
FEDERAL COURT OF AUSTRALIA
Ranbaxy Laboratories Limited v AstraZeneca AB [2013] FCA 368
IN THE FEDERAL COURT OF AUSTRALIA | |
| First Applicant RANBAXY AUSTRALIA PTY LTD Second Applicant ASTRAZENECA AB First Cross-Claimant ASTRAZENECA PTY LIMITED Second Cross-Claimant | |
AND: | First Respondent ASTRAZENECA PTY LIMITED Second Respondent RANBAXY LABORATORIES LIMITED First Cross-Respondent RANBAXY AUSTRALIA PTY LTD Second Cross-Respondent |
DATE OF ORDER: | |
WHERE MADE: |
THE COURT ORDERS THAT:
1. Pursuant to s 37AF of the Federal Court of Australia Act 1976 (Cth), and on the ground that the order is necessary to prevent prejudice to the proper administration of justice, the reasons for judgment be marked “CONFIDENTIAL” and until further order are not permitted to be received by any person other than the Australian external legal solicitors and barristers of each of the parties and those permitted or previously agreed to be permitted to receive confidential information about the Ranbaxy Products pursuant to paragraphs 2 and 3 and Schedule 2 of the Court’s order of 21 October 2011
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
VICTORIA DISTRICT REGISTRY | |
GENERAL DIVISION | VID 1008 of 2011 |
BETWEEN: | RANBAXY LABORATORIES LIMITED First Applicant RANBAXY AUSTRALIA PTY LTD Second Applicant ASTRAZENECA AB First Cross-Claimant ASTRAZENECA PTY LIMITED Second Cross-Claimant
|
AND: | ASTRAZENECA AB First Respondent ASTRAZENECA PTY LIMITED Second Respondent RANBAXY LABORATORIES LIMITED First Cross-Respondent RANBAXY AUSTRALIA PTY LTD Second Cross-Respondent
|
JUDGE: | MIDDLETON J |
DATE: | 23 APRIL 2013 |
PLACE: | MELBOURNE |
REASONS FOR JUDGMENT
INTRODUCTION
1 This proceeding concerns an application for revocation of a number of patents registered in the name of AstraZeneca AB, being one of the respondents and cross-claimants in the proceeding (all of whom will be referred to collectively in these reasons as ‘AstraZeneca’), and claims of infringement of those patents.
2 Subject to the outcome of this proceeding, the applicants and cross-respondents (who will be collectively referred to as ‘Ranbaxy’) intend to market in Australia a drug in tablet form, the active ingredient of which is the magnesium salt of the (-)-enantiomer of omeprazole (esomeprazole magnesium) (the ‘Ranbaxy Product’).
3 AstraZeneca allege infringement of three Australian patents:
(a) Australian patent number 676,337 (the ‘Purity Patent’), claim 1 of which claims an optically pure salt of the (-)-enantiomer of omeprazole, including the magnesium salt;
(b) Australian patent number 695,966 (being the Multiple Unit Tableted Dosage Form Patent, which was referred to by the parties in this proceeding as the ‘MUPS Patent’), claim 1 of which claims, amongst other things, omeprazole or one of its single enantiomers or an alkaline salt thereof, in a tablet form with defined characteristics; and
(c) Australian patent number 695,774 (the ‘774 Patent’), claim 1 of which claims an oral pharmaceutical dosage form comprising, amongst other things, a proton pump inhibitor (‘PPI’), including omeprazole or the (-)-enantiomer of omeprazole.
4 Omeprazole was the subject of an Australian patent filed by AstraZeneca on 11 April 1979 (Australian patent number 529,654), now expired. That patent has been followed by a series of Australian patents, including Australian patent number 563,842 for different salts of omeprazole, Australian patent number 640,246 for a process for the preparation of omeprazole, and Australian patent number 601,974 for an oral preparation, the active ingredient of which is omeprazole.
5 The Purity Patent, the MUPS Patent and the 774 Patent can be collectively referred to as ‘the Patents’. I will be dealing separately with each of the Patents in these reasons.
6 This proceeding, as will become apparent, involved a large number of expert witnesses, and extensive disputation between the parties on a number of issues. The parties, for commercial reasons (particularly Ranbaxy), desire a determination preferably by no later than 23 April 2013. The actual hearing, covering some weeks, concluded on 28 March 2013. Final written submissions were filed on 12 April 2013.
7 To accommodate the parties’ request in this regard, it is impossible to set out and survey all the evidence led by the parties. But nor is it necessary to do so. The task undertaken by the Court has been to seek to address the major determinative issues, and the essential areas of disputation between the experts.
RELEVANT LEGISLATION
8 By way of overview:
(a) The application for the Purity Patent was filed on 27 May 1994, and the Purity Patent was granted on 25 June 1997. The priority date for this patent is 28 May 1993.
(b) The application for the MUPS Patent was filed on 7 June 1995, and the MUPS Patent was granted on 10 December 1998. The priority date for this patent is 8 July 1994.
(c) The application for the 774 Patent was filed on 9 February 1996, and the 774 Patent was granted on 3 December 1998. The priority date for this patent is 9 February 1995.
9 The issue of infringement in relation to each of the Patents is to be determined under the Patents Act 1990 (Cth) (‘the Act’).
10 As for the issues of validity, s 7 of the Act and the definition of “prior art base” was amended by items 1 to 6 and 12 of Sch 1, Pt 1 of the Patents Amendment Act 2001 (Cth) (‘the Amendment Act’), which took effect on 1 April 2002. However, the amendments made by the Amendment Act apply only in relation to patents for which the complete applications are made on or after 1 April 2002 (item 13 of Sch 1, Pt 1 of the Amendment Act).
11 Accordingly, the issues of validity in relation to each of the Patents is to be determined under the Act in the form in which it existed prior to the Amendment Act.
BACKGROUND
12 It is useful to commence with a summary of some of the key scientific concepts relevant to the Patents and the issues raised in this proceeding.
Stereochemistry
13 As will shortly become apparent, the Purity Patent invokes certain stereochemical concepts, such as the concept of enantiomers, which may require explanation. I understand that these basic principles are uncontroversial as between the parties.
14 At 28 May 1993 (being the priority date of the Purity Patent, which was the earliest priority date in issue in this proceeding), it was known that enantiomers are isomers (being compounds that have the same molecular formula, but which differ in the way in which their atoms are arranged) with identical structural connectivity and physical properties, except the direction in which they rotate a plane of polarised light. But another key aspect of the definition of enantiomers is that they are, at the molecular level, non-superimposable mirror images of each other. This property is known as ‘chirality’.
15 In a molecule, a carbon atom with four different functional groups attached to it is known as the ‘stereogenic’ or ‘chiral’ centre. However, any atom that can form four or more bonds to other atoms can demonstrate chirality. Accordingly, other atoms (such as sulphur) can also behave as chiral centres.
16 Diastereomers (also known as diastereoisomers) are molecules that contain two or more chiral centres (and can also exist as pairs of enantiomers).
17 Enantiomers rotate the plane of polarised light in either a clockwise or counter-clockwise direction. Compounds that rotate the plane of polarised light in a clockwise direction are described as dextro-rotatory and are represented with a “(+)” symbol. Compounds that rotate the plane of polarised light in a counter-clockwise direction are described as levo-rotatory and are represented with a “(-)” symbol. Omeprazole has two enantiomers: (-)-omeprazole and (+)-omeprazole.
18 The absolute configuration of an enantiomer is characterised as “(S)” or “(R)” depending on the spatial arrangement of atoms around the chiral atom. It is now known that the (-)-enantiomer of omeprazole is the (S)-enantiomer of omeprazole.
19 When both enantiomers are present in a mixture in equal proportions (ie 1:1), this is known as a ‘racemic mixture’, or a ‘racemate’. A racemate does not rotate the plane of polarised light (and is therefore not optically active) because, the rotation caused by the (-)-enantiomer is cancelled out by the equal and opposite rotation of the (+)-enantiomer.
20 ‘Racemisation’ is a process whereby enantiomers interconvert to create a racemate (ie a 1:1 mixture of the two enantiomers just discussed), resulting in a complete loss of optical activity.
Omeprazole and acid secretion
21 Humans produce hydrochloric acid in the stomach, secreted by specialised cells known as ‘parietal cells’. These are generally located in the proximal two-thirds of the stomach lining, namely, in the part of the stomach lining that is closer to the mouth.
22 Acid secretion is regulated by a combination of hormones and nerves, working via three major pathways known as the paracrine, neurocrine and hormonal pathways. Stimulation of gastric acid secretion by all three pathways is primarily catalysed by a highly specialised enzyme located in the parietal cells known as hydrogen potassium adenosine triphosphatase (or H+/K+ ATPase). Its mechanism of action involves the transport (or ‘pumping’) of a hydrogen ion (ie a proton) out of the parietal cell and into the cavity of the stomach, in exchange for a potassium ion (which enters the parietal cell). Because it pumps protons out of the parietal cell, H+/K+ ATPase is known as a ‘proton pump’.
23 Omeprazole is a gastric proton pump inhibitor. It suppresses the secretion of gastric acid by binding to and inhibiting the H+/K+ ATPase enzyme in the parietal cell secretory membrane. For this reason, omeprazole has been used to treat gastrointestinal conditions (about which more will be said in due course).
24 Omeprazole is a chiral sulphoxide that exists as two enantiomers. Its chemical structure is as follows:
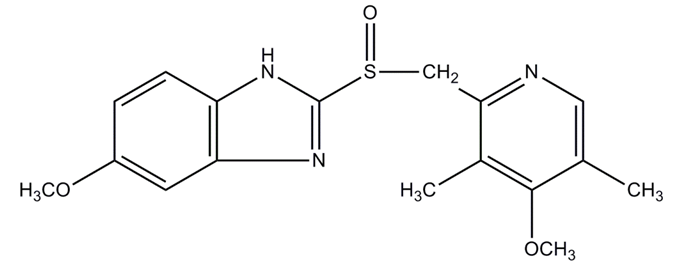
25 Omeprazole is described as being ‘acid labile’, meaning that it is sensitive to acid.
Pharmacodynamics
26 The field of pharmacodynamics examines the effect of a drug on a biological target or receptor. The therapeutic effect of a drug can result from the action of the unaltered drug molecule, or from one or more of its metabolites, or both.
27 Drugs taken orally are generally absorbed through the wall of the small intestine (the upper gastrointestinal tract) and then carried to the liver in the bloodstream. A significant fraction of a drug can be metabolised by enzymes in the wall of the small intestine and the liver. This is referred to as “first pass metabolism”. The remaining drug is then distributed to the ‘active site’ in the body by the systemic circulation.
28 In the case of omeprazole, the proportion of the drug that escapes first pass metabolism and reaches the systemic circulation unchanged is then available to act at the parietal cells of the stomach, where it is converted to the active species responsible for inhibiting the proton pumps responsible for gastric acid secretion. “Prodrugs” are drugs that are therapeutically inactive when introduced to the body, but that upon metabolism, produce a therapeutically active metabolite. They are designed with the goal of ensuring the delivery of the drug to the liver, plasma or other site where conversion to the active species occurs. Omeprazole is a prodrug. Both of its enantiomers are converted to the same achiral active species.
29 Interindividual variability (also referred to as ‘interindividual variation’) is the variability between different people in response to the same dose of a drug. In terms of pharmacokinetic properties, interindividual variability refers to differences in absorption, distribution and elimination of a drug between different people. This concept reflects the fact that there may be differences in the extent of metabolism of the same dosage of a drug between different people.
30 To this end, genetic differences in drug metabolism may manifest as different phenotypes (being the observable characteristics resulting from an individual’s genetic makeup), whereby individuals can be classified as “rapid”, “extensive” or “fast” metabolisers of a drug; or “poor” or “slow” metabolisers of a drug. That difference in metabolism is caused by genetic differences between individuals in the level of enzyme activity at the site of metabolism of a drug, resulting in variability in the metabolism of that drug by different individuals. This is an example of a genetic polymorphism (being the occurrence of two or more genetically determined phenotypes in a population in such proportions that the rarest phenotype cannot be maintained by mutation or migration alone).
31 Accordingly, the acknowledged desirability of a compound with improved metabolic properties is related to the objective of reducing interindividual variability. Assume, for example, a significant amount of a drug is converted by first pass metabolism into inactive metabolites, and the extent of that conversion varies between those who metabolise the drug more extensively and those who metabolise it less extensively. A modification which causes less first pass metabolism would increase the amount of drug available for its intended useful activity. The increase in the available amount of the drug would be greater for the more extensive metabolisers than the less extensive metabolisers. Hence, the amount of interindividual variability would be reduced.
WITNESSES
32 Ranbaxy relied on the following witnesses:
(a) Dr Phillip Reece, who swore affidavits dated 25 January 2012 and 12 October 2012. Dr Reece participated in the concurrent evidence session on pharmacology. Dr Reece holds a Bachelor of Science (Hons) Degree (1973) from the University of Adelaide, majoring in Organic Chemistry, and a PhD in Medical Chemistry (1976) from the John Curtin School of Medical Research at the Australian National University. Dr Reece worked as a hospital scientist, including as Chief Hospital Scientist, at the Queen Elizabeth Hospital in Adelaide between 1975 and 1986. Dr Reece then worked at AstraZeneca in Sydney as a Clinical Trials Manager in 1987 and 1988 before joining Parke-Davis in late 1988. From 1990 to 1993, Dr Reece was Director of Clinical Pharmacology at Parke-Davis’ research facility in Michigan, United States where he managed the transition of new pharmaceuticals from pre-clinical studies to clinical trials. He prepared the clinical pharmacology section of applications for certain types of new Parke-Davis drugs and liaised with regulatory authorities. Dr Reece currently works as a consultant to the biotechnology and pharmaceutical industries, advising on pharmaceutical drug development issues and clinical trials.
(b) Dr Graeme Irvine Stevenson, who swore an affidavit dated 12 October 2012. Dr Stevenson participated in the concurrent evidence session on organic chemistry. Dr Stevenson holds a Bachelor of Science Degree with First Class Honours in Chemistry (1984) and a PhD in Synthetic Organic Chemistry (1988) from the University of Edinburgh. At the time of swearing his affidavit, Dr Stevenson was Group Leader of Drug Discovery and Design at the Eskitis Institute for Cell and Molecular Therapies at Griffith University in Queensland. Between 1987 and 2001 Dr Stevenson worked in drug synthesis, medicinal chemistry and drug design in the United Kingdom for Merck & Co. Dr Stevenson commenced his employment at Merck as a research chemist and was promoted to Senior Research Chemist in 1989 and Research Fellow in 1996. His main responsibilities involved undertaking drug design and synthesis activities at an early stage of the drug discovery process.
(c) Dr Richard Allen Pyter, who swore affidavits dated 27 January 2012, 3 April 2012, 6 June 2012, 12 July 2012, 12 October 2012, 1 November 2012 and 5 February 2013. Dr Pyter participated in the concurrent evidence sessions on salts, non-TOF-SIMS experiments (being experiments not related to Time of Flight Secondary Ion Mass Spectrometry (‘TOF-SIMS’)), construction of the 774 Patent and infringement of the 774 Patent. Dr Pyter holds a Bachelor of Science Degree in Pharmacy (1973), a Masters of Science Degree in Pharmaceutics (1977) and a PhD in Pharmaceutics (1980) from the University of Wisconsin. Dr Pyter was employed at Abbott Laboratories in the United States from 1980 to 2007. Between 1992 and 1994 and also between 1998 and 2001, Dr Pyter was the Manager of Solid Products Development where he worked on the development of solid dosage forms for newly discovered and on-market molecules and provided technical support for manufacturing operations. In the intervening four years he was in different roles within Abbott Laboratories, including having responsibility for the conduct of stability studies and good management practice compliant excipient and drug storage facilities. Dr Pyter is currently a Pharmaceutical Product Development Consultant and he is the Adjunct Professor of Pharmaceutical Sciences at the University of Wisconsin.
(d) Dr Peter John Prichard, who swore an affidavit dated 11 October 2012. Dr Prichard participated in the concurrent evidence session on gastroenterology. Dr Prichard holds a Bachelor of Medicine, Bachelor of Surgery from Monash University (1977). Dr Prichard began his specialist training in gastroenterology in 1982 and qualified as a specialist in gastroenterology in 1985. Between 1987 and 1995, Dr Prichard worked at the Flinders Medical Centre in South Australia where he provided clinical treatment, attended outpatient clinics and undertook endoscopy sessions. Dr Prichard has been employed as a Senior Visiting Medical Specialist in Gastroenterology at the Royal Melbourne Hospital since 1997. He was the Acting Director of Gastroenterology at the Royal Melbourne Hospital from June 2001 until February 2002. Dr Prichard has also worked as a private medical consultant at the Epworth Hospital in Melbourne since 1997.
(e) Mr Rodney Ian Lindsay Cruise, who swore affidavits dated 30 January 2012 and 12 October 2012. Mr Cruise was not cross-examined. Mr Cruise is a patent attorney from Melbourne. In relation to the Purity Patent, Mr Cruise gave evidence as to the searches he completed which replicated searches that Dr Stevenson said he would have done on omeprazole in 1993 if he had been asked to prepare the enantiomers of omeprazole.
(f) Dr Walter Joseph Herzberg, who swore an affidavit dated 25 January 2012. Dr Herzberg was not cross-examined. Dr Herzberg is a translator from New Jersey, United States, who translated the document which is referred to in these reasons as “DE 455”, and related documents.
(g) Ms Janine Lois Krochmal, who swore an affidavit dated 4 April 2012. Ms Krochmal was not cross-examined. Ms Krochmal is a librarian from Monash University and gave evidence that certain publications were available in Australia at 28 May 1993.
(h) Dr Thomas Leo Reiland, who swore affidavits dated 27 January 2012, 22 March 2012, 16 July 2012, 10 October 2012 and 26 November 2012. Dr Reiland participated in the concurrent evidence session on the MUPS Patent. Dr Reiland holds a Bachelor of Science Degree in Pharmacy (1972) and a PhD in Pharmaceutical Chemistry (1981) from the College of Pharmacy at the University of Iowa. Dr Reiland worked for five years as a registered pharmacist in Iowa from 1972. Dr Reiland worked at Abbott Laboratories between 1981 and 2005 on drug delivery and drug formulation projects. Dr Reiland was the Manager of Preformulation and Drug Delivery at Abbott Laboratories between 1991 and 1994. This group was responsible for the detection of physical and chemical properties of potential pharmaceutical compounds. From 1994 to 1996, Dr Reiland managed the Liquid Products Development division at Abbott Laboratories and from 1996 to 1999 worked as the manager of the Formulation Development Centre. Since April 2005, Dr Reiland has worked as a consultant providing drug delivery and formulation development advice to clients.
(i) Mr Gilbert Siu-Chung Tsang, who swore an affidavit dated 30 January 2012. Mr Tsang was not cross-examined. Mr Tsang is a lawyer from King & Wood Mallesons, solicitors for Ranbaxy, in Melbourne. He gave evidence as to AU 222 and the identity of the Japanese priority document that it refers to.
(j) Dr Birgit Hagenhoff, who swore affidavits dated 6 June 2012, 25 June 2012 and 1 February 2013. Dr Hagenhoff participated in the concurrent evidence session on TOF-SIMS experiments. Dr Hagenhoff holds a Diploma in Physics (1987) from the University of Münster in Germany. During her studies she concurrently took courses at the University’s medical school as well as in mathematics at the University itself. Dr Hagenhoff also holds a PhD in Physics (1993) from the same University. Dr Hagenhoff was employed as a Project Scientist at that University in 1993 where she conducted and coordinated research into organic surface mass spectrometry. Dr Hagenhoff was a Senior Scientist and Deputy Group Leader of the Surface Analysis Group at the Philips Center for Manufacturing Technology in Eindhoven, The Netherlands between 1994 and 1997. She was responsible for conducting analytical services using techniques like TOF-SIMS. She founded and has operated a specialist TOF-SIMS analytical service since 1997, and has 27 years’ experience in conducting TOF-SIMS analysis.
(k) Mr Danish Bhutani, who swore an affidavit dated 28 June 2012. Mr Bhutani was not cross-examined. Mr Bhutani is a Senior Research Scientist employed by Ranbaxy in India. Mr Bhutani gave evidence of the pH of the Ranbaxy esomeprazole magnesium, which is the active pharmaceutical ingredient (‘API’) in the Ranbaxy Product.
(l) Mr Kiran Vasantrao Bachhav, who swore an affidavit dated 9 November 2012. Mr Bachhav was cross-examined. Mr Bachhav is a Pharmaceutical Scientist employed by Ranbaxy in India. He holds a Bachelor of Pharmacy and a Master of Pharmacy from the University of Pune, India.
33 AstraZeneca relied on the following witnesses:
(a) Mr Per Oskar Sverker von Unge, who swore an affidavit dated 5 June 2012. Mr von Unge was cross-examined. Mr von Unge, a Principal Scientist in the Medicinal Chemistry Department of AstraZeneca R&D Mölndal, Sweden, holds a Master of Science in Engineering, specialising in Organic Chemistry (1983), and a Licentiate of Engineering, specialising in Organic Chemistry (1988) from Chalmers University of Technology in Göteborg, Sweden. Mr von Unge commenced employment at AstraZeneca as an organic chemist in 1988 and has worked on PPIs since that time. Mr von Unge was promoted to Principal Scientist in 2007. In his time at AstraZeneca Mr von Unge has worked on numerous drug discovery projects, mainly in the gastrointestinal therapy area. Mr von Unge and his supervisor Dr Per Lindberg are the named inventors of the Purity Patent.
(b) Professor Allan Mark Evans, who swore affidavits dated 8 June 2012 and 1 February 2013. Professor Evans participated in the concurrent evidence session on pharmacology. Professor Evans is Pro Vice Chancellor and Professor of Pharmaceutics at the University of South Australia. Professor Evans holds a Bachelor of Pharmacy Degree (1982) from the South Australian Institute of Technology and a PhD from the department of Clinical and Experimental Pharmacology (1989) at the University of Adelaide. Professor Evans’ PhD research investigated the pharmacokinetics of the enantiomers of ibuprofen. He completed post-doctoral research as a research associate at the School of Pharmacy and Pharmaceutical Sciences at the University of Manchester (1989-91) and at the Department of Clinical & Experimental Pharmacology at the University of Adelaide (1992). In 1992 he began lecturing at the School of Pharmacy and Medical Sciences at the University of South Australia and, in 1993, was lecturing in pharmacokinetics, biopharmaceutics, clinical pharmacology and pharmacy practice. Professor Evans specialises in pharmaceutical science, particularly pharmacology, pharmacokinetics and formulation science.
(c) Professor Stephen Graham Davies, who swore affidavits dated 6 June 2012, 28 June 2012 and 1 February 2013. Professor Davies participated in the concurrent evidence sessions on organic chemistry and salts. Professor Davies is the Waynflete Professor of Chemistry at the University of Oxford. He holds a B.A. Degree (1973) and a D.Phil. Degree (1975), both in Chemistry, from the University of Oxford. He also holds a D.Sc in Chemistry (1980) from the University of Paris. Following his D.Sc., Professor Davies accepted a lectureship at the Dyson Perrins Laboratory at the University of Oxford where he remained until 2004 after becoming a Professor of Chemistry in 1996. In the late 1980s and early 1990s, Professor Davies consulted for scientists and pharmaceutical companies who sought his assistance in obtaining diastereomerically pure compounds of interest. In 1992, Professor Davies founded Oxford Asymmetry Ltd, which aimed to provide pharmaceutical companies with homochiral compounds of interest, and which focused on the preparation of compounds with high enantiomeric purities. In 1995, Professor Davies founded Oxford Diversity Ltd, which used combinatorial chemistry to produce large numbers of organic compounds for screening. Since 2004, he has continued to teach in the Department of Chemistry at the University of Oxford. In 2006, he became the Waynflete Professor of Chemistry and from 2006 to 2011 he was also the Chairman of the Department of Chemistry at Oxford.
(d) Professor John Dent, who swore affidavits dated 6 June 2012 and 30 January 2013. Professor Dent participated in the concurrent evidence session on gastroenterology. Professor Dent is a Clinical Professor of Medicine at the University of Adelaide. He holds a medical degree from the University of Cambridge (1968) and completed the clinical component of his degree at St Thomas’ Hospital in London (1965-68). Professor Dent migrated to Australia in 1972 and completed a PhD at Monash University (1978). Professor Dent was appointed Director of the Gastroenterology Unit at the Royal Adelaide Hospital in 1986 where he remained until 2004. He was involved in AstraZeneca’s clinical trials of omeprazole in the 1980s. In 1990, Professor Dent established a nerve-gut research laboratory at the Royal Adelaide Hospital, a facility of which he remains a member today. Professor Dent maintains broad expertise across the field of gastroenterology.
(e) Dr Louisa King, who swore an affidavit dated 20 April 2012. Dr King was not cross-examined. Dr King is a patent, design and literature searcher at KingSearch Pty Ltd and has been a patent, design and literature searcher since 2003. Dr King gave evidence as to the availability of a particular publication (which is referred to in these reasons as ‘Andersson 1992 Conference Abstract’) before 28 May 1993.
(f) Mr Mike James Henry Bull, who swore affidavits dated 30 January 2012 and 24 April 2012. Mr Bull was cross-examined. Mr Bull is Director of Primary Care (Gastrointestinal/Rheumatology, Cardiovascular and Respiratory Products) at AstraZeneca Pty Ltd and has been an employee of AstraZeneca since 1994. Mr Bull holds a Bachelor of Science (Honours) degree (1982) from Otago University. Mr Bull has held a number of sales and marketing roles within AstraZeneca since 1994. At the time of swearing his affidavits, Mr Bull had strategic and management control of AstraZeneca Australia’s Primary Care marketing and sales. Mr Bull gave evidence as to the sale of various AstraZeneca products in Australia.
(g) Mr Mark Donald Fladrich, who swore affidavits dated 30 January 2012 and 24 April 2012. Mr Fladrich was cross-examined. Mr Fladrich is the Managing Director of AstraZeneca Australia, a position he has held since 2011. Mr Fladrich was first employed by AstraZeneca in April 1993. He holds a degree in pharmacy from the South Australian Institute of Technology (1980) and a Master of Business Administration from Macquarie University (1995). He gave evidence as to worldwide sales figures of certain AstraZeneca products.
(h) Professor Roland Bodmeier, who swore affidavits dated 28 March 2012, 5 July 2012, 20 September 2012 and 20 December 2012. Professor Bodmeier participated in the concurrent evidence sessions on the MUPS Patent and construction and infringement of the 774 Patent. Professor Bodmeier holds a Bachelor of Science Degree in Pharmacy (1982) from the Ludwig-Maximilians-Universität in Munich, a PhD in Pharmaceutics from the University of Texas and an additional doctorate degree (habilitation - the highest academic qualification in Germany) (1993) from the Universität Regensburg in Germany. From 1986 to 1994, Professor Bodmeier worked first as an Assistant Professor and then as an Associate Professor at the University of Texas in Austin. His research focussed on controlled release polymeric drug delivery systems for oral use, microencapsulation, multiparticulate drug delivery systems and coating technology. Since June 1994, Professor Bodmeier has worked as a Professor at the College of Pharmacy, Freie Universität Berlin. The focus of Professor Bodmeier’s research has continued to be on polymeric drug delivery systems and, in particular, on the preparation of coated multiparticulate dosage forms. This research has received significant funding from public agencies and industrial partners, including major pharmaceutical companies and excipient suppliers.
(i) Dr Angelo Mario Morella, who swore affidavits dated 22 June 2012, 4 July 2012, 11 September 2012 and 21 September 2012. Dr Morella participated in the concurrent evidence sessions on the MUPS Patent and construction of the 774 Patent. Dr Morella holds a Bachelor of Science Degree with Honours in Organic Chemistry (1979) and a PhD in Organic Chemistry (1985) from the University of Adelaide. Dr Morella commenced work as a chemist at F.H. Faulding & Co. Limited in 1985 and later that year became an analytical scientist. The following year, Dr Morella took up a position as a formulator in Faulding’s Pellet Product Development Group. From 1988 to 1994, Dr Morella managed as well as actively participated as a formulator in Faulding’s research efforts in respect of the development of microencapsulated products, from initial concepts through to commercial products (“microencapsulated products” are coated particles that are less than 2mm in size and include pellets and powders). Dr Morella’s work at Faulding also involved the testing of pharmaceutical formulations using analytical and pharmacopoeial techniques. Dr Morella retired from Faulding in March 2012 and since that time has been a self-employed independent consultant to the pharmaceutical industry.
(j) Mr Mark Lee Nicholas, who swore affidavits dated 6 March 2012 and 4 December 2012. Mr Nicholas participated in the concurrent evidence sessions on TOF-SIMS and non-TOF-SIMS experiments. Mr Nicholas holds a Bachelor of Arts Degree in Chemistry (1975) from The College of Wooster in Ohio, United States. Mr Nicholas undertook a number of graduate chemistry and physics courses between 1978 and 1983. Between 1977 and 1992, he worked for The Standard Oil Company, in various research roles including one role involving the direct commercial precursor to TOF-SIMS. Mr Nicholas then held positions at the Chemistry Department of the University of North Carolina Chapel Hill as a researcher in TOF-SIMS of polymers and a researcher in Surface Mass Spectrometry of Combinatorial Libraries. From 1995 to 1999, Mr Nicholas worked for a contract surface laboratory in New Jersey as an analyst conducting TOF-SIMS analyses of polymer, metal, semiconductor, drug and biological surfaces. Since 1999, Mr Nicholas has been employed by AstraZeneca in various roles focussing on surface analysis techniques. He has been heavily involved in the analysis of drug substances and solid dosage formulations using a range of microanalytical and chemical imaging techniques, in particular TOF-SIMS. He has in total 19 years’ experience in conducting TOF-SIMS analysis (subsequent to earlier use since 1985 of other forms of time-of-flight mass spectrometry) and has specifically 14 years’ experience in conducting TOF-SIMS analysis of pharmaceutical samples.
(k) Dr Hanna Erika Matic, who swore affidavits dated 6 March 2012 and 30 November 2012. Dr Matic participated in the concurrent evidence session on non-TOF-SIMS experiments. Dr Matic holds a Master of Science Degree in Chemical Engineering (1996) and a PhD in Materials Science (2002) from Chalmers University of Technology, Gothenburg. At the time of swearing her affidavits, Dr Matic worked at AstraZeneca in Physical Science, Pharmaceutical Development. Dr Matic has worked at AstraZeneca since 2004. Dr Matic analyses pharmaceutical formulations for the development of new products and the improvement of existing products.
(l) Dr Thomas Nils Wännman, who swore an affidavit dated 7 March 2012. Dr Wännman was not cross-examined. Dr Wännman holds a Bachelor of Science Degree (1977) and a PhD in Analytical Chemistry (1985) from the University of Stockholm. At the time of swearing his affidavit, Dr Wännman was an analytical chemist in the Process Research and Development Department at AstraZeneca. He was employed by AstraZeneca from 1995 to 2012. Dr Wännman previously worked for IBM and has experience in the use of ICP-OES analysis (being Inductively-Coupled Plasma Optical Emissions Spectrometer analysis, which is intended to identify the presence of a specific element in a substance). He gave evidence as to the sodium content of the pharmaceutical excipient hydroxypropyl cellulose.
(m) Dr Shen Yung Luk, who swore affidavits dated 6 March 2012, 20 September 2012, and 20 December 2012. Dr Luk participated in the concurrent evidence sessions on TOF-SIMS and non-TOF-SIMS experiments. Dr Luk holds a Bachelor of Science Degree (1984) and a PhD in Physical Chemistry (1987) from the Institute of Science and Technology at the University of Manchester. After completing his PhD, Dr Luk worked as a Research Fellow at the same university, investigating the non-linear optical properties of ordered asymmetric bilayer films. From 1999 to 2000, he worked as a Research Scientist at Courtaulds Corporate Research in Coventry, where he was involved in optimising the manufacture of hydroxypropyl cellulose and other polymers. In 2000, Dr Luk began working for Molecular Profiles in Nottingham, United Kingdom, first as a Technical Director (from 2000 to 2005) and then as Chief Scientific Officer, the position that he held at the time of swearing his affidavits. As the Chief Scientific Officer, Dr Luk is responsible for the scientific and research divisions of the company. Dr Luk has experience in polymer synthesis, the characterisation of cellulosic polymers and understanding the concepts underpinning acid and base chemistry and its use in, among other, things, pharmaceutical product development.
34 The experts produced the following joint expert reports:
(a) “Joint Expert Report: Gastroenterology” dated 13 February 2013 (authors: Dr Dent and Dr Prichard);
(b) “Joint Expert Report: Pharmacology” dated 12 February 2013 (authors: Professor Evans and Dr Reece);
(c) “Joint Expert Report: Organic Chemistry” dated 12 February 2013 (authors: Professor Davies and Dr Stevenson);
(d) “Joint Expert Report: Salts” dated 13 February 2013 (authors: Professor Davies and Dr Pyter);
(e) “Joint Expert Report on AU 966 (MUPS)” dated 28 February 2013 (authors: Professor Bodmeier, Dr Morella and Dr Reiland);
(f) “Joint Expert Report: AU 774 – TOF-SIMS Analysis” dated 14 February 2013 (authors: Dr Luk, Dr Hagenhoff and Mr Nicholas);
(g) “Joint Expert Report: AU 774 – Non TOF-SIMS Experiments” dated 20 February 2013 (authors: Dr Luk, Dr Matic and Mr Levay);
(h) “Joint Expert Report: AU 774 – Construction” dated 5 March 2013 (authors: Professor Bodmeier, Dr Morella and Dr Pyter); and
(i) “Joint Expert Report: AU 774 – Infringement” dated 14 March 2013 (authors: Professor Bodmeier and Dr Pyter).
35 In addition to these reports, a document was produced by Dr Stevenson entitled “ORGANIC CHEMISTRY: Second meeting” (dated 22 February 2013). This document records the answers of Dr Stevenson to a number of additional questions that were put to both him and Professor Davies. AstraZeneca submitted that the document is neither an affidavit, a joint expert report nor an expert report that meets the requirements of r 23.13 of the Federal Court Rules 2011 (Cth). Accordingly, it was submitted, I should give it no weight. As will become apparent later in these reasons for judgment I do not accept this submission. However, I have not accepted the views expressed by Dr Stevenson in this document to the extent that they are inconsistent with his prior evidence and that of the other experts.
36 I will now make a number of general observations about the evidence of the various expert witnesses, and the comparative weight to be accorded to their evidence.
Comparative weight to be given to the testimony of expert witnesses
37 No issue of credit arises in respect of any expert witness being untruthful or not attempting to assist the Court in accordance with their obligations, although issues of advocacy or lack of expertise or practical experience arise. No party contended that any of the experts were incompetent or dishonest, but they did contend that the evidence of some witnesses should be preferred.
38 I do not consider I can say that I generally prefer any one expert in any given field in respect of all topics relevant to that field, although in certain respects I have adopted the evidence of a particular expert over another expert. In some instances, for instance, a witness would give positive and thoughtful answers, consistent with their own and other testimony, whilst on other matters the same witness would be uncertain or inconsistent. In some instances, an element of hindsight intruded, which affected the relative weight to be given to that witness’s evidence.
39 Expert evidence was presented by way of concurrent evidence, involving joint reports and oral “hot tub” testimony. This approach to the receiving of expert evidence proved effective, efficient and time-saving. It focussed on the real scientific issues, and enabled the expert witnesses to express their views openly and without inhibition. Discussion between the experts themselves and cross-examination in most instances assisted in the process of developing the position of each expert, particularly in areas of disagreement or qualification. Whilst I accept that over the course of giving evidence (including the preparation undertaken therefore), an expert becomes more informed, I do place significant weight on any agreement reached in a joint expert report. After all, such agreement was reached after discussion amongst the experts, in circumstances where proper reflection could be had outside an unfamiliar forensic setting, and where there was little or no influence of a third party. Nevertheless, in a contest such as that found in this proceeding, the Court must rely heavily on the expert testimony, and adjudicate on any differences of expert opinion based upon a consideration of the evidence as a whole, including the oral testimony provided during the hearing.
40 In light of these observations, the preferable approach is to consider the evidence of each witness on a topic by topic basis to the extent necessary, and consider the impact of each expert witness’s testimony in that context.
THE PURITY PATENT
41 I turn first to the Purity Patent.
Title of the Purity Patent and Field of Invention
42 The specification of the Purity Patent is entitled “Optically pure salts of pyridinylmethyl sulfinyl-IH-benzimidazole compounds”.
43 The field of the invention is described at p 1, line 5 as:
The present invention is directed to new compounds with high optical purity, their use in medicine, a process for their preparation and their use in the manufacture of pharmaceutical preparation. The invention also relates to novel intermediates in the preparation of the compounds of the invention.
Background to the invention
44 The Purity Patent begins at p 1, lines 15 to 18 by explaining that omeprazole and its alkaline salts are effective gastric acid secretion inhibitors that are useful as antiulcer agents, and that those compounds exist as two optical isomers (enantiomers).
45 At lines 18 to 22, it is said that:
It is desirable to obtain compounds with improved pharmacokinetic and metabolic properties which will give an improved therapeutic profile such as a lower degree of interindividual variation. The present invention provides such compounds, which are novel salts of the (-)-enantiomer of omeprazole.
46 The pharmacokinetic properties of a drug explain what happens to it once it is introduced into the body, generally according to three criteria: absorption, distribution and elimination. Elimination is the removal of a drug from the body via metabolism and excretion.
47 As noted above, the specification in its current form says that “novel salts of the (-)-enantiomer of omeprazole” are compounds with improved pharmacokinetic and metabolic properties which will give an improved therapeutic profile such as a lower degree of interindividual variation.
48 However, in the specification as filed, the invention was said to be “novel salts of single enantiomers of omeprazole”, that is, both the (-)-enantiomer and the (+)-enantiomer of omeprazole. Both enantiomers were said to have the improved pharmacokinetic and metabolic properties referred to in the specification. The specification was amended on 21 June 2000 to refer to the (-)-enantiomer of omeprazole only. This may explain why a number of the Examples in the Purity Patent do not relate to the compounds of the invention, but instead relate to the opposite enantiomer.
49 The specification acknowledges that the separation of the enantiomers of omeprazole has previously been described on both an analytical and preparative scale: p 1, lines 24 to 26.
50 The first publication referred to in the specification – “J. Chromatography, 532 (1990) 305-19” – is Erlandsson et al, “Resolution of the enantiomers of omeprazole and some of its analogues by liquid chromatography on a trisphenylcarbamoylcellulose-based stationary phase: The effect of the enantiomers of omeprazole on gastric glands”, 532 (1990) Journal of Chromatography 305-319 (‘Erlandsson 1990’). One author of Erlandsson 1990, Mr Per Lindberg, is an inventor of the Purity Patent. The specification identifies Erlandsson 1990 as a document describing the separation of the enantiomers of omeprazole on an “analytical” scale. More will be said about the scale on which separation of enantiomers is carried out in due course.
51 The second publication referred to in the specification – “DE 4035455” – is German patent application number DE 40 35 455 A1 (‘DE 455’). DE 455 describes, in terms, “optically pure compounds... or their salts with bases” including the (-)-enantiomer of omeprazole.
52 At p 2, lines 6 to 7 of the Purity Patent specification, it is said:
The present invention in a further aspect provides a novel method for preparing the novel compounds of the invention in large scale.
53 It is not contended by any party that any of the claims relating to the method or process for preparing the compounds of the invention is infringed or invalid.
54 At p 2, lines 11 to 13 the specification states:
There is no example known in the prior art of any isolated or characterized salt of optically pure omeprazole, i.e. single enantiomers of omeprazole, neither of any isolated or characterized salt of any optically pure omeprazole analogue.
Description of invention
55 A detailed description of the invention commences at p 2 of the specification. In one aspect, the invention is said, at lines 17 to 18, to provide Na+, Mg2+, Li+, K+, Ca2+ and N+(R)4 salts of the (-)-enantiomer of omeprazole, where R is an alkyl with 1-4 carbon atoms. The chemical name for the (-)-enantiomer of omeprazole is “(-)-5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazole”.
56 The most preferred salts according to the invention are said to be the optically pure Na+ salt of the (-)-enantiomer of omeprazole and the optically pure Mg2+ salt of the (-)-enantiomer of omeprazole: see p 3, lines 1 to 6.
57 Page 3, line 7 to p 4, line 8, describes a further aspect of the invention, being “a process for the preparation of compounds of the invention, ie, the salt of the (-)-enantiomer of omeprazole”. That process involves the separation of a diastereomeric mixture, followed by dissolving the separated diastereomer (comprising the (-)-enantiomer of omeprazole) in alkaline solution to give the “optically pure” (-)-enantiomer of omeprazole, which is then converted to the intended salt. That separation of diastereomeric mixture is exemplified in Examples 7 and 9. That dissolving of the separated diastereomer comprising the (-)-enantiomer of omeprazole in alkaline solution to give the (-)-enantiomer of omeprazole is exemplified in Example 10. The product of Example 10 has an enantiomeric excess of 94%.
58 At p 4, lines 10 to 21, it is said that in a further aspect the invention provides a process for the preparation of the (+)-enantiomer of omeprazole or the (-)-enantiomer of omeprazole, “to give the optically pure compound after neutralization with a neutralizing agent which can be an acid or an ester”. That step is described in Example 10 (as above) for the (-)-enantiomer of omeprazole and Example 11 for the (+)-enantiomer of omeprazole.
59 At p 4, lines 23 to 27, it is stated:
Thus, as indicated below, the direction of optical rotation of the enantiomers of omeprazole will change from (-) to (+) optical rotation and vice versa, from (+) to (-) optical rotation, when preparing the sodium salt from the neutral form (non-salt form) of omeprazole and when preparing the magnesium salt from the sodium salt of omeprazole.
60 The “(-)” rotation in the claims describes the rotation of the neutral (non-salt) form of the enantiomer of omeprazole. The invention is a salt of the neutral compound which has that (-) (namely, counter-clockwise) rotation, notwithstanding that the salt form may actually have a different rotation. For example, Example 1 describes the preparation of “(+)-5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazole sodium salt”. That compound is the sodium salt of the (-)-enantiomer of omeprazole.
61 At p 5, lines 1 to 11, the specification states:
With the expression “optically pure Na+ salts of omeprazole” is meant the Na-salt of the (-)-enantiomer of omeprazole essentially free of the Na-salt of (+)-enantiomer of omeprazole. Single enantiomers of omeprazole have hitherto only been obtained as syrups and not as crystalline products. By means of the novel specific method according to one aspect of the invention of preparing the single enantiomers of omeprazole, the salts defined by the present invention are easy to obtain. In addition, the salts, however not the neutral forms, are obtained as crystalline products. Because it is possible to purify optically impure salts of the enantiomers of omeprazole by crystallisation, they can be obtained in very high optical purity, namely ≥ 99.8% enantiomeric excess (e.e.) even from an optically contaminated preparation.
62 I interpolate to observe that the optical purity (or enantiomeric purity) of a mixture describes the relative proportion of each enantiomer present in the mixture. Optical purity can be expressed as enantiomeric excess (e.e.). Enantiomeric excess is the difference between the percentage amounts of two enantiomers in a mixture. For example, a mixture with an optical purity of 95% of the (S)-enantiomer and 5% of the (R)-enantiomer has a 90% enantiomeric excess (ie 90% e.e.).
63 The specification continues, at p 5, lines 11 to 16:
Moreover, the optically pure salts are stable towards racemization both in neutral pH and basic pH, which was surprising since the known deprotonation at the carbon atom between the pyridine ring and the chiral sulphur atom was expected to cause racemization under alkaline conditions. This high stability towards racemization makes it possible to use a salt of the (-)-enantiomer of omeprazole in therapy.
64 It is to be recalled that racemisation is a process where the enantiomers in an enantiomerically-enriched compound interconvert, creating a racemate (a 1:1 mixture of the two enantiomers) and thereby resulting in a loss of optical activity.
65 At p 5, line 21 to p 6, line 7, the specification states that the compounds according to the invention may (like omeprazole) be used for, amongst other things, inhibiting gastric acid secretion in mammals and man and the treatment of gastric acid-related diseases, gastrointestinal inflammatory diseases and other gastrointestinal disorders. One of those disorders is gastro-oesophageal reflux disease, sometimes referred to as ‘GORD’.
66 Page 6, lines 9 to 20 identifies an intermediate compound, said to be a further aspect of the invention, used in the method of preparation.
67 A process for the preparation of “optically pure compounds of the invention, ie the salts of the (-)-enantiomer” is then described from p 6, line 24.
68 This process first involves isolation of the (-)-enantiomer by resolution of the diastereomeric mixture. Examples 7 and 10 exemplify the process.
69 The specification goes on, at p 7, line 29 to p 8 line 6, to describe the preparation of the “optically pure Na+ salts of the invention, ie the Na+ salt of the (-)-enantiomer of omeprazole”. This is done, for example, by treating the (-)-enantiomer “with a base, such as NaOH, [being sodium hydroxide] in an aqueous or nonaqueous medium”. It is said that in order to obtain the crystalline form of the Na+ salt, “addition of NaOH in a non-aqueous medium such as a mixture of 2-butanone and toluene, is preferred”. That preparation is exemplified in Example 1.
70 The specification then describes (p 8, lines 8 to 16) the preparation of the Mg2+ (magnesium) salt of the (-)-enantiomer.
71 The compounds of the invention include “salts with Li+, K+, Ca2+ and N+(R)4, where R is an alkyl with 1-4 C-atoms”. The salts referred to are lithium, potassium, calcium and tetra alkyl ammonium salts: see p 8, lines 18 to 21, and claim 1. The preparation of those salts is not exemplified in the Purity Patent.
72 At p 8, lines 23 to 25, it is said that optically pure compounds of the invention are formulated into pharmaceutical formulations for oral, rectal, parenteral or other modes of administration. Such pharmaceutical formulations are described at p 8, line 25 to p 10, line 24.
73 At p 10, line 30, the specification states:
The invention is illustrated by the following examples, which describe processes in which optically pure salts of omeprazole are prepared.
74 In fact, only Examples 1, 5 and A exemplify the preparation of an “optically pure salt” of the (-)-enantiomer of omeprazole. Examples 2, 3 and 4 relate to preparations of salts of the (+)-enantiomer. Examples 6 and 8 describe the preparation of synthetic intermediates (diastereomers) used in the preparation of the compounds of the invention. As noted above, Examples 7 and 9 describe the chromatographic separation of the diastereomers. Example 10 describes the preparation, from one of those diastereomers, of the (-)-enantiomer of omeprazole as a colourless syrup, with an enantiomeric excess of 94%. Example 11 describes the preparation, from one of those diastereomers, of the (+)-enantiomer of omeprazole as a colourless syrup, with an enantiomeric excess of 98%.
75 The best mode of carrying out the invention is said to be the use of the compounds described in Example 1 and 2, being “the sodium salts of the optically pure compounds of the invention”: p 20, lines 15 to 17 (although it is noted that the compound prepared in accordance with Example 2 is not a compound of the invention – it is the opposite (namely, (+)) enantiomer of omeprazole).
76 The pharmaceutical formulations “containing the compounds of the invention as active ingredient” are illustrated by the formulations described at p 20, line 21 to p 24, line 4, which in each case involve an active ingredient “according to the invention”: p 20, line 26; p 21, lines 15 and 30; p 22, lines 18 and 30; and p 23, line 29.
77 The stability of “optically pure compounds of the invention towards racemization” is described by reference to two racemisation experiments at p 24, lines 6 to 24. In the first, the optically pure compound of the invention is the (-)-enantiomer of 5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl]sulfinyl]-1H-benzimidazole. Example 10 describes the preparation of that compound. The second refers to optically pure “(+)-isomer of 5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl]sulfinyl]-1H-benzimidazole sodium salt”, which is the sodium salt of the (-)-enantiomer of omeprazole. Example 1 describes the preparation of that compound.
78 On 13 January 1997, AstraZeneca lodged amendments to the specification to include the following passage, which now appears at p 24, line 24 and p 25:
Some evidence of the improved pharmacokinetic and metabolic properties of the optically pure salts of pyridinylmethyl sulfinyl-1H-benzimidazole compounds according to the invention, is presented in the data set out in the accompanying drawings, which shows the advantageous effects for a single enantiomer of omeprazole, a well known pyridinylmethyl-sulfinyl-1H-benzimidazole, i.e. the sodium salt of (-)-omeprazole (identified as neutral (-)-omeprazole), over the sodium salt of the racemic omeprazole (EPO 124 495), in being the closest prior art herein.
79 The accompanying drawings, Fig 1 and Fig 2, are at p 1/1 of the specification.
80 Figure 1 is described, at p 25, lines 6 to 15, as showing the mean plasma levels of omeprazole (the racemate) in “rapid metabolisers” (also known as ‘fast’ or ‘extensive’ metabolisers) compared to the mean plasma levels of the (-)-enantiomer of omeprazole in rapid metabolisers. At p 25 lines 9 to 11, it is said that in rapid metabolisers, the “mean AUC at steady state of (-)-omeprazole was almost 90% higher than that of omeprazole”.
81 The “AUC” or ‘area under curve’ is the area bound by the curve created by plotting plasma concentration of a drug against time (this is referred to as a “plasma concentration time profile”). The AUC represents the total amount of the drug that a patient is exposed to. The AUC for omeprazole (the racemate) is smaller in Fig 1 than the AUC for the (-)-enantiomer of omeprazole. This means that when the (-)-enantiomer of omeprazole is administered (as opposed to the racemate of omeprazole), the patient is exposed to more of the drug.
82 Figure 2 is described, at p 25, lines 17 to 21, as showing the mean plasma levels of omeprazole (the racemate) in slow metabolisers compared to the mean plasma levels of the (-)-enantiomer of omeprazole in slow metabolisers.
83 The specification then states, at p 25, lines 21 to 24:
Thus, after correction for different dose levels, the resulting difference in AUC between slow and rapid metabolisers was almost 9-fold for racemic omeprazole and only 3-fold for (-)-omeprazole.
84 That is, a comparison of the AUC in Figs 1 and 2 for both omeprazole (racemate) and the (-)-enantiomer of omeprazole in both slow and rapid metabolisers shows that there was less variability in the metabolism of the (-)-enantiomer of omeprazole between slow and rapid metabolisers (ie less ‘interindividual variability’).
85 The specification then says, at lines 25 to 28:
Thus, the present invention provides salts of the single enantiomers of specific pyridinylmethyl sulfinyl-1H-benzimidazole compounds, having improved antisecretory effect suitable for use in the treatment of gastric acid related diseases.
86 As already noted, although the specification refers to the “single enantiomers”, the invention as now claimed relates only to the (-)-enantiomer of omeprazole.
The invention claimed in the Purity Patent
87 The specification concludes at p 26 with 37 claims. Relevantly, for the purpose of infringement and validity:
(a) Claim 1 is an independent claim for an optically pure salt of the (-)-enantiomer of omeprazole, “said compound having improved pharmacokinetic and metabolic properties with respect to antisecretory effect in the treatment of gastric acid related diseases” (emphasis added). This compound is characterised in being selected from Na+, Mg2+, Li+, K+, Ca2+ and N+(R)4 salts of the (-)-enantiomer of omeprazole, wherein R is an alkyl with 1-4 carbon atoms. The italicised words were added to claim 1 by the 1997 amendments.
(b) Claims 2 and 3 are dependent claims to particular alkaline salts.
(c) Claim 18 is a claim to a pharmaceutical preparation comprising an optically pure compound according to, inter alia, any of claims 1 to 3 as an active ingredient, in combination with a pharmaceutically acceptable carrier. Claim 19 and 20 are dependent claims thereon.
(d) Claim 21 is a claim to use of an optically pure compound according to, inter alia, any of claims 1 to 3, in formulating a pharmaceutical preparation suitable for inhibiting gastric acid secretion or for the treatment of gastrointestinal inflammatory diseases.
(e) Claims 22 to 24 are claims to a method of treatment for inhibition of gastric acid secretion, comprising administration of an effective amount of an optically pure compound according to, inter alia, any of claims 1 to 3 or an effective amount of a preparation of any one of claims 18 to 20.
(f) Claims 25 to 27 are claims to a method for the treatment of gastrointestinal inflammatory diseases comprising administration of an effective amount of an optically pure compound according to, inter alia, any of claims 1 to 3, or an effective amount of a preparation of any one of claims 18 to 20.
88 Ranbaxy has made certain admissions to the effect that claims 1 to 3 and 18 to 27 of the Purity Patent are infringed, subject to the Court’s determination of the validity of those claims. As will be explored further in due course, Ranbaxy seeks to revoke those claims on the grounds of lack of novelty, lack of inventive step and lack of manner of manufacture.
89 It is necessary, before going to these specific grounds, to deal with the issue of the skilled addressee and the proper construction of the Purity Patent.
The skilled team and the evidence
Relevant principles and their application to this case
90 The parties accepted that the relevant principles regarding the identity of the skilled addressee in the context of the construction inquiry are as set out in my decision in Britax Childcare Pty Ltd v Infa-Secure Pty Ltd (2012) 96 IPR 1; [2012] FCA 467 at 43 to 45, [238]-[250]; and more generally in Eli Lilly and Company Ltd v Apotex Pty Ltd [2013] FCA 214 at [167] and following.
