
FEDERAL COURT OF AUSTRALIA
GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No. 2) Limited v Apotex Pty Ltd [2016] FCA 608
ORDERS
DATE OF ORDER: | 31 may 2016 |
THE COURT ORDERS THAT:
1. The applicants’ proceeding be dismissed.
2. The respondent’s cross-claim be dismissed.
3. The applicants on or before 14 June 2016 file and serve short written submissions (no more than five pages) on costs and any consequential orders sought.
4. The respondent on or before 28 June 2016 file and serve short written submissions (no more than five pages) on costs and any consequential orders sought.
5. The time for the filing of any notice of appeal or cross-appeal be extended until 21 days after all final orders, including on costs and ancillary orders, have been made.
6. Subject to further order, any undertakings and cross-undertakings given by a party on the applicants’ infringement case shall continue to have force and effect until 21 days after all final orders, including on costs and ancillary orders, have been made.
7. Liberty to apply.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
ORDERS
VID 638 of 2014 | ||
BETWEEN: | GLAXOSMITHKLINE CONSUMER HEALTHCARE INVESTMENTS (IRELAND) (NO. 2) LIMITED First Applicant GLAXOSMITHKLINE AUSTRALIA PTY LTD (ACN 100 162 481) Second Applicant | |
AND: | GENERIC PARTNERS PTY LTD (ACN 132 833 777) Respondent
| |
AND BETWEEN: | GENERIC PARTNERS PTY LTD (ACN 132 833 777) Cross-Claimant | |
AND: | GLAXOSMITHKLINE CONSUMER HEALTHCARE INVESTMENTS (IRELAND) (NO. 2) LIMITED First Cross-Respondent GLAXOSMITHKLINE AUSTRALIA PTY LTD (ACN 100 162 481) Second Cross-Respondent | |
JUDGE: | BEACH J |
DATE OF ORDER: | 31 may 2016 |
THE COURT ORDERS THAT:
1. The applicants’ proceeding be dismissed.
2. The respondent’s cross-claim be dismissed.
3. The applicants on or before 14 June 2016 file and serve short written submissions (no more than five pages) on costs and any consequential orders sought.
4. The respondent on or before 28 June 2016 file and serve short written submissions (no more than five pages) on costs and any consequential orders sought.
5. The time for the filing of any notice of appeal or cross-appeal be extended until 21 days after all final orders, including on costs and ancillary orders, have been made.
6. Subject to further order, any undertakings and cross-undertakings given by a party on the applicants’ infringement case shall continue to have force and effect until 21 days after all final orders, including on costs and ancillary orders, have been made.
7. Liberty to apply.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
BEACH J:
1 The first applicant, GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No. 2) Ltd, is the patentee of Australian standard patent AU 2001260212B2 (the Patent). The invention claimed in the Patent is for a sustained release paracetamol bilayer tablet with a specified in vitro dissolution profile.
2 The second applicant, GlaxoSmithKline Australia Pty Ltd (GSK Australia) is, as I have found, the exclusive licensee of the Patent. Since 2000 it has marketed, sold and supplied two sustained release paracetamol products in Australia, each of which is registered on the Australian Register of Therapeutic Goods (ARTG). These products are Panadol Back & Neck Long Lasting (ARTG 78493) and Panadol Osteo (ARTG 116619). The GSK products are commercial embodiments of the invention claimed in the Patent. For the moment, and unless otherwise specified, it is convenient to refer to the applicants collectively as GSK.
3 Apotex Pty Ltd (Apotex) is the Australian subsidiary of a Canadian group of companies. It markets and supplies in Australia, inter alia, generic pharmaceutical products.
4 Generic Partners Pty Ltd (Generic Partners) is an Australian company that manufactures and supplies only on a wholesale basis generic pharmaceutical products; its manufacturing function is sometimes outsourced to independent contractors. Generic Partners is the sponsor of 20 registrations on the ARTG, each for bilayer sustained release paracetamol tablets containing 665mg of paracetamol (the GP ARTG registrations).
5 Generic Partners intends to supply bilayer sustained release paracetamol tablets containing 665mg of paracetamol the subject of the GP ARTG registrations to Apotex for on-supply and sale in Australia (the alleged infringing products).
6 GSK commenced a proceeding against Apotex on 3 October 2014 (VID 571 of 2014) and a separate proceeding against Generic Partners on 29 October 2014 (VID 638 of 2014) for anticipated infringement of the claims of the Patent. GSK has sought injunctive relief to restrain the supply by Apotex and Generic Partners of the alleged infringing products which it is alleged would infringe claims 1 to 6, 8 to 11, 13 and 14 of the Patent.
7 Apotex and Generic Partners have brought cross-claims against GSK filed on 30 October 2014 and 25 November 2014 respectively seeking orders revoking claims 1 to 6, 8 to 11, 13 and 14 of the Patent on the following grounds:
lack of fair basis;
lack of sufficiency;
lack of clarity;
failure to define the invention;
failure to describe the best method;
lack of an inventive step.
8 Apotex and Generic Partners had also asserted a lack of utility as a ground of invalidity, but on 4 February 2016 that ground was abandoned.
9 As is typical of patent litigation, this case has raised a plethora of issues concerning the construction of the claims of the Patent, infringement and invalidity.
10 In summary, I have rejected both GSK’s infringement case and Apotex’s and Generic Partners’ invalidity assertions.
11 This case has raised numerous but interesting questions of pharmaceutical science; the length of these reasons is a reflection of that reality. Contrastingly, for the most part, only the application of well accepted legal principles has been necessary to dispose of the legal questions. But there is one legal issue that has troubled me, which has principally arisen on the case advanced by Ms Sophie Goddard SC for Apotex in defence of the infringement case.
12 I have found that the hypothetical skilled addressee is likely to have perceived that a mistake was made in claim 1 in identifying the relevant dissolution apparatus; “basket” should have read “cylinder”. The body of the complete specification also embodied that mistake. Should I construe the claim so as to re-write it to fit with that perception? Now “basket” has a clear and unambiguous meaning; I will elaborate later on why I say that. Moreover, there is nothing in the body of the specification, in either text or context that, in contradistinction to the term used in the claim, justifies re-conceptualising “basket”; the same mistake was made in the body of the specification as well. Further, the invention works whatever construction is used. Ms Goddard SC, astutely perceiving the risk that I might take a more free-wheeling construction approach more apposite to commercial contracts, cautioned me against such liberality. Her position was that I was not free to re-write the claim. Her point, however, was not to extol the virtue of some form of sclerotic intellectualisation engaged in by medieval scholastic types when construing a text. Rather, her point was that the specification was a public instrument and that I was not free to, in effect, amend the specification under the pretext or pretence of some construction exercise. Amendment was dealt with under a separate statutory mechanism.
13 Contrastingly, Mr David Shavin QC for GSK submitted that if the skilled addressee would have identified the reference to “basket” in the claim as a mistake and would have reinterpreted it to mean “cylinder”, then that was the end of the matter. He contended that I was constrained to read and construe “basket” in the claim as “cylinder”. But in my view, no case binding upon me has distorted the skilled addressee lens to that extent. This is not a case where an essential integer of a claim is ambiguous or uncertain. This is not a case where there is incongruence between a term used in the body of the specification and a term used in a claim; the same term is used throughout. This is not a case where the claim refers to or embodies an incorrect scientific theory (as distinct from being an essential integer) which can be put to one side. This is not a case where for one construction of an integer the invention works and for another construction it does not. This is not a case where the relevance of the mistake is being assessed only in a lack of sufficiency context.
14 No case expressly binds me to accept the result contended for by GSK. The hypothetical construct of the skilled addressee cannot be taken so far as to re-write or amend a claim of the specification. That conceptual tool has its limits. After all, the boundary constraint is that I am obliged to construe the claim as it is, rather than what it should have been. I accept Apotex’s contention. Accordingly, GSK must fail on infringement as claim 1 is the only independent claim. But Apotex and Generic Partners fail on invalidity.
15 For convenience, these reasons address the issues in the following sequence:
(a) Procedural history — [16] to [26];
(b) The Patent — [27] to [70];
(c) Arthritis and the use of paracetamol — [71] to [100];
(d) Basic principles of pharmaceutical formulations — [101] to [165];
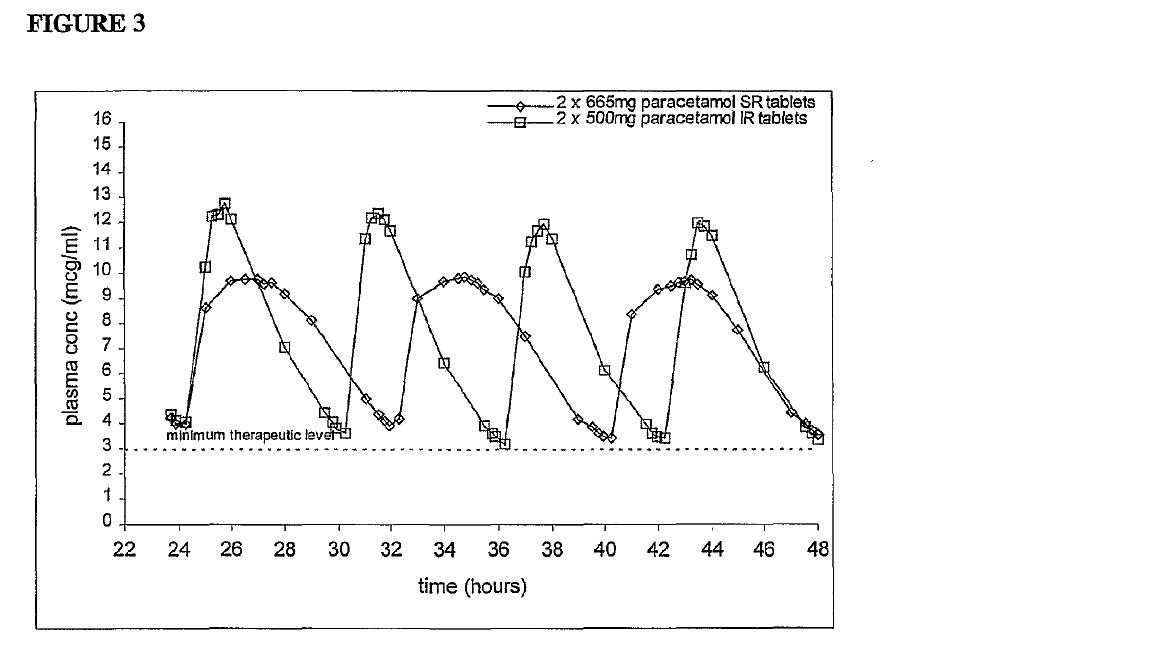
(e) Dissolution testing — [166] to [200];
(f) Approach to drug development as at April 2000 — [201] to [232];
(g) The expert witnesses — [233] to [266];
(h) Principles of construction — [267] to [280];
(i) Construction of claims 1 to 3: USP type III apparatus — [281] to [401];
(j) Construction of claims 8 to 11: matrix forming polymer — [402] to [494];
(k) Infringement case — [495] to [588];
(l) Exclusive licensee — [589] to [599];
(m) Fair basis — [600] to [682];
(n) Sufficiency — [683] to [720];
(o) Lack of clarity — [721] to [741];
(p) Failure to define the invention — [742] to [746];
(q) Failure to describe best method — [747] to [863];
(r) Lack of inventive step — [864] to [999];
(s) Conclusion — [1000].
procedural history
16 Before addressing the substance of the case it is necessary to make some observations on the procedural history.
17 Upon the return of GSK’s interlocutory injunction application on 8 October 2014, Apotex gave an undertaking that it would not offer to sell or supply the alleged infringing products pending the determination of the proceedings or further order. But it was foreshadowed at that hearing that Apotex would make an application to be released from its undertaking if a relevant competitor entered into the market in the meantime. It was anticipated that Apotex would also withdraw any application for listing of the alleged infringing products on the Schedule of Pharmaceutical Benefits under the Pharmaceutical Benefits Scheme (PBS) if its undertaking was not discharged. On 5 November 2014, Generic Partners gave similar interlocutory undertakings. GSK also gave a cross-undertaking that GSK would not offer to sell or supply any generic form of the GSK products whilst Apotex and Generic Partners were relevantly bound by their undertakings.
18 On 17 November 2014, Apotex advised that it had withdrawn its application to list the alleged infringing products on the PBS from 1 December 2014 but that it had made an application to list those products on the PBS from 1 January 2015.
19 On 5 December 2014, Apotex and Generic Partners each applied to be released from their undertakings, alternatively to amend their undertakings as to withdrawal of any application concerning the PBS. The apparent basis for their applications was that Pharmacor Pty Ltd had entered the market on 1 December 2014 with a relevant new generic product. By way of background, in GlaxoSmithKline Australia Pty Ltd v Pharmacor Pty Ltd [2014] FCA 1202 I made orders on GSK’s application for preliminary discovery against Pharmacor in respect of the latter’s generic extended release paracetamol products. But no further action was taken by GSK against Pharmacor to prevent Pharmacor from entering the relevant market for the supply of such generic products.
20 On 12 December 2014 I heard the applications of Apotex and Generic Partners to be released from their undertakings and reserved my decision until 15 December 2014. But on the morning of 15 December 2014, Generic Partners applied to re-open its case to raise a further ground of arguable invalidity. I heard the parties that morning on this issue. I then delivered judgment on 16 December 2014 (GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Ltd v Apotex Pty Ltd (2014) 109 IPR 492) leaving the restraints on Apotex and Generic Partners in place; but the undertakings dealing with Apotex and Generic Partners withdrawing their applications concerning the PBS were modified. The regime that was set in place at this time concerning the undertakings and cross-undertakings remains in place.
21 On 7 April 2015, I set both proceedings down for trial on 12 October 2015 on an estimate of 10 days. I also ordered that the two proceedings be heard concurrently and that evidence filed in each proceeding be treated as evidence filed in the other. The trial was to deal with all issues of liability on infringement and invalidity questions.
22 On 4 June 2015 and 3 July 2015, Apotex and Generic Partners each filed applications seeking leave to file and serve amended defences in order to withdraw various admissions. In particular, they sought to withdraw the admissions that the alleged infringing products had each of the essential integers of claims 1 and 8 to 11 of the Patent. On 24 July 2015, I heard the applications and adjourned both over for further hearing on 21 August 2015 after indicating my tentative views on how I might resolve such applications. On 20 August 2015, I made orders by consent that Apotex and Generic Partners have leave to withdraw the relevant admissions.
23 The trial proceeded for two weeks from 12 October 2015. GSK called Professor Dressman and Professor Davies, Apotex called Professor Fassihi and Dr Mooney and Generic Partners called Professor Tucker. On 15 October 2015 I was informed that Professor Davies had a personal difficulty and that it was uncertain when he would be in a position to travel from London to Melbourne to give evidence; given the importance of his evidence, it was not appropriate to take it on a video link. Tentatively, it was then anticipated that he could give evidence on 9 and 10 November 2015. But on 4 November 2015 I heard the parties as to the unavailability of Professor Davies to give evidence on 9 and 10 November 2015. In the circumstances and to meet the parties’ convenience and that of Professor Davies, I fixed a further five days for the trial from 1 February 2016 for Professor Davies’ evidence and closing submissions.
24 On 10 November 2015, Professor Tucker was also further cross-examined via video link from Dunedin, New Zealand.
25 The trial resumed on 1 and 2 February 2016 for Professor Davies to be cross-examined. Closing submissions were heard on 3, 4 and 8 February 2016.
26 Before proceeding further, it is worth identifying the applicable legislative framework. The Patents Amendment Act 2001 (Cth) amended inter alia s 7 of the Patents Act 1990 (Cth). But the amendments only apply in relation to patents for which the complete application was made on or after the day on which the amendments commenced, namely, 1 April 2002 (see Sch 1, part 1, item 13 of the Patents Amendment Act). The complete application for the Patent was filed prior to 1 April 2002 (12 April 2001). Accordingly, issues of infringement and validity are to be determined under the Patents Act 1990 (Cth) in the form in which it existed prior to the Patents Amendment Act 2001 (Cth) and, of course, prior to the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth).
THE PATENT
27 The Patent has a priority date of 19 April 2000.
28 The Patent claims a pharmaceutical composition containing paracetamol. It comprises “a bilayer tablet having an immediate release phase of paracetamol and a sustained release phase of paracetamol”. The following is stated in the complete specification (p 1 lines 3 to 23):
The present invention relates to pharmaceutical compositions containing N-acetyl-p-aminophenol, known by the generic names paracetamol, acetaminophen and APAP (hereinafter referred to as paracetamol). In particular, the invention relates to a sustained release paracetamol formulation having an advantageous pharmacokinetic profile.
Paracetamol is an analgesic and antipyretic agent which is widely used in prescription and non-prescription medicines, often in combination with other biologically active compounds.
The elimination half-life of paracetamol is reported to be in the range of 1.9 – 2.5 hours. Its absorption following oral doses of conventional immediate release tablets is characterised by passive absorption with high bioavailability (80%) and rapidly occurring maximum plasma concentration (tmax 30–90 min). These characteristics determine the conventional dosage regimen of 1000mg every 4 to 6 hours for the drug. Although this regimen is acceptable in the short-term treatment of acute pain, it becomes inconvenient in the context of long-term treatment of sub-chronic or chronic pain. Therefore, extended release paracetamol may improve patient’s quality of life by reducing the number of doses to be taken and providing steadier levels of the drug in the blood as determined by plasma or serum drug concentrations.
29 The Patent refers at p 1 lines 25 to 28 to the desirability of a t.i.d oral dosing paracetamol product (two tablets of 600 – 667mg paracetamol per dose); t.i.d is a reference to three times per day.
30 At p 1 lines 29 to 32 to p 2 line 3 the Patent refers to the McNeil Inc European patent EP-A-305051 (the McNeil patent) (a form of the 522 Patent that I will discuss later) which is said to disclose a sustained release bilayer tablet containing 650 or 667mg of paracetamol which contains an equal amount of paracetamol in the immediate and sustained release layers. Such a tablet is said to be marketed by McNeil Inc as Tylenol® Extended Relief. It is said that the sustained release layer is provided by a matrix comprising a mixture of hydroxyethylcellulose and polyvinyl-pyrrolidone.
31 The invention is then discussed by reference to the existing McNeil sustained release bilayer paracetamol tablet and the advantages that the invention provides over that tablet.
32 The Patent then discusses at pages 2 to 3 the ideal pharmacokinetic properties for a sustained release paracetamol product. At p 2 lines 5 to 7 the Patent notes that a sustained release paracetamol oral dosage form designed for t.i.d dosing should also provide all the benefits of immediate release paracetamol plus a sustained action.
33 The Patent notes that one potential disadvantage of a formulation containing more than the standard dose of paracetamol (500mg) is accidental or intentional overdose.
One potential disadvantage concerning a formulation containing more than the standard dose of paracetamol (500mg) is accidental or intentional overdose. In such circumstances more paracetamol will be ingested from an extended release formulation compared to a conventional immediate release formulation for any given number of unit doses such as tablets. This could have serious consequences for an overdose patient, especially if a large amount of the dose is absorbed before rescue therapy could be initiated. It would therefore be preferable if the unit dose (such as a tablet) was designed to limit the amount of paracetamol absorbed in the first few hours following dosing. An advantageous sustained release formulation should therefore demonstrate a lower mean Cmax (preferably at least 20% lower) than a conventional immediate release formulation which would be indicative of a lower initial exposure. (Patent p 2 lines 11 to 22)
34 Further, one possible consequence of formulating an oral paracetamol product designed to have a lower Cmax and slower rate of absorption is said to be:
that the extent of absorption may also be decreased, this could then lead to sub-therapeutic systemic levels of drug 6–8 hours following dosing thus leading to premature onset of pain before administration of a further dose. (Patent p 2 lines 24 to 28)
35 However, a further advantage for a product designed to have a lower Cmax and slower rate of absorption where the extent of absorption is essentially complete, as demonstrated by an equivalent dose corrected AUC (area under the concentration-time curve, a concept that I will elaborate on later) compared to the immediate release tablets, is said to be:
that it should have the advantage of maintaining therapeutic levels of paracetamol in plasma for extended periods following dosing and hence provide analgesia for longer than a conventional immediate release tablet or capsule. Furthermore as a result of a reduced Cmax, systemic levels of paracetamol are likely to remain at more constant levels, thus benefiting the patient. (Patent p 3 lines 1 to 5)
36 The Patent notes that it is desirable for plasma levels associated with a sustained release formulation to be maintained at therapeutic levels (>3mcg/ml) for longer than a conventional immediate release formulation:
Whilst such a formulation should have a lower Cmax compared to a conventional immediate formulation, it is still desirable to have a fast onset of action, therefore initial levels of drug in plasma should be rapidly attained (preferably within 30 minutes) and maintained at therapeutic levels of >3mcg/ml for at least 1.3 hours and preferably 1.5 hours longer than a standard immediate release tablet or capsule. In addition the extent of absorption should be equivalent to a conventional immediate release formulation.
Furthermore, upon multiple dosing of a sustained release formulation the steady state plasma levels of paracetamol should be more constant than those achieved following multiple dosing of a conventional immediate release formulation. A convenient measure of the fluctuation in plasma concentrations is the fluctuation index (FI) which is defined as (Cmax – Cmin) / Caverage. A low FI number (ie <1) is considered to be advantageous as it suggests a reduction in the variability of plasma concentrations indicative of a safer product. (Patent p 3 lines 7 to 21)
37 At p 3 line 23 to p 4 line 10 the Patent summarises the desirable attributes for a sustained release paracetamol product for oral administration to possess. It states:
In summary, an advantageous sustained release paracetamol product for oral administration should possess the following pharmacokinetic attributes:
(1) therapeutically active drug plasma concentrations should be attained rapidly.
(2) the mean maximum plasma concentration (Cmax) should be at least 20% lower compared to standard immediate release formulation;
(3) a mean plasma concentration of at least 3 mcg/ml should be maintained for at least 1.3 hours longer (preferably 1.5 hours longer) than a standard immediate release formulation;
(4) the extent of absorption should be equivalent to a conventional immediate release paracetamol;
(5) plasma levels of paracetamol following multiple dosing should be more constant compared to multiple dosing of an immediate release formulation as measured by a reduction in the fluctuation index.
38 At p 4 line 12 the specification states:
Surprisingly it has now been discovered that such an advantageous pharmacokinetic profile can be provided by a two phase (immediate release and sustained release) formulation of paracetamol which satisfied a unique in vitro dissolution profile.
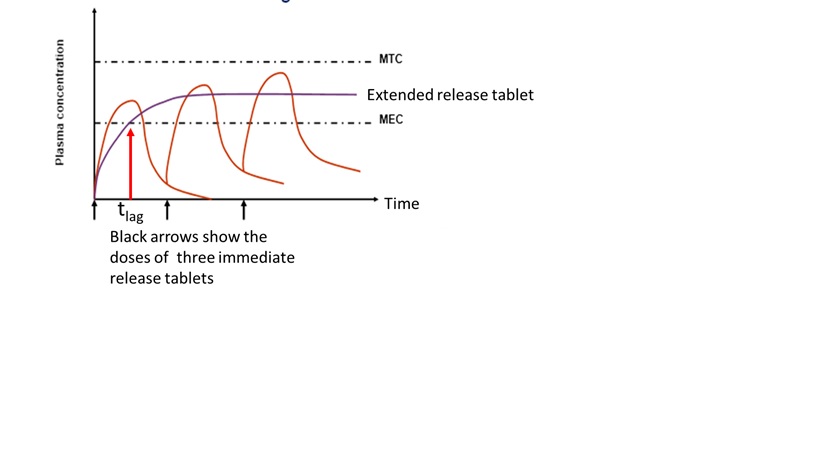
39 Pages 4 to 6 set out the various consistory clauses.
40 They begin with what is said at p 4 line 27 to p 4a line 8:
In one aspect, the present invention provides a pharmaceutical composition comprising a bilayer tablet having an immediate release phase of paracetamol and a sustained release phase of paracetamol,
the immediate release phase being in one layer and comprising from about 10 to 45% by weight of the total paracetamol; and
the sustained release phase being in the other layer and comprising from about 55% to 90% by weight of the total paracetamol in admixture with a matrix forming polymer or a mixture thereof;
said composition comprising from 600 to 700mg of paracetamol per unit dose and a pharmaceutically acceptable carrier, wherein said composition has an in vitro paracetamol dissolution profile (as determined by the USP type III apparatus, reciprocating basket, with 250ml of 0.1M HCl at 37C set at a cycle speed of 15 strokes/min) with the following constraints:
• 30 to 48% released after 15 minutes
• 56 to 75% released after 60 minutes
• >85% released after 180 minutes.
41 At p 5 lines 15 to 32 it is said:
The immediate release phase and the sustained release phase both contain paracetamol and a pharmaceutically acceptable carrier and are suitably combined together into a unit dose form. For example the immediate release phase and the sustained release phase can be separate blends, granules or pellets which can be mixed together before being compressed into a tablet or being filled into a capsule. A preferred unit dose form is a bilayer tablet having an immediate release layer of paracetamol and a sustained release layer of paracetamol.
Suitably the sustained release phase comprises a matrix-forming polymer to provide a sustained release of paracetamol.
Examples of matrix-forming polymers include both water soluble and water insoluble polymers or mixtures thereof, with soluble polymers being preferred. Examples of water soluble polymers include hydroxypropylmethylcellulose, hydroxyethylcellulose, carboxymethylcellulose, sodium carboxymethylcellulose, methacrylate hydrogels, polyethylene glycols and xanthan gum. An example of a water insoluble polymer is ethylcellulose. A preferred matrix-forming polymer is hydroxypropylmethylcellulose.
42 At p 6 lines 2 to 9 it is said:
The amount of matrix-forming polymer in the sustained release phase and the relative amounts of paracetamol in the sustained release and immediate release phases are selected so as to provide the desired in vitro dissolution rate as herein before described.
Thus, the matrix-forming polymer is suitably present in an amount from 0.5 to 10%, preferably from 1 to 6%, and more preferably from 2 to 4% by weight of the sustained release phase.
43 At p 6 lines 20 to 28 it is said:
Compositions of the present invention will generally contain at least one pharmaceutically acceptable carrier conventionally used in the art of tablet and/or capsule formulation. Suitable carriers which may be incorporated include lubricants, for example magnesium stearate and stearic acid; disintegrants, for example cellulose derivatives and starches; binders, for example modified starches, cellulose derivatives and polyvinylpyrrolidone; glidants, for example colloidol [sic] silicas; compression aids, for example cellulose derivatives; as well as preservatives, suspending agents, wetting agents, flavouring agents, bulking agents, adhesives, colouring agents, sweetening agents appropriate to their form.
44 The discovery is then illustrated by way of four example formulations (Formulations A to D), two of which are said to have an in vitro dissolution profile outside the scope of the invention (Formulations A and B). These are the subject of example 1. The other two formulations are said to have an in vitro profile falling within the scope of the invention (Formulations C and D).
45 The ingredients of Formulations A and B are listed in the table on p 9 of the Patent.
46 The paracetamol used in the immediate release layer of Formulations A and B is identified as DC90. The Patent notes at p 9 lines 30 to 32 that this is a commercially available directly compressible paracetamol granulation containing about 90% by weight of paracetamol together with pregelatinised starch, croscarmellose sodium, polyvinylpyrrolidone and stearic acid.
47 The release profiles for Formulations A and B, characterised using the described apparatus (I will return later to contentious aspects as to the description of the apparatus) with 250ml 0.1M HCl at 37°C set at a cycle speed of 15 strokes/min, are set out in Table 1 on p 10. It is not necessary for present purposes to reproduce Table 1 at this point.
48 Formulations A and B were assessed in a pharmacokinetic study in healthy fasted volunteers using 500mg immediate release paracetamol tablets as a control (p 10 lines 10 to 14). The results are presented graphically in Figure 1 on p 11. Again, it is not necessary to reproduce Figure 1.
49 The Patent notes that neither Formulation A nor B met the criterion of achieving a mean paracetamol plasma concentration of 3mcg/ml for at least 1.5 hours longer than the immediate release tablet (p 11 lines 5 to 7).
50 Thus far I have referred to Formulations A and B and example 1. It is appropriate to now discuss Formulation C and example 2.
51 Example 2 compares the properties of a commercially available immediate release 500mg paracetamol tablet with a sustained release bilayer tablet having an in vitro dissolution profile falling within the scope of the invention (Formulation C). The ingredients of Formulation C are set out in the table on p 12 of the Patent.
Example 2
This Example compares the properties of a commercially available immediate release 500mg paracetamol tablet with a sustained release bilayer tablet (Formulation C) having an in vitro dissolution profile falling within the scope of the present invention.
This advantageous bilayer tablet containing a total of 666.6mg of paracetamol was prepared from the following ingredients:
Ingredient | Tablet Formulation C | |
Sustained Release Layer | mg/tablet | % w/w |
Paracetamol | 473.57 | 64.39 |
High viscosity HPMC | 15.43 | 2.10 |
Pregelatinised Starch | 5.14 | 0.70 |
Polyvinylpyrrolidone | 10.28 | 1.40 |
Low viscosity HPMC | 8.23 | 1.12 |
Magnesium Stearate | 1.54 | 0.21 |
Immediate Release Layer | ||
Directly compressible paracetamol granulation DC90 | 214.92 | 29.22 |
(Paracetamol content in DC90) | (193.43) | (26.30) |
Film and Wax Coating | 6.305 | 0.86 |
Total | 735.42 | 100.00 |
% w/w SR:IR APAP | 71:29 | |
The release profile of test formulation C was characterised using the USP type III apparatus (reciprocating basket) as hereinbefore described and was found to have the following dissolution rate as detailed in table 2.
Table 2: Dissolution Profile for Formulation C
Time (minutes) | In-vitro release Results (% paracetamol released) |
15 60 120 180 | 39.4% 64.4% 89.0% 101.8% |
Formulation C was assessed in a pharmacokinetic study. The study design was a four-way crossover, using a panel of 26 healthy volunteers which compared the pharmacokinetics of paracetamol in serum in both fed and fasted states following a two tablet dose of the formula C and a two tablet dose of a currently marketed standard immediate release paracetamol 500 mg tablet. The mean pharmacokinetic profiles are shown in Figure 2.
FIGURE 2
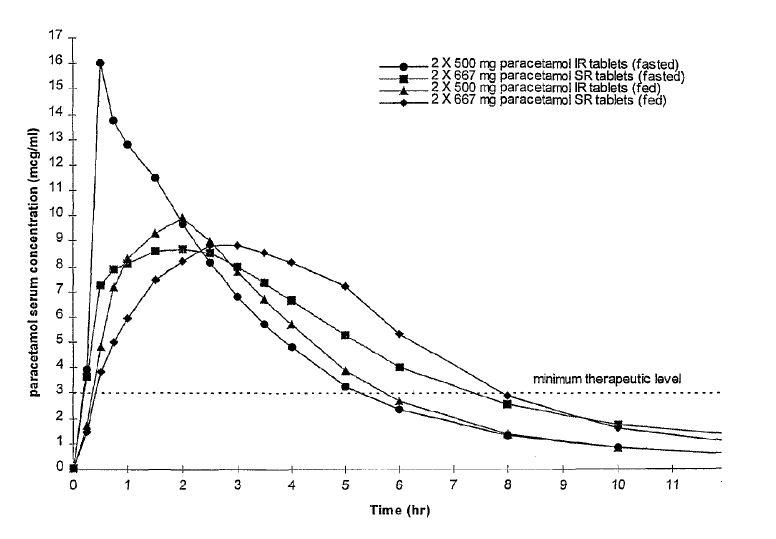
Formulation C met all of the pharmacokinetic criteria outlined above for an ideal sustained release paracetamol tablet. The pharmacokinetic analysis demonstrated that the Cmax was significantly lower for formulation C (mean value 10.1 mcg/ml) compared to the reference immediate release product (mean value 18.7 mcg/ml) (in the fasted state). In addition therapeutic serum concentrations were rapidly attained and mean serum levels of 3 mcg were maintained until 7.4 hours post dose compared to only 5.3 hours post dose for the 500mg immediate release tablet. The two formulae were bioequivalent with respect to AUC indicating that the extent of absorption was the same for formulation C as for conventional immediate release paracetamol.
These advantageous properties of formulation C are particularly surprising when compared with the plasma concentrations described in Example 1 of EP-A-305051 which suggests that the Cmax of the prior disclosed sustained release paracetamol formulation is as high as that observed for an immediate release formulation.
52 As stated in the specification, the release profile of Formulation C was characterised using the relevant apparatus and was found to have the dissolution profile as detailed in table 2. Formulation C was assessed in a pharmacokinetic study, using a panel of 26 healthy volunteers and a two tablet dose of a standard immediate release 500mg paracetamol tablet comparator. The results of the study were presented graphically as set out above. Formulation C was said to have met all of the pharmacokinetic criteria outlined above for an ideal sustained release paracetamol tablet. In particular, in comparison to the reference immediate release product, Formulation C was bioequivalent with respect to AUC, it had a significantly lower Cmax and mean plasma levels remained above the therapeutic level of 3mcg/ml for longer.
53 As indicated in the passages set out above, the Patent says (at p 15 lines 4 to 7) that the advantageous properties of Formulation C are particularly surprising when compared with the plasma concentrations described in example I of the McNeil patent which suggests that the Cmax of the prior disclosed sustained release paracetamol formulation is as high as that observed for an immediate release formulation. The Patent notes earlier at p 2 line 2 that McNeil markets a bilayer sustained release paracetamol tablet as Tylenol® Extended Relief. The Patent later highlights the advantages of Formulation D over the Tylenol® Extended Relief product at p 17. Let me turn to Formulation D and example 3.
54 Example 3 compares the properties of a commercially available immediate release 500mg paracetamol tablet with Formulation D, a sustained release bilayer tablet having an in vitro dissolution profile falling within the scope of the invention.
55 The bilayer tablet of Formulation D is said to be “essentially similar to Formulation C but contained a total of 665mg of paracetamol and had a slightly different ratio of sustained release to immediate release paracetamol (% w/w SR:IR APAP was 69:31)” (Patent p 15 lines 15 to 18).
56 The release profile of Formulation D characterised using the relevant apparatus was set out in Table 3 on p 15; it is unnecessary to reproduce this Table.
57 Formulation D was the subject of a further biostudy involving 27 subjects. There were two study sessions each consisting of two days’ dosing. The two study treatments were as follows:
(a) two bilayer sustained release (SR) tablets of Formulation D each containing 665mg given three times per day (every 8 hours); and
(b) two immediate release (IR) paracetamol 500mg tablets given four times per day (every 6 hours).
The study sessions were separated by 48 hours.
58 Pharmacokinetic analysis was conducted for the period of 24 hours — 48 hours following commencement of the dosing schedule. The results showed that the two treatments were bioequivalent with respect to AUC24–48 and Formulation D provided a lower Cmax, a higher Cmin and a substantially lower fluctuation index (FI) compared to the immediate release formulation. Mean plasma paracetamol concentrations versus time were shown in Figure 3. It is appropriate to reproduce Figure 3 at this point:
59 The Patent teaches that Tylenol Extended Relief does not have a lower FI than the immediate release tablet, stating at p 17 lines 7 to 13:
The substantially lower FI for the SR product is surprising considering previous reports for a steady state biostudy conducted with a 650mg bilayer tablet (Tylenol Extended Relief) which showed that the SR product had a numerically higher FI (of 1.49) compared to a reference 500mg IR tablet (of 1.44) as illustrated in Figure 4. Furthermore, Paracetamol plasma levels were maintained substantially above 3mcg/ml for the entire study period, which is in contrast to the steady state study reported for Tylenol® Extended relief.
60 The low FI number of <1 found for Formulation D is said to be “particularly advantageous” for a sustained release formulation as it indicates a reduction in the variability of plasma concentration suggesting a much safer and more reliable product (p 18 lines 5 to 7). In elaboration, there is a useful explanation of FI at p 3 lines 15 to 21:
Furthermore, upon multiple dosing of a sustained release formulation the steady state plasma levels of paracetamol should be more constant than those achieved following multiple dosing of a conventional immediate release formulation. A convenient measure of the fluctuation in plasma concentrations is the fluctuation index (FI) which is defined as (Cmax – Cmin) / Caverage. A low FI number (ie < 1) is considered to be advantageous as it suggests a reduction in the variability of plasma concentrations indicative of a safer product.
61 Figure 4 on p 18 compares the FI of Tylenol Extended Relief and an immediate release tablet based on a steady state biostudy of the two. It shows that the Tylenol Extended Relief had a numerically higher FI than the immediate release formulation. Figure 4 shows that sustained release formulations do not, as a matter of course, have a lower fluctuation index compared to a corresponding immediate release formulation. Figure 4 also illustrates that the plasma level of Tylenol Extended Relief drops below 3mcg/ml on a number of occasions. It is not necessary to reproduce Figure 4 for present purposes.
62 Example 4 compares the clinical properties of a commercially available paracetamol 500mg immediate release tablet with a sustained release bilayer tablet of Formulation D. The study was a multicentre, single dose, double-blind, double dummy, two armed parallel group efficacy study involving 510 patients with post-surgical dental pain following third molar extraction under general anaesthesia to compare the efficacy of a two tablet dose of either a sustained release tablet containing 665mg paracetamol per tablet (252 patients) or a two tablet dose of a commercially available tablet containing 500mg of immediate release paracetamol per tablet (258 patients). The Patent reports the results of the study at p 20 and says:
Based on the patient global assessment at 4 hours, the extended release product was shown to be equivalent or better than the immediate release product. A successful response was defined as a ‘very good’ or ‘excellent’ rating: 88 of 252 (35.1%) patients treated with the SR paracetamol formulation gave a successful response compared with 71 of 258 (27.7%) patients treated with standard IR paracetamol. Equivalence was concluded from the 90% confidence interval of the treatment difference (7.3% in favour of SR paracetamol) between the two formulations.
There was no significant difference between SR paracetamol and standard IR paracetamol in either development of analgesia (time to peak pain relief, time to peak pain intensity difference, total pain relief 1 hour after treatment) or peak analgesic effect (peak pain relief, peak pain intensity difference). However, the SR tablet was significantly more effective than standard IR paracetamol for the summed pain analogue intensity difference at 6 hours (p = 0.0344) and 8 hours (p = 0.0500). Furthermore, the median time to re-medication was longer for SR paracetamol (245 mins) compared with standard IR paracetamol (190 mins). Although this was not statistically significant, it was clear from the separation of the two curves on the Kaplan-Meier plot that a smaller proportion of patients treated with SR paracetamol re-medicated between approximately 3 and 6 hours compared with standard IR paracetamol.
63 The Patent concludes that the example 4 results indicate that the sustained release tablet gave rapid analgesia which was maintained for up to eight hours following dosing and the sustained release tablet had a longer duration of action than immediate release paracetamol (p 20 lines 27 to 30).
64 Let me now deal with the claims. The independent claim, claim 1, claims:
A pharmaceutical composition comprising
a bilayer tablet having an immediate release phase of paracetamol and a sustained release phase of paracetamol,
the immediate release phase being in one layer and comprising from about 10 to 45% by weight of the total paracetamol; and
the sustained release phase being in the other layer and comprising from about 55% to 90% by weight of the total paracetamol in admixture with a matrix forming polymer or a mixture thereof;
said composition comprising from 600 to 700mg of paracetamol per unit dose and a pharmaceutically acceptable carrier,
wherein said composition has an in vitro paracetamol dissolution profile (as determined by the USP type III apparatus, reciprocating basket, with 250ml of 0.1M HCl at 37C set at a cycle speed of 15 strokes/min) with the following constraints:
• 30 to 48% released after 15 minutes
• 56 to 75% released after 60 minutes
• >85% released after 180 minutes.
65 Claims 2 and 3 claim narrower dissolution ranges.
66 Claims 4 and 5 claim compositions in which the paracetamol is present in an amount of 630 to 680 mg and 650 to 667 mg respectively.
67 Claims 6, 8, 9, 10 and 11 are dependent claims as to the matrix forming polymer. They are expressed as follows:
6. A composition according to any one of claims 1 to 5 in which the matrix forming polymer is a water-soluble or a water-insoluble polymer or a mixture thereof.
7. …
8. A composition according to claim 6 in which the matrix forming water-soluble polymer is selected from hydroxypropylmethylcellulose, hydroxyethylcellulose, carboxymethylcellulose, sodium carboxymethylcellu-lose, methacrylate hydrogels, polyethylene glycols, xanthan gum or a mixture thereof.
9. A composition according to claim 8 in which the water-soluble matrix forming polymer is hydroxypropylmethylcellulose.
10. A composition according to any one of claims 1 to 9 in which the matrix forming polymer is present in an amount from 0.5 to 10% by weight of the sustained release phase.
11. A composition according to claim 10 in which the matrix forming polymer is present in an amount from 1 to 6% by weight of the sustained release phase.
68 Claims 7 and 12 can be put to one side. Claims 13 and 14 are dependent claims as to the percentages of total paracetamol in the immediate and extended release layers.
69 The dissolution profile in claim 1 (and accordingly incorporated in the other dependent claims) is defined by reference to being determined by “the USP type III apparatus, reciprocating basket”. Contrastingly, the text of the United States Pharmacopeia then current as at the priority date described the inner vessel of the USP type III apparatus as a “reciprocating cylinder”. One of the principal debates between the parties has been as to how the expression “the USP type III apparatus, reciprocating basket” would be read by the skilled addressee and how it should be construed. Is a reference to a “basket” a mistake? Should it be construed and would it be read and understood by the skilled addressee as meaning “cylinder”? Or should it be construed as it states, namely, a “basket”? I will return to this issue later. Apotex has taken the lead on this issue, which has many dimensions both for GSK’s infringement case and the respondents’ invalidity arguments. I have set out a diagram of what could be described as the standard or “complying” USP type III apparatus with reciprocating cylinder later in these reasons.
70 Before proceeding further I should also note one other construction question that has arisen concerning “matrix”, “matrix forming” and “matrix forming polymer”. Mr Tom Cordiner, counsel for Generic Partners, has had the principal carriage of this question on behalf of the respondents. I will also return to this issue later.
ARTHRITIS AND THE USE OF PARACETAMOL
71 It is appropriate to set the scene in terms of paracetamol and its primary use. In this section I have largely drawn upon the uncontested evidence of Professor Peter Brooks, a consultant rheumatologist and a professorial fellow at the Faculty of Medicine, Dentistry and Health Sciences at the University of Melbourne. From time to time in this section I have referred to April 2000; as I have said, the priority date of the Patent is 19 April 2000.
(a) Arthritis
72 Arthritis is a term which describes inflammation in a joint. The term derives from the Greek “arthro” meaning joint, and “itis” meaning inflammation. There are at least 150 different types of arthritis which have been generally divided into the following categories:
(a) the degenerative category;
(b) the inflammatory category;
(c) the gout category; and
(d) the infectious category.
73 The most common of these categories is the degenerative category, with the most common form being osteoarthritis. Osteoarthritis affects most people at some stage in their lives. It is a “wear and tear” condition. Osteoarthritis does not usually primarily involve inflammation, but there is an inflammatory component. Osteoarthritis occurs due to a degradation of cartilage, the “soft” material that covers the ends of bones. This causes rough surfaces to rub together which leads to intermittent mild inflammation. This is a major cause of pain in osteoarthritis.
74 The inflammatory category is a less common category of arthritis which affects only about 1 to 2% of the general population; it includes rheumatoid arthritis. Rheumatoid arthritis is a severe disease with affected persons being typically positive for the rheumatoid factor (RhF) in serological (blood) tests.
75 Another group of arthritic diseases within the inflammatory category are the seronegative arthropathies. Seronegative arthropathies include ankylosing spondylitis (a chronic, painful, degenerative inflammatory arthritis primarily affecting spine and sacroiliac joints causing eventual fusion of the spine) and psoriatic arthritis (associated with psoriasis, a skin rash). While seronegative arthropathies are inflammatory like rheumatoid arthritis, patients with seronegative arthritis are typically negative for RhF in blood tests.
76 Inflammation is the result of the body’s immune response. It is the way the body fights infection effectively and heals after injuries or operations. Inflammation involves a sequence of complex, interrelated events that ultimately brings plasma proteins, phagocytes (white blood cells that consume microbes and other foreign material) and other cellular elements to the injured area for the purpose of initiating tissue repair. Without inflammation, the common cold, for example, could result in death. The problem with inflammatory arthropathies such as rheumatoid arthritis and associated seronegative arthropathies is that the immune response does not know when to stop and “invades” the joints.
77 The gout and infectious categories are less common and can be put to one side for present purposes.
(b) Pain
78 Pain is the most common symptom of persons having arthritis, regardless of the type of arthritis. The pain experienced tends to be intermittent and not usually experienced at a constant level. With rheumatoid arthritis, the pain experienced is sometimes worse in the morning (morning stiffness) and then alleviates as the person is active during the day. With appropriate treatment, most pain associated with arthritis can be reasonably controlled within four to six weeks of treatment commencing.
79 Pain is usually categorised as either acute pain or chronic pain.
80 Acute pain appears suddenly and can be either mild or severe. Acute pain is usually associated with an event (e.g. childbirth) or an injury (e.g. a burn or broken limb) and tends to go away post-event or once the injury has healed or been treated. Acute pain can last for a short period of time, say an hour, or for weeks or months. However, acute pain does not usually last longer than six months. Examples of acute pain include the pain associated with a kidney stone or back pain say from collapsed vertebra caused by osteoporosis.
81 Contrastingly, chronic pain usually tends to endure for weeks, months or even for years. It is not usually experienced at a constant level but fluctuates over time, sometimes disappearing altogether for a few days. Examples of chronic pain include pain associated with arthritis, headaches and back pain say from a degenerative intervertebral disc.
82 Persons with pain associated with arthritis are usually (and were as at April 2000) under the care of their general practitioner. However, where the GP could not control the person’s pain, or the person needed specialist treatment or to be assessed for a joint replacement, they were generally referred to a rheumatologist.
83 The treatment of chronic pain associated with arthritis involved as at April 2000 (and indeed thereafter) a number of steps and considerations in addition to medication. For example, lifestyle changes to reduce weight and relaxation techniques could help to alleviate the pain or the perception of pain. The perception of pain is of course subjective. Back pain was prior to April 2000 (and still is) the most common pain.
(c) Paracetamol
84 Paracetamol has been marketed and used as an analgesic since the 1950s. It is appropriate to make a number of observations concerning its use in the period up to and including April 2000.
85 According to Professor Brooks, for a patient with pain associated with osteoarthritis, one would start them on a relatively mild analgesic, which was usually paracetamol, and monitor their response. Between 80% to 90% of patients were taking an analgesic. The most common analgesic was paracetamol. If a patient with arthritis had a very severe inflammatory process they may have had an increase in temperature that could be reduced by taking an antipyretic like paracetamol.
86 The November 1995 American College of Rheumatology Guidelines recommended paracetamol as a first line treatment for patients with osteoarthritis of the knee and of the hip, being the most common forms of osteoarthritis. The October 1993 British Society for Rheumatology Guidelines recommended paracetamol as the first line treatment for patients with osteoarthritis of the hip and knee. More generally, paracetamol was the first line treatment for patients with osteoarthritis anywhere in the body.
87 The use of paracetamol as a first line treatment for arthritis was also consistent with the principle that a person should be started on the drug that was least likely to cause side effects in the long term.
88 Some people experienced a skin rash as a side effect from using paracetamol, however the side effects were not usually as unpleasant as those from a non-steroidal anti-inflammatory drug (NSAID) or codeine. A skin rash was a common side effect of many drugs.
89 Paracetamol could be prescribed to treat the pain associated with the inflammatory forms of arthritis, such as rheumatoid arthritis. Paracetamol could also be prescribed for patients that did not have arthritis, for example those with back pain caused by non-arthritic conditions. Many patients took paracetamol for the long term treatment of chronic pain.
90 The available oral dosage forms of paracetamol were relatively fast-acting to alleviate pain. This was an advantage for patients. However, paracetamol had and has a relatively short half-life. The half-life refers to the time it takes for the drug concentration in the blood plasma to drop from peak concentration to 50%. The relatively short half-life of paracetamol meant that it had to be taken frequently to maintain pain relief. Typically a patient would take paracetamol four times a day. Often a person determined their own frequency based on the level of their pain. The level of pain could vary during the course of a day. As at April 2000 the recommended adult dose in Australia for use of paracetamol was 500mg to 1g every four to six hours, but not exceeding 4g a day. I will return to the significance of this later. The most commonly prescribed form of paracetamol in April 2000 was oral, immediate-release paracetamol 500mg tablets and capsules. By simple arithmetic, a person ought not to have had more than eight 500mg tablets a day of paracetamol.
91 Most patients were started on a dose of 2 to 3g of paracetamol per day. But if this was not providing enough pain relief, the dose of paracetamol could be increased to 4g a day before moving to another, stronger analgesic such as a combination of dextropropoxyphene hydrochloride and paracetamol, or dextropropoxyphene napsylate, or adding an NSAID. Patients were advised to take less paracetamol during periods when their pain was low and more paracetamol during periods when they were experiencing more pain.
92 Prior to 2000 in Australia in addition to the oral dosage forms, paracetamol was also available in other dosage modes. I do not need to elaborate for the moment.
93 In addition to immediate release forms of paracetamol, in the late 1990s there were slow-release paracetamol forms available.
94 A recognised potential problem with paracetamol was overdosing resulting in liver necrosis. Acute overdose was a problem for persons of all ages because paracetamol was (and still is) readily available in the home. It was also not recognised as being a dangerous drug. It was difficult to know exactly when toxicity issues might arise for an individual patient as there was not a good correlation between long term toxicity and liver function tests in somebody who might be developing liver toxicity from continuous regular usage, as opposed to an acute overdose. If a person took paracetamol regularly, then the level of toxicity in the liver could build up. The potential for liver toxicity was significant as many of the drugs prescribed to treat the inflammatory types of arthritis had liver toxicity as a side effect.
95 Persons suffering arthritis who took paracetamol and found it to be an effective treatment of pain relief generally needed to take paracetamol for life unless they were able to have surgery (such as a joint replacement) which sufficiently reduced or removed the pain. Persons with osteoarthritis also learnt to manage their pain by managing their medication.
96 A common problem with the treatment of chronic diseases was patient compliance in taking their medication. It was commonly known by medical prescribers that patient compliance with their medication decreased if the medication had to be taken multiple times a day.
97 According to Professor Brooks, the most desirable properties of an analgesic to treat chronic pain from arthritis were:
(a) simple dosage form, such as oral tablets or capsules;
(b) rapid relief from pain after dosing;
(c) the relief from pain to continue for as long as possible after dosing to reduce the number of doses required per day;
(d) no side effects; and
(e) low cost to the consumer.
98 Further, rapid relief from pain was desirable for any person in pain. A relatively long period before another dose was required was both advantageous and convenient for persons with chronic pain as it minimised the number of required doses per day of the analgesic. It also reduced the incidence of a person waking up during the night because of pain. For persons taking an analgesic for an extended period of time for chronic pain, the absence or minimisation of side effects and low cost were also important considerations.
99 Generally, in relation to the five properties set out above, the following observations can be made concerning paracetamol and its use as at April 2000:
(a) First, paracetamol was typically formulated in oral tablets, which met the first criterion;
(b) Second, paracetamol could be and usually was formulated to give rapid pain relief;
(c) Third, if formulated as an immediate-release product, the half-life of paracetamol was relatively low; patients would typically take paracetamol four times per day;
(d) Fourth, the side effects of paracetamol were well known and manageable; and
(e) Fifth, paracetamol was a relatively inexpensive drug.
100 I will expand further on some aspects of the above discussion when I come to deal with one of the respondents’ invalidity arguments concerning a lack of inventive step. For the moment it is appropriate to simply note that the respondents have contended that the principal property of paracetamol formulations that existed prior to April 2000 that would then have benefited from improvement was the duration of the pain relief provided, such that fewer doses were needed per day. Such a contention is supported by the evidence.
BASIC PRINCIPLES OF PHARMACEUTICAL FORMULATIONs
101 In what follows in this section, I will discuss some general concepts that would have been part of common general knowledge as at the priority date. In the following sections I will focus in on the approach to drug development as at April 2000. These are necessary background matters to my later discussion of inventive step.
102 The US Pharmacopeia 24 National Formulary 19 (1999) (USP) and the British Pharmacopeia (1993, 1998) are compendial references that were readily available before April 2000. The adjective “compendial” is referring to standards or compliance with standards. The USP and the British Pharmacopeia are books of pharmacopeial standards, including monographs for medicines, dosage forms and excipients. They also specify the tests and apparatus to be used to determine compliance with drug-release requirements in general and as particularly specified in individual monographs. I will elaborate on this later.
103 Literature in the field prior to the priority date also included the International Journal of Pharmaceutics, the Journal of Pharmacy and Pharmacology, the Journal of Pharmaceutical Sciences, Pharmaceutical Research and the Journal of Controlled Release.
(a) General
104 The parties have not put before me an agreed description of the basic chemistry and pharmacology. Accordingly, in what follows I have had to largely draw upon Professor Ian Tucker’s description of some of the background concepts that were not, as best as I can ascertain, in contention. But I have made some modifications in order to ensure that this section of my reasons does not trespass into contentious areas. Professor Tucker was called by Generic Partners. There were some specific aspects of his evidence that I have not been persuaded by as I discuss later, but his general discussion of the background theory was helpful. What is stated in this section are formulation concepts and practices as at the priority date.
105 Formulation is the process around converting a drug, the active ingredient, into the dosage form, which has certain properties. For example, aspirin is a drug and an aspirin tablet is a dosage form. A dosage form is required to be stable, aesthetically pleasing, and present the correct dose of drug in a convenient form for use. Types of dosage forms include tablets, capsules, solutions, linctuses, eye drops, nose drops, injections and so forth. About 80% of all medicines are taken orally, of which tablets are the most common dosage form. I will elaborate in more detail on dosage forms later.
106 The aim of drug delivery is to deliver the right drug, at the right dose, at the correct rate and to the correct site for the required time. For example, a simple immediate release dosage form such as an aspirin tablet releases all of its drug soon after it is swallowed. More sophisticated oral drug formulations might slow the release of the drug or attempt to target delivery to a particular region of the gastrointestinal tract.
107 The USP and the British Pharmacopeia divide solid dose formulations into immediate release (also referred to as conventional release and “IR”) and modified release. An immediate release formulation will deliver the drug dose immediately. Modified release means that the release of the drug is not immediate. I will elaborate in more detail on these aspects below, but it is useful to make some general observations at this point. There are two sub-categories of modified release:
(a) First, there is the sub-category of delayed release formulations. Delayed release means that the release does not occur immediately when the dosage form is swallowed. For example, an enteric coated tablet has a polymer coat that is insoluble in stomach acid but is soluble in the intestinal fluid. The polymer coat delays release of the drug until the tablet leaves the stomach;
(b) Second, there is the sub-category of extended release formulations. Extended release means that the drug is not released all at once, but is trickled out over a period of time, perhaps up to eight hours.
108 It is appropriate to elaborate further.
109 Delayed-release oral dosage forms may be used if irritation of the stomach is of concern, or to prevent the degradation of the drug substance in the acidic conditions of the stomach. Enteric coatings are pH-dependent and prevent the drug substance from being released in the acidic environment of the stomach. When the enteric coating is exposed to a higher pH (generally about pH 5.5 and above), the coating dissolves and the drug substance is released as an immediate-release dosage form. Before April 2000, enteric coatings typically involved the following types of excipients:
(a) Polymers — polymers form a film that are insoluble at acidic pH levels;
(b) Plasticizers — these are used in order for the coating to form a film. Examples include polyethylene glycols and diethyl phthalate;
(c) Colourants — these are used to colour the coating for marketing, identification purposes, or to stop light permeation. Examples include titanium dioxide and ferric oxide; and
(d) Anti-tacking agents — these are used to prevent the coating material from sticking together and causing imperfections in the coating that may compromise the delivery of the dosage.
110 In relation to extended release dosage forms, there were many on the market by April 2000. Apparently, they became popular in the 1950s. As a result, in the 1950–60s, drug companies tried to distinguish their extended release products by different branding, so that “extended release” became known as, for example, “slow release”, “prolonged release”, “sustained release” and “span release”. Generally speaking, one can refer to these formulations as extended release or “ER” or sustained release or “SR”. Concepts of extended release dosage forms were taught in pharmacy schools from the 1960s onwards. The Controlled Release Society was set up in the 1970s. The principles in relation to extended release dosage forms were well known before 2000.
111 There are a number of advantages of extended release formulations over immediate release formulations of the same drug.
112 Set out below is an example of plasma concentration time curves (shown in red) for three separate immediate release dosage forms. The graph also shows an “ideal” plasma concentration time curve (purple) for an extended release dosage form of the same drug. The minimum effective plasma concentration (MEC) of the drug and the minimum toxic plasma concentration (MTC) of the drug are shown by, respectively, the lower and upper broken lines; plasma is the liquid component of blood. Between those two lines is what is described as the “therapeutic window” for the drug.
113 The concentration of a drug in blood increases over time until a “steady” state is reached. The increase occurs because drug from the tablet just taken is added to drug remaining in the body from tablet(s) taken previously. A “steady” state is reached when the average drug intake rate equals the average rate at which drug is eliminated from the body.
114 The “ideal” plasma concentration time curve for the extended release dosage form shows:
(a) less fluctuation in drug levels in the blood over time, which may be associated with less side effects; and
(b) on the assumption that therapeutic effect is related to drug plasma concentration (which assumption holds good for many drugs), that a constant therapeutic effect is sustained for a longer period and there are no periods where the drug concentration is below the MEC.
115 There are, however, the following disadvantages associated with extended release formulations. First, there may be a longer onset time for therapeutic effect with an extended release dosage form. This is depicted above, where the purple curve exceeds the MEC at a time (“tlag”), which is later than the immediate release dosage form. Second, a single extended release dosage form (tablet) includes a quantum of drug equivalent to multiple doses. If the dosage form “dumps” all of those doses at once, the total drug concentration in the blood may be toxic (depending on the MTC for that drug). This may occur if the dosage form fails for some reason.
116 To address the problem of a slow onset time, a patient may be able to take an immediate release tablet and an extended release tablet at the same time. Some dosage forms include an immediate release component to give therapeutic effect quickly, and an extended release component to achieve a sustained therapeutic effect. An example of such a dosage form is a bilayer tablet, which consists of an immediate release layer and a second extended release layer. The immediate release layer disintegrates away releasing particles or granules (a particle or granule contains billions of drug molecules together with excipients). The drug then dissolves quickly from the particles or granules. Contrastingly, the extended release layer releases the drug slowly over time. Another example of such a dosage form is a capsule with both extended release and immediate release particles.
(b) Dissolution rate
117 An “ideal” extended release dosage form is a dosage form where the rate of release (rate of dissolution) of the drug is independent of the environmental conditions under which it is placed. What is meant by ideal is that the release rate is predictable because the release of the drug is controlled by the formulation, not by what is being done to the formulation by its environment.
118 But in reality all formulations change their release rate depending on the conditions of the environment, including the pH of the dissolution medium, anything that affects the hydrodynamics (flow) of that medium (e.g. the stirring rate, type of agitation, the shape of the vessel or the volume of liquid), temperature and the ionic strength of that medium.
119 The environmental conditions including the hydrodynamics of the dissolution medium can affect both “disintegration” of the dosage form to particles and “dissolution” of drug molecules from the particles; in these reasons, “particles” and “granules” can be treated as synonyms.
120 The rate of dissolution of a drug from a drug particle is predicted to some extent by the Noyes Whitney equation. But this is only a mathematical idealisation. I have previously observed (“Scientific Evidence: a Need for Caution in Decision-Making” (2010) 42 Australian Journal of Forensic Sciences 49 to 77) that there are the following limitations on using mathematical formulae in scientific empirical work:
(a) First, a formula is usually used to explain an apparent regularity between empirical observations. But a formula (and the underlying data) does not of itself demonstrate any necessary causal connection. From a correlation between the movement in two variables, sometimes such a link is inferred. But philosophically this is always contestable in a Humean sense. And practically may be contestable in many individual cases. The correlation may be due to a separate but common cause. Movement in one variable preceding movement in the other is only the starting point for any analysis.
(b) Second, a formula may only reflect a regularity observed from present or past data. And its function may simply be to explain that data rather than as a predictive tool. But even as an explanation for past data, the formula may have its limitations. Past data upon which the formula is built may only be a limited sub-set of the available data. The complete data set (if available) may change the apparent pattern of that regularity.
(c) Third, and relevantly to the predictive case, the formula may only be one of many ways to express the apparent regularity of the observed phenomena. So a formula may be a useful explanatory tool to explain observed phenomena, yet as a predictive tool it may be of little utility. For example, take 10 empirical observations which are plotted on a graph showing for each observation variables x and y measurements. Assume that this graphing shows that the 10 points can be connected by a straight line. You might conclude that there is a linear correlation between variables x and y and posit a formula for the straight line function (f(x) = ax + b), where f(x) = y. So you have a good explanation of the relationship between your 10 observations. But how good is the formula for predictions? Theoretically there are an infinite number of lines (including curved) that could have been drawn to join the 10 points, with correspondingly different formulae. And a further measurement (the 11th point) may show the function not to hold. For any set of empirical data, there are multiple potential theories/formulae to explain the same, hence what philosophers of science identify as the under-determination problem.
(d) Fourth, a mathematical formula takes its subject matter and their measurement as idealized and precise. But empirical data may be imprecise in quality or measurement. Indeed, statistical evaluation of primary data using stochastic equations and assumptions may be necessary to derive values for the variables to be used as inputs into the principal equation.
(e) Generally, mathematical equations should be seen in their limited context as imperfect tools. At most, any formula is only an idealized representation of the apparent regularity consistent with the model underpinning the theory. Their apparent elegance ought not to be taken as giving a greater air of verisimilitude to a scientific theory or its application than is warranted after considering all qualitative and quantitative assumptions and inputs.
121 According to the theory underpinning the Noyes Whitney equation, around each particle is an unstirred layer of liquid. The rate of diffusion of drug molecules across the unstirred layer controls the dissolution rate of drug. Molecules which diffuse across this unstirred (stagnant) layer are replaced “immediately” by molecules leaving the surface of the solid particle. Consequently, the solution which is in immediate contact with the particle surface is saturated with drug molecules and assumed to be at a constant concentration. The bulk solution in which the drug particle (and its unstirred layer) is suspended is assumed for the purpose of the equation to be uniformly mixed and of a uniform concentration. The rate of dissolution across the unstirred layer is determined by the concentration gradient across the unstirred layer. This is conveniently depicted below:
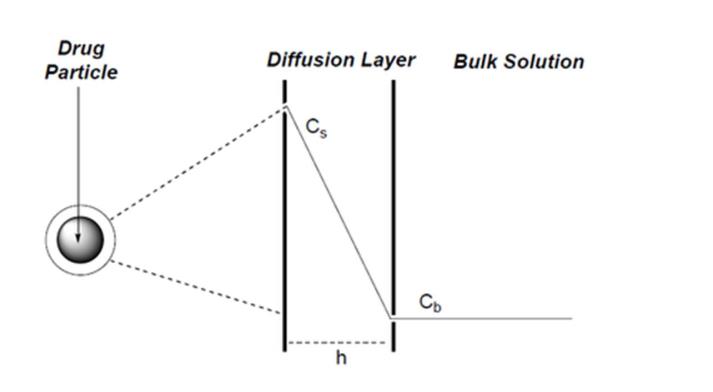
Noyes Whitney equation: rate of dissolution = |
|
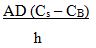
A = surface area of particle (or the sum of the surface areas of all particles).
D = diffusion coefficient for the drug molecules through the unstirred layer.
Cs = concentration of the drug at the particle surface (it is equal to the solubility of the drug in the medium).
CB = concentration of the drug in the bulk solution.
h = thickness of the unstirred layer (or the diffusional distance).
122 The effect of hydrodynamics on the rate of dissolution can be shown by reference to changes to the parameters in the Noyes Whitney equation. So, for example, the faster the solution is stirred, the smaller the thickness of the unstirred layer (“h”) becomes, so the rate of dissolution increases. Further, the stirring also increases the total surface area (“A”) of the particles. That is, by dispersing the particles through the medium, the total surface area is greater than if those particles were sitting at the bottom of the vessel. The greater “A”, the faster the dissolution rate.
123 Cs is determined by the solubility of the drug in the medium chosen. If a different medium is used, or a medium of a different pH is used, the Cs value may change. If a medium in which the drug is more soluble is selected, Cs becomes larger and the rate of dissolution faster.
124 The concentration of the drug in the bulk solution (CB) depends on the volume of the medium. As the drug is released from the particle over time, “CB” increases. The volume of solution is usually chosen so CB is in “sink conditions”. That is, CB is less than 10% of Cs. If a small volume of bulk solution is used, “CB” will increase over time at a faster rate. As a consequence, the rate of dissolution will slow because the difference between Cs and CB will decrease.
In vitro dissolution profile / in vivo dissolution profile
125 The in vitro dissolution profile of a formulation describes the dissolution of drug from the dosage form (e.g. a tablet) under specific prescribed conditions. The in vitro dissolution profile for a formulation shows the amount of a drug substance that is dissolved from a dosage form over time using an experimental apparatus. The drug concentration is measured in the dissolution medium at particular time intervals to produce the profile.
126 Obtaining an in vitro dissolution profile is simpler than obtaining an in vivo dissolution profile; patients are not required. It can provide information that may be useful for predicting the in vivo characteristics of a drug formulation, although in vivo measurements are usually required to confirm the performance of a formulation being studied or considered. But formulators make use of in vitro data to streamline formulations or identify formulations for further consideration or development. In vitro dissolution studies are used as a screening tool of candidate drug formulations. For example, based upon a number of formulations, one formulation may be identified as having a desirable in vitro release profile and, therefore, as having the best chance of working in vivo.
127 An in vitro dissolution profile can also be used as a quality control test to determine whether a particular batch of tablets was within the specification set down by the manufacturer or an official monograph in the USP or the British Pharmacopeia for that tablet.
128 Different apparatus can be used to measure dissolution profiles. Dissolution experiments must be carried out carefully in terms of technical work and the apparatus and the principal variables related to it defined. If any of these principal variables are substantially changed, it may significantly change the rate of release of drug from the formulation and therefore the in vitro dissolution profile of the dosage form.
129 The USP typically specifies the in vitro release profile under prescribed conditions using a prescribed apparatus for an extended release dosage form of a drug.
130 I will discuss dissolution testing in more detail in a separate section of my reasons.
131 In about the late 1970s, people started to think about whether in vitro dissolution profiles could be used to predict drug release in the body. Scientists working in this area developed computer models to attempt to predict in vivo behaviour from in vitro testing. But there is no guarantee that a dosage form with a particular in vitro dissolution profile will have the same in vivo dissolution profile (i.e. the same dissolution profile in the body). Drug release in the body is affected by many factors including differences in volume of fluid present in the in vivo environment, the degree of agitation and the pH the formulation is exposed to in the gut.
132 Some correlation work can, however, be undertaken. There are various possible levels of in vitro/in vivo correlation. The highest correlation is a point to point correlation between the in vitro and in vivo curves. If a high level of in vitro/in vivo correlation has been established for a specific formulation of a drug, it may be possible to predict that a specific formulation of that drug made in accordance with the relevant product specification or drug monograph with that same in vitro dissolution rate, will have that in vivo pharmacokinetic profile. Conversely, in theory it may also be possible to predict that a formulation of that drug made in accordance with the relevant product specification or drug monograph with the same in vivo pharmacokinetic profile, will have that in vitro dissolution profile. I will return to discuss these predictive and correlation questions later.
Dissolution rate from an extended release formulation
133 Matrices and reservoirs are two types of extended release dosage forms for controlling the rate of release of a drug from a solid oral dosage form (e.g. a tablet or pellet). For present purposes I do not need to discuss reservoirs further.
134 In one sense, a matrix can be thought of as a dispersion of a drug and excipient(s) through some other material(s). In that sense, a matrix system may be a tablet or a pellet in which drug particles and excipients are dispersed in a polymer or a lipid. Such a matrix (in that sense) will remain intact and extend the release of the drug from the matrix by eroding slowly or by controlling the release of the drug from a non-eroding matrix, or a combination of these processes. Polymers or lipids may be used for matrix materials. Polymers can be classified as water soluble, swellable in water or water insoluble. I will discuss the more contentious issues in a later section of these reasons concerning the different types of use of the concept or term “matrix” when I come to consider how the claims are to be construed and the meaning of the expression “matrix forming polymer”.
135 But just proceeding for the moment using “matrix” in this simplistic sense, it is appropriate to make the following observations. For such a “matrix”, release of drug generally occurs by the following steps: first, medium penetrates through the boundaries between the polymer granules; second, drug particles dissolve i.e. become drug molecules in the medium; third, these molecules diffuse out though the medium-filled channels or pathways. Such diffusion through channels is not straightforward as diffusion occurs through a complex pathway.
136 When a matrix is made from or formed by a gelling polymer (I do not need to discuss lipids further at this point given the issues that I need to address), the typical release process is as follows.
(a) First, water penetrates the outer layer of the tablet (or pellet).
(b) Second, the water reacts with the polymer causing it to swell, forming a gelatinous layer.
(c) Third, water transfers in through the gelatinous layer.
(d) Fourth, drug particles dissolve in the water and drug molecules diffuse out through the gelatinous layer. This gelatinous layer is a barrier to diffusion of the drug molecules and thereby slows and extends the period over which release occurs.
(e) Fifth, water continues to transfer into the tablet.
(f) Sixth, the above steps are repeated in a continuous process. At the same time erosion of the gelatinous layer is also occurring.
137 The rate at which drugs diffuse out of the tablet or pellet depends on the nature of the gelatinous layer. The nature of the gelatinous layer (including how quickly it is formed and its thickness, which is related to its ability to resist erosion) is determined by the type of polymer used in the formulation. Further, the hydrodynamic conditions and the type and concentration of excipients (I will explain excipients later) may also affect the thickness of the gelatinous layer and its ability to form a coherent gel layer.
138 Further, the choice of polymer can affect the size of the polymer granules which are compressed or formed into the matrix (used in the loose sense described earlier) and, therefore, the length of the pathway over which diffusion of drug molecules must occur. That then impacts on the diffusion rate. The size of the granules can also affect the rate at which the gelatinous membrane breaks off.
139 Further, the concentration of drug in the matrix can also affect the release rate from a matrix. For some matrices, if there is a low concentration of drug and no water-soluble excipients, the drug at the surface may release but the remaining drug may be trapped in the matrix. Contrastingly, if there is a high concentration of drug, the release profile may change because the water may follow the drug pathways into the matrix, resulting in faster drug release (and less drug being trapped in the matrix).
(c) Pharmacokinetics — in vivo concepts
140 I have touched upon some of these concepts earlier, but it is appropriate to elaborate further. Pharmacokinetics is the study of the change in drug concentration over time in the blood (pharmakon is Greek for drug, kinetics a time process). Pharmacokinetics concerns itself with what is referred to as ADME: absorption, distribution, metabolism, elimination. Distribution, metabolism and elimination constitute the disposition of the drug. One can look at these variables on a system wide basis or at a local level, say, a particular organ.
141 Pharmacodynamics is the dynamics (change) at the site of action or variable of interest in response to a drug over time. It is concerned with what is happening at the site of action or variable of interest by reason of the drug being in the body. For example, if a drug is meant to lower glucose levels in the blood, it would be possible to measure the concentration of drug in the blood (pharmacokinetics) or to measure what happens to glucose levels (pharmacodynamics).
142 I have discussed in vitro concepts above. It is appropriate to say something further on in vivo concepts.
143 The in vivo profile for a particular formulation looks at the concentration of the drug in a person’s bloodstream over time; it can also be looked at as an averaged concentration over a number of persons. Terms used to describe such a profile include:
(a) Cmax, the maximum concentration of drug that is measured in the blood;
(b) tmax, the time that the maximum concentration is reached; and
(c) AUC (area under the curve) which represents the total amount of drug that has been absorbed into the bloodstream over time; this can be compared with the quantity of drug administered to the person.
144 The following figure again shows the “therapeutic range” or the “therapeutic window” between the MEC and the MTC. Cmax, the maximum concentration of the drug in the blood, is estimated from a concentration time curve. A patient swallows the dose (e.g. a tablet) with a defined amount of water either in a fasting or non-fasting (“fed”) state. In a fasting experiment, a patient would typically be required to fast for eight to ten hours (since the night before). In a non-fasting experiment, a patient would typically eat a standard meal before the experiment. Blood is taken from the patient at specified times and analysed to determine the concentration of drug in the blood. This forms a profile for an individual. The following diagram is an “ideal” concentration/time curve for an immediate release dosage form.
145 But there are different approaches to determining that maximum. For example, in a bioequivalence study, the Cmax is the highest concentration datum, without interpolation. Other approaches involve interpolation between the two highest concentrations.
146 For some drugs, absorption is affected by whether the drug is taken with food or without food. Absorption of most drugs occurs in the intestine. If a non-disintegrating tablet is taken on a fasted stomach, the dosage form will most likely stay in the stomach until “house keeper” contractions occur (about every 90 minutes). If such a tablet is taken on a fed stomach (particularly after eating fatty foods), the tablet may stay in the stomach for a much long period. For this reason, the absorption rate of some drugs decreases if the drug is taken with food. The extent of absorption can also be affected by food for other reasons (e.g. binding of drug to food components). The fasted and fed states may have a different effect on the same drug in an immediate release dosage form as compared to an extended release dosage form.
147 Tmax is the time at which Cmax occurs. After Tmax, the rate of elimination exceeds the rate of absorption and so the blood concentration of drug falls. T1/2 is the elimination half-life. It is the time taken for the concentration of the drug in the blood to decrease by half, provided there is no drug still being absorbed. First order elimination means that the rate at which drug is eliminated is directly proportional to the plasma drug concentration (rate = constant x plasma concentration). Accordingly, the rate of drug elimination in first order elimination changes with concentration. Sometime after the Cmax it is assumed that absorption is complete and only elimination is occurring. The matter is represented diagrammatically below. The elimination rate constant (ke) represents the fraction of drug eliminated per unit of time. Elimination half-life t1/2 = 0.693/ke. However, this is more complex for extended release tablets because absorption may still be occurring during the time of the decay curve.
148 I have referred to AUC being the area underneath the concentration time curve. This is measured to tlast (time last) by the usual trapezoidal rule for approximating the region under the graph as a series of trapezoids. But the AUC can be calculated to t∞ (time infinity) by adding the area in the tail of the curve (after the last datum) to the AUC0-tlast. The AUC is related to the total amount of drug which the body receives as follows:
Dose x F (fraction of dose absorbed) = CL (clearance of drug) * AUC0-∞
149 If a drug is given intravenously, F necessarily = 1 (100% is “absorbed” by definition because the drug enters the bloodstream directly). This allows the clearance of the drug (CL) to be calculated. Assuming that CL is a constant (the same for a drug taken intravenously or orally), the fraction of the dose absorbed orally can then be back-solved from the AUC for the drug given orally, the dose and the clearance. One can then calculate the total amount of drug to which the body is exposed (the extent of absorption) when the dose is taken orally (dose x F).
(d) Dosage forms
150 It is appropriate at this point to elaborate in more detail on dosage forms.
151 Dr Brett Mooney, an expert in pharmaceutical formulation and called by Apotex, gave uncontroversial evidence on some background matters, some of which it is convenient to set out at this point. The statements that follow relate to dosage forms at and prior to April 2000.
152 Tablets were made by the compression of powders or particulate matter such as pellets, granules or beads. Tablets could be coated, including for functional reasons, such as an enteric coat, or for cosmetic reasons, such as a colour coat. Tablets could also be multilayer, with different layers having different properties. Most multilayer tablets had two layers and were called “bilayer” tablets. Bilayer tablets could be made to be biphasic, which meant that each layer had different release characteristics; for example one layer could have an immediate-release formulation and the other layer could have a modified-release formulation.
153 Capsules available by April 2000 consisted of either hard gelatin or soft gelatin. Hard gelatin capsules have a gelatin-based casing with a body and a cap that is loaded with materials, including one or more drug substance and excipients. These capsule shells were commonly coloured and printed for identification and marketing purposes. The material loaded into the hard gelatin capsules could be in the form of powder, granules/blend, beads or multiparticulates. Soft gelatin capsules are formed by injecting a liquid solution/suspension into a gelatine shell which seals during the process and is then dried.
154 The dosage forms prior to April 2000 included forms that differed in their release characteristics. As I have said, an immediate release formulation is a dosage form that is formulated to make the drug substance available for immediate dissolution after administration. For an immediate release formulation the drug substance is readily dissolved within a relatively short amount of time, such as no less than about 75% in less than about 30–40 minutes in a selected medium. I have explained above modified release forms including extended release or sustained release type formulations.
155 By April 2000 there were several different methods that could be used to formulate an extended or sustained release product, including use of a matrix formulation (diffusion-controlled or erosion), biphasic tablets or a formulation containing beads, pellets or multiparticulates.
Diffusion controlled matrix formulations
156 As I have explained, a diffusion controlled matrix formulation (using matrix in its simplistic sense) combines a polymer together with selected excipients and the drug substance. Examples of polymers that could be used for preparation prior to April 2000 included HPMC, ethylcellulose, alginates, hydroxypropyl cellulose and Eudragit (polymethacrylate-based copolymer). HPMC was commercially available in different molecular weights before April 2000. The molecular weight influences the viscosity of the polymer which affects its ability to control the release of the drug substance. For a specific drug substance high-molecular weight HPMC will have a higher viscosity and will generally provide a slower release as compared with low-molecular weight HPMC which will have a lower viscosity and will generally provide a faster release of the drug substance, ceteris paribus; of course, using the relative terms “slower” or “faster” says nothing about the absolute values or the real significance of any relative change.
157 Before April 2000 one could use a single polymer or a combination of polymers from the same or different families of polymers to achieve a target dissolution profile. Although it was preferable to have the simplest formulation consisting of only a single polymer, a combination of polymers could be necessary to achieve the desired dissolution profile. If a combination of polymers was used, the different polymers could be added to the dosage form at the same or different stages of the manufacturing process.
Erosion-controlled matrix formulations
158 For erosion-controlled matrix formulations (again using matrix in its simplistic sense), the matrix slowly erodes away, exposing the drug substance which then dissolves and diffuses out into the gastro-intestinal (GI) tract.
Biphasic tablets
159 Another form of sustained-release formulation before April 2000 was a biphasic tablet. An example is a tablet having an immediate-release layer and a sustained-release matrix-type layer. Each layer could contain the same, or a different, drug substance. These formulations were useful, for example, if it was necessary for some of the drug substance to be released immediately but also for the levels of drug substance in the blood to be maintained over a longer period of time.
Bead formulations
160 Sustained release formulations containing beads involve the use of spherical inert carriers, the surface of which can be loaded with drug substance and other excipients. A functional coat can then be applied to the beads. A formulation containing beads is generally more appropriate for use with low dosages of a drug substance, for example a dose of about 20mg or less (depending on the characteristics of the drug substance such as the solubility). If a large dosage is required, if one uses a sustained-release formulation based on beads this may increase the variability of the size of the beads in the dosage form, potentially making it more difficult to control the release profile of the dosage form. It may also take longer to manufacture the dosage form with more expense.
Pellet or multi-particulate formulations
161 Sustained release formulations can also be formulated that contain pellets or multiparticulates. In such a formulation, the individual particles can be formulated to create pellets or multiparticulates which can be used as is in a tablet or capsule that may be functionally coated. Alternatively, the particles can be coated with a functional coating and then used in tablets or capsules which again can be coated with a functional coat. The coating will be typically made from a polymer of the same types as described above.
(e) Manufacturing processes
162 The dosage forms available prior to April 2000 also differed by the way in which they were manufactured. Manufacturing in this context is the bringing together and combining as a dosage form, in a defined process, the drug substance and the excipients; I will provide a more detailed discussion later concerning excipients. Manufacturing techniques that were well-known and used before April 2000 for solid oral dosage forms included direct compression, dry granulation and wet granulation.
163 Direct compression involves blending the drug substance and excipients in a specific amount and order to create a blend. This blend is then compressed into tablets or encapsulated in capsules. The resulting tablet or capsule can be coated for cosmetic (if a tablet) or functional purposes. This is the simplest method of manufacture.
164 Dry granulation also involves dispensing the components of the drug product and then blending the drug substance with a selection of excipients in a specific order to form a powder blend. This blend is then consolidated by a process such as slugging or compaction, for example using roller compaction, which densifies the blend, which is then passed through a screen to produce small particles (this process is being “sized”), referred to as granules. This process of densification is referred to as dry granulation. The granules are then blended with any remaining excipients in a specific order to create a blend which is compressed to form the tablet or encapsulated into capsules, which can be coated for cosmetic (if a tablet) or functional purposes to produce the final product. This method is useful when handling poorly compressible or moisture-sensitive materials. The dry granulation process ensures the production of compressible material with homogenously distributed drug substance within the blend, which in turn ensures that there is consistency of the drug substance dose in individual tablets or capsules.
165 The process of wet granulation involves similar steps as for dry granulation. The tablet components are dispensed, select excipients are combined with or without the drug substance in a granulator and then a liquid solution is added to the powder blend. The liquid solution may consist of water or an organic solvent, with or without the drug substance and other excipients (for example a binder or surfactant). The granulation process can be performed using either a high shear mixer or fluid bed granulator. The wet granulated mass from the high shear mixer can be sized and dried or extruded, spheronised and dried, and if required sized and functionally coated. The wet mass from a fluid bed will be dried in situ and, if required, sized and functionally coated. In the case of functionally coated particles these can be blended with additional excipients as required and encapsulated or compressed into tablets and if required a functional coat applied. The resulting dried granules can be screened using a sieve and blended with any additional excipients in a specific order (for example fillers, disintegrants, glidants or a lubricant) before the mixture is compressed to form the tablet or encapsulated in a capsule, which can be coated for cosmetic (if a tablet) or functional purposes to form the final product.
DISSOLUTION TESTING
166 The rate at which a pharmaceutical formulation dissolves under certain conditions is identified by “dissolution testing”, usually in vitro testing. Dissolution testing is typically carried out in conditions which are designed to mimic the dissolution of the drug from the formulation in the body. Dissolution testing of pharmaceutical formulations is also typically carried out under standard conditions for purposes of quality control of formulations.
167 Dissolution testing is fundamental to the formulation development process. It is used to evaluate changes in formulation. It is also used to monitor and satisfy quality control and regulatory requirements, such as the US Department of Health and Human Services, Food and Drug Administration (FDA) requirements for a biowaiver. A biowaiver is where the efficacy and safety section of a drug application is approved by the FDA based on evidence of equivalence with an existing drug. A biowaiver also includes where a drug of a particular dosage strength is approved by the FDA based on the evidence of demonstrated bioequivalence or bioavailability data already provided for a higher dosage strength of that drug. Generally, comparative in vitro dissolution data is often required as part of an application for a biowaiver to support assertions as to equivalence.
168 One needs to evaluate dissolution testing parameters and the effect of these factors on dissolution, including:
(a) the effect of buffers (e.g. phosphate) and electrolytes (e.g. chloride ions);
(b) the hydrodynamics, i.e., the flow of the dissolution fluid within the dissolution vessel;
(c) the pH of the dissolution medium, which is usually considered within or across the physiological pH range of 1.2 to 7.5;
(d) the volume of the dissolution medium;
(e) the media surface tension and media viscosity;
(f) the dissolution medium (e.g. aqueous water or organic solvent with water);
(g) the temperature, which is usually 37°C, but can also be room temperature;
(h) the sink conditions; and
(i) the choice of the dissolution testing apparatus and components.
169 When considering the conditions for a reproducible and discriminatory dissolution procedure, it is important that the dissolution test is conducted under “sink conditions”. This refers to using a sufficient volume of dissolution medium in order for the drug to be freely dissolved and released from the formulation under test. To ensure that the dissolution test is conducted under sink conditions, the concentration of the total drug dissolved must not reach more than about 10 to 15% of its maximum solubility at any time during the dissolution procedure. If the dissolution test is not conducted under sink conditions, the drug release rate will be hindered and the dissolution medium around the drug can reach saturation solubility. Saturation solubility is the maximum amount of the drug that can be dissolved in the volume of the dissolution medium. If a dissolution study is performed and the drug is dissolved to a level of more than about 15% of its maximum solubility, it will not be under “sink conditions” and the dissolution rate would be significantly impacted by the solubility of the drug itself. When testing is under “sink conditions” the dissolution rate is principally impacted by the properties of the dosage form and not the drug’s solubility. For example, in the case of a sustained-release formulation, when sink conditions prevail, the properties of the matrix composition alone will control the rate of drug release from the dosage form (i.e. rate of release is limited only by the matrix polymer), rather than the combined effects of matrix composition and drug solubility limitation due to the absence of sink conditions.
170 It is important that all of the said parameters are well defined as part of the procedure including the type of dissolution testing apparatus that is used, so as to validate that the dissolution method works and is discriminatory in identifying release differences. This is important for reproducibility. Reproducibility is the ability of the dissolution test to be reproduced. It is also necessary to ensure that the test is discriminatory. A discriminatory dissolution test is sensitive to minor changes in a given formulation.
171 During the design of dissolution studies, it is also important to observe how the dosage form is moving within the dissolution apparatus. For example, if one observed a dosage form floating on the surface of the dissolution medium, one might decide to use a sinker to keep the dosage form on the bottom of the vessel below the stirring paddle used in USP type II apparatus. Any modifications to standard dissolution testing apparatus are based on the experience of the scientist, who has the purpose of optimising the method, with the aim of achieving reproducibility of results.
172 Standardised pharmaceutical dissolution testing apparatuses are described in the USP.
173 The United States Pharmacopeial Convention Inc is a scientific non-profit organisation that sets standards for drugs and excipients (these are referred to as “monographs”) and specifications for testing methods to evaluate drugs. That entity’s drug standards are enforceable in the United States by the FDA. Such standards are also used in many non-US countries such as Australia.
174 Such standards are continuously reviewed and updated based on new evidence and public health considerations. For such reviews and updates, the standards rely on volunteer experts who serve as decision makers on expert committees.
175 As at April 2000, the USP (the United States Pharmacopeia, US Pharmacopeia Convention, 24th review, 19th edition of the National Formulary, 1999 (USP 24 NF 19)) was the applicable standard for dissolution testing from 1 January 2000 through to about the end of 2001. The USP specifies seven apparatus for testing drug dissolution from a formulation. Four of these apparatus are designed primarily for oral solid dosage forms, being USP type I, II, III and IV apparatus. The remaining three apparatus are designed primarily for drug release testing of transdermal delivery systems, suppositories and topical products. The FDA would accept, as part of its regulatory approval of products, a modified apparatus standard where there was appropriate justification.
176 Prior to April 2000, there also existed the European Pharmacopeia, Council of Europe, 3rd Edition, 1996 (EP). The EP contained six apparatus for testing drug dissolution from a formulation. Three of these apparatus were designed primarily for oral solid dosage forms, being the paddle, basket and flow-through apparatus. The remaining three apparatus, being disk assembly, cell method and rotating cylinder, were designed primarily for drug-release testing of transdermal delivery patches.
177 USP type I apparatus is generally referred to by pharmaceutical scientists as a “basket”, USP type II apparatus as a “paddle”, USP type III apparatus as a “reciprocating cylinder” and USP type IV apparatus as a “flow through cell”. For convenience, in these reasons I will use Roman rather than Arabic notations for consistency.
178 USP type I apparatus and USP type II apparatus were first adopted by the USP in the 1970s. USP type III apparatus was adopted in the early 1990s.
179 Each of the four dissolution methods described in USP type I, II, III and IV apparatus provides different hydrodynamics. The differences in hydrodynamics have an impact on the dissolution results obtained. For example, if the same tablet is tested, the dissolution results from a test using the rotation of the paddle in USP type II apparatus will not be the same as the results when the rotating basket in USP type I apparatus is used. This is because each system creates a different fluid flow environment.
180 There is no established correlation between dissolution results measured using the different USP apparatus. Sometimes small differences are measured, other times the measured differences are large. Differences in measured dissolution profiles as between each of USP type I, II and III apparatus may not be predictable.
181 In a repeated dissolution study using any one of USP type I, II or III apparatus, variability in the measured dissolution profile for each sample of up to ±10% is considered normal and to be expected due to inherent analytical variability, and apparatus and sampling variability, although variability of ±5% is more ideal. It may be necessary to test at least 6 or 12 dosage units in a study using a particular USP apparatus, with well-defined and consistent testing parameters, in order to determine reproducibility with statistical reliability. The USP specifies the number of dosage units to test, which ranges from an initial 6 dosage units up to 24 dosage units.
182 Prior to undertaking a dissolution study, each USP apparatus has to be calibrated. Calibration involves making sure that all parts of the apparatus are in alignment and the equipment is functional with no mechanical failures. Reference standards can be purchased from the USP for calibration of a dissolution apparatus. These reference standards include disintegrating tablets, non-disintegrating tablets, extended-release tablets and extended-release beads, each of which were available prior to April 2000. The USP calibration standard specifies all parameters for the dissolution test as well as specific dissolution ranges for each measured time point. If the results fall within the dissolution ranges specified for the calibrator reference dosage form provided by the USP, it will indicate that the dissolution apparatus has been appropriately calibrated.
(a) USP type I apparatus
183 USP type I apparatus uses a basket. USP type I apparatus is used for immediate-release, delayed-release and sustained-release solid dosage forms. USP type I apparatus is comprised of a cylindrical mesh basket attached to a shaft that is placed inside a one litre round-bottom cylindrical vessel containing the dissolution medium. The dosage form being tested is placed inside the cylindrical basket which rotates inside the dissolution vessel. The speed at which the basket rotates (measured in rotations per minute) has an impact on the measured dissolution of the sample.
184 The USP specifies that the basket is made of stainless steel mesh. Prior to April 2000 and subsequently, different grades of mesh sizes were available. The USP specifies that 40 grade mesh be used unless otherwise specified in an applicable individual monograph.
185 The “grade” of mesh refers to the width of wire and of the openings. For example, a 40 grade mesh is specified in the USP to be a 40 x 40 mesh with 0.25mm wire diameter with wire openings of 0.40 ± 0.04mm. Where a 20 grade mesh is specified, a 20 x 20 mesh, 0.40mm wire diameter with wire openings of 0.90 ± 0.09mm is to be used.
186 The rotation speed (rpm) and grade of mesh used for the basket directly influences the hydrodynamics of the system and has a resulting impact on the rate of dissolution measured. The tighter the mesh, the more resistance there is to the dissolution medium passing through it. A 40 grade mesh is tighter than a 20 grade mesh and has a greater resistance to the passage of dissolution medium. The mesh grade used is important for reproducibility. Depending on the dosage form being tested, there may be a need to vary the mesh grade for a specific reason. For example, if the dosage form being tested included matrix-forming polymers that swell and gel (forming a gel layer around the tablet) in the dissolution medium, one would select a more open grade of mesh (e.g. 20 mesh) because the tablet is less likely to stick to a mesh with wider openings compared to a finer mesh, and so less variation should occur in the measured dissolution values. Pellet formulations might also require a larger opening in the mesh, as they may be comprised of gel and become sticky in the dissolution medium.
187 In assessing the parameters of the dissolution method to be used to create a set of reference data, it is sometimes necessary to modify the standard USP dissolution apparatus in order to achieve reproducible results. For biowaiver purposes, the FDA requires highly reliable, reproducible and discriminatory dissolution testing rather than mere adherence to the USP. The FDA provides guidance to guide pharmaceutical companies as to the dissolution testing required to obtain a biowaiver. Modifications to the apparatus to achieve the required reproducibility of results may be made, based on the experience of individual scientists and the facilities of the laboratory in which they work, but it is important that anyone seeking to determine whether a sample dosage form has the required dissolution profile uses the same apparatus and testing conditions.
(b) USP type II apparatus
188 USP type II apparatus uses a paddle. USP type II apparatus is comprised of a one litre round-bottom vessel containing the dissolution medium and a paddle attached to a shaft. The sample dosage form under test is added to the vessel and allowed to sink to the bottom, below the paddle. The paddle then rotates and stirs the dissolution fluid in the vessel.
189 USP type II apparatus is used for immediate-release, delayed-release and sustained-release solid dosage forms.
(c) USP type III apparatus
190 The USP type III apparatus was first included in the relevant standards in 1991, as an alternative to the basket (USP type I) and paddle (USP type II) apparatus for drug release testing. After increased interest in the apparatus in the 1980s, especially in light of the great interest in development of controlled release pharmaceutical formulations at that time, the apparatus was evaluated in 1990 and was then made official.
191 As at April 2000 the USP type III apparatus was attractive for the study of controlled release formulations, particularly since changing the media to simulate passage through the GI tract could be easily and reproducibly achieved if required. Borst, I, Ugwu, S and Beckett, AH “New and Extended Applications for USP Drug Release Apparatus 3” Dissolution Technologies (February 1997) 11 conclude at 15:
USP 3 should be considered the first line apparatus in product development of controlled release preparations, because of its usefulness and convenience in exposing products to mechanical as well as a variety of physicochemical conditions which may influence the release of products in the GI tract.
192 The USP type III apparatus assembly consists of:
(a) a set of cylindrical flat-bottomed glass vessels;
(b) a set of glass reciprocating cylinders;
(c) stainless steel fittings, and screens (made of suitable nonsorbing and nonreactive material) designed to fit in the bottom or top of the reciprocating cylinders;
(d) a motor and drive assembly to reciprocate or dip the cylinders vertically inside the vessels, and if desired, index the reciprocating cylinders horizontally to a different row of vessels; and
(e) a water bath which can be heated to the desired temperature.
193 A single test apparatus of the USP type III apparatus is shown below. This image has been taken from the USP with annotations added by Professor Dressman (a GSK expert witness):
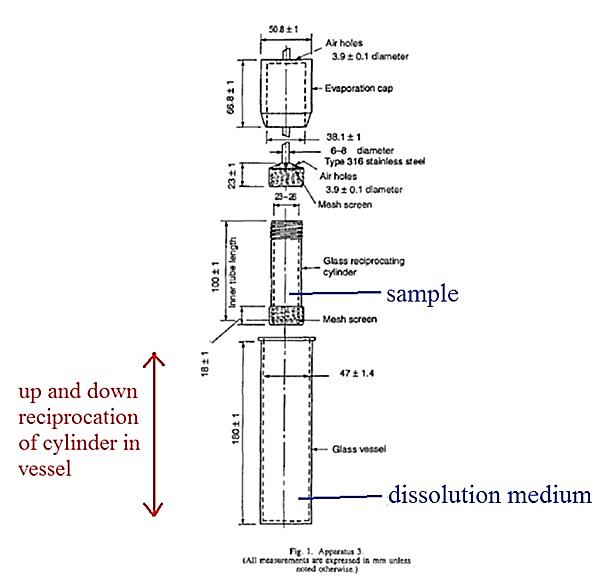
194 In general terms, a USP type III dissolution test is carried out in the following manner (for the moment just taking one of the set of vessels and cylinders):
(a) First, the dissolution medium is added to the glass vessel.
(b) Second, the sample is placed in the reciprocating cylinder and then the cylinder is screwed onto the dipping rod. In this “in use” position, the evaporation cap covers the cylinder.
(c) Third, the glass vessel is immersed in a suitable water bath of any convenient size that permits holding the temperature at 37 ± 0.5ºC during the test.
(d) Fourth, after about 15 minutes (the time needed for the medium temperature to reach 37 ± 0.5ºC) the apparatus is activated so that the dipping rod moves up and down at the prescribed rate to move the cylinder (containing the dosage form) up and down within the glass vessel containing the dissolution medium. During the “dipping” or reciprocation, the cylinder changes its position within the vessel, enabling the dosage form to interact with the dissolution medium. On the up stroke, the bottom mesh of the cylinder moves upwards to contact the dosage form and on the down stroke the dosage form leaves the mesh and floats freely within the cylinder. The interaction of the dissolution medium and the dosage form results in drug release.
(e) Fifth, at each of the sampling time points, a sample of fluid from the cylindrical glass vessel is removed, and then analysed to determine the percentage of the relevant active ingredient dissolved from the dosage form into the dissolution medium at that time point.
195 In USP type III apparatus, the mesh is inserted into the caps of the reciprocating cylinder. The purpose of the mesh is to allow adequate distribution of medium (and dissolved sample) into and out of the reciprocating cylinder, whilst retaining undissolved particles/dosage form within the reciprocating cylinder. As I have indicated, a mesh with larger holes may (undesirably) allow more undissolved particles to pass through the mesh into the lower part of the glass vessel, while a mesh with smaller holes may become blocked by undissolved particles, restricting flow of the medium into and out of the reciprocating cylinder. It is trite to observe that if either of these phenomena were observed during testing, the mesh size could be adjusted to optimise testing of the test dosage form.
196 The appropriate mesh size to use to carry out the dissolution testing using the USP type III apparatus on a paracetamol tablet is not specified in the USP. The protocol is silent on the mesh size to be used for USP type III apparatus. Moreover, the USP 24 NF 19 Official Monographs for acetaminophen (paracetamol) reveal that the dissolution test indicated for acetaminophen tablets does not use the USP type III apparatus. There is no guidance for mesh size in using the USP type III apparatus.
197 But notwithstanding that the USP is silent on which mesh size to use, one would normally select an appropriate mesh size to use. Professor Dressman said that selecting the mesh size to carry out testing using USP type III apparatus was a matter of routine as at April 2000 for those who had a knowledge of dissolution testing including using USP type III apparatus.
198 The mesh screens are sized to fit on the top and on the bottom of the reciprocating cylinder, but the USP does not specify that the mesh needs to be inserted at the top of the cylinder as well as to the bottom. Professor Dressman said that having a mesh at the top of the cylinder is optional. Further, she said that whether there was also a mesh at the top of the cylinder should generally have minimal influence on the dissolution results.
199 Finally, it is stated in the USP that:
Within the time interval specified, or at each of the times stated, raise the reciprocating cylinders and withdraw a portion of the solution under test for a zone midway between the surface of the Dissolution Medium and the bottom of each vessel.
200 The use of the word “zone” in the USP indicates that the withdrawal place is not a specific point within the vessel, but rather is a region. Professor Dressman said that the usual interpretation of a midway zone in a dissolution tester as at April 2000 (and still today) was and is that it is a zone which allows an accepted quantum of latitude in both the upwards and downwards directions. She said that that is how one would have taken a sample from USP type III apparatus in April 2000.
APPROACH TO DRUG DEVELOPMENT AS AT APRIL 2000
(a) General
201 At this point it is convenient to draw upon some of the uncontested evidence of Professor Martyn Davies, a professor of biomedical surface chemistry at the University of Nottingham (UK), an expert called by GSK.
202 The development of new drugs or new formulations of existing drugs within the pharmaceutical industry involves research and development (R&D). The R&D process is undertaken by an interdisciplinary team, typically made up of clinicians, marketing or business analysts, medicinal chemists, toxicologists, clinical pharmacologists and formulators. The various members of the R&D team become involved and undertake tasks at different stages of the R&D process. The R&D process typically starts by the identification of a clinical need by clinicians and marketing/business analysts within a pharmaceutical company. This need can either be a completely new therapy, a desire to improve existing therapies using new compounds, or a desire to improve existing therapies by providing an alternative formulation of an existing compound. The R&D team would also include analytical chemists, who were responsible for the testing of raw materials (that is, the drug substance and the excipients), testing of any reference drug formulation (for its dissolution characteristics and stability evaluation), in-process material testing (for example, testing the uniformity of the drug substance in a blend during tablet manufacture), finished product testing (that is, release and stability testing to evaluate the physical and chemical properties following manufacture and during storage), process validation, analytical method development and validation, cleaning validation testing and packaging material testing. The R&D team would also include regulatory affairs personnel, who were responsible for constructing the regulatory dossier or components of regulatory dossiers for local or overseas affiliates or regulators. There would also be a project manager to oversee the overall development process.
203 The process of pharmaceutical development endeavours to minimise risk. The R&D team, including the pharmaceutical formulator, would be highly aware that should any problem emerge with the drug or formulation during later stages of the development phase, such as during clinical trials, the R&D process would either have to be stopped or the drug reformulated, requiring all the work to be repeated. The R&D costs to bring a product to market are substantial and hence any delay or setback will cost time and money and significantly delay the launch of the product. At each stage of the drug development process, there will be a critical discussion between members of the R&D team to evaluate the risk and benefit for progression to the next phase.
204 The role of the formulator is to take the candidate compound and create a stable formulation which can be taken through the clinical trials and ultimately potentially into production and onto the market. The degree of responsibility of the formulator within the R&D team is high. A failure of a formulation during the clinical trials process can require a repeat of the clinical trials with very substantial cost and time consequences. The formulator is usually tasked with developing a drug formulation that:
(a) provides an accurate and safe dose of an active pharmaceutical ingredient (API);
(b) maintains the stability of the API within the formulation shelf-life and until it reaches the site of absorption in vivo;
(c) meets the target therapeutic effect; and
(d) delivers the drug in a suitable way such that it provides the best opportunity to be clinically effective.
205 In order for an orally administered drug to be effective, the drug must be “bioavailable”. That is, it must dissolve, but not degrade, in the GI tract so that it can be absorbed into the bloodstream and be available to act on the body and treat the patient. The amount of drug absorbed into the bloodstream is known as its bioavailability. The bioavailability of an API is influenced by its physicochemical properties, its route of administration and by the formulation used. Drug delivery and absorption of an oral dose is a complex biological process involving multiple biological systems and internal organs. In designing the optimal safe and effective dosage form, in addition to a host of other information and considerations, a formulator must be familiar with the physiology of the GI tract.
206 After a patient ingests an oral dosage form, it will rapidly travel down the food tract (oesophagus) and enter the stomach. The rate of dissolution of a drug into the stomach acid will depend in part on its formulation. As I have earlier indicated, an immediate release formulation will rapidly disintegrate into particles. On the other hand, certain oral dosage forms (e.g., those with enteric coatings) are specifically designed to resist the breakdown by gastric acid entirely and therefore to avoid dissolution in the stomach. The stomach contents empty into the upper part of the small intestine, which serves as the primary source of maximum absorption for food as well as orally administered drugs. Digested food and drugs are absorbed into the blood via the rich supply of blood vessels that line the walls of the intestine. Once in the bloodstream, a drug is transported to the liver where it may undergo some degree of metabolism. It is then distributed around the body to the tissues where it exerts its therapeutic effect. Eventually, the drug or its metabolite(s) will be excreted from the body.
207 As I have said, the study of the absorption, metabolism, distribution and excretion of a drug is known generally as the field of pharmacokinetics. The R&D team would also include pharmacokineticists. As earlier indicated, the study of the pharmacokinetics of a drug is undertaken by plotting the plasma concentration of the drug against time (see the diagram set out earlier). As I have earlier indicated, the values of Cmax, tmax and AUC are used in bioavailability studies to assess the rate and extent of absorption of a drug from a pharmaceutical formulation. The plasma concentration of the drug can be measured using several analytical methods. One common method is high performance liquid chromatography.
208 There are two commonly used measures of bioavailability. The term “relative bioavailability” is where the bioavailability of a test formulation using its AUC value is compared to another formulation of the same dose and route of administration of the drug using its AUC value. In contrast, the term “absolute bioavailability” is used to compare the bioavailability of a test formulation by a specific route of administration to that of the drug when administered intravenously, i.e. when there is no barrier to absorption. Absolute bioavailability therefore measures the amount of the drug that is absorbed into the systemic circulation and assesses the effect of a different route of administration on drug absorption.
(b) Developing a modified formulation of an existing formulation
209 Dr Mooney has given similar evidence to Professor Davies on the general approach to the research and development of a formulation, but with more of an emphasis on the approach one would have taken in April 2000 to developing a modified formulation of an existing formulation in order to change its properties. I generally accept his evidence on such general matters, although I have a difficulty with his specific evidence on inventive step which I will discuss as the final topic in my reasons.
210 Dr Mooney said that the R&D team would be assisted by:
(a) people from the patents group (who provided relevant information from patent searches and provided assistance with preparing the regulatory dossier);
(b) purchasing personnel, who were responsible for sourcing raw materials; and
(c) quality assurance personnel, who were responsible for certain material approvals, providing reports on excipients, packaging materials and various aspects concerning batch manufacturing documents.
211 He also explained that the role of a pharmaceutical formulator in a commercial setting was to develop a formulation for a particular drug substance that:
(a) consistently and reproducibly met appropriate physical quality limits and chemical quality properties, for example potency, dissolution where appropriate, purity and microbial contamination, as required by pharmacopeia and the regulatory authorities within the jurisdiction; the product had to remain stable within these specified parameters throughout the product’s shelf life;
(b) met regulatory requirements for marketing authorisation, (for example, if the formulation was a generic version of a reference formulation, the product had to be bioequivalent to the reference drug); and
(c) could successfully be scaled-up from pilot scale to commercial production.
212 Dr Mooney said that the first priority was the quality of the finished dosage form, but the R&D team was also mindful of cost considerations in all steps of the development process. Where opportunities for cost benefits or cost containment arose, these would be considered, including in relation to labour, choice of materials and process.
213 Dr Mooney said that for any formulation task, the formulation process could be divided into two stages: pre-formulation and formulation. Pre-formulation would be preceded by patent and literature searches. These searches might identify opportunities and information in relation to drug substances and potential products.
214 The pre-formulation process in the formulation of a generic version of a reference product involved:
(a) review of the development brief and Drug Master File (DMF) or technical package for the drug substance;
(b) identifying the physical and chemical properties of the drug substance, which might include experiments on the drug substance to be used;
(c) identifying excipients to use in the formulation; and
(d) further patent and literature searches (if necessary).
215 The formulation process for a generic version of a reference product involved:
(a) continued investigations of the physical and chemical properties of the drug substance;
(b) selecting the method of manufacture;
(c) excipient selection;
(d) analytical testing of the reference product, trial and scale-up batches;
(e) conducting stability testing and choosing appropriate packaging;
(f) bioequivalence studies; and
(g) completion of regulatory requirements for registration of the final product.
216 Dr Mooney said that prior to April 2000, there was certain information about the drug substance and reference product that the R&D team or the formulators would expect to receive before commencing the development of a generic version of a reference product. This information was usually provided in the form of a development brief. Such a brief typically included the name of the reference product, the target markets for the generic product and the name of the drug substance manufacturer.
217 After receiving a brief, he said that the team (or relevant member) would look up details of the reference product in the USP and British Pharmacopeia, Martindale: The Extra Pharmacopeia (Royal Pharmaceutical Society, 31st ed, 1996) (Martindale), The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals (Merck & Co Inc, 12th ed, 1996), US Physicians’ Desk Reference (Medical Economics Company, 53rd ed, 1999) (PDR), MIMS Annual (MediMedia Australia, 23rd ed, 1999) (MIMS) and the product information leaflet for the reference drug to obtain the following information (to the extent it was publicly available):
(a) chemical structure of the drug;
(b) physical properties of the drug;
(c) the route of administration (that is, the way the dosage form was administered to a patient, e.g. orally, by injection, by inhalation);
(d) qualitative formula of the reference drug (that is, a component list for the reference drug);
(e) the dose (that is, the amount of a drug substance in the dosage form);
(f) the dosage form (for example, tablet or capsule, if there were to be multiple strengths and whether or not a break line was required);
(g) Cmax and Tmax;
(h) the dissolution profile over a specified time period;
(i) for an oral formulation, whether the product was coated, and whether this was for functional or cosmetic purposes; and
(j) the packaging presentation.
218 The generic product’s dosage, dosage form, route of administration and release profile (immediate-release or sustained-release) would be selected to match those of the reference product.
219 The R&D team or the formulators would obtain a quantity of the drug substance (with the assistance of purchasing personnel) and samples of the reference product from markets where the reference drug was sold (from colleagues in the team). Samples of the reference product were required so that necessary tests, including in vitro dissolution tests, could be conducted.
220 In addition to the brief, the R&D team or formulators would either have been provided with or requested the DMF or technical package relevant to the drug substance. The DMF is a document prepared by the pharmaceutical company that has intellectual property rights to the synthetic process for the manufacture of the drug substance for the purpose of submission to a regulatory authority. The drug substance DMF contains information on the drug substance including, for example, information on the solubility of the drug. A DMF exists in both an open and a closed form. The open form contains quality information about the drug substance and may be requested from the company that submitted the DMF to the regulatory authority. The closed form contains quality information about the drug substance, as well as confidential details on the manufacturing process.
221 A technical package is typically a document produced by a drug substance manufacturer before a full DMF. It is less detailed than a DMF but contains quality information about the drug substance. The supplier of the drug substance may make a technical package available to potential purchasers.
222 It is necessary to understand the physical and chemical properties of the drug substance which will be used in a formulation. Details of the relevant physical and chemical properties of the drug substance were usually obtained. Where this information was not available, that data was generated by conducting or co-ordinating analytical testing of samples of the drug substance.
223 The relevant physical properties of a drug substance included:
(a) density — a numerical value of mass in a specific volume;
(b) particle size — a measure of the average diameter of particles of the drug substance;
(c) surface area — a measurement of the surface area of the particles of the drug substance in a specific weight;
(d) drug substance structure and structural features — the physical structure of the drug substance at the molecular level;
(e) crystallinity — whether the drug substance existed in a crystalline or amorphous state at relevant condition parameters;
(f) melting point — the temperature at which the drug substance changed state from a solid to a liquid; and
(g) optical rotation — the way the drug substance rotated light, being a reflection of its racemic purity if the drug substance existed as an isomer.
224 The relevant chemical properties, which encompassed both standard chemical properties and quality attributes, included:
(a) pH — the extent of acidity of the drug substance;
(b) stability — including both solid state stability (i.e. sensitivity to light, temperature, humidity) and solution stability (i.e. in an acid or alkali solution or under simulated oxidation conditions);
(c) solubility — the tendency of the drug substance to go from a solid phase into solution in a specific aqueous and organic media;
(d) potency — the amount of pure drug substance in a known weight of drug substance expressed as a percentage;
(e) purity — the amount of drug and non-drug substance in a known weight;
(f) isomeric purity — whether the drug substance existed as a cis- or trans- isomer or as a racemic mixture; and
(g) impurities — the amount and identity of non-drug substance in a known weight of drug substance.
225 Let me now elaborate further on the question of excipients, which also relates to the “matrix forming polymer” question that I will dwell on at some length later in these reasons.
226 As part of pre-formulation work, the development team or its formulators would identify the excipients to use in a formulation. Put simply for present purposes, the excipients are everything in the tablet or capsule other than the API. The excipients used in a formulation are based on the chemical properties of the drug substance (which influences its compatibility with excipients), the method of manufacture chosen and the desired dosage form. Compatibility studies can be used to test whether the drug substance interacts with any of the excipients, or any of the excipients interact with each other, in an undesirable way. The extent of any compatibility studies will be tailored referable to the chemical stability of the drug substance. For a generic product, if excipients are selected that are the same as those in the reference product and the drug substance is very stable, compatibility studies may be superfluous.
227 Before April 2000, common types of excipients that a formulator could use and that were well-known and used prior to April 2000 for oral dosage forms, included the following:
(a) Fillers — These fill the volume of the tablet not occupied by the drug substance and other excipients to provide a presentable, manageable dose. They can be used to form a tablet or capsule to give it sufficient bulk to enable it to be conveniently handled by a patient. Fillers also contribute to the flow of the blend and facilitate compression during manufacture of the dosage form. Fillers can also improve the solubilisation of the drug substance. Common examples of fillers were lactose, microcrystalline cellulose, maize starch, pregelatinised maize starch and dibasic calcium phosphate. Some fillers can also be used to perform other functions.
(b) Binding agents — These act to bind the drug substance with the excipients. Examples of binding agents included gelatin, maize starch, pregelatinised maize starch, povidone (a water-soluble polymer), low-molecular weight HPMC, hydroxypropylcellulose and ethylcellulose (water-insoluble). Again, any one of these binding agents could perform other functions.
(c) Glidants — These assist a blend of drug substance and excipients to flow through the compression/encapsulation process to facilitate consistent tablet or capsule weight. Examples of commonly used glidants included talc and colloidal anhydrous silica. Colloidal silicon dioxide has, of course, the empirical formula SiO2. Its small particle size and large specific surface area give it desirable flow characteristics.
(d) Lubricants — These ensure that the drug substance and excipients do not adhere to the punches and dies during the manufacture process. Prior to April 2000 the lubricant which was most commonly used was magnesium stearate (a salt formed from one magnesium cation with two anions from stearic acid, a fatty (octadecanoic) acid).
(e) Disintegrants — These assist in the rupture of the structure of the dosage form to release the drug substance to allow it to dissolve. Examples of commonly used disintegrants included maize starch, pregelatinised maize starch, sodium starch glycolate, sodium croscarmellose and povidone XL. As one can appreciate, a substance used as a disintegrant in one context can be used as a binding agent in another context or perhaps as a filler in yet another context.
(f) Antioxidants — These stop the oxidation of the drug substance. A well-known example was ascorbic acid.
(g) Colourants — These are used to give the final product colour. Colour may be added for identification, for example to identify different strengths of a product, or for marketing purposes. Commonly used examples included ferric oxide, aluminium lakes and titanium dioxide.
(h) Flavours — These are used to add flavour to the final product. Commonly used examples included spearmint and peppermint.
(i) Buffers — These assist in maintaining the pH of the dosage form in a range within which the drug substance is stable. An example of a commonly used buffer was citric acid.
228 In selecting between different excipients the R&D team or its formulators would initially ascertain what excipients were used in the reference product by referring to published information about the reference product, for example information contained in the PDR and MIMS. These documents would not usually include details of the quantitative formulation (that is, the amount of each excipient that had been used) or what manufacturing method was used to make the dosage form. However, one could usually identify from the documentation relating to the reference product the function of each excipient and one could generally infer the manufacturing method based on the excipients used. One generally assumed that because of the need to obtain regulatory approval, which included consideration of compatibility and stability data, the excipients used in the reference product were compatible with each other and the drug substance.
229 If the proposed generic formulation was a delayed-release formulation, a sustained-release formulation or a combination of both, then in addition to the excipients listed above, one would also consider the use of excipients such as matrix-forming polymers or enteric coating excipients that would be necessary to achieve the required release characteristics. But in choosing modified-release excipients, one would be guided by the excipients used for the reference product.
230 The final dosage form might also require a coating. As I have indicated earlier, a coating can have a number of purposes that may be functional or cosmetic. Functional coatings can be used to change the location or rate of the release of the drug substance within the body. They can also be used to protect the drug substance (for example from moisture penetration or to stop oxidation degradation), to reduce the odour of the drug substance or for ease of packaging (coated tablets can be packed into blister packs more readily). Cosmetic coatings can be used for identification or to increase patient compliance or patient acceptance (to make a dosage form look more appealing).
231 Further, as I have indicated, before the R&D team or its formulators were given a development brief, appropriate patent and literature searches would usually have been done on the drug substance, with formulations containing the drug substance and any relevant patents and literature highlighted for the team. This information would have been provided to and considered by the team in the formulation work.
232 Finally, the process steps undertaken to construct oral dosage forms varied depending on whether direct compression, dry granulation or wet granulation was used and whether the product was coated or uncoated. It was not expected to be known what manufacturing process was used to produce the reference product but to some extent this could be inferred from knowledge of the reference product’s excipients. The R&D team and the formulators would use their experience and manufacture trial formulations to establish the best process appropriate to the equipment available. The manufacturing technique adopted would depend upon factors such as quality (physical and chemical), ease of manufacturing and the suitability of the process to the particular formulation being developed.
THE expert WITNESSES
233 The expert witnesses each gave their evidence separately and were cross-examined in the classical style. In the circumstances of this case, I did not consider it desirable to have the experts give their evidence in the contemporary style of a concurrent evidence session, notwithstanding that, advantageously, prior expert conclaves had been convened culminating in a joint expert report (JER). As I have said, Professor Dressman and Professor Davies were called by GSK. Professor Fassihi and Dr Mooney were called by Apotex. Professor Tucker was called by Generic Partners. It is necessary to set out in a summary form at the outset some general observations on their background and my general perceptions of their evidence. This will inform the discussion which follows on the detailed technical issues.
(a) Professor Dressman
234 Professor Jennifer B Dressman completed her PhD in pharmaceutical chemistry in 1981 from the University of Kansas. She is a Professor of Pharmaceutical Technology and Director of the Institute of Pharmaceutical Technology at Johann Wolfgang Goethe University in Frankfurt, Germany. Prior to joining the University in 1994, Professor Dressman had been Associate Professor of Pharmaceutics at the University of Michigan and had also held various industry positions as a research chemist. She has published extensively in the field of pharmaceutical science including peer-reviewed articles, monographs, books and patents.
235 Professor Dressman is a talented and experienced dissolution expert with an extensive academic and practical background. More particularly, Professor Dressman had extensive practical knowledge and experience (both pre and post priority date) in conducting dissolution testing generally and using the USP type III apparatus. She also had considerable knowledge of and experience in dealing with sustained release oral dosage forms containing HPMC.
236 Professor Dressman’s evidence was neither selective nor given as an advocate for the party calling her. Moreover, she was able to answer detailed questions about dissolution testing and scientific publications without the need to constantly refresh her memory. Plainly she had the relevant technical knowledge at her fingertips and her technical literacy and conceptual understanding were impressive.
(b) Professor Davies
237 Professor Martyn Christopher Davies received his PhD for a thesis on aqueous film coatings in 1985 from what is now King’s College London. He was appointed in that year to a lectureship position at the University of Nottingham where he taught and still teaches pharmaceutical technologies. In 1996 he became Professor of Biomedical Surface Chemistry at that University. Professor Davies also co-founded a company in 1997 through which he provides pharmaceutical development and formulation characterisation services. He has also authored over 380 peer-reviewed papers and book chapters.
238 Professor Davies is a person skilled in the art of formulation. He is also a highly qualified academic with practical experience with HPMC and in formulating oral dosage forms containing HPMC.
(c) Dr Mooney
239 Dr Brett Antony Mooney obtained his PhD in synthetic organic chemistry in 1980 from the University of Adelaide. He has held a number of positions as a research and development chemist including as director of research and development at Alphapharm. Throughout his career he has been involved in the formulation of 130 oral solid dosage forms involving 150 drug substances. He is currently a consultant and director at BM Pharma Consulting Pty Ltd, a company he founded in December 2012 to provide advice in respect of oral solid dosage formulation and development.
240 Dr Mooney is an experienced and clever formulator. For most of the last 30 years he has been a successful formulator of generic drugs for Alphapharm. Dr Mooney gave evidence primarily on the issue of inventive step but he also expressed views on the question of matrix forming polymers.
241 GSK contended that Dr Mooney’s inventive nature and cleverness was well apparent when he was looking at the 522 and 194 Patents in the context of solving the problem articulated in the development brief. I am inclined to agree and I will elaborate on this later.
242 Dr Mooney’s formulation experience was gained in the context of making generic versions of innovator products. Moreover, as at 2000 he had no personal experience in formulating capsules using beads or pellets or with bilayer tablets. Generally, his only experience in making a sustained release product was making a generic version of a corresponding sustained release dosage form already on the market. He had never sought to create a completely new sustained release dosage form where he did not have access to another one already on the market. These matters justify circumspection in the weight to be given to some of his evidence.
243 Further, although Dr Mooney was familiar with looking at the results produced from dissolution testing using USP I and II type apparatus, he did not personally carry out such dissolution testing. Dr Mooney had no experience with dissolution testing conducted using the USP type III apparatus prior to 2000. Moreover, his employer, Alphapharm, did not have a USP type III apparatus at that time.
(d) Professor Fassihi
244 Professor Alireza Fassihi Dehkordi obtained his PhD in pharmaceutics in 1978 from Brighton University in England. Since 1986 he has been a consultant to pharmaceutical companies in South Africa, the United Kingdom and the USA on dissolution testing and manufacturing. In 1992 he became Professor of Biopharmaceutics and Industrial Pharmacy at Temple University in Philadelphia where he teaches pharmaceutical manufacturing and biopharmaceutics. Since 1994 he has also been a consultant to the United States Food and Drug Administration. He has published over 130 peer-reviewed papers. He uses Fassihi rather than Dehkordi for his surname.
245 Professor Fassihi’s area of expertise was in dissolution testing.
246 I must say at this point that it became apparent during the oral evidence that Professors Dressman and Fassihi were the only experts with any practical knowledge and experience of conducting dissolution testing particularly using the USP type III apparatus.
247 I agree with GSK that Professor Fassihi had a tendency to argue the case. A good example of such a tendency was his attempt to hold the patentee to a higher level of disclosure than he adopted in his own peer reviewed publications and in his own patent. In his written evidence, Professor Fassihi emphasised that without information as to inter alia the mesh material, the mesh size and the mesh placement, there was insufficient detail in the description of the dissolution test in the Patent for him to reproduce it. Contrastingly, in his own peer reviewed publications and patent, Professor Fassihi omitted parameters of the type for which he criticised the omission by the patentee. Each of Professor Fassihi’s papers (Pillay and Fassihi, “In vitro release modulation from crosslinked pellets for site-specific drug delivery to the gastrointestinal tract” Journal of Controlled Release 59 (1999) 229; Pillay and Fassihi, “In situ electrolyte interactions in a disk-compressed configuration system for up-curving and constant drug delivery” Journal of Controlled Release 67 (2000) 55; and Missaghi and Fassihi, “Release characterization of dimenhydrinate from an eroding and swelling matrix: selection of appropriate dissolution apparatus” International Journal of Pharmaceutics 293 (2005) 35) omitted details of the mesh size, mesh material and mesh placement in the descriptions of the dissolution tests using USP type III apparatus carried out. Professor Fassihi sought to justify the absence of details of mesh size in a number of ways: first, by asserting that he had always used size 20; second, by suggesting that reference could be had to the USP type I apparatus prescribed mesh sizes (20 and 40); and third, by asserting that it was not necessary to specify mesh size because “the editors, the reviewers, they were all familiar with my work”. Professor Fassihi then sought to justify the discrepancy between the levels of parameter detail given in his own articles and what should have been provided in the Patent by saying that a patent was not a peer reviewed publication. But his attempt to argue that patents should be held to a higher degree of disclosure than peer reviewed publications was doubtful given that his own patent (United States patent number 6,090,411 for a monolithic tablet for controlled drug release), like his articles, did not include any details of mesh size, mesh material or mesh placement. Apotex has asserted that the context of these papers was different and that “the reader would understand that the dissolution tests had been conducted using identical equipment, such that ‘like’ was being compared with ‘like’ …”. I am not convinced that this is a sufficient answer. Persons reading this literature might want to try and duplicate the results. It is apparent to me that, anomalously, the disclosures he made in his own publications did not meet his asserted threshold for the disclosure of the equipment and the parameters that he now asserts ought to have been made in the Patent. Yet skilled readers of his papers would have also needed sufficient disclosure in order to duplicate the results published in his literature. I must say that I have been a little sceptical of Professor Fassihi’s evidence on this issue.
248 More generally, I am concerned with other aspects of his evidence. On several occasions he sought to maintain problematic positions in the face of contrary documentary evidence. First, he refused to concede that the appendices to the Gray article were compendial (Gray, V A “Drug Release Calibrators for Apparatus 3 — Collaborative Study Results” Pharmacopeial Forum 20(1) Jan. to Feb. 1994, 6934); in my view there was no good reason to consider them to be anything other than compendial. Second, he sought to read “a selected row of vessels” as meaning all six rows of the USP type III apparatus:
MS ROFE: I suggest to you that you read that as a selected row of vessels and you use one row of them? --- Each vessel of selected rows. That means, whatever number of rows that you like to select. Generally there are, I think, seven or eight rows but you have to — depending on what conditions you want to use. Here they are using water, so they can have one row, two row, three rows. They can select, yes.
HIS HONOUR: It does say, Professor, “a selected row”. It doesn’t say “selected rows”. It says “a selected row” which suggests it’s in the singular. And you’re not changing the pH, so why would you have multiple rows? --- That is correct, yes. That could be the case here, yes. It could be one row, yes.
Third, he sought to maintain his position that a top mesh was essential in the face of USP documents showing an absence of a top mesh. Fourth, he refused to accept Professor Dressman’s solubility figures (20 to 23.7 mg/ml), notwithstanding that they were sourced from the peer reviewed literature published over several decades, for the solubility of paracetamol. Indeed and contrary to the other experts and the peer-reviewed literature cited by Professor Dressman, he sought to suggest that the solubility figure would vary according to polymorphs and particle size. Relatedly, he sought to depart from his and Professor Tucker’s evidence in the JER as to the relevant solubility being equilibrium solubility. I also enquired:
HIS HONOUR: Except, though, that all of the literature seems to be arranged at most between 20 and 23.7 milligrams a millilitre. So there seems to be general consensus on that as being the figure for 37 degrees Celsius. Or is that not how I should read this material? --- Again, your Honour, this is equilibrium solubility.
Yes? --- This is a little bit — you know, with this one — that’s why I’m saying that as a scientist, dissolution rate — rate is a dynamic phenomenon. This solubility is a static value. So if you’re measuring dissolution rate, of course, you need to be more careful.
Apotex has said that this factual issue did not relate to any “live issue”. I am not so sure, except that equilibrium solubility appeared to be agreed. Professor Fassihi seemed to seek to confuse this question with dynamic concepts that added little to the debate and in a manner I found unhelpful. Fifth, he raised for the first time in the JER the “percolation theory” that related to the use of HPMC in dosage forms that release active pharmaceutical ingredients over a period of 10 to 24 hours. Again, this was hardly of assistance to the issues in the case.
249 Professor Fassihi also found it difficult to give responsive answers to questions.
250 Further, another noticeable matter was Professor Fassihi’s need to frequently refresh his memory as to the contents of his own papers and those discussed in his affidavit. As I perceived it, this was not simply a manifestation of the usual caution to be expected from an expert witness seeking to be precise and accurate. To use the language submitted by GSK, it suggested that the dissolution issues on which he gave evidence were “not part of the working furniture of his mind central to his accumulated expertise”. The following was a typical example:
HIS HONOUR: Well, let’s take your own articles? --- All of them, they had sink conditions definitely, yes.
Do you report separately on equilibrium solubility, or dynamics or rates of solubility, as being a relevant metric? --- I don’t recall, your Honour, what I have said in there, but we definitely have done the calculation of sink conditions.
251 Generally, I have exercised caution in placing significant weight on his evidence concerning the use of the USP type III apparatus. To the extent that Professor Fassihi’s evidence differs from that of either Professor Dressman or Professor Davies on this topic, I am inclined to prefer the evidence of Professors Dressman and Davies.
252 Of course I accept that I should exercise caution in expressing observations on the reliability of evidence concerning expert witnesses. Nevertheless, where conflicting expert opinions have been expressed, I am bound to resolve any conflict where it goes to an issue in the case and to explain my reasons for preferring one view over another. After all, that is one of the tasks of a working trial judge mining facts from the coal face of forensic inquiry.
(e) Professor Tucker
253 Professor Ian Tucker obtained his PhD in pharmaceutical sciences in 1980 from the University of Queensland. In 1991, he was appointed Professor of Pharmaceutical Sciences at Otago University in New Zealand. He was the Dean of the School of Pharmacy at the University from 1999 to 2010. Since 2011, he has been the Associate Dean (Research Commercialisation) for the Division of Health Sciences at the University. Professor Tucker has also consulted to the pharmaceutical industry in relation to formulation development. He has published 161 peer-reviewed research articles.
254 Professor Tucker gave evidence on all issues in the proceeding including matters related to the conduct of dissolution tests. He gave particular evidence concerning the use of the USP type III apparatus and the choice of mesh size. He also gave evidence concerning oral dosage forms which included using HPMC. I must say at the outset, ceteris paribus, that given the breadth of the areas that he covered, the weight of his evidence in any one area was more difficult to justify as carrying the same (let alone greater) weight than the weight of evidence given by a more specialised expert in that area. For example, his evidence on dissolution testing and equipment carried less force with me than the evidence of Professor Dressman on that topic who had a more specialised focus.
255 Let me also say that, in my view, Professor Tucker in some aspects did not read the Patent in a practical and preferable fashion. In some aspects he read the Patent with a sceptical and critical eye and through a lens distorted such as to cast doubt upon the clarity and scope of the claimed invention. Further, he applied the same approach to articles relied upon by GSK and its experts. For example, when taken to Cao et al “Formulation, release characteristics and bioavailability of novel monolithic hydroxypropylmethylcellulose matrix tablets containing acetaminophen” Journal of Controlled Release 108 (2005) 351, the following exchange occurred during cross-examination:
MR SHAVIN: Yes. At the moment I’m asking you to take this paper simply as a peer review paper as if you were reading it as part of the literature that you’re reading.
HIS HONOUR: Well, putting it another way, take it at face value and assume that they’ve carried out properly conducted experiments and — do you follow what I mean? --- Yes.
In other words, you seem to be wanting to ---? --- Your Honour, I ---
--- find something missing or ---? --- I’m sorry, your Honour. I read scientific papers with a very critical and a very sceptical eye because there’s much in the literature which is not right and, therefore, I read it carefully. So I’m not trying to find holes here. If I’m going to be asked about water uptake, I need to know that that paper there, 13, has done it properly. That they’ve allowed for the loss of paracetamol from the tablet. I’ve done these water uptake studies myself.
256 Contrastingly, and as GSK has pointed out, Professor Tucker did not exercise the same critical approach to other evidence in support of the respondents’ case. For example, his hypothetical H2 “thought experiment” relied upon an uncritical reading of the unpublished paper T J Grattan et al “The use of in vitro – in vivo correlation to develop a sustained release formulation of paracetamol with a predefined plasma concentration-time profile: A report on a step-by-step progression” May 2000.
257 Professor Tucker’s approach to the Patent and its construction was typified by his extensive exploration of all the theoretical linguistic ways in which the word “after” in claim 1 could be construed. His construction was not constrained by practicality let alone by how a person skilled in the art would construe the specification.
258 Professor Tucker’s detailed exploration in his first affidavit of “after” included various figures that depicted sharp vertical jumps in release at various time points. But he neglected to explain that he never actually anticipated such sharp vertical jumps. Ultimately he agreed with the other experts in the JER that the releases he depicted were only achievable with systems consistent with dosage forms that were not contemplated in the Patent.
259 In the JER, Professor Tucker agreed with the other experts that “released after” meant “released at”:
… within the context of the patent we anticipate the profiles would be smooth curves which must fit within the boundaries specified in the patent. The reason we think this is that the patent does not mention pulsatile or coated systems which would be necessary for achieving the limits in Figure 10 of Tucker’s June affidavit.
260 Further, Professor Tucker sought to justify a greater level of disclosure from the patentee than in his own peer reviewed scientific articles. But in Krajacic and Tucker, “Matrix formation in sustained release tablets: possible mechanism of dose dumping” International Journal of Pharmaceutics 251 (2003) 67 there was no identification of sampling volume, sampling point or discussion of sample replacement. I refer to what I said earlier concerning Professor Fassihi’s evidence and a similar disconformity between his criticisms of the disclosures in the Patent and his own publications.
261 Further, some of Professor Tucker’s evidence was in the nature of a thought experiment; I do not mean by the use of that phrase the description given to the highly intellectual conceptualisations of a theoretical physicist. Further, some of his evidence was speculation based upon only his reading done in preparation for his evidence in this case rather than his existing knowledge or experience. Professor Tucker lacked hard-edged experience in the formulation of oral dosage forms containing HPMC and dissolution testing using the USP type III apparatus. Nevertheless, this did not constrain him from speculating broadly in these areas. Let me elaborate further on these aspects.
262 Professor Tucker expressed opinions on matters relating to HPMC and matrix forming polymers including HPMC and the formulation of HPMC containing oral dosage forms. As I say, he had little direct experience of such matters. He agreed that with respect to HPMC his opinions were principally derived from materials he had read for the purpose of this litigation. Prior to 2000 he had not published any research about the formulation of solid dosage forms. Moreover, his research had not concentrated on the application of HPMC in solid oral dosage forms; his work concerning analysing the release of sodium salicylate from HPMC matrices had little relevance. Further, he had not done any practical work on gel formed by HPMC and had not undertaken any research on the proportions of gel in relatively shorter acting extended release formulations as compared with longer acting extended release dosage forms with release over 24 hours.
263 Further, Professor Tucker expressed views on various matters relating to dissolution testing using USP type III apparatus. But Professor Tucker’s first affidavit referred only to his experience with USP type I and II apparatus. In his second affidavit, Professor Tucker suggested that he was familiar with how the USP type III apparatus worked because as he described it:
the School of Pharmacy at the University of Otago has a USP Type III apparatus and I supervised students using it before 2000.
264 But under cross-examination, it was apparent that as at the priority date, Professor Tucker had no direct experience using the USP type III apparatus in the course of his work. By reason of his position as Dean from 1999 to 2000, Professor Tucker’s teaching responsibilities during that time were very limited. Further, under cross-examination, his statement that he “supervised students” evolved into “supervision in the lab occasionally”. When pressed, this turned out to be one doctoral student who had used the USP type III apparatus in her work in Croatia before she came to the University of Otago. Moreover, that student did not go on to use the USP type III apparatus under his supervision at Otago. Further, and more generally, Professor Tucker agreed that a study of the USP type III apparatus and how it was described was not part of the skill set that he possessed as at 2000. He had not himself used the USP type III apparatus before or after the priority date. The set up of the USP type III apparatus in Professor Tucker’s department was done by other people. It also follows that he was not the person making decisions as to mesh size to be used in the USP type III apparatus as at the priority date or thereafter. Generally, he had no cause (prior to giving evidence in the present proceedings) to consider appropriate mesh size for use with the USP type III apparatus or to study its effect on the relevant hydrodynamics. Further, he was not familiar with the common literature on the USP type III apparatus as at the priority date.
265 GSK made the submission that in the areas that I have identified above, Professor Tucker was not qualified as a witness with “specialised knowledge” based on “training, study or experience” for the purposes of giving admissible opinion evidence under s 79(1) of the Evidence Act 1995 (Cth). It was said that his opinions on these issues were not based on his specialised knowledge. Accordingly, it was said that his evidence on these matters was inadmissible. But in my opinion Professor Tucker was at least formally qualified to express opinions on dissolution testing and dosage forms, notwithstanding that he lacked direct experience in the areas indicated. Accordingly his opinions are admissible under s 79 and I have treated them accordingly. Nevertheless, it is appropriate to observe that, in the circumstances, I have given his opinions on the following topics reduced weight:
(a) HPMC and matrix forming polymers;
(b) the formulation of oral dosage forms containing HPMC;
(c) the practicalities of dissolution testing such as the selection of appropriate mesh size, material and mesh placement in dissolution testing using the USP type III apparatus; and
(d) the terms used by persons skilled in the art to describe the USP type III apparatus and how they would understand the reference to “basket” in claim 1.
266 On such topics, the evidence of Professor Dressman and Professor Davies has carried greater weight and has been more persuasive because of their more targeted knowledge and expertise in these areas. Professor Fassihi’s evidence has been given diminished weight for the reasons that I have indicated.
principles of CONSTRUCTION
267 The principles of law governing the construction of patent specifications, including claims, are well established. I adopt the principles that I set out in Streetworx Pty Ltd v Artcraft Urban Group Pty Ltd (2014) 110 IPR 82 at [58] to [68]. For convenience I have reproduced them in what follows in this section. But I should say at the outset that the principles I have set out below are subject to the boundary constraint that they cannot be used to justify re-writing a claim or amending a mistake in an essential integer. After all, these principles have been developed to construe what is, rather than to construe what should have been. I will elaborate on this boundary constraint in a separate part of my reasons when discussing the construction of claim 1 and the dissolution testing apparatus description.
268 The proper construction of a claim is a question of law.
269 A claim is construed from the perspective of a person skilled in the relevant art as to how such a person would have understood the patentee to be using the words of the claim in the context in which they appear. Further, a claim is to be construed in the light of the common general knowledge including the art before the priority date.
270 A measure of common sense should be used. Further, ordinary words should be given their ordinary meaning unless a person skilled in the art would give them a technical meaning or the specification ascribes a special meaning (Kimberly-Clark Australia Pty Ltd v Multigate Medical Products Pty Ltd (2011) 92 IPR 21 at [39] per Greenwood and Nicholas JJ).
271 In terms of how the body of the specification may be used in construing a claim:
(a) The claim should be construed in the context of the specification as a whole even if there is no apparent ambiguity in the claim (Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 616);
(b) Nevertheless, it is not legitimate to narrow or expand the boundaries of the monopoly as fixed by the words of a claim by adding to these words glosses drawn from other parts of the specification (Jupiters Ltd v Neurizon Pty Ltd (2005) 65 IPR 86 at [67]; Kinabalu Investments Pty Ltd v Barron & Rawson Pty Ltd [2008] FCAFC 178 at [44]); and
(c) More particularly, if a claim is clear and unambiguous, to say that it is to be read in the context of the specification as a whole does not justify it being varied or made obscure by statements found in other parts of the specification.
272 Now the specification may stipulate the problem in the art before the priority date and the objects of the invention that are designed to address or ameliorate this. The specified objects may be useful in construing a claim in context. Further, the specified objects may be useful in considering any lack of utility argument (now abandoned by the respondents in the present case). Nevertheless, the specified objects are not controlling in terms of construing a claim; glosses cannot be drawn from the objects.
273 A claim should be given a “purposive” construction. To elaborate, words should be read in their proper context. Further, a too technical or narrow construction should be avoided. Further, the integers of a claim should not be considered individually and in isolation. Further, a construction according to which the invention will work is to be preferred to one in which it may not (Pfizer Overseas Pharmaceuticals v Eli Lilly & Co (2005) 68 IPR 1 at [250]). But to give a claim a “purposive” construction “does not involve extending or going beyond the definition of the technical matter for which the patentee seeks protection in the claims” (Sachtler GmbH & Co KG v RE Miller Pty Ltd (2005) 221 ALR 373 at [42] per Bennett J). To apply a “purposive” construction does not justify extending the patentee’s monopoly to the “ideas” disclosed in the specification (GlaxoSmithKline Australia Pty Ltd v Reckitt Benckiser Healthcare (UK) Ltd (2013) 305 ALR 363 at [60]).
274 I also adopt what was said in Artcraft Urban Group Pty Ltd v Streetworx Pty Ltd [2016] FCAFC 29 at [72] to [78] per Greenwood J (agreed to by Rares J at [142], [145] and [146]):
As already mentioned, s 40 of the Act provides that a complete specification for an invention must describe the invention fully including the best method known to the applicant of performing the invention and, so far as an application for an innovation patent is concerned, it must end with at least one claim (and no more than five claims) defining the invention.
In order to ascertain the invention “described and claimed”, it is necessary to refer to the specification as a whole. That was the course Menzies J took in Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at p 593 (“Welch Perrin v Worrel”). On appeal, Dixon CJ, Kitto and Windeyer JJ also observed at p 610 that the specification must be read as a whole and further observed that “[i]f it is impossible to ascertain what the invention is from a fair reading of the specification as a whole that, of course, is an end of the matter”.
The complete specification, which includes both the body and the claims (see Kimberley-Clark Australia Pty Limited v Arico Trading International Pty Limited and Others (2001) 207 CLR 1 at [14], per Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ), must be construed in the light of the common knowledge in the art before the priority date and must not be read in the abstract. The Court must place itself in the position of some person acquainted with the surrounding circumstances as to the state of the art and manufacture at the relevant time: Kimberley-Clark Australia Pty Limited v Arico Trading International Pty Limited and Others at [24], per Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ.
Because the patent instrument is not a document operating inter-parties, but rather a public instrument, it must define the monopoly in such a way that it is not reasonably capable of being misunderstood. Even though the specification must be read as a whole, it is necessarily made up of parts which have different functions. It is not legitimate to narrow or expand the boundaries of the monopoly, as defined by the claims, by adding to the language of the claims, glosses drawn from other parts of the specification. Further, if the language of a claim is clear, it is not to be rendered obscure simply because obscurities can be found in particular sentences in parts of the specification: Welch Perrin v Worrel at p 610, Dixon CJ, Kitto and Windeyer JJ.
A proper analysis of the specification may show that references to a particular embodiment are by way of “illustration and explanation of the invention” rather than a “definition of it”: Welch Perrin v Worrel at p 612, Dixon CJ, Kitto and Windeyer JJ.
In Interlego A.G. v Toltoys Pty Ltd (1973) 130 CLR 461 at p 477, Barwick CJ and Mason J approached the construction of the claims and identification of the invention by reading the specification as a whole. Their Honours observed at p 478, in the circumstances of the questions in issue in that case, that it would be unlikely that anyone would conclude on reading the specification as a whole that the invention “as described makes claim to a method of assembly” of a particular kind. Their Honours observed, however, adopting the propositions derived from Welch Perrin v Worrel (described at [75] of these reasons) that the settled rule is that in ascertaining the width of a particular claim (that is, the construction to be attributed to the words defining the monopoly), it is not permissible to vary or qualify the “plain and unambiguous meaning of the claim” by reference to the body of the specification.
However, the rational starting point is not to simply turn to the language of the claims and seek to attribute meaning to those words in the abstract. The body of the specification and the claims must be read together as a whole in an attempt to identify the invention and determine whether the meaning of the words used in the claim (that is, the construction of the claim in question) is truly “plain and unambiguous”. If, having examined the s 40(2)(a) description of the invention in the body of the specification and the s 40(2)(b) definition of the claims, an expression used in the claims is not clear, it is then permissible to return to the body of the specification to either “define or clarify” the meaning of words used in the claim (see Interlego A.G. v Toltoys Pty Ltd, p 479 per Barwick CJ and Mason J) without infringing the rule that clear and unambiguous words in the claim cannot be varied or qualified by reference to the body of the specification. See also, to the same effect, Kimberley-Clark Australia Pty Limited v Arico Trading International Pty Limited and Others at [15], per Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ.
275 How the stipulation of preferred embodiments interacts with questions of construction has also been analysed by Jessup J in Artcraft Urban Group at [181] to [186]. Further, I would also refer to Lord Hoffmann’s observations in Kirin-Amgen Inc v Hoechst Marion Roussel Ltd [2005] RPC 169 at [27] to [34] concerning a purposive approach to construction.
276 A claim is to be construed from the perspective of how a person skilled in the art would have understood the patentee to be using the words, informed by the notional skilled addressee’s general knowledge and what has been disclosed in the specification. But to consider such a perspective does not entail that the Court necessarily requires expert evidence to assist on construction. If it is clear that the claims are to be read according to their ordinary meaning with no special meaning given to any word or phrase, if the science or technical issues are easily comprehensible and if, more generally, the Court does not require expert assistance in understanding the context of the claims, then expert evidence on construction may not only be unnecessary, but unhelpful and distracting. The nature and complexity of the patent in suit and the issues raised will determine the utility or necessity for expert evidence on construction. But to say that expert evidence may not be required on construction does not entail that expert evidence may not in any event still be required on other issues such as novelty or innovative step, even if not necessary on construction per se.
277 The Court is to place itself in the position of a person acquainted with the state of the art and manufacture prior to the priority date. In terms of the skilled addressee, one is using a hypothetical construct. The following principles are applicable:
(a) First, to identify the characteristics of the skilled addressee, the field to which the invention relates must be identified.
(b) Second, the skilled addressee is taken to be a person of ordinary skill (as opposed to a leading expert) in that field and equipped with the relevant common general knowledge including the art before the priority date (Minnesota Mining & Manufacturing Co v Beiersdorf (Australia) Ltd (1980) 144 CLR 253 at 293; Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [24]).
(c) Third, the qualifications and experience of the skilled addressee will depend on the particular case, having regard to the nature of the invention and the relevant industry. Formal qualifications are not essential. Practical skill and experience in the field may suffice. A patent specification is addressed to those having a practical interest in the subject matter of the invention; such persons are those with practical knowledge and experience of the kind of work in which the invention is intended to be used.
(d) Fourth, the hypothetical person skilled in the art may possess an amalgam of attributes drawn from a team of persons whose combined skills, even if disparate, would normally be employed in interpreting and carrying into effect instructions such as those contained in the specification.
(e) Fifth, as the skilled addressee comes to a reading of the specification with the common general knowledge of persons skilled in the relevant art, they read it knowing that its purpose is to describe and demarcate an invention. But the person skilled in the art is not particularly imaginative or inventive.
(f) Sixth, the skilled addressee does not come to reading the specification seeking failure.
278 Generally, the Court is to place itself in the position of such a skilled addressee, using such expert evidence that is of assistance to the construction task.
279 As I have said, the legal construct may not be a single person but may be a team of persons whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those contained in the Patent. Where the art is a highly developed technology such as pharmaceutical science and the development and use of formulations, the hypothetical skilled addressee may be a team whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those which are contained in the Patent.
280 In the present case, the Patent is addressed essentially to a pharmaceutical formulator being a member of the wider R&D team that includes people with expertise in medicinal chemistry, analytical chemistry and in the field of pharmacokinetics, together with experience in pharmaceutical formulation development. The formulator would normally be educated to PhD level and have two to three years’ post-doctoral expertise. Alternatively, the formulator may not have a doctorate but would have five or more years’ experience working within the pharmaceutical industry and either an undergraduate or masters degree. In this case, a formulator would work with and seek guidance from a scientist with particular expertise in the dissolution testing of extended release oral dosage forms using USP apparatus, in particular the USP type III, to carry out the claimed invention.
Construction of claims 1 to 3: usp type iii apparatus
281 Apotex has contended that there are two alternative constructions of the words in claim 1 “the USP type III apparatus, reciprocating basket” being that the words:
(a) describe the USP type III apparatus modified to use a reciprocating basket instead of a reciprocating cylinder;
(b) alternatively, describe the USP type III apparatus where the word “basket” is erroneous and should read “cylinder”.
282 It contends that the first construction gives the words used a sensible construction that is supported by Professors Fassihi and Tucker and by Dr Mooney. It contends that adopting the second construction would involve the Court concluding instead that GSK made an error, the contention of GSK. In summary, in my view a mistake was made and the skilled addressee would so construe the relevant integer of claim 1.
283 Let me make several points at the outset on the mistake alternative. First, I accept that the cause of any mistake on the part of GSK is irrelevant. That is not the correct focus. Second, as Apotex rightly points out, GSK could have sought to amend the Patent to correct the mistake. But it has not done so, no doubt for what it perceives to be good reason. Third, and relatedly, the fact that the skilled addressee would so construe the relevant reference in claim 1 does not justify me in re-writing or in essence amending claim 1.
284 As to whether a mistake was made, Apotex contends that a conclusion that there is a mistake in the Patent should not readily be drawn for the following reasons in substance:
(a) First, it says that there are various factual considerations that mean that the use of a reciprocating basket makes practical sense. Modifications to USP apparatus were well known; indeed there had been a “proliferation” of such apparatus before the priority date. The word “basket” describes a known USP part (being a basket used with a USP type I apparatus). Further, attaching a USP basket to a USP type III apparatus was possible, and there would be practical reasons for doing so.
(b) Second, it says that there is nothing in the Patent itself including its repeated references to “reciprocating basket” that provides any support for the existence of a mistake.
(c) Third, it says that the language of the Patent being “words of the patentee’s own choosing” is to be construed more strictly than a document operating inter partes such as a contract. This means, so it is said, that I should be slow to construe a word in a claim as being erroneous, particularly when that word has been used by the patentee six times to describe the apparatus.
(d) Fourth, Apotex contends that the evidence suggests that an error was not made. It has referred to the fact that GSK by its legal representatives in a 7 May 2015 letter said that the words “reciprocating basket” were “synonymous” with “reciprocating cylinder”. Apotex asserts that I should assume that this explanation was given on instructions from GSK. I think that the reference to this letter is a jury point and a distraction. I have put it to one side.
285 In summary, and notwithstanding Apotex’s submissions, I have concluded that an error was made and that the skilled addressee would so construe claim 1. But as I have said, this does not avail GSK for reasons that I will later explain.
(a) The Patent and the USP
286 Before proceeding further it is appropriate to make some preliminary observations concerning the Patent and the USP. I should also say for completeness that although the specification has been amended in some aspects, I have taken the approach of reading the specification as if the amended language had been originally included in the specification.
The specification
287 Claim 1 uses the description “as determined by the USP type III apparatus, reciprocating basket”. Parameters are set for the dissolution medium (0.1M HCl), the temperature (37°C) and the cycle speed (15 strokes per minute).
288 As to the body of the specification, there is no express reference, apart from the word “basket”, to a non-compendial apparatus being used. At various points in the specification there is just repeated the phrase I have set out earlier, with no other elaboration; see for example p 4a (amended page, 4 September 2006) lines 3 and 4, lines 14 and 15, p 10 lines 1 and 2 (“the USP type III apparatus (reciprocating basket) …”), p 12 at foot (“the USP type III apparatus (reciprocating basket) …”), and p 15 lines 20 and 21 (“the USP type III apparatus (reciprocating basket) …”).
289 Indeed, the specification and claim 1 use the definite article “the USP type III apparatus”. One might have expected that if a non-compendial form was intended to be referred to that the indefinite article would have been used, so as to refer to “a USP type III apparatus”.
290 What also should be noted is that in some parts of the specification, the words “reciprocating basket” have been bracketed after the phrase “the USP type III apparatus”, perhaps suggesting that only an elaboration was being included rather than a non-compendial operation.
291 It is also to be noted that reference is made to a “reciprocating basket”. The word “reciprocating” is, of course, different to “rotating”. The latter refers to uniform circular motion about a centre or axis. The former refers, in this context, to an alternation or oscillation in movement or action; in the present case an up and down movement at a uniform speed. As at the priority date, and as I have found (as I will subsequently explain), part of common general knowledge was an understanding that when one was using a basket for compendial USP apparatus, one was contemplating a rotating basket (USP type I apparatus). The phrase “reciprocating basket” would either jar with the skilled addressee as being an incorrect reference or be taken as a reference to something non-compendial. I will return to this subject later.
292 It is also to be noted that there is no detail in the specification for the “basket” in terms of shape, size or material of manufacture (cf the detail given for a USP type I apparatus). The absence of such detail may possibly lead the skilled reader to the conclusion that the reference to “basket” was a mistake, although it may also be said that the reader, in the absence of any detail for “basket”, might simply conclude that what was intended was a “basket” with the parameters as set out for the USP type I apparatus.
The United States Pharmacopeia
293 Let me begin with USP 23 NF 18. This applied from 1 January 1995. It was superseded by USP 24 NF 19, which applied from 1 January 2000, and which I have referred to as the USP.
294 In both USP 23 NF 18 and the USP, the following may be noted:
(a) In section 711, there is a discussion of dissolution. There is a description of type I apparatus and type II apparatus.
(b) The description of type I apparatus refers to a cylindrical basket. The basket is to be fabricated of stainless steel to the specifications shown in figure 1. It is stated that unless otherwise specified in the individual monograph, use 40 mesh cloth. It is also said that a basket having a gold coating may be used. Further, there is a specification for the distance between the inside bottom of the vessel and the basket.
(c) Figure 1 is shown as follows:
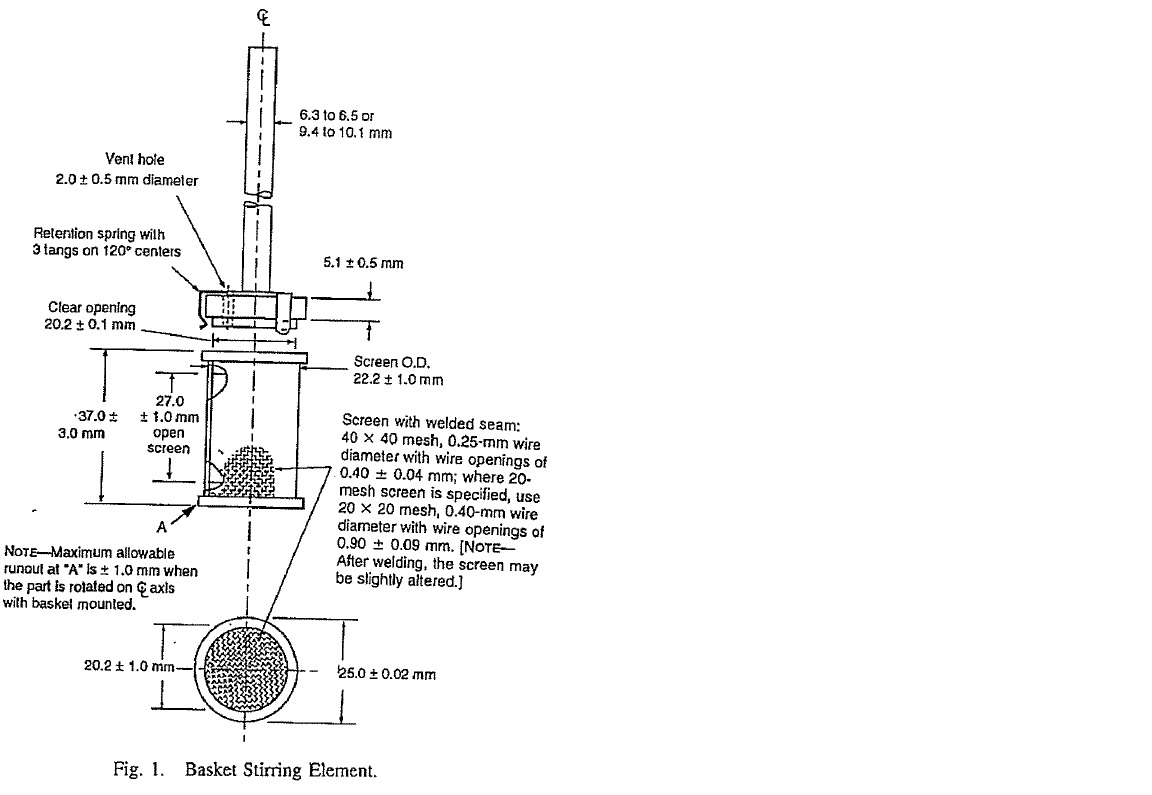
(d) There is also in section 711 a reference to type II apparatus, being the paddle. I do not need to discuss this further for the moment.
295 In both USP 23 NF 18 and the USP there is a further section 724 headed “Drug Release”. It is to be noted that some aspects of the setting out in USP 23 NF 18 were then altered in the USP. For example:
(a) In USP 23 NF 18, reference is made to types I and II apparatus before the heading “Extended-release Articles — General Drug Release Standard”. Type III apparatus is discussed under that heading. Contrastingly, in the USP, the heading appears before types I and II apparatus. There are also other textual differences concerning what is discussed under the heading type I apparatus and type II apparatus.
(b) In USP 23 NF 18 and the USP the headings differ. In USP 23 NF 18, the heading is “Apparatus 3 — ”. In the USP, the heading is “Apparatus 3 (Reciprocating Cylinder)”. That heading accentuates the principal mechanism and difference as compared with types I and II apparatus, namely, a “Reciprocating Cylinder”.
296 It is appropriate to set out fully what is said concerning type III apparatus in the USP:
Apparatus 3 (Reciprocating Cylinder)
Apparatus — The assembly consists of a set of cylindrical, flat-bottomed glass vessels; a set of glass reciprocating cylinders; stainless steel fittings (type 316 or equivalent) and screens that are made of suitable nonsorbing and nonreactive material and that are designed to fit the tops and bottoms of the reciprocating cylinders; and a motor and drive assembly to reciprocate the cylinders vertically inside the vessels and, if desired, index the reciprocating cylinders horizontally to a different row of vessels. The vessels are partially immersed in a suitable water bath of any convenient size that permits holding the temperature at 37 ± 0.5° during the test. No part of the assembly, including the environment in which the assembly is placed, contributes significant motion, agitation, or vibration beyond that due to the smooth, vertically reciprocating cylinder. A device is used that allows the reciprocation rate to be selected and maintained at the dip rate specified in the individual monograph, within ± 5%. An apparatus that permits observation of the specimens and reciprocating cylinders is preferable. The components conform to the dimensions shown in Figure 1 unless otherwise specified in the individual monograph.

Apparatus Suitability Test — Individually test 1 tablet of the USP Drug Release Calibrator Tablets (Single Unit) and a specified amount of beads of the USP Drug Release Calibrator Beads (Multiple Unit) according to the operation conditions specified. The apparatus is suitable if the results obtained are within the acceptable range stated in the certificate for that calibrator in the apparatus tested.
Dissolution Medium — Proceed as directed under Dissolution (711).
Procedure — Place the stated volume of the Dissolution Medium in each vessel of the apparatus, assemble the apparatus, equilibrate the Dissolution Medium to 37 ± 0.5°, and remove the thermometer. Place 1 dosage-form unit in each of the six reciprocating cylinders, taking care to exclude air bubbles from the surface of each dosage-form unit, and immediately operate the apparatus as specified in the individual monograph. During the upward and downward stroke, the reciprocating cylinder moves through a total distance of 9.9 to 10.1 cm. Within the time interval specified, or at each of the times stated, raise the reciprocating cylinders and withdraw a portion of the solution under test from a zone midway between the surface of the Dissolution Medium and the bottom of each vessel. Perform the analysis as directed in the individual monograph. If necessary, repeat the test with additional dosage-form units.
Where capsule shells interfere with the analysis, remove the contents of not less than 6 capsules as completely as possible, and dissolve the empty capsule shells in the specified volume of Dissolution Medium. Perform the analysis as directed in the individual monograph. Make any necessary correction. Correction factors greater than 25% of the labeled content are unacceptable.
Time and Interpretation — Proceed as directed under Apparatus 1 and 2.
297 I should note here a number of features. First, there is a reference to a top mesh and a bottom mesh. Second, there are only references to cylinders. There is no reference to baskets. Third, there is reference to withdrawing “a portion of the solution under test from a zone midway between the surface of the dissolution medium and the bottom of each vessel”. There is no specification for mesh size (cf the description for type I apparatus). Perhaps it might be inferred from its absence that, given the different mechanism (as compared with a rotating basket for type I apparatus), its specification was not as important.
(b) Relevant common general knowledge
298 I accept that the following common general knowledge of a skilled addressee of the Patent provides relevant factual context for construing the claim 1 dissolution test. The following propositions in my view would have been common general knowledge at the priority date.
General principles of dissolution testing
299 Claim 1 specifies a dissolution test for the purpose of defining what falls within the statutory monopoly.
300 In developing an in vitro dissolution test, a person skilled in the art of dissolution profiling would seek to develop a test that was reproducible and had good discrimination such that results were sensitive to changes in the formulation.
301 For a sustained-release formulation, the dissolution levels would usually be tested at a number of specified time points to produce a dissolution profile. But the dissolution profile is not an absolute property of the formulation. It is measured differently depending on the equipment and testing parameters used.
USP apparatus
302 To assist with the issue of reproducibility, there was a range of standardised dissolution testing apparatus available for use. The USP stipulated standardised sets of apparatus.
303 Standardised equipment permitted a skilled person to measure the dissolution profile for a formulation using the same testing conditions as those used by the person who specified any applicable dissolution profile criteria for that formulation. This facilitated reproducibility.
304 In addition to deciding which of the USP apparatus to specify, there could be further specified:
(a) the variables to use for the selected apparatus; and
(b) whether any modifications to the apparatus were to be made.
305 The variables that one could specify for use with a selected USP type III apparatus included:
(a) the type of dissolution medium (claim 1 of the Patent specified 0.1M HCl);
(b) the volume of dissolution medium (claim 1 of the Patent specified 250ml); and
(c) the speed of the moving component (in revolutions per minute for types I and II apparatus and strokes per minute for type III apparatus).
306 The skilled addressee would also have known that a change in any of these variables could have an impact upon the dissolution results that were measured.
Modifications to USP equipment
307 If one could not achieve a reproducible and discriminatory test method using standard USP apparatus, then one might explore using modifications to such equipment.
308 I accept that Professor Dressman, for example, was aware of the use prior to the priority date of modifications to USP equipment. She accepted that there had been use of such non-compendial apparatus before April 2000. She was familiar with the use of a “peak vessel” with the USP type II apparatus and a “stationary basket” with the USP type II apparatus. Moreover, Borst, I. et al “New and Extended Applications for USP Drug Release Apparatus 3” Dissolution Technologies (February 1997) 11 also revealed an early modification of the USP type III apparatus being a “specially designed and fabricated VanKel Type wider lower cap (Figure 6)”.
309 In relation to the question of a “basket” modification, I will deal with the VanKel documents and their relevance (if at all) separately.
The specification of the USP type III apparatus
310 As at April 2000, USP type I and USP type II apparatuses were more commonly used for conducting dissolution tests including for modified-release formulations.
311 The Guidance for Industry — Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations, U.S. Department of Health and Human Services, FDA, September 1997 indicated that USP type I and II apparatus were the preferred apparatus, particularly for tablets. USP type III apparatus was designed for use with dissolution media at different pH levels in order to simulate passage through the gastrointestinal tract. It was also recommended for use with beads. I interpolate here that Professor Dressman’s laboratory group only began using a USP type III apparatus in January 2000. I accept that the specification of a USP type III apparatus is an indication that a patentee had decided not to confine itself to the use of the more commonly used apparatus. Apotex has asserted that in those circumstances, the specification of a modification to the USP apparatus (as distinct from an error having been made) may be less surprising so far as the skilled addressee was concerned. But I consider Ms Goddard SC’s leveraged reasoning to push the envelope. That is a characterisation, not a criticism.
Use of a basket with the USP type III apparatus
312 Apotex has asserted that a basket as used with a USP type I apparatus would fit within a USP type III apparatus vessel and that this would have been known to the skilled addressee at the relevant time. I accept, as Apotex asserts, that the dimensions shown in the USP confirm that a USP type I apparatus basket would readily fit into a USP type III apparatus vessel and that this would have been known to the skilled addressee. This is because:
(a) the outer diameter of the basket is 25.0mm ± 0.02mm;
(b) the inner diameter of the USP type III apparatus vessel is 47 ± 1.4mm (and the outer diameter of a reciprocating cylinder is itself wider than a basket at 26mm);
(c) the length of the basket is 37 ± 3mm; and
(d) the length of the USP type III apparatus vessel is 180 ± 1mm (and the length of a reciprocating cylinder is longer than a basket at 100 ± 1mm).
313 Apotex asserts that the “basket adapter” product in the VanKel documents (I will deal with these in a moment) enabled a USP type I apparatus basket to be attached to the shaft of the USP type III apparatus. Apotex contends that the commercial availability of such a part, even if only available after the priority date, is evidence that supports the likelihood of a skilled addressee understanding such an arrangement to be practicable as at the priority date. Apotex also submits that the evidence shows that a suitable adapter was available at the priority date. For present purposes it is sufficient to say that I accept that it was part of common general knowledge as at the priority date that it may have been possible and potentially practicable to adapt USP type III apparatus to fit a basket (of the type used in USP type I apparatus) in substitute for a cylinder, but that as at the priority date it was not known that this had been done. I reject the assertion that it was common general knowledge that such an adapter was available at the priority date but I accept that it would have been known that one could have been made.
314 Apotex submits that a USP type III apparatus fitted with a basket instead of a cylinder would have had different hydrodynamics which were likely to be more aggressive. Apotex then asserts that one reason for the modification might have been to simulate the more aggressive conditions of the stomach. It is said that another reason might have been to prevent the tablet from adhering to the glass walls of the cylinder. I accept that these are all conjectured but realistic possibilities that would have been known by the skilled addressee. But I consider that the greater likelihood is that the skilled addressee would have perceived there to have been an error made.
The VanKel documents and their admissibility
315 Prior to seeing the VanKel documents, the evidence of both Professors Tucker and Fassihi was that they were unable to construe the dissolution test. But after seeing the documents, both came to the same construction as set out in [281(a)] above.
316 Professor Tucker in his first affidavit set out three possible interpretations of USP type III apparatus reciprocating basket. He then discussed two alternatives, the one he gave in the JER (see [281(a)] above) and the compendial USP type III apparatus with reciprocating cylinder. At that time, his evidence was that as at April 2000 a reciprocating basket did not exist and that he was “presently unaware of such an apparatus existing”. It is apparent that until Professor Tucker saw the VanKel documents he had not reached a concluded view as to a particular construction. Professor Tucker initially referred to locating the website of Agilent (one of the manufacturers of the USP type III apparatus):
The website talks about a “basket” option on a USP Type III apparatus.
317 Professor Tucker in his second affidavit then referred to a document provided to him by Generic Partners’ solicitors which he had not seen previously, which referred to rotating baskets for USP type I apparatus being available in volume ranges from 100ml to 4 litres. It was only after seeing such material that Professor Tucker then chose between his two alternatives and adopted the modified USP type III apparatus construction in the JER.
318 Professor Fassihi initially said that he was unable to understand the claimed dissolution test and did not proffer a construction of the claim. He was then directed to the Agilent website by Apotex’s solicitors where he had reference to:
(a) a 2014–15 document:
This page also referenced two types of basket adapters. … until reading these materials I was not aware that a basket option was available to purchase.
(b) a current presentation on the Agilent website:
I understand this to mean that a basket can be used to retain the product sample as a variation to the cylinders specified by USP Apparatus 3.
319 But prior to reading such materials, Professor Fassihi was not aware of a basket option for the USP type III apparatus. But after seeing the VanKel documents, Professor Fassihi also adopted the same construction as Professor Tucker in the JER.
320 But in my view, and for reasons that I will now elaborate on, the VanKel documents were not part of common general knowledge of skilled addressees at the priority date and the evidence of Professors Tucker and Fassihi has been infected with hindsight bias.
321 There is no evidence that a skilled person would have obtained the VanKel documents at the priority date, even if they were available, let alone that such information was part of the common general knowledge at the priority date. Even if there was public availability of such documents, that is not sufficient to establish that they were part of common general knowledge as at the priority date. At best, Professor Fassihi’s evidence was that he would have considered contacting a manufacturer of a USP type III apparatus. But Professor Fassihi gave no evidence of ever having conducted similar enquiries previously and made no such enquiries of VanKel in the course of preparing his evidence. Rather, he was directed to the current Agilent website by Apotex’s solicitors where he found Dissolution Testing and Analysis, Dissolution Systems Source Book 2014–2015 Edition.
322 It is now necessary to deal more rigorously with the admissibility of the VanKel documents and their use.
323 Apotex has sought to tender and rely upon:
(a) extracts from what purports to be the 1999 Buyer’s Guide produced by VanKel Technology Group;
(b) the ordering book from VanKel Technology Group (undated, but bearing a 2001 copyright statement);
(c) pages from Dissolution Testing and Analysis: Dissolution Systems Source Book 2014–2015 Ed (VanKel).
324 Now I accept that VanKel was a well-known manufacturer of USP apparatus well before 2000. Borst, I. et al “New and Extended Applications for USP Drug Release Apparatus 3” Dissolution Technologies (February 1997) 11 evidences this. Further, there are other references in the evidence to VanKel as a manufacturer of USP type apparatus as at the priority date.
325 Apotex asserts that the VanKel 1999 Buyer’s Guide should be accepted as proof that the modification to USP type III apparatus to use the basket attachment was available as at the priority date. It is said that it is apparent from the 1999 Buyer’s Guide that the basket was attached with the O-Ring arrangement. The “Dura-fit” attachment referenced in the extracts from the 1999 Buyer’s Guide produced by VanKel is the O-Ring attachment.
326 Apotex asserts that in relation to the 2001 VanKel ordering book, albeit a document post-dating the priority date of 19 April 2000, it contained a wide range of non-compendial variations to USP equipment. It is said that this indicates the existence of commercial demand for such products including before the priority date.
327 Apotex also submits that in any event, the correct date for construction of the Patent for the purposes of, inter alia, considering any s 40 ground is the date of filing of the complete specification, namely April 2001. I will address this argument in a moment.
328 Apotex also seeks to rely upon the 2014–2015 Source Book. Obviously that document cannot be directly used on any construction or invalidity question. It seemed, as I understood the argument, to be put on the basis of some “presumption of continuity” as to the commercial availability of the relevant modification attachments.
329 During the trial I held that the VanKel documents were provisionally relevant pursuant to s 57 of the Evidence Act and I stated that I would address and rule upon their admissibility in my reasons. The parties made submissions on the various possible options, being that such documents were admitted absolutely or ruled inadmissible on relevance grounds or otherwise excluded under s 135. Let me deal with the question first of their relevance (if any) to common general knowledge as at the priority date. I should say on this aspect that I am not satisfied that any of these documents can be used directly as part of common general knowledge as at the priority date. Let me first address Apotex’s shifting dates argument seeking to finesse into evidence the 2001 ordering book.
330 The patent specification is to be construed in light of the common general knowledge as at the priority date. This proposition applies whether one is considering infringement, s 18 validity issues or s 40 grounds. This is apparent from the statement of the High Court (Gleeson CJ, McHugh, Gummow, Hayne and Callinan JJ) in Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [24] (considering sufficiency) that:
It is well settled that the complete specification is not to be read in the abstract; here it is to be construed in the light of the common general knowledge and the art before 2 July 1984, the priority date; the court is to place itself “in the position of some person acquainted with the surrounding circumstances as to the state of [the] art and manufacture at the time”.
This was reinforced in Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (2004) 217 CLR 274 at [49] where it was said that in relation to the s 40 grounds:
[t]hey are commonly called “s 40 points”, and they do form a genus in that it is not necessary to look at common general knowledge at the priority date, except in construing the patent. (my emphasis)
Apotex’s shifting dates argument must be rejected.
331 Moreover, as indicated in Pfizer v Eli Lilly (2005) 68 IPR 1 it is apparent that whether the specification is sufficient is assessed at the date of grant. But the complete specification at the date of grant (including the claims) is construed in light of the common general knowledge at the priority date in order to assess sufficiency (see at [328]).
332 In my view, the 2001 ordering book cannot on any view constitute relevant common general knowledge at the priority date. The priority date of the Patent is 19 April 2000. The point applies with even greater force to the 2014–2015 Source Book. Accordingly, they are not relevant to directly establishing common general knowledge as at the priority date. I will address their admissibility in a moment, but first let me deal with the 1999 Buyer’s Guide.
333 The 1999 Buyer’s Guide has also not been proven to be part of common general knowledge at the priority date. Professor Fassihi could not recall any publication referring to a reciprocating basket and was not aware that a basket option was available to purchase. Professor Fassihi said that he would have “consider[ed] contacting the manufacturer of USP Apparatus 3” to determine what modifications were available. But such evidence of a possible enquiry which may or may not have found the 1999 Buyer’s Guide produced by VanKel Technology Group does not elevate the 1999 VanKel document to be part of the common general knowledge. Professor Tucker’s evidence was that to the best of his knowledge as at April 2000 the reciprocating basket apparatus did not exist. Certainly, he was not aware of reciprocating baskets in 2000. Further, he was not able to locate any information in relation to a reciprocating basket which was published before April 2000. Professor Dressman’s evidence was that as at 2000 and today she had never heard of a USP type I apparatus basket being used with the USP type III apparatus.
334 Generally, in my view, common general knowledge means only information which has become commonly known or used by persons skilled in the relevant field, not everything disclosed by the publicly available literature on the subject.
335 There is a distinction between publicly available information (even readily accessible) and common general knowledge. In Aktiebolaget Hässle v Alphapharm Pty Ltd (2002) 212 CLR 411 at [45] the Court said:
the respondent’s case in Minnesota Mining would have been no better off if the contents of the Patents Office library had been “online” and so even more readily available to search. The issue was not whether it was “obvious” to search there, but whether what a search would have disclosed had entered the body of common general knowledge.
336 I accept that common general knowledge is not limited to information that has been memorised but includes material that the skilled person knows exists and to which they would refer as a matter of course, such as standard texts and handbooks; see ICI Chemicals & Polymers Ltd v Lubrizol Corporation Inc (1999) 45 IPR 577 at [112] per Emmett J (and on appeal in (2000) 106 FCR 214 at [57]).
337 Apotex contends that the information about dissolution equipment in the 1999 Buyer’s Guide meets this description. VanKel was a well-known supplier of dissolution equipment. VanKel was also referenced in Rohrs, B.R. et al “USP Dissolution Apparatus 3 (Reciprocating Cylinder): Instrument Parameter Effects on Drug Release from Sustained Release Formulations” Journal of Pharmaceutical Sciences 84(8) 922 August 1995 and Borst, I. et al “New and Extended Applications for USP Drug Release Apparatus 3” Dissolution Technologies (February 1997) 11. Professor Dressman also had knowledge at the time of the existence of such catalogues from laboratory equipment suppliers. She agreed that such catalogues were commonly provided to commercial laboratories and university laboratories.
338 Apotex also submits that it would be a routine and logical step for the hypothetical skilled addressee to consult product catalogues from the principal suppliers of dissolution equipment in order to assist them to understand the meaning of an unfamiliar equipment description. Further, it is said that the nature of such a catalogue is such that if a supplier lists items of equipment in their catalogue, a person reading it would accept without question that that company supplied that equipment.
339 In my view, the evidence does not establish that the 1999 Buyer’s Guide formed part of the common general knowledge of the skilled person at the priority date. In my view, Apotex’s submission is too tenuous. True it is that someone making a targeted enquiry may have procured it, but to the hypothetical skilled addressee this was not material known to exist and referred to as a matter of course.
340 If the VanKel documents are not part of the common general knowledge as at the priority date, are they otherwise relevant? The VanKel documents as a necessary, but not sufficient, condition for admissibility, must be relevant (s 56 of the Evidence Act). GSK has asserted that they will only be relevant if they are proven to form part of the common general knowledge as at the priority date. But in my view that lens is too narrow.
341 Let me address some preliminary issues first. Section 58 of the Evidence Act provides that if a question arises as to the relevance of a document or thing, the Court may examine it and draw necessary inferences. Section 183 provides that if a question arises about the application of a provision of the Evidence Act in relation to a document or thing, the Court may examine the document or thing and draw any reasonable inferences from it as well as from other matters from which inferences may properly be drawn. Section 48 also permits proof of the contents of a document by tendering what purports to be a copy of the document in question.
342 GSK does not concede that s 183 permits me to draw sufficient inferences from the VanKel documents themselves to authenticate them as business records of Agilent (or VanKel). It asserts that s 183 does not generally dispense with the need to prove the authenticity of a document. GSK says that authentication cannot be proved solely by drawing inferences from the face of the document where there is no other evidence to indicate provenance. It is said that there is no admitted evidence as to the provenance of the document described as the 1999 VanKel Buyer’s Guide, the 2001 ordering book or the 2014–2015 Source Book. GSK also contends that I cannot be satisfied that the documents are business records. GSK notes that there is some doubt as to whether publications of a business can be classified as business records rather than products of a business in any case (see Roach v Page (No 15) [2003] NSWSC 939 at [4] to [6]). Let me say at this point that I am satisfied that each of the 1999 Buyer’s Guide, the 2001 ordering book and 2014–2015 Source Book are authentic copies of VanKel’s documents sourced from VanKel and that they are what they purport to be. I am satisfied that I can draw such inferences from the combination of ss 48, 58 and 183 of the Evidence Act. I am also satisfied, if I need to find this, that such documents are business records. But the use of these documents is another question. In my view they can only be relevant at most as providing some flimsy evidence that as at the priority date VanKel as a manufacturer offered a relevant basket modification to the USP type III apparatus and that it was representing to the reader that this was in its view a useful modification. At best that slightly enhances the probability that I can infer that a skilled addressee as at the priority date might also not have thought such a modification to be ridiculous, even if they were not aware of such documents or what VanKel was offering. Such documents can indirectly form part of a matrix which supports the drawing of such an inference. I will make a direction under s 136 that the VanKel documents are to be admitted into evidence and used for this limited purpose only. Further, by making such a direction I am thereby able to reject GSK’s s 135 application on the basis that condition (a) is removed and condition (b) is sufficiently ameliorated.
343 Finally on this aspect, GSK has complained that the respondents seek to draw inferences from the VanKel documents without calling a deponent to give evidence to the effect of the inferences sought to be drawn. It is said that the purported inferences are contrary to the evidence of the expert witnesses. It is said that GSK is prevented from testing the purported inferences by cross-examination. It is also said that GSK’s ability to respond effectively to the material has been severely restricted by the manner in which it has been sought to be tendered. In my view, even if any of these complaints had a scintilla of substance, and I do not need to linger on such questions, my s 136 direction should be adequate to ameliorate any perceived prejudice.
(c) Construction of the claim 1 dissolution test
344 Apotex contends that in light of the common general knowledge described earlier, the skilled addressee would approach construction of the claim 1 dissolution test with an awareness of the following matters:
(a) First, that there is a range of standardised testing apparatus specified in the USP;
(b) Second, that a selection can be made from those apparatus and variables chosen in order to have an appropriate test for a specific formulation that is reproducible and has good discrimination. Further, that these considerations are of particular importance for a patent claim since if the dissolution limits in the claim include non-useful subject matter it will be invalid;
(c) Third, that modifications can be made to standardised apparatus in order to seek to obtain a useful dissolution test in the sense of being reproducible and having good discrimination;
(d) Fourth, that it is important to accurately specify equipment for use in a dissolution test so that the reader can accurately reproduce the relevant dissolution test;
(e) Fifth, that the word “basket” has a clear and well-understood meaning in relation to USP dissolution testing, namely, a part of a USP type I apparatus;
(f) Sixth, that it would be sensible to attach a basket to a USP type III apparatus, for example, to simulate the more aggressive conditions of the stomach or to avoid any surface gel layer on the matrix tablet from adhering to the glass cylinder; and
(g) Seventh, that it would be practicable to attach a basket (of the type specified for USP type I apparatus) to a USP type III apparatus so that the reciprocating part is a basket instead of a cylinder.
345 I should say that I agree with points (a) to (e), but (f) and (g) have been overstated. I am only prepared to conclude that in light of common general knowledge, a skilled addressee would have seen these as theoretical possibilities that they would not have rejected out of hand.
346 Apotex stresses that the reference to “reciprocating basket” was not a singular reference within the Patent. The Patent repeats this reference at various places with no references to reciprocating cylinder. It is therefore said that there was nothing on the face of the Patent to suggest that the language used in claim 1 was in error and not chosen deliberately. I agree that there is nothing express other than the phraseology used in the claim, which is repeated. I will return to this.
347 Further, Apotex also says that Professor Fassihi explained that an awareness that the test specified in the Patent was directed to a relatively unusual formulation of paracetamol supported a conclusion that the claim was identifying a modified apparatus. It is said that the fact that the formulation had to dissolve relatively quickly so as to pass out of the stomach and be absorbed suggested to him that the dissolution test using the basket modification may have been trying to simulate the more aggressive hydrodynamics of the stomach; the pH of 0.1M HCl also simulated stomach conditions. This is a possible argument but not convincing.
348 Further, Apotex says that it was clear from Professor Dressman’s evidence that in forming her views on construction she placed considerable reliance on the fact that the dissolution test included the term “USP type III” in its description. She took the view that this was inherently inconsistent with the use of modified, non-compendial apparatus. And she formed this view notwithstanding that she accepted that it would be reasonable to understand the word “basket” as referring to a part for a USP type I apparatus. This resulted in her conclusion that this must have been an error. But Apotex contends that where the relevant apparatus is a modified version of a standardised USP apparatus, it is natural to identify the modified apparatus by reference to that standardised, unmodified form.
349 Further, Apotex has said that it is a natural use of the definite article “the” to refer to the particular standard apparatus to which the modification has been made. It is said that to construe the apparatus otherwise would give the definite article more work to do than the clearly denotative (and compendial) word “basket”.
350 Further, Apotex says that Professor Dressman’s actual difficulty with the claim 1 language was rather that any modification should be more clearly identified. But I have some sympathy for Professor Dressman’s view. If non-compendial was intended, it would have been reasonable to have expected that a further indication would have been given.
351 Further, Apotex says that Professor Dressman said that there was an additional issue in fitting a basket instead of a cylinder to the USP type III apparatus in that the location of the basket would need to be specified (but was not). However, in her affidavit she explained that a consideration for use of a cylinder would simply be that the cylinder remain “totally immersed when it was reciprocating down into the vessel”. Apotex says that the same consideration would apply to a basket.
352 Further, Apotex says that even on Professor Dressman’s preferred construction, there is still an omission to specify relevant testing details, namely, a failure to specify the mesh size for the cylinder ends. But, so Apotex says, if the word “basket” is understood to refer to a USP type I apparatus basket, which Professor Dressman accepted was reasonable, then no such issue would arise since a default mesh size of 40 is specified in the USP for a USP type I apparatus basket.
353 I reject Apotex’s arguments that a skilled addressee would construe claim 1 absent an error in the reference to “basket”. In my view, the skilled addressee would read claim 1 as referring to the compendial USP type III apparatus with reciprocating cylinder, understanding the reference to “basket” as being an error.
354 First, the dissolution profile in claim 1 is defined by reference to being determined by “the USP type III apparatus, reciprocating basket”. The description of the apparatus in claim 1 is preceded by the definite article “the”. This points against a non-compendial version. Claim 1 uses the definite article “the USP type III apparatus”, which directs the reader to the compendial USP III apparatus as described in the USP. The USP prescribes only one USP type III dissolution apparatus. That apparatus uses a reciprocating cylinder. As explained by Professor Dressman:
“There can be no deviation … given the USP definitions”; if a basket were to be used “it would no longer be a USP 3 apparatus”.
355 The USP type I apparatus, the only method described in the USP as using a basket, uses a cylindrical basket which rotates around a shaft. Further, claim 1 of the Patent requires a much smaller test volume (250ml) than is typical for testing using the USP type I apparatus (900ml or 1000ml) but which is consistent with the volumes used for USP type III apparatus.
356 Second, the text of the USP described the inner vessel of the USP type III apparatus as a “reciprocating cylinder” and used the designation “Apparatus 3 (Reciprocating Cylinder)”.
357 Third, the USP type III apparatus was an appropriate apparatus to use for testing of sustained release dosage forms comprising a matrix forming polymer (see, for example, Borst, I. et al “New and Extended Applications for USP Drug Release Apparatus 3” Dissolution Technologies (February 1997) 11; Rohrs, B.R. et al “USP Dissolution Apparatus 3 (Reciprocating Cylinder): Instrument Parameter Effects on Drug Release from Sustained Release Formulations” Journal of Pharmaceutical Sciences 84(8) 922 August 1995). The USP type I apparatus (basket) and the USP type II apparatus (paddle) were known to have problems with floating or sinking oral dosage forms. Further, in addition to being used to test formulations across a number of different pH values, the USP type III apparatus was also known to be used to test at a constant pH value (see, for example, the USP monograph for carbamazepine tablets).
358 Fourth, as Professor Dressman explained, at the priority date the FDA was “resistant” to the use of dissolution testing using modified non-compendial testing apparatus for companies seeking registration of dosage forms.
359 Fifth, it was known as at the priority date that modifications to compendial USP apparatuses were developed to overcome known issues encountered when testing formulations using USP type I apparatus or USP type II apparatus and, as explained by Professor Dressman, only accepted by regulators if the modified method was shown to be “superior to other established methods”.
360 Sixth, when modifications to compendial testing methods were made by those conducting dissolution tests, detailed descriptions of the modifications were included in the scientific literature to enable the modified dissolution testing to be reproduced by others. The existence of such modifications would be highlighted by, for example, use of the words “modified” or “non-compendial”.
361 Seventh, there would be no good scientific or technical reason to use a basket instead of a cylinder with a USP type III apparatus.
362 Eighth, the features of the dissolution test specified in the Patent (i.e. stroke rate, volume, temperature and dissolution medium) were consistent with what a person skilled in the art would expect for the USP type III apparatus.
363 Ninth, as at the priority date, none of the expert witnesses were aware of a modification of the USP type III apparatus. Further, none of the expert witnesses were aware of the possibility of a basket being fitted to be used with the USP type III apparatus.
364 Tenth, the absence of references to a USP type III apparatus reciprocating basket in the scientific literature prior to 2000 supports the conclusion that the availability of such a modification was not part of the common general knowledge of the skilled person at the priority date. The skilled addressee would understand the reference to “basket” in the context of “the USP type III apparatus” to be a mistake. GSK has pointed to the same mistake being made in an article published by the USP itself in 2014 (Brown and Marques “USP and Dissolution — 20 Years of Progress” Dissolution Technologies August 2014), although I consider this 2014 example to be another jury point.
365 Eleventh, I agree with GSK that nothing can be drawn from the fact that the Patent refers to “reciprocating basket” or “(reciprocating basket)” multiple times. The same single error has been made by the patentee and has been reproduced throughout the Patent. Further, the fact that the words “reciprocating basket” appear in parentheses in a number of places in the Patent emphasises that the words describe what comes before it, i.e., the USP type III apparatus, rather than to identify a modified non-compendial apparatus. The use of the parenthesis also accords with the description of the USP type III apparatus in the USP.
366 Twelfth, and as I have indicated, the views expressed by Professors Fassihi and Tucker that the apparatus referred to in the Patent is “a USP type III reciprocating dissolution apparatus with a reciprocating basket as a modification” have been tainted by those witnesses being influenced by post-priority date materials.
367 In summary, a mistake was made in claim 1, and the skilled addressee would have so perceived. But am I free to so construe claim 1 to in effect re-write or amend it? I do not think so. I can only use the tool of the hypothetical skilled addressee to construe a claim, but not to re-write or amend it.
(d) Legal principles applicable to construction of a claim involving an asserted error
368 A patent specification is a “unilateral document in words of the patentee’s own choosing” (Kirin-Amgen Inc v Hoechst at [34] per Lord Hoffmann). Further, a patent is not a document operating inter partes but is a public instrument which must define a monopoly in such a way that it is not reasonably capable of being misunderstood.
369 Apotex contends that while in construing a patent claim it may be permissible to determine that a patentee has used a word in an unusual manner, any “doubt” about the meaning of a term used in a claim is resolved either by considering technical evidence as to the specialised meaning of the word as used by the Patentee or by considering some specific teaching within the patent specification itself (Flexible Steel Lacing Company v Beltreco Ltd (2000) 49 IPR 331 at [74] per Hely J). But it is said that in the context of construing a patent claim, the Court cannot go further than this. Apotex says that it is not aware of any case in which as a matter of construction a court has determined that a claim has used a term in error and then justified re-writing it under the pretext of a construction exercise.
370 Further, Apotex contends that even if the Court were construing a contract operating inter partes, it would only be possible to construe that document as involving an error where the literal meaning produced absurdity and it was self-evident what the objective intention is to be taken to have been; see Mainteck Services Pty Ltd v Stein Heurtey SA (2014) 89 NSWLR 633 at [115] to [121] per Leeming JA and National Australia Bank Ltd v Clowes [2013] NSWCA 179 at [34] and [35] per Leeming JA. It says that, accordingly, if it were possible to apply the more flexible principles that have been developed for contractual construction, the word “basket” could still not be construed as meaning “cylinder” because there is an alternative and sensible construction available.
371 On the other hand, GSK has sought comfort from Product Management Group Pty Ltd v Blue Gentian LLC (2015) 116 IPR 54 (Kenny and Beach JJ, with Nicholas J dissenting on the construction case). In that case the Court recognised not only the importance of reading passages within a claim in their context, but in understanding the way in which a skilled addressee would approach the claim as a whole. The Court, in interpreting the claim, expressly relied upon expert evidence of incorrect use of language in the claim and as to how a person skilled in the art would look at the claims practically. GSK says that the above approach resonates with the evidence in the present case that no expert knew of the existence of or the possibility of using a basket with a USP type III apparatus. On the contrary, the expert evidence suggested that any non-compendial apparatus would be clearly distinguished from compendial apparatus and described in detail. In my view, Product Management Group can be put to one side. First, there were other aspects of the claim language itself that supported the construction. Second, the word “said” had a referential ambiguity. Third, the observations at [111] are not the present case. Fourth, the alternative reading of integer 1.8 (see at [112]) was also available.
372 GSK says that the skilled person would understand that the apparatus in claim 1 is the USP type III apparatus and the reference to “basket” in claim 1 (and in the specification) is an inadvertent error. I agree.
373 GSK says that the claim does not need to be re-written to be clear: the USP type III apparatus is clear to a skilled person and the error is superfluous to the meaning of the claim. I disagree. A re-writing is involved and I am not prepared to re-write the claim.
374 GSK says that it is not urging the Court to construe the apparatus in the way that it is understood by Professor Dressman simply because it is one of those exceptional cases where a skilled addressee would not construe a term in a claim in such a literal way. Further, GSK says that it is not seeking to “re-write” the claim. Rather, it says that this case concerns a “redundant error” and that I may take guidance from authorities which consider the impact of errors in claims. I will take guidance from the authorities. But contrary to GSK’s contentions, they do not support me in effect amending or re-writing claim 1.
375 GSK has made reference to Adhesives Pty Ltd v Aktieselskabet Dansk Gaerings-Industri (1935) 55 CLR 523 where Evatt J explained that an error carried into the claim does not vitiate the patent if otherwise valid, so long as from a practical point of view the public concerned are fairly given possession of the invention (at 546). But as Evatt J discussed at 545, the process described in claim 1 was not in any way dependent upon whether the alcohol produced in the process but not sought to be recovered was assimilated by the yeast. The assertion of the fact of actual assimilation was “only of theoretical and of no practical importance in enabling the public concerned to ascertain the territory covered by the claims”. It was an erroneous statement of theory. Evatt J at 545 cited Fletcher Moulton LJ in “Z” Electric Lamp Manufacturing Company Ld v Marples, Leach & Co Ld (1910) 27 RPC 737 who was commenting on a claim which (erroneously) stated that a manufacturing process needed to be “free from carbon”. This was an error because the claimed process was not free from carbon. Fletcher Moulton LJ at 746 said:
… I think that the erroneous view, from a chemical standpoint, was one into which a lamp-maker might naturally fall, and that it would not in the slightest degree diminish the completeness of the disclosure to the public of the invention, how to apply it, and what its practical consequences would be. Consequently I hold that, according to the English patent law, such an error is unimportant. The patentee’s obligation is not to be omniscient; the patentee’s obligation is to put the public in the possession of his invention, and if he does that bona fide, in such a way that they know its advantages practically, and they can obtain those advantages practically, the fact that he has formed an erroneous view in theory of that which procures those advantages, or the state of things in which those advantages occur, does not, in my opinion, militate against him.
376 Evatt J’s statement in Adhesives Pty Ltd v Aktieselskabet Dansk Gaerings-Industri was approved by the Full Federal Court in CCOM Pty Ltd v Jiejing Pty Ltd (1994) 51 FCR 260 at 282. But in CCOM, the error was not in a claim but the body of the specification; it was also dealing with fair basis.
377 I am not assisted by these cases. They involved erroneous statements of the underlying theory of the respective invention rather than an error embodied in an essential integer of a claim such that the public was not fairly given possession of the invention.
378 GSK also refer to No-Fume Ltd v Frank Pitchford & Co Ltd (1935) 52 RPC 231 where Romer LJ explained (at 243):
It is not necessary that he should describe in his specification the manner in which the invention is to be performed, with that wealth of detail with which the specification of the manufacturer of something is usually put before the workman who is engaged to manufacture it. Specifications very frequently contain mistakes; they also have omissions. But if a man skilled in the art can easily rectify the mistakes and can readily supply the omissions, the patent will not be held to be invalid. The test to be applied for the purpose of ascertaining whether a man skilled in the art can readily correct the mistakes or readily supply the omissions, has been stated to be this: Can he rectify the mistakes and supply the omissions without the exercise of any inventive faculty? If he can, then the description of the specification is sufficient. If he cannot, the patent will be void for insufficiency.
379 But this case also does not assist as it was dealing with an alleged lack of sufficiency; I will elaborate on this point in a moment.
380 GSK has contended that a skilled person reading the claim in a common sense fashion in the context of the other aspects of the claim (including the other test parameters) in light of the common general knowledge would clearly understand claim 1 of the Patent to be referring to the compendial USP type III apparatus. It is said that such a construction would not extend the patentee’s monopoly nor read into the claims words by reference to the purpose of the monopoly.
381 GSK has contended that the proper approach, consistently with both the authorities and the evidence, would be to construe the phrase in claim 1 “the USP type III apparatus, reciprocating basket” as meaning a compendial USP type III apparatus with a reciprocating cylinder.
382 I reject GSK’s arguments. In my view the claim means what it says notwithstanding that the skilled addressee would perceive an error to have been made. I am not able to re-write the claim under the guise of construction.
383 First, the present case is not one where an integer of a claim is ambiguous or uncertain.
384 Second, the present case is not one where there is an inconsistency between the body of the specification and a claim integer.
385 Third, if “basket” means “basket”, the invention still works.
386 Fourth, this is not a case of an erroneous stipulation of an underlying scientific theory upon which the invention proceeds.
387 Fifth, I am not construing a commercial contract with all the latitude and commercial massaging that such an exercise entails.
388 Sixth, I am not here to apply any rule of benevolent construction which would strive to construe the Patent in a way not claimed. I am here to construe what is claimed rather than what is not claimed (see generally Blanco White, TA “Patents for Inventions” (Stevens & Sons, 5th ed) at [2–104]). The universe of activity of construction must be bound by the claim. Construction that re-writes the claim is in effect construing what ought to have been rather than what is. I am not at liberty to adopt a method of construction that gives a patentee what it might have wished or intended to claim rather than what it did claim (H Lundbeck A/S v Alphapharm Pty Ltd (2009) 177 FCR 151 at [60]).
389 Mr Shavin QC has referred to my taking a purposive construction. But as is pointed out in “Terrell on the Law of Patents” (Sweet & Maxwell) at [6–68] (16th ed) and at [9-35] (17th ed):
Purposive construction is therefore a principle of construction of the claim language, and does not entitle the court to rewrite or amend the claim in the guise of construing it.
390 The Master of the Rolls in Norton and Gregory Ld v Jacobs (1937) 54 RPC 271 at 276 and 277 clearly recognised that the skilled addressee lens could not be used such as to justify acting “under the pretence of construing [a] claim” but “in reality seek[ing] to reform it”. Further, as was stated by Lord Sutherland (Outer House) in Conoco Specialty Products (Inc) v Merpro Montassa Ltd [1994] FSR 99 at 106:
While a purposive construction is necessary, it must be borne in mind that this is intended to be a method of construction and not an excuse for not construing the claims at all.
391 Bodkin, C “Patent Law in Australia” (Thomson Reuters, 2nd ed) at [5380] has suggested that “[n]evertheless, it is permissible for an obvious error in a claim to be corrected when construing the claim”. But the case cited in support of that proposition does not support the breadth of that statement. In Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479 at 488, Gummow J said:
I turn first to consider claim 12 and the dependent claims 13, 14 and 15. One preliminary point should be mentioned. It concerns the fifty-first word in claim 12. This reads “corners”. If the claim is read in this form it is clear that there is no infringement. At the trial, in the end it was accepted by all counsel that “corners” should read “sides”. If the significance of the discrepancy concerned no more than the parties presently before the court, then there might be scope for application of a principle that no formal amendment was necessary because the true meaning was apparent as a matter of construction; cf Fitzgerald v Masters (1956) 95 CLR 420 at 426–7; Watson v Phipps (1985) 63 ALR 321 at 322; 60 ALJR 1 at 3.
But, of course, the specification operates to confer rights in rem and the legislation makes specific provision for amendment. Further, I was told that infringement litigation concerning the patent is on foot in another court against the parties identified as Theta Developments Pty Ltd, Reinforcement Bar Spacer Pty Ltd and Podfix Pty Ltd.
Accordingly, on the last day of the trial, 2 September 1994, I gave directions for the advertisement of a motion seeking amendment of claim 12. The notice of motion will be made returnable no earlier than 14 days after delivery of these reasons for judgment. In the meantime, it was accepted that I should proceed to determine this case on the footing that claim 12 uses the term “sides” rather than “corners”.
392 But this all rather suggests that, in context, an error in a claim was to be corrected by amendment rather than by construction. His Honour appears to have been prepared to proceed on the basis as if amendment would occur.
393 Johnson, P, Roughton, A and Cook, T “The Modern Law of Patents” (LexisNexis, 3rd ed) at [6.49] states that:
Where a patent claim includes a “mistake”, and this is clear to the skilled reader, then it can be ignored. This might include the addition of a word, or simply using the wrong word. Again the touchstone is whether the skilled person would understand the word to be a mistake.
394 But only one case is cited in support of that proposition being Lizzanno Partitions (UK) Ltd v Interiors Manufacturing Ltd [2013] EWPCC 12, a decision of Judge Birss QC (as he then was) of the Patents County Court. But this case does not assist GSK. There was a mistake in the body of the specification which referred, in effect, to uPVC as a non-rigid material. But as his Honour said at [31], “no skilled person would call uPVC a ‘flexible plastics material’”. The mistake was not made in claim 1 (see at [34]) which did not refer to uPVC but referred to a “non-rigid material”. So, his Honour’s observations at [46] are to be read in that context. Accordingly, when his Honour was construing claim 1, the reference to uPVC in the body of the specification was put to one side when construing the term “non-rigid” in claim 1. But when his Honour dealt with the infringement case, his Honour did consider claim 10 which required the gasket to comprise of uPVC. This was a mistake in the claim. But in relation to the mistake in the claim itself, his Honour said at [78]:
The point on claim 10 is that this claim requires the gasket to comprise uPVC. [The claim] needs to be addressed because on any view the Lizzanno gasket is not made of uPVC. The argument is I think that because it is obvious that the references to uPVC in the specification are a mistake, the true interpretation of claim 10 is that it is a claim to PVC and not uPVC. I do not propose to spend much time on this. I do not agree. It is one thing to read the description fairly, realise that the reference to uPVC there must be the result of an error and so read the document as a whole bearing in mind that uPVC is not flexible plastic; it is quite another to come to a conclusion that a patent claim to uPVC actually means something else. For that to get off the ground the correction of the mistake would have to be clear as well but it is not. Perhaps the patentee genuinely thought uPVC was flexible and meant to claim it. This would still not affect the overall interpretation of the patent but it would not justify reading claim 10 as if it was a claim to PVC. I reject the allegation of infringement of claim 10.
395 His Honour did not correct the mistake, although perhaps suggested that in some circumstances he could. In my view the case does not sufficiently assist GSK such as to justify a re-writing of the mistake that I am dealing with.
396 His Honour at [46] also referred to Rediffusion Simulation Ltd v Link-Miles Ltd [1993] FSR 369 and Camco Inc v Whirlpool Corporation [2000] 2 SCR 1067. But neither case deals with the scenario of justifying the re-writing of a mistake made in an essential integer of a claim. In Camco, the dispute was one of the breadth of “vane” in the claims. The question was whether a broad or narrow interpretation should be given. Unsurprisingly it was held that one could look at the body of the specification to assist. The case does not directly address the issue I am dealing with. Rediffusion also does not assist. In that case, claim 11 contained an error, but it was corrected by amendment.
397 Further, I should say for completeness that cases dealing with mistakes and omissions in the body of a specification when dealing with a lack of sufficiency invalidity ground are not directly apposite to the present context. Accordingly, I have put to one side the observations in No-Fume Ltd v Frank Pitchford & Co Ltd at 243 and cases expressing similar sentiments (for example Milliken Denmark AS v Walk Off Mats Ltd [1996] FSR 292 at 303 per Jacob J). Importantly in the present context, I am not dealing with sufficiency, but rather the delimitation of the patentee’s monopoly which is a separate point (AMP Inc v Utilux Pty Ltd (1971) 45 ALJR 123 at 128 per McTiernan J). Considering the significance of errors in the body of the specification as distinct from an error in a claim when one is dealing with a lack of sufficiency argument is quite a different context. There is much more latitude to in effect overlook the error as one is not by doing so changing in any way the invention or the boundary limits or content of the monopoly as claimed.
398 Simpson v Holliday (1866) L R 1 H L 315 involved a mistake in the body of the specification rather than the claim. Moreover, the error was not saved even though it was not likely to mislead skilled workmen (at 321 per Lord Chelmsford and at 322 and 323 per Lord Cranworth). Simpson was applied in Vidal Dyes Syndicate Ld v Levinstein Ld (1912) 29 RPC 245 at 271 and 272; it was also noted in that case that errors in chemical theory do not make a patent invalid if the errors do not interfere with the sufficiency of the practical directions given.
399 The English Court of Appeal (sitting with a Scientific Adviser) in Valensi v British Radio Corporation [1972] FSR 273 was also dealing with an error in the context of a lack of sufficiency argument (at 307 to 314). The reference (at 310) to the proposition that the skilled addressee must be prepared to display a reasonable degree of skill and common knowledge of the art in making trials and to correct obvious errors in the specification if a means of correcting them can readily be found must be seen in that light (see also at 311 and 314). Plimpton v Malcolmson (1876) 3 Ch D 531 at 575 and 576 per the Master of the Rolls was also dealing with a lack of sufficiency argument.
400 The High Court of Eire in Farbwerke Hoechst Aktiengesellschaft v Intercontinental Pharmaceutical (Eire) Ltd [1968] FSR 187 at 189 per Kenny J dealt with a mistake in a claim (claim 8) and applied Simpson v Holliday to in effect re-write the claim. But as I have said, the observations in Simpson were more in the context of an insufficiency argument. I do not consider it appropriate to adopt the approach in Farbwerke.
401 Finally, for a separate reason, I am also not enamoured of the idea that, without amendment, construction can solve the problem of an obvious mistake. If the skilled addressee lens were to identify a mistake in an integer of a claim such as to be an “obvious mistake” for the purposes of s 102(3) or s 104(1)(b), what would be the purpose of that reference in s 102(3)(a) or s 104(1)(b) if one could simply sidestep the amendment process through the pretence or pretext of construction? Those statutory references implicitly proceed on the assumption that such mistakes cannot be cured or rectified through construction alone. Further, a fortiori, if a non-obvious mistake would have been identified by the skilled addressee. It would make even less sense for a construction exercise to be able to cure the non-obvious mistake, but not the obvious mistake. And indeed, a non-obvious mistake is not of itself a ground for amendment. Perhaps there is a sliver of an argument to the effect that the existence of the amendment power per se to amend an “obvious mistake” is not inconsistent with a construction exercise also being a cure. Perhaps it may be said that the amendment power exists, inter alia, to remove any risk or doubt, rather than leaving the matter to construction. But in my view, the existence of the amendment power and a trigger, viz., “obvious mistake” is a powerful indication of the limits of any construction exercise.
CONSTRUCTION of claims 8 to 11: matrix forming polymer
402 Given my findings on claim 1, which is the only independent claim, in one sense this next issue does not need to be addressed further at least in the infringement context. But in the circumstances it is appropriate to deal with it, particularly as some of the invalidity questions turn to some extent on this construction issue. The question of infringement of claims 8 to 11 turns on the meaning of the words “matrix forming polymer”.
403 This requires the relevant “matrix” to be identified. The parties have put the following competing constructions:
(a) The construction advanced by Generic Partners is that the “matrix” is the monolithic structure through which a drug is dispersed to sustain the release of a drug over time. This can be referred to as the tablet matrix. In the case of the bilayer tablet described in the Patent, this is a reference to the sustained release phase.
(b) The construction initially advanced by GSK was that the “matrix” is the matrix that forms when a polymer swells to form a gel in an aqueous environment. Of course, such a construction may be potentially viable for water soluble polymers, but is technically unviable for water insoluble polymers which the Patent also addresses. As a result, GSK by the time of its closing address put a more coherent construction as I will set out in a moment.
404 In simple terms, Generic Partners submits that a “matrix forming polymer” is any polymer that is responsible for forming a tablet matrix by contributing to its creation and integrity. It is said that it is the tablet matrix (not the individual polymer in its own right) that must provide for a sustained release of paracetamol. It says that on its construction the alleged infringing products will not infringe claims 8 to 11 of the Patent. I disagree with Generic Partners’ construction.
405 Contrastingly, on GSK’s ultimate construction, a “matrix forming polymer” means “the key polymer in a sustained release formulation that provides the sustained release” (GSK closing submissions). GSK further elaborated on its position by saying that the matrix must continue to be actively formed (e.g., upon exposure to water) and cannot include a static matrix formed, for example, upon compression. I agree with the former aspect, but disagree with the latter aspect.
406 Claim 1 of the Patent claims:
a bilayer tablet having an immediate release phase of paracetamol and a sustained release phase of paracetamol …
the sustained release phase … comprising from about 55% to 90% by weight of the total paracetamol in admixture with a matrix forming polymer or a mixture thereof …
407 Claims 8 to 11 are dependent claims. Claim 8 requires the matrix forming water soluble polymer to be selected from a group of compounds including HPMC but which, relevantly for infringement, do not include PVP, microcrystalline cellulose or pregelatinised starch.
408 Claim 9 encompasses a composition according to claim 8 in which the water soluble matrix forming polymer is HPMC. Claim 9 specifies “[a] composition according to claim 8 in which the water-soluble matrix forming polymer is hydroxypropylmethylcellulose”.
409 Claims 10 and 11 claim a composition in which the matrix forming polymer is present in a particular amount by weight of the sustained release phase. Claim 10 specifies “[a] composition according to any one of claims 1 to 9 in which the matrix forming polymer is present in an amount from 0.5 to 10% by weight of the sustained release phase”. Claim 11 claims a composition according to claim 10 where the matrix forming polymer is 1 to 6% by weight of the sustained release phase.
(a) The Patent and some literature
410 Before descending into the conceptual morass of the parties’ linguistic and technical arguments, it is appropriate that I introduce the topic with an analysis of, first, the patent specification and, second, the literature which used the expression “matrix” prior to the priority date.
The specification
411 First, there is no definition of “matrix” and “matrix forming polymer” in the specification.
412 Second, given how the expression “matrix forming polymer” has been used in context, it is apparent that the word “matrix” and the phrase “matrix forming polymer” have been used or are to be understood as having a technical meaning rather than an ordinary meaning. In my view, that is how the skilled addressee would so proceed.
413 But to say that “matrix” and “matrix forming polymer” are being used in a technical sense still leaves considerable scope for debate about the precise technical sense. Accordingly, context is all important.
414 Clearly, one is not concerned with linear algebra and its mathematical meaning dealing with a rectangular array of numbers or mathematical symbols arranged in columns and rows, where the array is treated as a single entity upon which various mathematical operations and manipulations can be performed, including computational functions with other arrays. Likewise, one is not dealing with matrix mechanics used in quantum mechanics where matrices are used to set out in an array, operators representing different types of physical co-ordinates and variables (space, momentum etc).
415 The Oxford English Dictionary gives various definitions of the noun form, including:
(a) a “supporting or enclosing structure”;
(b) a “place or medium in which something is originated, produced or developed; the environment in which a particular activity or process begins”;
(c) a “fine material used to bind together the coarser particles of a composite (usually artificial) substance”; so, for example, one might describe pebble stones as firmly fixed in a matrix of concrete;
(d) in the pharmacological context, a “material used to retain a drug for controlled release”; or
(e) the “elements which make up a particular system, regarded as an interconnecting network”; so, for example, one might describe a political matrix or social matrix.
416 Of course, the skilled addressee cannot be taken to be aware of the OED definitions, but in my view the skilled addressee, in terms of the noun use of matrix, is likely in the first instance to consider meanings analogous to (a), (b) and (d) in the pharmaceutical context generally. But that still leaves considerable room for what is meant by “matrix forming polymer”. Let me turn to the specification as, of course, the skilled addressee would consider the context in which both the word and the phrase were being used.
417 At p 1 line 32 to p 2 line 1, the word “matrix” is used as a noun. Moreover, it is used in the context of a discussion of a “sustained release layer”. Reference is made to a matrix comprising a mixture of hydroxyethylcellulose and polyvinyl-pyrrolidone (PVP). Just stopping here for the moment, hydroxyethylcellulose and PVP are polymers. It is apparent that meanings (a) and (d) are apposite, with meaning (b) less so unless one considers the matrix as something in which the activity or process of sustained release originates or is produced or developed. But what should be emphasised at this point is the significance of the reference to “comprising”. That word indicates that hydroxyethylcellulose and PVP are in the matrix rather than that hydroxyethylcellulose or PVP through some dynamic physical process by themselves or, say, through reaction with water, somehow form the matrix, which absent some dynamic physical process would not otherwise exist. This gives rise to the question of whether there is a distinction between the phrase “matrix forming polymer” and “matrix comprising a polymer(s)”. I will return to this later, but let me just flag the significance of this question. The respondents’ construction would, in effect, read “forming” as “comprising” so that matrix forming polymer is any polymer in the sustained release layer which layer takes the form of a matrix. Contrastingly, GSK would give an active operation to “forming” so that some dynamic physical process must occur involving the polymer to produce the matrix in order for the description “matrix forming polymer” to be apposite.
418 Before proceeding further, I note that on p 4 lines 23 to 25 there is an elaboration on “comprise” which tends to suggest that it is being looked at in terms of a substance being an ingredient without more.
419 At p 4 line 34, a consistory clause is set out which uses the expression “matrix forming polymer or a mixture thereof” without elaboration.
420 At p 5 line 23, there is the statement:
Suitably the sustained release phase comprises a matrix-forming polymer to provide a sustained release of paracetamol.
421 Now a number of important observations should be made at this point. First, whatever be the meaning of “matrix forming polymer”, what is apparent is that the specification is signifying its importance and significance to the dynamics of a process of providing the sustained release. In other words, it is not merely any polymer in the sustained release layer, but rather the polymer(s) that is there to provide directly the functionality of sustained release. No doubt a polymer in a sustained release layer could perform other functions, say, binder, disintegrant etc. And no doubt the combined effects of all excipients in a sustained release layer might ultimately work together to produce overall a sustained release. But the specification is clearly distinguishing such excipients in general in the sustained release layer (even though some may be polymers) and the primary agent directly producing sustained release, that is, the or a matrix forming polymer(s). Generic Partners would have one drop out the words “to provide a sustained release of paracetamol”. They would be superfluous on its construction. Clearly those words are setting out the functionality for the “matrix forming polymer”. It is not just any polymer in the sustained release phase. Indeed, if there was any doubt, it is removed by the passage on p 6 lines 2 to 4 which refers to “[t]he amount of matrix-forming polymer in the sustained release phase and the relative amounts of paracetamol … are selected so as to provide …”. Second, what is also significant about this phrase at p 5 line 23 is that it distinguishes “comprises” and “forming” by using the phrase “the sustained release phase comprises a matrix forming polymer”. So, this would suggest that “comprising” and “forming” may not be synonyms.
422 At p 5 lines 26 to 32, the following passage appears:
Examples of matrix-forming polymers include both water soluble and water insoluble polymers or mixtures thereof, with soluble polymers being preferred. Examples of water soluble polymers include hydroxypropylmethylcellulose, hydroxyethyl-cellulose, carboxymethylcellulose, sodium carboxymethylcellulose, methacrylate hydrogels, polyethylene glycols and xanthan gum. An example of a water insoluble polymer is ethylcellulose. A preferred matrix-forming polymer is hydroxypropyl-methylcellulose.
423 The specification gives examples of polymers as “matrix forming polymers”, but this is not to suggest that such polymers cannot also be used for their other properties. Further, that passage makes it apparent that a matrix forming polymer can be a water soluble polymer or a water insoluble polymer (or a mixture). But, whether one is dealing with a water soluble polymer or a water insoluble polymer, one must give the same meaning to “matrix forming polymer”, although different physical processes may be involved. As is known, a water soluble polymer such as HPMC will form a gel layer, whereas a water insoluble polymer will not (e.g. ethylcellulose). Clearly then, the word “matrix” does not mean gel layer per se, but must have a more generic meaning. GSK in its submissions seemed to flip flop on this question, but appears to have accepted that “matrix” cannot be confined by reference to gel layers given that the relevant term has to also embrace water insoluble polymers.
424 At p 6 lines 20 to 28, the following passage appears:
Compositions of the present invention will generally contain at least one pharmaceutically acceptable carrier conventionally used in the art of tablet and/or capsule formulation. Suitable carriers which may be incorporated include lubricants, for example magnesium stearate and stearic acid; disintegrants, for example cellulose derivatives and starches; binders, for example modified starches, cellulose derivatives and polyvinylpyrrolidone; glidants, for example colloidol [sic] silicas; compression aids, for example cellulose derivatives; as well as preservatives, suspending agents, wetting agents, flavouring agents, bulking agents, adhesives, colouring agents, sweetening agents appropriate to their form.
425 What is well apparent from this passage is that the specification is also distinguishing between the matrix forming polymer(s) and other excipients that might be in the sustained release layer. For example there is reference to disintegrants such as cellulose derivatives and starches; but these are polymers. Further, there is reference to binders, such as modified starches, cellulose derivatives and PVP; but these are all polymers. Likewise glidants in the form of cellulose derivatives.
426 At p 12, there is set out the tabulated contents for Example 2, Formulation C. Reference is made to various polymers in the sustained release layer being low viscosity and high viscosity HPMC, PVP and pregelatinised starch. But it is plain (see the passage at p 6 lines 20 to 28 set out above) that PVP and pregelatinised starch, although excipients, are not being referred to as the or a matrix forming polymer. Starch is a polymer and usually has the empirical formula (C6H10O5)n, where n = 300 to 1000. Pre-gelatinised starch is a starch that has been chemically or mechanically processed to rupture all or part of the starch granules. This is to render the starch flowable and directly compressible.
427 I have set out claims 1, 6, 8, 9, 10 and 11 earlier.
428 So what would the skilled addressee draw from the specification in terms of construing “matrix forming polymer”? In my view, the skilled addressee is likely to draw the following conclusions.
429 First, “matrix” does not simply mean the sustained release layer containing one or more polymers. But I do not need to linger on “matrix” as a standalone concept as I must construe the composite phrase.
430 Second, “matrix forming polymer” is not a label to be applied to any polymer in the sustained release layer, but to the polymer(s) that performs the primary and direct function of the sustained release property. In other words, it must operate as more than a binder or a disintegrant.
431 Third, for such a polymer so appropriately described, the mechanism for its function may operate differently depending upon whether it is water soluble (i.e. the formation of a gel layer) or water insoluble. I should say that the gel layer is not the matrix but may be said to form on the surface of the matrix when one is dealing with a water soluble polymer.
432 Fourth, the word “forming” is not a synonym for “comprising”, although the words are not unrelated. The polymer must somehow act to form the matrix, although query the precise mode of “form” when one is dealing with a water insoluble polymer. In that latter scenario one looks more at its primary and direct function of sustaining release.
433 Fifth, the specification, although not the claims, has a hyphen between “matrix” and “forming”. So, the property of “matrix-forming” is predicative for the polymer in question.
Some literature
434 The skilled addressee would also be aware of how literature in the field used the concept of matrix. Let me discuss some of this.
435 The following may be noted from Lordi, N.G.’s Chapter 14 “Sustained Release Dosage Forms” in Theory and Practice of Industrial Pharmacy by Lachman, L and Lieberman, H.A. (1986).
436 First, when one is referring to a matrix in the context of a sustained release formulation, one is looking at the context of an embedded matrix where the “drug is dispersed (embedded) in a matrix of retardant material” (my underlining) (p 443). Clearly, the matrix is not a gel layer which may form at the boundary where one is dealing with a hydrophilic polymer. It is the three dimensional space containing the principal retardant material. I emphasise that the focus is on the principal retardant material and not any or all excipients in the sustained release layer.
437 Second, at pp 453 to 455 there is a discussion of matrix tablets. What is well apparent is that matrix tablet formulations are characterised by reference to the retardant materials that they contain. And as so characterised, the matrix may have one of three characteristics:
(a) Insoluble, inert;
(b) Insoluble, erodable;
(c) Hydrophilic.
438 It is appropriate to set out relevant extracts:
Matrix Tablets
One of the least complicated approaches to the manufacture of sustained release dosage forms involves the direct compression of blends of drug, retardant material, and additives to form a tablet in which drug is embedded in a matrix core of the retardant. Alternately, retardant-drug blends may be granulated prior to compression. Table 14-7 identifies examples of the three classes of retardant material used to formulate matrix tablets, each class demonstrating a different approach to the matrix concept. The first class consists of retardants that form insoluble or “skeleton” matrices; the second class represents water-insoluble materials that are potentially erodable; and the third class consists of polymers that form hydrophilic matrices. Loading doses are best included as the second layer of a two-layer tablet or in a coating applied to the matrix core.
Insoluble, inert polymers such as polyethylene, polyvinyl chloride, and acrylate copolymers have been used as the basis for many marketed formulations. Tablets prepared from these materials are designed to be egested intact and not break apart in the GI tract. Tablets may be directly compressed from mixtures of drug and ground polymer; however, if ethyl cellulose is used as the matrix former, a wet granulation procedure using ethanol can be employed. The rate-limiting step in controlling release from these formulations is liquid penetration into the matrix unless channeling (wetting) agents are included to promote permeation of the polymer matrix by water, which allows drug dissolution and diffusion from the channels created in the matrix. Formulations should be designed so that pore diffusion becomes rate-controlling, release is defined by equation (16) or (17), and the release profile is represented by curve C (Fig. 14-10). …
Table 14-7. Materials Used as Retardants in Matrix Tablet Formulations | |
Matrix Characteristics | Material |
Insoluble, inert | Polyethylene Polyvinyl chloride Methyl acrylate-methacrylate copolymer Ethylcellulose |
Insoluble, erodable | Carnauba wax Stearyl alcohol Stearic acid Polyethylene glycol Castor wax Polyethylene glycol monostearate Triglycerides |
Hydrophilic | Methylcellulose (400 cps, 4000 cps) Hydroxyethylcellulose Hydroxypropylmethylcellulose (60 HG, 90 HG, 25 cps, 4000 cps, 15,000 cps) Sodium carboxymethylcellulose Carboxypolymethylene Galactomannose Sodium alginate |
…
The third group of matrix formers represents nondigestible materials that form gels in situ. Drug release is controlled by penetration of water through a gel layer produced by hydration of the polymer and diffusion of drug through the swollen, hydrated matrix, in addition to erosion of the gelled layer (curve D, Fig. 14-10). The extent to which diffusion or erosion controls release depends on the polymer selected for the formulation as well as on the drug:polymer ratio. Low-molecular-weight methylcelluloses release drug largely by attrition, since a significant intact hydrated layer is not maintained. Anionic polymers such as carboxymethyl cellulose and carpolene can interact with cationic drugs and show increased dissolution in intestinal fluid. Carboxypolymethylene does not hydrate in gastric fluid. The best matrix former in this group is hydroxymethylcellulose 90 HG 15,000 cps, an inert polymer that does not adversely interact with either acidic or basic drugs, and that on contact with water slowly forms a gel that is more resistant to attrition. Release rates can be adjusted for low-milligram-potency formulations by replacing polymer with lactose. High drug:polymer ratios result in formulations from which drug release is controlled by attrition.
439 One can see not only a focus on the type of retardant material, but also the use of the word “form”. So, some retardants form insoluble matrices. And, for example, “the third class consists of polymers that form hydrophilic matrices”, including for example HPMC. This appears to be distinguished from forming a gel layer.
440 Lordi therefore suggests that “form” is not used in terms of a dynamic active agent but rather in a more informal sense. But it does make plain that what one is identifying is the primary and direct retardant agent (in our case to be a polymer(s)) and not just any polymer in the sustained release layer acting as an excipient (Generic Partners’ simplistic position).
441 Other literature at the time used descriptions such as “hydrophilic polymer tablets” or “hydrating hydrophilic matrix pharmaceutical tablets”, “HPMC tablets”, “hydrating HPMC tablets” and the like (see, for example, R Bowtell et al “NMR Microscopy of Hydrating Hydrophilic Matrix Pharmaceutical Tablets” Journal of Magnetic Resonance Imaging 12(2), 361 to 364 (1994); A.R. Rajabi-Siahboomi et al “Structure and Behaviour in Hydrophilic Matrix Sustained Release Dosage Forms” Journal of Controlled Release 31, 121 to 128 (1994)).
442 Whether or not the skilled addressee had access to this literature, what is important to note is that “matrix forming polymer” is likely to have been understood by the skilled addressee as the principal retardant acting polymer(s) in the sustained release layer where the embedded matrix principle rather than the barrier principle applied to provide the sustained release.
443 Indeed, if fortification is needed for this position, Professor Fassihi’s patent application filed on 9 March 1998, US 6,090,411 referred to a “matrix composed of HPMC or polyethylene oxide”. It also referred to “One hydrophilic matrix material useful in the present invention is HPMC K4M”. In other words, one does not look at all excipients (whether polymers or not) but the principal retarding agent(s).
444 If there was a doubt about this, I should also note that Professor Fassihi gave the following evidence after my enquiry:
HIS HONOUR: Professor, can I just ask you, when you use the word or concept “matrix”, how are you using or defining it? Because there is an issue in this case about “matrix”. So how are you using that concept? --- Sure. Any structure can be called matrix, for example, even a single tablet is a matrix. However, within controlled release society and amongst scientists who design controlled release formulations — and I have designed many of them myself, maybe more than 50, for companies, for publications. So here the meaning of “matrix” is that a tablet that — especially hydrophilic matrix in this case — a tablet which has enough polymeric material in it that it can actually swell, make a gel, and keep the integrity of the system for whatever length of time it is designed for, 12 hours or 24 hours.
…
HIS HONOUR: Can I just ask you — so if you have your matrix-forming polymer HPMC but you have other polymers in there that assist to increase the viscosity? --- Yes.
Or otherwise assist in terms of a retardant re-action? --- That’s correct.
Are they considered matrix-forming polymers, or is it only the HPMC that’s considered the matrix-forming polymer? --- Your Honour, in general HPMC, when it is used in the large quantities beyond the threshold limit, would be regarded as the main player. However, when you don’t have that amount of polymer or you have other things which are present, they do their functions. So for example, we have polyvinylpyrrolidone, PVP. It’s a binder, so we know when you dissolve it in water it becomes viscous. So that viscosity would be imparted to the rest of the composition as well. So in a way it does contribute to also retardation. But it would be kind of indirect, not as its primary function but secondary function.
So when you talk about a matrix-forming polymer, you’re looking at a polymer that has its primary function to form the matrix rather than a polymer that might have a secondary or indirect ---? --- That is correct.
(b) “Matrix forming polymer”: Analysis
445 GSK contends that the phrase “matrix forming polymer” as used in claim 1 (and its dependent claims) is a technical term used in the art of formulating oral dosage forms to describe the key polymer in a sustained release formulation that provides the sustained release (see, for example, Gonçalves-Araújo et al, “Polymer Percolation Threshold in HPMC Extended Release Formulation of Carbamazepine and Verapamil HCl” AAPS PharmSciTech 11(2) (2010) 558 at 562 “It is important to note that in the case of the water-soluble drug, verapamil HCl, the matrix forming polymer (HPMC) was replaced in the formulations by a filler …”). It contends that this is the manner in which that term is used and is to be construed in the Patent. I generally agree for the reasons set out above when one considers the Patent and the literature.
446 It says that the alternative construction that the relevant matrix is the tablet matrix formed when the tablet ingredients are compressed during manufacture of the tablets and that all polymers present in that tablet matrix are “matrix forming polymers” is untenable. I also agree for such reasons.
447 GSK has referred to the Patent’s clear distinction between what the inventors considered to be matrix forming polymers and what they considered to be other formulation excipients or pharmaceutically acceptable carriers. It says that this is inconsistent with Generic Partners’ construction. It is said that such a construction requires an overlapping of those things expressly listed as pharmaceutically acceptable carriers with the list of example matrix forming polymers, from which list they are noticeably absent. I agree. The distinctions made in the specification do not support Generic Partners’ construction.
448 GSK says that on the respondents’ construction, the amount of matrix forming polymers in Formulations C and D would fall outside claim 11 and deprive the passage at page 6 lines 1 to 9 of the Patent of its obvious meaning and intent. On GSK’s construction Formulations C and D fall within claim 11 squarely. I also accept that argument.
449 Generally, I agree that the Patent makes it clear to a skilled formulator equipped with common general knowledge that a “matrix forming polymer” within the meaning of the Patent is the polymer(s) in the sustained release phase of the bilayer tablet that is primarily and directly responsible for slowing down or sustaining the release of paracetamol from the tablet. In other words, it is the polymer(s) that principally causes or provides the sustained release.
450 In my view, Generic Partners has erroneously equated compounds (such as PVP), which may affect the rate of release from the matrix, with “matrix forming polymers”.
451 Further, I agree with GSK that the passage on p 1 line 32 to p 2 line 2 of the Patent which refers to the McNeil patent (in essence to be equated with the 522 Patent) does not assist. That patent uses the phrase “Matrix Binding Agents” and, unlike the Patent, expressly defines that phrase as including PVP and hydroxyethylcellulose. Further, the 194 Patent refers to the 522 Patent and describes PVP as a granulating agent.
452 Let me discuss some of the expert evidence.
Professor Fassihi
453 Professor Fassihi’s experience with HPMC was in the context of extended release dosage forms designed to release over a prolonged timeframe (10 to 24 hour release). He acknowledged that these were different from the short time frame (two to three hour release) dosage forms of the Patent.
454 Professor Fassihi’s view of matrix formulations was influenced by his previous experience with extended release formulations. He did not regard a sustained release form (two to three hours) as a matrix formulation:
MS ROFE: And they’re intended to release over different periods of time? --- Well, you’re talking about matrix formulations. I have defined what matrix formulation is. I can assure you that matrix formulations are not designed for two hours or two and a half hours. That’s not the case. We don’t design sustained release formulations for two hours or three hours. We do not do that. So that’s not what I have done. I don’t regard that as a matrix formulation.
455 In my view, Professor Fassihi demonstrated that work with matrix formulations principally releasing over less than a 180 minute timeframe, as claimed in claims 1 to 3 of the Patent, was outside his experience.
456 In any event, Professor Fassihi embraced a number of propositions that suggested that he ultimately agreed that a high viscosity grade of HPMC was the only matrix forming polymer in the alleged infringing products. For example:
MS ROFE: So here a matrix-forming polymer is a polymer that swells and gels? --- Yes.
…
[with reference to exhibit G4 (Gonçalves-Araújo et al “Polymer Percolation Threshold in HPMC Extended Release Formulation of Carbamazepine and Verapamil HCl” AAPS PharmSciTech 11(2) (2010) 558)] MS ROFE: But in this article the matrix forming polymer is considered to be the HPMC? --- The matrix forming polymer is HPMC, and then you have contributory — there are other elements that contribute to retardation, and they have two clear example [sic] here: lactose does not actually contribute to formation of a matrix, because it’s soluble, it comes out. Microcrystalline cellulose strengthens the matrix and it slows down a drug — slow to — to diffuse out very slowly. So exactly that I what I have been saying in my affidavits [sic].
…
HIS HONOUR: So when you talk about a matrix-forming polymer, you’re looking at a polymer that has its primary function to form the matrix rather than a polymer that might have a secondary or indirect ---? --- That is correct.
…
HIS HONOUR: So you take the — what I will call as — the primary or the leading polymer and describe that as the type of matrix. So if you were using HPMC, it would be an HPMC matrix? --- Yes.
457 Further, Professor Fassihi’s initial evidence was that:
(a) “the type of formulation described in the ‘212 Patent (containing high viscosity … (HPMC)) … [has] a hydrophilic and swelling polymer as a main rate controlling excipient”; and
(b) “[a]s HPMC acts as the sustaining agent, having lower amounts of HPMC reduces the sustained release properties of the dosage form. I would consider a short duration sustained-release dosage form to be completely dissolved in vitro within 2 to 3 hours”.
458 Consequently, it would seem that Generic Partners’ construction of “matrix forming polymer” is principally founded on the evidence of Professor Tucker rather than that of Professor Fassihi.
Professor Tucker
459 As discussed earlier, Professor Tucker has no specialised skill or experience with respect to HPMC and the formulation of oral dosage forms containing HPMC. He conceded that the topic had not been a focus of his publications and that his opinions on the topic were predominantly derived from materials he had read for the purpose of this litigation. Although Professor Tucker is strictly qualified to give opinion evidence as a person skilled in the art on the construction of the term “matrix forming polymer” as used in the Patent, I have given his opinions less weight than I would otherwise, given his lack of direct experience on the topic.
460 Professor Tucker appears to have considered all the polymers in the sustained release layer to be “matrix forming polymers”. He considered that a matrix can be solid or semi-solid and can be formed upon compression of tablet ingredients or caused by the hydration of HPMC or a co-existence of the two.
461 In my view, Professor Tucker’s construction is misconceived for the following reasons.
462 First, whilst Professor Tucker acknowledges the existence of a gel matrix caused by the hydration of HPMC on contact with water and that the nature of the gel layer “is determined by the type of matrix forming polymer used in the formulation” (but may be “affected” by the “type and concentration of tablet excipients”), Professor Tucker has treated the sustained release layer of the alleged infringing products as a “matrix” which is formed on compression during tablet manufacture and identifies all of the polymeric excipients residing in that layer as matrix forming polymers. As GSK describes it, on Professor Tucker’s approach the polymers are matrix residing polymers, or tablet forming polymers, not matrix forming polymers.
463 A tablet or the sustained release layer of the tablet may be described as a sustained release “matrix tablet” or a sustained release “matrix layer” (as opposed to, for example, a “polymeric coated tablet”). However, a “matrix forming polymer” within the meaning of the Patent is not simply any excipient which resides in, forms or contributes to the formation or integrity of the sustained release layer of the tablet. As explained by Professor Dressman:
[M]atrix for pharmaceutical scientists could mean the composition of all of the ingredients together. And you have a tablet matrix. You could describe it that way. That’s all the excipients mixed together, granulated and then further excipients put into it and then compressed. You might speak about the matrix of the entire tablet. Or you could refer to the matrix in a functional way. And I believe that’s what [sic] meant here, where you have a matrix that provides a sustained release and that way, you may be more referring to what actually happens during the release of the tablet and how that ingredient would form — the matrix of that causes the sustained release. For example, there’s HPMC — while it’s in — so the tablet dry, it’s not sustaining the release. It’s only once it gets in touch with water and starts to swell so that it actually starts to form the matrix-forming pollen [sic] that actually causes a sustained release.
And by Dr Mooney:
MR SHAVIN: And then at the time it’s ingested, the hydration of the HPMC forming a gel and the gel inhibiting or controlling release of the API is, itself, a matrix, and that — in that context, the HPMC is called a matrix forming polymer, isn’t it, commonly? --- Yes.
And when one calls it a matrix forming polymer in that context, it’s not denying that the whole tablet is a matrix, but it’s identifying the gel-forming context, which is a gel in — through which things diffuse, and, in part, there’s an erosion effect as well? --- Yes. I mean — yes, but — but that swelling also is very depending on the amount of polymer.
464 I agree with GSK that Professor Tucker’s approach ignores the express teaching of the Patent to the effect that it is the “matrix forming polymer” and not the sustained release layer as a whole that is responsible for sustaining the release of paracetamol. In this respect, Professor Dressman explained:
MR CORDINER: So do you accept, Professor Dressman, that another way of reading a matrix-forming polymer to provide a sustained release of paracetamol is — in terms of functionality — is you look at the function of the matrix and whether or not that sustains the release of the polymer? --- No. Because it says the language is matrix-forming polymer to provide sustained release. It doesn’t say that the matrix is going to sustain a release. It says that the polymer is going to sustain release.
465 Second, on GSK’s construction, exemplary Formulations C and D both fall within the scope of claim 11. On Professor Tucker’s construction, exemplary Formulations C and D fall outside the scope of claim 11 of the Patent, a result which suggests that Professor Tucker’s construction is erroneous.
466 Third, Professor Tucker’s view appears to have been founded on his assertion that there is an insufficient amount of HPMC in the alleged infringing products to sustain the release of paracetamol, rather than to properly construe the claims:
MR SHAVIN: So we’re trying to understand in the context of the claim 8 to 11 the matrix forming water soluble polymer is either selected from the group in 8 or is HPMC in 9. Now, in the context of the patent, I put to you that a plain reading of the specification discloses that the references on page 5 and 6 to the amount of matrix forming polymer in the sustained release are plainly, in this context, a reference to the amount of HPMC and not a reference to the excipients that are referred to in lines 20 to 28 of that page? --- So I guess I have trouble with that. And particularly, I guess, with regard to claim 10, which makes me think about this the way I’m thinking about it.
What causes you a problem in claim 10? --- The very low concentrations of the matrix forming polymer or polymers which can be present.
You haven’t done any research, have you, which suggests that those concentrations of HPMC in a sustained release stage have no effect — are not of — I should rephrase that. I will rephrase the question. You have done no research, have you, which indicates that the presence of HPMC at those concentrations is ineffective to achieve a modified release of the type described in this patent? --- Do you mean experimental research?
Yes? --- No, I haven’t done experimental research.
467 Such an assertion is speculative.
468 Further and relatedly, to the extent that Professor Fassihi also said that the levels of HPMC were too low, such an opinion appears to have been based on Professor Fassihi’s experience with much longer lasting sustained or extended release products. Moreover, in his first affidavit, Professor Fassihi did not suggest that the level of HPMC in the alleged infringing products was insufficient to sustain release, instead saying:
Considering the relatively low amounts of high and low viscosity HPMC in the formulation, I expect that the dosage form is a relatively short duration sustained-release form. As HPMC acts as the sustaining agent, having lower amounts of HPMC reduces the sustained-release properties of the dosage form. I would consider a short duration sustained-release dosage form to be completely dissolved in vitro within 2 to 3 hours.
General
469 Let me further address some of Generic Partners’ other arguments.
470 First, it is said that a “matrix” is a three dimensional solid or semi-solid structure (“a monolith”) through which a drug is dispersed. All of the experts agreed that this was a reasonable interpretation of the word “matrix”. Now I accept that that is one meaning, but to accept that meaning does not answer what is meant by “matrix forming polymer”.
471 Dr Mooney explained it as follows:
HIS HONOUR: Can I just ask you, perhaps, also — because I’ve heard quite a bit of evidence over the last week or so on the meaning of “matrix” — how are you using the expression “matrix” and “matrix formulation”? --- Matrix — the way I would use “matrix” is the inclusion of the polymer within the materials as a single tablet. So the — the — the — the — the polymer and the other materials go together, either as a direct compression or as a mix — granulated and then compressed. So it’s a — the matrix is encompassing of the — the polymer and other materials — the active and other excipients.
In other words, the matrix is formed as soon as the tablet is produced, rather than some dynamic process involving hydration of HPMC? --- You — you — once you put it under compression ---
…
So you’re using “matrix” in a very broad sense as a three-dimensional structure within which you have the active pharmaceutical ingredient and all excipients. Is it any --- ? --- Yes.
Are you using it in any other sense than that? --- That’s the way I would use a matrix term, yes.
472 Professor Davies agreed that “matrix” is “often used by formulators” in this sense and he used it in that sense in his papers. He depicted this matrix in his affidavit. He described the stages “after ingestion of the matrix formulation” including:
The formation of the gel layer stops water percolating into the dry inner matrix of the tablet. …
473 He explained that in this context “the matrix contains the excipients and the drug”. Further, Professor Davies used the term “matrix” in the context of the entire matrix being the tablet or otherwise identified that he was talking about that part of the matrix which was hydrated, such as gel layer. He also referred to a paper on HPMC by J.E. Hogan “Hydroxypropylmethylcellulose Sustained Release Technology”, Drug Development and Industrial Pharmacy 15(6&7), 975–999 (1989) which stated:
In tablet matrix systems the tablet is in the form of a compressed compact containing an active ingredient, lubricant, excipient, filler or binder. The matrix may be tabletted from wet-massed granules or by direct compression.
The operative principle controlling drug release in matrix tablets is that on exposure to aqueous fluids the tablet surface becomes wet and the polymer starts to partially hydrate to form a gel layer.
474 Generic Partners contends that the sustained release phase of the bilayer tablet described in the Patent comprises a “matrix” in this sense (a tablet matrix). I agree with that submission in its generality, but as I have said it does not answer the question of what is meant by “matrix forming polymer”.
475 Second, Generic Partners has referred to page 1 line 29 to page 2 line 2 of the Patent which states in relation to the commercially available sustained release bilayer tablet of paracetamol (Tylenol Extended Relief) described in the McNeil patent:
The sustained release layer is provided by a matrix comprising a mixture of hydroxyethylcellulose and polyvinylpyrrolidone.
476 Generic Partners contends that “comprises” is not intended to exclude other additives. Accordingly, the “matrix” of the sustained release layer at least includes hydroxyethylcellulose and PVP. As I have said, PVP is a polymer. Hydroxyethylcellulose is also a polymer. PVP acts as a binder “sticking” powder together so that the monolithic “matrix” maintains its integrity. PVP does not form a gel “matrix”. It dissolves on contact with water. But none of this addresses what is the polymer(s) that is principally and directly providing the sustained release function.
477 Generic Partners says that Professor Davies agreed that reference to “matrix” at page 2 line 1 of the Patent is a reference to the “matrix” in the sense of the drug and a mixture of excipients. It is further said that his understanding of the word “matrix forming polymer” would require the “matrix” at page 1 line 29 to mean something different to the “matrix” referred to at page 5 line 23. I disagree.
478 Generic Partners also says that reference to the McNeil patent confirms that the “matrix” referred to at page 1 line 29 of the Patent means the tablet matrix created on compression. That may be so, but does not take the matter far.
479 Generic Partners says that the Patent does not use the term “matrix binding agents” like the McNeil patent but “matrix forming polymers”. But by using the term “matrix forming polymers” rather than “matrix binding agents”, it is said that the Patent makes clear that it is concerned with all of the polymers that assist in the formation of the matrix, not only binders. That argument is a non-sequitur.
480 Third, Generic Partners also seeks to draw comfort from the reference to ethylcellulose.
481 The Patent sets out a non-exhaustive list of matrix forming polymers at page 5 from line 26 to 32. The list of matrix forming polymers includes water insoluble matrix forming polymers, an example of which is ethylcellulose (p 5 lines 30 and 31). Ethylcellulose does not form a gel matrix on exposure to water. Ethylcellulose only forms a monolithic “matrix” on compression into the sustained release phase. I agree.
482 Professor Dressman’s evidence in this regard was as follows:
MR CORDINER: So if we’re talking — it gives us an example that you can have just an insoluble — water insoluble matrix-forming polymer and one of those is ethylcellulose. So if you’re comparing paracetamol with ethylcellulose there’s never going to be a gel formed, is there? We’re not talking about gel matrix? --- I think ethylcellulose doesn’t swell very much compared to, say, HPMC, for example, and it’s ---
No. So would it be more sensible to talk about the matrix which is formed in that regard is the matrix upon, as I said before, granulation, whether it’s wet or dry, and the compaction into a tablet, and I think you said before that one understanding of “matrix” is that? --- Yes. And then — so when the dosage form — if you’re talking about ethylcellulose comes into contact with water, then so the ethylcellulose has — is in place and has already forming the physical barrier but it doesn’t take effect and start to function until it gets into the gastrointestinal tract so it’s just a different mechanism of release, I feel.
HIS HONOUR: I was interested — what is that mechanism? In other words, if it’s water insoluble so it’s not forming the hydrates, when would it get — when ethylcellulose gets into the gastrointestinal tract, what is it about the chemistry — and you may not know this question, but what is it about the chemistry that starts to produce it acting as a retarding agent or having that function? --- Well, to draw an analogy, at one point the university made some — we retarded the release of cimetidine with some insoluble methacrylate types of polymer so in a way analogous to ethylcellulose but not exactly the same thing, obviously. It was methacrylate instead of an ethylcellulose but they’re both insoluble — and so at 7.5 per cent you didn’t see much retardation of the release; at 15 you saw some and at — we had to go up to about 26 per cent of the polymer in the matrix to actually retard the release of the cimetidine. Now, exactly how that functions, I’m not really sure. It’s still only one quarter of the excipients in the entire formulation but it has flattened out the release from that kind of a pattern to a linear pattern over several hours, so from experiences, the water-insoluble polymers, obviously, they have a different mechanism. Exactly what this is, I’m afraid I’m at a loss to explain.
483 Professor Tucker also gave the following evidence:
MR SHAVIN: This is something that’s quite differen[t], I suggest to you, from the polymers that sit passively in a tablet press and are compressed into the tablet as a whole. The matrix forming polymer — the words matrix forming are adjectives to describe the activity of the polymer, and I suggest to you that it was known in 2000 that HPMC, in the presence of water, hydrates to form a gel matrix, and that was known and well known by 2000? --- Yes, that was known, and if I look at the patent as a whole, it tells me I can use ethylcellulose as the matrix forming polymer.
Yes? --- And if I use ethylcellulose as the matrix forming polymer, there’s no gel layer formed here once I put it into water.
It doesn’t have to be a gel layer, it just has to be a matrix, doesn’t it? --- It has to ---
And in the cellulose, there will be a matrix, won’t there? --- In the cellulose.
You will have a stranded structure — fibrous structure? --- In the ethylcellulose, it will have to be compressed by the compression force, and the ethylcellulose will have had to have locked together — as I was saying for the methacrylate — into a coherent structure.
And in the presence of fluids in the digestive tract --- ? --- Yes.
--- there will be some small expansion into a matrix form? --- No, it already exists in the matrix form. What does the ethylcellulose do when you — once it’s in the water? No, it doesn’t form a matrix at that stage.
484 Professor Davies also gave evidence as to the sentence at page 5 line 23 of the Patent in the context of ethylcellulose as follows:
It doesn’t give it a function. It’s just simply a state of — well, I guess a formation of a tablet, whereas this is clearly describing — a matrix-forming polymer in the case of HPMC will form that gel there that controls the release of — in the case of ethylcellulose, it will form that matrix structure that will control the release — both form the matrix structures that control release.
485 Professor Davies explained that ethylcellulose worked to control and retard release because its water insolubility allowed the hydrophobicity of the molecules to slow down the penetration of water into the matrix.
486 I note that the fact that ethylcellulose forms an inert matrix upon compression is supported by the chapter by N. G. Lordi “Sustained Release Dosage Forms” from The Theory and Practice of Industrial Pharmacy by Lachman and Lieberman in the context of page 453 which explains:
Matrix Tablets
One of the least complicated approaches to the manufacture of sustained release dosage forms involves the direct compression of blends of drug, retardant material, and additives to form a tablet in which drug is embedded in a matrix core of the retardant. Alternately, retardant-drug blends may be granulated prior to compression. Table 14-7 identifies examples of the three classes of retardant material used to formulate matrix tablets, each class demonstrating a different approach to the matrix concept. The first class consists of retardants that form insoluble or “skeleton” matrices; the second class represents water-insoluble materials that are potentially erodable; and the third class consists of polymers that form hydrophilic matrices. Loading doses are best included as the second layer of a two-layer tablet or in a coating applied to the matrix core.
Insoluble, inert polymers such as polyethylene, polyvinyl chloride, and acrylate copolymers have been used as the basis for many marketed formulations. Tablets prepared from these materials are designed to be egested intact and not break apart in the GI tract. Tablets may be directly compressed from mixtures of drug and ground polymer; however, if ethyl cellulose is used as the matrix former, a wet granulation procedure using ethanol can be employed. The rate-limiting step in controlling release from these formulations is liquid penetration into the matrix unless channeling (wetting) agents are included to promote permeation of the polymer matrix by water, which allows drug dissolution and diffusion from the channels created in the matrix.
487 That is, the ethylcellulose forms an insoluble skeleton matrix as at the time of compression.
488 Generic Partners says that I must construe matrix forming polymer consistently for both soluble and insoluble polymers. I agree. It is said that the description of ethylcellulose as a matrix forming polymer is only consistent with the meaning of “matrix” in the sense advanced by Generic Partners, i.e. a monolithic matrix. It is said that there is no occasion to consider that ethylcellulose forms a matrix in an active sense after compression; it exists as a matrix only upon compression. That is true in one sense. But all this means is that ethylcellulose as the principal polymer in the sustained release layer is for relevant purposes the matrix forming polymer as it is the principal and direct retarding agent in the sustained release layer.
489 Fourth, Generic Partners says that “matrix forming polymer” is not a technical term used in the art. Professor Davies does not recall having used that expression in his work. In one sense this is true in relation to the composite phrase. But the individual words and their combination have and must be given a technical meaning.
490 Fifth, at page 5 lines 23 and 24, the Patent states:
Suitably the sustained release phase comprises a matrix-forming polymer to provide a sustained release of paracetamol.
491 Generic Partners would re-write it to say:
Suitably the sustained release phase comprises a polymer or mixture of polymers that form a matrix to provide a sustained release of paracetamol.
492 It is said that this necessary “re-writing” assists in understanding that it is the matrix that must provide the sustained release, not each individual component that makes up the matrix. In my view such a re-writing is impermissible.
493 Sixth, Generic Partners says that the use of the word “forming” in matrix forming polymer is broad and encapsulates any polymer that is responsible for forming the matrix. It is said that this does not require the matrix to be in a continual state of “forming” but the matrix forming polymers must be responsible for forming a matrix which can be static, such as a compressed tablet. It is said that this is the only sensible construction that the term can have once it is appreciated that that is the only way that ethylcellulose can be said to be a matrix forming polymer. I think that this is correct in one sense, but it fails to address the relevant feature which is to identify the polymer(s) which is the principal and direct retarding agent and performing that function in the sustained release layer.
494 Seventh, and generally, in my view Generic Partners’ construction in essence amounts to saying that all polymers in the sustained release layer are matrix forming polymers. Such a construction is not in accordance with the text or context of the Patent. Further, it is not consistent with the literature. Further, it gives too little work for “forming” to do. Generally its argument is little more than a linguistic word play leveraged off the usual and broad meaning to be given to “matrix”. It is quite clear, in my view, from the Patent that what is to be identified is the polymer(s) which acts as the principal and direct retarding agent in the sustained release layer, whether one is talking of a water soluble or water insoluble polymer.
Infringement case
495 If I am correct that “basket” in claim 1 means basket, then GSK accepts that it has failed to prove infringement concerning the alleged infringing products. It did not test the alleged infringing products using USP type III apparatus modified so as to use a basket. It tested using a reciprocating cylinder, i.e. it used compendial apparatus.
496 Strictly I do not need to go further on infringement questions. But just in case my view is ultimately displaced, it is appropriate that I set out my findings premised upon such a contingency. The following discussion therefore proceeds on the alternative view that “basket” in claim 1 means cylinder.
497 Each of the respondents has admitted that the alleged infringing products have each of the integers of claim 1 with the exception of the dissolution profile integer. As I have said, claim 1 claims a bilayer tablet having an in vitro paracetamol dissolution profile as determined by:
the USP type III apparatus, reciprocating basket, with 250ml of 0.1 M HCl at 37C set at a cycle speed of 15 strokes/min.
498 Accordingly, the alleged infringing products will only infringe claim 1 (and each of the dependent claims) if they have the dissolution profile integer as determined by the claimed dissolution test.
499 The respondents contend that even if I were to construe claim 1 as describing the USP type III apparatus with a reciprocating cylinder, GSK has not established that the alleged infringing products have a dissolution profile within the claim ranges. I disagree.
500 It is also said that GSK has also not established that the alleged infringing products have the integers of dependent claims 8 to 11 which involve greater definition of the matrix forming polymers in the formulation. I also disagree.
(a) The apparatus and technique used to measure the dissolution profile
501 A sample of 100 tablets from a commercial batch of the alleged infringing products was supplied to GSK’s solicitors on 2 April 2015. Apotex and Generic Partners each admit that the alleged infringing products have the following integers of the claims:
(a) a pharmaceutical composition comprising a bilayer tablet having an immediate release phase of paracetamol and a sustained release phase of paracetamol;
(b) the immediate release phase being in one layer and comprising from about 10 to 45% by weight of the total paracetamol;
(c) the sustained release phase being in the other layer and comprising from about 55% to 90% by weight of the total paracetamol in admixture with a matrix forming polymer or a mixture thereof; and
(d) said composition comprising from 600 to 700mg of paracetamol per unit dose and a pharmaceutically acceptable carrier.
502 Apotex and Generic Partners also admit that the alleged infringing products have each of the essential integers of claims 4, 5, 6, 13 and 14 considered alone (that is excluding the integers of the claims on which those claims are dependent).
503 Further, the authenticity of both Module 3.2.P Drug Product (including 3.2.P.2.2.1 Formulation development) and [Confidential] Process Validation Protocol (collectively for present purposes to be described as the Product Module) has not been in issue. The Product Module describes the alleged infringing products. The composition of the alleged infringing products is set out in the Product Module. Each product tablet contains 665mg of paracetamol per unit dose. Each of the ARTG Registrations is for 665mg paracetamol tablets. The amount of paracetamol in the alleged infringing products tablets is within the amount claimed in claims 1, 4 and 5 which claim respectively paracetamol in an amount of 600 to 700mg, 630 to 680mg, and 650 to 667mg per unit dose. The Product Module sets out a list of ingredients in the alleged infringing products. The Product Module provides that [Confidential]mg of paracetamol is in the IR layer and [Confidential]mg of paracetamol is in the SR layer. Accordingly, [Confidential]% by weight of the paracetamol is in the sustained release phase and [Confidential]% by weight is in the immediate release phase. These percentages fall within the ranges claimed in claims 1, 13 and 14.
504 Generally, the dissolution profiles of the claims in the Patent are specified in a conventional way “X to Y% [paracetamol] released after Z minutes”. Such profiles are expressed in a manner consistent with the guidance document published by the US FDA in 1997 entitled “Guidance for Industry — Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations”. The skilled addressee would be familiar with this document and would understand what was meant by the claimed dissolution ranges. The experts all now agree that “released after” means “released at”.
505 Dissolution testing in accordance with USP type III apparatus using a reciprocating cylinder was conducted on a sample of the alleged infringing products on each of:
(a) 9 and 10 May 2015; and
(b) 3 and 4 June 2015.
506 The dissolution testing was carried out by Mr Dayne Conroy-Southey in accordance with an experimental protocol and under the supervision of representatives of the parties. A video of the 3 and 4 June 2015 testing was made. Part of that video was displayed to me during the trial to show the apparatus in operation and the associated hydrodynamics created.
507 Professors Dressman, Fassihi and Tucker agree that the May and June 2015 testing was carried out in a USP type III apparatus (reciprocating cylinder), in 0.1M HCl at 37°C and at 15 strokes/minute, with an initial volume of 250ml and with sampling points at 15, 60, 120 and 180 minutes.
508 Both the May and June 2015 sets of testing used non-sorbing and non-reacting polypropylene mesh screens. The use of polypropylene is endorsed by USP III calibration methods.
509 The May and June 2015 testing differed in two respects:
(a) the presence of a top mesh; and
(b) the sampling point.
510 The May 2015 testing used a 40 mesh screen on the bottom and no mesh on the top, whilst the June 2015 testing was conducted using a 40 mesh screen on the top and bottom. Samples in the May 2015 testing were taken from the point of the sampling probe in the vessel. Samples in the June 2015 testing were taken from the midway zone.
511 It is apparent from the dissolution rates obtained in the May and June 2015 testing that there was no material difference in the results obtained from the two sets of testing. This is consistent with Professor Dressman’s evidence that the top mesh is optional for the dosage forms described in the Patent. Professor Dressman gave evidence that the appropriate way for the skilled person to compare the similarity of two dissolution rates was to calculate the “similarity factor” or “f2” value of the two profiles. Without getting too technical at this point, there are two mathematical approaches that can be used to compare dissolution profiles which use different factors, viz., f1 and f2. Shah, V et al “Dissolution Profile Comparison Using Similarity Factor, f2” (http://www.dissolutiontech.com/DTresour/899Art/DissProfile .html) have explained it in the following terms:
Among several methods investigated for dissolution profile comparison, f2 is the simplest. Moore and Flanner proposed a model independent mathematical approach to compare the dissolution profile using two factors, f1 and f2.
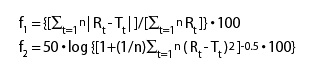
where Rt and Tt are the cumulative percentage dissolved at each of the selected n time points of the reference and test product respectively. The factor f1 is proportional to the average difference between the two profiles, where as factor f2 is inversely proportional to the average squared difference between the two profiles, with emphasis on the larger difference among all the time-points. The factor f2 measures the closeness between the two profiles. Because of the nature of measurement, f1 was described as difference factor, and f2 as similarity factor. In dissolution profile comparisons, especially to assure similarity in product performance, regulatory interest is in knowing how similar the two curves are, and to have a measure which is more sensitive to large differences at any particular time point. For this reason, the f2 comparison has been the focus in Agency guidances.
An f2 value of 100 entails that the dissolution profiles are identical. The FDA has set a standard of an f2 value between 50 to 100 to indicate similarity of profiles. Professor Dressman calculated the f2 value of the May and June 2015 dissolution profiles to be 73. She said that this result, and the fact that the tablets fell within the most stringent dissolution specifications set out in the Patent, demonstrated that the profiles in each set of testing were very similar. I have no reason to doubt her evidence or practical judgment on that question.
512 The results of the dissolution testing on 12 tablets on 9 and 10 May 2015 are set out below.
Tablet no. | 9 May | Tablet no. | 10 May | |
15 minutes (% paracetamol dissolved) | 1 | 41.18 | 7 | 42.35 |
2 | 40.56 | 8 | 41.52 | |
3 | 41.57 | 9 | 41.05 | |
4 | 41.61 | 10 | 43.43 | |
5 | 41.34 | 11 | 41.23 | |
6 | 40.64 | 12 | 40.95 | |
60 minutes (% paracetamol dissolved) | 1 | 66.65 | 7 | 68.58 |
2 | 66.80 | 8 | 68.21 | |
3 | 67.14 | 9 | 67.07 | |
4 | 67.71 | 10 | 69.66 | |
5 | 67.59 | 11 | 68.19 | |
6 | 66.00 | 12 | 66.87 | |
180 minutes (% paracetamol dissolved) | 1 | 99.10 | 7 | 100.38 |
2 | 101.33 | 8 | 99.69 | |
3 | 100.55 | 9 | 99.12 | |
4 | 100.08 | 10 | 97.32 | |
5 | 99.66 | 11 | 98.80 | |
6 | 100.14 | 12 | 100.38 |
513 The results of the dissolution testing on 12 tablets on 3 and 4 June 2015 are set out below.
Tablet no. | 3 June | Tablet no. | 4 June | |
15 minutes (% paracetamol dissolved) | 13 | 40.44 | 19 | 41.57 |
14 | 40.48 | 20 | 42.13 | |
15 | 40.47 | 21 | 40.48 | |
16 | 41.56 | 22 | 40.68 | |
17 | 42.08 | 23 | 40.72 | |
18 | 38.90 | 24 | 41.46 | |
60 minutes (% paracetamol dissolved) | 13 | 64.26 | 19 | 64.89 |
14 | 63.82 | 20 | 65.15 | |
15 | 64.66 | 21 | 64.44 | |
16 | 66.23 | 22 | 63.89 | |
17 | 65.97 | 23 | 63.48 | |
18 | 61.32 | 24 | 63.67 | |
180 minutes (% paracetamol dissolved) | 13 | 98.64 | 19 | 102.38 |
14 | 100.76 | 20 | 99.86 | |
15 | 99.57 | 21 | 101.57 | |
16 | 100.36 | 22 | 100.63 | |
17 | 100.11 | 23 | 101.08 | |
18 | 98.04 | 24 | 98.19 |
514 The average dissolution rates for each set of testing were:
Average of tablets 1 to 12 | Average of tablets 13 to 24 | |
15 minutes (% paracetamol dissolved) | 41.45 | 40.91 |
60 minutes (% paracetamol dissolved | 67.54 | 64.32 |
180 minutes (% paracetamol dissolved) | 99.71 | 100.10 |
515 All of the samples tested in May and June 2015 had a dissolution profile within the dissolution range claimed in claims 1 and 2.
516 23 of the 24 tablets tested in May and June 2015 fell within the dissolution ranges claimed in claim 3. Tablet 18 was just outside the claimed range at 60 minutes (61.32% paracetamol dissolved at 60 minutes cf 62 to 70% at 60 minutes). The average dissolution rate of all the tablets fell within claim 3. Taking into consideration the acceptance table in the USP, the sample batch fell within the dissolution range claimed in claim 3.
517 The respondents have contended that even if I were to find that the May and June 2015 tests had replicated in key respects the claimed dissolution test, there remained a number of issues as to the accuracy of the results reported. Generic Partners and Apotex have contended that the May tests were not carried out properly because they did not sample at the midway zone (as required by the USP) and they did not include a top mesh (as required by the USP). It is said that these errors had a marked impact on the dissolution results at 60 minutes. The mean dissolution at 60 minutes in the May tests was 65.17% (Day 1) and 66.26% (Day 2). The mean dissolution at 60 minutes in the June tests was 62.64% (Day 3) and 62.52% (Day 4). The protocol for the June tests was identical to that for the May tests apart from the fact it specified and involved a size 40 mesh screen at the bottom and top of each reciprocating cylinder and a sampling point about halfway between the top of the medium and the bottom of the vessel. Accordingly, in any event the June tests addressed these two issues with the May tests; for completeness I will deal with these issues later in any event.
518 But there is still a dispute as to whether the results of the June tests (and the May tests) were accurate given that there was a mean of approximately 6.7ml of dissolution medium missing at the end of the experiments. It is said that GSK’s calculation of the percentage of paracetamol released did not take into account this missing dissolution medium at all. In relation to the missing dissolution medium in the June tests, Professor Tucker was able to calculate a “corrected” percentage of paracetamol dissolved in each of the samples at each time point assuming that the dissolution medium evaporated at the outset of the experiments on each day of the June tests.
519 In my view, the June tests have replicated the claimed dissolution test. Accordingly, as I have said, I do not strictly need to resolve (although I will discuss these later) the disputes identified solely in relation to the May tests because Generic Partners and Apotex accept (subject to certain qualifications) that the corrected results of the June tests (corrected by Professor Tucker to allow for the missing dissolution medium) establish that the alleged infringing products have an average in vitro paracetamol dissolution profile as follows (that is, the alleged infringing products have this average in vitro paracetamol dissolution profile when measured using the USP type III apparatus, reciprocating cylinder, with a size 40 mesh top and bottom and a sample volume of 4ml (sampling without replacement)):
Day 3 Test
(a) 39.57% released at 15 minutes;
(b) 62.64% released at 60 minutes;
(c) 96.83% released at 180 minutes.
Day 4 Test
(a) 40.07% released at 15 minutes;
(b) 62.52% released at 60 minutes;
(c) 97.84% released at 180 minutes.
520 On this basis, the results of the June tests (corrected to allow for the missing dissolution medium) demonstrate that the alleged infringing products are within the scope of the constraints of claims 1 to 3 of the Patent (although only just within those constraints for claim 3 at the 60 minute time point on both June days).
521 Let me elaborate further on two matters and the respondents’ challenge that:
(a) first, the sampling procedure used in the GSK experiments was not appropriate as the sample volume withdrawn was not replaced during the course of the experiments; and
(b) second, in determining the dissolution profile of the alleged infringing products, GSK did not account for the observed loss of dissolution medium at the end of the experiments.
(b) Sampling volume and replacement
522 In Professor Dressman’s opinion, the sampling procedure used in the GSK experiments was appropriate and consistent with the USP, which simply stated that a “portion” of the solution under test was to be withdrawn at the specified time intervals: “USP 24 NF 19 does not direct the user to take a specific volume, nor to vary the volume of the sample solution”.
523 Professor Dressman also considered that sampling replacement would not have had an impact on the results of the experiments as long as a mathematical correction was made for the samples removed. As explained by Professor Dressman:
the sample volume specified in Protocol A and Protocol B is appropriate. Equations 1 to 4 (on pages 16–17, Annexure E of Protocol A and on pages 16–17, Annexure E of Protocol B) address effects induced by sampling without volume replacement. If other sample volumes had been taken, the effects of the other sample volumes could have been similarly corrected with an analogous set of equations so the results for percentage release would not be changed by changing the sample volume.
524 The equations referred to by Professor Dressman are as follows:

525 The sampling volume Y (4ml) is taken into account in the numerator of equations 2, 3 and 4.
526 Further, Professors Tucker and Fassihi did not say that the GSK sampling volume would lead to different results if another appropriate sampling volume was used or if volume was replaced. They did agree, however, that a skilled person at the priority date would be able to determine and use an appropriate sampling procedure.
527 Furthermore, in the JER the experts agreed that “sampling replacement” would only be relevant if the sampling meant that either sink conditions were not maintained and/or the cap of the reciprocating cylinder became exposed. As Professor Dressman explained:
If you have sink conditions for the drug that’s being dissolved, it doesn’t matter. I mean, our experience in the lab is that we usually don’t bother replacing the medium because we find that it makes no difference.
528 Professors Dressman and Tucker both agreed that based on simple arithmetic, using the equilibrium solubility data for paracetamol known at the priority date and the volume of media in the dissolution vessels, sink conditions were achieved and maintained in the experiments.
529 Professor Fassihi also originally agreed that sink conditions were maintained in the experiments. But in cross-examination he changed his evidence from that expressed in the JER, suggesting, contrary to his and Professor Tucker’s views in the JER, that he could not determine whether sink conditions were achieved in the experiments without ascertaining the “dynamic solubility” and particular crystalline form of the paracetamol used in the alleged infringing products at pH 1. But the peer-reviewed literature cited by Professor Dressman and adopted by Professor Tucker did not discriminate between any polymorphic forms of paracetamol for the purposes of solubility. Nor did it suggest that the solubility of paracetamol varies with pH. Indeed, Cao et al “Formulation, release characteristics and bioavailability of novel monolithic hydroxypropylmethyl-cellulose matrix tablets containing acetaminophen” Journal of Controlled Release 108 (2005) 351 explained at 359 that the solubility of paracetamol was “high and unaffected by pH”. Professors Tucker and Dressman did not suggest that the solubility of paracetamol was affected by its crystalline form or pH.
530 Further, Professor Fassihi’s oral evidence on the “sink conditions” issue appeared to be that he would have “preferred to use [a] larger volume” than 250ml. But in making this assertion, Professor Fassihi ignored the fact that 250ml is a typical volume for the USP type III apparatus. Further, Professor Dressman gave evidence that in her experience “if you have a volume of more than about 250 ml, something like that, you start to get splashing outside of the cylinder”. Further, Professor Fassihi agreed that he could have conducted his own experiments to work out the solubility of the paracetamol crystalline form used in the alleged infringing products but no such experiment was done.
531 Professor Fassihi’s change of approach was inconsistent with his earlier evidence in the JER and the evidence of Professors Tucker and Dressman. It was also unsupported by any cited literature.
532 Further, the experts also agreed that the cap of the reciprocating cylinder was at no stage exposed during the downstroke of the cylinder.
533 In summary, in the GSK experiments:
(a) A mathematical correction was made for sampling volume;
(b) Sink conditions were maintained; and
(c) The cap of the reciprocating cylinder was at no stage exposed during the downstroke of the cylinder.
534 Accordingly, the respondents’ sampling volume and replacement concerns are without any substance.
(c) Apparent loss of dissolution medium
535 The respondents raised an issue concerning loss of dissolution medium. Loss of dissolution medium volume due to factors other than the prescribed sampling volume is not routinely determined as it is not usual practice to measure the final volume of dissolution medium after sampling using the USP type III apparatus has been completed. Professor Fassihi did not routinely measure or report the final volume of dissolution medium at the completion of such testing. The respondents’ “loss of medium” criticism appears to arise from and is based on an experimental step that is not prescribed by any dissolution guidance documents nor taken by skilled persons in practice. This is another issue that has no substance.
536 Professor Fassihi appeared to tie his concerns about evaporation to the use of a paper towel during the experiments whilst the dissolution vessels were warming up. That the use of a paper towel would create an unusual amount of evaporation is speculative. The highest the respondents’ evidence reached on this point was that the use of a paper towel was “likely” to have been a “contributing factor” in the loss of volume. But as Professor Dressman noted, the loss of 6.7ml was about 2.7% of the total initial volume of 250ml of medium and was a small error margin.
537 Further, the respondents’ own evaporation analysis showed that the alleged infringing products fell squarely within the dissolution constraints of each of claims 1, 2 and 3 even if the “missing” media was taken into account.
538 I have put to one side Professor Tucker’s views on this aspect as he had no practical experience using USP type III apparatus.
(d) Sampling position in the May experiments
539 I will address this issue for the sake of completeness, even though the June tests “cured” any difficulty. Professor Dressman explained that:
It is stated in USP 24 NF 19, on page 1945 that
“Within the time interval specified, or at each of the times stated, raise the reciprocating cylinders and withdraw a portion of the solution under test from a zone midway between the surface of the Dissolution Medium and the bottom of each vessel”.
The use of the word “zone” in USP 24 NF 19 indicates that the withdrawal place is not a specific point within the vessel, but rather, is a region. The usual interpretation of a midway zone in a dissolution tester as at April 2000 (and still today) was and is that it is a zone which allows a latitude of several centimeters in both the upwards and downwards directions and that is how I would have taken a sample from USP 3 apparatus in April 2000, and today.
540 During the May experiments, samples were taken from the point of the sampling probe in the dissolution vessels, which is marginally higher than the precise midway point in the vessels.
541 Professor Dressman explained that:
The sampling probes are narrow and do not occupy a large volume of fluid. After considering the sampling probes … and bearing in mind that I expect that a reciprocation rate of 15 dips per minute will result in a homogenous distribution of dissolution fluid in the vessel, including at and around the sampling probe, I consider that taking a sample from “the point of the sampling probe in the vessel” would be highly unlikely to affect the dissolution results. I consider that the uniformity between the results of the May 2015 and June 2015 Experiments support my conclusion.
542 Whilst Professor Fassihi stated without any foundation that “it is important to extract samples from this midway zone to avoid issues such as possible lack of diffusion throughout the dissolution medium and to ensure reproducibility”, neither Professors Fassihi nor Tucker appeared to say that the sampling position rendered the May experiments inaccurate or unreliable. In cross-examination on this issue, Professor Tucker seemed to only be concerned that the sampling position was “standardised” to ensure reproducible results. There is no dispute that the samples were taken from the same position during the May experiments and that the sampling location was therefore standardised and reproducible.
543 But in any event, no issue has been raised by the respondents as to the sampling position adopted in the June experiments.
(e) Absence of top mesh in the May experiments
544 For completeness I will also deal with this issue even though it was also “cured” by the June tests. Professor Dressman considered the top mesh to be optional for the dosage forms described in the Patent. Professor Dressman’s view was supported by the findings reported both at col 2 on p 924 and Table 2 of Rohrs, B.R. et al “USP Dissolution Apparatus 3 (Reciprocating Cylinder): Instrument Parameter Effects on Drug Release from Sustained Release Formulations” Journal of Pharmaceutical Sciences 84(8) 922 August 1995 that showed that the top mesh only influenced the erosion controlled dosage forms and the official USP Drug Release Calibrator Single-Unit Type test for chlorpheniramine maleate extended release which said that there was “no screen on the top”. In cross-examination, Professor Fassihi appeared to discount some aspects of the latter on the basis that it related to calibration and was not an official monograph. But the official methods for calibration were promulgated by the USP itself and designed to ensure that the USP type III apparatus produced accurate and reliable results when other dosage forms were tested using the apparatus. Further, Professor Fassihi referred to and relied on a 2011 calibration certificate for chlorpheniramine maleate extended release tablets and said that the 2011 calibration document reflected “compendial testing conditions”.
545 In cross-examination, Professor Fassihi appeared to concede that an upper mesh was optional for tablet dosage forms as opposed to pellet or capsulated dosage forms.
546 Further, to the extent that Professors Tucker and Fassihi suggested that the “particles” seen at the bottom of the vessel in the May experiments suggested that a top mesh was required, such an argument is negated by the fact that particles were also observed in the June experiments (where a top mesh was used). Such an observation also accords with Professor Dressman’s evidence that she:
would also not consider it either necessary or appropriate to try to find and use a mesh at the top of the cylinder that could retain any fine particles which might be released from the tablet described in the Patent, even if such a fine mesh were available. The Rohrs 1995 article demonstrates (consistent with my understanding as a result of my experience described in my 15 June affidavit) that such a small top mesh would interrupt the hydrodynamics within the vessel and not be appropriate. Given this, and given that the cap on the cylinder prevents the tablet itself escaping out of the top of the cylinder, there is no advantage to using a top mesh for the matrix tablet dissolution testing described in the Patent.
547 As also explained by Professor Dressman:
The further Experiments performed on 3 and 4 June 2015 demonstrate that mesh at the top is optional, because according to the acceptance requirements in the USP … there was no material difference in the results and no difference in outcome according to the criteria in claims 2 and 3 of the Patent: both sets of data showed that the Apotex sample fits the requirements specified in claims 2 and 3 of the Patent.
548 In any event, a top mesh was used in the June experiments.
(f) Mesh size
549 Not content with all of their other criticisms, which in my view lacked force, the respondents also raised the question of mesh size. It is to be noted that mesh size is not an integer of claim 1.
550 It is clear from the evidence of Professors Dressman and Fassihi that there appears to be two commonly used mesh sizes (20 and 40) used in USP type III dissolution testings and that dissolution testers commonly have a preferred mesh size which they use unless it proves inappropriate for the dosage form being tested. Professor Dressman would start with a 40 mesh size and Professor Fassihi with a 20 mesh size. I should say that there was no evidence that a choice of either would produce materially different results.
551 Professor Dressman’s evidence was that she expected the hydrodynamics to be such that there would be no clogging of the 20 or 40 mesh and that either would be appropriate. She would not expect there to be an ascertainable difference in the results obtained using a 20 or 40 mesh.
552 Again, I have given Professor Tucker’s evidence little weight on this aspect. Professor Tucker had no specialised skill or experience as to the appropriate mesh size, mesh material or mesh configuration to use for a dissolution test using USP type III apparatus. Professor Tucker accepted that he had never in fact made a decision in respect of mesh size. Professor Tucker purported to suggest that the mesh size and configuration used in the May and June experiments was not appropriate because particles were observed at the bottom of the cylinders. But Professor Dressman explained, whose evidence I accept, that the fact that particles were observed in both the May and June experiments did not mean that the mesh sizes used in those experiments were inappropriate. Further, there was no guarantee that mesh sizes smaller than 40 would retain the fine white particles. Further, there was no probative evidence that the particles observed at the bottom of the vessels would have had any effect on the results or that no (or less) particles would have been observed if a different (i.e. smaller sized) mesh configuration was used.
553 Professor Tucker speculated that the particles could possibly contain some paracetamol and impact on the 15 minute sample results. Professor Tucker guessed that the particles may alter those results by about 1 to 2% but conceded that even if this factor was taken into account, it would not affect the outcome as the alleged infringing products would still have a dissolution profile falling with each of claims 1, 2 and 3.
554 Further, if the respondents wished to assert that a different experiment would yield a different result, they ought to have carried out such experiments. Instead, they chose not to conduct any experiments but preferred to rely on speculation only.
555 In any event, the following matters also demonstrate that the mesh configurations used in the GSK experiments were appropriate:
(a) The alleged infringing products did not stick to the mesh or block the mesh;
(b) The variation in the measured dissolution profiles within each experiment was very small;
(c) The f2 similarity factor of 73 that I have referred to earlier showed in accordance with the FDA Guidelines that the May and June results were similar.
(g) The respondents’ “affirmative mesh size case”
556 Related to the previous point, the respondents also deny infringement on the basis that GSK has not proved that the alleged infringing products would infringe using each possible USP type III apparatus mesh size. But I reject the respondents’ contention. There is no need for GSK to so prove.
557 First, “mesh size” and mesh configuration are not integers of the claims. Nor are they details commonly included in peer-reviewed reports of dissolution testing. GSK conducted the May and June experiments using appropriate mesh sizes and configurations and sampling procedures and the alleged infringing products were found to fall within the language of the dissolution test integers specified in each of claims 1, 2 and 3 on the basis of each of or both the May and June experiments.
558 Second, Bristol-Myers Squibb Co v Apotex Pty Ltd (2015) 228 FCR 1 at [51] has little to do with the case. That case was about whether directions in a piece of prior art would inevitably result in a notional infringement of the patent in suit for the purposes of novelty. Would a skilled person only choose a particular method that would infringe the patent, instead of another method also specified in the relevant piece of prior art, such that “infringement” would be inevitable? In other words, did the prior art contain clear and unmistakable directions to do what the patentee had invented? But such a context is irrelevant to my infringement enquiry which only requires a determination of whether the allegedly infringing product takes each of the essential features of the asserted claims.
559 Third, GSK has satisfied its onus of demonstrating that the alleged infringing products have a dissolution profile falling within the constraints set out in each of claims 1, 2 and 3 as determined by the claimed method.
560 But as mesh size, mesh configuration and sampling procedure are not essential integers of the asserted claims, GSK does not need to prove that the alleged infringing products infringe the claims using all possible mesh configurations or sampling procedures.
561 Further, I also agree with GSK’s contention that if the respondents wished to assert that under different experimental conditions the alleged infringing products did not infringe, they bore an evidentiary onus of establishing that the alleged infringing products would not fall within the scope of the claims using a different mesh size or a different sampling method once a prime facie case of infringement had been established.
562 The respondents could have conducted an experimental test on the alleged infringing products using a mesh configuration or sampling procedure that they claim would have produced a dissolution profile outside the scope of the asserted claims. An extra experiment could have been readily carried out at the time of the May or June experiments under supervision of the parties’ representatives. But the respondents chose not to take that course, no doubt for good reason.
563 The respondents have not adduced any experiment demonstrating that the alleged infringing products did not have a dissolution profile within the claimed constraints if a different mesh size or configuration had been applied. In any event, Professor Dressman said that the key factors that influenced the hydrodynamics in a vessel of the USP type III apparatus were the dip rate and dissolution medium volume, not mesh size or configuration (unless unrealistic options were chosen). There would not be a material difference across appropriate mesh sizes that a person skilled in the art would identify. As Professor Dressman explained:
If you choose an appropriate mesh size … then that choice between — in that range I don’t think would make any difference to the dissolution results. If you go and use a silly mesh size, like, an extremely fine one where you know it’s going to interfere with the hydrodynamics … then that may become the overriding factor. But you’ve got to use some commonsense.
564 Professor Dressman’s approach had an attraction over the respondents’ speculation on this particular aspect. The speculation that there may be a significant difference across mesh sizes is rejected if one confines the universe of choice to that sensibly indicated by Professor Dressman.
565 Nevertheless, the respondents assert that if it is possible for a finding of infringement to differ depending on what testing conditions are used, then that means that there has been a failure to properly define the statutory monopoly. It is said that:
(a) mesh sizes 20 and 78 are alternative mesh sizes to consider using with a USP type III apparatus reciprocating cylinder for the relevant formulation;
(b) changing the mesh size for a USP type III apparatus reciprocating cylinder can change the dissolution results;
(c) the extent to which the dissolution results change for a given formulation being tested can only be determined by experimentation; and
(d) if GSK contends that the alleged infringing products necessarily have a dissolution profile falling within the claim ranges, then it must establish that contention for every reasonable interpretation of the claim that by the patentee’s own drafting has been left open.
566 But I agree with GSK that the omission to specify mesh size is not fatal.
567 The respondents contend that:
(a) GSK cannot exclude the possibility that the patentee used a size 20 or a size 78 mesh as the basis for setting the limits in claims 1 to 3;
(b) GSK has led opinion evidence only as to a likelihood that size 20 mesh, but not size 78 mesh, would produce similar dissolution results;
(c) as to the size 20 mesh, that opinion is contradicted by Professor Fassihi who explains that the effect of a change of variable can only be known by conducting an experiment; and
(d) GSK could have readily incorporated experiments testing a size 20 mesh (and a size 78 mesh) in its experimental protocol.
568 The respondents’ assertion of the possible use of a size 78 mesh has an air of unreality to it. And I accept Professor Dressman’s evidence concerning the size 20 mesh that it would have made no difference. I have discussed the unreality of using a size 78 mesh in other parts of my reasons.
569 Generally, I reject the respondents’ contention that the failure of the patentee to specify the mesh size means that GSK needs to advance a positive case as to why that omission does not matter.
(h) The May results are accurate and reliable
570 In summary, the alleged infringing products are clearly covered by the language of the relevant claims of the Patent having regard to the May results considered alone, if one construes “basket” as cylinder (contrary to my conclusion on construction).
(i) The June Results are accurate and reliable
571 Further, the dissolution results obtained from the June experiments are accurate and reliable. They demonstrate that the alleged infringing products have a dissolution profile within the constraints set out in the relevant claims of the Patent “as determined by the USP type III apparatus, reciprocating basket, with 250ml of 0.1M HCl at 37C set at a cycle speed of 15 strokes/minute”, if one construes “basket” as cylinder.
(j) Infringement of claims 8 to 11
572 The polymer excipients in the sustained release phase of the alleged infringing products are:
(a) high viscosity HPMC;
(b) pregelatinised starch;
(c) PVP; and
(d) microcrystalline cellulose.
573 GSK’s infringement case in relation to claims 8 to 11 turns on the proposition that HPMC is the only matrix forming polymer in the sustained release phase of the alleged infringing products and that PVP, pregelatinised starch and microcrystalline cellulose in the alleged infringing products are not “matrix forming polymers”. On my construction of “matrix forming polymer” I would agree with that contention.
574 Generic Partners’ position is that each polymer must contribute to the formation and integrity of the matrix, which matrix must as a whole provide for a sustained release. It is said that by contributing to the integrity of the matrix, the polymers necessary influence the rate of release of paracetamol from it. On Generic Partners’ construction, it is only necessary that the polymers form the matrix and the matrix as a whole sustains release. On my construction of “matrix forming polymer” I would reject that contention for the reasons I have already given.
575 Generic Partners contends that even if I find that each polymer must also be said to act in some way to sustain the release of the paracetamol from the matrix, PVP, pregelatinised starch and microcrystalline cellulose in the alleged infringing products act in that way by being responsible for the integrity of the monolithic matrix which remains dry after ingestion. Were the tablet matrix to break up, it would necessarily release the drug more quickly. In my view this high level assertion does not accord with my construction of “matrix forming polymer”. It is HPMC that has the principal and direct function of sustaining release.
576 Generic Partners contends that Professor Tucker identified PVP, pregelatinised starch and microcrystalline cellulose in the alleged infringing products as “matrix forming polymers” on the basis that they were polymers and were the excipients that contribute to the formation and integrity of the extended release polymer matrix and influenced the release profile of paracetamol. Again, this high level assertion does not accord with my construction of “matrix forming polymer”. Moreover, for the reasons that I have previously given, I have given little weight to Professor Tucker’s evidence on this aspect.
577 Professor Davies agreed that each of PVP, pregelatinised starch and microcrystalline cellulose contributed to the structural integrity of the monolithic matrix and formed that matrix in the sense of the matrix being a mixture of the composition and excipients and drug. But as to whether these polymers were “forming that matrix”, Professor Davies’ evidence was as follows:
MR CORDINER: And then, in terms of the concept of a matrix, as I put it [as a matrix tablet not gel], being formed, these polymers are forming that matrix; they assist by forming the matrix by, in most cases, plastic deformation, bringing the — enabling the particles to come together in a more interlocked and stronger fashion? --- If you’re talking about forming the physical entity of the tablet ---
Yes? --- I would agree. But that’s very different to forming the matrix, the polymer matrix that controls the release ---
I appreciate that’s your ---? --- of the paracetamol.
578 Professor Davies agreed that a binder in the dry inner core was “holding together the tablets” but he would not accept that this could be described as sustaining release of the drug in the context of the Patent as understood by him.
579 Professor Fassihi said:
I consider hypromellose (low viscosity [sic] HPMC), pregelatinised starch and povidone (PVP) as each being capable of acting as a matrix-forming polymer and contributing to strengthening the matrix structure in the formulation of the modified-release layer described in part 2.3.P.3.2 for the reasons stated above at paragraph 58 in respect of Formulation C. I consider cellulose microcrystalline (also known as microcrystalline cellulose) to be capable of acting as a matrix-forming polymer especially when it is compressed along with other gel forming material, such as hypromellose, pregelatinised starch and PVP, as in the formulation in part 3.2.P.3.2. Cellulose microcrystalline can also act in other roles including as an insoluble diluent, and at low concentrations as a disintegrant.
580 But this evidence was couched as capability only. Moreover, it does not address the polymer with the relevant principal and direct function. Professor Dressman accepted that polymers that maintain the integrity of the matrix will have the effect of slowing release from it under reasonable conditions because the matrix is held together. But to accept that position in its generality does not support Generic Partners’ position. The principal or key polymer in question was HPMC.
581 Generic Partners contends that the alleged infringing products have [Confidential]mg (out of [Confidential]mg) or [Confidential]% w/w of matrix forming polymer (comprising microcrystalline cellulose, PVP, HPMC and pregelatinised starch) in the sustained release phase which is outside the limits claimed in claims 10 and 11 of the Patent.
582 Further, as I have said, Generic Partners has said that the matrix forming water soluble polymers in the alleged infringing products are:
(a) high viscosity HPMC;
(b) pregelatinised starch; and
(c) PVP.
583 It is also said that the matrix forming water soluble polymers in the alleged infringing products are not all selected from the group of compounds specified in claim 8 of the Patent.
584 Further, it is said that the alleged infringing products are therefore not compositions according to claim 9 of the Patent in which the water soluble matrix forming polymer is HPMC.
585 I reject Generic Partners’ contentions.
586 Accepting the construction of “matrix forming polymer” that I have given in a previous section, which is that it refers to the key or principal polymer(s) that directly provides the sustained release in the sustained release layer, then it is well apparent that HPMC performs that role in the alleged infringing products and not the other polymers identified such as microcrystalline cellulose, PVP or pre-gelatinised starch. Therefore the alleged infringing products fall within claims 8 and 9. Further, when one considers the proper focus of HPMC being the matrix forming polymer, the weight ranges in claims 10 and 11 are easily satisfied.
587 Further, if it is necessary to say so, the descriptions in the Product Module of the functions of the various polymers for the alleged infringing products hardly assists the respondents although I accept that the identification of one function for a polymer does not rule out another function. In the sustained release layer:
(a) PVP is described as a binder;
(b) Microcrystalline cellulose is described as a disintegrant; and
(c) Pre-gelatinised starch is described as an intragranular disintegrant and an extragranular disintegrant.
588 In summary, on my construction of “matrix forming polymer”, the alleged infringing products infringe claims 8 to 11.
EXCLUSIVE LICENSEE
589 If I am correct in the conclusion that GSK has failed on its infringement case, then the exclusive licensee issue for at least one purpose does not need to be resolved. But let me address the question.
590 Apotex contends that GSK Australia is not an exclusive licensee and that the licence granted to GSK Australia under the Exclusive Licence Agreement dated 7 October 2014 (the Licence) was not “granted by the patentee”.
591 “Exclusive licensee” is defined in s 3 and Schedule 1 to the Act to mean:
a licensee under a licence granted by the patentee and conferring on the licensee, or on the licensee and persons authorised by the licensee, the right to exploit the patented invention throughout the patent area to the exclusion of the patentee and all other persons.
592 It is appropriate to refer to the provisions of the Licence.
593 Under clause 2.1 of the Licence, the first applicant to these proceedings, the patentee (GSK Ireland), acknowledged and confirmed that it had granted to GlaxoSmithKline Consumer Healthcare Pte Ltd (GSK Singapore) “the exclusive right to Exploit the Australian Patent (including the Invention) in the Territory”, such rights being effective from 31 December 2013. The “Australian Patent” is the Patent and the term “Exploit” has the meaning under the Act (see clause 1). GSK Ireland also acknowledged that it retained no right to exploit the Patent on and from 31 December 2013 without GSK Singapore’s consent (clause 2.2). Accordingly, on and from 31 December 2013, GSK Singapore was the exclusive licensee under the Act. It had been granted a licence by GSK Ireland which conferred on GSK Singapore the right to exploit the patented invention throughout the patent area to the exclusion of GSK Ireland and all other persons.
594 Now the exclusive rights under a patent are personal property and are capable of assignment (Orion Corp v Actavis Pty Ltd (No 3) [2015] FCA 1373 at [61] per Rares J). I also consider that such assignability can necessarily be implied from s 13(2), although strictly it is dealing with the patentee’s exclusive rights rather than an exclusive licensee’s rights. The exclusive licence granted by GSK Ireland to GSK Singapore was therefore assignable.
595 Under clause 3.1, GSK Singapore granted to GSK Australia “its exclusive right to Exploit the Australian Patent (including the Invention) in the Territory”. GSK Ireland consented to the grant (clause 3.2). GSK Ireland and GSK Singapore both acknowledged that, on and from 7 October 2014, neither of them had any right to exploit the Patent without GSK Australia’s consent (clause 3.4). Consequently, the “licence” granted to GSK Australia amounted to an assignment by GSK Singapore to GSK Australia of the licence granted by GSK Ireland to GSK Singapore. This is notwithstanding that the text is structured as a sub-licence. If there was any doubt about the intention of the parties to the Licence to appoint GSK Australia as the exclusive licensee of the Patent for the purposes of the Act, clause 3.5 of the Licence expressly stated that the parties so intended. Other provisions of the Licence reinforce the point. For example, clause 5 and clause 12 are consistent more with the mechanism of assignment. Further, the language of trust in clauses 12.2 and 12.3 sits better with assignment than a sub-licence.
596 Further, if the grant by GSK Singapore to GSK Australia under clause 3.1 of the Licence was not sufficient to appoint GSK Australia as the exclusive licensee under the Act, then GSK Ireland directly granted GSK Australia the exclusive licence under the Patent (clause 3.3).
597 The operation of clause 3.3 is not unimportant. It provides:
If, for whatever reason, clause 3.1 does not effectively confer on GSK Australia the exclusive right to Exploit the Australian Patent (including the Invention) in the Territory, then GSK Ireland grants to GSK Australia, from the Effective Date, the exclusive right to Exploit the Australian Patent (including the Invention) in the Territory on the terms and conditions set out in this Deed.
598 Its evident purpose is to deal with the situation where the operation of clause 3.1 is not effective to place GSK Australia in the position of being the exclusive licensee under the Act by reason of the grant from GSK Singapore rather than GSK Ireland (i.e. Apotex’s contention). In that scenario, in other words accepting Apotex’s contention, the matter is then remedied by the direct grant from GSK Ireland. I accept though that the language of clause 3.3 does not say so directly. It uses the phrase “exclusive right to Exploit” (as defined in the Act), rather than the phrase “Exclusive Licensee” (as defined in the Act). But clause 3.5 makes it abundantly plain what is intended.
599 In summary, on and from 7 October 2014, GSK Australia was the exclusive licensee under the Act.
Fair Basis (section 40(3))
600 Section 40(3) requires a real and reasonably clear disclosure in the body of the specification of what is then claimed. The language of s 40(3) points to a comparison between the claims and what is described in the body of the specification only. As stated by Barwick CJ in Olin Corporation v Super Cartridge Co Pty Ltd (1977) 180 CLR 236 at 240:
the question is a narrow one, namely whether the claim to the product being new, useful and inventive, that is to say, the claim as expressed, travels beyond the matter disclosed in the specification.
601 Section 40(3) does not use the word “invention”, but it requires that the claims “be fairly based on the matter in it that discusses the ‘invention’”; Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 1) (2004) 217 CLR 274 at [53]. This is to be taken to mean the embodiment(s) which is described and around which the claims are drawn. But it does not mean the inventive step taken by the inventor or the advance in the art made by the inventor. Lockwood (No 1) described the test at [68] and [69] in the following terms:
Erroneous principles. The comparison which s 40(3) calls for is not analogous to that between a claim and an alleged anticipation or infringement. It is wrong to employ “an over meticulous verbal analysis”. It is wrong to seek to isolate in the body of the specification “essential integers” or “essential features” of an alleged invention and to ask whether they correspond with the essential integers of the claim in question.
“Real and reasonably clear disclosure”. Section 40(3) requires, in Fullagar J’s words, “a real and reasonably clear disclosure”. But those words, when used in connection with s 40(3), do not limit disclosures to preferred embodiments.
The circumstance that something is a requirement for the best method of performing an invention does not make it necessarily a requirement for all claims; likewise, the circumstance that material is part of the description of the invention does not mean that it must be included as an integer of each claim. Rather, the question is whether there is a real and reasonably clear disclosure in the body of the specification of what is then claimed, so that the alleged invention as claimed is broadly, that is to say in a general sense, described in the body of the specification.
Fullagar J’s phrase serves the function of compelling attention to the construction of the specification as a whole, putting aside particular parts which, although in isolation they might appear to point against the “real” disclosure, are in truth only loose or stray remarks. (footnotes omitted)
602 Clearly, one should not use an “over meticulous verbal analysis”. Further, the focus is not on an identity of language between the claims and the disclosure in the body of the specification. Rather, one is looking for a generalised disclosure in the body that provides support for the claims in substance. Moreover, it is inappropriate to isolate in the body of the specification “essential integers” or “essential features” of an alleged invention and to ask whether they correspond with the essential integers of the claim. See also Artcraft Urban Group Pty Ltd v Streetworx Pty Ltd [2016] FCAFC 29 at [105] to [114] per Greenwood J.
603 Fair basis can be established by a comparison with the consistory clause(s). However, even if a claim is based on and mirrors the form of the consistory clause(s), it will not be fairly based if other parts of the specification show that the invention is narrower than the consistory clause(s). What has been described as a “coincidence of language” between a claim and part of the body of a specification does not per se establish fair basing if that part of the language of the specification does not reflect the description of the invention in the light of the specification as a whole (Lockwood Security (No 1) at [87]).
604 Further, it is appropriate to restate that the complete specification is not to be read in the abstract, but is to be construed in the light of common general knowledge and the relevant art before the priority date.
605 In the present case, the relevant principles have not been in issue. Rather, the debate has concerned their application.
606 During the trial, the respondents advanced two arguments under the broad heading of lack of fair basis. Let me deal with each in turn.
(a) The respondents’ first fair basis case — claims 1, 2 and 3
607 The first case propounded by Apotex has asserted that the only disclosure in the Patent of formulations that met the advantageous attributes identified in the Patent were Formulations C and D. Formulation C comprised 667mg of paracetamol, had an SR:IR paracetamol ratio of 71:29 and released 39.4% of said paracetamol at 15 minutes, 64.4% at 60 minutes and 101.8% at 180 minutes. Formulation D comprised 665mg of paracetamol, had an SR:IR ratio of 69:31 and released 40.8% of said paracetamol at 15 minutes, 65.0% at 60 minutes and 101.8% at 180 minutes. Apotex asserts that claims 1, 2 and 3 travel beyond the aforementioned disclosure insofar as they claim a paracetamol content, IR:SR ratio and dissolution ranges that are not the same as or very close to those of Formulations C and D.
608 Apotex says that the sole relevant disclosure of a formulation in the Patent is Formulation C or D. Such a formulation has a dissolution profile broadly in the middle of the ranges of the claims. Its point is that the specification does not provide a “real and reasonably clear” disclosure of the wide range of formulations that are claimed in claims 1 to 3.
609 The specification refers on p 4 lines 12 to 14 to the “surprising” discovery that an “advantageous pharmacokinetic profile can be provided by a two-phase (immediate-release and sustained-release) formulation of paracetamol which satisfied a unique in vitro dissolution profile”.
610 It may be accepted, and Professor Davies agreed, that the Patent does not represent that the surprising connection has been established by a validated level A IVIVC. Professor Davies agreed that the Patent does not describe any internal validation of the type described in the Grattan paper or the FDA guideline. Further, he agreed that the Patent does not describe any external validation of the type described in the Grattan paper or the FDA guideline. Professor Davies did, however, say in cross-examination:
but the patent provides the relationship clearly between the in vitro and the pharmacokinetic [properties].
611 I raised some questions with Professor Davies on this topic:
HIS HONOUR: If it doesn’t maintain that there’s some sort of relationship, what’s the patent disclosing as to the basis of that relationship? Could you describe it as internal or how is the — remind me how is it ---? --- The patent is disclosing a relationship between the in vitro dissolution profile, as I read it, and the pharmacokinetic parameters that it has highlighted, in terms of the four key parameters that it has highlighted.
Yes. Yes? --- That’s how it’s describing that relationship.
But it’s not actually describing how that relationship is established, or is it? --- That’s correct. … Other than showing the individual examples and however it has come through, but it hasn’t described how it has been achieved. I agree with that.
On no view could you say it has been described in an external sense, but you could say it has been described in an internal sense or is that ---? --- I think it’s difficult to say that based on the definitions that’s provided in the FDA document, that it’s showing an internal — it’s just showing through examples how it has come to those — the relationship between the in vitro dissolution profile and showing that that will give you, with the other elements of the claim, the pharmacokinetic parameters of the patent.
…
MS GODDARD: So it would be correct then, wouldn’t it, Professor, that the specification of the patent is not representing a level A IVIVC correlation has been established? --- I think that’s correct. It’s not saying what form of the — from an FDA viewpoint the correlation is, but it’s just showing you that there is a relationship between the dissolution profile and those key pharmacokinetic parameters.
612 Apotex contends that the Patent does not purport to set out or describe any detailed basis or validation process for any predictive relationship between the dissolution parameters and the advantageous pharmacokinetic profile. Clearly, Formulations A and B are not reference formulations. The Patent describes essentially one formulation that satisfies the pharmacokinetic criteria, being Formulation C (or D).
613 Apotex then generally asserts that for dissolution tests to be used as a surrogate for bioequivalence, a level A IVIVC is required, subject to some limited exceptions. Clearly the Patent does not refer to any validated IVIVC as having been undertaken. Rather, it just asserts that there is the required favourable in vitro/in vivo relationship without more.
614 After I had enquired about the difference between a correlation and a relationship, Professor Davies said:
There’s a difference there. I’m using — I clearly think there is a relationship … between the in vitro and in vivo profile as the patent is teaching you. The patent comes to that view through some kind of correlation, but it doesn’t describe what that correlation is.
615 But as I say, it is apparent that the Patent does not describe the use of any or any validated IVIVC.
616 As I have earlier indicated, with a given percentage dissolution, tablets with different amounts of drug will give different amounts of drug in vivo. In short, Apotex contends that it is impossible to tell without an IVIVC whether all formulations within the Patent’s ranges will provide the advantageous pharmacokinetic profile. Apotex says that because there has been no disclosure of any or any validated IVIVC, the only disclosure in the Patent that can possibly provide a basis for the broad claims is:
(a) the results of Formulation C or D;
(b) the limited assertion of “a relationship” at p 4 lines 12 to 14; and
(c) the consistory clauses at p 4 line 26 to p 4a line 25.
617 As to the first aspect, Apotex submits that the results of Formulation C or D only provide a real and reasonably clear disclosure of those particular formulations. It contends that even taking into account the types of tolerances allowed by the FDA, the claimed formulations extend far beyond such tolerances.
618 Moreover, Apotex contends that any reference to the FDA’s “10 per cent” level does not justify the breadth of the ranges.
619 Further, it is said that even if an allowance of ± 10% were somehow to provide a fall back for fair basing, even the narrowest claim, claim 3, does not have all of the limits within a ± 10% range of Formulation C when dose is considered. For claim 3, at the 180 minute time point Formulation C had 101.8% of its 666.6 mg dissolved, whereas applying the 95% lower claim limit to the 600 mg lower dose limit gives 570 mg, which is 85.5% of 666.6 mg (i.e. more than 10% below).
620 Further, it is said that the claim 2 and claim 1 limits are broader and have more time points outside this 10% range when the impact of dose is also factored in. For claim 2 at 60 minutes, 58% of 600mg (the lower claim limits) is 348 mg, which is 52.2% of Formulation C’s 666.6mg, and 73% of 700mg (the upper claim limits) is 511 mg which is 76.7% of 666.6mg. Both are more than ± 10% of 64.4% from Table 2. Similarly at the 180 minute time point, the lower claim limits are 90% and 600mg, which gives 540mg which is 81% of 666.6 mg. Claim 1 is broader again.
621 Further, it is said that the skilled addressee knows that any assertion as to the provision of an equivalent in vivo effect for formulations falling outside a 10% range can only be supported by establishment of a validated IVIVC.
622 Apotex submits that the disclosure of the Patent amounts to no more than the particular disclosure of Formulation C or D. The Patent does not say that it arrives at the broad dissolution ranges by using the result for Formulation C or D +/– 10% for all compositions from 600–700 mg and 10%–45% IR: 55%–90% SR. It merely makes the assertion of “a relationship” or “a connection” at p 4 lines 12 to 14. It is said that this is insufficient to constitute a “real and reasonably clear disclosure”.
623 It is said that there are no particular passages where the Patent is teaching you that the pharmacokinetic profile will always be achieved by formulations within the broad ranges.
624 As to the second aspect, Apotex asserts that it is clear that the skilled addressee would not read the dissolution ranges as having been set by any reliance on a connection or relationship referred to at p 4 lines 12 to 14.
625 Apotex has referred to Professor Davies’ cross-examination:
MS GODDARD: [W]hat are the findings that you refer to specifically, if you wouldn’t mind? --- The findings are the — I’m — in that paragraph I’m talking in terms of the invention of the patent. I’m talking about in the concept of what have the inventors found, what are their findings. They found that there is this advantageous pharmacokinetic parameters with a particular dissolution profile for a bilayer tablet with a particular ratios of paracetamol with a polymer — a matrix-forming polymer.
So is that — can I clarify — are you referring to the passage on page 4, lines 12 to 14 or are you referring to the data for formulation C and the in vivo data for formulation D? --- I believe I’m referring to the entirety of the patent, that the formulations C and D are exemplary examples of what the patent describes, I believe. It also shows formulations that lie outside the invention in terms of A and B.
Where does the patent tell you that the finding that such formulations — this is the top of the joint expert report on page 19 — would have the advantageous pharmacokinetic profile? --- When one reads the entirety of the patent, equally when one — we’ve talked about before, it’s summarised in the fourth line — fourth paragraph, lines 12 to 14 of page 4 but then the patent goes on to discuss different aspects of the invention.
So more than that general reference, you can’t point to anything in particular upon which you rely? --- I don’t think that’s what I’ve just said. I think I’ve said that the patent is clearly teaching you what the invention is about.
626 Moreover, Apotex contends that merely to move from the assertion of p 4 lines 12 to 14 to the consistory clauses on pp 4 to 4a or to the matching claims does not provide any fair basis.
627 It is said that the claims are broader than the disclosure in Example 2 (Formulation C) because the skilled addressee understands that there is no scientific rationale to support the in vitro ranges as claimed. It is said that there is no “real and reasonably clear” disclosure of an invention having that scope and accordingly the claims are not fairly based on the description.
628 This applies to each of the claims. It is said that there is no basis for even the narrower ranges of claims 2 and 3.
629 As to the third aspect, it is said that a consistory clause will not necessarily provide fair basis for a claim. What is required is more than a mere “coincidence of language”, and other parts of the specification can, as they do here, show that what has been invented is of a narrower scope.
630 In my view, Apotex’s assertions should be rejected.
631 The ranges specified in the claims are meaningful in the context of the Patent. As Professor Davies explained:
in the context of the patent — the patent teaches you you can have a range. It would be impossible to make the same formulation over and over again with exactly the same percentage. That’s why you have these ranges. And that allows you to … understand that, you know, these formulations may release at different rates within the ranges of the patents but they will give you the pharmacokinetic attributes.
632 First, with respect to the paracetamol content issue, the following may be noted.
633 If one looks at an extract from the acetaminophen monograph from USP 24 NF 19, under the heading “Acetaminophen Tablets”, the document states that “Acetaminophen Tablets contain not less than 90.0 percent and not more than 110.0 percent of the labelled amount of C8H9NO2 (paracetamol)”. In cross-examination, Professor Fassihi explained:
MS ROFE: So, within one batch of acetaminophen tablets it’s allowable to have tablets containing 90 per cent and tablets containing 110 percent? --- Of the labelled … in a batch that is a acceptable range, yes, for the content uniformity.
634 Further, Dr Mooney agreed that it was impossible to produce tablets within or across batches with exactly the same content of active pharmaceutical ingredient. Moreover, he agreed that regulators such as the TGA and FDA permit a variance in the order of 10 to 20%.
635 Professor Davies also gave evidence that the content ranges specified in the claims were consistent with the content uniformity ranges allowed by regulatory authorities. Professor Davies explained that:
[T]he level of drug, six to 700, is well within the plus or minus 10 per cent allowed by regulatory authorities for the normal oral dose of formulations …
[B]ased on the teachings of the patent, a formulator would understand that the 600 milligrams is within 10 per cent of the label claim of the 650 that’s shown for Formulation C. …
Now, the second concept of plus and minus 10 per cent relates to the nominal amount of drug in a — allowed in a formulation and the regulatory authorities allow plus and minus 10 per cent of the labelled claimed amount of drug. …
HIS HONOUR: --- you could take Formulation C ---? --- Yes.
--- or somebody reading the specification would look at Formulation C ---? --- Yes.
--- and take plus or minus 10 per cent in terms of the total milligrams? --- Yes. And it’s within that range.
Yes. Yes? --- 600 to 700 is within the range of plus or minus 10 per cent of the labelled claim. … [T]his is the real world of pharma industry where they recognise that you cannot make a formulation of [650]; you would have to set a range and a specification.
MS GODDARD: And it depends how broad those ranges are allowed to be, of course? --- Yes, that’s correct, but plus or minus 10 per cent is typical and the ranges set in the patent is narrower than that.
636 Formulation D, being the subject of the clinical trials reported in the Patent, had a total paracetamol content of 665mg: 90% of 665 is 598.5mg and 110% of 665 is 731.5mg. The paracetamol limits specified in claim 1 were within those limits.
637 There is no substance to the respondents’ lack of fair basis complaint concerning the paracetamol content.
638 Second, the claims of the Patent are defined by the result to be achieved, namely, a bilayer tablet having a particular unique and advantageous in vitro dissolution profile. The Patent teaches that such a result may be achieved by varying the IR:SR ratio and the amount of matrix forming polymer in the formulation. The IR:SR ranges in the claims provide guidance to a skilled formulator as to the ratios (i.e. an unequal split of paracetamol across the IR and SR layers) that can produce the result claimed. The Patent teaches a skilled formulator that the IR:SR ratio parameter will not affect the in vivo performance of the formulation as long as the formulation satisfies the dissolution criteria of the claims.
639 In cross-examination, Professor Davies relevantly explained:
[T]his patent is teaching you that one can achieve that dissolution profile using the ranges of the paracetamol in the immediate release and sustained release phases of this bilayer tablet, and it’s — and using a matrix forming polymer. The very fact the ranges as such are set, ultimately it has to achieve that release profile. And if it achieves the release profile, that release profile will give you your pharmacokinetic parameters. That’s what the patent is teaching you. …
If it meets — I think if it meets the profile and it meets also the other elements of the claim, as formulator, based on the fact that certainly the dose is within the 10 per cent level, the percentage ranges are within the 10 per cent level as allowed by the FDA, this is teaching you that you can make the formulation, but it has got to achieve that particular release profile with the other elements of the claim. As a formulator, that’s how I see it’s teaching me.
640 There is no substance to the respondents’ lack of fair basis complaint concerning the IR:SR ratio. Moreover, in my view it is superfluous in one sense to enquire how the IR:SR ratio was arrived at in terms of a fair basis challenge, particularly given the dissolution profile constraint.
641 Third, with respect to the dissolution ratio, Professor Fassihi noted that “[i]n a repeated dissolution study, using any one of USP Apparatuses 1, 2 or 3, variability in the measured dissolution profile for each sample of up to ±10% is considered normal …” and explained that:
The dissolution ranges in Claim 1 are precise and not expressed with any qualification such as the use of the word “about”. I would normally expect the term “about” to be used, as dissolution involves an inherent level of ±10% variability. The patentee has been more strict in these claims, and I would expect that everything should fall within that range.
642 The USP monograph for acetaminophen extended release tablets identified a dissolution specification that was significantly broader than the specification set out in claims 1, 2 and 3 of the Patent, namely:
15 minutes: between 45% and 65%
60 minutes: between 60% and 85%
180 minutes: not less than 85%
643 Further, Dr Mooney understood that the width of the constraints included for the dissolution test claimed (e.g. 30 to 48% released after 15 minutes) were within the range of constraints typical for an in vitro quality control dissolution test, that the dissolution profile for claim 2 is narrower than for claim 1 and is narrower again in claim 3, and that the constraints included for the dissolution test in claims 2 and 3 (e.g. 38 to 44% released after 15 minutes) were more stringent than the range of constraints typical for an in vitro quality control dissolution test.
644 Professor Davies also explained:
I understand that the Patent, read in a common sense fashion, specifies in claims 1, 2 and 3, the dissolution profiles of the invention in a conventional way and as at April 2000, I would have no difficulty interpreting the dissolution specifications set out in the Patent.
The Patent, specifies the range of the amount of paracetamol that must be dissolved or released in vitro after or “at” an early, middle and late time point. This is consistent with, for example, the guidance document published by the FDA in 1997 entitled “Guidance for Industry — Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations” (the 1997 FDA Report), a copy of which is included in Tab 8 of Exhibit MCD–1. I was familiar with this document as at April 2000. At that time, the 1997 FDA Report would have been in my office either in hardcopy or saved electronically (or both).
Also consistent with the 1997 FDA Report:
(a) the last time point of each of the claims are the time points where at least 80% of drug has dissolved (see page 17 of the 1997 FDA Report); and
…
It is apparent from reading the Patent as a whole and having regard to the dissolution results reported for Formulations C and D, that the specified dissolution profile does not represent an arbitrary selection of dissolution values, but rather has been found to correlate with advantageous in vivo pharmacokinetic attributes, as described, for example, on page 3 lines 23–29 to page 4 lines 1 – 10 of the Patent.
…
I agree with Dr Mooney’s comment at paragraph 242 of his affidavit that the width of the ranges specified in each of claims 1, 2 and 3 of the Patent are typical.
The middle point figures are practically identical to the dissolution figures reported in the Patent for Formulations C and D. Formulation C is reported to release 39.4% of paracetamol at 15 minutes and 64.4% at 60 minutes (page 13 of the Patent). Formulation D is reported to release 40.8% of paracetamol at 15 minutes and 65.0% at 60 minutes (page 15 of the Patent).
The 1997 FDA Report also indicates, at page 7, that differences in pharmacokinetic properties may be observed when there is a 10 percentage point difference in release rates. Especially given that the range specified in each of the claims is less than 10 percentage points apart from the respective middle points, I would expect all manufacturable formulations falling within the claims to possess the advantageous pharmacokinetic properties referred to above. The 1997 FDA Report states, on page 17, that ± 10% is recommended for in vitro dissolution profiles and that broader specifications may also be acceptable in certain circumstances.
In my view, the ranges specified in the Patent have been reasonably set with reference to the in vitro data for Formulations A and B (which the Patent teaches do not work), the in vitro data for Formulations C and D (which do work) and the guidance issued by regulators such as the FDA, referred to above. I accordingly disagree with Professor Tucker’s comment at paragraph 232 of his affidavit that there is no scientific basis for the setting of the limits in the Patent.
645 The extract from the 1997 FDA Report referred to by Professor Davies provided that “The release rates, as measured by percent dissolved, for each formulation studied, should differ adequately (e.g., by 10%). This should result in in vivo profiles that show a comparable difference, for example, a 10% difference in the pharmacokinetic parameters of interest (Cmax or AUC) between each formulation”. Further, the 1997 FDA Report under the heading “Setting Dissolution Specifications Without an IVIVC” recognised that a variation of ±10 percentage points is acceptable even where no IVIVC has been established.
646 Further, Professor Davies in cross-examination confirmed his view that the ranges specified in the claims were reasonably set with reference to the FDA guidelines, an understanding of the relationship between the in vitro dissolution profile specified in the claims (with reference to Formulations A to D), and whether changes to the profile would have a meaningful effect within the context of the Patent. In cross-examination, Professor Davies variously explained:
The dissolution profiles as presented are well within the 10 per cent one would understand from an FDA perspective wouldn’t — plus or minus 10 per cent which would not impact the pharmacokinetic data. …
If they meet the patent — if they meet the elements of claims of the patent, I would believe, based on the reading of the patent, that it will be — it will meet the pharmacokinetic parameters. …
If you mean that it’s — the patent is teaching you that one can make formulations and if they fall within that range and they have the attributes as taught by the — the claim and the patent, then they should give you the [pharmacokinetic] attributes. … I’m relying on the whole patent. …
[T]he percentage ranges that are used within the 10 per cent again from a regulatory viewpoint that show that you don’t have changes in [pharmacokinetic] data. So I think the patent is teaching you that you can use such formulations and they will be useful. They will achieve the result. …
[I]t’s commonly understood that for such formulations plus the [sic] minus 10 per cent or beyond plus the [sic] minus 10 per cent will have an effect on the pharmacokinetic behaviour. That is at individual dissolution time points. … And that’s why in setting a dissolution and IVIVC in the … guidelines, they talk about a biobatch and then having — having a slower dissolution by at least 10 per cent — having a faster dissolution by at least 10 per cent will produce a change. …
[T]he patent dissolution ranges were clearly set based on an understanding the relationship between the in vitro dissolution profile and the pharmacokinetic parameters. …
The ranges were set with an understanding from my perspective that the plus or minus 10 per cent would affect the — you know, the relationship between the dissolution profile and the pharmacokinetic parameters. And the fact that they’re within that indicates to me that they have clearly been chosen around the mid points for C and D.
647 Further, the “10%” figures referred to in the 1997 FDA Report referred to “absolute” percentage points of total paracetamol released. In other words, if one was looking at 40% in vitro release after 15 minutes ± 10%, one would have a range of 30% to 50% (rather than a range of 36% to 44%).
648 In the JER, Professor Davies and Dr Mooney explained the relevance of the Formulations to the setting of the ranges and agreed that:
Based upon the pharmacokinetic behaviour of Formulations C and D, the % released values from the dissolution curves of C and D at 15 minutes and 60 minutes appear to have been chosen as the mid points of the ranges at 15 and 60 minutes. The third time point at 180 minutes is set at >85% to give a comfortable margin above that observed for Formulation B. The range at 15 minutes is set to exclude Formulation A and that at 60 minutes is set to exclude Formulation B and that at 180 minutes excludes Formulation B. In claim 1 we believe the first two windows were based on a +/–10% range as provided in guidance by the FDA.
649 Generally, the variance in paracetamol content and dissolution profile permitted by the claims and the regulatory guidelines were consistent with the skilled formulator’s recognition of inherent variability associated with manufacturing and dissolution testing processes.
650 Let me now deal with Apotex’s primary contention that the ranges in claim 1 were dependent upon a level A validated IVIVC, a contention that I also reject.
651 In my view, a skilled addressee would know that having regard to the accepted regulatory approach, formulations having a paracetamol content within ±10% of the ideal value or label claim and a dissolution profile within ±10 percentage points of the target, are likely to be for relevant purposes bioequivalent. Changes within these ranges are unlikely to have a significant clinical effect. Accordingly, even without an established level A IVIVC, there is a reasonable basis for the ranges specified in claim 1.
652 The dissolution specifications were set in accordance with FDA guidance. The relevant part of the FDA Report specifically concerns setting dissolution specifications “without an IVIVC”. Further, it is stated therein that “[t]his approach is based on the use of the in vitro dissolution test as a quality control test without any in vivo significance”.
653 In my view, the evidence demonstrates that a skilled formulator reading the Patent fairly with a view to making it work, with his common general knowledge (including relevant guidelines published by the FDA and regulatory practices) would understand that the patentee has, with reference to such common general knowledge, reasonably extrapolated from formulations shown to work (Formulations C and D) to specify the content, ratio and dissolution specifications in the claims. In my view, the respondents have not discharged their onus of establishing that there are formulations falling within the claims that do not meet the promise of the invention. They have only produced hypothetical speculations which were considered and rightly rejected by Professor Davies as not providing a basis to doubt the Patent’s teaching.
654 Further, as explained by Graham J in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd [1970] RPC 157 at 193:
Where, then, is the line to be drawn between a claim which goes beyond the consideration and one which equiparates with it? In my judgment this line was drawn properly by Sir Lionel when he very helpfully stated in the words quoted above that it depended upon whether or not it was possible to make a sound prediction. If it is possible for the patentee to make a sound prediction and to frame a claim which does not go beyond the limits within which the prediction remains sound, then he is entitled to do so. Of course, in so doing he takes the risk that a defendant may be able to show that his prediction is unsound or that some bodies falling within the words he has used have no utility or are old or obvious or that some promise he has made in his specification is false in a material respect; but if, when attacked, he survives this risk successfully, then his claim does not go beyond the consideration given by his disclosure, his claim is fairly based on such disclosure in these respects, and is valid.
655 Further, in Photocure ASA v Queen’s University at Kingston (2005) 216 ALR 41 at [137], Merkel J explained:
[T]here is no reason why the inclusion in claims of some embodiments which have not yet been proven to work would fall foul of s 40(3). Even if some such embodiments are subsequently shown not to work, that does not invalidate the claims in respect of those embodiments on the ground of lack of fair basis.
656 Similarly, in Asahi Kasei Kogyo Kabushiki Kaisha v W R Grace & Co (1991) 22 IPR 491, Heerey J explained at 519 and 520:
It was said that there was insufficient support in the body of the specification for the claim that a film gave “reliable seals even when contaminated” in that no method of testing such ability or reference point for comparison was given. But the specification clearly gives examples of the invention which achieve that object (paras (27) and (29)).
A further objection was that the accuracy of the upper and lower limits of the specified range are not shown for either butene or octene, so that the specified weight range of comonomer was merely speculative and lacked scientific foundation.
But the applicant has not shown that the claimed invention is in a field of science where relevant properties suddenly emerge or disappear as a precise numerical point is reached. The boundary marked out by the claims includes only films of the invention. If the test of the specification is satisfied — viz improved combination of properties — it is irrelevant that the claims might have been slightly broader or narrower.
657 Finally, Apotex has challenged Professor Davies’ evidence by asserting that the Patent does not itself disclose how the invention was arrived at or how or why the limits referred to above were set. But the Patent need only teach a skilled person how to perform the invention. Further, there is no need for the specification to identify the reasoning underpinning the selection of limits specified in the claims. As explained in NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1992) 24 IPR 1 at 27 per Wilcox J:
[I]t is not a legitimate objection that the specification does not disclose the experimental and statistical basis for the chosen STD and e.n values. It does not have to. The obligation of the patentee is to instruct the skilled addressee how to perform the invention. The patentee is not obliged to explain how the invention was made or the theoretical basis underlying any stipulated integer.
658 Apotex’s fair basis case amounts merely to speculation that the claims are likely to include within their scope formulations that do not possess the attributes identified in the Patent. Now, of course, such considerations might be relevant to any lack of utility allegation. But such an allegation, although initially made by the respondents, has now been abandoned, no doubt for good reason. It could not be proved. Indeed, it had no proper basis to be pursued once all the evidence was in. Apotex’s nuanced back door argument through an asserted lack of fair basis also fails. Finally, if it was necessary to say so, I would also hold that fair basis has been established on the basis of the matching with the text of the consistory clauses.
(b) The respondents’ second fair basis case — matrix forming polymer
659 Generic Partners says that the amount of matrix forming polymer in the sustained release phase is an important element of the invention. It says that claims 1 to 6, 8, 9, 13 and 14 are not limited as to a matrix forming polymer included so as to provide the desired in vitro dissolution rate. It also says that such claims are not limited as to the amount of matrix forming polymer in the sustained release layer. Accordingly, it is said that such claims are therefore not fairly based.
660 Further, it says that there is also no real and reasonably clear disclosure of formulations unlimited as to the nature of the “matrix-forming polymer or a mixture thereof” beyond the narrow disclosure of Formulation C. It is said that the thesis of the Patent is that “an advantageous pharmacokinetic profile can be provided by” a particular formulation, p 4 lines 12 to 14; p 1 lines 5 to 7. But differences in formulation give different results. Generic Partners says that the invention disclosed in the specification is “a sustained release paracetamol formulation” having an advantageous pharmacokinetic profile. It is said that the invention disclosed is a particular formulation of paracetamol necessarily having specific characteristics and having a specific dissolution profile. So, the Patent states:
The amount of matrix-forming polymer in the sustained release phase and the relative amounts of paracetamol in the sustained release and immediate release phases are selected so as to provide the desired in vitro dissolution rate as herein before described.
Thus, the matrix-forming polymer is suitably present in an amount from 0.5 to 10%, preferably from 1 to 6%, and more preferably from 2 to 4% by weight of the sustained release phase.
661 So, it is said that the invention is a two phase formulation of paracetamol where, inter alia, the amount of matrix-forming polymer in the sustained release phase must be selected to provide the desired in vitro paracetamol dissolution profile.
662 Generic Partners also says that the need to select the amount of matrix forming polymer in the sustained release phase is evident from the Examples in the Patent, each of which contains the same ingredients but in different amounts. Formulations A and B (outside the scope of the alleged invention) contain approximately 6% high viscosity HPMC and 7% pregelatinised starch in the sustained release layer, whereas Formulation C (within the scope of the alleged invention) contains about 3% high viscosity HPMC and 1% of pregelatinised starch in the sustained release layer.
663 Accordingly, so it is said, a comparison of Formulations A, B and C tells the skilled addressee that selecting the amount of matrix forming polymer (comprising high viscosity HPMC, PVP and pregelatinised starch) in the sustained release layer is important to achieving the desired and claimed in vitro dissolution rate.
664 Moreover, and I accept, Professor Davies agreed that the selection of the type and amount of matrix forming polymer in the sustained release phase was part of achieving the desired in vitro dissolution rate. Likewise, Professor Dressman accepted that the amount of matrix forming polymer was one of the two factors that were named necessary to achieve the desired in vitro dissolution profile. Further, in answer to the question “What, if anything, do pages 1 to 20 of the Patent disclose to you as to the amount of matrix forming polymer(s) in the sustained release phase of the invention described?” the experts agreed that:
the matrix forming polymer only relates to the sustained release phase. Page 6 lines 7–9 of the patent state that amount of matrix forming polymer in the sustained release phase is 0.5–10% by weight, preferably from 1–6% by weight and more preferably from 2–4% by weight.
665 Further, Generic Partners contends that the experts understood the term “suitably” at p 6 line 7 to indicate a requirement not a preference that the formulation contain 0.5–10% by weight matrix forming polymer. Whatever the experts may have said, in my view “suitably” as used on p 6 does not mean required in all cases; that is a reference to claims 10, 11 and 12. Such a submission ignores the first four lines on p 6 and that context.
666 Further, Generic Partners says that in the case of high viscosity HPMC, the experts disagreed as to how much high viscosity HPMC was required to achieve the sustained release described in formulations of the Patent. But Generic Partners says that it is apparent from this evidence that there is a minimum amount necessary to achieve a sustained release of paracetamol described in the Patent. Accordingly, it says that simply having an unspecified amount of high viscosity HPMC is not enough.
667 Accordingly, Generic Partners contends that the invention as disclosed in the specification requires a particular amount of matrix forming polymer to be present in the sustained release phase, selected so as to achieve the desired in vitro dissolution profile. Further, it says that the broadest disclosure of an amount of matrix forming polymer which has been selected to achieve the desired in vitro dissolution profile is an amount within the range 0.5 to 10% by weight.
668 Further, Generic Partners contends that the consistory clause does not assist GSK. It is said that a coincidence of language does not establish fair basing if that part of the language of the specification does not reflect the description of the invention in the light of the specification as a whole. Generic Partners says that the experts properly read the specification as a whole as describing a formulation where the amount of matrix forming polymer is a “crucial” characteristic of that formulation, which must be selected to achieve the desired in vitro dissolution profile. It is said that they each accepted that the amount must be between 0.5 and 10% by weight of the sustained release phase. It is said that the Patent does not teach that an amount outside that range would achieve the desired result. Nor does it disclose a formulation where the amount of matrix forming polymer has not been selected to provide the desired in vitro dissolution profile. Accordingly, so Generic Partners contends, the coincidence of language between claim 1 and the consistory clause at p 4 line 27 to p 4a line 8 of the Patent does not assist GSK.
669 Further, it is said that in any event, on 4 September 2006 the specification of the Patent was amended to add the consistory clause at p 4 line 27 to p 4a line 8 of the Patent and to change p 4a line 10 to refer to “another aspect” rather than the “first aspect” of the invention. Accordingly, it is said that in interpreting the specification as amended, the Court is entitled to refer to the specification prior to amendment. Furthermore, it is said that GSK has agreed that the addition of the consistory clause did not change the characterisation of the invention. So, the invention described in the body of the specification both before and after the 4 September 2006 amendment must be the same. Therefore, I should read the Patent without the amendment to determine whether it provides fair basis for claims which are not limited to a particular amount of matrix-forming polymer.
670 On that premise, Generic Partners then says that reference to the specification without amendment discloses the following. The original (now second) consistory clause (at p 4a line 10 to p 5 line 5) does not provide a real and reasonably clear disclosure of the invention now claimed in claims 1 to 6, 8, 9, 13 and 14. Specifically, the specification prior to amendment did not describe the relative amounts of paracetamol in the sustained release and immediate release phases. The only disclosure of the relative amounts of paracetamol in the sustained release and immediate release phases occurs in the paragraph starting at p 6 line 1, “The amount of matrix-forming polymer in the sustained release phase and the relative amounts of paracetamol in the sustained release and immediate release phases are selected so as to provide the desired in vitro dissolution rate as herein before described”. The specification describes the weight range for matrix forming polymer that must be selected from in order to provide the desired in vitro dissolution rate in the passage at p 6 lines 7 to 9. There is no broader disclosure in the Patent regarding the amount of matrix forming polymer that must be selected.
671 Accordingly, it is said that the unamended specification does not provide any support for a claim to a formulation having a sustained release phase comprising a matrix forming polymer in “an amount” or any amount. I must say that I found this argument based upon the unamended specification to be, in one sense, artificial given that I am construing the specification as if it had originally contained the amendment.
672 Further, Generic Partners says that the use of the word “suitably” on p 6 to describe the amount of matrix forming polymer to be at least between “0.5% to 10%” is “plainly” setting a requirement of the invention just as the use of the word “suitably” on p 5 describes the requirement for there to be a matrix forming polymer in the sustained release layer. The term “suitably” is used in the same way and is to be distinguished from the term “preferably” which describes a preference. I repeat what I have said earlier on “suitably”.
673 Generally, it is said that claims 1 to 6, 8, 9, 13 and 14 claim, inter alia, a bilayer tablet having an immediate release phase of paracetamol and a sustained release phase of paracetamol where the sustained release phase contains “a matrix forming polymer or a mixture thereof” and the bilayer tablet has an in vitro paracetamol dissolution profile as claimed. But the “amount” in those claims is not limited to what Generic Partners contends is the broadest disclosure in the Patent which is:
(a) an amount selected so as to provide the desired in vitro dissolution rate; and
(b) an amount from 0.5 to 10% by weight of the sustained release phase.
674 It is said by Generic Partners that, read as a whole, the invention disclosed in the body of the specification is a two phase (immediate release and sustained release) formulation of paracetamol where, inter alia, the amount of matrix-forming polymer in the sustained release phase is selected to provide a specified in vitro paracetamol dissolution profile and in an amount from 0.5 to 10% by weight of the sustained release phase. But the only relevant claims of the Patent that are limited to a particular amount of matrix-forming polymer in the sustained release phase to provide the specified in vitro paracetamol dissolution profile are claims 10 and 11 (claim 12 is not in issue). Claims 1 to 6, 8, 9, 13 and 14 are not so limited. Accordingly, by this omission, claims 1 to 6, 8, 9, 13 and 14 travel beyond the invention disclosed in the specification and are not fairly based.
675 I reject Generic Partners’ contentions generally and its conclusion on this aspect. In my view it has employed an overly meticulous verbal analysis of the specification and claims. Further, it has not undertaken the proper inquiry. It has sought to identify the features of the invention in the specification and to see whether they were present in the claims, rather than to look at the claims to see whether what was claimed was in substance disclosed in the specification. This is an inversion of the perspective which ought to have been used.
676 The specification sets out details of Formulations C and D which have certain paracetamol ratios in the IR and SR layers, an amount of high viscosity HPMC as the matrix forming polymer, and other conventional excipients all falling within the scope of claim 1 of the Patent (and the other asserted claims). Formulations C and D are example formulations. But the invention is not limited to matters discussed or described in connection with those examples.
677 The invention disclosed in the Patent is properly summarised on p 4 line 27 to p 4a line 8. This consistory clause identifies a bilayer tablet with a particular IR:SR paracetamol ratio, a matrix forming polymer (or a mixture thereof) and a unique in vitro dissolution profile. In the JER question 45(b), Dr Mooney and Professor Davies agreed that:
The patent teaches that it has surprisingly been discovered that such an advantageous pharmacokinetic profile can be provided by a two phase (immediate release and sustained release) formulation of paracetamol which satisfied a unique in vitro dissolution profile … . The patent teaches that the formulation contains a matrix forming polymer (or a mixture of matrix forming polymers) in the sustained release phase.
678 The invention disclosed did not limit the amount of matrix forming polymer present. At p 6 lines 1 to 5, the specification identified that the amount of matrix forming polymer in the sustained release phase was to be selected so as to provide the desired in vitro dissolution rate. The following passage at p 6 lines 7 to 9 then identified suitable amounts of matrix forming polymer. These lines provide support for claims 10 and 11 as the passage at p 5 lines 23 to 32 provides support for claims 6 to 9. These passages do not require a limitation to be present in claims 1 to 5.
679 Claims 1 to 3 simply require a matrix forming polymer. This is consistent with the disclosure in the specification at p 6 lines 1 to 5. Potential amounts of matrix forming polymer required to obtain the in vitro dissolution profile required by the claims are known to the skilled person or can be ascertained as the specification discloses. Further guidance as to the proportions of paracetamol and the amount of matrix forming polymer, although discussed in the body of the Patent specification at p 6 lines 1 to 9, does not form part of the essential characterisation of the invention disclosed.
680 Contrary to Generic Partners’ submission, claim 1 consistently with the disclosure in the specification as a whole, claims a unique dissolution profile and sets out the parameters necessary to produce that profile, relevantly including the relative amounts of paracetamol in the IR and SR layers and the inclusion of a matrix forming polymer in the sustained release layer. Moreover, because the dissolution profile is part of the essential characterisation of the invention, it is redundant for the amount of matrix forming polymer to be too. The skilled addressee understands that the amount of polymer is connected to the in vitro dissolution profile specified in the claims. It may be noted that in one sense the claim is not silent about the amount of matrix forming polymer required.
681 Finally, in relation to the consistory clause in the Patent that was added after the priority date, that clause did not change the nature of and is consistent with the invention disclosed in the body of the specification as a whole, including the original consistory clause. Further, the Patent explains at p 5 line 23 that “suitably, the sustained release phase comprises a matrix forming polymer” and provides examples of such polymers from p 5 lines 26 to 32. But there is nothing to suggest that the invention disclosed is narrower than the invention claimed.
682 In summary, I reject Generic Partners’ fair basis argument.
Sufficiency (section 40(2)(a))
683 The respondents have put their lack of sufficiency argument on three bases:
(a) First, that there was insufficient description in the Patent of any matrix-forming polymer other than HPMC in the sustained-release layer. Examples 2 and 3 (Formulations C and D) both use HPMC. It is said that there was no other disclosure of any other formulation, although the specification lists other matrix-forming polymers at p 5 lines 26 to 32.
(b) Second, that there was insufficient description of any formulation with an in vitro dissolution profile other than Formulations C and D. The in vitro dissolution profile for Formulation C was given at p 13 in Table 2 and Formulation D was at p 15, Table 3. The dissolution rates given there were precise figures, not expressed to be within a range. There was no description of how to make any other formulations encompassed by the claimed ranges. It is said that the broader ranges were selected simply on the basis of an “arbitrary” margin above and below these examples.
(c) Third, that there was insufficient description of the apparatus used to determine the in vitro dissolution profile. It is said that this failure went to the heart of the ability to work the patent. But if “basket” means basket, as I find (by reference to the USP type I apparatus basket), this contention is not made good.
684 Section 40(2)(a) of the Act in its then relevant form provided that a complete specification must “describe the invention fully including the best method known to the applicant of performing the invention”. The requirement that the invention be described “fully” imported the requirement of sufficiency of description.
685 The “invention” for the purposes of s 40(2)(a) is the embodiment(s) which is described and around which the claims are drawn. Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [19] and [21] approved that formulation and its application to s 40(2)(a) of the Act.
686 In SNF (Australia) Pty Ltd v Ciba Speciality Chemicals Water Treatments Ltd (2011) 92 IPR 46 at [234], Kenny J explained:
A specification is not insufficient merely because some experiment of a routine character (as distinct from prolonged study of matters presenting initial difficulty) is necessary in the particular case … Nor is a specification insufficient because it fails to give detailed instructions as to matters which a “practical person … would naturally settle, and expect to have to settle … himself”, provided he “would find no difficulty in so doing”.
687 There is no requirement for the patentee to provide an explanation of how the invention works. To this end, the Full Court in AstraZeneca AB v Apotex Pty Ltd (2014) 226 FCR 324 said at [205] per Besanko, Foster, Nicholas and Yates JJ:
although the complete specification must describe the invention fully, including the best method known to the inventor of performing the invention, it does not follow that the inventor must explain how he or she arrived at the invention. It is the invention itself that must be fully described, not the route that was travelled by the inventor to arrive at it. Again, the way in which the inventor came to the invention described and claimed may also have an evidentiary significance. However, an invention may be the result of chance or luck as much as long experiment and the question whether an invention, or an alleged invention, involves an inventive step is an objective one.
688 Only one embodiment within each claim need be enabled for sufficiency purposes. An invention is sufficiently disclosed if a skilled person could make a single embodiment of the invention which falls within the scope of the claims.
689 The test for sufficiency was formulated by the High Court in Kimberly-Clark at [25]:
The question is, will the disclosure enable the addressee of the specification to produce something within each claim without new inventions or additions or prolonged study of matters presenting initial difficulty?
690 In Lockwood (No 1), the High Court said at [60] citing its earlier decision in Kimberly-Clark (Kimberley-Clark at [25]):
For the purposes of s40(2)(a), it is not necessary for the inventor to disclose all the alternative means; it is enough that there is disclosure in the sense of enabling the addressee of the specification to produce something within each claim without new inventions or additions or prolonged study of matters presenting additional difficulty.
691 The submission of the respondents that the statement of principle in Kimberly-Clark should be understood as being limited to claims to a “single product with a particular feature” is in my view contrary to Kimberly-Clark and Lockwood (No 1). Those cases contain no recognition of such a limitation. The enablement of a single embodiment within each claim is sufficient for the purposes of s 40(2)(a).
692 Apotex has contended that Kimberly-Clark and Lockwood (No 1) each only concerned a simple product. It accepted that in such cases it may be sufficient for the patent to contain “such instructions as will enable all those to whom the specification is addressed to produce something within each claim”. However, it is said that the High Court did not consider the present question, where the claims encompass multiple formulations of the pharmaceutical composition, including as to the amount of paracetamol in each layer, the type and amount of matrix-forming polymer and as to the in vitro dissolution profile of the formulation when tested in accordance with the claimed dissolution apparatus at certain time intervals. It is said that each of these features is claimed across a broad range, so that the claims encompass multiple formulations.
693 In this context, Apotex has made reference to Tramanco Pty Ltd v BPW Transpec Pty Ltd (2014) 105 IPR 18. But the claims considered in Tramanco are different. The claims in Tramanco were to a method for logging the performance of a vehicle suspension and determining one or more parameters selected from the group consisting of A, B and C. The claim included methods for determining only one parameter out of A, B, C, combinations of two parameters A+B, A+C, B+C and all three parameters A+B+C. Contrastingly, the claims of the Patent are to ranges in a dissolution profile. They are not options of different parameters. The observations of Nicholas J at [207] as to the need to enable each of the options of parameters included within the claims in Tramanco are inapplicable. Claim 1 of the Patent relevantly refers to a dissolution profile, albeit that it is expressed in terms of ranges over the three time intervals.
694 The Patent provides sufficient information to enable a skilled formulator to produce a formulation with a profile which satisfied regulatory acceptance targets for replicating the profile of Formulation C (and Formulation D) without new inventions or additions or prolonged study of matters presenting additional difficulty.
695 Professors Davies and Tucker each accepted that the Patent provided enough information to allow them to make a Formulation C (and D) having the percentage (approximately) of paracetamol released at 15, 60, 120 and 180 minutes as Formulation C (and D) as specified in table 2 (and 3) of the Patent. Formulations C and D each fall within claims 1, 2 and 3 of the Patent. Accordingly, for that reason alone, the Patent satisfies the test for sufficiency.
696 But even if the test for sufficiency was to be constrained as asserted by the respondents, the evidence of both Professors Davies and Tucker is that the Patent provided enough information to allow them to make a formulation:
(a) having the percentage of paracetamol released at 15, 60 and 180 minutes as specified in claims 1, 2 and 3 of the Patent; and
(b) comprised of a matrix-forming polymer in the sustained release layer other than HPMC, having the percentage (approximately) of paracetamol released at 15, 60 and 180 minutes as specified in claims 1, 2 or 3 of the Patent.
697 In relation to the third ground, the respondents contend the following. I will park for the moment my conclusion that “basket” means basket.
698 It is said that a “peculiarity” of the invention as described and claimed is the inclusion of the in vitro dissolution profile in accordance with a particular test as an attribute or feature of the pharmaceutical composition. But, so it is said, the in vitro dissolution profile is not an intrinsic characteristic of the pharmaceutical composition. An intrinsic characteristic of the composition might be, for example, its solubility in the conditions of the stomach and then in the gut. However the in vitro dissolution profile claimed is simply one way of measuring the rate at which the composition dissolves under certain conditions, whether in a basket or cylinder with a particular grade of mesh to allow the solvent to pass through, that is agitated over time. But because the dissolution profile depends upon temperature, solvent, concentration of solvent and the hydrodynamics of the contact between the solute and the solvent, it is said that all these conditions must be specified with precision. Now as to the hydrodynamics, the Patent specifies “USP type III apparatus reciprocating basket, with 250 ml of 0.1M HCl at 37C set at a cycle speed of 15 strokes/min”. Generally, the dissolution profile values described are specific to dissolution values measured with a particular apparatus under particular conditions.
699 The rate of dissolution of the composition will be affected by the choice of test apparatus. The choice of apparatus will provide different hydrodynamics and these differences will have an impact on the dissolution results obtained. If the same tablet is tested, the dissolution results from a test with one apparatus will not be the same as the results from a different apparatus. There is no established correlation between dissolution tests measured using different USP apparatus. The differences can be small or large and are not predictable. Further, even within the one apparatus, variability in the measured profile of up to ±10% is considered normal, although ±5% is more ideal.
700 It is said that the claims, insofar as they concern the in vitro dissolution rate integer, are akin to a limitation by result claim. Accordingly, this puts the burden on the skilled addressee of seeking to make the composition in order to determine where the boundaries of the monopoly lie. In such a case, it is said that the specification must not only enable the addressee to carry out the invention claimed in the form of any particular embodiment but also enable the addressee of the Patent to ascertain whether the particular result concerned is or is not secured by forms of the invention lying near the boundaries of the claim. It is said that such a limitation is permissible only if it is sufficient to characterise the construction of the article claimed: Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 at 480 per Barwick CJ and Mason J. I accept these contentions in the generality with which they have been expressed.
701 In such a case, it is said that the specification cannot be sufficient unless it discloses the necessary information as to the type of departures from the preferred embodiment that will fall within or without the claims.
702 Generally, it is said that for a claim limited by result, particular care must be taken when claims of this sort are adopted to give in the specification all possible assistance in determining what does, and what does not, give the useful result concerned.
703 If my “basket” construction is correct, that is “basket” means basket and by reference to a USP type I apparatus basket, then the respondents’ sufficiency argument on the apparatus question fails in my view.
704 After the experts’ conclave, the expert opinions as to the identity of the test apparatus remained divided as follows:
(a) a standard USP type III apparatus reciprocating cylinder dissolution apparatus (Professors Dressman and Davies);
(b) a USP type III apparatus with a reciprocating basket as a modification (Professors Fassihi, Tucker and Dr Mooney).
705 The experts agreed that the Patent specification did not expressly disclose mesh size, mesh material or mesh position to carry out the claimed dissolution test. But if “basket” meant basket, the USP type I apparatus specification for basket would, for the skilled addressee, provide the source information for most of the variables.
706 Given the importance of the reproducibility of the dissolution test, I accept that in sufficiently characterising the construction of the composition claimed, certain features of the apparatus needed to be specified.
707 In Colgate-Palmolive Co v Cussons Pty Ltd (1993) 26 IPR 311 a claim that included an integer defining a detergent product by reference to the percentage of the “flowability of clean dry sand” failed for want of sufficiency. The instructions on how to perform this test were inadequate. The claim in suit was to a particulate composition said to have a “flowability at least 70% of that of clean dry sand”. The jars used for testing the flowability required a vent. The patentee’s expert’s evidence that an ordinary skilled worker would appreciate that a vented jar was required was substantially weakened by the fact that the patentee had supplied the expert who was to conduct the relevant flowability test with a non-vented jar. The trial judge found that the flowability test used by Colgate was well-known by those within Colgate, but not to the skilled worker outside Colgate. In addition, the flowability of dry sand varied with the sand used and a particular sand was not identified in the claim. The trial judge found that the ordinary skilled worker could not choose the appropriate sand without additional information. But those circumstances are not the present case. In the present case the skilled dissolution tester would be able to make an appropriate selection of the relevant parameters without new inventions or additions or prolonged study of matters presenting initial difficulty.
708 Let me elaborate further on the mesh question. If “basket” means basket (and by reference to USP type I apparatus), I do not need to go further. But let us assume for the sake of the argument that “basket” can be transmuted to cylinder.
709 The purpose of the mesh is to retain the dosage form within the reciprocating cylinder. The mesh configuration must also allow adequate distribution of the dissolution medium into and out of the reciprocating cylinder. A mesh with larger holes may (undesirably) allow more undissolved particles to pass through the mesh into the lower part of the glass vessel, whilst a mesh with smaller holes may become blocked by undissolved particles, restricting flow of the medium into and out of the reciprocating cylinder. But contrary to the respondents’ submissions, at the priority date it was known that mesh size could be identified by skilled dissolution technicians as a matter of routine and that there would not be a material difference in dissolution results across appropriate mesh sizes.
710 Rohrs, B.R. et al “USP Dissolution Apparatus 3 (Reciprocating Cylinder): Instrument Parameter Effects on Drug Release from Sustained Release Formulations” Journal of Pharmaceutical Sciences 84(8) 922 August 1995 studied the effects of various USP type III parameters on drug release from sustained release formulations, each of which were matrix formulations containing HPMC as a “rate controlling hydrophilic polymer”. The parameters studied included mesh size and placement, dissolution media composition and reciprocation (or dip rate). Rohrs et al also evaluated parameter effects including mesh size on drug release from sustained release formulations. The sustained release formulations tested “spanned the continuum of release mechanisms from pure erosion-controlled release [SR ibuprofen and SR flurbiprofen] to pure diffusion-controlled release”. SR alprazolam was said to have both erosion and diffusion components to drug release, with higher strength (3mg) formulations having significantly more erosion component than lower strength (0.5mg). 15mg SR adinozolam mesylate was said to have primarily diffusion controlled release and 7.5mg tablets were anticipated to also exhibit dissolution controlled release. A multiparticulate bead formulation (purely diffusion controlled release) was also tested. Rohrs et al noted that the USP dissolution apparatus 3 was supplied with four mesh sizes: 160, 100, 40 and 20. Rohrs et al reported that an initial experiment revealed that when the 160 and 100 sizes were used as the top screen the cylinders failed to drain and the 160 and 100 sizes were removed from further tests of top mesh. A first test was conducted on SR ibuprofen. It was chosen because drug release from an erosion controlled device would be more influenced than a diffusion controlled device by changes in hydrodynamic flow. On that basis Rohrs et al concluded that if a parameter had no effect on release from the erosion controlled device it could be eliminated from further study designs. The SR ibuprofen tests tested three parameters: three reciprocation rates, three top meshes (20, 40 and none) and four bottom meshes (20, 40, 100 and 160). The SR ibuprofen results were set out in figure 3. The authors concluded from the SR ibuprofen tests that the “[b]ottom mesh had no significant influence on the drug release profile”. The bottom mesh was held constant at 40 for subsequent trials. The results of the subsequent tests were set out in table 2. Table 2 shows that the top mesh size (20 or 40) did not affect the formulations with diffusion controlled release.
711 Moreover, Professor Dressman considered that mesh size selection was a matter of routine for an experienced dissolution tester. In the context of the dissolution claims in the Patent, she would have ruled out the smaller mesh sizes as they were likely to be so small that they risked becoming clogged up with undissolved particles and because flow of the dissolution medium through the mesh might be restricted. Professor Dressman would have chosen a 40 mesh screen, which she considered to be an intermediate screen, to carry out the claimed dissolution test. This was also the mesh size that her research group was using to carry out USP type III dissolution testing in 2000. Professor Dressman’s approach was supported by the findings reported by Rohrs et al.
712 Further, during cross-examination, Professor Dressman explained:
So that’s why most people say, “Okay, if I want to go with the mesh size that’s appropriate and I can use it for a tablet or a pellet then I will pick probably the 40”. But I really — for a tablet type of dosage form, I don’t see any difference between using a 20 and a 40.
713 Further, Professor Dressman believed that she had read Rohrs as at the priority date and in cross-examination said that:
Everybody looked at the Rohrs paper and said, “Okay. We had better go with the 40 mesh”.
714 Professor Fassihi, who had also read Rohrs at the priority date, also gave evidence on the topic:
MS ROFE: And similarly with mesh size, as we’ve discussed, you use commonly the 20 mesh and sometimes the 40 mesh? --- I have used number 20 and if, for example, I had difficulty, if it didn’t work for some reasons and the reasons are multiple, then we would change. We — we — obviously we have to optimise to get the right mesh size. Yes.
715 Professor Fassihi also noted with reference to S Missaghi and R Fassihi, “Release characterization of dimenhydrinate from an eroding and swelling matrix: selection of appropriate dissolution apparatus” International Journal of Pharmaceutics 293 (2005) 35:
PROFESSOR FASSIHI: The article says following USP apparatus 3 with all the details that are provided. So in that case, it is mesh number 20.
MS ROFE: So the person skilled in the art would know to start with mesh number 20? --- I believe so.
And if there was a problem with mesh number 20, then they could tweak it and try a different mesh? --- That is how experimentations need to be done.
716 More generally, Professor Dressman noted that of all the apparatus parameters considered in the Rohrs article (reciprocation rate, mesh size and configuration), reciprocation rate had the greatest influence on dissolution results. The claims prescribe the reciprocation rate as 15 strokes (dips) per minute, which Professor Dressman considered would provide a “robust interaction between the dosage form and the dissolution medium at an intermediate mesh size”. Professor Dressman also explained that “Once you set the dip rate, then you will have a defined hydrodynamic between the reciprocating cylinder and the vessel in the fluid”, as long as an appropriate volume of dissolution media is used. She also said that:
If you choose an appropriate mesh size … then that choice between — in that range I don’t think would make any difference to the dissolution results. If you go and use a silly mesh size, like, an extremely fine one where you know it’s going to interfere with the hydrodynamics … then that may become the overriding factor. But you’ve got to use some commonsense.
717 Professor Fassihi has only speculated that there may be a difference in dissolution results between each mesh size; I should note that in the following section dealing with lack of clarity I have disposed of some of his more extreme suggestions. Further, if there was a difference, that could readily have been established by experiment. But the respondents chose not to conduct such an experiment. Given the experiments reported upon by Rohrs, and the absence of any experiments, whether conducted by the respondents or reported in the literature, there is no basis upon which I can draw a conclusion on the balance of probabilities that the use of a 20 mesh may have had a material impact on the dissolution profile of the formulation claimed.
718 For completeness I have put to one side Professor Tucker’s comment that it “was not a matter of routine” for him to select an appropriate mesh. Professor Tucker had little if any experience as at the priority date, or indeed since, in selecting or making any decision as to the use of a mesh with a USP type III apparatus. He had to concede that he had never used the USP type III apparatus and never considered what mesh size might be appropriate to use with the USP type III apparatus.
719 In summary, a skilled addressee, in the light of Rohrs, the knowledge of the mesh to be used with the USP type III apparatus discussed above and with knowledge of the tablet to be tested (a bilayer sustained release paracetamol tablet with a matrix forming polymer in the sustained release layer) would have been able to make appropriate selections of mesh size as a matter of routine without new inventions or additions or prolonged study of matters presenting initial difficulty.
720 For the reasons set out above, the respondents’ sufficiency attack must fail in relation to mesh size assuming we are talking here of a reciprocating cylinder being used i.e. a compendial USP type III apparatus. As to the other matters i.e. mesh material and mesh position (and the absence of any specification thereof), the respondents’ complaints have no substance when one considers the text and diagram in the USP for USP type III apparatus that one would expect a skilled addressee to have regard to.
lack of Clarity (section 40(3))
(a) Legal principles
721 A valid claim is required to define with sufficient certainty the scope of the monopoly being claimed (s 40(3)). Given that a patent is a public instrument, it must define the claimed monopoly with sufficient precision and in such a way that it is not reasonably capable of being misunderstood. Members of the public must be able to know the “exact boundaries of the area within which they will be trespassers”: Electric & Musical Industries Ld v Lissen Ld (1939) 56 RPC 23 at 39 per Lord Russell of Killowen.
722 Now lack of precise definition will not be fatal to the validity of a claim as long as it provides a workable standard suitable to the intended use. But as stated in Kauzal v Lee (1936) 58 CLR 670 at 685 per Dixon and McTiernan JJ:
[v]agueness of description, want of particularity and evident indistinctiveness of thought may be the source of so much uncertainty as to the scope of the monopoly that the claim fails to fulfil the requirement of stating with definiteness to what the patentee is exclusively entitled.
723 A claim is clear if either there is no ambiguity therein or the ambiguity can be resolved by properly construing the claim applying the principles that I have set out earlier.
724 But I accept the proposition that a claim is bad if no reasonably certain construction can be given to it or it is fairly and equally open to diverse meanings. I also accept that a court is not bound to find a meaning for a claim nor to approach a claim with the conviction that its language is capable of a reasonable construction when carefully examined. A claim may be bad for uncertainty, even though a court could find its true meaning, if it is so obscure that its construction remains a matter of doubt.
725 In Flexible Steel Lacing Co v Beltreco Ltd (2000) 49 IPR 331, Hely J held that each of the method and product claims for pulling lagging used in conveyor systems was invalid for lack of clarity. In respect of the method claim, he said at [107]:
The method claim is fairly open to more than one meaning not because of grammatical problems but because, even to a skilled reader, it would not be clear which of two methods claim 13 describes.
726 In respect of the product claim, he held at [131]:
Thus, I conclude that the product claim is obscure; it is fairly and equally open to diverse meanings, namely that the sipes run at right angles across the strip, on the one hand, or that the sipes run along the length of the strip on the other. Another possibility is that the claim embraces both. Sometimes, ambiguity or insufficiency in description can be resolved by a skilled addressee through the application of commonsense and common knowledge: cf Innovative Agriculture Products Pty Ltd v Cranshaw (1996) 35 IPR 643 at 666. I do not think that this is such a case.
727 Accordingly, if the claim is open to more than one reasonable construction, including which of two dissolution test methods are described, it is bad for uncertainty.
728 Finally, if a claim is clear, it is not to be made obscure or treated as obscure by taking elements of a preferred embodiment not referred to in the claim and artificially creating obscurity.
(b) Application of principles
729 There are two issues to consider.
730 The first question to consider is the cylinder/basket question.
731 I have held that claim 1 means what it says. Accordingly, “basket” means basket. There is accordingly no lack of clarity. It seemed to be accepted by the respondents that “basket” is to be construed as a USP type I apparatus basket, which would be used in place of the cylinder. As I understood the respondents’ arguments, they did not suggest otherwise. In particular they did not submit, as I understood their case, that if I construed claim 1 to refer to “basket” that a s 40(3) issue would still arise. Rather, what they submitted was that if I could not resolve the question between basket and cylinder, then a lack of clarity would arise.
732 Accordingly, if “basket” is the correct construction, then no lack of clarity arises. The term “basket” means what it says, albeit that it was erroneous.
733 But let it be assumed that some polytropic fairy dust can be sprinkled over the word “basket” to transform it into “cylinder”. Then the respondents argue that there is a lack of clarity as there has been a failure to specify mesh size. I have dealt with these arguments earlier, but let me repeat some of them again including disposing of Professor Fassihi’s outlier possibilities.
734 Apotex has contended the following:
(a) For a USP type III apparatus, one of the variables that is left for the practitioner to specify is the mesh size for the ends of the reciprocating cylinder. In contrast, for a USP type I apparatus the USP specifies a default mesh size of 40 unless a different mesh size is specified.
(b) Mesh for a USP type III apparatus is available in a number of standard sizes, including 20, 40, 78 and 100.
(c) Mesh size is an important criterion in one sense for a USP type III apparatus because the size can affect the hydrodynamics of the fluid that moves past the tablet as the cylinder reciprocates up and down. Further, the size can affect whether drug particles clog up the mesh, affecting the hydrodynamics, or alternatively pass through the mesh and out of the reciprocating cylinder to interact with solution with different hydrodynamics.
(d) Professor Fassihi gave evidence that while mesh sizes 78 and 100 are not commonly used, it is possible that they would be appropriate for use with the relevant paracetamol tablets.
735 I must say that I found Professor Fassihi’s suggestion that mesh sizes 78 and 100 were realistic choices as entirely implausible. On any view, which a skilled addressee can be taken to have known, such smaller mesh sizes would have risked becoming clogged up with undissolved particles thereby restricting the free flow of the dissolution medium. Anybody knowing what they were doing would not have used mesh size 78, let alone mesh size 100.
736 In my view only mesh sizes 20 and 40 would have been considered appropriate by the skilled dissolution tester and both would be expected to produce similar results.
737 As is apparent from the evidence there appear to be two commonly used mesh sizes (20 and 40) used in testing using USP type III apparatus. Skilled dissolution testers using USP type III apparatus commonly have a preferred mesh size which they use unless it proves inappropriate for the dosage form being tested. Professor Dressman would start with 40, and Professor Fassihi would start with 20. There is no evidence that a choice of either would produce materially different results. In fact, in Professor Dressman’s opinion “it wouldn’t make any difference which one you use”. Further, it was Professor Dressman’s evidence that she “wouldn’t have gone to a 78 mesh size” and “[n]obody else did either”. As Professor Dressman said, reasonable mesh sizes (i.e. 20 or 40) would not:
make any difference to the dissolution results. If you go and use a silly mesh size, like, an extremely fine one where you know it’s going to interfere with the hydrodynamics … then that may become the overriding factor. But you’ve got to use some commonsense.
738 Further, the key aspects of the USP type III test that affect the hydrodynamics of a test are the dissolution media volume and the stroke rate. These features are specified in the claims of the Patent.
739 Professor Dressman’s evidence was that she expected the hydrodynamics to be such that there would be no clogging of the 20 or 40 mesh and that either would be appropriate as she would not expect there to be an ascertainable difference in the results obtained using the 20 or 40 mesh. To the extent that Professor Fassihi speculated that 20 rather than 40 mesh would make an appreciable difference, it was open to him and Apotex to test that speculation at the time of the May and/or June experiments. No such experiments were undertaken. As I have indicated, the respondents conducted no experiments to show that the mesh size selected by the skilled dissolution tester would lead to statistically significant different results, instead relying on speculation.
740 As GSK rightly contended, the Patent is addressed to the skilled addressee, not an uninformed “member of the public”. The skilled dissolution tester can select an appropriate mesh size without new invention or addition or prolonged study of matters presenting initial difficulty. The claims of the Patent provide a workable standard suitable to their intended use and are clear to the skilled dissolution tester to whom they are addressed.
741 The respondents’ lack of clarity arguments fail. Let me turn then to the argument of a failure to define the invention.
Failure to Define the invention (section 40(2)(b))
742 Section 40(2)(b) requires the complete specification “to end with a claim or claims defining the invention”.
743 In General Tire & Rubber Co v Firestone Tyre and Rubber Co Ltd (1971) 1A IPR 121, the Court stated at 167 that “the issue of definition is to be considered as a practical matter and little weight is to be given to puzzles set out at the edge of the claim which would not as a practical matter cause difficulty to a manufacturer wishing to satisfy himself that he is not infringing the patent”. The Court also observed that allowances should be made for any difficulties of the case, so that an alleged issue of want of definition should always be considered in relation to the particular facts. It concluded in the following terms (167 and 168):
It is clear in our judgment that the question whether the patentee has sufficiently defined the scope of his claims is to be considered in relation to the facts of each case, that allowance is to be made for any difficulties to which the circumstances give rise, and that all that is required of the patentee is to give as clear a definition as the subject matter admits of. It is also clear in our judgment that, while the court is to have regard to all the relevant facts, the issue of definition is to be considered as a practical matter and little weight is to be given to puzzles set out at the edge of the claim which would not as a practical matter cause difficulty to a manufacturer wishing to satisfy himself that he is not infringing the patent. We accept also that definition of the scope of a claim is not necessarily insufficient because cases may arise in which it is difficult to decide whether there has been infringement or not provided the question can be formulated which the court has to answer in the issue of infringement.
744 A claim will be bad if it fails to define the monopoly claimed so that the skilled addressee of the patent can know the exact boundaries of the area within which they will be trespassers.
745 Apotex submits that claim 1 and all dependent claims fail to define the invention by reason of the use of the words “USP type III apparatus, reciprocating basket …”. The addressee would not understand what dissolution test was specified and critically what apparatus was used. In addition, the test does not specify the materials and size of the mesh to be used, which is said to be “critical” in determining a particular dissolution profile.
746 As noted above, in my view “basket” means basket. And as I have said, “basket” should be taken as being a reference to the USP type I apparatus basket used as a substitute for the cylinder(s) in each case. As so construed therefore, there has been no failure to define the invention. Alternatively, if “basket” is to be taken as a reference to cylinder, then for the reasons that I have set out earlier I reject Apotex’s argument that the dissolution test method has not been adequately specified.
failure to describe Best Method (section 40(2)(a))
747 The best method attack has mutated during the course of these proceedings. Initially, so it was said, the patent applicant knew that Formulation D was better than Formulation C and that Formulation D had not been disclosed to the required level. That argument has been abandoned. It was without substance.
748 Now the respondents assert that each of the grade and specific viscosity of the high viscosity HPMC and the granule type was “a crucial part” of the best method of performing the invention and that the patent applicant had failed to disclose the same. This assertion was technically more nuanced, but it still fails.
749 The respondents contend that the complete specification failed to describe the best method known to the patent applicant and rely on the following chain of reasoning:
(a) First, the patent applicant used a particular grade and specific viscosity of HPMC and a particular granulation end point to make Formulations C and D. That proposition may be accepted.
(b) Second, Formulation D had an in vitro dissolution profile that very closely resembled what the patent applicant considered to be the “ideal” release profile intended to deliver the ideal in vivo pharmacokinetic properties. That proposition may also be accepted.
(c) Third, the patent applicant had tested its Formulations C and D in humans and found that those formulations worked. That proposition may also be accepted.
(d) Fourth, the method of making Formulations C and D disclosed in the complete specification described a method of making Formulations C and D using “high viscosity HPMC” and a “conventional” method of granulation only. That proposition is also correct.
(e) Accordingly, so it was concluded, the method known to the patent applicant of making Formulations C and D was better than that disclosed in the Patent because it eliminated experimentation involved in the skilled addressee trying a variety of possible grades of high viscosity HPMC and granulation end points in order to make Formulations C and D as made by the patent applicant, which was “guaranteed to work”. In one sense this conclusion is correct and flows from the above premises. But the lens itself is astigmatic in lacking the requisite focus.
750 The respondents’ best method attack fails in limine. The respondents have incorrectly identified the “invention” for the purposes of their best method argument. The invention is not Formulation C or D per se. The invention is defined by the claims. Formulations C and D were examples within the claims. It is their exposition in the body of the specification that satisfied the best method requirement.
751 In my view, the patent applicant disclosed the best method known to it of performing the invention by disclosing Formulations C and D in sufficient detail to enable a skilled addressee to make the same. Formulations C and D were themselves the best method to make or perform the invention known to the applicant. Not only that, but the experts in this case in fact agreed that they could make Formulations C and D from what had been disclosed in the specification. The respondents’ attack can be reduced to little more than saying that there has been a failure to disclose the best method of making the best method of performing the invention. That compounded idea and ideal is not the statutory requirement for disclosure.
752 But I also have other difficulties with the respondents’ arguments. First, the respondents seemed to confuse the distinction between the patent applicant’s work and its choices in making a commercial product designed for appropriate marketing and regulatory requirements and to reduce variability in commercial production. But I am dealing with the separate concept of what had to be disclosed to meet the statutory requirement of s 40(2)(a). It cannot reasonably be suggested that the detail embodied in the former design or functionality is the same detail that must be disclosed to meet the statutory requirement. No case has suggested as much. Moreover, to so suggest is conceptually confused and involves a categorical error. Second, the respondents trawled through the minutiae of the patent applicant’s technical working papers spanning some years in an attempt to make out its case for non-disclosure. This had various problematic aspects. The problem of attribution to the patent applicant of states of mind of individuals and the problem that some of what was set out in the early working papers did not necessarily demonstrate the patent applicant’s state of mind at the later relevant date were largely overlooked. Third, Mr Cordiner for Generic Partners put his own technical interpretation on some of the working papers that was in some respects contestable absent detailed expert evidence thereon, although I must say that for my part many of his interpretations were not incongruent with how I would also read this material. For my part, and for present purposes, I am prepared to pass over these second and third forensic difficulties, but the first point cannot be overlooked.
(a) The separate aspect of best method
753 Section 40(2)(a) at the relevant time required that a patent applicant describe the invention fully, including the best method known to it of performing the invention. This second element is separate to sufficiency. On the question of the requirement to disclose the best method, I am bound to follow what the Full Court recently said in Les Laboratoires Servier v Apotex Pty Ltd [2016] FCAFC 27 (Bennett, Besanko and Beach JJ). In particular, at [108] and [109] the Court said:
From the above authorities the following principles may be gleaned:
• Different policy reasons support the obligation to describe the invention fully and the obligation to provide the best method known to the patentee of performing the invention. The purpose of the former obligation is to circumscribe the monopoly granted to the patentee; the purpose of the latter is to allow the public the full benefit of that invention when the monopoly expires.
• Although a patentee might not be explicitly required to act in good faith, principles of good faith underlie the best method requirement.
• Even where legislation has not included an explicit “best method” requirement, courts have considered it to be a separate and additional requirement to the obligation to provide a sufficient description of the invention.
• The nature and extent of the disclosure required to satisfy the best method requirement will depend on the nature of the invention itself. Accordingly, a distinction between products and processes that ignores the specific features of the invention claimed is unhelpful.
It follows that the courts have recognised the necessity for a patentee to include in the specification not only sufficient instruction to work the invention but also the best method of performing the invention known to him, her or it. This requirement has been developed by the courts over time and has been reflected in statutory provisions, such as in s 40(2)(a). Where the best method question has been addressed by the courts, the separate or additional nature of the requirement has been confirmed, including by the Full Court. We see no reason to depart from this view.
754 By way of context, the Court also said at [103] to [106]:
The authorities that have dealt with s 40(2)(a), its precedents and equivalents, must be understood in context. The first, and most important, factor is the nature of the invention being described and claimed. Servier divides this simply into products and processes but that is not sufficient. It is necessary to understand the invention itself in order to appreciate what is required of an inventor by way of disclosure in the specification in order to secure a monopoly from the public. In some cases, the claim to a product will require no description of the method of obtaining it and it can be left to the skilled worker (as in AMP v Utilux). In other cases, the product claim, properly understood, will require sufficient directions in order to obtain the monopoly.
The Full Court in Firebelt observed that the statutory obligation was an obligation to disclose the best method of performing the invention. Their Honours also said (at [53]), referring to the requirement of s 40(2) of the Act (in its totality), that the patentee is required to give the best information in his (or now her) power as to how to carry out the invention (emphasis in the original). The Full Court was of the view that the requirement is ordinarily satisfied by including a detailed description of one or more preferred embodiments of the invention and concluded in that case that, by taking account of the rest of the specification together with the figures, an embodiment was depicted of the claimed device.
In Patent Law in Australia Dr Bodkin says (at [5270]) that the requirement to describe the best method known to the Patentee:
“is to supplement the necessity for a full description by requiring the patentee to disclose additional information which, if not available to potential users of the invention, could place the patentee in a stronger competitive position even though no patent protection existed [i.e. when the patent ceases to be in force]. It is included to help ensure good faith on behalf of the patentee”.
Citing Firebelt (at [48]), the author continues (at [5280]) to give the opinion that the patentee must disclose what it, subjectively, perceives to be the best embodiment of the invention known to it at the relevant time, whether or not a different embodiment is later shown to be better. Further, what must be disclosed is the best practical method of carrying out the invention as distinct from the best method in theory (citing Van Der Lely at 56). That is, the requirement is to disclose the most effective means of carrying out the invention known to the patentee at the relevant time. The view is also expressed that it appears not to be necessary to describe the best method for making an apparatus or the best method of using an apparatus (citing Illinois Tool Works and Van Der Lely).
755 Further, the best method was required to be disclosed in the complete specification at the time of filing, being the best method then known to the patent applicant. It appears that there are differences in the authorities concerning the date for disclosure and the date for when knowledge is to be assessed. But in the present case and by reason of the way that the respondents have put their arguments, these are all just academic points in the present case as it is sufficient to dispose of the matter by looking at knowledge as at the date of filing.
756 As to the question of what must be disclosed when dealing with a product claim, the Court’s observations at [123] to [129] are important:
Section 40(2)(a) requires that the best method of performing the invention be provided. Perform is relevantly defined in the Macquarie Dictionary to include: “to carry out; execute, do”; and “to carry into effect; fulfil”. The meanings of “perform” in the Shorter Oxford English Dictionary are relevantly “execute, accomplish, do, (any action, operation or process undertaken or ordered)” and “make or construct (an object)”.
The key to understanding the obligation of the patentee is to understand that the section is directed to the method of performance of the invention. The monopoly is circumscribed by the claims but the nature of the invention is as described in the whole of the specification. This approach accords with that adopted by Lord Nicholls in Van Der Lely and by the Full Court in Firebelt.
Section 40(2)(a) expressly uses the word “method”. Method is relevantly defined in the Macquarie Dictionary as: “a mode of procedure” and “a way of doing something”.
There is no distinction drawn in the language of the statute between a product and a process in providing for the obligation to provide the best method of performing the invention.
Servier knew of the 1986 and 1991 methods and the crystalline form obtained from those methods. It knew that this form was suitable for pharmaceutical compositions and that it was in fact used in the stability study referred to in the Patent from which the alleged advantages of the invention are derived.
Both parties accept that s 40(2)(a) must be judged through the lens of both claim 1 and claim 2. The use of the product, perindopril arginine, in a pharmaceutical composition was an important aspect of the invention claimed in the Patent. That is made clear in the description in the specification. Indeed it could be said to have been the “raison d’etre” of the need to find a more stable product over perindopril erbumine. This was recognised by the primary Judge at [182].
It can be accepted that there are cases where the claim is to a product or class of products and the best method requirement is satisfied by a description of the best embodiment known to the patentee at the relevant time. It can also be accepted that there are cases where the claim is to a product and there is no requirement to provide a method of using that product. It is also the case that there is no requirement actually to have carried out the best method and that a prediction will suffice (New England Biolabs, Inc v Hoffmann-La Roche AG (2004) 63 IPR 524 at [33]). However, it is necessary to understand the invention itself (Expo-Net). As was succinctly stated by Lord Hoffman in Kirin-Amgen Inc. v Hoechst Marion Roussel Limited [2005] RPC 9 at [104]:
“in order to decide whether the invention has been fully enabled, you first have to decide what the invention is”.
Lord Hoffman was there addressing the sufficiency requirement, but the observation applies equally to the best method requirement. The nature of the invention will determine what is “best” in the circumstances. (emphasis in original)
757 Further, in relation to the product claimed in that case, namely, perindopril arginine, the Court said the following at [133] to [137] and [141]:
In the present case, as the primary Judge recognised, the point of the invention was the storage ability of the compound. That storage ability can vary depending on the nature of the salt that is formed, for example, perindopril erbumine did not give satisfactory storage, and can vary with the form of a salt. Accordingly, as his Honour said, the particular salt formation and the methodology to get that salt formation have more importance than might be the case where the claimed invention was a product for which these characteristics were irrelevant.
Perindopril arginine is generally a more stable product than perindopril erbumine and the claim is not to a specific form of the arginine salt. If Servier knew of a method that provides a form of the salt with the characteristics exemplified in the Patent, which characteristics provided the stated advantages of the invention over the prior art, it was incumbent on it to provide that method. This would relieve the skilled worker from making the choices within those necessarily made or available in a classical salification. The disclosure of the method known to Servier would not only have relieved the skilled addressee of confronting blind alleys and pitfalls which may not be uncommon in a general sense but also, and importantly, would tell the skilled addressee the methodology to achieve the form that obtains the result which constitutes the invention, that is increased stability and storage length. While claim 1 does not refer to any particular form of perindopril arginine, crystalline or otherwise, if Servier had a method that produced a product that was at least sufficiently crystalline or in a sufficiently good form so that it could be used in the API for the tablets used in the stability study described in the specification, that is precisely what should have been disclosed.
Accepting that there was no lack of sufficiency, the mere fact that a complete specification described a method which conveyed sufficient information to a skilled addressee to enable him or her to work the invention does not necessarily satisfy the Patentee’s additional obligation to describe the best method. The patentee has an obligation to include aspects of the method of manufacture that are material to the advantages it is claimed the invention brings.
In the present circumstances, the inventor, Mr Damien, had not characterised the products of the two methods that he utilised but he did know that those methods created the arginine salt in a useable form and, as a person skilled in the art, he knew that there were many alternatives available from which to choose. As the skilled worker, he knew that the method of classical salification was sensitive to choices such as the choice of solvent. He knew that some were likely not to be as good as others. That is consistent with the expert evidence, although it was not specifically put to Mr Damien.
The method of making the perindopril arginine affects the form of that compound and that of the properties of the compound itself, including its stability and usability of formulation. Its making can also involve unnecessary choices and difficulties.
…
The Patent expressly asserts that different forms of perindopril arginine can exist (see page 2 of the specification); and they would be included within claim 1. It is relevant that a crystalline form will be preferable for the purposes of the use as described in the specification, by reason of its properties. Servier relies upon the breadth of claim 1 and the fact that no particular form is specified. However, when the claim is read in the context of the specification as a whole, it is clear that the claimed class of compounds are described as a patentable invention because of their properties, including the use, as set out in claim 2, as a pharmaceutical composition comprising the arginine salt of perindopril and its hydrates. In these circumstances, there is an obligation on the Patentee to provide the best method for producing a form of perindopril arginine that will best fulfil the promises of the invention.
758 I should make two other observations at this point. First, in terms of knowledge, it is the patent applicant’s relevant subjective state of mind that must be proved by the party seeking revocation. Second, the best method known to the patent applicant need not be described as such in the complete specification.
(b) What is the invention?
759 An assessment of best method starts with the proper identification of the invention described in the patent.
760 The invention claimed in the Patent is a pharmaceutical composition which has a dissolution profile within the ranges specified in claims 1, 2 and 3. Both Professor Davies and Dr Mooney have agreed that (JER, question 45(b)):
The patent teaches that it has surprisingly been discovered that such an advantageous pharmacokinetic profile can be provided by a two phase (immediate release and sustained release) formulation of paracetamol which satisfied a unique in vitro dissolution profile … . The patent teaches that the formulation contains a matrix forming polymer (or a mixture of matrix forming polymers) in the sustained release phase.
761 It is the possession of the unique dissolution profile which enables the pharmaceutical composition to achieve the advantageous pharmacokinetic profile.
762 The specification describes two formulations, C and D, which have dissolution profiles falling in the middle of the ranges of the claims and which are example compositions of the invention. In my view the best method of carrying out the invention known to the patent applicant at the time of filing the complete specification was Formulations C and D. Further, Professor Tucker and Professor Davies have agreed that Formulations C and D have equal merits. It is also not in dispute that there is sufficient information in the Patent to allow a skilled person to make Formulations C and D. In one sense, and as GSK would contend, that is sufficient to dismiss this ground of attack on validity.
763 Each of Professors Davies and Tucker have agreed that they could make:
(a) a formulation having the percentage of paracetamol released at 15, 60 and 180 minutes as specified in claims 1, 2 and 3 of the Patent;
(b) Formulation C matching as close as possible the dissolution profile specified in table 2 of the Patent; and
(c) Formulation D matching as close as possible the dissolution profile specified in table 3 of the Patent.
764 Clearly, the invention is not a manufacturing process for the commercial production of a particular formulation, let alone a formulation comprising a particular grade of HPMC or granulation characteristics. The invention is to formulations having a unique IR:SR paracetamol ratio, a matrix forming polymer and dissolution profile. Such formulations are said by the specification to have an advantageous pharmacokinetic profile.
765 In essence, as I have said, Generic Partners has wrongly asserted that the invention is Formulation C or Formulation D itself and that the patent applicant has omitted disclosure of its best method by omitting manufacturing optimisation details relating to Formulations C and D. But the invention is a formulation with broader specifications than Formulations C and D. The patent applicant has given the best method known to it of performing the invention by in fact giving details of Formulations C and D, including their precise dissolution profiles. Further, no detail necessary for the skilled addressee to perform that best method of the invention has been omitted. Each of Professors Davies and Tucker accepted that they could each carry out the teaching in the Patent to obtain Formulation C or D within realistic formulation variation limits. The present case is quite distinguishable from Servier where the Court in that case was not in doubt that important details had been impermissibly hidden under the superficial label “classical method of salification”, which in one sense was a chemical black box concealing the practice of a dark art once one moved beyond the superficiality of saying that you could make a salt by mixing equal molar amounts of an acid with a base. As the Court said in Servier at [120], this was “wholly inadequate to describe the substantive content for a particular classical method of which the Patentee knew”.
766 Generic Partners has asserted that disclosure is required of not just the best example of the invention, but of a product optimised to minimise batch variability for quality control purposes in the manufacture of a commercial product. But this is incorrect and has no support in the authorities. The patent applicant clearly disclosed the design or essential elements of Formulations C and D as well as the precise performance of Formulations C and D as embodied by the formulations’ respective in vitro dissolution profiles. I agree with GSK that the use of a particular grade of HPMC and granulation end point are matters of routine referable to the commercial manufacture of Formulations C and D. Their non-disclosure did not trigger non-compliance with s 40(2)(a).
767 Before delving into the facts, let me make one other point at this stage. Generic Partners has sought refuge in the argument that the party seeking revocation of a patent for failure to comply with the best method requirement typically does not have access to any primary source of information regarding the patent applicant’s knowledge, other than that obtained by compulsory processes such as discovery. So much may be accepted. Accordingly, it is said that if the patentee does not adduce evidence from any of its inventors or other persons who could give evidence regarding the patent applicant’s state of mind at the relevant date, then the Court should more readily draw adverse inferences available from the patent applicant’s documents. Perhaps that is so to a limited extent, but there is a limit to how far the respondents can go to use counsel’s technically perspicacious inferences (in this case Mr Cordiner’s attractive presentation of technical notes and other experimental incidentals) to plug evidentiary gaps of hard evidence. The respondents bear the legal and evidentiary onus on this aspect of the case.
(c) The facts
768 On 12 April 2001, SmithKline Beecham PLC (the patent applicant) filed an application for the Patent, which was granted on 5 January 2007. The first applicant is the successor in title to the patent applicant. The patent applicant assigned the Patent to GlaxoSmithKline Dungarvan Ltd by assignment dated 20 September 2013, who then assigned the Patent to the first applicant by a further assignment on that date.
769 The early development work from June 1994 for the invention appears to have been conducted by Sterling Winthrop Pty Ltd. By April 1995, it appears that the development work was being conducted by the patent applicant. I will proceed on the basis that the knowledge of Sterling Winthrop can be taken to be part of the knowledge of the patent applicant. The corporate relationship, if any, between Sterling Winthrop and the patent applicant is unknown, but I do not need to linger on that question.
770 In June 1994, McNeil (a subsidiary of Johnson & Johnson) announced that it had received marketing approval for and intended to market “Tylenol Extended Relief”, a bilayer formulation containing 650 mg of paracetamol to provide “up to eight hours” of pain relief. This sustained release formulation of paracetamol is described in the Patent. Around this time, the patent applicant had been involved in developing a sustained release 1,000 mg paracetamol tablet to replace an existing product marketed by Sterling Health/SB in Scandinavia.
771 In the course of developing the sustained release 1,000 mg paracetamol tablet, the patent applicant examined retarding agents including Metolose 60SH–4000F (a form of HPMC) and an acrylic polymer, Eudragit NE40D. Based on in vitro dissolution rates, the formula using Eudragit NE40D was chosen for a developmental biostudy of six volunteers. However, that formulation failed to release sufficient drug in the volunteers. That project was put on hold. It was later reported (facsimile from W.C. Walton of SmithKline Beecham to D. James dated 24 April 1995 with the subject “Panadol Retard — Formulations”) that:
Immediate release (dissolution) occurred and we are unable to account for this happening. It may well be due to the non formation of the film. Because of this I would be more inclined to continue development work using the HPMC grade of polymer. One of the reasons why this option was not followed was because of the size of tablet. A 1000mg paracetamol caplet becomes too large over 1200mg if using HPMC whereas using the acrylic polymers restricts the weight to about 1120mg.
772 As I have earlier indicated, HPMC is a polymer that can be used as an extended release agent. It is available in different grades that vary in viscosity and extent of substitution by methyl groups and hydroxypropyl groups added to the glucose monomer units. Such factors affect the gel forming behaviour associated with the relevant matrix. Grades of HPMC can be distinguished by adding to the product name a number indicative of the apparent viscosity in millipascal seconds (mPa s). Viscosity is also measured in the unit centipoise (cP or cps). One cP or cps is equal to one mPa s. All of this was known to the patent applicant.
773 In early developmental work, the patent applicant investigated the use of different types of HPMC. By 1 May 1995, it had selected a “very common and ‘middle of the road’ grade” of HPMC with a viscosity of 15,000 cps. This was explained in the patent applicant’s extended release paracetamol investigations (facsimile from David James of Sterling Winthrop to Rick Chan with the subject “Project Delay” dated 1 May 1995):
We have examined the effect of different types of HPMC, that is, substitution types and viscosities. The results we obtained (in a fairly basic study) agree with the literature results, that is there is no significant difference between viscosities of polymers (between 4000cps and 100000cps investigated), and the lower the hydroxypropyl content of the polymer the more difficult it is to sustain APAP (due to water permeation and polymer swelling rates). I realise this is a bit sketchy but it provides some justification for our current work being with HPMC – 90SH:15000cps, which is a very common and “middle of the road” grade to work with. We have achieved our best results with this polymer which is from Shinetsu, a similar grade from Colourcon – Methocel K15MCR has also sown [sic] good results.
774 An undated experimental report (D139.11789, described by Generic Partners as “research paper (estimated date approximately May 1995)”) also noted that “The polymer viscosity was chosen as 15000cps based in part on supplier recommendation and the fact that above a threshold value of approximately 4000 cps the viscosity has little affect [sic] on the release”. Generic Partners contends that the patent applicant later found that the viscosity of HPMC used, including changes in viscosity of HPMC above and around 4,000 cps, did have an effect on the release of paracetamol from the formulation. In one sense it is true, although the magnitude and contextual significance of the effect is another question. The patent applicant subsequently decided against using HPMC 90SH:15,000 cps.
775 The patent applicant also carried out experiments using HPMC with a considerably higher viscosity, HPMC SH 100,000 cps, but it concluded that the dissolution rate from using such a grade was very slow and that “a lower molecular weight HPMC eg HPMC SH 4,000 cps SR would be considered”. The patent applicant identified HPMC SH 4,000 cps as a variable to be investigated to speed up the dissolution rate. HPMC grade 90SH 4000 SR is known by the product name “Metolose 90SH 4000 SR”. The product specification for this grade of Metolose apparently specifies a viscosity of 3000 to 5600 cps.
776 In or around August 1995, an employee of the patent applicant, Tony Jones, performed simulations on a dosage form of sustained release paracetamol. Following this work, the patent applicant decided to progress development work on two formulations: a “Tylenol ER mimic” and a sustained release product designed to have immediate release and sustained release according to a computer simulation. The latter formulation was known as the “Tony Jones’s profile”.
777 On or about 27 October 1995, the patent applicant expected to finalise Tony Jones’ profile with a sustained release to immediate release paracetamol ratio of 60.5:39.5. Generic Partners says that Formulation B in the Patent was the formulation based on Tony Jones’ profile. It was described by the patent applicant as formulation 9464/19A/TJ or BT9464/19A/TJ. It would appear that GSK accepts this correspondence.
778 Further, the patent applicant expected to finalise a Tylenol equivalent with a sustained release to immediate release paracetamol ratio of 41.1:59.9. Generic Partners says that Formulation A in the Patent was designed to emulate Johnson & Johnson’s Tylenol ER. It has been variously described by the patent applicant as formulation 9464/19JJ and BT9464/19JJ (and sometimes 9464/19A/JJ). Again, GSK appears to accept this correspondence.
779 From 19 February 1996 to 23 February 1996, the patent applicant conducted a pilot biostudy to investigate the rate and extent of paracetamol absorption from these two formulations in six volunteers. The results from this biostudy indicated that “one formulation shows improved sustained release characteristics which have the potential to be enhanced further by reformulation or some minor formulation optimisation”.
780 In the Patent, the HPMC used in Formulations A and B is described as “high viscosity HPMC”, although it appears that the “high viscosity HPMC” was in fact Metolose 90SH 4000 SR. It is said that this can be inferred from the laboratory notebook that describes the development of Formulations A and B, which includes a certificate of analysis for Metolose 90SH 4000 SR. This seems correct to me from my reading. Of course, Metolose 90SH 4000 SR varies between 3000 and 5600 cps.
781 Around 25 March 1996, the patent applicant examined the effect of using different lots of the same grade of commercially available Metolose 90SH 4000 SR, with different viscosities, on the dissolution profile of sustained release granules. The patent applicant concluded that the use of different lots of the same grade of Metolose 90SH 4000 SR changed the release profile of a sustained release granule, with the higher viscosity material unsurprisingly decreasing the release rate. The question of course is the magnitude of the effect and its significance. The patent applicant later carried out further work regarding the use of Metolose 90SH 4000 SR in its optimal formulation akin to Formulation C.
782 For the purposes of the following discussion, I will treat formulation 9464/19A/TJ as akin, roughly, to Formulation B and 9464/19JJ as akin, roughly, to Formulation A.
783 On 8 May 1996, one of the named inventors, Shing Yue “Rick” Chan sent a message to the other two named inventors, Bounkhiene (“Ken”) Sengmanee and Timothy James Grattan and others asking (it would appear in relation to Formulations A and B):
What is the correlation between plasma levels and dissolution results? Would further modelling going to [sic] help us identify the “desired” dissolution profile and hence assist us in the formulation approach?
784 Minutes of a meeting on or about 15 May 1996 suggested that there was “team consensus” that an in vitro/in vivo deconvolution study on individual data (apparently for Formulations A and B) should be performed “to enable identification of the ‘ideal’ in vitro dissolution profile”.
785 It would appear that the deconvolution study was carried out by an external consultant, Geoff Tucker and his assistant who were not named as inventors in the Patent. Tucker was provided with the in vitro paracetamol dissolution profiles of Formulations A and B (as later set out in Table 1 at p 10 of the Patent). The deconvolution study performed by Tucker was subsequently described by the patent applicant in its May 2000 “Marketing Authorisation Application” in “Part IIA: Composition” (marketing application) as follows.
Results from the pilot pharmacokinetic study (5) demonstrated that neither of the two prototypes met the acceptance criteria with both products failing to achieve mean plasma levels of paracetamol of 4 mcg/ml beyond 5 hours. However these results were used to derive an appropriate absorption disposition model. Having identified a suitable model, deconvolution techniques were applied to identify the paracetamol absorption profiles from the in vivo studies. The in vivo absorption profiles were then compared with the respective in vitro dissolution profiles and a simple mapping technique was used to superimpose the in vitro data over the in vivo data. This in vivo/in vitro link was then applied to identify a dissolution profile which was intended to deliver the ideal in vivo pharmacokinetic profile. Full details of the methods used are detailed in the in vitro/in vivo correlation (IVIVC) report (6). [This appears to be and I will assume is the Grattan paper that I will refer to later.]
786 On 10 June 1996, Tucker presented results of his study to Grattan. Grattan’s notes of that meeting (attached to a memorandum from Grattan to Tucker of 11 June 1996) state:
Results of the deconvolution demonstrated that model 4 gave the best superimposition although it was necessary to apply a time multiplying factor of approximately 5 (ie in vivo release was 5 times slower than in vitro release). The inference from this observation is that both of the SB trial formulations had incomplete in vivo dissolution, and that an ideal product which is fully available would require a faster release rate such that 100% was released in vivo after 12 hours (maximum assumed “useful” gastric residence time), this would require 100% in vitro release at 2.4 hours.
787 If a formulator’s aim is to deliver 100% of the drug to the patient, this requires release and full absorption of the drug before the drug is voided. The time in which this must occur must be no longer than the gastrointestinal transit time for the drug. For a drug to be reliably absorbed it should be released in vivo within 12 hours. The time multiplying factor (of approximately 5 referred to in the above quote) is important to predicting, from the in vitro release at 2.4 hours, the extent of release in vivo at 12 hours (2.4 hours x 5).
788 The in vivo/in vitro link, identified by Tucker and his assistant, was then applied by Tucker to identify a preliminary in vitro paracetamol dissolution profile that was intended to deliver the “ideal” in vivo pharmacokinetic profile. Tucker also produced “worst case” simulated plasma concentration profiles. Worst case assumed 95% paracetamol release after 2.4 hours. As Grattan explained to Sengmanee and Chan (email dated 19 September 1996 with the subject “Extend — Formulation & process variation”):
If you remember his initial work showed that in vitro release was about 5 times faster than in vivo release (in vivo appearance in the blood stream is probably a better term) and that we assumed that 12 hours was the maximum residence time. Therefore % release at 2.4 hours becomes an important parameter (which perhaps we should measure in the future).
…
I know we are talking only small differences here, but you have to remember that small changes under go [sic] this 5X multiplication factor as you go from in vitro to in vivo, weird isn’t it?
789 On 12 June 1996, Grattan and Sengmanee held a meeting with others where it was estimated that it would take the patent applicant three months to re-formulate an existing formulation to achieve the Tucker ideal in vitro paracetamol dissolution profile.
790 The patent applicant commenced its reformulation work to mimic Tucker’s ideal in vitro paracetamol dissolution profile on or about 14 June 1996. After a series of reformulations, on about 28 August 1996 the patent applicant created formulation 9464/66 which had the ingredients, in roughly similar amounts, as Formulation C as described in Example 2 of the Patent (I should say at this point that Formulation C has been identified with 9464/72 that I discuss later). The laboratory notebook stated with respect to formulation 9464/66:
This is it!!! The levels of Starch, Povidone & HPMC 15 cps are all at their optimal levels. We can simply vary the level of HPMC 4000 to vary the dissolution rate.
791 The patent applicant continued to carry out further work in relation to the granule type (creating formulations 9464/68 and 9464/69) and the specific viscosity of HPMC (creating formulation 9464/70, formulation 9464/71 and formulation 9464/72 split into four sub-lots).
792 On 2 September 1996, after identification of formulation 9464/66, the patent applicant made Formulation C using the same grade of HPMC 90SH 4000 SR, but having a viscosity of 4100 cps, HPMC 4000 SR, but having a viscosity of 3280 cps and HPMC 4000 SR but having a viscosity of 3850 cps. Apparently, commercially available Metolose 90SH 4000 SR had viscosities within the ranges 3000 to 5600 cps. It is correct to say that increasing the HPMC viscosity is presented as having the effect of decreasing the dissolution rate. But I must say that when I looked at the graph (3 September 1996) presenting the results (D113.10865), the differences for “% dissolved” as a function of “HPMC Variation” were very modest and appear to have been exaggerated by Generic Partners. Further, there were few data points, which raises in my mind the statistical significance of the data presented. In a study titled “HPMC variation effect”, the patent applicant had concluded that the use of different lots of the same grade of HPMC 90SH 4000 SR, each lot being within specification but having different viscosities, gave different dissolution rates for that formulation and that, in particular, an “[i]ncrease in HPMC viscosity will decrease the dissolution rate”. But as I have said, the difference was modest. There was a difference of around 3% in the dissolution profile observed between 3280 and 4100 cps. Generic Partners says that the patent applicant would have known or at least been concerned that a difference of 2,500 cps in the viscosity of the commercially available product (3000 to 5600 cps) would cause a greater impact on dissolution profile.
793 On 10 September 1996, something close to Formulation C was said to be identified by the patent applicant, namely formulation 9464/72. Further experiments were conducted to 12 September 1996 in relation to that formulation looking at in vitro dissolution profiles of the sustained release layer only and the bilayer tablet and the effect of varying granule type from light to very heavy and granule size on those profiles.
794 By 21 March 1997, and after having conducted the HPMC variation effect study, the patent applicant had entered into an agreement with ShinEtsu, the manufacturer of Metolose 90 SH 4000 SR, for it to supply the patent applicant with material with a tighter viscosity range of 3500 to 4050 cps as compared with the ordinary specification for that commercially available grade of HPMC which was 3000 to 5600 cps; the email recording this refers to mPas which I have changed to cps. The range was 550 cps wide compared to that investigated in the HPMC variation effect study (820 cps) and the commercially available variation (2,500 cps). I should say that Generic Partners has dwelled on this arithmetic, but in terms of its ultimate significance relating to its effect on dissolution rate, there has been a tendency to give it disproportionate emphasis.
795 On 20 September 1996, Amin Rostami, Tucker’s assistant, wrote to Grattan explaining that individual differences in presumed population can affect the absorption rate of paracetamol. Rostami provided a graph which showed the pharmacokinetic profiles expected for three types of individuals resulting from “ideal release”. The three types of individuals were identified as persons that would absorb paracetamol at a fast rate, at a medium rate or at a slow rate. The graph showed expected differences between the three types of individuals with “ideal release”.
796 On 24 September 1996, Grattan sent to Tucker the complete release in vitro paracetamol dissolution profiles for formulation 9464/69 and two versions (9464/72/1 and 9464/72/2) of Formulation C, one with a very heavy granule and another with a normal light granule with a few lumps. Grattan asked Tucker to produce from those profiles simulated plasma concentration profiles for the three kinds of individuals. The upper and lower limits were then initially set at plus/minus 4% as practical limits. Grattan explained his thinking to the other inventors for setting the plus/minus 4% limit:
I am mindful of the fact that whatever pharmacopeas [sic] might say about recommended limits that if we stray only a little way from the ideal profile we are going to be in trouble when we do the biostudies, on the other hand there will inevitably be batch to batch variation in a production situation.
I have had to guess at what the practical limits would be … and I went for an arbitary [sic] +/– 4% of the stated dose for each time point, I know this may be difficult to achieve, but we know what the answer is going to be if the % release at 2.4 hours falls much below 95%, so I think that one has to be cast in stone. …
797 Generic Partners says that the patent applicant accordingly knew that it was desirable to achieve a narrow range around the ideal in vitro dissolution profile in order to avoid the possibility of individuals not achieving the desired or optimum pharmacokinetic profile or physiological benefit. Specifically, it is said that it knew that even within normal recommended limits around in vitro dissolution time points, the formulation might not achieve the advantageous pharmacokinetic properties. It is said that the patent applicant was also aware that achieving such a narrow range may be difficult due to batch to batch variations, but that was plainly a worthy goal. I am prepared to accept that the patent applicant had such knowledge.
798 On 1 October 1996, Tucker sent to Grattan updated calculations presented in graphical form in accordance with Grattan’s 24 September 1996 instructions. The graphs showed how using the plus/minus 4% limits (this may be more readily inferred from the 16 October 1996 table prepared by Grattan) the formulation would further broaden the pharmacokinetic profiles of the three types of individuals. On 16 October 1996, Grattan wrote to the other inventors noting that a further request had been sent to Tucker with slightly broader variations and noting that “We know we are right on the border line of acceptability anyway (i.e., on the assumption that the reg authorities accept that 4mg/L is the minimum therapeutic plasma level)”.
799 On 28 October 1996, Rostami sent to Grattan updated graphs for simulated plasma concentrations for Grattan’s revised lower and upper dissolution limits. Rostami explained that:
It appears fast absorbers may experience a period of concentration below 4 with new upper lower [sic] and upper limits. In the case of lower limit the period below 4 is longer and apparently there isn’t adequate dose in the immediate release part of formulation.
I think while the upper limit may serve your purpose well, the lower limit has gone to [sic] low and may need reconsideration.
800 These graphs showed how variability might have an effect not only on the average member of the population but also the fast and slow absorbers. This further work was provided by Grattan to the other inventors on 5 November 1996, explaining that the limits he set were +/–7.5% for the intermediate time points and +/–5.0% at the final time points. He also said that:
I think at this stage we have done all we can on the modelling side of things, and I suggest that we now proceed as planned. We can revisit the modelling following reciept [sic] of the results from the next biostudy.
801 On 13 November 1996, Grattan wrote, inter alia, to the other inventors noting that modelling for –4% to –5% variation at the intermediate time points looked better than –7.5%. Accordingly, his recommendation was to set a “tentative spec (to be revisited following the next biostudy results, further modelling work and further production experience) of –5% and +7.5% of target [sic] for the intermediate time points and –5% and +5% of target for the final time points …”.
802 Let me move to the question of granulation.
803 The type of granule depends on the ingredients used in the formulation as well as the granulation process.
804 By 8 March 1996, the patent applicant had concluded that the degree of granulation impacted on the release rate of the formulation it was then considering. But at that time the formulation had more pregelatinised starch in it than Formulation C or D.
805 On and after 28 August 1996, following the development of 9464/66 (close to Formulation C), formulations 9464/68 and 9464/69 were produced by the patent applicant to examine the impact of the granulation end point. On 29 August 1996, formulation 9464/68 was described by the patent applicant as “Over granulated to [see] the effect on release rate!!”. It appears to have had a heavy granule. Formulation 9464/69 appears to have been a batch with a normal/conventional type of granulation end pt and having a light granule. The patent applicant recorded (in various places) the mean dissolution rate of paracetamol of formulations 9464/66, 9464/67 (a repeat batch of 9464/66), 9464/68 and 9464/69 which can be tabulated as follows (ignoring the ranges for the standard deviations):
9464/66 (%) | 9464/67 (%) (repeat of batch 9464/66) | 9464/68 (%) (over granulation — heavy granule) | 9464/69 (%) (normal/conventional granulation — light granule) | |
15 min | 12.8 | 12.7 | 12.1 | 14.8 |
60 min | 42.6 | 42.3 | 40.2 | 45.6 |
120 min | 72.2 | 71.6 | 68.4 | 76.7 |
180 min | 92.6 | 91.6 | 88.4 | 96.9 |
806 The patent applicant appears to have concluded after testing 9464/68 that the granulation end point did not have a significant effect on release rate and after testing 9464/69 that “we have a very robust formulation here”. Further tests were done for formulations 9464/70 and 9464/71. The patent applicant then further examined the impact of granulation end point on formulation 9464/72 (now agreed by GSK to be Formulation C) by using a “[n]ormal light granule”. Formulation 9464/72 was divided into 9464/72/1 (light granule) and 9464/72/2 (heavy granule). The mean dissolution rates for the sustained release only part of formulations 9464/72/1 and 9464/72/2 can be tabulated as follows (ignoring the ranges for the standard deviations):
9464/72/1 (%) (normal light granule) | 9464/72/2 (%) (heavy granule) | |
15 min | 13.6 | 12.7 |
60 min | 44.4 | 42.8 |
120 min | 74.8 | 72.8 |
180 min | 95.1 | 93.6 |
807 In vitro dissolution profiles were also produced for bilayer tablets for batch 9464/72/1 and batch 9464/72/2 and were compared to “GT” (apparently the then proposed Tucker ideal dissolution profile). The results can be tabulated as follows (ignoring the ranges for the standard deviations):
9464/72/1 (%) (normal light granule) | 9464/72/2 (%) (heavy granule) | GT (%) (Geoff Tucker ideal) | |
15 min | 41 | 39.7 | 40 |
60 min | 66.4 | 65 | 62 |
120 min | 90.4 | 89.1 | 94 |
180 min | 100 | 100 | 100 |
808 Generic Partners rightly says that in each experiment, the “normal light” granule (9464/72/1) had a faster dissolution rate than the “heavy” granule (9464/72/2). But it exaggerates the effect. This can be easily appreciated by considering the graph that I have reproduced from the working notes (D115.11083):

809 Generic Partners asserts that Formulation C (9464/72) is ultimately described as having a “heavy granule” in a summary report for the project produced on or about 30 November 1996 (D115.11085, extract from laboratory notebook dated 10 September 1996 to 30 November 1996). It says that although the changes that were caused to the in vitro dissolution profile by differences in granule were small, a choice was made in the knowledge of these studies above to proceed with a particular granule for Formulation C. I must say that when one understands the data, Generic Partners’ position is quite exaggerated. Its point on granular end point and particle size lacks relevant significance.
810 Ultimately, the patent applicant selected Formulation D for commercialisation. The “application formula” in the marketing application is C317.7.003 which GSK agrees is Formulation D as described in Example 3 of the Patent. This formulation has the same dissolution profile as set out in the Patent for Formulation D. GSK has said that a change in paracetamol supplier necessitated the change from Formulation C to Formulation D. But Generic Partners has pointed out that further granulation end point work was done by the patent applicant with respect to Formulation D.
811 On 30 July 1999, one of the inventors, Sengmanee, approved a report and recommendation in the “Extend/9464” project arising from the patent applicant’s further granulation end point analysis with respect to batches M317.99.004 and M317.99.005, which it may be inferred have the same ingredients and amounts as for Formulation D but only differ in respect of variations in granulation method. The report stated:
The results for Batch No. M317.99 004 show faster release rate and a greater spread of results than Batch No. M317 99 005. This is consistent with the difference in granulation end point for Batch No M317 99 004 (15 0 kW) and for Batch No M317 99 004 (16.5kW).
It is recommended that a granulation end point of 15 5 – 16kW should be the target for Extend granulation applicable to use of the Gral 600 and Dungarvan and a batch size equal to that of Batch N’s M317 99 004 and M317 99 005.
812 The graph in that report shows a modest difference between the in vitro dissolution profiles of the sustained release phase of the formulation for each batch. That is confirmed by the mean data points given on the preceding page showing a difference of 2.4% at 15 minutes, 4.2% at 60 minutes, 5.1% at 120 minutes and 4.7% at 180 minutes. In the context of setting a specification with limits of 6% at 15, 60, 120 minutes and 5% at 180 minutes, this is a variation. Further, there was a further within batch variation recorded (relative standard deviation (RSD)) which is greater for one batch than the other, with the RSD for one batch (M317.99.004) at 15 minutes being a further 6.2%.
813 Accordingly, as at July 1999 I accept that the patent applicant was aware that optimising the granulation end point to between 15.5 to 16 kW would provide close to the ideal in vitro dissolution profile for a batch and less variability of in vitro dissolution profiles within the batch.
814 The marketing application dated May 2000 states:
Two distinct manufacturing trials have been carried out to evaluate and optimise the manufacturing process. Both trials consisted of two pilots (1 granulation mix) and one full scale (3 granulation mixes). The active is adequately controlled. Three different lots of actives produced acceptable granule with finished tablets of the desired dissolution characteristics (see Table IIA/7). The analytical file, which contained the results of analysis carried out on the batches produced in 1997, was misfiled and cannot be located. The homogeneity of the product was confirmed by the validation batches produced in March 2000, therefore the data originally generated is considered applicable.
Optimisation of granulation protocol
To achieve a granule, which compacted easily and had no visible lumps, the granulation fluid, speed of impellors/choppers and kW endpoint has been optimised. The granule obtained had good flow characteristics, did not segregate and performed well on the tablet press.
Granulation fluid was increased from 28 to 32 litres, which achieved a granule of the desired consistency. Water was added at approximately 5.8 litres/minute. The second manufacturing trial consistently used 32 litres and confirms this granulation fluid quantity and flow rate to consistently produce a granule of the desired characteristics, i.e., a copious quantity of small lumps within a base of fine granules.
Results from the first trial showed that an endpoint of between 15.3 kW and 18.1 kW was required to produce a granule of the desired characteristics and achieved tablets with the desired dissolution profile (see Table IIA/7). Following the second trial it was possible to refine the endpoint to within 15 kW and 16.5 kW with the optimum granulation endpoint identified at 15.5 kW. Dissolution studies, sieve analysis and consistent compression performance support this range.
815 The granulation end point optimisation work on Formulation D from the second trial is set out in Table IIA/7 of the marketing application. That table appears to be a summary of experimentation set out in the patent applicant’s “Evaluation Report No. 300317” prepared in January 1998 (concerning formulations M317.7.001 to 004 and C317.7.001 to 004) and what it may be inferred are original laboratory notebooks concerning experiments on formulations C317.99.001 to 003.
816 Accordingly, as at May 2000 I am also prepared to accept that the patent applicant was aware that for Formulation D the optimum granulation end point was 15.5 kW with a resulting base of fine granules with a copious quantity of small lumps and that changing the granulation end point would provide the desired in vitro dissolution profile and reduce the amount of variability of that profile within a batch.
817 The patent applicant set limits for the in vitro dissolution specification for the “ideal formulation” using the work by Grattan. The marketing application appended to it a report by the inventors, Tucker and others titled “The use of in vitro–in vivo correlation to develop a sustained release formulation of paracetamol with a predefined plasma concentration-time profile: A report on a step-by-step progression”. This report has been referred to in my reasons as the Grattan paper. The patent applicant also knew from this work that “if we stray only a little way from the ideal profile we are going to be in trouble when we do the biostudies”.
818 The patent applicant set the specification for the in vitro dissolution limits for Formulation D and explained that the specification allowed for a maximum change in AUC and Cmax of 20%. Generic Partners contends that the proper way to read the document is that the patent applicant anticipated that there could be up to a 20% change in the plasma profile for the formulation corresponding to a shift in dissolution profile of the specified limits. In my view that is a fair reading.
819 It may be inferred from the marketing application that the patent applicant knew that any change in the dissolution profile might change the pharmacokinetic attributes of the formulation. It would appear that the patent applicant was concerned to ensure that the in vitro dissolution profile did not move away from the target by more than 6%.
820 Ultimately, the in vitro dissolution profile for Formulation D, batch M317.7.003, as made by the patent applicant was as follows (the average figure matches the profile set out in the Patent for Formulation D):
(a) 15 minutes: average 40.8%, range 39.8 – 41.5%;
(b) 60 minutes: average 65.0%, range 63.2 – 66.8%;
(c) 120 minutes: average 90.2%, range 88.0 – 91.5%; and
(d) 180 minutes: average 101.8%, range 100.3 – 103.2%.
821 The pharmacokinetic study using that Formulation D demonstrated satisfactory pharmacokinetic profiles.
822 Finally, before discussing the arguments I should proffer a potential solution to one mystery. It seems that the patent applicant may have tested dissolution rates using neither a basket nor a cylinder, but rather a paddle. At all events, what it did in fact use does not speak to construction.
(d) Generic Partners’ contentions
823 In summary, Generic Partners contends the following in relation to the state of knowledge of the patent applicant as at the relevant date.
824 First, it had investigated the use of a number of different grades of HPMC (that is, substitution types and viscosities) and knew that the viscosity of the “high viscosity HPMC” affected the in vitro paracetamol dissolution. I agree with this contention.
825 Second, it was aware that even small increases in viscosity of the “high viscosity HPMC” would decrease the in vitro paracetamol dissolution rate of its formulation. I agree with this in its generality. The level and significance of the decrease is another matter.
826 Third, it had ultimately selected HPMC 90SH – 4000 SR. I agree with this contention.
827 Fourth, it was aware that even different lots of the same grade of commercially available HPMC 90SH – 4000 SR (with slightly different viscosities) (3000–5600 cps) gave different dissolution rates. I agree, but the level and significance of the difference is another matter.
828 Fifth, it had requested and obtained from the manufacturer of Metolose HPMC 90SH – 4000 SR an agreement that the manufacturer would only supply the patent applicant with material with a tighter viscosity range than was generally marketed by the manufacturer and the range that the patent applicant had tested to show the variation in in vitro dissolution profile inherent in the change in viscosity. I agree with this contention.
829 Sixth, it had used a grade of HPMC that was not “commercially available” to make Formulation D. I do not agree with this contention in the sense that any formulator seeking such a grade could have obtained it on request.
830 Seventh, it had conducted studies on granulation end point for Formulation C and Formulation D and had concluded, in particular for Formulation D, that a granulation end point had an impact on the in vitro dissolution rate and variation in the in vitro dissolution rate within a batch. I agree, but the level and significance of the impact is the real question.
831 Eighth, it had conducted trials and knew that a granulation end point of between 15.3 kW and 18.1 kW for Formulation D was required to produce a granule of its desired characteristics and to achieve its desired dissolution profile; after a second trial, it selected its optimum granulation endpoint of 15.5 kW. I agree.
832 Ninth, Generic Partners contends that the patent applicant identified Formulation D as the “optimised formulation” in its marketing application. It was also said to have been developed to give a release profile that very closely resembled the patent applicant’s ideal release profile and as having an in vitro dissolution profile closely approximating its ideal profile. Further, the patent applicant had made Formulation D with a very tight dissolution specification. I agree with these contentions.
833 Tenth, I accept that the complete specification only describes the high viscosity HPMC used as “high viscosity HPMC”. Further, as to granulation, it states that the compositions of the invention “can be formulated by conventional methods of admixture such as granulating, blending, filling and compressing”.
834 I also agree that the complete specification does not disclose:
(a) The grade and specific viscosity (within the commercial grade) of HPMC used by it to produce Formulations C and D.
(b) The granulation end point (or tight range of granulation end point) that was “required to produce a granule of the desired characteristics and achieved tablets with the desired dissolution profile” with as little variation as possible within a batch.
835 Eleventh, Generic Partners says that high viscosity HPMC could include HPMC with viscosities ranging from 40 to 100,000 and certainly from 3,000 to 15,000. I doubt this concerning the first-mentioned range.
836 Twelfth, Generic Partners contends that the method known to the patent applicant of making Formulation D and/or C with a particular grade and specific viscosity of HPMC and a particular granulation end point is better than that disclosed in the Patent (of making Formulation D and/or C with “high viscosity HPMC” and a “conventional” method of granulation) because it eliminated the “experimentation” involved in a skilled addressee trying a variety of possible grades of high viscosity HPMC and granulation end points in order to make Formulation D and/or C as made by the patent applicant and guaranteed to work. Generic Partners also contends that, in terms of the selection of parameters that could affect the performance of the best method, the complete specification must not leave the skilled addressee with choices requiring “any experimentation” (not just “undue experimentation or ingenuity”). But Generic Partners also says that this is not necessary to decide because the evidence is that undue experimentation or ingenuity would be required to arrive at the best method known to the patent applicant. I disagree that “undue experimentation or ingenuity” was involved. Moreover, I reject its “any experimentation” submission. In my view, such an absolute proposition does not accord with the authorities.
837 Now Professor Davies’ evidence is that it would be “practically impossible” to produce a batch of tablets with an average dissolution profile matching that reported for Formulation C. Nevertheless Generic Partners has contended that that does not relieve the patent applicant from the statutory requirement to disclose the best method of making the invention known to it. It is said that even accepting the inherent variability in the manufacture of a pharmaceutical formulation, the patent applicant did not disclose the specific ingredients and parameters that it knew were required to make Formulation D and/or C as made by it with the narrow variation in in vitro dissolution profile around the “ideal”. I accept that the closer a formulator wanted to get to the in vitro dissolution profile set out in the Patent for Formulation C, the more work that would be involved. Professor Davies accepted that he would have had to conduct a number of tests with different batches of HPMC and granulation end points in order to achieve the goal of a formulation as close to Formulation C or D as possible. Lordi, N.G.’s Chapter 14 “Sustained Release Dosage Forms” in The Theory and Practice of Industrial Pharmacy by Lachman and Lieberman (1986) p 453 table 14–7 suggests that numerous grades of HPMC are suitable as retardants in matrix tablet formulations. I also note that Professor Davies’ personal experience with K4MP HPMC led him to have a good feeling it would work and that he would start with that.
838 Generic Partners contends that the patent applicant was aware that the selection of the grade and viscosity of high viscosity HPMC and the granulation end point used to make Formulation D and/or C would affect the in vitro dissolution profile of the pharmaceutical composition, moving away from the “ideal release profile”. I accept that contention in the generality with which it has been expressed.
839 Generic Partners contends that the patent applicant knew that some high viscosity HPMC/granulation end points were not as good as others at least in the sense of achieving an in vitro dissolution profile that had little variation within and between batches and most closely aligned with the “ideal” in vitro profile. I accept that contention also.
840 Generic Partners contends that the patent applicant knew that the dissolution profile of the formulation affected its pharmacokinetic performance and that changes in the in vitro dissolution profile would affect its pharmacokinetic performance. I also accept this. But, as Professor Davies said, if the change in in vitro dissolution profile was less than 10% then you would not see a significant difference in the pharmacokinetic attributes. The plasma profile would be “bioequivalent” and the pharmacokinetic attributes would still be met.
841 Generic Partners contends that the patent applicant knew, in the words of Grattan, “…whatever pharmacopeas [sic] might say about recommended limits … if we stray only a little way from the ideal profile we are going to be in trouble when we do the biostudies” (email from Grattan to Chan and Sengmanee dated 24 September 1996); see also a facsimile from Rostami to Grattan dated 28 October 1996 and a facsimile from Grattan to Chan and Sengmanee dated 16 October 1996 which shows how the patent applicant appreciated that a change in in vitro dissolution profile within +/–4% would affect the in vivo dissolution profile.
(e) Analysis
842 A patent will satisfy the best method requirement if the skilled addressee can arrive at the best method known to the patent applicant of performing the invention by some “routine” experimentation, but without ingenuity or undue experimentation. A patent applicant does not need to disclose details of the method which would be well-known and understood by the skilled addressee such as well-known analytical agents, commonly used methods, well-known terms of art, or a description of machinery in standard use. But a complete specification must disclose each essential element or feature for performing the invention, even if a skilled addressee would know or could readily ascertain that element.
843 Where the best performance of the invention requires a step that is omitted by the complete specification, even if it could be readily ascertained by a skilled addressee, that will not meet the best method requirement.
844 If the skilled addressee is left to make a choice as to what analytical agent to use, what commonly used method to use or what machinery to use or is left in doubt as to what a term means, the best method will not have been described if that choice or uncertainty affects the performance of that method.
845 Professor Davies considered that he could make “tablets that were relevantly similar to Formulation C” within a dissolution profile within the claimed range. And as he said, which I accept, this was “routine” work.
Grade/viscosity of HPMC
846 The respondents’ assertion that the grade and specific viscosity of the high viscosity HPMC is a crucial part of the best method of performing the invention is in one sense contradicted by the evidence of Professor Tucker, who was confident that the Patent provided enough information for him to formulate Formulation C or D with a dissolution profile as close as possible to the dissolution profiles set out in tables 2 and 3 of the Patent for Formulations C and D. Professor Tucker’s view was that he would be able to make Formulations C or D “within +/– 5%”. This was a level of variability described in essence as satisfactory by Professor Fassihi and also narrower than the ranges permitted by the FDA.
847 Further, Professor Tucker agreed that a skilled formulator in 2000 who had knowledge of HPMC would have known that there would be batch-to-batch variation in any HPMC commercially acquired. Further, Professor Tucker also agreed that when a person skilled in the art was using a batch of HPMC, they would have known that they may have needed to have made adjustments to their composition to get a dissolution profile to take into account variations in the HPMC they received. Moreover, any such adjustment was routine trial-and-error optimisation by a person of ordinary skill in the art at the relevant time. Further, setting acceptable limits on the dissolution profile to accommodate changes in the viscosity to be supplied by the manufacturer was a matter of routine.
848 Dr Mooney also explained HPMC selection, in the context of formulation development, in the following exchange that I had with him:
HIS HONOUR: But if you’re thinking to yourself, “I need a high viscosity HPMC to form a matrix” — we’re talking about the gelatinous layers — would you go straight to the 4000, or would you — it would be a question of experimentation as to whether --- ? --- Would be — it would be an — taking into consideration the — the amount of active, the solubility of the active, the time over which I wanted the product to be released. So there’s the — the consideration, and you might — you might pick two or three as a starting point, just to — as a — as an initial small lab trial to see what the behaviour of this particular material was in the presence of those formulas, and then once you’ve got your — your profile, you then start to play with the other components to see which gives you the best quality tablet in terms of compressibility, in terms of flowability, and how you’ve constructed it.
And so, you would play around with the particular viscosity --- ? --- I — I believe back then we would have — I would have been using — evaluating at least two — a range to give you an idea of behaviour. (emphasis added)
849 Further, Professor Davies explained that he would start with “K4M” HPMC, which had a viscosity of between about 3000 to 5000 cps, if he was making Formulation C, given that he was aiming for a release profile over about three hours in vitro. Professor Davies had a “good feeling” that K4M HPMC would work to produce Formulation C. He noted that he had used higher viscosity HPMC grades in formulations which release over much longer time periods. Professor Davies explained that if K4M HPMC did not work, he would move to the next higher-viscosity grade, but “there are only a limited number of grades of these that will effectively work”. I accept Professor Davies’ evidence that selecting a grade of HPMC to make Formulation C was a matter of routine.
850 Further, the patent applicant’s documents showed that the variation in release profiles was just a few per cent between the batches and fell within the release profile ranges in claim 1. Further, they showed that for a formulation like Formulation C (with about three per cent high viscosity HPMC and one per cent pregelatinised starch by weight of the sustained release layer, and with a viscosity of HPMC within the ranges of 3280 cps and 4100 cps) different batches of HMPC had very little effect (about a few per cent) on the release profile of such a formulation. Moreover, the FDA recognised the inherent variability associated with the production of oral sustained release dosage forms, and allowed for such variation from a quality control perspective by permitting such a product to have a dissolution profile within a range of plus or minus 10 percentage points.
851 Further, Professor Tucker also accepted that for some of the formulations discussed in the patent applicant’s documents he had looked at, he could not know the impact of different viscosities of HPMC on Formulation C because he could not know the impact of other various factors that were in potential play as well. So he said:
MR SHAVIN: And you now accept that in looking at this data, we don’t know what the other — or the impact might be of the other factors. That is, more than double the HPMC that’s in formulation C and the very high proportion of pregelatinised starch, for example? --- Yes. And, indeed, I don’t know the impact if the manufacturing conditions changed or …
HIS HONOUR: So you would only really know the impact if you kept everything else equal ---? --- Yes.
852 The respondents have also asserted that the email from Sengmanee dated 21 March 1997 with the subject “Extend — scale-up batches” demonstrated that the patent applicant knew that a tighter-controlled viscosity grade of HPMC 4000 was used by the patent applicant to produce Formulation C. But as explained by Professor Davies:
The email suggests that, for reasons which are not made clear, after GSK had developed the formulation the subject of the email and were in the “scale-up” phase, they decided to request that the high viscosity HPMC be provided within a narrower viscosity range than the typical specification for the product.
Whilst it would always be of benefit to obtain ingredients for use in a pharmaceutical composition with as little variation as possible for quality control and manufacturing purposes, I do not consider, having regard to GSK’s findings reported in D113.10865 and my comments in paragraph 7 of this document above, that it would be necessary to obtain a high viscosity grade of HPMC with such narrow specifications to reproducibly produce Formulation C. As previously explained, in my experience, skilled formulators know that tolerances and variations in ingredients do not permit each tablet in a given batch to have a particular dissolution profile or for a given batch to match a dissolution profile previously produced using exactly the same ingredients and manufacturing equipment and method. Rather formulators and regulatory bodies accept there will be variation in the properties of the formulations within acceptable limits. For example, the specification sets forth the acceptable level of drug content as a range just as the dissolution profile has a range at each time point.
853 Furthermore, there was no evidence to establish that the patent applicant knew at the filing date of the complete specification that Formulation D could not be produced using other grades of high viscosity HPMC or with HPMC 4000 with narrower specifications.
854 Further, to the extent that Generic Partners asserts that the grade of HPMC used by the patent applicant was not commercially available, such submissions are misconceived. HPMC 4000 was a known and available grade and there is no evidence to suggest that a skilled person could not have obtained that grade, with or without a narrower specification than typically offered by a manufacturer, if the skilled person so desired.
Granule type
855 First, the experts acknowledged that they did not know if granule type would have had any significant impact on the dissolution profile of Formulations C or D.
856 Second, the evidence did not show that when a formulation contained as little as 1% pregelatinised starch (as Formulations C and D did) that granule type was relevant in making Formulation C or D. The extract from the laboratory notebook dated 10 September 1996 to 30 November 1996, “Investigation — Effects of granulation end point & particle size” showed dissolution curves for formulations which had 1% pregelatinised starch having a maximal difference in the dissolution profiles between the light and the heavy granule formulations of only about two percent. Indeed, in my view the difference might have merely been an artefact of the experiments. Further and in any event, those dissolution curves would have been regarded as equivalent according to a similarity factor f2 calculation; I have described such a metric elsewhere.
857 Third, Generic Partners has sought to rely upon other documents describing the optimisation of the granulation end point of Formulation D, namely, the Dissolution Report Extend/9464 dated 24 August 1999 and part IIA of the marketing application.
858 But the Dissolution Report Extend/9464 dated 24 August 1999 records experiments concerning tablets only comprising an SR layer. Further, the dissolution values obtained for the two experimental SR layer tablets are very similar. Further, the granulation end point recommendation made in the document was said to be “applicable to the use of the Gral 600 at Dungarvan” with a particular batch size.
859 As to part IIA of the marketing application, the batches described had similar dissolution profiles. The discussion of granulation optimisation again made clear that the patent applicant was at this stage concerned with optimising its particular commercial manufacturing method. It was optimising the flow and compaction procedures rather than improving the invention exemplified by Formulations C and D. Further, I agree with GSK that there was no evidence to suggest that the manufacturing method used by GSK to manufacture Formulation D on a commercial scale was anything other than conventional and routine.
General
860 It is readily apparent that GSK proceeded to optimise the manufacture of the formulation for their specific equipment and batch sizes. But GSK was not required to set out in the specification of the Patent manufacturing optimisation details developed with respect to its own particular equipment and manufacturing capabilities. In my view, such idiosyncratic detail is not relevant to the skilled person’s task of performing the invention claimed and is unlikely to assist a skilled person to perform the invention using his own particular excipients, manufacturing methods and equipment.
861 Furthermore, I am unable to conclude on the evidence that formulations made with grades of HPMC optimised for the patentee’s own equipment are any “better” than other formulations which a skilled person would make when reproducing Formulation C/D.
862 Finally, and as I will repeat, Professors Davies and Tucker considered that they could make Formulations C or D without undue experimentation. As discussed above, the Patent provided sufficient information for the skilled formulator to make formulations which acceptably replicated the dissolution profiles of both Formulations C and D. Accordingly the best method known by the patent applicant was disclosed in the Patent.
863 In summary, I do not consider that the respondents have made good their invalidity attack on this aspect.
lack of Inventive Step (section 18(1)(b)(ii))
864 Before descending into the detail of this ground on which Apotex took the lead, it is not inappropriate to observe that there was, on one view, a forensic tension between Generic Partners’ case on the lack of disclosure of the best method, which put into evidence considerable detail as to the evolution of the patent applicant’s experiments, and Apotex’s lack of inventive step case. But such impressionistic intuitions can be put to one side given the different legal contexts.
(a) The principles
865 The respondents bear the onus of establishing that the claimed invention lacks an inventive step over the prior art base. Pursuant to s 7(2) of the Act, an invention is to be taken to involve an inventive step when compared to the prior art base unless it would have been obvious to a person skilled in the relevant art in light of the common general knowledge as described by Aickin J in Minnesota Mining and Manufacturing Co v Beiersdorf (Australia) Ltd (1980) 144 CLR 253 at 292 as it existed in the patent area (the then provision) before the priority date of the relevant claim, whether that knowledge is considered separately or together with information of the kind in s 7(3) of the Act.
866 The term “obvious” means “very plain” (language endorsed in Aktiebolaget Hässle v Alphapharm Pty Ltd (2002) 212 CLR 411 at [34] per Gleeson CJ, Gaudron, Gummow and Hayne JJ (Alphapharm) and Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) (2007) 235 CLR 173 at [51] (Lockwood (No 2)). The inventive element needed to sustain a patent may be small. A “scintilla of inventiveness” will be sufficient and “no smallness or simplicity will prevent a patent being good” (Meyers Taylor Pty Ltd v Vicarr Industries Ltd (1977) 137 CLR 228 at 249 per Aickin J). Relevant content has been further given to “obvious” by Aickin J in The Wellcome Foundation Ltd v VR Laboratories (Aust) Pty Ltd (1981) 148 CLR 262 at 286 in stating:
whether the hypothetical addressee faced with the same problem would have taken as a matter of routine whatever steps might have led from the prior art to the invention, whether they be the steps of the inventor or not.
867 Further, in relation to experiments, his Honour at 280 and 281 said the following:
In the present case it was admitted by the respondent that the test of obviousness was an objective one, but it was argued that evidence of a subjective character was admissible. That is no doubt true in some cases because expert witnesses are often properly asked whether they found a particular invention “surprising” to them. That however does not answer the question whether evidence of the steps which the patentee took is relevant and therefore admissible. Evidence of what was in the patentee’s mind may be admissible as evidence of the state of the art, but could seldom be otherwise admissible. Evidence of what he did by way of experiment may be another matter. It might show that the experiments devised for the purpose were part of an inventive step. Alternatively it might show that the experiments were of a routine character which the uninventive worker in the field would try as a matter of course. The latter could be relevant though not decisive in every case. It may be that the perception of the true nature of the problem was the inventive step which, once taken, revealed that straightforward experiments will provide the solution. It will always be necessary to distinguish between experiments leading to an invention and subsequent experiments for checking and testing the product or process the subject of the invention. The latter would not be material to obviousness but might be material to the question of utility.
868 The question to be posed was whether putative experiments leading from the prior art to the invention as claimed were part of the inventive step or were “of a routine character” to be tried “as a matter of course” (Alphapharm at [52]). That question has an affinity with the “Cripps question” posed by Graham J in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd [1970] RPC 157. I reject Apotex’s submission that I am disabled from using a reformulated Cripps question. The Cripps question was paraphrased by French CJ in AstraZeneca AB v Apotex Pty Ltd (2015) 114 IPR 445 at [15] (AstraZeneca) as:
Would the notional research group at the relevant date, in all the circumstances, which include a knowledge of all the relevant prior art and of the facts of the nature and success of [the existing compound], directly be led as a matter of course to try [the claimed inventive step] in the expectation that it might well produce a useful alternative to or better drug than [the existing compound]?
869 But the question does not import as a criterion that the inventive step claimed would be perceived by the hypothetical addressee as “worth a try” or “obvious to try” (AstraZeneca per French CJ at [15]). Further, Alphapharm (at [72]) rejected an “obvious to try” or “worth a try” approach.
870 Section 7(2) first requires consideration of what would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed before the priority date (putting to one side for the moment s 7(3) information).
871 Common general knowledge is knowledge “generally known and accepted without question by the bulk of those who are engaged in the particular art” (British Acoustic Films Ld v Nettlefold Productions (1936) 53 RPC 221 at 250 per Luxmoore J).
872 Information cannot be treated as part of the common general knowledge unless there is evidence of its general acceptance and assimilation by persons skilled in the art. Information does not constitute common general knowledge merely because it might be found, say, in a journal, even if widely read by such persons. Further, as Luxmoore J said at 250:
In my judgment it is not sufficient to prove common general knowledge that a particular disclosure is made in an article, or series of articles, in a scientific journal, no matter how wide the circulation of that journal may be, in the absence of any evidence that the disclosure is accepted generally by those who are engaged in the art to which the disclosure relates. A piece of particular knowledge as disclosed in a scientific paper does not become common general knowledge merely because it is widely read, and still less because it is widely circulated. Such a piece of knowledge only becomes general knowledge when it is generally known and accepted without question by the bulk of those who are engaged in the particular art; in other words, when it becomes part of their common stock of knowledge relating to the art. Whatever else common general knowledge may be, it has never in my judgment included public knowledge of particular documents reports or scientific papers and the like. The knowledge of a number of individuals that a particular suggestion or particular suggestions has or have been made for the use of biasing in a particular apparatus, or a number of particular apparatus, cannot be held to be common general knowledge. It is certainly difficult to appreciate how the use of something which has in fact never been used in a particular art can ever be held to be common general knowledge in the art.
873 In addition to using common general knowledge on a standalone basis, common general knowledge can be aggregated with s 7(3) information.
874 Section 7(3) in its then applicable form relevantly provided:
For the purposes of subsection (2), the kinds of information are:
(a) prior art information made publicly available in a single document or through doing a single act; and
(b) prior art information made publicly available in 2 or more related documents, or through doing 2 or more related acts, if the relationship between the documents or acts is such that a person skilled in the relevant art in the patent area would treat them as a single source of that information;
being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant to work in the relevant art in the patent area.
875 The phrase “relevant to work in the relevant art” is directed to publicly available information, not part of the common general knowledge, which the skilled person could be expected to have regarded as relevant to solving a particular problem as the patentee claims to have done.
876 That part of the prior art base which is common general knowledge and the information referred to in s 7(3) are considered for the purpose of looking forward from the prior art base to see what the skilled person is likely to have done when faced with a particular problem.
877 In a case where the problem is known and is part of the common general knowledge, the problem will be similar to that which the patentee claims to have solved with the claimed invention. But where the problem addressed by the patentee does not form part of the common general knowledge, the relevant starting point is the prior art base, but not including the problem as identified in the patent specification. As the Full Federal Court noted in AstraZeneca AB v Apotex Pty Ltd (2014) 226 FCR 324 at [203]:
If the problem addressed by a patent specification is itself common general knowledge, or if knowledge of the problem is s7(3) information, then such knowledge or information will be attributed to the hypothetical person skilled in the art for the purpose of assessing obviousness. But if the problem cannot be attributed to the hypothetical person skilled in the art in either of these ways then it is not permissible to attribute a knowledge of the problem on the basis of the inventor’s “starting point” such as might be gleaned from a reading of the complete specification as a whole.
878 The purpose of the inquiry is to determine whether the invention is obvious to solve the perceived problem, looking forward from the prior art base. But of course this may not have been the patentee’s starting point.
879 GSK asserts that the obviousness inquiry also ends when the problem is solved. It is said that there is no motivation for the person skilled in the art to conduct further work once the solution to the problem is found. If further work is required to go beyond the solution to reach the claimed invention, it is said that such additional work is not work done as a matter of routine. Such further work constitutes a “voyage of discovery” to extend the field of human knowledge (Beecham Group Limited’s (Amoxycillin) Application [1980] RPC 261 at 296 per Buckley LJ). I will return to this question later as I do not accept this proposition in such absolute terms expounded by GSK. The fact that one problem is solved does not mean that another problem cannot also be solved. And if that other problem is solved as a “matter of routine”, and happens to be coincidentally the problem solved by the patentee, I do not see why a finding of a lack of inventive step would otherwise be foreclosed. Sections 7(2) and 7(3) do not enshrine GSK’s artificial boundary limit.
880 Finally, for the purposes of considering inventive step, the relevant skilled addressee in the present case is a team that includes someone with formulation skills. It also includes a clinician and a pharmacokineticist who would consider whether a formulation provided the required in vivo effects. Team members would also provide information about the API as well as feedback to the formulator for the purpose of making modifications to the formulation. In relation to expert evidence, the evidence of Dr Mooney and Professor Davies is relevant to assessing the knowledge and conduct of the hypothetical person skilled in the art.
(b) The impermissible use of hindsight
881 Hindsight or an ex post facto analysis should be avoided, to the extent possible, in deciding whether a claimed invention lacks an inventive step. Alphapharm warned against the clarity of hindsight and said at [21]:
The defendant to an infringement action who cross-claims for revocation on the ground of obviousness bears the onus of establishing that case. This obliges the defendant to lead evidence looking back to the priority date, sometimes, as here, many years before trial. In those circumstances, the warnings in the authorities against the misuse of hindsight are not to be repeated as but prefatory averments and statements of trite law. The danger of such misuse will be particularly acute where what is claimed is a new and inventive combination for the interaction of integers, some or all of which are known. It is worth repeating what was said by Lord Diplock in Technograph Printed Circuits Ltd v Mills & Rockley (Electronics) Ltd [[1972] RPC 346 at 362] … .
882 In my view, the respondents have engaged in an impermissible hindsight exercise.
883 For example, the respondents walked Professor Davies through, as GSK described it, a “step by step” tracking through GSK’s marketing application that summarised certain aspects of the lengthy research and development program that had led to the claimed invention. The respondents suggested that each step in the summary was “routine”. But in Technograph Printed Circuits Ltd v Mills & Rockley (Electronics) Ltd [1972] RPC 346 at 362, Lord Diplock criticised such an approach in the following terms:
I do not find it persuasive. Once an invention has been made it is generally possible to postulate a combination of steps by which the inventor might have arrived at the invention that he claims in his specification if he started from something that was already known. But it is only because the invention has been made and has proved successful that it is possible to postulate from what starting point and by what particular combination of steps the inventor could have arrived at his invention. It may be that taken in isolation none of the steps which it is now possible to postulate, if taken in isolation, appears to call for any inventive ingenuity. It is improbable that this reconstruction a posteriori represents the mental process by which the inventor in fact arrived at his invention, but, even it were, inventive ingenuity lay in perceiving that the final result which it was the object of the inventor to achieve was attainable from the particular starting point and in his selection of the particular combination of steps which would lead to that result.
884 Relying upon a summary of the development process compiled after the invention was made is impermissible hindsight. The marketing application summarised the development path taken to reach the invention. But it did not describe all of the problems, blind alleys and choices of options encountered by the patent applicant along the path to the invention. To present its evolution as some seamless and inevitable linear progression involving no more than the application of routine methods is to engage in some form of Whiggish ahistorical hindsight approach.
885 Further, the hindsight nature of the respondents’ case is also demonstrated by their reference to the Patent and GSK’s marketing application to support the choices made by Dr Mooney in postulating his formulations. It is trite to observe that once the claimed invention is known and has been proved successful and the ultimate pathway to reach the invention is laid out, it is easy to ex post facto reduce each step of that ultimate pathway to a trivial level or to assert it to be “routine” or obvious. But the statutory test looks forward from the prior art base with all the potential uncertainties. Apotex sought to trivialise each step of the drug development process, but Professor Davies resisted this. Professor Davies described the conduct of clinical trials as involving “intellectual input from a number of parties”. Apotex sought to diminish this to the proposition that the conduct of a clinical trial just involved taking small blood samples at various time points from a small number of volunteers. Professor Davies resisted the diminishment:
MS GODDARD: And you say that’s a complicated trial, do you? --- You’re trivialising it. It is much more complicated than that. It’s the — by the time you get to that point you’ve already manufactured the formulation, the formulation has to go for three months for stability before it actually gets to — before it’s allowed to be given to volunteers.
886 The formulator’s role in drug development was similarly sought to be diminished. The suggestion was put that following a clinical trial of a formulation that did not achieve the desired result, it would be a routine matter for the formulator to “improve something like bioavailability and perhaps chang[e] the release profile”. But that level of generality rather conceals the real point. It was more a question of what choices would need to be made; there would be a significant variety of different choices.
(c) The process of developing new sustained release formulation for paracetamol
887 I have set out in the earlier background sections of my reasons the general process used to develop a modified version of an existing formulation. It is now necessary to be more specific.
888 Paracetamol in its immediate release form was a well-known and widely used drug by April 2000. Paracetamol had been available commercially in Australia since 1956. The development brief an R&D team would have been given would have included the physiochemical properties of paracetamol, such as its solubility, stability, decomposition, absorption and metabolism under different conditions and whether there were any commercially available sustained release forms of paracetamol, including information as to whether there was anything wrong with such formulations. In the development brief would be pharmacokinetic data for the immediate release form of paracetamol, including minimum and maximum therapeutic blood plasma levels, rate of absorption, toxicity and overdosing levels and the elimination rate of paracetamol, which affected its duration of action. For example, drugs having elimination half-lives of one hour or less are more difficult to formulate as sustained release formulations because the high rates of elimination from the body are likely to mean that a large drug dose is required to meet the target sustained therapeutic effect.
889 If a complete suite of pharmacokinetic information was not provided with the brief, the clinician or pharmacokineticist in the R&D team would carry out a literature review to see whether they could find this information. If the information was not available in the literature, or perhaps was not available definitively, it would be important to establish such information through initial biostudies using the immediate release form of paracetamol at the outset of the project.
890 The development brief might also set a desired target of dosing frequency, which would be once, twice or three times daily dosing. One would also expect, even if not told, that the sustained release paracetamol formulation would be used to provide ongoing, continuous, pain relief, for example, for those suffering chronic pain.
891 Even if the physiochemical properties of paracetamol were identified in the development brief, one would carry out pre-formulation studies to confirm the fundamental physicochemical properties of the source and grade of paracetamol one was working with. Usually, formulators and the other members of the R&D team would not rely upon reports of the work of others, but would undertake their own pre-formulation studies on the particular source of the drug that the formulator was working with, even if there was published literature on the chemical and physical properties of the drug in question. Pre-formulation studies would allow the formulator to build up a detailed picture of the properties of the API and to plan the formulation strategy to try to anticipate and overcome any issues that might arise.
892 Further, at the outset the team would use pharmacokinetic data to determine a desired in vivo therapeutic target for the sustained release formulation. If there was no suitable existing sustained release formulation upon which to base the therapeutic level (or window), the therapeutic level for the new sustained release formulation would be determined from pharmacokinetic data for the immediate release form of paracetamol.
893 The R&D team would review the data generated from the pre-formulation studies, together with information concerning the anticipated dose, intended therapeutic use, route of administration and dosage form, which would identify any issues that needed to be addressed. The formulator(s) would be asked to attempt to address the issues that had been identified, by recommending a safe and effective dosage form. The appropriate formulation would depend on the physicochemical properties of paracetamol and the therapeutic target, as determined by conducting the routine pre-formulation studies and the initial pharmacokinetic investigations.
894 The pre-formulation work generally would have been likely to establish how much of the drug might be required to obtain the target therapeutic effect, but may not be known with certainty at this early stage. Immediate release forms of an API can have different relative bioavailability to sustained release forms.
895 Further, anticipating what the frequency of dosing might be to provide sustained pain relief would also be likely to have been identified by the pre-formulation work (assuming the team was not told in advance of the desired frequency of dosing). The frequency of dosing would be informed by factors such as how much of the drug was to be administered to obtain and maintain the therapeutic effect, and the impact of that dosage amount on the size of dosage form, any toxicity issues, and the elimination half-life of the drug.
896 It would be important to identify the therapeutic target, likely API amounts and likely frequency of dosing at the initial stages of the R&D project, so that informed choices about formulation options, such as its dosage form and its excipients, could be made early in the development project. Because of the significant time and costs associated with all stages of R&D projects, there would be little room within an R&D project to speculate about possible formulations which were not supported by pre-formulation and pharmacokinetic data.
897 The formulator would also need to consider the solubility and rate of dissolution of an API, which would be critical properties. I have explained dissolution tests in the earlier background sections.
898 Formulators would make use of in vitro data to streamline formulations for further development. For example, a poor dissolution rate in vitro could be predictive of a poor dissolution rate and hence poor absorption and bioavailability in vivo. If a formulator obtained a poor dissolution rate in vitro, they would not move to the next step of in vivo testing. The formulator would take steps to improve the dissolution rate of the API, and then repeat the in vitro dissolution tests to assess whether the dissolution rate of the API had been improved. This process would be designed to ensure that a formulation with the best possible release profile was taken into clinical trials. Similarly, a negative API stability result during in vitro stability tests could be predictive of a negative stability result in vivo. For example, if the API was shown to degrade under acidic conditions designed to mimic the conditions in the stomach, a formulator would have little expectation of a different outcome in vivo to the in vitro tests that had been carried out. Based on such results, the formulator would use an enteric coating for the formulation.
899 Although formulators would make use of in vitro data to streamline formulations or to identify a candidate formulation for further development, in vitro dissolution data would not be used by formulators to predict the in vivo efficacy of a formulation. The in vivo efficacy of a formulation had to be established by clinical trials. One would not expect that any in vitro profile would provide particular in vivo efficacy (save that a poor in vitro profile might entitle one to assume a lack of advantageous in vivo efficacy), nor would one anticipate that any particular in vivo profile would relate to a particular in vitro profile.
900 As the formulation progressed through the development process, the in vitro dissolution profile would often be used as a means of quality control of the reproducible manufacture of that product, to ensure that it was prepared in a consistent manner. It was important that the formulation was reproducible as it proceeded through the clinical trials and ultimately to a marketed product.
901 Once pre-formulation studies had been carried out and a therapeutic level (or window) had been determined, the team or formulators would consider options for formulating a sustained release formulation of paracetamol which met the desired therapeutic level. In general terms, the objectives for selecting a suitable type of formulation would include the following. First, to release the paracetamol in appropriate amounts at appropriate times to avoid “dose dumping” (the unintended release of a large portion of the drug). Given that the sustained release product might well contain more drug than an immediate release product, there would be a risk that if the formulation was not sufficiently robust, too much of the drug might be released over too short a time. Second, assuming the formulation was an oral formulation, to ensure the formulation was easily ingestible. Third, to understand and minimise side effects or other undesirable pharmacological responses to a sustained release form of the drug (for example, bioavailability might be decreased if the sustained release was longer than the gastrointestinal transit time). Fourth, to minimise costs associated with manufacturing the formulation. Fifth, whilst a formulator might have little, if any, idea of what a sustained release formulation’s in vitro release profile meant in terms of in vivo efficacy until in vivo clinical trial studies had been carried out on that formulation, an in vitro release profile somewhat slower than the immediate release formulation would initially be targeted.
902 There would be numerous different formulation starting points. Pharmaceutical companies would usually trust the technology they knew. A given company might have a favoured development path, for example, if they had experience in using specific excipients, and they already had the manufacturing equipment and formulation knowledge of a proven type.
903 Controlled release formulations could be in the form of tablets or capsules containing beads or pellets. Pellets may be designed to release the drug in a sustained release way either by using excipients within the pellets to retard release or coating them with film to either delay or retard release. Tablets or capsules could be formulated to delay release of a drug by utilising different mechanisms. First, by erosion, where the degradation of the surface of the formulation (principally by the ingress of gastrointestinal fluids) released the drug. Second, by diffusion from a matrix system, where drugs moved from a region of high concentration (in a tablet) to a lower concentration (in the gastrointestinal fluids). A problem with tablets which released drug by diffusion (only) was that they tended to be made from hydrophobic materials such that the tablets might not be formulated to readily degrade, leading to risk of slower gastric emptying within the GI tract. Third, by both erosion and diffusion. As at April 2000, the majority of sustained release tablets released drugs by a combination of erosion and diffusion. Fourth, by the use of a rate controlling membrane, where the drug was retained within a reservoir and was released in a controlled way through the coating membrane. Fifth, by ion exchange, which relied on the drug being mixed with a charged (either anionic or cationic) resin, with drug release occurring due to attraction to the oppositely charged contents of the gastrointestinal tract. Sixth, by osmotic pressure, for example by the osmotic delivery system tablet where a semipermeable membrane coated a matrix containing polymers which hydrated and swelled on exposure to water, with the membrane releasing the drug via a laser drilled hole within the membrane. A benefit of an osmotic delivery system was that it potentially provided a continuous release of drug.
(d) The problem to be solved
904 As at the priority date, paracetamol formed part of the common general knowledge, including its use as an analgesic and an antipyretic agent, its physicochemical properties and its useful dosages and immediate-release dosage regimes. Further, the dosage regime for immediate-release paracetamol was known. This was 500 mg to 1 g every 4 to 6 hours (i.e. 4 to 6 times per day), not exceeding 4 g a day. Further, it was known that it was more convenient for patients to take a medication fewer times per day, including reducing dosing from four times per day to three times per day. This convenience had the beneficial effect of increasing patient compliance. It was also known that paracetamol was a first-line treatment for patients with chronic pain, that patients taking paracetamol could wake during the night from pain because the analgesic effects ended before the patient had completed a night’s sleep and that available immediate-release paracetamol formulations had many of the desirable properties of an analgesic, including providing good immediate relief, but the duration of pain relief was relatively short.
905 It was also common general knowledge as at the priority date as to:
(a) the use of sustained-release formulations to extend the period over which an active pharmaceutical ingredient would be bioavailable;
(b) the use of matrix-forming polymers as part of a sustained-release formulation;
(c) the use of bilayer tablets comprising an immediate-release layer and a sustained-release layer to provide sustained-release formulations;
(d) the use of bilayer tablets in which the immediate-release layer provided initial rapid release of a therapeutic dose of the active pharmaceutical ingredient and the sustained-release layer provided delayed release of the active pharmaceutical ingredient over a longer period of time;
(e) the fact that a reduced fluctuation index was desirable; and
(f) the fact that paracetamol products presented a risk of overdosing.
906 GSK has submitted that the problem sought to be addressed by the Patent was to improve upon the extended release McNeil Tylenol Extended Relief product (ER Tylenol). But by this characterisation, Apotex contends that GSK has attempted to confine the case to one where the problem was not part of the common general knowledge. It is accepted by Apotex that neither the McNeil patent (and the associated 522 Patent) nor any issues with ER Tylenol, viz. its higher fluctuation index and problems with overdose, were part of the common general knowledge.
907 Apotex has contended that the approach of deriving a non common general knowledge starting point from the specification was expressly rejected by the Full Federal Court in AstraZeneca AB v Apotex Pty Ltd (2014) 226 FCR 324 at [203]. Further, it says that neither of the experts who gave evidence on inventive step agreed with GSK’s characterisation of the problem. Apotex says that both Dr Mooney and Professor Davies agreed in the JER that the problem addressed by the Patent was as follows:
As described by the patent, the problem is to develop an extended release paracetamol formulation that may improve patient’s [sic] quality of life by reducing the number of doses to be taken and in increasing the dose in the formulation, to address overdose concerns by limiting the amount of paracetamol absorbed in the first few hours but have rapid onset of action and prolonged therapeutic levels in the blood (compared to immediate release formulations) and providing steadier levels of the drug in blood. The inventors describe an advantageous pharmacokinetic profile for such a formulation (see pages 1–4 of the patent).
908 It is said that this description of the problem comes from p 2 of the Patent and is effectively what Dr Mooney was asked to address in the development brief. Accordingly, it says that the problem was common general knowledge.
909 Apotex says that the Patent itself does not pose the problem to be addressed in terms of improving upon the McNeil patent. It says that read in context, the reference to the McNeil patent in the specification is no more than a conventional description of the prior art with an express or implicit assertion of superiority over it.
910 Further, Apotex says that although this was not common general knowledge, asserted deficiencies with ER Tylenol were readily apparent to Dr Mooney upon his examination of the data in Tables 2a and 2b of the 522 Patent. I will return to this later as I do not agree that there were such deficiencies as asserted by Dr Mooney.
911 Contrastingly to Apotex’s approach, GSK has defined the problem differently. GSK contends that the problem articulated in the Patent is to improve upon ER Tylenol. The Patent at p 1 lines 29 to 32 to p 2 line 3 refers to the McNeil patent which is said to disclose a sustained release bilayer tablet containing 650 or 667 mg of paracetamol which contains an equal amount of paracetamol in the immediate and sustained release layers. Such a tablet is said to be marketed by McNeil Inc as ER Tylenol. The invention is then discussed by reference to the existing McNeil sustained release bilayer paracetamol tablet and the advantages it provides over that tablet. For example, at p 15 lines 4 to 7:
These advantageous properties of formulation C are particularly surprising when compared with the plasma concentrations described in Example 1 of [McNeil] which suggests that the Cmax of the prior disclosed sustained release paracetamol formulation is as high as that observed for an immediate release formulation.
and at p 17 lines 7 to 13:
The substantially lower FI for the SR product is surprising considering previous reports for a steady state biostudy conducted with a 650mg bilayer tablet (Tylenol Extended Relief) which showed that the SR product had a numerically higher FI (of 1.49) compared to a reference 500 mg IR tablet (of 1.44) as illustrated in Figure 4. Furthermore, Paracetamol plasma levels were maintained substantially above 3mcg/ml for the entire study period, which is in contrast to the steady state study reported for Tylenol ® Extended relief.
912 That ER Tylenol had problems such as accidental overdose and a higher fluctuation index than the immediate release paracetamol product is information which did not form part of the common general knowledge of the relevant skilled addressee.
913 It is said that it is clear from a reading of the Patent as a whole that GSK recognised these problems and ultimately arrived at an invention that surprisingly overcame such matters.
914 I am not at all convinced as to GSK’s articulation of the problem it was solving. True it is that it sought to develop its own product once it knew of ER Tylenol. True it is that once it developed its product it was able to extol its virtues and advantages over ER Tylenol. But I am not at all convinced that its perceived starting point was an identification of problems with ER Tylenol. I accept Apotex’s contentions and its identification of the problem as being part of common general knowledge. But even on that foundation Apotex has still failed to identify that there was a lack of inventive step.
(e) Common general knowledge alone
915 Let me first address the evidence of Dr Mooney.
916 Dr Mooney understood from the development brief that he should try to develop a formulation that would allow dosing three times per day, instead of four times per day, providing both rapid relief from pain as well as an extended duration of relief. There was substantial agreement between Dr Mooney and Professor Davies that:
(a) it would be possible to reformulate paracetamol in order to achieve rapid pain relief and an extended release profile, although Professor Davies would have first tried a single phase approach;
(b) there is more than one formulation option that could be used to achieve this;
(c) a bilayer tablet with an immediate-release layer and a sustained-release layer is one option; and
(d) the formulation could be varied to further change the in vivo efficacy of the formulation depending on the in vivo results that were obtained from testing in patients.
917 In response to the development brief, Dr Mooney proposed the essential elements of an initial formulation to satisfy the stated patient considerations in his affidavit. He did this before conducting any literature or patent searches. Dr Mooney chose a tablet. He did so because in his experience, patients preferred tablets and because in his experience they were easier and more cost effective to manufacture than other dosage forms. Further, in order to provide a formulation that provided both rapid and continued relief, apparently the first option that occurred to Dr Mooney was a bilayer, biphasic tablet, that is, a tablet with an immediate-release layer and a sustained-release layer. Apotex says that in criticising this choice, Professor Davies did not suggest that the use of a bilayer, biphasic tablet would have been an inappropriate option. And such a solution was common general knowledge. Apotex says that Professor Davies’ criticism of the choice of a bilayer tablet appears to be no more than an expression of his preference for using a capsule as part of a solution. But while he considered a bilayer tablet to be uncommon, he did not provide evidence that multiphase capsules were any more common. Apotex says that in any event, the existence of more than one solution to a problem does not prevent a conclusion of lack of inventive step. The fact that it may have been obvious to develop a formulation to meet the development brief using a capsule does not mean that it is not also obvious to use a biphasic tablet.
918 Dr Mooney also initially aimed to develop a product that could be dosed three times daily by taking one or two tablets at each dose. Understanding the risk of overdose, he said that he realised that he could not exceed the 4 g maximum dose of paracetamol in one day and that a dosage form that contained more than 1 g of paracetamol would be too large for patients to swallow. Accordingly, he arrived at a formulation of a daily tablet consisting of about 650 mg of paracetamol (noting that the actual figure was 667 mg but preferring to deal with units of 50). Thus, apparently by a simple process of mathematics based on available dosage information, he arrived at the dosage for a product that could be dosed three times daily by taking two tablets of 650 mg of paracetamol.
919 Dr Mooney (prior to seeing the 522 Patent) also initially proposed to develop and test a formulation containing between 200 mg and 300 mg of paracetamol in the immediate-release layer, noting that he could likely use less than 250 mg in the immediate-release layer and between 350 mg and 450 mg in the sustained-release layer. The proportion of paracetamol in each layer was arrived at by Dr Mooney based on what he said were the following factors. First, the need to develop a dose form containing less than 1 g of paracetamol that could be dosed on its own or as two tablets. Second, in determining the proportion in each layer, he was apparently guided by what dose of paracetamol was needed to achieve a blood concentration of paracetamol which was safe and therapeutic. Third, knowing that the immediate-release product contained 500 mg of paracetamol, he expected that a 500 mg dose would achieve a therapeutic effect. He believed two doses of 250 mg would achieve a therapeutic effect. Fourth, he also was aware that at the priority date there was available an immediate-release product of paracetamol that was scored and could be taken as a half dose of 250 mg.
920 In determining the proportions in each layer, Dr Mooney also said that he knew from experience that a product containing an immediate-release layer and a sustained-release layer would usually have a higher amount of the active pharmaceutical ingredient in the sustained-release layer.
921 Dr Mooney said that he expected that a dose of less than 250 mg in the immediate-release layer in combination with a sustained-release layer could achieve a therapeutic effect because, in a bilayer tablet formulation as he proposed, the paracetamol from the immediate-release layer would be supplemented by paracetamol in the sustained-release layer, which would start to be released from the sustained-release layer at a similar time to the immediate-release layer. Further, he said that two tablets would be taken, which in aggregate would be considered by reference to the known use of a 500 mg immediate-release tablet. Dr Mooney considered dosing two tablets, given the objective of delivery of 1300 mg three times per day up to a total maximum daily dosage of 4 g. As to whether there was inconsistency between Dr Mooney’s affidavit evidence and his evidence in the JER, Apotex says that I do not need to resolve this because the skilled person is a hypothetical person, whose views are informed by the evidence, rather than being an avatar for the views of the actual witnesses in court. Further, it is said that the position explained by Dr Mooney in the JER was sound and logical. Further, it is said that Professor Davies accepted that the minimum effective dose could be determined from the available immediate release product.
922 Accordingly, it is said that Dr Mooney had enough information about the likely dose distribution in each layer to propose an initial formulation that he believed would achieve the desired therapeutic effect.
923 In relation to the physical characteristics of the formulation proposed by Dr Mooney, he explained that the immediate-release layer of paracetamol would be easy to construct since he would use commercially available paracetamol in directly compressible form, which included excipients.
924 As for the sustained-release layer, Dr Mooney said that he immediately considered using a matrix-type formulation with an appropriate polymer because this was an efficient approach to formulating a high dose option for a sustained-release formulation. Dr Mooney said that such a matrix formulation would be appropriate, because he was aware at the priority date that this would allow a smaller amount of polymer to be used in the matrix and that this would be useful where he was developing a high dose of drug substance in the sustained-release layer (i.e. between 350 and 450 mg of paracetamol).
925 In relation to the choice of polymer, Dr Mooney initially said that he would use HPMC, Eudragit or ethylcellulose in common grades. In relation to the excipients, Dr Mooney said that he would use standard excipients.
926 Contrastingly and generally, GSK contends that the respondents’ approach to inventive step is flawed. It is said that the development brief and Dr Mooney’s unquestioning acceptance that it was all he had, is a highly unrealistic, artificial exercise, that Dr Mooney agreed bore little resemblance to the real world process of development of a new formulation at a pharmaceutical company.
927 I agree with GSK’s contentions.
928 First, Dr Mooney agreed that the development brief did not in relation to paracetamol give him the therapeutic window, the half life, the solubility or relevant Cmin and Cmax.
929 Second, as at April 2000, Dr Mooney had never been asked to develop a new solid oral dosage form with the little information that was available in the development brief. Further, I note that Dr Mooney had never developed a sustained release dosage form where there was not one already available.
930 Third, Dr Mooney agreed that given the lack of information in the development brief, his approach to his proposed formulation did not reflect the reality of a development process and was speculative. The steps taken by Dr Mooney were not those steps which a person of ordinary skill would take as a matter of routine from the prior art to the invention.
931 Fourth, the respondents’ case based upon common general knowledge alone was also dependent on the 250 mg dose therapeutic effect assumption. Dr Mooney’s justification for trying a formulation with less than 50% of the paracetamol in the immediate release layer was his earlier stated unproven assumption that 250 mg of paracetamol would achieve “a therapeutic effect”. The basis for this assumption was said by Dr Mooney to be that to the best of his knowledge before April 2000, there was a 500 mg immediate release paracetamol tablet that was scored “and could be taken as a half dose of 250 mg”. Dr Mooney concluded in his affidavit “I therefore expect that a dose of 250 mg [would] achieve a therapeutic effect”. Dr Mooney explained in his oral evidence that by referring to the dose achieving a “therapeutic effect”, he did not say it would achieve “therapeutic efficacy”, for example, it may not reach the Cmin. I agree with GSK that Dr Mooney’s assumption in his affidavit that a 250 mg dose of paracetamol would achieve a therapeutic effect was without foundation. Dr Mooney agreed under cross-examination that there were many reasons why a tablet may be scored; for example a child’s dose or to provide an intermediate 750 mg dose. Dr Mooney also agreed that he did not know if a 250 mg dose would provide the therapeutic effect from the information provided in the development brief.
932 Generally, in my view, Dr Mooney’s assumption as to the therapeutic effect of a 250 mg dose was speculative and did not reflect a typical drug development process.
933 There was also a shift in Dr Mooney’s evidence from his affidavit to his answer in the JER where he relied on dosing two tablets to provide 500 mg of immediate release paracetamol. In his affidavit, Dr Mooney did not rely on dosing with two tablets to achieve his “therapeutic effect”. Professor Brooks and the development brief gave the conventional IR dose as 500 to 1000 mg. Apotex has sought to ignore the shift in Dr Mooney’s evidence and to retrospectively justify Dr Mooney’s selection of 250 mg but in my view the shift in his evidence was significant and diminished the weight of his evidence.
934 Fifth, Dr Mooney immediately selected a bilayer tablet, rather than for example a capsule using beads or pellets whose technology was more developed and which were more common than bilayer tablets, or a single-layer tablet. Professor Davies explained that, in his experience, such formulations significantly release drug in the early phase thus avoiding the need for an immediate release layer.
935 Sixth, as at the priority date, Dr Mooney had never worked on a bilayer dosage form. Neither his employer, Alphapharm, nor anyone else in Australia had the plant and equipment necessary to produce a bilayer tablet and the only bilayer tablet he had heard of was one comprising two different active pharmaceutical ingredients (one that was enterically coated) that had been released in Europe and the United States. Further, it is implausible that an addressee would as a matter of routine move from the prior art to a formulation requiring the purchase of new equipment. On the one hand Dr Mooney confined himself to the equipment available at Alphapharm in April 2000, yet still said that for the purpose of developing his hypothetical formulation, he would buy a bilayer compression machine. I must say that I found that suggestion implausible.
936 Seventh, the respondents’ case relied on formulating a sustained release matrix which was not a typical matrix.
937 Eighth and generally, in my view Dr Mooney is an inventive and clever formulator. Generally, it would seem that thinking as creatively as he could as to how to deal with the challenge put to him, he immediately selected a bilayer two phase tablet. In the JER, Dr Mooney sought to defend his selection as being “based on experience with development of tablets as an oral dosage form”. But his experience with bilayer tablets was largely non-existent. Dr Mooney also properly conceded that his formulation decisions were artificially constrained and that his formulation selection was also artificially confined with reference to the particular equipment and expertise available at Alphapharm. I agree with GSK’s submissions that his selection was also made despite:
(a) knowing that sustained release dosage forms at 2000 tended to be single phase;
(b) knowing that modifying a monolithic sustained release profile was an option;
(c) knowing that dosage forms with more than one phase were uncommon, both in Australia and around the world;
(d) not having any experience preparing a dosage form with more than one phase prior to 2000;
(e) his only experience in making a sustained release tablet being to make a generic product, that is, when there was a corresponding sustained release dosage form in the market somewhere, and there was no immediate release phase in the dosage form;
(f) not having any experience prior to 2000 in formulating multilayer tablets;
(g) being aware that bilayer tablets require special equipment and being unaware of any bilayer machines in Australia at 2000;
(h) being only aware of one actual bilayer product at 2000; the product was not an immediate release/sustained release tablet, instead it had a first portion with an enteric coat which coated an immediate release form of a first active pharmaceutical ingredient and a second portion which comprised a mantle which comprised a second active pharmaceutical ingredient;
(i) being unaware of any bilayer tablet with sustained release and immediate release phases on the market in Australia at 2000;
(j) being aware that there was nothing typical about a bilayer tablet with immediate release and sustained release phases;
(k) being aware of the manufacture of beads, and being aware of a sustained release product on the market at 2000 which was an extended release capsule product;
(l) having not considered the results of any relevant literature (which he said in his affidavit would be the first step in a development process).
938 In my view, in April 2000 it was not a routine step to those skilled in the art in Australia to create a bilayer tablet for a sustained release product.
939 Dr Mooney acknowledged that his common general knowledge alone approach, based on simply looking at some immediate release paracetamol products and noting a score line, and relying on the development brief, did not reflect commercial reality. Further, his approach did not demonstrate a series of routine steps taken by a skilled but unimaginative worker.
940 Dr Mooney further agreed that there was no way to know whether any of his proposed formulations or in vitro profiles would meet the in vivo clinical targets. If one was trying to create for the first time a sustained release formulation one did not know the anticipated profile. Further, there was not necessarily a correlation between an in vitro profile of a drug and an in vivo consequence. Further, assuming that one was taking three doses a day, one did not know what the dissolution profile should be at two and a half hours or three and a half hours. Further, if one had a formulation with a desired in vitro profile, one tested it with clinical trials. If it did not give the desired pharmacokinetic properties, one would consider the formulation and data one had come up with and a new formulation and a new in vitro profile and try again.
941 Professor Davies considered that a more logical approach in answer to the development brief would be to first understand the suite of physicochemical and pharmacokinetic data and try to discern an optimal in vivo release profile. Once he had a target profile in mind, Professor Davies would consider whether a single system sustained release formulation might result in the advantageous pharmacokinetic profile or whether a more complicated formulation might be more desirable.
942 The respondents contend that the common general knowledge documents in evidence such as MIMS and Martindale contain much information that a person skilled in the art would readily have to hand. In particular, with respect to paracetamol this included the following:
(a) dosage and administration;
(b) maximum daily dose of 4 g;
(c) the elimination half life of paracetamol of 1 to 3 hours;
(d) the solubility of paracetamol (as sparingly soluble);
(e) the presentation of paracetamol, including scored tablets;
(f) that paracetamol is readily absorbed in 10 to 60 minutes;
(g) toxicity and overdose.
943 But I do not think that this carries the day.
944 In summary, in my view the evidence did not establish that a person skilled in the art would, by a matter of routine steps, move from the prior art in terms of common general knowledge to the invention claimed in each of the claims. To do so in the present case, Dr Mooney used ingenuity and cleverness well beyond the person of ordinary skill taking merely routine steps.
(f) The 522 Patent — Apotex’s principal case
945 It has been accepted by the parties that the 522 Patent is s 7(3) information that can be aggregated with common general knowledge.
946 Dr Mooney read the 522 Patent after he had proposed the basic elements of his initial formulation as set out earlier. The 522 Patent described a bilayer biphasic tablet of acetaminophen with an immediate-release and a sustained-release layer. The 522 Patent reported on a small-scale study of 12 adult male subjects, half of whom were given two 500 mg immediate-release tablets and half of whom were given two 650 mg bilayer biphasic tablets made using the formulation disclosed in the 522 Patent. Their blood concentrations were taken over nine time intervals and the averaged results provided in a table. The conclusion stated that the bilayer formulation of Example I (acetaminophen sustained release bilayer tablet) could extend the dosing interval to at least eight hours. Surprisingly in my view, Dr Mooney asserted that he did not agree that this was achieved. At this point I should note that Dr Mooney sought to second guess the results reported in the 522 Patent in a manner that was not justified in relation to Example I. I will return to this later. Apotex contends that the 522 Patent provided Dr Mooney with at least the following information:
(a) A recipe or list of components in a method to make an oral sustained-release form of acetaminophen including a bilayer tablet with an immediate-release and a sustained-release layer;
(b) The formulation contained a sustained release matrix which included a polymer, being hydroxyethylcellulose in combination with PVP;
(c) That it was beneficial to provide an acetaminophen product that extended the dosing interval beyond six hours while maintaining therapeutically effective levels of acetaminophen in the blood;
(d) The therapeutically effective levels of plasma concentration of paracetamol were likely to be about 2.9 mcg/ml, equivalent to the average plasma concentration level of the immediate-release product at six hours as shown in Table 2b (which required re-dosing at six hours). I should say that I disagree with this assertion. Table 2a shows 2.6 mcg/ml for the sustained release formulation at 8 hours and the 522 Patent in effect represents this to be an effective therapeutic level at 8 hours;
(e) The formulation could be dosed as two tablets each containing 650 mg of acetaminophen in a composition containing an IR and an SR layer;
(f) That the dosing intervals could be extended to eight hours (referring to Column 1, lines 53 and 54);
(g) That the relevant proportions of hydroxyethylcellulose and PVP were in the order of about 4% or less of the amount of acetaminophen (referring to Column 4, lines 4 to 9). While the small proportion of matrix binding agents was asserted to be a novel feature of the invention (at its priority date of 27 July 1987) and the preferred embodiment of the invention used only about 3% of hydroxyethylcellulose (Column 4, lines 40 to 42), Dr Mooney and Professor Davies were aware by the priority date of the Patent in suit of the ability to form a sustained-release matrix using a small amount of polymer;
(h) That the sustained-release matrix used well-known excipients such as disintegrants and water absorption promotion ingredients;
(i) The components of the method to construct a compressible bilayer tablet containing 325 mg in each tablet layer of paracetamol using wet granulation; and
(j) That an in vivo trial of the sustained-release formulation of Example 1, 2 tablets containing 650 mg each of paracetamol in each tablet, compared with the immediate-release paracetamol, 2 tablets containing 500 mg each of paracetamol in each tablet, showed a comparable extent of absorption measured by the AUC.
947 Surprisingly in my view, Dr Mooney did not agree that Example 1 demonstrated that eight hour dosing had been achieved for the sustained release formulations.
948 As concerns the choice of polymer, the binder used in the 522 Patent was a combination of hydroxyethylcellulose and PVP. Dr Mooney had already expressed a preference to use a polymer such as HPMC. He explained that he was not familiar with hydroxyethylcellulose as an excipient and he would use a more commonly used excipient from the cellulose family of polymers for creating a sustained-release matrix, including HPMC. Dr Mooney explained that he would not have used PVP because it is soluble and would not contribute to extending the release of paracetamol.
949 As concerns the proportion of paracetamol in each layer, apparently the information in the 522 Patent did not dissuade Dr Mooney from his earlier approach. His reasons for maintaining this view were said to be based on his reading of the data from the in vivo trials disclosed in tables 2a and 2b in the 522 Patent, as follows:
(a) That the time to Cmax (the Tmax value) was too rapid because the 522 formulation reached a Cmax of 12.8 mcg/ml at 1.5 hours after dosing, while the immediate-release formulation reached a Cmax of 12.1 mcg/ml 1 hour after dosing. In Dr Mooney’s experience, he would have expected a greater shift in Tmax than 30 minutes;
(b) The concentration levels of paracetamol in the in vivo study from the 522 formulation (measured in mcg/ml) at 1, 1.5, 2 and 3 hours in Example 1 were well in excess of the concentration levels of corresponding time points in the immediate-release tablets. This showed Dr Mooney, so it was said, that there was potential to extend the duration of release further by reducing those early concentrations;
(c) The concentration of paracetamol at 8 hours for the bilayer tablet of 2.6 mcg/ml was lower than the concentration at 6 hours of 2.9 mcg/ml for the immediate-release tablet (when re-dosing occurs).
950 For these reasons, Dr Mooney doubted, apparently, that Example 1 provided the required therapeutic levels of paracetamol and that genuine eight hour dosing was achieved, and he reasoned that it would be desirable to increase the concentration at eight hours. So he said, he saw the potential to improve the formulation by shifting the release of some of the paracetamol from a time when it significantly exceeded the minimum therapeutic level, to a later time when there was the potential for it to fall below such levels.
951 After reading the 522 Patent, Dr Mooney explained that he would conduct dissolution testing routinely as part of drug development to ensure that the formulation was pH independent and would release (i.e., dissolve) over a reasonable time frame. He initially proposed that the drug should have released by about 3 to 4 hours after ingestion. He believed that this time frame reflected the requirement that paracetamol be maintained in the blood at a level which would be therapeutic (i.e., at least 2.9 mcg/ml) for eight hours.
952 Dr Mooney proposed a dissolution profile that he would aim to achieve for his proposed formulation as follows:
30% to 60% at 30 minutes;
50% to 75% at 60 to 120 minutes (1 to 2 hours);
No less than 80% dissolved at 3 to 4 hours.
953 He explained that he would use USP type II apparatus, paddle at 50 rpm at 37C in 0.1 M HCL to test the dissolution profile. If the prototype floated (which the evidence revealed is a common problem with USP paddle apparatus) he would change to USP type I apparatus at 100 rpm in the same temperature and medium.
954 Dr Mooney’s evidence was that he would have tried to achieve a dissolution profile “similar to” the 522 Patent. His evidence was not that he was trying to match that profile precisely. He explained that similarity in this context means ±10%, which was in accordance with the FDA Guidelines.
955 Apotex contends that there was no inventiveness in the dissolution profiles proposed by Dr Mooney. As concerns the time points, the FDA recommended the use of three time points. The last time point should be at a point where 80% to 100% of the drug has dissolved. Given that Dr Mooney proposed that the paracetamol be dissolved preferably after three hours, this gives his final time point at 180 minutes. There should be an early time point when the paracetamol from the immediate-release layer and some from the sustained-release layer would have dissolved, namely, within 15 to 30 minutes. Finally there should be a time point somewhere in the middle. Thus Dr Mooney’s 60 minute time point simply reflects this middle point.
956 Apotex compared Dr Mooney’s proposed ranges with claims 1, 2 and 3 of the Patent. These showed the percentage of acetaminophen dissolved versus time. With respect to claim 1, Dr Mooney’s proposed ranges came within claim 1. With respect to the narrower claims, Dr Mooney’s ranges fell outside those ranges.
957 Further, Dr Mooney explained that his trial formulations would be revised depending on whether or not the in vivo studies showed that the formulations achieved the desired performance. In particular he would adjust the proportion of paracetamol used in the immediate-release and sustained-release phases of the tablet and he would adjust the excipients that affect the dissolution performance of the tablet, namely, the sustained-release polymer, including its amount and its molecular weight (which affects its viscosity) and other excipients that were related to dissolution rate, including disintegrant and wicking agents, by varying the excipient to polymer ratios. In so doing, Dr Mooney said that based on his experience as a formulator he would be confident of being able to make adjustments by “routine trial and error” so as to achieve a formulation that achieved the desired sustained-release performance for paracetamol, allowing 8-hourly dosing. Accordingly, Dr Mooney contemplated that his initial formulations would be revised, based on the in vivo pharmacokinetic data, following a trial in patients.
958 Further, while Dr Mooney considered his proposed formulations were good enough as a starting point, he also considered that something within his proposed range of trial formulations would be likely to achieve the in vivo profile he was aiming for. But if the formulation did not achieve the desired in vivo profile, he believed he would be able to revise the formulation to change the in vitro dissolution profile. For example, he could extend the time over which the acetaminophen was released from the dosage form by either increasing the amount of polymer, change to a polymer of higher molecular weight or vary the excipient to polymer ratio in relation to excipients that supported the sustained-release behaviour. Alternatively, he could do the opposite if he needed to speed up the release. His initial contemplated formulations also proposed to test different proportions in the immediate-release and sustained-release layer for 200/450 to 300/350.
959 Further, Dr Mooney said that in vivo clinical trials would need to be conducted to confirm whether or not any of his formulations achieved the desired in vivo targets. If the formulation did not do so, he said that he would use the results of the in vivo trials to guide modifications to his formulation within the ranges of his proposed formulation, for example by varying the grade of polymer.
960 Both Dr Mooney and Professor Davies agreed that clinical trials would conventionally be carried out. Apotex has submitted that the observations in AstraZeneca concerning the additional information as to dosages which was absent from the s 7(3) disclosure and the prior art, are applicable in this case (see at [44] per French CJ, [88] and [94] per Kiefel J and [116] per Gageler and Keane JJ). Further, Nettle J at [123] recognised that carrying out clinical trials was conventionally done and “fell within the concept of working towards an invention with an expectation of success and that was consistent with the conclusion that the invention was obvious”.
961 Finally, in response to GSK’s assertion that the 522 Patent solves the problem and that Dr Mooney should not have proceeded further, the respondents have advanced the following arguments.
962 It is said that GSK’s asserted requirement that the notional skilled person must stop all enquiries once any solution becomes apparent does not circumscribe what is, or is not, obvious within the meaning of s 7(2). It imposes a narrower limitation not found in the statutory language. The notional skilled addressee’s hypothetical response when equipped with the common general knowledge and then s 7(3) information is a matter of evidence. I agree with these submissions.
963 Further, it is said that GSK’s approach misstates the use which may be made of information in s 7(3) as explained by the High Court in AstraZeneca. Even if it were accepted that the 522 Patent solved the problem, it is impermissible to limit what use may be made of s 7(3) information in the way GSK suggests. I also agree with these contentions.
964 As Gageler and Keane JJ observed in AstraZeneca at [115]:
The question is not whether it would have been obvious to the skilled addressee to choose rosuvastatin over NK-104; rather, it is whether a person skilled in the art would, in light of the common general knowledge plus either the Watanabe article or the 471 patent, have been directly led as a matter of course to try rosuvastatin in the expectation that it might well produce a solution to the problem which existed in the common general knowledge. Section 7(2) does not contemplate that a choice between apparently effective solutions must be attributed to the notional skilled addressee, much less that the notional skilled addressee might be so befuddled by an embarrassment of choices as to cease pursuit of the solution.
965 I note that their Honours applied the reformulated Cripps question. Applying this to the present case, the question is whether the skilled person would, in light of the common general knowledge plus the information in the 522 Patent, have been directly led as a matter of course to try a formulation of paracetamol within the terms of claim 1 in the expectation that it might well produce a solution to the problem that existed in the common general knowledge. Section 7(3) information is not information that must directly lead to the invention. It is information that is relevant to addressing the problem. It is to be considered together with common general knowledge. In my view, to apply a test about whether or not there is motivation to improve upon a s 7(3) disclosure inappropriately confines the use of that information. The information in s 7(3) documents does not need to provide an alternative path directly leading to the invention.
(g) The 522 Patent — Analysis
966 In my view, Dr Mooney’s use of the 522 Patent does not support Apotex’s case on lack of inventive step.
967 First, Dr Mooney, as a highly skilled innovative formulator, looked at the 522 Patent not through the lens of the hypothetical non-inventive formulator in the field seeking to solve the problem in the development brief, but as someone bringing all of his creativity, innovation and lateral thinking experience to try to improve the 522 Patent to create a different product which gave a commercial opportunity or “the best” product.
968 Second, the 522 Patent did not provide any in vitro dissolution data for any formulations. It is difficult to see how Dr Mooney could in essence take Table 2a, in essence ignore its teaching for 8 hours, adjust the formulation to put more API in the sustained release layer and then speculate concerning both appropriate in vitro dissolution profiles and in vivo effects.
969 Third, Example I of the 522 Patent disclosed a bilayer tablet which contained 650 mg of paracetamol split equally between two layers (325 mg in an immediate release layer and 325 mg in a sustained release layer).
970 Dr Mooney agreed that the formulation of Example I of the 522 Patent disclosed an advantage, and that advantage was eight hour dosing (see col 8 lines 45 to 48). As Dr Mooney noted, “the formulation disclosed in the ‘522 Patent is stated to achieve the objective in the Development Brief of eight hourly dosing”.
971 Dr Mooney also agreed that the Cmax for Example I formulations was reached at a similar time to the immediate release formulation. Further, during cross-examination Professor Davies explained that the Cmax of Example I was comparable to or maybe slightly higher than the Cmax of the immediate release, and that it was not a general goal of a formulator to reduce the Cmax of an immediate release formulation when making a sustained release formulation. This was consistent with the disclosure in the 522 Patent itself which explained at col 8 lines 33 to 40:
The results show that two bi-layer tablets of Example I, when compared to two tablets of non-sustained release acetaminophen (1000 mg dose), achieve the following: comparable rate of absorption: comparable maximum plasma concentration; and comparable extent of absorption (AUC or area under the curve) when adjusted for dose.
972 Apotex’s assertion that the SR Cmax was “well in excess” of the Cmax of the IR is not supported by the evidence, especially at the early release times which would enable rapid pain relief.
973 Accordingly, the formulation of Example I of the 522 Patent provided the solution to the problems formulated by Apotex in the development brief. But I accept that Dr Mooney did not have to stop there.
974 Dr Mooney’s quest to revise the formulation beyond that of Example I to “potentially” provide lower early levels, whilst providing the same therapeutic efficacy, was undertaken by him not in response to the development brief and the problems set out therein, but in response to a perceived problem with the 522 Patent and involved, in my view, ingenuity and other than routine work.
975 Dr Mooney proceeded to revise the Example I formulation in a context where he did not know, and agreed that the development brief did not provide him with, information on:
(a) the therapeutic window of paracetamol or the therapeutic dose;
(b) the half life of paracetamol;
(c) the solubility of paracetamol;
(d) the Cmax of the immediate release;
(e) the maximum tolerated dose;
(f) any of the pre-formulation information and other matters identified in his affidavit.
976 Moreover, Dr Mooney’s approach was founded on the guess that 2.9 mcg/ml was the minimum therapeutic level for paracetamol and an unsupported assertion that the 522 Patent misrepresented that Example I would provide eight hourly dosing.
977 In my view, as I have already said, Dr Mooney ignored the express teaching of the 522 Patent of eight hourly pain relief to speculate that the 522 Patent formulation did not provide effective eight hourly dosing (i.e. 2.6 mcg/ml (at eight hours) was not therapeutically effective). The basis for Dr Mooney’s speculation (rejected by Professor Davies) was his assumption that the six hourly dose level of the immediate release product (2.9 mcg/ml) was close to the minimum therapeutic concentration. But in cross-examination, Dr Mooney conceded that at six hours the immediate release formulation could be above a minimum therapeutic effect but be below at eight hours (1.8 mcg/ml), meaning that the minimum therapeutic level may be in between 1.8 and 2.9 mcg/ml. Dr Mooney agreed that a tablet was not, in reality, going to be dosed at intervals longer than six hours but shorter than eight hours.
978 Generally, in this case, Dr Mooney had pharmacokinetic data for a suitable sustained release formulation (Example 1 of the 522 Patent) but chose to ignore or reject it (along with other statements made in the 522 Patent as to the efficacy of the Example 1 formulation), but at the same time to accept (and use as the foundation for his development project) pharmacokinetic information for an immediate release formulation provided in that patent. Dr Mooney’s approach was unrealistic. It could not reasonably be described as a series of steps taken as a “matter of routine”.
979 Fourth, Dr Mooney’s approach based on the 522 Patent was also founded on his assumption that a dose of 250 mg will give therapeutic efficacy. But this required him to assume that the 522 patentee’s statements at col 1 lines 34 and 35 and col 8 lines 45 to 48 were wrong. But this was contrary to the express teaching of the 522 Patent. It was also speculative.
980 Fifth, Dr Mooney said that in devising his proposed product, he would substitute the hydroxyethylcellulose specified in the 522 Patent for another matrix forming polymer (e.g. HPMC or ethylcellulose) simply on the basis of his peculiar lack of knowledge of hydroxyethylcellulose before the priority date. But hydroxyethylcellulose is said to be the preferred polymer matrix of the formulations disclosed in the 522 Patent. The use in the formulations of Example I of hydroxyethylcellulose did not routinely lead to the further work proposed by Dr Mooney. Hydroxyethylcellulose was at the priority date known to be suitable for use in a matrix for a sustained release formulation.
981 Sixth, and as I have said, the 522 Patent did not provide any in vitro dissolution data for any formulations. Dr Mooney acknowledged that because the 522 Patent only provided in vivo data, he had to intuitively work backwards to an in vitro profile. Dr Mooney conceded that his proposed formulation, having a predicted in vitro profile as set out in his affidavit, was only a prediction which was “something to investigate” and merely “a concept”. He was “not going to know about the in vivo performance until [he] test[ed] it”. His suggestion that the Tmax of a formulation might be extended by modifying the 522 Patent formulation was just a hypothesis to be explored with testing. Dr Mooney accepted in relation to his proposed formulation and its in vitro profile two qualifications. First, that there was no way to know whether his proposed profile would provide an effective formulation without carrying out in vivo testing of the subject formulation. Second, that he did not know if too much paracetamol might be released too early if 30 to 60 per cent of the paracetamol dissolved in vitro in the first 30 minutes, as he proposed.
982 I agree with GSK’s contention that given both the absence of foundation for the 250 mg dose therapeutic effect assumption and its centrality to the path of further work, and that Dr Mooney agreed that he could not predict the in vivo efficacy of his proposed in vitro profile, it is difficult to see how Dr Mooney or the hypothetical person skilled in the art could have sensibly embarked upon the further work with an expectation of success.
983 I accept Professor Davies’ evidence that:
Dr Mooney’s approach pre-supposes, without any basis in the data, that reducing the amount of active ingredient in the immediate release layer will provide a minimum therapeutically effective amount. It is improbable that a skilled formulator would make a guess of that nature. A formulator would not know whether reducing the amount of active ingredient in the immediate release layer might alter the part of the 522 patent formulation which enables that formulation both to give a high level of immediate pain relief in combination with longer lasting relief than is available from the immediate release tablet. Thus there could be no expectation that making the change Dr Mooney speculates upon would be successful in providing a formulation that would satisfy the requirements of the Development Brief.
984 Seventh, Dr Mooney’s approach was of course hypothetical. The respondents conducted no experiments, not even in vitro dissolution ones, notwithstanding that they assert that it is “inevitable” that Dr Mooney’s development work would have resulted in a formulation falling within claim 1 of the Patent.
985 In summary, I do not accept that Apotex has made out its case on lack of inventive step on the foundation of common general knowledge taken together with the 522 Patent.
(h) Combining the 522 Patent and the 194 Patent
986 An alternative basis that the respondents rely on is common general knowledge combined with Australian Patent No 751194 (the 194 Patent), which is accepted to be s 7(3) information, considered alone or with the 522 Patent. The 194 Patent cross-references the 522 Patent at p 3 line 4. It can be taken to incorporate the 522 Patent to that extent.
987 The 194 Patent was a further document that Dr Mooney identified as being relevant to making a formulation to meet the requirements of the development brief. The 194 Patent was published some ten years after the 522 Patent (1989) and shortly before the priority date.
988 Dr Mooney explained that in reading the 194 Patent he would have wanted to consider it together with the cross-referenced 522 Patent, in particular because he read in the 194 Patent that the 522 Patent involved use of a bilayer tablet, which was Dr Mooney’s first choice of a formulation method to address the development brief.
989 Dr Mooney explained that the 194 Patent provided additional useful information in developing a formulation in that it included a dissolution profile for Tylenol extended release paracetamol caplets, which would provide an initial in vitro dissolution profile to aim for (whether combined with the teaching in the 522 Patent or combined with Dr Mooney’s selection of a bilayer tablet using common general knowledge).
990 Professor Davies’ evidence about the disclosure in the 194 Patent was that:
I see no reason why a person skilled in the art in April 2000, having read the 194 patent, would want to make any change, let alone change the proportions of acetaminophen between the immediate release and the sustained release layers, based solely on the fact that one of the commercial immediate release tablets is scored (which appears to be the sole basis for Dr Mooney’s unequal split hypothesis in paragraphs 134, 135 and 187). In the absence of clinical trials demonstrating inadequate pain relief in the first hour, I would not have a basis in the data provided in the 194 patent to know whether a change in the proportions of paracetamol in the immediate release and sustained release layer would have an appropriate response and I note that there is no suggestion in the Development Brief that the conventional immediate release form fails to give adequate pain relief in the first hour.
991 The 194 Patent disclosed a sustained release paracetamol formulation made up of a blend of beads or particles of immediate release and sustained release paracetamol, which was delivered in a capsule or blister. Two example compositions were provided. The first appeared to comprise an equal amount of paracetamol in the immediate and sustained release bead material. The second comprised only extended release beads, with no immediate release paracetamol. The 194 Patent used ER Tylenol, known to be clinically effective at that time, as the bench mark for assessment of the 194 Patent formulations.
992 In vivo studies, reported in Figures 2 to 4, showed that the 194 Patent composition initially reduced fever at the same rate as immediate release paracetamol but continued to achieve temperature reduction for longer. A pain study reported that the 194 Patent composition provided equally effective pain relief over an eight hour period as ER Tylenol.
993 The formulation described in the 194 Patent was therapeutically effective in terms of providing rapid relief in the temperature study, matched the sustained release efficacy of ER Tylenol over eight hours in a pain study and had the same bioavailability as the Tylenol Immediate Release Elixir formulation.
994 But there was absolutely nothing in the 522 or 194 Patents which suggested that an equal amount of paracetamol in each of the two layers had failed to achieve an acceptable result. Quite the converse in each case. Dr Mooney had to concede that when he first read the 194 Patent he did not consider that it suggested an unequal split of paracetamol might be appropriate. Dr Mooney agreed in cross-examination that there was nothing in the 194 Patent claims which suggested an unequal split of paracetamol.
995 In my view, there was nothing in the combination of the 522 and 194 Patents which provided motivation for Dr Mooney to move beyond the solutions provided in both to undertake the further work proposed by Dr Mooney. And to the extent that he did so, the work was not routine, but rather involved ingenuity and cleverness on his part. Moreover, even if I were not to apply a reformulated Cripps question, but rather Apotex’s test taken from Lockwood (No 2), Apotex still fails on this ground.
(i) General
996 Further, there was no basis for saying that Dr Mooney’s proposed formulation would have a dissolution profile within the limits specified in claims 1, 2 or 3 of the Patent when tested under the conditions specified in those claims.
997 Further, having arrived at a prototype formulation and defined its theoretical dissolution profile, Dr Mooney explained that “the product would be evaluated in vivo to establish the pharmacokinetic behaviour and refinements to the formulation be made as required”. But even having devised a prototype formulation, there was no cogent evidence upon which to draw an inference that the prototype formulation would produce a therapeutic effect, let alone that such a formulation would have the particular advantageous attributes identified in the Patent.
998 Further, Dr Mooney did not provide any details of his prototype formulation, beyond possible ranges of paracetamol in each of the IR and SR layers.
999 In my view, the respondents’ case on lack of inventive step has not been made out. It has failed on the various permutations of the application of ss 7(2) and 7(3).
Conclusion
1000 None of the respondents’ arguments on invalidity have been made good. Accordingly, their cross-claims will be dismissed. But as I have said earlier, GSK has failed on its infringement case as “basket” in claim 1 means basket. Each of the parties has had a measure of success. My tentative view is to make no order for costs in favour of any party, but I will give the parties an opportunity to make submissions thereon including on any consequential orders.
I certify that the preceding one thousand (1000) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Beach. |
Associate:

