.
This would require a multidisciplinary team skilled in the art; inventive matters may arise at any stage.
268 Steps 1, 2, 4, 5 and 6 identified by the experts broadly reflect those previously discussed. The eighth step relates to Phase 4 “follow up” studies that occur after approval, or conditional approval, is obtained from the relevant regulatory authority. These are follow-up studies designed to monitor safety and effectiveness (ie. risks versus benefits) over the longer term.
269 The fact that steps 1 to 5 or 6 would involve complex, time consuming, and expensive work is not controversial. But that does not mean that the steps involved are not routine for the notional skilled team or that the work required to be performed to obtain regulatory approval cannot be performed without the need for invention.
270 No doubt it is conceivable that problems may arise during the course of the process that may require a creative or inventive solution, which the highly skilled but unimaginative notional team cannot deliver. However, for reasons that I will explain, there is no evidence to indicate that the notional skilled team would be confronted with any such problem when working with pregabalin.
271 There are two further points to make. First, it must be assumed, for the purpose of deciding whether the work required to perform an invention imposes some impermissible measure of difficulty, that the notional skilled team will possess the will and resources necessary to perform the invention. There are many inventions that are complex, time consuming and expensive to perform, but it does not follow that the description of the invention is therefore insufficient. Secondly, it is necessary to distinguish between work required to check or test an invention or obtain regulatory approval for its use, and work that is necessary to perform the invention. As the Full Court observed in Merck & Co Inc v Arrow Pharmaceuticals Ltd (2006) 154 FCR 31 at [108] in relation to pharmaceutical patents (though not in the context of sufficiency) “… it is a matter of notoriety that prolonged testing for the purpose of regulatory approval must occur between the stage of patent application and commercial marketing.”
272 Mr Catterns QC relied upon the words “inventive matters may arise at any stage” in the last sentence of the experts’ answer to question 3.6. However, as I read those words, they do no more than recognise that there is a possibility in the case of any new drug that is being evaluated for the purposes of obtaining regulatory approval, that the notional skilled team might be confronted at one or more stages of the process with a problem that cannot be solved without invention.
273 In cross-examination of Professor Christie, Mr Catterns QC referred to extracts from a textbook entitled “Drug Safety Assessment in Clinical Trials” (Ex 6) that included some observations concerning data analysis and interpretation in pre-clinical drug safety evaluations. Referring to the database of information that must be collected the authors stated (at p 17):
… the database created in a routine preclinical toxicology package is enormous by any standards. The organization, interpretation, integration, and summation of these data are the responsibility of the toxicologist. To adequately review all of the information collected, the toxicologist draws on a working understanding of pharmacology, physiology, biochemistry, pathology, pharmacokinetics, and other fields within the medical sciences.
274 Professor Christie was not familiar with the textbook but he did agree with this statement. He was then asked some questions concerning certain challenges referred to by the authors faced by the toxicologist that required the exercise of skill and judgment. He agreed that these included the integration of data into a single comprehensive statement on toxicity and the extrapolation of safety data between different test species. However, it was not suggested to him that this work would be anything other than routine to a person with relevant expertise. Nor was it suggested that, in making such judgments, the notional skilled team would be exercising skills or making judgments that involve creative or inventive reasoning or problem solving beyond the competence of the highly skilled, but unimaginative, expert working in the field.
275 Apotex relied upon various reports of studies (reproduced in Ex 1) conducted by or for Pfizer. These reports record the research and experimental work undertaken between July 1992 and December 2000 in relation to the use of CI-1008 (pregabalin) as a possible treatment for seizures or pain. There was almost no written or oral evidence specifically directed to these reports with the exception of Document 17 (which is a reproduction of the Field paper) and Apotex appears to have assumed that they would speak for themselves.
276 It is apparent from a reading of the documents in Ex 1 that they broadly reflect the course of research work that is usually undertaken for the purpose of obtaining regulatory approval from the FDA for the clinical use of a new drug.
277 The vast majority of the documents in Ex 1 record the results of pre-clinical evaluations of the toxicological and pharmacokinetic properties of CI-1008: see Documents 2-14, 20, 23-28, 30-31 and 38. In particular, these documents record the results of laboratory testing in mice, rats, monkeys, dogs and rabbits in relation to a wide range of matters including:
the bioavailability of CI-1008 as determined using mice, rats, monkeys and human plasma;
the stability of CI-1008 including the potential for degradation or racemization using mouse, rat, monkey and human plasma;
toxicity studies using rats, dogs and monkeys to determine metabolic profile and route of excretion of CI-1008;
toxicity tests using oral or intravenous administration of CI-1008 in rats and monkeys including escalating and continuous dosing with the compound;
toxicity studies using monkeys to determine effects of CI-1008 on (inter alia) blood pressure, heart rate and cardiac rhythm.
278 The other documents, excluding some summaries and memoranda prepared by or for the FDA, are reports of Phase 1, Phase 2/3 and Phase 3 clinical trials.
279 The first Phase 1 study in which CI-1008 was administered to healthy volunteers appears to have been conducted between February and June 1996: see Document 15. Doses administered were initially very small, but were slowly escalated in the absence of any significant adverse events. Some mild to moderate side effects were observed (eg. dizziness, headache, drowsiness and nausea). The drug was reported to be generally well tolerated at the doses tested. Other Phase 1 studies were conducted in 1996, 1997, 1999, 2000 and early 2001: see Documents 16, 18, 19, 32-33, 36 and 37.
280 Exhibit 1 contains two reports relating to what are described as Phase 2/3 studies: see Documents 22 and 29. The first of these was conducted between March 1998 and March 1999 and involved several hundred patients. The second report relates to another Phase 2/3 study conducted between October 1998 and July 1999 involving several hundred patients. Both studies appear to show that treatment with pregabalin was safe and well tolerated. Exhibit 1 also contains a report of a large Phase 3 multi-centre study of pregabalin that took place between September 1999 and December 2000. The study concluded (inter alia) that pregabalin was well-tolerated with no unexpected side effects.
281 The Ex 1 documents were relied by Apotex in support of two submissions. First, they were relied upon in support of the submission, previously discussed, that the notional skilled team would be confronted with an “undue burden” or, in the language of Kimberly-Clark, a need for “… new inventions or additions or prolonged study of matters presenting initial difficulty” when performing the invention. Secondly, the Ex 1 documents were relied upon to show that prior to the application date, Pfizer had engaged in a substantial amount of pre-clinical work and that it had by then already completed at least one Phase 1 study. Apotex submitted that it would not impose any “undue burden” on Pfizer to require it to include in the complete specification as originally filed some of the safety information thereby obtained. It is convenient to deal with this latter submission first.
282 The law of sufficiency is concerned with the “enablement” of the invention. In the United Kingdom what is sometimes referred to as “classical insufficiency” arises when the specification fails to provide the skilled addressee with the information required to enable the invention to be performed without “undue burden”. The term “undue burden” is used to describe the extent of the burden imposed upon the skilled addressee. Leaving aside the issue of “best method” (which is not relevant in this case) the question raised by a challenge to the sufficiency of the description of a claimed invention is not whether the patentee could have supplied the skilled addressee with more information than it did, but whether the skilled addressee has been supplied with enough information to enable the working of the invention.
283 During his opening Mr Catterns QC drew my attention to Document 9 which is a report entitled “Oral Toxicity Study of CI-1008 in Cynomolgus Monkeys” dated 14 November 1994. This report indicates that CI-1008 caused mortality (death), ataxia (loss of control of bodily movements) and hypoactivity (drop in activity) in a number of monkeys after doses of 1000 or 2000 mg/kg. The experiment was apparently terminated early because four moneys died after the first dose. The suggestion seemed to be, though it was never put in these terms, that I should conclude from this report and a few other animal studies in which some adverse events were noted, that the pre-clinical work undertaken by Pfizer would have presented the skilled addressee with problems that would be difficult for the notional skilled team to resolve and that would be beyond the competence of the notional skilled team engaged in routine testing. However, no such proposition was put to any expert witness and any submission to that effect based upon those particular reports, or any others included in Ex 1, cannot be sustained on the evidence.
284 The challenge to the validity of the Patent based upon any alleged insufficiency of description fails.
285 Section 138 of the Act relevantly provides:
138 Revocation of patents in other circumstances
(1) Subject to subsection (1A), the Minister or any other person may apply to a prescribed court for an order revoking a patent.
[…]
(3) After hearing the application, the court may, by order, revoke the patent, either wholly or so far as it relates to a claim, on one or more of the following grounds, but on no other ground:
(a) that the patentee is not entitled to the patent;
[…]
(4) A court must not make an order under subsection (3) on the ground that the patentee is not entitled to the patent unless the court is satisfied that, in all the circumstances, it is just and equitable to do so.
286 As to s 138(3)(a), Apotex’s pleaded case is that Warner-Lambert is not entitled to the Patent, because the inventor, Mr Singh, from whom Warner-Lambert purports to derive its title, did not invent the use of the racemate to treat pain. I do not think there is any substance to this contention.
287 A person may be the inventor of a patentable invention without reducing it to practice or proving that it actually works. Mr Singh will be the inventor of the invention described in the Patent by virtue of him having conceived that the compounds of Formula I (which include the racemate of the 3-amino compound) might be used in the manner described in the Patent in the treatment of pain. There is no evidence to suggest that he was not the person who conceived this invention. His status as the inventor does not depend upon him having used or tested any particular compound.
288 As to s 138(4), Apotex did not plead or submit (either in writing or orally) that in the circumstances of this case, if the Court was to hold that Warner-Lambert is not entitled to the Patent, it would be just and equitable to revoke the Patent or any one or more of its claims. As I pointed out at the commencement of these reasons, s 138(4) of the Act (in its current form) applies to the claims for relief made in these proceedings pursuant to s 138(3)(a) of the Act.
INFRINGEMENT
289 Apotex contends that it does not threaten to infringe any of the Swiss claims (claims 16-30 and 32) because the medicaments it threatens to import and supply in Australia will be made outside the “patent area” by a third party. It submits that the importation and sale in Australia of medicaments made outside the patent area cannot infringe a Swiss claim. GPPL relies on the same argument in relation to each of the Swiss claims relied upon against it (claims 16-30). For the following reasons I do not accept Apotex’s submission.
290 To understand Apotex’s argument it is necessary to refer to the relevant statutory provisions. Sections 12 and 13 of the Act provide:
This Act extends to:
(a) each external Territory; and
(b) the Australian continental shelf; and
(c) the waters above the Australian continental shelf; and
(d) the airspace above Australia, each external Territory and the Australian continental shelf.
13 Exclusive rights given by patent
(1) Subject to this Act, a patent gives the patentee the exclusive rights, during the term of the patent, to exploit the invention and to authorise another person to exploit the invention.
(2) The exclusive rights are personal property and are capable of assignment and of devolution by law.
(3) A patent has effect throughout the patent area.
The definition of “patent area” includes Australia and the other areas referred to in s 12. The term “exploit” is defined in Schedule 1 to mean:
exploit, in relation to an invention, includes:
(a) where the invention is a product–make, hire, sell or otherwise dispose of the product, offer to make, sell, hire or otherwise dispose of it, use or import it, or keep it for the purpose of doing any of those things; or
(b) where the invention is a method or process–use the method or process or do any act mentioned in paragraph (a) in respect of a product resulting from such use.
291 There are two points to note about the definition of exploit: first, it is inclusive; second, it does not include any express territorial limitation. Relevant express territorial limitations are found in s 12 and s 13. In particular, s 13 makes clear that the exclusive rights conferred on a patentee by the grant of a patent apply throughout the “patent area” and, by implication, not outside the “patent area”.
292 All parties approached the present question on the assumption that a Swiss claim is properly characterised as a method or process claim. This view of the Swiss claims accords with that expressed by Yates J in Otsuka Pharmaceutical Co Ltd v Generic Health Pty Ltd (No 4) (2015) 113 IPR 191 and the authorities referred to by his Honour at [117]-[121]. All parties accepted that it is appropriate to consider the question of infringement of the Swiss claims through the prism of para (b) of the definition of “exploit”.
293 Apotex submitted that para (a) of the definition should be interpreted as though it referred to a product made in the patent area and that para (b) should be given a corresponding interpretation so that “any act mentioned in paragraph (a)” will be taken to refer to an act that is done in respect of a product resulting from the use of a patented method in the patent area.
294 On Apotex’s construction, para (b) would include a sale of a product made by a patented method in Australia but not a sale of a product made by the patented method in another country. In support of this construction, Apotex relied upon some (but certainly not all) of Lindgren J’s reasoning in Alphapharm Pty Ltd v H Lundbeck A/S (2008) 76 IPR 618 (“Alphapharm”) at [691]-[694]. In that case Lindgren J held at [694] that the definition of “exploit” should be interpreted as follows:
exploit, in relation to an invention includes:
(a) where the invention is a product — [in Australia, or more accurately, in the patent area] make, hire, sell or otherwise dispose of the product, offer to make, sell, hire or otherwise dispose of it, use or import it, or keep it for the purpose of doing any of those things; or
(b) where the invention is a method or process — [in Australia, or more accurately, in the patent area] use the method or process or do any act mentioned in paragraph (a) in respect of a product resulting from [the use, anywhere, of the method or process].
(Emphasis added)
295 Apotex submits that Lindgren J should also have read into para (b) of the definition a further territorial limitation requiring that the product resulting from the use of the relevant method be one that results from the use of the method in the patent area.
296 The definition of “exploit” makes no reference to the patent area. As I have said, the express territorial limitation upon the patentee’s exclusive rights is found in s 12 and s 13. In my respectful view, there is therefore no reason to read down the words of either para (a) or para (b) of the definition of “exploit” to found any territorial limitation. This is because the Act expressly provides that a patent only has effect in the patent area: see also s 70 of the Patents Act 1952 (Cth).
297 Paragraph (b) of the definition of “exploit” refers to the doing of an act referred to in para (a) which includes to make or import a product. The patentee’s exclusive rights are infringed (subject to available defences) if another person does any such act within the patent area. The fact that the patented method is performed outside the patent area does not avoid infringement of a method claim (including a Swiss claim) if the product imported and sold in Australia was made using the patented method because the acts of importation and sale occur within the patent area. The relevant act of infringement is not the use of the method outside the patent area but the exploitation (by importation and sale) in Australia of a product made using the patented method.
298 In my respectful opinion, contrary to the approach taken by Lindgren J, the relevant territorial limitation is reflected in the language of s 12 and s 13(3) and there is therefore no justification for importing words of territorial limitation into the definition of “exploit”. It follows that I take a somewhat different approach to the construction of the definition of “exploit” to that taken by Lindgren J in Alphapharm, though I do not think the difference has any impact on whether or not Apotex threatens to infringe the Swiss claims in this case.
299 In opening Apotex also contended that it did not threaten to infringe claims 31 and 32 “because there were no relevant methods of treatment exemplified in the Patent.” This contention was not developed by Apotex in its closing submissions. In any event, so far as claim 31 is concerned, the point is not open to Apotex on the pleadings. As to claim 32, it seems to reflect a particularly poor attempt by the patentee to draft what was probably intended to be a claim for the use of a relevant compound as a medicament for the treatment of pain. Since the examples and accompanying figures referred to in claim 32 shows the use of a relevant compound in experiments on rats, I do not think it can be said that Apotex threatens to infringe claim 32.
300 I have already dealt with the relevance objection made by Pfizer based upon Apotex’s pleading at [171] to [185].
301 I admitted into evidence provisionally a bundle of copies of patent specifications (Ex 11) tendered by Apotex that relate to the use of various pharmaceutical compositions in the treatment of different medical conditions. These were said by Apotex to be relevant to the issue of sufficiency in that they revealed that other patentees included in their patent specification details of clinical trials in humans that were conducted prior to filing. In my view what other patentees may disclose in their patent specifications or at what point in their research and development they may choose to file for patent protection are not relevant to any fact in issue in these proceedings. Exhibit 11 is excluded from evidence on that basis.
302 I also admitted into evidence provisionally the Beach Report on the basis that it may be relevant to the issue of utility. Although I have not derived any assistance from the Beach Report, I consider that it is relevant and it is admitted into evidence unconditionally.
303 During the course of Professor Christie’s and Professor Easton’s cross-examinations Mr Bannon SC sought various orders pursuant to s 136 of the Evidence Act 1995 (Cth) on the basis that aspects of their evidence relating to the Carlton paper and the Jun paper might be unfairly prejudicial to Pfizer or misleading or confusing. Having considered their evidence in relation to those matters I am not satisfied that the requirements of s 136 have been satisfied. I therefore do not propose to make the orders sought.
304 The parties agreed on various other exclusions and limitations in relation to the evidence that are recorded in the transcript and documents marked for identification. It is not necessary for me to say anything more about those agreed rulings.
305 The challenges to the validity of claims 1 to 32 are rejected. Pfizer is entitled to quia timet injunctive relief against Apotex for threatened infringement of claims 1 to 31 and against GPPL for threatened infringement of claims 1 to 30.
306 Pfizer will be directed to file and serve a minute of the orders it seeks within 7 days. Both proceedings will be relisted on a date to be fixed for the purpose of making final orders.
I certify that the preceding three hundred and six (306) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Nicholas. |
Associate:
NSD 763 of 2013 | |
PF PRISM CV | |
Third Cross-Claimant: | PFIZER IRELAND PHARMACEUTICALS |
Fourth Cross-Claimant: | PFIZER ASIA PACIFIC PTE LTD |
Fifth Cross-Claimant: | PFIZER AUSTRALIA PTY LTD (ACN 008 422 348) |
NSD 251 of 2014 | |
PFIZER ASIA PACIFIC PTE LTE | |
Fifth Applicant: | PFIZER AUSTRALIA PTY LTD (ACN 008 422 348) |
Sixth Applicant: | GENERIC PARTNERS PTY LTD (ACN 132 833 777) |
PF PRISM CV | |
Third Cross-Respondent | PFIZER IRELAND PHARMACEUTICALS |
Fourth Cross-Respondent | PFIZER ASIA PACIFIC PTE LTD |
Fifth Cross-Respondent | PFIZER AUSTRALIA PTY LTD (ACN 008 422 348) |