91 The following recent statement from Jagot J in Apotex Pty Ltd v AstraZeneca AB (No 4) [2013] FCA 162 usefully summarises the position (at [94]):
… [I]t is important to understand that the skilled addressee is not a reference to a specific person but is a legal construct (Root Quality Pty Ltd v Root Control Technologies Pty Ltd (2000) 177 ALR 231; [2000] FCA 980 at [71]). The legal construct may not be a single person but may be a team of persons “whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those which are contained in the document to be construed” (General Tire & Rubber Co v Firestone Tyre & Rubber Co Ltd [1972] RPC 457 at 485) (General Tire). And as Watson and Ascent also said:
A patent specification is addressed to those likely to have a practical interest in the subject matter of the invention, and such persons are those with practical knowledge and experience of the kind of work in which the invention is intended to be used. The addressee comes to a reading of the specification with the common general knowledge of persons skilled in the relevant art, and they read it knowing that its purpose is to describe and demarcate an invention. The person skilled in the art is unimaginative without inventive capacity.
92 It is well-established that the hypothetical skilled addressee may be a team whose combined skills would normally be employed in that art in interpreting and carrying into effect instructions such as those which are contained in the document to be construed. It is accepted that the combined skills of a team would be required for the purposes of this proceeding.
93 The first step in identifying the membership of the skilled team is to identify the field of knowledge to which the invention relates.
94 In this proceeding, the relevant field is pharmaceuticals – specifically, those related to gastric acid-related diseases. The impugned claims of the Purity Patent are directed to certain identified optically pure salts of a single compound, and pharmaceutical preparations and treatment methods relating to those salts.
95 The skilled team for the Purity Patent is therefore a team of scientists working in the field of gastrointestinal disorders with a particular interest in the treatment of gastric acid-related diseases, involved in a research project to find a new pharmaceutical compound as at 28 May 1993, which (it will be recalled) is the priority date for the Purity Patent.
96 It seemed to be uncontroversial that the team would include:
(a) a medicinal chemist (ie an organic chemist);
(b) a pharmacologist; and
(c) a person familiar with the preparation of pharmaceutically acceptable salts.
97 Ranbaxy contended that the medicinal chemist would “normally be involved in the resolution of enantiomers and the testing of their properties”. Whether a medicinal chemist would “normally be involved” in such activity as at May 1993 is one of the many issues in contest in the proceeding. In AstraZeneca’s submission, the evidence does not support the proposition that a medicinal chemist on such a team would normally have had such involvement as at May 1993. In fact, it was submitted that the evidence shows that the standard approach to drug development was by the investigation of structure-activity relationships (described by Senior Counsel for Ranbaxy as the “analogue route”), and not by investigation of enantiomers. However, since the organic chemistry experts both have experience in the resolution and testing of enantiomers, this issue has no bearing on their capacity to give evidence as expert medicinal chemists, and their ability to assist on construction.
98 Ranbaxy also contended that the team would have included “a gastroenterologist (who would have clinical experience in the use of omeprazole)”. While AstraZeneca did not accept that the team would have included a gastroenterologist, they accepted that the team would have taken advice from gastroenterologists with clinical experience in the use of omeprazole from time to time. I think that very little rests on this distinction, although it may be significant insofar as it relates to “motivation” in the context of inventive step. I would include a gastroenterologist in the team. The extent of and timing of the contribution of this team member is a matter which may impact upon the team’s ultimate deliberations and its motivation to proceed in any particular direction. I will return to this issue later.
Construction
99 I now turn to the construction issues.
Summary of issues for the Court to determine
100 The key Purity Patent construction issue for determination is the meaning of the expression “optically pure salt of [(-)-omeprazole]” in claim 1. AstraZeneca contended that “optically pure” means greater than or equal to 98% enantiomeric excess (e.e.). That is to say, to be considered to be “optically pure” in this context, the preparations must contain at least 99% of the salt of the (-)-enantiomer of omeprazole, with no more than 1% of the (+)-enantiomer. In contrast, Ranbaxy contended that “optically pure” has its “ordinary meaning” – namely, it means greater than or equal to 90% e.e. (with the consequence that a salt of the (-)-enantiomer of omeprazole containing 5% of the (+) enantiomer would be “optically pure” for the purpose of this patent).
101 The issue is not relevant to infringement. Ranbaxy accepts that if the Purity Patent is held to be valid, then their products will infringe regardless of the outcome of this construction issue. No party contended that the claims lack clarity. Rather, the determination of the construction issue impacts only on the question of validity.
102 A further construction issue is the meaning to be given to the words “said compound having improved pharmacokinetic and metabolic properties with respect to antisecretory effect in the treatment of gastric acid related diseases” in claim 1.
Legal principles
103 The parties accepted the principles of construction reviewed and summarised in Eli Lilly [2013] FCA 214 at [139] to [144]. Nevertheless, certain points should be repeated, all of which are well settled.
104 The words of the patent should be read through the eyes of the skilled addressee in the context in which they appear. That is, the words are to be given the meaning which the person skilled in the art would attach to them, having regard to his or her own general knowledge and to what is disclosed in the body of the specification. The notional skilled addressee “is expected to read the specification on the assumption that its purpose is both to describe and to demarcate an invention”: see Dynamite Games Pty Ltd v Aruze Gaming Australia Pty Ltd [2013] FCA 163 at [192].
105 Of course, the Court has the ultimate responsibility for interpreting the patent: no one witness’s view however ‘skilled’ that witness may be, is to dictate the Court’s conclusion as to the proper interpretation of a patent.
106 In the case of terms with a scientific or industry meaning, evidence as to the meaning of such terms ought be received by the Court. If there is no such special meaning, then the Court can rely upon the ordinary meaning of the term or terms, albeit in the context of the science of the patent. A patent, of course, may provide its own dictionary of particular terms used in the patent and the claims.
107 Patents are to be given a purposive construction and the subject matter of a patent is not confined to the literal meaning of the claim.
108 Above all, the Court should approach the task of patent construction with a generous measure of common sense.
109 It is useful to set out the contentions of Ranbaxy on the first construction issue.
Ranbaxy’s submissions on construction
110 Ranbaxy contended that the Purity Patent does not describe the invention by reference to any specific level of optical purity. In particular, there was said to be “no indication in the Purity Patent that any level of optical purity is required to achieve the practical function of the invention”, namely, providing patients with a compound having improved pharmacokinetic and metabolic properties. As a result, it was submitted, “optically pure” should be given its ‘conventional meaning’.
111 Ranbaxy then contended that the accepted, conventional meaning of “optically pure” is 90% e.e. (which equates to 95% enantiomeric purity). In the abstract, Professor Davies and Dr Pyter were prepared to accept 90% e.e. as a ‘conventional’ meaning of “optically pure”. That level of purity was one which was said to permit statistically meaningful tests to be performed to determine the properties of enantiomers. In contrast, Dr Stevenson did not agree that there was any “conventional” meaning of this term at the priority date. However, despite these conclusions about what may constitute a conventional meaning of optically pure, the experts agreed that absent any indication of how optical purity is measured in a particular case, the expression has no meaning. I will return to this point later in these reasons for judgment.
112 The experts disagreed on the level of optical purity required by claim 1 of the Purity Patent. I consider there was a difference of approach: some of the experts seemed to proceed on the basis that “optically pure” must be defined with respect to a specific number in the patent specification. For example, Dr Stevenson said that the only number he could find for this purpose was 99.8%, which was found on p 5 at line 10. Professor Davies reviewed the Purity Patent and focused on the language at p 10 and the Examples, and ultimately adopted a figure of 98% e.e.. In contrast, Dr Pyter did not consider there to be a need to find a number in the patent specification, and interpreted claim 1 by reference to what he deemed to be the ‘conventional’ meaning of “optically pure”.
113 It was submitted by Ranbaxy that AstraZeneca’s construction of “optically pure”, namely, 98% e.e., should not be accepted because it represents the high or very high optical purity value that is shown to be achievable in examples of only two of the claimed salts. Although the expression “optically pure” is apt to include such a high e.e. value, Ranbaxy contended that it is used in the specification in such a way that makes it clear that the meaning is not limited to that high value.
114 To this end, it was contended by Ranbaxy that the specification distinguishes between “optically pure” compounds and compounds with “high optical purity” or “very high optical purity”: see, for example, p 1, line 5 and p 5, line 9 of the specification. The “very high” optical purity at p 5, line 9 is the product of crystallisation. The “optically pure” salts of claim 1 are not limited to salts in crystalline form. Rather, that limitation is introduced by dependent claims.
115 Ranbaxy then drew attention to the fact that the compounds of the invention are said to be “novel salts of the (-)-enantiomer of omeprazole”: at p 1, lines 20 to 22. Ranbaxy submitted that at p 2 lines 11 to 12, the specification refers to a:
salt of optically pure omeprazole, i.e. single enantiomers of omeprazole” (emphasis added) and at page 6, line 24 to “The optically pure compounds of the invention, i.e. the salts of the (-)-enantiomer…” (emphasis added)
116 It was argued that there was no suggestion here that the specification uses the term “optically pure” to mean high optical purity or very high optical purity, or in any way other than its ordinary meaning.
117 Ranbaxy then referred to the unamended specification, which at p 5 line 24 read: “The optically pure compounds of the invention, i.e. the single enantiomers…”. It was submitted that the expression “optically pure” had a broader meaning than the particular level of optical purity achieved in the examples of two of the salts of the single enantiomers set out elsewhere in the specification.
118 Ranbaxy further contended that the specification acknowledges that the separation of the enantiomers of omeprazole is described in the prior art, for example, in Erlandsson 1990 and DE 455. Erlandsson 1990 reports the separation of the (-)-enantiomer of omeprazole with an enantiomeric excess of 91.2%. DE 455 discloses, in terms, “optically pure compounds of Formula I… or their salts with bases”, including the (-)-enantiomer of omeprazole.
119 Ranbaxy argued that the specification distinguishes the invention in the Purity Patent by asserting, “[t]here is no example known in the prior art of any isolated or characterized salt of optically pure omeprazole…” (emphasis added): p 2, lines 11 to 14. It was then contended that this is not suggesting that the compounds obtained in Erlandsson 1990 (91.2% e.e.) and DE 455 (“optically pure”) were not optically pure in accordance with the way that term is used in the Purity Patent; just that those compounds were not in salt form.
120 Ranbaxy then went to the Examples. The invention is said to be “illustrated by” the Examples: see p 10, line 30. The Examples show a variety of measurements of optical purity ranging from 94% to 99.9% e.e.. As already alluded to, only three of those Examples (Examples 1, 5 and A) actually relate to the invention as claimed in claim 1 (namely, optically pure salts of the (-)-enantiomer of omeprazole).
121 Ranbaxy reminded the Court that it is not legitimate to read down the ordinary meaning of the term “optically pure” in the claims by drawing an impermissible gloss from particular Examples in the specification, or to use those Examples to introduce into the words of claim 1 a numerical qualification: Welch Perrin and Company v Worrel (1961) 106 CLR 588 at 610; Interlego AG v Toltoys Pty Ltd (1973) 130 CLR 461 at 478; Kimberly-Clark Australia Pty Limited v Arico Trading International Pty Limited (2001) 207 CLR 1 at 12 [15]; Pfizer Overseas Pharmaceuticals v Eli Lilly and Company (2005) 68 IPR 1; [2005] FCAFC 224 at 52 [247]; PAC Mining Pty Ltd v Esco Corp (2009) 80 IPR 1; [2009] FCAFC 18 at 14 [28]; and Britax Childcare (2012) 96 IPR 1; [2012] FCA 467 at 49 [272].
122 Ranbaxy then contended that the specification refers to “optically pure” compounds which are not the subject of any of the Examples in the Purity Patent. For example, “salts with Li+, K+, Ca2+ and N+(R)4 salts of the (-)-enantiomer of omeprazole, where R is an alkyl with 1-4 C-atoms” (p 8, lines 18 to 21 and claim 1), and the “optically pure Mg2+ salt... prepared by treating the (-)-enantiomer of omeprazole with a base… in a non-aqueous solvent”(at p 8, lines 11 to 13). There is no Example relating to the preparation of these compounds or, in the case of the “optically pure Mg2+ salt”, the preparation of this compound in this manner. These salts are, however, claimed in, inter alia, claims 1 and 15 as “optically pure” salts. It was then contended that there was no reason to suppose these salts of the invention have a particular optical purity greater than (as ordinarily understood) 90% e.e.. Therefore, Ranbaxy submitted, in the context of at least these salts, the skilled addressee can only sensibly assume that “optically pure” has its conventional meaning and is not to be limited to a particular numerical limit in a particular Example.
123 Ranbaxy further submitted that the pharmaceutical formulations referred to at pp 20 to 21 of the specification that contain “the compounds of the invention as active ingredient” are not fixed to the product of any Example, or otherwise limited to the (-)-enantiomer of omeprazole with a particular enantiomeric excess greater than “optically pure”.
124 Further, it was noted by Ranbaxy that the “evidence of the improved pharmacokinetic and metabolic properties of the optically pure salts of pyridinylmethyl sulfinyl-1H-benzimidazole compounds according to the invention” is not said to be limited to the salts of any particular (or indeed any) Example: see p 24.
125 Reference was also made by Ranbaxy to Example 10. It was submitted that “optically pure” is used to describe the product of Example 10, which has an enantiomeric excess of 94%. That description was contended to be consistent with the ordinary meaning of “optically pure”. The following examples in support were given (emphases as per Ranbaxy’s submissions):
(a) The process at p 4, lines 6 to 7 for the preparation of “optically pure (-)-5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazole compound” (the (-)-enantiomer) is exemplified in Example 10.
(b) The process at p 4, lines 19 to 20 for the preparation of the (+)-enantiomer of omeprazole or the (-)-enantiomer of omeprazole to give the “optically pure compound after neutralization with a neutralizing agent” is also exemplified in Example 10 (where that compound is the (-)-enantiomer of omeprazole).
(c) The stability of “optically pure compounds of the invention towards racemisation” is described by reference to an experiment with the (-)-isomer of 5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl]sulfinyl]-1H-benzimidazole. Example 10 describes the preparation of that compound.
(d) Claim 6 claims a process for the preparation of an optically pure compound according to claim 1, characterised in that a diastereomeric mixture of an ester is separated to obtain separated diastereomers, whereafter one of the diastereomers is dissolved in an alkaline solution to give “the optically pure (-)-5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl] sulfinyl]-1H-benzimidazole compound”. This is the product of Example 10.
(e) Claim 28 claims a process for the preparation of the (+)-enantiomer of omeprazole or the (-)-enantiomer of omeprazole to give the “optically pure compound after neutralization with a neutralizing agent”. That process, as it relates to the (-)-enantiomer of omeprazole, is exemplified in Example 10.
(f) Claim 35 claims as the invention the compound obtained by, inter alia, the process exemplified in claim 28, and thus claims the product of Example 10.
Construction – Analysis
First Issue: “Optically Pure”
126 As previously foreshadowed, Professor Davies, Dr Stevenson and Dr Pyter agreed that “optically pure” will normally take its meaning from the context in which it is used, including, in part, the nature and extent of identified measuring standards used to ascertain a particular level of optical purity.
127 Therefore, the Court, guided by evidence concerning the approach of the skilled addressee, will look for the context in which the term “optically pure” is used in the Purity Patent.
128 In my view, the Purity Patent has provided its own context or “internal dictionary” for the expression “optically pure”. At p 5, lines 1 to 11, it states:
With the expression “optically pure Na+ salts of omeprazole” is meant the Na-salt of the (-)-enantiomer of omeprazole essentially free of the Na-salt of (+)-enantiomer of omeprazole. Single enantiomers of omeprazole have hitherto only been obtained as syrups and not as crystalline products. By means of the novel specific method according to one aspect of the invention of preparing the single enantiomers of omeprazole, the salts defined by the present invention are easy to obtain. In addition, the salts, however not the neutral forms, are obtained as crystalline products. Because it is possible to purify optically impure salts of the enantiomers of omeprazole by crystallisation, they can be obtained in very high optical purity, namely ≥99.8% enantiomeric excess (e.e.) even from an optically contaminated preparation. Moreover, the optically pure salts are stable towards racemization both in neutral pH and basic pH, which was surprising since the known deprotonation at the carbon atom between the pyridine ring and the chiral sulphur atom was expected to cause racemization under alkaline conditions. This high stability towards racemization makes it possible to use a salt of the (-)-enantiomer of omeprazole in therapy.
129 I accept this as the “internal dictionary”, and I further accept that the passage draws a distinction between three levels of optical purity: first, “optically pure salts”, which are “essentially free” of the (+)-enantiomer; secondly, salts of a “very high optical purity”, namely ≥ 99.8% e.e.; and thirdly, an “optically contaminated” preparation. On this basis, and for the reasons that follow, I consider that the construction put forward by AstraZeneca is to be preferred – namely, that “optically pure” means ≥ 98% e.e..
130 The Examples in the Purity Patent are introduced by the words: “The invention is illustrated by the following examples, which describe processes in which optically pure salts of omeprazole are prepared” (p 10, lines 30 to 32; emphasis added). The only “salts of omeprazole” identified in the subsequent examples are those described in Examples 1 to 5, A and B. It follows that each of those salts of omeprazole may be characterised as “an optically pure salt”.
131 Further, each of the optically pure salts exemplified in Examples 1 to 5 and A and B has an enantiomeric excess of at least 98%. I find this fact to be very significant in determining this construction point.
132 Example 1 (p 11, line 7) refers to a preparation “contaminated with 3% of the (+)-isomer”, and Example 2 (p 11, line 20) refers to a preparation “contaminated with 3% of the (-)-isomer”. From each of those preparations, a sodium salt of an enantiomer of omeprazole with an optical purity greater than or equal to 99.8% e.e. is derived. A preparation “contaminated with 3%” of the other enantiomer has an enantiomeric excess of 94% (calculated by deducting 3% from 97%).
133 The preparations “contaminated with 3%” of the other enantiomer identified in Examples 1 and 2 are the preparations to which reference is made by the expression “an optically contaminated preparation” appearing on p 5, lines 10 to 11 of the Purity Patent. The use of this expression is in contradistinction to the “optically pure” salts of the invention also referred to in that passage on p 5.
134 Accordingly, the Purity Patent draws a distinction between:
(a) an “optically pure salt” of one enantiomer of omeprazole, being a salt (specifically a sodium salt, in the context of p 5 of the specification) which is “essentially free” of the other enantiomer; and
(b) a salt of an enantiomer that is “contaminated with 3%” of the other enantiomer.
135 Further, the proposition that an “optically pure salt of (-)-[omeprazole]” as referred to in claim 1 means a salt of 98% e.e. or more is supported by the following:
(a) the distinction drawn on p 5 between an “optically pure salt” that is “essentially free” of the (+)-enantiomer and a salt of “very high optical purity” (namely, ≥ 99.8% e.e.); and
(b) the inclusion, in the descriptive expression “[t]he invention is illustrated by the following examples, which describe processes in which optically pure salts of omeprazole are prepared” at p 10 at lines 30 to 31, of salts in the Examples with an optical purity of between 98% e.e. and 99.8% e.e..
136 None of the Examples expressly describes any salt of any enantiomer of omeprazole with an optical purity lower than 98% e.e. as being “optically pure”. Nor does any other part of the Purity Patent.
137 Even though only Examples 1, 5 and A exemplify the preparation of an optically pure salt of (-)-omeprazole, I accept that all of Examples 1 to 5, A and B are relevant to construing the meaning of the phrase “optically pure salt” in claim 1.
138 The “optically pure salts” of Examples 1 to 5, A and B are clearly differentiated from the compounds of Examples 6 to 11. First, Examples 6 to 11 are not salts. Secondly, Examples 6 to 11 are introduced by the words “[p]reparation of the synthetic intermediates according to the invention will be described in the following examples” (at p 15, lines 31 to 32). These are non-salt intermediate compounds, and I accept that their optical purities, to the extent they are reported, cannot be determinative or informative of the meaning of “optically pure salt” in the claims.
139 Thus, for the foregoing reasons, in the context of the Purity Patent, I find that the expression “optically pure” means at least 98% e.e..
140 The above approach leads to the conclusion I have just reached. However, it is appropriate to address a number of the specific arguments made by Ranbaxy in support of their contended construction of the meaning of “optically pure” as it appears in the Purity Patent – namely, that it means ≥ 90% e.e.
141 Ranbaxy relied on a statement in the specification that there is “no example known in the prior art of any isolated or characterized salt of optically pure omeprazole” (p 2, lines 11 to 12). This statement distinguishes the optically pure salts of either enantiomer of omeprazole as referred to in the Purity Patent (subject to what I have said already about the invention claimed being limited to the (-)-enantiomer) from the compounds of the prior art, including DE 455 and Erlandsson 1990. Ranbaxy emphasised the reference to a “salt” in this statement, and suggested that the non-salt material of DE 455 and Erlandsson 1990 is “optically pure” within the meaning used in the Purity Patent. I agree with AstraZeneca that this suggestion must be rejected for the following reasons:
(a) The specification of the Purity Patent does not describe the compounds of DE 455 or Erlandsson 1990 as “optically pure”. The statement in the Purity Patent relied upon by Ranbaxy is a composite expression, indicating that the levels of optical purity that are the subject of the Purity Patent are higher than anything previously identified, including in DE 455 and Erlandsson 1990.
(b) Even if it were permissible to pay regard to Erlandsson 1990 (which is not incorporated merely by the reference made to it in the Purity Patent), an inspection of that publication reveals (+)-omeprazole with an optical purity of 64% e.e., and (-)-omeprazole with an optical purity of 91.2% e.e.. The optical purity recorded for the (+)-enantiomer (64% e.e.) does not meet Ranbaxy’s proposed definition of at least 90% e.e..
(c) Even if it were permissible to pay regard to DE 455 (which is not incorporated merely by the reference made to it in the Purity Patent), an inspection of DE 455 shows that the (+)-omeprazole exemplified was an “amorphous solid”, and that the products of the DE 455 method are not “optically pure” in the same sense as that term is used in the Purity Patent. The experts agreed that the failure of DE 455 to identify any means of measurement of optical purity renders the expression “optically pure” as used in that publication devoid of relevant meaning to a skilled team.
142 Ranbaxy also argued that “optically pure” was used to describe the product of Example 10, which is (-)-omeprazole in neutral (non-salt) form, and is reported as having an enantiomeric excess of 94%. In opposing the construction of “optically pure” contended for by AstraZeneca (namely, 98% e.e.), Ranbaxy contended that an enantiomeric excess of 94% as reported in that example “is consistent with the ordinary meaning of optically pure”.
143 As I have said, the product of Example 10 is not described or characterised either as a salt or as “optically pure”. Rather, Examples 6 to 10 are collectively characterised as “synthetic intermediates”. They are non-salt intermediates and their optical purities, to the extent they are reported, cannot inform or be determinative of the meaning of “optically pure” in the claims.
144 It will be recalled that Ranbaxy also relied on the use of the phrase “optically pure” in certain parts of the Purity Patent. In addition to what I have said, including on the topic of Example 10, I address these in turn, adopting the submissions of AstraZeneca:
(a) Page 4, lines 6 to 7 and lines 19 to 20.
At lines 6 to 7, there is a reference to optically pure (-)-omeprazole in neutral form. At lines 19 to 20, there is a reference to optically pure (-)-omeprazole or (+)-omeprazole in neutral form. Ranbaxy’s submission relies on the reference to (-)-omeprazole in neutral form “as exemplified in Example 10”, which is said to have an e.e. of 94%, and which is said to be consistent with the conventional meaning contended for by Ranbaxy. However, I accept that this submission overlooks the reference to (+)-omeprazole in neutral form, which is the product of Example 11 and is reported as having an optical purity of 98% e.e.. The optical purity of 94% e.e. for (-)-omeprazole in neutral form as reported in Example 10 does not detract from the meaning provided by the rest of the document for “optically pure salt”.
(b) The reported stability towards racemisation of (-)-omeprazole in neutral form on p 24, lines 6 to 18.
These lines report the results of a racemisation experiment. The Purity Patent makes no express connection between this experiment and the product of Example 10. Accordingly, there is no reason for the skilled addressee to assume that the (-)-omeprazole tested in this experiment has the optical purity reported in Example 10.
(c) Claim 6, which claims a “[p]rocess for the preparation of an optically pure compound according to claim 1” and refers to the preparation of “optically pure” (-)-omeprazole in neutral form (p 27, lines 6 to 7).
Ranbaxy relies on the fact that the product referred to in claim 6 is the product of Example 10, which is expressed to have an optical purity of 94% e.e.. However, I note that claim 6 expressly claims a “[p]rocess for the preparation of an optically pure compound according to claim 1” (emphasis added). Accordingly, it cannot displace the meaning provided by the rest of the document for the definition of “optically pure salt” in claim 1.
(d) Claims 28 and 35, which claim a process for the preparation of (+)-omeprazole or (-)-omeprazole (each of which is described as an “optically pure compound”), and the product of that process.
Examples 10 and 11 are both relevant to interpreting these claims. However, I do not find that the optical purity of 94% e.e. reported in Example 10 for (-)-omeprazole in neutral form detracts from the meaning provided by the rest of the document for the definition of “optically pure salt”.
145 Finally, in respect of Ranbaxy’s argument relating to the absence of any examples illustrating the Li+, K+, Ca2+ and N+(R)4 salts of (-)-omeprazole claimed in claims 1 to 5 of the Purity Patent as being “optically pure” salts (and their contention that “[t]here is no reason to suppose these salts of the invention have a particular optical purity greater than (as ordinarily understood) 90% e.e.”), I note that equally, there is no suggestion in the Purity Patent – and no reason to suppose – that these salts have an optical purity less than the exemplified sodium and magnesium salts.
146 Therefore, none of contentions raised by Ranbaxy detracts from the ability to rely upon the “internal dictionary” approach I have taken, and my conclusion about the meaning of “optically pure” as used in the Purity Patent.
Expert Evidence on the Meaning of “Optically Pure”
147 Before leaving this topic, I note that there was a great deal of expert evidence on the meaning of “optically pure”. I think, in this context, there is much to be said in support of the comment made by Senior Counsel for Ranbaxy of the limits of the utility of asking experts about the specific meaning of terms in a patent.
148 Professor Davies had a consistent approach, maintaining throughout the proceeding that “optically pure” in the context of the Purity Patent meant ≥ 98% e.e.. I consider his evidence on this point to be persuasive, although it is always a matter for the Court to interpret a patent. Dr Stevenson and Dr Pyter did not seem to consider the context of the use of this expression in the Purity Patent, and clearly altered their positions during the course of the proceeding.
149 The steps I have taken to determine the meaning of “optically pure” have involved applying my understanding of the science to the terms of the Purity Patent. Even if a conventional meaning of “optically pure” exists (which the evidence suggests there does not, or at least, did not at 28 May 1993), the context of the Purity Patent shows that any such standard or conventional meaning was not adopted by the drafters of that patent, and therefore cannot be determinative of the construction of that term in context.
ADMISSIBILITY OF PATENT OFFICE FILE
150 Ranbaxy also sought to tender documents relating to the prosecution histories of the Purity Patent, the MUPS Patent and the 774 Patent. In the case of the Purity Patent, they sought to do so on the basis that they are relevant to construction, in particular to the meaning to be assigned to “optically pure”.
151 While s 116 of the Act permits the Court, in construing an amended patent, to refer to the specification without amendment, the section does not permit recourse to other documents on the Patent Office file.
152 In Prestige Group (Australia) Pty Ltd v Dart Industries Inc (1990) 26 FCR 197, when considering whether the United States doctrine of “file wrapper estoppel” could have any application in Australia, Gummow J said (at 211; citations omitted):
The patent specification is much more than a document setting down a commercial bargain. It is not “a written instrument operating inter partes, but a public instrument”: Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610. Provision for it is made in legislation permitting a special monopoly, and the English predecessor to the present standard patent, the issue of which is provided for in Pt VI of the Act, was Letters Patent issued by the Crown. The granting of the modern standard patent is an activity not contractual but quasi-legislative in character. Hence, one should have thought that the restrictions which apply to the admission of negotiations prior to contract as aids to construction … would apply a fortiori to dealings and procedures preceding the grant of patent protection. Learned Hand J was well aware of the difficulty, saying in Lyon v Boh, above at 50–1:
‘Ordinarily the final writing, which incorporates a solemn agreement, is taken by the courts as the parties want it taken; that is, it is the sole resort for ascertaining their intentions, for the excellent reason that the parties meant it to be such. All prior negotiations are disregarded, since otherwise the chief purpose of reducing the contract to writing would be frustrated. However, there is the exception, well settled and very recently confirmed by the Supreme Court [Weber Electric Co v Freeman Electric Co 256 US 668], that, if a patentee submits to the rejection of a claim while his application is in the Patent Office, he may not later insist that other claims which he does get are to be regarded as equivalent to that which was rejected.’
153 In Kirin-Amgen Inc v Hoechst Marion Roussel Ltd [2005] 1 All ER 667, Lord Hoffmann said, with the approval of the other members of the House of Lords (at 681-682 [35]):
The courts of the United Kingdom, the Netherlands and Germany certainly discourage, if they do not actually prohibit, use of the patent office file in aid of construction. There are good reasons: the meaning of the patent should not change according to whether or not the person skilled in the art has access to the file and in any case life is too short for the limited assistance which it can provide. It is however frequently impossible to know without access, not merely to the file but to the private thoughts of the patentee and his advisors as well, what the reason was for some apparently inexplicable limitation in the extent of the monopoly claimed. One possible explanation is that it does not represent what the patentee really meant to say. But another is that he did mean it, for reasons of his own; such as wanting to avoid arguments with the examiners over enablement or prior art and have his patent granted as soon as possible. This feature of the practical life of a patent agent reduces the scope for a conclusion that the patentee could not have meant what the words appear to be saying. It has been suggested that in the absence of any explanation for a restriction in the extent of protection claimed, it should be presumed that there was some good reason between the patentee and the patent office. I do not think that it is sensible to have presumptions about what people must be taken to have meant but a conclusion that they have departed from conventional usage obviously needs some rational basis.
154 These two cases were applied by Tamberlin J in Baygol Pty Ltd v Foamex Polystyrene Pty Ltd (2005) 64 IPR 437; [2005] FCA 145 in refusing to admit into evidence the relevant file of the Patent Office in that case. His Honour considered there was “great force” in Gummow J’s conclusion in Prestige (1990) 26 FCR 197 that “the restrictions which apply to the admission of negotiations as aids to the construction of contracts apply a fortiori to dealings and procedures preceding the grant of a patent protection”.
155 The same reasoning applies in this case.
156 The Patent Office’s file cannot assist in the construction of the patent. The file, apart from the unamended version of each patent, is irrelevant to that inquiry and to the other inquiries the subject of this proceeding.
Second Issue: Meaning of “Improved Pharmacokinetic and Metabolic Properties With Respect to Antisecretory Effect in the Treatment of Gastric Acid Related Diseases”
157 I now turn to the second construction issue. I do not consider that there is much difference between the parties on this issue, nor that any difference impacts upon the determination of this proceeding.
158 Claim 1 refers to an optically pure salt of (-)-omeprazole, “said compound having improved pharmacokinetic and metabolic properties with respect to antisecretory effect in the treatment of gastric acid related diseases”.
159 It is common ground that the point of comparison in this phrase is racemic omeprazole, and that the improved anti-secretory effect means increased anti-secretory effect at the same dose for extensive metabolisers, caused by an increase in AUC (that is, by altered pharmacokinetics).
160 Claim 1 was amended in 1997 to include this phrase. As I have said, data shows a greater AUC for rapid metabolisers when treated with the sodium salt of the (-)-enantiomer of omeprazole as compared to the sodium salt of racemic omeprazole. It also shows less interindividual variability in AUC between slow and rapid metabolisers when treated with the sodium salt of the (-)-enantiomer of omeprazole as compared to the sodium salt of omeprazole.
161 The specification including the claims does not suggest that a selection is made of certain salts of omeprazole which have the improved pharmacokinetic and metabolic properties from a number of optically pure salts of omeprazole. Rather, the salts of claim 1 are the only salts referred to in the specification, and the improved properties are said to apply to each of them. Accordingly, the evidence of improved pharmacokinetic and metabolic properties of the sodium salt of (-)-omeprazole over the sodium salt of omeprazole can be extrapolated to the enantiomers of all the claimed salts.
162 The issue of construction that is said to arise is whether the expression “improved pharmacokinetic and metabolic properties” operates to limit the invention claimed. Ranbaxy contended that it does not.
163 In my view, the expression merely describes a property claimed to be inherent in all of the claimed compounds of the invention. As Ranbaxy submitted, the claim could appropriately be re-written with that description in parentheses, as follows: “Optically pure salts of the (-)-enantiomer of omeprazole (which have improved pharmacokinetic and metabolic properties over omeprazole with respect to antisecretory effect in the treatment of gastric acid related diseases)…”.
164 This is consistent with the submissions of AstraZeneca to the effect that these properties are part of the inventive concept.
Novelty
165 Ranbaxy pleads that claims 1 to 3 and 18 to 27 of the Purity Patent lack novelty in light of the following prior art documents:
(a) the document referred to in these reasons for judgment as “DE 455”, being German patent application number DE 35 455 A1, published 14 May 1992; and
(b) International Patent Application PCT/EP1991/02096, which became Australian patent application number 88406/91 (‘AU 406’), published 29 May 1992.
166 Each document was filed in the name of Byk Gulden Lomberg Chemische Fabrik GmbH (‘Byk Gulden’), a company with no connection to AstraZeneca. AU 406 is a patent application in the same patent family as DE 455. The information contained in AU 406 generally corresponds to the information contained in DE 455. In the following analysis I will therefore refer only to DE 455, which effectively also deals with AU 406.
Legal principles
167 The ground of novelty arises under ss 18(1)(b)(i) and 138(3)(b) of the Act, and the question of novelty is to be assessed in accordance with the provisions of s 7(1). Under s 7(1) as it stood prior to amendment in 2002, a claimed invention is novel unless it is shown to be “not novel in the light of… information… made publicly available” in any prior disclosure. The principles to be applied are well-established.
168 The terms in the alleged anticipatory document “should be read through the eyes of the skilled addressee in the context in which they appear”. That is, the terms are to be given “the meaning which the person skilled in the art would attach to them, having regard to his or her own general knowledge and to what is disclosed in the body of the specification”: H Lundbeck A/S v Alphapharm Pty Ltd (2009) 177 FCR 151 at 179 [118] per Bennett J, with whom Middleton J agreed.
169 Thus, in the case of terms with a scientific meaning or an industry meaning used in the alleged anticipatory document, evidence as to the meaning of such terms ought to be received by the Court. I accept that the expression “optically pure” is such a term, although it may be a term specifically defined in a particular context as I have found in the Purity Patent itself.
170 The cases emphasise that for anticipation to be made out, a clear and precise disclosure of the subject matter of the claim is required. A prior publication will only anticipate if it discloses all of the essential integers of the claimed invention. It must contain “clear and unmistakable directions” to carry out the invention in order to render it not novel: see General Tire & Rubber Company v Firestone Tyre and Rubber Company Ltd (1971) 1A IPR 121 at 137-138. Thus, it has been said that “[a]nticipation is deadly but requires the accuracy of a sniper, not the firing of a 12 gauge shotgun”: Apotex Pty Ltd v Sanofi-Aventis (2008) 78 IPR 485; [2008] FCA 1194 at 524 [91], approved in H Lundbeck (2009) 177 FCR 151 at 189 [170]. Further, it is not sufficient for a prior publication to merely “include” or “encompass” the claimed invention – a broad disclosure will not necessarily anticipate a later, more specific claim: see eg Eli Lilly [2013] FCA 214 at [272] to [293] and the authorities cited therein.
171 In H Lundbeck (2009) 177 FCR 151, Bennett J (with whom Middleton J agreed) stated at 192 [180]:
Where the prior publication discloses exactly what is claimed, there is anticipation. This can be objectively determined and, apart from an understanding of terms of art, the evidence of the skilled addressee is not likely to be of much further assistance.
172 These principles were affirmed by Bennett and Middleton JJ in Apotex Pty Ltd v Sanofi-Aventis (2009) 82 IPR 416; [2009] FCAFC 134. The effect of 194 [190] of H Lundbeck was summarised by their Honours as follows (at 434 [204]):
The question is whether the disclosure is sufficient to enable the skilled addressee to perceive, understand and, where appropriate, apply the prior disclosure necessarily, but within the ordinary limits of trial and error, to obtain the invention: at [190].
173 Thus, want of novelty is not established if – with the assistance of expert evidence to understand “terms of art” – the Court cannot attribute to the words of the prior publication a meaning which discloses the claimed invention. I have already concluded that the expression “optically pure” is a term of art which requires an interpretation in context to determine whether it has any meaning at all, and if so, what that meaning is. In DE 455, it has no relevant meaning (and, for reasons which will shortly become apparent, certainly does not have a meaning that discloses the invention claimed in the Purity Patent).
Novelty - Analysis
DE 455 Does Not Disclose Exactly What is Claimed
174 DE 455 is directed towards a method for separating “chiral pyridyl methyl sulfinyl-1H-benzimidazols” into their enantiomers. These compounds are described as being of the type set out in Formula I, which is depicted on p 3 of the English translation as follows:
175 There are six variables (R1 to R6) in Formula I, each of which are located at differing positions on the relevant ring structure and each of which may be one of many different substituents. Accordingly, Formula I represents a very broad group of compounds.
176 Claim 1 of DE 455 claims the very broad group of compounds (“or their salts with bases”) falling within Formula I, only one of which is (-)-omeprazole. Claim 2 is dependent on claim 1. It is restricted to six neutral or non-salt compounds, including (-)-omeprazole, and encompasses both the compounds themselves “and their salts with bases”. It does not identify any particular salts or specify that a salt form is preferred over the non-salt form. Claims 3 and 4 of DE 455 are for methods of preparing the compounds described in claim 1.
177 Ranbaxy relies on the following passages in DE 455 in contending that claims 1 to 3 and 18 to 27 of the Purity Patent are invalid for lack of novelty:
(a) On p 2 at lines 7 and 8 of the English translation, DE 455 describes “pyridyl methyl sulfinyl-1H-benzimidazols” as having the property of “inhibiting the secretion of gastric juices”.
(b) On p 6 at lines 22 to 24 of the English translation, DE 455 gives “sodium, potassium, calcium, aluminium, magnesium, titanium, ammonium or guadinium salts” as examples of “basic salts” to be used in the method of preparing a diastereomeric intermediate.
(c) On p 10 at lines 2 to 10 of the English translation, DE 455 provides a list of compounds “the synthesis of which by the inventive method” is said to be “particularly preferred”. These are:
(i) (+)-5-difluormethoxy-2-[[(3,4-dimethoxy-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazol;
(ii) (-)-5-difluormethoxy-2-[[(3,4-dimethoxy-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazol;
(iii) (+)-5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazol;
(iv) (-)-5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazol;
(v) (+)-2-[[(3-methyl-4-(2,2,2-trifluorethoxy)-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazol; and
(vi) (-)-2-[[(3-methyl-4-(2,2,2-trifluorethoxy)-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazol;
“and their salts with bases”.
(a) On p 14 at lines 2 to 9 of the English translation, DE 455 asserts that “[p]yridylmethylsulfinyl-1H-benzimidazols can be split for the first time into their optical antipodes by the inventive method”, and that these compounds “are used as active ingredients in drugs for the treatment of gastrointestinal diseases”.
(b) On p 15 at line 2 and p 16 at lines 4 and 7 of the English translation, DE 455 claims “[c]onfigurationally uniform, optically pure… (-)-5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazol… and their salts with bases”.
178 The compound named in subparas (c)(iv) and (e) above is (-)-omeprazole.
179 Whilst a number of arguments were raised by AstraZeneca in support of the conclusion that DE 455 does not disclose the integers of the claims of the Patent, I focus on one in particular, which follows upon the construction I have put on the Purity Patent.
180 The expression “optically pure” is used on pp 4, 7 and 8 in the body of the English translation of the specification and in claims 1 and 3 of DE 455.
181 However, I consider that DE 455 does not disclose “optically pure” salts of (-)-omeprazole within the meaning of that expression as used in the relevant claims of the Purity Patent. Not only does the use of “optically pure” in DE 455 not convey ≥ 98% e.e., I find it conveys no level of optical purity at all. That is because, absent any disclosure of a method of measurement of optical purity in DE 455, the expression “optically pure” does not indicate to the skilled addressee any level of optical purity. There is no statement that any particular level of optical purity was either obtained or desired, and there is no indication of any means of testing or measurement by reference to which any conclusion as to levels of optical purity could be reached. As previously alluded to, both Professor Davies and Dr Stevenson expressed the opinion that, in the absence of any indication that the optical purity had been measured or an indication of the means of measurement, there was no meaning which could be given to this expression in this document, either conventionally or otherwise. I do not accept that the reference to “optically pure” has any conventional meaning applicable to DE 455 (even if such a “conventional meaning” exists).
182 Specifically, Professor Davies’ evidence was that, conventionally, one can attribute a meaning of “greater than or equal to 90% e.e.” to the expression “optically pure” provided that there is a method of measurement so one knows the context in which the expression is used. Absent that, the expression does not attract any meaning.
183 Professor Davies stated that he would generally understand “optically pure” in DE 455 to mean at least 90% e.e., but that he did not expect the compounds produced by the DE 455 method to be optically pure within that meaning. Professor Davies stated in his first affidavit:
“I understand that DE 455 describes the material produced by the DE 455 method as “optically pure”. Although I would generally understand a compound described as optically pure” (by an identified analytical method) as a compound of 90% or more enantiomeric excess, DE 455 does not identify any analytical method or technique. As such I do not have any confidence that the compounds produced by the DE 455 method achieve 90% or more enantiomeric excess. Such an assertion, for example, without any analytical support or context, would not be publishable in a reputable scientific journal.
184 Professor Davies said in the concurrent evidence session, in relation to the use of the term “optically pure” in DE 455:
It is just a throwaway phrase in the patent. There is nothing you can relate it to that would say - it doesn’t give you a method and it doesn’t give you a number, so it doesn’t have any meaning.
185 In that same concurrent evidence session, Dr Stevenson said:
I would agree with that. There is no evidence for the use of the phrase “optically pure” which needs to be defined by a number and it’s not there.
I would be immediately concerned about any document that measured – or claimed or indicated optical purity without supporting information such as, as Professor Davies has indicated, NMR or detailing some other technique to measure it, otherwise it becomes an abstract statement about optical purity. It can’t be defined.
187 In Dr Stevenson’s view, in reading a document such as DE 455, if the author had regarded the level of optical purity to be important, he would have expected it to have been defined.
188 Consistent with the foregoing, Professor Davies and Dr Stevenson recorded as their answer to question 20 in the Joint Expert Report on Organic Chemistry as follows: “We agree that “optically pure” in DE 455 has no meaning”. This statement was explored in the concurrent evidence session and it was clear that both Dr Stevenson and Professor Davies shared the view that they expressed in their report, and that Professor Davies expressed in his affidavit.
189 Senior Counsel for Ranbaxy asked Professor Davies why he would not take a claim to optical purity at face value and assume that it meant 90% e.e.. Professor Davies confirmed that “as chemists we don’t make that assumption”.
190 Ultimately, Dr Pyter gave evidence to the same effect as Professor Davies and Dr Stevenson on this issue. On 27 February 2013, he said that he agreed with the statement from Dr Stevenson’s oral testimony extracted above.
191 Therefore, I proceed on the basis that all of the relevant experts agreed that the person skilled in the art would not attribute any meaning to the references in DE 455 to “optically pure”.
192 I make a further observation. There is nothing in the specification of DE 455 to indicate that the authors of DE 455 ever isolated (-)-omeprazole at all, much less in an optically pure form. The only example relating to an enantiomer of omeprazole is Example 6, which relates to an attempted preparation of (+)-omeprazole. Example 6 states that the resulting compound was only obtained “in the form of an amorphous solid”, which indicates contamination with impurities and residual solvent. None of the compounds exemplified in DE 455 was produced as a salt.
193 Further, in his affidavit, Dr Stevenson said that “Example 6 does not contain sufficient information to enable me to establish the enantiomeric purity of the (+)-omeprazole recovered”. That was further developed in Dr Stevenson’s oral evidence, where he said in relation to Example 6 that “it’s actually impossible to say what the optical purity achieved in DE 455 is”.
194 In addition, the experts agreed that DE 455 does not disclose a workable method of resolving the enantiomers of omeprazole. A skilled addressee is thus even more unlikely to attribute any meaning to the words “optically pure”, where there is not only no reference point disclosed for measuring that quality, but there is also no disclosure of any separated enantiomer of omeprazole of any level of optical purity, and no disclosure of a workable separation method.
195 It may be, therefore, that DE 455 does not disclose “optically pure” salts of (-)-omeprazole within the meaning of claim 1 of the Purity Patent, regardless of whether “optically pure” is given the meaning for which AstraZeneca contends (98% e.e.) or the meaning for which Ranbaxy contends (90% e.e.). However, it is not necessary to ultimately determine that point. Based upon my construction of the Purity Patent and DE 455, Ranbaxy’s claim that the Purity Patent lacks novelty must fail. The DE 455 prior art which discloses in terms “optically pure” compounds does not disclose the “optically pure” compounds within the meaning of the Purity Patent.
Inventive Step
196 I now turn to the arguments relating to inventive step, based upon my construction of the Purity Patent.
197 In brief, Ranbaxy submitted that the invention the subject of the Purity Patent involved no inventive step, and was obvious in light of the common general knowledge alone, or in light of the common general knowledge considered together with one of a number of publications relied upon by Ranbaxy under s 7(3) of the Act.
198 It is convenient at this point to summarise the relevant legal principles.
Legal principles
199 Section 18(1) of the Act as in force immediately prior to the Amendment Act provided that:
Subject to subsection (2), an invention is a patentable invention for the purposes of a standard patent if the invention, so far as claimed in any claim:
…
(b) when compared with the prior art base as it existed before the priority date of that claim:
…
(ii) involves an inventive step…
200 Section 7 of the Act as in force immediately prior to the Amendment Act identified the nature of the inventive step inquiry. Section 7(2) is a deeming provision which operates directly on s 18(1)(b). At the relevant time, s 7(2) and (3) provided:
(2) For the purposes of this Act, an invention is to be taken to involve an inventive step when compared with the prior art base unless the invention would have been obvious to a person skilled in the relevant art in the light of the common general knowledge as it existed in the patent area before the priority date of the relevant claim, whether that knowledge is considered separately or together with either of the kinds of information mentioned in subsection (3), each of which must be considered separately.
(3) For the purposes of subsection (2), the kinds of information are:
(a) prior art information made publicly available in a single document or through doing a single act;
(b) prior art information made publicly available in 2 or more related documents, or through doing 2 or more related acts, if the relationship between the documents or acts is such that a person skilled in the relevant art in the patent area would treat them as a single source of that information;
being information that the skilled person mentioned in subsection (2) could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant to work in the relevant art in the patent area.
201 Neither “inventive step” nor “inventive” is defined in the Act. The definition of “prior art base” as in force immediately prior to the Amendment Act included, in relation to whether an invention does or does not involve an inventive step, information in a document that is publicly available, whether in or out of the patent area.
202 Therefore, as it stood prior to 1 April 2002, s 7(3) did not permit the skilled team to “combine” unrelated documents, even if those documents otherwise satisfied the requirements of the provision and it would have been obvious to the skilled team to combine them. Two (or more) documents could only be relied upon if the relationship between them was “such that the person skilled in the relevant art… would treat them as a single source of that information”. Accordingly, AstraZeneca submitted – and I accept – that it is not open for Ranbaxy to advance any case of obviousness based on a combination of information in the s 7(3) citations, unless that requirement is satisfied.
General Principles Regarding Inventive Step
203 The invention to be assessed in the inventive step inquiry under s 7 is the invention as claimed. As noted by Jagot J in Apotex Pty Ltd v AstraZeneca AB (No 4) [2013] FCA 162:
in Danisco at [326] one principle which Bennett J identified as orthodox having regard to the reasons “as enunciated by the High Court in Aktiebolaget, Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) (2007) 235 CLR 173 [(Lockwood v Doric (No 2)] and Wellcome and by the Full Court in Lundbeck [H Lundbeck A/S v Alphapharm Pty Ltd (2009) 177 FCR 151 ; [2009] FCAFC 70] and Apotex”, and which was not disturbed in Novozymes A/S v Danisco A/S [2013] FCAFC 6, is that:
In assessing obviousness, it is necessary first to determine the nature of the claimed invention and the inventive step described in the Patent. This may involve ascertaining the “starting point” of the inventive step, sometimes described in terms of an existing problem for which the inventor found a solution. The obviousness of the invention as claimed is then assessed by reference to common general knowledge in Australia at the priority date.
204 A key element of the test for inventive step as prescribed by the Act (prior to the enactment of the Amendment Act) is that the required comparison is between the relevant prior art base and the “invention, so far as claimed in any claim” (rather than each of the individual integers thereof). This was emphasised in Aktiebolaget Hässle v Alphapharm Pty Limited (2002) 212 CLR 411 at 441 [72] in the context of discussing the relevant section of the Patents Act 1952 (Cth) (this passage was cited in Eli Lilly [2013] FCA 214 at [460]):
[The relevant provision does not] direct an inquiry respecting each integer of the claimed combination. The paragraph asks whether “the invention … as claimed”, here the combination, was obvious, not each of its integers.
205 In the present case, the combination of integers of claim 1 is:
(a) a sodium, magnesium, lithium, potassium, calcium or alkyl ammonium salt;
(b) of (-)-omeprazole;
(c) that is “optically pure”; and
(d) has improved pharmacokinetic and metabolic properties with respect to antisecretory effect in the treatment of gastric acid related diseases.
206 It is the comparison between that combination of integers and the relevant prior art base which is required in testing the question of inventive step.
207 The cases emphasise that the test for inventive step under Australian law imposes a significant burden on the party challenging validity. The applicable principles were restated by the High Court in Aktiebolaget Hässle (2002) 212 CLR 411 and in Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (No 2) (2007) 235 CLR 173.
208 The test for obviousness (as applicable to the present context) was accepted by the majority in Aktiebolaget Hässle (2002) 212 CLR 411 as being as follows (at 433; emphasis removed):
Would the notional research group at the relevant date, in all the circumstances… directly be led as a matter of course to try [the invention claimed] in the expectation that it might well produce [a useful result]?
209 In Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth (2010) 88 IPR 459; [2010] FCA 1211, Jagot J said, after referring to this test:
The reference to “expectation” in the above passage means a reasonable expectation of success. Lindgren J observed in Alphapharm Pty Ltd v H Lundbeck A/S (2008) 76 IPR 618 ; [2008] FCA 559 at [180] (Alphapharm) that the High Court has “insisted on the two elements: (1) being led as a matter of routine to the desired result; and (2) having a reasonable expectation of achieving that result”.
210 In this proceeding, the skilled team must have had a reasonable expectation of successfully producing a compound with superior qualities to omeprazole in attempting to produce the claimed optically pure salts of (-)-omeprazole.
211 This test is sometimes also referred to as the ‘Cripps question’, as reformulated by Graham J in Olin Mathieson Chemical Corporation v Biorex Laboratories Ltd [1970] RPC 157.
212 Several important aspects of this inquiry have been highlighted in the cases. “Obvious”, in this context, means “very plain”: see Aktiebolaget Hässle (2002) 212 CLR 411 at 427 [34], citing General Tire (1971) 1A IPR 121 at 149. It is not legitimate just to ask whether the invention would have been “obvious to try”: see Aktiebolaget Hässle (2002) 212 CLR 411 at 441-442 [72]-[76], in particular, the following passage at 441 [72]:
In so expressing the critical question and then proceeding to answer it favourably to Alphapharm, the Full Court fell into various errors of law. Several points are to be made. First, the statute does not ask whether a particular avenue of research was obvious to try so that the result claimed therefore is obvious; the adoption of a criterion of validity expressed in terms of “worth a try” or “obvious to try” and the like begs the question presented by the statute. In a sense, any invention that would in fact have been obvious under the statute would also have been worth trying. Paragraph (e) of s 100(1) of the 1952 Act, applied to the present case, asks whether the combination claimed in claim 1 was obvious. The paragraph does not fix upon the direction to be taken in making efforts or attempts to reach that particular solution to the problem identified in the Patent. Nor does it direct an inquiry respecting each integer of the claimed combination. The paragraph asks whether ‘the invention … as claimed’, here the combination, was obvious, not each of its integers.
213 The inventive element needed to sustain a patent in Australia is very small: a mere “scintilla” of inventiveness is sufficient (Lockwood (No 2) (2007) 235 CLR 173 at 195 [52]). It is critical to guard against hindsight: see Aktiebolaget Hässle (2002) 212 CLR 411 at 423 [21]. It is wrong to characterise the variation of all parameters or the trying of all choices until one proves successful as being obvious: see Alphapharm Pty Ltd v H Lundbeck A/S (2008) 76 IPR 618 at 661 [183]. The particular steps taken by the inventor do not necessarily inform the question of obviousness. An invention may be “stumbled upon by accident” or “remembered from a dream” (The Wellcome Foundation Limited v V R Laboratories (Aust) Pty Ltd (1981) 148 CLR 262), and the inventor may be motivated by information, objectives or finances not shared by the hypothetical skilled team (Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth (2010) 88 IPR 459; [2010] FCA 1211 at 515 [257]). In Lockwood (No 2) (2007) 235 CLR 173, the High Court also emphasised the importance of secondary evidence such as subsequent commercial success in assessing whether a claimed invention was obvious.
Principles Relating to Common General Knowledge
214 As was made clear in Minnesota Mining and Manufacturing Co & 3M Australia Pty Ltd v Beiersdorf (Aust) Limited (1980) 144 CLR 253 at 292, the common general knowledge:
involves the use of that which is known or used by those in the relevant trade. It forms the background knowledge and experience which is available to all in the trade in considering the making of new products, or the making of improvements in old, and it must be treated as being used by an individual as a general body of knowledge.
215 Common general knowledge is knowledge actually known or used by skilled addressees generally, or accepted by “the bulk of those who are engaged in the particular art”: see British Acoustic Films Ltd v Nettlefold Productions (1936) 53 RPC 221 at 250. As the High Court emphasised in Aktiebolaget Hässle (2002) 212 CLR 411 at 426 [31], information cannot be treated as part of the common general knowledge unless there is “evidence of its general acceptance and assimilation” by persons skilled in the art.
216 In Aktiebolaget Hässle (2002) 212 CLR 411, the High Court expressly rejected the contention that the results of a routine literature search could be included within common general knowledge, absent proof that the results had become part of the common general knowledge (at 435-436 [57]):
There was no finding that what was disclosed by those documents had entered the common general knowledge of those in Australia experienced in the practical work of formulating drugs for therapeutic use. Rather, reliance was placed upon the notion, illegitimate after Minnesota Mining, of a ‘routine literature search’.
217 As I noted in Eli Lilly [2013] FCA 214, information does not constitute common general knowledge merely because it might be found, for example, in a journal, even if widely read by persons in the art: see Wake Forest University Health Sciences v Smith & Nephew Pty Ltd (No 2) (2011) 92 IPR 496; [2011] FCA 1002 at 514 [96], citing British Acoustic Films (1936) 53 RPC 221 at 250 (which was also affirmed in General Tire (1971) 1A IPR 121 at 135). Reference in this regard is made to the words of Luxmoore J in British Acoustic Films(1936) 53 RPC 221 at 250, cited by Lehane J in Aktiebolaget Hässle v Alphapharm Pty Ltd (1999) 44 IPR 593; [1999] FCA 628 at 605 [39]:
In my judgment it is not sufficient to prove common general knowledge that a particular disclosure is made in an article, or series of articles, in a scientific journal, no matter how wide the circulation of that journal may be, in the absence of any evidence that the disclosure is accepted generally by those who are engaged in the art to which the disclosure relates. A piece of particular knowledge as disclosed in a scientific paper does not become common general knowledge merely because it is widely read, and still less because it is widely circulated. Such a piece of knowledge only becomes general knowledge when it is generally known and accepted without question by the bulk of those who are engaged in the particular art; in other words, when it becomes part of their common stock of knowledge relating to the art.
218 In Alphapharm (2008) 76 IPR 618; [2008] FCA 559, Lindgren J observed that (at 667 [221]):
[I]t was held in Astra that information recorded in a document, even a document widely circulated within the art, is not part of general common knowledge merely because the skilled addressee could be expected to locate it. The question is whether it is “‘generally accepted without question”‘ or “‘generally regarded as a good basis for further action”‘ by the bulk of those in the art.
Inventive Step - Analysis
Summary of Ranbaxy’s Case on Inventive Step
219 Ranbaxy put their case on inventive step in at least four different ways and from three different “starting points”. The first way assumes as a starting point “the known enantiomers of omeprazole disclosed in Erlandsson and DE 455”. The second way begins with “the known omeprazole racemate”. The third way starts from “the desirability of obtaining compounds with improved pharmacokinetic and metabolic properties which will give an improved therapeutic profile such as a lower degree of interindividual variation compared with that provided by omeprazole and its salts”. The fourth way begins from the same point as the third.
220 Ranbaxy also put forward the modified Cripps Question in three ways:
(1) Would the notional research group at the relevant date in all the circumstances including knowledge that omeprazole was racemic and that its enantiomers had been disclosed directly be led as a matter of course to try the salts of the enantiomers of omeprazole in the expectation that they might well produce a useful alternative to or a better drug than omeprazole?
(2) Would the notional research group at the relevant date in all the circumstances including knowledge that omeprazole was racemic directly be led as a matter of course to try the salts of the enantiomers of omeprazole in the expectation that they might well produce a useful alternative to or a better drug than omeprazole?
(3) Would the notional research group at the relevant date in all the circumstances including knowledge in addition that it was desirable to obtain a compound with improved metabolic and pharmacokinetic properties which would give an improved therapeutic profile such as a lower degree of inter interindividual variation compared with that provided by omeprazole and its salts directly be led as a matter of course to try the salts of the enantiomers of omeprazole in the expectation that they might well produce a useful alternative to or a better drug than omeprazole?
221 In the Seventh Amended Particulars of Invalidity, Ranbaxy stated that their case on inventive step was based on information contained in a number of publications, which information formed part of the common general knowledge in Australia at the priority date of the Purity Patent alone. Further or alternatively, their case was said to be based on the common general knowledge considered together with the information made publicly available in the whole of any one or more of a number of s 7(3) publications. It was pleaded that the information contained in each of those publications:
is information which forms part of the common general knowledge in Australia at or before the Purity Priority Date and is also information which a person skilled in the art could, before the Purity Priority Date, be reasonably expected to have ascertained, understood and regarded as relevant to work in the relevant art in the patent area within the meaning of section 7(3) of the Patents Act.
222 Neither the pleading nor the submissions of Ranbaxy clearly explained how any of the s 7(3) publications identified in the Seventh Amended Particulars of Invalidity could be treated as a single source of relevant information, or how they could be relied upon in combination. Further, there seemed to be no evidence upon this issue, either directly or upon which inference could be drawn. However, I have not delayed to specifically address this issue, as I have reached the conclusion that Ranbaxy cannot succeed even on the combination of all the information they rely upon in this proceeding.
Relevant Common General Knowledge
223 Ranbaxy submitted that the relevant common general knowledge included the information in paragraphs (a) to (h) below, much of which comes from standard texts including the “MIMS Annual 1992” (1992, 16th ed), the “Physicians’ Desk Reference” (1992, 46th ed) (‘PDR’), “Goodman & Gilman’s The Pharmacological Basis of Therapeutics”, Alfred Gilman and Louis Goodman (1992, 8th ed), and “The Merck Index” (1989, 11th ed).
(a) Omeprazole was launched in Sweden in 1988, and in Australia as a capsule in 1990.
(b) It was used extensively in Australia prior to its launch by way of clinical trials and compassionate use from about the mid-1980s.
(c) Omeprazole is a chiral molecule.
(d) A substantial portion of (racemic) omeprazole is metabolised to inactive metabolites by first pass metabolism in the liver.
(e) Those inactive metabolites found in plasma include hydroxyomeprazole and omeprazole sulfone.
(f) The omeprazole which escapes first pass metabolism and reaches the systemic circulation is converted in the acid environment of the parietal cells in the stomach to the active chemical species.
(g) A major path by which omeprazole is metabolised is by hydroxylation, and the metabolite formed is hydroxyomeprazole. This can be depicted as follows.
(a) The metabolism of a drug may be stereoselective, ie the metabolism of one enantiomer in a racemic mixture occurs to a greater extent than the other. Indeed most chiral drugs that are metabolised undergo metabolism stereoselectively.
224 Further, Ranbaxy submitted that it was well known that there was clinically significant interindividual variability in clinical responses to omeprazole.
225 At a very general level, it can be accepted that the above information was common general knowledge. However, I note that (h) is a statement about the common general knowledge relating to the metabolism of drugs generally – it does not specifically relate to omeprazole.
226 Nevertheless, other information is important in assessing the inventive step. Significantly, omeprazole was not equally effective in all cases and this was evident in the first clinical trials that were conducted as early as 1986. As both AstraZeneca’s and Ranbaxy’s gastroenterologists acknowledge, a subset of patients had incomplete healing or inadequate relief of symptoms when treated with the recommended daily dose of 20 mg of omeprazole. There was a reported lack of responsiveness to that dose of up to 30% or 40% of patients with severe GORD, and there were approximately 10% to 20% of patients with GORD and some patients suffering from peptic ulcer disease who did not respond adequately to that recommended treatment. Professor Dent’s evidence in the concurrent evidence session was that the incomplete clinical response in a sub-set of patients was “very clear”, “well-documented in the medical literature” and was the “regular experience of practising gastroenterologists”.
227 However, Professor Dent and Dr Prichard both gave evidence that the reasons for the interindividual variability in clinical response to omeprazole were not known before 28 May 1993. An incomplete clinical response to omeprazole was thought to be associated with insufficient acid suppression, but this was not widely known to gastroenterologists as at 28 May 1993. Further, the underlying cause for such insufficient acid suppression was not then known, as can be seen from the following exchange between AstraZeneca’s Senior Counsel and Dr Prichard:
MR BANNON: So if I have it – do I understand what you’re saying there is that just why there is this variability in response, you didn’t think it was widely understood or even understood at all.
DR PRICHARD: It may not have been widely understood, correct. It was probably that there was less acid inhibition in those people who failed and therefore you increased the dose to achieve more acid inhibition and that was the standard approach, but why in an individual should there be less acid inhibition would not have been understood.
Assessing Inventiveness – Characterisation of the Invention and ‘Starting Point’
228 I now turn to the issue of inventiveness.
229 The invention to be assessed for obviousness is ascertained from the patent: see Apotex (2009) 82 IPR 416; [2009] FCAFC 134 at 442 to 443 [152] per Bennett and Middleton JJ. The inventions claimed in the Purity Patent are characterised as new compounds for the treatment of gastric acid related diseases which show improved pharmacokinetic and metabolic properties over omeprazole, consisting of optically pure salts of (-)-omeprazole having the combination of integers set out in the claims (and in particular, claim 1).
230 Information in the specification of a patent may be relevant to the question of “starting point” if it amounts to an admission as to the common general knowledge at the priority date; or assists in understanding the starting point and context of the invention described and claimed (see Apotex (2009) 82 IPR 416; [2009] FCAFC 134). But even then, it must be weighed against the other evidence: see Lockwood (No 2) (2007) 235 CLR 173 at 212 [109].
231 In this proceeding, to test for inventiveness, a comparison must be made between the combination of features set out in claim 1, and the state of the relevant art as at 28 May 1993.
232 The cases show that there is no general principle to be applied in determining the starting point for the assessment of inventive step. Further, while the “starting point” may include information that was not within the common general knowledge, this will only be for the purpose of defining the “problem” for the skilled addressee – it must not assist with the solution: see generally Apotex (2009) 82 IPR 416; [2009] FCAFC 134 at 442-443 [152]-[153]. Further, if the problem is faced by the patentee internally but is not appreciated by the ordinary skilled addressee, that is not a problem relevant to determining the inventive step involved in the invention. That is because the statute requires a comparison between the invention as claimed and the relevant prior art base. I noted in Eli Lilly [2013] FCA 214 at [478] that references in the authorities to the “starting point” and the “problem addressed in” a patent effectively relate to the same concept.
233 It is worthwhile to set out some of the relevant authorities on this issue.
234 In Aktiebolaget Hässle (2002) 212 CLR 411, the High Court considered the following comments made by Diplock LJ in Technograph Printed Circuits Ltd v Mills & Rockley (Electronics) Ltd [1972] RPC 346 at 362 to be worth repeating (at 423 to 424 [21]):
Once an invention has been made it is generally possible to postulate a combination of steps by which the inventor might have arrived at the invention that he claims in his specification if he started from something that was already known. But it is only because the invention has been made and has proved successful that it is possible to postulate from what starting point and by what particular combination of steps the inventor could have arrived at his invention. It may be that taken in isolation none of the steps which it is now possible to postulate, if taken in isolation, appears to call for any inventive ingenuity. It is improbable that this reconstruction a posteriori represents the mental process by which the inventor in fact arrived at his invention, but, even if it were, inventive ingenuity lay in perceiving that the final result which it was the object of the inventor to achieve was attainable from the particular starting point and in his selection of the particular combination of steps which would lead to that result.
235 In Lockwood (No 2) (2007) 235 CLR 173, the High Court said at 211 [105] (citations omitted):
While not every invention constitutes a solution to a problem, it is commonplace so to describe an invention where it is appropriate to do so. Admissions in a specification about any problem said to be overcome by an invention are made from the vantage point of knowing the solution. When used as evidence, they would always need to be weighed with evidence, if it exists, from persons skilled in the relevant art of their perception of any problem at the time before the priority date, before their exposure to any solution contained in the invention.
236 In Insta Image Pty Ltd v KD Kanopy Australasia Pty Ltd (2008) 78 IPR 20; [2008] FCAFC 139, the Full Court of the Federal Court of Australia said (at 38 [98], in relation to admissions made in a specification and the use that may be made of them) that “it is… necessary to pay close attention to the terms of the individual specification in which the admissions are said to be made”. The Full Court rejected the appellant’s contention that references to earlier patents in the specification in suit (and express identification of particular problems associated with the structures identified in those patents) amounted to admissions as to the state of the common general knowledge (at 38 to 39, [98]-[102]).
237 The Full Court said (at 39 [104]):
The present patent specification constitutes admissions by Mr Lynch as to his own familiarity with the inventions the subject of the earlier US patents mentioned and with the ongoing problem of instability of which he was aware, but does not constitute an admission by him as to the common general knowledge as it existed in the USA, let alone Australia, before the priority date.
238 In the light of other evidence, the Full Court upheld the primary judge’s conclusion that the structures described in the earlier patents “and their instability problem” had not become part of the common general knowledge at the priority date (at 40 [109]).
239 In Apotex (2009) 82 IPR 416; [2008] FCAFC 134, the majority of the Full Court said (at 443 to 444 [159]):
The question of obviousness is not confined to a problem/solution approach. It may not be appropriate or sufficient where, for example, no skilled person in the art had thought of a general idea or general method of solving a known difficulty with respect to a known product or where the appreciation that there was a problem with a known product was itself part of the inventive concept. As the High Court said [in Lockwood (No 2)] at [105], while not every invention constitutes a solution to a problem, it is commonplace so to describe an invention where it is appropriate to do so. However, admissions about a problem in a specification need to be weighed with evidence of the perception of any problem by the person of skill in the relevant art, before exposure to the solution contained in the invention.
240 In Danisco A/S v Novozymes A/S (No 2) (2011) 91 IPR 209; [2011] FCA 282, Bennett J rejected the proposition that an enzyme identified in the specification had to be considered part of the starting point for the assessment of obviousness. Her Honour said (at 282 [345]):
Even if the Poulsen enzyme was not part of the invention, the problem was not what to do with the Poulsen enzyme. The problem was the in situ creation of functional ingredients in the foodstuff so as to eliminate the need to list additives.
241 See also Novozymes A/S v Danisco A/S [2013] FCAFC 6, in which the Full Court of the Federal Court of Australia reversed some findings of the primary judge, but was not asked to consider the question of inventive step.
242 Justice Jagot considered this issue in Apotex (No 4) [2013] FCA 162. Her Honour noted at [212] that “[w]hether the particular starting point is or is not part of the invention cannot be considered in isolation from the terms of the particular patent”.
243 Applying the reasoning of the High Court in Lockwood (No 2) (2007) 235 CLR 173 and the Full Court in Apotex (2009) 82 IPR 416; [2009] FCAFC 134, her Honour said that it follows that (at [215]):
references to the correct ‘starting point’ and the ‘problem-solution’ approach, expressed at the level of principle, are not particularly helpful. As the terms of the statute and the cases disclose close attention to the terms of the specification is required in order to characterise the invention. The relevant comparison is between the invention and the prior art base in order to determine obviousness.
244 I also considered this issue in Eli Lilly [2013] FCA 214, which was decided by reference to the 1952 Act.
245 In that case, I noted at [534] that the “simple answer” to the dispute over the appropriate starting point is “to recall that the court is undertaking a comparison between the invention (as claimed) and the prior art, but only by reference to common general knowledge… An important part of the analysis involves the court identifying the invention, and the prior art, and then, with any necessary expert assistance, undertaking the comparison”.
246 I continued at [535]-[536]:
It is wrong to confine the question of obviousness to a “problem and solution” analysis, although such an analysis may be useful in certain contexts: see eg Lockwood No 2 (2007) 235 CLR 173 at 200 to 201 [65].
The approach I take is similar to the one adopted very recently by Jagot J in Apotex Pty Ltd v AstraZeneca AB (No 4) [2013] FCA 162 at [207]-[215]. It makes no difference for these purposes that her Honour was considering the 1990 Act.
THE ‘STARTING POINT’ DEBATE
247 On the subject of the appropriate starting point in respect of the Purity Patent, Ranbaxy asserted that the specification of the Purity Patent makes clear that neither the selection of omeprazole as the racemate to be resolved nor the resolution of it form any part of the invention as described.
248 The first part of Ranbaxy’s proposition “relating to the selection of omeprazole as the racemate to be resolved” effectively presupposes that an improvement in the relevant art must involve resolving the enantiomers of omeprazole. AstraZeneca submitted that there is no basis for such a presupposition in the Purity Patent or in the evidence. I accept this submission. On the evidence before me, the attempts to improve on the art were directed by AstraZeneca internally to the development of analogues of omeprazole. The idea of pursuing the resolution of the enantiomers of omeprazole was one of the aspects of the invention.
249 Similarly, the latter part of Ranbaxy’s proposition is that the resolution of omeprazole forms no part of the invention as claimed. Again, AstraZeneca submitted that there is no basis for such a presupposition in the Purity Patent, or in the evidence. The Purity Patent itself claims as an inventive process the means to resolve the enantiomers of omeprazole. There is no challenge to the validity of the process claims. I accept that the taking of the steps involved in those process claims a fortiori was not the taking of “routine steps”. The conception and execution of such steps after many failed attempts at different strategies was very much one of the aspects of the invention.
250 Accordingly, I accept AstraZeneca’s submissions that the specification of the Purity Patent goes no further than to admit that:
(a) omeprazole and its alkaline salts are effective gastric acid secretion inhibitors and are useful as antiulcer agents; and
(b) omeprazole is an asymmetric sulfoxide which exists as two enantiomers.
251 The decision to resolve the enantiomers of omeprazole and their actual resolution are elements of the invention.
252 Also on the subject of starting point, it was noted that the following statement appears on p 1 (lines 18 to 20) of the Purity Patent:
It is desirable to obtain compounds with improved pharmacokinetic and metabolic properties which will give an improved therapeutic profile such as a lower degree of interindividual variation.
253 Professor Evans and Dr Reece agreed that “an improved therapeutic profile such as a lower degree of interindividual variation” is referring to a clinical improvement, namely more patients experiencing efficacy, or fewer experiencing adverse events.
254 Ranbaxy regard this as one of several possible starting points for the assessment of obviousness. However, the statement is not in terms an admission that any such desirability formed part of the common general knowledge. The statement is not directed to omeprazole in terms. It refers to “compounds”. In particular the statement does not reflect any belief or wisdom at 28 May 1993 to the effect that it was desirable to improve the pharmacokinetic and metabolic properties of omeprazole to obtain an improvement in the clinical response to this drug. It does not assume that the cause of interindividual variation in clinical response to omeprazole was known, or that the common general knowledge before 28 May 1993 indicated that any interindividual variability in the pharmacokinetics of omeprazole was likely to be of any clinical significance. The statement may be seen as having been made in hindsight with knowledge of the particular improvement realised by the invention. The appreciation that such an improvement was desirable formed part of the invention. I will return later to the evidence on these matters.
255 Statements such as these are properly to be regarded as statements of the inventive journey and not statements as to the state of the common general knowledge: see for example: Insta Image (2008) 78 IPR 20; [2008] FCAFC 139 at 36 [84]-[85], and 39 to 40 [105], [108]-[109]; and Danisco (No 2) (2011) 91 IPR 209; [2011] FCA 282 at 278 to 279 [329], and 282 [345]-[346].
256 Further, I find that, contrary to Ranbaxy’s submissions, the specification of the Purity Patent does not contain any express admission that the common general knowledge included a desire to improve the metabolic or pharmacokinetic properties of omeprazole. In particular, the specification does not concede that there was any known problem with the pharmacokinetic and metabolic properties of omeprazole, or that there was any known desire to resolve the enantiomers of omeprazole. The specification merely states that it was “desirable” to improve these properties, and that the methods described in the prior art for resolving enantiomers are unsatisfactory.
257 I accept AstraZeneca’s submissions that both of these issues are described in the Purity Patent from the position of already knowing that the administration of optically pure salts of the (-)-enantiomer of omeprazole successfully achieved gastric acid suppression with improved metabolic and pharmacokinetic properties over omeprazole.
258 The problem for the skilled addressee was how to improve upon known antisecretory drugs, the most successful of which was omeprazole.
259 In the present case, the invention presupposes that the hypothetical worker was in possession of omeprazole in racemic form and the knowledge that it had acid-suppressing activity. It goes no further. It does not presuppose any desire to improve omeprazole’s pharmacokinetic or metabolic properties. That desire is properly regarded as part of the invention. For this reason, AstraZeneca submitted that the starting points for which Ranbaxy contends should not be accepted. Instead, it was contended, if a starting point is appropriate in this proceeding, it is simply the general desire to improve upon known antisecretory drugs, the most successful of which was omeprazole. I accept this submission.
260 I should make another observation following from my construction of the Purity Patent.
261 The ‘extent’ of the optical purity found to be required by the Purity Patent has an impact on inventive step, particularly when considered in the context of the quantity of material required by the skilled addressee at the priority date to carry out the testing necessary to the invention that is the subject of the Purity Patent. Putting aside motivation, I find that the task of obtaining enantiomers in general was difficult, but even more so to the extent of the optical purity required by the Purity Patent. So, even if motivation existed to go down the path contended for by Ranbaxy, the path would not have been taken because of the perceived obstacles in the way of actually achieving any success. This is the primary basis on which I find that Ranbaxy fails to demonstrate lack of inventiveness.
262 Against this background, I now turn to discuss the four ways in which Ranbaxy put their inventive step case.
Ranbaxy’s Case on Inventive Step: First Way
263 The first way in which Ranbaxy put their case on inventive step begins with the known enantiomers of omeprazole as disclosed in Erlandsson 1990 and DE 455.
264 In summary, Ranbaxy submitted that:
(a) At the priority date, the notional team would be able to separate the enantiomers of omeprazole.
(b) Erlandsson 1990 discloses the optically pure (-)-enantiomer of omeprazole (and a method for its production). It does not disclose salts. It would be routine to make pharmaceutically acceptable basic salts of the enantiomers of omeprazole. Common commercial basic salts included the sodium and magnesium salts. Standard techniques were used to make them.
(c) Further, many drugs are developed as a salt, due to the advantages they can provide (such as greater solubility in water, etc). Ranbaxy relied on the observations of Bennett and Middleton JJ in Apotex (2009) 82 IPR 416; [2009] FCAFC 134 regarding the findings of fact in that case about the formation of pharmaceutically acceptable salts of enantiomers. At the outset, I indicate that findings of fact made in another case cannot be relevant to my task in this proceeding.
(d) DE 455 discloses optically pure (-)-omeprazole and its “salts with bases”. There is no invention involved in separating the enantiomers and making the claimed salts of that known compound.
(e) Accordingly, the invention as claimed is obvious, as the step taken is not an inventive one.
265 I note that these submissions require determination of a number of further issues, such as whether, at the priority date, an attempted resolution of the enantiomers of omeprazole would have succeeded; and whether the skilled team, seeking an improvement in omeprazole at this time, would routinely have made the sodium or magnesium salts of the enantiomers of omeprazole. I will address these issues in turn.
ANALYSIS – ‘FIRST WAY’
266 The central planks of Ranbaxy’s argument in this regard are that, at the priority date, the skilled team would have been able to separate the enantiomers of omeprazole, and it would then have been routine to make pharmaceutically acceptable salts of those enantiomers.
267 As a preliminary matter, AstraZeneca submitted that Ranbaxy erred in taking “the known enantiomers of omeprazole” as the starting point for the first way in which they put their case. This submission is persuasive. I have already set out my conclusions as to what I consider the appropriate starting point of the inventive step inquiry to be (and what it is not). I am not prepared to accept on the evidence before me that the decision to resolve the enantiomers played no part in the actual invention that is the subject of the Purity Patent. Accordingly, I consider that Ranbaxy cannot succeed on the first way in which they put their inventive step case, by virtue of the starting point that they have selected.
268 However, even if this starting point as contended for by Ranbaxy were accepted, AstraZeneca submitted that the inventions claimed in the Purity Patent are not obvious, as Ranbaxy has failed to demonstrate that the skilled team would be directly led as a matter of course to any of the following:
(a) to identify the (-)-enantiomer of omeprazole as the preferred candidate drug;
(b) to resolve the enantiomers to the required levels of optical purity, and in sufficient quantities; and
(c) to make pharmaceutically acceptable salts of (-)-omeprazole.
269 Putting aside the identification of the (-)-enantiomer of omeprazole as the preferred candidate drug, the critical question which I find determinative is whether a notional skilled team would have been able to or would have expected to be able to separate the enantiomers of omeprazole at the priority date to the required levels of optical purity and in sufficient quantities. In my view, the answer to this question is ‘no’.
Likely success of an attempted resolution of the enantiomers of omeprazole at the priority date
270 Ranbaxy submitted that at the priority date, the skilled team would have succeeded in their attempt to resolve the enantiomers of omeprazole.
271 In the document entitled “ORGANIC CHEMISTRY: Second meeting”, Dr Stevenson stated:
At May 1993 I would have had an expectation of obtaining a single enantiomer of Omeprazole with an optical purity of 90% e.e. Erlandsson had already achieved this for (-) Omeprazole (91.2% e.e) and in my opinion could be improved to deliver (+) Omeprazole at a similar level. In addition Allenmark, Lindner and Marie had already demonstrated that Omeprazole was suitable for HPLC resolution.
The quantities delivered by the method of Erlandsson are sufficient (if repeated and batched) to complete primary and secondary in vitro analysis which would require around 3-5 mg of material.
272 Dr Stevenson also gave evidence that the chiral HPLC method (being chiral High Performance Liquid Chromatography) disclosed in Erlandsson 1990 was commonly used in the pharmaceutical industry before May 1993, and could be repeated and batched to obtain sufficient quantities for primary and secondary in vitro analysis to be performed. His evidence was that there were a number of chiral HPLC columns that were commercially available by 28 May 1993. Dr Stevenson elaborated on the routine nature of chiral HPLC, explaining the work actually done as reported in Erlandsson 1990, and suggested possible improvements to yield by techniques such as peak cutting. He also said that he would have considered the suitability of other columns that were then available, and would have been looking at possible homochiral synthesis or other methods of separation at the same time.
273 By contrast, Professor Davies was of the view that chiral HPLC was not commonly used or commonly available as at May 1993, and that there were few chiral HPLC columns that were commercially available. Ranbaxy submitted that Professor Davies’ view is to be contrasted with, for example:
(a) a publication by Einarsson (“Separation of Amino Acid Enantiomers and Chiral Amines Using Precolumn Derivatization with (+)-1-(9-Fluorenyl ethyl Chloroformate and Reversed-Phase Liquid Chromatography”, 59 (1987) Analytical Chemistry 1191) which stated that “a number of columns are commercially available” (the number of which Professor Davies acknowledged would have increased after 1987 up to May 1993) (‘Einarsson’); and
(b) a publication by Kinkel (“Preparative Resolution of Heterocyclic Acetals Derived from Glycine, Mercapto-acetic Acid, B-Alanine, and Formyl- or Acetylacetic Acid by Recycling Chromatography on Chiraspher and Temperature Dependence of Separation Factors” 74 (1991) Helvetica Chimica Acta 1622), which demonstrated that PhD students had successfully resolved 370 milligrams of material at 95% e.e. using “standard commercial HPLC equipment” (‘Kinkel’).
274 Professor Davies was also of the view that Erlandsson 1990 could not provide sufficient quantities for primary and secondary in vitro analysis to be performed. However, Ranbaxy pointed to the fact that AstraZeneca conducted in vitro tests on the enantiomers obtained by Erlandsson in 1987, and determined that the enantiomers had the same level of activity. Further, it was contended, Professor Davies has never personally performed chiral HPLC.
275 In making these submissions, Ranbaxy relied heavily on Dr Stevenson’s practical experience, and what was said “would have been done at any pharmaceutical company” at the priority date.
276 By contrast, AstraZeneca pointed to the general difficulty of resolving enantiomers at the priority date, as well as the particular difficulty associated with resolving the enantiomers of omeprazole.
277 There is a dispute between the parties as to the general level of difficulty of resolving enantiomers as at 28 May 1993. AstraZeneca contended that this issue is secondary to the primary, more specific issue of the level of difficulty of resolving enantiomers of omeprazole as at 28 May 1993.
278 Professor Davies’ evidence was that at the priority date, enantiomers could not easily be resolved from the racemate in which they are usually present. Resolution to high optical purities (such as optical purities greater than 99% (or even 98% e.e.)) further increases the difficulty. In his third affidavit, Professor Davies stated:
The task of obtaining single enantiomers on a preparative scale at a level of optical purity useful in drug development was a daunting one, widely known by medicinal chemists in Australia to be notoriously difficult and unpredictable, with failed experiments providing no guidance for subsequent attempts.
279 Professor Davies’ affidavit evidence on this issue was confirmed in his oral testimony. Professor Davies confirmed that the resolution of enantiomers before 28 May 1993 was an exercise “fraught with failure”, in that:
[y]ou would not expect to succeed. You wouldn’t have a great expectation of succeeding. You would expect to have to do a lot of work and then even at the end of the day you would not get the material.
280 I have found Professor Davies’ evidence in this regard very compelling. He is highly qualified, and emphasised the difficulties not really addressed by Dr Stevenson, namely, obtaining enantiomers on a preparative scale and at the required level of optical purity.
281 By contrast, Dr Stevenson’s affidavit evidence suggested that the single enantiomers of chiral molecules would “always be obtained” in order for advanced pharmacokinetic and toxicological profiling of each enantiomer to be carried out, that resolution of single enantiomers of high purities “would be very likely to be achieved”, and that separation “would be expected to be successful”.
282 AstraZeneca submitted that it is clear from Dr Stevenson’s oral evidence that his perspective on the resolution of single enantiomers was not that of the person of ordinary skill in the art. Dr Stevenson’s evidence was based upon his experiences at Merck from 1987 to 2001. Dr Stevenson stated that as at 28 May 1993, “time, resource and cash were not in any way in short supply within Merck for basic research”. He said that “[s]cientists such as myself were given absolute power to decide what was done… I would not have had to justify the resolution of any set of enantiomers to anybody or how much it cost”. This was in contrast to the comments made by some of the other expert witnesses such as Dr Pyter and Dr Reece, who spoke of commercial constraints and revenue-driven goals.
283 I consider that Dr Stevenson was very much influenced by his own particular experience at Merck. His approach involved not necessarily having any reasonable expectation of success, but a more free hand approach of trial and error. In other words, I consider he is not in the true category of the skilled addressee looking to see whether a particular journey would have a reasonable prospect of success.
284 I prefer the submissions of AstraZeneca and the evidence of Professor Davies on these issues. Professor Davies provided objective evidence as to the difficulties involved in separating enantiomers. I do not regard these difficulties as merely “theoretical”, or exaggerated. I accept the general difficulty involved in separating enantiomers at the priority date. In respect of the particular difficulty associated with resolving the enantiomers of omeprazole (even by chiral HPLC, as contended for by Dr Stevenson), it is true that AstraZeneca themselves were in fact able to resolve the enantiomers of omeprazole in the manner devised by Mr von Unge – but only after years of unsuccessful attempts. That method is the subject of process claims in the Purity Patent. I have already observed that there is no claim by Ranbaxy that the process claims are invalid (and in particular, no claim that they are not novel and do not involve an inventive step). The resolution of the enantiomers required the invention of a process. Such a process was not a routine step. As will be discussed in greater detail below, I do not accept that the modifications to Erlandsson 1990 proposed by Dr Stevenson were standard or routine.
285 In determining whether the skilled team would have succeeded in an attempted resolution of the enantiomers of omeprazole at the priority date, it is important to bear in mind the important difference between resolution on an analytical scale (which means “sufficient to detect a material (in the order of micrograms), but not sufficient to prepare or collect any material”) and resolution on a preparative scale (which means “a scale sufficient to produce a material that can be obtained or collected and used for further study”). AstraZeneca submitted that unless the skilled team could produce the optically pure enantiomers on a preparative scale, it would not have been able to conduct the relevant in vitro tests and in vivo tests to achieve the results that would indicate another step on the path to the invention. This ties in with one of the submissions made by AstraZeneca about the evidence of Dr Stevenson, namely, that one problem was that he did not, in his oral and affidavit evidence, always specify the level of optical purity or the scale of preparation of single enantiomers to which he was referring. This therefore made it difficult on occasion to determine the weight to be given to his statements about what would have occurred in industry as at the priority date.
286 I accept that by May 1993, no one had achieved resolution of the enantiomers of omeprazole at a preparative scale and at sufficient optical purity in order to conduct anything more than in vitro studies of racemisation and activity. As will be discussed in more detail in due course, the authors of Erlandsson 1990 had tried and failed. The authors of DE 455 had tried and failed. Further, as will shortly become apparent, the results of the tests reported in Erlandsson 1990 pointed away from the resolution of the enantiomers.
287 It is significant that Professor Davies and Dr Stevenson agreed in the Joint Expert Report on Organic Chemistry that the resolution of the enantiomers of omeprazole “at a preparative scale (> 3-5 mg) was not expected to be achievable” as at 28 May 1993.
288 Further, in response to a later question in the Joint Expert Report on Organic Chemistry, Professor Davies and Dr Stevenson agreed that in the case of omeprazole, there “would not have been a reasonable expectation to succeed in obtaining an optically pure salt of a single enantiomer on a preparative scale useful for the purposes of drug development”.
289 There was some further evidence given by Dr Stevenson in the course of the hearing. In particular, he gave evidence that another method of resolution would be found because:
The option in the pharmaceutical industry is not failure. You are given a task; you are expected to succeed.
290 But on this issue I prefer to accept the agreement reached in the Joint Report as a considered response that is consistent with other expert testimony.
291 Accordingly, I do not consider that Ranbaxy has discharged their burden of showing that the skilled team would have had any reasonable expectation of a successful resolution of the enantiomers of omeprazole at the priority date. This would have prevented them from pursuing this avenue of approach. This in itself disposes of Ranbaxy’s first way of putting their case on inventive step. However, for the sake of completeness, I proceed to the next issue raised.
292 This issue is whether the skilled team, as at the priority date, would have routinely made the sodium or magnesium salts of the enantiomers of omeprazole.
Whether it would have been routine for the skilled team to make the sodium or magnesium salts of the enantiomers of omeprazole at the priority date
293 Ranbaxy submitted that at the priority date, it would have been routine for the skilled team to make the sodium or magnesium salts of the enantiomers of omeprazole.
294 As a preliminary issue, there was a dispute between the parties in relation to how straightforward or routine the process of selecting a salt was at the priority date. Professor Davies’ affidavit evidence was that the process of selecting a salt is not straightforward. Dr Pyter’s affidavit evidence was that the process of screening salts is “systematic” and “well-defined”. However, in cross-examination, Dr Pyter accepted the accuracy as at 28 May 1993 of the following statements from the article written by Berge et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Sciences 66(1) (1977) 1-19 (an article which Dr Pyter himself described as “one of seminal references in the Field” prior to May 1993 regarding pharmaceutical salts (‘Berge’)):
(a) The chemical, biological, physical and economic characteristics of medicinal agents can be manipulated and, hence, often optimised by conversion to a salt form. Choosing the appropriate salt, however, can be a very difficult task, since each salt imparts unique properties to the parent compound.
(b) Salt-forming agents are often chosen empirically. Of the many salts synthesized, the preferred form is selected by pharmaceutical chemists primarily on a practical basis: cost of raw materials, ease of crystallization and percent yield.
(c) Other basic considerations include stability, hygroscopicity, and flowability of the resulting bulk drug.
(d) Unfortunately, there is no reliable way of predicting the influence of a particular salt species on the behavior of the parent compound.
(e) Furthermore, even after many salts of the same basic agent have been prepared, no efficient screening techniques exist to facilitate selection of the salt most likely to exhibit the desired pharmacokinetic, solubility, and formulation profiles.
(f) Theoretically, every compound that exhibits acid or base characteristics can participate in salt formation.
(g) The number of salt forms available to a chemist is large; surveys of patent literature show numerous new salts being synthesized annually.
295 Professor Davies also agreed with the statement set out in (d). In respect of (e), Dr Pyter stated that the technology was “improving” as at 28 May 1993, but still accepted that this statement was substantially true at that time. In respect of (g), Dr Pyter agreed that “if anything, the number that had been registered in various countries would have probably grown” between the publication of Berge in 1977 and May 1993.
296 Professor Davies gave evidence in the concurrent evidence session on salts (with which Dr Pyter agreed) that the potential number of basic salts that could be made with a single enantiomer of omeprazole would be “hundreds”. All of these could potentially be “salts with bases of a single enantiomer of omeprazole”.
297 Dr Pyter said:
DR PYTER: I mean – you know, I mean, I don’t know about ..... but there are many, many – this is one of my reasons for being optimistic about being able to make a salt. We have many options of counter ions that have very different chemical behaviour, will pack in a crystal differently so if we happen – if the most common ones don’t work out for us then we have a lot of alternatives of counter ions that have different properties. And that’s – you know, as [Professor] Davies said, I mean, there are many, many to choose from and many on the market so they have the regulatory pedigree.
298 As to how many of these would be used “in a practical sense”, Dr Pyter stated: “I would say, practically speaking, there’s probably between 30 and 60 or 70 or something like that”.
299 On this basis, AstraZeneca submitted that the Court should find that there is no way to predict the advantages or disadvantages of one salt over the neutral form, or over any other salt.
300 I accept that, as Ranbaxy submitted, at the priority date there were obvious advantages which could be obtained by making salts of a useful pharmaceutical compound. Both Dr Pyter and Professor Davies agreed with this. To this end, Professor Davies identified better stability characteristics and new formulations. Dr Pyter identified ease of crystallisation and purification, improved solubility or absorption and chemical and physical stability as well as the fact that different salts are often necessary to develop intravenous, paediatric and extended release products of an on-market compound. Professor Davies accepted that Dr Pyter’s list of advantages was accurate on the footing that the compound in question was not yet on the market, as was obviously the case for (-)-omeprazole at the priority date. Further, Mr von Unge conceded that there may be advantages in obtaining a salt, including improved chemical and physical stability and solubility. Mr von Unge also accepted that where possible, it would be a good idea to prepare the salt forms of the single enantiomers.
301 Ranbaxy submitted that each of the above provided a reason to attempt to make a salt of the enantiomers of omeprazole. However, the experts differed as to whether it would have been reasonably expected to obtain such a salt. Professor Davies referred to the difficulties with racemisation, and also to difficulties of resolution on a scale sufficiently large for investigating crystalline salt formation. However, in this regard Ranbaxy relied on Dr Pyter, who stated that one would approach the issue empirically without preconceived expectations. He agreed that resolution of the racemate would be the most challenging aspect, but once this was achieved, he believed that obtaining a crystalline salt would be likely. He stated in his first affidavit that most drugs are developed as a salt. In his fifth affidavit, he noted that in “27 years’ experience at Abbott, [he] can recall only one case in which [they] could not make a salt of a drug when this was attempted”. Ranbaxy submitted, therefore, that the evidence establishes that generally speaking, making salts was readily achievable. Although one could not say a priori whether a particular salt would form, the screening process was conducted using a variety of counter ions (being ions with an opposite charge to the acidic or basic drug candidate with which they are associated) in parallel.
302 Dr Pyter’s evidence was that sodium and magnesium would be amongst the most common counter ions to select. Professor Davies agreed that these would be included amongst the first choice of counter ions. Mr von Unge’s evidence was that “there are a couple of different salts that one can try, and sodium and magnesium were the ones that I tried” (emphasis added).
303 Dr Stevenson’s evidence in answer to question 30(b) of the document entitled “ORGANIC CHEMISTS: Second meeting” was as follows:
The role of the salt of a single enantiomer of Omeprazole would have been pursued for purely pharmacokinetic reasons. Different salts have different rates of dissolution in the stomach and digestive system and this can be used to influence both the point and time of absorption post ingestion. A specific salt may be chosen due to its ability to be re-crystallised to high e.e, although this salt may not be that chosen for the actual dosing formulation.
304 Ranbaxy submitted that making such salts requires basic conditions. Professor Davies observed that his expectation that the (-)-enantiomer of omeprazole would racemise under basic conditions would have provided a disincentive to attempting to make a basic salt of (-)-omeprazole. Ranbaxy contended that this disincentive effect was overstated by Professor Davies. Dr Pyter gave evidence that the skilled team would conduct routine pH rate profile experiments to ascertain whether, and if so at what pH, racemisation would occur, and select conditions which avoided that occurrence accordingly.
305 In summary, Ranbaxy contended that whatever the theoretical reasoning which might have suggested that making a basic salt would lead to racemisation, that did not in fact occur, as the person skilled in the art would have discovered once he or she performed these experiments. The experiments would have been performed because (a) that is ordinary scientific method; and (b) certain accepted advantages follow from obtaining a salt of a pharmaceutical compound, which would have encouraged its development.
306 I have reached the conclusion that despite the known advantages of salts at the priority date, the skilled addressee would not have been led to try Na+ and Mg2+ salts because of the difficulty involved in predicting the advantages or disadvantages of one particular salt over another. This difficulty arises because there is no reliable way of predicting the influence of a particular salt on the behaviour of the parent compound. This is the learning taught by Berge and others, specifically accepted by Dr Pyter. To be involved in determining the particular salt would thus require a level of inventiveness that would not be attributed to the skilled addressee as at the priority date.
307 Further, I accept that even if they had been led to try these salts, due to the risks of racemisation (which were alluded to above but are referred to in greater detail later in these reasons for judgment), the skilled addressee would not be expected to succeed in this endeavour.
308 For these reasons, I do not accept that even if the “known enantiomers” of omeprazole could permissibly be taken as the starting point for the inventive step inquiry, Ranbaxy has failed to demonstrate that the skilled team would either have succeeded in an attempted resolution of the enantiomers of omeprazole, or routinely made the sodium or magnesium salts of the enantiomers of omeprazole.
309 However, before considering the other ways in which Ranbaxy put their case on inventive step, it is appropriate to consider the disclosure claimed to be effected by DE 455, Erlandsson 1990 and European patent number 124495B1 (‘EP 495’), as these publications were relied upon by Ranbaxy in this regard. In doing so, however, I note that none of these publications alters the conclusion I have reached in respect of the first way in which Ranbaxy put their case.
DE 455
310 AstraZeneca submitted – and I accept – that the methodology set out in DE 455 requires fundamental change before it could be used to produce optically pure compounds. Notably, while Dr Stevenson consistently maintained his view that he would have been able to devise a method for the separation of the enantiomers of omeprazole, he conceded that any such method would have to be “fundamentally” different and “a new process” from that disclosed by DE 455.
311 I consider that the skilled team would not be able to perceive, understand and apply the method disclosed in DE 455 to obtain the invention claimed in the Purity Patent, working within the ordinary limits of trial and error and without inventive ingenuity or undue experimentation. DE 455 does not give the skilled team the ability to prepare the claimed compounds, or the means to do so.
312 The skilled addressee would not have regarded DE 455 as a workable proposal insofar as it purported to assert an “optically pure” salt of (-)-omeprazole.
313 In their response to question 16 of the Joint Expert Report on Organic Chemistry, Professor Davies and Dr Stevenson agreed that they “do not believe that any modifications to the DE 455 process would make it work satisfactorily”.
314 However, Dr Stevenson gave evidence that he would reasonably expect to succeed in obtaining optically pure (-)-omeprazole salts based on his common general knowledge together with the information in DE 455.
315 Dr Stevenson deposed that he would change the DE 455 method by using “a chiral auxiliary… that could be attached and removed without the use of acidic conditions”, one option being to “derivatise omeprazole with a base labile… carbamate group”, which could later be removed “under non-aqueous conditions using a very mild organic base”.
316 In response to Dr Stevenson’s affidavit evidence, Professor Davies explained in his third affidavit the reasons that “[n]obody would have modified the method described by DE 455 in the way that Dr Stevenson proposes” and, even if attempted, why it would not work. To this end, Professor Davies explained that nobody would have carried out Dr Stevenson’s proposed modifications because “[t]he use of a chiral auxiliary that can be removed under acidic conditions is an integral part of the teaching of DE 455”. He further explained that the basic conditions used in Dr Stevenson’s proposed method would be expected to cause racemisation. He explained that Dr Stevenson’s method would not work because the proposed chiral auxiliary would “produce an inherently unstable molecule”, it would require “extensive experimentation” using a vast number of potential chiral auxiliaries and possible base compounds, and it would not overcome the other limitations of the DE 455 method, such as the diastereomeric separation step.
317 In the document entitled “ORGANIC CHEMISTRY: Second meeting”, Dr Stevenson identified one particular potential chiral auxiliary that was not mentioned in his affidavit. He stated that he would change “the acid labile chiral auxiliary to a base labile one such as FLEC”. He did not identify the “suitable base” he would use to remove the chiral auxiliary.
318 Dr Stevenson accepted that the changes he was proposing were directly contrary to what DE 455 teaches. He said that he would be “fundamentally changing the process by using different reagents, applying a different strategy and completely developing what would be considered, in my opinion, a new process”. Professor Davies confirmed that the processes are fundamentally different. He said:
You have gone back to the drawing board… I wouldn’t say that’s routine at all, when you’re starting from nothing again.
319 It was also established by Dr Stevenson’s oral evidence that:
(a) FLEC was a “very exotic group”;
(b) Dr Stevenson had only used it on “five or six occasions”;
(c) it was “hard to come by and expensive to buy”; and
(d) it was unstable.
320 However, in the document entitled “ORGANIC CHEMISTRY: Second meeting”, Dr Stevenson described his fundamental changes to the DE 455 method as “routine” and having a reasonable chance of success. This was tested in cross-examination:
MR BANNON: By routine - do I understand this, by routine, it was a routine part of your life to undertake experiments, the outcome of which couldn’t be known?
DR STEVENSON: That’s correct.
MR BANNON: And this is an example of such an experiment?
DR STEVENSON: This would be a typical example, that’s correct.
321 Dr Stevenson accepted that the opinions he was expressing about matters of routine were not based on any personal experience with PPIs before 28 May 1993.
322 I conclude, on the basis of this evidence, that the changes Dr Stevenson would need to make to the DE 455 method could not be properly regarded as routine in the eyes of the skilled team. The changes he was proposing were directly contrary to what DE 455 teaches.
323 In his fifth affidavit, Dr Pyter stated that he would be able to optimise the conditions of DE 455 to minimise the racemisation and degradation of enantiomers of omeprazole in the DE 455 method. Dr Pyter did not elaborate on this. I am not sure I appreciate what exactly Dr Pyter had in mind. However, I note Dr Pyter’s acknowledgment in oral testimony that he had little or no personal involvement in separating enantiomers, and could not comment with the same volume of experience as Professor Davies on stereochemistry.
324 Accordingly, I do not consider that Dr Pyter’s views on this aspect in any way undermine the evidence of Professor Davies to the effect that the skilled team would not have reasonably expected to succeed in obtaining an optically pure (-)-omeprazole salt with the requisite improved pharmacokinetic and metabolic properties as a useful or improved alternative to omeprazole at the priority date, even taking into account their common general knowledge and the information contained in DE 455. I find that to accept Ranbaxy’s submissions on this point would require the skilled team to make too many fundamental changes to the teaching of DE 455, and engage in extensive experiments of uncertain outcome in a manner that cannot be said to be obvious. Accordingly, I do not accept that DE 455 assists Ranbaxy in their case on inventive step.
325 I now turn to Erlandsson 1990.
Erlandsson 1990
326 Ranbaxy also relied on Erlandsson 1990, which – it will be recalled – is the publication entitled “Resolution of the enantiomers of omeprazole and some of its analogues by liquid chromatography on a trisphenylcarbamoylcellulose-based stationary phase: The effect of the enantiomers of omeprazole on gastric glands”, published in the Journal of Chromatography in 1990 (vol 532, p 305).
327 Ranbaxy relied on this article on the basis that it shows “the resolution or separation of the enantiomers of omeprazole… on a preparative scale”. Professor Davies and Dr Reece both described the resolution achieved in Erlandsson 1990 as only “semi-preparative”. This was despite the fact that “[t]he people of the Erlandsson paper are probably of more than ordinary skill in the field of HPLC technology”. Erlandsson 1990 also does not disclose the preparation of salts of the enantiomers of omeprazole, and reports that the enantiomers of omeprazole have the same potency as the racemate.
328 I am inclined to view these factors as disincentives to the development of the enantiomers of omeprazole. Indeed, the authors of the Joint Expert Report on Organic Chemistry agreed that the “information contained in Erlandsson 1990 would not have provided motivation to pursue or consider pursuing either a salt or a single enantiomer of omeprazole or an optically pure (-)-omeprazole salt as a useful or improved alternative to omeprazole”.
329 In any event, even if Erlandsson 1990 did not teach away from the inventions of the Purity Patent, I find that it could not have assisted the skilled team in arriving at them. This is because not only does it not teach the production of sufficiently optically pure enantiomers; it does not teach the production of sufficient quantities of such enantiomers to enable further tests to be carried out. The evidence relating to this issue (on which I ultimately prefer the submissions of AstraZeneca) is set out in detail in the next section of these reasons for judgment. But it is convenient to provide an overview of that material as it relates to Erlandsson 1990 at this point.
330 First, I note that the method taught by Erlandsson 1990 only achieved 3 mg of 64% e.e. purity for the (+)-enantiomer, and 4 mg of 91.2% e.e. purity for the (-)-enantiomer. The organic chemistry experts agreed that this was the maximum optical purity obtainable by the use of this method.
331 Dr Stevenson said in his affidavit and in the Joint Expert Report: Organic Chemistry that he considered that the methods taught in Erlandsson 1990 could be adapted or improved “to isolate useful quantities of each enantiomer at 95% e.e.”. As foreshadowed above, in his oral evidence Dr Stevenson explained some of the steps he would have employed in seeking to improve upon the Erlandsson 1990 method. He indicated that this may involve the use of “our own knowledge and perhaps the knowledge of people that Erlandsson hasn’t had access to”. The steps that Dr Stevenson would have applied included heating the column. Dr Pyter subsequently gave evidence that this was likely to increase the rates of reaction. He suggested that temperature considerations related to trying to optimise separation.
332 In any event, Dr Stevenson accepted that Erlandsson 1990 (or his improvement upon it) “isn’t the method that you would use to progress the project… through to… experiments in vivo”, and that if more than 3-5 mg of optically pure material was required to be produced at a time, another method would have to be developed. He also accepted that his proposed improvement upon Erlandsson 1990 was “brutal, and cruel, and time consuming and costly”. By this, it seems he meant that it would be very low yielding and “time intensive, labour intensive [and] cost intensive”.
333 By contrast, Professor Davies was of the view that the authors of Erlandsson 1990 had changed all of the relevant parameters with a view to getting the best possible result. This included strategies such as recycling and peak-shaving. He considered that the authors of Erlandsson 1990 had gone to “a lot of trouble to… get these enantiomers”, without success. He pointed out that no matter how the method is manipulated, racemisation will be a constant problem. In his view, the method had already been optimised. Dr Stevenson did not agree that racemisation would be a constant problem. Dr Stevenson maintained his view that Erlandsson 1990 could be improved upon; although he agreed that the recycling (or repetition) method was “self-limiting in terms of enantiomeric purity”.
334 I prefer the evidence of Professor Davies. He showed a consistent approach. However, even accepting Dr Stevenson’s approach, one cannot conclude that the changes made to Erlandsson 1990 would enable the skilled addressee to reach the invention claimed in any routine way, and with any expectation of success.
335 For these reasons, I find that Erlandsson 1990 does not provide a method for the resolution of the enantiomers of omeprazole to the required level of optical purity, or in sufficient quantities to allow the necessary testing to reveal the metabolic advantages of (-)-omeprazole over the racemate and the (+)-enantiomer to take place.
336 Therefore, based on their common general knowledge in conjunction with the information in Erlandsson 1990, the skilled team would not have reasonably expected to succeed in obtaining an optically pure (-)-omeprazole salt as a useful or improved alternative to omeprazole.
EP 495
337 For the sake of completeness I mention EP 495, which, it will be recalled, is European Patent number 124495B1’. EP 495 relates only to salts of racemic omeprazole and not to salts of the individual enantiomers of omeprazole. The evidence before me indicates that the properties of the salts of racemates are not indicative of the properties of the salts of the individual enantiomers except in the unusual instance of a conglomerate – of which omeprazole is not one. Therefore, EP 495 cannot assist the skilled team in selecting a pharmaceutically acceptable salt form in which to produce (-)-omeprazole.
338 I now turn to the second way that Ranbaxy put their case on obviousness.
Ranbaxy’s Case on Inventive Step: Second Way
339 The second way begins with the known omeprazole racemate. Ranbaxy submitted that separating the enantiomers of a racemic drug to a sufficient level of optical purity, and testing them to see whether the enantiomers had different and more useful properties than the racemate (including superior metabolic properties), was done as a matter of course by May 1993. Ranbaxy submitted that the skilled addressee would have had reason to separate the enantiomers of omeprazole and test them in this way based on the common general knowledge.
340 This iteration of Ranbaxy’s argument on inventive step hinges on the conclusions reached in respect of two further questions, being:
(a) whether, at 28 May 1993, the notional skilled team (seeking an improvement in omeprazole) would routinely have attempted to separate the enantiomers of omeprazole in order to investigate their pharmacokinetic profiles; and
(b) what steps would have been taken, at 28 May 1993, to investigate whether the single enantiomers of omeprazole had different pharmacokinetic profiles.
WHETHER IT WOULD HAVE BEEN ROUTINE AT THE PRIORITY DATE FOR THE SKILLED TEAM TO ATTEMPT TO SEPARATE THE ENANTIOMERS OF OMEPRAZOLE TO INVESTIGATE THEIR PHARMACOKINETICS
Ranbaxy’s submissions
341 Ranbaxy submitted that the answer to this question is ‘yes’, as there was good reason to do so (and it had become routine to do so) for any racemate. Further, Ranbaxy submitted that there were “particular reasons” to do so for omeprazole.
342 Dr Stevenson’s answer to question 28 of the document entitled, “ORGANIC CHEMISTRY: Second meeting” (which asked how common it was at the priority date to separate the enantiomers of a racemate when dealing with a chiral molecule of interest) was as follows:
My experience at May 1993 was that separation of enantiomers was a routine part of the drug design process.
The reasons for pursuing single enantiomers can be found in the basis of drug design methods, namely that a successful drug has to be active, efficacious, and pharmacologically significant. This profile includes the relevant selectivity profile, toxicity profile and pharmacokinetic profile all of which enable the drug molecule to be delivered in vivo and demonstrate the desired therapeutic effect. The single enantiomers of a compound can have significantly different profiles in all of the above or only some of the above.
The only way to determine this is to engage in resolution and profile the individual enantiomers to look for evidence for these differences. Although in terms of primary activity there is only a potential twofold advantage (and in the specific case of Omeprazole no advantage in primary activity at all), this is not the case for the other parameters, which in some ways are the more difficult aspects of drug design. Therefore the use of single enantiomers was at May 1993 and is today an essential part of the drug design process.
343 Ranbaxy submitted that Dr Stevenson’s evidence in this regard should be accepted, as he had considerable practical experience to be able to say so, including the separations he personally conducted prior to 1993 whilst at Merck. Ranbaxy submitted that the ‘Second meeting’ document is consistent with his oral evidence and with his affidavit, and that Dr Stevenson was not squarely challenged on any of that evidence. As Dr Stevenson points out, there were three related pharmacological reasons for resolution of enantiomers, namely, the potential for improvements in efficacy (selectivity); the potential for improvements in safety (toxicity); and the potential for improvement in pharmacokinetic profile. The evidence of the pharmacologists, Professor Evans and Dr Reece, expanded on these reasons (this is addressed further below).
344 Further, Ranbaxy contended that Dr Stevenson’s evidence was consistent with the growing interest in the separation of enantiomers from the mid 1980s, reflected in the scientific literature including the 1992 Food and Drug Administration Guidance document entitled “Development of New Stereoisomeric Drugs” (‘1992 FDA Guidance document’).
345 Ranbaxy then relied upon a number of publications in support of their argument. I set out the main publications.
346 In 1984, an article written by Mr E J Ariens entitled “Stereochemistry, a Basis for Sophisticated Nonsense in Pharmacokinetics and Clinical Pharmacology” was published in the European Journal of Clinical Pharmacology (volume 26, p 663), in which he strongly advocated the investigation of the optically active forms of racemic drugs. He stated that “in clinical pharmacology, particularly in pharmacokinetics, neglect of stereo-selectivity in action leads to the performance of expensive “highly sophisticated scientific nonsense”.
347 Mr Ariens returned to the theme in 1987 in an article entitled “Bias in pharmacokinetics and clinical pharmacology”, published in Clinical Pharmacology & Therapeutics (volume 42(4), p 361), in which he stated that (at 363):
[a]s overwhelmingly documented, stereoisomers as a rule differ markedly in their pharmacodynamic and pharmacokinetic properties. From the point of view of biology, enantiomers are essentially different compounds.
348 Also in 1987, a paper co-written by Professor Davies entitled “The highly stereoselective conversion of N,N-dimethylamphetamine into N-methylpseudoephedrine; a mimic of the enzyme mediated stereospecific benzylic hydroxylation” was published in Tetrahedron (volume 43(19), p 4463). This publication referred to the “growing appreciation” of the different biological effects of enantiomeric molecules. He and his co-author said (at p 4463):
Whilst many pharmaceuticals are still produced in racemic form there is a growing appreciation of the different biological effects of enantiomeric molecules and as a result the preparation of optically pure β-amino-alchols [sic] has attracted considerable attention.
349 Professor Evans agreed with Mr Ariens’ views. Further, he made this fact known at various conferences at around that time, and co-authored a paper on the topic in 1988, entitled “Stereoselective drug disposition: potential for misinterpretation of drug disposition data”. It was published in the British Journal of Clinical Pharmacology (volume 26, p 771). He and his co-authors pointed out that:
Although enantiomers have essentially identical physico-chemical properties in a non-chiral environment, they may behave differently when exposed to an optically discriminating environment such as the human body. Consequently, they may differ in their pharmacodynamics, pharmacokinetics or both.
350 Reference was also made to the 1984 and 1987 Ariens’ articles, the latter of which was said to indicate “the need for an increased appreciation of stereoselectivity in the field of clinical pharmacology”.
351 Professor Evans and his co-authors conclude in that publication by expressing the hope that (at 778):
this paper will contribute to the recent move within the literature to promote awareness in clinical pharmacology of stereoselectivity. This awareness should be applied to re-evaluate the results of previous studies on chiral drugs which have failed to consider stereoselective drug disposition, and to anticipate potential complications of using non-stereoselective drug analysis in future studies.
352 In 1988, Wainer and Dreyer published a text book entitled “Drug Stereochemistry”, one chapter of which was devoted to the topic “Pharmacokinetic differences between drug enantiomers in man”. That book, which Professor Evans consulted regularly before the priority date, recalled Pasteur’s observation made over 100 years earlier that the essential products of life are asymmetric. They pointed out the pharmacokinetic as well as the pharmacodynamic reasons for separation of racemates and concluded that “many drug enantiomers have different pharmacological properties in man, and there seems to be substantial intersubject variation in the ratios of the concentration of the individual drug enantiomers in plasma in man”. The view was expressed that “it probably is a good idea for individual drug enantiomer plasma levels to be determined and correlated with effect to see if any extra clinical benefit is gained by this additional analytical step”.
353 The work sets out, in table 2, evidence for stereoselective biotransformation of drug enantiomers in man. ‘Biotransformation’ is another word for metabolism. The listed stereoselective drugs include mephenytoin (about which more will be said shortly), and it is stated that “in extensive metabolizers, the S-(+) enantiomer undergoes p-hydroxylation exclusively, while the R (-) enantiomer is biotransformed equally by p-hydroxylation and N-demethylation”.
354 In 1989, in a review article in Nature (volume 342, p 631) entitled “Chemical Asymmetric Synthesis”, Professor Davies wrote (at p 636):
The differing pharmacological effects of the two enantiomers of chiral molecules are now well documented. Even if the inactive enantiomer lacks undesirable side effects, administering the potent enantiomer alone halves the dose and alleviates stress on the patient’s already compromised metabolism. We are at the watershed of asymmetric synthesis – in the near future it will be common practice to synthesize all potential new drugs as single enantiomers and there is already pressure from regulatory agencies in this direction.
355 In an article entitled “Enantioselective pharmacodynamics and pharmacokinetics of chiral non-steroidal anti-inflammatory drugs” written in 1991 (but published in 1992 in the European Journal of Clinical Pharmacology, volume 42, p 237), Professor Evans wrote that (at 237):
it is becoming generally accepted that studies in which the complexities of chirality are neglected may be misinformative. At the same time substantial advances have been made in the technology required to resolve and quantify the enantiomers of chiral drugs and biological specimens. Because of these recent developments in attitudes and analytical capabilities, it is becoming more common for pharmacokinetic studies of racemic drugs to generate data for individual enantiomers rather than unresolved species.
356 Professor Evans confirmed the accuracy of these statements in his oral evidence. The article also stated:
[t]he consequences of using a chiral drug as a racemate can be established only after careful consideration of the pharmacodynamic and pharmacokinetic properties of its individual enantiomers.
357 Under the heading “Enantioselective Pharmacodynamics”, Professor Evans said that:
[p]harmacodynamic differences between enantiomers arise from enantioselective interactions with the chiral macromolecules which constitute the biological receptors. Typically, both enantiomers will elicit similar pharmacological effects but one will be more active than the other. In other cases, the individual enantiomers of a chiral drug may exhibit qualitatively different and sometimes opposing pharmacological properties.
358 Under the heading “Enantioselective Pharmacokinetics”, he said:
Interactions with chiral macromolecules may be involved in the absorption of a drug, its distribution throughout the body, its metabolism, and its excretion, and each process may be enantioselective.
359 Under the heading “Metabolism”, he said:
“It is well recognised that stereochemical factors play an important role in xenobiotic metabolism, and that enantioselective metabolism has important consequences for many chiral drugs.”
360 Xenobiotic refers to the introduction of foreign matter, such as a synthetic chemical, into the body.
361 In May 1992, the FDA published the Guidance document referred to above, that said:
To evaluate the pharmacokinetics of a single enantiomer or mixture of enantiomers, manufacturers should develop quantitative assays for individual enantiomers in in vivo samples early in drug development. This will allow assessment of the potential for interconversion and the absorption distribution, biotransformation, and excretion (ADBE) profile of the individual isomers. When the drug product is a racemate and the pharmacokinetic profiles of the isomers are different, manufacturers should monitor the enantiomers individually to determine such properties as dose linearity and the effects of altered metabolic or excretory function and drug-drug interactions.
362 It also stated, under the heading “Policy in General”:
FDA invites discussion with sponsors concerning whether to pursue development of the racemate or the individual enantiomer. All information developed by the sponsor or available from the literature that is relevant to the chemistry, pharmacology, toxicology, or clinical actions of the stereoisomers should be included in the IND and NDA submissions.
363 Further, under the heading “Pharmacology/Toxicology”, and the subheading “Pharmacokinetic Profile”, the document stated:
To monitor in vivo interconversion and disposition, the pharmacokinetic profile of each isomer should be characterized in animals and later compared to the clinical pharmacokinetic profile obtained in phase 1.
364 In his affidavits, Dr Reece drew attention to the following statement from the 1992 FDA Guidance document:
Where little difference is observed in activity and disposition of the enantiomers, the racemates may be developed.
365 His evidence on this point was that “in order to obtain regulatory approval for a chiral compound at May 1993, the individual enantiomers of the compound would normally be tested”. Professor Evans’ evidence was that it would be a brave (not to say foolhardy) drug developer who, after May of 1992, would pursue the development of a racemate without characterising the enantiomers, and that from May 1992 (12 months before the priority date of the Purity Patent) “any company submitting a registration dossier for a racemic drug would have been expected to comment on the activity of the enantiomers”.
366 The evidence of Dr Pyter, who had extensive experience with Abbott Laboratories, was that if a molecule contained a chiral centre, resolution to single enantiomers would normally take place during the discovery process.
367 The evidence of Dr Reece, who also had extensive experience in drug companies, was that prior to initiation of clinical trials, potential differences in metabolism of the enantiomers of a racemic drug candidate would be tested.
368 Ranbaxy submitted that the proposition that the skilled team would routinely have attempted to separate the enantiomers of omeprazole to investigate their pharmacokinetic profile is further supported by the evidence in relation to the gastroenterology and pharmacology of omeprazole, in that:
(a) Dr Prichard and Professor Dent both agreed that there was known to be a variability in clinical response to the recommended dose of 20 mg of omeprazole.
(b) A clinical trial conducted by Professor Dent in 1986 showed that a minority of patients did not respond, although his view was that at that time the mechanism for that lack of response was not well known. Extensive clinical trials in or around 1988 also disclosed variability of response to the recommended dose.
(c) Dr Prichard and Professor Dent agreed that the variability of effect of omeprazole 20 mg on acid secretion was “of considerable clinical importance, as it has been shown to be the most important underlying mechanism for failure of healing of oesophagitis by omeprazole and so the most important variable to tackle in order to improve the results of initial therapy”.
(d) The pharmacologists, Professor Evans and Dr Reece, also agreed in the Joint Expert Report: Pharmacology that it was known to be desirable to reduce interindividual variability in the pharmacokinetic properties of a drug “if that led to improved efficacy (or acceptable efficacy in a greater proportion of patients) or if it led to reduced toxicity or risk of toxicity, or if it simplified the use of the drug (eg making it easier to determine what dose to use in a specific patient)”.
(e) The pharmacologists also agreed in their Joint Expert Report at question 12 that there were a number of motivating factors to investigate and separate the enantiomers of omeprazole, and that none of the deterrents would have prevented them from pursuing the enantiomers. Professor Evans identified one deterrent (knowledge that the enantiomers of omeprazole are both converted to the same achiral active species) as having “extra significance”; however he agreed that this would not prevent him from separating the enantiomers if that was his intention. But Dr Reece pointed out that, regardless of whether the enantiomers were converted to the same active species, differences in the AUC of the enantiomers due to polymorphic drug metabolism would result in varying pharmacological effect.
369 On this basis, Ranbaxy submitted that this presents a compelling reason to resolve the enantiomers of omeprazole to test the extent to which the hydroxylation metabolism of omeprazole was stereoselective.
370 Ranbaxy submitted that at trial, the “lone voice” holding out against the separation of enantiomers being a routine part of the drug design process at least by 1993 was Professor Davies. Ranbaxy submitted that his evidence that it was not “common” by 1993 to resolve the enantiomers was not convincing, and he did not have the same level of practical experience as Dr Stevenson.
371 Ranbaxy further submitted that Professor Davies’ evidence that the skilled addressee would not be motivated to resolve the enantiomers of omeprazole was also unconvincing. They contended that he never squarely addressed the point that the desirability of investigating the pharmacokinetic profiles of the enantiomers supplied a reason to resolve them. Instead he kept resorting to the proposition that because the safety and efficacy of omeprazole was known and the enantiomers of omeprazole might racemise, there would be no motivation to resolve them. It was Ranbaxy’s case that this proposition “fails to meet all the reasons for resolution”, and that its constant repetition “suggested Professor Davies had (no doubt unconsciously) taken on the role of advocate” for AstraZeneca.
372 A similar argument was made in respect of the proposition that the enantiomers might racemise. Ranbaxy noted that Professor Davies’ initial evidence was that there was a risk of racemisation, but that this “quickly increased to an expectation”. They further submitted that despite acknowledging the experimental fact that the enantiomers do not racemise in alkaline conditions to any significant extent, his evidence then shifted and he asserted many times that racemisation was occurring under basic conditions. When challenged directly on this issue, it was said that he reverted to it being a risk only, before again shifting back to the stronger proposition. Again, Ranbaxy submitted that these “slides” suggest “an unfortunate lack of objectivity”.
373 Ranbaxy also noted the answer given by Professor Davies to question 3 in the Joint Expert Report: Organic Chemistry, where he added (in the context of speaking of expected racemisation under basic conditions) that “[t]his concurs with the inventors [sic] expectation described in AU 337 p 5 lines 11-14”. Ranbaxy submitted that this addition was unnecessary, and again suggests that Professor Davies had taken on the role of an advocate. A further reason for caution in relation to Professor Davies’ evidence was said to arise from his “enthusiasm” in suggesting that his own views would be shared by the “person of ordinary skill”. It was said that he used the expression many times when the question did not call for such a reference, although such usage declined following him being questioned about it.
374 Ranbaxy submitted that leaving aside the disagreement between the experts as to whether racemisation would be expected under certain conditions, the reality is that until separation was attempted, one could not predict with certainty whether racemisation would occur or, if it did, to what extent. In fact, Ranbaxy contended that the evidence of Dr Stevenson in particular suggests that the quite moderate rate of racemisation of the enantiomers of omeprazole in neutral conditions disclosed in Erlandsson 1990 (130 hours) would encourage rather than discourage resolution in light of the much shorter known time for absorption and elimination (four hours). They submitted that any expectation that it would occur could never, realistically, have caused a medicinal chemist not to attempt the separation. It did not deter Mr von Unge from doing so. In fact, he liberated the enantiomers under strongly basic conditions.
375 Ranbaxy also pointed to the fact that it was accepted by all of the experts in this case that racemisation is not inevitable. Rather, there is a “risk” or a “fear” that racemisation might occur, the likelihood of which is something upon which reasonable minds may (and do in fact) differ. Dr Stevenson, Dr Reece and Dr Pyter all considered that the risk of racemisation of omeprazole under basic conditions would be inconsequential.
376 Further, Ranbaxy contended, Dr Stevenson, Dr Reece and Dr Pyter all gave evidence that in the face of an apparent risk of racemisation, they would nevertheless take steps to attempt to resolve the enantiomers. Professor Evans also agreed in the Joint Expert Report: Pharmacology that he would still attempt to separate the enantiomers. Even Professor Davies acknowledged that if there was a sufficient motivation to resolve the material (ie putting aside the fact that omeprazole is a prodrug and “safe and efficacious”), the skilled team would try to resolve the enantiomers. Indeed, the co-inventor of the Purity Patent, Mr von Unge, “nevertheless went ahead and did the experiments”.
377 Ranbaxy also noted that in the book chapter entitled “An Innovative Asymmetric Sulfide Oxidation: The Process Development History Behind the New Antiulcer Agent Esomeprazole” written by the AstraZeneca employees Messrs Hans-Jürgen Federsel and Magnus Larsson (published in “Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions” (2004), H U Blaser and E Schmidt (eds)), there is no suggestion that an expectation that racemisation would occur was a reason for not having earlier resolved the enantiomers of omeprazole.
378 Finally, Ranbaxy asserted that the proposition that at May 1993 the skilled team would have routinely attempted to separate the enantiomers of omeprazole is supported by what AstraZeneca themselves did. They separated the enantiomers of omeprazole in 1987 and tested them. Those tests were done on gastric glands for differences in pharmacodynamics. Such confirmatory tests were performed, in spite of the fact that the mechanism of action of the enantiomers was known. The evidence does not disclose any tests carried out on liver microsomes for differences in pharmacokinetics at the priority date. Perhaps this was because of the small quantities of the enantiomers that had been obtained in 1987 (although only small amounts would be required to conduct the liver microsome experiments), or because 1987 was relatively early days in the development of knowledge about the desirability of doing such tests. However, by the early 1990s the desirability of doing such tests was apparent as is recognised in the book chapter written by the AstraZeneca employees Messrs Federsel and Larsson. At p 415 of the chapter, Federsel and Larsson refer (with what Ranbaxy described as “justified embarrassment”) to the strangeness of not having earlier investigated the pharmacokinetics of the enantiomers. They said (citations omitted):
Later on, one of the single enantiomers of omeprazole, esomeprazole, was selected as a CD [candidate drug]. This may seem strange and poses the question as to why had the single enantiomers not been investigated at an earlier stage? Omeprazole displays chirality by virtue of its sulfoxide functionality and a couple of analytical separations of the two isomers were performed during the 1980s. However, early on a single isomer was not considered for development. The reason being that the accepted mechanism of acid inhibition led to the prediction that both isomers of omeprazole would have exactly the same effect at the site of action because acid catalyzed conversion of either isomer to the same non-chiral sulphenamide species would occur at the same rate for both isomers. Thus while it was known that the isomers of chiral drugs often have different activities, the logical assumption was that this was not the case here. Furthermore, before 1990 only milligram amounts of the single isomers had been prepared, which was not enough for in vivo testing. For these reasons the “single-isomer concept” did not look very attractive. However, the Hässle scientists could not turn away from the idea of a possible difference in metabolism between the two isomers.
379 Later the authors say:
Switching back to the early days at the beginning of the 1990s when the hunt for a suitable omeprazole-successor was still vigorously being pursued, the pre-clinical evaluation including in vivo testing, required access to at least 100 mg of each investigated compound. In the case of the stereoisomers, the task was addressed in a way that must be regarded as most straightforward under the circumstances, since the racemate was abundantly available (commercial production on a multi-ton scale), namely by applying a conventional resolution procedure.
380 Ranbaxy contended that the “Two of a Kind” article of which Mr von Unge was a co-author (published in Chemistry in Britain in May 2002, p 118), explained that the availability of larger amounts of the pure isomers of omeprazole allowed testing in rats which demonstrated significant pharmacokinetic differences. This then led to testing in humans which identified the superiority of the (-)-enantiomer.
AstraZeneca’s submissions
381 AstraZeneca rejected the proposition that at the priority date, it was routine for the skilled team to attempt to separate the enantiomers of omeprazole to investigate their pharmacokinetic profiles.
382 At a general level, it was submitted that this assumption (and the assumption that at the priority date, the skilled team would have routinely tested the enantiomers to see whether they had different pharmacokinetic profiles) ignores the special reasons that the skilled team would have had not to consider the enantiomers of omeprazole, including:
(a) it was already highly successful, safe and efficacious;
(b) it was known to be a prodrug, with both enantiomers converted to the same achiral active species (so there could be no benefit in administering a single enantiomer); and
(c) it was expected to racemise, which would make separation difficult, and, to the extent that it occurred in vivo, this would undo any benefit in administering a single enantiomer.
383 At the general level they are put, I accept these arguments. There is no debate that omeprazole was highly successful, safe and generally efficacious. Undoubtedly, omeprazole was sought to be improved upon, but this does not detract from the conclusion that it was already considered successful, safe and generally efficacious. Some of the evidence relating to the risk of racemisation has already been referred to in the course of setting out Ranbaxy’s submissions. It does not appear to be in dispute that the enantiomers of omeprazole were expected to racemise under both acidic and neutral conditions at the priority date. The real issue was whether the skilled addressee would expect racemisation under all conditions. On this issue, I prefer the evidence of Professor Davies. As I have already indicated, Professor Davies is extraordinarily qualified. Whilst I accept he is largely of academic background, even Dr Stevenson accepted Professor Davies was someone of “extraordinary” skill in the field in 1993. On this issue Professor Davies gave compelling evidence, which I accept. Dr Stevenson, whilst qualified, was prone to inconsistency, and did not show the same depth of knowledge as Professor Davies.
384 There was much discussion between Professor Davies and Dr Stevenson regarding the expectation of racemisation in basic conditions and the relevant scientific principles (such as the pKa values to be ascribed to the protons of omeprazole). It is not necessary to reproduce that evidence at length here. I accept on the basis of all of the evidence before me that racemisation in a given case is rarely if ever inevitable – one cannot be certain about what will occur until one conducts experiments. However, on the basis of Professor Davies’ evidence in particular, I am satisfied that at the priority date, the skilled team would have been cognisant of the risk of racemisation of the enantiomers of omeprazole, and that this would have acted as a real deterrent to resolving the enantiomers of omeprazole.
385 In response to AstraZeneca’s argument that omeprazole’s status as a prodrug would be a disincentive to the skilled team to pursue the resolution of its enantiomers, Ranbaxy submitted that the “error” in this reasoning lies in the fact that differences in the extent to which the “prodrug” omeprazole is metabolised in the liver will give rise to differences in the amount of omeprazole available to be converted to the active species, depicted in the following diagram:
386 This shows that there will be a greater amount of unchanged (S)–omeprazole than of (R)–omeprazole available for conversion into the active species. It therefore makes no difference whether omeprazole is a prodrug or not.
387 Further, Ranbaxy concluded that the “pro drug justification” advanced in the Federsel and Larsson article (discussed above) for not having pursued the enantiomers of omeprazole earlier is in fact irrelevant. They contended that the fact that omeprazole is not stereoselective at its site of action in the parietal cell says nothing about whether it was being cleared stereoselectively by the liver before it could reach those cells.
388 I am not satisfied on the evidence presented by Ranbaxy that it was common general knowledge at the priority date that omeprazole was metabolised stereoselectively, and that more specifically, that this was clinically significant in terms of interindividual variability (a subject which I return to later in these reasons for judgment).
389 It is unnecessary, in light of other findings I make, to finally conclude on this aspect. I do find that omeprazole was known to be a prodrug. At the time of the inventive process, this may or may not have been a relevant consideration. It would appear on the evidence of Mr von Unge that it was not a relevant consideration (at least, in the follow up project investigation carried out within AstraZeneca to which I will later refer to).
390 Further, AstraZeneca submitted that Ranbaxy’s submissions appear to assume that there is no distinction between racemic compounds which were in the development phase, and racemic compounds which were hugely successful commercial products. AstraZeneca pointed out that no examples from prior to May 1993 have been cited to the Court of an existing, commercially-available racemic compound of which a single enantiomer was developed and marketed for the purpose of achieving improved metabolic or pharmacokinetic outcomes. This is persuasive, although not decisive.
391 In light of this evidence, I find that at the priority date, it was not routine to resolve the enantiomers of omeprazole.
392 Notwithstanding this conclusion, I consider that even if the skilled team were to decide to resolve the enantiomers of a chiral drug such as omeprazole at the priority date, the following further issues arise:
(a) the quantity and optical purity of enantiomers that the skilled team would have the objective of obtaining;
(b) the technique/s that would be employed to do so; and
(c) the degree of difficulty of the exercise.
393 These issues are interrelated. Even if it were assumed that it was routine to separate the enantiomers of a racemic drug as at the priority date, the second way in which Ranbaxy put their case also assumes that the skilled team would, at that time, have tested them to see whether the enantiomers had different and more useful properties than the racemate (including superior metabolic properties). If Ranbaxy cannot demonstrate that the skilled team could reasonably be expected to have been successful in having a method available to it that would have delivered the required quantity and optical purity of enantiomers for such testing, the second way in which they put their case on inventive step cannot succeed.
394 The final issue – degree of difficulty – has already been considered above, in the context of the first way in which Ranbaxy put their case. The remaining issues are considered in turn below.
Quantities of optically pure omeprazole enantiomers required by the skilled team
395 Claim 1 of the Purity Patent and its dependent claims are to an “optically pure salt” of (-)-omeprazole with “improved pharmacokinetic and metabolic properties”. The question is: would it have been obvious for the skilled team to obtain enantiomers of omeprazole to the level of optical purity required by claim 1, in sufficient quantities to identify that (-)-omeprazole had “improved pharmacokinetic and metabolic properties with respect to antisecretory effect in the treatment of gastric acid related diseases” compared with racemic omeprazole, and to have a reasonable expectation of successfully doing so?
396 I have already decided on the level of optical purity required by claim 1. To the extent that Ranbaxy contended that lower levels of optical purity are sufficient for the purposes of testing, this may be so, but I accept the agreement of the experts that until testing is actually carried at different levels of optical purity to confirm that this is the case, it is mere speculation.
397 In any event, I do not accept that an optical purity of, for example, around 90% (80% e.e.) would be sufficient to conduct in vitro microsome testing. First, it is only at this level of optical purity, according to Dr Reece, that “you would start to get useful information” (emphasis added). His evidence was that the “preference clearly is always for the most pure material you can get within practical limits”. Secondly, as I have indicated, both Dr Reece and Professor Evans agreed that whether the results from the material of lower optical purity will be the same as results from material which is optically pure “would be a speculation until you do the tests”. Thirdly, it is uncontroversial that unless sufficient quantities of both enantiomers are available at the requisite level of optical purity, the results of the testing are “unlikely to be helpful”, as acknowledged by Professor Davies and Dr Stevenson in their answer to question 3 of the Joint Expert Report: Organic Chemistry. AstraZeneca used both enantiomers to identify the improved pharmacokinetic and metabolic properties of optically pure (-)-omeprazole sodium at the relevant time.
398 Even assuming that it were possible to obtain enantiomers at the requisite level of purity, the skilled team would also need to have sufficient quantities of said enantiomers available for the “improved pharmacokinetic and metabolic properties” to be identified via testing and for the salts to be made. I consider that Ranbaxy has failed to demonstrate that this would be the case at the priority date.
399 In order to identify whether a single enantiomer of omeprazole has improved pharmacokinetic and metabolic properties compared with racemic omeprazole, it is uncontroversial that at least the following tests must be carried out:
(a) In vitro analysis, which includes:
(i) racemisation studies, such as those reported in Erlandsson 1990;
(ii) studies regarding pharmacodynamic activity, such as those reported in Erlandsson 1990; and
(iii) liver microsome experiments to investigate the pharmacokinetic profile.
(b) In vivo analysis, first in animals and then in humans. In vivo analysis is required because in vitro analysis is not always predictive of in vivo behaviour. Without in vivo testing in humans, the skilled team would not know if the improved pharmacokinetic and metabolic properties are useful in humans “with respect to antisecretory effect in the treatment of gastric acid related diseases”, as claimed by claim 1 and reported at pp 24 to 35 of the specification of the Purity Patent.
400 The quantities of material required for a number of these experiments were explained in Professor Davies and Dr Stevenson’s answer to question 3 on p 10 of the Joint Expert Report: Organic Chemistry. Professor Davies and Dr Stevenson reported their agreement that “on the assumption that optically pure (-)-omeprazole… was available, and that if ~10 mg was available, it would be reasonably expected to succeed in completing [the specified experiments]. However without optically pure (+)-omeprazole the results would be unlikely to be helpful”. In oral evidence, Dr Stevenson and Professor Davies confirmed that “around 10 mgs would allow you to do those preliminary experiments if you have 10 mgs of each enantiomer to compare to the racemate that would be fine”. Dr Stevenson said that the in vivo experiments being contemplated were in rats, not humans.
401 Professor Evans considered that 10 to 100 mg would be required for initial in vitro metabolic testing. He said he “certainly wouldn’t contemplate an in vivo experiment with that small amount”. One of the reasons Professor Evans gave for requiring at least 10 mg of material was the difficulty associated with measuring less than 10 mg, and the fact that using less could lead to “compromised experiments”.
402 Dr Reece’s oral evidence was unclear as to how much optically pure material would be required to conduct the relevant experiments, but he seemed to suggest that they could be done with microgram quantities. However, this view was not shared by Professor Evans, or any of the other experts, and appears to be inconsistent with Dr Reece’s second affidavit, which referred to “milligram quantities” in this context.
403 The quantities required for experiments in vivo in humans were not addressed in oral evidence. Professor Davies’ affidavit evidence is that “pre-clinical studies would require hundreds of grams of a drug candidate, whereas clinical trials generally require amounts in the multi-kilogram range”. Mr von Unge deposed that clinical trials run by AstraZeneca, which identified the improved pharmacokinetic and metabolic properties of optically pure (-)-omeprazole sodium, used at least 500 g of each of (-)-omeprazole sodium and (+)-omeprazole sodium. Dr Reece deposed that experiments in vivo in humans would require “scaled up synthesis” of the relevant enantiomer.
404 As for the quantity required to test the pharmaceutically acceptable salts, Dr Pyter confirmed in oral evidence that he would prefer “quantities at least in the order of 100 milligrams” to do the necessary testing.
405 On the basis of the foregoing, I find that:
(a) at least 10 mg of each optically pure enantiomer of omeprazole would be required for in vitro analysis, including liver microsome experiments, and preliminary in vivo analysis in animals;
(b) hundreds of grams of each optically pure enantiomer of omeprazole would be required for in vivo analysis in humans, including studies to identify the “improved pharmacokinetic and metabolic properties” referred to in claim 1 of the Purity Patent; and
(c) at least 100 mg of optically pure (-)-omeprazole would be required for salt screening.
Techniques of resolving enantiomers and chiral HPLC
406 Professor Davies deposed that it is difficult to design and implement methodologies to successfully resolve enantiomers from a racemate. In his first affidavit, Professor Davies set out 13 different possible methods he might have employed to resolve enantiomers at 28 May 1993, each of which “has many sub-methods” to choose from, and each sub-method of which has many experimental variables. In that same affidavit, Professor Davies stated:
The trouble is, it is impossible to know which, if any, method might work for obtaining the enantiomers of a given racemic compound, and whether and how that method needs to be modified to achieve separation. … In my experience, it is common never to succeed in separating enantiomers despite trying a large number of methods over many years.
407 The principal method proposed to be relied upon by Dr Stevenson for the purpose of resolving the enantiomers of omeprazole at the priority date was chiral HPLC. As previously foreshadowed, there is a dispute between the parties as to the general availability of chiral HPLC as at 28 May 1993 and whether or not medicinal chemists of ordinary skill would reasonably expect to successfully employ chiral HPLC to resolve enantiomers.
408 Medicinal chemists in pharmaceutical companies did not generally have access to chiral HPLC before 28 May 1993. This is established by Professor Davies’ affidavit evidence and oral testimony. I accept his evidence. Preparative columns in particular were “very few and far between”. On this issue, Dr Stevenson conceded:
(a) the HPLC method described by Erlandsson 1990 is “time intensive, labour intensive, [and] cost intensive”; and
(b) the chiral packings required for the process are “incredibly expensive even if you make them on your own”.
409 As previously noted, Ranbaxy relied on two papers which they say demonstrate the general use of HPLC before 28 May 1993, namely Einarsson and Kinkel, which respectively state that as of the publication dates of those articles, “a number of columns are commercially available”; and that “standard commercial HPLC equipment was employed”.
410 These papers show that HPLC was in use by May 1993 and that HPLC columns were commercially available for some purposes at that time. AstraZeneca did not contend otherwise. Instead, they contended these papers are not relevant to the use of chiral HPLC for the resolution of the enantiomers of omeprazole.
411 Professor Davies was shown these articles, or parts of them, in the course of the concurrent evidence session on organic chemistry. He confirmed the testimony he had given in his third affidavit:
The technique of chiral HPLC was at a relatively early stage in its development before the Relevant Date. There were few columns available, and those that were available were generally used by analytical chemists with specialist expertise in the use of these columns.
412 In relation to the Kinkel article, which purported to show the successful resolution of racemic compounds unrelated to omeprazole, Professor Davies explained that the authors had used “exactly the same techniques that Erlandsson was using, where he failed to get resolutions”.
413 Professor Davies and Dr Stevenson also agreed that the peak-shaving technique applied by Kinkel in order to isolate single enantiomers could not have been applied by Erlandsson to isolate both enantiomers of omeprazole. This is because while the chiral HPLC stationary phase used by Kinkel was available in both enantiomeric forms, the chiral HPLC stationary phase used by Erlandsson was not.
414 Dr Reece gave evidence that resolution of enantiomers was not as common as it might otherwise have been by 1993 due to the cost of the columns, “which were still expensive”.
415 For these reasons, I find that the common general knowledge did not include any method for the successful resolution of the enantiomers of omeprazole in quantities or at the level of optical purity required to identify differences in the metabolic properties of the enantiomers as compared with each other and the racemate. This conclusion alone is sufficient to dispose of this part of Ranbaxy’s case.
416 It will be recalled that the second assumption relied on by Ranbaxy in respect of the second way in which they put their case was that at the priority date, the skilled team would have tested the enantiomers of omeprazole to see whether they had different and more useful properties than the racemate. I consider that this question has already been adequately addressed, and I do not propose to repeat the parties’ submissions here (which significantly overlap with those previously considered).
Ranbaxy’s Case on Inventive Step: Third Way
417 The third, related way Ranbaxy put their case on inventive step begins with the starting point that Ranbaxy contended is identified in the specification “of the desirability of obtaining compounds with improved pharmacokinetic and metabolic properties which will give an improved therapeutic profile”, such as a lower degree of interindividual variation compared with that provided by racemic omeprazole and its salts.
418 Ranbaxy submitted that it follows from their arguments regarding common general knowledge set out at [223] to [224] above that administration of only one of the enantiomers of omeprazole might well provide improved metabolic and pharmacokinetic properties leading to greater bioavailability of the drug (where bioavailability is a measure of the fraction of an unchanged drug that is available to the systemic circulation, after administration by a route other than intravenous administration (the bioavailability of a drug administered intravenously is 100%)). The skilled addressee placed at that starting point would, therefore, have a particular motivation to separate the enantiomers of omeprazole and investigate whether interindividual variability is due to differences in the metabolism of the individual enantiomers. Those investigations involved standard studies. Again, Ranbaxy submitted that these steps would lead the skilled addressee to the invention.
419 Ranbaxy contended that genetic polymorphism in this context means that a reduction in the extent of first pass metabolism would be likely to reduce the degree of interindividual variability. Use of the enantiomer less prone to be metabolised to inactive metabolites would not only mean greater availability in patients of the active species of the drug, but the increase in availability could be expected to be greater for fast metabolisers than any increase for slow metabolisers.
420 Ranbaxy relied on the fact that one option for patients who did not respond to omeprazole was to increase the dose beyond the recommended dose and extend it for a long period of time. However, the Joint Expert Report: Pharmacology recorded the experts’ agreement that it was known to be desirable to reduce interindividual variability in the pharmacokinetic properties of a drug “if that led to improved efficacy (or acceptable efficacy in a greater proportion of patients) or if it led to reduced toxicity or risk of toxicity, or if it simplified the use of the drug (eg making it easier to determine what dose to use in a specific patient)”. Ranbaxy submitted that the first and third of those reasons apply here.
421 Moreover, as the supply of 40 mg of omeprazole was not funded by the Pharmaceutical Benefits Scheme, Ranbaxy submitted that the high cost associated with treatment with a higher dose of omeprazole meant that gastroenterologists would generally begin patients on the standard 20 mg dose and only later consider escalating the dose if a patient did not respond adequately after eight weeks. There were also concerns relating to prolonged acid suppression in patients that might arise from high doses and long-term treatment. Ranbaxy relied on evidence that, in 1984, the clinical development of omeprazole had been temporarily suspended after ECL (enterochromaffin-like) tumours were found in the stomachs of rats that had been treated with very high doses of omeprazole for an extended period of time.
422 Finally, Ranbaxy submitted that as at May 1993, the lack of long-term safety data available meant that there was still “room for question as to how safe long term PPI therapy was with regard to effects on gastric histology”. Whilst Dr Prichard indicated that as at May 1993 he “thought that it was probably not going to be in issue on a long term basis”, he also clarified that his view was “related partly to his “personal experience”, and as a result of his involvement in clinical studies, which may not have been generally published at the priority date. Professor Dent stated that “the amount of information that we had at the time was not sufficient to be dismissive of any concerns about this risk”. As a consequence, Ranbaxy submitted that both gastroenterologists agreed that gastroenterologists in Australia generally did have concerns about the risk of ECL tumours and long-term treatment with omeprazole as at May 1993. This was said to be supported by the warning about this that appeared in the MIMS Annual 1992 at the priority date.
423 It will be apparent from this summary that one of the assumptions underpinning this iteration of Ranbaxy’s case is that the skilled team would, as a matter of routine, have separated the enantiomers of omeprazole (and would be able to do that successfully). I have already concluded that I do not accept either of these propositions – I do not accept that it would have been routine at the priority date for the skilled team to separate the enantiomers of omeprazole, and I do not accept that the skilled team would be successful in doing so (or that they would be able to obtain enantiomers in sufficient quantities or optical purity to enable the necessary pharmacokinetic testing to be carried out). Ranbaxy’s submissions about particular pharmacokinetic motivations to do so do not affect my conclusion in this regard. In my view, this is sufficient to dispose of this iteration of Ranbaxy’s case on inventive step.
424 Ranbaxy relies in a number of ways on the motivation to separate enantiomers to investigate their pharmacokinetic profile. This is an important part of their case. However, even if there is this motivation, there is no reasonable chance of achieving the separation to the required level of optical purity, or sufficient quantities for metabolism studies in vitro or in vivo to be carried out. In the absence of such studies, I consider that the skilled team would not be led as a matter of course to the invention that is the subject of the Purity Patent. Therefore, even if motivation exists, Ranbaxy cannot succeed in their inventive step attack. On the basis of the findings I have reached so far, I consider that this is what has occurred in this proceeding.
425 Nevertheless, for the sake of completeness, I make the following observations on the question of motivation to pursue enantiomers of omeprazole for reasons of pharmacokinetics.
PHARMACOLOGISTS
426 Professor Evans stated that, at 28 May 1993, there was “a general disregard among pharmacologists towards the pharmacokinetics of individual enantiomers of racemic drugs”. This was from the perspective of someone who was “deeply involved in chiral molecules” at the time and publishing papers that advocated for pharmacological data about individual enantiomers to be considered. Dr Reece accepted that the investigation of enantiomers “was slower than, I think we all would have liked” and cited expense as a factor. Dr Reece accepted that there were many publications in journals that did not report on the pharmacokinetics of enantiomers of drugs, omeprazole included.
427 Dr Reece conceded that he did not pursue enantiomers of omeprazole, or suggest to others that they should be pursued, despite working for AstraZeneca at the priority date and being aware of the large interindividual variability in omeprazole AUC.
428 The Joint Expert Report: Pharmacology states that Professor Evans and Dr Reece agreed that:
pharmacologists with a specific interest in drug metabolism would have known that omeprazole… had large inter-subject variability in pharmacokinetics and this knowledge would have extended to a broader range of pharmacologists and gastroenterologists at the time.
429 In the concurrent evidence session, both Professor Evans and Dr Reece agreed that the source of knowledge of omeprazole’s “large intersubject variability” in pharmacokinetics would have been the literature. Dr Reece said in the concurrent evidence session that he personally knew about large interindividual variability in AUC of omeprazole through studies carried out at AstraZeneca, where he was employed in 1987 to 1988. He could not recall if he knew that information prior to joining AstraZeneca.
430 Dr Reece accepted that, if he were working as part of a drug development team and he was told at an “early stage it’s extremely difficult and time consuming to even get three to five milligrams” of single enantiomers, while he would not necessarily believe the advice of his colleagues, it “would be a disincentive if it was suggestive of a whole, you know, cascade of problems in the future”.
431 In questions 11 and 12 of the Joint Expert Report: Pharmacology, Professor Evans and Dr Reece were invited to nominate the courses of action they would have considered if instructed to develop a useful or improved alternative to omeprazole. I accept that Professor Evans’ responses to those questions and their exploration in the concurrent expert evidence at trial showed that he would not have been directly led as a matter of course to try the enantiomers of omeprazole. Dr Reece responded that he would have been directly led to try the enantiomers of omeprazole.
432 In the course of the concurrent evidence session on pharmacology, Professor Evans was shown the article written by Mr Ariens entitled “Stereochemistry, a Basis for Sophisticated Nonsense in Pharmacokinetics and Clinical Pharmacology”, which I have already noted was published in 1984. For the purpose of the following discussion, it is convenient to refer to this article as ‘Ariens 1’.
433 Professor Evans accepted that the “gist” of Ariens 1 is that “to present a racemate as a single compound and therefore to avoid characterising the two enantiomers which compose it is silly scientifically”.
434 Professor Evans was then taken to an article that he had co-authored, entitled “Stereoselective drug disposition: potential for misinterpretation of drug disposition data”, which, it will be recalled, was published in 1988. This publication referred to another article by Mr Ariens, entitled “Bias in pharmacokinetics and clinical pharmacology” and published in 1987 (‘Ariens 2’). Professor Evans was also taken to another of his own articles entitled “Enantioselective pharmacodynamics and pharmacokinetics of chiral non-steroidal anti-inflammatory drugs”, which it will be recalled was published in 1992.
435 Professor Evans accepted that his articles, like Ariens 1 and Ariens 2, advocated that enantiomers and their characteristics should be identified in the interpretation of pharmacological studies.
436 However, upon a proper reading of these articles, I accept the submission of AstraZeneca that the authors are calling upon their fellow scientists to change their behaviour and conduct pharmacological, toxicological and pharmacokinetic studies of separated enantiomers, and not just the racemate. As Dr Reece conceded:
It was slower than, I think we all would have liked because I worked on the propanolol separation in 1978 and here we are talking about ‘93. I agree, it should have happened a lot faster…
437 In fact, it is apparent from Dr Reece’s evidence that at the priority date Dr Reece himself was not personally involved in the studying of pharmocokinetics of a compound he knew to be chiral (such as omeprazole) and its enantiomers. Dr Reece did suggest that it was known that “it was relevant” to look at enantiomers but how far a clinical pharmacologist would go depended on commercial factors.
438 Importantly, Dr Reece accepted that there was only one PPI studied pharmaceutically at the priority date and that was omeprazole. However, when pressed, he acknowledged that he knew this by reference to publications he had no personal knowledge of at the priority date. These publications included publications referred to by Dr Reece as Allenmark, Marle, Erlandsson, Sohn and Andersson.
439 Whilst referring to Dr Reece I should say something about his evidence. He often failed to respond precisely to the question, and gave generalised statements. This made it difficult to determine precisely the extent of his agreement or disagreement with Professor Evans. Dr Reece did not display the same depth of knowledge of Professor Evans. Therefore, to the extent that there were discrepancies in the evidence of Dr Reece and Professor Evans, I prefer the approach of Professor Evans.
440 I now return to Professor Evans.
441 Professor Evans was also taken, by Senior Counsel for Ranbaxy, to the 1992 FDA Guidance document discussed above, which relates to the development of new stereoisomeric drugs. While this document makes certain recommendations in relation to the development of new compounds, it also states:
A relatively benign toxicologic profile using the racemate would ordinarily support further development without separate toxicologic evaluation of the individual enantiomers.
442 It is uncontroversial that racemic omeprazole has, and was known to have at 28 May 1992, a relatively benign toxicologic profile.
443 AstraZeneca submitted that the proper conclusion to draw from Ariens 1, Ariens 2 and the Evans papers referred to above is that, despite the calls by these authors, it was not common practice to resolve the enantiomers of racemic compounds in drug development. Indeed, it was not even common practice to do so for the purpose of conducting pharmacological and pharmacokinetic studies. There would be no need for such a call to action if it was already what the skilled worker in the field was doing “as a matter of course”.
444 Further, it was submitted that the 1992 FDA Guidance document is not relevant to omeprazole, because it was already known to be a safe and efficacious drug in racemic form. It had already been on the market for several years and was a very successful medication. For those reasons, it was submitted that Professor Evans’ oral evidence (which was relied upon by Ranbaxy) that “any company submitting a registration dossier for a racemic drug would have been expected to comment on the activity of the enantiomers” is not relevant to omeprazole.
445 I accept these submissions. Thus, neither the Ariens papers, the Evans papers nor the 1992 FDA Guidance document are of any assistance to Ranbaxy.
446 Finally, one has to consider the issue of the overall pharmacokinetics of omeprazole, and the move from a racemate to a single enantiomer.
447 Professor Evans said in the concurrent evidence session:
Now, when you move from a racemate to a single enantiomer, lots of things can change. The drug formulation may need to change, the absorption can change, the plasma protein binding can change, the metabolism can change, the interactions with other drugs, other dietary substituents can change and maybe when the racemate was used there was an interaction between the two enantiomers and that can change as well. So in moving from a racemate to a single isomer you may overcome one problem – and that is the co-segregation between extensive and poor metabolisers, but I think it’s many steps forward in terms of the assumptions and things that have to go right for you… also to solve the problem of the very extensive metabolisers.
448 Dr Reece accepted that experiments would need to be done in order to overcome the issues identified by Professor Evans. The subsequent discussion established that Dr Reece’s view was a result of his personal experience in successfully developing a single enantiomer of a racemic drug to treat epilepsy, in contrast to Professor Evans’ view which was informed by his experience of problems encountered in moving to a single enantiomer of ibuprofen.
449 Ultimately, Professor Evans’ evidence was that single enantiomers can behave differently from the racemate in all of these pharmacokinetic processes, and the effect of administering a single enantiomer simply cannot be predicted. Dr Reece’s personal view, informed by his recent successful development of a single enantiomer of a racemic drug, I do not take as representative of the skilled team.
Ranbaxy’s Case on Inventive Step: Fourth Way
450 The fourth (related) way of putting Ranbaxy’s obviousness case begins at the same starting point as the third, but relies as well on two s 7(3) publications, which Ranbaxy contended reveal an even clearer motivation for separation and testing of the enantiomers of omeprazole.
451 Ranbaxy submitted that it was well known that (S)-mephenytoin (an anti-convulsant drug) was an example of genetic polymorphism, and that people could be categorised as either “fast” or “slow” metabolisers of (S)-mephenytoin. Research had been done in relation to the metabolism of omeprazole which indicated that high interindividual variability in the AUC of omeprazole between slow and fast metabolisers co-segregated with the metabolism of (S)-mephenytoin. In other words, those people who were fast metabolisers of omeprazole were also fast metabolisers of (S)-mephenytoin; those that were slow metabolisers of omeprazole were also slow metabolisers of (S)-mephenytoin. Ranbaxy submitted that it was known that the enzyme responsible for polymorphic metabolism of (S)-mephenytoin operates stereoselectively (ie it metabolises (S)-mephenytoin in preference to (R)-mephenytoin). Co-segregation of the same metabolism rate of omeprazole with that for (S)-mephenytoin supported the hypothesis that omeprazole was being metabolised by the same enzyme.
452 Accordingly, Ranbaxy submitted, there was a good reason to think that the metabolism of omeprazole was stereoselective. The skilled addressee would therefore be even more motivated to resolve the enantiomers of the racemic omeprazole compound, and to test them to see whether there was a difference in the way the enantiomers are metabolised. That is, on Ranbaxy’s case, the skilled addressee would, in all the circumstances, directly be led as a matter of course to try the enantiomers of omeprazole in the expectation that one might well (indeed probably would) produce a drug (being a single enantiomer of omeprazole) less subject to polymorphic drug metabolism, and thus, produce a lower degree of interindividual variability.
453 The research relied upon by Ranbaxy to this end was:
(a) Sohn et al, “Disposition Kinetics and Metabolism of Omeprazole in Extensive and Poor Metabolizers of S-Mephenytoin 4’-Hydroxylation Recruited from an Oriental Population”, The Journal of Pharmacology and Experimental Therapeutics, 262(3) (1992) 1195-1202 (‘Sohn 1992’); and
(b) Andersson et al (February 1992), “Polymorphic Hydroxylation of S-mephenytoin and Omeprazole in Caucasian and Chinese subjects”, “Abstracts of papers, Ninety-third Annual Meeting, The Peabody Hotel Orlando, Florida, March 18–20, 1992”, American Society for Clinical Pharmacology and Therapeutics, 51(2) (1992) 121-194 at 178 (‘Andersson 1992 Conference Abstract’).
454 Ranbaxy submitted that each of those publications would disclose to the skilled addressee at least:
(a) the high interindividual variability in AUC of omeprazole between poor and fast metabolisers; and
(b) that the high interindividual variability in AUC of omeprazole co-segregates with metabolism of (S)-mephenytoin. That is, poor metabolisers of omeprazole were also poor metabolisers of (S)-mephenytoin, and extensive metabolisers of omeprazole were also extensive metabolisers of (S)-mephenytoin.
455 Ranbaxy submitted that there was ultimately not much difference between Dr Reece and Professor Evans regarding the extent of stereoselectivity that might be expected in the metabolism of the enantiomers of omeprazole. But regardless of whose evidence was ultimately preferred by the Court on this issue, Ranbaxy contended that each of Sohn 1992 and the Andersson 1992 Conference Abstract would provide a particular motivation to investigate the enantiomers of omeprazole with an expectation of a potentially useful difference in their metabolism.
456 By contrast, AstraZeneca contended that the skilled addressee would not attempt to resolve the enantiomers of omeprazole. Rather, they would prefer analogue-based drug design or reformulation. Ranbaxy contended that by May 1993, it was standard practice to resolve the enantiomers of a racemic drug, and cited in support of this proposition the potential advantages of one enantiomer over the other (and therefore over the racemate), including superior primary target affinity, superior selectivity profile and superior metabolic stability. Ranbaxy pointed to DE 455 and Erlandsson 1990 as referred to in the Purity Patent specification at pp 1 to 2. Accordingly, Ranbaxy submitted, the skilled addressee would regard separation and testing of enantiomers as more attractive than the unpredictable and high risk route of analogue-based drug design in which they would not know whether the analogue had any efficacy or whether the analogue would be safe, let alone whether it had improved pharmacokinetic properties in comparison to omeprazole until testing was carried out.
457 Putting aside the evidence of Dr Reece, the objective evidence indicates that as at May 1993, there was no publication or part of the common general knowledge that spoke about the pharmacokinetics of the enantiomers of omeprazole.
458 In light of my conclusions already reached in these reasons, I do not consider that the Ranbaxy arguments relating to pharmacokinetics, even if a motivating factor, get over the chemistry issues which would need to be confronted by the skilled team.
459 In any event, I find that there were no clear pharmacokinetic reasons to pursue the resolution of optically pure enantiomers of omeprazole in a search for a useful or improved alternative to omeprazole.
460 If anything, the reasons why (-)-omeprazole has improved pharmacokinetic and metabolic properties over the racemate were the subject of study by AstraZeneca and were revealed well after 28 May 1993.
461 As to the specific publications relied upon by Ranbaxy, namely Sohn 1992 and the Andersson 1992 Conference Abstract, I make these observations.
Sohn 1992
462 Sohn 1992 does not assist the skilled addressee in arriving at the invention claimed in the Purity Patent. Sohn 1992 expressly points the reader away from the invention by stating that:
it seems unlikely that treatment with therapeutically recommended doses of omeprazole will result in a clinically significant implication for patients with the different phenotypes of S-mephenytoin 4’-hydroxylation who receive this proton pump inhibitor in the repetitive doses.
463 The conclusion of Sohn 1992 is that the implications of the genetic polymorphism of omeprazole “remain totally obscure at present”.
464 In his second affidavit, Dr Reece referred to the information in Sohn 1992 to support his opinion that the interindividual variability in AUC is in some way related to the “issue” of interindividual variability in clinical response. Dr Reece referred to the following statement in Sohn 1992 as support for this:
the steady-state inhibition of acid secretion during treatment with the same daily dose of 20 mg of omeprazole has been reported to vary widely from person to person: ranges of 35 to 65%, based on measuring acid secretion 24 hr after drug administration…
465 However, AstraZeneca submitted – and I accept – that this statement relates to interindividual variability in pharmacodynamics, ie the degree of acid suppression – not interindividual variability in clinical response. In fact, Sohn 1992 provides no information about clinical response. This is established by Professor Evans’ evidence:
It is not even clear from the statement referred to by Dr Reece, or the other information in Sohn 1992, that a range of 35% to 65% in the rate of acid inhibition (measured 24 hours after administration) is clinically significant. There is no data in Sohn 1992 correlating the rate of acid inhibition with the clinical endpoints relevant to the condition(s) being treated.
466 The authors of Sohn 1992 state that they are “tempted to consider” the polymorphism as “one of the confounding factors” possibly influencing the interindividual variability in the pharmacodynamic responsiveness to omeprazole observed in clinical practice, but they did not elaborate on this matter further. Professor Evans’ evidence is that at the priority date, he would not have considered it further. Professor Evans pointed to the fact that Sohn 1992 was a single dose study, and that the article expressly states that AUC variability is expected to decrease with multiple dosing. Omeprazole is a drug that is taken in multiple doses.
467 Further, Professor Davies gave evidence that Sohn 1992 “does not… provide any information that would motivate me to seek either an alternative or an improvement to omeprazole”.
468 In my view, the information in Sohn 1992 cannot assist Ranbaxy’s case. Sohn 1992 expressly states that the interindividual variability in AUC is unlikely to have any clinical significance. It does not suggest that the interindividual variability in AUC is in any way linked to the interindividual variability in clinical response.
ANDERSSON 1992 CONFERENCE ABSTRACT
469 The Andersson 1992 Conference Abstract is a very brief document, concluding that “a major metabolic pathway of omeprazole, the formation of hydroxyomeprazole, is correlated to the polymorphic metabolism of S-mephenytoin in both Caucasian and Chinese”.
470 This document, as with the others relied upon by Ranbaxy, concerns racemic omeprazole.
471 There is no discussion of the enantiomers of omeprazole or their pharmacokinetics, or the possibility that administration of either enantiomer might produce a drug with improved pharmacokinetic or metabolic properties.
472 The Andersson 1992 Conference Abstract does disclose to the skilled addressee:
(a) large interindividual variability in the AUC of patients taking omeprazole; and
(b) that the large interindividual variability in AUC was caused by genetic polymorphism of the (S)-mephenytoin type.
473 However, this does not disclose or provide any support for the proposition that the administration of an optically pure (-)-omeprazole salt would, or would be likely to, produce metabolic or pharmacokinetic advantages over the administration of the racemate. There is no suggestion that there is, or is likely to be, any clinical advantage in the administration of a salt of either of omeprazole’s enantiomers over the administration of the racemate.
474 I conclude that the information in Andersson 1992 Conference Abstract cannot assist Ranbaxy’s case for many of the same reasons as Sohn 1992.
Inventive Journey of AstraZeneca
475 The above conclusions indicate that the attack by Ranbaxy on validity based on lack of inventive step fails. This conclusion is supported also by an analysis of the inventive journey undertaken by AstraZeneca.
476 I interpolate to note that the basic facts relevant to the inventive journey undertaken by Mr von Unge, one of the two inventors named in the Purity Patent, were not challenged. I have accepted the evidence of Mr Von Unge, even where he gives evidence upon matters in some contention. I have accepted that between 1988 and 1994, pursuant to what was referred to as ‘the Omeprazole Follow-up Project’, AstraZeneca synthesised and tested racemic analogues of omeprazole within the class of substituted benzimidazoles (pyridinylmethylsulphinyl-1H-benzimidazoles) useful for inhibiting gastric acid production. This had nothing to do with single enantiomers of omeprazole. However, during this time, Mr von Unge himself did work in an attempt to resolve the enantiomers of omeprazole, purely for academic interest.
Work Done Outside of the Omeprazole Follow-up Project
477 Mr von Unge’s first attempts to create and separate diastereomeric derivatives of omeprazole and its analogues were carried out in 1989 using a hydroxymethyl derivative of omeprazole. Mr von Unge made a mandelic acid ester and attempted to separate the diastereomers using normal phase flash chromatography. This attempted separation failed, and at the time Mr von Unge did not understand why.
478 Mr von Unge discussed his experiment with Dr Lindberg, his co-inventor of the Purity Patent. Through that discussion, Mr von Unge learned of the work of Messrs Per Erlandsson and Roland Isaksson at Lund University who, in 1987, had attempted to separate a quantity of racemic omeprazole using chiral HPLC. The work of Messrs Erlandsson and Isaksson was later reported in the Erlandsson 1990 publication. Dr Lindberg regarded the work of Messrs Erlandsson and Isaksson as a failure, and communicated to Mr von Unge his expectation that Mr von Unge would not succeed in separating the enantiomers of omeprazole.
479 Dr Lindberg’s insistence that separating the enantiomers of omeprazole would be difficult spurred on Mr von Unge, who became determined to prove Dr Lindberg and others within AstraZeneca wrong, although there was no commercial reason to do so at the time.
480 The task of separating the enantiomers of omeprazole proved to be difficult. Over a period of two years, Mr von Unge tried between eight and ten strategies, and multiple attempts, with no success.
481 It was not until November 1991, more than two years after his initial attempt, that Mr von Unge finally succeeded. The method used by Mr von Unge that was ultimately successful enabled the production of 100 mg of each enantiomer of omeprazole. That method, and compounds produced by it, are exemplified and claimed in the Purity Patent.
Racemisation
482 After Mr von Unge had separated the enantiomers of omeprazole, Mr von Unge and Dr Lindberg asked AstraZeneca’s analytical laboratory to test whether racemisation occurred under various conditions, and if so, at what rate. Tests conducted by AstraZeneca’s analytical laboratory in December 1991 found that the enantiomers of omeprazole did not racemise, even under alkaline conditions. This was contrary to Mr von Unge and Dr Lindberg’s expectation at the time.
483 There was an attack upon Mr von Unge regarding the expectation of racemisation. In particular, Mr von Unge was cross-examined about his expressed surprised at discovering that the separated enantiomers did not racemise under basic conditions, given that the method of preparing the isolated enantiomers that was ultimately successful used sodium hydroxide (a basic substance) to hydrolyse diastereomeric derivatives of omeprazole. However, I consider that this attack was not successful, particularly in light of the following evidence (which was not undermined during cross-examination).
484 In his affidavit, Mr von Unge stated:
There was a general consensus among the chemists on the [Omeprazole Follow-up Project], all of whom where experienced scientists and many of whom were involved in the discovery and development of omeprazole, that the enantiomers of omeprazole would racemise. My colleagues and I considered that omeprazole would racemise under acidic conditions because the very mechanism of action of omeprazole (discussed in the paper by Dr Lindberg in Annexure SVU-4) involves the reversible protonation of omeprazole under acidic conditions. My colleagues and I also considered that omeprazole would racemise under basic conditions because the methylene group adjacent to the sulphur atom would be deprotonated and cause pyramidal inversion of the sulphoxide group.
485 Mr von Unge explained that his belief of a “general consensus” was based on discussions with his colleagues after his first attempts to separate diastereomers in 1989, including his conversation with Dr Lindberg referred to above. Mr von Unge also referred in cross-examination to a conversation he had with Dr Arne Brändström regarding racemisation after 1989. Dr Brändström and six colleagues at AstraZeneca wrote the article tendered by Ranbaxy as Exhibit R1, being “Chemical Reactions of Omeprazole and Omeprazole Analogues III”, which was published in Acta Chemica Scandinavica 1989 (vol 43, p 569). This article was put to Mr von Unge and, consistent with Professor Davies’ evidence, Mr von Unge confirmed the teaching of the article that the protons on the CH2 group within the omeprazole molecule would undergo proton exchange under alkaline conditions, which was expected to lead to racemisation. Based at least on his discussions with Dr Lindberg and Dr Brändström, at the relevant time, Mr von Unge reasonably believed that senior chemists at AstraZeneca expected omeprazole to racemise, at least by the time the Omeprazole Follow-up Project had commenced.
486 In his affidavit, Mr von Unge referred to the publication of Erlandsson 1990 and stated that it “confirmed my and my colleagues[’] lack of interest in developing the enantiomers of omeprazole because my colleagues and I considered that any racemisation is problematic when developing a drug” (emphasis in original).
487 Mr von Unge explained that he personally reached the conclusion that omeprazole would racemise as soon as he learned the mechanism of action of omeprazole shortly after joining AstraZeneca in 1988. He learned of the experimental proof that omeprazole racemised under neutral conditions, that was later reported in Erlandsson 1990, through discussions with Dr Lindberg in 1989.
488 The mere fact that Mr von Unge carried out the experimentation does not in itself show he did not hold the belief he said he had about racemisation. Mr von Unge in fact took measures to minimise the racemisation he expected to occur, so was at least mindful of the problem.
489 The evidence of both Dr Stevenson and Professor Davies is that the experiment performed by Mr von Unge would have been counterintuitive at least in strongly basic conditions.
490 Further, the Purity Patent states in terms that the racemisation in alkaline conditions was expected. Accordingly, I accept Mr von Unge’s evidence regarding expectation of racemisation at the relevant time.
Motivations of the Omeprazole Follow-up Project
491 Further, the hypothesis motivating the Omeprazole Follow-up Project team (namely, the hypothesis set out at paragraph 16(a)(i) of Mr von Unge’s affidavit, that some patients were so effective at metabolising omeprazole that it was largely cleared by the liver before it could reach the parietal cells in the stomach) concerned the metabolism of “omeprazole”. Omeprazole is a racemic compound. The hypothesis had nothing to do with the enantiomers of omeprazole. That the hypothesis was limited to racemic omeprazole is clear from the terms of paragraph 16(a)(i) of Mr von Unge’s affidavit, which does not state that “some patients were so effective at metabolising one of the enantiomers of omeprazole” (emphasis added). That the hypothesis was so limited was made clear by Mr von Unge in re-examination:
MR BANNON: Now, the last sentence of that paragraph refers to the hypothesis in paragraph 16(a)(i). Could you just explain what the relevance of the – changing the methyl group had to do with that hypothesis?
MR VON UNGE: Correct.
MR BANNON: Explain what the relevance was?
MR VON UNGE: The relevance – I mean, we – the assumption was that we thought that it was a problem with the methyl group of the pyridine ring of the omeprazole racemic structure. So – and we believed that reducing – removing that methyl group could provide an improvement of the drug by improving the availability through decreasing the metabolism.
…
MR BANNON: If the idea which is explained behind the 16(a)(i) hypothesis, which you’ve just described, was the matter you just referred to, in what way, or can you explain in what way the successful synthesis of the enantiomers and whatever flowed from that proved that hypothesis correct?
MR VON UNGE: As far as metabolism is being concerned, so that’s correct. Yes, we were right in thinking about the metabolism or one of the factors, however, we were dealing with that in the project by synthesising racemic new analogues with a structure lacking this method. We didn’t think about stereoisomers could do any improvement on that.
492 That the hypothesis that was the focus of the Omeprazole Follow-up Project was limited to the metabolism of racemic omeprazole is also borne out by what AstraZeneca in fact did, none of which was challenged by Ranbaxy. Mr von Unge explained in his affidavit that, through a process of synthesising racemic analogues of omeprazole and developing structure activity relationships, the scientists working on the Omeprazole Follow-up Project had concluded that a methyl group at position ‘5’ of the pyridine ring was involved in the formation of 5-hydroxyomeprazole, one of the metabolites of omeprazole. In other words, AstraZeneca hypothesised that there was a problem with one of the substituents on the chemical structure of omeprazole, which could only be solved by changing the chemical structure. AstraZeneca’s hypothesis is the antithesis of an assumption that the problem lay with one of the individual enantiomers, because each enantiomer has the same substituents as the other.
The Relevance of Omeprazole Being a Prodrug
493 A further issue was whether the fact that omeprazole was a prodrug was relevant to AstraZeneca’s lack of desire to pursue enantiomers. I do not consider that, even if this is not a matter that was considered by AstraZeneca to be relevant, this impacts on the approach otherwise taken at AstraZeneca. I accept the evidence of Mr von Unge as what in fact occurred during the Omeprazole Follow-up Project. The focus was to test racemic analogues of omeprazole to identify a new PPI.
AstraZeneca Internal Documents
494 I make one final observation about Mr von Unge’s evidence. Ranbaxy tendered through the cross-examination of Mr von Unge certain internal documents of AstraZeneca. None of the documents contradicted the evidence of Mr von Unge that at least before he synthesised the enantiomers of omeprazole, he was not aware of any person in AstraZeneca having an interest in the enantiomers of omeprazole.
495 Based on Mr von Unge’s affidavit and oral evidence, I find that nobody within AstraZeneca had an interest in the enantiomers of omeprazole as part of the search for a useful or improved alternative for omeprazole that was occurring in the context of the Omeprazole Follow-up Project. There are no documents that suggest or indicate that AstraZeneca had an interest in developing the enantiomers of omeprazole as new drugs if no other reason that the enantiomers of omeprazole were expected to racemise.
496 I note that there was research into and consideration of the enantiomers of omeprazole undertaken by a group at Lund University at the relevant time, and that this group included AstraZeneca employees. To the extent that partial separation of the enantiomers of omeprazole was carried out, Mr von Unge indicated (and I accept) that the work was carried out entirely by the Lund University scientists, and not by AstraZeneca or at their request. Mr von Unge was quite clear and consistent in his evidence on this matter throughout.
497 Further, none of the documentary evidence shows work being done by AstraZeneca on the process of separation itself, as distinct from conducting tests on partially separated enantiomers. Further, there was some evidence that one of the scientists at Lund University, and an employee of AstraZeneca (Mr Sam Larsson), determined optical purity of omeprazole in 1987 – but as Mr von Unge explained in cross-examination, this was using analytical HPLC, and did not involve the separation of enantiomers using preparative scale HPLC. There was also no indication that there was any sufficient quantity obtained, at a high enough purity for further investigation.
498 Therefore, to the extent the inventive journey is relevant to, but not decisive, of the question of obviousness, I accept that Mr von Unge’s evidence reflects the inventiveness of the final invention.
Secondary Indicators
499 In further support of their case on lack of obviousness of the Purity Patent, AstraZeneca relies on the fact that other pharmaceutical companies at the priority date were not seeking to resolve the enantiomers of omeprazole or its analogues. This is essentially correct, although not completely. Ranbaxy pointed to the fact that Byk Gulden – the pharmaceutical company selling pantoprazole, a competitor to omeprazole – published in its patent application DE 455 that it was resolving the enantiomers of benzimidazoles including omeprazole.
500 Notwithstanding this example, there is a body of evidence to suggest that others failed to arrive at the invention, and the reference to Byk Gulden does not detract from the evidence in this regard.
501 Pharmaceutical companies are constantly searching for improvements to existing medications.
502 A number of patents were tendered by AstraZeneca to demonstrate that other companies were also working to find improvements on omeprazole. AstraZeneca submitted that those companies’ failure to come up with (-)-omeprazole is strongly indicative that the claims of the Purity Patent were not obvious to the skilled team.
503 It is known that prior to May 1993, patent applications were filed by other companies for racemic omeprazole analogues, including:
(a) Patent specification 573,744 by The Upjohn Company, dated July 1983;
(b) Patent specification 570,130 by Takeda Chemical Industries Ltd, dated 1984;
(c) Patent specification 601,396 by Tokyo Tanabe Company Limited, dated 1986;
(d) Patent specification 594,836 by Eisai Co Ltd, dated 1986;
(e) Patent specification 610,585 by Shionogi & Co Ltd, dated 1987;
(f) Patent specification 615,933 by Chiesi Farmaceutici SpA, dated 1987; and
(g) Patent specification EP 0 481 764 B1 by Takeda Chemical Industries Ltd, dated 1990.
504 AstraZeneca submitted that these earlier patent publications are admissible as evidence of attempts by others to solve the “problem” of improving upon omeprazole. In The Wellcome Foundation (1981) 148 CLR 262, Aickin J said (at 287):
Evidence is admissible of previous workers having failed to solve the relevant problem or having produced solutions which turn out to be failures, whether by reference to prior specifications which have failed to work or by other evidence.
...
If evidence of the failure of other attempts to solve a well-known problem, i.e. to satisfy a “long-felt want”, is admissible in support of the inventive nature of a successful solution to such a problem, as it undoubtedly is, it is difficult to see why the patentee’s own lack of success in earlier attempts would not also be admissible for the same purpose.
505 It was accepted by Dr Reece that at least some of these patents were by major pharmaceutical companies, for compounds that were direct competitors of omeprazole.
506 Further, Mr von Unge gave evidence that he was aware “very soon after I joined AstraZeneca” that their competitors were developing racemic analogues of omeprazole.
507 Dr Stevenson gave evidence that his employer at the relevant time, Merck, was working with PPIs and with enantiomers but, as far as he was aware, no one at Merck was working on resolving the enantiomers of omeprazole in 1993. The following exchange took place in the course of the concurrent evidence session:
MR BANNON: Dr Stevenson, in relation to your agreement to the proposition that, given that omeprazole was a safe and efficacious drug, there would be no motivation to resolve the enantiomers. Could I just ask you a couple more questions about a matter I raised with you before lunch, namely, I think you agreed that, as far as you were aware, there was no resolution of enantiomers going on in Merck as at 1993?
DR STEVENSON: There was no resolution of omeprazole going on. There was resolution of enantiomers going on for sure.
MR BANNON: I’m sorry. Yes, thank you. No resolution of omeprazole into enantiomers at Merck; correct?
DR STEVENSON: That’s correct.
MR BANNON: But I think you indicated Merck was working on proton-pump inhibitors.
DR STEVENSON: I believe they were, yes.
508 Byk Gulden’s resolving of the enantiomers of benzimidazoles as exemplified in DE 455 does not detract from the overwhelming evidence of a general approach taken by pharmaceutical companies at the relevant time. This approach was to develop racemic analogues of omeprazole. At best, the experience of Byk Gulden was but one isolated example which does not reflect general experience.
509 Therefore, I accept that there is evidence that supports the contention that pharmaceutical companies in general had not considered the single enantiomer of any PPI for pharmacokinetic, metabolic or any other reasons at the priority date. This also supports the proposition that to do so was not obvious.
510 In response, Ranbaxy contended that it is not surprising that no one sought to exploit the enantiomers of omeprazole, as AstraZeneca had the benefit of a monopoly by virtue of Australian patent number 529,654. That patent taught the skilled addressee to separate the compounds that exist as racemates by ‘conventional methods’. Ranbaxy submitted that there was a risk that this patent claimed each of the enantiomers. There was also a risk that if the enantiomers racemised in the body, their use would be caught by this patent. It was said that it is obvious that this would have represented a significant commercial deterrent to competitors, to make and use either of the optical isomers of omeprazole prior to 11 April 1999 when Australian patent number 529,654 expired.
511 Ranbaxy further submitted that this disincentive to competitors might also have encouraged AstraZeneca to pursue an analogue program, which might yield a whole new field of patent protection. It was said to be clear from AstraZeneca’s own documents that they were strongly motivated by a desire for extended patent protection. In this regard, Ranbaxy relied on documents discovered by AstraZeneca’s that showed that AstraZeneca assessed potential drug candidates in the Omeprazole Follow-up Project on the basis of their patentability.
512 However, I do not accept these arguments, even if established by evidence.
513 As I said in Eli Lilly [2013] FCA 214 at [677]-[678]:
Assuming the genus patents did monopolise as alleged, I do not accept that there was no incentive for another player to undertake research and try to enter the market. Nor do I accept that such a state of monopoly would mean that I cannot conclude that there was a long-felt need which led to commercial success.
I take the view that if there is a potential drug within a genus that is possibly valuable, then there is an incentive to seek to develop it. Opportunities may arise upon its development at the end of the already existing patents.
Commercial Success of NEXIUM
514 AstraZeneca relied on the commercial success of the invention as secondary evidence of its inventiveness.
515 Ranbaxy contended that AstraZeneca has not demonstrated the relevant nexus between its commercial success and the merits of the invention. On this issue: see eg Eli Lilly [2013] FCA 214 at [669].
516 Ranbaxy made the following submissions which, as far as they go, I accept. In the present case the power of the patentee, AstraZeneca, was substantial when they released the product called “NEXIUM” in Australia. Their existing product, “LOSEC” (the active pharmaceutical ingredient of which is racemic omeprazole), was the first PPI to be prescribed in Australia. LOSEC first received approval to be registered on the Australian Register of Therapeutic Goods on 8 May 1990. LOSEC was superior to previous medications for treating GORD and other gastric acid related conditions and by reason of that superiority it was a runaway success in the market.
517 Unit sales of LOSEC at mid-2002 were 3.5 million units. Australian patent number 529,654 expired in 1999. AstraZeneca then had limited patent protection for their LOSEC market in the form of the MUPS patent and various other secondary patents, but – in Ranbaxy’s submission – they also had a strategy to hold onto their large customer base of LOSEC users, notwithstanding the generic competition which began to enter the market after the expiry of the compound patent in 1999 (and which would increase on the expiry of the secondary patents at various times up until 2007). The strategy involved resolving the enantiomers of omeprazole to produce NEXIUM (which, by the Purity Patent, would provide further protection) but the further step was to then switch the LOSEC customer base to NEXIUM. The strategy was successful. LOSEC sales declined rapidly in the second half of 2002. That is because, in August 2002, NEXIUM was listed under the Pharmaceutical Benefits Scheme, and from that point AstraZeneca marketed NEXIUM at the expense of LOSEC.
518 Ranbaxy put it in these terms:
This was the pharmaceutical equivalent to selling a new version of Windows: the customer base is there; its loyalty is already established; small changes will reap massive commercial benefits. That is exactly what happened.
519 Mr Bull gave this evidence:
MR RYAN SC: So you weren’t just expecting and trying to get new patients onto NEXIUM; you were expecting and trying and succeeding in getting existing LOSEC patients to switch to NEXIUM?
MR BULL: I think both those dynamics are at play, clearly.
…
MR RYAN SC: What I’m suggesting to you is that the most significant reason for that decrease [in sales of LOSEC] is that patients who would otherwise have taken LOSEC began to take NEXIUM?
MR BULL: I agree. I think that is an element of the overall dynamic of NEXIUM growth.
520 I also accept that the campaign conducted by AstraZeneca to effect the switch is exemplified by the large, glossy advertisements published in the Australian Family Physician, which directly pitted NEXIUM against omeprazole. The campaign was backed up by sales representatives visiting physicians.
521 Ranbaxy, therefore, contended that the success and acceptance of NEXIUM does not support the conclusion that the invention claimed by the Purity Patent is not obvious.
522 Whilst accepting the many matters submitted by Ranbaxy, I do not accept this conclusion.
523 NEXIUM has been a very successful product for AstraZeneca. So much is not in contention. However, I find that this success is primarily due to the claimed therapeutic advantages of the drug. AstraZeneca submitted that this supports the conclusion that the Purity Patent involved an inventive step.
524 The parties’ gastroenterologists agreed that NEXIUM represents a significant advance over omeprazole, which was in itself considered to be a revolutionary drug in its class. In particular, Professor Dent and Dr Prichard agreed that:
(a) NEXIUM provides improved bioavailability in patients as compared to omeprazole;
(b) a 20 mg dose of NEXIUM provides almost double the bioavailability offered by a 20 mg dose of omeprazole;
(c) a 40 mg dose of NEXIUM offers an AUC that is up to five times greater than that offered by a 20 mg dose of omeprazole;
(d) NEXIUM provides more effective suppression of gastric acid secretion than approved healing doses of omeprazole;
(e) NEXIUM provides superior healing rates for erosive oesophagitis as compared to omeprazole;
(f) NEXIUM provides a higher degree of complete resolution of symptoms of heartburn as compared to omeprazole;
(g) NEXIUM provides improved healing of severe (Los Angeles grade C and D) erosive oesophagitis as compared to omeprazole;
(h) NEXIUM provides less interpatient variability in bioavailability and gastric acid suppression than omeprazole;
(i) NEXIUM provides a significantly more effective treatment, and is indeed the best possible treatment, for reflux oesophagitis patients presenting with severe erosive gastro-oesophageal reflux disease symptoms for whom conventional therapy (such as other PPIs at their recommended approved initial healing doses) simply does not work effectively enough to give complete symptomatic relief; and
(j) for those patients with severe reflux oesophagitis who are healed by treatment with NEXIUM, the alleviation of their symptoms represents a significant improvement in their quality of life.
525 However, Ranbaxy contended that the commercial success of NEXIUM was caused primarily by marketing activity rather than as a result of NEXIUM’s clinical superiority over its competitors.
526 In my view, the answer to the submissions of Ranbaxy was given by Mr Bull, in the context of the evidence of Dr Prichard and Professor Dent as to the attributes of NEXIUM (as set out above). Mr Bull was AstraZeneca Pty Ltd’s Director of Primary Care (Gastrointestinal, Rheumatology, Cardiovascular and Respiratory Products) at the relevant time. He gave evidence that although the advertising of NEXIUM was intended to increase prescribing of the drug by clinicians, it ultimately could not have such an effect unless the clinicians were persuaded of the clinical benefit. The advertising served the purpose of gaining the attention of the clinician and communicating that clinical benefit. Mr Bull said that in the absence of a clinical benefit, “showing an image of a football player or a conductor would [not] have any hope of selling the drug to a GP”.
527 So, even accepting the advertising campaign, and other issues raised by Ranbaxy, the continued success of NEXIUM can properly be attributed to the clinical benefit. I appreciate that Mr Bull made comparisons of different amounts of NEXIUM and omeprazole. Nevertheless, I accept that NEXIUM had a margin of superiority over omeprazole, even if only in patients suffering from severe reflux. In addition, as I have indicated, the experts agreed as to the advance of NEXIUM over omeprazole.
528 In these circumstances, it is proper to conclude that the commercial success of NEXIUM can be attributed to its clinical superiority, and not just by the advertisements it used to communicate that message to prescribers or by any other reason. Undoubtedly the initial springboard into the market was primarily driven by the advertising. However, in light of the evidence of the experts, I readily infer that clinicians must be assumed to have continued to use NEXIUM due to clinical benefit.
Manner of manufacture
529 It was acknowledged by Senior Counsel for Ranbaxy that if Ranbaxy failed on their other invalidity arguments, they could not independently succeed in their case concerning manner of manufacture. Accordingly, in light of the conclusions I have reached on Ranbaxy’s invalidity case, I find it unnecessary to consider the arguments on manner of manufacture.
Purity Patent – Conclusion
530 For the foregoing reasons, the Purity Patent is valid.
THE MUPS PATENT
531 I now turn to the MUPS Patent.
532 Ranbaxy admitted infringement of certain claims of the MUPS Patent, but pleaded that the Ranbaxy Product does not have a core material “comprising alkaline compounds”.
533 The relevant claims of the MUPS Patent refer to core material “optionally” comprising alkaline compounds. It would seem on this basis that this integer is not essential to the relevant claims.
534 In any event, during the hearing and in written submissions, Ranbaxy did not raise this issue (or any other) as being relevant to infringement. Accordingly, I proceed on the basis that the admissions on infringement stand, and the focus of the dispute between the parties was on the validity of the MUPS Patent.
Introduction
535 The specification of the MUPS Patent is entitled “Multiple unit tableted dosage form I”. The invention is directed to pharmaceutical preparations in the form of a multiunit tableted dosage form comprising omeprazole or one of its enantiomers or alkaline salts thereof. A multiunit tableted dosage form is a dosage form with multiple units formulated into a tablet.
536 It is convenient to commence by setting out the matters of common general knowledge that are not in dispute, as this serves as a useful primer of the key concepts relevant to the MUPS Patent. Some of these matters have already been referred to in the context of discussing the Purity Patent.
537 I note that most of these principles are taken from the evidence of Professor Bodmeier.
Pharmaceutical formulation
538 Active substances (also called drug substances, drugs or active pharmaceutical ingredients (which, it will be recalled, are also referred to as ‘APIs’)) in medications must generally be formulated with other ingredients (constituting a drug delivery system) in order to achieve a safe, efficacious and reliable drug product. Each active substance presents unique pharmaceutical formulation challenges. For example, even structurally similar related compounds, such as erythromycin and clarithromycin, behave differently and therefore require different formulations.
539 There are many different routes of administration available to deliver an active substance to a patient (eg oral, parenteral, topical or ophthalmic). The choice of method of delivery is significantly influenced by the bulk properties, physicochemical properties, stability properties and pharmacokinetic properties of the active substance in question.
540 After the route of administration has been selected, a decision is made between many possible dosage forms for the selected route of administration. The dosage form in which the active substance is to be delivered could be solid (eg a tablet, capsule or powder for oral administration), semi-solid (eg a cream for topical application or dry powder for inhalation) or liquid (for injection, oral administration or topical application).
541 Different considerations apply in relation to the formulation and processing of the different dosage forms. Formulation considerations relate to the form, amounts and structure of product components (being the active substance(s) and excipients (being pharmacologically inactive substances)). Processing considerations relate to the development of suitable manufacturing processes to obtain the desired final dosage form, ie how the components are put together into a final dosage form.
Oral route solid dosage forms
542 The starting materials for formulating an oral solid dosage form include pure powders of the active substance or small agglomerates containing active substance (variously referred to as “granules”, “beads” or “pellets”), and excipients. These starting materials can be presented directly to the patient (eg powders filled into a plastic container) or they can be subject to further processing (eg powders compressed into tablets or filled into capsules).
543 A “multiple unit”, “multiparticulate” or “multiunit” (these terms are used interchangeably) oral dosage form is made from multiple units, which are designed to act as individual units in the body. The individual units may be formulated into capsules, oral sachets (eg for dispersing in water before being consumed) or tablets.
544 The formulator must choose excipients to enable the active substance to be formulated into an administrable dosage form. The formulator must also choose the method of processing the active substance and excipients into the desired form. The processing method will vary depending upon the dosage form. Processing can involve numerous steps, each of which can affect the efficacy, safety, and reliability of the drug product.
Formulation of pellets and single-unit tablets
545 A single-unit tablet is made by compressing the active substance and tablet excipients into a compact shape, which may or may not then be coated. In order to make a tablet compact, high compression forces must generally be used.
546 Pellets may be made by extruding a mixture of the active substance and excipients through an extruder (making spaghetti-like strings) and then spheronizing the extruded mass into spheres. Alternatively, pellets can be made by layering the active substance, together with excipients, onto spherical seeds, eg sugar spheres.
547 The formulation of pellets and single-unit tablets involves the use of various classes of excipients. The classes are defined by the function that the excipients perform. Excipients can perform more than one function.
Coating of single-unit tablets and pellets
548 Single-unit tablets and pellets may be coated. Common objectives of coating are to modify the release of the active substance, protect the active substance from part of the gastrointestinal tract (for example, stomach acid), or protect part of the gastrointestinal tract from the active substance.
549 The types of materials used to coat single-unit tablets and pellets include film-forming or coating polymers, plasticizers, anti-tacking agents, colorants, opacifiers and solvents. Each coating polymer has different properties and, therefore, requires different coating formulations and processes.
550 Due to the complexity of the interactions between the coating and the single-unit tablet or pellet being coated, problems can arise during the coating process. For example, the coating solution may dry before it reaches the tablets or pellets (called “spray drying”), or the tablets or pellets may be over-wet, causing them to stick together. Variations in formulation or process parameters can cause film defects. These defects include sticking and picking, roughness, peeling, chipping and cracking.
Enteric coating of single-unit tablets and pellets
551 A delayed release dosage form is a dosage form in which release of the active substance is delayed, rather than released immediately in the stomach. This is to be distinguished from a sustained release formulation, which releases the active substance gradually. An enteric coating is one type of delayed release system, which can be applied to single-unit tablets and to pellets. An enteric coating delays the release of the active substance until after it has passed through the stomach. The enteric coating does not dissolve in the acidic gastric fluid (which has a pH of 1 to 3.5), but dissolves further down the gastrointestinal tract, eg in the small intestine so that the drug is released and can be absorbed.
552 One standard in vitro test for enteric coated formulations is set out in Edition 23 of the United States Pharmacopeia (‘USP’). This is an in vitro measure of acid resistance and dissolution rate. In the acid stage of the USP test, an enteric coated dosage form is placed in an acidic medium, 0.1 N HCl (to simulate the pH of the stomach), for two hours. The amount of active substance released into this acidic medium must not be more than 10%. In the buffer stage of the test, the pH of the fluid is then raised to pH 6.8 (to simulate the pH of the small intestine). After raising the pH level, in general, at least 75% of the active substance must be released within 45 minutes.
553 Figure 1 below illustrates the hypothetical release profile of an enteric coated dosage form.
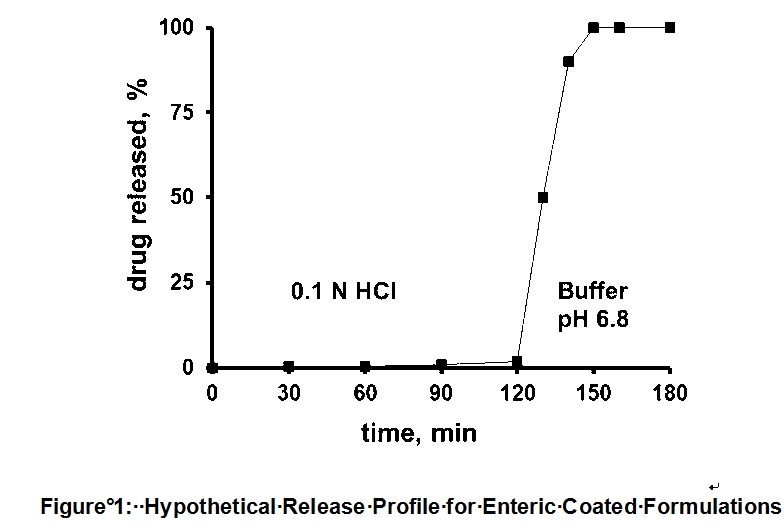
Multiparticulate tablets
554 “Multiparticulates” are multiunit dosage forms that consist of numerous particles (pellets, beads or granules). Multiparticulates can be made into capsules, oral sachets and tablets. In tablet form, the excipient material surrounding the pellets is generally referred to as the “tablet matrix”. Different considerations apply to single-unit as opposed to multiunit tablets.
Components available for use in enteric coatings
555 The components available for use in enteric coatings are:
(a) enteric coating material (polymer or shellac);
(b) plasticizers;
(c) anti-tacking agents;
(d) solvents (or water);
(e) opacifiers; and
(f) colourants.
Tableting excipients available for use by the formulator
556 The tableting excipients available for use by the formulator are:
(a) diluents or fillers, which add bulk to the tablet and ideally enhance compressibility;
(b) binders, which improve the flow of powders via granulation processes, and give strength to the tablet;
(c) disintegrants, which promote disintegration of the tablet after intake, generally in the stomach;
(d) lubricants, which reduce friction between the tablet and the die wall upon ejection of the tablet from the die cavity during tableting; and
(e) glidants, which improve the flow of powders or granules during tableting.
557 In the Joint Expert Report on AU 966, Professor Bodmeier, Dr Morella and Dr Reiland broadly agreed with these components and their functions, although Dr Reiland referred to “cushioning agents” rather than “fillers” (and occasionally sought to define the function of some excipients slightly differently to the other experts). But I consider that little if anything turns on this.
558 At the priority date omeprazole and the capsule form in which it was sold were well known. The solid oral dosage form LOSEC was on the market in Australia. This consisted of enteric coated pellets containing the active ingredient filled in a capsule. The pellets were coated with an enteric coating to protect the active ingredient from degradation in the acid environment of the stomach until it reached the intestine. The recommended dose was 20 mg once daily.
559 However, there is no evidence that would demonstrate that a formulator would have knowledge of the properties of omeprazole as at the priority date.
The formulator’s knowledge of omeprazole
560 Question 5(a) of the Joint Expert Report on AU 966 asked the experts to comment on what was known in relation to omeprazole and solid oral dose formulations of omeprazole and their components as at 8 July 1994. Professor Bodmeier responded that he had heard of omeprazole but was not aware of its properties or dosage form. Dr Morella responded that he cannot recall being aware of omeprazole or formulations thereof. In oral evidence, Dr Reiland said that he did not recall whether he was aware of omeprazole as at 8 July 1994.
561 Dr Reiland nevertheless asserted, in responding to question 5(a), that certain matters about omeprazole were known to “someone skilled in the art”. In his first affidavit, Dr Reiland referred to reviewing the entries for omeprazole in Goodman & Gilman, The Merck Index and the PDR to ascertain “what is known about the API omeprazole”. AstraZeneca submitted – and I accept – that information of this nature cannot be treated as part of the common general knowledge unless there is evidence of its general acceptance and assimilation by persons skilled in the art.
562 I do accept that the compound omeprazole and its acid labile character were known to the skilled addressee at the priority date. However, beyond that, I find that Ranbaxy has failed to prove that any other matters relating to omeprazole and solid oral dose formulations of omeprazole and their components were part of the common general knowledge of the skilled addressee as at 8 July 1994.
563 As for the common general knowledge in relation to the tableting of multiunit enteric coated pellets, I accept that the enteric coated pellets were known to have advantages over the use of an enteric coated whole tablet, because they could be expected to pass from the stomach into the intestine to release the active ingredient in a shorter period of time and in a multitude of small units.
Background to the invention
564 The MUPS Patent states at p 1, lines 22 to p 2, line 1, that omeprazole or one of its single enantiomers or alkaline salts thereof (referred to collectively herein as “omeprazole” in the context of the MUPS Patent) is useful for inhibiting gastric acid secretion in mammals and man, and is thus useful for the prevention and treatment of gastric acid related diseases in mammals and man.
565 At p 2, lines 7 to 11, it is recorded that omeprazole is “susceptible to degradation / transformation in acidic and neutral media” and is “stabilized in mixtures with alkaline compounds”.
566 It follows, it is said, at lines 14 to 18, that “it is obvious that omeprazole in an oral solid dosage form must be protected from contact with the acidic gastric juice and the active substance must be transferred in intact form to that part of the gastrointestinal tract where pH is near neutral and where rapid absorption of omeprazole can occur”.
567 The specification records that a pharmaceutical dosage form of omeprazole is best protected from contact with acidic gastric juice by an enteric coating layer: see p 2, lines 20 to 21. The specification states, at lines 21 to 24:
In US-A 4,786,505 such an enteric coated omeprazole preparation is described. Said omeprazole preparation contains an alkaline core comprising omeprazole, a separating layer and an enteric coating layer.
568 At p 2, lines 27 to 28, reference is made to the AstraZeneca commercial product, described as “hard gelatine capsules containing an enteric coated pellet formulation of omeprazole”.
569 The specification then refers to the desirability of a tablet containing enteric coating layered multiunit preparations by reference to the advantages of suitability for blister packaging, divisibility, dispersibility (which I understand to mean dispersibility in liquid for ease of administration), and dispersibility in the stomach into a multitude of small units. It states at p 2, line 27 to p 3, line 9:
The hard gelatine capsules containing an enteric coated pellet formulation of omeprazole marketed by the Applicant today, are not suitable for press-through blister packages. Thus, there has been a demand for development of new enteric coating layered multiple unit preparations of omeprazole with good chemical stability as well as improved mechanical stability making it possible to produce well functioning and patient-friendly packages. Furthermore, there is a demand for omeprazole formulations having improved patient acceptance, such as divisible and/or dispersible tablets.
An improved mechanical stability can be obtained with the enteric coating layered tablet …. However, only an enteric coating layered multiple units tablet can be made divisible and dispersible. A further advantage of a multiple unit dosage form is that it disperses into a multitude of small units in the stomach upon administration.
570 At p 3, lines 11 to 25, the specification refers to the prior art, which it states “discloses many different types of multiple unit dosage forms”. It states that:
Typically, the multiple unit formulation may be a tablet which disintegrates in the stomach to make available a multitude of coated units, or pellets filled in a capsule. (See for example EP 0 080 341 and US-A 4,786,505).
571 These examples of prior art are referred to in these reasons for judgment as ‘EP 341’ and ‘US 505’, respectively.
572 EP 341 is an example of a controlled release formulation (that is, one that controls the gradual release of the active ingredient over a period of time). US 505 is an example of a delayed release formulation.
573 US 505 describes at column 1, lines 6 to 7, a “new stable pharmaceutical preparation containing omeprazole”. The final dosage form of that preparation includes, “in the case of enteric coated pellets... pellets formulated into tablets” (column 5, lines 61 to 63).
574 At p 3, lines 18 to 25, the specification refers to another example of a controlled released dosage form. The specification states that the mechanical properties of “such multiple units formulated into tablets are reported in Pharmaceutical Research, 10 (1993), p.S-274”. This is a publication by AstraZeneca (then known as Astra Hässle AB). It deals with both controlled and delayed release formulations. This publication states that, inter alia:
the introduction of fast disintegrating tablets containing coated pellets meets the demand for tablets, designed to release the drug at some finite time or rate after administration. The coating of these pellets provides a physical protection of the pellet core which must remain intact and have suitable mechanical properties in order to be resistant to fragmentation during compaction of the tablet...
The composition of the polymeric membrane was the most important factor and fragmentation of the pellets increased with increasing film hardness. A vickershardness [sic] of 6-8 gave pellet films with a minimum of fragmentation during compaction.
575 At p 3, line 27 to p 4, line 5, the specification of the MUPS Patent continues:
Even if the specification of US-A 4,786,505 under the subtitle Final dosage form mentions that the manufactured pellets may be formulated into tablets there are no examples describing any compositions of such a tablet formulation or a technique to manufacture such a formulation. In practice, problems arise when enteric coating layered pellets containing acidic susceptible benzimidazoles as active substance are compressed into tablets. If the enteric coating layer does not withstand the compression of the pellets into a tablet the susceptible active substance will be destroyed by penetrating acidic gastric juice, i.e. the acid resistance of the enteric coating layer of the pellets will not be sufficient in the tablet after compression.
576 At p 4, lines 8 to 22, the specification describes another publication, being “Fast Disintegrating Controlled Release Tablets from Coated Particles” authored by Mr K Lehmann et al (published in Drugs Made In Germany 37(2) (1994) 53, and referred to by the parties in this proceeding as the “Drugs Made in Germany” article). This publication concerns a suitable combination of coating polymers for “enteric coated particles compressed into tablets”. The active ingredient (indomethacin) is not identified in the specification. The specification continues, at lines 13 to 15, to the effect that this combination of coating polymers is not suitable:
when formulating multiple unit tableted dosage forms of the acidic susceptible substance omeprazole. The acid resistance of the pellets compressed into a tablet is too low.
577 It then claims to have found that pellets coated in one of the polymers used in that publication, a methacrylic acid copolymer, could be satisfactorily compressed into a tablet.
578 The specification then states, at p 4, lines 24 to 26:
The Applicant is not aware of any working example in the prior art of a multiple unit tableted dosage form comprising an acidic susceptible benzimidazole compound, such as omeprazole.
The invention described in the specification of the MUPS Patent
579 The invention is identified at p 4, line 28 to p 5, line 5 of the specification:
The Applicant has now surprisingly found that tablets according to the present invention comprising enteric coating layered units containing an acidic susceptible substance in the form of omeprazole or one of its single enantiomers or an alkaline salt thereof can be manufactured by compressing said units into tablets without significantly affecting the properties of the enteric coating layer. As explained above, if the enteric coating layer is damaged during compression of the enteric coating layered units, the acid resistance of said enteric coating layer in the manufactured tablet will not be sufficient, and the manufactured tablets will not fulfil standard requirements on enteric coated articles, such as those defined in the United States Pharmacopeia, Edition 23...
580 The invention is said to be to provide a multiunit tableted dosage form comprising omeprazole, in which the active substance is in the form of individually enteric coating layered units compressed into a tablet: p 5, lines 10 to 14. The specification states, at p 5, lines 14 to 17:
The enteric coating layer(s) covering the individual units of active substance has properties such that the compression of the units into a tablet does not significantly affect the acid resistance of the individually enteric coating layered units.
581 The other objects of the invention are said to be to provide such a multiunit tableted dosage form which is suitable for press-through blister packages and which also has an improved patient acceptance (p 5, lines 21 to 25), and which is also divisible and easy to handle (p 5, lines 29 to 30).
582 A detailed description of the invention commences at p 6A, line 20. It is said that the “novel multiple unit tableted dosage form comprising omeprazole” is characterised in that individually enteric coating layered units containing omeprazole, and optionally alkaline substances, are mixed with tablet excipients and compressed into multiunit tableted dosage forms. The specification continues, at p 6A, line 29 to p 7, line 4:
The compaction process (compression) for formulating the multiple unit tableted dosage form must not significantly affect the acid resistance of the enteric coating layered pellets. In other words the mechanical properties, such as flexibility and hardness as well as the thickness, of the enteric coating layer(s) generally meet the requirements on enteric coated articles in the United States Pharmacopeia, Edition 23, with the acid resistance preferably not decreasing more than 10% during the compression of pellets into tablets.
583 It is then said, at p 7, lines 5 to 9:
The flexibility/hardness of enteric coating layers can be characterized for instance as Vickers hardness measured with a Shimadzu micro hardness indentation tester type HMV 2 000. The present enteric coating layer(s) preferably has a Vickers hardness value of less than 8.
584 The core material for the individually enteric coating layered pellets is described at p 7, line 20 to p 9, line 29. Seeds layered with omeprazole, optionally mixed with alkaline reacting compounds, can be used as the core material.
585 The specification then states that separating layer(s) can be applied to the core material to separate the core material from the enteric coating layer. Such separating layer(s) are described at p 10, line 1 to p 11, line 11. Enteric coating layers generally comprise compounds which contain acidic groups. A separating layer acts to prevent contact between an acid labile compound like omeprazole and those acidic groups, which could lead to degradation of the omeprazole.
586 The specification states that one or more enteric coating layer(s) are applied “onto the core material or onto the core material covered with separating layer(s)”: p 11, lines 13 to 14. The specification gives examples of typical enteric coating polymers: p 11, lines 17 to 22.
587 At p 11, lines 24 to 26, the specification states:
The enteric coating layers contain pharmaceutically acceptable plasticizers to obtain the desired mechanical properties, such as flexibility and hardness of the enteric coating layers.
588 The specification then gives examples of typical plasticizers: lines 26 to 28. The specification continues, at line 30 to p 12, line 11:
The amount of plasticizer is optimized for each enteric coating layer formula, in relation to selected enteric coating layer polymer(s), selected plasticizer(s) and the applied amount of said polymer(s), in such a way that the mechanical properties, i.e. flexibility and hardness of the enteric coating layer(s), for instance exemplified as Vickers hardness, are adjusted so that the acid resistance of the pellets covered with enteric coating layer(s) does not decrease significantly during the compression of pellets into tablets. The amount of plasticizer is usually above 10% by weight of the enteric coating layer polymer(s), preferably 15 - 50%, and more preferably 20 - 50% .... Other compounds may be added to increase film thickness and to decrease diffusion of acidic gastric juices into the acid susceptible material.
589 At p 12, lines 13 to 19, it is said:
To protect an acidic susceptible substance in the form of omeprazole... and to obtain an acceptable acid resistance of the multiple unit tableted dosage form according to the invention, the enteric coating layer(s) constitutes a thickness preferably of approximately at least 10 µm, more preferably exceeding 20 µm. The maximum thickness of the applied enteric coating layer(s) is normally only limited by processing conditions.
590 The over-coating layer(s) which can be applied to the enteric coated pellets are described at p 12, line 24 to p 13, line 9.
591 Enteric coated pellets (with or without an over-coating layer) are then mixed with tablet excipients and compressed into a multiunit tableted dosage form: p 13, lines 14 to 29.
592 At p 14, lines 1 to 2, the specification states:
The mechanical properties, i.e. the flexibility and hardness of the enteric coating layer are essential for the acid resistance of the multiple unit tableted dosage form.
593 The specification says that the “flexibility / hardness of the enteric coating layer surface” may be characterised as a “preliminary process parameter” in the form of Vickers hardness: p 14, lines 3 to 5. The specification continues, at lines 8 to 14:
The ability of the enteric coating layer(s) to withstand compression into tablets is, of course, a function of both the amount of applied coating layer and the mechanical properties of said coating material. To obtain well functioning enteric coating layered pellets with a reasonable amount of enteric coating layer material and which pellets can be compressed into tablets without significantly affecting the acid resistance, an enteric coating layer surface with a Vickers hardness of less than 8 is preferred.
594 At p 15, lines 8 to 9, it is said the process for the manufacture of the multiunit tableted dosage form represents a further aspect of the invention.
595 The invention is illustrated by Examples 1 to 17 (pp 16 to 30). In each case the enteric coating layer comprises a standard polymer or copolymer and standard plasticizers. In each case the standard cushioning excipient, microcrystalline cellulose, is used as a tableting excipient. Three Reference Examples (at pp 31 to 33) are said to show that the enteric coating layer of three pellets (two from marketed capsule products and one described in the “Drugs Made in Germany” article) do not possess the mechanical properties required to withstand compression of pellets into tablets.
596 The preparation of omeprazole and its salts, and salts of single enantiomers of omeprazole, is described at pp 34 to 36.
597 The specification concludes at p 37 with 38 claims.
598 Claim 1 is an independent claim. It provides as follows:
1. An oral pharmaceutical multiple unit tableted dosage form suitable for use in therapy, in particular for the treatment and prevention of gastric acid related diseases in mammals (including man), comprising pharmaceutically acceptable tablet excipients and individually enteric coating layered units of a core material containing active substance in the form of omeprazole or one of its single enantiomers or an alkaline salt of omeprazole or one of its single enantiomers, the core material being covered with one or more enteric coating layers and optionally comprising alkaline compounds and optionally covered with a separating layer situated between the core material and the enteric coating layer(s), characterized in that at least one of said enteric coating layers is a polymeric enteric coating layer comprising an effective amount of a plasticizing compound, and that the enteric coating layer(s) has mechanical properties which preclude significantly adverse affect essentially on the gastric acid resistance of the individually enteric coating layered units upon the compression of those individual units mixed with said tablet excipients, into the multiple unit tableted dosage form.
599 There are then a series of dependent claims, which I address later in the reasons.
600 AstraZeneca alleges infringement of claims 1 to 7, 10, 12 and 14 to 28 of the MUPS Patent. As I have already indicated, Ranbaxy has made certain admissions as to infringement.
601 Ranbaxy seeks to revoke those claims, and also claim 13, on the grounds of lack of novelty, lack of inventive step and lack of manner of manufacture. Ranbaxy also contended on the basis of additional particulars that claim 10 is invalid, on the grounds of lack of inventive step, lack of manner of manufacture and lack of definition.
The person skilled in the art
602 It is necessary to address the notional person skilled in the art as this concept applies to the MUPS Patent.
603 It appears to be common ground that the hypothetical person to whom the MUPS Patent specification is addressed is a pharmaceutical formulation scientist working in the field of solid oral dosage formulation and design.
604 Ranbaxy’s expert was Dr Reiland as a formulation scientist with experience in this role in a pharmaceutical company. AstraZeneca’s witnesses were Dr Morella as a formulation scientist with experience in this role in a pharmaceutical company, and Professor Bodmeier as an academic who has experience interacting with the pharmaceutical industry, but has not worked on a day to day basis in a pharmaceutical company.
Ranbaxy’s criticisms of AstraZeneca’s witnesses
605 I note that in particular, Ranbaxy suggested that Professor Bodmeier lacked the requisite industry experience. I do not accept this suggestion. Professor Bodmeier established significant exposure to industry in his affidavits. Professor Bodmeier also exhibited a strong technical understanding of all aspects of formulation. The following exchange that occurred during the concurrent evidence session clearly indicates that Professor Bodmeier has worked closely with industry on developing formulations:
MR MACAW: How many occasions did you consult on an actual project before 1994 which involved the preparation of enteric-coated products?
PROF BODMEIER: We have worked, for example, with a company called Moreflex, which is the leading company on plasticisers. We have worked with FMC and with Rhom Pharma, which is the supplier of enteric polymers and we have worked with pharmaceutical companies on coating of dosage forms with enteric and non-enteric polymers.
MR MACAW: When you say, “We,” what do you mean?
PROF BODMEIER: Myself and my research group or my research group and myself.
MR MACAW: Yes. Had they called you in to participate in the formulation or to solve problems that had been encountered in the course of their own formulation?
PROF BODMEIER: Both.
MR MACAW: So you actually participated, did you, in the construction of the formulation?
PROF BODMEIER: Yes.
MR MACAW: On three occasions?
PROF BODMEIER: What do you mean “On three occasions”?
MR MACAW: Well, you gave three examples, I think.
PROF BODMEIER: I said I worked with companies which made the polymers, with companies which make the plasticisers and then with several pharmaceutical companies which make that product.
606 In addition, Professor Bodmeier gave evidence that he was “quite familiar with issues in pharmaceutical companies”.
607 In fact, both Professor Bodmeier and Dr Morella provided evidence that was technically detailed and generally consistent. They are both highly qualified, and have had substantial involvement with polymer coated dosage forms.
608 In contrast, I do have some concerns in respect of Dr Reiland’s evidence. I perceive that Dr Reiland’s evidence has been affected by hindsight.
609 One example in particular was relied upon by AstraZeneca in their submissions to this effect. In his first affidavit, Dr Reiland described the development of Abbott Laboratories’ PCE 333 product (which will be described further in due course). In doing so, he specifically noted the use of excipients to bind and cushion the particles during compression, “so that the particles held together and the enteric coating did not crack”. However, in the concurrent evidence session, Dr Reiland agreed that, in listing the relevant features of PCE 333 in his first affidavit, he made no mention of Abbott Laboratories adjusting the amount of plasticizer or the thickness of the enteric coating to ensure that the particles did not crack on compression (but on which he later sought to rely, both in subsequent affidavit evidence and during the concurrent evidence session).
610 This evidence in his first affidavit was given before he saw the MUPS Patent. However, later in his first affidavit (after he had seen the MUPS Patent), Dr Reiland stated that:
Indeed, this same formulation approach (i.e. enteric coated particles compressed into a tablet and the use of a plasticizer in the enteric coating of the particles) was used in the development of Abbott’s PCE product in the mid-1980s.
611 Just before the trial, in the Joint Expert Report on AU 966, Dr Reiland responded to question 20(f) (which asked whether US 505 would have caused the skilled addressee to vary the amount of film-forming agent in any applied coating formulations at the priority date):
Definitely would consider as varying the amount of film-forming agent along with sufficient plasticisers was key to making the original PCE 333 able to maintain sufficient acid resistance …
(emphasis added)
612 AstraZeneca submitted that Dr Reiland’s understanding of the development of Abbott Laboratories’ PCE products was coloured by his reading of the MUPS Patent. Before seeing the MUPS Patent, Dr Reiland saw the binder and cushioning agent as key to preventing the enteric coated particles from cracking in the PCE 333 product. I accept that this interpretation is supported by the patents covering the PCE 333 product (which make no mention of modifying the quantity of plasticizer or the thickness of the enteric coating, and to which I return later), and the lack of any documentary evidence showing that Abbott Laboratories had experimented with increased plasticizer or the thickness of the enteric coating in making the PCE products. Further, I was not persuaded by his explanation for the absence of any mention of the plasticizing of the enteric coating in the patents relating to the PCE products (his explanation being that the failure to disclose a feature that he claimed was critical to the formulation working was based on patent strategy, and the fact that it was a trade secret of Abbott Laboratories). Accordingly, I decline to place much weight on Dr Reiland’s evidence about the Abbott Laboratories’ PCE products and the conclusions he draws from that experience.
Construction
613 A number of issues of construction of the MUPS Patent arose for determination by the Court. Before going to these issues specifically, I make the following general observations.
614 The MUPS Patent achieves its objects by a particular combination of integers, which characteristically include at least one enteric coating layer having an effective amount of a plasticizing compound, and mechanical properties that mean the units can be compressed into a tablet without compromising their gastric acid resistance. The MUPS Patent teaches that the compressibility of the pellets arises from the use of an effective amount of a plasticizing compound and the enteric coating layer(s) with specific mechanical properties, rather than any excipients external to the pellets (including microcrystalline cellulose). Example 7 of the MUPS Patent uses calcium phosphate anhydrous which is crystalline and does not provide cushioning on compression. These pellets showed good acid resistance after compression (Table 1 of the MUPS Patent). Reference Example II uses lansoprazole with 644 g of microcrystalline cellulose, but does not have the modified enteric coating taught by the Patent. It shows very poor (25%) acid resistance (Table 2).
615 All of these characteristic features are interrelated. Page 11, line 30 to p 12, line 5 of the MUPS Patent discloses that the amount of plasticizer in the enteric coating layer(s) is optimised in relation to:
(a) the selected enteric coating layer polymer(s);
(a) the selected plasticizer(s); and
(b) the applied amount of said polymer(s);
in such a way that the mechanical properties are adjusted so that the acid resistance of the pellets covered with the enteric coating layer(s) does not decrease significantly during the compression of pellets into tablets.
616 As to the specific issue of construction, there remains a dispute between the parties as to the proper construction of the integers “the mechanical properties” and the “gastric acid resistance”. Ultimately, the dispute between the parties in respect of the integer “effective amount of a plasticizing compound” appears to have been resolved during the concurrent evidence session. Nevertheless, I will address the concept of “effective amount of a plasticizing compound” because it is relevant to the interpretation of the term “mechanical properties”.
617 I now turn to the specific issues of construction.
Claim 1
“Effective Amount of a Plasticizing Compound”
618 The expert witnesses for the parties agreed that an “effective amount of a plasticizing compound” means an amount effective to provide the mechanical properties required to achieve the desired purpose of the MUPS Patent (namely, acid resistance) in the final dosage form, without compromising the performance of the formulated product in therapy (eg by its effect on stability, rate of dissolution etc).
619 Claim 1 does not prescribe any numerical limits as to an “effective amount of a plasticizing compound”. The skilled addressee would understand that this is because the amount required will depend on a number of matters that may be adjusted in the optimisation process, including the identity and amount of applied polymer(s), the identity of the plasticizer and the amount of cushioning excipients. As “effective amount” is not specifically delineated in claim 1 with any numerical limit, it is not linked to a “high” or “extraordinarily high” amount of plasticizer. This can be contrasted with claim 8 (wherein the amount of a plasticizing compound is expressed to be more than 20% and less than 50%). That claim is not said to be infringed by the Ranbaxy Product.
620 Issues still arise between the parties as to whether the use of an effective amount of plasticizing compound was a standard way to work with polymers and plasticizers, and the range of plasticizer that would be understood by the skilled addressee to be “effective”. These issues are relevant to the inventive step inquiry, and will be addressed in due course in the context of the common general knowledge of the skilled formulator in relation to the use of plasticizers.
“Mechanical Properties”
621 There is a dispute as to the meaning of “mechanical properties” in claim 1. The ordinary meaning of “mechanical properties” relates to properties involving mechanical or movement-related measurement. The experts agreed that the ordinary meaning of “mechanical properties” includes flexibility and hardness. The experts also agreed that as a general principle, thickness is not a mechanical property in accordance with the ordinary meaning of that term.
622 The specification uses the term “mechanical properties” in a number of contexts:
(a) page 11, lines 25 to 26 (“...the desired mechanical properties, such as flexibility and hardness of the enteric coating layers”);
(b) page 12, lines 1 to 2 (“the mechanical properties, i.e. flexibility and hardness of the enteric coating layer(s)”);
(c) page 12, lines 1 to 11, which draws a distinction between “film thickness” and the “mechanical properties” of the enteric coating layer;
(d) page 14, lines 1 to 2 (“the mechanical properties, i.e. the flexibility and hardness of the enteric coating layer are essential for the acid resistance”); and
(e) page 14, lines 9 to 10, which distinguishes between “the amount of applied coating layer and the mechanical properties of said coating material”. I accept that the amount of applied coating layer is an indirect reference to its thickness.
623 AstraZeneca contended that “mechanical properties” as it is used in the MUPS Patent includes thickness. To this end, AstraZeneca relied on p 6A, lines 31 to 32 of the specification (“mechanical properties, such as the flexibility and hardness as well as the thickness, of the enteric coating layer(s)”). In my view, that passage is entirely consistent with the passages describing the mechanical properties as flexibility and hardness, with thickness being a different physical property.
624 However, I do not consider that the difference between the parties on this construction issue impacts on the ultimate determination of this proceeding. Based upon the evidence, whether the Court construes thickness as part of the “mechanical properties” or not, the MUPS Patent teaches the skilled formulator that the thickness of the enteric coating layer(s), an effective amount of a plasticizing compound and the mechanical properties are ultimately all interrelated. Ranbaxy acknowledged this in their opening submissions, saying: “[t]rue it is [thickness] might affect the acid resistance of the product, because the more you put on, the less chance of it not passing the acid resistance test”. As explained by Professor Bodmeier in the concurrent evidence session:
Within the context of the patent thickness has a very important contribution to the mechanical properties of the enteric-coating and as you can ..... it should make sense the thicker the coating more molecularly stronger the coating is, and that’s why I think the two components discussed in the patent: one, the plasticiser, which we have discussed already, which affects the flexibility and the hardness and the second point is the thickness of the coating which also contributes significantly to the mechanical strengths of the coating to resist fracture during compression. And if you look at the examples in the patent and they summarise the amounts of coating polymer, the amount of coating polymer applied transfers into thickness. So the more amount you apply the thicker the coating, and these amounts go up to 100 per cent and this is far beyond what is normally used.
625 It is not in dispute that the mechanical properties of the enteric coating layer(s) include its flexibility and its hardness. Professor Bodmeier explained that the MUPS Patent discloses that the type of polymer, the type of plasticizer and the amounts of the plasticizer and the coating formulation that are applied all interact to produce the properties of the final coating layer. Professor Bodmeier gave evidence that:
this is in my view is the invention here that here you have an interplay of the plasticiser together with the thickness. Either one on its own would not work, and if you look at the examples you have higher than normal or usually used plasticiser levels and you have higher than usually used coating thicknesses.
626 In my view, this evidence of Professor Bodmeier is compelling, and I accept it.
627 I now turn to the phrase “gastric acid resistance”.
“Gastric Acid Resistance”
628 The MUPS Patent states:
acid resistance is defined as the amount of active substance in tablets or pellets after being exposed to simulated gastric fluid, USP, or to 0.1 M HCl(aq) relative to that of unexposed tablets or pellets, respectively.
629 The method for performing the acid resistance test is set out in the rest of the paragraph.
630 The MUPS Patent also refers to another acid resistance test, being the general test documented in the USP. Professor Bodmeier gave evidence, with which Dr Morella agreed, that (emphases in original):
The acid resistance of omeprazole is defined as the amount of active omeprazole remaining within the coated pellets after exposure to the simulated gastric fluid as set out in the USP or to 0.1 M HCI(aq). This is different from the standard USP test for acid resistance … which tests for release of the active substance. In the USP test, there could be much more active substance that, although not released, is degraded within the coated pellet and no longer active. This is because some of the simulated gastric fluid may enter into the dosage form (eg, pellet), causing unacceptable degradation of the omeprazole inside. The amount of degraded active substance would therefore not be reflected by the results of the standard USP test.
631 Dr Reiland understood the term “gastric acid resistance” in claim 1 to refer to the standard USP test for acid resistance. According to Professor Bodmeier, the standard USP test does not provide a complete picture of the acid resistance of the final dosage form of omeprazole. I accept this evidence. The standard USP test is a general guideline for formulators to use unless there is a specific monograph for the compound in question. In the case of erythromycin, the active ingredient in the PCE products, there is a specific monograph, with a different and less rigorous test.
632 Dr Reiland contended that “in the real world”, formulators will use the standard USP test. But Dr Reiland also gave evidence that if there are specific characteristics of a particular active substance that make the standard USP test not suitable, an alternative test will be developed and used. Omeprazole is such a case.
633 In my view, the MUPS Patent specifically defines a test for acid resistance. The term “gastric acid resistance” in claim 1 of the MUPS Patent and related claims therefore refers to the test for acid resistance defined at p 7 of the MUPS Patent. This is consistent with the evidence of Professor Bodmeier and Dr Morella. It is also consistent with Dr Reiland’s evidence where he accepted that, in certain circumstances, the standard USP test is not suitable (for example, where the active substances makes the USP test not suitable).
Other claims of the MUPS Patent
634 I now discuss the other relevant claims of the MUPS Patent and address the remaining construction issues.
635 Claims 2 to 16, which claim tableted dosage forms, are dependent on claim 1 (either directly or through claims dependent on claim 1), and add various limitations, as follows.
Claim 2
636 Under claim 2, the mechanical properties of the enteric coating layer(s) preclude significantly adverse effect on the degradation and dissolution of the active substance when exposed to simulated gastric acid and intestinal media respectively. The fact that these features are specifically claimed distinguishes claim 2 from claim 1. Thickness, not a mechanical property, will (as I have indicated already) impact on the mechanical strength of the enteric coating.
Claim 3
637 Under claim 3, a separating layer is situated between the core material and the enteric coating layer(s).
Claim 4
638 Under claim 4, the separating layer referred to in claim 3 comprises pharmaceutically acceptable excipients which are soluble, or insoluble but disintegrate in water, and optionally alkaline compounds.
Claim 5
639 Under claim 5, the individually enteric coating layered units are each further covered with an over-coating layer comprising pharmaceutically acceptable excipients. The over-coating layer may further prevent potential agglomeration of enteric coating layered pellets, further protect the enteric coating layer towards cracking during the compaction process and enhance the tableting process (p 13, lines 5 to 8 of the MUPS Patent).
Claim 6
640 Ranbaxy contended that claim 6 does not add anything to claim 1. Claim 6 would therefore be redundant. AstraZeneca submitted that this contention is based on a construction of “gastric acid resistance” as it appears in claim 1 as meaning passing the general USP test. As already explained, “gastric acid resistance” in claim 1 refers to passing the acid resistance test as specifically defined in the MUPS Patent. In claim 6, the additional specific limitation imposed is that the gastric acid resistance of the individually enteric coating layered units is also “in coherence” with the requirements on enteric coated articles defined in the USP, which is incorporated as a whole by reference (p 5, lines 5 to 6 of the MUPS Patent). As was explained in Professor Bodmeier’s second affidavit:
In the acid stage of the USP general drug release test, an enteric coated dosage form is placed in an acidic medium, 0.1 N HCl (to simulate the pH of the stomach), for two hours. The amount of active substance released into this acidic medium must not be more than 10%. In the buffer stage of the test, the pH of the fluid is then raised to pH 6.8 (to simulate the pH of the small intestine). After raising the pH level, in general at least 75% of the active substance must be released within 45 minutes, unless otherwise prescribed in the relevant individual monograph.
641 The words “in coherence” in claim 6 indicate that the acid resistance should “generally meet the requirements on enteric coated articles” (p 7, lines 1 to 2) in the USP test, in the sense that the acid resistance does not decrease more than 10% after two hours in an acidic medium. In any view, this additional limitation imposed by claim 6 does not change the meaning of “gastric acid resistance” in claim 1 (where it is determined according to a different test).
Claim 7
642 Similarly, Ranbaxy contended that claim 7 does not add anything to claim 1, such that it is also redundant. Claim 7 adds the specific limitation that the gastric acid resistance of the individually enteric coating layered units does not decrease more than 10% during the compression of the individual units into the multiunit tableted dosage form. Upon a proper construction, this limitation requires a comparison between the acid resistance of uncompressed and compressed pellets. The acid resistance of the enteric coating layered pellets must not decrease “more than 10% during the compression of pellets into tablets” (p 7, lines 2 to 5 of the MUPS Patent). It is therefore not correct to say that this claim does not add anything to claim 1.
Claim 8
643 Under claim 8, the amount of plasticizing compound is more than 20% and less than 50% by weight of the enteric coating layer polymer covering the individual units.
Claim 10
644 Under claim 10, the polymeric enteric coating layer covering the individual units has a thickness of at least 10 μm. Thickness is important to the invention. As explained before, mechanical properties are, in part, a function of thickness. Claim 10 sets out a particular thickness for the enteric coating layer of the invention. I will return to claim 10 later in these reasons.
Claim 12
645 Under claim 12, the active substance is an alkaline salt of (+)-omeprazole or (-)-omeprazole, preferably a magnesium salt.
Claim 13
646 Under claim 13, the dosage form is divisible.
Claim 14
647 Claim 14 was in dispute.
648 Under claim 14, the dosage form is “dispersible to a suspension of individually enteric coating layered units in an aqueous liquid”. Claim 14 is designed to answer the need for omeprazole formulations with improved patient acceptance, such as dispersible tablets. Dr Morella’s evidence was that dispersibility is an additional product performance criterion that could lead to problems with other performance criteria. Dr Morella’s evidence was that there is a distinction between “disintegration” and “dispersibility”. Dr Reiland did not draw this distinction.
649 AstraZeneca submitted that dispersibility should be distinguished from disintegration. For example, dispersibility requires that a suspension form when a tablet is placed in a glass of water at room temperature, whereas “disintegration” simply requires that a tablet ingested disintegrates in the acid environment of the stomach at body temperature.
650 In Ranbaxy’s submissions on this point, they say:
AstraZeneca appears to contend that claim 14 requires something more than disintegration into a suspended form for a time sufficient to allow administration and that it calls for something which is not an inherent property of all pellets formulated into tablets but requires additional development work.
651 I am not entirely sure the distinction sought to be drawn by the parties has any significance to the outcome of this proceeding. Nevertheless, I accept that there is a distinction between the two concepts, and accept the submissions of AstraZeneca on this point.
Claim 15
652 Under claim 15, the core material is a seed layered with active substance.
Claim 16
653 Under claim 16, the seeds have a size of 0.1 to 2 mm.
Claim 17
654 Claim 17 claims a process for the manufacture of the oral pharmaceutical multiunit tableted dosage form described in claim 1.
655 Claims 18 to 21 are dependent on claim 17 (either directly or through claims dependent on claim 17), and add various limitations.
Claim 18
656 Under claim 18, mechanical properties of the enteric coating layer(s) preclude significantly adverse effect on the degradation and dissolution of the active substance when exposed to simulated gastric acid and intestinal media respectively.
Claim 19
657 Under claim 19, the core material of step (a) in claim 17 (being the formation of “a multiple of a core material comprising the active substance and optionally comprising alkaline compound(s)”) is covered with a separating layer.
Claim 20
658 Under claim 20, the separating layer comprises pharmaceutically acceptable excipients which are soluble, or insoluble but disintegrate in water, and optionally alkaline compounds.
Claim 21
659 Under claim 21, the individually enteric coating layered units each are further coated with an over-coating layer before the individual units, together with tablet excipients, are compressed into the multiunit tableted dosage form.
660 Claims 22 to 25 claim tableted dosage forms according to claim 1 or one of the dependent claims 2 to 16 with further limitations.
Claim 22
661 Claim 22 claims a tableted dosage form according to any one of claims 1 to 16, when produced according to the process set out in any one of claims 17 to 21.
Claim 23
662 Claim 23 claims a tableted dosage form according to any one of claims 1 to 16 and 22, which has been formulated into a particular form appropriate for a given therapeutic use.
Claim 24
663 Claim 24 claims a tableted dosage form according to any one of claims 1 to 16 and 22, which has been formulated for use in inhibiting gastric acid secretion in mammals (including man).
Claim 25
664 Claim 25 claims a tableted dosage form according to any one of claims 1 to 16 and 22, which has been formulated for use in the treatment of gastrointestinal inflammatory diseases in mammals (including man).
Claim 26
665 Claim 26 is for a press-through blister package comprising a pharmaceutical multiunit tableted dosage form according to any one of claims 1 to 16 and 22.
666 Claims 27 to 28 are method claims.
Claim 27
667 Claim 27 claims a method for inhibiting gastric acid secretion in mammals (including man) by administering to a host in need thereof a therapeutically effective dose of a multiunit tableted dosage form according to any one of claims 1 to 16 and 22.
Claim 28
668 Claim 28 claims a method for the treatment of gastrointestinal inflammatory diseases in mammals (including man) by administering to a host in need thereof a therapeutically effective dose of a multiunit tableted dosage form according to any one of claims 1 to 16 and 22.
669 Apart from the construction issues detailed above, there was no other substantive dispute between the parties as to the construction of the claims.
Novelty
670 I now turn to novelty. The relevant principles are as set out above in respect of the Purity Patent.
Ranbaxy’s pleaded case
671 Ranbaxy relied on the following documents:
(a) US Patent No. 4,786,505 (which has already been defined in these reasons as ‘US 505’); and
(b) Australian Patent No. AU 78222/94 (‘AU 222’).
672 Ranbaxy alleges claims 1 to 7, 10, and 12 to 28 of the MUPS Patent lack novelty in light of AstraZeneca’s patent, US 505. US 505 is entitled “New Pharmaceutical Preparation For Oral Use”. It was published on 22 November 1988. Kurt Lovgren is an inventor of both US 505 and the MUPS Patent. He was not called to give evidence.
673 US 505 is primarily directed to a pharmaceutical preparation containing omeprazole or its alkaline salts. US 505 observes, at column 1, lines 36 to 39, that “it is obvious that an oral dosage form of omeprazole must be protected from contact with the acid reacting gastric juice in order to reach the small intestine without degradation”.
674 The specification states at column 3, lines 14 to 28:
The object of the present invention is to provide an enteric coated dosage form of omeprazole, which is resistant to dissolution in acid media and which dissolves rapidly in neutral to alkaline media and which has a good stability during long-term storage. … Cores containing omeprazole mixed with alkaline compounds or an alkaline salt of omeprazole optionally mixed with an alkaline compound are coated with two or more layers, whereby the first layer/layers … separates/separate the alkaline core material from the outer layer, which is an enteric coating.
675 Alkaline reacting salts of omeprazole include sodium, potassium, magnesium and calcium: see column 3, lines 61 to 62.
676 The enteric coating layer applied to the pellets “can optionally contain a pharmaceutically acceptable plasticizer such as, for instance, cetanol, triacetin, citric acid esters such as, for instance, those known under the trade name Citroflex® (Pfizer), phthalic acid esters, dibutyl succinate or similar plasticizers” (see column 5, lines 8 to 13). At lines 13 to 16, the specification states:
The amount of plasticizer is usually optimized for each enteric coating polymer(s) and is usually in the range of 1-20% of the enteric coating polymer(s).
677 At column 5, lines 60 to 63, US 505 states:
The final dosage form is either an enteric coated tablet or capsule or in the case of enteric coated pellets, pellets dispensed in hard gelatin capsules or sachets or pellets formulated into tablets.
(Emphasis added)
678 The specification continues, at column 6, lines 12 to 13:
The preparation according to the invention is especially advantageous in reducing gastric acid secretion...
US 505
679 Ranbaxy contended that US 505 discloses an oral pharmaceutical multiunit tableted dosage form (ie pellets formulated into tablets) suitable for use in therapy, comprising omeprazole or an alkaline salt thereof, characterised in that the enteric coating layers on the pellets comprise an “optimised” (ie effective) amount of plasticizing compound, and the enteric coating layer has appropriate mechanical properties for such a pharmaceutical multiunit tableted dosage form (including, necessarily, gastric acid resistance upon compression of the pellets into tablets). In essence, Ranbaxy contended that the protection of omeprazole from contact with acid is the whole purpose of the invention described in US 505.
680 The specification of the MUPS Patent admits the disclosure by US 505 of enteric coated pellets formulated into tablets (see p 3, lines 11 to 16 and 26 to 27). But the MUPS specification also states that there are “no examples describing any compositions of such a tablet formulation or a technique to manufacture such a formulation”.
681 However, Ranbaxy submitted that the absence of an example or specific instructions as to how to manufacture pellets formulated into tablets in US 505 cannot save the relevant claims of the MUPS Patent from anticipation. In this regard they relied upon the decision of Jessup J in Albany Molecular Research Inc v Alphapharm Pty Ltd (2011) 90 IPR 457 at 474; [2011] FCA 120 (see discussion at 465 [14] to [36]; following H Lundbeck (2009) 177 FCR 151 and Apotex (2009) 82 IPR 416; [2009] FCAFC 134). On Ranbaxy’s case, an example is unnecessary, given the assumption in US 505 that the addressee knows how to optimise the amount of plasticizer and the instruction given at column 5 lines 13 to 16 to do so. Further, and in any event, Ranbaxy submitted that the skilled addressee would expect, with some routine trial, to be able to make that pharmaceutical dosage form.
682 Effectively, Ranbaxy contended that the disclosure of pellets in tablets in US 505 is clear, and that there is no ambiguity in the words used in that patent. In response to AstraZeneca’s submissions, Ranbaxy contended that any alleged difficulty in making a tablet and the absence of tablet examples cannot create such ambiguity that the anticipation effected by US 505 is vitiated.
683 It was also contended by Ranbaxy (in response to AstraZeneca’s submissions that US 505 does not disclose that the enteric coating has the mechanical properties referred to in the MUPS Patent) that the skilled addressee would understand that optimisation of the plasticizer would provide appropriate flexibility for the coating.
684 Focussing on claim 1 and its alleged anticipation, I do not accept that US 505 discloses a multiunit tableted dosage form, nor does it direct, recommend or suggest such a thing. Rather, US 505 directs, recommends or suggests that omeprazole should be formulated into a capsule. US 505 exemplifies single-unit tablets, multiunit capsules and multiunit oral sachets, but not multiunit tablets. US 505 contains no examples of, no claims to, and no description of methods for compressing enteric coated pellets into tablets.
685 I do not accept that the very brief reference to “pellets formulated into tablets” appearing at column 5, lines 59 to 63 of that document (as set out at 677 above) amounts to “clear and unmistakeable directions” so as to anticipate the multiunit tableted dosage form that is the subject of the MUPS Patent claims. The whole context of the US 505 patent must be considered. It is not at all directed to multiunit tablets.
686 I accept that the skilled addressee would assume that the pellets disclosed by US 505 cannot be compressed into tablets, due to the known difficulties of compressing enteric coated pellets into tablets, and the absence of any exemplification in US 505 of enteric coated pellets compressed into tablets. I consider that this was accepted by Dr Reiland, who described the task of developing the new formulation for compression as “challenging”. I accept the evidence of the experts that taking the pellets disclosed by US 505 and attempting to compress them into tablets would require a new formulation development program, one which Dr Morella commented would have low prospects of success. Thus, just looking at what US 505 discloses (putting aside any issue of enablement), the skilled addressee would not view US 505 as directed to tablets at all.
687 Therefore, the mere incidental reference to “pellets” referred to by Ranbaxy in US 505, whilst clear in its terms, is not clear in its teaching of all of the combination of integers claimed in the MUPS Patent. In other words, I do not consider that US 505 discloses exactly what is claimed in claim 1 of the MUPS Patent when US 505 is read by the skilled addressee.
688 In light of my findings that US 505 does not disclose multiunit tablets, I do not consider that Ranbaxy is aided by the decision of Jessup J in Albany Molecular Research (2011) 90 IPR 457; [2011] FCA 120 (and accordingly, I do not consider it necessary to recite the arguments on this subject).
689 I also conclude that US 505 does not anticipate the multiunit tableted dosage forms of claims 2 to 8, 10 and 12 to 16, the manufacturing processes of claims 17 to 21, the tableted dosage forms of claims 22 to 25, the press-through blister pack of claim 26 or the method of use claims of claims 27 to 28.
690 In this regard, I note that all three experts agreed that US 505 does not disclose all of the integers of claims 5, 6, 7, 8, 10, 16 and 21 of the MUPS Patent.
Australian Patent Application Number 78222/94
691 I now turn to AU 222.
692 Ranbaxy alleged that claims 1 to 7, 10 and 12 to 28 of the MUPS Patent lack novelty in light of AU 222.
693 The complete specification for AU 222 was filed on 6 October 1994 and published on 22 June 1995. AU 222 is entitled “Enteric Granule-Containing Tablets”. The claims of AU 222 each claim a priority date of 12 October 1993, claiming priority from Japanese patent application 254049/1993 (‘JP 049’). This date is earlier than the MUPS priority date (8 July 1994), but AU 222 was not published until after that MUPS priority date.
694 Section 7(1)(c) as it appeared in the Act prior to the enactment of the Amendment Act provided that an invention is taken to be novel when compared with the prior art base, unless it is not novel in light of (among other things) prior art information contained in a single specification of the kind mentioned in subpara (b)(ii) of the definition of ‘prior art base’.
695 The definition of “prior art base” for the purpose of novelty included, at para (b):
(ii) information contained in a published specification filed in respect of a complete application where:
(A) if the information is, or were to be, the subject of a claim of the specification, the claim has, or would have, a priority date earlier than that of the claim under consideration; and
(B) the specification was published after the priority date of the claim under consideration; and
(C) the information was contained in the specification on its filing date and when it was published.”
696 This is a limited statutory exception to the fundamental principle that only information made publicly available before the priority date is relevant to assessing novelty: see eg Alcatel NV v Commissioner of Patents (1996) 68 FCR 8 at 10-11. Bennett J made the following comments about the operation of these provisions in Danisco (No 2) (2011) 91 IPR 209; [2011] FCA 282 at 272 [298] (I note that these comments were not disturbed on appeal: see Novozymes [2013] FCAFC 6):
The introduction of s 7(1) and the definition of “prior art base” into the Act expanded the concept of novelty to include some aspects of prior claiming, such that the doctrine of prior claiming was subsumed into novelty: see Alcatel NV v Commissioner of Patents (1996) 68 FCR 8 at 10–1 ; 138 ALR 504 at 506–7 ; 35 IPR 255 at 257–8 (Alcatel). The intention, as revealed in the IPAC report (Industrial Property Advisory Committee, Patents, Innovation and Competition in Australia (Industrial Property Advisory Committee, Canberra, 1984)) to which Burchett J referred in Alcatel, was to introduce a “whole of contents” approach such that novelty was judged by reference to the whole of a specification filed but not published before the priority date and not just by the claims of that specification. The IPAC report refers at 7.3 to “any disclosure contained in an earlier specification”. That does not assist in an understanding of the scope of “information” in para (b)(ii) of the definition of “prior art base”. It may refer to the disclosure of the whole of the specification or, as Novozymes has accepted, require identified information, such as the passages relied on by Novozymes below.
697 Justice Emmett concluded in EI Du Pont de Nemours & Co v ICI Chemicals & Polymers Ltd (2005) 66 IPR 462; [2005] FCA 892 at 482-483 [80]-[85] that it is necessary first to identify the information in the prior-filed patent, and then to determine whether that information either is or could be the subject of a valid notional claim of that prior-filed patent.
Submissions of Ranbaxy
698 In order to make it clear that the notional claim is fairly based on JP 049 and thus entitled to the earlier priority date, Ranbaxy bases their notional claim on the information contained in JP 049, which they submitted is equally disclosed in the published specification of AU 222. The relevant parts of JP 049 relied upon by Ranbaxy are as follows:
(a) JP 049 says (at [0001]) that the invention relates to multiunit tablets and “[m]ore specifically, it relates to multiple-unit tablets whose units are enteric granules”.
(b) At [0002], it refers to the need for multiunit enteric tablets.
(c) At [0003] it says:
Enteric granule-containing tablets are prepared by adding substances to the enteric coating which soften it and, after preparation of the granules, adding an excipient for tabletting…
Ranbaxy contended that the addressee would understand that the reference in this passage to the substances added to the enteric coating to soften it would include a plasticizer.
(d) It also says at [0003]:
[I]n order to prevent damage to the coating, excipients are sometimes laminated over the granules prior to compression moulding, or a large amount of an excipient is added for the tabletting.
(e) At [0004], it says:
Multiple-unit enteric tablets must have acid resistance in the stomach, and deterioration of their acid resistance by crushing of the granules during tabletting has been found to have an adverse effect on their bioavailability. Furthermore, the large amounts of excipients added in addition to the granules to prevent crushing of the granules during tablet preparation result in relatively large tablets which are difficult to administer, and this influences patient compliance.
(f) At [0005], it is said that the invention “provides tablets which adequately maintain acid resistance in the stomach while having a high granule content”.
(g) At [0006], reference is made to the object of “obtaining granule-containing tablets having granules coated with an enteric coating which are compression molded without undergoing damage”. It is said that “according to the present invention, tablets with satisfactory acid resistance and with a high granule content may be prepared by compression molding a mixture containing [new tableting excipients] as an additive or additives with the coated granules”.
(h) At [0007], the active agent is referred to. It is said that it is “not particularly restricted so long as it is a [proton pump inhibitor], or is an agent which is not properly released in the stomach by acid degradation, drug stimulation, etc”.
Ranbaxy contended that the chemical compounds named in JP 049 are proton pump inhibitors. Ranbaxy further contended that the reference to an agent which is not properly released in the stomach by acid degradation would be understood to include other agents (including other proton pump inhibitors) which should not be released in the stomach because, if they were, they would suffer acid degradation (and that is the “whole point” of an enteric coating).
(i) At [0008], plasticizers are referred to. It is said that “a plasticizer is preferably added to 15-40 wt% during formulation of the coating to be coated on the granules”.
(j) At [0009], the new tableting additives are referred to, as is their function of providing “sufficient hardness under low tabletting pressure”.
699 Examples are then given in JP 049. They disclose tablets with a high granule content compared to the tableting excipient content. This is 80% granules and 20% excipients in Example 1, and 85% granules and 15% excipients in Example 2 (this may be compared to the proportions in the MUPS Patent of up to 70% excipients, eg in example 2). Ranbaxy contended that the skilled addressee would understand that the new tableting excipients are useful to allow a tablet which has a high proportion of granule content successfully to achieve acid resistance for the enteric coating layer.
700 Ranbaxy further contended that the abovementioned information in JP 049 is equally disclosed in the published specification of AU 222. AU 222 further names omeprazole on p 2 as an example of an active agent.
701 Ranbaxy submitted that AU 222 therefore discloses a tablet containing enteric coated pellets, which successfully avoids loss of acid resistance in the stomach by a combination of an appropriate amount of plasticizer to soften the enteric coating layer and particular pharmaceutically acceptable tableting excipients designed to allow a high proportion of granule content to tableting excipient content. That is, all the features of the MUPS Patent claims are disclosed and in addition, new tableting excipients are disclosed which are useful to obtain that high granule content.
702 Ranbaxy further contended that this information could be the subject of a claim of the AU 222 specification as follows:
A multiple unit tablet whose units are enteric coated granules, for the treatment and prevention of gastric acid related diseases, said tablet comprising pharmaceutically acceptable excipients selected from the group consisting of synthetic hydrotalcite, dried aluminium hydroxide gel, aluminum hydroxide/sodium hydrogen carbonate coprecipitate, alumina hydroxide/magnesium, synthetic aluminium silicate and dihydroxyaluminumamino acetate, and optionally appropriate binders, disintegrators or lubricants, wherein the enteric coated granules comprise an active ingredient which is an agent which is not properly released in the stomach due to acid degradation, such as a proton pump inhibitor, and an enteric coating comprising a polymer selected from the group consisting of methacrylic acid copolymer LD, cellulose acetate phthalate, hydroxypropylmethylcellulose phthalate or hydroxypropylmethyl cellulose acetate succinate, and an amount of plasticizer added to the enteric coating so as to soften it in order to prevent damage to the coating so that the difference in acid resistance before and after tableting is minimal.
703 Ranbaxy contended that the information that supports the notional claim falls within the definition of “prior art base” and anticipates the relevant claims of the MUPS Patent.
CONSIDERATION
704 For the purpose of determining this issue, I will assume that the limited statutory exception set out in s 7(1)(c) as it existed prior to the enactment of the Amendment Act applies (although AstraZeneca contended it did not apply on the facts of this case).
705 To the extent that AU 222 is relevant to novelty on the basis that it claims priority from JP 049, there is a significant amount of information disclosed in AU 222 that is not disclosed in JP 049. AU 222 discloses benzimidazole-based antiulcer agents such as omeprazole, but JP 049 does not refer to omeprazole at all. Neither of the compounds “2-[[(2-pyridyl)methyl]sulfinyl]-imidazo[4,5-b]pyridine-based drug” nor TU-199, referred to in [0007] of JP 049, is omeprazole. They are in a different class of chemical compounds.
706 JP 049 also refers to “an agent which is not properly released in the stomach by acid degradation, drug stimulation, etc”. In the context of having considered AU 222 and JP 049 together, Dr Reiland considered that this statement in JP 049 could cover compounds which were either degraded by gastric acid, or a different class of compounds which stimulated or irritated the stomach. During oral closing submissions, Senior Counsel for Ranbaxy accepted that it was necessary to “add” common general knowledge to this statement in JP 049 to give it the scope contended for by Ranbaxy, as follows:
Our proposition is that in the context of an enteric coated pellet formulation, which this is - bearing in mind the purposes of the enteric coating – it must be a reference to an agent which you don’t want to have degraded release in the stomach. You don’t want it to be released in the stomach, because it will degrade in the acid environment of the stomach. If you add that bit of practical common general knowledge to the statement there, there will be no doubt in our submission, that although inelegantly expressed no doubt, it’s a reference to other agents than the named proton-pump inhibitors that you don’t want to have released in the stomach, and to include, therefore, other proton-pump inhibitors such as omeprazole.
707 However, the common general knowledge cannot be added to the prior art disclosure in order to establish that all the essential integers of the claim are disclosed: see eg ICI Chemicals & Polymers Ltd v The Lubrizol Corporation Inc (2000) 106 FCR 214 at 228 [43].
708 Ranbaxy has failed to adduce evidence that the notional claim they have proposed could be fairly based on either JP 049 or AU 222. There is no evidence that Dr Reiland has considered the claim formulated by Ranbaxy. It was not put to any experts in the concurrent evidence session.
709 In any event, I find that neither AU 222 nor JP 049 disclose a polymeric enteric coating comprising an “effective amount of a plasticizing compound” as used in the MUPS Patent. Upon reading both AU 222 and JP 049, they teach that the amount of plasticizer is not relevant to the acid resistance of the formulation. Table 1 of both documents compares formulations which all have the same amount of plasticizer (including comparative example 1). The only variable is that comparative example 1 did not contain the antacid in the tableting matrix. The results indicate that it is the absence of the antacid that resulted in the significant reduction in acid resistance and that the presence and quantity of plasticizer was irrelevant. Accordingly, AstraZeneca submitted – and I accept – that the last integer of the notional Ranbaxy claim to an “amount of plasticizer added to the enteric coating so as to soften it in order to prevent damage to the coating so that the difference in acid resistance before and after tableting is minimal” is inconsistent with the disclosures in both AU 222 and JP 049.
710 Further, neither JP 049 nor AU 222 disclose the gastric acid resistance that is a component of the invention disclosed by the MUPS Patent. The acid resistance test in these documents is from the Japanese Pharmacopeia. There is no indication that this test is the same as the acid resistance test described on p 7, lines 10 to 18 of the MUPS Patent.
711 To the extent that it may be relevant prior art, AU 222 discloses an invention, the key aspect of which is the addition of specific antacid excipients in order to prepare granule-containing tablets with acceptable acid resistance, elution rate, disintegration time, hardness and abrasiveness values as well as lower variation in tablet weight during tableting. The invention is an alternative approach to achieving acid resistance (and other relevant properties) to the invention disclosed in the MUPS Patent. This was confirmed by Dr Reiland in the concurrent evidence session when he referred to the advance disclosed in AU 222 as being the surprising use of an antacid as a cushioning agent.
712 In my view, neither AU 222 nor JP 049 disclose all the essential integers of the invention as claimed in claim 1 of the MUPS Patent. Therefore, neither discloses the tableted dosage form claims dependent on claim 1, being claims 2 to 7, 10, 12 to 16 and 22 to 25, and the blister pack and method of use claims dependent on claim 1, being claim 26 and method of use claims 27 to 28. Further, neither AU 222 nor JP 049 discloses all the essential integers of the invention as claimed in claim 17, and therefore neither discloses the process of manufacture claims dependent on claim 17, being claims 18 to 21.
713 I make this further observation. Ranbaxy’s closing submissions alleged that claims 1 to 7, 10 and 12 to 28 of the MUPS Patent lack novelty in light of AU 222. Further, Ranbaxy’s Seventh Amended Particulars of Invalidity dated 25 March 2013 alleged that claims 1 to 8, 10 and 12 to 28 of the MUPS Patent lack novelty in light of AU 222. The reason for this inconsistency is unclear. However, Ranbaxy’s evidence and the Joint Expert Report on AU 966 only relate to whether claims 1 to 2, 5 to 8, 12, 15, 17, 18, 21 to 25 and 27 to 28 of the MUPS Patent lack novelty in light of AU 222. Accordingly, I accept AstraZeneca’s submissions that the allegations that claims 3, 4, 10, 13, 14, 16, 19, 20 and 26 of the MUPS Patent lack novelty in light of AU 222 cannot be maintained.
714 I also observe that neither AU 222 nor JP 049 disclose omeprazole’s single enantiomers, or an alkaline salt of one of omeprazole’s single enantiomers, as required by claim 12. Further, neither AU 222 nor JP 049 disclose the use of a separating layer between the core material and the enteric coating layer as required by claim 4.
715 For all of these reasons, I find that AU 222 does not anticipate any of the claims of the MUPS Patent.
Inventive step
716 I now turn to inventive step. The relevant principles to be applied are as set out above in respect of the Purity Patent.
Ranbaxy’s submissions
717 Ranbaxy made the following general submissions as to inventive step.
718 Ranbaxy contended that the statements in the “Background of the invention” section of the MUPS Patent admit that the common general knowledge included omeprazole and its characteristics, the commercial capsule-filled enteric coated pellet formulation, and the advantages which would be achieved by a multiunit tablet formulation. It was said that in doing so, the specification identifies “the starting point and objective of the addressee” for the purpose of testing inventive step. Whether this is the correct ‘starting point’ (and its relevance) will depend on the extent to which the matters identified by Ranbaxy as common general knowledge are in fact so.
719 Ranbaxy submitted that a skilled addressee beginning at that starting point and with that objective would routinely take the steps which would produce a satisfactory tableted formulation. They would appreciate that more plasticizer may be necessary to make the enteric coating layer softer and more flexible so as to resist cracking during compaction. They would optimise the identity and amount of the plasticizer by reference to an appropriate amount of an appropriate enteric coating polymer(s) and other standard ingredients (including standard tableting excipients) to produce such a product. They would screen selected components of enteric coatings to investigate how the choice of components in the enteric coatings changed the properties of the formulation. By this process, Ranbaxy contended that the skilled addressee would identify and select a sufficient amount of plasticizer to provide the enteric coating with the necessary flexibility to avoid cracking during compression.
720 Ranbaxy contended that the passage spanning pp 11 and 12 of the specification admits that such an optimisation process is within the ordinary skill of the skilled addressee, and that this admission gives the lie to the ritual incantation of “surprise” appearing in the MUPS Patent at p 4, line 28.
721 It was then contended that there could be no suggestion that any of the matters by which the dependent claims are limited involve anything other than a routine choice of available materials or features that are inherent in a tableted dosage form.
722 Accordingly, on Ranbaxy’s case, the claimed invention is obvious in the light of the common general knowledge, and also when resort is had to the s 7(3) documents relied upon by Ranbaxy (considered separately as required by statute).
723 At the outset, I make this observation. I consider that the complete answer to the contentions of Ranbaxy comes down to the degree of difficulty involved and the problems confronting the skilled addressee in attempting to design a multiunit tablet containing coated pellets of omeprazole. Even if there was some motivation to try, there were known difficulties which would dull the enthusiasm of the skilled addressee to proceed to such an extent that I consider that they would not proceed with any reasonable prospect of success.
724 Put another way, to the person skilled in the art at the priority date, with the complexities to be confronted, the solution found in the MUPS Patent would not have been seen as one worthy of attempting.
725 In addition to the above general submissions, Ranbaxy specifically made submissions on whether the claims of the MUPS Patent involve anything more than a routine selection of ingredients for an oral dosage formulation for omeprazole as an alternative to LOSEC. Obviously, Ranbaxy contends for a negative answer.
726 Ranbaxy put their primary position, supported by other factors, as follows:
(a) The known advantages of a tableted multiunit pellet system meant that it was a desirable alternative to the LOSEC capsule.
(b) It was known that the pellets will require a degree of flexibility in order to withstand the compression forces involved in tableting and still pass the acid resistance test.
(c) It was known that a suitable plasticizer could be used in a suitable amount to make the enteric coating appropriately flexible.
(d) It was routine for an experienced formulator to experiment with different amounts of different polymers, plasticizers and known tableting excipients to find a combination that would meet the required performance criteria.
(e) Those experiments would yield a workable tablet as defined in the claims, including an appropriate amount of plasticizer to make the pellet coating suitably flexible.
(f) The above propositions reflect the common general knowledge.
727 It was said that these propositions were supported by Dr Reiland’s evidence about what he would have done had he been asked what course he would have taken at July 1994 to develop an alternative solid oral dosage form of omeprazole:
Based on the information in Goodman & Gilman and the PDR… [t]here are three options for solid oral dosage formulations that I would initially consider: an enteric coated tablet, enteric coated particles in a capsule, or enteric coated particles in a tablet. I would focus primarily on enteric coated particles… and would be particularly interested in enteric coated particles in a tablet. Given the ability to break such a tablet in half and the ease of swallowing, enteric coated particles in a tablet provides an attractive alternative to enteric coated particles in a capsule. The stated recommended dose of omeprazole (20 mg) is low enough such that I would expect to be able to develop a tablet containing enteric coated particles.
728 Ranbaxy submitted that this evidence was given before Dr Reiland was shown the MUPS Patent or prior art. It reflected the practical approach of an experienced industrial formulator. Ranbaxy submitted that it was not challenged in cross-examination, and should be accepted.
729 It was further submitted that the evidence of Dr Reiland about the way in which the hypothetical project would be approached was confirmed by the way in which AstraZeneca themselves pursued the MUPS project (evidenced by certain internal documents tendered in evidence in this proceeding). Ranbaxy asserted that the objectives of this project were largely commercial. There was no suggestion of any expected difficulty in formulating the tablet. Nor was there any expression of surprise that it proved possible. The standard experimental approach was adopted and it worked.
730 It was further submitted by Ranbaxy that the suggestions by Dr Morella and Professor Bodmeier that a formulator would not have succeeded in making a workable tablet at the priority date were unconvincing, for the following reasons.
731 First, Ranbaxy contended that this position was at odds with the practical approach an experienced industrial formulator would take. Professor Bodmeier’s second affidavit was said to have used the word “problem” 46 times. Dr Morella’s second affidavit was similarly said to have focused attention on difficulties and complexity. It was submitted that unlike Dr Morella and Professor Bodmeier, an experienced industrial formulator would not regard the kinds of problems that might be experienced as insurmountably complex, but rather, as problems to avoid or overcome by known methods and the application of ordinary skill, including routine experimentation.
732 It was further contended in this context that Professor Bodmeier had very limited practical industry experience. In summary, Ranbaxy submitted that Professor Bodmeier may well have the scientific knowledge of a notional skilled addressee, but he has neither their practical experience nor the attitude that the skilled addressee would necessarily bring to the task of formulating an appropriate dosage form for the purposes of commercial development. I have already rejected this submission.
733 Likewise, it was submitted, the experienced industrial formulator would not feel constrained (as it was said Dr Morella did) to retain the polymer and plasticizer in the amounts used in a previous capsule product, when attempting to make a multiunit tablet. Two of the three unsuccessful attempts with which Dr Morella was involved before the priority date used commercially available pellets designed for capsules and did not consider changes to the pellet coatings. No changes were made to the enteric coating formulation in either instance. On Dr Reiland’s evidence, it is entirely unsurprising that such pellets failed to withstand the more rigorous forces involved in tablet preparation, as they had not been optimised for this task.
734 Nor would, in Ranbaxy’s submission, the experienced industrial formulator feel constrained (as each of Professor Bodmeier and Dr Morella said they would be) by manufacturers’ recommendations, for example as to the amount of plasticizer that might appropriately be used.
735 To this end, Ranbaxy contended that Dr Morella and Professor Bodmeier’s evidence that the notional skilled addressee would be limited by the manufacturers’ recommendations regarding the amount of plasticizer to be used was not consistent with the examples included in Dr Morella’s patent applications. Dr Morella indicated that he would not use more than 12-15% plasticizer in a product, but three separate patent applications of which Dr Morella was listed as one of the inventors were tendered in evidence, and they each referred to amounts of up to 50% plasticizer. One gave examples which used 20% plasticizer in an enteric coating formulation of erythromycin, which is an acid labile drug (like omeprazole).
736 Ranbaxy submitted that Professor Bodmeier suggested that one reason for the formulator limiting himself to the manufacturers’ recommendations was that otherwise it was “a task which could not be managed within a reasonable time”. Ranbaxy contended that this was a matter that neither Dr Reiland nor Dr Morella raised as a concern, and that as between Dr Reiland and Dr Morella (both of whom had worked in pharmaceutical companies all their professional lives), the debate focused more on what was it possible to achieve, rather than any time constraints.
737 Ranbaxy also relied on Dr Reiland’s evidence that the manufacturers’ recommendations will be for particular uses, and there were no recommendations for use in multiparticulate tablets at the priority date.
738 However, Ranbaxy noted in their submissions that the disagreement between the experts concerning the breadth of the range of experimentation (including whether an experienced formulator would try ingredients at levels beyond those recommended by an excipient manufacturer) may ultimately be irrelevant, as the Ranbaxy Product uses only 13% plasticizer. Ranbaxy submitted that this percentage is, on the evidence, likely to be within the manufacturers’ recommended range and is an effective amount within the scope of claim 1.
739 It was separately submitted that the commercially available PCE tableted products demonstrate that any suggestion by Dr Morella and Professor Bodmeier that an experienced industrial formulator would not succeed in making a workable tablet containing multiunit enteric coated pellets was untenable.
740 The details of these products are discussed later in these reasons. It is sufficient for present purposes to note Ranbaxy’s argument that those products had a significantly higher amount of active ingredient in the tablet: 333 mg in one and 500 mg in the other. It was the need to deal with a large amount of active ingredient which gave rise to the discovery of the hitherto unknown ability of microcrystalline cellulose to protect the pellets from fracture in the tableting process (see United States Patent number 4,863,741, “Tablet Composition for Drug Combinations” (‘US 741’)). It was the need to deal with an even larger amount of active ingredient in the 500 mg tablet that led to the discovery that a new compression-enhancing coating could be used (see United States Patent number 5,009,897, “Pharmaceutical Granules and Tablets Made Therefrom” (‘US 897’)).
741 In the case of omeprazole, the recommended dose was only 20 mg. Accordingly, Ranbaxy submitted that (based on the evidence of Dr Reiland) there was significantly greater scope to use (amongst other things) substantial amounts of a cushioning agent such as microcrystalline cellulose, and to apply a thicker enteric coating in this product. It was submitted that knowledge that the PCE multiunit tablet had successfully been formulated would have encouraged the experienced formulator to think that a successful multiunit tablet form of omeprazole could be achieved.
742 On this issue, Ranbaxy further submitted that it is a mark of the “remoteness” of Professor Bodmeier from the work of an experienced industrial formulator that he was unaware of the PCE products. Further, “a significant pillar in his negative approach” to the prospects of success associated with formulating the invention that is the subject of the MUPS Patent at the priority date was that he was “not aware of any pharmaceutically acceptable, marketed enteric coated multiparticulate tablets”. Ranbaxy contended that the clear inference to be drawn from this is that knowledge of a marketed multiparticulate tablet, especially one of an acid labile drug, would have substantially altered his expectation of success.
743 Further, the insistence of Dr Morella and Professor Bodmeier that a formulator would not expect to be able to make a workable tablet formulation of multiunit enteric coated pellets was said to be inconsistent with the position taken in Dr Morella’s own patents (mentioned above). The 1988 patent (being Australian Patent number 29,522, “Tetracycline Dosage Form”) stated that “[a]n encapsulated form or tableted pellets may be used” and referred to the release of the pellets from the dosage form upon disintegration of the tablet. Similarly, the 1989 patent (being Australian Patent number 47,732, “Sustained Release Pharmaceutical Composition”) said: “the pharmaceutical pellet composition may be provided in a pellet or tableted pellet form. A tablet may be formed by compression of the pellets optionally with the addition of suitable excipients”.
744 Ranbaxy contended that there were additional reasons to treat the evidence of Dr Morella and Professor Bodmeier with caution. It was said that each of them was enthusiastic to embrace the suggestion that the publication referred to as the “Drugs Made in Germany” article taught away from the claimed invention. Ranbaxy submitted that the article was not shown to have been published before the priority date, still less to have formed part of common general knowledge. In Dr Morella’s case, the explanation for not having made the point in his second affidavit (namely, that the article dealt with things that did not work) was said to be “unconvincing” and “extraordinary”. Further, Dr Morella’s resistance to the proposition that one purpose of a plasticizer was to improve flexibility was said to be difficult to follow, especially given what he had said in his second affidavit (namely, that plasticizers function by interacting with coating polymers and lowering the glass transition temperature of the polymer (being the temperature below which the polymer is hard, and above which it is flexible)). It was submitted that his further evidence that he had to read the MUPS Patent to understand that the plasticizer improved flexibility is also difficult to follow and should not be accepted.
745 Ranbaxy also submitted that Professor Bodmeier’s position that all enteric coating polymers are brittle, and thus would only have limited additional flexibility upon addition of a plasticizer, misses the point. Clearly, enteric coating polymers are used in enteric coated formulations and have some degree of robustness. The text by Leon Lachman, “The Theory and Practice of Industrial Pharmacy” (1986, 3rd ed) (‘Lachman’) was said to teach that plasticizers are used to address the brittleness of enteric coatings and that the amounts of plasticizers which can be used are up to 50%.
746 Further, Ranbaxy referred to the fact that Professor Bodmeier interpreted claim 1 of the MUPS Patent to involve the use of “extraordinarily high amounts” of plasticizer (and Dr Morella reached a similar conclusion). However, Ranbaxy contended that claim 1 merely refers to an “effective amount” and does not provide any indication of a specific value. Dr Reiland’s evidence on point did not specify that a high amount of plasticizer would need to be used. Dr Reiland merely indicated that the amount of plasticizer might be more than that required for pellets filled into capsules. Ranbaxy emphasised that Dr Reiland made this comment before being asked about whether the claims of the MUPS Patent involve anything more than a routine selection of ingredients, or being provided with the MUPS Patent.
747 These matters will be considered in further detail below to the extent necessary to determine the question of inventive step. But before going to the evidence on these issues, I set out my findings on the aspects of the common general knowledge of the skilled addressee that were in dispute between the parties.
Common general knowledge
Matters in Dispute
748 The common general knowledge that is not in dispute in this proceeding has already been set out at the commencement of these reasons relating to the MUPS Patent.
749 The following paragraphs relate to the common general knowledge as at 8 July 1994 relating to the tableting of multiunit enteric coated pellets that was controversial in this proceeding. I consider the following topics:
(a) Plasticizers;
(b) Binders, diluents, glidants, lubricants and disintegrants;
(c) Overall experimental approach;
(d) Investigation of formulation problems;
(e) The quantity of enteric coating used by the formulator;
(f) The degree to which enteric coated multiunit tablets were common, and the Abbott Laboratories PCE products;
(g) The degree to which enteric coated multiunit tablets are advantageous compared with capsules;
(h) Tablets containing compressed enteric coated particles;
(i) The difficulty of compressing enteric coated particles into multiunit tablets; and
(j) The formulator’s approach to enantiomers and racemates.
750 In the course of dealing with each of these topics, some of the general complexities and difficulties I have already referred to which would arise in any attempt to design a multiunit tablet containing coated pellets of omeprazole will be considered. It will shortly become apparent that it is the combination of these complexities and difficulties that has caused me to reject the attack on inventiveness by Ranbaxy, to which I will return.
Plasticizers
751 One question arose as to the use and function of plasticizer in enteric coatings. Ranbaxy submitted that one purpose of the plasticizer is to make the enteric coating softer and more flexible. Dr Morella and Professor Bodmeier agreed that this is one purpose of a plasticizer. Some degree of flexibility is required to avoid cracking of the enteric coating. Cracking would, of course, defeat the purpose of providing acid protection. Plasticizers work by modifying polymer to polymer molecular binding. A consequence of this, in addition to increased softness and flexibility, may be some reduction in acid resistance.
752 Dr Reiland stated in his evidence (on which he was not cross-examined):
If the enteric coated particles are to be compressed to form a tablet, this may impact the amount of plasticizer in the enteric coating depending on the other excipients used in the tablet. That is, if the enteric coated particles are to be compressed into a tablet, then more plasticizer could be required in the coating formulation to provide the coating with the necessary flexibility to avoid cracking during the compression. This is because compression will normally cause drug granules/particles to deform to some extent. Excipients would typically also be needed to provide cushioning during the compression stage. Cracking is an example of a significant defect in the coating which would lead to failure of routine performance testing.
753 Reference was also made to the textbooks “Remington’s on Pharmaceutical Sciences” by Alfonso Gennaro (ed) (1990, 18th ed) (‘Remington’s’) and Lachman, regarded as standard textbooks in the field. Dr Morella gave evidence that he was familiar with these books and had referred to them before. Professor Bodmeier gave similar evidence that he was familiar with Remington’s. Lachman was said to be a more practical text for industrial formulators. I accept the information in these texts to be common general knowledge at the priority date.
754 Remington’s at p 1669 states:
The incorporation of a plasticizer into the formulation improves the flexibility of the coating, reduces the risk of the film cracking and possibly improves adhesion of the film to the substrate. To ensure that these benefits are achieved, the plasticizer must show a high degree of compatibility with the polymer, and a degree of permanence, if the properties of the coating are to be stable on storage.
755 Lachman at p 363 states:
Coating compositions that yield brittle films must be plasticized to obtain a more flexible film that is acceptable for tablet coating.
756 Further, at p 368 it states:
The quality of a film can be modified by the use of “internal” or “external” plasticizing techniques. … Most often, the formulator uses external plasticizers as additives to the coating solution formula so that the desired effects are achieved for the film. An external plasticizer can be a nonvolatile liquid or another polymer, which when incorporated with the primary polymeric film former, changes the flexibility, tensile strength, or adhesion properties of the resulting film.
757 Dr Reiland provided the evidence referred to above before he saw the MUPS Patent. However, as I have indicated, I find this approach was infected by his recollection (far from exact) of how Abbott Laboratories developed the PCE 333 formulation. This is one matter which causes me to prefer to rely upon the evidence of Professor Bodmeier and Dr Morella. In addition, as indicated, I have generally found the evidence of Professor Bodmeier and Dr Morella compelling.
758 Therefore, I accept the following conclusions reached by those witnesses. Professor Bodmeier and Dr Morella both stated that enteric coating polymers are brittle and that plasticizers cannot be added to the extent that they are no longer brittle, and certainly not to the extent that they would permit enteric coated particles to be compressed into a tablet. Professor Bodmeier said that enteric polymers are inflexible polymers, which are known to be very brittle and whose elongation (a measure of flexibility) is only improved to a very limited extent by the addition of a plasticizer. He gave evidence that in enteric coating formulations, plasticizers are present not to improve flexibility, but just to allow film formation (and thereby enhance gastric acid resistance of the enteric coating). Dr Morella said that plasticizers imparted flexibility but only in the sense of reducing the likelihood of coatings cracking, like house paint cracking if it dries too quickly. He gave evidence that in this sense, plasticizers enhanced stability of the coating. I accept that this is different to the problems of compaction caused by the compression of enteric coated pellets into a multiunit tablet.
759 In my view, whatever general approach is taken by Dr Reiland, Remington’s and Lachman, the use of plasticizers to achieve sufficient flexibility in enteric coating of pellets to enable compression into tablets is a matter which was not routine at the priority date.
Binders, diluents, glidants, lubricants and disintegrants
760 Ranbaxy submitted that Remington’s shows that these components and their functions were well known. As previously noted, the experts broadly agreed on the components and their functions in the Joint Expert Report on AU 966. Ranbaxy contended that it was well known that the right quantities of these ingredients had to be used in a coating formulation, and indeed, that “most excipients used in formulating tablets… have many uses, and a thorough understanding of their properties and limitations is necessary in order to use them rationally”. Ranbaxy relied on the following statements from Remington’s:
(a) For example, in relation to binders, it was stated that “[t]he quantity of binder used has considerable influence on the characteristics of the compressed tablets. The use of too much binder or too strong a binder will make a hard tablet which will not disintegrate easily and which will cause excessive wear of punches and dies”.
(b) In relation to lubricants, it was stated that “[i]n selecting a lubricant, proper attention must be given to its compatibility with the drug agent”.
(c) In relation to glidants, it was noted that “[i]t is especially important to optimize the order of addition and the mixing process for these materials in order to maximize their effect and to make sure that their influence on the lubricant(s) is minimized”.
(d) In relation to disintegrants, it was stated that:
[f]actors other than the presence of disintegrants can affect significantly the disintegration time of compressed tablets. The binder, tablet hardness and the lubricant have been shown to influence the disintegration time. Thus, when the formulator is faced with a problem concerning the disintegration of a compressed tablet, the answer may not lie in the selection and quantity of the disintegrating agent alone.
761 However, all this shows is that there is a variety of choices available to the skilled formulator. I accept the evidence that the formulator works within established ranges, based on prior experience, manufacturers’ recommendations and literature when developing a pharmaceutical formulation, including when selecting excipients. Too much or too little of a particular excipient is a concern, and usually a formulator uses as few excipients as possible. At a very general level a formulator may go beyond these ‘constraints’. But even if, as Dr Reiland suggested, the only real concern is toxicology issues, this does not mean that care need not be taken in selecting excipients.
762 This is yet another matter the formulator must consider in determining the way forward, which has its own complexities and difficulties.
Overall experimental approach
763 Looking more generally at the experimental approach to be adopted by the skilled formulator at the priority date, I am satisfied that at this time, all of the enteric coating components were known to affect the final performance of the coating. Undoubtedly it was standard practice to undertake a systematic experimental approach to selecting the final film coating ingredients and their quantities.
764 The approach adopted by Ranbaxy was explained by Dr Reiland:
“systematic”… mean[s] the step-wise, logical approach to formulation development … “experimental” mean[s] the routine process in which a series of formulations are made with different levels of ingredients (excipient identity and amount) or process parameters. Routine performance tests are conducted on the formulations, and the data from these tests are gathered and studied to assess the effect of the specific levels of ingredients or process parameters on the performance of the formulation. For instance, with an enteric coated product, this would involve making a series of formulations in which the levels of polymer, plasticizer, opacifier and other excipients including solvents are varied, and different process parameters are studied.
765 I accept that optimisation of the identity and quantities of the ingredients of the enteric coating, according to the context of the final oral solid dosage form, was part of the standard experimental approach of any solid oral dose formulator to find a combination of all the ingredients of the tablet which would meet its performance requirements.
766 For example, Lachman at p 362 states:
An experienced formulator usually takes the pragmatic approach and develops a coating formulation as a modification of one that has performed well in the past. The inexperienced coater or the formulator seeking a better coating system needs to start from a more basic position and essentially builds his coating composition from a primary film former. The effect of the addition of plasticizers, opaquants, colorants, and the solvent system can then be individually and collectively assessed.
767 Lachman then states at p 364:
Optimization is usually associated with minor modifications in a basic formula. As discussed earlier, the basic or starting formula is obtained from past experience or from various sources in the literature. Modifications on this basic formula may be necessary to improve … any property of the coating that the formulator deems deficient. … Changes of the polymer(s)-to-plasticizer ratio, however, or the addition of different plasticizers or polymers, are common modifications made in optimization of the coating.
768 Specifically in relation to plasticizers, Lachman states at p 369:
The choice of plasticizer depends upon the ability of plasticizer material to solvate the polymer and alter the polymer-polymer interactions. When used in correct proportion to the polymer, these materials impart flexibility by relieving the molecular rigidity. The type of plasticizer(s) and its ratio to the polymer can be optimized to achieve the desired film properties. One should also consider the viscosity of the plasticizer; its influence on the final coating solution; its effect on film permeability, tackiness, flexibility, solubility, and taste; and its toxicity, compatibility with other coating solution components, and stability of the film and the final coating product.
A combination of plasticizers may be needed to achieve the desired effect. The concentration of the plasticizers depends on many factors, including the polymer chemistry, method of application, and the other components present in the system.
…
The amount and type of plasticizers to be used for any given polymer can be based on the polymer manufacturer’s recommendations. Optimization of the plasticizer concentration must be based on the presence of the other additives. Concentration of a plasticizer is expressed in relation to the polymer being plasticized. Recommended levels of plasticizers range from 1 to 50% by weight of the film former.
769 All three expert witnesses agreed that the selection of suitable ingredients in appropriate amounts is part of the normal task of the experienced formulator to whom the MUPS Patent is addressed.
770 To this end, Dr Morella agreed with Dr Reiland in question 5(d) of the Joint Expert Report on AU 966 that the nature of the component and how much was needed to achieve the intended purpose were factors which would be taken into account in the selection of components for use in an enteric coating.
771 I accept that the formulator would then conduct some routine tests to assess the relative properties of the selected polymers and plasticizers in the various chosen combinations. Ranbaxy further submitted that based on the test results, the formulator would adapt the formulation(s) that worked best to further optimise and prepare the final formulation.
772 In so submitting, Ranbaxy referred to a number of AstraZeneca’s own internal documents as a “real world” example of “a systematic experimental approach put into practice, so as to develop a commercial MUPS omeprazole product”.
773 Ranbaxy further relied on the fact that the MUPS Patent specification itself recognises that it is within the skill of the experienced industrial formulator to select the appropriate excipients and their amounts, when it records that:
[T]he active substance is mixed with pharmaceutical constituents to obtain preferred handling and processing properties and a suitable concentration of the active substance in the final mixture. Pharmaceutical constituents such as fillers, binders, lubricants, disintegrating agents, surfactants and other pharmaceutically acceptable additives, can be used.
774 In relation to plasticizers, I accept that the MUPS Patent specification particularly recognises that optimisation is within the ordinary skill of the formulator when it states that:
[T]he amount of plasticizer is optimized for each enteric coating layer formula, in relation to selected coating layer polymer(s), selected plasticizer(s) and the applied amount of said polymer(s), in such a way that the mechanical properties… are adjusted so that the acid resistance of the pellets covered with enteric coating layer(s) does not decrease significantly during the compression of pellets into tablets.
775 However, these general observations do not necessarily lead to the conclusion that there is no inventive step. They do not take into account the various difficulties that could arise in selection of suitable ingredients in appropriate amounts, and the general difficulties otherwise referred to by Professor Bodmeier and Dr Morella.
776 For instance, Professor Bodmeier and Dr Morella’s evidence was that too much or too little of a particular excipient will generally cause problems, including a final dosage form that fails to deliver the full dose of the active substance to the patient in the desired way. Further, a formulator cannot necessarily predict in advance whether a particular excipient will cause problems. For example, an excipient could cause the active substance to degrade, or it could alter the drug release profile of the dosage form.
777 Professor Bodmeier and Dr Morella agreed that modifying one ingredient in a pharmaceutical formulation in an attempt to meet one performance criterion (eg acid resistance) will often cause problems (both expected and unexpected) in relation to another criterion (eg dissolution rate). For example, adding too little enteric coating polymer could result in the final dosage form having inadequate acid resistance. Adding too much enteric coating polymer could result in undesirable interactions between components of the final dosage form, or give rise to processing or other problems, or compromise the dissolution rate of the dosage form (making it less likely that the buffer stage of the USP test for enteric coated formulations would be satisfied). Dr Reiland acknowledged that “every combination of ingredients and processing parameters can behave differently” and that “all coating components can affect the final performance of the barrier film formed”.
778 I accept that because a modification to one aspect of the pharmaceutical formulation almost always results in changes to other aspects, in selecting excipients and process parameters, the formulator generally works within established ranges. This is particularly the case with tablets and with complicated dosage forms, such as modified release multiparticulate dosage forms. Even for active substances, whose properties make them suitable for formulating into tablet form (eg where they have good compressibility, dissolution, and bioavailability properties), it can be difficult to design pharmaceutically acceptable single-unit tablets, let alone multiparticulate tablets.
779 Professor Bodmeier gave evidence that departing from the established ranges for excipients used in tablets and pellets will cause formulation problems. In many cases, an increase in the concentration of an excipient does not improve, but rather worsens, its desired effect. Professor Bodmeier gave the example of the common lubricant magnesium stearate (used in many commercial solid oral dosage forms to reduce friction during tablet ejection between the die cavity walls and the tablet) which has a recommended range of 0.5% to 1% as a percentage of all tableting excipients. If higher lubricant concentrations are used (eg 2% or more), problems may arise, including the tablet becoming too soft or disintegrating too slowly. This prevents the active substance from being released in the desired way. Conversely, too little lubricant can cause the tablets to stick to the die cavity walls, which means that the formulation cannot be processed.
Investigation of formulation problems
780 Professor Bodmeier and Dr Morella gave evidence that the formulator investigates the causes of problems (eg not passing quality control tests such as the USP test) to understand how to resolve them. The potential causes of such a problem include things like imperfections in the coating, the amount of enteric coating polymer applied, and coating composition. Further, Professor Bodmeier and Dr Morella agreed that identifying the causes of problems is difficult as they are manifold. During cross-examination, Professor Bodmeier stated:
Because when a formulator formulates and he sees a problem, he doesn’t know the solution. He doesn’t know if – for example, if there are problems with gastric-acid resistance that this comes from the plasticiser and when we focus here always on the plasticiser, but when a formulator works and sees a problem, he doesn’t know where to start. It could be any excipient, it could be the process, and I highlighted here – highlighted here – the problems you could run into with a plasticiser. Okay? And this is already a multitude of problems you can have with a plasticiser only – but you don’t know if this problem comes from the plasticiser.
781 Dr Reiland’s evidence on this issue was that a formulator does not investigate the causes of every problem; rather, the formulator “would take note of the particular combination of ingredients or process variables that resulted in the failure and move on to try a different combination of ingredients or process variables”. He said that it was normally sufficient for a formulator to know that a particular combination of variables did not work, without diverting one’s attention to exploring in detail the cause of why it did not work. However, Dr Reiland also accepted that, for example, “[t]o satisfy the performance criteria plasticizers would be extensively studied via formulation trials”. In respect of US 505 and whether it would have caused him at the priority date to vary the compression parameters, he accepted that “[s]ystematic changes would need to be applied to give the components in the formulation a chance to show they could be optimized to success because every combination of ingredients and processing parameters can behave differently”.
782 I consider that a formulator would be interested in investigating the causes of problems, as this may lead to a solution. In doing so, the formulator necessarily must undergo further experimentation, which in all probability will produce a slower and more laborious approach. This is relevant in determining the extent to which the formulator will carry out experimentation with any reasonable expectation of success, and the extent of the task of experimentation that would need to be undertaken. Dr Reiland’s position, even if accepted (involving as it does formulation trials and the application of systematic changes), would not necessarily be routine in the overall context of determining the way forward.
The quantity of enteric coating used by the formulator
783 As previously noted, it is uncontroversial that all components within the enteric coating can affect the performance of the final dosage form. One controversy before me was the extent to which the quantity of the enteric coating can affect the performance of the final dosage form.
784 AstraZeneca submitted that the skilled formulator would not seek to increase the quantity of enteric coating beyond established ranges. Ranbaxy’s arguments on this issue have already been set out in these reasons for judgment. Professor Bodmeier and Dr Morella agreed that increasing the quantity of enteric coating can compromise the rate of dissolution of the dosage form in the intestine, among other potential problems.
785 Professor Bodmeier gave affidavit evidence that commonly used enteric coating amounts by weight of the pellet substrate to be enteric coated are 10% to 30%. In the Joint Expert Report on AU 966, Dr Morella stated that the factors which would be taken into account in the selection of enteric coating components include “[c]ost of manufacture which includes using enteric coat weights of less than about 15% to avoid long manufacturing and contain the cost of excipients”.
786 Further, Professor Bodmeier and Dr Morella agreed that, even if an enteric coated dosage form meets the requirements of the in vitro USP test, it may not achieve adequate bioavailability in vivo. Professor Bodmeier explained that this is due to factors that complicate absorption of the active substance, including:
(a) the fact that the pH of gastric juices can range from 1.5 to 4.0;
(b) the fact that the amount of gastric fluid may vary between patients, and for the same patient at different times; and
(c) the fact that “gastric residence time of the enteric coated dosage form” can vary depending on the time of administration of the pharmaceutical preparation and whether it is taken with food.
787 Professor Bodmeier and Dr Morella both considered that the thickness of the enteric coating layer will affect the rate of dissolution of the enteric coating layer in the intestine. Professor Bodmeier explained this during the concurrent evidence session:
So if the enteric coating, for example, is too thick or is very thick, you have good protection in the stomach, so the drug does not degrade, but when this formulation then gets in the intestine, it takes a long time for the enteric coating to dissolve and the drug then actually – like a drug like omeprazole is not absorbed sufficiently.
788 Dr Morella agreed with this explanation. As Professor Bodmeier explained, while enteric coating polymers dissolve in intestinal fluid, “the question then is the rate, how quickly do they dissolve, and it takes time for a coating to dissolve”.
789 Dr Reiland, by contrast, maintained that increasing the amount of enteric coating would not be expected to compromise the dissolution rate. However, Dr Reiland’s evidence accepted that he would “make a series of formulations with higher enteric coating amounts and collect and assess the USP Enteric Coated Standard data to assure the dissolution rate was not significantly affected”. In cross-examination, Dr Reiland stated that “with a slight difference in the thickness of the coating, [he] would not expect to have any substantial effect on releasing the core”, but also acknowledged that “significantly large changes” in thickness of an enteric coating would make a difference.
790 In the examples, the MUPS Patent teaches the skilled addressee to add at least 40% and up to 100% enteric coating polymer (ie film forming agent) (% w/w based on core). Professor Bodmeier gave evidence that these weights of enteric coating are extraordinary. Dr Reiland agreed that 100% is unusual. AstraZeneca submitted that it was “telling” that Dr Reiland gave no evidence of the weights of enteric coating he commonly applied at the priority date. Further, Dr Reiland’s comments regarding dissolution were limited to what happens in a test apparatus, where the pH is substantially above the pH where the active substance is supposed to be released. AstraZeneca submitted that this was a further example of Dr Reiland’s concern with what happens in vitro, rather than in vivo.
791 Again, I accept the evidence of Professor Bodmeier and Dr Morella. This is yet another example of a difficulty that needs to be dealt with by the skilled formulator along the way to arriving at the invention.
The degree to which enteric coated multiunit tablets were common, and the Abbott Laboratories PCE products
792 Professor Bodmeier gave evidence that, as at 8 July 1994, while modified release multiunit capsules and oral sachets were relatively common, modified release multiunit tablets were much less common. The modified release multiunit tablets that did exist were generally prolonged release (ie sustained release), and not enteric coated formulations. As previously foreshadowed, Professor Bodmeier’s evidence was that, as at 8 July 1994, he was not aware of any commercially marketed multiunit tablets containing enteric coated pellets. Remington’s and Lachman make no reference to enteric coated multiunit tablets.
793 Ranbaxy has provided no evidence of the existence of multiunit tablets containing delayed release coated pellets at the priority date. Dr Reiland gave evidence that he was only aware of one example of an active substance that had been formulated into a successful multiunit tablet containing enteric coated particles that was marketed before 8 July 1994: namely, Abbott Laboratories’ PCE products, over which it had a number of patents. These products were erythromycin (an antibiotic) formulated into enteric coated particles compressed into a tablet. As a result, they are relevant to a number of different issues relating to the MUPS Patent. Broadly, I understand that there were two stages to the development of Abbott Laboratories’ PCE products – the first stage involved the development of a 333 mg dose of erythromycin (previously described in these reasons as ‘PCE 333’). Dr Reiland was not directly involved in this process. The second stage involved the development of a 500 mg dose of erythromycin (previously described in these reasons as ‘PCE 500’). Dr Reiland was involved in this process.
794 Dr Reiland was aware of these products because of his role at Abbott Laboratories (being the developer of PCE). Dr Reiland’s statement in response to question 5(a) of the Joint Expert Report on AU 966, that “[i]t was also known at July 1994 that enteric coated particles had been successfully compressed into tablets several years earlier”, refers to his first affidavit, in which Dr Reiland discussed his personal experience with PCE as a result of his role at Abbott Laboratories. Dr Reiland admitted that his evidence as to how he would have solved the “inventive step” question posed to him by King & Wood Mallesons was informed in part by what he knew about the PCE products, and that this knowledge was “very instrumental”. Dr Reiland also admitted that he was not involved in the formulation of the enteric film of the PCE pellets, and did not know what was in the film formulation.
795 As at 8 July 1994, Professor Bodmeier was not aware of Abbott Laboratories’ PCE formulation. Professor Bodmeier’s 1997 review article entitled “Tableting of coated pellets”, published in the European Journal of Pharmaceutics and Biopharmaceutics (volume 43, p 1), does not refer to Abbott Laboratories’ PCE products (however, footnote 29 of that article does refer to one patent related to Abbott Laboratories’ PCE 333 formulation, being US Patent No 4,874,614 (‘US 614’)).
796 According to Dr Morella, the PCE formulation was the only marketed multiunit tablet containing enteric coated pellets of which he was aware before 8 July 1994. Dr Morella was aware of Abbott Laboratories’ PCE formulation only because he worked on a specific project attempting to compress enteric coated erythromycin pellets into a tablet. As a related issue, I note that the two projects that Dr Morella worked on before 8 July 1994 which attempted to compress enteric coated particles into a tablet both failed.
797 AstraZeneca submitted that, given that Professor Bodmeier, a university academic with a specialist interest in the field, was not aware of PCE, and that Dr Reiland and Dr Morella were only aware of PCE because of their personal experience working on erythromycin formulations, the evidence is not sufficient to prove that an awareness of Abbott Laboratories’ PCE formulation was part of the common general knowledge of the skilled addressee (let alone any knowledge of the particular manner in which Abbott Laboratories’ PCE formulation had been formulated).
798 For the same reasons, it was submitted that the information about PCE in the PDR (being the Physicians’ Desk Reference, an American publication related to products available in the USA, referred to hereafter as ‘PDR PCE’) was not part the common general knowledge either. In any event, the very limited nature of that information was explained by Professor Bodmeier and Dr Morella in their evidence. Professor Bodmeier stated that “[t]he PDR provides a list of inactive ingredients but does not identify the coating polymer nor the structural composition of the granules nor how the PCE tablets are manufactured”. Dr Morella stated that “PDR PCE does not disclose any information about the identity of the enteric coating polymer (the general expression ‘cellulosic polymers’ is used), nor the relative quantities of excipients used, nor where in the formulation (eg in which layer) each of those excipients is used”.
799 I accept these submissions. I do not think that the PCE products (or their corresponding PDR entry) formed part of the common general knowledge at the priority date. I consider that there is no evidence before me to conclude that any formulator outside of the United States as at 8 July 1994 should have been aware of the PCE products as commercially available. But even if I am mistaken about that, I do not consider that the PCE products (or the corresponding patents) were sufficient to point the skilled addressee to the invention that is the subject of the MUPS Patent. The patents are dealt with further below.
The degree to which enteric coated multiunit tablets are advantageous compared with capsules
800 Question 5(b) of the Joint Expert Report on AU 966 invited the experts to comment on what was known as at 8 July 1994 in relation to any advantages of making tablet forms of multiunit enteric coated particle formulations. In their responses, the experts referred to a number of advantages of tablets compared with capsules, such as:
(a) reduced risk of tampering, and divisibility (Professor Bodmeier);
(b) allowing a combination of drugs to be included (Dr Morella); and
(c) ease of swallowing and push-through blister packaging (Dr Reiland).
801 Question 5(b) did not invite the experts to comment on any disadvantages of tablets compared with capsules. However, Dr Reiland in his first affidavit referred to a number of reasons why capsules might be preferred, such as aesthetics, identification and accommodating sprinkle dosage forms.
802 In oral evidence, Dr Morella noted some advantages of tablets over capsules, including that they could be cheaper to manufacture, and are more compact. Dr Morella also noted advantages of capsules over tablets, including that they have the advantage of already being coated by hard gelatine which will mask flavour, and that capsules are easier to administer. Dr Morella identified the following attributes as being applicable to both tablets and capsules:
(a) simplicity (although he noted that one of the simplest dosage forms, in his experience, was a powder-filled capsule, which avoids “many of the difficulties that you have in blending powder, and getting adequate powder flow from powders that have to be tabletted”);
(b) stability (although he noted that in his experience, “putting products into a tablet created stability problems, whereas they were stable in capsule product”);
(c) convenience and packaging;
(d) shipping and dispensing; and
(e) accuracy of dosage.
803 In oral evidence, Professor Bodmeier said:
So it really depends on the particular drug on the particular problem you are doing and then I agree with Dr Morella, actually, the first dosage form when you go in humans, is formulating the drug into capsules because it’s the simplest formulation. You don’t need to worry about compressibility. In tablets, you need to add other excipients that could cause stability problems. Here in capsules, you have - you don’t really need fillers. You don’t have to worry about bindings, so capsule formulation is, in general, a simpler than tablet formulation. And again, you will find in textbooks, advantages of tablets and then a few chapters later on, you find the same advantages for capsules.
804 Remington’s at p 1633 states:
Tablets remain popular as a dosage form because of the advantages afforded both to the manufacturer (eg, simplicity and economy of preparation, stability and convenience in packaging, shipping and dispensing) and the patient (eg, accuracy of dosage, compactness, portability, blandness of taste and ease of administration).
805 At p 1658, Remington’s lists a number of advantages of capsules, including that “[s]ome patients find it easier to swallow capsules than tablets, therefore preferring to take this form when possible”. Remington’s goes on to state that “[t]his preference has prompted pharmaceutical manufacturers to market the product in capsule form even though the product has already been produced in tablet form”.
806 Further, Ranbaxy submitted that AstraZeneca’s own internal documents characterised the “marketing advantages” of tablets containing compressed enteric coated particles over capsules in the following terms:
• better chemical and mechanical stability than capsules → patient-friendly packages e.g. press-through [sic] blisters;
• dispersable;
• brand strengthening;
• better patient-acceptance than capsules;
• probably cheaper to produce than capsules;
• lactose and gelatin free.
807 In light of the foregoing evidence, particularly the oral evidence of Professor Bodmeier referred to above, I find capsules and tablets each have different advantages over the other depending on the objectives that the formulator is trying to achieve, and that neither dosage form was clearly known to be superior to the other in all circumstances at the priority date. However, I note that Dr Reiland agreed that in 1994, capsules were the conventional dosage form for enteric coated particles.
THE DIFFICULTY OF COMPRESSING ENTERIC COATED PARTICLES INTO MULTIUNIT TABLETS
808 Professor Bodmeier and Dr Morella gave evidence that multiunit tablets containing enteric coated pellets are highly complex. This is significant evidence, which I have accepted. The compression of enteric coated pellets into multiunit tablets, which, ideally, have the same modified release properties as the individual coated pellets, is problematic. The formulation of each active substance into an enteric coated multiunit tablet presents challenges, which often cannot be overcome, even with the best know-how and resources. I accept this evidence.
809 Professor Bodmeier’s evidence is that the potential difficulties faced by a formulator attempting to design a multiunit tablet containing coated pellets include:
(a) damage to or changes to the properties of the pellet coating during compression, including changes to the thickness, uniformity and permeability of the pellet coating;
(b) damage to the pellet beneath the coating during compression;
(c) fusion of pellets during compression;
(d) incompatibilities between the pellet coating and the tablet excipients;
(e) problems with the tablet disintegrating (eg the tablet disintegrates after compression or does not disintegrate in the stomach); and
(f) lack of content uniformity because of de-mixing and flow problems of pellets and other excipients.
810 Professor Bodmeier’s evidence was that, if it were absolutely necessary to develop multiunit tablets of omeprazole, the formulator could attempt to overcome such difficulties by adjusting one or more of the following variables:
(a) the process parameters, eg the compression and coating parameters; and/or
(b) the tablet matrix (the material surrounding the pellets), eg by modifying its composition, concentration or ratio of matrix to number of pellets, including inert granules in the matrix; and/or
(c) the pellet substrate beneath the coating, eg, modifying its size, hardness, composition, porosity; and/or
(d) the coating, eg using a different polymer; or modifying the amount of polymer or the composition of the coating formulation by reference to the coating polymer manufacturer’s recommendations, the formulator’s experience with the polymer and the literature.
811 However, Professor Bodmeier’s evidence was that modifying these variables in a way that successfully overcomes the difficulties described above is highly complicated. These variables are interrelated and as I have previously noted, a modification to one aspect almost always results in changes to other aspects, often in ways that are unpredictable and that can give rise to further problems that need to be solved. Professor Bodmeier’s 1997 review article supports the position that compressing coated pellets into tablets involves an extremely complex and also time consuming process. The conclusion of the article is that a multiparticulate system involving the use of matrix-type pellets (which involve the active substance being homogenously dispersed throughout the polymeric carrier) showed potentially fewer problems under compaction than coated multiparticulate systems. However, matrix-type pellets are used for prolonged release, not enteric coated, formulations.
812 Professor Bodmeier gave evidence that despite publication of this article in 1997, he considered that sufficient information was available as at 8 July 1994 to provide a basis for the same conclusions to be drawn by the skilled addressee. I accept that evidence.
813 In summary, Professor Bodmeier’s evidence was that the potential difficulties referred to above make formulating modified release multiparticulate tablets a much less attractive option than other modified release multiparticulate formulations, such as capsules or oral sachets. This accorded with Dr Morella’s personal experience that, prior to 1995, the vast majority of pellet products that he worked on developing were pellets formulated into capsules.
814 Dr Morella also set out the large number of variables that would be investigated by the skilled addressee in trying to compress enteric coated pellets containing omeprazole into a multiunit tablet. Dr Reiland accepted these variables as relevant to be considered by the formulator.
815 However, Dr Reiland relied on his “systematic experimental approach” and his knowledge of Abbott Laboratories’ PCE formulation to overcome the difficulties of compressing enteric coated pellets into a tablet. In the concurrent evidence session, Dr Reiland said:
a logical approach that a formulator would take would be to take what’s known that a compression event is much more stressful and they would naturally want to use a more plasticised formulation and use more coating to allow that to withstand the compression force and then the obvious thing that everyone seems to acknowledge is they would additionally use cushioning agents to attempt success.
[It]… is very well recognised that a formulator would take a logical approach and two things he can do is he can plasticise the formulation to make the particles more flexible and he can add tableting excipients
…
[the developers of PCE 333] took the – the standard industrial formulation of approach knowing that plasticisers affected flexibility and hardness, and because they were making a tablet they would need to use higher levels of plasticiser to withstand this compression event, because the previous product that they were attempting to improve on didn’t have to go through that. So they were just doing the logical thing, and they were using plasticisers to plasticise, which didn’t seem anything out of the ordinary. That’s what formulators do.
816 I accept that Dr Reiland’s evidence on this aspect is affected by hindsight, and is very much based on his own experience and recollections. I do not regard this as the approach of the skilled addressee in 1994. I accept that the large number of variables set out by both Professor Bodmeier and Dr Morella include so many possibilities confronting the formulator that he or she would not have been led to try the invention with any expectation of success.
The PCE patents
817 A separate but related issue is whether the patents relating to the PCE products teach away from the invention claimed in the MUPS Patent.
818 AstraZeneca submitted that even if the PCE patents were somehow relevant to the inventive step inquiry, they lead away from the MUPS Formulation.
819 In the concurrent evidence session, US 614 and US 741 were put to Dr Reiland. Dr Reiland agreed that:
(a) these patents covered the PCE 333 product that made it to market; and
(b) the only PCE 333 product that made it to market used microcrystalline cellulose as a cushioning agent.
820 Dr Reiland’s position was that a formulator would be led to add more plasticizer and increase the thickness of the enteric coating because Abbott Laboratories had already solved the problem with erythromycin. However, Abbott Laboratories solved the problem in a different way, which would not lead to the MUPS invention.
821 In oral evidence, Dr Reiland agreed that the purpose of US 614 and US 741 was to illustrate that the problem of enteric coated particles crushing when compressed could be solved by adding microcrystalline cellulose as a cushioning agent. It was undisputed that microcrystalline cellulose is a binder that provides cushioning for the coated particles and is not a plasticizer. Dr Morella and Professor Bodmeier both gave evidence that, in the PCE patents, microcrystalline cellulose is not being used to change the enteric coating of the particles.
822 In cross examination, Dr Reiland conceded that there is nothing in either of US 614 or US 741 that teaches that the way to solve the problem of enteric coated particles fracturing under compression is to adjust levels of plasticizer or adjust the thickness of the coating. Dr Morella and Professor Bodmeier agreed.
823 Dr Reiland’s position is that, despite the absence of any reference to increasing plasticizer or the thickness of the enteric coating in US 614 and US 741, Abbott Laboratories did nonetheless modify the enteric coating of its PCE 333 product, and there was nothing special in this process that needed mentioning in the patents. I cannot accept Dr Reiland’s recollection or view in this regard. Dr Reiland was not involved in the development of the PCE 333 formulation and cannot recall the formulation. Rather, Dr Reiland’s knowledge of the formulation of the PCE 333 product is primarily based on his recollections of discussions between colleagues at meetings approximately thirty years ago.
824 Further, the failure of the patents to mention the role of a plasticizer would be remarkable, if, as Dr Reiland contended, the role of plasticizer was to make the particles more flexible.
825 US 897, on which Dr Reiland is named a co-inventor, underpins or reflects the PCE 500 formulation. As with the previous PCE patents there is nothing in US 897 to suggest that the enteric coat should be modified. Example 3 of US 897 discloses a product that has erythromycin alone as the active ingredient (like PCE 333 and PCE 500), and the level of plasticizer used is 12%. Example 1 uses the same (enteric coat) polymer as example 3 but a different plasticizer (and the level of plasticizer is 10%). There is no disclosure of an unusually thick enteric coat. Dr Reiland indicated that he started his PCE 500 project using the pellets made for PCE 333. Dr Reiland accepted that these pellets were friable.
826 I accept that to the extent that the PCE patents are relevant, the lower dosage amount required for omeprazole compared to erythromycin would have suggested adding microcrystalline cellulose as a tableting excipient rather than modifying the enteric coating. Furthermore, omeprazole is an entirely different substance to erythromycin. I have previously noted that I accept the evidence before me that even related compounds can present different challenges to a formulator.
THE FORMULATOR’S APPROACH TO ENANTIOMERS AND RACEMATES
827 Dr Morella and Professor Evans gave evidence that, because the physicochemical properties of enantiomers (and salts thereof) are expected to be different from the physicochemical properties of the racemate (and salts thereof), a formulation of a racemate is not expected to work for its enantiomers (and vice versa). Further, in cross-examination, Ranbaxy’s witness, Dr Pyter, agreed that for a racemic compound, it is not possible to predict the physical and chemical properties of one of its enantiomers.
Analysis of Common General Knowledge and Section 7(3) Documents
828 It is convenient here to refer to a number of documents relied upon by Ranbaxy as common general knowledge or s 7(3) documents:
(a) an extract from The Merck Index (1989, 11th ed);
(b) an extract from Goodman & Gilman’s (1992, 8th ed);
(c) extracts from Physicians’ Desk Reference (1992, 46th ed) relating to Prilosec® (‘PDR Prilosec’);
(d) extracts from the PDR relating to Particle Coated Erythromycin Dispertab® tablets (previously referred to in these reasons as ‘PDR PCE’); and
(e) US 505.
829 As I will later discuss, I do not consider the information contained in these documents assists Ranbaxy even if part of common general knowledge or if treated as separate s 7(3) documents. Nevertheless, I make some observations in relation to certain contentions made by the parties as to the ability of Ranbaxy to rely on these documents.
The Merck Index 1989, Goodman & Gilman and PDR Prilosec
830 The first contention arises in relation to Ranbaxy’s reliance on the information in The Merck Index 1989, Goodman & Gilman and PDR Prilosec. Dr Reiland’s affidavit evidence was that at the priority date, the course of action he would have taken to develop an alternative solid oral dosage formulation of omeprazole would have commenced with an initial review of these documents in order to ascertain “what is known about the API omeprazole, in particular the physicochemical properties of the API, its formulation, bioavailability and pharmacokinetics”. As I have already discussed in respect of the Purity Patent, the inclusion of information in a document, however widely circulated, does not of itself make that information part of the common general knowledge. Ranbaxy has presented no evidence that all of the information in each of these three documents is “‘generally accepted without question’ or ‘generally regarded as a good basis for further action’ by the bulk of those in the art” (Alphapharm (2008) 76 IPR 618; [2008] FCA 559 at 667 [221]). In my view, Ranbaxy has failed to prove that the information relied upon in each of The Merck Index 1989, Goodman & Gilman and PDR Prilosec is common general knowledge.
831 Because Ranbaxy has failed to prove that the information in each of The Merck Index 1989, Goodman & Gilman and PDR Prilosec relied upon by Dr Reiland is common general knowledge, each document must be considered as a separate s 7(3) document. On this basis, AstraZeneca took issue with the evidence of Dr Reiland, who – it was said – impermissibly approached the inventive step inquiry based on all of the information in all three documents combined. Accordingly, it was submitted, Dr Reiland’s evidence cannot be relied upon.
832 However, in my view, a preliminary issue arises.
833 Section 7(3) information must be information that the person skilled in the relevant art “could, before the priority date of the relevant claim, be reasonably expected to have ascertained, understood and regarded as relevant”. As Lockwood (No 2) (2007) 235 CLR 173 at 223 [153] confirms, this must be established by evidence. Relevantly, the Court in that case held that “ascertained” (as used in s 7(3)) means discovered, or found out: Lockwood (No 2) (2007) 235 CLR 173 at 219 [132]. Accordingly, it must be shown by appropriate evidence that the information relied upon could reasonably be expected to have been discovered or found out by the skilled addressee, without hindsight and in the ordinary course of events. Similarly, it must be established that the skilled addressee could reasonably be expected to have regarded the information as relevant: Lockwood (No 2) (2007) 235 CLR 173 at 222-223 [152].
834 There is no evidence to conclude that the three documents are s 7(3) documents. In particular, there is no evidence that the person skilled in the relevant art would reasonably be expected to treat each document, in itself, as relevant. On this basis, the three documents cannot be relied upon by Ranbaxy.
PDR PCE
835 I have already concluded that the existence of the PCE products, and the information about the PCE formulation contained in PDR PCE, was not part of the common general knowledge of the skilled formulator at the priority date.
836 Ranbaxy sought to rely upon this information as s 7(3) information. I conclude that it is not s 7(3) information. In his first affidavit, Dr Reiland was asked what course of action he would have taken at July 1994 to develop an alternative solid oral dosage form of omeprazole (assuming that he was part of a formulation development team at a pharmaceutical company at the time). Dr Reiland stated that he would “initially review the entries for omeprazole in… the PDR”. Dr Reiland does not state that he would ascertain, understand and regard as relevant the information in PDR PCE to a formulation of omeprazole. Also in his first affidavit, Dr Reiland describes the literature search that he would do as part of the inventive step inquiry. The results of that literature search (which are extracted at annexure TLR-11 to that affidavit) do not include PDR PCE or any other information about PCE. The PDR PCE entry is separately annexed to that affidavit, but in the context of Dr Reiland explaining his own personal experience with PCE.
837 I should also indicate that even if the skilled formulator ascertained information about PCE in the course of developing a formulation of omeprazole, or even if that information was part of the skilled formulator’s common general knowledge, they would not have regarded that information as relevant. The active substances are different, and hence, as I have earlier accepted, their properties (which will affect the way in which delivery of the active substance is to be effected) are likely to be different.
838 Further, PDR PCE does not contain sufficient meaningful information about the way PCE is formulated to be of any assistance to the skilled formulator. Professor Bodmeier stated that “[t]he PDR provides a list of inactive ingredients but does not identify the coating polymer nor the structural composition of the granules nor how the PCE tablets are manufactured”. Dr Morella stated that “PDR PCE does not disclose any information about the identity of the enteric coating polymer (the general expression “cellulosic polymers” is used), nor the relative quantities of excipients used, nor where in the formulation (e.g. in which layer) each of those excipients is used”.
839 In addition, the information contained in PDR PCE suggests that the PCE formulation would not be suitable for omeprazole. PDR states that the polymer coating “allows for minimal release of erythromycin in acidic environments”. Professor Bodmeier understood from this statement that “the coating on the granules in the PCE tablets loses some of its function following compression into tablet form”, which Professor Bodmeier regards as unacceptable for a highly acid sensitive compound such as omeprazole.
840 It is on the basis of this evidence that I conclude that PDR PCE is not s 7(3) information.
REMINGTON’S AND LACHMAN
841 Ranbaxy also relied on selected extracts from Remington’s and Lachman to establish common general knowledge. Neither Remington’s nor Lachman was ever pleaded by Ranbaxy as being representative of the common general knowledge, or as a s 7(3) document. However, whatever their status, there is no reference therein to the compression of enteric coated pellets into multiunit tablets, and I find that the textbooks did not teach it. Remington’s and Lachman relate to single-unit tablets. Whilst the extracts I have referred to deal with ‘general’ matters relevant to this proceeding, they do not specifically deal with the issues dealt with by the witnesses as to the compression of enteric coated pellets into multiunit tablets.
842 In light of my findings on the common general knowledge of the skilled addressee at the priority date and the evidence as to the problems in the path of a formulator, I now proceed to consider the analysis of inventive step, as informed by the reformulated Cripps question.
The Cripps Question
843 It will be apparent from the foregoing that I accept that the specification of the MUPS Patent goes no further than to admit what AstraZeneca contended is part of the common general knowledge, namely that:
(a) omeprazole and its alkaline salts are effective gastric acid secretion inhibitors and are useful as antiulcer agents; and
(b) omeprazole is acid labile, and must therefore be administered as an enteric coated formulation.
844 I accept that the decision to formulate a multiunit tablet of omeprazole is an aspect of the invention. The common general knowledge included knowledge of the advantages of multiunit tablets and capsules, although with a preference for capsules, depending on the circumstances. Accordingly, the skilled formulator would not necessarily consider it desirable to develop a multiunit tablet, particularly given the skilled formulator’s common general knowledge of the great difficulty of doing so.
845 While there are references in the specification of the MUPS Patent to the desirability of formulating multiunit tablets containing omeprazole, these statements go no higher than expressions of a desire to improve upon the status quo. They are made in hindsight with knowledge of the particular improvement realised by the invention. The desire for a multiunit tablet containing omeprazole is not the starting point for the assessment of inventive step. Rather, the decision to formulate a multiunit tablet containing omeprazole (as opposed to some other formulation) was part of the invention.
846 The “step” which must be tested for inventiveness is the step of developing a useful or improved solid oral dosage formulation of omeprazole. The invention of the MUPS Patent is a combination of integers, including an “effective amount of a plasticizing compound”, the “mechanical properties” and the “gastric acid resistance” (referred to by AstraZeneca as “the MUPS Formulation”). Each dependent claim adds specific integers to the MUPS Formulation.
847 The invention claimed in each of the relevant claims of the MUPS Patent is taken to involve an inventive step when compared with the prior art base, unless the invention would have been obvious to a skilled formulator in the light of the common general knowledge, whether that knowledge is considered separately or together with a s 7(3) document. In AstraZeneca’s submission, for the purpose of determining whether the MUPS Formulation involved an inventive step, the Cripps question (formulated in terms appropriate for the MUPS Patent) is:
Would the notional research group at the relevant date, based on their common general knowledge, directly be led as a matter of course to try the MUPS Formulation, in the expectation that they might well produce a useful alternative to or a better formulation than existing formulations of omeprazole?
848 There are two limbs to the Cripps question, namely, (i) whether the skilled formulator would have been directly led to try the invention of the MUPS Patent; and, if so, (ii) whether they would have reasonably expected to succeed. It will be appreciated from the foregoing that I consider the answer to these questions is ‘no’.
849 Ranbaxy has not provided any compelling evidence as to why the formulator, faced with that practical disincentive, would nevertheless be directly led as a matter of course to try a multiunit tablet containing omeprazole. I have found that there were advantages to multiunit tablet formulations, but there were also advantages to multiunit capsule formulations. The evidence is against the proposition that in the specific case of omeprazole, the skilled formulator would be directly led as a matter of course to try a multiunit tablet, let alone the MUPS Formulation. There was not a generally recognised problem with an existing omeprazole formulation as at 8 July 1994. Dr Reiland’s evidence in his first affidavit about developing an alternative oral solid dosage formulation of omeprazole was asserted to be based on information in Goodman & Gilman and the PDR. However, I have already indicated that I accept that his approach was strongly influenced by his personal experience at Abbott Laboratories, which included information confidential to Abbott Laboratories.
850 While I am satisfied that there is evidence before the Court of some advantages of multiunit tablets, there is no evidence before the Court as to why the skilled formulator may consider that those advantages justify the development of a multiunit tablet containing omeprazole. For example, one potential advantage of a multiunit tablet relied upon by Ranbaxy is divisibility. But there was no evidence of a need for a dosage form of omeprazole that is divisible (and indeed, as AstraZeneca noted in their submissions, the tablets comprising the Ranbaxy Product are not divisible). In the concurrent evidence session, Dr Morella said that he “hadn’t considered that as a desirable attribute at the time”. Even if one were to accept that the information in PDR Prilosec was part of the common general knowledge, that document only discloses that the recommended dose of omeprazole is 20 mg and that it is sold in capsules containing 20 mg of omeprazole. PDR Prilosec does not disclose a desirability for a 20 mg dose that can be divided in two.
851 As previously foreshadowed, another potential advantage of a multiunit tablet relied upon by Ranbaxy is ease of swallowing. Accepting that it is generally advantageous to have a dosage form that is easier to swallow, there is again no evidence before the Court that such a need would motivate or justify the research and development program required to formulate a multiunit tablet containing omeprazole. In the concurrent evidence session, Dr Morella said that in his experience “capsules were described as being easier to administer than tablets”. Remington’s indicates a consumer preference for capsules. In the concurrent evidence session, Dr Reiland agreed that capsules could be easier to administer:
The intent purpose of Depakote Sprinkle was to provide an easy to administer taste mask particle coated dosage form that mothers could pull apart and sprinkle. You can’t pull apart a tablet, so that is definitely why we chose to use a capsule. We wanted to have a dosage form to allow the mother’s [sic] to administer the epileptic medication to their children without them tasting it and the easiest way we could see to do that was to take advantage of the advantage of a capsule, that it could be easily pulled apart, taste mask particles could be sprinkled on food which we couldn’t do with the tablet.
852 Further, I have already indicated that because of the known difficulties with compressing enteric coated pellets into tablets, the skilled formulator would not be directly led as a matter of course to try to compress enteric coated pellets containing omeprazole into a tablet. Professor Bodmeier explained in detail that a number of problems can arise when compressing enteric coated pellets into tablets.
853 Professor Bodmeier states that “[t]hese difficulties make formulating modified release multiparticulate tablets a much less attractive option than other modified release multiparticulate formulations, such as capsules”. Dr Morella agrees that “compressing enteric coated pellets into a tablet, in a manner that meets performance criteria such as acid resistance, acceptable dissolution rate and stability, is a very challenging task”. It will be recalled that I have accepted the evidence before me about the problems likely to be experienced during the tableting process, the difficulty of determining the causes of those problems and the large number of possible variables that might need to be explored in an attempt to overcome those problems.
854 Accordingly, I am satisfied that the degree of difficulty of compressing enteric coated pellets containing omeprazole into a tablet would, in the absence of any problems or ‘needs’ identified in the common general knowledge at the priority date in respect of omeprazole, lead the formulator towards an easier formulation, such as enteric coated pellets formulated into capsules or oral sachets.
855 I note that it is, in any event, insufficient for Ranbaxy to prove that the skilled formulator would be directly led as a matter of course to try a multiunit tablet containing omeprazole. Ranbaxy must prove that the skilled formulator would be directly led as a matter of course to try the particular combination of integers claimed in the MUPS Patent, including an enteric coating layer with an effective amount of a plasticizing compound, the mechanical properties and the gastric acid resistance. I do not consider that Ranbaxy gets over either of these hurdles.
856 AstraZeneca submitted that Ranbaxy’s evidence based on Dr Reiland’s “systematic experimental approach” is insufficient to prove as a matter of fact that the skilled formulator would be directly led as a matter of course to try the invention of the MUPS Patent. In particular, they asserted that his evidence was that he would not have investigated the composition of the enteric coating layer in preference to any other variables. He would not have been directly led to try the combination of integers claimed in the MUPS Patent. This was said to be illustrated by the following passage from Dr Reiland’s affidavit evidence:
Both Dr Morella and Professor Bodmeier have commented that I have an apparent ‘focus’ on the amount of plasticizer in the enteric coating, and both state that I have focused on the amount of plasticizer in preference to other variables in the formulation. More generally, they also characterize my approach as investigating the composition of the enteric coating layer in preference to other aspects of the formulation. I do not consider that this accurately reflects my approach.
The approach that I described in my First Affidavit in response to the Question began with making rapidly dissolving particles, exploring the need or benefit for a subcoating layer, selecting the enteric coating polymer, moving to consider the identity and amount of plasticizer and systematically varying the other components of the formulation. I reiterate that this is a logical approach that an industrial formulator would use to formulate an enteric coated product of the kind I identified in paragraphs 7.5(b), 7.20 and 7.30 of my First Affidavit.
The investigation of the composition of the enteric coating layer in response to the Question is entirely logical, given the knowledge that omeprazole is an acid labile drug. This investigation is not done in preference to, or at the expense of, other aspects of the formulation.
(emphasis added)
857 AstraZeneca submitted that Dr Reiland acknowledged that there are any number of variables in a formulation, and that his “systematic experimental approach” would not have focused on the particular variables that are the subject of the MUPS Patent. AstraZeneca further pointed to the fact that Dr Reiland did not adjust the enteric coating composition to develop the PCE 500 product.
858 Although Dr Reiland asserted in the Joint Expert Report on AU 966 that he would have optimised the amount of coating material applied to pellets, the process Dr Reiland described is different from the process described and claimed in the MUPS Patent. The process in the MUPS Patent includes adjusting the amount of plasticizer and the amount of coating material applied (for a particular polymer-plasticizer combination) to achieve mechanical properties sufficient to ensure the gastric acid resistance of the pellets during compression into tablets. The MUPS Patent exemplifies a number of plasticizer amount and applied coating amount combinations that achieve the desired result.
859 However, in one answer in the Joint Expert Report on AU 966, Dr Reiland referred back to his third affidavit, where he described a process whereby a range of pellets with different levels of coating material applied could be generated and then tested for compliance with the standard USP drug release test. Dr Reiland gave evidence that he would then select a level of coating material somewhat greater than that required for uncompressed pellets to pass the standard USP drug release test, as a safety buffer. The pellets would not be compressed before making the selection. In other words, in the course of carrying out his “systematic experimental approach”, Dr Reiland would not investigate how combinations of enteric coating polymer amount and plasticizer concentration could be adjusted to examine the effect of such combinations on the ability to successfully compress enteric coated pellets of omeprazole into tablets. AstraZeneca submitted that he therefore would not generate any data on the basis of which he would be directly led as a matter of course to the MUPS Formulation.
860 AstraZeneca submitted that the significance of this issue is that a formulator, following the series of steps set out by Dr Reiland in his third affidavit (ie the “experimental approach that [he] would adopt at July 1994 to determine an alternative solid oral dosage formulation of omeprazole”), would not be led directly as a matter of course to the MUPS Formulation (in comparison to, for example, a cushioning excipient solution, or a compression enhancing coating solution). Rather, the experimental approach suggested by Dr Reiland involves an unpredictable process involving what may be extensive trial and error.
861 As previously foreshadowed during my discussion of the common general knowledge, Professor Bodmeier and Dr Morella gave evidence that a large number of variables would be investigated by the skilled addressee trying to compress enteric coated pellets into a multiunit tablet. Dr Morella explained:
(a) the quantity and size of the coated pellets to be tableted. Smaller pellets may assist in achieving content uniformity between tablets. However, smaller pellets may be more difficult to compress into a hard, non-friable tablet (a non-friable tablet is one that is resistant to abrasion and chipping on handling);
(b) the composition of the uncoated core of the pellets to be tableted. A more robust uncoated core may survive the tableting process without significant deformation and breakage. However, a more robust uncoated core may not disintegrate or dissolve quickly enough. The composition of the uncoated core will also affect the performance of coatings applied to it;
(c) the composition of the coating layer(s) within the coated pellets. The identity and quantity of each ingredient in the coating layer(s) can have a different effect on the desired performance criteria. For example, increasing the amount of enteric coating polymer to improve acid resistance may have a detrimental effect on the rate of dissolution…
(d) the presence or absence of compression enhancing layer(s) on the pellets. I note that Dr Reiland added a compression enhancing layer in the course of developing his PCE product… While at Faulding, my colleagues and I experimented with a compression enhancing layer in the project relating to nifedipine, to which I refer in paragraph 16(c) of my First Affidavit;
(e) the tableting excipients. The types of tableting excipients include fillers, binders, disintegrants, glidants and lubricants and each excipient has different effects on the desired performance criteria. Fillers that deform plastically permit lower compression forces, which may help to retain acid resistance. However, fillers that deform plastically may contain water, which can affect stability. The solubility of the excipients may affect how the product disintegrates;
(f) the processing conditions, such as the compression forces applied on the tablet punches. Weaker compression forces may help to maintain acid resistance. However, weaker compression forces may result in a friable tablet which does not survive packaging and transport.
862 Dr Reiland stated that he agreed that those variables referred to by Dr Morella and Professor Bodmeier “are all relevant for a formulator to consider in the context of compressing enteric coated pellets”, but that they are variables that an industrial formulator would be trained and well equipped to accommodate in a systematic and logical way.
863 However, be this as it may, accepting the evidence of Professor Bodmeier and Dr Morella as I do, Dr Reiland’s evidence in this regard does not enable the Court to conclude that the formulator would be directly led as a matter of course to try the invention claimed in the MUPS Patent from among the many possible variables and avenues of experimentation available to the formulator.
Information Contained in The Merck Index, Goodman & Gilman, PDR Prilosec and PDR PCE
864 I should say something more specifically about the above documents relied upon by Ranbaxy, to which I have already referred to in the context of considering Ranbaxy’s ability to rely upon such documents in this proceeding. Even if, contrary to my view, these documents could be relied upon, I accept AstraZeneca’s submissions that the information in these documents would not directly lead the formulator as a matter of course to try the invention of the MUPS Patent.
The Merck Index 1989, Goodman & Gilman and PDR Prilosec
865 The Merck Index 1989, Goodman & Gilman and PDR Prilosec supply only basic information about some of the properties and commercial formulations of omeprazole. I do not consider that the information in each of those documents is, on its own, a sufficient basis on which to develop a new formulation of omeprazole.
866 Professor Bodmeier summarises the information in The Merck Index 1989, Goodman & Gilman and PDR Prilosec as follows:
(a) omeprazole can exist in a crystalline form, which has a melting point of 155°C or 156°C;
(b) it is rapidly degraded in acid media;
(c) it undergoes a saturable first-pass effect;
(d) it is freely soluble in ethanol and methanol, slightly soluble in acetone and isopropanol and very slightly soluble in water;
(e) it is available in 20 mg capsules containing enteric coated granules, which should be stored in tight containers protected from light and moisture;
(f) its absorption begins only after granules leave the stomach;
(g) its absorption is rapid, with peak plasma levels of omeprazole occurring within 0.5 to 3.0 hours; and
(h) its bioavailability increases slightly upon repeated administration.
867 Even if one were to impermissibly consider this information collectively (namely, contrary to the manner prescribed by s 7 of the Act), Professor Bodmeier explained that more information would be required to embark upon a formulation development program, including:
(a) more information concerning the bulk properties (for example, size, density and surface morphology) of omeprazole;
(b) more accurate information concerning the solubility of omeprazole in organic and aqueous solvents;
(c) more information concerning the stability properties of omeprazole, for example, stability in solid phase and degree of moisture sensitivity; and
(d) more information concerning the formulation and processing of the Prilosec enteric coated granules.
868 I accept that the above information, in combination with the common general knowledge, would not lead a skilled addressee to attempt to compress enteric coated pellets into tablets, let alone to the particular solution taught by the MUPS Patent. Further, I accept that the acid sensitivity issues and the stability issues would turn the skilled addressee away from multiunit tablets and toward capsules, as (in the words of Professor Bodmeier), “when coated pellets are compressed into a tablet, there is almost always at least some loss of acid resistance”.
PDR PCE
869 The information in PDR PCE, in combination with the common general knowledge, would also not directly lead the formulator to the invention in the MUPS Patent.
870 Professor Bodmeier explains that PDR PCE teaches him that:
(a) PCE Dispertab® tablets contain high doses, 333 mg or 500 mg (column 3 on page 549);
(b) the PCE product contains specially coated erythromycin particles in tablets for oral administration (columns 2 and 3 on page 549);
(c) the coating protects the antibiotic from the inactivating effects of gastric acidity and permits efficient absorption of the antibiotic in the small intestine;
(d) the erythromycin particles in PCE tablets are coated with a polymer whose dissolution is pH dependent. This coating allows for minimal release of erythromycin in acidic environments, for example, the stomach. This delivery system is designed for optimal drug release and absorption in the small intestine (column 1 on page 550); and
(e) the PCE product is supplied as unscored, ovaloid, Disperstab® [sic] tablets in bottles (column 2 on page 551).
871 I accept that in the absence of information in PDR PCE identifying the coating polymer, the structural composition of the granules or how the PCE tablets are manufactured, there is nothing at all that leads or points to the MUPS Patent invention.
872 At best for Ranbaxy, the information in the PDR PCE entry could be said to show that one compound, in a completely different chemical and therapeutic class (a macrolide antibiotic as opposed to a proton pump inhibitor), could be formulated into multiunit tablets. But without more, this does not lead to the MUPS Patent invention.
US 505
873 Another document relied upon by Ranbaxy is US 505, which I have already discussed in the context of novelty. I will assume it is either part of the common general knowledge or a s 7(3) document. I have already made findings about what US 505 teaches.
874 However, US 505 does not provide information which, when combined with the common general knowledge, makes the invention disclosed in the MUPS Patent obvious to a skilled addressee. AstraZeneca submitted that in fact, the opposite is true – the more that is disclosed about the properties of omeprazole (including its acid labile character), the less likely the skilled addressee would be to even consider compressing enteric coated particles containing omeprazole into tablets, due to the risk of compression compromising gastric acid resistance.
875 To this end, US 505 teaches the formulator that omeprazole:
(a) is highly sensitive to acid and requires an enteric coating that prevents not only release of the active substance in gastric acid, but also penetration of gastric acid through the enteric coating;
(b) must be released rapidly in the duodenum; and
(c) is affected by moisture and organic solvents.
876 US 505 teaches the skilled addressee how to formulate enteric coated pellets containing omeprazole that overcome these issues and have good acid resistance, the right dissolution profile and good long-term stability characteristics.
877 Dr Reiland gave the following evidence in the context of US 505:
My strategy for developing an alternative solid oral dosage formulation of omeprazole would be to focus on making acid protected (ie enteric coated) particles of omeprazole that are compressed into a tablet or filled into a capsule final dosage form.
878 While Dr Reiland stated that his “preference” would be to make enteric coated particles compressed into a tablet (for the reasons already discussed), the above extract demonstrates that Dr Reiland clearly contemplated two courses of action: capsules and tablets. That is not being “directly led” to the invention of the MUPS Patent by US 505 for the purposes of answering the Cripps question.
879 US 505 provides no motivation for the skilled addressee to attempt to develop a multiunit tablet containing omeprazole. I accept Professor Bodmeier’s evidence that given the acid sensitivity of omeprazole taught by US 505, the requirement that omeprazole be rapidly released in the duodenum, and omeprazole’s sensitivity to moisture and organic solvents, a skilled formulator reading US 505 would have no motivation to depart from the capsule formulation. Further, I accept that if the skilled formulator were required to make an “alternative” to what US 505 teaches, he or she would attempt to modify the capsule formulation taught in US 505, but would not depart from the capsule dosage form. If a skilled formulator were required to develop a multiunit tablet containing omeprazole using the information in US 505, Professor Bodmeier gave evidence (that I accept) that they would recognise that the US 505 pellets are unsuitable for this purpose (due to acid sensitivity and stability issues specific to omeprazole). As previously noted, Dr Morella gave evidence that such an endeavour would require a new formulation development program that would have low prospects of success. I am not persuaded to the contrary by either Dr Reiland’s evidence on this issue, or the terms of US 505 itself.
Reasonable Expectation of Success
880 It was further submitted by AstraZeneca that even if the skilled formulator would be directly led as a matter of course to try the invention in the MUPS Patent, Ranbaxy’s evidence falls well short of establishing that the skilled formulator would have reasonably expected to succeed.
881 Ranbaxy’s evidence of a reasonable expectation of success of arriving at the invention in the MUPS Patent rests on Dr Reiland’s “systematic experimental approach”. I have already alluded to this approach in these reasons, being what Dr Reiland suggested was a standard systematic approach that an experienced formulator would use to formulate an enteric coated product.
882 Its features were described as follows:
(a) “a series of formulations are made with different levels of ingredients (excipient, identity and amount) or process parameters”;
(b) “[r]outine performance tests [that] are conducted on the formulations, and the data from these tests are gathered and studied to assess the effect of the specific levels of ingredients or process parameters on the performance of the formulation”;
(c) “[a]fter specific data has been collected, the industrial formulator selects a formulation or series of formulations with which to begin their formulation trials, and further trials are then guided by the data obtained”; and
(d) “[t]ypically, the industrial formulator will systematically change variables on the basis of data that leads them in a positive direction”.
883 It will be recalled that part of Dr Reiland’s “systematic experimental approach” had the skilled industrial formulator reviewing all available information they can obtain about the API and formulation at the beginning to identify a course of action. The examples referred to in this regard include all of the s 7(3) documents pleaded by Ranbaxy, together with a number of other publications identified by Dr Reiland following a literature search. AstraZeneca submitted that the implications of this are that Dr Reiland’s “systematic experimental approach” is not based on common general knowledge alone or combined with each of the s 7(3) documents pleaded by Ranbaxy, considered in isolation from the other documents as required by the statutory provisions. I have already dealt with this issue.
884 However, even assuming that the approach taken is based on common general knowledge and a s 7(3) document, I note that Dr Reiland summarised his “systematic experimental approach” in the following manner:
In summary, producing a successful dosage formulation in light of a number of interrelated variables in the formulation is part and parcel of what I, and any experienced industrial formulator, is trained and required to do. The number of interrelated variables is why a systematic experimental approach (designed to continually add data to guide the formulator towards achieving their formulation goal) is typically practised by industrial formulators.
885 AstraZeneca submitted that Dr Reiland seemed to adopt the approach that an industrial formulator does not have an expectation of failure. AstraZeneca acknowledged that some formulation projects are more challenging than others, but submitted that Dr Reiland’s evidence does not seem to discriminate between easier and more difficult tasks. Hence, it was contended, it does not present a balanced view of the practical difficulties faced by the skilled formulator.
886 Further, it was submitted that Dr Reiland’s expectation of success must be understood as the product of his personal knowledge and experience with the PCE products, which involved over ten years of development work at Abbott Laboratories. I have already concluded that such experience is not part of the common general knowledge of the skilled formulator.
887 I accept these submissions.
888 Dr Reiland’s expectation of success is not shared by Professor Bodmeier and Dr Morella. As will be apparent from the foregoing, their evidence is that the task of compressing enteric coated pellets containing omeprazole into a multiunit tablet is a very challenging one, fundamentally due to the problems that can arise, the difficulty of determining the causes of those problems and the large number of possible variables that can be explored in an attempt to overcome those problems. It is even more challenging in the case of omeprazole because of its acid labile character, making it more difficult to produce a multiunit tablet formulation with an enteric coating that maintains the required acid resistance.
889 Ranbaxy criticised Professor Bodmeier and Dr Morella as foreseeing “nothing but onerous experimentation, complexities and problems”. In response, AstraZeneca contended that Dr Reiland went the other way in his optimism.
890 I find that Dr Reiland’s “systematic experimental approach” is essentially trial and error. I note that the Abbott Laboratories’ formulators who successfully formulated PCE 333 tried to make PCE 500 and failed. The project was then given to a different group (Dr Reiland’s group) who then ultimately succeeded and obtained patent protection. As Dr Reiland stated:
That is what happened with PCE 500. The people that who tried to make PCE 333 couldn’t get it to work so they gave it to my group and we tried something different that they hadn’t thought of. So there’s a whole bunch of options formulators have and they keep pursuing those options until they find success and, of course, somebody else tries something else and finds success. That’s how I would describe it.
891 As I have already noted, Dr Reiland accepted that there are a large number of variables that an industrial formulator would need to explore. He accepted that problems would be experienced along the way, but it will be recalled that he gave evidence that “no industrial formulator would have time to explore every possible combination of variables”, and that to be as effective as possible, he would simply “aim… to carry out as many formulation trials as [he] can to find the combination of variables that achieves the formulation development goal”. I consider that this approach minimises the challenges and difficulties associated with the relevant formulation task to an unacceptable extent.
892 On this issue, I prefer the evidence of Professor Bodmeier and Dr Morella regarding the practical realities of the formulation process and the specific problems that arise when attempting to compress enteric coated pellets containing omeprazole into a tablet. When asked to comment on Dr Reiland’s systematic experimental approach during the concurrent evidence session, Professor Bodmeier stated:
No, I don’t agree on this because if I interpret this statement correctly that would mean you pick a polymer and then you have to decide which plasticiser you choose and, as I said before, you would go by the information you are provided by the excipient manufacturer. They recommend the plasticisers, they recommend the range and they would go by these ranges. And to put this in a bigger picture, we have a pharmaceutical formulation here – now, with hindsight, we focus everything on the plasticiser and on an enteric polymer. But this formulation consists of many excipients. So for us – if you do this exercise with each excipient you had in your formulation that would take a long time. That’s why a formulator goes by the recommendations of the excipient supplier and stays within the recommended – it’s plasticisers or enteric polymers and the recommended ranges because otherwise it’s not just you need to optimise this. You need to optimise all other ingredients, too.
893 When Senior Counsel for Ranbaxy put it to Professor Bodmeier that “the experienced industrial formulator wouldn’t regard these things as problems rather they’re the kinds of things that he bears in mind in seeking to achieve the performance characteristics that he wants”, Professor Bodmeier responded:
No, I strongly disagree because – because of these problems, the formulator would really stick with the recommendations he gets from the manufacturer of the excipients and of the polymers … So you start with a basic formula and this basic formula normally comes from an excipient manufacturer, who again has the most experience with these excipients and then they want to sell it. And then you make minor modifications to this basic formula. You don’t go up and down – and this plasticiser and now I try this one because 10 per cent to 50 per cent that’s all – in theory, you can write a lot, but in practice there is no time for a formulator to do this. They really go with basic formulas, formulas - they are recommended to them by the excipient manufacturer and then they play around, if they do it, all within these ranges.
894 Finally, I consider that it makes no difference whether or not Professor Bodmeier and Dr Morella were exposed to both the prior art and the solution provided by the MUPS Patent when giving their evidence about inventive step. The fact that they were exposed to that information and nevertheless consider the MUPS Patent to be inventive makes their evidence even more compelling.
895 Overall, in my view, the “systematic experimental approach” of Dr Reiland cannot be said (without more) to lead directly to the invention that is the subject of the MUPS Patent.
896 For these reasons, Ranbaxy’s contention that the skilled formulator would have tried the invention of the MUPS Patent with a reasonable expectation of success cannot succeed.
The commercial success of the MUPS Formulation
897 Finally, I come to the question of commercial success.
898 I have already discussed this issue in relation to the Purity Patent.
899 Ranbaxy tendered a number of AstraZeneca’s discovery documents to the Court relating to the development of the invention of the MUPS Patent. These documents contain statements about AstraZeneca’s development of a MUPS formulation of omeprazole as part of a “post-patent strategy”, referring to the expiry of the omeprazole compound patent in most countries in or around 1999.
900 I consider that the documents tendered by Ranbaxy in no way undermine AstraZeneca’s case on inventive step.
901 Whatever may have been the effect of the initial marketing campaign, the continued commercial success of the LOSEC tablets and of the NEXIUM tablets which both embody the MUPS Formulation, indicates that the commercial success must, at least in significant part, be attributed to the advantages of the MUPS Formulation and not solely to other possible sources of success (such as the marketing, price and the cost of the drug).
Specific issues regarding dependent claims of the MUPS Patent
902 In light of the foregoing conclusions, I need not address any specific issues that may arise in respect of any other claims. The parties did not specifically engage on any new limitations introduced by these claims over claim 1.
Manner of manufacture
Ranbaxy’s “manner of manufacture” pleading
903 Ranbaxy contended that the invention as claimed in each of claims 1 to 8, 10 and 12 to 28 of the MUPS Patent is not a patentable invention within the meaning of s 18(1)(a) of the Act, in that it is not an “invention” or, further or alternatively, is not a “manner of manufacture” within the meaning of s 6 of the Statute of Monopolies. This was claimed on the following bases, as set out in the Seventh Amended Particulars of Invalidity:
(a) On the face of the specification of the MUPS Patent, the subject matter claimed in each and every claim does not pass the threshold requirement in order to be the proper subject of Letters Patent within the meaning of s 6 of the Statute of Monopolies.
(b) The specification of the MUPS Patent discloses nothing more than the mere use of known compounds (namely, plasticizers) to perform a function for which their known properties make them suitable and for which they were being used (namely to improve the mechanical properties, namely flexibility, of enteric coatings).
904 The specific further allegations made in respect of claim 10 will be addressed in due course.
Summary of AstraZeneca’s submissions
905 In summary, AstraZeneca’s main submission was that the MUPS Patent claims a “manner of new manufacture”, as is apparent from the specification itself.
Analysis
906 It is only when the absence of inventiveness is clearly revealed on the face of the specification that an objection that an invention is not a new manner of manufacture can succeed (see eg Commissioner of Patents v Microcell Limited (1959) 102 CLR 232 at 246). In such limited circumstances, as was also noted by the High Court in NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1995) 183 CLR 655, the need to go further and consider the conventional grounds of novelty and inventive step are obviated. Once the inquiry travels outside the specification, one impermissibly trespasses onto the ground of inventive step.
907 Accordingly, an objection to validity of this kind applies where the specification (properly construed), in effect, admits that the invention lacks inventiveness. Only in such a case can it properly be said there is no manner of new manufacture such that it is not necessary to go on to consider the merits of the grounds of novelty or inventive step.
The MUPS Patent
908 In this case, it cannot be said that the specification ‘on its face’ shows the invention claimed is not a manner of new manufacture. The specification does not admit that the combinations, processes or methods of use claimed in the MUPS Patent were not new or inventive, and nor does it support the drawing of any inference to that effect. The invention as claimed is for a combination of integers, and not merely for the use of plasticizers.
909 Further, I am satisfied the specification reveals the new qualities of the invention and positively asserts that there is an invention. Ranbaxy may not “go behind” these statements, and rely on extrinsic evidence.
910 In particular, the MUPS Patent at the very least states the following.
(a) The invention is related to new pharmaceutical preparations in the form of a multiunit tableted dosage form comprising omeprazole or one of its single enantiomers or an alkaline salt of omeprazole or one of its single enantiomers.
(b) The novel dosage form is intended for oral use.
(c) Even if the specification of US 505 under the subtitle “Final dosage form” mentions that the manufactured pellets may be formulated into tablets, there are no examples in that document describing any composition of such a tablet formulation or a technique to manufacture such a formulation. This statement informs the skilled addressee that the specification of US 505 is ambiguous, and not of assistance in making a multiunit tablet of omeprazole.
(d) The Applicant for the MUPS Patent is not aware of any working example in the prior art of a multiunit tableted dosage form comprising an acidic susceptible benzimidazole compound, such as omeprazole.
(e) The Applicant for the MUPS Patent has now surprisingly found that tablets according to the invention comprising enteric coating layered units containing omeprazole can be manufactured by compressing the units into tablets without significantly affecting the properties of the enteric coating layer.
911 Whatever else Ranbaxy may refer to in the MUPS Patent, the above references adequately describe, in their terms, a new invention. This is sufficient to dispose of the arguments of Ranbaxy on the ‘no manner of new manufacture’ ground.
Claim 10
912 AstraZeneca objected to Ranbaxy’s proposed amendment to the pleadings filed with the Court on 25 March 2012 (being the Seventh Amended Particulars of Invalidity), insofar as it raised new grounds of attack in relation to claim 10.
913 It will be recalled that claim 10 relates to a tableted dosage form wherein the enteric coating has a thickness of at least 10 μm.
914 The proposed further particulars raise a range of new issues relating to lack of definition, manner of manufacture and inventive step in relation to claim 10.
915 I propose to allow the amendment. In my view, it just involves the application of the law to established facts before the Court already, upon which the parties have presented argument.
916 Ranbaxy plead that claim 10 is not a manner of manufacture, is a mere collocation and lacks definition. These allegations can be dealt with compendiously. As Ranbaxy plead, the preclusion of a significantly adverse effect on the gastric acid resistance of coated pellets upon compression is a function of the mechanical properties of the enteric coating layer. They pleaded that the thickness of the enteric coating layer does not contribute to the “desired result”, does not interact with the other integers of the invention, and that there is no working relationship between it and the requirements of the claims on which claim 10 is dependent.
917 As explained in the MUPS Patent and by Professor Bodmeier, the amount of coating applied, and hence the thickness of the coat, makes “a very important contribution to the mechanical properties of the enteric coating”. Professor Bodmeier further explained that the plasticizer “affects the flexibility and the hardness” and that the thickness of the coating “contributes significantly to the mechanical strengths of the coating to resist fracture during compression”. Professor Bodmeier gave evidence that:
this in my view is the invention here, that you have an interplay of the plasticizer together with the thickness. Either one on its own would not work.
918 Therefore, the amount of plasticizer added and the thickness of the enteric coat interact to produce the mechanical properties, and there is a working relationship between the two factors in producing the mechanical properties and the desired result. This builds on the conclusions already reached in the foregoing reasons.
919 Claim 1 does not limit either the level of plasticizer or the thickness of the enteric coat to any particular figure, as long as the mechanical properties are attained. The mechanical properties may be obtained by a combination of a particular level of plasticizer and an enteric coat thickness of less than 10 μm. Claim 10 merely limits the claim to the subset of claim 1 formulations that achieve the mechanical properties and where the enteric coat is at least 10 μm thick. This is not an “otiose parameter that has nothing to do with the alleged invention”, but a measure of a parameter inextricably involved in the invention. The fact that there are other measures related to thickness, such as the percentage of coating solution applied, does not make a direct measure of thickness otiose.
920 These conclusions result in each of Ranbaxy’s objections to claim 10 being rejected.
THE 774 PATENT
Introduction
921 I now turn to the 774 Patent.
922 The principal issues concerning the 774 Patent relate to construction and infringement. The validity allegations of Ranbaxy fall away if the construction contended for by those parties is successful.
923 Extensive evidence was led as to infringement, involving conflicting views of various expert witnesses. To a large extent the evidence was of a highly technical nature.
924 I have now come to a very firm view on two principal construction issues relating to the 774 Patent raised by the parties, and have found in favour of Ranbaxy. Those two principal construction issues relate to the interpretation of the terms “a core material” and “separating layer thereon” in the context of claim 1. It has been accepted that in the event of my finding in favour of Ranbaxy on these two principal construction issues, the infringement case brought by AstraZeneca cannot succeed.
925 As the parties are desirous of a decision being made no later than 23 April 2013, in the circumstances, I propose to confine myself to those principal construction issues.
926 To adequately consider and determine the very many technical issues raised by the parties relating to the 774 Patent will take many months. If I am right on construction, this task is unnecessary. By confining myself to the two principal construction issues, the parties are now provided with the Court’s ultimate decision. The parties can then consider their position. If my construction of the 774 Patent is incorrect, and the question of infringement needs determination, it may then need to be considered at a later time. This is a risk that is worth taking in the interests of the expeditious disposal of the proceeding, and to facilitate the just resolution of this dispute as quickly, inexpensively and efficiently as possible.
Background
927 The 774 Patent is entitled “New pharmaceutical formulation and process”.
928 The field of the invention relates to:
(a) new pharmaceutical formulations for oral use comprising acid labile PPIs (which, it will be recalled, stands for ‘proton pump inhibitors’);
(b) a new method for the manufacture of these new formulations; and
(c) the use of these new formulations in medicine.
929 The relevant science of the invention, for the purposes of disposing of the two principal issues of construction, is not in dispute and is not complicated. The 774 Patent is directed to a process by which two functionally different layers (namely, an enteric coating layer and a separating layer) form in one manufacturing step. That step involves the enteric coating of a pellet or tablet core. During the enteric coating process, a separating layer is formed in situ. The separating layer comprises a water soluble salt of the enteric coating polymer(s). The separating layer is required between the core material (containing the acid labile PPI)) and the enteric coating layer (which ordinarily comprises compounds containing acidic groups) because otherwise, the active compound will degrade, and the acid labile substance may also discolour. The separating layer has the function of reducing the extent of any reaction between the PPI and the enteric coating polymer(s) as part of a stable pharmaceutical formulation.
930 The two principal construction issues in dispute between the parties arise by reference to the integers of claim 1 of the 774 Patent, parsed and relevantly emphasised as follows:
(a) an oral pharmaceutical dosage form:
(i) comprising a core material that contains …
(A) a PPI;
(B) one or more alkaline reacting compound(s); and
(C) optionally pharmaceutically acceptable excipients;
(ii) said core material having a water soluble separating layer thereon;
(iii) with an enteric coating layer thereupon;
(a) characterised in that:
(i) the core material is alkaline reacting; and
(ii) that the separating layer:
(A) comprises a water soluble salt of the enteric coating polymer(s); and
(B) is formed in situ during the enteric coating.
Construction principles
931 The principles of construction have been referred to earlier in this judgment, in the context of the Purity Patent.
932 AstraZeneca, in the context of the construction debate relating to the 774 Patent, submitted that the Court should not adopt a narrow, non-purposive interpretation of the key integers, or impose limitations on those integers. This was the way they described the approach taken by Ranbaxy to the construction issues.
933 It was also said that Ranbaxy’s approach to the construction of the claims of the 774 Patent was misconceived and contrary to the accepted principle explained by Bennett J in Sachtler GmbH & Co KG v RE Miller Pty Ltd (2005) 65 IPR 605; [2005] FCA 788 at 613 [42] that:
Although the claims are construed in the context of the specification as a whole, it is not legitimate to narrow or expand the boundaries of monopoly, as fixed by the words of a claim, by adding to those words glosses drawn from other parts of the specification. If a claim is clear and unambiguous, it is not to be varied, qualified or made obscure by statements found in other parts of the document.
934 In guiding the Court as to the proper approach to construction, Ranbaxy also referred to the comments made in Populin v HB Nominees Pty Ltd (1982) 41 ALR 471, where the Full Court of the Federal Court (Bowen CJ, Deane and Ellicott JJ), said at p 476:
The essential features of the product or process for which it claims a monopoly are to be determined not as a matter of abstract uninformed construction but by a common sense assessment of what the words used convey in the context of then-existing published knowledge.
First principal construction issue
935 Of the principal construction issues, the central debate is about the term “core material”. Of course, the term must be read in context of claim 1, and the specification as a whole.
936 There is no scientific or industry meaning of “core material”. It is an expression of ordinary English, although it may be used in various ways in the context of the 774 Patent.
937 It is necessary to look at the whole specification in some detail to assist in determining the meaning of the term “core material” in claim 1. As I go through the specification, I will emphasise the word “core”, and the relevant phrases which include the words “core” or “core material”. This will help the reader follow the various ways in which the word “core” is used. In each context, the various references I make support the conclusion I have reached as to the meaning of “core material” in claim 1.
938 The conclusion I have reached is as follows.
939 In essence, in the context of claim 1, the dosage form comprises “a core material”, with “said core material” “having” a water soluble separating layer “thereon”. There is a specific reference to core material, specifically defined as having a specific water soluble separating layer thereon. The dosage form is then “characterized” as referring to the “core material” being alkaline reacting, with the separating layer described as to its composition and formation. There is a very clear distinction drawn between core material and coating layers (enteric or separating). Therefore, the reference to core material does not include a coating layer.
940 This clear description in claim 1 has no ambiguity, and is consistent with the concept of the invention.
941 Claim 1 of the 774 Patent does not describe an oral pharmaceutical dosage form with an applied separating layer being part of the core material. In the context of the 774 Patent, “core material” is defined in claim 1 according to what it actually contains. In this sense, it means the material in the centre of the dosage form (containing the API, alkaline reacting compound and any pharmaceutically acceptable excipients), on which the functioning layers may be subsequently formed. This definition of “core material” necessarily excludes any layers applied to the core material as defined, including coating layers that do not contain API.
942 I now go to the specification, upon which both sides relied for their own interpretation of claim 1.
The invention as described in the patent
943 The 774 Patent begins on pp 1 to 6 by identifying certain PPIs. The specification states, at p 6, lines 5 to 10:
The proton pump inhibitors used in the dosage forms of the invention may be used in neutral form or in the form of an alkaline salt, such as for instance the Mg2+, Ca2+, Na+, K+ or Li+ salts, preferably the Mg2+ salts. Further where applicable, the compounds listed above may be used in racemic form or in the form of a substantially pure enantiomer thereof, or alkaline salts of the racemates or the single enantiomers.
944 At p 7, lines 9 to 11 it is recorded that these PPIs are susceptible to degradation or transformation in acidic reacting and neutral media:
The degradation is catalyzed by acidic reacting compounds and the proton pump inhibitors are usually stabilized in mixtures with alkaline reacting compounds.
945 It follows, it is said, at lines 13 to 15, that “it is obvious that a proton pump inhibitor in an oral solid dosage form must be protected from contact with the acidic reacting gastric juice”. The skilled addressee would understand that, otherwise, the PPI will be degraded in the acidic environment in the stomach before it reaches the part of the gastrointestinal tract where absorption of the PPI can occur.
946 The specification records that a pharmaceutical dosage form of such PPIs is best protected from contact with acidic gastric juice by an enteric coating layer: see p 7, lines 20 to 21. An example is given of such “enteric coated preparations” at lines 21 to 22, and it is then stated that (emphasis added):
Said preparations contain an alkaline core material comprising the active substance, a separating layer and an enteric coating layer.
947 Three separate items are addressed here, as emphasised (namely, the alkaline core material, the separating layer, and the enteric coating layer).
948 However, the problem is that the PPI might also react with some acidic compound in the enteric coating layer itself. At p 7, lines 26 to 31, the specification states (emphasis added):
Ordinary enteric coating layers, however, comprise compounds which contain acidic groups. If covered with such an enteric coating layer, the acid labile substance may rapidly decompose by direct or indirect contact with the acidic groups resulting in discoloration of the content and loss in content of the active compound with the passage of time. The discoloration can be avoided by applying some type of separating layer between the core material comprising the susceptible proton pump inhibitor and the enteric coating layer.
949 Three separate items are again addressed here, as emphasised.
950 The specification states that the prior art pharmaceutical dosage forms included a separating layer between “a core material” and an enteric coating layer: p 8, lines 1 to 3 (emphasis added):
Thus, there are a lot of patent applications describing such a separating layer between a core material comprising the pharmaceutically active substance and an enteric coating layer. See for instance, US-A 4,786,505, EP 0,277,741 and EP 0,342,522.
951 Again, three separate items are referred to here.
952 “US-A 4,786,505” is a reference to US 505 (being AstraZeneca’s patent dated 22 November 1988, that has already been the subject of discussion in these reasons).
953 The specification further states, at p 8, lines 3 to 7 (emphasis added):
The prior art techniques to apply at least two different layers on a pellet core or a tablet comprising an acid labile compound is rather complicated and there is a demand for finding new processes and formulations to simplify the manufacturing of such enteric coated articles comprising acid labile substances.
954 This passage indicates that the terms “pellet core” and “tablet” are interchangeable. The reference here to “pellet core” as a tablet is only in the context of discussing techniques to apply at least two different layers, and the complications involved. This obviously follows on from the earlier reference in the very same paragraph to the prior art patent applications, which I have already noted draws a clear distinction between the separating layer, core material, and enteric coating layer.
Aspects of the invention
955 There are said to be two aspects of the invention. Under the heading “Summary of the Invention” at p 8, the first aspect of the invention is said to be (at p 8, lines 9 to 16; emphasis added):
…an oral pharmaceutical dosage form comprising a core material that contains a proton pump inhibitor, one or more alkaline reacting compound(s) and optionally pharmaceutically acceptable excipients, said core material having a water soluble separating layer thereon, with an enteric coating layer thereupon, characterized in that the core material is alkaline reacting and that the separating layer comprises a water soluble salt of an enteric coating polymer(s) and is formed in situ during the enteric coating.
956 It is said, at p 8, lines 17 to 20, “[t]he separating layer between the alkaline reacting core material comprising the indicated pharmaceutically active acid labile substance and the enteric coating layer, importantly comprises a water soluble salt of an enteric coating polymer”.
957 In relation to the second aspect of the invention, it is said at p 8, lines 24 to 27:
…the present invention provides a process for the manufacture of two functionally different layers in one manufacturing step. By such a process a separating layer comprising a water soluble salt of an enteric coating polymer is obtained, as well as the enteric coating layer itself.
958 As previously noted, the complete specification describes the invention as simplifying the preparation of enteric coated pharmaceutical dosage forms by providing a process for the manufacture of two functionally different layers in one manufacturing step: p 8, lines 28 to 30.
959 By this process, it would appear that the core or core material is the base upon which to work and build up on in the preparation of the invention.
960 Importantly, the 774 Patent goes on to say (emphasis added):
Thus, the present invention simplifies the preparation of enteric coated articles comprising a separating layer between a core material and an enteric coating layer by providing a new process for the manufacture of such dosage forms. According to said process the separating layer is formed by an in situ reaction between the enteric coating polymer and the alkaline core material comprising the pharmaceutically active substance.
961 Again, the distinction between the three separate items (core material, separating layer and enteric coating layer) is clear. The in situ reaction occurs between a specifically defined core material, and the enteric coating polymer.
962 As recorded on p 9, lines 1 to 2, Fig 1 in the 77 Patent is a photograph showing a cross-section of a tablet manufactured according to the invention. Then (again of some significance), the 774 Patent describes Fig 2 (being a schematic drawing of the photograph disclosed in Fig 1) as follows (emphasis added):
The tablet has an enteric coating layer (3), which has been applied on an alkaline core material (1) comprising the pharmaceutically active substance. Between the enteric coating layer (3) and the core material (1) there is a separating layer (2) shown.
963 The distinction is again clear: there is an enteric coating layer “which has been applied on an alkaline core material”, which comprises the active substance. Then there is the separating layer. Figures 1 and 2 of the 774 Patent are set out in “Annexure A” to these reasons.
964 A detailed description of the invention commences at p 9 of the specification.
965 At p 9, line 28 to p 10, line 5, it is said that (emphasis added):
Compacted tablets or individual cores (in the form of small tablets, small beads, granules or pellets) contain the proton pump inhibitor in the form of a racemate or one of its single enantiomers or an alkaline salt of said compound or one of its single enantiomers. The tablets or individual cores, that also comprise one or more alkaline reacting compound(s) which is in the position to form a water soluble salt by a reaction with an enteric coating material, are coated with one or more enteric coating layers.
966 This is introduced as the further characterisation of the new dosage form, and is not meant to change any of the basic tenets of the invention.
967 In fact, the 774 Patent goes on to say immediately after this reference, at p 10, lines 6 to 8 (emphasis added):
The separating layer is formed in situ by a reaction between the enteric coating polymer(s) and the alkaline reacting compound(s) in the core material during the enteric coating process.
968 The “core material” (so specifically mentioned) can be prepared according to two main processes, set out at p 10, lines 12 to 15 (emphasis added):
Firstly, seeds can be layered with the proton pump inhibitor, alkaline reacting compound(s) and necessary excipients to give an alkaline reacting core material, or the alkaline reacting core material can be prepared as substantially homogeneous cores or tablets comprising the proton pump inhibitor and the alkaline reacting compound(s).
969 Layered seeds are a particle or substrate (eg a sugar sphere) upon which one or more layers of active substances are applied. Homogeneous cores involve the preparation of the core material by mixing the active substance with alkaline compounds and suitable constituents (without a seed).
970 Then the 774 Patent continues at p 10, lines 16 to 18 (emphasis added):
The alkaline reacting compound(s) in the core material or tablet cores, necessary for an in situ reaction with the enteric coating polymer, is a substance in the position to form a water soluble salt with an enteric coating polymer.
971 This shows that in this patent, core material and tablet cores are treated for all practical purposes as the same thing.
972 Then in the same context, it is also said, at p 10, lines 29 to 31, that (emphasis added):
The core material as such should be an alkaline reacting core material, i.e. the amount of alkaline reacting compound(s) available in the core material should be enough to form a salt between the enteric coating polymer(s) and the alkaline reacting compound(s).
973 That is, there must be enough alkaline reacting compound(s) in the core material to form the claimed separating layer in situ by a reaction between the enteric coating polymer(s) on the one hand, and the alkaline reacting compounds of the core material on the other.
974 The concentration of alkaline reacting compounds in the core material is described at p 11, lines 1 to 16. It is said (emphasis added):
Thus, the concentration of alkaline reacting compound(s) in the core material (before applying the enteric coating polymer) is from approximately 0.1 mmol/g dry ingredients in the alkali containing part of the core material up to approximately 15 mmol/g, preferably the concentration shall be more than 0.3 mmol/g dry ingredients in the alkaline part of the core material.
975 It is then said, at lines 8 to 13 (emphasis added):
The upper limit range is only restricted by the need to include a pharmaceutically active ingredient and excipients such as binders etc in the alkaline core material. The concentration of alkaline reacting compound(s) may be illustrated as follows. For a core material where, for instance, 10% w/w of a proton pump inhibitor and 5% w/w of excipients (binders, surfactants etc) are to be included, 85% w/w remains to possible disposition to the alkaline reacting compound(s)…
976 It is then said, at p 11, line 18, that “[o]ne or more enteric coating layers are applied onto the prepared core material or tablets…” (emphasis added).
977 The preparation of the core material is described at p 12 line 6 to p 13 line 25 of the specification (emphasis added):
The preparation of the core material containing the proton pump inhibitor and alkaline reacting compound(s) is described more in detail below. The individually enteric coated cores can be constituted according to different principles.
The active substance, the proton pump inhibitor, used as a racemate or one of its single enantiomers or an alkaline salt of said compound or one of its single enantiomers, mixed with the alkaline reacting compound(s) is applied on seeds and are used for further processing.
The seeds, which are to be layered with the active substances, can be water insoluble seeds comprising different oxides, celluloses, organic polymers and other materials, alone or in mixtures or water soluble seeds comprising different inorganic salts, sugars, non-pareils and other materials, alone or in mixtures. Further, the seeds may comprise active substance in the form of crystals, agglomerates, compacts etc. The size of the seeds is not essential for the present invention but may vary between approximately 0.1 and 2 mm. The seeds layered with active substance are produced either by power or solution/suspension layering using for instance granulating or spray coating/layering equipment.
Before the seeds are layered, the active substance is mixed with alkaline reacting compound(s) and further components to obtain preferred handling and processing properties and suitable concentration of the active substance. Pharmaceutical constituents such as fillers, binders, lubricants, disintegrating agents, surfactants and other pharmaceutically acceptable additives, can be used. Binders are for example celluloses such as hydroxypropyl methylcellulose, hydroxypropyl cellulose and carboxymethylcellulose sodium, polyvinylpyrrolidone, sugars, starches and other pharmaceutically acceptable substances with cohesive properties. Suitable surfactants are found in the groups of pharmaceutically acceptable non-ionic or ionic surfactants such as a for instance sodium lauryl sulphate or polysorbates.
Alternatively, the active substance mixed with alkaline compound(s) and further mixed with suitable constituents can be formulated into tablets or individual cores. Said tablets or cores may be produced by compression/extrusion/spheronization or balling utilizing different processing equipments. The manufactured tablets or cores can further be layered with additional ingredients comprising active substance and alkaline reacting compound(s) and/or be used for further processing.
The active substance may optionally be mixed with alkaline pharmaceutically acceptable substance (or substances) for further stabilisation. Such substances can be chosen among, but are not restricted to, substances such as for instance the above mentioned alkaline reacting compounds or other alkaline reacting substances known by the skilled person in the art to be useful as stablizers for acidic susceptible substances.
Alternatively, the aforementioned alkaline reacting core material can be prepared by the use of spray drying or spray congealing technique.
The prepared alkaline reacting core material in the form of tablets or pellets are spray coated with an aqueous enteric coating polymer dispersion/solution. The process parameters such as inlet air temperature, air flow, atomizer air flow and spraying rate are adjusted with respect to the equipment used for the process as well as the specific enteric coating polymer(s). The inlet air temperature must not be such that the enteric coating polymer(s) will block in the spraying nozzles.
978 This demonstrates the nature of the process. The reference to “tablets or individual cores” relates to the preparation of the “core material”. In one instance, the tablets or pellets are the form of the prepared alkaline reacting core material before they are spray coated.
979 The invention as described in the examples set out in the specification describes the formulation of the “core material”. Undoubtedly, there are a variety of words or terms used in these examples, such as “cores”, “core formation”, “sugar cores”. But these terms are all used in the context of referring to the formation of the “core material”.
980 The layered seeds referred to in the 774 Patent involve the application of a layer or even layers of active substance – that is, material containing the PPI. Some pharmaceutically acceptable excipients are exemplified at p 12, line 28 to p 13, line 2, including (at p 12 line 29) binders such as hydroxypropyl cellulose (‘HPC’). The layered seeds described in the specification do not in and of themselves include any layers that do not include the active substance.
981 Further, the homogeneous core material referred to at p 13, lines 4 to 9, may be layered with additional “active substance and alkaline reacting compound(s)”. That is, further active drug layers are added to the homogeneous cores containing the active substance. Again, the homogeneous cores as described in the specification do not in and of themselves include any layers that do not include the active substance.
982 Therefore, as is evident from a reading of the 774 Patent, but as was also stated by Dr Pyter (whose evidence on this aspect I accept), the specification does not describe core material that is “layered” or “further layered” with an applied separating layer or any layer not including an active substance.
The invention claimed
983 The specification concludes at p 28 with 25 claims. Claim 1 is an independent claim for an oral pharmaceutical dosage form. Claims 2 to 20 are dependent claims. Claim 21 is an independent claim to a process for the preparation of an oral, enteric coated, pharmaceutical dosage form. Claim 21 refers specifically to a core material in a similar manner to claim 1. Claim 22 is a ‘product by process’ claim. Claim 23 is a claim to a method for inhibiting gastric acid secretion by administering a dosage form as claimed in any one of claims 1 to 20 and 22. Claims 24 and 25 are claims to use of the claimed oral pharmaceutical dosage form.
984 I make specific mention of claim 9, as it (like claim 21 and claim 1) specifically refers to “core material”, as follows (emphasis added):
9. A dosage form according to any one of claims 1 to 8, wherein the alkaline reacting compounds are present in a concentration of more than 0.1 mmol/g dry ingredients in the alkaline part of the core material.
The person skilled in the art
985 Before going any further in analysing the construction of the 774 Patent, I should briefly discuss the skilled addressee and the evidence of the experts. The 774 Patent relates to solid oral dosage formulation and design. It appears to be common ground that the hypothetical person to whom the 774 Patent specification is addressed is a pharmaceutical formulation scientist working in the field of solid oral dosage formulation and design. Dr Pyter, Dr Morella and Professor Bodmeier were put forward by the parties as being representative of the notional skilled addressee.
986 I have already described each of these witnesses.
987 Ranbaxy’s expert was Dr Pyter as a formulation scientist with experience in this role in a pharmaceutical company. AstraZeneca’s witnesses were Dr Morella as a formulation scientist with experience in this role in a pharmaceutical company, and Professor Bodmeier as an academic who has experience interacting with the pharmaceutical industry.
988 The evidence of the other expert witnesses relevant to the 774 Patent (Dr Hagenhoff, Dr Luk, Dr Matic, Mr Nicholas and Dr Wännman) was not evidence of the skilled addressee of the 774 Patent, or put forward as being evidence of persons who could assist the Court on construction. Their role was properly confined to assisting the Court in interpreting the experimental results.
989 I should say something more about the evidence led on construction and its relevance to the Court’s task. It was necessary for the Court to be informed as to the invention of the 774 Patent and the science relating to it. The concept underpinning the 774 Patent is relatively straight-forward as described previously – namely, a process by which two functionally different layers form in one manufacturing step. However, the terms “core”, “core material”, “separating layer” and “thereon” are not technical terms, and will be read as expressions of ordinary English, albeit in the scientific context of the 774 Patent.
990 Both sides attacked the evidence of the opposing experts, and sought to support their own.
991 I have not found this debate helpful on the question of construction. Each witness gave his view as to the interpretation of claim 1, and referred to different parts of the 774 Patent in support of that view. They referred also to the main purpose of the invention and to the relevant content of the prior art. The Court has now been informed of each of these matters, and can use that information to interpret claim 1. There was no dispute as to the purpose of the invention, nor relating to the reference to and content of the prior art in this regard. As I have said, the invention simplifies the preparation of the enteric coating by forming two layers in the one step. The question then is not what “core material” or “core” may mean in a general sense, but what it means in the context of claim 1.
AstraZeneca’s submissions and analysis
992 Apart from stressing the principles of construction referred to above, AstraZeneca’s position was as follows.
993 In the context of the claim, “core material” is to be understood by reference to the dynamic process by which the claimed in situ layer forms. The manner in which the dynamic process informs the construction of the integer is illustrated by the fact that the enteric coating is applied to the alkaline reacting core material, but due to the formation of the in situ separating layer, the enteric coat is not adjacent to the alkaline reacting core material. The integer “core material” means material that is beneath the water soluble separating layer (which layer forms during the enteric coating process). This meaning is informed by the nature of the invention claimed, which involves an alkaline reacting compound(s) from the core material reacting with the enteric coating polymer, by which process the water soluble separating layer (comprising a water soluble salt of the enteric coating polymer) is formed in situ during the enteric coating.
994 It is then submitted by AstraZeneca that the term “core material” must take its meaning from the claims which define the separating layer forming on the core material. In the context of the invention, the skilled addressee would understand this to mean that the core material is everything beneath the separating layer once it has formed. What is required is that the alkaline reacting compounds react with the enteric coating polymers to form the separating layer. When this occurs, everything beneath the separating layer is core material.
995 AstraZeneca submitted that the 774 Patent refers to, and exemplifies, multi-layered cores. For example, it is contended, in discussing the prior art, it refers to “at least two different layers” (emphasis added), which – it was said – clearly contemplates multi-layered cores.
996 AstraZeneca further submitted that the expression “core” can only be understood by reference to something which surrounds or is intended to surround that “core”. It takes its meaning from the functional layer being considered.
997 Put another way, AstraZeneca submitted that the phrase “core material” is used in two interrelated senses. First, in a functional sense to refer to material that is capable of participating in a reaction whereby two functional layers are formed in one step. To this end, AstraZeneca pointed to p 10, lines 8 to 29, and p 13, line 20. Secondly, in a situational sense to refer to “the relation of the core material to the separating layer”. In support of this contention, AstraZeneca pointed to p 7, line 30, and p 8, lines 1 to 2.
998 In my view, this is a distinction of no significance in the task of interpreting claim 1. By looking at the references in the 774 Patent that I have already emphasised in these reasons, it is clear that the reference to “core material” is a separate distinct concept to any subsequent layers. Take one of the examples given by AstraZeneca, located at p 10 line 8 of the 774 Patent. The subject matter is the defined “core material”. There is a reaction between the alkaline reacting compound(s) and the enteric coating polymer(s) so that the separating layer is formed.
999 To take another example relied upon by AstraZeneca, at p 8 lines 28 to 32 (emphasis added):
Thus, the present invention simplifies the preparation of enteric coated articles comprising a separating layer between a core material and an enteric coating layer by providing a new process for the manufacture of such dosage forms. According to said process the separating layer is formed by an in situ reaction between the enteric coating polymer and the alkaline core material comprising the pharmaceutically active substance.
1000 Here one of the specifically identified reference points in both the prior art techniques and the new process is the core material. Figure 2 referred to at p 9 makes this clear insofar as it differentiates between the alkaline core material and the separating layer, as do the examples referred to in the specification.
1001 Therefore, I do not accept AstraZeneca’s argument that the expression “core” or “core material” can only be understood by reference to something which surrounds or is intended to support the “core”.
1002 In my view, “a core material” means just that – the material specifically described in claim 1 and throughout the specification of the 774 Patent. This interpretation will exclude any applied sub-coats on the core material. But contrary to what was submitted by AstraZeneca, this is not a “limitation” imposed other than as a consequence of the proper reading of the phrase “core material” in the context of claim 1 and the specification as a whole.
1003 Of course, it is impermissible to look to the allegedly infringing article to determine the scope of the claim. This has been made clear in a number of cases. In Nobel’s Explosives Company Ltd v Anderson (1894) 11 RPC 519, Lord Esher MR observed (at 523):
...in construing this patent, one has no right to ... construe the patent by considering what the Defendant has done. I adjure that altogether, and I say we are bound to construe the patent as if we had to construe it before the Defendant was born, if the patent was before that time.
1004 His Lordship’s comments are apposite in this proceeding.
1005 However, in many ways, this appears to have been the approach of AstraZeneca to the construction of the 774 Patent. AstraZeneca attempted to formulate the so-called “functional” approach to overcome the fact that Ranbaxy’s allegedly infringing product has an applied barrier coating.
1006 The foregoing discussion has focussed a great deal on the specification, and not the wording of claim 1. It is claim 1 that must be interpreted. Focussing on the terms of claim 1 the conclusion I have reached seems equally clear. You have an oral pharmaceutical dosage form. It comprises “a core material”. For present purposes, it does not matter whether “comprises” means “includes”; or whether it is exhaustive. “A core material” contains (a term which I take to be exhaustive, but again, this does not matter) a PPI and one or more alkaline reacting compound(s), and optionally pharmaceutically acceptable excipients. This is the “core material”. The claim then goes on to refer to “said” core material having thereon a separating layer, with the enteric coating layer thereupon. Claim 1 goes on to say that “the core material” (previously defined) is alkaline reacting.
1007 Recalling that the expression “core material” is not a technical or scientific term, and is one which must be read in the context of the 774 Patent, and keeping in mind the purpose of the invention claimed, the term “core material” as defined in claim 1 should be interpreted as I have concluded above. This is consistent with the aim and purpose of the invention, and the way in which the term is used in the specification.
1008 It is also important to remember that the phrase “a core material” and “thereon” inter-relate, and when read together in context, support the view that I have taken as to the interpretation and ambit of claim 1.
1009 If one returns to the experts called by AstraZeneca, they had no answer to the contention put by Senior Counsel for Ranbaxy that the expression “core material” has its own separate and contained meaning. Both experts seemed to have some difficulty in considering the issue other than by returning to the so-called “functional” point of view. Their evidence, like AstraZeneca’s submissions, was infected by construing claim 1 with the Ranbaxy Product (and its separately applied barrier coating) in mind. The full exchange between the experts and Senior Counsel is instructive, and for this reason, the relevant parts of the transcript are set out in “Annexure B” to these reasons.
1010 The question of construction, as I have said, is one for the Court. However, the debate between Senior Counsel for Ranbaxy and the experts exposed, in my mind, that both witnesses failed to properly consider the actual wording of claim 1 and the context in which the term “core material” is used throughout the specification.
1011 I do not accept that the fact that the nature of the invention claimed (which involves an alkaline reacting compound from the core material reacting with the enteric coating polymer, by which process the separating layer is formed in situ during the enteric coating) means that Dr Morella and Professor Bodmeier are correct in their approach to the construction of claim 1 (exemplified by the exchange set out in Annexure B). The “core material” is as defined, and the invention is still of utility adopting such an interpretation. There is no limitation introduced upon claim 1, unless one views the construction of claim 1 by reference to the allegedly infringing Ranbaxy Product. This, of course, cannot be done.
Second Principal Construction Issue
1012 The second principal construction issue, which was dependent on the construction of “core material”, was the term “thereon”. As I have alluded to, this term must be read in the context of the use of the phrase “a core material”, as that phrase also must be read in the context of the term “thereon”.
1013 I do not think there was any real debate that “thereon” means directly thereon.
1014 The word “thereon” is not a term of art. Its ordinary meaning would indicate a direct relationship. The invention is characterised in that the enteric coating is applied onto the core material to form the separating layer. The claimed separating layer formed by a reaction between the enteric coating layer and the core material must be directly on the core material. The photo in Fig 1 and the schematic drawing in Fig 2 (both set out in Annexure A) confirm this understanding.
1015 Therefore, I accept the arguments put forward by Ranbaxy on the two principal construction issues, which means that there is no infringement of the 774 Patent by the Ranbaxy Product.
OVERALL DISPOSITION OF THIS PROCEEDING
1016 The parties are provided with these reasons for decision on a confidential basis as contemplated by the Orders of the Court made on 23 February 2012.
1017 The parties should now confer and draft minutes of orders reflecting the outcome of the proceedings, and provide them to the Court no later than 4:00pm on Monday 29 April 2013.
I certify that the preceding one thousand and seventeen (1017) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Middleton. |
Associate:
ANNEXURE ‘A’
ANNEXURE ‘B’









