
FEDERAL COURT OF AUSTRALIA
Apotex Pty Ltd v Les Laboratoires Servier (No 4)
[2015] FCA 104
IN THE FEDERAL COURT OF AUSTRALIA | |
APOTEX PTY LTD ACN 096 916 148 Applicant | |
AND: | First Respondent SERVIER LABORATORIES (AUST) PTY LTD Second Respondent ACTAVIS PTY LIMITED ACN 003 854 626 Opponent |
DATE OF ORDER: | |
WHERE MADE: |
THE COURT ORDERS THAT:
1. Until further order the publication of the reasons for judgment published in this matter today (the reasons) (but not these orders) be limited to:
(a) the respondents;
(b) the legal representatives of the applicant and the opponent who acted on the amendment application.
2. On or before 25 February 2015 each party identify any information in the reasons that is confidential, or that the party contends is confidential, and inform the associate to Rares J thereof.
3. The parties bring in short minutes of order on or before 25 February 2015 to give effect to these reasons.
4. The proceedings stand over for directions to 9.30 am on 12 March 2015.
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
NEW SOUTH WALES DISTRICT REGISTRY | |
GENERAL DIVISION | NSD 51 of 2012 |
BETWEEN: | APOTEX PTY LTD ACN 096 916 148 Applicant |
AND: | LES LABORATOIRES SERVIER First Respondent SERVIER LABORATORIES (AUST) PTY LTD Second Respondent ACTAVIS PTY LIMITED ACN 003 854 626 Opponent |
JUDGE: | RARES J |
DATE: | 23 FEBRUARY 2015 |
PLACE: | SYDNEY |
REASONS FOR JUDGMENT
1 This is yet another round in the war on many fronts between Apotex Pty Ltd and its related companies, on one side, and Les Laboratoires Servier and its related companies, including the second respondent, Servier Laboratories (Aust) Pty Ltd, on the other side, over the medicinal compound perindopril. That compound is used for lowering blood pressure in patients with hypertension (high blood pressure), in contrast to the effect on the parties of the efflorescence of litigation over almost every conceivable issue that the invention, or recognition of the beneficial properties, of perindopril has spawned: see, e.g., the recent decision of the Supreme Court of the United Kingdom in Les Laboratoires Servier v Apotex Inc [2014] 3 WLR 1257. Servier markets its products containing perindopril under the brand name Coversyl.
2 The occasion for this battle is Servier’s application to amend the complete specification for Australian Patent AU 2003200700 (the patent). The patent claimed the arginine salt of perindopril (perindopril arginine) (claim 1), various pharmaceutical compositions comprising perindopril arginine as the active pharmaceutical ingredient (claims 2-5), a method of treatment (claim 6) and a use of the salt (claim 7).
3 Following a three week long trial in late 2013, I found that the complete specification did not comply with the requirement of s 40(2)(a) of the Patents Act 1990 (Cth) that it describe the invention fully, including the best method of performing the invention known to the applicant for the patent: Apotex Pty Ltd v Les Laboratoires Servier [2013] FCA 1426. That was because the complete specification merely stated that the L-arginine (or naturally occurring amino acid) salt used in the study it referred to had “been prepared according to a classical method of salification of organic chemistry” (page 3 lines 22-23).
4 I found that, on the expert evidence, that description was too generalised and comprehended many possible means of performance of the invention that could involve considerable trial and error, when Servier knew of two particular methods, that it had used in 1986 and 1991 (the 1986 and 1991 methods), that had produced an arginine salt that could be used in a pharmaceutical composition (Apotex [2013] FCA 1426 at [182]-[187]). Those methods were only used on a laboratory scale. They each produced a form of the salt that was a white crystalline product that Servier then used in making tablets. As the patent disclosed, Servier conducted tests on those tablets to ascertain the stability of the active pharmaceutical ingredient (API), namely perindopril arginine, in packaging in three different conditions of temperature and humidity. The results of those tests showed that the new salt had better stability in two of those three conditions than Servier’s existing commercialised patented salt, perindopril tert-butylamine, also called perindopril erbumine, when both sets of tablets used the same excipients (Apotex [2013] FCA 1426 at [225], [226], [263]). This offered the advantage of cheaper packaging with a three year shelf life for pharmaceutical compositions made with the new salt, over those made with perindopril erbumine.
5 Thus, I held that while the patent claimed the new salt, Servier had failed to comply with s 40(2) of the Act. That was because Servier knew of, but did not disclose in the complete specification, the two particular methods of classical salifaction to produce the new salt that yielded a crystalline substance, that could be used to make tablets – i.e. pharmaceutical compositions – with the characteristics that the stability studies referred to in the patent confirmed.
6 On 21 March 2014, after publication of my reasons for holding that the complete specification did not comply with the requirement in s 40(2)(a) to describe the best method in Apotex [2013] FCA 1426, Servier applied to amend the complete specification under s 105 of the Act to include a description of each of the 1986 and 1991 methods. Subsequently, Servier twice added to its proposed amendment, first, on 6 June 2014 to add to its initial description of the 1986 and 1991 methods that each had produced a white crystalline substance, so that the description would accord with the declaration I had made on 29 May 2014 and, secondly, on 8 October 2014, to describe an industrial scale method that Servier had used in late 2002 to manufacture perindopril arginine tablets (the 2002 method). Servier made that latter addition as a result of a contention in the grounds of opposition to the proposed amendment that relied on the previously undisclosed 2002 method as being a better method than that disclosed in the patent or the 1986 or 1991 methods.
7 Apotex, together with Actavis Pty Ltd, another generic manufacturer who appeared under r 34.41(1)(d) of the Federal Court Rules 2011 (Cth), relied on, among many other grounds, the existence of the 2002 method to oppose the amendment of the patent. I will refer to Apotex and Actavis as the opponents where their interests or submissions were the same or similar, as was largely the case.
8 I will explain the detail of these matters later in these reasons. The hearing of the amendment application took five days. The grounds of opposition were over 50 pages long and there were over 240 pages of closing written submissions. To aid understanding of these reasons, I will identify the 4 substantive issues, set out the relevant legislative scheme, describe the facts, summarise the relevant submissions on each issue and, finally, give my reasons to resolve each issue. These reasons should be understood in the context of what I found in Apotex [2013] FCA 1426; and I will use the same terms and expressions and refer to the witnesses and other evidence described in those earlier reasons generally, without repeating those matters.
The issues
9 The amendment application requires the following substantive issues to be resolved:
(1) what is the proper construction of s 105 of the Act and the nature and scope of the discretion that it confers on the Court to grant an amendment (the construction issue)?
(2) was the amendment application futile because it was not possible to amend a complete specification after a patent has been granted (the futility issue)?
(3) what was Servier’s knowledge and state of mind in relation to the requirement to disclose the best method known to it of performing the invention during the period up to the grant of the patent (the Servier’s knowledge issue)?
(4) should the amendment be granted (the discretion issue)?
The legislative scheme
10 As I explained in Apotex [2013] FCA 1426 at [29], the parties agreed that the relevant provisions of the Act were those in force as at 27 February 2003, when Servier filed the complete specification. For present purposes, the Act provided:
40 Specifications
(2) A complete specification must:
(a) describe the invention fully, including the best method known to the applicant of performing the invention;
Chapter 10 – Amendments
Part 1 – Amendments that are not allowable
102 What amendments are not allowable?
Amendment of complete specification not allowable if amended specification would claim matter not in substance disclosed in the filed specification
(1) An amendment of a complete specification is not allowable if, as a result of the amendment, the specification would claim matter not in substance disclosed in the specification as filed.
Certain amendments of complete specification are not allowable after relevant time
(2) An amendment of a complete specification is not allowable after the relevant time if, as a result of the amendment:
(a) a claim of the specification would not in substance fall within the scope of the claims of the specification before amendment; or
(b) the specification would not comply with subsection 40(2) or (3).
Meaning of relevant time [This heading has been placed in the correct position.]
(2A) For the purposes of subsection (2), relevant time means:
(a) in relation to an amendment proposed to a complete specification relating to a standard patent – after the specification has been accepted;
…
(3) This section does not apply to an amendment for the purpose of correcting a clerical error or an obvious mistake made in, or in relation to, a complete specification.
Part 2 – Amendments of patent requests, specifications and other filed documents
104 Amendments by applicants and patentees
(1) An applicant for a patent or a patentee, may, subject to this Act, and subject to and in accordance with the regulations, ask the Commissioner for leave to amend the relevant patent request or complete specification, or any other filed document, for any purpose including either or both of the following:
(a) removing a lawful ground of objection to the request or specification, whether that objection is raised in the course of an examination or re-examination or otherwise;
(b) correcting a clerical error or an obvious mistake.
(2) Where an applicant or patentee asks for leave to amend a patent request or complete specification, or any other filed document, the Commissioner must consider and deal with the request in accordance with the regulations.
(3) Subject to the regulations, the Commissioner may allow an amendment subject to conditions.
(4) The Minister or any other person may, subject to and in accordance with the regulations, oppose allowing an amendment.
(5) The Commissioner must not allow an amendment that is not allowable under section 102.
(6) On the allowance of an amendment, the amendment is to be taken to have been made.
…
105 Amendments directed by court
(1) In any relevant proceedings in relation to a patent, the court may, on the application of the patentee, by order direct the amendment of the patent, the patent request or the complete specification in the manner specified in the order.
(2) An order may be made subject to such terms (if any) as to costs, advertisements or otherwise, as the court thinks fit.
(3) The patentee must give notice of an application for an order to the Commissioner, who is entitled to appear and be heard, and must appear if the court directs.
(4) A court is not to direct an amendment that is not allowable under section 102.
(5) The patentee must file a copy of an order within the prescribed period.
(6) On the filing of a copy of an order, the patent, patent request or complete specification is to be taken to have been amended in the manner specified in the order.
138 Revocation of patents in other circumstances
(3) After hearing the application, the court may, by order, revoke the patent, either wholly or so far as it relates to a claim, on one or more of the following grounds, but on no other ground:
….
(f) that the specification does not comply with subsection 40(2) or (3).
Schedule 1 – Dictionary
Section 3
In this Act, unless the contrary intention appears:
complete specification means (other than in section 116) a specification filed in respect of a complete patent application or, if the specification has been amended, the complete specification as amended.
relevant proceedings, in relation to a patent, means court proceedings:
(a) for infringement of the patent; or
(b) for revocation of the patent; or
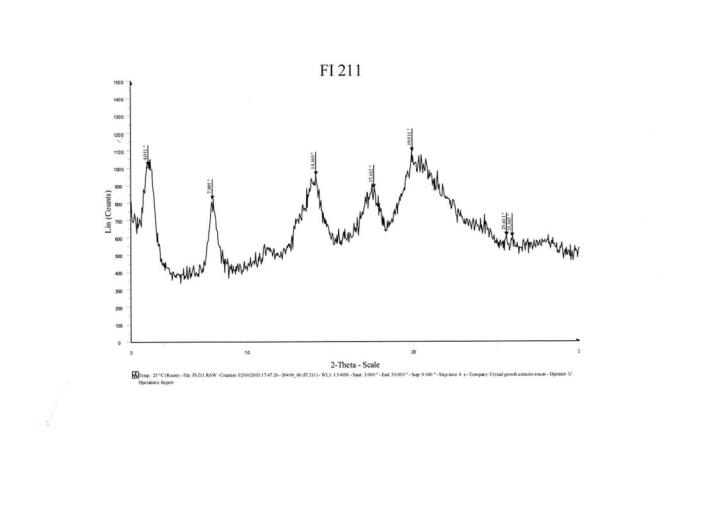
(c) in which the validity of the patent, or of a claim, is in dispute.
The Patent
11 I described the patent in detail in Apotex [2013] FCA 1426 at [34]-[41] and, for ease of understanding these reasons, I will now repeat that description in [12]-[18] below.
12 The specification stated that perindopril had previously been described in a European patent (EP 0 049 658) that had mentioned, as was usual, that compounds of the invention might be presented in the form of addition salts with a pharmaceutically acceptable, mineral or organic, base or acid. It stated that the compounds described in the European patent were in a non-salt form and “primarily, when addition salts with a pharmaceutically acceptable base or acid are mentioned by way of example, the sodium salt or the maleate are given” (p 1 lines 12-14). The specification continued:
“In the development of that product, however, it has proved very difficult to find a pharmaceutically acceptable salt having not only good bioavailability but also adequate stability to be suitable for the preparation and storage of pharmaceutical compositions.” (p 1 lines 15-17)
13 The specification noted that the non-salt form had been studied, as well as the maleate and the sodium salt, and that in the course of temperature and humidity stability studies these had been found not to be suitable because the sodium salt was immediately converted into an oil on contact with the atmosphere and the non-salt form and maleate degraded rapidly under higher temperature conditions. The specification continued:
“The tert-butylamine salt was thus alone in exhibiting the best stability compared to the other forms studied. However, in view of the intrinsic fragility of perindopril, the tert-butylamine salt has not been capable of providing a complete solution to the problems of the product’s stability to heat and humidity. Indeed, for marketing, tablets of perindopril tert-butylamine salt must, in certain countries, be protected with additional packaging measures. Moreover, even for temperate-climate countries, that instability has made it impossible to obtain a shelf-life of more than 2 years for the tablets. Finally, for marketing of the tablets, they have to be marked “to be stored at a temperature less than or equal to 30 degrees”.
These constraints are, of course, onerous especially in terms of organisation and cost, and it has appeared especially useful to try to develop a new perindopril salt in order to reduce the constraints due to the tert-butylamine salt.” (p 1 line 23-p 2 line 9)
14 The specification then said that numerous salts had been studied and that those customarily used in the pharmaceutical sector had proved to be unusable and continued:
“On the other hand, and in surprising manner, it has been found that the arginine salt of perindopril, besides being new, has entirely unexpected advantages over all the other salts studied and, more especially, over the tert-butylamine salt of perindopril.” (p 2 lines 12-14)
15 The specification then said that the present invention related to the arginine salt, its hydrates and also to the pharmaceutical compositions comprising it. It identified the preferential form of the salt as being natural arginine (L-arginine) and said that the pharmaceutical compositions according to the invention comprised the arginine salt together with one or more, non-toxic, pharmaceutically acceptable and appropriate excipients. The specification then identified a variety of possible pharmaceutical compositions and various administration and dosage methods. It stated that the pharmaceutical compositions according to the invention would preferably be immediate release tablets and that the useful dosage varied according to the age and weight of the patient, the nature and severity of the disorder and to the administration route (p 2 line 15, p 3 line 5).
16 The specification identified the amount of arginine salt contained in the compositions according to the invention, including the preferred quantities between 1 to 10 mg and that those compositions were of use in the treatment of hypertension and heart failure. It continued:
“The basic characteristics of this salt are very great stability to heat and to humidity compared to the tert-butylamine salt.
Long-term stability studies carried out under very precise temperature and humidity conditions have yielded the results indicated in the Table hereinbelow.
In that study, perindopril was assayed by inverse-phase high-pressure liquid chromatography using, as eluant, an aqueous phase (comprising sodium heptane-sulphonate, and the pH of which is 2) and acetonitrile (67/33). Detection of the product was carried out by UV (215 nm).
The study was carried out using immediate-release tablets containing either 2.4 mg of the arginine salt of perindopril or 2.0 mg of the tert-butylamine salt of perindopril (each of the two tablets containing 1.7 mg of perindopril). The tablets were assayed 6 months after the start of storage of the tablets at different temperatures and different relative humidities (% R.H.).
The arginine salt used in this study is the L-arginine salt. It has been prepared according to a classical method of salification of organic chemistry.
Conditions 6 months | tert-Butylamine salt of perindopril Percentage remaining (%) | Arginine salt of perindopril Percentage remaining (%) |
25°C 60% R.H. | 101.0 | 99.5 |
30 °C 60% R.H. | 94.4 | 98.1 |
40°C 75% R.H. | 67.2 | 98.6 |
The results presented in the Table above show extremely clearly the very great stability of the arginine salt compared to the tert-butylamine salt. Indeed, after 6 months, practically no degradation of the arginine salt has taken place whereas the tert-butylamine salt exhibits a degradation rate of approximately 33%.” (p 3 line 9-p 4 line 5)
17 The specification then asserted that the results were entirely unexpected and could not have been deduced from or suggested by the teaching of the literature on the product and concluded by stating:
“The results allow us to consider less onerous constraints with respect to the packaging of the pharmaceutical compositions and also to obtain a shelf-life of at least three years for our pharmaceutical compositions.” (p 4 lines 8-10)
18 The patent asserted 11 claims defining the invention but … only the following five are relevant:
“1. The arginine salt of perindopril and its hydrates.
2. Pharmaceutical composition comprising, as active ingredient, the arginine salt of perindopril and its hydrates, in combination with one or more pharmaceutically acceptable excipients.
3. Pharmaceutical composition according to claim 2, characterised in that it is presented in the form of an immediate-release tablet.
4. Pharmaceutical composition according to either claim 2 or claim 3, characterised in that it contains from 0.2 to 10 mg of the arginine salt of perindopril.
…
6. A method of treatment or prophylaxis of hypertension and heart failure comprising administering to a patient in need of such treatment or prophylaxis an efficacious amount of the arginine salt of perindopril and its hydrates of claim 1, or a pharmaceutical composition as claimed in any one of claims 2 to 4.”
19 Claims 8-11 referred to descriptions of the invention in “the Examples”, but there were no examples set out anywhere in the patent. The opponents did not oppose Servier’s application to delete claims 8-11 which I had found to be otiose. Those claims should be deleted: Apotex [2013] FCA 1426 at [41].
The history of the best method issue
20 Apotex commenced these proceedings on 16 January 2012. On 27 January 2012, Servier promptly filed a defence and cross-claim alleging infringement. The proceedings were case managed by Bennett J and the parties filed evidence and gave discovery during the course of 2012. On 6 March 2013, Apotex’ solicitors wrote to Servier’s solicitors and, for the first time, made the assertion that the complete specification did not comply with the best method requirement in s 40(2)(a) because, the letter asserted, it did not disclose “the means of formulating Perindopril Arginine so as to achieve the results of the long term stability study set out at page 3 line 11 to page 4 line 5”. Apotex’ solicitors followed this up in a letter on 12 March 2013 foreshadowing that they would amend the particulars of invalidity to rely on this new ground.
21 On 18 March 2013, Apotex provided Servier with a draft of its amended pleadings that alleged that the failure to disclose the best method rendered the patent invalid. The next day, Bennett J ordered discovery by categories on the existing pleaded issues, including in respect of a category concerning the stability study referred to in the patent. On 28 March 2013, her Honour ordered Apotex to file and serve, on or before 21 May 2013, its evidence in support of the new allegations in its recent draft pleadings. On 16 April 2013, Servier gave discovery of the above category of documents concerning the study.
22 On 24 April 2013, Servier filed its evidence in chief on the issue of inventive step that included the following statement by Prof Byrn in an affidavit:
Unless it was already known that a particular compound could form crystals (for example from published literature stating that a crystalline substance is obtainable), I could not predict in advance whether a particular compound would form crystals, nor which combination of methods and variables would be more or less likely to form crystals for a particular compound.
23 On 4 June 2013, Apotex served a further amended version of its particulars of invalidity in which it alleged that the best method requirement was not satisfied because the specification “does not describe which form or forms of perindopril arginine to make so as to obtain the alleged benefits of the invention”. The parties then debated, in correspondence, their rival contentions over Apotex’ proposed amendments.
24 On 18 June 2013, Apotex filed an interlocutory application seeking leave to file and serve its proposed further amended particulars of invalidity. In the meantime, on about 17 July 2013 and unbeknownst to Apotex, senior counsel for Servier prepared a note of advice on the best method issue, that he emailed to Servier’s solicitors. Servier waived privilege and tendered the note on this application. It was common ground that Servier’s solicitors did not forward the note to their client. Senior counsel’s advice was to the effect that in a product claim, as he said was the present case, “the requirement is simply to describe the best form of the invention, irrespective of how it may in fact have been made: Illinois Tool Works Inc v Autobars Co (Services) Ltd [1972] FSR 67 at 71-72”. He advised that in Servier’s case the ground could only be made out if there were evidence that one form of the invention were demonstrated to work better than another form and that the patentee was aware of that matter. Senior counsel advised that there was no such evidence in the then present case.
25 On 26 July 2013, Servier served its evidence in answer to the amendment application that was to be heard by Bennett J on 2 August 2013. However, the parties agreed on consent orders, that her Honour made on 2 August 2013, permitting the best method issue to be raised in the limited manner that was confined to the 1986 and 1991 methods that I described in Apotex [2013] FCA 1426 at [145] as follows:
Apotex had obtained leave to amend its particulars of invalidity to rely on the allegation that the complete specification did not describe the best method known to the patentee on the basis that it could not rely at the hearing on evidence other than the face of the specification and the evidence of Servier’s internal methods [ie: the 1986 and 1991 methods]. That is why Apotex pleaded that the specification failed to describe, first, either of those two methods to prepare the invention each of which, clearly enough, was a method known to the patentee and, secondly, a means to prepare a pharmaceutical composition containing perindopril arginine (including which form or forms to use in that composition) so as to achieve the results of the long term stability study described in the table in the specification.
26 The trial was fixed to commence on 8 October 2013 before me. Apotex filed its further amended particulars of invalidity on 19 August 2013.
The factual basis for Servier’s amendment
27 Dr Sylvie Jaguelin has been the patent director for Servier since September 2001. She obtained the degree of doctor of philosophy in organic chemistry in 1984 and began working for the Servier group in 1985. In 1995 she completed an international degree of industrial property, patents – trademarks and designs at the Centre d′Etudes Internationales de la Propriété Intellectuelle (CEIPI) in Strasbourg, France. As part of her course, Dr Jaguelin had to study the Patent Cooperation Treaty 1970 (PCT), European and international patent law as well as some national patent laws, including those of France and the United States of America, but not those of Australia.
28 Dr Jaguelin’s responsibilities as Servier’s patent director included what she termed “managing the lifecycle of Servier’s patent files”, which involved drafting, filing, prosecution, maintenance and enforcement of Servier’s patents. As at March 2014, she was managing about 250 patent families for Servier.
29 Dr Jaguelin made affidavits in support of Servier’s amendment application on 20 March 2014 and 7 October 2014, and gave oral evidence, largely through an interpreter, over the course of three days. The opponents did not raise any serious challenge to Dr Jaguelin’s credibility in giving her evidence. In any event, I found her evidence to be generally honest and reliable. Given the passage of many years since most of the events in issue, much of what occurred is best discerned from, or in light of, contemporaneous documents.
30 Dr Jaguelin said that although she had not been able to attend the trial, due to a broken tibia, she had reviewed the evidence and transcript, including the joint experts’ report. She said that based on her understanding of the best method requirement, the pleadings and the evidence at the trial, from the time that Apotex had first raised the issue in early March 2013 until my reasons were delivered on 24 December 2013, she believed that the patent in suit had satisfied the best method requirement and that there was no need to amend the patent. She said that she first contemplated whether to apply to amend the patent only after delivery of my reasons in Apotex [2013] FCA 1426.
31 Dr Jaguelin explained that the reason why Servier applied on 21 March 2014 to amend the patent to add descriptions of the 1986 and 1991 methods was to overcome the findings in Apotex [2013] FCA 1426 [187] and [41], that the patent did not comply with s 40(2)(a) and that claims 8-11 had no basis since there were no examples given in the complete specification.
32 Dr Jaguelin then explained her understanding as at the time she drafted the complete specification for the patent. She began that process in late 2001 or early 2002 by working on the version of the arginine patent application to be filed in France after she had had a discussion with the inventor, Mr Damien. She understood that, in Europe, an applicant had an obligation to disclose the invention in a manner sufficiently clear and complete for it to be carried out by a person skilled in the art. Her approach to drafting complete specifications for the purpose of applications in foreign jurisdictions in which Servier regularly filed, was to disclose all information that she understood was required, including in respect of those jurisdictions that still required disclosure of a best mode or method. Nonetheless, she also drafted specifications on the basis that they should not contain unnecessary detail, in accordance with the style that she had been taught when at the CEIPI.
33 Dr Jaguelin said that while there was no requirement to disclose a best mode or method in Europe, she was aware of the best mode requirement in the United States of America. She understood that the United States law required:
the specification to disclose the best mode contemplated by the inventor of carrying out the invention;
the disclosure of more than what was necessary for the skilled addressee, using his or her normal skill and knowledge, to make a product or to use a process that the patent claimed;
no further disclosure than that of performing the invention claimed in the patent;
in the case of a claim to a method of making a salt, disclosure of the best mode of making the salt.
34 However, Dr Jaguelin said that she understood that where the patent simply claimed a salt, United States’ law did not require the specification to disclose a best mode of making the salt. Additionally, she understood that it was not necessary to disclose information that was routine or well-known to a person skilled in the art.
35 She did not now recall whether, when she discussed matters with Mr Damien in late 2001 or early 2002, he had told her that a routine method of salification had been used or she concluded that this was so after he had described the 1986 and 1991 methods. She said that her usual practice was that, if she had not been given information about a method, she would have asked whether there was any particular difficulty involved in making the salt concerned or whether it was routine.
36 Dr Jaguelin said that looking now at what was involved in the 1986 and 1991 methods, had she been aware of them at the time, she would have concluded that each was a simple and routine classical salification and that there was no special information necessary to make the arginine salt. Indeed for her, it was “a particularly simple example.” Dr Jaguelin said that, based on her usual practice, Mr Damien would have approved the draft specification before it was filed. She made the first filing in France on 18 April 2002 under the Paris Convention for the Protection of Industrial Property. She decided to proceed with filings for the arginine patent in other jurisdictions under, variously, the PCT, the Convention or directly under a local law.
37 In the meantime, without reference to Dr Jaguelin and unknown to her, by 2002 Servier was preparing to commercialise perindopril arginine so as to exploit it under the new monopolies that the patent applications Dr Jaguelin was preparing and filing in various jurisdictions, if granted, would create. In that way, as the patents in various countries for perindopril erbumine expired, Servier would have a new product to market as Coversyl. Internally within Servier, others were engaged simultaneously (but separately from Dr Jaguelin’s work in securing patents) in the search for useful and possibly patentable crystalline forms of the salt and in the development of an industrial process of salification, the conduct of stability testing and satisfaction of other regulatory approval requirements. This activity led to the development of the 2002 method and, in later years, of several crystalline forms of perindopril arginine that I will detail later in these reasons, after first dealing with Dr Jaguelin’s work and state of mind. She was unaware of those matters at all times before the filing of the complete specification.
38 The majority of Servier’s applications for patents making the same, or similar, claims to the patent in suit were filed on 17 February 2003 or shortly after. On 17 February 2003, Dr Jaguelin wrote to Servier’s then Australian patent attorneys, Watermark Intellectual Asset Management, asking them to file here a new patent application for perindopril arginine based on the French priority document that had been filed on 18 April 2002. Watermark filed the application for the patent on 27 February 2003, including the complete specification, and on 12 March 2003 wrote advising Dr Jaguelin of this.
39 Dr Jaguelin said that for the purposes of meeting the disclosure requirements of foreign jurisdictions under the PCT and the Convention, she included in her native French, what in translation is the following English wording that appears in the Australian patent, namely:
“The arginine salt of perindopril is preferentially the salt of natural arginine (L- arginine).” (p 2 line 17)
“The arginine salt used in this study is the L-arginine salt. It has been prepared according to a classical method of salification of organic chemistry.” (p 3 lines 22-23)
40 When she included the original French words for the English expression “a classical method of salification of organic chemistry”, Dr Jaguelin intended, as she said, “to teach the skilled person that the salt is easy to make by routine means which was my understanding of the true position”. Next, Dr Jaguelin said:
I also understood that the best mode requirement was directed to the mode of performing the invention. In this case, I understood the invention to be the arginine salt of perindopril itself. There is no claim in the patent for a process for making the salt. If the invention had been a process for making the salt I would have included a detailed description of the method of making the salt.
I now appreciate that the Court has found my understanding of the requirements to disclose best mode/best method to be wrong insofar as the principles apply in Australia, but that was, unfortunately, the basis on which I proceeded at the time. (original emphasis)
41 In cross-examination, Dr Jaguelin gave this evidence:
THE INTERPRETER: I added the words that the salt can be prepared according to a classical method of salification of organic chemistry in order to fulfil the requirement for sufficient description, which is the standard in all types of patents.
MR CATTERNS: Including description as to the method?
THE INTERPRETER: I didn’t put the method deliberately because I consider that a salt is made according to classical chemistry processes. (emphasis added)
42 She also said that “at school, making [a] salt is something that’s as easy as can be”. She said that the patent was not one about a process and that what she wanted the patent to protect was the arginine salt and nothing else. She was concerned to protect the new salt in the patent, not a method of making it. She considered that the benefit of the salt itself that merited protection was that Servier could sell it all over the world in one form of packaging, using one production line and not two as had been the case with perindopril erbumine. The complete specification explained that the invention of the new salt offered a promise of less onerous packaging constraints for pharmaceutical compositions than those made with perindopril erbumine (patent p 2 lines 1-3, 7-9, p 3 line 9, p 4 line 10) as I found was the case: Apotex [2013] FCA 1426 at [38]-[39], [290]-[294].
43 Dr Jaguelin said that Servier had not tried to withhold or hide anything, including the 1986 and 1991 methods, in the way in which she had worded the complete specification. She said that it was now making the amendment application so as to meet the requirements of s 40(2)(a) of the Act, and not just in response to the findings that I had made in Apotex [2013] FCA 1426. Dr Jaguelin said:
THE INTERPRETER: What convinced me were the result of the stability study that demonstrated to me that this product was capable of being patented. And it’s only in Australia that people talk to me about a best method. It has not been mentioned anywhere else in the world.
MR CATTERNS: It’s mentioned in America, isn’t it?
THE INTERPRETER: It is mentioned in the law, but I wrote the US patent and it’s exactly the same as the Australian one. And I thought that it was sufficient. (emphasis added)
44 Dr Jaguelin accepted that Mr Damien’s notes of the use of the 1991 method recorded that it produced a form of the arginine salt that was a white crystallised product, but she thought that what she had written about the best method in the patent was sufficient. She accepted that, when making tablets, a crystalline or powdery form of a salt was much less difficult to use than a glassy or viscous one. She said that claim 1 in the patent was about a product that covered all forms of the salt and that, in chemistry, where one described a product, one described a structure, not a form. She said that patents claiming a salt never described the method of making that salt. Dr Jaguelin denied that she had omitted from the patent any details of a method and the fact that a crystalline product had been obtained in order to leave open the possibility of subsequently filing patents claiming methods or crystalline forms of the salt. As I have noted above, I accept her evidence on these matters and generally on the matters which I have set out in these reasons.
45 At the trial, the experts examined the full versions of the data of Servier’s studies in 1998 and 2000 of the comparative stability of tablets containing perindopril erbumine and perindopril arginine. I found that it was likely that Mr Damien had used the 1991 method to make the perindopril arginine used in those studies and that the tablets using two different salts had been made with exactly the same excipients and in the same quantities as were used in the commercial perindopril erbumine Coversyl product: Apotex [2013] FCA 1426 at [152], [216], [255]-[264.]
46 At the time of filing of the patent, Dr Jaguelin was not aware of any x-ray diffraction or x-ray powder diffraction (XRPD) analysis of the crystalline powder produced by the 1986 or 1991 methods. She said that the form of the arginine salt that Servier now uses in its tablets was poorly or weakly crystallised. Dr Jaguelin distinguished the crystalline or powdery form of the arginine salt that Mr Damien recorded had been produced using the 1986 and 1991 methods from the nature of a crystal that is identified through the use of crystallography.
47 In February 2003, Dr Jaguelin was not aware of any decision that Servier was then moving to market perindopril arginine, although she was aware of internal discussions about that possibility based on the advantages of using a single production line and one form of packaging for its manufacture. She was also then conscious that the European patents for perindopril erbumine were going to expire in June 2003 and that seeking a patent for the new salt was “part of what we call the management of the life cycle of the [perindopril] product and for reasons of profitability as well”. She was not aware at that time of any further patent applications that might be made relating to perindopril arginine.
Ms Harris’ recommendation to Servier
48 On 17 August 2004, Dr Jaguelin wrote to Carolyn Harris, who was the patent attorney at Watermark with carriage of the file for the application for the patent in suit. In the letter, Dr Jaguelin informed Ms Harris that she had received Watermark’s letter of 12 March 2003, and that Servier wanted to request the Commissioner to expedite the examination of the application. Dr Jaguelin wrote that the corresponding patents had been granted in the United States of America and the European Union, adding “we do not expect major problems with this patent application in your country”. She understood that the application complied with the Australian requirements and expected that the Commissioner would grant it. On 19 August 2004, Ms Harris responded by informing Dr Jaguelin that she had requested that examination of the application be expedited.
49 On 3 September 2004, Ms Harris forwarded to Dr Jaguelin, with her letter of that date, a copy of the first examiner’s report that remarked on the then absence of any notice of entitlement on the file. Ms Harris wrote the following in her covering letter:
You will see that no substantive objections have been raised by the Examiner.
Now is the time to make voluntary amendments to the specification to correct any defects or to add further examples.
We recommend an amendment to the present claims to add “Swiss-style” and “method of treatment” claims. Moreover, we suggest the addition of omnibus claims directed to the subject matter of the examples as a final fall-back position.
In order to support proposed claims 8, 10 and 11, we recommend the addition of further examples, if possible. These would be, for example, invitro/invivo data that would normally be submitted with an application for regulatory approval. In addition, we recommend that a method for manufacture of the arginine salt of perindopril be inserted into the description, even if the manufacturing method is well known in the art, to satisfy the written description requirements.
In the absence of examples for the use of the arginine salt of perindopril to treat hypertension and heart failure, we would delete proposed claims 8, 10 and 11.
We enclose a draft response and claim set for your approval.
(emphasis added)
50 Dr Jaguelin replied to Ms Harris on 20 December 2004 as follows:
Further to your letter of September 3, 2004 concerning the first examination report about the above mentioned case, we understand that no substantive objections have been raised by the Examiner. We also understand that you have sent to the Australian Patent Office on September 3 a new set of claims including addition of “Method of treatment” “Swiss-style” and “omnibus claims”: we agree with these amendments.
In order to support claims 8, 10 and 11 you suggest to us giving supplementary data: you will find enclosed (only by mail) a part of our bioequivalence study. As it is a confidential document and I am not sure it is a good idea at this stage to give such document to the Patent Office.
I would prefer wait Examiner's comments about this new set of claims.
Concerning the manufacture of the arginine salt of perindopril, we have mentioned in the description page 3, lines 22-23 that this salt is prepared according to a classical method of salification of organic chemistry: we do not think it is necessary to add more detail for the moment: we will see later.
Finally, concerning the search reports, I hope that you have received them with our letter of September 7.
For your information, European and US patent have been granted. (emphasis added)
51 Dr Jaguelin gave evidence that she meant by “we will see later”, in the emphasised passage above from her letter, that she would wait for the examiner’s comments on the topic of the descriptions of the manufacture of the salt, just as she had stated earlier in the same letter that she would await the examiner’s comments on the new claims. The opponents accepted that explanation and did not cross-examine Dr Jaguelin about it.
52 The examiner did not make any further comments and on 2 February 2005 Watermark informed Dr Jaguelin that the application had been advertised as accepted on 20 January 2005. No oppositions were filed and, on 17 May 2005, Dr Jaguelin received the sealed deed of letters patent granting a term of 20 years from 27 February 2003.
The 2002 method
53 In effect, the 1986 and 1991 methods offered a proof of concept that it was possible to make a useful crystalline substance comprising perindopril arginine that could be used in tablet production. However, those were both laboratory methods and not suitable for use on an industrial production scale. Servier’s management had decided at some point before October 2002 to seek to exploit perindopril arginine commercially. That objective required not only patent protection, if it were available, but also the development of a salt form that could be reliably reproduced in industrial quantities so as to meet regulatory requirements.
54 There is no challenge to the accuracy of the description of the 2002 method that Servier seeks to insert into the complete specification, namely:
The arginine salt may also be made in the following manner. Load 15 kg of perindopril tert-butylamine and 32.52 kg of toluene in a reactor. Add, under stirring, 8.473 kg (33.96 mol HCl) of a prepared solution of concentrated hydrochloric acid and 9 kg of permuted water. Separate the organic phase and then extract twice the aqueous phase with 13 kg of toluene each time. Load, under stirring, the combined organic phases in a further reactor containing a prepared aqueous solution of 5.325 kg of L arginine. Separate the aqueous phase. Wash the organic phase with 7.5 kg of permuted water and filter the combined aqueous phases. Cool the solution to about 3°C, and add 385.14 kg of filtered dimethylsulfoxide to the combined aqueous phases without allowing the temperature of the reaction mass to exceed 16°C. Collect the precipitated solid on a closed filter and wash the cake twice with 71.5 kg of acetone each time and then once with 69.3 kg of methylcyclohexane. Dry the product at 65°C for at least 36 hours. This produces approximately 13.481 kg of dried perindopril arginine.
55 There is little evidence as to how or when the 2002 method was developed, but before 27 February 2003, Servier had manufactured a total of about 23.1 million tablet doses of 2.5 mg, 5 mg and 10 mg using perindopril arginine produced by that method. In October 2002 it made a total of 2.9 million film-coated tablets in the three dosages for the purpose of pharmacokinetic and bioequivalence studies. In late October and November 2002 Servier manufactured pilot and development batches in the three dosages, totalling 6.3 million tablets. On 23 January 2003, Servier manufactured in industrial scale-up batches, 2 million 10 mg dosage tablets, 4 million 5 mg dosage tablets and 8 million 2.5 mg dosage tablets. A pilot batch uses about 15 kg of the API and an industrial batch uses about 75 kg of the AP1.
56 The pilot and development batches were in the nature of experiments to test a particular method of tablet formulation to determine whether it could be reproduced consistently, that Servier ultimately decided to use on an industrial scale. Tablets produced in a pilot scale batch were used in the pharmacokinetic and stability studies. By 23 January 2003, Servier had used the method to produce the industrial batches employing the different equipment and scales of production at its two factories in Gidy, France and Arklow, Ireland. Those pilot and industrial scale batches complied with the specifications identified in the regulatory dossier that Servier had submitted to the Therapeutic Goods Administration (TGA) on 30 March 2004.
57 During the course of the interlocutory processes in 2014 to prepare Servier’s amendment application for hearing, I granted the opponents access to the regulatory dossier: Apotex Pty Ltd v Les Laboratoires Servier (No 3) [2014] FCA 1029. The dossier included detailed results of testing of perindopril arginine tablets that had been manufactured in late 2002 using the 2002 method. Some regulatory testing of those tablets began around late October 2002, and regulatory stability tests began on 19 November 2002, but that testing not completed until after Servier filed the complete specification on 27 February 2003.
58 Despite Servier not knowing the results of those tests (including stability tests that were capable of comparison to the six month trials on tablets produced using the 1991 method described in the complete specification) at the time of filing the complete specification, the opponents argued that Servier somehow knew, as at 27 February 2003, that the 2002 method was better than the 1986 and 1991 method.
59 In the event, in about May 2003 Servier received the results of the stability studies of perindopril arginine tablets made using the 2002 method. Servier included the 2002 method study results in the 30 March 2004 regulatory dossier lodged with the TGA.
60 As shown in the table below, those results were directly comparable with the results of studies done in 1998 and 2000 and recorded in the complete specification for the measures of 25oC at 60% relative humidity (25oC/60%RH) and 40oC/75%RH. However, the two studies used two differing relative humidities at 30oC, so that the results for 60%RH and 70%RH at that temperature respectively are not directly comparable.
Perindopril arginine after six months storage
25°C/60%RH | 30°C/60%RH | 30 °C/70%RH | 40 °C/75%RH | |
1991 method 2.4 mg tablets | 99.5% | 98.1% | N/A | 98.6% |
2002 method 2.5 mg tablets | 98.01% | N/A | 98.41% | 96.81% |
61 The two directly comparable sets of results showed that the tablets manufactured using perindopril arginine made with the 1991 method retained a greater percentage of the salt assay and so were more stable than those made with the 2002 method. Nonetheless, the later results were still sufficiently stable for the purposes of achieving regulatory approval.
62 As Dr Jaguelin said, the results showed that the tablets made with the 2002 method were slightly less stable than those made with the 1991 method, but the difference was small.
63 The stability studies that Servier did for regulatory purposes using the 2002 method also tested 5 mg and 10 mg tablets. Dr Jaguelin said that the stability results for those weights would not alter her opinion that the tablets produced with perindopril arginine made by the 2002 method were slightly less stable than those made with the 1991 method. She said that even if she had been aware at 27 February 2003 of the subsequently obtained 2002 method stability study results, she would not have been in a position to conclude that the 2002 method was preferable to the 1986 or 1991 methods for the purpose of producing more stable perindopril arginine tablets.
64 When she prepared the complete specification, and caused it to be filed, Dr Jaguelin did not enquire, and had no information, about any studies other than the 1998 and 2000 ones, whence the data in that specification came. She said that “before carrying out a study for regulatory purposes you always want to check whether the product is stable” and that if it were not stable “you’re not going to go for the regulatory study”.
65 Actavis used that thin reed of evidence to contend that Servier had not disclosed stability studies of perindopril arginine produced using the 2002 method that, Actavis asserted, Servier must have undertaken before October 2002. That was because, Actavis argued, Servier would not otherwise have manufactured pilot, development and industrial scale batches of over 23 million tablets, some of which were used later in regulatory studies, including the regulatory stability studies that commenced on 19 November 2002. I reject that argument. Dr Jaguelin, in that evidence, was referring to the general stability of the salt, perindopril arginine, and not to any particular product of a method of salification of it.
66 Dr Jaguelin was responsible for the applications and subsequent grants in 36 other jurisdictions of the equivalent of the Australian arginine patent. She was not aware of any objections or oppositions to those 36 applications based on the relevant foreign requirements as to sufficiency or best mode or method. She said that Australia is the only jurisdiction in which there has been an application for revocation of the arginine patent.
67 I am satisfied by Dr Jaguelin’s evidence that, at 27 February 2003, first, neither she, nor anyone else in Servier, knew of the stability study results for tablets containing perindopril arginine made by the 2002 method because those tests required a period of six months to produce results and began only on 19 November 2002. Thus, the results of those studies could not have been known until at least later in May 2003. Secondly, it is not possible to say that the 2002 method produced perindopril arginine with better stability than the 1986 or 1991 methods.
68 In order to commence the pharmacokinetic study on humans using the tablets produced in October 2002, Servier had first to obtain ethics committee approval, develop study protocols, recruit patients and obtain regulatory documentation, as Dr Jaguelin confirmed. Self-evidently, Servier’s decision-maker responsible for the decision(s) to manufacture the 23.1 million perindopril arginine tablets using the 2002 method, must have believed that the 2002 method would result in a successful product.
69 However, belief falls short of knowledge, including for the purposes of s 40(2)(a): cf George v Rockett (1990) 170 CLR 104 at 112-113, 115-116 per Mason CJ, Brennan, Deane, Dawson, Toohey, Gaudron and McHugh JJ. This distinction between belief and other lesser states of mind that fall short of knowledge was made clear by the following evidence that Dr Jaguelin gave in cross-examination:
MR COOKE: Yes. And, Dr Jaguelin, you would accept as a general proposition that pharmaceutical companies such as Servier only undertake such clinical studies when they’re confident that the product is likely to be successful.
THE INTERPRETER: Pharmaceutical companies actually undertake many studies, but three-quarters of them do not lead to a commercial product.
…
MR COOKE: Do you agree that when Servier undertook those clinical studies, it believed that the
product administered would be successful?
THE INTERPRETER: You know, whether you’re talking about the pharmaceutical industry or the automotive industry, when you launch into a study you believe that it’s going to be successful. It’s very rare that you would launch into a study thinking it won’t be successful. (emphasis added)
70 She also explained that, in her experience, projects had failed at all stages including when providing material to regulatory authorities. She said that studies that Servier carried out in late 2002 and following could be used if the product continued to the regulatory stage.
71 The fact that, in late January 2003, Servier produced 14 million tablets on an industrial scale using the 2002 method does not take its belief in that method to the point of knowledge. Those tablets could be sold if regulatory approval were later granted, but there was a not insignificant risk that it would be refused. There was no evidence as to the cost of that production run or whether its occurrence was outside of Servier’s ordinary processes for the commercialisation of a new product, other than that Servier sets aside for research and development about 25% of its €4 billion annual turnover. Indeed, as was common ground at the trial, Coversyl made using perindopril arginine was first launched in Poland only in April 2006. Thus, the three year shelf life of all of the batches manufactured in late 2002 and early 2003 had expired before that launch.
72 I am also not satisfied that Servier had any obligation under s 40(2)(a) of the Act to disclose the 2002 method in the complete specification. However, there is no reason why Servier cannot include the description of the 2002 method, if it is otherwise entitled to amend the complete specification.
XRPD data and characterisation of crystalline forms
73 Professor Stephen Byrn, who had already given expert evidence at the trial (see Apotex [2013] FCA 1426 at [49]), prepared a joint report and gave concurrent oral evidence, with Professor Stuart Batten, whom Apotex called on the amendment application in relation to the interpretation of XRPD data.
74 Professor Batten was a professor in the School of Chemistry at Monash University where he has spent most of his academic career since 1995. Between 2006 and 2011, he was the manager of the X-ray Diffraction Facility at Monash’s School of Chemistry. He had taught crystallography, including XRPD, to undergraduate and postgraduate students since 2000 and was the current president of the Society of Crystallographers in Australia and New Zealand and Chair of the International Union of Crystallography Commission on Structural Chemistry.
75 XRPD is an analysis technique used to reveal the fundamental physical properties of a material. It uses a beam of x-rays of a known wavelength to generate a diffraction pattern when it passes through a sample of crystalline material that has been ground to a fine powder. The crystal structure of every different form of a crystalline substance produces a unique diffraction pattern when the x-ray passes through it.
76 The experts agreed in the joint report that XRPD patterns can be used in the same way as finger prints. Thus, like finger prints, if one pattern is different from another and the difference is not attributable to experimental variations, the crystal forms, whence each pattern is produced, must be different. If different materials having the same elemental analysis produce significantly different XRPD patterns, they are polymorphs. Hydrates and solvates are sometimes called pseudopolymorphs or, loosely, polymorphs. However, because they have different compositions, they are different compounds. A hydrate is a solvate in which water is the solvent. Different hydrates and solvates may differ because solvent is included within channels in its crystal structure or because the molecules have to rearrange themselves to accommodate the solvent. Such materials may produce a range of similar, but nonetheless different, XRPD patterns.
77 The experts also agreed that, in XRPD analysis results, the way in which the measuring instrument is set up can lead to a small amount of variability in the accuracy and precision of the measurement of the position of a peak. Variations in such measurement results can occur because of factors such as calibration, the filtering of the x-rays, the way in which the particular computer program used selects the peaks and determines their position, sample height, properties of the holder of the sample and the way in which the sample was prepared.
78 The experts said that XRPD peaks would not be expected to change their positions as a sample loses crystallinity, but rather those peaks would be expected to broaden and decrease in intensity.
79 The particular shape of a crystal can have an effect on each of the solubility and absorbability of that crystalline form. Pharmaceutical companies need to devise a process to reproduce a particular crystalline form of an API with reliable accuracy and consistency on an industrial scale. Part of the standard international regulatory process for approval of new pharmaceutical products involves the demonstration of the nature of the crystalline form of the API by reference to various matters, including XRPD data, stability studies and the accuracy and consistency of that form when produced in industrial sized batches. A particular crystalline form is characterised by XRPD data analysis, using specific measured peaks of a substance to ascribe unique points of identity to it. Thus, XRPD analysis focuses on whether the uniquely characteristic peaks of a particular crystalline form are present in each later attempt to reproduce that substance (Joint report Q 1).
80 On 1 March 2001, the European Agency for the Evaluation of Medicinal Products published a note for guidance on process validation that came into operation in September 2001. It is common ground that that Agency’s guidelines, including the note, were applicable in Australia. The note commenced by explaining that process validation was the means of ensuring and providing documentary evidence that processes, within their specified design parameters, were capable of consistently producing a finished product of the required quality. The note stated that it applied to pharmaceutical process validation that was used in a regulatory dossier. The note also said that:
It is recognised that at the time of submission of a marketing authorisation dossier [ie like the regulatory dossier for perindopril arginine] manufacturers may not have completed formal validation studies on production scale batches. (comment in square brackets and emphasis added)
81 The characterisation using XRPD analysis of the crystalline form of a drug substance is part of the validation of the proposed manufacturing process for regulatory purposes.
82 Servier provided the TGA with the following information about the form of the crystalline substance, being perindopril arginine (described in the dossier as S 9490-6), for which it sought approval in part 3.2.S.3.1 of the regulatory dossier (that part was prepared on 2 September 2003):
Solid state study:
Though S 9490-6 is very soluble in water at all the physiological pH values, a study of polymorphism was performed. Three potential different forms of the solid state have been identified and characterized by X ray powder diffraction, thermal analysis and IR spectroscopy: two crystalline forms (⍺ and β) and amorphous S 9490-6. The industrial process described in 3.2.S.2 always leads to the ⍺ form, which is an hydrated form with a relatively low level of crystallinity. It is difficult to attribute a stoechiometry to this hydrate because of its hygroscopicity and the relatively low level of crystallinity. The β form is an anhydrous form which is rapidly transformed into ⍺ form under relative humidity. The ⍺ form is the stable form under ambient conditions.
All the batches obtained by precipitation in water/dimethylsulfoxide correspond to the ⍺ form. (emphasis added)
83 The industrial process in part 3.2.S.2 that the regulatory dossier referred to in the passage above was the 2002 method. The dossier noted that the reproducibility of the 2002 process had been evaluated on one pilot batch that used 14 kg of perindopril erbumine as the starting material and two industrial batches that had used 75 kg of the same starting material.
84 The regulatory dossier explained that an early process (being the 1991 method) had involved freeze drying but that Servier had preferred to use a solvent precipitation process for industrial reasons. The drug substance and drug stability studies relied on for the dossier used perindopril arginine obtained by the precipitation process.
85 The opponents argued that the statements in part 3.2.S.1 of the regulatory dossier as to the then solid state study (set out at [82] above) concerning the ⍺ and β forms showed that by 27 February 2003, the filing date of the complete specification, Servier knew that the 2002 method produced the “hydrated alpha” [⍺] form. They contended that it was inconceivable that that form had not been characterised before Servier embarked on manufacturing 23 million tablets using the 2002 method to produce that crystalline form.
86 On 2 June 2003, Servier conducted XRPD analysis of an analytical reference batch of perindopril arginine prepared by the 2002 method, labelled batch F I 211. The diffractogram produced by that analysis is below (the 2003 graph):
The 2003 graph
87 The 2003 graph was included in 3.2.S.3 of the regulatory dossier that was entitled “Characterisation”. There, Servier stated that in order for the substance to be well defined, it had undertaken a study of the polymorphism of perindopril arginine involving “the characterisation of the industrial solid form (⍺ form)” (being, at that time, the way in which the crystalline form produced by the 2002 method was styled. It is not the ⍺ form claimed in the alpha crystalline patent) and “screening of potential polymorphic forms designed to isolate, identify and characterize any new solid form”. The screening protocol resulted in the isolation and identification of 3 forms, the hydrated ⍺ form, an anhydrous β form and an amorphous state. From late April 2003, Servier conducted XRPD studies to characterise those 3 forms.
88 Servier also characterised industrial batch 58 211 made with the 2002 method and the 2003 graph recorded the XRPD analysis. In Part 3.2.S.3.1 of the regulatory dossier, Servier reproduced the 2003 graph stating that “[s]ome characteristic lines have been identified from the [XRPD] pattern at the following angles: 4.01o, 7.91o, 14.16o, 17.61o and 19.91o. However, these lines are broad and weak, indicating a low level of crystallinity”. The dossier also noted that all batches prepared using the 2002 method had the same behaviour as batch 58 211.
89 The dossier reported that Servier had conducted some crystallisation experiments using a wide range of solvents and that it had performed some XRPD studies on those samples. The regulatory dossier did not state at what dates any of the XRPD or other characterisation analysis occurred.
90 The opponents accepted that the 2003 graph was created on 2 June 2003, the date it bore in its original form that Dr Jaguelin annexed to her second affidavit. That document came from the file that Mr Damien maintained at his principal place of work in Orleans. At the trial, he gave evidence that he and his team had done “some late studies on the physical properties of the active ingredient just before submitting the file” for the purposes of bringing perindopril arginine to market. He denied that those studies occurred before the filing of the patent. I accept that evidence. The XRPD graph of the amorphous form of perindopril arginine produced using lyophilisation or freeze drying (i.e with the 1986 or 1991 method) bore the date 22 May 2003 and the XRPD graph for the β form bore the date 29 April 2003.
91 In 2007 and 2012, Servier filed applications for patents relating to perindopril arginine, that were granted subsequently by the Commissioner, as appears in the table below:
Patent No | Patent title | Date of filing |
AU 2007220435 B2 (the alpha crystalline patent) | Alpha crystalline form of the arginine salt of perindopril, process for preparing it, and pharmaceutical compositions comprising it | 26 February 2007 |
AU 2007220434 B2 | Beta-crystalline form of perindopril arginine salt, method for making same, and pharmaceutical compositions containing same | 26 February 2007 |
AU2012268815 B2 | Delta Crystalline Form of the Arginine Salt of Perindopril, a Process For Its Preparation, and Pharmaceutical Compositions Thereof | 20 December 2012 |
AU 2012268816 A1 | Process For the Preparation of the L Arginine Salt of Perindopril | 20 December 2012 |
92 Relevantly, the alpha crystalline patent made claims for a form of crystal characterised by x-ray diffraction or XRPD peaks. Claims 1-3 were as follows:
CLAIMS
1. α crystalline form of the arginine salt of perindopril of formula (I):
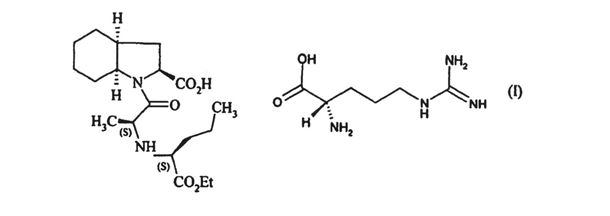
characterised by the following powder X-ray diffraction peaks, measured using a diffractometer with a copper anti-cathode and expressed in terms of Bragg’s angle 2 theta (°): 4.5, 7.9 and 13.5.
2. α crystalline form of the compound of formula (I) according to claim 1, characterised by the following powder X-ray diffraction peaks, measured using a diffractometer with a copper anti-cathode and expressed in terms of Bragg’s angle 2 theta (°): 4.5, 7.9, 13.5, 17.5 and 20.6.
3. α crystalline form of the compound of formula (I) according to claim 1, characterised by the following powder X-ray diffraction diagram measured using a diffactometer (copper anti-cathode) and expressed in terms of inter-planar distance d, Bragg’s angle 2 theta, intensity and relative intensity (expressed as a percentage of the most intense line):
Angle 2 theta (°) | Inter-planar distance d (Å) | Intensity | Relative intensity (%) |
4.52 | 19.53 | 2211 | 88.7 |
7.94 | 11.12 | 2080 | 83.5 |
12.152 | 7.277 | 682 | 27.4 |
13.480 | 6.563 | 2492 | 100.0 |
14.029 | 6.308 | 422 | 16.9 |
14.948 | 5.922 | 552 | 22.1 |
15.873 | 5.579 | 493 | 19.8 |
17.531 | 5.055 | 1600 | 64.2 |
18.787 | 4.719 | 363 | 14.5 |
19.579 | 4.530 | 1078 | 43.3 |
20.635 | 4.301 | 1794 | 72.0 |
22.616 | 3.928 | 798 | 32.0 |
23.367 | 3.804 | 473 | 19.0 |
23.807 | 3.735 | 362 | 14.5 |
24.434 | 3.640 | 409 | 16.4 |
27.148 | 3.282 | 450 | 18.1 |
28.214 | 3.160 | 417 | 16.7 |
Did the 2002 method produce the same crystalline form as the alpha-crystalline form?
93 Profs Batten and Byrn agreed that the methods used to produce a crystalline form on an industrial scale are quite likely to be different from those used on a laboratory scale. They said that often when the production process is scaled up from laboratory to industrial scale, the crystallinity of the resulting product is harder to control, but that, ordinarily, the industrial purpose will still be satisfied provided that the process is efficient, the product is reproducible and is useful for the intended purpose. In other words, so long as the industrial process generates an acceptable and reproducible crystalline form, the manufacturer will not, or may not, need to pursue another method that might produce a better quality crystalline form. As Prof Byrn put it, “the goal is to get a form that you can reproducibly make and submit that to the regulatory authorities”.
94 Prof Byrn said that ordinarily a manufacturer will try to characterise the solid form of a crystalline drug substance before selecting its final solid form and doing stability clinical and polymorph studies, prior to entering into commercial production, although not everyone did so. He said, however, that typically, the manufacturer will characterise the drug substance before putting it into tablets for production on a pilot or industrial scale.
95 By late 2002 and early 2003, Servier had an obvious commercial imperative to secure patent protection for perindopril arginine and to commercialise its production as quickly as possible. There is no evidence to suggest that persons within Servier, outside Mr Damien’s team, had done any work on characterising the crystalline forms of perindopril arginine produced by any method, prior to the time that Mr Damien had identified, namely close to the submission of the regulatory dossier. That timing is consistent with the 3 dated XRPD patterns in evidence that were found on Mr Damien’s file, produced between late April and early June 2003. It is also consistent with the guidance note issued by the Agency (see [80] above).
96 Accordingly, I am not satisfied that Servier had characterised the crystalline form produced by the 2002 method before the complete specification was filed. Even if it had, that would not have sufficed to create a state of mind in Servier that it knew that the 2002 method was as good as, or better than, the 1986 and 1991 methods. That is because no results of the stability tests on the API in the tablets produced using the 2002 method were available until after 27 February 2003.
97 The 2003 graph includes 7 arrows marking peaks at angle 2 theta (o) measurements of 4.011o, 7.905o, 14.160o, 17.612o, 19.911o, 25.611o and 25.947o. There was no diffractogram in evidence of the 17 peaks recorded in the left hand column of claim 3 (the claim 3 peaks) in the alpha-crystalline patent, but the measurements in that column could be represented on such a diffractogram in a similar way to the 7 peaks depicted on the 2003 graph.
98 The opponents contended that locations of the significant peaks in the 2003 graph measured using the angle 2 theta technique corresponded to substantively the same counterparts in claim 3. The opponents argued that this correspondence indicated that the 2002 method produced a crystalline form of perindopril arginine that was the same as that produced by the method claimed for the alpha-crystalline form in the alpha crystalline patent.
99 It is difficult to deconvolute overlapped peaks because neither the number of peaks that underlie a final average peak nor their individual intensities is known. However, the experts agreed that they did not expect that any differences in temperature, strain, the wavelength of the x-rays or the degree to which they were monochromatic would have influenced significantly the XRPD patterns in the 2003 graph and the characteristics of the claim 3 peaks that they considered for the purposes of their evidence.
100 The experts agreed that the XRPD pattern of the substance measured in the 2003 graph was poorly crystalline. The specification in the alpha-crystalline patent described the substance claimed there as having a well-defined crystalline form (p 2 lines 4, 8-9). The experts disagreed about the degree to which XRPD peaks should match between samples of the same material.
101 I found Prof Byrn’s evidence generally to be more persuasive where he differed from Prof Batten. This was because, first, Prof Byrn had a wealth of experience in dealing with the use of XRPD analysis in relation to pharmaceutical substances. In contrast, Prof Batten had none at all in that field and had never dealt with the TGA or any requirement to identify different crystalline forms of a pharmaceutical product. Also, Prof Batten was not aware of the United States Pharmacopeia or the standard that it prescribed for acceptable deviations in peak measurements between samples on which Prof Byrn had relied, namely:
Agreement between sample and reference should be within the calibrated precision of the diffractometer for diffraction angle (2 theta values should typically be reproducible to ±0.10 degrees).
102 Prof Byrn explained that this meant that the diffraction angles of the same peak in two different samples should be within 0.20 degrees. He said that the principles outlined in the United States Pharmacopeia related to the position of peaks are commonly applied in the pharmaceutical industry.
103 Secondly, Prof Batten used a larger margin of error than 0.2 degrees when he reached his conclusion that the claim 3 peaks were also present in the 2003 graph. He assumed both that the locations of the 5 broad peaks using three decimal places in the 2003 graph had not been well chosen and, critically, that the correct position for each of those 5 peaks was in fact that of one of the claim 3 peaks. That is, Prof Batten assumed that each respective position of 5 of the claim 3 peaks, being angle 2 thetas 4.52o, 7.94o, 13.480o, 17.531o and, collectively, those at 19.579o, 20.635o, and also possibly 22.616o, was the true position for a perfect monohydrate. Based on that assumption, he reasoned that, after allowing for his broader margin of error than 0.2o, the angle 2 theta peaks in the 2003 graph recorded as being at 4.011o, 7.905o, 14.160o, 17.612o and 19.911o were respectively the same as the first 5 of those claim 3 peaks.
104 Prof Batten accepted that, if he had not had the benefit of seeing the alpha crystalline patent when interpreting the position of the 5 angle 2 theta peaks in the 2003 graph, he would have been just as likely to have read, as an example, the recording of the peak at 4.011o as identifying a peak located within a margin of 0.4o on either side of the recorded measured position. He gave this evidence of his reasoning process:
HIS HONOUR: But if you have that difference going on, why is it that you identify them as being really referable to the same substance in the alpha crystalline patent than saying you just don’t know? I just don’t understand why you’re saying it’s the same.
PROF BATTEN: Because I well, it basically comes down to that I’ve used a larger margin of error to account for the fact that it’s a broad peak, a broad spectrum, and that the peak positions are not necessarily well chosen and certainly not to three decimal places in the [2003 graph]. (emphasis added)
105 Prof Byrn explained that it was not possible to predict whether a peak would shift or what direction it might take. He accepted that in the 2003 graph:
the peak at 7.905o was within plus or minus 0.1o of the claim 3 peak of 7.94o, and so was capable of being classified as measuring the same feature in both substances;
the peak at 14.16o was also within 0.2o of the claim 3 peak at 14.029o and so could be a match, but he considered that it probably was not a match;
the peak at 17.612o was capable of being matched to the claim 3 peak at 17.513o.
106 Prof Byrn said that 4 of the 7 peaks in the 2003 graph, namely, those at 4.011o, 19.911o, 25.611o and 25.947o, were not within plus or minus 0.1o of the claim 3 peaks. Accordingly, he concluded that the form of the substance analysed in the 2003 graph was not the same as that in claim 3.
107 Prof Batten argued that those four recordings were not of peaks but of noise in the diffraction process and could be ignored. He said that he “would not expect the same sort of matching to 0.2o with something that’s poorly crystalline and badly affected by noise” and that the quality of the spectrum of the 2003 graph results made it difficult to use the 0.2o standard in the United States Pharmacopeia as the yardstick. Prof Batten agreed that if peaks in the 2003 graph were eliminated as noise, there would be fewer peaks to use in matching those results to the fingerprint characterising the claim 3 peaks. The experts then gave this evidence:
HIS HONOUR: And therefore make it less likely that they’re the same crystal?
PROF BATTEN: No, it doesn’t make it less likely. It makes it … more difficult to be certain one way or the other. It doesn’t make it less likely that they match.
HIS HONOUR: Right. Yes, well, do you want to say anything there, Professor Byrn?
PROF BYRN: Yes. I think since the peak picking routine picked them, we have to use them in our analysis. … it seems too arbitrary to me to just say they’re noise. And I do agree with your Honour. If you did agree that those were noise, then that means that there’s five peaks in the [2003 graph] pattern and there’s 17 peaks in the [claim 3 peaks] pattern. And somehow Professor Batten, somehow to me, if there’s five peaks in one pattern and 17 in the other, that’s just, you just can’t get agreement with those two. Those aren’t the same samples. Those aren’t the same forms. (emphasis added)
108 Notably, the peaks at 4.5o and 13.5o in claims 1 and 2 of the alpha-crystalline patent were not present on the 2003 graph within the 0.2o margin of error. As I have indicated above, I accept Prof Byrn’s evidence and the persuasive logic of his reasoning. In the passage I have just emphasised, Prof Batten acknowledged that the elimination of peaks that can be used from the 2003 graph made it more difficult to be certain that the two forms were matches. In my opinion, this was another example of his arguing, subconsciously, from a conclusion that the two forms matched and any differences between their XRPD analysis results could be explained as inconsequential, rather than approaching the analysis without such a predisposition.
109 However, I find, as Prof Byrn reasoned in the passage of evidence I have just quoted, that the differences were too great and that the crystalline forms in the 2003 graph and the alpha-crystalline patent were not the same.
Was the 2002 method the same or strikingly similar to the method claimed in the alpha-crystalline patent?
110 The opponents argued that the 2002 method was, if not the same as, strikingly similar to, the method for producing the form of perindopril arginine claimed in the alpha-crystalline patent (the ⍺ method). The 2002 method is set out at [54] above. Relevantly, the alpha-crystalline patent claimed a method for preparing the alpha-crystalline form in claims 5, 6 and 7 as follows:
5. A process for the preparation of the ⍺ crystalline form of the L-arginine salt of perindopril of formula (l) according to any one of claims 1 to 3, wherein perindopril is dissolved in water with L-arginine, and then an apolar solvent and a polar solvent are added and the crystals obtained are filtered off, washed and then dried.
6. A process according to claim 5, wherein the apolar solvent is selected from … and toluene.
7. A process according to either claim 5 or claim 6, wherein the polar solvent is selected from dimethyl sulphoxide (DMSO). …
111 The opponents submitted that their asserted degree of striking similarity was evident from what they identified as the following common features of the two methods, namely:
(1) perindopril and arginine are mixed with water and toluene, which is a two phase mixture. The perindopril arginine in that mixture is in an aqueous phase;
(2) DMSO is added to the mixture causing the precipitation of the perindopril arginine salt that is in the aqueous phase;
(3) the precipitated crystals of the salt are filtered off, washed and then dried.
112 The opponents said that they accepted that the descriptions of the 2002 method and the ⍺ method had several differences including the following:
(a) the 2002 method begins with a salt break of perindopril erbumine, while the ⍺ method simply begins with perindopril without explaining how it is obtained;
(b) in the 2002 method, the aqueous phase, containing the perindopril arginine, is first separated from the organic phase (in the two phase mixture referred to in step (1) above) and only after that separation is DMSO added to the aqueous phase. The ⍺ method does not specify whether or not the phases are separated at the time DMSO is added;
(c) the 2002 method provides precise details as to the temperature of cooling, the solvents used to wash the crystals and the temperature and duration of drying the crystals, whereas the ⍺ method does not specify those details;
(d) the 2002 method specifies the quantities of reagents and solvents to be used whereas the ⍺ method does not specify those matters.
113 Only Dr Jaguelin gave scientific evidence, in cross-examination and re-examination, on the features, similarities and differences between the 2002 and ⍺ methods. She said, and I find, that the two methods were very different. That was principally because of the following:
the starting materials for each method were different. The 2002 method commences with perindopril erbumine to which the apolar solvent, toluene, is added before the salt break occurs. The now separated perindopril is in the organic phase because of the continuing presence of toluene when an aqueous solution, containing L-arginine, is added to the mixture. In contrast, the ⍺ method begins with perindopril being dissolved in water with L-arginine after which toluene is added;
in the 2002 method, after the perindopril arginine in the aqueous phase has been extracted from the toluene in the organic phase, the salt in the organic phase alone (i.e. without the presence of the toluene) is then precipitated by adding the polar solvent, DMSO, to the mixture. In the ⍺ method, the DMSO is added to the mixture of the two phases comprising the perindopril arginine in the aqueous phase which has been extracted from the toluene but which is still present in the organic phase. In other words, in the 2002 method, the polar solvent, DMSO, is added solely to an aqueous phase solution only after the toluene has been extracted, while in the ⍺ method DMSO is added to the two phase solution in which the toluene (in the organic phase) is present with the phase containing the perindopril arginine.
114 In a letter to the European Patent Office dated 3 December 2009, one of Dr Jaguelin’s colleagues explained why Servier maintained that it ought to be granted the European equivalent of the alpha-crystalline patent and the importance of being able to obtain a well-defined crystalline form of perindopril arginine. It explained why the addition of an organic solvent to an aqueous solution of perindopril and arginine had not led to a well-crystallised solid. The letter contrasted that approach with how the alpha-crystalline form had been achieved by adding a polar solvent and an apolar solvent to the aqueous solution.
115 The regulatory dossier described the substance produced by the 2002 method as “poorly crystalline” whereas the alpha-crystalline patent described the promise of the ⍺ method claimed as enabling perindopril arginine to “be obtained in a well-defined crystalline form” (p 2 lines 8-9). Those descriptions conform to Prof Byrn’s interpretation of the XRPD data for the two forms. I infer that the differences between the 2002 and ⍺ methods result in the poorly crystalline form of perindopril arginine being obtained from the former and the well-defined form being obtained from the latter. The absence of any expert evidence to the contrary led by either opponent is telling.
116 For these reasons, I am not satisfied that the 2002 and ⍺ methods are similar or strikingly similar.
Mr Caine’s evidence
117 Servier led expert evidence from Michael Caine, an experienced Australian patent attorney, as to his understanding of the requirements of s 40(2)(a) of the Act to describe the best method in the period before my decision in Apotex [2013] FCA 1426. Mr Caine had been awarded the degree of Bachelor of Science (Organic Chemistry) with Honours by the University of Melbourne in 1987. He had worked as technical assistant to a patent attorney for five years before himself becoming registered as a patent attorney in 1994. At the time of the hearing, Mr Caine had been a fellow since 1994, and council member since 2001, of the Institute of Patent Attorneys of Australia and the convenor of its Patents Legislation Committee. He was also the Chair of the International Patents Study Group (CET-3) of the Fédération Internationale des Conseils en Propriété Intellectuelle. He has held a number of representative positions that have required him to address existing and proposed Australian and international patent laws and treaties.
118 Mr Caine explained in his affidavit that he had understood, previously to my decision, that s 40(2)(a) required a patentee to disclose the best embodiment of the invention of which the patentee was aware at the time of filing of the application. He understood that if the invention were a product, the obligation was to disclose the best embodiment, if any were known to be better than others, of the invention and added:
However, if the patentee was aware of a particular best method of making the product, that method did not need to be disclosed, as long as the best embodiment of the product was disclosed. If a process for making the product was also claimed then the best process known to the patentee at the time of filing the application should also be disclosed. I understood the same principle to apply to claims to chemical compounds. That is, the best chemical compound needed to be disclosed, but there was no requirement to disclose the best way of making the compound. (emphasis added)
119 In his affidavit, Mr Caine drew on two sources to support his previous understanding, namely, a statement in TA Blanco White : Patents for Inventions (5th ed, 1983) at [4-515] and a statement in IP Australia’s Patent Manual of Practice & Procedure at [2.11.3.17]. The passage from Blanco White’s text that he set out omitted the footnotes that I have included in brackets below:
There would seem to be no obligation under this provision to include information not strictly relating to “the invention,” however necessary to anyone needing to work the invention. Thus it would seem unnecessary to disclose how starting materials for a process are to be obtained (American Cynamid (Dann) [1971] RPC 425, ante [4-506]); whilst it has been held that the patentee of a new article need not disclose the best method of making it, "performing" here going only to the design of the article and not to techniques for manufacturing it (Illinois Tool Works v Autobars [1972] FSR 67 at 71-72. But cf. ante [4-502]). (emphasis added)
The full passage, applicable to patent applications filed prior to 15 April 2013, from [2.11.3.17] of the Patent Manual (to which I have added a portion of the original text in square brackets that was not included by Mr Caine) was:
The specification, in addition to fully describing the invention, must include ‘the best method of performing’ the invention. In American Cyanamid Company v Ethicon Limited (1979) RPC 215 at page 269, it was stated:
“The Act is intending to protect the public against a patentee who deliberately keeps to himself something novel and not previously published which he knows of or has found out gives the best results, with a view to getting the benefit of a monopoly without giving to the public the corresponding consideration of knowledge of the best method of performing the invention.”
Consequently, even if a manner of performing an invention is self-evident, applicants are nevertheless required to set out the best method of performing the invention known to them.
[The best method requirement is assessed on the basis of the applicant’s knowledge at the time of filing the complete specification (Rescare Ltd. v Anaesthetic Supplies Pty. Ltd., 25 IPR 119). If the applicant identifies a better method at a time subsequent to filing, there is no obligation to amend the specification to include that method. In addition, if the specification does not include the best method, it can be amended to include the best method (as known to the applicant at the time of filing), at least until the time of grant (Pfizer Overseas Pharmaceuticals v Eli Lilly [2005] FCAFC 224).]
While a specification must include a best method of performance, the specification does not need to recite the words “the best method known to the applicant of performing the invention is ...”. There is also no requirement to provide a ‘best method of performance’ that differs in any way from that which is otherwise provided when fully describing the invention. Note that a ‘best method of performance’ need not be a specific exemplification of the invention, simply sufficient instruction for the skilled worker to put the claimed invention into effect. (emphasis added)
120 Materially, Blanco White at [4-502] stated (in addition to the identical passage that I set out in Apotex [2013] FCA 1426 at [166] from the 4th edition):
The specification is required to explain the nature of the invention, as well as the manner of performing it, and these are separate requirements, although the description meeting them need not necessarily be separate [Proctor & Gamble [1982] RPC 473 at 495 (CA)]. The necessary instructions to enable a skilled man to carry out the invention may or may not, it would seem, suffice to make its nature clear. Somehow or other, however, the skilled man wishing to work the invention must be told what it is he is trying to do.
The idea that a specification ought to render the working of the invention simple, not merely render it possible without further invention, has a long history. It goes with the idea – see the next paragraph – that the specification must be addressed not to the fully qualified research man who is likely to have made the invention, but to his technician: obviously, a man writing instructions for his technicians should not require too much from them in the way of thought. But his sort of approach does not lead to a rational doctrine of sufficiency. If the description in the specification is to be of practical value, it must teach other research teams what they want to know, not merely give detailed directions for working some embodiment that will rarely in the end be of industrial significance. Some matters, after all, are difficult, and do call for prolonged thought.
It is not entirely clear, whether or to what extent there is an obligation to give directions as to techniques of manufacture where the invention claimed is only the finished article [Held in Illinois Tool Works v Autobars [1972] FSR 67 at 71 (so far as can be ascertained from a very scrappy report) that the obligation to describe “the method by which” the invention “is to be performed” really only amounts to giving full enough details of the article, and is not concerned with methods of manufacture. It seems however long to have been taken for granted that an inventor must disclose how his article can be made; and this point was not in issue (see at 72; apparently, those in the art could at the date have made the thing) – the actual issue was whether the obligation to describe the best method of performance extended to the best technique of manufacture. The words used by the Act are, however, the same in the two cases. Probably this point must remain uncertain until settled in the higher courts.] (emphasis added)
121 In cross-examination, Mr Caine accepted that the long footnote in square brackets in the last quotation, discussing Illinois Tool [1972] FSR at 71, in [4-502] of Blanco White emphasised in the quotation above, to which he had not referred when he considered [4-515] of that text, rendered the position “not quite as certain as was suggested in the paragraph that I referred to”.
122 Mr Caine considered that the best method of performing the invention claimed in the patent in suit was the L-arginine salt because that was the best salt and that this had been disclosed. In cross-examination, he accepted that his understanding quoted above had not come from the extract of the Patent Manual that he had set out in his affidavit, also quoted above. And, as I have already noted, he had not considered Blanco White’s discussion at [4-502] of Illinois Tool [1972] FSR at 71-72.
123 Accordingly, I am not satisfied that Mr Caine’s opinion was supported by the two sources that he referred to, although I do not doubt that what he said represented his honestly held belief. Nor am I persuaded on the evidence that his opinion was one widely accepted by professional patent attorneys practising in Australia as at February 2003 as a reasonable opinion that a competent patent attorney could have held.
124 Indeed, Ms Harris, whom Servier did not call, had expressly recommended to Dr Jaguelin in her letter of 3 September 2004 that a method of manufacture of the new salt be inserted into the complete specification, even if it were “well known in the art, to satisfy the written description requirements”. Servier offered no explanation for its failure to call Ms Harris. I infer that any evidence that Ms Harris may have given concerning each of her recommendation and the then state of reasonable professional judgment of patent attorneys as to compliance with the best method requirement in s 40(2)(a) would not have assisted Servier’s case: cf Australian Securities & Investments Commission v Hellicar (2012) 247 CLR 345 at 412-414 [165]-[169] per French CJ, Gummow, Hayne, Crennan, Kiefel and Bell JJ; Jones v Dunkel (1959) 101 CLR 298 at 308 per Kitto J, 312 per Menzies J and 320-321 per Windeyer J.
125 Servier also relied, for its previous understanding of the best method and best mode requirement, on the decision of the United States Court of Appeals for the Federal Circuit in Eli Lilly & Co v Barr Laboratories Inc 222 F 3d 973 (2000). However, that decision did not support its position for there, the Court of Appeals said (222 F 3d at 982):
To be sure, if the best mode for carrying out a claimed invention involves novel subject matter, then an inventor must disclose a method for obtaining that subject matter even if it is unclaimed. (emphasis added)
(1) and (2) The construction and futility issues – the opponents’ submissions
126 The opponents argued that any failure to comply with the requirement in s 40(2)(a) to describe the best method known to the patentee at the time of filing the complete specification was incurable by any subsequent amendment. They contended that the scheme of Act made clear that this was so and that s 40(2)(a) expressed a temporal requirement that had to be met by the complete specification once for all at the moment it was filed. They argued that if a complete specification, when filed under s 29(4) (that required a patent request to be accompanied by a complete specification), did not then comply with s 40(2)(a), it was incurably invalid and could never be amended to correct any error or omission. The opponents submitted that for the purposes of ss 40(2)(a) and 138(3)(f), the definition of “complete specification” in the Dictionary in Sch 1 was qualified by the chapeau “unless the contrary intention appears”. That was, so the argument ran, because a failure to comply with the patentee’s duty of good faith at the time of filing could never be cured by a subsequent amendment seeking to fix a deficiency in a filed complete specification. The opponents argued that this construction was supported by what French and Lindgren JJ, with whom Crennan J agreed, had held in Pfizer Overseas Pharmaceuticals v Eli Lilly & Co (2005) 68 IPR 1 at 78 [379].
127 They also submitted that a complete specification could be amended by narrowing or deleting a claim but not in a way that negated “the date-specific fundamental statutory bargain” of full disclosure. They contended that the amendment of the definition of “complete specification” by the Patents Amendment (Innovation Patents) Act 2000 (Cth) to add the words “or, if the specification has been amended, the complete specification as amended” was not intended to have any effect on the meaning of s 40(2)(a). The opponents submitted that their construction of the once for all nature of the time of filing of the complete specification as the absolute cut-off date for compliance with s 40(2)(a) was supported by the requirement in s 42(1)(b). The latter section required, in the case of an invention involving a micro-organism to which s 41(2)(a) applied, the relevant micro-organism to have been reasonably available to a skilled addressee “at the date of filing of the complete specification”. They submitted that what the Full Court had held in Pfizer 68 IPR at 80 [390]-[391] was wrong or distinguishable. They argued in the alternative, that if what French and Lindgren JJ said in Pfizer 68 IPR at 80 [390]-[391] were correct, the latest date on which the complete specification could be amended was the date of grant of the patent or the date of commencement of proceedings.
(1) The construction issue – consideration
128 The task of statutory construction must begin with a consideration of the statutory text, considered in its context, and, where relevant, policy: Alphapharm Pty Ltd v H Lundbeck A/S (2014) 314 ALR 182 at 193 [42] per Crennan, Bell and Gageler JJ; Alcan (NT) Alumina Pty Ltd v Commissioner of Territory Revenue (2009) 239 CLR 27 at 46-47 [47] per Hayne, Heydon, Crennan and Kiefel JJ; Federal Commissioner of Taxation v Consolidated Media Holdings Ltd (2012) 250 CLR 503 at 519 [39] per French CJ, Hayne, Crennan, Bell and Gageler JJ.
129 In Project Blue Sky Inc v Australian Broadcasting Authority (1998) 194 CLR 355 at 381-382 [69]-[71] McHugh, Gummow, Kirby and Hayne JJ emphasised that the context of the provision being construed must be examined. They applied what Dixon CJ had said in Commissioner for Railways (NSW) v Agalianos (1955) 92 CLR 390 at 397, namely that “the context, the general purpose and policy of a provision and its consistency and fairness are surer guides to its meaning than the logic with which it is constructed”. They also explained that a legislative instrument must be construed on the prima facie basis that its provisions are intended to give effect to harmonious goals. And Gleeson CJ, Gaudron, Gummow, Hayne and Callinan JJ said in Australian Securities and Investments Commission v DB Management Pty Ltd (2000) 199 CLR 321 at 338 [34]-[35]:
In Project Blue Sky Inc v Australian Broadcasting Authority [(1998) 194 CLR 355 at 384, per McHugh, Gummow, Kirby and Hayne JJ], after pointing out that the duty of a court is to give the words of a statutory provision the meaning that the legislature is taken to have intended them to have, the majority said:
“Ordinarily, that meaning (the legal meaning) will correspond with the grammatical meaning of the provision. But not always. The context of the words, the consequences of a literal or grammatical construction, the purpose of the statute or the canons of construction may require the words of a legislative provision to be read in a way that does not correspond with the literal or grammatical meaning.”
It may be added that, if a party contends that a provision, by reason of such considerations, should not be given its literal meaning, then such a contention may lack force unless accompanied by some plausible formulation of an alternative legal meaning.
130 Moreover, it is inappropriate to read provisions conferring jurisdiction or granting powers to a court by making implications or imposing limitations which are not found in the express words: Owners of the ship “Shin Kobe Maru” v Empire Shipping Co Inc (1994) 181 CLR 404 at 421 per Mason CJ, Brennan, Deane, Dawson, Toohey, Gaudron and McHugh JJ; Campbell v Backoffice Investments Pty Ltd (2009) 238 CLR 304 at 361 [178] per Gummow, Hayne, Heydon and Kiefel JJ; Weinstock v Beck (2013) 251 CLR 396 at 419-420 [55] per Hayne, Crennan and Kiefel JJ; see too at 416 [43] per French CJ.
131 Chapter 10 of the Act deals with amendments of patents. The Parliament specified in Pt 1 of Ch 10 those particular amendments that were not allowable. It is common ground that Servier’s proposed amendments are not excluded by s 102 or otherwise under Pt 1 of Ch 10. The Parliament precluded the amendment (other than to correct a clerical error or obvious mistake) of a complete specification, relevantly, in s 102 on two grounds. First, if as a result of the amendment the specification would claim matter not already disclosed in the complete specification as filed and, secondly, if at a time after a specification relating to a standard patent had been accepted, either a claim would not in substance fall within the scope of the pre-amendment claims or the specification would not comply with s 40(2) or (3).
132 An applicant for a patent or a patentee could apply, subject to the Act, to the Commissioner under s 104 for leave to amend, relevantly, a complete specification “for any purpose” including the removal of a lawful ground of objection however raised (s 104(1)). However, such an application was subject to, and had to comply with, the regulations, and could not be allowed if it were precluded by s 102 (s 104(1), (5)). The potential scope of amendments that were allowable under s 104 was very broad. The power that s 104(1) conferred on the Commissioner would permit her to grant an amendment not just in respect of a patent request, but also of a granted patent, at any time for any purpose except to do what s 102 precluded.
133 In contrast to the Commissioner’s power to grant an amendment under s 104, the Act conferred on the Court a power to amend a granted patent in proceedings relating to its infringement, revocation or validity, or the validity of a claim (s 105(1)). Again, s 105(4) precluded the grant of any amendment that was not allowable under s 102. Notably, s 105(1) can only apply in respect of, first, a patent that has been granted, and, secondly, in proceedings concerning its enforceability. The Court can grant amendment not only of the granted patent itself, but also of the patent request as well as the complete specification.
134 The natural and ordinary meaning of s 105(1) is that, subject to the constraints in s 102, the Court may grant any amendment to a patent, including a complete specification, in proceedings where the subject-matter of the proposed amendment, if granted, was relevant to the enforceability or validity of the patent or a claim. Importantly, s 105(1) was available to the patentee to enable it to cure any ground of revocation specified in s 138(3) on which a challenger relied, subject to s 102 and to the patentee persuading the Court to exercise its discretion to grant an amendment.
135 I reject the opponents’ argument that the description, including of the best method, required by s 40(2)(a) cannot be changed by an amendment under s 105(1). That argument is irreconcilable with the natural and ordinary meaning of s 105(1) and its evident purpose of enabling the Court to grant an amendment, in revocation proceedings, that could save the patent, including expressly, the specification, from revocation on the ground specified in s 138(3)(f) namely that “the specification does not comply with subsection 40(2) or (3)”.
136 The specific power to amend a complete specification that s 105(1) expressly conferred on the Court could only be exercised in respect of a patent that had been granted. Moreover, s 138(3)(f) created a discretion in the Court to order revocation of a patent if the complete specification did not comply with s 40(2) or (3). That is why the Parliament conferred on the Court the discretionary power in s 105(1) to grant an amendment to a complete specification in revocation proceedings so as to save a patent from revocation.
137 The power in s 105(1) was not subject to any textual or other limitation in the Act other than that in s 102 which had no application to the amendment sought by Servier. The literal meaning of s 105(1) accorded with the evident facultative purpose of conferring power on the Court in proceedings in which the validity, enforceability or efficacy of the patent were in issue, to grant an amendment that related, or could relate, to its validity, enforceability or efficacy. The Parliament intended by the words of s 105(1) to confer power on the Court to grant an amendment to a patent to remedy the failure to include a, or a full or accurate, description of the invention, including the best method known to the applicant of performing it, that complied with s 40(2)(a): DB Management 199 CLR at 338 [34]-[35]; Agalianos 92 CLR at 397; Alphapharm 314 ALR at 193 [42].
(2) The futility issue
138 I reject the opponents’ argument that the description of best method cannot be amended under s 105(1) after the date of filing of the complete specification or, at latest, after the grant of the patent. First, acceptance of that argument would both extend the range of amendments that were not allowable beyond those that the Parliament specifically identified in s 102 and narrow the otherwise plenary nature of the power conferred by s 105(1) to grant an amendment in, relevantly, revocation proceedings. The opponents’ construction would impose an unexpressed limitation on a broad remedial power, conferred on the Court contrary to ordinary principles of statutory construction: Shin Kobe Maru 181 CLR at 421; DB Management 199 CLR at 338 [34]-[35].
139 Secondly, the use of the present tense in s 138(3)(f) does not negate the availability of the express power in s 105(1) to grant an amendment to a specification that “does not comply with subsection 40(2) or (3)”. To the contrary, the Parliament conferred on the Court the power to amend a complete specification in proceedings for revocation of a patent. The circumstance specified in s 138(3)(f) was a ground for the exercise of the Court’s discretion to revoke. An amendment of the specification can be made in “relevant proceedings”, such as these, at any time before the making of a final order in the exercise of the discretion to revoke under s 138(3). If the complete specification were amended under s 105(1) before exercising the power to make an order under s 138(3)(f), so that the specification after amendment did then comply with s 40(2) and (3), the precondition for the exercise of the power to revoke under s 138(3)(f) would no longer exist. Such a construction is harmonious with the remedial purpose of s 105(1) and its express availability in proceedings for revocation. In contrast, the opponents’ construction is strained, requires the implication of a further unexpressed limitation on the plenary power in s 105(1) (beyond those expressed in s 102) and fails to recognise that s 105(1) actually applies only in proceedings where the validity of the patent is in peril: DB Management 199 CLR at 338 [34]-[35].
140 Thirdly, in Pfizer 68 IPR at 80 [390]-[391] French and Lindgren JJ, with whom Crennan J agreed on this issue (see 68 IPR at 83 [408], 87 [424]) held, in obiter dicta, that the question of whether s 138(3)(f) permitted an order for revocation depended on whether the complete specification as amended (in that case to change the description of the best method) complied with s 40(2) at the time when the Court had to decide whether to order revocation. I am of opinion that their Honours were correct in that construction of the power in s 105(1).
141 Fourthly, the deeming provisions of s 41 in dealing with inventions of, or involving, micro-organisms do not require a departure from the natural and ordinary meaning of the power to amend conferred by s 105. That is because s 41 and the deposit requirements defined in s 6 create a self-contained regime for satisfying the requirement in s 40(2)(a) to describe fully an invention of a micro-organism or involving the use, modification or cultivation of a micro-organism. The construction of ss 6 and 41 involves different considerations specific to micro-organisms, in contrast to those of s 40(2)(a) that apply to the general requirements for all patents.
142 Fifthly, the full description of the best method was an included, but not exclusive, requirement of the full description of the invention that s 40(2)(a) required. If the opponents’ argument that s 105(1) did not permit the description of best method to be amended were correct, then no part of the complete specification could be amended, despite the express words of s 105(1). The opponents’ argument is thus directly in the teeth of the express words of s 105(1) and would deny that provision any operation with respect to an amendment of the complete specification in relevant proceedings, as defined: DB Management 199 CLR at 338 [34]-[35].
143 Sixthly, the opponents’ contention that in Pfizer 68 IPR at 78 [379], French and Lindgren JJ somehow contradicted what they subsequently said about the power to amend the description of best method (at 80 [390]-[391]) is without substance. Their Honours said (at [379]), that s 40(2)(a) required an applicant for a patent to describe the best method known to that applicant at the date of filing of the complete specification, no doubt because that was what s 40(2)(a) required. The subsection also required the applicant for a patent otherwise to describe the invention fully. Later in their reasons, French and Lindgren JJ explained that, if the applicant had failed correctly to describe the best method in accordance with s 40(2)(a) on the first attempt, s 104 permitted the applicant to obtain an amendment from the Commissioner to correct the error. They were discussing (in [379]), the latest date as at which the complete specification could describe the applicant’s or patentee’s knowledge of the best method, not (as the opponents’ incorrectly asserted) the latest date on which an amendment could be made. In other words, their Honours were seeking to clarify in [379] that when the complete specification described the best method known to the applicant, the description had to be of the method known at no later than the date of filing, even if the description of that knowledge was inserted at a later date. A person cannot backdate his or her knowledge of something, such as a best method, but the person can alter retrospectively his or her previous description of that knowledge as at a particular earlier point in time of that matter.
144 In [390] and [391], French and Lindgren JJ were dealing with the latter, namely they held that the power to amend a description of the best method in the complete specification in s 40(2)(a) could be used after the filing of that document but not so as to add knowledge of a method that was gained at a time later than the date of the original filing. That is, the description could be amended, but not to add knowledge that the applicant only gained at a later time than the original filing. And, this is also the context in which they discussed, but did not resolve, the question of whether an applicant could amend a complete specification to include a best method that became known to that applicant between the initial filing and the cut-off date of the grant of the patent.
145 Self-evidently, a person could not include a description of a best method of performing an invention for which a patent was sought that he, she or it did know of until after the patent had been granted. The requirement in s 40(2)(a) was to fully describe the fact of the applicant’s knowledge of the best method of performing the invention at the time of the application for the patent. However, the requirement was not designed to preclude the capacity of a patentee to apply under ss 104 or 105, to amend an erroneous or incomplete description of his, her or its knowledge at the time that the complete specification was first filed. There was no inconsistency in their Honours’ reasons, only logic that the opponents’ submissions eschewed.
146 Of course, the nature of the original error or omission in the complete specification, the circumstances in which that occurred and the reasons why the amendment is later sought will all be material to the exercise of the Court’s discretion in its consideration of whether the amendment ought be granted. An amendment to correct a deliberately false statement in a complete specification is likely to have a different fate to one designed to correct an accidental oversight.
147 For these reasons, I am of opinion that, ordinarily (without deciding the position in relation to micro-organisms), the Court has power under s 105(1) to grant an amendment of any part of the complete specification that is not prohibited by ss 102 and 105(4).
(3) The Servier’s knowledge issue – Consideration
148 The opponents have not established that as at 27 February 2003, being the date that Servier filed the complete specification, Servier knew of any matter that affected the correctness or completeness of my findings in Apotex [2013] FCA 1426 at [183]-[187]. I found there that Servier knew that the 1986 and 1991 methods were better than what it disclosed in the patent as the best method that it knew of for performing the invention – namely a classical method of salification. Those findings were the foundation of the declaration to that effect that I made on 29 May 2014: Apotex (No 3) [2014] FCA 751.
149 I am of opinion that that declaration was a final, not interlocutory, order, but even if it were not, I have no power to revisit the facts or legal basis on which I made it: Bass v Permanent Trustee Co Ltd (1999) 198 CLR 334 at 360 [57] per Gleeson CJ, Gaudron, McHugh, Gummow, Hayne and Callinan JJ.
150 Nonetheless, the opponents adduced a raft of fresh grounds, facts and arguments concerning the 2002 method in opposition to the amendment application that, if I had accepted any of them, would, or may, have required me to make findings that contradicted the essential foundation on which I had decided the best method issue at the trial. Late during the hearing of the amendment application, Apotex withdrew its application to vary the declaration.
151 The consequence is that the rights of the parties (Apotex and Servier), in respect of the litigated issue of whether the complete specification failed to comply with s 40(2)(a) only because it did not include the 1986 and 1991 methods, have been finally decided (subject, of course, to any appeal). An issue estoppel in terms of the declaration binds both Apotex and Servier at this point in this litigation. That is because neither party can deny the resolution by the declaration of the issue that they litigated at the trial for the reasons Dixon J lucidly explained in Blair v Curran (1939) 62 CLR 464 at 531-532 as follows:
A judicial determination directly involving an issue of fact or of law disposes once for all of the issue, so that it cannot afterwards be raised between the same parties or their privies. The estoppel covers only those matters which the prior judgment, decree or order necessarily established as the legal foundation or justification of its conclusion, whether that conclusion is that a money sum be recovered or that the doing of an act be commanded or be restrained or that rights be declared. The distinction between res judicata and issue-estoppel is that in the first the very right or cause of action claimed or put in suit has in the former proceedings passed into judgment, so that it is merged and has no longer an independent existence, while in the second, for the purpose of some other claim or cause of action, a state of fact or law is alleged or denied the existence of which is a matter necessarily decided by the prior judgment, decree or order.
Nothing but what is legally indispensable to the conclusion is thus finally closed or precluded. In matters of fact the issue-estoppel is confined to those ultimate facts which form the ingredients in the cause of action, that is, the title to the right established.
152 Here, one ultimate issue remains to be decided: namely whether I make an order revoking the patent under s 138(3)(f) or enforcing it by enjoining Apotex’ threatened infringement. The resolution of that issue depends on the fate of the amendment application.
153 Actavis was, and is, not a party to the proceedings because it chose not to be joined as a party and instead exercised its limited right to oppose the amendment sought by Servier. Actavis cannot contest or ignore the preclusive effect of the issue estoppel in the declaration, any more than could Apotex or Servier. Nonetheless, Apotex and Actavis were entitled to oppose Servier’s application on substantive and discretionary bases, including that any amendment might be futile.
154 I have set out above my detailed findings as to Servier’s state of mind. Those findings are relevant to how I should determine the exercise of the discretion under s 105(1), to which I now turn.
155 There was no evidence that Servier knew, as opposed to believed, perhaps very strongly, that the 2002 method was better than the 1986 or 1991 methods. As I found in my principal reasons, Servier knew that, when perindopril arginine was used in the tablets studied, the 1986 and 1991 methods worked and produced the results that were set out in the patent. As I have explained above, Servier could not have known, as at 27 February 2003, whether the 2002 method produced perindopril arginine that, when used in tablets that were packaged and tested for stability, would have the same or better results than those given in the patent, because the six months necessary for the stability study of those tablets began in November 2002 and did not elapse until May 2003 – well after the date of filing of the complete specification.
156 Knowledge is a different state of mind than belief: George 170 CLR at 112-113, 115-116. Hindsight is not a substitute for earlier knowledge. On the evidence, minute analysis of the performance or characteristics of the perindopril arginine produced by the 2002 method, its uses in tablets, and the performance and characteristics of the salt produced by the method in the alpha-crystalline patent only occurred after 27 February 2003. That later analysis does not have any relevance to the question of Servier’s state of mind as to whether, at 27 February 2003, it knew that the 2002 method was as good as or better than the 1986 and 1991 methods that it had proved worked in the analytical studies referred to in the patent in suit.
157 I find that, to the extent that I can revisit the declaration made on 29 May 2014, on the evidence adduced in the amendment application the declaration correctly expressed why the complete specification did not comply with s 40(2)(a). I am not satisfied that Servier knew at the date of filing of the complete specification that the 2002 method was as good as, or better than the 1986 and 1991 methods.
(4) The discretion issue – the relevant arguments
158 The opponents argued that, although they did not directly apply to the consideration of this amendment application, the 2012 amendments to the Act indicated a legislative intention to prohibit amendments to a complete specification after it has been filed. They submitted that the amendments were intended to correct the decision in Pfizer 68 IPR 1. They asserted that the amendment should be refused on five grounds: first, it was made after the trial had concluded when Servier was on notice of the ground for revocation under s 138(3)(f) (for failure to comply with s 40(2)(a)) on which it failed, secondly, Servier had not complied in the course of the amendment application with its duty of full disclosure in respect of its knowledge of what was the best method known to it at the filing date of 27 February 2003, thirdly, Servier had delayed in seeking to amend and had not explained satisfactorily why it had delayed, fourthly, Servier had attempted to gain an advantage from its original decision to file the complete specification without disclosing the 1986 and 1991 methods, and, fifthly, Servier ignored Ms Harris’ advice in 2004.
159 Servier contended, in response to the fifth of the opponents’ arguments, that Dr Jaguelin had considered that Ms Harris’ recommendation went to sufficiency, rather than best method, and that there was no basis for any concern as to sufficiency. Indeed, as it noted, in Europe an opposition based on sufficiency grounds had been dismissed.
160 The opponents argued that, not only had Servier done nothing to seek an amendment after Apotex had first raised its best method ground in mid-March 2013 until my principal reasons were delivered on 24 December 2013, but it had delayed filing its application for some time after that decision and, initially, had not sought to include in the amendment the statement that each of the 1986 and 1991 methods had resulted in a white crystalline product. They submitted that Servier had caused disadvantage to the public by depriving it of the knowledge of the 1986, 1991 and 2002 methods for the period between the filing of the complete specification until late 2013, during which time Servier had subsequently filed the patents claiming various crystalline forms and methods for producing them referred to in [82] above. Moreover, the opponents argued that Servier had not put on evidence that its proposed amendments would actually disclose the best method known to it as at 27 February 2003, as opposed to that disclosure simply amounting to what Servier had chosen to reveal to deal with what Servier was aware that the opponents knew from the forensic process.
(4) The discretion issue – the scope of the discretion under s 105(1) – consideration
161 It is common ground that Servier’s proposed amendments are not precluded from allowance by ss 102 and 105(4). As I have explained above, the discretion conferred by s 105(1) is unconfined by implications or limitations that are not part of the grant of power when read with s 102. Nonetheless, in exercising the discretion under s 105(1) the Court must act judicially, mindful that the grant or rejection of an amendment may affect not just the rights and liabilities of the patentee and the other parties in the relevant litigation, but also those of the public at large.
162 The words and other descriptions used in a patent are chosen by the original applicant who, generally, will become the patentee. The choices of those words and descriptions do not involve a process such as that in the negotiation of a contract in which two or more parties express what is ultimately a common outcome, usually reached after compromises of various positions. Thus, when a patentee applies to amend the terms of the document originally put forward as the basis for the State granting the patentee a limited monopoly, the circumstances surrounding the patentee’s decision to make the application in that form, ordinarily, will be relevant to the Court’s consideration of whether the patentee should be allowed to depart from what he, she or it originally wrote to express the extent of the monopoly sought.
163 The Act does not evince any particular approach or predisposition as to how the Court should exercise its discretion to permit an amendment to a patent that is otherwise allowable, except to the extent that the occasion for the exercise of the power in s 105 necessarily will be in proceedings in which questions of revocation, infringement or validity of the patent are in issue.
164 That being so, I am of opinion that the power in s 105(1) exists for the purpose of enabling the Court to have an unfettered discretion to permit or refuse such amendments as are both allowable and may make the patent valid or enforceable in respects that, without amendment, it would not be. Nonetheless, the ordinary principle in litigation ought apply that the patentee must make out a case in favour of the exercise of the discretion in the circumstances. As Yates J observed in Bayer Pharma Aktiengesellschaft v Generic Health Pty Ltd (2012) 99 IPR 59 at 87 [161], drawing on what Emmett J had said in Les Laboratoires Servier v Apotex Pty Ltd (2010) 89 IPR 219 at 231 [59], the power under s 105 exists for the benefit of the patentee to validate what would otherwise be an invalid, or partly invalid, patent.
165 Kenny and Stone JJ said in Servier 89 IPR at 235 [76] that the remarks of Aldous J in Smith Kline & French Laboratories Ltd v Evans Medical Ltd [1989] 1 FSR 561 at 569 offered guidelines, that have often been relied on in the exercise of the discretion to grant or refuse amendments under s 105 and its analogues. His Lordship said there:
The discretion as to whether or not to allow amendment is a wide one and the cases illustrate some principles which are applicable to the present case. First, the onus to establish that amendment should be allowed is upon the patentee and full disclosure must be made of all relevant matters. If there is a failure to disclose all the relevant matters, amendment will be refused. Secondly, amendment will be allowed provided the amendments are permitted under the Act and no circumstances arise which would lead the court to refuse the amendment. Thirdly, it is in the public interest that amendment is sought promptly. Thus, in cases where a patentee delays for an unreasonable period before seeking amendment, it will not be allowed unless the patentee shows reasonable grounds for his delay. Such includes cases where a patentee believed that amendment was not necessary and had reasonable grounds for that belief. Fourthly, a patentee who seeks to obtain an unfair advantage from a patent, which he knows or should have known should be amended, will not be allowed to amend. Such a case is where a patentee threatens an infringer with his unamended patent after he knows or should have known of the need to amend. Fifthly, the court is concerned with the conduct of the patentee and not with the merit of the invention. (emphasis added)
166 Subsequently, in Oxford Gene Technology Ltd v Affymetrix Inc (No 2) [2001] RPC 18 (commencing at page 310) at 317 [18]-[20] Aldous LJ, (with the agreement of Brooke LJ at 327 [58], and Sedley LJ at 328 [64]) explained that under an analogue of s 105 (s 75 of the Patents Act 1977 (UK)) the Court had the power to refuse an amendment that sought to validate a patent or to impose conditions to safeguard the public in cases where the patentee had abused the monopoly, particularly when the amendment operated retrospectively. He said that all types of abuse could result in a refusal to exercise the discretion favourably to the patentee, but that each case depended on its own facts. Aldous LJ said that his remarks from Smith Kline [1989] 1 FSR at 569 quoted above instanced the most common situations, but then went on to say ([2001] RPC at 317 [19]-[20]):
It is also not surprising that when a patentee seeks amendment, the court requires him to place before it the relevant facts and matters upon which it is to exercise its discretion. That is particularly appropriate where some amendment proceedings are conducted without the presence of an opponent. The obligation is akin to that required by a party seeking an ex parte injunction or Mareva relief. That can be illustrated by taking a typical case where a patentee seeks to amend under section 75 to strengthen his patent against an attack based upon a particular piece of prior art. Thus his statement of reasons will give that as the reason for amendment. It follows that the court is concerned with whether to exercise its discretion to allow amendment for that reason and the patentee must turn his mind to that issue so as to be able to inform the court of the relevant facts. Any disclosure should be limited to that issue and only ordered if necessary. An opponent may raise other grounds of abuse, but they should be properly particularised before wider disclosure becomes potentially necessary.
… it seems that practitioners believe there to be an obligation upon a patentee to trawl through his documents to see whether they are relevant to the exercise of the discretion, whatever the reason put forward for the amendment. That results in considerable expense and is not required under modern principles. The obligation of good faith requires the patentee to put forward correct reasons for the amendment. If there be facts relevant to the exercise of the discretion for those reasons then those facts need to be put before the court. (emphasis added)
167 The parties debated what Aldous J had said at first instance and in the Court of Appeal in the passages quoted above as if his Lordship’s remarks were the words of a statute, rather than the explanation by the judge of generalised factors that he considered may be relevant to the exercise of an unfettered judicial discretion. The focal point of their debate concerned the subject matter of what Aldous J had meant when he said that the patentee had to make full disclosure of all relevant matters: Smith Kline [1989] 1 FSR at 569.
168 As Jessup J observed in CSL Ltd v Novo Nordisk Pharmaceuticals Pty Ltd (No 2) (2010) 190 FCR 522 at 539 [56], his Lordship’s observation related to the difficulty for a Court to deal appropriately with an application to amend if relevant facts were not before it. Jessup J held that, even if some material facts only came to light during, or as a result of, the forensic process, the Court should proceed to determine the application on its merits. That view is consistent with the explanation given by Aldous LJ in Oxford Gene [2001] RPC at 317 [19] that the patentee has an obligation of candour that has particular importance as to the patentee’s reasons for seeking the amendment as well as on an ex parte application. Pertinently, his Lordship said that that obligation “requires the patentee to put forward correct reasons for the amendment” but did not impose a general obligation to “trawl through”, and I would add produce, documents to see whether they are relevant to the exercise of the discretion on bases other than the disclosure of the patentee’s reasons for seeking to amend ([2001] RPC at 317 [20]).
169 Accordingly, I have made findings of fact in these reasons on the basis that Servier had an obligation of candour as to its reasons for seeking the amendments (which I have found Servier properly met) and an onus to establish that the discretion be exercised in its favour. The opponents carried an onus of proof on the other matters that they sought to advance in the grounds of opposition, such as those concerning the 2002 method and the alpha-crystalline patent (on which, as I explain in these reasons, they failed to satisfy me). An amendment application is not a trial on the validity of the patent were the amendment granted.
170 I reject the opponents’ argument that the subsequent amendments to the Act in the Intellectual Property Laws Amendment (Raising the Bar) Act 2012 (Cth) are relevant to the exercise of the discretion. Those amendments operated prospectively, not retrospectively, on existing rights. The Explanatory Memorandum for the Bill that became that Act accepted the interpretation of s 105 by the Full Court in Pfizer 68 IPR 1 seven years earlier, and explained that the Parliament intended to change the law as to amendment of patents in order to bring it into conformity with that of other countries. The 2012 amendments, and the secondary materials associated with them, do not assist in the interpretation of s 105 as it operated in the Act in respect of Servier’s accrued rights to seek an amendment the subject of the present application: cf: Allina Pty Ltd v Commissioner of Taxation (1991) 28 FCR 203 at 212 per Lockhart, Burchett and Gummow JJ.
171 Here, Servier’s reason for the amendment is not far to seek. Self-evidently, it was to correct Servier’s failure to comply with s 40(2)(a), because the complete specification did not include the 1986 and 1991 methods, as Apotex had pleaded and proved. That is what Dr Jaguelin said and I find.
172 The trial had proceeded on the basis that the relevant controversy that Apotex had raised was about the failure of the complete specification to disclose the best method known to Servier of performing the invention, being the relevantly indistinguishable 1986 and 1991 methods. Apotex conducted its case at the trial in such a way that it relied only on the failure of the patentee to disclose the 1986 and 1991 methods. Apotex’ decision to confine its case amounted to a conscious abandonment of any other basis on which it could seek revocation of the patent under s 138(3)(f), in respect of any failure by Servier to disclose the best method known to it of performing the invention as required by s 40(2)(a), as I explained in Apotex Pty Ltd v Les Laboratoires Servier (No 2) [2014] FCA 751 at [3]-[14]. Having lost that battle, Servier sought an amendment, initially on a more limited basis, and ultimately in a form that would include in the complete specification the description of the 1986 and 1991 methods as expressed in the declaration that I made on 29 May 2004 (see: Apotex (No 2) [2014] FCA 751).
173 First, there is some force in the opponents’ argument that Servier was on notice about the possibility of the need to seek an amendment to the patent after Apotex had been given leave to amend on 2 August 2013 to rely on the asserted failure to disclose the best method. However, both Apotex and Servier wished to preserve the hearing that had been fixed to commence on 8 October 2013. That was why Apotex had confined its case on failure to disclose the best method to just the 1986 and 1991 methods. From about 17 July 2013, Servier’s solicitors had senior counsel’s advice in his note (see [24] above) and, no doubt, formed the view based on that advice that there was no need to seek an amendment. Both senior counsel and Servier’s solicitors are well experienced in this area of law. I am satisfied that any failure of Servier to seek an amendment prior to the trial was an error reasonably made in good faith on the basis of that advice: Smith Kline [1989] 1 FSR at 569.
174 Had Servier applied to amend, the fixture could not have proceeded. The amendment application had to be advertised and third parties had to be given an opportunity to appear and to oppose the amendment. As it was, that process took some time in 2014. After the advertisement appeared in late March 2014, Actavis only appeared in late May 2014. Thereafter, Servier and the opponents had to formulate the grounds for the amendment and opposition. Servier had provided affidavit evidence as early as Dr Jaguelin’s 7 March 2014 affidavit. But in the end, both it and the opponents relied on more documentary and expert evidence.
175 I would not have allowed the hearing date to remain because the whole of the issues would have been unclear, including the issues that the experts had to address. In closing address senior counsel for Apotex submitted that he would have sought an adjournment of the trial date if Servier had applied to amend the complete specification before the trial. While I accept that that submission was honestly made, both parties made tactical choices as to how they would deal with matters. Shortly before 2 August 2013, Servier had opposed Apotex’ original attempt to plead that Servier had not described the best method known to it without any limitation. Servier based that opposition on its solicitor’s evidence that extensive discovery and evidence would be necessary to meet such a claim. That was the context in which Apotex agreed to limit its claim under s 40(2)(a) to a failure to disclose only the 1986 and 1991 methods and so preserve the hearing dates.
176 And Apotex had every reason to maintain the hearing date because it was restrained from launching its generic perindopril arginine product. In those circumstances, having regard to the interests of the parties in a prompt trial, the consequences of an adjournment and in light of the long period since the grant of the patent, I am unpersuaded that Servier’s failure to seek an amendment earlier than it did has any substantive impact on the exercise of the discretion under s 105(1).
177 Secondly, for the reasons I have given, Servier’s obligation of disclosure was confined to explaining why it sought the amendment: Oxford Gene [2001] RPC 18 at 317 [19]. I reject the opponents’ arguments that that obligation was more extensive. The consequence is that the opponents carried the onus of proof on the issues that they raised about the 2002 method and all of the other respects in which they challenged the grant of the amendment.
178 I have accepted Dr Jaguelin’s evidence as to Servier’s reasons for seeking the amendment (see [67] and [171] above). I have also found that the opponents have not established that:
Servier knew, at 27 February 2003, that the 2002 method was as good as, or better than, the 1986 and 1991 methods, or that the 2002 method had to be disclosed in the complete specification ([69] –[72]);
the 2002 method was, or produced a crystalline form that was, similar or strikingly similar to the ⍺ method or alpha-crystalline form (see [116]);
Servier had characterised any form of perindopril arginine ([96]);
Dr Jaguelin, as the relevant person in Servier, intended to gain some advantage by the non-disclosure of the 1986 and 1991 methods. (Rather, she thought, correctly, that based on what she knew was involved in those two methods, each was a simple and routine classical salification ([36]).)
179 Thirdly, I do not accept that Servier has been guilty of any relevant delay causing prejudice to anyone in applying for the amendment since I published my reasons on 24 December 2013. From that time, the reasons for judgment provided the public with notice of the detail of the 1986 and 1991 methods and, no doubt, those in the pharmaceutical industry with any interest in the matter were than able to understand how to make a useful form of the salt.
180 Last, however, I am of opinion that Dr Jaguelin’s response of 20 December 2004 to Ms Harris’ recommendation to include a method of manufacture of the new salt in the complete specification cannot be gainsaid, as Servier argued, as being directed only to sufficiency. I accept that Dr Jaguelin did not think that such a disclosure was necessary (see [33], [43]), but that was an error on her part. Ms Harris’ recommendation was clear. Dr Jaguelin, on behalf of Servier, chose not to amend the complete specification to add a description of either of the 1986 or 1991 methods that Servier knew, at 27 February 2003, would enable a skilled addressee to make a form of salt that could be used in pharmaceutical compositions.
181 As I have found, it was possible to amend the complete specification before the grant to add the best method. In substance, Dr Jaguelin’s decision not to act on Ms Harris’ recommendation but to “see later” was a calculated risk about a potentially very valuable asset, were the patent granted without the recommended amendment. Although Dr Jaguelin honestly did not consider it necessary to act on the recommendation, she did not suggest that, had she done so, Servier would have suffered any disadvantage. The amendment would have been simple and straightforward. I am not satisfied that it was reasonable for Dr Jaguelin to decide not to act on Ms Harris’ recommendation. In Smith Kline [1989] 1 FSR at 577, Aldous J said, in a passage applied by Merkel J in Novartis AG v Bausch & Lomb (Australia) Pty Ltd (2004) 62 IPR 71 at 106-107 [135]:
Although I accept that nobody suffered any detriment by the delay in seeking amendment, I do not believe that the plaintiffs ever considered whether or not their inaction would cause detriment. At all times they were concerned to enforce their patent rights, and they have not established that they ever considered whether others would suffer detriment by any failure to amend. The plaintiffs never considered the position of others except with a view to preventing them marketing cimetidine.
The fact that nobody has suffered detriment due to the control of cimetidine by the generic and master patents is not, in my view, a ground for failing to amend for eight years. That fact was not the reason why the plaintiffs did not seek to amend; it was the consequence of the ambit of the generic and master patents. If there be delay in amending by a patentee who knows or ought to know of the need to amend, as is the position in this case, then he must establish a reason for his decision not to amend or to do nothing and also that the reason was reasonable. It is not sufficient to show that nobody has been hurt by the delay, as that is not a reason for the delay. Even if it be assumed that it be a reason, it is not a reasonable reason in that it disregards the public interest in ensuring that patents are amended promptly when the need arises, so that the public can rely on patents as representing that to which the patentee believes he is entitled. Further, if the plaintiffs had decided not to amend earlier because they had concluded that nobody would suffer damage by the delay, that would not have been a reasonable ground for the delay as it disregarded the public interest. (emphasis added)
182 Similar considerations apply here. The policy of the requirement in s 40(2)(a) is that the patentee disclose, as part of the statutory bargain to secure the monopoly conferred by the patent, the best method of performing the invention known to him, her or it. I found in Apotex [2014] FCA 1426 at [185] (and see too at [177]-[187]:
Servier had used a particular method or methods of classical salification and parameters that produced a guaranteed result. The complete specification described a broad and very general method of performing the invention that left to chance whether the skilled addressee would choose, from among the very large range of variables identified by the first joint expert report, the method (or one of the 1986 or 1991 methods) that the patentee knew actually worked to enable the API to be used in a tablet form. (emphasis added)
183 I am of opinion that the damage to the public interest involved in Servier’s failure to disclose the 1986 and 1991 methods in the complete specification, even after Ms Harris’ recommendation, was not reasonable and outweighs the proprietorial interest of Servier in being able to save the patent from revocation under s 138(3)(f).
Conclusion
184 For these reasons, Servier has failed on its substantive application to amend the patent. I will make orders that the patent be amended by deleting claims 8 to 11 but that the application for amendment be otherwise dismissed. As a result, pursuant to s 138(3)(f), the patent will need to be revoked on the ground that the specification does not comply with s 40(2)(a) of the Act. The parties should bring in short minutes of orders to give effect to these conclusions.
185 The parties asked to be heard on costs. That is appropriate. As was the case in the trial, Servier has lost on a narrow ground. The hearing of the amendment application was prolix and the opponents failed on almost every issue that they raised and pursued. That outcome should be reflected in an order for costs. The amendment application should have been capable of being heard in one day, not five. I will direct the parties and the opponent to make brief written submissions as to costs of the trial and amendment application.
I certify that the preceding one hundred and eighty-five (185) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Rares. |
Associate:

